User login
Managing bloodstream infections
To the Editor: I congratulate Drs. O’Grady and Chertow for their excellent review on bloodstream infections.1 I just want to call attention to one aspect that the authors forgot. In Figure 1, they classified patients as being mildly or moderately ill if they had no hypotension or organ failure, and subdivided this group into those having or not having high-risk factors. The high-risk factors included evidence of severe sepsis, which by definition needs dysfunction or failure of one or more organs.2
As has been demonstrated by epidemiologic studies, severe sepsis is associated with a high risk of death,3 twice as high as in patients with only catheter-related bloodstream infection.4 So, according to the joint guidelines of the American College of Chest Physicians and the Society of Critical Care Medicine,2 severe sepsis implies dysfunction or failure of at least one organ. I believe that patients with severe sepsis should be classified in the group of seriously ill.
- O’Grady NP, Chertow DS. Managing bloodstream infections in patients who have short-term central venous catheters. Clev Clin J Med 2011; 78:10–17.
- Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992; 101:1644–1655.
- Vincent J-L, Sakr Y, Sprung CL, et al; Sepsis Occurrence in Acutely Ill Patients Investigators. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Zias N, Chroneou A, Beamis JF, Craven DE. Vascular catheter-related bloodstream infections. In:O’Donnell JM, Nácul FE, editors. Surgical Intensive Care, 2nd Edition. New York: Springer, 2010:311–324.
To the Editor: I congratulate Drs. O’Grady and Chertow for their excellent review on bloodstream infections.1 I just want to call attention to one aspect that the authors forgot. In Figure 1, they classified patients as being mildly or moderately ill if they had no hypotension or organ failure, and subdivided this group into those having or not having high-risk factors. The high-risk factors included evidence of severe sepsis, which by definition needs dysfunction or failure of one or more organs.2
As has been demonstrated by epidemiologic studies, severe sepsis is associated with a high risk of death,3 twice as high as in patients with only catheter-related bloodstream infection.4 So, according to the joint guidelines of the American College of Chest Physicians and the Society of Critical Care Medicine,2 severe sepsis implies dysfunction or failure of at least one organ. I believe that patients with severe sepsis should be classified in the group of seriously ill.
To the Editor: I congratulate Drs. O’Grady and Chertow for their excellent review on bloodstream infections.1 I just want to call attention to one aspect that the authors forgot. In Figure 1, they classified patients as being mildly or moderately ill if they had no hypotension or organ failure, and subdivided this group into those having or not having high-risk factors. The high-risk factors included evidence of severe sepsis, which by definition needs dysfunction or failure of one or more organs.2
As has been demonstrated by epidemiologic studies, severe sepsis is associated with a high risk of death,3 twice as high as in patients with only catheter-related bloodstream infection.4 So, according to the joint guidelines of the American College of Chest Physicians and the Society of Critical Care Medicine,2 severe sepsis implies dysfunction or failure of at least one organ. I believe that patients with severe sepsis should be classified in the group of seriously ill.
- O’Grady NP, Chertow DS. Managing bloodstream infections in patients who have short-term central venous catheters. Clev Clin J Med 2011; 78:10–17.
- Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992; 101:1644–1655.
- Vincent J-L, Sakr Y, Sprung CL, et al; Sepsis Occurrence in Acutely Ill Patients Investigators. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Zias N, Chroneou A, Beamis JF, Craven DE. Vascular catheter-related bloodstream infections. In:O’Donnell JM, Nácul FE, editors. Surgical Intensive Care, 2nd Edition. New York: Springer, 2010:311–324.
- O’Grady NP, Chertow DS. Managing bloodstream infections in patients who have short-term central venous catheters. Clev Clin J Med 2011; 78:10–17.
- Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992; 101:1644–1655.
- Vincent J-L, Sakr Y, Sprung CL, et al; Sepsis Occurrence in Acutely Ill Patients Investigators. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 2006; 34:344–353.
- Zias N, Chroneou A, Beamis JF, Craven DE. Vascular catheter-related bloodstream infections. In:O’Donnell JM, Nácul FE, editors. Surgical Intensive Care, 2nd Edition. New York: Springer, 2010:311–324.
In reply: Managing bloodstream infections
In Reply: We thank Dr. Dias for his careful read of our article, “Managing bloodstream infections in patients who have short-term central venous catheters,” and we acknowledge that he is correct to point out that, by definition, severe sepsis is sepsis associated with organ dysfunction, hypoperfusion, or hypotension. Given this, he is correct that patients with severe sepsis should be categorized in the “seriously ill” patient group in our Figure 1.
In effect, however, the recommendations for patients in the “high-risk-factor” group are the same as the recommendations for the “seriously ill” patient group, which are to remove the catheter, draw at least two sets of blood cultures with at least one from a peripheral vein, and start empiric antibiotic therapy.
In Reply: We thank Dr. Dias for his careful read of our article, “Managing bloodstream infections in patients who have short-term central venous catheters,” and we acknowledge that he is correct to point out that, by definition, severe sepsis is sepsis associated with organ dysfunction, hypoperfusion, or hypotension. Given this, he is correct that patients with severe sepsis should be categorized in the “seriously ill” patient group in our Figure 1.
In effect, however, the recommendations for patients in the “high-risk-factor” group are the same as the recommendations for the “seriously ill” patient group, which are to remove the catheter, draw at least two sets of blood cultures with at least one from a peripheral vein, and start empiric antibiotic therapy.
In Reply: We thank Dr. Dias for his careful read of our article, “Managing bloodstream infections in patients who have short-term central venous catheters,” and we acknowledge that he is correct to point out that, by definition, severe sepsis is sepsis associated with organ dysfunction, hypoperfusion, or hypotension. Given this, he is correct that patients with severe sepsis should be categorized in the “seriously ill” patient group in our Figure 1.
In effect, however, the recommendations for patients in the “high-risk-factor” group are the same as the recommendations for the “seriously ill” patient group, which are to remove the catheter, draw at least two sets of blood cultures with at least one from a peripheral vein, and start empiric antibiotic therapy.
ST-segment depression and T-wave inversion: Classification, differential diagnosis, and caveats
Depression of the ST segment and inversion of the T wave are common electrocardiographic abnormalities. Knowing the various ischemic and nonischemic morphologic features is critical for a timely diagnosis of high-risk myocardial ischemia and electrolyte- or drug-related abnormalities. Moreover, it is important to recognize that true posterior infarction or subtle ST-segment elevation infarction may masquerade as ST-segment depression ischemia, and that pulmonary embolism may masquerade as anterior ischemia. These common electrocardiographic abnormalities are summarized in Table 1.
THE ST SEGMENT AND THE T WAVE: A PRIMER

The ST segment corresponds to the plateau phase of ventricular repolarization (phase 2 of the action potential), while the T wave corresponds to the phase of rapid ventricular repolarization (phase 3). ST-segment or T-wave changes may be secondary to abnormalities of depolarization, ie, pre-excitation or abnormalities of QRS voltage or duration.
On the other hand, ST-segment and T-wave abnormalities may be unrelated to any QRS abnormality, in which case they are called primary repolarization abnormalities. These are caused by ischemia, pericarditis, myocarditis, drugs (digoxin, antiarrhythmic drugs), and electrolyte abnormalities, particularly potassium abnormalities.
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 But some prefer to measure the magnitude of the ST-segment deviation 40 to 80 ms after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment; at the very onset of repolarization, small differences in membrane potential may normally be seen and may cause deviation of the J point and of the early portion of the ST segment.2
Although a diagnosis of ST-segment elevation myocardial infarction (STEMI) that mandates emergency reperfusion therapy requires ST-segment elevation greater than 1 mm in at least two contiguous leads,3 any ST-segment depression or elevation (≥ 0.5 mm, using the usual standard of 1.0 mV = 10 mm) may be abnormal, particularly when the clinical context or the shape of the ST segment suggests ischemia, or when other ischemic signs such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are concomitantly present. On the other hand, ST-segment depression of up to 0.5 mm in leads V2 and V3 and 1 mm in the other leads may be normal.1
In adults, the T wave normally is inverted in lead aVR; is upright or inverted in leads aVL, III, and V1; and is upright in leads I, II, aVF, and V2 through V6. The T wave is considered inverted when it is deeper than 1 mm; it is considered flat when its peak amplitude is between 1.0 mm and −1.0 mm.1
As we will discuss, certain features allow the various causes of ST-segment and T-wave abnormalities to be distinguished from one another.
SECONDARY ST-SEGMENT AND T-WAVE ABNORMALITIES
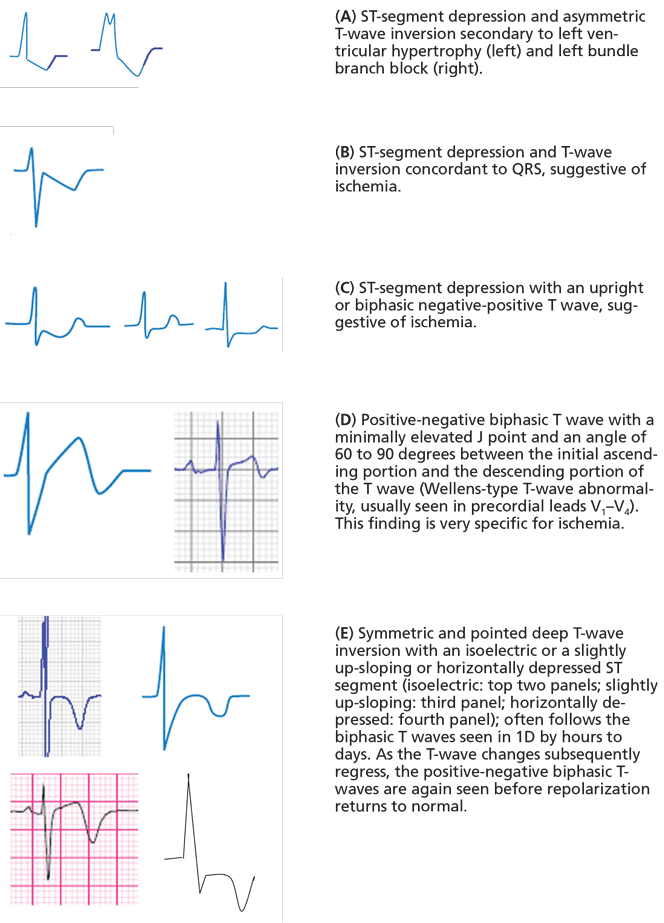
- The ST segment and T wave are directed opposite to the QRS: this is called discordance between the QRS complex and the ST-T abnormalities. In the case of right bundle branch block, the ST and T are directed opposite to the terminal portion of the QRS, ie, the part of the QRS deformed by the conduction abnormality.
- The ST segment and T wave are both abnormal and deviate in the same direction, ie, the ST segment is down-sloping and the T wave is inverted in leads with an upright QRS complex, which gives the ST-T complex a “reverse checkmark” asymmetric morphology.
- The ST and T abnormalities are not dynamic, ie, they do not change in the course of several hours to several days.
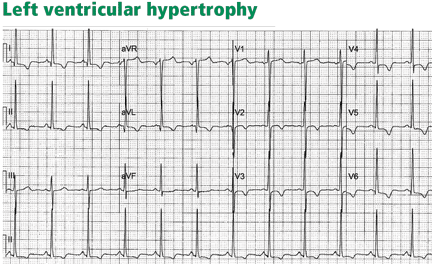
Thus, in cases of left ventricular hypertrophy or left bundle branch block, since the QRS complex is upright in the left lateral leads I, aVL, V5, and V6, the ST segment is characteristically depressed and the T wave is inverted in these leads (Figure 2). In cases of right ventricular hypertrophy or right bundle branch block, T waves are characteristically inverted in the right precordial leads V1, V2, and V3.
Left bundle branch block is always associated with secondary ST-T abnormalities, the absence of which suggests associated ischemia. Left and right ventricular hypertrophy, on the other hand, are not always associated with ST-T abnormalities, but when these are present, they correlate with more severe hypertrophy or ventricular systolic dysfunction,4 and have been called strain pattern. In addition, while these morphologic features are consistent with secondary abnormalities, they do not rule out ischemia in a patient with angina.
Some exceptions to these typical morphologic features:
- Right ventricular hypertrophy and right bundle branch block may be associated with isolated T-wave inversion without ST-segment depression in precordial leads V1, V2, and V3.
- Left ventricular hypertrophy may be associated with symmetric T-wave inversion without ST-segment depression or with a horizontally depressed ST segment. This may be the case in up to one-third of ST-T abnormalities secondary to left ventricular hypertrophy and is seen in hypertrophic cardiomyopathy, particularly the apical variant, in leads V3 through V6.5
ISCHEMIC ST-SEGMENT DEPRESSION, T-WAVE INVERSION, OR BOTH
ST-segment depression or T-wave inversion is consistent with ischemia if any of the following is true:
- The ST-segment depression or T-wave inversion is directed in the same direction as the QRS complex: this is called concordance between the QRS complex and the ST or T abnormality (Figure 1B).
- The ST segment is depressed but the T wave is upright (Figure 1C).
- The T wave has a positive-negative biphasic pattern (Figure 1D).
- The T wave is symmetrically inverted and has a pointed configuration, while the ST segment is not deviated or is upwardly bowed (coved) or horizontally depressed (Figure 1E).
- The magnitude of ST-segment depression progresses or regresses on serial tracings, or ST-segment depression progresses to T-wave abnormality during ischemia-free intervals (dynamic ST-segment depression).
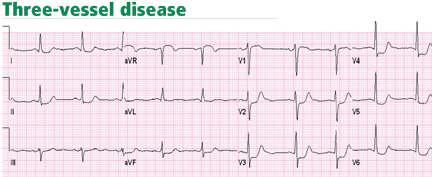
Unlike ST-segment elevation, ST-segment depression does not localize ischemia.6 However, the extent and the magnitude of ST-segment depression correlate with the extent and the severity of ischemia. In fact, ST-segment depression in eight or more leads, combined with ST-segment elevation in leads aVR and V1 and occurring during ischemic pain, is associated with a 75% predictive accuracy for left main coronary artery or three-vessel disease (Figure 3).7,8 This finding may also be seen in cases of tight proximal stenosis of the left anterior descending coronary artery.9
Wellens syndrome
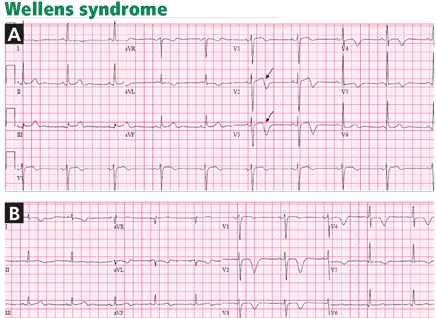
Wellens and his colleagues showed that 75% of patients who developed these T-wave abnormalities and who were treated medically without angiographic investigation went on to develop extensive anterior wall myocardial infarction within a mean of 8.5 days.10
In a later investigation of 1,260 patients presenting with unstable angina, 180 patients (14%) had this characteristic T-wave pattern.11 All of the latter patients had stenosis of 50% or more in the proximal left anterior descending artery, and 18% had total occlusion of the left anterior descending artery.
Thus, although medical management may provide symptomatic improvement at first, early coronary angiography and revascularization should be strongly considered in anyone with Wellens syndrome because it usually predicts impending anterior myocardial infarction.
Wellens syndrome is characterized by two patterns of T-wave changes. In 75% of cases, T waves are deeply (≥ 5 mm) and symmetrically inverted in leads V2 through V4 (Figures 1E, 4B). In 25% of cases, the T wave has a characteristic positive-negative biphasic morphology in leads V2 through V4 (Figures 1D, 4A).10 In both patterns, the ST segment is isoelectric or minimally elevated (< 1 mm) with a straight or convex morphology, the down-slope of the T wave is sharp, and the QT interval is often prolonged. These abnormalities are characteristically seen hours to days after the ischemic chest pain resolves. In fact, the ischemic episode is usually associated with transient ST-segment elevation or depression that progresses to the T-wave abnormality after the pain subsides.11
In Wellens’ original description, only 12% of patients had increases in their creatine kinase levels, and these were small. Therefore, the electrocardiogram may be the only indication of an impending large anterior infarction in a chest-pain-free patient.12
T waves that are symmetrically but less deeply inverted than Wellens-type T waves may still represent ischemia. However, this finding is less specific for ischemia and is associated with better outcomes than Wellens syndrome or ST-segment deviation, particularly when the T wave is less than 3 mm deep.14 In fact, one prospective cohort study found that isolated mild T-wave inversion in patients presenting with acute coronary syndrome is associated with a favorable long-term outcome, similar to that in patients with no electrocardiographic changes.15
FREQUENTLY MISSED DIAGNOSES MANIFESTING AS ST-SEGMENT DEPRESSION OR T-WAVE INVERSION
True posterior ST-segment elevation myocardial infarction
When accompanied by inferior STEMI, posterior infarction is easily recognized, but it can be difficult to diagnose when it occurs alone, the so-called true posterior STEMI.
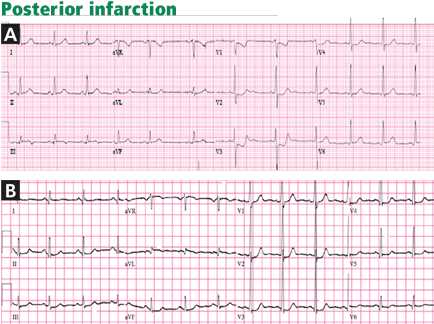
In most cases of posterior infarction, the posterior chest leads V7, V8, and V9 reveal ST-segment elevation.19 One study found that ST-segment depression in the anterior precordial leads was as sensitive as ST-segment elevation in leads V7 through V9 in identifying posterior myocardial infarction (sensitivity 80%),20 while other studies found that ST-segment deviation on standard 12-lead electrocardiography has a lower sensitivity (about 60%) in identifying posterior infarction.18,21
Tall or wide (≥ 0.04-s) R waves in leads V1 or V2, particularly when associated with upright T waves, suggest posterior infarction and may further corroborate this diagnosis, but this finding may take up to 24 hours to manifest and is seen in only about 50% of patients with posterior infarction.21
Studies have shown that ST-segment elevation on standard 12-lead electrocardiography is found in fewer than 50% of patients with acute left circumflex occlusion and inferoposterior infarction,18 yet these are cases of “missed” STEMI that indeed benefit from emergency angiography and reperfusion. In addition, studies of non–ST-segment elevation acute coronary syndrome consistently identify patients who have epicardial vessel occlusion (about 15%–20% of cases),18 yet their initial angiography is usually delayed for hours or days after the initial presentation.
A subgroup analysis from TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel Thrombolysis in Myocardial Infarction 38) evaluated patients with isolated anterior ST-segment depression. An occluded “culprit” artery was found 26% of the time, most often the left circumflex artery. Moreover, those patients had a significantly higher rate of death or myocardial infarction at 30-day follow-up than patients without a culprit artery, probably related to delayed revascularization.22
Recognizing that ST-segment depression that is greatest in leads V1, V2, or V3 represents posterior infarction helps identify a portion of the missed STEMIs in a timely fashion. In addition, in cases of anterior ST-segment depression and in cases of chest pain with nondiagnostic electrocardiography, the recording of ST elevation in leads V7, V8, and V9 is highly sensitive for detecting a true posterior injury.
Acute pulmonary embolism
An anterior ischemic pattern of symmetric T-wave inversion in the precordial leads V1 through V4 may also be a sign of acute or chronic right ventricular strain, particularly acute pulmonary embolism. Sinus tachycardia is usually present, but other signs of pulmonary embolism, such as right ventricular hypertrophy and right bundle branch block, may be absent. In fact, T-wave inversion in leads V1 through V4 is noted in 19% of patients with nonmassive pulmonary embolism and in 85% of patients with massive pulmonary embolism, and is the most sensitive and specific electrocardiographic finding in massive pulmonary embolism.23
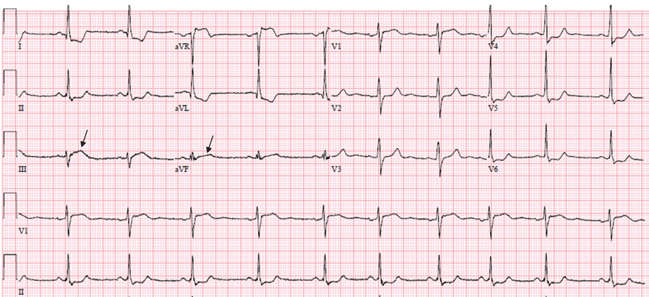
In addition, acute pulmonary embolism may be associated with T-wave inversion in leads III and aVF,24 and changes of concomitant anterior and inferior ischemia should always raise the question of this diagnosis.
In one retrospective study of patients with acute pulmonary embolism, nonspecific ST-segment or T-wave changes were the most common finding on electrocardiography, noted in 49%.25 Rapid regression of these changes on serial tracings favors pulmonary embolism rather than myocardial infarction.
ST-segment depression reciprocal to a subtle ST-segment elevation
When ST-segment elevation occurs in two contiguous standard leads while ST-segment depression occurs in other leads, and when the ST-segment and T-wave abnormalities are ischemic rather than secondary to depolarization abnormalities, ST-segment elevation is considered the primary ischemic abnormality whereas ST-segment depression is often considered a reciprocal “mirror image” change. This “reciprocal” change may also represent remote ischemia in a distant territory in patients with multivessel coronary disease.26,27
Reciprocal ST-segment depression is present in all patients with inferior myocardial infarction and in 70% of patients with anterior myocardial infarction.28
Hypokalemia and digitalis effect
DIFFUSE (GLOBAL) T-WAVE INVERSION
Walder and Spodick36 have found this pattern to be caused most often by myocardial ischemia or neurologic events, particularly intracranial hemorrhage, and it seems more prevalent in women. Other causes include hypertrophic cardiomyopathy, stress-induced cardiomyopathy (takotsubo cardiomyopathy), cocaine abuse, pericarditis, pulmonary embolism, and advanced or complete atrioventricular block.36,37
The prognosis in patients with global T-wave inversion is determined by the underlying disease, and the striking T-wave changes per se do not imply a poor prognosis.38
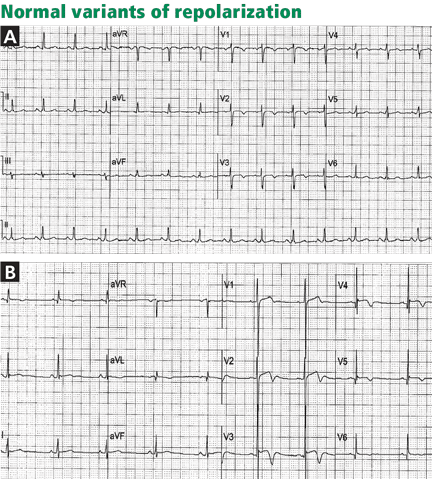
OTHER CAUSES OF T-WAVE INVERSION OR ST-SEGMENT DEPRESSION
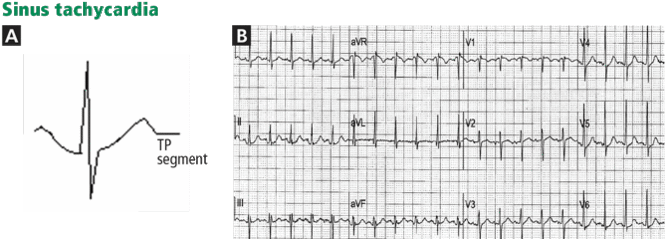
Various other entities may cause T-wave inversion, notably acute pericarditis or myocarditis, 41,42 memory T-wave phenomenon,43,44 and normal variants of repolarization (Table 1, Figure 9).45 Additionally, a nonpathologic junctional ST-segment depression may be seen in tachycardia (Figure 10).
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Surawicz B, Knilans TK. Non-Q wave myocardial infarction, unstable angina pectoris, myocardial ischemia. In: Chou's Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia: WB Saunders; 2001:194–207.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Okin PM, Devereux RB, Nieminen MS, et al; LIFE Study Investigators. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 2006; 113:67–73.
- Huwez FU, Pringle SD, Macfarlane PW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J 1992; 67:304–307.
- Li D, Li CY, Yong AC, Kilpatrick D. Source of electrocardiographic ST changes in subendocardial ischemia. Circ Res 1998; 82:957–970.
- Gorgels AP, Vos MA, Mulleneers R, de Zwaan C, Bär FW, Wellens HJ. Value of the electrocardiogram in diagnosing the number of severely narrowed coronary arteries in rest angina pectoris. Am J Cardiol 1993; 72:999–1003.
- Glancy DL. Electrocardiographic diagnosis of acute myocardial infarction. J La State Med Soc 2002; 154:66–75.
- Yamaji H, Iwasaki K, Kusachi S, et al. Prediction of acute left main coronary artery obstruction by 12-lead electrocardiography. ST segment elevation in lead aVR with less ST segment elevation in lead V(1). J Am Coll Cardiol 2001; 38:1348–1354.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- Lilaonitkul M, Robinson K, Roberts M. Wellens’ syndrome: significance of ECG pattern recognition in the emergency department. Emerg Med J 2009; 26:750–751.
- Glancy DL, Khuri B, Cospolich B. Heed the warning: Wellens’ type T-wave inversion is caused by proximal left anterior descending lesion. Proc (Bayl Univ Med Cent) 2000; 13:416–418.
- Savonitto S, Ardissino D, Granger CB, et al. Prognostic value of the admission electrocardiogram in acute coronary syndromes. JAMA 1999; 281:707–713.
- Mueller C, Neumann FJ, Perach W, Perruchoud AP, Buettner HJ. Prognostic value of the admission electrocardiogram in patients with unstable angina/non-ST-segment elevation myocardial infarction treated with very early revascularization. Am J Med 2004; 117:145–150.
- Boden WE, Spodick DH. Diagnostic significance of precordial ST-segment depression. Am J Cardiol 1989; 63:358–361.
- Shah A, Wagner GS, Green CL, et al. Electrocardiographic differentiation of the ST-segment depression of acute myocardial injury due to the left circumflex artery occlusion from that of myocardial ischemia of nonocclusive etiologies. Am J Cardiol 1997; 80:512–513.
- Krishnaswamy A, Lincoff AM, Menon V. Magnitude and consequences of missing the acute infarct-related circumflex artery. Am Heart J 2009; 158:706–712.
- Matetzky S, Freimark D, Feinberg MS, et al. Acute myocardial infarction with isolated ST-segment elevation in posterior chest leads V7-9: “hidden” ST-segment elevations revealing acute posterior infarction. J Am Coll Cardiol 1999; 34:748–753.
- Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
- Huey BL, Beller GA, Kaiser DL, Gibson RS. A comprehensive analysis of myocardial infarction due to left circumflex artery occlusion: comparison with infarction due to right coronary artery and left anterior descending artery occlusion. J Am Coll Cardiol 1988; 12:1156–1166.
- Gibson CM, Pride YB, Mohanavelu S, Wiviott SD, Antman EM, Braunwald E. Abstract 1999: Angiographic and clinical outcomes among patients with acute coronary syndrome presenting with isolated anterior ST-segment depressions. Circulation 2008; 118:S–654.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Norell MS, Lyons JP, Gardener JE, Layton CA, Balcon R. Significance of “reciprocal” ST segment depression: left ventriculographic observations during left anterior descending coronary angioplasty. J Am Coll Cardiol 1989; 13:1270–1274.
- Haraphongse M, Tanomsup S, Jugdutt BI. Inferior ST segment depression during acute anterior myocardial infarction: clinical and angiographic correlations. J Am Coll Cardiol 1984; 4:467–476.
- Surawicz B, Knilans TK. Acute ischemia: electrocardiographic patterns. In: Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th edition. Philadelphia: WB Saunders; 2001:122–153.
- Wagner GS, Macfarlane P, Wellens H, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:1003–1011.
- Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med 2002; 20:35–38.
- Surawicz B. Electrolytes and the electrocardiogram. Postgrad Med 1974; 55:123–129.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Glancy DL, Wang WL. ECG of the month. Abnormal electrocardiogram in a woman with a urinary tract infection. Sinus rhythm, rate 82/minute. Sagging ST segments, low T waves, and prominent U waves suggest hypokalemia. J La State Med Soc 2007; 159:5–7.
- Surawicz B, Braun HA, Crum WB, Kemp RL, Wagner S, Bellet S. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation 1957; 16:750–763.
- Glancy DL, Rochon BJ, Ilie CC, Parker JM, Jones MB, Atluri P. Global T-wave inversion in a 77-year-old woman. Proc (Bayl Univ Med Cent) 2009; 22:81–82.
- Walder LA, Spodick DH. Global T wave inversion. J Am Coll Cardiol 1991; 17:1479–1485.
- Lui CY. Acute pulmonary embolism as the cause of global T wave inversion and QT prolongation. A case report. J Electrocardiol 1993; 26:91–95.
- Walder LA, Spodick DH. Global T wave inversion: long-term followup. J Am Coll Cardiol 1993; 21:1652–1656.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Spodick DH. Electrocardiogram in acute pericarditis. Distributions of morphologic and axial changes by stages. Am J Cardiol 1974; 33:470–474.
- Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Rosenbaum MB, Blanco HH, Elizari MV, Lázzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50:213–222.
- Paparella N, Ouyang F, Fuca G, Kuck KH, Cappato R, Alboni P. Significance of newly acquired negative T waves after interruption of paroxysmal reentrant supraventricular tachycardia with narrow QRS complex. Am J Cardiol 2000; 85:261–263.
- Kaid KA, Maqsood A, Cohen M, Rothfeld E. Further characterization of the “persistent juvenile T-wave pattern” in adults. J Electrocardiol 2008; 41:644–645.
Depression of the ST segment and inversion of the T wave are common electrocardiographic abnormalities. Knowing the various ischemic and nonischemic morphologic features is critical for a timely diagnosis of high-risk myocardial ischemia and electrolyte- or drug-related abnormalities. Moreover, it is important to recognize that true posterior infarction or subtle ST-segment elevation infarction may masquerade as ST-segment depression ischemia, and that pulmonary embolism may masquerade as anterior ischemia. These common electrocardiographic abnormalities are summarized in Table 1.
THE ST SEGMENT AND THE T WAVE: A PRIMER
The ST segment corresponds to the plateau phase of ventricular repolarization (phase 2 of the action potential), while the T wave corresponds to the phase of rapid ventricular repolarization (phase 3). ST-segment or T-wave changes may be secondary to abnormalities of depolarization, ie, pre-excitation or abnormalities of QRS voltage or duration.
On the other hand, ST-segment and T-wave abnormalities may be unrelated to any QRS abnormality, in which case they are called primary repolarization abnormalities. These are caused by ischemia, pericarditis, myocarditis, drugs (digoxin, antiarrhythmic drugs), and electrolyte abnormalities, particularly potassium abnormalities.
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 But some prefer to measure the magnitude of the ST-segment deviation 40 to 80 ms after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment; at the very onset of repolarization, small differences in membrane potential may normally be seen and may cause deviation of the J point and of the early portion of the ST segment.2
Although a diagnosis of ST-segment elevation myocardial infarction (STEMI) that mandates emergency reperfusion therapy requires ST-segment elevation greater than 1 mm in at least two contiguous leads,3 any ST-segment depression or elevation (≥ 0.5 mm, using the usual standard of 1.0 mV = 10 mm) may be abnormal, particularly when the clinical context or the shape of the ST segment suggests ischemia, or when other ischemic signs such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are concomitantly present. On the other hand, ST-segment depression of up to 0.5 mm in leads V2 and V3 and 1 mm in the other leads may be normal.1
In adults, the T wave normally is inverted in lead aVR; is upright or inverted in leads aVL, III, and V1; and is upright in leads I, II, aVF, and V2 through V6. The T wave is considered inverted when it is deeper than 1 mm; it is considered flat when its peak amplitude is between 1.0 mm and −1.0 mm.1
As we will discuss, certain features allow the various causes of ST-segment and T-wave abnormalities to be distinguished from one another.
SECONDARY ST-SEGMENT AND T-WAVE ABNORMALITIES
- The ST segment and T wave are directed opposite to the QRS: this is called discordance between the QRS complex and the ST-T abnormalities. In the case of right bundle branch block, the ST and T are directed opposite to the terminal portion of the QRS, ie, the part of the QRS deformed by the conduction abnormality.
- The ST segment and T wave are both abnormal and deviate in the same direction, ie, the ST segment is down-sloping and the T wave is inverted in leads with an upright QRS complex, which gives the ST-T complex a “reverse checkmark” asymmetric morphology.
- The ST and T abnormalities are not dynamic, ie, they do not change in the course of several hours to several days.
Thus, in cases of left ventricular hypertrophy or left bundle branch block, since the QRS complex is upright in the left lateral leads I, aVL, V5, and V6, the ST segment is characteristically depressed and the T wave is inverted in these leads (Figure 2). In cases of right ventricular hypertrophy or right bundle branch block, T waves are characteristically inverted in the right precordial leads V1, V2, and V3.
Left bundle branch block is always associated with secondary ST-T abnormalities, the absence of which suggests associated ischemia. Left and right ventricular hypertrophy, on the other hand, are not always associated with ST-T abnormalities, but when these are present, they correlate with more severe hypertrophy or ventricular systolic dysfunction,4 and have been called strain pattern. In addition, while these morphologic features are consistent with secondary abnormalities, they do not rule out ischemia in a patient with angina.
Some exceptions to these typical morphologic features:
- Right ventricular hypertrophy and right bundle branch block may be associated with isolated T-wave inversion without ST-segment depression in precordial leads V1, V2, and V3.
- Left ventricular hypertrophy may be associated with symmetric T-wave inversion without ST-segment depression or with a horizontally depressed ST segment. This may be the case in up to one-third of ST-T abnormalities secondary to left ventricular hypertrophy and is seen in hypertrophic cardiomyopathy, particularly the apical variant, in leads V3 through V6.5
ISCHEMIC ST-SEGMENT DEPRESSION, T-WAVE INVERSION, OR BOTH
ST-segment depression or T-wave inversion is consistent with ischemia if any of the following is true:
- The ST-segment depression or T-wave inversion is directed in the same direction as the QRS complex: this is called concordance between the QRS complex and the ST or T abnormality (Figure 1B).
- The ST segment is depressed but the T wave is upright (Figure 1C).
- The T wave has a positive-negative biphasic pattern (Figure 1D).
- The T wave is symmetrically inverted and has a pointed configuration, while the ST segment is not deviated or is upwardly bowed (coved) or horizontally depressed (Figure 1E).
- The magnitude of ST-segment depression progresses or regresses on serial tracings, or ST-segment depression progresses to T-wave abnormality during ischemia-free intervals (dynamic ST-segment depression).
Unlike ST-segment elevation, ST-segment depression does not localize ischemia.6 However, the extent and the magnitude of ST-segment depression correlate with the extent and the severity of ischemia. In fact, ST-segment depression in eight or more leads, combined with ST-segment elevation in leads aVR and V1 and occurring during ischemic pain, is associated with a 75% predictive accuracy for left main coronary artery or three-vessel disease (Figure 3).7,8 This finding may also be seen in cases of tight proximal stenosis of the left anterior descending coronary artery.9
Wellens syndrome
Wellens and his colleagues showed that 75% of patients who developed these T-wave abnormalities and who were treated medically without angiographic investigation went on to develop extensive anterior wall myocardial infarction within a mean of 8.5 days.10
In a later investigation of 1,260 patients presenting with unstable angina, 180 patients (14%) had this characteristic T-wave pattern.11 All of the latter patients had stenosis of 50% or more in the proximal left anterior descending artery, and 18% had total occlusion of the left anterior descending artery.
Thus, although medical management may provide symptomatic improvement at first, early coronary angiography and revascularization should be strongly considered in anyone with Wellens syndrome because it usually predicts impending anterior myocardial infarction.
Wellens syndrome is characterized by two patterns of T-wave changes. In 75% of cases, T waves are deeply (≥ 5 mm) and symmetrically inverted in leads V2 through V4 (Figures 1E, 4B). In 25% of cases, the T wave has a characteristic positive-negative biphasic morphology in leads V2 through V4 (Figures 1D, 4A).10 In both patterns, the ST segment is isoelectric or minimally elevated (< 1 mm) with a straight or convex morphology, the down-slope of the T wave is sharp, and the QT interval is often prolonged. These abnormalities are characteristically seen hours to days after the ischemic chest pain resolves. In fact, the ischemic episode is usually associated with transient ST-segment elevation or depression that progresses to the T-wave abnormality after the pain subsides.11
In Wellens’ original description, only 12% of patients had increases in their creatine kinase levels, and these were small. Therefore, the electrocardiogram may be the only indication of an impending large anterior infarction in a chest-pain-free patient.12
T waves that are symmetrically but less deeply inverted than Wellens-type T waves may still represent ischemia. However, this finding is less specific for ischemia and is associated with better outcomes than Wellens syndrome or ST-segment deviation, particularly when the T wave is less than 3 mm deep.14 In fact, one prospective cohort study found that isolated mild T-wave inversion in patients presenting with acute coronary syndrome is associated with a favorable long-term outcome, similar to that in patients with no electrocardiographic changes.15
FREQUENTLY MISSED DIAGNOSES MANIFESTING AS ST-SEGMENT DEPRESSION OR T-WAVE INVERSION
True posterior ST-segment elevation myocardial infarction
When accompanied by inferior STEMI, posterior infarction is easily recognized, but it can be difficult to diagnose when it occurs alone, the so-called true posterior STEMI.
In most cases of posterior infarction, the posterior chest leads V7, V8, and V9 reveal ST-segment elevation.19 One study found that ST-segment depression in the anterior precordial leads was as sensitive as ST-segment elevation in leads V7 through V9 in identifying posterior myocardial infarction (sensitivity 80%),20 while other studies found that ST-segment deviation on standard 12-lead electrocardiography has a lower sensitivity (about 60%) in identifying posterior infarction.18,21
Tall or wide (≥ 0.04-s) R waves in leads V1 or V2, particularly when associated with upright T waves, suggest posterior infarction and may further corroborate this diagnosis, but this finding may take up to 24 hours to manifest and is seen in only about 50% of patients with posterior infarction.21
Studies have shown that ST-segment elevation on standard 12-lead electrocardiography is found in fewer than 50% of patients with acute left circumflex occlusion and inferoposterior infarction,18 yet these are cases of “missed” STEMI that indeed benefit from emergency angiography and reperfusion. In addition, studies of non–ST-segment elevation acute coronary syndrome consistently identify patients who have epicardial vessel occlusion (about 15%–20% of cases),18 yet their initial angiography is usually delayed for hours or days after the initial presentation.
A subgroup analysis from TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel Thrombolysis in Myocardial Infarction 38) evaluated patients with isolated anterior ST-segment depression. An occluded “culprit” artery was found 26% of the time, most often the left circumflex artery. Moreover, those patients had a significantly higher rate of death or myocardial infarction at 30-day follow-up than patients without a culprit artery, probably related to delayed revascularization.22
Recognizing that ST-segment depression that is greatest in leads V1, V2, or V3 represents posterior infarction helps identify a portion of the missed STEMIs in a timely fashion. In addition, in cases of anterior ST-segment depression and in cases of chest pain with nondiagnostic electrocardiography, the recording of ST elevation in leads V7, V8, and V9 is highly sensitive for detecting a true posterior injury.
Acute pulmonary embolism
An anterior ischemic pattern of symmetric T-wave inversion in the precordial leads V1 through V4 may also be a sign of acute or chronic right ventricular strain, particularly acute pulmonary embolism. Sinus tachycardia is usually present, but other signs of pulmonary embolism, such as right ventricular hypertrophy and right bundle branch block, may be absent. In fact, T-wave inversion in leads V1 through V4 is noted in 19% of patients with nonmassive pulmonary embolism and in 85% of patients with massive pulmonary embolism, and is the most sensitive and specific electrocardiographic finding in massive pulmonary embolism.23
In addition, acute pulmonary embolism may be associated with T-wave inversion in leads III and aVF,24 and changes of concomitant anterior and inferior ischemia should always raise the question of this diagnosis.
In one retrospective study of patients with acute pulmonary embolism, nonspecific ST-segment or T-wave changes were the most common finding on electrocardiography, noted in 49%.25 Rapid regression of these changes on serial tracings favors pulmonary embolism rather than myocardial infarction.
ST-segment depression reciprocal to a subtle ST-segment elevation
When ST-segment elevation occurs in two contiguous standard leads while ST-segment depression occurs in other leads, and when the ST-segment and T-wave abnormalities are ischemic rather than secondary to depolarization abnormalities, ST-segment elevation is considered the primary ischemic abnormality whereas ST-segment depression is often considered a reciprocal “mirror image” change. This “reciprocal” change may also represent remote ischemia in a distant territory in patients with multivessel coronary disease.26,27
Reciprocal ST-segment depression is present in all patients with inferior myocardial infarction and in 70% of patients with anterior myocardial infarction.28
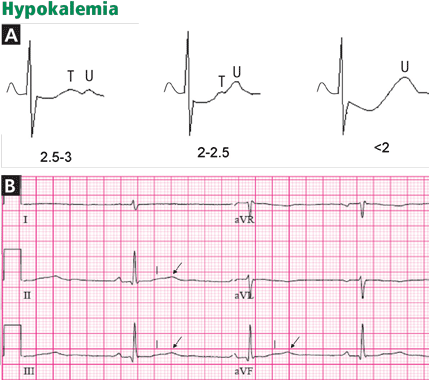
Hypokalemia and digitalis effect
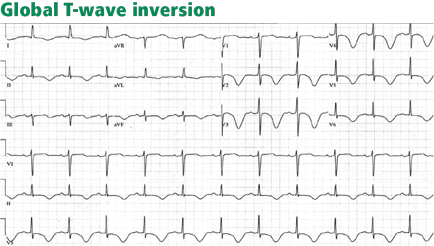
DIFFUSE (GLOBAL) T-WAVE INVERSION
Walder and Spodick36 have found this pattern to be caused most often by myocardial ischemia or neurologic events, particularly intracranial hemorrhage, and it seems more prevalent in women. Other causes include hypertrophic cardiomyopathy, stress-induced cardiomyopathy (takotsubo cardiomyopathy), cocaine abuse, pericarditis, pulmonary embolism, and advanced or complete atrioventricular block.36,37
The prognosis in patients with global T-wave inversion is determined by the underlying disease, and the striking T-wave changes per se do not imply a poor prognosis.38
OTHER CAUSES OF T-WAVE INVERSION OR ST-SEGMENT DEPRESSION
Various other entities may cause T-wave inversion, notably acute pericarditis or myocarditis, 41,42 memory T-wave phenomenon,43,44 and normal variants of repolarization (Table 1, Figure 9).45 Additionally, a nonpathologic junctional ST-segment depression may be seen in tachycardia (Figure 10).
Depression of the ST segment and inversion of the T wave are common electrocardiographic abnormalities. Knowing the various ischemic and nonischemic morphologic features is critical for a timely diagnosis of high-risk myocardial ischemia and electrolyte- or drug-related abnormalities. Moreover, it is important to recognize that true posterior infarction or subtle ST-segment elevation infarction may masquerade as ST-segment depression ischemia, and that pulmonary embolism may masquerade as anterior ischemia. These common electrocardiographic abnormalities are summarized in Table 1.
THE ST SEGMENT AND THE T WAVE: A PRIMER
The ST segment corresponds to the plateau phase of ventricular repolarization (phase 2 of the action potential), while the T wave corresponds to the phase of rapid ventricular repolarization (phase 3). ST-segment or T-wave changes may be secondary to abnormalities of depolarization, ie, pre-excitation or abnormalities of QRS voltage or duration.
On the other hand, ST-segment and T-wave abnormalities may be unrelated to any QRS abnormality, in which case they are called primary repolarization abnormalities. These are caused by ischemia, pericarditis, myocarditis, drugs (digoxin, antiarrhythmic drugs), and electrolyte abnormalities, particularly potassium abnormalities.
ST-segment deviation is usually measured at its junction with the end of the QRS complex, ie, the J point, and is referenced against the TP or PR segment.1 But some prefer to measure the magnitude of the ST-segment deviation 40 to 80 ms after the J point, when all myocardial fibers are expected to have reached the same level of membrane potential and to form an isoelectric ST segment; at the very onset of repolarization, small differences in membrane potential may normally be seen and may cause deviation of the J point and of the early portion of the ST segment.2
Although a diagnosis of ST-segment elevation myocardial infarction (STEMI) that mandates emergency reperfusion therapy requires ST-segment elevation greater than 1 mm in at least two contiguous leads,3 any ST-segment depression or elevation (≥ 0.5 mm, using the usual standard of 1.0 mV = 10 mm) may be abnormal, particularly when the clinical context or the shape of the ST segment suggests ischemia, or when other ischemic signs such as T-wave abnormalities, Q waves, or reciprocal ST-segment changes are concomitantly present. On the other hand, ST-segment depression of up to 0.5 mm in leads V2 and V3 and 1 mm in the other leads may be normal.1
In adults, the T wave normally is inverted in lead aVR; is upright or inverted in leads aVL, III, and V1; and is upright in leads I, II, aVF, and V2 through V6. The T wave is considered inverted when it is deeper than 1 mm; it is considered flat when its peak amplitude is between 1.0 mm and −1.0 mm.1
As we will discuss, certain features allow the various causes of ST-segment and T-wave abnormalities to be distinguished from one another.
SECONDARY ST-SEGMENT AND T-WAVE ABNORMALITIES
- The ST segment and T wave are directed opposite to the QRS: this is called discordance between the QRS complex and the ST-T abnormalities. In the case of right bundle branch block, the ST and T are directed opposite to the terminal portion of the QRS, ie, the part of the QRS deformed by the conduction abnormality.
- The ST segment and T wave are both abnormal and deviate in the same direction, ie, the ST segment is down-sloping and the T wave is inverted in leads with an upright QRS complex, which gives the ST-T complex a “reverse checkmark” asymmetric morphology.
- The ST and T abnormalities are not dynamic, ie, they do not change in the course of several hours to several days.
Thus, in cases of left ventricular hypertrophy or left bundle branch block, since the QRS complex is upright in the left lateral leads I, aVL, V5, and V6, the ST segment is characteristically depressed and the T wave is inverted in these leads (Figure 2). In cases of right ventricular hypertrophy or right bundle branch block, T waves are characteristically inverted in the right precordial leads V1, V2, and V3.
Left bundle branch block is always associated with secondary ST-T abnormalities, the absence of which suggests associated ischemia. Left and right ventricular hypertrophy, on the other hand, are not always associated with ST-T abnormalities, but when these are present, they correlate with more severe hypertrophy or ventricular systolic dysfunction,4 and have been called strain pattern. In addition, while these morphologic features are consistent with secondary abnormalities, they do not rule out ischemia in a patient with angina.
Some exceptions to these typical morphologic features:
- Right ventricular hypertrophy and right bundle branch block may be associated with isolated T-wave inversion without ST-segment depression in precordial leads V1, V2, and V3.
- Left ventricular hypertrophy may be associated with symmetric T-wave inversion without ST-segment depression or with a horizontally depressed ST segment. This may be the case in up to one-third of ST-T abnormalities secondary to left ventricular hypertrophy and is seen in hypertrophic cardiomyopathy, particularly the apical variant, in leads V3 through V6.5
ISCHEMIC ST-SEGMENT DEPRESSION, T-WAVE INVERSION, OR BOTH
ST-segment depression or T-wave inversion is consistent with ischemia if any of the following is true:
- The ST-segment depression or T-wave inversion is directed in the same direction as the QRS complex: this is called concordance between the QRS complex and the ST or T abnormality (Figure 1B).
- The ST segment is depressed but the T wave is upright (Figure 1C).
- The T wave has a positive-negative biphasic pattern (Figure 1D).
- The T wave is symmetrically inverted and has a pointed configuration, while the ST segment is not deviated or is upwardly bowed (coved) or horizontally depressed (Figure 1E).
- The magnitude of ST-segment depression progresses or regresses on serial tracings, or ST-segment depression progresses to T-wave abnormality during ischemia-free intervals (dynamic ST-segment depression).
Unlike ST-segment elevation, ST-segment depression does not localize ischemia.6 However, the extent and the magnitude of ST-segment depression correlate with the extent and the severity of ischemia. In fact, ST-segment depression in eight or more leads, combined with ST-segment elevation in leads aVR and V1 and occurring during ischemic pain, is associated with a 75% predictive accuracy for left main coronary artery or three-vessel disease (Figure 3).7,8 This finding may also be seen in cases of tight proximal stenosis of the left anterior descending coronary artery.9
Wellens syndrome
Wellens and his colleagues showed that 75% of patients who developed these T-wave abnormalities and who were treated medically without angiographic investigation went on to develop extensive anterior wall myocardial infarction within a mean of 8.5 days.10
In a later investigation of 1,260 patients presenting with unstable angina, 180 patients (14%) had this characteristic T-wave pattern.11 All of the latter patients had stenosis of 50% or more in the proximal left anterior descending artery, and 18% had total occlusion of the left anterior descending artery.
Thus, although medical management may provide symptomatic improvement at first, early coronary angiography and revascularization should be strongly considered in anyone with Wellens syndrome because it usually predicts impending anterior myocardial infarction.
Wellens syndrome is characterized by two patterns of T-wave changes. In 75% of cases, T waves are deeply (≥ 5 mm) and symmetrically inverted in leads V2 through V4 (Figures 1E, 4B). In 25% of cases, the T wave has a characteristic positive-negative biphasic morphology in leads V2 through V4 (Figures 1D, 4A).10 In both patterns, the ST segment is isoelectric or minimally elevated (< 1 mm) with a straight or convex morphology, the down-slope of the T wave is sharp, and the QT interval is often prolonged. These abnormalities are characteristically seen hours to days after the ischemic chest pain resolves. In fact, the ischemic episode is usually associated with transient ST-segment elevation or depression that progresses to the T-wave abnormality after the pain subsides.11
In Wellens’ original description, only 12% of patients had increases in their creatine kinase levels, and these were small. Therefore, the electrocardiogram may be the only indication of an impending large anterior infarction in a chest-pain-free patient.12
T waves that are symmetrically but less deeply inverted than Wellens-type T waves may still represent ischemia. However, this finding is less specific for ischemia and is associated with better outcomes than Wellens syndrome or ST-segment deviation, particularly when the T wave is less than 3 mm deep.14 In fact, one prospective cohort study found that isolated mild T-wave inversion in patients presenting with acute coronary syndrome is associated with a favorable long-term outcome, similar to that in patients with no electrocardiographic changes.15
FREQUENTLY MISSED DIAGNOSES MANIFESTING AS ST-SEGMENT DEPRESSION OR T-WAVE INVERSION
True posterior ST-segment elevation myocardial infarction
When accompanied by inferior STEMI, posterior infarction is easily recognized, but it can be difficult to diagnose when it occurs alone, the so-called true posterior STEMI.
In most cases of posterior infarction, the posterior chest leads V7, V8, and V9 reveal ST-segment elevation.19 One study found that ST-segment depression in the anterior precordial leads was as sensitive as ST-segment elevation in leads V7 through V9 in identifying posterior myocardial infarction (sensitivity 80%),20 while other studies found that ST-segment deviation on standard 12-lead electrocardiography has a lower sensitivity (about 60%) in identifying posterior infarction.18,21
Tall or wide (≥ 0.04-s) R waves in leads V1 or V2, particularly when associated with upright T waves, suggest posterior infarction and may further corroborate this diagnosis, but this finding may take up to 24 hours to manifest and is seen in only about 50% of patients with posterior infarction.21
Studies have shown that ST-segment elevation on standard 12-lead electrocardiography is found in fewer than 50% of patients with acute left circumflex occlusion and inferoposterior infarction,18 yet these are cases of “missed” STEMI that indeed benefit from emergency angiography and reperfusion. In addition, studies of non–ST-segment elevation acute coronary syndrome consistently identify patients who have epicardial vessel occlusion (about 15%–20% of cases),18 yet their initial angiography is usually delayed for hours or days after the initial presentation.
A subgroup analysis from TRITON–TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel Thrombolysis in Myocardial Infarction 38) evaluated patients with isolated anterior ST-segment depression. An occluded “culprit” artery was found 26% of the time, most often the left circumflex artery. Moreover, those patients had a significantly higher rate of death or myocardial infarction at 30-day follow-up than patients without a culprit artery, probably related to delayed revascularization.22
Recognizing that ST-segment depression that is greatest in leads V1, V2, or V3 represents posterior infarction helps identify a portion of the missed STEMIs in a timely fashion. In addition, in cases of anterior ST-segment depression and in cases of chest pain with nondiagnostic electrocardiography, the recording of ST elevation in leads V7, V8, and V9 is highly sensitive for detecting a true posterior injury.
Acute pulmonary embolism
An anterior ischemic pattern of symmetric T-wave inversion in the precordial leads V1 through V4 may also be a sign of acute or chronic right ventricular strain, particularly acute pulmonary embolism. Sinus tachycardia is usually present, but other signs of pulmonary embolism, such as right ventricular hypertrophy and right bundle branch block, may be absent. In fact, T-wave inversion in leads V1 through V4 is noted in 19% of patients with nonmassive pulmonary embolism and in 85% of patients with massive pulmonary embolism, and is the most sensitive and specific electrocardiographic finding in massive pulmonary embolism.23
In addition, acute pulmonary embolism may be associated with T-wave inversion in leads III and aVF,24 and changes of concomitant anterior and inferior ischemia should always raise the question of this diagnosis.
In one retrospective study of patients with acute pulmonary embolism, nonspecific ST-segment or T-wave changes were the most common finding on electrocardiography, noted in 49%.25 Rapid regression of these changes on serial tracings favors pulmonary embolism rather than myocardial infarction.
ST-segment depression reciprocal to a subtle ST-segment elevation
When ST-segment elevation occurs in two contiguous standard leads while ST-segment depression occurs in other leads, and when the ST-segment and T-wave abnormalities are ischemic rather than secondary to depolarization abnormalities, ST-segment elevation is considered the primary ischemic abnormality whereas ST-segment depression is often considered a reciprocal “mirror image” change. This “reciprocal” change may also represent remote ischemia in a distant territory in patients with multivessel coronary disease.26,27
Reciprocal ST-segment depression is present in all patients with inferior myocardial infarction and in 70% of patients with anterior myocardial infarction.28
Hypokalemia and digitalis effect
DIFFUSE (GLOBAL) T-WAVE INVERSION
Walder and Spodick36 have found this pattern to be caused most often by myocardial ischemia or neurologic events, particularly intracranial hemorrhage, and it seems more prevalent in women. Other causes include hypertrophic cardiomyopathy, stress-induced cardiomyopathy (takotsubo cardiomyopathy), cocaine abuse, pericarditis, pulmonary embolism, and advanced or complete atrioventricular block.36,37
The prognosis in patients with global T-wave inversion is determined by the underlying disease, and the striking T-wave changes per se do not imply a poor prognosis.38
OTHER CAUSES OF T-WAVE INVERSION OR ST-SEGMENT DEPRESSION
Various other entities may cause T-wave inversion, notably acute pericarditis or myocarditis, 41,42 memory T-wave phenomenon,43,44 and normal variants of repolarization (Table 1, Figure 9).45 Additionally, a nonpathologic junctional ST-segment depression may be seen in tachycardia (Figure 10).
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Surawicz B, Knilans TK. Non-Q wave myocardial infarction, unstable angina pectoris, myocardial ischemia. In: Chou's Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia: WB Saunders; 2001:194–207.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Okin PM, Devereux RB, Nieminen MS, et al; LIFE Study Investigators. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 2006; 113:67–73.
