User login
Identification of Cutaneous Warts: Cryotherapy-Induced Acetowhitelike Epithelium
To the Editor:
Cutaneous warts are benign proliferations of the epidermis that occur secondary to human papillomavirus (HPV) infection. The diagnosis of cutaneous warts is generally based on clinical appearance. Occasionally subtle lesions, particularly those of verruca plana, escape clinical identification leading to incomplete treatment and spreading. The acetic acid test (sometimes called the acetic acid visual inspection) causes epithelial whitening of HPV-infected areas after application of a 3% to 5% aqueous solution of acetic acid and has been used to detect subclinical HPV infection.1 Although the acetic acid test can support the diagnosis of cutaneous warts, it is more effective at detecting hyperplastic rather than flat warts and may be cumbersome to use routinely.2 We describe a simple clinical maneuver to help confirm the presence of subtle warts using gentle liquid nitrogen cryotherapy to induce epithelial whitening in areas of HPV infection.
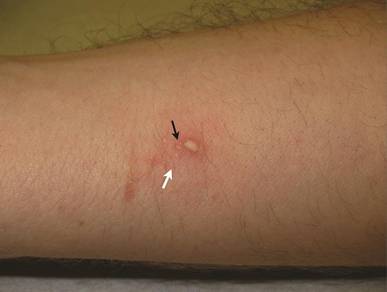
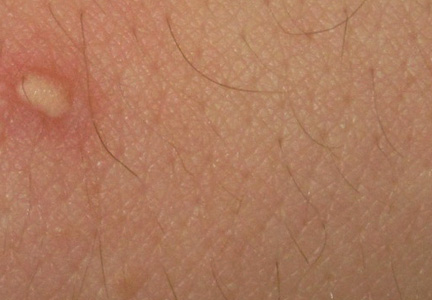
A 22-year-old man presented for evaluation of a 5-mm verrucous papule on the right wrist. He was diagnosed with verruca vulgaris. During treatment, small satellite verrucous papules were visualized by differential whitening from the surrounding uninfected skin (Figure). A brief light spray of liquid nitrogen cryotherapy (-196°C) was applied over areas containing suspicious lesions for confirmation. This acetowhitelike change from indirect collateral cryotherapy allowed for identification and treatment of these subtle warts.
Cutaneous warts represent foci of epithelial proliferation, and acetowhite changes are thought to occur from extravasation of intracellular water with subsequent tissue whitening in areas of high nuclear density.3 Acetowhite epithelium also has been reported after other ablative wart therapies.4 Similarly, acetowhitelike changes after cryotherapy may be secondary to cellular dehydration from ice crystal formation,5 with HPV-infected areas demonstrating increased susceptibility to freezing because of increased cellular water content in areas of hyperkeratosis. In addition, it has been demonstrated that cryotherapy alters the composition of the epithelium by destroying neutral and acidic mucopolysaccharides, which may subsequently induce the characteristic acetowhitelike changes in the epithelium of cutaneous warts.6
We propose that gentle painless sprays of liquid nitrogen to areas with suspicious lesions can help confirm the presence of subtle warts through cryotherapy-induced epithelial whitening. Although this test is a valuable diagnostic pearl, it should be noted that cryotherapy may accentuate an area of hyperkeratosis from causes other than an HPV infection. As such, clinical judgment is required.
1. Allan BM. Acetowhite epithelium. Gynecol Oncol. 2004;95:691-694.
2. Kumar B, Gupta S. The acetowhite test in genital human papillomavirus infection in men: what does it add? J Eur Acad Dermatol Venereol. 2001;15:27-29.
3. O’Connor DM. A tissue basis for colposcopic findings. Obstet Gynecol Clin North Am. 2008;35:565-582.
4. MacLean AB. Healing of the cervical epithelium after laser treatment of cervical intraepithelial neoplasia. Br J Obstet Gynaecol. 1984;91:697-706.
5. Gage AA, Baust J. Mechanisms of tissue injury in cryosurgery. Cryobiology. 1998;37:171-186.
6. Ciecierski L. Histochemical studies on acid and neutral mucopolysaccharides in the acanthotic epidermis of warts before and after cryotherapy with liquid nitrogen. Przegl Dermatol. 1970;57:11-15.
To the Editor:
Cutaneous warts are benign proliferations of the epidermis that occur secondary to human papillomavirus (HPV) infection. The diagnosis of cutaneous warts is generally based on clinical appearance. Occasionally subtle lesions, particularly those of verruca plana, escape clinical identification leading to incomplete treatment and spreading. The acetic acid test (sometimes called the acetic acid visual inspection) causes epithelial whitening of HPV-infected areas after application of a 3% to 5% aqueous solution of acetic acid and has been used to detect subclinical HPV infection.1 Although the acetic acid test can support the diagnosis of cutaneous warts, it is more effective at detecting hyperplastic rather than flat warts and may be cumbersome to use routinely.2 We describe a simple clinical maneuver to help confirm the presence of subtle warts using gentle liquid nitrogen cryotherapy to induce epithelial whitening in areas of HPV infection.
A 22-year-old man presented for evaluation of a 5-mm verrucous papule on the right wrist. He was diagnosed with verruca vulgaris. During treatment, small satellite verrucous papules were visualized by differential whitening from the surrounding uninfected skin (Figure). A brief light spray of liquid nitrogen cryotherapy (-196°C) was applied over areas containing suspicious lesions for confirmation. This acetowhitelike change from indirect collateral cryotherapy allowed for identification and treatment of these subtle warts.
Cutaneous warts represent foci of epithelial proliferation, and acetowhite changes are thought to occur from extravasation of intracellular water with subsequent tissue whitening in areas of high nuclear density.3 Acetowhite epithelium also has been reported after other ablative wart therapies.4 Similarly, acetowhitelike changes after cryotherapy may be secondary to cellular dehydration from ice crystal formation,5 with HPV-infected areas demonstrating increased susceptibility to freezing because of increased cellular water content in areas of hyperkeratosis. In addition, it has been demonstrated that cryotherapy alters the composition of the epithelium by destroying neutral and acidic mucopolysaccharides, which may subsequently induce the characteristic acetowhitelike changes in the epithelium of cutaneous warts.6
We propose that gentle painless sprays of liquid nitrogen to areas with suspicious lesions can help confirm the presence of subtle warts through cryotherapy-induced epithelial whitening. Although this test is a valuable diagnostic pearl, it should be noted that cryotherapy may accentuate an area of hyperkeratosis from causes other than an HPV infection. As such, clinical judgment is required.
To the Editor:
Cutaneous warts are benign proliferations of the epidermis that occur secondary to human papillomavirus (HPV) infection. The diagnosis of cutaneous warts is generally based on clinical appearance. Occasionally subtle lesions, particularly those of verruca plana, escape clinical identification leading to incomplete treatment and spreading. The acetic acid test (sometimes called the acetic acid visual inspection) causes epithelial whitening of HPV-infected areas after application of a 3% to 5% aqueous solution of acetic acid and has been used to detect subclinical HPV infection.1 Although the acetic acid test can support the diagnosis of cutaneous warts, it is more effective at detecting hyperplastic rather than flat warts and may be cumbersome to use routinely.2 We describe a simple clinical maneuver to help confirm the presence of subtle warts using gentle liquid nitrogen cryotherapy to induce epithelial whitening in areas of HPV infection.
A 22-year-old man presented for evaluation of a 5-mm verrucous papule on the right wrist. He was diagnosed with verruca vulgaris. During treatment, small satellite verrucous papules were visualized by differential whitening from the surrounding uninfected skin (Figure). A brief light spray of liquid nitrogen cryotherapy (-196°C) was applied over areas containing suspicious lesions for confirmation. This acetowhitelike change from indirect collateral cryotherapy allowed for identification and treatment of these subtle warts.
Cutaneous warts represent foci of epithelial proliferation, and acetowhite changes are thought to occur from extravasation of intracellular water with subsequent tissue whitening in areas of high nuclear density.3 Acetowhite epithelium also has been reported after other ablative wart therapies.4 Similarly, acetowhitelike changes after cryotherapy may be secondary to cellular dehydration from ice crystal formation,5 with HPV-infected areas demonstrating increased susceptibility to freezing because of increased cellular water content in areas of hyperkeratosis. In addition, it has been demonstrated that cryotherapy alters the composition of the epithelium by destroying neutral and acidic mucopolysaccharides, which may subsequently induce the characteristic acetowhitelike changes in the epithelium of cutaneous warts.6
We propose that gentle painless sprays of liquid nitrogen to areas with suspicious lesions can help confirm the presence of subtle warts through cryotherapy-induced epithelial whitening. Although this test is a valuable diagnostic pearl, it should be noted that cryotherapy may accentuate an area of hyperkeratosis from causes other than an HPV infection. As such, clinical judgment is required.
1. Allan BM. Acetowhite epithelium. Gynecol Oncol. 2004;95:691-694.
2. Kumar B, Gupta S. The acetowhite test in genital human papillomavirus infection in men: what does it add? J Eur Acad Dermatol Venereol. 2001;15:27-29.
3. O’Connor DM. A tissue basis for colposcopic findings. Obstet Gynecol Clin North Am. 2008;35:565-582.
4. MacLean AB. Healing of the cervical epithelium after laser treatment of cervical intraepithelial neoplasia. Br J Obstet Gynaecol. 1984;91:697-706.
5. Gage AA, Baust J. Mechanisms of tissue injury in cryosurgery. Cryobiology. 1998;37:171-186.
6. Ciecierski L. Histochemical studies on acid and neutral mucopolysaccharides in the acanthotic epidermis of warts before and after cryotherapy with liquid nitrogen. Przegl Dermatol. 1970;57:11-15.
1. Allan BM. Acetowhite epithelium. Gynecol Oncol. 2004;95:691-694.
2. Kumar B, Gupta S. The acetowhite test in genital human papillomavirus infection in men: what does it add? J Eur Acad Dermatol Venereol. 2001;15:27-29.
3. O’Connor DM. A tissue basis for colposcopic findings. Obstet Gynecol Clin North Am. 2008;35:565-582.
4. MacLean AB. Healing of the cervical epithelium after laser treatment of cervical intraepithelial neoplasia. Br J Obstet Gynaecol. 1984;91:697-706.
5. Gage AA, Baust J. Mechanisms of tissue injury in cryosurgery. Cryobiology. 1998;37:171-186.
6. Ciecierski L. Histochemical studies on acid and neutral mucopolysaccharides in the acanthotic epidermis of warts before and after cryotherapy with liquid nitrogen. Przegl Dermatol. 1970;57:11-15.
Subcutaneous Sarcoidosis on Ultrasonography
To the Editor:
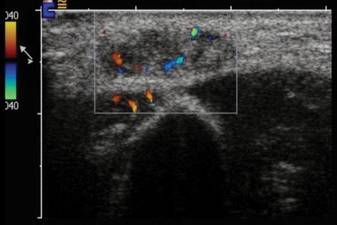
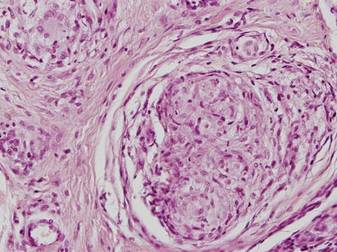
A 54-year-old woman presented with painless, firm, flesh-colored nodules measuring 1.0 to 1.5 cm in diameter on the extensor surface of the left forearm (Figure 1) and on the distal phalanx of the left thumb of 3 months’ duration. No other signs and symptoms were present. A detailed clinical examination revealed a slightly elevated erythrocyte sedimentation rate (24 mm/h [reference range, 0–20 mm/h]) and a high antinuclear antibody titer (1:3200 [reference range, <1:100])(anti–Sjögren syndrome anti-gen A, anti–Sjögren syndrome antigen B, anti-Ro52). Complete blood cell count, basic metabolic panel, liver function tests, urinalysis, pulmonary function tests, chest radiograph, and chest computed tomography all were normal. Hepatitis B antigen and antibody tests; hepatitis C antibody tests; and tuberculin test all were negative. An ophthalmic examination revealed no abnormalities. Ultrasonography of the nodules was performed with a system using an 8- to 12-MHz linear transducer and revealed 4 heterogenous hypoechoic lesions measuring up to 1.5 cm in size. Color Doppler images showed moderate hypervascularity (Figure 2). The largest nodule was excised. Histologic examination revealed noncaseating granulomas; special stains for microorganisms were negative. The histopathologic findings confirmed a diagnosis of sarcoidosis (Figure 3). The patient refused any medication. The nodules were stable at 6-month follow-up, then spontaneously resolved.
|
Subcutaneous sarcoidosis (SS) is a rare cutaneous expression of systemic sarcoidosis. The entity was first described by French physicians Darier and Roussy in 1904 as granulomatous panniculitis. Although their original study referred to a case of tuberculosis, the term Darier-Roussy sarcoid was coined and had been applied to a true sarcoid as well as to a variety of other forms of granulomatous panniculitis including those of infectious origin. A more accurate term subcutaneous sarcoidosis was established in 1984 by Vainsencher and Winkelmann.1
The most characteristic clinical picture of this disorder consists of the presence of multiple painless, firm, mobile nodules located on the extremities, most frequently the arms. However, other sites such as the trunk, buttocks, groin, head, face, and neck also have been reported.2,3
Marcoval et al2 demonstrated SS in only 2.1% of 480 patients with systemic sarcoidosis (10 patients). In the majority of these patients, subcutaneous nodules were the initial presentation of the disease.2 Ahmed and Harshad3 reported evidence of systemic involvement in 84.9% (45/53) of patients with SS. Chest involvement was the most common finding (eg, hilar lymphadenopathy, mediastinal adenopathy, interstitial pulmonary infiltration).3 Parotitis, uveitis, neuritis, and hepatosplenomegaly also have been noted systemically.4 The vast majority of reviews have suggested that SS has a relatively good prognosis. Ahmed and Harshad3 reported a satisfactory response to steroid treatment in all patients who received corticosteroids as the primary treatment. Subcutaneous sarcoidosis usually does not herald severe systemic involvement or chronic systemic complications. Both subcutaneous granulomas and hilar adenopathy may spontaneously resolve.
