User login
Evaluating the Clinical and Demographic Features of Extrafacial Granuloma Faciale
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.

Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
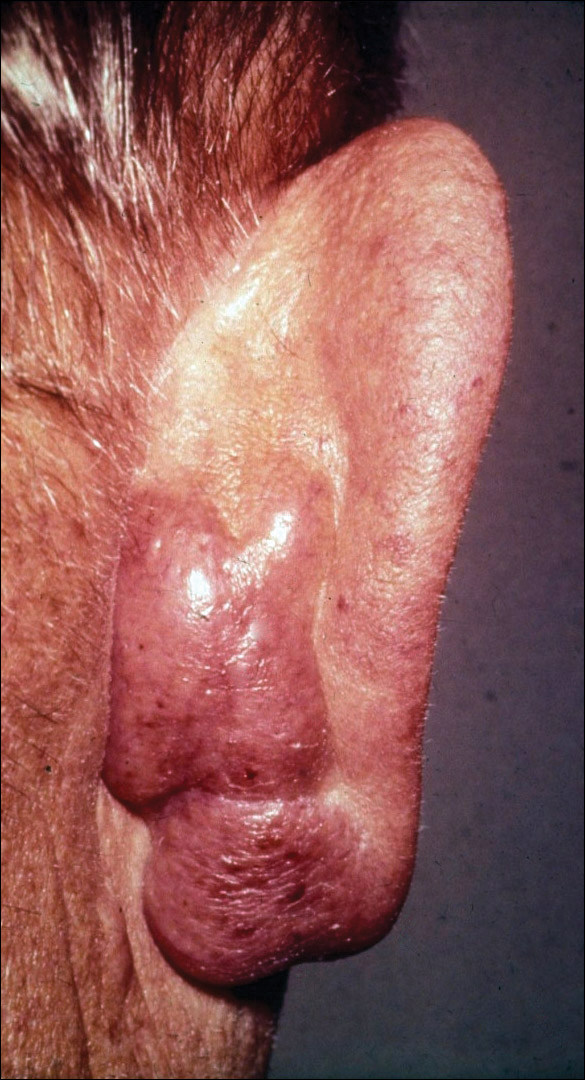
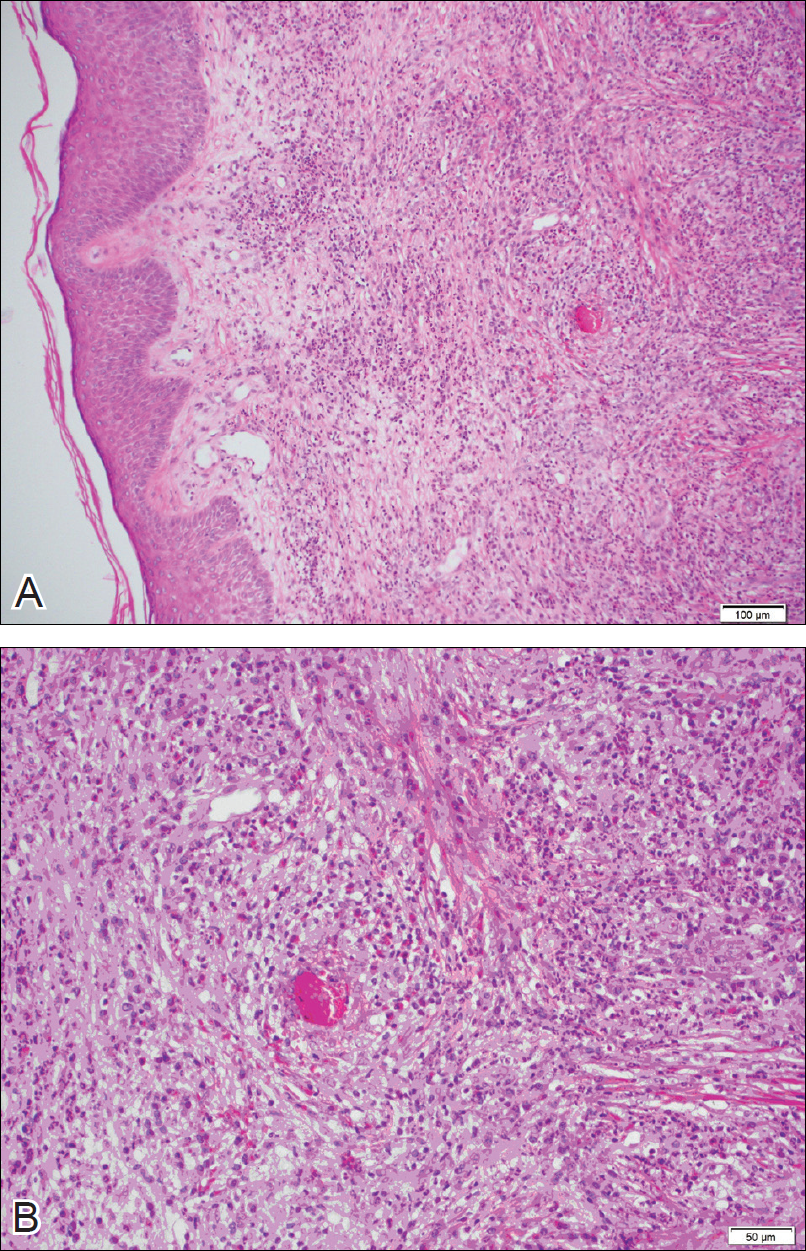
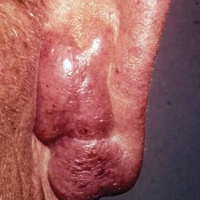
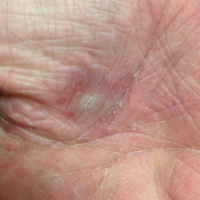
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.
Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.
Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
Practice Points
- Extrafacial lesions are rare in granuloma faciale (GF).
- Extrafacial GF should be included in the differential diagnosis of well-demarcated plaques and nodules found on the trunk or extremities.
- Diagnosis of extrafacial GF is based on the presence of distinct histologic features identical to GF.
- Granuloma faciale is a chronic benign leukocytoclastic vasculitis that can be difficult to treat.
Rowell Syndrome: Targeting a True Definition
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
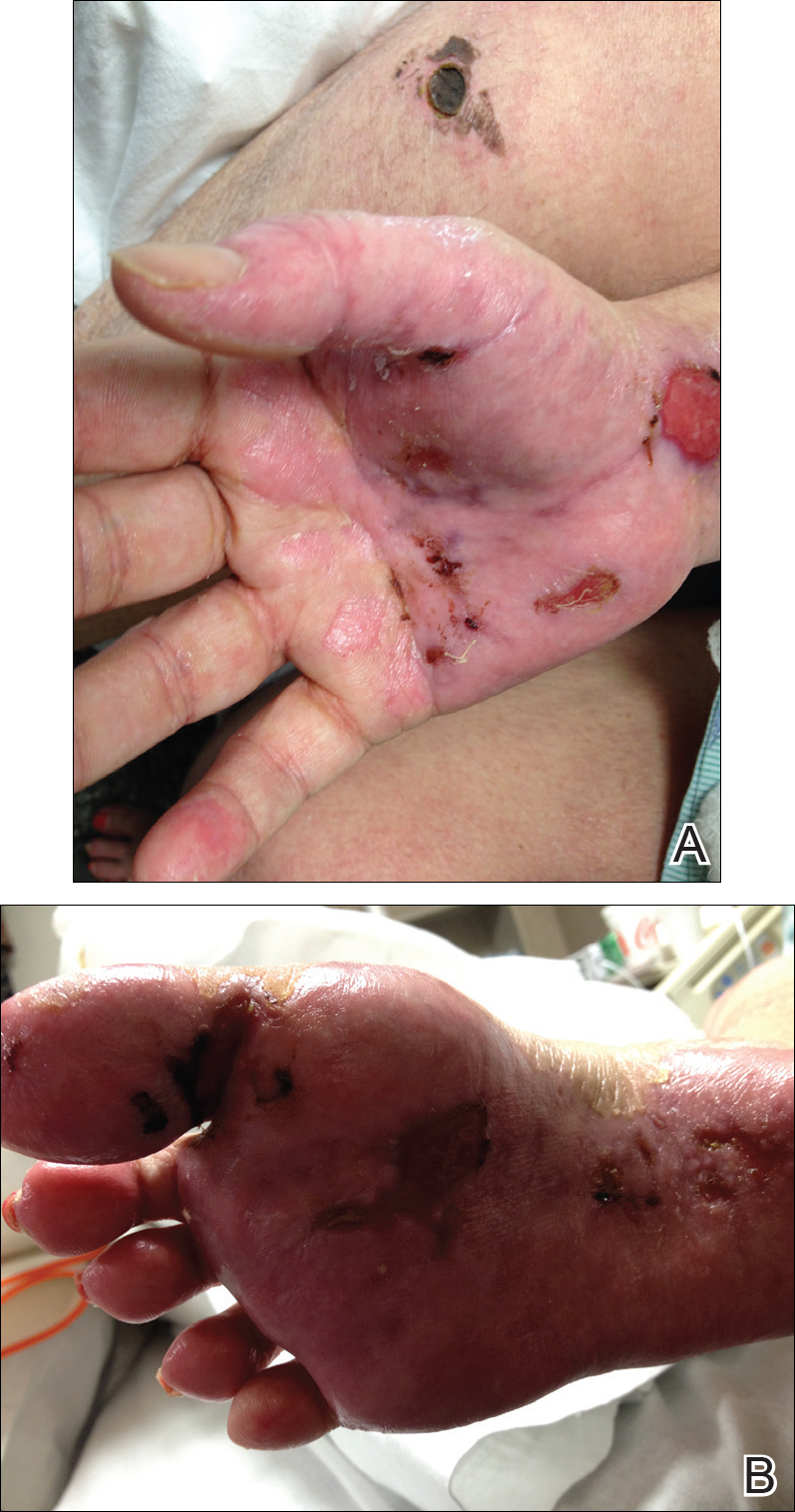
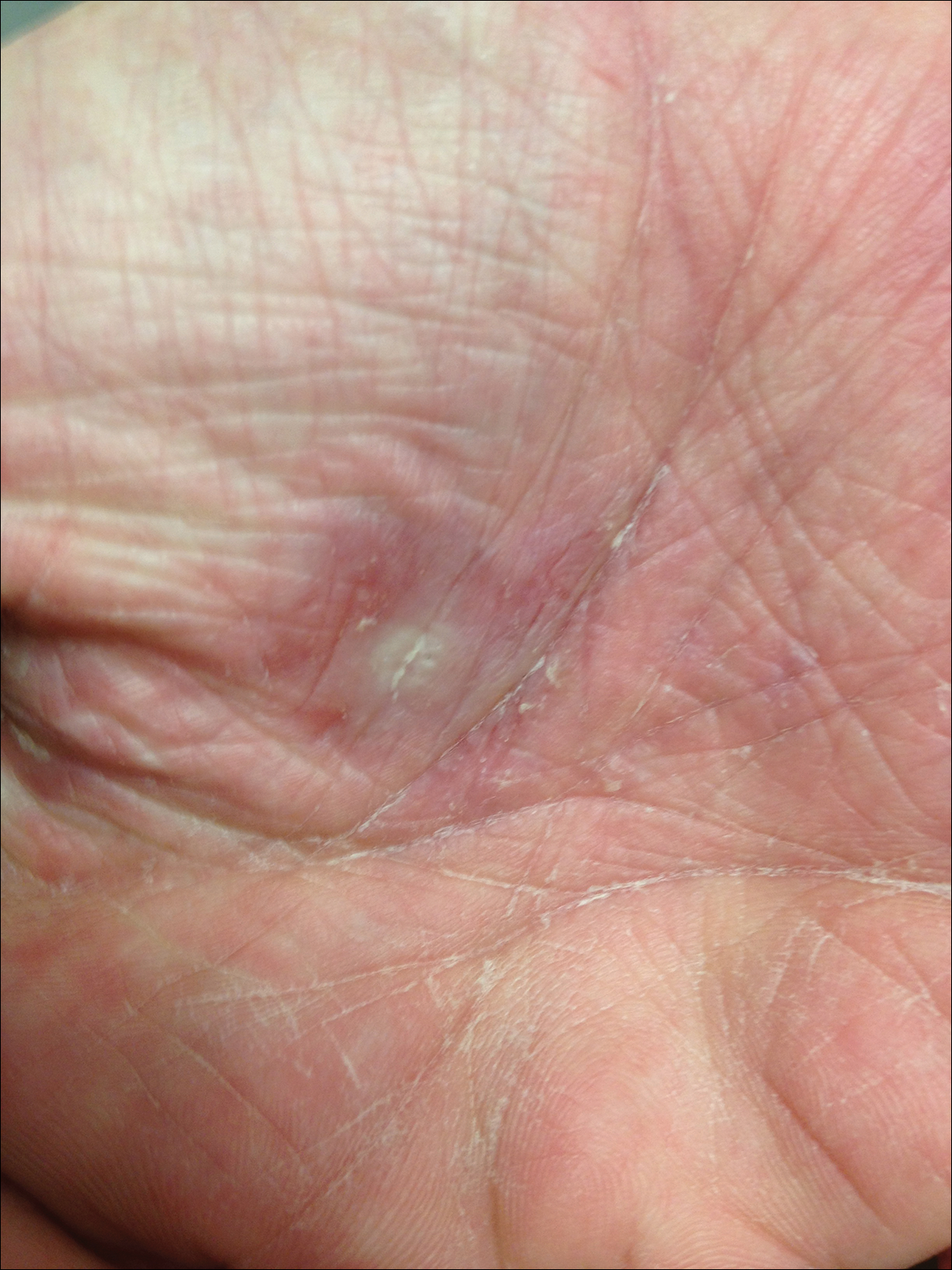
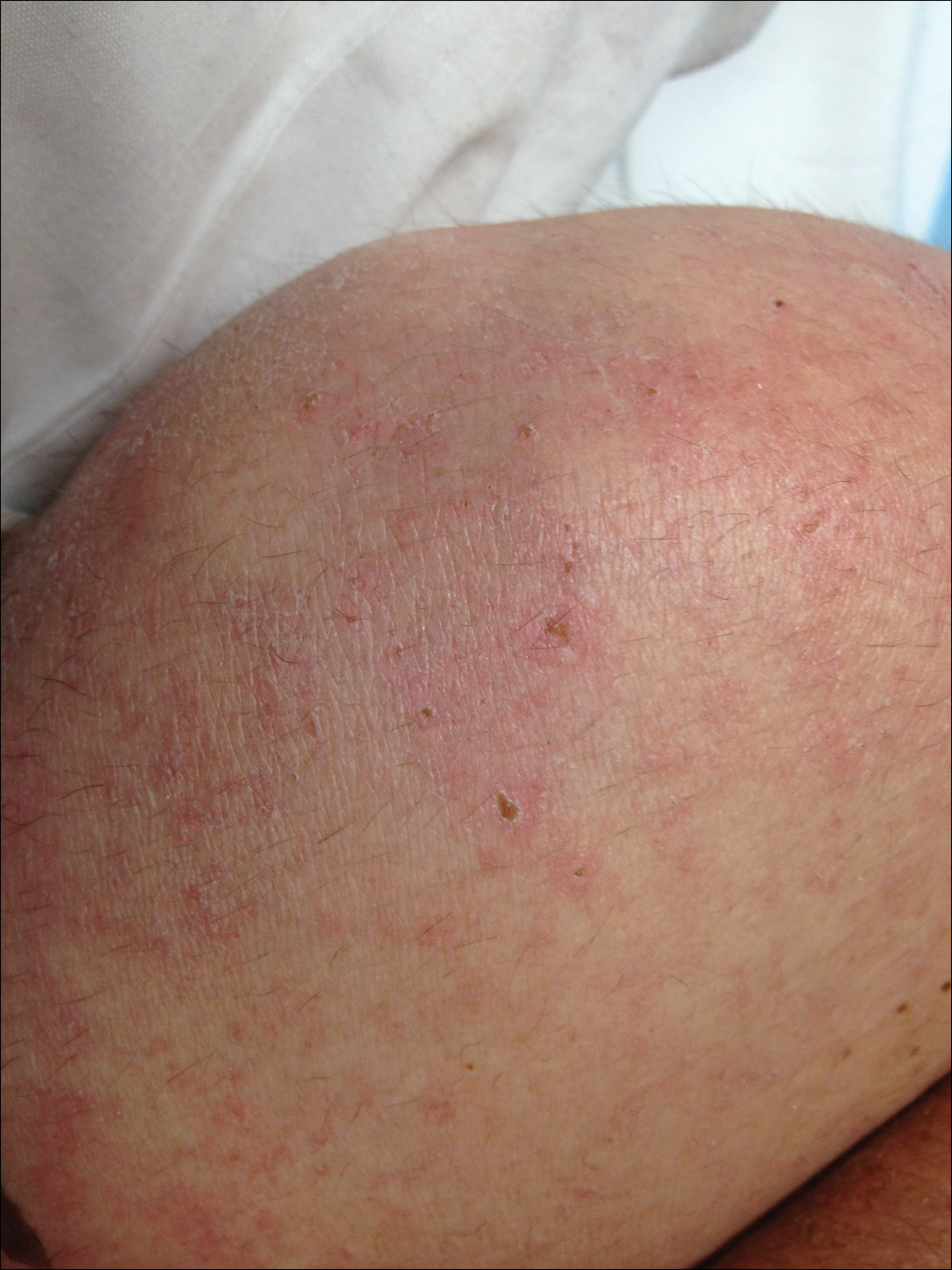
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
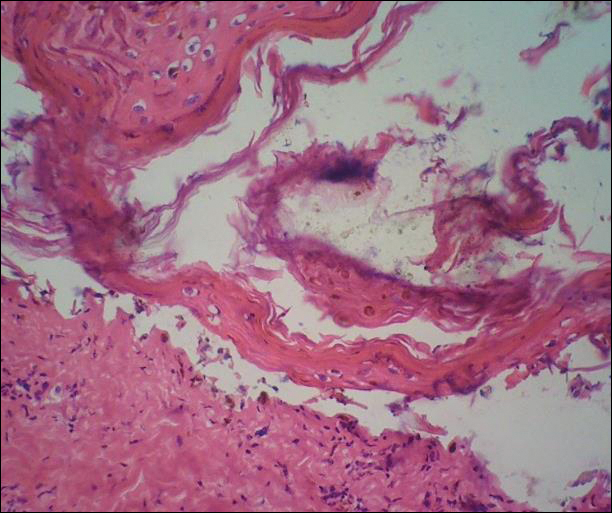
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.

Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Practice Points
- Rowell syndrome (RS) is an often unrecognized unique presentation of lupus erythematosus.
- There have been a variety of historical criteria that have sought to characterize RS.
Lower Limb Morel-Lavallée Lesion Treated With Short-Stretch Compression Bandaging
Take-Home Points
- Have a high-index of suspicion for MLLs and initiate treatment early.
- Compression needs to occur through short-stretch bandaging over a conventional Ace wrap in order to be successful.
- Apply the short-stretch compression with care to avoid shearing underlying tissue.
- Nonoperative treatment modalities require high patient compliance.
- MLLs need close monitoring until final healing occurs.
Morel-Lavallée lesions (MLLs) are traumatic degloving injuries resulting from separation of subcutaneous fat from underlying fascia. MLLs occur in association with acetabular fractures and are also associated with low-velocity crush injuries.1,2 Shearing creates a “false” space that is filled with hemorrhaged blood, fat, and lymphatic tissue.3 Disruption of the lymphatics leads to cavity formation and, eventually, a fibrotic pseudocapsule.4The pseudocapsule prevents resorption, leading to a chronic fluid collection, which potentiates the risk of infection or tissue necrosis.3,5,6 Skin necrosis may occur through direct-pressure compromise of the dermal vascular plexus.4 Necrotic skin may require multiple débridements, negative-pressure wound therapy or soft-tissue coverage, and may ultimately result in infection. MLLs classically occur in the greater trochanteric region, lateral thigh, buttocks, and back but also appear in the prepatellar region.1,3 Patients present with soft-tissue swelling, bruising, bulging, decreased cutaneous sensation over the region, and a palpable, fluctuant subcutaneous fluid collection with mobile skin.2,4,7 The mechanism of injury may cause a concomitant fracture. Magnetic resonance imaging (MRI), the preferred imaging modality, shows a discrete fluid collection between subcutaneous fat and underlying fascia. Ultrasonography may reveal a thickened capsule surrounding either a hypoechoic area or an anechoic area but its accuracy is user-dependent.7
Large MLLs may be treated with open serial débridement and healing by secondary intention; infection rates, however, are high. Authors have described several other treatment modalities, including percutaneous débridement with a brush followed by use of a large-bore drain and antibiotics; open débridement with meticulous dead-space closure; elastic compression bandaging; aspiration; and doxycycline sclerodesis.1,5,6,8,9 Modifications of short-stretch compression bandaging were recently described in edema control for hindfoot trauma, ankle trauma, and total ankle arthroplasty, but not for MLLs.10,11 Nickerson and colleagues4 retrospectively reviewed 87 MLLs, found that fluid aspirate of >50 mL predicted recurrence and failure with conservative measures, and recommended operative intervention for any MLL with >50 mL of fluid aspirated.
We report the case of an MLL that occurred in an unusual anatomical region, and we describe a novel application of a conservative treatment, which was selected on the basis of its success in lymphedema management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 66-year-old man was injured when a parked vehicle began moving, pulled him under, and ran over his lower right leg. In the emergency department, no fractures or major injuries were noted (Figures 1A, 1B), and the patient was discharged.
About 10 days after injury, profuse ecchymosis and swelling were noted running from the distal medial thigh to the proximal medial calf (Figures 2A-2C).
Given the size of the MLL, the fluid collection reaccumulated. The patient was evaluated by an orthopedic traumatologist 3 days after the aspiration (17 days after injury).
Another orthopedic traumatologist confirmed the low likelihood that compression would resolve the MLL, given its size (Figures 4A, 4B). 

After the second orthopedic consultation, the patient saw a physical therapist trained in complete decongestive therapy. The therapist suggested placing short-stretch bandage wraps over the conventional long-stretch Ace bandage currently being used—a treatment common in lymphedema. The patient was wrapped from toe to groin without an initial layer of padding (Figures 6A, 6B), and the response was immediate. 
Nine weeks after injury, the leg was significantly improved, and clinical signs resolved (Figure 7). 
Discussion
Short-stretch bandaging has been performed mainly in lymphedema and ulcer management. 
Compression bandaging reduces volume in lymphedematous limbs by reducing capillary filtration, shifting fluid into noncompressed parts of the body, increasing lymphatic reabsorption and lymphatic transport stimulation, improving venous pumping, and breaking down fibrosclerotic tissue.15 We think containment, improved venous flow, and enhanced muscle contraction contributed to the effectiveness of short-stretch bandaging as treatment for our patient’s MLL. Because MLLs also contain disrupted lymphatics, lymphedema management strategies (eg, short-stretch bandages) can be used. Our patient rapidly improved after conversion to short-stretch bandages.
These bandages are applied with 50% overlap to ensure even pressures throughout.16 Multiple layers are applied using a combination of spiral and figure-of-8 techniques, first clockwise and then counterclockwise, to avoid shearing underlying tissue.17 This method is very important in MLL treatment, given the degloving involved and the highly mobile skin and subcutaneous fat.
In standard lymphedema management, a foam padding layer is applied before the short-stretch bandage in order to reshape the limb and avoid proximal constrictions.13 In our patient’s case, the short-stretch wrap was applied without padding. Because his condition was acute, and the limb contour was preserved, limb reshaping and thus padding were not necessary.
Given the rapid, high-volume reduction that occurs within the first 1 to 2 weeks, bandages are reapplied daily to effectively adjust for the decreased swelling and altered limb shape.17 Most improvement is expected within the first few weeks—consistent with our patient’s case. Bandages usually are applied to the entire limb. For partial cases, the bandaging must extend past the area of swelling and incorporate the knee to prevent displacement of fluid into the joint.17 Feet and ankles are bandaged in dorsiflexion.17Several factors must be considered with short-stretch wraps. For example, pressure may need to be adjusted in patients with peripheral vascular disease. In patients with ankle-brachial indexes >0.5, it is safe to apply pressure up to 40 mm Hg.12 Reduced pressure is recommended for patients with arterial disease, sensory disturbance, lipoedema, poor mobility, frailty, or palliative needs.13The unusual location of our patient’s MLL accounts for the delay in diagnosis. To our knowledge, no other authors have reported such a large MLL in this location. A few series and case reports have listed MLLs in the calf near the gastrocnemius muscle, in the ankle, in the prepatellar area, and in the suprapatellar region, including the thigh,1,3,18-20 but there are no reports of MLLs running from medial thigh to proximal calf. MLLs of this size classically are treated surgically, but our patient selected nonoperative management.
To our knowledge, there are no earlier reports of using this nonoperative technique to treat MLLs. Conservative treatment with compression has been discussed, but no case involved short-stretch bandages. Large MLLs are thought to require surgery plus some type of drainage. The success of using short-stretch bandages in our patient’s case should prompt further investigation of use in adherent patients—which could ultimately result in reduced surgical needs, improved wound care (surgery is avoided), and a maintained low risk of infection. Although more work is needed to come to a more definitive verdict on this treatment method, it is a promising option that warrants consideration.
