User login
Radial Shaft Stress Fracture in a Major League Pitcher
Take-Home Points
- Stress fractures should always be considered when dealing with overuse injuries.
- Radial shaft stress fractures in overhead throwing athletes are rare.
- Stress fractures can occur anywhere increased muscular forces exceed the bone’s ability to remodel.
- Proper imaging is necessary to make the diagnosis of a stress fracture.
- Nonoperative management of radial shaft stress fractures is an effective treatment.
In athletes, the incidence of stress fractures has been reported to be 1.4% to 4.4%.1 Stress fractures of the upper extremity are less common and not as well described as lower extremity stress fractures. Although data is lacking, stress fractures involving the upper extremity appear to account for <6% of all stress fractures.2 Stress fractures of the upper extremity, though rare, are being recognized more often in overhead athletes.3-6 In baseball pitchers, stress fractures most commonly occur in the olecranon but have also been found in the ribs, clavicle, humerus, and ulnar shaft.2,4,7-10 Stress fractures of the radius are a rare cause of forearm pain in athletes, and there are only a few case reports involving overhead athletes.4,11-15 To our knowledge, a stress fracture of the radial shaft has not been reported in a throwing athlete. Currently, there are no reports on stress fractures of the proximal radial shaft.16-18
In this article, we report the case of a radial shaft stress fracture that was causing forearm pain in a Major League Baseball (MLB) pitcher. We also discuss the etiology, diagnosis, and management of stress fractures of the upper extremity of overhead throwing athletes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 28-year-old right-hand-dominant MLB pitcher presented to the clinic with a 4-week history of right dorsal forearm pain that was refractory to a period of rest and physical therapy modalities. The pain radiated to the wrist and along the dorsal forearm. The pain started after the man attempted to develop a new pitch that required a significant amount of supination. The pain prevented him from pitching competitively. Indomethacin, diclofenac sodium topical gel, and methylprednisolone (Medrol Dosepak) reduced his symptoms only slightly.
Physical examination of the right elbow showed mild range of motion deficits; about 5° of extension and 5° of flexion were lacking. The patient had full pronation and supination. Palpation of the dorsal aspect of the forearm revealed marked tenderness in the area of the proximal radius. There was no tenderness over the posterior olecranon or the ulnar collateral ligament, and a moving valgus stress test was negative. No pain was elicited by resisted extension of the wrist or fingers. Motor innervation from the posterior interosseous nerve, anterior interosseous nerve, and ulnar nerve was intact with 5/5 strength, and there were no sensory deficits in the distribution of the radial, median, or ulnar nerves.
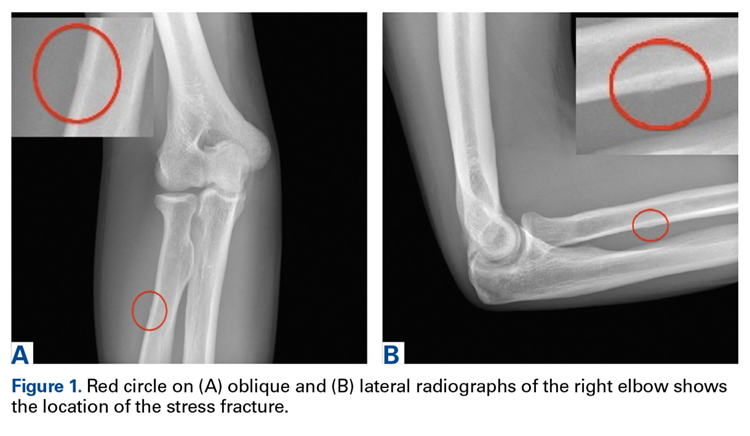
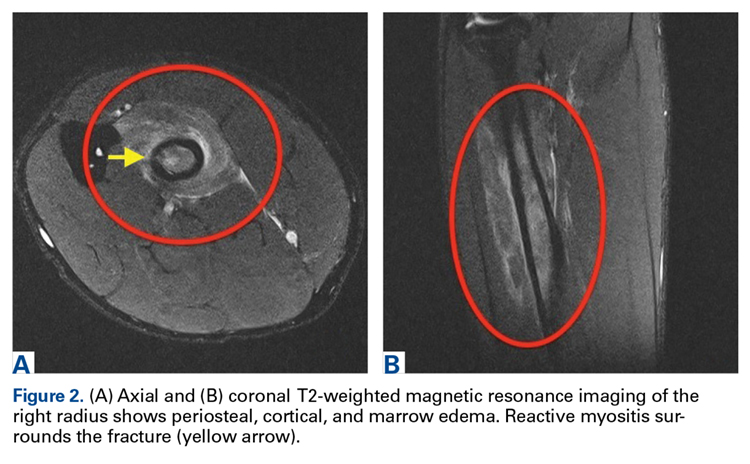
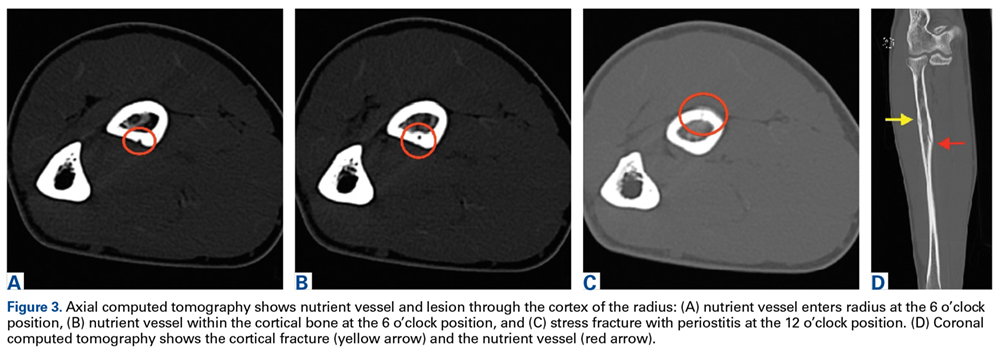
Discussion
Stress fractures account for 0.7% to 20% of sports medicine clinic injuries; <10% of all stress fractures involve the rib or upper extremity.4,6 When the intensity or frequency of physical activity is increased, as with overuse, bone resorption surpasses bone production, locally weakening the bone and making it prone to mechanical failure. Failure is thought to be induced by a combination of contractile muscular forces across damaged bone and increased mechanical loading caused by fatigue of supporting structures.5,6,19 These forces may have contributed to our baseball pitcher’s development of a stress fracture near the insertion of the supinator muscle in his throwing arm.
Given the insidious nature of stress fractures, the evaluating physician must have a high index of suspicion. Early recognition of a stress fracture is important in preventing further injury and allowing for early intervention, which is associated with faster healing.6,20 The clinical history often involves a change in training regimen within the weeks before pain onset. Furthermore, understanding the type of pitches used and the mechanics of each pitch can help with diagnosis. Often, pain increases as the inciting activity continues, and relief comes with rest. In an upper extremity examination, it is important to recall the usual stress fracture locations in throwers—the ribs, clavicle, humerus, ulnar shaft, and most often the olecranon—though the patient’s history often narrows the anatomical region of suspicion.2,4,7-10 Examination begins with inspection of the skin and soft tissues. Range of motion and strength testing results likely are normal throughout the upper extremity.3 Palpation over the suspected injury location often elicits pain and indicates further imaging is needed.6 The tuning fork test or the 3-point fulcrum test may elicit symptoms in occult fractures.3 Completing the assessment is a thorough neurovascular examination.
Insidious forearm pain requires a broad differential, including flexor-pronator mass or distal biceps injury, chronic exertional compartment syndrome, radial tunnel syndrome, intersection syndrome, pronator teres syndrome, anterior interosseous syndrome, thoracic outlet syndrome, musculocutaneous nerve compression, deep vein thrombosis of ulnar vein, and periostitis. Stress fractures distal to the elbow more commonly occur in weight-bearing athletes, though as this case shows it is important to consider stress fractures of the radius and ulna when evaluating forearm pain in a throwing athlete.21
The first imaging examination for a suspected stress fracture is a radiograph, which can be normal in up to 90% of patients, as it initially was in our athlete’s case.22 Often, radiographic evidence takes 2 to 12 weeks to appear.5 Even then, radiographs may be positive in only 50% of cases.19 CT, often regarded as insensitive during the early stages, is useful in visualizing fracture lines in a suspicious location.19,22 Radionuclide uptake scanning is highly sensitive during the early stages of stress injury but is nonspecific and may indicate neoplasm or infection; in addition, up to 46% of abnormal foci are asymptomatic.19 MRI has sensitivity comparable to that of radionuclide scanning but also many advantages, including lack of ionizing radiation, improved spatial resolution, and ability to image bone and soft tissue simultaneously.19 In our patient’s case, the unusual stress fracture location potentially could have hindered identification of the cause of injury. The lesion was just distal to the field of view of a normal elbow MRI and was not detected until a dedicated forearm MRI was examined. Both MRI and CT helped in identifying the stress fracture, and CT was used to follow interval healing.
In baseball players, upper extremity stress fractures are often nonoperatively treated with throwing cessation for 4 to 6 weeks followed by participation in a structured rehabilitation program.4,5 The throwing program that we suggest, and that was used in this case, has 21 stages of progression in duration, distance, and velocity of throwing. The athlete advances from each stage on the basis of symptoms.23 Other issues that may be addressed are vitamin D and calcium status and any flawed throwing mechanics that may have predisposed the athlete to injury. Such mechanics are gradually corrected.
The literature suggests that appropriate nonoperative management of stress fractures allows for return to sport in 8 to 10 weeks. It is important to note that most of the literature on stress fractures involves the lower extremity, and that treatment and time to return to play are therefore better described for such fractures.6 More study and evaluation of upper extremity stress fractures are needed to make return-to-sport predictions more reliable and successful treatment modalities more unified for this patient population. Last, it is imperative that clinical examination and symptoms be correlated with serial imaging when deciding on return to play. Our patient took 12 weeks to return to high-level sport. He progressed pain-free through the throwing program and showed radiographic evidence of healing on follow-up CT.
Conclusion
Radial shaft stress fractures are rare in throwing athletes. However, with a thorough history, a physical examination, and appropriate imaging, the correct diagnosis can be made early on, and proper treatment can be started to facilitate return to sport. To our knowledge, this is the first report of a stress fracture in the radial shaft of a MLB pitcher. Although the radial shaft is an uncommon location for stress fractures, we should keep in mind that they can occur wherever increased muscular forces exceed the ability of native bone to remodel. After diagnosis, the fracture usually heals with nonoperative treatment, and healing is confirmed with follow-up imaging, as was done in our patient’s case. Improved prediction of time to return to play for upper extremity fractures, such as the radial stress fracture described in this article, requires more study.
1. Monteleone GP Jr. Stress fractures in the athlete. Orthop Clin North Am. 1995;26(3):423-432.
2. Iwamoto J, Takeda T. Stress fractures in athletes: review of 196 cases. J Orthop Sci. 2003;8(3):273-278.
3. Miller TL, Kaeding CC. Upper-extremity stress fractures: distribution and causative activities in 70 patients. Orthopedics. 2012;35(9):789-793.
4. Jones GL. Upper extremity stress fractures. Clin Sports Med. 2006;25(1):159-174.
5. Brooks AA. Stress fractures of the upper extremity. Clin Sports Med. 2001;20(3):613-620.
6. Fredericson M, Jennings F, Beaulieu C, Matheson GO. Stress fractures in athletes. Top Magn Reson Imaging. 2006;17(5):309-325.
7. Gurtler R, Pavlov H, Torg JS. Stress fracture of the ipsilateral first rib in a pitcher. Am J Sports Med. 1985;13(4):277-279.
8. Polu KR, Schenck RC Jr, Wirth MA, Greeson J, Cone RO 3rd, Rockwood CA Jr. Stress fracture of the humerus in a collegiate baseball pitcher. A case report. Am J Sports Med. 1999;27(6):813-816.
9. Wu C, Chen Y. Stress fracture of the clavicle in a professional baseball player. J Shoulder Elbow Surg. 1998;7(2):164-167.
10. Schickendantz MS, Ho CP, Koh J. Stress injury of the proximal ulna in professional baseball players. Am J Sports Med. 2002;30(5):737-741.
11. Loosli AR, Leslie M. Stress fractures of the distal radius. A case report. Am J Sports Med. 1991;19(5):523-524.
12. Inagaki H, Inoue G. Stress fracture of the scaphoid combined with the distal radial epiphysiolysis. Br J Sports Med. 1997;31(3):256-257.
13. Read MT. Stress fractures of the distal radius in adolescent gymnasts. Br J Sports Med. 1981;15(4):272-276.
14. Orloff AS, Resnick D. Fatigue fracture of the distal part of the radius in a pool player. Injury. 1986;17(6):418-419.
15. Eisenberg D, Kirchner SG, Green NE. Stress fracture of the distal radius caused by “wheelies.” South Med J. 1986;79(7):918-919.
16. Brukner P. Stress fractures of the upper limb. Sports Med. 1998;26(6):415-424.
17. Farquharson-Roberts MA, Fulford PC. Stress fracture of the radius. J Bone Joint Surg Br. 1980;62(2):194-195.
18. Orloff AS, Resnick D. Fatigue fracture of the distal part of the radius in a pool player. Injury. 1986;17(6):418-419.
19. Anderson MW. Imaging of upper extremity stress fractures in the athlete. Clin Sports Med. 2006;25(3):489-504.
20. Bennell K, Brukner P. Preventing and managing stress fractures in athletes. Phys Ther Sport. 2005;6(4):171-180.
21. Sinha AK, Kaeding CC, Wadley GM. Upper extremity stress fractures in athletes: clinical features of 44 cases. Clin J Sport Med. 1999;9(4):199-202.
22. Matheson GO, Clement DB, McKenzie DC, Taunton JE, Lloyd-Smith DR, MacIntyre JG. Stress fractures in athletes. A study of 320 cases. Am J Sports Med. 1987;15(1):46-58.
23. Kaplan L, Lesniak B, Baraga M, et al. Throwing program for baseball players. 2009. http://uhealthsportsmedicine.com/documents/UHealth_Throwing_Program.pdf. Accessed May 24, 2016.
Take-Home Points
- Stress fractures should always be considered when dealing with overuse injuries.
- Radial shaft stress fractures in overhead throwing athletes are rare.
- Stress fractures can occur anywhere increased muscular forces exceed the bone’s ability to remodel.
- Proper imaging is necessary to make the diagnosis of a stress fracture.
- Nonoperative management of radial shaft stress fractures is an effective treatment.
In athletes, the incidence of stress fractures has been reported to be 1.4% to 4.4%.1 Stress fractures of the upper extremity are less common and not as well described as lower extremity stress fractures. Although data is lacking, stress fractures involving the upper extremity appear to account for <6% of all stress fractures.2 Stress fractures of the upper extremity, though rare, are being recognized more often in overhead athletes.3-6 In baseball pitchers, stress fractures most commonly occur in the olecranon but have also been found in the ribs, clavicle, humerus, and ulnar shaft.2,4,7-10 Stress fractures of the radius are a rare cause of forearm pain in athletes, and there are only a few case reports involving overhead athletes.4,11-15 To our knowledge, a stress fracture of the radial shaft has not been reported in a throwing athlete. Currently, there are no reports on stress fractures of the proximal radial shaft.16-18
In this article, we report the case of a radial shaft stress fracture that was causing forearm pain in a Major League Baseball (MLB) pitcher. We also discuss the etiology, diagnosis, and management of stress fractures of the upper extremity of overhead throwing athletes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 28-year-old right-hand-dominant MLB pitcher presented to the clinic with a 4-week history of right dorsal forearm pain that was refractory to a period of rest and physical therapy modalities. The pain radiated to the wrist and along the dorsal forearm. The pain started after the man attempted to develop a new pitch that required a significant amount of supination. The pain prevented him from pitching competitively. Indomethacin, diclofenac sodium topical gel, and methylprednisolone (Medrol Dosepak) reduced his symptoms only slightly.
Physical examination of the right elbow showed mild range of motion deficits; about 5° of extension and 5° of flexion were lacking. The patient had full pronation and supination. Palpation of the dorsal aspect of the forearm revealed marked tenderness in the area of the proximal radius. There was no tenderness over the posterior olecranon or the ulnar collateral ligament, and a moving valgus stress test was negative. No pain was elicited by resisted extension of the wrist or fingers. Motor innervation from the posterior interosseous nerve, anterior interosseous nerve, and ulnar nerve was intact with 5/5 strength, and there were no sensory deficits in the distribution of the radial, median, or ulnar nerves.
Discussion
Stress fractures account for 0.7% to 20% of sports medicine clinic injuries; <10% of all stress fractures involve the rib or upper extremity.4,6 When the intensity or frequency of physical activity is increased, as with overuse, bone resorption surpasses bone production, locally weakening the bone and making it prone to mechanical failure. Failure is thought to be induced by a combination of contractile muscular forces across damaged bone and increased mechanical loading caused by fatigue of supporting structures.5,6,19 These forces may have contributed to our baseball pitcher’s development of a stress fracture near the insertion of the supinator muscle in his throwing arm.
Given the insidious nature of stress fractures, the evaluating physician must have a high index of suspicion. Early recognition of a stress fracture is important in preventing further injury and allowing for early intervention, which is associated with faster healing.6,20 The clinical history often involves a change in training regimen within the weeks before pain onset. Furthermore, understanding the type of pitches used and the mechanics of each pitch can help with diagnosis. Often, pain increases as the inciting activity continues, and relief comes with rest. In an upper extremity examination, it is important to recall the usual stress fracture locations in throwers—the ribs, clavicle, humerus, ulnar shaft, and most often the olecranon—though the patient’s history often narrows the anatomical region of suspicion.2,4,7-10 Examination begins with inspection of the skin and soft tissues. Range of motion and strength testing results likely are normal throughout the upper extremity.3 Palpation over the suspected injury location often elicits pain and indicates further imaging is needed.6 The tuning fork test or the 3-point fulcrum test may elicit symptoms in occult fractures.3 Completing the assessment is a thorough neurovascular examination.
Insidious forearm pain requires a broad differential, including flexor-pronator mass or distal biceps injury, chronic exertional compartment syndrome, radial tunnel syndrome, intersection syndrome, pronator teres syndrome, anterior interosseous syndrome, thoracic outlet syndrome, musculocutaneous nerve compression, deep vein thrombosis of ulnar vein, and periostitis. Stress fractures distal to the elbow more commonly occur in weight-bearing athletes, though as this case shows it is important to consider stress fractures of the radius and ulna when evaluating forearm pain in a throwing athlete.21
The first imaging examination for a suspected stress fracture is a radiograph, which can be normal in up to 90% of patients, as it initially was in our athlete’s case.22 Often, radiographic evidence takes 2 to 12 weeks to appear.5 Even then, radiographs may be positive in only 50% of cases.19 CT, often regarded as insensitive during the early stages, is useful in visualizing fracture lines in a suspicious location.19,22 Radionuclide uptake scanning is highly sensitive during the early stages of stress injury but is nonspecific and may indicate neoplasm or infection; in addition, up to 46% of abnormal foci are asymptomatic.19 MRI has sensitivity comparable to that of radionuclide scanning but also many advantages, including lack of ionizing radiation, improved spatial resolution, and ability to image bone and soft tissue simultaneously.19 In our patient’s case, the unusual stress fracture location potentially could have hindered identification of the cause of injury. The lesion was just distal to the field of view of a normal elbow MRI and was not detected until a dedicated forearm MRI was examined. Both MRI and CT helped in identifying the stress fracture, and CT was used to follow interval healing.
In baseball players, upper extremity stress fractures are often nonoperatively treated with throwing cessation for 4 to 6 weeks followed by participation in a structured rehabilitation program.4,5 The throwing program that we suggest, and that was used in this case, has 21 stages of progression in duration, distance, and velocity of throwing. The athlete advances from each stage on the basis of symptoms.23 Other issues that may be addressed are vitamin D and calcium status and any flawed throwing mechanics that may have predisposed the athlete to injury. Such mechanics are gradually corrected.
The literature suggests that appropriate nonoperative management of stress fractures allows for return to sport in 8 to 10 weeks. It is important to note that most of the literature on stress fractures involves the lower extremity, and that treatment and time to return to play are therefore better described for such fractures.6 More study and evaluation of upper extremity stress fractures are needed to make return-to-sport predictions more reliable and successful treatment modalities more unified for this patient population. Last, it is imperative that clinical examination and symptoms be correlated with serial imaging when deciding on return to play. Our patient took 12 weeks to return to high-level sport. He progressed pain-free through the throwing program and showed radiographic evidence of healing on follow-up CT.
Conclusion
Radial shaft stress fractures are rare in throwing athletes. However, with a thorough history, a physical examination, and appropriate imaging, the correct diagnosis can be made early on, and proper treatment can be started to facilitate return to sport. To our knowledge, this is the first report of a stress fracture in the radial shaft of a MLB pitcher. Although the radial shaft is an uncommon location for stress fractures, we should keep in mind that they can occur wherever increased muscular forces exceed the ability of native bone to remodel. After diagnosis, the fracture usually heals with nonoperative treatment, and healing is confirmed with follow-up imaging, as was done in our patient’s case. Improved prediction of time to return to play for upper extremity fractures, such as the radial stress fracture described in this article, requires more study.
Take-Home Points
- Stress fractures should always be considered when dealing with overuse injuries.
- Radial shaft stress fractures in overhead throwing athletes are rare.
- Stress fractures can occur anywhere increased muscular forces exceed the bone’s ability to remodel.
- Proper imaging is necessary to make the diagnosis of a stress fracture.
- Nonoperative management of radial shaft stress fractures is an effective treatment.
In athletes, the incidence of stress fractures has been reported to be 1.4% to 4.4%.1 Stress fractures of the upper extremity are less common and not as well described as lower extremity stress fractures. Although data is lacking, stress fractures involving the upper extremity appear to account for <6% of all stress fractures.2 Stress fractures of the upper extremity, though rare, are being recognized more often in overhead athletes.3-6 In baseball pitchers, stress fractures most commonly occur in the olecranon but have also been found in the ribs, clavicle, humerus, and ulnar shaft.2,4,7-10 Stress fractures of the radius are a rare cause of forearm pain in athletes, and there are only a few case reports involving overhead athletes.4,11-15 To our knowledge, a stress fracture of the radial shaft has not been reported in a throwing athlete. Currently, there are no reports on stress fractures of the proximal radial shaft.16-18
In this article, we report the case of a radial shaft stress fracture that was causing forearm pain in a Major League Baseball (MLB) pitcher. We also discuss the etiology, diagnosis, and management of stress fractures of the upper extremity of overhead throwing athletes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 28-year-old right-hand-dominant MLB pitcher presented to the clinic with a 4-week history of right dorsal forearm pain that was refractory to a period of rest and physical therapy modalities. The pain radiated to the wrist and along the dorsal forearm. The pain started after the man attempted to develop a new pitch that required a significant amount of supination. The pain prevented him from pitching competitively. Indomethacin, diclofenac sodium topical gel, and methylprednisolone (Medrol Dosepak) reduced his symptoms only slightly.
Physical examination of the right elbow showed mild range of motion deficits; about 5° of extension and 5° of flexion were lacking. The patient had full pronation and supination. Palpation of the dorsal aspect of the forearm revealed marked tenderness in the area of the proximal radius. There was no tenderness over the posterior olecranon or the ulnar collateral ligament, and a moving valgus stress test was negative. No pain was elicited by resisted extension of the wrist or fingers. Motor innervation from the posterior interosseous nerve, anterior interosseous nerve, and ulnar nerve was intact with 5/5 strength, and there were no sensory deficits in the distribution of the radial, median, or ulnar nerves.
Discussion
Stress fractures account for 0.7% to 20% of sports medicine clinic injuries; <10% of all stress fractures involve the rib or upper extremity.4,6 When the intensity or frequency of physical activity is increased, as with overuse, bone resorption surpasses bone production, locally weakening the bone and making it prone to mechanical failure. Failure is thought to be induced by a combination of contractile muscular forces across damaged bone and increased mechanical loading caused by fatigue of supporting structures.5,6,19 These forces may have contributed to our baseball pitcher’s development of a stress fracture near the insertion of the supinator muscle in his throwing arm.
Given the insidious nature of stress fractures, the evaluating physician must have a high index of suspicion. Early recognition of a stress fracture is important in preventing further injury and allowing for early intervention, which is associated with faster healing.6,20 The clinical history often involves a change in training regimen within the weeks before pain onset. Furthermore, understanding the type of pitches used and the mechanics of each pitch can help with diagnosis. Often, pain increases as the inciting activity continues, and relief comes with rest. In an upper extremity examination, it is important to recall the usual stress fracture locations in throwers—the ribs, clavicle, humerus, ulnar shaft, and most often the olecranon—though the patient’s history often narrows the anatomical region of suspicion.2,4,7-10 Examination begins with inspection of the skin and soft tissues. Range of motion and strength testing results likely are normal throughout the upper extremity.3 Palpation over the suspected injury location often elicits pain and indicates further imaging is needed.6 The tuning fork test or the 3-point fulcrum test may elicit symptoms in occult fractures.3 Completing the assessment is a thorough neurovascular examination.
Insidious forearm pain requires a broad differential, including flexor-pronator mass or distal biceps injury, chronic exertional compartment syndrome, radial tunnel syndrome, intersection syndrome, pronator teres syndrome, anterior interosseous syndrome, thoracic outlet syndrome, musculocutaneous nerve compression, deep vein thrombosis of ulnar vein, and periostitis. Stress fractures distal to the elbow more commonly occur in weight-bearing athletes, though as this case shows it is important to consider stress fractures of the radius and ulna when evaluating forearm pain in a throwing athlete.21
The first imaging examination for a suspected stress fracture is a radiograph, which can be normal in up to 90% of patients, as it initially was in our athlete’s case.22 Often, radiographic evidence takes 2 to 12 weeks to appear.5 Even then, radiographs may be positive in only 50% of cases.19 CT, often regarded as insensitive during the early stages, is useful in visualizing fracture lines in a suspicious location.19,22 Radionuclide uptake scanning is highly sensitive during the early stages of stress injury but is nonspecific and may indicate neoplasm or infection; in addition, up to 46% of abnormal foci are asymptomatic.19 MRI has sensitivity comparable to that of radionuclide scanning but also many advantages, including lack of ionizing radiation, improved spatial resolution, and ability to image bone and soft tissue simultaneously.19 In our patient’s case, the unusual stress fracture location potentially could have hindered identification of the cause of injury. The lesion was just distal to the field of view of a normal elbow MRI and was not detected until a dedicated forearm MRI was examined. Both MRI and CT helped in identifying the stress fracture, and CT was used to follow interval healing.
In baseball players, upper extremity stress fractures are often nonoperatively treated with throwing cessation for 4 to 6 weeks followed by participation in a structured rehabilitation program.4,5 The throwing program that we suggest, and that was used in this case, has 21 stages of progression in duration, distance, and velocity of throwing. The athlete advances from each stage on the basis of symptoms.23 Other issues that may be addressed are vitamin D and calcium status and any flawed throwing mechanics that may have predisposed the athlete to injury. Such mechanics are gradually corrected.
The literature suggests that appropriate nonoperative management of stress fractures allows for return to sport in 8 to 10 weeks. It is important to note that most of the literature on stress fractures involves the lower extremity, and that treatment and time to return to play are therefore better described for such fractures.6 More study and evaluation of upper extremity stress fractures are needed to make return-to-sport predictions more reliable and successful treatment modalities more unified for this patient population. Last, it is imperative that clinical examination and symptoms be correlated with serial imaging when deciding on return to play. Our patient took 12 weeks to return to high-level sport. He progressed pain-free through the throwing program and showed radiographic evidence of healing on follow-up CT.
Conclusion
Radial shaft stress fractures are rare in throwing athletes. However, with a thorough history, a physical examination, and appropriate imaging, the correct diagnosis can be made early on, and proper treatment can be started to facilitate return to sport. To our knowledge, this is the first report of a stress fracture in the radial shaft of a MLB pitcher. Although the radial shaft is an uncommon location for stress fractures, we should keep in mind that they can occur wherever increased muscular forces exceed the ability of native bone to remodel. After diagnosis, the fracture usually heals with nonoperative treatment, and healing is confirmed with follow-up imaging, as was done in our patient’s case. Improved prediction of time to return to play for upper extremity fractures, such as the radial stress fracture described in this article, requires more study.
1. Monteleone GP Jr. Stress fractures in the athlete. Orthop Clin North Am. 1995;26(3):423-432.
2. Iwamoto J, Takeda T. Stress fractures in athletes: review of 196 cases. J Orthop Sci. 2003;8(3):273-278.
3. Miller TL, Kaeding CC. Upper-extremity stress fractures: distribution and causative activities in 70 patients. Orthopedics. 2012;35(9):789-793.
4. Jones GL. Upper extremity stress fractures. Clin Sports Med. 2006;25(1):159-174.
5. Brooks AA. Stress fractures of the upper extremity. Clin Sports Med. 2001;20(3):613-620.
6. Fredericson M, Jennings F, Beaulieu C, Matheson GO. Stress fractures in athletes. Top Magn Reson Imaging. 2006;17(5):309-325.
7. Gurtler R, Pavlov H, Torg JS. Stress fracture of the ipsilateral first rib in a pitcher. Am J Sports Med. 1985;13(4):277-279.
8. Polu KR, Schenck RC Jr, Wirth MA, Greeson J, Cone RO 3rd, Rockwood CA Jr. Stress fracture of the humerus in a collegiate baseball pitcher. A case report. Am J Sports Med. 1999;27(6):813-816.
9. Wu C, Chen Y. Stress fracture of the clavicle in a professional baseball player. J Shoulder Elbow Surg. 1998;7(2):164-167.
10. Schickendantz MS, Ho CP, Koh J. Stress injury of the proximal ulna in professional baseball players. Am J Sports Med. 2002;30(5):737-741.
11. Loosli AR, Leslie M. Stress fractures of the distal radius. A case report. Am J Sports Med. 1991;19(5):523-524.
12. Inagaki H, Inoue G. Stress fracture of the scaphoid combined with the distal radial epiphysiolysis. Br J Sports Med. 1997;31(3):256-257.
13. Read MT. Stress fractures of the distal radius in adolescent gymnasts. Br J Sports Med. 1981;15(4):272-276.
14. Orloff AS, Resnick D. Fatigue fracture of the distal part of the radius in a pool player. Injury. 1986;17(6):418-419.
15. Eisenberg D, Kirchner SG, Green NE. Stress fracture of the distal radius caused by “wheelies.” South Med J. 1986;79(7):918-919.
16. Brukner P. Stress fractures of the upper limb. Sports Med. 1998;26(6):415-424.
17. Farquharson-Roberts MA, Fulford PC. Stress fracture of the radius. J Bone Joint Surg Br. 1980;62(2):194-195.
18. Orloff AS, Resnick D. Fatigue fracture of the distal part of the radius in a pool player. Injury. 1986;17(6):418-419.
19. Anderson MW. Imaging of upper extremity stress fractures in the athlete. Clin Sports Med. 2006;25(3):489-504.
20. Bennell K, Brukner P. Preventing and managing stress fractures in athletes. Phys Ther Sport. 2005;6(4):171-180.
21. Sinha AK, Kaeding CC, Wadley GM. Upper extremity stress fractures in athletes: clinical features of 44 cases. Clin J Sport Med. 1999;9(4):199-202.
22. Matheson GO, Clement DB, McKenzie DC, Taunton JE, Lloyd-Smith DR, MacIntyre JG. Stress fractures in athletes. A study of 320 cases. Am J Sports Med. 1987;15(1):46-58.
23. Kaplan L, Lesniak B, Baraga M, et al. Throwing program for baseball players. 2009. http://uhealthsportsmedicine.com/documents/UHealth_Throwing_Program.pdf. Accessed May 24, 2016.
1. Monteleone GP Jr. Stress fractures in the athlete. Orthop Clin North Am. 1995;26(3):423-432.
2. Iwamoto J, Takeda T. Stress fractures in athletes: review of 196 cases. J Orthop Sci. 2003;8(3):273-278.
3. Miller TL, Kaeding CC. Upper-extremity stress fractures: distribution and causative activities in 70 patients. Orthopedics. 2012;35(9):789-793.
4. Jones GL. Upper extremity stress fractures. Clin Sports Med. 2006;25(1):159-174.
5. Brooks AA. Stress fractures of the upper extremity. Clin Sports Med. 2001;20(3):613-620.
6. Fredericson M, Jennings F, Beaulieu C, Matheson GO. Stress fractures in athletes. Top Magn Reson Imaging. 2006;17(5):309-325.
7. Gurtler R, Pavlov H, Torg JS. Stress fracture of the ipsilateral first rib in a pitcher. Am J Sports Med. 1985;13(4):277-279.
8. Polu KR, Schenck RC Jr, Wirth MA, Greeson J, Cone RO 3rd, Rockwood CA Jr. Stress fracture of the humerus in a collegiate baseball pitcher. A case report. Am J Sports Med. 1999;27(6):813-816.
9. Wu C, Chen Y. Stress fracture of the clavicle in a professional baseball player. J Shoulder Elbow Surg. 1998;7(2):164-167.
10. Schickendantz MS, Ho CP, Koh J. Stress injury of the proximal ulna in professional baseball players. Am J Sports Med. 2002;30(5):737-741.
11. Loosli AR, Leslie M. Stress fractures of the distal radius. A case report. Am J Sports Med. 1991;19(5):523-524.
12. Inagaki H, Inoue G. Stress fracture of the scaphoid combined with the distal radial epiphysiolysis. Br J Sports Med. 1997;31(3):256-257.
13. Read MT. Stress fractures of the distal radius in adolescent gymnasts. Br J Sports Med. 1981;15(4):272-276.
14. Orloff AS, Resnick D. Fatigue fracture of the distal part of the radius in a pool player. Injury. 1986;17(6):418-419.
15. Eisenberg D, Kirchner SG, Green NE. Stress fracture of the distal radius caused by “wheelies.” South Med J. 1986;79(7):918-919.
16. Brukner P. Stress fractures of the upper limb. Sports Med. 1998;26(6):415-424.
17. Farquharson-Roberts MA, Fulford PC. Stress fracture of the radius. J Bone Joint Surg Br. 1980;62(2):194-195.
18. Orloff AS, Resnick D. Fatigue fracture of the distal part of the radius in a pool player. Injury. 1986;17(6):418-419.
19. Anderson MW. Imaging of upper extremity stress fractures in the athlete. Clin Sports Med. 2006;25(3):489-504.
20. Bennell K, Brukner P. Preventing and managing stress fractures in athletes. Phys Ther Sport. 2005;6(4):171-180.
21. Sinha AK, Kaeding CC, Wadley GM. Upper extremity stress fractures in athletes: clinical features of 44 cases. Clin J Sport Med. 1999;9(4):199-202.
22. Matheson GO, Clement DB, McKenzie DC, Taunton JE, Lloyd-Smith DR, MacIntyre JG. Stress fractures in athletes. A study of 320 cases. Am J Sports Med. 1987;15(1):46-58.
23. Kaplan L, Lesniak B, Baraga M, et al. Throwing program for baseball players. 2009. http://uhealthsportsmedicine.com/documents/UHealth_Throwing_Program.pdf. Accessed May 24, 2016.
A Case of Streptococcus pyogenes Sepsis of Possible Oral Origin
Sepsis can be the result of single or multiple factors and sources of infection. Oral sources of sepsis and systemic infection are not commonly considered as the first potential source of infection when evaluating a septic patient. Oral infections of odontogenic or periodontal origin are frequently associated with localized or diffuse cellulitis of the head and neck region.1 The patient’s health status and complicating problems, such as an immunocompromising condition, can further reduce the immune response for controlling chronic sources of infection. This in turn, can lead to acute manifestations such as cellulitis, sepsis, or necrotizing fasciitis. Necrotizing fasciitis is caused by a polymicrobial or mixed aerobic-anaerobic infection from a variety of sources, including Streptococcus pyogenes (S pyogenes).
Case Presentation
A 57-year-old female with a history of major depressive disorder, paroxysmal atrial fibrillation, and opioid dependence that was in remission for more than 3 years was brought to the emergency department (ED) by a family member after the patient developed confusion and lethargy. She was primarily experiencing right breast pain and swelling. The breast pain was associated with high fevers, nausea, vomiting, and chills. On examination the patient was noted to have a fever of 104° F, heart rate of 160 bpm, respirations of 22 breaths per minute, blood pressure (BP) 109/58, and a white blood cell count (WBC) of 8.7 X 103. There was a noted skin abrasion on her right hand. She was lethargic and confused. Blood cultures were positive for S pyogenes, and a swab of the right breast was negative for bacterial growth.
The patient was admitted to the medical intensive care unit (MICU) and placed on 2 vasopressors for control of low BP and assistance with low urine output. After a 6 L fluid resuscitation, the patient was started on vancomycin and piperacillin/tazobactam for possible cellulitis causing sepsis. An echocardiogram was negative for endocarditis. The patient continued to decline the following day with continuing tachycardia and tachypnea with hypotension and was intubated. Pulmonology was consulted for possible acute respiratory distress syndrome secondary to sepsis. General surgery was consulted for possible necrotizing fasciitis of the chest wall, and cardiology was consulted for low cardiac output.
On day 4 of her hospitalization, the patient was taken to surgery for exploration, drainage, and debridement of the right axilla and breast; cultures with lack of organism growth was noted. While in the MICU, she was followed by the Infectious Disease service as her WBC remained elevated and peaking at 32.6 X 103, while blood cultures were negative for bacterial growth. The dental service was consulted on day 5 to evaluate for other possible sources of infection.
The patient’s oral condition was noted as having advanced chronic periodontal disease that required full mouth extraction. The patient remained hemodynamically unstable with platelet counts below 50,000 until day 7, at which time she was taken for surgery for full mouth extraction and associated alveoloplasty. On extraction the patient continued to improve and was extubated on day 11 with platelets and WBC returning to normal levels by day 13 of her hospital stay. The patient remained hospitalized for a total MICU stay of 20 days and rehabilitation stay of more than 2 weeks.
Discussion
Oral infections most often present with acute onset and noted oral-facial cellulitis or abscess. Oral source of septicemia often are considered after ruling out most other potential sources. Although it is not certain that this case is directly related to the advanced chronic periodontal disease, S pyogenes has been noted to be a pathogen in periodontal disease progression.
According to the American Dental Association in 2012, dental visits to the ED cost the U.S. health care system $1.6 billion and an average cost of $749 per visit. There are more than 2 million ED visits each year for dental pain and infection, and 39% return due to nonresolution of the dental problem. Patients return to the ED due to lack of access and resources to routine and emergent dental care.2 The average daily cost of an MICU stay with mechanical ventilation was $2,193 in 2002. This particular case consisted of 11 days of mechanical ventilation, 20 MICU days, and an additional 20 days of inpatient rehabilitation which resulted in costs that exceeded $50,000.3
Conclusion
This case demonstrates the successful collaboration of dentistry for the overall medical management of the patient. An integrated approach highlights the need for and the value of integrating dental programs within large tertiary hospital systems. Such integration will likely improve earlier recognition and better management of oral infections resulting in systemic illness and improve patient outcomes, reduced length of hospital stay, and reduction of overall costs.
1. Krishnan V, Johnson JV, Helfric JF. Management of maxillofacial infections: a review of 50 cases. J Oral Maxillofac Surg. 1993; 51(8):868-873.
2. Wall T, Vujicic M. Emergency department use for dental conditions continues to increase. American Dental Association: Health Policy Institute. http://www.ada.org/~/media/ADA/Science%20and%20Research/HPI/Files/HPIBrief_0415_2.ashx. Published April 2015. Accessed September 5, 2017.
3. Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med. 2005;33(6):1266-1271.
Sepsis can be the result of single or multiple factors and sources of infection. Oral sources of sepsis and systemic infection are not commonly considered as the first potential source of infection when evaluating a septic patient. Oral infections of odontogenic or periodontal origin are frequently associated with localized or diffuse cellulitis of the head and neck region.1 The patient’s health status and complicating problems, such as an immunocompromising condition, can further reduce the immune response for controlling chronic sources of infection. This in turn, can lead to acute manifestations such as cellulitis, sepsis, or necrotizing fasciitis. Necrotizing fasciitis is caused by a polymicrobial or mixed aerobic-anaerobic infection from a variety of sources, including Streptococcus pyogenes (S pyogenes).
Case Presentation
A 57-year-old female with a history of major depressive disorder, paroxysmal atrial fibrillation, and opioid dependence that was in remission for more than 3 years was brought to the emergency department (ED) by a family member after the patient developed confusion and lethargy. She was primarily experiencing right breast pain and swelling. The breast pain was associated with high fevers, nausea, vomiting, and chills. On examination the patient was noted to have a fever of 104° F, heart rate of 160 bpm, respirations of 22 breaths per minute, blood pressure (BP) 109/58, and a white blood cell count (WBC) of 8.7 X 103. There was a noted skin abrasion on her right hand. She was lethargic and confused. Blood cultures were positive for S pyogenes, and a swab of the right breast was negative for bacterial growth.
The patient was admitted to the medical intensive care unit (MICU) and placed on 2 vasopressors for control of low BP and assistance with low urine output. After a 6 L fluid resuscitation, the patient was started on vancomycin and piperacillin/tazobactam for possible cellulitis causing sepsis. An echocardiogram was negative for endocarditis. The patient continued to decline the following day with continuing tachycardia and tachypnea with hypotension and was intubated. Pulmonology was consulted for possible acute respiratory distress syndrome secondary to sepsis. General surgery was consulted for possible necrotizing fasciitis of the chest wall, and cardiology was consulted for low cardiac output.
On day 4 of her hospitalization, the patient was taken to surgery for exploration, drainage, and debridement of the right axilla and breast; cultures with lack of organism growth was noted. While in the MICU, she was followed by the Infectious Disease service as her WBC remained elevated and peaking at 32.6 X 103, while blood cultures were negative for bacterial growth. The dental service was consulted on day 5 to evaluate for other possible sources of infection.
