User login
Increased syncopal episodes post surgery • Dx?
THE CASE
A 58-year-old woman sought care at our clinic for recurrent syncopal and near-syncopal events following surgical repair of a left hip fracture. The first syncopal event occurred one day post-surgery shortly after standing and was attributed to orthostatic hypotension. Subsequently, the patient experienced 2 events during her hospital stay. Both events occurred in the upright position and were preceded by lightheadedness, warmth, and diaphoresis. They were short in duration (<30 seconds) with spontaneous and complete recovery. The patient had no associated chest pain or palpitations.
The patient’s past medical history included osteopenia, dyslipidemia, and vasovagal syncope, averaging one to 2 events per year. Given her past history, the physicians caring for her assumed that she was having recurrences of her vasovagal syncope. She was discharged home on fludrocortisone 0.1 mg/d, sodium chloride 1 g tid, enoxaparin 40 mg/d, and acetaminophen and oxycodone as needed for pain.
One week later, the patient experienced another syncopal event at home, prompting her to visit our clinic for further evaluation. On arrival, her vital signs were stable. Her oxygen saturation level was 98%, she was not orthostatic, and her physical exam and blood studies were unremarkable. An echocardiogram showed preserved left ventricular function with no evidence of right ventricular dilatation or strain.
THE DIAGNOSIS
The patient’s revised Geneva Score for pulmonary embolism (PE) was 2 to 5 depending on the heart rate used (66-80 beats per minute), putting her in a low-to-intermediate risk group with an estimated PE prevalence between 8% and 28%.1 Given her recent surgery and the increase in the frequency of her vasovagal events, a computed tomography pulmonary angiogram (CT-PA) was performed. The CT-PA showed a PE in the lateral and posterior basal subsegmental branches of the right lower lobe. Doppler ultrasound revealed no evidence of acute deep vein thrombosis.
DISCUSSION
Syncope may develop in 9% to 19% of patients with PE.2-6 While syncope in patients with PE is often attributed to reduced cardiac filling secondary to massive emboli, it is important to recognize that patients can also present with vasovagal syncope in the absence of massive emboli.
One mechanism for the development of syncope is right ventricular failure with subsequent impairment of left ventricular filling, leading to arterial hypotension. Indeed, the majority of patients with PE and syncope have a massive embolism defined as greater than a 50% reduction in the pulmonary circulation.7 In one study, 60% of patients with PE who presented with syncope had a massive PE compared to 39% of patients presenting without syncope (P=.036).8
Another reported mechanism for syncope in a patient with PE is transient high-degree atrioventricular (AV) block.9 Sudden increases in right-sided pressure can lead to transient right bundle branch block, which may result in complete heart block in the setting of baseline left bundle branch block.
Lastly, patients with PE may develop a vasovagal-like reaction, such as the Bezold-Jarisch reflex, which results in transient arterial hypotension and cerebral hypoperfusion.10 In such instances, the postulated mechanism is activation of cardiac vagal afferents, which results in an increase in vagal tone and peripheral sympathetic withdrawal leading to hypotension and syncope. It is important to note that this mechanism can occur in the absence of massive PE. In one study, up to 40% of patients with PE and syncope did not have a massive PE, and almost 6% had thrombi only in small branches of the pulmonary artery.8
This patient had isolated subsegmental defects, identified on the CT-PA. The sensitivity of CT-PA to detect subsegmental PE ranges from 53% to 100%.11 While this test has its limitations, the introduction of the multi-detector CT technique has significantly increased the rate of detection with a specificity of 96%.12,13
Was PE the cause of the syncope, or just an incidental finding?
In this case, we believe the CT-PA findings were diagnostic for PE. What is less clear is whether the PE was the cause of the syncope.
Asymptomatic post-operative PE with isolated subsegmental defects has been reported.14-16 When compared to patients with a defect at a segmental or more proximal level, these patients often have less dyspnea, are less likely to be classified as having a high clinical probability of PE, and have a lower prevalence of proximal deep vein thrombosis (3.3% vs 43.8%; P<.0001).17 Therefore, one could argue that the PE finding in our case was incidental. While this is a possibility, we believe the patient’s syncope was due to PE for the following reasons.
First, several investigators have reported transient increases in vagal tone and syncope following PE consistent with a vasovagal-like response.7,18 Therefore, it is possible that the reduction in preload associated with PE triggered a Bezold-Jarisch-like reflex leading to syncope. The patient’s history of vasovagal syncope was certainly indicative of increased susceptibility to reflex-mediated events, thus supporting our hypothesis.
Second, our patient had a cluster of events following surgery compared to the one to 2 events she experienced per year prior to surgery. The increased incidence of events would be an unusual progression of her syncope in the absence of clear triggers, again rendering our hypothesis more plausible.
The patient was admitted to our hospital and started on a higher dose of enoxaparin (60 mg twice daily). She was subsequently discharged home on rivaroxaban 15 mg twice daily and midodrine 2.5 mg twice daily in addition to the medications she was already taking. At her 6-week follow-up visit, she reported no recurrences.
THE TAKEAWAY
This case demonstrates that non-massive PE can present as vasovagal syncope. Recognizing that PE could lead to reflex-mediated syncope in the absence of massive emboli, it is important to rule it out in the evaluation of patients with vasovagal syncope when risk factors for PE are present.
1. Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006;144:165-171.
2. Calvo-Romero JM, Pérez-Miranda M, Bureo-Dacal P. Syncope in acute pulmonary embolism. Eur J Emerg Med. 2004;11:208-209.
3. Castelli R, Tarsia P, Tantardini C, et al. Syncope in patients with pulmonary embolism: comparison between patients with syncope as the presenting symptom of pulmonary embolism and patients with pulmonary embolism without syncope. Vasc Med. 2003;8:257-261.
4. Kasper W, Konstantinides S, Geibel A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol. 1997;30:1165-1171.
5. Koutkia P, Wachtel TJ. Pulmonary embolism presenting as syncope: case report and review of the literature. Heart Lung. 1999;28:342-347.
6. Torbicki A, Perrier A, Konstantinides S, et al; ESC Committee for Practice Guidelines (CPG). Guidelines on the diagnosis and management of acute pulmonary embolism: the Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2008;29:2276-2315.
7. Thames MD, Alpert JS, Dalen JE. Syncope in patients with pulmonary embolism. JAMA. 1977;238:2509-2511.
8. Duplyakov D, Kurakina E, Pavlova T, et al. Value of syncope in patients with high-to-intermediate risk pulmonary artery embolism. Eur Heart J Acute Cardiovasc Care. 2015;4:353-358.
9. Wilner C, Garnier-Crussard JP, Huygue De Mahenge A, et al. [Paroxysmal atrioventricular block, cause of syncope in pulmonary embolism. 2 cases]. Presse Med. 1983;12:2987-2989.
10. Frink RJ, James TN. Intracardiac route of the Bezold-Jarisch reflex. Am J Physiol. 1971;221:1464-1469.
11. Rathbun SW, Raskob GE, Whitsett TL. Sensitivity and specificity of helical computed tomography in the diagnosis of pulmonary embolism: A systematic review. Ann Intern Med. 2000;132:227-232.
12. Stein PD, Fowler SE, Goodman LR, et al; PIOPED II Investigators. Multidetector computed tomography for acute pulmonary embolism. N Engl J Med. 2006;354:2317-2327.
13. Vedovati MC, Becattini C, Agnelli G, et al. Multidetector CT scan for acute pulmonary embolism: embolic burden and clinical outcome. Chest. 2012;142:1417-1424.
14. Musset D, Parent F, Meyer G, et al; Evaluation du Scanner Spiralé dans l’Embolie Pulmonaire study group. Diagnostic strategy for patients with suspected pulmonary embolism: a prospective multicentre outcome study. Lancet. 2002;360:1914-1920.
15. Simpson RJ Jr, Podolak R, Mangano CA Jr, et al. Vagal syncope during recurrent pulmonary embolism. JAMA. 1983;249:390-393.
16. Perrier A, Roy PM, Sanchez O, et al. Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med. 2005;352:1760-1768.
17. Le Gal G, Righini M, Parent F, et al. Diagnosis and management of subsegmental pulmonary embolism. J Thromb Haemost. 2006;4:724-731.
18. Eldadah ZA, Najjar SS, Ziegelstein RC. A patient with syncope, only “vagally” related to the heart. Chest. 2000;117:1801-1803.
THE CASE
A 58-year-old woman sought care at our clinic for recurrent syncopal and near-syncopal events following surgical repair of a left hip fracture. The first syncopal event occurred one day post-surgery shortly after standing and was attributed to orthostatic hypotension. Subsequently, the patient experienced 2 events during her hospital stay. Both events occurred in the upright position and were preceded by lightheadedness, warmth, and diaphoresis. They were short in duration (<30 seconds) with spontaneous and complete recovery. The patient had no associated chest pain or palpitations.
The patient’s past medical history included osteopenia, dyslipidemia, and vasovagal syncope, averaging one to 2 events per year. Given her past history, the physicians caring for her assumed that she was having recurrences of her vasovagal syncope. She was discharged home on fludrocortisone 0.1 mg/d, sodium chloride 1 g tid, enoxaparin 40 mg/d, and acetaminophen and oxycodone as needed for pain.
One week later, the patient experienced another syncopal event at home, prompting her to visit our clinic for further evaluation. On arrival, her vital signs were stable. Her oxygen saturation level was 98%, she was not orthostatic, and her physical exam and blood studies were unremarkable. An echocardiogram showed preserved left ventricular function with no evidence of right ventricular dilatation or strain.
THE DIAGNOSIS
The patient’s revised Geneva Score for pulmonary embolism (PE) was 2 to 5 depending on the heart rate used (66-80 beats per minute), putting her in a low-to-intermediate risk group with an estimated PE prevalence between 8% and 28%.1 Given her recent surgery and the increase in the frequency of her vasovagal events, a computed tomography pulmonary angiogram (CT-PA) was performed. The CT-PA showed a PE in the lateral and posterior basal subsegmental branches of the right lower lobe. Doppler ultrasound revealed no evidence of acute deep vein thrombosis.
DISCUSSION
Syncope may develop in 9% to 19% of patients with PE.2-6 While syncope in patients with PE is often attributed to reduced cardiac filling secondary to massive emboli, it is important to recognize that patients can also present with vasovagal syncope in the absence of massive emboli.
One mechanism for the development of syncope is right ventricular failure with subsequent impairment of left ventricular filling, leading to arterial hypotension. Indeed, the majority of patients with PE and syncope have a massive embolism defined as greater than a 50% reduction in the pulmonary circulation.7 In one study, 60% of patients with PE who presented with syncope had a massive PE compared to 39% of patients presenting without syncope (P=.036).8
Another reported mechanism for syncope in a patient with PE is transient high-degree atrioventricular (AV) block.9 Sudden increases in right-sided pressure can lead to transient right bundle branch block, which may result in complete heart block in the setting of baseline left bundle branch block.
Lastly, patients with PE may develop a vasovagal-like reaction, such as the Bezold-Jarisch reflex, which results in transient arterial hypotension and cerebral hypoperfusion.10 In such instances, the postulated mechanism is activation of cardiac vagal afferents, which results in an increase in vagal tone and peripheral sympathetic withdrawal leading to hypotension and syncope. It is important to note that this mechanism can occur in the absence of massive PE. In one study, up to 40% of patients with PE and syncope did not have a massive PE, and almost 6% had thrombi only in small branches of the pulmonary artery.8
This patient had isolated subsegmental defects, identified on the CT-PA. The sensitivity of CT-PA to detect subsegmental PE ranges from 53% to 100%.11 While this test has its limitations, the introduction of the multi-detector CT technique has significantly increased the rate of detection with a specificity of 96%.12,13
Was PE the cause of the syncope, or just an incidental finding?
In this case, we believe the CT-PA findings were diagnostic for PE. What is less clear is whether the PE was the cause of the syncope.
Asymptomatic post-operative PE with isolated subsegmental defects has been reported.14-16 When compared to patients with a defect at a segmental or more proximal level, these patients often have less dyspnea, are less likely to be classified as having a high clinical probability of PE, and have a lower prevalence of proximal deep vein thrombosis (3.3% vs 43.8%; P<.0001).17 Therefore, one could argue that the PE finding in our case was incidental. While this is a possibility, we believe the patient’s syncope was due to PE for the following reasons.
First, several investigators have reported transient increases in vagal tone and syncope following PE consistent with a vasovagal-like response.7,18 Therefore, it is possible that the reduction in preload associated with PE triggered a Bezold-Jarisch-like reflex leading to syncope. The patient’s history of vasovagal syncope was certainly indicative of increased susceptibility to reflex-mediated events, thus supporting our hypothesis.
Second, our patient had a cluster of events following surgery compared to the one to 2 events she experienced per year prior to surgery. The increased incidence of events would be an unusual progression of her syncope in the absence of clear triggers, again rendering our hypothesis more plausible.
The patient was admitted to our hospital and started on a higher dose of enoxaparin (60 mg twice daily). She was subsequently discharged home on rivaroxaban 15 mg twice daily and midodrine 2.5 mg twice daily in addition to the medications she was already taking. At her 6-week follow-up visit, she reported no recurrences.
THE TAKEAWAY
This case demonstrates that non-massive PE can present as vasovagal syncope. Recognizing that PE could lead to reflex-mediated syncope in the absence of massive emboli, it is important to rule it out in the evaluation of patients with vasovagal syncope when risk factors for PE are present.
THE CASE
A 58-year-old woman sought care at our clinic for recurrent syncopal and near-syncopal events following surgical repair of a left hip fracture. The first syncopal event occurred one day post-surgery shortly after standing and was attributed to orthostatic hypotension. Subsequently, the patient experienced 2 events during her hospital stay. Both events occurred in the upright position and were preceded by lightheadedness, warmth, and diaphoresis. They were short in duration (<30 seconds) with spontaneous and complete recovery. The patient had no associated chest pain or palpitations.
The patient’s past medical history included osteopenia, dyslipidemia, and vasovagal syncope, averaging one to 2 events per year. Given her past history, the physicians caring for her assumed that she was having recurrences of her vasovagal syncope. She was discharged home on fludrocortisone 0.1 mg/d, sodium chloride 1 g tid, enoxaparin 40 mg/d, and acetaminophen and oxycodone as needed for pain.
One week later, the patient experienced another syncopal event at home, prompting her to visit our clinic for further evaluation. On arrival, her vital signs were stable. Her oxygen saturation level was 98%, she was not orthostatic, and her physical exam and blood studies were unremarkable. An echocardiogram showed preserved left ventricular function with no evidence of right ventricular dilatation or strain.
THE DIAGNOSIS
The patient’s revised Geneva Score for pulmonary embolism (PE) was 2 to 5 depending on the heart rate used (66-80 beats per minute), putting her in a low-to-intermediate risk group with an estimated PE prevalence between 8% and 28%.1 Given her recent surgery and the increase in the frequency of her vasovagal events, a computed tomography pulmonary angiogram (CT-PA) was performed. The CT-PA showed a PE in the lateral and posterior basal subsegmental branches of the right lower lobe. Doppler ultrasound revealed no evidence of acute deep vein thrombosis.
DISCUSSION
Syncope may develop in 9% to 19% of patients with PE.2-6 While syncope in patients with PE is often attributed to reduced cardiac filling secondary to massive emboli, it is important to recognize that patients can also present with vasovagal syncope in the absence of massive emboli.
One mechanism for the development of syncope is right ventricular failure with subsequent impairment of left ventricular filling, leading to arterial hypotension. Indeed, the majority of patients with PE and syncope have a massive embolism defined as greater than a 50% reduction in the pulmonary circulation.7 In one study, 60% of patients with PE who presented with syncope had a massive PE compared to 39% of patients presenting without syncope (P=.036).8
Another reported mechanism for syncope in a patient with PE is transient high-degree atrioventricular (AV) block.9 Sudden increases in right-sided pressure can lead to transient right bundle branch block, which may result in complete heart block in the setting of baseline left bundle branch block.
Lastly, patients with PE may develop a vasovagal-like reaction, such as the Bezold-Jarisch reflex, which results in transient arterial hypotension and cerebral hypoperfusion.10 In such instances, the postulated mechanism is activation of cardiac vagal afferents, which results in an increase in vagal tone and peripheral sympathetic withdrawal leading to hypotension and syncope. It is important to note that this mechanism can occur in the absence of massive PE. In one study, up to 40% of patients with PE and syncope did not have a massive PE, and almost 6% had thrombi only in small branches of the pulmonary artery.8
This patient had isolated subsegmental defects, identified on the CT-PA. The sensitivity of CT-PA to detect subsegmental PE ranges from 53% to 100%.11 While this test has its limitations, the introduction of the multi-detector CT technique has significantly increased the rate of detection with a specificity of 96%.12,13
Was PE the cause of the syncope, or just an incidental finding?
In this case, we believe the CT-PA findings were diagnostic for PE. What is less clear is whether the PE was the cause of the syncope.
Asymptomatic post-operative PE with isolated subsegmental defects has been reported.14-16 When compared to patients with a defect at a segmental or more proximal level, these patients often have less dyspnea, are less likely to be classified as having a high clinical probability of PE, and have a lower prevalence of proximal deep vein thrombosis (3.3% vs 43.8%; P<.0001).17 Therefore, one could argue that the PE finding in our case was incidental. While this is a possibility, we believe the patient’s syncope was due to PE for the following reasons.
First, several investigators have reported transient increases in vagal tone and syncope following PE consistent with a vasovagal-like response.7,18 Therefore, it is possible that the reduction in preload associated with PE triggered a Bezold-Jarisch-like reflex leading to syncope. The patient’s history of vasovagal syncope was certainly indicative of increased susceptibility to reflex-mediated events, thus supporting our hypothesis.
Second, our patient had a cluster of events following surgery compared to the one to 2 events she experienced per year prior to surgery. The increased incidence of events would be an unusual progression of her syncope in the absence of clear triggers, again rendering our hypothesis more plausible.
The patient was admitted to our hospital and started on a higher dose of enoxaparin (60 mg twice daily). She was subsequently discharged home on rivaroxaban 15 mg twice daily and midodrine 2.5 mg twice daily in addition to the medications she was already taking. At her 6-week follow-up visit, she reported no recurrences.
THE TAKEAWAY
This case demonstrates that non-massive PE can present as vasovagal syncope. Recognizing that PE could lead to reflex-mediated syncope in the absence of massive emboli, it is important to rule it out in the evaluation of patients with vasovagal syncope when risk factors for PE are present.
1. Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006;144:165-171.
2. Calvo-Romero JM, Pérez-Miranda M, Bureo-Dacal P. Syncope in acute pulmonary embolism. Eur J Emerg Med. 2004;11:208-209.
3. Castelli R, Tarsia P, Tantardini C, et al. Syncope in patients with pulmonary embolism: comparison between patients with syncope as the presenting symptom of pulmonary embolism and patients with pulmonary embolism without syncope. Vasc Med. 2003;8:257-261.
4. Kasper W, Konstantinides S, Geibel A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol. 1997;30:1165-1171.
5. Koutkia P, Wachtel TJ. Pulmonary embolism presenting as syncope: case report and review of the literature. Heart Lung. 1999;28:342-347.
6. Torbicki A, Perrier A, Konstantinides S, et al; ESC Committee for Practice Guidelines (CPG). Guidelines on the diagnosis and management of acute pulmonary embolism: the Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2008;29:2276-2315.
7. Thames MD, Alpert JS, Dalen JE. Syncope in patients with pulmonary embolism. JAMA. 1977;238:2509-2511.
8. Duplyakov D, Kurakina E, Pavlova T, et al. Value of syncope in patients with high-to-intermediate risk pulmonary artery embolism. Eur Heart J Acute Cardiovasc Care. 2015;4:353-358.
9. Wilner C, Garnier-Crussard JP, Huygue De Mahenge A, et al. [Paroxysmal atrioventricular block, cause of syncope in pulmonary embolism. 2 cases]. Presse Med. 1983;12:2987-2989.
10. Frink RJ, James TN. Intracardiac route of the Bezold-Jarisch reflex. Am J Physiol. 1971;221:1464-1469.
11. Rathbun SW, Raskob GE, Whitsett TL. Sensitivity and specificity of helical computed tomography in the diagnosis of pulmonary embolism: A systematic review. Ann Intern Med. 2000;132:227-232.
12. Stein PD, Fowler SE, Goodman LR, et al; PIOPED II Investigators. Multidetector computed tomography for acute pulmonary embolism. N Engl J Med. 2006;354:2317-2327.
13. Vedovati MC, Becattini C, Agnelli G, et al. Multidetector CT scan for acute pulmonary embolism: embolic burden and clinical outcome. Chest. 2012;142:1417-1424.
14. Musset D, Parent F, Meyer G, et al; Evaluation du Scanner Spiralé dans l’Embolie Pulmonaire study group. Diagnostic strategy for patients with suspected pulmonary embolism: a prospective multicentre outcome study. Lancet. 2002;360:1914-1920.
15. Simpson RJ Jr, Podolak R, Mangano CA Jr, et al. Vagal syncope during recurrent pulmonary embolism. JAMA. 1983;249:390-393.
16. Perrier A, Roy PM, Sanchez O, et al. Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med. 2005;352:1760-1768.
17. Le Gal G, Righini M, Parent F, et al. Diagnosis and management of subsegmental pulmonary embolism. J Thromb Haemost. 2006;4:724-731.
18. Eldadah ZA, Najjar SS, Ziegelstein RC. A patient with syncope, only “vagally” related to the heart. Chest. 2000;117:1801-1803.
1. Le Gal G, Righini M, Roy PM, et al. Prediction of pulmonary embolism in the emergency department: the revised Geneva score. Ann Intern Med. 2006;144:165-171.
2. Calvo-Romero JM, Pérez-Miranda M, Bureo-Dacal P. Syncope in acute pulmonary embolism. Eur J Emerg Med. 2004;11:208-209.
3. Castelli R, Tarsia P, Tantardini C, et al. Syncope in patients with pulmonary embolism: comparison between patients with syncope as the presenting symptom of pulmonary embolism and patients with pulmonary embolism without syncope. Vasc Med. 2003;8:257-261.
4. Kasper W, Konstantinides S, Geibel A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: results of a multicenter registry. J Am Coll Cardiol. 1997;30:1165-1171.
5. Koutkia P, Wachtel TJ. Pulmonary embolism presenting as syncope: case report and review of the literature. Heart Lung. 1999;28:342-347.
6. Torbicki A, Perrier A, Konstantinides S, et al; ESC Committee for Practice Guidelines (CPG). Guidelines on the diagnosis and management of acute pulmonary embolism: the Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2008;29:2276-2315.
7. Thames MD, Alpert JS, Dalen JE. Syncope in patients with pulmonary embolism. JAMA. 1977;238:2509-2511.
8. Duplyakov D, Kurakina E, Pavlova T, et al. Value of syncope in patients with high-to-intermediate risk pulmonary artery embolism. Eur Heart J Acute Cardiovasc Care. 2015;4:353-358.
9. Wilner C, Garnier-Crussard JP, Huygue De Mahenge A, et al. [Paroxysmal atrioventricular block, cause of syncope in pulmonary embolism. 2 cases]. Presse Med. 1983;12:2987-2989.
10. Frink RJ, James TN. Intracardiac route of the Bezold-Jarisch reflex. Am J Physiol. 1971;221:1464-1469.
11. Rathbun SW, Raskob GE, Whitsett TL. Sensitivity and specificity of helical computed tomography in the diagnosis of pulmonary embolism: A systematic review. Ann Intern Med. 2000;132:227-232.
12. Stein PD, Fowler SE, Goodman LR, et al; PIOPED II Investigators. Multidetector computed tomography for acute pulmonary embolism. N Engl J Med. 2006;354:2317-2327.
13. Vedovati MC, Becattini C, Agnelli G, et al. Multidetector CT scan for acute pulmonary embolism: embolic burden and clinical outcome. Chest. 2012;142:1417-1424.
14. Musset D, Parent F, Meyer G, et al; Evaluation du Scanner Spiralé dans l’Embolie Pulmonaire study group. Diagnostic strategy for patients with suspected pulmonary embolism: a prospective multicentre outcome study. Lancet. 2002;360:1914-1920.
15. Simpson RJ Jr, Podolak R, Mangano CA Jr, et al. Vagal syncope during recurrent pulmonary embolism. JAMA. 1983;249:390-393.
16. Perrier A, Roy PM, Sanchez O, et al. Multidetector-row computed tomography in suspected pulmonary embolism. N Engl J Med. 2005;352:1760-1768.