Am J Orthop. 2017;46(4):E213-E218. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
2. Tsur A, Galin A, Kogan L, Loberant N. Morel-Lavallee syndrome after crush injury [in Hebrew]. Harefuah. 2006;145(2):111-113.
3. Ciaschini M, Sundaram M. Radiologic case study. Prepatellar Morel-Lavallée lesion. Orthopedics. 2008;31(7):626, 719-721.
4. Nickerson TP, Zielinski MD, Jenkins DH, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014;76(2):493-497.
5. Bansal A, Bhatia N, Singh A, Singh AK. Doxycycline sclerodesis as a treatment option for persistent Morel-Lavallée lesions. Injury. 2013;44(1):66-69.
6. Carlson DA, Simmons J, Sando W, Weber T, Clements B. Morel-Lavalée lesions treated with debridement and meticulous dead space closure: surgical technique. J Orthop Trauma. 2007;21(2):140-144.
7. Miller J, Daggett J, Ambay R, Payne WG. Morel-Lavallée lesion. Eplasty. 2014;14:ic12.
8. Tseng S, Tornetta P 3rd. Percutaneous management of Morel-Lavallee lesions. J Bone Joint Surg Am. 2006;88(1):92-96.
9. Harma A, Inan M, Ertem K. The Morel-Lavallée lesion: a conservative approach to closed degloving injuries [in Turkish]. Acta Orthop Traumatol Turc. 2004;38(4):270-273.
10. Hsu A, Franceschina D, Haddad SL. A novel method of postoperative wound care following total ankle arthroplasty. Foot Ankle Int. 2014;35(7):719-724.
11. Rohner-Spengler M, Frotzler A, Honigmann P, Babst R. Effective treatment of posttraumatic and postoperative edema in patients with ankle and hindfoot fractures: a randomized controlled trial comparing multilayer compression therapy and intermittent impulse compression with the standard treatment with ice. J Bone Joint Surg Am. 2014;96(15):1263-1271.
12. Bjork R. The long and short of it: understanding compression bandaging. Wound Care Advisor. 2013;2(6):12-15.
13. Partsch H. Assessing the effectiveness of multilayer inelastic bandaging. J Lymphoedema. 2007;2(2):55-61.
14. Hafner J, Botonakis I, Burg G. A comparison of multilayer bandage systems during rest, exercise, and over 2 days of wear time. Arch Dermatol. 2000;136(7):857-863.
15. Földi E, Jünger M, Partsch H. The science of lymphoedema bandaging. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:2-4.
16. King TI, Droessler JL. Physical properties of short-stretch compression bandages used to treat lymphedema. Am J Occup Ther. 2001;55(5):573-576.
17. Williams AF, Keller M. Practical guidance on lymphoedema bandaging of the upper and lower limbs. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:10-14.
18. Moriarty JM, Borrero CG, Kavanagh EC. A rare cause of calf swelling: the Morel-Lavallee lesion. Ir J Med Sci. 2011;180(1):265-268.
19. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Knee Morel-Lavallee lesion after a football injury in an 11-year-old boy: case report and review of the literature. Univ Pa Orthop J. 2011;21:56-58.
20. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
Take-Home Points
- Have a high-index of suspicion for MLLs and initiate treatment early.
- Compression needs to occur through short-stretch bandaging over a conventional Ace wrap in order to be successful.
- Apply the short-stretch compression with care to avoid shearing underlying tissue.
- Nonoperative treatment modalities require high patient compliance.
- MLLs need close monitoring until final healing occurs.
Morel-Lavallée lesions (MLLs) are traumatic degloving injuries resulting from separation of subcutaneous fat from underlying fascia. MLLs occur in association with acetabular fractures and are also associated with low-velocity crush injuries.1,2 Shearing creates a “false” space that is filled with hemorrhaged blood, fat, and lymphatic tissue.3 Disruption of the lymphatics leads to cavity formation and, eventually, a fibrotic pseudocapsule.4The pseudocapsule prevents resorption, leading to a chronic fluid collection, which potentiates the risk of infection or tissue necrosis.3,5,6 Skin necrosis may occur through direct-pressure compromise of the dermal vascular plexus.4 Necrotic skin may require multiple débridements, negative-pressure wound therapy or soft-tissue coverage, and may ultimately result in infection. MLLs classically occur in the greater trochanteric region, lateral thigh, buttocks, and back but also appear in the prepatellar region.1,3 Patients present with soft-tissue swelling, bruising, bulging, decreased cutaneous sensation over the region, and a palpable, fluctuant subcutaneous fluid collection with mobile skin.2,4,7 The mechanism of injury may cause a concomitant fracture. Magnetic resonance imaging (MRI), the preferred imaging modality, shows a discrete fluid collection between subcutaneous fat and underlying fascia. Ultrasonography may reveal a thickened capsule surrounding either a hypoechoic area or an anechoic area but its accuracy is user-dependent.7
Large MLLs may be treated with open serial débridement and healing by secondary intention; infection rates, however, are high. Authors have described several other treatment modalities, including percutaneous débridement with a brush followed by use of a large-bore drain and antibiotics; open débridement with meticulous dead-space closure; elastic compression bandaging; aspiration; and doxycycline sclerodesis.1,5,6,8,9 Modifications of short-stretch compression bandaging were recently described in edema control for hindfoot trauma, ankle trauma, and total ankle arthroplasty, but not for MLLs.10,11 Nickerson and colleagues4 retrospectively reviewed 87 MLLs, found that fluid aspirate of >50 mL predicted recurrence and failure with conservative measures, and recommended operative intervention for any MLL with >50 mL of fluid aspirated.
We report the case of an MLL that occurred in an unusual anatomical region, and we describe a novel application of a conservative treatment, which was selected on the basis of its success in lymphedema management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 66-year-old man was injured when a parked vehicle began moving, pulled him under, and ran over his lower right leg. In the emergency department, no fractures or major injuries were noted (Figures 1A, 1B), and the patient was discharged.
About 10 days after injury, profuse ecchymosis and swelling were noted running from the distal medial thigh to the proximal medial calf (Figures 2A-2C).
Given the size of the MLL, the fluid collection reaccumulated. The patient was evaluated by an orthopedic traumatologist 3 days after the aspiration (17 days after injury).
Another orthopedic traumatologist confirmed the low likelihood that compression would resolve the MLL, given its size (Figures 4A, 4B).
After the second orthopedic consultation, the patient saw a physical therapist trained in complete decongestive therapy. The therapist suggested placing short-stretch bandage wraps over the conventional long-stretch Ace bandage currently being used—a treatment common in lymphedema. The patient was wrapped from toe to groin without an initial layer of padding (Figures 6A, 6B), and the response was immediate.
Nine weeks after injury, the leg was significantly improved, and clinical signs resolved (Figure 7).
Discussion
Short-stretch bandaging has been performed mainly in lymphedema and ulcer management.
Compression bandaging reduces volume in lymphedematous limbs by reducing capillary filtration, shifting fluid into noncompressed parts of the body, increasing lymphatic reabsorption and lymphatic transport stimulation, improving venous pumping, and breaking down fibrosclerotic tissue.15 We think containment, improved venous flow, and enhanced muscle contraction contributed to the effectiveness of short-stretch bandaging as treatment for our patient’s MLL. Because MLLs also contain disrupted lymphatics, lymphedema management strategies (eg, short-stretch bandages) can be used. Our patient rapidly improved after conversion to short-stretch bandages.
These bandages are applied with 50% overlap to ensure even pressures throughout.16 Multiple layers are applied using a combination of spiral and figure-of-8 techniques, first clockwise and then counterclockwise, to avoid shearing underlying tissue.17 This method is very important in MLL treatment, given the degloving involved and the highly mobile skin and subcutaneous fat.
In standard lymphedema management, a foam padding layer is applied before the short-stretch bandage in order to reshape the limb and avoid proximal constrictions.13 In our patient’s case, the short-stretch wrap was applied without padding. Because his condition was acute, and the limb contour was preserved, limb reshaping and thus padding were not necessary.
Given the rapid, high-volume reduction that occurs within the first 1 to 2 weeks, bandages are reapplied daily to effectively adjust for the decreased swelling and altered limb shape.17 Most improvement is expected within the first few weeks—consistent with our patient’s case. Bandages usually are applied to the entire limb. For partial cases, the bandaging must extend past the area of swelling and incorporate the knee to prevent displacement of fluid into the joint.17 Feet and ankles are bandaged in dorsiflexion.17Several factors must be considered with short-stretch wraps. For example, pressure may need to be adjusted in patients with peripheral vascular disease. In patients with ankle-brachial indexes >0.5, it is safe to apply pressure up to 40 mm Hg.12 Reduced pressure is recommended for patients with arterial disease, sensory disturbance, lipoedema, poor mobility, frailty, or palliative needs.13The unusual location of our patient’s MLL accounts for the delay in diagnosis. To our knowledge, no other authors have reported such a large MLL in this location. A few series and case reports have listed MLLs in the calf near the gastrocnemius muscle, in the ankle, in the prepatellar area, and in the suprapatellar region, including the thigh,1,3,18-20 but there are no reports of MLLs running from medial thigh to proximal calf. MLLs of this size classically are treated surgically, but our patient selected nonoperative management.
To our knowledge, there are no earlier reports of using this nonoperative technique to treat MLLs. Conservative treatment with compression has been discussed, but no case involved short-stretch bandages. Large MLLs are thought to require surgery plus some type of drainage. The success of using short-stretch bandages in our patient’s case should prompt further investigation of use in adherent patients—which could ultimately result in reduced surgical needs, improved wound care (surgery is avoided), and a maintained low risk of infection. Although more work is needed to come to a more definitive verdict on this treatment method, it is a promising option that warrants consideration.
Am J Orthop. 2017;46(4):E213-E218. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Have a high-index of suspicion for MLLs and initiate treatment early.
- Compression needs to occur through short-stretch bandaging over a conventional Ace wrap in order to be successful.
- Apply the short-stretch compression with care to avoid shearing underlying tissue.
- Nonoperative treatment modalities require high patient compliance.
- MLLs need close monitoring until final healing occurs.
Morel-Lavallée lesions (MLLs) are traumatic degloving injuries resulting from separation of subcutaneous fat from underlying fascia. MLLs occur in association with acetabular fractures and are also associated with low-velocity crush injuries.1,2 Shearing creates a “false” space that is filled with hemorrhaged blood, fat, and lymphatic tissue.3 Disruption of the lymphatics leads to cavity formation and, eventually, a fibrotic pseudocapsule.4The pseudocapsule prevents resorption, leading to a chronic fluid collection, which potentiates the risk of infection or tissue necrosis.3,5,6 Skin necrosis may occur through direct-pressure compromise of the dermal vascular plexus.4 Necrotic skin may require multiple débridements, negative-pressure wound therapy or soft-tissue coverage, and may ultimately result in infection. MLLs classically occur in the greater trochanteric region, lateral thigh, buttocks, and back but also appear in the prepatellar region.1,3 Patients present with soft-tissue swelling, bruising, bulging, decreased cutaneous sensation over the region, and a palpable, fluctuant subcutaneous fluid collection with mobile skin.2,4,7 The mechanism of injury may cause a concomitant fracture. Magnetic resonance imaging (MRI), the preferred imaging modality, shows a discrete fluid collection between subcutaneous fat and underlying fascia. Ultrasonography may reveal a thickened capsule surrounding either a hypoechoic area or an anechoic area but its accuracy is user-dependent.7
Large MLLs may be treated with open serial débridement and healing by secondary intention; infection rates, however, are high. Authors have described several other treatment modalities, including percutaneous débridement with a brush followed by use of a large-bore drain and antibiotics; open débridement with meticulous dead-space closure; elastic compression bandaging; aspiration; and doxycycline sclerodesis.1,5,6,8,9 Modifications of short-stretch compression bandaging were recently described in edema control for hindfoot trauma, ankle trauma, and total ankle arthroplasty, but not for MLLs.10,11 Nickerson and colleagues4 retrospectively reviewed 87 MLLs, found that fluid aspirate of >50 mL predicted recurrence and failure with conservative measures, and recommended operative intervention for any MLL with >50 mL of fluid aspirated.
We report the case of an MLL that occurred in an unusual anatomical region, and we describe a novel application of a conservative treatment, which was selected on the basis of its success in lymphedema management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 66-year-old man was injured when a parked vehicle began moving, pulled him under, and ran over his lower right leg. In the emergency department, no fractures or major injuries were noted (Figures 1A, 1B), and the patient was discharged.
About 10 days after injury, profuse ecchymosis and swelling were noted running from the distal medial thigh to the proximal medial calf (Figures 2A-2C).
Given the size of the MLL, the fluid collection reaccumulated. The patient was evaluated by an orthopedic traumatologist 3 days after the aspiration (17 days after injury).
Another orthopedic traumatologist confirmed the low likelihood that compression would resolve the MLL, given its size (Figures 4A, 4B).
After the second orthopedic consultation, the patient saw a physical therapist trained in complete decongestive therapy. The therapist suggested placing short-stretch bandage wraps over the conventional long-stretch Ace bandage currently being used—a treatment common in lymphedema. The patient was wrapped from toe to groin without an initial layer of padding (Figures 6A, 6B), and the response was immediate.
Nine weeks after injury, the leg was significantly improved, and clinical signs resolved (Figure 7).
Discussion
Short-stretch bandaging has been performed mainly in lymphedema and ulcer management.
Compression bandaging reduces volume in lymphedematous limbs by reducing capillary filtration, shifting fluid into noncompressed parts of the body, increasing lymphatic reabsorption and lymphatic transport stimulation, improving venous pumping, and breaking down fibrosclerotic tissue.15 We think containment, improved venous flow, and enhanced muscle contraction contributed to the effectiveness of short-stretch bandaging as treatment for our patient’s MLL. Because MLLs also contain disrupted lymphatics, lymphedema management strategies (eg, short-stretch bandages) can be used. Our patient rapidly improved after conversion to short-stretch bandages.
These bandages are applied with 50% overlap to ensure even pressures throughout.16 Multiple layers are applied using a combination of spiral and figure-of-8 techniques, first clockwise and then counterclockwise, to avoid shearing underlying tissue.17 This method is very important in MLL treatment, given the degloving involved and the highly mobile skin and subcutaneous fat.
In standard lymphedema management, a foam padding layer is applied before the short-stretch bandage in order to reshape the limb and avoid proximal constrictions.13 In our patient’s case, the short-stretch wrap was applied without padding. Because his condition was acute, and the limb contour was preserved, limb reshaping and thus padding were not necessary.
Given the rapid, high-volume reduction that occurs within the first 1 to 2 weeks, bandages are reapplied daily to effectively adjust for the decreased swelling and altered limb shape.17 Most improvement is expected within the first few weeks—consistent with our patient’s case. Bandages usually are applied to the entire limb. For partial cases, the bandaging must extend past the area of swelling and incorporate the knee to prevent displacement of fluid into the joint.17 Feet and ankles are bandaged in dorsiflexion.17Several factors must be considered with short-stretch wraps. For example, pressure may need to be adjusted in patients with peripheral vascular disease. In patients with ankle-brachial indexes >0.5, it is safe to apply pressure up to 40 mm Hg.12 Reduced pressure is recommended for patients with arterial disease, sensory disturbance, lipoedema, poor mobility, frailty, or palliative needs.13The unusual location of our patient’s MLL accounts for the delay in diagnosis. To our knowledge, no other authors have reported such a large MLL in this location. A few series and case reports have listed MLLs in the calf near the gastrocnemius muscle, in the ankle, in the prepatellar area, and in the suprapatellar region, including the thigh,1,3,18-20 but there are no reports of MLLs running from medial thigh to proximal calf. MLLs of this size classically are treated surgically, but our patient selected nonoperative management.
To our knowledge, there are no earlier reports of using this nonoperative technique to treat MLLs. Conservative treatment with compression has been discussed, but no case involved short-stretch bandages. Large MLLs are thought to require surgery plus some type of drainage. The success of using short-stretch bandages in our patient’s case should prompt further investigation of use in adherent patients—which could ultimately result in reduced surgical needs, improved wound care (surgery is avoided), and a maintained low risk of infection. Although more work is needed to come to a more definitive verdict on this treatment method, it is a promising option that warrants consideration.
Am J Orthop. 2017;46(4):E213-E218. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
2. Tsur A, Galin A, Kogan L, Loberant N. Morel-Lavallee syndrome after crush injury [in Hebrew]. Harefuah. 2006;145(2):111-113.
3. Ciaschini M, Sundaram M. Radiologic case study. Prepatellar Morel-Lavallée lesion. Orthopedics. 2008;31(7):626, 719-721.
4. Nickerson TP, Zielinski MD, Jenkins DH, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014;76(2):493-497.
5. Bansal A, Bhatia N, Singh A, Singh AK. Doxycycline sclerodesis as a treatment option for persistent Morel-Lavallée lesions. Injury. 2013;44(1):66-69.
6. Carlson DA, Simmons J, Sando W, Weber T, Clements B. Morel-Lavalée lesions treated with debridement and meticulous dead space closure: surgical technique. J Orthop Trauma. 2007;21(2):140-144.
7. Miller J, Daggett J, Ambay R, Payne WG. Morel-Lavallée lesion. Eplasty. 2014;14:ic12.
8. Tseng S, Tornetta P 3rd. Percutaneous management of Morel-Lavallee lesions. J Bone Joint Surg Am. 2006;88(1):92-96.
9. Harma A, Inan M, Ertem K. The Morel-Lavallée lesion: a conservative approach to closed degloving injuries [in Turkish]. Acta Orthop Traumatol Turc. 2004;38(4):270-273.
10. Hsu A, Franceschina D, Haddad SL. A novel method of postoperative wound care following total ankle arthroplasty. Foot Ankle Int. 2014;35(7):719-724.
11. Rohner-Spengler M, Frotzler A, Honigmann P, Babst R. Effective treatment of posttraumatic and postoperative edema in patients with ankle and hindfoot fractures: a randomized controlled trial comparing multilayer compression therapy and intermittent impulse compression with the standard treatment with ice. J Bone Joint Surg Am. 2014;96(15):1263-1271.
12. Bjork R. The long and short of it: understanding compression bandaging. Wound Care Advisor. 2013;2(6):12-15.
13. Partsch H. Assessing the effectiveness of multilayer inelastic bandaging. J Lymphoedema. 2007;2(2):55-61.
14. Hafner J, Botonakis I, Burg G. A comparison of multilayer bandage systems during rest, exercise, and over 2 days of wear time. Arch Dermatol. 2000;136(7):857-863.
15. Földi E, Jünger M, Partsch H. The science of lymphoedema bandaging. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:2-4.
16. King TI, Droessler JL. Physical properties of short-stretch compression bandages used to treat lymphedema. Am J Occup Ther. 2001;55(5):573-576.
17. Williams AF, Keller M. Practical guidance on lymphoedema bandaging of the upper and lower limbs. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:10-14.
18. Moriarty JM, Borrero CG, Kavanagh EC. A rare cause of calf swelling: the Morel-Lavallee lesion. Ir J Med Sci. 2011;180(1):265-268.
19. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Knee Morel-Lavallee lesion after a football injury in an 11-year-old boy: case report and review of the literature. Univ Pa Orthop J. 2011;21:56-58.
20. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
1. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
2. Tsur A, Galin A, Kogan L, Loberant N. Morel-Lavallee syndrome after crush injury [in Hebrew]. Harefuah. 2006;145(2):111-113.
3. Ciaschini M, Sundaram M. Radiologic case study. Prepatellar Morel-Lavallée lesion. Orthopedics. 2008;31(7):626, 719-721.
4. Nickerson TP, Zielinski MD, Jenkins DH, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014;76(2):493-497.
5. Bansal A, Bhatia N, Singh A, Singh AK. Doxycycline sclerodesis as a treatment option for persistent Morel-Lavallée lesions. Injury. 2013;44(1):66-69.
6. Carlson DA, Simmons J, Sando W, Weber T, Clements B. Morel-Lavalée lesions treated with debridement and meticulous dead space closure: surgical technique. J Orthop Trauma. 2007;21(2):140-144.
7. Miller J, Daggett J, Ambay R, Payne WG. Morel-Lavallée lesion. Eplasty. 2014;14:ic12.
8. Tseng S, Tornetta P 3rd. Percutaneous management of Morel-Lavallee lesions. J Bone Joint Surg Am. 2006;88(1):92-96.
9. Harma A, Inan M, Ertem K. The Morel-Lavallée lesion: a conservative approach to closed degloving injuries [in Turkish]. Acta Orthop Traumatol Turc. 2004;38(4):270-273.
10. Hsu A, Franceschina D, Haddad SL. A novel method of postoperative wound care following total ankle arthroplasty. Foot Ankle Int. 2014;35(7):719-724.
11. Rohner-Spengler M, Frotzler A, Honigmann P, Babst R. Effective treatment of posttraumatic and postoperative edema in patients with ankle and hindfoot fractures: a randomized controlled trial comparing multilayer compression therapy and intermittent impulse compression with the standard treatment with ice. J Bone Joint Surg Am. 2014;96(15):1263-1271.
12. Bjork R. The long and short of it: understanding compression bandaging. Wound Care Advisor. 2013;2(6):12-15.
13. Partsch H. Assessing the effectiveness of multilayer inelastic bandaging. J Lymphoedema. 2007;2(2):55-61.
14. Hafner J, Botonakis I, Burg G. A comparison of multilayer bandage systems during rest, exercise, and over 2 days of wear time. Arch Dermatol. 2000;136(7):857-863.
15. Földi E, Jünger M, Partsch H. The science of lymphoedema bandaging. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:2-4.
16. King TI, Droessler JL. Physical properties of short-stretch compression bandages used to treat lymphedema. Am J Occup Ther. 2001;55(5):573-576.
17. Williams AF, Keller M. Practical guidance on lymphoedema bandaging of the upper and lower limbs. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:10-14.
18. Moriarty JM, Borrero CG, Kavanagh EC. A rare cause of calf swelling: the Morel-Lavallee lesion. Ir J Med Sci. 2011;180(1):265-268.
19. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Knee Morel-Lavallee lesion after a football injury in an 11-year-old boy: case report and review of the literature. Univ Pa Orthop J. 2011;21:56-58.
20. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
Symptoms Mimicking Those of Hypokalemic Periodic Paralysis Induced by Soluble Barium Poisoning
Hypokalemic periodic paralysis (HPP) is a relatively common and potentially life-threating condition that can be either sporadic or recurring and has both inherited and acquired causes.1 Familial HPP, on the other hand, is a rare condition (1:100,000) caused by loss of function mutations leading to the disruption of membrane potential consequently making them inexcitable.2 Appearance of symptoms is typically in the first or second decade of life (60% of cases have onset aged < 16 years) with susceptible individuals experiencing sudden onset of perioral numbness; weakness; centrifugal paralysis, often with nausea; vomiting and diarrhea; and prostration, usually triggered by highcarbohydrate meals and rest following sustained muscle-group use.3
These symptoms are common to all forms of HPP, making the differential diagnosis wide and confusing. Rhabdomyolysis is occasionally associated with many severe hypokalemic episodes.4 Myopathy and permanent muscle weakness have been reported in HPP.5,6 Other reported inciting factors include a drop in serum potassium caused by β-adrenergic bronchodilator treatment.7 Clinical attacks also have been associated with diabetic ketoacidosis and combined hypokalemia and hypophosphatemia.8 Thyrotoxicosis also causes similar muscle action potential changes but only when hyperthyroidism is uncorrected. 9-12 Less commonly, hypothyroidism has been reported to be associated with hypokalemic paralysis.3
Pa Ping, a condition involving hypokalemic paralysis of uncertain etiology, is geographically centered in the Szechuan region of China.13 Cases of Bartter, Liddle, and Gitelman syndromes also have been associated with hypokalemic paralysis.3,14 There is an association with malignant hyperthermia following or during systemic anesthesia. Patients presenting as Guillain-Barré syndrome have been found to have periodic paralysis triggered by hypokalemia from any cause.15 Sjögren syndrome and renal tubular acidosis also are reported to have triggered symptoms of hypokalemic paralysis.16,17
True type 1 HPP is caused by channelopathies resulting from mutations in the calcium channel gene CACN1AS (HypoPP1), which accounts for 70% of the cases, whereas type 2 HPP is cause by sodium channel gene SCN4A (HypoPP2) mutations, which accounts for 10% to 20% of cases.18,19 An association with a voltage-gated potassium channel KCNE3 mutation has been made but is disputed.20,21 Females typically have less severe and less frequent attacks, and attacks lessen or disappear during pregnancy.22
In a small controlled trial, acetazolamide has been reported to have prophylactic benefit, although a more powerful carbonic anhydrase inhibitor, dichlorophenamide, was reported to be effective in a study after acetazolamide had become ineffective.23,24 These treatments would not be expected to be of clinical use in hypokalemia due to barium poisoning.
