User login
Positively False
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A51‐year‐old woman presented after 5 days of fever, rigors, anorexia, right‐sided abdominal pain, nausea, and dizziness. She had 2 loose stools the day before admission, without blood or mucus, but otherwise had recently been constipated. She denied cough, shortness of breath, chest pain, headache, sore throat, rash, arthritis, or dysuria.
In a 51‐year‐old woman with right‐sided abdominal pain and systemic symptoms, major concerns include biliary disease, liver abscess, or appendicitis. Right‐sided diverticulitis would be more unusual. Pyelonephritis infrequently presents with epigastric and lower quadrant pain, instead of flank pain. Basilar pneumonia may present with abdominal pain, but this is less likely in the absence of respiratory symptoms.
The patient used an albuterol inhaler for mild asthma and had experienced an episode of herpes zoster 7 years prior, but was otherwise well. Her surgical history was notable for a remote appendectomy. She was a native of the Dominican Republic who had lived in the United States for the past 20 years. She visited the Dominican Republic for 3 weeks every year, with her last visit occurring about 10 months before. She was a cleaning and maintenance worker. She had 2 adult children in good health, was divorced from her husband, and had not been sexually active for the past 8 years. The patient had no pets or other animal exposures. She did not smoke, drink alcohol, or use intravenous drugs.
The remote episode of shingles makes me a bit worried about chronic human immunodeficiency virus (HIV) infection. As a native of and traveler to the Dominican Republic, she is at risk for a variety of tropical pathogens. Hyperinfection syndrome from strongyloides can cause fever and bacteremia, but this is almost always associated with significant immunosuppression. Dengue fever has become very common in the Caribbean, but should occur within 2 weeks of travel. Her work in cleaning and maintenance might bring her into contact with rats and mice, putting her at risk for leptospirosis. This can present as a fairly nonspecific febrile syndrome, but this is unlikely without a major complaint of headache.
The patient appeared fatigued. Her temperature was 39.7C, her heart rate was 110 beats per minute, and her blood pressure 80/62 mm Hg. The oropharynx was normal. Mild cervical lymphadenopathy was present (less than 1 cm in diameter). The chest was clear and the cardiac examination unremarkable. Bowel sounds were present. Moderate right‐sided abdominal tenderness was noted, somewhat more marked in the right lower quadrant, without guarding or rebound. There was no hepatosplenomegaly. There was no rash. A bedside right upper quadrant ultrasound was negative for gallstones.
Her low blood pressure is concerning for bacterial sepsis. The negative right upper quadrant ultrasound makes cholecystitis or cholangitis less likely, but does not exclude diverticulitis or pelvic inflammatory disease. She lacks peritoneal signs, but they may be absent in these conditions. Another worrisome finding on her physical examination is cervical lymphadenopathy. In an older patient, this raises the specter of malignancy. In a younger patient, it could suggest a mononucleosis syndrome from Epstein‐Barr virus (EBV) or cytomegalovirus (CMV). In addition, HIV must be considered in any patient with unexplained lymphadenopathy.
She received intravenous levofloxacin and 1 L of intravenous normal saline, with a rise in her blood pressure to 100/59 mm Hg. Her white blood cell count was 5.0, with 71% polys and 9% bands. The hematocrit was 32%, with a normal mean corpuscular volume. The erythrocyte sedimentation rate (ESR) was 109 mm/hour. The platelet count, serum electrolytes, creatinine, aminotransferases, alkaline phosphatase, bilirubin, amylase, and lipase were normal. The serum level of lactate dehydrogenase (LDH) was 498 unit/L (normal range, 107231 units/L). Computed tomography (CT) scan of the abdomen showed multiple enlarged lymph nodes up to 1.2 cm in size along the gastrohepatic ligament and the mesentery, with mild associated fat stranding, consistent with mesenteric lymphadenitis.
Her blood pressure has responded to fluids; perhaps she was just volume‐depleted from not eating for several days. She has bandemia, which is consistent with acute bacterial infection, but might also signify a stress response. The low hematocrit and high ESR raise the possibility of anemia of chronic disease; perhaps her illness is more longstanding than her presentation suggests. Mesenteric lymphadenitis may be related to EBV and HIV, but in a patient originally from the Caribbean, it raises the possibility of gastrointestinal tuberculosis or histoplasmosis. Human T‐cell lymphotropic virus type 1 (HTLV‐1) is also endemic in the Caribbean, and may cause adult T‐cell leukemia/lymphoma (ATLL), which could explain her lymphadenopathy and elevated LDH. Mesenteric lymphadenitis is also characteristic of several bacterial infections, especially Yersinia, Salmonella, and Bartonella. The lack of diarrhea makes yersiniosis doubtful, and the absence of cat exposure makes bartonellosis unlikely. Salmonella infection is also associated with diarrhea, except for typhoid fever, in which patients have diarrhea, constipation, or normal stools.
A urine culture and 3 sets of blood cultures obtained prior to the initiation of antibiotics were negative. A blood smear showed no malaria or babesia parasites. The patient's fever continued for the first 2 days of her hospitalization, but subsequently abated. The patient had no loose stools during her hospitalization. Serologies for EBV, hepatitis A, CMV, and toxoplasmosis were indicative of remote infection. Serum rapid plasma reagin (RPR), Bartonella antibodies, antinuclear antibodies, hepatitis B surface antigen, and hepatitis C antibody were negative. Tuberculin skin testing and urinary histoplasma antigen were negative. By the fifth hospital day, she had been afebrile for over 48 hours, and her abdominal pain had improved, though it had not completely resolved. She was discharged to complete a 10‐day course of levofloxacin.
It is not clear to me whether she has just experienced a spontaneous remission in her illness, or whether her disease course has truly been modified with antibiotics. Could she have typhoid fever or another salmonellosis? The patient has not traveled abroad in several months. If she had typhoid fever, the source of infection would have to be imported food, or exposure to a family member who was a carrier. The negative tuberculin skin test makes tuberculosis less likely, but does not exclude it entirely. Similarly, while a negative histoplasma urinary antigen essentially rules out acute disseminated histoplasmosis, as would be seen in acquired immune deficiency syndrome (AIDS), it is less sensitive for more chronic forms of disseminated histoplasmosis, including gastrointestinal involvement.
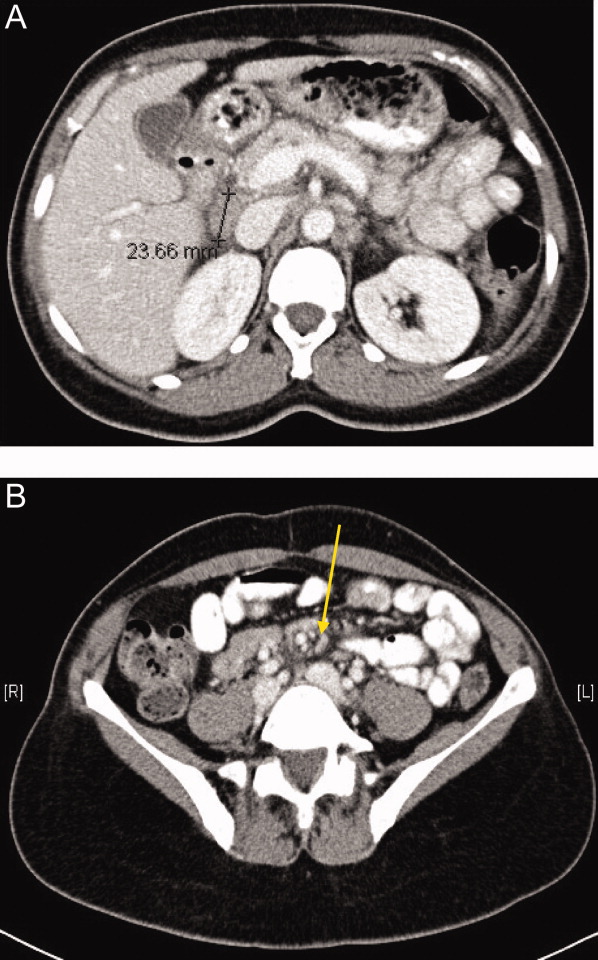
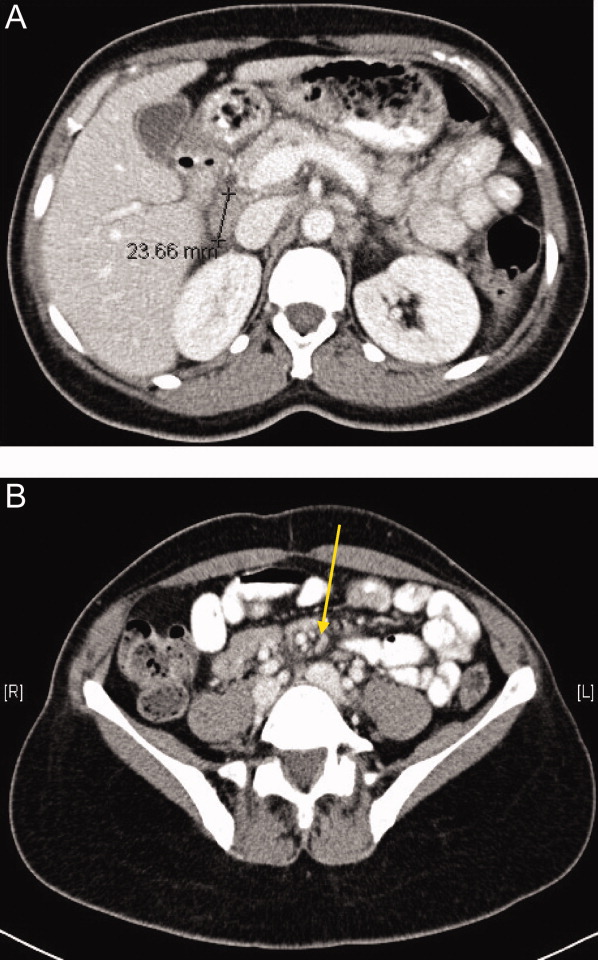
One week later, the patient returned to the emergency department with recurrent abdominal pain, anorexia, fever, night sweats, and increased swelling of the lymph nodes in her neck. She had lost a total of 4.4 kg (10 lb) since the onset of her illness. Physical examination revealed moderate (up to 2 cm), slightly tender cervical and inguinal lymphadenopathy, and continued moderate right‐sided abdominal tenderness. Mesenteric, retroperitoneal, and inguinal lymphadenopathy was more prominent than on the prior CT scan (Figure 1).She was told by the emergency room physicians that the HIV test obtained during the prior hospitalization was positive. Further questioning elicited that her estranged husband had been promiscuous prior to their separation. She was transferred to a second hospital for further care. Blood cultures were sent for fungi and acid‐fast bacilli.
Everyone with fever of unknown origin (FUO) deserves an HIV test. Is this just lymphadenopathy from HIV, or is she suffering from an opportunistic infection? Mycobacterium avium infection is an attractive explanation for her fever, abdominal pain, and lymphadenopathy, but I would hold off on empiric treatment until the results of a CD4+ cell count were available. Could she have a secondary, HIV‐related lymphoma?
Full review of records from the outside hospital showed that the patient had a positive HIV enzyme immunoassay, with an indeterminate HIV Western blot. The patient's CD4 cell count was 313/cm3, with a CD4/CD8 ratio within the normal range. Her HIV enzyme immunoassay was repeated and found to be negative, and the HIV viral load was undetectable.
The HIV enzyme immunoassay is only a screening test, and must be confirmed with a positive HIV Western blot. (In most clinical laboratories, this is done automatically before the test is reported.) Indeterminate HIV Western blots are common in acute HIV infection, but outside of this setting, most patients with indeterminate HIV Western blots turn out not to have HIV infection. I am still very concerned about lymphoma, and would pursue a lymph node biopsy. Disseminated tuberculosis is still possible.
Biopsy of a right inguinal lymph node was performed on the third day of the second hospitalization. The patient was persistently febrile, developed swelling of the knees, ankles, and interphalangeal joints of the second and third digits of the left hand, and complained of pruritus. Anti‐double‐stranded DNA antibody, antineutrophilic cytoplasmic antibody, and Brucella antibody were negative. Antibody against cyclic citrullinated peptide (CCP) was weakly positive.
Now she has a more florid syndrome, with arthritic symptoms. Sarcoidosis is an attractive explanation for her fever, lymphadenopathy, and arthritis. Lupus could explain some features of her presentation, but the negative serology makes it unlikely. Antibody to cyclic citrullinated peptide is a newer diagnostic test for rheumatoid arthritis. Although anti‐CCP is more specific than rheumatoid factor for the diagnosis of rheumatoid arthritis, this result could still be a false positive, particularly given the low titer. As well, the patient has more impressive lymphadenopathy than is usual for rheumatoid arthritis. Reactive arthritis can follow enteric infection with Campylobacter, Salmonella, Yersinia, and Shigella, but lymphadenopathy is not a feature. Arthritis may be prominent in parvovirus B19, rubella, disseminated gonococcal infection, and Lyme disease, but mesenteric lymphadenitis and a prolonged, waxing and waning course would not be expected with any of these. I worry that the arthritis and pruritus are paraneoplastic manifestations of lymphoma.
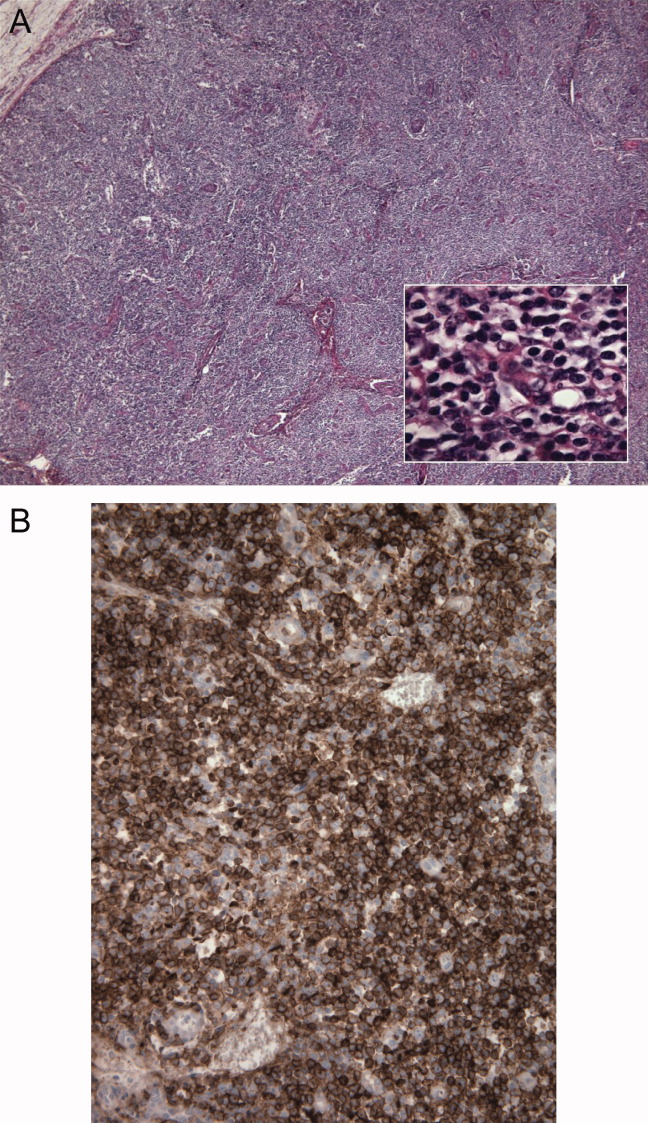
The inguinal lymph node biopsy showed markedly distorted nodal architecture with an atypical proliferation of small‐sized to large‐sized lymphoid cells, with predominantly round nuclei, vesicular chromatin, small nucleoli, and variable amounts of clear to eosinophilic cytoplasm (Figure 2). Residual follicles and occasional apoptotic bodies and mitoses were seen. Large B‐cells were frequently present, which stained positive for EBV‐associated mRNA by in situ hybridization studies. Immunoperoxidase staining revealed that the atypical cell population was largely composed of CD4+ T‐cells. T‐cell receptor gene rearrangement studies demonstrated that the T‐cell population was monoclonal. These results were consistent with angioimmunoblastic T‐cell lymphoma (AITL). Positron emission tomography (PET) scanning was performed, showing diffuse fluorodeoxyglucose (FDG)‐avid lymphadenopathy involving cervical, axillary, mediastinal, retroperitoneal, mesenteric, and inguinal lymph nodes, up to 2 cm in diameter. The patient completed 6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), as well as experimental treatment with denileukin diftitox. Her fever and arthritis subsided quickly, and she was clinically well and free of disease by CT scans and PET scans 1 year after diagnosis.
COMMENTARY
The differential diagnosis of FUO is one of the largest in medicine, encompassing a dizzying range of infectious, inflammatory, and neoplastic conditions. This has made the development of standardized diagnostic algorithms difficult.1 The workup of patients with FUO begins with a detailed history and physical examination, followed by a core set of microbiology cultures, imaging studies, and blood tests on all patients. Further testing is individualized, based on key clinical findings, also known as pivot points.2 Unfortunately, many of the diseases presenting as FUO have overlapping symptoms and signs, somewhat limiting the utility of pivot points. In a prospective study, 81% of these potentially diagnostic clues were misleading, although 19% of them contributed to the final diagnosis.3 Key clinical findings in patients with FUO may trigger a barrage of diagnostic tests, often leading to a large number of false‐positive results.4 Clinicians who investigate patients with FUO must remember that many clues are diagnostic dead ends, and should be wary of drawing positively‐false conclusions.
Fever pattern is usually not helpful in the diagnosis of FUO, with occasional exceptions, such as the tertian and quartan fevers in some forms of malaria. A minority of patients with Hodgkin's disease have Pel‐Ebstein fevers, in which 1 to 2 weeks of fever alternate with an afebrile period of similar or longer duration. More often, fever in lymphoma waxes and wanes unpredictably,5 a circumstance that may lead to the mistaken impression of response to antibiotics, as in this case.
Tissue biopsies are helpful in FUO when suggestive findings are present on physical examination or imaging studies. In patients with lymphadenopathy and FUO, lymph node biopsy is a high‐yield procedure, contributing to the final diagnosis 46% of the time. This is exceeded only by biopsies of skin lesions, which have a 63% diagnostic yield in FUO.3 In older patients with FUO, temporal artery biopsies have a significant diagnostic yield, in the range of 16% to 17%. Liver biopsies have a similar yield (14‐17%), but a higher risk of complications. Bone marrow biopsies have a low yield in most FUO patients.1
AITL makes up 1% of all non‐Hodgkin's lymphomas. AITL was once known as angioimmunoblastic lymphadenopathy, and was thought to be either a disorder of immune regulation or a premalignant lymphoid disease. However, molecular diagnostic techniques have established that monoclonal T‐cell populations and cytogenetic abnormalities are usually present at the time of diagnosis.6, 7 The prognosis in AITL is unfavorable. Disease is usually widespread at the time of diagnosis. Most patients achieve complete remission with anthracycline‐based chemotherapy, such as CHOP, but the duration of remission is often brief, and median survival after diagnosis is only 3 years.6 Novel treatments under investigation include denileukin diftitox,8 a fusion protein of interleukin‐2 (IL‐2) conjugated to diphtheria toxin, which leads to apoptosis of cells expressing the IL‐2 receptor; rituximab, which targets the reactive population of B‐cells in AITL, rather than the malignant clone of T‐cells; and antiangiogenic therapy, such as thalidomide.9
The diagnosis of AITL is usually elusive, and the average patient has symptoms for 4 months prior to diagnosis.6 This patient's clinical presentation, while certainly not specific, was typical of AITL. AITL usually presents as fever of unknown origin with generalized, nonbulky lymphadenopathy. Fever is present in 57% of patients with AITL, and up to 2% of FUO is caused by AITL.6, 10 Other features of this patient's illness, such as night sweats, weight loss, pruritus, and arthritis, are common in AITL.
T‐cell depletion and immune dysregulation are frequent in AITL, explaining why AITL shares a number of clinical features with HIV disease. These include a high incidence of drug rashes, immune thrombocytopenic purpura, polyclonal hypergammaglobulinemia, and autoantibodies.6, 7 As with HIV patients, death in AITL is often due to opportunistic infections or diffuse large B‐cell lymphomas.7 Over 95% of patients with AITL display a proliferation of EBV‐infected B cells, presumably from immune dysregulation, and the B‐cell lymphomas in these patients are usually EBV‐positive.11
Because AITL is a proinflammatory state, false‐positive antibody tests are fairly common. The occasional occurrence of positive HIV enzyme immunoassays, with indeterminate Western blot results, may be a particular source of diagnostic confusion.12, 13 The significance of indeterminate Western blots is often erroneously communicated to patients, as happened here. Indeterminate HIV Western blots may be seen in HIV seroconversion, HIV‐2 infection, or advanced HIV disease with loss of core antibody. They may also result from laboratory error, multiparity, syphilis, malaria, or cross‐reacting antibodies in autoimmune diseases. Indeterminate HIV Western blots in low‐risk patients usually do not represent true HIV infection.14
KEY POINTS FOR HOSPITALISTS
-
FUO is one of the most challenging diagnoses faced by hospitalists. Biopsies of new skin lesions and enlarged lymph nodes are particularly high‐yield diagnostic procedures in FUO. Fever pattern is generally not helpful in the diagnosis of FUO, with few exceptions, such as the tertian fevers of malaria.
-
Misleading and false‐positive tests results often occur in the course of FUO evaluation, due to the sheer number of tests ordered, and the higher likelihood of false‐positive serologic tests in the setting of inflammatory states.
-
AITL may be an elusive diagnosis in FUO with diverse clinical features, including weight loss, night sweats, rashes, arthritis, autoantibodies, immune dysregulation, and opportunistic infections. The prognosis traditionally has been guarded, but may be more hopeful in an era of emerging molecular therapies.
- ,,.A comprehensive evidence‐based approach to fever of unknown origin.Arch Intern Med.2003;163:545–551.
- ,.The art of diagnosis: solving the clinicopathological exercise.N Engl J Med.1982;306:1263–1268.
- ,,, et al.A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol.Medicine (Baltimore).2007;86:26–38.
- ,,.Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:401–414.
- ,.Neoplastic diseases. In:Murray HW, ed.FUO: Fever of Undetermined Origin.New York, NY:Futura Publishing;1983:39–48.
- ,,, et al.Angioimmunoblastic T‐cell lymphoma: clinical and laboratory features at diagnosis in 77 patients.Medicine (Baltimore).2007;86:282–292.
- ,,.Angioimmunoblastic T‐cell lymphoma.Br J Haematol.2003;121:681–691.
- ,,, et al.Phase II trial of denileukin diftitox for relapsed/refractory T‐cell non‐Hodgkin lymphoma.Br J Haematol.2007;136:439–447.
- ,.Angioimmunoblastic T‐cell lymphoma: still a dismal prognosis with current treatment approaches.Leuk Lymphoma.2007;48:645–646.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:392–400.
- ,,, et al.Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression.Am J Surg Pathol.2007;31:1077–1088.
- ,,,.Angioimmunoblastic lymphadenopathy, immunoblastic lymphoma, and false‐positive seroconversion for human immunodeficiency virus.Ann Intern Med.1987;107:114.
- ,.Angioimmunoblastic T‐cell lymphoma associated with an antibody to human immunodeficiency virus protein.Int J Hematol.2003;78:160–162.
- ,,.Communicating indeterminate HIV Western blot test results to clients: an observational study of three community testing sites.AIDS Patient Care STDS.2006;20:620–627.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A51‐year‐old woman presented after 5 days of fever, rigors, anorexia, right‐sided abdominal pain, nausea, and dizziness. She had 2 loose stools the day before admission, without blood or mucus, but otherwise had recently been constipated. She denied cough, shortness of breath, chest pain, headache, sore throat, rash, arthritis, or dysuria.
In a 51‐year‐old woman with right‐sided abdominal pain and systemic symptoms, major concerns include biliary disease, liver abscess, or appendicitis. Right‐sided diverticulitis would be more unusual. Pyelonephritis infrequently presents with epigastric and lower quadrant pain, instead of flank pain. Basilar pneumonia may present with abdominal pain, but this is less likely in the absence of respiratory symptoms.
The patient used an albuterol inhaler for mild asthma and had experienced an episode of herpes zoster 7 years prior, but was otherwise well. Her surgical history was notable for a remote appendectomy. She was a native of the Dominican Republic who had lived in the United States for the past 20 years. She visited the Dominican Republic for 3 weeks every year, with her last visit occurring about 10 months before. She was a cleaning and maintenance worker. She had 2 adult children in good health, was divorced from her husband, and had not been sexually active for the past 8 years. The patient had no pets or other animal exposures. She did not smoke, drink alcohol, or use intravenous drugs.
The remote episode of shingles makes me a bit worried about chronic human immunodeficiency virus (HIV) infection. As a native of and traveler to the Dominican Republic, she is at risk for a variety of tropical pathogens. Hyperinfection syndrome from strongyloides can cause fever and bacteremia, but this is almost always associated with significant immunosuppression. Dengue fever has become very common in the Caribbean, but should occur within 2 weeks of travel. Her work in cleaning and maintenance might bring her into contact with rats and mice, putting her at risk for leptospirosis. This can present as a fairly nonspecific febrile syndrome, but this is unlikely without a major complaint of headache.