Interestingly, various autoimmune disease associations were seen in 6 of 21 patients (29%) in the study by Ahmed and Harshad3 including Hashimoto thyroiditis, rheumatoid arthritis, ulcerative colitis, systemic lupus erythematosus, and sicca syndrome. Barnadas et al5 reported a case of SS associated with vitiligo, pernicious anemia, and Hashimoto thyroiditis. Although our patient was not diagnosed with any particular autoimmune disease, an antinuclear antibody test was positive at a titer of 1:3200.
Our case is interesting for 2 reasons. First, it is a rare case of isolated SS. Thorough systemic evaluation showed no evidence of extracutaneous involvement. The literature only provides a few instances of isolated SS.6,7 Second, the sonographic appearance of SS is rare.8,9 Chen et al9 reported that gray-scale sonography revealed heterogenous, hypoechoic, well-demarcated plaquelike lesions with an intensive vascular pattern indicating Doppler hypervascularization. We obtained similar findings.
It has been widely acknowledged that sonographic findings of subcutaneous nodules tend to be nonspecific and overlapping. Color Doppler examination may show internal vessels both in malignant soft-tissue masses (eg, lymphoma, synovial sarcoma, liposarcoma, malignant fibrohistocytoma, metastases) and in benign lesions (eg, schwannoma, hemangioma, fibromatosis). However, the application of Doppler ultrasonography may restrict the diagnostic field, as it excludes nonvascularized benign masses such as lipomas as well as ganglion or epidermoid cysts. The ultimate diagnosis can only be made based on histopathology.
1. Vainsencher D, Winkelmann RK. Subcutaneous sarcoidosis. Arch Dermatol. 1984;120:1028-1031.
2. Marcoval J, Maña J, Moreno A, et al. Subcutaneous sarcoidosis—clinicopathological study of 10 cases. Br J Dermatol. 2005;153:790-794.
3. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease [published online ahead of print December 2, 2005]? J Am Acad Dermatol. 2006;54:55-60.
4. Dalle Vedove C, Colato C, Girolomoni G. Subcutaneous sarcoidosis: report of two cases and review of the literature [published online ahead of print April 2, 2011]. Clin Rheumatol. 2011;30:1123-1128.
5. Barnadas MA, Rodríguez-Arias JM, Alomar A. Subcutaneous sarcoidosis associated with vitiligo, pernicious anaemia and autoimmune thyroiditis. Clin Exp Dermatol. 2000;25:55-56.
6. Higgins EM, Salisbury JR, Du Vivier AW. Subcutaneous sarcoidosis. Clin Exp Dermatol. 1993;18:65-66.
7. Heller M, Soldano AC. Sarcoidosis with subcutaneous lesions. Dermatol Online J. 2008;14:1.
8. Bosni´c D, Baresi´c M, Bagatin D, et al. Subcutaneous sarcoidosis of the face [published online ahead of print March 15, 2010]. Intern Med. 2010;49:589-592.
9. Chen HH, Chen YM, Lan HH, et al. Sonographic appearance of subcutaneous sarcoidosis. J Ultrasound Med. 2009;28:813-816.
To the Editor:
A 54-year-old woman presented with painless, firm, flesh-colored nodules measuring 1.0 to 1.5 cm in diameter on the extensor surface of the left forearm (Figure 1) and on the distal phalanx of the left thumb of 3 months’ duration. No other signs and symptoms were present. A detailed clinical examination revealed a slightly elevated erythrocyte sedimentation rate (24 mm/h [reference range, 0–20 mm/h]) and a high antinuclear antibody titer (1:3200 [reference range, <1:100])(anti–Sjögren syndrome anti-gen A, anti–Sjögren syndrome antigen B, anti-Ro52). Complete blood cell count, basic metabolic panel, liver function tests, urinalysis, pulmonary function tests, chest radiograph, and chest computed tomography all were normal. Hepatitis B antigen and antibody tests; hepatitis C antibody tests; and tuberculin test all were negative. An ophthalmic examination revealed no abnormalities. Ultrasonography of the nodules was performed with a system using an 8- to 12-MHz linear transducer and revealed 4 heterogenous hypoechoic lesions measuring up to 1.5 cm in size. Color Doppler images showed moderate hypervascularity (Figure 2). The largest nodule was excised. Histologic examination revealed noncaseating granulomas; special stains for microorganisms were negative. The histopathologic findings confirmed a diagnosis of sarcoidosis (Figure 3). The patient refused any medication. The nodules were stable at 6-month follow-up, then spontaneously resolved.
|
Subcutaneous sarcoidosis (SS) is a rare cutaneous expression of systemic sarcoidosis. The entity was first described by French physicians Darier and Roussy in 1904 as granulomatous panniculitis. Although their original study referred to a case of tuberculosis, the term Darier-Roussy sarcoid was coined and had been applied to a true sarcoid as well as to a variety of other forms of granulomatous panniculitis including those of infectious origin. A more accurate term subcutaneous sarcoidosis was established in 1984 by Vainsencher and Winkelmann.1
The most characteristic clinical picture of this disorder consists of the presence of multiple painless, firm, mobile nodules located on the extremities, most frequently the arms. However, other sites such as the trunk, buttocks, groin, head, face, and neck also have been reported.2,3
Marcoval et al2 demonstrated SS in only 2.1% of 480 patients with systemic sarcoidosis (10 patients). In the majority of these patients, subcutaneous nodules were the initial presentation of the disease.2 Ahmed and Harshad3 reported evidence of systemic involvement in 84.9% (45/53) of patients with SS. Chest involvement was the most common finding (eg, hilar lymphadenopathy, mediastinal adenopathy, interstitial pulmonary infiltration).3 Parotitis, uveitis, neuritis, and hepatosplenomegaly also have been noted systemically.4 The vast majority of reviews have suggested that SS has a relatively good prognosis. Ahmed and Harshad3 reported a satisfactory response to steroid treatment in all patients who received corticosteroids as the primary treatment. Subcutaneous sarcoidosis usually does not herald severe systemic involvement or chronic systemic complications. Both subcutaneous granulomas and hilar adenopathy may spontaneously resolve.
Interestingly, various autoimmune disease associations were seen in 6 of 21 patients (29%) in the study by Ahmed and Harshad3 including Hashimoto thyroiditis, rheumatoid arthritis, ulcerative colitis, systemic lupus erythematosus, and sicca syndrome. Barnadas et al5 reported a case of SS associated with vitiligo, pernicious anemia, and Hashimoto thyroiditis. Although our patient was not diagnosed with any particular autoimmune disease, an antinuclear antibody test was positive at a titer of 1:3200.
Our case is interesting for 2 reasons. First, it is a rare case of isolated SS. Thorough systemic evaluation showed no evidence of extracutaneous involvement. The literature only provides a few instances of isolated SS.6,7 Second, the sonographic appearance of SS is rare.8,9 Chen et al9 reported that gray-scale sonography revealed heterogenous, hypoechoic, well-demarcated plaquelike lesions with an intensive vascular pattern indicating Doppler hypervascularization. We obtained similar findings.
It has been widely acknowledged that sonographic findings of subcutaneous nodules tend to be nonspecific and overlapping. Color Doppler examination may show internal vessels both in malignant soft-tissue masses (eg, lymphoma, synovial sarcoma, liposarcoma, malignant fibrohistocytoma, metastases) and in benign lesions (eg, schwannoma, hemangioma, fibromatosis). However, the application of Doppler ultrasonography may restrict the diagnostic field, as it excludes nonvascularized benign masses such as lipomas as well as ganglion or epidermoid cysts. The ultimate diagnosis can only be made based on histopathology.
To the Editor:
A 54-year-old woman presented with painless, firm, flesh-colored nodules measuring 1.0 to 1.5 cm in diameter on the extensor surface of the left forearm (Figure 1) and on the distal phalanx of the left thumb of 3 months’ duration. No other signs and symptoms were present. A detailed clinical examination revealed a slightly elevated erythrocyte sedimentation rate (24 mm/h [reference range, 0–20 mm/h]) and a high antinuclear antibody titer (1:3200 [reference range, <1:100])(anti–Sjögren syndrome anti-gen A, anti–Sjögren syndrome antigen B, anti-Ro52). Complete blood cell count, basic metabolic panel, liver function tests, urinalysis, pulmonary function tests, chest radiograph, and chest computed tomography all were normal. Hepatitis B antigen and antibody tests; hepatitis C antibody tests; and tuberculin test all were negative. An ophthalmic examination revealed no abnormalities. Ultrasonography of the nodules was performed with a system using an 8- to 12-MHz linear transducer and revealed 4 heterogenous hypoechoic lesions measuring up to 1.5 cm in size. Color Doppler images showed moderate hypervascularity (Figure 2). The largest nodule was excised. Histologic examination revealed noncaseating granulomas; special stains for microorganisms were negative. The histopathologic findings confirmed a diagnosis of sarcoidosis (Figure 3). The patient refused any medication. The nodules were stable at 6-month follow-up, then spontaneously resolved.
|
Subcutaneous sarcoidosis (SS) is a rare cutaneous expression of systemic sarcoidosis. The entity was first described by French physicians Darier and Roussy in 1904 as granulomatous panniculitis. Although their original study referred to a case of tuberculosis, the term Darier-Roussy sarcoid was coined and had been applied to a true sarcoid as well as to a variety of other forms of granulomatous panniculitis including those of infectious origin. A more accurate term subcutaneous sarcoidosis was established in 1984 by Vainsencher and Winkelmann.1
The most characteristic clinical picture of this disorder consists of the presence of multiple painless, firm, mobile nodules located on the extremities, most frequently the arms. However, other sites such as the trunk, buttocks, groin, head, face, and neck also have been reported.2,3
Marcoval et al2 demonstrated SS in only 2.1% of 480 patients with systemic sarcoidosis (10 patients). In the majority of these patients, subcutaneous nodules were the initial presentation of the disease.2 Ahmed and Harshad3 reported evidence of systemic involvement in 84.9% (45/53) of patients with SS. Chest involvement was the most common finding (eg, hilar lymphadenopathy, mediastinal adenopathy, interstitial pulmonary infiltration).3 Parotitis, uveitis, neuritis, and hepatosplenomegaly also have been noted systemically.4 The vast majority of reviews have suggested that SS has a relatively good prognosis. Ahmed and Harshad3 reported a satisfactory response to steroid treatment in all patients who received corticosteroids as the primary treatment. Subcutaneous sarcoidosis usually does not herald severe systemic involvement or chronic systemic complications. Both subcutaneous granulomas and hilar adenopathy may spontaneously resolve.
Interestingly, various autoimmune disease associations were seen in 6 of 21 patients (29%) in the study by Ahmed and Harshad3 including Hashimoto thyroiditis, rheumatoid arthritis, ulcerative colitis, systemic lupus erythematosus, and sicca syndrome. Barnadas et al5 reported a case of SS associated with vitiligo, pernicious anemia, and Hashimoto thyroiditis. Although our patient was not diagnosed with any particular autoimmune disease, an antinuclear antibody test was positive at a titer of 1:3200.
