User login
Total Hip Arthroplasty After Proximal Femoral Osteotomy: A Technique That Can Be Used to Address Presence of a Retained Intracortical Plate
Total hip arthroplasty (THA) is an effective treatment for advanced hip arthritis from a variety of causes, including osteoarthritis, inflammatory arthritis, posttraumatic arthritis, and sequelae of developmental disorders. It is not uncommon to perform THA in the presence of a previous proximal femoral osteotomy that may have been performed for slipped capital femoral epiphysis (SCFE), Legg-Calvé-Perthes disease, or developmental dysplasia of the hip, among other conditions. These osteotomies are commonly combined with internal fixation, a plate-and-screw device. These patients are at risk for developing degenerative arthritis at an earlier age than patients with other types of arthritis and subsequently may undergo THA at a younger age.1-3 Presence of a plate can pose a technical challenge during THA surgery. THA performed after intertrochanteric osteotomy has higher rates of perioperative and postoperative complications.4 Ferguson and colleagues4 noted difficulty during hardware removal in 24% of cases. Among the complications encountered were broken hardware, stripped screws, greater trochanteric fracture, stress risers from previous screw holes, canal narrowing from endosteal hypertrophy around hardware, and lateral cortical deficiency after removal of the side plate. As intertrochanteric osteotomies are often performed in patients who have yet to reach skeletal maturity, cortical hypertrophy can lead to complete coverage of the side plate and an “intracortical” position.
This article reports on 2 THA cases in which a technique was used to avoid intracortical plate removal and the resulting problems of lateral cortical deficiency. During each THA, the plate was left in place to avoid compromise of the lateral femoral cortex. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
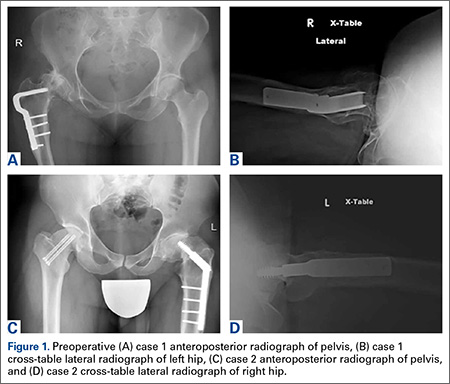
An adolescent with bilateral SCFE was treated first with internal fixation of the right hip and subsequently with left proximal femoral osteotomy with internal fixation. He did well until age 31 years, when he developed progressively worsening pain about the left hip. Clinical findings and imaging studies were consistent with advanced degenerative arthritis of the left hip. Radiographs showed a sliding hip screw in place, with proximal femoral deformity consisting of femoral neck shortening and posterior angulation (Figures 1A, 1B). Preoperative Harris Hip Score was 54.5.
Case 2
A 51-year-old woman presented with a history of right hip problems dating back to age 13 years, when she sustained a fracture of the right hip and was treated with internal fixation. At age 15 years, she underwent proximal femoral osteotomy to correct residual deformity. She did well until age 45 years, when she developed worsening hip symptoms. Clinical findings and imaging studies were consistent with advanced degenerative arthritis of the right hip. Radiographs showed a fixed-angle blade plate in the proximal femur, with significant proximal femoral deformity (Figures 1C, 1D). Preoperative Harris Hip Score was 53.6.
Surgical Technique
In both cases, a standard series of radiographs was obtained—an anteroposterior (AP) radiograph of the pelvis and AP and cross-table lateral radiographs of the operative hip (Figure 1). Computed tomography (CT) with a metal-artifact-reducing technique may be useful in determining amount of cortical bone remaining under the plate. CT showed limited lateral cortex beneath the side plate and bony overgrowth covering the side plate. Preoperative templating was performed using previously described techniques.5
During THA, before removing any portion of any retained hardware, the surgeon should perform 3 important actions: Dislocate the hip, perform all appropriate capsular releases, and reduce the hip. Dislocating the hip before hardware removal significantly decreases the risk for fracture caused by stress risers, as the force required for dislocation is much more controlled because of the capsular releases. After hardware removal, the hip can be easily redislocated, and the femoral neck osteotomy can be performed.
When plate and screws are in an intracortical position, the screws can be removed only after removing the small shell of cortical bone covering them. The amount of bone to be removed is minimal. After the screws are removed, the plate remains in place. A motorized device with a metal-cutting attachment is used to transect the construct at the junction of the plate and barrel (case 1) or at the bend of a fixed-angle device (case 2). Laparotomy sponges are placed around the proximal femur to minimize the amount of soft tissue that could be exposed to metal shavings. Copious irrigation is used throughout this part of the procedure. Osteotomes are used to elevate the proximal portion of the plate and the barrel, preserving the distal portion of the plate on the lateral cortex of the femoral shaft.
After the head is removed, the rest of the THA can be performed using standard press-fit insertion technique (Figures 2A-2D). Care must be taken to ensure that the distal aspect of the femoral stem bypasses the most distal screw hole by at least 2 cortical diameters in order to reduce the risk for periprosthetic fracture.
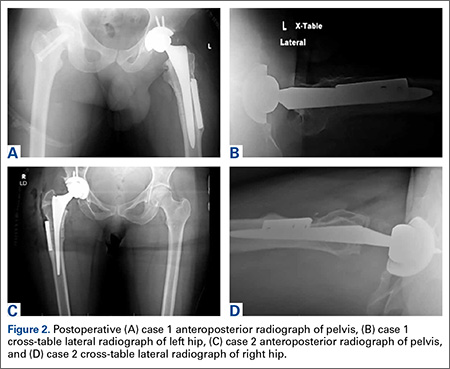
By 2-year follow-up, both patients had regained excellent range of motion, ambulation, and overall function. Postoperative Harris Hip Scores were 86.6 and 83.8, respectively. There were no radiographic signs of complications.
Discussion
THA can be challenging in the setting of previously placed internal fixation devices, particularly devices inserted during a patient’s adolescence, as significant bony overgrowth can occur. The standard approach has been to remove the internal fixation device and then perform the THA. In most cases, and particularly when the internal fixation device is in an intracortical position, the result is significant compromise of bone. This article describes a technique in which a portion of the hardware is retained to avoid compromise of the lateral femoral cortex, thereby allowing insertion of a noncemented femoral component.
THA is the most effective procedure for reducing hip pain and disability in the setting of degenerative changes.6 Patients with SCFE, Legg-Calvé-Perthes disease, or developmental dysplasia of the hip generally are younger at the time they may be sufficiently symptomatic to consider THA.7,8 Many have had previous surgery using internal fixation devices. THAs after previous osteotomies with internal fixation devices are more technically demanding, require more operative time, are subject to more blood loss, and have a higher rate of complications, including femoral fracture. Ferguson and colleagues4 and Boos and colleagues9 found these surgeries were more difficult 33.8% and 36.8% of the time, respectively. For these reasons, some authors have recommended removing the internal fixation device as soon as the osteotomy is healed.4 However, this has not become the standard of care, and surgeons continue to perform THAs in the presence of a previous osteotomy with an internal fixation device in place.
The technique described in this article was used successfully in 2 cases. In each case, leaving the intracortical plate in place avoided compromise of the lateral femoral cortex and allowed insertion of a noncemented femoral component without complication. Of course, with the screw holes representing stress risers, careful insertion of the femoral component was required. Retaining the intracortical plate allowed it to function as part of the lateral femoral cortex, thereby maintaining the structural integrity of the femoral canal. As has been described for the 2 cases, a blade plate and plate and barrel were converted to a limited intracortical plate by removing the proximal portion of the plates—a modification that could be applied to other types of internal fixation devices that extend into the femoral neck as long as appropriate cutting tools are available.
Conclusion
THA in the setting of a retained internal fixation device is relatively common. This article describes a technique that can be used when a plate applied to the lateral femoral cortex has become intracortical as a result of extensive bony overgrowth. In using this technique to avoid plate removal, the surgeon eliminates the need for more extensive procedures aimed at compensating for deficiency of the femoral cortex in the area of plate removal. Although only 2 cases are presented here, this technique potentially can be used more broadly in these specific clinical situations.
1. Engesæter LB, Engesæter IØ, Fenstad AM, et al. Low revision rate after total hip arthroplasty in patients with pediatric hip diseases. Acta Orthop. 2012;83(5):436-441.
2. Froberg L, Christensen F, Pedersen NW, Overgaard S. The need for total hip arthroplasty in Perthes disease: a long-term study. Clin Orthop Relat Res. 2011;469(4):1134-1140.
3. Furnes O, Lie SA, Espehaug B, Vollset SE, Engesæter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586.
4. Ferguson GM, Cabanela ME, Ilstrup DM. Total hip arthroplasty after failed intertrochanteric osteotomy. J Bone Joint Surg Br. 1994;76(2):252-257.
5. Scheerlinck T. Primary hip arthroplasty templating on standard radiographs. A stepwise approach. Acta Orthop Belg. 2010;76(4):432-442.
6. Wroblewski BM, Siney PD. Charnley low-friction arthroplasty of the hip. Long-term results. Clin Orthop Relat Res. 1993;(292):191-201.
7. Chandler HP, Reineck FT, Wixson RL, McCarthy JC. Total hip replacement in patients younger than thirty years old. A five-year follow-up study. J Bone Joint Surg Am. 1981;63(9):1426-1434.
8. Dorr LD, Luckett M, Conaty JP. Total hip arthroplasties in patients younger than 45 years. A nine- to ten-year follow-up study. Clin Orthop Relat Res. 1990;(260):215-219.
9. Boos N, Krushell R, Ganz R, Müller ME. Total hip arthroplasty after previous proximal femoral osteotomy. J Bone Joint Surg Br. 1997;79(2):247-253.
Total hip arthroplasty (THA) is an effective treatment for advanced hip arthritis from a variety of causes, including osteoarthritis, inflammatory arthritis, posttraumatic arthritis, and sequelae of developmental disorders. It is not uncommon to perform THA in the presence of a previous proximal femoral osteotomy that may have been performed for slipped capital femoral epiphysis (SCFE), Legg-Calvé-Perthes disease, or developmental dysplasia of the hip, among other conditions. These osteotomies are commonly combined with internal fixation, a plate-and-screw device. These patients are at risk for developing degenerative arthritis at an earlier age than patients with other types of arthritis and subsequently may undergo THA at a younger age.1-3 Presence of a plate can pose a technical challenge during THA surgery. THA performed after intertrochanteric osteotomy has higher rates of perioperative and postoperative complications.4 Ferguson and colleagues4 noted difficulty during hardware removal in 24% of cases. Among the complications encountered were broken hardware, stripped screws, greater trochanteric fracture, stress risers from previous screw holes, canal narrowing from endosteal hypertrophy around hardware, and lateral cortical deficiency after removal of the side plate. As intertrochanteric osteotomies are often performed in patients who have yet to reach skeletal maturity, cortical hypertrophy can lead to complete coverage of the side plate and an “intracortical” position.
This article reports on 2 THA cases in which a technique was used to avoid intracortical plate removal and the resulting problems of lateral cortical deficiency. During each THA, the plate was left in place to avoid compromise of the lateral femoral cortex. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
An adolescent with bilateral SCFE was treated first with internal fixation of the right hip and subsequently with left proximal femoral osteotomy with internal fixation. He did well until age 31 years, when he developed progressively worsening pain about the left hip. Clinical findings and imaging studies were consistent with advanced degenerative arthritis of the left hip. Radiographs showed a sliding hip screw in place, with proximal femoral deformity consisting of femoral neck shortening and posterior angulation (Figures 1A, 1B). Preoperative Harris Hip Score was 54.5.
Case 2
A 51-year-old woman presented with a history of right hip problems dating back to age 13 years, when she sustained a fracture of the right hip and was treated with internal fixation. At age 15 years, she underwent proximal femoral osteotomy to correct residual deformity. She did well until age 45 years, when she developed worsening hip symptoms. Clinical findings and imaging studies were consistent with advanced degenerative arthritis of the right hip. Radiographs showed a fixed-angle blade plate in the proximal femur, with significant proximal femoral deformity (Figures 1C, 1D). Preoperative Harris Hip Score was 53.6.
