User login
Terra Firma-Forme Dermatosis Mimicking Livedo Racemosa
To the Editor:
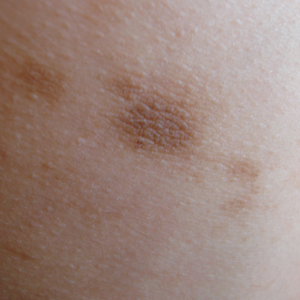
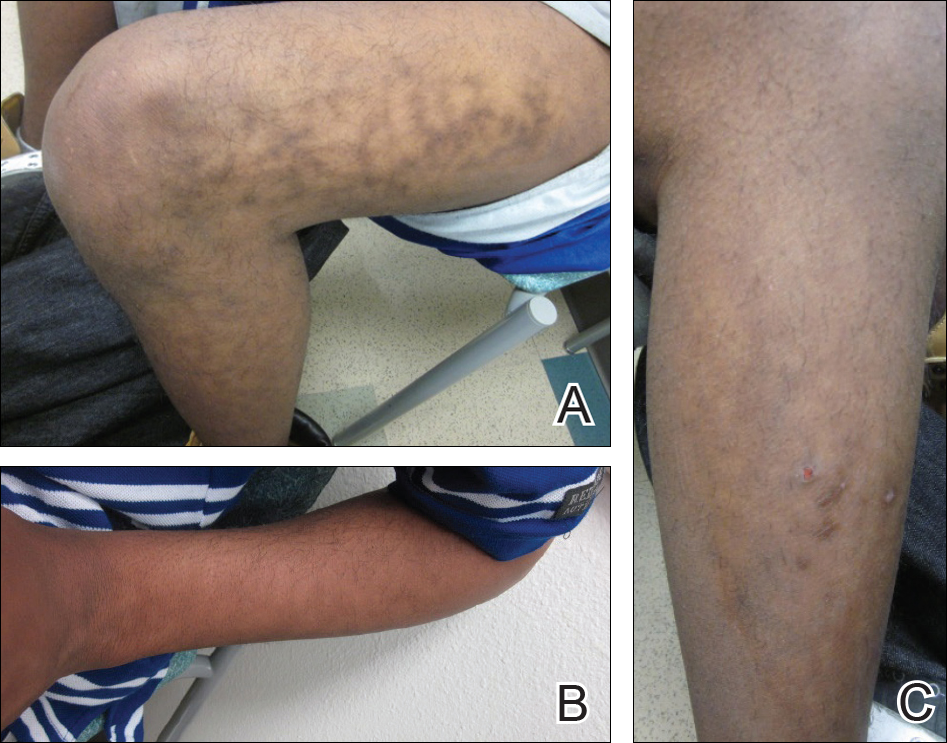
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
Practice Points
- Clinicians should include terra firma-forme dermatosis in the differential diagnosis of any hyperpigmented condition, regardless of pattern of presentation.
- Clean the skin with an alcohol wipe to rule out a diagnosis of terra firma-forme dermatosis.
Microcystic Adnexal Carcinoma of the External Auditory Canal
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
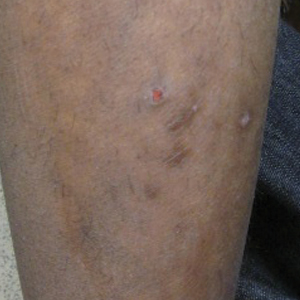
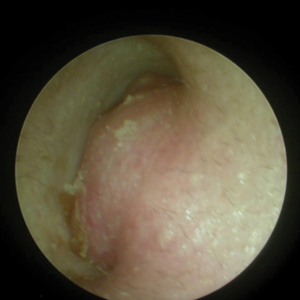
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.

