User login
Back to Basics: An Uncommon, Unrelated Presentation of a Common Disease
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
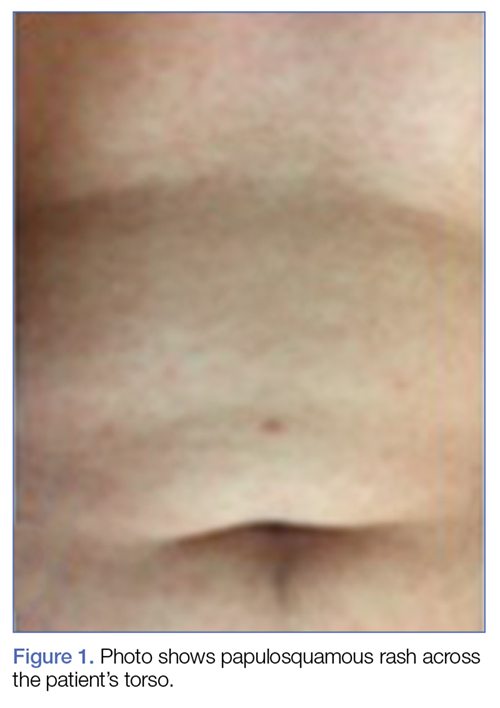
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
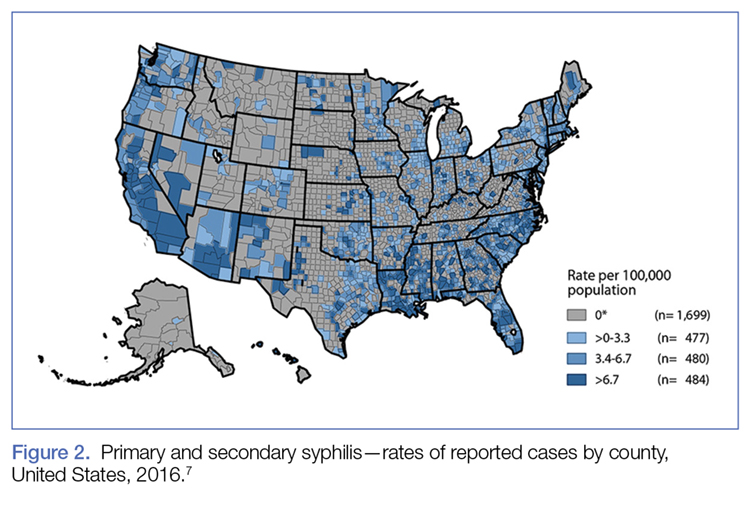
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
The early initial ulcerative lesion (chancre) caused by Treponema pallidum infection, has a median incubation period of 21 days (primary syphilis). When untreated, secondary syphilis will develop within weeks to months and is characterized by generalized symptoms such as malaise, fevers, headaches, sore throat, and myalgia. However, the most characteristic finding in secondary syphilis remains a rash that is classically identified as symmetric, macular, or papular, and involving the entire trunk and extremities, including the palms and soles.
When secondary syphilis is left untreated, late syphilis or tertiary syphilis can develop, which is characterized by cardiovascular involvement, including aortitis, gummatous syphilis (granulomatous nodules in a variety of organs but typically the skin and bones), or central nervous system involvement.1-3 The following case describes a patient with nondescript symptoms, including malaise and cough, who had a characteristic rash of secondary syphilis that was diagnosed and treated in our Houston-area community hospital.
Case
In late autumn, a 30-year-old man presented to our community ED for evaluation of a cough productive of green sputum along with mild chest discomfort, malaise, and generalized myalgia, which were intermittent over the course of the past month. The patient denied rhinorrhea, fevers, chills, dyspnea, or any other systemic complaints. He also denied any sick contacts, but noted that his influenza vaccine was not up to date.
The patient denied any remote or recent medical or surgical history. He further denied taking any medications, and noted that his only medical allergy was to penicillin. His family history was noncontributory. Regarding his social history, the patient admitted to smoking one pack of cigarettes per day and to a daily alcohol intake of approximately one 6-pack of beer. He also admitted to frequently smoking crystal methamphetamine, which he stated he had last used 2 days prior to presentation. The patient said his current chest pain was similar to prior episodes, noting that when the pain occurred, he would temporarily stop smoking crystal methamphetamine.
Plain chest radiography, electrocardiogram, complete metabolic panel, complete blood count, B-natriuretic peptide, and troponin levels were all unremarkable. Due to the presence and nature of the patient’s rash, a rapid plasma reagin (RPR) screen was also taken, the results of which were reactive.
On further questioning, the patient admitted to having multiple female sexual partners with whom he used barrier protection sporadically. A more detailed physical examination revealed multiple painless ulcerations/chancres over the penile shaft and scrotum, without urethral drainage or inguinal lymphadenopathy. The patient denied dysuria or hematuria.
Since the patient was allergic to penicillin, he was given a single oral dose of azithromycin 2 g, and started on a 2-week course of oral doxycycline 100 mg. Further laboratory studies included gonorrhea and chlamydia cultures, both of which were negative. He was instructed to follow-up with his primary care physician for extended sexually transmitted infection (STI) panel-testing, including HIV, hepatitis, and confirmatory syphilis testing. Unfortunately, it is not known whether the patient complied with discharge instructions as he was lost to follow-up.
Discussion
Diagnostic algorithms for syphilis, one of the best studied STIs, have changed with technological advancement, but diagnosis and treatment for the most part has remained mostly the same. The uniqueness of this case is really focused around the patient’s chief complaint. While it is classic to present with malaise, headache, and rash, our patient complained of cough productive of sputum with chest pain—a rare presentation of secondary syphilis. The fortuitous key finding of the truncal rash directed the emergency physician toward the appropriate diagnosis.
Diagnosis
In the ED, where patients such as the one in our case are often lost to follow-up, and consistent infectious disease and primary care follow-up is unavailable, prompt treatment based on history and physical examination alone is recommended. Patients should be tested for syphilis, as well as other STIs including chlamydia, gonorrhea, hepatitis, and HIV as an outpatient. In addition, any partners with whom the patient has had sexual contact within the last 90 days should also undergo STI testing; sexual partners from over 90 days should be notified of the patient’s status and evaluated with testing as indicated.4 All positive test results should be reported to the Centers for Disease Control and Prevention (CDC).5
Nontreponemal and Treponemal Testing
For patients with clinical signs and symptoms of syphilis, recommended laboratory evaluation includes both nontreponemal and treponemal testing. Nontreponemal tests include RPR, venereal disease research laboratory test, and toluidine red unheated serum test. Treponemal tests include fluorescent treponemal antibody absorption, microhemagglutination test for antibodies to T pallidum, T pallidum particle agglutination assay, T pallidum enzyme immunoassay, and chemiluminescence immunoassay. Patients who test positive for treponemal antibody will typically remain reactive for life.5,6
In the setting of discordant test results, patients with a nonreactive treponemal result are generally considered to be negative for syphilis. Discordant results with a negative nontreponemal test are more complicated, and recommendations are based on symptomatology and repeat testing.5
Treatment
When a patient has a positive nontreponemal and treponemal test, treatment is usually indicated. As with the patient in this case, treatment is always indicated for patients who have no prior history of syphilis. For patients who have a history of treated syphilis, attention must be given to titer levels on previous testing and to patient symptomatology.
The treatment for early (primary and secondary) syphilis in patients with no penicillin allergy is a single dose of penicillin G benzathine intramuscularly, at a dose of 2.4 million U. Alternative regimens include doxycycline 100 mg orally twice daily for 14 days, and azithromycin 2 g orally as a single dose; however, there is an association of treatment failure with azithromycin due to macrolide resistance.5 The patient in this case received empiric treatment targeting syphilis, gonorrhea, and chlamydia.
Conclusion
Ten years ago, the rates of primary and secondary syphilis were low, leading the infectious disease community to believe that preventive efforts had been effective. According to the CDC, however, “[current] rates…are the highest they have been in more than 20 years.”5Figure 2 demonstrates the geographic distribution of syphilis cases in the United States in 2016.7
Heightened concern has prompted the CDC to promote the theme “Syphilis Strikes Back” in April 2017, which was STI Awareness Month.8 Identification of disease is critical in the ED, especially when a previously common disease has become uncommon, like the resurgence of syphilis we are now seeing.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
1. Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: An epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
2. Rockwell DH, Yobs AR, Moore MB Jr. The Tuskegee study of untreated syphilis; the 30th year of observation. Arch Intern Med. 1964;114:792-798.
3. Sparling PF, Swartz MN, Musher DM, Healy BP. Clinical manifestations of syphilis. In: Holmes KK, Sparling PF, Stamm WE, et al, eds. Sexually Transmitted Diseases. 4th ed. New York, NY: McGraw-Hill; 1999:661-684.
4. Birnbaumer DM. Sexually transmitted diseases. In: Marx JA, Hockberger RS, Walls RM, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Vol 2. 8th ed. Philadelphia, PA: Saunders; 2014:1312-1325.
5. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
6. Larsen SA. Syphilis. Clin Lab Med. 1989;9(3):545-557.
7. Centers for Disease Control Prevention. Primary and secondary syphilis—rates of reported cases by county, United States, 2016. https://www.cdc.gov/std/stats16/figures/33.htm. Updated September 26, 2017. Accessed October 31 2017.]
8. Centers for Disease Control and Prevention. STD Awareness Month. Syphilis Strikes Back. https://www.cdc.gov/std/sam/index.htm?s_cid=tw_SAM_17001. Updated April 6, 2017. Accessed October 31, 2017.
Duodenal Perforation After Endoscopic Procedure
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure). 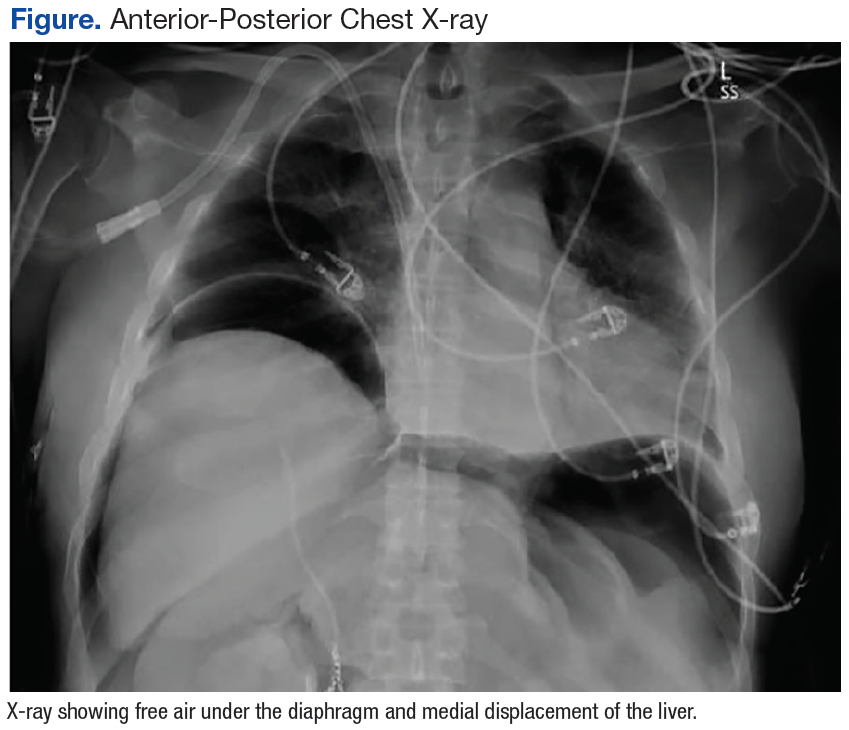
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure).
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
Tension pneumoperitoneum (TPP), also known as hyperacute abdominal compartment syndrome or abdominal tamponade, is a rare condition most commonly associated with gastrointestinal (GI) perforation during endoscopy and iatrogenic insufflation of gas into the peritoneal cavity.1 Other reported causes of TPP include gastric rupture after cardiopulmonary resuscitation, barotrauma during scuba diving, positive pressure ventilation through pleural-peritoneal channels, and spontaneous TPP of uncertain mechanism.1-4
Case Presentation
A 76-year-old male with a history of ischemic cardiomyopathy, hypertension, and diabetes mellitus presented to the VA Puget Sound Health Care System in Seattle, Washington emergency department with painless jaundice, hematemesis, melena, and acute renal failure. On esophagogastroduodenoscopy (EGD), he was found to have an ulcer on the posterior wall of the duodenal bulb. The ulcer was coagulated and injected with epinephrine. The patient’s subsequent hospital course was complicated by worsening liver function, the need for renal replacement therapy, and recurrence of upper GI bleeding that required a transcatheter embolization of 2 separate superior pancreaticoduodenal arteries (SPDA) and the inferior pancreaticoduodenal artery (IPDA).
Once clinically stable, an endoscopic retrograde cholangiopancreatography (ERCP) was performed to evaluate for cholangiocarcinoma. A stricture was discovered in the common hepatic duct, brushings were taken, and a 15 cm, 7 Fr stent was placed in the common hepatic duct. The procedure was performed with an Olympus TJF Type Q180V duodenovideoscope (Tokyo, Japan) with an external diameter of 13.7 mm. The patient became hypotensive during the procedure and was treated with phenylephrine and ephedrine boluses. There was no endoscopic evidence of bleeding or bowel trauma.
After completion of the procedure, in the recovery area the patient became severely hypotensive and unresponsive. The physical examination was noteworthy for gross abdominal distention. Arterial blood gas analysis revealed severe metabolic and respiratory acidosis. Chest radiography demonstrated massive pneumoperitoneum, low lung volumes, and diaphragmatic compression (Figure).
A diagnosis of tension pneumoperitoneum was made, and as the patient was transported to the operating room he became bradycardic without a pulse, requiring initiation of cardiopulmonary resuscitation. The abdomen was decompressed with a 14-gauge needle, followed by insertion of a laparoscopic trocar as a decompressive maneuver. This procedure resulted in return of spontaneous circulation.
An exploratory laparotomy was performed, and a massive rush of air was noted on opening the peritoneum. A pinhole perforation of the anterior wall of the second portion of the duodenum was found along with large-volume bilious ascites. This perforation was repaired with a Graham patch, and the patient was taken to the intensive care unit. Postoperatively, the patient developed disseminated intravascular coagulation, shock liver, and acute respiratory distress syndrome, expiring 10 days later from sequelae of multiorgan failure.
Discussion
In relation to upper GI endoscopic procedures, TPP has been reported after diagnostic EGD, endoscopic sphincterotomy, and submucosal tumor dissection.5-7 During these interventions, clinically apparent or overt iatrogenic perforations can occur either in the stomach or duodenum. These perforations may function as one-way valves that cause massive air accumulation and marked elevation of the diaphragm, which severely decreases lung volumes, pulmonary compliance, and limits gas exchange. Hemodynamically, compression of the inferior vena cava restricts venous return to the heart, resulting in decreased cardiac output.8
Patients with TPP present in acute distress with dyspnea, abdominal pain, and shock. On physical examination the abdomen is markedly distended, tympanic, and rigid. Rectal prolapse and subcutaneous emphysema also may be present.9 Roentographic features of TPP include findings of intraperitoneal air with elevation of the diaphragm, medial displacement of the liver (saddlebag sign), and juxtaposition of air in visceral interfaces, making intra-abdominal structures (spleen and gallbladder) appear more discrete.10 Abdominal computer tomography may show massive pneumoperitoneum with bowel loop compression and centralization of abdominal organs.4
Treatment strategies include emergent decompression either with percutaneous catheter insertion or abdominal drain placement followed by a definitive surgical repair. As with management of tension pneumothorax, treatment should not be delayed while awaiting confirmatory radiologic studies.9 When percutaneous needle decompression is undertaken, it is preferable to use a large bore (14-gauge venous catheter) and to advance a catheter over a needle to minimize the risk of visceral injury with egress of air and return of abdominal organs to their normal anatomical positions. The needle should be inserted directly above or below the umbilicus or in the left or right lower quadrants to avoid solid organ (ie, liver or spleen) injury.
Etiologic possibilities for the duodenal perforation in this case include mechanical trauma from the endoscope and duodenal tissue infarction after embolization of a bleeding duodenal ulcer. The duodenum and pancreatic head have a dual blood supply from the SPDA, a branch of the gastroduodenal artery, and the IPDA, a branch of the superior mesenteric artery.11 After failed endoscopic management of persistent duodenal hemorrhage, the patient underwent synchronous embolization of 2 separate SPDAs and the IPDA. This might have put the first 2 segments of the duodenum at risk for ischemic damage and caused it to perforate at some point during the patient’s hospitalization (as evidenced by the bilious ascitis) or rendered them vulnerable to perforation from intraluminal insufflation during endoscopy.12
During the laparotomy, a pinhole-sized perforation was noted in the anterior wall of the second part of the duodenum, distinct form the duodenal ulcer present on the posterior wall. This perforation likely provided a pathway for the intraluminal gas to escape into the peritoneal cavity, culminating in abdominal tamponade, cardiopulmonary deterioration, and arrest. Needle decompression of the abdominal cavity provided an effective, though temporizing relief of this pressure, enabling return of spontaneous circulation.
This case highlights the need for a high index of suspicion for TPP in a patient with cardiopulmonary compromise and abdominal distension after upper GI endoscopic procedures even in the absence of identifiable perforations. Close coordination among gastroenterologists, anesthesiologists, and surgeons is key in prevention, recognition, and management of this rare but catastrophic complication.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
1. Bunni J, Bryson PJ, Higgs SM. Abdominal compartment syndrome caused by tension pneumoperitoneum in a scuba diver. Ann R Coll Surg Engl. 2012;94(8):e237-e239.
2. Cameron PA, Rosengarten PL, Johnson WR, Dziukas L. Tension pneumoperitoneum after cardiopulmonary resuscitation. Med J Aust. 1991;155(1):44-47.
3. Burdett-Smith P, Jaffey L. Tension pneumoperitoneum. J Accid Emerg Med. 1996;13(3):220-221.
4. Joshi D, Ganai B. Radiological features of tension pneumoperitoneum. BMJ Case Rep. 2015;2015.
5. Rai A, Iftikhar S. Tension pneumothorax complicating diagnostic upper endoscopy: a case report. Am J Gastroenterol. 1999;94(3):845-847.
6. Iyilikci L, Akarsu M, Duran E, et al. Duodenal perforation and bilateral tension pneumothorax following endoscopic sphincterotomy. J Anesth. 2009;23(1):164-165.
7. Siboni S, Bona D, Abate E, Bonavina L. Tension pneumoperitoneum following endoscopic submucosal dissection of leiomyoma of the cardia. Endoscopy. 2010;42(suppl 2):E152.
8. Deenichin GP. Abdominal compartment syndrome. Surg Today. 2008;38(1):5-19.
9. Chiapponi C, Stocker U, Korner M, et al. Emergency percutaneous needle decompression for tension pneumoperitoneum. BMC Gastroenterol. 2011;11:48.
10. Lin BW, Thanassi W. Tension pneumoperitoneum. J Emerg Med. 2010;38(1):57-59.
11. Bell SD, Lau KY, Sniderman KW. Synchronous embolization of the gastroduodenal artery and the inferior pancreaticoduodenal artery in patients with massive duodenal hemorrhage. J Vasc Interv Radiol. 1995;6(4):531-536.
12. Wang YL, Cheng YS, Liu LZ, He ZH, Ding KH. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18(34):4765-4770.
A Case of Leprosy in Central Florida
Case Report
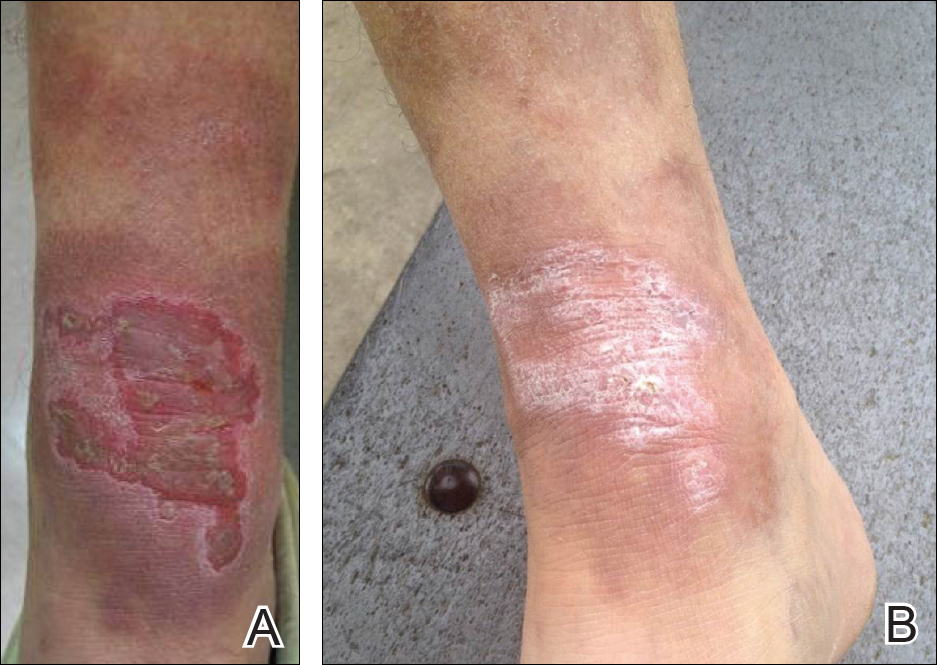
A 65-year-old man presented with multiple anesthetic, annular, erythematous, scaly plaques with a raised border of 6 weeks’ duration that were unresponsive to topical steroid therapy. Four plaques were noted on the lower back ranging from 2 to 4 cm in diameter as well as a fifth plaque on the anterior portion of the right ankle that was approximately 6×6 cm. He denied fever, malaise, muscle weakness, changes in vision, or sensory deficits outside of the lesions themselves. The patient also denied any recent travel to endemic areas or exposure to armadillos.
Biopsies were taken from lesions on the lumbar back and anterior aspect of the right ankle (Figure 1A). Hematoxylin and eosin staining revealed a granulomatous infiltrate spreading along neurovascular structures (Figure 2). Granulomas also were identified in the dermal interstitium exhibiting partial necrosis (Figure 2 inset). Conspicuous distension of lymphovascular and perineural areas also was noted. Immunohistochemical studies with S-100 and neurofilament stains allowed insight into the pathomechanism of the clinically observed anesthesia, as nerve fibers were identified showing different stages of damage elicited by the granulomatous inflammatory infiltrate (Figure 3). Fite staining was positive for occasional bacilli within histiocytes (Figure 3A inset). Despite the clinical, histologic, and immunohistochemical evidence, the patient had no known exposure to leprosy; consequently, a polymerase chain reaction (PCR) assay was ordered for confirmation of the diagnosis. Surprisingly, the PCR was positive for Mycobacterium leprae DNA. These findings were consistent with borderline tuberculoid leprosy.
The case was reported to the National Hansen’s Disease Program (Baton Rouge, Louisiana). The patient was started on rifampicin 600 mg once monthly and dapsone 100 mg once daily for 6 months. The lesions exhibited marked improvement after completion of therapy (Figure 1B).
Comment
Disease Transmission
Hansen disease, also known as leprosy, is a chronic granulomatous infectious disease that is caused by M leprae, an obligate intracellular bacillus aerobe.1 The mechanism of spread of M leprae is not clear. It is thought to be transmitted via respiratory droplets, though it may occur through injured skin.2 Studies have suggested that in addition to humans, nine-banded armadillos are a source of infection.2,3 Exposure to infected individuals, particularly multibacillary patients, increases the likelihood of contracting leprosy.2
According to the Centers for Disease Control and Prevention, 81 cases of Hansen disease were diagnosed in the United States in 2013,4 compared to 178 cases registered in 2015.5 Cases from Hawaii, Texas, California, Louisiana, New York, and Florida made up 72% (129/178) of the reported cases. There was an increase from 34 cases to 49 cases in Florida from 2014 to 2015.5 The spread of leprosy throughout Florida may be from the merge of 2 armadillo populations, an M leprae–infected population migrating east from Texas and one from south central Florida that historically had not been infected with M leprae until recently.3,6 Our patient did not have any known exposures to armadillos.
Classification and Presentation
The clinical presentation of Hansen disease is widely variable, as it can present at any point along a spectrum ranging from tuberculoid leprosy to lepromatous leprosy with borderline conditions in between, according to the Ridley-Jopling critera.7 The World Health Organization also classifies leprosy based on the number of acid-fast bacilli seen in a skin smear as either paucibacillary or multibacillary.2 The paucibacillary classification correlates with tuberculoid, borderline tuberculoid, and indeterminate leprosy, and multibacillary correlates with borderline lepromatous and lepromatous leprosy. Paucibacillary leprosy usually presents with a less dramatic clinical picture than multibacillary leprosy. The clinical presentation is dependent on the magnitude of immune response to M leprae.2
Paucibacillary infection occurs when the body generates a strong cell-mediated immune response against the bacteria,8 which causes the activation and proliferation of CD4 and CD8 T cells, limiting the spread of the mycobacterium. Subsequently, the patient typically presents with a mild clinical picture with few skin lesions and limited nerve involvement.8 The skin lesions are papules or plaques with raised borders that are usually hypopigmented on dark skin and erythematous on light skin. Nerve involvement in paucibacillary forms of leprosy include sensory impairment and anhidrosis within the lesions. Nerve enlargement usually affects superficial nerves, with the posterior tibial nerve being most commonly affected.
Multibacillary leprosy presents with systemic involvement due to a weak cell-mediated immune response. Patients generally present with diffuse, poorly defined nodules; greater nerve impairment; and other systemic symptoms such as blindness, swelling of the fingers and toes, and testicular atrophy (in men). Additionally, enlargement of the earlobes and widening of the nasal bridge may contribute to the appearance of leonine facies. Nerve impairment in multibacillary forms of leprosy may be more severe, including more diffuse sensory involvement (eg, stocking glove–pattern neuropathy, nerve-trunk palsies), which ultimately may lead to foot drop, claw toe, and lagophthalmos.8
In addition to the clinical presentation, the histology of the paucibacillary and multibacillary types differ. Multibacillary leprosy shows diffuse histiocytes without granulomas and multiple bacilli seen on Fite staining.8 In the paucibacillary form, there are well-formed granulomas with Langerhans giant cells and a perineural lymphocytic infiltrate seen on hematoxylin and eosin staining with rare acid-fast bacilli seen on Fite staining.
To diagnose leprosy, at least one of the following 3 clinical signs must be present: (1) a hypopigmented or erythematous lesion with loss of sensation, (2) thickened peripheral nerve, or (3) acid-fast bacilli on slit-skin smear.2
Management
The World Health Organization guidelines involve multidrug therapy over an extended period of time.2 For adults, the paucibacillary regimen includes rifampicin 600 mg once monthly and dapsone 100 mg once daily for 6 months. The adult regimen for multibacillary leprosy includes clofazimine 300 mg once monthly and 50 mg once daily, in addition to rifampicin 600 mg once monthly and dapsone 100 mg once daily for 12 months. If classification cannot be determined, it is recommended the patient be treated for multibacillary disease.2
Reversal Reactions
During the course of the disease, patients may upgrade (to a less severe form) or downgrade (to a more severe form) between the tuberculoid, borderline, and lepromatous forms.8 The patient’s clinical picture also may change with complications of leprosy, which include type 1 and type 2 reactions. Type 1 reaction is a reversal reaction seen in 15% to 30% of patients at risk, usually those with borderline forms of leprosy.9 Reversal reactions usually manifest as erythema and edema of current skin lesions, formation of new tumid lesions, and tenderness of peripheral nerves with loss of nerve function.8 Treatment of reversal reactions involves systemic corticosteroids.10 Type 2 reaction is classified as erythema nodosum leprosum. It presents within the first 2 years of treatment in approximately 20% of lepromatous patients and approximately 10% of borderline lepromatous patients but is rare in paucibacillary infections.11 It presents with fever and crops of pink nodules and may include iritis, neuritis, lymphadenitis, orchitis, dactylitis, arthritis, and proteinuria.8 Treatment options for erythema nodosum leprosum include corticosteroids, clofazimine, and thalidomide.12,13
Conclusion
Hansen disease is a rare condition in the United States. This case is unique because, to our knowledge, it is the first known PCR-confirmed case of Hansen disease in Okeechobee County, Florida. Additionally, the patient had no known exposure to M leprae. Exposure is increasing due to the increased geographical range of infected armadillos. Infection rates also may rise due to travel to endemic countries. Initially lesions may appear as innocuous erythematous plaques. When they do not respond to standard therapy, infectious agents such as M leprae should be part of the differential diagnosis. Because hematoxylin and eosin staining does not always yield results, if clinical suspicion is present, PCR should be performed. If a patient meets the clinical and histological diagnosis, the case should be reported to the National Hansen’s Disease Program.
After completion of treatment, our patient has shown excellent results. He has not yet demonstrated a reversal reaction; however, he may still be at risk, as it most commonly presents 2 months after starting treatment but can present years after treatment has been initiated.8 Cutaneous leprosy must be considered in the differential diagnosis for steroid-nonresponsive skin lesions, particularly in states such as Florida with a documented increase in incidence.
Acknowledgment
We thank Sharon Barrineau, ARNP (Okeechobee, Florida), for her acumen, contributions, and support on this case.
- Britton WJ, Lockwood DN. Leprosy. Lancet. 2004;363:1209-1219.
- World Health Organization. WHO Expert Committee on Leprosy, 8th Report. Geneva, Switzerland: World Health Organization; 2010.
- Truman RW, Singh P, Sharma R, et al. Probable zoonotic leprosy in the southern United States. N Engl J Med. 2011;364:1626-1633.
- Adams DA, Fullerton K, Jajosky R, et al; Division of Notifiable Diseases and Healthcare Information, Office of Surveillance, Epidemiology, and Laboratory Services, CDC. Summary of notifiable diseases—United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;62:1-122.
- A summary of Hansen’s disease in the United States—2015. Department of Health and Human Services, Health Resources and Services Administration, National Hansen’s Disease Program. https://www.hrsa.gov/sites/default/files/hansensdisease/pdfs/hansens2015report.pdf. Accessed October 23, 2017.
- Loughry WJ, Truman RW, McDonough CM, et al. Is leprosy spreading among nine-banded armadillos in the southeastern United States? J Wildl Dis. 2009;45:144-152.
- Ridley DS, Jopling WH. Classification of leprosy according to immunity: a five group system. Int J Lepr. 1966;34:225-273.
- Lee DJ, Rea TH, Modlin RL. Leprosy. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381.
- Britton WJ. The management of leprosy reversal reactions. Lepr Rev. 1998;69:225-234.
- Manandhar R, LeMaster JW, Roche PW. Risk factors for erythema nodosum leprosum. Int J Lepr Other Mycobact Dis. 1999;67:270-278.
- Lockwood DN. The management of erythema nodosum leprosum: current and future options. Lepr Rev. 1996;67:253-259.
- Jakeman P, Smith WC. Thalidomide in leprosy reaction. Lancet. 1994;343:432-433.
Case Report
A 65-year-old man presented with multiple anesthetic, annular, erythematous, scaly plaques with a raised border of 6 weeks’ duration that were unresponsive to topical steroid therapy. Four plaques were noted on the lower back ranging from 2 to 4 cm in diameter as well as a fifth plaque on the anterior portion of the right ankle that was approximately 6×6 cm. He denied fever, malaise, muscle weakness, changes in vision, or sensory deficits outside of the lesions themselves. The patient also denied any recent travel to endemic areas or exposure to armadillos.
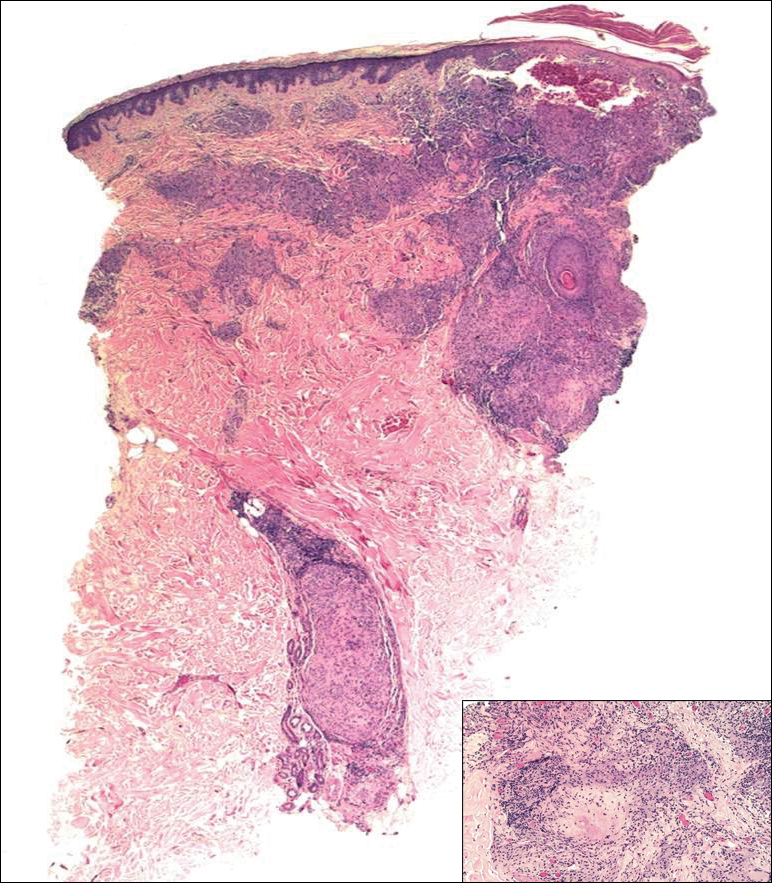
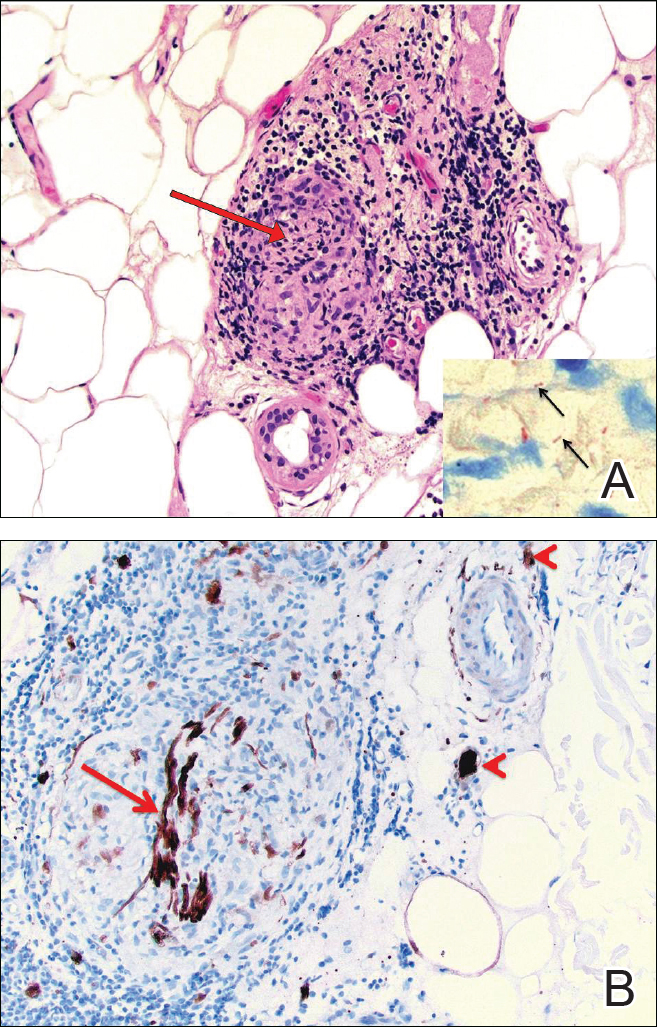
Biopsies were taken from lesions on the lumbar back and anterior aspect of the right ankle (Figure 1A). Hematoxylin and eosin staining revealed a granulomatous infiltrate spreading along neurovascular structures (Figure 2). Granulomas also were identified in the dermal interstitium exhibiting partial necrosis (Figure 2 inset). Conspicuous distension of lymphovascular and perineural areas also was noted. Immunohistochemical studies with S-100 and neurofilament stains allowed insight into the pathomechanism of the clinically observed anesthesia, as nerve fibers were identified showing different stages of damage elicited by the granulomatous inflammatory infiltrate (Figure 3). Fite staining was positive for occasional bacilli within histiocytes (Figure 3A inset). Despite the clinical, histologic, and immunohistochemical evidence, the patient had no known exposure to leprosy; consequently, a polymerase chain reaction (PCR) assay was ordered for confirmation of the diagnosis. Surprisingly, the PCR was positive for Mycobacterium leprae DNA. These findings were consistent with borderline tuberculoid leprosy.
