User login
Fungal Melanonychia Caused by Trichophyton rubrum and the Value of Dermoscopy
To the Editor:
Longitudinal melanonychia encompasses a broad spectrum of diseases and often is a complex diagnostic problem. Differential diagnoses include ethnic-type nail pigmentation, which is more frequently seen in darker-skinned individuals; drug-induced pigmentation; subungual hemorrhage; fungal or bacterial infection; nevus; and melanoma.1,2 Fungal melan-onychia is an uncommon presentation of onychomycosis. Dermoscopy can assist in the evaluation of nail pigmentation caused by fungi to avoid unnecessary nail biopsies.
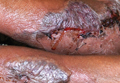
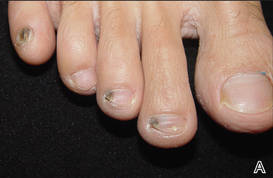
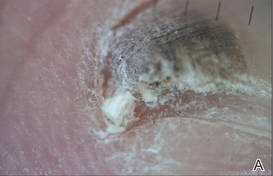
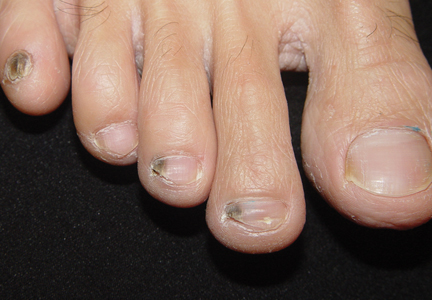
A 39-year-old man visited the dermatology clinic with a concern for melanoma because of blackish pigmentation of the toenails of 1 month’s duration. He denied history of trauma and was not taking any medications. On physical examination the second and third toenails revealed a 2-mm longitudinal band of black pigment on the lateral side; the fifth toenail showed diffuse black pigment (Figure 1A). The nail plates were thickened. Dermoscopy revealed prominent subungual hyperkeratosis, a homogeneous brown-black band with wide yellow streaks that were wider in the distal ends, and some focal reddish hue. No visible melanin inclusions were observed (Figure 2). These findings were suggestive of fungal infection. Cultures from the diseased nail grew a fungus identified as Trichophyton rubrum. The patient was treated with itraconazole 200 mg daily for 3 months. Clinical cure with disappearance of pigment was obtained at 5-month follow-up (Figure 1B).
 |
 |
| Figure 1. Blackish discoloration of the right second, third, and fifth toenails (A). Resolution of pigmentation and subungual hyperkeratosis was achieved at 5-month follow-up after treatment (B). |
 |
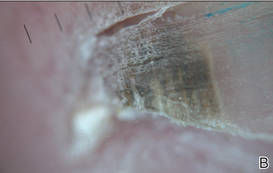 |
| Figure 2. Top view (A) and front view (B) of a homogeneous brown-black band with subungual hyperkeratosis and wide intervening yellow streaks. Each ruler mark denotes 1 mm. |
Our patient illustrates the value of dermoscopy in evaluating melanonychia. The pigmentation of adult-onset melanonychia involving multiple fingers can be divided into nonmelanocytic or melanocytic origin. Causes of the former include subungual hematoma, fungal or bacterial infection, and exogenous pigmentation. The nonmelanocytic pigment often is homogeneously distributed without melanin inclusions under the dermoscope.2
On the contrary, melanin inclusions can be detected as fine granules in pigmentation of melanocytic origin, either from focal melanocytic activation or melanocyte proliferation. Causes of focal melanocytic activation include ethnic-type nail hyperpigmentation; inflammatory nail diseases; or drug-, radiation-, and friction-induced hyperpigmentation. The characteristic dermoscopic features are thin longitudinal gray lines with regular thickness and spacing in a grayish background.1-3
Melanocyte proliferation can result in a nevus or melanoma of the nail apparatus. Both share dermoscopic features of brown-black longitudinal lines in a brown background. However, the longitudinal lines in melanoma are irregular in coloration, spacing, thickness, and parallelism, in contrast with the regular pattern of a nevus.1-4 Although patients often are concerned about melanoma, involvement of multiple fingers at the same time is less likely.
In our patient, the homogeneous deep brown color without melanin inclusions favored a nonmel-anocytic origin. The distally wider pigmentation suggested fungal infection because most ungual infections extend from the distal to the proximal part of the nail.5,6 The focal reddish hue may be related with traumatic hemorrhage from subungual hyperkeratosis.
Cases of fungal melanonychia are being reported at an increasing rate. Some fungal strains are capable of synthesizing melanin, which is associated with virulence and acts as a fungal armor against toxic insults.5 In T rubrum, the melanoid variant, the diffusible black pigment infiltrates the nail plate and attributes to the black nail clinically.4 The most frequently isolated fungi in fungal melanonychia are T rubrum and Scytalidium dimidiatum6; however, Candida species,7,8 dematiaceous fungus,9 and other dermatophytes such as Trichophyton soudanense10 have been reported to be the cause.5
Our patient presented with fungal melanonychia due to T rubrum with dermoscopic features. The prominent subungual hyperkeratosis, distally wider homogeneous brown-black pigmented band, and wide yellow streaks with focal reddish hue all suggested fungal melanonychia. The diagnosis was further confirmed by a good response to antifungal agents.
1. Ronger S, Touzet S, Ligeron C, et al. Dermoscopic examination of nail pigmentation. Arch Dermatol. 2002;138:1327-1333.
2. Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations [published online ahead of print February 22, 2007]. J Am Acad Dermatol. 2007;56:835-847.
3. Koga H, Saida T, Uhara H. Key point in dermoscopic differentiation between early nail apparatus melanoma and benign longitudinal melanonychia. J Dermatol. 2011;38:45-52.
4. Phan A, Dalle S, Touzet S, et al. Dermoscopic features of acral lentiginous melanoma in a large series of 110 cases in a white population [published online ahead of print November 18, 2009]. Br J Dermatol. 2010;162:765-771.
5. Finch J, Arenas R, Baran R. Fungal melanonychia [published online ahead of print January 17, 2012]. J Am Acad Dermatol. 2012;66:830-841.
6. Lee SW, Kim YC, Kim DK, et al. Fungal melanonychia. J Dermatol. 2004;31:904-909.
7. Parlak AH, Goksugur N, Karabay O. A case of melanonychia due to Candida albicans. Clin Exp Dermatol. 2006;31:398-400.
8. Gautret P, Rodier MH, Kauffmann-Lacroix C, et al. Case report and review. onychomycosis due to Candida parapsilosis. Mycoses. 2000;43:433-435.
9. Barua P, Barua S, Borkakoty B, et al. Onychomycosis by Scytalidium dimidiatum in green tea leaf pluckers: report of two cases [published online ahead of print July 20, 2007]. Mycopathologia. 2007;164:193-195.
10. Ricci C, Monod M, Baudraz-Rosselet F. Onychomycosis due to Trichophyton soudanense in Switzerland. Dermatology. 1998;197:297-298.
To the Editor:
Longitudinal melanonychia encompasses a broad spectrum of diseases and often is a complex diagnostic problem. Differential diagnoses include ethnic-type nail pigmentation, which is more frequently seen in darker-skinned individuals; drug-induced pigmentation; subungual hemorrhage; fungal or bacterial infection; nevus; and melanoma.1,2 Fungal melan-onychia is an uncommon presentation of onychomycosis. Dermoscopy can assist in the evaluation of nail pigmentation caused by fungi to avoid unnecessary nail biopsies.
A 39-year-old man visited the dermatology clinic with a concern for melanoma because of blackish pigmentation of the toenails of 1 month’s duration. He denied history of trauma and was not taking any medications. On physical examination the second and third toenails revealed a 2-mm longitudinal band of black pigment on the lateral side; the fifth toenail showed diffuse black pigment (Figure 1A). The nail plates were thickened. Dermoscopy revealed prominent subungual hyperkeratosis, a homogeneous brown-black band with wide yellow streaks that were wider in the distal ends, and some focal reddish hue. No visible melanin inclusions were observed (Figure 2). These findings were suggestive of fungal infection. Cultures from the diseased nail grew a fungus identified as Trichophyton rubrum. The patient was treated with itraconazole 200 mg daily for 3 months. Clinical cure with disappearance of pigment was obtained at 5-month follow-up (Figure 1B).
|
|
| Figure 1. Blackish discoloration of the right second, third, and fifth toenails (A). Resolution of pigmentation and subungual hyperkeratosis was achieved at 5-month follow-up after treatment (B). |
|
|
| Figure 2. Top view (A) and front view (B) of a homogeneous brown-black band with subungual hyperkeratosis and wide intervening yellow streaks. Each ruler mark denotes 1 mm. |
Our patient illustrates the value of dermoscopy in evaluating melanonychia. The pigmentation of adult-onset melanonychia involving multiple fingers can be divided into nonmelanocytic or melanocytic origin. Causes of the former include subungual hematoma, fungal or bacterial infection, and exogenous pigmentation. The nonmelanocytic pigment often is homogeneously distributed without melanin inclusions under the dermoscope.2
On the contrary, melanin inclusions can be detected as fine granules in pigmentation of melanocytic origin, either from focal melanocytic activation or melanocyte proliferation. Causes of focal melanocytic activation include ethnic-type nail hyperpigmentation; inflammatory nail diseases; or drug-, radiation-, and friction-induced hyperpigmentation. The characteristic dermoscopic features are thin longitudinal gray lines with regular thickness and spacing in a grayish background.1-3
Melanocyte proliferation can result in a nevus or melanoma of the nail apparatus. Both share dermoscopic features of brown-black longitudinal lines in a brown background. However, the longitudinal lines in melanoma are irregular in coloration, spacing, thickness, and parallelism, in contrast with the regular pattern of a nevus.1-4 Although patients often are concerned about melanoma, involvement of multiple fingers at the same time is less likely.
In our patient, the homogeneous deep brown color without melanin inclusions favored a nonmel-anocytic origin. The distally wider pigmentation suggested fungal infection because most ungual infections extend from the distal to the proximal part of the nail.5,6 The focal reddish hue may be related with traumatic hemorrhage from subungual hyperkeratosis.
Cases of fungal melanonychia are being reported at an increasing rate. Some fungal strains are capable of synthesizing melanin, which is associated with virulence and acts as a fungal armor against toxic insults.5 In T rubrum, the melanoid variant, the diffusible black pigment infiltrates the nail plate and attributes to the black nail clinically.4 The most frequently isolated fungi in fungal melanonychia are T rubrum and Scytalidium dimidiatum6; however, Candida species,7,8 dematiaceous fungus,9 and other dermatophytes such as Trichophyton soudanense10 have been reported to be the cause.5
Our patient presented with fungal melanonychia due to T rubrum with dermoscopic features. The prominent subungual hyperkeratosis, distally wider homogeneous brown-black pigmented band, and wide yellow streaks with focal reddish hue all suggested fungal melanonychia. The diagnosis was further confirmed by a good response to antifungal agents.
To the Editor:
Longitudinal melanonychia encompasses a broad spectrum of diseases and often is a complex diagnostic problem. Differential diagnoses include ethnic-type nail pigmentation, which is more frequently seen in darker-skinned individuals; drug-induced pigmentation; subungual hemorrhage; fungal or bacterial infection; nevus; and melanoma.1,2 Fungal melan-onychia is an uncommon presentation of onychomycosis. Dermoscopy can assist in the evaluation of nail pigmentation caused by fungi to avoid unnecessary nail biopsies.
A 39-year-old man visited the dermatology clinic with a concern for melanoma because of blackish pigmentation of the toenails of 1 month’s duration. He denied history of trauma and was not taking any medications. On physical examination the second and third toenails revealed a 2-mm longitudinal band of black pigment on the lateral side; the fifth toenail showed diffuse black pigment (Figure 1A). The nail plates were thickened. Dermoscopy revealed prominent subungual hyperkeratosis, a homogeneous brown-black band with wide yellow streaks that were wider in the distal ends, and some focal reddish hue. No visible melanin inclusions were observed (Figure 2). These findings were suggestive of fungal infection. Cultures from the diseased nail grew a fungus identified as Trichophyton rubrum. The patient was treated with itraconazole 200 mg daily for 3 months. Clinical cure with disappearance of pigment was obtained at 5-month follow-up (Figure 1B).
|
|
| Figure 1. Blackish discoloration of the right second, third, and fifth toenails (A). Resolution of pigmentation and subungual hyperkeratosis was achieved at 5-month follow-up after treatment (B). |
|
|
| Figure 2. Top view (A) and front view (B) of a homogeneous brown-black band with subungual hyperkeratosis and wide intervening yellow streaks. Each ruler mark denotes 1 mm. |
Our patient illustrates the value of dermoscopy in evaluating melanonychia. The pigmentation of adult-onset melanonychia involving multiple fingers can be divided into nonmelanocytic or melanocytic origin. Causes of the former include subungual hematoma, fungal or bacterial infection, and exogenous pigmentation. The nonmelanocytic pigment often is homogeneously distributed without melanin inclusions under the dermoscope.2
On the contrary, melanin inclusions can be detected as fine granules in pigmentation of melanocytic origin, either from focal melanocytic activation or melanocyte proliferation. Causes of focal melanocytic activation include ethnic-type nail hyperpigmentation; inflammatory nail diseases; or drug-, radiation-, and friction-induced hyperpigmentation. The characteristic dermoscopic features are thin longitudinal gray lines with regular thickness and spacing in a grayish background.1-3
Melanocyte proliferation can result in a nevus or melanoma of the nail apparatus. Both share dermoscopic features of brown-black longitudinal lines in a brown background. However, the longitudinal lines in melanoma are irregular in coloration, spacing, thickness, and parallelism, in contrast with the regular pattern of a nevus.1-4 Although patients often are concerned about melanoma, involvement of multiple fingers at the same time is less likely.
In our patient, the homogeneous deep brown color without melanin inclusions favored a nonmel-anocytic origin. The distally wider pigmentation suggested fungal infection because most ungual infections extend from the distal to the proximal part of the nail.5,6 The focal reddish hue may be related with traumatic hemorrhage from subungual hyperkeratosis.
Cases of fungal melanonychia are being reported at an increasing rate. Some fungal strains are capable of synthesizing melanin, which is associated with virulence and acts as a fungal armor against toxic insults.5 In T rubrum, the melanoid variant, the diffusible black pigment infiltrates the nail plate and attributes to the black nail clinically.4 The most frequently isolated fungi in fungal melanonychia are T rubrum and Scytalidium dimidiatum6; however, Candida species,7,8 dematiaceous fungus,9 and other dermatophytes such as Trichophyton soudanense10 have been reported to be the cause.5
Our patient presented with fungal melanonychia due to T rubrum with dermoscopic features. The prominent subungual hyperkeratosis, distally wider homogeneous brown-black pigmented band, and wide yellow streaks with focal reddish hue all suggested fungal melanonychia. The diagnosis was further confirmed by a good response to antifungal agents.
1. Ronger S, Touzet S, Ligeron C, et al. Dermoscopic examination of nail pigmentation. Arch Dermatol. 2002;138:1327-1333.
2. Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations [published online ahead of print February 22, 2007]. J Am Acad Dermatol. 2007;56:835-847.
3. Koga H, Saida T, Uhara H. Key point in dermoscopic differentiation between early nail apparatus melanoma and benign longitudinal melanonychia. J Dermatol. 2011;38:45-52.
4. Phan A, Dalle S, Touzet S, et al. Dermoscopic features of acral lentiginous melanoma in a large series of 110 cases in a white population [published online ahead of print November 18, 2009]. Br J Dermatol. 2010;162:765-771.
5. Finch J, Arenas R, Baran R. Fungal melanonychia [published online ahead of print January 17, 2012]. J Am Acad Dermatol. 2012;66:830-841.
6. Lee SW, Kim YC, Kim DK, et al. Fungal melanonychia. J Dermatol. 2004;31:904-909.
7. Parlak AH, Goksugur N, Karabay O. A case of melanonychia due to Candida albicans. Clin Exp Dermatol. 2006;31:398-400.
8. Gautret P, Rodier MH, Kauffmann-Lacroix C, et al. Case report and review. onychomycosis due to Candida parapsilosis. Mycoses. 2000;43:433-435.