Our case is interesting for 2 reasons. First, it is a rare case of isolated SS. Thorough systemic evaluation showed no evidence of extracutaneous involvement. The literature only provides a few instances of isolated SS.6,7 Second, the sonographic appearance of SS is rare.8,9 Chen et al9 reported that gray-scale sonography revealed heterogenous, hypoechoic, well-demarcated plaquelike lesions with an intensive vascular pattern indicating Doppler hypervascularization. We obtained similar findings.
It has been widely acknowledged that sonographic findings of subcutaneous nodules tend to be nonspecific and overlapping. Color Doppler examination may show internal vessels both in malignant soft-tissue masses (eg, lymphoma, synovial sarcoma, liposarcoma, malignant fibrohistocytoma, metastases) and in benign lesions (eg, schwannoma, hemangioma, fibromatosis). However, the application of Doppler ultrasonography may restrict the diagnostic field, as it excludes nonvascularized benign masses such as lipomas as well as ganglion or epidermoid cysts. The ultimate diagnosis can only be made based on histopathology.
1. Vainsencher D, Winkelmann RK. Subcutaneous sarcoidosis. Arch Dermatol. 1984;120:1028-1031.
2. Marcoval J, Maña J, Moreno A, et al. Subcutaneous sarcoidosis—clinicopathological study of 10 cases. Br J Dermatol. 2005;153:790-794.
3. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease [published online ahead of print December 2, 2005]? J Am Acad Dermatol. 2006;54:55-60.
4. Dalle Vedove C, Colato C, Girolomoni G. Subcutaneous sarcoidosis: report of two cases and review of the literature [published online ahead of print April 2, 2011]. Clin Rheumatol. 2011;30:1123-1128.
5. Barnadas MA, Rodríguez-Arias JM, Alomar A. Subcutaneous sarcoidosis associated with vitiligo, pernicious anaemia and autoimmune thyroiditis. Clin Exp Dermatol. 2000;25:55-56.
6. Higgins EM, Salisbury JR, Du Vivier AW. Subcutaneous sarcoidosis. Clin Exp Dermatol. 1993;18:65-66.
7. Heller M, Soldano AC. Sarcoidosis with subcutaneous lesions. Dermatol Online J. 2008;14:1.
8. Bosni´c D, Baresi´c M, Bagatin D, et al. Subcutaneous sarcoidosis of the face [published online ahead of print March 15, 2010]. Intern Med. 2010;49:589-592.
9. Chen HH, Chen YM, Lan HH, et al. Sonographic appearance of subcutaneous sarcoidosis. J Ultrasound Med. 2009;28:813-816.
1. Vainsencher D, Winkelmann RK. Subcutaneous sarcoidosis. Arch Dermatol. 1984;120:1028-1031.
2. Marcoval J, Maña J, Moreno A, et al. Subcutaneous sarcoidosis—clinicopathological study of 10 cases. Br J Dermatol. 2005;153:790-794.
3. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease [published online ahead of print December 2, 2005]? J Am Acad Dermatol. 2006;54:55-60.
4. Dalle Vedove C, Colato C, Girolomoni G. Subcutaneous sarcoidosis: report of two cases and review of the literature [published online ahead of print April 2, 2011]. Clin Rheumatol. 2011;30:1123-1128.
5. Barnadas MA, Rodríguez-Arias JM, Alomar A. Subcutaneous sarcoidosis associated with vitiligo, pernicious anaemia and autoimmune thyroiditis. Clin Exp Dermatol. 2000;25:55-56.
6. Higgins EM, Salisbury JR, Du Vivier AW. Subcutaneous sarcoidosis. Clin Exp Dermatol. 1993;18:65-66.
7. Heller M, Soldano AC. Sarcoidosis with subcutaneous lesions. Dermatol Online J. 2008;14:1.
8. Bosni´c D, Baresi´c M, Bagatin D, et al. Subcutaneous sarcoidosis of the face [published online ahead of print March 15, 2010]. Intern Med. 2010;49:589-592.
9. Chen HH, Chen YM, Lan HH, et al. Sonographic appearance of subcutaneous sarcoidosis. J Ultrasound Med. 2009;28:813-816.
Inability to Grow Long Hair: A Presentation of Trichorrhexis Nodosa
To the Editor:
First identified by Samuel Wilks in 1852, trichorrhexis nodosa (TN) is a congenital or acquired hair shaft disorder that is characterized by fragile and easily broken hair.1 Congenital TN is rare and can occur in syndromes such as pseudomonilethrix, Netherton syndrome, pili annulati,2 argininosuccinic aciduria,3 trichothiodystrophy,4 Menkes syndrome,5 and trichohepatoenteric syndrome.6 The primary congenital form of TN is inherited as an autosomal-dominant trait in some families. Acquired TN is the most common hair shaft abnormality and often is overlooked. It is provoked by hair injury, usually mechanical or physical, or chemical trauma.7,8
Chemical trauma is caused by the use of permanent hair liquids or dyes. Mechanical injuries are the result of frequent brushing, scalp massage, or lengthy backcombing, and physical damage includes excessive UV exposure or repeated application of heat. Habit tics, trichotillomania, and the scratching and pulling associated with pruritic dermatoses also can result in sufficient damage to provoke TN. Furthermore, this acquired disorder may develop from malnutrition, particularly iron deficiency, or endocrinopathy such as hypothyroidism.9 Seasonal recurrence of TN has been reported from the cumulative effect of repeated soaking in salt water and exposure to UV light. Macroscopically, hair shafts affected by TN contain small white nodes at irregular intervals throughout the length of the hair shaft. These nodes represent areas of cuticular cell disruption, which allows the underlying cortical fibers to separate and fray and gives the node the microscopic appearance of 2 brooms or paintbrushes thrusting together end-to-end by the bristles. The classic description is known as paintbrush fracture.10 Generally, complete breakage occurs at these nodes.
A 21-year-old white woman presented to our clinic with hair fragility and inability to grow long hair of 2 years’ duration. The hair was lusterless and dry. Dermoscopic examination revealed broken blunt-ended hair of uneven length with minute pinpoint grayish white nodules (Figure 1). Small fragments could be easily broken off with gentle tugging on the distal ends. She reported a history of severe sunlight and seawater exposure during the last 2 summers and the continuous use of a flat iron in the last year. Microscopic examination of hair samples with a scanning electron microscope showed the characteristic paintbrush fracture (Figure 2). She had no history of diseases, and blood examinations including complete blood cell count, thyroid function test, and iron levels were within reference range.

|

We hypothesize that the seasonal damage caused by exposure to UV light and salt water with repeated trauma from the heat of the flat iron caused distal TN. The patient was given an explanation about the diagnosis of TN and was instructed to avoid the practices that were suspected causes of the condition. Use of a gentle shampoo and conditioner also was recommended. At 6-month follow-up, we noticed an improvement of the quality of hair with a reduction in the whitish nodules and a revival of hair growth.
Acquired TN has been classified into 3 clinical forms: proximal, distal, and localized.1 Proximal TN is common in black individuals who use caustic chemicals when styling the hair. The involved hairs develop the characteristic nodes that break within a few centimeters from the scalp, especially in areas subject to friction from combing or sleeping. Distal TN primarily occurs in white or Asian individuals. In this disorder, nodes and breakage occur near the ends of the hairs that appear dull, dry, and uneven. Breakage commonly is associated with trichoptilosis, or longitudinal splitting, commonly referred to as split ends. This breakage may reflect frequent use of shampoo or heat treatments. The distal acquired form may simulate dandruff or pediculosis and the detection of this hair defect often is casual.
Localized TN, described by Raymond Sabouraud in 1921, is a rare disorder. It occurs in a patch that is usually a few centimeters long. It generally is accompanied by a pruritic dermatosis, such as circumscribed neurodermatitis, contact dermatitis, or atopic dermatitis. Scratching and rubbing most likely are the ultimate causes.
Trichorrhexis nodosa can spontaneously resolve. In all cases, diagnosis depends on careful microscopy examination and, if possible, scanning electron microscopy. Treatment is aimed at minimizing mechanical and physical injury, and chemical trauma. Excessive brushing, hot-combing, permanent waving, and other harsh hair treatments should be avoided. If the hair is long and the damage is distal, it may be sufficient to cut the distal fraction and to change cosmetic practices to prevent relapse.
Dermatologists who see patients with hair fragility and inability to grow long hair should consider the diagnosis of TN. Acquired TN often is reversible. Complete resolution may take 2 to 4 years depending on the growth of new anagen hairs. All patients with a history of white flecking on the scalp, abnormal fragility of the hair, and failure to attain normal hair length should be questioned about their routine hair care habits as well as environmental or chemical exposures to determine and remove the source of physical or chemical trauma.
1. Whiting DA. Structural abnormalities of hair shaft. J Am Acad Dermatol. 1987;16(1, pt 1):1-25.
2. Leider M. Multiple simultaneous anomalies of the hair; report of a case exhibiting trichorrhexis nodosa, pili annulati and trichostasis spinulosa. AMA Arch Derm Syphilol. 1950;62:510-514.
3. Allan JD, Cusworth DC, Dent CE, et al. A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet. 1958;1:182-187.
4. Liang C, Morris A, Schlücker S, et al. Structural and molecular hair abnormalities in trichothiodystrophy [published online ahead of print May 25, 2006]. J Invest Dermatol. 2006;126:2210-2216.
5. Taylor CJ, Green SH. Menkes’ syndrome (trichopoliodystrophy): use of scanning electron-microscope in diagnosis and carrier identification. Dev Med Child Neurol. 1981;23:361-368.
6. Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy)[published online ahead of print February 20, 2010]. Gastroenterology. 2010;138:2388-2398.
7. Chernosky ME, Owens DW. Trichorrhexis nodosa. clinical and investigative studies. Arch Dermatol. 1966;94:577-585.
8. Owens DW, Chernosky ME. Trichorrhexis nodosa; in vitro reproduction. Arch Dermatol. 1966;94:586-588.
9. Lurie R, Hodak E, Ginzburg A, et al. Trichorrhexis nodosa: a manifestation of hypothyroidism. Cutis. 1996;57:358-359.
10. Miyamoto M, Tsuboi R, Oh-I T. Case of acquired trichorrhexis nodosa: scanning electron microscopic observation. J Dermatol. 2009;36:109-110.
To the Editor:
First identified by Samuel Wilks in 1852, trichorrhexis nodosa (TN) is a congenital or acquired hair shaft disorder that is characterized by fragile and easily broken hair.1 Congenital TN is rare and can occur in syndromes such as pseudomonilethrix, Netherton syndrome, pili annulati,2 argininosuccinic aciduria,3 trichothiodystrophy,4 Menkes syndrome,5 and trichohepatoenteric syndrome.6 The primary congenital form of TN is inherited as an autosomal-dominant trait in some families. Acquired TN is the most common hair shaft abnormality and often is overlooked. It is provoked by hair injury, usually mechanical or physical, or chemical trauma.7,8
Chemical trauma is caused by the use of permanent hair liquids or dyes. Mechanical injuries are the result of frequent brushing, scalp massage, or lengthy backcombing, and physical damage includes excessive UV exposure or repeated application of heat. Habit tics, trichotillomania, and the scratching and pulling associated with pruritic dermatoses also can result in sufficient damage to provoke TN. Furthermore, this acquired disorder may develop from malnutrition, particularly iron deficiency, or endocrinopathy such as hypothyroidism.9 Seasonal recurrence of TN has been reported from the cumulative effect of repeated soaking in salt water and exposure to UV light. Macroscopically, hair shafts affected by TN contain small white nodes at irregular intervals throughout the length of the hair shaft. These nodes represent areas of cuticular cell disruption, which allows the underlying cortical fibers to separate and fray and gives the node the microscopic appearance of 2 brooms or paintbrushes thrusting together end-to-end by the bristles. The classic description is known as paintbrush fracture.10 Generally, complete breakage occurs at these nodes.