The case was reported to the National Hansen’s Disease Program (Baton Rouge, Louisiana). The patient was started on rifampicin 600 mg once monthly and dapsone 100 mg once daily for 6 months. The lesions exhibited marked improvement after completion of therapy (Figure 1B).
Comment
Disease Transmission
Hansen disease, also known as leprosy, is a chronic granulomatous infectious disease that is caused by M leprae, an obligate intracellular bacillus aerobe.1 The mechanism of spread of M leprae is not clear. It is thought to be transmitted via respiratory droplets, though it may occur through injured skin.2 Studies have suggested that in addition to humans, nine-banded armadillos are a source of infection.2,3 Exposure to infected individuals, particularly multibacillary patients, increases the likelihood of contracting leprosy.2
According to the Centers for Disease Control and Prevention, 81 cases of Hansen disease were diagnosed in the United States in 2013,4 compared to 178 cases registered in 2015.5 Cases from Hawaii, Texas, California, Louisiana, New York, and Florida made up 72% (129/178) of the reported cases. There was an increase from 34 cases to 49 cases in Florida from 2014 to 2015.5 The spread of leprosy throughout Florida may be from the merge of 2 armadillo populations, an M leprae–infected population migrating east from Texas and one from south central Florida that historically had not been infected with M leprae until recently.3,6 Our patient did not have any known exposures to armadillos.
Classification and Presentation
The clinical presentation of Hansen disease is widely variable, as it can present at any point along a spectrum ranging from tuberculoid leprosy to lepromatous leprosy with borderline conditions in between, according to the Ridley-Jopling critera.7 The World Health Organization also classifies leprosy based on the number of acid-fast bacilli seen in a skin smear as either paucibacillary or multibacillary.2 The paucibacillary classification correlates with tuberculoid, borderline tuberculoid, and indeterminate leprosy, and multibacillary correlates with borderline lepromatous and lepromatous leprosy. Paucibacillary leprosy usually presents with a less dramatic clinical picture than multibacillary leprosy. The clinical presentation is dependent on the magnitude of immune response to M leprae.2
Paucibacillary infection occurs when the body generates a strong cell-mediated immune response against the bacteria,8 which causes the activation and proliferation of CD4 and CD8 T cells, limiting the spread of the mycobacterium. Subsequently, the patient typically presents with a mild clinical picture with few skin lesions and limited nerve involvement.8 The skin lesions are papules or plaques with raised borders that are usually hypopigmented on dark skin and erythematous on light skin. Nerve involvement in paucibacillary forms of leprosy include sensory impairment and anhidrosis within the lesions. Nerve enlargement usually affects superficial nerves, with the posterior tibial nerve being most commonly affected.
Multibacillary leprosy presents with systemic involvement due to a weak cell-mediated immune response. Patients generally present with diffuse, poorly defined nodules; greater nerve impairment; and other systemic symptoms such as blindness, swelling of the fingers and toes, and testicular atrophy (in men). Additionally, enlargement of the earlobes and widening of the nasal bridge may contribute to the appearance of leonine facies. Nerve impairment in multibacillary forms of leprosy may be more severe, including more diffuse sensory involvement (eg, stocking glove–pattern neuropathy, nerve-trunk palsies), which ultimately may lead to foot drop, claw toe, and lagophthalmos.8
In addition to the clinical presentation, the histology of the paucibacillary and multibacillary types differ. Multibacillary leprosy shows diffuse histiocytes without granulomas and multiple bacilli seen on Fite staining.8 In the paucibacillary form, there are well-formed granulomas with Langerhans giant cells and a perineural lymphocytic infiltrate seen on hematoxylin and eosin staining with rare acid-fast bacilli seen on Fite staining.
To diagnose leprosy, at least one of the following 3 clinical signs must be present: (1) a hypopigmented or erythematous lesion with loss of sensation, (2) thickened peripheral nerve, or (3) acid-fast bacilli on slit-skin smear.2
Management
The World Health Organization guidelines involve multidrug therapy over an extended period of time.2 For adults, the paucibacillary regimen includes rifampicin 600 mg once monthly and dapsone 100 mg once daily for 6 months. The adult regimen for multibacillary leprosy includes clofazimine 300 mg once monthly and 50 mg once daily, in addition to rifampicin 600 mg once monthly and dapsone 100 mg once daily for 12 months. If classification cannot be determined, it is recommended the patient be treated for multibacillary disease.2
Reversal Reactions
During the course of the disease, patients may upgrade (to a less severe form) or downgrade (to a more severe form) between the tuberculoid, borderline, and lepromatous forms.8 The patient’s clinical picture also may change with complications of leprosy, which include type 1 and type 2 reactions. Type 1 reaction is a reversal reaction seen in 15% to 30% of patients at risk, usually those with borderline forms of leprosy.9 Reversal reactions usually manifest as erythema and edema of current skin lesions, formation of new tumid lesions, and tenderness of peripheral nerves with loss of nerve function.8 Treatment of reversal reactions involves systemic corticosteroids.10 Type 2 reaction is classified as erythema nodosum leprosum. It presents within the first 2 years of treatment in approximately 20% of lepromatous patients and approximately 10% of borderline lepromatous patients but is rare in paucibacillary infections.11 It presents with fever and crops of pink nodules and may include iritis, neuritis, lymphadenitis, orchitis, dactylitis, arthritis, and proteinuria.8 Treatment options for erythema nodosum leprosum include corticosteroids, clofazimine, and thalidomide.12,13
Conclusion
Hansen disease is a rare condition in the United States. This case is unique because, to our knowledge, it is the first known PCR-confirmed case of Hansen disease in Okeechobee County, Florida. Additionally, the patient had no known exposure to M leprae. Exposure is increasing due to the increased geographical range of infected armadillos. Infection rates also may rise due to travel to endemic countries. Initially lesions may appear as innocuous erythematous plaques. When they do not respond to standard therapy, infectious agents such as M leprae should be part of the differential diagnosis. Because hematoxylin and eosin staining does not always yield results, if clinical suspicion is present, PCR should be performed. If a patient meets the clinical and histological diagnosis, the case should be reported to the National Hansen’s Disease Program.
After completion of treatment, our patient has shown excellent results. He has not yet demonstrated a reversal reaction; however, he may still be at risk, as it most commonly presents 2 months after starting treatment but can present years after treatment has been initiated.8 Cutaneous leprosy must be considered in the differential diagnosis for steroid-nonresponsive skin lesions, particularly in states such as Florida with a documented increase in incidence.
Acknowledgment
We thank Sharon Barrineau, ARNP (Okeechobee, Florida), for her acumen, contributions, and support on this case.
Case Report
A 65-year-old man presented with multiple anesthetic, annular, erythematous, scaly plaques with a raised border of 6 weeks’ duration that were unresponsive to topical steroid therapy. Four plaques were noted on the lower back ranging from 2 to 4 cm in diameter as well as a fifth plaque on the anterior portion of the right ankle that was approximately 6×6 cm. He denied fever, malaise, muscle weakness, changes in vision, or sensory deficits outside of the lesions themselves. The patient also denied any recent travel to endemic areas or exposure to armadillos.
Biopsies were taken from lesions on the lumbar back and anterior aspect of the right ankle (Figure 1A). Hematoxylin and eosin staining revealed a granulomatous infiltrate spreading along neurovascular structures (Figure 2). Granulomas also were identified in the dermal interstitium exhibiting partial necrosis (Figure 2 inset). Conspicuous distension of lymphovascular and perineural areas also was noted. Immunohistochemical studies with S-100 and neurofilament stains allowed insight into the pathomechanism of the clinically observed anesthesia, as nerve fibers were identified showing different stages of damage elicited by the granulomatous inflammatory infiltrate (Figure 3). Fite staining was positive for occasional bacilli within histiocytes (Figure 3A inset). Despite the clinical, histologic, and immunohistochemical evidence, the patient had no known exposure to leprosy; consequently, a polymerase chain reaction (PCR) assay was ordered for confirmation of the diagnosis. Surprisingly, the PCR was positive for Mycobacterium leprae DNA. These findings were consistent with borderline tuberculoid leprosy.
The case was reported to the National Hansen’s Disease Program (Baton Rouge, Louisiana). The patient was started on rifampicin 600 mg once monthly and dapsone 100 mg once daily for 6 months. The lesions exhibited marked improvement after completion of therapy (Figure 1B).
Comment
Disease Transmission
Hansen disease, also known as leprosy, is a chronic granulomatous infectious disease that is caused by M leprae, an obligate intracellular bacillus aerobe.1 The mechanism of spread of M leprae is not clear. It is thought to be transmitted via respiratory droplets, though it may occur through injured skin.2 Studies have suggested that in addition to humans, nine-banded armadillos are a source of infection.2,3 Exposure to infected individuals, particularly multibacillary patients, increases the likelihood of contracting leprosy.2
According to the Centers for Disease Control and Prevention, 81 cases of Hansen disease were diagnosed in the United States in 2013,4 compared to 178 cases registered in 2015.5 Cases from Hawaii, Texas, California, Louisiana, New York, and Florida made up 72% (129/178) of the reported cases. There was an increase from 34 cases to 49 cases in Florida from 2014 to 2015.5 The spread of leprosy throughout Florida may be from the merge of 2 armadillo populations, an M leprae–infected population migrating east from Texas and one from south central Florida that historically had not been infected with M leprae until recently.3,6 Our patient did not have any known exposures to armadillos.
Classification and Presentation
The clinical presentation of Hansen disease is widely variable, as it can present at any point along a spectrum ranging from tuberculoid leprosy to lepromatous leprosy with borderline conditions in between, according to the Ridley-Jopling critera.7 The World Health Organization also classifies leprosy based on the number of acid-fast bacilli seen in a skin smear as either paucibacillary or multibacillary.2 The paucibacillary classification correlates with tuberculoid, borderline tuberculoid, and indeterminate leprosy, and multibacillary correlates with borderline lepromatous and lepromatous leprosy. Paucibacillary leprosy usually presents with a less dramatic clinical picture than multibacillary leprosy. The clinical presentation is dependent on the magnitude of immune response to M leprae.2
Paucibacillary infection occurs when the body generates a strong cell-mediated immune response against the bacteria,8 which causes the activation and proliferation of CD4 and CD8 T cells, limiting the spread of the mycobacterium. Subsequently, the patient typically presents with a mild clinical picture with few skin lesions and limited nerve involvement.8 The skin lesions are papules or plaques with raised borders that are usually hypopigmented on dark skin and erythematous on light skin. Nerve involvement in paucibacillary forms of leprosy include sensory impairment and anhidrosis within the lesions. Nerve enlargement usually affects superficial nerves, with the posterior tibial nerve being most commonly affected.
Multibacillary leprosy presents with systemic involvement due to a weak cell-mediated immune response. Patients generally present with diffuse, poorly defined nodules; greater nerve impairment; and other systemic symptoms such as blindness, swelling of the fingers and toes, and testicular atrophy (in men). Additionally, enlargement of the earlobes and widening of the nasal bridge may contribute to the appearance of leonine facies. Nerve impairment in multibacillary forms of leprosy may be more severe, including more diffuse sensory involvement (eg, stocking glove–pattern neuropathy, nerve-trunk palsies), which ultimately may lead to foot drop, claw toe, and lagophthalmos.8
In addition to the clinical presentation, the histology of the paucibacillary and multibacillary types differ. Multibacillary leprosy shows diffuse histiocytes without granulomas and multiple bacilli seen on Fite staining.8 In the paucibacillary form, there are well-formed granulomas with Langerhans giant cells and a perineural lymphocytic infiltrate seen on hematoxylin and eosin staining with rare acid-fast bacilli seen on Fite staining.
To diagnose leprosy, at least one of the following 3 clinical signs must be present: (1) a hypopigmented or erythematous lesion with loss of sensation, (2) thickened peripheral nerve, or (3) acid-fast bacilli on slit-skin smear.2
Management
The World Health Organization guidelines involve multidrug therapy over an extended period of time.2 For adults, the paucibacillary regimen includes rifampicin 600 mg once monthly and dapsone 100 mg once daily for 6 months. The adult regimen for multibacillary leprosy includes clofazimine 300 mg once monthly and 50 mg once daily, in addition to rifampicin 600 mg once monthly and dapsone 100 mg once daily for 12 months. If classification cannot be determined, it is recommended the patient be treated for multibacillary disease.2
Reversal Reactions
During the course of the disease, patients may upgrade (to a less severe form) or downgrade (to a more severe form) between the tuberculoid, borderline, and lepromatous forms.8 The patient’s clinical picture also may change with complications of leprosy, which include type 1 and type 2 reactions. Type 1 reaction is a reversal reaction seen in 15% to 30% of patients at risk, usually those with borderline forms of leprosy.9 Reversal reactions usually manifest as erythema and edema of current skin lesions, formation of new tumid lesions, and tenderness of peripheral nerves with loss of nerve function.8 Treatment of reversal reactions involves systemic corticosteroids.10 Type 2 reaction is classified as erythema nodosum leprosum. It presents within the first 2 years of treatment in approximately 20% of lepromatous patients and approximately 10% of borderline lepromatous patients but is rare in paucibacillary infections.11 It presents with fever and crops of pink nodules and may include iritis, neuritis, lymphadenitis, orchitis, dactylitis, arthritis, and proteinuria.8 Treatment options for erythema nodosum leprosum include corticosteroids, clofazimine, and thalidomide.12,13
Conclusion
Hansen disease is a rare condition in the United States. This case is unique because, to our knowledge, it is the first known PCR-confirmed case of Hansen disease in Okeechobee County, Florida. Additionally, the patient had no known exposure to M leprae. Exposure is increasing due to the increased geographical range of infected armadillos. Infection rates also may rise due to travel to endemic countries. Initially lesions may appear as innocuous erythematous plaques. When they do not respond to standard therapy, infectious agents such as M leprae should be part of the differential diagnosis. Because hematoxylin and eosin staining does not always yield results, if clinical suspicion is present, PCR should be performed. If a patient meets the clinical and histological diagnosis, the case should be reported to the National Hansen’s Disease Program.
After completion of treatment, our patient has shown excellent results. He has not yet demonstrated a reversal reaction; however, he may still be at risk, as it most commonly presents 2 months after starting treatment but can present years after treatment has been initiated.8 Cutaneous leprosy must be considered in the differential diagnosis for steroid-nonresponsive skin lesions, particularly in states such as Florida with a documented increase in incidence.
Acknowledgment
We thank Sharon Barrineau, ARNP (Okeechobee, Florida), for her acumen, contributions, and support on this case.
- Britton WJ, Lockwood DN. Leprosy. Lancet. 2004;363:1209-1219.
- World Health Organization. WHO Expert Committee on Leprosy, 8th Report. Geneva, Switzerland: World Health Organization; 2010.
- Truman RW, Singh P, Sharma R, et al. Probable zoonotic leprosy in the southern United States. N Engl J Med. 2011;364:1626-1633.
- Adams DA, Fullerton K, Jajosky R, et al; Division of Notifiable Diseases and Healthcare Information, Office of Surveillance, Epidemiology, and Laboratory Services, CDC. Summary of notifiable diseases—United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;62:1-122.
- A summary of Hansen’s disease in the United States—2015. Department of Health and Human Services, Health Resources and Services Administration, National Hansen’s Disease Program. https://www.hrsa.gov/sites/default/files/hansensdisease/pdfs/hansens2015report.pdf. Accessed October 23, 2017.
- Loughry WJ, Truman RW, McDonough CM, et al. Is leprosy spreading among nine-banded armadillos in the southeastern United States? J Wildl Dis. 2009;45:144-152.
- Ridley DS, Jopling WH. Classification of leprosy according to immunity: a five group system. Int J Lepr. 1966;34:225-273.
- Lee DJ, Rea TH, Modlin RL. Leprosy. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381.
- Britton WJ. The management of leprosy reversal reactions. Lepr Rev. 1998;69:225-234.
- Manandhar R, LeMaster JW, Roche PW. Risk factors for erythema nodosum leprosum. Int J Lepr Other Mycobact Dis. 1999;67:270-278.
- Lockwood DN. The management of erythema nodosum leprosum: current and future options. Lepr Rev. 1996;67:253-259.
- Jakeman P, Smith WC. Thalidomide in leprosy reaction. Lancet. 1994;343:432-433.
- Britton WJ, Lockwood DN. Leprosy. Lancet. 2004;363:1209-1219.
- World Health Organization. WHO Expert Committee on Leprosy, 8th Report. Geneva, Switzerland: World Health Organization; 2010.
- Truman RW, Singh P, Sharma R, et al. Probable zoonotic leprosy in the southern United States. N Engl J Med. 2011;364:1626-1633.
- Adams DA, Fullerton K, Jajosky R, et al; Division of Notifiable Diseases and Healthcare Information, Office of Surveillance, Epidemiology, and Laboratory Services, CDC. Summary of notifiable diseases—United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;62:1-122.
- A summary of Hansen’s disease in the United States—2015. Department of Health and Human Services, Health Resources and Services Administration, National Hansen’s Disease Program. https://www.hrsa.gov/sites/default/files/hansensdisease/pdfs/hansens2015report.pdf. Accessed October 23, 2017.
- Loughry WJ, Truman RW, McDonough CM, et al. Is leprosy spreading among nine-banded armadillos in the southeastern United States? J Wildl Dis. 2009;45:144-152.
- Ridley DS, Jopling WH. Classification of leprosy according to immunity: a five group system. Int J Lepr. 1966;34:225-273.
- Lee DJ, Rea TH, Modlin RL. Leprosy. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381.
- Britton WJ. The management of leprosy reversal reactions. Lepr Rev. 1998;69:225-234.
- Manandhar R, LeMaster JW, Roche PW. Risk factors for erythema nodosum leprosum. Int J Lepr Other Mycobact Dis. 1999;67:270-278.
- Lockwood DN. The management of erythema nodosum leprosum: current and future options. Lepr Rev. 1996;67:253-259.
- Jakeman P, Smith WC. Thalidomide in leprosy reaction. Lancet. 1994;343:432-433.
Practice Points
- A majority of leprosy cases in the United States have been reported in Florida, California, Texas, Louisiana, Hawaii, and New York.
- Leprosy should be included in the differential diagnosis for annular plaques, particularly those not responding to traditional treatment.
Atypical Disseminated Herpes Zoster: Management Guidelines in Immunocompromised Patients
Well-known for its typical presentation, classic herpes zoster (HZ) presents as a dermatomal eruption of painful erythematous papules that evolve into grouped vesicles or bullae.1,2 Thereafter, the lesions can become pustular or hemorrhagic.1 Although the diagnosis most often is made clinically, confirmatory techniques for diagnosis include viral culture, direct fluorescent antibody testing, or polymerase chain reaction (PCR) assay.1,3
The main risk factor for HZ is advanced age, most commonly affecting elderly patients.4 It is hypothesized that a physiological decline in varicella-zoster virus (VZV)–specific cell-mediated immunity among elderly individuals helps trigger reactivation of the virus within the dorsal root ganglion.1,5 Similarly affected are immunocompromised individuals, including those with human immunodeficiency virus (HIV) infection, due to suppression of T cells immune to VZV,1,5 as well as immunosuppressed transplant recipients who have diminished VZV-specific cellular responses and VZV IgG antibody avidity.6
Secondary complications of VZV infection (eg, postherpetic neuralgia, bacterial superinfection progressing to cellulitis) lead to increased morbidity.7,8 Disseminated cutaneous HZ is another grave complication of VZV infection and almost exclusively occurs with immunosuppression.1,8 It manifests as an eruption of at least 20 widespread vesiculobullous lesions outside the primary and adjacent dermatomes.6 Immunocompromised patients also are at increased risk for visceral involvement of VZV infection, which may affect vital organs such as the brain, liver, or lungs.7,8 Given the atypical presentation of VZV infection among some immunocompromised individuals, these patients are at increased risk for diagnostic delay and morbidity in the absence of high clinical suspicion for disseminated HZ.
Case Reports
Patient 1
A 52-year-old man developed a painless nonpruritic rash on the left leg of 4 days’ duration. It initially appeared as an erythematous maculopapular rash on the medial aspect of the left knee without any prodromal symptoms. Over the next 4 days, erythematous vesicles developed that progressed to pustules, and the rash spread both proximally and distally along the left leg. Shortly following hospital admission, he developed a fever (temperature, 38.4°C). His medical history included alcoholic liver cirrhosis and AIDS, with a CD4 count of 174 cells/µL (reference range, 500–1500 cells/µL). He had been taking antiretroviral therapy (abacavir-lamivudine and dolutegravir) and prophylaxis against opportunistic infections (dapsone and itraconazole).
Physical examination was remarkable for an extensive rash consisting of multiple 1-cm clusters of approximately 40 pustules each scattered in a nondermatomal distribution along the left leg (Figure 1). Many of the vesicles were confluent with an erythematous base and were in different stages of evolution with some crusted and others emanating a thin liquid exudate. The lesions were nontender and without notable induration. The leg was warm and edematous.
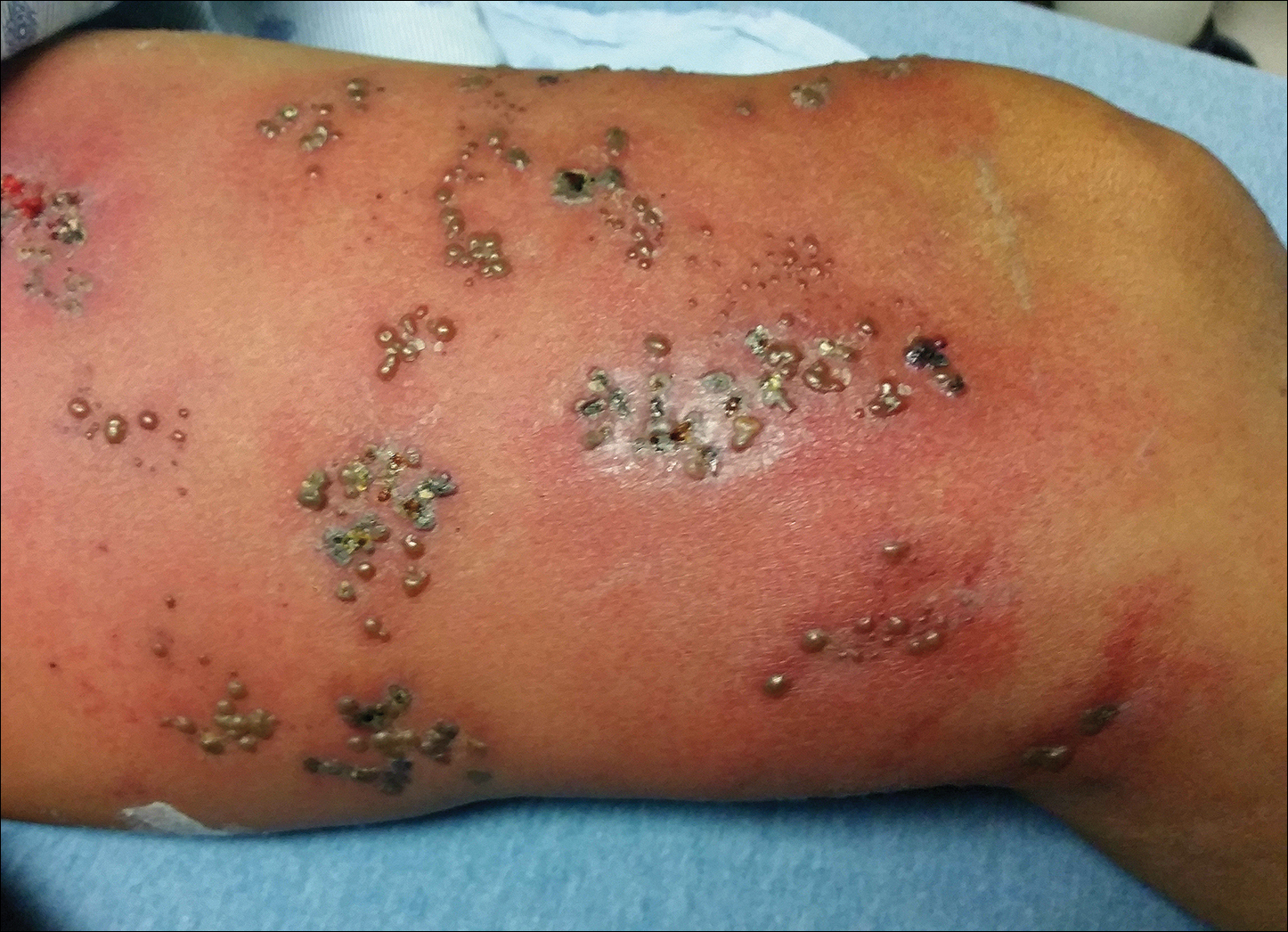
Clinically, the differential diagnosis included disseminated HZ with bacterial superinfection, Vibrio vulnificus infection, and herpes simplex virus (HSV) infection. The patient was treated with intravenous vancomycin, levofloxacin, and acyclovir, and no new lesions developed throughout the course of treatment. On this regimen, his fever resolved after 1 day, the active lesions began to crust, and the edema and erythema diminished. Results of bacterial cultures and plasma PCR and IgM for HSV types 1 and 2 were negative. Viral culture results were negative, but a PCR assay for VZV was positive, reflective of acute reactivation of VZV.
Patient 2
A 63-year-old man developed a pruritic burning rash involving the face, trunk, arms, and legs of 6 days’ duration. His medical history included a heart transplant 6 months prior to presentation, type 2 diabetes mellitus, and chronic kidney disease. He was taking antirejection therapy with mycophenolate mofetil (MMF), prednisone, and tacrolimus.
Physical examination was remarkable for an extensive rash consisting of clusters of 1- to 2-mm vesicles scattered in a nondermatomal pattern. Isolated vesicles involved the forehead, nose, and left ear, and diffuse vesicles with a relatively symmetric distribution were scattered across the back, chest, and proximal and distal arms and legs (Figure 2). Many of the vesicles had an associated overlying crust with hemorrhage. Some of the vesicles coalesced with central necrotic plaques.
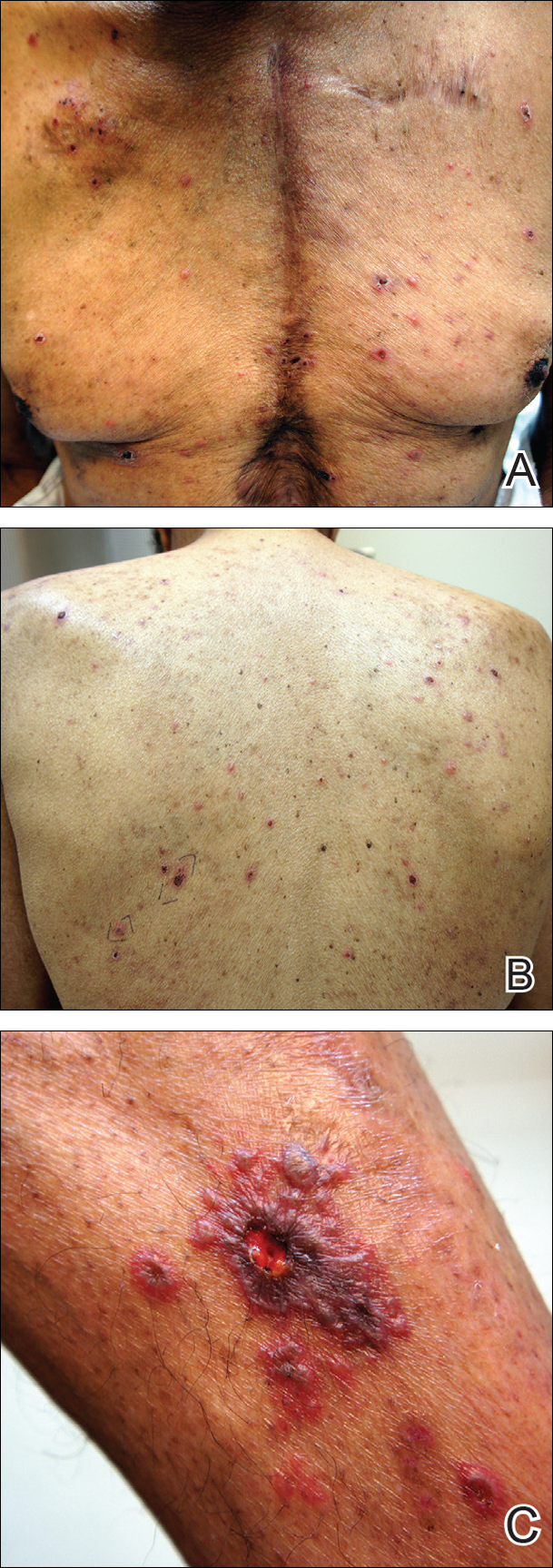
Given a clinical suspicion for disseminated HZ, therapy with oral valacyclovir was initiated. Two punch biopsies were consistent with herpesvirus cytopathic changes. Multiple sections demonstrated ulceration as well as acantholysis and necrosis of keratinocytes with multinucleation and margination of chromatin. There was an intense lichenoid and perivascular lymphocytic infiltrate in the dermis. Immunohistochemistry staining was positive for VZV and negative for HSV, indicating acute reactivation of VZV (Figure 3). Upon completion of an antiviral regimen, the patient returned to clinic with healed crusted lesions.
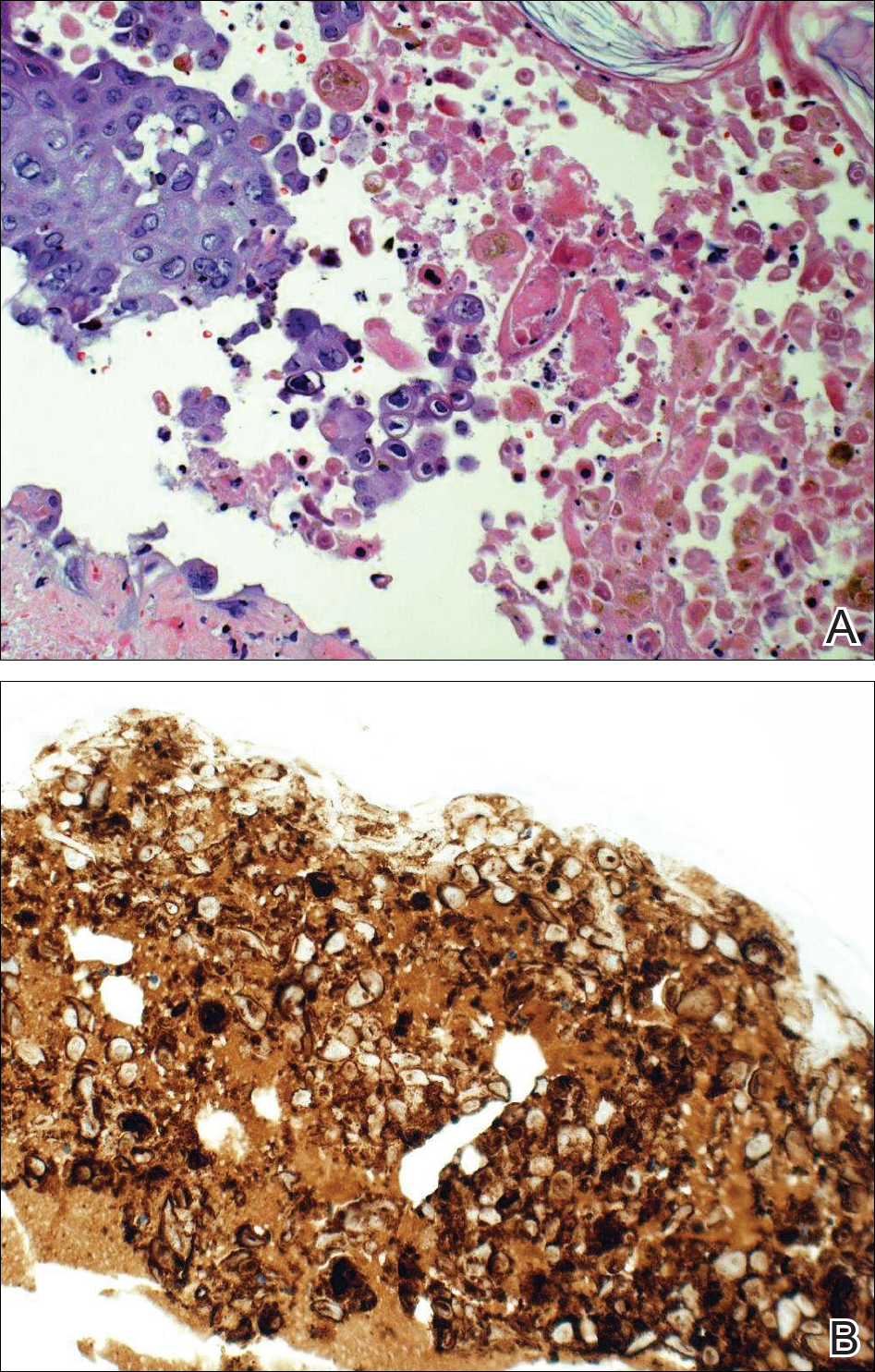
Comment
Frequently, the clinical features of HZ in immunocompromised patients mirror those in immunocompetent hosts.8 However, each of our 2 patients developed an unusual presentation of atypical generalized HZ.7 In this clinical variant, lesions develop along a single dermatome, then a diffuse vesicular eruption subsequently develops without dermatomal localization. These lesions can be chronic, persisting for months or years.7
The classic clinical presentation of HZ is distinct and often is readily diagnosed by visual inspection.7 However, atypical presentations and their associated complications can pose diagnostic and therapeutic challenges.7 Painless HZ lesions in a nondermatomal pattern were described in a patient who also had AIDS.9 Interestingly, multiple reports have found that patients with a severe but painless rash are less likely to have experienced a viral prodrome consisting of hyperesthesia, paresthesia, or pruritus.2,10 This observation suggests that lack of a prodrome, as in the case of patient 1 in our report, may aid in the recognition of painless HZ. Because of these atypical presentations, laboratory testing is even more important than in immunocompetent hosts, as diagnosis may be more difficult to establish on clinical presentation alone.
Several studies11-32 have evaluated modalities for treatment and prophylaxis for disseminated HZ in immunocompromised hosts, given its increased risk and potentially fatal complications in this population. The current guidelines in patients with HIV/AIDS, solid organ transplantation (SOT), and hematopoietic stem cell transplantation (HSCT) are summarized in the eTable.