17. Le Gal G, Righini M, Parent F, et al. Diagnosis and management of subsegmental pulmonary embolism. J Thromb Haemost. 2006;4:724-731.
18. Eldadah ZA, Najjar SS, Ziegelstein RC. A patient with syncope, only “vagally” related to the heart. Chest. 2000;117:1801-1803.

Congenital Self-healing Reticulohistiocytosis: An Underreported Entity
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a general term that describes a group of rare disorders characterized by the proliferation of Langerhans cells.1 Central to immune surveillance and the elimination of foreign substances from the body, Langerhans cells are derived from bone marrow progenitor cells and found in the epidermis but are capable of migrating from the skin to the lymph nodes. In LCH, these cells congregate on bone tissue, particularly in the head and neck region, causing a multitude of problems.2
The spectrum of LCH includes 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma (also known as pulmonary histiocytosis X)(Table). Despite the various clinical presentations and levels of severity, all variants are caused by the proliferation of Langerhans cells. We present a case of CSHR in a 6-month-old male infant that was initially diagnosed as molluscum contagiosum. We believe the actual incidence of CSHR may be underreported due to its spontaneous regression and low rate of clinical recognition.
Case Report
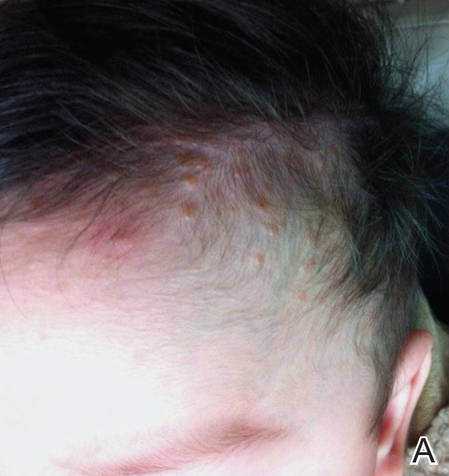
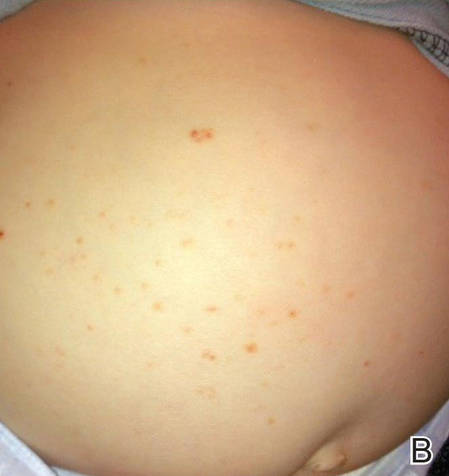
A 6-month-old male infant was referred to our clinic by his pediatrician with a generalized cutaneous eruption of 3 weeks’ duration. The eruption, which followed a recent viral upper respiratory tract infection, was characterized by multiple flesh-colored to erythematous, umbilicated papules distributed along the postauricular region, scalp (Figure 1A), abdomen (Figure 1B), and anterior aspect of the neck. Due to his recent illness, the patient was diagnosed with molluscum contagiosum by the referring pediatrician that was treated symptomatically with hydrocortisone lotion, Schamberg’s cream formulated in our office (a compound mixture of zinc oxide, menthol, calcium hydroxide solution, and olive oil), and pediatric diphenhydramine as needed. During a subsequent visit 2 weeks later, a more potent topical corticosteroid and a low-dose systemic corticosteroid was prescribed for 1 week due to development of new lesions and exacerbation of existing lesions. On follow-up 1 week later, the lesions on the trunk had improved, but the patient had developed new lesions on the scalp that differed from prior findings in that they were darker (more erythematous to brown) and firmer (papules and nodules).
| 
| |
Figure 1. Multiple fleshcolored to erythematous, umbilicated papules on the frontal scalp (A) and erythematous papules on the abdomen (B). | ||
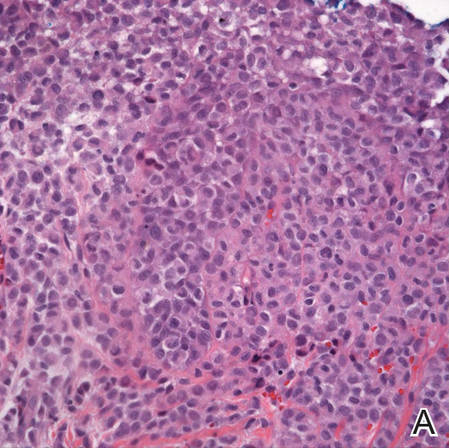
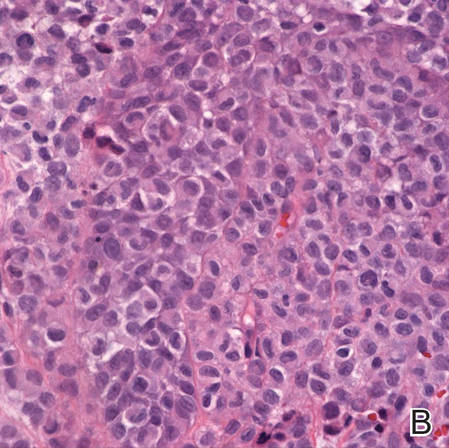
A shave biopsy was obtained from the frontal scalp to rule out LCH. Histologic examination and culture of the biopsy specimen revealed an atypical cellular infiltrate effacing the dermoepidermal junction and extensive epidermotropism. Focal erosion of the epidermis and an acute inflammatory exudate were visible. The nuclei of the cellular infiltrate were enlarged and hyperchromatic with a characteristic reniform appearance and indistinct nucleoli (Figure 2). The cells were admixed with scattered eosinophils and extravasated red blood cells.
| 
| |
Figure 2. Low-power view of dermal mononuclear cells with reniform nuclei (A)(H&E, original magnification ×100), and high-power view of enlarged and hyperchromatic nuclei with a characteristic reniform appearance admixed with eosinophils and extravasated red blood cells (B) (H&E, original magnification ×400). | ||
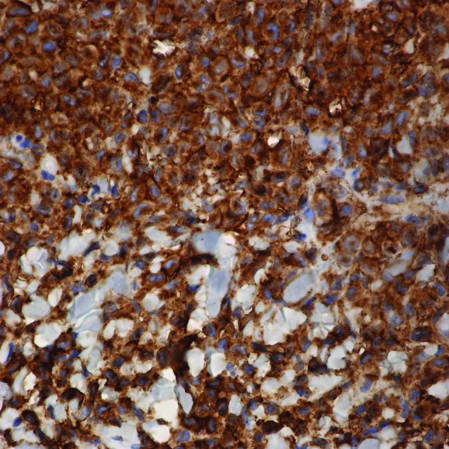
Immunohistochemical staining of the biopsy specimen was strongly positive for both CD1a and S-100 expression (Figure 3). Histopathologic findings were consistent with LCH. Clinicopathologic correlation strongly favored the diagnosis of CSHR.
Comment
Congenital self-healing reticulohistiocytosis is a rare, benign, congenital variant of LCH that spontaneously resolves with no systemic involvement. The more aggressive forms typically manifest at birth or during the first 2 months of life and regress within 3 to 4 months.5 Since CSHR was first described in 1973 by Hashimoto and Pritzker,5 more than 100 cases have been reported, but the true incidence is believed to be higher than reported given the high rate of spontaneous resolution and the low rate of clinical recognition.2 The first reported case of CSHR occurred in a female infant who presented at birth with multiple, diffusely distributed, red-brown papules that were 2 to 4 mm in diameter. Although the patient received no treatment, the exanthem completely resolved within 3.5 months without recurrence at 14-year follow-up.5 Most often, CSHR presents as multiple papules or nodules with occasional disseminated crusting and is followed within a few months by a dramatic and spontaneous regression. Lesions may heal with mild postinflammatory hyperpigmentation. Pseudo-Darier sign, the propensity to urticate from physical manipulation, has been reported in some lesions with an increased number of mast cells.6 Extensive superficial nasal and oral mucosal erosions have been reported in 2 cases.7 Solitary lesions have been reported in 25% of cases.8
The etiology of CSHR remains unknown, though neoplastic, viral, and immunologic origins have been suggested. There have been reports that human herpesvirus 6 may contribute to the development of LCH.9 It may be postulated that our patient’s presentation of CSHR was potentiated by his recent upper respiratory tract illness. In the literature, CSHR is distributed equally among males and females. Prevalence is higher in the white population than in other racial groups.5
Although CSHR is a benign cutaneous variant of LCH, there have been reports of patients with disseminated and extracutaneous involvement. In 1 rare case, CSHR reportedly involved the eyes, producing multiple, bilateral, well-circumscribed, diffuse, yellow-white lesions of the retinal pigment epithelium throughout the posterior pole of the eyes.10 The retinal lesions spontaneously regressed along with the skin manifestations. Additionally, it was reported that a neonate in Thailand presented with CSHR at birth and 1 month later developed multiple lung cysts that had completely regressed 11 months later.11 One study reported that initial diagnoses of LCH in 18 patients with only cutaneous involvement eventually progressed to systemic LCH, requiring further management.12 When LCH is suspected, a thorough physical examination, including hematologic and coagulation evaluation, liver function tests, musculoskeletal examination, and consultation with specialists if necessary, is recommended.13
There are 3 additional variants of LCH. Letterer-Siwe disease is an acute form of LCH that accounts for 10% of all LCH cases and typically presents in children younger than 2 years. It involves multiple organs, including the bones, lungs, liver, and lymph nodes.14 Affected patients usually present with fever; hepatosplenomegaly; anemia; lymphadenopathy; extensive lytic skull lesions; and a generalized cutaneous eruption, appearing as a maculopapular scaling rash with underlying purpura on the scalp, neck, axilla, and trunk.3 Letterer-Siwe disease is inherited in an autosomal-recessive pattern. Diagnosis is confirmed by skin biopsy demonstrating a thinning of the epidermis and a collection of reticulum cells in the dermis.3 Letterer-Siwe disease is treated with radiation and chemotherapy; if left untreated, the disease is fatal.4
Hand-Schüller-Christian disease, a chronic form of LCH, is most commonly seen in children aged 2 to 6 years and accounts for 15% to 20% of all LCH cases. This LCH variant presents with a classic triad of diabetes insipidus (resulting from erosion into the sella turcica), lytic bone lesions, and exophthalmos.15 Hand-Schüller-Christian disease also affects the oral cavity, producing nodular ulcerations of the hard palate, trouble swallowing, and halitosis.4 The involvement of lytic bone lesions of the mastoid process and petrous portions of the temporal bones may cause recurrent or chronic otitis media and otitis externa. Hand-Schüller-Christian disease is treated with a combination of chemotherapy, radiation, and surgical excision. The mortality rate is 30%.4
Eosinophilic granuloma is the most prevalent variant of LCH, accounting for 60% to 80% of all cases. Characterized by Langerhans cell granulomatous infiltration of the lungs and painful cystic bone lesions, eosinophilic granuloma primarily presents in the third or fourth decades of life.16 Some studies suggest an epidemiologic association with tobacco use.17 In the preliminary stages of this disease, Langerhans cells, eosinophils, lymphocytes, and fibroblasts infiltrate and form nodules on the terminal bronchioles in the upper and middle lung zones, damaging the airway walls.18 Fibrotic scarring progresses, ultimately resulting in alveolar destruction.10 The common signs and symptoms of eosinophilic granuloma are a nonproductive cough, dyspnea, weight loss, spontaneous pneumothorax, fever, peripheral edema, and a tricuspid regurgitation murmur.14 The prognosis of eosinophilic granuloma is variable. Although some patients progress to end-stage fibrotic lung disease requiring lung transplant, there have been reports of complete remission following cessation of cigarette smoking.17
Langerhans cells travel from the bone marrow to the epidermis where they express the CD1a protein on the surface of the antigen-presenting cell. Elevated levels of cytokines, such as tumor necrosis factor α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and interleukins have been seen in patients with LCH.1 Their role in the pathogenesis of this disease remains unknown, but the elevated levels of cytokines may indicate the lack of an efficient immune system.
Histologically, hematoxylin and eosin–stained sections demonstrate an infiltrate of histiocytes, neutrophils, eosinophils, and an increased number of mast cells involving the papillary and reticular dermis. Infiltrating Langerhans cells have concave reniform nuclei18 and stain positive for CD1a, S-100, and CD68 antigens.15 In 10% to 30% of CSHR cases, Birbeck granules can be seen on electron microscopy and tend to transform into laminated dense bodies, signifying the degenerative changes seen in CSHR.15 The various forms of LCH exhibit no significant differences in the expression of the epithelial cadherin, the phosphorylated histone H3, and the Ki-67 proteins, indicating that they are simply different forms of the same disease represented on a spectrum.15
Conclusion
The actual incidence of CSHR may be notably underreported due to its spontaneous regression and low rate of clinical recognition. A subtype of LCH, CSHR is a diagnosis of exclusion. Although CSHR generally follows a benign clinical course, a thorough workup and evaluation for systemic disease with close follow-up is recommended after diagnosis due to the potential of LCH to involve multiple organs and to relapse at a later date after apparent regression.
1. Hussein MR. Skin-limited Langerhans’ cell histiocytosis in children. Cancer Invest. 2009;27:504-511.
2. Nakahigashi K, Ohta M, Sakai R, et al. Late-onset self-healing reticulohistiocytosis: pediatric case of Hashimoto-Pritzker type Langerhans cell histiocytosis. J Dermatol. 2007;34:205-209.
3. Pant C, Madonia P, Bahna SL, et al. Langerhans cell histiocytosis, a case of Letterer Siwe disease. J La State Med Soc. 2009;161:211-212.
4. Ferreira LM, Emerich PS, Diniz LM, et al. Langerhans cell histiocytosis: Letterer-Siwe disease–the importance of dermatological diagnosis in two cases [in Portuguese]. An Bras Dermatol. 2009;84:405-409.
5. Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. an unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol. 1973;107:263-270.
6. Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol. 2007;56:290-294.
7. Le Bidre E, Lorette G, Delage M, et al. Extensive, erosive congenital self-healing cell histiocytosis [published online December 22, 2008]. J Eur Acad Dermatol Venereol. 2009;23:835-836.
8. Weiss T, Weber L, Scharffetter-Kochanek K, et al. Solitary cutaneous dendritic cell tumor in a child: role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J Am Acad Dermatol. 2005;53:838-844.
9. Csire M, Mikala G, Jákó J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with Langerhans cell histiocytosis [published online July 3, 2007]. Pathol Oncol Res. 2007;13:157-160.
10. Zaenglein AL, Steele MA, Kamino H, et al. Congenital self-healing reticulohistiocytosis with eye involvement. Pediatr Dermatol. 2001;18:135-137.
11. Chunharas A, Pabunruang W, Hongeng S. Congenital self-healing Langerhans cell histiocytosis with pulmonary involvement: spontaneous regression. J Med Assoc Thai. 2002;85(suppl 4):S1309-S1313.
12. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802-807.
13. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-295.
14. Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax: a potential diagnostic pitfall [published online June 27, 2009]. Virchows Arch. 2009;455:187-190.
15. Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature [published online October 10, 2003]. Eur Arch Otorhinolaryngol. 2004;261:326-330.
16. Noonan V, Kabani S, Alibhai K. Langerhans cell histiocytosis (eosinophilic granuloma). J Mass Dent Soc. 2011;60:35.
17. Podbielski FJ, Worley TA, Korn JM, et al. Eosinophilic granuloma of the lung and rib. Asian Cardiovasc Thorac Ann. 2009;17:194-195.
18. Rosso DA, Ripoli MF, Roy A, et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480-483.
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a general term that describes a group of rare disorders characterized by the proliferation of Langerhans cells.1 Central to immune surveillance and the elimination of foreign substances from the body, Langerhans cells are derived from bone marrow progenitor cells and found in the epidermis but are capable of migrating from the skin to the lymph nodes. In LCH, these cells congregate on bone tissue, particularly in the head and neck region, causing a multitude of problems.2
The spectrum of LCH includes 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma (also known as pulmonary histiocytosis X)(Table). Despite the various clinical presentations and levels of severity, all variants are caused by the proliferation of Langerhans cells. We present a case of CSHR in a 6-month-old male infant that was initially diagnosed as molluscum contagiosum. We believe the actual incidence of CSHR may be underreported due to its spontaneous regression and low rate of clinical recognition.
Case Report
A 6-month-old male infant was referred to our clinic by his pediatrician with a generalized cutaneous eruption of 3 weeks’ duration. The eruption, which followed a recent viral upper respiratory tract infection, was characterized by multiple flesh-colored to erythematous, umbilicated papules distributed along the postauricular region, scalp (Figure 1A), abdomen (Figure 1B), and anterior aspect of the neck. Due to his recent illness, the patient was diagnosed with molluscum contagiosum by the referring pediatrician that was treated symptomatically with hydrocortisone lotion, Schamberg’s cream formulated in our office (a compound mixture of zinc oxide, menthol, calcium hydroxide solution, and olive oil), and pediatric diphenhydramine as needed. During a subsequent visit 2 weeks later, a more potent topical corticosteroid and a low-dose systemic corticosteroid was prescribed for 1 week due to development of new lesions and exacerbation of existing lesions. On follow-up 1 week later, the lesions on the trunk had improved, but the patient had developed new lesions on the scalp that differed from prior findings in that they were darker (more erythematous to brown) and firmer (papules and nodules).
|
|
| |
Figure 1. Multiple fleshcolored to erythematous, umbilicated papules on the frontal scalp (A) and erythematous papules on the abdomen (B). | ||
A shave biopsy was obtained from the frontal scalp to rule out LCH. Histologic examination and culture of the biopsy specimen revealed an atypical cellular infiltrate effacing the dermoepidermal junction and extensive epidermotropism. Focal erosion of the epidermis and an acute inflammatory exudate were visible. The nuclei of the cellular infiltrate were enlarged and hyperchromatic with a characteristic reniform appearance and indistinct nucleoli (Figure 2). The cells were admixed with scattered eosinophils and extravasated red blood cells.
|
|
| |
Figure 2. Low-power view of dermal mononuclear cells with reniform nuclei (A)(H&E, original magnification ×100), and high-power view of enlarged and hyperchromatic nuclei with a characteristic reniform appearance admixed with eosinophils and extravasated red blood cells (B) (H&E, original magnification ×400). | ||
Immunohistochemical staining of the biopsy specimen was strongly positive for both CD1a and S-100 expression (Figure 3). Histopathologic findings were consistent with LCH. Clinicopathologic correlation strongly favored the diagnosis of CSHR.
Comment
Congenital self-healing reticulohistiocytosis is a rare, benign, congenital variant of LCH that spontaneously resolves with no systemic involvement. The more aggressive forms typically manifest at birth or during the first 2 months of life and regress within 3 to 4 months.5 Since CSHR was first described in 1973 by Hashimoto and Pritzker,5 more than 100 cases have been reported, but the true incidence is believed to be higher than reported given the high rate of spontaneous resolution and the low rate of clinical recognition.2 The first reported case of CSHR occurred in a female infant who presented at birth with multiple, diffusely distributed, red-brown papules that were 2 to 4 mm in diameter. Although the patient received no treatment, the exanthem completely resolved within 3.5 months without recurrence at 14-year follow-up.5 Most often, CSHR presents as multiple papules or nodules with occasional disseminated crusting and is followed within a few months by a dramatic and spontaneous regression. Lesions may heal with mild postinflammatory hyperpigmentation. Pseudo-Darier sign, the propensity to urticate from physical manipulation, has been reported in some lesions with an increased number of mast cells.6 Extensive superficial nasal and oral mucosal erosions have been reported in 2 cases.7 Solitary lesions have been reported in 25% of cases.8
The etiology of CSHR remains unknown, though neoplastic, viral, and immunologic origins have been suggested. There have been reports that human herpesvirus 6 may contribute to the development of LCH.9 It may be postulated that our patient’s presentation of CSHR was potentiated by his recent upper respiratory tract illness. In the literature, CSHR is distributed equally among males and females. Prevalence is higher in the white population than in other racial groups.5
Although CSHR is a benign cutaneous variant of LCH, there have been reports of patients with disseminated and extracutaneous involvement. In 1 rare case, CSHR reportedly involved the eyes, producing multiple, bilateral, well-circumscribed, diffuse, yellow-white lesions of the retinal pigment epithelium throughout the posterior pole of the eyes.10 The retinal lesions spontaneously regressed along with the skin manifestations. Additionally, it was reported that a neonate in Thailand presented with CSHR at birth and 1 month later developed multiple lung cysts that had completely regressed 11 months later.11 One study reported that initial diagnoses of LCH in 18 patients with only cutaneous involvement eventually progressed to systemic LCH, requiring further management.12 When LCH is suspected, a thorough physical examination, including hematologic and coagulation evaluation, liver function tests, musculoskeletal examination, and consultation with specialists if necessary, is recommended.13
There are 3 additional variants of LCH. Letterer-Siwe disease is an acute form of LCH that accounts for 10% of all LCH cases and typically presents in children younger than 2 years. It involves multiple organs, including the bones, lungs, liver, and lymph nodes.14 Affected patients usually present with fever; hepatosplenomegaly; anemia; lymphadenopathy; extensive lytic skull lesions; and a generalized cutaneous eruption, appearing as a maculopapular scaling rash with underlying purpura on the scalp, neck, axilla, and trunk.3 Letterer-Siwe disease is inherited in an autosomal-recessive pattern. Diagnosis is confirmed by skin biopsy demonstrating a thinning of the epidermis and a collection of reticulum cells in the dermis.3 Letterer-Siwe disease is treated with radiation and chemotherapy; if left untreated, the disease is fatal.4
Hand-Schüller-Christian disease, a chronic form of LCH, is most commonly seen in children aged 2 to 6 years and accounts for 15% to 20% of all LCH cases. This LCH variant presents with a classic triad of diabetes insipidus (resulting from erosion into the sella turcica), lytic bone lesions, and exophthalmos.15 Hand-Schüller-Christian disease also affects the oral cavity, producing nodular ulcerations of the hard palate, trouble swallowing, and halitosis.4 The involvement of lytic bone lesions of the mastoid process and petrous portions of the temporal bones may cause recurrent or chronic otitis media and otitis externa. Hand-Schüller-Christian disease is treated with a combination of chemotherapy, radiation, and surgical excision. The mortality rate is 30%.4
Eosinophilic granuloma is the most prevalent variant of LCH, accounting for 60% to 80% of all cases. Characterized by Langerhans cell granulomatous infiltration of the lungs and painful cystic bone lesions, eosinophilic granuloma primarily presents in the third or fourth decades of life.16 Some studies suggest an epidemiologic association with tobacco use.17 In the preliminary stages of this disease, Langerhans cells, eosinophils, lymphocytes, and fibroblasts infiltrate and form nodules on the terminal bronchioles in the upper and middle lung zones, damaging the airway walls.18 Fibrotic scarring progresses, ultimately resulting in alveolar destruction.10 The common signs and symptoms of eosinophilic granuloma are a nonproductive cough, dyspnea, weight loss, spontaneous pneumothorax, fever, peripheral edema, and a tricuspid regurgitation murmur.14 The prognosis of eosinophilic granuloma is variable. Although some patients progress to end-stage fibrotic lung disease requiring lung transplant, there have been reports of complete remission following cessation of cigarette smoking.17
Langerhans cells travel from the bone marrow to the epidermis where they express the CD1a protein on the surface of the antigen-presenting cell. Elevated levels of cytokines, such as tumor necrosis factor α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and interleukins have been seen in patients with LCH.1 Their role in the pathogenesis of this disease remains unknown, but the elevated levels of cytokines may indicate the lack of an efficient immune system.
Histologically, hematoxylin and eosin–stained sections demonstrate an infiltrate of histiocytes, neutrophils, eosinophils, and an increased number of mast cells involving the papillary and reticular dermis. Infiltrating Langerhans cells have concave reniform nuclei18 and stain positive for CD1a, S-100, and CD68 antigens.15 In 10% to 30% of CSHR cases, Birbeck granules can be seen on electron microscopy and tend to transform into laminated dense bodies, signifying the degenerative changes seen in CSHR.15 The various forms of LCH exhibit no significant differences in the expression of the epithelial cadherin, the phosphorylated histone H3, and the Ki-67 proteins, indicating that they are simply different forms of the same disease represented on a spectrum.15
Conclusion
The actual incidence of CSHR may be notably underreported due to its spontaneous regression and low rate of clinical recognition. A subtype of LCH, CSHR is a diagnosis of exclusion. Although CSHR generally follows a benign clinical course, a thorough workup and evaluation for systemic disease with close follow-up is recommended after diagnosis due to the potential of LCH to involve multiple organs and to relapse at a later date after apparent regression.