9. Barua P, Barua S, Borkakoty B, et al. Onychomycosis by Scytalidium dimidiatum in green tea leaf pluckers: report of two cases [published online ahead of print July 20, 2007]. Mycopathologia. 2007;164:193-195.
10. Ricci C, Monod M, Baudraz-Rosselet F. Onychomycosis due to Trichophyton soudanense in Switzerland. Dermatology. 1998;197:297-298.
1. Ronger S, Touzet S, Ligeron C, et al. Dermoscopic examination of nail pigmentation. Arch Dermatol. 2002;138:1327-1333.
2. Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations [published online ahead of print February 22, 2007]. J Am Acad Dermatol. 2007;56:835-847.
3. Koga H, Saida T, Uhara H. Key point in dermoscopic differentiation between early nail apparatus melanoma and benign longitudinal melanonychia. J Dermatol. 2011;38:45-52.
4. Phan A, Dalle S, Touzet S, et al. Dermoscopic features of acral lentiginous melanoma in a large series of 110 cases in a white population [published online ahead of print November 18, 2009]. Br J Dermatol. 2010;162:765-771.
5. Finch J, Arenas R, Baran R. Fungal melanonychia [published online ahead of print January 17, 2012]. J Am Acad Dermatol. 2012;66:830-841.
6. Lee SW, Kim YC, Kim DK, et al. Fungal melanonychia. J Dermatol. 2004;31:904-909.
7. Parlak AH, Goksugur N, Karabay O. A case of melanonychia due to Candida albicans. Clin Exp Dermatol. 2006;31:398-400.
8. Gautret P, Rodier MH, Kauffmann-Lacroix C, et al. Case report and review. onychomycosis due to Candida parapsilosis. Mycoses. 2000;43:433-435.
9. Barua P, Barua S, Borkakoty B, et al. Onychomycosis by Scytalidium dimidiatum in green tea leaf pluckers: report of two cases [published online ahead of print July 20, 2007]. Mycopathologia. 2007;164:193-195.
10. Ricci C, Monod M, Baudraz-Rosselet F. Onychomycosis due to Trichophyton soudanense in Switzerland. Dermatology. 1998;197:297-298.
Narrow-Toed Shoes and the Toe-to-Toe Sign
To the Editor:
Macro- or microtrauma to nails can cause and/or exacerbate chronic diseases such as ingrown nails, onycholysis, onychauxis, onychogryposis, and hallux valgus (bunions). This trauma also can break the anatomic barrier of the hyponychium, thereby creating a portal for dermatophytes and other organisms to penetrate the nail apparatus.1
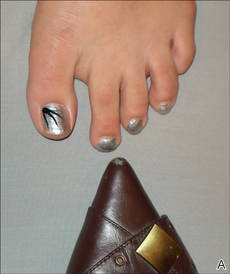
For many years, one author (C.R.D.) has used the following demonstration to communicate to patients how improper shoe fit may cause toenail trauma. The patient’s shoe is flipped 180° and placed toe-to-toe with the patient’s foot (Figure). Most patients can comprehend the relationship between their shoe fit and their toenail disease when they see this demonstration. We have termed it toe-to-toe sign.
 |
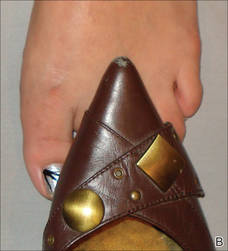 |
| The foot and shoe are toe-to-toe, demonstrating the narrow toe box in comparison to the actual toes (A). This comparison is accentuated when the shoe is placed over the foot with toes splayed from standing (B). |
Narrow toe box is the usual culprit. The toe-to-toe sign best emphasizes this relationship. This sign serves as a powerful tool when demonstrating how much the foot widens when bearing full weight, and how forces of ambulation and foot strike can damage the nails with narrow-toed footwear. This trauma is compounded with high-heeled shoes that force the toes forward. However, proper shoe fit may compete with idealized shoe style. It is not until patients realize the relationship between improper shoe fit, foot strike, and toe/toenail trauma that they can make long-term decisions that favorably impact their toes and toenails.
Reference
1. Daniel CR 3rd, Jellinek NJ. The pedal fungus reservoir. Arch Dermatol. 2006;142:1344-1346.
To the Editor:
Macro- or microtrauma to nails can cause and/or exacerbate chronic diseases such as ingrown nails, onycholysis, onychauxis, onychogryposis, and hallux valgus (bunions). This trauma also can break the anatomic barrier of the hyponychium, thereby creating a portal for dermatophytes and other organisms to penetrate the nail apparatus.1
For many years, one author (C.R.D.) has used the following demonstration to communicate to patients how improper shoe fit may cause toenail trauma. The patient’s shoe is flipped 180° and placed toe-to-toe with the patient’s foot (Figure). Most patients can comprehend the relationship between their shoe fit and their toenail disease when they see this demonstration. We have termed it toe-to-toe sign.
|
|
| The foot and shoe are toe-to-toe, demonstrating the narrow toe box in comparison to the actual toes (A). This comparison is accentuated when the shoe is placed over the foot with toes splayed from standing (B). |
Narrow toe box is the usual culprit. The toe-to-toe sign best emphasizes this relationship. This sign serves as a powerful tool when demonstrating how much the foot widens when bearing full weight, and how forces of ambulation and foot strike can damage the nails with narrow-toed footwear. This trauma is compounded with high-heeled shoes that force the toes forward. However, proper shoe fit may compete with idealized shoe style. It is not until patients realize the relationship between improper shoe fit, foot strike, and toe/toenail trauma that they can make long-term decisions that favorably impact their toes and toenails.
To the Editor:
Macro- or microtrauma to nails can cause and/or exacerbate chronic diseases such as ingrown nails, onycholysis, onychauxis, onychogryposis, and hallux valgus (bunions). This trauma also can break the anatomic barrier of the hyponychium, thereby creating a portal for dermatophytes and other organisms to penetrate the nail apparatus.1
For many years, one author (C.R.D.) has used the following demonstration to communicate to patients how improper shoe fit may cause toenail trauma. The patient’s shoe is flipped 180° and placed toe-to-toe with the patient’s foot (Figure). Most patients can comprehend the relationship between their shoe fit and their toenail disease when they see this demonstration. We have termed it toe-to-toe sign.
|
|
| The foot and shoe are toe-to-toe, demonstrating the narrow toe box in comparison to the actual toes (A). This comparison is accentuated when the shoe is placed over the foot with toes splayed from standing (B). |
Narrow toe box is the usual culprit. The toe-to-toe sign best emphasizes this relationship. This sign serves as a powerful tool when demonstrating how much the foot widens when bearing full weight, and how forces of ambulation and foot strike can damage the nails with narrow-toed footwear. This trauma is compounded with high-heeled shoes that force the toes forward. However, proper shoe fit may compete with idealized shoe style. It is not until patients realize the relationship between improper shoe fit, foot strike, and toe/toenail trauma that they can make long-term decisions that favorably impact their toes and toenails.
Reference
1. Daniel CR 3rd, Jellinek NJ. The pedal fungus reservoir. Arch Dermatol. 2006;142:1344-1346.
Reference
1. Daniel CR 3rd, Jellinek NJ. The pedal fungus reservoir. Arch Dermatol. 2006;142:1344-1346.

Plaques: A Rare Presentation of Acrokeratoelastoidosis
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
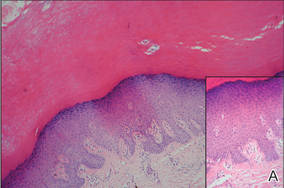
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
 |
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
 |
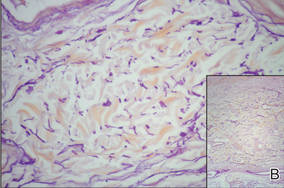 |
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
To the Editor:
Acrokeratoelastoidosis (AKE) is a rare disease first described by Costa1 in 1953. Typically it is only a cosmetic nuisance in the majority of patients and presents as asymptomatic, small, firm, flesh-colored to yellowish, round to polygonal papules with occasional keratosis or umbilication on the radial and ulnar margins of the hands and/or feet.1-3 In some cases, the lesions occur on the anterior aspects of the wrists, fingers, or lower legs.1 The lesions are always bilaterally distributed. Acrokeratoelastoidosis is a chronic skin disorder that commonly presents during childhood or adolescence, but presentation in adulthood also has been described.3 Histologically, AKE always shows hyperkeratosis, acanthosis, decrease of elastic tissue, and elastorrhexis of remaining elastic fibers. Plaque-type lesions are rare. We describe a patient who presented with plaques on the radial and ulnar margins of the hands.
A 36-year-old Chinese woman presented with asymptomatic, small, firm papules of 6 months’ duration that initially developed on the hands and gradually increased in number, coalescing into plaques. The feet were spared. She had no medical history of hyperhidrosis, chronic trauma, friction, or excessive sun exposure, and no family history of similar symptoms. No prior therapy had been attempted.
Physical examination showed nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules with wavy edges that were symmetrically distributed on the radial and ulnar margins of the hands; some papules had coalesced into plaques (Figure 1). A biopsy specimen taken from a plaque on the hypothenar eminence of the right hand revealed focal hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation with hematoxylin and eosin stain (Figure 2A). Aldehyde fuchsin staining showed fragmented and rarefied elastic fibers in the reticular dermis (Figure 2B). The patient was diagnosed with AKE. Oral tretinoin 10 mg twice daily was initiated and resulted in an evident response after 2 weeks of treatment. However, the patient stopped taking the medication because of pruritus and dry skin and the lesions then reappeared.
|
| Figure 1. Nonconfluent, firm, flesh-colored to yellowish, translucent, smooth papules distributed on the radial margin of the hand; some of papules coalesced into plaques. |
|
|
| Figure 2. Histopathology revealed hyperkeratosis, hypergranulosis, acanthosis, and mild chronic inflammation (A) (H&E, original magnification ×250 [inset, original magnification ×400]), as well as fragmented elastic fibers in the reticular dermis (B) (Aldehyde fuchsin, original magnification ×400 [inset, original magnification ×100]). |
Acrokeratoelastoidosis is a rare keratotic disorder. It seems to have no racial or ethnic predilection and occurs more frequently in women.4,5 It also is rare in China, with few cases reported, all women.5 The reason for the gender predilection in China remains unknown. The course is chronic, but it may rapidly progress during pregnancy.6
The pathogenesis of AKE is still unresolved.2,3 Although many cases are sporadic,5 it appears to be inherited in an autosomal-dominant fashion, most likely related to chromosome 2.7 Typically, AKE presents as papules that are discrete and bilaterally distributed in the palmoplantar margins,2,3 but some of the papules in our patient coalesced into plaques, which is unique. The histologic hallmarks indicated that the lesions were AKE.
The differential diagnosis of AKE includes hereditary papulotranslucent acrokeratoderma, focal acral hyperkeratosis, and keratoelastoidosis marginalis.8 Hereditary papulotranslucent acrokeratoderma also is inherited in an autosomal-dominant fashion and shares similar acral, translucent, keratotic papules with AKE, but there is no chronic inflammatory cell infiltrate, degeneration of collagenous fibers, or fragmentation of elastic fibers. The clinical appearance of focal acral hyperkeratosis is similar to AKE, but no changes are revealed in the elastic tissue.9 Because AKE, focal acral hyperkeratosis, and hereditary papulotranslucent acrokeratoderma have similar lesions and overlapping histologic changes, they may be considered variants of the same entity.4 Keratoelastoidosis marginalis, also called degenerative collagenous plaques of the hand, mainly affects white individuals aged 40 to 60 years with a history of prolonged sun exposure. Papules often are distributed over the junction of the dorsal and palmar skin and less often on the ulnar sides of the hands. The clinical lesions are similar to those in our patient, but histopathology of keratoelastoidosis marginalis shows amorphous, basophilic, elastotic masses and thickened, fragmented, calcified elastic fibers in the upper and mid dermis.
Therapies including liquid nitrogen, topical salicylic acid, methotrexate, dapsone, tar, cryotherapy, systemic prednisone, retinoic acid, clobetasone cream,5 and erbium:YAG laser10 have been applied. Thus far, no optimal treatment has been recommended and no tendency of spontaneous resolution has been previously reported in the literature. Our patient responded to tretinoin, but the lesions recurred after withdrawal of the medication; therefore, tretinoin may not be an optimal treatment option. Because the lesions are limited to the skin and AKE is only considered a cosmetic problem with a good prognosis, we recommend a wait-and-watch approach.
Acknowledgment—We thank Rashmi Sarkar, MD, New Delhi, India, for her assistance.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.
1. Costa OG. Akrokerato-elastoidosis; a hitherto undescribed skin disease. Dermatologica. 1953;107:164-168.
2. Bogle MA, Huang LY, Tschen JA. Acrokeratoelastoidosis. J Am Acad Dermatol. 2002;47:448-451.
3. Highet AS, Rook A, Anderson JR. Acrokeratoelastoidosis. Br J Dermatol. 1982;106:337-344.
4. Abulafia J, Vignale RA. Degenerative collagenous
plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
5. Luo DQ, Zhang B, Huang YB, et al. Papules on a young woman’s hands and feet. Clin Exp Dermatol. 2010;35:451-452.
6. Nelson-Adesokan P, Mallory SB, Lombardi C, et al. Acrokeratoelastoidosis of Costa [published correction appears in Int J Dermatol. 1996;35:380]. Int J Dermatol. 1995;34:431-433.
7. Greiner J, Krüger J, Palden L, et al. A linkage study of acrokeratoelastoidosis. possible mapping to chromosome 2. Hum Genet. 1983;63:222-227.
8. Hu W, Cook TF, Vicki GJ, et al. Acrokeratoelastoidosis. Pediatr Dermatol. 2002;19:320-322.
9. Dowd PM, Harman RR, Black MM. Focal acral hyperkeratosis. Br J Dermatol. 1983;109:97-103.
10. Erbil AH, Sezer E, Koç E, et al. Acrokeratoelastoidosis treated with the erbium:YAG laser [published online ahead of print November 3, 2007]. Clin Exp Dermatol. 2008;33:30-31.

Primary Mucinous Carcinoma of the Skin
To the Editor:
A 41-year-old man presented to our dermatology clinic with a recurrent asymptomatic nodule on the right cheek that had gradually increased in size over 1 year. The patient underwent laser excision at an outside facility 1 year after the first presentation. Pathology reports and tissue cultures from the excision were not available. The lesion recurred in the same location 6 months following excision. The patient reported no history of pain, fever, cough, weight loss, or loss of appetite, and he denied any trauma or radiation to the affected area. Dermatologic examination revealed a 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek (Figure 1). There was no tenderness or pruritus. Clinical examination and extensive radiographic studies revealed no primary disease. A complete blood cell count, biochemical tests, and serous tumor markers were within reference range. The lesion was resected with a 2-mm margin. The margins were free of tumor cells. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa and scattered floating islands of tumor cells in the dermis. Small-sized glands were organized in some areas of the tumor cells (Figure 2). Epithelial tumor cell islands contained uniform oval nuclei and focally vacuolated cytoplasm. The tumor cells were positive for cytokeratin 7 (CK7) (Figure 3) and negative for cytokeratin 20 (CK20), carcinoembryonic antigen, homeobox transcription factor (CDX2), villin, mucus-associated peptides of the thyroid transcription factor (TTF1), and prostate-specific antigen, all indicating a diagnosis of primary mucinous carcinoma of the skin (PMCS). No local recurrence, regional lymph node involvement, or distant metastasis was observed after resection.
 |
| Figure 1. A 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek. |
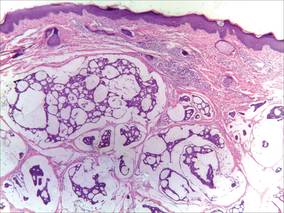 |
| Figure 2. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa that contained scattered floating islands of tumor cells in the dermis (H&E, original magnification ×40). |
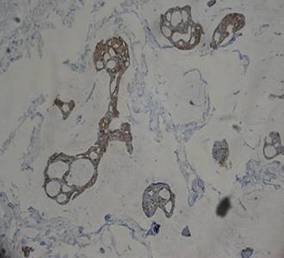 |
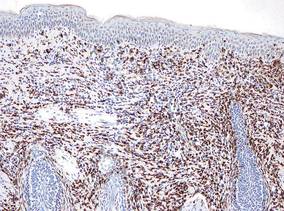
| Figure 3. The tumor cells were positive for cytokeratin 7 (original magnification ×400). |
Primary mucinous carcinoma of the skin is a rare malignant adnexal tumor. It was first described by Lennox et al1 in 1952 and was later given its name by Mendoza and Helwig2 through summarizing clinical and histochemical characteristics based on the observation of 14 cases. A PubMed search of articles indexed for MEDLINE using the search terms adenocarcinoma, mucinous (Medical Subject Headings); mucinous adenocarcinoma (title/abstract); colloid carcinoma* (title/abstract); skin neoplasms (Medical Subject Headings); skin neoplasm (title/abstract); skin cancer* (title/abstract), and primary (title/abstract) revealed more than 200 reported cases of PMCS. It usually is located in the head and neck region, with the eyelids being the most common site of presentation, but rare presentations in the gluteal region, axillae, hemithoraxes, vulva, arms, and legs also have been reported.3-6 Primary mucinous carcinoma of the skin mainly affects elderly patients (age range, 61–80 years), with men being affected more often than women. It often presents as a painless, solitary, nontender, sometimes ulcerated, translucent, white or reddish nodule. The tumors generally grow slowly with high rates of local recurrence and rare chances of distant metastasis.7 Most metastases involve the regional lymph nodes and lungs, but rare cases of bone marrow and parotid gland metastases also have been reported.8 Because PMCS is an indolent tumor, which may be mistaken for a benign tumor, it is not always histologically examined on initial presentation.