Surgical Technique
In both cases, a standard series of radiographs was obtained—an anteroposterior (AP) radiograph of the pelvis and AP and cross-table lateral radiographs of the operative hip (Figure 1). Computed tomography (CT) with a metal-artifact-reducing technique may be useful in determining amount of cortical bone remaining under the plate. CT showed limited lateral cortex beneath the side plate and bony overgrowth covering the side plate. Preoperative templating was performed using previously described techniques.5
During THA, before removing any portion of any retained hardware, the surgeon should perform 3 important actions: Dislocate the hip, perform all appropriate capsular releases, and reduce the hip. Dislocating the hip before hardware removal significantly decreases the risk for fracture caused by stress risers, as the force required for dislocation is much more controlled because of the capsular releases. After hardware removal, the hip can be easily redislocated, and the femoral neck osteotomy can be performed.
When plate and screws are in an intracortical position, the screws can be removed only after removing the small shell of cortical bone covering them. The amount of bone to be removed is minimal. After the screws are removed, the plate remains in place. A motorized device with a metal-cutting attachment is used to transect the construct at the junction of the plate and barrel (case 1) or at the bend of a fixed-angle device (case 2). Laparotomy sponges are placed around the proximal femur to minimize the amount of soft tissue that could be exposed to metal shavings. Copious irrigation is used throughout this part of the procedure. Osteotomes are used to elevate the proximal portion of the plate and the barrel, preserving the distal portion of the plate on the lateral cortex of the femoral shaft.
After the head is removed, the rest of the THA can be performed using standard press-fit insertion technique (Figures 2A-2D). Care must be taken to ensure that the distal aspect of the femoral stem bypasses the most distal screw hole by at least 2 cortical diameters in order to reduce the risk for periprosthetic fracture.
By 2-year follow-up, both patients had regained excellent range of motion, ambulation, and overall function. Postoperative Harris Hip Scores were 86.6 and 83.8, respectively. There were no radiographic signs of complications.
Discussion
THA can be challenging in the setting of previously placed internal fixation devices, particularly devices inserted during a patient’s adolescence, as significant bony overgrowth can occur. The standard approach has been to remove the internal fixation device and then perform the THA. In most cases, and particularly when the internal fixation device is in an intracortical position, the result is significant compromise of bone. This article describes a technique in which a portion of the hardware is retained to avoid compromise of the lateral femoral cortex, thereby allowing insertion of a noncemented femoral component.
THA is the most effective procedure for reducing hip pain and disability in the setting of degenerative changes.6 Patients with SCFE, Legg-Calvé-Perthes disease, or developmental dysplasia of the hip generally are younger at the time they may be sufficiently symptomatic to consider THA.7,8 Many have had previous surgery using internal fixation devices. THAs after previous osteotomies with internal fixation devices are more technically demanding, require more operative time, are subject to more blood loss, and have a higher rate of complications, including femoral fracture. Ferguson and colleagues4 and Boos and colleagues9 found these surgeries were more difficult 33.8% and 36.8% of the time, respectively. For these reasons, some authors have recommended removing the internal fixation device as soon as the osteotomy is healed.4 However, this has not become the standard of care, and surgeons continue to perform THAs in the presence of a previous osteotomy with an internal fixation device in place.
The technique described in this article was used successfully in 2 cases. In each case, leaving the intracortical plate in place avoided compromise of the lateral femoral cortex and allowed insertion of a noncemented femoral component without complication. Of course, with the screw holes representing stress risers, careful insertion of the femoral component was required. Retaining the intracortical plate allowed it to function as part of the lateral femoral cortex, thereby maintaining the structural integrity of the femoral canal. As has been described for the 2 cases, a blade plate and plate and barrel were converted to a limited intracortical plate by removing the proximal portion of the plates—a modification that could be applied to other types of internal fixation devices that extend into the femoral neck as long as appropriate cutting tools are available.
Conclusion
THA in the setting of a retained internal fixation device is relatively common. This article describes a technique that can be used when a plate applied to the lateral femoral cortex has become intracortical as a result of extensive bony overgrowth. In using this technique to avoid plate removal, the surgeon eliminates the need for more extensive procedures aimed at compensating for deficiency of the femoral cortex in the area of plate removal. Although only 2 cases are presented here, this technique potentially can be used more broadly in these specific clinical situations.
Total hip arthroplasty (THA) is an effective treatment for advanced hip arthritis from a variety of causes, including osteoarthritis, inflammatory arthritis, posttraumatic arthritis, and sequelae of developmental disorders. It is not uncommon to perform THA in the presence of a previous proximal femoral osteotomy that may have been performed for slipped capital femoral epiphysis (SCFE), Legg-Calvé-Perthes disease, or developmental dysplasia of the hip, among other conditions. These osteotomies are commonly combined with internal fixation, a plate-and-screw device. These patients are at risk for developing degenerative arthritis at an earlier age than patients with other types of arthritis and subsequently may undergo THA at a younger age.1-3 Presence of a plate can pose a technical challenge during THA surgery. THA performed after intertrochanteric osteotomy has higher rates of perioperative and postoperative complications.4 Ferguson and colleagues4 noted difficulty during hardware removal in 24% of cases. Among the complications encountered were broken hardware, stripped screws, greater trochanteric fracture, stress risers from previous screw holes, canal narrowing from endosteal hypertrophy around hardware, and lateral cortical deficiency after removal of the side plate. As intertrochanteric osteotomies are often performed in patients who have yet to reach skeletal maturity, cortical hypertrophy can lead to complete coverage of the side plate and an “intracortical” position.
This article reports on 2 THA cases in which a technique was used to avoid intracortical plate removal and the resulting problems of lateral cortical deficiency. During each THA, the plate was left in place to avoid compromise of the lateral femoral cortex. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
An adolescent with bilateral SCFE was treated first with internal fixation of the right hip and subsequently with left proximal femoral osteotomy with internal fixation. He did well until age 31 years, when he developed progressively worsening pain about the left hip. Clinical findings and imaging studies were consistent with advanced degenerative arthritis of the left hip. Radiographs showed a sliding hip screw in place, with proximal femoral deformity consisting of femoral neck shortening and posterior angulation (Figures 1A, 1B). Preoperative Harris Hip Score was 54.5.
Case 2
A 51-year-old woman presented with a history of right hip problems dating back to age 13 years, when she sustained a fracture of the right hip and was treated with internal fixation. At age 15 years, she underwent proximal femoral osteotomy to correct residual deformity. She did well until age 45 years, when she developed worsening hip symptoms. Clinical findings and imaging studies were consistent with advanced degenerative arthritis of the right hip. Radiographs showed a fixed-angle blade plate in the proximal femur, with significant proximal femoral deformity (Figures 1C, 1D). Preoperative Harris Hip Score was 53.6.
Surgical Technique
In both cases, a standard series of radiographs was obtained—an anteroposterior (AP) radiograph of the pelvis and AP and cross-table lateral radiographs of the operative hip (Figure 1). Computed tomography (CT) with a metal-artifact-reducing technique may be useful in determining amount of cortical bone remaining under the plate. CT showed limited lateral cortex beneath the side plate and bony overgrowth covering the side plate. Preoperative templating was performed using previously described techniques.5
During THA, before removing any portion of any retained hardware, the surgeon should perform 3 important actions: Dislocate the hip, perform all appropriate capsular releases, and reduce the hip. Dislocating the hip before hardware removal significantly decreases the risk for fracture caused by stress risers, as the force required for dislocation is much more controlled because of the capsular releases. After hardware removal, the hip can be easily redislocated, and the femoral neck osteotomy can be performed.
When plate and screws are in an intracortical position, the screws can be removed only after removing the small shell of cortical bone covering them. The amount of bone to be removed is minimal. After the screws are removed, the plate remains in place. A motorized device with a metal-cutting attachment is used to transect the construct at the junction of the plate and barrel (case 1) or at the bend of a fixed-angle device (case 2). Laparotomy sponges are placed around the proximal femur to minimize the amount of soft tissue that could be exposed to metal shavings. Copious irrigation is used throughout this part of the procedure. Osteotomes are used to elevate the proximal portion of the plate and the barrel, preserving the distal portion of the plate on the lateral cortex of the femoral shaft.
After the head is removed, the rest of the THA can be performed using standard press-fit insertion technique (Figures 2A-2D). Care must be taken to ensure that the distal aspect of the femoral stem bypasses the most distal screw hole by at least 2 cortical diameters in order to reduce the risk for periprosthetic fracture.
By 2-year follow-up, both patients had regained excellent range of motion, ambulation, and overall function. Postoperative Harris Hip Scores were 86.6 and 83.8, respectively. There were no radiographic signs of complications.
Discussion
THA can be challenging in the setting of previously placed internal fixation devices, particularly devices inserted during a patient’s adolescence, as significant bony overgrowth can occur. The standard approach has been to remove the internal fixation device and then perform the THA. In most cases, and particularly when the internal fixation device is in an intracortical position, the result is significant compromise of bone. This article describes a technique in which a portion of the hardware is retained to avoid compromise of the lateral femoral cortex, thereby allowing insertion of a noncemented femoral component.
THA is the most effective procedure for reducing hip pain and disability in the setting of degenerative changes.6 Patients with SCFE, Legg-Calvé-Perthes disease, or developmental dysplasia of the hip generally are younger at the time they may be sufficiently symptomatic to consider THA.7,8 Many have had previous surgery using internal fixation devices. THAs after previous osteotomies with internal fixation devices are more technically demanding, require more operative time, are subject to more blood loss, and have a higher rate of complications, including femoral fracture. Ferguson and colleagues4 and Boos and colleagues9 found these surgeries were more difficult 33.8% and 36.8% of the time, respectively. For these reasons, some authors have recommended removing the internal fixation device as soon as the osteotomy is healed.4 However, this has not become the standard of care, and surgeons continue to perform THAs in the presence of a previous osteotomy with an internal fixation device in place.
The technique described in this article was used successfully in 2 cases. In each case, leaving the intracortical plate in place avoided compromise of the lateral femoral cortex and allowed insertion of a noncemented femoral component without complication. Of course, with the screw holes representing stress risers, careful insertion of the femoral component was required. Retaining the intracortical plate allowed it to function as part of the lateral femoral cortex, thereby maintaining the structural integrity of the femoral canal. As has been described for the 2 cases, a blade plate and plate and barrel were converted to a limited intracortical plate by removing the proximal portion of the plates—a modification that could be applied to other types of internal fixation devices that extend into the femoral neck as long as appropriate cutting tools are available.
Conclusion
THA in the setting of a retained internal fixation device is relatively common. This article describes a technique that can be used when a plate applied to the lateral femoral cortex has become intracortical as a result of extensive bony overgrowth. In using this technique to avoid plate removal, the surgeon eliminates the need for more extensive procedures aimed at compensating for deficiency of the femoral cortex in the area of plate removal. Although only 2 cases are presented here, this technique potentially can be used more broadly in these specific clinical situations.
1. Engesæter LB, Engesæter IØ, Fenstad AM, et al. Low revision rate after total hip arthroplasty in patients with pediatric hip diseases. Acta Orthop. 2012;83(5):436-441.
2. Froberg L, Christensen F, Pedersen NW, Overgaard S. The need for total hip arthroplasty in Perthes disease: a long-term study. Clin Orthop Relat Res. 2011;469(4):1134-1140.
3. Furnes O, Lie SA, Espehaug B, Vollset SE, Engesæter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586.
4. Ferguson GM, Cabanela ME, Ilstrup DM. Total hip arthroplasty after failed intertrochanteric osteotomy. J Bone Joint Surg Br. 1994;76(2):252-257.
5. Scheerlinck T. Primary hip arthroplasty templating on standard radiographs. A stepwise approach. Acta Orthop Belg. 2010;76(4):432-442.
6. Wroblewski BM, Siney PD. Charnley low-friction arthroplasty of the hip. Long-term results. Clin Orthop Relat Res. 1993;(292):191-201.
7. Chandler HP, Reineck FT, Wixson RL, McCarthy JC. Total hip replacement in patients younger than thirty years old. A five-year follow-up study. J Bone Joint Surg Am. 1981;63(9):1426-1434.
8. Dorr LD, Luckett M, Conaty JP. Total hip arthroplasties in patients younger than 45 years. A nine- to ten-year follow-up study. Clin Orthop Relat Res. 1990;(260):215-219.
9. Boos N, Krushell R, Ganz R, Müller ME. Total hip arthroplasty after previous proximal femoral osteotomy. J Bone Joint Surg Br. 1997;79(2):247-253.