A 21-year-old white woman presented to our clinic with hair fragility and inability to grow long hair of 2 years’ duration. The hair was lusterless and dry. Dermoscopic examination revealed broken blunt-ended hair of uneven length with minute pinpoint grayish white nodules (Figure 1). Small fragments could be easily broken off with gentle tugging on the distal ends. She reported a history of severe sunlight and seawater exposure during the last 2 summers and the continuous use of a flat iron in the last year. Microscopic examination of hair samples with a scanning electron microscope showed the characteristic paintbrush fracture (Figure 2). She had no history of diseases, and blood examinations including complete blood cell count, thyroid function test, and iron levels were within reference range.
|
We hypothesize that the seasonal damage caused by exposure to UV light and salt water with repeated trauma from the heat of the flat iron caused distal TN. The patient was given an explanation about the diagnosis of TN and was instructed to avoid the practices that were suspected causes of the condition. Use of a gentle shampoo and conditioner also was recommended. At 6-month follow-up, we noticed an improvement of the quality of hair with a reduction in the whitish nodules and a revival of hair growth.
Acquired TN has been classified into 3 clinical forms: proximal, distal, and localized.1 Proximal TN is common in black individuals who use caustic chemicals when styling the hair. The involved hairs develop the characteristic nodes that break within a few centimeters from the scalp, especially in areas subject to friction from combing or sleeping. Distal TN primarily occurs in white or Asian individuals. In this disorder, nodes and breakage occur near the ends of the hairs that appear dull, dry, and uneven. Breakage commonly is associated with trichoptilosis, or longitudinal splitting, commonly referred to as split ends. This breakage may reflect frequent use of shampoo or heat treatments. The distal acquired form may simulate dandruff or pediculosis and the detection of this hair defect often is casual.
Localized TN, described by Raymond Sabouraud in 1921, is a rare disorder. It occurs in a patch that is usually a few centimeters long. It generally is accompanied by a pruritic dermatosis, such as circumscribed neurodermatitis, contact dermatitis, or atopic dermatitis. Scratching and rubbing most likely are the ultimate causes.
Trichorrhexis nodosa can spontaneously resolve. In all cases, diagnosis depends on careful microscopy examination and, if possible, scanning electron microscopy. Treatment is aimed at minimizing mechanical and physical injury, and chemical trauma. Excessive brushing, hot-combing, permanent waving, and other harsh hair treatments should be avoided. If the hair is long and the damage is distal, it may be sufficient to cut the distal fraction and to change cosmetic practices to prevent relapse.
Dermatologists who see patients with hair fragility and inability to grow long hair should consider the diagnosis of TN. Acquired TN often is reversible. Complete resolution may take 2 to 4 years depending on the growth of new anagen hairs. All patients with a history of white flecking on the scalp, abnormal fragility of the hair, and failure to attain normal hair length should be questioned about their routine hair care habits as well as environmental or chemical exposures to determine and remove the source of physical or chemical trauma.
To the Editor:
First identified by Samuel Wilks in 1852, trichorrhexis nodosa (TN) is a congenital or acquired hair shaft disorder that is characterized by fragile and easily broken hair.1 Congenital TN is rare and can occur in syndromes such as pseudomonilethrix, Netherton syndrome, pili annulati,2 argininosuccinic aciduria,3 trichothiodystrophy,4 Menkes syndrome,5 and trichohepatoenteric syndrome.6 The primary congenital form of TN is inherited as an autosomal-dominant trait in some families. Acquired TN is the most common hair shaft abnormality and often is overlooked. It is provoked by hair injury, usually mechanical or physical, or chemical trauma.7,8
Chemical trauma is caused by the use of permanent hair liquids or dyes. Mechanical injuries are the result of frequent brushing, scalp massage, or lengthy backcombing, and physical damage includes excessive UV exposure or repeated application of heat. Habit tics, trichotillomania, and the scratching and pulling associated with pruritic dermatoses also can result in sufficient damage to provoke TN. Furthermore, this acquired disorder may develop from malnutrition, particularly iron deficiency, or endocrinopathy such as hypothyroidism.9 Seasonal recurrence of TN has been reported from the cumulative effect of repeated soaking in salt water and exposure to UV light. Macroscopically, hair shafts affected by TN contain small white nodes at irregular intervals throughout the length of the hair shaft. These nodes represent areas of cuticular cell disruption, which allows the underlying cortical fibers to separate and fray and gives the node the microscopic appearance of 2 brooms or paintbrushes thrusting together end-to-end by the bristles. The classic description is known as paintbrush fracture.10 Generally, complete breakage occurs at these nodes.
A 21-year-old white woman presented to our clinic with hair fragility and inability to grow long hair of 2 years’ duration. The hair was lusterless and dry. Dermoscopic examination revealed broken blunt-ended hair of uneven length with minute pinpoint grayish white nodules (Figure 1). Small fragments could be easily broken off with gentle tugging on the distal ends. She reported a history of severe sunlight and seawater exposure during the last 2 summers and the continuous use of a flat iron in the last year. Microscopic examination of hair samples with a scanning electron microscope showed the characteristic paintbrush fracture (Figure 2). She had no history of diseases, and blood examinations including complete blood cell count, thyroid function test, and iron levels were within reference range.
|
We hypothesize that the seasonal damage caused by exposure to UV light and salt water with repeated trauma from the heat of the flat iron caused distal TN. The patient was given an explanation about the diagnosis of TN and was instructed to avoid the practices that were suspected causes of the condition. Use of a gentle shampoo and conditioner also was recommended. At 6-month follow-up, we noticed an improvement of the quality of hair with a reduction in the whitish nodules and a revival of hair growth.
Acquired TN has been classified into 3 clinical forms: proximal, distal, and localized.1 Proximal TN is common in black individuals who use caustic chemicals when styling the hair. The involved hairs develop the characteristic nodes that break within a few centimeters from the scalp, especially in areas subject to friction from combing or sleeping. Distal TN primarily occurs in white or Asian individuals. In this disorder, nodes and breakage occur near the ends of the hairs that appear dull, dry, and uneven. Breakage commonly is associated with trichoptilosis, or longitudinal splitting, commonly referred to as split ends. This breakage may reflect frequent use of shampoo or heat treatments. The distal acquired form may simulate dandruff or pediculosis and the detection of this hair defect often is casual.
Localized TN, described by Raymond Sabouraud in 1921, is a rare disorder. It occurs in a patch that is usually a few centimeters long. It generally is accompanied by a pruritic dermatosis, such as circumscribed neurodermatitis, contact dermatitis, or atopic dermatitis. Scratching and rubbing most likely are the ultimate causes.
Trichorrhexis nodosa can spontaneously resolve. In all cases, diagnosis depends on careful microscopy examination and, if possible, scanning electron microscopy. Treatment is aimed at minimizing mechanical and physical injury, and chemical trauma. Excessive brushing, hot-combing, permanent waving, and other harsh hair treatments should be avoided. If the hair is long and the damage is distal, it may be sufficient to cut the distal fraction and to change cosmetic practices to prevent relapse.
Dermatologists who see patients with hair fragility and inability to grow long hair should consider the diagnosis of TN. Acquired TN often is reversible. Complete resolution may take 2 to 4 years depending on the growth of new anagen hairs. All patients with a history of white flecking on the scalp, abnormal fragility of the hair, and failure to attain normal hair length should be questioned about their routine hair care habits as well as environmental or chemical exposures to determine and remove the source of physical or chemical trauma.
1. Whiting DA. Structural abnormalities of hair shaft. J Am Acad Dermatol. 1987;16(1, pt 1):1-25.
2. Leider M. Multiple simultaneous anomalies of the hair; report of a case exhibiting trichorrhexis nodosa, pili annulati and trichostasis spinulosa. AMA Arch Derm Syphilol. 1950;62:510-514.
3. Allan JD, Cusworth DC, Dent CE, et al. A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet. 1958;1:182-187.
4. Liang C, Morris A, Schlücker S, et al. Structural and molecular hair abnormalities in trichothiodystrophy [published online ahead of print May 25, 2006]. J Invest Dermatol. 2006;126:2210-2216.
5. Taylor CJ, Green SH. Menkes’ syndrome (trichopoliodystrophy): use of scanning electron-microscope in diagnosis and carrier identification. Dev Med Child Neurol. 1981;23:361-368.
6. Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy)[published online ahead of print February 20, 2010]. Gastroenterology. 2010;138:2388-2398.
7. Chernosky ME, Owens DW. Trichorrhexis nodosa. clinical and investigative studies. Arch Dermatol. 1966;94:577-585.
8. Owens DW, Chernosky ME. Trichorrhexis nodosa; in vitro reproduction. Arch Dermatol. 1966;94:586-588.
9. Lurie R, Hodak E, Ginzburg A, et al. Trichorrhexis nodosa: a manifestation of hypothyroidism. Cutis. 1996;57:358-359.
10. Miyamoto M, Tsuboi R, Oh-I T. Case of acquired trichorrhexis nodosa: scanning electron microscopic observation. J Dermatol. 2009;36:109-110.
1. Whiting DA. Structural abnormalities of hair shaft. J Am Acad Dermatol. 1987;16(1, pt 1):1-25.
2. Leider M. Multiple simultaneous anomalies of the hair; report of a case exhibiting trichorrhexis nodosa, pili annulati and trichostasis spinulosa. AMA Arch Derm Syphilol. 1950;62:510-514.
3. Allan JD, Cusworth DC, Dent CE, et al. A disease, probably hereditary characterised by severe mental deficiency and a constant gross abnormality of aminoacid metabolism. Lancet. 1958;1:182-187.
4. Liang C, Morris A, Schlücker S, et al. Structural and molecular hair abnormalities in trichothiodystrophy [published online ahead of print May 25, 2006]. J Invest Dermatol. 2006;126:2210-2216.
5. Taylor CJ, Green SH. Menkes’ syndrome (trichopoliodystrophy): use of scanning electron-microscope in diagnosis and carrier identification. Dev Med Child Neurol. 1981;23:361-368.
6. Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy)[published online ahead of print February 20, 2010]. Gastroenterology. 2010;138:2388-2398.
7. Chernosky ME, Owens DW. Trichorrhexis nodosa. clinical and investigative studies. Arch Dermatol. 1966;94:577-585.
8. Owens DW, Chernosky ME. Trichorrhexis nodosa; in vitro reproduction. Arch Dermatol. 1966;94:586-588.
9. Lurie R, Hodak E, Ginzburg A, et al. Trichorrhexis nodosa: a manifestation of hypothyroidism. Cutis. 1996;57:358-359.
10. Miyamoto M, Tsuboi R, Oh-I T. Case of acquired trichorrhexis nodosa: scanning electron microscopic observation. J Dermatol. 2009;36:109-110.

Dermatologic Toxicity in a Patient Receiving Liposomal Doxorubicin
To the Editor:
Liposomal doxorubicin hydrochloride is an anthracycline topoisomerase inhibitor indicated for ovarian cancer, AIDS-related Kaposi sarcoma, and multiple myeloma.1 It also has been used with limited success in a clinical trial of previously treated patients with endometrial cancer.2 The most common adverse reactions include asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-and-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia.1
A 58-year-old woman with a history of stage IIIA endometrial cancer underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy soon after diagnosis. She then completed 5 high-dose-rate brachytherapy treatments and 6 cycles of paclitaxel and carboplatin. Follow-up imaging revealed pulmonary metastasis. The patient was then enrolled in a clinical trial but was switched to 40 mg/m2 liposomal doxorubicin given once every 28 days for 5 cycles after progression of disease.
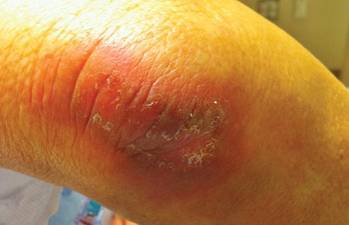
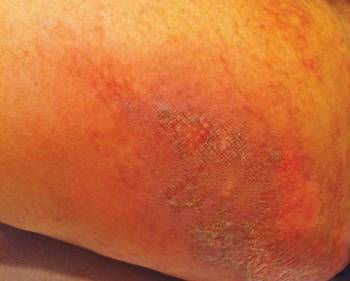
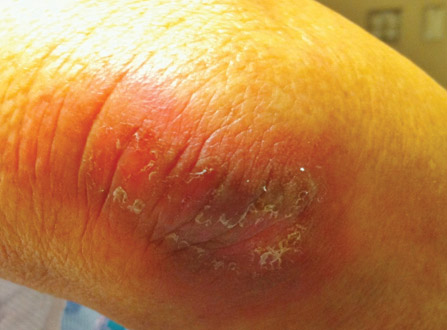
After each dose of doxorubicin, she developed redness of the palms and soles. Following the third cycle of doxorubicin, a painful rash involving the thighs and axilla appeared with some desquamation in the left axilla. Three weeks after the fourth dose of doxorubicin, she presented with severe worsening of the rash to involve the extensor elbows (Figure 1), back, and lower legs with bilateral axillary desquamation. The bilateral medial thighs were erythematous with maceration that was tender and blanchable (Figure 2). The total affected body surface area was 10% to 15%. There was no involvement of the mucosa. She was treated with hydrogel sheet dressings and silver sulfadiazine cream 1%.
|
|
The patient’s rash was thought to be due to doxorubicin toxicity; however, a 4-mm punch biopsy specimen from the left thigh was taken for culture and hemotoxylin and eosin stain to rule out other possibilities. Biopsy was consistent with a drug reaction, revealing superficial perivascular dermatitis with keratinocyte atypia of the epidermis. Doxorubicin was discontinued and the rash resolved completely within 2 weeks, except for some thickening of the skin on the palms, soles, and thighs. After a delay of approximately 1 week, doxorubicin was resumed at a lower dose of 30 mg/m2. No dermatologic symptoms followed treatment at this dose.