Primary mucinous carcinoma of the skin is characterized histopathologically by floating epithelial cell nests in mucinous lakes. It is unknown if this neoplasm shows eccrine- or apocrine-type differentiation. S-100 protein seems to indicate eccrine rather than apocrine differentiation.9 Misdiagnosis of PMCS is common, as it has an uncharacteristic gross appearance and may microscopically resemble cutaneous metastasis from a mucinous carcinoma of the breast, gastrointestinal tract, lungs, ovaries, or prostate. It is important to distinguish between metastatic tumors and PMCS because PMCS generally is more benign and has a better prognosis. A variety of stains have been reported as helpful in the diagnosis of PMCS, including CK7 and CK20, D2-40, p63, and others. In some reports, labeling for CK7 and CK20 was helpful in differentiating colon carcinoma from breast or skin malignancies but did not differentiate primary skin lesions from metastasis of breast malignancies to the skin.10-13 Paradela et al5 proposed D2-40 as a reliable immunostain to detect lymphatic invasion in node-negative primary tumors. Kalebi and Hale11 and Ivan et al12 highlighted the importance of p63 in excluding metastasis to the skin, particularly from the gastrointestinal tract, pancreaticobiliary tree, and lungs.
Even though these factors may help in establishing the primary site of the tumor, a final diagnosis only can be drawn when the patient has been subjected to a thorough investigation. In our patient, clinical workup did not reveal other possible primary sites in other organs or evidence of metastasis from the skin lesion to other sites. Based on the histologic findings, the diagnosis of PMCS was made.
The recommended treatment of PMCS varies from standard excision to wide local excision including dissection of the regional lymph nodes. Alam et al14 reported that Mohs micrographic surgery may be appropriate for first-line therapy because the procedure gives complete margin control and spares tissue. Immunohistochemistry should be used as an adjunct to routine hematoxylin and eosin staining to aid in ensuring negative margins.15 Considering a high potential for recurrence following surgical excision, it is important to detect hormone receptors in this tumor because patients may be treated using antiestrogenic drugs such as tamoxifen. Moreover, recurrent or metastatic PMCS is both resistant to radiotherapy and unresponsive to chemotherapy.3 Annual follow-up is recommended due to the potential for recurrence or metastasis.
1. Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin, with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-890.
2. Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
3. Breiting L, Dahlstrøm K, Christensen L, et al. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2007;29:595-596.
4. Krishnamurthy J, Saba F, Sunila. Primary mucinous carcinoma of the skin: a rare tumor in the gluteal region. Indian J Pathol Microbiol. 2009;52:225-227.
5. Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
6. Laco J, Simáková E, Svobodová J, et al. Recurrent mucinous carcinoma of skin mimicking primary mucinous carcinoma of parotid gland: a diagnostic pitfall. Cesk Patol. 2009;45:79-82.
7. Breiting L, Christensen L, Dahlstrøm K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
8. Ajithkumar TV, Nileena N, Abraham EK, et al. Bone marrow relapse in primary mucinous carcinoma of skin. Am J Clin Oncol. 1999;22:303-304.
9. Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
10. Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin [published online ahead of print September 24, 2009]. J Cutan Pathol. 2010;37:411-415.
11. Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
12. Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34:474-480.
13. Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
14. Alam M, Trela R, Kim N, et al. Treatment of primary mucinous carcinoma of the skin: meta-analysis of 189 cases. J Invest Dermatol. 2009;129:382.
15. Stranahan D, Cherpelis BS, Glass LF, et al. Immunohistochemical stains in Mohs surgery: a review [published online ahead of print April 16, 2009]. Dermatol Surg. 2009;35:1023-1034.
To the Editor:
A 41-year-old man presented to our dermatology clinic with a recurrent asymptomatic nodule on the right cheek that had gradually increased in size over 1 year. The patient underwent laser excision at an outside facility 1 year after the first presentation. Pathology reports and tissue cultures from the excision were not available. The lesion recurred in the same location 6 months following excision. The patient reported no history of pain, fever, cough, weight loss, or loss of appetite, and he denied any trauma or radiation to the affected area. Dermatologic examination revealed a 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek (Figure 1). There was no tenderness or pruritus. Clinical examination and extensive radiographic studies revealed no primary disease. A complete blood cell count, biochemical tests, and serous tumor markers were within reference range. The lesion was resected with a 2-mm margin. The margins were free of tumor cells. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa and scattered floating islands of tumor cells in the dermis. Small-sized glands were organized in some areas of the tumor cells (Figure 2). Epithelial tumor cell islands contained uniform oval nuclei and focally vacuolated cytoplasm. The tumor cells were positive for cytokeratin 7 (CK7) (Figure 3) and negative for cytokeratin 20 (CK20), carcinoembryonic antigen, homeobox transcription factor (CDX2), villin, mucus-associated peptides of the thyroid transcription factor (TTF1), and prostate-specific antigen, all indicating a diagnosis of primary mucinous carcinoma of the skin (PMCS). No local recurrence, regional lymph node involvement, or distant metastasis was observed after resection.
|
| Figure 1. A 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek. |
|
| Figure 2. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa that contained scattered floating islands of tumor cells in the dermis (H&E, original magnification ×40). |
|
| Figure 3. The tumor cells were positive for cytokeratin 7 (original magnification ×400). |
Primary mucinous carcinoma of the skin is a rare malignant adnexal tumor. It was first described by Lennox et al1 in 1952 and was later given its name by Mendoza and Helwig2 through summarizing clinical and histochemical characteristics based on the observation of 14 cases. A PubMed search of articles indexed for MEDLINE using the search terms adenocarcinoma, mucinous (Medical Subject Headings); mucinous adenocarcinoma (title/abstract); colloid carcinoma* (title/abstract); skin neoplasms (Medical Subject Headings); skin neoplasm (title/abstract); skin cancer* (title/abstract), and primary (title/abstract) revealed more than 200 reported cases of PMCS. It usually is located in the head and neck region, with the eyelids being the most common site of presentation, but rare presentations in the gluteal region, axillae, hemithoraxes, vulva, arms, and legs also have been reported.3-6 Primary mucinous carcinoma of the skin mainly affects elderly patients (age range, 61–80 years), with men being affected more often than women. It often presents as a painless, solitary, nontender, sometimes ulcerated, translucent, white or reddish nodule. The tumors generally grow slowly with high rates of local recurrence and rare chances of distant metastasis.7 Most metastases involve the regional lymph nodes and lungs, but rare cases of bone marrow and parotid gland metastases also have been reported.8 Because PMCS is an indolent tumor, which may be mistaken for a benign tumor, it is not always histologically examined on initial presentation.
Primary mucinous carcinoma of the skin is characterized histopathologically by floating epithelial cell nests in mucinous lakes. It is unknown if this neoplasm shows eccrine- or apocrine-type differentiation. S-100 protein seems to indicate eccrine rather than apocrine differentiation.9 Misdiagnosis of PMCS is common, as it has an uncharacteristic gross appearance and may microscopically resemble cutaneous metastasis from a mucinous carcinoma of the breast, gastrointestinal tract, lungs, ovaries, or prostate. It is important to distinguish between metastatic tumors and PMCS because PMCS generally is more benign and has a better prognosis. A variety of stains have been reported as helpful in the diagnosis of PMCS, including CK7 and CK20, D2-40, p63, and others. In some reports, labeling for CK7 and CK20 was helpful in differentiating colon carcinoma from breast or skin malignancies but did not differentiate primary skin lesions from metastasis of breast malignancies to the skin.10-13 Paradela et al5 proposed D2-40 as a reliable immunostain to detect lymphatic invasion in node-negative primary tumors. Kalebi and Hale11 and Ivan et al12 highlighted the importance of p63 in excluding metastasis to the skin, particularly from the gastrointestinal tract, pancreaticobiliary tree, and lungs.
Even though these factors may help in establishing the primary site of the tumor, a final diagnosis only can be drawn when the patient has been subjected to a thorough investigation. In our patient, clinical workup did not reveal other possible primary sites in other organs or evidence of metastasis from the skin lesion to other sites. Based on the histologic findings, the diagnosis of PMCS was made.
The recommended treatment of PMCS varies from standard excision to wide local excision including dissection of the regional lymph nodes. Alam et al14 reported that Mohs micrographic surgery may be appropriate for first-line therapy because the procedure gives complete margin control and spares tissue. Immunohistochemistry should be used as an adjunct to routine hematoxylin and eosin staining to aid in ensuring negative margins.15 Considering a high potential for recurrence following surgical excision, it is important to detect hormone receptors in this tumor because patients may be treated using antiestrogenic drugs such as tamoxifen. Moreover, recurrent or metastatic PMCS is both resistant to radiotherapy and unresponsive to chemotherapy.3 Annual follow-up is recommended due to the potential for recurrence or metastasis.
To the Editor:
A 41-year-old man presented to our dermatology clinic with a recurrent asymptomatic nodule on the right cheek that had gradually increased in size over 1 year. The patient underwent laser excision at an outside facility 1 year after the first presentation. Pathology reports and tissue cultures from the excision were not available. The lesion recurred in the same location 6 months following excision. The patient reported no history of pain, fever, cough, weight loss, or loss of appetite, and he denied any trauma or radiation to the affected area. Dermatologic examination revealed a 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek (Figure 1). There was no tenderness or pruritus. Clinical examination and extensive radiographic studies revealed no primary disease. A complete blood cell count, biochemical tests, and serous tumor markers were within reference range. The lesion was resected with a 2-mm margin. The margins were free of tumor cells. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa and scattered floating islands of tumor cells in the dermis. Small-sized glands were organized in some areas of the tumor cells (Figure 2). Epithelial tumor cell islands contained uniform oval nuclei and focally vacuolated cytoplasm. The tumor cells were positive for cytokeratin 7 (CK7) (Figure 3) and negative for cytokeratin 20 (CK20), carcinoembryonic antigen, homeobox transcription factor (CDX2), villin, mucus-associated peptides of the thyroid transcription factor (TTF1), and prostate-specific antigen, all indicating a diagnosis of primary mucinous carcinoma of the skin (PMCS). No local recurrence, regional lymph node involvement, or distant metastasis was observed after resection.
|
| Figure 1. A 24×18-mm, slightly elevated, dome-shaped, translucent, pink-colored tumor with telangiectasia on the right cheek. |
|
| Figure 2. Histopathology showed a circumscribed tumor with large amounts of mucin compartmentalized by fibrous septa that contained scattered floating islands of tumor cells in the dermis (H&E, original magnification ×40). |
|
| Figure 3. The tumor cells were positive for cytokeratin 7 (original magnification ×400). |
Primary mucinous carcinoma of the skin is a rare malignant adnexal tumor. It was first described by Lennox et al1 in 1952 and was later given its name by Mendoza and Helwig2 through summarizing clinical and histochemical characteristics based on the observation of 14 cases. A PubMed search of articles indexed for MEDLINE using the search terms adenocarcinoma, mucinous (Medical Subject Headings); mucinous adenocarcinoma (title/abstract); colloid carcinoma* (title/abstract); skin neoplasms (Medical Subject Headings); skin neoplasm (title/abstract); skin cancer* (title/abstract), and primary (title/abstract) revealed more than 200 reported cases of PMCS. It usually is located in the head and neck region, with the eyelids being the most common site of presentation, but rare presentations in the gluteal region, axillae, hemithoraxes, vulva, arms, and legs also have been reported.3-6 Primary mucinous carcinoma of the skin mainly affects elderly patients (age range, 61–80 years), with men being affected more often than women. It often presents as a painless, solitary, nontender, sometimes ulcerated, translucent, white or reddish nodule. The tumors generally grow slowly with high rates of local recurrence and rare chances of distant metastasis.7 Most metastases involve the regional lymph nodes and lungs, but rare cases of bone marrow and parotid gland metastases also have been reported.8 Because PMCS is an indolent tumor, which may be mistaken for a benign tumor, it is not always histologically examined on initial presentation.
Primary mucinous carcinoma of the skin is characterized histopathologically by floating epithelial cell nests in mucinous lakes. It is unknown if this neoplasm shows eccrine- or apocrine-type differentiation. S-100 protein seems to indicate eccrine rather than apocrine differentiation.9 Misdiagnosis of PMCS is common, as it has an uncharacteristic gross appearance and may microscopically resemble cutaneous metastasis from a mucinous carcinoma of the breast, gastrointestinal tract, lungs, ovaries, or prostate. It is important to distinguish between metastatic tumors and PMCS because PMCS generally is more benign and has a better prognosis. A variety of stains have been reported as helpful in the diagnosis of PMCS, including CK7 and CK20, D2-40, p63, and others. In some reports, labeling for CK7 and CK20 was helpful in differentiating colon carcinoma from breast or skin malignancies but did not differentiate primary skin lesions from metastasis of breast malignancies to the skin.10-13 Paradela et al5 proposed D2-40 as a reliable immunostain to detect lymphatic invasion in node-negative primary tumors. Kalebi and Hale11 and Ivan et al12 highlighted the importance of p63 in excluding metastasis to the skin, particularly from the gastrointestinal tract, pancreaticobiliary tree, and lungs.
Even though these factors may help in establishing the primary site of the tumor, a final diagnosis only can be drawn when the patient has been subjected to a thorough investigation. In our patient, clinical workup did not reveal other possible primary sites in other organs or evidence of metastasis from the skin lesion to other sites. Based on the histologic findings, the diagnosis of PMCS was made.
The recommended treatment of PMCS varies from standard excision to wide local excision including dissection of the regional lymph nodes. Alam et al14 reported that Mohs micrographic surgery may be appropriate for first-line therapy because the procedure gives complete margin control and spares tissue. Immunohistochemistry should be used as an adjunct to routine hematoxylin and eosin staining to aid in ensuring negative margins.15 Considering a high potential for recurrence following surgical excision, it is important to detect hormone receptors in this tumor because patients may be treated using antiestrogenic drugs such as tamoxifen. Moreover, recurrent or metastatic PMCS is both resistant to radiotherapy and unresponsive to chemotherapy.3 Annual follow-up is recommended due to the potential for recurrence or metastasis.
1. Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin, with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-890.
2. Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
3. Breiting L, Dahlstrøm K, Christensen L, et al. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2007;29:595-596.
4. Krishnamurthy J, Saba F, Sunila. Primary mucinous carcinoma of the skin: a rare tumor in the gluteal region. Indian J Pathol Microbiol. 2009;52:225-227.
5. Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
6. Laco J, Simáková E, Svobodová J, et al. Recurrent mucinous carcinoma of skin mimicking primary mucinous carcinoma of parotid gland: a diagnostic pitfall. Cesk Patol. 2009;45:79-82.