- Huwez FU, Pringle SD, Macfarlane PW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J 1992; 67:304–307.
- Li D, Li CY, Yong AC, Kilpatrick D. Source of electrocardiographic ST changes in subendocardial ischemia. Circ Res 1998; 82:957–970.
- Gorgels AP, Vos MA, Mulleneers R, de Zwaan C, Bär FW, Wellens HJ. Value of the electrocardiogram in diagnosing the number of severely narrowed coronary arteries in rest angina pectoris. Am J Cardiol 1993; 72:999–1003.
- Glancy DL. Electrocardiographic diagnosis of acute myocardial infarction. J La State Med Soc 2002; 154:66–75.
- Yamaji H, Iwasaki K, Kusachi S, et al. Prediction of acute left main coronary artery obstruction by 12-lead electrocardiography. ST segment elevation in lead aVR with less ST segment elevation in lead V(1). J Am Coll Cardiol 2001; 38:1348–1354.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- Lilaonitkul M, Robinson K, Roberts M. Wellens’ syndrome: significance of ECG pattern recognition in the emergency department. Emerg Med J 2009; 26:750–751.
- Glancy DL, Khuri B, Cospolich B. Heed the warning: Wellens’ type T-wave inversion is caused by proximal left anterior descending lesion. Proc (Bayl Univ Med Cent) 2000; 13:416–418.
- Savonitto S, Ardissino D, Granger CB, et al. Prognostic value of the admission electrocardiogram in acute coronary syndromes. JAMA 1999; 281:707–713.
- Mueller C, Neumann FJ, Perach W, Perruchoud AP, Buettner HJ. Prognostic value of the admission electrocardiogram in patients with unstable angina/non-ST-segment elevation myocardial infarction treated with very early revascularization. Am J Med 2004; 117:145–150.
- Boden WE, Spodick DH. Diagnostic significance of precordial ST-segment depression. Am J Cardiol 1989; 63:358–361.
- Shah A, Wagner GS, Green CL, et al. Electrocardiographic differentiation of the ST-segment depression of acute myocardial injury due to the left circumflex artery occlusion from that of myocardial ischemia of nonocclusive etiologies. Am J Cardiol 1997; 80:512–513.
- Krishnaswamy A, Lincoff AM, Menon V. Magnitude and consequences of missing the acute infarct-related circumflex artery. Am Heart J 2009; 158:706–712.
- Matetzky S, Freimark D, Feinberg MS, et al. Acute myocardial infarction with isolated ST-segment elevation in posterior chest leads V7-9: “hidden” ST-segment elevations revealing acute posterior infarction. J Am Coll Cardiol 1999; 34:748–753.
- Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
- Huey BL, Beller GA, Kaiser DL, Gibson RS. A comprehensive analysis of myocardial infarction due to left circumflex artery occlusion: comparison with infarction due to right coronary artery and left anterior descending artery occlusion. J Am Coll Cardiol 1988; 12:1156–1166.
- Gibson CM, Pride YB, Mohanavelu S, Wiviott SD, Antman EM, Braunwald E. Abstract 1999: Angiographic and clinical outcomes among patients with acute coronary syndrome presenting with isolated anterior ST-segment depressions. Circulation 2008; 118:S–654.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Norell MS, Lyons JP, Gardener JE, Layton CA, Balcon R. Significance of “reciprocal” ST segment depression: left ventriculographic observations during left anterior descending coronary angioplasty. J Am Coll Cardiol 1989; 13:1270–1274.
- Haraphongse M, Tanomsup S, Jugdutt BI. Inferior ST segment depression during acute anterior myocardial infarction: clinical and angiographic correlations. J Am Coll Cardiol 1984; 4:467–476.
- Surawicz B, Knilans TK. Acute ischemia: electrocardiographic patterns. In: Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th edition. Philadelphia: WB Saunders; 2001:122–153.
- Wagner GS, Macfarlane P, Wellens H, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:1003–1011.
- Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med 2002; 20:35–38.
- Surawicz B. Electrolytes and the electrocardiogram. Postgrad Med 1974; 55:123–129.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Glancy DL, Wang WL. ECG of the month. Abnormal electrocardiogram in a woman with a urinary tract infection. Sinus rhythm, rate 82/minute. Sagging ST segments, low T waves, and prominent U waves suggest hypokalemia. J La State Med Soc 2007; 159:5–7.
- Surawicz B, Braun HA, Crum WB, Kemp RL, Wagner S, Bellet S. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation 1957; 16:750–763.
- Glancy DL, Rochon BJ, Ilie CC, Parker JM, Jones MB, Atluri P. Global T-wave inversion in a 77-year-old woman. Proc (Bayl Univ Med Cent) 2009; 22:81–82.
- Walder LA, Spodick DH. Global T wave inversion. J Am Coll Cardiol 1991; 17:1479–1485.
- Lui CY. Acute pulmonary embolism as the cause of global T wave inversion and QT prolongation. A case report. J Electrocardiol 1993; 26:91–95.
- Walder LA, Spodick DH. Global T wave inversion: long-term followup. J Am Coll Cardiol 1993; 21:1652–1656.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Spodick DH. Electrocardiogram in acute pericarditis. Distributions of morphologic and axial changes by stages. Am J Cardiol 1974; 33:470–474.
- Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Rosenbaum MB, Blanco HH, Elizari MV, Lázzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50:213–222.
- Paparella N, Ouyang F, Fuca G, Kuck KH, Cappato R, Alboni P. Significance of newly acquired negative T waves after interruption of paroxysmal reentrant supraventricular tachycardia with narrow QRS complex. Am J Cardiol 2000; 85:261–263.
- Kaid KA, Maqsood A, Cohen M, Rothfeld E. Further characterization of the “persistent juvenile T-wave pattern” in adults. J Electrocardiol 2008; 41:644–645.
- Rautaharju PM, Surawicz B, Gettes LS, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part IV: the ST segment, T and U waves, and the QT interval: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:982–991.
- Surawicz B, Knilans TK. Non-Q wave myocardial infarction, unstable angina pectoris, myocardial ischemia. In: Chou's Electrocardiography in Clinical Practice: Adult and Pediatric. 5th ed. Philadelphia: WB Saunders; 2001:194–207.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Okin PM, Devereux RB, Nieminen MS, et al; LIFE Study Investigators. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 2006; 113:67–73.
- Huwez FU, Pringle SD, Macfarlane PW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J 1992; 67:304–307.
- Li D, Li CY, Yong AC, Kilpatrick D. Source of electrocardiographic ST changes in subendocardial ischemia. Circ Res 1998; 82:957–970.
- Gorgels AP, Vos MA, Mulleneers R, de Zwaan C, Bär FW, Wellens HJ. Value of the electrocardiogram in diagnosing the number of severely narrowed coronary arteries in rest angina pectoris. Am J Cardiol 1993; 72:999–1003.
- Glancy DL. Electrocardiographic diagnosis of acute myocardial infarction. J La State Med Soc 2002; 154:66–75.
- Yamaji H, Iwasaki K, Kusachi S, et al. Prediction of acute left main coronary artery obstruction by 12-lead electrocardiography. ST segment elevation in lead aVR with less ST segment elevation in lead V(1). J Am Coll Cardiol 2001; 38:1348–1354.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- Lilaonitkul M, Robinson K, Roberts M. Wellens’ syndrome: significance of ECG pattern recognition in the emergency department. Emerg Med J 2009; 26:750–751.
- Glancy DL, Khuri B, Cospolich B. Heed the warning: Wellens’ type T-wave inversion is caused by proximal left anterior descending lesion. Proc (Bayl Univ Med Cent) 2000; 13:416–418.
- Savonitto S, Ardissino D, Granger CB, et al. Prognostic value of the admission electrocardiogram in acute coronary syndromes. JAMA 1999; 281:707–713.
- Mueller C, Neumann FJ, Perach W, Perruchoud AP, Buettner HJ. Prognostic value of the admission electrocardiogram in patients with unstable angina/non-ST-segment elevation myocardial infarction treated with very early revascularization. Am J Med 2004; 117:145–150.
- Boden WE, Spodick DH. Diagnostic significance of precordial ST-segment depression. Am J Cardiol 1989; 63:358–361.
- Shah A, Wagner GS, Green CL, et al. Electrocardiographic differentiation of the ST-segment depression of acute myocardial injury due to the left circumflex artery occlusion from that of myocardial ischemia of nonocclusive etiologies. Am J Cardiol 1997; 80:512–513.
- Krishnaswamy A, Lincoff AM, Menon V. Magnitude and consequences of missing the acute infarct-related circumflex artery. Am Heart J 2009; 158:706–712.
- Matetzky S, Freimark D, Feinberg MS, et al. Acute myocardial infarction with isolated ST-segment elevation in posterior chest leads V7-9: “hidden” ST-segment elevations revealing acute posterior infarction. J Am Coll Cardiol 1999; 34:748–753.
- Matetzky S, Freimark D, Chouraqui P, et al. Significance of ST segment elevations in posterior chest leads (V7 to V9) in patients with acute inferior myocardial infarction: application for thrombolytic therapy. J Am Coll Cardiol 1998; 31:506–511.
- Huey BL, Beller GA, Kaiser DL, Gibson RS. A comprehensive analysis of myocardial infarction due to left circumflex artery occlusion: comparison with infarction due to right coronary artery and left anterior descending artery occlusion. J Am Coll Cardiol 1988; 12:1156–1166.
- Gibson CM, Pride YB, Mohanavelu S, Wiviott SD, Antman EM, Braunwald E. Abstract 1999: Angiographic and clinical outcomes among patients with acute coronary syndrome presenting with isolated anterior ST-segment depressions. Circulation 2008; 118:S–654.
- Ferrari E, Imbert A, Chevalier T, Mihoubi A, Morand P, Baudouy M. The ECG in pulmonary embolism. Predictive value of negative T waves in precordial leads—80 case reports. Chest 1997; 111:537–543.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100:598–603.
- Norell MS, Lyons JP, Gardener JE, Layton CA, Balcon R. Significance of “reciprocal” ST segment depression: left ventriculographic observations during left anterior descending coronary angioplasty. J Am Coll Cardiol 1989; 13:1270–1274.
- Haraphongse M, Tanomsup S, Jugdutt BI. Inferior ST segment depression during acute anterior myocardial infarction: clinical and angiographic correlations. J Am Coll Cardiol 1984; 4:467–476.
- Surawicz B, Knilans TK. Acute ischemia: electrocardiographic patterns. In: Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric. 5th edition. Philadelphia: WB Saunders; 2001:122–153.
- Wagner GS, Macfarlane P, Wellens H, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part VI: acute ischemia/infarction: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:1003–1011.
- Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med 2002; 20:35–38.
- Surawicz B. Electrolytes and the electrocardiogram. Postgrad Med 1974; 55:123–129.
- Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med 2004; 27:153–160.
- Glancy DL, Wang WL. ECG of the month. Abnormal electrocardiogram in a woman with a urinary tract infection. Sinus rhythm, rate 82/minute. Sagging ST segments, low T waves, and prominent U waves suggest hypokalemia. J La State Med Soc 2007; 159:5–7.
- Surawicz B, Braun HA, Crum WB, Kemp RL, Wagner S, Bellet S. Quantitative analysis of the electrocardiographic pattern of hypopotassemia. Circulation 1957; 16:750–763.
- Glancy DL, Rochon BJ, Ilie CC, Parker JM, Jones MB, Atluri P. Global T-wave inversion in a 77-year-old woman. Proc (Bayl Univ Med Cent) 2009; 22:81–82.
- Walder LA, Spodick DH. Global T wave inversion. J Am Coll Cardiol 1991; 17:1479–1485.
- Lui CY. Acute pulmonary embolism as the cause of global T wave inversion and QT prolongation. A case report. J Electrocardiol 1993; 26:91–95.
- Walder LA, Spodick DH. Global T wave inversion: long-term followup. J Am Coll Cardiol 1993; 21:1652–1656.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Wittstein IS, Thiemann DR, Lima JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 2005; 352:539–548.
- Spodick DH. Electrocardiogram in acute pericarditis. Distributions of morphologic and axial changes by stages. Am J Cardiol 1974; 33:470–474.
- Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Rosenbaum MB, Blanco HH, Elizari MV, Lázzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50:213–222.
- Paparella N, Ouyang F, Fuca G, Kuck KH, Cappato R, Alboni P. Significance of newly acquired negative T waves after interruption of paroxysmal reentrant supraventricular tachycardia with narrow QRS complex. Am J Cardiol 2000; 85:261–263.
- Kaid KA, Maqsood A, Cohen M, Rothfeld E. Further characterization of the “persistent juvenile T-wave pattern” in adults. J Electrocardiol 2008; 41:644–645.
KEY POINTS
- ST-T abnormalities concordant to the QRS complex suggest ischemia.
- Deep T-wave inversion or positive-negative biphasic T waves in the anterior precordial leads reflect severe left anterior descending coronary artery stenosis.
- Two particular patterns of ST-segment depression reflect ST-segment elevation myocardial infarction rather than non–ST-segment elevation acute coronary syndrome: ST-segment depression that is reciprocal to a subtle and sometimes overlooked ST-segment elevation, and ST-segment depression that is maximal in leads V1–V3, suggesting true posterior infarction.
- T-wave inversion in the anterior precordial leads may be seen in cases of acute pulmonary embolism, while flattened T waves with prominent U waves and ST-segment depression may reflect hypokalemia or digitalis therapy.
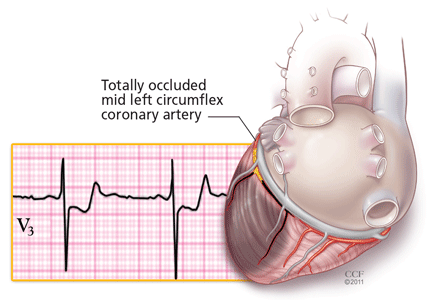
Progressive muscle weakness: More there than meets the eye
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
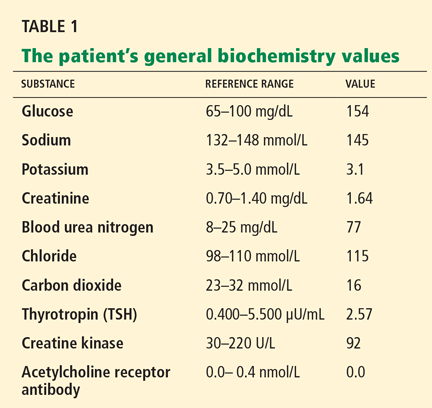
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
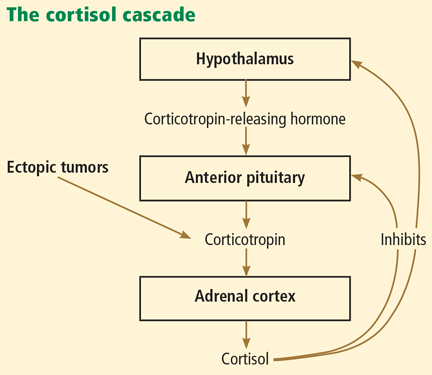
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
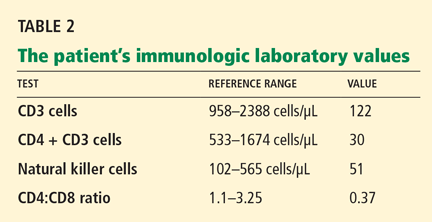
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.

Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.
Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.
Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
How to manage type 2 diabetes in medical and surgical patients in the hospital
Hyperglycemia and diabetes mellitus are very common in hospitalized patients. Although more data are available on the prevalence of this problem and on how to manage it in the intensive care unit (ICU) than on regular hospital floors, the situation is changing. Information is emerging on the prevalence and impact of hyperglycemia and diabetes in the non-ICU setting, which is the focus of this paper.
HYPERGLYCEMIA IS COMMON AND PREDICTS POOR OUTCOMES
Cook et al,1 in a survey of 126 US hospitals, found that the prevalence of hyperglycemia (blood glucose > 180 mg/dL) was 46% in the ICU and 32% in regular wards.
Kosiborod et al2 reported that hyperglycemia (blood glucose > 140 mg/dL) was present in 78% of diabetic patients hospitalized with acute coronary syndrome and 26% of similar hospitalized nondiabetic patients.
Hyperglycemia is a common comorbidity in medical-surgical patients in community hospitals. Our group3 found that, in our hospital, 62% of patients were normoglycemic (ie, had a fasting blood glucose < 126 mg/dL or a random blood glucose < 200 mg/dL on two occasions), 26% had known diabetes, and 12% had new hyperglycemia. Further, new hyperglycemia was associated with a higher in-hospital death rate than the other two conditions.
Failure to identify diabetes is a predictor of rehospitalization. Robbins and Webb4 reported that 30.6% of those who had diabetes that was missed during hospitalization were readmitted within 30 days, compared with 9.4% of patients with diabetes first diagnosed during hospitalization.
WHAT DIAGNOSTIC CRITERIA SHOULD WE USE?
Blood glucose greater than 140 mg/dL
A consensus statement from the American Association of Clinical Endocrinologists (ACE) and the American Diabetes Association (ADA)5 defines in-hospital hyperglycemia as a blood glucose level greater than 140 mg/dL on admission or in the hospital. If the blood glucose is higher than this, the question arises as to whether the patient has preexisting diabetes or has stress hyperglycemia.
Hemoglobin A1c of 6.5% or higher
In view of the uncertainty as to whether a patient with an elevated blood glucose level has preexisting diabetes or stress hyperglycemia, upcoming guidelines will recommend measuring the hemoglobin A1c level if the blood glucose level is higher than 140 mg/dL.
A patient with an elevated blood glucose level (>140 mg/dL) whose hemoglobin A1c level is 6.5% or higher can be identified as having diabetes that preceded the hospitalization. Hemoglobin A1c testing can also be useful to assess glycemic control before admission and in designing an optional regimen at the time of discharge. In patients with newly recognized hyperglycemia, a hemoglobin A1c measurement can help differentiate patients with previously undiagnosed diabetes from those with stress-induced hyperglycemia.
Clinicians should keep in mind that a hemoglobin A1c cutoff of 6.5% identifies fewer cases of undiagnosed diabetes than does a high fasting glucose concentration, and that a level less than 6.5% does not rule out the diagnosis of diabetes. Several epidemiologic studies6 have reported a low sensitivity (44% to 66%) but a high specificity (76% to 99%) for hemoglobin A1c values higher than 6.5% in an outpatient population. The high specificity therefore supports the use of hemoglobin A1c to confirm the diagnosis of diabetes in patients with hyperglycemia, but the low sensitivity indicates that this test should not be used for universal screening in the hospital.
Many factors can influence the hemoglobin A1c level, such as anemia, iron deficiency, blood transfusions, hemolytic anemia, and renal failure.
Until now, if patients had hyperglycemia but no prior diagnosis of diabetes, the recommendation was for an oral 2-hour glucose tolerance test shortly after discharge to confirm the diagnosis of diabetes. Norhammar et al7 performed oral glucose tolerance tests in patients admitted with acute myocardial infarction, and Matz et al8 performed glucose tolerance tests in patients with acute stroke. They found that impaired glucose tolerance and undiagnosed type 2 diabetes were very common in these two groups. However, physicians rarely order oral glucose tolerance tests. We believe that hemoglobin A1c will be a better tool than an oral glucose tolerance test to confirm diabetes in hyperglycemic patients in the hospital setting.

WHAT IS THE ASSOCIATION BETWEEN HYPERGLYCEMIA AND OUTCOMES?
In 2,471 patients admitted to the hospital with community-acquired pneumonia, McAlister et al10 found that the rates of hospital complications and of death rose with blood glucose levels.
Falguera et al11 found that, in 660 episodes of community-acquired pneumonia, the rates of hospitalization, death, pleural effusion, and concomitant illnesses were all significantly higher in diabetic patients than in nondiabetic patients.
Noordzij et al12 performed a case-control study of 108,593 patients who underwent noncardiac surgery. The odds ratio for perioperative death was 1.19 (95% confidence interval [CI] 1.1–1.3) for every 1-mmol/L increase in the glucose level.
Frisch et al,13 in patients undergoing noncardiac surgery, found that the 30-day rates of death and of in-hospital complications were all higher in patients with diabetes than without diabetes.
Our group3 identified hyperglycemia as an independent marker of in-hospital death in patients with undiagnosed diabetes. The rates of death were 1.7% in those with normoglycemia, 3.0% in those with known diabetes, and 16.0% (P < .01) in those with new hyperglycemia.
The ACE/ADA consensus panel14 set the following glucose targets for patients in the non-ICU setting:
- Pre-meal blood glucose < 140 mg/dL
- Random blood glucose < 180 mg/dL.
On the other hand, hypoglycemia is also associated with adverse outcomes. Therefore, to avoid hypoglycemia, the insulin regimen should be reassessed if blood glucose levels fall below 100 mg/dL. New guidelines will suggest keeping the blood glucose between 100 and 140 mg/dL.
HOW SHOULD WE MANAGE HYPERGLYCEMIA IN THE NON-ICU SETTING?
The ACE/ADA guidelines recommend subcutaneous insulin therapy for most medical-surgical patients with diabetes, reserving intravenous insulin therapy for hyperglycemic crises and uncontrolled hyperglycemia.14
Oral antidiabetic agents are not generally recommended, as we have no data to support their use in the hospital. Another argument against using noninsulin therapies in the hospital is that sulfonylureas, especially glyburide (Diabeta, Micronase) are a major cause of hypoglycemia. Metformin (Glucophage) is contraindicated in decreased renal function, in hemodynamic instability, in surgical patients, and with the use of iodinated contrast dye. Thiazolidinediones are associated with edema and congestive heart failure, and they take up to 12 weeks to lower blood glucose levels. Alpha-glucosidase inhibitors are weak glucose-lowering agents. Also, therapies directed at glucagon-like-protein 1 can cause nausea and have a greater effect on postprandial glucose.14
The two main options for managing hyperglycemia and diabetes in the non-ICU setting are short-acting insulin on a sliding scale and basal-bolus therapy, the latter with either NPH plus regular insulin or long-acting plus rapid-acting insulin analogues.
Basal-bolus vs sliding scale insulin: The RABBIT-2 trial
In the RABBIT 2 trial (Randomized Basal Bolus Versus Sliding Scale Regular Insulin in Patients With Type 2 Diabetes Mellitus),15 our group compared the efficacy and safety of a basal-bolus regimen and a sliding-scale regimen in 130 hospitalized patients with type 2 diabetes treated with diet, with oral hypoglycemic agents, or with both. Oral antidiabetic drugs were discontinued on admission, and patients were randomized to one of the treatment groups.
In the basal-bolus group, the starting total daily dose was 0.4 U/kg/day if the blood glucose level on admission was between 140 and 200 mg/dL, or 0.5 U/kg/day if the glucose level was between 201 and 400 mg/dL. Half of the total daily dose was given as insulin glargine (Lantus) once daily, and the other half was given as insulin glulisine (Apidra) before meals. These doses were adjusted if the patient’s fasting or pre-meal blood glucose levels rose above 140 mg/dL or fell below 70 mg/dL.
The sliding-scale group received regular insulin four times daily (before meals and at bedtime) for glucose levels higher than 140 mg/dL; the higher the level, the more they got.
The basal-bolus regimen was better than sliding-scale regular insulin. At admission, the mean glucose values and hemoglobin A1c values were similar in both groups, but the mean glucose level on therapy was significantly lower in the basal-bolus group than in the sliding-scale group, 166 ± 32 mg/dL vs 193 ± 54 mg/dL, P < .001). About two-thirds of the basal-bolus group achieved a blood glucose target of less than 140 mg/dL, compared with only about one-third of the sliding-scale group. The basal-bolus group received more insulin, a mean of 42 units per day vs 12.5 units per day in the sliding-scale group. Yet the incidence of hypoglycemia was 3% in both groups.
NPH plus regular vs detemir plus aspart: The DEAN trial
Several long-acting insulin analogues are available and have a longer duration of action than NPH. Similarly, several newer rapid-acting analogues act more rapidly than regular insulin. Do these pharmacokinetic advantages matter? And do they justify the higher costs of the newer agents?
In the randomized Insulin Detemir Versus NPH Insulin in Hospitalized Patients With Diabetes (DEAN) trial,16 we compared two regimens: detemir plus aspart in a basal-bolus regimen, and NPH plus regular insulin in two divided doses, two-thirds of the total daily dose in the morning before breakfast and one-third before dinner, both doses in a ratio of two-thirds NPH and one-third regular, mixed in the same syringe. We recruited 130 patients with type 2 diabetes mellitus who were on oral hypoglycemic agents or insulin therapy.
NPH plus regular was just as good as detemir plus aspart in improving glycemic control. Blood glucose levels fell during the first day of therapy and were similar in both groups throughout the trial, as measured before breakfast, lunch, and dinner and at bedtime. The mean total daily insulin dose was not significantly different between treatment groups: 56 ± 45 units in the basal-bolus detemir-aspart group and 45 ± 32 units in the NPH-regular group. However, the basal-bolus group received significantly more short-acting insulin: 27 ± 20 units a day of aspart vs 18 ± 14 units of regular.
Somewhat fewer patients in the NPH-regular group had episodes of hypoglycemia, although the difference between groups was not statistically significant.
In a univariate analysis of the RABBIT-2 and DEAN trials,17 factors that predicted a blood glucose level less than 60 mg/dL were older age, lower body weight, higher serum creatinine level, and previous insulin therapy. Factors that were not predictive were the hemoglobin A1c level and the enrollment blood glucose level. Based on these data, we believe that to reduce the rate of hypoglycemia, lower insulin doses are needed in elderly patients and patients with renal impairment, and that if patients have been taking insulin before they come to the hospital, the dose should be cut back by about 25% while they are hospitalized.
Basal-bolus vs sliding-scale insulin for surgical patients: The RABBIT 2 Surgery trial
Does better glucose control in surgical patients affect outcomes in patients undergoing general surgery? To find out, we performed a prospective, multicenter, randomized, open-label trial in general surgery patients not in the ICU.18 We recruited and randomized 211 patients with type 2 diabetes who were on diet therapy or oral hypoglycemic agents or insulin in low doses (< 0.4 U/kg/day).
Oral drugs were discontinued on admission, and patients were randomized to receive either a basal-bolus regimen of glargine plus glulisine or regular insulin on a sliding scale. The basal-bolus group got 0.5 U/kg/day, half of it as glargine once daily and half as glulisine before meals. The total daily dose was reduced to 0.3 U/kg/day in patients age 70 and older or who had a serum creatinine level of 2.0 mg/dL or higher.
The goal was to maintain fasting and pre-meal glucose concentrations between 100 and 140 mg/dL. The total daily dose was raised by 10% (mostly in the glargine dose) if the blood glucose level was in the range of 141 to 180 mg/dL, and by 20% if the glucose level was higher than 181 mg/dL. The dose was decreased by 10% for glucose levels between 70 and 99 mg/dL, was decreased by 20% if the glucose level was between 40 and 69, and was held if the glucose level was lower than 40 mg/dL. If a patient was not able to eat, insulin glulisine was held until meals were resumed.
The sliding-scale group received regular insulin four times a day for blood glucose levels higher than 140 mg/dL.
The primary outcomes measured were the difference between groups in mean daily blood glucose concentration and a composite of hospital complications including postoperative wound infection, pneumonia, respiratory failure, acute renal failure, and bacteremia. Secondary outcomes were differences between groups in mean fasting and pre-meal blood glucose, number of hypoglycemic episodes (blood glucose < 70 mg/dL), hyperglycemic episodes (blood glucose > 200 mg/dL), length of hospital stay, need for intensive care, and rate of complications including wound infection, pneumonia, acute renal failure, and death.
