User login
Benign Cephalic Histiocytosis
To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
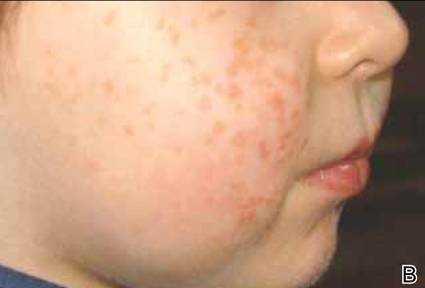
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |

Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4
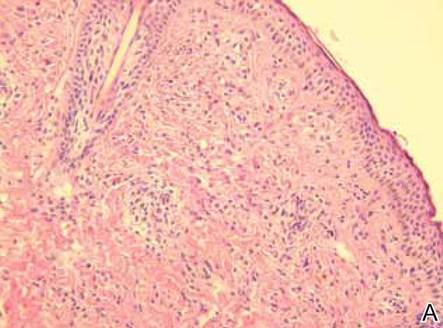
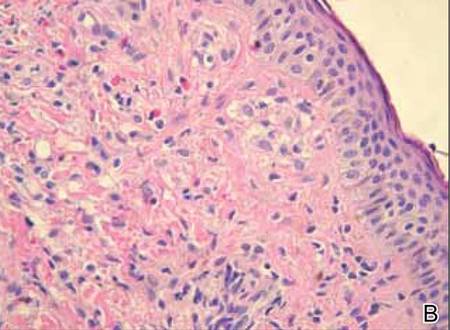
Figure 2. Skin section showing the upper and mid dermis infiltrated with slightly pleomorphic epithelioid histiocytic cells with clear cytoplasm and vesicular nuclei. Few accompanying lymphocytes and eosinophils were visible (H&E, original magnifications ×200 and ×400). |
A close histologic relationship and presence of overlapping symptoms observed among BCH, GEH, and juvenile xanthogranuloma indicate that these entities fall into a spectrum of the same disorder. However, the presence of a uniform infiltrate of large foamy histiocytes readily distinguishes xanthomas from BCH.4 In some unusual clinical presentations of CM or in cases of the nodular form of the condition, there is a need to distinguish between non-LCH and CM, as in our patient. Darier sign, consisting of urtication and erythema appearing after mechanical irritation of the skin lesion, is pathognomonic for CM. Nevertheless, Darier sign is not sufficient to confirm CM when it is not pronounced. Therefore, histologic examination with the use of immunostaining plays a key role in the differential diagnosis of these disorders in children.15 Treatment of BCH is not recommended because of spontaneous remission of the disease.1-5
Benign cephalic histiocytosis is a rare clinical form of non-LCH. No systemic or mucosal involvement has been described. Lesions often are confused with plane warts, but a biopsy is definitive. Therapy is not effective but fortunately none is necessary.
1. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13:383-404.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Dadzie O, Hopster D, Cerio R, et al. Benign cephalic histiocytosis in a British-African child. Pediatr Dermatol. 2005;22:444-446.
4. Goodman WT, Barret TL. Histiocytoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Hong Kong, China: Elsevier Saunders; 2012:1527-1546.
5. Gianotti F, Caputo R, Ermacora E. Singular infantile histiocytosis with cells with intracytoplasmic vermiform particles [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
6. Barsky B, Lao I, Barsky S, et al. Benign cephalic histiocytosis. Arch Dermatol. 1984;120:650-655.
7. Gianotti F, Caputo R, Ermacora E, et al. Benign cephalic histiocytosis. Arch Dermatol. 1986;122:1038-1043.
8. Zelger BW, Sidoroff A, Orchard G, et al. Non-Langerhans cell histiocytosis. a new unifying concept. Am J Dermatopathol. 1996;18:490-504.
9. Hasegawa S, Deguchi M, Chiba-Okada S, et al. Japanese case of benign cephalic histiocytosis. J Dermatol. 2009;36:69-71.
10. Weston WL, Travers SH, Mierau GW, et al. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol. 2000;17:296-298.
11. Gianotti R, Alessi E, Caputo R. Benign cephalic histiocytosis: a distinct entity or a part of a wide spectrum of histiocytic proliferative disorders of children? a histopathological study. Am J Dermatopathol. 1993;15:315-319.
12. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
13. Sidwell RU, Francis N, Slater DN, et al. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-43.
14. Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S17-S23.
15. Heide R, Beishuizen A, De Groot H, et al. Mastocytosis in children: a protocol for management. Pediatr Dermatol. 2008;25:493-500.
To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |
Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4
|
Figure 2. Skin section showing the upper and mid dermis infiltrated with slightly pleomorphic epithelioid histiocytic cells with clear cytoplasm and vesicular nuclei. Few accompanying lymphocytes and eosinophils were visible (H&E, original magnifications ×200 and ×400). |
A close histologic relationship and presence of overlapping symptoms observed among BCH, GEH, and juvenile xanthogranuloma indicate that these entities fall into a spectrum of the same disorder. However, the presence of a uniform infiltrate of large foamy histiocytes readily distinguishes xanthomas from BCH.4 In some unusual clinical presentations of CM or in cases of the nodular form of the condition, there is a need to distinguish between non-LCH and CM, as in our patient. Darier sign, consisting of urtication and erythema appearing after mechanical irritation of the skin lesion, is pathognomonic for CM. Nevertheless, Darier sign is not sufficient to confirm CM when it is not pronounced. Therefore, histologic examination with the use of immunostaining plays a key role in the differential diagnosis of these disorders in children.15 Treatment of BCH is not recommended because of spontaneous remission of the disease.1-5
Benign cephalic histiocytosis is a rare clinical form of non-LCH. No systemic or mucosal involvement has been described. Lesions often are confused with plane warts, but a biopsy is definitive. Therapy is not effective but fortunately none is necessary.
To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |
Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4
|
Figure 2. Skin section showing the upper and mid dermis infiltrated with slightly pleomorphic epithelioid histiocytic cells with clear cytoplasm and vesicular nuclei. Few accompanying lymphocytes and eosinophils were visible (H&E, original magnifications ×200 and ×400). |
A close histologic relationship and presence of overlapping symptoms observed among BCH, GEH, and juvenile xanthogranuloma indicate that these entities fall into a spectrum of the same disorder. However, the presence of a uniform infiltrate of large foamy histiocytes readily distinguishes xanthomas from BCH.4 In some unusual clinical presentations of CM or in cases of the nodular form of the condition, there is a need to distinguish between non-LCH and CM, as in our patient. Darier sign, consisting of urtication and erythema appearing after mechanical irritation of the skin lesion, is pathognomonic for CM. Nevertheless, Darier sign is not sufficient to confirm CM when it is not pronounced. Therefore, histologic examination with the use of immunostaining plays a key role in the differential diagnosis of these disorders in children.15 Treatment of BCH is not recommended because of spontaneous remission of the disease.1-5
Benign cephalic histiocytosis is a rare clinical form of non-LCH. No systemic or mucosal involvement has been described. Lesions often are confused with plane warts, but a biopsy is definitive. Therapy is not effective but fortunately none is necessary.
1. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13:383-404.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Dadzie O, Hopster D, Cerio R, et al. Benign cephalic histiocytosis in a British-African child. Pediatr Dermatol. 2005;22:444-446.
4. Goodman WT, Barret TL. Histiocytoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Hong Kong, China: Elsevier Saunders; 2012:1527-1546.
5. Gianotti F, Caputo R, Ermacora E. Singular infantile histiocytosis with cells with intracytoplasmic vermiform particles [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
6. Barsky B, Lao I, Barsky S, et al. Benign cephalic histiocytosis. Arch Dermatol. 1984;120:650-655.
7. Gianotti F, Caputo R, Ermacora E, et al. Benign cephalic histiocytosis. Arch Dermatol. 1986;122:1038-1043.
8. Zelger BW, Sidoroff A, Orchard G, et al. Non-Langerhans cell histiocytosis. a new unifying concept. Am J Dermatopathol. 1996;18:490-504.
9. Hasegawa S, Deguchi M, Chiba-Okada S, et al. Japanese case of benign cephalic histiocytosis. J Dermatol. 2009;36:69-71.
10. Weston WL, Travers SH, Mierau GW, et al. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol. 2000;17:296-298.
11. Gianotti R, Alessi E, Caputo R. Benign cephalic histiocytosis: a distinct entity or a part of a wide spectrum of histiocytic proliferative disorders of children? a histopathological study. Am J Dermatopathol. 1993;15:315-319.
12. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
13. Sidwell RU, Francis N, Slater DN, et al. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-43.
14. Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S17-S23.
15. Heide R, Beishuizen A, De Groot H, et al. Mastocytosis in children: a protocol for management. Pediatr Dermatol. 2008;25:493-500.
1. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13:383-404.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Dadzie O, Hopster D, Cerio R, et al. Benign cephalic histiocytosis in a British-African child. Pediatr Dermatol. 2005;22:444-446.
4. Goodman WT, Barret TL. Histiocytoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Hong Kong, China: Elsevier Saunders; 2012:1527-1546.
5. Gianotti F, Caputo R, Ermacora E. Singular infantile histiocytosis with cells with intracytoplasmic vermiform particles [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
6. Barsky B, Lao I, Barsky S, et al. Benign cephalic histiocytosis. Arch Dermatol. 1984;120:650-655.
7. Gianotti F, Caputo R, Ermacora E, et al. Benign cephalic histiocytosis. Arch Dermatol. 1986;122:1038-1043.
8. Zelger BW, Sidoroff A, Orchard G, et al. Non-Langerhans cell histiocytosis. a new unifying concept. Am J Dermatopathol. 1996;18:490-504.
9. Hasegawa S, Deguchi M, Chiba-Okada S, et al. Japanese case of benign cephalic histiocytosis. J Dermatol. 2009;36:69-71.
10. Weston WL, Travers SH, Mierau GW, et al. Benign cephalic histiocytosis with diabetes insipidus. Pediatr Dermatol. 2000;17:296-298.
11. Gianotti R, Alessi E, Caputo R. Benign cephalic histiocytosis: a distinct entity or a part of a wide spectrum of histiocytic proliferative disorders of children? a histopathological study. Am J Dermatopathol. 1993;15:315-319.
12. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
13. Sidwell RU, Francis N, Slater DN, et al. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol. 2005;22:40-43.
14. Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S17-S23.
15. Heide R, Beishuizen A, De Groot H, et al. Mastocytosis in children: a protocol for management. Pediatr Dermatol. 2008;25:493-500.
Mycoplasma pneumoniae Infection in Adults With Acute and Chronic Urticaria
To the Editor:
Mycoplasma pneumoniae has been implicated as a cause of acute urticaria (AU) in children,1 but its role in adults with AU is unknown. The aim of this retrospective study was to compare the incidence of acute M pneumoniae infection in adults with AU and chronic urticaria (CU).
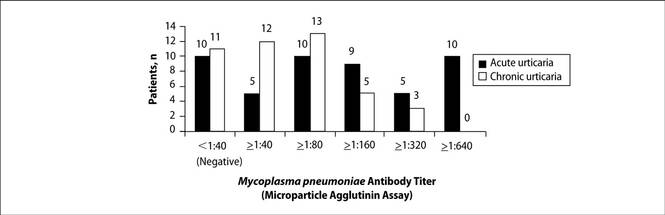
A chart review was performed on adult patients with AU and CU who presented at a private dermatology practice in Singapore. Acute M pneumoniae infection was diagnosed on the basis of a single indirect microparticle agglutinin assay (MAA) titer of 1:320 or higher. All statistical tests were performed using SPSS version 13.0. P=.05 was regarded as significant. Data from 49 adults with AU and 44 adults with CU were analyzed. The distribution of MAA titers in adults with AU and CU are shown in the Figure. Microparticle agglutinin assay was negative in 10 (20.4%) of 49 adults with AU. Fifteen (30.6%) of 49 adults with AU had evidence of acute M pneumoniae infection, as indicated by an MAA titer of 1:320 or higher. The remaining 24 (49.0%) had evidence of prior infection as indicated by titers above the manufacturer’s cutoff of 1:402 and below our cutoff for acute infection of 1:320 or higher. Microparticle agglutinin assay was negative in 11 (25%) of 44 adults with CU. Three (6.8%) adults with CU were diagnosed with acute M pneumoniae infection and 30 (68.2%) were diagnosed with prior infection. The incidence of acute M pneumoniae infection was 30.6% in adults with AU compared to 6.8% in adults with CU, and the difference was statistically significant (P=.004).
Extrapulmonary complications of M pneumoniae involving practically every organ system have been described and 25% of patients develop cutaneous symptoms3 including AU. In 2007, Kano et al4 reported M pneumoniae infection-induced erythema nodosum, anaphylactoid purpura, and AU in a family of 3. This report was interesting because it showed that M pneumoniae had the ability to elicit different cutaneous reactions depending on the maturity of the adaptive immunity of a host, even among individuals of a common genetic background. A Taiwanese study found that 21 (32%) of 65 children with AU had M pneumoniae infection as determined by a positive Mycoplasma IgM test or an equivocal Mycoplasma IgM coupled with positive cold agglutinin test results.1
In our study, we found serological evidence of acute M pneumoniae infection in 15 (30.6%) of 49 adults with AU compared to 3 (6.8%) of 44 adults with CU (P=.004), suggesting that M pneumoniae also may play a role in the etiology of adult AU. Diagnosis of acute M pneumoniae infection is challenging, as it is often impossible to obtain convalescent serum that will show the 4-fold rise in titer. Single MAA titers of 1:160 or higher have been recommended for diagnosis of acute infection,5 but because of higher background activity in Singapore, we used a higher titer (>1:320). However, in doing so, we could be underestimating the true incidence of acute M pneumoniae infections.
The role of M pneumoniae in CU is uncertain. A Thai study reported that 55% of 38 children with CU had elevated M pneumoniae titers but did not provide details of actual titers or define what they meant by elevated titers.6 The incidence of acute and prior infection in our patients with CU was 6.8% and 68.2%, respectively. Unfortunately, we cannot determine the significance of the 68.2% incidence rate of prior infection in the absence of a normal control population of patients without urticaria. Another limitation of this study is that we compared M pneumoniae infection rate in AU with CU on the assumption that infection is not likely to play a significant role in CU, which may not necessarily be the case. Tests for other etiologic agents, including viruses, also were not performed. Not withstanding these limitations, this study suggests that acute M pneumoniae infection is significantly more common in adults with AU than in adults with CU.
This study showed that M pneumoniae might play a role in the etiology of AU in adults and our findings need to be confirmed by prospective studies. Several more questions must be answered before deciding whether the current practice of treating AU symptomatically without investigation needs to be changed. First, does treatment of M pneumoniae infection have any influence on the course of AU? The fact that AU usually is self-limiting suggests that treatment may not influence the disease course. Second, does treatment of underlying M pneumoniae infection shorten the course of AU? Third, do AU patients with untreated M pneumoniae infection face a higher risk for developing CU? This question is intriguing for the following reasons: (1) 30% to 50% of CU cases are autoimmune in etiology7; (2) antibodies to galactocerebroside that cross-react with glycolipids on M pneumoniae have been detected in patients with M pneumoniae–associated Guillain-Barré syndrome, suggesting a form of molecular mimicry8; and (3) antinuclear antibody also has occasionally been detected in sera of patients with M pneumoniae.9 It would be interesting to test patients with autoimmune and nonautoimmune CU for evidence of M pneumoniae serology infection.
We hope that prospective studies will be conducted in the future to confirm our findings and answer some of the questions raised.
1. Wu CC, Kuo HC, Yu HR, et al. Association of acute urticaria with Mycoplasma pneumoniae infection in hospitalized children. Ann Allergy Asthma Immunol. 2009;103:134-139.
2. Serodia-Myco II [package insert]. Tokyo, Japan: Fujirebo; 2013.
3. Murray HW, Masur H, Senterfit LB, et al. The protean manifestations of Mycoplasma pneumoniae infection in adults. Am J Med. 1975;58:229-242.
4. Kano Y, Mitsuyama Y, Hirahara K, et al. Mycoplasma pneumoniae infection-induced erythema nodosum, anaphylactoid purpura, and acute urticaria in 3 people in a single family. J Am Acad Dermatol. 2007;57(suppl 2):S33-S35.
5. Daxboeck F, Krause R, Wenisch C. Laboratory diagnosis of Mycoplasma pneumoniae infection. Clin Microbiol Infect. 2003;9:263-273.