7. Breiting L, Christensen L, Dahlstrøm K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
8. Ajithkumar TV, Nileena N, Abraham EK, et al. Bone marrow relapse in primary mucinous carcinoma of skin. Am J Clin Oncol. 1999;22:303-304.
9. Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
10. Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin [published online ahead of print September 24, 2009]. J Cutan Pathol. 2010;37:411-415.
11. Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
12. Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34:474-480.
13. Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
14. Alam M, Trela R, Kim N, et al. Treatment of primary mucinous carcinoma of the skin: meta-analysis of 189 cases. J Invest Dermatol. 2009;129:382.
15. Stranahan D, Cherpelis BS, Glass LF, et al. Immunohistochemical stains in Mohs surgery: a review [published online ahead of print April 16, 2009]. Dermatol Surg. 2009;35:1023-1034.
1. Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin, with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-890.
2. Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
3. Breiting L, Dahlstrøm K, Christensen L, et al. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2007;29:595-596.
4. Krishnamurthy J, Saba F, Sunila. Primary mucinous carcinoma of the skin: a rare tumor in the gluteal region. Indian J Pathol Microbiol. 2009;52:225-227.
5. Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
6. Laco J, Simáková E, Svobodová J, et al. Recurrent mucinous carcinoma of skin mimicking primary mucinous carcinoma of parotid gland: a diagnostic pitfall. Cesk Patol. 2009;45:79-82.
7. Breiting L, Christensen L, Dahlstrøm K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
8. Ajithkumar TV, Nileena N, Abraham EK, et al. Bone marrow relapse in primary mucinous carcinoma of skin. Am J Clin Oncol. 1999;22:303-304.
9. Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
10. Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin [published online ahead of print September 24, 2009]. J Cutan Pathol. 2010;37:411-415.
11. Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
12. Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34:474-480.
13. Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
14. Alam M, Trela R, Kim N, et al. Treatment of primary mucinous carcinoma of the skin: meta-analysis of 189 cases. J Invest Dermatol. 2009;129:382.
15. Stranahan D, Cherpelis BS, Glass LF, et al. Immunohistochemical stains in Mohs surgery: a review [published online ahead of print April 16, 2009]. Dermatol Surg. 2009;35:1023-1034.
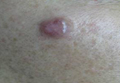
Large Plaque-Type Benign Cephalic Histiocytosis Showing Rapid Aggravation Following Vaccination
To the Editor:
Benign cephalic histiocytosis (BCH) is a rare benign dermatosis in which self-healing papular eruptions develop. This condition is classified as non-X histiocytosis and behaves as a benign histiocytic proliferation. Since the first report by Gianotti et al1 in 1971, the etiology and pathophysiology of this condition have not been elucidated. The clinical and histologic features of BCH overlap with juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis.2 Rodriguez-Jurado et al3 reported that BCH showed clinical evolution to JXG with varicella-zoster infection, suggesting that BCH is a part of a spectrum of diseases that non–Langerhans cell histiocytosis encompasses. We present a case of plaque-type BCH in an infant.
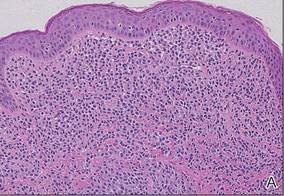
An 11-month-old male infant presented with asymptomatic, ill-defined, infiltrated plaques on the cheeks of 1 month’s duration (Figure 1). Similar red plaques were present on both ears, the left forearm, and the right middle finger. One month following the initial onset as tiny red papules, the lesions had become larger and more prominent after an episode of fever (temperature, 39°C) and immunization against rubella virus and Haemophilus influenzae type b. No therapeutic effect was observed with the application of a topical steroid at the time of presentation. A skin biopsy specimen was taken from a flat plaque on the right cheek. Histopathologic findings revealed dense cell infiltrates predominated by histiocytic cells with some vacuolization in the papillary and upper reticular dermis (Figure 2). Neither foamy histiocytic cells nor giant cells were seen. Immunohistochemical analysis revealed that the cells were CD68+ (Figure 3) and negative for S-100. Following 2 months of observation without any treatment, the plaques on the ears, right forearm, and right middle finger had begun to spontaneously subside. Overall, the remaining lesions showed complete remission by 21 months of age.
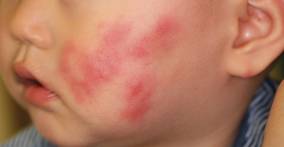 |
| Figure 1. Asymptomatic red, infiltrated, flat plaques on the left cheek. |
 |
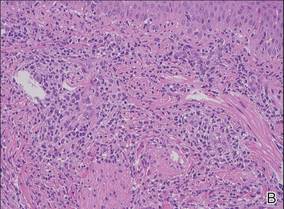 |
| Figure 2. A biopsy revealed an infiltrate of histiocytes with vacuolization of cells in the papillary and upper reticular dermis (A and B) (both H&E, original magnifications ×50 and ×200). |
 |
| Figure 3. Immunohistochemistry revealed histiocytic infiltrate cells that were CD68+ (original magnification ×50). |
Benign cephalic histiocytosis commonly presents as smooth, round, or flat-topped papules with a diameter of 1 to 8 mm and color ranging from tan-yellow to brown-red.2 The etiology of BCH is unknown. Plaque-type BCH represented by this patient is a rare feature, suggesting sarcoidosis, perniosis, or Sjögren syndrome because the lesions are comprised of infiltrated erythematous plaques rather than papules. Rodriguez-Jurado et al3 described a transitional form of BCH to JXG in a 2-year-old girl with facial plaques after an episode of varicella-zoster virus infection. Vasconcelos et al4 also indicated that JXG is associated with cytomegalovirus infection. These findings support the notion that BCH and non–Langerhans cell histiocytosis are on the same spectrum of diseases, representing a reactive histiocytic process to an infectious agent.
The difference in clinical manifestation of each non–Langerhans cell histiocytosis subgroup may reflect the degree or stage of infection. T lymphocytes may release different cytokines and activate macrophage migration when sensitized by a virus or vaccine. As a result, BCH may show various clinical features even when confirmed by histological analysis. Our patient might have had an early stage of BCH that would transform into JXG.
1. Gianotti F, Caputo R, Ermacora E. Singular “infantile histiocytosis with cells with intracytoplasmic vermiform particles” [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
4. Vasconcelos FO, Oliveira LA, Naves MD, et al. Juvenile xanthogranuloma: case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci. 2001;43:21-25.
To the Editor:
Benign cephalic histiocytosis (BCH) is a rare benign dermatosis in which self-healing papular eruptions develop. This condition is classified as non-X histiocytosis and behaves as a benign histiocytic proliferation. Since the first report by Gianotti et al1 in 1971, the etiology and pathophysiology of this condition have not been elucidated. The clinical and histologic features of BCH overlap with juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis.2 Rodriguez-Jurado et al3 reported that BCH showed clinical evolution to JXG with varicella-zoster infection, suggesting that BCH is a part of a spectrum of diseases that non–Langerhans cell histiocytosis encompasses. We present a case of plaque-type BCH in an infant.
An 11-month-old male infant presented with asymptomatic, ill-defined, infiltrated plaques on the cheeks of 1 month’s duration (Figure 1). Similar red plaques were present on both ears, the left forearm, and the right middle finger. One month following the initial onset as tiny red papules, the lesions had become larger and more prominent after an episode of fever (temperature, 39°C) and immunization against rubella virus and Haemophilus influenzae type b. No therapeutic effect was observed with the application of a topical steroid at the time of presentation. A skin biopsy specimen was taken from a flat plaque on the right cheek. Histopathologic findings revealed dense cell infiltrates predominated by histiocytic cells with some vacuolization in the papillary and upper reticular dermis (Figure 2). Neither foamy histiocytic cells nor giant cells were seen. Immunohistochemical analysis revealed that the cells were CD68+ (Figure 3) and negative for S-100. Following 2 months of observation without any treatment, the plaques on the ears, right forearm, and right middle finger had begun to spontaneously subside. Overall, the remaining lesions showed complete remission by 21 months of age.
|
| Figure 1. Asymptomatic red, infiltrated, flat plaques on the left cheek. |
|
|
| Figure 2. A biopsy revealed an infiltrate of histiocytes with vacuolization of cells in the papillary and upper reticular dermis (A and B) (both H&E, original magnifications ×50 and ×200). |
|
| Figure 3. Immunohistochemistry revealed histiocytic infiltrate cells that were CD68+ (original magnification ×50). |
Benign cephalic histiocytosis commonly presents as smooth, round, or flat-topped papules with a diameter of 1 to 8 mm and color ranging from tan-yellow to brown-red.2 The etiology of BCH is unknown. Plaque-type BCH represented by this patient is a rare feature, suggesting sarcoidosis, perniosis, or Sjögren syndrome because the lesions are comprised of infiltrated erythematous plaques rather than papules. Rodriguez-Jurado et al3 described a transitional form of BCH to JXG in a 2-year-old girl with facial plaques after an episode of varicella-zoster virus infection. Vasconcelos et al4 also indicated that JXG is associated with cytomegalovirus infection. These findings support the notion that BCH and non–Langerhans cell histiocytosis are on the same spectrum of diseases, representing a reactive histiocytic process to an infectious agent.
The difference in clinical manifestation of each non–Langerhans cell histiocytosis subgroup may reflect the degree or stage of infection. T lymphocytes may release different cytokines and activate macrophage migration when sensitized by a virus or vaccine. As a result, BCH may show various clinical features even when confirmed by histological analysis. Our patient might have had an early stage of BCH that would transform into JXG.
To the Editor:
Benign cephalic histiocytosis (BCH) is a rare benign dermatosis in which self-healing papular eruptions develop. This condition is classified as non-X histiocytosis and behaves as a benign histiocytic proliferation. Since the first report by Gianotti et al1 in 1971, the etiology and pathophysiology of this condition have not been elucidated. The clinical and histologic features of BCH overlap with juvenile xanthogranuloma (JXG) and generalized eruptive histiocytosis.2 Rodriguez-Jurado et al3 reported that BCH showed clinical evolution to JXG with varicella-zoster infection, suggesting that BCH is a part of a spectrum of diseases that non–Langerhans cell histiocytosis encompasses. We present a case of plaque-type BCH in an infant.
An 11-month-old male infant presented with asymptomatic, ill-defined, infiltrated plaques on the cheeks of 1 month’s duration (Figure 1). Similar red plaques were present on both ears, the left forearm, and the right middle finger. One month following the initial onset as tiny red papules, the lesions had become larger and more prominent after an episode of fever (temperature, 39°C) and immunization against rubella virus and Haemophilus influenzae type b. No therapeutic effect was observed with the application of a topical steroid at the time of presentation. A skin biopsy specimen was taken from a flat plaque on the right cheek. Histopathologic findings revealed dense cell infiltrates predominated by histiocytic cells with some vacuolization in the papillary and upper reticular dermis (Figure 2). Neither foamy histiocytic cells nor giant cells were seen. Immunohistochemical analysis revealed that the cells were CD68+ (Figure 3) and negative for S-100. Following 2 months of observation without any treatment, the plaques on the ears, right forearm, and right middle finger had begun to spontaneously subside. Overall, the remaining lesions showed complete remission by 21 months of age.
|
| Figure 1. Asymptomatic red, infiltrated, flat plaques on the left cheek. |
|
|
| Figure 2. A biopsy revealed an infiltrate of histiocytes with vacuolization of cells in the papillary and upper reticular dermis (A and B) (both H&E, original magnifications ×50 and ×200). |
|
| Figure 3. Immunohistochemistry revealed histiocytic infiltrate cells that were CD68+ (original magnification ×50). |
Benign cephalic histiocytosis commonly presents as smooth, round, or flat-topped papules with a diameter of 1 to 8 mm and color ranging from tan-yellow to brown-red.2 The etiology of BCH is unknown. Plaque-type BCH represented by this patient is a rare feature, suggesting sarcoidosis, perniosis, or Sjögren syndrome because the lesions are comprised of infiltrated erythematous plaques rather than papules. Rodriguez-Jurado et al3 described a transitional form of BCH to JXG in a 2-year-old girl with facial plaques after an episode of varicella-zoster virus infection. Vasconcelos et al4 also indicated that JXG is associated with cytomegalovirus infection. These findings support the notion that BCH and non–Langerhans cell histiocytosis are on the same spectrum of diseases, representing a reactive histiocytic process to an infectious agent.
The difference in clinical manifestation of each non–Langerhans cell histiocytosis subgroup may reflect the degree or stage of infection. T lymphocytes may release different cytokines and activate macrophage migration when sensitized by a virus or vaccine. As a result, BCH may show various clinical features even when confirmed by histological analysis. Our patient might have had an early stage of BCH that would transform into JXG.
1. Gianotti F, Caputo R, Ermacora E. Singular “infantile histiocytosis with cells with intracytoplasmic vermiform particles” [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
4. Vasconcelos FO, Oliveira LA, Naves MD, et al. Juvenile xanthogranuloma: case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci. 2001;43:21-25.
1. Gianotti F, Caputo R, Ermacora E. Singular “infantile histiocytosis with cells with intracytoplasmic vermiform particles” [in French]. Bull Soc Fr Dermatol Syphiligr. 1971;78:232-233.
2. Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol. 2002;47:908-913.
3. Rodriguez-Jurado R, Duran-McKinster C, Ruiz-Maldonado R. Benign cephalic histiocytosis progressing into juvenile xanthogranuloma: a non-Langerhans cell histiocytosis transforming under the influence of a virus? Am J Dermatopathol. 2000;22:70-74.
4. Vasconcelos FO, Oliveira LA, Naves MD, et al. Juvenile xanthogranuloma: case report with immunohistochemical identification of early and late cytomegalovirus antigens. J Oral Sci. 2001;43:21-25.
Pedunculated Scrotal Nodule in an Elderly Male: A Rare Presentation of Trichoblastoma
To the Editor:
A 78-year-old man with no relevant dermatologic history presented for a routine skin examination. He had a large pedunculated nodule on the right scrotum that had been stable in size for more than 40 years. He reported the lesion had been diagnosed as a skin tag by his urologist 20 years prior and had remained asymptomatic in nature. On examination, a 2.5×1-cm, pedunculated, firm, rubbery nodule protruded from the anterior aspect of the right scrotal skin (Figure 1). Prominent telangiectases were noted on the surface of the lesion along with an irregular pigmented macule on the tip. There was no inguinal lymphadenopathy. Given the pigmentation at the tip of the lesion, he was advised to have it completely excised to rule out melanocytic atypia. The lesion subsequently was excised under local anesthesia and the resulting defect was primarily closed. A group of basaloid lobules within the dermis with scant stroma was noted (Figure 2). There was no epidermal connection and retraction artifact was absent. The epidermis appeared normal, except for basilar pigmentation corresponding to the pigmented macule at the tip of the lesion. Higher magnification of one of the basaloid islands demonstrated a papillary mesenchymal body (Figure 3). Immunohistochemistry with cytokeratin 20 showed positive staining of Merkel cells colonizing the basaloid islands (Figure 4). A diagnosis of trichoblastoma was rendered. Two nodular basal cell carcinomas (BCCs) were seen on initial presentation, but he remained free of recurrence of the scrotal tumor at 2-year follow-up.