1. Engesæter LB, Engesæter IØ, Fenstad AM, et al. Low revision rate after total hip arthroplasty in patients with pediatric hip diseases. Acta Orthop. 2012;83(5):436-441.
2. Froberg L, Christensen F, Pedersen NW, Overgaard S. The need for total hip arthroplasty in Perthes disease: a long-term study. Clin Orthop Relat Res. 2011;469(4):1134-1140.
3. Furnes O, Lie SA, Espehaug B, Vollset SE, Engesæter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586.
4. Ferguson GM, Cabanela ME, Ilstrup DM. Total hip arthroplasty after failed intertrochanteric osteotomy. J Bone Joint Surg Br. 1994;76(2):252-257.
5. Scheerlinck T. Primary hip arthroplasty templating on standard radiographs. A stepwise approach. Acta Orthop Belg. 2010;76(4):432-442.
6. Wroblewski BM, Siney PD. Charnley low-friction arthroplasty of the hip. Long-term results. Clin Orthop Relat Res. 1993;(292):191-201.
7. Chandler HP, Reineck FT, Wixson RL, McCarthy JC. Total hip replacement in patients younger than thirty years old. A five-year follow-up study. J Bone Joint Surg Am. 1981;63(9):1426-1434.
8. Dorr LD, Luckett M, Conaty JP. Total hip arthroplasties in patients younger than 45 years. A nine- to ten-year follow-up study. Clin Orthop Relat Res. 1990;(260):215-219.
9. Boos N, Krushell R, Ganz R, Müller ME. Total hip arthroplasty after previous proximal femoral osteotomy. J Bone Joint Surg Br. 1997;79(2):247-253.
Merkel Cell Carcinoma in a Vein Graft Donor Site
Case Report
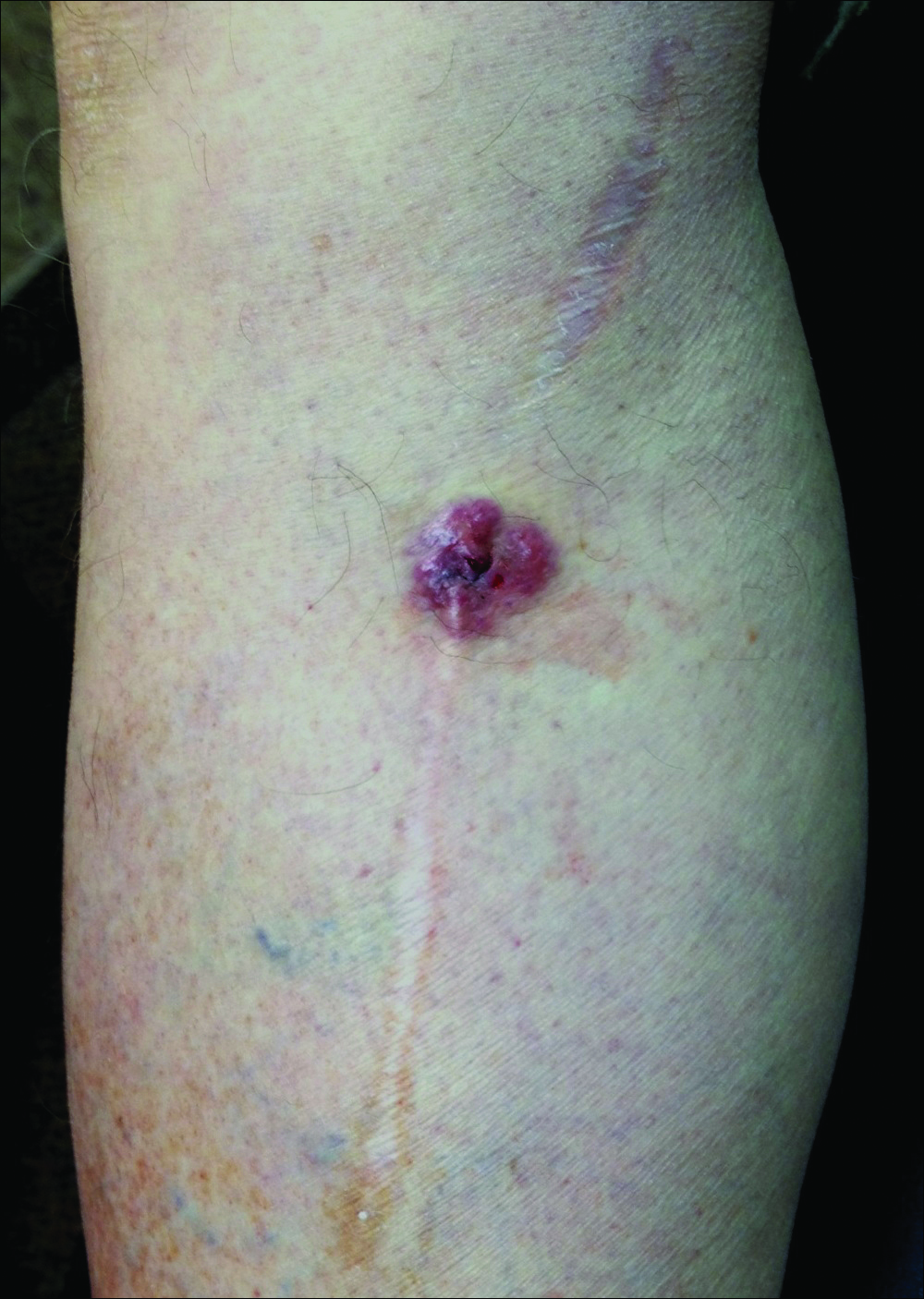
A 70-year-old man with history of coronary artery disease presented with a growing lesion on the right leg of 1 year’s duration. The lesion developed at a vein graft donor site for a coronary artery bypass that had been performed 18 years prior to presentation. The patient reported that the lesion was sensitive to touch. Physical examination revealed a 27-mm, firm, violaceous plaque on the medial aspect of the right upper shin (Figure 1). Mild pitting edema also was noted on both lower legs but was more prominent on the right leg. A 6-mm punch biopsy was performed.
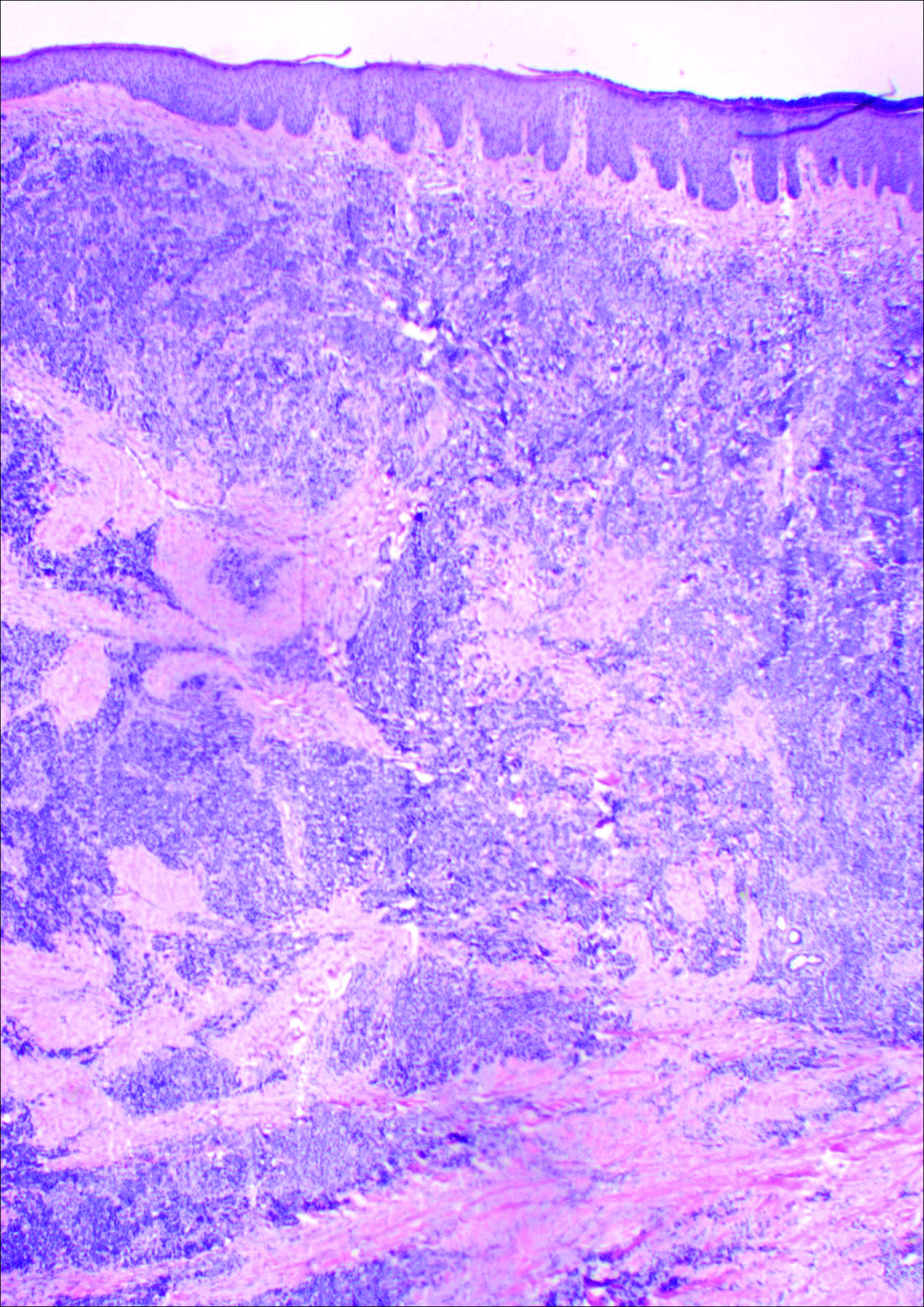
Histology showed diffuse infiltration of the dermis and subcutaneous fat by intermediate-sized atypical blue cells with scant cytoplasm (Figure 2). The tumor exhibited moderate cytologic atypia with occasional mitotic figures, and lymphovascular invasion was present. Staining for CD3 was negative within the tumor, but a few reactive lymphocytes were highlighted at the periphery. Staining for CD20 and CD30 was negative. Strong and diffuse staining for cyto-keratin 20 and pan-cytokeratin was noted within the tumor with the distinctive perinuclear pattern characteristic of Merkel cell carcinoma (MCC). Staining for cytokeratin 7 was negative. Synaptophysin and chromogranin were strongly and diffusely positive within the tumor, consistent with a diagnosis of MCC.
The patient was found to have stage IIA (T2N0M0) MCC. Computed tomography completed for staging showed no evidence of metastasis. Wide local excision of the lesion was performed. Margins were negative, as was a right inguinal sentinel lymph node dissection. Because of the size of the tumor and the presence of lymphovascular invasion, radiation therapy at the primary tumor site was recommended. Local radiation treatment (200 cGy daily) was administered for a total dose of 5000 cGy over 5 weeks. The patient currently is free of recurrence or metastases and is being followed by the oncology, surgery, and dermatology departments.
Comment
Merkel cell carcinoma is a rare aggressive cutaneous malignancy. The exact pathogenesis is unknown, but immunosuppression and UV radiation, possibly through its immunosuppressive effects, appear to be contributing factors. More recently, the Merkel cell polyomavirus has been linked to MCC in approximately 80% of cases.1,2
Merkel cell carcinoma is more common in individuals with fair skin, and the average age at diagnosis is 69 years.1 Patients typically present with an asymptomatic, firm, erythematous or violaceous, dome-shaped nodule or a small indurated plaque, most commonly on sun-exposed areas of the head and neck followed by the upper and lower extremities including the hands, feet, ankles, and wrists. Fifteen percent to 20% of MCCs develop on the legs and feet.1 Our patient presented with an MCC that developed on the right shin at a vein graft donor site.