The patient’s oral condition was noted as having advanced chronic periodontal disease that required full mouth extraction. The patient remained hemodynamically unstable with platelet counts below 50,000 until day 7, at which time she was taken for surgery for full mouth extraction and associated alveoloplasty. On extraction the patient continued to improve and was extubated on day 11 with platelets and WBC returning to normal levels by day 13 of her hospital stay. The patient remained hospitalized for a total MICU stay of 20 days and rehabilitation stay of more than 2 weeks.
Discussion
Oral infections most often present with acute onset and noted oral-facial cellulitis or abscess. Oral source of septicemia often are considered after ruling out most other potential sources. Although it is not certain that this case is directly related to the advanced chronic periodontal disease, S pyogenes has been noted to be a pathogen in periodontal disease progression.
According to the American Dental Association in 2012, dental visits to the ED cost the U.S. health care system $1.6 billion and an average cost of $749 per visit. There are more than 2 million ED visits each year for dental pain and infection, and 39% return due to nonresolution of the dental problem. Patients return to the ED due to lack of access and resources to routine and emergent dental care.2 The average daily cost of an MICU stay with mechanical ventilation was $2,193 in 2002. This particular case consisted of 11 days of mechanical ventilation, 20 MICU days, and an additional 20 days of inpatient rehabilitation which resulted in costs that exceeded $50,000.3
Conclusion
This case demonstrates the successful collaboration of dentistry for the overall medical management of the patient. An integrated approach highlights the need for and the value of integrating dental programs within large tertiary hospital systems. Such integration will likely improve earlier recognition and better management of oral infections resulting in systemic illness and improve patient outcomes, reduced length of hospital stay, and reduction of overall costs.
Sepsis can be the result of single or multiple factors and sources of infection. Oral sources of sepsis and systemic infection are not commonly considered as the first potential source of infection when evaluating a septic patient. Oral infections of odontogenic or periodontal origin are frequently associated with localized or diffuse cellulitis of the head and neck region.1 The patient’s health status and complicating problems, such as an immunocompromising condition, can further reduce the immune response for controlling chronic sources of infection. This in turn, can lead to acute manifestations such as cellulitis, sepsis, or necrotizing fasciitis. Necrotizing fasciitis is caused by a polymicrobial or mixed aerobic-anaerobic infection from a variety of sources, including Streptococcus pyogenes (S pyogenes).
Case Presentation
A 57-year-old female with a history of major depressive disorder, paroxysmal atrial fibrillation, and opioid dependence that was in remission for more than 3 years was brought to the emergency department (ED) by a family member after the patient developed confusion and lethargy. She was primarily experiencing right breast pain and swelling. The breast pain was associated with high fevers, nausea, vomiting, and chills. On examination the patient was noted to have a fever of 104° F, heart rate of 160 bpm, respirations of 22 breaths per minute, blood pressure (BP) 109/58, and a white blood cell count (WBC) of 8.7 X 103. There was a noted skin abrasion on her right hand. She was lethargic and confused. Blood cultures were positive for S pyogenes, and a swab of the right breast was negative for bacterial growth.
The patient was admitted to the medical intensive care unit (MICU) and placed on 2 vasopressors for control of low BP and assistance with low urine output. After a 6 L fluid resuscitation, the patient was started on vancomycin and piperacillin/tazobactam for possible cellulitis causing sepsis. An echocardiogram was negative for endocarditis. The patient continued to decline the following day with continuing tachycardia and tachypnea with hypotension and was intubated. Pulmonology was consulted for possible acute respiratory distress syndrome secondary to sepsis. General surgery was consulted for possible necrotizing fasciitis of the chest wall, and cardiology was consulted for low cardiac output.
On day 4 of her hospitalization, the patient was taken to surgery for exploration, drainage, and debridement of the right axilla and breast; cultures with lack of organism growth was noted. While in the MICU, she was followed by the Infectious Disease service as her WBC remained elevated and peaking at 32.6 X 103, while blood cultures were negative for bacterial growth. The dental service was consulted on day 5 to evaluate for other possible sources of infection.
The patient’s oral condition was noted as having advanced chronic periodontal disease that required full mouth extraction. The patient remained hemodynamically unstable with platelet counts below 50,000 until day 7, at which time she was taken for surgery for full mouth extraction and associated alveoloplasty. On extraction the patient continued to improve and was extubated on day 11 with platelets and WBC returning to normal levels by day 13 of her hospital stay. The patient remained hospitalized for a total MICU stay of 20 days and rehabilitation stay of more than 2 weeks.
Discussion
Oral infections most often present with acute onset and noted oral-facial cellulitis or abscess. Oral source of septicemia often are considered after ruling out most other potential sources. Although it is not certain that this case is directly related to the advanced chronic periodontal disease, S pyogenes has been noted to be a pathogen in periodontal disease progression.
According to the American Dental Association in 2012, dental visits to the ED cost the U.S. health care system $1.6 billion and an average cost of $749 per visit. There are more than 2 million ED visits each year for dental pain and infection, and 39% return due to nonresolution of the dental problem. Patients return to the ED due to lack of access and resources to routine and emergent dental care.2 The average daily cost of an MICU stay with mechanical ventilation was $2,193 in 2002. This particular case consisted of 11 days of mechanical ventilation, 20 MICU days, and an additional 20 days of inpatient rehabilitation which resulted in costs that exceeded $50,000.3
Conclusion
This case demonstrates the successful collaboration of dentistry for the overall medical management of the patient. An integrated approach highlights the need for and the value of integrating dental programs within large tertiary hospital systems. Such integration will likely improve earlier recognition and better management of oral infections resulting in systemic illness and improve patient outcomes, reduced length of hospital stay, and reduction of overall costs.
1. Krishnan V, Johnson JV, Helfric JF. Management of maxillofacial infections: a review of 50 cases. J Oral Maxillofac Surg. 1993; 51(8):868-873.
2. Wall T, Vujicic M. Emergency department use for dental conditions continues to increase. American Dental Association: Health Policy Institute. http://www.ada.org/~/media/ADA/Science%20and%20Research/HPI/Files/HPIBrief_0415_2.ashx. Published April 2015. Accessed September 5, 2017.
3. Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med. 2005;33(6):1266-1271.
1. Krishnan V, Johnson JV, Helfric JF. Management of maxillofacial infections: a review of 50 cases. J Oral Maxillofac Surg. 1993; 51(8):868-873.
2. Wall T, Vujicic M. Emergency department use for dental conditions continues to increase. American Dental Association: Health Policy Institute. http://www.ada.org/~/media/ADA/Science%20and%20Research/HPI/Files/HPIBrief_0415_2.ashx. Published April 2015. Accessed September 5, 2017.
3. Dasta JF, McLaughlin TP, Mody SH, Piech CT. Daily cost of an intensive care unit day: the contribution of mechanical ventilation. Crit Care Med. 2005;33(6):1266-1271.
A Severe Case of Paliperidone Palmitate-Induced Parkinsonism Leading to Prolonged Hospitalization: Opportunities for Improvement
Many patients with psychiatric illness have difficulty with medication adherence. Patients with impaired reality testing especially are at risk.
Keck and McElroy evaluated 141 patients who were initially hospitalized for bipolar disorder prospectively over 1 year to assess adherence with medication. During the follow-up period, 71 patients (51%) were partially or totally nonadherent with medication as prescribed. The most commonly cited reason for nonadherence was denial of need.1
Clinicians and patients face additional challenges due to the deleterious effects of relapse in the setting of both schizophrenia and bipolar disorder. Almost all oral antipsychotic or mood stabilizer medications require a minimum dosing schedule to effectively treat these disorders, and some of these oral medications require regular laboratory monitoring. Moreover, some of the agents can have serious adverse effects (AEs), such as seizure or withdrawal, if stopped abruptly. Social support from family or friends may improve adherence, but many psychiatric outpatients have a smaller social support network than do patients without psychiatric illnesses.2
Long-acting injectable (LAI) antipsychotics have been available for the past 40 years. These medications have provided clinicians with an additional option for patients with schizophrenia or bipolar disorder who are nonadherent to their medication treatment plans or who desire an administration choice that is more convenient than daily oral pills.3-7 Some clinical practice guidelines recommend considering LAIs as a maintenance treatment for schizophrenia.5 Like the rest of the pharmacopoeia, these formulations have AEs, such as extrapyramidal symptoms (EPS), weight gain, and metabolic syndrome.1 The longer half-life of these drugs may make such effects difficult to reverse.
This article presents a case of the use of depot formulation paliperidone palmitate in an elderly patient with bipolar disorder who was previously on high-dose oral second generation antipsychotics. He developed severe parkinsonism during a protracted hospitalization that ended in death.
Case Presentation
Mr. W was a 68-year-old homeless white male with a history of coronary artery disease status-post coronary artery bypass surgery, obstructive sleep apnea, and bipolar 1 disorder who presented to a large rural VAMC emergency department (ED) as a transfer from an outside hospital (OSH). He originally presented at the OSH for vomiting and diarrhea, but while there, he was placed under involuntary psychiatric commitment. The involuntary commitment form noted him to be tangential and disorganized; he was found walking about the ED without clothes. During the initial psychiatry interview, the clinician noted a disorganized thought process. When asked about circumstances leading to admission, he stated he was “a scuba diver, pilot, actor, submarine commander.” He also reported he had given “seminars to 6,000 people,” he held a psychology degree, and he came from a family that owned part of the island of Kodiak, Alaska. Mr. W stated he had no mental health history and believed psychiatry was witchcraft. He reported having no hallucinations and stated he heard the voice of god. He also reported to have met god multiple times and to have been married to a supermodel.
Mr. W’s chart demonstrated a history of mental illness over 30 years and that he previously was prescribed psychiatric medications. He had multiple inpatient psychiatric admissions with grandiose ideations, disorganized behaviors, and hypersexuality. He had been prescribed quetiapine, divalproex, lithium, carbamazepine, and lorazepam. He was formally diagnosed in the past with bipolar 1 disorder. There also was a family history of psychiatric illness. His mother had received electroconvulsive therapy, and both parents had alcohol substance use disorder.
Mr. W had been homeless for 20 years and had several psychiatric admissions during this period. Mr. W also had chronic difficulty with obtaining food and taking medications as prescribed. Sometimes he would only be able to eat 1 to 2 meals per day. He often changed location and had lived in at least 7 different states. Currently, he was estranged from his family. About 19 years ago, his sister reported that the veteran had told her that he was Jesus Christ, per clinical records. His estranged sister’s statement was corroborated by past psychology consult records citing episodes of the patient hearing god 30 and 26 years before the current admission. His second ex-wife cited inappropriate sexual behavior in front of their children. He had difficulty in school, failed at least 2 grades, and joined the U.S. Navy in tenth grade. A Neurobehavioral Cognitive Status Examination given 19 years ago showed mild impairment on attention and severe impairment in memory.
The physical examination on presentation to the OSH was unremarkable. Mr. W did not cooperate with formal neurocognitive testing, and he consistently made errors during orientation testing. Complete blood count from a OSH ED laboratory test was remarkable for a mild pancytopenia with a leukocyte count of 3,100 cells/mcL, hemoglobin 13.1 g/dL, and hematocrit 38.4%. Red cell distribution width was within normal limits at 13.5%. Stool cultures showed normal fecal flora and no salmonella, shigella, or campylobacter. Thyroid-stimulating hormone (TSH) was slightly elevated at 5.32 U/mL. An electrocardiogram showed a QTc interval of 412 ms. A computerized tomography scan of his head showed no acute intracranial abnormality along with chronic ischemic changes in the brain (Table 1). Presumed cause of his nausea and diarrhea was viral gastroenteritis likely acquired at a homeless shelter.
Once stabilized, Mr. W was admitted to the VA hospital inpatient psychiatry unit under involuntary commitment for acute mania. Risperidone 0.25 mg orally twice a day was started for mood stabilization and psychosis along with trazodone 50 mg orally as needed for insomnia. Despite upward titration and change in frequency of the risperidone dose, Mr. W’s manic episode persisted. He remained on the psychiatric floor for 2 months (Figure). His TSH and free T4 were monitored during his stay, and levothyroxine was started. Risperidone was titrated to 8 mg/d. Mr. W’s Young Mania Rating Scale (YMRS) score decreased from 30 to 24. Mr. W had a mild improvement in irritability and speech rate but little change in elevated mood and delusional content.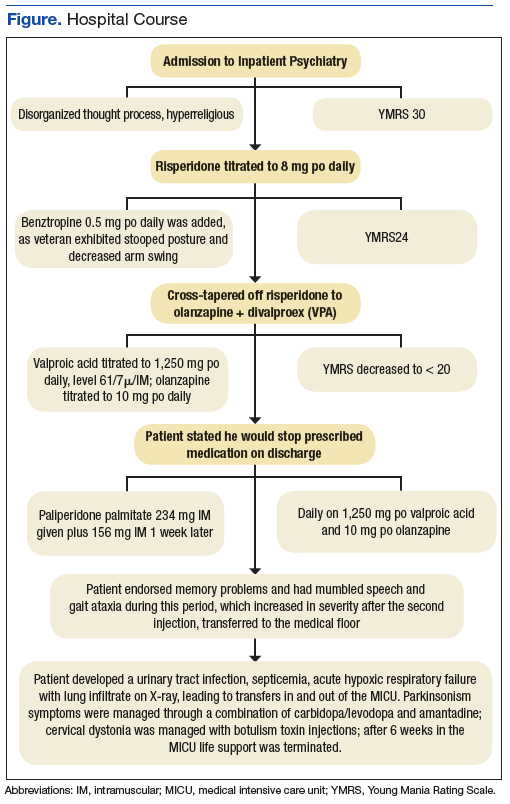
He continued to endorse “speaking to god 16 times” even at the highest risperidone dose. The treatment team prescribed dissolvable risperidone tablets secondary to diversion concerns. In addition, the team added benztropine 0.5 mg once a day after observing a stooped posture and decreased arm swing. Mr. W noted risperidone made him “lethargic” and that his “body did not need” it. After 1 month of treatment with risperidone, the treatment team decided to cross taper the veteran from risperidone to a combination of olanzapine and divalproex secondary to inadequate treatment response.
The inpatient team started Mr. W on oral disintegrating tablets of olanzapine 5 mg once a day, and oral divalproex 1,000 mg once a day. An intramuscular backup of olanzapine was made available if oral medication was refused. Divalproex was titrated to 1,250 mg once a day to target a serum level of 61.7 µg/mL, and olanzapine was titrated to 10 mg once a day. After 9 days, the veteran showed moderate improvement in mania symptoms with a YMRS score < 20, indicating the absence of mania. However, the veteran made it very clear that he would stop taking the prescribed medication on discharge. The team elected to initiate a LAI.
The veteran received his first injection of the LAI psychiatric medication paliperidone palmitate 234 mg and a second 156-mg injection of the same medication 1 week later as per loading protocol. He was concurrently on daily oral divalproex 1,250 mg and olanzapine 10 mg. Mr. W continued to note he felt sedated during this period; his sedation worsened after the second injection. He also began to forget the location of his room and developed mumbled speech. His gait deteriorated to where he required a walker 6 days after injection and a wheelchair 3 days later. He became incontinent of urine and feces. Mr. W exhibited masked facies with severe drooling. This eventually progressed to difficulty swallowing. At the advice of speech pathology, he was downgraded to a pureed and nectar-thick liquid diet. He required assistance with meals.
Because of his sedation and parkinsonism symptoms, he was tapered off both olanzapine and divalproex. His last dose of olanzapine was on the date of his first injection and last dose of divalproex was 15 days after the second injection. The benztropine, which was originally given to counteract the effects of risperidone monotherapy, was discontinued over concern of anticholinergic load and sedation. The neurology consultant recommended carbidopa 25 mg and levodopa 100 mg 3 times per day for treatment of parkinsonism symptoms. Mr. W was only able to take 1 dose because of trouble swallowing. Twenty days after his second injection, a rapid response team (local clinical team 1 step below a code team) was called as Mr. W was unusually lethargic and unable to eat. He was then transferred to the medical floor.
While on the medical floor, dobhoff tube access was established for nutrition and to allow administration of carbidopa and levodopa. Mr. W could still speak at this time and was distraught. He stated, “I don’t know why god would do this to me.” Further workup was performed to look for other etiologies of the patient’s change in status. Creatinine kinase testing, lumbar puncture with cerebral spinal fluid (CSF) bacterial culture, CSF cryptococcal testing, and syphilis antigens were all negative. Magnetic resonance imaging of the brain demonstrated diffuse cerebral atrophy with widened cistern and sulci resulting in ex vacuo dilatation.
Neurology thought that the ventriculomegaly did not have features of normal pressure hydrocephalus and was secondary to chronic ischemic demyelination caused by chronic malnutrition. During follow-up visits, the veteran was less and less verbal. It progressed to where he answered questions only in grunts. Eight days after transfer to the medical floor, Mr. W was noted to have his neck locked in a laterally rotated position with clonus of the sternocleidomastoid. Due to concern about possibility of neck dystonia and the poor adherence of the patient with carbidopa and levodopa given orally, the psychiatric team made the recommendation to start benztropine 1 mg given twice a day, delivered via the dobhoff tube to treat both the parkinsonism and dystonia. The following day Mr. W failed a repeat swallow study and was no longer allowed to receive anything orally.
Mild icterus and jaundice were noted on physical examination along with transaminitis and elevated bilirubin. He developed a fever. Thirteen days after transfer to the medical floor, blood cultures revealed Klebsiella septicemia. Benztropine was discontinued at this time because of concern the medication was causing or exacerbating the fever. While being treated for Klebsiella sepsis, the psychiatry team addressed his continued hypophonia, inability to ambulate, masked facies, and neck dystonia with diphenhydramine 50 mg given intramuscular (IM) twice per day.
Mr. W developed several more iatrogenic complications near this time, including urinary tract infection septicemia and acute hypoxic respiratory failure with lung infiltrate on X-ray, requiring ventilator support. His clinical status led to a number of transfers in and out of the medical intensive care unit (MICU). During this time, his parkinsonism symptoms were managed through a combination of carbidopa and levodopa and amantadine. Cervical dystonia was managed with botulism toxin injections. Mr. W spent 6 weeks in the MICU until the decision was made to terminate life support, and he was taken off the ventilator. He died shortly thereafter. Autopsy findings suggested that Mr. W had severe Alzheimer disease.
Discussion
Following the IM injection of paliperidone palmitate, Mr. W had a complicated hospital stay resulting in his demise from sepsis and multiorgan failure. Severe immobilization, rigidity, and dystonia prevented Mr. W from conducting activities of daily living, which resulted in invasive interventions, such as continued foley catheterization. His sepsis was likely secondary to aspiration, catheterization, and eventual ventilation—all iatrogenic complications. Previous estimates in the U.S. have suggested a total of 225,000 deaths per year from iatrogenic causes.8
There are several areas of concern. Clearly, Mr. W had severe illness that greatly affected his life. He was estranged from family and had endured a 2-decade period of homelessness. He deserved effective treatment for his psychiatric illness to relieve his suffering. His long period of mental illness without effective treatment very likely biased the initial treatment team toward an aggressive approach.
Fragmented Care
The prolonged hospital stay and multiple complications directly led to fragmentation in Mr. W’s care. He was hospitalized for months on 3 different main services: psychiatry, medicine, and the MICU. Even when he remained on the same service, the primary members of his treatment team changed every few weeks. Many different specialties were consulted and reconsulted. Members of the specialty consult teams changed throughout the hospitalization as well. Given the nature of the local clinical administration, Mr. W likely received the most consistent team members from the attendings on the psychiatry consult-liaison service (who do not rotate) and from a local subspecialty delirium consult team (all members stay consistent except pharmacy residents).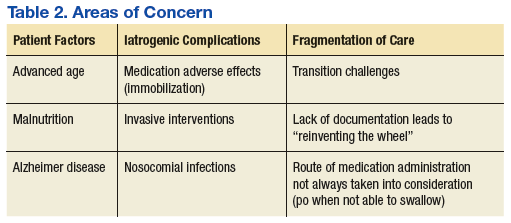
Documentation of clinical reasoning behind treatment decisions was not ideal and occasionally lacking. This led to a tendency to “reinvent the wheel” with Mr. W’s treatment approach every few weeks. It was not until Mr. W had spent a significant amount of time on the medical service that an interdisciplinary treatment team meeting involving medicine, psychiatry, nursing, delirium, and neurology experts occurred. Although the interdisciplinary meeting helped by reviewing the hospital course, agreeing on a likely cause of the symptoms, and creating a treatment plan going forward, Mr. W was not able to recover.
Even when team members were stable, communication in a timely fashion did not always occur. At several points, expert recommendations were delayed by a day or more. Difficulties in treatment implementation were not communicated back to the specialty teams. The most significant example was a delay in recognition when Mr. W could no longer take oral pills secondary to the parkinsonism. Many days passed before an alternative liquid or dissolved medication was recommended on 2 separate occasions.
Subspecialty Consult
Addressing these documentation, communication, and transition challenges is neither easy nor unique to this large rural VA medical center. The authors have attempted to address this in the local system with the creation of a delirium team subspecialty consult service. Team members do not rotate and are able to follow patients throughout their hospital course. At the time of Mr. W’s hospitalization, the team included representatives from nursing, psychiatry, and occasionally pharmacy. Since then, it has expanded to include geriatrics and medicine. In addition to delirium being a marker for complex patients at risk for hospital complications, medical reasons for an extended length of stay could serve as a trigger for a referral to such a team of experts. In Mr. W’s case, that could have led to interdisciplinary consultation up to 2 months before it occurred. This may have led to a much better outcome.
Secondary parkinsonism is most notable with the typical antipychotics. The prevalence can vary between 50% and 75% and may be higher within the elderly population. However, all antipsychotics have a chance of demonstrating EPS. Risperidone has a low incidence at low doses; studies have shown dose-related parkinsonism at doses of 2 to 6 mg/d. Significant risk of parkinsonism is further exacerbated when drug-drug interactions are considered.9 Concurrently receiving 2 antipsychotics, olanzapine and paliperidone, initially caused the EPS. The veteran’s cerebral atrophy from significant malnutrition related to chronic homelessness, and the presence of Alzheimer disease only identified postmortem exacerbated this AE. Further complicating the management of the EPS, paliperidone palmitate has a long half-life of 25 to 49 days.9 Simply discontinuing the medication did not remove it from Mr. W’s system. Paliperidone would have continued to be present for months.
Conclusion
In this case, aggressive changes in the antipsychotic medications in a short period led to Mr. W effectively having 3 different agents in his system at the same time. This significantly elevated his risk of AEs, including parkinsonism. The clinician must be vigilant to further recognize the initial symptoms of parkinsonism on clinical presentation. Administration of clinical scales, such as the Simpson-Angus Extrapyramidal Side Effect, can help in these situations.10 Malnutrition and increased age can predispose patients to neurolepticAEs, so treatment teams should exercise caution when administering antipsychotics in such a population. Pharmacokinetic changes in all major organ systems from aging result in higher and more variable drug concentrations. This leads to an increased sensitivity to drugs and AEs.9
Given the increasing geriatric patient population in the U.S., treating mania in the elderly will become more common. Providers should carefully consider the risks vs benefit ratio for each individual because a serious adverse reaction may result in detrimental consequences. Even with severe symptoms leading to a bias toward an aggressive approach, it may be better to “start low and go slow.” Early inclusion of interdisciplinary expertise should be sought in complex cases.
1. Keck PE Jr, McElroy SL, Strakowski SM, Bourne ML, West SA. Compliance with maintenance treatment in bipolar disorder. Psychopharmacol Bull. 1997;33(1):87-91.
2. Henderson S, Duncan-Jones P, McAuley H, Ritchie K. The patient’s primary group. Br J Psychiatry. 1978;132:74-86.
3. Buoli M, Ciappolino V, Altamura AC. Paliperidone palmitate depot in the long-term treatment of psychotic bipolar disorder: a case series. Clin Neuropharmacol. 2015;38(5):209-211.
4. Chou YH, Chu PC, Wu SW, et al. A systematic review and experts’ consensus for long-acting injectable antipsychotics in bipolar disorder. Clin Psychopharmacol Neurosci. 2015;13(2):121-128.
5. Kishi T, Oya K, Iwata N. Long-acting injectable antipsychotics for prevention of relapse in bipolar disorder: a systematic review and meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2016;19(9):1-10.
6. Llorca PM, Abbar M, Courtet P, Guillaume S, Lancrenon S, Samalin L. Guidelines for the use and management of long-acting injectable antipsychotics in serious mental illness. BMC Psychiatry. 2013;13:340.
7. Spanarello S, La Ferla T. The pharmacokinetics of long-acting antipsychotic medications. Curr Clin Pharmacol. 2014;9(3):310-317.
8. Starfield B. Is US health really the best in the world? JAMA. 2000;284(4):483-485.
9. Labbate LA, Fava M, Rosenbaum JF, Arana GW. Handbook of Psychiatric Drug Therapy. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.
10. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11-19.
Many patients with psychiatric illness have difficulty with medication adherence. Patients with impaired reality testing especially are at risk.
Keck and McElroy evaluated 141 patients who were initially hospitalized for bipolar disorder prospectively over 1 year to assess adherence with medication. During the follow-up period, 71 patients (51%) were partially or totally nonadherent with medication as prescribed. The most commonly cited reason for nonadherence was denial of need.1
Clinicians and patients face additional challenges due to the deleterious effects of relapse in the setting of both schizophrenia and bipolar disorder. Almost all oral antipsychotic or mood stabilizer medications require a minimum dosing schedule to effectively treat these disorders, and some of these oral medications require regular laboratory monitoring. Moreover, some of the agents can have serious adverse effects (AEs), such as seizure or withdrawal, if stopped abruptly. Social support from family or friends may improve adherence, but many psychiatric outpatients have a smaller social support network than do patients without psychiatric illnesses.2
Long-acting injectable (LAI) antipsychotics have been available for the past 40 years. These medications have provided clinicians with an additional option for patients with schizophrenia or bipolar disorder who are nonadherent to their medication treatment plans or who desire an administration choice that is more convenient than daily oral pills.3-7 Some clinical practice guidelines recommend considering LAIs as a maintenance treatment for schizophrenia.5 Like the rest of the pharmacopoeia, these formulations have AEs, such as extrapyramidal symptoms (EPS), weight gain, and metabolic syndrome.1 The longer half-life of these drugs may make such effects difficult to reverse.
This article presents a case of the use of depot formulation paliperidone palmitate in an elderly patient with bipolar disorder who was previously on high-dose oral second generation antipsychotics. He developed severe parkinsonism during a protracted hospitalization that ended in death.
Case Presentation
Mr. W was a 68-year-old homeless white male with a history of coronary artery disease status-post coronary artery bypass surgery, obstructive sleep apnea, and bipolar 1 disorder who presented to a large rural VAMC emergency department (ED) as a transfer from an outside hospital (OSH). He originally presented at the OSH for vomiting and diarrhea, but while there, he was placed under involuntary psychiatric commitment. The involuntary commitment form noted him to be tangential and disorganized; he was found walking about the ED without clothes. During the initial psychiatry interview, the clinician noted a disorganized thought process. When asked about circumstances leading to admission, he stated he was “a scuba diver, pilot, actor, submarine commander.” He also reported he had given “seminars to 6,000 people,” he held a psychology degree, and he came from a family that owned part of the island of Kodiak, Alaska. Mr. W stated he had no mental health history and believed psychiatry was witchcraft. He reported having no hallucinations and stated he heard the voice of god. He also reported to have met god multiple times and to have been married to a supermodel.
Mr. W’s chart demonstrated a history of mental illness over 30 years and that he previously was prescribed psychiatric medications. He had multiple inpatient psychiatric admissions with grandiose ideations, disorganized behaviors, and hypersexuality. He had been prescribed quetiapine, divalproex, lithium, carbamazepine, and lorazepam. He was formally diagnosed in the past with bipolar 1 disorder. There also was a family history of psychiatric illness. His mother had received electroconvulsive therapy, and both parents had alcohol substance use disorder.
Mr. W had been homeless for 20 years and had several psychiatric admissions during this period. Mr. W also had chronic difficulty with obtaining food and taking medications as prescribed. Sometimes he would only be able to eat 1 to 2 meals per day. He often changed location and had lived in at least 7 different states. Currently, he was estranged from his family. About 19 years ago, his sister reported that the veteran had told her that he was Jesus Christ, per clinical records. His estranged sister’s statement was corroborated by past psychology consult records citing episodes of the patient hearing god 30 and 26 years before the current admission. His second ex-wife cited inappropriate sexual behavior in front of their children. He had difficulty in school, failed at least 2 grades, and joined the U.S. Navy in tenth grade. A Neurobehavioral Cognitive Status Examination given 19 years ago showed mild impairment on attention and severe impairment in memory.
The physical examination on presentation to the OSH was unremarkable. Mr. W did not cooperate with formal neurocognitive testing, and he consistently made errors during orientation testing. Complete blood count from a OSH ED laboratory test was remarkable for a mild pancytopenia with a leukocyte count of 3,100 cells/mcL, hemoglobin 13.1 g/dL, and hematocrit 38.4%. Red cell distribution width was within normal limits at 13.5%. Stool cultures showed normal fecal flora and no salmonella, shigella, or campylobacter. Thyroid-stimulating hormone (TSH) was slightly elevated at 5.32 U/mL. An electrocardiogram showed a QTc interval of 412 ms. A computerized tomography scan of his head showed no acute intracranial abnormality along with chronic ischemic changes in the brain (Table 1). Presumed cause of his nausea and diarrhea was viral gastroenteritis likely acquired at a homeless shelter.
Once stabilized, Mr. W was admitted to the VA hospital inpatient psychiatry unit under involuntary commitment for acute mania. Risperidone 0.25 mg orally twice a day was started for mood stabilization and psychosis along with trazodone 50 mg orally as needed for insomnia. Despite upward titration and change in frequency of the risperidone dose, Mr. W’s manic episode persisted. He remained on the psychiatric floor for 2 months (Figure). His TSH and free T4 were monitored during his stay, and levothyroxine was started. Risperidone was titrated to 8 mg/d. Mr. W’s Young Mania Rating Scale (YMRS) score decreased from 30 to 24. Mr. W had a mild improvement in irritability and speech rate but little change in elevated mood and delusional content.
He continued to endorse “speaking to god 16 times” even at the highest risperidone dose. The treatment team prescribed dissolvable risperidone tablets secondary to diversion concerns. In addition, the team added benztropine 0.5 mg once a day after observing a stooped posture and decreased arm swing. Mr. W noted risperidone made him “lethargic” and that his “body did not need” it. After 1 month of treatment with risperidone, the treatment team decided to cross taper the veteran from risperidone to a combination of olanzapine and divalproex secondary to inadequate treatment response.
The inpatient team started Mr. W on oral disintegrating tablets of olanzapine 5 mg once a day, and oral divalproex 1,000 mg once a day. An intramuscular backup of olanzapine was made available if oral medication was refused. Divalproex was titrated to 1,250 mg once a day to target a serum level of 61.7 µg/mL, and olanzapine was titrated to 10 mg once a day. After 9 days, the veteran showed moderate improvement in mania symptoms with a YMRS score < 20, indicating the absence of mania. However, the veteran made it very clear that he would stop taking the prescribed medication on discharge. The team elected to initiate a LAI.
The veteran received his first injection of the LAI psychiatric medication paliperidone palmitate 234 mg and a second 156-mg injection of the same medication 1 week later as per loading protocol. He was concurrently on daily oral divalproex 1,250 mg and olanzapine 10 mg. Mr. W continued to note he felt sedated during this period; his sedation worsened after the second injection. He also began to forget the location of his room and developed mumbled speech. His gait deteriorated to where he required a walker 6 days after injection and a wheelchair 3 days later. He became incontinent of urine and feces. Mr. W exhibited masked facies with severe drooling. This eventually progressed to difficulty swallowing. At the advice of speech pathology, he was downgraded to a pureed and nectar-thick liquid diet. He required assistance with meals.
Because of his sedation and parkinsonism symptoms, he was tapered off both olanzapine and divalproex. His last dose of olanzapine was on the date of his first injection and last dose of divalproex was 15 days after the second injection. The benztropine, which was originally given to counteract the effects of risperidone monotherapy, was discontinued over concern of anticholinergic load and sedation. The neurology consultant recommended carbidopa 25 mg and levodopa 100 mg 3 times per day for treatment of parkinsonism symptoms. Mr. W was only able to take 1 dose because of trouble swallowing. Twenty days after his second injection, a rapid response team (local clinical team 1 step below a code team) was called as Mr. W was unusually lethargic and unable to eat. He was then transferred to the medical floor.
While on the medical floor, dobhoff tube access was established for nutrition and to allow administration of carbidopa and levodopa. Mr. W could still speak at this time and was distraught. He stated, “I don’t know why god would do this to me.” Further workup was performed to look for other etiologies of the patient’s change in status. Creatinine kinase testing, lumbar puncture with cerebral spinal fluid (CSF) bacterial culture, CSF cryptococcal testing, and syphilis antigens were all negative. Magnetic resonance imaging of the brain demonstrated diffuse cerebral atrophy with widened cistern and sulci resulting in ex vacuo dilatation.
Neurology thought that the ventriculomegaly did not have features of normal pressure hydrocephalus and was secondary to chronic ischemic demyelination caused by chronic malnutrition. During follow-up visits, the veteran was less and less verbal. It progressed to where he answered questions only in grunts. Eight days after transfer to the medical floor, Mr. W was noted to have his neck locked in a laterally rotated position with clonus of the sternocleidomastoid. Due to concern about possibility of neck dystonia and the poor adherence of the patient with carbidopa and levodopa given orally, the psychiatric team made the recommendation to start benztropine 1 mg given twice a day, delivered via the dobhoff tube to treat both the parkinsonism and dystonia. The following day Mr. W failed a repeat swallow study and was no longer allowed to receive anything orally.
Mild icterus and jaundice were noted on physical examination along with transaminitis and elevated bilirubin. He developed a fever. Thirteen days after transfer to the medical floor, blood cultures revealed Klebsiella septicemia. Benztropine was discontinued at this time because of concern the medication was causing or exacerbating the fever. While being treated for Klebsiella sepsis, the psychiatry team addressed his continued hypophonia, inability to ambulate, masked facies, and neck dystonia with diphenhydramine 50 mg given intramuscular (IM) twice per day.
Mr. W developed several more iatrogenic complications near this time, including urinary tract infection septicemia and acute hypoxic respiratory failure with lung infiltrate on X-ray, requiring ventilator support. His clinical status led to a number of transfers in and out of the medical intensive care unit (MICU). During this time, his parkinsonism symptoms were managed through a combination of carbidopa and levodopa and amantadine. Cervical dystonia was managed with botulism toxin injections. Mr. W spent 6 weeks in the MICU until the decision was made to terminate life support, and he was taken off the ventilator. He died shortly thereafter. Autopsy findings suggested that Mr. W had severe Alzheimer disease.
Discussion
Following the IM injection of paliperidone palmitate, Mr. W had a complicated hospital stay resulting in his demise from sepsis and multiorgan failure. Severe immobilization, rigidity, and dystonia prevented Mr. W from conducting activities of daily living, which resulted in invasive interventions, such as continued foley catheterization. His sepsis was likely secondary to aspiration, catheterization, and eventual ventilation—all iatrogenic complications. Previous estimates in the U.S. have suggested a total of 225,000 deaths per year from iatrogenic causes.8
There are several areas of concern. Clearly, Mr. W had severe illness that greatly affected his life. He was estranged from family and had endured a 2-decade period of homelessness. He deserved effective treatment for his psychiatric illness to relieve his suffering. His long period of mental illness without effective treatment very likely biased the initial treatment team toward an aggressive approach.
Fragmented Care
The prolonged hospital stay and multiple complications directly led to fragmentation in Mr. W’s care. He was hospitalized for months on 3 different main services: psychiatry, medicine, and the MICU. Even when he remained on the same service, the primary members of his treatment team changed every few weeks. Many different specialties were consulted and reconsulted. Members of the specialty consult teams changed throughout the hospitalization as well. Given the nature of the local clinical administration, Mr. W likely received the most consistent team members from the attendings on the psychiatry consult-liaison service (who do not rotate) and from a local subspecialty delirium consult team (all members stay consistent except pharmacy residents).
Documentation of clinical reasoning behind treatment decisions was not ideal and occasionally lacking. This led to a tendency to “reinvent the wheel” with Mr. W’s treatment approach every few weeks. It was not until Mr. W had spent a significant amount of time on the medical service that an interdisciplinary treatment team meeting involving medicine, psychiatry, nursing, delirium, and neurology experts occurred. Although the interdisciplinary meeting helped by reviewing the hospital course, agreeing on a likely cause of the symptoms, and creating a treatment plan going forward, Mr. W was not able to recover.
Even when team members were stable, communication in a timely fashion did not always occur. At several points, expert recommendations were delayed by a day or more. Difficulties in treatment implementation were not communicated back to the specialty teams. The most significant example was a delay in recognition when Mr. W could no longer take oral pills secondary to the parkinsonism. Many days passed before an alternative liquid or dissolved medication was recommended on 2 separate occasions.
Subspecialty Consult
Addressing these documentation, communication, and transition challenges is neither easy nor unique to this large rural VA medical center. The authors have attempted to address this in the local system with the creation of a delirium team subspecialty consult service. Team members do not rotate and are able to follow patients throughout their hospital course. At the time of Mr. W’s hospitalization, the team included representatives from nursing, psychiatry, and occasionally pharmacy. Since then, it has expanded to include geriatrics and medicine. In addition to delirium being a marker for complex patients at risk for hospital complications, medical reasons for an extended length of stay could serve as a trigger for a referral to such a team of experts. In Mr. W’s case, that could have led to interdisciplinary consultation up to 2 months before it occurred. This may have led to a much better outcome.
Secondary parkinsonism is most notable with the typical antipychotics. The prevalence can vary between 50% and 75% and may be higher within the elderly population. However, all antipsychotics have a chance of demonstrating EPS. Risperidone has a low incidence at low doses; studies have shown dose-related parkinsonism at doses of 2 to 6 mg/d. Significant risk of parkinsonism is further exacerbated when drug-drug interactions are considered.9 Concurrently receiving 2 antipsychotics, olanzapine and paliperidone, initially caused the EPS. The veteran’s cerebral atrophy from significant malnutrition related to chronic homelessness, and the presence of Alzheimer disease only identified postmortem exacerbated this AE. Further complicating the management of the EPS, paliperidone palmitate has a long half-life of 25 to 49 days.9 Simply discontinuing the medication did not remove it from Mr. W’s system. Paliperidone would have continued to be present for months.
Conclusion
In this case, aggressive changes in the antipsychotic medications in a short period led to Mr. W effectively having 3 different agents in his system at the same time. This significantly elevated his risk of AEs, including parkinsonism. The clinician must be vigilant to further recognize the initial symptoms of parkinsonism on clinical presentation. Administration of clinical scales, such as the Simpson-Angus Extrapyramidal Side Effect, can help in these situations.10 Malnutrition and increased age can predispose patients to neurolepticAEs, so treatment teams should exercise caution when administering antipsychotics in such a population. Pharmacokinetic changes in all major organ systems from aging result in higher and more variable drug concentrations. This leads to an increased sensitivity to drugs and AEs.9
Given the increasing geriatric patient population in the U.S., treating mania in the elderly will become more common. Providers should carefully consider the risks vs benefit ratio for each individual because a serious adverse reaction may result in detrimental consequences. Even with severe symptoms leading to a bias toward an aggressive approach, it may be better to “start low and go slow.” Early inclusion of interdisciplinary expertise should be sought in complex cases.
Many patients with psychiatric illness have difficulty with medication adherence. Patients with impaired reality testing especially are at risk.
Keck and McElroy evaluated 141 patients who were initially hospitalized for bipolar disorder prospectively over 1 year to assess adherence with medication. During the follow-up period, 71 patients (51%) were partially or totally nonadherent with medication as prescribed. The most commonly cited reason for nonadherence was denial of need.1
Clinicians and patients face additional challenges due to the deleterious effects of relapse in the setting of both schizophrenia and bipolar disorder. Almost all oral antipsychotic or mood stabilizer medications require a minimum dosing schedule to effectively treat these disorders, and some of these oral medications require regular laboratory monitoring. Moreover, some of the agents can have serious adverse effects (AEs), such as seizure or withdrawal, if stopped abruptly. Social support from family or friends may improve adherence, but many psychiatric outpatients have a smaller social support network than do patients without psychiatric illnesses.2
Long-acting injectable (LAI) antipsychotics have been available for the past 40 years. These medications have provided clinicians with an additional option for patients with schizophrenia or bipolar disorder who are nonadherent to their medication treatment plans or who desire an administration choice that is more convenient than daily oral pills.3-7 Some clinical practice guidelines recommend considering LAIs as a maintenance treatment for schizophrenia.5 Like the rest of the pharmacopoeia, these formulations have AEs, such as extrapyramidal symptoms (EPS), weight gain, and metabolic syndrome.1 The longer half-life of these drugs may make such effects difficult to reverse.
This article presents a case of the use of depot formulation paliperidone palmitate in an elderly patient with bipolar disorder who was previously on high-dose oral second generation antipsychotics. He developed severe parkinsonism during a protracted hospitalization that ended in death.
Case Presentation
Mr. W was a 68-year-old homeless white male with a history of coronary artery disease status-post coronary artery bypass surgery, obstructive sleep apnea, and bipolar 1 disorder who presented to a large rural VAMC emergency department (ED) as a transfer from an outside hospital (OSH). He originally presented at the OSH for vomiting and diarrhea, but while there, he was placed under involuntary psychiatric commitment. The involuntary commitment form noted him to be tangential and disorganized; he was found walking about the ED without clothes. During the initial psychiatry interview, the clinician noted a disorganized thought process. When asked about circumstances leading to admission, he stated he was “a scuba diver, pilot, actor, submarine commander.” He also reported he had given “seminars to 6,000 people,” he held a psychology degree, and he came from a family that owned part of the island of Kodiak, Alaska. Mr. W stated he had no mental health history and believed psychiatry was witchcraft. He reported having no hallucinations and stated he heard the voice of god. He also reported to have met god multiple times and to have been married to a supermodel.