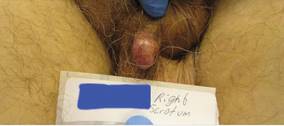 |
Figure 1. Firm, pedunculated, flesh-colored nodule on the scrotum. |
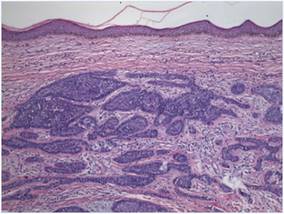 |
Figure 2. Basaloid tumor islands within the dermis with no epidermal connection (H&E, original magnification ×20). |
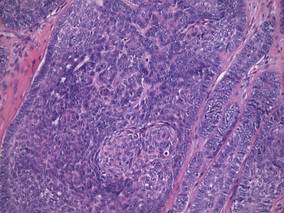 |
Figure 3. Papillary mesenchymal body adjacent to several dermal basaloid islands (H&E, original magnification ×40). |
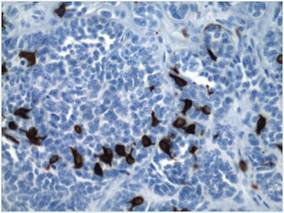 |
Figure 4. Cytokeratin 20 immunohistochemical staining indicated Merkel cells within the basaloid islands (original magnification ×40). |
Trichoblastoma is a rare benign tumor with differentiation toward the primitive hair follicle. The lesion typically presents as a slow-growing, solitary, well-circumscribed nodule that typically measures up to 3 cm and is predominantly located on the head and neck with a predilection for the scalp. Rarely the lesion will appear on the trunk, proximal extremities, or perianal and genital regions.1 Trichoblastomas show no gender predilection and predominantly affect adults between the fifth and seventh decades of life. In children it is the most common neoplasm arising in preexisting nevus sebaceous (Jadassohn nevus).2 Several clinical variants of trichoblastoma have been described, including giant trichoblastoma, pigmented trichoblastoma, and subcutaneous trichoblastoma, among others.3
Histologically, these tumors are characterized by well-circumscribed basaloid islands in the dermis with peripheral palisading closely resembling BCCs and trichoepitheliomas. Unlike BCCs, retraction artifact in trichoblastoma is a rare finding, while papillary mesenchymal bodies and an increased number of Merkel cells characterize both trichoblastomas and trichoepitheliomas, as in our patient.4 Other histologic features include lack of epidermal connection and location in the deeper dermis and/or subcutis. There also are reported cases with sebaceous, pigmented, rippled patterns and clear cell differentiation on histology.3,5
The genetic basis for trichoblastoma remains unclear. Although these primitive hair follicle tumors share histologic features with BCC and trichoepithelioma, which have both demonstrated PTCH (patched) mutations, no classic mutations in the PTCH gene were seen in 11 cases of trichoblastoma.6 In contrast, there have been reports of trichoblastoma occurring in Brooke-Spiegler syndrome and a case of a somatic CYLD (cylindromatosis) frameshift mutation in a trichoblastoma in a patient with Brooke-Spiegler syndrome7; however, the significance of CYLD mutation in thepathogenesis of trichoblastoma has not been fully elucidated.8
Trichoblastomas generally behave in an indolent asymptomatic manner, with malignant transformation being a rare occurrence.9 However, complete excision of these tumors is recommended given the potential for continued slow growth if incompletely removed. In the case of our patient, the lesion was completely excised at the outset. We plan to clinically monitor for evidence of disease recurrence as part of an annual full-body examination and have noted no evidence of recurrence in more than 2 years.
1. Brenn T, McKee PH. Tumors of the hair follicle. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. London, England: Elsevier Mosby; 2005:1552-1557.
2. Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceous of Jadassohn: a clinicopathologic study of series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
3. Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:e37-e40.
4. Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
5. Swick BL, Baum CL, Walling HW. Rippled-patterntrichoblastoma with apocrine differentiation arising in a nevus sebaceus: report of a case and review of the literature. J Cutan Pathol. 2009;36:1200-1205.
6. Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas [published online ahead of print June 26, 2007]. Hum Pathol. 2007;38:1496-1500.
7. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
8. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors [published online ahead of print February 4, 2010]. J Cutan Pathol. 2010;37:886-890.
9. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16
To the Editor:
A 78-year-old man with no relevant dermatologic history presented for a routine skin examination. He had a large pedunculated nodule on the right scrotum that had been stable in size for more than 40 years. He reported the lesion had been diagnosed as a skin tag by his urologist 20 years prior and had remained asymptomatic in nature. On examination, a 2.5×1-cm, pedunculated, firm, rubbery nodule protruded from the anterior aspect of the right scrotal skin (Figure 1). Prominent telangiectases were noted on the surface of the lesion along with an irregular pigmented macule on the tip. There was no inguinal lymphadenopathy. Given the pigmentation at the tip of the lesion, he was advised to have it completely excised to rule out melanocytic atypia. The lesion subsequently was excised under local anesthesia and the resulting defect was primarily closed. A group of basaloid lobules within the dermis with scant stroma was noted (Figure 2). There was no epidermal connection and retraction artifact was absent. The epidermis appeared normal, except for basilar pigmentation corresponding to the pigmented macule at the tip of the lesion. Higher magnification of one of the basaloid islands demonstrated a papillary mesenchymal body (Figure 3). Immunohistochemistry with cytokeratin 20 showed positive staining of Merkel cells colonizing the basaloid islands (Figure 4). A diagnosis of trichoblastoma was rendered. Two nodular basal cell carcinomas (BCCs) were seen on initial presentation, but he remained free of recurrence of the scrotal tumor at 2-year follow-up.
|
Figure 1. Firm, pedunculated, flesh-colored nodule on the scrotum. |
|
Figure 2. Basaloid tumor islands within the dermis with no epidermal connection (H&E, original magnification ×20). |
|
Figure 3. Papillary mesenchymal body adjacent to several dermal basaloid islands (H&E, original magnification ×40). |
|
Figure 4. Cytokeratin 20 immunohistochemical staining indicated Merkel cells within the basaloid islands (original magnification ×40). |
Trichoblastoma is a rare benign tumor with differentiation toward the primitive hair follicle. The lesion typically presents as a slow-growing, solitary, well-circumscribed nodule that typically measures up to 3 cm and is predominantly located on the head and neck with a predilection for the scalp. Rarely the lesion will appear on the trunk, proximal extremities, or perianal and genital regions.1 Trichoblastomas show no gender predilection and predominantly affect adults between the fifth and seventh decades of life. In children it is the most common neoplasm arising in preexisting nevus sebaceous (Jadassohn nevus).2 Several clinical variants of trichoblastoma have been described, including giant trichoblastoma, pigmented trichoblastoma, and subcutaneous trichoblastoma, among others.3
Histologically, these tumors are characterized by well-circumscribed basaloid islands in the dermis with peripheral palisading closely resembling BCCs and trichoepitheliomas. Unlike BCCs, retraction artifact in trichoblastoma is a rare finding, while papillary mesenchymal bodies and an increased number of Merkel cells characterize both trichoblastomas and trichoepitheliomas, as in our patient.4 Other histologic features include lack of epidermal connection and location in the deeper dermis and/or subcutis. There also are reported cases with sebaceous, pigmented, rippled patterns and clear cell differentiation on histology.3,5
The genetic basis for trichoblastoma remains unclear. Although these primitive hair follicle tumors share histologic features with BCC and trichoepithelioma, which have both demonstrated PTCH (patched) mutations, no classic mutations in the PTCH gene were seen in 11 cases of trichoblastoma.6 In contrast, there have been reports of trichoblastoma occurring in Brooke-Spiegler syndrome and a case of a somatic CYLD (cylindromatosis) frameshift mutation in a trichoblastoma in a patient with Brooke-Spiegler syndrome7; however, the significance of CYLD mutation in thepathogenesis of trichoblastoma has not been fully elucidated.8
Trichoblastomas generally behave in an indolent asymptomatic manner, with malignant transformation being a rare occurrence.9 However, complete excision of these tumors is recommended given the potential for continued slow growth if incompletely removed. In the case of our patient, the lesion was completely excised at the outset. We plan to clinically monitor for evidence of disease recurrence as part of an annual full-body examination and have noted no evidence of recurrence in more than 2 years.
To the Editor:
A 78-year-old man with no relevant dermatologic history presented for a routine skin examination. He had a large pedunculated nodule on the right scrotum that had been stable in size for more than 40 years. He reported the lesion had been diagnosed as a skin tag by his urologist 20 years prior and had remained asymptomatic in nature. On examination, a 2.5×1-cm, pedunculated, firm, rubbery nodule protruded from the anterior aspect of the right scrotal skin (Figure 1). Prominent telangiectases were noted on the surface of the lesion along with an irregular pigmented macule on the tip. There was no inguinal lymphadenopathy. Given the pigmentation at the tip of the lesion, he was advised to have it completely excised to rule out melanocytic atypia. The lesion subsequently was excised under local anesthesia and the resulting defect was primarily closed. A group of basaloid lobules within the dermis with scant stroma was noted (Figure 2). There was no epidermal connection and retraction artifact was absent. The epidermis appeared normal, except for basilar pigmentation corresponding to the pigmented macule at the tip of the lesion. Higher magnification of one of the basaloid islands demonstrated a papillary mesenchymal body (Figure 3). Immunohistochemistry with cytokeratin 20 showed positive staining of Merkel cells colonizing the basaloid islands (Figure 4). A diagnosis of trichoblastoma was rendered. Two nodular basal cell carcinomas (BCCs) were seen on initial presentation, but he remained free of recurrence of the scrotal tumor at 2-year follow-up.
|
Figure 1. Firm, pedunculated, flesh-colored nodule on the scrotum. |
|
Figure 2. Basaloid tumor islands within the dermis with no epidermal connection (H&E, original magnification ×20). |
|
Figure 3. Papillary mesenchymal body adjacent to several dermal basaloid islands (H&E, original magnification ×40). |
|
Figure 4. Cytokeratin 20 immunohistochemical staining indicated Merkel cells within the basaloid islands (original magnification ×40). |
Trichoblastoma is a rare benign tumor with differentiation toward the primitive hair follicle. The lesion typically presents as a slow-growing, solitary, well-circumscribed nodule that typically measures up to 3 cm and is predominantly located on the head and neck with a predilection for the scalp. Rarely the lesion will appear on the trunk, proximal extremities, or perianal and genital regions.1 Trichoblastomas show no gender predilection and predominantly affect adults between the fifth and seventh decades of life. In children it is the most common neoplasm arising in preexisting nevus sebaceous (Jadassohn nevus).2 Several clinical variants of trichoblastoma have been described, including giant trichoblastoma, pigmented trichoblastoma, and subcutaneous trichoblastoma, among others.3
Histologically, these tumors are characterized by well-circumscribed basaloid islands in the dermis with peripheral palisading closely resembling BCCs and trichoepitheliomas. Unlike BCCs, retraction artifact in trichoblastoma is a rare finding, while papillary mesenchymal bodies and an increased number of Merkel cells characterize both trichoblastomas and trichoepitheliomas, as in our patient.4 Other histologic features include lack of epidermal connection and location in the deeper dermis and/or subcutis. There also are reported cases with sebaceous, pigmented, rippled patterns and clear cell differentiation on histology.3,5
The genetic basis for trichoblastoma remains unclear. Although these primitive hair follicle tumors share histologic features with BCC and trichoepithelioma, which have both demonstrated PTCH (patched) mutations, no classic mutations in the PTCH gene were seen in 11 cases of trichoblastoma.6 In contrast, there have been reports of trichoblastoma occurring in Brooke-Spiegler syndrome and a case of a somatic CYLD (cylindromatosis) frameshift mutation in a trichoblastoma in a patient with Brooke-Spiegler syndrome7; however, the significance of CYLD mutation in thepathogenesis of trichoblastoma has not been fully elucidated.8
Trichoblastomas generally behave in an indolent asymptomatic manner, with malignant transformation being a rare occurrence.9 However, complete excision of these tumors is recommended given the potential for continued slow growth if incompletely removed. In the case of our patient, the lesion was completely excised at the outset. We plan to clinically monitor for evidence of disease recurrence as part of an annual full-body examination and have noted no evidence of recurrence in more than 2 years.
1. Brenn T, McKee PH. Tumors of the hair follicle. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. London, England: Elsevier Mosby; 2005:1552-1557.
2. Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceous of Jadassohn: a clinicopathologic study of series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
3. Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:e37-e40.
4. Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
5. Swick BL, Baum CL, Walling HW. Rippled-patterntrichoblastoma with apocrine differentiation arising in a nevus sebaceus: report of a case and review of the literature. J Cutan Pathol. 2009;36:1200-1205.
6. Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas [published online ahead of print June 26, 2007]. Hum Pathol. 2007;38:1496-1500.
7. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
8. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors [published online ahead of print February 4, 2010]. J Cutan Pathol. 2010;37:886-890.
9. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16
1. Brenn T, McKee PH. Tumors of the hair follicle. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. London, England: Elsevier Mosby; 2005:1552-1557.
2. Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceous of Jadassohn: a clinicopathologic study of series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
3. Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:e37-e40.
4. Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinomas but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
5. Swick BL, Baum CL, Walling HW. Rippled-patterntrichoblastoma with apocrine differentiation arising in a nevus sebaceus: report of a case and review of the literature. J Cutan Pathol. 2009;36:1200-1205.
6. Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas [published online ahead of print June 26, 2007]. Hum Pathol. 2007;38:1496-1500.
7. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
8. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors [published online ahead of print February 4, 2010]. J Cutan Pathol. 2010;37:886-890.
9. Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblastic carcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16
Lepromatous Leprosy Associated With Erythema Nodosum Leprosum
To the Editor:
A 41-year-old man presented to our clinic with concerns of ulceration, skin thickening, and loss of eyebrows. The ulceration developed on the knees approximately 1 year prior to presentation; however, he reported loss of sensation in the knees after an accident involving his back 7 years prior. Loss of the eyebrows and skin thickening on the abdomen occurred within the same time frame of the knee ulceration. He also experienced generalized loss of skin sensation. The patient had a history of hunting and fishing but denied any contact with an armadillo.
On clinical examination thin yellow plaques were present on both cheeks, as well as typical leonine facies, a saddle nose deformity, loss of bilateral eyebrows, slate gray color on the face, and infiltrated plaques with telangiectases on the nose. Subcutaneous nodules developed on the arms, chest, and abdomen. He also had angulated ulcers with granulation tissue on the knees.
Laboratory data collected included a complete metabolic panel and complete blood cell count with differential. The complete metabolic panel was within reference range excluding a moderately high glucose level of 114 mg/dL (reference range, 70–100 mg/dL) and a low creatinine level of 0.7 mg/dL (reference range, 0.5–1.2 mg/dL). The complete blood cell count showed mild anemia with a hemoglobin to hematocrit ratio of 11.0 g/dL (reference range, 12.0–16.0 g/dL) to 32.6% (reference range, 37.0%–48.5%) and a nonspecific elevated mean corpuscular volume of 94.4 fL (reference range, 82–95 fL). The differential revealed a low percentage of lymphocytes (11.4% [reference range, 21%–44%]) and a high percentage of granulocytes (85.6% [reference range, 38%–73%]).
A total of 3 biopsies were taken from the abdomen, upper chest, and right wrist. Histology showed a diffuse infiltrate of epithelioid histiocytes forming ill-defined nodules in the dermis, extending into the represented subcutis (Figure 1). Many of the histiocytes had prominent foamy cytoplasms (Figure 2). Scattered lymphocytes and plasma cells also were present. Fite-modified acid-fast staining was performed on all of the biopsies and yielded large numbers of acid-fast bacilli dispersed throughout the specimens (Figure 3), which confirmed the diagnosis of lepromatous leprosy. The patient was referred to the National Hansen’s Disease (Leprosy) Program in Baton Rouge, Louisiana. The physician team started him on high-dose clofazimine, dapsone, and rifampin. He also was diagnosed with erythema nodosum leprosum for which he was given thalidomide.
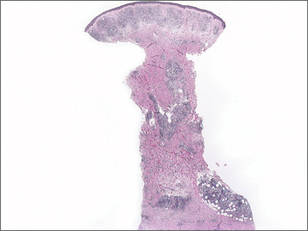
Hansen disease, also known as leprosy, is a chronic inflammatory disease caused by Mycobacterium leprae and Mycobacterium lepromatosis. The mode of transmission is postulated to be through respiratory droplets and nasal secretions. Armadillo exposure, poor sanitary conditions, endemic location, and infected family members are considered risk factors for the development of leprosy. The incubation period is approximately 5 years, with symptoms arising up to 20 years after acquisition of the bacillus. Replication tends to begin in cooler regions of the body. The disease spectrum ranges from lepromatous to tuberculoid.1-3 Lepromatous leprosy consists of an uncontrolled high-titer replication of the mycobacteria, resulting mostly in cutaneous changes and late nerve damage. Histologically, foamy or undifferentiated macrophages predominate the field; they often include many bacilli that can be demonstrated by the modified Fite-Faraco method, a clinically relevant carbol-fuchsin stain.3-5 Acid-fast organisms stain red. A Ziehl-Neelsen stain and Harada modified Allochrome method also may be used.