HIV/AIDS Patients
Given their efficacy and low rate of toxicity, oral acyclovir, valacyclovir, and famciclovir are recommended treatment options for HIV patients with localized, mild, dermatomal HZ.11 Two exceptions include HZ ophthalmicus and Ramsay Hunt syndrome for which some experts recommend intravenous acyclovir given the risk for vision loss and facial palsy, respectively. Intravenous acyclovir often is the drug of choice for treating complicated, disseminated, or severe HZ in HIV-infected patients, though prospective efficacy data remain limited.11
With regard to prevention of infection, a large randomized trial in 2016 found that acyclovir prophylaxis resulted in a 68% reduction in HZ over 2 years among HIV patients.12 Despite data that acyclovir may be effective for this purpose, long-term antiviral prophylaxis is not routinely recommended for HZ,11,13 as it has been linked to rare cases of acyclovir-resistant HZ in HIV patients.14,15 However, antiviral prophylaxis against HSV type 2 reactivation in HIV patients also confers protection against VZV reactivation.11,12
Solid Organ Transplantation
Localized, mild to moderately severe dermatomal HZ can be treated with oral acyclovir, valacyclovir, or famciclovir. As in HIV patients, SOT patients with severe, disseminated, or complicated HZ should receive IV acyclovir.11 In the first 3 to 6 months following the procedure, SOT patients receive cytomegalovirus prophylaxis with ganciclovir or valgan-ciclovir, which also provides protection against HZ.13-18 For patients not receiving cytomegalovirus prophylaxis, HSV prophylaxis with oral acyclovir or valacyclovir is given for at least the first month after transplantation, which also confers protection against HZ.16,19 Antiviral therapy is critical during the early posttransplantation period when patients are most severely immunosuppressed and thus have the highest risk for VZV-associated complications.20 Although immunosuppression is lifelong in most SOT recipients, there is insufficient evidence for extending prophylaxis beyond 6 months.16,21
As a possible risk factor for HZ,22 MMF use is another consideration among SOT patients, similar to patient 2 in our report. A 2003 observational study supported withdrawal of MMF therapy during active VZV infection due to clinical observation of an association with HZ.23 However, a multicenter, randomized, controlled trial reported no cases of HZ in renal transplant recipients on MMF.24 Additionally, MMF has been observed to enhance the antiviral activity of acyclovir, at least in vitro.25 Given the lack of evidence of MMF as a risk factor for HZ, there is insufficient evidence for cessation of use during VZVreactivation in SOT patients.
Hematopoietic Stem Cell Transplantation
The preferred agents for treatment of localized mild dermatomal HZ are oral acyclovir or valacyclovir, as data on the safety and efficacy of famciclovir among HSCT recipients are limited.13,26 Patients should receive antiviral prophylaxis with one of these agents during the first year following allogeneic or autologous HSCT. This 1-year course has proven highly effective in reducing HZ in the first year following transplantation when most severe cases occur,21,26-29 and it has been associated with a persistently decreased risk for HZ even after discontinuation.21 Prophylaxis may be continued beyond 1 year in allogeneic HSCT recipients experiencing graft-versus-host disease who should receive acyclovir until 6 months after the end of immunosuppressive therapy.21,26
Vaccination remains a potential strategy to reduce the incidence of HZ in this patient population. A heat-inactivated vaccine administered within the first 3 months after the procedure has been shown to be safe among autologous and allogeneic HSCT patients.30,31 The vaccine notably reduced the incidence of HZ in patients who underwent autologous HSCT,32 but no known data are available on its clinical efficacy in allogeneic HSCT patients. Accordingly, there are no known official recommendations to date regarding vaccine use in these patient populations.26
Conclusion
It is incumbent upon clinicians to recognize the spectrum of atypical presentations of HZ and maintain a low threshold for performing appropriate diagnostic or confirmatory studies among at-risk patients with impaired immune function. Disseminated HZ can have potentially life-threatening visceral complications such as encephalitis, hepatitis, or pneumonitis.7,8 As such, an understanding of prevention and treatment modalities for VZV infection among immunocompromised patients is critical. Because the morbidity associated with complications of VZV infection is substantial and the risks associated with antiviral agents are minimal, antiviral prophylaxis is recommended for 6 months following SOT or 1 year following HSCT, and prompt treatment is warranted in cases of reasonable clinical suspicion for HZ.
Acknowledgment
The authors gratefully acknowledge the generosity of our patients in permitting photography of their skin findings for the furthering of medical education.
- McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol. 1999;41:1-16.
- Nagasako EM, Johnson RW, Griffin DR, et al. Rash severity in herpes zoster: correlates and relationship to postherpetic neuralgia. J Am Acad Dermatol. 2002;46:834-839.
- Leung J, Harpaz R, Baughman AL, et al. Evaluation of laboratory methods for diagnosis of varicella. Clin Infect Dis. 2010;51:23-32.
- Herpes Zoster and Functional Decline Consortium. Functional decline and herpes zoster in older people: an interplay of multiple factors. Aging Clin Exp Res. 2015;27:757-765.
- Weinberg A, Levin MJ. VZV T cell-mediated immunity. Curr Top Microbiol Immunol. 2010;342:341-357.
- Prelog M, Schonlaub J, Jeller V, et al. Reduced varicella-zoster-virus (VZV)-specific lymphocytes and IgG antibody avidity in solid organ transplant recipients. Vaccine. 2013;31:2420-2426.
- Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
- Glesby MJ, Moore RD, Chaisson RE. Clinical spectrum of herpes zoster in adults infected with human immunodeficiency virus. Clin Infect Dis. 1995;21:370-375.
- Blankenship W, Herchline T, Hockley A. Asymptomatic vesicles in a patient with the acquired immunodeficiency syndrome. disseminated varicella-zoster virus (VZV) infection. Arch Dermatol. 1994;130:1193, 1196.
- Katz J, Cooper EM, Walther RR, et al. Acute pain in herpes zoster and its impact on health-related quality of life. Clin Infect Dis. 2004;39:342-348.
- Gnann JW. Antiviral therapy of varicella-zoster virus infections. In: Arvin A, Campadelli-Fiume G, Mocarski E, et al, eds. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge, United Kingdom: Cambridge University Press; 2007:1175-1191.
- Barnabas RV, Baeten JM, Lingappa JR, et al. Acyclovir prophylaxis reduces the incidence of herpes zoster among HIV-infected individuals: results of a randomized clinical trial. J Infect Dis. 2016;213:551-555.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
- Jacobson MA, Berger TG, Fikrig S, et al. Acyclovir-resistant varicella zoster virus infection after chronic oral acyclovir therapy in patients with the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1990;112:187-191.
- Linnemann CC Jr, Biron KK, Hoppenjans WG, et al. Emergence of acyclovir-resistant varicella zoster virus in an AIDS patient on prolonged acyclovir therapy. AIDS. 1990;4:577-579.
- Pergam SA, Limaye AP; AST Infectious Diseases Community of Practice. Varicella zoster virus (VZV) in solid organ transplant recipients. Am J Transplant. 2009;9(suppl 4):S108-S115.
- Preiksaitis JK, Brennan DC, Fishman J, et al. Canadian society of transplantation consensus workshop on cytomegalovirus management in solid organ transplantation final report. Am J Transplant. 2005;5:218-227.
- Fishman JA, Doran MT, Volpicelli SA, et al. Dosing of intravenous ganciclovir for the prophylaxis and treatment of cytomegalovirus infection in solid organ transplant recipients. Transplantation. 2000;69:389-394.
- Zuckerman R, Wald A; AST Infectious Diseases Community of Practice. Herpes simplex virus infections in solid organ transplant recipients. Am J Transplant. 2009;9(suppl 4):S104-S107.
- Arness T, Pedersen R, Dierkhising R, et al. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis. 2008;10:260-268.
- Erard V, Guthrie KA, Varley C, et al. One-year acyclovir prophylaxis for preventing varicella-zoster virus disease after hematopoietic cell transplantation: no evidence of rebound varicella-zoster virus disease after drug discontinuation. Blood. 2007;110:3071-3077.
- Rothwell WS, Gloor JM, Morgenstern BZ, et al. Disseminated varicella infection in pediatric renal transplant recipients treated with mycophenolate mofetil. Transplantation. 1999;68:158-161.
- Lauzurica R, Bayés B, Frías C, et al. Disseminated varicella infection in adult renal allograft recipients: role of mycophenolate mofetil. Transplant Proc. 2003;35:1758-1759.
- A blinded, randomized clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation. TheTricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation. 1996;61:1029-1037.
- Neyts J, De Clercq E. Mycophenolate mofetil strongly potentiates the anti-herpesvirus activity of acyclovir. Antiviral Res. 1998;40:53-56.
- Tomblyn M, Chiller T, Einsele H, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15:1143-1238.
- Boeckh M, Kim HW, Flowers ME, et al. Long-term acyclovir for prevention of varicella zoster virus disease after allogeneic hematopoietic cell transplantation—a randomized double-blind placebo-controlled study. Blood. 2006;107:1800-1805.
- Kawamura K, Hayakawa J, Akahoshi Y, et al. Low-dose acyclovir prophylaxis for the prevention of herpes simplex virus and varicella zoster virus diseases after autologous hematopoietic stem cell transplantation. Int J Hematol. 2015;102:230-237.
- Fred Hutchinson Cancer Research Center/Seattle Cancer Care Alliance. Long-term follow-up after hematopoietic stem cell transplant general guidelines for referring physicians. Fred Hutchinson Cancer Research Center website. https://www.fredhutch.org/content/dam/public/Treatment-Suport/Long-Term-Follow-Up/physician.pdf. Published July 17, 2014. Accessed October 19, 2017.
- Kussmaul SC, Horn BN, Dvorak CC, et al. Safety of the live, attenuated varicella vaccine in pediatric recipients of hematopoietic SCTs. Bone Marrow Transplant. 2010;45:1602-1606.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med. 2002;347:26-34.
- Issa NC, Marty FM, Leblebjian H, et al. Live attenuated varicella-zoster vaccine in hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant. 2014;20:285-287.
Well-known for its typical presentation, classic herpes zoster (HZ) presents as a dermatomal eruption of painful erythematous papules that evolve into grouped vesicles or bullae.1,2 Thereafter, the lesions can become pustular or hemorrhagic.1 Although the diagnosis most often is made clinically, confirmatory techniques for diagnosis include viral culture, direct fluorescent antibody testing, or polymerase chain reaction (PCR) assay.1,3
The main risk factor for HZ is advanced age, most commonly affecting elderly patients.4 It is hypothesized that a physiological decline in varicella-zoster virus (VZV)–specific cell-mediated immunity among elderly individuals helps trigger reactivation of the virus within the dorsal root ganglion.1,5 Similarly affected are immunocompromised individuals, including those with human immunodeficiency virus (HIV) infection, due to suppression of T cells immune to VZV,1,5 as well as immunosuppressed transplant recipients who have diminished VZV-specific cellular responses and VZV IgG antibody avidity.6
Secondary complications of VZV infection (eg, postherpetic neuralgia, bacterial superinfection progressing to cellulitis) lead to increased morbidity.7,8 Disseminated cutaneous HZ is another grave complication of VZV infection and almost exclusively occurs with immunosuppression.1,8 It manifests as an eruption of at least 20 widespread vesiculobullous lesions outside the primary and adjacent dermatomes.6 Immunocompromised patients also are at increased risk for visceral involvement of VZV infection, which may affect vital organs such as the brain, liver, or lungs.7,8 Given the atypical presentation of VZV infection among some immunocompromised individuals, these patients are at increased risk for diagnostic delay and morbidity in the absence of high clinical suspicion for disseminated HZ.
Case Reports
Patient 1
A 52-year-old man developed a painless nonpruritic rash on the left leg of 4 days’ duration. It initially appeared as an erythematous maculopapular rash on the medial aspect of the left knee without any prodromal symptoms. Over the next 4 days, erythematous vesicles developed that progressed to pustules, and the rash spread both proximally and distally along the left leg. Shortly following hospital admission, he developed a fever (temperature, 38.4°C). His medical history included alcoholic liver cirrhosis and AIDS, with a CD4 count of 174 cells/µL (reference range, 500–1500 cells/µL). He had been taking antiretroviral therapy (abacavir-lamivudine and dolutegravir) and prophylaxis against opportunistic infections (dapsone and itraconazole).
Physical examination was remarkable for an extensive rash consisting of multiple 1-cm clusters of approximately 40 pustules each scattered in a nondermatomal distribution along the left leg (Figure 1). Many of the vesicles were confluent with an erythematous base and were in different stages of evolution with some crusted and others emanating a thin liquid exudate. The lesions were nontender and without notable induration. The leg was warm and edematous.
Clinically, the differential diagnosis included disseminated HZ with bacterial superinfection, Vibrio vulnificus infection, and herpes simplex virus (HSV) infection. The patient was treated with intravenous vancomycin, levofloxacin, and acyclovir, and no new lesions developed throughout the course of treatment. On this regimen, his fever resolved after 1 day, the active lesions began to crust, and the edema and erythema diminished. Results of bacterial cultures and plasma PCR and IgM for HSV types 1 and 2 were negative. Viral culture results were negative, but a PCR assay for VZV was positive, reflective of acute reactivation of VZV.
Patient 2
A 63-year-old man developed a pruritic burning rash involving the face, trunk, arms, and legs of 6 days’ duration. His medical history included a heart transplant 6 months prior to presentation, type 2 diabetes mellitus, and chronic kidney disease. He was taking antirejection therapy with mycophenolate mofetil (MMF), prednisone, and tacrolimus.
Physical examination was remarkable for an extensive rash consisting of clusters of 1- to 2-mm vesicles scattered in a nondermatomal pattern. Isolated vesicles involved the forehead, nose, and left ear, and diffuse vesicles with a relatively symmetric distribution were scattered across the back, chest, and proximal and distal arms and legs (Figure 2). Many of the vesicles had an associated overlying crust with hemorrhage. Some of the vesicles coalesced with central necrotic plaques.
Given a clinical suspicion for disseminated HZ, therapy with oral valacyclovir was initiated. Two punch biopsies were consistent with herpesvirus cytopathic changes. Multiple sections demonstrated ulceration as well as acantholysis and necrosis of keratinocytes with multinucleation and margination of chromatin. There was an intense lichenoid and perivascular lymphocytic infiltrate in the dermis. Immunohistochemistry staining was positive for VZV and negative for HSV, indicating acute reactivation of VZV (Figure 3). Upon completion of an antiviral regimen, the patient returned to clinic with healed crusted lesions.
Comment
Frequently, the clinical features of HZ in immunocompromised patients mirror those in immunocompetent hosts.8 However, each of our 2 patients developed an unusual presentation of atypical generalized HZ.7 In this clinical variant, lesions develop along a single dermatome, then a diffuse vesicular eruption subsequently develops without dermatomal localization. These lesions can be chronic, persisting for months or years.7
The classic clinical presentation of HZ is distinct and often is readily diagnosed by visual inspection.7 However, atypical presentations and their associated complications can pose diagnostic and therapeutic challenges.7 Painless HZ lesions in a nondermatomal pattern were described in a patient who also had AIDS.9 Interestingly, multiple reports have found that patients with a severe but painless rash are less likely to have experienced a viral prodrome consisting of hyperesthesia, paresthesia, or pruritus.2,10 This observation suggests that lack of a prodrome, as in the case of patient 1 in our report, may aid in the recognition of painless HZ. Because of these atypical presentations, laboratory testing is even more important than in immunocompetent hosts, as diagnosis may be more difficult to establish on clinical presentation alone.
Several studies11-32 have evaluated modalities for treatment and prophylaxis for disseminated HZ in immunocompromised hosts, given its increased risk and potentially fatal complications in this population. The current guidelines in patients with HIV/AIDS, solid organ transplantation (SOT), and hematopoietic stem cell transplantation (HSCT) are summarized in the eTable.
HIV/AIDS Patients
Given their efficacy and low rate of toxicity, oral acyclovir, valacyclovir, and famciclovir are recommended treatment options for HIV patients with localized, mild, dermatomal HZ.11 Two exceptions include HZ ophthalmicus and Ramsay Hunt syndrome for which some experts recommend intravenous acyclovir given the risk for vision loss and facial palsy, respectively. Intravenous acyclovir often is the drug of choice for treating complicated, disseminated, or severe HZ in HIV-infected patients, though prospective efficacy data remain limited.11
With regard to prevention of infection, a large randomized trial in 2016 found that acyclovir prophylaxis resulted in a 68% reduction in HZ over 2 years among HIV patients.12 Despite data that acyclovir may be effective for this purpose, long-term antiviral prophylaxis is not routinely recommended for HZ,11,13 as it has been linked to rare cases of acyclovir-resistant HZ in HIV patients.14,15 However, antiviral prophylaxis against HSV type 2 reactivation in HIV patients also confers protection against VZV reactivation.11,12
Solid Organ Transplantation
Localized, mild to moderately severe dermatomal HZ can be treated with oral acyclovir, valacyclovir, or famciclovir. As in HIV patients, SOT patients with severe, disseminated, or complicated HZ should receive IV acyclovir.11 In the first 3 to 6 months following the procedure, SOT patients receive cytomegalovirus prophylaxis with ganciclovir or valgan-ciclovir, which also provides protection against HZ.13-18 For patients not receiving cytomegalovirus prophylaxis, HSV prophylaxis with oral acyclovir or valacyclovir is given for at least the first month after transplantation, which also confers protection against HZ.16,19 Antiviral therapy is critical during the early posttransplantation period when patients are most severely immunosuppressed and thus have the highest risk for VZV-associated complications.20 Although immunosuppression is lifelong in most SOT recipients, there is insufficient evidence for extending prophylaxis beyond 6 months.16,21
As a possible risk factor for HZ,22 MMF use is another consideration among SOT patients, similar to patient 2 in our report. A 2003 observational study supported withdrawal of MMF therapy during active VZV infection due to clinical observation of an association with HZ.23 However, a multicenter, randomized, controlled trial reported no cases of HZ in renal transplant recipients on MMF.24 Additionally, MMF has been observed to enhance the antiviral activity of acyclovir, at least in vitro.25 Given the lack of evidence of MMF as a risk factor for HZ, there is insufficient evidence for cessation of use during VZVreactivation in SOT patients.
Hematopoietic Stem Cell Transplantation
The preferred agents for treatment of localized mild dermatomal HZ are oral acyclovir or valacyclovir, as data on the safety and efficacy of famciclovir among HSCT recipients are limited.13,26 Patients should receive antiviral prophylaxis with one of these agents during the first year following allogeneic or autologous HSCT. This 1-year course has proven highly effective in reducing HZ in the first year following transplantation when most severe cases occur,21,26-29 and it has been associated with a persistently decreased risk for HZ even after discontinuation.21 Prophylaxis may be continued beyond 1 year in allogeneic HSCT recipients experiencing graft-versus-host disease who should receive acyclovir until 6 months after the end of immunosuppressive therapy.21,26
Vaccination remains a potential strategy to reduce the incidence of HZ in this patient population. A heat-inactivated vaccine administered within the first 3 months after the procedure has been shown to be safe among autologous and allogeneic HSCT patients.30,31 The vaccine notably reduced the incidence of HZ in patients who underwent autologous HSCT,32 but no known data are available on its clinical efficacy in allogeneic HSCT patients. Accordingly, there are no known official recommendations to date regarding vaccine use in these patient populations.26
Conclusion
It is incumbent upon clinicians to recognize the spectrum of atypical presentations of HZ and maintain a low threshold for performing appropriate diagnostic or confirmatory studies among at-risk patients with impaired immune function. Disseminated HZ can have potentially life-threatening visceral complications such as encephalitis, hepatitis, or pneumonitis.7,8 As such, an understanding of prevention and treatment modalities for VZV infection among immunocompromised patients is critical. Because the morbidity associated with complications of VZV infection is substantial and the risks associated with antiviral agents are minimal, antiviral prophylaxis is recommended for 6 months following SOT or 1 year following HSCT, and prompt treatment is warranted in cases of reasonable clinical suspicion for HZ.
Acknowledgment
The authors gratefully acknowledge the generosity of our patients in permitting photography of their skin findings for the furthering of medical education.
Well-known for its typical presentation, classic herpes zoster (HZ) presents as a dermatomal eruption of painful erythematous papules that evolve into grouped vesicles or bullae.1,2 Thereafter, the lesions can become pustular or hemorrhagic.1 Although the diagnosis most often is made clinically, confirmatory techniques for diagnosis include viral culture, direct fluorescent antibody testing, or polymerase chain reaction (PCR) assay.1,3
The main risk factor for HZ is advanced age, most commonly affecting elderly patients.4 It is hypothesized that a physiological decline in varicella-zoster virus (VZV)–specific cell-mediated immunity among elderly individuals helps trigger reactivation of the virus within the dorsal root ganglion.1,5 Similarly affected are immunocompromised individuals, including those with human immunodeficiency virus (HIV) infection, due to suppression of T cells immune to VZV,1,5 as well as immunosuppressed transplant recipients who have diminished VZV-specific cellular responses and VZV IgG antibody avidity.6
Secondary complications of VZV infection (eg, postherpetic neuralgia, bacterial superinfection progressing to cellulitis) lead to increased morbidity.7,8 Disseminated cutaneous HZ is another grave complication of VZV infection and almost exclusively occurs with immunosuppression.1,8 It manifests as an eruption of at least 20 widespread vesiculobullous lesions outside the primary and adjacent dermatomes.6 Immunocompromised patients also are at increased risk for visceral involvement of VZV infection, which may affect vital organs such as the brain, liver, or lungs.7,8 Given the atypical presentation of VZV infection among some immunocompromised individuals, these patients are at increased risk for diagnostic delay and morbidity in the absence of high clinical suspicion for disseminated HZ.
Case Reports
Patient 1
A 52-year-old man developed a painless nonpruritic rash on the left leg of 4 days’ duration. It initially appeared as an erythematous maculopapular rash on the medial aspect of the left knee without any prodromal symptoms. Over the next 4 days, erythematous vesicles developed that progressed to pustules, and the rash spread both proximally and distally along the left leg. Shortly following hospital admission, he developed a fever (temperature, 38.4°C). His medical history included alcoholic liver cirrhosis and AIDS, with a CD4 count of 174 cells/µL (reference range, 500–1500 cells/µL). He had been taking antiretroviral therapy (abacavir-lamivudine and dolutegravir) and prophylaxis against opportunistic infections (dapsone and itraconazole).
Physical examination was remarkable for an extensive rash consisting of multiple 1-cm clusters of approximately 40 pustules each scattered in a nondermatomal distribution along the left leg (Figure 1). Many of the vesicles were confluent with an erythematous base and were in different stages of evolution with some crusted and others emanating a thin liquid exudate. The lesions were nontender and without notable induration. The leg was warm and edematous.
Clinically, the differential diagnosis included disseminated HZ with bacterial superinfection, Vibrio vulnificus infection, and herpes simplex virus (HSV) infection. The patient was treated with intravenous vancomycin, levofloxacin, and acyclovir, and no new lesions developed throughout the course of treatment. On this regimen, his fever resolved after 1 day, the active lesions began to crust, and the edema and erythema diminished. Results of bacterial cultures and plasma PCR and IgM for HSV types 1 and 2 were negative. Viral culture results were negative, but a PCR assay for VZV was positive, reflective of acute reactivation of VZV.
Patient 2
A 63-year-old man developed a pruritic burning rash involving the face, trunk, arms, and legs of 6 days’ duration. His medical history included a heart transplant 6 months prior to presentation, type 2 diabetes mellitus, and chronic kidney disease. He was taking antirejection therapy with mycophenolate mofetil (MMF), prednisone, and tacrolimus.
Physical examination was remarkable for an extensive rash consisting of clusters of 1- to 2-mm vesicles scattered in a nondermatomal pattern. Isolated vesicles involved the forehead, nose, and left ear, and diffuse vesicles with a relatively symmetric distribution were scattered across the back, chest, and proximal and distal arms and legs (Figure 2). Many of the vesicles had an associated overlying crust with hemorrhage. Some of the vesicles coalesced with central necrotic plaques.
Given a clinical suspicion for disseminated HZ, therapy with oral valacyclovir was initiated. Two punch biopsies were consistent with herpesvirus cytopathic changes. Multiple sections demonstrated ulceration as well as acantholysis and necrosis of keratinocytes with multinucleation and margination of chromatin. There was an intense lichenoid and perivascular lymphocytic infiltrate in the dermis. Immunohistochemistry staining was positive for VZV and negative for HSV, indicating acute reactivation of VZV (Figure 3). Upon completion of an antiviral regimen, the patient returned to clinic with healed crusted lesions.
Comment
Frequently, the clinical features of HZ in immunocompromised patients mirror those in immunocompetent hosts.8 However, each of our 2 patients developed an unusual presentation of atypical generalized HZ.7 In this clinical variant, lesions develop along a single dermatome, then a diffuse vesicular eruption subsequently develops without dermatomal localization. These lesions can be chronic, persisting for months or years.7
The classic clinical presentation of HZ is distinct and often is readily diagnosed by visual inspection.7 However, atypical presentations and their associated complications can pose diagnostic and therapeutic challenges.7 Painless HZ lesions in a nondermatomal pattern were described in a patient who also had AIDS.9 Interestingly, multiple reports have found that patients with a severe but painless rash are less likely to have experienced a viral prodrome consisting of hyperesthesia, paresthesia, or pruritus.2,10 This observation suggests that lack of a prodrome, as in the case of patient 1 in our report, may aid in the recognition of painless HZ. Because of these atypical presentations, laboratory testing is even more important than in immunocompetent hosts, as diagnosis may be more difficult to establish on clinical presentation alone.
Several studies11-32 have evaluated modalities for treatment and prophylaxis for disseminated HZ in immunocompromised hosts, given its increased risk and potentially fatal complications in this population. The current guidelines in patients with HIV/AIDS, solid organ transplantation (SOT), and hematopoietic stem cell transplantation (HSCT) are summarized in the eTable.
HIV/AIDS Patients
Given their efficacy and low rate of toxicity, oral acyclovir, valacyclovir, and famciclovir are recommended treatment options for HIV patients with localized, mild, dermatomal HZ.11 Two exceptions include HZ ophthalmicus and Ramsay Hunt syndrome for which some experts recommend intravenous acyclovir given the risk for vision loss and facial palsy, respectively. Intravenous acyclovir often is the drug of choice for treating complicated, disseminated, or severe HZ in HIV-infected patients, though prospective efficacy data remain limited.11
With regard to prevention of infection, a large randomized trial in 2016 found that acyclovir prophylaxis resulted in a 68% reduction in HZ over 2 years among HIV patients.12 Despite data that acyclovir may be effective for this purpose, long-term antiviral prophylaxis is not routinely recommended for HZ,11,13 as it has been linked to rare cases of acyclovir-resistant HZ in HIV patients.14,15 However, antiviral prophylaxis against HSV type 2 reactivation in HIV patients also confers protection against VZV reactivation.11,12
Solid Organ Transplantation
Localized, mild to moderately severe dermatomal HZ can be treated with oral acyclovir, valacyclovir, or famciclovir. As in HIV patients, SOT patients with severe, disseminated, or complicated HZ should receive IV acyclovir.11 In the first 3 to 6 months following the procedure, SOT patients receive cytomegalovirus prophylaxis with ganciclovir or valgan-ciclovir, which also provides protection against HZ.13-18 For patients not receiving cytomegalovirus prophylaxis, HSV prophylaxis with oral acyclovir or valacyclovir is given for at least the first month after transplantation, which also confers protection against HZ.16,19 Antiviral therapy is critical during the early posttransplantation period when patients are most severely immunosuppressed and thus have the highest risk for VZV-associated complications.20 Although immunosuppression is lifelong in most SOT recipients, there is insufficient evidence for extending prophylaxis beyond 6 months.16,21
As a possible risk factor for HZ,22 MMF use is another consideration among SOT patients, similar to patient 2 in our report. A 2003 observational study supported withdrawal of MMF therapy during active VZV infection due to clinical observation of an association with HZ.23 However, a multicenter, randomized, controlled trial reported no cases of HZ in renal transplant recipients on MMF.24 Additionally, MMF has been observed to enhance the antiviral activity of acyclovir, at least in vitro.25 Given the lack of evidence of MMF as a risk factor for HZ, there is insufficient evidence for cessation of use during VZVreactivation in SOT patients.
Hematopoietic Stem Cell Transplantation
The preferred agents for treatment of localized mild dermatomal HZ are oral acyclovir or valacyclovir, as data on the safety and efficacy of famciclovir among HSCT recipients are limited.13,26 Patients should receive antiviral prophylaxis with one of these agents during the first year following allogeneic or autologous HSCT. This 1-year course has proven highly effective in reducing HZ in the first year following transplantation when most severe cases occur,21,26-29 and it has been associated with a persistently decreased risk for HZ even after discontinuation.21 Prophylaxis may be continued beyond 1 year in allogeneic HSCT recipients experiencing graft-versus-host disease who should receive acyclovir until 6 months after the end of immunosuppressive therapy.21,26
Vaccination remains a potential strategy to reduce the incidence of HZ in this patient population. A heat-inactivated vaccine administered within the first 3 months after the procedure has been shown to be safe among autologous and allogeneic HSCT patients.30,31 The vaccine notably reduced the incidence of HZ in patients who underwent autologous HSCT,32 but no known data are available on its clinical efficacy in allogeneic HSCT patients. Accordingly, there are no known official recommendations to date regarding vaccine use in these patient populations.26
Conclusion
It is incumbent upon clinicians to recognize the spectrum of atypical presentations of HZ and maintain a low threshold for performing appropriate diagnostic or confirmatory studies among at-risk patients with impaired immune function. Disseminated HZ can have potentially life-threatening visceral complications such as encephalitis, hepatitis, or pneumonitis.7,8 As such, an understanding of prevention and treatment modalities for VZV infection among immunocompromised patients is critical. Because the morbidity associated with complications of VZV infection is substantial and the risks associated with antiviral agents are minimal, antiviral prophylaxis is recommended for 6 months following SOT or 1 year following HSCT, and prompt treatment is warranted in cases of reasonable clinical suspicion for HZ.
Acknowledgment
The authors gratefully acknowledge the generosity of our patients in permitting photography of their skin findings for the furthering of medical education.
- McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol. 1999;41:1-16.
- Nagasako EM, Johnson RW, Griffin DR, et al. Rash severity in herpes zoster: correlates and relationship to postherpetic neuralgia. J Am Acad Dermatol. 2002;46:834-839.
- Leung J, Harpaz R, Baughman AL, et al. Evaluation of laboratory methods for diagnosis of varicella. Clin Infect Dis. 2010;51:23-32.
- Herpes Zoster and Functional Decline Consortium. Functional decline and herpes zoster in older people: an interplay of multiple factors. Aging Clin Exp Res. 2015;27:757-765.
- Weinberg A, Levin MJ. VZV T cell-mediated immunity. Curr Top Microbiol Immunol. 2010;342:341-357.
- Prelog M, Schonlaub J, Jeller V, et al. Reduced varicella-zoster-virus (VZV)-specific lymphocytes and IgG antibody avidity in solid organ transplant recipients. Vaccine. 2013;31:2420-2426.
- Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
- Glesby MJ, Moore RD, Chaisson RE. Clinical spectrum of herpes zoster in adults infected with human immunodeficiency virus. Clin Infect Dis. 1995;21:370-375.
- Blankenship W, Herchline T, Hockley A. Asymptomatic vesicles in a patient with the acquired immunodeficiency syndrome. disseminated varicella-zoster virus (VZV) infection. Arch Dermatol. 1994;130:1193, 1196.
- Katz J, Cooper EM, Walther RR, et al. Acute pain in herpes zoster and its impact on health-related quality of life. Clin Infect Dis. 2004;39:342-348.
- Gnann JW. Antiviral therapy of varicella-zoster virus infections. In: Arvin A, Campadelli-Fiume G, Mocarski E, et al, eds. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge, United Kingdom: Cambridge University Press; 2007:1175-1191.
- Barnabas RV, Baeten JM, Lingappa JR, et al. Acyclovir prophylaxis reduces the incidence of herpes zoster among HIV-infected individuals: results of a randomized clinical trial. J Infect Dis. 2016;213:551-555.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
- Jacobson MA, Berger TG, Fikrig S, et al. Acyclovir-resistant varicella zoster virus infection after chronic oral acyclovir therapy in patients with the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1990;112:187-191.
- Linnemann CC Jr, Biron KK, Hoppenjans WG, et al. Emergence of acyclovir-resistant varicella zoster virus in an AIDS patient on prolonged acyclovir therapy. AIDS. 1990;4:577-579.
- Pergam SA, Limaye AP; AST Infectious Diseases Community of Practice. Varicella zoster virus (VZV) in solid organ transplant recipients. Am J Transplant. 2009;9(suppl 4):S108-S115.
- Preiksaitis JK, Brennan DC, Fishman J, et al. Canadian society of transplantation consensus workshop on cytomegalovirus management in solid organ transplantation final report. Am J Transplant. 2005;5:218-227.
- Fishman JA, Doran MT, Volpicelli SA, et al. Dosing of intravenous ganciclovir for the prophylaxis and treatment of cytomegalovirus infection in solid organ transplant recipients. Transplantation. 2000;69:389-394.
- Zuckerman R, Wald A; AST Infectious Diseases Community of Practice. Herpes simplex virus infections in solid organ transplant recipients. Am J Transplant. 2009;9(suppl 4):S104-S107.
- Arness T, Pedersen R, Dierkhising R, et al. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis. 2008;10:260-268.
- Erard V, Guthrie KA, Varley C, et al. One-year acyclovir prophylaxis for preventing varicella-zoster virus disease after hematopoietic cell transplantation: no evidence of rebound varicella-zoster virus disease after drug discontinuation. Blood. 2007;110:3071-3077.
- Rothwell WS, Gloor JM, Morgenstern BZ, et al. Disseminated varicella infection in pediatric renal transplant recipients treated with mycophenolate mofetil. Transplantation. 1999;68:158-161.
- Lauzurica R, Bayés B, Frías C, et al. Disseminated varicella infection in adult renal allograft recipients: role of mycophenolate mofetil. Transplant Proc. 2003;35:1758-1759.
- A blinded, randomized clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation. TheTricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation. 1996;61:1029-1037.
- Neyts J, De Clercq E. Mycophenolate mofetil strongly potentiates the anti-herpesvirus activity of acyclovir. Antiviral Res. 1998;40:53-56.
- Tomblyn M, Chiller T, Einsele H, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15:1143-1238.
- Boeckh M, Kim HW, Flowers ME, et al. Long-term acyclovir for prevention of varicella zoster virus disease after allogeneic hematopoietic cell transplantation—a randomized double-blind placebo-controlled study. Blood. 2006;107:1800-1805.
- Kawamura K, Hayakawa J, Akahoshi Y, et al. Low-dose acyclovir prophylaxis for the prevention of herpes simplex virus and varicella zoster virus diseases after autologous hematopoietic stem cell transplantation. Int J Hematol. 2015;102:230-237.
- Fred Hutchinson Cancer Research Center/Seattle Cancer Care Alliance. Long-term follow-up after hematopoietic stem cell transplant general guidelines for referring physicians. Fred Hutchinson Cancer Research Center website. https://www.fredhutch.org/content/dam/public/Treatment-Suport/Long-Term-Follow-Up/physician.pdf. Published July 17, 2014. Accessed October 19, 2017.
- Kussmaul SC, Horn BN, Dvorak CC, et al. Safety of the live, attenuated varicella vaccine in pediatric recipients of hematopoietic SCTs. Bone Marrow Transplant. 2010;45:1602-1606.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med. 2002;347:26-34.
- Issa NC, Marty FM, Leblebjian H, et al. Live attenuated varicella-zoster vaccine in hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant. 2014;20:285-287.
- McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol. 1999;41:1-16.
- Nagasako EM, Johnson RW, Griffin DR, et al. Rash severity in herpes zoster: correlates and relationship to postherpetic neuralgia. J Am Acad Dermatol. 2002;46:834-839.
- Leung J, Harpaz R, Baughman AL, et al. Evaluation of laboratory methods for diagnosis of varicella. Clin Infect Dis. 2010;51:23-32.
- Herpes Zoster and Functional Decline Consortium. Functional decline and herpes zoster in older people: an interplay of multiple factors. Aging Clin Exp Res. 2015;27:757-765.
- Weinberg A, Levin MJ. VZV T cell-mediated immunity. Curr Top Microbiol Immunol. 2010;342:341-357.
- Prelog M, Schonlaub J, Jeller V, et al. Reduced varicella-zoster-virus (VZV)-specific lymphocytes and IgG antibody avidity in solid organ transplant recipients. Vaccine. 2013;31:2420-2426.
- Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
- Glesby MJ, Moore RD, Chaisson RE. Clinical spectrum of herpes zoster in adults infected with human immunodeficiency virus. Clin Infect Dis. 1995;21:370-375.