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a general term that describes a group of rare disorders characterized by the proliferation of Langerhans cells.1 Central to immune surveillance and the elimination of foreign substances from the body, Langerhans cells are derived from bone marrow progenitor cells and found in the epidermis but are capable of migrating from the skin to the lymph nodes. In LCH, these cells congregate on bone tissue, particularly in the head and neck region, causing a multitude of problems.2
The spectrum of LCH includes 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma (also known as pulmonary histiocytosis X)(Table). Despite the various clinical presentations and levels of severity, all variants are caused by the proliferation of Langerhans cells. We present a case of CSHR in a 6-month-old male infant that was initially diagnosed as molluscum contagiosum. We believe the actual incidence of CSHR may be underreported due to its spontaneous regression and low rate of clinical recognition.
Case Report
A 6-month-old male infant was referred to our clinic by his pediatrician with a generalized cutaneous eruption of 3 weeks’ duration. The eruption, which followed a recent viral upper respiratory tract infection, was characterized by multiple flesh-colored to erythematous, umbilicated papules distributed along the postauricular region, scalp (Figure 1A), abdomen (Figure 1B), and anterior aspect of the neck. Due to his recent illness, the patient was diagnosed with molluscum contagiosum by the referring pediatrician that was treated symptomatically with hydrocortisone lotion, Schamberg’s cream formulated in our office (a compound mixture of zinc oxide, menthol, calcium hydroxide solution, and olive oil), and pediatric diphenhydramine as needed. During a subsequent visit 2 weeks later, a more potent topical corticosteroid and a low-dose systemic corticosteroid was prescribed for 1 week due to development of new lesions and exacerbation of existing lesions. On follow-up 1 week later, the lesions on the trunk had improved, but the patient had developed new lesions on the scalp that differed from prior findings in that they were darker (more erythematous to brown) and firmer (papules and nodules).
|
|
| |
Figure 1. Multiple fleshcolored to erythematous, umbilicated papules on the frontal scalp (A) and erythematous papules on the abdomen (B). | ||
A shave biopsy was obtained from the frontal scalp to rule out LCH. Histologic examination and culture of the biopsy specimen revealed an atypical cellular infiltrate effacing the dermoepidermal junction and extensive epidermotropism. Focal erosion of the epidermis and an acute inflammatory exudate were visible. The nuclei of the cellular infiltrate were enlarged and hyperchromatic with a characteristic reniform appearance and indistinct nucleoli (Figure 2). The cells were admixed with scattered eosinophils and extravasated red blood cells.
|
|
| |
Figure 2. Low-power view of dermal mononuclear cells with reniform nuclei (A)(H&E, original magnification ×100), and high-power view of enlarged and hyperchromatic nuclei with a characteristic reniform appearance admixed with eosinophils and extravasated red blood cells (B) (H&E, original magnification ×400). | ||
Immunohistochemical staining of the biopsy specimen was strongly positive for both CD1a and S-100 expression (Figure 3). Histopathologic findings were consistent with LCH. Clinicopathologic correlation strongly favored the diagnosis of CSHR.
Comment
Congenital self-healing reticulohistiocytosis is a rare, benign, congenital variant of LCH that spontaneously resolves with no systemic involvement. The more aggressive forms typically manifest at birth or during the first 2 months of life and regress within 3 to 4 months.5 Since CSHR was first described in 1973 by Hashimoto and Pritzker,5 more than 100 cases have been reported, but the true incidence is believed to be higher than reported given the high rate of spontaneous resolution and the low rate of clinical recognition.2 The first reported case of CSHR occurred in a female infant who presented at birth with multiple, diffusely distributed, red-brown papules that were 2 to 4 mm in diameter. Although the patient received no treatment, the exanthem completely resolved within 3.5 months without recurrence at 14-year follow-up.5 Most often, CSHR presents as multiple papules or nodules with occasional disseminated crusting and is followed within a few months by a dramatic and spontaneous regression. Lesions may heal with mild postinflammatory hyperpigmentation. Pseudo-Darier sign, the propensity to urticate from physical manipulation, has been reported in some lesions with an increased number of mast cells.6 Extensive superficial nasal and oral mucosal erosions have been reported in 2 cases.7 Solitary lesions have been reported in 25% of cases.8
The etiology of CSHR remains unknown, though neoplastic, viral, and immunologic origins have been suggested. There have been reports that human herpesvirus 6 may contribute to the development of LCH.9 It may be postulated that our patient’s presentation of CSHR was potentiated by his recent upper respiratory tract illness. In the literature, CSHR is distributed equally among males and females. Prevalence is higher in the white population than in other racial groups.5
Although CSHR is a benign cutaneous variant of LCH, there have been reports of patients with disseminated and extracutaneous involvement. In 1 rare case, CSHR reportedly involved the eyes, producing multiple, bilateral, well-circumscribed, diffuse, yellow-white lesions of the retinal pigment epithelium throughout the posterior pole of the eyes.10 The retinal lesions spontaneously regressed along with the skin manifestations. Additionally, it was reported that a neonate in Thailand presented with CSHR at birth and 1 month later developed multiple lung cysts that had completely regressed 11 months later.11 One study reported that initial diagnoses of LCH in 18 patients with only cutaneous involvement eventually progressed to systemic LCH, requiring further management.12 When LCH is suspected, a thorough physical examination, including hematologic and coagulation evaluation, liver function tests, musculoskeletal examination, and consultation with specialists if necessary, is recommended.13
There are 3 additional variants of LCH. Letterer-Siwe disease is an acute form of LCH that accounts for 10% of all LCH cases and typically presents in children younger than 2 years. It involves multiple organs, including the bones, lungs, liver, and lymph nodes.14 Affected patients usually present with fever; hepatosplenomegaly; anemia; lymphadenopathy; extensive lytic skull lesions; and a generalized cutaneous eruption, appearing as a maculopapular scaling rash with underlying purpura on the scalp, neck, axilla, and trunk.3 Letterer-Siwe disease is inherited in an autosomal-recessive pattern. Diagnosis is confirmed by skin biopsy demonstrating a thinning of the epidermis and a collection of reticulum cells in the dermis.3 Letterer-Siwe disease is treated with radiation and chemotherapy; if left untreated, the disease is fatal.4
Hand-Schüller-Christian disease, a chronic form of LCH, is most commonly seen in children aged 2 to 6 years and accounts for 15% to 20% of all LCH cases. This LCH variant presents with a classic triad of diabetes insipidus (resulting from erosion into the sella turcica), lytic bone lesions, and exophthalmos.15 Hand-Schüller-Christian disease also affects the oral cavity, producing nodular ulcerations of the hard palate, trouble swallowing, and halitosis.4 The involvement of lytic bone lesions of the mastoid process and petrous portions of the temporal bones may cause recurrent or chronic otitis media and otitis externa. Hand-Schüller-Christian disease is treated with a combination of chemotherapy, radiation, and surgical excision. The mortality rate is 30%.4
Eosinophilic granuloma is the most prevalent variant of LCH, accounting for 60% to 80% of all cases. Characterized by Langerhans cell granulomatous infiltration of the lungs and painful cystic bone lesions, eosinophilic granuloma primarily presents in the third or fourth decades of life.16 Some studies suggest an epidemiologic association with tobacco use.17 In the preliminary stages of this disease, Langerhans cells, eosinophils, lymphocytes, and fibroblasts infiltrate and form nodules on the terminal bronchioles in the upper and middle lung zones, damaging the airway walls.18 Fibrotic scarring progresses, ultimately resulting in alveolar destruction.10 The common signs and symptoms of eosinophilic granuloma are a nonproductive cough, dyspnea, weight loss, spontaneous pneumothorax, fever, peripheral edema, and a tricuspid regurgitation murmur.14 The prognosis of eosinophilic granuloma is variable. Although some patients progress to end-stage fibrotic lung disease requiring lung transplant, there have been reports of complete remission following cessation of cigarette smoking.17
Langerhans cells travel from the bone marrow to the epidermis where they express the CD1a protein on the surface of the antigen-presenting cell. Elevated levels of cytokines, such as tumor necrosis factor α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and interleukins have been seen in patients with LCH.1 Their role in the pathogenesis of this disease remains unknown, but the elevated levels of cytokines may indicate the lack of an efficient immune system.
Histologically, hematoxylin and eosin–stained sections demonstrate an infiltrate of histiocytes, neutrophils, eosinophils, and an increased number of mast cells involving the papillary and reticular dermis. Infiltrating Langerhans cells have concave reniform nuclei18 and stain positive for CD1a, S-100, and CD68 antigens.15 In 10% to 30% of CSHR cases, Birbeck granules can be seen on electron microscopy and tend to transform into laminated dense bodies, signifying the degenerative changes seen in CSHR.15 The various forms of LCH exhibit no significant differences in the expression of the epithelial cadherin, the phosphorylated histone H3, and the Ki-67 proteins, indicating that they are simply different forms of the same disease represented on a spectrum.15
Conclusion
The actual incidence of CSHR may be notably underreported due to its spontaneous regression and low rate of clinical recognition. A subtype of LCH, CSHR is a diagnosis of exclusion. Although CSHR generally follows a benign clinical course, a thorough workup and evaluation for systemic disease with close follow-up is recommended after diagnosis due to the potential of LCH to involve multiple organs and to relapse at a later date after apparent regression.
1. Hussein MR. Skin-limited Langerhans’ cell histiocytosis in children. Cancer Invest. 2009;27:504-511.
2. Nakahigashi K, Ohta M, Sakai R, et al. Late-onset self-healing reticulohistiocytosis: pediatric case of Hashimoto-Pritzker type Langerhans cell histiocytosis. J Dermatol. 2007;34:205-209.
3. Pant C, Madonia P, Bahna SL, et al. Langerhans cell histiocytosis, a case of Letterer Siwe disease. J La State Med Soc. 2009;161:211-212.
4. Ferreira LM, Emerich PS, Diniz LM, et al. Langerhans cell histiocytosis: Letterer-Siwe disease–the importance of dermatological diagnosis in two cases [in Portuguese]. An Bras Dermatol. 2009;84:405-409.
5. Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. an unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol. 1973;107:263-270.
6. Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol. 2007;56:290-294.
7. Le Bidre E, Lorette G, Delage M, et al. Extensive, erosive congenital self-healing cell histiocytosis [published online December 22, 2008]. J Eur Acad Dermatol Venereol. 2009;23:835-836.
8. Weiss T, Weber L, Scharffetter-Kochanek K, et al. Solitary cutaneous dendritic cell tumor in a child: role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J Am Acad Dermatol. 2005;53:838-844.
9. Csire M, Mikala G, Jákó J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with Langerhans cell histiocytosis [published online July 3, 2007]. Pathol Oncol Res. 2007;13:157-160.
10. Zaenglein AL, Steele MA, Kamino H, et al. Congenital self-healing reticulohistiocytosis with eye involvement. Pediatr Dermatol. 2001;18:135-137.
11. Chunharas A, Pabunruang W, Hongeng S. Congenital self-healing Langerhans cell histiocytosis with pulmonary involvement: spontaneous regression. J Med Assoc Thai. 2002;85(suppl 4):S1309-S1313.
12. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802-807.
13. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-295.
14. Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax: a potential diagnostic pitfall [published online June 27, 2009]. Virchows Arch. 2009;455:187-190.
15. Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature [published online October 10, 2003]. Eur Arch Otorhinolaryngol. 2004;261:326-330.
16. Noonan V, Kabani S, Alibhai K. Langerhans cell histiocytosis (eosinophilic granuloma). J Mass Dent Soc. 2011;60:35.
17. Podbielski FJ, Worley TA, Korn JM, et al. Eosinophilic granuloma of the lung and rib. Asian Cardiovasc Thorac Ann. 2009;17:194-195.
18. Rosso DA, Ripoli MF, Roy A, et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480-483.
1. Hussein MR. Skin-limited Langerhans’ cell histiocytosis in children. Cancer Invest. 2009;27:504-511.
2. Nakahigashi K, Ohta M, Sakai R, et al. Late-onset self-healing reticulohistiocytosis: pediatric case of Hashimoto-Pritzker type Langerhans cell histiocytosis. J Dermatol. 2007;34:205-209.
3. Pant C, Madonia P, Bahna SL, et al. Langerhans cell histiocytosis, a case of Letterer Siwe disease. J La State Med Soc. 2009;161:211-212.
4. Ferreira LM, Emerich PS, Diniz LM, et al. Langerhans cell histiocytosis: Letterer-Siwe disease–the importance of dermatological diagnosis in two cases [in Portuguese]. An Bras Dermatol. 2009;84:405-409.
5. Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. an unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol. 1973;107:263-270.
6. Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol. 2007;56:290-294.
7. Le Bidre E, Lorette G, Delage M, et al. Extensive, erosive congenital self-healing cell histiocytosis [published online December 22, 2008]. J Eur Acad Dermatol Venereol. 2009;23:835-836.
8. Weiss T, Weber L, Scharffetter-Kochanek K, et al. Solitary cutaneous dendritic cell tumor in a child: role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J Am Acad Dermatol. 2005;53:838-844.
9. Csire M, Mikala G, Jákó J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with Langerhans cell histiocytosis [published online July 3, 2007]. Pathol Oncol Res. 2007;13:157-160.
10. Zaenglein AL, Steele MA, Kamino H, et al. Congenital self-healing reticulohistiocytosis with eye involvement. Pediatr Dermatol. 2001;18:135-137.
11. Chunharas A, Pabunruang W, Hongeng S. Congenital self-healing Langerhans cell histiocytosis with pulmonary involvement: spontaneous regression. J Med Assoc Thai. 2002;85(suppl 4):S1309-S1313.
12. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802-807.
13. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-295.
14. Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax: a potential diagnostic pitfall [published online June 27, 2009]. Virchows Arch. 2009;455:187-190.
15. Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature [published online October 10, 2003]. Eur Arch Otorhinolaryngol. 2004;261:326-330.
16. Noonan V, Kabani S, Alibhai K. Langerhans cell histiocytosis (eosinophilic granuloma). J Mass Dent Soc. 2011;60:35.
17. Podbielski FJ, Worley TA, Korn JM, et al. Eosinophilic granuloma of the lung and rib. Asian Cardiovasc Thorac Ann. 2009;17:194-195.
18. Rosso DA, Ripoli MF, Roy A, et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480-483.
Practice Points
- Langerhans cell histiocytosis (LCH) is believed to occur in 1:200,000 children and tends to be underdiagnosed, as some patients may have no symptoms while others have symptoms that are misdiagnosed as other conditions.
- Patients with LCH usually should have long-term follow-up care to detect progression or complications of the disease or treatment.
Recurrent Abdominal Pain and Bowel Edema in a Middle-Aged Woman
A 34-year-old African American woman presented to the emergency department (ED) after several hours of sharp lower abdominal pain and cramping followed by nausea and vomiting. The pain initially began in the periumbilical region and migrated to the bilateral lower quadrants. The patient reported no fevers, chills, diarrhea, hematemesis, or hematochezia associated with these symptoms. She also reported no unusual food exposures or sick contacts.
The patient’s medical history was notable only for hypertension; her surgical history included 2 cesarean section births several years prior to presentation. Her father was diagnosed with stomach cancer in his 40s. The patient’s only medications were an oral contraceptive, lisinopril, and an antihistamine taken as needed for seasonal allergies. She had no history of tobacco, alcohol, or illicit drug use and no known drug allergies.
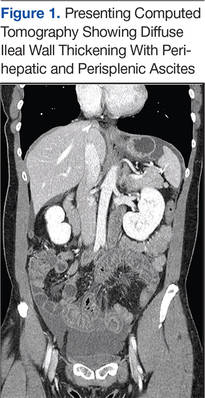
While in the ED, the patient’s physical exam revealed mild tachycardia (104 bpm on arrival, which improved with fluid resuscitation) and diffuse abdominal tenderness. Laboratory evaluation revealed a mild leukocytosis (10.7 x 103/L) but normal liver-associated enzymes, lipase, and urinalysis. A computed tomography (CT) scan of the abdomen and pelvis with oral and IV contrast revealed diffuse ileal wall thickening with significant perihepatic and perisplenic ascites with pelvic free fluid suspicious for an inflammatory vs infectious enteritis (Figure 1).
The patient was treated supportively with IV fluids, antiemetics, and pain medication. Her symptoms generally improved over several days, though she did develop loose stools that prompted infectious stool studies, which were negative for typical pathogens. Follow-up laboratory testing revealed resolution of her leukocytosis.
About 2 weeks later, the patient had another acute attack of abdominal pain, again associated with nausea and vomiting.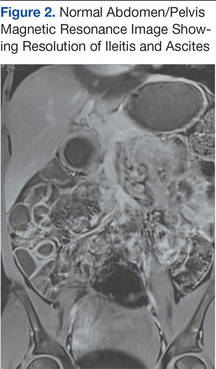

Six weeks after her initial presentation, the patient presented to the ED for the third time with the same symptoms. A CT scan again displayed diffuse ileal wall thickening with significant ascites; slightly worse than the image from initial presentation (Figure 3).
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Diagnosis
This patient presented with recurrent episodes of diffuse small bowel wall thickening and ascites associated with diffuse abdominal pain, nausea, and vomiting. Her symptoms resolved spontaneously, correlating with normalization of bowel wall appearance on imaging studies. Initially, the patient’s symptoms were most concerning for infectious or inflammatory enteritis. Infection became lower on the differential as the patient’s symptoms continued to recur and then resolve spontaneously without antiviral or antibiotic treatment. She also had no fevers, and stools samples were negative for infectious causes of her symptoms.
Inflammatory bowel disease (IBD) was considered, but direct visualization of the small bowel and colonic mucosa was unremarkable. In addition, the sporadic nature of her symptoms did not fit the typical IBD presentation. The patient had no risk factors or history that would suggest ischemic disease, vasculitis, or radiation-induced enteritis. Hereditary angioedema and acquired C1 esterase deficiency were considered given the intermittent nature and characteristic quality of her symptoms. However, serum C4 and C1 esterase inhibitor levels returned within normal limits when measured during these episodes. Finally, visceral angioedema was considered.
Visceral angioedema may be related to medications and is specifically associated with angiotensin-converting-enzyme (ACE) inhibitors as well as β-lactams and high doses of nonsteroidal anti-inflammatory drugs (NSAIDs). Given the characteristic presentation with no other inciting cause, the patient’s lisinopril was felt to be the causative agent and was discontinued. Her symptoms resolved completely and never returned. The patient’s final diagnosis was ACE inhibitor-induced visceral angioedema.
About This Condition
Angiotensin-converting enzyme inhibitors were first introduced in the early 1980s and have been prescribed more frequently as the indications for their use have increased. Some estimate that ACE inhibitors are used by more than 40 million people worldwide.1 Angioedema has been reported to occur in 0.1% to 0.2% of patients taking ACE inhibitorsand accounts for 20% to 30% of all angioedema cases presenting to EDs.2,3 However, ACE inhibitors recently have been recognized as a rare cause of angioedema of the gastrointestinal tract. One of the largest literature reviews on ACE inhibitor-induced gastrointestinal angioedema describes only 27 cases.3
Prevalence seems to be highest among middle-aged or older women, particularly among African Americans.3 The interval between medication initiation and onset of symptoms can vary, ranging from 24 hours to 9 years.3,4 In many cases, lack of recognition of this condition early in the disease course led to costly and invasive procedures, such as abdominal laparotomy, before reaching a diagnosis. Other literature reviews report similar patient characteristics and initial disease manifestations: female predominance, often middle-aged, presenting with abdominal pain and emesis associated with bowel wall thickening and ascites on CT.4,5 Additionally, in the majority of cases, visceral angioedema occurred in the absence of oropharyngeal angioedema. Unlike allergic angioedema or NSAID-induced angioedema, ACE inhibitor-induced angioedema is not associated with urticaria.6
The exact pathway of ACE inhibitor-induced angioedema is not completely understood but is thought to be bradykinin mediated. Angiotensin-converting enzyme inhibitors decrease the degradation of bradykinin, which ultimately leads to an increase in vascular permeability and results in an increased plasma extravasation into the interstitial space of subcutaneous or submucosal tissue.1 However, many experts believe that the exclusive role of bradykinin is unlikely. Some suggest that patients with ACE inhibitor-induced angioedema are more likely to have decreased levels or defects in other enzymes such as carboxypeptidase N and aminopeptidase P, which are involved in the breakdown of bradykinin and its metabolites.6 Given the female predominance in this patient population, it also seems reasonable to consider the role of estrogens in the pathogenesis of this disease, although none have been identified to the knowledge of the authors.
Treatment of ACE inhibitor-induced angioedema is largely supportive following discontinuation of the offending medication. There have been case reports of infrequent, mild, recurrent episodes of angioedema, even after ACE inhibitor discontinuation, so these should be anticipated.7
Conclusions
Angiotensin-converting enzyme inhibitor-induced gastrointestinal angioedema is a rare condition. It generally presents as recurrent abdominal pain and nausea with CT findings of intestinal edema and ascites. It is more common among the middle-aged, women, and minorities. ACE inhibitor-induced angioedema should be kept on the differential for patients with the aforementioned characteristics, especially if infection, inflammatory bowel disease, ischemic disease, or vasculitis is deemed unlikely. Identifying this condition early can save patients from unnecessary hospitalizations, physical and emotional discomfort, and further health care costs.
1. Campo P, Fernandez TD, Canto G, Mayorga C. Angioedema induced by angiotensin-converting enzyme inhibitor. Curr Opin Allergy Clin Immunol. 2013;13(4):337-344.
2. Chase MP, Fiarman GS, Scholz FJ, MacDermott RP. Angioedema of the small bowel due to an angiotensin-converting enzyme inhibitor. J Clin Gastroenterol. 2000;31(3):254-257.
3. Benson BC, Smith C, Laczek JT. Angiotensin converting enzyme inhibitor-induced gastrointestinal angioedema: a case series and literature review. J Clin Gastroenterol. 2013;47(10):844-849.
4. Schmidt TD, McGrath KM. Angiotensin-converting enzyme inhibitor angioedema of the intestine: a case report and review of the literature. Am J Med Sci. 2002;324(2):106-108.
5. Scheirey CD, Scholz FJ, Shortsleeve MJ, Katz DS. Angiotensin-converting enzyme inhibitor-induced small-bowel angioedema: clinical and imaging findings in 20 patients. AJR Am R Roentgenol. 2011;197(2):393-398.
6. Inomata N. Recent advances in drug-induced angioedema. Allergol Int. 2012;61(4):545-557.
7. Cicardi MC, Zingale LC, Bergamaschini L, Agostoni A. Angioedema associated with angiotensin-converting enzyme inhibitor use: outcome after switching to a different treatment. Arch Intern Med. 2004;164(8):910-913.
A 34-year-old African American woman presented to the emergency department (ED) after several hours of sharp lower abdominal pain and cramping followed by nausea and vomiting. The pain initially began in the periumbilical region and migrated to the bilateral lower quadrants. The patient reported no fevers, chills, diarrhea, hematemesis, or hematochezia associated with these symptoms. She also reported no unusual food exposures or sick contacts.
The patient’s medical history was notable only for hypertension; her surgical history included 2 cesarean section births several years prior to presentation. Her father was diagnosed with stomach cancer in his 40s. The patient’s only medications were an oral contraceptive, lisinopril, and an antihistamine taken as needed for seasonal allergies. She had no history of tobacco, alcohol, or illicit drug use and no known drug allergies.
While in the ED, the patient’s physical exam revealed mild tachycardia (104 bpm on arrival, which improved with fluid resuscitation) and diffuse abdominal tenderness. Laboratory evaluation revealed a mild leukocytosis (10.7 x 103/L) but normal liver-associated enzymes, lipase, and urinalysis. A computed tomography (CT) scan of the abdomen and pelvis with oral and IV contrast revealed diffuse ileal wall thickening with significant perihepatic and perisplenic ascites with pelvic free fluid suspicious for an inflammatory vs infectious enteritis (Figure 1).
The patient was treated supportively with IV fluids, antiemetics, and pain medication. Her symptoms generally improved over several days, though she did develop loose stools that prompted infectious stool studies, which were negative for typical pathogens. Follow-up laboratory testing revealed resolution of her leukocytosis.
About 2 weeks later, the patient had another acute attack of abdominal pain, again associated with nausea and vomiting.