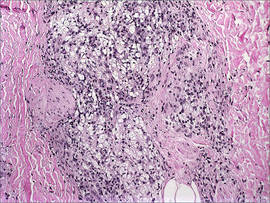
The damage to peripheral nerves and the cutis can cause substantial loss of function and lead to painless ulceration of the skin. Changes in the skin may include nodules, hypopigmented macules, sores, and skin thickening. Leonine facies and disfigurement also may be observed. A type III hypersensitivity reaction may occur, resulting in erythema nodosum leprosum, a type II lepra reaction characterized by elevated tumor necrosis factor α.6
Treatment requires multidrug therapy involving dapsone, rifampin, and clofazimine.4 The World Health Organization is targeted at eliminating the disease and providing free multidrug therapy to patients.7

- Leprosy (Hansen’s disease): Technical Information. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nczved/divisions/dfbmd/diseases/hansens_disease/technical.html/. Updated May 17, 2010. Accessed July 2, 2014.
- Esfandbod M. Images in clinical medicine. tuberculoid leprosy. N Engl J Med. 2011;364:1657.
- Robati RM, Rahimi H, Asadi-Kani Z, et al. Photoclinic. lepromatous leprosy. Arch Iran Med. 2010;13:443-444.
- Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
- Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25:165-172.
- World Health Organization Committee on Leprosy. WHO Expert Committee on Leprosy: Seventh Report. Geneva, Switzerland: World Health Organization Committee on Leprosy; 1997.
To the Editor:
A 41-year-old man presented to our clinic with concerns of ulceration, skin thickening, and loss of eyebrows. The ulceration developed on the knees approximately 1 year prior to presentation; however, he reported loss of sensation in the knees after an accident involving his back 7 years prior. Loss of the eyebrows and skin thickening on the abdomen occurred within the same time frame of the knee ulceration. He also experienced generalized loss of skin sensation. The patient had a history of hunting and fishing but denied any contact with an armadillo.
On clinical examination thin yellow plaques were present on both cheeks, as well as typical leonine facies, a saddle nose deformity, loss of bilateral eyebrows, slate gray color on the face, and infiltrated plaques with telangiectases on the nose. Subcutaneous nodules developed on the arms, chest, and abdomen. He also had angulated ulcers with granulation tissue on the knees.
Laboratory data collected included a complete metabolic panel and complete blood cell count with differential. The complete metabolic panel was within reference range excluding a moderately high glucose level of 114 mg/dL (reference range, 70–100 mg/dL) and a low creatinine level of 0.7 mg/dL (reference range, 0.5–1.2 mg/dL). The complete blood cell count showed mild anemia with a hemoglobin to hematocrit ratio of 11.0 g/dL (reference range, 12.0–16.0 g/dL) to 32.6% (reference range, 37.0%–48.5%) and a nonspecific elevated mean corpuscular volume of 94.4 fL (reference range, 82–95 fL). The differential revealed a low percentage of lymphocytes (11.4% [reference range, 21%–44%]) and a high percentage of granulocytes (85.6% [reference range, 38%–73%]).
A total of 3 biopsies were taken from the abdomen, upper chest, and right wrist. Histology showed a diffuse infiltrate of epithelioid histiocytes forming ill-defined nodules in the dermis, extending into the represented subcutis (Figure 1). Many of the histiocytes had prominent foamy cytoplasms (Figure 2). Scattered lymphocytes and plasma cells also were present. Fite-modified acid-fast staining was performed on all of the biopsies and yielded large numbers of acid-fast bacilli dispersed throughout the specimens (Figure 3), which confirmed the diagnosis of lepromatous leprosy. The patient was referred to the National Hansen’s Disease (Leprosy) Program in Baton Rouge, Louisiana. The physician team started him on high-dose clofazimine, dapsone, and rifampin. He also was diagnosed with erythema nodosum leprosum for which he was given thalidomide.
Hansen disease, also known as leprosy, is a chronic inflammatory disease caused by Mycobacterium leprae and Mycobacterium lepromatosis. The mode of transmission is postulated to be through respiratory droplets and nasal secretions. Armadillo exposure, poor sanitary conditions, endemic location, and infected family members are considered risk factors for the development of leprosy. The incubation period is approximately 5 years, with symptoms arising up to 20 years after acquisition of the bacillus. Replication tends to begin in cooler regions of the body. The disease spectrum ranges from lepromatous to tuberculoid.1-3 Lepromatous leprosy consists of an uncontrolled high-titer replication of the mycobacteria, resulting mostly in cutaneous changes and late nerve damage. Histologically, foamy or undifferentiated macrophages predominate the field; they often include many bacilli that can be demonstrated by the modified Fite-Faraco method, a clinically relevant carbol-fuchsin stain.3-5 Acid-fast organisms stain red. A Ziehl-Neelsen stain and Harada modified Allochrome method also may be used.
The damage to peripheral nerves and the cutis can cause substantial loss of function and lead to painless ulceration of the skin. Changes in the skin may include nodules, hypopigmented macules, sores, and skin thickening. Leonine facies and disfigurement also may be observed. A type III hypersensitivity reaction may occur, resulting in erythema nodosum leprosum, a type II lepra reaction characterized by elevated tumor necrosis factor α.6
Treatment requires multidrug therapy involving dapsone, rifampin, and clofazimine.4 The World Health Organization is targeted at eliminating the disease and providing free multidrug therapy to patients.7
To the Editor:
A 41-year-old man presented to our clinic with concerns of ulceration, skin thickening, and loss of eyebrows. The ulceration developed on the knees approximately 1 year prior to presentation; however, he reported loss of sensation in the knees after an accident involving his back 7 years prior. Loss of the eyebrows and skin thickening on the abdomen occurred within the same time frame of the knee ulceration. He also experienced generalized loss of skin sensation. The patient had a history of hunting and fishing but denied any contact with an armadillo.
On clinical examination thin yellow plaques were present on both cheeks, as well as typical leonine facies, a saddle nose deformity, loss of bilateral eyebrows, slate gray color on the face, and infiltrated plaques with telangiectases on the nose. Subcutaneous nodules developed on the arms, chest, and abdomen. He also had angulated ulcers with granulation tissue on the knees.
Laboratory data collected included a complete metabolic panel and complete blood cell count with differential. The complete metabolic panel was within reference range excluding a moderately high glucose level of 114 mg/dL (reference range, 70–100 mg/dL) and a low creatinine level of 0.7 mg/dL (reference range, 0.5–1.2 mg/dL). The complete blood cell count showed mild anemia with a hemoglobin to hematocrit ratio of 11.0 g/dL (reference range, 12.0–16.0 g/dL) to 32.6% (reference range, 37.0%–48.5%) and a nonspecific elevated mean corpuscular volume of 94.4 fL (reference range, 82–95 fL). The differential revealed a low percentage of lymphocytes (11.4% [reference range, 21%–44%]) and a high percentage of granulocytes (85.6% [reference range, 38%–73%]).
A total of 3 biopsies were taken from the abdomen, upper chest, and right wrist. Histology showed a diffuse infiltrate of epithelioid histiocytes forming ill-defined nodules in the dermis, extending into the represented subcutis (Figure 1). Many of the histiocytes had prominent foamy cytoplasms (Figure 2). Scattered lymphocytes and plasma cells also were present. Fite-modified acid-fast staining was performed on all of the biopsies and yielded large numbers of acid-fast bacilli dispersed throughout the specimens (Figure 3), which confirmed the diagnosis of lepromatous leprosy. The patient was referred to the National Hansen’s Disease (Leprosy) Program in Baton Rouge, Louisiana. The physician team started him on high-dose clofazimine, dapsone, and rifampin. He also was diagnosed with erythema nodosum leprosum for which he was given thalidomide.
Hansen disease, also known as leprosy, is a chronic inflammatory disease caused by Mycobacterium leprae and Mycobacterium lepromatosis. The mode of transmission is postulated to be through respiratory droplets and nasal secretions. Armadillo exposure, poor sanitary conditions, endemic location, and infected family members are considered risk factors for the development of leprosy. The incubation period is approximately 5 years, with symptoms arising up to 20 years after acquisition of the bacillus. Replication tends to begin in cooler regions of the body. The disease spectrum ranges from lepromatous to tuberculoid.1-3 Lepromatous leprosy consists of an uncontrolled high-titer replication of the mycobacteria, resulting mostly in cutaneous changes and late nerve damage. Histologically, foamy or undifferentiated macrophages predominate the field; they often include many bacilli that can be demonstrated by the modified Fite-Faraco method, a clinically relevant carbol-fuchsin stain.3-5 Acid-fast organisms stain red. A Ziehl-Neelsen stain and Harada modified Allochrome method also may be used.
The damage to peripheral nerves and the cutis can cause substantial loss of function and lead to painless ulceration of the skin. Changes in the skin may include nodules, hypopigmented macules, sores, and skin thickening. Leonine facies and disfigurement also may be observed. A type III hypersensitivity reaction may occur, resulting in erythema nodosum leprosum, a type II lepra reaction characterized by elevated tumor necrosis factor α.6
Treatment requires multidrug therapy involving dapsone, rifampin, and clofazimine.4 The World Health Organization is targeted at eliminating the disease and providing free multidrug therapy to patients.7
- Leprosy (Hansen’s disease): Technical Information. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nczved/divisions/dfbmd/diseases/hansens_disease/technical.html/. Updated May 17, 2010. Accessed July 2, 2014.
- Esfandbod M. Images in clinical medicine. tuberculoid leprosy. N Engl J Med. 2011;364:1657.
- Robati RM, Rahimi H, Asadi-Kani Z, et al. Photoclinic. lepromatous leprosy. Arch Iran Med. 2010;13:443-444.
- Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
- Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25:165-172.
- World Health Organization Committee on Leprosy. WHO Expert Committee on Leprosy: Seventh Report. Geneva, Switzerland: World Health Organization Committee on Leprosy; 1997.
- Leprosy (Hansen’s disease): Technical Information. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nczved/divisions/dfbmd/diseases/hansens_disease/technical.html/. Updated May 17, 2010. Accessed July 2, 2014.
- Esfandbod M. Images in clinical medicine. tuberculoid leprosy. N Engl J Med. 2011;364:1657.
- Robati RM, Rahimi H, Asadi-Kani Z, et al. Photoclinic. lepromatous leprosy. Arch Iran Med. 2010;13:443-444.
- Moschella SL. An update on the diagnosis and treatment of leprosy. J Am Acad Dermatol. 2004;51:417-426.
- Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Walker SL, Lockwood DN. Leprosy. Clin Dermatol. 2007;25:165-172.
- World Health Organization Committee on Leprosy. WHO Expert Committee on Leprosy: Seventh Report. Geneva, Switzerland: World Health Organization Committee on Leprosy; 1997.
Frostbite on the Hand of a Homeless Man
To the Editor:
A 58-year-old homeless man presented to the emergency department after being found wandering in the middle of winter in Detroit, Michigan, with altered mental status. A workup for his mental incapacitation uncovered severe electrolyte disturbances, hyperglycemia, and acute renal failure, as well as both alcohol and drug intoxication. After 1 day of admission the patient reported progressive swelling, blistering, and pain in the right hand. The pain was stabbing in nature, worse with movement, and graded 10 of 10 (1=minimal; 10=severe). His medical history was notable for diabetes mellitus with peripheral neuropathy, hypertension, hyperlipidemia, and alcohol and drug abuse. The patient was not taking any medications for these conditions.
Physical examination revealed 2+ moderate pitting edema in all distal extremities, with increased edema of the dorsal aspect of the right hand. The right hand also demonstrated patchy erythema and was warm to touch. The dorsal aspect of the right ring finger had a dusky tip and was studded with several tense blisters (Figure). Vital signs were stable. Based on the patient’s history and physical examination findings, a diagnosis of frostbite was made. Our treatment process involved several modalities including immersion of the affected site in a warm water bath, surgical debridement of blistered sites, tetanus toxoid, penicillin to prevent infection, and oral ibuprofen for pain management. At 3-day follow-up, the patient’s condition substantially improved with a decreased amount of erythema, edema, and pain. All affected sites were successfully preserved with no evidence of focal, motor, or sensory impairment.

Frostbite is a form of localized tissue injury due to extreme cold that most commonly affects the hands and feet, with the greatest incidence occurring in adults aged 30 to 49 years.1,2 Other sites commonly affected include the ears, nose, cheeks, and penis. Frostbite injuries can be categorized into 4 degrees of severity that correlate with the clinical presentation.1,3 Rewarming the affected site is necessary to properly classify the injury, as the initial appearance may be similar among the different degrees of injury. A first-degree injury classically shows a central white plaque with peripheral erythema and is extremely cold to touch. Second-degree injuries display tense blisters filled with clear or milky fluid surrounded by erythema and edema within the first 24 hours. Third-degree injuries are associated with hemorrhagic blisters. Fourth-degree injuries involve complete tissue loss and necrosis.1 Frostbite injuries also may be classified as superficial or deep; the former affects skin and subcutaneous tissue, while the latter affects bones, joints, and tendons.3,4 The superficial form exhibits clear blisters, whereas hemorrhagic blisters demonstrate deep frostbite.
Factors such as the surrounding temperature, length of exposure, and alcohol consumption may exacerbate frostbite injuries.1 Conditions such as atherosclerosis and diabetes mellitus, which can cause neuropathy and peripheral vascular disease, also are potential risks. Psychiatric patients also are at risk for frostbite given the propensity for eccentric behavior as well as the homeless due to inadequate clothing or shelter. Diagnosis often can be made based on medical history and physical examination, though techniques such as radiography, angiography, digital plethysmography, Doppler ultrasonography, and bone scintigraphy (technetium-99) also have been utilized to determine severity and prognosis.2 Differential diagnoses of frostbite are listed in the Table.
Frostbite treatment begins with removal of wet clothing and region protection. Rewarming the site should not begin until refreezing is unlikely to occur and involves placing the injured area in water (temperature, 40°C–42°C) for 15 to 30 minutes to minimize tissue loss.1,2 Analgesics, tetanus toxoid, oral ibuprofen, and benzylpenicillin also are indicated, along with daily hydrotherapy.1,2 White blisters should be debrided, while hemorrhagic blisters should be left intact. Amputation and aggressive debridement typically are delayed until complete ischemia occurs and final demarcation is determined, usually over 1 to 3 months.1 Combination therapy allowed for a positive outcome in our patient.
Frostnip is a mild form of cold injury characterized by localized pain, pallor, and possible numbness.3 Warming the cold area restores the function and sensation with no loss of tissue. Chilblain or pernio refers to a localized cold injury that typically presents as pruritic red-purple papules, macules, plaques, or nodules on the face, anterior tibial surface, or dorsum and tips of the hands and feet.3 The primary cause is repeated exposure to cold, not freezing, temperatures.
Trench foot or cold immersion foot (or hand) is a nonfreezing injury to the hands or feet caused by chronic exposure to wet conditions and temperatures above freezing.3 Painful burning and dysesthesia as well as tissue damage involving edema, blistering, redness, ecchymosis, and ulceration are common. Cellulitis is a bacterial infection of the skin and underlying tissues that can occur anywhere on the body, but the legs are most commonly affected. Typical presentation involves erythema, warmth, swelling, and pain in the infected area.
Although the conditions described above may be considered in the differential diagnosis, physical examination and the patient’s clinical history typically will allow for the distinction of frostbite from these other disease processes.
- Petrone P, Kuncir EJ, Asensio JA. Surgical management and strategies in the treatment of hypothermia and cold injury. Emerg Med Clin North Am. 2003;21:1165-1178.
- Reamy BV. Frostbite: review and current concepts. J Am Board Fam Pract. 1998;11:34-40.
- Jurkovich GJ. Environmental cold-induced injury. Surg Clin North Am. 2007;87:247-267, viii.
- Biem J, Koehncke N, Classen D, et al. Out of the cold: management of hypothermia and frostbite. CMAJ. 2003;168:305-311.