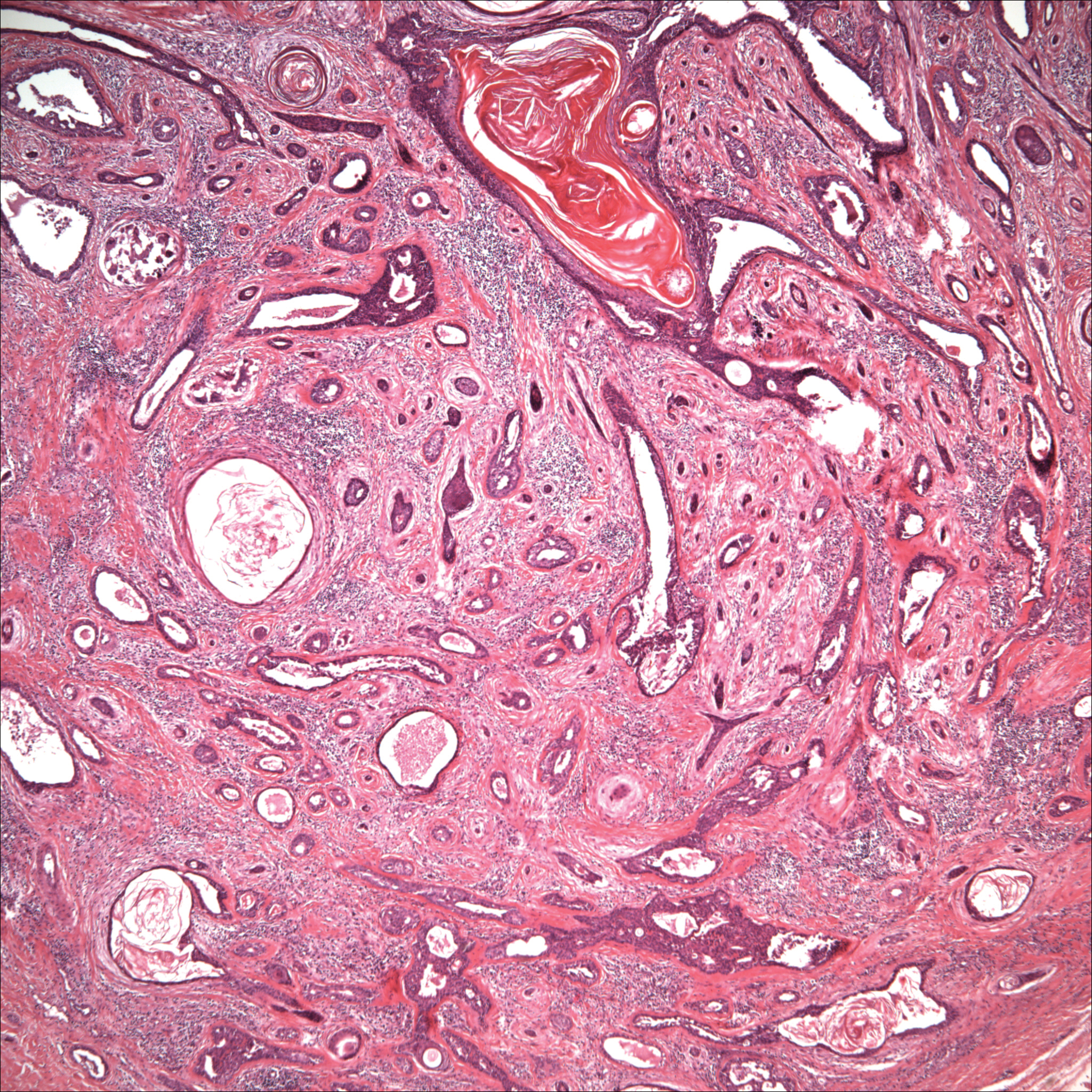
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
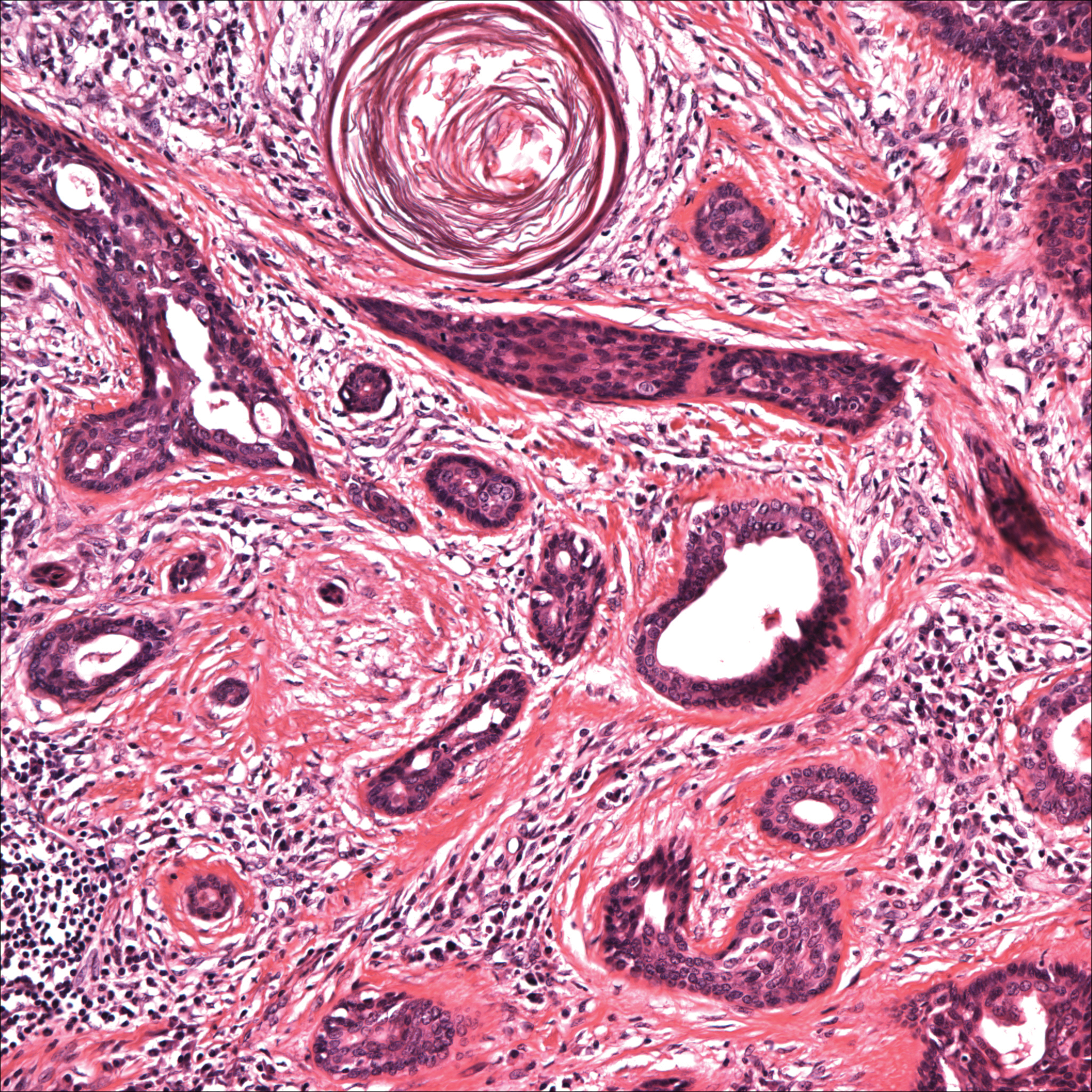
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
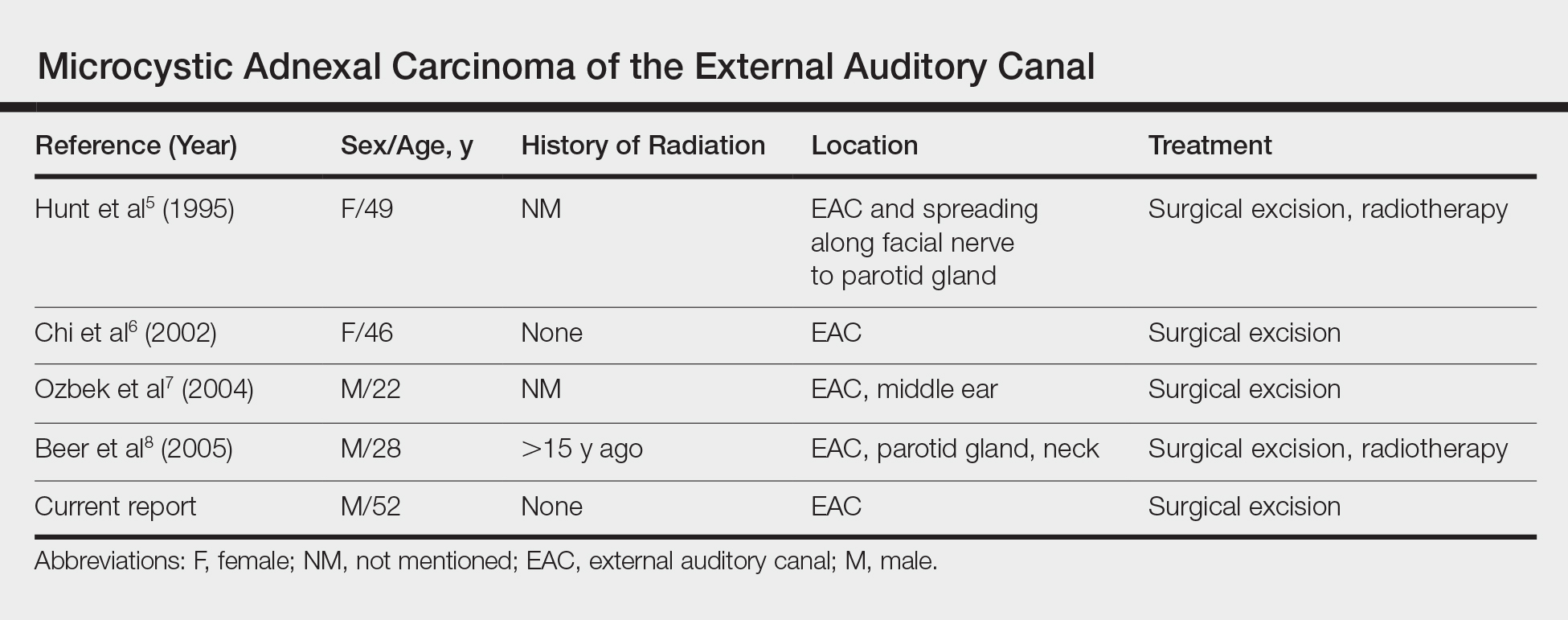
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
Practice Points
- Microcystic adnexal carcinoma is a locally aggressive malignant adnexal tumor with a high potential for local recurrence.
- The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.
- Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence is essential.
Atrophodermalike Guttate Morphea
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
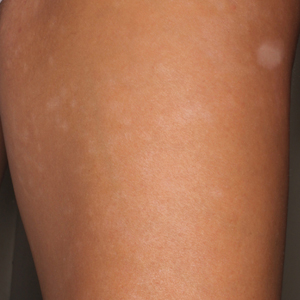
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
 as well as hypopigmented macules and a minimally depressed patch on the left thigh (B).")
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
(H&E, original magnification ×40)...")
(Verhoeff-van Gieson, original magnification ×40). Normal elastin network in the lower reticular dermis; note the normal size of elastin fibers (B)(Verhoeff-van Gieson, original magnification ×200).")
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
Practice Points
- Atrophodermalike guttate morphea is a potentially underreported or undescribed entity consisting of a combination of clinicopathologic features.
- Widespread hypopigmented macules on the trunk and extremities marked by thinned collagen, fibroplasia, and altered fragmented elastin in the papillary dermis and upper reticular dermis are the key features.