Mr. W’s chart demonstrated a history of mental illness over 30 years and that he previously was prescribed psychiatric medications. He had multiple inpatient psychiatric admissions with grandiose ideations, disorganized behaviors, and hypersexuality. He had been prescribed quetiapine, divalproex, lithium, carbamazepine, and lorazepam. He was formally diagnosed in the past with bipolar 1 disorder. There also was a family history of psychiatric illness. His mother had received electroconvulsive therapy, and both parents had alcohol substance use disorder.
Mr. W had been homeless for 20 years and had several psychiatric admissions during this period. Mr. W also had chronic difficulty with obtaining food and taking medications as prescribed. Sometimes he would only be able to eat 1 to 2 meals per day. He often changed location and had lived in at least 7 different states. Currently, he was estranged from his family. About 19 years ago, his sister reported that the veteran had told her that he was Jesus Christ, per clinical records. His estranged sister’s statement was corroborated by past psychology consult records citing episodes of the patient hearing god 30 and 26 years before the current admission. His second ex-wife cited inappropriate sexual behavior in front of their children. He had difficulty in school, failed at least 2 grades, and joined the U.S. Navy in tenth grade. A Neurobehavioral Cognitive Status Examination given 19 years ago showed mild impairment on attention and severe impairment in memory.
The physical examination on presentation to the OSH was unremarkable. Mr. W did not cooperate with formal neurocognitive testing, and he consistently made errors during orientation testing. Complete blood count from a OSH ED laboratory test was remarkable for a mild pancytopenia with a leukocyte count of 3,100 cells/mcL, hemoglobin 13.1 g/dL, and hematocrit 38.4%. Red cell distribution width was within normal limits at 13.5%. Stool cultures showed normal fecal flora and no salmonella, shigella, or campylobacter. Thyroid-stimulating hormone (TSH) was slightly elevated at 5.32 U/mL. An electrocardiogram showed a QTc interval of 412 ms. A computerized tomography scan of his head showed no acute intracranial abnormality along with chronic ischemic changes in the brain (Table 1). Presumed cause of his nausea and diarrhea was viral gastroenteritis likely acquired at a homeless shelter.
Once stabilized, Mr. W was admitted to the VA hospital inpatient psychiatry unit under involuntary commitment for acute mania. Risperidone 0.25 mg orally twice a day was started for mood stabilization and psychosis along with trazodone 50 mg orally as needed for insomnia. Despite upward titration and change in frequency of the risperidone dose, Mr. W’s manic episode persisted. He remained on the psychiatric floor for 2 months (Figure). His TSH and free T4 were monitored during his stay, and levothyroxine was started. Risperidone was titrated to 8 mg/d. Mr. W’s Young Mania Rating Scale (YMRS) score decreased from 30 to 24. Mr. W had a mild improvement in irritability and speech rate but little change in elevated mood and delusional content.
He continued to endorse “speaking to god 16 times” even at the highest risperidone dose. The treatment team prescribed dissolvable risperidone tablets secondary to diversion concerns. In addition, the team added benztropine 0.5 mg once a day after observing a stooped posture and decreased arm swing. Mr. W noted risperidone made him “lethargic” and that his “body did not need” it. After 1 month of treatment with risperidone, the treatment team decided to cross taper the veteran from risperidone to a combination of olanzapine and divalproex secondary to inadequate treatment response.
The inpatient team started Mr. W on oral disintegrating tablets of olanzapine 5 mg once a day, and oral divalproex 1,000 mg once a day. An intramuscular backup of olanzapine was made available if oral medication was refused. Divalproex was titrated to 1,250 mg once a day to target a serum level of 61.7 µg/mL, and olanzapine was titrated to 10 mg once a day. After 9 days, the veteran showed moderate improvement in mania symptoms with a YMRS score < 20, indicating the absence of mania. However, the veteran made it very clear that he would stop taking the prescribed medication on discharge. The team elected to initiate a LAI.
The veteran received his first injection of the LAI psychiatric medication paliperidone palmitate 234 mg and a second 156-mg injection of the same medication 1 week later as per loading protocol. He was concurrently on daily oral divalproex 1,250 mg and olanzapine 10 mg. Mr. W continued to note he felt sedated during this period; his sedation worsened after the second injection. He also began to forget the location of his room and developed mumbled speech. His gait deteriorated to where he required a walker 6 days after injection and a wheelchair 3 days later. He became incontinent of urine and feces. Mr. W exhibited masked facies with severe drooling. This eventually progressed to difficulty swallowing. At the advice of speech pathology, he was downgraded to a pureed and nectar-thick liquid diet. He required assistance with meals.
Because of his sedation and parkinsonism symptoms, he was tapered off both olanzapine and divalproex. His last dose of olanzapine was on the date of his first injection and last dose of divalproex was 15 days after the second injection. The benztropine, which was originally given to counteract the effects of risperidone monotherapy, was discontinued over concern of anticholinergic load and sedation. The neurology consultant recommended carbidopa 25 mg and levodopa 100 mg 3 times per day for treatment of parkinsonism symptoms. Mr. W was only able to take 1 dose because of trouble swallowing. Twenty days after his second injection, a rapid response team (local clinical team 1 step below a code team) was called as Mr. W was unusually lethargic and unable to eat. He was then transferred to the medical floor.
While on the medical floor, dobhoff tube access was established for nutrition and to allow administration of carbidopa and levodopa. Mr. W could still speak at this time and was distraught. He stated, “I don’t know why god would do this to me.” Further workup was performed to look for other etiologies of the patient’s change in status. Creatinine kinase testing, lumbar puncture with cerebral spinal fluid (CSF) bacterial culture, CSF cryptococcal testing, and syphilis antigens were all negative. Magnetic resonance imaging of the brain demonstrated diffuse cerebral atrophy with widened cistern and sulci resulting in ex vacuo dilatation.
Neurology thought that the ventriculomegaly did not have features of normal pressure hydrocephalus and was secondary to chronic ischemic demyelination caused by chronic malnutrition. During follow-up visits, the veteran was less and less verbal. It progressed to where he answered questions only in grunts. Eight days after transfer to the medical floor, Mr. W was noted to have his neck locked in a laterally rotated position with clonus of the sternocleidomastoid. Due to concern about possibility of neck dystonia and the poor adherence of the patient with carbidopa and levodopa given orally, the psychiatric team made the recommendation to start benztropine 1 mg given twice a day, delivered via the dobhoff tube to treat both the parkinsonism and dystonia. The following day Mr. W failed a repeat swallow study and was no longer allowed to receive anything orally.
Mild icterus and jaundice were noted on physical examination along with transaminitis and elevated bilirubin. He developed a fever. Thirteen days after transfer to the medical floor, blood cultures revealed Klebsiella septicemia. Benztropine was discontinued at this time because of concern the medication was causing or exacerbating the fever. While being treated for Klebsiella sepsis, the psychiatry team addressed his continued hypophonia, inability to ambulate, masked facies, and neck dystonia with diphenhydramine 50 mg given intramuscular (IM) twice per day.
Mr. W developed several more iatrogenic complications near this time, including urinary tract infection septicemia and acute hypoxic respiratory failure with lung infiltrate on X-ray, requiring ventilator support. His clinical status led to a number of transfers in and out of the medical intensive care unit (MICU). During this time, his parkinsonism symptoms were managed through a combination of carbidopa and levodopa and amantadine. Cervical dystonia was managed with botulism toxin injections. Mr. W spent 6 weeks in the MICU until the decision was made to terminate life support, and he was taken off the ventilator. He died shortly thereafter. Autopsy findings suggested that Mr. W had severe Alzheimer disease.
Discussion
Following the IM injection of paliperidone palmitate, Mr. W had a complicated hospital stay resulting in his demise from sepsis and multiorgan failure. Severe immobilization, rigidity, and dystonia prevented Mr. W from conducting activities of daily living, which resulted in invasive interventions, such as continued foley catheterization. His sepsis was likely secondary to aspiration, catheterization, and eventual ventilation—all iatrogenic complications. Previous estimates in the U.S. have suggested a total of 225,000 deaths per year from iatrogenic causes.8
There are several areas of concern. Clearly, Mr. W had severe illness that greatly affected his life. He was estranged from family and had endured a 2-decade period of homelessness. He deserved effective treatment for his psychiatric illness to relieve his suffering. His long period of mental illness without effective treatment very likely biased the initial treatment team toward an aggressive approach.
Fragmented Care
The prolonged hospital stay and multiple complications directly led to fragmentation in Mr. W’s care. He was hospitalized for months on 3 different main services: psychiatry, medicine, and the MICU. Even when he remained on the same service, the primary members of his treatment team changed every few weeks. Many different specialties were consulted and reconsulted. Members of the specialty consult teams changed throughout the hospitalization as well. Given the nature of the local clinical administration, Mr. W likely received the most consistent team members from the attendings on the psychiatry consult-liaison service (who do not rotate) and from a local subspecialty delirium consult team (all members stay consistent except pharmacy residents).
Documentation of clinical reasoning behind treatment decisions was not ideal and occasionally lacking. This led to a tendency to “reinvent the wheel” with Mr. W’s treatment approach every few weeks. It was not until Mr. W had spent a significant amount of time on the medical service that an interdisciplinary treatment team meeting involving medicine, psychiatry, nursing, delirium, and neurology experts occurred. Although the interdisciplinary meeting helped by reviewing the hospital course, agreeing on a likely cause of the symptoms, and creating a treatment plan going forward, Mr. W was not able to recover.
Even when team members were stable, communication in a timely fashion did not always occur. At several points, expert recommendations were delayed by a day or more. Difficulties in treatment implementation were not communicated back to the specialty teams. The most significant example was a delay in recognition when Mr. W could no longer take oral pills secondary to the parkinsonism. Many days passed before an alternative liquid or dissolved medication was recommended on 2 separate occasions.
Subspecialty Consult
Addressing these documentation, communication, and transition challenges is neither easy nor unique to this large rural VA medical center. The authors have attempted to address this in the local system with the creation of a delirium team subspecialty consult service. Team members do not rotate and are able to follow patients throughout their hospital course. At the time of Mr. W’s hospitalization, the team included representatives from nursing, psychiatry, and occasionally pharmacy. Since then, it has expanded to include geriatrics and medicine. In addition to delirium being a marker for complex patients at risk for hospital complications, medical reasons for an extended length of stay could serve as a trigger for a referral to such a team of experts. In Mr. W’s case, that could have led to interdisciplinary consultation up to 2 months before it occurred. This may have led to a much better outcome.
Secondary parkinsonism is most notable with the typical antipychotics. The prevalence can vary between 50% and 75% and may be higher within the elderly population. However, all antipsychotics have a chance of demonstrating EPS. Risperidone has a low incidence at low doses; studies have shown dose-related parkinsonism at doses of 2 to 6 mg/d. Significant risk of parkinsonism is further exacerbated when drug-drug interactions are considered.9 Concurrently receiving 2 antipsychotics, olanzapine and paliperidone, initially caused the EPS. The veteran’s cerebral atrophy from significant malnutrition related to chronic homelessness, and the presence of Alzheimer disease only identified postmortem exacerbated this AE. Further complicating the management of the EPS, paliperidone palmitate has a long half-life of 25 to 49 days.9 Simply discontinuing the medication did not remove it from Mr. W’s system. Paliperidone would have continued to be present for months.
Conclusion
In this case, aggressive changes in the antipsychotic medications in a short period led to Mr. W effectively having 3 different agents in his system at the same time. This significantly elevated his risk of AEs, including parkinsonism. The clinician must be vigilant to further recognize the initial symptoms of parkinsonism on clinical presentation. Administration of clinical scales, such as the Simpson-Angus Extrapyramidal Side Effect, can help in these situations.10 Malnutrition and increased age can predispose patients to neurolepticAEs, so treatment teams should exercise caution when administering antipsychotics in such a population. Pharmacokinetic changes in all major organ systems from aging result in higher and more variable drug concentrations. This leads to an increased sensitivity to drugs and AEs.9
Given the increasing geriatric patient population in the U.S., treating mania in the elderly will become more common. Providers should carefully consider the risks vs benefit ratio for each individual because a serious adverse reaction may result in detrimental consequences. Even with severe symptoms leading to a bias toward an aggressive approach, it may be better to “start low and go slow.” Early inclusion of interdisciplinary expertise should be sought in complex cases.
1. Keck PE Jr, McElroy SL, Strakowski SM, Bourne ML, West SA. Compliance with maintenance treatment in bipolar disorder. Psychopharmacol Bull. 1997;33(1):87-91.
2. Henderson S, Duncan-Jones P, McAuley H, Ritchie K. The patient’s primary group. Br J Psychiatry. 1978;132:74-86.
3. Buoli M, Ciappolino V, Altamura AC. Paliperidone palmitate depot in the long-term treatment of psychotic bipolar disorder: a case series. Clin Neuropharmacol. 2015;38(5):209-211.
4. Chou YH, Chu PC, Wu SW, et al. A systematic review and experts’ consensus for long-acting injectable antipsychotics in bipolar disorder. Clin Psychopharmacol Neurosci. 2015;13(2):121-128.
5. Kishi T, Oya K, Iwata N. Long-acting injectable antipsychotics for prevention of relapse in bipolar disorder: a systematic review and meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2016;19(9):1-10.
6. Llorca PM, Abbar M, Courtet P, Guillaume S, Lancrenon S, Samalin L. Guidelines for the use and management of long-acting injectable antipsychotics in serious mental illness. BMC Psychiatry. 2013;13:340.
7. Spanarello S, La Ferla T. The pharmacokinetics of long-acting antipsychotic medications. Curr Clin Pharmacol. 2014;9(3):310-317.
8. Starfield B. Is US health really the best in the world? JAMA. 2000;284(4):483-485.
9. Labbate LA, Fava M, Rosenbaum JF, Arana GW. Handbook of Psychiatric Drug Therapy. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.
10. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11-19.
1. Keck PE Jr, McElroy SL, Strakowski SM, Bourne ML, West SA. Compliance with maintenance treatment in bipolar disorder. Psychopharmacol Bull. 1997;33(1):87-91.
2. Henderson S, Duncan-Jones P, McAuley H, Ritchie K. The patient’s primary group. Br J Psychiatry. 1978;132:74-86.
3. Buoli M, Ciappolino V, Altamura AC. Paliperidone palmitate depot in the long-term treatment of psychotic bipolar disorder: a case series. Clin Neuropharmacol. 2015;38(5):209-211.
4. Chou YH, Chu PC, Wu SW, et al. A systematic review and experts’ consensus for long-acting injectable antipsychotics in bipolar disorder. Clin Psychopharmacol Neurosci. 2015;13(2):121-128.
5. Kishi T, Oya K, Iwata N. Long-acting injectable antipsychotics for prevention of relapse in bipolar disorder: a systematic review and meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2016;19(9):1-10.
6. Llorca PM, Abbar M, Courtet P, Guillaume S, Lancrenon S, Samalin L. Guidelines for the use and management of long-acting injectable antipsychotics in serious mental illness. BMC Psychiatry. 2013;13:340.
7. Spanarello S, La Ferla T. The pharmacokinetics of long-acting antipsychotic medications. Curr Clin Pharmacol. 2014;9(3):310-317.
8. Starfield B. Is US health really the best in the world? JAMA. 2000;284(4):483-485.
9. Labbate LA, Fava M, Rosenbaum JF, Arana GW. Handbook of Psychiatric Drug Therapy. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010.
10. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11-19.
Emergency Imaging: Left Periorbital Swelling
Case
A 3-year-old boy was brought to the ED by his parents for evaluation of left periorbital swelling. A few days prior to presentation, the child was seen at an outpatient center where he was diagnosed with preseptal cellulitis and given an oral antibiotic. However, even after receiving three doses of the antibiotic, the periorbital swelling and redness around the child’s eye worsened, prompting this visit to the ED.
Physical examination revealed edema and erythema both above and below the left eye, with associated tenderness to palpation. A contrast-enhanced maxillofacial computed tomography (CT) scan, with special attention to the orbits, was ordered; representative images are shown (Figure 1a-1c).
What is the diagnosis?
Answer
The CT images of the orbits demonstrated edema in the superficial left eyelid (white arrows, Figure 2a and 2b) and left deep orbital septum (red arrows, Figure 2a-2c). A peripherally enhancing fluid collection centered in the left nasolacrimal gland was present (red asterisks, Figure 2b and 2c) with mild mass effect on the left globe. Opacification was also noted within the paranasal sinuses (white asterisks, Figure 2a-2c). Together these findings indicated sinusitis with dacryocystitis and orbital cellulitis.
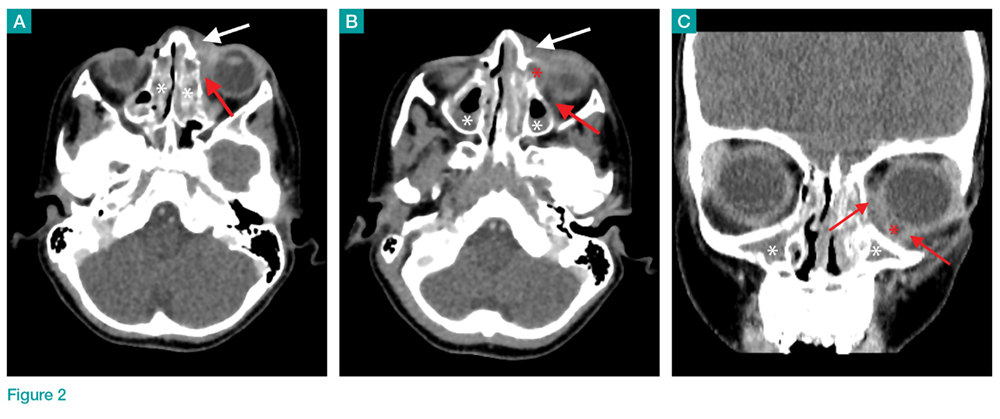
Dacryocystitis
Dacryocystitis is an infection or inflammation of the lacrimal sac, usually developing secondary to blockage of the nasolacrimal duct. Orbital cellulitis is an infection involving the contents of the orbit, including the fat and ocular muscles. Orbital cellulitis should not be confused with preseptal cellulitis, which is an infection involving the eyelid occurring posterior to the orbital septum. While both of these conditions are more common in children than in adults, preseptal cellulitis is much more common than orbital cellulitis.
Preseptal Cellulitis
Preseptal cellulitis is typically due to local trauma, local skin infection, or dacryocystitis.1 Preseptal cellulitis rarely extends into the orbit, though a minority of cases have been reported in patients with concomitant dacryocystitis.2 Orbital cellulitis most commonly results from paranasal sinus disease, particularly of the ethmoid sinus, which is only separated from the orbit by the thin lamina papyracea.3 While both preseptal cellulitis and orbital cellulitis can cause eyelid swelling and erythema, preseptal cellulitis is typically a mild condition. Orbital cellulitis, however, is a serious medical emergency that requires prompt diagnosis and treatment to avoid loss of vision and intracranial complications, such as venous thrombosis and empyema.3
Imaging Studies
Although the clinical features of orbital cellulitis (eg, proptosis, ophthalmoplegia, pain with ocular movement) can sometimes distinguish it from preseptal cellulitis, imaging studies are helpful to confirm the diagnosis.4 As previously noted, prompt recognition, diagnosis, and treatment of orbital cellulitis are essential to avoid serious complications.
Computed tomography has a high specificity and sensitivity in detecting the extension of infection into the orbit and associated complications such as subperiosteal or intracranial abscess. For patients in whom intravenous (IV) contrast is contraindicated or who wish to avoid ionizing radiation, magnetic resonance imaging is a useful alternate modality, and diffusion-weighted imaging is particularly sensitive in diagnosing abscess.5
Treatment
1. Baring DE, Hilmi OJ. An evidence based review of periorbital cellulitis. Clin Otolaryngol. 2011;36(1):57-64. doi:10.1111/j.1749-4486.2011.02258.x.
2. Kikkawa DO, Heinz GW, Martin RT, Nunery WN, Eiseman AS. Orbital cellulitis and abscess secondary to dacryocystitis. Arch Ophthalmol. 2002;120(8):1096-1099.
3. Mathew AV, Craig E, Al-Mahmoud R, et al. Paediatric post-septal and pre-septal cellulitis: 10 years’ experience at a tertiary-level children’s hospital. Br J Radiol. 2014;87(1033):20130503. doi:10.1259/bjr.20130503.
4. Rudloe TF, Harper MB, Prabhu SP, Rahbar R, Vanderveen D, Kimia AA. Acute periorbital infections: who needs emergent imaging? Pediatrics. 2010;125(4):e719-e726. doi:10.1542/peds.2009-1709.
5. Sepahdari AR, Aakalu VK, Kapur R, et al. MRI of orbital cellulitis and orbital abscess: the role of diffusion-weighted imaging. AJR Am J Roentgenol. 2009;193(3):W244-W250. doi:10.2214/AJR.08.1838.
6. Ho CF, Huang YC, Wang CJ, Chiu CH, Lin TY. Clinical analysis of computed tomography-staged orbital cellulitis in children. J Microbiol Immunol Infect. 2007;40(6):518-524.
Case
A 3-year-old boy was brought to the ED by his parents for evaluation of left periorbital swelling. A few days prior to presentation, the child was seen at an outpatient center where he was diagnosed with preseptal cellulitis and given an oral antibiotic. However, even after receiving three doses of the antibiotic, the periorbital swelling and redness around the child’s eye worsened, prompting this visit to the ED.
Physical examination revealed edema and erythema both above and below the left eye, with associated tenderness to palpation. A contrast-enhanced maxillofacial computed tomography (CT) scan, with special attention to the orbits, was ordered; representative images are shown (Figure 1a-1c).
What is the diagnosis?
Answer
The CT images of the orbits demonstrated edema in the superficial left eyelid (white arrows, Figure 2a and 2b) and left deep orbital septum (red arrows, Figure 2a-2c). A peripherally enhancing fluid collection centered in the left nasolacrimal gland was present (red asterisks, Figure 2b and 2c) with mild mass effect on the left globe. Opacification was also noted within the paranasal sinuses (white asterisks, Figure 2a-2c). Together these findings indicated sinusitis with dacryocystitis and orbital cellulitis.
Dacryocystitis
Dacryocystitis is an infection or inflammation of the lacrimal sac, usually developing secondary to blockage of the nasolacrimal duct. Orbital cellulitis is an infection involving the contents of the orbit, including the fat and ocular muscles. Orbital cellulitis should not be confused with preseptal cellulitis, which is an infection involving the eyelid occurring posterior to the orbital septum. While both of these conditions are more common in children than in adults, preseptal cellulitis is much more common than orbital cellulitis.
Preseptal Cellulitis
Preseptal cellulitis is typically due to local trauma, local skin infection, or dacryocystitis.1 Preseptal cellulitis rarely extends into the orbit, though a minority of cases have been reported in patients with concomitant dacryocystitis.2 Orbital cellulitis most commonly results from paranasal sinus disease, particularly of the ethmoid sinus, which is only separated from the orbit by the thin lamina papyracea.3 While both preseptal cellulitis and orbital cellulitis can cause eyelid swelling and erythema, preseptal cellulitis is typically a mild condition. Orbital cellulitis, however, is a serious medical emergency that requires prompt diagnosis and treatment to avoid loss of vision and intracranial complications, such as venous thrombosis and empyema.3
Imaging Studies
Although the clinical features of orbital cellulitis (eg, proptosis, ophthalmoplegia, pain with ocular movement) can sometimes distinguish it from preseptal cellulitis, imaging studies are helpful to confirm the diagnosis.4 As previously noted, prompt recognition, diagnosis, and treatment of orbital cellulitis are essential to avoid serious complications.
Computed tomography has a high specificity and sensitivity in detecting the extension of infection into the orbit and associated complications such as subperiosteal or intracranial abscess. For patients in whom intravenous (IV) contrast is contraindicated or who wish to avoid ionizing radiation, magnetic resonance imaging is a useful alternate modality, and diffusion-weighted imaging is particularly sensitive in diagnosing abscess.5
Treatment
Case
A 3-year-old boy was brought to the ED by his parents for evaluation of left periorbital swelling. A few days prior to presentation, the child was seen at an outpatient center where he was diagnosed with preseptal cellulitis and given an oral antibiotic. However, even after receiving three doses of the antibiotic, the periorbital swelling and redness around the child’s eye worsened, prompting this visit to the ED.
Physical examination revealed edema and erythema both above and below the left eye, with associated tenderness to palpation. A contrast-enhanced maxillofacial computed tomography (CT) scan, with special attention to the orbits, was ordered; representative images are shown (Figure 1a-1c).
What is the diagnosis?
Answer
The CT images of the orbits demonstrated edema in the superficial left eyelid (white arrows, Figure 2a and 2b) and left deep orbital septum (red arrows, Figure 2a-2c). A peripherally enhancing fluid collection centered in the left nasolacrimal gland was present (red asterisks, Figure 2b and 2c) with mild mass effect on the left globe. Opacification was also noted within the paranasal sinuses (white asterisks, Figure 2a-2c). Together these findings indicated sinusitis with dacryocystitis and orbital cellulitis.
Dacryocystitis
Dacryocystitis is an infection or inflammation of the lacrimal sac, usually developing secondary to blockage of the nasolacrimal duct. Orbital cellulitis is an infection involving the contents of the orbit, including the fat and ocular muscles. Orbital cellulitis should not be confused with preseptal cellulitis, which is an infection involving the eyelid occurring posterior to the orbital septum. While both of these conditions are more common in children than in adults, preseptal cellulitis is much more common than orbital cellulitis.
Preseptal Cellulitis
Preseptal cellulitis is typically due to local trauma, local skin infection, or dacryocystitis.1 Preseptal cellulitis rarely extends into the orbit, though a minority of cases have been reported in patients with concomitant dacryocystitis.2 Orbital cellulitis most commonly results from paranasal sinus disease, particularly of the ethmoid sinus, which is only separated from the orbit by the thin lamina papyracea.3 While both preseptal cellulitis and orbital cellulitis can cause eyelid swelling and erythema, preseptal cellulitis is typically a mild condition. Orbital cellulitis, however, is a serious medical emergency that requires prompt diagnosis and treatment to avoid loss of vision and intracranial complications, such as venous thrombosis and empyema.3
Imaging Studies
Although the clinical features of orbital cellulitis (eg, proptosis, ophthalmoplegia, pain with ocular movement) can sometimes distinguish it from preseptal cellulitis, imaging studies are helpful to confirm the diagnosis.4 As previously noted, prompt recognition, diagnosis, and treatment of orbital cellulitis are essential to avoid serious complications.
Computed tomography has a high specificity and sensitivity in detecting the extension of infection into the orbit and associated complications such as subperiosteal or intracranial abscess. For patients in whom intravenous (IV) contrast is contraindicated or who wish to avoid ionizing radiation, magnetic resonance imaging is a useful alternate modality, and diffusion-weighted imaging is particularly sensitive in diagnosing abscess.5
Treatment
1. Baring DE, Hilmi OJ. An evidence based review of periorbital cellulitis. Clin Otolaryngol. 2011;36(1):57-64. doi:10.1111/j.1749-4486.2011.02258.x.
2. Kikkawa DO, Heinz GW, Martin RT, Nunery WN, Eiseman AS. Orbital cellulitis and abscess secondary to dacryocystitis. Arch Ophthalmol. 2002;120(8):1096-1099.
3. Mathew AV, Craig E, Al-Mahmoud R, et al. Paediatric post-septal and pre-septal cellulitis: 10 years’ experience at a tertiary-level children’s hospital. Br J Radiol. 2014;87(1033):20130503. doi:10.1259/bjr.20130503.
4. Rudloe TF, Harper MB, Prabhu SP, Rahbar R, Vanderveen D, Kimia AA. Acute periorbital infections: who needs emergent imaging? Pediatrics. 2010;125(4):e719-e726. doi:10.1542/peds.2009-1709.
5. Sepahdari AR, Aakalu VK, Kapur R, et al. MRI of orbital cellulitis and orbital abscess: the role of diffusion-weighted imaging. AJR Am J Roentgenol. 2009;193(3):W244-W250. doi:10.2214/AJR.08.1838.
6. Ho CF, Huang YC, Wang CJ, Chiu CH, Lin TY. Clinical analysis of computed tomography-staged orbital cellulitis in children. J Microbiol Immunol Infect. 2007;40(6):518-524.
1. Baring DE, Hilmi OJ. An evidence based review of periorbital cellulitis. Clin Otolaryngol. 2011;36(1):57-64. doi:10.1111/j.1749-4486.2011.02258.x.
2. Kikkawa DO, Heinz GW, Martin RT, Nunery WN, Eiseman AS. Orbital cellulitis and abscess secondary to dacryocystitis. Arch Ophthalmol. 2002;120(8):1096-1099.
3. Mathew AV, Craig E, Al-Mahmoud R, et al. Paediatric post-septal and pre-septal cellulitis: 10 years’ experience at a tertiary-level children’s hospital. Br J Radiol. 2014;87(1033):20130503. doi:10.1259/bjr.20130503.
4. Rudloe TF, Harper MB, Prabhu SP, Rahbar R, Vanderveen D, Kimia AA. Acute periorbital infections: who needs emergent imaging? Pediatrics. 2010;125(4):e719-e726. doi:10.1542/peds.2009-1709.
5. Sepahdari AR, Aakalu VK, Kapur R, et al. MRI of orbital cellulitis and orbital abscess: the role of diffusion-weighted imaging. AJR Am J Roentgenol. 2009;193(3):W244-W250. doi:10.2214/AJR.08.1838.
6. Ho CF, Huang YC, Wang CJ, Chiu CH, Lin TY. Clinical analysis of computed tomography-staged orbital cellulitis in children. J Microbiol Immunol Infect. 2007;40(6):518-524.
Case Studies in Toxicology: Always Cook Your Boba
Case
A 45-year-old Chinese man with no known medical history presented to the ED with right-sided facial spasm and cheek swelling, which began immediately after he bit into a piece of taro root, approximately 2 hours prior to presentation. The patient stated that the root was an ingredient in a soup that a relative had made. According to the patient, after biting into the root, he immediately experienced a burning pain on the right side of his mouth. He further noted that he swallowed less than two bites of the root and stopped eating because the act of chewing was too painful.
Initial vital signs at presentation were: blood pressure, 140/100 mm Hg; heart rate, 84 beats/min; respiratory rate, 14 beats/min; and temperature, 97.6°F. Oxygen saturation was 98% on room air. The patient’s physical examination was remarkable for pain upon opening the mouth, as well as right-sided cheek and lip swelling and tenderness. The tongue and oropharynx were not erythematous or swollen. The patient was only able to speak in short sentences, secondary to oropharyngeal pain, but he was in no respiratory distress. No urticaria, pruritus, wheezing, or stridor was present.
During the patient’s workup, his 40-year-old wife also presented to the same ED for evaluation of burning pain and spasm on the left side of her mouth, which she stated also developed immediately after she bit into a piece of taro root contained in the same soup as that ingested by the patient.
The wife’s vital signs were unremarkable, and she was in no respiratory distress. Her physical examination was remarkable only for left-sided cheek and lip swelling and tenderness, associated with an erythematous oropharynx and pain with speaking.
What is taro? What are the manifestations of taro toxicity?
Taro commonly refers to plants from the Araceae family, usually Colocasia esculenta.1 Taro is ubiquitous in Southern Asia and Southeast India. It is a widely naturalized and perennial tropical plant primarily grown as a root vegetable, and is a common flavor in boba (bubble) tea. All members of Araceae contain calcium oxalate crystals in the form of raphides, sharp needle-shaped crystals packaged in idioblasts and contained within the waxy leaf.2 Pressure on the idioblasts, such as from mastication, triggers the release of the raphides. The needles pierce the surface of any tissue with which they come into contact, creating a gateway for proteolytic enzymes to enter the consumer.3 The leaves and root of Araceae must be cooked before eating to inactivate the raphides.
Oral exposure to uncooked taro leaves or taro root can result in mouth irritation and swelling that can progress to angioedema and airway obstruction. Although the traditional method of removing taro raphides is to soak the root in cold water overnight,4,5 this does not fully remove all of the raphides. Instead, taro root should be thoroughly cooked in boiling water to draw-out oxalates from the root into the cooking water, which must then be discarded. Consuming taro with warm milk also reduces the effect of the oxalates by about 80%.6
Many other plants of the Araceae family, such as Dieffenbachia (dumbcane), share similar toxicity and are commonly kept in the home and office.
Patients with oral exposure to taro may experience a delayed (also termed biphasic) anaphylactic reaction, ie, the development of anaphylactic symptoms more than 4 hours after the inciting event. Delayed anaphylaxis is distinct from delayed hypersensitivity, though both may be immunoglobulin E-mediated. Delayed hypersensitivity presents later (2-14 days) and with less immediately life-threatening effects, most commonly dermatitis (eg, poison ivy dermatitis).
While both of the patients in this case presented with mild symptoms, life-threatening angioedema of the oropharynx, anaphylaxis, and hypocalcemia have been reported7,8 and should be considered in any symptomatic patient with exposure to taro.
What is the differential diagnosis of plant-related mouth pain?
The oral mucosa is composed of superficial layers of mucin and epithelial cells that lie over the dermis and connective tissue. Local immune cells, including mast cells and Langerhans cells, reside in the deeper layers. The differential diagnosis of plant-based mouth pain can be divided into mechanical, chemical, and thermal causes.
Mechanical Causes. Causes of mechanical plant-based oral pain include structural damage when foreign matter, such as barbs, sharp leaves, or hard seeds, pierce the layers of the oral mucosa.
Chemical Causes. Chemical-related causes of oral pain include caustic ingestion, for example from detergents or cleaning agents that contaminate the broth. Araceae, such as taro or arum, have sharp calcium oxalate crystals tipped with phospholipases and proteases that cause mechanical pain on piercing mucous membranes, and chemical pain by enzymatically degrading epithelium and mucosa. Both chemical and mechanical irritation can lead to an inflammatory response. Raw taro can cause irritant contact stomatitis as the raphides pierce the oral mucosa. It can also cause allergic stomatitis if antigens related to the phospholipases or proteases are presented to Langerhans cells.9
Thermal Causes. The hot temperature of the ingested broth could cause thermal injury, but the injury is likely to be more diffuse.
How common is taro exposure, and how is it treated?
From 1995 to 1999, 15 cases of taro poisoning were reported to the Drug and Toxicology Information service in Zimbabwe.10 From 2005 to 2009, 21 out of 31 cases reported to the Hong Kong Poison Control Center involving gastrointestinal irritation involved the consumption of Colocasia fallax, a form of taro more common in Tibet, the Himalayas, and northern Indochina.7 Of the 31 cases, six patients were treated with diphenhydramine, epinephrine, and dexamethasone for angioedema.
From 2011 to 2013, two cases of mouth irritation and swelling after eating raw taro leaves were reported to the British Columbia Poison Control Center.11 Those two patients were observed for 6 hours without specific treatment and discharged.
Case Conclusion
Due to concerns of the potential for anaphylaxis, both patients were treated intravenously with 50 mg diphenhydramine and 10 mg dexamethasone. The husband was also given 650 mg acetaminophen orally for pain relief; his wife declined pain medication. Laboratory evaluation, including a complete blood count, basic metabolic panel, liver function panel, and urinalysis were ordered for both patients; all results were within normal limits for both patients.
After an uneventful 6-hour observation period, both patients were discharged home with instructions to return to the ED if they develop any signs of allergic reaction and to call emergency medical services for any sign of anaphylaxis.
1. Rao RV, Matthews PJ, Eyzaguirre PB, Hunter D, eds. 2010. The Global Diversity of Taro: Ethnobotany and Conservation. Rome, Italy; Biouniversity International; 2010. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/1402.pdf#page=11. Accessed September 15, 2017.
2. Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005;56:41-71. doi:10.1146/annurev.arplant.56.032604.144106.
3. Herbert DA. Stinging crystals in plants. Science. 1924;60(1548):204-205. doi:10.1126/science.60.1548.204-a.
4. Njintang YN, Mbofung CMF. Effect of precooking time and drying temperature on the physico-chemical characteristics and in-vitro carbohydrate digestibility of taro flour. LWT – Food Sci and Tech. 2006;39(6):684-691. doi.org/10.1016/j.lwt.2005.03.022.
5. Savage GP, Dubois M. The effect of soaking and cooking on the oxalate content of taro leaves. Int J Food Sci Nutr. 2006;57(5-6):376-381. doi:10.1080/09637480600855239.
6. Oscarsson, KV. Savage GP. Composition and availability of soluble and insoluble oxalates in raw and cooked taro (Colocasia esculenta var. Schott) leaves. Food Chem 101. 2007;101(2):559-562. doi:10.1016/j.foodchem.2006.02.014.
7. Pang CT, Ng HW, Lau FL. Oral mucosal irritating plant ingestion in Hong Kong, epidemiology and its clinical presentation. Hong Kong J Emerg Med. 2010;17(5):477-481.
8. Yuen E. Upper airway obstruction as a presentation of Taro poisoning. Hong Kong J Emerg Med. 2001;8(3):163-165.
9. Davis CC, Squier CA, Lilly GE. Irritant contact stomatitis: a review of the condition. J Periodontol. 1998;69(6):620-631. doi:10.1902/jop.1998.69.6.620.
10 Tagwireyi D, Ball DE. The management of Elephant’s Ear poisoning. Hum Exp Toxicol. 2001;20(4):189-192. doi:10.1191/096032701678766822.
11. Omura JD, Blake C, McIntyre L, Li D, Kosatsky T. Two cases of poisoning by raw taro leaf and how a poison control centre, food safety inspectors, and a specialty supermarket chain found a solution.” Environ Health Rev. 2014;57(3):59-64. doi.org/10.5864/d2014-027.
Case
A 45-year-old Chinese man with no known medical history presented to the ED with right-sided facial spasm and cheek swelling, which began immediately after he bit into a piece of taro root, approximately 2 hours prior to presentation. The patient stated that the root was an ingredient in a soup that a relative had made. According to the patient, after biting into the root, he immediately experienced a burning pain on the right side of his mouth. He further noted that he swallowed less than two bites of the root and stopped eating because the act of chewing was too painful.
Initial vital signs at presentation were: blood pressure, 140/100 mm Hg; heart rate, 84 beats/min; respiratory rate, 14 beats/min; and temperature, 97.6°F. Oxygen saturation was 98% on room air. The patient’s physical examination was remarkable for pain upon opening the mouth, as well as right-sided cheek and lip swelling and tenderness. The tongue and oropharynx were not erythematous or swollen. The patient was only able to speak in short sentences, secondary to oropharyngeal pain, but he was in no respiratory distress. No urticaria, pruritus, wheezing, or stridor was present.
During the patient’s workup, his 40-year-old wife also presented to the same ED for evaluation of burning pain and spasm on the left side of her mouth, which she stated also developed immediately after she bit into a piece of taro root contained in the same soup as that ingested by the patient.
The wife’s vital signs were unremarkable, and she was in no respiratory distress. Her physical examination was remarkable only for left-sided cheek and lip swelling and tenderness, associated with an erythematous oropharynx and pain with speaking.
What is taro? What are the manifestations of taro toxicity?
Taro commonly refers to plants from the Araceae family, usually Colocasia esculenta.1 Taro is ubiquitous in Southern Asia and Southeast India. It is a widely naturalized and perennial tropical plant primarily grown as a root vegetable, and is a common flavor in boba (bubble) tea. All members of Araceae contain calcium oxalate crystals in the form of raphides, sharp needle-shaped crystals packaged in idioblasts and contained within the waxy leaf.2 Pressure on the idioblasts, such as from mastication, triggers the release of the raphides. The needles pierce the surface of any tissue with which they come into contact, creating a gateway for proteolytic enzymes to enter the consumer.3 The leaves and root of Araceae must be cooked before eating to inactivate the raphides.
Oral exposure to uncooked taro leaves or taro root can result in mouth irritation and swelling that can progress to angioedema and airway obstruction. Although the traditional method of removing taro raphides is to soak the root in cold water overnight,4,5 this does not fully remove all of the raphides. Instead, taro root should be thoroughly cooked in boiling water to draw-out oxalates from the root into the cooking water, which must then be discarded. Consuming taro with warm milk also reduces the effect of the oxalates by about 80%.6
Many other plants of the Araceae family, such as Dieffenbachia (dumbcane), share similar toxicity and are commonly kept in the home and office.
Patients with oral exposure to taro may experience a delayed (also termed biphasic) anaphylactic reaction, ie, the development of anaphylactic symptoms more than 4 hours after the inciting event. Delayed anaphylaxis is distinct from delayed hypersensitivity, though both may be immunoglobulin E-mediated. Delayed hypersensitivity presents later (2-14 days) and with less immediately life-threatening effects, most commonly dermatitis (eg, poison ivy dermatitis).
While both of the patients in this case presented with mild symptoms, life-threatening angioedema of the oropharynx, anaphylaxis, and hypocalcemia have been reported7,8 and should be considered in any symptomatic patient with exposure to taro.
What is the differential diagnosis of plant-related mouth pain?
The oral mucosa is composed of superficial layers of mucin and epithelial cells that lie over the dermis and connective tissue. Local immune cells, including mast cells and Langerhans cells, reside in the deeper layers. The differential diagnosis of plant-based mouth pain can be divided into mechanical, chemical, and thermal causes.
Mechanical Causes. Causes of mechanical plant-based oral pain include structural damage when foreign matter, such as barbs, sharp leaves, or hard seeds, pierce the layers of the oral mucosa.
Chemical Causes. Chemical-related causes of oral pain include caustic ingestion, for example from detergents or cleaning agents that contaminate the broth. Araceae, such as taro or arum, have sharp calcium oxalate crystals tipped with phospholipases and proteases that cause mechanical pain on piercing mucous membranes, and chemical pain by enzymatically degrading epithelium and mucosa. Both chemical and mechanical irritation can lead to an inflammatory response. Raw taro can cause irritant contact stomatitis as the raphides pierce the oral mucosa. It can also cause allergic stomatitis if antigens related to the phospholipases or proteases are presented to Langerhans cells.9
Thermal Causes. The hot temperature of the ingested broth could cause thermal injury, but the injury is likely to be more diffuse.
How common is taro exposure, and how is it treated?