To the Editor:
A 58-year-old homeless man presented to the emergency department after being found wandering in the middle of winter in Detroit, Michigan, with altered mental status. A workup for his mental incapacitation uncovered severe electrolyte disturbances, hyperglycemia, and acute renal failure, as well as both alcohol and drug intoxication. After 1 day of admission the patient reported progressive swelling, blistering, and pain in the right hand. The pain was stabbing in nature, worse with movement, and graded 10 of 10 (1=minimal; 10=severe). His medical history was notable for diabetes mellitus with peripheral neuropathy, hypertension, hyperlipidemia, and alcohol and drug abuse. The patient was not taking any medications for these conditions.
Physical examination revealed 2+ moderate pitting edema in all distal extremities, with increased edema of the dorsal aspect of the right hand. The right hand also demonstrated patchy erythema and was warm to touch. The dorsal aspect of the right ring finger had a dusky tip and was studded with several tense blisters (Figure). Vital signs were stable. Based on the patient’s history and physical examination findings, a diagnosis of frostbite was made. Our treatment process involved several modalities including immersion of the affected site in a warm water bath, surgical debridement of blistered sites, tetanus toxoid, penicillin to prevent infection, and oral ibuprofen for pain management. At 3-day follow-up, the patient’s condition substantially improved with a decreased amount of erythema, edema, and pain. All affected sites were successfully preserved with no evidence of focal, motor, or sensory impairment.
Frostbite is a form of localized tissue injury due to extreme cold that most commonly affects the hands and feet, with the greatest incidence occurring in adults aged 30 to 49 years.1,2 Other sites commonly affected include the ears, nose, cheeks, and penis. Frostbite injuries can be categorized into 4 degrees of severity that correlate with the clinical presentation.1,3 Rewarming the affected site is necessary to properly classify the injury, as the initial appearance may be similar among the different degrees of injury. A first-degree injury classically shows a central white plaque with peripheral erythema and is extremely cold to touch. Second-degree injuries display tense blisters filled with clear or milky fluid surrounded by erythema and edema within the first 24 hours. Third-degree injuries are associated with hemorrhagic blisters. Fourth-degree injuries involve complete tissue loss and necrosis.1 Frostbite injuries also may be classified as superficial or deep; the former affects skin and subcutaneous tissue, while the latter affects bones, joints, and tendons.3,4 The superficial form exhibits clear blisters, whereas hemorrhagic blisters demonstrate deep frostbite.
Factors such as the surrounding temperature, length of exposure, and alcohol consumption may exacerbate frostbite injuries.1 Conditions such as atherosclerosis and diabetes mellitus, which can cause neuropathy and peripheral vascular disease, also are potential risks. Psychiatric patients also are at risk for frostbite given the propensity for eccentric behavior as well as the homeless due to inadequate clothing or shelter. Diagnosis often can be made based on medical history and physical examination, though techniques such as radiography, angiography, digital plethysmography, Doppler ultrasonography, and bone scintigraphy (technetium-99) also have been utilized to determine severity and prognosis.2 Differential diagnoses of frostbite are listed in the Table.
Frostbite treatment begins with removal of wet clothing and region protection. Rewarming the site should not begin until refreezing is unlikely to occur and involves placing the injured area in water (temperature, 40°C–42°C) for 15 to 30 minutes to minimize tissue loss.1,2 Analgesics, tetanus toxoid, oral ibuprofen, and benzylpenicillin also are indicated, along with daily hydrotherapy.1,2 White blisters should be debrided, while hemorrhagic blisters should be left intact. Amputation and aggressive debridement typically are delayed until complete ischemia occurs and final demarcation is determined, usually over 1 to 3 months.1 Combination therapy allowed for a positive outcome in our patient.
Frostnip is a mild form of cold injury characterized by localized pain, pallor, and possible numbness.3 Warming the cold area restores the function and sensation with no loss of tissue. Chilblain or pernio refers to a localized cold injury that typically presents as pruritic red-purple papules, macules, plaques, or nodules on the face, anterior tibial surface, or dorsum and tips of the hands and feet.3 The primary cause is repeated exposure to cold, not freezing, temperatures.
Trench foot or cold immersion foot (or hand) is a nonfreezing injury to the hands or feet caused by chronic exposure to wet conditions and temperatures above freezing.3 Painful burning and dysesthesia as well as tissue damage involving edema, blistering, redness, ecchymosis, and ulceration are common. Cellulitis is a bacterial infection of the skin and underlying tissues that can occur anywhere on the body, but the legs are most commonly affected. Typical presentation involves erythema, warmth, swelling, and pain in the infected area.
Although the conditions described above may be considered in the differential diagnosis, physical examination and the patient’s clinical history typically will allow for the distinction of frostbite from these other disease processes.
To the Editor:
A 58-year-old homeless man presented to the emergency department after being found wandering in the middle of winter in Detroit, Michigan, with altered mental status. A workup for his mental incapacitation uncovered severe electrolyte disturbances, hyperglycemia, and acute renal failure, as well as both alcohol and drug intoxication. After 1 day of admission the patient reported progressive swelling, blistering, and pain in the right hand. The pain was stabbing in nature, worse with movement, and graded 10 of 10 (1=minimal; 10=severe). His medical history was notable for diabetes mellitus with peripheral neuropathy, hypertension, hyperlipidemia, and alcohol and drug abuse. The patient was not taking any medications for these conditions.
Physical examination revealed 2+ moderate pitting edema in all distal extremities, with increased edema of the dorsal aspect of the right hand. The right hand also demonstrated patchy erythema and was warm to touch. The dorsal aspect of the right ring finger had a dusky tip and was studded with several tense blisters (Figure). Vital signs were stable. Based on the patient’s history and physical examination findings, a diagnosis of frostbite was made. Our treatment process involved several modalities including immersion of the affected site in a warm water bath, surgical debridement of blistered sites, tetanus toxoid, penicillin to prevent infection, and oral ibuprofen for pain management. At 3-day follow-up, the patient’s condition substantially improved with a decreased amount of erythema, edema, and pain. All affected sites were successfully preserved with no evidence of focal, motor, or sensory impairment.
Frostbite is a form of localized tissue injury due to extreme cold that most commonly affects the hands and feet, with the greatest incidence occurring in adults aged 30 to 49 years.1,2 Other sites commonly affected include the ears, nose, cheeks, and penis. Frostbite injuries can be categorized into 4 degrees of severity that correlate with the clinical presentation.1,3 Rewarming the affected site is necessary to properly classify the injury, as the initial appearance may be similar among the different degrees of injury. A first-degree injury classically shows a central white plaque with peripheral erythema and is extremely cold to touch. Second-degree injuries display tense blisters filled with clear or milky fluid surrounded by erythema and edema within the first 24 hours. Third-degree injuries are associated with hemorrhagic blisters. Fourth-degree injuries involve complete tissue loss and necrosis.1 Frostbite injuries also may be classified as superficial or deep; the former affects skin and subcutaneous tissue, while the latter affects bones, joints, and tendons.3,4 The superficial form exhibits clear blisters, whereas hemorrhagic blisters demonstrate deep frostbite.
Factors such as the surrounding temperature, length of exposure, and alcohol consumption may exacerbate frostbite injuries.1 Conditions such as atherosclerosis and diabetes mellitus, which can cause neuropathy and peripheral vascular disease, also are potential risks. Psychiatric patients also are at risk for frostbite given the propensity for eccentric behavior as well as the homeless due to inadequate clothing or shelter. Diagnosis often can be made based on medical history and physical examination, though techniques such as radiography, angiography, digital plethysmography, Doppler ultrasonography, and bone scintigraphy (technetium-99) also have been utilized to determine severity and prognosis.2 Differential diagnoses of frostbite are listed in the Table.
Frostbite treatment begins with removal of wet clothing and region protection. Rewarming the site should not begin until refreezing is unlikely to occur and involves placing the injured area in water (temperature, 40°C–42°C) for 15 to 30 minutes to minimize tissue loss.1,2 Analgesics, tetanus toxoid, oral ibuprofen, and benzylpenicillin also are indicated, along with daily hydrotherapy.1,2 White blisters should be debrided, while hemorrhagic blisters should be left intact. Amputation and aggressive debridement typically are delayed until complete ischemia occurs and final demarcation is determined, usually over 1 to 3 months.1 Combination therapy allowed for a positive outcome in our patient.
Frostnip is a mild form of cold injury characterized by localized pain, pallor, and possible numbness.3 Warming the cold area restores the function and sensation with no loss of tissue. Chilblain or pernio refers to a localized cold injury that typically presents as pruritic red-purple papules, macules, plaques, or nodules on the face, anterior tibial surface, or dorsum and tips of the hands and feet.3 The primary cause is repeated exposure to cold, not freezing, temperatures.
Trench foot or cold immersion foot (or hand) is a nonfreezing injury to the hands or feet caused by chronic exposure to wet conditions and temperatures above freezing.3 Painful burning and dysesthesia as well as tissue damage involving edema, blistering, redness, ecchymosis, and ulceration are common. Cellulitis is a bacterial infection of the skin and underlying tissues that can occur anywhere on the body, but the legs are most commonly affected. Typical presentation involves erythema, warmth, swelling, and pain in the infected area.
Although the conditions described above may be considered in the differential diagnosis, physical examination and the patient’s clinical history typically will allow for the distinction of frostbite from these other disease processes.
- Petrone P, Kuncir EJ, Asensio JA. Surgical management and strategies in the treatment of hypothermia and cold injury. Emerg Med Clin North Am. 2003;21:1165-1178.
- Reamy BV. Frostbite: review and current concepts. J Am Board Fam Pract. 1998;11:34-40.
- Jurkovich GJ. Environmental cold-induced injury. Surg Clin North Am. 2007;87:247-267, viii.
- Biem J, Koehncke N, Classen D, et al. Out of the cold: management of hypothermia and frostbite. CMAJ. 2003;168:305-311.
- Petrone P, Kuncir EJ, Asensio JA. Surgical management and strategies in the treatment of hypothermia and cold injury. Emerg Med Clin North Am. 2003;21:1165-1178.
- Reamy BV. Frostbite: review and current concepts. J Am Board Fam Pract. 1998;11:34-40.
- Jurkovich GJ. Environmental cold-induced injury. Surg Clin North Am. 2007;87:247-267, viii.
- Biem J, Koehncke N, Classen D, et al. Out of the cold: management of hypothermia and frostbite. CMAJ. 2003;168:305-311.
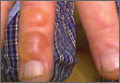
Disseminated Waxy Papules: A Sign of Systemic Disease
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
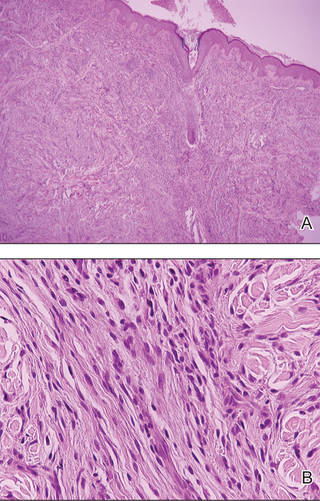
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
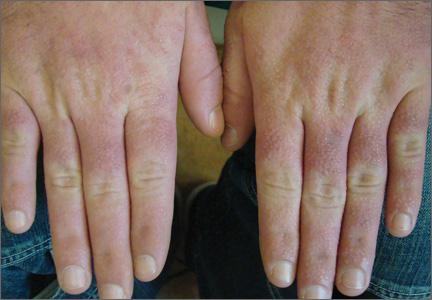
Photoinduced Classic Sweet Syndrome Presenting as Hemorrhagic Bullae
To the Editor:
Sweet syndrome (SS) is characterized by fever; acute onset of painful erythematous papules, plaques, or nodules; peripheral neutrophilic leukocytosis; and histologic findings of a dense neutrophilic infiltrate without evidence of primary vasculitis.1 We report a rare case of classic SS presenting with hemorrhagic bullae over photoexposed areas. Our case is notable because of the unusual nature of the clinical manifestation.
A 45-year-old woman presented with painful, fluid-filled lesions on the upper extremities of 1 week’s duration. Lesions were acute in onset and associated with fever. The patient had a history of diabetes mellitus and hypertension, which were well controlled. She also had a history of minimal itching on sun exposure as well as an upper respiratory tract infection 2 months prior to presentation. There was no history of muscle weakness, pain and/or discoloration of the fingertips, or treatment with topical or systemic agents.
On physical examination, the patient was well nourished with an average build. She was febrile (temperature, 38.5°C) with a pulse of 82 beats per minute, a blood pressure of 130/80, and a respiratory rate of 14 breaths per minute. On cutaneous examination multiple erythematous plaques with central large hemorrhagic bullae were present on the extensor aspect of the forearms and dorsum of the left hand. The smallest plaque measured 4×8 cm and the largest measured 8×15 cm (Figure 1). The lesions were tender, and Nikolsky sign was negative. Considering the clinical features, a differential diagnosis of bullous systemic lupus erythematosus, polymorphic light eruption, Jessner lymphocytic infiltrate, and SS were considered. Complete blood cell count demonstrated a hemoglobin level of 12.1 g/dL (reference range, 14.0–17.5 g/dL), total leukocyte count of 13,280/μL (reference range, 4500–11,000/μL), neutrophil count of 80% (reference range, 56%), lymphocyte count of 13% (reference range, 34%), monocyte count of 8% (reference range, 4%), and an erythrocyte sedimentation rate of 40 mm/h (reference range, 0–20 mm/h). Serum creatinine levels were 0.9 mg/dL (reference range, 0.6–1.2 mg/dL) and urea nitrogen levels were 26 mg/dL (reference range, 8–23 mg/dL). C-reactive protein was positive, antinuclear antibody was negative, and double-stranded DNA was negative. Ultrasonography of the abdomen and pelvis and a chest radiograph revealed no abnormalities.
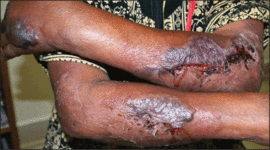
Histopathology from a lesion on the forearm revealed a dense, predominantly neutrophilic infiltrate located in the superficial dermis as well as prominent papillary dermal edema infiltration in the dermis with vasodilatation in some areas without leukocytoclastic vasculitis features (Figures 2 and 3). Considering the clinical and histopathologic features, a diagnosis of SS was made, and the patient was started on intravenous dexamethasone (4 mg twice daily) with a dramatic response, as the lesions almost cleared within 4 to 5 days of treatment (Figure 4).
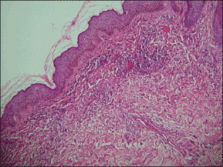
Sweet syndrome was first described in 1964 as acute febrile neutrophilic dermatosis.2 Sweet syndrome can be subdivided into 3 groups depending on the clinical setting: classic or idiopathic, malignancy associated, and drug induced.3 Classic or idiopathic SS typically affects women in the third to fifth decades of life,4 as seen in our case. The proposed diagnostic criteria for SS state that patients must meet both of the 2 major criteria and 2 of 4 minor criteria for the diagnosis.3 The major criteria include acute onset of typical skin lesions and histopathologic findings consistent with SS. The minor criteria include fever (temperature >38°C) or general malaise; association with malignancy, inflammatory disease, pregnancy, or antecedent respiratory or gastrointestinal tract infection; excellent response to treatment with systemic corticosteroids or potassium iodide; and abnormal laboratory values at presentation (3 of 4 required: erythrocyte sedimentation rate >20 mm/h; leukocyte count >8000/μL; neutrophil count >70%; positive C-reactive protein). Our patient fulfilled both the major criteria and 3 of 4 minor criteria. Although the exact etiology of SS is unknown, it is widely believed that SS may be a hypersensitivity response to underlying bacterial infections such as Yersinia enterocolitica, viral infections, or tumors.5
Cytokine dysregulation also has been indicated in the pathogenesis of SS, an imbalance of cytokine secretion from helper T cells such as IL-2 and IFN-γ, which may stimulate the cytokine cascade leading to activation of neutrophils and release of toxic metabolites.6 The cutaneous manifestations of SS consist of erythematous to violaceous tender papules or nodules that often coalesce to form irregular plaques.7 Rare clinical manifestations include bullous lesions; oral involvement; glomerulonephritis; myositis; and ocular manifestations including conjunctivitis, episcleritis, and iridocyclitis.5,8 Photoaggravated and photoinduced syndromes also have been reported.9
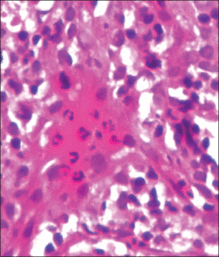
Bullous-type SS has been reported, but all the known cases were secondary to malignancy or were drug induced8,10; they did not present in a classic or idiopathic variant. Our case is unique in that it is a report of the classic SS variant with lesions including bullae over photoexposed areas, possibly indicating a causal association of sun exposure and the development of SS.