Barium poisoning has been reported as a result of accidental contamination of foodstuffs with soluble barium.25 Onset of symptoms is rapid, with nausea, vomiting, diarrhea, and malaise followed rapidly by weakness, which can include the muscles of respiration. This littleconsidered but rapidly lethal poisoning event can be accidental as a result of environmental exposure due to unintentional ingestion of the toxin or deliberate criminal poisoning as in this case. Because deliberate poisoning rarely crosses the mind of the clinician, awareness of the potential similarity of barium poisoning to other forms of HPP and even familial HPP is important.
Case Presentation
A male veteran aged 45 years when treated by the authors was well until moving into a new rural home when he began to experience acute episodes of variable perioral numbness, diarrhea, paresthesias, abdominal cramping, and weakness, which ranged from mild, self-terminating extremity weakness to 3 episodes of respiratory failure that required intubation and mechanical ventilation.
All episodes were accompanied by hypokalemia in the range of 2 to 3 mEq/L, but levels varied erratically during admissions from severe hypokalemia to normo- and hyperkalemia. Over 3 years, the patient was admitted to the hospital 19 times, underwent extensive workup, and was referred to endocrinology services at Duke University, Vanderbilt University, and the Cleveland Clinic. Diagnostic efforts centered on establishing whether he had a latepresenting variant of familial HPP.
Genetic evaluations could not identify known single-nucleotide polymorphisms associated with that condition. The consensus was that he had a potassium leak somewhere between his kidneys and bladder. Recommended management was a high baseline oral potassium supplementation and spironolactone. He had a brief period of improvement after moving to a different house, but the episodes returned once he moved back to his old house despite adherence to recommended treatment. In December 2012, he experienced his worst episode, with potassium 1.8 mEq/L on admission, resulting in admission to the intensive care unit (ICU).
Following a precipitous clinical decline, the patient was intubated and mechanically ventilated. Nephrology was consulted and given the recurrent life-threatening pattern, an intensive chart review was undertaken. It was noted that a urine arsenic level that had been normal several admissions previously at 18 μg/L was elevated during a subsequent admission at 59 μg/L, and several weeks later during a later admission the level had fallen to 15 μg/L. Urine lead was undetectable on 3 occasions, and urine mercury was within normal limits.
Arsenic toxicity did not match the patient’s clinical syndrome, but the pattern seemed to be consistent with the possibility of unexplained toxic exposure and subsequent clearance. Therefore, an intensive literature search for syndromes of environmental exposure or poisoning resembling HPP was undertaken. The search revealed several references in the literature to paralysis similar to HPP that involved ingestion of hair-removing soap and rat poison containing barium sulfide and carbonate. References also pointed to the similarity of the symptoms to Guillain-Barre syndrome.
As a result of that literature search, a blood barium level was collected in the ICU that revealed 14,550 ng/mL. A scalp hair sample showed 6.1 μg barium per gram of hair (reference, 0.53 μg/g to 2 μg/g). Neither the patient nor his wife reported being involved in painting, ceramic work, decorating glassware or fabric with dyes, working with stained glass, smelting, metal welding, or use of vermicides.
A U.S. Environmental Protection Agency team was sent to the house, and a detailed toxic survey of the house and the surrounding grounds was conducted with no excess barium found. Barium levels were checked by a private physician on the wife and 2 minor children. The wife’s barium levels came back undetectable in a blood sample and elevated in a hair sample. One child had a very low detected level in her blood and slightly elevated in her hair, and the other child had a low level in her blood and her hair. Because the circumstances of the wife’s and children’s exposure could not be explained environmentally nor could the veteran’s exposure source be identified, the VA Police Service contacted the Tennessee Bureau of Investigation, and they questioned the veteran and his wife.
Shortly after that the veteran received a paralyzing gunshot wound to the back, and the ensuing investigation resulted in incarceration of his wife for both attempted murder by firearm and serial poisoning after soluble barium-containing materials were found hidden in the house.
Discussion
Human barium poisoning is a rarely reported toxic exposure that results in rapid onset of nausea, vomiting, diarrhea, progressive weakness that may end in respiratory paralysis and death if intubation and mechanical ventilation are not promptly initiated. Although the barium found in radiographic contrast media is highly insoluble, ingested barium carbonate and sulfide are rapidly absorbed into the bloodstream, reaching high levels quickly and altering the conductance of potassium channels. The result is erratic variation in blood potassium and prolonged paralysis unless it is immediately suspected and hemodialysis is initiated. In this case, the suspicion level at the time of intubation was insufficient to justify initiating acute hemodialysis.
Soluble barium is available from a number of open sources. Depilatory powders and several rat poisons list barium sulfide or carbonate, both soluble forms of barium rapidly absorbed through the gastrointestinal mucosa, as a major ingredient. One celebrated 2012 case in a city near Chattanooga, Tennessee, involved allegations of barium carbonate poisoning involving rat poison mixed into coffee creamer, but no charges could be filed because the sample handling precluded definitive linkage. Another deliberate toxic poisoning in Texas was traced to soluble barium introduced into a father’s food by his daughter.
The patient reported here experienced 3 years and 19 admissions with 3 episodes of mechanical intubation before his suspected variant HPP was recognized as actually being due to soluble barium poisoning.
Barium does not appear in usual heavy metal urine and blood screens and as a result may not be asked for if not thought of in the differential diagnosis. Physicians dealing with instances of recurrent suspected HPP that do not fit usual age and clinical characteristics for HPP, lack the single-nucleotide polymorphisms associated with the disease, and are not associated with other conditions causing severe hypokalemia, such as renal tubular acidosis, Bartter, Liddle or Gitelman syndrome or severe diuretic or licorice-induced hypokalemia should have soluble barium poisoning included in the differential diagnosis. Appropriately drawn blood specimens in special metal-free sampling tubes and hair barium levels should be included in the diagnostic workup. If poisoning is suspected, a chain of evidence should be obtained to protect possible future criminal investigation against compromise.
Acknowledgments
The authors thanks Tennessee 2nd District Attorney General Barry P. Staubus, 2nd District Assistant Attorney General Teresa A. Nelson, the VA Police Service, and the Tennessee Bureau of Investigation for their help.
1. Ahlawat SK, Sachdev A. Hypokalaemic paralysis. Postgrad Med J. 1999;75(882):193-197.
2. Fontaine B. Periodic paralysis. Adv Genet.2008;63:3-23.
3. Kayal AK, Goswami M, Das M, Jain R. Clinical and biochemical spectrum of hypokalemic paralysis in North: East India. Ann Indian Acad Neurol.2013;16(2):211-217.
4. Johnson CH, VanTassell VJ. Acute barium poisoning with respiratory failure and rhabdomyolysis. Ann Emerg Med. 1991;20(10):1138-1142.
5. Gold R, Reichmann H. Muscle pathology correlates with permanent weakness in hypokalemic periodic paralysis: a case report. Acta Neuropathol. 1992;84(2):202-206.
6. Links TP, Zwarts MJ, Wilmink JT, Molenaar WM, Oosterhuis HJ. Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. 1990;113(pt 6):1873-1889.
7. Tucker C, Villanueva L. Acute hypokalemic periodic paralysis possibly precipitated by albuterol. Am J Health Syst Pharm. 2013;70(18):1588-1591.
8. Liu PY, Jeng CY. Severe hypophosphatemia in a patient with diabetic ketoacidosis and acute respiratory failure. J Chin Med Assoc. 2004;67(7):355-359.
9. Sigue G, Gamble L, Pelitere M, et al. From profound hypokalemia to life-threatening hyperkalemia: a case of barium sulfide poisoning. Arch Intern Med. 2000;160(4):548-541.
10. Kuntzer T, Flocard F, Vial C, et al. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000;23(7):1089-1094.
11. Tengan CH, Antunes AC, Gabbai AA, Manzano GM. The exercise test as a monitor of disease status in hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry. 2004;75(3):497-499.
12. McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986;9(8):704-710.
13. Huang K-W. Pa ping (transient paralysis simulating family periodic paralysis). Chin Med J. 1943;61(4):305-312.
14. Ng HY, Lin SH, Hsu CY, Tsai YZ, Chen HC, Lee CT. Hypokalemic paralysis due to Gitelman syndrome:a family study. Neurology. 2006;67(6):1080-1082.
15. Mohta M, Kalra B, Shukla R, Sethi AK. An unusual presentation of hypokalemia. J Anesth Clin Res. 2014;5(3):389.
16. Fujimoto T, Shiiki H, Takahi Y, Dohi K. Primary Sjögren’s Syndrome presenting as hypokalaemic periodic paralysis and respiratory arrest. Clin Rheumatol. 2001;20(5):365-368.
17. Chang YC, Huang CC, Chiou YY, Yu CY. Renal tubular acidosis complicated with hypokalemic periodic paralysis. Pediatr Neurol. 1995;13(1):52-54.
18. Lehmann-Horn F, Jurkat-Rott K, Rüdel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep. 2002;2(1):61-69.
19. Venance SL, Cannon SC, Fialho D, et al; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129(pt 1):8-17.
20. Sharma C, Nath K, Parekh J. Reversible electrophysiological abnormalities in hypokalemic paralysis: case report of two cases. Ann Indian Acad Neurol. 2014;17(1):100-102.
21. Sternberg D, Tabti N, Fournier E, Hainque B, Fontaine B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology. 2003;61(6):857-859.
22. Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. 2013;47(1):41-45.
23. Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of hypokalemic periodic paralysis. Prevention of attacks and improvement of persistent weakness. Ann Intern Med. 1970;73(1):39-48.
24. Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve. 1983;6(3):182-186.
25. Ghose A, Sayeed AA, Hossain A, Rahman R, Faiz A, Haque G. Mass barium carbonate poisoning with fatal outcome, lessons learned: a case series. Cases J. 2009;2:9327.
Hypokalemic periodic paralysis (HPP) is a relatively common and potentially life-threating condition that can be either sporadic or recurring and has both inherited and acquired causes.1 Familial HPP, on the other hand, is a rare condition (1:100,000) caused by loss of function mutations leading to the disruption of membrane potential consequently making them inexcitable.2 Appearance of symptoms is typically in the first or second decade of life (60% of cases have onset aged < 16 years) with susceptible individuals experiencing sudden onset of perioral numbness; weakness; centrifugal paralysis, often with nausea; vomiting and diarrhea; and prostration, usually triggered by highcarbohydrate meals and rest following sustained muscle-group use.3
These symptoms are common to all forms of HPP, making the differential diagnosis wide and confusing. Rhabdomyolysis is occasionally associated with many severe hypokalemic episodes.4 Myopathy and permanent muscle weakness have been reported in HPP.5,6 Other reported inciting factors include a drop in serum potassium caused by β-adrenergic bronchodilator treatment.7 Clinical attacks also have been associated with diabetic ketoacidosis and combined hypokalemia and hypophosphatemia.8 Thyrotoxicosis also causes similar muscle action potential changes but only when hyperthyroidism is uncorrected. 9-12 Less commonly, hypothyroidism has been reported to be associated with hypokalemic paralysis.3
Pa Ping, a condition involving hypokalemic paralysis of uncertain etiology, is geographically centered in the Szechuan region of China.13 Cases of Bartter, Liddle, and Gitelman syndromes also have been associated with hypokalemic paralysis.3,14 There is an association with malignant hyperthermia following or during systemic anesthesia. Patients presenting as Guillain-Barré syndrome have been found to have periodic paralysis triggered by hypokalemia from any cause.15 Sjögren syndrome and renal tubular acidosis also are reported to have triggered symptoms of hypokalemic paralysis.16,17
True type 1 HPP is caused by channelopathies resulting from mutations in the calcium channel gene CACN1AS (HypoPP1), which accounts for 70% of the cases, whereas type 2 HPP is cause by sodium channel gene SCN4A (HypoPP2) mutations, which accounts for 10% to 20% of cases.18,19 An association with a voltage-gated potassium channel KCNE3 mutation has been made but is disputed.20,21 Females typically have less severe and less frequent attacks, and attacks lessen or disappear during pregnancy.22
In a small controlled trial, acetazolamide has been reported to have prophylactic benefit, although a more powerful carbonic anhydrase inhibitor, dichlorophenamide, was reported to be effective in a study after acetazolamide had become ineffective.23,24 These treatments would not be expected to be of clinical use in hypokalemia due to barium poisoning.
Barium poisoning has been reported as a result of accidental contamination of foodstuffs with soluble barium.25 Onset of symptoms is rapid, with nausea, vomiting, diarrhea, and malaise followed rapidly by weakness, which can include the muscles of respiration. This littleconsidered but rapidly lethal poisoning event can be accidental as a result of environmental exposure due to unintentional ingestion of the toxin or deliberate criminal poisoning as in this case. Because deliberate poisoning rarely crosses the mind of the clinician, awareness of the potential similarity of barium poisoning to other forms of HPP and even familial HPP is important.
Case Presentation
A male veteran aged 45 years when treated by the authors was well until moving into a new rural home when he began to experience acute episodes of variable perioral numbness, diarrhea, paresthesias, abdominal cramping, and weakness, which ranged from mild, self-terminating extremity weakness to 3 episodes of respiratory failure that required intubation and mechanical ventilation.
All episodes were accompanied by hypokalemia in the range of 2 to 3 mEq/L, but levels varied erratically during admissions from severe hypokalemia to normo- and hyperkalemia. Over 3 years, the patient was admitted to the hospital 19 times, underwent extensive workup, and was referred to endocrinology services at Duke University, Vanderbilt University, and the Cleveland Clinic. Diagnostic efforts centered on establishing whether he had a latepresenting variant of familial HPP.
Genetic evaluations could not identify known single-nucleotide polymorphisms associated with that condition. The consensus was that he had a potassium leak somewhere between his kidneys and bladder. Recommended management was a high baseline oral potassium supplementation and spironolactone. He had a brief period of improvement after moving to a different house, but the episodes returned once he moved back to his old house despite adherence to recommended treatment. In December 2012, he experienced his worst episode, with potassium 1.8 mEq/L on admission, resulting in admission to the intensive care unit (ICU).
Following a precipitous clinical decline, the patient was intubated and mechanically ventilated. Nephrology was consulted and given the recurrent life-threatening pattern, an intensive chart review was undertaken. It was noted that a urine arsenic level that had been normal several admissions previously at 18 μg/L was elevated during a subsequent admission at 59 μg/L, and several weeks later during a later admission the level had fallen to 15 μg/L. Urine lead was undetectable on 3 occasions, and urine mercury was within normal limits.
Arsenic toxicity did not match the patient’s clinical syndrome, but the pattern seemed to be consistent with the possibility of unexplained toxic exposure and subsequent clearance. Therefore, an intensive literature search for syndromes of environmental exposure or poisoning resembling HPP was undertaken. The search revealed several references in the literature to paralysis similar to HPP that involved ingestion of hair-removing soap and rat poison containing barium sulfide and carbonate. References also pointed to the similarity of the symptoms to Guillain-Barre syndrome.
As a result of that literature search, a blood barium level was collected in the ICU that revealed 14,550 ng/mL. A scalp hair sample showed 6.1 μg barium per gram of hair (reference, 0.53 μg/g to 2 μg/g). Neither the patient nor his wife reported being involved in painting, ceramic work, decorating glassware or fabric with dyes, working with stained glass, smelting, metal welding, or use of vermicides.
A U.S. Environmental Protection Agency team was sent to the house, and a detailed toxic survey of the house and the surrounding grounds was conducted with no excess barium found. Barium levels were checked by a private physician on the wife and 2 minor children. The wife’s barium levels came back undetectable in a blood sample and elevated in a hair sample. One child had a very low detected level in her blood and slightly elevated in her hair, and the other child had a low level in her blood and her hair. Because the circumstances of the wife’s and children’s exposure could not be explained environmentally nor could the veteran’s exposure source be identified, the VA Police Service contacted the Tennessee Bureau of Investigation, and they questioned the veteran and his wife.
Shortly after that the veteran received a paralyzing gunshot wound to the back, and the ensuing investigation resulted in incarceration of his wife for both attempted murder by firearm and serial poisoning after soluble barium-containing materials were found hidden in the house.
Discussion
Human barium poisoning is a rarely reported toxic exposure that results in rapid onset of nausea, vomiting, diarrhea, progressive weakness that may end in respiratory paralysis and death if intubation and mechanical ventilation are not promptly initiated. Although the barium found in radiographic contrast media is highly insoluble, ingested barium carbonate and sulfide are rapidly absorbed into the bloodstream, reaching high levels quickly and altering the conductance of potassium channels. The result is erratic variation in blood potassium and prolonged paralysis unless it is immediately suspected and hemodialysis is initiated. In this case, the suspicion level at the time of intubation was insufficient to justify initiating acute hemodialysis.
Soluble barium is available from a number of open sources. Depilatory powders and several rat poisons list barium sulfide or carbonate, both soluble forms of barium rapidly absorbed through the gastrointestinal mucosa, as a major ingredient. One celebrated 2012 case in a city near Chattanooga, Tennessee, involved allegations of barium carbonate poisoning involving rat poison mixed into coffee creamer, but no charges could be filed because the sample handling precluded definitive linkage. Another deliberate toxic poisoning in Texas was traced to soluble barium introduced into a father’s food by his daughter.
The patient reported here experienced 3 years and 19 admissions with 3 episodes of mechanical intubation before his suspected variant HPP was recognized as actually being due to soluble barium poisoning.
Barium does not appear in usual heavy metal urine and blood screens and as a result may not be asked for if not thought of in the differential diagnosis. Physicians dealing with instances of recurrent suspected HPP that do not fit usual age and clinical characteristics for HPP, lack the single-nucleotide polymorphisms associated with the disease, and are not associated with other conditions causing severe hypokalemia, such as renal tubular acidosis, Bartter, Liddle or Gitelman syndrome or severe diuretic or licorice-induced hypokalemia should have soluble barium poisoning included in the differential diagnosis. Appropriately drawn blood specimens in special metal-free sampling tubes and hair barium levels should be included in the diagnostic workup. If poisoning is suspected, a chain of evidence should be obtained to protect possible future criminal investigation against compromise.
Acknowledgments
The authors thanks Tennessee 2nd District Attorney General Barry P. Staubus, 2nd District Assistant Attorney General Teresa A. Nelson, the VA Police Service, and the Tennessee Bureau of Investigation for their help.
Hypokalemic periodic paralysis (HPP) is a relatively common and potentially life-threating condition that can be either sporadic or recurring and has both inherited and acquired causes.1 Familial HPP, on the other hand, is a rare condition (1:100,000) caused by loss of function mutations leading to the disruption of membrane potential consequently making them inexcitable.2 Appearance of symptoms is typically in the first or second decade of life (60% of cases have onset aged < 16 years) with susceptible individuals experiencing sudden onset of perioral numbness; weakness; centrifugal paralysis, often with nausea; vomiting and diarrhea; and prostration, usually triggered by highcarbohydrate meals and rest following sustained muscle-group use.3
These symptoms are common to all forms of HPP, making the differential diagnosis wide and confusing. Rhabdomyolysis is occasionally associated with many severe hypokalemic episodes.4 Myopathy and permanent muscle weakness have been reported in HPP.5,6 Other reported inciting factors include a drop in serum potassium caused by β-adrenergic bronchodilator treatment.7 Clinical attacks also have been associated with diabetic ketoacidosis and combined hypokalemia and hypophosphatemia.8 Thyrotoxicosis also causes similar muscle action potential changes but only when hyperthyroidism is uncorrected. 9-12 Less commonly, hypothyroidism has been reported to be associated with hypokalemic paralysis.3
Pa Ping, a condition involving hypokalemic paralysis of uncertain etiology, is geographically centered in the Szechuan region of China.13 Cases of Bartter, Liddle, and Gitelman syndromes also have been associated with hypokalemic paralysis.3,14 There is an association with malignant hyperthermia following or during systemic anesthesia. Patients presenting as Guillain-Barré syndrome have been found to have periodic paralysis triggered by hypokalemia from any cause.15 Sjögren syndrome and renal tubular acidosis also are reported to have triggered symptoms of hypokalemic paralysis.16,17
True type 1 HPP is caused by channelopathies resulting from mutations in the calcium channel gene CACN1AS (HypoPP1), which accounts for 70% of the cases, whereas type 2 HPP is cause by sodium channel gene SCN4A (HypoPP2) mutations, which accounts for 10% to 20% of cases.18,19 An association with a voltage-gated potassium channel KCNE3 mutation has been made but is disputed.20,21 Females typically have less severe and less frequent attacks, and attacks lessen or disappear during pregnancy.22
In a small controlled trial, acetazolamide has been reported to have prophylactic benefit, although a more powerful carbonic anhydrase inhibitor, dichlorophenamide, was reported to be effective in a study after acetazolamide had become ineffective.23,24 These treatments would not be expected to be of clinical use in hypokalemia due to barium poisoning.
Barium poisoning has been reported as a result of accidental contamination of foodstuffs with soluble barium.25 Onset of symptoms is rapid, with nausea, vomiting, diarrhea, and malaise followed rapidly by weakness, which can include the muscles of respiration. This littleconsidered but rapidly lethal poisoning event can be accidental as a result of environmental exposure due to unintentional ingestion of the toxin or deliberate criminal poisoning as in this case. Because deliberate poisoning rarely crosses the mind of the clinician, awareness of the potential similarity of barium poisoning to other forms of HPP and even familial HPP is important.
Case Presentation
A male veteran aged 45 years when treated by the authors was well until moving into a new rural home when he began to experience acute episodes of variable perioral numbness, diarrhea, paresthesias, abdominal cramping, and weakness, which ranged from mild, self-terminating extremity weakness to 3 episodes of respiratory failure that required intubation and mechanical ventilation.
All episodes were accompanied by hypokalemia in the range of 2 to 3 mEq/L, but levels varied erratically during admissions from severe hypokalemia to normo- and hyperkalemia. Over 3 years, the patient was admitted to the hospital 19 times, underwent extensive workup, and was referred to endocrinology services at Duke University, Vanderbilt University, and the Cleveland Clinic. Diagnostic efforts centered on establishing whether he had a latepresenting variant of familial HPP.
Genetic evaluations could not identify known single-nucleotide polymorphisms associated with that condition. The consensus was that he had a potassium leak somewhere between his kidneys and bladder. Recommended management was a high baseline oral potassium supplementation and spironolactone. He had a brief period of improvement after moving to a different house, but the episodes returned once he moved back to his old house despite adherence to recommended treatment. In December 2012, he experienced his worst episode, with potassium 1.8 mEq/L on admission, resulting in admission to the intensive care unit (ICU).
Following a precipitous clinical decline, the patient was intubated and mechanically ventilated. Nephrology was consulted and given the recurrent life-threatening pattern, an intensive chart review was undertaken. It was noted that a urine arsenic level that had been normal several admissions previously at 18 μg/L was elevated during a subsequent admission at 59 μg/L, and several weeks later during a later admission the level had fallen to 15 μg/L. Urine lead was undetectable on 3 occasions, and urine mercury was within normal limits.
Arsenic toxicity did not match the patient’s clinical syndrome, but the pattern seemed to be consistent with the possibility of unexplained toxic exposure and subsequent clearance. Therefore, an intensive literature search for syndromes of environmental exposure or poisoning resembling HPP was undertaken. The search revealed several references in the literature to paralysis similar to HPP that involved ingestion of hair-removing soap and rat poison containing barium sulfide and carbonate. References also pointed to the similarity of the symptoms to Guillain-Barre syndrome.
As a result of that literature search, a blood barium level was collected in the ICU that revealed 14,550 ng/mL. A scalp hair sample showed 6.1 μg barium per gram of hair (reference, 0.53 μg/g to 2 μg/g). Neither the patient nor his wife reported being involved in painting, ceramic work, decorating glassware or fabric with dyes, working with stained glass, smelting, metal welding, or use of vermicides.
A U.S. Environmental Protection Agency team was sent to the house, and a detailed toxic survey of the house and the surrounding grounds was conducted with no excess barium found. Barium levels were checked by a private physician on the wife and 2 minor children. The wife’s barium levels came back undetectable in a blood sample and elevated in a hair sample. One child had a very low detected level in her blood and slightly elevated in her hair, and the other child had a low level in her blood and her hair. Because the circumstances of the wife’s and children’s exposure could not be explained environmentally nor could the veteran’s exposure source be identified, the VA Police Service contacted the Tennessee Bureau of Investigation, and they questioned the veteran and his wife.
Shortly after that the veteran received a paralyzing gunshot wound to the back, and the ensuing investigation resulted in incarceration of his wife for both attempted murder by firearm and serial poisoning after soluble barium-containing materials were found hidden in the house.
Discussion
Human barium poisoning is a rarely reported toxic exposure that results in rapid onset of nausea, vomiting, diarrhea, progressive weakness that may end in respiratory paralysis and death if intubation and mechanical ventilation are not promptly initiated. Although the barium found in radiographic contrast media is highly insoluble, ingested barium carbonate and sulfide are rapidly absorbed into the bloodstream, reaching high levels quickly and altering the conductance of potassium channels. The result is erratic variation in blood potassium and prolonged paralysis unless it is immediately suspected and hemodialysis is initiated. In this case, the suspicion level at the time of intubation was insufficient to justify initiating acute hemodialysis.