The patient appeared fatigued. Her temperature was 39.7C, her heart rate was 110 beats per minute, and her blood pressure 80/62 mm Hg. The oropharynx was normal. Mild cervical lymphadenopathy was present (less than 1 cm in diameter). The chest was clear and the cardiac examination unremarkable. Bowel sounds were present. Moderate right‐sided abdominal tenderness was noted, somewhat more marked in the right lower quadrant, without guarding or rebound. There was no hepatosplenomegaly. There was no rash. A bedside right upper quadrant ultrasound was negative for gallstones.
Her low blood pressure is concerning for bacterial sepsis. The negative right upper quadrant ultrasound makes cholecystitis or cholangitis less likely, but does not exclude diverticulitis or pelvic inflammatory disease. She lacks peritoneal signs, but they may be absent in these conditions. Another worrisome finding on her physical examination is cervical lymphadenopathy. In an older patient, this raises the specter of malignancy. In a younger patient, it could suggest a mononucleosis syndrome from Epstein‐Barr virus (EBV) or cytomegalovirus (CMV). In addition, HIV must be considered in any patient with unexplained lymphadenopathy.
She received intravenous levofloxacin and 1 L of intravenous normal saline, with a rise in her blood pressure to 100/59 mm Hg. Her white blood cell count was 5.0, with 71% polys and 9% bands. The hematocrit was 32%, with a normal mean corpuscular volume. The erythrocyte sedimentation rate (ESR) was 109 mm/hour. The platelet count, serum electrolytes, creatinine, aminotransferases, alkaline phosphatase, bilirubin, amylase, and lipase were normal. The serum level of lactate dehydrogenase (LDH) was 498 unit/L (normal range, 107231 units/L). Computed tomography (CT) scan of the abdomen showed multiple enlarged lymph nodes up to 1.2 cm in size along the gastrohepatic ligament and the mesentery, with mild associated fat stranding, consistent with mesenteric lymphadenitis.
Her blood pressure has responded to fluids; perhaps she was just volume‐depleted from not eating for several days. She has bandemia, which is consistent with acute bacterial infection, but might also signify a stress response. The low hematocrit and high ESR raise the possibility of anemia of chronic disease; perhaps her illness is more longstanding than her presentation suggests. Mesenteric lymphadenitis may be related to EBV and HIV, but in a patient originally from the Caribbean, it raises the possibility of gastrointestinal tuberculosis or histoplasmosis. Human T‐cell lymphotropic virus type 1 (HTLV‐1) is also endemic in the Caribbean, and may cause adult T‐cell leukemia/lymphoma (ATLL), which could explain her lymphadenopathy and elevated LDH. Mesenteric lymphadenitis is also characteristic of several bacterial infections, especially Yersinia, Salmonella, and Bartonella. The lack of diarrhea makes yersiniosis doubtful, and the absence of cat exposure makes bartonellosis unlikely. Salmonella infection is also associated with diarrhea, except for typhoid fever, in which patients have diarrhea, constipation, or normal stools.
A urine culture and 3 sets of blood cultures obtained prior to the initiation of antibiotics were negative. A blood smear showed no malaria or babesia parasites. The patient's fever continued for the first 2 days of her hospitalization, but subsequently abated. The patient had no loose stools during her hospitalization. Serologies for EBV, hepatitis A, CMV, and toxoplasmosis were indicative of remote infection. Serum rapid plasma reagin (RPR), Bartonella antibodies, antinuclear antibodies, hepatitis B surface antigen, and hepatitis C antibody were negative. Tuberculin skin testing and urinary histoplasma antigen were negative. By the fifth hospital day, she had been afebrile for over 48 hours, and her abdominal pain had improved, though it had not completely resolved. She was discharged to complete a 10‐day course of levofloxacin.
It is not clear to me whether she has just experienced a spontaneous remission in her illness, or whether her disease course has truly been modified with antibiotics. Could she have typhoid fever or another salmonellosis? The patient has not traveled abroad in several months. If she had typhoid fever, the source of infection would have to be imported food, or exposure to a family member who was a carrier. The negative tuberculin skin test makes tuberculosis less likely, but does not exclude it entirely. Similarly, while a negative histoplasma urinary antigen essentially rules out acute disseminated histoplasmosis, as would be seen in acquired immune deficiency syndrome (AIDS), it is less sensitive for more chronic forms of disseminated histoplasmosis, including gastrointestinal involvement.
One week later, the patient returned to the emergency department with recurrent abdominal pain, anorexia, fever, night sweats, and increased swelling of the lymph nodes in her neck. She had lost a total of 4.4 kg (10 lb) since the onset of her illness. Physical examination revealed moderate (up to 2 cm), slightly tender cervical and inguinal lymphadenopathy, and continued moderate right‐sided abdominal tenderness. Mesenteric, retroperitoneal, and inguinal lymphadenopathy was more prominent than on the prior CT scan (Figure 1).She was told by the emergency room physicians that the HIV test obtained during the prior hospitalization was positive. Further questioning elicited that her estranged husband had been promiscuous prior to their separation. She was transferred to a second hospital for further care. Blood cultures were sent for fungi and acid‐fast bacilli.
Everyone with fever of unknown origin (FUO) deserves an HIV test. Is this just lymphadenopathy from HIV, or is she suffering from an opportunistic infection? Mycobacterium avium infection is an attractive explanation for her fever, abdominal pain, and lymphadenopathy, but I would hold off on empiric treatment until the results of a CD4+ cell count were available. Could she have a secondary, HIV‐related lymphoma?
Full review of records from the outside hospital showed that the patient had a positive HIV enzyme immunoassay, with an indeterminate HIV Western blot. The patient's CD4 cell count was 313/cm3, with a CD4/CD8 ratio within the normal range. Her HIV enzyme immunoassay was repeated and found to be negative, and the HIV viral load was undetectable.
The HIV enzyme immunoassay is only a screening test, and must be confirmed with a positive HIV Western blot. (In most clinical laboratories, this is done automatically before the test is reported.) Indeterminate HIV Western blots are common in acute HIV infection, but outside of this setting, most patients with indeterminate HIV Western blots turn out not to have HIV infection. I am still very concerned about lymphoma, and would pursue a lymph node biopsy. Disseminated tuberculosis is still possible.
Biopsy of a right inguinal lymph node was performed on the third day of the second hospitalization. The patient was persistently febrile, developed swelling of the knees, ankles, and interphalangeal joints of the second and third digits of the left hand, and complained of pruritus. Anti‐double‐stranded DNA antibody, antineutrophilic cytoplasmic antibody, and Brucella antibody were negative. Antibody against cyclic citrullinated peptide (CCP) was weakly positive.
Now she has a more florid syndrome, with arthritic symptoms. Sarcoidosis is an attractive explanation for her fever, lymphadenopathy, and arthritis. Lupus could explain some features of her presentation, but the negative serology makes it unlikely. Antibody to cyclic citrullinated peptide is a newer diagnostic test for rheumatoid arthritis. Although anti‐CCP is more specific than rheumatoid factor for the diagnosis of rheumatoid arthritis, this result could still be a false positive, particularly given the low titer. As well, the patient has more impressive lymphadenopathy than is usual for rheumatoid arthritis. Reactive arthritis can follow enteric infection with Campylobacter, Salmonella, Yersinia, and Shigella, but lymphadenopathy is not a feature. Arthritis may be prominent in parvovirus B19, rubella, disseminated gonococcal infection, and Lyme disease, but mesenteric lymphadenitis and a prolonged, waxing and waning course would not be expected with any of these. I worry that the arthritis and pruritus are paraneoplastic manifestations of lymphoma.
The inguinal lymph node biopsy showed markedly distorted nodal architecture with an atypical proliferation of small‐sized to large‐sized lymphoid cells, with predominantly round nuclei, vesicular chromatin, small nucleoli, and variable amounts of clear to eosinophilic cytoplasm (Figure 2). Residual follicles and occasional apoptotic bodies and mitoses were seen. Large B‐cells were frequently present, which stained positive for EBV‐associated mRNA by in situ hybridization studies. Immunoperoxidase staining revealed that the atypical cell population was largely composed of CD4+ T‐cells. T‐cell receptor gene rearrangement studies demonstrated that the T‐cell population was monoclonal. These results were consistent with angioimmunoblastic T‐cell lymphoma (AITL). Positron emission tomography (PET) scanning was performed, showing diffuse fluorodeoxyglucose (FDG)‐avid lymphadenopathy involving cervical, axillary, mediastinal, retroperitoneal, mesenteric, and inguinal lymph nodes, up to 2 cm in diameter. The patient completed 6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), as well as experimental treatment with denileukin diftitox. Her fever and arthritis subsided quickly, and she was clinically well and free of disease by CT scans and PET scans 1 year after diagnosis.
COMMENTARY
The differential diagnosis of FUO is one of the largest in medicine, encompassing a dizzying range of infectious, inflammatory, and neoplastic conditions. This has made the development of standardized diagnostic algorithms difficult.1 The workup of patients with FUO begins with a detailed history and physical examination, followed by a core set of microbiology cultures, imaging studies, and blood tests on all patients. Further testing is individualized, based on key clinical findings, also known as pivot points.2 Unfortunately, many of the diseases presenting as FUO have overlapping symptoms and signs, somewhat limiting the utility of pivot points. In a prospective study, 81% of these potentially diagnostic clues were misleading, although 19% of them contributed to the final diagnosis.3 Key clinical findings in patients with FUO may trigger a barrage of diagnostic tests, often leading to a large number of false‐positive results.4 Clinicians who investigate patients with FUO must remember that many clues are diagnostic dead ends, and should be wary of drawing positively‐false conclusions.
Fever pattern is usually not helpful in the diagnosis of FUO, with occasional exceptions, such as the tertian and quartan fevers in some forms of malaria. A minority of patients with Hodgkin's disease have Pel‐Ebstein fevers, in which 1 to 2 weeks of fever alternate with an afebrile period of similar or longer duration. More often, fever in lymphoma waxes and wanes unpredictably,5 a circumstance that may lead to the mistaken impression of response to antibiotics, as in this case.
Tissue biopsies are helpful in FUO when suggestive findings are present on physical examination or imaging studies. In patients with lymphadenopathy and FUO, lymph node biopsy is a high‐yield procedure, contributing to the final diagnosis 46% of the time. This is exceeded only by biopsies of skin lesions, which have a 63% diagnostic yield in FUO.3 In older patients with FUO, temporal artery biopsies have a significant diagnostic yield, in the range of 16% to 17%. Liver biopsies have a similar yield (14‐17%), but a higher risk of complications. Bone marrow biopsies have a low yield in most FUO patients.1
AITL makes up 1% of all non‐Hodgkin's lymphomas. AITL was once known as angioimmunoblastic lymphadenopathy, and was thought to be either a disorder of immune regulation or a premalignant lymphoid disease. However, molecular diagnostic techniques have established that monoclonal T‐cell populations and cytogenetic abnormalities are usually present at the time of diagnosis.6, 7 The prognosis in AITL is unfavorable. Disease is usually widespread at the time of diagnosis. Most patients achieve complete remission with anthracycline‐based chemotherapy, such as CHOP, but the duration of remission is often brief, and median survival after diagnosis is only 3 years.6 Novel treatments under investigation include denileukin diftitox,8 a fusion protein of interleukin‐2 (IL‐2) conjugated to diphtheria toxin, which leads to apoptosis of cells expressing the IL‐2 receptor; rituximab, which targets the reactive population of B‐cells in AITL, rather than the malignant clone of T‐cells; and antiangiogenic therapy, such as thalidomide.9
The diagnosis of AITL is usually elusive, and the average patient has symptoms for 4 months prior to diagnosis.6 This patient's clinical presentation, while certainly not specific, was typical of AITL. AITL usually presents as fever of unknown origin with generalized, nonbulky lymphadenopathy. Fever is present in 57% of patients with AITL, and up to 2% of FUO is caused by AITL.6, 10 Other features of this patient's illness, such as night sweats, weight loss, pruritus, and arthritis, are common in AITL.
T‐cell depletion and immune dysregulation are frequent in AITL, explaining why AITL shares a number of clinical features with HIV disease. These include a high incidence of drug rashes, immune thrombocytopenic purpura, polyclonal hypergammaglobulinemia, and autoantibodies.6, 7 As with HIV patients, death in AITL is often due to opportunistic infections or diffuse large B‐cell lymphomas.7 Over 95% of patients with AITL display a proliferation of EBV‐infected B cells, presumably from immune dysregulation, and the B‐cell lymphomas in these patients are usually EBV‐positive.11
Because AITL is a proinflammatory state, false‐positive antibody tests are fairly common. The occasional occurrence of positive HIV enzyme immunoassays, with indeterminate Western blot results, may be a particular source of diagnostic confusion.12, 13 The significance of indeterminate Western blots is often erroneously communicated to patients, as happened here. Indeterminate HIV Western blots may be seen in HIV seroconversion, HIV‐2 infection, or advanced HIV disease with loss of core antibody. They may also result from laboratory error, multiparity, syphilis, malaria, or cross‐reacting antibodies in autoimmune diseases. Indeterminate HIV Western blots in low‐risk patients usually do not represent true HIV infection.14
KEY POINTS FOR HOSPITALISTS
-
FUO is one of the most challenging diagnoses faced by hospitalists. Biopsies of new skin lesions and enlarged lymph nodes are particularly high‐yield diagnostic procedures in FUO. Fever pattern is generally not helpful in the diagnosis of FUO, with few exceptions, such as the tertian fevers of malaria.
-
Misleading and false‐positive tests results often occur in the course of FUO evaluation, due to the sheer number of tests ordered, and the higher likelihood of false‐positive serologic tests in the setting of inflammatory states.
-
AITL may be an elusive diagnosis in FUO with diverse clinical features, including weight loss, night sweats, rashes, arthritis, autoantibodies, immune dysregulation, and opportunistic infections. The prognosis traditionally has been guarded, but may be more hopeful in an era of emerging molecular therapies.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A51‐year‐old woman presented after 5 days of fever, rigors, anorexia, right‐sided abdominal pain, nausea, and dizziness. She had 2 loose stools the day before admission, without blood or mucus, but otherwise had recently been constipated. She denied cough, shortness of breath, chest pain, headache, sore throat, rash, arthritis, or dysuria.
In a 51‐year‐old woman with right‐sided abdominal pain and systemic symptoms, major concerns include biliary disease, liver abscess, or appendicitis. Right‐sided diverticulitis would be more unusual. Pyelonephritis infrequently presents with epigastric and lower quadrant pain, instead of flank pain. Basilar pneumonia may present with abdominal pain, but this is less likely in the absence of respiratory symptoms.
The patient used an albuterol inhaler for mild asthma and had experienced an episode of herpes zoster 7 years prior, but was otherwise well. Her surgical history was notable for a remote appendectomy. She was a native of the Dominican Republic who had lived in the United States for the past 20 years. She visited the Dominican Republic for 3 weeks every year, with her last visit occurring about 10 months before. She was a cleaning and maintenance worker. She had 2 adult children in good health, was divorced from her husband, and had not been sexually active for the past 8 years. The patient had no pets or other animal exposures. She did not smoke, drink alcohol, or use intravenous drugs.
The remote episode of shingles makes me a bit worried about chronic human immunodeficiency virus (HIV) infection. As a native of and traveler to the Dominican Republic, she is at risk for a variety of tropical pathogens. Hyperinfection syndrome from strongyloides can cause fever and bacteremia, but this is almost always associated with significant immunosuppression. Dengue fever has become very common in the Caribbean, but should occur within 2 weeks of travel. Her work in cleaning and maintenance might bring her into contact with rats and mice, putting her at risk for leptospirosis. This can present as a fairly nonspecific febrile syndrome, but this is unlikely without a major complaint of headache.
The patient appeared fatigued. Her temperature was 39.7C, her heart rate was 110 beats per minute, and her blood pressure 80/62 mm Hg. The oropharynx was normal. Mild cervical lymphadenopathy was present (less than 1 cm in diameter). The chest was clear and the cardiac examination unremarkable. Bowel sounds were present. Moderate right‐sided abdominal tenderness was noted, somewhat more marked in the right lower quadrant, without guarding or rebound. There was no hepatosplenomegaly. There was no rash. A bedside right upper quadrant ultrasound was negative for gallstones.
Her low blood pressure is concerning for bacterial sepsis. The negative right upper quadrant ultrasound makes cholecystitis or cholangitis less likely, but does not exclude diverticulitis or pelvic inflammatory disease. She lacks peritoneal signs, but they may be absent in these conditions. Another worrisome finding on her physical examination is cervical lymphadenopathy. In an older patient, this raises the specter of malignancy. In a younger patient, it could suggest a mononucleosis syndrome from Epstein‐Barr virus (EBV) or cytomegalovirus (CMV). In addition, HIV must be considered in any patient with unexplained lymphadenopathy.
She received intravenous levofloxacin and 1 L of intravenous normal saline, with a rise in her blood pressure to 100/59 mm Hg. Her white blood cell count was 5.0, with 71% polys and 9% bands. The hematocrit was 32%, with a normal mean corpuscular volume. The erythrocyte sedimentation rate (ESR) was 109 mm/hour. The platelet count, serum electrolytes, creatinine, aminotransferases, alkaline phosphatase, bilirubin, amylase, and lipase were normal. The serum level of lactate dehydrogenase (LDH) was 498 unit/L (normal range, 107231 units/L). Computed tomography (CT) scan of the abdomen showed multiple enlarged lymph nodes up to 1.2 cm in size along the gastrohepatic ligament and the mesentery, with mild associated fat stranding, consistent with mesenteric lymphadenitis.
Her blood pressure has responded to fluids; perhaps she was just volume‐depleted from not eating for several days. She has bandemia, which is consistent with acute bacterial infection, but might also signify a stress response. The low hematocrit and high ESR raise the possibility of anemia of chronic disease; perhaps her illness is more longstanding than her presentation suggests. Mesenteric lymphadenitis may be related to EBV and HIV, but in a patient originally from the Caribbean, it raises the possibility of gastrointestinal tuberculosis or histoplasmosis. Human T‐cell lymphotropic virus type 1 (HTLV‐1) is also endemic in the Caribbean, and may cause adult T‐cell leukemia/lymphoma (ATLL), which could explain her lymphadenopathy and elevated LDH. Mesenteric lymphadenitis is also characteristic of several bacterial infections, especially Yersinia, Salmonella, and Bartonella. The lack of diarrhea makes yersiniosis doubtful, and the absence of cat exposure makes bartonellosis unlikely. Salmonella infection is also associated with diarrhea, except for typhoid fever, in which patients have diarrhea, constipation, or normal stools.
A urine culture and 3 sets of blood cultures obtained prior to the initiation of antibiotics were negative. A blood smear showed no malaria or babesia parasites. The patient's fever continued for the first 2 days of her hospitalization, but subsequently abated. The patient had no loose stools during her hospitalization. Serologies for EBV, hepatitis A, CMV, and toxoplasmosis were indicative of remote infection. Serum rapid plasma reagin (RPR), Bartonella antibodies, antinuclear antibodies, hepatitis B surface antigen, and hepatitis C antibody were negative. Tuberculin skin testing and urinary histoplasma antigen were negative. By the fifth hospital day, she had been afebrile for over 48 hours, and her abdominal pain had improved, though it had not completely resolved. She was discharged to complete a 10‐day course of levofloxacin.
It is not clear to me whether she has just experienced a spontaneous remission in her illness, or whether her disease course has truly been modified with antibiotics. Could she have typhoid fever or another salmonellosis? The patient has not traveled abroad in several months. If she had typhoid fever, the source of infection would have to be imported food, or exposure to a family member who was a carrier. The negative tuberculin skin test makes tuberculosis less likely, but does not exclude it entirely. Similarly, while a negative histoplasma urinary antigen essentially rules out acute disseminated histoplasmosis, as would be seen in acquired immune deficiency syndrome (AIDS), it is less sensitive for more chronic forms of disseminated histoplasmosis, including gastrointestinal involvement.
One week later, the patient returned to the emergency department with recurrent abdominal pain, anorexia, fever, night sweats, and increased swelling of the lymph nodes in her neck. She had lost a total of 4.4 kg (10 lb) since the onset of her illness. Physical examination revealed moderate (up to 2 cm), slightly tender cervical and inguinal lymphadenopathy, and continued moderate right‐sided abdominal tenderness. Mesenteric, retroperitoneal, and inguinal lymphadenopathy was more prominent than on the prior CT scan (Figure 1).She was told by the emergency room physicians that the HIV test obtained during the prior hospitalization was positive. Further questioning elicited that her estranged husband had been promiscuous prior to their separation. She was transferred to a second hospital for further care. Blood cultures were sent for fungi and acid‐fast bacilli.
Everyone with fever of unknown origin (FUO) deserves an HIV test. Is this just lymphadenopathy from HIV, or is she suffering from an opportunistic infection? Mycobacterium avium infection is an attractive explanation for her fever, abdominal pain, and lymphadenopathy, but I would hold off on empiric treatment until the results of a CD4+ cell count were available. Could she have a secondary, HIV‐related lymphoma?
Full review of records from the outside hospital showed that the patient had a positive HIV enzyme immunoassay, with an indeterminate HIV Western blot. The patient's CD4 cell count was 313/cm3, with a CD4/CD8 ratio within the normal range. Her HIV enzyme immunoassay was repeated and found to be negative, and the HIV viral load was undetectable.
The HIV enzyme immunoassay is only a screening test, and must be confirmed with a positive HIV Western blot. (In most clinical laboratories, this is done automatically before the test is reported.) Indeterminate HIV Western blots are common in acute HIV infection, but outside of this setting, most patients with indeterminate HIV Western blots turn out not to have HIV infection. I am still very concerned about lymphoma, and would pursue a lymph node biopsy. Disseminated tuberculosis is still possible.
Biopsy of a right inguinal lymph node was performed on the third day of the second hospitalization. The patient was persistently febrile, developed swelling of the knees, ankles, and interphalangeal joints of the second and third digits of the left hand, and complained of pruritus. Anti‐double‐stranded DNA antibody, antineutrophilic cytoplasmic antibody, and Brucella antibody were negative. Antibody against cyclic citrullinated peptide (CCP) was weakly positive.
Now she has a more florid syndrome, with arthritic symptoms. Sarcoidosis is an attractive explanation for her fever, lymphadenopathy, and arthritis. Lupus could explain some features of her presentation, but the negative serology makes it unlikely. Antibody to cyclic citrullinated peptide is a newer diagnostic test for rheumatoid arthritis. Although anti‐CCP is more specific than rheumatoid factor for the diagnosis of rheumatoid arthritis, this result could still be a false positive, particularly given the low titer. As well, the patient has more impressive lymphadenopathy than is usual for rheumatoid arthritis. Reactive arthritis can follow enteric infection with Campylobacter, Salmonella, Yersinia, and Shigella, but lymphadenopathy is not a feature. Arthritis may be prominent in parvovirus B19, rubella, disseminated gonococcal infection, and Lyme disease, but mesenteric lymphadenitis and a prolonged, waxing and waning course would not be expected with any of these. I worry that the arthritis and pruritus are paraneoplastic manifestations of lymphoma.
The inguinal lymph node biopsy showed markedly distorted nodal architecture with an atypical proliferation of small‐sized to large‐sized lymphoid cells, with predominantly round nuclei, vesicular chromatin, small nucleoli, and variable amounts of clear to eosinophilic cytoplasm (Figure 2). Residual follicles and occasional apoptotic bodies and mitoses were seen. Large B‐cells were frequently present, which stained positive for EBV‐associated mRNA by in situ hybridization studies. Immunoperoxidase staining revealed that the atypical cell population was largely composed of CD4+ T‐cells. T‐cell receptor gene rearrangement studies demonstrated that the T‐cell population was monoclonal. These results were consistent with angioimmunoblastic T‐cell lymphoma (AITL). Positron emission tomography (PET) scanning was performed, showing diffuse fluorodeoxyglucose (FDG)‐avid lymphadenopathy involving cervical, axillary, mediastinal, retroperitoneal, mesenteric, and inguinal lymph nodes, up to 2 cm in diameter. The patient completed 6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), as well as experimental treatment with denileukin diftitox. Her fever and arthritis subsided quickly, and she was clinically well and free of disease by CT scans and PET scans 1 year after diagnosis.
COMMENTARY
The differential diagnosis of FUO is one of the largest in medicine, encompassing a dizzying range of infectious, inflammatory, and neoplastic conditions. This has made the development of standardized diagnostic algorithms difficult.1 The workup of patients with FUO begins with a detailed history and physical examination, followed by a core set of microbiology cultures, imaging studies, and blood tests on all patients. Further testing is individualized, based on key clinical findings, also known as pivot points.2 Unfortunately, many of the diseases presenting as FUO have overlapping symptoms and signs, somewhat limiting the utility of pivot points. In a prospective study, 81% of these potentially diagnostic clues were misleading, although 19% of them contributed to the final diagnosis.3 Key clinical findings in patients with FUO may trigger a barrage of diagnostic tests, often leading to a large number of false‐positive results.4 Clinicians who investigate patients with FUO must remember that many clues are diagnostic dead ends, and should be wary of drawing positively‐false conclusions.