Four clinical patterns of doxorubicin toxicity are recognized. The most common pattern is acral erythema, also known as hand-and-foot syndrome, which is followed by desquamation of the palms and soles, occurring in approximately 50% of patients. Ten percent of patients experience a diffuse follicular rash with mild, diffuse, scaly erythema and follicular accentuation that often occurs over the lateral limbs but also may occur over the trunk. New melanotic macules may appear on the trunk or extremities including palms and soles.3 Finally, an intertrigolike eruption exacerbated by friction with erythematous patches over skin folds or in areas of friction also has been described.3-5 Our patient presented with a combination of dermatologic toxicities including acral erythema and intertrigolike eruption. Acral erythema occurred in 24 of 60 patients and intertrigolike eruption occurred in 5 of 60 patients in one study.3 Another report documented both occurring together.5
Treatment of doxorubicin skin toxicity consists of reduction of the dose of doxorubicin, supportive care, and patient education. Specific treatments include topical wound care, emollient creams, and pain management with analgesics. Other interventions include wearing loose clothing, avoiding vigorous exercise, and sitting on padded surfaces.6
Doxorubicin skin toxicity presents in several clinical patterns. Although acral erythema is the most common pattern, severe intertrigolike eruptions similar to our case may occur. Physicians caring for patients receiving doxorubicin should be aware of the variety of presentations of skin toxicity and the possible need for dose reduction to decrease symptoms.
1. Doxil [package insert]. Horsham, PA: Janssen Products, LP; 2014.
2. Muggia FM, Blessing JA, Sorosky J, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2002;20:2360-2364.
3. Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475-1480.
4. Korver GE, Ronald H, Petersen MJ. An intertrigo-like eruption from pegylated liposomal doxorubicin. J Drugs Dermatol. 2006;5:901-902.
5. Sánchez Henarejos P, Ros Martinez S, Marín Zafra GR,
et al. Intertrigo-like eruption caused by pegylated liposomal doxorubicin (PLD). Clin Transl Oncol. 2009;11:486-487.
6. von Moos R, Thuerlimann BJ, Aapro M, et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: recommendations of an international panel of experts [published online ahead of print March 10, 2008]. Eur J Cancer. 2008;44:781-790.
To the Editor:
Liposomal doxorubicin hydrochloride is an anthracycline topoisomerase inhibitor indicated for ovarian cancer, AIDS-related Kaposi sarcoma, and multiple myeloma.1 It also has been used with limited success in a clinical trial of previously treated patients with endometrial cancer.2 The most common adverse reactions include asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-and-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia.1
A 58-year-old woman with a history of stage IIIA endometrial cancer underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy soon after diagnosis. She then completed 5 high-dose-rate brachytherapy treatments and 6 cycles of paclitaxel and carboplatin. Follow-up imaging revealed pulmonary metastasis. The patient was then enrolled in a clinical trial but was switched to 40 mg/m2 liposomal doxorubicin given once every 28 days for 5 cycles after progression of disease.
After each dose of doxorubicin, she developed redness of the palms and soles. Following the third cycle of doxorubicin, a painful rash involving the thighs and axilla appeared with some desquamation in the left axilla. Three weeks after the fourth dose of doxorubicin, she presented with severe worsening of the rash to involve the extensor elbows (Figure 1), back, and lower legs with bilateral axillary desquamation. The bilateral medial thighs were erythematous with maceration that was tender and blanchable (Figure 2). The total affected body surface area was 10% to 15%. There was no involvement of the mucosa. She was treated with hydrogel sheet dressings and silver sulfadiazine cream 1%.
|
|
|
|
The patient’s rash was thought to be due to doxorubicin toxicity; however, a 4-mm punch biopsy specimen from the left thigh was taken for culture and hemotoxylin and eosin stain to rule out other possibilities. Biopsy was consistent with a drug reaction, revealing superficial perivascular dermatitis with keratinocyte atypia of the epidermis. Doxorubicin was discontinued and the rash resolved completely within 2 weeks, except for some thickening of the skin on the palms, soles, and thighs. After a delay of approximately 1 week, doxorubicin was resumed at a lower dose of 30 mg/m2. No dermatologic symptoms followed treatment at this dose.
Four clinical patterns of doxorubicin toxicity are recognized. The most common pattern is acral erythema, also known as hand-and-foot syndrome, which is followed by desquamation of the palms and soles, occurring in approximately 50% of patients. Ten percent of patients experience a diffuse follicular rash with mild, diffuse, scaly erythema and follicular accentuation that often occurs over the lateral limbs but also may occur over the trunk. New melanotic macules may appear on the trunk or extremities including palms and soles.3 Finally, an intertrigolike eruption exacerbated by friction with erythematous patches over skin folds or in areas of friction also has been described.3-5 Our patient presented with a combination of dermatologic toxicities including acral erythema and intertrigolike eruption. Acral erythema occurred in 24 of 60 patients and intertrigolike eruption occurred in 5 of 60 patients in one study.3 Another report documented both occurring together.5
Treatment of doxorubicin skin toxicity consists of reduction of the dose of doxorubicin, supportive care, and patient education. Specific treatments include topical wound care, emollient creams, and pain management with analgesics. Other interventions include wearing loose clothing, avoiding vigorous exercise, and sitting on padded surfaces.6
Doxorubicin skin toxicity presents in several clinical patterns. Although acral erythema is the most common pattern, severe intertrigolike eruptions similar to our case may occur. Physicians caring for patients receiving doxorubicin should be aware of the variety of presentations of skin toxicity and the possible need for dose reduction to decrease symptoms.
To the Editor:
Liposomal doxorubicin hydrochloride is an anthracycline topoisomerase inhibitor indicated for ovarian cancer, AIDS-related Kaposi sarcoma, and multiple myeloma.1 It also has been used with limited success in a clinical trial of previously treated patients with endometrial cancer.2 The most common adverse reactions include asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-and-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia.1
A 58-year-old woman with a history of stage IIIA endometrial cancer underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy soon after diagnosis. She then completed 5 high-dose-rate brachytherapy treatments and 6 cycles of paclitaxel and carboplatin. Follow-up imaging revealed pulmonary metastasis. The patient was then enrolled in a clinical trial but was switched to 40 mg/m2 liposomal doxorubicin given once every 28 days for 5 cycles after progression of disease.
After each dose of doxorubicin, she developed redness of the palms and soles. Following the third cycle of doxorubicin, a painful rash involving the thighs and axilla appeared with some desquamation in the left axilla. Three weeks after the fourth dose of doxorubicin, she presented with severe worsening of the rash to involve the extensor elbows (Figure 1), back, and lower legs with bilateral axillary desquamation. The bilateral medial thighs were erythematous with maceration that was tender and blanchable (Figure 2). The total affected body surface area was 10% to 15%. There was no involvement of the mucosa. She was treated with hydrogel sheet dressings and silver sulfadiazine cream 1%.
|
|
|
|
The patient’s rash was thought to be due to doxorubicin toxicity; however, a 4-mm punch biopsy specimen from the left thigh was taken for culture and hemotoxylin and eosin stain to rule out other possibilities. Biopsy was consistent with a drug reaction, revealing superficial perivascular dermatitis with keratinocyte atypia of the epidermis. Doxorubicin was discontinued and the rash resolved completely within 2 weeks, except for some thickening of the skin on the palms, soles, and thighs. After a delay of approximately 1 week, doxorubicin was resumed at a lower dose of 30 mg/m2. No dermatologic symptoms followed treatment at this dose.
Four clinical patterns of doxorubicin toxicity are recognized. The most common pattern is acral erythema, also known as hand-and-foot syndrome, which is followed by desquamation of the palms and soles, occurring in approximately 50% of patients. Ten percent of patients experience a diffuse follicular rash with mild, diffuse, scaly erythema and follicular accentuation that often occurs over the lateral limbs but also may occur over the trunk. New melanotic macules may appear on the trunk or extremities including palms and soles.3 Finally, an intertrigolike eruption exacerbated by friction with erythematous patches over skin folds or in areas of friction also has been described.3-5 Our patient presented with a combination of dermatologic toxicities including acral erythema and intertrigolike eruption. Acral erythema occurred in 24 of 60 patients and intertrigolike eruption occurred in 5 of 60 patients in one study.3 Another report documented both occurring together.5
Treatment of doxorubicin skin toxicity consists of reduction of the dose of doxorubicin, supportive care, and patient education. Specific treatments include topical wound care, emollient creams, and pain management with analgesics. Other interventions include wearing loose clothing, avoiding vigorous exercise, and sitting on padded surfaces.6
Doxorubicin skin toxicity presents in several clinical patterns. Although acral erythema is the most common pattern, severe intertrigolike eruptions similar to our case may occur. Physicians caring for patients receiving doxorubicin should be aware of the variety of presentations of skin toxicity and the possible need for dose reduction to decrease symptoms.
1. Doxil [package insert]. Horsham, PA: Janssen Products, LP; 2014.
2. Muggia FM, Blessing JA, Sorosky J, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2002;20:2360-2364.
3. Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475-1480.
4. Korver GE, Ronald H, Petersen MJ. An intertrigo-like eruption from pegylated liposomal doxorubicin. J Drugs Dermatol. 2006;5:901-902.
5. Sánchez Henarejos P, Ros Martinez S, Marín Zafra GR,
et al. Intertrigo-like eruption caused by pegylated liposomal doxorubicin (PLD). Clin Transl Oncol. 2009;11:486-487.
6. von Moos R, Thuerlimann BJ, Aapro M, et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: recommendations of an international panel of experts [published online ahead of print March 10, 2008]. Eur J Cancer. 2008;44:781-790.
1. Doxil [package insert]. Horsham, PA: Janssen Products, LP; 2014.
2. Muggia FM, Blessing JA, Sorosky J, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2002;20:2360-2364.
3. Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475-1480.
4. Korver GE, Ronald H, Petersen MJ. An intertrigo-like eruption from pegylated liposomal doxorubicin. J Drugs Dermatol. 2006;5:901-902.
5. Sánchez Henarejos P, Ros Martinez S, Marín Zafra GR,
et al. Intertrigo-like eruption caused by pegylated liposomal doxorubicin (PLD). Clin Transl Oncol. 2009;11:486-487.
6. von Moos R, Thuerlimann BJ, Aapro M, et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: recommendations of an international panel of experts [published online ahead of print March 10, 2008]. Eur J Cancer. 2008;44:781-790.
An Unusual Case of Sporadic Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
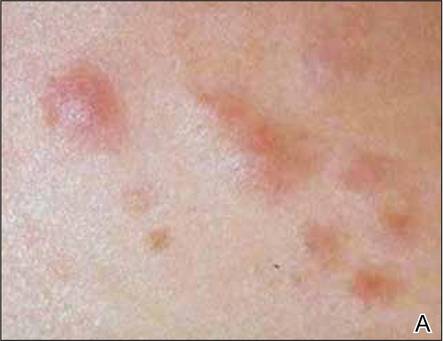

Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.