Soluble barium is available from a number of open sources. Depilatory powders and several rat poisons list barium sulfide or carbonate, both soluble forms of barium rapidly absorbed through the gastrointestinal mucosa, as a major ingredient. One celebrated 2012 case in a city near Chattanooga, Tennessee, involved allegations of barium carbonate poisoning involving rat poison mixed into coffee creamer, but no charges could be filed because the sample handling precluded definitive linkage. Another deliberate toxic poisoning in Texas was traced to soluble barium introduced into a father’s food by his daughter.
The patient reported here experienced 3 years and 19 admissions with 3 episodes of mechanical intubation before his suspected variant HPP was recognized as actually being due to soluble barium poisoning.
Barium does not appear in usual heavy metal urine and blood screens and as a result may not be asked for if not thought of in the differential diagnosis. Physicians dealing with instances of recurrent suspected HPP that do not fit usual age and clinical characteristics for HPP, lack the single-nucleotide polymorphisms associated with the disease, and are not associated with other conditions causing severe hypokalemia, such as renal tubular acidosis, Bartter, Liddle or Gitelman syndrome or severe diuretic or licorice-induced hypokalemia should have soluble barium poisoning included in the differential diagnosis. Appropriately drawn blood specimens in special metal-free sampling tubes and hair barium levels should be included in the diagnostic workup. If poisoning is suspected, a chain of evidence should be obtained to protect possible future criminal investigation against compromise.
Acknowledgments
The authors thanks Tennessee 2nd District Attorney General Barry P. Staubus, 2nd District Assistant Attorney General Teresa A. Nelson, the VA Police Service, and the Tennessee Bureau of Investigation for their help.
1. Ahlawat SK, Sachdev A. Hypokalaemic paralysis. Postgrad Med J. 1999;75(882):193-197.
2. Fontaine B. Periodic paralysis. Adv Genet.2008;63:3-23.
3. Kayal AK, Goswami M, Das M, Jain R. Clinical and biochemical spectrum of hypokalemic paralysis in North: East India. Ann Indian Acad Neurol.2013;16(2):211-217.
4. Johnson CH, VanTassell VJ. Acute barium poisoning with respiratory failure and rhabdomyolysis. Ann Emerg Med. 1991;20(10):1138-1142.
5. Gold R, Reichmann H. Muscle pathology correlates with permanent weakness in hypokalemic periodic paralysis: a case report. Acta Neuropathol. 1992;84(2):202-206.
6. Links TP, Zwarts MJ, Wilmink JT, Molenaar WM, Oosterhuis HJ. Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. 1990;113(pt 6):1873-1889.
7. Tucker C, Villanueva L. Acute hypokalemic periodic paralysis possibly precipitated by albuterol. Am J Health Syst Pharm. 2013;70(18):1588-1591.
8. Liu PY, Jeng CY. Severe hypophosphatemia in a patient with diabetic ketoacidosis and acute respiratory failure. J Chin Med Assoc. 2004;67(7):355-359.
9. Sigue G, Gamble L, Pelitere M, et al. From profound hypokalemia to life-threatening hyperkalemia: a case of barium sulfide poisoning. Arch Intern Med. 2000;160(4):548-541.
10. Kuntzer T, Flocard F, Vial C, et al. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000;23(7):1089-1094.
11. Tengan CH, Antunes AC, Gabbai AA, Manzano GM. The exercise test as a monitor of disease status in hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry. 2004;75(3):497-499.
12. McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986;9(8):704-710.
13. Huang K-W. Pa ping (transient paralysis simulating family periodic paralysis). Chin Med J. 1943;61(4):305-312.
14. Ng HY, Lin SH, Hsu CY, Tsai YZ, Chen HC, Lee CT. Hypokalemic paralysis due to Gitelman syndrome:a family study. Neurology. 2006;67(6):1080-1082.
15. Mohta M, Kalra B, Shukla R, Sethi AK. An unusual presentation of hypokalemia. J Anesth Clin Res. 2014;5(3):389.
16. Fujimoto T, Shiiki H, Takahi Y, Dohi K. Primary Sjögren’s Syndrome presenting as hypokalaemic periodic paralysis and respiratory arrest. Clin Rheumatol. 2001;20(5):365-368.
17. Chang YC, Huang CC, Chiou YY, Yu CY. Renal tubular acidosis complicated with hypokalemic periodic paralysis. Pediatr Neurol. 1995;13(1):52-54.
18. Lehmann-Horn F, Jurkat-Rott K, Rüdel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep. 2002;2(1):61-69.
19. Venance SL, Cannon SC, Fialho D, et al; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129(pt 1):8-17.
20. Sharma C, Nath K, Parekh J. Reversible electrophysiological abnormalities in hypokalemic paralysis: case report of two cases. Ann Indian Acad Neurol. 2014;17(1):100-102.
21. Sternberg D, Tabti N, Fournier E, Hainque B, Fontaine B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology. 2003;61(6):857-859.
22. Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. 2013;47(1):41-45.
23. Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of hypokalemic periodic paralysis. Prevention of attacks and improvement of persistent weakness. Ann Intern Med. 1970;73(1):39-48.
24. Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve. 1983;6(3):182-186.
25. Ghose A, Sayeed AA, Hossain A, Rahman R, Faiz A, Haque G. Mass barium carbonate poisoning with fatal outcome, lessons learned: a case series. Cases J. 2009;2:9327.
1. Ahlawat SK, Sachdev A. Hypokalaemic paralysis. Postgrad Med J. 1999;75(882):193-197.
2. Fontaine B. Periodic paralysis. Adv Genet.2008;63:3-23.
3. Kayal AK, Goswami M, Das M, Jain R. Clinical and biochemical spectrum of hypokalemic paralysis in North: East India. Ann Indian Acad Neurol.2013;16(2):211-217.
4. Johnson CH, VanTassell VJ. Acute barium poisoning with respiratory failure and rhabdomyolysis. Ann Emerg Med. 1991;20(10):1138-1142.
5. Gold R, Reichmann H. Muscle pathology correlates with permanent weakness in hypokalemic periodic paralysis: a case report. Acta Neuropathol. 1992;84(2):202-206.
6. Links TP, Zwarts MJ, Wilmink JT, Molenaar WM, Oosterhuis HJ. Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. 1990;113(pt 6):1873-1889.
7. Tucker C, Villanueva L. Acute hypokalemic periodic paralysis possibly precipitated by albuterol. Am J Health Syst Pharm. 2013;70(18):1588-1591.
8. Liu PY, Jeng CY. Severe hypophosphatemia in a patient with diabetic ketoacidosis and acute respiratory failure. J Chin Med Assoc. 2004;67(7):355-359.
9. Sigue G, Gamble L, Pelitere M, et al. From profound hypokalemia to life-threatening hyperkalemia: a case of barium sulfide poisoning. Arch Intern Med. 2000;160(4):548-541.
10. Kuntzer T, Flocard F, Vial C, et al. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000;23(7):1089-1094.
11. Tengan CH, Antunes AC, Gabbai AA, Manzano GM. The exercise test as a monitor of disease status in hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry. 2004;75(3):497-499.
12. McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986;9(8):704-710.
13. Huang K-W. Pa ping (transient paralysis simulating family periodic paralysis). Chin Med J. 1943;61(4):305-312.
14. Ng HY, Lin SH, Hsu CY, Tsai YZ, Chen HC, Lee CT. Hypokalemic paralysis due to Gitelman syndrome:a family study. Neurology. 2006;67(6):1080-1082.
15. Mohta M, Kalra B, Shukla R, Sethi AK. An unusual presentation of hypokalemia. J Anesth Clin Res. 2014;5(3):389.
16. Fujimoto T, Shiiki H, Takahi Y, Dohi K. Primary Sjögren’s Syndrome presenting as hypokalaemic periodic paralysis and respiratory arrest. Clin Rheumatol. 2001;20(5):365-368.
17. Chang YC, Huang CC, Chiou YY, Yu CY. Renal tubular acidosis complicated with hypokalemic periodic paralysis. Pediatr Neurol. 1995;13(1):52-54.
18. Lehmann-Horn F, Jurkat-Rott K, Rüdel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep. 2002;2(1):61-69.
19. Venance SL, Cannon SC, Fialho D, et al; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129(pt 1):8-17.
20. Sharma C, Nath K, Parekh J. Reversible electrophysiological abnormalities in hypokalemic paralysis: case report of two cases. Ann Indian Acad Neurol. 2014;17(1):100-102.
21. Sternberg D, Tabti N, Fournier E, Hainque B, Fontaine B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology. 2003;61(6):857-859.
22. Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. 2013;47(1):41-45.
23. Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of hypokalemic periodic paralysis. Prevention of attacks and improvement of persistent weakness. Ann Intern Med. 1970;73(1):39-48.
24. Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve. 1983;6(3):182-186.
25. Ghose A, Sayeed AA, Hossain A, Rahman R, Faiz A, Haque G. Mass barium carbonate poisoning with fatal outcome, lessons learned: a case series. Cases J. 2009;2:9327.
Cerebral Venous Thrombosis
Cerebral venous thrombosis (CVT) is a rare cerebrovascular disease that affects about 5 in 1 million people each year and accounts for 0.5% of all strokes.1 Previously it was thought to be caused most commonly by infections (eg, mastoiditis, otitis, meningitis) affecting the superior sagittal sinus and often resulting in focal neurologic deficits, seizures, coma, and death. Although local and systemic infections are still prominent risk factors in its development, CVT is now primarily recognized as a nonseptic disease with a wide spectrum of clinical presentations.
Cerebral venous thrombosis causes reduced outflow of blood and cerebrospinal fluid, which in half of affected patients leads to significant venous infarct. As opposed to arterial infarctions, CVT mainly affects children and young adults; it is an important cause of stroke in the younger population.2 There is significant overlap of the many risk factors for CVT and those for venous thromboembolism (VTE): cancer, obesity, genetic thrombophilia, trauma, infection, and prior neurosurgery. However, the most common acquired risk factors for CVT are oral contraceptive use and pregnancy, which explains why CVT is 3 times more likely to occur in young and middle-aged women.3
Cerebral venous thrombosis was first recognized by a French physician in the 19th century, a time when the condition was diagnosed at autopsy, which typically showed hemorrhagic lesions at the thrombus site.4 For many years heparin was contraindicated in the treatment of CVT, and only within the past 25 years did advances in neuroimaging allow for earlier diagnosis and change perspectives on the management of this disease.
Cerebral venous thrombosis occurs from formation of a thrombus within the cerebral venous sinuses, leading to elevated intracranial pressure and eventually ischemia or intracranial hemorrhage. Improved imaging techniques, notably magnetic resonance imaging (MRI) and computed tomography (CT) venography, allow physicians to identify thrombus formation earlier and begin anticoagulation therapy with heparin before infarction. A meta-analysis of studies found that heparin was safer and more efficacious in treating CVT compared with placebo.5 Furthermore, several small randomized trials found treatment with unfractionated heparin (UFH) or low-molecularweight heparin (LMWH) was not associated with higher risk of hemorrhagic stroke in these patients.6-8
Despite improvements in imaging modalities, in many cases diagnosis is delayed, as most patients with CVT have a wide spectrum of presentations with nonspecific symptoms, such as headache, seizure, focal sensorimotor deficits, and papilledema.9 Clinical presentations of CVT depend on a variety of factors, including age, time of onset, CVT location, and presence of parenchymal lesions. Isolated headache is the most common initial symptom, and in many cases is the only presenting manifestation of CVT.1 Encephalopathy with multifocal signs, delirium, or dysfunction in executive functions most commonly occurs in elderly populations.
Cavernous sinus thrombosis most commonly produces generalized headaches, orbital pain, proptosis, and diplopia, whereas cortical vein thrombosis often produces seizures and focal sensorimotor deficits. Aphasia may be present in patients with isolated left transverse sinus thrombosis. In the presence of deep cerebral venous occlusion, patients can present in coma or with severe cognitive deficits and widespread paresis.10 Thrombosis of different veins and sinuses results in a wide spectrum of diverse clinical pictures, posing a diagnostic challenge and affecting clinical outcomes.
Given the variable and often nonspecific clinical presentations of these cases, unenhanced CT typically is the first imaging study ordered. According to the literature, noncontrast CT is not sensitive (25%-56%) in detecting CVT, and findings are normal in up to 26% of patients, rarely providing a specific diagnosis.11 Furthermore, visualization on MRI can be difficult during the acute phase of CVT, as the thrombus initially appears isointense on T1-weighted images and gradually becomes hyperintense over the course of the disease process.12 These difficulties with the usual first-choice imaging examinations often result in a delay in diagnosing CVT. However, several points on close examination of these imaging studies may help physicians establish a high index of clinical suspicion and order the appropriate follow-up studies for CVT.
The authors report the case of a patient who presented with a 1-week history of confusion, headaches, and dizziness. His nonspecific presentation along with the absence of obvious signs of CVT on imaging prolonged his diagnosis and the initiation of an appropriate treatment plan.
Case Report
A 57-year-old white air-conditioning mechanic presented to the emergency department (ED) with a 1-week history of gradual-onset confusion, severe headaches, dizziness, light-headedness, poor memory, and increased sleep. Two days earlier, he presented with similar symptoms to an outside facility, where he was admitted and underwent a workup for stroke, hemorrhage, and cardiac abnormalities—including noncontrast CT of the head. With nothing of clinical significance found, the patient was discharged and was advised to follow up on an outpatient basis.
Persisting symptoms brought the patient to the ED 1 day later, and he was admitted. He described severe, progressive, generalized headaches that were more severe when he was lying down at night and waking in the morning. He did not report that the headaches worsened with coughing or straining, and he reported no history of trauma, neck stiffness, vision change, seizures, and migraines. His medical history was significant for hypertension, dyslipidemia, and in 2011, an unprovoked deep vein thrombosis (DVT) in the right leg. He reported no history of tobacco, alcohol, or illicit drug use. He had served in the U.S. Navy, working in electronics, intelligence, data systems, and satellite communications.
On initial physical examination, the patient was afebrile and lethargic, and his blood pressure was mildly elevated (144/83 mm Hg). Cardiopulmonary examination was normal. Neurologic examination revealed no severe focal deficits, and cranial nerves II to XII were intact. Funduscopic examination was normal, with no papilledema noted. Motor strength was 5/5 bilaterally in the deltoids, biceps, triceps, radial and ulnar wrist extensors, iliopsoas, quadriceps, hamstrings, tibialis anterior and posterior, fibulares, and gastrocnemius. Pinprick sensation and light-touch sensation were decreased within the lateral bicipital region of the left upper extremity. Pinprick sensation was intact bilaterally in 1-inch increments at all other distributions along the medial and lateral aspects of the upper and lower extremities. Muscle tone and bulk were normal in all extremities. Reflexes were 2+ bilaterally in the biceps, brachioradialis, triceps, quadriceps, and Achilles. The Babinski sign was absent bilaterally, the finger-tonose and heel-to-shin tests were normal, the Romberg sign was absent, and there was no evidence of pronator drift. Laboratory test results were normal except for slightly elevated hemoglobin (17.5 g/dL) and D-dimer (588 ng/mL) levels.
Although noncontrast CT of the head initially showed no acute intracranial abnormalities, retrospective close comparison with the arterial system revealed slightly increased attenuation in the superior sagittal sinus, straight sinus, vein of Galen, and internal cerebral veins (Figures 1A and 1B) relative to the arterial carotid anterior circulation (Figures 2A and 2B).

Subsequent brain MRI without contrast showed a hyperintense T1 signal involving the superior sagittal sinus (Figures 3A and 3B), extending into the straight sinus and the vein of Galen. Magnetic resonance imaging with contrast demonstrated a prominent filling defect primarily in the superior sagittal sinus, in the right transverse sinus, and in the vein of Galen. Diffusion-weighted brain MRI sequence showed restricted diffusion localized to the right thalamic area (Figure 4) and no evidence of hemorrhage.
Treatment
International guidelines recommend using heparin to achieve rapid anticoagulation, stop the thrombotic process, and prevent extension of the thrombus.13 Theoretically, more rapid recanalization may have been achieved by performing endovascular thrombectomy in the present case. However, severe bleeding complications, combined with higher cost and the limited availability of clinicians experienced in treating this rare disease, convince physicians to rely on heparin as first-line treatment for CVT.14 A small randomized clinical trial found LMWH safer and more efficacious than UFH in treating CVT.15 After stabilization, oral anticoagulation therapy is used to maintain an international normalized ratio (INR) between 2.0 and 3.0 for at least 3 months.14
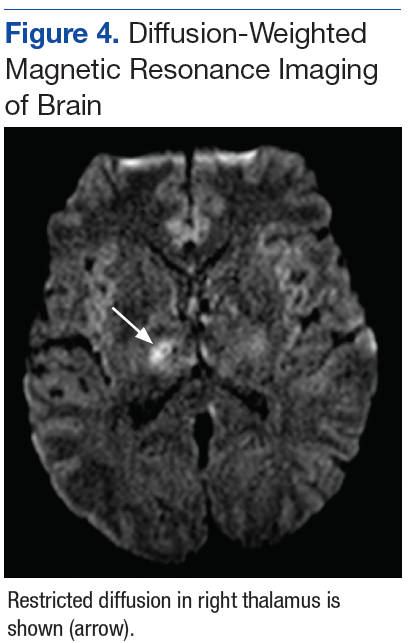
Given these findings, the patient was initially treated with LMWH. Eventually he was switched to oral warfarin and showed signs of clinical improvement. A hypercoagulability state workup revealed that the patient was heterozygous for the prothrombin G20210A mutation, and he was discharged and instructed to continue the oral warfarin therapy.
On follow-up, the hematology and neurology team initiated indefinite treatment with warfarin for his genetic hypercoagulability state. Monitoring of the dose of anticoagulation therapy was started to maintain INR between 2.0 and 3.0. The patient began coming to the office for INR monitoring every 2 to 3 weeks, and his most recent INR, in May 2017, was 2.66. He is taking 7.5 mg of warfarin on Wednesdays and Sundays and 5 mg on all other days and currently does not report any progressive neurologic deficits.
Discussion
The clinical findings of CVT and the hypercoagulability state workup revealed that the patient was heterozygous for the prothrombin G20210A mutation. Prothrombin is the precursor to thrombin, which is a key regulator of the clotting cascade and a promoter of coagulation. Carriers of the mutation have elevated levels of blood plasma prothrombin and have been associated with a 4 times higher risk for VTE.16
Several large studies and systematic reviews have confirmed that the prothrombin G20210A mutation is associated with higher rates of VTE, leading to an increased risk for DVT of the leg or pulmonary embolism.17-19 More specifically, a metaanalysis of 15 case–control studies found strong associations between the mutation and CVT.20 Despite this significant association, studies are inconclusive about whether heterozygosity for the mutation is associated with increased rates of recurrent CVT or other VTE in the absence of other risk factors, such as oral contraceptive use, trauma, malignancy, and infection.21-23 Therefore, the optimal duration of anticoagulation therapy for CVT is not well established in patients with the mutation. However, the present patient was started on indefinite anticoagulation therapy because the underlying etiology of the CVT was not reversible or transient, and this CVT was his second episode of VTE, following a 2011 DVT in the right leg.
The case discussed here illustrates the clinical presentation and diagnostic complexities of CVT. Two days before coming to the ED, the patient presented to an outside facility and underwent a workup for nonspecific symptoms (eg, confusion, headaches). Due to the nonspecific presentation associated with CVT, a detailed history is imperative to distinguish symptoms suggesting increased intracranial pressure, such as headaches worse when lying down or present in the morning, with a high clinical suspicion of CVT. The ability to attain these specific details leads clinicians toward obtaining the necessary imaging studies for potential CVT patients, and may prevent delay in diagnosis and treatment. The thrombus in CVT initially consists of deoxyhemoglobin and appears on MRI as an isointense signal on T1-weighted images and a hypointense signal on T2-weighted images; over subsequent days, the thrombus changes to methemoglobin and appears as a slightly hyperintense signal on both T1- and T2-weighted images.24
During this phase, there are some false negatives, as the thrombus can be mistaken for imaging artifacts, hematocrit elevations, or low flow of normal venous blood. Given the clinical findings and imaging studies, it is essential to distinguish CVT from other benign etiologies. Earlier diagnosis and initiation of anticoagulation therapy may have precluded the small amount of localized ischemic changes in this patient’s right thalamus, thus preventing the mild sensory loss in the left upper extremity. With the variable and nonspecific clinical presentations and the difficulties in identifying CVT with first-line imaging, progression of thrombus formation may lead to severe focal neurologic deficits, coma, or death.
Using CT imaging studies to compare the blood in the draining cerebral sinuses with the blood in the arterial system can help distinguish CVT from other etiologies of hyperdense abnormalities, such as increased hematocrit or decreased flow. Retrospective close examination of the present patient’s noncontrast CT images of the head and brain revealed slight hyperdensity in the cerebral sinuses compared with the arterial blood, suggesting the presence of thrombus formation in the cerebral veins. As CT is often the first study used to evaluate the nonspecific clinical presentations of these patients, identifying subtle signaldensity differences between the arterial and venous systems could guide physicians in identifying CVT earlier.
The authors reiterate the importance of meticulous imaging interpretation in light of the entire clinical picture: In these patients, it is imperative to have a high index of clinical suspicion for CVT in order to prevent more serious complications, such as ischemic or hemorrhagic stroke.
1. Bousser MG, Ferro JM. Cerebral venous thrombosis: an update. Lancet Neurol. 2007;6(2):162-170.
2. Coutinho JM. Cerebral venous thrombosis. J Thromb Haemost. 2015;13(suppl 1):S238-S244.
3. Coutinho JM, Ferro JM, Canhão P, et al. Cerebral venous and sinus thrombosis in women. Stroke. 2009;40(7):2356-2361.
4. Zuurbier SM, Coutinho JM. Cerebral venous thrombosis. Adv Exp Med Biol. 2017;906:183-193.
5. Einhäupl KM, Villringer A, Meister W, et al. Heparin treatment in sinus venous thrombosis. Lancet. 1991;338(8767):597-600.
6. de Bruijn SF, Stam J. Randomized, placebocontrolled trial of anticoagulant treatment with lowmolecular-weight heparin for cerebral sinus thrombosis. Stroke. 1999;30(3):484-488.
7. Nagaraja D, Haridas T, Taly AB, Veerendrakumar M, SubbuKrishna DK. Puerperal cerebral venous thrombosis: therapeutic benefit of low dose heparin. Neurol India. 1999;47(1):43-46.
8. Coutinho JM, de Bruijn SF, deVeber G, Stam J. Anticoagulation for cerebral venous sinus thrombosis. Stroke. 2012;43(4):e41-e42.
9. Sassi SB, Touati N, Baccouche H, Drissi C, Romdhane NB, Hentati F. Cerebral venous thrombosis. Clin Appl Thromb Hemost. 2016:1076029616665168. [Epub ahead of print.]
10. Ferro JM, Canhão P. Cerebral venous sinus thrombosis: update on diagnosis and management. Curr Cardiol Rep. 2014;16(9):523.
11. Albright KC, Freeman WD, Kruse BT. Cerebral venous thrombosis. J Emerg Med. 2010;38(2):238-239.
12. Lafitte F, Boukobza M, Guichard JP, et al. MRI and MRA for diagnosis and follow-up of cerebral venous thrombosis (CVT). Clin Radiol. 1997;52(9):672-679.
13. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192.
14. Coutinho JM, Middeldorp S, Stam J. Advances in the treatment of cerebral venous thrombosis. Curr Treat Options Neurol. 2014;16(7):299.
15. Coutinho JM, Ferro JM, Canhão P, Barinagarrementeria F, Bousser MG, Stam J; ISCVT Investigators. Unfractionated or low-molecular weight heparin for the treatment of cerebral venous thrombosis. Stroke. 2010;41(11):2575-2580.
16. Rosendaal FR. Venous thrombosis: the role of genes, environment, and behavior. Hematology Am Soc Hematol Educ Program. 2005:1-12.
17. Dentali F, Crowther M, Ageno W. Thrombophilic abnormalities, oral contraceptives, and risk of cerebral vein thrombosis: a meta-analysis. Blood.2006;107(7):2766-2773.
18. Salomon O, Steinberg DM, Zivelin A, et al. Single and combined prothrombotic factors in patients with idiopathic venous thromboembolism: prevalence and risk assessment. Arterioscler Thromb Vasc Biol. 1999;19(3):511-518.
19. Emmerich J, Rosendaal FR, Cattaneo M, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism— pooled analysis of 8 case–control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost. 2001;86(3):809-816.
20. Lauw MN, Barco S, Coutinho JM, Middeldorp S. Cerebral venous thrombosis and thrombophilia: a systematic review and meta-analysis. Semin Thromb Hemost. 2013;39(8):913-927.
21. Dentali F, Poli D, Scoditti U, et al. Long-term outcomes of patients with cerebral vein thrombosis: a multicenter study. J Thromb Haemost. 2012;10(7):1297-1302.
22. Martinelli I, Bucciarelli P, Passamonti SM, Battaglioli T, Previtali E, Mannucci PM. Long-term evaluation of the risk of recurrence after cerebral sinus-venous thrombosis. Circulation. 2010;121(25):2740-2746.