- Blankenship W, Herchline T, Hockley A. Asymptomatic vesicles in a patient with the acquired immunodeficiency syndrome. disseminated varicella-zoster virus (VZV) infection. Arch Dermatol. 1994;130:1193, 1196.
- Katz J, Cooper EM, Walther RR, et al. Acute pain in herpes zoster and its impact on health-related quality of life. Clin Infect Dis. 2004;39:342-348.
- Gnann JW. Antiviral therapy of varicella-zoster virus infections. In: Arvin A, Campadelli-Fiume G, Mocarski E, et al, eds. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge, United Kingdom: Cambridge University Press; 2007:1175-1191.
- Barnabas RV, Baeten JM, Lingappa JR, et al. Acyclovir prophylaxis reduces the incidence of herpes zoster among HIV-infected individuals: results of a randomized clinical trial. J Infect Dis. 2016;213:551-555.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
- Jacobson MA, Berger TG, Fikrig S, et al. Acyclovir-resistant varicella zoster virus infection after chronic oral acyclovir therapy in patients with the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1990;112:187-191.
- Linnemann CC Jr, Biron KK, Hoppenjans WG, et al. Emergence of acyclovir-resistant varicella zoster virus in an AIDS patient on prolonged acyclovir therapy. AIDS. 1990;4:577-579.
- Pergam SA, Limaye AP; AST Infectious Diseases Community of Practice. Varicella zoster virus (VZV) in solid organ transplant recipients. Am J Transplant. 2009;9(suppl 4):S108-S115.
- Preiksaitis JK, Brennan DC, Fishman J, et al. Canadian society of transplantation consensus workshop on cytomegalovirus management in solid organ transplantation final report. Am J Transplant. 2005;5:218-227.
- Fishman JA, Doran MT, Volpicelli SA, et al. Dosing of intravenous ganciclovir for the prophylaxis and treatment of cytomegalovirus infection in solid organ transplant recipients. Transplantation. 2000;69:389-394.
- Zuckerman R, Wald A; AST Infectious Diseases Community of Practice. Herpes simplex virus infections in solid organ transplant recipients. Am J Transplant. 2009;9(suppl 4):S104-S107.
- Arness T, Pedersen R, Dierkhising R, et al. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis. 2008;10:260-268.
- Erard V, Guthrie KA, Varley C, et al. One-year acyclovir prophylaxis for preventing varicella-zoster virus disease after hematopoietic cell transplantation: no evidence of rebound varicella-zoster virus disease after drug discontinuation. Blood. 2007;110:3071-3077.
- Rothwell WS, Gloor JM, Morgenstern BZ, et al. Disseminated varicella infection in pediatric renal transplant recipients treated with mycophenolate mofetil. Transplantation. 1999;68:158-161.
- Lauzurica R, Bayés B, Frías C, et al. Disseminated varicella infection in adult renal allograft recipients: role of mycophenolate mofetil. Transplant Proc. 2003;35:1758-1759.
- A blinded, randomized clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation. TheTricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation. 1996;61:1029-1037.
- Neyts J, De Clercq E. Mycophenolate mofetil strongly potentiates the anti-herpesvirus activity of acyclovir. Antiviral Res. 1998;40:53-56.
- Tomblyn M, Chiller T, Einsele H, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15:1143-1238.
- Boeckh M, Kim HW, Flowers ME, et al. Long-term acyclovir for prevention of varicella zoster virus disease after allogeneic hematopoietic cell transplantation—a randomized double-blind placebo-controlled study. Blood. 2006;107:1800-1805.
- Kawamura K, Hayakawa J, Akahoshi Y, et al. Low-dose acyclovir prophylaxis for the prevention of herpes simplex virus and varicella zoster virus diseases after autologous hematopoietic stem cell transplantation. Int J Hematol. 2015;102:230-237.
- Fred Hutchinson Cancer Research Center/Seattle Cancer Care Alliance. Long-term follow-up after hematopoietic stem cell transplant general guidelines for referring physicians. Fred Hutchinson Cancer Research Center website. https://www.fredhutch.org/content/dam/public/Treatment-Suport/Long-Term-Follow-Up/physician.pdf. Published July 17, 2014. Accessed October 19, 2017.
- Kussmaul SC, Horn BN, Dvorak CC, et al. Safety of the live, attenuated varicella vaccine in pediatric recipients of hematopoietic SCTs. Bone Marrow Transplant. 2010;45:1602-1606.
- Hata A, Asanuma H, Rinki M, et al. Use of an inactivated varicella vaccine in recipients of hematopoietic-cell transplants. N Engl J Med. 2002;347:26-34.
- Issa NC, Marty FM, Leblebjian H, et al. Live attenuated varicella-zoster vaccine in hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant. 2014;20:285-287.
Practice Points
- Clinician awareness of management guidelines for the prevention and treatment of varicella-zoster virus infection in immunocompromised individuals is critical to minimize the risk for disease and associated morbidity.
- Antiviral prophylaxis is recommended for 6 months following solid organ transplantation or 1 year following hematopoietic stem cell transplantation, and prompt treatment is warranted in cases of reasonable clinical suspicion for herpes zoster.
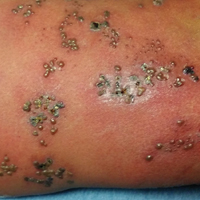
Ulcerative Sarcoidosis: A Prototypical Presentation and Review
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that primarily affects the lungs and lymphatic system but also may involve the skin, eyes, liver, spleen, muscles, bones, and nervous system.1 Cutaneous symptoms of sarcoidosis occur in approximately 25% of patients and are classified as specific and nonspecific, with specific lesions demonstrating noncaseating granuloma formation, which is typical of sarcoidosis.2 Nonspecific lesions primarily include erythema nodosum and calcinosis cutis. Specific lesions commonly present as reddish brown infiltrated plaques that may be annular, polycyclic, or serpiginous.1,3 They also may appear as yellowish brown or violaceous maculopapular lesions. However, specific lesions may present in a wide variety of morphologies, most often papules, nodules, subcutaneous infiltrates, and lupus pernio.4 Additionally, atypical cutaneous manifestations of sarcoidosis include erythroderma; scarring alopecia; nail dystrophy; and verrucous, ichthyosiform, psoriasiform, hypopigmented, or ulcerative skin lesions.3-5 Among these many potential clinical presentations, ulcerative sarcoidosis is quite uncommon.
We report a case of a patient who presented with classic clinical and histopathological findings of ulcerative sarcoidosis to highlight the prototypical presentation of a rare condition. We also review 34 additional cases of ulcerative sarcoidosis published in the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid.4-32 Analyzing this historical information, the scope of this unusual form of cutaneous sarcoidosis can be better understood, recognized, and treated. Although current standard-of-care treatments are most often successful, there is a paucity of definitive clinical trials to justify and verify comparative therapeutic efficacy.
Case Report
A 49-year-old black man with known pulmonary sarcoidosis, idiopathic (human immunodeficiency virus–negative) CD4 depletion syndrome, and chronic kidney disease presented with persistent bilateral ulcers of the legs of 1 month’s duration. The lesions first appeared as multiple “dark spots” on the legs. After the patient applied homemade aloe vera extract under occlusion for 1 to 2 days, the lesions became painful and began to ulcerate approximately 3 months prior to presentation. The patient applied a combination of a topical first aid antibiotic ointment, Epsom salts, and hydrogen peroxide without any improvement. A current review of systems was negative.
The patient’s medical history was notable for sarcoidosis diagnosed more than 10 years prior. During this time, he had intermittently been treated elsewhere with low-dose oral prednisone (5 mg once daily), hydroxychloroquine (200 mg twice daily), and an inhaled steroid as needed. He had a history of human immunodeficiency virus–negative, idiopathic CD4 depletion syndrome, which had been complicated by cryptococcal meningitis 7 years prior to presentation. He also had renal insufficiency, with baseline creatinine levels ranging from 1.4 to 1.7 mg/dL (reference range, 0.6–1.2 mg/dL). There was no personal or family history of known or suspected inflammatory bowel disease.
On physical examination, numerous discrete, coalescing, punched out–appearing ulcerations with foul-smelling, greenish yellow, purulent drainage were present bilaterally on the legs (Figure 1). The ulcers had a rolled border with a moderate amount of seemingly nonviable necrotic tissue. A number of hyperpigmented round papules, patches, and plaques also were present on the proximal legs. Laboratory evaluation revealed a CD4 count of 151 cc/mm3 (reference range, 500–1600 cc/mm3) and mildly elevated calcium of 10.7 mg/dL (reference range, 8.2–10.2 mg/dL).
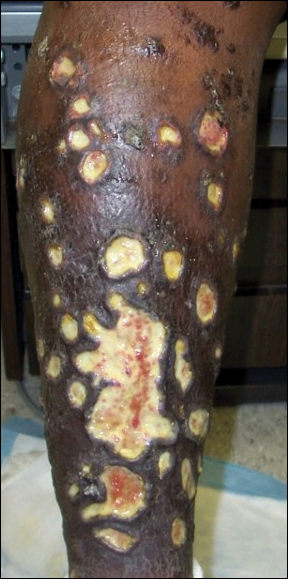
Aerobic, anaerobic, mycobacterial, and fungal cultures of the purulent exudate were obtained. Given a high suspicion for secondary infection of the exogenous wound sites, doxycycline (100 mg twice daily) and topical mupir-ocin were initiated. Gram stain revealed few to moderate polymorphonuclear cells and many gram-positive cocci in pairs, chains, and clusters, along with many gram-negative rods. Bacterial culture grew Pseudomonas aeruginosa, Enterococcus species group G streptococci, and methicillin-resistant Staphylococcus aureus–positive staphylococci. Ciprofloxacin (500 mg twice daily) was then initiated, but the ulcers showed absolutely no clinical improvement and in fact worsened both in number and depth (Figure 2) over subsequent clinic visits during the next 3 months, even after amoxicillin (500 mg 3 times daily) was added. The patient was admitted for treatment with intravenous antibiotics after additional wound cultures revealed fluoroquinolone-resistant Pseudomonas.
Punch biopsies of the ulcers showed nonspecific acute inflammation and tissue necrosis in the active ulcers with nonnecrotizing granulomatous inflammation extending into the deep dermis, with many Langerhans-type giant cells present in the palpable ulcer borders (Figure 3). Neither birefringent particles nor asteroid bodies were observed. Tissue Gram stains did not reveal evidence of bacterial infection. Special stains for acid-fast and fungal organisms (ie, periodic acid–Schiff, Gomori methenamine-silver, Fite, acid-fast bacilli) were similarly negative. Tissue cultures obtained on deep biopsy revealed only rare colonies of P aeruginosa and no isolates on anaerobic, mycobacterial, or fungal cultures. Polymerase chain reaction for mycobacteria and common endemic fungi also was negative. In the absence of infection and considering his history of known sarcoidosis, these histologic features were consistent with ulcerative sarcoidosis. The patient was started on prednisone (60 mg once daily) and hydroxychloroquine (200 mg twice daily). The prednisone was tapered to 20 mg once daily over a 2-year period, at which point 90% of the ulcers had healed. He was continued on hydroxychloroquine at the initial dose, and at a 3-year follow-up his ulcers had healed completely without relapse.
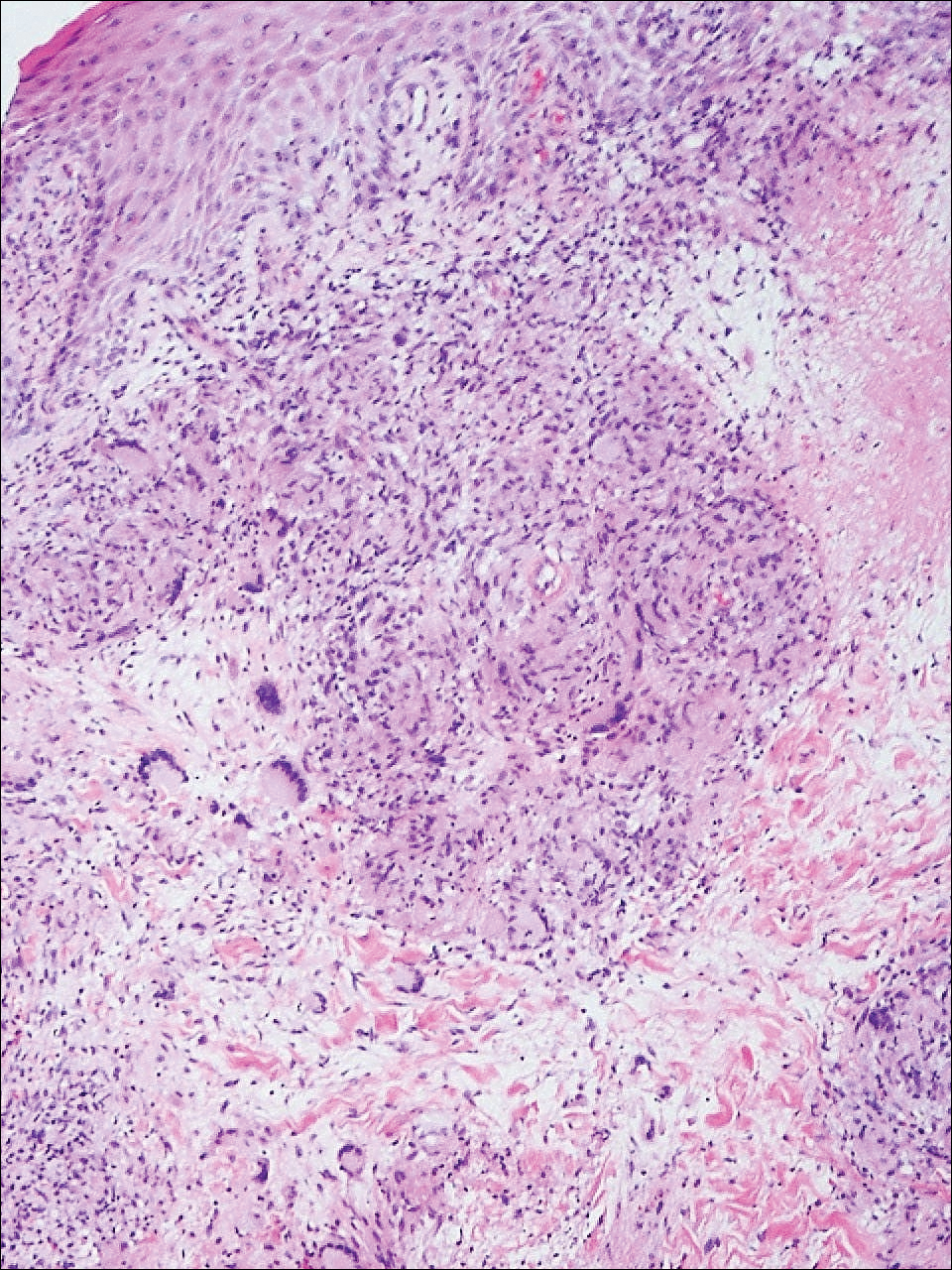
Comment
Ulcerative sarcoidosis is rare, seen worldwide in only 5% of patients with cutaneous sarcoidosis.33 However, cases have been encountered worldwide, with reports emanating from Japan, China, Germany, France, and Russia, among others.6,34-55 We reviewed 34 cases from the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid and examined patient demographics, clinical presentation, histological findings, treatment type, and outcome. Key references are presented in the Table. Disease prevalence previously has been estimated as being 3-times more common in women than men1; in our literature review, we found a female to male ratio of 3.25 to 1. Additionally, ulcerative sarcoidosis is reported to be twice as common in black versus white individuals.33 In our literature review, when race was reported, 66% (21/32) of patients were black. Disease prevalence has been reported to peak at 20 to 40 years of age.3 In this review, the average age of presentation was 45 years (age range, 24–79 years).
Ulceration may arise de novo but more commonly arises in preexisting scars or cutaneous lesions. There are 2 distinct patterns seen in ulcerative sarcoidosis.4 The first is characterized by ulceration within necrotic yellow plaques.2 The second pattern is characterized by violaceous nodules arising in an annular confluent pattern that eventually ulcerate.4 This presentation commonly mimics or may be mimicked by multiple disease states, including sporotrichosis, tuberculosis, stasis dermatitis with venous ulceration, and even metastatic breast cancer.7,46,55,56 Regardless of presentation, the legs are the most common location of ulcer formation.1,33 In our review, 85% (29/34) of cases presented with involvement of the legs, including our own case. Other locations of ulcer formation have included the face, arms, trunk, and genital area.
On histologic examination of ulcerative sarcoidosis, epithelioid granulomas composed of multinucleated giant cells, histiocytes, and scant numbers of lymphocytes are present.1,3 These formations are the noncaseating granulomas typical of sarcoidosis (Table). All of the cases in our review of the literature were described as either a collection of epithelioid granulomas with giant cell formation or noncaseating granulomas. There also have been reports of atypical features including necrotizing granulomas and granulomatous vasculitis.4,8,9,50 The histologic differential diagnosis in this case also would primarily include an infectious granulomatous process and less so an id reaction, rosacea, a paraneoplastic phenomenon, foreign body granulomas, and metastatic Crohn disease. The presence of ulceration, the large number of lesions, and the anatomic distribution help rule out most of these alternate diagnostic considerations. Diligent extensive workup was done in our patient to insure it was not an infection.
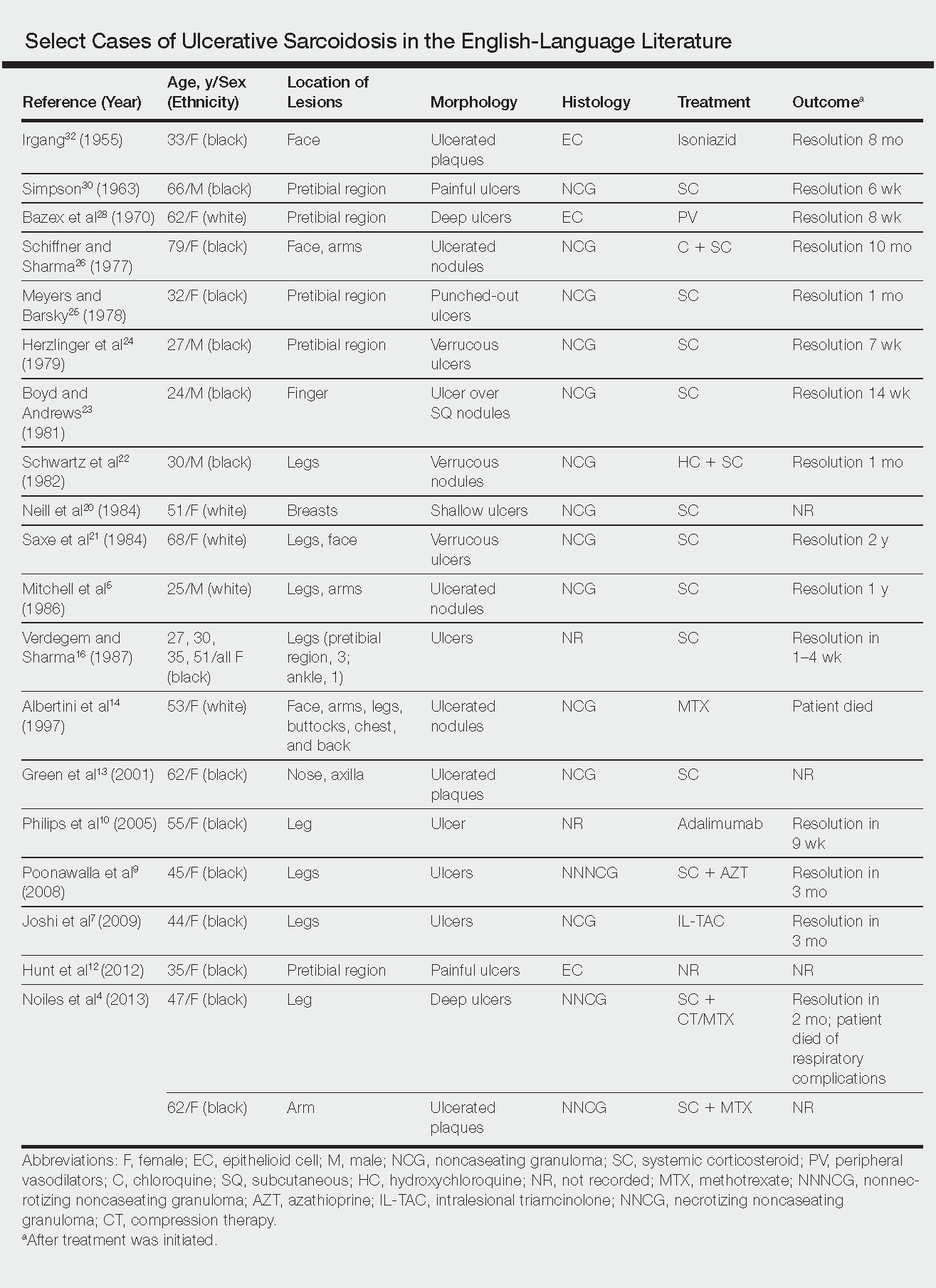
The goals of treatment include symptomatic relief, improvement in objective parameters of disease activity, and prevention of disease progression and subsequent disability.33,57 Fortunately, the majority of sarcoidosis patients with cutaneous symptoms achieve full recovery within months to years.33 Our literature review indicated that 81% (22/27) of patients with ulcerative lesions experienced full resolution within 1 year of treatment. Of those that did not (19% [5/27]), the patients were either lost to follow-up or died from other complications of sarcoidosis.
The widely accepted standard therapy for cutaneous sarcoidosis includes topical, intralesional, and systemic corticosteroids; antimalarials; and methotrexate.33,57 Steroids and methotrexate act by suppressing granuloma formation, while antimalarials prevent antigen presentation (presumably part of the pathogenesis).33 For mild to moderate disease, topical and intralesional steroids may be all that is necessary.33,57 Systemic steroids are used for disfiguring, destructive, and widespread lesions that have been refractory to local and other systemic therapies.33,57 Steroids are tapered gradually depending on the patient’s response, as it is common for patients to relapse below a certain dose.33,57 Antimalarials (chloroquine or hydroxychloroquine) and methotrexate are considered adjunct treatments for patients who are either steroid unresponsive or who are unable to tolerate corticosteroid treatment due to adverse events.33,57
Standard therapy is complicated by the side effects of treatment. Use of corticosteroids may lead to gastrointestinal tract upset, increased appetite, mood disturbances, impaired wound healing, hyperglycemia, hypertension, cushingoid features, and acne.57 Antimalarials can cause nausea, anorexia, and agranulocytosis, and chloroquine therapy in particular can lead to blurred vision, corneal deposits, and central retinopathy.33,57 Methotrexate is associated with hematologic, gastrointestinal tract, pulmonary, and hepatic toxicities well known to most practitioners.
Because of the variable clinical response of patients to standard therapy and their associated toxicities, other treatment options have been used including pentoxifylline, tetracyclines, isotretinoin, leflunomide, thalidomide, infliximab, adalimumab, allopurinol, and the pulsed dye or CO2 laser.10,33,57 In nonhealing ulcers, split-thickness grafting and a bilayered bioengineered skin substitute have been used with good results in conjunction with ongoing systemic therapy.11,47 Additionally, nanoparticle silver burn paste has been used successfully, with resolution of ulcers within 2 weeks in the Chinese literature.53
All of these treatment recommendations are based on historically accepted modalities. Controlled trials with longitudinal follow-up are needed to provide justification for the current standard of care.34
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1421-1435.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361-1383.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Noiles K, Beleznay K, Crawford RI, et al. Sarcoidosis can present with necrotizing granulomas histologically: two cases of ulcerated sarcoidosis and review of the literature. J Cutan Med Surg. 2013;17:377-378.
- Mitchell IC, Sweatman MC, Rustin MH, et al. Ulcerative and hypopigmented sarcoidosis. J Am Acad Dermatol. 1986;15:1062-1065.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Joshi SS, Romanelli R, Kirsner RS. Sarcoidosis mimicking a venous ulcer: a case report. Ostomy Wound Manage. 2009;55:46-48.
- Petri M, Barr E, Cho K, et al. Overlap of granulomatous vasculitis and sarcoidosis: presentation with uveitis, eosinophilia, leg ulcers, sinusitis and past foot drop. J Rheumatol. 1988;15:1171-1173.
- Poonawalla T, Colome-Grimmer MI, Kelly B. Ulcerative sarcoidosis in the legs with granulomatous vasculitis. Clin Exp Dermatol. 2008;33:282-286.
- Philips MA, Lynch J, Azmi FH. Ulcerative sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917.
- Collison DW, Novice F, Banse L, et al. Split-thickness skin grafting in extensive ulcerative sarcoidosis. J Dermatol Surg Oncol. 1989;15:679-683.
- Hunt RD, Gonzalez ME, Robinson M, et al. Ulcerative sarcoidosis. Dermatol Online J. 2012;18:29.
- Green JJ, Lawrence N, Heymann WR. Generalized ulcerative sarcoidosis induced by therapy with the flashlamp-pumped pulsed dye. Arch Dermatol. 2001;137:507-508.
- Albertini JG, Tyler W, Miller OF. Ulcerative sarcoidosis. case report and review of the literature. Arch Dermatol. 1997;133:215-219.
- Thomas J, Williams DW. Peritoneal involvement and ulcerative skin plaques in sarcoidosis: a case report. Sarcoidosis. 1989;6:161-162.
- Verdegem TD, Sharma OP. Cutaneous ulcers in sarcoidosis. Arch Dermatol. 1987;123:1531-1534.
- Gupta AK, Haberman HF, From GL, et al. Sarcoidosis with extensive cutaneous ulceration. unusual clinical presentation. Dermatologica. 1987;174:135-139.
- Hruza GJ, Kerdel FA. Generalized atrophic sarcoidosis with ulcerations. Arch Dermatol. 1986;122:320-322.
- Muhlemann MF, Walker NP, Tan LB, et al. Elephantine sarcoidosis presenting as ulcerating lymphoedema. J R Soc Med. 1985;78:260-261.
- Neill SM, Smith NP, Eady RA. Ulcerative sarcoidosis: a rare manifestation of a common disease. Clin Exp Dermatol. 1984;9:277-279.
- Saxe N, Benatar SR, Bok L, et al. Sarcoidosis with leg ulcers and annular facial lesions. Arch Dermatol. 1984;120:93-96.
- Schwartz RA, Robertson DB, Tierney LM, et al. Generalized ulcerative sarcoidosis. Arch Dermatol. 1982;118:931-933.
- Boyd RE, Andrews BS. Sarcoidosis presenting as cutaneous ulceration, subcutaneous nodules and chronic arthritis. J Rheumatol. 1981;8:311-316.
- Herzlinger DC, Marland AM, Barr RJ. Verrucous ulcerative skin lesions in sarcoidosis. an unusual clinical presentation. Cutis. 1979;23:569-572.
- Meyers M, Barsky S. Ulcerative sarcoidosis. Arch Dermatol. 1978;114:447.
- Schiffner J, Sharma OP. Ulcerative sarcoidosis. report of an unusual case. Arch Dermatol. 1977;113:676-677.
- Williamson DM. Sarcoidosis with atrophic lesions and ulcers of the legs. Br J Dermatol. 1971;84:92-93.
- Bazex A, Dupre A, Christol B, et al. Sarcoidosis with atrophic lesions and ulcers and the presence in some sarcoid granulomata of orceinophil fibres. Br J Dermatol. 1970;83:255-262.
- Brodkin RH. Leg ulcers. a report of two cases caused by sarcoidosis. Acta Derm Venereol. 1969;49:584-587.
- Simpson JR. Sarcoidosis with erythrodermia and ulceration. Br J Dermatol. 1963;75:193-198.
- Irgang S. Ulcerative cutaneous lesion in sarcoidosis; report of a case with clinical resemblance to lupus vulgaris. Harlem Hosp Bull. 1956;8:134-139.
- Irgang S. Ulcerative cutaneous lesions in sarcoidosis; report of a case with clinical resemblance to papulonecrotic tuberculide. Br J Dermatol. 1955;67:255-260.
- Hoffman MD. Atypical ulcers. Dermatol Ther. 2013;26:222-235.
- Hopf B, Krebs A. Ulcera cruris as a rare manifestation of sarcoidosis. Dermatologica. 1974;113:55-62.
- Metz J, Hartmann A, Hautkr Z. Ulcerative form of skin sarcoidosis. Z Hautkr. 1977;52:890-896.
- Berenbeĭn BA, Malygina LA, Tiutiunnikova IA. Ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1984;4:50-53.
- Takahashi N, Hoshino M, Takase T, et al. A case of ulcerative sarcoidosis [in Japanese]. Nihon Hifuka Gakkai Zasshi. 1985;95:1049-1054.
- Schamroth JM. Sarcoidosis with severe extensive skin ulceration. Int J Dermatol. 1985;24:451-452.
- Porteau L, Dromer C, Le Guennec P, et al. Ulcer lesions in sarcoidosis: apropos of a case [in French]. Ann Med Interne (Paris). 1997;148:105-106.
- de La Blanchardière A, Bachmeyer C, Toutous L, et al. Cutaneous ulcerations in sarcoidosis [in French]. Rev Med Interne. 1995;16:927-929.
- Mitsuishi T, Nogita T, Kawashima M. Psoriasiform sarcoidosis with ulceration. Int J Dermatol. 1992;31:339-340.
- Rodionov AN, Samtsov AV. The ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1990;7:68-71.
- Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol. 1999;38:841-845.
- Gungor E, Artuz F, Alli N, et al. Ulcerative sarcoidosis. J Eur Acad Dermatol Venereol. 1999;12:78-79.
- Schleinitz N, Luc M, Genot S, et al. Ulcerative cutaneous lesions: a rare manifestation of sarcoidosis [in French]. Rev Med Interne. 2005;26:758-759.
- Klocker J, Duckers J, Morse R, et al. Ulcerative cutaneous sarcoidosis masquerading as metastatic carcinoma of the breast. Age Ageing. 2002;31:77-79.
- Streit M, Bohlen LM, Braathen LR. Ulcerative sarcoidosis successfully treated with apligraf. Dermatology. 2001;202:367-370.
- Ichiki Y, Kitajima Y. Ulcerative sarcoidosis: case report and review of the Japanese literature. Acta Derm Venereol. 2008;88:526-528
- Meyersburg D, Schön MP, Bertsch HP, et al. Uncommon cutaneous ulcerative and systemic sarcoidosis. successful treatment with hydroxychloroquine and compression therapy [in German]. Hautarzt. 2011;62:691-695.
- Wei CH, Huang YH, Shih YC, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Kluger N, Girard C, Durand L, et al. Leg ulcers revealing systemic sarcoidosis with splenomegaly and thrombocytopenia. Int J Dermatol. 2013;52:1425-1427.
- Jun L, Jia-Wei L, Hong-Zhong J. Ulcerative sarcoidosis. Int J Dermatol. 2014;53:E315-E316.
- Chen JH, Wang TT, Lin ZQ. Successful application of a novel dressing for the treatment of ulcerative cutaneous sarcoidosis. Chin Med J. 2013;126:3400.
- Ri G, Yoshikawa E, Shigekiyo T, et al. Takayasu artertitis and ulcerative sarcoidosis. Intern Med. 2015;54:1075-1080.
- Spiliopoulou I, Foka A, Bounas A, et al. Mycobacterium kansasii cutaneous infection in a patient with sarcoidosis treated with anti-TNF agents. Acta Clin Belg. 2014;69:229-231.
- Yang DJ, Krishnan RS, Guillen DR, et al. Disseminated sporotrichosis mimicking sarcoidosis. Int J Dermatol. 2006;45:450-453.
- Badgwell C, Rosen T. Cutaneous sarcoidosis therapy updated. J Am Acad Dermatol. 2007;56:69-83.
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that primarily affects the lungs and lymphatic system but also may involve the skin, eyes, liver, spleen, muscles, bones, and nervous system.1 Cutaneous symptoms of sarcoidosis occur in approximately 25% of patients and are classified as specific and nonspecific, with specific lesions demonstrating noncaseating granuloma formation, which is typical of sarcoidosis.2 Nonspecific lesions primarily include erythema nodosum and calcinosis cutis. Specific lesions commonly present as reddish brown infiltrated plaques that may be annular, polycyclic, or serpiginous.1,3 They also may appear as yellowish brown or violaceous maculopapular lesions. However, specific lesions may present in a wide variety of morphologies, most often papules, nodules, subcutaneous infiltrates, and lupus pernio.4 Additionally, atypical cutaneous manifestations of sarcoidosis include erythroderma; scarring alopecia; nail dystrophy; and verrucous, ichthyosiform, psoriasiform, hypopigmented, or ulcerative skin lesions.3-5 Among these many potential clinical presentations, ulcerative sarcoidosis is quite uncommon.
We report a case of a patient who presented with classic clinical and histopathological findings of ulcerative sarcoidosis to highlight the prototypical presentation of a rare condition. We also review 34 additional cases of ulcerative sarcoidosis published in the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid.4-32 Analyzing this historical information, the scope of this unusual form of cutaneous sarcoidosis can be better understood, recognized, and treated. Although current standard-of-care treatments are most often successful, there is a paucity of definitive clinical trials to justify and verify comparative therapeutic efficacy.
Case Report
A 49-year-old black man with known pulmonary sarcoidosis, idiopathic (human immunodeficiency virus–negative) CD4 depletion syndrome, and chronic kidney disease presented with persistent bilateral ulcers of the legs of 1 month’s duration. The lesions first appeared as multiple “dark spots” on the legs. After the patient applied homemade aloe vera extract under occlusion for 1 to 2 days, the lesions became painful and began to ulcerate approximately 3 months prior to presentation. The patient applied a combination of a topical first aid antibiotic ointment, Epsom salts, and hydrogen peroxide without any improvement. A current review of systems was negative.
The patient’s medical history was notable for sarcoidosis diagnosed more than 10 years prior. During this time, he had intermittently been treated elsewhere with low-dose oral prednisone (5 mg once daily), hydroxychloroquine (200 mg twice daily), and an inhaled steroid as needed. He had a history of human immunodeficiency virus–negative, idiopathic CD4 depletion syndrome, which had been complicated by cryptococcal meningitis 7 years prior to presentation. He also had renal insufficiency, with baseline creatinine levels ranging from 1.4 to 1.7 mg/dL (reference range, 0.6–1.2 mg/dL). There was no personal or family history of known or suspected inflammatory bowel disease.
On physical examination, numerous discrete, coalescing, punched out–appearing ulcerations with foul-smelling, greenish yellow, purulent drainage were present bilaterally on the legs (Figure 1). The ulcers had a rolled border with a moderate amount of seemingly nonviable necrotic tissue. A number of hyperpigmented round papules, patches, and plaques also were present on the proximal legs. Laboratory evaluation revealed a CD4 count of 151 cc/mm3 (reference range, 500–1600 cc/mm3) and mildly elevated calcium of 10.7 mg/dL (reference range, 8.2–10.2 mg/dL).
Aerobic, anaerobic, mycobacterial, and fungal cultures of the purulent exudate were obtained. Given a high suspicion for secondary infection of the exogenous wound sites, doxycycline (100 mg twice daily) and topical mupir-ocin were initiated. Gram stain revealed few to moderate polymorphonuclear cells and many gram-positive cocci in pairs, chains, and clusters, along with many gram-negative rods. Bacterial culture grew Pseudomonas aeruginosa, Enterococcus species group G streptococci, and methicillin-resistant Staphylococcus aureus–positive staphylococci. Ciprofloxacin (500 mg twice daily) was then initiated, but the ulcers showed absolutely no clinical improvement and in fact worsened both in number and depth (Figure 2) over subsequent clinic visits during the next 3 months, even after amoxicillin (500 mg 3 times daily) was added. The patient was admitted for treatment with intravenous antibiotics after additional wound cultures revealed fluoroquinolone-resistant Pseudomonas.