The development of a cutaneous malignancy in a chronic wound (also known as a Marjolin ulcer) is a rare but well-recognized process. These malignancies occur in previously traumatized or chronically inflamed wounds and have been found to occur most commonly in chronic burn wounds, especially in ungrafted full-thickness burns. Squamous cell carcinomas (SCCs) are the most common malignancies to arise in chronic wounds, but basal cell carcinomas, adenocarcinomas, melanomas, malignant fibrous histiocytomas, adenoacanthomas, liposarcomas, and osteosarcomas also have been reported.3 There also have been a few reports of MCC associated with Bowen disease that developed in burn wounds.4 These malignancies generally occur years after injury (average, 35.5 years), but there have been reports of keratoacanthomas developing as early as 3 weeks after injury.5,6
In some reports, malignancies in skin graft donor sites are differentiated from Marjolin ulcers, as the former appear in healed surgical wounds rather than in chronic unstable wounds and tend to occur sooner (ie, in weeks to months after graft harvesting).7,8 The development of these malignancies in graft donor sites is not as well recognized and has been reported in donor sites for split-thickness skin grafts (STSGs), full-thickness skin grafts, tendon grafts, and bone grafts. In addition to malignancies that arise de novo, some develop due to metastatic and iatrogenic spread. The majority of reported malignancies in tendon and bone graft donor sites have been due to metastasis or iatrogenic spread.9-14
Iatrogenic implantation of tumor cells is a well-recognized phenomenon. Hussain et al10 reported a case of implantation of SCC in an STSG donor site, most likely due to direct seeding from a hollow needle used to infiltrate local anesthetic in the tumor area and the STSG. In this case, metastasis could not be completely ruled out.10 There also have been reports of osteosarcoma, ameloblastoma, scirrhous carcinoma of the breast, and malignant fibrous histiocytoma thought to be implanted at bone graft donor sites.14-17 Iatrogenic spread of malignancies can occur through seeding from contaminated gloves or instruments such as hollow bore needles or trocar placement in laparoscopic surgery.11 Airborne spread also may be possible, as viable melanoma cells have been detected in electrocautery plume in mice.13
Metastatic malignancies including metastases from SCC, adenocarcinoma, melanoma, malignant fibrous histiocytoma, angiosarcoma, and osteosarcoma also have been reported to develop in graft donor sites.11,13,18,19 Many malignancies thought to have developed from iatrogenic seeding may actually be from metastasis either by hematogenous or lymphatic spread. A possible contributing factor may be surgery-induced immunosuppression, which has been linked to increased tumor metastasis formation.20 Surgery or trauma have been shown to have an effect on cellular components of the immune system, causing changes such as a shift in T lymphocytes toward immune-suppressive T lymphocytes and impaired function of natural killer cells, neutrophils, and macrophages.20 The suppression of cell-mediated immunity has been shown to decrease over days to weeks in the postoperative period.21 In addition to surgery- or trauma-induced immunosuppression, the risk for metastasis may increase due to increased vascular, including lymphatic, flow toward a skin graft donor site.13,16 Furthermore, trauma predisposes areas to a hypercoagulable state with increased sludging as well as increased platelet counts and fibrinogen levels, which may lead to localization of metastatic lesions.22 All of these factors could potentially work simultaneously to induce the development of metastasis in graft donor sites.
We found that SCCs and keratoacanthomas, which may be a variant of SCC, are among the only primary malignancies that have been reported to develop in skin graft donor sites.6-8 Malignancies in these donor sites appear to develop sooner than those found in chronic wounds and are reported to develop within weeks to several months postoperatively, even in as few as 2 weeks.6,8 Tamir et al6 reported 2 keratoacanthomas that developed simultaneously in a burn scar and STSG donor site. The investigators believed it could be a sign of reduced immune surveillance in the 2 affected areas.6 It has been hypothesized that one cause of local immune suppression in Marjolin ulcers could be due to poor lymphatic regeneration in scar tissue, which would prevent delivery of antigens and stimulated lymphocytes.23 Haik et al7 considered this possibility when discussing a case of SCC that developed at the site of an STSG. The authors did not feel it applied, however, as the donor site had only undergone a single skin harvesting procedure.7 Ponnuvelu et al8 felt that inflammation was the underlying etiology behind the 2 cases they reported of SCCs that developed in STSG donor sites. The inflammation associated with tumors has many of the same processes involved in wound healing (eg, cellular proliferation, angiogenesis). Ponnuvelu et al8 hypothesized that the local inflammation caused by graft harvesting produced an ideal environment for early carcinogenesis. Although in chronic wounds it is believed that continual repair and regeneration in recurrent ulceration contributes to neoplastic initiation, it is thought that even a single injury may lead to malignant change, which may be because prior actinic damage or another cause has made the area more susceptible to these changes.24,25 Surgery-induced immunosuppression also may play a role in development of primary malignancies in graft donor sites.
There have been a few reports of SCCs and basal cell carcinomas occurring in other surgical scars that healed without complications.24,26-28 Similar to the malignancies in graft donor sites, some authors differentiate malignancies that occur in surgical scars that heal without complications from Marjolin ulcers, as they do not occur in chronically irritated wounds. These malignancies have been reported in scars from sternotomies, an infertility procedure, hair transplantation, thyroidectomy, colostomy, cleft lip repair, inguinal hernia repair, and paraumbilical laparoscopic port site. The time between surgery and diagnosis of malignancy ranged from 9 months to 67 years.24,26-28 The development of malignancies in these surgical scars may be due to local immunosuppression, possibly from decreased lymphatic flow; additionally, the inflammation in wound healing may provide the ideal environment for carcinogenesis. Trauma in areas already susceptible to malignant change could be a contributing factor.
Conclusion
Our patient developed an MCC in a vein graft donor site 18 years after vein harvesting. It was likely a primary tumor, as vein harvesting was done for coronary artery bypass graft. There was no evidence of any other lesions on physical examination or computed tomography, making it doubtful that an MCC serving as a primary lesion for seeding or metastasis was present. If such a lesion had been present at that time, it would likely have spread well before the time of presentation to our clinic due to the fast doubling time and high rate of metastasis characteristic of MCCs, further lessening the possibility of metastasis or implantation.
The extended length of time from procedure to lesion development in our patient is much longer than for other reported malignancies in graft donor sites, but the reported time for malignancies in other postsurgical scars is more varied. Regardless of whether the MCC in our patient is classified as a Marjolin ulcer, the pathogenesis is unclear. It is thought that a single injury could lead to malignant change in predisposed skin. Our patient’s legs did not have any evidence of prior actinic damage; however, it is likely that he had local immune suppression, which may have made him more susceptible to these changes. It is unlikely that surgery-induced immunosuppression played a role in our patient, as specific cellular components of the immune system only appear to be affected over days to weeks in the postoperative period. Although poor lymphatic regeneration in scar tissue leading to decreased immune surveillance is not generally thought to contribute to malignancies in most surgical scars, our patient underwent vein harvesting. Chronic edema commonly occurs after vein harvesting and is believed to be due to trauma to the lymphatics. Local immune suppression also may have led to increased susceptibility to infection by the MCC polyomavirus, which has been found to be associated with many MCCs. In addition, the area may have been more susceptible to carcinogenesis due to changes from inflammation from wound healing. We suspect together these factors contributed to the development of our patient’s MCC. Although rare, graft donor sites should be examined periodically for the development of malignancy.
- Swann MH, Yoon J. Merkel cell carcinoma. Semin Oncol. 2007;34:51-56.
- Schrama D, Ugurel S, Becker JC. Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol. 2012;24:141-149.
- Kadir AR. Burn scar neoplasm. Ann Burns Fire Disasters. 2007;20:185-188.
- Walsh NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol. 2001;32:680-689.
- Guenther N, Menenakos C, Braumann C, et al. Squamous cell carcinoma arising on a skin graft 64 years after primary injury. Dermatol Online J. 2007;13:27.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;40:870-871.
- Haik J, Georgiou I, Farber N, et al. Squamous cell carcinoma arising in a split-thickness skin graft donor site. Burns. 2008;34:891-893.
- Ponnuvelu G, Ng MF, Connolly CM, et al. Inflammation to skin malignancy, time to rethink the link: SCC in skin graft donor sites. Surgeon. 2011;9:168-169.
- Bekar A, Kahveci R, Tolunay S, et al. Metastatic gliosarcoma mass extension to a donor fascia lata graft harvest site by tumor cell contamination. World Neurosurg. 2010;73:719-721.
- Hussain A, Ekwobi C, Watson S. Metastatic implantation squamous cell carcinoma in a split-thickness skin graft donor site. J Plast Reconstr Aesthet Surg. 2011;64:690-692.
- May JT, Patil YJ. Keratoacanthoma-type squamous cell carcinoma developing in a skin graft donor site after tumor extirpation at a distant site. Ear Nose Throat J. 2010;89:E11-E13.
- Serrano-Ortega S, Buendia-Eisman A, Ortega del Olmo RM, et al. Melanoma metastasis in donor site of full-thickness skin graft. Dermatology. 2000;201:377-378.
- Wright H, McKinnell TH, Dunkin C. Recurrence of cutaneous squamous cell carcinoma at remote limb donor site. J Plast Reconstr Aesthet Surg. 2012;65:1265-1266.
- Yip KM, Lin J, Kumta SM. A pelvic osteosarcoma with metastasis to the donor site of the bone graft. a case report. Int Orthop. 1996;20:389-391.
- Dias RG, Abudu A, Carter SR, et al. Tumour transfer to bone graft donor site: a case report and review of the literature of the mechanism of seeding. Sarcoma. 2000;4:57-59.
- Neilson D, Emerson DJ, Dunn L. Squamous cell carcinoma of skin developing in a skin graft donor site. Br J Plast Surg. 1988;41:417-419.
- Singh C, Ibrahim S, Pang KS, et al. Implantation metastasis in a 13-year-old girl: a case report. J Orthop Surg (Hong Kong). 2003;11:94-96.
- Enion DS, Scott MJ, Gouldesbrough D. Cutaneous metastasis from a malignant fibrous histiocytoma to a limb skin graft donor site. Br J Surg. 1993;80:366.
- Yamasaki O, Terao K, Asagoe K, et al. Koebner phenomenon on skin graft donor site in cutaneous angiosarcoma. Eur J Dermatol. 2001;11:584-586.
- Hogan BV, Peter MB, Shenoy HG, et al. Surgery induced immunosuppression. Surgeon. 2011;9:38-43.
- Neeman E, Ben-Eliyahu S. The perioperative period and promotion of cancer metastasis: new outlooks on mediating mechanisms and immune involvement. Brain Behav Immun. 2013;30(suppl):32-40.
- Agostino D, Cliffton EE. Trauma as a cause of localization of blood-borne metastases: preventive effect of heparin and fibrinolysin. Ann Surg. 1965;161:97-102.
- Hammond JS, Thomsen S, Ward CG. Scar carcinoma arising acutely in a skin graft donor site. J Trauma. 1987;27:681-683.
- Korula R, Hughes CF. Squamous cell carcinoma arising in a sternotomy scar. Ann Thorac Surg. 1991;51:667-669.
- Kennedy CTC, Burd DAR, Creamer D. Mechanical and thermal injury. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 2. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:28.1-28.94.
- Durrani AJ, Miller RJ, Davies M. Basal cell carcinoma arising in a laparoscopic port site scar at the umbilicus. Plast Reconstr Surg. 2005;116:348-350.
- Kotwal S, Madaan S, Prescott S, et al. Unusual squamous cell carcinoma of the scrotum arising from a well healed, innocuous scar of an infertility procedure: a case report. Ann R Coll Surg Engl. 2007;89:17-19.
- Ozyazgan I, Kontas O. Previous injuries or scars as risk factors for the development of basal cell carcinoma. Scand J Plast Reconstr Surg Hand Surg. 2004;38:11-15.
Case Report
A 70-year-old man with history of coronary artery disease presented with a growing lesion on the right leg of 1 year’s duration. The lesion developed at a vein graft donor site for a coronary artery bypass that had been performed 18 years prior to presentation. The patient reported that the lesion was sensitive to touch. Physical examination revealed a 27-mm, firm, violaceous plaque on the medial aspect of the right upper shin (Figure 1). Mild pitting edema also was noted on both lower legs but was more prominent on the right leg. A 6-mm punch biopsy was performed.
Histology showed diffuse infiltration of the dermis and subcutaneous fat by intermediate-sized atypical blue cells with scant cytoplasm (Figure 2). The tumor exhibited moderate cytologic atypia with occasional mitotic figures, and lymphovascular invasion was present. Staining for CD3 was negative within the tumor, but a few reactive lymphocytes were highlighted at the periphery. Staining for CD20 and CD30 was negative. Strong and diffuse staining for cyto-keratin 20 and pan-cytokeratin was noted within the tumor with the distinctive perinuclear pattern characteristic of Merkel cell carcinoma (MCC). Staining for cytokeratin 7 was negative. Synaptophysin and chromogranin were strongly and diffusely positive within the tumor, consistent with a diagnosis of MCC.