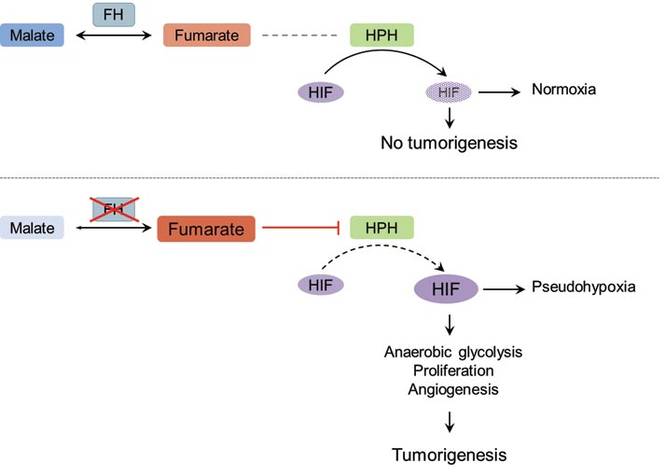
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
|
Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
|
Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
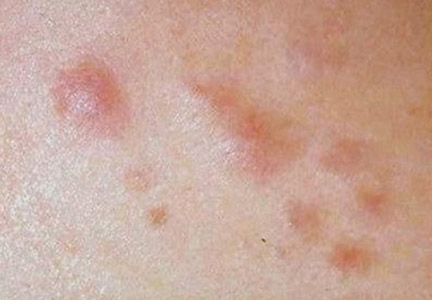
Tuberculosis Cutis Orificialis in an Immunocompetent Patient
To the Editor:
Orificial tuberculosis (OT) constitutes 2% of cutaneous tuberculosis cases and 0.01% to 1% of all clinical presentations of tuberculosis.1 It is clinically classified as primary or secondary OT. In primary OT, the oral mucosa is the initial site of the infection without any internal organ involvement.2 This form is more prevalent among men and young adults.1,3 Secondary OT is the cutaneous tuberculosis type that occurs in patients with internal organ tuberculosis from autoinoculation of bacilli to the orificial area. It is more common and usually affects elderly patients.2-4 We present the development of primary OT in an immunocompetent woman.
A 51-year-old woman was admitted with painful enlarging oral ulcers of 1 year’s duration. There was no history of tuberculosis infection, dental trauma, or smoking habit prior to the development of oral ulcers, and no family history of tuberculosis. On dermatological examination white-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate and gingiva were observed (Figures 1A and 1B). Oral hygiene was good. There was no regional lym-phadenopathy on palpation. Physical findings were normal. The histopathology of the biopsy from the gingival ulcer revealed noncaseating granulomatous inflammation in the dermis (Figure 2). Ziehl-Neelsen and periodic acid–Schiff stains were negative for acid-fast bacilli and fungi, respectively.
| 
| |

| 
| |
| Figure 1. White-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate (A) and gingiva (B). Resolution of lesions on the hard palate (C) and the gingiva (D) after 9 months of antituberculosis therapy. | ||
Laboratory results from blood chemistry, complete blood cell count, erythrocyte sedimentation rate, C-reactive protein level, and urine analysis, as well as titers of serum immunoglobulin, antineutrophil cytoplasmic antibodies, and antinuclear antibodies, were within reference range. Human immunodeficiency virus serology was negative. Chest radiography and ultrasonography of the abdomen revealed no abnormalities. A purified protein derivative (tuberculin) test showed an induration of 20 mm. Mycobacterium tuberculosis grew on the culture of the tissue specimen.
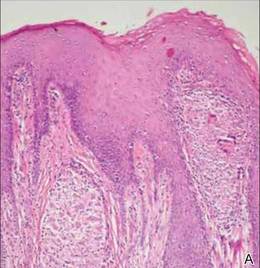
|
|
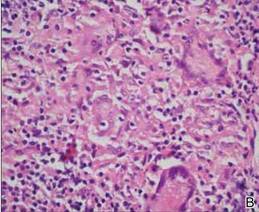
The patient was diagnosed with primary OT and treated with isoniazid (300 mg daily), rifampin (600 mg daily), ethambutol (1500 mg daily), and pyrazinamide (2000 mg daily). At 2 months of therapy the lesions started to heal and showed complete resolution at the end of 9 months of treatment (Figures 1C and 1D). There was no recurrence at 2-year follow-up.
Orificial tuberculosis is a rare form of cutaneous tuberculosis. Therefore, it is regarded as a “forgotten disease” in the literature.5 The pathogenesis of OT has not been clearly defined. The intact mucous membrane, thickened squamous epithelium, cleansing and antibacterial function of saliva, and presence of saprophytes act as protective mechanisms against penetration of mycobacteria. Some factors such as poor oral hygiene, smoking, local trauma, or presence of a dental cyst or an abscess may predispose the direct mucosal inoculation of the tuberculosis bacilli.1,2,6 Orificial tuberculosis may develop as an opportunistic infection in 1.33% of human immunodeficiency virus patients.7 However, our patient had no prior trauma or known predisposing factors.
The common presentation of OT is an ulcerative lesion with irregular and well-delineated margins with a yellowish granular base. A vesicle, papule, or nodule may precede the ulcers. The presence of yellow satellite nodules around the lesion is characteristic for OT.1,2,6 Although there were no satellite nodules around the ulcerations in our case, the lesions had a yellowish granular base and irregular, well-delineated margins.
The tongue, gingiva, lips, tonsils, and epiglottis are the most common sites of involvement.2,5,8 Hard palate involvement rarely has been reported, even in immunosuppressed patients.7 Enlarged painful cervical lymph nodes may accompany the disease. Most patients with OT have been reported to have active pulmonary tuberculosis at the time of diagnosis.6,8 Thus, internal organ involvement, particularly the pulmonary system, should be checked in patients with OT. In our patient, the hard palate was involved together with the gingiva despite the absence of immunosuppression. No internal organ involvement was found in the systemic evaluation.
Orificial tuberculosis is a form of cutaneous tuberculosis that is difficult to diagnose because of the varying nature of clinical features, failure of growth of M tuberculosis on culture, and rarity of the disease.1,2,6 As in our case, biopsies may not always exhibit a caseous necrosis, which is specific to tuberculosis.6,7 Thus, it may be difficult to distinguish oral cavity tuberculosis from conditions demonstrating oral ulcers such as bullous diseases, trauma, fungal diseases, syphilis, sarcoidosis, or squamous cell carcinoma by evaluating only signs and symptoms.6 Clinical suspicion is the first and foremost step in the diagnostic process of OT. In our patient, OT was suspected in the differential diagnosis because the resistant oral ulcerations showed the most common presentation of OT: irregular and well-delineated margins and a yellowish granular base. By considering tuberculosis within the differential diagnosis in our patient, microbiologic cultivation was performed from the oral mucosa and accurate diagnosis was established by determination of the pathogen’s growth in the culture.
Because of the increased incidence of tuberculosis and unusual manifestations, clinicians may easily overlook OT. It should be considered in the differential diagnosis of resistant nodules or ulcers of the oral cavity.
1. Kiliç A, Gül U, Gönül M, et al. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol. 2009;48:178-180.
2. Ito FA, de Andrade CR, Vargas PA, et al. Primary tuberculosis of the oral cavity. Oral Dis. 2005;11:50-53.
3. Dixit R, Sharma S, Nuwal P. Tuberculosis of oral cavity. Indian J Tuberc. 2008;55:51-53.
4. Smolka W, Burger H, Iizuka T, et al. Primary tuberculosis of the oral cavity in an elderly nonimmunosuppressed patient: case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2008;134:1107-1109.
5. Rodrigues G, Carnelio S, Valliathan M. Primary isolated gingival tuberculosis. Braz J Infect Dis. 2007;11:172-173.
6. Vilar FC, de Souza A, Moya MJ, et al. Atypical oral lesion in a patient with pulmonary tuberculosis. Int J Dermatol. 2009;48:910-912.
7. Kakisi OK, Kechagia AS, Kakisis IK, et al. Tuberculosis of the oral cavity: a systematic review. Eur J Oral Sci. 2010;118:103-109.
8. Eng HL, Lu SY, Yang CH, et al. Oral tuberculosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:415-420.
To the Editor:
Orificial tuberculosis (OT) constitutes 2% of cutaneous tuberculosis cases and 0.01% to 1% of all clinical presentations of tuberculosis.1 It is clinically classified as primary or secondary OT. In primary OT, the oral mucosa is the initial site of the infection without any internal organ involvement.2 This form is more prevalent among men and young adults.1,3 Secondary OT is the cutaneous tuberculosis type that occurs in patients with internal organ tuberculosis from autoinoculation of bacilli to the orificial area. It is more common and usually affects elderly patients.2-4 We present the development of primary OT in an immunocompetent woman.
A 51-year-old woman was admitted with painful enlarging oral ulcers of 1 year’s duration. There was no history of tuberculosis infection, dental trauma, or smoking habit prior to the development of oral ulcers, and no family history of tuberculosis. On dermatological examination white-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate and gingiva were observed (Figures 1A and 1B). Oral hygiene was good. There was no regional lym-phadenopathy on palpation. Physical findings were normal. The histopathology of the biopsy from the gingival ulcer revealed noncaseating granulomatous inflammation in the dermis (Figure 2). Ziehl-Neelsen and periodic acid–Schiff stains were negative for acid-fast bacilli and fungi, respectively.
|
|
| |
|
|
| |
| Figure 1. White-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate (A) and gingiva (B). Resolution of lesions on the hard palate (C) and the gingiva (D) after 9 months of antituberculosis therapy. | ||
Laboratory results from blood chemistry, complete blood cell count, erythrocyte sedimentation rate, C-reactive protein level, and urine analysis, as well as titers of serum immunoglobulin, antineutrophil cytoplasmic antibodies, and antinuclear antibodies, were within reference range. Human immunodeficiency virus serology was negative. Chest radiography and ultrasonography of the abdomen revealed no abnormalities. A purified protein derivative (tuberculin) test showed an induration of 20 mm. Mycobacterium tuberculosis grew on the culture of the tissue specimen.
|
|
|
The patient was diagnosed with primary OT and treated with isoniazid (300 mg daily), rifampin (600 mg daily), ethambutol (1500 mg daily), and pyrazinamide (2000 mg daily). At 2 months of therapy the lesions started to heal and showed complete resolution at the end of 9 months of treatment (Figures 1C and 1D). There was no recurrence at 2-year follow-up.
Orificial tuberculosis is a rare form of cutaneous tuberculosis. Therefore, it is regarded as a “forgotten disease” in the literature.5 The pathogenesis of OT has not been clearly defined. The intact mucous membrane, thickened squamous epithelium, cleansing and antibacterial function of saliva, and presence of saprophytes act as protective mechanisms against penetration of mycobacteria. Some factors such as poor oral hygiene, smoking, local trauma, or presence of a dental cyst or an abscess may predispose the direct mucosal inoculation of the tuberculosis bacilli.1,2,6 Orificial tuberculosis may develop as an opportunistic infection in 1.33% of human immunodeficiency virus patients.7 However, our patient had no prior trauma or known predisposing factors.
The common presentation of OT is an ulcerative lesion with irregular and well-delineated margins with a yellowish granular base. A vesicle, papule, or nodule may precede the ulcers. The presence of yellow satellite nodules around the lesion is characteristic for OT.1,2,6 Although there were no satellite nodules around the ulcerations in our case, the lesions had a yellowish granular base and irregular, well-delineated margins.
The tongue, gingiva, lips, tonsils, and epiglottis are the most common sites of involvement.2,5,8 Hard palate involvement rarely has been reported, even in immunosuppressed patients.7 Enlarged painful cervical lymph nodes may accompany the disease. Most patients with OT have been reported to have active pulmonary tuberculosis at the time of diagnosis.6,8 Thus, internal organ involvement, particularly the pulmonary system, should be checked in patients with OT. In our patient, the hard palate was involved together with the gingiva despite the absence of immunosuppression. No internal organ involvement was found in the systemic evaluation.
Orificial tuberculosis is a form of cutaneous tuberculosis that is difficult to diagnose because of the varying nature of clinical features, failure of growth of M tuberculosis on culture, and rarity of the disease.1,2,6 As in our case, biopsies may not always exhibit a caseous necrosis, which is specific to tuberculosis.6,7 Thus, it may be difficult to distinguish oral cavity tuberculosis from conditions demonstrating oral ulcers such as bullous diseases, trauma, fungal diseases, syphilis, sarcoidosis, or squamous cell carcinoma by evaluating only signs and symptoms.6 Clinical suspicion is the first and foremost step in the diagnostic process of OT. In our patient, OT was suspected in the differential diagnosis because the resistant oral ulcerations showed the most common presentation of OT: irregular and well-delineated margins and a yellowish granular base. By considering tuberculosis within the differential diagnosis in our patient, microbiologic cultivation was performed from the oral mucosa and accurate diagnosis was established by determination of the pathogen’s growth in the culture.
Because of the increased incidence of tuberculosis and unusual manifestations, clinicians may easily overlook OT. It should be considered in the differential diagnosis of resistant nodules or ulcers of the oral cavity.