From 1995 to 1999, 15 cases of taro poisoning were reported to the Drug and Toxicology Information service in Zimbabwe.10 From 2005 to 2009, 21 out of 31 cases reported to the Hong Kong Poison Control Center involving gastrointestinal irritation involved the consumption of Colocasia fallax, a form of taro more common in Tibet, the Himalayas, and northern Indochina.7 Of the 31 cases, six patients were treated with diphenhydramine, epinephrine, and dexamethasone for angioedema.
From 2011 to 2013, two cases of mouth irritation and swelling after eating raw taro leaves were reported to the British Columbia Poison Control Center.11 Those two patients were observed for 6 hours without specific treatment and discharged.
Case Conclusion
Due to concerns of the potential for anaphylaxis, both patients were treated intravenously with 50 mg diphenhydramine and 10 mg dexamethasone. The husband was also given 650 mg acetaminophen orally for pain relief; his wife declined pain medication. Laboratory evaluation, including a complete blood count, basic metabolic panel, liver function panel, and urinalysis were ordered for both patients; all results were within normal limits for both patients.
After an uneventful 6-hour observation period, both patients were discharged home with instructions to return to the ED if they develop any signs of allergic reaction and to call emergency medical services for any sign of anaphylaxis.
Case
A 45-year-old Chinese man with no known medical history presented to the ED with right-sided facial spasm and cheek swelling, which began immediately after he bit into a piece of taro root, approximately 2 hours prior to presentation. The patient stated that the root was an ingredient in a soup that a relative had made. According to the patient, after biting into the root, he immediately experienced a burning pain on the right side of his mouth. He further noted that he swallowed less than two bites of the root and stopped eating because the act of chewing was too painful.
Initial vital signs at presentation were: blood pressure, 140/100 mm Hg; heart rate, 84 beats/min; respiratory rate, 14 beats/min; and temperature, 97.6°F. Oxygen saturation was 98% on room air. The patient’s physical examination was remarkable for pain upon opening the mouth, as well as right-sided cheek and lip swelling and tenderness. The tongue and oropharynx were not erythematous or swollen. The patient was only able to speak in short sentences, secondary to oropharyngeal pain, but he was in no respiratory distress. No urticaria, pruritus, wheezing, or stridor was present.
During the patient’s workup, his 40-year-old wife also presented to the same ED for evaluation of burning pain and spasm on the left side of her mouth, which she stated also developed immediately after she bit into a piece of taro root contained in the same soup as that ingested by the patient.
The wife’s vital signs were unremarkable, and she was in no respiratory distress. Her physical examination was remarkable only for left-sided cheek and lip swelling and tenderness, associated with an erythematous oropharynx and pain with speaking.
What is taro? What are the manifestations of taro toxicity?
Taro commonly refers to plants from the Araceae family, usually Colocasia esculenta.1 Taro is ubiquitous in Southern Asia and Southeast India. It is a widely naturalized and perennial tropical plant primarily grown as a root vegetable, and is a common flavor in boba (bubble) tea. All members of Araceae contain calcium oxalate crystals in the form of raphides, sharp needle-shaped crystals packaged in idioblasts and contained within the waxy leaf.2 Pressure on the idioblasts, such as from mastication, triggers the release of the raphides. The needles pierce the surface of any tissue with which they come into contact, creating a gateway for proteolytic enzymes to enter the consumer.3 The leaves and root of Araceae must be cooked before eating to inactivate the raphides.
Oral exposure to uncooked taro leaves or taro root can result in mouth irritation and swelling that can progress to angioedema and airway obstruction. Although the traditional method of removing taro raphides is to soak the root in cold water overnight,4,5 this does not fully remove all of the raphides. Instead, taro root should be thoroughly cooked in boiling water to draw-out oxalates from the root into the cooking water, which must then be discarded. Consuming taro with warm milk also reduces the effect of the oxalates by about 80%.6
Many other plants of the Araceae family, such as Dieffenbachia (dumbcane), share similar toxicity and are commonly kept in the home and office.
Patients with oral exposure to taro may experience a delayed (also termed biphasic) anaphylactic reaction, ie, the development of anaphylactic symptoms more than 4 hours after the inciting event. Delayed anaphylaxis is distinct from delayed hypersensitivity, though both may be immunoglobulin E-mediated. Delayed hypersensitivity presents later (2-14 days) and with less immediately life-threatening effects, most commonly dermatitis (eg, poison ivy dermatitis).
While both of the patients in this case presented with mild symptoms, life-threatening angioedema of the oropharynx, anaphylaxis, and hypocalcemia have been reported7,8 and should be considered in any symptomatic patient with exposure to taro.
What is the differential diagnosis of plant-related mouth pain?
The oral mucosa is composed of superficial layers of mucin and epithelial cells that lie over the dermis and connective tissue. Local immune cells, including mast cells and Langerhans cells, reside in the deeper layers. The differential diagnosis of plant-based mouth pain can be divided into mechanical, chemical, and thermal causes.
Mechanical Causes. Causes of mechanical plant-based oral pain include structural damage when foreign matter, such as barbs, sharp leaves, or hard seeds, pierce the layers of the oral mucosa.
Chemical Causes. Chemical-related causes of oral pain include caustic ingestion, for example from detergents or cleaning agents that contaminate the broth. Araceae, such as taro or arum, have sharp calcium oxalate crystals tipped with phospholipases and proteases that cause mechanical pain on piercing mucous membranes, and chemical pain by enzymatically degrading epithelium and mucosa. Both chemical and mechanical irritation can lead to an inflammatory response. Raw taro can cause irritant contact stomatitis as the raphides pierce the oral mucosa. It can also cause allergic stomatitis if antigens related to the phospholipases or proteases are presented to Langerhans cells.9
Thermal Causes. The hot temperature of the ingested broth could cause thermal injury, but the injury is likely to be more diffuse.
How common is taro exposure, and how is it treated?
From 1995 to 1999, 15 cases of taro poisoning were reported to the Drug and Toxicology Information service in Zimbabwe.10 From 2005 to 2009, 21 out of 31 cases reported to the Hong Kong Poison Control Center involving gastrointestinal irritation involved the consumption of Colocasia fallax, a form of taro more common in Tibet, the Himalayas, and northern Indochina.7 Of the 31 cases, six patients were treated with diphenhydramine, epinephrine, and dexamethasone for angioedema.
From 2011 to 2013, two cases of mouth irritation and swelling after eating raw taro leaves were reported to the British Columbia Poison Control Center.11 Those two patients were observed for 6 hours without specific treatment and discharged.
Case Conclusion
Due to concerns of the potential for anaphylaxis, both patients were treated intravenously with 50 mg diphenhydramine and 10 mg dexamethasone. The husband was also given 650 mg acetaminophen orally for pain relief; his wife declined pain medication. Laboratory evaluation, including a complete blood count, basic metabolic panel, liver function panel, and urinalysis were ordered for both patients; all results were within normal limits for both patients.
After an uneventful 6-hour observation period, both patients were discharged home with instructions to return to the ED if they develop any signs of allergic reaction and to call emergency medical services for any sign of anaphylaxis.
1. Rao RV, Matthews PJ, Eyzaguirre PB, Hunter D, eds. 2010. The Global Diversity of Taro: Ethnobotany and Conservation. Rome, Italy; Biouniversity International; 2010. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/1402.pdf#page=11. Accessed September 15, 2017.
2. Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005;56:41-71. doi:10.1146/annurev.arplant.56.032604.144106.
3. Herbert DA. Stinging crystals in plants. Science. 1924;60(1548):204-205. doi:10.1126/science.60.1548.204-a.
4. Njintang YN, Mbofung CMF. Effect of precooking time and drying temperature on the physico-chemical characteristics and in-vitro carbohydrate digestibility of taro flour. LWT – Food Sci and Tech. 2006;39(6):684-691. doi.org/10.1016/j.lwt.2005.03.022.
5. Savage GP, Dubois M. The effect of soaking and cooking on the oxalate content of taro leaves. Int J Food Sci Nutr. 2006;57(5-6):376-381. doi:10.1080/09637480600855239.
6. Oscarsson, KV. Savage GP. Composition and availability of soluble and insoluble oxalates in raw and cooked taro (Colocasia esculenta var. Schott) leaves. Food Chem 101. 2007;101(2):559-562. doi:10.1016/j.foodchem.2006.02.014.
7. Pang CT, Ng HW, Lau FL. Oral mucosal irritating plant ingestion in Hong Kong, epidemiology and its clinical presentation. Hong Kong J Emerg Med. 2010;17(5):477-481.
8. Yuen E. Upper airway obstruction as a presentation of Taro poisoning. Hong Kong J Emerg Med. 2001;8(3):163-165.
9. Davis CC, Squier CA, Lilly GE. Irritant contact stomatitis: a review of the condition. J Periodontol. 1998;69(6):620-631. doi:10.1902/jop.1998.69.6.620.
10 Tagwireyi D, Ball DE. The management of Elephant’s Ear poisoning. Hum Exp Toxicol. 2001;20(4):189-192. doi:10.1191/096032701678766822.
11. Omura JD, Blake C, McIntyre L, Li D, Kosatsky T. Two cases of poisoning by raw taro leaf and how a poison control centre, food safety inspectors, and a specialty supermarket chain found a solution.” Environ Health Rev. 2014;57(3):59-64. doi.org/10.5864/d2014-027.
1. Rao RV, Matthews PJ, Eyzaguirre PB, Hunter D, eds. 2010. The Global Diversity of Taro: Ethnobotany and Conservation. Rome, Italy; Biouniversity International; 2010. http://www.bioversityinternational.org/fileadmin/user_upload/online_library/publications/pdfs/1402.pdf#page=11. Accessed September 15, 2017.
2. Franceschi VR, Nakata PA. Calcium oxalate in plants: formation and function. Annu Rev Plant Biol. 2005;56:41-71. doi:10.1146/annurev.arplant.56.032604.144106.
3. Herbert DA. Stinging crystals in plants. Science. 1924;60(1548):204-205. doi:10.1126/science.60.1548.204-a.
4. Njintang YN, Mbofung CMF. Effect of precooking time and drying temperature on the physico-chemical characteristics and in-vitro carbohydrate digestibility of taro flour. LWT – Food Sci and Tech. 2006;39(6):684-691. doi.org/10.1016/j.lwt.2005.03.022.
5. Savage GP, Dubois M. The effect of soaking and cooking on the oxalate content of taro leaves. Int J Food Sci Nutr. 2006;57(5-6):376-381. doi:10.1080/09637480600855239.
6. Oscarsson, KV. Savage GP. Composition and availability of soluble and insoluble oxalates in raw and cooked taro (Colocasia esculenta var. Schott) leaves. Food Chem 101. 2007;101(2):559-562. doi:10.1016/j.foodchem.2006.02.014.
7. Pang CT, Ng HW, Lau FL. Oral mucosal irritating plant ingestion in Hong Kong, epidemiology and its clinical presentation. Hong Kong J Emerg Med. 2010;17(5):477-481.
8. Yuen E. Upper airway obstruction as a presentation of Taro poisoning. Hong Kong J Emerg Med. 2001;8(3):163-165.
9. Davis CC, Squier CA, Lilly GE. Irritant contact stomatitis: a review of the condition. J Periodontol. 1998;69(6):620-631. doi:10.1902/jop.1998.69.6.620.
10 Tagwireyi D, Ball DE. The management of Elephant’s Ear poisoning. Hum Exp Toxicol. 2001;20(4):189-192. doi:10.1191/096032701678766822.
11. Omura JD, Blake C, McIntyre L, Li D, Kosatsky T. Two cases of poisoning by raw taro leaf and how a poison control centre, food safety inspectors, and a specialty supermarket chain found a solution.” Environ Health Rev. 2014;57(3):59-64. doi.org/10.5864/d2014-027.

Sweet Syndrome Induced by Oral Acetaminophen-Codeine Following Repair of a Facial Fracture
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
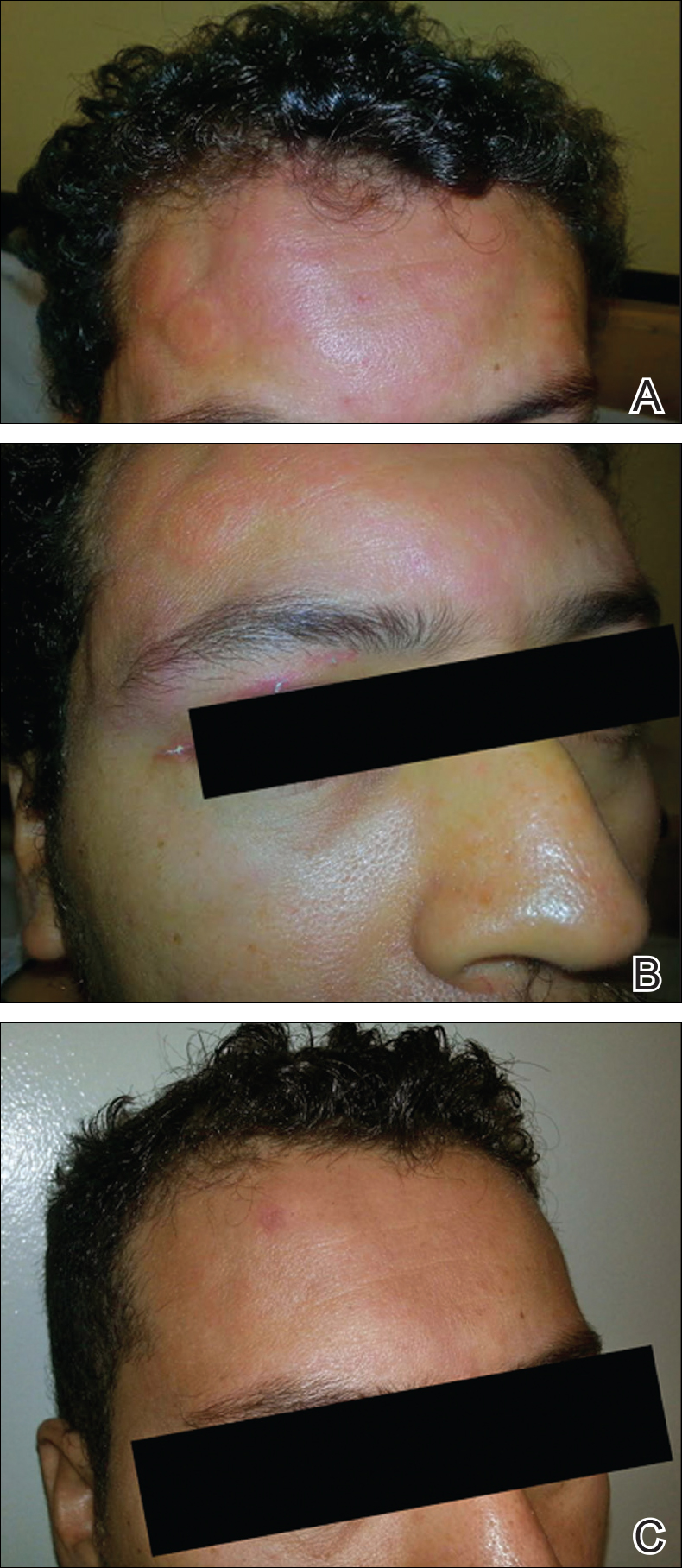
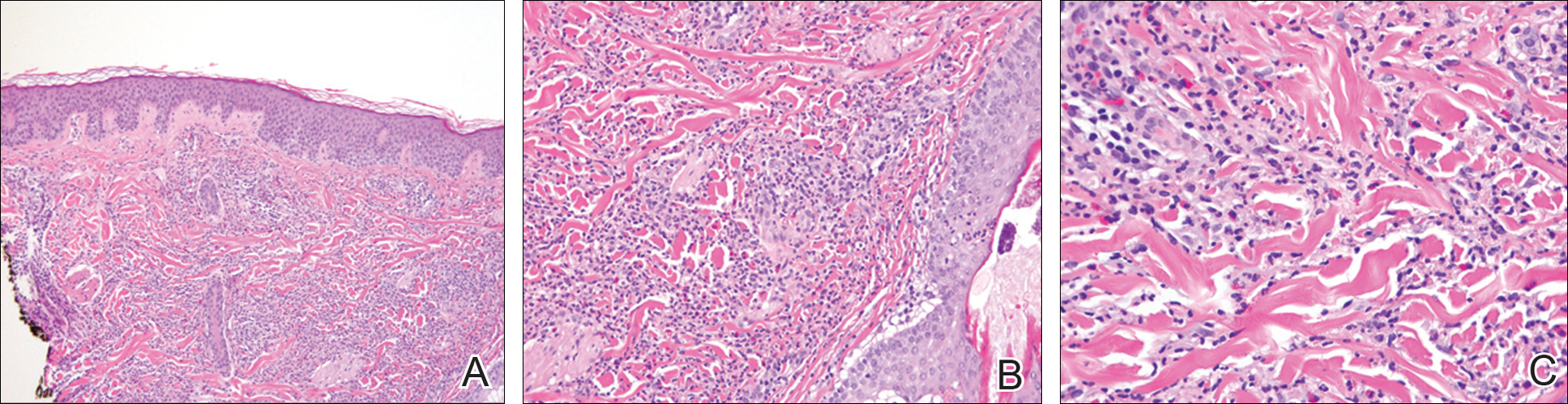
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
Practice Points
- The rate of medication-induced Sweet syndrome is on the rise.
- Oral acetaminophen-codeine may induce Sweet syndrome.

Melanotrichoblastoma: A Rare Pigmented Variant of Trichoblastoma
Trichoblastomas are rare cutaneous tumors that recapitulate the germinative hair bulb and the surrounding mesenchyme. Although benign, they can present diagnostic difficulties for both the clinician and pathologist because of their rarity and overlap both clinically and microscopically with other follicular neoplasms as well as basal cell carcinoma (BCC). Several classification schemes for hair follicle neoplasms have been established based on the relative proportions of epithelial and mesenchymal components as well as stromal inductive change, but nomenclature continues to be problematic, as individual neoplasms show varying degrees of differentiation that do not always uniformly fit within these categories.1,2 One of these established categories is a pigmented trichoblastoma.3 An exceedingly rare variant of a pigmented trichoblastoma referred to as melanotrichoblastoma was first described in 20024 and has only been documented in 3 cases, according to a PubMed search of articles indexed for MEDLINE using the term melanotrichoblastoma.4-6 We report another case of this rare tumor and review the literature on this unique group of tumors.
Case Report
A 25-year-old white woman with a medical history of chronic migraines, myofascial syndrome, and Arnold-Chiari malformation type I presented to dermatology with a 1.5-cm, pedunculated, well-circumscribed tumor on the left side of the scalp (Figure 1). The tumor was grossly flesh colored with heterogeneous areas of dark pigmentation. Microscopic examination demonstrated that within the superficial and deep dermis were variable-sized nests of basaloid cells. Some of the nests had large central cystic spaces with brown pigment within some of these spaces and focal pigmentation of the basaloid cells (Figure 2A). Focal areas of keratinization were present. Mitotic figures were easily identified; however, no atypical mitotic figures were present. Areas of peripheral palisading were present but there was no retraction artifact. Connection to the overlying epidermis was not identified. Surrounding the basaloid nodules was a mildly cellular proliferation of cytologically bland spindle cells. Occasional pigment-laden macrophages were present in the dermis. Focal areas suggestive of papillary mesenchymal body formation were present (Figure 2B). Immunohistochemical staining for Melan-A was performed and demonstrated the presence of a prominent number of melanocytes in some of the nests (Figure 3) and minimal to no melanocytes in other nests. There was no evidence of a melanocytic lesion involving the overlying epidermis. Features of nevus sebaceus were not present. Immunohistochemical staining for cytokeratin (CK) 20 was performed and demonstrated no notable number of Merkel cells within the lesion.
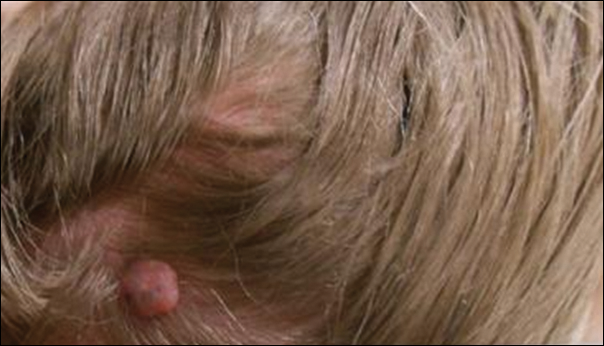
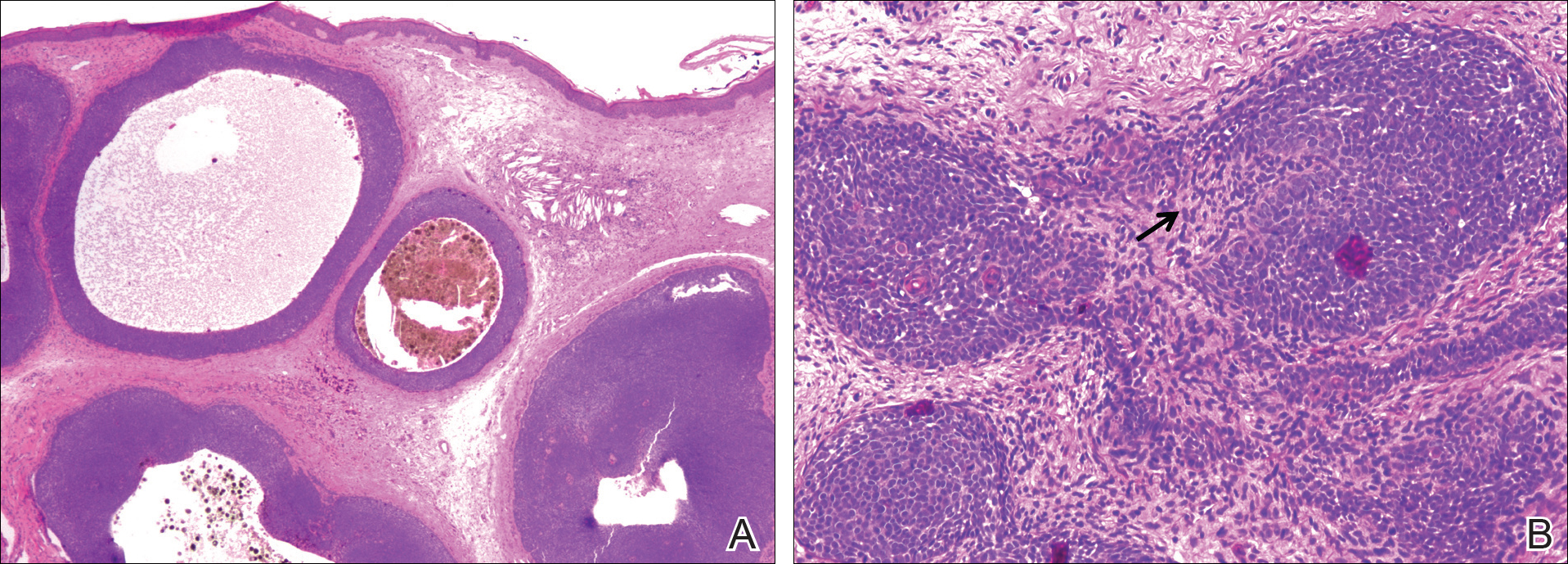
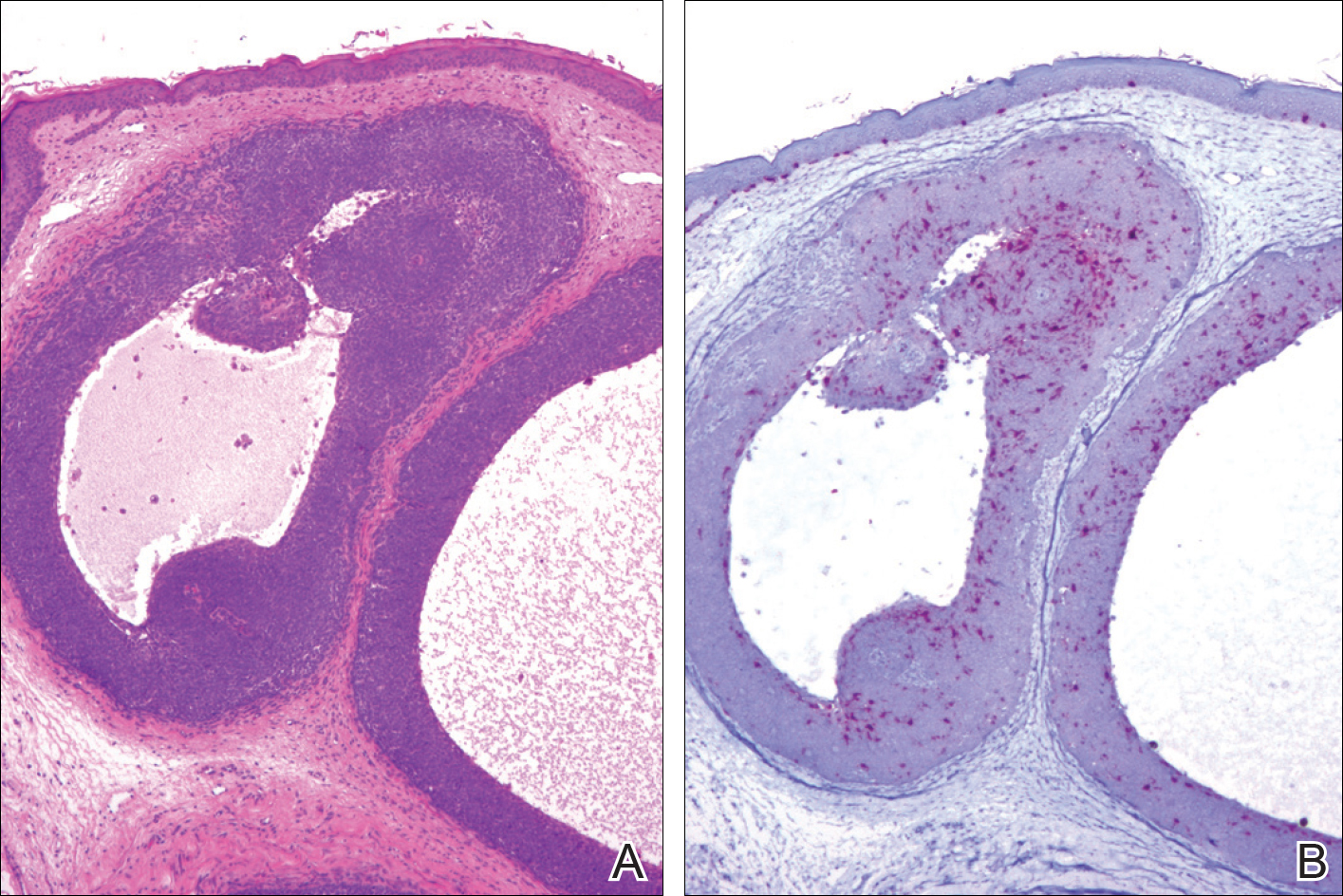
Comment
Overview of Trichoblastomas
Trichoblastomas most often present as solitary, flesh-colored, well-circumscribed, slow-growing tumors that usually progress in size over months to years. Although they may be present at any age, they most commonly occur in adults in the fifth to seventh decades of life and are equally distributed between males and females.7,8 They most often occur on the head and neck with a predilection for the scalp. Although they behave in a benign fashion, cases of malignant trichoblastomas have been reported.9
Histopathology
Histologically, these tumors are well circumscribed but unencapsulated and usually located within the deep dermis, often with extension into the subcutaneous tissue. An epidermal connection is not identified. The tumor typically is composed of variable-sized nests of basaloid cells surrounded by a variable cellular stromal component. Although peripheral palisading is present in the basaloid component, retraction artifact is not present. Several histologic variants of trichoblastomas have been reported including cribriform, racemiform, retiform, pigmented, giant, subcutaneous, rippled pattern, and clear cell.5 Pigmented trichoblastomas are histologically similar to typical trichoblastomas, except for the presence of large amounts of melanin deposited within and around the tumor nests.6 A melanotrichoblastoma is a rare variant of a pigmented trichoblastoma; pigment is present in the lesion and melanocytes are identified within the basaloid nests.
The stromal component of trichoblastomas may show areas of condensation associated with some of the basaloid cells, resembling an attempt at hair bulb formation. Staining for CD10 will be positive in these areas of papillary mesenchymal bodies.10
In an immunohistochemical study of 13 cases of trichoblastomas, there was diffuse positive staining for CK14 and CK17 in all cases (similar to BCC) and positive staining for CK19 in 70% (9/13) of cases compared to 21% (4/19) of BCC cases. Staining for CK8 and CK20 demonstrated the presence of numerous Merkel cells in all trichoblastomas but in none of the 19 cases of BCC tested.11 However, other studies have reported the presence of Merkel cells in only 42% to 70% of trichoblastomas.12,13 Despite the lack of Merkel cells in our case, the lesion was interpreted as a melanotrichoblastoma based on the histologic features in conjunction with the presence of the melanocytes.
Differential Diagnosis
The clinical and histologic differential diagnosis of trichoblastomas includes both trichoepithelioma and BCC. Clinically, all 3 lesions often are slow growing, dome shaped, and small in size (several millimeters), and are observed in the same anatomic distribution of the head and neck region. Furthermore, they often affect middle-aged to older individuals and those of Caucasian descent, though other ethnicities can be affected. Histologic evaluation often is necessary to differentiate between these 3 entities.
Histologically, trichoepitheliomas are composed of nodules of basaloid cells encircled by stromal spindle cells. Although there can be histologic overlap between trichoepitheliomas and trichoblastoma, trichoepitheliomas typically will display obvious features of hair follicle differentiation with the presence of small keratinous cysts and hair bulb structures, while trichoblastomas tend to display minimal changes suggestive of its hair follicle origin. Similar to trichoblastomas, BCC is composed of nests of basaloid cells; however, BCCs often demonstrate retraction artifact and connection to the overlying epidermis. In addition, BCCs typically demonstrate a fibromucinous stromal component that is distinct from the cellular stroma of trichoblastic tumors. Immunoperoxidase staining for androgen receptors has been reported to be positive in 78% (25/32) of BCCs and negative in trichoblastic tumors.14
Melanotrichoblastoma Differentiating Characteristics
An exceedingly rare variant of pigmented trichoblastoma is the melanotrichoblastoma. There are clinical and histologic similarities and differences between the reported cases. The first case, described by Kanitakis et al,4 reported a 32-year-old black woman with a 2-cm scalp mass that slowly enlarged over the course of 2 years. The second case, presented by Kim et al,5 described a 51-year-old Korean man with a subcutaneous 6-cm mass on the back that had been present and slowly enlarging over the course of 5 years. The third case, reported by Hung et al,6 described a 34-year-old Taiwanese man with a 1-cm, left-sided, temporal scalp mass present for 3 years, arising from a nevus sebaceous. Comparing these clinical findings with our case of a 25-year-old white woman with a 1.5-cm mass on the left side of the scalp, melanotrichoblastomas demonstrate a relatively similar age of onset in the early to middle-aged adult years. All 4 tumors were slow growing. Additionally, 3 of 4 cases demonstrated a predilection for the head, particularly the scalp, and grossly showed well-circumscribed lesions with notable pigmentation. Although age, size, location, and gross appearance were similar, a comparable ethnic and gender demographic was not identified.
Microscopic similarities between the 4 cases were present. Each case was characterized by a large, well-circumscribed, unencapsulated, basaloid tumor present in the lower dermis, with only 1 case having tumor cells occasionally reaching the undersurface of the epidermis. The tumor cells were monomorphic round-ovoid in appearance with scant cytoplasm. There was melanin pigment in the basaloid nests. The basaloid nests were surrounded by a proliferation of stromal cells. The mitotic rate was sparse in 2 cases, brisk in 1 case, and not discussed in 1 case. Melanocytes were identified in the basaloid nests in all 4 cases; however, in the current case, the melanocytes were seen in only some of the nests. None of the cases exhibited an overlying junctional melanocytic lesion, which would argue against a possible collision tumor or colonization of an epithelial lesion by a melanocytic lesion.
Although the histologic features of our cases are consistent with prior reports of melanotrichoblastoma, there is some question as to whether it represents a true variant of a pigmented trichoblastoma. There are relatively few articles in the literature that describe pigmented trichoblastomas, and of those, immunohistochemistry staining for melanocytes is uncommon. In one of the earliest descriptions of a pigmented trichoblastoma, dendritic melanocytes were present within the tumor lobules; however, the lesion was reported as a pigmented trichoblastoma and not a melanotrichoblastoma.3 It is possible that all pigmented trichoblastomas may contain some number of dendritic melanocytes, thus negating the existence of a melanotrichoblastoma as a true subtype of pigmented trichoblastomas. Additional study looking at multiple examples of pigmented trichoblastomas would be required to more definitively classify melanotrichoblastomas. It is important to appreciate that at least some cases of pigmented trichoblastomas may contain melanocytes and not to confuse the lesion as representing an example of colonization or collision tumor. A rare case of melanoma possibly arising from these dendritic melanocytes has been reported.15
Conclusion
Trichoblastomas are uncommon tumors of germinative hair bulb origin that can have several histologic variants. A well-documented subtype of trichoblastoma characterized by melanin deposits within and around tumor nests has been identified and classified as a pigmented trichoblastoma. Four cases of melanotrichoblastoma have been reported and represent a variant of a pigmented trichoblastoma characterized by the presence of melanocytes within the lesion. Whether they represent a true variant is of some debate and additional study is required. Although these tumors are exceedingly rare, it is important for the clinician and pathologist to be aware of this entity to prevent confusion with other similarly appearing follicular lesions, most notably BCCs, because of the difference in treatment and follow-up.
- Headington JT. Tumors of the hair follicle: a review. Am J Pathol. 1976; 85 : 479- 514 .
- Wong TY, Reed JA, Suster S, et al. Benign trichogenic tumors: a report of two cases supporting a simplified nomenclature. Histopathology. 1993;22:575-580.
- Aloi F, Tomasini C, Pippione M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14:345-349.
- Kanitakis J, Brutzkus A, Butnaru AC, et al. Melanotrichoblastoma: immunohistochemical study of a variant of pigmented trichoblastoma. Am J Dermatopathol. 2002;24:498-501.
- Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:E37-E40.
- Hung CT, Chiang CP, Gao HW, et al. Ripple-pattern melanotrichoblastoma arising within nevus sebaceous. Indian J Dermatol Venereol Leprol. 2012;78:665.
- Sau P, Lupton GP, Graham JH. Trichogerminoma: report of 14 cases. J Cutan Pathol. 1992;19:357-365.
- Johnson TV, Wojno TH, Grossniklaus HE. Trichoblastoma of the eyelid. Ophthal Plast Reconstr Surg. 2011;27:E148-E149.
- Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblasticcarcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16.
- Aslani FS, Akbarzadeh-Jahromi M, Jowkar F. Value of CD10 expression in differentiating cutaneous basal from squamous cell carcinomas and basal cell carcinoma from trichoepithelioma. Iran J Med Sci. 2013;38:100-106.
- Kurzen H, Esposito L, Langbein L, et al. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23:501-509.
- Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinoma but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
- McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26:119-124.
- Izikson L, Bhan A, Zembowicz A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am J Dermatopathol. 2005;27:91-95.
- Benaim G, Castillo C, Houang M, et al. Melanoma arising from a long standing pigmented trichoblastoma: clinicopathologic study with complementary aCGH/mutation analysis. Am J Dermatopathol. 2014;36:E146-E151.
Trichoblastomas are rare cutaneous tumors that recapitulate the germinative hair bulb and the surrounding mesenchyme. Although benign, they can present diagnostic difficulties for both the clinician and pathologist because of their rarity and overlap both clinically and microscopically with other follicular neoplasms as well as basal cell carcinoma (BCC). Several classification schemes for hair follicle neoplasms have been established based on the relative proportions of epithelial and mesenchymal components as well as stromal inductive change, but nomenclature continues to be problematic, as individual neoplasms show varying degrees of differentiation that do not always uniformly fit within these categories.1,2 One of these established categories is a pigmented trichoblastoma.3 An exceedingly rare variant of a pigmented trichoblastoma referred to as melanotrichoblastoma was first described in 20024 and has only been documented in 3 cases, according to a PubMed search of articles indexed for MEDLINE using the term melanotrichoblastoma.4-6 We report another case of this rare tumor and review the literature on this unique group of tumors.
Case Report
A 25-year-old white woman with a medical history of chronic migraines, myofascial syndrome, and Arnold-Chiari malformation type I presented to dermatology with a 1.5-cm, pedunculated, well-circumscribed tumor on the left side of the scalp (Figure 1). The tumor was grossly flesh colored with heterogeneous areas of dark pigmentation. Microscopic examination demonstrated that within the superficial and deep dermis were variable-sized nests of basaloid cells. Some of the nests had large central cystic spaces with brown pigment within some of these spaces and focal pigmentation of the basaloid cells (Figure 2A). Focal areas of keratinization were present. Mitotic figures were easily identified; however, no atypical mitotic figures were present. Areas of peripheral palisading were present but there was no retraction artifact. Connection to the overlying epidermis was not identified. Surrounding the basaloid nodules was a mildly cellular proliferation of cytologically bland spindle cells. Occasional pigment-laden macrophages were present in the dermis. Focal areas suggestive of papillary mesenchymal body formation were present (Figure 2B). Immunohistochemical staining for Melan-A was performed and demonstrated the presence of a prominent number of melanocytes in some of the nests (Figure 3) and minimal to no melanocytes in other nests. There was no evidence of a melanocytic lesion involving the overlying epidermis. Features of nevus sebaceus were not present. Immunohistochemical staining for cytokeratin (CK) 20 was performed and demonstrated no notable number of Merkel cells within the lesion.
Comment
Overview of Trichoblastomas
Trichoblastomas most often present as solitary, flesh-colored, well-circumscribed, slow-growing tumors that usually progress in size over months to years. Although they may be present at any age, they most commonly occur in adults in the fifth to seventh decades of life and are equally distributed between males and females.7,8 They most often occur on the head and neck with a predilection for the scalp. Although they behave in a benign fashion, cases of malignant trichoblastomas have been reported.9
Histopathology
Histologically, these tumors are well circumscribed but unencapsulated and usually located within the deep dermis, often with extension into the subcutaneous tissue. An epidermal connection is not identified. The tumor typically is composed of variable-sized nests of basaloid cells surrounded by a variable cellular stromal component. Although peripheral palisading is present in the basaloid component, retraction artifact is not present. Several histologic variants of trichoblastomas have been reported including cribriform, racemiform, retiform, pigmented, giant, subcutaneous, rippled pattern, and clear cell.5 Pigmented trichoblastomas are histologically similar to typical trichoblastomas, except for the presence of large amounts of melanin deposited within and around the tumor nests.6 A melanotrichoblastoma is a rare variant of a pigmented trichoblastoma; pigment is present in the lesion and melanocytes are identified within the basaloid nests.
The stromal component of trichoblastomas may show areas of condensation associated with some of the basaloid cells, resembling an attempt at hair bulb formation. Staining for CD10 will be positive in these areas of papillary mesenchymal bodies.10
In an immunohistochemical study of 13 cases of trichoblastomas, there was diffuse positive staining for CK14 and CK17 in all cases (similar to BCC) and positive staining for CK19 in 70% (9/13) of cases compared to 21% (4/19) of BCC cases. Staining for CK8 and CK20 demonstrated the presence of numerous Merkel cells in all trichoblastomas but in none of the 19 cases of BCC tested.11 However, other studies have reported the presence of Merkel cells in only 42% to 70% of trichoblastomas.12,13 Despite the lack of Merkel cells in our case, the lesion was interpreted as a melanotrichoblastoma based on the histologic features in conjunction with the presence of the melanocytes.
Differential Diagnosis
The clinical and histologic differential diagnosis of trichoblastomas includes both trichoepithelioma and BCC. Clinically, all 3 lesions often are slow growing, dome shaped, and small in size (several millimeters), and are observed in the same anatomic distribution of the head and neck region. Furthermore, they often affect middle-aged to older individuals and those of Caucasian descent, though other ethnicities can be affected. Histologic evaluation often is necessary to differentiate between these 3 entities.
Histologically, trichoepitheliomas are composed of nodules of basaloid cells encircled by stromal spindle cells. Although there can be histologic overlap between trichoepitheliomas and trichoblastoma, trichoepitheliomas typically will display obvious features of hair follicle differentiation with the presence of small keratinous cysts and hair bulb structures, while trichoblastomas tend to display minimal changes suggestive of its hair follicle origin. Similar to trichoblastomas, BCC is composed of nests of basaloid cells; however, BCCs often demonstrate retraction artifact and connection to the overlying epidermis. In addition, BCCs typically demonstrate a fibromucinous stromal component that is distinct from the cellular stroma of trichoblastic tumors. Immunoperoxidase staining for androgen receptors has been reported to be positive in 78% (25/32) of BCCs and negative in trichoblastic tumors.14
Melanotrichoblastoma Differentiating Characteristics
An exceedingly rare variant of pigmented trichoblastoma is the melanotrichoblastoma. There are clinical and histologic similarities and differences between the reported cases. The first case, described by Kanitakis et al,4 reported a 32-year-old black woman with a 2-cm scalp mass that slowly enlarged over the course of 2 years. The second case, presented by Kim et al,5 described a 51-year-old Korean man with a subcutaneous 6-cm mass on the back that had been present and slowly enlarging over the course of 5 years. The third case, reported by Hung et al,6 described a 34-year-old Taiwanese man with a 1-cm, left-sided, temporal scalp mass present for 3 years, arising from a nevus sebaceous. Comparing these clinical findings with our case of a 25-year-old white woman with a 1.5-cm mass on the left side of the scalp, melanotrichoblastomas demonstrate a relatively similar age of onset in the early to middle-aged adult years. All 4 tumors were slow growing. Additionally, 3 of 4 cases demonstrated a predilection for the head, particularly the scalp, and grossly showed well-circumscribed lesions with notable pigmentation. Although age, size, location, and gross appearance were similar, a comparable ethnic and gender demographic was not identified.