23. Gosk-Bierska I, Wysokinski W, Brown RD Jr, et al. Cerebral venous sinus thrombosis: incidence of venous thrombosis recurrence and survival. Neurology. 2006;67(5):814-819.
24. Galidie G, Le Gall R, Cordoliani YS, Pharaboz C, Le Marec E, Cosnard G. Thrombosis of the cerebral veins. X-ray computed tomography and MRI imaging. 11 cases [in French]. J Radiol. 1992;73(3):175-190
Cerebral venous thrombosis (CVT) is a rare cerebrovascular disease that affects about 5 in 1 million people each year and accounts for 0.5% of all strokes.1 Previously it was thought to be caused most commonly by infections (eg, mastoiditis, otitis, meningitis) affecting the superior sagittal sinus and often resulting in focal neurologic deficits, seizures, coma, and death. Although local and systemic infections are still prominent risk factors in its development, CVT is now primarily recognized as a nonseptic disease with a wide spectrum of clinical presentations.
Cerebral venous thrombosis causes reduced outflow of blood and cerebrospinal fluid, which in half of affected patients leads to significant venous infarct. As opposed to arterial infarctions, CVT mainly affects children and young adults; it is an important cause of stroke in the younger population.2 There is significant overlap of the many risk factors for CVT and those for venous thromboembolism (VTE): cancer, obesity, genetic thrombophilia, trauma, infection, and prior neurosurgery. However, the most common acquired risk factors for CVT are oral contraceptive use and pregnancy, which explains why CVT is 3 times more likely to occur in young and middle-aged women.3
Cerebral venous thrombosis was first recognized by a French physician in the 19th century, a time when the condition was diagnosed at autopsy, which typically showed hemorrhagic lesions at the thrombus site.4 For many years heparin was contraindicated in the treatment of CVT, and only within the past 25 years did advances in neuroimaging allow for earlier diagnosis and change perspectives on the management of this disease.
Cerebral venous thrombosis occurs from formation of a thrombus within the cerebral venous sinuses, leading to elevated intracranial pressure and eventually ischemia or intracranial hemorrhage. Improved imaging techniques, notably magnetic resonance imaging (MRI) and computed tomography (CT) venography, allow physicians to identify thrombus formation earlier and begin anticoagulation therapy with heparin before infarction. A meta-analysis of studies found that heparin was safer and more efficacious in treating CVT compared with placebo.5 Furthermore, several small randomized trials found treatment with unfractionated heparin (UFH) or low-molecularweight heparin (LMWH) was not associated with higher risk of hemorrhagic stroke in these patients.6-8
Despite improvements in imaging modalities, in many cases diagnosis is delayed, as most patients with CVT have a wide spectrum of presentations with nonspecific symptoms, such as headache, seizure, focal sensorimotor deficits, and papilledema.9 Clinical presentations of CVT depend on a variety of factors, including age, time of onset, CVT location, and presence of parenchymal lesions. Isolated headache is the most common initial symptom, and in many cases is the only presenting manifestation of CVT.1 Encephalopathy with multifocal signs, delirium, or dysfunction in executive functions most commonly occurs in elderly populations.
Cavernous sinus thrombosis most commonly produces generalized headaches, orbital pain, proptosis, and diplopia, whereas cortical vein thrombosis often produces seizures and focal sensorimotor deficits. Aphasia may be present in patients with isolated left transverse sinus thrombosis. In the presence of deep cerebral venous occlusion, patients can present in coma or with severe cognitive deficits and widespread paresis.10 Thrombosis of different veins and sinuses results in a wide spectrum of diverse clinical pictures, posing a diagnostic challenge and affecting clinical outcomes.
Given the variable and often nonspecific clinical presentations of these cases, unenhanced CT typically is the first imaging study ordered. According to the literature, noncontrast CT is not sensitive (25%-56%) in detecting CVT, and findings are normal in up to 26% of patients, rarely providing a specific diagnosis.11 Furthermore, visualization on MRI can be difficult during the acute phase of CVT, as the thrombus initially appears isointense on T1-weighted images and gradually becomes hyperintense over the course of the disease process.12 These difficulties with the usual first-choice imaging examinations often result in a delay in diagnosing CVT. However, several points on close examination of these imaging studies may help physicians establish a high index of clinical suspicion and order the appropriate follow-up studies for CVT.
The authors report the case of a patient who presented with a 1-week history of confusion, headaches, and dizziness. His nonspecific presentation along with the absence of obvious signs of CVT on imaging prolonged his diagnosis and the initiation of an appropriate treatment plan.
Case Report
A 57-year-old white air-conditioning mechanic presented to the emergency department (ED) with a 1-week history of gradual-onset confusion, severe headaches, dizziness, light-headedness, poor memory, and increased sleep. Two days earlier, he presented with similar symptoms to an outside facility, where he was admitted and underwent a workup for stroke, hemorrhage, and cardiac abnormalities—including noncontrast CT of the head. With nothing of clinical significance found, the patient was discharged and was advised to follow up on an outpatient basis.
Persisting symptoms brought the patient to the ED 1 day later, and he was admitted. He described severe, progressive, generalized headaches that were more severe when he was lying down at night and waking in the morning. He did not report that the headaches worsened with coughing or straining, and he reported no history of trauma, neck stiffness, vision change, seizures, and migraines. His medical history was significant for hypertension, dyslipidemia, and in 2011, an unprovoked deep vein thrombosis (DVT) in the right leg. He reported no history of tobacco, alcohol, or illicit drug use. He had served in the U.S. Navy, working in electronics, intelligence, data systems, and satellite communications.
On initial physical examination, the patient was afebrile and lethargic, and his blood pressure was mildly elevated (144/83 mm Hg). Cardiopulmonary examination was normal. Neurologic examination revealed no severe focal deficits, and cranial nerves II to XII were intact. Funduscopic examination was normal, with no papilledema noted. Motor strength was 5/5 bilaterally in the deltoids, biceps, triceps, radial and ulnar wrist extensors, iliopsoas, quadriceps, hamstrings, tibialis anterior and posterior, fibulares, and gastrocnemius. Pinprick sensation and light-touch sensation were decreased within the lateral bicipital region of the left upper extremity. Pinprick sensation was intact bilaterally in 1-inch increments at all other distributions along the medial and lateral aspects of the upper and lower extremities. Muscle tone and bulk were normal in all extremities. Reflexes were 2+ bilaterally in the biceps, brachioradialis, triceps, quadriceps, and Achilles. The Babinski sign was absent bilaterally, the finger-tonose and heel-to-shin tests were normal, the Romberg sign was absent, and there was no evidence of pronator drift. Laboratory test results were normal except for slightly elevated hemoglobin (17.5 g/dL) and D-dimer (588 ng/mL) levels.
Although noncontrast CT of the head initially showed no acute intracranial abnormalities, retrospective close comparison with the arterial system revealed slightly increased attenuation in the superior sagittal sinus, straight sinus, vein of Galen, and internal cerebral veins (Figures 1A and 1B) relative to the arterial carotid anterior circulation (Figures 2A and 2B).
Subsequent brain MRI without contrast showed a hyperintense T1 signal involving the superior sagittal sinus (Figures 3A and 3B), extending into the straight sinus and the vein of Galen. Magnetic resonance imaging with contrast demonstrated a prominent filling defect primarily in the superior sagittal sinus, in the right transverse sinus, and in the vein of Galen. Diffusion-weighted brain MRI sequence showed restricted diffusion localized to the right thalamic area (Figure 4) and no evidence of hemorrhage.
Treatment
International guidelines recommend using heparin to achieve rapid anticoagulation, stop the thrombotic process, and prevent extension of the thrombus.13 Theoretically, more rapid recanalization may have been achieved by performing endovascular thrombectomy in the present case. However, severe bleeding complications, combined with higher cost and the limited availability of clinicians experienced in treating this rare disease, convince physicians to rely on heparin as first-line treatment for CVT.14 A small randomized clinical trial found LMWH safer and more efficacious than UFH in treating CVT.15 After stabilization, oral anticoagulation therapy is used to maintain an international normalized ratio (INR) between 2.0 and 3.0 for at least 3 months.14
Given these findings, the patient was initially treated with LMWH. Eventually he was switched to oral warfarin and showed signs of clinical improvement. A hypercoagulability state workup revealed that the patient was heterozygous for the prothrombin G20210A mutation, and he was discharged and instructed to continue the oral warfarin therapy.
On follow-up, the hematology and neurology team initiated indefinite treatment with warfarin for his genetic hypercoagulability state. Monitoring of the dose of anticoagulation therapy was started to maintain INR between 2.0 and 3.0. The patient began coming to the office for INR monitoring every 2 to 3 weeks, and his most recent INR, in May 2017, was 2.66. He is taking 7.5 mg of warfarin on Wednesdays and Sundays and 5 mg on all other days and currently does not report any progressive neurologic deficits.
Discussion
The clinical findings of CVT and the hypercoagulability state workup revealed that the patient was heterozygous for the prothrombin G20210A mutation. Prothrombin is the precursor to thrombin, which is a key regulator of the clotting cascade and a promoter of coagulation. Carriers of the mutation have elevated levels of blood plasma prothrombin and have been associated with a 4 times higher risk for VTE.16
Several large studies and systematic reviews have confirmed that the prothrombin G20210A mutation is associated with higher rates of VTE, leading to an increased risk for DVT of the leg or pulmonary embolism.17-19 More specifically, a metaanalysis of 15 case–control studies found strong associations between the mutation and CVT.20 Despite this significant association, studies are inconclusive about whether heterozygosity for the mutation is associated with increased rates of recurrent CVT or other VTE in the absence of other risk factors, such as oral contraceptive use, trauma, malignancy, and infection.21-23 Therefore, the optimal duration of anticoagulation therapy for CVT is not well established in patients with the mutation. However, the present patient was started on indefinite anticoagulation therapy because the underlying etiology of the CVT was not reversible or transient, and this CVT was his second episode of VTE, following a 2011 DVT in the right leg.
The case discussed here illustrates the clinical presentation and diagnostic complexities of CVT. Two days before coming to the ED, the patient presented to an outside facility and underwent a workup for nonspecific symptoms (eg, confusion, headaches). Due to the nonspecific presentation associated with CVT, a detailed history is imperative to distinguish symptoms suggesting increased intracranial pressure, such as headaches worse when lying down or present in the morning, with a high clinical suspicion of CVT. The ability to attain these specific details leads clinicians toward obtaining the necessary imaging studies for potential CVT patients, and may prevent delay in diagnosis and treatment. The thrombus in CVT initially consists of deoxyhemoglobin and appears on MRI as an isointense signal on T1-weighted images and a hypointense signal on T2-weighted images; over subsequent days, the thrombus changes to methemoglobin and appears as a slightly hyperintense signal on both T1- and T2-weighted images.24
During this phase, there are some false negatives, as the thrombus can be mistaken for imaging artifacts, hematocrit elevations, or low flow of normal venous blood. Given the clinical findings and imaging studies, it is essential to distinguish CVT from other benign etiologies. Earlier diagnosis and initiation of anticoagulation therapy may have precluded the small amount of localized ischemic changes in this patient’s right thalamus, thus preventing the mild sensory loss in the left upper extremity. With the variable and nonspecific clinical presentations and the difficulties in identifying CVT with first-line imaging, progression of thrombus formation may lead to severe focal neurologic deficits, coma, or death.
Using CT imaging studies to compare the blood in the draining cerebral sinuses with the blood in the arterial system can help distinguish CVT from other etiologies of hyperdense abnormalities, such as increased hematocrit or decreased flow. Retrospective close examination of the present patient’s noncontrast CT images of the head and brain revealed slight hyperdensity in the cerebral sinuses compared with the arterial blood, suggesting the presence of thrombus formation in the cerebral veins. As CT is often the first study used to evaluate the nonspecific clinical presentations of these patients, identifying subtle signaldensity differences between the arterial and venous systems could guide physicians in identifying CVT earlier.
The authors reiterate the importance of meticulous imaging interpretation in light of the entire clinical picture: In these patients, it is imperative to have a high index of clinical suspicion for CVT in order to prevent more serious complications, such as ischemic or hemorrhagic stroke.
Cerebral venous thrombosis (CVT) is a rare cerebrovascular disease that affects about 5 in 1 million people each year and accounts for 0.5% of all strokes.1 Previously it was thought to be caused most commonly by infections (eg, mastoiditis, otitis, meningitis) affecting the superior sagittal sinus and often resulting in focal neurologic deficits, seizures, coma, and death. Although local and systemic infections are still prominent risk factors in its development, CVT is now primarily recognized as a nonseptic disease with a wide spectrum of clinical presentations.
Cerebral venous thrombosis causes reduced outflow of blood and cerebrospinal fluid, which in half of affected patients leads to significant venous infarct. As opposed to arterial infarctions, CVT mainly affects children and young adults; it is an important cause of stroke in the younger population.2 There is significant overlap of the many risk factors for CVT and those for venous thromboembolism (VTE): cancer, obesity, genetic thrombophilia, trauma, infection, and prior neurosurgery. However, the most common acquired risk factors for CVT are oral contraceptive use and pregnancy, which explains why CVT is 3 times more likely to occur in young and middle-aged women.3
Cerebral venous thrombosis was first recognized by a French physician in the 19th century, a time when the condition was diagnosed at autopsy, which typically showed hemorrhagic lesions at the thrombus site.4 For many years heparin was contraindicated in the treatment of CVT, and only within the past 25 years did advances in neuroimaging allow for earlier diagnosis and change perspectives on the management of this disease.
Cerebral venous thrombosis occurs from formation of a thrombus within the cerebral venous sinuses, leading to elevated intracranial pressure and eventually ischemia or intracranial hemorrhage. Improved imaging techniques, notably magnetic resonance imaging (MRI) and computed tomography (CT) venography, allow physicians to identify thrombus formation earlier and begin anticoagulation therapy with heparin before infarction. A meta-analysis of studies found that heparin was safer and more efficacious in treating CVT compared with placebo.5 Furthermore, several small randomized trials found treatment with unfractionated heparin (UFH) or low-molecularweight heparin (LMWH) was not associated with higher risk of hemorrhagic stroke in these patients.6-8
Despite improvements in imaging modalities, in many cases diagnosis is delayed, as most patients with CVT have a wide spectrum of presentations with nonspecific symptoms, such as headache, seizure, focal sensorimotor deficits, and papilledema.9 Clinical presentations of CVT depend on a variety of factors, including age, time of onset, CVT location, and presence of parenchymal lesions. Isolated headache is the most common initial symptom, and in many cases is the only presenting manifestation of CVT.1 Encephalopathy with multifocal signs, delirium, or dysfunction in executive functions most commonly occurs in elderly populations.
Cavernous sinus thrombosis most commonly produces generalized headaches, orbital pain, proptosis, and diplopia, whereas cortical vein thrombosis often produces seizures and focal sensorimotor deficits. Aphasia may be present in patients with isolated left transverse sinus thrombosis. In the presence of deep cerebral venous occlusion, patients can present in coma or with severe cognitive deficits and widespread paresis.10 Thrombosis of different veins and sinuses results in a wide spectrum of diverse clinical pictures, posing a diagnostic challenge and affecting clinical outcomes.
Given the variable and often nonspecific clinical presentations of these cases, unenhanced CT typically is the first imaging study ordered. According to the literature, noncontrast CT is not sensitive (25%-56%) in detecting CVT, and findings are normal in up to 26% of patients, rarely providing a specific diagnosis.11 Furthermore, visualization on MRI can be difficult during the acute phase of CVT, as the thrombus initially appears isointense on T1-weighted images and gradually becomes hyperintense over the course of the disease process.12 These difficulties with the usual first-choice imaging examinations often result in a delay in diagnosing CVT. However, several points on close examination of these imaging studies may help physicians establish a high index of clinical suspicion and order the appropriate follow-up studies for CVT.
The authors report the case of a patient who presented with a 1-week history of confusion, headaches, and dizziness. His nonspecific presentation along with the absence of obvious signs of CVT on imaging prolonged his diagnosis and the initiation of an appropriate treatment plan.
Case Report
A 57-year-old white air-conditioning mechanic presented to the emergency department (ED) with a 1-week history of gradual-onset confusion, severe headaches, dizziness, light-headedness, poor memory, and increased sleep. Two days earlier, he presented with similar symptoms to an outside facility, where he was admitted and underwent a workup for stroke, hemorrhage, and cardiac abnormalities—including noncontrast CT of the head. With nothing of clinical significance found, the patient was discharged and was advised to follow up on an outpatient basis.
Persisting symptoms brought the patient to the ED 1 day later, and he was admitted. He described severe, progressive, generalized headaches that were more severe when he was lying down at night and waking in the morning. He did not report that the headaches worsened with coughing or straining, and he reported no history of trauma, neck stiffness, vision change, seizures, and migraines. His medical history was significant for hypertension, dyslipidemia, and in 2011, an unprovoked deep vein thrombosis (DVT) in the right leg. He reported no history of tobacco, alcohol, or illicit drug use. He had served in the U.S. Navy, working in electronics, intelligence, data systems, and satellite communications.
On initial physical examination, the patient was afebrile and lethargic, and his blood pressure was mildly elevated (144/83 mm Hg). Cardiopulmonary examination was normal. Neurologic examination revealed no severe focal deficits, and cranial nerves II to XII were intact. Funduscopic examination was normal, with no papilledema noted. Motor strength was 5/5 bilaterally in the deltoids, biceps, triceps, radial and ulnar wrist extensors, iliopsoas, quadriceps, hamstrings, tibialis anterior and posterior, fibulares, and gastrocnemius. Pinprick sensation and light-touch sensation were decreased within the lateral bicipital region of the left upper extremity. Pinprick sensation was intact bilaterally in 1-inch increments at all other distributions along the medial and lateral aspects of the upper and lower extremities. Muscle tone and bulk were normal in all extremities. Reflexes were 2+ bilaterally in the biceps, brachioradialis, triceps, quadriceps, and Achilles. The Babinski sign was absent bilaterally, the finger-tonose and heel-to-shin tests were normal, the Romberg sign was absent, and there was no evidence of pronator drift. Laboratory test results were normal except for slightly elevated hemoglobin (17.5 g/dL) and D-dimer (588 ng/mL) levels.
Although noncontrast CT of the head initially showed no acute intracranial abnormalities, retrospective close comparison with the arterial system revealed slightly increased attenuation in the superior sagittal sinus, straight sinus, vein of Galen, and internal cerebral veins (Figures 1A and 1B) relative to the arterial carotid anterior circulation (Figures 2A and 2B).
Subsequent brain MRI without contrast showed a hyperintense T1 signal involving the superior sagittal sinus (Figures 3A and 3B), extending into the straight sinus and the vein of Galen. Magnetic resonance imaging with contrast demonstrated a prominent filling defect primarily in the superior sagittal sinus, in the right transverse sinus, and in the vein of Galen. Diffusion-weighted brain MRI sequence showed restricted diffusion localized to the right thalamic area (Figure 4) and no evidence of hemorrhage.
Treatment
International guidelines recommend using heparin to achieve rapid anticoagulation, stop the thrombotic process, and prevent extension of the thrombus.13 Theoretically, more rapid recanalization may have been achieved by performing endovascular thrombectomy in the present case. However, severe bleeding complications, combined with higher cost and the limited availability of clinicians experienced in treating this rare disease, convince physicians to rely on heparin as first-line treatment for CVT.14 A small randomized clinical trial found LMWH safer and more efficacious than UFH in treating CVT.15 After stabilization, oral anticoagulation therapy is used to maintain an international normalized ratio (INR) between 2.0 and 3.0 for at least 3 months.14
Given these findings, the patient was initially treated with LMWH. Eventually he was switched to oral warfarin and showed signs of clinical improvement. A hypercoagulability state workup revealed that the patient was heterozygous for the prothrombin G20210A mutation, and he was discharged and instructed to continue the oral warfarin therapy.
On follow-up, the hematology and neurology team initiated indefinite treatment with warfarin for his genetic hypercoagulability state. Monitoring of the dose of anticoagulation therapy was started to maintain INR between 2.0 and 3.0. The patient began coming to the office for INR monitoring every 2 to 3 weeks, and his most recent INR, in May 2017, was 2.66. He is taking 7.5 mg of warfarin on Wednesdays and Sundays and 5 mg on all other days and currently does not report any progressive neurologic deficits.
Discussion
The clinical findings of CVT and the hypercoagulability state workup revealed that the patient was heterozygous for the prothrombin G20210A mutation. Prothrombin is the precursor to thrombin, which is a key regulator of the clotting cascade and a promoter of coagulation. Carriers of the mutation have elevated levels of blood plasma prothrombin and have been associated with a 4 times higher risk for VTE.16
Several large studies and systematic reviews have confirmed that the prothrombin G20210A mutation is associated with higher rates of VTE, leading to an increased risk for DVT of the leg or pulmonary embolism.17-19 More specifically, a metaanalysis of 15 case–control studies found strong associations between the mutation and CVT.20 Despite this significant association, studies are inconclusive about whether heterozygosity for the mutation is associated with increased rates of recurrent CVT or other VTE in the absence of other risk factors, such as oral contraceptive use, trauma, malignancy, and infection.21-23 Therefore, the optimal duration of anticoagulation therapy for CVT is not well established in patients with the mutation. However, the present patient was started on indefinite anticoagulation therapy because the underlying etiology of the CVT was not reversible or transient, and this CVT was his second episode of VTE, following a 2011 DVT in the right leg.
The case discussed here illustrates the clinical presentation and diagnostic complexities of CVT. Two days before coming to the ED, the patient presented to an outside facility and underwent a workup for nonspecific symptoms (eg, confusion, headaches). Due to the nonspecific presentation associated with CVT, a detailed history is imperative to distinguish symptoms suggesting increased intracranial pressure, such as headaches worse when lying down or present in the morning, with a high clinical suspicion of CVT. The ability to attain these specific details leads clinicians toward obtaining the necessary imaging studies for potential CVT patients, and may prevent delay in diagnosis and treatment. The thrombus in CVT initially consists of deoxyhemoglobin and appears on MRI as an isointense signal on T1-weighted images and a hypointense signal on T2-weighted images; over subsequent days, the thrombus changes to methemoglobin and appears as a slightly hyperintense signal on both T1- and T2-weighted images.24
During this phase, there are some false negatives, as the thrombus can be mistaken for imaging artifacts, hematocrit elevations, or low flow of normal venous blood. Given the clinical findings and imaging studies, it is essential to distinguish CVT from other benign etiologies. Earlier diagnosis and initiation of anticoagulation therapy may have precluded the small amount of localized ischemic changes in this patient’s right thalamus, thus preventing the mild sensory loss in the left upper extremity. With the variable and nonspecific clinical presentations and the difficulties in identifying CVT with first-line imaging, progression of thrombus formation may lead to severe focal neurologic deficits, coma, or death.
Using CT imaging studies to compare the blood in the draining cerebral sinuses with the blood in the arterial system can help distinguish CVT from other etiologies of hyperdense abnormalities, such as increased hematocrit or decreased flow. Retrospective close examination of the present patient’s noncontrast CT images of the head and brain revealed slight hyperdensity in the cerebral sinuses compared with the arterial blood, suggesting the presence of thrombus formation in the cerebral veins. As CT is often the first study used to evaluate the nonspecific clinical presentations of these patients, identifying subtle signaldensity differences between the arterial and venous systems could guide physicians in identifying CVT earlier.
The authors reiterate the importance of meticulous imaging interpretation in light of the entire clinical picture: In these patients, it is imperative to have a high index of clinical suspicion for CVT in order to prevent more serious complications, such as ischemic or hemorrhagic stroke.
1. Bousser MG, Ferro JM. Cerebral venous thrombosis: an update. Lancet Neurol. 2007;6(2):162-170.
2. Coutinho JM. Cerebral venous thrombosis. J Thromb Haemost. 2015;13(suppl 1):S238-S244.
3. Coutinho JM, Ferro JM, Canhão P, et al. Cerebral venous and sinus thrombosis in women. Stroke. 2009;40(7):2356-2361.
4. Zuurbier SM, Coutinho JM. Cerebral venous thrombosis. Adv Exp Med Biol. 2017;906:183-193.
5. Einhäupl KM, Villringer A, Meister W, et al. Heparin treatment in sinus venous thrombosis. Lancet. 1991;338(8767):597-600.
6. de Bruijn SF, Stam J. Randomized, placebocontrolled trial of anticoagulant treatment with lowmolecular-weight heparin for cerebral sinus thrombosis. Stroke. 1999;30(3):484-488.
7. Nagaraja D, Haridas T, Taly AB, Veerendrakumar M, SubbuKrishna DK. Puerperal cerebral venous thrombosis: therapeutic benefit of low dose heparin. Neurol India. 1999;47(1):43-46.
8. Coutinho JM, de Bruijn SF, deVeber G, Stam J. Anticoagulation for cerebral venous sinus thrombosis. Stroke. 2012;43(4):e41-e42.
9. Sassi SB, Touati N, Baccouche H, Drissi C, Romdhane NB, Hentati F. Cerebral venous thrombosis. Clin Appl Thromb Hemost. 2016:1076029616665168. [Epub ahead of print.]