- Atrophoderma, morphea, and lichen sclerosus et atrophicus should be ruled out during clinical workup.
Gottron Papules Mimicking Dermatomyositis: An Unusual Manifestation of Systemic Lupus Erythematosus
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
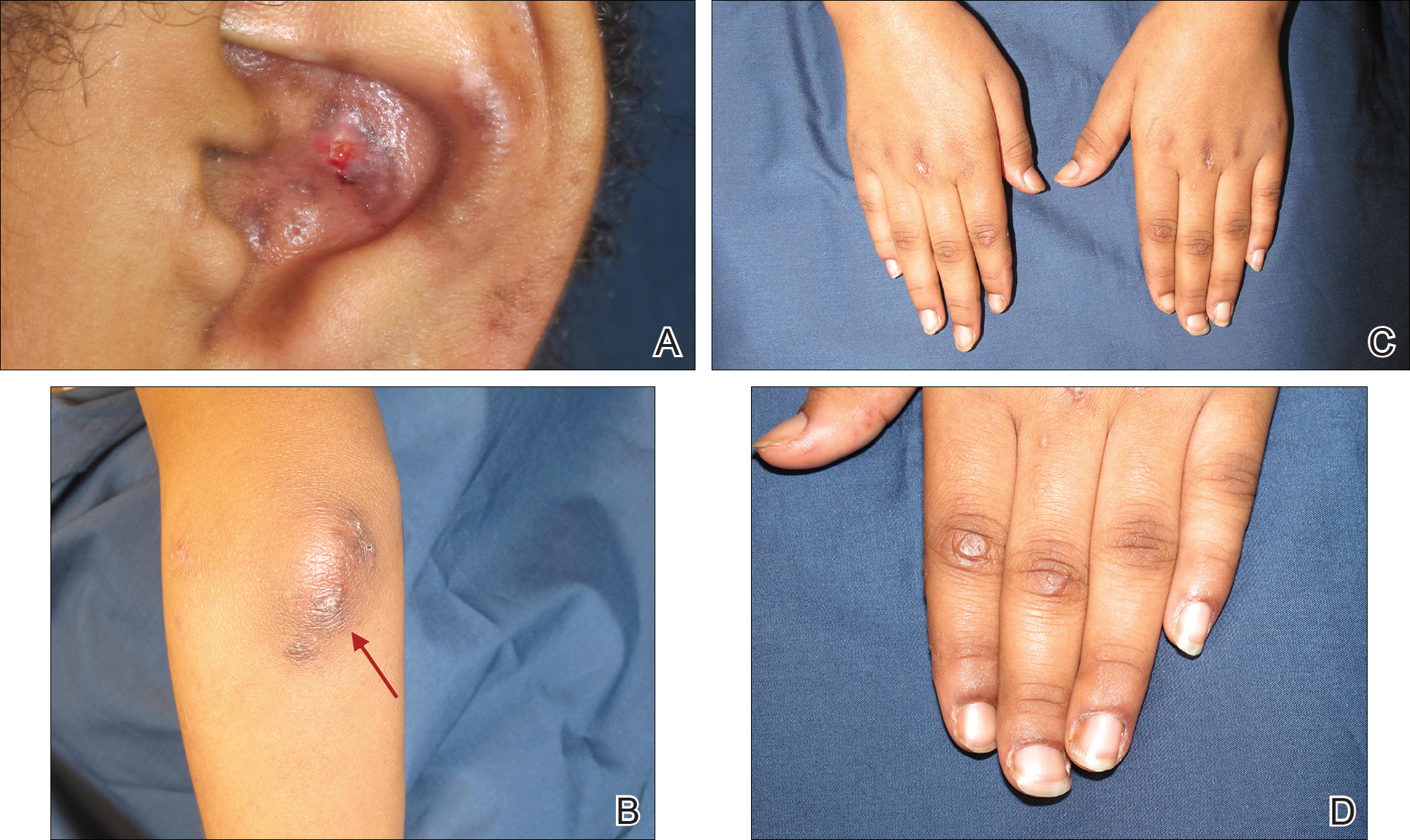
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
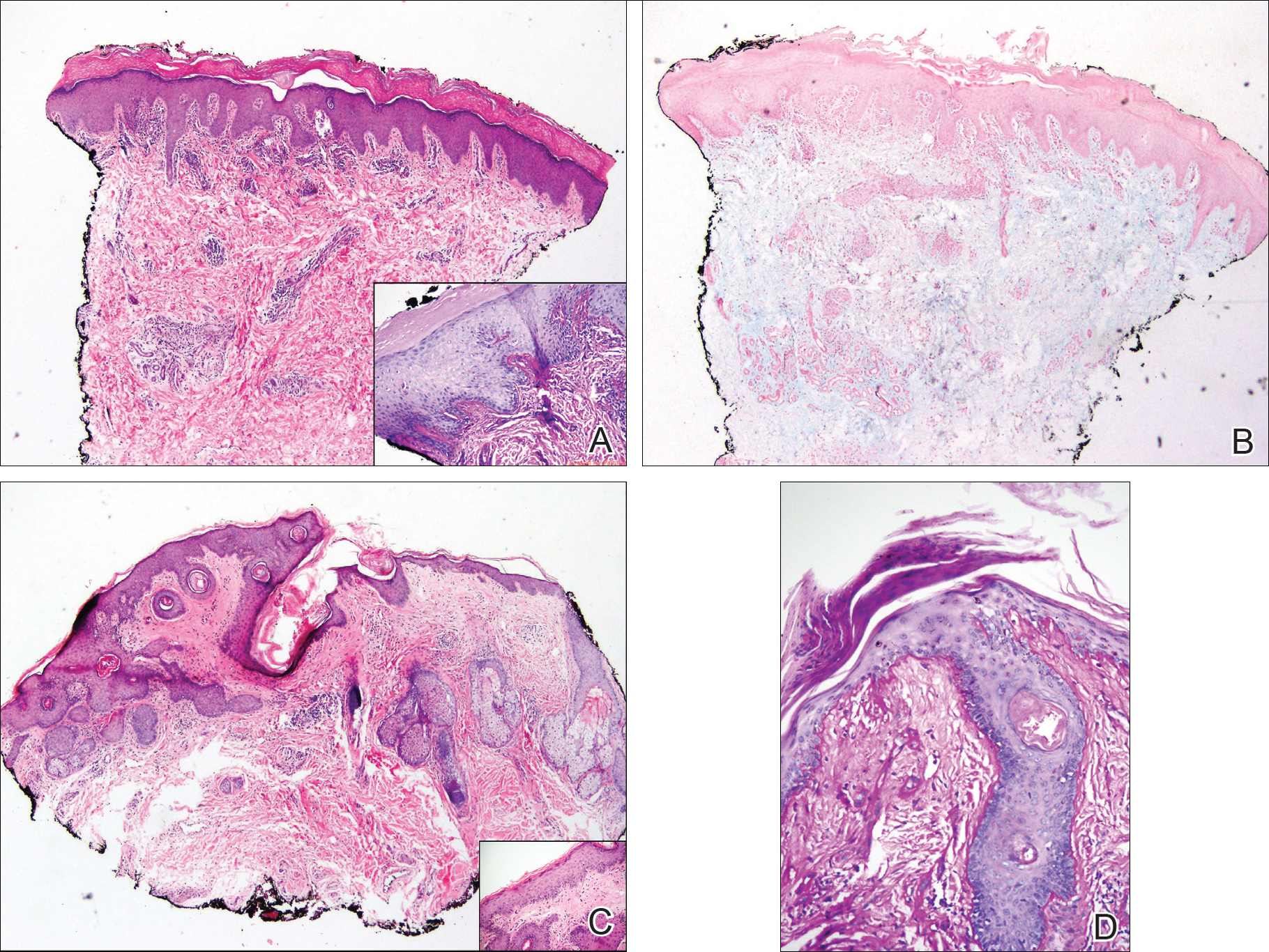
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
Practice Points
- Gottron-like papules can be a dermatologic presentation of lupus erythematosus.