The patient was found to have stage IIA (T2N0M0) MCC. Computed tomography completed for staging showed no evidence of metastasis. Wide local excision of the lesion was performed. Margins were negative, as was a right inguinal sentinel lymph node dissection. Because of the size of the tumor and the presence of lymphovascular invasion, radiation therapy at the primary tumor site was recommended. Local radiation treatment (200 cGy daily) was administered for a total dose of 5000 cGy over 5 weeks. The patient currently is free of recurrence or metastases and is being followed by the oncology, surgery, and dermatology departments.
Comment
Merkel cell carcinoma is a rare aggressive cutaneous malignancy. The exact pathogenesis is unknown, but immunosuppression and UV radiation, possibly through its immunosuppressive effects, appear to be contributing factors. More recently, the Merkel cell polyomavirus has been linked to MCC in approximately 80% of cases.1,2
Merkel cell carcinoma is more common in individuals with fair skin, and the average age at diagnosis is 69 years.1 Patients typically present with an asymptomatic, firm, erythematous or violaceous, dome-shaped nodule or a small indurated plaque, most commonly on sun-exposed areas of the head and neck followed by the upper and lower extremities including the hands, feet, ankles, and wrists. Fifteen percent to 20% of MCCs develop on the legs and feet.1 Our patient presented with an MCC that developed on the right shin at a vein graft donor site.
The development of a cutaneous malignancy in a chronic wound (also known as a Marjolin ulcer) is a rare but well-recognized process. These malignancies occur in previously traumatized or chronically inflamed wounds and have been found to occur most commonly in chronic burn wounds, especially in ungrafted full-thickness burns. Squamous cell carcinomas (SCCs) are the most common malignancies to arise in chronic wounds, but basal cell carcinomas, adenocarcinomas, melanomas, malignant fibrous histiocytomas, adenoacanthomas, liposarcomas, and osteosarcomas also have been reported.3 There also have been a few reports of MCC associated with Bowen disease that developed in burn wounds.4 These malignancies generally occur years after injury (average, 35.5 years), but there have been reports of keratoacanthomas developing as early as 3 weeks after injury.5,6
In some reports, malignancies in skin graft donor sites are differentiated from Marjolin ulcers, as the former appear in healed surgical wounds rather than in chronic unstable wounds and tend to occur sooner (ie, in weeks to months after graft harvesting).7,8 The development of these malignancies in graft donor sites is not as well recognized and has been reported in donor sites for split-thickness skin grafts (STSGs), full-thickness skin grafts, tendon grafts, and bone grafts. In addition to malignancies that arise de novo, some develop due to metastatic and iatrogenic spread. The majority of reported malignancies in tendon and bone graft donor sites have been due to metastasis or iatrogenic spread.9-14
Iatrogenic implantation of tumor cells is a well-recognized phenomenon. Hussain et al10 reported a case of implantation of SCC in an STSG donor site, most likely due to direct seeding from a hollow needle used to infiltrate local anesthetic in the tumor area and the STSG. In this case, metastasis could not be completely ruled out.10 There also have been reports of osteosarcoma, ameloblastoma, scirrhous carcinoma of the breast, and malignant fibrous histiocytoma thought to be implanted at bone graft donor sites.14-17 Iatrogenic spread of malignancies can occur through seeding from contaminated gloves or instruments such as hollow bore needles or trocar placement in laparoscopic surgery.11 Airborne spread also may be possible, as viable melanoma cells have been detected in electrocautery plume in mice.13
Metastatic malignancies including metastases from SCC, adenocarcinoma, melanoma, malignant fibrous histiocytoma, angiosarcoma, and osteosarcoma also have been reported to develop in graft donor sites.11,13,18,19 Many malignancies thought to have developed from iatrogenic seeding may actually be from metastasis either by hematogenous or lymphatic spread. A possible contributing factor may be surgery-induced immunosuppression, which has been linked to increased tumor metastasis formation.20 Surgery or trauma have been shown to have an effect on cellular components of the immune system, causing changes such as a shift in T lymphocytes toward immune-suppressive T lymphocytes and impaired function of natural killer cells, neutrophils, and macrophages.20 The suppression of cell-mediated immunity has been shown to decrease over days to weeks in the postoperative period.21 In addition to surgery- or trauma-induced immunosuppression, the risk for metastasis may increase due to increased vascular, including lymphatic, flow toward a skin graft donor site.13,16 Furthermore, trauma predisposes areas to a hypercoagulable state with increased sludging as well as increased platelet counts and fibrinogen levels, which may lead to localization of metastatic lesions.22 All of these factors could potentially work simultaneously to induce the development of metastasis in graft donor sites.
We found that SCCs and keratoacanthomas, which may be a variant of SCC, are among the only primary malignancies that have been reported to develop in skin graft donor sites.6-8 Malignancies in these donor sites appear to develop sooner than those found in chronic wounds and are reported to develop within weeks to several months postoperatively, even in as few as 2 weeks.6,8 Tamir et al6 reported 2 keratoacanthomas that developed simultaneously in a burn scar and STSG donor site. The investigators believed it could be a sign of reduced immune surveillance in the 2 affected areas.6 It has been hypothesized that one cause of local immune suppression in Marjolin ulcers could be due to poor lymphatic regeneration in scar tissue, which would prevent delivery of antigens and stimulated lymphocytes.23 Haik et al7 considered this possibility when discussing a case of SCC that developed at the site of an STSG. The authors did not feel it applied, however, as the donor site had only undergone a single skin harvesting procedure.7 Ponnuvelu et al8 felt that inflammation was the underlying etiology behind the 2 cases they reported of SCCs that developed in STSG donor sites. The inflammation associated with tumors has many of the same processes involved in wound healing (eg, cellular proliferation, angiogenesis). Ponnuvelu et al8 hypothesized that the local inflammation caused by graft harvesting produced an ideal environment for early carcinogenesis. Although in chronic wounds it is believed that continual repair and regeneration in recurrent ulceration contributes to neoplastic initiation, it is thought that even a single injury may lead to malignant change, which may be because prior actinic damage or another cause has made the area more susceptible to these changes.24,25 Surgery-induced immunosuppression also may play a role in development of primary malignancies in graft donor sites.
There have been a few reports of SCCs and basal cell carcinomas occurring in other surgical scars that healed without complications.24,26-28 Similar to the malignancies in graft donor sites, some authors differentiate malignancies that occur in surgical scars that heal without complications from Marjolin ulcers, as they do not occur in chronically irritated wounds. These malignancies have been reported in scars from sternotomies, an infertility procedure, hair transplantation, thyroidectomy, colostomy, cleft lip repair, inguinal hernia repair, and paraumbilical laparoscopic port site. The time between surgery and diagnosis of malignancy ranged from 9 months to 67 years.24,26-28 The development of malignancies in these surgical scars may be due to local immunosuppression, possibly from decreased lymphatic flow; additionally, the inflammation in wound healing may provide the ideal environment for carcinogenesis. Trauma in areas already susceptible to malignant change could be a contributing factor.
Conclusion
Our patient developed an MCC in a vein graft donor site 18 years after vein harvesting. It was likely a primary tumor, as vein harvesting was done for coronary artery bypass graft. There was no evidence of any other lesions on physical examination or computed tomography, making it doubtful that an MCC serving as a primary lesion for seeding or metastasis was present. If such a lesion had been present at that time, it would likely have spread well before the time of presentation to our clinic due to the fast doubling time and high rate of metastasis characteristic of MCCs, further lessening the possibility of metastasis or implantation.
The extended length of time from procedure to lesion development in our patient is much longer than for other reported malignancies in graft donor sites, but the reported time for malignancies in other postsurgical scars is more varied. Regardless of whether the MCC in our patient is classified as a Marjolin ulcer, the pathogenesis is unclear. It is thought that a single injury could lead to malignant change in predisposed skin. Our patient’s legs did not have any evidence of prior actinic damage; however, it is likely that he had local immune suppression, which may have made him more susceptible to these changes. It is unlikely that surgery-induced immunosuppression played a role in our patient, as specific cellular components of the immune system only appear to be affected over days to weeks in the postoperative period. Although poor lymphatic regeneration in scar tissue leading to decreased immune surveillance is not generally thought to contribute to malignancies in most surgical scars, our patient underwent vein harvesting. Chronic edema commonly occurs after vein harvesting and is believed to be due to trauma to the lymphatics. Local immune suppression also may have led to increased susceptibility to infection by the MCC polyomavirus, which has been found to be associated with many MCCs. In addition, the area may have been more susceptible to carcinogenesis due to changes from inflammation from wound healing. We suspect together these factors contributed to the development of our patient’s MCC. Although rare, graft donor sites should be examined periodically for the development of malignancy.
Case Report
A 70-year-old man with history of coronary artery disease presented with a growing lesion on the right leg of 1 year’s duration. The lesion developed at a vein graft donor site for a coronary artery bypass that had been performed 18 years prior to presentation. The patient reported that the lesion was sensitive to touch. Physical examination revealed a 27-mm, firm, violaceous plaque on the medial aspect of the right upper shin (Figure 1). Mild pitting edema also was noted on both lower legs but was more prominent on the right leg. A 6-mm punch biopsy was performed.
Histology showed diffuse infiltration of the dermis and subcutaneous fat by intermediate-sized atypical blue cells with scant cytoplasm (Figure 2). The tumor exhibited moderate cytologic atypia with occasional mitotic figures, and lymphovascular invasion was present. Staining for CD3 was negative within the tumor, but a few reactive lymphocytes were highlighted at the periphery. Staining for CD20 and CD30 was negative. Strong and diffuse staining for cyto-keratin 20 and pan-cytokeratin was noted within the tumor with the distinctive perinuclear pattern characteristic of Merkel cell carcinoma (MCC). Staining for cytokeratin 7 was negative. Synaptophysin and chromogranin were strongly and diffusely positive within the tumor, consistent with a diagnosis of MCC.
The patient was found to have stage IIA (T2N0M0) MCC. Computed tomography completed for staging showed no evidence of metastasis. Wide local excision of the lesion was performed. Margins were negative, as was a right inguinal sentinel lymph node dissection. Because of the size of the tumor and the presence of lymphovascular invasion, radiation therapy at the primary tumor site was recommended. Local radiation treatment (200 cGy daily) was administered for a total dose of 5000 cGy over 5 weeks. The patient currently is free of recurrence or metastases and is being followed by the oncology, surgery, and dermatology departments.
Comment
Merkel cell carcinoma is a rare aggressive cutaneous malignancy. The exact pathogenesis is unknown, but immunosuppression and UV radiation, possibly through its immunosuppressive effects, appear to be contributing factors. More recently, the Merkel cell polyomavirus has been linked to MCC in approximately 80% of cases.1,2
Merkel cell carcinoma is more common in individuals with fair skin, and the average age at diagnosis is 69 years.1 Patients typically present with an asymptomatic, firm, erythematous or violaceous, dome-shaped nodule or a small indurated plaque, most commonly on sun-exposed areas of the head and neck followed by the upper and lower extremities including the hands, feet, ankles, and wrists. Fifteen percent to 20% of MCCs develop on the legs and feet.1 Our patient presented with an MCC that developed on the right shin at a vein graft donor site.