10. Ferro JM, Canhão P. Cerebral venous sinus thrombosis: update on diagnosis and management. Curr Cardiol Rep. 2014;16(9):523.
11. Albright KC, Freeman WD, Kruse BT. Cerebral venous thrombosis. J Emerg Med. 2010;38(2):238-239.
12. Lafitte F, Boukobza M, Guichard JP, et al. MRI and MRA for diagnosis and follow-up of cerebral venous thrombosis (CVT). Clin Radiol. 1997;52(9):672-679.
13. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192.
14. Coutinho JM, Middeldorp S, Stam J. Advances in the treatment of cerebral venous thrombosis. Curr Treat Options Neurol. 2014;16(7):299.
15. Coutinho JM, Ferro JM, Canhão P, Barinagarrementeria F, Bousser MG, Stam J; ISCVT Investigators. Unfractionated or low-molecular weight heparin for the treatment of cerebral venous thrombosis. Stroke. 2010;41(11):2575-2580.
16. Rosendaal FR. Venous thrombosis: the role of genes, environment, and behavior. Hematology Am Soc Hematol Educ Program. 2005:1-12.
17. Dentali F, Crowther M, Ageno W. Thrombophilic abnormalities, oral contraceptives, and risk of cerebral vein thrombosis: a meta-analysis. Blood.2006;107(7):2766-2773.
18. Salomon O, Steinberg DM, Zivelin A, et al. Single and combined prothrombotic factors in patients with idiopathic venous thromboembolism: prevalence and risk assessment. Arterioscler Thromb Vasc Biol. 1999;19(3):511-518.
19. Emmerich J, Rosendaal FR, Cattaneo M, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism— pooled analysis of 8 case–control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost. 2001;86(3):809-816.
20. Lauw MN, Barco S, Coutinho JM, Middeldorp S. Cerebral venous thrombosis and thrombophilia: a systematic review and meta-analysis. Semin Thromb Hemost. 2013;39(8):913-927.
21. Dentali F, Poli D, Scoditti U, et al. Long-term outcomes of patients with cerebral vein thrombosis: a multicenter study. J Thromb Haemost. 2012;10(7):1297-1302.
22. Martinelli I, Bucciarelli P, Passamonti SM, Battaglioli T, Previtali E, Mannucci PM. Long-term evaluation of the risk of recurrence after cerebral sinus-venous thrombosis. Circulation. 2010;121(25):2740-2746.
23. Gosk-Bierska I, Wysokinski W, Brown RD Jr, et al. Cerebral venous sinus thrombosis: incidence of venous thrombosis recurrence and survival. Neurology. 2006;67(5):814-819.
24. Galidie G, Le Gall R, Cordoliani YS, Pharaboz C, Le Marec E, Cosnard G. Thrombosis of the cerebral veins. X-ray computed tomography and MRI imaging. 11 cases [in French]. J Radiol. 1992;73(3):175-190
1. Bousser MG, Ferro JM. Cerebral venous thrombosis: an update. Lancet Neurol. 2007;6(2):162-170.
2. Coutinho JM. Cerebral venous thrombosis. J Thromb Haemost. 2015;13(suppl 1):S238-S244.
3. Coutinho JM, Ferro JM, Canhão P, et al. Cerebral venous and sinus thrombosis in women. Stroke. 2009;40(7):2356-2361.
4. Zuurbier SM, Coutinho JM. Cerebral venous thrombosis. Adv Exp Med Biol. 2017;906:183-193.
5. Einhäupl KM, Villringer A, Meister W, et al. Heparin treatment in sinus venous thrombosis. Lancet. 1991;338(8767):597-600.
6. de Bruijn SF, Stam J. Randomized, placebocontrolled trial of anticoagulant treatment with lowmolecular-weight heparin for cerebral sinus thrombosis. Stroke. 1999;30(3):484-488.
7. Nagaraja D, Haridas T, Taly AB, Veerendrakumar M, SubbuKrishna DK. Puerperal cerebral venous thrombosis: therapeutic benefit of low dose heparin. Neurol India. 1999;47(1):43-46.
8. Coutinho JM, de Bruijn SF, deVeber G, Stam J. Anticoagulation for cerebral venous sinus thrombosis. Stroke. 2012;43(4):e41-e42.
9. Sassi SB, Touati N, Baccouche H, Drissi C, Romdhane NB, Hentati F. Cerebral venous thrombosis. Clin Appl Thromb Hemost. 2016:1076029616665168. [Epub ahead of print.]
10. Ferro JM, Canhão P. Cerebral venous sinus thrombosis: update on diagnosis and management. Curr Cardiol Rep. 2014;16(9):523.
11. Albright KC, Freeman WD, Kruse BT. Cerebral venous thrombosis. J Emerg Med. 2010;38(2):238-239.
12. Lafitte F, Boukobza M, Guichard JP, et al. MRI and MRA for diagnosis and follow-up of cerebral venous thrombosis (CVT). Clin Radiol. 1997;52(9):672-679.
13. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192.
14. Coutinho JM, Middeldorp S, Stam J. Advances in the treatment of cerebral venous thrombosis. Curr Treat Options Neurol. 2014;16(7):299.
15. Coutinho JM, Ferro JM, Canhão P, Barinagarrementeria F, Bousser MG, Stam J; ISCVT Investigators. Unfractionated or low-molecular weight heparin for the treatment of cerebral venous thrombosis. Stroke. 2010;41(11):2575-2580.
16. Rosendaal FR. Venous thrombosis: the role of genes, environment, and behavior. Hematology Am Soc Hematol Educ Program. 2005:1-12.
17. Dentali F, Crowther M, Ageno W. Thrombophilic abnormalities, oral contraceptives, and risk of cerebral vein thrombosis: a meta-analysis. Blood.2006;107(7):2766-2773.
18. Salomon O, Steinberg DM, Zivelin A, et al. Single and combined prothrombotic factors in patients with idiopathic venous thromboembolism: prevalence and risk assessment. Arterioscler Thromb Vasc Biol. 1999;19(3):511-518.
19. Emmerich J, Rosendaal FR, Cattaneo M, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism— pooled analysis of 8 case–control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost. 2001;86(3):809-816.
20. Lauw MN, Barco S, Coutinho JM, Middeldorp S. Cerebral venous thrombosis and thrombophilia: a systematic review and meta-analysis. Semin Thromb Hemost. 2013;39(8):913-927.
21. Dentali F, Poli D, Scoditti U, et al. Long-term outcomes of patients with cerebral vein thrombosis: a multicenter study. J Thromb Haemost. 2012;10(7):1297-1302.
22. Martinelli I, Bucciarelli P, Passamonti SM, Battaglioli T, Previtali E, Mannucci PM. Long-term evaluation of the risk of recurrence after cerebral sinus-venous thrombosis. Circulation. 2010;121(25):2740-2746.
23. Gosk-Bierska I, Wysokinski W, Brown RD Jr, et al. Cerebral venous sinus thrombosis: incidence of venous thrombosis recurrence and survival. Neurology. 2006;67(5):814-819.
24. Galidie G, Le Gall R, Cordoliani YS, Pharaboz C, Le Marec E, Cosnard G. Thrombosis of the cerebral veins. X-ray computed tomography and MRI imaging. 11 cases [in French]. J Radiol. 1992;73(3):175-190
Case Studies in Toxicology: An Unlikely Cause of Paralysis
Case
An Asian man in his third decade, with a medical history of hypertension and hyperlipidemia, and who had recently been involved in a motor vehicle collision (MVC), presented to the ED with a chief complaint of severe bilateral upper and lower extremity weakness. The patient noted that the weakness had begun the previous evening and became progressively worse throughout the night, to the point that he was unable to move any of his extremities on the morning of presentation.
Upon arrival at the ED, the patient was awake, alert, and oriented to self, time, and place; he also spoke in full sentences without distress. He denied fever, chills, difficulty breathing, or preceding viral illness. The patient stated that he was not taking any medications and denied a history of alcohol, tobacco, or drug abuse.
Initial vital signs at presentation were: blood pressure, 141/50 mm Hg; heart rate, 90 beats/min; respiratory rate, 16 breaths/min; and temperature, 97.4°F. Oxygen saturation was 100% on room air. On physical examination, the patient was in no acute distress and had a normal mental status. His pupils were normally reactive and his other cranial nerves were normal. Muscle strength in the upper and lower extremities was 1/5 with 1+ reflexes bilaterally, and there was no sensory deficit. The patient was placed on continuous cardiac monitoring with pulse oximetry.
What is the differential diagnosis for acute extremity weakness or paralysis?
The differential diagnosis for acute symmetrical extremity weakness or paralysis is broad and includes conditions of neurological, inflammatory, and toxic/metabolic etiologies.1 Neurological diagnoses to consider include acute stroke, specifically of the anterior cerebral or middle cerebral artery territories; Guillain-Barré syndrome; myasthenia gravis; spinal cord compression; and tick paralysis. Acute ischemic or hemorrhagic stroke most frequently presents with unilateral upper or lower extremity weakness accompanied by garbled speech and sensory deficits. Patients who have suffered a brainstem or cerebellar stroke commonly present with alterations of consciousness, visual changes, and ataxia. Posterior circulation strokes are also characterized by crossed neurological deficits, such as motor deficits on one side of the body and sensory deficits on the other.
Spinal Cord Pathology. Signs and symptoms of spinal cord compression or inflammation vary widely depending on the level affected. Motor and sensory findings of spinal cord pathology include muscle weakness, spasticity, hyper- or hyporeflexia, and a discrete level below which sensation is absent or reduced.
Guillain-Barré Syndrome. Patients who have Guillain-Barré syndrome (a disease of the myelin sheaths of the peripheral nerves) often present with complaints of numbness or paresthesias in the extremities.2 The condition is characterized by progressive symmetric muscle weakness accompanied by absent or depressed deep tendon reflexes and is typically associated with a recent exposure to an infectious agent such as a viral upper respiratory infection, bacterial infection, or vaccine.
Myasthenia Gravis. Myasthenia gravis is a disease of the neuromuscular junction. It presents with weakness in any muscle group, and the muscles are easily fatigued by repetitive use.
Toxic Exposures. Toxins, such as botulinum, ixovotoxin, nicotine, succinylcholine, and tetrodotoxin, are prominent, though less common, causes of muscular weakness or paralysis. Botulinum toxin acts at the neuromuscular junction. Patients with botulism typically present with a gastrointestinal prodrome of nausea, vomiting, and diarrhea followed by cranial nerve dysfunction and descending muscle weakness.3
Tetrodotoxin, nicotine, and curare-like paralytics act at the motor end plate of the neuromuscular junction to produce neuromuscular blockade with subsequent muscular weakness or paralysis. Similarly, ixovotoxin, the toxin responsible for tick paralysis, causes ascending flaccid paralysis by decreasing the release of acetylcholine at the neuromuscular junction.3
Metabolic and Endocrine Disorders. Conditions such as hypokalemia, hypomagnesemia, and periodic paralysis can also present with neurological complaints such as generalized weakness and paresthesias. Of note, it is important to differentiate true neuromuscular weakness from weakness secondary to limited effort.
Case Continuation
Because of the patient’s history of an MVC, cervical cord compression was considered concerning enough to require exclusion through magnetic resonance imaging (MRI) of the cervical spine. However, upon arrival at the MRI suite, the patient became severely tachypneic and tachycardic, and was unable to tolerate lying flat. He was intubated for impending respiratory failure. Laboratory results from blood drawn prior to transport to MRI were reported immediately after the resuscitation and were notable for the following: potassium, <1.5 mEq/L; bicarbonate, 20 mEq/L; creatine kinase, 889 U/L; ethanol, not detected.
What is hypokalemic periodic paralysis?
Hypokalemic periodic paralysis (HypoKPP) is a syndrome of episodic muscle weakness with concomitant hypokalemia. Familial forms of HypoKPP have been attributed to mutations in genes coding for either calcium or sodium channels.
The nonfamilial form of HypoKPP is attributed to hyperthyroidism and is most often seen in Asian men in the second and third decades of life. The disorder is characterized by acute onset hypokalemia and extremity paralysis with simultaneous hyperthyroid state. It is believed that hypokalemia occurs as a result of intracellular shift of potassium from thyroid-induced hormone sensitization of the Na+/K+-ATPase rather than a depletion of total body potassium. Acute episodes of paralysis are triggered by high-carbohydrate meals, alcohol consumption, emotional stress, and infection. Paralysis can last from 3 to 96 hours and is accompanied by decreased or absent deep tendon reflexes with normal sensation and mental status.
In the nonfamilial form of HypoKPP, signs of thyrotoxicosis are often present and include tachycardia, moist skin, and hyperthermia, but it may be difficult to specifically recognize this etiology given the patient’s grave clinical condition.4 Similar to many significant metabolic and electrolyte disturbances, complications of HypoKPP include dysrhythmia, respiratory failure, and sometimes death.5
How should HypoKPP be managed in the ED?
Management of HypoKPP begins with careful assessment of the patient’s airway, breathing, and circulation. Once the patient is stabilized, management of consequential effects of hypokalemia, such as respiratory distress and muscular paralysis, should focus on correcting the electrolyte and endocrine derangements.
Propranolol. If the patient exhibits signs of thyrotoxicosis, initial treatment includes propranolol, a nonselective beta-blocker, which both prevents the intracellular shift of potassium and assists in correcting the underlying hyperthyroid and hypermetabolic state. Although there is no standard propranolol dosing protocol for HypoKPP, some authors suggest that an aggressive dose of 2 mg intravenously (IV) every 10 minutes can shorten the patient’s episode of paralysis to 6 hours.6
Potassium Chloride. Administration of potassium chloride to raise the serum potassium to life-sustaining concentrations should be done cautiously through IV infusion of standard doses.7 In correcting hypokalemia with potassium, care should be taken to avoid overcorrection, which may subsequently result in rebound hyperkalemia as the total body potassium redistributes. Lower doses of potassium (ie, <50 mEq per dose), are preferred to achieve adequate repletion while avoiding rebound hyperkalemia.8
Case Conclusion
The results of thyroid studies that had been added on to the original set of laboratory studies revealed profound hyperthyroidism, with an essentially absent concentration of thyroid-stimulating hormone.
1. Morchi RS. Weakness. In: Rosen P, ed. Rosen’s Emergency Medicine. 8th ed. Philadelphia, PA: Elsevier; 2014:124-128.
2. McGillicuddy DC, Walker O, Shapiro NI, Edlow JA. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393. doi:10.1016/j.annemergmed.2005.05.008.
3. Rao RB. Neurological principles. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:315-323.
4. Lam L, Nair RJ, Tingle L. Thyrotoxic periodic paralysis. Proc (Bayl Univ Med Cent). 2006;19(2):126-129.
5. Li X, Yao S, Xiang Y, et al. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurol. 2015;15:38. doi:10.1186/s12883-015-0290-8.
6. Birkhahn RH, Gaeta TJ, Melniker L. Thyrotoxic periodic paralysis and intravenous propranolol in the emergency setting. J Emerg Med. 2000;18(2):199-202.
7. Lu KC, Hsu YJ, Chiu JS, Hsu YD, Lin SH. Effects of potassium supplementation on the recovery of thyrotoxic periodic paralysis. Am J Emerg Med. 2004;22(7):544-547.
8. Tassone H, Moulin A, Henderson SO. The pitfalls of potassium replacement in thyrotoxic periodic paralysis: a case report and review of the literature. J Emerg Med. 2004;26(2):157-161. doi:10.1016/j.jemermed.2003.05.004.
Case
An Asian man in his third decade, with a medical history of hypertension and hyperlipidemia, and who had recently been involved in a motor vehicle collision (MVC), presented to the ED with a chief complaint of severe bilateral upper and lower extremity weakness. The patient noted that the weakness had begun the previous evening and became progressively worse throughout the night, to the point that he was unable to move any of his extremities on the morning of presentation.
Upon arrival at the ED, the patient was awake, alert, and oriented to self, time, and place; he also spoke in full sentences without distress. He denied fever, chills, difficulty breathing, or preceding viral illness. The patient stated that he was not taking any medications and denied a history of alcohol, tobacco, or drug abuse.
Initial vital signs at presentation were: blood pressure, 141/50 mm Hg; heart rate, 90 beats/min; respiratory rate, 16 breaths/min; and temperature, 97.4°F. Oxygen saturation was 100% on room air. On physical examination, the patient was in no acute distress and had a normal mental status. His pupils were normally reactive and his other cranial nerves were normal. Muscle strength in the upper and lower extremities was 1/5 with 1+ reflexes bilaterally, and there was no sensory deficit. The patient was placed on continuous cardiac monitoring with pulse oximetry.
What is the differential diagnosis for acute extremity weakness or paralysis?
The differential diagnosis for acute symmetrical extremity weakness or paralysis is broad and includes conditions of neurological, inflammatory, and toxic/metabolic etiologies.1 Neurological diagnoses to consider include acute stroke, specifically of the anterior cerebral or middle cerebral artery territories; Guillain-Barré syndrome; myasthenia gravis; spinal cord compression; and tick paralysis. Acute ischemic or hemorrhagic stroke most frequently presents with unilateral upper or lower extremity weakness accompanied by garbled speech and sensory deficits. Patients who have suffered a brainstem or cerebellar stroke commonly present with alterations of consciousness, visual changes, and ataxia. Posterior circulation strokes are also characterized by crossed neurological deficits, such as motor deficits on one side of the body and sensory deficits on the other.
Spinal Cord Pathology. Signs and symptoms of spinal cord compression or inflammation vary widely depending on the level affected. Motor and sensory findings of spinal cord pathology include muscle weakness, spasticity, hyper- or hyporeflexia, and a discrete level below which sensation is absent or reduced.
Guillain-Barré Syndrome. Patients who have Guillain-Barré syndrome (a disease of the myelin sheaths of the peripheral nerves) often present with complaints of numbness or paresthesias in the extremities.2 The condition is characterized by progressive symmetric muscle weakness accompanied by absent or depressed deep tendon reflexes and is typically associated with a recent exposure to an infectious agent such as a viral upper respiratory infection, bacterial infection, or vaccine.
Myasthenia Gravis. Myasthenia gravis is a disease of the neuromuscular junction. It presents with weakness in any muscle group, and the muscles are easily fatigued by repetitive use.
Toxic Exposures. Toxins, such as botulinum, ixovotoxin, nicotine, succinylcholine, and tetrodotoxin, are prominent, though less common, causes of muscular weakness or paralysis. Botulinum toxin acts at the neuromuscular junction. Patients with botulism typically present with a gastrointestinal prodrome of nausea, vomiting, and diarrhea followed by cranial nerve dysfunction and descending muscle weakness.3
Tetrodotoxin, nicotine, and curare-like paralytics act at the motor end plate of the neuromuscular junction to produce neuromuscular blockade with subsequent muscular weakness or paralysis. Similarly, ixovotoxin, the toxin responsible for tick paralysis, causes ascending flaccid paralysis by decreasing the release of acetylcholine at the neuromuscular junction.3
Metabolic and Endocrine Disorders. Conditions such as hypokalemia, hypomagnesemia, and periodic paralysis can also present with neurological complaints such as generalized weakness and paresthesias. Of note, it is important to differentiate true neuromuscular weakness from weakness secondary to limited effort.
Case Continuation
Because of the patient’s history of an MVC, cervical cord compression was considered concerning enough to require exclusion through magnetic resonance imaging (MRI) of the cervical spine. However, upon arrival at the MRI suite, the patient became severely tachypneic and tachycardic, and was unable to tolerate lying flat. He was intubated for impending respiratory failure. Laboratory results from blood drawn prior to transport to MRI were reported immediately after the resuscitation and were notable for the following: potassium, <1.5 mEq/L; bicarbonate, 20 mEq/L; creatine kinase, 889 U/L; ethanol, not detected.
What is hypokalemic periodic paralysis?
Hypokalemic periodic paralysis (HypoKPP) is a syndrome of episodic muscle weakness with concomitant hypokalemia. Familial forms of HypoKPP have been attributed to mutations in genes coding for either calcium or sodium channels.
The nonfamilial form of HypoKPP is attributed to hyperthyroidism and is most often seen in Asian men in the second and third decades of life. The disorder is characterized by acute onset hypokalemia and extremity paralysis with simultaneous hyperthyroid state. It is believed that hypokalemia occurs as a result of intracellular shift of potassium from thyroid-induced hormone sensitization of the Na+/K+-ATPase rather than a depletion of total body potassium. Acute episodes of paralysis are triggered by high-carbohydrate meals, alcohol consumption, emotional stress, and infection. Paralysis can last from 3 to 96 hours and is accompanied by decreased or absent deep tendon reflexes with normal sensation and mental status.
In the nonfamilial form of HypoKPP, signs of thyrotoxicosis are often present and include tachycardia, moist skin, and hyperthermia, but it may be difficult to specifically recognize this etiology given the patient’s grave clinical condition.4 Similar to many significant metabolic and electrolyte disturbances, complications of HypoKPP include dysrhythmia, respiratory failure, and sometimes death.5
How should HypoKPP be managed in the ED?
Management of HypoKPP begins with careful assessment of the patient’s airway, breathing, and circulation. Once the patient is stabilized, management of consequential effects of hypokalemia, such as respiratory distress and muscular paralysis, should focus on correcting the electrolyte and endocrine derangements.
Propranolol. If the patient exhibits signs of thyrotoxicosis, initial treatment includes propranolol, a nonselective beta-blocker, which both prevents the intracellular shift of potassium and assists in correcting the underlying hyperthyroid and hypermetabolic state. Although there is no standard propranolol dosing protocol for HypoKPP, some authors suggest that an aggressive dose of 2 mg intravenously (IV) every 10 minutes can shorten the patient’s episode of paralysis to 6 hours.6
Potassium Chloride. Administration of potassium chloride to raise the serum potassium to life-sustaining concentrations should be done cautiously through IV infusion of standard doses.7 In correcting hypokalemia with potassium, care should be taken to avoid overcorrection, which may subsequently result in rebound hyperkalemia as the total body potassium redistributes. Lower doses of potassium (ie, <50 mEq per dose), are preferred to achieve adequate repletion while avoiding rebound hyperkalemia.8
Case Conclusion
The results of thyroid studies that had been added on to the original set of laboratory studies revealed profound hyperthyroidism, with an essentially absent concentration of thyroid-stimulating hormone.
Case
An Asian man in his third decade, with a medical history of hypertension and hyperlipidemia, and who had recently been involved in a motor vehicle collision (MVC), presented to the ED with a chief complaint of severe bilateral upper and lower extremity weakness. The patient noted that the weakness had begun the previous evening and became progressively worse throughout the night, to the point that he was unable to move any of his extremities on the morning of presentation.
Upon arrival at the ED, the patient was awake, alert, and oriented to self, time, and place; he also spoke in full sentences without distress. He denied fever, chills, difficulty breathing, or preceding viral illness. The patient stated that he was not taking any medications and denied a history of alcohol, tobacco, or drug abuse.
Initial vital signs at presentation were: blood pressure, 141/50 mm Hg; heart rate, 90 beats/min; respiratory rate, 16 breaths/min; and temperature, 97.4°F. Oxygen saturation was 100% on room air. On physical examination, the patient was in no acute distress and had a normal mental status. His pupils were normally reactive and his other cranial nerves were normal. Muscle strength in the upper and lower extremities was 1/5 with 1+ reflexes bilaterally, and there was no sensory deficit. The patient was placed on continuous cardiac monitoring with pulse oximetry.
What is the differential diagnosis for acute extremity weakness or paralysis?
The differential diagnosis for acute symmetrical extremity weakness or paralysis is broad and includes conditions of neurological, inflammatory, and toxic/metabolic etiologies.1 Neurological diagnoses to consider include acute stroke, specifically of the anterior cerebral or middle cerebral artery territories; Guillain-Barré syndrome; myasthenia gravis; spinal cord compression; and tick paralysis. Acute ischemic or hemorrhagic stroke most frequently presents with unilateral upper or lower extremity weakness accompanied by garbled speech and sensory deficits. Patients who have suffered a brainstem or cerebellar stroke commonly present with alterations of consciousness, visual changes, and ataxia. Posterior circulation strokes are also characterized by crossed neurological deficits, such as motor deficits on one side of the body and sensory deficits on the other.
Spinal Cord Pathology. Signs and symptoms of spinal cord compression or inflammation vary widely depending on the level affected. Motor and sensory findings of spinal cord pathology include muscle weakness, spasticity, hyper- or hyporeflexia, and a discrete level below which sensation is absent or reduced.
Guillain-Barré Syndrome. Patients who have Guillain-Barré syndrome (a disease of the myelin sheaths of the peripheral nerves) often present with complaints of numbness or paresthesias in the extremities.2 The condition is characterized by progressive symmetric muscle weakness accompanied by absent or depressed deep tendon reflexes and is typically associated with a recent exposure to an infectious agent such as a viral upper respiratory infection, bacterial infection, or vaccine.
Myasthenia Gravis. Myasthenia gravis is a disease of the neuromuscular junction. It presents with weakness in any muscle group, and the muscles are easily fatigued by repetitive use.