Six weeks after her initial presentation, the patient presented to the ED for the third time with the same symptoms. A CT scan again displayed diffuse ileal wall thickening with significant ascites; slightly worse than the image from initial presentation (Figure 3).
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Diagnosis
This patient presented with recurrent episodes of diffuse small bowel wall thickening and ascites associated with diffuse abdominal pain, nausea, and vomiting. Her symptoms resolved spontaneously, correlating with normalization of bowel wall appearance on imaging studies. Initially, the patient’s symptoms were most concerning for infectious or inflammatory enteritis. Infection became lower on the differential as the patient’s symptoms continued to recur and then resolve spontaneously without antiviral or antibiotic treatment. She also had no fevers, and stools samples were negative for infectious causes of her symptoms.
Inflammatory bowel disease (IBD) was considered, but direct visualization of the small bowel and colonic mucosa was unremarkable. In addition, the sporadic nature of her symptoms did not fit the typical IBD presentation. The patient had no risk factors or history that would suggest ischemic disease, vasculitis, or radiation-induced enteritis. Hereditary angioedema and acquired C1 esterase deficiency were considered given the intermittent nature and characteristic quality of her symptoms. However, serum C4 and C1 esterase inhibitor levels returned within normal limits when measured during these episodes. Finally, visceral angioedema was considered.
Visceral angioedema may be related to medications and is specifically associated with angiotensin-converting-enzyme (ACE) inhibitors as well as β-lactams and high doses of nonsteroidal anti-inflammatory drugs (NSAIDs). Given the characteristic presentation with no other inciting cause, the patient’s lisinopril was felt to be the causative agent and was discontinued. Her symptoms resolved completely and never returned. The patient’s final diagnosis was ACE inhibitor-induced visceral angioedema.
About This Condition
Angiotensin-converting enzyme inhibitors were first introduced in the early 1980s and have been prescribed more frequently as the indications for their use have increased. Some estimate that ACE inhibitors are used by more than 40 million people worldwide.1 Angioedema has been reported to occur in 0.1% to 0.2% of patients taking ACE inhibitorsand accounts for 20% to 30% of all angioedema cases presenting to EDs.2,3 However, ACE inhibitors recently have been recognized as a rare cause of angioedema of the gastrointestinal tract. One of the largest literature reviews on ACE inhibitor-induced gastrointestinal angioedema describes only 27 cases.3
Prevalence seems to be highest among middle-aged or older women, particularly among African Americans.3 The interval between medication initiation and onset of symptoms can vary, ranging from 24 hours to 9 years.3,4 In many cases, lack of recognition of this condition early in the disease course led to costly and invasive procedures, such as abdominal laparotomy, before reaching a diagnosis. Other literature reviews report similar patient characteristics and initial disease manifestations: female predominance, often middle-aged, presenting with abdominal pain and emesis associated with bowel wall thickening and ascites on CT.4,5 Additionally, in the majority of cases, visceral angioedema occurred in the absence of oropharyngeal angioedema. Unlike allergic angioedema or NSAID-induced angioedema, ACE inhibitor-induced angioedema is not associated with urticaria.6
The exact pathway of ACE inhibitor-induced angioedema is not completely understood but is thought to be bradykinin mediated. Angiotensin-converting enzyme inhibitors decrease the degradation of bradykinin, which ultimately leads to an increase in vascular permeability and results in an increased plasma extravasation into the interstitial space of subcutaneous or submucosal tissue.1 However, many experts believe that the exclusive role of bradykinin is unlikely. Some suggest that patients with ACE inhibitor-induced angioedema are more likely to have decreased levels or defects in other enzymes such as carboxypeptidase N and aminopeptidase P, which are involved in the breakdown of bradykinin and its metabolites.6 Given the female predominance in this patient population, it also seems reasonable to consider the role of estrogens in the pathogenesis of this disease, although none have been identified to the knowledge of the authors.
Treatment of ACE inhibitor-induced angioedema is largely supportive following discontinuation of the offending medication. There have been case reports of infrequent, mild, recurrent episodes of angioedema, even after ACE inhibitor discontinuation, so these should be anticipated.7
Conclusions
Angiotensin-converting enzyme inhibitor-induced gastrointestinal angioedema is a rare condition. It generally presents as recurrent abdominal pain and nausea with CT findings of intestinal edema and ascites. It is more common among the middle-aged, women, and minorities. ACE inhibitor-induced angioedema should be kept on the differential for patients with the aforementioned characteristics, especially if infection, inflammatory bowel disease, ischemic disease, or vasculitis is deemed unlikely. Identifying this condition early can save patients from unnecessary hospitalizations, physical and emotional discomfort, and further health care costs.
A 34-year-old African American woman presented to the emergency department (ED) after several hours of sharp lower abdominal pain and cramping followed by nausea and vomiting. The pain initially began in the periumbilical region and migrated to the bilateral lower quadrants. The patient reported no fevers, chills, diarrhea, hematemesis, or hematochezia associated with these symptoms. She also reported no unusual food exposures or sick contacts.
The patient’s medical history was notable only for hypertension; her surgical history included 2 cesarean section births several years prior to presentation. Her father was diagnosed with stomach cancer in his 40s. The patient’s only medications were an oral contraceptive, lisinopril, and an antihistamine taken as needed for seasonal allergies. She had no history of tobacco, alcohol, or illicit drug use and no known drug allergies.
While in the ED, the patient’s physical exam revealed mild tachycardia (104 bpm on arrival, which improved with fluid resuscitation) and diffuse abdominal tenderness. Laboratory evaluation revealed a mild leukocytosis (10.7 x 103/L) but normal liver-associated enzymes, lipase, and urinalysis. A computed tomography (CT) scan of the abdomen and pelvis with oral and IV contrast revealed diffuse ileal wall thickening with significant perihepatic and perisplenic ascites with pelvic free fluid suspicious for an inflammatory vs infectious enteritis (Figure 1).
The patient was treated supportively with IV fluids, antiemetics, and pain medication. Her symptoms generally improved over several days, though she did develop loose stools that prompted infectious stool studies, which were negative for typical pathogens. Follow-up laboratory testing revealed resolution of her leukocytosis.
About 2 weeks later, the patient had another acute attack of abdominal pain, again associated with nausea and vomiting.
Six weeks after her initial presentation, the patient presented to the ED for the third time with the same symptoms. A CT scan again displayed diffuse ileal wall thickening with significant ascites; slightly worse than the image from initial presentation (Figure 3).
- What is your diagnosis?
- How would you treat this patient?
[Click through to the next page to see the answer.]
Diagnosis
This patient presented with recurrent episodes of diffuse small bowel wall thickening and ascites associated with diffuse abdominal pain, nausea, and vomiting. Her symptoms resolved spontaneously, correlating with normalization of bowel wall appearance on imaging studies. Initially, the patient’s symptoms were most concerning for infectious or inflammatory enteritis. Infection became lower on the differential as the patient’s symptoms continued to recur and then resolve spontaneously without antiviral or antibiotic treatment. She also had no fevers, and stools samples were negative for infectious causes of her symptoms.
Inflammatory bowel disease (IBD) was considered, but direct visualization of the small bowel and colonic mucosa was unremarkable. In addition, the sporadic nature of her symptoms did not fit the typical IBD presentation. The patient had no risk factors or history that would suggest ischemic disease, vasculitis, or radiation-induced enteritis. Hereditary angioedema and acquired C1 esterase deficiency were considered given the intermittent nature and characteristic quality of her symptoms. However, serum C4 and C1 esterase inhibitor levels returned within normal limits when measured during these episodes. Finally, visceral angioedema was considered.
Visceral angioedema may be related to medications and is specifically associated with angiotensin-converting-enzyme (ACE) inhibitors as well as β-lactams and high doses of nonsteroidal anti-inflammatory drugs (NSAIDs). Given the characteristic presentation with no other inciting cause, the patient’s lisinopril was felt to be the causative agent and was discontinued. Her symptoms resolved completely and never returned. The patient’s final diagnosis was ACE inhibitor-induced visceral angioedema.
About This Condition
Angiotensin-converting enzyme inhibitors were first introduced in the early 1980s and have been prescribed more frequently as the indications for their use have increased. Some estimate that ACE inhibitors are used by more than 40 million people worldwide.1 Angioedema has been reported to occur in 0.1% to 0.2% of patients taking ACE inhibitorsand accounts for 20% to 30% of all angioedema cases presenting to EDs.2,3 However, ACE inhibitors recently have been recognized as a rare cause of angioedema of the gastrointestinal tract. One of the largest literature reviews on ACE inhibitor-induced gastrointestinal angioedema describes only 27 cases.3
Prevalence seems to be highest among middle-aged or older women, particularly among African Americans.3 The interval between medication initiation and onset of symptoms can vary, ranging from 24 hours to 9 years.3,4 In many cases, lack of recognition of this condition early in the disease course led to costly and invasive procedures, such as abdominal laparotomy, before reaching a diagnosis. Other literature reviews report similar patient characteristics and initial disease manifestations: female predominance, often middle-aged, presenting with abdominal pain and emesis associated with bowel wall thickening and ascites on CT.4,5 Additionally, in the majority of cases, visceral angioedema occurred in the absence of oropharyngeal angioedema. Unlike allergic angioedema or NSAID-induced angioedema, ACE inhibitor-induced angioedema is not associated with urticaria.6
The exact pathway of ACE inhibitor-induced angioedema is not completely understood but is thought to be bradykinin mediated. Angiotensin-converting enzyme inhibitors decrease the degradation of bradykinin, which ultimately leads to an increase in vascular permeability and results in an increased plasma extravasation into the interstitial space of subcutaneous or submucosal tissue.1 However, many experts believe that the exclusive role of bradykinin is unlikely. Some suggest that patients with ACE inhibitor-induced angioedema are more likely to have decreased levels or defects in other enzymes such as carboxypeptidase N and aminopeptidase P, which are involved in the breakdown of bradykinin and its metabolites.6 Given the female predominance in this patient population, it also seems reasonable to consider the role of estrogens in the pathogenesis of this disease, although none have been identified to the knowledge of the authors.
Treatment of ACE inhibitor-induced angioedema is largely supportive following discontinuation of the offending medication. There have been case reports of infrequent, mild, recurrent episodes of angioedema, even after ACE inhibitor discontinuation, so these should be anticipated.7
Conclusions
Angiotensin-converting enzyme inhibitor-induced gastrointestinal angioedema is a rare condition. It generally presents as recurrent abdominal pain and nausea with CT findings of intestinal edema and ascites. It is more common among the middle-aged, women, and minorities. ACE inhibitor-induced angioedema should be kept on the differential for patients with the aforementioned characteristics, especially if infection, inflammatory bowel disease, ischemic disease, or vasculitis is deemed unlikely. Identifying this condition early can save patients from unnecessary hospitalizations, physical and emotional discomfort, and further health care costs.
1. Campo P, Fernandez TD, Canto G, Mayorga C. Angioedema induced by angiotensin-converting enzyme inhibitor. Curr Opin Allergy Clin Immunol. 2013;13(4):337-344.
2. Chase MP, Fiarman GS, Scholz FJ, MacDermott RP. Angioedema of the small bowel due to an angiotensin-converting enzyme inhibitor. J Clin Gastroenterol. 2000;31(3):254-257.
3. Benson BC, Smith C, Laczek JT. Angiotensin converting enzyme inhibitor-induced gastrointestinal angioedema: a case series and literature review. J Clin Gastroenterol. 2013;47(10):844-849.
4. Schmidt TD, McGrath KM. Angiotensin-converting enzyme inhibitor angioedema of the intestine: a case report and review of the literature. Am J Med Sci. 2002;324(2):106-108.
5. Scheirey CD, Scholz FJ, Shortsleeve MJ, Katz DS. Angiotensin-converting enzyme inhibitor-induced small-bowel angioedema: clinical and imaging findings in 20 patients. AJR Am R Roentgenol. 2011;197(2):393-398.
6. Inomata N. Recent advances in drug-induced angioedema. Allergol Int. 2012;61(4):545-557.
7. Cicardi MC, Zingale LC, Bergamaschini L, Agostoni A. Angioedema associated with angiotensin-converting enzyme inhibitor use: outcome after switching to a different treatment. Arch Intern Med. 2004;164(8):910-913.
1. Campo P, Fernandez TD, Canto G, Mayorga C. Angioedema induced by angiotensin-converting enzyme inhibitor. Curr Opin Allergy Clin Immunol. 2013;13(4):337-344.
2. Chase MP, Fiarman GS, Scholz FJ, MacDermott RP. Angioedema of the small bowel due to an angiotensin-converting enzyme inhibitor. J Clin Gastroenterol. 2000;31(3):254-257.
3. Benson BC, Smith C, Laczek JT. Angiotensin converting enzyme inhibitor-induced gastrointestinal angioedema: a case series and literature review. J Clin Gastroenterol. 2013;47(10):844-849.
4. Schmidt TD, McGrath KM. Angiotensin-converting enzyme inhibitor angioedema of the intestine: a case report and review of the literature. Am J Med Sci. 2002;324(2):106-108.
5. Scheirey CD, Scholz FJ, Shortsleeve MJ, Katz DS. Angiotensin-converting enzyme inhibitor-induced small-bowel angioedema: clinical and imaging findings in 20 patients. AJR Am R Roentgenol. 2011;197(2):393-398.
6. Inomata N. Recent advances in drug-induced angioedema. Allergol Int. 2012;61(4):545-557.
7. Cicardi MC, Zingale LC, Bergamaschini L, Agostoni A. Angioedema associated with angiotensin-converting enzyme inhibitor use: outcome after switching to a different treatment. Arch Intern Med. 2004;164(8):910-913.
Possible Simeprevir/Sofosbuvir-Induced Hepatic Decompensation With Acute Kidney Failure
The emergence of hepatitis C (HCV) treatment regimens in the past 5 years has resulted in a major paradigm shift in the management of those infected with this virus. The 2011 approval of boceprevir and telaprevir was associated with a higher virologic response (50%-75%) and a shorter length of therapy depending on the patient population. Despite these gains, first generation direct-acting antivirals (DAAs) required multiple doses, had a higher pill burden with numerous drug interactions, and adverse effects (AEs). In addition, viral resistance limited the full use of the first generation DAAs for all genotypes.
Sofosbuvir, simeprevir, and ledipasvir-sofosbuvir (second generation DAAs) boast even higher (> 90%) sustained virologic response rates (SVR) and more tolerable AE profiles especially anemia, depression, and gastrointestinal symptoms compared with the first generation DAAs. At the time of treatment for this case study, sofosbuvir/ledipasvir was not commercially available. Sofosbuvir in combination with simeprevir with or without ribavirin was one of the preferred treatment options for chronic HCV.1
Unlike the first generation DAAs, which have been associated with a decline in renal function compared with conventional pegylated interferon and ribavirin, sofosbuvir is extensively renally eliminated by glomerular filtration and active tubular secretion as the metabolite GS-331007. On the other hand, simeprevir is hepatically metabolized.
A PubMed literature search for reports of “simeprevir-induced” or “sofosbuvir-induced with hepatic, renal failure, acute kidney injury” yielded only 1 published case of hepatic decompensation likely related to simeprevir, but no case report of simeprevir and sofosbuvir associated with hepatic decompensation and acute kidney injury.4 In this article, the authors describe a case of hepatic decompensation and acute kidney injury caused by simeprevir/sofosbuvir initiation for chronic HCV that required intensive care and dialysis.
Case Report
The patient was a 62-year-old African American man with chronic HCV, genotype 1b, TT IL28B, and 4,980,000 IU baseline viral load. He was treatment naïve with biopsy proven compensated cirrhosis, and Child-Turcotte-Pugh class A with a pretreatment model for end-stage liver disease score of 12. His past medical history included hypertension, chronic kidney disease (CKD) (baseline serum creatinine [SCr] 1.4-1.8 mg/dL), benign prostatic hypertrophy, depression, obesity (30.6 body mass index, 246 lb), and psoriasis. In addition, the patient was on the following maintenance medications: allopurinol, bupropion, diltiazem, sustained-release and immediate-release morphine, sennosides, and terazosin.
In September 2014, the patient was diagnosed with biopsy-confirmed hepatocellular carcinoma (HCC) Barcelona clinic liver cancer stage B T3aN0M0 stage III. He was considered for transarterial chemoembolization (TACE), but treatment was withheld due to subsequent increase in liver function tests (LFTs) with total bilirubin (TB) 2.9 mg/dL, direct bilirubin (DB) 1.8 mg/dL, aspartate aminotransferase test (AST) 130 U/L, and alanine aminotransferase test (ALT) 188 U/L (baseline: TB 1.1 mg/dL, AST 69 U/L, and ALT 76 U/L). These results were thought to be the result of worsening hepatic function from untreated HCV, therefore, treatment was initiated.
The patient was started on simeprevir 150 mg orally daily and sofosbuvir 400 mg orally daily with an estimated baseline creatinine clearance of 67 mL/min per Cockcroft-Gault equation.5 Two days after therapy initiation, the patient presented to the emergency department with the following symptoms: hiccups, nausea, vomiting, and abdominal pain. Laboratory results showed 10.85 mg/dL SCr and 91 mg/dL blood urea nitrogen (BUN), TB increased to 14.6 mg/dL with AST of 325 U/L and ALT 277 U/L. The patient reported no use of acetaminophen, alcohol, nonsteroidal anti-inflammatory drugs, or other nephrotoxic agents.
Upon admission, the patient was diagnosed with drug-induced hepatitis and acute kidney injury (AKI). Simeprevir/sofosbuvir was discontinued along with allopurinol, bupropion, lisinopril, and morphine. An abdominal ultrasound was negative for obstructive uropathy. The patient did not respond to fluid boluses. A nephrologist was consulted, and dialysis was initiated. The patient underwent dialysis for 3 days and his LFTs and SCr levels started trending downward (Figures 1 to 5).
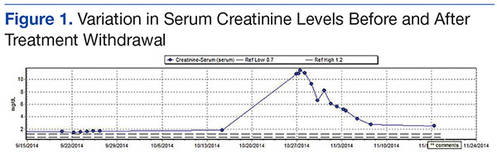
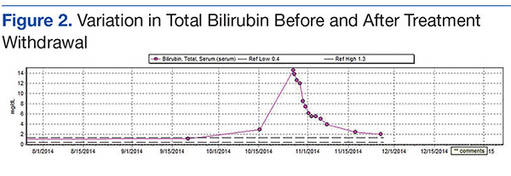
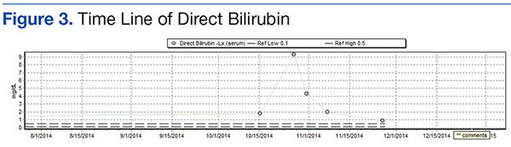
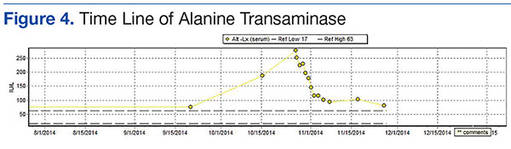
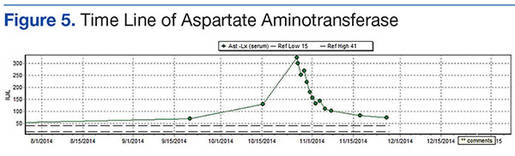
The patient was discharged after 8 days. After 3 weeks, the SCr decreased to 2.29 mg/dL, BUN was 26 mg/dL, TB was 2 mg/dL, DB was 0.9 mg/dL, AST was 73 U/L, and ALT was 81 U/L. Weekly laboratory values continued to improve following discharge but did not return to baseline levels. The patient remained off HCV treatment.
Discussion
The patient had baseline CKD with SCr > 1.5 mg/dL; however, the significant decline in renal function and worsening hepatic function were thought to be the result of external factors. Although hepatorenal syndrome was considered, the authors suspected that the AKI and hepatic decompensation were related to simeprevir/sofosbuvir regimens due to their presumed relationship and probability analysis. Osinusi and colleagues noted a decline in renal function in a patient who received ledipasvir/sofosbuvir for 6 weeks in an open-label pilot study.6 Stine and colleagues also reported on cases of simeprevir-related hepatic decompensation.4
In this case, the authors employed the Naranjo algorithm adverse drug reaction probability scale to assess whether there was a causal relationship between this event and initiation of simeprevir/sofosbuvir regimen.7 The Naranjo score was 4, indicating a possible link between simeprevir/sofosbuvir initiation and hepatic decompensation and AKI. This case may be the first postmarketing report of significant hepatic decompensation and AKI related to simeprevir/sofosbuvir.
Unlike simeprevir, which undergoes extensive oxidative metabolism by CYP3A in the liver and has negligible renal clearance with < 1% of the dose recovered in the urine, sofosbuvir is extensively metabolized by the kidneys with an active metabolite, GS-331007, and about 80% of the dose is recovered in urine (78% as GS-331007; 3.5% as sofosbuvir).8,9 The potential for drug-drug interaction also was assessed because simeprevir is extensively metabolized by the hepatic cytochrome CYP34 system and possibly CYP2C8 and CYP2C19. Clinically significant interactions could have occurred with diltiazem and morphine, because the coadministration of these medications along with simeprevir, an inhibitor of P-glycoprotein (P-gp), and intestinal CYP3A4, may result in increased diltiazem and morphine plasma concentrations.
Of note, because sofosbuvir is a substrate of P-gp, it may have its serum concentration increased by simeprevir. Inducers and inhibitors of P-gp may alter the plasma concentration of sofosbuvir. The major metabolite, GS-331007, is not a substrate of P-gp. Drugs that induce P-gp may reduce the therapeutic effect of sofosbuvir; however, the FDA-labeling suggests that inhibitors of P-gp may be coadministered with sofosbuvir.
According to simeprevir prescribing information, drug interaction studies have demonstrated that moderate CYP3A4 inhibitors, such as diltiazem (although coadministration have not been studied), increased the maximum serum concentration (Cmax), minumum serum concentration (Cmin), and AUC of simeprevir.7 As a result, concurrent use of simeprevir with a moderate CYP3A4 inhibitors is not recommended. Morphine and simeprevir interaction also is possible via the P-gp inhibition of simeprevir. Morphine concentration may have increased and metabolites may have accumulated, leading to urinary retention and elevated creatinine. In addition, decreased oral intake and subsequent nausea/vomiting may have compounded the renal insult.
Conclusion
Given that updated HCV treatment guidelines include simeprevir/sofosbuvir as an alternative treatment option, clinicians should be aware of hepatic decompensation with markedly elevated bilirubin and AKI during simeprevir and sofosbuvir treatment. Careful consideration is needed prior to the initiation of simeprevir/sofosbuvir, particularly in patients with advanced liver disease or known HCC and baseline renal impairment.
1. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C: Initial Treatment of HCV. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America Website. http://www.hcvguidelines.org. Accessed February 8, 2016.
2. Mauss S, Hueppe D, Alshuth U. Renal impairment is frequent in chronic hepatitis C patient under triple therapy with telaprevir or boceprevir. Hepatology. 2014;59(1):46-48.
3. Virlogeux V, Pradat P, Bailly F, et al. Boceprevir and telaprevir-based triple therapy for chronic hepatitis C: virolgical efficacy and impact on kidney function and model for end-stage liver disease score. J Viral Hepat. 2014;21(9):e98-e107.
4. Stine JG, Intagliata N, Shah L, et al. Hepatic decompensation likely attributable to simeprevir in patients with advanced cirrhosis. Dig Dis Sci. 2015;60(4):1031-1035.
5. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
6. Osinusi A, Kohli A, Marti MM, et al. Re-treamtent of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634-638.
7. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
8. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2014.
9. Sovaldi (sofosbuvir) [package insert]. Foster City, CA: Gilead Sciences, Inc; 2014.
The emergence of hepatitis C (HCV) treatment regimens in the past 5 years has resulted in a major paradigm shift in the management of those infected with this virus. The 2011 approval of boceprevir and telaprevir was associated with a higher virologic response (50%-75%) and a shorter length of therapy depending on the patient population. Despite these gains, first generation direct-acting antivirals (DAAs) required multiple doses, had a higher pill burden with numerous drug interactions, and adverse effects (AEs). In addition, viral resistance limited the full use of the first generation DAAs for all genotypes.