6. Pongpreuksa S, Boochoo S, Kulthanan K, et al. Chronic urticaria: what is worth doing in pediatric population? J Allergy Clin Immunol. 2004:S134.
7. Grattan CE. Autoimmune urticaria. Immunol Allergy Clin North Am. 2004;24:163-181.
8. Kusunoki S, Shiina M, Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. 2001;57:736-738.
9. Arikan S, Ergüven S, Ustaçelebi S, et al. Detection of antinuclear antibody (ANA) in patients with Mycoplasmal Pneumonia. Turk J Med Sci. 1998;28:97-98.
To the Editor:
Mycoplasma pneumoniae has been implicated as a cause of acute urticaria (AU) in children,1 but its role in adults with AU is unknown. The aim of this retrospective study was to compare the incidence of acute M pneumoniae infection in adults with AU and chronic urticaria (CU).
A chart review was performed on adult patients with AU and CU who presented at a private dermatology practice in Singapore. Acute M pneumoniae infection was diagnosed on the basis of a single indirect microparticle agglutinin assay (MAA) titer of 1:320 or higher. All statistical tests were performed using SPSS version 13.0. P=.05 was regarded as significant. Data from 49 adults with AU and 44 adults with CU were analyzed. The distribution of MAA titers in adults with AU and CU are shown in the Figure. Microparticle agglutinin assay was negative in 10 (20.4%) of 49 adults with AU. Fifteen (30.6%) of 49 adults with AU had evidence of acute M pneumoniae infection, as indicated by an MAA titer of 1:320 or higher. The remaining 24 (49.0%) had evidence of prior infection as indicated by titers above the manufacturer’s cutoff of 1:402 and below our cutoff for acute infection of 1:320 or higher. Microparticle agglutinin assay was negative in 11 (25%) of 44 adults with CU. Three (6.8%) adults with CU were diagnosed with acute M pneumoniae infection and 30 (68.2%) were diagnosed with prior infection. The incidence of acute M pneumoniae infection was 30.6% in adults with AU compared to 6.8% in adults with CU, and the difference was statistically significant (P=.004).
Extrapulmonary complications of M pneumoniae involving practically every organ system have been described and 25% of patients develop cutaneous symptoms3 including AU. In 2007, Kano et al4 reported M pneumoniae infection-induced erythema nodosum, anaphylactoid purpura, and AU in a family of 3. This report was interesting because it showed that M pneumoniae had the ability to elicit different cutaneous reactions depending on the maturity of the adaptive immunity of a host, even among individuals of a common genetic background. A Taiwanese study found that 21 (32%) of 65 children with AU had M pneumoniae infection as determined by a positive Mycoplasma IgM test or an equivocal Mycoplasma IgM coupled with positive cold agglutinin test results.1
In our study, we found serological evidence of acute M pneumoniae infection in 15 (30.6%) of 49 adults with AU compared to 3 (6.8%) of 44 adults with CU (P=.004), suggesting that M pneumoniae also may play a role in the etiology of adult AU. Diagnosis of acute M pneumoniae infection is challenging, as it is often impossible to obtain convalescent serum that will show the 4-fold rise in titer. Single MAA titers of 1:160 or higher have been recommended for diagnosis of acute infection,5 but because of higher background activity in Singapore, we used a higher titer (>1:320). However, in doing so, we could be underestimating the true incidence of acute M pneumoniae infections.
The role of M pneumoniae in CU is uncertain. A Thai study reported that 55% of 38 children with CU had elevated M pneumoniae titers but did not provide details of actual titers or define what they meant by elevated titers.6 The incidence of acute and prior infection in our patients with CU was 6.8% and 68.2%, respectively. Unfortunately, we cannot determine the significance of the 68.2% incidence rate of prior infection in the absence of a normal control population of patients without urticaria. Another limitation of this study is that we compared M pneumoniae infection rate in AU with CU on the assumption that infection is not likely to play a significant role in CU, which may not necessarily be the case. Tests for other etiologic agents, including viruses, also were not performed. Not withstanding these limitations, this study suggests that acute M pneumoniae infection is significantly more common in adults with AU than in adults with CU.
This study showed that M pneumoniae might play a role in the etiology of AU in adults and our findings need to be confirmed by prospective studies. Several more questions must be answered before deciding whether the current practice of treating AU symptomatically without investigation needs to be changed. First, does treatment of M pneumoniae infection have any influence on the course of AU? The fact that AU usually is self-limiting suggests that treatment may not influence the disease course. Second, does treatment of underlying M pneumoniae infection shorten the course of AU? Third, do AU patients with untreated M pneumoniae infection face a higher risk for developing CU? This question is intriguing for the following reasons: (1) 30% to 50% of CU cases are autoimmune in etiology7; (2) antibodies to galactocerebroside that cross-react with glycolipids on M pneumoniae have been detected in patients with M pneumoniae–associated Guillain-Barré syndrome, suggesting a form of molecular mimicry8; and (3) antinuclear antibody also has occasionally been detected in sera of patients with M pneumoniae.9 It would be interesting to test patients with autoimmune and nonautoimmune CU for evidence of M pneumoniae serology infection.
We hope that prospective studies will be conducted in the future to confirm our findings and answer some of the questions raised.
To the Editor:
Mycoplasma pneumoniae has been implicated as a cause of acute urticaria (AU) in children,1 but its role in adults with AU is unknown. The aim of this retrospective study was to compare the incidence of acute M pneumoniae infection in adults with AU and chronic urticaria (CU).
A chart review was performed on adult patients with AU and CU who presented at a private dermatology practice in Singapore. Acute M pneumoniae infection was diagnosed on the basis of a single indirect microparticle agglutinin assay (MAA) titer of 1:320 or higher. All statistical tests were performed using SPSS version 13.0. P=.05 was regarded as significant. Data from 49 adults with AU and 44 adults with CU were analyzed. The distribution of MAA titers in adults with AU and CU are shown in the Figure. Microparticle agglutinin assay was negative in 10 (20.4%) of 49 adults with AU. Fifteen (30.6%) of 49 adults with AU had evidence of acute M pneumoniae infection, as indicated by an MAA titer of 1:320 or higher. The remaining 24 (49.0%) had evidence of prior infection as indicated by titers above the manufacturer’s cutoff of 1:402 and below our cutoff for acute infection of 1:320 or higher. Microparticle agglutinin assay was negative in 11 (25%) of 44 adults with CU. Three (6.8%) adults with CU were diagnosed with acute M pneumoniae infection and 30 (68.2%) were diagnosed with prior infection. The incidence of acute M pneumoniae infection was 30.6% in adults with AU compared to 6.8% in adults with CU, and the difference was statistically significant (P=.004).
Extrapulmonary complications of M pneumoniae involving practically every organ system have been described and 25% of patients develop cutaneous symptoms3 including AU. In 2007, Kano et al4 reported M pneumoniae infection-induced erythema nodosum, anaphylactoid purpura, and AU in a family of 3. This report was interesting because it showed that M pneumoniae had the ability to elicit different cutaneous reactions depending on the maturity of the adaptive immunity of a host, even among individuals of a common genetic background. A Taiwanese study found that 21 (32%) of 65 children with AU had M pneumoniae infection as determined by a positive Mycoplasma IgM test or an equivocal Mycoplasma IgM coupled with positive cold agglutinin test results.1
In our study, we found serological evidence of acute M pneumoniae infection in 15 (30.6%) of 49 adults with AU compared to 3 (6.8%) of 44 adults with CU (P=.004), suggesting that M pneumoniae also may play a role in the etiology of adult AU. Diagnosis of acute M pneumoniae infection is challenging, as it is often impossible to obtain convalescent serum that will show the 4-fold rise in titer. Single MAA titers of 1:160 or higher have been recommended for diagnosis of acute infection,5 but because of higher background activity in Singapore, we used a higher titer (>1:320). However, in doing so, we could be underestimating the true incidence of acute M pneumoniae infections.
The role of M pneumoniae in CU is uncertain. A Thai study reported that 55% of 38 children with CU had elevated M pneumoniae titers but did not provide details of actual titers or define what they meant by elevated titers.6 The incidence of acute and prior infection in our patients with CU was 6.8% and 68.2%, respectively. Unfortunately, we cannot determine the significance of the 68.2% incidence rate of prior infection in the absence of a normal control population of patients without urticaria. Another limitation of this study is that we compared M pneumoniae infection rate in AU with CU on the assumption that infection is not likely to play a significant role in CU, which may not necessarily be the case. Tests for other etiologic agents, including viruses, also were not performed. Not withstanding these limitations, this study suggests that acute M pneumoniae infection is significantly more common in adults with AU than in adults with CU.
This study showed that M pneumoniae might play a role in the etiology of AU in adults and our findings need to be confirmed by prospective studies. Several more questions must be answered before deciding whether the current practice of treating AU symptomatically without investigation needs to be changed. First, does treatment of M pneumoniae infection have any influence on the course of AU? The fact that AU usually is self-limiting suggests that treatment may not influence the disease course. Second, does treatment of underlying M pneumoniae infection shorten the course of AU? Third, do AU patients with untreated M pneumoniae infection face a higher risk for developing CU? This question is intriguing for the following reasons: (1) 30% to 50% of CU cases are autoimmune in etiology7; (2) antibodies to galactocerebroside that cross-react with glycolipids on M pneumoniae have been detected in patients with M pneumoniae–associated Guillain-Barré syndrome, suggesting a form of molecular mimicry8; and (3) antinuclear antibody also has occasionally been detected in sera of patients with M pneumoniae.9 It would be interesting to test patients with autoimmune and nonautoimmune CU for evidence of M pneumoniae serology infection.
We hope that prospective studies will be conducted in the future to confirm our findings and answer some of the questions raised.
1. Wu CC, Kuo HC, Yu HR, et al. Association of acute urticaria with Mycoplasma pneumoniae infection in hospitalized children. Ann Allergy Asthma Immunol. 2009;103:134-139.
2. Serodia-Myco II [package insert]. Tokyo, Japan: Fujirebo; 2013.
3. Murray HW, Masur H, Senterfit LB, et al. The protean manifestations of Mycoplasma pneumoniae infection in adults. Am J Med. 1975;58:229-242.
4. Kano Y, Mitsuyama Y, Hirahara K, et al. Mycoplasma pneumoniae infection-induced erythema nodosum, anaphylactoid purpura, and acute urticaria in 3 people in a single family. J Am Acad Dermatol. 2007;57(suppl 2):S33-S35.
5. Daxboeck F, Krause R, Wenisch C. Laboratory diagnosis of Mycoplasma pneumoniae infection. Clin Microbiol Infect. 2003;9:263-273.
6. Pongpreuksa S, Boochoo S, Kulthanan K, et al. Chronic urticaria: what is worth doing in pediatric population? J Allergy Clin Immunol. 2004:S134.
7. Grattan CE. Autoimmune urticaria. Immunol Allergy Clin North Am. 2004;24:163-181.
8. Kusunoki S, Shiina M, Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. 2001;57:736-738.
9. Arikan S, Ergüven S, Ustaçelebi S, et al. Detection of antinuclear antibody (ANA) in patients with Mycoplasmal Pneumonia. Turk J Med Sci. 1998;28:97-98.
1. Wu CC, Kuo HC, Yu HR, et al. Association of acute urticaria with Mycoplasma pneumoniae infection in hospitalized children. Ann Allergy Asthma Immunol. 2009;103:134-139.
2. Serodia-Myco II [package insert]. Tokyo, Japan: Fujirebo; 2013.
3. Murray HW, Masur H, Senterfit LB, et al. The protean manifestations of Mycoplasma pneumoniae infection in adults. Am J Med. 1975;58:229-242.
4. Kano Y, Mitsuyama Y, Hirahara K, et al. Mycoplasma pneumoniae infection-induced erythema nodosum, anaphylactoid purpura, and acute urticaria in 3 people in a single family. J Am Acad Dermatol. 2007;57(suppl 2):S33-S35.
5. Daxboeck F, Krause R, Wenisch C. Laboratory diagnosis of Mycoplasma pneumoniae infection. Clin Microbiol Infect. 2003;9:263-273.
6. Pongpreuksa S, Boochoo S, Kulthanan K, et al. Chronic urticaria: what is worth doing in pediatric population? J Allergy Clin Immunol. 2004:S134.
7. Grattan CE. Autoimmune urticaria. Immunol Allergy Clin North Am. 2004;24:163-181.
8. Kusunoki S, Shiina M, Kanazawa I. Anti-Gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. 2001;57:736-738.
9. Arikan S, Ergüven S, Ustaçelebi S, et al. Detection of antinuclear antibody (ANA) in patients with Mycoplasmal Pneumonia. Turk J Med Sci. 1998;28:97-98.
Lupus-like Rash of Chronic Granulomatous Disease Effectively Treated With Hydroxychloroquine
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
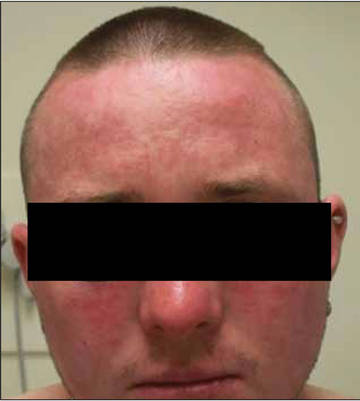
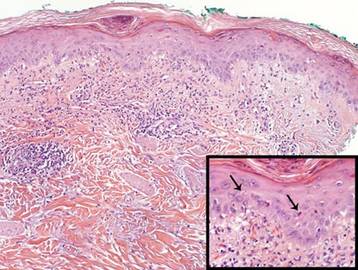
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
Shiitake Mushroom Dermatitis
To the Editor:
The shiitake mushroom (Lentinula edodes) is a popular Asian food and represents the second most consumed mushroom in the world. It is known for having a range of strong health benefits including antihypertensive, anti-inflammatory, and immunomodulatory effects. Especially in Asia, this mushroom has been used in patients with cancers of the gastrointestinal tract and also may be helpful in the treatment of human immunodeficiency virus.1,2 The source of these effects is lentinan, a polysaccharide in the mushroom. However, lentinan also can cause a toxic reaction of the skin when the mushrooms are eaten raw or undercooked. These reactions are mainly reported in Asia, but more cases have been published in the last decade in Europe and the United States, evidence that the incidence of this adverse effect has increased in the Western world.
A 65-year-old woman with no notable medical history presented to our outpatient practice with sudden onset of a pruritic, erythematous, papular eruption on the neck. The eruption began that morning. The diagnosis of eczematous dermatitis was made and hydrocortisone cream 2.5% was started. Three days later, she returned with spread of the rash to the trunk, arms, and legs despite the topical treatment. She denied fevers, chills, or constitutional symptoms. The patient also denied recent travel or bug bites. However, she reported that she recently had started using raw shiitake mushrooms in her salad; the first time was 3 days before the symptoms appeared. Physical examination revealed erythematous skin with long flagellate streaks composed of petechiae, papules, and vesicles involving the trunk, arms, and legs (Figure). Oral and nasal mucosae were uninvolved. Dermatographism was negative. The diagnosis of flagellate dermatitis from shiitake mushrooms was made given the patient’s history and the unique clinical findings of the skin. Blood work and a biopsy were not performed. Instead, the patient was advised to avoid shiitake mushrooms and use clobetasol propionate cream 0.05% twice daily for 2 weeks on the affected areas. The symptoms resolved within 10 days.
|

The first known case of toxicoderma to shiitake mushrooms was reported in Japan by Nakamura3 in 1977. Since this seminal report, numerous cases have followed. This disorder is mainly seen in Asia.
Patients usually present with linear groups of pruritic, papular, petechial, and vesicular lesions in a flagellate pattern, most commonly localized on the trunk, arms, and legs. Oral and nasal mucosae usually are not involved, and fever and malaise may be associated. All symptoms typically occur 1 to 2 days after ingestion of the mushrooms. The patient’s history and typical clinical findings lead to a diagnosis; however, blood tests may show inflammation with leukocytosis and elevated C-reactive protein levels. Biopsy of the skin shows lymphocytic dermal infiltrates with spongiosis and necrotic cells within the epidermis.4
Differential diagnoses include flagellate dermatitis associated with bleomycin, a glycopeptide antibiotic produced by the bacterium Streptomyces verticillus. Because it causes breaks in the DNA, bleomycin is commonly used as a chemotherapeutic agent in treating Hodgkin lymphoma and other malignancies. It presents with linear postinflammatory hyperpigmentation of the skin. However, unlike shiitake dermatitis, there is a lack of papules. Another differential diagnosis includes herpes zoster virus, which should be ruled out clinically.