Punch biopsies of the ulcers showed nonspecific acute inflammation and tissue necrosis in the active ulcers with nonnecrotizing granulomatous inflammation extending into the deep dermis, with many Langerhans-type giant cells present in the palpable ulcer borders (Figure 3). Neither birefringent particles nor asteroid bodies were observed. Tissue Gram stains did not reveal evidence of bacterial infection. Special stains for acid-fast and fungal organisms (ie, periodic acid–Schiff, Gomori methenamine-silver, Fite, acid-fast bacilli) were similarly negative. Tissue cultures obtained on deep biopsy revealed only rare colonies of P aeruginosa and no isolates on anaerobic, mycobacterial, or fungal cultures. Polymerase chain reaction for mycobacteria and common endemic fungi also was negative. In the absence of infection and considering his history of known sarcoidosis, these histologic features were consistent with ulcerative sarcoidosis. The patient was started on prednisone (60 mg once daily) and hydroxychloroquine (200 mg twice daily). The prednisone was tapered to 20 mg once daily over a 2-year period, at which point 90% of the ulcers had healed. He was continued on hydroxychloroquine at the initial dose, and at a 3-year follow-up his ulcers had healed completely without relapse.
Comment
Ulcerative sarcoidosis is rare, seen worldwide in only 5% of patients with cutaneous sarcoidosis.33 However, cases have been encountered worldwide, with reports emanating from Japan, China, Germany, France, and Russia, among others.6,34-55 We reviewed 34 cases from the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid and examined patient demographics, clinical presentation, histological findings, treatment type, and outcome. Key references are presented in the Table. Disease prevalence previously has been estimated as being 3-times more common in women than men1; in our literature review, we found a female to male ratio of 3.25 to 1. Additionally, ulcerative sarcoidosis is reported to be twice as common in black versus white individuals.33 In our literature review, when race was reported, 66% (21/32) of patients were black. Disease prevalence has been reported to peak at 20 to 40 years of age.3 In this review, the average age of presentation was 45 years (age range, 24–79 years).
Ulceration may arise de novo but more commonly arises in preexisting scars or cutaneous lesions. There are 2 distinct patterns seen in ulcerative sarcoidosis.4 The first is characterized by ulceration within necrotic yellow plaques.2 The second pattern is characterized by violaceous nodules arising in an annular confluent pattern that eventually ulcerate.4 This presentation commonly mimics or may be mimicked by multiple disease states, including sporotrichosis, tuberculosis, stasis dermatitis with venous ulceration, and even metastatic breast cancer.7,46,55,56 Regardless of presentation, the legs are the most common location of ulcer formation.1,33 In our review, 85% (29/34) of cases presented with involvement of the legs, including our own case. Other locations of ulcer formation have included the face, arms, trunk, and genital area.
On histologic examination of ulcerative sarcoidosis, epithelioid granulomas composed of multinucleated giant cells, histiocytes, and scant numbers of lymphocytes are present.1,3 These formations are the noncaseating granulomas typical of sarcoidosis (Table). All of the cases in our review of the literature were described as either a collection of epithelioid granulomas with giant cell formation or noncaseating granulomas. There also have been reports of atypical features including necrotizing granulomas and granulomatous vasculitis.4,8,9,50 The histologic differential diagnosis in this case also would primarily include an infectious granulomatous process and less so an id reaction, rosacea, a paraneoplastic phenomenon, foreign body granulomas, and metastatic Crohn disease. The presence of ulceration, the large number of lesions, and the anatomic distribution help rule out most of these alternate diagnostic considerations. Diligent extensive workup was done in our patient to insure it was not an infection.
The goals of treatment include symptomatic relief, improvement in objective parameters of disease activity, and prevention of disease progression and subsequent disability.33,57 Fortunately, the majority of sarcoidosis patients with cutaneous symptoms achieve full recovery within months to years.33 Our literature review indicated that 81% (22/27) of patients with ulcerative lesions experienced full resolution within 1 year of treatment. Of those that did not (19% [5/27]), the patients were either lost to follow-up or died from other complications of sarcoidosis.
The widely accepted standard therapy for cutaneous sarcoidosis includes topical, intralesional, and systemic corticosteroids; antimalarials; and methotrexate.33,57 Steroids and methotrexate act by suppressing granuloma formation, while antimalarials prevent antigen presentation (presumably part of the pathogenesis).33 For mild to moderate disease, topical and intralesional steroids may be all that is necessary.33,57 Systemic steroids are used for disfiguring, destructive, and widespread lesions that have been refractory to local and other systemic therapies.33,57 Steroids are tapered gradually depending on the patient’s response, as it is common for patients to relapse below a certain dose.33,57 Antimalarials (chloroquine or hydroxychloroquine) and methotrexate are considered adjunct treatments for patients who are either steroid unresponsive or who are unable to tolerate corticosteroid treatment due to adverse events.33,57
Standard therapy is complicated by the side effects of treatment. Use of corticosteroids may lead to gastrointestinal tract upset, increased appetite, mood disturbances, impaired wound healing, hyperglycemia, hypertension, cushingoid features, and acne.57 Antimalarials can cause nausea, anorexia, and agranulocytosis, and chloroquine therapy in particular can lead to blurred vision, corneal deposits, and central retinopathy.33,57 Methotrexate is associated with hematologic, gastrointestinal tract, pulmonary, and hepatic toxicities well known to most practitioners.
Because of the variable clinical response of patients to standard therapy and their associated toxicities, other treatment options have been used including pentoxifylline, tetracyclines, isotretinoin, leflunomide, thalidomide, infliximab, adalimumab, allopurinol, and the pulsed dye or CO2 laser.10,33,57 In nonhealing ulcers, split-thickness grafting and a bilayered bioengineered skin substitute have been used with good results in conjunction with ongoing systemic therapy.11,47 Additionally, nanoparticle silver burn paste has been used successfully, with resolution of ulcers within 2 weeks in the Chinese literature.53
All of these treatment recommendations are based on historically accepted modalities. Controlled trials with longitudinal follow-up are needed to provide justification for the current standard of care.34
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that primarily affects the lungs and lymphatic system but also may involve the skin, eyes, liver, spleen, muscles, bones, and nervous system.1 Cutaneous symptoms of sarcoidosis occur in approximately 25% of patients and are classified as specific and nonspecific, with specific lesions demonstrating noncaseating granuloma formation, which is typical of sarcoidosis.2 Nonspecific lesions primarily include erythema nodosum and calcinosis cutis. Specific lesions commonly present as reddish brown infiltrated plaques that may be annular, polycyclic, or serpiginous.1,3 They also may appear as yellowish brown or violaceous maculopapular lesions. However, specific lesions may present in a wide variety of morphologies, most often papules, nodules, subcutaneous infiltrates, and lupus pernio.4 Additionally, atypical cutaneous manifestations of sarcoidosis include erythroderma; scarring alopecia; nail dystrophy; and verrucous, ichthyosiform, psoriasiform, hypopigmented, or ulcerative skin lesions.3-5 Among these many potential clinical presentations, ulcerative sarcoidosis is quite uncommon.
We report a case of a patient who presented with classic clinical and histopathological findings of ulcerative sarcoidosis to highlight the prototypical presentation of a rare condition. We also review 34 additional cases of ulcerative sarcoidosis published in the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid.4-32 Analyzing this historical information, the scope of this unusual form of cutaneous sarcoidosis can be better understood, recognized, and treated. Although current standard-of-care treatments are most often successful, there is a paucity of definitive clinical trials to justify and verify comparative therapeutic efficacy.
Case Report
A 49-year-old black man with known pulmonary sarcoidosis, idiopathic (human immunodeficiency virus–negative) CD4 depletion syndrome, and chronic kidney disease presented with persistent bilateral ulcers of the legs of 1 month’s duration. The lesions first appeared as multiple “dark spots” on the legs. After the patient applied homemade aloe vera extract under occlusion for 1 to 2 days, the lesions became painful and began to ulcerate approximately 3 months prior to presentation. The patient applied a combination of a topical first aid antibiotic ointment, Epsom salts, and hydrogen peroxide without any improvement. A current review of systems was negative.
The patient’s medical history was notable for sarcoidosis diagnosed more than 10 years prior. During this time, he had intermittently been treated elsewhere with low-dose oral prednisone (5 mg once daily), hydroxychloroquine (200 mg twice daily), and an inhaled steroid as needed. He had a history of human immunodeficiency virus–negative, idiopathic CD4 depletion syndrome, which had been complicated by cryptococcal meningitis 7 years prior to presentation. He also had renal insufficiency, with baseline creatinine levels ranging from 1.4 to 1.7 mg/dL (reference range, 0.6–1.2 mg/dL). There was no personal or family history of known or suspected inflammatory bowel disease.
On physical examination, numerous discrete, coalescing, punched out–appearing ulcerations with foul-smelling, greenish yellow, purulent drainage were present bilaterally on the legs (Figure 1). The ulcers had a rolled border with a moderate amount of seemingly nonviable necrotic tissue. A number of hyperpigmented round papules, patches, and plaques also were present on the proximal legs. Laboratory evaluation revealed a CD4 count of 151 cc/mm3 (reference range, 500–1600 cc/mm3) and mildly elevated calcium of 10.7 mg/dL (reference range, 8.2–10.2 mg/dL).
Aerobic, anaerobic, mycobacterial, and fungal cultures of the purulent exudate were obtained. Given a high suspicion for secondary infection of the exogenous wound sites, doxycycline (100 mg twice daily) and topical mupir-ocin were initiated. Gram stain revealed few to moderate polymorphonuclear cells and many gram-positive cocci in pairs, chains, and clusters, along with many gram-negative rods. Bacterial culture grew Pseudomonas aeruginosa, Enterococcus species group G streptococci, and methicillin-resistant Staphylococcus aureus–positive staphylococci. Ciprofloxacin (500 mg twice daily) was then initiated, but the ulcers showed absolutely no clinical improvement and in fact worsened both in number and depth (Figure 2) over subsequent clinic visits during the next 3 months, even after amoxicillin (500 mg 3 times daily) was added. The patient was admitted for treatment with intravenous antibiotics after additional wound cultures revealed fluoroquinolone-resistant Pseudomonas.
Punch biopsies of the ulcers showed nonspecific acute inflammation and tissue necrosis in the active ulcers with nonnecrotizing granulomatous inflammation extending into the deep dermis, with many Langerhans-type giant cells present in the palpable ulcer borders (Figure 3). Neither birefringent particles nor asteroid bodies were observed. Tissue Gram stains did not reveal evidence of bacterial infection. Special stains for acid-fast and fungal organisms (ie, periodic acid–Schiff, Gomori methenamine-silver, Fite, acid-fast bacilli) were similarly negative. Tissue cultures obtained on deep biopsy revealed only rare colonies of P aeruginosa and no isolates on anaerobic, mycobacterial, or fungal cultures. Polymerase chain reaction for mycobacteria and common endemic fungi also was negative. In the absence of infection and considering his history of known sarcoidosis, these histologic features were consistent with ulcerative sarcoidosis. The patient was started on prednisone (60 mg once daily) and hydroxychloroquine (200 mg twice daily). The prednisone was tapered to 20 mg once daily over a 2-year period, at which point 90% of the ulcers had healed. He was continued on hydroxychloroquine at the initial dose, and at a 3-year follow-up his ulcers had healed completely without relapse.
Comment
Ulcerative sarcoidosis is rare, seen worldwide in only 5% of patients with cutaneous sarcoidosis.33 However, cases have been encountered worldwide, with reports emanating from Japan, China, Germany, France, and Russia, among others.6,34-55 We reviewed 34 cases from the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid and examined patient demographics, clinical presentation, histological findings, treatment type, and outcome. Key references are presented in the Table. Disease prevalence previously has been estimated as being 3-times more common in women than men1; in our literature review, we found a female to male ratio of 3.25 to 1. Additionally, ulcerative sarcoidosis is reported to be twice as common in black versus white individuals.33 In our literature review, when race was reported, 66% (21/32) of patients were black. Disease prevalence has been reported to peak at 20 to 40 years of age.3 In this review, the average age of presentation was 45 years (age range, 24–79 years).
Ulceration may arise de novo but more commonly arises in preexisting scars or cutaneous lesions. There are 2 distinct patterns seen in ulcerative sarcoidosis.4 The first is characterized by ulceration within necrotic yellow plaques.2 The second pattern is characterized by violaceous nodules arising in an annular confluent pattern that eventually ulcerate.4 This presentation commonly mimics or may be mimicked by multiple disease states, including sporotrichosis, tuberculosis, stasis dermatitis with venous ulceration, and even metastatic breast cancer.7,46,55,56 Regardless of presentation, the legs are the most common location of ulcer formation.1,33 In our review, 85% (29/34) of cases presented with involvement of the legs, including our own case. Other locations of ulcer formation have included the face, arms, trunk, and genital area.
On histologic examination of ulcerative sarcoidosis, epithelioid granulomas composed of multinucleated giant cells, histiocytes, and scant numbers of lymphocytes are present.1,3 These formations are the noncaseating granulomas typical of sarcoidosis (Table). All of the cases in our review of the literature were described as either a collection of epithelioid granulomas with giant cell formation or noncaseating granulomas. There also have been reports of atypical features including necrotizing granulomas and granulomatous vasculitis.4,8,9,50 The histologic differential diagnosis in this case also would primarily include an infectious granulomatous process and less so an id reaction, rosacea, a paraneoplastic phenomenon, foreign body granulomas, and metastatic Crohn disease. The presence of ulceration, the large number of lesions, and the anatomic distribution help rule out most of these alternate diagnostic considerations. Diligent extensive workup was done in our patient to insure it was not an infection.
The goals of treatment include symptomatic relief, improvement in objective parameters of disease activity, and prevention of disease progression and subsequent disability.33,57 Fortunately, the majority of sarcoidosis patients with cutaneous symptoms achieve full recovery within months to years.33 Our literature review indicated that 81% (22/27) of patients with ulcerative lesions experienced full resolution within 1 year of treatment. Of those that did not (19% [5/27]), the patients were either lost to follow-up or died from other complications of sarcoidosis.
The widely accepted standard therapy for cutaneous sarcoidosis includes topical, intralesional, and systemic corticosteroids; antimalarials; and methotrexate.33,57 Steroids and methotrexate act by suppressing granuloma formation, while antimalarials prevent antigen presentation (presumably part of the pathogenesis).33 For mild to moderate disease, topical and intralesional steroids may be all that is necessary.33,57 Systemic steroids are used for disfiguring, destructive, and widespread lesions that have been refractory to local and other systemic therapies.33,57 Steroids are tapered gradually depending on the patient’s response, as it is common for patients to relapse below a certain dose.33,57 Antimalarials (chloroquine or hydroxychloroquine) and methotrexate are considered adjunct treatments for patients who are either steroid unresponsive or who are unable to tolerate corticosteroid treatment due to adverse events.33,57
Standard therapy is complicated by the side effects of treatment. Use of corticosteroids may lead to gastrointestinal tract upset, increased appetite, mood disturbances, impaired wound healing, hyperglycemia, hypertension, cushingoid features, and acne.57 Antimalarials can cause nausea, anorexia, and agranulocytosis, and chloroquine therapy in particular can lead to blurred vision, corneal deposits, and central retinopathy.33,57 Methotrexate is associated with hematologic, gastrointestinal tract, pulmonary, and hepatic toxicities well known to most practitioners.
Because of the variable clinical response of patients to standard therapy and their associated toxicities, other treatment options have been used including pentoxifylline, tetracyclines, isotretinoin, leflunomide, thalidomide, infliximab, adalimumab, allopurinol, and the pulsed dye or CO2 laser.10,33,57 In nonhealing ulcers, split-thickness grafting and a bilayered bioengineered skin substitute have been used with good results in conjunction with ongoing systemic therapy.11,47 Additionally, nanoparticle silver burn paste has been used successfully, with resolution of ulcers within 2 weeks in the Chinese literature.53
All of these treatment recommendations are based on historically accepted modalities. Controlled trials with longitudinal follow-up are needed to provide justification for the current standard of care.34
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1421-1435.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361-1383.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Noiles K, Beleznay K, Crawford RI, et al. Sarcoidosis can present with necrotizing granulomas histologically: two cases of ulcerated sarcoidosis and review of the literature. J Cutan Med Surg. 2013;17:377-378.
- Mitchell IC, Sweatman MC, Rustin MH, et al. Ulcerative and hypopigmented sarcoidosis. J Am Acad Dermatol. 1986;15:1062-1065.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Joshi SS, Romanelli R, Kirsner RS. Sarcoidosis mimicking a venous ulcer: a case report. Ostomy Wound Manage. 2009;55:46-48.
- Petri M, Barr E, Cho K, et al. Overlap of granulomatous vasculitis and sarcoidosis: presentation with uveitis, eosinophilia, leg ulcers, sinusitis and past foot drop. J Rheumatol. 1988;15:1171-1173.
- Poonawalla T, Colome-Grimmer MI, Kelly B. Ulcerative sarcoidosis in the legs with granulomatous vasculitis. Clin Exp Dermatol. 2008;33:282-286.
- Philips MA, Lynch J, Azmi FH. Ulcerative sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917.
- Collison DW, Novice F, Banse L, et al. Split-thickness skin grafting in extensive ulcerative sarcoidosis. J Dermatol Surg Oncol. 1989;15:679-683.
- Hunt RD, Gonzalez ME, Robinson M, et al. Ulcerative sarcoidosis. Dermatol Online J. 2012;18:29.
- Green JJ, Lawrence N, Heymann WR. Generalized ulcerative sarcoidosis induced by therapy with the flashlamp-pumped pulsed dye. Arch Dermatol. 2001;137:507-508.
- Albertini JG, Tyler W, Miller OF. Ulcerative sarcoidosis. case report and review of the literature. Arch Dermatol. 1997;133:215-219.
- Thomas J, Williams DW. Peritoneal involvement and ulcerative skin plaques in sarcoidosis: a case report. Sarcoidosis. 1989;6:161-162.
- Verdegem TD, Sharma OP. Cutaneous ulcers in sarcoidosis. Arch Dermatol. 1987;123:1531-1534.
- Gupta AK, Haberman HF, From GL, et al. Sarcoidosis with extensive cutaneous ulceration. unusual clinical presentation. Dermatologica. 1987;174:135-139.
- Hruza GJ, Kerdel FA. Generalized atrophic sarcoidosis with ulcerations. Arch Dermatol. 1986;122:320-322.
- Muhlemann MF, Walker NP, Tan LB, et al. Elephantine sarcoidosis presenting as ulcerating lymphoedema. J R Soc Med. 1985;78:260-261.
- Neill SM, Smith NP, Eady RA. Ulcerative sarcoidosis: a rare manifestation of a common disease. Clin Exp Dermatol. 1984;9:277-279.
- Saxe N, Benatar SR, Bok L, et al. Sarcoidosis with leg ulcers and annular facial lesions. Arch Dermatol. 1984;120:93-96.
- Schwartz RA, Robertson DB, Tierney LM, et al. Generalized ulcerative sarcoidosis. Arch Dermatol. 1982;118:931-933.
- Boyd RE, Andrews BS. Sarcoidosis presenting as cutaneous ulceration, subcutaneous nodules and chronic arthritis. J Rheumatol. 1981;8:311-316.
- Herzlinger DC, Marland AM, Barr RJ. Verrucous ulcerative skin lesions in sarcoidosis. an unusual clinical presentation. Cutis. 1979;23:569-572.
- Meyers M, Barsky S. Ulcerative sarcoidosis. Arch Dermatol. 1978;114:447.
- Schiffner J, Sharma OP. Ulcerative sarcoidosis. report of an unusual case. Arch Dermatol. 1977;113:676-677.
- Williamson DM. Sarcoidosis with atrophic lesions and ulcers of the legs. Br J Dermatol. 1971;84:92-93.
- Bazex A, Dupre A, Christol B, et al. Sarcoidosis with atrophic lesions and ulcers and the presence in some sarcoid granulomata of orceinophil fibres. Br J Dermatol. 1970;83:255-262.
- Brodkin RH. Leg ulcers. a report of two cases caused by sarcoidosis. Acta Derm Venereol. 1969;49:584-587.
- Simpson JR. Sarcoidosis with erythrodermia and ulceration. Br J Dermatol. 1963;75:193-198.
- Irgang S. Ulcerative cutaneous lesion in sarcoidosis; report of a case with clinical resemblance to lupus vulgaris. Harlem Hosp Bull. 1956;8:134-139.
- Irgang S. Ulcerative cutaneous lesions in sarcoidosis; report of a case with clinical resemblance to papulonecrotic tuberculide. Br J Dermatol. 1955;67:255-260.
- Hoffman MD. Atypical ulcers. Dermatol Ther. 2013;26:222-235.
- Hopf B, Krebs A. Ulcera cruris as a rare manifestation of sarcoidosis. Dermatologica. 1974;113:55-62.
- Metz J, Hartmann A, Hautkr Z. Ulcerative form of skin sarcoidosis. Z Hautkr. 1977;52:890-896.
- Berenbeĭn BA, Malygina LA, Tiutiunnikova IA. Ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1984;4:50-53.
- Takahashi N, Hoshino M, Takase T, et al. A case of ulcerative sarcoidosis [in Japanese]. Nihon Hifuka Gakkai Zasshi. 1985;95:1049-1054.
- Schamroth JM. Sarcoidosis with severe extensive skin ulceration. Int J Dermatol. 1985;24:451-452.
- Porteau L, Dromer C, Le Guennec P, et al. Ulcer lesions in sarcoidosis: apropos of a case [in French]. Ann Med Interne (Paris). 1997;148:105-106.
- de La Blanchardière A, Bachmeyer C, Toutous L, et al. Cutaneous ulcerations in sarcoidosis [in French]. Rev Med Interne. 1995;16:927-929.
- Mitsuishi T, Nogita T, Kawashima M. Psoriasiform sarcoidosis with ulceration. Int J Dermatol. 1992;31:339-340.
- Rodionov AN, Samtsov AV. The ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1990;7:68-71.
- Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol. 1999;38:841-845.
- Gungor E, Artuz F, Alli N, et al. Ulcerative sarcoidosis. J Eur Acad Dermatol Venereol. 1999;12:78-79.
- Schleinitz N, Luc M, Genot S, et al. Ulcerative cutaneous lesions: a rare manifestation of sarcoidosis [in French]. Rev Med Interne. 2005;26:758-759.
- Klocker J, Duckers J, Morse R, et al. Ulcerative cutaneous sarcoidosis masquerading as metastatic carcinoma of the breast. Age Ageing. 2002;31:77-79.
- Streit M, Bohlen LM, Braathen LR. Ulcerative sarcoidosis successfully treated with apligraf. Dermatology. 2001;202:367-370.
- Ichiki Y, Kitajima Y. Ulcerative sarcoidosis: case report and review of the Japanese literature. Acta Derm Venereol. 2008;88:526-528
- Meyersburg D, Schön MP, Bertsch HP, et al. Uncommon cutaneous ulcerative and systemic sarcoidosis. successful treatment with hydroxychloroquine and compression therapy [in German]. Hautarzt. 2011;62:691-695.
- Wei CH, Huang YH, Shih YC, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Kluger N, Girard C, Durand L, et al. Leg ulcers revealing systemic sarcoidosis with splenomegaly and thrombocytopenia. Int J Dermatol. 2013;52:1425-1427.
- Jun L, Jia-Wei L, Hong-Zhong J. Ulcerative sarcoidosis. Int J Dermatol. 2014;53:E315-E316.
- Chen JH, Wang TT, Lin ZQ. Successful application of a novel dressing for the treatment of ulcerative cutaneous sarcoidosis. Chin Med J. 2013;126:3400.
- Ri G, Yoshikawa E, Shigekiyo T, et al. Takayasu artertitis and ulcerative sarcoidosis. Intern Med. 2015;54:1075-1080.
- Spiliopoulou I, Foka A, Bounas A, et al. Mycobacterium kansasii cutaneous infection in a patient with sarcoidosis treated with anti-TNF agents. Acta Clin Belg. 2014;69:229-231.
- Yang DJ, Krishnan RS, Guillen DR, et al. Disseminated sporotrichosis mimicking sarcoidosis. Int J Dermatol. 2006;45:450-453.
- Badgwell C, Rosen T. Cutaneous sarcoidosis therapy updated. J Am Acad Dermatol. 2007;56:69-83.
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1421-1435.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361-1383.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Noiles K, Beleznay K, Crawford RI, et al. Sarcoidosis can present with necrotizing granulomas histologically: two cases of ulcerated sarcoidosis and review of the literature. J Cutan Med Surg. 2013;17:377-378.
- Mitchell IC, Sweatman MC, Rustin MH, et al. Ulcerative and hypopigmented sarcoidosis. J Am Acad Dermatol. 1986;15:1062-1065.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Joshi SS, Romanelli R, Kirsner RS. Sarcoidosis mimicking a venous ulcer: a case report. Ostomy Wound Manage. 2009;55:46-48.
- Petri M, Barr E, Cho K, et al. Overlap of granulomatous vasculitis and sarcoidosis: presentation with uveitis, eosinophilia, leg ulcers, sinusitis and past foot drop. J Rheumatol. 1988;15:1171-1173.
- Poonawalla T, Colome-Grimmer MI, Kelly B. Ulcerative sarcoidosis in the legs with granulomatous vasculitis. Clin Exp Dermatol. 2008;33:282-286.
- Philips MA, Lynch J, Azmi FH. Ulcerative sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917.
- Collison DW, Novice F, Banse L, et al. Split-thickness skin grafting in extensive ulcerative sarcoidosis. J Dermatol Surg Oncol. 1989;15:679-683.
- Hunt RD, Gonzalez ME, Robinson M, et al. Ulcerative sarcoidosis. Dermatol Online J. 2012;18:29.
- Green JJ, Lawrence N, Heymann WR. Generalized ulcerative sarcoidosis induced by therapy with the flashlamp-pumped pulsed dye. Arch Dermatol. 2001;137:507-508.
- Albertini JG, Tyler W, Miller OF. Ulcerative sarcoidosis. case report and review of the literature. Arch Dermatol. 1997;133:215-219.
- Thomas J, Williams DW. Peritoneal involvement and ulcerative skin plaques in sarcoidosis: a case report. Sarcoidosis. 1989;6:161-162.
- Verdegem TD, Sharma OP. Cutaneous ulcers in sarcoidosis. Arch Dermatol. 1987;123:1531-1534.
- Gupta AK, Haberman HF, From GL, et al. Sarcoidosis with extensive cutaneous ulceration. unusual clinical presentation. Dermatologica. 1987;174:135-139.
- Hruza GJ, Kerdel FA. Generalized atrophic sarcoidosis with ulcerations. Arch Dermatol. 1986;122:320-322.
- Muhlemann MF, Walker NP, Tan LB, et al. Elephantine sarcoidosis presenting as ulcerating lymphoedema. J R Soc Med. 1985;78:260-261.
- Neill SM, Smith NP, Eady RA. Ulcerative sarcoidosis: a rare manifestation of a common disease. Clin Exp Dermatol. 1984;9:277-279.
- Saxe N, Benatar SR, Bok L, et al. Sarcoidosis with leg ulcers and annular facial lesions. Arch Dermatol. 1984;120:93-96.
- Schwartz RA, Robertson DB, Tierney LM, et al. Generalized ulcerative sarcoidosis. Arch Dermatol. 1982;118:931-933.
- Boyd RE, Andrews BS. Sarcoidosis presenting as cutaneous ulceration, subcutaneous nodules and chronic arthritis. J Rheumatol. 1981;8:311-316.
- Herzlinger DC, Marland AM, Barr RJ. Verrucous ulcerative skin lesions in sarcoidosis. an unusual clinical presentation. Cutis. 1979;23:569-572.
- Meyers M, Barsky S. Ulcerative sarcoidosis. Arch Dermatol. 1978;114:447.
- Schiffner J, Sharma OP. Ulcerative sarcoidosis. report of an unusual case. Arch Dermatol. 1977;113:676-677.
- Williamson DM. Sarcoidosis with atrophic lesions and ulcers of the legs. Br J Dermatol. 1971;84:92-93.
- Bazex A, Dupre A, Christol B, et al. Sarcoidosis with atrophic lesions and ulcers and the presence in some sarcoid granulomata of orceinophil fibres. Br J Dermatol. 1970;83:255-262.
- Brodkin RH. Leg ulcers. a report of two cases caused by sarcoidosis. Acta Derm Venereol. 1969;49:584-587.
- Simpson JR. Sarcoidosis with erythrodermia and ulceration. Br J Dermatol. 1963;75:193-198.
- Irgang S. Ulcerative cutaneous lesion in sarcoidosis; report of a case with clinical resemblance to lupus vulgaris. Harlem Hosp Bull. 1956;8:134-139.
- Irgang S. Ulcerative cutaneous lesions in sarcoidosis; report of a case with clinical resemblance to papulonecrotic tuberculide. Br J Dermatol. 1955;67:255-260.
- Hoffman MD. Atypical ulcers. Dermatol Ther. 2013;26:222-235.
- Hopf B, Krebs A. Ulcera cruris as a rare manifestation of sarcoidosis. Dermatologica. 1974;113:55-62.
- Metz J, Hartmann A, Hautkr Z. Ulcerative form of skin sarcoidosis. Z Hautkr. 1977;52:890-896.
- Berenbeĭn BA, Malygina LA, Tiutiunnikova IA. Ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1984;4:50-53.
- Takahashi N, Hoshino M, Takase T, et al. A case of ulcerative sarcoidosis [in Japanese]. Nihon Hifuka Gakkai Zasshi. 1985;95:1049-1054.
- Schamroth JM. Sarcoidosis with severe extensive skin ulceration. Int J Dermatol. 1985;24:451-452.
- Porteau L, Dromer C, Le Guennec P, et al. Ulcer lesions in sarcoidosis: apropos of a case [in French]. Ann Med Interne (Paris). 1997;148:105-106.
- de La Blanchardière A, Bachmeyer C, Toutous L, et al. Cutaneous ulcerations in sarcoidosis [in French]. Rev Med Interne. 1995;16:927-929.
- Mitsuishi T, Nogita T, Kawashima M. Psoriasiform sarcoidosis with ulceration. Int J Dermatol. 1992;31:339-340.
- Rodionov AN, Samtsov AV. The ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1990;7:68-71.
- Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol. 1999;38:841-845.
- Gungor E, Artuz F, Alli N, et al. Ulcerative sarcoidosis. J Eur Acad Dermatol Venereol. 1999;12:78-79.
- Schleinitz N, Luc M, Genot S, et al. Ulcerative cutaneous lesions: a rare manifestation of sarcoidosis [in French]. Rev Med Interne. 2005;26:758-759.
- Klocker J, Duckers J, Morse R, et al. Ulcerative cutaneous sarcoidosis masquerading as metastatic carcinoma of the breast. Age Ageing. 2002;31:77-79.
- Streit M, Bohlen LM, Braathen LR. Ulcerative sarcoidosis successfully treated with apligraf. Dermatology. 2001;202:367-370.
- Ichiki Y, Kitajima Y. Ulcerative sarcoidosis: case report and review of the Japanese literature. Acta Derm Venereol. 2008;88:526-528
- Meyersburg D, Schön MP, Bertsch HP, et al. Uncommon cutaneous ulcerative and systemic sarcoidosis. successful treatment with hydroxychloroquine and compression therapy [in German]. Hautarzt. 2011;62:691-695.
- Wei CH, Huang YH, Shih YC, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Kluger N, Girard C, Durand L, et al. Leg ulcers revealing systemic sarcoidosis with splenomegaly and thrombocytopenia. Int J Dermatol. 2013;52:1425-1427.
- Jun L, Jia-Wei L, Hong-Zhong J. Ulcerative sarcoidosis. Int J Dermatol. 2014;53:E315-E316.
- Chen JH, Wang TT, Lin ZQ. Successful application of a novel dressing for the treatment of ulcerative cutaneous sarcoidosis. Chin Med J. 2013;126:3400.
- Ri G, Yoshikawa E, Shigekiyo T, et al. Takayasu artertitis and ulcerative sarcoidosis. Intern Med. 2015;54:1075-1080.
- Spiliopoulou I, Foka A, Bounas A, et al. Mycobacterium kansasii cutaneous infection in a patient with sarcoidosis treated with anti-TNF agents. Acta Clin Belg. 2014;69:229-231.
- Yang DJ, Krishnan RS, Guillen DR, et al. Disseminated sporotrichosis mimicking sarcoidosis. Int J Dermatol. 2006;45:450-453.
- Badgwell C, Rosen T. Cutaneous sarcoidosis therapy updated. J Am Acad Dermatol. 2007;56:69-83.
Practice Points
- Sarcoidosis can present as a primary ulcerative disease.
- Suspect ulcerative sarcoidosis when ulcerations are seen on the leg.
- Systemic corticosteroids may be the most effective treatment of ulcerative sarcoidosis.
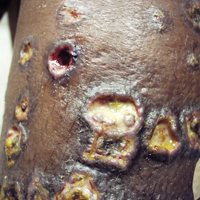
Atypical Herpes Zoster Presentation in a Healthy Vaccinated Pediatric Patient
Varicella-zoster virus (VZV) is a neurotropic human herpesvirus that causes varicella (chicken pox) and herpes zoster (shingles). During infection, the virus invades the dorsal root ganglia and establishes permanent latency. It can later reactivate and travel through sensory nerves to the skin where localized viral replication causes herpes zoster (HZ), which manifests with pain in a unilateral dermatomal distribution followed closely by an eruption of grouped macules and papules that evolve into vesicles on an erythematous base.1 These lesions form pustules and crusts over 7 to 10 days and heal completely within 4 weeks. Although postherpetic neuralgia is rare in children, the pain associated with HZ can last months or years.1,2
Universal childhood vaccination against VZV has existed in the United States since 1995, with a 2-dose vaccine regimen recommended by the CDC since 2007. Consequently, primary varicella infection in children is uncommon, and the majority of cases now occur in the vaccinated population.3 However, breakthrough varicella infection and postvaccination HZ are rare due to the long-lasting immunity and low virulence of the attenuated vaccine strain. We recount the case of a 6-year-old vaccinated girl with a unique presentation of HZ with no known primary varicella infection.
Case Report
A healthy 6-year-old girl presented with a stabbing burning pain in the left thigh extending down the calf of 4 days’ duration. The intense pain made walking difficult and responded minimally to ibuprofen and naproxen. Poor appetite, nausea, colicky abdominal pain, and fever (temperature, 38°C) accompanied the pain. Three days after the pain began she developed a pruritic rash on the same leg. Notably, she reported falling on a rosebush and sustaining a thorn prick in the left thigh 3 days prior to the onset of pain. Before presenting to our dermatology clinic, she was seen by a pediatrician, an emergency department physician, and an infectious disease specialist. The initial workup included a complete blood cell count, C-reactive protein test, erythrocyte sedimentation rate test, and hip and femur radiograph, which were all unremarkable. She was referred to dermatology with a differential diagnosis of sporotrichosis, contact dermatitis, reactive arthritis, viral myalgia, and Legg-Calvé-Perthes disease.
Physical examination revealed a well-appearing child with pink eczematous patches and plaques extending from the left side of the lower back to the mid shin in an L5 distribution (Figure). The left thigh was tender to palpation, and nontender left inguinal lymphadenopathy was present. A single isolated 2-mm vesicle was found on the anterior aspect of the left lower leg. Direct fluorescent antibody testing of vesicle fluid was positive for VZV antigen, confirming the diagnosis of HZ.
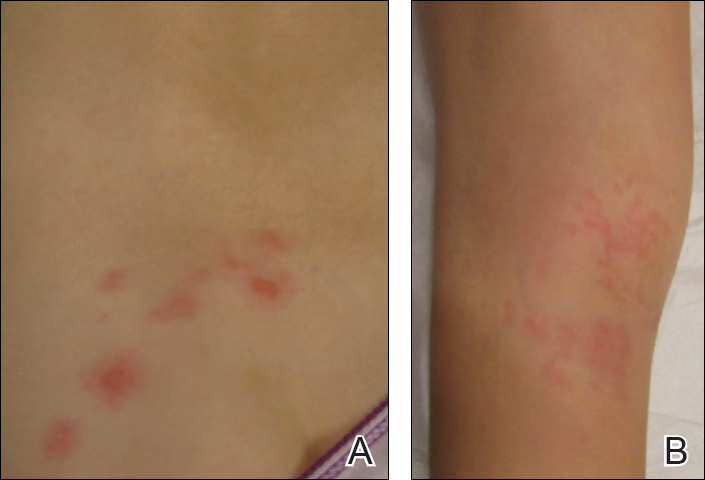
The patient’s mother confirmed that she had no obvious history of VZV. She had received VZV vaccinations in the left leg and arm at 1 and 4 years of age, respectively. She was treated with acyclovir (80 mg/kg daily at 6-hour intervals for 5 days) with immediate improvement in symptoms and resolution of the rash by day 5 of treatment. She experienced intermittent burning pain in the leg throughout the course of treatment, which resolved shortly thereafter.