Sofosbuvir, simeprevir, and ledipasvir-sofosbuvir (second generation DAAs) boast even higher (> 90%) sustained virologic response rates (SVR) and more tolerable AE profiles especially anemia, depression, and gastrointestinal symptoms compared with the first generation DAAs. At the time of treatment for this case study, sofosbuvir/ledipasvir was not commercially available. Sofosbuvir in combination with simeprevir with or without ribavirin was one of the preferred treatment options for chronic HCV.1
Unlike the first generation DAAs, which have been associated with a decline in renal function compared with conventional pegylated interferon and ribavirin, sofosbuvir is extensively renally eliminated by glomerular filtration and active tubular secretion as the metabolite GS-331007. On the other hand, simeprevir is hepatically metabolized.
A PubMed literature search for reports of “simeprevir-induced” or “sofosbuvir-induced with hepatic, renal failure, acute kidney injury” yielded only 1 published case of hepatic decompensation likely related to simeprevir, but no case report of simeprevir and sofosbuvir associated with hepatic decompensation and acute kidney injury.4 In this article, the authors describe a case of hepatic decompensation and acute kidney injury caused by simeprevir/sofosbuvir initiation for chronic HCV that required intensive care and dialysis.
Case Report
The patient was a 62-year-old African American man with chronic HCV, genotype 1b, TT IL28B, and 4,980,000 IU baseline viral load. He was treatment naïve with biopsy proven compensated cirrhosis, and Child-Turcotte-Pugh class A with a pretreatment model for end-stage liver disease score of 12. His past medical history included hypertension, chronic kidney disease (CKD) (baseline serum creatinine [SCr] 1.4-1.8 mg/dL), benign prostatic hypertrophy, depression, obesity (30.6 body mass index, 246 lb), and psoriasis. In addition, the patient was on the following maintenance medications: allopurinol, bupropion, diltiazem, sustained-release and immediate-release morphine, sennosides, and terazosin.
In September 2014, the patient was diagnosed with biopsy-confirmed hepatocellular carcinoma (HCC) Barcelona clinic liver cancer stage B T3aN0M0 stage III. He was considered for transarterial chemoembolization (TACE), but treatment was withheld due to subsequent increase in liver function tests (LFTs) with total bilirubin (TB) 2.9 mg/dL, direct bilirubin (DB) 1.8 mg/dL, aspartate aminotransferase test (AST) 130 U/L, and alanine aminotransferase test (ALT) 188 U/L (baseline: TB 1.1 mg/dL, AST 69 U/L, and ALT 76 U/L). These results were thought to be the result of worsening hepatic function from untreated HCV, therefore, treatment was initiated.
The patient was started on simeprevir 150 mg orally daily and sofosbuvir 400 mg orally daily with an estimated baseline creatinine clearance of 67 mL/min per Cockcroft-Gault equation.5 Two days after therapy initiation, the patient presented to the emergency department with the following symptoms: hiccups, nausea, vomiting, and abdominal pain. Laboratory results showed 10.85 mg/dL SCr and 91 mg/dL blood urea nitrogen (BUN), TB increased to 14.6 mg/dL with AST of 325 U/L and ALT 277 U/L. The patient reported no use of acetaminophen, alcohol, nonsteroidal anti-inflammatory drugs, or other nephrotoxic agents.
Upon admission, the patient was diagnosed with drug-induced hepatitis and acute kidney injury (AKI). Simeprevir/sofosbuvir was discontinued along with allopurinol, bupropion, lisinopril, and morphine. An abdominal ultrasound was negative for obstructive uropathy. The patient did not respond to fluid boluses. A nephrologist was consulted, and dialysis was initiated. The patient underwent dialysis for 3 days and his LFTs and SCr levels started trending downward (Figures 1 to 5).
The patient was discharged after 8 days. After 3 weeks, the SCr decreased to 2.29 mg/dL, BUN was 26 mg/dL, TB was 2 mg/dL, DB was 0.9 mg/dL, AST was 73 U/L, and ALT was 81 U/L. Weekly laboratory values continued to improve following discharge but did not return to baseline levels. The patient remained off HCV treatment.
Discussion
The patient had baseline CKD with SCr > 1.5 mg/dL; however, the significant decline in renal function and worsening hepatic function were thought to be the result of external factors. Although hepatorenal syndrome was considered, the authors suspected that the AKI and hepatic decompensation were related to simeprevir/sofosbuvir regimens due to their presumed relationship and probability analysis. Osinusi and colleagues noted a decline in renal function in a patient who received ledipasvir/sofosbuvir for 6 weeks in an open-label pilot study.6 Stine and colleagues also reported on cases of simeprevir-related hepatic decompensation.4
In this case, the authors employed the Naranjo algorithm adverse drug reaction probability scale to assess whether there was a causal relationship between this event and initiation of simeprevir/sofosbuvir regimen.7 The Naranjo score was 4, indicating a possible link between simeprevir/sofosbuvir initiation and hepatic decompensation and AKI. This case may be the first postmarketing report of significant hepatic decompensation and AKI related to simeprevir/sofosbuvir.
Unlike simeprevir, which undergoes extensive oxidative metabolism by CYP3A in the liver and has negligible renal clearance with < 1% of the dose recovered in the urine, sofosbuvir is extensively metabolized by the kidneys with an active metabolite, GS-331007, and about 80% of the dose is recovered in urine (78% as GS-331007; 3.5% as sofosbuvir).8,9 The potential for drug-drug interaction also was assessed because simeprevir is extensively metabolized by the hepatic cytochrome CYP34 system and possibly CYP2C8 and CYP2C19. Clinically significant interactions could have occurred with diltiazem and morphine, because the coadministration of these medications along with simeprevir, an inhibitor of P-glycoprotein (P-gp), and intestinal CYP3A4, may result in increased diltiazem and morphine plasma concentrations.
Of note, because sofosbuvir is a substrate of P-gp, it may have its serum concentration increased by simeprevir. Inducers and inhibitors of P-gp may alter the plasma concentration of sofosbuvir. The major metabolite, GS-331007, is not a substrate of P-gp. Drugs that induce P-gp may reduce the therapeutic effect of sofosbuvir; however, the FDA-labeling suggests that inhibitors of P-gp may be coadministered with sofosbuvir.
According to simeprevir prescribing information, drug interaction studies have demonstrated that moderate CYP3A4 inhibitors, such as diltiazem (although coadministration have not been studied), increased the maximum serum concentration (Cmax), minumum serum concentration (Cmin), and AUC of simeprevir.7 As a result, concurrent use of simeprevir with a moderate CYP3A4 inhibitors is not recommended. Morphine and simeprevir interaction also is possible via the P-gp inhibition of simeprevir. Morphine concentration may have increased and metabolites may have accumulated, leading to urinary retention and elevated creatinine. In addition, decreased oral intake and subsequent nausea/vomiting may have compounded the renal insult.
Conclusion
Given that updated HCV treatment guidelines include simeprevir/sofosbuvir as an alternative treatment option, clinicians should be aware of hepatic decompensation with markedly elevated bilirubin and AKI during simeprevir and sofosbuvir treatment. Careful consideration is needed prior to the initiation of simeprevir/sofosbuvir, particularly in patients with advanced liver disease or known HCC and baseline renal impairment.
The emergence of hepatitis C (HCV) treatment regimens in the past 5 years has resulted in a major paradigm shift in the management of those infected with this virus. The 2011 approval of boceprevir and telaprevir was associated with a higher virologic response (50%-75%) and a shorter length of therapy depending on the patient population. Despite these gains, first generation direct-acting antivirals (DAAs) required multiple doses, had a higher pill burden with numerous drug interactions, and adverse effects (AEs). In addition, viral resistance limited the full use of the first generation DAAs for all genotypes.
Sofosbuvir, simeprevir, and ledipasvir-sofosbuvir (second generation DAAs) boast even higher (> 90%) sustained virologic response rates (SVR) and more tolerable AE profiles especially anemia, depression, and gastrointestinal symptoms compared with the first generation DAAs. At the time of treatment for this case study, sofosbuvir/ledipasvir was not commercially available. Sofosbuvir in combination with simeprevir with or without ribavirin was one of the preferred treatment options for chronic HCV.1
Unlike the first generation DAAs, which have been associated with a decline in renal function compared with conventional pegylated interferon and ribavirin, sofosbuvir is extensively renally eliminated by glomerular filtration and active tubular secretion as the metabolite GS-331007. On the other hand, simeprevir is hepatically metabolized.
A PubMed literature search for reports of “simeprevir-induced” or “sofosbuvir-induced with hepatic, renal failure, acute kidney injury” yielded only 1 published case of hepatic decompensation likely related to simeprevir, but no case report of simeprevir and sofosbuvir associated with hepatic decompensation and acute kidney injury.4 In this article, the authors describe a case of hepatic decompensation and acute kidney injury caused by simeprevir/sofosbuvir initiation for chronic HCV that required intensive care and dialysis.
Case Report
The patient was a 62-year-old African American man with chronic HCV, genotype 1b, TT IL28B, and 4,980,000 IU baseline viral load. He was treatment naïve with biopsy proven compensated cirrhosis, and Child-Turcotte-Pugh class A with a pretreatment model for end-stage liver disease score of 12. His past medical history included hypertension, chronic kidney disease (CKD) (baseline serum creatinine [SCr] 1.4-1.8 mg/dL), benign prostatic hypertrophy, depression, obesity (30.6 body mass index, 246 lb), and psoriasis. In addition, the patient was on the following maintenance medications: allopurinol, bupropion, diltiazem, sustained-release and immediate-release morphine, sennosides, and terazosin.
In September 2014, the patient was diagnosed with biopsy-confirmed hepatocellular carcinoma (HCC) Barcelona clinic liver cancer stage B T3aN0M0 stage III. He was considered for transarterial chemoembolization (TACE), but treatment was withheld due to subsequent increase in liver function tests (LFTs) with total bilirubin (TB) 2.9 mg/dL, direct bilirubin (DB) 1.8 mg/dL, aspartate aminotransferase test (AST) 130 U/L, and alanine aminotransferase test (ALT) 188 U/L (baseline: TB 1.1 mg/dL, AST 69 U/L, and ALT 76 U/L). These results were thought to be the result of worsening hepatic function from untreated HCV, therefore, treatment was initiated.
The patient was started on simeprevir 150 mg orally daily and sofosbuvir 400 mg orally daily with an estimated baseline creatinine clearance of 67 mL/min per Cockcroft-Gault equation.5 Two days after therapy initiation, the patient presented to the emergency department with the following symptoms: hiccups, nausea, vomiting, and abdominal pain. Laboratory results showed 10.85 mg/dL SCr and 91 mg/dL blood urea nitrogen (BUN), TB increased to 14.6 mg/dL with AST of 325 U/L and ALT 277 U/L. The patient reported no use of acetaminophen, alcohol, nonsteroidal anti-inflammatory drugs, or other nephrotoxic agents.
Upon admission, the patient was diagnosed with drug-induced hepatitis and acute kidney injury (AKI). Simeprevir/sofosbuvir was discontinued along with allopurinol, bupropion, lisinopril, and morphine. An abdominal ultrasound was negative for obstructive uropathy. The patient did not respond to fluid boluses. A nephrologist was consulted, and dialysis was initiated. The patient underwent dialysis for 3 days and his LFTs and SCr levels started trending downward (Figures 1 to 5).
The patient was discharged after 8 days. After 3 weeks, the SCr decreased to 2.29 mg/dL, BUN was 26 mg/dL, TB was 2 mg/dL, DB was 0.9 mg/dL, AST was 73 U/L, and ALT was 81 U/L. Weekly laboratory values continued to improve following discharge but did not return to baseline levels. The patient remained off HCV treatment.
Discussion
The patient had baseline CKD with SCr > 1.5 mg/dL; however, the significant decline in renal function and worsening hepatic function were thought to be the result of external factors. Although hepatorenal syndrome was considered, the authors suspected that the AKI and hepatic decompensation were related to simeprevir/sofosbuvir regimens due to their presumed relationship and probability analysis. Osinusi and colleagues noted a decline in renal function in a patient who received ledipasvir/sofosbuvir for 6 weeks in an open-label pilot study.6 Stine and colleagues also reported on cases of simeprevir-related hepatic decompensation.4
In this case, the authors employed the Naranjo algorithm adverse drug reaction probability scale to assess whether there was a causal relationship between this event and initiation of simeprevir/sofosbuvir regimen.7 The Naranjo score was 4, indicating a possible link between simeprevir/sofosbuvir initiation and hepatic decompensation and AKI. This case may be the first postmarketing report of significant hepatic decompensation and AKI related to simeprevir/sofosbuvir.
Unlike simeprevir, which undergoes extensive oxidative metabolism by CYP3A in the liver and has negligible renal clearance with < 1% of the dose recovered in the urine, sofosbuvir is extensively metabolized by the kidneys with an active metabolite, GS-331007, and about 80% of the dose is recovered in urine (78% as GS-331007; 3.5% as sofosbuvir).8,9 The potential for drug-drug interaction also was assessed because simeprevir is extensively metabolized by the hepatic cytochrome CYP34 system and possibly CYP2C8 and CYP2C19. Clinically significant interactions could have occurred with diltiazem and morphine, because the coadministration of these medications along with simeprevir, an inhibitor of P-glycoprotein (P-gp), and intestinal CYP3A4, may result in increased diltiazem and morphine plasma concentrations.
Of note, because sofosbuvir is a substrate of P-gp, it may have its serum concentration increased by simeprevir. Inducers and inhibitors of P-gp may alter the plasma concentration of sofosbuvir. The major metabolite, GS-331007, is not a substrate of P-gp. Drugs that induce P-gp may reduce the therapeutic effect of sofosbuvir; however, the FDA-labeling suggests that inhibitors of P-gp may be coadministered with sofosbuvir.
According to simeprevir prescribing information, drug interaction studies have demonstrated that moderate CYP3A4 inhibitors, such as diltiazem (although coadministration have not been studied), increased the maximum serum concentration (Cmax), minumum serum concentration (Cmin), and AUC of simeprevir.7 As a result, concurrent use of simeprevir with a moderate CYP3A4 inhibitors is not recommended. Morphine and simeprevir interaction also is possible via the P-gp inhibition of simeprevir. Morphine concentration may have increased and metabolites may have accumulated, leading to urinary retention and elevated creatinine. In addition, decreased oral intake and subsequent nausea/vomiting may have compounded the renal insult.
Conclusion
Given that updated HCV treatment guidelines include simeprevir/sofosbuvir as an alternative treatment option, clinicians should be aware of hepatic decompensation with markedly elevated bilirubin and AKI during simeprevir and sofosbuvir treatment. Careful consideration is needed prior to the initiation of simeprevir/sofosbuvir, particularly in patients with advanced liver disease or known HCC and baseline renal impairment.
1. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C: Initial Treatment of HCV. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America Website. http://www.hcvguidelines.org. Accessed February 8, 2016.
2. Mauss S, Hueppe D, Alshuth U. Renal impairment is frequent in chronic hepatitis C patient under triple therapy with telaprevir or boceprevir. Hepatology. 2014;59(1):46-48.
3. Virlogeux V, Pradat P, Bailly F, et al. Boceprevir and telaprevir-based triple therapy for chronic hepatitis C: virolgical efficacy and impact on kidney function and model for end-stage liver disease score. J Viral Hepat. 2014;21(9):e98-e107.
4. Stine JG, Intagliata N, Shah L, et al. Hepatic decompensation likely attributable to simeprevir in patients with advanced cirrhosis. Dig Dis Sci. 2015;60(4):1031-1035.
5. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
6. Osinusi A, Kohli A, Marti MM, et al. Re-treamtent of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634-638.
7. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
8. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2014.
9. Sovaldi (sofosbuvir) [package insert]. Foster City, CA: Gilead Sciences, Inc; 2014.
1. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C: Initial Treatment of HCV. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America Website. http://www.hcvguidelines.org. Accessed February 8, 2016.
2. Mauss S, Hueppe D, Alshuth U. Renal impairment is frequent in chronic hepatitis C patient under triple therapy with telaprevir or boceprevir. Hepatology. 2014;59(1):46-48.
3. Virlogeux V, Pradat P, Bailly F, et al. Boceprevir and telaprevir-based triple therapy for chronic hepatitis C: virolgical efficacy and impact on kidney function and model for end-stage liver disease score. J Viral Hepat. 2014;21(9):e98-e107.
4. Stine JG, Intagliata N, Shah L, et al. Hepatic decompensation likely attributable to simeprevir in patients with advanced cirrhosis. Dig Dis Sci. 2015;60(4):1031-1035.
5. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
6. Osinusi A, Kohli A, Marti MM, et al. Re-treamtent of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634-638.
7. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
8. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2014.
9. Sovaldi (sofosbuvir) [package insert]. Foster City, CA: Gilead Sciences, Inc; 2014.
Onychomatricoma: A Rare Case of Unguioblastic Fibroma of the Fingernail Associated With Trauma
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
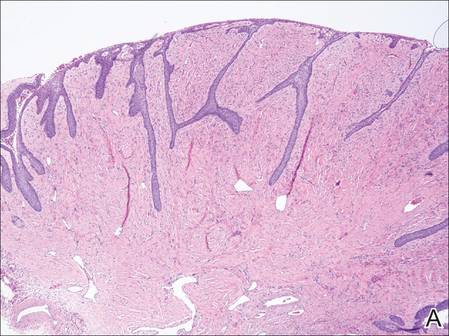
| 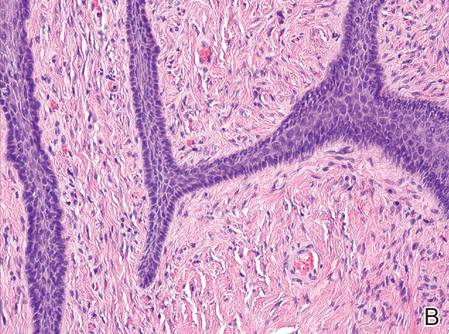
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
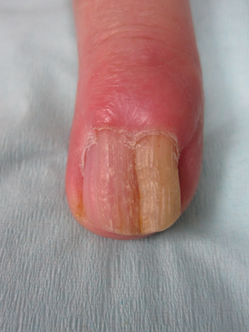
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
|
|
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
|
|
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
Practice Points
- Onychomatricoma is a rare benign neoplasm of the nail matrix that actively produces a nail plate.
- Onychomatricoma should be in the differential diagnosis of a thickened discolored nail plate with transverse overcurvature.
- Onychomatricoma has been associated with onychomycosis and trauma to the nail apparatus.
Angioedema Following tPA Administration for Acute Cerebrovascular Accident
The use of thrombolytic medications for the treatment of acute ischemic cerebral infarctions has dynamically altered stroke care. However, there are both major and minor side effects associated with its use—most notably major bleeding, which led to strict inclusion and exclusion criteria governing the administration of this medication class. One less recognized but potentially serious complication is angioedema secondary to tissue plasminogen activator (tPA). Our case emphasizes the importance of early recognition of this clinical syndrome as it relates to airway compromise and potential respiratory failure in patients who are treated with tPA.
Case
A 70-year-old woman with a history of diabetes and hypertension and a remote history of breast cancer, nonhemiplegic migraines, and hypothyroidism presented to the ED with complaints of aphasia and right-sided paralysis, with onset 2 hours prior. Regarding the patient’s medication history, she had been taking lisinopril for hypertension.
Upon assessment, the patient was awake and alert and her vital signs were normal and stable, but she was aphasic, unable to accurately phonate, and was not able to move her right arm or leg against gravity. Her sensation appeared intact, and she had mild facial asymmetry with inability to raise the right corner of her mouth; her tongue had midline protrusion.
An emergent computed tomography (CT) scan of the head demonstrated mild brain atrophy and minimal low attenuation within the cerebral hemispheric white matter—most noticeably within the subcortical region of the left frontal lobe, consistent with small vessel ischemia. There was no evidence of acute intracranial hemorrhage, midline shift, or focal mass effect, and no convincing CT evidence for acute large vessel, cortical-based infarction.
The patient was determined to be an appropriate candidate for tPA, and was consented in the usual fashion. Within 15 minutes of administration of intravenous (IV) tPA, her symptoms improved, the aphasia resolved, and she was able to lift her right arm and leg against gravity and verbally communicate. Approximately 30 minutes following the resolution of her neurological symptoms, however, the patient was noted to have bleeding around a tooth socket, which was controlled with gauze and pressure. She subsequently began to complain of swelling on her right inferior lip without acute airway compromise. Over the next 10 to 15 minutes, she began to develop tongue swelling and feelings of dyspnea without wheezing.
The patient’s airway was reassessed and was classified as a Mallampati class IV. Anesthesia services were consulted for an emergent, awake intubation for airway protection. She was medicated with midazolam IV, as well as atomized lidocaine and lidocaine gargle for local anesthesia. The patient was successfully intubated awake using a flexible fiber optic technique. She was admitted to the medical intensive care unit for further monitoring, where she was treated with IV methylprednisolone, famotidine, and diphenhydramine. She was extubated the following day, had a relatively uncomplicated hospital course, and was discharged on hospital day 5 with improvement in her speech and right-sided weakness.
Discussion
The risk of angioedema associated with tPA administration has been previously described, with an estimated rate of 1.3 to 5.1%.1-3 Studies have shown the risk of developing angioedema is significantly increased in the setting of concomitant use of an angiotensin converting enzyme inhibitor (ACE-I); CT studies have also shown evidence of frontal and insular ischemia, with an odds radio of 13.6 and 9.1, respectively.2 Our patient was on lisinopril and had early signs of ischemia in the frontal lobe on initial CT scan, which likely increased her risk for angioedema.
How tPA Can Trigger Angioedema
The development of angioedema after administration of tPA has a well-described biochemical basis. Angioedema has been linked to the local vasodilatory effects of bradykinin, mast cell degranulation, and histamine release from activation of the complement pathway.4 Tissue plasminogen activator may trigger both of these pathways. It is a serine protease that cleaves plasminogen to plasmin; the plasmin in turn cleaves fibrin, resulting in the desired thrombolytic effects.5 Plasmin can cause mast cell degranulation through conversion of C3 to C3a and through activation of the complement pathway through conversion of C1 to C1a.6
Studies have shown tPA to have low antigenicity, and activation of this pathway is most likely secondary to direct proteolytic effects as opposed to antibody complexes.7 In a study by Bennett et al,6 tPA was shown to significantly increase C3a, C4a, and C5a serum levels when given in the setting of myocardial infarction (MI). It has also been shown to activate and increase serum kallikrein, which cleaves high-molecular weight kininogen to bradykinin, a potent vasodilator.8,9
Since bradykinin is broken down by several enzymes, including ACE, degradation is therefore delayed in patients on ACE-I.10 The alternate pathway for bradykinin degradation in the absence of ACE may also result in formation of des-Arg bradykinin, another similar active metabolite that mimics the effects of bradykinins.9 The formation of bradykinin through the proteolytic effects of tPA, in combination with the delayed breakdown in patient’s taking an ACE-I, likely plays a significant role in the development of angioedema.
In addition to the direct proteolytic effect of tPA resulting in angioedema, the underlying ischemic insult may also predispose patients to angioedema. As was the case with our patient, angioedema preferentially affects the ipsilateral side of the patient’s deficit.2,11,12 Theories suggest this is due to the lack of autonomic compensatory responses in the setting of ischemic insult.2 Interestingly, the development of angioedema in relation to the use of recombinant-tPA (eg, alteplase) in the setting of MI has not been as well described and may be related to the effect of central nervous system insult.3
Treatment
Although hemorrhagic complications of tPA therapy for cerebrovascular accident are well known, the risk for angioedema as a complication is less recognized. In most cases, angioedema is transient, and very few patients require aggressive support.3,12 Treatments that have previously been described include antihistamines and steroids.1,11,13 Epinephrine has been reported in one case study as an adjunct treatment of tPA-induced angioedema; however, it was given in combination with steroids and antihistimines.14 Therefore, caution should be taken regarding the use of epinephrine in this setting as there may be a theoretical precipitation of intracranial hypertension or hemorrhage.2
Given the likely significant role of the bradykinin-mediated pathway in tPA-induced angioedema, the true efficacy of these agents is unknown. Our patient had significant labial and lingual involvement, and given the concern for impending airway compromise, fiber optic intubation was performed. The decision to intubate and the technique employed must be carefully considered as a failed airway and need for a surgical airway is a concerning prospect in the setting of fibrinolytics. Successful cricothyroidotomy without significant complications has been described in the setting of streptokinase-induced angioedema when given for MI.15
Conclusion
The use of tPA for the treatment of ischemic stroke has been increasing over the last decade.16,17 Given the high prevalence of ACE-I use in patients who are also at risk for ischemic stroke, physicians administering tPA must be aware of the risk of tPA-associated angioedema. Patients with a known history of angioedema or anaphylaxis to tPA should be counseled on these risks and should not be given this medication, but rather considered for potential endovascular or mechanical clot retrieval therapy if they meet inclusion criteria for its use.