All symptoms in shiitake dermatitis usually resolve within 1 to 8 weeks of avoidance of the culprit food. Topical steroids and antihistamines can be given.
The underlying pathology is a toxic reaction to the polysaccharide lentinan in the mushrooms, which is known as a thermolabile agent.5 Therefore, it may only cause a toxic reaction when the mushrooms are consumed raw or undercooked. Prick testing is usually negative in these patients, which suggests a toxic and not an immunologic reaction of the human body.6 Other forms of reaction to shiitake mushrooms include contact dermatitis after skin contact and allergic alveolitis after occupational exposure to mushroom spores, mainly in individuals cultivating shiitake mushrooms (mushroom worker’s lung). In these forms of the disease, prick testing may be positive.7,8
Flagellate dermatitis caused by shiitake mushrooms is still an uncommon dermatologic phenomenon in the Western world. Future studies and cases should be reported to increase the awareness of this disorder. Although the patients present with typical clinical findings, the diagnosis can be missed if history is not carefully considered.
1. Ng ML, Yap AT. Inhibition of human colon carcinoma development by lentinan from shiitake mushrooms (Lentinus edodes). J Altern Complement Med. 2002;8:581-589.
2. Gordon M, Bihari B, Goosby E, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29:305-330.
3. Nakamura T. Toxicoderma caused by shiitake. Jpn J Clin Dermatol. 1977;31:65-68.
4. Hanada K, Hashimoto I. Flagellate mushroom (shiitake) dermatitis and photosensitivity. Dermatology. 1998;197:255-257.
5. Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
6. Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:6570.
7. Ueda A, Obama K, Aoyama K, et al. Allergic contact dermatitis in shiitake (Lentinus edodes (Berk) Sing) growers. Contact Dermatitis. 1992;26:228-233.
8. Ampere A, Delhaes L, Soots J, et al. Hypersensitivity pneumonitis induced by shiitake mushroom spores. Med Mycol. 2012;50:654-657.
To the Editor:
The shiitake mushroom (Lentinula edodes) is a popular Asian food and represents the second most consumed mushroom in the world. It is known for having a range of strong health benefits including antihypertensive, anti-inflammatory, and immunomodulatory effects. Especially in Asia, this mushroom has been used in patients with cancers of the gastrointestinal tract and also may be helpful in the treatment of human immunodeficiency virus.1,2 The source of these effects is lentinan, a polysaccharide in the mushroom. However, lentinan also can cause a toxic reaction of the skin when the mushrooms are eaten raw or undercooked. These reactions are mainly reported in Asia, but more cases have been published in the last decade in Europe and the United States, evidence that the incidence of this adverse effect has increased in the Western world.
A 65-year-old woman with no notable medical history presented to our outpatient practice with sudden onset of a pruritic, erythematous, papular eruption on the neck. The eruption began that morning. The diagnosis of eczematous dermatitis was made and hydrocortisone cream 2.5% was started. Three days later, she returned with spread of the rash to the trunk, arms, and legs despite the topical treatment. She denied fevers, chills, or constitutional symptoms. The patient also denied recent travel or bug bites. However, she reported that she recently had started using raw shiitake mushrooms in her salad; the first time was 3 days before the symptoms appeared. Physical examination revealed erythematous skin with long flagellate streaks composed of petechiae, papules, and vesicles involving the trunk, arms, and legs (Figure). Oral and nasal mucosae were uninvolved. Dermatographism was negative. The diagnosis of flagellate dermatitis from shiitake mushrooms was made given the patient’s history and the unique clinical findings of the skin. Blood work and a biopsy were not performed. Instead, the patient was advised to avoid shiitake mushrooms and use clobetasol propionate cream 0.05% twice daily for 2 weeks on the affected areas. The symptoms resolved within 10 days.
|
The first known case of toxicoderma to shiitake mushrooms was reported in Japan by Nakamura3 in 1977. Since this seminal report, numerous cases have followed. This disorder is mainly seen in Asia.
Patients usually present with linear groups of pruritic, papular, petechial, and vesicular lesions in a flagellate pattern, most commonly localized on the trunk, arms, and legs. Oral and nasal mucosae usually are not involved, and fever and malaise may be associated. All symptoms typically occur 1 to 2 days after ingestion of the mushrooms. The patient’s history and typical clinical findings lead to a diagnosis; however, blood tests may show inflammation with leukocytosis and elevated C-reactive protein levels. Biopsy of the skin shows lymphocytic dermal infiltrates with spongiosis and necrotic cells within the epidermis.4
Differential diagnoses include flagellate dermatitis associated with bleomycin, a glycopeptide antibiotic produced by the bacterium Streptomyces verticillus. Because it causes breaks in the DNA, bleomycin is commonly used as a chemotherapeutic agent in treating Hodgkin lymphoma and other malignancies. It presents with linear postinflammatory hyperpigmentation of the skin. However, unlike shiitake dermatitis, there is a lack of papules. Another differential diagnosis includes herpes zoster virus, which should be ruled out clinically.
All symptoms in shiitake dermatitis usually resolve within 1 to 8 weeks of avoidance of the culprit food. Topical steroids and antihistamines can be given.
The underlying pathology is a toxic reaction to the polysaccharide lentinan in the mushrooms, which is known as a thermolabile agent.5 Therefore, it may only cause a toxic reaction when the mushrooms are consumed raw or undercooked. Prick testing is usually negative in these patients, which suggests a toxic and not an immunologic reaction of the human body.6 Other forms of reaction to shiitake mushrooms include contact dermatitis after skin contact and allergic alveolitis after occupational exposure to mushroom spores, mainly in individuals cultivating shiitake mushrooms (mushroom worker’s lung). In these forms of the disease, prick testing may be positive.7,8
Flagellate dermatitis caused by shiitake mushrooms is still an uncommon dermatologic phenomenon in the Western world. Future studies and cases should be reported to increase the awareness of this disorder. Although the patients present with typical clinical findings, the diagnosis can be missed if history is not carefully considered.
To the Editor:
The shiitake mushroom (Lentinula edodes) is a popular Asian food and represents the second most consumed mushroom in the world. It is known for having a range of strong health benefits including antihypertensive, anti-inflammatory, and immunomodulatory effects. Especially in Asia, this mushroom has been used in patients with cancers of the gastrointestinal tract and also may be helpful in the treatment of human immunodeficiency virus.1,2 The source of these effects is lentinan, a polysaccharide in the mushroom. However, lentinan also can cause a toxic reaction of the skin when the mushrooms are eaten raw or undercooked. These reactions are mainly reported in Asia, but more cases have been published in the last decade in Europe and the United States, evidence that the incidence of this adverse effect has increased in the Western world.
A 65-year-old woman with no notable medical history presented to our outpatient practice with sudden onset of a pruritic, erythematous, papular eruption on the neck. The eruption began that morning. The diagnosis of eczematous dermatitis was made and hydrocortisone cream 2.5% was started. Three days later, she returned with spread of the rash to the trunk, arms, and legs despite the topical treatment. She denied fevers, chills, or constitutional symptoms. The patient also denied recent travel or bug bites. However, she reported that she recently had started using raw shiitake mushrooms in her salad; the first time was 3 days before the symptoms appeared. Physical examination revealed erythematous skin with long flagellate streaks composed of petechiae, papules, and vesicles involving the trunk, arms, and legs (Figure). Oral and nasal mucosae were uninvolved. Dermatographism was negative. The diagnosis of flagellate dermatitis from shiitake mushrooms was made given the patient’s history and the unique clinical findings of the skin. Blood work and a biopsy were not performed. Instead, the patient was advised to avoid shiitake mushrooms and use clobetasol propionate cream 0.05% twice daily for 2 weeks on the affected areas. The symptoms resolved within 10 days.
|
The first known case of toxicoderma to shiitake mushrooms was reported in Japan by Nakamura3 in 1977. Since this seminal report, numerous cases have followed. This disorder is mainly seen in Asia.
Patients usually present with linear groups of pruritic, papular, petechial, and vesicular lesions in a flagellate pattern, most commonly localized on the trunk, arms, and legs. Oral and nasal mucosae usually are not involved, and fever and malaise may be associated. All symptoms typically occur 1 to 2 days after ingestion of the mushrooms. The patient’s history and typical clinical findings lead to a diagnosis; however, blood tests may show inflammation with leukocytosis and elevated C-reactive protein levels. Biopsy of the skin shows lymphocytic dermal infiltrates with spongiosis and necrotic cells within the epidermis.4
Differential diagnoses include flagellate dermatitis associated with bleomycin, a glycopeptide antibiotic produced by the bacterium Streptomyces verticillus. Because it causes breaks in the DNA, bleomycin is commonly used as a chemotherapeutic agent in treating Hodgkin lymphoma and other malignancies. It presents with linear postinflammatory hyperpigmentation of the skin. However, unlike shiitake dermatitis, there is a lack of papules. Another differential diagnosis includes herpes zoster virus, which should be ruled out clinically.
All symptoms in shiitake dermatitis usually resolve within 1 to 8 weeks of avoidance of the culprit food. Topical steroids and antihistamines can be given.
The underlying pathology is a toxic reaction to the polysaccharide lentinan in the mushrooms, which is known as a thermolabile agent.5 Therefore, it may only cause a toxic reaction when the mushrooms are consumed raw or undercooked. Prick testing is usually negative in these patients, which suggests a toxic and not an immunologic reaction of the human body.6 Other forms of reaction to shiitake mushrooms include contact dermatitis after skin contact and allergic alveolitis after occupational exposure to mushroom spores, mainly in individuals cultivating shiitake mushrooms (mushroom worker’s lung). In these forms of the disease, prick testing may be positive.7,8
Flagellate dermatitis caused by shiitake mushrooms is still an uncommon dermatologic phenomenon in the Western world. Future studies and cases should be reported to increase the awareness of this disorder. Although the patients present with typical clinical findings, the diagnosis can be missed if history is not carefully considered.
1. Ng ML, Yap AT. Inhibition of human colon carcinoma development by lentinan from shiitake mushrooms (Lentinus edodes). J Altern Complement Med. 2002;8:581-589.
2. Gordon M, Bihari B, Goosby E, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29:305-330.
3. Nakamura T. Toxicoderma caused by shiitake. Jpn J Clin Dermatol. 1977;31:65-68.
4. Hanada K, Hashimoto I. Flagellate mushroom (shiitake) dermatitis and photosensitivity. Dermatology. 1998;197:255-257.
5. Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
6. Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:6570.
7. Ueda A, Obama K, Aoyama K, et al. Allergic contact dermatitis in shiitake (Lentinus edodes (Berk) Sing) growers. Contact Dermatitis. 1992;26:228-233.
8. Ampere A, Delhaes L, Soots J, et al. Hypersensitivity pneumonitis induced by shiitake mushroom spores. Med Mycol. 2012;50:654-657.
1. Ng ML, Yap AT. Inhibition of human colon carcinoma development by lentinan from shiitake mushrooms (Lentinus edodes). J Altern Complement Med. 2002;8:581-589.
2. Gordon M, Bihari B, Goosby E, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29:305-330.
3. Nakamura T. Toxicoderma caused by shiitake. Jpn J Clin Dermatol. 1977;31:65-68.
4. Hanada K, Hashimoto I. Flagellate mushroom (shiitake) dermatitis and photosensitivity. Dermatology. 1998;197:255-257.
5. Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
6. Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:6570.
7. Ueda A, Obama K, Aoyama K, et al. Allergic contact dermatitis in shiitake (Lentinus edodes (Berk) Sing) growers. Contact Dermatitis. 1992;26:228-233.
8. Ampere A, Delhaes L, Soots J, et al. Hypersensitivity pneumonitis induced by shiitake mushroom spores. Med Mycol. 2012;50:654-657.
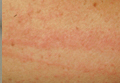
Marjolin Ulcer in a Surgical Scar
To the Editor:
Marjolin ulcers are malignancies arising in nonhealing cutaneous wounds. Although burn wounds are the most common type of cutaneous trauma associated with this entity, there are a multitude of possible lesions that may initiate this disease process including traumatic wounds, venous stasis ulcers, and vaccination sites.1,2 The most common type of malignancy reported in a Marjolin ulcer is an aggressive squamous cell carcinoma (SCC).1-3 Less commonly, basal cell carcinoma (BCC) also has been reported.1,3,4 However, cases of BCCs developing in surgical scars are exceedingly rare. We describe a case of a morphoeic BCC in a long-standing surgical scar in a 50-year-old woman with Crohn disease.
A 50-year-old woman presented with an intermittent ulceration within a horizontal surgical scar on the right side of the upper abdomen of 2 years’ duration that had not healed over the course of the last 6 months. The scar was present from surgeries conducted while she was a teenager for complications associated with Crohn disease. She underwent her first abdominal surgery for a partial gastric resection at 16 years of age, followed by multiple laparotomies from a perforated bile duct that occurred during the first surgery. The original incision created for the partial gastric resection was used for all subsequent surgeries.
The patient’s medical history was notable for central nervous system vasculitis with vision loss, chronic pancreatitis, Crohn disease, arthritis, multiple superficial BCCs on the back that were successfully treated with imiquimod cream, a nodular BCC on the neck that was surgically removed, and facial actinic keratoses treated with liquid nitrogen. She had Fitzpatrick skin type I. She grew up in a residential area in Southern Ontario and did not have a history of heavy sun exposure. She did not receive notable radiation from treatment of Crohn disease, and she usually wore a 1-piece bathing suit when swimming outdoors. According to the patient, she had never been exposed to arsenic. The patient’s family history was negative for skin cancer and she was a nonsmoker. She was taking methotrexate, prednisone, folic acid, pentazocine, and vitamin B12 injections at the time of presentation for the aforementioned conditions.
On physical examination a 1.5-cm honey-crusted ulcer with surrounding violaceous erythema in a long-standing surgical scar was observed (Figure 1). There was no palpable adenopathy in the inguinal or axillary regions. The suspected diagnosis prebiopsy was an SCC developing within the scar tissue. On histologic examination sections of small nests and strands of basal cell infiltrating thick collagen bundles were visualized. The appearance was consistent with a morphoeic BCC. The pathologist’s interpretation indicated that the lesion appeared to be a morphoeic BCC within the scar as opposed to a BCC that appeared morphoeic because of background scarring. Histologic images stained with hematoxylin and eosin showed small nests and strands of basal cells penetrating thick collagen bundles (Figure 2).

|
|
|

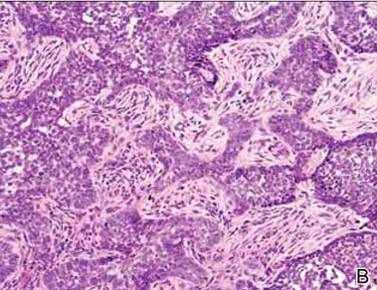
Marjolin ulcer was first described in 1828 by the French surgeon Jean-Nicolas Marjolin who published 4 cases of ulcers arising from scar tissue but did not appreciate their malignant capacity. However, the term Marjolin ulcer is now widely accepted as meaning any malignant tumor occurring within scar tissue or a chronic nonhealing wound.1,2
The exact incidence of malignant transformation in cutaneous wounds remains unknown, but this phenomenon can occur in individuals of all races and across all age groups.1,5-7 The most prevalent malignancy identified on biopsy is SCC, followed by BCC, melanoma, osteosarcoma, fibrosarcoma, and liposarcoma.1,2,6,7 With this entity infrequently occurring in the clinical setting, it often is overlooked or misdiagnosed.2 In addition, malignancies presenting as Marjolin ulcers have a greater tendency to metastasize and are reported to have a higher associated fatality rate.2,8 Thus, early recognition is essential, as a delay in diagnosis can potentially allow the tumor to progress to a life-threatening stage. In our patient, malignancy was clinically suspected based on the presence of an ulcer that was not healing despite adequate wound care and the location in a scar that was present for more than 30 years. The surgical scar had been a place of repeated trauma given the number of surgical procedures and the perforated bile duct, which can increase the potential for malignant transformation. Furthermore, the patient also was on immunosuppressive therapy for an extended period of time, possibly contributing to the development of this cancerous lesion and prior cutaneous malignancies.