Microscopic similarities between the 4 cases were present. Each case was characterized by a large, well-circumscribed, unencapsulated, basaloid tumor present in the lower dermis, with only 1 case having tumor cells occasionally reaching the undersurface of the epidermis. The tumor cells were monomorphic round-ovoid in appearance with scant cytoplasm. There was melanin pigment in the basaloid nests. The basaloid nests were surrounded by a proliferation of stromal cells. The mitotic rate was sparse in 2 cases, brisk in 1 case, and not discussed in 1 case. Melanocytes were identified in the basaloid nests in all 4 cases; however, in the current case, the melanocytes were seen in only some of the nests. None of the cases exhibited an overlying junctional melanocytic lesion, which would argue against a possible collision tumor or colonization of an epithelial lesion by a melanocytic lesion.
Although the histologic features of our cases are consistent with prior reports of melanotrichoblastoma, there is some question as to whether it represents a true variant of a pigmented trichoblastoma. There are relatively few articles in the literature that describe pigmented trichoblastomas, and of those, immunohistochemistry staining for melanocytes is uncommon. In one of the earliest descriptions of a pigmented trichoblastoma, dendritic melanocytes were present within the tumor lobules; however, the lesion was reported as a pigmented trichoblastoma and not a melanotrichoblastoma.3 It is possible that all pigmented trichoblastomas may contain some number of dendritic melanocytes, thus negating the existence of a melanotrichoblastoma as a true subtype of pigmented trichoblastomas. Additional study looking at multiple examples of pigmented trichoblastomas would be required to more definitively classify melanotrichoblastomas. It is important to appreciate that at least some cases of pigmented trichoblastomas may contain melanocytes and not to confuse the lesion as representing an example of colonization or collision tumor. A rare case of melanoma possibly arising from these dendritic melanocytes has been reported.15
Conclusion
Trichoblastomas are uncommon tumors of germinative hair bulb origin that can have several histologic variants. A well-documented subtype of trichoblastoma characterized by melanin deposits within and around tumor nests has been identified and classified as a pigmented trichoblastoma. Four cases of melanotrichoblastoma have been reported and represent a variant of a pigmented trichoblastoma characterized by the presence of melanocytes within the lesion. Whether they represent a true variant is of some debate and additional study is required. Although these tumors are exceedingly rare, it is important for the clinician and pathologist to be aware of this entity to prevent confusion with other similarly appearing follicular lesions, most notably BCCs, because of the difference in treatment and follow-up.
Trichoblastomas are rare cutaneous tumors that recapitulate the germinative hair bulb and the surrounding mesenchyme. Although benign, they can present diagnostic difficulties for both the clinician and pathologist because of their rarity and overlap both clinically and microscopically with other follicular neoplasms as well as basal cell carcinoma (BCC). Several classification schemes for hair follicle neoplasms have been established based on the relative proportions of epithelial and mesenchymal components as well as stromal inductive change, but nomenclature continues to be problematic, as individual neoplasms show varying degrees of differentiation that do not always uniformly fit within these categories.1,2 One of these established categories is a pigmented trichoblastoma.3 An exceedingly rare variant of a pigmented trichoblastoma referred to as melanotrichoblastoma was first described in 20024 and has only been documented in 3 cases, according to a PubMed search of articles indexed for MEDLINE using the term melanotrichoblastoma.4-6 We report another case of this rare tumor and review the literature on this unique group of tumors.
Case Report
A 25-year-old white woman with a medical history of chronic migraines, myofascial syndrome, and Arnold-Chiari malformation type I presented to dermatology with a 1.5-cm, pedunculated, well-circumscribed tumor on the left side of the scalp (Figure 1). The tumor was grossly flesh colored with heterogeneous areas of dark pigmentation. Microscopic examination demonstrated that within the superficial and deep dermis were variable-sized nests of basaloid cells. Some of the nests had large central cystic spaces with brown pigment within some of these spaces and focal pigmentation of the basaloid cells (Figure 2A). Focal areas of keratinization were present. Mitotic figures were easily identified; however, no atypical mitotic figures were present. Areas of peripheral palisading were present but there was no retraction artifact. Connection to the overlying epidermis was not identified. Surrounding the basaloid nodules was a mildly cellular proliferation of cytologically bland spindle cells. Occasional pigment-laden macrophages were present in the dermis. Focal areas suggestive of papillary mesenchymal body formation were present (Figure 2B). Immunohistochemical staining for Melan-A was performed and demonstrated the presence of a prominent number of melanocytes in some of the nests (Figure 3) and minimal to no melanocytes in other nests. There was no evidence of a melanocytic lesion involving the overlying epidermis. Features of nevus sebaceus were not present. Immunohistochemical staining for cytokeratin (CK) 20 was performed and demonstrated no notable number of Merkel cells within the lesion.
Comment
Overview of Trichoblastomas
Trichoblastomas most often present as solitary, flesh-colored, well-circumscribed, slow-growing tumors that usually progress in size over months to years. Although they may be present at any age, they most commonly occur in adults in the fifth to seventh decades of life and are equally distributed between males and females.7,8 They most often occur on the head and neck with a predilection for the scalp. Although they behave in a benign fashion, cases of malignant trichoblastomas have been reported.9
Histopathology
Histologically, these tumors are well circumscribed but unencapsulated and usually located within the deep dermis, often with extension into the subcutaneous tissue. An epidermal connection is not identified. The tumor typically is composed of variable-sized nests of basaloid cells surrounded by a variable cellular stromal component. Although peripheral palisading is present in the basaloid component, retraction artifact is not present. Several histologic variants of trichoblastomas have been reported including cribriform, racemiform, retiform, pigmented, giant, subcutaneous, rippled pattern, and clear cell.5 Pigmented trichoblastomas are histologically similar to typical trichoblastomas, except for the presence of large amounts of melanin deposited within and around the tumor nests.6 A melanotrichoblastoma is a rare variant of a pigmented trichoblastoma; pigment is present in the lesion and melanocytes are identified within the basaloid nests.
The stromal component of trichoblastomas may show areas of condensation associated with some of the basaloid cells, resembling an attempt at hair bulb formation. Staining for CD10 will be positive in these areas of papillary mesenchymal bodies.10
In an immunohistochemical study of 13 cases of trichoblastomas, there was diffuse positive staining for CK14 and CK17 in all cases (similar to BCC) and positive staining for CK19 in 70% (9/13) of cases compared to 21% (4/19) of BCC cases. Staining for CK8 and CK20 demonstrated the presence of numerous Merkel cells in all trichoblastomas but in none of the 19 cases of BCC tested.11 However, other studies have reported the presence of Merkel cells in only 42% to 70% of trichoblastomas.12,13 Despite the lack of Merkel cells in our case, the lesion was interpreted as a melanotrichoblastoma based on the histologic features in conjunction with the presence of the melanocytes.
Differential Diagnosis
The clinical and histologic differential diagnosis of trichoblastomas includes both trichoepithelioma and BCC. Clinically, all 3 lesions often are slow growing, dome shaped, and small in size (several millimeters), and are observed in the same anatomic distribution of the head and neck region. Furthermore, they often affect middle-aged to older individuals and those of Caucasian descent, though other ethnicities can be affected. Histologic evaluation often is necessary to differentiate between these 3 entities.
Histologically, trichoepitheliomas are composed of nodules of basaloid cells encircled by stromal spindle cells. Although there can be histologic overlap between trichoepitheliomas and trichoblastoma, trichoepitheliomas typically will display obvious features of hair follicle differentiation with the presence of small keratinous cysts and hair bulb structures, while trichoblastomas tend to display minimal changes suggestive of its hair follicle origin. Similar to trichoblastomas, BCC is composed of nests of basaloid cells; however, BCCs often demonstrate retraction artifact and connection to the overlying epidermis. In addition, BCCs typically demonstrate a fibromucinous stromal component that is distinct from the cellular stroma of trichoblastic tumors. Immunoperoxidase staining for androgen receptors has been reported to be positive in 78% (25/32) of BCCs and negative in trichoblastic tumors.14
Melanotrichoblastoma Differentiating Characteristics
An exceedingly rare variant of pigmented trichoblastoma is the melanotrichoblastoma. There are clinical and histologic similarities and differences between the reported cases. The first case, described by Kanitakis et al,4 reported a 32-year-old black woman with a 2-cm scalp mass that slowly enlarged over the course of 2 years. The second case, presented by Kim et al,5 described a 51-year-old Korean man with a subcutaneous 6-cm mass on the back that had been present and slowly enlarging over the course of 5 years. The third case, reported by Hung et al,6 described a 34-year-old Taiwanese man with a 1-cm, left-sided, temporal scalp mass present for 3 years, arising from a nevus sebaceous. Comparing these clinical findings with our case of a 25-year-old white woman with a 1.5-cm mass on the left side of the scalp, melanotrichoblastomas demonstrate a relatively similar age of onset in the early to middle-aged adult years. All 4 tumors were slow growing. Additionally, 3 of 4 cases demonstrated a predilection for the head, particularly the scalp, and grossly showed well-circumscribed lesions with notable pigmentation. Although age, size, location, and gross appearance were similar, a comparable ethnic and gender demographic was not identified.
Microscopic similarities between the 4 cases were present. Each case was characterized by a large, well-circumscribed, unencapsulated, basaloid tumor present in the lower dermis, with only 1 case having tumor cells occasionally reaching the undersurface of the epidermis. The tumor cells were monomorphic round-ovoid in appearance with scant cytoplasm. There was melanin pigment in the basaloid nests. The basaloid nests were surrounded by a proliferation of stromal cells. The mitotic rate was sparse in 2 cases, brisk in 1 case, and not discussed in 1 case. Melanocytes were identified in the basaloid nests in all 4 cases; however, in the current case, the melanocytes were seen in only some of the nests. None of the cases exhibited an overlying junctional melanocytic lesion, which would argue against a possible collision tumor or colonization of an epithelial lesion by a melanocytic lesion.
Although the histologic features of our cases are consistent with prior reports of melanotrichoblastoma, there is some question as to whether it represents a true variant of a pigmented trichoblastoma. There are relatively few articles in the literature that describe pigmented trichoblastomas, and of those, immunohistochemistry staining for melanocytes is uncommon. In one of the earliest descriptions of a pigmented trichoblastoma, dendritic melanocytes were present within the tumor lobules; however, the lesion was reported as a pigmented trichoblastoma and not a melanotrichoblastoma.3 It is possible that all pigmented trichoblastomas may contain some number of dendritic melanocytes, thus negating the existence of a melanotrichoblastoma as a true subtype of pigmented trichoblastomas. Additional study looking at multiple examples of pigmented trichoblastomas would be required to more definitively classify melanotrichoblastomas. It is important to appreciate that at least some cases of pigmented trichoblastomas may contain melanocytes and not to confuse the lesion as representing an example of colonization or collision tumor. A rare case of melanoma possibly arising from these dendritic melanocytes has been reported.15
Conclusion
Trichoblastomas are uncommon tumors of germinative hair bulb origin that can have several histologic variants. A well-documented subtype of trichoblastoma characterized by melanin deposits within and around tumor nests has been identified and classified as a pigmented trichoblastoma. Four cases of melanotrichoblastoma have been reported and represent a variant of a pigmented trichoblastoma characterized by the presence of melanocytes within the lesion. Whether they represent a true variant is of some debate and additional study is required. Although these tumors are exceedingly rare, it is important for the clinician and pathologist to be aware of this entity to prevent confusion with other similarly appearing follicular lesions, most notably BCCs, because of the difference in treatment and follow-up.
- Headington JT. Tumors of the hair follicle: a review. Am J Pathol. 1976; 85 : 479- 514 .
- Wong TY, Reed JA, Suster S, et al. Benign trichogenic tumors: a report of two cases supporting a simplified nomenclature. Histopathology. 1993;22:575-580.
- Aloi F, Tomasini C, Pippione M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14:345-349.
- Kanitakis J, Brutzkus A, Butnaru AC, et al. Melanotrichoblastoma: immunohistochemical study of a variant of pigmented trichoblastoma. Am J Dermatopathol. 2002;24:498-501.
- Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:E37-E40.
- Hung CT, Chiang CP, Gao HW, et al. Ripple-pattern melanotrichoblastoma arising within nevus sebaceous. Indian J Dermatol Venereol Leprol. 2012;78:665.
- Sau P, Lupton GP, Graham JH. Trichogerminoma: report of 14 cases. J Cutan Pathol. 1992;19:357-365.
- Johnson TV, Wojno TH, Grossniklaus HE. Trichoblastoma of the eyelid. Ophthal Plast Reconstr Surg. 2011;27:E148-E149.
- Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblasticcarcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16.
- Aslani FS, Akbarzadeh-Jahromi M, Jowkar F. Value of CD10 expression in differentiating cutaneous basal from squamous cell carcinomas and basal cell carcinoma from trichoepithelioma. Iran J Med Sci. 2013;38:100-106.
- Kurzen H, Esposito L, Langbein L, et al. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23:501-509.
- Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinoma but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
- McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26:119-124.
- Izikson L, Bhan A, Zembowicz A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am J Dermatopathol. 2005;27:91-95.
- Benaim G, Castillo C, Houang M, et al. Melanoma arising from a long standing pigmented trichoblastoma: clinicopathologic study with complementary aCGH/mutation analysis. Am J Dermatopathol. 2014;36:E146-E151.
- Headington JT. Tumors of the hair follicle: a review. Am J Pathol. 1976; 85 : 479- 514 .
- Wong TY, Reed JA, Suster S, et al. Benign trichogenic tumors: a report of two cases supporting a simplified nomenclature. Histopathology. 1993;22:575-580.
- Aloi F, Tomasini C, Pippione M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14:345-349.
- Kanitakis J, Brutzkus A, Butnaru AC, et al. Melanotrichoblastoma: immunohistochemical study of a variant of pigmented trichoblastoma. Am J Dermatopathol. 2002;24:498-501.
- Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:E37-E40.
- Hung CT, Chiang CP, Gao HW, et al. Ripple-pattern melanotrichoblastoma arising within nevus sebaceous. Indian J Dermatol Venereol Leprol. 2012;78:665.
- Sau P, Lupton GP, Graham JH. Trichogerminoma: report of 14 cases. J Cutan Pathol. 1992;19:357-365.
- Johnson TV, Wojno TH, Grossniklaus HE. Trichoblastoma of the eyelid. Ophthal Plast Reconstr Surg. 2011;27:E148-E149.
- Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblasticcarcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16.
- Aslani FS, Akbarzadeh-Jahromi M, Jowkar F. Value of CD10 expression in differentiating cutaneous basal from squamous cell carcinomas and basal cell carcinoma from trichoepithelioma. Iran J Med Sci. 2013;38:100-106.
- Kurzen H, Esposito L, Langbein L, et al. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23:501-509.
- Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinoma but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
- McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26:119-124.
- Izikson L, Bhan A, Zembowicz A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am J Dermatopathol. 2005;27:91-95.
- Benaim G, Castillo C, Houang M, et al. Melanoma arising from a long standing pigmented trichoblastoma: clinicopathologic study with complementary aCGH/mutation analysis. Am J Dermatopathol. 2014;36:E146-E151.
Practice Points
- Pigmented trichoblastoma is a histologic variant of trichoblastoma characterized by the presence of melanin pigment.
- At least some pigmented trichoblastomas contain melanocytes and have been referred to as melanotrichoblastomas.
- The presence of melanocytes within pigmented trichoblastomas should not be confused as representing an example of colonization or a collision tumor.
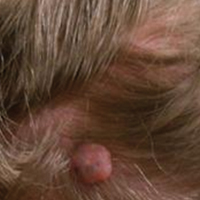
Granulomatous Pigmented Purpuric Dermatosis
Pigmented purpuric dermatoses (PPDs) are a spectrum of chronic disorders that present as speckled brown to purpuric lesions and orange-brown discoloration of the skin.1 Eruptions generally occur in middle-aged to elderly patients and commonly follow a chronic waxing and waning course.2 Lesions usually are found in a localized distribution on the legs. Histologically, PPD presents with perivascular infiltrates of lymphocytes and macrophages centered around the superficial small blood vessels with narrowing of the lumina. Extravasation of red blood cells and hemosiderin deposition are commonly seen in the absence of vasculitis.
The etiology of PPD is unknown; however, important cofactors include venous hypertension, exercise and gravitational dependency, capillary fragility, focal infections, and chemical ingestions.1 Drugs are the most important provoking factors, including acetaminophen, aspirin, adalin, carbromal, chlordiazepoxide, glipizide, glybuzole, hydralazine, meprobamate, dipyridamole, reserpine, thiamine, and interferon-alfa, as well as medroxyprogesterone acetate injection. Other phenomena include contact allergy and alcohol ingestion.1
Although the diagnosis often is made clinically, many forms of PPD exist. The 4 main forms include Schaumberg disease, purpura annularis telangiectaticum of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, and eczematoidlike purpura of Doucas and Kapetanakis. Less common variants include itching purpura of Lowenthal, lichen purpuricus, lichen aureus, granulomatous pigmented purpura, transitory pigmented purpuric dermatosis, and linear pigmented purpura.1Granulomatous PPD (GPPD) is a rare histologic variant of PPD. Clinically, it is indistinguishable from other forms of PPD but reveals itself histologically with granulomatous infiltrates superimposed on classic PPD. We report a case of GPPD and provide a thorough literature review focusing on epidemiology, clinical symptoms, and treatment.2-17 The eTable summarizes all reported cases of GPPD.
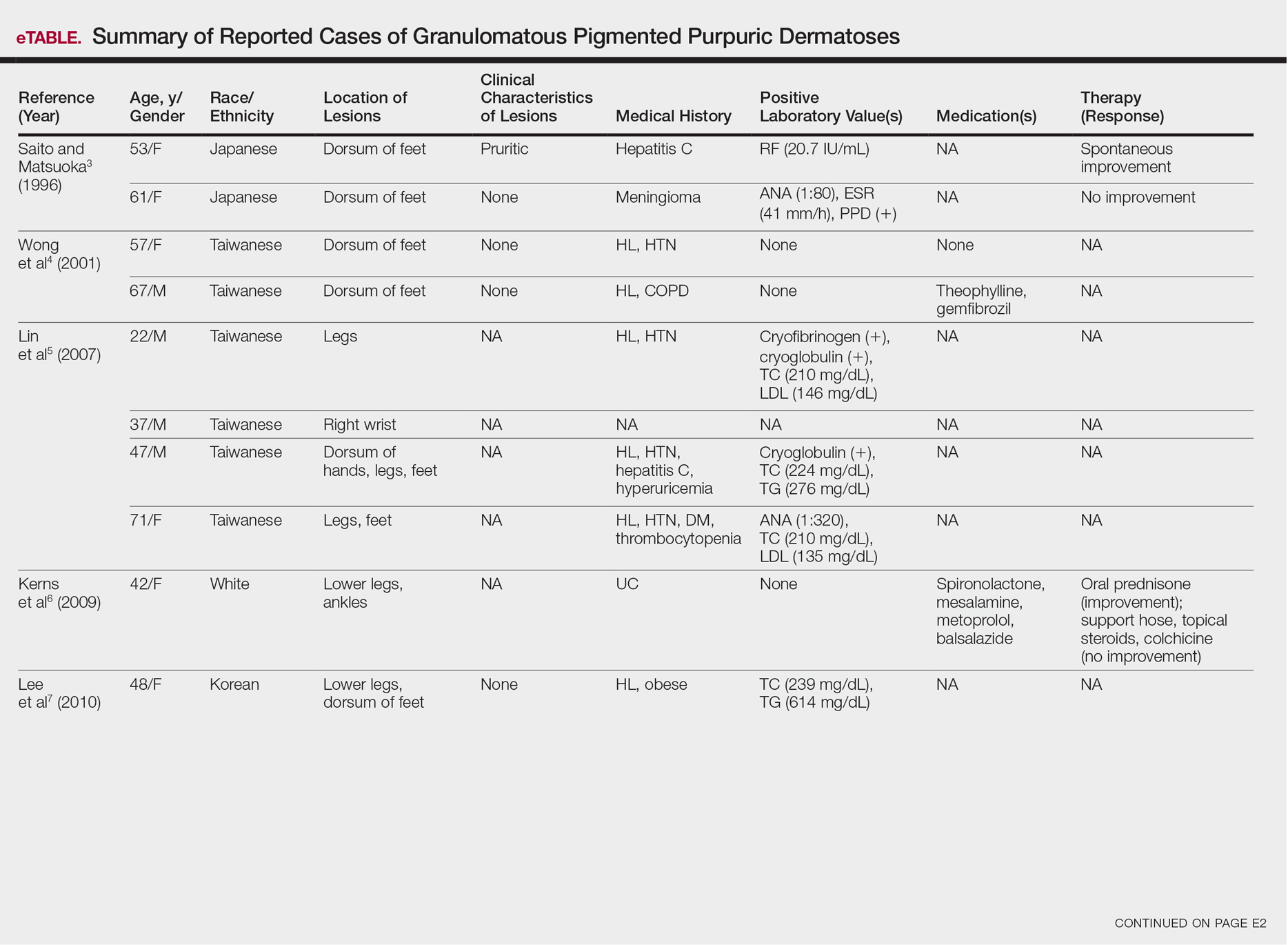
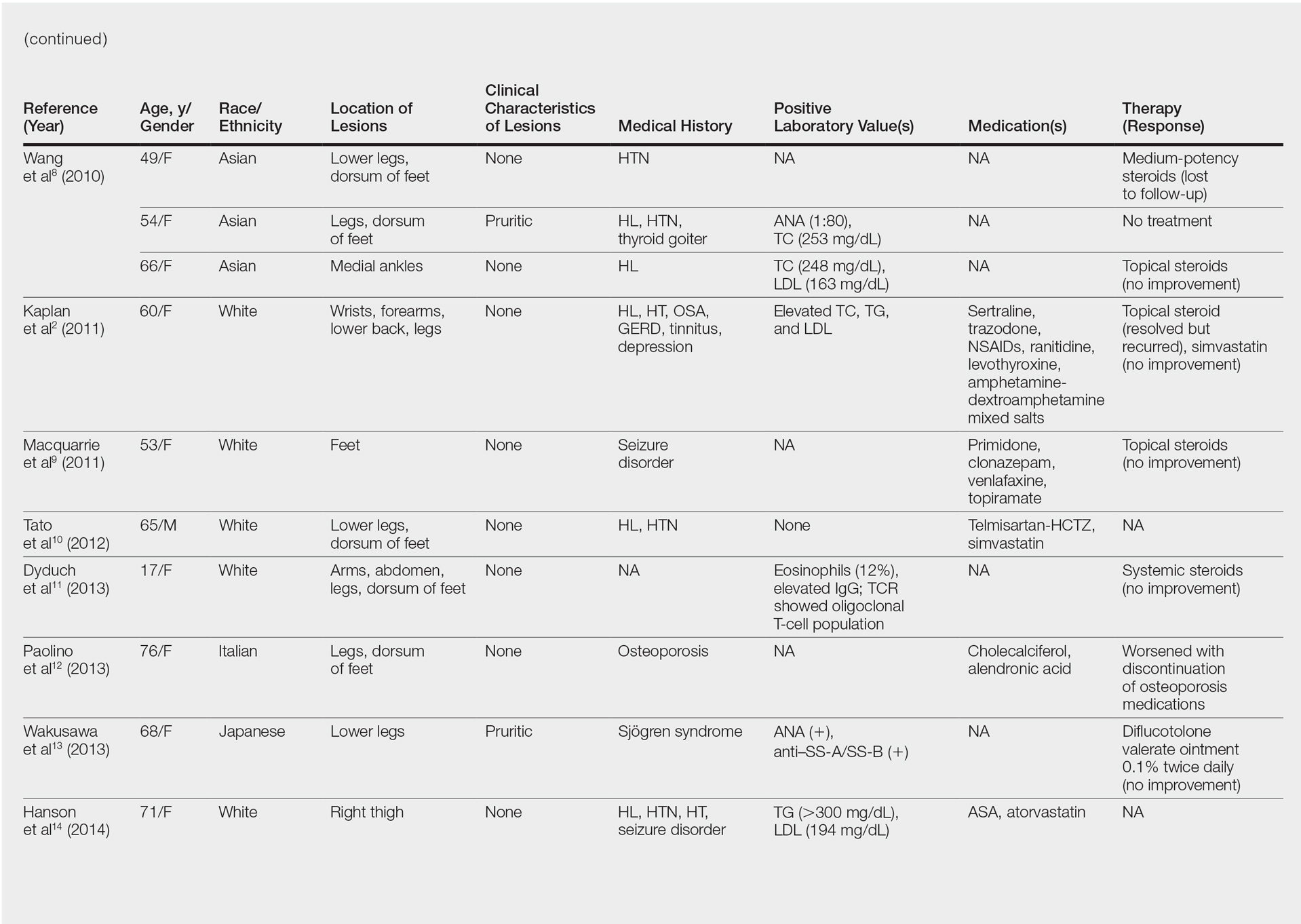
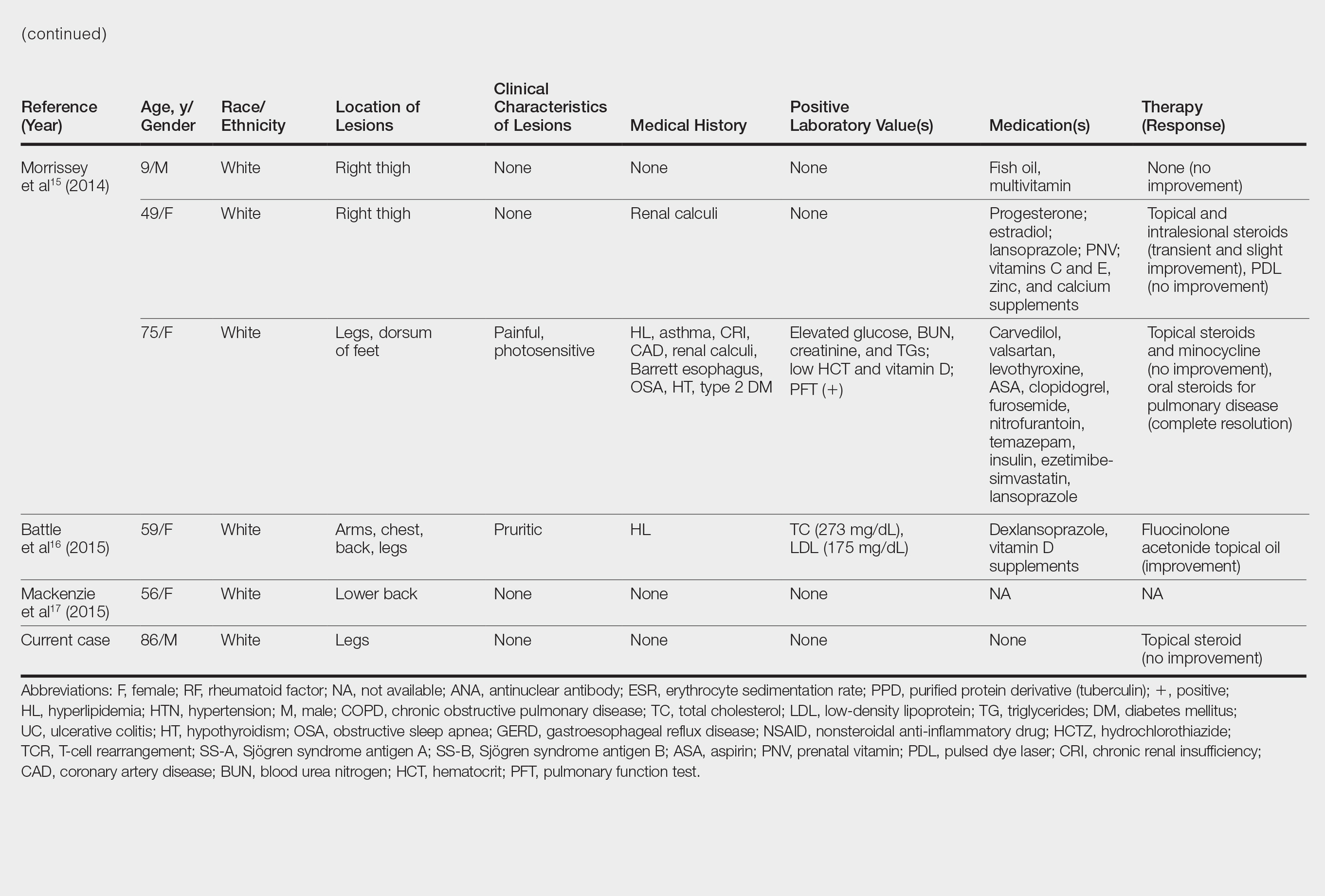
Case Report
An 86-year-old white man with no remarkable medical history presented with an asymptomatic eruption over the bilateral shins extending up both thighs of 6 years’ duration (Figure 1). It began as a 15-cm patch on the right medial thigh that rapidly spread over 1 year to involve the majority of the legs. Physical examination revealed scattered 1- to 2-mm brown macules coalescing into patches on both legs. The patches increased in density distally and extended from the bilateral thighs to the ankles. Edema of the legs was absent, and lesions were nonblanchable and without scale or induration. The differential diagnoses included stasis dermatitis, vasculitis, and PPD. All laboratory values were within reference range, including complete blood cell count, comprehensive metabolic panel, urine analysis, and lipid profile.
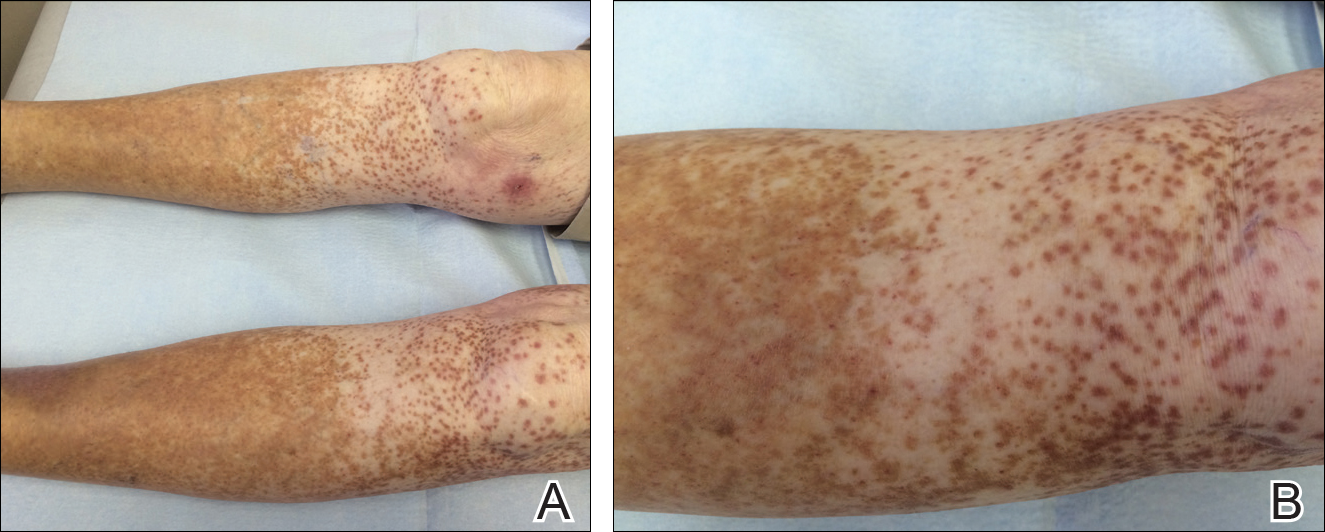
A punch biopsy from the distal right thigh revealed a superficial to mid dermal perivascular lymphocyte-predominant infiltrate with associated siderophages and a focal granulomatous infiltrate comprised of histiocytes (Figure 2). Periodic acid–Schiff, acid-fast bacilli, and Fite stains were negative for microorganisms. No eosinophils or leukocytoclasia were seen. The patient applied betamethasone dipropionate cream 0.05% twice daily for several weeks without improvement. Because the lesions were asymptomatic, he discontinued the topical medication.
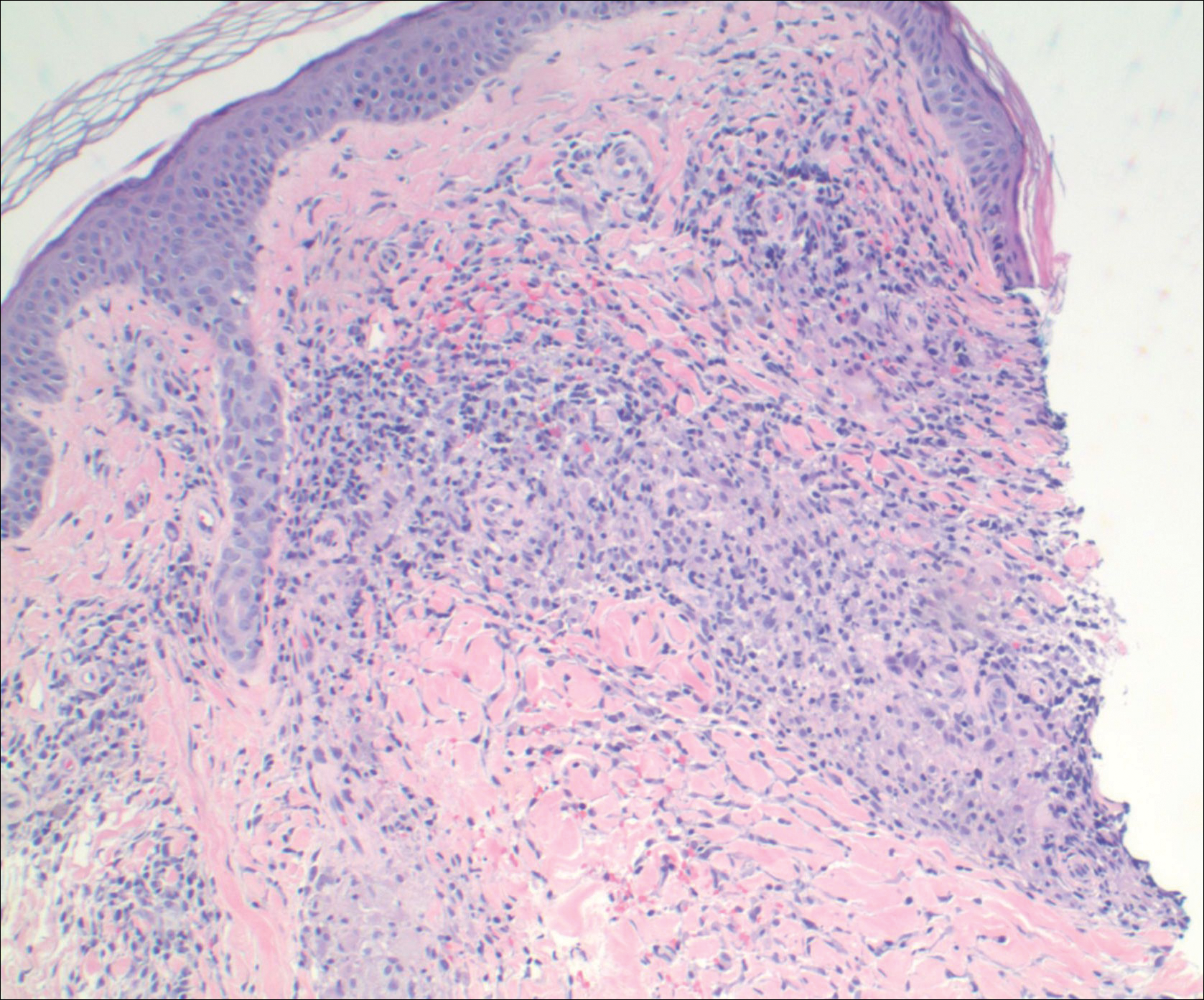
Comment
Pathogenesis/Etiology of GPPD
Granulomatous PPD is a rare histological variant of PPD, which was first reported in 1996 by Saito and Matsuoka.3 Originally, GPPD was mainly thought to affect individuals in the Far East and be associated with the hepatitis C virus, antinuclear antibodies, or rheumatoid factor.3 Since its initial description, GPPD continues to predominantly be seen on the distal legs. According to a PubMed search of articles indexed for MEDLINE and the Michigan State University library database using the terms granulomatous pigmented purpuric dermatosis and pigmented purpuric dermatosis, 26 known cases including the current case (Asian, n=13; white, n=13) have been reported. The mean age of onset was 54.5 years and the female to male ratio was 2.5 to 1.
Currently, the etiology of GPPD is unknown; however, 13 reported cases have been associated with hyperlipidemia,2,4,5,7,8,10,14-16 which has led to the speculation that they may be related. Previous investigators have postulated that the granulomatous infiltrate is a response to lipid deposition in the endothelial cells or that the elevated lipid levels launch an incompetent helper T cell (TH1) response, leading to granuloma formation.5,7,8 Currently, hyperlipidemia is present in 50% of patients and appears to be trending downward as more cases present in the literature.
Medications have been implicated in the pathogenesis of PPD and may have a possible role in the development of the granulomatous variant.9 One case report noted preceding medication changes, alluding to the possibility of aminosalicylates being the culprit.6
Another case described GPPD appearing after an upper respiratory tract infection.11 Comorbidities are not uncommon in patients presenting with GPPD. Although the majority of cases are single reports, they include systemic derangements such as hepatitis C,3,5 Sjögren syndrome,13 hypertension,2,4,5,8,10,14,15 seizure disorder,9,14 ulcerative colitis,6 diabetes mellitus,5,15 meningioma,3 renal calculi,15 thrombocytopenia,5 chronic obstructive pulmonary disease,4 thyroid goiter,8 obstructive sleep apnea,2,15 osteoporosis,12 asthma,15 gastroesophageal reflux disease/Barrett esophagus,2,15 hypothyroidism,2,14,15 and hyperuricemia.5
Clinical Presentation
Clinically, GPPD commonly presents as asymptomatic petechiae and bronze discoloration of the lower legs. The clinical presentation can vary from a solitary lesion to a localized eruption typically on the lower legs or rarely a widespread eruption. A review of the literature revealed 5 cases presenting on the upper arms2,5,11,16 and 4 on the trunk.2,11,16,17 Four patients presented with pruritus3,8,13,16 and 1 described pain and photosensitive lesions.15 No other clinical signs of hyperlipidemia were described (eg, xanthomas). The duration of the disease has a wide spectrum, ranging from 3 weeks to 20 years.4,16
Histopathology
With the increasing trend toward dermatoscopic evaluation, 2 reviews evaluated dermatoscopic features of GPPD. These reports described scattered, round to oval, red dots, globules, and patches with a diffuse red-brown or coppery background of pigmentation.14,17
The granulomatous variant of PPD is characterized histopathologically by ill-defined, nonnecrotizing granulomas admixed with a lymphocytic infiltrate. Commonly, erythrocyte extravasation and hemosiderin are seen with granulomas superimposed on classic changes of PPD.15 Vasculitis features including endothelial swelling, fibrinoid necrosis, and leukocytoclasia are absent. Rarely, eosinophils are seen.6 Mild epidermal spongiosis and exocytosis of lymphocytes may be seen in all variants of PPD, except lichen aureus.1 This exocytosis was observed focally in one case of GPPD.4 Although loosely formed granulomas in the papillary dermis are characteristic, 7 cases have had a concomitant lichenoid infiltrate.2,9-11,15,16
Kaplan et al2 reported granulomatous and nongranulomatous PPD occurring together in different areas of the body. A new granulomatous variant was proposed in a 2015 report that revealed 2 patients with granulomatous infiltrates in the mid to deep dermis rather than the classic superficial dermis.15 One case of GPPD was suspicious for progression into mycosis fungoides (MF) and described a lichenoid infiltrate with mild atypical and small lymphocytes migrating into the epidermis.11 Follow-up biopsy lacked epidermotropism and quantitative representation of T-cell subsets. The diagnosis of early-phase MF was based on the progressive clinical course rather than immunohistologic and molecular findings.11 One other case exhibited minimal epidermotropism.15
Management of GPPD should require a lipid profile with other tests to assess cardiovascular risk.10 A thorough medication review and a punch rather than a shave biopsy should be performed, especially because granulomatous infiltrates have been found in the mid to deep dermis.15 With the lack of rebiopsies documented, follow-up and rebiopsy has been suggested if there is suspicion of MF; however, we favor rebiopsy at a later time to help reveal the course of this disease and rule out progression into MF.
Therapy
Thus far, therapy has mostly been with oral and topical steroids. Five case reports noted improvement,2,3,6,15,16 2 with oral and 3 with topical steroids. However, therapy has been discouraging, with clinical improvement being transient in most treatment-responsive patients. One case spontaneously resolved.3 Ten cases did not document therapy or follow-up.4,5,7,10,14,17 Only 1 case reported follow-up after treatment with simvastatin; unfortunately, the patient had no improvement.2 Our case revealed no improvement with topical steroids.
Conclusion
The exact pathogenesis of GPPD is unknown. The initial impression that GPPD was a disease in Far East Asians and patients with hyperlipidemia is becoming less clear. Based on the current literature including the addition of our case, the prevalence appears to be equal among white individuals and Asians, possibly due to increased awareness of this condition and documentation in the literature. Correlation with systemic disorders such as hyperlipidemia and hypertensive medications needs further review. Eight cases reported a medical history of hypertension.4,5,8,10,14 With antihypertensive medications being a potential culprit of PPD, this etiology should not be overlooked. A punch biopsy should be performed, especially because granulomatous infiltrates may be lurking in the mid to deep dermis.15 Granulomatous PPD has a chronic course with a disappointing response to therapy but appears to be benign in nature.12 A rebiopsy is recommended if MF is suspected. Evaluation of GPPD following therapy for hyperlipidemia is not well documented and should be pursued. Clinicians and pathologists should be aware of the suspected associations and consider this variant when dermal granulomatous infiltrates are present with a background of PPD.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Kaplan J, Burgin S, Sepehr A. Granulomatous pigmented purpura: report of a case and review of the literature. J Cutan Pathol. 2011;38:984-989.
- Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
- Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
- Lin WL, Kuo TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia. Clin Exp Dermatol. 2007;32:513-515.
- Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
- Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidaemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
- Wang J, Wu Y, Hsiao P, et al. Granulomatous pigmented purpuric dermatoses: report of three cases and review of the literature. Dermatologica Sinica. 2010;28:77-81.