Fever pattern is usually not helpful in the diagnosis of FUO, with occasional exceptions, such as the tertian and quartan fevers in some forms of malaria. A minority of patients with Hodgkin's disease have Pel‐Ebstein fevers, in which 1 to 2 weeks of fever alternate with an afebrile period of similar or longer duration. More often, fever in lymphoma waxes and wanes unpredictably,5 a circumstance that may lead to the mistaken impression of response to antibiotics, as in this case.
Tissue biopsies are helpful in FUO when suggestive findings are present on physical examination or imaging studies. In patients with lymphadenopathy and FUO, lymph node biopsy is a high‐yield procedure, contributing to the final diagnosis 46% of the time. This is exceeded only by biopsies of skin lesions, which have a 63% diagnostic yield in FUO.3 In older patients with FUO, temporal artery biopsies have a significant diagnostic yield, in the range of 16% to 17%. Liver biopsies have a similar yield (14‐17%), but a higher risk of complications. Bone marrow biopsies have a low yield in most FUO patients.1
AITL makes up 1% of all non‐Hodgkin's lymphomas. AITL was once known as angioimmunoblastic lymphadenopathy, and was thought to be either a disorder of immune regulation or a premalignant lymphoid disease. However, molecular diagnostic techniques have established that monoclonal T‐cell populations and cytogenetic abnormalities are usually present at the time of diagnosis.6, 7 The prognosis in AITL is unfavorable. Disease is usually widespread at the time of diagnosis. Most patients achieve complete remission with anthracycline‐based chemotherapy, such as CHOP, but the duration of remission is often brief, and median survival after diagnosis is only 3 years.6 Novel treatments under investigation include denileukin diftitox,8 a fusion protein of interleukin‐2 (IL‐2) conjugated to diphtheria toxin, which leads to apoptosis of cells expressing the IL‐2 receptor; rituximab, which targets the reactive population of B‐cells in AITL, rather than the malignant clone of T‐cells; and antiangiogenic therapy, such as thalidomide.9
The diagnosis of AITL is usually elusive, and the average patient has symptoms for 4 months prior to diagnosis.6 This patient's clinical presentation, while certainly not specific, was typical of AITL. AITL usually presents as fever of unknown origin with generalized, nonbulky lymphadenopathy. Fever is present in 57% of patients with AITL, and up to 2% of FUO is caused by AITL.6, 10 Other features of this patient's illness, such as night sweats, weight loss, pruritus, and arthritis, are common in AITL.
T‐cell depletion and immune dysregulation are frequent in AITL, explaining why AITL shares a number of clinical features with HIV disease. These include a high incidence of drug rashes, immune thrombocytopenic purpura, polyclonal hypergammaglobulinemia, and autoantibodies.6, 7 As with HIV patients, death in AITL is often due to opportunistic infections or diffuse large B‐cell lymphomas.7 Over 95% of patients with AITL display a proliferation of EBV‐infected B cells, presumably from immune dysregulation, and the B‐cell lymphomas in these patients are usually EBV‐positive.11
Because AITL is a proinflammatory state, false‐positive antibody tests are fairly common. The occasional occurrence of positive HIV enzyme immunoassays, with indeterminate Western blot results, may be a particular source of diagnostic confusion.12, 13 The significance of indeterminate Western blots is often erroneously communicated to patients, as happened here. Indeterminate HIV Western blots may be seen in HIV seroconversion, HIV‐2 infection, or advanced HIV disease with loss of core antibody. They may also result from laboratory error, multiparity, syphilis, malaria, or cross‐reacting antibodies in autoimmune diseases. Indeterminate HIV Western blots in low‐risk patients usually do not represent true HIV infection.14
KEY POINTS FOR HOSPITALISTS
-
FUO is one of the most challenging diagnoses faced by hospitalists. Biopsies of new skin lesions and enlarged lymph nodes are particularly high‐yield diagnostic procedures in FUO. Fever pattern is generally not helpful in the diagnosis of FUO, with few exceptions, such as the tertian fevers of malaria.
-
Misleading and false‐positive tests results often occur in the course of FUO evaluation, due to the sheer number of tests ordered, and the higher likelihood of false‐positive serologic tests in the setting of inflammatory states.
-
AITL may be an elusive diagnosis in FUO with diverse clinical features, including weight loss, night sweats, rashes, arthritis, autoantibodies, immune dysregulation, and opportunistic infections. The prognosis traditionally has been guarded, but may be more hopeful in an era of emerging molecular therapies.
- ,,.A comprehensive evidence‐based approach to fever of unknown origin.Arch Intern Med.2003;163:545–551.
- ,.The art of diagnosis: solving the clinicopathological exercise.N Engl J Med.1982;306:1263–1268.
- ,,, et al.A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol.Medicine (Baltimore).2007;86:26–38.
- ,,.Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:401–414.
- ,.Neoplastic diseases. In:Murray HW, ed.FUO: Fever of Undetermined Origin.New York, NY:Futura Publishing;1983:39–48.
- ,,, et al.Angioimmunoblastic T‐cell lymphoma: clinical and laboratory features at diagnosis in 77 patients.Medicine (Baltimore).2007;86:282–292.
- ,,.Angioimmunoblastic T‐cell lymphoma.Br J Haematol.2003;121:681–691.
- ,,, et al.Phase II trial of denileukin diftitox for relapsed/refractory T‐cell non‐Hodgkin lymphoma.Br J Haematol.2007;136:439–447.
- ,.Angioimmunoblastic T‐cell lymphoma: still a dismal prognosis with current treatment approaches.Leuk Lymphoma.2007;48:645–646.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:392–400.
- ,,, et al.Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression.Am J Surg Pathol.2007;31:1077–1088.
- ,,,.Angioimmunoblastic lymphadenopathy, immunoblastic lymphoma, and false‐positive seroconversion for human immunodeficiency virus.Ann Intern Med.1987;107:114.
- ,.Angioimmunoblastic T‐cell lymphoma associated with an antibody to human immunodeficiency virus protein.Int J Hematol.2003;78:160–162.
- ,,.Communicating indeterminate HIV Western blot test results to clients: an observational study of three community testing sites.AIDS Patient Care STDS.2006;20:620–627.
- ,,.A comprehensive evidence‐based approach to fever of unknown origin.Arch Intern Med.2003;163:545–551.
- ,.The art of diagnosis: solving the clinicopathological exercise.N Engl J Med.1982;306:1263–1268.
- ,,, et al.A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol.Medicine (Baltimore).2007;86:26–38.
- ,,.Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:401–414.
- ,.Neoplastic diseases. In:Murray HW, ed.FUO: Fever of Undetermined Origin.New York, NY:Futura Publishing;1983:39–48.
- ,,, et al.Angioimmunoblastic T‐cell lymphoma: clinical and laboratory features at diagnosis in 77 patients.Medicine (Baltimore).2007;86:282–292.
- ,,.Angioimmunoblastic T‐cell lymphoma.Br J Haematol.2003;121:681–691.
- ,,, et al.Phase II trial of denileukin diftitox for relapsed/refractory T‐cell non‐Hodgkin lymphoma.Br J Haematol.2007;136:439–447.
- ,.Angioimmunoblastic T‐cell lymphoma: still a dismal prognosis with current treatment approaches.Leuk Lymphoma.2007;48:645–646.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:392–400.
- ,,, et al.Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression.Am J Surg Pathol.2007;31:1077–1088.
- ,,,.Angioimmunoblastic lymphadenopathy, immunoblastic lymphoma, and false‐positive seroconversion for human immunodeficiency virus.Ann Intern Med.1987;107:114.
- ,.Angioimmunoblastic T‐cell lymphoma associated with an antibody to human immunodeficiency virus protein.Int J Hematol.2003;78:160–162.
- ,,.Communicating indeterminate HIV Western blot test results to clients: an observational study of three community testing sites.AIDS Patient Care STDS.2006;20:620–627.
Research Roundup
Question: Does gentamicin use affect clinical outcomes and prognosis or just creatinine clearance?
Background: Impaired kidney function in patients with endocarditis predicts worse outcomes in both morbidity and mortality. Given that the aminoglycosides can be nephrotoxic, it has been debated whether physicians should abandon its use in these patients.
Study design: Prospective, observational, cohort study.
Setting: Two tertiary-care hospitals serving Copenhagen, Denmark, from 2002-2007.
Synopsis: The study identified 373 patients as having definite or probable infective endocarditis. ("Probable" meant patients underwent the same treatment for endocarditis as those with confirmed disease.) Gentamicin treatment decreased estimated creatinine clearance (CrCl) by 0.5% per day of treatment, with more significant decreases in CrCl associated with length of treatment and age. It did not increase the risk of in-hospital or post-discharge mortality, nor did it increase the need for dialysis. The mean duration of follow up was 562 days.
Bottom line: With appropriate monitoring, gentamicin is a reasonable treatment choice for patients with endocarditis when clinically indicated. Patient-centered outcomes are not negatively impacted by its use.
Citation: Buchholtz K, Larsen CT, Hassager C, Bruun NE. Severity of gentamicin’s nephrotoxic effect on patients with infective endocarditis: a prospective observational cohort study of 373 patients. Clin Infect Dis. 2009;48:65-71.
In the Literature: For the latest HM literature reviews, visit www.the-hospitalist.org and search "literature."
—Reviewed by Michael Kedansky, MD, Victor Weaver, MD, Michael Goldman, MD, Lisa Gushwa, MD, Paul Hicks, MD, Barbara Eckstein, MD, and Christine Kneisel, MD, Department of Family and Community Medicine, University of Arizona
Question: Does gentamicin use affect clinical outcomes and prognosis or just creatinine clearance?
Background: Impaired kidney function in patients with endocarditis predicts worse outcomes in both morbidity and mortality. Given that the aminoglycosides can be nephrotoxic, it has been debated whether physicians should abandon its use in these patients.
Study design: Prospective, observational, cohort study.
Setting: Two tertiary-care hospitals serving Copenhagen, Denmark, from 2002-2007.
Synopsis: The study identified 373 patients as having definite or probable infective endocarditis. ("Probable" meant patients underwent the same treatment for endocarditis as those with confirmed disease.) Gentamicin treatment decreased estimated creatinine clearance (CrCl) by 0.5% per day of treatment, with more significant decreases in CrCl associated with length of treatment and age. It did not increase the risk of in-hospital or post-discharge mortality, nor did it increase the need for dialysis. The mean duration of follow up was 562 days.
Bottom line: With appropriate monitoring, gentamicin is a reasonable treatment choice for patients with endocarditis when clinically indicated. Patient-centered outcomes are not negatively impacted by its use.
Citation: Buchholtz K, Larsen CT, Hassager C, Bruun NE. Severity of gentamicin’s nephrotoxic effect on patients with infective endocarditis: a prospective observational cohort study of 373 patients. Clin Infect Dis. 2009;48:65-71.
In the Literature: For the latest HM literature reviews, visit www.the-hospitalist.org and search "literature."
—Reviewed by Michael Kedansky, MD, Victor Weaver, MD, Michael Goldman, MD, Lisa Gushwa, MD, Paul Hicks, MD, Barbara Eckstein, MD, and Christine Kneisel, MD, Department of Family and Community Medicine, University of Arizona
Question: Does gentamicin use affect clinical outcomes and prognosis or just creatinine clearance?
Background: Impaired kidney function in patients with endocarditis predicts worse outcomes in both morbidity and mortality. Given that the aminoglycosides can be nephrotoxic, it has been debated whether physicians should abandon its use in these patients.
Study design: Prospective, observational, cohort study.
Setting: Two tertiary-care hospitals serving Copenhagen, Denmark, from 2002-2007.
Synopsis: The study identified 373 patients as having definite or probable infective endocarditis. ("Probable" meant patients underwent the same treatment for endocarditis as those with confirmed disease.) Gentamicin treatment decreased estimated creatinine clearance (CrCl) by 0.5% per day of treatment, with more significant decreases in CrCl associated with length of treatment and age. It did not increase the risk of in-hospital or post-discharge mortality, nor did it increase the need for dialysis. The mean duration of follow up was 562 days.
Bottom line: With appropriate monitoring, gentamicin is a reasonable treatment choice for patients with endocarditis when clinically indicated. Patient-centered outcomes are not negatively impacted by its use.
Citation: Buchholtz K, Larsen CT, Hassager C, Bruun NE. Severity of gentamicin’s nephrotoxic effect on patients with infective endocarditis: a prospective observational cohort study of 373 patients. Clin Infect Dis. 2009;48:65-71.
In the Literature: For the latest HM literature reviews, visit www.the-hospitalist.org and search "literature."
—Reviewed by Michael Kedansky, MD, Victor Weaver, MD, Michael Goldman, MD, Lisa Gushwa, MD, Paul Hicks, MD, Barbara Eckstein, MD, and Christine Kneisel, MD, Department of Family and Community Medicine, University of Arizona
Head of the Class
Brian Tyson, MD, was named medical director of the hospitalist program at St. Bernardine Medical Center in San Bernardino, Calif., about a year ago, but he still can't learn enough about business drivers, communication, and leadership skills. That's why he attended SHM's Leadership Academy last week in Honolulu.
"I needed some things to work on. ...I think we all need to," says Dr. Tyson, whose HM program is operated by Cogent Healthcare. "This is that opportunity. When people have done things and run things, you don't need to reinvent the wheel. It's easier sometimes to get the keys to success from someone who’s been there."
More than a hundred hospitalists apparently agreed, joining Dr. Tyson at the four-day session. Dr. Tyson found the insights into business particularly helpful, especially because fiscal matters are not a major focus of medical school. Analyzing his managerial personality and identifying his program's strengths and weaknesses were "eye opening," as was the chance to discuss staffing and budget issues with HM directors from different parts of the country.
The leadership program offers two tracks, and the first course must be completed before taking the advanced level. A first-time attendee, Dr. Tyson says he is looking forward to completing the second part of the academy.
"Medicine is changing so much, this helps us manage our programs," he says. "Having the tools to be able to do that is absolutely necessary."
Brian Tyson, MD, was named medical director of the hospitalist program at St. Bernardine Medical Center in San Bernardino, Calif., about a year ago, but he still can't learn enough about business drivers, communication, and leadership skills. That's why he attended SHM's Leadership Academy last week in Honolulu.
"I needed some things to work on. ...I think we all need to," says Dr. Tyson, whose HM program is operated by Cogent Healthcare. "This is that opportunity. When people have done things and run things, you don't need to reinvent the wheel. It's easier sometimes to get the keys to success from someone who’s been there."
More than a hundred hospitalists apparently agreed, joining Dr. Tyson at the four-day session. Dr. Tyson found the insights into business particularly helpful, especially because fiscal matters are not a major focus of medical school. Analyzing his managerial personality and identifying his program's strengths and weaknesses were "eye opening," as was the chance to discuss staffing and budget issues with HM directors from different parts of the country.
The leadership program offers two tracks, and the first course must be completed before taking the advanced level. A first-time attendee, Dr. Tyson says he is looking forward to completing the second part of the academy.
"Medicine is changing so much, this helps us manage our programs," he says. "Having the tools to be able to do that is absolutely necessary."
Brian Tyson, MD, was named medical director of the hospitalist program at St. Bernardine Medical Center in San Bernardino, Calif., about a year ago, but he still can't learn enough about business drivers, communication, and leadership skills. That's why he attended SHM's Leadership Academy last week in Honolulu.
"I needed some things to work on. ...I think we all need to," says Dr. Tyson, whose HM program is operated by Cogent Healthcare. "This is that opportunity. When people have done things and run things, you don't need to reinvent the wheel. It's easier sometimes to get the keys to success from someone who’s been there."
More than a hundred hospitalists apparently agreed, joining Dr. Tyson at the four-day session. Dr. Tyson found the insights into business particularly helpful, especially because fiscal matters are not a major focus of medical school. Analyzing his managerial personality and identifying his program's strengths and weaknesses were "eye opening," as was the chance to discuss staffing and budget issues with HM directors from different parts of the country.
The leadership program offers two tracks, and the first course must be completed before taking the advanced level. A first-time attendee, Dr. Tyson says he is looking forward to completing the second part of the academy.
"Medicine is changing so much, this helps us manage our programs," he says. "Having the tools to be able to do that is absolutely necessary."
Team Effort to Combat Fractures
A joint venture between a hospitalist and an orthopedist in rural Minnesota is targeting geriatric fracture patients, and aims to reduce lengths of stay and morbidity.
The program, created by Northern Orthopedics and St. Joseph's Medical Center in Brainerd, Minn., has developed a toolkit for elderly fracture patients. The kit includes a pictorial guidebook, postoperative instructions and rehabilitation information, and early discharge planning. Only a half-dozen or so patients have used the service since its January launch, but developers say the purpose is to pair a surgeon with a hospitalist soon after a patient's admission to ensure that other medical problems are treated alongside any fractures.
"The fracture is not really the biggest problem," says Ben Robertson, MD, a surgeon with Northern Orthopedics. "The surgeon can deal with that. These patients, after we fix their hip fracture ... there's a whole host of medical problems that can happen."
That's where St. Joseph's hospitalist Jim Baumgartner, MD, steps in. The early introduction of a hospitalist allows Dr. Baumgartner to know a patient's treatment history, keep an eye out for medical-related complications, and set up corresponding treatment programs. Dr. Baumgartner says the Minnesota program—modeled after one at the University of Rochester—could be replicated at other hospitals to produce better functional outcomes.
"Everybody works together from the beginning," Dr. Baumgartner says. "The results are patients getting better faster, shorter lengths of stay, and much more efficient resource utilization. This is what hospitalists are geared for."
A joint venture between a hospitalist and an orthopedist in rural Minnesota is targeting geriatric fracture patients, and aims to reduce lengths of stay and morbidity.
The program, created by Northern Orthopedics and St. Joseph's Medical Center in Brainerd, Minn., has developed a toolkit for elderly fracture patients. The kit includes a pictorial guidebook, postoperative instructions and rehabilitation information, and early discharge planning. Only a half-dozen or so patients have used the service since its January launch, but developers say the purpose is to pair a surgeon with a hospitalist soon after a patient's admission to ensure that other medical problems are treated alongside any fractures.
"The fracture is not really the biggest problem," says Ben Robertson, MD, a surgeon with Northern Orthopedics. "The surgeon can deal with that. These patients, after we fix their hip fracture ... there's a whole host of medical problems that can happen."
That's where St. Joseph's hospitalist Jim Baumgartner, MD, steps in. The early introduction of a hospitalist allows Dr. Baumgartner to know a patient's treatment history, keep an eye out for medical-related complications, and set up corresponding treatment programs. Dr. Baumgartner says the Minnesota program—modeled after one at the University of Rochester—could be replicated at other hospitals to produce better functional outcomes.
"Everybody works together from the beginning," Dr. Baumgartner says. "The results are patients getting better faster, shorter lengths of stay, and much more efficient resource utilization. This is what hospitalists are geared for."
A joint venture between a hospitalist and an orthopedist in rural Minnesota is targeting geriatric fracture patients, and aims to reduce lengths of stay and morbidity.
The program, created by Northern Orthopedics and St. Joseph's Medical Center in Brainerd, Minn., has developed a toolkit for elderly fracture patients. The kit includes a pictorial guidebook, postoperative instructions and rehabilitation information, and early discharge planning. Only a half-dozen or so patients have used the service since its January launch, but developers say the purpose is to pair a surgeon with a hospitalist soon after a patient's admission to ensure that other medical problems are treated alongside any fractures.
"The fracture is not really the biggest problem," says Ben Robertson, MD, a surgeon with Northern Orthopedics. "The surgeon can deal with that. These patients, after we fix their hip fracture ... there's a whole host of medical problems that can happen."
That's where St. Joseph's hospitalist Jim Baumgartner, MD, steps in. The early introduction of a hospitalist allows Dr. Baumgartner to know a patient's treatment history, keep an eye out for medical-related complications, and set up corresponding treatment programs. Dr. Baumgartner says the Minnesota program—modeled after one at the University of Rochester—could be replicated at other hospitals to produce better functional outcomes.
"Everybody works together from the beginning," Dr. Baumgartner says. "The results are patients getting better faster, shorter lengths of stay, and much more efficient resource utilization. This is what hospitalists are geared for."
Capitol Investment
Members of SHM's Public Policy Committee traveled to Capitol Hill for their fourth annual legislative visit last week and they found congressional representatives and staff more eager than ever to listen.
"With healthcare at the forefront of what's happening [in Congress] right now, the members of congress and their staffs were particularly interested in what we had to say," says committee member Felix Aguirre, MD, vice president of medical affairs for IPC: The Hospitalist Company in San Antonio. Specifically, the 13 committee members attended 35 Capitol Hill meetings and touted support for increased access to healthcare; delivery models to improve care coordination; and changes in payment methodologies that improve quality and value of healthcare, including consideration of alternative models, such as bundling payments for select conditions in hospitalized patients and physician value-based purchasing.
"The part that was noticeable [this year] was the elevation of the level of discussions we had," says committee member Gregory Seymann, MD, a hospitalist and associate clinical professor at the University of California San Diego School of Medicine. "We are now positioned to be influential … in healthcare matters that are obviously a big priority."
One of SHM's priorities is care transitions, and the committee brought legislators’ attention to SHM's Project BOOST (Better Outcomes for Older adults through Safe Transitions), a program designed to optimize transitions from the hospital to the home.
Mark V. Williams, MD, the project’s principal investigator, joined the committee and met with staff of key congressional committees, including the House Energy and Commerce Committee; the Senate Committee on Health, Education, Labor and Pensions; and the Senate Finance Committee. “I was impressed. They were all aware of the President's budget proposal to reduce rehospitalizations and understood how Project BOOST could help," Dr. Williams says. "They are excited about it and interested in data regarding its impact."
For more information about SHM’s public policy efforts, visit www.hospitalmedicine.org/Advocacy.
Members of SHM's Public Policy Committee traveled to Capitol Hill for their fourth annual legislative visit last week and they found congressional representatives and staff more eager than ever to listen.
"With healthcare at the forefront of what's happening [in Congress] right now, the members of congress and their staffs were particularly interested in what we had to say," says committee member Felix Aguirre, MD, vice president of medical affairs for IPC: The Hospitalist Company in San Antonio. Specifically, the 13 committee members attended 35 Capitol Hill meetings and touted support for increased access to healthcare; delivery models to improve care coordination; and changes in payment methodologies that improve quality and value of healthcare, including consideration of alternative models, such as bundling payments for select conditions in hospitalized patients and physician value-based purchasing.
"The part that was noticeable [this year] was the elevation of the level of discussions we had," says committee member Gregory Seymann, MD, a hospitalist and associate clinical professor at the University of California San Diego School of Medicine. "We are now positioned to be influential … in healthcare matters that are obviously a big priority."
One of SHM's priorities is care transitions, and the committee brought legislators’ attention to SHM's Project BOOST (Better Outcomes for Older adults through Safe Transitions), a program designed to optimize transitions from the hospital to the home.
Mark V. Williams, MD, the project’s principal investigator, joined the committee and met with staff of key congressional committees, including the House Energy and Commerce Committee; the Senate Committee on Health, Education, Labor and Pensions; and the Senate Finance Committee. “I was impressed. They were all aware of the President's budget proposal to reduce rehospitalizations and understood how Project BOOST could help," Dr. Williams says. "They are excited about it and interested in data regarding its impact."
For more information about SHM’s public policy efforts, visit www.hospitalmedicine.org/Advocacy.
Members of SHM's Public Policy Committee traveled to Capitol Hill for their fourth annual legislative visit last week and they found congressional representatives and staff more eager than ever to listen.
"With healthcare at the forefront of what's happening [in Congress] right now, the members of congress and their staffs were particularly interested in what we had to say," says committee member Felix Aguirre, MD, vice president of medical affairs for IPC: The Hospitalist Company in San Antonio. Specifically, the 13 committee members attended 35 Capitol Hill meetings and touted support for increased access to healthcare; delivery models to improve care coordination; and changes in payment methodologies that improve quality and value of healthcare, including consideration of alternative models, such as bundling payments for select conditions in hospitalized patients and physician value-based purchasing.
"The part that was noticeable [this year] was the elevation of the level of discussions we had," says committee member Gregory Seymann, MD, a hospitalist and associate clinical professor at the University of California San Diego School of Medicine. "We are now positioned to be influential … in healthcare matters that are obviously a big priority."
One of SHM's priorities is care transitions, and the committee brought legislators’ attention to SHM's Project BOOST (Better Outcomes for Older adults through Safe Transitions), a program designed to optimize transitions from the hospital to the home.
Mark V. Williams, MD, the project’s principal investigator, joined the committee and met with staff of key congressional committees, including the House Energy and Commerce Committee; the Senate Committee on Health, Education, Labor and Pensions; and the Senate Finance Committee. “I was impressed. They were all aware of the President's budget proposal to reduce rehospitalizations and understood how Project BOOST could help," Dr. Williams says. "They are excited about it and interested in data regarding its impact."