The pathogenesis of a Marjolin ulcer is unclear, though many hypotheses have been suggested.1,2,6,9 Theories investigating decreased vascularity, lowered immune surveillance, decreased regenerative capacity, genetic mutations, and injury-related release of toxins have all been postulated as possible explanations for the increase in potential of malignant transformation.1-3,6,9 However, despite the pathogenesis, the mainstay of treatment remains wide local excision with at least 2-cm margins.1-3,10 Alternatively, Mohs micrographic surgery can be considered for Marjolin ulcers, but it is less frequently conducted in comparison to wide local excision. Radiation therapy often follows excision as adjuvant therapy, depending on the type of tumor.2,10 Prophylactic lymph node dissection is not indicated in most cases, but regional node dissection is suggested when palpable lymphadenopathy is present.1,2,10 Moreover, amputation is indicated with deep bone or joint involvement.1-3,10 Recurrence rates are high, ranging from 20% to 50%, and metastases to the brain, liver, lungs, kidneys, and lymph nodes have been reported.1,3 The prognosis of the cutaneous malignancy in this setting is not as favorable, and the 5-year survival rate is cited at approximately 60%.3 Overall prognosis depends on several factors including location, type of malignancy, immune status, progression of disease, and lymph node metastasis. Our patient’s presentation with a BCC instead of the more common SCC should carry a good overall prognosis, though she will need to be closely followed for recurrence after wide local excision.
This novel presentation of a morpheaform BCC in a surgical scar may serve as a reminder to consider this diagnosis and biopsy nonhealing ulcers within any type of chronic wound or scar.
1. Daya M, Balakrishan T. Advanced Marjolin’s ulcer of the scalp in a 13-year-old boy treated by excision and free tissue transfer: case report and review of literature. Indian J Plast Surg. 2009;42:106-111.
2. Pavlovic S, Wiley E, Guzman G, et al. Marjolin ulcer: an overlooked entity [published online ahead of print May 17, 2011]. Int Wound J. 2011;8:419-424.
3. Asuquo M, Ugare G, Ebughe G, et al. Marjolin’s ulcer: the importance of surgical management of chronic cutaneous ulcers. Int J Dermatol. 2007;46(suppl 2):S29-S32.
4. Ogawa B, Chen M, Margolis J, et al. Marjolin’s ulcer arising at the elbow: a case report and literature review. Hand (N Y). 2006;1:89-93.
5. Dupree MT, Boyer JD, Cobb MW. Marjolin’s ulcer arising in a burn scar. Cutis. 1998;62:49-51.
6. Er-Fan X, Li AO, Shi-ling W, et al. Burn scar carcinoma: case reports and review of the literature. Ann MBC. 1992;5:2.
7. Malheiro E, Pinto A, Choupina M, et al. Marjolin’s ulcer of the scalp: case report and literature review. Ann Burns and Fire Disasters. 2001;14:115-118.
8. Ozek C, Celik N, Bilkay U, et al. Marjolin’s ulcer of the scalp: report of 5 cases and review of the literature. J Burn Care Rehabil. 2001;22:65-69.
9. Thio D, Clarkson JH, Misra A, et al. Malignant change after 18 months in a lower limb ulcer: acute Marjolin’s revisited. Br J Plast Surg. 2003;56:825-828.
10. Aydogdu E, Yildirim S, Aköz T. Is surgery an effective and adequate treatmegnt in advanced Marjolin’s ulcer [published online ahead of print April 1, 2005]? Burns. 2005;31:421-431.
To the Editor:
Marjolin ulcers are malignancies arising in nonhealing cutaneous wounds. Although burn wounds are the most common type of cutaneous trauma associated with this entity, there are a multitude of possible lesions that may initiate this disease process including traumatic wounds, venous stasis ulcers, and vaccination sites.1,2 The most common type of malignancy reported in a Marjolin ulcer is an aggressive squamous cell carcinoma (SCC).1-3 Less commonly, basal cell carcinoma (BCC) also has been reported.1,3,4 However, cases of BCCs developing in surgical scars are exceedingly rare. We describe a case of a morphoeic BCC in a long-standing surgical scar in a 50-year-old woman with Crohn disease.
A 50-year-old woman presented with an intermittent ulceration within a horizontal surgical scar on the right side of the upper abdomen of 2 years’ duration that had not healed over the course of the last 6 months. The scar was present from surgeries conducted while she was a teenager for complications associated with Crohn disease. She underwent her first abdominal surgery for a partial gastric resection at 16 years of age, followed by multiple laparotomies from a perforated bile duct that occurred during the first surgery. The original incision created for the partial gastric resection was used for all subsequent surgeries.
The patient’s medical history was notable for central nervous system vasculitis with vision loss, chronic pancreatitis, Crohn disease, arthritis, multiple superficial BCCs on the back that were successfully treated with imiquimod cream, a nodular BCC on the neck that was surgically removed, and facial actinic keratoses treated with liquid nitrogen. She had Fitzpatrick skin type I. She grew up in a residential area in Southern Ontario and did not have a history of heavy sun exposure. She did not receive notable radiation from treatment of Crohn disease, and she usually wore a 1-piece bathing suit when swimming outdoors. According to the patient, she had never been exposed to arsenic. The patient’s family history was negative for skin cancer and she was a nonsmoker. She was taking methotrexate, prednisone, folic acid, pentazocine, and vitamin B12 injections at the time of presentation for the aforementioned conditions.
On physical examination a 1.5-cm honey-crusted ulcer with surrounding violaceous erythema in a long-standing surgical scar was observed (Figure 1). There was no palpable adenopathy in the inguinal or axillary regions. The suspected diagnosis prebiopsy was an SCC developing within the scar tissue. On histologic examination sections of small nests and strands of basal cell infiltrating thick collagen bundles were visualized. The appearance was consistent with a morphoeic BCC. The pathologist’s interpretation indicated that the lesion appeared to be a morphoeic BCC within the scar as opposed to a BCC that appeared morphoeic because of background scarring. Histologic images stained with hematoxylin and eosin showed small nests and strands of basal cells penetrating thick collagen bundles (Figure 2).
|
|
|
|
Marjolin ulcer was first described in 1828 by the French surgeon Jean-Nicolas Marjolin who published 4 cases of ulcers arising from scar tissue but did not appreciate their malignant capacity. However, the term Marjolin ulcer is now widely accepted as meaning any malignant tumor occurring within scar tissue or a chronic nonhealing wound.1,2
The exact incidence of malignant transformation in cutaneous wounds remains unknown, but this phenomenon can occur in individuals of all races and across all age groups.1,5-7 The most prevalent malignancy identified on biopsy is SCC, followed by BCC, melanoma, osteosarcoma, fibrosarcoma, and liposarcoma.1,2,6,7 With this entity infrequently occurring in the clinical setting, it often is overlooked or misdiagnosed.2 In addition, malignancies presenting as Marjolin ulcers have a greater tendency to metastasize and are reported to have a higher associated fatality rate.2,8 Thus, early recognition is essential, as a delay in diagnosis can potentially allow the tumor to progress to a life-threatening stage. In our patient, malignancy was clinically suspected based on the presence of an ulcer that was not healing despite adequate wound care and the location in a scar that was present for more than 30 years. The surgical scar had been a place of repeated trauma given the number of surgical procedures and the perforated bile duct, which can increase the potential for malignant transformation. Furthermore, the patient also was on immunosuppressive therapy for an extended period of time, possibly contributing to the development of this cancerous lesion and prior cutaneous malignancies.
The pathogenesis of a Marjolin ulcer is unclear, though many hypotheses have been suggested.1,2,6,9 Theories investigating decreased vascularity, lowered immune surveillance, decreased regenerative capacity, genetic mutations, and injury-related release of toxins have all been postulated as possible explanations for the increase in potential of malignant transformation.1-3,6,9 However, despite the pathogenesis, the mainstay of treatment remains wide local excision with at least 2-cm margins.1-3,10 Alternatively, Mohs micrographic surgery can be considered for Marjolin ulcers, but it is less frequently conducted in comparison to wide local excision. Radiation therapy often follows excision as adjuvant therapy, depending on the type of tumor.2,10 Prophylactic lymph node dissection is not indicated in most cases, but regional node dissection is suggested when palpable lymphadenopathy is present.1,2,10 Moreover, amputation is indicated with deep bone or joint involvement.1-3,10 Recurrence rates are high, ranging from 20% to 50%, and metastases to the brain, liver, lungs, kidneys, and lymph nodes have been reported.1,3 The prognosis of the cutaneous malignancy in this setting is not as favorable, and the 5-year survival rate is cited at approximately 60%.3 Overall prognosis depends on several factors including location, type of malignancy, immune status, progression of disease, and lymph node metastasis. Our patient’s presentation with a BCC instead of the more common SCC should carry a good overall prognosis, though she will need to be closely followed for recurrence after wide local excision.
This novel presentation of a morpheaform BCC in a surgical scar may serve as a reminder to consider this diagnosis and biopsy nonhealing ulcers within any type of chronic wound or scar.
To the Editor:
Marjolin ulcers are malignancies arising in nonhealing cutaneous wounds. Although burn wounds are the most common type of cutaneous trauma associated with this entity, there are a multitude of possible lesions that may initiate this disease process including traumatic wounds, venous stasis ulcers, and vaccination sites.1,2 The most common type of malignancy reported in a Marjolin ulcer is an aggressive squamous cell carcinoma (SCC).1-3 Less commonly, basal cell carcinoma (BCC) also has been reported.1,3,4 However, cases of BCCs developing in surgical scars are exceedingly rare. We describe a case of a morphoeic BCC in a long-standing surgical scar in a 50-year-old woman with Crohn disease.
A 50-year-old woman presented with an intermittent ulceration within a horizontal surgical scar on the right side of the upper abdomen of 2 years’ duration that had not healed over the course of the last 6 months. The scar was present from surgeries conducted while she was a teenager for complications associated with Crohn disease. She underwent her first abdominal surgery for a partial gastric resection at 16 years of age, followed by multiple laparotomies from a perforated bile duct that occurred during the first surgery. The original incision created for the partial gastric resection was used for all subsequent surgeries.
The patient’s medical history was notable for central nervous system vasculitis with vision loss, chronic pancreatitis, Crohn disease, arthritis, multiple superficial BCCs on the back that were successfully treated with imiquimod cream, a nodular BCC on the neck that was surgically removed, and facial actinic keratoses treated with liquid nitrogen. She had Fitzpatrick skin type I. She grew up in a residential area in Southern Ontario and did not have a history of heavy sun exposure. She did not receive notable radiation from treatment of Crohn disease, and she usually wore a 1-piece bathing suit when swimming outdoors. According to the patient, she had never been exposed to arsenic. The patient’s family history was negative for skin cancer and she was a nonsmoker. She was taking methotrexate, prednisone, folic acid, pentazocine, and vitamin B12 injections at the time of presentation for the aforementioned conditions.
On physical examination a 1.5-cm honey-crusted ulcer with surrounding violaceous erythema in a long-standing surgical scar was observed (Figure 1). There was no palpable adenopathy in the inguinal or axillary regions. The suspected diagnosis prebiopsy was an SCC developing within the scar tissue. On histologic examination sections of small nests and strands of basal cell infiltrating thick collagen bundles were visualized. The appearance was consistent with a morphoeic BCC. The pathologist’s interpretation indicated that the lesion appeared to be a morphoeic BCC within the scar as opposed to a BCC that appeared morphoeic because of background scarring. Histologic images stained with hematoxylin and eosin showed small nests and strands of basal cells penetrating thick collagen bundles (Figure 2).
|
|
|
|
Marjolin ulcer was first described in 1828 by the French surgeon Jean-Nicolas Marjolin who published 4 cases of ulcers arising from scar tissue but did not appreciate their malignant capacity. However, the term Marjolin ulcer is now widely accepted as meaning any malignant tumor occurring within scar tissue or a chronic nonhealing wound.1,2
The exact incidence of malignant transformation in cutaneous wounds remains unknown, but this phenomenon can occur in individuals of all races and across all age groups.1,5-7 The most prevalent malignancy identified on biopsy is SCC, followed by BCC, melanoma, osteosarcoma, fibrosarcoma, and liposarcoma.1,2,6,7 With this entity infrequently occurring in the clinical setting, it often is overlooked or misdiagnosed.2 In addition, malignancies presenting as Marjolin ulcers have a greater tendency to metastasize and are reported to have a higher associated fatality rate.2,8 Thus, early recognition is essential, as a delay in diagnosis can potentially allow the tumor to progress to a life-threatening stage. In our patient, malignancy was clinically suspected based on the presence of an ulcer that was not healing despite adequate wound care and the location in a scar that was present for more than 30 years. The surgical scar had been a place of repeated trauma given the number of surgical procedures and the perforated bile duct, which can increase the potential for malignant transformation. Furthermore, the patient also was on immunosuppressive therapy for an extended period of time, possibly contributing to the development of this cancerous lesion and prior cutaneous malignancies.
The pathogenesis of a Marjolin ulcer is unclear, though many hypotheses have been suggested.1,2,6,9 Theories investigating decreased vascularity, lowered immune surveillance, decreased regenerative capacity, genetic mutations, and injury-related release of toxins have all been postulated as possible explanations for the increase in potential of malignant transformation.1-3,6,9 However, despite the pathogenesis, the mainstay of treatment remains wide local excision with at least 2-cm margins.1-3,10 Alternatively, Mohs micrographic surgery can be considered for Marjolin ulcers, but it is less frequently conducted in comparison to wide local excision. Radiation therapy often follows excision as adjuvant therapy, depending on the type of tumor.2,10 Prophylactic lymph node dissection is not indicated in most cases, but regional node dissection is suggested when palpable lymphadenopathy is present.1,2,10 Moreover, amputation is indicated with deep bone or joint involvement.1-3,10 Recurrence rates are high, ranging from 20% to 50%, and metastases to the brain, liver, lungs, kidneys, and lymph nodes have been reported.1,3 The prognosis of the cutaneous malignancy in this setting is not as favorable, and the 5-year survival rate is cited at approximately 60%.3 Overall prognosis depends on several factors including location, type of malignancy, immune status, progression of disease, and lymph node metastasis. Our patient’s presentation with a BCC instead of the more common SCC should carry a good overall prognosis, though she will need to be closely followed for recurrence after wide local excision.
This novel presentation of a morpheaform BCC in a surgical scar may serve as a reminder to consider this diagnosis and biopsy nonhealing ulcers within any type of chronic wound or scar.
1. Daya M, Balakrishan T. Advanced Marjolin’s ulcer of the scalp in a 13-year-old boy treated by excision and free tissue transfer: case report and review of literature. Indian J Plast Surg. 2009;42:106-111.
2. Pavlovic S, Wiley E, Guzman G, et al. Marjolin ulcer: an overlooked entity [published online ahead of print May 17, 2011]. Int Wound J. 2011;8:419-424.
3. Asuquo M, Ugare G, Ebughe G, et al. Marjolin’s ulcer: the importance of surgical management of chronic cutaneous ulcers. Int J Dermatol. 2007;46(suppl 2):S29-S32.
4. Ogawa B, Chen M, Margolis J, et al. Marjolin’s ulcer arising at the elbow: a case report and literature review. Hand (N Y). 2006;1:89-93.
5. Dupree MT, Boyer JD, Cobb MW. Marjolin’s ulcer arising in a burn scar. Cutis. 1998;62:49-51.
6. Er-Fan X, Li AO, Shi-ling W, et al. Burn scar carcinoma: case reports and review of the literature. Ann MBC. 1992;5:2.
7. Malheiro E, Pinto A, Choupina M, et al. Marjolin’s ulcer of the scalp: case report and literature review. Ann Burns and Fire Disasters. 2001;14:115-118.
8. Ozek C, Celik N, Bilkay U, et al. Marjolin’s ulcer of the scalp: report of 5 cases and review of the literature. J Burn Care Rehabil. 2001;22:65-69.
9. Thio D, Clarkson JH, Misra A, et al. Malignant change after 18 months in a lower limb ulcer: acute Marjolin’s revisited. Br J Plast Surg. 2003;56:825-828.
10. Aydogdu E, Yildirim S, Aköz T. Is surgery an effective and adequate treatmegnt in advanced Marjolin’s ulcer [published online ahead of print April 1, 2005]? Burns. 2005;31:421-431.
1. Daya M, Balakrishan T. Advanced Marjolin’s ulcer of the scalp in a 13-year-old boy treated by excision and free tissue transfer: case report and review of literature. Indian J Plast Surg. 2009;42:106-111.
2. Pavlovic S, Wiley E, Guzman G, et al. Marjolin ulcer: an overlooked entity [published online ahead of print May 17, 2011]. Int Wound J. 2011;8:419-424.
3. Asuquo M, Ugare G, Ebughe G, et al. Marjolin’s ulcer: the importance of surgical management of chronic cutaneous ulcers. Int J Dermatol. 2007;46(suppl 2):S29-S32.