- Macquarrie EK, Pasternak S, Torok M, et al. Persistent pigmented purpuric dermatitis: granulomatous variant. J Cutan Pathol. 2011;38:979-983.
- Tato BP, Marinero Escobedo S, Pérez González YC, et al. Granulomatous variant of pigmented purpuric dermatosis. Am J Dermatopathol. 2012;34:746-748.
- Dyduch G, Zuber Z, Turowska-Heydel D, et al. Granulomatous pigmented purpura in an adolescent girl: a precursor of mycosis fungoides? Pol J Pathol. 2013;64:157-159; answer 160.
- Paolino S, Cinotti E, Merlo V, et al. Progressive petechial and pigmented macules and papules on the lower extremities. Am J Dermatopathol. 2013;35:370, 388.
- Wakusawa C, Fujimura T, Haga T, et al. Granulomatous pigmented purpuric dermatitis associated with primary Sjögren’s syndrome. Acta Derm Venereol. 2013;93:95-96.
- Hanson C, Fischer R, Fraga G, et al. Granulomatous pigmented purpuric dermatosis: an unusual variant associated with hyperlipidemia. Dermatol Online J. 2014;21. pii:13030/qt0tp272d1.
- Morrissey K, Rosenbach M, DeHoratius D, et al. Granulomatous changes associated with pigmented purpuric dermatosis. Cutis. 2014;94:197-202.
- Battle LR, Shalin SC, Gao L. Granulomatous pigmented purpuric dermatosis [published online December 18, 2014]. Clin Exp Dermatol. 2015;40:387-390.
- Mackenzie AI, Biswas A. Granulomatous pigmented purpuric dermatosis: report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-314.
Pigmented purpuric dermatoses (PPDs) are a spectrum of chronic disorders that present as speckled brown to purpuric lesions and orange-brown discoloration of the skin.1 Eruptions generally occur in middle-aged to elderly patients and commonly follow a chronic waxing and waning course.2 Lesions usually are found in a localized distribution on the legs. Histologically, PPD presents with perivascular infiltrates of lymphocytes and macrophages centered around the superficial small blood vessels with narrowing of the lumina. Extravasation of red blood cells and hemosiderin deposition are commonly seen in the absence of vasculitis.
The etiology of PPD is unknown; however, important cofactors include venous hypertension, exercise and gravitational dependency, capillary fragility, focal infections, and chemical ingestions.1 Drugs are the most important provoking factors, including acetaminophen, aspirin, adalin, carbromal, chlordiazepoxide, glipizide, glybuzole, hydralazine, meprobamate, dipyridamole, reserpine, thiamine, and interferon-alfa, as well as medroxyprogesterone acetate injection. Other phenomena include contact allergy and alcohol ingestion.1
Although the diagnosis often is made clinically, many forms of PPD exist. The 4 main forms include Schaumberg disease, purpura annularis telangiectaticum of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, and eczematoidlike purpura of Doucas and Kapetanakis. Less common variants include itching purpura of Lowenthal, lichen purpuricus, lichen aureus, granulomatous pigmented purpura, transitory pigmented purpuric dermatosis, and linear pigmented purpura.1Granulomatous PPD (GPPD) is a rare histologic variant of PPD. Clinically, it is indistinguishable from other forms of PPD but reveals itself histologically with granulomatous infiltrates superimposed on classic PPD. We report a case of GPPD and provide a thorough literature review focusing on epidemiology, clinical symptoms, and treatment.2-17 The eTable summarizes all reported cases of GPPD.
Case Report
An 86-year-old white man with no remarkable medical history presented with an asymptomatic eruption over the bilateral shins extending up both thighs of 6 years’ duration (Figure 1). It began as a 15-cm patch on the right medial thigh that rapidly spread over 1 year to involve the majority of the legs. Physical examination revealed scattered 1- to 2-mm brown macules coalescing into patches on both legs. The patches increased in density distally and extended from the bilateral thighs to the ankles. Edema of the legs was absent, and lesions were nonblanchable and without scale or induration. The differential diagnoses included stasis dermatitis, vasculitis, and PPD. All laboratory values were within reference range, including complete blood cell count, comprehensive metabolic panel, urine analysis, and lipid profile.
A punch biopsy from the distal right thigh revealed a superficial to mid dermal perivascular lymphocyte-predominant infiltrate with associated siderophages and a focal granulomatous infiltrate comprised of histiocytes (Figure 2). Periodic acid–Schiff, acid-fast bacilli, and Fite stains were negative for microorganisms. No eosinophils or leukocytoclasia were seen. The patient applied betamethasone dipropionate cream 0.05% twice daily for several weeks without improvement. Because the lesions were asymptomatic, he discontinued the topical medication.
Comment
Pathogenesis/Etiology of GPPD
Granulomatous PPD is a rare histological variant of PPD, which was first reported in 1996 by Saito and Matsuoka.3 Originally, GPPD was mainly thought to affect individuals in the Far East and be associated with the hepatitis C virus, antinuclear antibodies, or rheumatoid factor.3 Since its initial description, GPPD continues to predominantly be seen on the distal legs. According to a PubMed search of articles indexed for MEDLINE and the Michigan State University library database using the terms granulomatous pigmented purpuric dermatosis and pigmented purpuric dermatosis, 26 known cases including the current case (Asian, n=13; white, n=13) have been reported. The mean age of onset was 54.5 years and the female to male ratio was 2.5 to 1.
Currently, the etiology of GPPD is unknown; however, 13 reported cases have been associated with hyperlipidemia,2,4,5,7,8,10,14-16 which has led to the speculation that they may be related. Previous investigators have postulated that the granulomatous infiltrate is a response to lipid deposition in the endothelial cells or that the elevated lipid levels launch an incompetent helper T cell (TH1) response, leading to granuloma formation.5,7,8 Currently, hyperlipidemia is present in 50% of patients and appears to be trending downward as more cases present in the literature.
Medications have been implicated in the pathogenesis of PPD and may have a possible role in the development of the granulomatous variant.9 One case report noted preceding medication changes, alluding to the possibility of aminosalicylates being the culprit.6
Another case described GPPD appearing after an upper respiratory tract infection.11 Comorbidities are not uncommon in patients presenting with GPPD. Although the majority of cases are single reports, they include systemic derangements such as hepatitis C,3,5 Sjögren syndrome,13 hypertension,2,4,5,8,10,14,15 seizure disorder,9,14 ulcerative colitis,6 diabetes mellitus,5,15 meningioma,3 renal calculi,15 thrombocytopenia,5 chronic obstructive pulmonary disease,4 thyroid goiter,8 obstructive sleep apnea,2,15 osteoporosis,12 asthma,15 gastroesophageal reflux disease/Barrett esophagus,2,15 hypothyroidism,2,14,15 and hyperuricemia.5
Clinical Presentation
Clinically, GPPD commonly presents as asymptomatic petechiae and bronze discoloration of the lower legs. The clinical presentation can vary from a solitary lesion to a localized eruption typically on the lower legs or rarely a widespread eruption. A review of the literature revealed 5 cases presenting on the upper arms2,5,11,16 and 4 on the trunk.2,11,16,17 Four patients presented with pruritus3,8,13,16 and 1 described pain and photosensitive lesions.15 No other clinical signs of hyperlipidemia were described (eg, xanthomas). The duration of the disease has a wide spectrum, ranging from 3 weeks to 20 years.4,16
Histopathology
With the increasing trend toward dermatoscopic evaluation, 2 reviews evaluated dermatoscopic features of GPPD. These reports described scattered, round to oval, red dots, globules, and patches with a diffuse red-brown or coppery background of pigmentation.14,17
The granulomatous variant of PPD is characterized histopathologically by ill-defined, nonnecrotizing granulomas admixed with a lymphocytic infiltrate. Commonly, erythrocyte extravasation and hemosiderin are seen with granulomas superimposed on classic changes of PPD.15 Vasculitis features including endothelial swelling, fibrinoid necrosis, and leukocytoclasia are absent. Rarely, eosinophils are seen.6 Mild epidermal spongiosis and exocytosis of lymphocytes may be seen in all variants of PPD, except lichen aureus.1 This exocytosis was observed focally in one case of GPPD.4 Although loosely formed granulomas in the papillary dermis are characteristic, 7 cases have had a concomitant lichenoid infiltrate.2,9-11,15,16
Kaplan et al2 reported granulomatous and nongranulomatous PPD occurring together in different areas of the body. A new granulomatous variant was proposed in a 2015 report that revealed 2 patients with granulomatous infiltrates in the mid to deep dermis rather than the classic superficial dermis.15 One case of GPPD was suspicious for progression into mycosis fungoides (MF) and described a lichenoid infiltrate with mild atypical and small lymphocytes migrating into the epidermis.11 Follow-up biopsy lacked epidermotropism and quantitative representation of T-cell subsets. The diagnosis of early-phase MF was based on the progressive clinical course rather than immunohistologic and molecular findings.11 One other case exhibited minimal epidermotropism.15
Management of GPPD should require a lipid profile with other tests to assess cardiovascular risk.10 A thorough medication review and a punch rather than a shave biopsy should be performed, especially because granulomatous infiltrates have been found in the mid to deep dermis.15 With the lack of rebiopsies documented, follow-up and rebiopsy has been suggested if there is suspicion of MF; however, we favor rebiopsy at a later time to help reveal the course of this disease and rule out progression into MF.
Therapy
Thus far, therapy has mostly been with oral and topical steroids. Five case reports noted improvement,2,3,6,15,16 2 with oral and 3 with topical steroids. However, therapy has been discouraging, with clinical improvement being transient in most treatment-responsive patients. One case spontaneously resolved.3 Ten cases did not document therapy or follow-up.4,5,7,10,14,17 Only 1 case reported follow-up after treatment with simvastatin; unfortunately, the patient had no improvement.2 Our case revealed no improvement with topical steroids.
Conclusion
The exact pathogenesis of GPPD is unknown. The initial impression that GPPD was a disease in Far East Asians and patients with hyperlipidemia is becoming less clear. Based on the current literature including the addition of our case, the prevalence appears to be equal among white individuals and Asians, possibly due to increased awareness of this condition and documentation in the literature. Correlation with systemic disorders such as hyperlipidemia and hypertensive medications needs further review. Eight cases reported a medical history of hypertension.4,5,8,10,14 With antihypertensive medications being a potential culprit of PPD, this etiology should not be overlooked. A punch biopsy should be performed, especially because granulomatous infiltrates may be lurking in the mid to deep dermis.15 Granulomatous PPD has a chronic course with a disappointing response to therapy but appears to be benign in nature.12 A rebiopsy is recommended if MF is suspected. Evaluation of GPPD following therapy for hyperlipidemia is not well documented and should be pursued. Clinicians and pathologists should be aware of the suspected associations and consider this variant when dermal granulomatous infiltrates are present with a background of PPD.
Pigmented purpuric dermatoses (PPDs) are a spectrum of chronic disorders that present as speckled brown to purpuric lesions and orange-brown discoloration of the skin.1 Eruptions generally occur in middle-aged to elderly patients and commonly follow a chronic waxing and waning course.2 Lesions usually are found in a localized distribution on the legs. Histologically, PPD presents with perivascular infiltrates of lymphocytes and macrophages centered around the superficial small blood vessels with narrowing of the lumina. Extravasation of red blood cells and hemosiderin deposition are commonly seen in the absence of vasculitis.
The etiology of PPD is unknown; however, important cofactors include venous hypertension, exercise and gravitational dependency, capillary fragility, focal infections, and chemical ingestions.1 Drugs are the most important provoking factors, including acetaminophen, aspirin, adalin, carbromal, chlordiazepoxide, glipizide, glybuzole, hydralazine, meprobamate, dipyridamole, reserpine, thiamine, and interferon-alfa, as well as medroxyprogesterone acetate injection. Other phenomena include contact allergy and alcohol ingestion.1
Although the diagnosis often is made clinically, many forms of PPD exist. The 4 main forms include Schaumberg disease, purpura annularis telangiectaticum of Majocchi, pigmented purpuric lichenoid dermatitis of Gougerot and Blum, and eczematoidlike purpura of Doucas and Kapetanakis. Less common variants include itching purpura of Lowenthal, lichen purpuricus, lichen aureus, granulomatous pigmented purpura, transitory pigmented purpuric dermatosis, and linear pigmented purpura.1Granulomatous PPD (GPPD) is a rare histologic variant of PPD. Clinically, it is indistinguishable from other forms of PPD but reveals itself histologically with granulomatous infiltrates superimposed on classic PPD. We report a case of GPPD and provide a thorough literature review focusing on epidemiology, clinical symptoms, and treatment.2-17 The eTable summarizes all reported cases of GPPD.
Case Report
An 86-year-old white man with no remarkable medical history presented with an asymptomatic eruption over the bilateral shins extending up both thighs of 6 years’ duration (Figure 1). It began as a 15-cm patch on the right medial thigh that rapidly spread over 1 year to involve the majority of the legs. Physical examination revealed scattered 1- to 2-mm brown macules coalescing into patches on both legs. The patches increased in density distally and extended from the bilateral thighs to the ankles. Edema of the legs was absent, and lesions were nonblanchable and without scale or induration. The differential diagnoses included stasis dermatitis, vasculitis, and PPD. All laboratory values were within reference range, including complete blood cell count, comprehensive metabolic panel, urine analysis, and lipid profile.
A punch biopsy from the distal right thigh revealed a superficial to mid dermal perivascular lymphocyte-predominant infiltrate with associated siderophages and a focal granulomatous infiltrate comprised of histiocytes (Figure 2). Periodic acid–Schiff, acid-fast bacilli, and Fite stains were negative for microorganisms. No eosinophils or leukocytoclasia were seen. The patient applied betamethasone dipropionate cream 0.05% twice daily for several weeks without improvement. Because the lesions were asymptomatic, he discontinued the topical medication.
Comment
Pathogenesis/Etiology of GPPD
Granulomatous PPD is a rare histological variant of PPD, which was first reported in 1996 by Saito and Matsuoka.3 Originally, GPPD was mainly thought to affect individuals in the Far East and be associated with the hepatitis C virus, antinuclear antibodies, or rheumatoid factor.3 Since its initial description, GPPD continues to predominantly be seen on the distal legs. According to a PubMed search of articles indexed for MEDLINE and the Michigan State University library database using the terms granulomatous pigmented purpuric dermatosis and pigmented purpuric dermatosis, 26 known cases including the current case (Asian, n=13; white, n=13) have been reported. The mean age of onset was 54.5 years and the female to male ratio was 2.5 to 1.
Currently, the etiology of GPPD is unknown; however, 13 reported cases have been associated with hyperlipidemia,2,4,5,7,8,10,14-16 which has led to the speculation that they may be related. Previous investigators have postulated that the granulomatous infiltrate is a response to lipid deposition in the endothelial cells or that the elevated lipid levels launch an incompetent helper T cell (TH1) response, leading to granuloma formation.5,7,8 Currently, hyperlipidemia is present in 50% of patients and appears to be trending downward as more cases present in the literature.
Medications have been implicated in the pathogenesis of PPD and may have a possible role in the development of the granulomatous variant.9 One case report noted preceding medication changes, alluding to the possibility of aminosalicylates being the culprit.6
Another case described GPPD appearing after an upper respiratory tract infection.11 Comorbidities are not uncommon in patients presenting with GPPD. Although the majority of cases are single reports, they include systemic derangements such as hepatitis C,3,5 Sjögren syndrome,13 hypertension,2,4,5,8,10,14,15 seizure disorder,9,14 ulcerative colitis,6 diabetes mellitus,5,15 meningioma,3 renal calculi,15 thrombocytopenia,5 chronic obstructive pulmonary disease,4 thyroid goiter,8 obstructive sleep apnea,2,15 osteoporosis,12 asthma,15 gastroesophageal reflux disease/Barrett esophagus,2,15 hypothyroidism,2,14,15 and hyperuricemia.5
Clinical Presentation
Clinically, GPPD commonly presents as asymptomatic petechiae and bronze discoloration of the lower legs. The clinical presentation can vary from a solitary lesion to a localized eruption typically on the lower legs or rarely a widespread eruption. A review of the literature revealed 5 cases presenting on the upper arms2,5,11,16 and 4 on the trunk.2,11,16,17 Four patients presented with pruritus3,8,13,16 and 1 described pain and photosensitive lesions.15 No other clinical signs of hyperlipidemia were described (eg, xanthomas). The duration of the disease has a wide spectrum, ranging from 3 weeks to 20 years.4,16
Histopathology
With the increasing trend toward dermatoscopic evaluation, 2 reviews evaluated dermatoscopic features of GPPD. These reports described scattered, round to oval, red dots, globules, and patches with a diffuse red-brown or coppery background of pigmentation.14,17
The granulomatous variant of PPD is characterized histopathologically by ill-defined, nonnecrotizing granulomas admixed with a lymphocytic infiltrate. Commonly, erythrocyte extravasation and hemosiderin are seen with granulomas superimposed on classic changes of PPD.15 Vasculitis features including endothelial swelling, fibrinoid necrosis, and leukocytoclasia are absent. Rarely, eosinophils are seen.6 Mild epidermal spongiosis and exocytosis of lymphocytes may be seen in all variants of PPD, except lichen aureus.1 This exocytosis was observed focally in one case of GPPD.4 Although loosely formed granulomas in the papillary dermis are characteristic, 7 cases have had a concomitant lichenoid infiltrate.2,9-11,15,16
Kaplan et al2 reported granulomatous and nongranulomatous PPD occurring together in different areas of the body. A new granulomatous variant was proposed in a 2015 report that revealed 2 patients with granulomatous infiltrates in the mid to deep dermis rather than the classic superficial dermis.15 One case of GPPD was suspicious for progression into mycosis fungoides (MF) and described a lichenoid infiltrate with mild atypical and small lymphocytes migrating into the epidermis.11 Follow-up biopsy lacked epidermotropism and quantitative representation of T-cell subsets. The diagnosis of early-phase MF was based on the progressive clinical course rather than immunohistologic and molecular findings.11 One other case exhibited minimal epidermotropism.15
Management of GPPD should require a lipid profile with other tests to assess cardiovascular risk.10 A thorough medication review and a punch rather than a shave biopsy should be performed, especially because granulomatous infiltrates have been found in the mid to deep dermis.15 With the lack of rebiopsies documented, follow-up and rebiopsy has been suggested if there is suspicion of MF; however, we favor rebiopsy at a later time to help reveal the course of this disease and rule out progression into MF.
Therapy
Thus far, therapy has mostly been with oral and topical steroids. Five case reports noted improvement,2,3,6,15,16 2 with oral and 3 with topical steroids. However, therapy has been discouraging, with clinical improvement being transient in most treatment-responsive patients. One case spontaneously resolved.3 Ten cases did not document therapy or follow-up.4,5,7,10,14,17 Only 1 case reported follow-up after treatment with simvastatin; unfortunately, the patient had no improvement.2 Our case revealed no improvement with topical steroids.
Conclusion
The exact pathogenesis of GPPD is unknown. The initial impression that GPPD was a disease in Far East Asians and patients with hyperlipidemia is becoming less clear. Based on the current literature including the addition of our case, the prevalence appears to be equal among white individuals and Asians, possibly due to increased awareness of this condition and documentation in the literature. Correlation with systemic disorders such as hyperlipidemia and hypertensive medications needs further review. Eight cases reported a medical history of hypertension.4,5,8,10,14 With antihypertensive medications being a potential culprit of PPD, this etiology should not be overlooked. A punch biopsy should be performed, especially because granulomatous infiltrates may be lurking in the mid to deep dermis.15 Granulomatous PPD has a chronic course with a disappointing response to therapy but appears to be benign in nature.12 A rebiopsy is recommended if MF is suspected. Evaluation of GPPD following therapy for hyperlipidemia is not well documented and should be pursued. Clinicians and pathologists should be aware of the suspected associations and consider this variant when dermal granulomatous infiltrates are present with a background of PPD.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Kaplan J, Burgin S, Sepehr A. Granulomatous pigmented purpura: report of a case and review of the literature. J Cutan Pathol. 2011;38:984-989.
- Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
- Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
- Lin WL, Kuo TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia. Clin Exp Dermatol. 2007;32:513-515.
- Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
- Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidaemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
- Wang J, Wu Y, Hsiao P, et al. Granulomatous pigmented purpuric dermatoses: report of three cases and review of the literature. Dermatologica Sinica. 2010;28:77-81.
- Macquarrie EK, Pasternak S, Torok M, et al. Persistent pigmented purpuric dermatitis: granulomatous variant. J Cutan Pathol. 2011;38:979-983.
- Tato BP, Marinero Escobedo S, Pérez González YC, et al. Granulomatous variant of pigmented purpuric dermatosis. Am J Dermatopathol. 2012;34:746-748.
- Dyduch G, Zuber Z, Turowska-Heydel D, et al. Granulomatous pigmented purpura in an adolescent girl: a precursor of mycosis fungoides? Pol J Pathol. 2013;64:157-159; answer 160.
- Paolino S, Cinotti E, Merlo V, et al. Progressive petechial and pigmented macules and papules on the lower extremities. Am J Dermatopathol. 2013;35:370, 388.
- Wakusawa C, Fujimura T, Haga T, et al. Granulomatous pigmented purpuric dermatitis associated with primary Sjögren’s syndrome. Acta Derm Venereol. 2013;93:95-96.
- Hanson C, Fischer R, Fraga G, et al. Granulomatous pigmented purpuric dermatosis: an unusual variant associated with hyperlipidemia. Dermatol Online J. 2014;21. pii:13030/qt0tp272d1.
- Morrissey K, Rosenbach M, DeHoratius D, et al. Granulomatous changes associated with pigmented purpuric dermatosis. Cutis. 2014;94:197-202.
- Battle LR, Shalin SC, Gao L. Granulomatous pigmented purpuric dermatosis [published online December 18, 2014]. Clin Exp Dermatol. 2015;40:387-390.
- Mackenzie AI, Biswas A. Granulomatous pigmented purpuric dermatosis: report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-314.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Kaplan J, Burgin S, Sepehr A. Granulomatous pigmented purpura: report of a case and review of the literature. J Cutan Pathol. 2011;38:984-989.
- Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
- Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
- Lin WL, Kuo TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia. Clin Exp Dermatol. 2007;32:513-515.
- Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
- Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidaemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
- Wang J, Wu Y, Hsiao P, et al. Granulomatous pigmented purpuric dermatoses: report of three cases and review of the literature. Dermatologica Sinica. 2010;28:77-81.
- Macquarrie EK, Pasternak S, Torok M, et al. Persistent pigmented purpuric dermatitis: granulomatous variant. J Cutan Pathol. 2011;38:979-983.
- Tato BP, Marinero Escobedo S, Pérez González YC, et al. Granulomatous variant of pigmented purpuric dermatosis. Am J Dermatopathol. 2012;34:746-748.
- Dyduch G, Zuber Z, Turowska-Heydel D, et al. Granulomatous pigmented purpura in an adolescent girl: a precursor of mycosis fungoides? Pol J Pathol. 2013;64:157-159; answer 160.
- Paolino S, Cinotti E, Merlo V, et al. Progressive petechial and pigmented macules and papules on the lower extremities. Am J Dermatopathol. 2013;35:370, 388.
- Wakusawa C, Fujimura T, Haga T, et al. Granulomatous pigmented purpuric dermatitis associated with primary Sjögren’s syndrome. Acta Derm Venereol. 2013;93:95-96.
- Hanson C, Fischer R, Fraga G, et al. Granulomatous pigmented purpuric dermatosis: an unusual variant associated with hyperlipidemia. Dermatol Online J. 2014;21. pii:13030/qt0tp272d1.
- Morrissey K, Rosenbach M, DeHoratius D, et al. Granulomatous changes associated with pigmented purpuric dermatosis. Cutis. 2014;94:197-202.
- Battle LR, Shalin SC, Gao L. Granulomatous pigmented purpuric dermatosis [published online December 18, 2014]. Clin Exp Dermatol. 2015;40:387-390.
- Mackenzie AI, Biswas A. Granulomatous pigmented purpuric dermatosis: report of a case with atypical clinical presentation including dermoscopic findings. Am J Dermatopathol. 2015;37:311-314.
Practice Points
- Granulomatous pigmented purpuric dermatosis is not only seen in Far East Asians and patients with hyperlipidemia.
- Suspected pigmented purpuric dermatoses should be managed with a punch biopsy to exclude the granulomatous variant.
Paraskiing Crash and Knee Dislocation With Multiligament Reconstruction and Iliotibial Band Repair
Take-Home Points
- Reconstruction of a torn ITB is important in restoration of native anatomy and function given its properties in anterolateral stabilization and resistance to varus stress and internal tibial rotation.
- Restoration of posterolateral instability primarily involves reconstructing the FCL, PLT, and popliteofibular ligament.
- For combined PLC injuries, concurrent reconstruction of the cruciate ligaments in one stage is highly recommended.
- Post-surgery, a 6-week non-weight-bearing, limited flexion rehab protocol utilizing a dynamic PCL brace, such as the PCL Rebound brace, is recommended to prevent posterior tibial sag.
- Arthrofibrosis and decreased ROM can be seen following a violent knee injury which requires extensive multiligament reconstruction surgeries, occasionally requiring a secondary surgery for further restoration of knee motion.
Tibiofemoral knee dislocations are uncommon injuries that have devastating complications and potentially result in complex surgeries.1 Knee dislocations (KDs) can be classified with the Schenck system.2 KD-I is a multiligament injury involving the anterior cruciate ligament (ACL) or the posterior cruciate ligament (PCL), and the scale increases in severity/number of ligaments involved, with KD-V being a multiligament injury with periarticular fracture.2
In this article, we report the case of a complex multiligament knee reconstruction performed with a midsubstance iliotibial band (ITB) repair. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 27-year-old man presented 12 days after a paraskiing crash in which he collided with a tree at 45 mph and fell 40 feet before hitting snow. Physical examination revealed a large hemarthrosis of the left lower extremity and ecchymosis about the posterolateral aspect of the knee and popliteal fossa. Range of motion (ROM) was limited from 5° of hyperextension to 90° of flexion. Additional motion was deferred secondary to pain. Varus stress testing at 0° and 30° of knee flexion demonstrated significant side-to-side differences. The Lachman test, posterior drawer test, and posterolateral drawer test were all 3+. The dial test was 3 to 4+ compared with the contralateral knee. Valgus stress testing at 0° and 30° of flexion did not reveal any side-to-side laxity. The calf was nontender, and all compartments were soft. The patient reported no neurovascular symptoms and had no neuromotor deficits other than mild common peroneal nerve dysesthesias.
Varus stress radiographs showed increased side-to-side gapping (8 mm) of the lateral compartment of the injured knee. Kneeling posterior stress radiographs, limited because of the patient’s inability to apply full stress on the injured knee secondary to pain, showed a difference of 6 mm in increased posterior translation on the uninjured leg (Figures 1A-1D).
First Surgery
1. PLC Approach. A lateral hockey-stick skin incision was made along the ITB and extended distally between the fibular head and the Gerdy tubercle. The subcutaneous tissue was then dissected, and a posteriorly based flap was developed for preservation of vascular support to the superficial tissues. The ITB and the lateral capsule had completely torn off of the femur, allowing exposure directly into the joint. The long and short heads of the biceps femoris were exposed, with about 50% of the biceps attachment torn. The FCL was torn midsubstance, and the PLT had no remnant attachment left on the femur.
2. ITB and Lateral Capsule Tag Stitched. The torn ends of the ITB were dissected and tag stitches placed in each end. Tag stitches were also placed in the lateral capsule in preparation for a direct repair.
3. Neurolysis. The common peroneal nerve was found encased in a significant amount of scar tissue, and extensive neurolysis was required. Slow, methodical dissection was performed under the partially torn long head of the biceps femoris and was continued through the scar tissue and adhesions. Distally, 5 mm to 7 mm of the peroneus longus fascia was incised as part of the neurolysis in order to prevent nerve irritation or foot drop caused by postoperative swelling.
4. PLC Tunnels. The margin between the lateral gastrocnemius tendon and the soleus muscle was identified by blunt dissection that allowed palpation of the posteromedial aspect of the fibular styloid and the popliteus musculotendinous junction. The underlying biceps bursa was incised in order to locate the midportion of the FCL remnant, which typically is tag-stitched with No. 2 FiberWire to help identify the femoral attachment (this was not done because of the complete tear at the midsubstance of the FCL).
Subperiosteal dissection of the lateral aspect of the fibular head was performed anterior to posterior and distally extended to the champagne-glass drop-off of the fibular head. Continuing the dissection distally beyond this point can endanger the common peroneal nerve. A small sulcus can be palpated where the distal FCL inserts on the fibular head. Posteriorly, a small elevator was used to dissect the soleus muscle off of the posteromedial aspect of the fibular head, where the fibular tunnel would later be created.
A Chandler retractor was placed posterior to the fibular head to protect the neurovascular bundle. With the aid of a collateral ligament aiming device, a guide pin was drilled from the lateral aspect of the fibular head (FCL attachment) to the posteromedial downslope of the fibular styloid (popliteofibular ligament attachment). The entry point of the guide pin was immediately above the champagne- glass drop-off, at the distal insertion site of the FCL, which was described as being 28.4 mm from the styloid tip and 8.2 mm posterior to the anterior margin of the fibular head.3 Care should be taken not to ream the tunnel too proximal, as doing so increases the risk of iatrogenic fracture. A 7-mm reamer was then used to drill the fibular tunnel. To facilitate later passage of the graft, a passing suture was placed through the tunnel, leaving the loop anterolateral.
Next, the starting point for the tibial tunnel was located on the flat spot of the anterolateral tibia distal and medial to the Gerdy tubercle, just lateral to the tibial tubercle. The tibial popliteal sulcus was identified by palpation of the posterolateral tibial plateau to localize the site of the popliteus musculotendinous junction, which is the ideal location of the posterior aperture of the tibial tunnel. This point is 1 cm proximal and 1 cm medial to the posteromedial exit of the fibular tunnel. A Chandler retractor was placed anterior to the lateral gastrocnemius to protect the neurovascular bundle. In the locations described earlier, a cruciate aiming device was used to place a guide pin anterior to posterior. A 9-mm tunnel was overreamed and a passing suture placed, leaving the loop posterior to facilitate graft passage.
The femoral insertions of the FCL and the PLT were then identified. ITB splitting was not necessary, given the complete midsubstance tear of this structure. The FCL attachment was identified 1.4 mm proximal and 3.1 mm posterior to the lateral epicondyle.3 Sharp dissection was performed in this location, proximal to distal, exposing the lateral epicondyle and the small sulcus at the FCL attachment site. A collateral ligament reconstruction aiming sleeve was used to drill a guide pin over the FCL femoral attachment site and out the medial aspect of the distal thigh, about 5 cm proximal and anterior to the adductor tubercle.
The femoral attachment of the PLT was reported located 18.5 mm anterior to the FCL insertion, in the anterior fifth of the popliteal sulcus.3 Although arthrotomy is usually required in order to access the PLT attachment, it was not necessary in this case, given the lateral capsule tear. A guide pin was inserted at the PLT attachment site, parallel to the FCL pin. After proper placement was verified, a 9-mm reamer was used to drill the FCL and PLT tunnels to a depth of 25 mm (socket), and a passing suture was placed into each tunnel to facilitate graft passage.
5. ACL Graft Harvest. The central third of the ipsilateral patellar tendon was harvested for use in the ACL reconstruction. Included were a 10-mm × 20-mm bone plug from the patella and a 10-mm × 25-mm bone plug from the tibial tubercle. The patella defect was then bone-grafted, and the patellar tendon closed side-to-side.
6. Graft Preparation. For the PLC, we used a split Achilles tendon allograft that had two 9-mm × 25-mm bone plugs proximally and were tubularized distally. For the PCL, we used an anterolateral bundle (ALB), which consisted of an Achilles tendon allograft that had an 11-mm × 25-mm bone plug proximally and was tubularized distally, and a posteromedial bundle (PMB), which consisted of a tibialis anterior allograft that was tubularized at both ends. For the ACL, we used a bone–patellar tendon–bone autograft 10 mm in diameter with a 20-mm femoral bone plug and a 25-mm tibial bone plug distally.
7. Arthroscopy. We created standard anterolateral and anteromedial parapatellar portals and performed arthroscopy, including lysis of adhesions. Cartilage and menisci were lesion-free.
8. PCL Femoral Tunnels. The ALB attachment was identified and outlined with a coagulator between the trochlear point and the medial arch point, adjacent to the edge of the articular cartilage. Similarly, the PMB attachment was marked about 8 mm or 9 mm posterior to the edge of the articular cartilage of the medial femoral condyle and slightly posterior to the ALB tunnel.4
In the anterolateral tunnel, an acorn reamer 11 mm in diameter was used to score the entry point of the ALB femoral tunnel. An eyelet pin was then drilled through the reamer anteromedially out the knee. Then a closed socket tunnel was reamed over the eyelet pin to a depth of 25 mm. A passing suture was pulled through the tunnel in preparation for graft passage.
With use of the same technique, a 7-mm reamer was placed against the outline of the PMB attachment site, and an eyelet pin was drilled through this reamer and out the anteromedial aspect of the knee. Again, a 25-mm deep closed socket was reamed. A bone bridge distance of 2 mm was maintained between the 2 femoral PCL bundle tunnels.
9. ACL Femoral Tunnel. The femoral ACL attachment was identified and outlined. An over-the-top guide was used to determine proper placement of the 10-mm low-profile reamer. A guide pin was drilled through the center of the reamer. The reamer was used to create a 25-mm deep closed socket tunnel, and a passing stitch was placed.
10. PCL Tibial Tunnel. With use of a 70° arthroscope for visualization, a posteromedial arthroscopic portal was created, and a shaver and a coagulator were used to identify the tibial PCL attachment, located distally along the PCL facet, until the proximal aspect of the popliteus muscle fibers were visualized. A guide pin was drilled starting at the anteromedial aspect of the tibia, about 6 cm distal to the joint line and centered between the anterior tibial crest and the medial tibial border. The pin exited posteriorly at the center of the PCL tibial attachment along the PCL bundle ridge, which was reported located between the ALB and the PMB on the tibia.5 Pin placement was verified with intraoperative lateral and anteroposterior radiographs. On the lateral radiograph, the pin should be about 6 mm or 7 mm proximal to the champagne-glass drop-off at the PCL facet on the posterior aspect of the tibia. On the anteroposterior radiograph, the pin should be 1 mm to 2 mm distal to the joint line and at the medial aspect of the lateral tibial eminence. A large curette was passed through the posteromedial arthroscopic portal both to retract the posterior tissues away from the reamer and to protect against guide-pin protrusion The guide pin was then overreamed with a 12-mm acorn reamer.
A large smoother was passed proximally up the tibial tunnel and then pulled out the anteromedial portal with a grasper. The smoother was gently cycled to smooth the intra-articular tibial tunnel aperture to remove any bony spicules that could interfere with graft passage. The smoother was then pulled back into the joint, passed out the anterolateral arthroscopic portal, and secured with a small clamp.4
11. ACL Tibial Tunnel. The ACL tibial attachment site was identified and cleaned of soft tissue. A guide pin was placed and then overreamed with a 10-mm acorn reamer.
12. PCL Femoral Fixation. The PMB graft was passed into its tunnel and secured with a 7-mm × 23-mm titanium screw. Next, the ALB was secured to the femur with a 7-mm × 20-mm titanium screw. The smoother was used to pull both grafts down through the tibial tunnel.
13. ACL Femoral Fixation. A 7-mm × 20-mm titanium screw was then used to fix the ACL autograft inside the femur. Traction was applied to the 3 cruciate grafts. There was no sign of impingement.
14. PLC Femoral Fixation. The FCL and the popliteus bone plugs were passed into their respective femoral sockets and secured with 7-mm × 20-mm titanium screws.
15. Lateral Capsule Femoral Anchors. Two suture anchors were placed into the femur, and the sutures were passed through the femoral portion of the lateral capsule for later repair.
16. PCL Tibial Fixation. Both grafts were fixed with a fully threaded bicortical 6.5-mm × 40-mm cannulated cancellous screw and an 18-mm spiked washer. The ALB was fixed first, with the knee flexed to 90°, traction on the graft, and the tibia in neutral rotation. Restoration of the normal tibiofemoral step-off was verified. The PMB was then fixed with the knee in full extension. A posterior drawer test was performed to verify restoration of stability.
17. PLC Fibula Fixation. The PLT graft was passed down the popliteal hiatus, and the FCL graft was passed under the remnant of the biceps bursa on the fibular head and then through the fibular head, anterolateral to posteromedial. The FCL graft was fixed in the fibular tunnel with the knee in 20° of flexion, a slight valgus reduction force, the tibia in neutral rotation, and traction on the graft. A 7-mm × 23-mm bioabsorbable screw was used.
18. Lateral Capsular Repair. The lateral capsule was directly repaired with the previously placed sutures. The sutures were tied with the knee in 20° of flexion.
19. PLC Tibial Fixation. The grafts were passed together, posterior to anterior, through the tibial tunnel. The knee was cycled several times through complete flexion/extension ROM. A 9-mm × 23-mm bioabsorbable screw was then used to fix the grafts to the tibia. During this fixation, the knee was kept in 60° of flexion and neutral rotation while traction was being applied to the distal end of both grafts.
20. ACL Tibial Fixation. A 9-mm × 20-mm titanium screw was used to fix the ACL graft with the knee in full extension. The graft was then viewed intra-articularly to confirm there was no impingement. The Lachman, posterior drawer, posterolateral drawer, dial, and varus stress tests were performed to ensure restoration of stability.
21. ITB Repair. A portion of the remaining Achilles tendon allograft was used to perform ITB reconstruction (reconstitution of the gaped portion of the ITB). Orthocord (DePuy Synthes) and Vicryl (Ethicon) sutures were used for this reconstruction. Knee stability was deemed restored, and the incisions were closed in standard layered fashion.
First Surgery: Postoperative Management
The patient remained non-weight-bearing the first 6 weeks after surgery, with prone knee flexion limited (0°-90°) the first 2 weeks. In addition, a PCL Jack brace (Albrecht) was placed 1 week after surgery and was to be worn at all times to decrease stress on the PCL grafts.
As ROM was not progressing as expected, the patient was instructed to use a continuous passive motion (CPM) machine 2 hours 3 times a day. About 4 weeks after surgery, with ROM still not progressing, the frequency of use of this machine was increased.
Despite continued physical therapy, use of the CPM machine, and pain management, ROM was limited (11°-90° of flexion) 5.5 months after left knee multiligament reconstruction. However, stress radiographs showed excellent stability. Varus stress radiographs showed a side-to-side difference of 0.3 mm less on the left (injured) knee, and kneeling PCL stress radiographs showed a side-to-side difference of 1.3 mm more on the left knee (Figures 3A-3D).
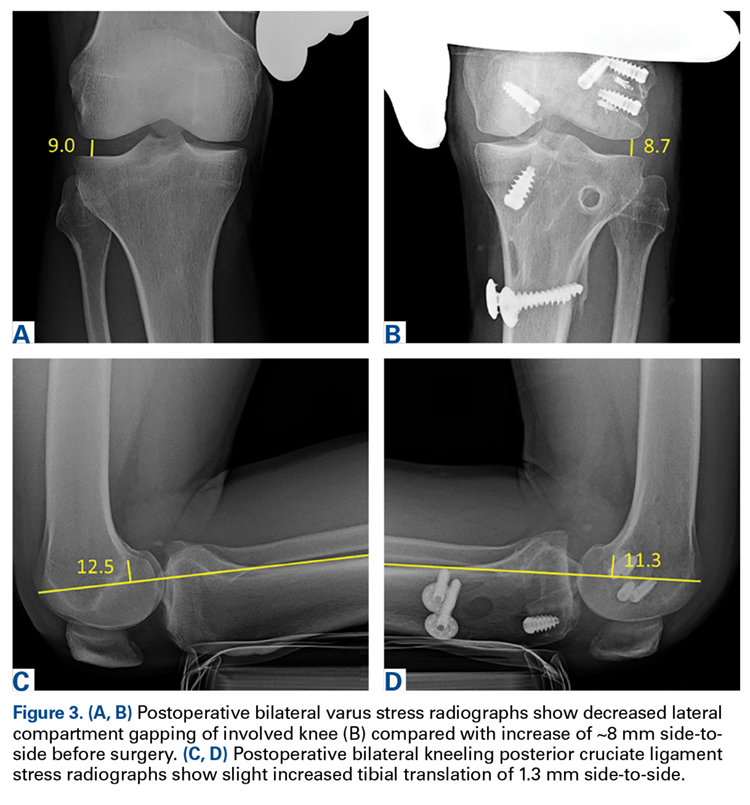
Second Surgery and Postoperative Management
As gentle manipulation under anesthesia was unsuccessful, the patient underwent knee arthroscopy, including 4-compartment lysis of adhesions, arthroscopically assisted posteromedial capsular release, and post-débridement manipulation under anesthesia. During manipulation, full extension and knee flexion up to 135° were achieved. ACL, PCL, and popliteus grafts were visualized and confirmed to be intact.
After this second surgery, the patient was to resume physical therapy and begin weight- bearing as tolerated. Active ROM was prioritized in an attempt to reach full ROM. In addition, a CPM machine was to be used from 0° to 135° of knee flexion 4 hours 3 times a day for 6 weeks.
Two weeks after surgery, the patient had continued pain, and extracapsular swelling in the left knee. However, ROM (0°-115° of flexion) was improved relative to before surgery (11°-90° of flexion), though it remained below the range on the contralateral side. Of note, the patient reported having a flexion contracture (~10°) in the immediate postoperative period. He had woken up with it after sleeping with the CPM machine the night before. The contracture delayed his physical therapy for several hours and resulted in a redesign of his therapy protocol to emphasize full, active knee extension and patellar mobilization, as well as discontinuation of use of the CPM machine. Corticosteroids were initiated to help with the extracapsular swelling, and the new therapy regimen brought adequate progress in ROM. Four months after the second surgery, the patient had full extension and 135° of flexion and was transitioned into wearing the PCL Rebound brace.