The development of a cutaneous malignancy in a chronic wound (also known as a Marjolin ulcer) is a rare but well-recognized process. These malignancies occur in previously traumatized or chronically inflamed wounds and have been found to occur most commonly in chronic burn wounds, especially in ungrafted full-thickness burns. Squamous cell carcinomas (SCCs) are the most common malignancies to arise in chronic wounds, but basal cell carcinomas, adenocarcinomas, melanomas, malignant fibrous histiocytomas, adenoacanthomas, liposarcomas, and osteosarcomas also have been reported.3 There also have been a few reports of MCC associated with Bowen disease that developed in burn wounds.4 These malignancies generally occur years after injury (average, 35.5 years), but there have been reports of keratoacanthomas developing as early as 3 weeks after injury.5,6
In some reports, malignancies in skin graft donor sites are differentiated from Marjolin ulcers, as the former appear in healed surgical wounds rather than in chronic unstable wounds and tend to occur sooner (ie, in weeks to months after graft harvesting).7,8 The development of these malignancies in graft donor sites is not as well recognized and has been reported in donor sites for split-thickness skin grafts (STSGs), full-thickness skin grafts, tendon grafts, and bone grafts. In addition to malignancies that arise de novo, some develop due to metastatic and iatrogenic spread. The majority of reported malignancies in tendon and bone graft donor sites have been due to metastasis or iatrogenic spread.9-14
Iatrogenic implantation of tumor cells is a well-recognized phenomenon. Hussain et al10 reported a case of implantation of SCC in an STSG donor site, most likely due to direct seeding from a hollow needle used to infiltrate local anesthetic in the tumor area and the STSG. In this case, metastasis could not be completely ruled out.10 There also have been reports of osteosarcoma, ameloblastoma, scirrhous carcinoma of the breast, and malignant fibrous histiocytoma thought to be implanted at bone graft donor sites.14-17 Iatrogenic spread of malignancies can occur through seeding from contaminated gloves or instruments such as hollow bore needles or trocar placement in laparoscopic surgery.11 Airborne spread also may be possible, as viable melanoma cells have been detected in electrocautery plume in mice.13
Metastatic malignancies including metastases from SCC, adenocarcinoma, melanoma, malignant fibrous histiocytoma, angiosarcoma, and osteosarcoma also have been reported to develop in graft donor sites.11,13,18,19 Many malignancies thought to have developed from iatrogenic seeding may actually be from metastasis either by hematogenous or lymphatic spread. A possible contributing factor may be surgery-induced immunosuppression, which has been linked to increased tumor metastasis formation.20 Surgery or trauma have been shown to have an effect on cellular components of the immune system, causing changes such as a shift in T lymphocytes toward immune-suppressive T lymphocytes and impaired function of natural killer cells, neutrophils, and macrophages.20 The suppression of cell-mediated immunity has been shown to decrease over days to weeks in the postoperative period.21 In addition to surgery- or trauma-induced immunosuppression, the risk for metastasis may increase due to increased vascular, including lymphatic, flow toward a skin graft donor site.13,16 Furthermore, trauma predisposes areas to a hypercoagulable state with increased sludging as well as increased platelet counts and fibrinogen levels, which may lead to localization of metastatic lesions.22 All of these factors could potentially work simultaneously to induce the development of metastasis in graft donor sites.
We found that SCCs and keratoacanthomas, which may be a variant of SCC, are among the only primary malignancies that have been reported to develop in skin graft donor sites.6-8 Malignancies in these donor sites appear to develop sooner than those found in chronic wounds and are reported to develop within weeks to several months postoperatively, even in as few as 2 weeks.6,8 Tamir et al6 reported 2 keratoacanthomas that developed simultaneously in a burn scar and STSG donor site. The investigators believed it could be a sign of reduced immune surveillance in the 2 affected areas.6 It has been hypothesized that one cause of local immune suppression in Marjolin ulcers could be due to poor lymphatic regeneration in scar tissue, which would prevent delivery of antigens and stimulated lymphocytes.23 Haik et al7 considered this possibility when discussing a case of SCC that developed at the site of an STSG. The authors did not feel it applied, however, as the donor site had only undergone a single skin harvesting procedure.7 Ponnuvelu et al8 felt that inflammation was the underlying etiology behind the 2 cases they reported of SCCs that developed in STSG donor sites. The inflammation associated with tumors has many of the same processes involved in wound healing (eg, cellular proliferation, angiogenesis). Ponnuvelu et al8 hypothesized that the local inflammation caused by graft harvesting produced an ideal environment for early carcinogenesis. Although in chronic wounds it is believed that continual repair and regeneration in recurrent ulceration contributes to neoplastic initiation, it is thought that even a single injury may lead to malignant change, which may be because prior actinic damage or another cause has made the area more susceptible to these changes.24,25 Surgery-induced immunosuppression also may play a role in development of primary malignancies in graft donor sites.
There have been a few reports of SCCs and basal cell carcinomas occurring in other surgical scars that healed without complications.24,26-28 Similar to the malignancies in graft donor sites, some authors differentiate malignancies that occur in surgical scars that heal without complications from Marjolin ulcers, as they do not occur in chronically irritated wounds. These malignancies have been reported in scars from sternotomies, an infertility procedure, hair transplantation, thyroidectomy, colostomy, cleft lip repair, inguinal hernia repair, and paraumbilical laparoscopic port site. The time between surgery and diagnosis of malignancy ranged from 9 months to 67 years.24,26-28 The development of malignancies in these surgical scars may be due to local immunosuppression, possibly from decreased lymphatic flow; additionally, the inflammation in wound healing may provide the ideal environment for carcinogenesis. Trauma in areas already susceptible to malignant change could be a contributing factor.
Conclusion
Our patient developed an MCC in a vein graft donor site 18 years after vein harvesting. It was likely a primary tumor, as vein harvesting was done for coronary artery bypass graft. There was no evidence of any other lesions on physical examination or computed tomography, making it doubtful that an MCC serving as a primary lesion for seeding or metastasis was present. If such a lesion had been present at that time, it would likely have spread well before the time of presentation to our clinic due to the fast doubling time and high rate of metastasis characteristic of MCCs, further lessening the possibility of metastasis or implantation.
The extended length of time from procedure to lesion development in our patient is much longer than for other reported malignancies in graft donor sites, but the reported time for malignancies in other postsurgical scars is more varied. Regardless of whether the MCC in our patient is classified as a Marjolin ulcer, the pathogenesis is unclear. It is thought that a single injury could lead to malignant change in predisposed skin. Our patient’s legs did not have any evidence of prior actinic damage; however, it is likely that he had local immune suppression, which may have made him more susceptible to these changes. It is unlikely that surgery-induced immunosuppression played a role in our patient, as specific cellular components of the immune system only appear to be affected over days to weeks in the postoperative period. Although poor lymphatic regeneration in scar tissue leading to decreased immune surveillance is not generally thought to contribute to malignancies in most surgical scars, our patient underwent vein harvesting. Chronic edema commonly occurs after vein harvesting and is believed to be due to trauma to the lymphatics. Local immune suppression also may have led to increased susceptibility to infection by the MCC polyomavirus, which has been found to be associated with many MCCs. In addition, the area may have been more susceptible to carcinogenesis due to changes from inflammation from wound healing. We suspect together these factors contributed to the development of our patient’s MCC. Although rare, graft donor sites should be examined periodically for the development of malignancy.
- Swann MH, Yoon J. Merkel cell carcinoma. Semin Oncol. 2007;34:51-56.
- Schrama D, Ugurel S, Becker JC. Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol. 2012;24:141-149.
- Kadir AR. Burn scar neoplasm. Ann Burns Fire Disasters. 2007;20:185-188.
- Walsh NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol. 2001;32:680-689.
- Guenther N, Menenakos C, Braumann C, et al. Squamous cell carcinoma arising on a skin graft 64 years after primary injury. Dermatol Online J. 2007;13:27.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;40:870-871.
- Haik J, Georgiou I, Farber N, et al. Squamous cell carcinoma arising in a split-thickness skin graft donor site. Burns. 2008;34:891-893.
- Ponnuvelu G, Ng MF, Connolly CM, et al. Inflammation to skin malignancy, time to rethink the link: SCC in skin graft donor sites. Surgeon. 2011;9:168-169.
- Bekar A, Kahveci R, Tolunay S, et al. Metastatic gliosarcoma mass extension to a donor fascia lata graft harvest site by tumor cell contamination. World Neurosurg. 2010;73:719-721.
- Hussain A, Ekwobi C, Watson S. Metastatic implantation squamous cell carcinoma in a split-thickness skin graft donor site. J Plast Reconstr Aesthet Surg. 2011;64:690-692.
- May JT, Patil YJ. Keratoacanthoma-type squamous cell carcinoma developing in a skin graft donor site after tumor extirpation at a distant site. Ear Nose Throat J. 2010;89:E11-E13.
- Serrano-Ortega S, Buendia-Eisman A, Ortega del Olmo RM, et al. Melanoma metastasis in donor site of full-thickness skin graft. Dermatology. 2000;201:377-378.
- Wright H, McKinnell TH, Dunkin C. Recurrence of cutaneous squamous cell carcinoma at remote limb donor site. J Plast Reconstr Aesthet Surg. 2012;65:1265-1266.
- Yip KM, Lin J, Kumta SM. A pelvic osteosarcoma with metastasis to the donor site of the bone graft. a case report. Int Orthop. 1996;20:389-391.
- Dias RG, Abudu A, Carter SR, et al. Tumour transfer to bone graft donor site: a case report and review of the literature of the mechanism of seeding. Sarcoma. 2000;4:57-59.
- Neilson D, Emerson DJ, Dunn L. Squamous cell carcinoma of skin developing in a skin graft donor site. Br J Plast Surg. 1988;41:417-419.
- Singh C, Ibrahim S, Pang KS, et al. Implantation metastasis in a 13-year-old girl: a case report. J Orthop Surg (Hong Kong). 2003;11:94-96.
- Enion DS, Scott MJ, Gouldesbrough D. Cutaneous metastasis from a malignant fibrous histiocytoma to a limb skin graft donor site. Br J Surg. 1993;80:366.
- Yamasaki O, Terao K, Asagoe K, et al. Koebner phenomenon on skin graft donor site in cutaneous angiosarcoma. Eur J Dermatol. 2001;11:584-586.
- Hogan BV, Peter MB, Shenoy HG, et al. Surgery induced immunosuppression. Surgeon. 2011;9:38-43.
- Neeman E, Ben-Eliyahu S. The perioperative period and promotion of cancer metastasis: new outlooks on mediating mechanisms and immune involvement. Brain Behav Immun. 2013;30(suppl):32-40.
- Agostino D, Cliffton EE. Trauma as a cause of localization of blood-borne metastases: preventive effect of heparin and fibrinolysin. Ann Surg. 1965;161:97-102.
- Hammond JS, Thomsen S, Ward CG. Scar carcinoma arising acutely in a skin graft donor site. J Trauma. 1987;27:681-683.
- Korula R, Hughes CF. Squamous cell carcinoma arising in a sternotomy scar. Ann Thorac Surg. 1991;51:667-669.
- Kennedy CTC, Burd DAR, Creamer D. Mechanical and thermal injury. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 2. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:28.1-28.94.
- Durrani AJ, Miller RJ, Davies M. Basal cell carcinoma arising in a laparoscopic port site scar at the umbilicus. Plast Reconstr Surg. 2005;116:348-350.
- Kotwal S, Madaan S, Prescott S, et al. Unusual squamous cell carcinoma of the scrotum arising from a well healed, innocuous scar of an infertility procedure: a case report. Ann R Coll Surg Engl. 2007;89:17-19.
- Ozyazgan I, Kontas O. Previous injuries or scars as risk factors for the development of basal cell carcinoma. Scand J Plast Reconstr Surg Hand Surg. 2004;38:11-15.
- Swann MH, Yoon J. Merkel cell carcinoma. Semin Oncol. 2007;34:51-56.
- Schrama D, Ugurel S, Becker JC. Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol. 2012;24:141-149.
- Kadir AR. Burn scar neoplasm. Ann Burns Fire Disasters. 2007;20:185-188.
- Walsh NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol. 2001;32:680-689.
- Guenther N, Menenakos C, Braumann C, et al. Squamous cell carcinoma arising on a skin graft 64 years after primary injury. Dermatol Online J. 2007;13:27.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;40:870-871.
- Haik J, Georgiou I, Farber N, et al. Squamous cell carcinoma arising in a split-thickness skin graft donor site. Burns. 2008;34:891-893.
- Ponnuvelu G, Ng MF, Connolly CM, et al. Inflammation to skin malignancy, time to rethink the link: SCC in skin graft donor sites. Surgeon. 2011;9:168-169.
- Bekar A, Kahveci R, Tolunay S, et al. Metastatic gliosarcoma mass extension to a donor fascia lata graft harvest site by tumor cell contamination. World Neurosurg. 2010;73:719-721.
- Hussain A, Ekwobi C, Watson S. Metastatic implantation squamous cell carcinoma in a split-thickness skin graft donor site. J Plast Reconstr Aesthet Surg. 2011;64:690-692.
- May JT, Patil YJ. Keratoacanthoma-type squamous cell carcinoma developing in a skin graft donor site after tumor extirpation at a distant site. Ear Nose Throat J. 2010;89:E11-E13.