1. Hill MD, Barber PA, Takahashi J, Demchuk AM, Feasby TE, Buchan AM. Anaphylactoid reactions and angioedema during alteplase treatment of acute ischemic stroke. CMAJ. 2000;162(9):1281-1284.
2. Hill MD, Lye T, Moss H, et al. Hemi-orolingual angioedema and ACE inhibition after alteplase treatment of stroke. Neurology. 2003;60(9):1525-1527.
3. Hill MD, Buchan AM; Canadian Alteplase for Stroke Effectiveness Study (CASES) Investigators. Thrombolysis for acute ischemic stroke: results of the Canadian Alteplase for Stroke Effectiveness Study. CMAJ. 2005;172(10):1307-1312.
4. Lewis LM. Angioedema: etiology, pathophysiology, current and emerging therapies. J Emerg Med. 2013;45(5):789-796.
5. Loscalzo J, Braunwald E. Tissue plasminogen activator. N Engl J Med. 1988;319(14):935-931.
6. Bennett WR, Yawn DH, Migliore PJ, et al. Activation of the complement system by recombinant tissue plasminogen activator. J Am Coll Cardiol. 1987;10(3):627-632.
7. Reed BR, Chen AB, Tanswell P, et al. Low incidence of antibodies to recombinant human tissue-type plasminogen activator in treated patients. Thromb Haemost. 1990;64(2):276-280.
8. Hoffmeister HM, Szabo S, Kastner C, et al. Thrombolytic therapy in acute myocardial infarction: comparison of procoagulant effects of streptokinase and alteplase regimens with focus on the kallikrein system and plasmin. Circulation. 1998;98(23):2527-2533.
9. Molinaro G, Gervais N, Adam A. Biochemical basis of angioedema associated with recombinant tissue plasminogen activator treatment: an in vitro experimental approach. Stroke. 2002;33(6):1712-1716.
10. Bezalel S, Mahlab-Guri K, Asher I, Werner B, Sthoeger ZM. Angiotensin-converting enzyme inhibitor-induced angioedema. Am J Med. 2015;128(2):120-125.
11. Pancioli A, Brott T, Donaldson V, Miller R. Asymmetric angioneurotic edema associated with thrombolysis for acute stroke. Ann Emerg Med. 1997;30(2):227-229.
12. Correia AS, Matias G, Calado S, Lourenço A, Viana-Baptista M. Orolingual angiodema associated with alteplase treatment of acute stroke: a reappraisal. J Stroke Cerebrovasc Dis. 2015;24(1):31-40.
13. Maertins M, Wol R, Swider M. Angioedema after administration of tPA for ischemic stroke: case report. Air Med J. 2011;30(5):276-278.
14. Fugate JE, Kalimullah EA, Wijdicks EF. Angioedema after tPA: what neurointensivists should know. Neurocrit Care. 2012;16(3):440-443.
15. Walls RM, Pollack CV Jr. Successful cricothyrotomy after thrombolytic therapy for acute myocardial infarction: a report of two cases. Ann Emerg Med. 2000;35(2):188-191.
16. Lichtman JH, Watanabe E, Allen NB, Jones SB, Dostal J, Goldstein LB. Hospital arrival time and intravenous t-PA use in US Academic Medical Centers, 2001-2004. Stroke. 2009;40(12):3845-3850.
17. Schwamm LH, Ali SF, Reeves MJ, et al. Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With the Guidelines-Stroke hospitals. Circ Cardiovasc Qual Outcomes. 2013;6(5):543-549.
The use of thrombolytic medications for the treatment of acute ischemic cerebral infarctions has dynamically altered stroke care. However, there are both major and minor side effects associated with its use—most notably major bleeding, which led to strict inclusion and exclusion criteria governing the administration of this medication class. One less recognized but potentially serious complication is angioedema secondary to tissue plasminogen activator (tPA). Our case emphasizes the importance of early recognition of this clinical syndrome as it relates to airway compromise and potential respiratory failure in patients who are treated with tPA.
Case
A 70-year-old woman with a history of diabetes and hypertension and a remote history of breast cancer, nonhemiplegic migraines, and hypothyroidism presented to the ED with complaints of aphasia and right-sided paralysis, with onset 2 hours prior. Regarding the patient’s medication history, she had been taking lisinopril for hypertension.
Upon assessment, the patient was awake and alert and her vital signs were normal and stable, but she was aphasic, unable to accurately phonate, and was not able to move her right arm or leg against gravity. Her sensation appeared intact, and she had mild facial asymmetry with inability to raise the right corner of her mouth; her tongue had midline protrusion.
An emergent computed tomography (CT) scan of the head demonstrated mild brain atrophy and minimal low attenuation within the cerebral hemispheric white matter—most noticeably within the subcortical region of the left frontal lobe, consistent with small vessel ischemia. There was no evidence of acute intracranial hemorrhage, midline shift, or focal mass effect, and no convincing CT evidence for acute large vessel, cortical-based infarction.
The patient was determined to be an appropriate candidate for tPA, and was consented in the usual fashion. Within 15 minutes of administration of intravenous (IV) tPA, her symptoms improved, the aphasia resolved, and she was able to lift her right arm and leg against gravity and verbally communicate. Approximately 30 minutes following the resolution of her neurological symptoms, however, the patient was noted to have bleeding around a tooth socket, which was controlled with gauze and pressure. She subsequently began to complain of swelling on her right inferior lip without acute airway compromise. Over the next 10 to 15 minutes, she began to develop tongue swelling and feelings of dyspnea without wheezing.
The patient’s airway was reassessed and was classified as a Mallampati class IV. Anesthesia services were consulted for an emergent, awake intubation for airway protection. She was medicated with midazolam IV, as well as atomized lidocaine and lidocaine gargle for local anesthesia. The patient was successfully intubated awake using a flexible fiber optic technique. She was admitted to the medical intensive care unit for further monitoring, where she was treated with IV methylprednisolone, famotidine, and diphenhydramine. She was extubated the following day, had a relatively uncomplicated hospital course, and was discharged on hospital day 5 with improvement in her speech and right-sided weakness.
Discussion
The risk of angioedema associated with tPA administration has been previously described, with an estimated rate of 1.3 to 5.1%.1-3 Studies have shown the risk of developing angioedema is significantly increased in the setting of concomitant use of an angiotensin converting enzyme inhibitor (ACE-I); CT studies have also shown evidence of frontal and insular ischemia, with an odds radio of 13.6 and 9.1, respectively.2 Our patient was on lisinopril and had early signs of ischemia in the frontal lobe on initial CT scan, which likely increased her risk for angioedema.
How tPA Can Trigger Angioedema
The development of angioedema after administration of tPA has a well-described biochemical basis. Angioedema has been linked to the local vasodilatory effects of bradykinin, mast cell degranulation, and histamine release from activation of the complement pathway.4 Tissue plasminogen activator may trigger both of these pathways. It is a serine protease that cleaves plasminogen to plasmin; the plasmin in turn cleaves fibrin, resulting in the desired thrombolytic effects.5 Plasmin can cause mast cell degranulation through conversion of C3 to C3a and through activation of the complement pathway through conversion of C1 to C1a.6
Studies have shown tPA to have low antigenicity, and activation of this pathway is most likely secondary to direct proteolytic effects as opposed to antibody complexes.7 In a study by Bennett et al,6 tPA was shown to significantly increase C3a, C4a, and C5a serum levels when given in the setting of myocardial infarction (MI). It has also been shown to activate and increase serum kallikrein, which cleaves high-molecular weight kininogen to bradykinin, a potent vasodilator.8,9
Since bradykinin is broken down by several enzymes, including ACE, degradation is therefore delayed in patients on ACE-I.10 The alternate pathway for bradykinin degradation in the absence of ACE may also result in formation of des-Arg bradykinin, another similar active metabolite that mimics the effects of bradykinins.9 The formation of bradykinin through the proteolytic effects of tPA, in combination with the delayed breakdown in patient’s taking an ACE-I, likely plays a significant role in the development of angioedema.
In addition to the direct proteolytic effect of tPA resulting in angioedema, the underlying ischemic insult may also predispose patients to angioedema. As was the case with our patient, angioedema preferentially affects the ipsilateral side of the patient’s deficit.2,11,12 Theories suggest this is due to the lack of autonomic compensatory responses in the setting of ischemic insult.2 Interestingly, the development of angioedema in relation to the use of recombinant-tPA (eg, alteplase) in the setting of MI has not been as well described and may be related to the effect of central nervous system insult.3
Treatment
Although hemorrhagic complications of tPA therapy for cerebrovascular accident are well known, the risk for angioedema as a complication is less recognized. In most cases, angioedema is transient, and very few patients require aggressive support.3,12 Treatments that have previously been described include antihistamines and steroids.1,11,13 Epinephrine has been reported in one case study as an adjunct treatment of tPA-induced angioedema; however, it was given in combination with steroids and antihistimines.14 Therefore, caution should be taken regarding the use of epinephrine in this setting as there may be a theoretical precipitation of intracranial hypertension or hemorrhage.2
Given the likely significant role of the bradykinin-mediated pathway in tPA-induced angioedema, the true efficacy of these agents is unknown. Our patient had significant labial and lingual involvement, and given the concern for impending airway compromise, fiber optic intubation was performed. The decision to intubate and the technique employed must be carefully considered as a failed airway and need for a surgical airway is a concerning prospect in the setting of fibrinolytics. Successful cricothyroidotomy without significant complications has been described in the setting of streptokinase-induced angioedema when given for MI.15
Conclusion
The use of tPA for the treatment of ischemic stroke has been increasing over the last decade.16,17 Given the high prevalence of ACE-I use in patients who are also at risk for ischemic stroke, physicians administering tPA must be aware of the risk of tPA-associated angioedema. Patients with a known history of angioedema or anaphylaxis to tPA should be counseled on these risks and should not be given this medication, but rather considered for potential endovascular or mechanical clot retrieval therapy if they meet inclusion criteria for its use.
The use of thrombolytic medications for the treatment of acute ischemic cerebral infarctions has dynamically altered stroke care. However, there are both major and minor side effects associated with its use—most notably major bleeding, which led to strict inclusion and exclusion criteria governing the administration of this medication class. One less recognized but potentially serious complication is angioedema secondary to tissue plasminogen activator (tPA). Our case emphasizes the importance of early recognition of this clinical syndrome as it relates to airway compromise and potential respiratory failure in patients who are treated with tPA.
Case
A 70-year-old woman with a history of diabetes and hypertension and a remote history of breast cancer, nonhemiplegic migraines, and hypothyroidism presented to the ED with complaints of aphasia and right-sided paralysis, with onset 2 hours prior. Regarding the patient’s medication history, she had been taking lisinopril for hypertension.
Upon assessment, the patient was awake and alert and her vital signs were normal and stable, but she was aphasic, unable to accurately phonate, and was not able to move her right arm or leg against gravity. Her sensation appeared intact, and she had mild facial asymmetry with inability to raise the right corner of her mouth; her tongue had midline protrusion.
An emergent computed tomography (CT) scan of the head demonstrated mild brain atrophy and minimal low attenuation within the cerebral hemispheric white matter—most noticeably within the subcortical region of the left frontal lobe, consistent with small vessel ischemia. There was no evidence of acute intracranial hemorrhage, midline shift, or focal mass effect, and no convincing CT evidence for acute large vessel, cortical-based infarction.
The patient was determined to be an appropriate candidate for tPA, and was consented in the usual fashion. Within 15 minutes of administration of intravenous (IV) tPA, her symptoms improved, the aphasia resolved, and she was able to lift her right arm and leg against gravity and verbally communicate. Approximately 30 minutes following the resolution of her neurological symptoms, however, the patient was noted to have bleeding around a tooth socket, which was controlled with gauze and pressure. She subsequently began to complain of swelling on her right inferior lip without acute airway compromise. Over the next 10 to 15 minutes, she began to develop tongue swelling and feelings of dyspnea without wheezing.
The patient’s airway was reassessed and was classified as a Mallampati class IV. Anesthesia services were consulted for an emergent, awake intubation for airway protection. She was medicated with midazolam IV, as well as atomized lidocaine and lidocaine gargle for local anesthesia. The patient was successfully intubated awake using a flexible fiber optic technique. She was admitted to the medical intensive care unit for further monitoring, where she was treated with IV methylprednisolone, famotidine, and diphenhydramine. She was extubated the following day, had a relatively uncomplicated hospital course, and was discharged on hospital day 5 with improvement in her speech and right-sided weakness.
Discussion
The risk of angioedema associated with tPA administration has been previously described, with an estimated rate of 1.3 to 5.1%.1-3 Studies have shown the risk of developing angioedema is significantly increased in the setting of concomitant use of an angiotensin converting enzyme inhibitor (ACE-I); CT studies have also shown evidence of frontal and insular ischemia, with an odds radio of 13.6 and 9.1, respectively.2 Our patient was on lisinopril and had early signs of ischemia in the frontal lobe on initial CT scan, which likely increased her risk for angioedema.
How tPA Can Trigger Angioedema
The development of angioedema after administration of tPA has a well-described biochemical basis. Angioedema has been linked to the local vasodilatory effects of bradykinin, mast cell degranulation, and histamine release from activation of the complement pathway.4 Tissue plasminogen activator may trigger both of these pathways. It is a serine protease that cleaves plasminogen to plasmin; the plasmin in turn cleaves fibrin, resulting in the desired thrombolytic effects.5 Plasmin can cause mast cell degranulation through conversion of C3 to C3a and through activation of the complement pathway through conversion of C1 to C1a.6
Studies have shown tPA to have low antigenicity, and activation of this pathway is most likely secondary to direct proteolytic effects as opposed to antibody complexes.7 In a study by Bennett et al,6 tPA was shown to significantly increase C3a, C4a, and C5a serum levels when given in the setting of myocardial infarction (MI). It has also been shown to activate and increase serum kallikrein, which cleaves high-molecular weight kininogen to bradykinin, a potent vasodilator.8,9
Since bradykinin is broken down by several enzymes, including ACE, degradation is therefore delayed in patients on ACE-I.10 The alternate pathway for bradykinin degradation in the absence of ACE may also result in formation of des-Arg bradykinin, another similar active metabolite that mimics the effects of bradykinins.9 The formation of bradykinin through the proteolytic effects of tPA, in combination with the delayed breakdown in patient’s taking an ACE-I, likely plays a significant role in the development of angioedema.
In addition to the direct proteolytic effect of tPA resulting in angioedema, the underlying ischemic insult may also predispose patients to angioedema. As was the case with our patient, angioedema preferentially affects the ipsilateral side of the patient’s deficit.2,11,12 Theories suggest this is due to the lack of autonomic compensatory responses in the setting of ischemic insult.2 Interestingly, the development of angioedema in relation to the use of recombinant-tPA (eg, alteplase) in the setting of MI has not been as well described and may be related to the effect of central nervous system insult.3
Treatment
Although hemorrhagic complications of tPA therapy for cerebrovascular accident are well known, the risk for angioedema as a complication is less recognized. In most cases, angioedema is transient, and very few patients require aggressive support.3,12 Treatments that have previously been described include antihistamines and steroids.1,11,13 Epinephrine has been reported in one case study as an adjunct treatment of tPA-induced angioedema; however, it was given in combination with steroids and antihistimines.14 Therefore, caution should be taken regarding the use of epinephrine in this setting as there may be a theoretical precipitation of intracranial hypertension or hemorrhage.2
Given the likely significant role of the bradykinin-mediated pathway in tPA-induced angioedema, the true efficacy of these agents is unknown. Our patient had significant labial and lingual involvement, and given the concern for impending airway compromise, fiber optic intubation was performed. The decision to intubate and the technique employed must be carefully considered as a failed airway and need for a surgical airway is a concerning prospect in the setting of fibrinolytics. Successful cricothyroidotomy without significant complications has been described in the setting of streptokinase-induced angioedema when given for MI.15
Conclusion
The use of tPA for the treatment of ischemic stroke has been increasing over the last decade.16,17 Given the high prevalence of ACE-I use in patients who are also at risk for ischemic stroke, physicians administering tPA must be aware of the risk of tPA-associated angioedema. Patients with a known history of angioedema or anaphylaxis to tPA should be counseled on these risks and should not be given this medication, but rather considered for potential endovascular or mechanical clot retrieval therapy if they meet inclusion criteria for its use.
1. Hill MD, Barber PA, Takahashi J, Demchuk AM, Feasby TE, Buchan AM. Anaphylactoid reactions and angioedema during alteplase treatment of acute ischemic stroke. CMAJ. 2000;162(9):1281-1284.
2. Hill MD, Lye T, Moss H, et al. Hemi-orolingual angioedema and ACE inhibition after alteplase treatment of stroke. Neurology. 2003;60(9):1525-1527.
3. Hill MD, Buchan AM; Canadian Alteplase for Stroke Effectiveness Study (CASES) Investigators. Thrombolysis for acute ischemic stroke: results of the Canadian Alteplase for Stroke Effectiveness Study. CMAJ. 2005;172(10):1307-1312.
4. Lewis LM. Angioedema: etiology, pathophysiology, current and emerging therapies. J Emerg Med. 2013;45(5):789-796.
5. Loscalzo J, Braunwald E. Tissue plasminogen activator. N Engl J Med. 1988;319(14):935-931.
6. Bennett WR, Yawn DH, Migliore PJ, et al. Activation of the complement system by recombinant tissue plasminogen activator. J Am Coll Cardiol. 1987;10(3):627-632.
7. Reed BR, Chen AB, Tanswell P, et al. Low incidence of antibodies to recombinant human tissue-type plasminogen activator in treated patients. Thromb Haemost. 1990;64(2):276-280.
8. Hoffmeister HM, Szabo S, Kastner C, et al. Thrombolytic therapy in acute myocardial infarction: comparison of procoagulant effects of streptokinase and alteplase regimens with focus on the kallikrein system and plasmin. Circulation. 1998;98(23):2527-2533.
9. Molinaro G, Gervais N, Adam A. Biochemical basis of angioedema associated with recombinant tissue plasminogen activator treatment: an in vitro experimental approach. Stroke. 2002;33(6):1712-1716.
10. Bezalel S, Mahlab-Guri K, Asher I, Werner B, Sthoeger ZM. Angiotensin-converting enzyme inhibitor-induced angioedema. Am J Med. 2015;128(2):120-125.
11. Pancioli A, Brott T, Donaldson V, Miller R. Asymmetric angioneurotic edema associated with thrombolysis for acute stroke. Ann Emerg Med. 1997;30(2):227-229.
12. Correia AS, Matias G, Calado S, Lourenço A, Viana-Baptista M. Orolingual angiodema associated with alteplase treatment of acute stroke: a reappraisal. J Stroke Cerebrovasc Dis. 2015;24(1):31-40.
13. Maertins M, Wol R, Swider M. Angioedema after administration of tPA for ischemic stroke: case report. Air Med J. 2011;30(5):276-278.
14. Fugate JE, Kalimullah EA, Wijdicks EF. Angioedema after tPA: what neurointensivists should know. Neurocrit Care. 2012;16(3):440-443.
15. Walls RM, Pollack CV Jr. Successful cricothyrotomy after thrombolytic therapy for acute myocardial infarction: a report of two cases. Ann Emerg Med. 2000;35(2):188-191.
16. Lichtman JH, Watanabe E, Allen NB, Jones SB, Dostal J, Goldstein LB. Hospital arrival time and intravenous t-PA use in US Academic Medical Centers, 2001-2004. Stroke. 2009;40(12):3845-3850.
17. Schwamm LH, Ali SF, Reeves MJ, et al. Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With the Guidelines-Stroke hospitals. Circ Cardiovasc Qual Outcomes. 2013;6(5):543-549.
1. Hill MD, Barber PA, Takahashi J, Demchuk AM, Feasby TE, Buchan AM. Anaphylactoid reactions and angioedema during alteplase treatment of acute ischemic stroke. CMAJ. 2000;162(9):1281-1284.
2. Hill MD, Lye T, Moss H, et al. Hemi-orolingual angioedema and ACE inhibition after alteplase treatment of stroke. Neurology. 2003;60(9):1525-1527.
3. Hill MD, Buchan AM; Canadian Alteplase for Stroke Effectiveness Study (CASES) Investigators. Thrombolysis for acute ischemic stroke: results of the Canadian Alteplase for Stroke Effectiveness Study. CMAJ. 2005;172(10):1307-1312.
4. Lewis LM. Angioedema: etiology, pathophysiology, current and emerging therapies. J Emerg Med. 2013;45(5):789-796.
5. Loscalzo J, Braunwald E. Tissue plasminogen activator. N Engl J Med. 1988;319(14):935-931.
6. Bennett WR, Yawn DH, Migliore PJ, et al. Activation of the complement system by recombinant tissue plasminogen activator. J Am Coll Cardiol. 1987;10(3):627-632.
7. Reed BR, Chen AB, Tanswell P, et al. Low incidence of antibodies to recombinant human tissue-type plasminogen activator in treated patients. Thromb Haemost. 1990;64(2):276-280.
8. Hoffmeister HM, Szabo S, Kastner C, et al. Thrombolytic therapy in acute myocardial infarction: comparison of procoagulant effects of streptokinase and alteplase regimens with focus on the kallikrein system and plasmin. Circulation. 1998;98(23):2527-2533.
9. Molinaro G, Gervais N, Adam A. Biochemical basis of angioedema associated with recombinant tissue plasminogen activator treatment: an in vitro experimental approach. Stroke. 2002;33(6):1712-1716.
10. Bezalel S, Mahlab-Guri K, Asher I, Werner B, Sthoeger ZM. Angiotensin-converting enzyme inhibitor-induced angioedema. Am J Med. 2015;128(2):120-125.
11. Pancioli A, Brott T, Donaldson V, Miller R. Asymmetric angioneurotic edema associated with thrombolysis for acute stroke. Ann Emerg Med. 1997;30(2):227-229.
12. Correia AS, Matias G, Calado S, Lourenço A, Viana-Baptista M. Orolingual angiodema associated with alteplase treatment of acute stroke: a reappraisal. J Stroke Cerebrovasc Dis. 2015;24(1):31-40.
13. Maertins M, Wol R, Swider M. Angioedema after administration of tPA for ischemic stroke: case report. Air Med J. 2011;30(5):276-278.
14. Fugate JE, Kalimullah EA, Wijdicks EF. Angioedema after tPA: what neurointensivists should know. Neurocrit Care. 2012;16(3):440-443.
15. Walls RM, Pollack CV Jr. Successful cricothyrotomy after thrombolytic therapy for acute myocardial infarction: a report of two cases. Ann Emerg Med. 2000;35(2):188-191.
16. Lichtman JH, Watanabe E, Allen NB, Jones SB, Dostal J, Goldstein LB. Hospital arrival time and intravenous t-PA use in US Academic Medical Centers, 2001-2004. Stroke. 2009;40(12):3845-3850.
17. Schwamm LH, Ali SF, Reeves MJ, et al. Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With the Guidelines-Stroke hospitals. Circ Cardiovasc Qual Outcomes. 2013;6(5):543-549.
An unconventional approach to chest wall pain
THE CASE
A 45-year-old airman presented to our medical group with acute onset of sharp, positional left lateral chest wall pain that he’d had for 2 days. The pain began after an extreme core body workout. Treatment with ibuprofen 800 mg and local electrical stimulation one day prior provided no benefit. The patient reported the pain to be a 6 out of 10 when still and a 9 to 10 when sitting for more than a few minutes, turning, or taking a medium to deep breath. The patient felt “dangerously distracted by the pain” while driving in for his appointment.
We noted focal left lower lateral intercostal muscle tenderness without trigger point-like thickness or spasm. The patient also had restricted inspiration, secondary to the severe pain, and decreased left lower field breath sounds. His vital signs were normal, as was his cardiac exam.
THE DIAGNOSIS
While awaiting a chest x-ray, the patient was offered and opted to try acupuncture for pain relief. (We have medical acupuncturists on staff.) Analgesics had already been used, but had provided little relief.
We identified 4 acupuncture sites in the ear: 2 were battlefield acupuncture (BFA) points (more on this in a bit) and 2 points were deemed active by a skin conductance point finder (a handheld device that assesses changes in electrical skin resistance at auricular acupuncture points). The left ear points that were treated included the cingulate gyrus (intertragic notch), Shen Men (triangular fossa), and chest and abdomen regional points (antihelix) (FIGURE 1).