Comment
Herpes zoster is rare in young healthy children, and its incidence has decreased since the introduction of universal varicella vaccination.4 Reported incidence rates in vaccinated children vary from approximately 15 to 93 per 100,000 person-years,5,6 and the reported relative risk is 0.08 to 0.36 in vaccinated compared to unvaccinated children.6,7 No correlations with gender, race, or ethnicity and postvaccination HZ have been observed.5,8 Reported intervals between vaccination and HZ presentation are as short as 3 months and as long as 11 years.9 Although HZ is uncommon in immunocompetent children, the diagnosis of HZ itself is not an indication for formal workup for an underlying immunodeficiency or malignancy.10
Both wild-type and vaccine-strain VZV establish latent infection and can cause HZ in vaccinated children. Direct fluorescent antibody testing or polymerase chain reaction of HZ lesions can be used to identify VZV. Genotyping can distinguish the wild-type versus the vaccine strain but is not required for clinical management.3 In previously vaccinated children with HZ, approximately half present with wild-type and half with vaccine-strain VZV. In approximately half of wild-type cases, prior clinical varicella infection also occurred.8
Regardless of virus strain, vaccinated children typically present with the characteristic painful, vesicular, dermatomal HZ rash.8,9 This presentation can be milder with less pain and fewer vesicles than with unvaccinated cases.6 When vaccine-strain HZ occurs, the rash often presents at or near the site of initial vaccination, which typically is the arm or thigh.3,4,6,9 The vaccine strain has lower virulence than the wild-type virus. Eight cases of vaccine-strain zoster severe enough to cause neurological complications such as meningitis or encephalitis have been reported in children, with 6 cases reported in healthy children.9,11-17 Antiviral drugs hasten the healing of the HZ rash and shorten the duration of associated pain.1
Although pediatric HZ is uncommon, all physicians should be aware of possible atypical presentations in healthy vaccinated children to appropriately and quickly manage treatment.
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84:274-280.
- Hillebrand K, Bricout H, Schulze-Rath R, et al. Incidence of herpes zoster and its complications in Germany, 2005-2009. J Infect. 2015;70:178-186.
- Lopez A, Schmid S, Bialek S. Varicella. In: Centers for Disease Control and Prevention. Manual for the Surveillance of Vaccine-Preventable Diseases. 5th ed. 2011:1-16.
- Tanuseputroa P, Zagorskia B, Chanc KJ, et al. Population-based incidence of herpes zoster after introduction of a publicly funded varicella vaccination program. Vaccine. 2011;29:8580- 8584.
- Wen SY, Liu WL. Epidemiology of pediatric herpes zoster after varicella infection: a population-based study. Pediatrics. 2015;135:565-571.
- Civen R, Chaves SS, Jumaan A, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
- Stein M, Cohen R, Bromberg M, et al. Herpes zoster in a partially vaccinated pediatric population in Central Israel. Pediatr Infect Dis J. 2012;31:906-909.
- Weinmann S, Chun C, Schmid DS, et al. Incidence and clinical characteristics of herpes zoster among children in the varicella vaccine era, 2005-2009. J Infect Dis. 2013;208:1859-1868.
- Horien C, Grose C. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin Pediatr Neurol. 2012;19:124-129.
- Petursson G, Helgason S, Gudmundsson S, et al. Herpes zoster in children and adolescents. Pediatr Infect Dis J. 1998;17:905-908.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus in an immunosuppressed child. J Infect Dis. 2003;188:954-959.
- Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995-2005. J Infect Dis. 2008;197(suppl 2):S170-S177.
- Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella zoster virus vaccine. J Infect Dis. 2008;198:1444-1447.
- Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792-795.
- Chouliaras G, Spoulou V, Quinlivan M, et al. Vaccine-associated herpes zoster ophthalmicus and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969-e972.
- Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316-323.
- Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266-268.
Varicella-zoster virus (VZV) is a neurotropic human herpesvirus that causes varicella (chicken pox) and herpes zoster (shingles). During infection, the virus invades the dorsal root ganglia and establishes permanent latency. It can later reactivate and travel through sensory nerves to the skin where localized viral replication causes herpes zoster (HZ), which manifests with pain in a unilateral dermatomal distribution followed closely by an eruption of grouped macules and papules that evolve into vesicles on an erythematous base.1 These lesions form pustules and crusts over 7 to 10 days and heal completely within 4 weeks. Although postherpetic neuralgia is rare in children, the pain associated with HZ can last months or years.1,2
Universal childhood vaccination against VZV has existed in the United States since 1995, with a 2-dose vaccine regimen recommended by the CDC since 2007. Consequently, primary varicella infection in children is uncommon, and the majority of cases now occur in the vaccinated population.3 However, breakthrough varicella infection and postvaccination HZ are rare due to the long-lasting immunity and low virulence of the attenuated vaccine strain. We recount the case of a 6-year-old vaccinated girl with a unique presentation of HZ with no known primary varicella infection.
Case Report
A healthy 6-year-old girl presented with a stabbing burning pain in the left thigh extending down the calf of 4 days’ duration. The intense pain made walking difficult and responded minimally to ibuprofen and naproxen. Poor appetite, nausea, colicky abdominal pain, and fever (temperature, 38°C) accompanied the pain. Three days after the pain began she developed a pruritic rash on the same leg. Notably, she reported falling on a rosebush and sustaining a thorn prick in the left thigh 3 days prior to the onset of pain. Before presenting to our dermatology clinic, she was seen by a pediatrician, an emergency department physician, and an infectious disease specialist. The initial workup included a complete blood cell count, C-reactive protein test, erythrocyte sedimentation rate test, and hip and femur radiograph, which were all unremarkable. She was referred to dermatology with a differential diagnosis of sporotrichosis, contact dermatitis, reactive arthritis, viral myalgia, and Legg-Calvé-Perthes disease.
Physical examination revealed a well-appearing child with pink eczematous patches and plaques extending from the left side of the lower back to the mid shin in an L5 distribution (Figure). The left thigh was tender to palpation, and nontender left inguinal lymphadenopathy was present. A single isolated 2-mm vesicle was found on the anterior aspect of the left lower leg. Direct fluorescent antibody testing of vesicle fluid was positive for VZV antigen, confirming the diagnosis of HZ.
The patient’s mother confirmed that she had no obvious history of VZV. She had received VZV vaccinations in the left leg and arm at 1 and 4 years of age, respectively. She was treated with acyclovir (80 mg/kg daily at 6-hour intervals for 5 days) with immediate improvement in symptoms and resolution of the rash by day 5 of treatment. She experienced intermittent burning pain in the leg throughout the course of treatment, which resolved shortly thereafter.
Comment
Herpes zoster is rare in young healthy children, and its incidence has decreased since the introduction of universal varicella vaccination.4 Reported incidence rates in vaccinated children vary from approximately 15 to 93 per 100,000 person-years,5,6 and the reported relative risk is 0.08 to 0.36 in vaccinated compared to unvaccinated children.6,7 No correlations with gender, race, or ethnicity and postvaccination HZ have been observed.5,8 Reported intervals between vaccination and HZ presentation are as short as 3 months and as long as 11 years.9 Although HZ is uncommon in immunocompetent children, the diagnosis of HZ itself is not an indication for formal workup for an underlying immunodeficiency or malignancy.10
Both wild-type and vaccine-strain VZV establish latent infection and can cause HZ in vaccinated children. Direct fluorescent antibody testing or polymerase chain reaction of HZ lesions can be used to identify VZV. Genotyping can distinguish the wild-type versus the vaccine strain but is not required for clinical management.3 In previously vaccinated children with HZ, approximately half present with wild-type and half with vaccine-strain VZV. In approximately half of wild-type cases, prior clinical varicella infection also occurred.8
Regardless of virus strain, vaccinated children typically present with the characteristic painful, vesicular, dermatomal HZ rash.8,9 This presentation can be milder with less pain and fewer vesicles than with unvaccinated cases.6 When vaccine-strain HZ occurs, the rash often presents at or near the site of initial vaccination, which typically is the arm or thigh.3,4,6,9 The vaccine strain has lower virulence than the wild-type virus. Eight cases of vaccine-strain zoster severe enough to cause neurological complications such as meningitis or encephalitis have been reported in children, with 6 cases reported in healthy children.9,11-17 Antiviral drugs hasten the healing of the HZ rash and shorten the duration of associated pain.1
Although pediatric HZ is uncommon, all physicians should be aware of possible atypical presentations in healthy vaccinated children to appropriately and quickly manage treatment.
Varicella-zoster virus (VZV) is a neurotropic human herpesvirus that causes varicella (chicken pox) and herpes zoster (shingles). During infection, the virus invades the dorsal root ganglia and establishes permanent latency. It can later reactivate and travel through sensory nerves to the skin where localized viral replication causes herpes zoster (HZ), which manifests with pain in a unilateral dermatomal distribution followed closely by an eruption of grouped macules and papules that evolve into vesicles on an erythematous base.1 These lesions form pustules and crusts over 7 to 10 days and heal completely within 4 weeks. Although postherpetic neuralgia is rare in children, the pain associated with HZ can last months or years.1,2
Universal childhood vaccination against VZV has existed in the United States since 1995, with a 2-dose vaccine regimen recommended by the CDC since 2007. Consequently, primary varicella infection in children is uncommon, and the majority of cases now occur in the vaccinated population.3 However, breakthrough varicella infection and postvaccination HZ are rare due to the long-lasting immunity and low virulence of the attenuated vaccine strain. We recount the case of a 6-year-old vaccinated girl with a unique presentation of HZ with no known primary varicella infection.
Case Report
A healthy 6-year-old girl presented with a stabbing burning pain in the left thigh extending down the calf of 4 days’ duration. The intense pain made walking difficult and responded minimally to ibuprofen and naproxen. Poor appetite, nausea, colicky abdominal pain, and fever (temperature, 38°C) accompanied the pain. Three days after the pain began she developed a pruritic rash on the same leg. Notably, she reported falling on a rosebush and sustaining a thorn prick in the left thigh 3 days prior to the onset of pain. Before presenting to our dermatology clinic, she was seen by a pediatrician, an emergency department physician, and an infectious disease specialist. The initial workup included a complete blood cell count, C-reactive protein test, erythrocyte sedimentation rate test, and hip and femur radiograph, which were all unremarkable. She was referred to dermatology with a differential diagnosis of sporotrichosis, contact dermatitis, reactive arthritis, viral myalgia, and Legg-Calvé-Perthes disease.
Physical examination revealed a well-appearing child with pink eczematous patches and plaques extending from the left side of the lower back to the mid shin in an L5 distribution (Figure). The left thigh was tender to palpation, and nontender left inguinal lymphadenopathy was present. A single isolated 2-mm vesicle was found on the anterior aspect of the left lower leg. Direct fluorescent antibody testing of vesicle fluid was positive for VZV antigen, confirming the diagnosis of HZ.
The patient’s mother confirmed that she had no obvious history of VZV. She had received VZV vaccinations in the left leg and arm at 1 and 4 years of age, respectively. She was treated with acyclovir (80 mg/kg daily at 6-hour intervals for 5 days) with immediate improvement in symptoms and resolution of the rash by day 5 of treatment. She experienced intermittent burning pain in the leg throughout the course of treatment, which resolved shortly thereafter.
Comment
Herpes zoster is rare in young healthy children, and its incidence has decreased since the introduction of universal varicella vaccination.4 Reported incidence rates in vaccinated children vary from approximately 15 to 93 per 100,000 person-years,5,6 and the reported relative risk is 0.08 to 0.36 in vaccinated compared to unvaccinated children.6,7 No correlations with gender, race, or ethnicity and postvaccination HZ have been observed.5,8 Reported intervals between vaccination and HZ presentation are as short as 3 months and as long as 11 years.9 Although HZ is uncommon in immunocompetent children, the diagnosis of HZ itself is not an indication for formal workup for an underlying immunodeficiency or malignancy.10
Both wild-type and vaccine-strain VZV establish latent infection and can cause HZ in vaccinated children. Direct fluorescent antibody testing or polymerase chain reaction of HZ lesions can be used to identify VZV. Genotyping can distinguish the wild-type versus the vaccine strain but is not required for clinical management.3 In previously vaccinated children with HZ, approximately half present with wild-type and half with vaccine-strain VZV. In approximately half of wild-type cases, prior clinical varicella infection also occurred.8
Regardless of virus strain, vaccinated children typically present with the characteristic painful, vesicular, dermatomal HZ rash.8,9 This presentation can be milder with less pain and fewer vesicles than with unvaccinated cases.6 When vaccine-strain HZ occurs, the rash often presents at or near the site of initial vaccination, which typically is the arm or thigh.3,4,6,9 The vaccine strain has lower virulence than the wild-type virus. Eight cases of vaccine-strain zoster severe enough to cause neurological complications such as meningitis or encephalitis have been reported in children, with 6 cases reported in healthy children.9,11-17 Antiviral drugs hasten the healing of the HZ rash and shorten the duration of associated pain.1
Although pediatric HZ is uncommon, all physicians should be aware of possible atypical presentations in healthy vaccinated children to appropriately and quickly manage treatment.
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84:274-280.
- Hillebrand K, Bricout H, Schulze-Rath R, et al. Incidence of herpes zoster and its complications in Germany, 2005-2009. J Infect. 2015;70:178-186.
- Lopez A, Schmid S, Bialek S. Varicella. In: Centers for Disease Control and Prevention. Manual for the Surveillance of Vaccine-Preventable Diseases. 5th ed. 2011:1-16.
- Tanuseputroa P, Zagorskia B, Chanc KJ, et al. Population-based incidence of herpes zoster after introduction of a publicly funded varicella vaccination program. Vaccine. 2011;29:8580- 8584.
- Wen SY, Liu WL. Epidemiology of pediatric herpes zoster after varicella infection: a population-based study. Pediatrics. 2015;135:565-571.
- Civen R, Chaves SS, Jumaan A, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
- Stein M, Cohen R, Bromberg M, et al. Herpes zoster in a partially vaccinated pediatric population in Central Israel. Pediatr Infect Dis J. 2012;31:906-909.
- Weinmann S, Chun C, Schmid DS, et al. Incidence and clinical characteristics of herpes zoster among children in the varicella vaccine era, 2005-2009. J Infect Dis. 2013;208:1859-1868.
- Horien C, Grose C. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin Pediatr Neurol. 2012;19:124-129.
- Petursson G, Helgason S, Gudmundsson S, et al. Herpes zoster in children and adolescents. Pediatr Infect Dis J. 1998;17:905-908.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus in an immunosuppressed child. J Infect Dis. 2003;188:954-959.
- Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995-2005. J Infect Dis. 2008;197(suppl 2):S170-S177.
- Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella zoster virus vaccine. J Infect Dis. 2008;198:1444-1447.
- Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792-795.
- Chouliaras G, Spoulou V, Quinlivan M, et al. Vaccine-associated herpes zoster ophthalmicus and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969-e972.
- Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316-323.
- Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266-268.
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84:274-280.
- Hillebrand K, Bricout H, Schulze-Rath R, et al. Incidence of herpes zoster and its complications in Germany, 2005-2009. J Infect. 2015;70:178-186.
- Lopez A, Schmid S, Bialek S. Varicella. In: Centers for Disease Control and Prevention. Manual for the Surveillance of Vaccine-Preventable Diseases. 5th ed. 2011:1-16.
- Tanuseputroa P, Zagorskia B, Chanc KJ, et al. Population-based incidence of herpes zoster after introduction of a publicly funded varicella vaccination program. Vaccine. 2011;29:8580- 8584.
- Wen SY, Liu WL. Epidemiology of pediatric herpes zoster after varicella infection: a population-based study. Pediatrics. 2015;135:565-571.
- Civen R, Chaves SS, Jumaan A, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
- Stein M, Cohen R, Bromberg M, et al. Herpes zoster in a partially vaccinated pediatric population in Central Israel. Pediatr Infect Dis J. 2012;31:906-909.
- Weinmann S, Chun C, Schmid DS, et al. Incidence and clinical characteristics of herpes zoster among children in the varicella vaccine era, 2005-2009. J Infect Dis. 2013;208:1859-1868.
- Horien C, Grose C. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin Pediatr Neurol. 2012;19:124-129.
- Petursson G, Helgason S, Gudmundsson S, et al. Herpes zoster in children and adolescents. Pediatr Infect Dis J. 1998;17:905-908.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus in an immunosuppressed child. J Infect Dis. 2003;188:954-959.
- Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995-2005. J Infect Dis. 2008;197(suppl 2):S170-S177.
- Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella zoster virus vaccine. J Infect Dis. 2008;198:1444-1447.
- Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792-795.
- Chouliaras G, Spoulou V, Quinlivan M, et al. Vaccine-associated herpes zoster ophthalmicus and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969-e972.
- Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316-323.
- Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266-268.
Practice Points
- Both wild-type and vaccine-strain varicella-zoster virus (VZV) can establish latency in dorsal root ganglia and can cause herpes zoster (HZ) in vaccinated children.
- When HZ due to a vaccine strain of VZV occurs, the rash often presents near the site of initial vaccination.
- Although most cases of HZ in vaccinated children present with a characteristic HZ rash, physicians should be aware of the possibility for atypical presentations.
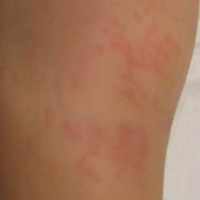
Hip pain • difficulty walking • tenderness along the anteromedial thigh and groin • Dx?
THE CASE
A 14-year-old Caucasian boy presented to our clinic with a complaint of left anterior hip pain. The patient had been running during a flag football match when he suddenly developed a sharp, stabbing pain in his left hip. He said he felt a “pop” in his left groin while his left foot was planted and he was cutting to the right. The patient said this was followed by worsening pain with ambulation and hip flexion.
The patient had considerable difficulty walking into the exam room. On physical examination, he had significant tenderness to palpation along the anteromedial thigh and groin. The patient’s strength was 1/5 with left hip flexion. There was apparent muscle firing, but no significant leg movement. He had full passive range of motion and there was no soft-tissue swelling, erythema, or other integumentary changes.
THE DIAGNOSIS
Plain radiographs revealed a lesser trochanter avulsion fracture with a 2-cm displacement (FIGURE 1).
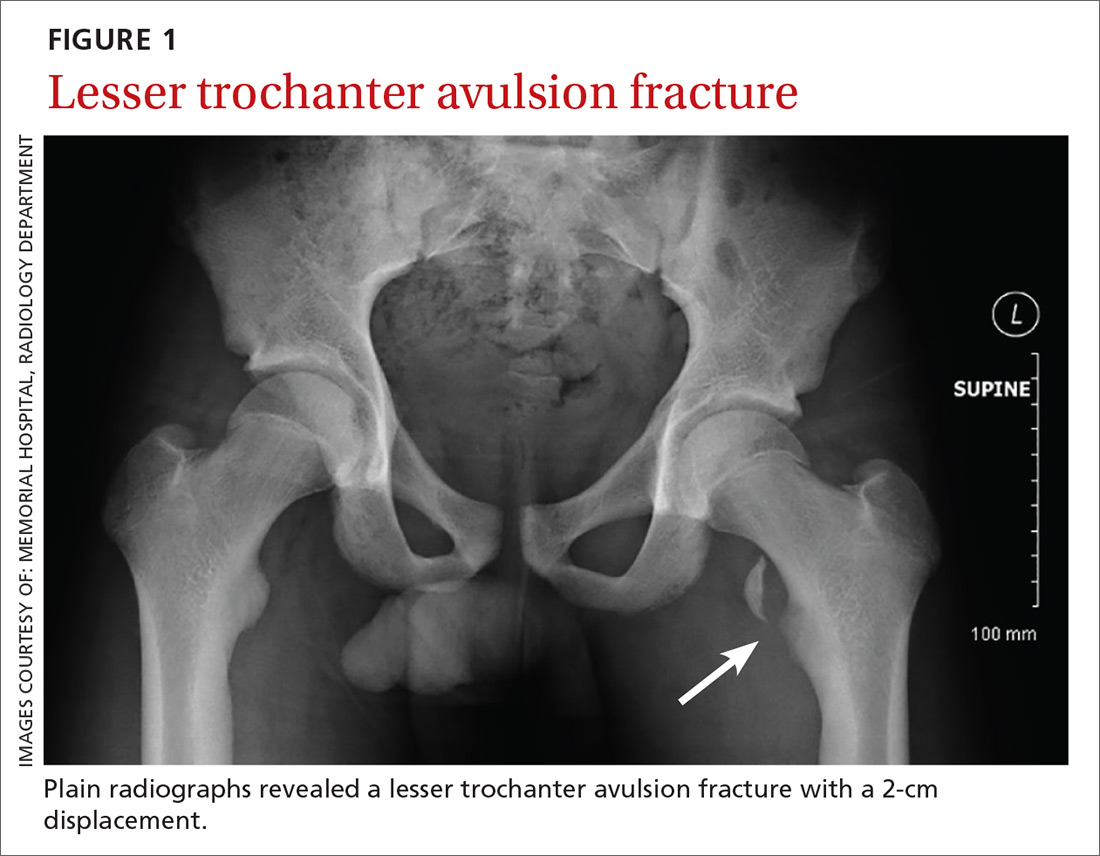
DISCUSSION
Pelvic and proximal femur avulsion fractures tend to occur during the second decade of life.1,2 They’re more frequently seen in boys and adolescent athletes, especially those involved in soccer and gymnastics.3,4
Anterior superior iliac spine (ASIS), ischial tuberosity (IT), and anterior inferior iliac spine (AIIS) avulsion fractures are more prevalent,4 while lesser trochanter avulsion fractures are more rare. In one review of 1126 children with femoral neck and proximal 1/3 femoral shaft fractures, only 3 of them had lesser trochanter avulsion fractures.5
Clinical presentation. Presenting symptoms of lesser trochanter avulsion fractures can be vague, but are usually localized to the groin and medial hip region. Patients will demonstrate pain and weakness with hip flexion.3,6 There may be signs of inflammation, tenderness, and ecchymosis near the site of injury.
On physical exam, a positive Ludloff sign helps localize the injury to the iliopsoas muscle, which inserts at the lesser trochanter and is involved in hip flexion.3,6,7 The Ludloff test is performed by flexing the patient’s hip while he/she is in a seated position.
BIOMECHANICS OF AVULSION FRACTURES
Perhaps surprisingly, the majority of avulsion injuries in children and adolescents are the result of non-contact athletic movement and indirect trauma.4 In children, muscles and tendons are often stronger than their bones,7 and physes—structurally weak regions—are particularly predisposed to fractures.2,4,6
The mechanism of injury in children and adolescents is commonly a sudden, forceful contraction of the iliopsoas muscle.6,7 While similar movement in adults will produce tendon sprains and muscle strains, children often experience a complete avulsion fracture.7 So uncommon are these fractures among adults that an adult patient presenting with one should receive further work-up for underlying pathology such as malignancy.8,9
While other hip and femur avulsion fractures in children and adolescents involve different muscle groups, the etiologic mechanism—forceful muscle contraction—is usually the same.2,4,7 IT injuries are often seen with sudden, aggressive lengthening of the hamstring muscles, whereas injuries to the ASIS and AIIS are the result of abrupt eccentric contraction of hip extensor muscles while the knee is flexed.4
DIFFERENTIAL DIAGNOSIS
There are several entities that can mimic a lesser trochanter avulsion fracture including Legg-Calve-Perthes disease (LCPD), slipped capital femoral epiphysis (SCFE), snapping hip with the iliofemoral ligament, iliopsoas tendonitis, referred pain from the gastrointestinal region, and a genito-urologic etiology.1,7,10
Diagnostic studies. Physical exam findings of severe pain and reduced strength are clear indications for obtaining baseline imaging. Baseline radiographs are key to the diagnosis of avulsion fractures. They help differentiate between more benign fractures, such as a nondisplaced avulsion fracture, and more substantial conditions, such as LCPD and SCFE, which require significantly different approaches to treatment and follow-up.1,7
Anteroposterior, oblique, and axial views of the pelvis all assist in assessing avulsion fractures radiographically.3,4,7 In the event that an avulsion fracture is not radiographically visible, but is still suspected, additional imaging should be obtained.10 A computerized tomography (CT) scan is an appropriate follow-up, given its meticulous detail of bony anatomy.3,10 Alternatively, if physes have yet to ossify or there are concerns about soft tissue injury, magnetic resonance imaging can be useful.3,7,10
MANAGEMENT
The majority of lesser trochanter avulsion fractures are managed conservatively with rest, nonsteroidal anti-inflammatory drugs (NSAIDs), and physical therapy. Patients are often placed on non-weight bearing activity for up to 6 weeks while the fracture repairs and forms a new union.7 Current management strategies have moved away from immobilization with splints and braces.
In rare instances when the fragment is displaced >2 cm, or there is inadequate healing or pain relief after 3 months of supportive care, surgery may be required.1 With appropriate diagnosis and medical care, the injured athlete should fully recover with no impairment or chronic pain.2
Our patient was placed on non-weight-bearing activity and treated with NSAIDs and acetaminophen. We advanced him to weight-bearing activities 4 weeks after injury. After 8 weeks of conservative management, he returned to competitive play with no further complications (FIGURE 2).
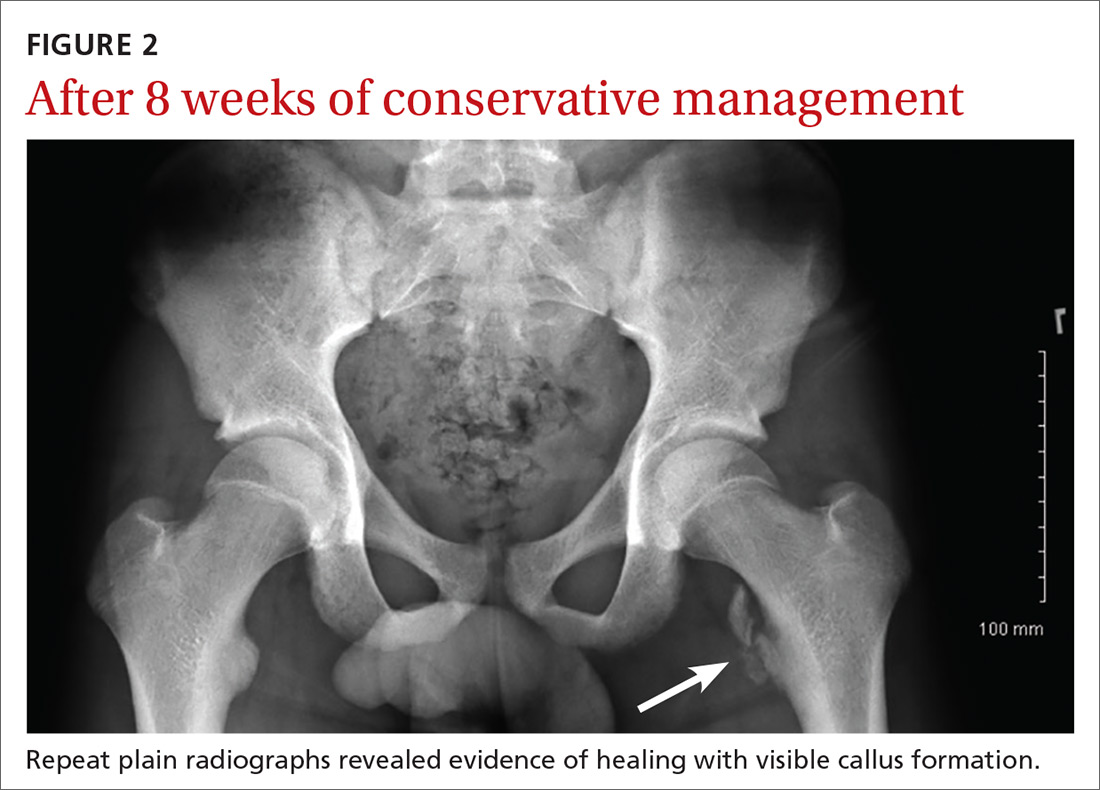
THE TAKEAWAY
Pelvic and proximal femur avulsion fractures occur more often in child and adolescent athletes. As this population becomes increasingly competitive in athletics, the risk of injury increases. Infrequent fractures such as lesser trochanter avulsion fractures may become more common, as well. The majority of avulsion fractures don’t require surgical intervention, but it’s important to obtain baseline radiographs to rule out other injuries or pathologies that may lead to poor prognoses if they are left untreated.
1. Byrne A, Reidy D. Acute groin pain in an adolescent sprinter: a case report. Int J Clin Pediatr. 2012;1:46-48.
2. Fernbach SK, Wilkinson RH. Avulsion injuries of the pelvis and proximal femur. AJR Am J Roentgenol. 1981;137:581-584.
3. McKinney BI, Nelson C, Carrion W. Apophyseal avulsion fractures of the hip and pelvis. Orthopedics. 2009;32:42.
4. Rossi F, Dragoni S. Acute avulsion fractures of the pelvis in adolescent competitive athletes: prevalence, location and sports distribution of 203 cases collected. Skeletal Radiol. 2001;30:127-131.
5. Theologis TN, Epps H, Latz K, et al. Isolated fractures of the lesser trochanter in children. Injury. 1997;28:363-364.
6. Paluska SA. An overview of hip injuries in running. Sports Med. 2005;35:991-1014.
7. Vazquez E, Kim TY, Young TP. Avulsion fracture of the lesser trochanter: an unusual cause of hip pain in an adolescent. CJEM. 2013;15:123-125.
8. Afra R, Boardman DL, Kabo JM, et al. Avulsion fracture of the lesser trochanter as a result of a preliminary malignant tumor of bone. A report of four cases. J Bone Joint Surg Am. 1999;81:1299-1304.
9. DePasse JM, Varner K, Cosculluela P, et al. Atraumatic avulsion of the distal iliopsoas tendon: an unusual cause of hip pain. Orthopedics. 2010;33.
10. Suarez JC, Ely EE, Mutnal AB, et al. Comprehensive approach to the evaluation of groin pain. J Am Acad Orthop Surg. 2013;21:558-570.
THE CASE
A 14-year-old Caucasian boy presented to our clinic with a complaint of left anterior hip pain. The patient had been running during a flag football match when he suddenly developed a sharp, stabbing pain in his left hip. He said he felt a “pop” in his left groin while his left foot was planted and he was cutting to the right. The patient said this was followed by worsening pain with ambulation and hip flexion.
The patient had considerable difficulty walking into the exam room. On physical examination, he had significant tenderness to palpation along the anteromedial thigh and groin. The patient’s strength was 1/5 with left hip flexion. There was apparent muscle firing, but no significant leg movement. He had full passive range of motion and there was no soft-tissue swelling, erythema, or other integumentary changes.
THE DIAGNOSIS
Plain radiographs revealed a lesser trochanter avulsion fracture with a 2-cm displacement (FIGURE 1).
DISCUSSION
Pelvic and proximal femur avulsion fractures tend to occur during the second decade of life.1,2 They’re more frequently seen in boys and adolescent athletes, especially those involved in soccer and gymnastics.3,4
Anterior superior iliac spine (ASIS), ischial tuberosity (IT), and anterior inferior iliac spine (AIIS) avulsion fractures are more prevalent,4 while lesser trochanter avulsion fractures are more rare. In one review of 1126 children with femoral neck and proximal 1/3 femoral shaft fractures, only 3 of them had lesser trochanter avulsion fractures.5
Clinical presentation. Presenting symptoms of lesser trochanter avulsion fractures can be vague, but are usually localized to the groin and medial hip region. Patients will demonstrate pain and weakness with hip flexion.3,6 There may be signs of inflammation, tenderness, and ecchymosis near the site of injury.
On physical exam, a positive Ludloff sign helps localize the injury to the iliopsoas muscle, which inserts at the lesser trochanter and is involved in hip flexion.3,6,7 The Ludloff test is performed by flexing the patient’s hip while he/she is in a seated position.
BIOMECHANICS OF AVULSION FRACTURES
Perhaps surprisingly, the majority of avulsion injuries in children and adolescents are the result of non-contact athletic movement and indirect trauma.4 In children, muscles and tendons are often stronger than their bones,7 and physes—structurally weak regions—are particularly predisposed to fractures.2,4,6
The mechanism of injury in children and adolescents is commonly a sudden, forceful contraction of the iliopsoas muscle.6,7 While similar movement in adults will produce tendon sprains and muscle strains, children often experience a complete avulsion fracture.7 So uncommon are these fractures among adults that an adult patient presenting with one should receive further work-up for underlying pathology such as malignancy.8,9
While other hip and femur avulsion fractures in children and adolescents involve different muscle groups, the etiologic mechanism—forceful muscle contraction—is usually the same.2,4,7 IT injuries are often seen with sudden, aggressive lengthening of the hamstring muscles, whereas injuries to the ASIS and AIIS are the result of abrupt eccentric contraction of hip extensor muscles while the knee is flexed.4
DIFFERENTIAL DIAGNOSIS
There are several entities that can mimic a lesser trochanter avulsion fracture including Legg-Calve-Perthes disease (LCPD), slipped capital femoral epiphysis (SCFE), snapping hip with the iliofemoral ligament, iliopsoas tendonitis, referred pain from the gastrointestinal region, and a genito-urologic etiology.1,7,10
Diagnostic studies. Physical exam findings of severe pain and reduced strength are clear indications for obtaining baseline imaging. Baseline radiographs are key to the diagnosis of avulsion fractures. They help differentiate between more benign fractures, such as a nondisplaced avulsion fracture, and more substantial conditions, such as LCPD and SCFE, which require significantly different approaches to treatment and follow-up.1,7
Anteroposterior, oblique, and axial views of the pelvis all assist in assessing avulsion fractures radiographically.3,4,7 In the event that an avulsion fracture is not radiographically visible, but is still suspected, additional imaging should be obtained.10 A computerized tomography (CT) scan is an appropriate follow-up, given its meticulous detail of bony anatomy.3,10 Alternatively, if physes have yet to ossify or there are concerns about soft tissue injury, magnetic resonance imaging can be useful.3,7,10
MANAGEMENT
The majority of lesser trochanter avulsion fractures are managed conservatively with rest, nonsteroidal anti-inflammatory drugs (NSAIDs), and physical therapy. Patients are often placed on non-weight bearing activity for up to 6 weeks while the fracture repairs and forms a new union.7 Current management strategies have moved away from immobilization with splints and braces.
In rare instances when the fragment is displaced >2 cm, or there is inadequate healing or pain relief after 3 months of supportive care, surgery may be required.1 With appropriate diagnosis and medical care, the injured athlete should fully recover with no impairment or chronic pain.2
Our patient was placed on non-weight-bearing activity and treated with NSAIDs and acetaminophen. We advanced him to weight-bearing activities 4 weeks after injury. After 8 weeks of conservative management, he returned to competitive play with no further complications (FIGURE 2).
THE TAKEAWAY
Pelvic and proximal femur avulsion fractures occur more often in child and adolescent athletes. As this population becomes increasingly competitive in athletics, the risk of injury increases. Infrequent fractures such as lesser trochanter avulsion fractures may become more common, as well. The majority of avulsion fractures don’t require surgical intervention, but it’s important to obtain baseline radiographs to rule out other injuries or pathologies that may lead to poor prognoses if they are left untreated.
THE CASE
A 14-year-old Caucasian boy presented to our clinic with a complaint of left anterior hip pain. The patient had been running during a flag football match when he suddenly developed a sharp, stabbing pain in his left hip. He said he felt a “pop” in his left groin while his left foot was planted and he was cutting to the right. The patient said this was followed by worsening pain with ambulation and hip flexion.