For more information about SHM’s public policy efforts, visit www.hospitalmedicine.org/Advocacy.
Potential cause of R-CHOP failure ruled out

Researchers say they have come one step closer to determining the cause of R-CHOP failure in diffuse large B-cell lymphoma (DLBCL) by ruling out a potential cause.
Randy D. Gascoyne, MD, of the British Columbia Cancer Agency, and colleagues found that CD20 mutations involving the rituximab epitope are not the source of R-CHOP resistance. In fact, the mutations are rare in both de novo and relapsed DLBCL.
The rituximab epitope is located in exon 5 of the MS4A1 gene, so Dr Gascoyne and colleagues sequenced this region in DLBCL samples taken at diagnosis and relapse (1 month after completion of 6 cycles of R-CHOP). The team successfully sequenced 264 diagnostic samples and 15 relapsed samples.
The samples could be considered representative of the DLBCL population in British Columbia because clinical characteristics were similar to those observed in previous studies, according to the researchers. In addition, most of the patients had nodal disease with a minimum of 80% tumor, which was sufficient for detecting mutations.
Only 1 of 264 diagnostic samples showed a CD20 mutation involving the rituximab epitope—a 13 base pair heterozygous deletion at position IVS5(+8) in intron 5. Dr Gascoyne and colleagues were unable to determine if this was a polymorphism or a somatic mutation.
This patient achieved a complete response to R-CHOP and is still in remission more than 2 years after diagnosis. This outcome rules out the possibility of mutation-induced rituximab resistance.
As with the diagnostic samples, only 1 of the relapsed samples showed a CD20 mutation involving the rituximab epitope. This was a heterozygous 4 base pair deletion (TAAT) at nucleotide position 353-356, which predicted for a premature termination at amino acid position 121, well before the critical ANPS binding site.
The researchers were unable to establish whether this mutation was present at diagnosis, but they did determine there were no single nucleotide polymorphisms in exon 5 of the CD20 gene.
The rarity of mutations in the rituximab epitope observed in this study suggests these mutations cannot be responsible for the majority of R-CHOP treatment failures. However, Dr Gascoyne and colleagues said they cannot exclude the possibility that R-CHOP resistance might result from mutations at other sites in MS4A1, as these sites were not evaluated.
These findings appear in the March issue of haematologica. ![]()
Researchers say they have come one step closer to determining the cause of R-CHOP failure in diffuse large B-cell lymphoma (DLBCL) by ruling out a potential cause.
Randy D. Gascoyne, MD, of the British Columbia Cancer Agency, and colleagues found that CD20 mutations involving the rituximab epitope are not the source of R-CHOP resistance. In fact, the mutations are rare in both de novo and relapsed DLBCL.
The rituximab epitope is located in exon 5 of the MS4A1 gene, so Dr Gascoyne and colleagues sequenced this region in DLBCL samples taken at diagnosis and relapse (1 month after completion of 6 cycles of R-CHOP). The team successfully sequenced 264 diagnostic samples and 15 relapsed samples.
The samples could be considered representative of the DLBCL population in British Columbia because clinical characteristics were similar to those observed in previous studies, according to the researchers. In addition, most of the patients had nodal disease with a minimum of 80% tumor, which was sufficient for detecting mutations.
Only 1 of 264 diagnostic samples showed a CD20 mutation involving the rituximab epitope—a 13 base pair heterozygous deletion at position IVS5(+8) in intron 5. Dr Gascoyne and colleagues were unable to determine if this was a polymorphism or a somatic mutation.
This patient achieved a complete response to R-CHOP and is still in remission more than 2 years after diagnosis. This outcome rules out the possibility of mutation-induced rituximab resistance.
As with the diagnostic samples, only 1 of the relapsed samples showed a CD20 mutation involving the rituximab epitope. This was a heterozygous 4 base pair deletion (TAAT) at nucleotide position 353-356, which predicted for a premature termination at amino acid position 121, well before the critical ANPS binding site.
The researchers were unable to establish whether this mutation was present at diagnosis, but they did determine there were no single nucleotide polymorphisms in exon 5 of the CD20 gene.
The rarity of mutations in the rituximab epitope observed in this study suggests these mutations cannot be responsible for the majority of R-CHOP treatment failures. However, Dr Gascoyne and colleagues said they cannot exclude the possibility that R-CHOP resistance might result from mutations at other sites in MS4A1, as these sites were not evaluated.
These findings appear in the March issue of haematologica. ![]()
Researchers say they have come one step closer to determining the cause of R-CHOP failure in diffuse large B-cell lymphoma (DLBCL) by ruling out a potential cause.
Randy D. Gascoyne, MD, of the British Columbia Cancer Agency, and colleagues found that CD20 mutations involving the rituximab epitope are not the source of R-CHOP resistance. In fact, the mutations are rare in both de novo and relapsed DLBCL.
The rituximab epitope is located in exon 5 of the MS4A1 gene, so Dr Gascoyne and colleagues sequenced this region in DLBCL samples taken at diagnosis and relapse (1 month after completion of 6 cycles of R-CHOP). The team successfully sequenced 264 diagnostic samples and 15 relapsed samples.
The samples could be considered representative of the DLBCL population in British Columbia because clinical characteristics were similar to those observed in previous studies, according to the researchers. In addition, most of the patients had nodal disease with a minimum of 80% tumor, which was sufficient for detecting mutations.
Only 1 of 264 diagnostic samples showed a CD20 mutation involving the rituximab epitope—a 13 base pair heterozygous deletion at position IVS5(+8) in intron 5. Dr Gascoyne and colleagues were unable to determine if this was a polymorphism or a somatic mutation.
This patient achieved a complete response to R-CHOP and is still in remission more than 2 years after diagnosis. This outcome rules out the possibility of mutation-induced rituximab resistance.
As with the diagnostic samples, only 1 of the relapsed samples showed a CD20 mutation involving the rituximab epitope. This was a heterozygous 4 base pair deletion (TAAT) at nucleotide position 353-356, which predicted for a premature termination at amino acid position 121, well before the critical ANPS binding site.
The researchers were unable to establish whether this mutation was present at diagnosis, but they did determine there were no single nucleotide polymorphisms in exon 5 of the CD20 gene.
The rarity of mutations in the rituximab epitope observed in this study suggests these mutations cannot be responsible for the majority of R-CHOP treatment failures. However, Dr Gascoyne and colleagues said they cannot exclude the possibility that R-CHOP resistance might result from mutations at other sites in MS4A1, as these sites were not evaluated.
These findings appear in the March issue of haematologica. ![]()
Discharge Summary Improvement
Preventable or ameliorable adverse events have been reported to occur in 12% of patients in the period immediately following hospital discharge.1, 2 A potential contributor to this is the inadequate transfer of clinical information at hospital discharge. The discharge summary comprises a vital component of the information transfer between the inpatient and outpatient settings. Unfortunately, discharge summaries are often unavailable at the time of follow‐up care and often lack important content.37
A growing number of hospitals are implementing electronic medical records (EMR). This creates the opportunity to standardize the content of clinical documentation and creates the potential to assemble, immediately at the time of hospital discharge, major components of a discharge summary. With enhanced communication systems, this information can be delivered in a variety of ways with minimal delay. Previously, we reported the results of a survey of medicine faculty at an urban academic medical center evaluating the timeliness and quality of discharge summaries, the perceived incidence of preventable adverse events related to suboptimal information transfer at discharge, and a needs assessment for an electronically generated discharge summary that we planned to design.8 We now report the results of the follow‐up survey of outpatient physicians and an evaluation of the quality and timeliness of the electronic discharge summary we created.
Materials and Methods
Design
We conducted a pre‐post evaluation of the quality and timeliness of discharge summaries. In the initial phase of the study, we convened an advisory board comprised of 16 Department of Medicine physicians. The advisory board gave input on needs assessment and helped to create a survey to be administered to all medicine faculty with an outpatient practice. All respondents who had at least 1 patient admitted to the hospital within the 6 months prior to the survey were eligible. The results of the initial survey were reviewed with the advisory board and an electronic discharge summary was created with their input. To evaluate its impact, we conducted a repeat survey of all medicine faculty with an outpatient practice approximately 1 year after implementation of the electronic discharge summary.
To complement data received from the outpatient physician survey, a randomly selected sample of discharge summaries from general medical services during the same 3 month period before and after implementation of the electronic discharge summary were rated by 1 of 3 board‐certified internists (D.B.E., N.K., or M.P.L.).
Setting and Participants
The study was conducted at Northwestern Memorial Hospital, a 753‐bed hospital in Chicago, IL. The study was approved by the Institutional Review Board of the Northwestern University Feinberg School of Medicine. General medical patients were admitted to 1 of 2 primary physician services during the study period: a teaching service or a nonteaching hospitalist service. Discharge summaries had traditionally been dictated by inpatient physicians and delivered to outpatient physicians by both mail and facsimile via the medical record department. A recommended template for dictated discharge summaries was provided in the paper paging directory distributed yearly to inpatient physicians.
The hospital implemented an EMR and computerized physician order entry (CPOE) system (PowerChart Millennium; Cerner Corporation, Kansas City, MO) in August 2004. Although all history and physicals and progress notes were documented in the EMR, the system did not provide a method for delivering discharge summaries performed within the EMR to outpatient physician offices. Because of this, inpatient physicians were instructed to continue to dictate discharge summaries during the initial phase of the study.
Approximately 65% of outpatient physicians at the study site used an EMR in their offices during the study. Approximately 10% of outpatient physicians used the same EMR the hospital uses, while approximately 55% used a different EMR (EPIC Hyperspace; EPIC Systems Corporation, Verona, WI). The remaining physicians did not use an EMR in their offices.
Intervention: The Electronic Discharge Summary
A draft electronic discharge summary template was created by including elements ranked as highly important by outpatient physicians in our initial survey8 and elements required by The Joint Commission.9 The draft electronic discharge summary template was reviewed by the advisory board and modifications were made with their input. We automated the insertion of specific patient data elements, such as listed allergies and home medications, into the discharge summary template. We also created an electronic reminder system to inpatient physicians for summaries not completed 24 hours after discharge.
Because the majority of physicians in our initial survey preferred discharge summaries to be delivered either by facsimile or via an EMR, we concentrated our efforts on creating reliable systems for delivery by those routes. We created logic that queried the primary care physician field within the EMR at the time the discharge summary was electronically signed. An automated process then sent the discharge summary via electronic fax to the physician listed in the primary care physician field. Because a large number of outpatient physicians used an EMR different from the hospital's, we also created a process that sent discharge summaries from the hospital EMR into patient charts within this separate EMR.
The draft electronic discharge summary template was available for use in the EMR beginning in July 2005. The final electronic discharge summary, including automated content, physician reminder for incomplete summaries, and delivery systems as described above was implemented in June 2006. Upon implementation, inpatient physicians were instructed via email announcements and group meetings to begin completing electronic discharge summaries using the EMR. Beyond these announcements, inpatient physicians did not receive any specific training with regard to the new discharge summary process. An example of the final electronic discharge summary product is available in the Appendix.
Outpatient Physician Survey
Satisfaction with timeliness and quality of discharge summaries was assessed using a 5‐point Likert scale, where 5 represented very satisfied and 1 represented very dissatisfied. We also asked respondents to estimate the number of their patients who had sustained a preventable adverse event or near miss related to suboptimal transfer of information at discharge. We defined a preventable adverse event as a preventable medical problem or worsening of an existing problem and near miss as an error that did not result in patient harm but easily could have.
The preimplementation survey, accompanied by a cover letter signed by the hospital's chief of staff, was sent out in March 2005. A postcard reminder was sent approximately 2 weeks after the initial mail survey. A second survey was sent to nonresponders 6 weeks after the initial survey. Simultaneously, the survey was also sent in web‐based format to nonresponders via email. The postimplementation survey was sent out in February 2007 using a similar survey process.
Discharge Summary Review
A random sample of discharge summaries completed before and after the implementation of the electronic discharge summary was selected for review. The sample universe consisted of all general medicine service discharges between August and November 2005, before the electronic discharge summary was implemented, and August to November 2006, after implementation. To provide a balanced comparison, the sample was further limited to only the first chronological (index) discharge of a unique patient to home self‐care or home health nursing, with length of stay between 3 and 14 days. A total of 2232 discharges in 2005 and 2570 discharges in 2006 met these criteria. The discharge summary review sample was designed to randomly select approximately 100 discharge summaries meeting the criteria above within each study year, to produce an approximate 200‐record analysis sample. Each of the 3 physician reviewers was assigned to complete an approximately equal number of the 200 primary reviews.
Physician reviewers recorded whether the discharge summary was dictated versus done electronically, the length of the discharge summary (in words), the number of days from discharge to discharge summary completion, the type of service the patient was discharged from, and the author type (medical student, intern, resident, or attending). Physicians reviewers also assessed the overall clarity of discharge summaries using a 5‐point ordinal scale (1 = unintelligible; 2 = hard to read; 3 = neutral; 4 = understandable; and 5 = lucid).
Prior studies have evaluated the quality of discharge summaries using scoring tools created by the investigators.10, 11 We created our own discharge summary scoring tool based on these prior studies, recommendations from the literature,12 and the findings from our initial survey.8 We pilot‐tested the scoring tool and made minor revisions prior to the study. The final scoring tool consisted of 16 essential elements. Reviewers assessed whether each of the 16 essential elements was present, absent, or not applicable. A Discharge Summary Completeness Score was calculated by the number of the 16 essential elements that were rated as present divided by the number of applicable elements for each discharge summary, multiplied by 100 to produce a completeness percentage.
To assess interrater reliability, reviewers were assigned to independently complete second, duplicate reviews of approximately 90 summaries (30 per reviewer). The duplicate review sample was designed to produce approximately 45 paired re‐reviews in each year for reliability assessment. A final sample of 196 available summaries was completed for the main analysis and 174 primary and duplicate reviews were used to establish interrater reliability across 87 reviewer pairs.
Data Analysis
Physician characteristics, including specialty, faculty appointment type, and year of medical school graduation were provided by the hospital's medical staff office. Physician characteristics from before and after the implementation of the electronic discharge summary were compared using chi‐square tests. Likert scale ratings of physician satisfaction with the timeliness and quality of discharge summaries were compared using t‐tests. The proportion of physicians reporting 1 or more preventable adverse event or near miss before the implementation of the electronic discharge summary was compared to postimplementation proportions using chi‐square tests. In addition, we performed multivariate logistic regression to examine the likelihood of physicians reporting any preventable adverse event or near miss related to suboptimal information transfer. The regression models tested the likelihood of 1 or more preventable adverse event or near miss before versus after the implementation of the electronic discharge summary, controlling for physician characteristics and their number of hospitalized patients in the previous 6 months.
The proportions of discharge summary elements found to be present, the proportion of discharge summaries completed within 3 days, and discharge summary readability ratings before and after the implementation of the electronic discharge summary were compared using chi‐square tests; length in words was compared using t‐tests. Preimplementation and postimplementation Discharge Summary Completeness Scores were compared using the Mann‐Whitney U test. Discharge summary score interrater reliability was assessed using the Brennan‐Prediger Kappa for individual elements.13
Results
Outpatient Physician Survey
Physician Characteristics
Two hundred and twenty‐six of 416 (54%) eligible outpatient physicians completed the baseline survey and 256 of 397 (64%) completed the postimplementation survey. As shown in Table 1, there were no significant differences in specialty, faculty appointment type, or number of patients hospitalized between respondents to the survey before compared to respondents after the implementation of the electronic discharge summary. The number of respondents graduating medical school in 1990 or later was higher after implementation of the electronic discharge summary; however, this result was of borderline statistical significance.
| Preelectronic Discharge Summary (n = 226) | Postelectronic Discharge Summary (n = 256) | P Value | |
|---|---|---|---|
| |||
| Practice Type | 0.23 | ||
| Generalist, n (%) | 127 (56.2) | 130 (50.8) | |
| Specialist, n (%) | 99 (43.8) | 126 (49.2) | |
| Faculty Appointment | 0.38 | ||
| Full‐time, n (%) | 104 (46.0) | 128 (50.0) | |
| Affiliated, n (%) | 122 (54.0) | 128 (50.0) | |
| Year of medical school graduation* | 0.06 | ||
| Before 1990, n (%) | 128 (57.4) | 124 (48.8) | |
| 1990 or later, n (%) | 95 (42.6) | 130 (51.2) | |
| Number of patients hospitalized (last 6 months) | 0.56 | ||
| 1‐4, n (%) | 15 (7.9) | 24 (12.0) | |
| 5‐10, n (%) | 62 (32.5) | 66 (33.0) | |
| 11‐19, n (%) | 35 (18.3) | 33 (16.5) | |
| 20 or more, n (%) | 79 (41.4) | 77 (38.5) | |
Timeliness and Content
Changes in outpatient physician satisfaction with the timeliness and quality of discharge summaries are summarized in Table 2. Satisfaction with the timeliness and quality of discharge summarizes improved significantly after the implementation of the electronic discharge summary (mean standard deviation [SD] timeliness rating, 2.59 1.02 versus 3.34 1.09; P < 0.001, mean quality rating 3.04 0.93 versus 3.64 0.99; P < 0.001).
| Likert Scale Mean Score (SD)* | |||
|---|---|---|---|
| Preelectronic Discharge Summary | Postelectronic Discharge Summary | P Value | |
| |||
| Timeliness of the discharge summary | 2.59 (1.02) | 3.34 (1.09) | <0.001 |
| Quality of the discharge summary | 3.04 (0.93) | 3.64 (0.99) | <0.001 |
Medical Error
The effect of the electronic discharge summary on perceived near misses and preventable adverse events is summarized in Table 3. Fewer outpatient physicians felt that 1 or more of their patients hospitalized in the preceding 6 months sustained a near miss due to suboptimal transfer of information after the implementation of the electronic discharge summary (65.7% vs. 52.9%, P = 0.008). Similarly, fewer outpatient physicians felt that 1 or more of their patients hospitalized in the preceding 6 months sustained a preventable adverse event due to suboptimal transfer of information after the implementation of the electronic discharge summary (40.7% vs. 30.2%, P = 0.02). In multivariate logistic regression analyses controlling for physician characteristics and their number of hospitalized patients in the previous 6 months, there was a statistically significant 40% reduction in the odds of a reported near miss (adjusted odds ratio [OR] = 0.60, P = 0.02). Although not quite statistically significant, there was a 33% reduction in the odds of a reported preventable adverse event (OR = 0.67, P = 0.08) after the implementation of the electronic discharge summary.
| Preelectronic Discharge Summary | Postelectronic Discharge Summary | P Value | |
|---|---|---|---|
| |||
| Near miss* | |||
| Number (%) reporting 1 | 142 (65.7) | 108 (52.9) | |
| Crude odds ratio | Ref. | 0.57 | 0.008 |
| Adjusted odds ratio | Ref. | 0.60 | 0.02 |
| Preventable adverse event | |||
| Number (%) reporting 1 | 88 (40.7) | 62 (30.2) | |
| Crude odds ratio | Ref. | 0.63 | 0.03 |
| Adjusted odds ratio | Ref. | 0.67 | 0.08 |
Discharge Summary Review
Discharge Summary Characteristics
One hundred and one discharge summaries before implementation of the electronic discharge summary were compared to 95 discharge summaries produced the following year. Characteristics of discharge summaries before and after the implementation of the electronic discharge summary are summarized in Table 4. A large number of discharge summaries (52.5%) were already being typed into the EMR in 2005, prior to the implementation of our final electronic discharge summary product. The number of dictated discharge summaries decreased from 47.5% to 10.5% after implementation of the final electronic discharge summary product (P < 0.001). Discharge summaries were similar in length before and after the implementation of the electronic discharge summary. A higher percentage of discharge summaries were completed within 3 days of discharge after implementation of the electronic discharge summary; however, this result was of borderline statistical significance (59.4% vs. 72.6%; P = 0.05). The type of service from which patients were discharged and the distribution of author types were similar after the implementation of the electronic discharge summary.
| Number (%) or MeanSD | P Value | ||
|---|---|---|---|
| Preelectronic Discharge Summary (n = 101) | Postelectronic Discharge Summary (n = 95) | ||
| Dictated, n (%) | 48 (47.5) | 10 (10.5) | <0.001 |
| Length in words, mean SD | 785 407 | 830 389 | 0.43 |
| Completed within 3 days, n (%) | 60 (59.4) | 69 (72.6) | 0.05 |
| Type of service, n (%) | 0.29 | ||
| Teaching service | 63 (62.4) | 66 (69.5) | |
| Nonteaching hospitalist service | 38 (37.6) | 29 (30.5) | |
| Author type, n (%) | 0.62 | ||
| Fourth year medical student | 13 (12.9) | 13 (13.7) | |
| Intern | 31 (30.7) | 37 (38.9) | |
| Resident | 19 (18.8) | 15 (15.8) | |
| Attending | 38 (37.6) | 30 (31.6) | |
Because a large percentage of discharge summaries were already being done electronically in 2005, we evaluated the timeliness of dictated discharge summaries compared to electronic discharge summaries across both periods combined (preimplementation and postimplementation of the electronic discharge summary). A higher percentage of electronic discharge summaries were completed within 3 days of discharge as compared to dictated discharge summaries (44.8% versus 74.1%; P < 0.001).
Discharge Summary Completeness Score
The presence or absence of discharge summary elements before and after the implementation of the electronic discharge summary is shown in Table 5. Several elements of the discharge summary were present more often after the implementation of the electronic discharge summary. Specific improvements included discussion of follow‐up issues (52.0% versus 75.8%; P = 0.001, = 0.78), pending test results (13.9% vs. 46.3%; P < 0.001, = 0.92), and information provided to the patient and/or family (85.1% vs. 95.8%; P = 0.01, = 0.91). Significant laboratory findings were present less often after implementation of the electronic discharge summary (66.0% versus 51.1%; P = 0.04, = 0.84). The Discharge Summary Completeness Score was higher after the implementation of the electronic discharge summary (mean 74.1 versus 80.3, P = 0.007). Dictated discharge summaries had a significantly lower Discharge Summary Completeness Score compared to discharge summaries done electronically (71.3 vs. 79.6, P = 0.002) across both periods combined.
| Number (%) of Content Items Present* | P Value | Brennan‐Prediger Kappa | ||
|---|---|---|---|---|
| Preelectronic Discharge Summary (n = 101) | Postelectronic Discharge Summary (n = 95) | |||
| ||||
| Dates of admission and discharge | 96 (95.0) | 94 (98.9) | 0.11 | 1.0 |
| Reason for hospitalization | 100 (99.0) | 94 (100) | 0.33 | 1.0 |
| Significant findings from history and exam | 78 (77.2) | 65 (68.4) | 0.16 | 0.26 |
| Significant laboratory findings | 64 (66.0) | 47 (51.1) | 0.04 | 0.84 |
| Significant radiological findings | 67 (75.3) | 71 (81.6) | 0.31 | 0.89 |
| Significant findings from other tests | 41 (63.1) | 40 (71.4) | 0.33 | 0.88 |
| List of procedures performed | 45 (81.8) | 35 (77.8) | 0.77 | 0.99 |
| Procedure report findings | 49 (80.3) | 43 (78.2) | 0.61 | 0.92 |
| Stress test report findings | 7 (100) | 3 (100) | N/A | 1.0 |
| Pathology report findings | 11 (39.3) | 3 (30.0) | 0.60 | 0.91 |
| Discharge diagnosis | 89 (88.1) | 86 (93.5) | 0.20 | 0.86 |
| Condition at discharge | 81 (81.0) | 80 (85.1) | 0.45 | 0.76 |
| Discharge medications | 88 (87.1) | 88 (93.6) | 0.13 | 0.79 |
| Follow‐up issues | 52 (52.0) | 72 (75.8) | 0.001 | 0.78 |
| Pending test results | 14 (13.9) | 44 (46.3) | <0.001 | 0.92 |
| Information provided to patient and/or family, as appropriate | 86 (85.1) | 91 (95.8) | 0.01 | 0.91 |
| Discharge Summary Completeness Score (percent present all applicable items) | 74.1 | 80.3 | 0.007 | |
Significantly more discharge summaries were rated as understandable or lucid after the implementation of the electronic discharge summary (41.6% vs. 59.0%; P = 0.02). In both periods combined, dictated discharge summaries were rated as understandable or lucid less often than electronic discharge summaries (34.5% vs. 56.5%; P < 0.001).
Discussion
Our study found that an electronic discharge summary was well accepted by inpatient physicians and significantly improved the quality and timeliness of discharge summaries. Prior studies have shown that the use of electronically entered discharge summaries improved the timeliness of discharge summaries.1416 However, the discharge summaries used in these studies required manual input of data into a computer system separate from the patient's medical record. To our knowledge, this is the first study to report the impact of discharge summaries generated from an EMR. Leveraging the EMR, we were able to automate the insertion of specific patient data elements, streamline delivery, and create an electronic reminder system to inpatient physicians for summaries not completed 24 hours after discharge.