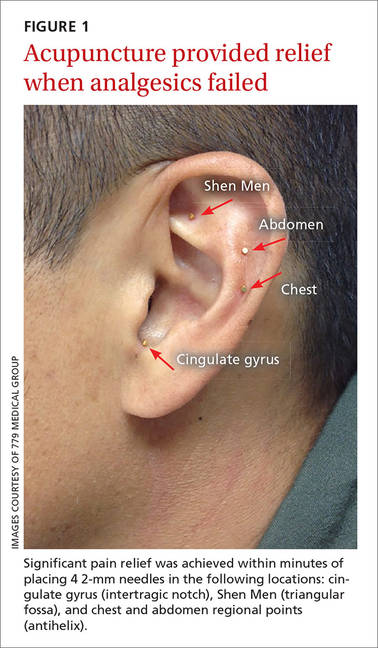
Within 15 minutes, the patient reported significant pain relief and was able to inspire deeply without pain. The patient also underwent a chest x-ray, which revealed atelectasis of the left lower lobe (FIGURE 2A) caused by pain-induced hypoventilation.
Because the patient’s pain was so well controlled, he returned to work immediately after the appointment. At the end of his shift 6 hours later he returned, unscheduled, to report pain at a level of one out of 10 and said he was able to breathe normally. In addition, lung auscultation was normal and a repeat chest x-ray revealed that the atelectasis had almost completely resolved (FIGURE 2B). This occurred without medication or other therapy. The pain did not return.
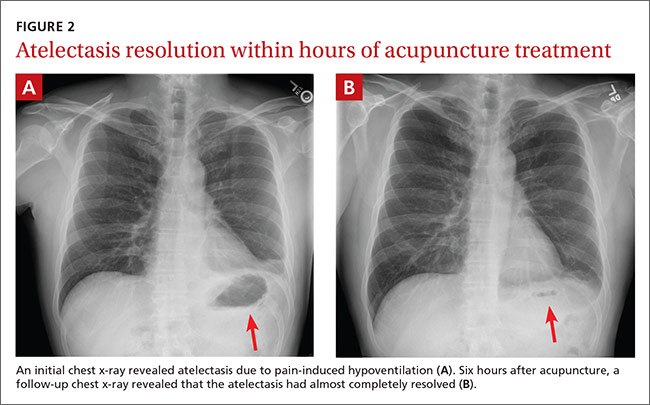
DISCUSSION
Although acupuncture is over 2000 years old, it has been largely disregarded in the United States due to a lack of mainstream evidence supporting its efficacy. Research is hindered by significant variation in approach between providers, the difficulties inherent to blinding patients and providers to treatment vs placebo, and poor insurance coverage and reimbursement.
Acupuncture research is burgeoning. A 2012 meta-analysis concluded that patients receiving acupuncture had less pain than those receiving sham or no acupuncture for several pain conditions. Specifically, scores for back and neck pain, osteoarthritis, and chronic headache were 0.23, 0.16, and 0.15 standard deviations (SDs), respectively, lower for patients receiving acupuncture than for those who got sham acupuncture. The effect sizes for acupuncture patients compared to no acupuncture controls were 0.55, 0.57, and 0.42 respectively (all P<.001).1
Several theories explain how auricular acupuncture may work. Paul Nogier, MD, noted that the ear is composed of ectodermal, mesodermal, and endodermal tissues, and mapped the “inverted fetus” homunculus in the ear, which corresponds to specific body points.2 Functional magnetic resonance imaging has demonstrated increased brain activity in the cingulate gyrus and thalamic regions in response to a painful stimulus, as well as attenuation of this activity after the placement of needles in corresponding auricular cingulate gyrus and thalamus points.3 In addition, research has confirmed that acupuncture raises serum and cerebrospinal levels of endorphins and enkephalins.4
Battlefield acupuncture (BFA) was developed by Richard Niemtzow, MD, and has been used for acute injuries in the front lines of battle as well as for many health conditions. BFA treats pain using a sequence of 5 predetermined auricular acupuncture points.5 Onset and duration of pain relief vary depending on the location and nature of the pathology. We’ve noted that BFA for chronic pain has a shorter duration of benefit and is more likely to need to be repeated.
One randomized pilot study involving 87 patients presenting to the emergency room blinded emergency health care providers to the inclusion of the first 2 BFA points in their otherwise usual care of acute pain patients. Participants in the acupuncture group experienced a 23% reduction in pain before discharge compared to no change in the standard care group (P<.0005).6
Our patient. We inserted semi-permanent needles with a needle length of 2 mm into 4 locations on the ear. (These needles can remain in the ear for several days and fall out on their own or they may be removed by pulling the stud ends.) As noted earlier, our patient reported pain relief within 15 minutes and was pain free by the next day.
THE TAKEAWAY
Auricular acupuncture can treat acute and chronic pain. As proof, the BFA technique is widely used by health care providers throughout the US military and Department of Veterans Affairs. In this case, the immediate pain relief and x-ray documentation of atelectasis resolution within 6 hours of treatment provide support that auricular acupuncture was beneficial in reversing the cause of this atelectasis, which was pain-induced hypoventilation.
While the acute pain control observed with this patient is not unusual in our experience, what is unusual is the rare visual confirmation of the striking degree of pain reduction possible with auricular acupuncture.
1. Vickers AJ, Cronin AM, Maschino AC, et al. Acupuncture Trialists’ Collaboration. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172:1444-1453.
2. Oleson T. Auriculotherapy Manual: Chinese and Western Systems of Ear Acupuncture. 4th ed. Los Angeles: Churchill Livingstone; 2014.
3. Sjölund B, Eriksson M. Electro-acupunture and endogenous morphines. Lancet. 1976;2:1085.
4. Cho ZH, Chung SC, Jones JP, et al. New findings of the correlation between acupoints and corresponding brain cortices using functional MRI. Proc Natl Acad Sci U S A. 1998;95:2670-2673.
5. Niemtzow RC. Battlefield acupuncture: Update. Medical Acupuncture. 2007;19:225-228.
6.Goertz CM, Niemtzow R, Burns SM, et al. Auricular acupuncture in the treatment of acute pain syndromes: A pilot study. Mil Med. 2006;171:1010-1014.
THE CASE
A 45-year-old airman presented to our medical group with acute onset of sharp, positional left lateral chest wall pain that he’d had for 2 days. The pain began after an extreme core body workout. Treatment with ibuprofen 800 mg and local electrical stimulation one day prior provided no benefit. The patient reported the pain to be a 6 out of 10 when still and a 9 to 10 when sitting for more than a few minutes, turning, or taking a medium to deep breath. The patient felt “dangerously distracted by the pain” while driving in for his appointment.
We noted focal left lower lateral intercostal muscle tenderness without trigger point-like thickness or spasm. The patient also had restricted inspiration, secondary to the severe pain, and decreased left lower field breath sounds. His vital signs were normal, as was his cardiac exam.
THE DIAGNOSIS
While awaiting a chest x-ray, the patient was offered and opted to try acupuncture for pain relief. (We have medical acupuncturists on staff.) Analgesics had already been used, but had provided little relief.
We identified 4 acupuncture sites in the ear: 2 were battlefield acupuncture (BFA) points (more on this in a bit) and 2 points were deemed active by a skin conductance point finder (a handheld device that assesses changes in electrical skin resistance at auricular acupuncture points). The left ear points that were treated included the cingulate gyrus (intertragic notch), Shen Men (triangular fossa), and chest and abdomen regional points (antihelix) (FIGURE 1).
Within 15 minutes, the patient reported significant pain relief and was able to inspire deeply without pain. The patient also underwent a chest x-ray, which revealed atelectasis of the left lower lobe (FIGURE 2A) caused by pain-induced hypoventilation.
Because the patient’s pain was so well controlled, he returned to work immediately after the appointment. At the end of his shift 6 hours later he returned, unscheduled, to report pain at a level of one out of 10 and said he was able to breathe normally. In addition, lung auscultation was normal and a repeat chest x-ray revealed that the atelectasis had almost completely resolved (FIGURE 2B). This occurred without medication or other therapy. The pain did not return.
DISCUSSION
Although acupuncture is over 2000 years old, it has been largely disregarded in the United States due to a lack of mainstream evidence supporting its efficacy. Research is hindered by significant variation in approach between providers, the difficulties inherent to blinding patients and providers to treatment vs placebo, and poor insurance coverage and reimbursement.
Acupuncture research is burgeoning. A 2012 meta-analysis concluded that patients receiving acupuncture had less pain than those receiving sham or no acupuncture for several pain conditions. Specifically, scores for back and neck pain, osteoarthritis, and chronic headache were 0.23, 0.16, and 0.15 standard deviations (SDs), respectively, lower for patients receiving acupuncture than for those who got sham acupuncture. The effect sizes for acupuncture patients compared to no acupuncture controls were 0.55, 0.57, and 0.42 respectively (all P<.001).1
Several theories explain how auricular acupuncture may work. Paul Nogier, MD, noted that the ear is composed of ectodermal, mesodermal, and endodermal tissues, and mapped the “inverted fetus” homunculus in the ear, which corresponds to specific body points.2 Functional magnetic resonance imaging has demonstrated increased brain activity in the cingulate gyrus and thalamic regions in response to a painful stimulus, as well as attenuation of this activity after the placement of needles in corresponding auricular cingulate gyrus and thalamus points.3 In addition, research has confirmed that acupuncture raises serum and cerebrospinal levels of endorphins and enkephalins.4
Battlefield acupuncture (BFA) was developed by Richard Niemtzow, MD, and has been used for acute injuries in the front lines of battle as well as for many health conditions. BFA treats pain using a sequence of 5 predetermined auricular acupuncture points.5 Onset and duration of pain relief vary depending on the location and nature of the pathology. We’ve noted that BFA for chronic pain has a shorter duration of benefit and is more likely to need to be repeated.
One randomized pilot study involving 87 patients presenting to the emergency room blinded emergency health care providers to the inclusion of the first 2 BFA points in their otherwise usual care of acute pain patients. Participants in the acupuncture group experienced a 23% reduction in pain before discharge compared to no change in the standard care group (P<.0005).6
Our patient. We inserted semi-permanent needles with a needle length of 2 mm into 4 locations on the ear. (These needles can remain in the ear for several days and fall out on their own or they may be removed by pulling the stud ends.) As noted earlier, our patient reported pain relief within 15 minutes and was pain free by the next day.
THE TAKEAWAY
Auricular acupuncture can treat acute and chronic pain. As proof, the BFA technique is widely used by health care providers throughout the US military and Department of Veterans Affairs. In this case, the immediate pain relief and x-ray documentation of atelectasis resolution within 6 hours of treatment provide support that auricular acupuncture was beneficial in reversing the cause of this atelectasis, which was pain-induced hypoventilation.
While the acute pain control observed with this patient is not unusual in our experience, what is unusual is the rare visual confirmation of the striking degree of pain reduction possible with auricular acupuncture.
THE CASE
A 45-year-old airman presented to our medical group with acute onset of sharp, positional left lateral chest wall pain that he’d had for 2 days. The pain began after an extreme core body workout. Treatment with ibuprofen 800 mg and local electrical stimulation one day prior provided no benefit. The patient reported the pain to be a 6 out of 10 when still and a 9 to 10 when sitting for more than a few minutes, turning, or taking a medium to deep breath. The patient felt “dangerously distracted by the pain” while driving in for his appointment.
We noted focal left lower lateral intercostal muscle tenderness without trigger point-like thickness or spasm. The patient also had restricted inspiration, secondary to the severe pain, and decreased left lower field breath sounds. His vital signs were normal, as was his cardiac exam.
THE DIAGNOSIS
While awaiting a chest x-ray, the patient was offered and opted to try acupuncture for pain relief. (We have medical acupuncturists on staff.) Analgesics had already been used, but had provided little relief.
We identified 4 acupuncture sites in the ear: 2 were battlefield acupuncture (BFA) points (more on this in a bit) and 2 points were deemed active by a skin conductance point finder (a handheld device that assesses changes in electrical skin resistance at auricular acupuncture points). The left ear points that were treated included the cingulate gyrus (intertragic notch), Shen Men (triangular fossa), and chest and abdomen regional points (antihelix) (FIGURE 1).
Within 15 minutes, the patient reported significant pain relief and was able to inspire deeply without pain. The patient also underwent a chest x-ray, which revealed atelectasis of the left lower lobe (FIGURE 2A) caused by pain-induced hypoventilation.
Because the patient’s pain was so well controlled, he returned to work immediately after the appointment. At the end of his shift 6 hours later he returned, unscheduled, to report pain at a level of one out of 10 and said he was able to breathe normally. In addition, lung auscultation was normal and a repeat chest x-ray revealed that the atelectasis had almost completely resolved (FIGURE 2B). This occurred without medication or other therapy. The pain did not return.
DISCUSSION
Although acupuncture is over 2000 years old, it has been largely disregarded in the United States due to a lack of mainstream evidence supporting its efficacy. Research is hindered by significant variation in approach between providers, the difficulties inherent to blinding patients and providers to treatment vs placebo, and poor insurance coverage and reimbursement.
Acupuncture research is burgeoning. A 2012 meta-analysis concluded that patients receiving acupuncture had less pain than those receiving sham or no acupuncture for several pain conditions. Specifically, scores for back and neck pain, osteoarthritis, and chronic headache were 0.23, 0.16, and 0.15 standard deviations (SDs), respectively, lower for patients receiving acupuncture than for those who got sham acupuncture. The effect sizes for acupuncture patients compared to no acupuncture controls were 0.55, 0.57, and 0.42 respectively (all P<.001).1
Several theories explain how auricular acupuncture may work. Paul Nogier, MD, noted that the ear is composed of ectodermal, mesodermal, and endodermal tissues, and mapped the “inverted fetus” homunculus in the ear, which corresponds to specific body points.2 Functional magnetic resonance imaging has demonstrated increased brain activity in the cingulate gyrus and thalamic regions in response to a painful stimulus, as well as attenuation of this activity after the placement of needles in corresponding auricular cingulate gyrus and thalamus points.3 In addition, research has confirmed that acupuncture raises serum and cerebrospinal levels of endorphins and enkephalins.4
Battlefield acupuncture (BFA) was developed by Richard Niemtzow, MD, and has been used for acute injuries in the front lines of battle as well as for many health conditions. BFA treats pain using a sequence of 5 predetermined auricular acupuncture points.5 Onset and duration of pain relief vary depending on the location and nature of the pathology. We’ve noted that BFA for chronic pain has a shorter duration of benefit and is more likely to need to be repeated.
One randomized pilot study involving 87 patients presenting to the emergency room blinded emergency health care providers to the inclusion of the first 2 BFA points in their otherwise usual care of acute pain patients. Participants in the acupuncture group experienced a 23% reduction in pain before discharge compared to no change in the standard care group (P<.0005).6
Our patient. We inserted semi-permanent needles with a needle length of 2 mm into 4 locations on the ear. (These needles can remain in the ear for several days and fall out on their own or they may be removed by pulling the stud ends.) As noted earlier, our patient reported pain relief within 15 minutes and was pain free by the next day.
THE TAKEAWAY
Auricular acupuncture can treat acute and chronic pain. As proof, the BFA technique is widely used by health care providers throughout the US military and Department of Veterans Affairs. In this case, the immediate pain relief and x-ray documentation of atelectasis resolution within 6 hours of treatment provide support that auricular acupuncture was beneficial in reversing the cause of this atelectasis, which was pain-induced hypoventilation.
While the acute pain control observed with this patient is not unusual in our experience, what is unusual is the rare visual confirmation of the striking degree of pain reduction possible with auricular acupuncture.
1. Vickers AJ, Cronin AM, Maschino AC, et al. Acupuncture Trialists’ Collaboration. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172:1444-1453.
2. Oleson T. Auriculotherapy Manual: Chinese and Western Systems of Ear Acupuncture. 4th ed. Los Angeles: Churchill Livingstone; 2014.
3. Sjölund B, Eriksson M. Electro-acupunture and endogenous morphines. Lancet. 1976;2:1085.
4. Cho ZH, Chung SC, Jones JP, et al. New findings of the correlation between acupoints and corresponding brain cortices using functional MRI. Proc Natl Acad Sci U S A. 1998;95:2670-2673.
5. Niemtzow RC. Battlefield acupuncture: Update. Medical Acupuncture. 2007;19:225-228.
6.Goertz CM, Niemtzow R, Burns SM, et al. Auricular acupuncture in the treatment of acute pain syndromes: A pilot study. Mil Med. 2006;171:1010-1014.
1. Vickers AJ, Cronin AM, Maschino AC, et al. Acupuncture Trialists’ Collaboration. Acupuncture for chronic pain: individual patient data meta-analysis. Arch Intern Med. 2012;172:1444-1453.
2. Oleson T. Auriculotherapy Manual: Chinese and Western Systems of Ear Acupuncture. 4th ed. Los Angeles: Churchill Livingstone; 2014.
3. Sjölund B, Eriksson M. Electro-acupunture and endogenous morphines. Lancet. 1976;2:1085.
4. Cho ZH, Chung SC, Jones JP, et al. New findings of the correlation between acupoints and corresponding brain cortices using functional MRI. Proc Natl Acad Sci U S A. 1998;95:2670-2673.
5. Niemtzow RC. Battlefield acupuncture: Update. Medical Acupuncture. 2007;19:225-228.
6.Goertz CM, Niemtzow R, Burns SM, et al. Auricular acupuncture in the treatment of acute pain syndromes: A pilot study. Mil Med. 2006;171:1010-1014.

Uninjured athlete with edematous arm • Dx?
THE CASE
A 16-year-old boy presented to the emergency room (ER) with pain, redness, and swelling of his right upper arm that had been bothering him for 2 days. He was the quarterback of his high school football team, a sport that he’d been playing since he was 8 years old. He indicated that his football training—which involved repetitive throwing with his right arm—had intensified over the previous 2 months.
Prior to the ER visit, the patient was healthy and active with no significant medical history. He’d had no shoulder trauma and there was no family history of any coagulopathies, venous thrombosis, or pulmonary embolism. He denied chest pain, shortness of breath, palpitations, and fever, and said that he did not smoke cigarettes or drink alcohol.
On physical examination, his blood pressure was 118/70 mm Hg and his heart rate was 74 beats per minute. He had nonpitting edema and erythema of his right upper arm. His radial and brachial pulses were strong and equal in both arms. Assessment of neurologic and vascular integrity produced positive Wright’s and Adson’s tests, but a negative Halstead’s test. (For more on these tests, see: Wright’s test, Adson’s test, and Halstead’s test.) The circumference of the patient’s right upper arm was 2.5 cm greater than the left upper arm. The remainder of the physical exam was normal.
THE DIAGNOSIS
A duplex ultrasound of the right upper arm revealed an acute occlusive thrombus in the axillary vein. We started the patient on intravenous heparin. A venogram confirmed thrombosis of the axillary-subclavian vein (FIGURE 1A). Based on the patient’s clinical presentation and the results of the venogram, we diagnosed Paget-Schroetter syndrome. The venogram was followed by thrombolysis with alteplase (FIGURE 1B) and a balloon angioplasty (FIGURE 1C). One week later, a repeat venogram demonstrated partial removal of the thrombus and an area of compression on the inferior aspect of the subclavian vein due to a cervical band (FIGURE 1D).
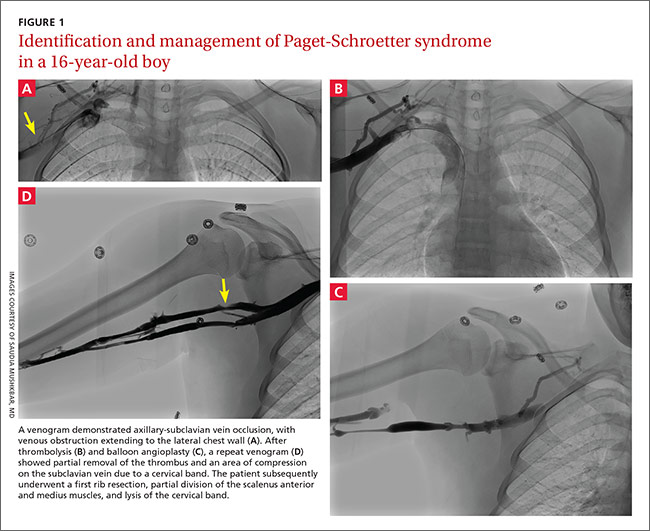
DISCUSSION
Paget-Schroetter syndrome (PSS), or effort thrombosis of the upper extremities, is defined as spontaneous thrombus in the axillary and subclavian veins that occurs as a consequence of strenuous upper-extremity activity. It is a rare condition with an incidence of one to 2 cases per 100,000 people per year, and represents 1% to 4% of all cases of deep vein thrombosis (DVT).1
Spontaneous thrombosis of the upper extremities typically presents in young, otherwise healthy individuals. It has been described in athletes who are involved in ball games, games with rackets or clubs, aquatic sports, combatant sports, and in violin players.2 The repetitive movements used in these activities can lead to compression of the axillary and subclavian veins by hypertrophied muscles. Repetitive trauma causes intimal damage and thrombogenesis.3
PSS is characterized by the abrupt, spontaneous swelling of the entire arm, cyanosis, and pain that occurs with use or overhead positioning. Enlarged subcutaneous veins are present in the upper arm, around the shoulder, or in the upper anterior chest wall (Urschel’s sign). The classic presentation is acute onset of upper extremity pain and swelling in the dominant arm following a particularly strenuous activity.4 A low-grade fever, superficial thrombophlebitis, or neurologic symptoms may coexist. Certain provocative maneuvers can help reproduce the symptoms (TABLE 15,6). Complications of PSS include pulmonary embolism, postthrombotic syndrome (pain, heaviness, and swelling), and recurrent thrombosis.7
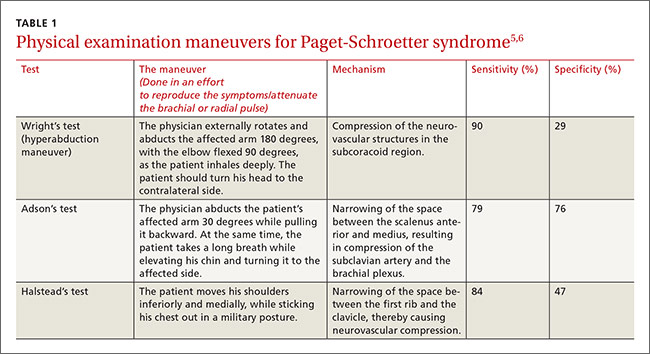
Contrast venography best shows the extent of thrombosis
Duplex ultrasound, with its high sensitivity and specificity, is the initial, noninvasive test of choice (TABLE 24,8-11). However, duplex ultrasound has a false-negative rate of 30% because it is highly technician-dependent and can be complicated by acoustic shadows from the clavicle or sternum.8
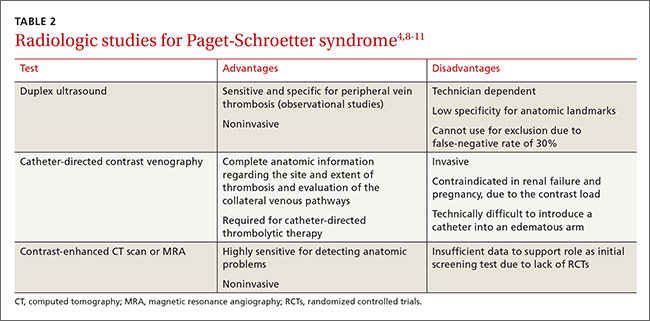
The most direct and definitive means to confirm the diagnosis of PSS is catheter-directed contrast venography.9 This method provides complete anatomic information regarding the site and extent of thrombosis, allows definitive evaluation of the collateral venous pathways, and is a necessary step toward the use of thrombolytic therapy. Contrast load, however, contraindicates the procedure in patients with renal failure and in those who are pregnant.
Contrast-enhanced computed tomography (CT) and magnetic resonance angiography (MRA) are also highly sensitive for detecting focal stenosis at the level of the first rib, the presence or absence of enlarged collateral veins, and the chronicity of any thrombus present. However, the usefulness of CT and magnetic resonance venography in initial screening is unclear, due to a lack of randomized controlled trials.