To the Editor:
Orificial tuberculosis (OT) constitutes 2% of cutaneous tuberculosis cases and 0.01% to 1% of all clinical presentations of tuberculosis.1 It is clinically classified as primary or secondary OT. In primary OT, the oral mucosa is the initial site of the infection without any internal organ involvement.2 This form is more prevalent among men and young adults.1,3 Secondary OT is the cutaneous tuberculosis type that occurs in patients with internal organ tuberculosis from autoinoculation of bacilli to the orificial area. It is more common and usually affects elderly patients.2-4 We present the development of primary OT in an immunocompetent woman.
A 51-year-old woman was admitted with painful enlarging oral ulcers of 1 year’s duration. There was no history of tuberculosis infection, dental trauma, or smoking habit prior to the development of oral ulcers, and no family history of tuberculosis. On dermatological examination white-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate and gingiva were observed (Figures 1A and 1B). Oral hygiene was good. There was no regional lym-phadenopathy on palpation. Physical findings were normal. The histopathology of the biopsy from the gingival ulcer revealed noncaseating granulomatous inflammation in the dermis (Figure 2). Ziehl-Neelsen and periodic acid–Schiff stains were negative for acid-fast bacilli and fungi, respectively.
|
|
| |
|
|
| |
| Figure 1. White-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate (A) and gingiva (B). Resolution of lesions on the hard palate (C) and the gingiva (D) after 9 months of antituberculosis therapy. | ||
Laboratory results from blood chemistry, complete blood cell count, erythrocyte sedimentation rate, C-reactive protein level, and urine analysis, as well as titers of serum immunoglobulin, antineutrophil cytoplasmic antibodies, and antinuclear antibodies, were within reference range. Human immunodeficiency virus serology was negative. Chest radiography and ultrasonography of the abdomen revealed no abnormalities. A purified protein derivative (tuberculin) test showed an induration of 20 mm. Mycobacterium tuberculosis grew on the culture of the tissue specimen.
|
|
|
The patient was diagnosed with primary OT and treated with isoniazid (300 mg daily), rifampin (600 mg daily), ethambutol (1500 mg daily), and pyrazinamide (2000 mg daily). At 2 months of therapy the lesions started to heal and showed complete resolution at the end of 9 months of treatment (Figures 1C and 1D). There was no recurrence at 2-year follow-up.
Orificial tuberculosis is a rare form of cutaneous tuberculosis. Therefore, it is regarded as a “forgotten disease” in the literature.5 The pathogenesis of OT has not been clearly defined. The intact mucous membrane, thickened squamous epithelium, cleansing and antibacterial function of saliva, and presence of saprophytes act as protective mechanisms against penetration of mycobacteria. Some factors such as poor oral hygiene, smoking, local trauma, or presence of a dental cyst or an abscess may predispose the direct mucosal inoculation of the tuberculosis bacilli.1,2,6 Orificial tuberculosis may develop as an opportunistic infection in 1.33% of human immunodeficiency virus patients.7 However, our patient had no prior trauma or known predisposing factors.
The common presentation of OT is an ulcerative lesion with irregular and well-delineated margins with a yellowish granular base. A vesicle, papule, or nodule may precede the ulcers. The presence of yellow satellite nodules around the lesion is characteristic for OT.1,2,6 Although there were no satellite nodules around the ulcerations in our case, the lesions had a yellowish granular base and irregular, well-delineated margins.
The tongue, gingiva, lips, tonsils, and epiglottis are the most common sites of involvement.2,5,8 Hard palate involvement rarely has been reported, even in immunosuppressed patients.7 Enlarged painful cervical lymph nodes may accompany the disease. Most patients with OT have been reported to have active pulmonary tuberculosis at the time of diagnosis.6,8 Thus, internal organ involvement, particularly the pulmonary system, should be checked in patients with OT. In our patient, the hard palate was involved together with the gingiva despite the absence of immunosuppression. No internal organ involvement was found in the systemic evaluation.
Orificial tuberculosis is a form of cutaneous tuberculosis that is difficult to diagnose because of the varying nature of clinical features, failure of growth of M tuberculosis on culture, and rarity of the disease.1,2,6 As in our case, biopsies may not always exhibit a caseous necrosis, which is specific to tuberculosis.6,7 Thus, it may be difficult to distinguish oral cavity tuberculosis from conditions demonstrating oral ulcers such as bullous diseases, trauma, fungal diseases, syphilis, sarcoidosis, or squamous cell carcinoma by evaluating only signs and symptoms.6 Clinical suspicion is the first and foremost step in the diagnostic process of OT. In our patient, OT was suspected in the differential diagnosis because the resistant oral ulcerations showed the most common presentation of OT: irregular and well-delineated margins and a yellowish granular base. By considering tuberculosis within the differential diagnosis in our patient, microbiologic cultivation was performed from the oral mucosa and accurate diagnosis was established by determination of the pathogen’s growth in the culture.
Because of the increased incidence of tuberculosis and unusual manifestations, clinicians may easily overlook OT. It should be considered in the differential diagnosis of resistant nodules or ulcers of the oral cavity.
1. Kiliç A, Gül U, Gönül M, et al. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol. 2009;48:178-180.
2. Ito FA, de Andrade CR, Vargas PA, et al. Primary tuberculosis of the oral cavity. Oral Dis. 2005;11:50-53.
3. Dixit R, Sharma S, Nuwal P. Tuberculosis of oral cavity. Indian J Tuberc. 2008;55:51-53.
4. Smolka W, Burger H, Iizuka T, et al. Primary tuberculosis of the oral cavity in an elderly nonimmunosuppressed patient: case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2008;134:1107-1109.
5. Rodrigues G, Carnelio S, Valliathan M. Primary isolated gingival tuberculosis. Braz J Infect Dis. 2007;11:172-173.
6. Vilar FC, de Souza A, Moya MJ, et al. Atypical oral lesion in a patient with pulmonary tuberculosis. Int J Dermatol. 2009;48:910-912.
7. Kakisi OK, Kechagia AS, Kakisis IK, et al. Tuberculosis of the oral cavity: a systematic review. Eur J Oral Sci. 2010;118:103-109.
8. Eng HL, Lu SY, Yang CH, et al. Oral tuberculosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:415-420.
1. Kiliç A, Gül U, Gönül M, et al. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol. 2009;48:178-180.
2. Ito FA, de Andrade CR, Vargas PA, et al. Primary tuberculosis of the oral cavity. Oral Dis. 2005;11:50-53.
3. Dixit R, Sharma S, Nuwal P. Tuberculosis of oral cavity. Indian J Tuberc. 2008;55:51-53.
4. Smolka W, Burger H, Iizuka T, et al. Primary tuberculosis of the oral cavity in an elderly nonimmunosuppressed patient: case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2008;134:1107-1109.
5. Rodrigues G, Carnelio S, Valliathan M. Primary isolated gingival tuberculosis. Braz J Infect Dis. 2007;11:172-173.
6. Vilar FC, de Souza A, Moya MJ, et al. Atypical oral lesion in a patient with pulmonary tuberculosis. Int J Dermatol. 2009;48:910-912.
7. Kakisi OK, Kechagia AS, Kakisis IK, et al. Tuberculosis of the oral cavity: a systematic review. Eur J Oral Sci. 2010;118:103-109.
8. Eng HL, Lu SY, Yang CH, et al. Oral tuberculosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:415-420.
Cutaneous Metastasis of Gastric Adenocarcinoma at the Site of a Traumatic Ecchymosis
To the Editor:
In a recent Cutis® article, Cesaretti et al1 reported a case of cutaneous metastasis from primary gastric cancer that appeared on a resection scar 6 years after remission and without any relapse of the primary tumor. We report a case of a 68-year-old man who was referred to the dermatology clinic with a 15×20-cm nonpruritic, nonscaly, bruiselike lesion on the right forearm of 1 month’s duration. Approximately 1.5 years prior to presentation, the patient was diagnosed with gastric adenocarcinoma (stage IV: T4N3M1) with hepatic and lung metastasis. Following 6 months of chemotherapy with cisplatin and 5-fluorouracil, a positron emission tomography–computed tomography scan was performed and showed a reduction in metastasis but growth of the primitive tumor. After 1 year of chemotherapy, the new positron emission tomography–computed tomography scan showed no metastases. However, the primitive tumor had increased in size.
One month prior to presentation to the dermatology department, a traumatic blood sample on the right forearm left the patient with a persistent ecchymosis. The lesion was thought to be a healing ecchymosis and no biopsy was performed. One month later, the skin lesion had become much thicker and more erythematous (Figure) but not larger. A skin biopsy of this well-defined plaque was performed. Histologic examination showed neovascularization, proliferative epithelial cells, and cytokeratin markers AE1/AE3 and CK20, leading to the diagnosis of skin metastasis of the gastric adenocarcinoma. Chemotherapy was discontinued because of the patient’s altered general status and palliative care was given until he died the following month (2 months after presentation).
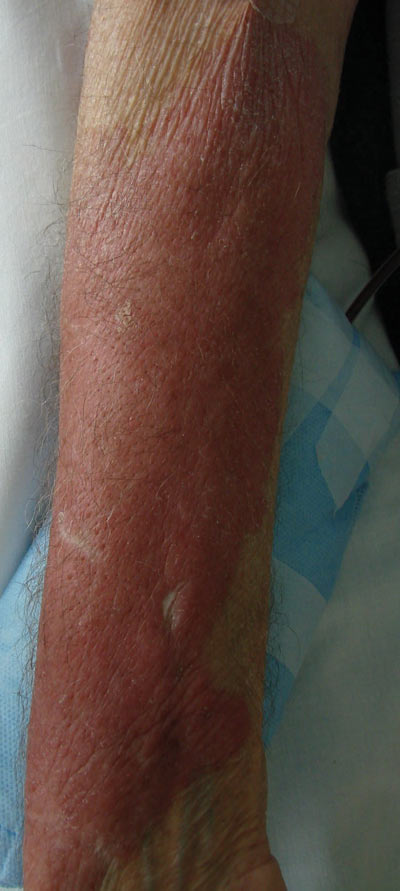
Iatrogenic dissemination of cancer cells has been described often on scars of tumor surgery,2 and in malignant melanoma, bruises and hematoma revealing preexisting metastases have been reported.3,4 Our report of secondary metastasis on an ecchymosis suggests that the traumatic blood sample performed before the development of the metastasis caused circulating tumor cells in the skin, which led to their local proliferation. The skin metastasis was the first sign of relapse and was followed by alteration of the general status and death.
Our patient is an example of the “soil and seed”5 hypothesis. Our case illustrates the abilities of tumor cells to colonize the skin under favorable conditions and emphasizes the importance of minimizing bleeding events and iatrogenic seeding of internal neoplasms in daily practice.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:e9-e13.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
- Pham-ledard A, Taieb A, Vergier B, et al. Metastatic cutaneous hematoma variant from melanoma: five cases. Bull Cancer. 2011;98:108-112.
- Connolly CM, Soldin M, Dawson A, et al. Metastatic malignant melanoma presenting with a bruise. Br J Plast Surg. 2003;56:725.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101.
To the Editor:
In a recent Cutis® article, Cesaretti et al1 reported a case of cutaneous metastasis from primary gastric cancer that appeared on a resection scar 6 years after remission and without any relapse of the primary tumor. We report a case of a 68-year-old man who was referred to the dermatology clinic with a 15×20-cm nonpruritic, nonscaly, bruiselike lesion on the right forearm of 1 month’s duration. Approximately 1.5 years prior to presentation, the patient was diagnosed with gastric adenocarcinoma (stage IV: T4N3M1) with hepatic and lung metastasis. Following 6 months of chemotherapy with cisplatin and 5-fluorouracil, a positron emission tomography–computed tomography scan was performed and showed a reduction in metastasis but growth of the primitive tumor. After 1 year of chemotherapy, the new positron emission tomography–computed tomography scan showed no metastases. However, the primitive tumor had increased in size.
One month prior to presentation to the dermatology department, a traumatic blood sample on the right forearm left the patient with a persistent ecchymosis. The lesion was thought to be a healing ecchymosis and no biopsy was performed. One month later, the skin lesion had become much thicker and more erythematous (Figure) but not larger. A skin biopsy of this well-defined plaque was performed. Histologic examination showed neovascularization, proliferative epithelial cells, and cytokeratin markers AE1/AE3 and CK20, leading to the diagnosis of skin metastasis of the gastric adenocarcinoma. Chemotherapy was discontinued because of the patient’s altered general status and palliative care was given until he died the following month (2 months after presentation).