The patient had considerable difficulty walking into the exam room. On physical examination, he had significant tenderness to palpation along the anteromedial thigh and groin. The patient’s strength was 1/5 with left hip flexion. There was apparent muscle firing, but no significant leg movement. He had full passive range of motion and there was no soft-tissue swelling, erythema, or other integumentary changes.
THE DIAGNOSIS
Plain radiographs revealed a lesser trochanter avulsion fracture with a 2-cm displacement (FIGURE 1).
DISCUSSION
Pelvic and proximal femur avulsion fractures tend to occur during the second decade of life.1,2 They’re more frequently seen in boys and adolescent athletes, especially those involved in soccer and gymnastics.3,4
Anterior superior iliac spine (ASIS), ischial tuberosity (IT), and anterior inferior iliac spine (AIIS) avulsion fractures are more prevalent,4 while lesser trochanter avulsion fractures are more rare. In one review of 1126 children with femoral neck and proximal 1/3 femoral shaft fractures, only 3 of them had lesser trochanter avulsion fractures.5
Clinical presentation. Presenting symptoms of lesser trochanter avulsion fractures can be vague, but are usually localized to the groin and medial hip region. Patients will demonstrate pain and weakness with hip flexion.3,6 There may be signs of inflammation, tenderness, and ecchymosis near the site of injury.
On physical exam, a positive Ludloff sign helps localize the injury to the iliopsoas muscle, which inserts at the lesser trochanter and is involved in hip flexion.3,6,7 The Ludloff test is performed by flexing the patient’s hip while he/she is in a seated position.
BIOMECHANICS OF AVULSION FRACTURES
Perhaps surprisingly, the majority of avulsion injuries in children and adolescents are the result of non-contact athletic movement and indirect trauma.4 In children, muscles and tendons are often stronger than their bones,7 and physes—structurally weak regions—are particularly predisposed to fractures.2,4,6
The mechanism of injury in children and adolescents is commonly a sudden, forceful contraction of the iliopsoas muscle.6,7 While similar movement in adults will produce tendon sprains and muscle strains, children often experience a complete avulsion fracture.7 So uncommon are these fractures among adults that an adult patient presenting with one should receive further work-up for underlying pathology such as malignancy.8,9
While other hip and femur avulsion fractures in children and adolescents involve different muscle groups, the etiologic mechanism—forceful muscle contraction—is usually the same.2,4,7 IT injuries are often seen with sudden, aggressive lengthening of the hamstring muscles, whereas injuries to the ASIS and AIIS are the result of abrupt eccentric contraction of hip extensor muscles while the knee is flexed.4
DIFFERENTIAL DIAGNOSIS
There are several entities that can mimic a lesser trochanter avulsion fracture including Legg-Calve-Perthes disease (LCPD), slipped capital femoral epiphysis (SCFE), snapping hip with the iliofemoral ligament, iliopsoas tendonitis, referred pain from the gastrointestinal region, and a genito-urologic etiology.1,7,10
Diagnostic studies. Physical exam findings of severe pain and reduced strength are clear indications for obtaining baseline imaging. Baseline radiographs are key to the diagnosis of avulsion fractures. They help differentiate between more benign fractures, such as a nondisplaced avulsion fracture, and more substantial conditions, such as LCPD and SCFE, which require significantly different approaches to treatment and follow-up.1,7
Anteroposterior, oblique, and axial views of the pelvis all assist in assessing avulsion fractures radiographically.3,4,7 In the event that an avulsion fracture is not radiographically visible, but is still suspected, additional imaging should be obtained.10 A computerized tomography (CT) scan is an appropriate follow-up, given its meticulous detail of bony anatomy.3,10 Alternatively, if physes have yet to ossify or there are concerns about soft tissue injury, magnetic resonance imaging can be useful.3,7,10
MANAGEMENT
The majority of lesser trochanter avulsion fractures are managed conservatively with rest, nonsteroidal anti-inflammatory drugs (NSAIDs), and physical therapy. Patients are often placed on non-weight bearing activity for up to 6 weeks while the fracture repairs and forms a new union.7 Current management strategies have moved away from immobilization with splints and braces.
In rare instances when the fragment is displaced >2 cm, or there is inadequate healing or pain relief after 3 months of supportive care, surgery may be required.1 With appropriate diagnosis and medical care, the injured athlete should fully recover with no impairment or chronic pain.2
Our patient was placed on non-weight-bearing activity and treated with NSAIDs and acetaminophen. We advanced him to weight-bearing activities 4 weeks after injury. After 8 weeks of conservative management, he returned to competitive play with no further complications (FIGURE 2).
THE TAKEAWAY
Pelvic and proximal femur avulsion fractures occur more often in child and adolescent athletes. As this population becomes increasingly competitive in athletics, the risk of injury increases. Infrequent fractures such as lesser trochanter avulsion fractures may become more common, as well. The majority of avulsion fractures don’t require surgical intervention, but it’s important to obtain baseline radiographs to rule out other injuries or pathologies that may lead to poor prognoses if they are left untreated.
1. Byrne A, Reidy D. Acute groin pain in an adolescent sprinter: a case report. Int J Clin Pediatr. 2012;1:46-48.
2. Fernbach SK, Wilkinson RH. Avulsion injuries of the pelvis and proximal femur. AJR Am J Roentgenol. 1981;137:581-584.
3. McKinney BI, Nelson C, Carrion W. Apophyseal avulsion fractures of the hip and pelvis. Orthopedics. 2009;32:42.
4. Rossi F, Dragoni S. Acute avulsion fractures of the pelvis in adolescent competitive athletes: prevalence, location and sports distribution of 203 cases collected. Skeletal Radiol. 2001;30:127-131.
5. Theologis TN, Epps H, Latz K, et al. Isolated fractures of the lesser trochanter in children. Injury. 1997;28:363-364.
6. Paluska SA. An overview of hip injuries in running. Sports Med. 2005;35:991-1014.
7. Vazquez E, Kim TY, Young TP. Avulsion fracture of the lesser trochanter: an unusual cause of hip pain in an adolescent. CJEM. 2013;15:123-125.
8. Afra R, Boardman DL, Kabo JM, et al. Avulsion fracture of the lesser trochanter as a result of a preliminary malignant tumor of bone. A report of four cases. J Bone Joint Surg Am. 1999;81:1299-1304.
9. DePasse JM, Varner K, Cosculluela P, et al. Atraumatic avulsion of the distal iliopsoas tendon: an unusual cause of hip pain. Orthopedics. 2010;33.
10. Suarez JC, Ely EE, Mutnal AB, et al. Comprehensive approach to the evaluation of groin pain. J Am Acad Orthop Surg. 2013;21:558-570.
1. Byrne A, Reidy D. Acute groin pain in an adolescent sprinter: a case report. Int J Clin Pediatr. 2012;1:46-48.
2. Fernbach SK, Wilkinson RH. Avulsion injuries of the pelvis and proximal femur. AJR Am J Roentgenol. 1981;137:581-584.
3. McKinney BI, Nelson C, Carrion W. Apophyseal avulsion fractures of the hip and pelvis. Orthopedics. 2009;32:42.
4. Rossi F, Dragoni S. Acute avulsion fractures of the pelvis in adolescent competitive athletes: prevalence, location and sports distribution of 203 cases collected. Skeletal Radiol. 2001;30:127-131.
5. Theologis TN, Epps H, Latz K, et al. Isolated fractures of the lesser trochanter in children. Injury. 1997;28:363-364.
6. Paluska SA. An overview of hip injuries in running. Sports Med. 2005;35:991-1014.
7. Vazquez E, Kim TY, Young TP. Avulsion fracture of the lesser trochanter: an unusual cause of hip pain in an adolescent. CJEM. 2013;15:123-125.
8. Afra R, Boardman DL, Kabo JM, et al. Avulsion fracture of the lesser trochanter as a result of a preliminary malignant tumor of bone. A report of four cases. J Bone Joint Surg Am. 1999;81:1299-1304.
9. DePasse JM, Varner K, Cosculluela P, et al. Atraumatic avulsion of the distal iliopsoas tendon: an unusual cause of hip pain. Orthopedics. 2010;33.
10. Suarez JC, Ely EE, Mutnal AB, et al. Comprehensive approach to the evaluation of groin pain. J Am Acad Orthop Surg. 2013;21:558-570.
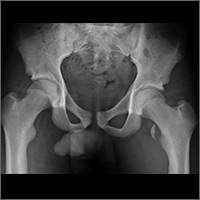
Unsuspected Lymphomatoid Granulomatosis in a Patient With Antisynthetase Syndrome
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
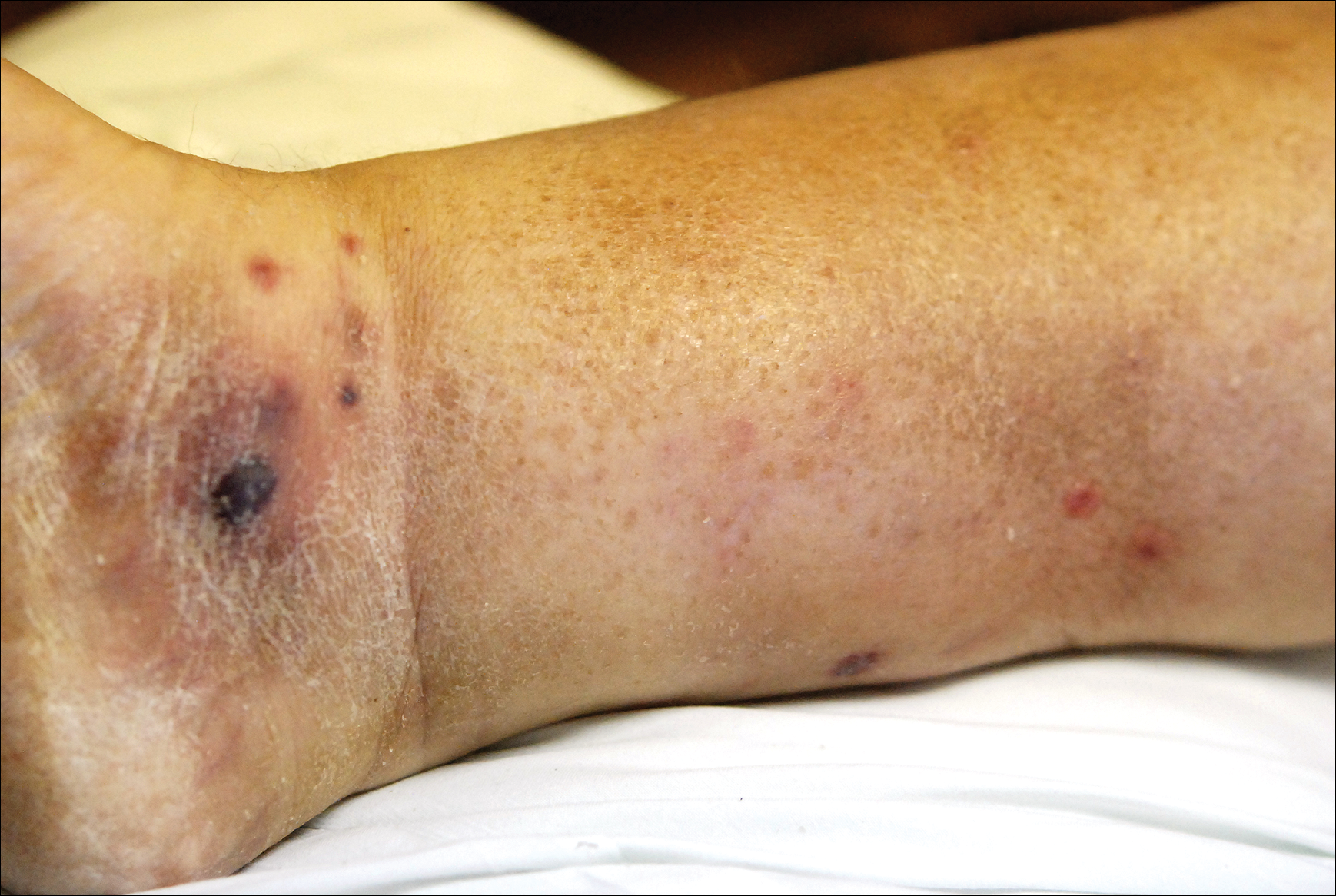
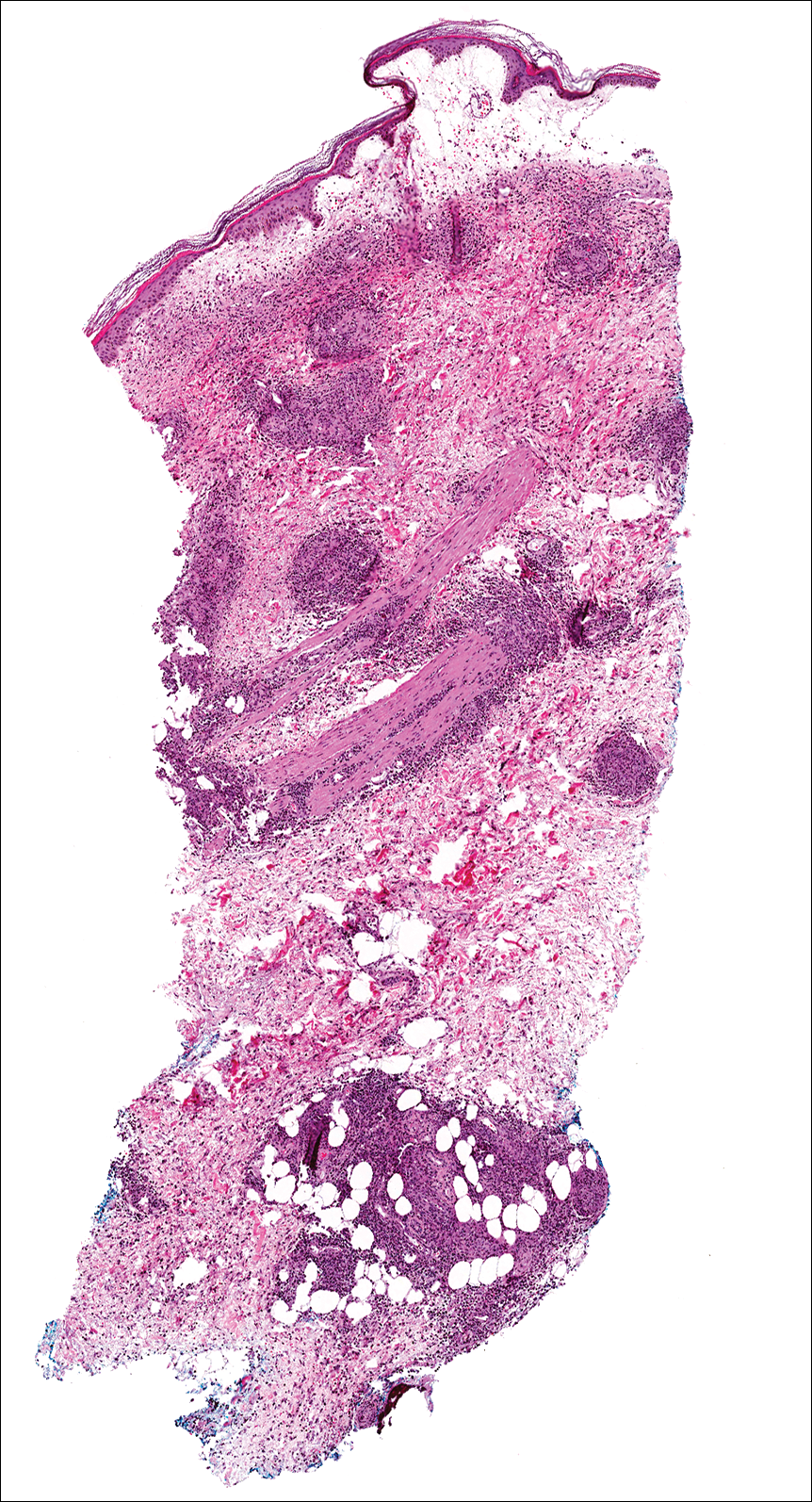
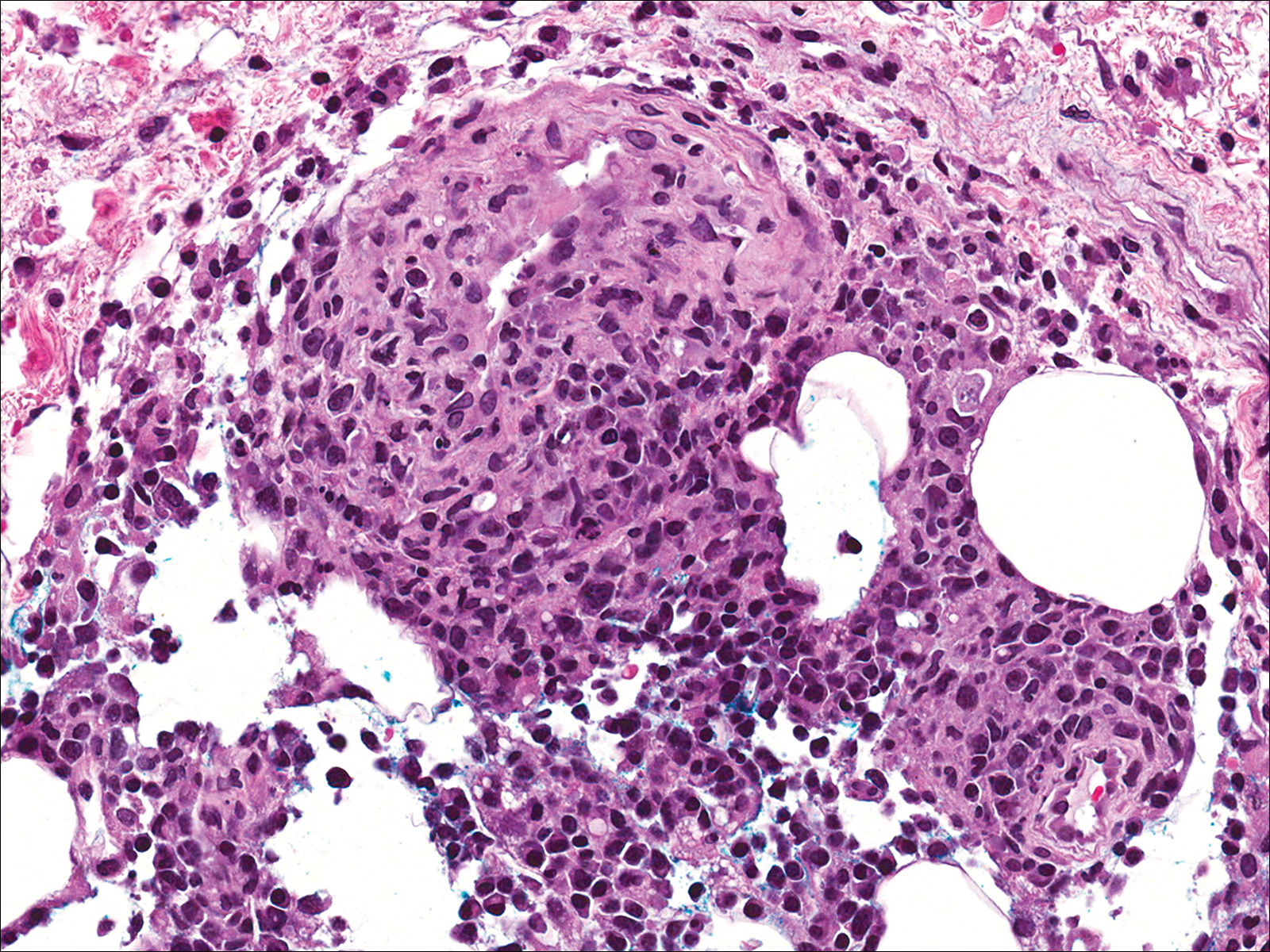
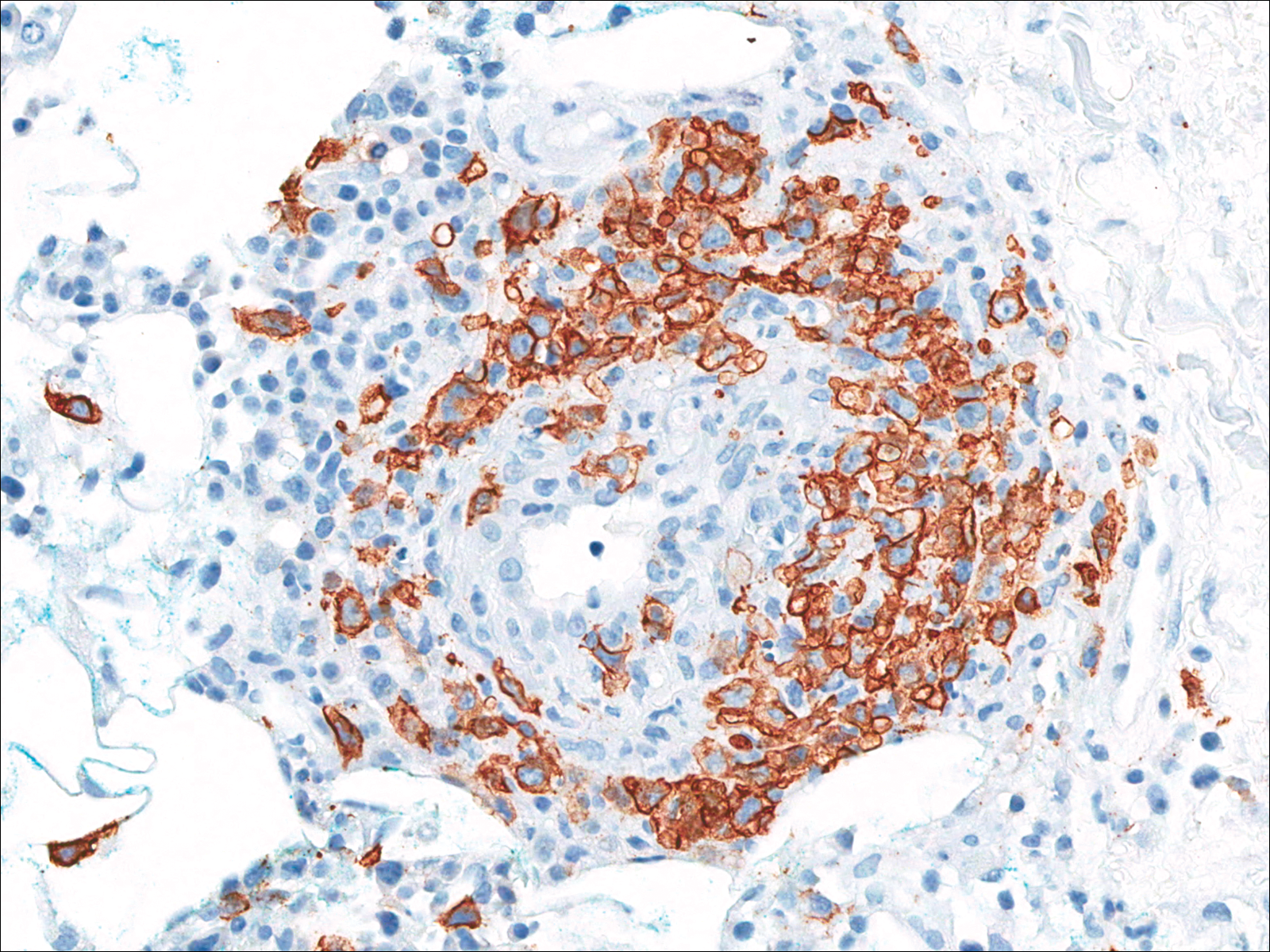
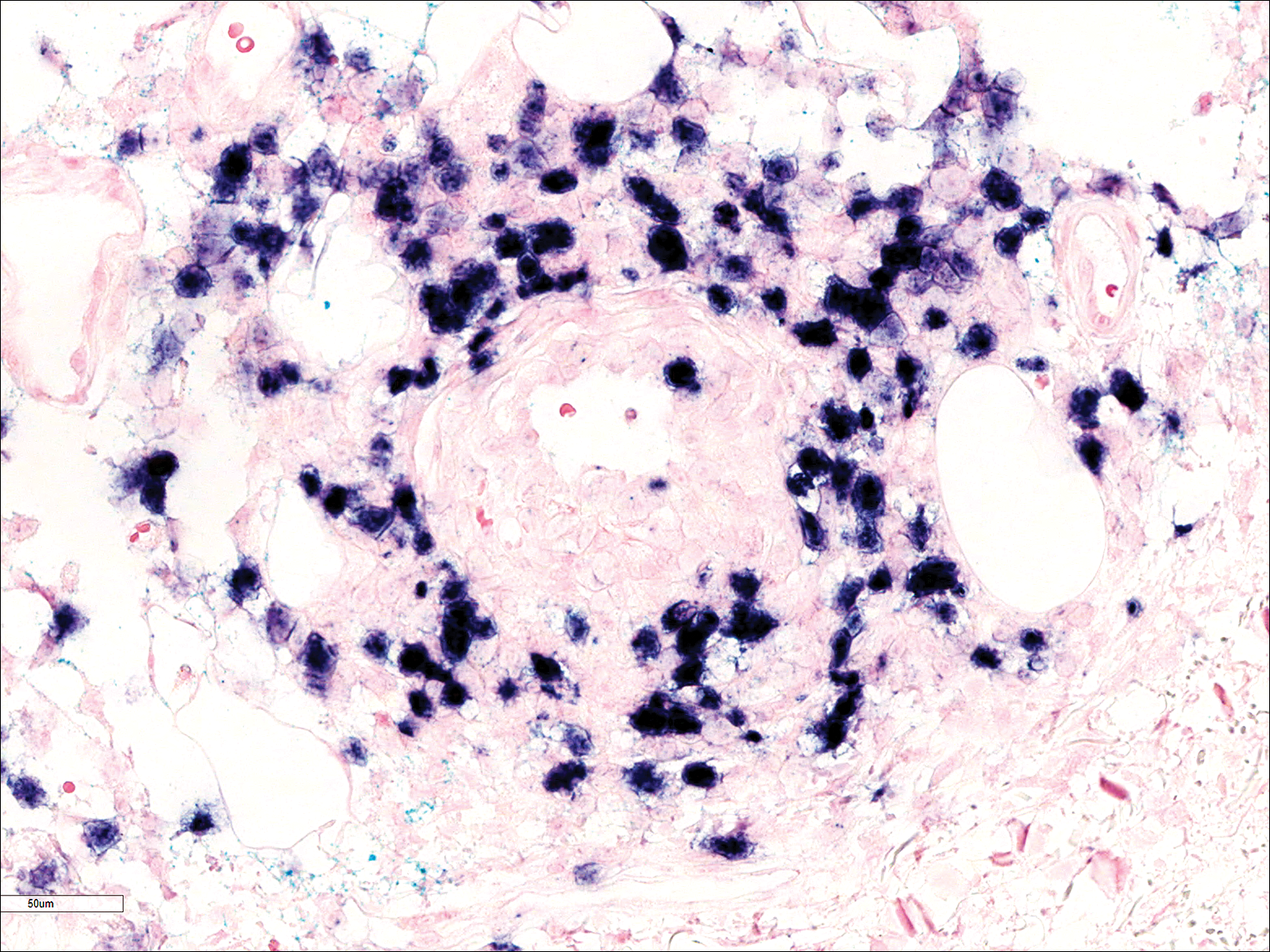
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
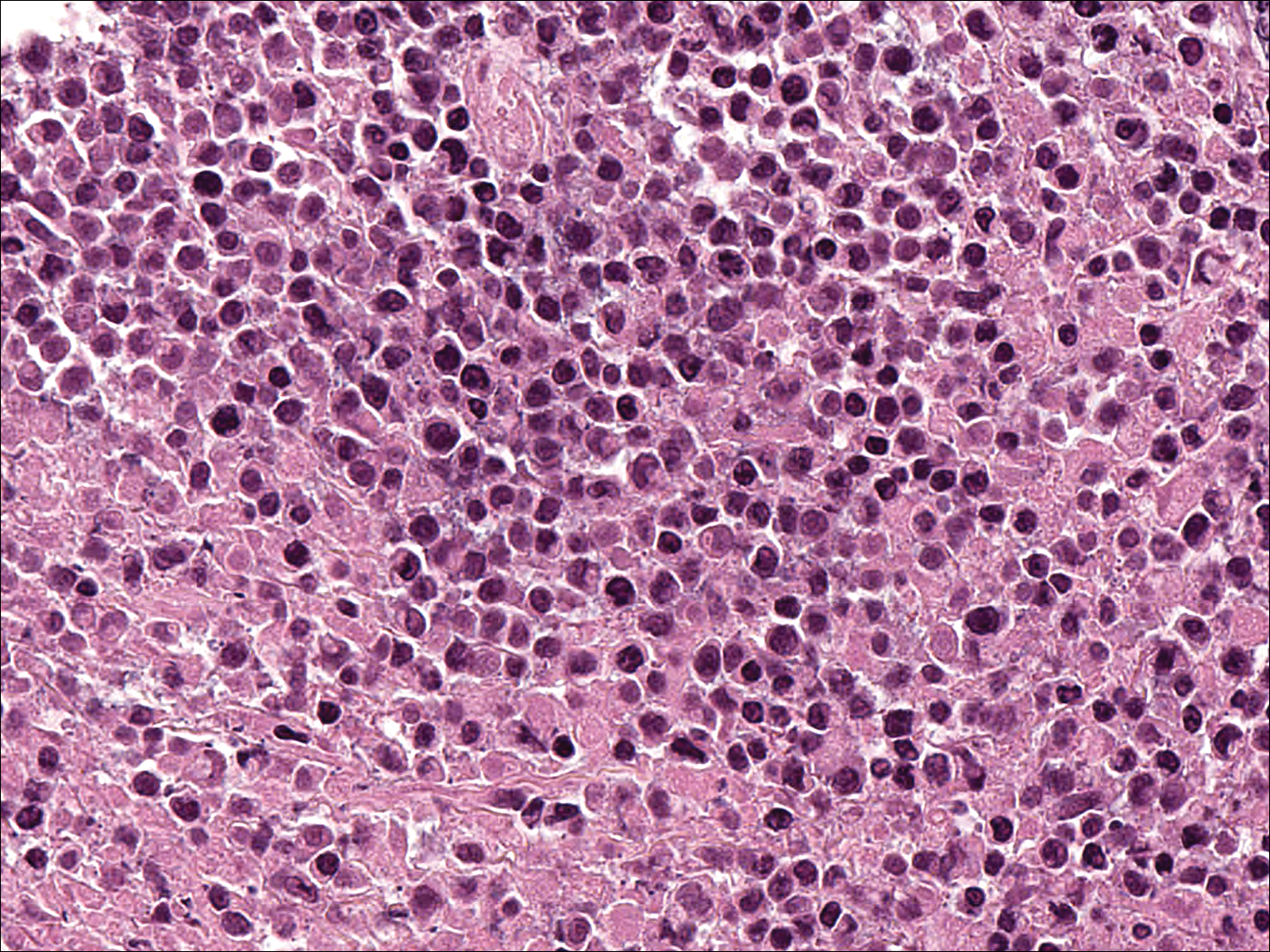
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.
Comment
Lymphomatoid granulomatosis is a rare lymphoproliferative disorder associated with various immunocompromised states including primary immunodeficiency disorders, human immunodeficiency virus infection, and immunosuppression for organ transplantation and autoimmune diseases. Our patient was receiving azathioprine for antisynthetase syndrome, which put her at risk for EBV infection and LYG. Azathioprine rarely has been reported as a possible culprit of LYG,3,4 but there are no known reported cases that were related to antisynthetase syndrome. There are multiple reports of development of LYG in patients receiving methotrexate for rheumatoid arthritis.5-10 Other iatrogenic causes reported in the literature include thiopurines11,12 and imatinib.13,14
The clinical diagnosis of our patient was particularly challenging given her complicated medical history including interstitial lung disease, predisposition to infection secondary to immunosuppression, and recent radiation therapy to the chest. This case illustrates the importance of maintaining a high index of suspicion for LYG in immunosuppressed patients presenting with lung infiltrates.
Presentation
Radiologically, LYG typically manifests as nodular densities accentuated in the lower lung lobes, which may become confluent.15 Because the nodular pattern in LYG is nonspecific and may mimic sarcoidosis, hypersensitivity pneumonitis, vasculitis, and infectious and neoplastic diseases,16 open lung biopsy often is required to establish the diagnosis in the absence of more accessible lesions.
Cutaneous lesions are seen in 40% to 50% of patients2 and may be the presenting sign of LYG. In a retrospective study, 16% (3/19) of LYG patients presented with cutaneous lesions months before diagnostic pulmonary lesions were identified.17 The skin is the most accessible site for biopsy, allowing definitive tissue diagnosis even when the condition is not clinically suspected. Therefore, dermatologists and dermatopathologists should be aware of this rare entity.
The clinical morphologies of the skin lesions are nonspecific, ranging from erythematous papules and subcutaneous nodules to indurated plaques. Ulceration may be present. The lesions may be widely disseminated or limited to the arms and legs. Our patient presented with erythematous and hemorrhagic papules, macules, and blisters on the lower leg. The hemorrhagic and blistering nature of some of these lesions in our patient may be attributable to thrombocytopenia and lymphedema in addition to LYG.
Histopathology and Differential
The skin biopsy from our patient demonstrated typical features of LYG, namely EBV-positive neoplastic large B cells in a background of predominating reactive T cells.18 The neoplastic large cells frequently invade blood vessels, leading to luminal narrowing without necrosis of the vessel walls. Grading is based on the density of EBV-positive large B cells: grade 1 is defined as fewer than 5 cells per HPF; grade 2, 5 to 50 cells per HPF; and grade 3, more than 50 cells per HPF.18 Grade 2 or 3 disease predicts worse outcome,2 as observed in our case. It is important for pathologists and clinicians to be aware that the proportion of EBV-positive large B cells is variable even within a single lesion; therefore, more than 1 biopsy may be necessary for appropriate grading and management.1,17 Additionally, skin biopsy may have a lower sensitivity for detecting EBV-positive B cells compared to lung biopsy, possibly due to sampling error in small biopsies.17
The histopathologic features of LYG frequently overlap with other lymphomas. Due to the abundance of T cells, LYG may be misclassified as T-cell/histiocyte-rich large B-cell lymphoma.19 Because the latter is not associated with EBV, EBER in situ hybridization is helpful in distinguishing the 2 conditions. On the other hand, EBER in situ hybridization has no value in discriminating LYG and extranodal natural killer (NK)/T-cell lymphoma, as both are EBV driven. Unlike LYG, the neoplastic EBV-positive cells in extranodal NK/T-cell lymphoma make up the majority of the infiltrate and exhibit an NK-cell immunophenotype (positive CD56 and cytoplasmic CD3 epsilon).20 Pulmonary involvement also is uncommon in NK/T-cell lymphoma.
Aside from lymphomas, LYG also resembles granulomatosis with polyangiitis (GPA)(formerly known as Wegener granulomatosis). Clinically, both LYG and GPA can present with constitutional symptoms, as well as lung, kidney, and skin lesions. The 2 conditions differ microscopically, with leukocytoclastic vasculitis and necrotizing granulomatous inflammation being characteristic of GPA but absent in LYG.1,21 Neutrophils and eosinophils are much more likely to be present in GPA.22,23
Disease Progression
Although LYG is an extranodal disease, there is a 7% to 45% risk of progression to nodal lymphoma in patients with high-grade disease.2,22,24 Our patient progressed to nodal large B-cell lymphoma shortly after the diagnosis of high-grade LYG. She developed additional lesions in the liver, spleen, and kidneys, and ultimately succumbed to the disease. Prior studies have shown higher mortality in patients with bilateral lung involvement and neurologic abnormalities, whereas cutaneous involvement does not affect outcome.2
Treatment
A prospective study used an initial treatment regimen of cyclophosphamide and prednisone but mortality was high.24 More recently, chemotherapy regimens including CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), CVP or CHOP combined with rituximab, C-MOPP (cyclophosphamide, vincristine, prednisone, and procarbazine), EPOCH, and rituximab with high-dose cytarabine have been used with variable success for grades 2 and 3 LYG.17,23,25,26 Antiviral and immunomodulatory (interferon alfa) therapy has been used to induce remission in a majority of patients with grades 1 or 2 LYG.3,17,27,28 There is a report of successful treatment of relapsed LYG with the retinoid agent bexarotene.29 Autologous or allogeneic stem cell transplantation was effective for some patients with refractory or relapsed LYG.30 Further studies are needed to clarify optimal treatment of LYG, especially high-grade disease.