Prior research has shown that the quality of discharges summaries is improved with the use of standardized content.10, 17 Using a standardized template for the electronic discharge summary, we likewise demonstrated improved quality of discharge summaries. Key discharge summary elements, specifically discussion of follow‐up issues, pending test results, and information provided to the patient and/or family, were present more reliably after the implementation of the electronic discharge summary. The importance of identifying pending test results is underscored by a recent study showing that many patients are discharged from hospitals with test results still pending, and that physicians are often unaware when results are abnormal.18 One discharge summary element, significant laboratory findings, was present less often after the implementation of the electronic discharge summary. Our template did not designate significant laboratory findings under a separate heading. Instead, we used a heading entitled Key Results (labs, imaging, pathology). Physicians completing the discharge summaries may have prioritized the report of imaging and pathology results in this section. A simple revision of our discharge summary template to include a separate heading for significant laboratory findings may result in improvement in this regard.
Timeliness of discharge summaries was improved in our study, but remained less than optimal. Although nearly three‐quarters of electronic discharge summaries were completed within 3 days of discharge, our ultimate goal is to have 100% of discharge summaries completed within 3 days. This is especially important for complicated patients requiring outpatient follow‐up soon after discharge. We are currently in the process of designing further modifications to the electronic discharge summary completion process. One modification that may be beneficial is the automation of additional patient specific data elements into the discharge summary. We also plan to link performance of medication reconciliation, completion of patient discharge instructions, and completion of the discharge summary into an integrated set of activities performed in the EMR prior to patient discharge.
We found that fewer outpatient physicians reported 1 or more of their patients having a preventable adverse event or near miss as a result of suboptimal transfer of information at discharge after the implementation of the electronic discharge summary. Although we did not measure preventable adverse events directly in our study, this is an important finding in light of the large number of patients who sustain preventable adverse events after hospital discharge1, 2 and prior research showing that the absence of discharge summaries at postdischarge follow‐up visits increased the risk for hospital readmission.19
We had wondered what effect the electronic discharge summary would have on the length and clarity of discharge summaries. A published commentary suggested that notes performed in EMRs were inordinately long and often difficult to read.20 We were pleased to discover that electronic discharge summaries were similar in length to previous discharge summaries and were rated higher with regard to clarity.
Our study has several limitations. First, many inpatient physicians began to use electronic discharge summaries prior to our creation of the final electronic discharge summary product. We had explicitly instructed physicians to continue to dictate discharge summaries in the first phase of our study. The fact that physicians quickly adopted the practice of completing discharge summaries electronically suggests that they preferred this method for completion and may help to explain the improvement in timeliness. A second limitation, as previously mentioned, is that our study did not measure adverse events directly. Instead, we asked outpatient physicians to estimate the number of their patients discharged in the last 6 months who had sustained a preventable adverse event or near miss related to suboptimal information transfer at discharge. We had limited space in the survey to define the meaning of a preventable adverse event; therefore, the description in the survey does not exactly match previous definitions.1, 2 Finally, the ordinal scale used to assess clarity of discharge summaries has not been previously validated.
In conclusion, the use of an electronic discharge summary significantly improved the quality and timeliness of discharge summaries. The discharge summary comprises a vital component of the information transfer between the inpatient and outpatient settings during the vulnerable period following hospital discharge. As hospitals expand their use of EMRs, they should take advantage of opportunities to leverage functionality to improve quality and timeliness of discharge summaries.
- ,,, et al.Adverse events among medical patient after hospital discharge.CMAJ.2004;170:345–349.
- ,,,,.The incidence and severity of adverse events affecting patients after discharge from the hospital.Ann Intern Med.2003;138:161–167.
- ,,.Dissemination of discharge summaries. Not reaching follow‐up physicians.Can Fam Physician.2002;48:737–742.
- ,,,.Effect of discharge summary availability during post‐discharge visits on hospital readmission.J Gen Intern Med.2002;17:186–192.
- ,,,.General practitioner‐hospital communications: a review of discharge summaries.J Qual Clin Practice.2001;21:104–108.
- ,,,,.Quality assessment of discharge letters in a French university hospital.Int J Health Care Qual Assur.1998;11:90–95.
- ,,,,,.Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care.JAMA.2007;297:831–841.
- ,,,,.Outpatient physicians' satisfaction with discharge summaries and perceived need for an electronic discharge summary.J Hosp Med.2006;1:317–320.
- Standard IM.6.10: Hospital accreditation standards.Oakbrook Terrace, IL:Joint Commission on Accreditation of Healthcare Organizations;2006:338–340.
- ,,,,.Assessing quality and efficiency of discharge summaries.Am J Med Qual.2005;20:337–343.
- ,,,,,.Are discharge summaries teachable? The effects of a discharge summary curriculum on the quality of discharge summaries in an internal medicine residency program.Acad Med.2006;81(10 Suppl):S5–S8.
- ,,, et al.Transition of care for hospitalized elderly patients–development of a discharge checklist for hospitalists.J Hosp Med.2006;1:354–360.
- ,.Coefficient kappa: some uses, misuses, and alternatives.Educ Psychol Meas.1981;41:687–699.
- ,,,.Dictated versus database‐generated discharge summaries: a randomized clinical trial.CMAJ.1999;160:319–326.
- ,,,.Evaluation of computer generated neonatal discharge summaries.Arch Dis Child.1991;66:433–436.
- ,,,,,.Evaluation of a computer‐generated discharge summary for patients with acute coronary syndromes.Br J Gen Pract.1998;48:1163–1164.
- ,,,.Standardized or narrative discharge summaries: Which do family physicians prefer?Can Fam Phys.1998;44:62–69.
- ,,, et al.Patient safety concerns arising from test results that return after hospital discharge.Ann Intern Med.2005;143:121–128.
- ,,,Effect of discharge summary availability during post‐discharge visits on hospital readmission.J Gen Intern Med.2002:17;186–192.
- .A piece of my mind. Copy‐and‐paste.JAMA.2006;295:2335–2336.
Preventable or ameliorable adverse events have been reported to occur in 12% of patients in the period immediately following hospital discharge.1, 2 A potential contributor to this is the inadequate transfer of clinical information at hospital discharge. The discharge summary comprises a vital component of the information transfer between the inpatient and outpatient settings. Unfortunately, discharge summaries are often unavailable at the time of follow‐up care and often lack important content.37
A growing number of hospitals are implementing electronic medical records (EMR). This creates the opportunity to standardize the content of clinical documentation and creates the potential to assemble, immediately at the time of hospital discharge, major components of a discharge summary. With enhanced communication systems, this information can be delivered in a variety of ways with minimal delay. Previously, we reported the results of a survey of medicine faculty at an urban academic medical center evaluating the timeliness and quality of discharge summaries, the perceived incidence of preventable adverse events related to suboptimal information transfer at discharge, and a needs assessment for an electronically generated discharge summary that we planned to design.8 We now report the results of the follow‐up survey of outpatient physicians and an evaluation of the quality and timeliness of the electronic discharge summary we created.
Materials and Methods
Design
We conducted a pre‐post evaluation of the quality and timeliness of discharge summaries. In the initial phase of the study, we convened an advisory board comprised of 16 Department of Medicine physicians. The advisory board gave input on needs assessment and helped to create a survey to be administered to all medicine faculty with an outpatient practice. All respondents who had at least 1 patient admitted to the hospital within the 6 months prior to the survey were eligible. The results of the initial survey were reviewed with the advisory board and an electronic discharge summary was created with their input. To evaluate its impact, we conducted a repeat survey of all medicine faculty with an outpatient practice approximately 1 year after implementation of the electronic discharge summary.
To complement data received from the outpatient physician survey, a randomly selected sample of discharge summaries from general medical services during the same 3 month period before and after implementation of the electronic discharge summary were rated by 1 of 3 board‐certified internists (D.B.E., N.K., or M.P.L.).
Setting and Participants
The study was conducted at Northwestern Memorial Hospital, a 753‐bed hospital in Chicago, IL. The study was approved by the Institutional Review Board of the Northwestern University Feinberg School of Medicine. General medical patients were admitted to 1 of 2 primary physician services during the study period: a teaching service or a nonteaching hospitalist service. Discharge summaries had traditionally been dictated by inpatient physicians and delivered to outpatient physicians by both mail and facsimile via the medical record department. A recommended template for dictated discharge summaries was provided in the paper paging directory distributed yearly to inpatient physicians.
The hospital implemented an EMR and computerized physician order entry (CPOE) system (PowerChart Millennium; Cerner Corporation, Kansas City, MO) in August 2004. Although all history and physicals and progress notes were documented in the EMR, the system did not provide a method for delivering discharge summaries performed within the EMR to outpatient physician offices. Because of this, inpatient physicians were instructed to continue to dictate discharge summaries during the initial phase of the study.
Approximately 65% of outpatient physicians at the study site used an EMR in their offices during the study. Approximately 10% of outpatient physicians used the same EMR the hospital uses, while approximately 55% used a different EMR (EPIC Hyperspace; EPIC Systems Corporation, Verona, WI). The remaining physicians did not use an EMR in their offices.
Intervention: The Electronic Discharge Summary
A draft electronic discharge summary template was created by including elements ranked as highly important by outpatient physicians in our initial survey8 and elements required by The Joint Commission.9 The draft electronic discharge summary template was reviewed by the advisory board and modifications were made with their input. We automated the insertion of specific patient data elements, such as listed allergies and home medications, into the discharge summary template. We also created an electronic reminder system to inpatient physicians for summaries not completed 24 hours after discharge.
Because the majority of physicians in our initial survey preferred discharge summaries to be delivered either by facsimile or via an EMR, we concentrated our efforts on creating reliable systems for delivery by those routes. We created logic that queried the primary care physician field within the EMR at the time the discharge summary was electronically signed. An automated process then sent the discharge summary via electronic fax to the physician listed in the primary care physician field. Because a large number of outpatient physicians used an EMR different from the hospital's, we also created a process that sent discharge summaries from the hospital EMR into patient charts within this separate EMR.
The draft electronic discharge summary template was available for use in the EMR beginning in July 2005. The final electronic discharge summary, including automated content, physician reminder for incomplete summaries, and delivery systems as described above was implemented in June 2006. Upon implementation, inpatient physicians were instructed via email announcements and group meetings to begin completing electronic discharge summaries using the EMR. Beyond these announcements, inpatient physicians did not receive any specific training with regard to the new discharge summary process. An example of the final electronic discharge summary product is available in the Appendix.
Outpatient Physician Survey
Satisfaction with timeliness and quality of discharge summaries was assessed using a 5‐point Likert scale, where 5 represented very satisfied and 1 represented very dissatisfied. We also asked respondents to estimate the number of their patients who had sustained a preventable adverse event or near miss related to suboptimal transfer of information at discharge. We defined a preventable adverse event as a preventable medical problem or worsening of an existing problem and near miss as an error that did not result in patient harm but easily could have.
The preimplementation survey, accompanied by a cover letter signed by the hospital's chief of staff, was sent out in March 2005. A postcard reminder was sent approximately 2 weeks after the initial mail survey. A second survey was sent to nonresponders 6 weeks after the initial survey. Simultaneously, the survey was also sent in web‐based format to nonresponders via email. The postimplementation survey was sent out in February 2007 using a similar survey process.
Discharge Summary Review
A random sample of discharge summaries completed before and after the implementation of the electronic discharge summary was selected for review. The sample universe consisted of all general medicine service discharges between August and November 2005, before the electronic discharge summary was implemented, and August to November 2006, after implementation. To provide a balanced comparison, the sample was further limited to only the first chronological (index) discharge of a unique patient to home self‐care or home health nursing, with length of stay between 3 and 14 days. A total of 2232 discharges in 2005 and 2570 discharges in 2006 met these criteria. The discharge summary review sample was designed to randomly select approximately 100 discharge summaries meeting the criteria above within each study year, to produce an approximate 200‐record analysis sample. Each of the 3 physician reviewers was assigned to complete an approximately equal number of the 200 primary reviews.
Physician reviewers recorded whether the discharge summary was dictated versus done electronically, the length of the discharge summary (in words), the number of days from discharge to discharge summary completion, the type of service the patient was discharged from, and the author type (medical student, intern, resident, or attending). Physicians reviewers also assessed the overall clarity of discharge summaries using a 5‐point ordinal scale (1 = unintelligible; 2 = hard to read; 3 = neutral; 4 = understandable; and 5 = lucid).
Prior studies have evaluated the quality of discharge summaries using scoring tools created by the investigators.10, 11 We created our own discharge summary scoring tool based on these prior studies, recommendations from the literature,12 and the findings from our initial survey.8 We pilot‐tested the scoring tool and made minor revisions prior to the study. The final scoring tool consisted of 16 essential elements. Reviewers assessed whether each of the 16 essential elements was present, absent, or not applicable. A Discharge Summary Completeness Score was calculated by the number of the 16 essential elements that were rated as present divided by the number of applicable elements for each discharge summary, multiplied by 100 to produce a completeness percentage.
To assess interrater reliability, reviewers were assigned to independently complete second, duplicate reviews of approximately 90 summaries (30 per reviewer). The duplicate review sample was designed to produce approximately 45 paired re‐reviews in each year for reliability assessment. A final sample of 196 available summaries was completed for the main analysis and 174 primary and duplicate reviews were used to establish interrater reliability across 87 reviewer pairs.
Data Analysis
Physician characteristics, including specialty, faculty appointment type, and year of medical school graduation were provided by the hospital's medical staff office. Physician characteristics from before and after the implementation of the electronic discharge summary were compared using chi‐square tests. Likert scale ratings of physician satisfaction with the timeliness and quality of discharge summaries were compared using t‐tests. The proportion of physicians reporting 1 or more preventable adverse event or near miss before the implementation of the electronic discharge summary was compared to postimplementation proportions using chi‐square tests. In addition, we performed multivariate logistic regression to examine the likelihood of physicians reporting any preventable adverse event or near miss related to suboptimal information transfer. The regression models tested the likelihood of 1 or more preventable adverse event or near miss before versus after the implementation of the electronic discharge summary, controlling for physician characteristics and their number of hospitalized patients in the previous 6 months.
The proportions of discharge summary elements found to be present, the proportion of discharge summaries completed within 3 days, and discharge summary readability ratings before and after the implementation of the electronic discharge summary were compared using chi‐square tests; length in words was compared using t‐tests. Preimplementation and postimplementation Discharge Summary Completeness Scores were compared using the Mann‐Whitney U test. Discharge summary score interrater reliability was assessed using the Brennan‐Prediger Kappa for individual elements.13
Results
Outpatient Physician Survey
Physician Characteristics
Two hundred and twenty‐six of 416 (54%) eligible outpatient physicians completed the baseline survey and 256 of 397 (64%) completed the postimplementation survey. As shown in Table 1, there were no significant differences in specialty, faculty appointment type, or number of patients hospitalized between respondents to the survey before compared to respondents after the implementation of the electronic discharge summary. The number of respondents graduating medical school in 1990 or later was higher after implementation of the electronic discharge summary; however, this result was of borderline statistical significance.
| Preelectronic Discharge Summary (n = 226) | Postelectronic Discharge Summary (n = 256) | P Value | |
|---|---|---|---|
| |||
| Practice Type | 0.23 | ||
| Generalist, n (%) | 127 (56.2) | 130 (50.8) | |
| Specialist, n (%) | 99 (43.8) | 126 (49.2) | |
| Faculty Appointment | 0.38 | ||
| Full‐time, n (%) | 104 (46.0) | 128 (50.0) | |
| Affiliated, n (%) | 122 (54.0) | 128 (50.0) | |
| Year of medical school graduation* | 0.06 | ||
| Before 1990, n (%) | 128 (57.4) | 124 (48.8) | |
| 1990 or later, n (%) | 95 (42.6) | 130 (51.2) | |
| Number of patients hospitalized (last 6 months) | 0.56 | ||
| 1‐4, n (%) | 15 (7.9) | 24 (12.0) | |
| 5‐10, n (%) | 62 (32.5) | 66 (33.0) | |
| 11‐19, n (%) | 35 (18.3) | 33 (16.5) | |
| 20 or more, n (%) | 79 (41.4) | 77 (38.5) | |
Timeliness and Content
Changes in outpatient physician satisfaction with the timeliness and quality of discharge summaries are summarized in Table 2. Satisfaction with the timeliness and quality of discharge summarizes improved significantly after the implementation of the electronic discharge summary (mean standard deviation [SD] timeliness rating, 2.59 1.02 versus 3.34 1.09; P < 0.001, mean quality rating 3.04 0.93 versus 3.64 0.99; P < 0.001).
| Likert Scale Mean Score (SD)* | |||
|---|---|---|---|
| Preelectronic Discharge Summary | Postelectronic Discharge Summary | P Value | |
| |||
| Timeliness of the discharge summary | 2.59 (1.02) | 3.34 (1.09) | <0.001 |
| Quality of the discharge summary | 3.04 (0.93) | 3.64 (0.99) | <0.001 |
Medical Error
The effect of the electronic discharge summary on perceived near misses and preventable adverse events is summarized in Table 3. Fewer outpatient physicians felt that 1 or more of their patients hospitalized in the preceding 6 months sustained a near miss due to suboptimal transfer of information after the implementation of the electronic discharge summary (65.7% vs. 52.9%, P = 0.008). Similarly, fewer outpatient physicians felt that 1 or more of their patients hospitalized in the preceding 6 months sustained a preventable adverse event due to suboptimal transfer of information after the implementation of the electronic discharge summary (40.7% vs. 30.2%, P = 0.02). In multivariate logistic regression analyses controlling for physician characteristics and their number of hospitalized patients in the previous 6 months, there was a statistically significant 40% reduction in the odds of a reported near miss (adjusted odds ratio [OR] = 0.60, P = 0.02). Although not quite statistically significant, there was a 33% reduction in the odds of a reported preventable adverse event (OR = 0.67, P = 0.08) after the implementation of the electronic discharge summary.
| Preelectronic Discharge Summary | Postelectronic Discharge Summary | P Value | |
|---|---|---|---|
| |||
| Near miss* | |||
| Number (%) reporting 1 | 142 (65.7) | 108 (52.9) | |
| Crude odds ratio | Ref. | 0.57 | 0.008 |
| Adjusted odds ratio | Ref. | 0.60 | 0.02 |
| Preventable adverse event | |||
| Number (%) reporting 1 | 88 (40.7) | 62 (30.2) | |
| Crude odds ratio | Ref. | 0.63 | 0.03 |
| Adjusted odds ratio | Ref. | 0.67 | 0.08 |
Discharge Summary Review
Discharge Summary Characteristics
One hundred and one discharge summaries before implementation of the electronic discharge summary were compared to 95 discharge summaries produced the following year. Characteristics of discharge summaries before and after the implementation of the electronic discharge summary are summarized in Table 4. A large number of discharge summaries (52.5%) were already being typed into the EMR in 2005, prior to the implementation of our final electronic discharge summary product. The number of dictated discharge summaries decreased from 47.5% to 10.5% after implementation of the final electronic discharge summary product (P < 0.001). Discharge summaries were similar in length before and after the implementation of the electronic discharge summary. A higher percentage of discharge summaries were completed within 3 days of discharge after implementation of the electronic discharge summary; however, this result was of borderline statistical significance (59.4% vs. 72.6%; P = 0.05). The type of service from which patients were discharged and the distribution of author types were similar after the implementation of the electronic discharge summary.
| Number (%) or MeanSD | P Value | ||
|---|---|---|---|
| Preelectronic Discharge Summary (n = 101) | Postelectronic Discharge Summary (n = 95) | ||
| Dictated, n (%) | 48 (47.5) | 10 (10.5) | <0.001 |
| Length in words, mean SD | 785 407 | 830 389 | 0.43 |
| Completed within 3 days, n (%) | 60 (59.4) | 69 (72.6) | 0.05 |
| Type of service, n (%) | 0.29 | ||
| Teaching service | 63 (62.4) | 66 (69.5) | |
| Nonteaching hospitalist service | 38 (37.6) | 29 (30.5) | |
| Author type, n (%) | 0.62 | ||
| Fourth year medical student | 13 (12.9) | 13 (13.7) | |
| Intern | 31 (30.7) | 37 (38.9) | |
| Resident | 19 (18.8) | 15 (15.8) | |
| Attending | 38 (37.6) | 30 (31.6) | |
Because a large percentage of discharge summaries were already being done electronically in 2005, we evaluated the timeliness of dictated discharge summaries compared to electronic discharge summaries across both periods combined (preimplementation and postimplementation of the electronic discharge summary). A higher percentage of electronic discharge summaries were completed within 3 days of discharge as compared to dictated discharge summaries (44.8% versus 74.1%; P < 0.001).
Discharge Summary Completeness Score
The presence or absence of discharge summary elements before and after the implementation of the electronic discharge summary is shown in Table 5. Several elements of the discharge summary were present more often after the implementation of the electronic discharge summary. Specific improvements included discussion of follow‐up issues (52.0% versus 75.8%; P = 0.001, = 0.78), pending test results (13.9% vs. 46.3%; P < 0.001, = 0.92), and information provided to the patient and/or family (85.1% vs. 95.8%; P = 0.01, = 0.91). Significant laboratory findings were present less often after implementation of the electronic discharge summary (66.0% versus 51.1%; P = 0.04, = 0.84). The Discharge Summary Completeness Score was higher after the implementation of the electronic discharge summary (mean 74.1 versus 80.3, P = 0.007). Dictated discharge summaries had a significantly lower Discharge Summary Completeness Score compared to discharge summaries done electronically (71.3 vs. 79.6, P = 0.002) across both periods combined.
| Number (%) of Content Items Present* | P Value | Brennan‐Prediger Kappa | ||
|---|---|---|---|---|
| Preelectronic Discharge Summary (n = 101) | Postelectronic Discharge Summary (n = 95) | |||
| ||||
| Dates of admission and discharge | 96 (95.0) | 94 (98.9) | 0.11 | 1.0 |
| Reason for hospitalization | 100 (99.0) | 94 (100) | 0.33 | 1.0 |
| Significant findings from history and exam | 78 (77.2) | 65 (68.4) | 0.16 | 0.26 |
| Significant laboratory findings | 64 (66.0) | 47 (51.1) | 0.04 | 0.84 |
| Significant radiological findings | 67 (75.3) | 71 (81.6) | 0.31 | 0.89 |
| Significant findings from other tests | 41 (63.1) | 40 (71.4) | 0.33 | 0.88 |
| List of procedures performed | 45 (81.8) | 35 (77.8) | 0.77 | 0.99 |
| Procedure report findings | 49 (80.3) | 43 (78.2) | 0.61 | 0.92 |
| Stress test report findings | 7 (100) | 3 (100) | N/A | 1.0 |
| Pathology report findings | 11 (39.3) | 3 (30.0) | 0.60 | 0.91 |
| Discharge diagnosis | 89 (88.1) | 86 (93.5) | 0.20 | 0.86 |
| Condition at discharge | 81 (81.0) | 80 (85.1) | 0.45 | 0.76 |
| Discharge medications | 88 (87.1) | 88 (93.6) | 0.13 | 0.79 |
| Follow‐up issues | 52 (52.0) | 72 (75.8) | 0.001 | 0.78 |
| Pending test results | 14 (13.9) | 44 (46.3) | <0.001 | 0.92 |
| Information provided to patient and/or family, as appropriate | 86 (85.1) | 91 (95.8) | 0.01 | 0.91 |
| Discharge Summary Completeness Score (percent present all applicable items) | 74.1 | 80.3 | 0.007 | |
Significantly more discharge summaries were rated as understandable or lucid after the implementation of the electronic discharge summary (41.6% vs. 59.0%; P = 0.02). In both periods combined, dictated discharge summaries were rated as understandable or lucid less often than electronic discharge summaries (34.5% vs. 56.5%; P < 0.001).
Discussion
Our study found that an electronic discharge summary was well accepted by inpatient physicians and significantly improved the quality and timeliness of discharge summaries. Prior studies have shown that the use of electronically entered discharge summaries improved the timeliness of discharge summaries.1416 However, the discharge summaries used in these studies required manual input of data into a computer system separate from the patient's medical record. To our knowledge, this is the first study to report the impact of discharge summaries generated from an EMR. Leveraging the EMR, we were able to automate the insertion of specific patient data elements, streamline delivery, and create an electronic reminder system to inpatient physicians for summaries not completed 24 hours after discharge.
Prior research has shown that the quality of discharges summaries is improved with the use of standardized content.10, 17 Using a standardized template for the electronic discharge summary, we likewise demonstrated improved quality of discharge summaries. Key discharge summary elements, specifically discussion of follow‐up issues, pending test results, and information provided to the patient and/or family, were present more reliably after the implementation of the electronic discharge summary. The importance of identifying pending test results is underscored by a recent study showing that many patients are discharged from hospitals with test results still pending, and that physicians are often unaware when results are abnormal.18 One discharge summary element, significant laboratory findings, was present less often after the implementation of the electronic discharge summary. Our template did not designate significant laboratory findings under a separate heading. Instead, we used a heading entitled Key Results (labs, imaging, pathology). Physicians completing the discharge summaries may have prioritized the report of imaging and pathology results in this section. A simple revision of our discharge summary template to include a separate heading for significant laboratory findings may result in improvement in this regard.