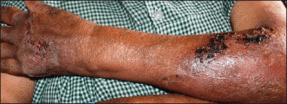
The diagnostic histopathologic features of SS include a dense, predominantly neutrophilic infiltrate located in the superficial dermis as well as prominent papillary dermal edema, which occasionally may lead to subepidermal vesiculation.11,12 The epidermis often is normal but spongiosis may be present, and rarely neutrophils may extend into the epidermis to form subcorneal pustules.13 Severe edema in the papillary dermis may cause subepidermal blistering and bullous lesions.10 Systemic steroids are the therapeutic mainstay in SS. Other treatment options include methylprednisolone, potassium iodide, colchicine, indomethacin, cyclosporine, and dapsone.
1. Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;74:349-356.
3. Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
4. Cohen PR. Pregnancy-associated Sweet’s syndrome: world literature review. Obstet Gynecol Surv. 1993;48:584-587.
5. Cohen PR, Hönigsmann H, Kurzrock R. Acute febrile neutrophilic dermatosis (Sweet syndrome). In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008:289-295.
6. Giasuddin AS, El-Orfi AH, Ziu MM, et al. Sweet’s syndrome: is the pathogenesis mediated by helper T cell type 1 cytokines? J Am Acad Dermatol. 1998;39:940-943.
7. Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
8. Lund JJ, Stratman EJ, Jose D, et al. Drug-induced bullous Sweet syndrome with multiple autoimmune features. Autoimmune Dis. 2010;2011:176749.
9. Bessis D, Dereure O, Peyron JL, et al. Photoinduced Sweet syndrome. Arch Dermatol. 2003;137:1106-1108.
10. Bielsa S, Baradad M, Martí RM, et al. Sweet’s syndrome with bullous lesions [in Spanish]. Actas Dermosifiliogr. 2005;96:315-316.
11. Jordaan HF. Acute febrile neutrophilic dermatosis: a histopathological study of 37 cases and a review of the literature. Am J Dermatopathol. 1989;11:99-111.
12. Kemmett D, Hunter JA. Sweet’s syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23(3, pt 1):503-507.
13. Wallach D. Neutrophilic disease [in French]. Rev Prat. 1999;49:356-358.
To the Editor:
Sweet syndrome (SS) is characterized by fever; acute onset of painful erythematous papules, plaques, or nodules; peripheral neutrophilic leukocytosis; and histologic findings of a dense neutrophilic infiltrate without evidence of primary vasculitis.1 We report a rare case of classic SS presenting with hemorrhagic bullae over photoexposed areas. Our case is notable because of the unusual nature of the clinical manifestation.
A 45-year-old woman presented with painful, fluid-filled lesions on the upper extremities of 1 week’s duration. Lesions were acute in onset and associated with fever. The patient had a history of diabetes mellitus and hypertension, which were well controlled. She also had a history of minimal itching on sun exposure as well as an upper respiratory tract infection 2 months prior to presentation. There was no history of muscle weakness, pain and/or discoloration of the fingertips, or treatment with topical or systemic agents.
On physical examination, the patient was well nourished with an average build. She was febrile (temperature, 38.5°C) with a pulse of 82 beats per minute, a blood pressure of 130/80, and a respiratory rate of 14 breaths per minute. On cutaneous examination multiple erythematous plaques with central large hemorrhagic bullae were present on the extensor aspect of the forearms and dorsum of the left hand. The smallest plaque measured 4×8 cm and the largest measured 8×15 cm (Figure 1). The lesions were tender, and Nikolsky sign was negative. Considering the clinical features, a differential diagnosis of bullous systemic lupus erythematosus, polymorphic light eruption, Jessner lymphocytic infiltrate, and SS were considered. Complete blood cell count demonstrated a hemoglobin level of 12.1 g/dL (reference range, 14.0–17.5 g/dL), total leukocyte count of 13,280/μL (reference range, 4500–11,000/μL), neutrophil count of 80% (reference range, 56%), lymphocyte count of 13% (reference range, 34%), monocyte count of 8% (reference range, 4%), and an erythrocyte sedimentation rate of 40 mm/h (reference range, 0–20 mm/h). Serum creatinine levels were 0.9 mg/dL (reference range, 0.6–1.2 mg/dL) and urea nitrogen levels were 26 mg/dL (reference range, 8–23 mg/dL). C-reactive protein was positive, antinuclear antibody was negative, and double-stranded DNA was negative. Ultrasonography of the abdomen and pelvis and a chest radiograph revealed no abnormalities.
Histopathology from a lesion on the forearm revealed a dense, predominantly neutrophilic infiltrate located in the superficial dermis as well as prominent papillary dermal edema infiltration in the dermis with vasodilatation in some areas without leukocytoclastic vasculitis features (Figures 2 and 3). Considering the clinical and histopathologic features, a diagnosis of SS was made, and the patient was started on intravenous dexamethasone (4 mg twice daily) with a dramatic response, as the lesions almost cleared within 4 to 5 days of treatment (Figure 4).
Sweet syndrome was first described in 1964 as acute febrile neutrophilic dermatosis.2 Sweet syndrome can be subdivided into 3 groups depending on the clinical setting: classic or idiopathic, malignancy associated, and drug induced.3 Classic or idiopathic SS typically affects women in the third to fifth decades of life,4 as seen in our case. The proposed diagnostic criteria for SS state that patients must meet both of the 2 major criteria and 2 of 4 minor criteria for the diagnosis.3 The major criteria include acute onset of typical skin lesions and histopathologic findings consistent with SS. The minor criteria include fever (temperature >38°C) or general malaise; association with malignancy, inflammatory disease, pregnancy, or antecedent respiratory or gastrointestinal tract infection; excellent response to treatment with systemic corticosteroids or potassium iodide; and abnormal laboratory values at presentation (3 of 4 required: erythrocyte sedimentation rate >20 mm/h; leukocyte count >8000/μL; neutrophil count >70%; positive C-reactive protein). Our patient fulfilled both the major criteria and 3 of 4 minor criteria. Although the exact etiology of SS is unknown, it is widely believed that SS may be a hypersensitivity response to underlying bacterial infections such as Yersinia enterocolitica, viral infections, or tumors.5
Cytokine dysregulation also has been indicated in the pathogenesis of SS, an imbalance of cytokine secretion from helper T cells such as IL-2 and IFN-γ, which may stimulate the cytokine cascade leading to activation of neutrophils and release of toxic metabolites.6 The cutaneous manifestations of SS consist of erythematous to violaceous tender papules or nodules that often coalesce to form irregular plaques.7 Rare clinical manifestations include bullous lesions; oral involvement; glomerulonephritis; myositis; and ocular manifestations including conjunctivitis, episcleritis, and iridocyclitis.5,8 Photoaggravated and photoinduced syndromes also have been reported.9
Bullous-type SS has been reported, but all the known cases were secondary to malignancy or were drug induced8,10; they did not present in a classic or idiopathic variant. Our case is unique in that it is a report of the classic SS variant with lesions including bullae over photoexposed areas, possibly indicating a causal association of sun exposure and the development of SS.
The diagnostic histopathologic features of SS include a dense, predominantly neutrophilic infiltrate located in the superficial dermis as well as prominent papillary dermal edema, which occasionally may lead to subepidermal vesiculation.11,12 The epidermis often is normal but spongiosis may be present, and rarely neutrophils may extend into the epidermis to form subcorneal pustules.13 Severe edema in the papillary dermis may cause subepidermal blistering and bullous lesions.10 Systemic steroids are the therapeutic mainstay in SS. Other treatment options include methylprednisolone, potassium iodide, colchicine, indomethacin, cyclosporine, and dapsone.
To the Editor:
Sweet syndrome (SS) is characterized by fever; acute onset of painful erythematous papules, plaques, or nodules; peripheral neutrophilic leukocytosis; and histologic findings of a dense neutrophilic infiltrate without evidence of primary vasculitis.1 We report a rare case of classic SS presenting with hemorrhagic bullae over photoexposed areas. Our case is notable because of the unusual nature of the clinical manifestation.
A 45-year-old woman presented with painful, fluid-filled lesions on the upper extremities of 1 week’s duration. Lesions were acute in onset and associated with fever. The patient had a history of diabetes mellitus and hypertension, which were well controlled. She also had a history of minimal itching on sun exposure as well as an upper respiratory tract infection 2 months prior to presentation. There was no history of muscle weakness, pain and/or discoloration of the fingertips, or treatment with topical or systemic agents.
On physical examination, the patient was well nourished with an average build. She was febrile (temperature, 38.5°C) with a pulse of 82 beats per minute, a blood pressure of 130/80, and a respiratory rate of 14 breaths per minute. On cutaneous examination multiple erythematous plaques with central large hemorrhagic bullae were present on the extensor aspect of the forearms and dorsum of the left hand. The smallest plaque measured 4×8 cm and the largest measured 8×15 cm (Figure 1). The lesions were tender, and Nikolsky sign was negative. Considering the clinical features, a differential diagnosis of bullous systemic lupus erythematosus, polymorphic light eruption, Jessner lymphocytic infiltrate, and SS were considered. Complete blood cell count demonstrated a hemoglobin level of 12.1 g/dL (reference range, 14.0–17.5 g/dL), total leukocyte count of 13,280/μL (reference range, 4500–11,000/μL), neutrophil count of 80% (reference range, 56%), lymphocyte count of 13% (reference range, 34%), monocyte count of 8% (reference range, 4%), and an erythrocyte sedimentation rate of 40 mm/h (reference range, 0–20 mm/h). Serum creatinine levels were 0.9 mg/dL (reference range, 0.6–1.2 mg/dL) and urea nitrogen levels were 26 mg/dL (reference range, 8–23 mg/dL). C-reactive protein was positive, antinuclear antibody was negative, and double-stranded DNA was negative. Ultrasonography of the abdomen and pelvis and a chest radiograph revealed no abnormalities.
Histopathology from a lesion on the forearm revealed a dense, predominantly neutrophilic infiltrate located in the superficial dermis as well as prominent papillary dermal edema infiltration in the dermis with vasodilatation in some areas without leukocytoclastic vasculitis features (Figures 2 and 3). Considering the clinical and histopathologic features, a diagnosis of SS was made, and the patient was started on intravenous dexamethasone (4 mg twice daily) with a dramatic response, as the lesions almost cleared within 4 to 5 days of treatment (Figure 4).
Sweet syndrome was first described in 1964 as acute febrile neutrophilic dermatosis.2 Sweet syndrome can be subdivided into 3 groups depending on the clinical setting: classic or idiopathic, malignancy associated, and drug induced.3 Classic or idiopathic SS typically affects women in the third to fifth decades of life,4 as seen in our case. The proposed diagnostic criteria for SS state that patients must meet both of the 2 major criteria and 2 of 4 minor criteria for the diagnosis.3 The major criteria include acute onset of typical skin lesions and histopathologic findings consistent with SS. The minor criteria include fever (temperature >38°C) or general malaise; association with malignancy, inflammatory disease, pregnancy, or antecedent respiratory or gastrointestinal tract infection; excellent response to treatment with systemic corticosteroids or potassium iodide; and abnormal laboratory values at presentation (3 of 4 required: erythrocyte sedimentation rate >20 mm/h; leukocyte count >8000/μL; neutrophil count >70%; positive C-reactive protein). Our patient fulfilled both the major criteria and 3 of 4 minor criteria. Although the exact etiology of SS is unknown, it is widely believed that SS may be a hypersensitivity response to underlying bacterial infections such as Yersinia enterocolitica, viral infections, or tumors.5
Cytokine dysregulation also has been indicated in the pathogenesis of SS, an imbalance of cytokine secretion from helper T cells such as IL-2 and IFN-γ, which may stimulate the cytokine cascade leading to activation of neutrophils and release of toxic metabolites.6 The cutaneous manifestations of SS consist of erythematous to violaceous tender papules or nodules that often coalesce to form irregular plaques.7 Rare clinical manifestations include bullous lesions; oral involvement; glomerulonephritis; myositis; and ocular manifestations including conjunctivitis, episcleritis, and iridocyclitis.5,8 Photoaggravated and photoinduced syndromes also have been reported.9
Bullous-type SS has been reported, but all the known cases were secondary to malignancy or were drug induced8,10; they did not present in a classic or idiopathic variant. Our case is unique in that it is a report of the classic SS variant with lesions including bullae over photoexposed areas, possibly indicating a causal association of sun exposure and the development of SS.
The diagnostic histopathologic features of SS include a dense, predominantly neutrophilic infiltrate located in the superficial dermis as well as prominent papillary dermal edema, which occasionally may lead to subepidermal vesiculation.11,12 The epidermis often is normal but spongiosis may be present, and rarely neutrophils may extend into the epidermis to form subcorneal pustules.13 Severe edema in the papillary dermis may cause subepidermal blistering and bullous lesions.10 Systemic steroids are the therapeutic mainstay in SS. Other treatment options include methylprednisolone, potassium iodide, colchicine, indomethacin, cyclosporine, and dapsone.
1. Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;74:349-356.
3. Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
4. Cohen PR. Pregnancy-associated Sweet’s syndrome: world literature review. Obstet Gynecol Surv. 1993;48:584-587.
5. Cohen PR, Hönigsmann H, Kurzrock R. Acute febrile neutrophilic dermatosis (Sweet syndrome). In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008:289-295.
6. Giasuddin AS, El-Orfi AH, Ziu MM, et al. Sweet’s syndrome: is the pathogenesis mediated by helper T cell type 1 cytokines? J Am Acad Dermatol. 1998;39:940-943.
7. Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
8. Lund JJ, Stratman EJ, Jose D, et al. Drug-induced bullous Sweet syndrome with multiple autoimmune features. Autoimmune Dis. 2010;2011:176749.
9. Bessis D, Dereure O, Peyron JL, et al. Photoinduced Sweet syndrome. Arch Dermatol. 2003;137:1106-1108.
10. Bielsa S, Baradad M, Martí RM, et al. Sweet’s syndrome with bullous lesions [in Spanish]. Actas Dermosifiliogr. 2005;96:315-316.
11. Jordaan HF. Acute febrile neutrophilic dermatosis: a histopathological study of 37 cases and a review of the literature. Am J Dermatopathol. 1989;11:99-111.
12. Kemmett D, Hunter JA. Sweet’s syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23(3, pt 1):503-507.
13. Wallach D. Neutrophilic disease [in French]. Rev Prat. 1999;49:356-358.
1. Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;74:349-356.
3. Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
4. Cohen PR. Pregnancy-associated Sweet’s syndrome: world literature review. Obstet Gynecol Surv. 1993;48:584-587.
5. Cohen PR, Hönigsmann H, Kurzrock R. Acute febrile neutrophilic dermatosis (Sweet syndrome). In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008:289-295.
6. Giasuddin AS, El-Orfi AH, Ziu MM, et al. Sweet’s syndrome: is the pathogenesis mediated by helper T cell type 1 cytokines? J Am Acad Dermatol. 1998;39:940-943.
7. Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
8. Lund JJ, Stratman EJ, Jose D, et al. Drug-induced bullous Sweet syndrome with multiple autoimmune features. Autoimmune Dis. 2010;2011:176749.
9. Bessis D, Dereure O, Peyron JL, et al. Photoinduced Sweet syndrome. Arch Dermatol. 2003;137:1106-1108.
10. Bielsa S, Baradad M, Martí RM, et al. Sweet’s syndrome with bullous lesions [in Spanish]. Actas Dermosifiliogr. 2005;96:315-316.
11. Jordaan HF. Acute febrile neutrophilic dermatosis: a histopathological study of 37 cases and a review of the literature. Am J Dermatopathol. 1989;11:99-111.
12. Kemmett D, Hunter JA. Sweet’s syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23(3, pt 1):503-507.
13. Wallach D. Neutrophilic disease [in French]. Rev Prat. 1999;49:356-358.