Discussion
This case was unique because of the midsubstance ITB tear and simultaneous multiligament injury caused by a KD-IIIL, a KD involving the ACL, the PCL, and the PLC with the medial side intact. There is limited research on ITB repair generally, with or without KD involvement. In a retrospective review of acute knee trauma cases, ITB pathologies were seen on 45% of reviewed MRI scans, and only 3% of the injuries were grade III; in addition, only 9 (5%) of the 200 cases involved both ITB and multiligament (ACL, PCL) knee injuries.6
After our patient’s ACL, PCL, and PLC were reconstructed, a fan piece of the Achilles tendon allograft from the PLC reconstruction was used to repair the ITB. The graft was used to reconstitute the torn gapped portion of the band in multiple locations, and this repair helped restore stability. The literature has reported numerous surgical uses for a portion of the ITB but few studies on repairing this anatomical structure. Preservation of the ITB is important to restoration of native anatomy and function. The ITB helps with anterolateral stabilization of the knee and with resistance of varus stress and internal tibial rotation.
The PLC reconstruction used in this case has been biomechanically validated as restoring the knee to near native stability through anatomical reconstruction of the PLC’s 3 main static stabilizers: the FCL, the PLT, and the popliteofibular ligament.7-9 First described in 2004,7 this anatomical PLC reconstruction technique has improved subjective and objective patient outcomes.10,11 For combined PLC injuries (eg, our patient’s injuries), Geeslin and LaPrade10 recommended concurrent reconstruction of the cruciate ligaments. In addition to the PLC reconstruction, the anatomical double-bundle PCL reconstruction used in this case has demonstrated significant improvements in subjective and objective outcome scores and objective knee stability.12
Although the stability and anatomy of this patient’s injured knee were reestablished, his development of arthrofibrosis is important. Many have discussed the commonality of arthrofibrosis or decreased ROM after extensive multiligament reconstruction surgeries.13,14 One study involving surgical management and outcomes of multiligament knee injuries found that, in more than half of its cases, restoration of full ROM required at least one operation after the initial one.13 Therefore, it is not unusual that our patient required a second operation for decreased ROM.
Conclusion
After surgery, excellent stabilization was achieved. Although the patient had setbacks related to pain and decreased ROM, his second surgery and continued physical therapy likely will help him return to his preoperative recreational activity levels.
1. Delos D, Warren RF, Marx RG. Multiligament knee injuries and their treatment. Oper Tech Sports Med. 2010;18(4):219-226.
2. Hobby B, Treme G, Wascher DC, Schenck RC. How I manage knee dislocations. Oper Tech Sports Med. 2010;18(4):227-234.
3. LaPrade RF, Ly TV, Wentorf FA, Engebretsen L. The posterolateral attachments of the knee: a qualitative and quantitative morphologic analysis of the fibular collateral ligament, popliteus tendon, popliteofibular ligament, and lateral gastrocnemius tendon. Am J Sports Med. 2003;31(6):854-860.
4. Chahla J, Nitri M, Civitarese D, Dean CS, Moulton SG, LaPrade RF. Anatomic double-bundle posterior cruciate ligament reconstruction. Arthrosc Tech. 2016;5(1):e149-e156.
5. Anderson CJ, Ziegler CG, Wijdicks CA, Engebretsen L, LaPrade RF. Arthroscopically pertinent anatomy of the anterolateral and posteromedial bundles of the posterior cruciate ligament. J Bone Joint Surg Am. 2012;94(21):1936-1945.
6. Mansour R, Yoong P, McKean D, Teh JL. The iliotibial band in acute knee trauma: patterns of injury on MR imaging. Skeletal Radiol. 2014;43(10):1369-1375.
7. LaPrade RF, Johansen S, Wentorf FA, Engebretsen L, Esterberg JL, Tso A. An analysis of an anatomical posterolateral knee reconstruction: an in vitro biomechanical study and development of a surgical technique. Am J Sports Med. 2004;32(6):1405-1414.
8. McCarthy M, Camarda L, Wijdicks CA, Johansen S, Engebretsen L, LaPrade RF. Anatomic posterolateral knee reconstructions require a popliteofibular ligament reconstruction through a tibial tunnel. Am J Sports Med. 2010;38(8):1674-1681.
9. LaPrade RF, Wozniczka JK, Stellmaker MP, Wijdicks CA. Analysis of the static function of the popliteus tendon and evaluation of an anatomic reconstruction: the “fifth ligament” of the knee. Am J Sports Med. 2010;38(3):543-549.
10. Geeslin AG, LaPrade RF. Outcomes of treatment of acute grade-III isolated and combined posterolateral knee injuries: a prospective case series and surgical technique. J Bone Joint Surg Am. 2011;93(18):1672-1683.
11. LaPrade RF, Johansen S, Agel J, Risberg MA, Moksnes H, Engebretsen L. Outcomes of an anatomic posterolateral knee reconstruction. J Bone Joint Surg Am. 2010;92(1):16-22.
12. Spiridonov SI, Slinkard NJ, LaPrade RF. Isolated and combined grade-III posterior cruciate ligament tears treated with double-bundle reconstruction with use of endoscopically placed femoral tunnels and grafts: operative technique and clinical outcomes. J Bone Joint Surg Am. 2011;93(19):1773-1780.
13. Noyes FR, Barber-Westin SD. Reconstruction of the anterior and posterior cruciate ligaments after knee dislocation. Use of early protected postoperative motion to decrease arthrofibrosis. Am J Sports Med. 1997;25(6):769-778.
14. Yenchak AJ, Wilk KE, Arrigo CA, Simpson CD, Andrews JR. Criteria-based management of an acute multistructure knee injury in a professional football player: a case report. J Orthop Sports Phys Ther. 2011;41(9):675-686.
Take-Home Points
- Reconstruction of a torn ITB is important in restoration of native anatomy and function given its properties in anterolateral stabilization and resistance to varus stress and internal tibial rotation.
- Restoration of posterolateral instability primarily involves reconstructing the FCL, PLT, and popliteofibular ligament.
- For combined PLC injuries, concurrent reconstruction of the cruciate ligaments in one stage is highly recommended.
- Post-surgery, a 6-week non-weight-bearing, limited flexion rehab protocol utilizing a dynamic PCL brace, such as the PCL Rebound brace, is recommended to prevent posterior tibial sag.
- Arthrofibrosis and decreased ROM can be seen following a violent knee injury which requires extensive multiligament reconstruction surgeries, occasionally requiring a secondary surgery for further restoration of knee motion.
Tibiofemoral knee dislocations are uncommon injuries that have devastating complications and potentially result in complex surgeries.1 Knee dislocations (KDs) can be classified with the Schenck system.2 KD-I is a multiligament injury involving the anterior cruciate ligament (ACL) or the posterior cruciate ligament (PCL), and the scale increases in severity/number of ligaments involved, with KD-V being a multiligament injury with periarticular fracture.2
In this article, we report the case of a complex multiligament knee reconstruction performed with a midsubstance iliotibial band (ITB) repair. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 27-year-old man presented 12 days after a paraskiing crash in which he collided with a tree at 45 mph and fell 40 feet before hitting snow. Physical examination revealed a large hemarthrosis of the left lower extremity and ecchymosis about the posterolateral aspect of the knee and popliteal fossa. Range of motion (ROM) was limited from 5° of hyperextension to 90° of flexion. Additional motion was deferred secondary to pain. Varus stress testing at 0° and 30° of knee flexion demonstrated significant side-to-side differences. The Lachman test, posterior drawer test, and posterolateral drawer test were all 3+. The dial test was 3 to 4+ compared with the contralateral knee. Valgus stress testing at 0° and 30° of flexion did not reveal any side-to-side laxity. The calf was nontender, and all compartments were soft. The patient reported no neurovascular symptoms and had no neuromotor deficits other than mild common peroneal nerve dysesthesias.
Varus stress radiographs showed increased side-to-side gapping (8 mm) of the lateral compartment of the injured knee. Kneeling posterior stress radiographs, limited because of the patient’s inability to apply full stress on the injured knee secondary to pain, showed a difference of 6 mm in increased posterior translation on the uninjured leg (Figures 1A-1D).
First Surgery
1. PLC Approach. A lateral hockey-stick skin incision was made along the ITB and extended distally between the fibular head and the Gerdy tubercle. The subcutaneous tissue was then dissected, and a posteriorly based flap was developed for preservation of vascular support to the superficial tissues. The ITB and the lateral capsule had completely torn off of the femur, allowing exposure directly into the joint. The long and short heads of the biceps femoris were exposed, with about 50% of the biceps attachment torn. The FCL was torn midsubstance, and the PLT had no remnant attachment left on the femur.
2. ITB and Lateral Capsule Tag Stitched. The torn ends of the ITB were dissected and tag stitches placed in each end. Tag stitches were also placed in the lateral capsule in preparation for a direct repair.
3. Neurolysis. The common peroneal nerve was found encased in a significant amount of scar tissue, and extensive neurolysis was required. Slow, methodical dissection was performed under the partially torn long head of the biceps femoris and was continued through the scar tissue and adhesions. Distally, 5 mm to 7 mm of the peroneus longus fascia was incised as part of the neurolysis in order to prevent nerve irritation or foot drop caused by postoperative swelling.
4. PLC Tunnels. The margin between the lateral gastrocnemius tendon and the soleus muscle was identified by blunt dissection that allowed palpation of the posteromedial aspect of the fibular styloid and the popliteus musculotendinous junction. The underlying biceps bursa was incised in order to locate the midportion of the FCL remnant, which typically is tag-stitched with No. 2 FiberWire to help identify the femoral attachment (this was not done because of the complete tear at the midsubstance of the FCL).
Subperiosteal dissection of the lateral aspect of the fibular head was performed anterior to posterior and distally extended to the champagne-glass drop-off of the fibular head. Continuing the dissection distally beyond this point can endanger the common peroneal nerve. A small sulcus can be palpated where the distal FCL inserts on the fibular head. Posteriorly, a small elevator was used to dissect the soleus muscle off of the posteromedial aspect of the fibular head, where the fibular tunnel would later be created.
A Chandler retractor was placed posterior to the fibular head to protect the neurovascular bundle. With the aid of a collateral ligament aiming device, a guide pin was drilled from the lateral aspect of the fibular head (FCL attachment) to the posteromedial downslope of the fibular styloid (popliteofibular ligament attachment). The entry point of the guide pin was immediately above the champagne- glass drop-off, at the distal insertion site of the FCL, which was described as being 28.4 mm from the styloid tip and 8.2 mm posterior to the anterior margin of the fibular head.3 Care should be taken not to ream the tunnel too proximal, as doing so increases the risk of iatrogenic fracture. A 7-mm reamer was then used to drill the fibular tunnel. To facilitate later passage of the graft, a passing suture was placed through the tunnel, leaving the loop anterolateral.
Next, the starting point for the tibial tunnel was located on the flat spot of the anterolateral tibia distal and medial to the Gerdy tubercle, just lateral to the tibial tubercle. The tibial popliteal sulcus was identified by palpation of the posterolateral tibial plateau to localize the site of the popliteus musculotendinous junction, which is the ideal location of the posterior aperture of the tibial tunnel. This point is 1 cm proximal and 1 cm medial to the posteromedial exit of the fibular tunnel. A Chandler retractor was placed anterior to the lateral gastrocnemius to protect the neurovascular bundle. In the locations described earlier, a cruciate aiming device was used to place a guide pin anterior to posterior. A 9-mm tunnel was overreamed and a passing suture placed, leaving the loop posterior to facilitate graft passage.
The femoral insertions of the FCL and the PLT were then identified. ITB splitting was not necessary, given the complete midsubstance tear of this structure. The FCL attachment was identified 1.4 mm proximal and 3.1 mm posterior to the lateral epicondyle.3 Sharp dissection was performed in this location, proximal to distal, exposing the lateral epicondyle and the small sulcus at the FCL attachment site. A collateral ligament reconstruction aiming sleeve was used to drill a guide pin over the FCL femoral attachment site and out the medial aspect of the distal thigh, about 5 cm proximal and anterior to the adductor tubercle.
The femoral attachment of the PLT was reported located 18.5 mm anterior to the FCL insertion, in the anterior fifth of the popliteal sulcus.3 Although arthrotomy is usually required in order to access the PLT attachment, it was not necessary in this case, given the lateral capsule tear. A guide pin was inserted at the PLT attachment site, parallel to the FCL pin. After proper placement was verified, a 9-mm reamer was used to drill the FCL and PLT tunnels to a depth of 25 mm (socket), and a passing suture was placed into each tunnel to facilitate graft passage.
5. ACL Graft Harvest. The central third of the ipsilateral patellar tendon was harvested for use in the ACL reconstruction. Included were a 10-mm × 20-mm bone plug from the patella and a 10-mm × 25-mm bone plug from the tibial tubercle. The patella defect was then bone-grafted, and the patellar tendon closed side-to-side.
6. Graft Preparation. For the PLC, we used a split Achilles tendon allograft that had two 9-mm × 25-mm bone plugs proximally and were tubularized distally. For the PCL, we used an anterolateral bundle (ALB), which consisted of an Achilles tendon allograft that had an 11-mm × 25-mm bone plug proximally and was tubularized distally, and a posteromedial bundle (PMB), which consisted of a tibialis anterior allograft that was tubularized at both ends. For the ACL, we used a bone–patellar tendon–bone autograft 10 mm in diameter with a 20-mm femoral bone plug and a 25-mm tibial bone plug distally.
7. Arthroscopy. We created standard anterolateral and anteromedial parapatellar portals and performed arthroscopy, including lysis of adhesions. Cartilage and menisci were lesion-free.
8. PCL Femoral Tunnels. The ALB attachment was identified and outlined with a coagulator between the trochlear point and the medial arch point, adjacent to the edge of the articular cartilage. Similarly, the PMB attachment was marked about 8 mm or 9 mm posterior to the edge of the articular cartilage of the medial femoral condyle and slightly posterior to the ALB tunnel.4
In the anterolateral tunnel, an acorn reamer 11 mm in diameter was used to score the entry point of the ALB femoral tunnel. An eyelet pin was then drilled through the reamer anteromedially out the knee. Then a closed socket tunnel was reamed over the eyelet pin to a depth of 25 mm. A passing suture was pulled through the tunnel in preparation for graft passage.
With use of the same technique, a 7-mm reamer was placed against the outline of the PMB attachment site, and an eyelet pin was drilled through this reamer and out the anteromedial aspect of the knee. Again, a 25-mm deep closed socket was reamed. A bone bridge distance of 2 mm was maintained between the 2 femoral PCL bundle tunnels.
9. ACL Femoral Tunnel. The femoral ACL attachment was identified and outlined. An over-the-top guide was used to determine proper placement of the 10-mm low-profile reamer. A guide pin was drilled through the center of the reamer. The reamer was used to create a 25-mm deep closed socket tunnel, and a passing stitch was placed.
10. PCL Tibial Tunnel. With use of a 70° arthroscope for visualization, a posteromedial arthroscopic portal was created, and a shaver and a coagulator were used to identify the tibial PCL attachment, located distally along the PCL facet, until the proximal aspect of the popliteus muscle fibers were visualized. A guide pin was drilled starting at the anteromedial aspect of the tibia, about 6 cm distal to the joint line and centered between the anterior tibial crest and the medial tibial border. The pin exited posteriorly at the center of the PCL tibial attachment along the PCL bundle ridge, which was reported located between the ALB and the PMB on the tibia.5 Pin placement was verified with intraoperative lateral and anteroposterior radiographs. On the lateral radiograph, the pin should be about 6 mm or 7 mm proximal to the champagne-glass drop-off at the PCL facet on the posterior aspect of the tibia. On the anteroposterior radiograph, the pin should be 1 mm to 2 mm distal to the joint line and at the medial aspect of the lateral tibial eminence. A large curette was passed through the posteromedial arthroscopic portal both to retract the posterior tissues away from the reamer and to protect against guide-pin protrusion The guide pin was then overreamed with a 12-mm acorn reamer.
A large smoother was passed proximally up the tibial tunnel and then pulled out the anteromedial portal with a grasper. The smoother was gently cycled to smooth the intra-articular tibial tunnel aperture to remove any bony spicules that could interfere with graft passage. The smoother was then pulled back into the joint, passed out the anterolateral arthroscopic portal, and secured with a small clamp.4
11. ACL Tibial Tunnel. The ACL tibial attachment site was identified and cleaned of soft tissue. A guide pin was placed and then overreamed with a 10-mm acorn reamer.
12. PCL Femoral Fixation. The PMB graft was passed into its tunnel and secured with a 7-mm × 23-mm titanium screw. Next, the ALB was secured to the femur with a 7-mm × 20-mm titanium screw. The smoother was used to pull both grafts down through the tibial tunnel.
13. ACL Femoral Fixation. A 7-mm × 20-mm titanium screw was then used to fix the ACL autograft inside the femur. Traction was applied to the 3 cruciate grafts. There was no sign of impingement.
14. PLC Femoral Fixation. The FCL and the popliteus bone plugs were passed into their respective femoral sockets and secured with 7-mm × 20-mm titanium screws.
15. Lateral Capsule Femoral Anchors. Two suture anchors were placed into the femur, and the sutures were passed through the femoral portion of the lateral capsule for later repair.
16. PCL Tibial Fixation. Both grafts were fixed with a fully threaded bicortical 6.5-mm × 40-mm cannulated cancellous screw and an 18-mm spiked washer. The ALB was fixed first, with the knee flexed to 90°, traction on the graft, and the tibia in neutral rotation. Restoration of the normal tibiofemoral step-off was verified. The PMB was then fixed with the knee in full extension. A posterior drawer test was performed to verify restoration of stability.
17. PLC Fibula Fixation. The PLT graft was passed down the popliteal hiatus, and the FCL graft was passed under the remnant of the biceps bursa on the fibular head and then through the fibular head, anterolateral to posteromedial. The FCL graft was fixed in the fibular tunnel with the knee in 20° of flexion, a slight valgus reduction force, the tibia in neutral rotation, and traction on the graft. A 7-mm × 23-mm bioabsorbable screw was used.
18. Lateral Capsular Repair. The lateral capsule was directly repaired with the previously placed sutures. The sutures were tied with the knee in 20° of flexion.
19. PLC Tibial Fixation. The grafts were passed together, posterior to anterior, through the tibial tunnel. The knee was cycled several times through complete flexion/extension ROM. A 9-mm × 23-mm bioabsorbable screw was then used to fix the grafts to the tibia. During this fixation, the knee was kept in 60° of flexion and neutral rotation while traction was being applied to the distal end of both grafts.
20. ACL Tibial Fixation. A 9-mm × 20-mm titanium screw was used to fix the ACL graft with the knee in full extension. The graft was then viewed intra-articularly to confirm there was no impingement. The Lachman, posterior drawer, posterolateral drawer, dial, and varus stress tests were performed to ensure restoration of stability.
21. ITB Repair. A portion of the remaining Achilles tendon allograft was used to perform ITB reconstruction (reconstitution of the gaped portion of the ITB). Orthocord (DePuy Synthes) and Vicryl (Ethicon) sutures were used for this reconstruction. Knee stability was deemed restored, and the incisions were closed in standard layered fashion.
First Surgery: Postoperative Management
The patient remained non-weight-bearing the first 6 weeks after surgery, with prone knee flexion limited (0°-90°) the first 2 weeks. In addition, a PCL Jack brace (Albrecht) was placed 1 week after surgery and was to be worn at all times to decrease stress on the PCL grafts.
As ROM was not progressing as expected, the patient was instructed to use a continuous passive motion (CPM) machine 2 hours 3 times a day. About 4 weeks after surgery, with ROM still not progressing, the frequency of use of this machine was increased.
Despite continued physical therapy, use of the CPM machine, and pain management, ROM was limited (11°-90° of flexion) 5.5 months after left knee multiligament reconstruction. However, stress radiographs showed excellent stability. Varus stress radiographs showed a side-to-side difference of 0.3 mm less on the left (injured) knee, and kneeling PCL stress radiographs showed a side-to-side difference of 1.3 mm more on the left knee (Figures 3A-3D).
Second Surgery and Postoperative Management
As gentle manipulation under anesthesia was unsuccessful, the patient underwent knee arthroscopy, including 4-compartment lysis of adhesions, arthroscopically assisted posteromedial capsular release, and post-débridement manipulation under anesthesia. During manipulation, full extension and knee flexion up to 135° were achieved. ACL, PCL, and popliteus grafts were visualized and confirmed to be intact.
After this second surgery, the patient was to resume physical therapy and begin weight- bearing as tolerated. Active ROM was prioritized in an attempt to reach full ROM. In addition, a CPM machine was to be used from 0° to 135° of knee flexion 4 hours 3 times a day for 6 weeks.
Two weeks after surgery, the patient had continued pain, and extracapsular swelling in the left knee. However, ROM (0°-115° of flexion) was improved relative to before surgery (11°-90° of flexion), though it remained below the range on the contralateral side. Of note, the patient reported having a flexion contracture (~10°) in the immediate postoperative period. He had woken up with it after sleeping with the CPM machine the night before. The contracture delayed his physical therapy for several hours and resulted in a redesign of his therapy protocol to emphasize full, active knee extension and patellar mobilization, as well as discontinuation of use of the CPM machine. Corticosteroids were initiated to help with the extracapsular swelling, and the new therapy regimen brought adequate progress in ROM. Four months after the second surgery, the patient had full extension and 135° of flexion and was transitioned into wearing the PCL Rebound brace.
Discussion
This case was unique because of the midsubstance ITB tear and simultaneous multiligament injury caused by a KD-IIIL, a KD involving the ACL, the PCL, and the PLC with the medial side intact. There is limited research on ITB repair generally, with or without KD involvement. In a retrospective review of acute knee trauma cases, ITB pathologies were seen on 45% of reviewed MRI scans, and only 3% of the injuries were grade III; in addition, only 9 (5%) of the 200 cases involved both ITB and multiligament (ACL, PCL) knee injuries.6
After our patient’s ACL, PCL, and PLC were reconstructed, a fan piece of the Achilles tendon allograft from the PLC reconstruction was used to repair the ITB. The graft was used to reconstitute the torn gapped portion of the band in multiple locations, and this repair helped restore stability. The literature has reported numerous surgical uses for a portion of the ITB but few studies on repairing this anatomical structure. Preservation of the ITB is important to restoration of native anatomy and function. The ITB helps with anterolateral stabilization of the knee and with resistance of varus stress and internal tibial rotation.
The PLC reconstruction used in this case has been biomechanically validated as restoring the knee to near native stability through anatomical reconstruction of the PLC’s 3 main static stabilizers: the FCL, the PLT, and the popliteofibular ligament.7-9 First described in 2004,7 this anatomical PLC reconstruction technique has improved subjective and objective patient outcomes.10,11 For combined PLC injuries (eg, our patient’s injuries), Geeslin and LaPrade10 recommended concurrent reconstruction of the cruciate ligaments. In addition to the PLC reconstruction, the anatomical double-bundle PCL reconstruction used in this case has demonstrated significant improvements in subjective and objective outcome scores and objective knee stability.12
Although the stability and anatomy of this patient’s injured knee were reestablished, his development of arthrofibrosis is important. Many have discussed the commonality of arthrofibrosis or decreased ROM after extensive multiligament reconstruction surgeries.13,14 One study involving surgical management and outcomes of multiligament knee injuries found that, in more than half of its cases, restoration of full ROM required at least one operation after the initial one.13 Therefore, it is not unusual that our patient required a second operation for decreased ROM.
Conclusion
After surgery, excellent stabilization was achieved. Although the patient had setbacks related to pain and decreased ROM, his second surgery and continued physical therapy likely will help him return to his preoperative recreational activity levels.
Take-Home Points
- Reconstruction of a torn ITB is important in restoration of native anatomy and function given its properties in anterolateral stabilization and resistance to varus stress and internal tibial rotation.
- Restoration of posterolateral instability primarily involves reconstructing the FCL, PLT, and popliteofibular ligament.
- For combined PLC injuries, concurrent reconstruction of the cruciate ligaments in one stage is highly recommended.
- Post-surgery, a 6-week non-weight-bearing, limited flexion rehab protocol utilizing a dynamic PCL brace, such as the PCL Rebound brace, is recommended to prevent posterior tibial sag.
- Arthrofibrosis and decreased ROM can be seen following a violent knee injury which requires extensive multiligament reconstruction surgeries, occasionally requiring a secondary surgery for further restoration of knee motion.
Tibiofemoral knee dislocations are uncommon injuries that have devastating complications and potentially result in complex surgeries.1 Knee dislocations (KDs) can be classified with the Schenck system.2 KD-I is a multiligament injury involving the anterior cruciate ligament (ACL) or the posterior cruciate ligament (PCL), and the scale increases in severity/number of ligaments involved, with KD-V being a multiligament injury with periarticular fracture.2
In this article, we report the case of a complex multiligament knee reconstruction performed with a midsubstance iliotibial band (ITB) repair. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 27-year-old man presented 12 days after a paraskiing crash in which he collided with a tree at 45 mph and fell 40 feet before hitting snow. Physical examination revealed a large hemarthrosis of the left lower extremity and ecchymosis about the posterolateral aspect of the knee and popliteal fossa. Range of motion (ROM) was limited from 5° of hyperextension to 90° of flexion. Additional motion was deferred secondary to pain. Varus stress testing at 0° and 30° of knee flexion demonstrated significant side-to-side differences. The Lachman test, posterior drawer test, and posterolateral drawer test were all 3+. The dial test was 3 to 4+ compared with the contralateral knee. Valgus stress testing at 0° and 30° of flexion did not reveal any side-to-side laxity. The calf was nontender, and all compartments were soft. The patient reported no neurovascular symptoms and had no neuromotor deficits other than mild common peroneal nerve dysesthesias.
Varus stress radiographs showed increased side-to-side gapping (8 mm) of the lateral compartment of the injured knee. Kneeling posterior stress radiographs, limited because of the patient’s inability to apply full stress on the injured knee secondary to pain, showed a difference of 6 mm in increased posterior translation on the uninjured leg (Figures 1A-1D).
First Surgery
1. PLC Approach. A lateral hockey-stick skin incision was made along the ITB and extended distally between the fibular head and the Gerdy tubercle. The subcutaneous tissue was then dissected, and a posteriorly based flap was developed for preservation of vascular support to the superficial tissues. The ITB and the lateral capsule had completely torn off of the femur, allowing exposure directly into the joint. The long and short heads of the biceps femoris were exposed, with about 50% of the biceps attachment torn. The FCL was torn midsubstance, and the PLT had no remnant attachment left on the femur.
2. ITB and Lateral Capsule Tag Stitched. The torn ends of the ITB were dissected and tag stitches placed in each end. Tag stitches were also placed in the lateral capsule in preparation for a direct repair.
3. Neurolysis. The common peroneal nerve was found encased in a significant amount of scar tissue, and extensive neurolysis was required. Slow, methodical dissection was performed under the partially torn long head of the biceps femoris and was continued through the scar tissue and adhesions. Distally, 5 mm to 7 mm of the peroneus longus fascia was incised as part of the neurolysis in order to prevent nerve irritation or foot drop caused by postoperative swelling.
4. PLC Tunnels. The margin between the lateral gastrocnemius tendon and the soleus muscle was identified by blunt dissection that allowed palpation of the posteromedial aspect of the fibular styloid and the popliteus musculotendinous junction. The underlying biceps bursa was incised in order to locate the midportion of the FCL remnant, which typically is tag-stitched with No. 2 FiberWire to help identify the femoral attachment (this was not done because of the complete tear at the midsubstance of the FCL).
Subperiosteal dissection of the lateral aspect of the fibular head was performed anterior to posterior and distally extended to the champagne-glass drop-off of the fibular head. Continuing the dissection distally beyond this point can endanger the common peroneal nerve. A small sulcus can be palpated where the distal FCL inserts on the fibular head. Posteriorly, a small elevator was used to dissect the soleus muscle off of the posteromedial aspect of the fibular head, where the fibular tunnel would later be created.
A Chandler retractor was placed posterior to the fibular head to protect the neurovascular bundle. With the aid of a collateral ligament aiming device, a guide pin was drilled from the lateral aspect of the fibular head (FCL attachment) to the posteromedial downslope of the fibular styloid (popliteofibular ligament attachment). The entry point of the guide pin was immediately above the champagne- glass drop-off, at the distal insertion site of the FCL, which was described as being 28.4 mm from the styloid tip and 8.2 mm posterior to the anterior margin of the fibular head.3 Care should be taken not to ream the tunnel too proximal, as doing so increases the risk of iatrogenic fracture. A 7-mm reamer was then used to drill the fibular tunnel. To facilitate later passage of the graft, a passing suture was placed through the tunnel, leaving the loop anterolateral.
Next, the starting point for the tibial tunnel was located on the flat spot of the anterolateral tibia distal and medial to the Gerdy tubercle, just lateral to the tibial tubercle. The tibial popliteal sulcus was identified by palpation of the posterolateral tibial plateau to localize the site of the popliteus musculotendinous junction, which is the ideal location of the posterior aperture of the tibial tunnel. This point is 1 cm proximal and 1 cm medial to the posteromedial exit of the fibular tunnel. A Chandler retractor was placed anterior to the lateral gastrocnemius to protect the neurovascular bundle. In the locations described earlier, a cruciate aiming device was used to place a guide pin anterior to posterior. A 9-mm tunnel was overreamed and a passing suture placed, leaving the loop posterior to facilitate graft passage.
The femoral insertions of the FCL and the PLT were then identified. ITB splitting was not necessary, given the complete midsubstance tear of this structure. The FCL attachment was identified 1.4 mm proximal and 3.1 mm posterior to the lateral epicondyle.3 Sharp dissection was performed in this location, proximal to distal, exposing the lateral epicondyle and the small sulcus at the FCL attachment site. A collateral ligament reconstruction aiming sleeve was used to drill a guide pin over the FCL femoral attachment site and out the medial aspect of the distal thigh, about 5 cm proximal and anterior to the adductor tubercle.
The femoral attachment of the PLT was reported located 18.5 mm anterior to the FCL insertion, in the anterior fifth of the popliteal sulcus.3 Although arthrotomy is usually required in order to access the PLT attachment, it was not necessary in this case, given the lateral capsule tear. A guide pin was inserted at the PLT attachment site, parallel to the FCL pin. After proper placement was verified, a 9-mm reamer was used to drill the FCL and PLT tunnels to a depth of 25 mm (socket), and a passing suture was placed into each tunnel to facilitate graft passage.
5. ACL Graft Harvest. The central third of the ipsilateral patellar tendon was harvested for use in the ACL reconstruction. Included were a 10-mm × 20-mm bone plug from the patella and a 10-mm × 25-mm bone plug from the tibial tubercle. The patella defect was then bone-grafted, and the patellar tendon closed side-to-side.
6. Graft Preparation. For the PLC, we used a split Achilles tendon allograft that had two 9-mm × 25-mm bone plugs proximally and were tubularized distally. For the PCL, we used an anterolateral bundle (ALB), which consisted of an Achilles tendon allograft that had an 11-mm × 25-mm bone plug proximally and was tubularized distally, and a posteromedial bundle (PMB), which consisted of a tibialis anterior allograft that was tubularized at both ends. For the ACL, we used a bone–patellar tendon–bone autograft 10 mm in diameter with a 20-mm femoral bone plug and a 25-mm tibial bone plug distally.
7. Arthroscopy. We created standard anterolateral and anteromedial parapatellar portals and performed arthroscopy, including lysis of adhesions. Cartilage and menisci were lesion-free.
8. PCL Femoral Tunnels. The ALB attachment was identified and outlined with a coagulator between the trochlear point and the medial arch point, adjacent to the edge of the articular cartilage. Similarly, the PMB attachment was marked about 8 mm or 9 mm posterior to the edge of the articular cartilage of the medial femoral condyle and slightly posterior to the ALB tunnel.4
In the anterolateral tunnel, an acorn reamer 11 mm in diameter was used to score the entry point of the ALB femoral tunnel. An eyelet pin was then drilled through the reamer anteromedially out the knee. Then a closed socket tunnel was reamed over the eyelet pin to a depth of 25 mm. A passing suture was pulled through the tunnel in preparation for graft passage.
With use of the same technique, a 7-mm reamer was placed against the outline of the PMB attachment site, and an eyelet pin was drilled through this reamer and out the anteromedial aspect of the knee. Again, a 25-mm deep closed socket was reamed. A bone bridge distance of 2 mm was maintained between the 2 femoral PCL bundle tunnels.
9. ACL Femoral Tunnel. The femoral ACL attachment was identified and outlined. An over-the-top guide was used to determine proper placement of the 10-mm low-profile reamer. A guide pin was drilled through the center of the reamer. The reamer was used to create a 25-mm deep closed socket tunnel, and a passing stitch was placed.
10. PCL Tibial Tunnel. With use of a 70° arthroscope for visualization, a posteromedial arthroscopic portal was created, and a shaver and a coagulator were used to identify the tibial PCL attachment, located distally along the PCL facet, until the proximal aspect of the popliteus muscle fibers were visualized. A guide pin was drilled starting at the anteromedial aspect of the tibia, about 6 cm distal to the joint line and centered between the anterior tibial crest and the medial tibial border. The pin exited posteriorly at the center of the PCL tibial attachment along the PCL bundle ridge, which was reported located between the ALB and the PMB on the tibia.5 Pin placement was verified with intraoperative lateral and anteroposterior radiographs. On the lateral radiograph, the pin should be about 6 mm or 7 mm proximal to the champagne-glass drop-off at the PCL facet on the posterior aspect of the tibia. On the anteroposterior radiograph, the pin should be 1 mm to 2 mm distal to the joint line and at the medial aspect of the lateral tibial eminence. A large curette was passed through the posteromedial arthroscopic portal both to retract the posterior tissues away from the reamer and to protect against guide-pin protrusion The guide pin was then overreamed with a 12-mm acorn reamer.
A large smoother was passed proximally up the tibial tunnel and then pulled out the anteromedial portal with a grasper. The smoother was gently cycled to smooth the intra-articular tibial tunnel aperture to remove any bony spicules that could interfere with graft passage. The smoother was then pulled back into the joint, passed out the anterolateral arthroscopic portal, and secured with a small clamp.4
11. ACL Tibial Tunnel. The ACL tibial attachment site was identified and cleaned of soft tissue. A guide pin was placed and then overreamed with a 10-mm acorn reamer.
12. PCL Femoral Fixation. The PMB graft was passed into its tunnel and secured with a 7-mm × 23-mm titanium screw. Next, the ALB was secured to the femur with a 7-mm × 20-mm titanium screw. The smoother was used to pull both grafts down through the tibial tunnel.
13. ACL Femoral Fixation. A 7-mm × 20-mm titanium screw was then used to fix the ACL autograft inside the femur. Traction was applied to the 3 cruciate grafts. There was no sign of impingement.
14. PLC Femoral Fixation. The FCL and the popliteus bone plugs were passed into their respective femoral sockets and secured with 7-mm × 20-mm titanium screws.
15. Lateral Capsule Femoral Anchors. Two suture anchors were placed into the femur, and the sutures were passed through the femoral portion of the lateral capsule for later repair.
16. PCL Tibial Fixation. Both grafts were fixed with a fully threaded bicortical 6.5-mm × 40-mm cannulated cancellous screw and an 18-mm spiked washer. The ALB was fixed first, with the knee flexed to 90°, traction on the graft, and the tibia in neutral rotation. Restoration of the normal tibiofemoral step-off was verified. The PMB was then fixed with the knee in full extension. A posterior drawer test was performed to verify restoration of stability.
17. PLC Fibula Fixation. The PLT graft was passed down the popliteal hiatus, and the FCL graft was passed under the remnant of the biceps bursa on the fibular head and then through the fibular head, anterolateral to posteromedial. The FCL graft was fixed in the fibular tunnel with the knee in 20° of flexion, a slight valgus reduction force, the tibia in neutral rotation, and traction on the graft. A 7-mm × 23-mm bioabsorbable screw was used.
18. Lateral Capsular Repair. The lateral capsule was directly repaired with the previously placed sutures. The sutures were tied with the knee in 20° of flexion.
19. PLC Tibial Fixation. The grafts were passed together, posterior to anterior, through the tibial tunnel. The knee was cycled several times through complete flexion/extension ROM. A 9-mm × 23-mm bioabsorbable screw was then used to fix the grafts to the tibia. During this fixation, the knee was kept in 60° of flexion and neutral rotation while traction was being applied to the distal end of both grafts.
20. ACL Tibial Fixation. A 9-mm × 20-mm titanium screw was used to fix the ACL graft with the knee in full extension. The graft was then viewed intra-articularly to confirm there was no impingement. The Lachman, posterior drawer, posterolateral drawer, dial, and varus stress tests were performed to ensure restoration of stability.
21. ITB Repair. A portion of the remaining Achilles tendon allograft was used to perform ITB reconstruction (reconstitution of the gaped portion of the ITB). Orthocord (DePuy Synthes) and Vicryl (Ethicon) sutures were used for this reconstruction. Knee stability was deemed restored, and the incisions were closed in standard layered fashion.
First Surgery: Postoperative Management
The patient remained non-weight-bearing the first 6 weeks after surgery, with prone knee flexion limited (0°-90°) the first 2 weeks. In addition, a PCL Jack brace (Albrecht) was placed 1 week after surgery and was to be worn at all times to decrease stress on the PCL grafts.
As ROM was not progressing as expected, the patient was instructed to use a continuous passive motion (CPM) machine 2 hours 3 times a day. About 4 weeks after surgery, with ROM still not progressing, the frequency of use of this machine was increased.
Despite continued physical therapy, use of the CPM machine, and pain management, ROM was limited (11°-90° of flexion) 5.5 months after left knee multiligament reconstruction. However, stress radiographs showed excellent stability. Varus stress radiographs showed a side-to-side difference of 0.3 mm less on the left (injured) knee, and kneeling PCL stress radiographs showed a side-to-side difference of 1.3 mm more on the left knee (Figures 3A-3D).
Second Surgery and Postoperative Management
As gentle manipulation under anesthesia was unsuccessful, the patient underwent knee arthroscopy, including 4-compartment lysis of adhesions, arthroscopically assisted posteromedial capsular release, and post-débridement manipulation under anesthesia. During manipulation, full extension and knee flexion up to 135° were achieved. ACL, PCL, and popliteus grafts were visualized and confirmed to be intact.
After this second surgery, the patient was to resume physical therapy and begin weight- bearing as tolerated. Active ROM was prioritized in an attempt to reach full ROM. In addition, a CPM machine was to be used from 0° to 135° of knee flexion 4 hours 3 times a day for 6 weeks.
Two weeks after surgery, the patient had continued pain, and extracapsular swelling in the left knee. However, ROM (0°-115° of flexion) was improved relative to before surgery (11°-90° of flexion), though it remained below the range on the contralateral side. Of note, the patient reported having a flexion contracture (~10°) in the immediate postoperative period. He had woken up with it after sleeping with the CPM machine the night before. The contracture delayed his physical therapy for several hours and resulted in a redesign of his therapy protocol to emphasize full, active knee extension and patellar mobilization, as well as discontinuation of use of the CPM machine. Corticosteroids were initiated to help with the extracapsular swelling, and the new therapy regimen brought adequate progress in ROM. Four months after the second surgery, the patient had full extension and 135° of flexion and was transitioned into wearing the PCL Rebound brace.
Discussion
This case was unique because of the midsubstance ITB tear and simultaneous multiligament injury caused by a KD-IIIL, a KD involving the ACL, the PCL, and the PLC with the medial side intact. There is limited research on ITB repair generally, with or without KD involvement. In a retrospective review of acute knee trauma cases, ITB pathologies were seen on 45% of reviewed MRI scans, and only 3% of the injuries were grade III; in addition, only 9 (5%) of the 200 cases involved both ITB and multiligament (ACL, PCL) knee injuries.6
After our patient’s ACL, PCL, and PLC were reconstructed, a fan piece of the Achilles tendon allograft from the PLC reconstruction was used to repair the ITB. The graft was used to reconstitute the torn gapped portion of the band in multiple locations, and this repair helped restore stability. The literature has reported numerous surgical uses for a portion of the ITB but few studies on repairing this anatomical structure. Preservation of the ITB is important to restoration of native anatomy and function. The ITB helps with anterolateral stabilization of the knee and with resistance of varus stress and internal tibial rotation.
The PLC reconstruction used in this case has been biomechanically validated as restoring the knee to near native stability through anatomical reconstruction of the PLC’s 3 main static stabilizers: the FCL, the PLT, and the popliteofibular ligament.7-9 First described in 2004,7 this anatomical PLC reconstruction technique has improved subjective and objective patient outcomes.10,11 For combined PLC injuries (eg, our patient’s injuries), Geeslin and LaPrade10 recommended concurrent reconstruction of the cruciate ligaments. In addition to the PLC reconstruction, the anatomical double-bundle PCL reconstruction used in this case has demonstrated significant improvements in subjective and objective outcome scores and objective knee stability.12
Although the stability and anatomy of this patient’s injured knee were reestablished, his development of arthrofibrosis is important. Many have discussed the commonality of arthrofibrosis or decreased ROM after extensive multiligament reconstruction surgeries.13,14 One study involving surgical management and outcomes of multiligament knee injuries found that, in more than half of its cases, restoration of full ROM required at least one operation after the initial one.13 Therefore, it is not unusual that our patient required a second operation for decreased ROM.
Conclusion
After surgery, excellent stabilization was achieved. Although the patient had setbacks related to pain and decreased ROM, his second surgery and continued physical therapy likely will help him return to his preoperative recreational activity levels.
1. Delos D, Warren RF, Marx RG. Multiligament knee injuries and their treatment. Oper Tech Sports Med. 2010;18(4):219-226.
2. Hobby B, Treme G, Wascher DC, Schenck RC. How I manage knee dislocations. Oper Tech Sports Med. 2010;18(4):227-234.
3. LaPrade RF, Ly TV, Wentorf FA, Engebretsen L. The posterolateral attachments of the knee: a qualitative and quantitative morphologic analysis of the fibular collateral ligament, popliteus tendon, popliteofibular ligament, and lateral gastrocnemius tendon. Am J Sports Med. 2003;31(6):854-860.
4. Chahla J, Nitri M, Civitarese D, Dean CS, Moulton SG, LaPrade RF. Anatomic double-bundle posterior cruciate ligament reconstruction. Arthrosc Tech. 2016;5(1):e149-e156.