Iatrogenic dissemination of cancer cells has been described often on scars of tumor surgery,2 and in malignant melanoma, bruises and hematoma revealing preexisting metastases have been reported.3,4 Our report of secondary metastasis on an ecchymosis suggests that the traumatic blood sample performed before the development of the metastasis caused circulating tumor cells in the skin, which led to their local proliferation. The skin metastasis was the first sign of relapse and was followed by alteration of the general status and death.
Our patient is an example of the “soil and seed”5 hypothesis. Our case illustrates the abilities of tumor cells to colonize the skin under favorable conditions and emphasizes the importance of minimizing bleeding events and iatrogenic seeding of internal neoplasms in daily practice.
To the Editor:
In a recent Cutis® article, Cesaretti et al1 reported a case of cutaneous metastasis from primary gastric cancer that appeared on a resection scar 6 years after remission and without any relapse of the primary tumor. We report a case of a 68-year-old man who was referred to the dermatology clinic with a 15×20-cm nonpruritic, nonscaly, bruiselike lesion on the right forearm of 1 month’s duration. Approximately 1.5 years prior to presentation, the patient was diagnosed with gastric adenocarcinoma (stage IV: T4N3M1) with hepatic and lung metastasis. Following 6 months of chemotherapy with cisplatin and 5-fluorouracil, a positron emission tomography–computed tomography scan was performed and showed a reduction in metastasis but growth of the primitive tumor. After 1 year of chemotherapy, the new positron emission tomography–computed tomography scan showed no metastases. However, the primitive tumor had increased in size.
One month prior to presentation to the dermatology department, a traumatic blood sample on the right forearm left the patient with a persistent ecchymosis. The lesion was thought to be a healing ecchymosis and no biopsy was performed. One month later, the skin lesion had become much thicker and more erythematous (Figure) but not larger. A skin biopsy of this well-defined plaque was performed. Histologic examination showed neovascularization, proliferative epithelial cells, and cytokeratin markers AE1/AE3 and CK20, leading to the diagnosis of skin metastasis of the gastric adenocarcinoma. Chemotherapy was discontinued because of the patient’s altered general status and palliative care was given until he died the following month (2 months after presentation).
Iatrogenic dissemination of cancer cells has been described often on scars of tumor surgery,2 and in malignant melanoma, bruises and hematoma revealing preexisting metastases have been reported.3,4 Our report of secondary metastasis on an ecchymosis suggests that the traumatic blood sample performed before the development of the metastasis caused circulating tumor cells in the skin, which led to their local proliferation. The skin metastasis was the first sign of relapse and was followed by alteration of the general status and death.
Our patient is an example of the “soil and seed”5 hypothesis. Our case illustrates the abilities of tumor cells to colonize the skin under favorable conditions and emphasizes the importance of minimizing bleeding events and iatrogenic seeding of internal neoplasms in daily practice.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:e9-e13.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
- Pham-ledard A, Taieb A, Vergier B, et al. Metastatic cutaneous hematoma variant from melanoma: five cases. Bull Cancer. 2011;98:108-112.
- Connolly CM, Soldin M, Dawson A, et al. Metastatic malignant melanoma presenting with a bruise. Br J Plast Surg. 2003;56:725.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:e9-e13.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
- Pham-ledard A, Taieb A, Vergier B, et al. Metastatic cutaneous hematoma variant from melanoma: five cases. Bull Cancer. 2011;98:108-112.
- Connolly CM, Soldin M, Dawson A, et al. Metastatic malignant melanoma presenting with a bruise. Br J Plast Surg. 2003;56:725.
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98-101.
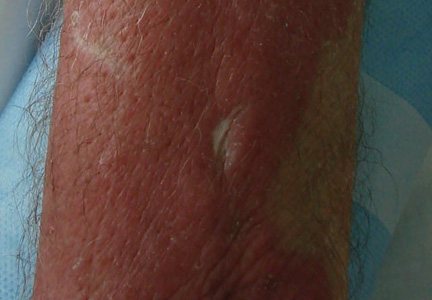
Paclitaxel-Associated Melanonychia
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).

| 
| |
| 
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
|
|
| |
|
|
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
|
|
| |
|
|
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
Verrucous Kaposi Sarcoma in an HIV-Positive Man
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
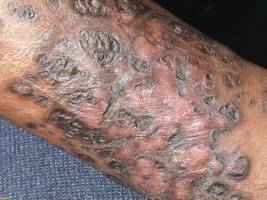
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
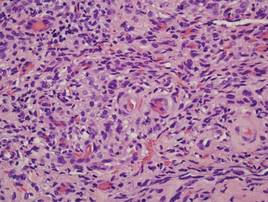
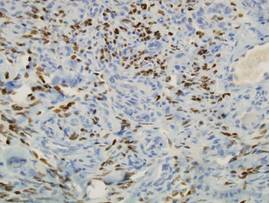
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
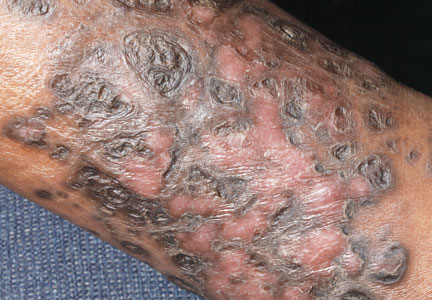
Aspergillus nidulans Causing Primary Cutaneous Aspergillosis in an Immunocompetent Patient
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
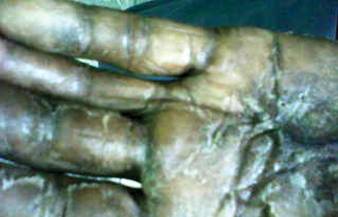

The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
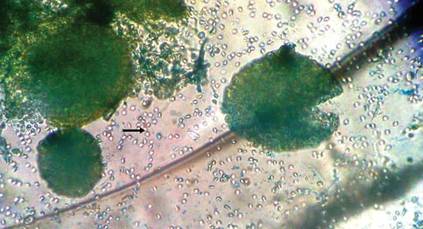
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
To the Editor:
Cutaneous aspergillosis mostly has been reported in immunosuppressed hosts and usually is caused by Aspergillus flavus or Aspergillus fumigatus. We report the occurrence of primary cutaneous aspergillosis (PCA) caused by a relatively rare species, Aspergillus nidulans, in a middle-aged patient without overt immunosuppression or history of trauma.
A 57-year-old woman was referred to the dermatology outpatient department for evaluation of a lesion on the right hand of 1 month's duration. On examination the lesion measured approximately 4×3 cm with central necrosis (Figure 1). Her medical history was unremarkable and routine laboratory test results were within reference range.
The patient was an agricultural worker with no history of trauma. Her history was unremarkable. A 20% potassium hydroxide mount of the tissue revealed septate, branched, hyaline hyphae. A soft, wooly, greenish brown growth was observed after 3 days of incubation on Sabouraud dextrose agar (Figure 2). No growth was observed on dermatophyte test medium. A lactophenol cotton blue mount revealed columnar conidial heads with brown, short, smooth-walled conidiophores (Figures 3–6). Vesicles were hemispheric and small (8–12 µm in diameter), with metulae and phialides occurring in the upper portion. Conidia were globose (3–4 µm) and rough. Based on these findings the fungus was identified as A nidulans. The patient did not respond to daily oral ketoconazole, and after 1 month of therapy the lesion did not regress. She was eventually treated with oral itraconazole and the lesion completely healed within 15 weeks.
An overwhelming majority of the cases of cutaneous aspergillosis have been reported either in immunocompromised hosts (ie, leukemia, cutaneous T-cell lymphoma, Hodgkin disease, human immunodeficiency virus/AIDS, solid-organ or hematopoietic stem cell transplant recipients) or in patients with contributing risk factors (ie, severe burns, diabetes mellitus, preterm or underweight neonates, elderly patients). Two outbreaks of this condition have been reported in neonatal intensive care units, with the source of contamination being linked to nonsterile disposable gloves, incubators, and humidity chambers.1,2 However, PCA is a relatively rare condition and often is associated with disruption of dermal integrity by trauma or maceration, followed by colonization of the wound by Aspergillus spores that are ubiquitously present in soil and decomposed vegetation.3-5 Our case was remarkable, as the patient was not immunosuppressed and did not have a history of trauma. However, we surmise that fungal inoculation might have inadvertently occurred through some trivial trauma sustained through her professional work.
The 2 species that have most commonly been associated with PCA are A flavus and A fumigatus.6,7 There have been isolated reports of PCA caused by other organisms such as Aspergillus niger,8,9 Aspergillus terreus,10Aspergillus ustus,11 or Aspergillus calidoustus.12 In a report of a neutropenic 56-year-old patient suffering from acute myeloblastic leukemia, PCA developed in association with a double-lumen Hickman catheter after a period of prolonged hospitalization.13 A study by the National Institutes of Health (1976-1997) revealed 6 life-threatening cases of A nidulans infection in patients with chronic granulomatous disease.14
We did not perform antifungal susceptibility testing on the isolate in our patient. However, we observed disease that was refractory to ketoconazole therapy but successfully resolved with oral itraconazole. Antifungal susceptibility was noted in a large number of reported cases of Aspergillus infections that were resistant to conventional treatment, such as voriconazole, itraconazole, and amphotericin B.15 Thus antifungal susceptibility testing is necessary before starting treatment. There also have been reports of recurrence of cutaneous aspergillosis following incomplete and irregular treatment.16 Our case of PCA also failed to respond to ketoconazole therapy, thus stressing the need for thorough mycological characterization, including the determination of an antifungal susceptibility profile, for successful and complete management of this condition.
Acknowledgment
The authors would like to thank Arunaloke Chakraborti, MD, Chandigarh, India, for the help extended for identification of the fungus.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.
- Stock C, Veyrier M, Raberin H, et al. Severe cutaneous aspergillosis in a premature neonate linked to nonsterile disposable glove contamination [published online ahead of print August 31, 2011]? Am J Infect Control. 2012;40:465-467.
- Etienne KA, Subudhi CP, Chadwick PR, et al. Investigation of a cluster of cutaneous aspergillosis in a neonatal intensive care unit [published online ahead of print August 12, 2011]. J Hosp Infect. 2011;79:344-348.
- Isaac M. Cutaneous aspergillosis. Dermatol Clin. 1996;14:137-140.
- Cahill KM, Mofty AM, Kawaguchi TP. Primary cutaneous aspergillosis. Arch Dermatol. 1967;96:545-547.
- Carlile JR, Millet RE, Cho CT, et al. Primary cutaneous aspergillosis in a leukemic child. Arch Dermatol. 1978;114:78-80.
- John PU, Shadomy HJ. Deep fungal infections. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. New York, NY: McGraw Hill; 1987:2266-2268.
- Chakrabarti A, Gupta V, Biswas G, et al. Primary cutaneous aspergillosis: our experience in 10 years. J Infect. 1998;37:24-27.
- Robinson A, Fien S, Grassi MA. Nonhealing scalp wound infected with Aspergillus niger in an elderly patient. Cutis. 2011;87:197-200.
- Thomas LM, Rand HK, Miller JL, et al. Primary cutaneous aspergillosis in a patient with a solid organ transplant: case report and review of the literature. Cutis. 2008;81:127-130.
- Yuanjie Z, Jingxia D, Hai W, et al. Primary cutaneous aspergillosis in a patient with cutaneous T-cell lymphoma [published online ahead of print October 22, 2008]. Mycoses. 2009;52:462-464.
- Krishnan-Natesan S, Chandrasekar PH, Manavathu EK, et al. Successful treatment of primary cutaneous Aspergillus ustus infection with surgical debridement and a combination of voriconazole and terbinafine [published online ahead of print October 7, 2008]. Diagn Microbiol Infect Dis. 2008;62:443-446.
- Sato Y, Suzino K, Suzuki A, et al. Case of primary cutaneous Aspergillus calidoustus infection caused by nerve block therapy [in Japanese]. Med Mycol J. 2011;52:239-244.
- Lucas GM, Tucker P, Merz WG. Primary cutaneous Aspergillus nidulans infection associated with a Hickman catheter in a patient with neutropenia. Clin Infect Dis. 1999;29:1594-1596.
- Segal BH, DeCarlo ES, Kwon-Chung KJ, et al. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore). 1998;77:345-354.
- Woodruff CA, Hebert AA. Neonatal primary cutaneous aspergillosis: case report and review of the literature. Pediatr Dermatol. 2002;19:439-444.
- Mohapatra S, Xess I, Swetha JV, et al. Primary cutaneous aspergillosis due to Aspergillus niger in an immunocompetent patient. Indian J Med Microbiol. 2009;27:367-370.