4. Ogawa B, Chen M, Margolis J, et al. Marjolin’s ulcer arising at the elbow: a case report and literature review. Hand (N Y). 2006;1:89-93.
5. Dupree MT, Boyer JD, Cobb MW. Marjolin’s ulcer arising in a burn scar. Cutis. 1998;62:49-51.
6. Er-Fan X, Li AO, Shi-ling W, et al. Burn scar carcinoma: case reports and review of the literature. Ann MBC. 1992;5:2.
7. Malheiro E, Pinto A, Choupina M, et al. Marjolin’s ulcer of the scalp: case report and literature review. Ann Burns and Fire Disasters. 2001;14:115-118.
8. Ozek C, Celik N, Bilkay U, et al. Marjolin’s ulcer of the scalp: report of 5 cases and review of the literature. J Burn Care Rehabil. 2001;22:65-69.
9. Thio D, Clarkson JH, Misra A, et al. Malignant change after 18 months in a lower limb ulcer: acute Marjolin’s revisited. Br J Plast Surg. 2003;56:825-828.
10. Aydogdu E, Yildirim S, Aköz T. Is surgery an effective and adequate treatmegnt in advanced Marjolin’s ulcer [published online ahead of print April 1, 2005]? Burns. 2005;31:421-431.
Sports Purpura From Floorball, Indoor Climbing, and Archery
To the Editor:
Sports purpura can be broken down into different types including traumatic purpura,1 exercise-induced cutaneous vasculitis,2 occurrence of coincidental systemic purpura,3 and other conditions.4-6 Traumatic purpura results from brutal contact with an opponent, the court, the equipment, or the ball. Three cases of sports purpura related to equipment and balls are reported.
An otherwise healthy 27-year-old woman presented with multiple ecchymotic round patches on her legs. The largest patch was 70 mm and displayed a heterogeneous Swiss cheese–like pattern with discrete whiter round areas within the patch (Figure 1). She reported that she played as a defender in a second division floorball team weekly, acknowledging frequent body contacts and being hit on the legs with the sticks and balls. Purpura was diagnosed due to hits from the floorball.
A 32-year-old healthy man presented with purpuric petechiae of the left palm after indoor climbing. He had been regularly climbing indoors for 3 years and denied a history of similar eruptions. The lesions were painless, noninfiltrated, and did not disappear after pressure (Figure 2). Lesions presumably were due to repeated friction on the climbing hold. Petechiae took a transiently golden hue before resolving within a week.
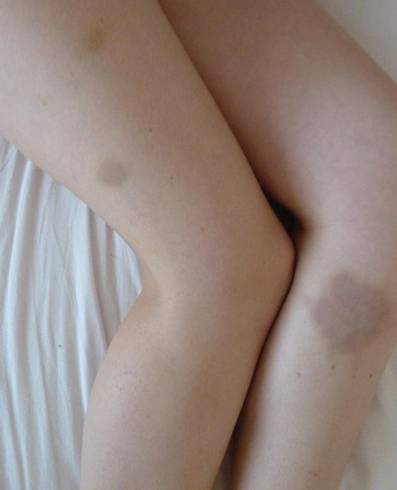
| 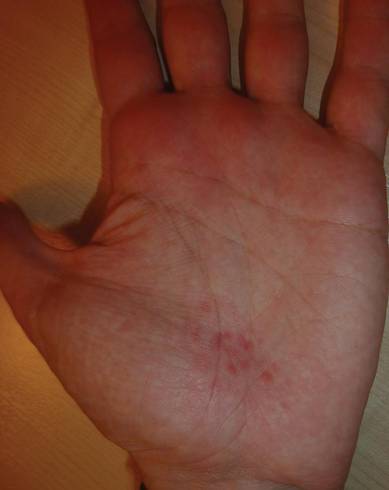
|
A 26-year-old right-handed woman injured the left forearm while practicing target archery. She was not wearing an arm guard at the time of the injury. Once released, the bowstring scraped the volar aspect of the forearm, causing a painful warm ecchymotic and swollen plaque. She denied neurologic or vascular symptoms. The hematoma rapidly evolved from red to blue (Figure 3) and spontaneously resolved within weeks.
|
|

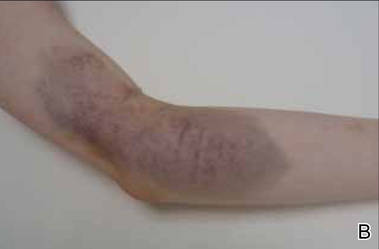
Purpura related to the high-velocity impact of sport balls has been previously reported with ping-pong,7 paintball,8,9 racquetball, squash,10 and baseball. Floorball, one of the most popular team sports in Finland, is played indoors and resembles ice hockey. The players use graphite compound sticks and a light hollow plastic ball. Except for the goalkeeper, players do not wear specific protective gear. Accidental body contact, including a direct hit from the floorball stick or ball, are frequent.11 The ball weighs 23 g, measures 72 mm in diameter, and has 26 holes that are 11 mm in diameter. The fastest shot was recorded at 127 miles per hour.12 The cutaneous imprint from the ball impact on bare skin, as shown with patient 1, initially is annular,8-10 but the bruise later takes an unusual design due to the peculiar shape of the ball. This complication is no stranger to floorball players but has been rarely reported. The diagnosis is easy, the condition is benign and asymptomatic, and it resolves when the season is over; therefore, players commonly will not seek medical attention. Of note, lower limb injuries, including joint sprains, muscle strains, and soft-tissue contusions, are frequent in female athletes.11 Additional causes of purpura include collision with another player or with boards and stick hits.
Palmar petechiae from indoor climbing is similar to black palm from weight lifting.13 Although the typical black discoloration is absent, the mechanisms of friction and brutal trauma, clinical presentation, and evolution are similar.
Lastly, archery-induced hematomas are caused by the absence of an arm guard, which protects the wrist and forearm when the string snaps back.14 This complication is not often reported but is known by archers. Because archers usually wear protective gear, these injuries are expected to occur in novices or when safety measures are not respected.
1. Aguayo-Leiva I, Vano-Galvan S, Arrazola JM. A purpuric rash. Aust Fam Physician. 2009;38:889-890.
2. Ramelet AA. Exercise-induced vasculitis. J Eur Acad Dermatol Venereol. 2006;20:423-427.
3. Leonard JC, Rieger M. Idiopathic thrombocytopenic purpura presenting in a high school football player: a case report. J Athl Train. 1998;33:269-270.
4. Nordlind K, Bondesson L, Johansson SG, et al. Purpura provoked by cold exposure in a skier. Dermatologica. 1983;167:101-103.
5. Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
6. Allan SJ, Humphreys F, Buxton PK. Annular purpura and step aerobics. Clin Exp Dermatol. 1994;19:418.
7. Scott MJ Jr, Scott MJ 3rd. Ping pong patches. Cutis. 1989;43:363-364.
8. Aboutalebi S, Stetson CL. Paintball purpura. J Am Acad Dermatol. 2005;53:901-902.
9. Levsky ME, Crowe M. What is your diagnosis? paintball purpura. Cutis. 2005;75:148, 157-158.
10. Barazi H, Adams BB. Sports purpura. Int J Dermatol. 2006;45:1443.
11. Pasanen K, Parkkari J, Kannus P, et al. Injury risk in female floorball: a prospective one-season follow-up [published online ahead of print May 9, 2007]. Scand J Med Sci Sports. 2008;18:49-54.
12. New world record. Floorball Central Web site. http://www.floorballcentral.com/2010/11/new-world -record.html. Published November 5, 2010. Accessed April 8, 2015.
13. Izumi AK. Letter: pigmented palmar petechiae (black palm). Arch Dermatol. 1974;109:261.
14. Rayan GM. Archery-related injuries of the hand, forearm, and elbow. South Med J. 1992;85:961-964.
To the Editor:
Sports purpura can be broken down into different types including traumatic purpura,1 exercise-induced cutaneous vasculitis,2 occurrence of coincidental systemic purpura,3 and other conditions.4-6 Traumatic purpura results from brutal contact with an opponent, the court, the equipment, or the ball. Three cases of sports purpura related to equipment and balls are reported.
An otherwise healthy 27-year-old woman presented with multiple ecchymotic round patches on her legs. The largest patch was 70 mm and displayed a heterogeneous Swiss cheese–like pattern with discrete whiter round areas within the patch (Figure 1). She reported that she played as a defender in a second division floorball team weekly, acknowledging frequent body contacts and being hit on the legs with the sticks and balls. Purpura was diagnosed due to hits from the floorball.
A 32-year-old healthy man presented with purpuric petechiae of the left palm after indoor climbing. He had been regularly climbing indoors for 3 years and denied a history of similar eruptions. The lesions were painless, noninfiltrated, and did not disappear after pressure (Figure 2). Lesions presumably were due to repeated friction on the climbing hold. Petechiae took a transiently golden hue before resolving within a week.
|
|
|
A 26-year-old right-handed woman injured the left forearm while practicing target archery. She was not wearing an arm guard at the time of the injury. Once released, the bowstring scraped the volar aspect of the forearm, causing a painful warm ecchymotic and swollen plaque. She denied neurologic or vascular symptoms. The hematoma rapidly evolved from red to blue (Figure 3) and spontaneously resolved within weeks.
|
|
Purpura related to the high-velocity impact of sport balls has been previously reported with ping-pong,7 paintball,8,9 racquetball, squash,10 and baseball. Floorball, one of the most popular team sports in Finland, is played indoors and resembles ice hockey. The players use graphite compound sticks and a light hollow plastic ball. Except for the goalkeeper, players do not wear specific protective gear. Accidental body contact, including a direct hit from the floorball stick or ball, are frequent.11 The ball weighs 23 g, measures 72 mm in diameter, and has 26 holes that are 11 mm in diameter. The fastest shot was recorded at 127 miles per hour.12 The cutaneous imprint from the ball impact on bare skin, as shown with patient 1, initially is annular,8-10 but the bruise later takes an unusual design due to the peculiar shape of the ball. This complication is no stranger to floorball players but has been rarely reported. The diagnosis is easy, the condition is benign and asymptomatic, and it resolves when the season is over; therefore, players commonly will not seek medical attention. Of note, lower limb injuries, including joint sprains, muscle strains, and soft-tissue contusions, are frequent in female athletes.11 Additional causes of purpura include collision with another player or with boards and stick hits.
Palmar petechiae from indoor climbing is similar to black palm from weight lifting.13 Although the typical black discoloration is absent, the mechanisms of friction and brutal trauma, clinical presentation, and evolution are similar.
Lastly, archery-induced hematomas are caused by the absence of an arm guard, which protects the wrist and forearm when the string snaps back.14 This complication is not often reported but is known by archers. Because archers usually wear protective gear, these injuries are expected to occur in novices or when safety measures are not respected.
To the Editor:
Sports purpura can be broken down into different types including traumatic purpura,1 exercise-induced cutaneous vasculitis,2 occurrence of coincidental systemic purpura,3 and other conditions.4-6 Traumatic purpura results from brutal contact with an opponent, the court, the equipment, or the ball. Three cases of sports purpura related to equipment and balls are reported.
An otherwise healthy 27-year-old woman presented with multiple ecchymotic round patches on her legs. The largest patch was 70 mm and displayed a heterogeneous Swiss cheese–like pattern with discrete whiter round areas within the patch (Figure 1). She reported that she played as a defender in a second division floorball team weekly, acknowledging frequent body contacts and being hit on the legs with the sticks and balls. Purpura was diagnosed due to hits from the floorball.
A 32-year-old healthy man presented with purpuric petechiae of the left palm after indoor climbing. He had been regularly climbing indoors for 3 years and denied a history of similar eruptions. The lesions were painless, noninfiltrated, and did not disappear after pressure (Figure 2). Lesions presumably were due to repeated friction on the climbing hold. Petechiae took a transiently golden hue before resolving within a week.
|
|
|
A 26-year-old right-handed woman injured the left forearm while practicing target archery. She was not wearing an arm guard at the time of the injury. Once released, the bowstring scraped the volar aspect of the forearm, causing a painful warm ecchymotic and swollen plaque. She denied neurologic or vascular symptoms. The hematoma rapidly evolved from red to blue (Figure 3) and spontaneously resolved within weeks.
|
|
Purpura related to the high-velocity impact of sport balls has been previously reported with ping-pong,7 paintball,8,9 racquetball, squash,10 and baseball. Floorball, one of the most popular team sports in Finland, is played indoors and resembles ice hockey. The players use graphite compound sticks and a light hollow plastic ball. Except for the goalkeeper, players do not wear specific protective gear. Accidental body contact, including a direct hit from the floorball stick or ball, are frequent.11 The ball weighs 23 g, measures 72 mm in diameter, and has 26 holes that are 11 mm in diameter. The fastest shot was recorded at 127 miles per hour.12 The cutaneous imprint from the ball impact on bare skin, as shown with patient 1, initially is annular,8-10 but the bruise later takes an unusual design due to the peculiar shape of the ball. This complication is no stranger to floorball players but has been rarely reported. The diagnosis is easy, the condition is benign and asymptomatic, and it resolves when the season is over; therefore, players commonly will not seek medical attention. Of note, lower limb injuries, including joint sprains, muscle strains, and soft-tissue contusions, are frequent in female athletes.11 Additional causes of purpura include collision with another player or with boards and stick hits.
Palmar petechiae from indoor climbing is similar to black palm from weight lifting.13 Although the typical black discoloration is absent, the mechanisms of friction and brutal trauma, clinical presentation, and evolution are similar.
Lastly, archery-induced hematomas are caused by the absence of an arm guard, which protects the wrist and forearm when the string snaps back.14 This complication is not often reported but is known by archers. Because archers usually wear protective gear, these injuries are expected to occur in novices or when safety measures are not respected.
1. Aguayo-Leiva I, Vano-Galvan S, Arrazola JM. A purpuric rash. Aust Fam Physician. 2009;38:889-890.
2. Ramelet AA. Exercise-induced vasculitis. J Eur Acad Dermatol Venereol. 2006;20:423-427.
3. Leonard JC, Rieger M. Idiopathic thrombocytopenic purpura presenting in a high school football player: a case report. J Athl Train. 1998;33:269-270.
4. Nordlind K, Bondesson L, Johansson SG, et al. Purpura provoked by cold exposure in a skier. Dermatologica. 1983;167:101-103.
5. Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
6. Allan SJ, Humphreys F, Buxton PK. Annular purpura and step aerobics. Clin Exp Dermatol. 1994;19:418.
7. Scott MJ Jr, Scott MJ 3rd. Ping pong patches. Cutis. 1989;43:363-364.
8. Aboutalebi S, Stetson CL. Paintball purpura. J Am Acad Dermatol. 2005;53:901-902.
9. Levsky ME, Crowe M. What is your diagnosis? paintball purpura. Cutis. 2005;75:148, 157-158.
10. Barazi H, Adams BB. Sports purpura. Int J Dermatol. 2006;45:1443.
11. Pasanen K, Parkkari J, Kannus P, et al. Injury risk in female floorball: a prospective one-season follow-up [published online ahead of print May 9, 2007]. Scand J Med Sci Sports. 2008;18:49-54.
12. New world record. Floorball Central Web site. http://www.floorballcentral.com/2010/11/new-world -record.html. Published November 5, 2010. Accessed April 8, 2015.
13. Izumi AK. Letter: pigmented palmar petechiae (black palm). Arch Dermatol. 1974;109:261.
14. Rayan GM. Archery-related injuries of the hand, forearm, and elbow. South Med J. 1992;85:961-964.
1. Aguayo-Leiva I, Vano-Galvan S, Arrazola JM. A purpuric rash. Aust Fam Physician. 2009;38:889-890.
2. Ramelet AA. Exercise-induced vasculitis. J Eur Acad Dermatol Venereol. 2006;20:423-427.
3. Leonard JC, Rieger M. Idiopathic thrombocytopenic purpura presenting in a high school football player: a case report. J Athl Train. 1998;33:269-270.
4. Nordlind K, Bondesson L, Johansson SG, et al. Purpura provoked by cold exposure in a skier. Dermatologica. 1983;167:101-103.
5. Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
6. Allan SJ, Humphreys F, Buxton PK. Annular purpura and step aerobics. Clin Exp Dermatol. 1994;19:418.
7. Scott MJ Jr, Scott MJ 3rd. Ping pong patches. Cutis. 1989;43:363-364.
8. Aboutalebi S, Stetson CL. Paintball purpura. J Am Acad Dermatol. 2005;53:901-902.
9. Levsky ME, Crowe M. What is your diagnosis? paintball purpura. Cutis. 2005;75:148, 157-158.
10. Barazi H, Adams BB. Sports purpura. Int J Dermatol. 2006;45:1443.
11. Pasanen K, Parkkari J, Kannus P, et al. Injury risk in female floorball: a prospective one-season follow-up [published online ahead of print May 9, 2007]. Scand J Med Sci Sports. 2008;18:49-54.
12. New world record. Floorball Central Web site. http://www.floorballcentral.com/2010/11/new-world -record.html. Published November 5, 2010. Accessed April 8, 2015.
13. Izumi AK. Letter: pigmented palmar petechiae (black palm). Arch Dermatol. 1974;109:261.
14. Rayan GM. Archery-related injuries of the hand, forearm, and elbow. South Med J. 1992;85:961-964.
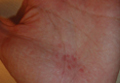
In Vivo Confocal Microscopy in the Diagnosis of Onychomycosis
To the Editor:
Onychomycosis is a common nail disease that frequently is caused by dermatophytes and is diagnosed by direct microscopy. Conventional diagnostic methods are often time consuming and can produce false-positive or false-negative results. We report a case of onychomycosis diagnosed by confocal microscopy and confirmed with routine potassium hydroxide (KOH) examination and fungal culture. Confocal microscopy is a reliable, practical, and noninvasive technique in the diagnosis of onychomycosis.
A 46-year-old woman presented with yellow-brown discoloration and dystrophy of the toenails (Figure 1) that had become worse over a 5-year period. She was otherwise healthy and had no other dermatologic problems. Examination revealed yellow-brown discoloration, subungual hyperkeratosis, and onycholysis of the toenails. Clinically, a diagnosis of onychomycosis was made. Potassium hydroxide examination of a scraping from the subungual region showed fungal elements. Trichophyton rubrum on Sabouraud dextrose agar was determined.
|
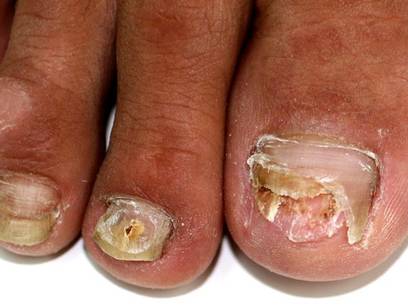

We performed both in vivo and in vitro confocal laser scanning microscopic examination of the nail of the right great toe (Figure 2). For the diagnosis of onychomycosis in our case, we used a multilaser reflectance confocal microscope (RCM) with a wavelength of 786 nm. In vivo confocal microscopy of the nail revealed branching hyphae just below the surface of the nail plate. Hyphae were seen as refractile, bright, linear structures along the laminates of the nail.
Onychomycosis is a common condition affecting 5.5% of the population worldwide and representing 20% to 40% of all onychopathies and approximately 30% of cutaneous mycotic infections.1,2 There are many methods available to confirm the clinical diagnosis of onychomycosis by detecting the causative organisms. Direct microscopic examination of the scraping with a KOH culture, histopathologic assessment with periodic acid–Schiff staining, immunofluorescence analysis with calcofluor white staining, enzyme analysis, and polymerase chain reaction can be used for diagnosis of fungal infections. The most frequently used diagnostic method for onychomycosis is KOH examination of the scraping; however, fungal culture and histopathologic examination also can be used in cases having diagnostic difficulties.1,3,4 There are many studies comparing the efficacies of these methods in the literature.5-9
The causative fungal agent should be determined with at least 1 laboratory method due to the high cost, long duration, and serious potential adverse effects of systemic antifungal treatment. Direct microscopic examination with KOH in the diagnosis of onychomycosis is simple, fast, and inexpensive. However, inadequate material, using crystallized KOH for hydrolysis, insufficient or too much hydrolysis of scrapings, inappropriate staining, and not scanning all areas in the microscopy produce false-negative results. Similarly, secondary contamination of hair, cotton, yarn, or air bubbles mimicking fungal structures can cause false-positive results.9,10
Fungal culture is another diagnostic method that is accepted as the gold standard for diagnosis of onychomycosis.9 However, fungal cultures were positive in only 43% to 50% of all cases of onychomycosis that were diagnosed with other methods,11,12 which may be due to the loss of viability and ability of the fungi to grow in culture media during the transport. A major advantage of fungal culture is that the fungal agent can be classified as dermatophyte, nondermatophyte, mold, or yeast. However, culture does determine if the growing fungi is contamination or the real pathogen. Moreover, it is necessary to wait 3 to 4 weeks for culture results. For nondermatophyte fungi this time may be much longer.12
In vivo RCM is a noninvasive imaging method that allows optical en face sectioning of the living tissue with high resolution. Currently, RCM has a wide range of applications, such as the evaluation of both benign and malignant skin lesions in clinical dermatology.13
In vivo RCM was used first by Hongcharu et al.14 The diagnoses of onychomycosis and fungal hyphae were shown both in vivo and in vitro.14 The sensitivity and specificity of confocal examination in the diagnosis of onychomycosis is not known yet. Large clinical trials are needed to assess the sensitivity and specificity of this method in diagnosing fungal infections.
Onychomycosis is a contagious infectious disease characterized by hyphae proliferation in the nail plate. Definitive diagnosis is necessary before treatment because onychomycosis can be mistaken for many infectious or noninfectious skin diseases with nail involvement. Conventional methods are time consuming, laborious, and less reliable. Instead of high-cost procedures, in vivo confocal microscopic examination can be a rapid and reliable diagnostic method for onychomycosis in the near future.
1. Singal A, Khanna D. Onychomycosis: diagnosis and management. Indian J Dermatol Venereol Leprol. 2011;77:659-672.
2. Kaur R, Kashyap B, Bhalla P. Onychomycosis—epidemiology, diagnosis and management. Indian J Med Microbiol. 2008;26:108-116.
3. Richardson MD. Diagnosis and pathogenesis of dermatophyte infections. Br J Clin Pract Suppl. 1990;71:98-102.
4. Jensen RH, Arendrup MC. Molecular diagnosis of dermato-phyte infections. Curr Opin Infect Dis. 2012;25:126-134.
5. Weinberg JM, Koestenblatt EK, Tutrone WD, et al. Comparison of diagnostic methods in the evaluation of onychomycosis. J Am Acad Dermatol. 2003;49:193-197.
6. Gianni C, Morelli V, Cerri A, et al. Usefulness of histological examination for the diagnosis of onychomycosis. Dermatology. 2001;202:283-288.
7. Machler BC, Kirsner RS, Elgart GW. Routine histologic examination for the diagnosis of onychomycosis: an evaluation of sensitivity and specificity. Cutis. 1998;61:217-219.
8. Wilsmann-Theis D, Sareika F, Bieber T, et al. New reasons for histopathological nail-clipping examination in the diagnosis of onychomycosis. J Eur Acad Dermatol Venereol. 2011;25:235-237.
9. Reisberger EM, Abels C, Landthaler M, et al. Histopathological diagnosis of onychomycosis by periodic acid-Schiff-stained nail clippings. Br J Dermatol. 2003;148:749-754.
10. Shemer A, Trau H, Davidovici B, et al. Collection of fungi samples from nails: comparative study of curettage and drilling techniques. J Eur Acad Dermatol Venereol. 2008;22:182-185.
11. Daniel CR 3rd, Elewski BE. The diagnosis of nail fungus infection revisited. Arch Dermatol. 2000;136:1162-1164.
12. Borkowski P, Williams M, Holewinski J, et al. Onychomycosis: an analysis of 50 cases and a comparison of diagnostic techniques. J Am Podiatr Med Assoc. 2001;91:351-355.
13. Rajadhyaksha M, Gonzalez S, Zavislan JM, et al. In vivo confocal scanning laser microscopy of human skin II: advances in instrumentation and comparison with histology. J Invest Dermatol. 1999;113:293-303.
14. Hongcharu W, Dwyer P, Gonzalez S, et al. Confirmation of onychomycosis by in vivo confocal microscopy. J Am Acad Dermatol. 2000;42(2, pt 1):214-216.
To the Editor:
Onychomycosis is a common nail disease that frequently is caused by dermatophytes and is diagnosed by direct microscopy. Conventional diagnostic methods are often time consuming and can produce false-positive or false-negative results. We report a case of onychomycosis diagnosed by confocal microscopy and confirmed with routine potassium hydroxide (KOH) examination and fungal culture. Confocal microscopy is a reliable, practical, and noninvasive technique in the diagnosis of onychomycosis.
A 46-year-old woman presented with yellow-brown discoloration and dystrophy of the toenails (Figure 1) that had become worse over a 5-year period. She was otherwise healthy and had no other dermatologic problems. Examination revealed yellow-brown discoloration, subungual hyperkeratosis, and onycholysis of the toenails. Clinically, a diagnosis of onychomycosis was made. Potassium hydroxide examination of a scraping from the subungual region showed fungal elements. Trichophyton rubrum on Sabouraud dextrose agar was determined.
|
We performed both in vivo and in vitro confocal laser scanning microscopic examination of the nail of the right great toe (Figure 2). For the diagnosis of onychomycosis in our case, we used a multilaser reflectance confocal microscope (RCM) with a wavelength of 786 nm. In vivo confocal microscopy of the nail revealed branching hyphae just below the surface of the nail plate. Hyphae were seen as refractile, bright, linear structures along the laminates of the nail.
Onychomycosis is a common condition affecting 5.5% of the population worldwide and representing 20% to 40% of all onychopathies and approximately 30% of cutaneous mycotic infections.1,2 There are many methods available to confirm the clinical diagnosis of onychomycosis by detecting the causative organisms. Direct microscopic examination of the scraping with a KOH culture, histopathologic assessment with periodic acid–Schiff staining, immunofluorescence analysis with calcofluor white staining, enzyme analysis, and polymerase chain reaction can be used for diagnosis of fungal infections. The most frequently used diagnostic method for onychomycosis is KOH examination of the scraping; however, fungal culture and histopathologic examination also can be used in cases having diagnostic difficulties.1,3,4 There are many studies comparing the efficacies of these methods in the literature.5-9
The causative fungal agent should be determined with at least 1 laboratory method due to the high cost, long duration, and serious potential adverse effects of systemic antifungal treatment. Direct microscopic examination with KOH in the diagnosis of onychomycosis is simple, fast, and inexpensive. However, inadequate material, using crystallized KOH for hydrolysis, insufficient or too much hydrolysis of scrapings, inappropriate staining, and not scanning all areas in the microscopy produce false-negative results. Similarly, secondary contamination of hair, cotton, yarn, or air bubbles mimicking fungal structures can cause false-positive results.9,10
Fungal culture is another diagnostic method that is accepted as the gold standard for diagnosis of onychomycosis.9 However, fungal cultures were positive in only 43% to 50% of all cases of onychomycosis that were diagnosed with other methods,11,12 which may be due to the loss of viability and ability of the fungi to grow in culture media during the transport. A major advantage of fungal culture is that the fungal agent can be classified as dermatophyte, nondermatophyte, mold, or yeast. However, culture does determine if the growing fungi is contamination or the real pathogen. Moreover, it is necessary to wait 3 to 4 weeks for culture results. For nondermatophyte fungi this time may be much longer.12
In vivo RCM is a noninvasive imaging method that allows optical en face sectioning of the living tissue with high resolution. Currently, RCM has a wide range of applications, such as the evaluation of both benign and malignant skin lesions in clinical dermatology.13
In vivo RCM was used first by Hongcharu et al.14 The diagnoses of onychomycosis and fungal hyphae were shown both in vivo and in vitro.14 The sensitivity and specificity of confocal examination in the diagnosis of onychomycosis is not known yet. Large clinical trials are needed to assess the sensitivity and specificity of this method in diagnosing fungal infections.
Onychomycosis is a contagious infectious disease characterized by hyphae proliferation in the nail plate. Definitive diagnosis is necessary before treatment because onychomycosis can be mistaken for many infectious or noninfectious skin diseases with nail involvement. Conventional methods are time consuming, laborious, and less reliable. Instead of high-cost procedures, in vivo confocal microscopic examination can be a rapid and reliable diagnostic method for onychomycosis in the near future.
To the Editor:
Onychomycosis is a common nail disease that frequently is caused by dermatophytes and is diagnosed by direct microscopy. Conventional diagnostic methods are often time consuming and can produce false-positive or false-negative results. We report a case of onychomycosis diagnosed by confocal microscopy and confirmed with routine potassium hydroxide (KOH) examination and fungal culture. Confocal microscopy is a reliable, practical, and noninvasive technique in the diagnosis of onychomycosis.
A 46-year-old woman presented with yellow-brown discoloration and dystrophy of the toenails (Figure 1) that had become worse over a 5-year period. She was otherwise healthy and had no other dermatologic problems. Examination revealed yellow-brown discoloration, subungual hyperkeratosis, and onycholysis of the toenails. Clinically, a diagnosis of onychomycosis was made. Potassium hydroxide examination of a scraping from the subungual region showed fungal elements. Trichophyton rubrum on Sabouraud dextrose agar was determined.
|
We performed both in vivo and in vitro confocal laser scanning microscopic examination of the nail of the right great toe (Figure 2). For the diagnosis of onychomycosis in our case, we used a multilaser reflectance confocal microscope (RCM) with a wavelength of 786 nm. In vivo confocal microscopy of the nail revealed branching hyphae just below the surface of the nail plate. Hyphae were seen as refractile, bright, linear structures along the laminates of the nail.
Onychomycosis is a common condition affecting 5.5% of the population worldwide and representing 20% to 40% of all onychopathies and approximately 30% of cutaneous mycotic infections.1,2 There are many methods available to confirm the clinical diagnosis of onychomycosis by detecting the causative organisms. Direct microscopic examination of the scraping with a KOH culture, histopathologic assessment with periodic acid–Schiff staining, immunofluorescence analysis with calcofluor white staining, enzyme analysis, and polymerase chain reaction can be used for diagnosis of fungal infections. The most frequently used diagnostic method for onychomycosis is KOH examination of the scraping; however, fungal culture and histopathologic examination also can be used in cases having diagnostic difficulties.1,3,4 There are many studies comparing the efficacies of these methods in the literature.5-9
The causative fungal agent should be determined with at least 1 laboratory method due to the high cost, long duration, and serious potential adverse effects of systemic antifungal treatment. Direct microscopic examination with KOH in the diagnosis of onychomycosis is simple, fast, and inexpensive. However, inadequate material, using crystallized KOH for hydrolysis, insufficient or too much hydrolysis of scrapings, inappropriate staining, and not scanning all areas in the microscopy produce false-negative results. Similarly, secondary contamination of hair, cotton, yarn, or air bubbles mimicking fungal structures can cause false-positive results.9,10
Fungal culture is another diagnostic method that is accepted as the gold standard for diagnosis of onychomycosis.9 However, fungal cultures were positive in only 43% to 50% of all cases of onychomycosis that were diagnosed with other methods,11,12 which may be due to the loss of viability and ability of the fungi to grow in culture media during the transport. A major advantage of fungal culture is that the fungal agent can be classified as dermatophyte, nondermatophyte, mold, or yeast. However, culture does determine if the growing fungi is contamination or the real pathogen. Moreover, it is necessary to wait 3 to 4 weeks for culture results. For nondermatophyte fungi this time may be much longer.12
In vivo RCM is a noninvasive imaging method that allows optical en face sectioning of the living tissue with high resolution. Currently, RCM has a wide range of applications, such as the evaluation of both benign and malignant skin lesions in clinical dermatology.13
In vivo RCM was used first by Hongcharu et al.14 The diagnoses of onychomycosis and fungal hyphae were shown both in vivo and in vitro.14 The sensitivity and specificity of confocal examination in the diagnosis of onychomycosis is not known yet. Large clinical trials are needed to assess the sensitivity and specificity of this method in diagnosing fungal infections.
Onychomycosis is a contagious infectious disease characterized by hyphae proliferation in the nail plate. Definitive diagnosis is necessary before treatment because onychomycosis can be mistaken for many infectious or noninfectious skin diseases with nail involvement. Conventional methods are time consuming, laborious, and less reliable. Instead of high-cost procedures, in vivo confocal microscopic examination can be a rapid and reliable diagnostic method for onychomycosis in the near future.
1. Singal A, Khanna D. Onychomycosis: diagnosis and management. Indian J Dermatol Venereol Leprol. 2011;77:659-672.
2. Kaur R, Kashyap B, Bhalla P. Onychomycosis—epidemiology, diagnosis and management. Indian J Med Microbiol. 2008;26:108-116.