Timeliness of discharge summaries was improved in our study, but remained less than optimal. Although nearly three‐quarters of electronic discharge summaries were completed within 3 days of discharge, our ultimate goal is to have 100% of discharge summaries completed within 3 days. This is especially important for complicated patients requiring outpatient follow‐up soon after discharge. We are currently in the process of designing further modifications to the electronic discharge summary completion process. One modification that may be beneficial is the automation of additional patient specific data elements into the discharge summary. We also plan to link performance of medication reconciliation, completion of patient discharge instructions, and completion of the discharge summary into an integrated set of activities performed in the EMR prior to patient discharge.
We found that fewer outpatient physicians reported 1 or more of their patients having a preventable adverse event or near miss as a result of suboptimal transfer of information at discharge after the implementation of the electronic discharge summary. Although we did not measure preventable adverse events directly in our study, this is an important finding in light of the large number of patients who sustain preventable adverse events after hospital discharge1, 2 and prior research showing that the absence of discharge summaries at postdischarge follow‐up visits increased the risk for hospital readmission.19
We had wondered what effect the electronic discharge summary would have on the length and clarity of discharge summaries. A published commentary suggested that notes performed in EMRs were inordinately long and often difficult to read.20 We were pleased to discover that electronic discharge summaries were similar in length to previous discharge summaries and were rated higher with regard to clarity.
Our study has several limitations. First, many inpatient physicians began to use electronic discharge summaries prior to our creation of the final electronic discharge summary product. We had explicitly instructed physicians to continue to dictate discharge summaries in the first phase of our study. The fact that physicians quickly adopted the practice of completing discharge summaries electronically suggests that they preferred this method for completion and may help to explain the improvement in timeliness. A second limitation, as previously mentioned, is that our study did not measure adverse events directly. Instead, we asked outpatient physicians to estimate the number of their patients discharged in the last 6 months who had sustained a preventable adverse event or near miss related to suboptimal information transfer at discharge. We had limited space in the survey to define the meaning of a preventable adverse event; therefore, the description in the survey does not exactly match previous definitions.1, 2 Finally, the ordinal scale used to assess clarity of discharge summaries has not been previously validated.
In conclusion, the use of an electronic discharge summary significantly improved the quality and timeliness of discharge summaries. The discharge summary comprises a vital component of the information transfer between the inpatient and outpatient settings during the vulnerable period following hospital discharge. As hospitals expand their use of EMRs, they should take advantage of opportunities to leverage functionality to improve quality and timeliness of discharge summaries.
Preventable or ameliorable adverse events have been reported to occur in 12% of patients in the period immediately following hospital discharge.1, 2 A potential contributor to this is the inadequate transfer of clinical information at hospital discharge. The discharge summary comprises a vital component of the information transfer between the inpatient and outpatient settings. Unfortunately, discharge summaries are often unavailable at the time of follow‐up care and often lack important content.37
A growing number of hospitals are implementing electronic medical records (EMR). This creates the opportunity to standardize the content of clinical documentation and creates the potential to assemble, immediately at the time of hospital discharge, major components of a discharge summary. With enhanced communication systems, this information can be delivered in a variety of ways with minimal delay. Previously, we reported the results of a survey of medicine faculty at an urban academic medical center evaluating the timeliness and quality of discharge summaries, the perceived incidence of preventable adverse events related to suboptimal information transfer at discharge, and a needs assessment for an electronically generated discharge summary that we planned to design.8 We now report the results of the follow‐up survey of outpatient physicians and an evaluation of the quality and timeliness of the electronic discharge summary we created.
Materials and Methods
Design
We conducted a pre‐post evaluation of the quality and timeliness of discharge summaries. In the initial phase of the study, we convened an advisory board comprised of 16 Department of Medicine physicians. The advisory board gave input on needs assessment and helped to create a survey to be administered to all medicine faculty with an outpatient practice. All respondents who had at least 1 patient admitted to the hospital within the 6 months prior to the survey were eligible. The results of the initial survey were reviewed with the advisory board and an electronic discharge summary was created with their input. To evaluate its impact, we conducted a repeat survey of all medicine faculty with an outpatient practice approximately 1 year after implementation of the electronic discharge summary.
To complement data received from the outpatient physician survey, a randomly selected sample of discharge summaries from general medical services during the same 3 month period before and after implementation of the electronic discharge summary were rated by 1 of 3 board‐certified internists (D.B.E., N.K., or M.P.L.).
Setting and Participants
The study was conducted at Northwestern Memorial Hospital, a 753‐bed hospital in Chicago, IL. The study was approved by the Institutional Review Board of the Northwestern University Feinberg School of Medicine. General medical patients were admitted to 1 of 2 primary physician services during the study period: a teaching service or a nonteaching hospitalist service. Discharge summaries had traditionally been dictated by inpatient physicians and delivered to outpatient physicians by both mail and facsimile via the medical record department. A recommended template for dictated discharge summaries was provided in the paper paging directory distributed yearly to inpatient physicians.
The hospital implemented an EMR and computerized physician order entry (CPOE) system (PowerChart Millennium; Cerner Corporation, Kansas City, MO) in August 2004. Although all history and physicals and progress notes were documented in the EMR, the system did not provide a method for delivering discharge summaries performed within the EMR to outpatient physician offices. Because of this, inpatient physicians were instructed to continue to dictate discharge summaries during the initial phase of the study.
Approximately 65% of outpatient physicians at the study site used an EMR in their offices during the study. Approximately 10% of outpatient physicians used the same EMR the hospital uses, while approximately 55% used a different EMR (EPIC Hyperspace; EPIC Systems Corporation, Verona, WI). The remaining physicians did not use an EMR in their offices.
Intervention: The Electronic Discharge Summary
A draft electronic discharge summary template was created by including elements ranked as highly important by outpatient physicians in our initial survey8 and elements required by The Joint Commission.9 The draft electronic discharge summary template was reviewed by the advisory board and modifications were made with their input. We automated the insertion of specific patient data elements, such as listed allergies and home medications, into the discharge summary template. We also created an electronic reminder system to inpatient physicians for summaries not completed 24 hours after discharge.
Because the majority of physicians in our initial survey preferred discharge summaries to be delivered either by facsimile or via an EMR, we concentrated our efforts on creating reliable systems for delivery by those routes. We created logic that queried the primary care physician field within the EMR at the time the discharge summary was electronically signed. An automated process then sent the discharge summary via electronic fax to the physician listed in the primary care physician field. Because a large number of outpatient physicians used an EMR different from the hospital's, we also created a process that sent discharge summaries from the hospital EMR into patient charts within this separate EMR.
The draft electronic discharge summary template was available for use in the EMR beginning in July 2005. The final electronic discharge summary, including automated content, physician reminder for incomplete summaries, and delivery systems as described above was implemented in June 2006. Upon implementation, inpatient physicians were instructed via email announcements and group meetings to begin completing electronic discharge summaries using the EMR. Beyond these announcements, inpatient physicians did not receive any specific training with regard to the new discharge summary process. An example of the final electronic discharge summary product is available in the Appendix.
Outpatient Physician Survey
Satisfaction with timeliness and quality of discharge summaries was assessed using a 5‐point Likert scale, where 5 represented very satisfied and 1 represented very dissatisfied. We also asked respondents to estimate the number of their patients who had sustained a preventable adverse event or near miss related to suboptimal transfer of information at discharge. We defined a preventable adverse event as a preventable medical problem or worsening of an existing problem and near miss as an error that did not result in patient harm but easily could have.
The preimplementation survey, accompanied by a cover letter signed by the hospital's chief of staff, was sent out in March 2005. A postcard reminder was sent approximately 2 weeks after the initial mail survey. A second survey was sent to nonresponders 6 weeks after the initial survey. Simultaneously, the survey was also sent in web‐based format to nonresponders via email. The postimplementation survey was sent out in February 2007 using a similar survey process.
Discharge Summary Review
A random sample of discharge summaries completed before and after the implementation of the electronic discharge summary was selected for review. The sample universe consisted of all general medicine service discharges between August and November 2005, before the electronic discharge summary was implemented, and August to November 2006, after implementation. To provide a balanced comparison, the sample was further limited to only the first chronological (index) discharge of a unique patient to home self‐care or home health nursing, with length of stay between 3 and 14 days. A total of 2232 discharges in 2005 and 2570 discharges in 2006 met these criteria. The discharge summary review sample was designed to randomly select approximately 100 discharge summaries meeting the criteria above within each study year, to produce an approximate 200‐record analysis sample. Each of the 3 physician reviewers was assigned to complete an approximately equal number of the 200 primary reviews.
Physician reviewers recorded whether the discharge summary was dictated versus done electronically, the length of the discharge summary (in words), the number of days from discharge to discharge summary completion, the type of service the patient was discharged from, and the author type (medical student, intern, resident, or attending). Physicians reviewers also assessed the overall clarity of discharge summaries using a 5‐point ordinal scale (1 = unintelligible; 2 = hard to read; 3 = neutral; 4 = understandable; and 5 = lucid).
Prior studies have evaluated the quality of discharge summaries using scoring tools created by the investigators.10, 11 We created our own discharge summary scoring tool based on these prior studies, recommendations from the literature,12 and the findings from our initial survey.8 We pilot‐tested the scoring tool and made minor revisions prior to the study. The final scoring tool consisted of 16 essential elements. Reviewers assessed whether each of the 16 essential elements was present, absent, or not applicable. A Discharge Summary Completeness Score was calculated by the number of the 16 essential elements that were rated as present divided by the number of applicable elements for each discharge summary, multiplied by 100 to produce a completeness percentage.
To assess interrater reliability, reviewers were assigned to independently complete second, duplicate reviews of approximately 90 summaries (30 per reviewer). The duplicate review sample was designed to produce approximately 45 paired re‐reviews in each year for reliability assessment. A final sample of 196 available summaries was completed for the main analysis and 174 primary and duplicate reviews were used to establish interrater reliability across 87 reviewer pairs.
Data Analysis
Physician characteristics, including specialty, faculty appointment type, and year of medical school graduation were provided by the hospital's medical staff office. Physician characteristics from before and after the implementation of the electronic discharge summary were compared using chi‐square tests. Likert scale ratings of physician satisfaction with the timeliness and quality of discharge summaries were compared using t‐tests. The proportion of physicians reporting 1 or more preventable adverse event or near miss before the implementation of the electronic discharge summary was compared to postimplementation proportions using chi‐square tests. In addition, we performed multivariate logistic regression to examine the likelihood of physicians reporting any preventable adverse event or near miss related to suboptimal information transfer. The regression models tested the likelihood of 1 or more preventable adverse event or near miss before versus after the implementation of the electronic discharge summary, controlling for physician characteristics and their number of hospitalized patients in the previous 6 months.
The proportions of discharge summary elements found to be present, the proportion of discharge summaries completed within 3 days, and discharge summary readability ratings before and after the implementation of the electronic discharge summary were compared using chi‐square tests; length in words was compared using t‐tests. Preimplementation and postimplementation Discharge Summary Completeness Scores were compared using the Mann‐Whitney U test. Discharge summary score interrater reliability was assessed using the Brennan‐Prediger Kappa for individual elements.13
Results
Outpatient Physician Survey
Physician Characteristics
Two hundred and twenty‐six of 416 (54%) eligible outpatient physicians completed the baseline survey and 256 of 397 (64%) completed the postimplementation survey. As shown in Table 1, there were no significant differences in specialty, faculty appointment type, or number of patients hospitalized between respondents to the survey before compared to respondents after the implementation of the electronic discharge summary. The number of respondents graduating medical school in 1990 or later was higher after implementation of the electronic discharge summary; however, this result was of borderline statistical significance.
| Preelectronic Discharge Summary (n = 226) | Postelectronic Discharge Summary (n = 256) | P Value | |
|---|---|---|---|
| |||
| Practice Type | 0.23 | ||
| Generalist, n (%) | 127 (56.2) | 130 (50.8) | |
| Specialist, n (%) | 99 (43.8) | 126 (49.2) | |
| Faculty Appointment | 0.38 | ||
| Full‐time, n (%) | 104 (46.0) | 128 (50.0) | |
| Affiliated, n (%) | 122 (54.0) | 128 (50.0) | |
| Year of medical school graduation* | 0.06 | ||
| Before 1990, n (%) | 128 (57.4) | 124 (48.8) | |
| 1990 or later, n (%) | 95 (42.6) | 130 (51.2) | |
| Number of patients hospitalized (last 6 months) | 0.56 | ||
| 1‐4, n (%) | 15 (7.9) | 24 (12.0) | |
| 5‐10, n (%) | 62 (32.5) | 66 (33.0) | |
| 11‐19, n (%) | 35 (18.3) | 33 (16.5) | |
| 20 or more, n (%) | 79 (41.4) | 77 (38.5) | |
Timeliness and Content
Changes in outpatient physician satisfaction with the timeliness and quality of discharge summaries are summarized in Table 2. Satisfaction with the timeliness and quality of discharge summarizes improved significantly after the implementation of the electronic discharge summary (mean standard deviation [SD] timeliness rating, 2.59 1.02 versus 3.34 1.09; P < 0.001, mean quality rating 3.04 0.93 versus 3.64 0.99; P < 0.001).
| Likert Scale Mean Score (SD)* | |||
|---|---|---|---|
| Preelectronic Discharge Summary | Postelectronic Discharge Summary | P Value | |
| |||
| Timeliness of the discharge summary | 2.59 (1.02) | 3.34 (1.09) | <0.001 |
| Quality of the discharge summary | 3.04 (0.93) | 3.64 (0.99) | <0.001 |
Medical Error
The effect of the electronic discharge summary on perceived near misses and preventable adverse events is summarized in Table 3. Fewer outpatient physicians felt that 1 or more of their patients hospitalized in the preceding 6 months sustained a near miss due to suboptimal transfer of information after the implementation of the electronic discharge summary (65.7% vs. 52.9%, P = 0.008). Similarly, fewer outpatient physicians felt that 1 or more of their patients hospitalized in the preceding 6 months sustained a preventable adverse event due to suboptimal transfer of information after the implementation of the electronic discharge summary (40.7% vs. 30.2%, P = 0.02). In multivariate logistic regression analyses controlling for physician characteristics and their number of hospitalized patients in the previous 6 months, there was a statistically significant 40% reduction in the odds of a reported near miss (adjusted odds ratio [OR] = 0.60, P = 0.02). Although not quite statistically significant, there was a 33% reduction in the odds of a reported preventable adverse event (OR = 0.67, P = 0.08) after the implementation of the electronic discharge summary.
| Preelectronic Discharge Summary | Postelectronic Discharge Summary | P Value | |
|---|---|---|---|
| |||
| Near miss* | |||
| Number (%) reporting 1 | 142 (65.7) | 108 (52.9) | |
| Crude odds ratio | Ref. | 0.57 | 0.008 |
| Adjusted odds ratio | Ref. | 0.60 | 0.02 |
| Preventable adverse event | |||
| Number (%) reporting 1 | 88 (40.7) | 62 (30.2) | |
| Crude odds ratio | Ref. | 0.63 | 0.03 |
| Adjusted odds ratio | Ref. | 0.67 | 0.08 |
Discharge Summary Review
Discharge Summary Characteristics
One hundred and one discharge summaries before implementation of the electronic discharge summary were compared to 95 discharge summaries produced the following year. Characteristics of discharge summaries before and after the implementation of the electronic discharge summary are summarized in Table 4. A large number of discharge summaries (52.5%) were already being typed into the EMR in 2005, prior to the implementation of our final electronic discharge summary product. The number of dictated discharge summaries decreased from 47.5% to 10.5% after implementation of the final electronic discharge summary product (P < 0.001). Discharge summaries were similar in length before and after the implementation of the electronic discharge summary. A higher percentage of discharge summaries were completed within 3 days of discharge after implementation of the electronic discharge summary; however, this result was of borderline statistical significance (59.4% vs. 72.6%; P = 0.05). The type of service from which patients were discharged and the distribution of author types were similar after the implementation of the electronic discharge summary.
| Number (%) or MeanSD | P Value | ||
|---|---|---|---|
| Preelectronic Discharge Summary (n = 101) | Postelectronic Discharge Summary (n = 95) | ||
| Dictated, n (%) | 48 (47.5) | 10 (10.5) | <0.001 |
| Length in words, mean SD | 785 407 | 830 389 | 0.43 |
| Completed within 3 days, n (%) | 60 (59.4) | 69 (72.6) | 0.05 |
| Type of service, n (%) | 0.29 | ||
| Teaching service | 63 (62.4) | 66 (69.5) | |
| Nonteaching hospitalist service | 38 (37.6) | 29 (30.5) | |
| Author type, n (%) | 0.62 | ||
| Fourth year medical student | 13 (12.9) | 13 (13.7) | |
| Intern | 31 (30.7) | 37 (38.9) | |
| Resident | 19 (18.8) | 15 (15.8) | |
| Attending | 38 (37.6) | 30 (31.6) | |
Because a large percentage of discharge summaries were already being done electronically in 2005, we evaluated the timeliness of dictated discharge summaries compared to electronic discharge summaries across both periods combined (preimplementation and postimplementation of the electronic discharge summary). A higher percentage of electronic discharge summaries were completed within 3 days of discharge as compared to dictated discharge summaries (44.8% versus 74.1%; P < 0.001).
Discharge Summary Completeness Score
The presence or absence of discharge summary elements before and after the implementation of the electronic discharge summary is shown in Table 5. Several elements of the discharge summary were present more often after the implementation of the electronic discharge summary. Specific improvements included discussion of follow‐up issues (52.0% versus 75.8%; P = 0.001, = 0.78), pending test results (13.9% vs. 46.3%; P < 0.001, = 0.92), and information provided to the patient and/or family (85.1% vs. 95.8%; P = 0.01, = 0.91). Significant laboratory findings were present less often after implementation of the electronic discharge summary (66.0% versus 51.1%; P = 0.04, = 0.84). The Discharge Summary Completeness Score was higher after the implementation of the electronic discharge summary (mean 74.1 versus 80.3, P = 0.007). Dictated discharge summaries had a significantly lower Discharge Summary Completeness Score compared to discharge summaries done electronically (71.3 vs. 79.6, P = 0.002) across both periods combined.
| Number (%) of Content Items Present* | P Value | Brennan‐Prediger Kappa | ||
|---|---|---|---|---|
| Preelectronic Discharge Summary (n = 101) | Postelectronic Discharge Summary (n = 95) | |||
| ||||
| Dates of admission and discharge | 96 (95.0) | 94 (98.9) | 0.11 | 1.0 |
| Reason for hospitalization | 100 (99.0) | 94 (100) | 0.33 | 1.0 |
| Significant findings from history and exam | 78 (77.2) | 65 (68.4) | 0.16 | 0.26 |
| Significant laboratory findings | 64 (66.0) | 47 (51.1) | 0.04 | 0.84 |
| Significant radiological findings | 67 (75.3) | 71 (81.6) | 0.31 | 0.89 |
| Significant findings from other tests | 41 (63.1) | 40 (71.4) | 0.33 | 0.88 |
| List of procedures performed | 45 (81.8) | 35 (77.8) | 0.77 | 0.99 |
| Procedure report findings | 49 (80.3) | 43 (78.2) | 0.61 | 0.92 |
| Stress test report findings | 7 (100) | 3 (100) | N/A | 1.0 |
| Pathology report findings | 11 (39.3) | 3 (30.0) | 0.60 | 0.91 |
| Discharge diagnosis | 89 (88.1) | 86 (93.5) | 0.20 | 0.86 |
| Condition at discharge | 81 (81.0) | 80 (85.1) | 0.45 | 0.76 |
| Discharge medications | 88 (87.1) | 88 (93.6) | 0.13 | 0.79 |
| Follow‐up issues | 52 (52.0) | 72 (75.8) | 0.001 | 0.78 |
| Pending test results | 14 (13.9) | 44 (46.3) | <0.001 | 0.92 |
| Information provided to patient and/or family, as appropriate | 86 (85.1) | 91 (95.8) | 0.01 | 0.91 |
| Discharge Summary Completeness Score (percent present all applicable items) | 74.1 | 80.3 | 0.007 | |
Significantly more discharge summaries were rated as understandable or lucid after the implementation of the electronic discharge summary (41.6% vs. 59.0%; P = 0.02). In both periods combined, dictated discharge summaries were rated as understandable or lucid less often than electronic discharge summaries (34.5% vs. 56.5%; P < 0.001).
Discussion
Our study found that an electronic discharge summary was well accepted by inpatient physicians and significantly improved the quality and timeliness of discharge summaries. Prior studies have shown that the use of electronically entered discharge summaries improved the timeliness of discharge summaries.1416 However, the discharge summaries used in these studies required manual input of data into a computer system separate from the patient's medical record. To our knowledge, this is the first study to report the impact of discharge summaries generated from an EMR. Leveraging the EMR, we were able to automate the insertion of specific patient data elements, streamline delivery, and create an electronic reminder system to inpatient physicians for summaries not completed 24 hours after discharge.
Prior research has shown that the quality of discharges summaries is improved with the use of standardized content.10, 17 Using a standardized template for the electronic discharge summary, we likewise demonstrated improved quality of discharge summaries. Key discharge summary elements, specifically discussion of follow‐up issues, pending test results, and information provided to the patient and/or family, were present more reliably after the implementation of the electronic discharge summary. The importance of identifying pending test results is underscored by a recent study showing that many patients are discharged from hospitals with test results still pending, and that physicians are often unaware when results are abnormal.18 One discharge summary element, significant laboratory findings, was present less often after the implementation of the electronic discharge summary. Our template did not designate significant laboratory findings under a separate heading. Instead, we used a heading entitled Key Results (labs, imaging, pathology). Physicians completing the discharge summaries may have prioritized the report of imaging and pathology results in this section. A simple revision of our discharge summary template to include a separate heading for significant laboratory findings may result in improvement in this regard.
Timeliness of discharge summaries was improved in our study, but remained less than optimal. Although nearly three‐quarters of electronic discharge summaries were completed within 3 days of discharge, our ultimate goal is to have 100% of discharge summaries completed within 3 days. This is especially important for complicated patients requiring outpatient follow‐up soon after discharge. We are currently in the process of designing further modifications to the electronic discharge summary completion process. One modification that may be beneficial is the automation of additional patient specific data elements into the discharge summary. We also plan to link performance of medication reconciliation, completion of patient discharge instructions, and completion of the discharge summary into an integrated set of activities performed in the EMR prior to patient discharge.
We found that fewer outpatient physicians reported 1 or more of their patients having a preventable adverse event or near miss as a result of suboptimal transfer of information at discharge after the implementation of the electronic discharge summary. Although we did not measure preventable adverse events directly in our study, this is an important finding in light of the large number of patients who sustain preventable adverse events after hospital discharge1, 2 and prior research showing that the absence of discharge summaries at postdischarge follow‐up visits increased the risk for hospital readmission.19
We had wondered what effect the electronic discharge summary would have on the length and clarity of discharge summaries. A published commentary suggested that notes performed in EMRs were inordinately long and often difficult to read.20 We were pleased to discover that electronic discharge summaries were similar in length to previous discharge summaries and were rated higher with regard to clarity.
Our study has several limitations. First, many inpatient physicians began to use electronic discharge summaries prior to our creation of the final electronic discharge summary product. We had explicitly instructed physicians to continue to dictate discharge summaries in the first phase of our study. The fact that physicians quickly adopted the practice of completing discharge summaries electronically suggests that they preferred this method for completion and may help to explain the improvement in timeliness. A second limitation, as previously mentioned, is that our study did not measure adverse events directly. Instead, we asked outpatient physicians to estimate the number of their patients discharged in the last 6 months who had sustained a preventable adverse event or near miss related to suboptimal information transfer at discharge. We had limited space in the survey to define the meaning of a preventable adverse event; therefore, the description in the survey does not exactly match previous definitions.1, 2 Finally, the ordinal scale used to assess clarity of discharge summaries has not been previously validated.
In conclusion, the use of an electronic discharge summary significantly improved the quality and timeliness of discharge summaries. The discharge summary comprises a vital component of the information transfer between the inpatient and outpatient settings during the vulnerable period following hospital discharge. As hospitals expand their use of EMRs, they should take advantage of opportunities to leverage functionality to improve quality and timeliness of discharge summaries.
- ,,, et al.Adverse events among medical patient after hospital discharge.CMAJ.2004;170:345–349.
- ,,,,.The incidence and severity of adverse events affecting patients after discharge from the hospital.Ann Intern Med.2003;138:161–167.
- ,,.Dissemination of discharge summaries. Not reaching follow‐up physicians.Can Fam Physician.2002;48:737–742.
- ,,,.Effect of discharge summary availability during post‐discharge visits on hospital readmission.J Gen Intern Med.2002;17:186–192.
- ,,,.General practitioner‐hospital communications: a review of discharge summaries.J Qual Clin Practice.2001;21:104–108.
- ,,,,.Quality assessment of discharge letters in a French university hospital.Int J Health Care Qual Assur.1998;11:90–95.