Treatment involves anticoagulants, thrombolytics, and possibly surgery
Prompt use of anticoagulation is indicated in PSS. Initial anticoagulation with low molecular weight unfractionated heparin or a direct thrombin inhibitor followed by warfarin for a minimum of 3 to 6 months is recommended.12
Patients treated with anticoagulation alone have a higher incidence of long-term residual symptoms, disability, and recurrent thrombosis.7 As a result, a more aggressive approach with the use of thrombolytic therapy is indicated, especially in young, active patients, to minimize long-term consequences. Alteplase or reteplase are used for this purpose. Thrombolysis is less likely to be beneficial if the thrombus is more than 2 weeks old or if there are inflammatory changes in the vein. The use of catheter-directed thrombolysis minimizes the risk of systemic adverse effects and achieves higher clot resolution rates.13
Because PSS is caused by compression of the vein, rather than a disorder of blood clotting, there is still a 50% to 70% risk of recurrent thrombosis despite thrombolysis and anticoagulation.14 Therefore, the most definitive management approach remains surgical treatment. Patients with recent thrombosis who are within the first several weeks of undergoing successful thrombolytic therapy are excellent candidates for surgery. Operative treatment for PSS includes first rib resection, scalene muscle removal, or subclavius muscle removal, along with removal of constricting scar tissue from around the vein.7
THE TAKEAWAY
PSS is characterized by upper-extremity DVT resulting from repetitive trauma to the subclavian-axillary vein. Early diagnosis of PSS with contrast venography and prompt use of anticoagulation can effectively restore venous patency, reduce the risk of rethrombosis, and return the patient to normal function. Primary care physicians should be aware of this condition, because delayed recognition in a high-functioning person can be potentially disabling.
Our patient had a first rib resection, partial division of the scalenus anterior and medius muscles, and lysis of the cervical band. Follow-up venography confirmed resolution of thrombosis without any complications. The patient was continued on anticoagulation with warfarin for 3 months.
1. Isma N, Svensson PJ, Gottsäter A, et al. Upper extremity deep venous thrombosis in the population-based Malmö thrombophilia study (MATS). Epidemiology, risk factors, recurrence risk, and mortality. Thromb Res. 2010;125:e335-e338.
2. DiFelice GS, Paletta GA Jr, Phillips BB, et al. Effort thrombosis in the elite throwing athlete. Am J Sports Med. 2002;30:708-712.
3. Thompson JF, Winterborn RJ, Bays S, et al. Venous thoracic outlet compression and the Paget-Schroetter syndrome: a review and recommendations for management. Cardiovasc Intervent Radiol. 2011;34:903-910.
4. Joffe HV, Kucher N, Tapson VF, et al; Deep vein thrombosis (DVT) FREE steering committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605-1611.
5. Osterman AL, Lincoski C. Thoracic outlet syndrome. In: Skirven TM, Osterman AL, Fedorczyk JM, et al, eds. Rehabilitation of the Hand and Upper Extremity. 6th ed. Philadelphia, Pa: Mosby, Inc; 2011:723-732.
6. Laker S, Sullivan WJ, Whitehill TA. Thoracic outlet syndrome. In: Akuthota V, Herring SA, eds. Nerve and vascular injuries in sports medicine. New York, NY: Springer; 2009:117.
7. Urschel HC Jr, Patel AN. Surgery remains the most effective treatment for Paget-Schroetter syndrome: 50 years’ experience. Ann Thorac Surg. 2008;86:254-260; discussion 260.
8. Melby SJ, Vedantham S, Narra VR, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809-820; discussion 821.
9. Di Nisio M, Van Sluis GL, Bossuyt PM, et al. Accuracy of diagnostic tests for clinically suspected upper extremity deep vein thrombosis: a systematic review. J Thromb Haemost. 2010;8:684-692.
10. Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol. 2012;29:44-51.
11. Desjardins B, Rybicki FJ, Kim HS, et al. ACR Appropriateness Criteria® Suspected upper extremity deep vein thrombosis. J Am Coll Radiol. 2012;9:613-619.
12. Savage KJ, Wells PS, Schulz V, et al. Outpatient use of low molecular weight heparin (Dalteparin) for the treatment of deep vein thrombosis of the upper extremity. Thromb Haemost. 1999;82:1008-1010.
13. Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315; discussion 316-317.
14. Thomas IH, Zierler BK. An integrative review of outcomes in patients with acute primary upper extremity deep venous thrombosis following no treatment or treatment with anticoagulation, thrombolysis, or surgical algorithms. Vasc Endovascular Surg. 2005;39:163-174.
THE CASE
A 16-year-old boy presented to the emergency room (ER) with pain, redness, and swelling of his right upper arm that had been bothering him for 2 days. He was the quarterback of his high school football team, a sport that he’d been playing since he was 8 years old. He indicated that his football training—which involved repetitive throwing with his right arm—had intensified over the previous 2 months.
Prior to the ER visit, the patient was healthy and active with no significant medical history. He’d had no shoulder trauma and there was no family history of any coagulopathies, venous thrombosis, or pulmonary embolism. He denied chest pain, shortness of breath, palpitations, and fever, and said that he did not smoke cigarettes or drink alcohol.
On physical examination, his blood pressure was 118/70 mm Hg and his heart rate was 74 beats per minute. He had nonpitting edema and erythema of his right upper arm. His radial and brachial pulses were strong and equal in both arms. Assessment of neurologic and vascular integrity produced positive Wright’s and Adson’s tests, but a negative Halstead’s test. (For more on these tests, see: Wright’s test, Adson’s test, and Halstead’s test.) The circumference of the patient’s right upper arm was 2.5 cm greater than the left upper arm. The remainder of the physical exam was normal.
THE DIAGNOSIS
A duplex ultrasound of the right upper arm revealed an acute occlusive thrombus in the axillary vein. We started the patient on intravenous heparin. A venogram confirmed thrombosis of the axillary-subclavian vein (FIGURE 1A). Based on the patient’s clinical presentation and the results of the venogram, we diagnosed Paget-Schroetter syndrome. The venogram was followed by thrombolysis with alteplase (FIGURE 1B) and a balloon angioplasty (FIGURE 1C). One week later, a repeat venogram demonstrated partial removal of the thrombus and an area of compression on the inferior aspect of the subclavian vein due to a cervical band (FIGURE 1D).
DISCUSSION
Paget-Schroetter syndrome (PSS), or effort thrombosis of the upper extremities, is defined as spontaneous thrombus in the axillary and subclavian veins that occurs as a consequence of strenuous upper-extremity activity. It is a rare condition with an incidence of one to 2 cases per 100,000 people per year, and represents 1% to 4% of all cases of deep vein thrombosis (DVT).1
Spontaneous thrombosis of the upper extremities typically presents in young, otherwise healthy individuals. It has been described in athletes who are involved in ball games, games with rackets or clubs, aquatic sports, combatant sports, and in violin players.2 The repetitive movements used in these activities can lead to compression of the axillary and subclavian veins by hypertrophied muscles. Repetitive trauma causes intimal damage and thrombogenesis.3
PSS is characterized by the abrupt, spontaneous swelling of the entire arm, cyanosis, and pain that occurs with use or overhead positioning. Enlarged subcutaneous veins are present in the upper arm, around the shoulder, or in the upper anterior chest wall (Urschel’s sign). The classic presentation is acute onset of upper extremity pain and swelling in the dominant arm following a particularly strenuous activity.4 A low-grade fever, superficial thrombophlebitis, or neurologic symptoms may coexist. Certain provocative maneuvers can help reproduce the symptoms (TABLE 15,6). Complications of PSS include pulmonary embolism, postthrombotic syndrome (pain, heaviness, and swelling), and recurrent thrombosis.7
Contrast venography best shows the extent of thrombosis
Duplex ultrasound, with its high sensitivity and specificity, is the initial, noninvasive test of choice (TABLE 24,8-11). However, duplex ultrasound has a false-negative rate of 30% because it is highly technician-dependent and can be complicated by acoustic shadows from the clavicle or sternum.8
The most direct and definitive means to confirm the diagnosis of PSS is catheter-directed contrast venography.9 This method provides complete anatomic information regarding the site and extent of thrombosis, allows definitive evaluation of the collateral venous pathways, and is a necessary step toward the use of thrombolytic therapy. Contrast load, however, contraindicates the procedure in patients with renal failure and in those who are pregnant.
Contrast-enhanced computed tomography (CT) and magnetic resonance angiography (MRA) are also highly sensitive for detecting focal stenosis at the level of the first rib, the presence or absence of enlarged collateral veins, and the chronicity of any thrombus present. However, the usefulness of CT and magnetic resonance venography in initial screening is unclear, due to a lack of randomized controlled trials.
Treatment involves anticoagulants, thrombolytics, and possibly surgery
Prompt use of anticoagulation is indicated in PSS. Initial anticoagulation with low molecular weight unfractionated heparin or a direct thrombin inhibitor followed by warfarin for a minimum of 3 to 6 months is recommended.12
Patients treated with anticoagulation alone have a higher incidence of long-term residual symptoms, disability, and recurrent thrombosis.7 As a result, a more aggressive approach with the use of thrombolytic therapy is indicated, especially in young, active patients, to minimize long-term consequences. Alteplase or reteplase are used for this purpose. Thrombolysis is less likely to be beneficial if the thrombus is more than 2 weeks old or if there are inflammatory changes in the vein. The use of catheter-directed thrombolysis minimizes the risk of systemic adverse effects and achieves higher clot resolution rates.13
Because PSS is caused by compression of the vein, rather than a disorder of blood clotting, there is still a 50% to 70% risk of recurrent thrombosis despite thrombolysis and anticoagulation.14 Therefore, the most definitive management approach remains surgical treatment. Patients with recent thrombosis who are within the first several weeks of undergoing successful thrombolytic therapy are excellent candidates for surgery. Operative treatment for PSS includes first rib resection, scalene muscle removal, or subclavius muscle removal, along with removal of constricting scar tissue from around the vein.7
THE TAKEAWAY
PSS is characterized by upper-extremity DVT resulting from repetitive trauma to the subclavian-axillary vein. Early diagnosis of PSS with contrast venography and prompt use of anticoagulation can effectively restore venous patency, reduce the risk of rethrombosis, and return the patient to normal function. Primary care physicians should be aware of this condition, because delayed recognition in a high-functioning person can be potentially disabling.
Our patient had a first rib resection, partial division of the scalenus anterior and medius muscles, and lysis of the cervical band. Follow-up venography confirmed resolution of thrombosis without any complications. The patient was continued on anticoagulation with warfarin for 3 months.
THE CASE
A 16-year-old boy presented to the emergency room (ER) with pain, redness, and swelling of his right upper arm that had been bothering him for 2 days. He was the quarterback of his high school football team, a sport that he’d been playing since he was 8 years old. He indicated that his football training—which involved repetitive throwing with his right arm—had intensified over the previous 2 months.
Prior to the ER visit, the patient was healthy and active with no significant medical history. He’d had no shoulder trauma and there was no family history of any coagulopathies, venous thrombosis, or pulmonary embolism. He denied chest pain, shortness of breath, palpitations, and fever, and said that he did not smoke cigarettes or drink alcohol.
On physical examination, his blood pressure was 118/70 mm Hg and his heart rate was 74 beats per minute. He had nonpitting edema and erythema of his right upper arm. His radial and brachial pulses were strong and equal in both arms. Assessment of neurologic and vascular integrity produced positive Wright’s and Adson’s tests, but a negative Halstead’s test. (For more on these tests, see: Wright’s test, Adson’s test, and Halstead’s test.) The circumference of the patient’s right upper arm was 2.5 cm greater than the left upper arm. The remainder of the physical exam was normal.
THE DIAGNOSIS
A duplex ultrasound of the right upper arm revealed an acute occlusive thrombus in the axillary vein. We started the patient on intravenous heparin. A venogram confirmed thrombosis of the axillary-subclavian vein (FIGURE 1A). Based on the patient’s clinical presentation and the results of the venogram, we diagnosed Paget-Schroetter syndrome. The venogram was followed by thrombolysis with alteplase (FIGURE 1B) and a balloon angioplasty (FIGURE 1C). One week later, a repeat venogram demonstrated partial removal of the thrombus and an area of compression on the inferior aspect of the subclavian vein due to a cervical band (FIGURE 1D).
DISCUSSION
Paget-Schroetter syndrome (PSS), or effort thrombosis of the upper extremities, is defined as spontaneous thrombus in the axillary and subclavian veins that occurs as a consequence of strenuous upper-extremity activity. It is a rare condition with an incidence of one to 2 cases per 100,000 people per year, and represents 1% to 4% of all cases of deep vein thrombosis (DVT).1
Spontaneous thrombosis of the upper extremities typically presents in young, otherwise healthy individuals. It has been described in athletes who are involved in ball games, games with rackets or clubs, aquatic sports, combatant sports, and in violin players.2 The repetitive movements used in these activities can lead to compression of the axillary and subclavian veins by hypertrophied muscles. Repetitive trauma causes intimal damage and thrombogenesis.3
PSS is characterized by the abrupt, spontaneous swelling of the entire arm, cyanosis, and pain that occurs with use or overhead positioning. Enlarged subcutaneous veins are present in the upper arm, around the shoulder, or in the upper anterior chest wall (Urschel’s sign). The classic presentation is acute onset of upper extremity pain and swelling in the dominant arm following a particularly strenuous activity.4 A low-grade fever, superficial thrombophlebitis, or neurologic symptoms may coexist. Certain provocative maneuvers can help reproduce the symptoms (TABLE 15,6). Complications of PSS include pulmonary embolism, postthrombotic syndrome (pain, heaviness, and swelling), and recurrent thrombosis.7
Contrast venography best shows the extent of thrombosis
Duplex ultrasound, with its high sensitivity and specificity, is the initial, noninvasive test of choice (TABLE 24,8-11). However, duplex ultrasound has a false-negative rate of 30% because it is highly technician-dependent and can be complicated by acoustic shadows from the clavicle or sternum.8
The most direct and definitive means to confirm the diagnosis of PSS is catheter-directed contrast venography.9 This method provides complete anatomic information regarding the site and extent of thrombosis, allows definitive evaluation of the collateral venous pathways, and is a necessary step toward the use of thrombolytic therapy. Contrast load, however, contraindicates the procedure in patients with renal failure and in those who are pregnant.
Contrast-enhanced computed tomography (CT) and magnetic resonance angiography (MRA) are also highly sensitive for detecting focal stenosis at the level of the first rib, the presence or absence of enlarged collateral veins, and the chronicity of any thrombus present. However, the usefulness of CT and magnetic resonance venography in initial screening is unclear, due to a lack of randomized controlled trials.
Treatment involves anticoagulants, thrombolytics, and possibly surgery
Prompt use of anticoagulation is indicated in PSS. Initial anticoagulation with low molecular weight unfractionated heparin or a direct thrombin inhibitor followed by warfarin for a minimum of 3 to 6 months is recommended.12
Patients treated with anticoagulation alone have a higher incidence of long-term residual symptoms, disability, and recurrent thrombosis.7 As a result, a more aggressive approach with the use of thrombolytic therapy is indicated, especially in young, active patients, to minimize long-term consequences. Alteplase or reteplase are used for this purpose. Thrombolysis is less likely to be beneficial if the thrombus is more than 2 weeks old or if there are inflammatory changes in the vein. The use of catheter-directed thrombolysis minimizes the risk of systemic adverse effects and achieves higher clot resolution rates.13
Because PSS is caused by compression of the vein, rather than a disorder of blood clotting, there is still a 50% to 70% risk of recurrent thrombosis despite thrombolysis and anticoagulation.14 Therefore, the most definitive management approach remains surgical treatment. Patients with recent thrombosis who are within the first several weeks of undergoing successful thrombolytic therapy are excellent candidates for surgery. Operative treatment for PSS includes first rib resection, scalene muscle removal, or subclavius muscle removal, along with removal of constricting scar tissue from around the vein.7
THE TAKEAWAY
PSS is characterized by upper-extremity DVT resulting from repetitive trauma to the subclavian-axillary vein. Early diagnosis of PSS with contrast venography and prompt use of anticoagulation can effectively restore venous patency, reduce the risk of rethrombosis, and return the patient to normal function. Primary care physicians should be aware of this condition, because delayed recognition in a high-functioning person can be potentially disabling.
Our patient had a first rib resection, partial division of the scalenus anterior and medius muscles, and lysis of the cervical band. Follow-up venography confirmed resolution of thrombosis without any complications. The patient was continued on anticoagulation with warfarin for 3 months.
1. Isma N, Svensson PJ, Gottsäter A, et al. Upper extremity deep venous thrombosis in the population-based Malmö thrombophilia study (MATS). Epidemiology, risk factors, recurrence risk, and mortality. Thromb Res. 2010;125:e335-e338.
2. DiFelice GS, Paletta GA Jr, Phillips BB, et al. Effort thrombosis in the elite throwing athlete. Am J Sports Med. 2002;30:708-712.
3. Thompson JF, Winterborn RJ, Bays S, et al. Venous thoracic outlet compression and the Paget-Schroetter syndrome: a review and recommendations for management. Cardiovasc Intervent Radiol. 2011;34:903-910.
4. Joffe HV, Kucher N, Tapson VF, et al; Deep vein thrombosis (DVT) FREE steering committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605-1611.
5. Osterman AL, Lincoski C. Thoracic outlet syndrome. In: Skirven TM, Osterman AL, Fedorczyk JM, et al, eds. Rehabilitation of the Hand and Upper Extremity. 6th ed. Philadelphia, Pa: Mosby, Inc; 2011:723-732.
6. Laker S, Sullivan WJ, Whitehill TA. Thoracic outlet syndrome. In: Akuthota V, Herring SA, eds. Nerve and vascular injuries in sports medicine. New York, NY: Springer; 2009:117.
7. Urschel HC Jr, Patel AN. Surgery remains the most effective treatment for Paget-Schroetter syndrome: 50 years’ experience. Ann Thorac Surg. 2008;86:254-260; discussion 260.
8. Melby SJ, Vedantham S, Narra VR, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809-820; discussion 821.
9. Di Nisio M, Van Sluis GL, Bossuyt PM, et al. Accuracy of diagnostic tests for clinically suspected upper extremity deep vein thrombosis: a systematic review. J Thromb Haemost. 2010;8:684-692.
10. Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol. 2012;29:44-51.
11. Desjardins B, Rybicki FJ, Kim HS, et al. ACR Appropriateness Criteria® Suspected upper extremity deep vein thrombosis. J Am Coll Radiol. 2012;9:613-619.
12. Savage KJ, Wells PS, Schulz V, et al. Outpatient use of low molecular weight heparin (Dalteparin) for the treatment of deep vein thrombosis of the upper extremity. Thromb Haemost. 1999;82:1008-1010.
13. Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315; discussion 316-317.
14. Thomas IH, Zierler BK. An integrative review of outcomes in patients with acute primary upper extremity deep venous thrombosis following no treatment or treatment with anticoagulation, thrombolysis, or surgical algorithms. Vasc Endovascular Surg. 2005;39:163-174.
1. Isma N, Svensson PJ, Gottsäter A, et al. Upper extremity deep venous thrombosis in the population-based Malmö thrombophilia study (MATS). Epidemiology, risk factors, recurrence risk, and mortality. Thromb Res. 2010;125:e335-e338.
2. DiFelice GS, Paletta GA Jr, Phillips BB, et al. Effort thrombosis in the elite throwing athlete. Am J Sports Med. 2002;30:708-712.
3. Thompson JF, Winterborn RJ, Bays S, et al. Venous thoracic outlet compression and the Paget-Schroetter syndrome: a review and recommendations for management. Cardiovasc Intervent Radiol. 2011;34:903-910.
4. Joffe HV, Kucher N, Tapson VF, et al; Deep vein thrombosis (DVT) FREE steering committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605-1611.
5. Osterman AL, Lincoski C. Thoracic outlet syndrome. In: Skirven TM, Osterman AL, Fedorczyk JM, et al, eds. Rehabilitation of the Hand and Upper Extremity. 6th ed. Philadelphia, Pa: Mosby, Inc; 2011:723-732.
6. Laker S, Sullivan WJ, Whitehill TA. Thoracic outlet syndrome. In: Akuthota V, Herring SA, eds. Nerve and vascular injuries in sports medicine. New York, NY: Springer; 2009:117.
7. Urschel HC Jr, Patel AN. Surgery remains the most effective treatment for Paget-Schroetter syndrome: 50 years’ experience. Ann Thorac Surg. 2008;86:254-260; discussion 260.
8. Melby SJ, Vedantham S, Narra VR, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809-820; discussion 821.
9. Di Nisio M, Van Sluis GL, Bossuyt PM, et al. Accuracy of diagnostic tests for clinically suspected upper extremity deep vein thrombosis: a systematic review. J Thromb Haemost. 2010;8:684-692.
10. Thompson RW. Comprehensive management of subclavian vein effort thrombosis. Semin Intervent Radiol. 2012;29:44-51.
11. Desjardins B, Rybicki FJ, Kim HS, et al. ACR Appropriateness Criteria® Suspected upper extremity deep vein thrombosis. J Am Coll Radiol. 2012;9:613-619.
12. Savage KJ, Wells PS, Schulz V, et al. Outpatient use of low molecular weight heparin (Dalteparin) for the treatment of deep vein thrombosis of the upper extremity. Thromb Haemost. 1999;82:1008-1010.
13. Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315; discussion 316-317.
14. Thomas IH, Zierler BK. An integrative review of outcomes in patients with acute primary upper extremity deep venous thrombosis following no treatment or treatment with anticoagulation, thrombolysis, or surgical algorithms. Vasc Endovascular Surg. 2005;39:163-174.

Acute promyelocytic leukemia presenting as a paraspinal mass
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)], which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor α (RARA) genes.1,2 Historically, APL was a fatal disease because of the high relapse rates with cytotoxic chemotherapy alone and a significant bleeding risk secondary to disseminated intravascular coagulation (DIC).
Click on the PDF icon at the top of this introduction to read the full article.
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)], which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor α (RARA) genes.1,2 Historically, APL was a fatal disease because of the high relapse rates with cytotoxic chemotherapy alone and a significant bleeding risk secondary to disseminated intravascular coagulation (DIC).
Click on the PDF icon at the top of this introduction to read the full article.
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)], which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor α (RARA) genes.1,2 Historically, APL was a fatal disease because of the high relapse rates with cytotoxic chemotherapy alone and a significant bleeding risk secondary to disseminated intravascular coagulation (DIC).
Click on the PDF icon at the top of this introduction to read the full article.
An uncommon presentation of non-small-cell lung cancer with acrometastases to the great toe and index finger
Case presentation and summary
A 71-year-old white woman was referred to the emergency department by her primary care physician for necrosis and swelling of the left great toe for work-up of possible osteomyelitis (Figure 1). Before she presented to her physician, she had been complaining of severe pain, swelling, and erythema of the left great toe that had lasted for 1-2 months. Infection was initially suspected. She completed 2 courses of oral antibiotics with no improvement. She was also complaining of similar symptoms on the left index finger and attributed her symptoms to an injury a month earlier (Figure 2). The pain was so severe that she was not able to bear weight on her left foot. An outpatient X-ray of her left great toe raised her physician’s concerns that it might be osteomyelitis so she was referred to the emergency department.
Click on the PDF icon at the top of this introduction to read the full article.
Case presentation and summary
A 71-year-old white woman was referred to the emergency department by her primary care physician for necrosis and swelling of the left great toe for work-up of possible osteomyelitis (Figure 1). Before she presented to her physician, she had been complaining of severe pain, swelling, and erythema of the left great toe that had lasted for 1-2 months. Infection was initially suspected. She completed 2 courses of oral antibiotics with no improvement. She was also complaining of similar symptoms on the left index finger and attributed her symptoms to an injury a month earlier (Figure 2). The pain was so severe that she was not able to bear weight on her left foot. An outpatient X-ray of her left great toe raised her physician’s concerns that it might be osteomyelitis so she was referred to the emergency department.
Click on the PDF icon at the top of this introduction to read the full article.
Case presentation and summary
A 71-year-old white woman was referred to the emergency department by her primary care physician for necrosis and swelling of the left great toe for work-up of possible osteomyelitis (Figure 1). Before she presented to her physician, she had been complaining of severe pain, swelling, and erythema of the left great toe that had lasted for 1-2 months. Infection was initially suspected. She completed 2 courses of oral antibiotics with no improvement. She was also complaining of similar symptoms on the left index finger and attributed her symptoms to an injury a month earlier (Figure 2). The pain was so severe that she was not able to bear weight on her left foot. An outpatient X-ray of her left great toe raised her physician’s concerns that it might be osteomyelitis so she was referred to the emergency department.
Click on the PDF icon at the top of this introduction to read the full article.