Toxic Exposures. Toxins, such as botulinum, ixovotoxin, nicotine, succinylcholine, and tetrodotoxin, are prominent, though less common, causes of muscular weakness or paralysis. Botulinum toxin acts at the neuromuscular junction. Patients with botulism typically present with a gastrointestinal prodrome of nausea, vomiting, and diarrhea followed by cranial nerve dysfunction and descending muscle weakness.3
Tetrodotoxin, nicotine, and curare-like paralytics act at the motor end plate of the neuromuscular junction to produce neuromuscular blockade with subsequent muscular weakness or paralysis. Similarly, ixovotoxin, the toxin responsible for tick paralysis, causes ascending flaccid paralysis by decreasing the release of acetylcholine at the neuromuscular junction.3
Metabolic and Endocrine Disorders. Conditions such as hypokalemia, hypomagnesemia, and periodic paralysis can also present with neurological complaints such as generalized weakness and paresthesias. Of note, it is important to differentiate true neuromuscular weakness from weakness secondary to limited effort.
Case Continuation
Because of the patient’s history of an MVC, cervical cord compression was considered concerning enough to require exclusion through magnetic resonance imaging (MRI) of the cervical spine. However, upon arrival at the MRI suite, the patient became severely tachypneic and tachycardic, and was unable to tolerate lying flat. He was intubated for impending respiratory failure. Laboratory results from blood drawn prior to transport to MRI were reported immediately after the resuscitation and were notable for the following: potassium, <1.5 mEq/L; bicarbonate, 20 mEq/L; creatine kinase, 889 U/L; ethanol, not detected.
What is hypokalemic periodic paralysis?
Hypokalemic periodic paralysis (HypoKPP) is a syndrome of episodic muscle weakness with concomitant hypokalemia. Familial forms of HypoKPP have been attributed to mutations in genes coding for either calcium or sodium channels.
The nonfamilial form of HypoKPP is attributed to hyperthyroidism and is most often seen in Asian men in the second and third decades of life. The disorder is characterized by acute onset hypokalemia and extremity paralysis with simultaneous hyperthyroid state. It is believed that hypokalemia occurs as a result of intracellular shift of potassium from thyroid-induced hormone sensitization of the Na+/K+-ATPase rather than a depletion of total body potassium. Acute episodes of paralysis are triggered by high-carbohydrate meals, alcohol consumption, emotional stress, and infection. Paralysis can last from 3 to 96 hours and is accompanied by decreased or absent deep tendon reflexes with normal sensation and mental status.
In the nonfamilial form of HypoKPP, signs of thyrotoxicosis are often present and include tachycardia, moist skin, and hyperthermia, but it may be difficult to specifically recognize this etiology given the patient’s grave clinical condition.4 Similar to many significant metabolic and electrolyte disturbances, complications of HypoKPP include dysrhythmia, respiratory failure, and sometimes death.5
How should HypoKPP be managed in the ED?
Management of HypoKPP begins with careful assessment of the patient’s airway, breathing, and circulation. Once the patient is stabilized, management of consequential effects of hypokalemia, such as respiratory distress and muscular paralysis, should focus on correcting the electrolyte and endocrine derangements.
Propranolol. If the patient exhibits signs of thyrotoxicosis, initial treatment includes propranolol, a nonselective beta-blocker, which both prevents the intracellular shift of potassium and assists in correcting the underlying hyperthyroid and hypermetabolic state. Although there is no standard propranolol dosing protocol for HypoKPP, some authors suggest that an aggressive dose of 2 mg intravenously (IV) every 10 minutes can shorten the patient’s episode of paralysis to 6 hours.6
Potassium Chloride. Administration of potassium chloride to raise the serum potassium to life-sustaining concentrations should be done cautiously through IV infusion of standard doses.7 In correcting hypokalemia with potassium, care should be taken to avoid overcorrection, which may subsequently result in rebound hyperkalemia as the total body potassium redistributes. Lower doses of potassium (ie, <50 mEq per dose), are preferred to achieve adequate repletion while avoiding rebound hyperkalemia.8
Case Conclusion
The results of thyroid studies that had been added on to the original set of laboratory studies revealed profound hyperthyroidism, with an essentially absent concentration of thyroid-stimulating hormone.
1. Morchi RS. Weakness. In: Rosen P, ed. Rosen’s Emergency Medicine. 8th ed. Philadelphia, PA: Elsevier; 2014:124-128.
2. McGillicuddy DC, Walker O, Shapiro NI, Edlow JA. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393. doi:10.1016/j.annemergmed.2005.05.008.
3. Rao RB. Neurological principles. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:315-323.
4. Lam L, Nair RJ, Tingle L. Thyrotoxic periodic paralysis. Proc (Bayl Univ Med Cent). 2006;19(2):126-129.
5. Li X, Yao S, Xiang Y, et al. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurol. 2015;15:38. doi:10.1186/s12883-015-0290-8.
6. Birkhahn RH, Gaeta TJ, Melniker L. Thyrotoxic periodic paralysis and intravenous propranolol in the emergency setting. J Emerg Med. 2000;18(2):199-202.
7. Lu KC, Hsu YJ, Chiu JS, Hsu YD, Lin SH. Effects of potassium supplementation on the recovery of thyrotoxic periodic paralysis. Am J Emerg Med. 2004;22(7):544-547.
8. Tassone H, Moulin A, Henderson SO. The pitfalls of potassium replacement in thyrotoxic periodic paralysis: a case report and review of the literature. J Emerg Med. 2004;26(2):157-161. doi:10.1016/j.jemermed.2003.05.004.
1. Morchi RS. Weakness. In: Rosen P, ed. Rosen’s Emergency Medicine. 8th ed. Philadelphia, PA: Elsevier; 2014:124-128.
2. McGillicuddy DC, Walker O, Shapiro NI, Edlow JA. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393. doi:10.1016/j.annemergmed.2005.05.008.
3. Rao RB. Neurological principles. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:315-323.
4. Lam L, Nair RJ, Tingle L. Thyrotoxic periodic paralysis. Proc (Bayl Univ Med Cent). 2006;19(2):126-129.
5. Li X, Yao S, Xiang Y, et al. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurol. 2015;15:38. doi:10.1186/s12883-015-0290-8.
6. Birkhahn RH, Gaeta TJ, Melniker L. Thyrotoxic periodic paralysis and intravenous propranolol in the emergency setting. J Emerg Med. 2000;18(2):199-202.
7. Lu KC, Hsu YJ, Chiu JS, Hsu YD, Lin SH. Effects of potassium supplementation on the recovery of thyrotoxic periodic paralysis. Am J Emerg Med. 2004;22(7):544-547.
8. Tassone H, Moulin A, Henderson SO. The pitfalls of potassium replacement in thyrotoxic periodic paralysis: a case report and review of the literature. J Emerg Med. 2004;26(2):157-161. doi:10.1016/j.jemermed.2003.05.004.

Vascular Injury Following a Fall Onto an Outstretched Hand
A 46-year-old man with a remote history of general tonic-clonic seizures, for which he was taking phenytoin, presented to the ED 30 minutes after sustaining a witnessed mechanical fall. The patient had fallen onto his nondominant left hand, which resulted in an injury to his elbow. He reported neither losing consciousness nor experiencing any seizures following the incident. He denied dislocating the joint or sustaining any other injuries from the fall. He also denied a history of past left elbow injury.
The patient was alert, oriented, and provided a full history of the incident. Regarding medical history, he stated that his last seizure had occurred 10 years prior. Except for the left elbow pain, a review of his systems was negative. The patient appeared in no acute distress, and supported his left upper extremity with a bandana and his right hand.
The patient’s vital signs were normal. The physical examination was negative except for the left elbow, which had significant swelling and limited range of motion without skin break, leading to suspicion for a prehospital dislocation with self-reduction. The joints above, below, and at the injury site were assessed for neurovascular injury.

Computed tomography angiography of the left upper extremity showed a brachial artery occlusion above the elbow, with reconstitution below the joint (Figure 2).

Discussion
There is a paucity of information on vascular injury from elbow dislocation in the emergency medicine literature. A recent literature search referenced orthopedic pitfalls in the ED,1 but most data appear in the orthopedic and vascular literature. A case report from the orthopedic literature in Brazil cites a vascular injury after ED relocation of a dislocated elbow following an assault.2
The elbow is the second most commonly dislocated joint (not including the patella) after the shoulder.3 Posterior dislocations make up the majority of these injuries. Simple versus complex injuries can be differentiated by the presence or absence of fracture.4 Simple complications include stiffness; loss of mobility, especially with full extension; neurovascular injuries; and compartment syndrome. Complex injuries involve fractures and potential neurovascular injuries, stiffness, pain, and loss of mobility.
Soft tissue injuries, fractures, and neurovascular complaints represent the majority of ED encounters, and are commonly related to falls. The elbow is the articulation of the humerus, ulna, and radius bones. Range of motion includes, but is not limited to, flexion, extension, supination, and pronation. Tears in the lateral ulnar ligament, joint capsule, and medial collateral ligament lead to instability of the joint and increase risk of dislocation.
Fractures make up to 20% of injuries to the elbow. These include fractures of the radial head and neck (most common), olecranon, and distal humerus.5 Open elbow fractures are rare, as are vascular injuries (5%-13% of cases).6 When present, vascular elbow injuries usually involve the brachial artery, and display abnormal palpable and Doppler assessment of the brachial and radial arteries.6
Nerve injuries may include injury to the radial nerve. Manifestations of radial nerve injury include abnormal sensation to the dorsum of the hand, trouble straightening the arm, and wrist-drop. Ulnar nerve injury typically presents with abnormal sensation to the fourth and fifth digits and decreased grip strength.
Conclusion
Vascular abnormalities are rare complications following elbow injuries. Our patient sustained a lacerated brachial artery, which was repaired via saphenous graft; brachial and basilic vein lacerations, which were ligated; and an avulsion fracture with an unstable joint, which was stabilized with external fixation and stabilization. He was discharged the following day with full neurovascular function.
A methodical approach to assessing patients presenting with elbow injury is essential to making the correct diagnosis. This should include a careful evaluation of the joints above and below the area of injury, as well as attention to the neurovascular examination, with a heightened suspicion for a vascular abnormality in complex injuries. Doppler and ultrasound evaluation with multiple rechecks can assist with the diagnosis. Our patient was rapidly assessed with a concern for a vascular injury, and was emergently referred to vascular surgery for repair of the brachial artery and stabilization of the joint.
1. Carter SJ, Germann CA, Dacus AA, Sweeney TW, Perron AD. Orthopedic pitfalls in the ED: neurovascular injury associated with posterior elbow dislocations. Am J Emerg Med. 2010;28(8):960-965. doi:10.1016/j.ajem.2009.05.024.
2. Miyazaki AN, Fregoneze M, Santos PD, do Val Sella G, Checchia CS, Checchia SL. Brachial artery injury due to closed posterior elbow dislocation: case report. Rev Bras Ortop. 2016;51(2):239-243. doi:10.1016/j.rboe.2016.02.007.
3. Beingessner J, Pollock W, King GJW. Elbow fractures and dislocations. In: Court-Brown CM, Heckman JD, McQueen MM, Ricci WM, Tornetta P, eds. Rockwood and Green’s Fractures in Adults. Vol 1. 8th ed. Philadelphia, PA: Wolters Kluwer Health; 2015:1179-1228.
4. McCabe MP, Savoie FH 3rd. Simple elbow dislocations: evaluation, management, and outcomes. Phys Sportsmed. 2012;40(1):62-71. doi:10.3810/psm.2012.02.1952.
5. Jungbluth P, Hakimi M, Linhart W, Windolf J. Current concepts: simple and complex elbow dislocations—acute and definitive treatment. Eur J Trauma Emerg Surg. 2008;34(2):120-130. doi:10.1007/s00068-008-8033-9.
6. Marcheix B, Chaufour X, Ayel J, et al. Transection of the brachial artery after closed posterior elbow dislocation. J Vasc Surg. 2005;42(6):1230-1232. doi:10.1016/j.jvs.2005.07.046.
A 46-year-old man with a remote history of general tonic-clonic seizures, for which he was taking phenytoin, presented to the ED 30 minutes after sustaining a witnessed mechanical fall. The patient had fallen onto his nondominant left hand, which resulted in an injury to his elbow. He reported neither losing consciousness nor experiencing any seizures following the incident. He denied dislocating the joint or sustaining any other injuries from the fall. He also denied a history of past left elbow injury.
The patient was alert, oriented, and provided a full history of the incident. Regarding medical history, he stated that his last seizure had occurred 10 years prior. Except for the left elbow pain, a review of his systems was negative. The patient appeared in no acute distress, and supported his left upper extremity with a bandana and his right hand.
The patient’s vital signs were normal. The physical examination was negative except for the left elbow, which had significant swelling and limited range of motion without skin break, leading to suspicion for a prehospital dislocation with self-reduction. The joints above, below, and at the injury site were assessed for neurovascular injury.
Computed tomography angiography of the left upper extremity showed a brachial artery occlusion above the elbow, with reconstitution below the joint (Figure 2).
Discussion
There is a paucity of information on vascular injury from elbow dislocation in the emergency medicine literature. A recent literature search referenced orthopedic pitfalls in the ED,1 but most data appear in the orthopedic and vascular literature. A case report from the orthopedic literature in Brazil cites a vascular injury after ED relocation of a dislocated elbow following an assault.2
The elbow is the second most commonly dislocated joint (not including the patella) after the shoulder.3 Posterior dislocations make up the majority of these injuries. Simple versus complex injuries can be differentiated by the presence or absence of fracture.4 Simple complications include stiffness; loss of mobility, especially with full extension; neurovascular injuries; and compartment syndrome. Complex injuries involve fractures and potential neurovascular injuries, stiffness, pain, and loss of mobility.
Soft tissue injuries, fractures, and neurovascular complaints represent the majority of ED encounters, and are commonly related to falls. The elbow is the articulation of the humerus, ulna, and radius bones. Range of motion includes, but is not limited to, flexion, extension, supination, and pronation. Tears in the lateral ulnar ligament, joint capsule, and medial collateral ligament lead to instability of the joint and increase risk of dislocation.
Fractures make up to 20% of injuries to the elbow. These include fractures of the radial head and neck (most common), olecranon, and distal humerus.5 Open elbow fractures are rare, as are vascular injuries (5%-13% of cases).6 When present, vascular elbow injuries usually involve the brachial artery, and display abnormal palpable and Doppler assessment of the brachial and radial arteries.6
Nerve injuries may include injury to the radial nerve. Manifestations of radial nerve injury include abnormal sensation to the dorsum of the hand, trouble straightening the arm, and wrist-drop. Ulnar nerve injury typically presents with abnormal sensation to the fourth and fifth digits and decreased grip strength.
Conclusion
Vascular abnormalities are rare complications following elbow injuries. Our patient sustained a lacerated brachial artery, which was repaired via saphenous graft; brachial and basilic vein lacerations, which were ligated; and an avulsion fracture with an unstable joint, which was stabilized with external fixation and stabilization. He was discharged the following day with full neurovascular function.
A methodical approach to assessing patients presenting with elbow injury is essential to making the correct diagnosis. This should include a careful evaluation of the joints above and below the area of injury, as well as attention to the neurovascular examination, with a heightened suspicion for a vascular abnormality in complex injuries. Doppler and ultrasound evaluation with multiple rechecks can assist with the diagnosis. Our patient was rapidly assessed with a concern for a vascular injury, and was emergently referred to vascular surgery for repair of the brachial artery and stabilization of the joint.
A 46-year-old man with a remote history of general tonic-clonic seizures, for which he was taking phenytoin, presented to the ED 30 minutes after sustaining a witnessed mechanical fall. The patient had fallen onto his nondominant left hand, which resulted in an injury to his elbow. He reported neither losing consciousness nor experiencing any seizures following the incident. He denied dislocating the joint or sustaining any other injuries from the fall. He also denied a history of past left elbow injury.
The patient was alert, oriented, and provided a full history of the incident. Regarding medical history, he stated that his last seizure had occurred 10 years prior. Except for the left elbow pain, a review of his systems was negative. The patient appeared in no acute distress, and supported his left upper extremity with a bandana and his right hand.
The patient’s vital signs were normal. The physical examination was negative except for the left elbow, which had significant swelling and limited range of motion without skin break, leading to suspicion for a prehospital dislocation with self-reduction. The joints above, below, and at the injury site were assessed for neurovascular injury.
Computed tomography angiography of the left upper extremity showed a brachial artery occlusion above the elbow, with reconstitution below the joint (Figure 2).
Discussion
There is a paucity of information on vascular injury from elbow dislocation in the emergency medicine literature. A recent literature search referenced orthopedic pitfalls in the ED,1 but most data appear in the orthopedic and vascular literature. A case report from the orthopedic literature in Brazil cites a vascular injury after ED relocation of a dislocated elbow following an assault.2
The elbow is the second most commonly dislocated joint (not including the patella) after the shoulder.3 Posterior dislocations make up the majority of these injuries. Simple versus complex injuries can be differentiated by the presence or absence of fracture.4 Simple complications include stiffness; loss of mobility, especially with full extension; neurovascular injuries; and compartment syndrome. Complex injuries involve fractures and potential neurovascular injuries, stiffness, pain, and loss of mobility.
Soft tissue injuries, fractures, and neurovascular complaints represent the majority of ED encounters, and are commonly related to falls. The elbow is the articulation of the humerus, ulna, and radius bones. Range of motion includes, but is not limited to, flexion, extension, supination, and pronation. Tears in the lateral ulnar ligament, joint capsule, and medial collateral ligament lead to instability of the joint and increase risk of dislocation.
Fractures make up to 20% of injuries to the elbow. These include fractures of the radial head and neck (most common), olecranon, and distal humerus.5 Open elbow fractures are rare, as are vascular injuries (5%-13% of cases).6 When present, vascular elbow injuries usually involve the brachial artery, and display abnormal palpable and Doppler assessment of the brachial and radial arteries.6
Nerve injuries may include injury to the radial nerve. Manifestations of radial nerve injury include abnormal sensation to the dorsum of the hand, trouble straightening the arm, and wrist-drop. Ulnar nerve injury typically presents with abnormal sensation to the fourth and fifth digits and decreased grip strength.
Conclusion
Vascular abnormalities are rare complications following elbow injuries. Our patient sustained a lacerated brachial artery, which was repaired via saphenous graft; brachial and basilic vein lacerations, which were ligated; and an avulsion fracture with an unstable joint, which was stabilized with external fixation and stabilization. He was discharged the following day with full neurovascular function.
A methodical approach to assessing patients presenting with elbow injury is essential to making the correct diagnosis. This should include a careful evaluation of the joints above and below the area of injury, as well as attention to the neurovascular examination, with a heightened suspicion for a vascular abnormality in complex injuries. Doppler and ultrasound evaluation with multiple rechecks can assist with the diagnosis. Our patient was rapidly assessed with a concern for a vascular injury, and was emergently referred to vascular surgery for repair of the brachial artery and stabilization of the joint.
1. Carter SJ, Germann CA, Dacus AA, Sweeney TW, Perron AD. Orthopedic pitfalls in the ED: neurovascular injury associated with posterior elbow dislocations. Am J Emerg Med. 2010;28(8):960-965. doi:10.1016/j.ajem.2009.05.024.
2. Miyazaki AN, Fregoneze M, Santos PD, do Val Sella G, Checchia CS, Checchia SL. Brachial artery injury due to closed posterior elbow dislocation: case report. Rev Bras Ortop. 2016;51(2):239-243. doi:10.1016/j.rboe.2016.02.007.
3. Beingessner J, Pollock W, King GJW. Elbow fractures and dislocations. In: Court-Brown CM, Heckman JD, McQueen MM, Ricci WM, Tornetta P, eds. Rockwood and Green’s Fractures in Adults. Vol 1. 8th ed. Philadelphia, PA: Wolters Kluwer Health; 2015:1179-1228.
4. McCabe MP, Savoie FH 3rd. Simple elbow dislocations: evaluation, management, and outcomes. Phys Sportsmed. 2012;40(1):62-71. doi:10.3810/psm.2012.02.1952.
5. Jungbluth P, Hakimi M, Linhart W, Windolf J. Current concepts: simple and complex elbow dislocations—acute and definitive treatment. Eur J Trauma Emerg Surg. 2008;34(2):120-130. doi:10.1007/s00068-008-8033-9.
6. Marcheix B, Chaufour X, Ayel J, et al. Transection of the brachial artery after closed posterior elbow dislocation. J Vasc Surg. 2005;42(6):1230-1232. doi:10.1016/j.jvs.2005.07.046.
1. Carter SJ, Germann CA, Dacus AA, Sweeney TW, Perron AD. Orthopedic pitfalls in the ED: neurovascular injury associated with posterior elbow dislocations. Am J Emerg Med. 2010;28(8):960-965. doi:10.1016/j.ajem.2009.05.024.
2. Miyazaki AN, Fregoneze M, Santos PD, do Val Sella G, Checchia CS, Checchia SL. Brachial artery injury due to closed posterior elbow dislocation: case report. Rev Bras Ortop. 2016;51(2):239-243. doi:10.1016/j.rboe.2016.02.007.
3. Beingessner J, Pollock W, King GJW. Elbow fractures and dislocations. In: Court-Brown CM, Heckman JD, McQueen MM, Ricci WM, Tornetta P, eds. Rockwood and Green’s Fractures in Adults. Vol 1. 8th ed. Philadelphia, PA: Wolters Kluwer Health; 2015:1179-1228.
4. McCabe MP, Savoie FH 3rd. Simple elbow dislocations: evaluation, management, and outcomes. Phys Sportsmed. 2012;40(1):62-71. doi:10.3810/psm.2012.02.1952.
5. Jungbluth P, Hakimi M, Linhart W, Windolf J. Current concepts: simple and complex elbow dislocations—acute and definitive treatment. Eur J Trauma Emerg Surg. 2008;34(2):120-130. doi:10.1007/s00068-008-8033-9.
6. Marcheix B, Chaufour X, Ayel J, et al. Transection of the brachial artery after closed posterior elbow dislocation. J Vasc Surg. 2005;42(6):1230-1232. doi:10.1016/j.jvs.2005.07.046.

Elevated levels of AST, ALT, and CPK • no family history of liver disease • Dx?
THE CASE
A 26-year-old healthy male veteran with bipolar disorder and post-traumatic stress disorder was referred for a gastroenterology consultation after a routine laboratory evaluation revealed elevated levels of aspartate aminotransferase (AST), 1040 IU/L (normal range, 10-40 IU/L), and alanine aminotransferase (ALT), 334 IU/L (normal range, 7-56 IU/L). He had been taking divalproex and ziprasidone for the previous 2 years, during which time liver test results had been normal.
The patient reported no symptoms in the course of a detailed history. He had no family history of liver disease, drank alcohol infrequently, and didn’t use tobacco. He hadn’t received any blood transfusions and didn’t have tattoos.
The patient indicated that he had recently returned from military deployment and that a week before his laboratory tests, he’d resumed weight training. To boost his workout, he’d begun taking a nutritional supplement supplied by a friend. Further questioning revealed that the supplement was MuscleMeds’ Code Red, which contains 1,3-dimethylamylamine (DMAA). He denied using any other dietary supplements.
The physical examination was unremarkable and additional lab work was unrevealing. Lab results included normal levels of ceruloplasmin, alpha-1 antitrypsin, ferritin, iron, and transferrin. Viral hepatitis serologies revealed immunity to the hepatitis A and B virus. The patient tested negative for Epstein-Barr virus, cytomegalovirus, herpes simplex virus, human immunodeficiency virus, antinuclear antibody, anti-smooth muscle antibody, and antimitochondrial antibody. A toxicology screen was remarkable for cannabinoids. The remainder of the basic metabolic panel and complete blood count were within normal limits.
THE DIAGNOSIS
The patient’s AST and ALT levels prompted measurement of creatine phosphokinase (CPK), which was elevated at 34,270 IU/L (normal range, 22-198 IU/L). We diagnosed rhabdomyolysis in this patient, which can be associated with elevated levels of AST and ALT. When we contacted the patient about the diagnosis, he reported no muscle aches or pains, or other symptoms.
We instructed the patient to increase his fluid intake and refrain from further use of Code Red. Repeat liver tests one month after the initial consultation revealed significant improvement in AST (29 IU/L) and ALT (68 IU/L), as well as a decline in CPK to 743 IU/L.