- Serrano-Ortega S, Buendia-Eisman A, Ortega del Olmo RM, et al. Melanoma metastasis in donor site of full-thickness skin graft. Dermatology. 2000;201:377-378.
- Wright H, McKinnell TH, Dunkin C. Recurrence of cutaneous squamous cell carcinoma at remote limb donor site. J Plast Reconstr Aesthet Surg. 2012;65:1265-1266.
- Yip KM, Lin J, Kumta SM. A pelvic osteosarcoma with metastasis to the donor site of the bone graft. a case report. Int Orthop. 1996;20:389-391.
- Dias RG, Abudu A, Carter SR, et al. Tumour transfer to bone graft donor site: a case report and review of the literature of the mechanism of seeding. Sarcoma. 2000;4:57-59.
- Neilson D, Emerson DJ, Dunn L. Squamous cell carcinoma of skin developing in a skin graft donor site. Br J Plast Surg. 1988;41:417-419.
- Singh C, Ibrahim S, Pang KS, et al. Implantation metastasis in a 13-year-old girl: a case report. J Orthop Surg (Hong Kong). 2003;11:94-96.
- Enion DS, Scott MJ, Gouldesbrough D. Cutaneous metastasis from a malignant fibrous histiocytoma to a limb skin graft donor site. Br J Surg. 1993;80:366.
- Yamasaki O, Terao K, Asagoe K, et al. Koebner phenomenon on skin graft donor site in cutaneous angiosarcoma. Eur J Dermatol. 2001;11:584-586.
- Hogan BV, Peter MB, Shenoy HG, et al. Surgery induced immunosuppression. Surgeon. 2011;9:38-43.
- Neeman E, Ben-Eliyahu S. The perioperative period and promotion of cancer metastasis: new outlooks on mediating mechanisms and immune involvement. Brain Behav Immun. 2013;30(suppl):32-40.
- Agostino D, Cliffton EE. Trauma as a cause of localization of blood-borne metastases: preventive effect of heparin and fibrinolysin. Ann Surg. 1965;161:97-102.
- Hammond JS, Thomsen S, Ward CG. Scar carcinoma arising acutely in a skin graft donor site. J Trauma. 1987;27:681-683.
- Korula R, Hughes CF. Squamous cell carcinoma arising in a sternotomy scar. Ann Thorac Surg. 1991;51:667-669.
- Kennedy CTC, Burd DAR, Creamer D. Mechanical and thermal injury. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 2. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:28.1-28.94.
- Durrani AJ, Miller RJ, Davies M. Basal cell carcinoma arising in a laparoscopic port site scar at the umbilicus. Plast Reconstr Surg. 2005;116:348-350.
- Kotwal S, Madaan S, Prescott S, et al. Unusual squamous cell carcinoma of the scrotum arising from a well healed, innocuous scar of an infertility procedure: a case report. Ann R Coll Surg Engl. 2007;89:17-19.
- Ozyazgan I, Kontas O. Previous injuries or scars as risk factors for the development of basal cell carcinoma. Scand J Plast Reconstr Surg Hand Surg. 2004;38:11-15.
Practice Points
- Malignancies (both primary and metastatic) can develop in graft donor sites including donor sites for split-thickness skin, full-thickness skin, tendon, bone, and vein grafts.
- Primary malignancies that develop in graft donor sites may be distinct from malignancies that develop in chronic wounds, as the former occur in healed surgical wounds and tend to occur sooner after injury (ie, weeks to months after graft harvesting versus years).
- Although the occurrence is rare, graft donor sites should be examined periodically for development of malignancies.
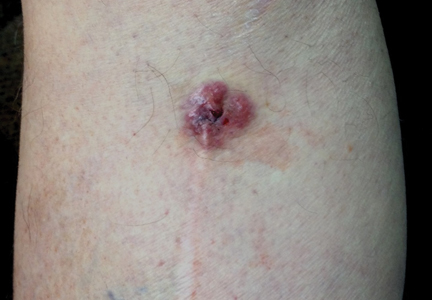
Regional Lymphomatoid Papulosis of the Breast Restricted to an Area of Prior Radiotherapy
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.

The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.
The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.
The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
Practice Points
- Cutaneous lesions of lymphomatoid papulosis (LyP) sometimes are confined to only one area of the skin, which is known as regional LyP.
- Patients with regional LyP have the same prognosis as those with widespread LyP, and no specific association has been reported with this clinical variant.
- Lesions of regional LyP respond to the same treatments as widespread LyP.

Thrower’s Fracture of the Humerus
Case
An otherwise healthy 29-year-old man presented to the ED for evaluation of right arm pain. He had been throwing a baseball when he felt acute onset of severe pain in his right shoulder and became unable to use his arm. Radiographs of the humerus were obtained (Figure a and b).
Fracture of the Humerus
A thrower’s fracture is a rare fracture pattern characterized by a spontaneous fracture of the mid to distal third of the humeral diaphysis during an attempted throwing motion. It was first described by Wilmoth in a case report published in 1930.1 Understanding the proposed mechanism and complications of injury are important for proper work-up and management in the ED.
Fractures of the humerus in young adults are typically the result of high-energy direct trauma. So how does the humerus fracture from throwing a baseball? The most commonly proposed mechanism is an excessive torque during the cocking and acceleration phases of the throwing motion.2-5 This can be visualized as a pitcher’s arm maximally cocked back prior to forward acceleration. During the transition into the acceleration phase, internal rotation is abruptly initiated by the subscapularis, pectoralis major, and latissimus dorsi.6,7 The distal humerus continues to externally rotate due to the momentum generated by the cocking phase, while the proximal humerus violently internally rotates, creating a torsional force on the humerus at the insertion of these muscles and a fulcrum for potential fracture.8 Spiral fractures are the most commonly seen fracture pattern, which correlates with this proposed mechanism.9
Thrower’s fractures are most commonly reported in men in their 20s and 30s who are less seasoned athletes.10,11 These individuals are potentially at greater risk due to the lack of compensatory humeral cortical hypertrophy from repetitive throwing10,12 coupled with a less refined throwing motion.13 Additionally, up to 75% of patients experience prodromal throwing pain at the impending fracture site,11 which suggests that a primary insult such as a stress fracture may also predispose patients to this fracture pattern.
Once a fracture is suspected, a neurovascular assessment should immediately be performed, because concurrent radial nerve injuries have been reported in an average of 11.8% of mid-distal humeral fractures.14 Fractures with associated radial nerve deficits should not be reduced without an orthopedic consultation. Most radial nerve injuries are the result of neuropraxia, which usually resolves spontaneously, and attempted reduction may result in worsening nerve damage.14,15 Additionally, the orthopedist may consider late exploration if no spontaneous nerve recovery occurs within 3 to 6 months.16 Thrower’s fractures with or without associated radial nerve palsies are typically treated conservatively with a hanging cast, which has shown similar results to orthopedic fixation.10,17 The emergency physician should feel comfortable not ordering additional imaging to search for a pathological fracture, unless plain films suggest otherwise.
1. Wilmoth CL. Recurrent fracture of the humerus due to sudden extreme muscular action. J Bone Joint Surg.1930;12(1):168-169.
2. Miller A, Dodson CC, Ilyas AM. Thrower’s fracture of the humerus. Orthop Clin North Am. 2014;45(4):565-569.
3. Weseley MS, Barenfeld PA. Ball throwers’ fracture of the humerus. Six case reports. Clin Orthop Relat Res. 1969;64:153-156.
4. Chao SL, Miller M,Teng SW. A mechanism of spiral fracture of the humerus: a report of 129 cases following the throwing of hand grenades. J Trauma. 1971;11(7):602-605.
5. Polu KR, Schenck RC Jr, Wirth MA, Greeson J, Cone RO 3rd, Rockwood CA Jr. Stress fracture of the humerus in a collegiate baseball pitcher. A case report. Am J Sports Med. 1999;27(6):813-816.
6. Jobe FW, Moynes DR, Tibone JE, Perry J. An EMG analysis of the shoulder in pitching. A second report. Am J Sports Med. 1984;12(3):218-220.
7. Pappas AM, Zawacki RM, Sullivan TJ. Biomechanics of baseball pitching. A preliminary report. Am J Sports Med. 1985;13(4):216-222.
8. Sabick MB, Torry MR, Kim YK, Hawkins RJ. Humeral torque in professional baseball pitchers. Am J Sports Med. 2004;32(4):892-898.
9. Klenerman L. Fractures of the shaft of the humerus. J Bone Joint Surg Br. 1966;48(1):105-111.
10. Ogawa K, Yoshida A. Throwing fracture of the humeral shaft. An analysis of 90 patients. Am J Sports Med. 1998;26(2):242-246.
11. Branch T, Partin C, Chamberland P, Emeterio E, Sabetelle M. Spontaneous fractures of the humerus during pitching. A series of 12 cases. Am J Sports Med. 1992;20(4):468-470.
12. Tullos HS, Erwin WD, Woods GW, Wukasch DC, Cooley DA, King JW. Unusual lesions of the pitching arm. Clin Orthop Relat Res. 1972;88:169-182.
13. Bingham EL. Fractures of the humerus from muscular violence. U S Armed Forces Med J. 1959;10(1):22-25.
14. Shao YC, Harwood P, Grotz MR, Limb D, Giannoudis PV. Radial nerve palsy associated with fractures of the shaft of the humerus: a systematic review. J Bone Joint Surg Br. 2005;87(12):1647-1652.
15. Bishop J, Ring D. Management of radial nerve palsy associated with humeral shaft fracture: a decision analysis model. J Hand Surg Am. 2009;34(6)991-996.
16. Niver GE, Ilyas AM. Management of radial nerve palsy following fractures of the humerus. Orthop Clin North Am. 2013;44(3):419-424.
17. Kaplan H, Kiral A, Kuskucu M, Arpacioglu MO, Sarioglu A, Rodop O. Report of eight cases of humeral fracture following the throwing of hand grenades. Arch Orthop Trauma Surg. 1998;117(1-2):50-52.
Case
An otherwise healthy 29-year-old man presented to the ED for evaluation of right arm pain. He had been throwing a baseball when he felt acute onset of severe pain in his right shoulder and became unable to use his arm. Radiographs of the humerus were obtained (Figure a and b).
Fracture of the Humerus
A thrower’s fracture is a rare fracture pattern characterized by a spontaneous fracture of the mid to distal third of the humeral diaphysis during an attempted throwing motion. It was first described by Wilmoth in a case report published in 1930.1 Understanding the proposed mechanism and complications of injury are important for proper work-up and management in the ED.
Fractures of the humerus in young adults are typically the result of high-energy direct trauma. So how does the humerus fracture from throwing a baseball? The most commonly proposed mechanism is an excessive torque during the cocking and acceleration phases of the throwing motion.2-5 This can be visualized as a pitcher’s arm maximally cocked back prior to forward acceleration. During the transition into the acceleration phase, internal rotation is abruptly initiated by the subscapularis, pectoralis major, and latissimus dorsi.6,7 The distal humerus continues to externally rotate due to the momentum generated by the cocking phase, while the proximal humerus violently internally rotates, creating a torsional force on the humerus at the insertion of these muscles and a fulcrum for potential fracture.8 Spiral fractures are the most commonly seen fracture pattern, which correlates with this proposed mechanism.9
Thrower’s fractures are most commonly reported in men in their 20s and 30s who are less seasoned athletes.10,11 These individuals are potentially at greater risk due to the lack of compensatory humeral cortical hypertrophy from repetitive throwing10,12 coupled with a less refined throwing motion.13 Additionally, up to 75% of patients experience prodromal throwing pain at the impending fracture site,11 which suggests that a primary insult such as a stress fracture may also predispose patients to this fracture pattern.
Once a fracture is suspected, a neurovascular assessment should immediately be performed, because concurrent radial nerve injuries have been reported in an average of 11.8% of mid-distal humeral fractures.14 Fractures with associated radial nerve deficits should not be reduced without an orthopedic consultation. Most radial nerve injuries are the result of neuropraxia, which usually resolves spontaneously, and attempted reduction may result in worsening nerve damage.14,15 Additionally, the orthopedist may consider late exploration if no spontaneous nerve recovery occurs within 3 to 6 months.16 Thrower’s fractures with or without associated radial nerve palsies are typically treated conservatively with a hanging cast, which has shown similar results to orthopedic fixation.10,17 The emergency physician should feel comfortable not ordering additional imaging to search for a pathological fracture, unless plain films suggest otherwise.