- ,,,,,.Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care.JAMA.2007;297:831–841.
- ,,,,.Outpatient physicians' satisfaction with discharge summaries and perceived need for an electronic discharge summary.J Hosp Med.2006;1:317–320.
- Standard IM.6.10: Hospital accreditation standards.Oakbrook Terrace, IL:Joint Commission on Accreditation of Healthcare Organizations;2006:338–340.
- ,,,,.Assessing quality and efficiency of discharge summaries.Am J Med Qual.2005;20:337–343.
- ,,,,,.Are discharge summaries teachable? The effects of a discharge summary curriculum on the quality of discharge summaries in an internal medicine residency program.Acad Med.2006;81(10 Suppl):S5–S8.
- ,,, et al.Transition of care for hospitalized elderly patients–development of a discharge checklist for hospitalists.J Hosp Med.2006;1:354–360.
- ,.Coefficient kappa: some uses, misuses, and alternatives.Educ Psychol Meas.1981;41:687–699.
- ,,,.Dictated versus database‐generated discharge summaries: a randomized clinical trial.CMAJ.1999;160:319–326.
- ,,,.Evaluation of computer generated neonatal discharge summaries.Arch Dis Child.1991;66:433–436.
- ,,,,,.Evaluation of a computer‐generated discharge summary for patients with acute coronary syndromes.Br J Gen Pract.1998;48:1163–1164.
- ,,,.Standardized or narrative discharge summaries: Which do family physicians prefer?Can Fam Phys.1998;44:62–69.
- ,,, et al.Patient safety concerns arising from test results that return after hospital discharge.Ann Intern Med.2005;143:121–128.
- ,,,Effect of discharge summary availability during post‐discharge visits on hospital readmission.J Gen Intern Med.2002:17;186–192.
- .A piece of my mind. Copy‐and‐paste.JAMA.2006;295:2335–2336.
- ,,, et al.Adverse events among medical patient after hospital discharge.CMAJ.2004;170:345–349.
- ,,,,.The incidence and severity of adverse events affecting patients after discharge from the hospital.Ann Intern Med.2003;138:161–167.
- ,,.Dissemination of discharge summaries. Not reaching follow‐up physicians.Can Fam Physician.2002;48:737–742.
- ,,,.Effect of discharge summary availability during post‐discharge visits on hospital readmission.J Gen Intern Med.2002;17:186–192.
- ,,,.General practitioner‐hospital communications: a review of discharge summaries.J Qual Clin Practice.2001;21:104–108.
- ,,,,.Quality assessment of discharge letters in a French university hospital.Int J Health Care Qual Assur.1998;11:90–95.
- ,,,,,.Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care.JAMA.2007;297:831–841.
- ,,,,.Outpatient physicians' satisfaction with discharge summaries and perceived need for an electronic discharge summary.J Hosp Med.2006;1:317–320.
- Standard IM.6.10: Hospital accreditation standards.Oakbrook Terrace, IL:Joint Commission on Accreditation of Healthcare Organizations;2006:338–340.
- ,,,,.Assessing quality and efficiency of discharge summaries.Am J Med Qual.2005;20:337–343.
- ,,,,,.Are discharge summaries teachable? The effects of a discharge summary curriculum on the quality of discharge summaries in an internal medicine residency program.Acad Med.2006;81(10 Suppl):S5–S8.
- ,,, et al.Transition of care for hospitalized elderly patients–development of a discharge checklist for hospitalists.J Hosp Med.2006;1:354–360.
- ,.Coefficient kappa: some uses, misuses, and alternatives.Educ Psychol Meas.1981;41:687–699.
- ,,,.Dictated versus database‐generated discharge summaries: a randomized clinical trial.CMAJ.1999;160:319–326.
- ,,,.Evaluation of computer generated neonatal discharge summaries.Arch Dis Child.1991;66:433–436.
- ,,,,,.Evaluation of a computer‐generated discharge summary for patients with acute coronary syndromes.Br J Gen Pract.1998;48:1163–1164.
- ,,,.Standardized or narrative discharge summaries: Which do family physicians prefer?Can Fam Phys.1998;44:62–69.
- ,,, et al.Patient safety concerns arising from test results that return after hospital discharge.Ann Intern Med.2005;143:121–128.
- ,,,Effect of discharge summary availability during post‐discharge visits on hospital readmission.J Gen Intern Med.2002:17;186–192.
- .A piece of my mind. Copy‐and‐paste.JAMA.2006;295:2335–2336.
Copyright © 2009 Society of Hospital Medicine
Thrombolytics for VTE: Current Practice
More than a decade ago, we surveyed a group of practicing pulmonologists to determine their attitudes regarding the use of thrombolytic therapy in various settings of acute venous thromboembolism (VTE).1 Since that time, the literature regarding the treatment of acute VTE has grown dramatically.214 However, despite the available evidence, there remains considerable controversy regarding the appropriate setting for thrombolysis in acute pulmonary embolism (PE) or deep‐vein thrombosis (DVT). We therefore sought to better describe the current patterns of thrombolytic use among practicing pulmonologists and to determine if these patterns have changed over the last decade.
Methods
Five‐hundred and ten physicians in the southeastern US were selected from the American Thoracic Society (ATS) membership roster and were e‐mailed a link to an online questionnaire. The roster was searched for physicians who described their subspecialty as pulmonary disease or pulmonary and critical care.
Participants were asked background information and questions regarding hypothetical clinical scenarios. All participants were offered a $50 stipend, and to further improve the response rate, 2 reminder e‐mail messages were sent 30 days and 45 days after the initial request.
Baseline findings of the survey were summarized using descriptive statistics. Differences among participants and their responses were determined by Fisher's exact test. Analyses were performed using SAS E‐Guide Version 3.0 for Windows (SAS Institute, Cary, NC) with 2‐sided P values at the standard 0.05 level used to determine statistical significance.
Results
Baseline Characteristics
Eighty‐one physicians completed the questionnaire; their baseline characteristics are shown in Table 1. During the previous 2 years, all physicians surveyed had treated at least 1 patient with acute PE and all but 1 had treated at least 1 patient with DVT. Also, 68 respondents reported that they had used thrombolytic therapy in at least 1 case of PE in the past 2 years.
| |
| Age, mean (years) | 45.6 |
| Training completed, n (%) | |
| 1980‐1989 | 28 (34.5) |
| 1990‐1999 | 25 (31.0) |
| 2000‐2007 | 28 (34.5) |
| Practice type n (%) | |
| Academic | 35 (43) |
| Private practice | 37 (46) |
| Private practice with academic appointment | 6 (7) |
| Other | 3 (4) |
| Practice setting, n (%) | |
| Predominantly outpatient | 8 (10) |
| Predominantly inpatient | 29 (36) |
| Equal inpatient and outpatient | 44 (54) |
| Hospital size (beds), n (%) | |
| 50 | 1 (1) |
| 50‐100 | 1 (1) |
| 100‐300 | 20 (25) |
| 300‐500 | 22 (27) |
| >500 | 37 (46) |
| Number of patients treated with PE in the past 2 years, n (%) | |
| 0 | 0 (0) |
| 1‐5 | 3 (4) |
| 6‐10 | 14 (17) |
| 11‐15 | 12 (15) |
| 16‐20 | 17 (21) |
| >20 | 35 (43) |
| Number of patients treated with DVT in the past 2 years, n (%) | |
| 0 | 1 (1) |
| 1‐5 | 3 (4) |
| 6‐10 | 7 (9) |
| 11‐15 | 16 (20) |
| 16‐20 | 11 (14) |
| >20 | 43 (53) |
| Number of patients with PE treated with thrombolysis, n (%) | |
| 0 | 13 (16) |
| 1‐5 | 53 (65) |
| 6‐10 | 11 (14) |
| 11‐15 | 1 (1) |
| 16‐20 | 2 (2) |
| >20 | 1 (1) |
Use of Thrombolytic Therapy in Various Scenarios
The responses for the 8 clinical scenarios are shown in Table 2. Approximately equal numbers of academic and private practice physicians completed the questionnaire, and comparison between these groups showed no significant differences in decision‐making for each of the case scenarios. Less experienced physicians (>10 cases treated versus 10 cases treated) were more likely to consider thrombolytic therapy in a patient with a smaller PE but with poor cardiopulmonary reserve (P = 0.001), and with proximal symptomatic DVT of any size present less than 7 days (P = 0.047).
| Scenario | Current Study (%) | Previous Study1 (%) | P |
|---|---|---|---|
| |||
| Massive PE with hypotension | 80 (99) | 56 (100) | NS |
| Large PE with hypoxemia | 67 (83) | 41 (73) | NS |
| PE with RV strain or failure | 50 (62) | 31 (55) | NS |
| Large PE without hypotension, hypoxemia, or RV strain | 9 (11) | 6 (11) | NS |
| Smaller PE in a patient with poor cardiopulmonary reserve | 11 (14) | ||
| Massive symptomatic DVT, 7 days | 41 (51) | 33 (59) | NS |
| Massive symptomatic DVT, >7 days | 14 (17) | ||
| Proximal DVT, any size, 7 days | 6 (7) | 7 (13) | NS |
Use of Thrombolytic Therapy When Contraindications Exist
The vast majority of respondents reported that they would consider giving thrombolytic therapy to a patient with massive PE and hypotension requiring vasopressor therapy despite having a traditional contraindication (relative or absolute) to thrombolysis (Table 3). Most respondents would consider giving thrombolytic therapy to postoperative orthopedic, abdominal, or thoracic surgery patients if they were more than 2 weeks postoperation, and very few would give thrombolytic therapy to patients who were less than 2 days postoperation. Many respondents would also consider giving thrombolytic therapy to a patient with a massive PE and with a history of major gastrointestinal (GI) bleeding (requiring blood transfusion) if the bleed was more than 4 weeks prior to the embolism (Figure 1).

| Condition | Number of Physicians (%) |
|---|---|
| |
| Age >75 years | 58 (72) |
| Guaiac + stool | 54 (67) |
| CPR in past 10 days | 39 (48) |
| History of ischemic stroke | 37 (46) |
| Recent venipuncture of a noncompressible vessel | 33 (41) |
| History of ICH | 6 (7) |
| Brain tumor | 6 (7) |
| Would never use thrombolytics in these scenarios | 7 (9) |
Discussion
Given the paucity of data from randomized controlled trials, there remains considerable controversy regarding the indications for thrombolytic therapy. It may be difficult to define those patients in whom the benefit of a rapid reduction in clot burden outweighs the increased hemorrhagic risk. The case for thrombolysis is the strongest in patients with massive PE complicated by hypotension, in whom the mortality rate may be 30%.15 Our survey confirms that the vast majority of practicing pulmonologists would strongly consider systemic thrombolysis in this clinical setting, which is in accordance with current guidelines and with our previous survey results.1, 5, 10, 12
No clinical trial has specifically evaluated thrombolytic therapy in patients with large PE and hypoxemia but without hypotension, and it is interesting that so many physicians would consider thrombolytic therapy in this scenario. As right heart failure is the cause of death in PE, the absence of significant hypotension would imply less cardiovascular risk and thrombolytic use would seemingly be less justifiable from a physiologic point of view. It may be that further study and education is warranted in this area.
Many patients who present with acute, life‐threatening PE have contraindications or relative contraindications to systemic thrombolysis. Our study suggests that most practicing pulmonologists would consider giving thrombolytic therapy in some of these situations, such as if the patient was more than 2 weeks postoperative from major thoracic or abdominal surgery (or even a few days following orthopedic surgery), or in the setting of advanced age or guaiac positive stools. Physicians were appropriately very reluctant to use thrombolytic therapy in the setting of a brain tumor or prior intracranial hemorrhage. These scenarios emphasize the vagaries of the current guidelines and real‐world complexities of considering thrombolytic therapy in clinical practice, in which the risks and benefits must be weighed on a case‐by‐case basis.
One major difference between our current and past findings is the general experience with thrombolytic therapy in acute PE. In our first study, only 54% of physicians queried had employed systemic thrombolysis for acute PE. Our current findings were that 84% of physicians had used thrombolysis for acute PE within the last 2 years, perhaps suggesting a greater comfort with this therapy.
Response bias is a major limitation of our study. We sought to keep questions short and clear, and offered a small stipend to improve the return rate. Despite these measures, only 81 of 510 questionnaires were completed. We selected our list of participants from the ATS roster and by geographic location. As suggested by our findings, the results may have been different had we focused solely on VTE experts or those treating large numbers of VTE patients. One strength of this study is that our sample had approximately even numbers of academic and private practice physicians, and that we could compare current results with our prior findings.
In conclusion, practicing pulmonologists generally agreed that in the absence of contraindications, thrombolytic therapy should be considered in patients with massive PE and hypotension, which is in accordance with current guidelines. Furthermore, a majority would still consider thrombolytic therapy in this scenario even if certain contraindications were present. Although there is less agreement in other scenarios, a majority of physicians would consider using thrombolytics in patients with PE and severe hypoxemia or right ventricular (RV) dysfunction. Despite the evolving data and guidelines, our findings are similar to prior survey results, with the notable exception that more physicians reported thrombolytic therapy use in acute PE in the current study. This emphasizes the need for further physician education and future randomized clinical trials to delineate and unify therapeutic strategies in cases of VTE.
- ,,.Thrombolytic therapy for venous thromboembolism. Utilization by practicing pulmonologists.Arch Intern Med.1994;154:1601–1604.
- ,,, et al.Streptokinase vs alteplase in massive pulmonary embolism. A randomized trial assessing right heart haemodynamics and pulmonary vascular obstruction.Eur Heart J.1997;18:1141–1148.
- ,,, et al.Comparative efficacy of a two‐hour regimen of streptokinase versus alteplase in acute massive pulmonary embolism: immediate clinical and hemodynamic outcome and one‐year follow‐up.J Am Coll Cardiol.1998;31:1057–1063.
- ,,.Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER).Lancet.1999;353:1386–1389.
- ,,, et al.Guidelines on diagnosis and management of acute pulmonary embolism. Task Force on Pulmonary Embolism, European Society of Cardiology.Eur Heart J.2000;21:1301–1336.
- ,,,.Long‐term benefit of thrombolytic therapy in patients with pulmonary embolism.Vasc Med.2000;5:91–95.
- ,,, et al.Thrombolytic therapy of pulmonary embolism: a meta‐analysis.J Am Coll Cardiol.2002;40:1660–1667.
- ,,, et al.Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism.N Engl J Med.2002;347:1143–1150.
- ,,.Thrombolysis vs heparin in the treatment of pulmonary embolism: a clinical outcome‐based meta‐analysis.Arch Intern Med.2002;162:2537–2541.
- ,,, et al.British Thoracic Society guidelines for the management of suspected acute pulmonary embolism.Thorax.2003;58:470–483.
- ,.Thrombolysis for acute deep vein thrombosis.Cochrane Database Syst Rev.2004;CD002783.
- ,,, et al.Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.Chest.2004;126:401S–428S.
- ,,, et al.Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: a meta‐analysis of the randomized controlled trials.Circulation.2004;110:744–749.
- ,,, et al.Thrombolytic therapy for pulmonary embolism.Cochrane Database Syst Rev.2006;CD004437.
- ,,Thrombolytic therapy for pulmonary embolism: is it effective? Is it safe? When is it indicated?Arch Intern Med.1997;157:2550–2556.
More than a decade ago, we surveyed a group of practicing pulmonologists to determine their attitudes regarding the use of thrombolytic therapy in various settings of acute venous thromboembolism (VTE).1 Since that time, the literature regarding the treatment of acute VTE has grown dramatically.214 However, despite the available evidence, there remains considerable controversy regarding the appropriate setting for thrombolysis in acute pulmonary embolism (PE) or deep‐vein thrombosis (DVT). We therefore sought to better describe the current patterns of thrombolytic use among practicing pulmonologists and to determine if these patterns have changed over the last decade.
Methods
Five‐hundred and ten physicians in the southeastern US were selected from the American Thoracic Society (ATS) membership roster and were e‐mailed a link to an online questionnaire. The roster was searched for physicians who described their subspecialty as pulmonary disease or pulmonary and critical care.
Participants were asked background information and questions regarding hypothetical clinical scenarios. All participants were offered a $50 stipend, and to further improve the response rate, 2 reminder e‐mail messages were sent 30 days and 45 days after the initial request.
Baseline findings of the survey were summarized using descriptive statistics. Differences among participants and their responses were determined by Fisher's exact test. Analyses were performed using SAS E‐Guide Version 3.0 for Windows (SAS Institute, Cary, NC) with 2‐sided P values at the standard 0.05 level used to determine statistical significance.
Results
Baseline Characteristics
Eighty‐one physicians completed the questionnaire; their baseline characteristics are shown in Table 1. During the previous 2 years, all physicians surveyed had treated at least 1 patient with acute PE and all but 1 had treated at least 1 patient with DVT. Also, 68 respondents reported that they had used thrombolytic therapy in at least 1 case of PE in the past 2 years.
| |
| Age, mean (years) | 45.6 |
| Training completed, n (%) | |
| 1980‐1989 | 28 (34.5) |
| 1990‐1999 | 25 (31.0) |
| 2000‐2007 | 28 (34.5) |
| Practice type n (%) | |
| Academic | 35 (43) |
| Private practice | 37 (46) |
| Private practice with academic appointment | 6 (7) |
| Other | 3 (4) |
| Practice setting, n (%) | |
| Predominantly outpatient | 8 (10) |
| Predominantly inpatient | 29 (36) |
| Equal inpatient and outpatient | 44 (54) |
| Hospital size (beds), n (%) | |
| 50 | 1 (1) |
| 50‐100 | 1 (1) |
| 100‐300 | 20 (25) |
| 300‐500 | 22 (27) |
| >500 | 37 (46) |
| Number of patients treated with PE in the past 2 years, n (%) | |
| 0 | 0 (0) |
| 1‐5 | 3 (4) |
| 6‐10 | 14 (17) |
| 11‐15 | 12 (15) |
| 16‐20 | 17 (21) |
| >20 | 35 (43) |
| Number of patients treated with DVT in the past 2 years, n (%) | |
| 0 | 1 (1) |
| 1‐5 | 3 (4) |
| 6‐10 | 7 (9) |
| 11‐15 | 16 (20) |
| 16‐20 | 11 (14) |
| >20 | 43 (53) |
| Number of patients with PE treated with thrombolysis, n (%) | |
| 0 | 13 (16) |
| 1‐5 | 53 (65) |
| 6‐10 | 11 (14) |
| 11‐15 | 1 (1) |
| 16‐20 | 2 (2) |
| >20 | 1 (1) |
Use of Thrombolytic Therapy in Various Scenarios
The responses for the 8 clinical scenarios are shown in Table 2. Approximately equal numbers of academic and private practice physicians completed the questionnaire, and comparison between these groups showed no significant differences in decision‐making for each of the case scenarios. Less experienced physicians (>10 cases treated versus 10 cases treated) were more likely to consider thrombolytic therapy in a patient with a smaller PE but with poor cardiopulmonary reserve (P = 0.001), and with proximal symptomatic DVT of any size present less than 7 days (P = 0.047).
| Scenario | Current Study (%) | Previous Study1 (%) | P |
|---|---|---|---|
| |||
| Massive PE with hypotension | 80 (99) | 56 (100) | NS |
| Large PE with hypoxemia | 67 (83) | 41 (73) | NS |
| PE with RV strain or failure | 50 (62) | 31 (55) | NS |
| Large PE without hypotension, hypoxemia, or RV strain | 9 (11) | 6 (11) | NS |
| Smaller PE in a patient with poor cardiopulmonary reserve | 11 (14) | ||
| Massive symptomatic DVT, 7 days | 41 (51) | 33 (59) | NS |
| Massive symptomatic DVT, >7 days | 14 (17) | ||
| Proximal DVT, any size, 7 days | 6 (7) | 7 (13) | NS |
Use of Thrombolytic Therapy When Contraindications Exist
The vast majority of respondents reported that they would consider giving thrombolytic therapy to a patient with massive PE and hypotension requiring vasopressor therapy despite having a traditional contraindication (relative or absolute) to thrombolysis (Table 3). Most respondents would consider giving thrombolytic therapy to postoperative orthopedic, abdominal, or thoracic surgery patients if they were more than 2 weeks postoperation, and very few would give thrombolytic therapy to patients who were less than 2 days postoperation. Many respondents would also consider giving thrombolytic therapy to a patient with a massive PE and with a history of major gastrointestinal (GI) bleeding (requiring blood transfusion) if the bleed was more than 4 weeks prior to the embolism (Figure 1).
| Condition | Number of Physicians (%) |
|---|---|
| |
| Age >75 years | 58 (72) |
| Guaiac + stool | 54 (67) |
| CPR in past 10 days | 39 (48) |
| History of ischemic stroke | 37 (46) |
| Recent venipuncture of a noncompressible vessel | 33 (41) |
| History of ICH | 6 (7) |
| Brain tumor | 6 (7) |
| Would never use thrombolytics in these scenarios | 7 (9) |
Discussion
Given the paucity of data from randomized controlled trials, there remains considerable controversy regarding the indications for thrombolytic therapy. It may be difficult to define those patients in whom the benefit of a rapid reduction in clot burden outweighs the increased hemorrhagic risk. The case for thrombolysis is the strongest in patients with massive PE complicated by hypotension, in whom the mortality rate may be 30%.15 Our survey confirms that the vast majority of practicing pulmonologists would strongly consider systemic thrombolysis in this clinical setting, which is in accordance with current guidelines and with our previous survey results.1, 5, 10, 12
No clinical trial has specifically evaluated thrombolytic therapy in patients with large PE and hypoxemia but without hypotension, and it is interesting that so many physicians would consider thrombolytic therapy in this scenario. As right heart failure is the cause of death in PE, the absence of significant hypotension would imply less cardiovascular risk and thrombolytic use would seemingly be less justifiable from a physiologic point of view. It may be that further study and education is warranted in this area.
Many patients who present with acute, life‐threatening PE have contraindications or relative contraindications to systemic thrombolysis. Our study suggests that most practicing pulmonologists would consider giving thrombolytic therapy in some of these situations, such as if the patient was more than 2 weeks postoperative from major thoracic or abdominal surgery (or even a few days following orthopedic surgery), or in the setting of advanced age or guaiac positive stools. Physicians were appropriately very reluctant to use thrombolytic therapy in the setting of a brain tumor or prior intracranial hemorrhage. These scenarios emphasize the vagaries of the current guidelines and real‐world complexities of considering thrombolytic therapy in clinical practice, in which the risks and benefits must be weighed on a case‐by‐case basis.
One major difference between our current and past findings is the general experience with thrombolytic therapy in acute PE. In our first study, only 54% of physicians queried had employed systemic thrombolysis for acute PE. Our current findings were that 84% of physicians had used thrombolysis for acute PE within the last 2 years, perhaps suggesting a greater comfort with this therapy.
Response bias is a major limitation of our study. We sought to keep questions short and clear, and offered a small stipend to improve the return rate. Despite these measures, only 81 of 510 questionnaires were completed. We selected our list of participants from the ATS roster and by geographic location. As suggested by our findings, the results may have been different had we focused solely on VTE experts or those treating large numbers of VTE patients. One strength of this study is that our sample had approximately even numbers of academic and private practice physicians, and that we could compare current results with our prior findings.
In conclusion, practicing pulmonologists generally agreed that in the absence of contraindications, thrombolytic therapy should be considered in patients with massive PE and hypotension, which is in accordance with current guidelines. Furthermore, a majority would still consider thrombolytic therapy in this scenario even if certain contraindications were present. Although there is less agreement in other scenarios, a majority of physicians would consider using thrombolytics in patients with PE and severe hypoxemia or right ventricular (RV) dysfunction. Despite the evolving data and guidelines, our findings are similar to prior survey results, with the notable exception that more physicians reported thrombolytic therapy use in acute PE in the current study. This emphasizes the need for further physician education and future randomized clinical trials to delineate and unify therapeutic strategies in cases of VTE.
More than a decade ago, we surveyed a group of practicing pulmonologists to determine their attitudes regarding the use of thrombolytic therapy in various settings of acute venous thromboembolism (VTE).1 Since that time, the literature regarding the treatment of acute VTE has grown dramatically.214 However, despite the available evidence, there remains considerable controversy regarding the appropriate setting for thrombolysis in acute pulmonary embolism (PE) or deep‐vein thrombosis (DVT). We therefore sought to better describe the current patterns of thrombolytic use among practicing pulmonologists and to determine if these patterns have changed over the last decade.
Methods
Five‐hundred and ten physicians in the southeastern US were selected from the American Thoracic Society (ATS) membership roster and were e‐mailed a link to an online questionnaire. The roster was searched for physicians who described their subspecialty as pulmonary disease or pulmonary and critical care.
Participants were asked background information and questions regarding hypothetical clinical scenarios. All participants were offered a $50 stipend, and to further improve the response rate, 2 reminder e‐mail messages were sent 30 days and 45 days after the initial request.