- When present along with other findings of lupus erythematosus without any clinical manifestations of dermatomyositis, Gottron-like papules can be thought of as a manifestation of lupus erythematosus rather than dermatomyositis.

Ecthyma Gangrenosum Due to Pseudomonas fluorescens
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
...")
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
(H&E, original magnification ×100); biopsy from the periphery of the lesion showed leukocytoclastic vasculitis (B)...")
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
Practice Points
- Immunocompromised patients with a high exposure to agricultural products may be at increased risk for systemic infection by Pseudomonas fluorescens.
- Pseudomonas fluorescens is an opportunistic pathogen that can cause ecthyma gangrenosum, which necessitates rapid diagnosis and treatment to prevent mortality.
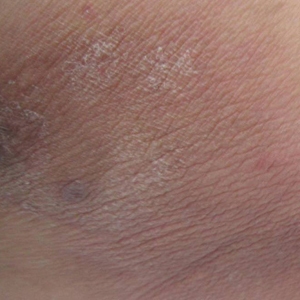
Copresentation of Common Variable Immune Deficiency and Sweet Syndrome
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.


Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
Practice Points
- Suggested workup for Sweet syndrome includes ruling out connective tissue disorders and malignancies.
- Familial common variable immune deficiency is rare and can first manifest in adulthood.
Chilblain Lupus Erythematosus Presenting With Bilateral Hemorrhagic Bullae of Distal Halluces
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.

(H&E, original magnification ×400) with deep periadnexal mucin deposition (B)(colloidal iron, original magnification ×40).")
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
Practice Points
- Up to 20% of patients with chilblain lupus erythematosus (CHLE) will develop systemic lupus erythematosus (SLE), necessitating close long-term follow-up.
- Medications such as antihypertensives, antifungals, chemotherapeutic agents, and tumor necrosis factor 11α inhibitors have been reported to trigger CHLE.
- Chilblain lupus erythematosus is less responsive to traditional antimalarial agents commonly used to treat SLE.
- Management of CHLE includes physical protection from cold environments, calcium channel blockers, topical and systemic steroids, and pentoxifylline, among other treatment modalities.
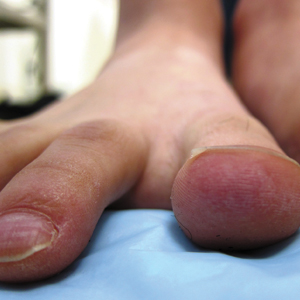
Epidermolysis Bullosa Acquisita in Association With Mantle Cell Lymphoma
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
 and multiple bullae on the sole (B).")
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
.")
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
Practice Points
- Epidermolysis bullosa acquisita (EBA) is an uncommon blistering disorder and few cases have been associated with malignancy.
- Diagnosis of EBA is challenging and requires exclusion of other blistering diseases.
Polypoid Melanoma: An Aggressive Variant of Nodular Melanoma
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.


.")
 immunostain (original magnification ×200).")
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
Practice Points
- The differential diagnosis of polypoid melanoma includes pyogenic granuloma and squamous cell carcinoma.
- Polypoid melanoma has a poor prognosis because of its thickness and ulceration at the time of diagnosis and the risk of vascular embolization.
Idiopathic Eruptive Macular Pigmentation With Papillomatosis
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).

.")
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
Practice Points
- Idiopathic eruptive macular pigmentation with papillomatosis is a rare disorder that most frequently affects children and young adults.
- Idiopathic eruptive macular pigmentation with papillomatosis is characterized by asymptomatic, brownish, hyperpigmented macules involving the neck, trunk, arms, and legs.
- The disorder is important to consider in the differential diagnosis of asymptomatic pigmentary disorders to avoid unnecessary treatment because the disease is self-limiting and resolves over weeks to years.