Blood glucose levels were significantly lower in the basal-bolus group through the first 7 days after randomization, as measured before breakfast, lunch, and dinner, and at bedtime, and then they converged.
More patients in the sliding-scale group had hospital complications, 26 vs 9, P = .003. On the other hand, more patients in the basal-bolus group had episodes of hypoglycemia: 24 (23%) vs 5 (4.7%) had episodes of less than 70 mg/dL (P < .001), 12 (12%) vs 2 (1.9%) had episodes of less than 60 mg/dL (P = .005), and 4 (3.8%) vs 0 had episodes of less than 40 mg/dL (P = .057). The mean total daily dose of insulin was 33.4 units in the basal-bolus group and 12.3 units in the sliding-scale group.
WHAT HAVE WE LEARNED?
Don’t use a sliding-scale regimen as a single agent in patients with diabetes. Glycemic control is better with a basal-bolus regimen than with a sliding-scale regimen, and a basal-bolus insulin regimen is preferred for most patients with hyperglycemia.
The old human insulins (ie, regular and NPH) are still good and improve glycemic control as well as the new basal insulin analogues (detemir and aspart) do.
Improved control may reduce the rate of hospital complications, according to preliminary evidence. More studies are under way.
One size does not fit all. Those who are elderly or who have impaired renal function should receive lower doses of insulin, eg, 0.3 U/kg/day instead of 0.5 U/kg/day. Those who are on insulin should have their dose decreased when they are admitted to the hospital. Perhaps lean patients with type 2 diabetes should also have a lower dose.
Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of NPH plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
Patients treated with insulin at home should continue to receive insulin therapy in the hospital. However, the insulin dosage should be reduced by about 25% to allow for lower food intake.
QUESTIONS FOR FURTHER STUDY
Should we modify the standard basal-bolus regimen?
In a typical basal-bolus regimen, patients get 50% of their total daily insulin dose in the form of a basal injection and 50% in the form of rapid-acting boluses before meals. However, for a variety of reasons, hospitalized patients do not eat very much. Thus, a 50-50 basal-bolus regimen may not be ideal for patients with poor oral intake.
In the Basal-PLUS trial, currently under way, we are comparing the safety and efficacy of a daily dose of basal insulin (glargine) plus correction doses of a rapid-acting insulin analogue (glulisine) on a sliding scale and a standard basal-bolus regimen in medical and surgical patients.
Does one glycemic target fit all patients?
Falciglia et al19 found an association between hyperglycemia and death in patients with unstable angina, arrhythmias, stroke, pneumonia, gastrointestinal bleeding, respiratory failure, sepsis, acute renal failure, and congestive heart failure. However, they found no such association in patients with chronic obstructive pulmonary disease, liver failure, diabetic ketoacidosis, gastrointestinal neoplasm, musculoskeletal disease, peripheral vascular disease with bypass, hip fracture, amputation due to peripheral vascular disease, or prostate surgery. Should patients in this second group be treated with a less-intensive insulin regimen?
What is the best regimen after hospital discharge?
We are conducting a prospective clinical trial to assess the impact of insulin after hospital discharge. Our current practice when a patient is discharged from the hospital is as follows:
- If the admission hemoglobin A1c level is less than 7%, we restart the previous outpatient treatment regimen of oral antidiabetic agents, or insulin, or both.
- If the admission hemoglobin A1c is between 7% and 9%, we restart the outpatient oral agents and continue glargine once daily at 50% to 80% of the hospital dose.
- If the hemoglobin A1c level is higher than 9%, we discharge the patient on a basal-bolus regimen at the same dosage as in the hospital. As an alternative, we could restart the oral agents and add glargine once daily at 80% of the hospital dose.
- Cook CB, Kongable GL, Potter DJ, Abad VJ, Leija DE, Anderson M. Inpatient glucose control: a glycemic survey of 126 U.S. hospitals. J Hosp Med 2009; 4:E7–E14.
- Kosiborod M, Inzucchi S, Clark B, et al. National patterns of glucose control among patients hospitalized with acute myocardial infarction [abstract]. J Am Coll Cardiol 2007; 49:283A–284A.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Robbins JM, Webb DA. Diagnosing diabetes and preventing rehospitalizations: the urban diabetes study. Med Care 2006; 44:292–296.
- Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Saudek D, Herman WH, Sacks DB, Bergenstal RM, Edelman D, Davidson MB. A new look at screening and diagnosing diabetes mellitus. J Clin Endocrinol Metab 2008; 93:2447–2453.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 2002; 359:2140–2144.
- Matz K, Keresztes K, Tatschl C, et al. Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 2006; 29:792–797.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33(suppl 1):S62–S69.
- McAlister FA, Majumdar SR, Blitz S, Rowe BH, Romney J, Marrie TJ. The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community-acquired pneumonia. Diabetes Care 2005; 28:810–815.
- Falguera M, Pifarre R, Martin A, Sheikh A, Moreno A. Etiology and outcome of community-acquired pneumonia in patients with diabetes mellitus. Chest 2005; 128:3233–3239.
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol 2007; 156:137–142.
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care 2010; 33:1783–1788.
- Moghissi ES, Korythowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocrine Pract 2009; 15:1–17.
- Umpierrrez GE, Smiley D, Zisman A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care 2007; 30:2181–2186.
- Umpierrez GE, Hor T, Smiley D, et al. Comparison of inpatient insulin regimens with detemir plus aspart versus neutral protamine Hagedorn plus regular in medical patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:564–569.
- Umpierrez GE, Smiley D, Umpierrez D, Ceron M, Temponi A. Hypoglycemic events during subcutaneous insulin therapy in type 2 diabetes (abstract). Presented at American Diabetes Association 69th Scientific Sessions, New Orleans, LA, June 5–9, 2009.
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care 2011; 34:256–261.
- Falciglia M, Freyberg RW, Almenoff PL, D’Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009; 37:3001–3009.
Hyperglycemia and diabetes mellitus are very common in hospitalized patients. Although more data are available on the prevalence of this problem and on how to manage it in the intensive care unit (ICU) than on regular hospital floors, the situation is changing. Information is emerging on the prevalence and impact of hyperglycemia and diabetes in the non-ICU setting, which is the focus of this paper.
HYPERGLYCEMIA IS COMMON AND PREDICTS POOR OUTCOMES
Cook et al,1 in a survey of 126 US hospitals, found that the prevalence of hyperglycemia (blood glucose > 180 mg/dL) was 46% in the ICU and 32% in regular wards.
Kosiborod et al2 reported that hyperglycemia (blood glucose > 140 mg/dL) was present in 78% of diabetic patients hospitalized with acute coronary syndrome and 26% of similar hospitalized nondiabetic patients.
Hyperglycemia is a common comorbidity in medical-surgical patients in community hospitals. Our group3 found that, in our hospital, 62% of patients were normoglycemic (ie, had a fasting blood glucose < 126 mg/dL or a random blood glucose < 200 mg/dL on two occasions), 26% had known diabetes, and 12% had new hyperglycemia. Further, new hyperglycemia was associated with a higher in-hospital death rate than the other two conditions.
Failure to identify diabetes is a predictor of rehospitalization. Robbins and Webb4 reported that 30.6% of those who had diabetes that was missed during hospitalization were readmitted within 30 days, compared with 9.4% of patients with diabetes first diagnosed during hospitalization.
WHAT DIAGNOSTIC CRITERIA SHOULD WE USE?
Blood glucose greater than 140 mg/dL
A consensus statement from the American Association of Clinical Endocrinologists (ACE) and the American Diabetes Association (ADA)5 defines in-hospital hyperglycemia as a blood glucose level greater than 140 mg/dL on admission or in the hospital. If the blood glucose is higher than this, the question arises as to whether the patient has preexisting diabetes or has stress hyperglycemia.
Hemoglobin A1c of 6.5% or higher
In view of the uncertainty as to whether a patient with an elevated blood glucose level has preexisting diabetes or stress hyperglycemia, upcoming guidelines will recommend measuring the hemoglobin A1c level if the blood glucose level is higher than 140 mg/dL.
A patient with an elevated blood glucose level (>140 mg/dL) whose hemoglobin A1c level is 6.5% or higher can be identified as having diabetes that preceded the hospitalization. Hemoglobin A1c testing can also be useful to assess glycemic control before admission and in designing an optional regimen at the time of discharge. In patients with newly recognized hyperglycemia, a hemoglobin A1c measurement can help differentiate patients with previously undiagnosed diabetes from those with stress-induced hyperglycemia.
Clinicians should keep in mind that a hemoglobin A1c cutoff of 6.5% identifies fewer cases of undiagnosed diabetes than does a high fasting glucose concentration, and that a level less than 6.5% does not rule out the diagnosis of diabetes. Several epidemiologic studies6 have reported a low sensitivity (44% to 66%) but a high specificity (76% to 99%) for hemoglobin A1c values higher than 6.5% in an outpatient population. The high specificity therefore supports the use of hemoglobin A1c to confirm the diagnosis of diabetes in patients with hyperglycemia, but the low sensitivity indicates that this test should not be used for universal screening in the hospital.
Many factors can influence the hemoglobin A1c level, such as anemia, iron deficiency, blood transfusions, hemolytic anemia, and renal failure.
Until now, if patients had hyperglycemia but no prior diagnosis of diabetes, the recommendation was for an oral 2-hour glucose tolerance test shortly after discharge to confirm the diagnosis of diabetes. Norhammar et al7 performed oral glucose tolerance tests in patients admitted with acute myocardial infarction, and Matz et al8 performed glucose tolerance tests in patients with acute stroke. They found that impaired glucose tolerance and undiagnosed type 2 diabetes were very common in these two groups. However, physicians rarely order oral glucose tolerance tests. We believe that hemoglobin A1c will be a better tool than an oral glucose tolerance test to confirm diabetes in hyperglycemic patients in the hospital setting.
WHAT IS THE ASSOCIATION BETWEEN HYPERGLYCEMIA AND OUTCOMES?
In 2,471 patients admitted to the hospital with community-acquired pneumonia, McAlister et al10 found that the rates of hospital complications and of death rose with blood glucose levels.
Falguera et al11 found that, in 660 episodes of community-acquired pneumonia, the rates of hospitalization, death, pleural effusion, and concomitant illnesses were all significantly higher in diabetic patients than in nondiabetic patients.
Noordzij et al12 performed a case-control study of 108,593 patients who underwent noncardiac surgery. The odds ratio for perioperative death was 1.19 (95% confidence interval [CI] 1.1–1.3) for every 1-mmol/L increase in the glucose level.
Frisch et al,13 in patients undergoing noncardiac surgery, found that the 30-day rates of death and of in-hospital complications were all higher in patients with diabetes than without diabetes.
Our group3 identified hyperglycemia as an independent marker of in-hospital death in patients with undiagnosed diabetes. The rates of death were 1.7% in those with normoglycemia, 3.0% in those with known diabetes, and 16.0% (P < .01) in those with new hyperglycemia.
The ACE/ADA consensus panel14 set the following glucose targets for patients in the non-ICU setting:
- Pre-meal blood glucose < 140 mg/dL
- Random blood glucose < 180 mg/dL.
On the other hand, hypoglycemia is also associated with adverse outcomes. Therefore, to avoid hypoglycemia, the insulin regimen should be reassessed if blood glucose levels fall below 100 mg/dL. New guidelines will suggest keeping the blood glucose between 100 and 140 mg/dL.
HOW SHOULD WE MANAGE HYPERGLYCEMIA IN THE NON-ICU SETTING?
The ACE/ADA guidelines recommend subcutaneous insulin therapy for most medical-surgical patients with diabetes, reserving intravenous insulin therapy for hyperglycemic crises and uncontrolled hyperglycemia.14
Oral antidiabetic agents are not generally recommended, as we have no data to support their use in the hospital. Another argument against using noninsulin therapies in the hospital is that sulfonylureas, especially glyburide (Diabeta, Micronase) are a major cause of hypoglycemia. Metformin (Glucophage) is contraindicated in decreased renal function, in hemodynamic instability, in surgical patients, and with the use of iodinated contrast dye. Thiazolidinediones are associated with edema and congestive heart failure, and they take up to 12 weeks to lower blood glucose levels. Alpha-glucosidase inhibitors are weak glucose-lowering agents. Also, therapies directed at glucagon-like-protein 1 can cause nausea and have a greater effect on postprandial glucose.14
The two main options for managing hyperglycemia and diabetes in the non-ICU setting are short-acting insulin on a sliding scale and basal-bolus therapy, the latter with either NPH plus regular insulin or long-acting plus rapid-acting insulin analogues.
Basal-bolus vs sliding scale insulin: The RABBIT-2 trial
In the RABBIT 2 trial (Randomized Basal Bolus Versus Sliding Scale Regular Insulin in Patients With Type 2 Diabetes Mellitus),15 our group compared the efficacy and safety of a basal-bolus regimen and a sliding-scale regimen in 130 hospitalized patients with type 2 diabetes treated with diet, with oral hypoglycemic agents, or with both. Oral antidiabetic drugs were discontinued on admission, and patients were randomized to one of the treatment groups.
In the basal-bolus group, the starting total daily dose was 0.4 U/kg/day if the blood glucose level on admission was between 140 and 200 mg/dL, or 0.5 U/kg/day if the glucose level was between 201 and 400 mg/dL. Half of the total daily dose was given as insulin glargine (Lantus) once daily, and the other half was given as insulin glulisine (Apidra) before meals. These doses were adjusted if the patient’s fasting or pre-meal blood glucose levels rose above 140 mg/dL or fell below 70 mg/dL.
The sliding-scale group received regular insulin four times daily (before meals and at bedtime) for glucose levels higher than 140 mg/dL; the higher the level, the more they got.
The basal-bolus regimen was better than sliding-scale regular insulin. At admission, the mean glucose values and hemoglobin A1c values were similar in both groups, but the mean glucose level on therapy was significantly lower in the basal-bolus group than in the sliding-scale group, 166 ± 32 mg/dL vs 193 ± 54 mg/dL, P < .001). About two-thirds of the basal-bolus group achieved a blood glucose target of less than 140 mg/dL, compared with only about one-third of the sliding-scale group. The basal-bolus group received more insulin, a mean of 42 units per day vs 12.5 units per day in the sliding-scale group. Yet the incidence of hypoglycemia was 3% in both groups.
NPH plus regular vs detemir plus aspart: The DEAN trial
Several long-acting insulin analogues are available and have a longer duration of action than NPH. Similarly, several newer rapid-acting analogues act more rapidly than regular insulin. Do these pharmacokinetic advantages matter? And do they justify the higher costs of the newer agents?
In the randomized Insulin Detemir Versus NPH Insulin in Hospitalized Patients With Diabetes (DEAN) trial,16 we compared two regimens: detemir plus aspart in a basal-bolus regimen, and NPH plus regular insulin in two divided doses, two-thirds of the total daily dose in the morning before breakfast and one-third before dinner, both doses in a ratio of two-thirds NPH and one-third regular, mixed in the same syringe. We recruited 130 patients with type 2 diabetes mellitus who were on oral hypoglycemic agents or insulin therapy.
NPH plus regular was just as good as detemir plus aspart in improving glycemic control. Blood glucose levels fell during the first day of therapy and were similar in both groups throughout the trial, as measured before breakfast, lunch, and dinner and at bedtime. The mean total daily insulin dose was not significantly different between treatment groups: 56 ± 45 units in the basal-bolus detemir-aspart group and 45 ± 32 units in the NPH-regular group. However, the basal-bolus group received significantly more short-acting insulin: 27 ± 20 units a day of aspart vs 18 ± 14 units of regular.
Somewhat fewer patients in the NPH-regular group had episodes of hypoglycemia, although the difference between groups was not statistically significant.
In a univariate analysis of the RABBIT-2 and DEAN trials,17 factors that predicted a blood glucose level less than 60 mg/dL were older age, lower body weight, higher serum creatinine level, and previous insulin therapy. Factors that were not predictive were the hemoglobin A1c level and the enrollment blood glucose level. Based on these data, we believe that to reduce the rate of hypoglycemia, lower insulin doses are needed in elderly patients and patients with renal impairment, and that if patients have been taking insulin before they come to the hospital, the dose should be cut back by about 25% while they are hospitalized.
Basal-bolus vs sliding-scale insulin for surgical patients: The RABBIT 2 Surgery trial
Does better glucose control in surgical patients affect outcomes in patients undergoing general surgery? To find out, we performed a prospective, multicenter, randomized, open-label trial in general surgery patients not in the ICU.18 We recruited and randomized 211 patients with type 2 diabetes who were on diet therapy or oral hypoglycemic agents or insulin in low doses (< 0.4 U/kg/day).
Oral drugs were discontinued on admission, and patients were randomized to receive either a basal-bolus regimen of glargine plus glulisine or regular insulin on a sliding scale. The basal-bolus group got 0.5 U/kg/day, half of it as glargine once daily and half as glulisine before meals. The total daily dose was reduced to 0.3 U/kg/day in patients age 70 and older or who had a serum creatinine level of 2.0 mg/dL or higher.
The goal was to maintain fasting and pre-meal glucose concentrations between 100 and 140 mg/dL. The total daily dose was raised by 10% (mostly in the glargine dose) if the blood glucose level was in the range of 141 to 180 mg/dL, and by 20% if the glucose level was higher than 181 mg/dL. The dose was decreased by 10% for glucose levels between 70 and 99 mg/dL, was decreased by 20% if the glucose level was between 40 and 69, and was held if the glucose level was lower than 40 mg/dL. If a patient was not able to eat, insulin glulisine was held until meals were resumed.
The sliding-scale group received regular insulin four times a day for blood glucose levels higher than 140 mg/dL.
The primary outcomes measured were the difference between groups in mean daily blood glucose concentration and a composite of hospital complications including postoperative wound infection, pneumonia, respiratory failure, acute renal failure, and bacteremia. Secondary outcomes were differences between groups in mean fasting and pre-meal blood glucose, number of hypoglycemic episodes (blood glucose < 70 mg/dL), hyperglycemic episodes (blood glucose > 200 mg/dL), length of hospital stay, need for intensive care, and rate of complications including wound infection, pneumonia, acute renal failure, and death.
Blood glucose levels were significantly lower in the basal-bolus group through the first 7 days after randomization, as measured before breakfast, lunch, and dinner, and at bedtime, and then they converged.
More patients in the sliding-scale group had hospital complications, 26 vs 9, P = .003. On the other hand, more patients in the basal-bolus group had episodes of hypoglycemia: 24 (23%) vs 5 (4.7%) had episodes of less than 70 mg/dL (P < .001), 12 (12%) vs 2 (1.9%) had episodes of less than 60 mg/dL (P = .005), and 4 (3.8%) vs 0 had episodes of less than 40 mg/dL (P = .057). The mean total daily dose of insulin was 33.4 units in the basal-bolus group and 12.3 units in the sliding-scale group.
WHAT HAVE WE LEARNED?
Don’t use a sliding-scale regimen as a single agent in patients with diabetes. Glycemic control is better with a basal-bolus regimen than with a sliding-scale regimen, and a basal-bolus insulin regimen is preferred for most patients with hyperglycemia.
The old human insulins (ie, regular and NPH) are still good and improve glycemic control as well as the new basal insulin analogues (detemir and aspart) do.
Improved control may reduce the rate of hospital complications, according to preliminary evidence. More studies are under way.
One size does not fit all. Those who are elderly or who have impaired renal function should receive lower doses of insulin, eg, 0.3 U/kg/day instead of 0.5 U/kg/day. Those who are on insulin should have their dose decreased when they are admitted to the hospital. Perhaps lean patients with type 2 diabetes should also have a lower dose.
Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of NPH plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
Patients treated with insulin at home should continue to receive insulin therapy in the hospital. However, the insulin dosage should be reduced by about 25% to allow for lower food intake.
QUESTIONS FOR FURTHER STUDY
Should we modify the standard basal-bolus regimen?
In a typical basal-bolus regimen, patients get 50% of their total daily insulin dose in the form of a basal injection and 50% in the form of rapid-acting boluses before meals. However, for a variety of reasons, hospitalized patients do not eat very much. Thus, a 50-50 basal-bolus regimen may not be ideal for patients with poor oral intake.
In the Basal-PLUS trial, currently under way, we are comparing the safety and efficacy of a daily dose of basal insulin (glargine) plus correction doses of a rapid-acting insulin analogue (glulisine) on a sliding scale and a standard basal-bolus regimen in medical and surgical patients.
Does one glycemic target fit all patients?
Falciglia et al19 found an association between hyperglycemia and death in patients with unstable angina, arrhythmias, stroke, pneumonia, gastrointestinal bleeding, respiratory failure, sepsis, acute renal failure, and congestive heart failure. However, they found no such association in patients with chronic obstructive pulmonary disease, liver failure, diabetic ketoacidosis, gastrointestinal neoplasm, musculoskeletal disease, peripheral vascular disease with bypass, hip fracture, amputation due to peripheral vascular disease, or prostate surgery. Should patients in this second group be treated with a less-intensive insulin regimen?
What is the best regimen after hospital discharge?
We are conducting a prospective clinical trial to assess the impact of insulin after hospital discharge. Our current practice when a patient is discharged from the hospital is as follows:
- If the admission hemoglobin A1c level is less than 7%, we restart the previous outpatient treatment regimen of oral antidiabetic agents, or insulin, or both.
- If the admission hemoglobin A1c is between 7% and 9%, we restart the outpatient oral agents and continue glargine once daily at 50% to 80% of the hospital dose.
- If the hemoglobin A1c level is higher than 9%, we discharge the patient on a basal-bolus regimen at the same dosage as in the hospital. As an alternative, we could restart the oral agents and add glargine once daily at 80% of the hospital dose.
Hyperglycemia and diabetes mellitus are very common in hospitalized patients. Although more data are available on the prevalence of this problem and on how to manage it in the intensive care unit (ICU) than on regular hospital floors, the situation is changing. Information is emerging on the prevalence and impact of hyperglycemia and diabetes in the non-ICU setting, which is the focus of this paper.
HYPERGLYCEMIA IS COMMON AND PREDICTS POOR OUTCOMES
Cook et al,1 in a survey of 126 US hospitals, found that the prevalence of hyperglycemia (blood glucose > 180 mg/dL) was 46% in the ICU and 32% in regular wards.
Kosiborod et al2 reported that hyperglycemia (blood glucose > 140 mg/dL) was present in 78% of diabetic patients hospitalized with acute coronary syndrome and 26% of similar hospitalized nondiabetic patients.
Hyperglycemia is a common comorbidity in medical-surgical patients in community hospitals. Our group3 found that, in our hospital, 62% of patients were normoglycemic (ie, had a fasting blood glucose < 126 mg/dL or a random blood glucose < 200 mg/dL on two occasions), 26% had known diabetes, and 12% had new hyperglycemia. Further, new hyperglycemia was associated with a higher in-hospital death rate than the other two conditions.
Failure to identify diabetes is a predictor of rehospitalization. Robbins and Webb4 reported that 30.6% of those who had diabetes that was missed during hospitalization were readmitted within 30 days, compared with 9.4% of patients with diabetes first diagnosed during hospitalization.
WHAT DIAGNOSTIC CRITERIA SHOULD WE USE?
Blood glucose greater than 140 mg/dL
A consensus statement from the American Association of Clinical Endocrinologists (ACE) and the American Diabetes Association (ADA)5 defines in-hospital hyperglycemia as a blood glucose level greater than 140 mg/dL on admission or in the hospital. If the blood glucose is higher than this, the question arises as to whether the patient has preexisting diabetes or has stress hyperglycemia.
Hemoglobin A1c of 6.5% or higher
In view of the uncertainty as to whether a patient with an elevated blood glucose level has preexisting diabetes or stress hyperglycemia, upcoming guidelines will recommend measuring the hemoglobin A1c level if the blood glucose level is higher than 140 mg/dL.
A patient with an elevated blood glucose level (>140 mg/dL) whose hemoglobin A1c level is 6.5% or higher can be identified as having diabetes that preceded the hospitalization. Hemoglobin A1c testing can also be useful to assess glycemic control before admission and in designing an optional regimen at the time of discharge. In patients with newly recognized hyperglycemia, a hemoglobin A1c measurement can help differentiate patients with previously undiagnosed diabetes from those with stress-induced hyperglycemia.
Clinicians should keep in mind that a hemoglobin A1c cutoff of 6.5% identifies fewer cases of undiagnosed diabetes than does a high fasting glucose concentration, and that a level less than 6.5% does not rule out the diagnosis of diabetes. Several epidemiologic studies6 have reported a low sensitivity (44% to 66%) but a high specificity (76% to 99%) for hemoglobin A1c values higher than 6.5% in an outpatient population. The high specificity therefore supports the use of hemoglobin A1c to confirm the diagnosis of diabetes in patients with hyperglycemia, but the low sensitivity indicates that this test should not be used for universal screening in the hospital.
Many factors can influence the hemoglobin A1c level, such as anemia, iron deficiency, blood transfusions, hemolytic anemia, and renal failure.
Until now, if patients had hyperglycemia but no prior diagnosis of diabetes, the recommendation was for an oral 2-hour glucose tolerance test shortly after discharge to confirm the diagnosis of diabetes. Norhammar et al7 performed oral glucose tolerance tests in patients admitted with acute myocardial infarction, and Matz et al8 performed glucose tolerance tests in patients with acute stroke. They found that impaired glucose tolerance and undiagnosed type 2 diabetes were very common in these two groups. However, physicians rarely order oral glucose tolerance tests. We believe that hemoglobin A1c will be a better tool than an oral glucose tolerance test to confirm diabetes in hyperglycemic patients in the hospital setting.
WHAT IS THE ASSOCIATION BETWEEN HYPERGLYCEMIA AND OUTCOMES?
In 2,471 patients admitted to the hospital with community-acquired pneumonia, McAlister et al10 found that the rates of hospital complications and of death rose with blood glucose levels.
Falguera et al11 found that, in 660 episodes of community-acquired pneumonia, the rates of hospitalization, death, pleural effusion, and concomitant illnesses were all significantly higher in diabetic patients than in nondiabetic patients.
Noordzij et al12 performed a case-control study of 108,593 patients who underwent noncardiac surgery. The odds ratio for perioperative death was 1.19 (95% confidence interval [CI] 1.1–1.3) for every 1-mmol/L increase in the glucose level.
Frisch et al,13 in patients undergoing noncardiac surgery, found that the 30-day rates of death and of in-hospital complications were all higher in patients with diabetes than without diabetes.
Our group3 identified hyperglycemia as an independent marker of in-hospital death in patients with undiagnosed diabetes. The rates of death were 1.7% in those with normoglycemia, 3.0% in those with known diabetes, and 16.0% (P < .01) in those with new hyperglycemia.
The ACE/ADA consensus panel14 set the following glucose targets for patients in the non-ICU setting:
- Pre-meal blood glucose < 140 mg/dL
- Random blood glucose < 180 mg/dL.
On the other hand, hypoglycemia is also associated with adverse outcomes. Therefore, to avoid hypoglycemia, the insulin regimen should be reassessed if blood glucose levels fall below 100 mg/dL. New guidelines will suggest keeping the blood glucose between 100 and 140 mg/dL.
HOW SHOULD WE MANAGE HYPERGLYCEMIA IN THE NON-ICU SETTING?
The ACE/ADA guidelines recommend subcutaneous insulin therapy for most medical-surgical patients with diabetes, reserving intravenous insulin therapy for hyperglycemic crises and uncontrolled hyperglycemia.14
Oral antidiabetic agents are not generally recommended, as we have no data to support their use in the hospital. Another argument against using noninsulin therapies in the hospital is that sulfonylureas, especially glyburide (Diabeta, Micronase) are a major cause of hypoglycemia. Metformin (Glucophage) is contraindicated in decreased renal function, in hemodynamic instability, in surgical patients, and with the use of iodinated contrast dye. Thiazolidinediones are associated with edema and congestive heart failure, and they take up to 12 weeks to lower blood glucose levels. Alpha-glucosidase inhibitors are weak glucose-lowering agents. Also, therapies directed at glucagon-like-protein 1 can cause nausea and have a greater effect on postprandial glucose.14
The two main options for managing hyperglycemia and diabetes in the non-ICU setting are short-acting insulin on a sliding scale and basal-bolus therapy, the latter with either NPH plus regular insulin or long-acting plus rapid-acting insulin analogues.