Baseline findings of the survey were summarized using descriptive statistics. Differences among participants and their responses were determined by Fisher's exact test. Analyses were performed using SAS E‐Guide Version 3.0 for Windows (SAS Institute, Cary, NC) with 2‐sided P values at the standard 0.05 level used to determine statistical significance.
Results
Baseline Characteristics
Eighty‐one physicians completed the questionnaire; their baseline characteristics are shown in Table 1. During the previous 2 years, all physicians surveyed had treated at least 1 patient with acute PE and all but 1 had treated at least 1 patient with DVT. Also, 68 respondents reported that they had used thrombolytic therapy in at least 1 case of PE in the past 2 years.
| |
| Age, mean (years) | 45.6 |
| Training completed, n (%) | |
| 1980‐1989 | 28 (34.5) |
| 1990‐1999 | 25 (31.0) |
| 2000‐2007 | 28 (34.5) |
| Practice type n (%) | |
| Academic | 35 (43) |
| Private practice | 37 (46) |
| Private practice with academic appointment | 6 (7) |
| Other | 3 (4) |
| Practice setting, n (%) | |
| Predominantly outpatient | 8 (10) |
| Predominantly inpatient | 29 (36) |
| Equal inpatient and outpatient | 44 (54) |
| Hospital size (beds), n (%) | |
| 50 | 1 (1) |
| 50‐100 | 1 (1) |
| 100‐300 | 20 (25) |
| 300‐500 | 22 (27) |
| >500 | 37 (46) |
| Number of patients treated with PE in the past 2 years, n (%) | |
| 0 | 0 (0) |
| 1‐5 | 3 (4) |
| 6‐10 | 14 (17) |
| 11‐15 | 12 (15) |
| 16‐20 | 17 (21) |
| >20 | 35 (43) |
| Number of patients treated with DVT in the past 2 years, n (%) | |
| 0 | 1 (1) |
| 1‐5 | 3 (4) |
| 6‐10 | 7 (9) |
| 11‐15 | 16 (20) |
| 16‐20 | 11 (14) |
| >20 | 43 (53) |
| Number of patients with PE treated with thrombolysis, n (%) | |
| 0 | 13 (16) |
| 1‐5 | 53 (65) |
| 6‐10 | 11 (14) |
| 11‐15 | 1 (1) |
| 16‐20 | 2 (2) |
| >20 | 1 (1) |
Use of Thrombolytic Therapy in Various Scenarios
The responses for the 8 clinical scenarios are shown in Table 2. Approximately equal numbers of academic and private practice physicians completed the questionnaire, and comparison between these groups showed no significant differences in decision‐making for each of the case scenarios. Less experienced physicians (>10 cases treated versus 10 cases treated) were more likely to consider thrombolytic therapy in a patient with a smaller PE but with poor cardiopulmonary reserve (P = 0.001), and with proximal symptomatic DVT of any size present less than 7 days (P = 0.047).
| Scenario | Current Study (%) | Previous Study1 (%) | P |
|---|---|---|---|
| |||
| Massive PE with hypotension | 80 (99) | 56 (100) | NS |
| Large PE with hypoxemia | 67 (83) | 41 (73) | NS |
| PE with RV strain or failure | 50 (62) | 31 (55) | NS |
| Large PE without hypotension, hypoxemia, or RV strain | 9 (11) | 6 (11) | NS |
| Smaller PE in a patient with poor cardiopulmonary reserve | 11 (14) | ||
| Massive symptomatic DVT, 7 days | 41 (51) | 33 (59) | NS |
| Massive symptomatic DVT, >7 days | 14 (17) | ||
| Proximal DVT, any size, 7 days | 6 (7) | 7 (13) | NS |
Use of Thrombolytic Therapy When Contraindications Exist
The vast majority of respondents reported that they would consider giving thrombolytic therapy to a patient with massive PE and hypotension requiring vasopressor therapy despite having a traditional contraindication (relative or absolute) to thrombolysis (Table 3). Most respondents would consider giving thrombolytic therapy to postoperative orthopedic, abdominal, or thoracic surgery patients if they were more than 2 weeks postoperation, and very few would give thrombolytic therapy to patients who were less than 2 days postoperation. Many respondents would also consider giving thrombolytic therapy to a patient with a massive PE and with a history of major gastrointestinal (GI) bleeding (requiring blood transfusion) if the bleed was more than 4 weeks prior to the embolism (Figure 1).
| Condition | Number of Physicians (%) |
|---|---|
| |
| Age >75 years | 58 (72) |
| Guaiac + stool | 54 (67) |
| CPR in past 10 days | 39 (48) |
| History of ischemic stroke | 37 (46) |
| Recent venipuncture of a noncompressible vessel | 33 (41) |
| History of ICH | 6 (7) |
| Brain tumor | 6 (7) |
| Would never use thrombolytics in these scenarios | 7 (9) |
Discussion
Given the paucity of data from randomized controlled trials, there remains considerable controversy regarding the indications for thrombolytic therapy. It may be difficult to define those patients in whom the benefit of a rapid reduction in clot burden outweighs the increased hemorrhagic risk. The case for thrombolysis is the strongest in patients with massive PE complicated by hypotension, in whom the mortality rate may be 30%.15 Our survey confirms that the vast majority of practicing pulmonologists would strongly consider systemic thrombolysis in this clinical setting, which is in accordance with current guidelines and with our previous survey results.1, 5, 10, 12
No clinical trial has specifically evaluated thrombolytic therapy in patients with large PE and hypoxemia but without hypotension, and it is interesting that so many physicians would consider thrombolytic therapy in this scenario. As right heart failure is the cause of death in PE, the absence of significant hypotension would imply less cardiovascular risk and thrombolytic use would seemingly be less justifiable from a physiologic point of view. It may be that further study and education is warranted in this area.
Many patients who present with acute, life‐threatening PE have contraindications or relative contraindications to systemic thrombolysis. Our study suggests that most practicing pulmonologists would consider giving thrombolytic therapy in some of these situations, such as if the patient was more than 2 weeks postoperative from major thoracic or abdominal surgery (or even a few days following orthopedic surgery), or in the setting of advanced age or guaiac positive stools. Physicians were appropriately very reluctant to use thrombolytic therapy in the setting of a brain tumor or prior intracranial hemorrhage. These scenarios emphasize the vagaries of the current guidelines and real‐world complexities of considering thrombolytic therapy in clinical practice, in which the risks and benefits must be weighed on a case‐by‐case basis.
One major difference between our current and past findings is the general experience with thrombolytic therapy in acute PE. In our first study, only 54% of physicians queried had employed systemic thrombolysis for acute PE. Our current findings were that 84% of physicians had used thrombolysis for acute PE within the last 2 years, perhaps suggesting a greater comfort with this therapy.
Response bias is a major limitation of our study. We sought to keep questions short and clear, and offered a small stipend to improve the return rate. Despite these measures, only 81 of 510 questionnaires were completed. We selected our list of participants from the ATS roster and by geographic location. As suggested by our findings, the results may have been different had we focused solely on VTE experts or those treating large numbers of VTE patients. One strength of this study is that our sample had approximately even numbers of academic and private practice physicians, and that we could compare current results with our prior findings.
In conclusion, practicing pulmonologists generally agreed that in the absence of contraindications, thrombolytic therapy should be considered in patients with massive PE and hypotension, which is in accordance with current guidelines. Furthermore, a majority would still consider thrombolytic therapy in this scenario even if certain contraindications were present. Although there is less agreement in other scenarios, a majority of physicians would consider using thrombolytics in patients with PE and severe hypoxemia or right ventricular (RV) dysfunction. Despite the evolving data and guidelines, our findings are similar to prior survey results, with the notable exception that more physicians reported thrombolytic therapy use in acute PE in the current study. This emphasizes the need for further physician education and future randomized clinical trials to delineate and unify therapeutic strategies in cases of VTE.
- ,,.Thrombolytic therapy for venous thromboembolism. Utilization by practicing pulmonologists.Arch Intern Med.1994;154:1601–1604.
- ,,, et al.Streptokinase vs alteplase in massive pulmonary embolism. A randomized trial assessing right heart haemodynamics and pulmonary vascular obstruction.Eur Heart J.1997;18:1141–1148.
- ,,, et al.Comparative efficacy of a two‐hour regimen of streptokinase versus alteplase in acute massive pulmonary embolism: immediate clinical and hemodynamic outcome and one‐year follow‐up.J Am Coll Cardiol.1998;31:1057–1063.
- ,,.Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER).Lancet.1999;353:1386–1389.
- ,,, et al.Guidelines on diagnosis and management of acute pulmonary embolism. Task Force on Pulmonary Embolism, European Society of Cardiology.Eur Heart J.2000;21:1301–1336.
- ,,,.Long‐term benefit of thrombolytic therapy in patients with pulmonary embolism.Vasc Med.2000;5:91–95.
- ,,, et al.Thrombolytic therapy of pulmonary embolism: a meta‐analysis.J Am Coll Cardiol.2002;40:1660–1667.
- ,,, et al.Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism.N Engl J Med.2002;347:1143–1150.
- ,,.Thrombolysis vs heparin in the treatment of pulmonary embolism: a clinical outcome‐based meta‐analysis.Arch Intern Med.2002;162:2537–2541.
- ,,, et al.British Thoracic Society guidelines for the management of suspected acute pulmonary embolism.Thorax.2003;58:470–483.
- ,.Thrombolysis for acute deep vein thrombosis.Cochrane Database Syst Rev.2004;CD002783.
- ,,, et al.Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.Chest.2004;126:401S–428S.
- ,,, et al.Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: a meta‐analysis of the randomized controlled trials.Circulation.2004;110:744–749.
- ,,, et al.Thrombolytic therapy for pulmonary embolism.Cochrane Database Syst Rev.2006;CD004437.
- ,,Thrombolytic therapy for pulmonary embolism: is it effective? Is it safe? When is it indicated?Arch Intern Med.1997;157:2550–2556.
- ,,.Thrombolytic therapy for venous thromboembolism. Utilization by practicing pulmonologists.Arch Intern Med.1994;154:1601–1604.
- ,,, et al.Streptokinase vs alteplase in massive pulmonary embolism. A randomized trial assessing right heart haemodynamics and pulmonary vascular obstruction.Eur Heart J.1997;18:1141–1148.
- ,,, et al.Comparative efficacy of a two‐hour regimen of streptokinase versus alteplase in acute massive pulmonary embolism: immediate clinical and hemodynamic outcome and one‐year follow‐up.J Am Coll Cardiol.1998;31:1057–1063.
- ,,.Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER).Lancet.1999;353:1386–1389.
- ,,, et al.Guidelines on diagnosis and management of acute pulmonary embolism. Task Force on Pulmonary Embolism, European Society of Cardiology.Eur Heart J.2000;21:1301–1336.
- ,,,.Long‐term benefit of thrombolytic therapy in patients with pulmonary embolism.Vasc Med.2000;5:91–95.
- ,,, et al.Thrombolytic therapy of pulmonary embolism: a meta‐analysis.J Am Coll Cardiol.2002;40:1660–1667.
- ,,, et al.Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism.N Engl J Med.2002;347:1143–1150.
- ,,.Thrombolysis vs heparin in the treatment of pulmonary embolism: a clinical outcome‐based meta‐analysis.Arch Intern Med.2002;162:2537–2541.
- ,,, et al.British Thoracic Society guidelines for the management of suspected acute pulmonary embolism.Thorax.2003;58:470–483.
- ,.Thrombolysis for acute deep vein thrombosis.Cochrane Database Syst Rev.2004;CD002783.
- ,,, et al.Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.Chest.2004;126:401S–428S.
- ,,, et al.Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: a meta‐analysis of the randomized controlled trials.Circulation.2004;110:744–749.
- ,,, et al.Thrombolytic therapy for pulmonary embolism.Cochrane Database Syst Rev.2006;CD004437.
- ,,Thrombolytic therapy for pulmonary embolism: is it effective? Is it safe? When is it indicated?Arch Intern Med.1997;157:2550–2556.
A “Routine” Electrocardiogram (ECG)
Patient Presentation
A 52‐year‐old woman with cirrhosis presented with a large‐volume upper gastrointestinal (GI) bleed. After receiving massive volume and blood product resuscitation (18 U packed red blood cells [PRBCs], 17 U fresh frozen plasma [FFP], 2 U cryoprecipitate, 1 U platelets, and 14 L normal saline), a routine admission electrocardiogram (ECG) was obtained (Figure 1).
Discussion
The ECG shows normal sinus rhythm with a prolonged QT interval (specifically due to a long ST segment). The differential diagnosis of this pattern of QT prolongation includes hypocalcemia, long QT syndrome (variant 3), and hypothermia. Massive transfusion can result in the chelation of calcium by citrate resulting in hypocalcemia. This patient's serum calcium was 7 mg/dL (8.9‐10.2) (ionized 0.21 mmol/L; 1.18‐1.38), her pH was 7.03. There were no overt signs or symptoms of hypocalcemia (paresthesias, twitching, tetany, or seizures.) and with aggressive replacement the calcium and the ECG normalized. Transfusions are an uncommon cause of hypocalcemia, with hypomagnesemia or hypermagnesemia, acute pancreatitis, rhabdomyolysis, tumor lysis syndrome, renal failure, vitamin D deficiency, pseudoparathyroidism and hypoparathyroidism being the more common disorders caused.1, 2 This patient's magnesium was normal on admission (2.1 mg/dL) and she did not have any of the other conditions mentioned. It is possible that hemodilution may also have played a role.
Genetic long QT syndrome (variant 3) can manifest with a similar ECG pattern, although the T‐wave is sometimes peaked or biphasic in that condition. Our patient had no personal or family history of syncope or sudden death, and her ECG normalized with calcium replacement.3
Hypothermia can also present with a long QT interval secondary to a long ST segment. Our patient was normothermic, and had no other ECG findings suggestive of hypothermia (Osborn waves: notching of terminal portion of QRS), shivering artifacts, sinus bradycardia, atrial fibrillation, QRS prolongation, or prolongation of the PR interval.4
The differential diagnosis of a long QT interval with a long ST segment is short, and the clinical scenario will provide the etiology in most cases.
- .Hypercalcemia and hypocalcemia. In:Fauci AS,Braunwald E,Kasper DL, et al., eds.Harrison's Principles of Internal Medicine. Available at: http://www.accessmedicine.com/content.aspx?aID=2864354.
- ,,.Massive transfusion.Crit Care Clin.1986;2:791–805.
- ,,,.Novel insights in the congenital long QT syndrome.Ann Intern Med.2002;137:981–992.
- ,,.The clinical value of the ECG in noncardiac conditions.Chest.2004;125:1561–1576.
Patient Presentation
A 52‐year‐old woman with cirrhosis presented with a large‐volume upper gastrointestinal (GI) bleed. After receiving massive volume and blood product resuscitation (18 U packed red blood cells [PRBCs], 17 U fresh frozen plasma [FFP], 2 U cryoprecipitate, 1 U platelets, and 14 L normal saline), a routine admission electrocardiogram (ECG) was obtained (Figure 1).
Discussion
The ECG shows normal sinus rhythm with a prolonged QT interval (specifically due to a long ST segment). The differential diagnosis of this pattern of QT prolongation includes hypocalcemia, long QT syndrome (variant 3), and hypothermia. Massive transfusion can result in the chelation of calcium by citrate resulting in hypocalcemia. This patient's serum calcium was 7 mg/dL (8.9‐10.2) (ionized 0.21 mmol/L; 1.18‐1.38), her pH was 7.03. There were no overt signs or symptoms of hypocalcemia (paresthesias, twitching, tetany, or seizures.) and with aggressive replacement the calcium and the ECG normalized. Transfusions are an uncommon cause of hypocalcemia, with hypomagnesemia or hypermagnesemia, acute pancreatitis, rhabdomyolysis, tumor lysis syndrome, renal failure, vitamin D deficiency, pseudoparathyroidism and hypoparathyroidism being the more common disorders caused.1, 2 This patient's magnesium was normal on admission (2.1 mg/dL) and she did not have any of the other conditions mentioned. It is possible that hemodilution may also have played a role.
Genetic long QT syndrome (variant 3) can manifest with a similar ECG pattern, although the T‐wave is sometimes peaked or biphasic in that condition. Our patient had no personal or family history of syncope or sudden death, and her ECG normalized with calcium replacement.3
Hypothermia can also present with a long QT interval secondary to a long ST segment. Our patient was normothermic, and had no other ECG findings suggestive of hypothermia (Osborn waves: notching of terminal portion of QRS), shivering artifacts, sinus bradycardia, atrial fibrillation, QRS prolongation, or prolongation of the PR interval.4
The differential diagnosis of a long QT interval with a long ST segment is short, and the clinical scenario will provide the etiology in most cases.
Patient Presentation
A 52‐year‐old woman with cirrhosis presented with a large‐volume upper gastrointestinal (GI) bleed. After receiving massive volume and blood product resuscitation (18 U packed red blood cells [PRBCs], 17 U fresh frozen plasma [FFP], 2 U cryoprecipitate, 1 U platelets, and 14 L normal saline), a routine admission electrocardiogram (ECG) was obtained (Figure 1).
Discussion
The ECG shows normal sinus rhythm with a prolonged QT interval (specifically due to a long ST segment). The differential diagnosis of this pattern of QT prolongation includes hypocalcemia, long QT syndrome (variant 3), and hypothermia. Massive transfusion can result in the chelation of calcium by citrate resulting in hypocalcemia. This patient's serum calcium was 7 mg/dL (8.9‐10.2) (ionized 0.21 mmol/L; 1.18‐1.38), her pH was 7.03. There were no overt signs or symptoms of hypocalcemia (paresthesias, twitching, tetany, or seizures.) and with aggressive replacement the calcium and the ECG normalized. Transfusions are an uncommon cause of hypocalcemia, with hypomagnesemia or hypermagnesemia, acute pancreatitis, rhabdomyolysis, tumor lysis syndrome, renal failure, vitamin D deficiency, pseudoparathyroidism and hypoparathyroidism being the more common disorders caused.1, 2 This patient's magnesium was normal on admission (2.1 mg/dL) and she did not have any of the other conditions mentioned. It is possible that hemodilution may also have played a role.
Genetic long QT syndrome (variant 3) can manifest with a similar ECG pattern, although the T‐wave is sometimes peaked or biphasic in that condition. Our patient had no personal or family history of syncope or sudden death, and her ECG normalized with calcium replacement.3
Hypothermia can also present with a long QT interval secondary to a long ST segment. Our patient was normothermic, and had no other ECG findings suggestive of hypothermia (Osborn waves: notching of terminal portion of QRS), shivering artifacts, sinus bradycardia, atrial fibrillation, QRS prolongation, or prolongation of the PR interval.4
The differential diagnosis of a long QT interval with a long ST segment is short, and the clinical scenario will provide the etiology in most cases.
- .Hypercalcemia and hypocalcemia. In:Fauci AS,Braunwald E,Kasper DL, et al., eds.Harrison's Principles of Internal Medicine. Available at: http://www.accessmedicine.com/content.aspx?aID=2864354.
- ,,.Massive transfusion.Crit Care Clin.1986;2:791–805.
- ,,,.Novel insights in the congenital long QT syndrome.Ann Intern Med.2002;137:981–992.
- ,,.The clinical value of the ECG in noncardiac conditions.Chest.2004;125:1561–1576.
- .Hypercalcemia and hypocalcemia. In:Fauci AS,Braunwald E,Kasper DL, et al., eds.Harrison's Principles of Internal Medicine. Available at: http://www.accessmedicine.com/content.aspx?aID=2864354.
- ,,.Massive transfusion.Crit Care Clin.1986;2:791–805.
- ,,,.Novel insights in the congenital long QT syndrome.Ann Intern Med.2002;137:981–992.
- ,,.The clinical value of the ECG in noncardiac conditions.Chest.2004;125:1561–1576.
The Blog Rounds
It seems appropriate that in the same month President Obama signed a $787 billion economic stimulus package, the preferred topic of choice in the blogosphere would focus on money—or, more specifically, lack of it.
On his blog "Dr. Wes," Westby G. Fisher, MD, an internist, cardiologist, and cardiac electrophysiologist, warns about the looming Medicare physician fee cuts that Congress might not prevent this year as pressure increases to trim healthcare costs. Physicians should plan to work with the American Medical Association (AMA) to prevent the cuts, he writes.
"Do we honestly think that our individual subspecialty societies for cardiologists, internists, surgeons, hospitalists, or even newer, heavily promoted doctors' Internet sites will hold a policy-making candle to the AMA's lobbying stature on the Hill? No way."
Agree or disagree? Post your comment.
Speaking of healthcare spending, The Wall Street Journal Health Blog writer Jacob Goldstein reports startling figures recently published in Health Affairs: a 5.5% projected rise in U.S. healthcare spending this year and healthcare projecting to make up 17.6% of total GDP, compared with 16.6% last year.
Commenting on Goldstein’s post, urologist James G. Knight, MD, CEO of Consumer Directed Health Care Inc., had the following to say: "While everyone should have major medical insurance that protects them from financial ruin, each person should be financially responsible for their day-to-day care. This gives people who smoke, are overweight, or who have other behavior-related illnesses a financial incentive to manage their personal health."
FridaWrites of Hospitalist With a View offers another option to lower health spending: "Jumbo serving sizes are the enemy—in our house, half a bagel is enough and we split restaurant orders and enjoy leftovers; this also makes eating out on occasion more affordable. In some coffee shops, we've seen that one muffin or cookie in the display is large enough to serve a whole family. Teaching people to measure portions and count calories seems to be important—and this goes for hospital meals, too, where I've also seen surprisingly large portion sizes for people who don't need to gain weight."
It seems appropriate that in the same month President Obama signed a $787 billion economic stimulus package, the preferred topic of choice in the blogosphere would focus on money—or, more specifically, lack of it.
On his blog "Dr. Wes," Westby G. Fisher, MD, an internist, cardiologist, and cardiac electrophysiologist, warns about the looming Medicare physician fee cuts that Congress might not prevent this year as pressure increases to trim healthcare costs. Physicians should plan to work with the American Medical Association (AMA) to prevent the cuts, he writes.
"Do we honestly think that our individual subspecialty societies for cardiologists, internists, surgeons, hospitalists, or even newer, heavily promoted doctors' Internet sites will hold a policy-making candle to the AMA's lobbying stature on the Hill? No way."
Agree or disagree? Post your comment.
Speaking of healthcare spending, The Wall Street Journal Health Blog writer Jacob Goldstein reports startling figures recently published in Health Affairs: a 5.5% projected rise in U.S. healthcare spending this year and healthcare projecting to make up 17.6% of total GDP, compared with 16.6% last year.
Commenting on Goldstein’s post, urologist James G. Knight, MD, CEO of Consumer Directed Health Care Inc., had the following to say: "While everyone should have major medical insurance that protects them from financial ruin, each person should be financially responsible for their day-to-day care. This gives people who smoke, are overweight, or who have other behavior-related illnesses a financial incentive to manage their personal health."
FridaWrites of Hospitalist With a View offers another option to lower health spending: "Jumbo serving sizes are the enemy—in our house, half a bagel is enough and we split restaurant orders and enjoy leftovers; this also makes eating out on occasion more affordable. In some coffee shops, we've seen that one muffin or cookie in the display is large enough to serve a whole family. Teaching people to measure portions and count calories seems to be important—and this goes for hospital meals, too, where I've also seen surprisingly large portion sizes for people who don't need to gain weight."
It seems appropriate that in the same month President Obama signed a $787 billion economic stimulus package, the preferred topic of choice in the blogosphere would focus on money—or, more specifically, lack of it.
On his blog "Dr. Wes," Westby G. Fisher, MD, an internist, cardiologist, and cardiac electrophysiologist, warns about the looming Medicare physician fee cuts that Congress might not prevent this year as pressure increases to trim healthcare costs. Physicians should plan to work with the American Medical Association (AMA) to prevent the cuts, he writes.
"Do we honestly think that our individual subspecialty societies for cardiologists, internists, surgeons, hospitalists, or even newer, heavily promoted doctors' Internet sites will hold a policy-making candle to the AMA's lobbying stature on the Hill? No way."
Agree or disagree? Post your comment.
Speaking of healthcare spending, The Wall Street Journal Health Blog writer Jacob Goldstein reports startling figures recently published in Health Affairs: a 5.5% projected rise in U.S. healthcare spending this year and healthcare projecting to make up 17.6% of total GDP, compared with 16.6% last year.
Commenting on Goldstein’s post, urologist James G. Knight, MD, CEO of Consumer Directed Health Care Inc., had the following to say: "While everyone should have major medical insurance that protects them from financial ruin, each person should be financially responsible for their day-to-day care. This gives people who smoke, are overweight, or who have other behavior-related illnesses a financial incentive to manage their personal health."
FridaWrites of Hospitalist With a View offers another option to lower health spending: "Jumbo serving sizes are the enemy—in our house, half a bagel is enough and we split restaurant orders and enjoy leftovers; this also makes eating out on occasion more affordable. In some coffee shops, we've seen that one muffin or cookie in the display is large enough to serve a whole family. Teaching people to measure portions and count calories seems to be important—and this goes for hospital meals, too, where I've also seen surprisingly large portion sizes for people who don't need to gain weight."