Conclusion
We report a rare case of LYG in a patient with antisynthetase syndrome, which highlights the critical role of skin biopsy in establishing the diagnosis of LYG when the clinical and radiologic presentations are obscured by other comorbidities. Dermatologists should be familiar with this rare disease and maintain a low threshold for biopsy in immunocompromised patients presenting with nodular lung infiltrates and/or nonspecific skin lesions.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
- Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. 2010;34:E35-E48.
- Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. 1979;43:360-373.
- Connors W, Griffiths C, Patel J, et al. Lymphomatoid granulomatosis associated with azathioprine therapy in Crohn disease. BMC Gastroenterol. 2014;14:127.
- Katherine Martin L, Porcu P, Baiocchi RA, et al. Primary central nervous system lymphomatoid granulomatosis in a patient receiving azathioprine therapy. Clin Adv Hematol Oncol. 2009;7:65-68.
- Barakat A, Grover K, Peshin R. Rituximab for pulmonary lymphomatoid granulomatosis which developed as a complication of methotrexate and azathioprine therapy for rheumatoid arthritis. Springerplus. 2014;3:751.
- Kobayashi S, Kikuchi Y, Sato K, et al. Reversible iatrogenic, MTX-associated EBV-driven lymphoproliferation with histopathological features of a lymphomatoid granulomatosis in a patient with rheumatoid arthritis. Ann Hematol. 2013;92:1561-1564.
- Kameda H, Okuyama A, Tamaru J, et al. Lymphomatoid granulomatosis and diffuse alveolar damage associated with methotrexate therapy in a patient with rheumatoid arthritis. Clin Rheumatol. 2007;26:1585-1589.
- Oiwa H, Mihara K, Kan T, et al. Grade 3 lymphomatoid granulomatosis in a patient receiving methotrexate therapy for rheumatoid arthritis. Intern Med. 2014;53:1873-1875.
- Blanchart K, Paciencia M, Seguin A, et al. Fatal pulmonary lymphomatoid granulomatosis in a patient taking methotrexate for rheumatoid arthritis. Minerva Anestesiol. 2014;80:119-120.
- Schalk E, Krogel C, Scheinpflug K, et al. Lymphomatoid granulomatosis in a patient with rheumatoid arthritis receiving methotrexate: successful treatment with the anti-CD20 antibody mabthera. Onkologie. 2009;32:440-441.
- Subramaniam K, Cherian M, Jain S, et al. Two rare cases of Epstein-Barr virus-associated lymphoproliferative disorders in inflammatory bowel disease patients on thiopurines and other immunosuppressive medications. Intern Med J. 2013;43:1339-1342.
- Destombe S, Bouron-DalSoglio D, Rougemont AL, et al. Lymphomatoid granulomatosis: a unique complication of Crohn disease and its treatment in pediatrics. J Pediatr Gastroenterol Nutr. 2010;50:559-561.
- Yazdi AS, Metzler G, Weyrauch S, et al. Lymphomatoid granulomatosis induced by imatinib treatment. Arch Dermatol. 2007;143:1222-1223.
- Salmons N, Gregg RJ, Pallalau A, et al. Lymphomatoid granulomatosis in a patient previously diagnosed with a gastrointestinal stromal tumour and treated with imatinib. J Clin Pathol. 2007;60:199-201.
- Dee PM, Arora NS, Innes DJ Jr. The pulmonary manifestations of lymphomatoid granulomatosis. Radiology. 1982;143:613-618.
- Rezai P, Hart EM, Patel SK. Case 169: lymphomatoid granulomatosis. Radiology. 2011;259:604-609.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol. 2001;25:1111-1120.
- Pittaluga S, Wilson WH, Jaffe E. Lymphomatoid granulomatosis. In: Swerdlow S, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:247-249.
- Abramson JS. T-cell/histiocyte-rich B-cell lymphoma: biology, diagnosis, and management. Oncologist. 2006;11:384-392.
- Jaffe E. Nasal and nasal-type T/NK cell lymphoma: a unique form of lymphoma associated with the Epstein-Barr virus. Histopathology. 1995;27:581-583.
- Barksdale SK, Hallahan CW, Kerr GS, et al. Cutaneous pathology in Wegener’s granulomatosis: a clinicopathologic study of 74 biopsies in 46 patients. Am J Surg Pathol. 1995;19:161-172.
- Koss MN, Hochholzer L, Langloss JM, et al. Lymphomatoid granulomatosis: a clinicopathologic study of 42 patients. Pathology. 1986;18:283-288.
- Aoki T, Harada Y, Matsubara E, et al. Long-term remission after multiple relapses in an elderly patient with lymphomatoid granulomatosis after rituximab and high-dose cytarabine chemotherapy without stem-cell transplantation. J Clin Oncol. 2013;31:E390-E393.
- Fauci AS, Haynes BF, Costa J, et al. Lymphomatoid granulomatosis: prospective clinical and therapeutic experience over 10 years. N Engl J Med. 1982;306:68-74.
- Jung KH, Sung HJ, Lee JH, et al. A case of pulmonary lymphomatoid granulomatosis successfully treated by combination chemotherapy with rituximab. Chemotherapy. 2009;55:386-390.
- Hernandez-Marques C, Lassaletta A, Torrelo A, et al. Rituximab in lymphomatoid granulomatosis. J Pediatr Hematol Oncol. 2014;36:E69-E74.
- Wilson WH, Gutierrez M, Raffeld M, et al. Lymphomatoid granulomatosis: phase 2 study of dose-adjusted interferon-alfa or EPOCH chemotherapy. Blood. 1999;94:599A.
- Wilson WH, Kingma DW, Raffeld M, et al. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87:4531-4537.
- Berg SE, Downs LH, Torigian DA, et al. Successful treatment of relapsed lymphomatoid granulomatosis with bexarotene. Cancer Biol Ther. 2008;7:1544-1546.
- Siegloch K, Schmitz N, Wu HS, et al. Hematopoietic stem cell transplantation in patients with lymphomatoid granulomatosis: a European group for blood and marrow transplantation report. Biol Blood Marrow Transplant. 2013;19:1522-1525.
Practice Points
- Lymphomatoid granulomatosis (LYG) is a rare extranodal angiocentric large B-cell lymphoma driven by the Epstein-Barr virus.
- Lymphomatoid granulomatosis should be suspected when immunocompromised patients present with nodular lung infiltrates and/or nonspecific skin lesions.
- Skin biopsy serves a critical role in establishing the diagnosis of LYG, especially when clinical and radiologic findings are obscured by other comorbidities.
Cannabinoid Hyperemesis Syndrome
Given the recent rise in marijuana legalization efforts and an overall increase in the prevalence of marijuana use, it is becoming increasingly important to recognize conditions that are associated with its use. Data obtained from the National Survey on Drug Use and Health show the prevalence of marijuana use within the past month among those surveyed was 8.4% in 2014. This represents a 35% increase from the same study in 2002. Based on this survey, 2.5 million people (or ~7,000 per day) used marijuana for the first time.1
Following the liberalization of marijuana in Colorado, the prevalence of presentation to the emergency department (ED) for cyclic vomiting nearly doubled.2 During the 2016 election season, several states included legislation that increased access to marijuana on the ballot, most of which passed. There are now 28 states plus the District of Columbia that permit medical marijuana usage, and 8 of those states and the District of Columbia have laws allowing for recreational use of marijuana.3
First described in a case series by Allen and colleagues in 2004, cannabinoid hyperemesis syndrome (CHS) is indicated by recurrent episodes of nausea and vomiting with vague abdominal pain and compulsive hot bathing in the setting of chronic, often daily, cannabis use.4 A case of a middle-aged veteran with chronic marijuana use and recurrent, self-limited nausea and vomiting is presented here.
Case Presentation
A 45-year-old man presented to the ED with a 5-day history of persistent nausea and vomiting that began abruptly. The symptoms had been constant since onset, resulting in very little oral intake. The patient reported no hematemesis or coffee ground emesis. He noted a drop in his urine output over the previous 2 days. He also reported abdominal pain associated with the nausea. The patient characterized his pain as “dull and achy” diffuse pain that was partially relieved with emesis. His bowel movements had been regular, and he reported no diarrhea, fever, chills, or other constitutional symptoms. Additional 10-point review of systems was otherwise negative. The patient reported smoking marijuana multiple times daily for many years. The patient reported he had not used alcohol for several months.
A physical exam showed a pale and diaphoretic patient. Vital signs were significant for mild hypertension (150/75), but the patient was afebrile with a normal heart rate. An abdominal exam revealed a nontender, nondistended abdomen with no signs of rebound or guarding. The remainder of the examination was unremarkable. An initial workup showed a mild elevation of serum creatinine to 1.36 mg/dL (baseline is 1.10 mg/dL). Other workups, including complete blood count (CBC) with differential, complete metabolic panel, lipase, amylase, and urine analysis, were all unremarkable.
The patient’s urine drug screen (UDS) was positive for tetrahydrocannabinol (THC). A computed tomography (CT) scan of his abdomen and pelvis with contrast was unremarkable. The patient was admitted for his inability to tolerate oral intake and dehydration and treated supportively with IV fluids and antiemetics.
Overnight, the nursing staff reported that the patient took multiple, prolonged hot showers. Upon further questioning, he reported the hot showers significantly helped the nausea and abdominal pain. He had learned this behavior after experiencing previous episodes of self-limited nausea, vomiting, and abdominal pain.
Extensive review of his medical record revealed that the patient had, in fact, presented to the ED with similar symptoms 11 times in the prior 8 years. He was admitted on 8 occasions over that time frame. The typical hospital course included supportive care with antiemetics and IV fluids. The patient’s symptoms typically resolved within 24 to 72 hours of hospitalization. Previous evaluations included additional unremarkable CT imaging of the abdomen and pelvis. The patient also had received 2 esophagogastroduodenoscopies (EGDs), one 2 years prior and the other 5 years prior. Both EGDs showed only mild gastritis. On every check during the previous 8 years, the patient’s UDS was positive for THC.
Most of his previous admissions were attributed to viral gastroenteritis due to the self-limited nature of the symptoms. Other admissions were attributed to alcohol-induced gastritis. However, after abstaining from alcohol for long periods, the patient had continued recurrence of the symptoms and increased frequency of presentations to the ED.
The characteristics, signs, and symptoms of CHS were discussed with the patient. The patient strongly felt as though these symptoms aligned with his clinical course over the prior 8 years. At time of writing, the patient had gone 20 months without requiring hospitalization; however, he had a recent relapse of marijuana use and subsequently required hospitalization.
Discussion
As in this case, CHS often presents with refractory, self-limited nausea and vomiting with vague abdominal pain that is temporarily relieved by hot baths or showers. In the largest case series, it was noted the average age was 32 years, and the majority of subjects used marijuana at least weekly for > 2 years.5 Many studies categorize CHS into 3 phases: prodromal, hyperemetic, and recovery.
The prodromal, or preemetic phase, is characterized by early morning nausea without emesis and abdominal discomfort. The hyperemetic phase begins when the patient accesses the health care system via either the ED or primary care physician. This phase is characterized by intractable nausea and vomiting and may be associated with mild diffuse abdominal pain. The nausea and vomiting typically do not respond to antiemetic medications. Patients in this stage also develop a compulsive behavior of hot showers that temporarily relieve the symptoms. These behaviors are thought to be learned through their cyclical periods of emesis and may not be present during the first few hyperemetic phases. During the recovery phase, the patient returns to a baseline state of health and often ceases utilizing the hot shower. The recovery phase can last weeks to months despite continued cannabis use prior to returning to the hyperemetic phase (Figure).6,7
Simonetto and colleagues proposed clinical criteria for the diagnosis of CHS based on their case series as well as on previously proposed criteria presented by Sontineni and colleagues.5,8 Long-term cannabis use is required for the diagnosis. In the Simonetto and colleagues case series, the majority of patients developed symptoms within the first 5 years of cannabis use; however, Soriano and colleagues conducted a smaller case series that showed that the majority of subjects used marijuana for roughly 16 years prior to the onset of vomiting.5,7
The major CHS features that suggest the diagnosis are severe cyclic nausea and vomiting, relief of symptoms with abstinence from cannabis, temporary symptom relief with hot bathing, abdominal pain, and at least weekly use of marijuana. Other supportive features include aged < 50 years, weight loss > 5 kg, symptoms that are worse in the morning, normal bowel habits, and negative evaluation, including laboratory, radiography, and endoscopy (Table).5
Treatment often is supportive with emphasis placed on marijuana cessation. Intravenous fluids often are used due to dehydration from the emesis. The use of antiemetics, such as 5-HT3 (eg, ondansetron), D2 (eg, prochlorperazine), H1 (eg, promethazine), or neurokinin-1 receptor antagonists (eg, aprepitant) can be tried, but these therapies often are ineffective. Diet can be advanced as the patient tolerates. Given that many patients are found to have a mild gastritis, H2 blockers or proton pump inhibitors may be used. Extensive counseling on marijuana cessation is needed as it is the only therapy shown to have prolonged relief of the hyperemetic phase.6 The length of cessation from marijuana for resolution of the cyclical hyperemesis varies from 1 to 3 months. Returning to marijuana use often results in the returning of CHS.5
The pathophysiology of CHS is largely unknown; however, there are several hypothesized mechanisms. Many theorize that due to the lipophilicity and long half-life of THC, a primary compound in marijuana, it accumulates in the body over time.4,6 It is thought that this accumulation may cause toxicity in both the gastrointestinal tract as well as in the brain. Central effects on the hypothalamic-pituitary axis may play a major role, and the reason for the symptom relief of hot baths is due to a change in thermoregulation in the hypothalamus.5 One interesting mechanism relates to CB1 receptor activation and vasodilation within the gastrointestinal tract due to chronic THC accumulation. The relief of the abdominal pain, nausea, and vomiting with hot showers can be secondary to the vasodilation of the skin, causing a redistribution from the gut. This theorized mechanism has been referred to as “cutaneous steal.”9
Conclusion
With the increased prevalence of marijuana use in the U.S. over the past decade and reform in legislation taking place over the next couple of years, it is increasingly important to be able to recognize CHS to avoid frequent hospital utilization and repeated costly evaluations. Cannabinoid hyperemesis syndrome is recognized by the triad of chronic cannabis use, cyclical hyperemesis, and compulsive hot bathing.4
The syndrome has 3 phases. In the prodromal phase the patient has morning predominance of nausea, usually without emesis. This is followed by the hyperemesis phase, which is characterized by hyperemesis, vague abdominal pain, and learned compulsive hot bathing.
The third phase is the recovery phase, which is a return to normal behavior. During the recovery phase, if patients cease marijuana use, they remain asymptomatic; however, if patients continue to use marijuana, they often have recurrence of the hyperemesis phase.5 The diagnosis of cannabinoid hyperemesis syndrome is difficult as it is a diagnosis of exclusion. Patients may present to the ED many times prior to diagnosis. With the changing climate of marijuana laws, it is an important condition to consider when establishing a differential. More studies will be required to evaluate the overall prevalence of this condition as well as if there are any changes following the liberalization of marijuana laws in many states.
1. Azofeifa A, Mattson ME, Schauer G, McAfee T, Grant A, Lyerla R. National estimates of marijuana use and related indicators - National Survey on Drug Use and Health, United States, 2002-2014. MMWR Surveill Summ. 2016;65(11):1-28.
2. Kim HS, Anderson JD, Saghafi O, Heard KJ, Monte AA. Cyclic vomiting presentations following marijuana liberalization in Colorado. Acad Emerg Med. 2015;22(6):694-699.
3. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated July 7, 2017. Accessed August 3, 2017.
4. Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut. 2004;53(11):1566-1570.
5. Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc. 2012;87(2):114-119.
6. Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev. 2011;4(4):241-249.
7. Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci. 2010;55(11):3113-3119.
8. Sontineni SP, Chaudhary S, Sontineni V, Lanspa SJ. Cannabinoid hyperemesis syndrome: clinical diagnosis of an underrecognised manifestation of chronic cannabis abuse. World J Gastroenterol. 2009;15(10):1264-1266.
9. Patterson DA, Smith E, Monahan M, et al. Cannabinoid hyperemesis and compulsive bathing: a case series and paradoxical pathophysiological explanation. J Am Board Fam Med. 2010;23(6):790-793
Given the recent rise in marijuana legalization efforts and an overall increase in the prevalence of marijuana use, it is becoming increasingly important to recognize conditions that are associated with its use. Data obtained from the National Survey on Drug Use and Health show the prevalence of marijuana use within the past month among those surveyed was 8.4% in 2014. This represents a 35% increase from the same study in 2002. Based on this survey, 2.5 million people (or ~7,000 per day) used marijuana for the first time.1
Following the liberalization of marijuana in Colorado, the prevalence of presentation to the emergency department (ED) for cyclic vomiting nearly doubled.2 During the 2016 election season, several states included legislation that increased access to marijuana on the ballot, most of which passed. There are now 28 states plus the District of Columbia that permit medical marijuana usage, and 8 of those states and the District of Columbia have laws allowing for recreational use of marijuana.3
First described in a case series by Allen and colleagues in 2004, cannabinoid hyperemesis syndrome (CHS) is indicated by recurrent episodes of nausea and vomiting with vague abdominal pain and compulsive hot bathing in the setting of chronic, often daily, cannabis use.4 A case of a middle-aged veteran with chronic marijuana use and recurrent, self-limited nausea and vomiting is presented here.
Case Presentation
A 45-year-old man presented to the ED with a 5-day history of persistent nausea and vomiting that began abruptly. The symptoms had been constant since onset, resulting in very little oral intake. The patient reported no hematemesis or coffee ground emesis. He noted a drop in his urine output over the previous 2 days. He also reported abdominal pain associated with the nausea. The patient characterized his pain as “dull and achy” diffuse pain that was partially relieved with emesis. His bowel movements had been regular, and he reported no diarrhea, fever, chills, or other constitutional symptoms. Additional 10-point review of systems was otherwise negative. The patient reported smoking marijuana multiple times daily for many years. The patient reported he had not used alcohol for several months.
A physical exam showed a pale and diaphoretic patient. Vital signs were significant for mild hypertension (150/75), but the patient was afebrile with a normal heart rate. An abdominal exam revealed a nontender, nondistended abdomen with no signs of rebound or guarding. The remainder of the examination was unremarkable. An initial workup showed a mild elevation of serum creatinine to 1.36 mg/dL (baseline is 1.10 mg/dL). Other workups, including complete blood count (CBC) with differential, complete metabolic panel, lipase, amylase, and urine analysis, were all unremarkable.
The patient’s urine drug screen (UDS) was positive for tetrahydrocannabinol (THC). A computed tomography (CT) scan of his abdomen and pelvis with contrast was unremarkable. The patient was admitted for his inability to tolerate oral intake and dehydration and treated supportively with IV fluids and antiemetics.
Overnight, the nursing staff reported that the patient took multiple, prolonged hot showers. Upon further questioning, he reported the hot showers significantly helped the nausea and abdominal pain. He had learned this behavior after experiencing previous episodes of self-limited nausea, vomiting, and abdominal pain.
Extensive review of his medical record revealed that the patient had, in fact, presented to the ED with similar symptoms 11 times in the prior 8 years. He was admitted on 8 occasions over that time frame. The typical hospital course included supportive care with antiemetics and IV fluids. The patient’s symptoms typically resolved within 24 to 72 hours of hospitalization. Previous evaluations included additional unremarkable CT imaging of the abdomen and pelvis. The patient also had received 2 esophagogastroduodenoscopies (EGDs), one 2 years prior and the other 5 years prior. Both EGDs showed only mild gastritis. On every check during the previous 8 years, the patient’s UDS was positive for THC.
Most of his previous admissions were attributed to viral gastroenteritis due to the self-limited nature of the symptoms. Other admissions were attributed to alcohol-induced gastritis. However, after abstaining from alcohol for long periods, the patient had continued recurrence of the symptoms and increased frequency of presentations to the ED.
The characteristics, signs, and symptoms of CHS were discussed with the patient. The patient strongly felt as though these symptoms aligned with his clinical course over the prior 8 years. At time of writing, the patient had gone 20 months without requiring hospitalization; however, he had a recent relapse of marijuana use and subsequently required hospitalization.
Discussion
As in this case, CHS often presents with refractory, self-limited nausea and vomiting with vague abdominal pain that is temporarily relieved by hot baths or showers. In the largest case series, it was noted the average age was 32 years, and the majority of subjects used marijuana at least weekly for > 2 years.5 Many studies categorize CHS into 3 phases: prodromal, hyperemetic, and recovery.
The prodromal, or preemetic phase, is characterized by early morning nausea without emesis and abdominal discomfort. The hyperemetic phase begins when the patient accesses the health care system via either the ED or primary care physician. This phase is characterized by intractable nausea and vomiting and may be associated with mild diffuse abdominal pain. The nausea and vomiting typically do not respond to antiemetic medications. Patients in this stage also develop a compulsive behavior of hot showers that temporarily relieve the symptoms. These behaviors are thought to be learned through their cyclical periods of emesis and may not be present during the first few hyperemetic phases. During the recovery phase, the patient returns to a baseline state of health and often ceases utilizing the hot shower. The recovery phase can last weeks to months despite continued cannabis use prior to returning to the hyperemetic phase (Figure).6,7
Simonetto and colleagues proposed clinical criteria for the diagnosis of CHS based on their case series as well as on previously proposed criteria presented by Sontineni and colleagues.5,8 Long-term cannabis use is required for the diagnosis. In the Simonetto and colleagues case series, the majority of patients developed symptoms within the first 5 years of cannabis use; however, Soriano and colleagues conducted a smaller case series that showed that the majority of subjects used marijuana for roughly 16 years prior to the onset of vomiting.5,7
The major CHS features that suggest the diagnosis are severe cyclic nausea and vomiting, relief of symptoms with abstinence from cannabis, temporary symptom relief with hot bathing, abdominal pain, and at least weekly use of marijuana. Other supportive features include aged < 50 years, weight loss > 5 kg, symptoms that are worse in the morning, normal bowel habits, and negative evaluation, including laboratory, radiography, and endoscopy (Table).5
Treatment often is supportive with emphasis placed on marijuana cessation. Intravenous fluids often are used due to dehydration from the emesis. The use of antiemetics, such as 5-HT3 (eg, ondansetron), D2 (eg, prochlorperazine), H1 (eg, promethazine), or neurokinin-1 receptor antagonists (eg, aprepitant) can be tried, but these therapies often are ineffective. Diet can be advanced as the patient tolerates. Given that many patients are found to have a mild gastritis, H2 blockers or proton pump inhibitors may be used. Extensive counseling on marijuana cessation is needed as it is the only therapy shown to have prolonged relief of the hyperemetic phase.6 The length of cessation from marijuana for resolution of the cyclical hyperemesis varies from 1 to 3 months. Returning to marijuana use often results in the returning of CHS.5
The pathophysiology of CHS is largely unknown; however, there are several hypothesized mechanisms. Many theorize that due to the lipophilicity and long half-life of THC, a primary compound in marijuana, it accumulates in the body over time.4,6 It is thought that this accumulation may cause toxicity in both the gastrointestinal tract as well as in the brain. Central effects on the hypothalamic-pituitary axis may play a major role, and the reason for the symptom relief of hot baths is due to a change in thermoregulation in the hypothalamus.5 One interesting mechanism relates to CB1 receptor activation and vasodilation within the gastrointestinal tract due to chronic THC accumulation. The relief of the abdominal pain, nausea, and vomiting with hot showers can be secondary to the vasodilation of the skin, causing a redistribution from the gut. This theorized mechanism has been referred to as “cutaneous steal.”9
Conclusion
With the increased prevalence of marijuana use in the U.S. over the past decade and reform in legislation taking place over the next couple of years, it is increasingly important to be able to recognize CHS to avoid frequent hospital utilization and repeated costly evaluations. Cannabinoid hyperemesis syndrome is recognized by the triad of chronic cannabis use, cyclical hyperemesis, and compulsive hot bathing.4
The syndrome has 3 phases. In the prodromal phase the patient has morning predominance of nausea, usually without emesis. This is followed by the hyperemesis phase, which is characterized by hyperemesis, vague abdominal pain, and learned compulsive hot bathing.
The third phase is the recovery phase, which is a return to normal behavior. During the recovery phase, if patients cease marijuana use, they remain asymptomatic; however, if patients continue to use marijuana, they often have recurrence of the hyperemesis phase.5 The diagnosis of cannabinoid hyperemesis syndrome is difficult as it is a diagnosis of exclusion. Patients may present to the ED many times prior to diagnosis. With the changing climate of marijuana laws, it is an important condition to consider when establishing a differential. More studies will be required to evaluate the overall prevalence of this condition as well as if there are any changes following the liberalization of marijuana laws in many states.
Given the recent rise in marijuana legalization efforts and an overall increase in the prevalence of marijuana use, it is becoming increasingly important to recognize conditions that are associated with its use. Data obtained from the National Survey on Drug Use and Health show the prevalence of marijuana use within the past month among those surveyed was 8.4% in 2014. This represents a 35% increase from the same study in 2002. Based on this survey, 2.5 million people (or ~7,000 per day) used marijuana for the first time.1
Following the liberalization of marijuana in Colorado, the prevalence of presentation to the emergency department (ED) for cyclic vomiting nearly doubled.2 During the 2016 election season, several states included legislation that increased access to marijuana on the ballot, most of which passed. There are now 28 states plus the District of Columbia that permit medical marijuana usage, and 8 of those states and the District of Columbia have laws allowing for recreational use of marijuana.3
First described in a case series by Allen and colleagues in 2004, cannabinoid hyperemesis syndrome (CHS) is indicated by recurrent episodes of nausea and vomiting with vague abdominal pain and compulsive hot bathing in the setting of chronic, often daily, cannabis use.4 A case of a middle-aged veteran with chronic marijuana use and recurrent, self-limited nausea and vomiting is presented here.
Case Presentation
A 45-year-old man presented to the ED with a 5-day history of persistent nausea and vomiting that began abruptly. The symptoms had been constant since onset, resulting in very little oral intake. The patient reported no hematemesis or coffee ground emesis. He noted a drop in his urine output over the previous 2 days. He also reported abdominal pain associated with the nausea. The patient characterized his pain as “dull and achy” diffuse pain that was partially relieved with emesis. His bowel movements had been regular, and he reported no diarrhea, fever, chills, or other constitutional symptoms. Additional 10-point review of systems was otherwise negative. The patient reported smoking marijuana multiple times daily for many years. The patient reported he had not used alcohol for several months.
A physical exam showed a pale and diaphoretic patient. Vital signs were significant for mild hypertension (150/75), but the patient was afebrile with a normal heart rate. An abdominal exam revealed a nontender, nondistended abdomen with no signs of rebound or guarding. The remainder of the examination was unremarkable. An initial workup showed a mild elevation of serum creatinine to 1.36 mg/dL (baseline is 1.10 mg/dL). Other workups, including complete blood count (CBC) with differential, complete metabolic panel, lipase, amylase, and urine analysis, were all unremarkable.
The patient’s urine drug screen (UDS) was positive for tetrahydrocannabinol (THC). A computed tomography (CT) scan of his abdomen and pelvis with contrast was unremarkable. The patient was admitted for his inability to tolerate oral intake and dehydration and treated supportively with IV fluids and antiemetics.
Overnight, the nursing staff reported that the patient took multiple, prolonged hot showers. Upon further questioning, he reported the hot showers significantly helped the nausea and abdominal pain. He had learned this behavior after experiencing previous episodes of self-limited nausea, vomiting, and abdominal pain.
Extensive review of his medical record revealed that the patient had, in fact, presented to the ED with similar symptoms 11 times in the prior 8 years. He was admitted on 8 occasions over that time frame. The typical hospital course included supportive care with antiemetics and IV fluids. The patient’s symptoms typically resolved within 24 to 72 hours of hospitalization. Previous evaluations included additional unremarkable CT imaging of the abdomen and pelvis. The patient also had received 2 esophagogastroduodenoscopies (EGDs), one 2 years prior and the other 5 years prior. Both EGDs showed only mild gastritis. On every check during the previous 8 years, the patient’s UDS was positive for THC.
Most of his previous admissions were attributed to viral gastroenteritis due to the self-limited nature of the symptoms. Other admissions were attributed to alcohol-induced gastritis. However, after abstaining from alcohol for long periods, the patient had continued recurrence of the symptoms and increased frequency of presentations to the ED.
The characteristics, signs, and symptoms of CHS were discussed with the patient. The patient strongly felt as though these symptoms aligned with his clinical course over the prior 8 years. At time of writing, the patient had gone 20 months without requiring hospitalization; however, he had a recent relapse of marijuana use and subsequently required hospitalization.
Discussion
As in this case, CHS often presents with refractory, self-limited nausea and vomiting with vague abdominal pain that is temporarily relieved by hot baths or showers. In the largest case series, it was noted the average age was 32 years, and the majority of subjects used marijuana at least weekly for > 2 years.5 Many studies categorize CHS into 3 phases: prodromal, hyperemetic, and recovery.
The prodromal, or preemetic phase, is characterized by early morning nausea without emesis and abdominal discomfort. The hyperemetic phase begins when the patient accesses the health care system via either the ED or primary care physician. This phase is characterized by intractable nausea and vomiting and may be associated with mild diffuse abdominal pain. The nausea and vomiting typically do not respond to antiemetic medications. Patients in this stage also develop a compulsive behavior of hot showers that temporarily relieve the symptoms. These behaviors are thought to be learned through their cyclical periods of emesis and may not be present during the first few hyperemetic phases. During the recovery phase, the patient returns to a baseline state of health and often ceases utilizing the hot shower. The recovery phase can last weeks to months despite continued cannabis use prior to returning to the hyperemetic phase (Figure).6,7
Simonetto and colleagues proposed clinical criteria for the diagnosis of CHS based on their case series as well as on previously proposed criteria presented by Sontineni and colleagues.5,8 Long-term cannabis use is required for the diagnosis. In the Simonetto and colleagues case series, the majority of patients developed symptoms within the first 5 years of cannabis use; however, Soriano and colleagues conducted a smaller case series that showed that the majority of subjects used marijuana for roughly 16 years prior to the onset of vomiting.5,7
The major CHS features that suggest the diagnosis are severe cyclic nausea and vomiting, relief of symptoms with abstinence from cannabis, temporary symptom relief with hot bathing, abdominal pain, and at least weekly use of marijuana. Other supportive features include aged < 50 years, weight loss > 5 kg, symptoms that are worse in the morning, normal bowel habits, and negative evaluation, including laboratory, radiography, and endoscopy (Table).5
Treatment often is supportive with emphasis placed on marijuana cessation. Intravenous fluids often are used due to dehydration from the emesis. The use of antiemetics, such as 5-HT3 (eg, ondansetron), D2 (eg, prochlorperazine), H1 (eg, promethazine), or neurokinin-1 receptor antagonists (eg, aprepitant) can be tried, but these therapies often are ineffective. Diet can be advanced as the patient tolerates. Given that many patients are found to have a mild gastritis, H2 blockers or proton pump inhibitors may be used. Extensive counseling on marijuana cessation is needed as it is the only therapy shown to have prolonged relief of the hyperemetic phase.6 The length of cessation from marijuana for resolution of the cyclical hyperemesis varies from 1 to 3 months. Returning to marijuana use often results in the returning of CHS.5
The pathophysiology of CHS is largely unknown; however, there are several hypothesized mechanisms. Many theorize that due to the lipophilicity and long half-life of THC, a primary compound in marijuana, it accumulates in the body over time.4,6 It is thought that this accumulation may cause toxicity in both the gastrointestinal tract as well as in the brain. Central effects on the hypothalamic-pituitary axis may play a major role, and the reason for the symptom relief of hot baths is due to a change in thermoregulation in the hypothalamus.5 One interesting mechanism relates to CB1 receptor activation and vasodilation within the gastrointestinal tract due to chronic THC accumulation. The relief of the abdominal pain, nausea, and vomiting with hot showers can be secondary to the vasodilation of the skin, causing a redistribution from the gut. This theorized mechanism has been referred to as “cutaneous steal.”9
Conclusion
With the increased prevalence of marijuana use in the U.S. over the past decade and reform in legislation taking place over the next couple of years, it is increasingly important to be able to recognize CHS to avoid frequent hospital utilization and repeated costly evaluations. Cannabinoid hyperemesis syndrome is recognized by the triad of chronic cannabis use, cyclical hyperemesis, and compulsive hot bathing.4
The syndrome has 3 phases. In the prodromal phase the patient has morning predominance of nausea, usually without emesis. This is followed by the hyperemesis phase, which is characterized by hyperemesis, vague abdominal pain, and learned compulsive hot bathing.
The third phase is the recovery phase, which is a return to normal behavior. During the recovery phase, if patients cease marijuana use, they remain asymptomatic; however, if patients continue to use marijuana, they often have recurrence of the hyperemesis phase.5 The diagnosis of cannabinoid hyperemesis syndrome is difficult as it is a diagnosis of exclusion. Patients may present to the ED many times prior to diagnosis. With the changing climate of marijuana laws, it is an important condition to consider when establishing a differential. More studies will be required to evaluate the overall prevalence of this condition as well as if there are any changes following the liberalization of marijuana laws in many states.
1. Azofeifa A, Mattson ME, Schauer G, McAfee T, Grant A, Lyerla R. National estimates of marijuana use and related indicators - National Survey on Drug Use and Health, United States, 2002-2014. MMWR Surveill Summ. 2016;65(11):1-28.
2. Kim HS, Anderson JD, Saghafi O, Heard KJ, Monte AA. Cyclic vomiting presentations following marijuana liberalization in Colorado. Acad Emerg Med. 2015;22(6):694-699.
3. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated July 7, 2017. Accessed August 3, 2017.
4. Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut. 2004;53(11):1566-1570.
5. Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc. 2012;87(2):114-119.
6. Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev. 2011;4(4):241-249.
7. Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci. 2010;55(11):3113-3119.
8. Sontineni SP, Chaudhary S, Sontineni V, Lanspa SJ. Cannabinoid hyperemesis syndrome: clinical diagnosis of an underrecognised manifestation of chronic cannabis abuse. World J Gastroenterol. 2009;15(10):1264-1266.
9. Patterson DA, Smith E, Monahan M, et al. Cannabinoid hyperemesis and compulsive bathing: a case series and paradoxical pathophysiological explanation. J Am Board Fam Med. 2010;23(6):790-793
1. Azofeifa A, Mattson ME, Schauer G, McAfee T, Grant A, Lyerla R. National estimates of marijuana use and related indicators - National Survey on Drug Use and Health, United States, 2002-2014. MMWR Surveill Summ. 2016;65(11):1-28.
2. Kim HS, Anderson JD, Saghafi O, Heard KJ, Monte AA. Cyclic vomiting presentations following marijuana liberalization in Colorado. Acad Emerg Med. 2015;22(6):694-699.
3. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated July 7, 2017. Accessed August 3, 2017.
4. Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut. 2004;53(11):1566-1570.
5. Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc. 2012;87(2):114-119.
6. Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev. 2011;4(4):241-249.
7. Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci. 2010;55(11):3113-3119.
8. Sontineni SP, Chaudhary S, Sontineni V, Lanspa SJ. Cannabinoid hyperemesis syndrome: clinical diagnosis of an underrecognised manifestation of chronic cannabis abuse. World J Gastroenterol. 2009;15(10):1264-1266.
9. Patterson DA, Smith E, Monahan M, et al. Cannabinoid hyperemesis and compulsive bathing: a case series and paradoxical pathophysiological explanation. J Am Board Fam Med. 2010;23(6):790-793