Basal-bolus vs sliding scale insulin: The RABBIT-2 trial
In the RABBIT 2 trial (Randomized Basal Bolus Versus Sliding Scale Regular Insulin in Patients With Type 2 Diabetes Mellitus),15 our group compared the efficacy and safety of a basal-bolus regimen and a sliding-scale regimen in 130 hospitalized patients with type 2 diabetes treated with diet, with oral hypoglycemic agents, or with both. Oral antidiabetic drugs were discontinued on admission, and patients were randomized to one of the treatment groups.
In the basal-bolus group, the starting total daily dose was 0.4 U/kg/day if the blood glucose level on admission was between 140 and 200 mg/dL, or 0.5 U/kg/day if the glucose level was between 201 and 400 mg/dL. Half of the total daily dose was given as insulin glargine (Lantus) once daily, and the other half was given as insulin glulisine (Apidra) before meals. These doses were adjusted if the patient’s fasting or pre-meal blood glucose levels rose above 140 mg/dL or fell below 70 mg/dL.
The sliding-scale group received regular insulin four times daily (before meals and at bedtime) for glucose levels higher than 140 mg/dL; the higher the level, the more they got.
The basal-bolus regimen was better than sliding-scale regular insulin. At admission, the mean glucose values and hemoglobin A1c values were similar in both groups, but the mean glucose level on therapy was significantly lower in the basal-bolus group than in the sliding-scale group, 166 ± 32 mg/dL vs 193 ± 54 mg/dL, P < .001). About two-thirds of the basal-bolus group achieved a blood glucose target of less than 140 mg/dL, compared with only about one-third of the sliding-scale group. The basal-bolus group received more insulin, a mean of 42 units per day vs 12.5 units per day in the sliding-scale group. Yet the incidence of hypoglycemia was 3% in both groups.
NPH plus regular vs detemir plus aspart: The DEAN trial
Several long-acting insulin analogues are available and have a longer duration of action than NPH. Similarly, several newer rapid-acting analogues act more rapidly than regular insulin. Do these pharmacokinetic advantages matter? And do they justify the higher costs of the newer agents?
In the randomized Insulin Detemir Versus NPH Insulin in Hospitalized Patients With Diabetes (DEAN) trial,16 we compared two regimens: detemir plus aspart in a basal-bolus regimen, and NPH plus regular insulin in two divided doses, two-thirds of the total daily dose in the morning before breakfast and one-third before dinner, both doses in a ratio of two-thirds NPH and one-third regular, mixed in the same syringe. We recruited 130 patients with type 2 diabetes mellitus who were on oral hypoglycemic agents or insulin therapy.
NPH plus regular was just as good as detemir plus aspart in improving glycemic control. Blood glucose levels fell during the first day of therapy and were similar in both groups throughout the trial, as measured before breakfast, lunch, and dinner and at bedtime. The mean total daily insulin dose was not significantly different between treatment groups: 56 ± 45 units in the basal-bolus detemir-aspart group and 45 ± 32 units in the NPH-regular group. However, the basal-bolus group received significantly more short-acting insulin: 27 ± 20 units a day of aspart vs 18 ± 14 units of regular.
Somewhat fewer patients in the NPH-regular group had episodes of hypoglycemia, although the difference between groups was not statistically significant.
In a univariate analysis of the RABBIT-2 and DEAN trials,17 factors that predicted a blood glucose level less than 60 mg/dL were older age, lower body weight, higher serum creatinine level, and previous insulin therapy. Factors that were not predictive were the hemoglobin A1c level and the enrollment blood glucose level. Based on these data, we believe that to reduce the rate of hypoglycemia, lower insulin doses are needed in elderly patients and patients with renal impairment, and that if patients have been taking insulin before they come to the hospital, the dose should be cut back by about 25% while they are hospitalized.
Basal-bolus vs sliding-scale insulin for surgical patients: The RABBIT 2 Surgery trial
Does better glucose control in surgical patients affect outcomes in patients undergoing general surgery? To find out, we performed a prospective, multicenter, randomized, open-label trial in general surgery patients not in the ICU.18 We recruited and randomized 211 patients with type 2 diabetes who were on diet therapy or oral hypoglycemic agents or insulin in low doses (< 0.4 U/kg/day).
Oral drugs were discontinued on admission, and patients were randomized to receive either a basal-bolus regimen of glargine plus glulisine or regular insulin on a sliding scale. The basal-bolus group got 0.5 U/kg/day, half of it as glargine once daily and half as glulisine before meals. The total daily dose was reduced to 0.3 U/kg/day in patients age 70 and older or who had a serum creatinine level of 2.0 mg/dL or higher.
The goal was to maintain fasting and pre-meal glucose concentrations between 100 and 140 mg/dL. The total daily dose was raised by 10% (mostly in the glargine dose) if the blood glucose level was in the range of 141 to 180 mg/dL, and by 20% if the glucose level was higher than 181 mg/dL. The dose was decreased by 10% for glucose levels between 70 and 99 mg/dL, was decreased by 20% if the glucose level was between 40 and 69, and was held if the glucose level was lower than 40 mg/dL. If a patient was not able to eat, insulin glulisine was held until meals were resumed.
The sliding-scale group received regular insulin four times a day for blood glucose levels higher than 140 mg/dL.
The primary outcomes measured were the difference between groups in mean daily blood glucose concentration and a composite of hospital complications including postoperative wound infection, pneumonia, respiratory failure, acute renal failure, and bacteremia. Secondary outcomes were differences between groups in mean fasting and pre-meal blood glucose, number of hypoglycemic episodes (blood glucose < 70 mg/dL), hyperglycemic episodes (blood glucose > 200 mg/dL), length of hospital stay, need for intensive care, and rate of complications including wound infection, pneumonia, acute renal failure, and death.
Blood glucose levels were significantly lower in the basal-bolus group through the first 7 days after randomization, as measured before breakfast, lunch, and dinner, and at bedtime, and then they converged.
More patients in the sliding-scale group had hospital complications, 26 vs 9, P = .003. On the other hand, more patients in the basal-bolus group had episodes of hypoglycemia: 24 (23%) vs 5 (4.7%) had episodes of less than 70 mg/dL (P < .001), 12 (12%) vs 2 (1.9%) had episodes of less than 60 mg/dL (P = .005), and 4 (3.8%) vs 0 had episodes of less than 40 mg/dL (P = .057). The mean total daily dose of insulin was 33.4 units in the basal-bolus group and 12.3 units in the sliding-scale group.
WHAT HAVE WE LEARNED?
Don’t use a sliding-scale regimen as a single agent in patients with diabetes. Glycemic control is better with a basal-bolus regimen than with a sliding-scale regimen, and a basal-bolus insulin regimen is preferred for most patients with hyperglycemia.
The old human insulins (ie, regular and NPH) are still good and improve glycemic control as well as the new basal insulin analogues (detemir and aspart) do.
Improved control may reduce the rate of hospital complications, according to preliminary evidence. More studies are under way.
One size does not fit all. Those who are elderly or who have impaired renal function should receive lower doses of insulin, eg, 0.3 U/kg/day instead of 0.5 U/kg/day. Those who are on insulin should have their dose decreased when they are admitted to the hospital. Perhaps lean patients with type 2 diabetes should also have a lower dose.
Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of NPH plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
Patients treated with insulin at home should continue to receive insulin therapy in the hospital. However, the insulin dosage should be reduced by about 25% to allow for lower food intake.
QUESTIONS FOR FURTHER STUDY
Should we modify the standard basal-bolus regimen?
In a typical basal-bolus regimen, patients get 50% of their total daily insulin dose in the form of a basal injection and 50% in the form of rapid-acting boluses before meals. However, for a variety of reasons, hospitalized patients do not eat very much. Thus, a 50-50 basal-bolus regimen may not be ideal for patients with poor oral intake.
In the Basal-PLUS trial, currently under way, we are comparing the safety and efficacy of a daily dose of basal insulin (glargine) plus correction doses of a rapid-acting insulin analogue (glulisine) on a sliding scale and a standard basal-bolus regimen in medical and surgical patients.
Does one glycemic target fit all patients?
Falciglia et al19 found an association between hyperglycemia and death in patients with unstable angina, arrhythmias, stroke, pneumonia, gastrointestinal bleeding, respiratory failure, sepsis, acute renal failure, and congestive heart failure. However, they found no such association in patients with chronic obstructive pulmonary disease, liver failure, diabetic ketoacidosis, gastrointestinal neoplasm, musculoskeletal disease, peripheral vascular disease with bypass, hip fracture, amputation due to peripheral vascular disease, or prostate surgery. Should patients in this second group be treated with a less-intensive insulin regimen?
What is the best regimen after hospital discharge?
We are conducting a prospective clinical trial to assess the impact of insulin after hospital discharge. Our current practice when a patient is discharged from the hospital is as follows:
- If the admission hemoglobin A1c level is less than 7%, we restart the previous outpatient treatment regimen of oral antidiabetic agents, or insulin, or both.
- If the admission hemoglobin A1c is between 7% and 9%, we restart the outpatient oral agents and continue glargine once daily at 50% to 80% of the hospital dose.
- If the hemoglobin A1c level is higher than 9%, we discharge the patient on a basal-bolus regimen at the same dosage as in the hospital. As an alternative, we could restart the oral agents and add glargine once daily at 80% of the hospital dose.
- Cook CB, Kongable GL, Potter DJ, Abad VJ, Leija DE, Anderson M. Inpatient glucose control: a glycemic survey of 126 U.S. hospitals. J Hosp Med 2009; 4:E7–E14.
- Kosiborod M, Inzucchi S, Clark B, et al. National patterns of glucose control among patients hospitalized with acute myocardial infarction [abstract]. J Am Coll Cardiol 2007; 49:283A–284A.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Robbins JM, Webb DA. Diagnosing diabetes and preventing rehospitalizations: the urban diabetes study. Med Care 2006; 44:292–296.
- Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Saudek D, Herman WH, Sacks DB, Bergenstal RM, Edelman D, Davidson MB. A new look at screening and diagnosing diabetes mellitus. J Clin Endocrinol Metab 2008; 93:2447–2453.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 2002; 359:2140–2144.
- Matz K, Keresztes K, Tatschl C, et al. Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 2006; 29:792–797.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33(suppl 1):S62–S69.
- McAlister FA, Majumdar SR, Blitz S, Rowe BH, Romney J, Marrie TJ. The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community-acquired pneumonia. Diabetes Care 2005; 28:810–815.
- Falguera M, Pifarre R, Martin A, Sheikh A, Moreno A. Etiology and outcome of community-acquired pneumonia in patients with diabetes mellitus. Chest 2005; 128:3233–3239.
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol 2007; 156:137–142.
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care 2010; 33:1783–1788.
- Moghissi ES, Korythowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocrine Pract 2009; 15:1–17.
- Umpierrrez GE, Smiley D, Zisman A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care 2007; 30:2181–2186.
- Umpierrez GE, Hor T, Smiley D, et al. Comparison of inpatient insulin regimens with detemir plus aspart versus neutral protamine Hagedorn plus regular in medical patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:564–569.
- Umpierrez GE, Smiley D, Umpierrez D, Ceron M, Temponi A. Hypoglycemic events during subcutaneous insulin therapy in type 2 diabetes (abstract). Presented at American Diabetes Association 69th Scientific Sessions, New Orleans, LA, June 5–9, 2009.
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care 2011; 34:256–261.
- Falciglia M, Freyberg RW, Almenoff PL, D’Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009; 37:3001–3009.
- Cook CB, Kongable GL, Potter DJ, Abad VJ, Leija DE, Anderson M. Inpatient glucose control: a glycemic survey of 126 U.S. hospitals. J Hosp Med 2009; 4:E7–E14.
- Kosiborod M, Inzucchi S, Clark B, et al. National patterns of glucose control among patients hospitalized with acute myocardial infarction [abstract]. J Am Coll Cardiol 2007; 49:283A–284A.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Robbins JM, Webb DA. Diagnosing diabetes and preventing rehospitalizations: the urban diabetes study. Med Care 2006; 44:292–296.
- Moghissi ES, Korytkowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Saudek D, Herman WH, Sacks DB, Bergenstal RM, Edelman D, Davidson MB. A new look at screening and diagnosing diabetes mellitus. J Clin Endocrinol Metab 2008; 93:2447–2453.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 2002; 359:2140–2144.
- Matz K, Keresztes K, Tatschl C, et al. Disorders of glucose metabolism in acute stroke patients: an underrecognized problem. Diabetes Care 2006; 29:792–797.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33(suppl 1):S62–S69.
- McAlister FA, Majumdar SR, Blitz S, Rowe BH, Romney J, Marrie TJ. The relation between hyperglycemia and outcomes in 2,471 patients admitted to the hospital with community-acquired pneumonia. Diabetes Care 2005; 28:810–815.
- Falguera M, Pifarre R, Martin A, Sheikh A, Moreno A. Etiology and outcome of community-acquired pneumonia in patients with diabetes mellitus. Chest 2005; 128:3233–3239.
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol 2007; 156:137–142.
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care 2010; 33:1783–1788.
- Moghissi ES, Korythowski MT, DiNardo M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Endocrine Pract 2009; 15:1–17.
- Umpierrrez GE, Smiley D, Zisman A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care 2007; 30:2181–2186.
- Umpierrez GE, Hor T, Smiley D, et al. Comparison of inpatient insulin regimens with detemir plus aspart versus neutral protamine Hagedorn plus regular in medical patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:564–569.
- Umpierrez GE, Smiley D, Umpierrez D, Ceron M, Temponi A. Hypoglycemic events during subcutaneous insulin therapy in type 2 diabetes (abstract). Presented at American Diabetes Association 69th Scientific Sessions, New Orleans, LA, June 5–9, 2009.
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care 2011; 34:256–261.
- Falciglia M, Freyberg RW, Almenoff PL, D’Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009; 37:3001–3009.
KEY POINTS
- Hyperglycemia and undiagnosed diabetes are very common in hospitalized patients and are associated with poorer outcomes.
- Hospitalized patients should be screened for diabetes with a blood glucose measurement. Those who have a value of 140 mg/dL or higher should be tested for hemoglobin A1c. A value higher than 6.5% is very specific for diabetes, although not very sensitive for it.
- Most hospitalized patients with diabetes and elevated blood glucose values (or hyperglycemia) should receive subcutaneous insulin treatment with a basal-bolus regimen or a multidose combination of neutral protamine Hagedorn (NPH) plus regular insulin. Selected patients with severe insulin resistance and persistent hyperglycemia despite subcutaneous insulin may benefit from continuous intravenous insulin infusion.
- Sliding-scale insulin as a single form of therapy in patients with diabetes is undesirable.
Subphrenic abscess from a perforated duodenal ulcer
A 55-year-old man presented after 3 weeks of sharp epigastric pain radiating to the right upper quadrant, fever, and generalized weakness. He had a history of significant heroin and cocaine abuse and was currently in a methadone maintenance program. He was not taking any nonsteroidal anti-inflammatory drugs.
His temperature was 38.7°C (101.7°F). He had tenderness in the right upper quadrant but no rebound tenderness.
Laboratory studies revealed an elevated white blood cell count of 14.9 × 109/L (reference range 4.5–11.0) with 13% band cells, a normal lipase level, and normal liver function tests. Urine toxicology testing was positive for cocaine.
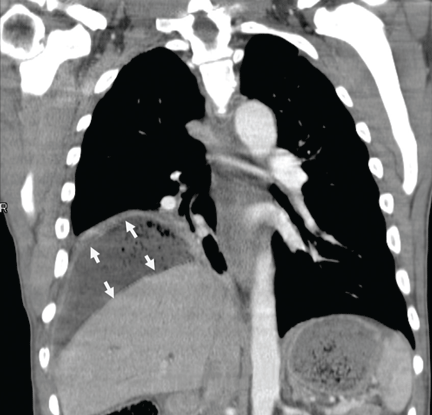
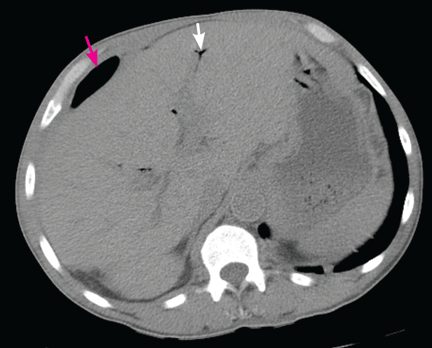
To investigate the cause of the abscess, we obtained an upper gastrointestinal radiographic series with oral contrast. This showed leaking of contrast from a perforated ulcer in the proximal duodenum (Figure 3).
TREATING PERFORATED ULCER AND ITS COMPLICATIONS
Perforated ulcers usually present with an acute abdomen and are life-threatening unless immediately recognized and treated. A more insidious presentation, as reported by Wong et al1 and as seen in our patient, presents additional diagnostic challenges. Our patient’s presentation may have been insidious and relatively benign because the perforation was not a free perforation (ie, the bowel contents were not freely spilling into the abdominal cavity), and because the spillage and inflammation were confined to an abscess cavity. Also, he was taking methadone 60 mg per day, which may have further masked the symptoms.
Perforated duodenal ulcers are usually treated surgically. Nonoperative management—ie, fluid resuscitation, nasogastric tube aspiration, and intravenous antibiotics—has been described,2 but the hemodynamic status and abdominal findings need to be continually monitored to detect any deterioration in the patient’s condition.
Initial management of the abscess with early percutaneous drainage and empiric intravenous antibiotics to cover enteric flora and anaerobes may suffice if there is no fistula.3 Also, in a series of 62 patients with subphrenic abscess managed by percutaneous drainage, Mueller et al4 found that this method was successful in most of the patients, noting that small-bowel perforation required the longest drainage period, generally more than 10 days.4
Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs are responsible for the vast majority of duodenal ulcers. Serum H pylori antibody testing should be done, and if it is positive, treatment should be started. This patient did not have H pylori infection as an inpatient. He received metronidazole to cover the anaerobes and a proton pump inhibitor to treat the ulcer.
Although upper endoscopy was contraindicated in our patient because of his perforation, it was warranted for follow-up. Medical management with a proton pump inhibitor in high doses helped ulcer healing by reducing acid and gastric secretions. Octreotide had similar benefits, including reducing pancreatic and biliary secretion. This conservative management resulted in healing of the ulcer and early closure of the enteric fistula.
- Wong CH, Chow PK, Ong HS, Chan WH, Khin LW, Soo KC. Posterior perforation of peptic ulcers: presentation and outcome of an uncommon surgical emergency. Surgery 2004; 135:321–325.
- Berne TV, Donovan AJ. Nonoperative treatment of perforated duodenal ulcer. Arch Surg 1989; 124:830–832.
- Flancbaum L, Nosher JL, Brolin RE. Percutaneous catheter drainage of abdominal abscesses associated with perforated viscus. Am Surg 1990; 56:52–56.
- Mueller PR, Simeone JF, Butch RJ, et al. Percutaneous drainage of subphrenic abscess: a review of 62 patients. AJR Am J Roentgenol 1986; 147:1237–1240.
A 55-year-old man presented after 3 weeks of sharp epigastric pain radiating to the right upper quadrant, fever, and generalized weakness. He had a history of significant heroin and cocaine abuse and was currently in a methadone maintenance program. He was not taking any nonsteroidal anti-inflammatory drugs.
His temperature was 38.7°C (101.7°F). He had tenderness in the right upper quadrant but no rebound tenderness.
Laboratory studies revealed an elevated white blood cell count of 14.9 × 109/L (reference range 4.5–11.0) with 13% band cells, a normal lipase level, and normal liver function tests. Urine toxicology testing was positive for cocaine.
To investigate the cause of the abscess, we obtained an upper gastrointestinal radiographic series with oral contrast. This showed leaking of contrast from a perforated ulcer in the proximal duodenum (Figure 3).
TREATING PERFORATED ULCER AND ITS COMPLICATIONS
Perforated ulcers usually present with an acute abdomen and are life-threatening unless immediately recognized and treated. A more insidious presentation, as reported by Wong et al1 and as seen in our patient, presents additional diagnostic challenges. Our patient’s presentation may have been insidious and relatively benign because the perforation was not a free perforation (ie, the bowel contents were not freely spilling into the abdominal cavity), and because the spillage and inflammation were confined to an abscess cavity. Also, he was taking methadone 60 mg per day, which may have further masked the symptoms.
Perforated duodenal ulcers are usually treated surgically. Nonoperative management—ie, fluid resuscitation, nasogastric tube aspiration, and intravenous antibiotics—has been described,2 but the hemodynamic status and abdominal findings need to be continually monitored to detect any deterioration in the patient’s condition.
Initial management of the abscess with early percutaneous drainage and empiric intravenous antibiotics to cover enteric flora and anaerobes may suffice if there is no fistula.3 Also, in a series of 62 patients with subphrenic abscess managed by percutaneous drainage, Mueller et al4 found that this method was successful in most of the patients, noting that small-bowel perforation required the longest drainage period, generally more than 10 days.4
Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs are responsible for the vast majority of duodenal ulcers. Serum H pylori antibody testing should be done, and if it is positive, treatment should be started. This patient did not have H pylori infection as an inpatient. He received metronidazole to cover the anaerobes and a proton pump inhibitor to treat the ulcer.
Although upper endoscopy was contraindicated in our patient because of his perforation, it was warranted for follow-up. Medical management with a proton pump inhibitor in high doses helped ulcer healing by reducing acid and gastric secretions. Octreotide had similar benefits, including reducing pancreatic and biliary secretion. This conservative management resulted in healing of the ulcer and early closure of the enteric fistula.
A 55-year-old man presented after 3 weeks of sharp epigastric pain radiating to the right upper quadrant, fever, and generalized weakness. He had a history of significant heroin and cocaine abuse and was currently in a methadone maintenance program. He was not taking any nonsteroidal anti-inflammatory drugs.
His temperature was 38.7°C (101.7°F). He had tenderness in the right upper quadrant but no rebound tenderness.
Laboratory studies revealed an elevated white blood cell count of 14.9 × 109/L (reference range 4.5–11.0) with 13% band cells, a normal lipase level, and normal liver function tests. Urine toxicology testing was positive for cocaine.
To investigate the cause of the abscess, we obtained an upper gastrointestinal radiographic series with oral contrast. This showed leaking of contrast from a perforated ulcer in the proximal duodenum (Figure 3).
TREATING PERFORATED ULCER AND ITS COMPLICATIONS
Perforated ulcers usually present with an acute abdomen and are life-threatening unless immediately recognized and treated. A more insidious presentation, as reported by Wong et al1 and as seen in our patient, presents additional diagnostic challenges. Our patient’s presentation may have been insidious and relatively benign because the perforation was not a free perforation (ie, the bowel contents were not freely spilling into the abdominal cavity), and because the spillage and inflammation were confined to an abscess cavity. Also, he was taking methadone 60 mg per day, which may have further masked the symptoms.
Perforated duodenal ulcers are usually treated surgically. Nonoperative management—ie, fluid resuscitation, nasogastric tube aspiration, and intravenous antibiotics—has been described,2 but the hemodynamic status and abdominal findings need to be continually monitored to detect any deterioration in the patient’s condition.
Initial management of the abscess with early percutaneous drainage and empiric intravenous antibiotics to cover enteric flora and anaerobes may suffice if there is no fistula.3 Also, in a series of 62 patients with subphrenic abscess managed by percutaneous drainage, Mueller et al4 found that this method was successful in most of the patients, noting that small-bowel perforation required the longest drainage period, generally more than 10 days.4
Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs are responsible for the vast majority of duodenal ulcers. Serum H pylori antibody testing should be done, and if it is positive, treatment should be started. This patient did not have H pylori infection as an inpatient. He received metronidazole to cover the anaerobes and a proton pump inhibitor to treat the ulcer.
Although upper endoscopy was contraindicated in our patient because of his perforation, it was warranted for follow-up. Medical management with a proton pump inhibitor in high doses helped ulcer healing by reducing acid and gastric secretions. Octreotide had similar benefits, including reducing pancreatic and biliary secretion. This conservative management resulted in healing of the ulcer and early closure of the enteric fistula.
- Wong CH, Chow PK, Ong HS, Chan WH, Khin LW, Soo KC. Posterior perforation of peptic ulcers: presentation and outcome of an uncommon surgical emergency. Surgery 2004; 135:321–325.
- Berne TV, Donovan AJ. Nonoperative treatment of perforated duodenal ulcer. Arch Surg 1989; 124:830–832.
- Flancbaum L, Nosher JL, Brolin RE. Percutaneous catheter drainage of abdominal abscesses associated with perforated viscus. Am Surg 1990; 56:52–56.
- Mueller PR, Simeone JF, Butch RJ, et al. Percutaneous drainage of subphrenic abscess: a review of 62 patients. AJR Am J Roentgenol 1986; 147:1237–1240.
- Wong CH, Chow PK, Ong HS, Chan WH, Khin LW, Soo KC. Posterior perforation of peptic ulcers: presentation and outcome of an uncommon surgical emergency. Surgery 2004; 135:321–325.
- Berne TV, Donovan AJ. Nonoperative treatment of perforated duodenal ulcer. Arch Surg 1989; 124:830–832.
- Flancbaum L, Nosher JL, Brolin RE. Percutaneous catheter drainage of abdominal abscesses associated with perforated viscus. Am Surg 1990; 56:52–56.
- Mueller PR, Simeone JF, Butch RJ, et al. Percutaneous drainage of subphrenic abscess: a review of 62 patients. AJR Am J Roentgenol 1986; 147:1237–1240.
Immune thrombocytopenia: No longer ‘idiopathic’
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.

FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
KEY POINTS
- ITP is defined as an isolated platelet count of less than 100 × 109/L (100,000/μL) and usually presents without symptoms.
- Patients without symptoms who have a platelet count above 30 × 109/L should generally not be treated unless they have an increased risk of bleeding.
- Recent studies suggest that viruses and other pathogens play an important role in secondary ITP.
- Initially, corticosteroids are usually given as prednisone (1–2 mg/kg/day, then tapered), though recent studies suggest that dexamethasone pulses (40 mg/day for 4 days) may provide more durable responses when used in this setting.
- Thrombopoietic agents are important new treatments, although their place in the overall therapy of ITP has not been established.
The pain of cholesterol-lowering therapy

Given the perception that statin myopathy is common, the incidence of significant myalgias in clinical trials is surprisingly low and similar to that with placebo (generally less than 5%), and that of myositis or rhabdomyolysis is much rarer.
In this issue of the Journal, Dr. Genaro Fernandez and colleagues discuss possible reasons for the discrepancy between the prevalence of statin-associated myalgias in clinical trials vs real practice. They suggest that patients more likely to develop myalgias are weeded out in the screening phase of clinical trials and that the trials may be too small and too short to capture this information. Yet in practice, many patients develop muscle pain shortly after starting statin therapy. I suggest another explanation—ie, that volunteers in clinical trials want to take the medication, while in the clinic my patients are reluctant to take one more medication and have trepidations about starting one that they “know” causes muscle pain.
But I don’t think all statin myopathy is due to the power of suggestion. Some patients clearly have drug-elicited elevations in creatine kinase (CK), and others (including me) experience significant myalgias with one statin but can tolerate another.