3. Richardson MD. Diagnosis and pathogenesis of dermatophyte infections. Br J Clin Pract Suppl. 1990;71:98-102.
4. Jensen RH, Arendrup MC. Molecular diagnosis of dermato-phyte infections. Curr Opin Infect Dis. 2012;25:126-134.
5. Weinberg JM, Koestenblatt EK, Tutrone WD, et al. Comparison of diagnostic methods in the evaluation of onychomycosis. J Am Acad Dermatol. 2003;49:193-197.
6. Gianni C, Morelli V, Cerri A, et al. Usefulness of histological examination for the diagnosis of onychomycosis. Dermatology. 2001;202:283-288.
7. Machler BC, Kirsner RS, Elgart GW. Routine histologic examination for the diagnosis of onychomycosis: an evaluation of sensitivity and specificity. Cutis. 1998;61:217-219.
8. Wilsmann-Theis D, Sareika F, Bieber T, et al. New reasons for histopathological nail-clipping examination in the diagnosis of onychomycosis. J Eur Acad Dermatol Venereol. 2011;25:235-237.
9. Reisberger EM, Abels C, Landthaler M, et al. Histopathological diagnosis of onychomycosis by periodic acid-Schiff-stained nail clippings. Br J Dermatol. 2003;148:749-754.
10. Shemer A, Trau H, Davidovici B, et al. Collection of fungi samples from nails: comparative study of curettage and drilling techniques. J Eur Acad Dermatol Venereol. 2008;22:182-185.
11. Daniel CR 3rd, Elewski BE. The diagnosis of nail fungus infection revisited. Arch Dermatol. 2000;136:1162-1164.
12. Borkowski P, Williams M, Holewinski J, et al. Onychomycosis: an analysis of 50 cases and a comparison of diagnostic techniques. J Am Podiatr Med Assoc. 2001;91:351-355.
13. Rajadhyaksha M, Gonzalez S, Zavislan JM, et al. In vivo confocal scanning laser microscopy of human skin II: advances in instrumentation and comparison with histology. J Invest Dermatol. 1999;113:293-303.
14. Hongcharu W, Dwyer P, Gonzalez S, et al. Confirmation of onychomycosis by in vivo confocal microscopy. J Am Acad Dermatol. 2000;42(2, pt 1):214-216.
1. Singal A, Khanna D. Onychomycosis: diagnosis and management. Indian J Dermatol Venereol Leprol. 2011;77:659-672.
2. Kaur R, Kashyap B, Bhalla P. Onychomycosis—epidemiology, diagnosis and management. Indian J Med Microbiol. 2008;26:108-116.
3. Richardson MD. Diagnosis and pathogenesis of dermatophyte infections. Br J Clin Pract Suppl. 1990;71:98-102.
4. Jensen RH, Arendrup MC. Molecular diagnosis of dermato-phyte infections. Curr Opin Infect Dis. 2012;25:126-134.
5. Weinberg JM, Koestenblatt EK, Tutrone WD, et al. Comparison of diagnostic methods in the evaluation of onychomycosis. J Am Acad Dermatol. 2003;49:193-197.
6. Gianni C, Morelli V, Cerri A, et al. Usefulness of histological examination for the diagnosis of onychomycosis. Dermatology. 2001;202:283-288.
7. Machler BC, Kirsner RS, Elgart GW. Routine histologic examination for the diagnosis of onychomycosis: an evaluation of sensitivity and specificity. Cutis. 1998;61:217-219.
8. Wilsmann-Theis D, Sareika F, Bieber T, et al. New reasons for histopathological nail-clipping examination in the diagnosis of onychomycosis. J Eur Acad Dermatol Venereol. 2011;25:235-237.
9. Reisberger EM, Abels C, Landthaler M, et al. Histopathological diagnosis of onychomycosis by periodic acid-Schiff-stained nail clippings. Br J Dermatol. 2003;148:749-754.
10. Shemer A, Trau H, Davidovici B, et al. Collection of fungi samples from nails: comparative study of curettage and drilling techniques. J Eur Acad Dermatol Venereol. 2008;22:182-185.
11. Daniel CR 3rd, Elewski BE. The diagnosis of nail fungus infection revisited. Arch Dermatol. 2000;136:1162-1164.
12. Borkowski P, Williams M, Holewinski J, et al. Onychomycosis: an analysis of 50 cases and a comparison of diagnostic techniques. J Am Podiatr Med Assoc. 2001;91:351-355.
13. Rajadhyaksha M, Gonzalez S, Zavislan JM, et al. In vivo confocal scanning laser microscopy of human skin II: advances in instrumentation and comparison with histology. J Invest Dermatol. 1999;113:293-303.
14. Hongcharu W, Dwyer P, Gonzalez S, et al. Confirmation of onychomycosis by in vivo confocal microscopy. J Am Acad Dermatol. 2000;42(2, pt 1):214-216.
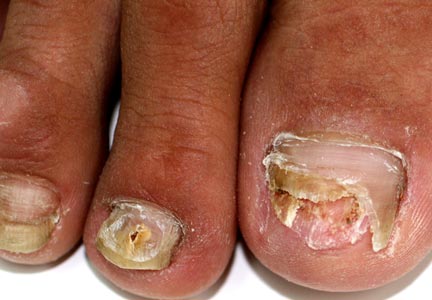
Pneumonic Tularemia Presenting With a Vesicular Eruption
To the Editor:
A 38-year-old microbiologist presented to a primary care physician with fevers, night sweats, myalgia, and headaches of 2 weeks’ duration. She was treated for a presumed viral illness with antipyretics and fluids. The patient subsequently developed a persistent nonproductive cough and chest pain as well as painful nodules of the lower legs and a vesicular rash over the trunk and arms. The patient worked closely with Yersinia pestis and Francisella tularensis and because of the occupational exposure had a thorough evaluation. An increased bacterial agglutinin titer for F tularensis from 1:40 to 1:1280 was noted during repeat testing over a 1-week period and a polymerase chain reaction test of sputum was positive for F tularensis. Chest radiography revealed right lower lobe pneumonia and adenopathy. The patient was admitted to the hospital for pneumonic tularemia and was treated with streptomycin with rapid improvement; however, after development of tinnitus and vertigo she was switched to ciprofloxacin.
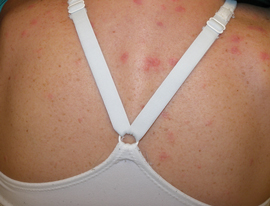
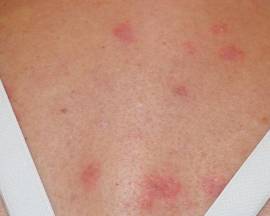
|
Dermatology was consulted to evaluate the patient’s nonpruritic vesicular rash that had been present for 6 days. Examination revealed multiple erythematous papules and plaques with vesicles rimming the periphery or studded throughout the lesions (Figures 1 and 2). Tender ecchymotic subcutaneous nodules of the lower extremities consistent with erythema nodosum also were present. Punch biopsies taken from vesicular papules of the back showed superficial perivascular inflammation and vesiculation within the epidermis (Figure 3). Polymerase chain reaction analysis revealed F tularensis. The patient was discharged with continued improvement after completion of the 1-month antibiotic regimen.
Vesicular papules and plaques are uncommon cutaneous manifestations of tularemia with few reports since the first documented cases of vesicular tularemia.1,2 A summary of 654 cases of tularemia in 1928 revealed 1 case with a vesicular rash.3 A retrospective review of 234 cases in Sweden in 2007 noted a vesicular rash in 7 patients (3.0%),4 and 2 subsequent cases of vesicular skin lesions in children with culture-positive tularemia initially were misdiagnosed as herpes simplex virus or varicella-zoster virus.5
Fewer than 200 cases of tularemia are reported to the Centers for Disease Control and Prevention annually, yet outbreaks do occur.5 Tularemia is primarily contracted through contact with infected animals (eg, rabbits) or vector insects (eg, deer flies, Dermacentor ticks). However, the disease remains a concern as a potential bioweapon via inhalation of aerosolized particles. A victim of bioterrorism may present in a manner similar to our patient. Although the threat of bioterrorism and incidence of tularemia in the United States is low, vesicular papules may be a presentation of tularemia and should be considered in the evaluation of a vesicular rash.
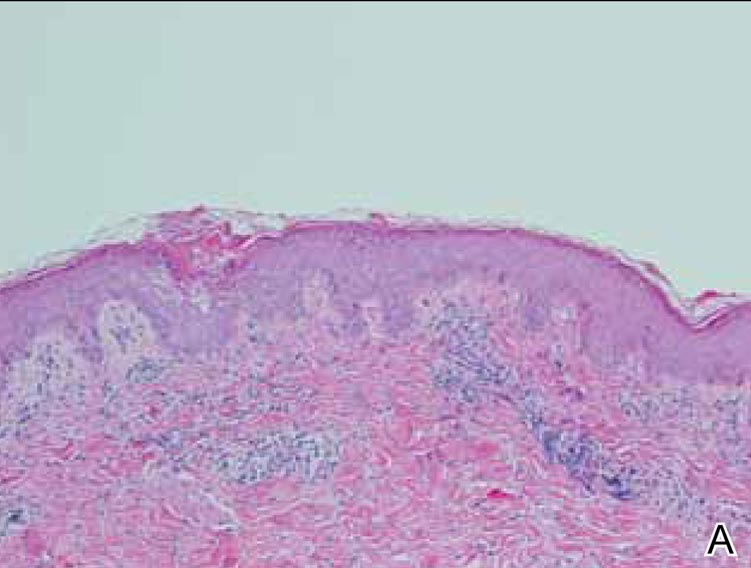
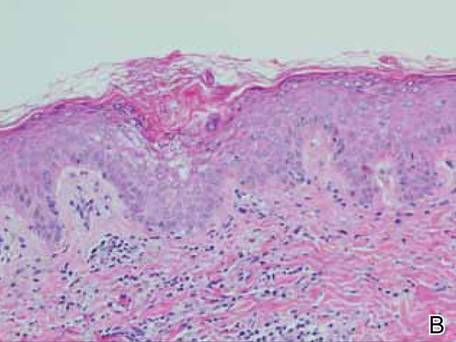
|
1. Pearse R. Insect bites. Northwest Med. 1911;3:81-82.
2. Francis E. The occurrence of tularemia in nature as a disease of man. Pub Health Rep. 1921;36:1731-1746.
3. Francis E. A summary of present knowledge of tularemia. Medicine. 1928;7:411-432.
4. Eliasson H, Bäck E. Tularaemia in an emergent area in Sweden: an analysis of 234 cases in five years. Scand J Infect Dis. 2007;39:880-889.
5. Byington C, Bender J, Ampofo K, et al. Tularemia with vesicular skin lesions may be mistaken for infection with herpes viruses. Clin Infect Dis. 2008;47:e4-e6.
To the Editor:
A 38-year-old microbiologist presented to a primary care physician with fevers, night sweats, myalgia, and headaches of 2 weeks’ duration. She was treated for a presumed viral illness with antipyretics and fluids. The patient subsequently developed a persistent nonproductive cough and chest pain as well as painful nodules of the lower legs and a vesicular rash over the trunk and arms. The patient worked closely with Yersinia pestis and Francisella tularensis and because of the occupational exposure had a thorough evaluation. An increased bacterial agglutinin titer for F tularensis from 1:40 to 1:1280 was noted during repeat testing over a 1-week period and a polymerase chain reaction test of sputum was positive for F tularensis. Chest radiography revealed right lower lobe pneumonia and adenopathy. The patient was admitted to the hospital for pneumonic tularemia and was treated with streptomycin with rapid improvement; however, after development of tinnitus and vertigo she was switched to ciprofloxacin.
|
|
Dermatology was consulted to evaluate the patient’s nonpruritic vesicular rash that had been present for 6 days. Examination revealed multiple erythematous papules and plaques with vesicles rimming the periphery or studded throughout the lesions (Figures 1 and 2). Tender ecchymotic subcutaneous nodules of the lower extremities consistent with erythema nodosum also were present. Punch biopsies taken from vesicular papules of the back showed superficial perivascular inflammation and vesiculation within the epidermis (Figure 3). Polymerase chain reaction analysis revealed F tularensis. The patient was discharged with continued improvement after completion of the 1-month antibiotic regimen.
Vesicular papules and plaques are uncommon cutaneous manifestations of tularemia with few reports since the first documented cases of vesicular tularemia.1,2 A summary of 654 cases of tularemia in 1928 revealed 1 case with a vesicular rash.3 A retrospective review of 234 cases in Sweden in 2007 noted a vesicular rash in 7 patients (3.0%),4 and 2 subsequent cases of vesicular skin lesions in children with culture-positive tularemia initially were misdiagnosed as herpes simplex virus or varicella-zoster virus.5
Fewer than 200 cases of tularemia are reported to the Centers for Disease Control and Prevention annually, yet outbreaks do occur.5 Tularemia is primarily contracted through contact with infected animals (eg, rabbits) or vector insects (eg, deer flies, Dermacentor ticks). However, the disease remains a concern as a potential bioweapon via inhalation of aerosolized particles. A victim of bioterrorism may present in a manner similar to our patient. Although the threat of bioterrorism and incidence of tularemia in the United States is low, vesicular papules may be a presentation of tularemia and should be considered in the evaluation of a vesicular rash.
|
|
To the Editor:
A 38-year-old microbiologist presented to a primary care physician with fevers, night sweats, myalgia, and headaches of 2 weeks’ duration. She was treated for a presumed viral illness with antipyretics and fluids. The patient subsequently developed a persistent nonproductive cough and chest pain as well as painful nodules of the lower legs and a vesicular rash over the trunk and arms. The patient worked closely with Yersinia pestis and Francisella tularensis and because of the occupational exposure had a thorough evaluation. An increased bacterial agglutinin titer for F tularensis from 1:40 to 1:1280 was noted during repeat testing over a 1-week period and a polymerase chain reaction test of sputum was positive for F tularensis. Chest radiography revealed right lower lobe pneumonia and adenopathy. The patient was admitted to the hospital for pneumonic tularemia and was treated with streptomycin with rapid improvement; however, after development of tinnitus and vertigo she was switched to ciprofloxacin.
|
|
Dermatology was consulted to evaluate the patient’s nonpruritic vesicular rash that had been present for 6 days. Examination revealed multiple erythematous papules and plaques with vesicles rimming the periphery or studded throughout the lesions (Figures 1 and 2). Tender ecchymotic subcutaneous nodules of the lower extremities consistent with erythema nodosum also were present. Punch biopsies taken from vesicular papules of the back showed superficial perivascular inflammation and vesiculation within the epidermis (Figure 3). Polymerase chain reaction analysis revealed F tularensis. The patient was discharged with continued improvement after completion of the 1-month antibiotic regimen.
Vesicular papules and plaques are uncommon cutaneous manifestations of tularemia with few reports since the first documented cases of vesicular tularemia.1,2 A summary of 654 cases of tularemia in 1928 revealed 1 case with a vesicular rash.3 A retrospective review of 234 cases in Sweden in 2007 noted a vesicular rash in 7 patients (3.0%),4 and 2 subsequent cases of vesicular skin lesions in children with culture-positive tularemia initially were misdiagnosed as herpes simplex virus or varicella-zoster virus.5
Fewer than 200 cases of tularemia are reported to the Centers for Disease Control and Prevention annually, yet outbreaks do occur.5 Tularemia is primarily contracted through contact with infected animals (eg, rabbits) or vector insects (eg, deer flies, Dermacentor ticks). However, the disease remains a concern as a potential bioweapon via inhalation of aerosolized particles. A victim of bioterrorism may present in a manner similar to our patient. Although the threat of bioterrorism and incidence of tularemia in the United States is low, vesicular papules may be a presentation of tularemia and should be considered in the evaluation of a vesicular rash.
|
|
1. Pearse R. Insect bites. Northwest Med. 1911;3:81-82.
2. Francis E. The occurrence of tularemia in nature as a disease of man. Pub Health Rep. 1921;36:1731-1746.
3. Francis E. A summary of present knowledge of tularemia. Medicine. 1928;7:411-432.
4. Eliasson H, Bäck E. Tularaemia in an emergent area in Sweden: an analysis of 234 cases in five years. Scand J Infect Dis. 2007;39:880-889.
5. Byington C, Bender J, Ampofo K, et al. Tularemia with vesicular skin lesions may be mistaken for infection with herpes viruses. Clin Infect Dis. 2008;47:e4-e6.