5. Anderson CJ, Ziegler CG, Wijdicks CA, Engebretsen L, LaPrade RF. Arthroscopically pertinent anatomy of the anterolateral and posteromedial bundles of the posterior cruciate ligament. J Bone Joint Surg Am. 2012;94(21):1936-1945.
6. Mansour R, Yoong P, McKean D, Teh JL. The iliotibial band in acute knee trauma: patterns of injury on MR imaging. Skeletal Radiol. 2014;43(10):1369-1375.
7. LaPrade RF, Johansen S, Wentorf FA, Engebretsen L, Esterberg JL, Tso A. An analysis of an anatomical posterolateral knee reconstruction: an in vitro biomechanical study and development of a surgical technique. Am J Sports Med. 2004;32(6):1405-1414.
8. McCarthy M, Camarda L, Wijdicks CA, Johansen S, Engebretsen L, LaPrade RF. Anatomic posterolateral knee reconstructions require a popliteofibular ligament reconstruction through a tibial tunnel. Am J Sports Med. 2010;38(8):1674-1681.
9. LaPrade RF, Wozniczka JK, Stellmaker MP, Wijdicks CA. Analysis of the static function of the popliteus tendon and evaluation of an anatomic reconstruction: the “fifth ligament” of the knee. Am J Sports Med. 2010;38(3):543-549.
10. Geeslin AG, LaPrade RF. Outcomes of treatment of acute grade-III isolated and combined posterolateral knee injuries: a prospective case series and surgical technique. J Bone Joint Surg Am. 2011;93(18):1672-1683.
11. LaPrade RF, Johansen S, Agel J, Risberg MA, Moksnes H, Engebretsen L. Outcomes of an anatomic posterolateral knee reconstruction. J Bone Joint Surg Am. 2010;92(1):16-22.
12. Spiridonov SI, Slinkard NJ, LaPrade RF. Isolated and combined grade-III posterior cruciate ligament tears treated with double-bundle reconstruction with use of endoscopically placed femoral tunnels and grafts: operative technique and clinical outcomes. J Bone Joint Surg Am. 2011;93(19):1773-1780.
13. Noyes FR, Barber-Westin SD. Reconstruction of the anterior and posterior cruciate ligaments after knee dislocation. Use of early protected postoperative motion to decrease arthrofibrosis. Am J Sports Med. 1997;25(6):769-778.
14. Yenchak AJ, Wilk KE, Arrigo CA, Simpson CD, Andrews JR. Criteria-based management of an acute multistructure knee injury in a professional football player: a case report. J Orthop Sports Phys Ther. 2011;41(9):675-686.
1. Delos D, Warren RF, Marx RG. Multiligament knee injuries and their treatment. Oper Tech Sports Med. 2010;18(4):219-226.
2. Hobby B, Treme G, Wascher DC, Schenck RC. How I manage knee dislocations. Oper Tech Sports Med. 2010;18(4):227-234.
3. LaPrade RF, Ly TV, Wentorf FA, Engebretsen L. The posterolateral attachments of the knee: a qualitative and quantitative morphologic analysis of the fibular collateral ligament, popliteus tendon, popliteofibular ligament, and lateral gastrocnemius tendon. Am J Sports Med. 2003;31(6):854-860.
4. Chahla J, Nitri M, Civitarese D, Dean CS, Moulton SG, LaPrade RF. Anatomic double-bundle posterior cruciate ligament reconstruction. Arthrosc Tech. 2016;5(1):e149-e156.
5. Anderson CJ, Ziegler CG, Wijdicks CA, Engebretsen L, LaPrade RF. Arthroscopically pertinent anatomy of the anterolateral and posteromedial bundles of the posterior cruciate ligament. J Bone Joint Surg Am. 2012;94(21):1936-1945.
6. Mansour R, Yoong P, McKean D, Teh JL. The iliotibial band in acute knee trauma: patterns of injury on MR imaging. Skeletal Radiol. 2014;43(10):1369-1375.
7. LaPrade RF, Johansen S, Wentorf FA, Engebretsen L, Esterberg JL, Tso A. An analysis of an anatomical posterolateral knee reconstruction: an in vitro biomechanical study and development of a surgical technique. Am J Sports Med. 2004;32(6):1405-1414.
8. McCarthy M, Camarda L, Wijdicks CA, Johansen S, Engebretsen L, LaPrade RF. Anatomic posterolateral knee reconstructions require a popliteofibular ligament reconstruction through a tibial tunnel. Am J Sports Med. 2010;38(8):1674-1681.
9. LaPrade RF, Wozniczka JK, Stellmaker MP, Wijdicks CA. Analysis of the static function of the popliteus tendon and evaluation of an anatomic reconstruction: the “fifth ligament” of the knee. Am J Sports Med. 2010;38(3):543-549.
10. Geeslin AG, LaPrade RF. Outcomes of treatment of acute grade-III isolated and combined posterolateral knee injuries: a prospective case series and surgical technique. J Bone Joint Surg Am. 2011;93(18):1672-1683.
11. LaPrade RF, Johansen S, Agel J, Risberg MA, Moksnes H, Engebretsen L. Outcomes of an anatomic posterolateral knee reconstruction. J Bone Joint Surg Am. 2010;92(1):16-22.
12. Spiridonov SI, Slinkard NJ, LaPrade RF. Isolated and combined grade-III posterior cruciate ligament tears treated with double-bundle reconstruction with use of endoscopically placed femoral tunnels and grafts: operative technique and clinical outcomes. J Bone Joint Surg Am. 2011;93(19):1773-1780.
13. Noyes FR, Barber-Westin SD. Reconstruction of the anterior and posterior cruciate ligaments after knee dislocation. Use of early protected postoperative motion to decrease arthrofibrosis. Am J Sports Med. 1997;25(6):769-778.
14. Yenchak AJ, Wilk KE, Arrigo CA, Simpson CD, Andrews JR. Criteria-based management of an acute multistructure knee injury in a professional football player: a case report. J Orthop Sports Phys Ther. 2011;41(9):675-686.
Fever, rash, and leukopenia in a 32-year-old man • Dx?
THE CASE
A 32-year-old man was admitted to our hospital with fever, chills, malaise, leukopenia, and a rash. About 3 weeks earlier, he’d had oral maxillofacial surgery and started a 10-day course of prophylactic amoxicillin/clavulanic acid. Fifteen days after the surgery, he developed a fever (temperature, 103˚ F), chills, arthralgia, myalgia, cough, diarrhea, and malaise. He was seen by his physician, who obtained a chest x-ray showing a lingular infiltrate. The physician diagnosed influenza and pneumonia in this patient, and prescribed oseltamivir, azithromycin, and an additional course of amoxicillin/clavulanic acid.
Upon admission to the hospital, laboratory tests revealed a white blood cell count (WBC) of 3.1 k/mcL (normal: 3.2-10.8 k/mcL). The patient’s physical examination was notable for lip edema, white mucous membrane plaques, submandibular and inguinal lymphadenopathy, and a morbilliform rash across his chest (FIGURE 1). Broad-spectrum antibiotics were initiated for presumed sepsis.
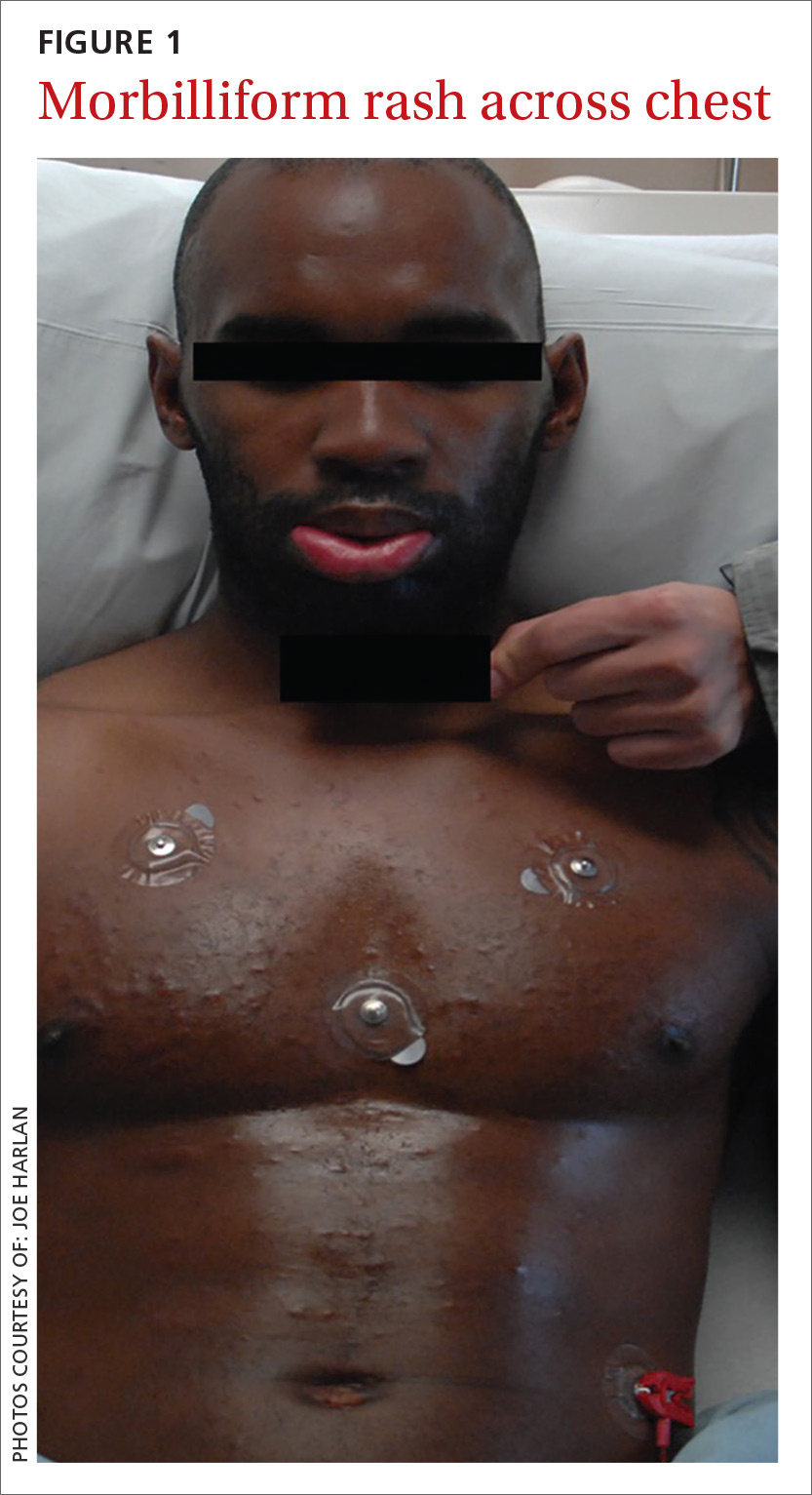
On hospital day (HD) 1, tests revealed a WBC count of 1.8 k/mcL, an erythrocyte sedimentation rate of 53 mm/hr (normal: 20-30 mm/hr for women, 15-20 mm/hr for men), and a C-reactive protein level of 6.7 mg/dL (normal: <0.5 mg/dL). A repeat chest x-ray and orofacial computerized tomography scan were normal.
By HD 3, all bacterial cultures were negative, but the patient was positive for human herpesvirus (HHV)-6 on viral cultures. His leukopenia persisted and he had elevated levels of alanine transaminase ranging from 40 to 73 U/L (normal: 6-43 U/L) and aspartate aminotransferase ranging from 66 to 108 U/L (normal range: 10-40 U/L), both downtrending during his hospitalization. He also had elevated levels of antinuclear antibodies (ANAs) and anti-Smith (Sm) antibody titers.
A posterior-auricular biopsy was consistent with lymphocytic perivasculitis. The rash continued to progress, involving his chest, abdomen, and face (FIGURE 2). Bacterial and viral cultures remained negative and on HD 4, broad-spectrum antibiotics were discontinued.
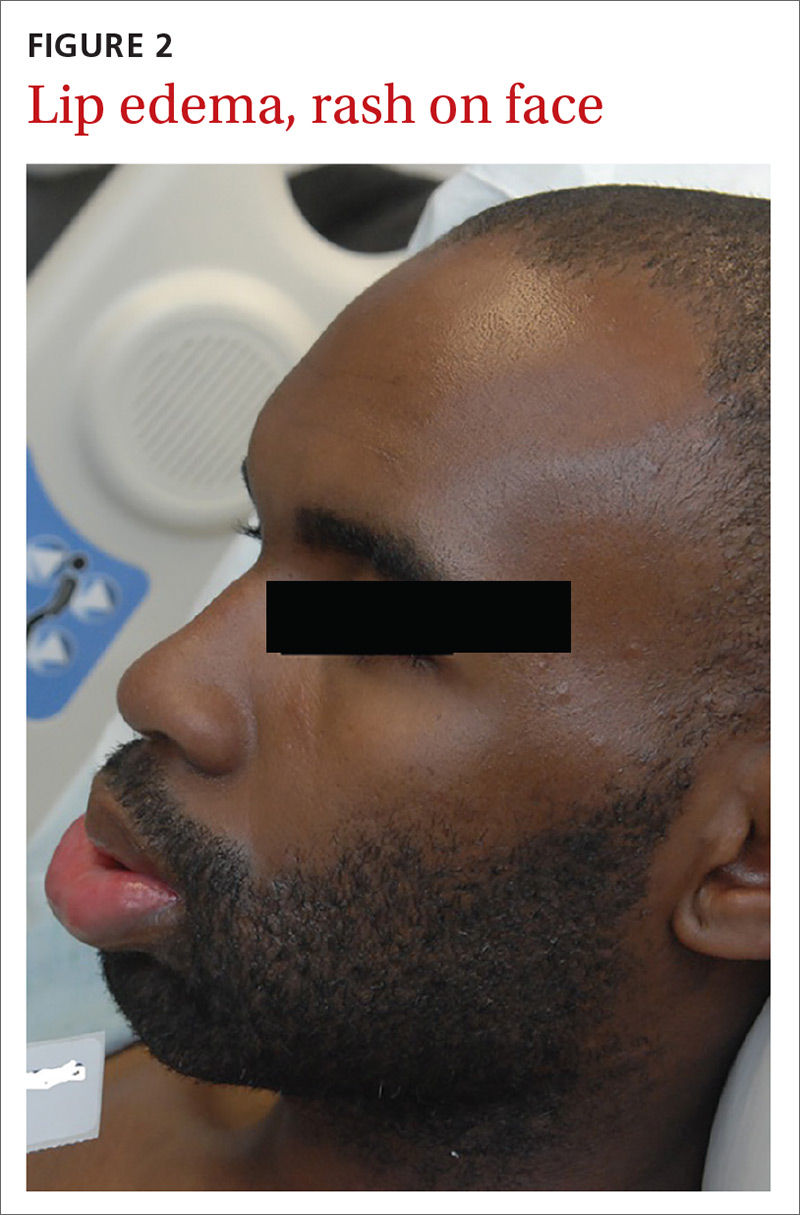
THE DIAGNOSIS
We diagnosed the patient with DRESS (drug reaction with eosinophilia and systemic symptoms) based on persistent fever, onset of cutaneous manifestations (facial edema and morbilliform eruption), lymphadenopathy, increased liver function tests, and recent exposure to an offending drug. The patient did not have eosinophilia; however, atypical lymphocytes were present on his peripheral smear.
DISCUSSION
DRESS is typically characterized by fever, rash, eosinophilia, atypical lymphocytes, lymphadenopathy, and organ involvement (primarily liver, but multiple organ systems can be affected).1 Patients with severe symptoms have renal involvement, anemia, respiratory and cardiac symptoms (chest pain, tachycardia, and myocarditis), and transaminase levels up to 5 times greater than normal.1-3 Anticonvulsants and antibiotics are the most common offending classes among the medications that are associated with DRESS (TABLE 1).2,4
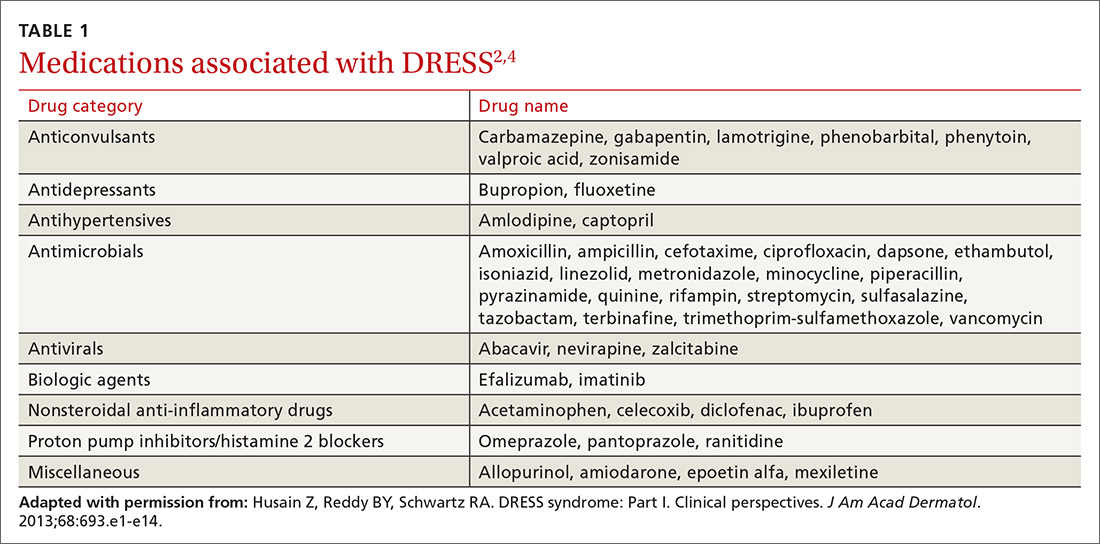
The reported incidence of DRESS is between one in 1000 and one in 10,000 drug exposures.1 Due to the broad presentation and a lack of established diagnostic criteria associated with DRESS, this number may be even higher. DRESS has a 10% mortality rate,1 and hepatic necrosis is the most common cause of death.2
Certain people may be more prone to DRESS. People with certain gene mutations that code for drug detoxification enzymes have shown a greater incidence of DRESS.5 Viral reactivation, commonly of HHV-6, has also been shown to have an effect on the pathogenesis of DRESS. Additionally, genetic predisposition involving specific human leukocyte antigens (HLAs) makes certain people more prone to the development of DRESS (TABLE 2).2,5
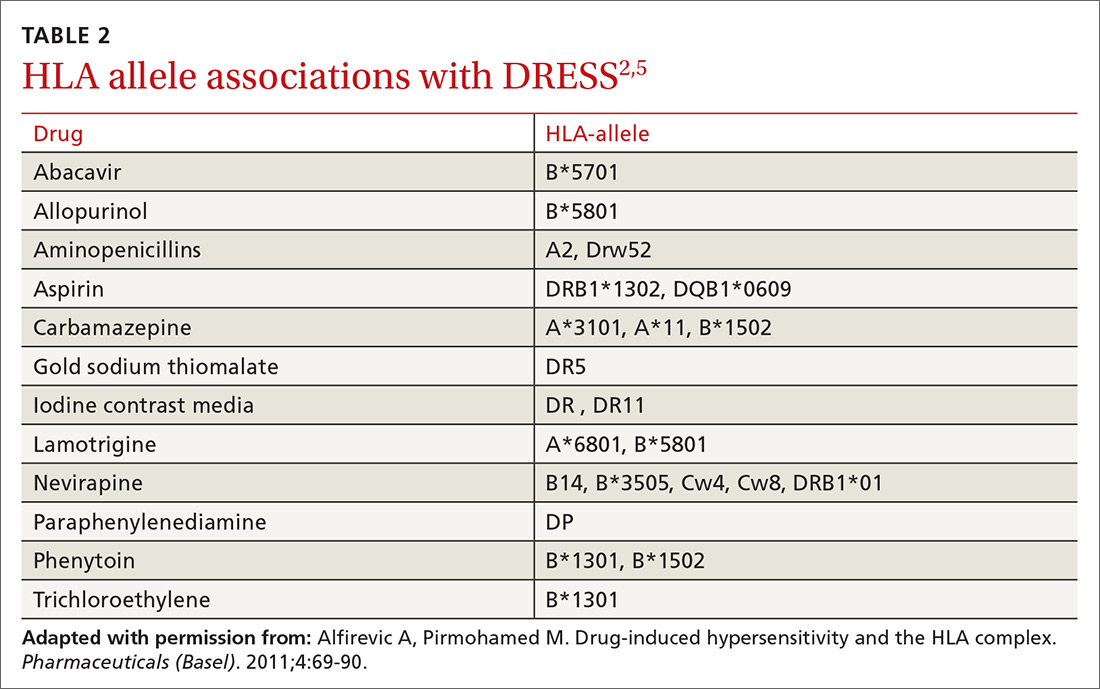
Case reports have demonstrated a link between certain autoimmune syndromes and DRESS, specifically Grave’s disease and type 1 diabetes mellitus.2
A unique finding of this case was the presence of elevated ANA and anti-Sm antibody titers at initial presentation, with spontaneous negative seroconversion 2 months later. Because of these 2 findings, as well as the patient’s leukopenia and rash, he briefly met 4 of the 11 criteria set forth by the American College of Rheumatology for a diagnosis of systemic lupus erythematosus (SLE).6 It is unclear whether the transiently elevated anti-Sm antibody titers were an acute phase reactant due to DRESS, a viral illness, or an evolving autoimmune process.
The false-positive rate for anti-Sm antibodies in association with DRESS has not been previously reported.
MAKING THE DIAGNOSIS
Distinguishing DRESS from other life-threatening cutaneous drug reactions, particularly Stevens-Johnson syndrome and toxic epidermal necrolysis, can be difficult. Likewise, acute bacterial/viral infections, autoimmune syndromes, vasculitis, and hematologic diseases can mimic DRESS.7 Exposure to an offending drug 2 to 6 weeks prior to the onset of symptoms is supportive of DRESS.
This scoring system can help. The RegiSCAR (Registry of Severe Cutaneous Adverse Reaction) has developed a scoring system to aid in the accurate diagnosis of DRESS.1,8 The scoring consists of 8 categories: fever, eosinophilia, enlarged lymph nodes, atypical lymphocytes, skin involvement, organ involvement, time of resolution, and the evaluation of other potential causes.1 Each category is graded a number from -1 (not supportive of DRESS) to 2 (highly supportive of DRESS) based on the patient’s presentation. The total score grades potential cases as “no,” “possible,” “probable,” or “definite.”1,8 In one review, cases classified as “probable” or “definite” by the RegiSCAR scoring system constituted 88% of the cases reported in the literature.1
Two tests that can also aid in the diagnosis of DRESS include patch testing (exposing the skin to a diluted version of the suspected offending drug and observing for a local reaction) and lymphocyte transformation tests. The latter are a better method of diagnosing drug-induced DRESS, with a sensitivity of 60% to 70%, and a specificity of 85%.9 However, this testing is not readily available.
Once DRESS is diagnosed, the offending drug should be immediately discontinued. For mild cases, supportive treatment is recommended. For more severe cases, the use of corticosteroids tapered over several months is the treatment of choice.10 Further studies are needed to determine the optimal type of corticosteroids, as well as the dose, route, and duration of therapy. Immunotherapy, plasmapheresis, and antivirals have been used with mixed results.10,11
Our patient was started on topical and systemic oral corticosteroids. Within 24 hours, his fever resolved and his rash improved. By HD 7, his laboratory values were normal and he was discharged.
The patient was advised that in the future, he should avoid exposure to the penicillin class of medication.
THE TAKEAWAY
The presence of rash, fever, lymphadenopathy, eosinophilia, atypical lymphocytes, liver involvement, and HHV-6 reactivation in the absence of sepsis should raise suspicion for DRESS. Early diagnosis, discontinuation of the culprit drug, and timely treatment are imperative in the management of the condition. Due to a potential genetic predisposition to DRESS, clinicians should use caution when treating first-degree family members with the same class of medication that was problematic for their relative. Long-term sequelae, such as Grave’s disease and diabetes mellitus, have been reported following DRESS. Therefore, long-term monitoring with appropriate testing is recommended.
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: Part I. Clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-e14.
3. Bourgeois GP, Cafardi JA, Groysman V, et al. Fulminant myocarditis as a late sequelae of DRESS-2 cases. J Am Acad Dermatol. 2011;65:889-890.
4. Cho YT, Yang CW, Chu CY. Drug reaction with eosinophilia and systemic symptoms (DRESS): an interplay among drugs, viruses, and immune system. Int J Mol Sci. 2017;18:1-21.
5. Alfirevic A, Pirmohamed M. Drug-induced hypersensitivity and the HLA complex. Pharmaceuticals (Basel). 2011;4:69-90.
6. American College of Rheumatology. 1997 Update of the 1982 American College of Rheumatology Revised Criteria for Classification of Systemic Lupus Erythematosus. Available at: https://www.rheumatology.org/Portals/0/Files/1982%20SLE%20Classification_Excerpt.pdf. Accessed August 30, 2017.
7. Descamps V, Ben Saïd B, Sassolas B, et al. Management of drug reaction with eosinophilia and systemic symptoms (DRESS). Ann Dermatol Venereol. 2010;137:703-708.
8. Peyrière H, Dereure O, Breton H, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2006;155:422-428.
9. Pichler WJ, Tilch J. The lymphocyte transformation test in the diagnosis of drug hypersensitivity. Allergy. 2004;59:809-820.
10. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome part II: management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-e9.
11. Funck-Brentano E, Duong TA, Bouvresses S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72:246-252.
THE CASE
A 32-year-old man was admitted to our hospital with fever, chills, malaise, leukopenia, and a rash. About 3 weeks earlier, he’d had oral maxillofacial surgery and started a 10-day course of prophylactic amoxicillin/clavulanic acid. Fifteen days after the surgery, he developed a fever (temperature, 103˚ F), chills, arthralgia, myalgia, cough, diarrhea, and malaise. He was seen by his physician, who obtained a chest x-ray showing a lingular infiltrate. The physician diagnosed influenza and pneumonia in this patient, and prescribed oseltamivir, azithromycin, and an additional course of amoxicillin/clavulanic acid.
Upon admission to the hospital, laboratory tests revealed a white blood cell count (WBC) of 3.1 k/mcL (normal: 3.2-10.8 k/mcL). The patient’s physical examination was notable for lip edema, white mucous membrane plaques, submandibular and inguinal lymphadenopathy, and a morbilliform rash across his chest (FIGURE 1). Broad-spectrum antibiotics were initiated for presumed sepsis.
On hospital day (HD) 1, tests revealed a WBC count of 1.8 k/mcL, an erythrocyte sedimentation rate of 53 mm/hr (normal: 20-30 mm/hr for women, 15-20 mm/hr for men), and a C-reactive protein level of 6.7 mg/dL (normal: <0.5 mg/dL). A repeat chest x-ray and orofacial computerized tomography scan were normal.
By HD 3, all bacterial cultures were negative, but the patient was positive for human herpesvirus (HHV)-6 on viral cultures. His leukopenia persisted and he had elevated levels of alanine transaminase ranging from 40 to 73 U/L (normal: 6-43 U/L) and aspartate aminotransferase ranging from 66 to 108 U/L (normal range: 10-40 U/L), both downtrending during his hospitalization. He also had elevated levels of antinuclear antibodies (ANAs) and anti-Smith (Sm) antibody titers.
A posterior-auricular biopsy was consistent with lymphocytic perivasculitis. The rash continued to progress, involving his chest, abdomen, and face (FIGURE 2). Bacterial and viral cultures remained negative and on HD 4, broad-spectrum antibiotics were discontinued.
THE DIAGNOSIS
We diagnosed the patient with DRESS (drug reaction with eosinophilia and systemic symptoms) based on persistent fever, onset of cutaneous manifestations (facial edema and morbilliform eruption), lymphadenopathy, increased liver function tests, and recent exposure to an offending drug. The patient did not have eosinophilia; however, atypical lymphocytes were present on his peripheral smear.
DISCUSSION
DRESS is typically characterized by fever, rash, eosinophilia, atypical lymphocytes, lymphadenopathy, and organ involvement (primarily liver, but multiple organ systems can be affected).1 Patients with severe symptoms have renal involvement, anemia, respiratory and cardiac symptoms (chest pain, tachycardia, and myocarditis), and transaminase levels up to 5 times greater than normal.1-3 Anticonvulsants and antibiotics are the most common offending classes among the medications that are associated with DRESS (TABLE 1).2,4
The reported incidence of DRESS is between one in 1000 and one in 10,000 drug exposures.1 Due to the broad presentation and a lack of established diagnostic criteria associated with DRESS, this number may be even higher. DRESS has a 10% mortality rate,1 and hepatic necrosis is the most common cause of death.2
Certain people may be more prone to DRESS. People with certain gene mutations that code for drug detoxification enzymes have shown a greater incidence of DRESS.5 Viral reactivation, commonly of HHV-6, has also been shown to have an effect on the pathogenesis of DRESS. Additionally, genetic predisposition involving specific human leukocyte antigens (HLAs) makes certain people more prone to the development of DRESS (TABLE 2).2,5
Case reports have demonstrated a link between certain autoimmune syndromes and DRESS, specifically Grave’s disease and type 1 diabetes mellitus.2
A unique finding of this case was the presence of elevated ANA and anti-Sm antibody titers at initial presentation, with spontaneous negative seroconversion 2 months later. Because of these 2 findings, as well as the patient’s leukopenia and rash, he briefly met 4 of the 11 criteria set forth by the American College of Rheumatology for a diagnosis of systemic lupus erythematosus (SLE).6 It is unclear whether the transiently elevated anti-Sm antibody titers were an acute phase reactant due to DRESS, a viral illness, or an evolving autoimmune process.
The false-positive rate for anti-Sm antibodies in association with DRESS has not been previously reported.
MAKING THE DIAGNOSIS
Distinguishing DRESS from other life-threatening cutaneous drug reactions, particularly Stevens-Johnson syndrome and toxic epidermal necrolysis, can be difficult. Likewise, acute bacterial/viral infections, autoimmune syndromes, vasculitis, and hematologic diseases can mimic DRESS.7 Exposure to an offending drug 2 to 6 weeks prior to the onset of symptoms is supportive of DRESS.
This scoring system can help. The RegiSCAR (Registry of Severe Cutaneous Adverse Reaction) has developed a scoring system to aid in the accurate diagnosis of DRESS.1,8 The scoring consists of 8 categories: fever, eosinophilia, enlarged lymph nodes, atypical lymphocytes, skin involvement, organ involvement, time of resolution, and the evaluation of other potential causes.1 Each category is graded a number from -1 (not supportive of DRESS) to 2 (highly supportive of DRESS) based on the patient’s presentation. The total score grades potential cases as “no,” “possible,” “probable,” or “definite.”1,8 In one review, cases classified as “probable” or “definite” by the RegiSCAR scoring system constituted 88% of the cases reported in the literature.1
Two tests that can also aid in the diagnosis of DRESS include patch testing (exposing the skin to a diluted version of the suspected offending drug and observing for a local reaction) and lymphocyte transformation tests. The latter are a better method of diagnosing drug-induced DRESS, with a sensitivity of 60% to 70%, and a specificity of 85%.9 However, this testing is not readily available.
Once DRESS is diagnosed, the offending drug should be immediately discontinued. For mild cases, supportive treatment is recommended. For more severe cases, the use of corticosteroids tapered over several months is the treatment of choice.10 Further studies are needed to determine the optimal type of corticosteroids, as well as the dose, route, and duration of therapy. Immunotherapy, plasmapheresis, and antivirals have been used with mixed results.10,11
Our patient was started on topical and systemic oral corticosteroids. Within 24 hours, his fever resolved and his rash improved. By HD 7, his laboratory values were normal and he was discharged.
The patient was advised that in the future, he should avoid exposure to the penicillin class of medication.
THE TAKEAWAY
The presence of rash, fever, lymphadenopathy, eosinophilia, atypical lymphocytes, liver involvement, and HHV-6 reactivation in the absence of sepsis should raise suspicion for DRESS. Early diagnosis, discontinuation of the culprit drug, and timely treatment are imperative in the management of the condition. Due to a potential genetic predisposition to DRESS, clinicians should use caution when treating first-degree family members with the same class of medication that was problematic for their relative. Long-term sequelae, such as Grave’s disease and diabetes mellitus, have been reported following DRESS. Therefore, long-term monitoring with appropriate testing is recommended.
THE CASE
A 32-year-old man was admitted to our hospital with fever, chills, malaise, leukopenia, and a rash. About 3 weeks earlier, he’d had oral maxillofacial surgery and started a 10-day course of prophylactic amoxicillin/clavulanic acid. Fifteen days after the surgery, he developed a fever (temperature, 103˚ F), chills, arthralgia, myalgia, cough, diarrhea, and malaise. He was seen by his physician, who obtained a chest x-ray showing a lingular infiltrate. The physician diagnosed influenza and pneumonia in this patient, and prescribed oseltamivir, azithromycin, and an additional course of amoxicillin/clavulanic acid.
Upon admission to the hospital, laboratory tests revealed a white blood cell count (WBC) of 3.1 k/mcL (normal: 3.2-10.8 k/mcL). The patient’s physical examination was notable for lip edema, white mucous membrane plaques, submandibular and inguinal lymphadenopathy, and a morbilliform rash across his chest (FIGURE 1). Broad-spectrum antibiotics were initiated for presumed sepsis.
On hospital day (HD) 1, tests revealed a WBC count of 1.8 k/mcL, an erythrocyte sedimentation rate of 53 mm/hr (normal: 20-30 mm/hr for women, 15-20 mm/hr for men), and a C-reactive protein level of 6.7 mg/dL (normal: <0.5 mg/dL). A repeat chest x-ray and orofacial computerized tomography scan were normal.
By HD 3, all bacterial cultures were negative, but the patient was positive for human herpesvirus (HHV)-6 on viral cultures. His leukopenia persisted and he had elevated levels of alanine transaminase ranging from 40 to 73 U/L (normal: 6-43 U/L) and aspartate aminotransferase ranging from 66 to 108 U/L (normal range: 10-40 U/L), both downtrending during his hospitalization. He also had elevated levels of antinuclear antibodies (ANAs) and anti-Smith (Sm) antibody titers.
A posterior-auricular biopsy was consistent with lymphocytic perivasculitis. The rash continued to progress, involving his chest, abdomen, and face (FIGURE 2). Bacterial and viral cultures remained negative and on HD 4, broad-spectrum antibiotics were discontinued.
THE DIAGNOSIS
We diagnosed the patient with DRESS (drug reaction with eosinophilia and systemic symptoms) based on persistent fever, onset of cutaneous manifestations (facial edema and morbilliform eruption), lymphadenopathy, increased liver function tests, and recent exposure to an offending drug. The patient did not have eosinophilia; however, atypical lymphocytes were present on his peripheral smear.
DISCUSSION
DRESS is typically characterized by fever, rash, eosinophilia, atypical lymphocytes, lymphadenopathy, and organ involvement (primarily liver, but multiple organ systems can be affected).1 Patients with severe symptoms have renal involvement, anemia, respiratory and cardiac symptoms (chest pain, tachycardia, and myocarditis), and transaminase levels up to 5 times greater than normal.1-3 Anticonvulsants and antibiotics are the most common offending classes among the medications that are associated with DRESS (TABLE 1).2,4
The reported incidence of DRESS is between one in 1000 and one in 10,000 drug exposures.1 Due to the broad presentation and a lack of established diagnostic criteria associated with DRESS, this number may be even higher. DRESS has a 10% mortality rate,1 and hepatic necrosis is the most common cause of death.2
Certain people may be more prone to DRESS. People with certain gene mutations that code for drug detoxification enzymes have shown a greater incidence of DRESS.5 Viral reactivation, commonly of HHV-6, has also been shown to have an effect on the pathogenesis of DRESS. Additionally, genetic predisposition involving specific human leukocyte antigens (HLAs) makes certain people more prone to the development of DRESS (TABLE 2).2,5
Case reports have demonstrated a link between certain autoimmune syndromes and DRESS, specifically Grave’s disease and type 1 diabetes mellitus.2
A unique finding of this case was the presence of elevated ANA and anti-Sm antibody titers at initial presentation, with spontaneous negative seroconversion 2 months later. Because of these 2 findings, as well as the patient’s leukopenia and rash, he briefly met 4 of the 11 criteria set forth by the American College of Rheumatology for a diagnosis of systemic lupus erythematosus (SLE).6 It is unclear whether the transiently elevated anti-Sm antibody titers were an acute phase reactant due to DRESS, a viral illness, or an evolving autoimmune process.
The false-positive rate for anti-Sm antibodies in association with DRESS has not been previously reported.
MAKING THE DIAGNOSIS
Distinguishing DRESS from other life-threatening cutaneous drug reactions, particularly Stevens-Johnson syndrome and toxic epidermal necrolysis, can be difficult. Likewise, acute bacterial/viral infections, autoimmune syndromes, vasculitis, and hematologic diseases can mimic DRESS.7 Exposure to an offending drug 2 to 6 weeks prior to the onset of symptoms is supportive of DRESS.
This scoring system can help. The RegiSCAR (Registry of Severe Cutaneous Adverse Reaction) has developed a scoring system to aid in the accurate diagnosis of DRESS.1,8 The scoring consists of 8 categories: fever, eosinophilia, enlarged lymph nodes, atypical lymphocytes, skin involvement, organ involvement, time of resolution, and the evaluation of other potential causes.1 Each category is graded a number from -1 (not supportive of DRESS) to 2 (highly supportive of DRESS) based on the patient’s presentation. The total score grades potential cases as “no,” “possible,” “probable,” or “definite.”1,8 In one review, cases classified as “probable” or “definite” by the RegiSCAR scoring system constituted 88% of the cases reported in the literature.1
Two tests that can also aid in the diagnosis of DRESS include patch testing (exposing the skin to a diluted version of the suspected offending drug and observing for a local reaction) and lymphocyte transformation tests. The latter are a better method of diagnosing drug-induced DRESS, with a sensitivity of 60% to 70%, and a specificity of 85%.9 However, this testing is not readily available.
Once DRESS is diagnosed, the offending drug should be immediately discontinued. For mild cases, supportive treatment is recommended. For more severe cases, the use of corticosteroids tapered over several months is the treatment of choice.10 Further studies are needed to determine the optimal type of corticosteroids, as well as the dose, route, and duration of therapy. Immunotherapy, plasmapheresis, and antivirals have been used with mixed results.10,11
Our patient was started on topical and systemic oral corticosteroids. Within 24 hours, his fever resolved and his rash improved. By HD 7, his laboratory values were normal and he was discharged.
The patient was advised that in the future, he should avoid exposure to the penicillin class of medication.
THE TAKEAWAY
The presence of rash, fever, lymphadenopathy, eosinophilia, atypical lymphocytes, liver involvement, and HHV-6 reactivation in the absence of sepsis should raise suspicion for DRESS. Early diagnosis, discontinuation of the culprit drug, and timely treatment are imperative in the management of the condition. Due to a potential genetic predisposition to DRESS, clinicians should use caution when treating first-degree family members with the same class of medication that was problematic for their relative. Long-term sequelae, such as Grave’s disease and diabetes mellitus, have been reported following DRESS. Therefore, long-term monitoring with appropriate testing is recommended.
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: Part I. Clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-e14.
3. Bourgeois GP, Cafardi JA, Groysman V, et al. Fulminant myocarditis as a late sequelae of DRESS-2 cases. J Am Acad Dermatol. 2011;65:889-890.
4. Cho YT, Yang CW, Chu CY. Drug reaction with eosinophilia and systemic symptoms (DRESS): an interplay among drugs, viruses, and immune system. Int J Mol Sci. 2017;18:1-21.
5. Alfirevic A, Pirmohamed M. Drug-induced hypersensitivity and the HLA complex. Pharmaceuticals (Basel). 2011;4:69-90.
6. American College of Rheumatology. 1997 Update of the 1982 American College of Rheumatology Revised Criteria for Classification of Systemic Lupus Erythematosus. Available at: https://www.rheumatology.org/Portals/0/Files/1982%20SLE%20Classification_Excerpt.pdf. Accessed August 30, 2017.
7. Descamps V, Ben Saïd B, Sassolas B, et al. Management of drug reaction with eosinophilia and systemic symptoms (DRESS). Ann Dermatol Venereol. 2010;137:703-708.
8. Peyrière H, Dereure O, Breton H, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2006;155:422-428.
9. Pichler WJ, Tilch J. The lymphocyte transformation test in the diagnosis of drug hypersensitivity. Allergy. 2004;59:809-820.
10. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome part II: management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-e9.
11. Funck-Brentano E, Duong TA, Bouvresses S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72:246-252.
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: Part I. Clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-e14.
3. Bourgeois GP, Cafardi JA, Groysman V, et al. Fulminant myocarditis as a late sequelae of DRESS-2 cases. J Am Acad Dermatol. 2011;65:889-890.
4. Cho YT, Yang CW, Chu CY. Drug reaction with eosinophilia and systemic symptoms (DRESS): an interplay among drugs, viruses, and immune system. Int J Mol Sci. 2017;18:1-21.
5. Alfirevic A, Pirmohamed M. Drug-induced hypersensitivity and the HLA complex. Pharmaceuticals (Basel). 2011;4:69-90.
6. American College of Rheumatology. 1997 Update of the 1982 American College of Rheumatology Revised Criteria for Classification of Systemic Lupus Erythematosus. Available at: https://www.rheumatology.org/Portals/0/Files/1982%20SLE%20Classification_Excerpt.pdf. Accessed August 30, 2017.
7. Descamps V, Ben Saïd B, Sassolas B, et al. Management of drug reaction with eosinophilia and systemic symptoms (DRESS). Ann Dermatol Venereol. 2010;137:703-708.
8. Peyrière H, Dereure O, Breton H, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2006;155:422-428.
9. Pichler WJ, Tilch J. The lymphocyte transformation test in the diagnosis of drug hypersensitivity. Allergy. 2004;59:809-820.
10. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome part II: management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-e9.
11. Funck-Brentano E, Duong TA, Bouvresses S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72:246-252.