A challenge in my rheumatology clinic is distinguishing statin myopathy from other underlying problems in patients referred for evaluation of pain, weakness, or elevated CK. I first establish a temporal relationship between the drug initiation and the start of symptoms, and I look for other drugs or possible drug interactions that could be causing the problem, such as colchicine vacuolar myopathy in the setting of newly initiated statin therapy. I look for an alternative explanation for the pain syndrome, such as upper-arm pain and physical findings that suggest rotator cuff disease, or lateral hip-area pain due to bursitis. In some patients, statins may pose a metabolic challenge that unmasks (or brings to the physician’s attention) an underlying biochemical disorder of the muscle, such as myotonic dystrophy or even polymyositis.
Dr. Fernandez et al offer sound advice, as they suggest keeping an open diagnostic mind when evaluating patients with apparent statin myopathy. In particular, with these authors, I urge you to perform a careful personal and family history and a focused examination, ask about vigorous physical exercise, check the CK, and withhold and then rechallenge with the statin before ordering a slew of serologic and metabolic tests.
Given the perception that statin myopathy is common, the incidence of significant myalgias in clinical trials is surprisingly low and similar to that with placebo (generally less than 5%), and that of myositis or rhabdomyolysis is much rarer.
In this issue of the Journal, Dr. Genaro Fernandez and colleagues discuss possible reasons for the discrepancy between the prevalence of statin-associated myalgias in clinical trials vs real practice. They suggest that patients more likely to develop myalgias are weeded out in the screening phase of clinical trials and that the trials may be too small and too short to capture this information. Yet in practice, many patients develop muscle pain shortly after starting statin therapy. I suggest another explanation—ie, that volunteers in clinical trials want to take the medication, while in the clinic my patients are reluctant to take one more medication and have trepidations about starting one that they “know” causes muscle pain.
But I don’t think all statin myopathy is due to the power of suggestion. Some patients clearly have drug-elicited elevations in creatine kinase (CK), and others (including me) experience significant myalgias with one statin but can tolerate another.
A challenge in my rheumatology clinic is distinguishing statin myopathy from other underlying problems in patients referred for evaluation of pain, weakness, or elevated CK. I first establish a temporal relationship between the drug initiation and the start of symptoms, and I look for other drugs or possible drug interactions that could be causing the problem, such as colchicine vacuolar myopathy in the setting of newly initiated statin therapy. I look for an alternative explanation for the pain syndrome, such as upper-arm pain and physical findings that suggest rotator cuff disease, or lateral hip-area pain due to bursitis. In some patients, statins may pose a metabolic challenge that unmasks (or brings to the physician’s attention) an underlying biochemical disorder of the muscle, such as myotonic dystrophy or even polymyositis.
Dr. Fernandez et al offer sound advice, as they suggest keeping an open diagnostic mind when evaluating patients with apparent statin myopathy. In particular, with these authors, I urge you to perform a careful personal and family history and a focused examination, ask about vigorous physical exercise, check the CK, and withhold and then rechallenge with the statin before ordering a slew of serologic and metabolic tests.
Given the perception that statin myopathy is common, the incidence of significant myalgias in clinical trials is surprisingly low and similar to that with placebo (generally less than 5%), and that of myositis or rhabdomyolysis is much rarer.
In this issue of the Journal, Dr. Genaro Fernandez and colleagues discuss possible reasons for the discrepancy between the prevalence of statin-associated myalgias in clinical trials vs real practice. They suggest that patients more likely to develop myalgias are weeded out in the screening phase of clinical trials and that the trials may be too small and too short to capture this information. Yet in practice, many patients develop muscle pain shortly after starting statin therapy. I suggest another explanation—ie, that volunteers in clinical trials want to take the medication, while in the clinic my patients are reluctant to take one more medication and have trepidations about starting one that they “know” causes muscle pain.
But I don’t think all statin myopathy is due to the power of suggestion. Some patients clearly have drug-elicited elevations in creatine kinase (CK), and others (including me) experience significant myalgias with one statin but can tolerate another.
A challenge in my rheumatology clinic is distinguishing statin myopathy from other underlying problems in patients referred for evaluation of pain, weakness, or elevated CK. I first establish a temporal relationship between the drug initiation and the start of symptoms, and I look for other drugs or possible drug interactions that could be causing the problem, such as colchicine vacuolar myopathy in the setting of newly initiated statin therapy. I look for an alternative explanation for the pain syndrome, such as upper-arm pain and physical findings that suggest rotator cuff disease, or lateral hip-area pain due to bursitis. In some patients, statins may pose a metabolic challenge that unmasks (or brings to the physician’s attention) an underlying biochemical disorder of the muscle, such as myotonic dystrophy or even polymyositis.
Dr. Fernandez et al offer sound advice, as they suggest keeping an open diagnostic mind when evaluating patients with apparent statin myopathy. In particular, with these authors, I urge you to perform a careful personal and family history and a focused examination, ask about vigorous physical exercise, check the CK, and withhold and then rechallenge with the statin before ordering a slew of serologic and metabolic tests.
Statin myopathy: A common dilemma not reflected in clinical trials
When a patient taking a statin complains of muscle aches, is he or she experiencing statin-induced myopathy or some other problem? Should statin therapy be discontinued?
Statins have proven efficacy in preventing heart attacks and death,1 and they are the most widely prescribed drugs worldwide. Nevertheless, they remain underused, with only 50% of those who would benefit from being on a statin receiving one.2,3 In addition, at least 25% of adults who start taking statins stop taking them by 6 months, and up to 60% stop by 2 years.4
Patient and physician fears about myopathy remain a key reason for stopping. Myopathy, a known side effect of statins, is rare in randomized controlled trials, but less so in observational studies and clinical experience. This discrepancy between clinical trials and clinical experience reduces confidence in lipid-lowering therapy and contributes to its underuse.
This review emphasizes clinical aspects of statin myopathy that are important to the practicing physician. We will define myopathy, review its purported mechanisms, and describe a clinical approach to patients with possible toxicity, including risk factors, physical findings, and consideration of alternate diagnoses. Since there is no single test to diagnose statin-induced myopathy, we offer a framework to aid clinicians in stratifying patients based on the likelihood that their symptoms are due to statin toxicity weighed against the likelihood that they will benefit from statin therapy.
DEFINITIONS DIFFER
Little consensus exists on how to define the adverse muscle effects of statins,5 which may contribute to the underdiagnosis of this complication. The magnitude of creatine kinase (CK) elevation required to define rhabdomyolysis has increased from 500 IU/L in 1982,6 to 1,000 IU/L in 1988,7 to 50 times the upper limit of normal in one current definition.8 The American Heart Association, the American College of Cardiology, the National Heart Lung and Blood Institute,9 the National Lipid Association,8 and the US Food and Drug Administration10 all differ in their definitions.
Our definitions
For the purpose of this article, we offer the following definitions:
Myalgia—muscle weakness, soreness, tenderness, stiffness, cramping, or aching, either at rest or with exertion, without any elevation in CK.
Myositis—elevated CK with or without muscle symptoms. The “-itis” suffix is unfortunate since myositis does not correspond to inflammation on biopsy.
Rhabdomyolysis—muscle symptoms with a CK level 10 times the upper limit of normal or higher. Evidence of renal dysfunction is not required for the diagnosis, as preexisting renal disease and hydration status are more closely related to kidney damage than the degree of muscle injury.11
STATIN MYOPATHY IS MORE COMMON IN THE REAL WORLD THAN IN TRIALS
The incidence of statin-induced myopathy is significantly lower in randomized controlled trials of statin efficacy than in observational studies of real-world patients. In randomized clinical trials, myalgia was reported in 1% to 5% of patients in the statin groups and placebo groups alike,9,12 whereas clinical practice would suggest it is more common.
Why is statin-induced myopathy so uncommon in clinical trials?
A reason may be that patients in clinical trials are carefully screened. To minimize toxicity, the clinical trials of statins excluded patients with renal insufficiency, hepatic insufficiency, a history of muscular complaints, and poorly controlled diabetes, as well as patients taking drugs with possible interactions. Large efficacy trials have excluded up to 30% of the participants in active prerandomization phases.13,14
Another reason is that these trials were designed to assess the efficacy of statins and were not sensitive to adverse effects like muscle pain. When they looked at myopathy, they focused on rhabdomyolysis—the most severe form—rather than on myalgia, fatigue, or other minor muscle complaints.15 Additionally, most trials enrolled too few patients and did not have long enough follow-up to reveal infrequent toxicities.
Despite the strict criteria, a significant number of trial patients discontinued statin therapy during the study period. In the Treat to New Targets (TNT) trial, 5% of patients in both the high- and low-dose atorvastatin (Lipitor) groups experienced muscle toxicity, even though 35% of eligible patients had been excluded during the open-label run-in phase.14
Also, physicians may overlook and patients may fail to report symptoms such as fatigue, malaise, or dyspnea that are not commonly accepted as signs of statin toxicity.16
Findings from observational studies
Observational studies in nonselected outpatients show a higher frequency of muscle complaints in the statin groups than in the control groups. These studies suggest the frequency of statin myopathy is 9% to 20%.17–19
The Prediction of Muscular Risk in Observational Conditions (PRIMO) study20 was one of the largest and best-defined observational studies of muscular symptoms in an unselected population. It included 7,924 French outpatients with hypercholesterolemia, ages 18 to 75 years, on high-dose statins for 3 or more months before the study. Daily statin regimens included atorvastatin 40 to 80 mg, fluvastatin (Lescol) 80 mg, pravastatin (Pravachol) 40 mg, and simvastatin (Zocor) 40 to 80 mg. In this study, 10.5% of patients reported muscle-related symptoms.
Buettner et al,21 in another cross-sectional study, interviewed and examined 3,580 adults over age 40. Of those taking statins, 22% reported having had musculoskeletal pain in at least one anatomic region in the last 30 days, compared with 16.7% of those not taking a statin.
In the United States, where an estimated 33 million adults use statins, musculoskeletal pain can be expected to occur in 7 million people, likely induced by statin therapy in 25% of cases.22
WHAT CAUSES STATIN MYOPATHY?
The causes of statin-induced myopathy are poorly understood.
Historically, statin-induced toxicity was thought to be caused by inhibition of the synthesis of mevalonate, leading to depletion of its metabolites, such as cholesterol, isoprenoids, and ubiquinone (coenzyme Q10). Depletion of intracellular cholesterol may lead to abnormal membrane behaviors; depletion of isoprenoids may affect intracellular signaling; depletion of coenzyme Q10 may in turn reduce mitochondrial respiratory function. Genetic factors may also play a role, contributing to pharmacokinetics and predisposing metabolic muscle disorders.23,24
Statin-induced myopathy seems to be different in randomized efficacy trials than in the clinical setting. In randomized trials, the mechanism appears to involve abnormal pharmacokinetics. The participants are carefully selected to have a low risk of muscle toxicity, but some develop toxicity when statin levels are elevated because of reduced drug breakdown and metabolism. However, in clinical practice, where a much larger group of unselected patients are exposed to statins, toxicity appears to be more related to a metabolic predisposition.25–27
Multiple minor metabolic abnormalities have been described in the muscles of patients with statin-induced muscle toxicity, suggesting that some patients have a predisposition for muscle complaints.23 About 25% of patients with recurrent rhabdomyolysis irrespective of lipid-lowering therapy have an underlying metabolic muscle disorder.28 In these vulnerable patients, minor metabolic defects are exacerbated by any agent that reduces the delivery of fat substrate to muscle, leading to muscle starvation.
This concept explains why patients may develop the same muscle complaint on different lipid-lowering agents.29,30 It also may explain why rhabdomyolysis can occur after apheresis of lipids, when no drugs have been given.31 In vulnerable patients, muscle toxicity may result from any agent that lessens the lipid substrate available to muscle rather than from the reduction of products downstream from mevalonate by statins.
SOME STATINS MAY BE LESS TOXIC
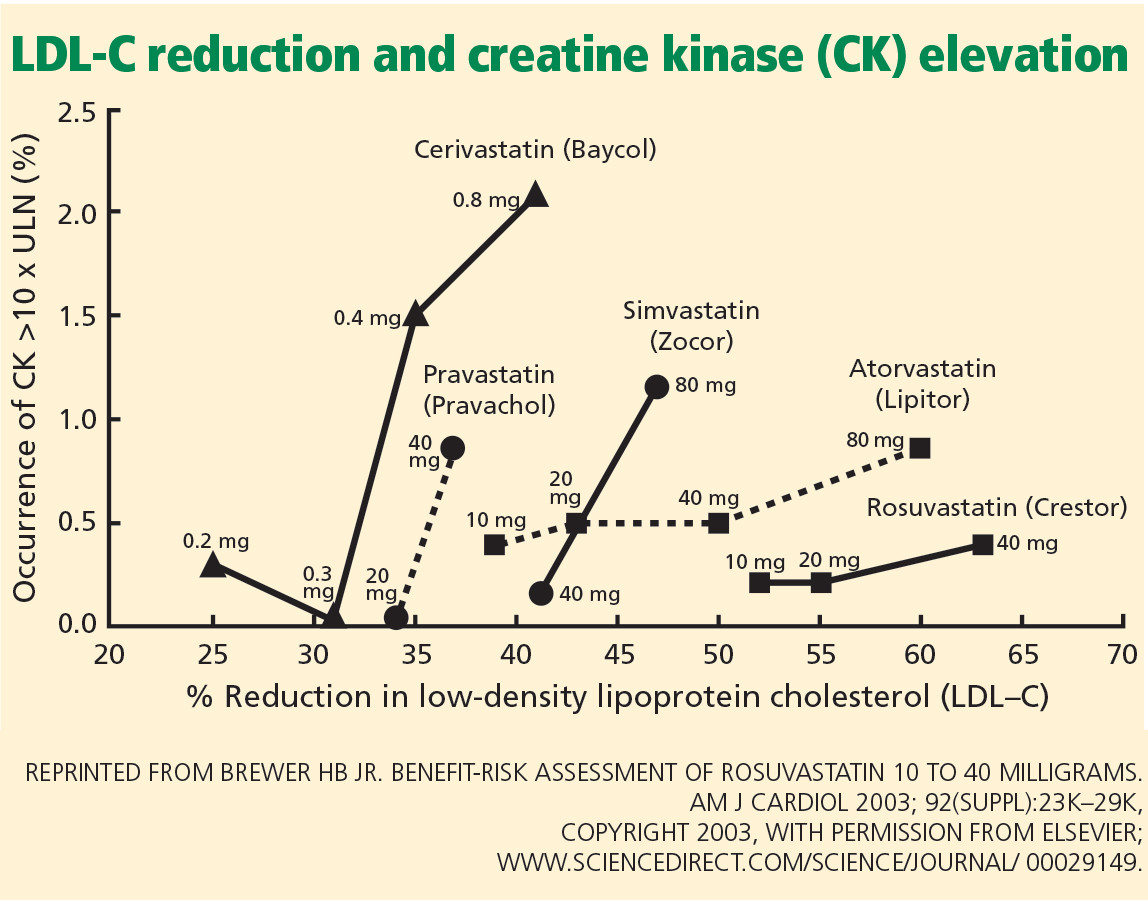
In the PRIMO study, muscle-related symptoms occurred with the various regimens as follows:
- Fluvastatin XL 40 mg—5.1%
- Pravastatin 40 mg—10.9%
- Atorvastatin 40 to 80 mg—14.9%
- Simvastatin 40 to 80 mg—18.2%.
Others have also shown that fluvastatin contributed to the smallest number of reported cases of rhabdomyolysis among statins: 55 (1.6%) of 3,339 cases.34
More recent studies indicate that rosuvastatin (Crestor), the most hydrophilic statin, may be well tolerated in those who do not tolerate other statins,35–37 though no head-to-head trial has been done.
RISK FACTORS FOR STATIN-INDUCED MYOPATHY

APPROACH TO SUSPECTED STATIN-INDUCED MYOPATHY
The recommendations that follow are based on observations from our statin myopathy clinic in more than 650 patients, of whom more than 60 have suffered rhabdomyolysis. This experience is largely anecdotal, since such patients have not been well studied in controlled trials.
Are the symptoms due to the statin?
In our experience, most patients who develop significant weakness and pain on statin therapy have normal CK levels.40
Since there is as yet no test to confirm or reject the diagnosis of statin toxicity, the first objective is to determine the likelihood that the muscle complaint is being caused by statin therapy. Factors that make statin toxicity more likely must be weighed against any features that are atypical or that favor an alternate diagnosis.
The final decision about future statin treatment depends on the balance between the expected benefit of statin therapy for each individual and the likelihood that the symptoms are due to statin therapy. Below, we provide an algorithmic approach based on history, physical examination, and laboratory findings.
Findings from the history that implicate statins
Many muscle symptoms resolve within 2 weeks of starting statin therapy. Therefore, if patients have a normal CK level and can tolerate the symptoms, we ask them to continue therapy and see if their symptoms resolve with continued use.
Symptoms that persist beyond the first 2 weeks of therapy are likely due to the statin. These include symmetric burning or pain in the large muscles during exercise that was not present before lipid-lowering therapy. Any symptom that reproducibly recurs with statin rechallenge and disappears within 2 weeks of discontinuing therapy is more likely to be caused by the statin.
Findings of the PRIMO study are representative of typical statin-induced symptoms that we see in our clinic.20
- Most patients did not identify a trigger, but the 40% who did had engaged in unusual physical exertion or had received a new drug in addition to the statin.
- Heaviness, stiffness, or cramps predominated in 70%, with only a quarter noting weakness and another quarter suffering myalgias during exercise. Pain was diffuse in 60% and more common in the lower extremities than the upper extremities.
- Physically active patients were more likely to suffer muscle symptoms than sedentary patients, echoing the observation by Sinzinger and O’Grady that athletes are especially intolerant of lipid-lowering therapy.41
- Patients who have had muscle complaints with other drug therapies such as bisphosphonates, 42 raloxifene (Evista),43 or diuretics may be having the same provocation of an underlying muscle predisposition by the statin.
- A personal or family history of muscle complaints predisposed patients to statin-induced myopathy.20
Finally, we have repeatedly found that when dyspnea and fatigue are associated with the muscle complaint, they are more likely to be caused by statins.44
Statins can unveil underlying musculoskeletal disorders

Atypical complaints require us to search for an alternate diagnosis before dismissing them as not statin-induced.
Previous episodes of myoglobinuria (dark urine after exertion) would raise the possibility of a metabolic myopathy. Frequent muscle cramps raise the possibility of a metabolic myopathy or motor neuron disease.
Asymmetric pain or pain involving joints and ligaments is less likely to be statin-related but in our experience occasionally occurs.
Some patients with underlying degenerative arthritis or tendinitis repeatedly develop worsening symptoms each time they take statins, perhaps because muscle weakness exacerbates the arthropathy or tendinopathy.45
Although statin-related muscle complaints are almost always symmetric, many patients with underlying peripheral vascular disease have asymmetric pain in the limb with poor vascular supply; the pain is reproducible by statin rechallenge.
In our experience, many patients whose chronic low back pain is due to a lumbar radiculopathy experience exacerbations of that pain whenever they start statins.
Weakness preceding the use of the statins or a family history of neuromuscular disorders may indicate a neurodegenerative disorder and warrants consideration of early neurological consultation.
Although these rules are not absolute, they are helpful in the initial evaluation, which must exclude alternative diagnoses.
Is the patient a vegetarian? A drinker? Taking supplements?
Taking a careful history of diet and supplement use is important to find exposures that may increase the risk of statin-related muscle complaints. Vegetarians may develop carnitine or vitamin B12 deficiencies. Alcohol and vitamin E and other supplements are occasional causes of muscle symptoms falsely attributed to statin therapy. It is also important to remember that red yeast rice contains lovastatin, which can exacerbate myopathy, especially when taken in conjunction with another statin.
Physical examination
The examination of patients with possible statin-induced myopathy begins with a general assessment for signs of hypothyroidism or excess alcohol consumption.
Ankle-brachial indices are used to exclude significant peripheral vascular disease.
The musculoskeletal examination focuses on muscle atrophy, tone, and strength but also excludes tendinopathies, arthropathies, and myofascial pain syndromes, which are often confused with muscle pain.
We conduct quantitative dynamometry, measuring handgrip with a Jamar dynamometer and hip abduction with a Nicholas Manual Muscle Tester. Precise dynamometric measurements are tracked at subsequent visits and are helpful in following recovery from myopathy as well as in tracking strength during subsequent statin rechallenges.
We routinely look for hyperreflexia, fasciculations, extensor-plantar responses, and decreased heel-to-shin movement, which would suggest myelopathy. Reflexes and a sensory examination including vibration and temperature sensation help exclude radiculopathy and peripheral neuropathy.
Laboratory evaluation
In every patient with possibly statin myopathy, the primary care physician should measure:
- The serum CK level (preferably more than 72 hours after exercise)
- The 25-hydroxy vitamin level
- The thyroid-stimulating hormone level.
Further laboratory evaluation depends on the findings and will often be directed by subspecialists. For example, we assess the sedimentation rate, anti-Ro and anti-La antibodies, and the myositis panel in patients with elevated CK whose other findings suggest an autoimmune or inflammatory process. We test serum carnitine levels (free, total, and esterified), fasting serum lactate levels, and serum cortisol in those with findings suggestive of metabolic myopathy. We order electromyography and nerve conduction studies in patients with possible myelopathy, peripheral neuropathy, or inflammatory myopathy.
Ultimately, a muscle biopsy may be necessary to exclude inflammatory or necrotizing myopathies in patients whose CK remains elevated despite withdrawal of statins. It may also be helpful when other findings suggest a metabolic myopathy. When a biopsy is needed, magnetic resonance imaging of the affected limb may identify an affected muscle for biopsy.
MANAGEMENT
Reassess the lipid goal

If the source of a complaint remains unclear after challenge and rechallenge with alternate statins, we generally recommend restarting therapy and trying to achieve the LDL-C goal. If the workup suggests a neurologic or rheumatolic etiology, a referral to a specialist is indicated. However, if the evaluation leads to a diagnosis of statin-induced myopathy, the next task is to reassess the lipid treatment goals.
The Adult Treatment Panel (ATP) III guidelines should be used to assess the patient’s risk of having major coronary heart disease in the next 10 years, and to determine appropriate LDL-C goals.46 Some patients with suspected toxicity may have been treated to more aggressive LDL-C levels than recommended by these guidelines. The first step in these patients is to reduce or discontinue the unnecessary statin, even if there is no clear evidence of toxicity.
For the rest of patients with suspected toxicity, the decision to discontinue statin therapy must be weighed against the estimated reduction in risk associated with taking a statin medication.
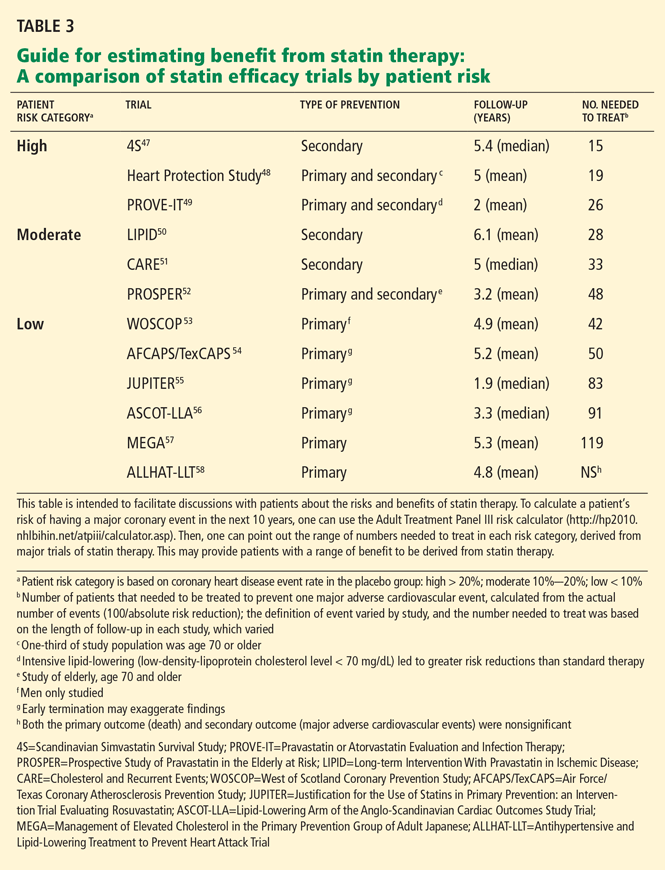
Prescribe a 6-week ‘statin holiday’ and see if symptoms resolve
In patients whose evaluation suggests statin myopathy, we stop all lipid-lowering therapy for 6 weeks and see if symptoms resolve and if grip and hip strength increase by dynamometry.
We often give these patient supplements of 600 mg daily of a bioavailable source of coenzyme Q10 and fish oil during this statin holiday. The data supporting the use of these supplements are mixed but the risks are minimal.5 In patients whose evaluation suggests a primary disorder in fatty acid oxidation, we add a trial of l-carnitine supplementation if symptoms do not resolve after a 6-week course of the coenzyme Q10 and fish oil.
If symptoms persist or if resolution is unclear at 6 weeks, we extend the holiday for an additional 6 weeks, except in patients with recent unstable coronary disease: for these patients, unless there is evidence of rhabdomyolysis, we believe that the benefits of continued statin therapy exceed the risks.
If the initial evaluation is consistent with statin myopathy and the neuromuscular symptoms (myalgias and weakness) do not respond within a few months of statin withdrawal, neurologic consultation is indicated to evaluate for an underlying neurologic disorder that has become symptomatic during statin therapy but whose existence is independent of the statin therapy. In some cases, the preexisting neurologic disorder may become symptomatic because of the statin therapy and remain symptomatic despite discontinuation of the statin therapy.
Restarting lipid-lowering therapy
Once the myopathy symptoms have abated or are controlled, a rechallenge of statin therapy is in order for those whose risk profile suggests greater benefit from statin therapy.
We consider the complete statin exposure history and any concomitant therapy that may have been competing with cytochrome P450 (CYP) metabolism of statins in designing an alternate lipid-lowering plan.
For patients with known coronary or vascular disease, in whom the survival benefit of statins is greatest, we generally try to find a statin regimen that is tolerable. Long-acting fluvastatin or a statin with less CYP dependence, such as pravastatin, is often successful.59 For patients whose myopathy has recurred with multiple statin rechallenges or whose lipid-lowering goal requires a more potent therapy, rosuvastatin in alternate-day or once- or twice-a-week schedules is efficacious and well tolerated in many patients.36,37,60 Of note, however, although such alternate-day therapies may produce excellent reductions in cholesterol levels, these regimens have not been proven to reduce cardiovascular end points.
Alternative lipid-lowering therapy. Occasionally, a patient cannot tolerate even intermittent rosuvastatin. In these cases, we prescribe resin therapy, which is well tolerated in those with recurrent statin myopathy.61
Although some believe that ezetimibe (Zetia) is an option for these patients, we do not agree, since it often causes similar muscle complaints in the most sensitive statin myopathy patients.30 Furthermore, ezetimibe has not been shown to improve cardiac end points.
Red yeast rice is also not a safe alternative in these patients, in whom muscle complaints and CK elevations frequently develop anew on this unregulated supplement despite its low lovastatin equivalence, 6 mg a day.62 A recent study showed that there is wide variability in the amount of lovastatin in over-the-counter red yeast rice; the median dose was 6 mg, and the maximum dose was 14.5 mg.63
The ultimate lipid-lowering plan for most of these patients will require a compromise between the ideal LDL-C goal and the LDL-C level that is achievable with these alternate attempts at lipid lowering.
While combination therapy may be attractive in patients with combined lipid disorders and no muscle complaints, fibrates are more likely to cause muscle toxicity per dose prescribed than statins, and the addition of fibrates to statin therapy increases the risks of muscle reactions.64,65 The evidence that fibrates reduce cardiovascular end points is much less robust than that for statins, which further reduces enthusiasm for combination therapy in patients with statin muscle toxicity.66,67
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Foley KA, Simpson RJ, Crouse JR, et al. Effectiveness of statin titration on low-density lipoprotein cholesterol goal attainment in patients at high risk of atherogenic events. Am J Cardiol 2003; 92:79–81.