1. Pearse R. Insect bites. Northwest Med. 1911;3:81-82.
2. Francis E. The occurrence of tularemia in nature as a disease of man. Pub Health Rep. 1921;36:1731-1746.
3. Francis E. A summary of present knowledge of tularemia. Medicine. 1928;7:411-432.
4. Eliasson H, Bäck E. Tularaemia in an emergent area in Sweden: an analysis of 234 cases in five years. Scand J Infect Dis. 2007;39:880-889.
5. Byington C, Bender J, Ampofo K, et al. Tularemia with vesicular skin lesions may be mistaken for infection with herpes viruses. Clin Infect Dis. 2008;47:e4-e6.
Rapidly Recurring Keratoacanthoma
To the Editor:
A 61-year-old man with a medical history of type 2 diabetes mellitus presented to us with a 2.5×3.0-cm erythematous, ulcerated, and exophytic tumor on the right dorsal forearm that had rapidly developed over 2 weeks. A tangential biopsy was performed followed by treatment with electrodesiccation and curettage (ED&C). Histology revealed a squamous cell carcinoma (SCC), keratoacanthoma (KA) type. Over the next 11 days the lesion rapidly recurred and the patient returned with his own daily photodocumentation of the KA’s progression (Figure). The lesion was re-excised with 5-mm margins; histology again revealed SCC, KA type, with deep margin involvement. Chest radiograph revealed findings suspicious for metastatic lesions in the right lung. He was referred to oncology for metastatic workup; positron emission tomography was negative and ultimately the lung lesion was found to be benign. The patient underwent adjuvant radia-tion to the KA resection bed and lymph nodes with minimal side effects. The patient has remained cancer free to date.
Keratoacanthomas are rapidly growing, typically painless, cutaneous neoplasms that often develop on sun-exposed areas. They can occur spontaneously or following trauma and have the propensity to regress with time.1-3 They are described as progressing through 3 clinical stages: rapid proliferation, mature/stable, and involution. However, KAs can be aggressive, becoming locally destructive; therefore, KAs are typically treated to avoid further morbidity. Keratoacanthomas may be considered a subtype of SCC, as some have the potential to become locally destructive and metastasize.3-5 There are reports of spontaneous resolution of KAs over weeks to months, though surgical excision is the gold standard of treatment.3,5
Reactive KA is a subtype that is thought to develop at the site of prior trauma, representing a sort of Köbner phenomenon.3,4 We demonstrated a case of a recurrent KA in the setting of recent ED&C. Several reports describe KAs developing after dermatologic surgery, including Mohs micrographic surgery, laser resurfacing, radiation therapy, and after skin grafting.3,4,6 Trauma-induced epidermal injury and dermal inflammation may play a role in postoperative KA formation or recurrence.6
Keratoacanthoma recurrence has been reported in 3% to 8% of cases within a few weeks after treatment, as seen in our current patient.3,5 In our case, the patient photodocumented the regrowth of his lesion (Figure). Treatment of reactive KAs may be therapeutically challenging, as they can form or worsen with repeated surgeries and may require several treatment modalities to eradicate them.4 Treatment options include observation, ED&C, excision, Mohs micrographic surgery, radiation, cryosurgery, laser, isotretinoin, acitretin, imiquimod, 5-fluorouracil, methotrexate, interferon alfa-2b, or bleomycin, to name a few.3,4,7
Combination therapy should be considered in the presence of recurrent and/or aggressive KAs, such as in our case. Our patient has remained disease free after a combination of surgical excision with radiation therapy.
1. Schwartz R. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
2. Kingman J. Keratoacanthoma. Arch Dermatol. 1984;20:736-740.
3. Goldberg L, Silapunt S, Beyrau K, et al. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
4. Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
5. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46:671-678.
6. Chesnut GT, Maggio KL, Turiansky GW. Letter: re: case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2011;37:884-885.
7. Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
To the Editor:
A 61-year-old man with a medical history of type 2 diabetes mellitus presented to us with a 2.5×3.0-cm erythematous, ulcerated, and exophytic tumor on the right dorsal forearm that had rapidly developed over 2 weeks. A tangential biopsy was performed followed by treatment with electrodesiccation and curettage (ED&C). Histology revealed a squamous cell carcinoma (SCC), keratoacanthoma (KA) type. Over the next 11 days the lesion rapidly recurred and the patient returned with his own daily photodocumentation of the KA’s progression (Figure). The lesion was re-excised with 5-mm margins; histology again revealed SCC, KA type, with deep margin involvement. Chest radiograph revealed findings suspicious for metastatic lesions in the right lung. He was referred to oncology for metastatic workup; positron emission tomography was negative and ultimately the lung lesion was found to be benign. The patient underwent adjuvant radia-tion to the KA resection bed and lymph nodes with minimal side effects. The patient has remained cancer free to date.
Keratoacanthomas are rapidly growing, typically painless, cutaneous neoplasms that often develop on sun-exposed areas. They can occur spontaneously or following trauma and have the propensity to regress with time.1-3 They are described as progressing through 3 clinical stages: rapid proliferation, mature/stable, and involution. However, KAs can be aggressive, becoming locally destructive; therefore, KAs are typically treated to avoid further morbidity. Keratoacanthomas may be considered a subtype of SCC, as some have the potential to become locally destructive and metastasize.3-5 There are reports of spontaneous resolution of KAs over weeks to months, though surgical excision is the gold standard of treatment.3,5
Reactive KA is a subtype that is thought to develop at the site of prior trauma, representing a sort of Köbner phenomenon.3,4 We demonstrated a case of a recurrent KA in the setting of recent ED&C. Several reports describe KAs developing after dermatologic surgery, including Mohs micrographic surgery, laser resurfacing, radiation therapy, and after skin grafting.3,4,6 Trauma-induced epidermal injury and dermal inflammation may play a role in postoperative KA formation or recurrence.6
Keratoacanthoma recurrence has been reported in 3% to 8% of cases within a few weeks after treatment, as seen in our current patient.3,5 In our case, the patient photodocumented the regrowth of his lesion (Figure). Treatment of reactive KAs may be therapeutically challenging, as they can form or worsen with repeated surgeries and may require several treatment modalities to eradicate them.4 Treatment options include observation, ED&C, excision, Mohs micrographic surgery, radiation, cryosurgery, laser, isotretinoin, acitretin, imiquimod, 5-fluorouracil, methotrexate, interferon alfa-2b, or bleomycin, to name a few.3,4,7
Combination therapy should be considered in the presence of recurrent and/or aggressive KAs, such as in our case. Our patient has remained disease free after a combination of surgical excision with radiation therapy.
To the Editor:
A 61-year-old man with a medical history of type 2 diabetes mellitus presented to us with a 2.5×3.0-cm erythematous, ulcerated, and exophytic tumor on the right dorsal forearm that had rapidly developed over 2 weeks. A tangential biopsy was performed followed by treatment with electrodesiccation and curettage (ED&C). Histology revealed a squamous cell carcinoma (SCC), keratoacanthoma (KA) type. Over the next 11 days the lesion rapidly recurred and the patient returned with his own daily photodocumentation of the KA’s progression (Figure). The lesion was re-excised with 5-mm margins; histology again revealed SCC, KA type, with deep margin involvement. Chest radiograph revealed findings suspicious for metastatic lesions in the right lung. He was referred to oncology for metastatic workup; positron emission tomography was negative and ultimately the lung lesion was found to be benign. The patient underwent adjuvant radia-tion to the KA resection bed and lymph nodes with minimal side effects. The patient has remained cancer free to date.
Keratoacanthomas are rapidly growing, typically painless, cutaneous neoplasms that often develop on sun-exposed areas. They can occur spontaneously or following trauma and have the propensity to regress with time.1-3 They are described as progressing through 3 clinical stages: rapid proliferation, mature/stable, and involution. However, KAs can be aggressive, becoming locally destructive; therefore, KAs are typically treated to avoid further morbidity. Keratoacanthomas may be considered a subtype of SCC, as some have the potential to become locally destructive and metastasize.3-5 There are reports of spontaneous resolution of KAs over weeks to months, though surgical excision is the gold standard of treatment.3,5
Reactive KA is a subtype that is thought to develop at the site of prior trauma, representing a sort of Köbner phenomenon.3,4 We demonstrated a case of a recurrent KA in the setting of recent ED&C. Several reports describe KAs developing after dermatologic surgery, including Mohs micrographic surgery, laser resurfacing, radiation therapy, and after skin grafting.3,4,6 Trauma-induced epidermal injury and dermal inflammation may play a role in postoperative KA formation or recurrence.6
Keratoacanthoma recurrence has been reported in 3% to 8% of cases within a few weeks after treatment, as seen in our current patient.3,5 In our case, the patient photodocumented the regrowth of his lesion (Figure). Treatment of reactive KAs may be therapeutically challenging, as they can form or worsen with repeated surgeries and may require several treatment modalities to eradicate them.4 Treatment options include observation, ED&C, excision, Mohs micrographic surgery, radiation, cryosurgery, laser, isotretinoin, acitretin, imiquimod, 5-fluorouracil, methotrexate, interferon alfa-2b, or bleomycin, to name a few.3,4,7
Combination therapy should be considered in the presence of recurrent and/or aggressive KAs, such as in our case. Our patient has remained disease free after a combination of surgical excision with radiation therapy.
1. Schwartz R. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
2. Kingman J. Keratoacanthoma. Arch Dermatol. 1984;20:736-740.
3. Goldberg L, Silapunt S, Beyrau K, et al. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
4. Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
5. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46:671-678.
6. Chesnut GT, Maggio KL, Turiansky GW. Letter: re: case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2011;37:884-885.
7. Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
1. Schwartz R. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
2. Kingman J. Keratoacanthoma. Arch Dermatol. 1984;20:736-740.
3. Goldberg L, Silapunt S, Beyrau K, et al. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
4. Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
5. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46:671-678.
6. Chesnut GT, Maggio KL, Turiansky GW. Letter: re: case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2011;37:884-885.
7. Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
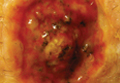
A Case of Morfan Syndrome
To the Editor:
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
|
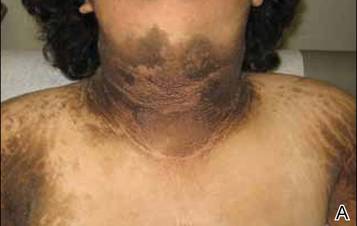

First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.
1. Seemanová E, Rüdiger HW, Dreyer M. Morfan: a new syndrome characterized by mental retardation, pre- and postnatal overgrowth, remarkable face and acanthosis nigricans in 5-year-old boy. Am J Med Genet. 1993;45:525-528.
2. Rendon MI, Cruz PD Jr, Sontheimer RD, et al. Acanthosis nigricans: a cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol. 1989;21:461-469.
3. Unna PG, Morris M, Besnier E, eds. International Atlas of Rare Skin Diseases. London, England: HK Lewis & Co; 1890.
4. Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31:1-19.
5. Inamdar AC, Palit A. Generalized acanthosis nigricans in childhood. Pediatr Dermatol. 2004;21:277-279.
6. Cruz PD, Hud JA. Excess insulin binding to insulin-like growth factor receptors: proposed mechanism for acanthosis nigricans. J Invest Dermatol. 1992;98:82-85.
7. Krawczyk M, Mykała-Cies´la J, Kołodziej-Jaskuła A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
8. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 1995;11:462-464.
9. Moller DE, Cohen O, Yamaguchi Y, et al. Prevalence of mutations in the insulin receptor gene in subjects with features of the type A syndrome of insulin resistance. Diabetes. 1994;43:247-255.
10. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nature Genet. 1996;13:492-494.
11. Anderson JL, Khan M, David WS, et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet. 1992;82:161-165.
12. Simha V, Agarwal AK, Aronin PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet. 2008;146A:2318-2326.
13. Kahn CR, Flier JS, Bar RS, et al. the syndromes of insulin resistance and acanthosis nigricans. insulin-receptor disorders in man. N Engl J Med. 1976;294:739-745.
14. Brickman WWJ, Huang J, Silverman BL, et al. Acanthosis nigricans identifies youth at high risk for metabolic abnormalities. J Pediatr. 2010;156:87-92.
15. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. insulin-like growth factors.
N Engl J Med. 1997;336:633-640.
16. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818-835.
17. Berk DR, Spector EB, Bayliss SJ. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch Dermatol. 2007;143:1153-1156.
To the Editor:
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
|
First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.
To the Editor:
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
|
First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.
1. Seemanová E, Rüdiger HW, Dreyer M. Morfan: a new syndrome characterized by mental retardation, pre- and postnatal overgrowth, remarkable face and acanthosis nigricans in 5-year-old boy. Am J Med Genet. 1993;45:525-528.
2. Rendon MI, Cruz PD Jr, Sontheimer RD, et al. Acanthosis nigricans: a cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol. 1989;21:461-469.
3. Unna PG, Morris M, Besnier E, eds. International Atlas of Rare Skin Diseases. London, England: HK Lewis & Co; 1890.
4. Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31:1-19.
5. Inamdar AC, Palit A. Generalized acanthosis nigricans in childhood. Pediatr Dermatol. 2004;21:277-279.
6. Cruz PD, Hud JA. Excess insulin binding to insulin-like growth factor receptors: proposed mechanism for acanthosis nigricans. J Invest Dermatol. 1992;98:82-85.
7. Krawczyk M, Mykała-Cies´la J, Kołodziej-Jaskuła A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
8. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 1995;11:462-464.
9. Moller DE, Cohen O, Yamaguchi Y, et al. Prevalence of mutations in the insulin receptor gene in subjects with features of the type A syndrome of insulin resistance. Diabetes. 1994;43:247-255.
10. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nature Genet. 1996;13:492-494.
11. Anderson JL, Khan M, David WS, et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet. 1992;82:161-165.
12. Simha V, Agarwal AK, Aronin PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet. 2008;146A:2318-2326.
13. Kahn CR, Flier JS, Bar RS, et al. the syndromes of insulin resistance and acanthosis nigricans. insulin-receptor disorders in man. N Engl J Med. 1976;294:739-745.
14. Brickman WWJ, Huang J, Silverman BL, et al. Acanthosis nigricans identifies youth at high risk for metabolic abnormalities. J Pediatr. 2010;156:87-92.
15. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. insulin-like growth factors.
N Engl J Med. 1997;336:633-640.
16. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818-835.
17. Berk DR, Spector EB, Bayliss SJ. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch Dermatol. 2007;143:1153-1156.
1. Seemanová E, Rüdiger HW, Dreyer M. Morfan: a new syndrome characterized by mental retardation, pre- and postnatal overgrowth, remarkable face and acanthosis nigricans in 5-year-old boy. Am J Med Genet. 1993;45:525-528.
2. Rendon MI, Cruz PD Jr, Sontheimer RD, et al. Acanthosis nigricans: a cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol. 1989;21:461-469.
3. Unna PG, Morris M, Besnier E, eds. International Atlas of Rare Skin Diseases. London, England: HK Lewis & Co; 1890.
4. Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31:1-19.
5. Inamdar AC, Palit A. Generalized acanthosis nigricans in childhood. Pediatr Dermatol. 2004;21:277-279.
6. Cruz PD, Hud JA. Excess insulin binding to insulin-like growth factor receptors: proposed mechanism for acanthosis nigricans. J Invest Dermatol. 1992;98:82-85.
7. Krawczyk M, Mykała-Cies´la J, Kołodziej-Jaskuła A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
8. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 1995;11:462-464.
9. Moller DE, Cohen O, Yamaguchi Y, et al. Prevalence of mutations in the insulin receptor gene in subjects with features of the type A syndrome of insulin resistance. Diabetes. 1994;43:247-255.
10. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nature Genet. 1996;13:492-494.
11. Anderson JL, Khan M, David WS, et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet. 1992;82:161-165.
12. Simha V, Agarwal AK, Aronin PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet. 2008;146A:2318-2326.
13. Kahn CR, Flier JS, Bar RS, et al. the syndromes of insulin resistance and acanthosis nigricans. insulin-receptor disorders in man. N Engl J Med. 1976;294:739-745.
14. Brickman WWJ, Huang J, Silverman BL, et al. Acanthosis nigricans identifies youth at high risk for metabolic abnormalities. J Pediatr. 2010;156:87-92.
15. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. insulin-like growth factors.
N Engl J Med. 1997;336:633-640.
16. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818-835.
17. Berk DR, Spector EB, Bayliss SJ. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch Dermatol. 2007;143:1153-1156.