DISCUSSION
Much debate has surrounded the safety and use of DMAA, also known as methylhexamine or Geranamine, in dietary supplements such as Code Red. Eli Lilly and Company developed and patented DMAA in the 1940s, then trademarked it under the name Forthane as an inhaled nasal decongestant in 1971.1-3 United States Food and Drug Administration (FDA) approval for Forthane was withdrawn in 1983 at Lilly’s request.4 DMAA was reintroduced as a dietary supplement more than a decade ago after the FDA, in 2004, banned supplements containing ephedrine alkaloids, which have effects similar to DMAA.5
DMAA has been used to increase muscle mass, promote weight loss, and improve physical performance; it’s also been used as a recreational drug.6-8 Several case reports have described poor outcomes in patients who consumed DMAA products. In 2012, the deaths of 2 military personnel who used DMAA prompted the FDA to warn manufacturers of DMAA-containing supplements to stop production, but such supplements remain easily available in the United States.6
DMAA’s validity as a dietary supplement is controversial. The claim that DMAA is naturally present in geraniums hasn’t been verified, leading some to question whether an inaccurate description of DMAA as a natural substance was employed to justify its use as a nutritional supplement.9 No published evidence exists to establish DMAA as a dietary ingredient.10,11
A long list of potential adverse effects
DMAA is an indirect sympathomimetic with vasoconstricting and cardiovascular effects.12 Animal studies have shown effects similar to ephedrine and amphetamines.12-15 Marsh and colleagues reported that a single oral dose of 3 mg/kg in a human (210 mg/70 kg) moderately increases heart rate and blood pressure and can lead to confusion and concentration problems.16
Oral intake of DMAA affects the lungs at doses above 4 to 15 mg, the heart after 50 to 75 mg, and blood pressure after 100 mg.17 Because of the drug’s long half-life—24 hours based on urinary excretion rates—Venhuis and Kaste reported that there is a risk from repeated doses within 24 to 36 hours that can lead to steadily stronger pharmacologic effects.17
The use of DMAA has been cited in 5 cases of hemorrhagic stroke, a case of acute heart failure, and the deaths of 2 military personnel who experienced asystole during aerobic exercise.7,8,18-20 These individuals ranged in age from 22 to 41 years.
Initial symptoms included severe headaches, palpitations, dizziness, twitching of extremities, nausea, vomiting, confusion, agitation, and chest pain. The 2 military personnel suffered leg cramps and dyspnea followed by loss of consciousness. Several individuals were hypertensive on presentation to the emergency department with blood pressures as high as 240/120 mm Hg.
THE TAKEAWAY
Our patient presented with transaminitis and was found to have rhabdomyolysis after using DMAA. A few case reports have associated rhabdomyolysis with elevated liver function tests.21,22 We suspect that DMAA use, which has been linked to adverse effects such as hypertension, tachycardia, and muscle aches, may also cause leakage of muscle enzymes and the development of rhabdomyolysis.
Although a single instance can’t prove causation, this case may illustrate additional adverse effects of DMAA beyond the already long list of risks, including hypertension, seizures, cerebral hemorrhage, arrhythmias, myocardial infarction, cardiomyopathy, and death.7,8,18-20,23 It’s important for physicians to recognize that their patients may be using dietary supplements to increase strength, energy, or weight loss and to be aware of the potential adverse effects.
1. Shonle HA, Rohrmann E, inventors; Eli Lilly and Company, assignee. Aminoalkanes. Patent US2350318A. May 30, 1944.
2. Shonle HA, Rohrmann E, inventors; Eli Lilly and Company, assignee. Carbonates of 1-R-1 aminoethanes. Patent US2386273. October 9, 1945.
3. Eli Lilly and Company. Forthane. Registration 0925396, February 1, 1971. United States Patent and Trademark Office.
4. Federal Register. Vol. 48, No. 218/Notices. November 9, 1983.
5. Shipley A. Chemist’s new product contains hidden substance. Washington Post. May 8, 2006:Sports. Available at: http://www.washingtonpost.com/wp-dyn/content/article/2006/05/07/AR2006050700913.html. Accessed June 5, 2017.
6. Gregory PJ. Availability of DMAA supplements despite US Food and Drug Administration action. JAMA Intern Med. 2013;173:164-165.
7. Gee P, Jackson S, Easton J. Another bitter pill: a case of toxicity from DMAA party pills. N Z Med J. 2010;123:124-127.
8. Gee P, Tallon C, Long N, et al. Use of recreational drug 1,3 Dimethylamylamine (DMAA) [corrected] associated with cerebral hemorrhage. Ann Emerg Med. 2012;60:431-434.
9. Ping Z, Jun Q, Qing L. A study on the chemical constituents of geranium oil. Journal of Guizhou Institute of Technology. 1996;25:82-85.
10. Lisi A, Hasick N, Kazlauskas R, et al. Studies of methylhexaneamine in supplements and geranium oil. Drug Test Anal. 2011;3:873-876.
11. Elsohly MA, Gul W, Elsohly KM, et al. Pelargonium oil and methyl hexaneamine (MHA): analytical approaches supporting the absence of MHA in authenticated Pelargonium graveolens plant material and oil. J Anal Toxicol. 2012;36:457-471.
12. Charlier R. [Pharmacology of 2-amino-4-methylhexane]. Arch Int Pharmacodyn Ther. 1950;83:573-584.
13. Ahlquist R. A contribution to the pharmacology of the aliphatic amines. J Pharmacol Exp Ther. 1944;81:235-239.
14. Swanson EE, Chen KK. Comparison of pressor action of aliphatic amines. J Pharmacol Exp Ther. 1946;88:10-13.
15. Swanson EE, Chen KK. Comparison of pressor action of alicyclic derivatives of aliphatic amines. J Pharmacol Exp Ther. 1948;93:423-429.
16. Marsh DF, Howard A, Herring DA. The comparative pharmacology of the isomeric nitrogen methyl substituted heptylamines. J Pharmacol Exp Ther. 1951;103:325-329.
17. Venhuis BJ, Kaste D. Scientific opinion on the regulatory status of 1,3-dimethylamylamine (DMAA). European Journal of Food Research and Review. 2012;2:93-100.
18. Eliason MJ, Eichner A, Cancio A, et al. Case reports: Death of active duty soldiers following ingestion of dietary supplements containing 1,3-dimethylamylamine (DMAA). Mil Med. 2012;177:1455-1459.
19. Young C, Oladipo O, Frasier S, et al. Hemorrhagic stroke in young healthy male following use of sports supplement Jack3d. Mil Med. 2012;177:1450-1454.
20. Salinger L, Daniels B, Sangalli B, et al. Recreational use of a bodybuilding supplement resulting in severe cardiotoxicity. Clin Toxicol (Philadelphia). 2011;49:573-574.
21. Lee GY, Lee H, Kim YJ. Rhabdomyolysis recognized after elevation of liver enzymes following prolonged urologic surgery with lateral decubitus position: a case report. Korean J Anesthesiol. 2011;61:341-343.
22. Karcher C, Dieterich HJ, Schroeder TH. Rhabdomyolysis in an obese patient after total knee arthroplasty. Br J Anaesth. 2006;97:822-824.
23. Karnatovskaia LV, Leoni JC, Freeman ML. Cardiac arrest in a 21-year-old man after ingestion of 1,3-DMAA-containing workout supplement. Clin J Sport Med. 2015;25:e23-e25.
THE CASE
A 26-year-old healthy male veteran with bipolar disorder and post-traumatic stress disorder was referred for a gastroenterology consultation after a routine laboratory evaluation revealed elevated levels of aspartate aminotransferase (AST), 1040 IU/L (normal range, 10-40 IU/L), and alanine aminotransferase (ALT), 334 IU/L (normal range, 7-56 IU/L). He had been taking divalproex and ziprasidone for the previous 2 years, during which time liver test results had been normal.
The patient reported no symptoms in the course of a detailed history. He had no family history of liver disease, drank alcohol infrequently, and didn’t use tobacco. He hadn’t received any blood transfusions and didn’t have tattoos.
The patient indicated that he had recently returned from military deployment and that a week before his laboratory tests, he’d resumed weight training. To boost his workout, he’d begun taking a nutritional supplement supplied by a friend. Further questioning revealed that the supplement was MuscleMeds’ Code Red, which contains 1,3-dimethylamylamine (DMAA). He denied using any other dietary supplements.
The physical examination was unremarkable and additional lab work was unrevealing. Lab results included normal levels of ceruloplasmin, alpha-1 antitrypsin, ferritin, iron, and transferrin. Viral hepatitis serologies revealed immunity to the hepatitis A and B virus. The patient tested negative for Epstein-Barr virus, cytomegalovirus, herpes simplex virus, human immunodeficiency virus, antinuclear antibody, anti-smooth muscle antibody, and antimitochondrial antibody. A toxicology screen was remarkable for cannabinoids. The remainder of the basic metabolic panel and complete blood count were within normal limits.
THE DIAGNOSIS
The patient’s AST and ALT levels prompted measurement of creatine phosphokinase (CPK), which was elevated at 34,270 IU/L (normal range, 22-198 IU/L). We diagnosed rhabdomyolysis in this patient, which can be associated with elevated levels of AST and ALT. When we contacted the patient about the diagnosis, he reported no muscle aches or pains, or other symptoms.
We instructed the patient to increase his fluid intake and refrain from further use of Code Red. Repeat liver tests one month after the initial consultation revealed significant improvement in AST (29 IU/L) and ALT (68 IU/L), as well as a decline in CPK to 743 IU/L.
DISCUSSION
Much debate has surrounded the safety and use of DMAA, also known as methylhexamine or Geranamine, in dietary supplements such as Code Red. Eli Lilly and Company developed and patented DMAA in the 1940s, then trademarked it under the name Forthane as an inhaled nasal decongestant in 1971.1-3 United States Food and Drug Administration (FDA) approval for Forthane was withdrawn in 1983 at Lilly’s request.4 DMAA was reintroduced as a dietary supplement more than a decade ago after the FDA, in 2004, banned supplements containing ephedrine alkaloids, which have effects similar to DMAA.5
DMAA has been used to increase muscle mass, promote weight loss, and improve physical performance; it’s also been used as a recreational drug.6-8 Several case reports have described poor outcomes in patients who consumed DMAA products. In 2012, the deaths of 2 military personnel who used DMAA prompted the FDA to warn manufacturers of DMAA-containing supplements to stop production, but such supplements remain easily available in the United States.6
DMAA’s validity as a dietary supplement is controversial. The claim that DMAA is naturally present in geraniums hasn’t been verified, leading some to question whether an inaccurate description of DMAA as a natural substance was employed to justify its use as a nutritional supplement.9 No published evidence exists to establish DMAA as a dietary ingredient.10,11
A long list of potential adverse effects
DMAA is an indirect sympathomimetic with vasoconstricting and cardiovascular effects.12 Animal studies have shown effects similar to ephedrine and amphetamines.12-15 Marsh and colleagues reported that a single oral dose of 3 mg/kg in a human (210 mg/70 kg) moderately increases heart rate and blood pressure and can lead to confusion and concentration problems.16
Oral intake of DMAA affects the lungs at doses above 4 to 15 mg, the heart after 50 to 75 mg, and blood pressure after 100 mg.17 Because of the drug’s long half-life—24 hours based on urinary excretion rates—Venhuis and Kaste reported that there is a risk from repeated doses within 24 to 36 hours that can lead to steadily stronger pharmacologic effects.17
The use of DMAA has been cited in 5 cases of hemorrhagic stroke, a case of acute heart failure, and the deaths of 2 military personnel who experienced asystole during aerobic exercise.7,8,18-20 These individuals ranged in age from 22 to 41 years.
Initial symptoms included severe headaches, palpitations, dizziness, twitching of extremities, nausea, vomiting, confusion, agitation, and chest pain. The 2 military personnel suffered leg cramps and dyspnea followed by loss of consciousness. Several individuals were hypertensive on presentation to the emergency department with blood pressures as high as 240/120 mm Hg.
THE TAKEAWAY
Our patient presented with transaminitis and was found to have rhabdomyolysis after using DMAA. A few case reports have associated rhabdomyolysis with elevated liver function tests.21,22 We suspect that DMAA use, which has been linked to adverse effects such as hypertension, tachycardia, and muscle aches, may also cause leakage of muscle enzymes and the development of rhabdomyolysis.
Although a single instance can’t prove causation, this case may illustrate additional adverse effects of DMAA beyond the already long list of risks, including hypertension, seizures, cerebral hemorrhage, arrhythmias, myocardial infarction, cardiomyopathy, and death.7,8,18-20,23 It’s important for physicians to recognize that their patients may be using dietary supplements to increase strength, energy, or weight loss and to be aware of the potential adverse effects.
THE CASE
A 26-year-old healthy male veteran with bipolar disorder and post-traumatic stress disorder was referred for a gastroenterology consultation after a routine laboratory evaluation revealed elevated levels of aspartate aminotransferase (AST), 1040 IU/L (normal range, 10-40 IU/L), and alanine aminotransferase (ALT), 334 IU/L (normal range, 7-56 IU/L). He had been taking divalproex and ziprasidone for the previous 2 years, during which time liver test results had been normal.
The patient reported no symptoms in the course of a detailed history. He had no family history of liver disease, drank alcohol infrequently, and didn’t use tobacco. He hadn’t received any blood transfusions and didn’t have tattoos.
The patient indicated that he had recently returned from military deployment and that a week before his laboratory tests, he’d resumed weight training. To boost his workout, he’d begun taking a nutritional supplement supplied by a friend. Further questioning revealed that the supplement was MuscleMeds’ Code Red, which contains 1,3-dimethylamylamine (DMAA). He denied using any other dietary supplements.
The physical examination was unremarkable and additional lab work was unrevealing. Lab results included normal levels of ceruloplasmin, alpha-1 antitrypsin, ferritin, iron, and transferrin. Viral hepatitis serologies revealed immunity to the hepatitis A and B virus. The patient tested negative for Epstein-Barr virus, cytomegalovirus, herpes simplex virus, human immunodeficiency virus, antinuclear antibody, anti-smooth muscle antibody, and antimitochondrial antibody. A toxicology screen was remarkable for cannabinoids. The remainder of the basic metabolic panel and complete blood count were within normal limits.
THE DIAGNOSIS
The patient’s AST and ALT levels prompted measurement of creatine phosphokinase (CPK), which was elevated at 34,270 IU/L (normal range, 22-198 IU/L). We diagnosed rhabdomyolysis in this patient, which can be associated with elevated levels of AST and ALT. When we contacted the patient about the diagnosis, he reported no muscle aches or pains, or other symptoms.
We instructed the patient to increase his fluid intake and refrain from further use of Code Red. Repeat liver tests one month after the initial consultation revealed significant improvement in AST (29 IU/L) and ALT (68 IU/L), as well as a decline in CPK to 743 IU/L.
DISCUSSION
Much debate has surrounded the safety and use of DMAA, also known as methylhexamine or Geranamine, in dietary supplements such as Code Red. Eli Lilly and Company developed and patented DMAA in the 1940s, then trademarked it under the name Forthane as an inhaled nasal decongestant in 1971.1-3 United States Food and Drug Administration (FDA) approval for Forthane was withdrawn in 1983 at Lilly’s request.4 DMAA was reintroduced as a dietary supplement more than a decade ago after the FDA, in 2004, banned supplements containing ephedrine alkaloids, which have effects similar to DMAA.5
DMAA has been used to increase muscle mass, promote weight loss, and improve physical performance; it’s also been used as a recreational drug.6-8 Several case reports have described poor outcomes in patients who consumed DMAA products. In 2012, the deaths of 2 military personnel who used DMAA prompted the FDA to warn manufacturers of DMAA-containing supplements to stop production, but such supplements remain easily available in the United States.6
DMAA’s validity as a dietary supplement is controversial. The claim that DMAA is naturally present in geraniums hasn’t been verified, leading some to question whether an inaccurate description of DMAA as a natural substance was employed to justify its use as a nutritional supplement.9 No published evidence exists to establish DMAA as a dietary ingredient.10,11
A long list of potential adverse effects
DMAA is an indirect sympathomimetic with vasoconstricting and cardiovascular effects.12 Animal studies have shown effects similar to ephedrine and amphetamines.12-15 Marsh and colleagues reported that a single oral dose of 3 mg/kg in a human (210 mg/70 kg) moderately increases heart rate and blood pressure and can lead to confusion and concentration problems.16
Oral intake of DMAA affects the lungs at doses above 4 to 15 mg, the heart after 50 to 75 mg, and blood pressure after 100 mg.17 Because of the drug’s long half-life—24 hours based on urinary excretion rates—Venhuis and Kaste reported that there is a risk from repeated doses within 24 to 36 hours that can lead to steadily stronger pharmacologic effects.17
The use of DMAA has been cited in 5 cases of hemorrhagic stroke, a case of acute heart failure, and the deaths of 2 military personnel who experienced asystole during aerobic exercise.7,8,18-20 These individuals ranged in age from 22 to 41 years.
Initial symptoms included severe headaches, palpitations, dizziness, twitching of extremities, nausea, vomiting, confusion, agitation, and chest pain. The 2 military personnel suffered leg cramps and dyspnea followed by loss of consciousness. Several individuals were hypertensive on presentation to the emergency department with blood pressures as high as 240/120 mm Hg.
THE TAKEAWAY
Our patient presented with transaminitis and was found to have rhabdomyolysis after using DMAA. A few case reports have associated rhabdomyolysis with elevated liver function tests.21,22 We suspect that DMAA use, which has been linked to adverse effects such as hypertension, tachycardia, and muscle aches, may also cause leakage of muscle enzymes and the development of rhabdomyolysis.
Although a single instance can’t prove causation, this case may illustrate additional adverse effects of DMAA beyond the already long list of risks, including hypertension, seizures, cerebral hemorrhage, arrhythmias, myocardial infarction, cardiomyopathy, and death.7,8,18-20,23 It’s important for physicians to recognize that their patients may be using dietary supplements to increase strength, energy, or weight loss and to be aware of the potential adverse effects.
1. Shonle HA, Rohrmann E, inventors; Eli Lilly and Company, assignee. Aminoalkanes. Patent US2350318A. May 30, 1944.
2. Shonle HA, Rohrmann E, inventors; Eli Lilly and Company, assignee. Carbonates of 1-R-1 aminoethanes. Patent US2386273. October 9, 1945.
3. Eli Lilly and Company. Forthane. Registration 0925396, February 1, 1971. United States Patent and Trademark Office.
4. Federal Register. Vol. 48, No. 218/Notices. November 9, 1983.
5. Shipley A. Chemist’s new product contains hidden substance. Washington Post. May 8, 2006:Sports. Available at: http://www.washingtonpost.com/wp-dyn/content/article/2006/05/07/AR2006050700913.html. Accessed June 5, 2017.
6. Gregory PJ. Availability of DMAA supplements despite US Food and Drug Administration action. JAMA Intern Med. 2013;173:164-165.
7. Gee P, Jackson S, Easton J. Another bitter pill: a case of toxicity from DMAA party pills. N Z Med J. 2010;123:124-127.
8. Gee P, Tallon C, Long N, et al. Use of recreational drug 1,3 Dimethylamylamine (DMAA) [corrected] associated with cerebral hemorrhage. Ann Emerg Med. 2012;60:431-434.
9. Ping Z, Jun Q, Qing L. A study on the chemical constituents of geranium oil. Journal of Guizhou Institute of Technology. 1996;25:82-85.
10. Lisi A, Hasick N, Kazlauskas R, et al. Studies of methylhexaneamine in supplements and geranium oil. Drug Test Anal. 2011;3:873-876.
11. Elsohly MA, Gul W, Elsohly KM, et al. Pelargonium oil and methyl hexaneamine (MHA): analytical approaches supporting the absence of MHA in authenticated Pelargonium graveolens plant material and oil. J Anal Toxicol. 2012;36:457-471.
12. Charlier R. [Pharmacology of 2-amino-4-methylhexane]. Arch Int Pharmacodyn Ther. 1950;83:573-584.
13. Ahlquist R. A contribution to the pharmacology of the aliphatic amines. J Pharmacol Exp Ther. 1944;81:235-239.
14. Swanson EE, Chen KK. Comparison of pressor action of aliphatic amines. J Pharmacol Exp Ther. 1946;88:10-13.
15. Swanson EE, Chen KK. Comparison of pressor action of alicyclic derivatives of aliphatic amines. J Pharmacol Exp Ther. 1948;93:423-429.
16. Marsh DF, Howard A, Herring DA. The comparative pharmacology of the isomeric nitrogen methyl substituted heptylamines. J Pharmacol Exp Ther. 1951;103:325-329.
17. Venhuis BJ, Kaste D. Scientific opinion on the regulatory status of 1,3-dimethylamylamine (DMAA). European Journal of Food Research and Review. 2012;2:93-100.
18. Eliason MJ, Eichner A, Cancio A, et al. Case reports: Death of active duty soldiers following ingestion of dietary supplements containing 1,3-dimethylamylamine (DMAA). Mil Med. 2012;177:1455-1459.
19. Young C, Oladipo O, Frasier S, et al. Hemorrhagic stroke in young healthy male following use of sports supplement Jack3d. Mil Med. 2012;177:1450-1454.
20. Salinger L, Daniels B, Sangalli B, et al. Recreational use of a bodybuilding supplement resulting in severe cardiotoxicity. Clin Toxicol (Philadelphia). 2011;49:573-574.
21. Lee GY, Lee H, Kim YJ. Rhabdomyolysis recognized after elevation of liver enzymes following prolonged urologic surgery with lateral decubitus position: a case report. Korean J Anesthesiol. 2011;61:341-343.
22. Karcher C, Dieterich HJ, Schroeder TH. Rhabdomyolysis in an obese patient after total knee arthroplasty. Br J Anaesth. 2006;97:822-824.
23. Karnatovskaia LV, Leoni JC, Freeman ML. Cardiac arrest in a 21-year-old man after ingestion of 1,3-DMAA-containing workout supplement. Clin J Sport Med. 2015;25:e23-e25.
1. Shonle HA, Rohrmann E, inventors; Eli Lilly and Company, assignee. Aminoalkanes. Patent US2350318A. May 30, 1944.
2. Shonle HA, Rohrmann E, inventors; Eli Lilly and Company, assignee. Carbonates of 1-R-1 aminoethanes. Patent US2386273. October 9, 1945.
3. Eli Lilly and Company. Forthane. Registration 0925396, February 1, 1971. United States Patent and Trademark Office.
4. Federal Register. Vol. 48, No. 218/Notices. November 9, 1983.
5. Shipley A. Chemist’s new product contains hidden substance. Washington Post. May 8, 2006:Sports. Available at: http://www.washingtonpost.com/wp-dyn/content/article/2006/05/07/AR2006050700913.html. Accessed June 5, 2017.
6. Gregory PJ. Availability of DMAA supplements despite US Food and Drug Administration action. JAMA Intern Med. 2013;173:164-165.
7. Gee P, Jackson S, Easton J. Another bitter pill: a case of toxicity from DMAA party pills. N Z Med J. 2010;123:124-127.
8. Gee P, Tallon C, Long N, et al. Use of recreational drug 1,3 Dimethylamylamine (DMAA) [corrected] associated with cerebral hemorrhage. Ann Emerg Med. 2012;60:431-434.
9. Ping Z, Jun Q, Qing L. A study on the chemical constituents of geranium oil. Journal of Guizhou Institute of Technology. 1996;25:82-85.
10. Lisi A, Hasick N, Kazlauskas R, et al. Studies of methylhexaneamine in supplements and geranium oil. Drug Test Anal. 2011;3:873-876.
11. Elsohly MA, Gul W, Elsohly KM, et al. Pelargonium oil and methyl hexaneamine (MHA): analytical approaches supporting the absence of MHA in authenticated Pelargonium graveolens plant material and oil. J Anal Toxicol. 2012;36:457-471.
12. Charlier R. [Pharmacology of 2-amino-4-methylhexane]. Arch Int Pharmacodyn Ther. 1950;83:573-584.
13. Ahlquist R. A contribution to the pharmacology of the aliphatic amines. J Pharmacol Exp Ther. 1944;81:235-239.
14. Swanson EE, Chen KK. Comparison of pressor action of aliphatic amines. J Pharmacol Exp Ther. 1946;88:10-13.
15. Swanson EE, Chen KK. Comparison of pressor action of alicyclic derivatives of aliphatic amines. J Pharmacol Exp Ther. 1948;93:423-429.
16. Marsh DF, Howard A, Herring DA. The comparative pharmacology of the isomeric nitrogen methyl substituted heptylamines. J Pharmacol Exp Ther. 1951;103:325-329.
17. Venhuis BJ, Kaste D. Scientific opinion on the regulatory status of 1,3-dimethylamylamine (DMAA). European Journal of Food Research and Review. 2012;2:93-100.
18. Eliason MJ, Eichner A, Cancio A, et al. Case reports: Death of active duty soldiers following ingestion of dietary supplements containing 1,3-dimethylamylamine (DMAA). Mil Med. 2012;177:1455-1459.
19. Young C, Oladipo O, Frasier S, et al. Hemorrhagic stroke in young healthy male following use of sports supplement Jack3d. Mil Med. 2012;177:1450-1454.
20. Salinger L, Daniels B, Sangalli B, et al. Recreational use of a bodybuilding supplement resulting in severe cardiotoxicity. Clin Toxicol (Philadelphia). 2011;49:573-574.
21. Lee GY, Lee H, Kim YJ. Rhabdomyolysis recognized after elevation of liver enzymes following prolonged urologic surgery with lateral decubitus position: a case report. Korean J Anesthesiol. 2011;61:341-343.
22. Karcher C, Dieterich HJ, Schroeder TH. Rhabdomyolysis in an obese patient after total knee arthroplasty. Br J Anaesth. 2006;97:822-824.
23. Karnatovskaia LV, Leoni JC, Freeman ML. Cardiac arrest in a 21-year-old man after ingestion of 1,3-DMAA-containing workout supplement. Clin J Sport Med. 2015;25:e23-e25.