- O’Meara JG, Kardia SL, Armon JJ, et al. Ethnic and sex differences in the prevalence, treatment, and control of dyslipidemia among hypertensive adults in the GENOA study. Arch Intern Med 2004; 164:1313–1318.
- Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002; 288:462–467.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore) 1982; 61:141–152.
- Ward MM. Factors predictive of acute renal failure in rhabdomyolysis. Arch Intern Med 1988; 148:1553–1557.
- McKenney JM, Davidson MH, Jacobson TA, et al. National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–84C.
- Pasternak RC, Smith SC, Bairey-Merz CN, et al. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol 2002; 40:567–572.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Linares LA, Golomb BA, Jaojoco JA, et al. The modern spectrum of rhabdomyolysis: drug toxicity revealed by creatine kinase screening. Curr Drug Saf 2009; 4:181–187.
- Armitage J. The safety of statins in clinical practice. Lancet 2007; 370:1781–1790.
- Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Sinzinger H, Wolfram R, Peskar BA. Muscular side effects of statins. J Cardiovasc Pharmacol 2002; 40:163–171.
- de Sauvage Nolting PR, Buirma RJ, et al. Two-year efficacy and safety of simvastatin 80 mg in familial hypercholesterolemia (the Examination of Probands and Relatives in Statin Studies With Familial Hypercholesterolemia [ExPRESS FH]). Am J Cardiol 2002; 90:181–184.
- Franc S, Dejager S, Bruckert E, et al. A comprehensive description of muscle symptoms associated with lipid-lowering drugs. Cardiovasc Drugs Ther 2003; 17:459–465.
- Kashani A, Phillips CO, Foody JM, et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation 2006; 114:2788–2797.
- Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Spatz ES, Canavan ME, Desai MM. From here to JUPITER: identifying new patients for statin therapy using data from the 1999–2004 National Health and Nutrition Examination Survey. Circ Cardiovasc Qual Outcomes 2009; 2:41–48.
- Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006; 34:153–162.
- Vladutiu GD. Genetic predisposition to statin myopathy. Curr Opin Rheumatol 2008; 20:648–655.
- Phillips PS, Phillips CT, Sullivan MJ, et al. Statin myotoxicity is associated with changes in the cardiopulmonary function. Atherosclerosis 2004; 177:183–188.
- Phillips PS, Ciaraldi TP, Kim DL, et al. Myotoxic reactions to lipidlowering therapy are associated with altered oxidation of fatty acids. Endocrine 2009; 35:38–46.
- Phillips PS, Haas RH. Statin myopathy as a metabolic muscle disease. Expert Rev Cardiovasc Ther 2008; 6:971–978.
- Löfberg M, Jänkälä H, Paetau A, et al. Metabolic causes of recurrent rhabdomyolysis. Acta Neurol Scand 1998; 98:268–275.
- Havranek JM, Wolfsen A, Warnke GA, et al. Monotherapy with Ezetimibe Causing Myopathy. Am J Med 2006; 119:285–286.
- Phillips PS. Ezetimibe and statin-associated myopathy. Ann Intern Med 2004; 141:649.
- Durst R, Rund D, Schurr D, et al. One year experience with a low density lipoprotein apheresis system. Isr Med Assoc J 2002; 4:677–680.
- Roberts WC. The rule of 5 and the rule of 7 in lipid-lowering by statin drugs. Am J Cardiol 1997; 80:106–107.
- Brewer HB. Benefit-risk assessment of rosuvastatin 10 to 40 milligrams. Am J Cardiol 2003; 92:23K–29K.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther 2006; 28:933–942.
- Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every-other-day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother 2008; 42:341–346.
- Backes JM, Moriarty PM, Ruisinger JF, et al. Effects of once weekly rosuvastatin among patients with a prior statin intolerance. Am J Cardiol 2007; 100:554–555.
- Antons KA, Williams CD, Baker SK, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med 2006; 119:400–409.
- Jacobson TA. Toward “pain-free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc 2008; 83:687–700.
- Phillips PS, Haas RH, Bannykh S, et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 2002; 137:581–585.
- Sinzinger H, O’Grady J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br J Clin Pharmacol 2004; 57:525–528.
- Wysowski DK, Chang JT. Alendronate and risedronate: reports of severe bone, joint, and muscle pain. Arch Intern Med 2005; 165:346–347.
- Martino S, Disch D, Dowsett SA, et al. Safety assessment of raloxifene over eight years in a clinical trial setting. Curr Med Res Opin 2005; 21:1441–1452.
- Phillips PS, Kimura BJ, Kelley R, et al. Feasibility of using the internet to perform medical research: observations from a statin-myopathy public information web site. Circulation 2003; 107:e7001–e7039.
- Chazerain P, Hayem G, Hamza S, et al. Four cases of tendinopathy in patients on statin therapy. Joint Bone Spine 2001; 68:430–433.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004; 44:720–732.
- Pedersen TR, Olsson AG, Faergeman O, et al. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S). Circulation 1998; 97:1453–1460.
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20, 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med 1996; 335:1001–1009.
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 1995; 333:1301–1307.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHATLLT). JAMA 2002; 288:2998–3007.
- Stein EA, Ballantyne CM, Windler E, et al. Efficacy and tolerability of fluvastatin XL 80 mg alone, ezetimibe alone, and the combination of fluvastatin XL 80 mg with ezetimibe in patients with a history of muscle-related side effects with other statins. Am J Cardiol 2008; 101:490–496.
- Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther 2006; 28:933–942.
- Phillips PS, Gray NL, McDonald FG, et al. Colesevelam is safe and effective in patients with statin myotoxicity. Arterioscler Thromb Vasc Biol 2005 25:E-97.
- Phillips PS. Balancing randomized trials with anecdote. Ann Intern Med 2009; 150:885–886.
- Gordon RY, Cooperman T, Obermeyer W, et al. Marked variability of monacolin levels in commercial red yeast rice products: buyer beware! Arch Intern Med 2010; 170:1722–1727.
- Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004; 292:2585–2590.
- Gaist D, Rodríguez LA, Huerta C, et al. Lipid-lowering drugs and risk of myopathy: a population-based follow-up study. Epidemiology 2001; 12:565–569.
- Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005; 366:1849–1861.
- ACCORD Study Group; Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
When a patient taking a statin complains of muscle aches, is he or she experiencing statin-induced myopathy or some other problem? Should statin therapy be discontinued?
Statins have proven efficacy in preventing heart attacks and death,1 and they are the most widely prescribed drugs worldwide. Nevertheless, they remain underused, with only 50% of those who would benefit from being on a statin receiving one.2,3 In addition, at least 25% of adults who start taking statins stop taking them by 6 months, and up to 60% stop by 2 years.4
Patient and physician fears about myopathy remain a key reason for stopping. Myopathy, a known side effect of statins, is rare in randomized controlled trials, but less so in observational studies and clinical experience. This discrepancy between clinical trials and clinical experience reduces confidence in lipid-lowering therapy and contributes to its underuse.
This review emphasizes clinical aspects of statin myopathy that are important to the practicing physician. We will define myopathy, review its purported mechanisms, and describe a clinical approach to patients with possible toxicity, including risk factors, physical findings, and consideration of alternate diagnoses. Since there is no single test to diagnose statin-induced myopathy, we offer a framework to aid clinicians in stratifying patients based on the likelihood that their symptoms are due to statin toxicity weighed against the likelihood that they will benefit from statin therapy.
DEFINITIONS DIFFER
Little consensus exists on how to define the adverse muscle effects of statins,5 which may contribute to the underdiagnosis of this complication. The magnitude of creatine kinase (CK) elevation required to define rhabdomyolysis has increased from 500 IU/L in 1982,6 to 1,000 IU/L in 1988,7 to 50 times the upper limit of normal in one current definition.8 The American Heart Association, the American College of Cardiology, the National Heart Lung and Blood Institute,9 the National Lipid Association,8 and the US Food and Drug Administration10 all differ in their definitions.
Our definitions
For the purpose of this article, we offer the following definitions:
Myalgia—muscle weakness, soreness, tenderness, stiffness, cramping, or aching, either at rest or with exertion, without any elevation in CK.
Myositis—elevated CK with or without muscle symptoms. The “-itis” suffix is unfortunate since myositis does not correspond to inflammation on biopsy.
Rhabdomyolysis—muscle symptoms with a CK level 10 times the upper limit of normal or higher. Evidence of renal dysfunction is not required for the diagnosis, as preexisting renal disease and hydration status are more closely related to kidney damage than the degree of muscle injury.11
STATIN MYOPATHY IS MORE COMMON IN THE REAL WORLD THAN IN TRIALS
The incidence of statin-induced myopathy is significantly lower in randomized controlled trials of statin efficacy than in observational studies of real-world patients. In randomized clinical trials, myalgia was reported in 1% to 5% of patients in the statin groups and placebo groups alike,9,12 whereas clinical practice would suggest it is more common.
Why is statin-induced myopathy so uncommon in clinical trials?
A reason may be that patients in clinical trials are carefully screened. To minimize toxicity, the clinical trials of statins excluded patients with renal insufficiency, hepatic insufficiency, a history of muscular complaints, and poorly controlled diabetes, as well as patients taking drugs with possible interactions. Large efficacy trials have excluded up to 30% of the participants in active prerandomization phases.13,14
Another reason is that these trials were designed to assess the efficacy of statins and were not sensitive to adverse effects like muscle pain. When they looked at myopathy, they focused on rhabdomyolysis—the most severe form—rather than on myalgia, fatigue, or other minor muscle complaints.15 Additionally, most trials enrolled too few patients and did not have long enough follow-up to reveal infrequent toxicities.
Despite the strict criteria, a significant number of trial patients discontinued statin therapy during the study period. In the Treat to New Targets (TNT) trial, 5% of patients in both the high- and low-dose atorvastatin (Lipitor) groups experienced muscle toxicity, even though 35% of eligible patients had been excluded during the open-label run-in phase.14
Also, physicians may overlook and patients may fail to report symptoms such as fatigue, malaise, or dyspnea that are not commonly accepted as signs of statin toxicity.16
Findings from observational studies
Observational studies in nonselected outpatients show a higher frequency of muscle complaints in the statin groups than in the control groups. These studies suggest the frequency of statin myopathy is 9% to 20%.17–19
The Prediction of Muscular Risk in Observational Conditions (PRIMO) study20 was one of the largest and best-defined observational studies of muscular symptoms in an unselected population. It included 7,924 French outpatients with hypercholesterolemia, ages 18 to 75 years, on high-dose statins for 3 or more months before the study. Daily statin regimens included atorvastatin 40 to 80 mg, fluvastatin (Lescol) 80 mg, pravastatin (Pravachol) 40 mg, and simvastatin (Zocor) 40 to 80 mg. In this study, 10.5% of patients reported muscle-related symptoms.
Buettner et al,21 in another cross-sectional study, interviewed and examined 3,580 adults over age 40. Of those taking statins, 22% reported having had musculoskeletal pain in at least one anatomic region in the last 30 days, compared with 16.7% of those not taking a statin.
In the United States, where an estimated 33 million adults use statins, musculoskeletal pain can be expected to occur in 7 million people, likely induced by statin therapy in 25% of cases.22
WHAT CAUSES STATIN MYOPATHY?
The causes of statin-induced myopathy are poorly understood.
Historically, statin-induced toxicity was thought to be caused by inhibition of the synthesis of mevalonate, leading to depletion of its metabolites, such as cholesterol, isoprenoids, and ubiquinone (coenzyme Q10). Depletion of intracellular cholesterol may lead to abnormal membrane behaviors; depletion of isoprenoids may affect intracellular signaling; depletion of coenzyme Q10 may in turn reduce mitochondrial respiratory function. Genetic factors may also play a role, contributing to pharmacokinetics and predisposing metabolic muscle disorders.23,24
Statin-induced myopathy seems to be different in randomized efficacy trials than in the clinical setting. In randomized trials, the mechanism appears to involve abnormal pharmacokinetics. The participants are carefully selected to have a low risk of muscle toxicity, but some develop toxicity when statin levels are elevated because of reduced drug breakdown and metabolism. However, in clinical practice, where a much larger group of unselected patients are exposed to statins, toxicity appears to be more related to a metabolic predisposition.25–27
Multiple minor metabolic abnormalities have been described in the muscles of patients with statin-induced muscle toxicity, suggesting that some patients have a predisposition for muscle complaints.23 About 25% of patients with recurrent rhabdomyolysis irrespective of lipid-lowering therapy have an underlying metabolic muscle disorder.28 In these vulnerable patients, minor metabolic defects are exacerbated by any agent that reduces the delivery of fat substrate to muscle, leading to muscle starvation.
This concept explains why patients may develop the same muscle complaint on different lipid-lowering agents.29,30 It also may explain why rhabdomyolysis can occur after apheresis of lipids, when no drugs have been given.31 In vulnerable patients, muscle toxicity may result from any agent that lessens the lipid substrate available to muscle rather than from the reduction of products downstream from mevalonate by statins.
SOME STATINS MAY BE LESS TOXIC
In the PRIMO study, muscle-related symptoms occurred with the various regimens as follows:
- Fluvastatin XL 40 mg—5.1%
- Pravastatin 40 mg—10.9%
- Atorvastatin 40 to 80 mg—14.9%
- Simvastatin 40 to 80 mg—18.2%.
Others have also shown that fluvastatin contributed to the smallest number of reported cases of rhabdomyolysis among statins: 55 (1.6%) of 3,339 cases.34
More recent studies indicate that rosuvastatin (Crestor), the most hydrophilic statin, may be well tolerated in those who do not tolerate other statins,35–37 though no head-to-head trial has been done.
RISK FACTORS FOR STATIN-INDUCED MYOPATHY
APPROACH TO SUSPECTED STATIN-INDUCED MYOPATHY
The recommendations that follow are based on observations from our statin myopathy clinic in more than 650 patients, of whom more than 60 have suffered rhabdomyolysis. This experience is largely anecdotal, since such patients have not been well studied in controlled trials.
Are the symptoms due to the statin?
In our experience, most patients who develop significant weakness and pain on statin therapy have normal CK levels.40
Since there is as yet no test to confirm or reject the diagnosis of statin toxicity, the first objective is to determine the likelihood that the muscle complaint is being caused by statin therapy. Factors that make statin toxicity more likely must be weighed against any features that are atypical or that favor an alternate diagnosis.
The final decision about future statin treatment depends on the balance between the expected benefit of statin therapy for each individual and the likelihood that the symptoms are due to statin therapy. Below, we provide an algorithmic approach based on history, physical examination, and laboratory findings.
Findings from the history that implicate statins
Many muscle symptoms resolve within 2 weeks of starting statin therapy. Therefore, if patients have a normal CK level and can tolerate the symptoms, we ask them to continue therapy and see if their symptoms resolve with continued use.
Symptoms that persist beyond the first 2 weeks of therapy are likely due to the statin. These include symmetric burning or pain in the large muscles during exercise that was not present before lipid-lowering therapy. Any symptom that reproducibly recurs with statin rechallenge and disappears within 2 weeks of discontinuing therapy is more likely to be caused by the statin.
Findings of the PRIMO study are representative of typical statin-induced symptoms that we see in our clinic.20
- Most patients did not identify a trigger, but the 40% who did had engaged in unusual physical exertion or had received a new drug in addition to the statin.
- Heaviness, stiffness, or cramps predominated in 70%, with only a quarter noting weakness and another quarter suffering myalgias during exercise. Pain was diffuse in 60% and more common in the lower extremities than the upper extremities.
- Physically active patients were more likely to suffer muscle symptoms than sedentary patients, echoing the observation by Sinzinger and O’Grady that athletes are especially intolerant of lipid-lowering therapy.41
- Patients who have had muscle complaints with other drug therapies such as bisphosphonates, 42 raloxifene (Evista),43 or diuretics may be having the same provocation of an underlying muscle predisposition by the statin.
- A personal or family history of muscle complaints predisposed patients to statin-induced myopathy.20
Finally, we have repeatedly found that when dyspnea and fatigue are associated with the muscle complaint, they are more likely to be caused by statins.44
Statins can unveil underlying musculoskeletal disorders
Atypical complaints require us to search for an alternate diagnosis before dismissing them as not statin-induced.
Previous episodes of myoglobinuria (dark urine after exertion) would raise the possibility of a metabolic myopathy. Frequent muscle cramps raise the possibility of a metabolic myopathy or motor neuron disease.
Asymmetric pain or pain involving joints and ligaments is less likely to be statin-related but in our experience occasionally occurs.
Some patients with underlying degenerative arthritis or tendinitis repeatedly develop worsening symptoms each time they take statins, perhaps because muscle weakness exacerbates the arthropathy or tendinopathy.45
Although statin-related muscle complaints are almost always symmetric, many patients with underlying peripheral vascular disease have asymmetric pain in the limb with poor vascular supply; the pain is reproducible by statin rechallenge.
In our experience, many patients whose chronic low back pain is due to a lumbar radiculopathy experience exacerbations of that pain whenever they start statins.
Weakness preceding the use of the statins or a family history of neuromuscular disorders may indicate a neurodegenerative disorder and warrants consideration of early neurological consultation.
Although these rules are not absolute, they are helpful in the initial evaluation, which must exclude alternative diagnoses.
Is the patient a vegetarian? A drinker? Taking supplements?
Taking a careful history of diet and supplement use is important to find exposures that may increase the risk of statin-related muscle complaints. Vegetarians may develop carnitine or vitamin B12 deficiencies. Alcohol and vitamin E and other supplements are occasional causes of muscle symptoms falsely attributed to statin therapy. It is also important to remember that red yeast rice contains lovastatin, which can exacerbate myopathy, especially when taken in conjunction with another statin.
Physical examination
The examination of patients with possible statin-induced myopathy begins with a general assessment for signs of hypothyroidism or excess alcohol consumption.
Ankle-brachial indices are used to exclude significant peripheral vascular disease.
The musculoskeletal examination focuses on muscle atrophy, tone, and strength but also excludes tendinopathies, arthropathies, and myofascial pain syndromes, which are often confused with muscle pain.
We conduct quantitative dynamometry, measuring handgrip with a Jamar dynamometer and hip abduction with a Nicholas Manual Muscle Tester. Precise dynamometric measurements are tracked at subsequent visits and are helpful in following recovery from myopathy as well as in tracking strength during subsequent statin rechallenges.
We routinely look for hyperreflexia, fasciculations, extensor-plantar responses, and decreased heel-to-shin movement, which would suggest myelopathy. Reflexes and a sensory examination including vibration and temperature sensation help exclude radiculopathy and peripheral neuropathy.
Laboratory evaluation
In every patient with possibly statin myopathy, the primary care physician should measure:
- The serum CK level (preferably more than 72 hours after exercise)
- The 25-hydroxy vitamin level
- The thyroid-stimulating hormone level.
Further laboratory evaluation depends on the findings and will often be directed by subspecialists. For example, we assess the sedimentation rate, anti-Ro and anti-La antibodies, and the myositis panel in patients with elevated CK whose other findings suggest an autoimmune or inflammatory process. We test serum carnitine levels (free, total, and esterified), fasting serum lactate levels, and serum cortisol in those with findings suggestive of metabolic myopathy. We order electromyography and nerve conduction studies in patients with possible myelopathy, peripheral neuropathy, or inflammatory myopathy.
Ultimately, a muscle biopsy may be necessary to exclude inflammatory or necrotizing myopathies in patients whose CK remains elevated despite withdrawal of statins. It may also be helpful when other findings suggest a metabolic myopathy. When a biopsy is needed, magnetic resonance imaging of the affected limb may identify an affected muscle for biopsy.
MANAGEMENT
Reassess the lipid goal
If the source of a complaint remains unclear after challenge and rechallenge with alternate statins, we generally recommend restarting therapy and trying to achieve the LDL-C goal. If the workup suggests a neurologic or rheumatolic etiology, a referral to a specialist is indicated. However, if the evaluation leads to a diagnosis of statin-induced myopathy, the next task is to reassess the lipid treatment goals.
The Adult Treatment Panel (ATP) III guidelines should be used to assess the patient’s risk of having major coronary heart disease in the next 10 years, and to determine appropriate LDL-C goals.46 Some patients with suspected toxicity may have been treated to more aggressive LDL-C levels than recommended by these guidelines. The first step in these patients is to reduce or discontinue the unnecessary statin, even if there is no clear evidence of toxicity.
For the rest of patients with suspected toxicity, the decision to discontinue statin therapy must be weighed against the estimated reduction in risk associated with taking a statin medication.
Prescribe a 6-week ‘statin holiday’ and see if symptoms resolve
In patients whose evaluation suggests statin myopathy, we stop all lipid-lowering therapy for 6 weeks and see if symptoms resolve and if grip and hip strength increase by dynamometry.
We often give these patient supplements of 600 mg daily of a bioavailable source of coenzyme Q10 and fish oil during this statin holiday. The data supporting the use of these supplements are mixed but the risks are minimal.5 In patients whose evaluation suggests a primary disorder in fatty acid oxidation, we add a trial of l-carnitine supplementation if symptoms do not resolve after a 6-week course of the coenzyme Q10 and fish oil.
If symptoms persist or if resolution is unclear at 6 weeks, we extend the holiday for an additional 6 weeks, except in patients with recent unstable coronary disease: for these patients, unless there is evidence of rhabdomyolysis, we believe that the benefits of continued statin therapy exceed the risks.
If the initial evaluation is consistent with statin myopathy and the neuromuscular symptoms (myalgias and weakness) do not respond within a few months of statin withdrawal, neurologic consultation is indicated to evaluate for an underlying neurologic disorder that has become symptomatic during statin therapy but whose existence is independent of the statin therapy. In some cases, the preexisting neurologic disorder may become symptomatic because of the statin therapy and remain symptomatic despite discontinuation of the statin therapy.
Restarting lipid-lowering therapy
Once the myopathy symptoms have abated or are controlled, a rechallenge of statin therapy is in order for those whose risk profile suggests greater benefit from statin therapy.
We consider the complete statin exposure history and any concomitant therapy that may have been competing with cytochrome P450 (CYP) metabolism of statins in designing an alternate lipid-lowering plan.
For patients with known coronary or vascular disease, in whom the survival benefit of statins is greatest, we generally try to find a statin regimen that is tolerable. Long-acting fluvastatin or a statin with less CYP dependence, such as pravastatin, is often successful.59 For patients whose myopathy has recurred with multiple statin rechallenges or whose lipid-lowering goal requires a more potent therapy, rosuvastatin in alternate-day or once- or twice-a-week schedules is efficacious and well tolerated in many patients.36,37,60 Of note, however, although such alternate-day therapies may produce excellent reductions in cholesterol levels, these regimens have not been proven to reduce cardiovascular end points.
Alternative lipid-lowering therapy. Occasionally, a patient cannot tolerate even intermittent rosuvastatin. In these cases, we prescribe resin therapy, which is well tolerated in those with recurrent statin myopathy.61
Although some believe that ezetimibe (Zetia) is an option for these patients, we do not agree, since it often causes similar muscle complaints in the most sensitive statin myopathy patients.30 Furthermore, ezetimibe has not been shown to improve cardiac end points.
Red yeast rice is also not a safe alternative in these patients, in whom muscle complaints and CK elevations frequently develop anew on this unregulated supplement despite its low lovastatin equivalence, 6 mg a day.62 A recent study showed that there is wide variability in the amount of lovastatin in over-the-counter red yeast rice; the median dose was 6 mg, and the maximum dose was 14.5 mg.63
The ultimate lipid-lowering plan for most of these patients will require a compromise between the ideal LDL-C goal and the LDL-C level that is achievable with these alternate attempts at lipid lowering.
While combination therapy may be attractive in patients with combined lipid disorders and no muscle complaints, fibrates are more likely to cause muscle toxicity per dose prescribed than statins, and the addition of fibrates to statin therapy increases the risks of muscle reactions.64,65 The evidence that fibrates reduce cardiovascular end points is much less robust than that for statins, which further reduces enthusiasm for combination therapy in patients with statin muscle toxicity.66,67
When a patient taking a statin complains of muscle aches, is he or she experiencing statin-induced myopathy or some other problem? Should statin therapy be discontinued?
Statins have proven efficacy in preventing heart attacks and death,1 and they are the most widely prescribed drugs worldwide. Nevertheless, they remain underused, with only 50% of those who would benefit from being on a statin receiving one.2,3 In addition, at least 25% of adults who start taking statins stop taking them by 6 months, and up to 60% stop by 2 years.4
Patient and physician fears about myopathy remain a key reason for stopping. Myopathy, a known side effect of statins, is rare in randomized controlled trials, but less so in observational studies and clinical experience. This discrepancy between clinical trials and clinical experience reduces confidence in lipid-lowering therapy and contributes to its underuse.
This review emphasizes clinical aspects of statin myopathy that are important to the practicing physician. We will define myopathy, review its purported mechanisms, and describe a clinical approach to patients with possible toxicity, including risk factors, physical findings, and consideration of alternate diagnoses. Since there is no single test to diagnose statin-induced myopathy, we offer a framework to aid clinicians in stratifying patients based on the likelihood that their symptoms are due to statin toxicity weighed against the likelihood that they will benefit from statin therapy.
DEFINITIONS DIFFER
Little consensus exists on how to define the adverse muscle effects of statins,5 which may contribute to the underdiagnosis of this complication. The magnitude of creatine kinase (CK) elevation required to define rhabdomyolysis has increased from 500 IU/L in 1982,6 to 1,000 IU/L in 1988,7 to 50 times the upper limit of normal in one current definition.8 The American Heart Association, the American College of Cardiology, the National Heart Lung and Blood Institute,9 the National Lipid Association,8 and the US Food and Drug Administration10 all differ in their definitions.
Our definitions
For the purpose of this article, we offer the following definitions:
Myalgia—muscle weakness, soreness, tenderness, stiffness, cramping, or aching, either at rest or with exertion, without any elevation in CK.
Myositis—elevated CK with or without muscle symptoms. The “-itis” suffix is unfortunate since myositis does not correspond to inflammation on biopsy.
Rhabdomyolysis—muscle symptoms with a CK level 10 times the upper limit of normal or higher. Evidence of renal dysfunction is not required for the diagnosis, as preexisting renal disease and hydration status are more closely related to kidney damage than the degree of muscle injury.11
STATIN MYOPATHY IS MORE COMMON IN THE REAL WORLD THAN IN TRIALS
The incidence of statin-induced myopathy is significantly lower in randomized controlled trials of statin efficacy than in observational studies of real-world patients. In randomized clinical trials, myalgia was reported in 1% to 5% of patients in the statin groups and placebo groups alike,9,12 whereas clinical practice would suggest it is more common.
Why is statin-induced myopathy so uncommon in clinical trials?
A reason may be that patients in clinical trials are carefully screened. To minimize toxicity, the clinical trials of statins excluded patients with renal insufficiency, hepatic insufficiency, a history of muscular complaints, and poorly controlled diabetes, as well as patients taking drugs with possible interactions. Large efficacy trials have excluded up to 30% of the participants in active prerandomization phases.13,14
Another reason is that these trials were designed to assess the efficacy of statins and were not sensitive to adverse effects like muscle pain. When they looked at myopathy, they focused on rhabdomyolysis—the most severe form—rather than on myalgia, fatigue, or other minor muscle complaints.15 Additionally, most trials enrolled too few patients and did not have long enough follow-up to reveal infrequent toxicities.
Despite the strict criteria, a significant number of trial patients discontinued statin therapy during the study period. In the Treat to New Targets (TNT) trial, 5% of patients in both the high- and low-dose atorvastatin (Lipitor) groups experienced muscle toxicity, even though 35% of eligible patients had been excluded during the open-label run-in phase.14
Also, physicians may overlook and patients may fail to report symptoms such as fatigue, malaise, or dyspnea that are not commonly accepted as signs of statin toxicity.16
Findings from observational studies
Observational studies in nonselected outpatients show a higher frequency of muscle complaints in the statin groups than in the control groups. These studies suggest the frequency of statin myopathy is 9% to 20%.17–19
The Prediction of Muscular Risk in Observational Conditions (PRIMO) study20 was one of the largest and best-defined observational studies of muscular symptoms in an unselected population. It included 7,924 French outpatients with hypercholesterolemia, ages 18 to 75 years, on high-dose statins for 3 or more months before the study. Daily statin regimens included atorvastatin 40 to 80 mg, fluvastatin (Lescol) 80 mg, pravastatin (Pravachol) 40 mg, and simvastatin (Zocor) 40 to 80 mg. In this study, 10.5% of patients reported muscle-related symptoms.
Buettner et al,21 in another cross-sectional study, interviewed and examined 3,580 adults over age 40. Of those taking statins, 22% reported having had musculoskeletal pain in at least one anatomic region in the last 30 days, compared with 16.7% of those not taking a statin.
In the United States, where an estimated 33 million adults use statins, musculoskeletal pain can be expected to occur in 7 million people, likely induced by statin therapy in 25% of cases.22
WHAT CAUSES STATIN MYOPATHY?
The causes of statin-induced myopathy are poorly understood.
Historically, statin-induced toxicity was thought to be caused by inhibition of the synthesis of mevalonate, leading to depletion of its metabolites, such as cholesterol, isoprenoids, and ubiquinone (coenzyme Q10). Depletion of intracellular cholesterol may lead to abnormal membrane behaviors; depletion of isoprenoids may affect intracellular signaling; depletion of coenzyme Q10 may in turn reduce mitochondrial respiratory function. Genetic factors may also play a role, contributing to pharmacokinetics and predisposing metabolic muscle disorders.23,24
Statin-induced myopathy seems to be different in randomized efficacy trials than in the clinical setting. In randomized trials, the mechanism appears to involve abnormal pharmacokinetics. The participants are carefully selected to have a low risk of muscle toxicity, but some develop toxicity when statin levels are elevated because of reduced drug breakdown and metabolism. However, in clinical practice, where a much larger group of unselected patients are exposed to statins, toxicity appears to be more related to a metabolic predisposition.25–27
Multiple minor metabolic abnormalities have been described in the muscles of patients with statin-induced muscle toxicity, suggesting that some patients have a predisposition for muscle complaints.23 About 25% of patients with recurrent rhabdomyolysis irrespective of lipid-lowering therapy have an underlying metabolic muscle disorder.28 In these vulnerable patients, minor metabolic defects are exacerbated by any agent that reduces the delivery of fat substrate to muscle, leading to muscle starvation.
This concept explains why patients may develop the same muscle complaint on different lipid-lowering agents.29,30 It also may explain why rhabdomyolysis can occur after apheresis of lipids, when no drugs have been given.31 In vulnerable patients, muscle toxicity may result from any agent that lessens the lipid substrate available to muscle rather than from the reduction of products downstream from mevalonate by statins.
SOME STATINS MAY BE LESS TOXIC
In the PRIMO study, muscle-related symptoms occurred with the various regimens as follows:
- Fluvastatin XL 40 mg—5.1%
- Pravastatin 40 mg—10.9%
- Atorvastatin 40 to 80 mg—14.9%
- Simvastatin 40 to 80 mg—18.2%.
Others have also shown that fluvastatin contributed to the smallest number of reported cases of rhabdomyolysis among statins: 55 (1.6%) of 3,339 cases.34
More recent studies indicate that rosuvastatin (Crestor), the most hydrophilic statin, may be well tolerated in those who do not tolerate other statins,35–37 though no head-to-head trial has been done.
RISK FACTORS FOR STATIN-INDUCED MYOPATHY
APPROACH TO SUSPECTED STATIN-INDUCED MYOPATHY
The recommendations that follow are based on observations from our statin myopathy clinic in more than 650 patients, of whom more than 60 have suffered rhabdomyolysis. This experience is largely anecdotal, since such patients have not been well studied in controlled trials.
Are the symptoms due to the statin?
In our experience, most patients who develop significant weakness and pain on statin therapy have normal CK levels.40
Since there is as yet no test to confirm or reject the diagnosis of statin toxicity, the first objective is to determine the likelihood that the muscle complaint is being caused by statin therapy. Factors that make statin toxicity more likely must be weighed against any features that are atypical or that favor an alternate diagnosis.
The final decision about future statin treatment depends on the balance between the expected benefit of statin therapy for each individual and the likelihood that the symptoms are due to statin therapy. Below, we provide an algorithmic approach based on history, physical examination, and laboratory findings.
Findings from the history that implicate statins
Many muscle symptoms resolve within 2 weeks of starting statin therapy. Therefore, if patients have a normal CK level and can tolerate the symptoms, we ask them to continue therapy and see if their symptoms resolve with continued use.
Symptoms that persist beyond the first 2 weeks of therapy are likely due to the statin. These include symmetric burning or pain in the large muscles during exercise that was not present before lipid-lowering therapy. Any symptom that reproducibly recurs with statin rechallenge and disappears within 2 weeks of discontinuing therapy is more likely to be caused by the statin.
Findings of the PRIMO study are representative of typical statin-induced symptoms that we see in our clinic.20
- Most patients did not identify a trigger, but the 40% who did had engaged in unusual physical exertion or had received a new drug in addition to the statin.
- Heaviness, stiffness, or cramps predominated in 70%, with only a quarter noting weakness and another quarter suffering myalgias during exercise. Pain was diffuse in 60% and more common in the lower extremities than the upper extremities.
- Physically active patients were more likely to suffer muscle symptoms than sedentary patients, echoing the observation by Sinzinger and O’Grady that athletes are especially intolerant of lipid-lowering therapy.41
- Patients who have had muscle complaints with other drug therapies such as bisphosphonates, 42 raloxifene (Evista),43 or diuretics may be having the same provocation of an underlying muscle predisposition by the statin.
- A personal or family history of muscle complaints predisposed patients to statin-induced myopathy.20
Finally, we have repeatedly found that when dyspnea and fatigue are associated with the muscle complaint, they are more likely to be caused by statins.44
Statins can unveil underlying musculoskeletal disorders
Atypical complaints require us to search for an alternate diagnosis before dismissing them as not statin-induced.
Previous episodes of myoglobinuria (dark urine after exertion) would raise the possibility of a metabolic myopathy. Frequent muscle cramps raise the possibility of a metabolic myopathy or motor neuron disease.
Asymmetric pain or pain involving joints and ligaments is less likely to be statin-related but in our experience occasionally occurs.
Some patients with underlying degenerative arthritis or tendinitis repeatedly develop worsening symptoms each time they take statins, perhaps because muscle weakness exacerbates the arthropathy or tendinopathy.45
Although statin-related muscle complaints are almost always symmetric, many patients with underlying peripheral vascular disease have asymmetric pain in the limb with poor vascular supply; the pain is reproducible by statin rechallenge.
In our experience, many patients whose chronic low back pain is due to a lumbar radiculopathy experience exacerbations of that pain whenever they start statins.
Weakness preceding the use of the statins or a family history of neuromuscular disorders may indicate a neurodegenerative disorder and warrants consideration of early neurological consultation.
Although these rules are not absolute, they are helpful in the initial evaluation, which must exclude alternative diagnoses.
Is the patient a vegetarian? A drinker? Taking supplements?
Taking a careful history of diet and supplement use is important to find exposures that may increase the risk of statin-related muscle complaints. Vegetarians may develop carnitine or vitamin B12 deficiencies. Alcohol and vitamin E and other supplements are occasional causes of muscle symptoms falsely attributed to statin therapy. It is also important to remember that red yeast rice contains lovastatin, which can exacerbate myopathy, especially when taken in conjunction with another statin.
Physical examination
The examination of patients with possible statin-induced myopathy begins with a general assessment for signs of hypothyroidism or excess alcohol consumption.
Ankle-brachial indices are used to exclude significant peripheral vascular disease.
The musculoskeletal examination focuses on muscle atrophy, tone, and strength but also excludes tendinopathies, arthropathies, and myofascial pain syndromes, which are often confused with muscle pain.
We conduct quantitative dynamometry, measuring handgrip with a Jamar dynamometer and hip abduction with a Nicholas Manual Muscle Tester. Precise dynamometric measurements are tracked at subsequent visits and are helpful in following recovery from myopathy as well as in tracking strength during subsequent statin rechallenges.
We routinely look for hyperreflexia, fasciculations, extensor-plantar responses, and decreased heel-to-shin movement, which would suggest myelopathy. Reflexes and a sensory examination including vibration and temperature sensation help exclude radiculopathy and peripheral neuropathy.
Laboratory evaluation
In every patient with possibly statin myopathy, the primary care physician should measure:
- The serum CK level (preferably more than 72 hours after exercise)
- The 25-hydroxy vitamin level
- The thyroid-stimulating hormone level.
Further laboratory evaluation depends on the findings and will often be directed by subspecialists. For example, we assess the sedimentation rate, anti-Ro and anti-La antibodies, and the myositis panel in patients with elevated CK whose other findings suggest an autoimmune or inflammatory process. We test serum carnitine levels (free, total, and esterified), fasting serum lactate levels, and serum cortisol in those with findings suggestive of metabolic myopathy. We order electromyography and nerve conduction studies in patients with possible myelopathy, peripheral neuropathy, or inflammatory myopathy.
Ultimately, a muscle biopsy may be necessary to exclude inflammatory or necrotizing myopathies in patients whose CK remains elevated despite withdrawal of statins. It may also be helpful when other findings suggest a metabolic myopathy. When a biopsy is needed, magnetic resonance imaging of the affected limb may identify an affected muscle for biopsy.
MANAGEMENT
Reassess the lipid goal
If the source of a complaint remains unclear after challenge and rechallenge with alternate statins, we generally recommend restarting therapy and trying to achieve the LDL-C goal. If the workup suggests a neurologic or rheumatolic etiology, a referral to a specialist is indicated. However, if the evaluation leads to a diagnosis of statin-induced myopathy, the next task is to reassess the lipid treatment goals.
The Adult Treatment Panel (ATP) III guidelines should be used to assess the patient’s risk of having major coronary heart disease in the next 10 years, and to determine appropriate LDL-C goals.46 Some patients with suspected toxicity may have been treated to more aggressive LDL-C levels than recommended by these guidelines. The first step in these patients is to reduce or discontinue the unnecessary statin, even if there is no clear evidence of toxicity.
For the rest of patients with suspected toxicity, the decision to discontinue statin therapy must be weighed against the estimated reduction in risk associated with taking a statin medication.
Prescribe a 6-week ‘statin holiday’ and see if symptoms resolve
In patients whose evaluation suggests statin myopathy, we stop all lipid-lowering therapy for 6 weeks and see if symptoms resolve and if grip and hip strength increase by dynamometry.
We often give these patient supplements of 600 mg daily of a bioavailable source of coenzyme Q10 and fish oil during this statin holiday. The data supporting the use of these supplements are mixed but the risks are minimal.5 In patients whose evaluation suggests a primary disorder in fatty acid oxidation, we add a trial of l-carnitine supplementation if symptoms do not resolve after a 6-week course of the coenzyme Q10 and fish oil.
If symptoms persist or if resolution is unclear at 6 weeks, we extend the holiday for an additional 6 weeks, except in patients with recent unstable coronary disease: for these patients, unless there is evidence of rhabdomyolysis, we believe that the benefits of continued statin therapy exceed the risks.
If the initial evaluation is consistent with statin myopathy and the neuromuscular symptoms (myalgias and weakness) do not respond within a few months of statin withdrawal, neurologic consultation is indicated to evaluate for an underlying neurologic disorder that has become symptomatic during statin therapy but whose existence is independent of the statin therapy. In some cases, the preexisting neurologic disorder may become symptomatic because of the statin therapy and remain symptomatic despite discontinuation of the statin therapy.
Restarting lipid-lowering therapy
Once the myopathy symptoms have abated or are controlled, a rechallenge of statin therapy is in order for those whose risk profile suggests greater benefit from statin therapy.
We consider the complete statin exposure history and any concomitant therapy that may have been competing with cytochrome P450 (CYP) metabolism of statins in designing an alternate lipid-lowering plan.
For patients with known coronary or vascular disease, in whom the survival benefit of statins is greatest, we generally try to find a statin regimen that is tolerable. Long-acting fluvastatin or a statin with less CYP dependence, such as pravastatin, is often successful.59 For patients whose myopathy has recurred with multiple statin rechallenges or whose lipid-lowering goal requires a more potent therapy, rosuvastatin in alternate-day or once- or twice-a-week schedules is efficacious and well tolerated in many patients.36,37,60 Of note, however, although such alternate-day therapies may produce excellent reductions in cholesterol levels, these regimens have not been proven to reduce cardiovascular end points.
Alternative lipid-lowering therapy. Occasionally, a patient cannot tolerate even intermittent rosuvastatin. In these cases, we prescribe resin therapy, which is well tolerated in those with recurrent statin myopathy.61
Although some believe that ezetimibe (Zetia) is an option for these patients, we do not agree, since it often causes similar muscle complaints in the most sensitive statin myopathy patients.30 Furthermore, ezetimibe has not been shown to improve cardiac end points.
Red yeast rice is also not a safe alternative in these patients, in whom muscle complaints and CK elevations frequently develop anew on this unregulated supplement despite its low lovastatin equivalence, 6 mg a day.62 A recent study showed that there is wide variability in the amount of lovastatin in over-the-counter red yeast rice; the median dose was 6 mg, and the maximum dose was 14.5 mg.63
The ultimate lipid-lowering plan for most of these patients will require a compromise between the ideal LDL-C goal and the LDL-C level that is achievable with these alternate attempts at lipid lowering.
While combination therapy may be attractive in patients with combined lipid disorders and no muscle complaints, fibrates are more likely to cause muscle toxicity per dose prescribed than statins, and the addition of fibrates to statin therapy increases the risks of muscle reactions.64,65 The evidence that fibrates reduce cardiovascular end points is much less robust than that for statins, which further reduces enthusiasm for combination therapy in patients with statin muscle toxicity.66,67
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Foley KA, Simpson RJ, Crouse JR, et al. Effectiveness of statin titration on low-density lipoprotein cholesterol goal attainment in patients at high risk of atherogenic events. Am J Cardiol 2003; 92:79–81.
- O’Meara JG, Kardia SL, Armon JJ, et al. Ethnic and sex differences in the prevalence, treatment, and control of dyslipidemia among hypertensive adults in the GENOA study. Arch Intern Med 2004; 164:1313–1318.
- Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002; 288:462–467.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore) 1982; 61:141–152.
- Ward MM. Factors predictive of acute renal failure in rhabdomyolysis. Arch Intern Med 1988; 148:1553–1557.
- McKenney JM, Davidson MH, Jacobson TA, et al. National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–84C.
- Pasternak RC, Smith SC, Bairey-Merz CN, et al. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol 2002; 40:567–572.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Linares LA, Golomb BA, Jaojoco JA, et al. The modern spectrum of rhabdomyolysis: drug toxicity revealed by creatine kinase screening. Curr Drug Saf 2009; 4:181–187.
- Armitage J. The safety of statins in clinical practice. Lancet 2007; 370:1781–1790.
- Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Sinzinger H, Wolfram R, Peskar BA. Muscular side effects of statins. J Cardiovasc Pharmacol 2002; 40:163–171.
- de Sauvage Nolting PR, Buirma RJ, et al. Two-year efficacy and safety of simvastatin 80 mg in familial hypercholesterolemia (the Examination of Probands and Relatives in Statin Studies With Familial Hypercholesterolemia [ExPRESS FH]). Am J Cardiol 2002; 90:181–184.
- Franc S, Dejager S, Bruckert E, et al. A comprehensive description of muscle symptoms associated with lipid-lowering drugs. Cardiovasc Drugs Ther 2003; 17:459–465.
- Kashani A, Phillips CO, Foody JM, et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation 2006; 114:2788–2797.
- Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Spatz ES, Canavan ME, Desai MM. From here to JUPITER: identifying new patients for statin therapy using data from the 1999–2004 National Health and Nutrition Examination Survey. Circ Cardiovasc Qual Outcomes 2009; 2:41–48.
- Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006; 34:153–162.
- Vladutiu GD. Genetic predisposition to statin myopathy. Curr Opin Rheumatol 2008; 20:648–655.
- Phillips PS, Phillips CT, Sullivan MJ, et al. Statin myotoxicity is associated with changes in the cardiopulmonary function. Atherosclerosis 2004; 177:183–188.
- Phillips PS, Ciaraldi TP, Kim DL, et al. Myotoxic reactions to lipidlowering therapy are associated with altered oxidation of fatty acids. Endocrine 2009; 35:38–46.
- Phillips PS, Haas RH. Statin myopathy as a metabolic muscle disease. Expert Rev Cardiovasc Ther 2008; 6:971–978.
- Löfberg M, Jänkälä H, Paetau A, et al. Metabolic causes of recurrent rhabdomyolysis. Acta Neurol Scand 1998; 98:268–275.
- Havranek JM, Wolfsen A, Warnke GA, et al. Monotherapy with Ezetimibe Causing Myopathy. Am J Med 2006; 119:285–286.
- Phillips PS. Ezetimibe and statin-associated myopathy. Ann Intern Med 2004; 141:649.
- Durst R, Rund D, Schurr D, et al. One year experience with a low density lipoprotein apheresis system. Isr Med Assoc J 2002; 4:677–680.
- Roberts WC. The rule of 5 and the rule of 7 in lipid-lowering by statin drugs. Am J Cardiol 1997; 80:106–107.
- Brewer HB. Benefit-risk assessment of rosuvastatin 10 to 40 milligrams. Am J Cardiol 2003; 92:23K–29K.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther 2006; 28:933–942.
- Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every-other-day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother 2008; 42:341–346.
- Backes JM, Moriarty PM, Ruisinger JF, et al. Effects of once weekly rosuvastatin among patients with a prior statin intolerance. Am J Cardiol 2007; 100:554–555.
- Antons KA, Williams CD, Baker SK, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med 2006; 119:400–409.
- Jacobson TA. Toward “pain-free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc 2008; 83:687–700.
- Phillips PS, Haas RH, Bannykh S, et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 2002; 137:581–585.
- Sinzinger H, O’Grady J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br J Clin Pharmacol 2004; 57:525–528.
- Wysowski DK, Chang JT. Alendronate and risedronate: reports of severe bone, joint, and muscle pain. Arch Intern Med 2005; 165:346–347.
- Martino S, Disch D, Dowsett SA, et al. Safety assessment of raloxifene over eight years in a clinical trial setting. Curr Med Res Opin 2005; 21:1441–1452.
- Phillips PS, Kimura BJ, Kelley R, et al. Feasibility of using the internet to perform medical research: observations from a statin-myopathy public information web site. Circulation 2003; 107:e7001–e7039.
- Chazerain P, Hayem G, Hamza S, et al. Four cases of tendinopathy in patients on statin therapy. Joint Bone Spine 2001; 68:430–433.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004; 44:720–732.
- Pedersen TR, Olsson AG, Faergeman O, et al. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S). Circulation 1998; 97:1453–1460.
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20, 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med 1996; 335:1001–1009.
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 1995; 333:1301–1307.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHATLLT). JAMA 2002; 288:2998–3007.
- Stein EA, Ballantyne CM, Windler E, et al. Efficacy and tolerability of fluvastatin XL 80 mg alone, ezetimibe alone, and the combination of fluvastatin XL 80 mg with ezetimibe in patients with a history of muscle-related side effects with other statins. Am J Cardiol 2008; 101:490–496.
- Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther 2006; 28:933–942.
- Phillips PS, Gray NL, McDonald FG, et al. Colesevelam is safe and effective in patients with statin myotoxicity. Arterioscler Thromb Vasc Biol 2005 25:E-97.
- Phillips PS. Balancing randomized trials with anecdote. Ann Intern Med 2009; 150:885–886.
- Gordon RY, Cooperman T, Obermeyer W, et al. Marked variability of monacolin levels in commercial red yeast rice products: buyer beware! Arch Intern Med 2010; 170:1722–1727.
- Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004; 292:2585–2590.
- Gaist D, Rodríguez LA, Huerta C, et al. Lipid-lowering drugs and risk of myopathy: a population-based follow-up study. Epidemiology 2001; 12:565–569.
- Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005; 366:1849–1861.
- ACCORD Study Group; Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Foley KA, Simpson RJ, Crouse JR, et al. Effectiveness of statin titration on low-density lipoprotein cholesterol goal attainment in patients at high risk of atherogenic events. Am J Cardiol 2003; 92:79–81.
- O’Meara JG, Kardia SL, Armon JJ, et al. Ethnic and sex differences in the prevalence, treatment, and control of dyslipidemia among hypertensive adults in the GENOA study. Arch Intern Med 2004; 164:1313–1318.
- Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002; 288:462–467.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore) 1982; 61:141–152.
- Ward MM. Factors predictive of acute renal failure in rhabdomyolysis. Arch Intern Med 1988; 148:1553–1557.
- McKenney JM, Davidson MH, Jacobson TA, et al. National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–84C.
- Pasternak RC, Smith SC, Bairey-Merz CN, et al. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol 2002; 40:567–572.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Linares LA, Golomb BA, Jaojoco JA, et al. The modern spectrum of rhabdomyolysis: drug toxicity revealed by creatine kinase screening. Curr Drug Saf 2009; 4:181–187.
- Armitage J. The safety of statins in clinical practice. Lancet 2007; 370:1781–1790.
- Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Sinzinger H, Wolfram R, Peskar BA. Muscular side effects of statins. J Cardiovasc Pharmacol 2002; 40:163–171.
- de Sauvage Nolting PR, Buirma RJ, et al. Two-year efficacy and safety of simvastatin 80 mg in familial hypercholesterolemia (the Examination of Probands and Relatives in Statin Studies With Familial Hypercholesterolemia [ExPRESS FH]). Am J Cardiol 2002; 90:181–184.
- Franc S, Dejager S, Bruckert E, et al. A comprehensive description of muscle symptoms associated with lipid-lowering drugs. Cardiovasc Drugs Ther 2003; 17:459–465.
- Kashani A, Phillips CO, Foody JM, et al. Risks associated with statin therapy: a systematic overview of randomized clinical trials. Circulation 2006; 114:2788–2797.
- Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med 2008; 23:1182–1186.
- Spatz ES, Canavan ME, Desai MM. From here to JUPITER: identifying new patients for statin therapy using data from the 1999–2004 National Health and Nutrition Examination Survey. Circ Cardiovasc Qual Outcomes 2009; 2:41–48.
- Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006; 34:153–162.
- Vladutiu GD. Genetic predisposition to statin myopathy. Curr Opin Rheumatol 2008; 20:648–655.
- Phillips PS, Phillips CT, Sullivan MJ, et al. Statin myotoxicity is associated with changes in the cardiopulmonary function. Atherosclerosis 2004; 177:183–188.
- Phillips PS, Ciaraldi TP, Kim DL, et al. Myotoxic reactions to lipidlowering therapy are associated with altered oxidation of fatty acids. Endocrine 2009; 35:38–46.
- Phillips PS, Haas RH. Statin myopathy as a metabolic muscle disease. Expert Rev Cardiovasc Ther 2008; 6:971–978.
- Löfberg M, Jänkälä H, Paetau A, et al. Metabolic causes of recurrent rhabdomyolysis. Acta Neurol Scand 1998; 98:268–275.
- Havranek JM, Wolfsen A, Warnke GA, et al. Monotherapy with Ezetimibe Causing Myopathy. Am J Med 2006; 119:285–286.
- Phillips PS. Ezetimibe and statin-associated myopathy. Ann Intern Med 2004; 141:649.
- Durst R, Rund D, Schurr D, et al. One year experience with a low density lipoprotein apheresis system. Isr Med Assoc J 2002; 4:677–680.
- Roberts WC. The rule of 5 and the rule of 7 in lipid-lowering by statin drugs. Am J Cardiol 1997; 80:106–107.
- Brewer HB. Benefit-risk assessment of rosuvastatin 10 to 40 milligrams. Am J Cardiol 2003; 92:23K–29K.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther 2006; 28:933–942.
- Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every-other-day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother 2008; 42:341–346.
- Backes JM, Moriarty PM, Ruisinger JF, et al. Effects of once weekly rosuvastatin among patients with a prior statin intolerance. Am J Cardiol 2007; 100:554–555.
- Antons KA, Williams CD, Baker SK, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med 2006; 119:400–409.
- Jacobson TA. Toward “pain-free” statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc 2008; 83:687–700.
- Phillips PS, Haas RH, Bannykh S, et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 2002; 137:581–585.
- Sinzinger H, O’Grady J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br J Clin Pharmacol 2004; 57:525–528.
- Wysowski DK, Chang JT. Alendronate and risedronate: reports of severe bone, joint, and muscle pain. Arch Intern Med 2005; 165:346–347.
- Martino S, Disch D, Dowsett SA, et al. Safety assessment of raloxifene over eight years in a clinical trial setting. Curr Med Res Opin 2005; 21:1441–1452.
- Phillips PS, Kimura BJ, Kelley R, et al. Feasibility of using the internet to perform medical research: observations from a statin-myopathy public information web site. Circulation 2003; 107:e7001–e7039.
- Chazerain P, Hayem G, Hamza S, et al. Four cases of tendinopathy in patients on statin therapy. Joint Bone Spine 2001; 68:430–433.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004; 44:720–732.
- Pedersen TR, Olsson AG, Faergeman O, et al. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S). Circulation 1998; 97:1453–1460.
- MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20, 536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998; 339:1349–1357.
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med 1996; 335:1001–1009.
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 1995; 333:1301–1307.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHATLLT). JAMA 2002; 288:2998–3007.
- Stein EA, Ballantyne CM, Windler E, et al. Efficacy and tolerability of fluvastatin XL 80 mg alone, ezetimibe alone, and the combination of fluvastatin XL 80 mg with ezetimibe in patients with a history of muscle-related side effects with other statins. Am J Cardiol 2008; 101:490–496.
- Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther 2006; 28:933–942.
- Phillips PS, Gray NL, McDonald FG, et al. Colesevelam is safe and effective in patients with statin myotoxicity. Arterioscler Thromb Vasc Biol 2005 25:E-97.
- Phillips PS. Balancing randomized trials with anecdote. Ann Intern Med 2009; 150:885–886.
- Gordon RY, Cooperman T, Obermeyer W, et al. Marked variability of monacolin levels in commercial red yeast rice products: buyer beware! Arch Intern Med 2010; 170:1722–1727.
- Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004; 292:2585–2590.
- Gaist D, Rodríguez LA, Huerta C, et al. Lipid-lowering drugs and risk of myopathy: a population-based follow-up study. Epidemiology 2001; 12:565–569.
- Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005; 366:1849–1861.
- ACCORD Study Group; Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574.
KEY POINTS
- There is little consensus on the definition of statin-induced myopathy, and it is underdiagnosed. The incidence of statin-induced muscle toxicity in randomized controlled trials is lower than in clinical practice.
- Abnormal pharmacokinetic activity contributes to toxicity, but some patients may be predisposed by underlying metabolic muscle disorders.
- A focused history and neuromusculoskeletal examination are important in the evaluation of muscle complaints that may be induced by statins.
- In patients with possible statin-induced myopathy, assessing the risks and benefits of statin therapy is essential.
- For patients who cannot tolerate statin therapy, alternatives include a “statin holiday” followed by a rechallenge with a different statin, intermittent rosuvastatin (Crestor), or resin therapy. Sometimes the best alternative is a compromise between the goal level for low-density-lipoprotein cholesterol and the level achievable with alternative therapy.
Correction: Giant cell arteritis
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.