Case
An otherwise healthy 29-year-old man presented to the ED for evaluation of right arm pain. He had been throwing a baseball when he felt acute onset of severe pain in his right shoulder and became unable to use his arm. Radiographs of the humerus were obtained (Figure a and b).
Fracture of the Humerus
A thrower’s fracture is a rare fracture pattern characterized by a spontaneous fracture of the mid to distal third of the humeral diaphysis during an attempted throwing motion. It was first described by Wilmoth in a case report published in 1930.1 Understanding the proposed mechanism and complications of injury are important for proper work-up and management in the ED.
Fractures of the humerus in young adults are typically the result of high-energy direct trauma. So how does the humerus fracture from throwing a baseball? The most commonly proposed mechanism is an excessive torque during the cocking and acceleration phases of the throwing motion.2-5 This can be visualized as a pitcher’s arm maximally cocked back prior to forward acceleration. During the transition into the acceleration phase, internal rotation is abruptly initiated by the subscapularis, pectoralis major, and latissimus dorsi.6,7 The distal humerus continues to externally rotate due to the momentum generated by the cocking phase, while the proximal humerus violently internally rotates, creating a torsional force on the humerus at the insertion of these muscles and a fulcrum for potential fracture.8 Spiral fractures are the most commonly seen fracture pattern, which correlates with this proposed mechanism.9
Thrower’s fractures are most commonly reported in men in their 20s and 30s who are less seasoned athletes.10,11 These individuals are potentially at greater risk due to the lack of compensatory humeral cortical hypertrophy from repetitive throwing10,12 coupled with a less refined throwing motion.13 Additionally, up to 75% of patients experience prodromal throwing pain at the impending fracture site,11 which suggests that a primary insult such as a stress fracture may also predispose patients to this fracture pattern.
Once a fracture is suspected, a neurovascular assessment should immediately be performed, because concurrent radial nerve injuries have been reported in an average of 11.8% of mid-distal humeral fractures.14 Fractures with associated radial nerve deficits should not be reduced without an orthopedic consultation. Most radial nerve injuries are the result of neuropraxia, which usually resolves spontaneously, and attempted reduction may result in worsening nerve damage.14,15 Additionally, the orthopedist may consider late exploration if no spontaneous nerve recovery occurs within 3 to 6 months.16 Thrower’s fractures with or without associated radial nerve palsies are typically treated conservatively with a hanging cast, which has shown similar results to orthopedic fixation.10,17 The emergency physician should feel comfortable not ordering additional imaging to search for a pathological fracture, unless plain films suggest otherwise.
1. Wilmoth CL. Recurrent fracture of the humerus due to sudden extreme muscular action. J Bone Joint Surg.1930;12(1):168-169.
2. Miller A, Dodson CC, Ilyas AM. Thrower’s fracture of the humerus. Orthop Clin North Am. 2014;45(4):565-569.
3. Weseley MS, Barenfeld PA. Ball throwers’ fracture of the humerus. Six case reports. Clin Orthop Relat Res. 1969;64:153-156.
4. Chao SL, Miller M,Teng SW. A mechanism of spiral fracture of the humerus: a report of 129 cases following the throwing of hand grenades. J Trauma. 1971;11(7):602-605.
5. Polu KR, Schenck RC Jr, Wirth MA, Greeson J, Cone RO 3rd, Rockwood CA Jr. Stress fracture of the humerus in a collegiate baseball pitcher. A case report. Am J Sports Med. 1999;27(6):813-816.
6. Jobe FW, Moynes DR, Tibone JE, Perry J. An EMG analysis of the shoulder in pitching. A second report. Am J Sports Med. 1984;12(3):218-220.
7. Pappas AM, Zawacki RM, Sullivan TJ. Biomechanics of baseball pitching. A preliminary report. Am J Sports Med. 1985;13(4):216-222.
8. Sabick MB, Torry MR, Kim YK, Hawkins RJ. Humeral torque in professional baseball pitchers. Am J Sports Med. 2004;32(4):892-898.
9. Klenerman L. Fractures of the shaft of the humerus. J Bone Joint Surg Br. 1966;48(1):105-111.
10. Ogawa K, Yoshida A. Throwing fracture of the humeral shaft. An analysis of 90 patients. Am J Sports Med. 1998;26(2):242-246.
11. Branch T, Partin C, Chamberland P, Emeterio E, Sabetelle M. Spontaneous fractures of the humerus during pitching. A series of 12 cases. Am J Sports Med. 1992;20(4):468-470.
12. Tullos HS, Erwin WD, Woods GW, Wukasch DC, Cooley DA, King JW. Unusual lesions of the pitching arm. Clin Orthop Relat Res. 1972;88:169-182.
13. Bingham EL. Fractures of the humerus from muscular violence. U S Armed Forces Med J. 1959;10(1):22-25.
14. Shao YC, Harwood P, Grotz MR, Limb D, Giannoudis PV. Radial nerve palsy associated with fractures of the shaft of the humerus: a systematic review. J Bone Joint Surg Br. 2005;87(12):1647-1652.
15. Bishop J, Ring D. Management of radial nerve palsy associated with humeral shaft fracture: a decision analysis model. J Hand Surg Am. 2009;34(6)991-996.
16. Niver GE, Ilyas AM. Management of radial nerve palsy following fractures of the humerus. Orthop Clin North Am. 2013;44(3):419-424.
17. Kaplan H, Kiral A, Kuskucu M, Arpacioglu MO, Sarioglu A, Rodop O. Report of eight cases of humeral fracture following the throwing of hand grenades. Arch Orthop Trauma Surg. 1998;117(1-2):50-52.
1. Wilmoth CL. Recurrent fracture of the humerus due to sudden extreme muscular action. J Bone Joint Surg.1930;12(1):168-169.
2. Miller A, Dodson CC, Ilyas AM. Thrower’s fracture of the humerus. Orthop Clin North Am. 2014;45(4):565-569.
3. Weseley MS, Barenfeld PA. Ball throwers’ fracture of the humerus. Six case reports. Clin Orthop Relat Res. 1969;64:153-156.
4. Chao SL, Miller M,Teng SW. A mechanism of spiral fracture of the humerus: a report of 129 cases following the throwing of hand grenades. J Trauma. 1971;11(7):602-605.
5. Polu KR, Schenck RC Jr, Wirth MA, Greeson J, Cone RO 3rd, Rockwood CA Jr. Stress fracture of the humerus in a collegiate baseball pitcher. A case report. Am J Sports Med. 1999;27(6):813-816.
6. Jobe FW, Moynes DR, Tibone JE, Perry J. An EMG analysis of the shoulder in pitching. A second report. Am J Sports Med. 1984;12(3):218-220.
7. Pappas AM, Zawacki RM, Sullivan TJ. Biomechanics of baseball pitching. A preliminary report. Am J Sports Med. 1985;13(4):216-222.
8. Sabick MB, Torry MR, Kim YK, Hawkins RJ. Humeral torque in professional baseball pitchers. Am J Sports Med. 2004;32(4):892-898.
9. Klenerman L. Fractures of the shaft of the humerus. J Bone Joint Surg Br. 1966;48(1):105-111.
10. Ogawa K, Yoshida A. Throwing fracture of the humeral shaft. An analysis of 90 patients. Am J Sports Med. 1998;26(2):242-246.
11. Branch T, Partin C, Chamberland P, Emeterio E, Sabetelle M. Spontaneous fractures of the humerus during pitching. A series of 12 cases. Am J Sports Med. 1992;20(4):468-470.
12. Tullos HS, Erwin WD, Woods GW, Wukasch DC, Cooley DA, King JW. Unusual lesions of the pitching arm. Clin Orthop Relat Res. 1972;88:169-182.
13. Bingham EL. Fractures of the humerus from muscular violence. U S Armed Forces Med J. 1959;10(1):22-25.
14. Shao YC, Harwood P, Grotz MR, Limb D, Giannoudis PV. Radial nerve palsy associated with fractures of the shaft of the humerus: a systematic review. J Bone Joint Surg Br. 2005;87(12):1647-1652.
15. Bishop J, Ring D. Management of radial nerve palsy associated with humeral shaft fracture: a decision analysis model. J Hand Surg Am. 2009;34(6)991-996.
16. Niver GE, Ilyas AM. Management of radial nerve palsy following fractures of the humerus. Orthop Clin North Am. 2013;44(3):419-424.
17. Kaplan H, Kiral A, Kuskucu M, Arpacioglu MO, Sarioglu A, Rodop O. Report of eight cases of humeral fracture following the throwing of hand grenades. Arch Orthop Trauma Surg. 1998;117(1-2):50-52.
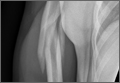
Spontaneous Cervical Spinal Epidural Hematoma
Unilateral weakness is a common ED presentation with a diverse etiology, including stroke.1,2 Studies have reported a misdiagnosis rate of stroke and transient ischemic attack of approximately 10%.3 This case presents an unusual stroke mimic where treatment with an anticoagulant could have led to adverse outcomes. It also highlights the importance of considering a spontaneous cervical spinal epidural hematoma (SCSEH) as a stroke mimic. This is especially pertinent when a patient’s symptoms are not fully consistent with an acute stroke, in order to avoid potentially dangerous anticoagulation and allow for prompt treatment of the hematoma.
Case
A 68-year-old woman presented to the ED via ambulance with the complaint of stroke-like symptoms. She had a 2-hour history of sudden-onset right arm and leg weakness with loss of sensation, and right-sided neck pain. She also complained of a mild right-sided headache, which had an insidious onset and had been present for the past 2 days. She had recently finished a course of antibiotics for a urinary tract infection. There was no history of trauma. The patient had no significant medical history, was taking no medications, and was a nonsmoker with a normal body mass index. Her family history was significant for cerebral vascular accidents.
On arrival at the ED, the patient had a blood pressure of 179/95 mm Hg; her other vital signs were normal. On examination, she had a right-sided hemiplegia, with a 0/5 power grading observed for motor strength for both her right arm and leg. She reported paresthesia in dermatomes C4 to C5 and L3 to L5. There was extreme tenderness when her right trapezius and upper paraspinal muscles were palpated, but she had no midline cervical spine tenderness and had full, though painful, range of movement of her neck. Her left side was unaffected. She had normal cranial nerves, no higher cortical dysfunction, a Glasgow Coma Scale (GCS) score of 15, and complete control of her bladder and bowel.
A computed tomography angiogram (CTA) of the carotid arteries and a CT scan of the head were ordered to rule out acute stroke and carotid artery dissection. The CT scan of the head showed no acute bleeding or evidence of infarction. However, the CTA raised suspicion of an SCSEH.
Subsequent magnetic resonance imaging showed a right posterior epidural lesion measuring 3 to 3.5 cm in length and 8 to 9 mm in maximal thickness, extending from the midpoint of C2 to the C3-C4 disc level. The lesion compressed the spinal cord, and this stenosis was made worse by posterior disc osteophytes. There was a single strongly enhancing vessel projecting within the hematoma, raising the suspicion of an active bleed (Figure).
Blood tests, including a coagulation screen, were normal and an electrocardiogram showed normal sinus rhythm at 57 beats/minute.
The patient was transferred to the neurosurgery team at the tertiary center, where she underwent an emergency C2-C3 laminectomy that day. She was discharged 6 days later and has made a full recovery with physiotherapy, regaining full function of her right arm and leg.
Discussion
Spontaneous cervical spinal epidural hematomas are rare, with an estimated annual incidence of 0.1 cases per 100,000 people.4 The etiology is largely unknown, but SCSEH has been attributed to a venous source.5 Multiple predisposing factors have been reported, including coagulopathies, anticoagulation, disc herniation, vascular malformations, neoplasms, and idiopathic causes.3,4
The most common presentation of an SCSEH is sudden onset of interscapular or cervical pain with paresthesia and even paralysis. Paraplegia and quadriplegia are common; however, hemiplegia as a presentation of an SCSEH is rarely reported in the literature.6-8
In a case report, Wang et al6 described a similar presentation and management of an SCSEH in the ED as our case. However, the patient in that report was initially treated for a stroke with heparin, and the authors commented that the unnecessary anticoagulation could have caused expansion of the hematoma and subsequent spinal cord compression.6
Both our patient and the patient described by Wang et al6 presented with hemiplegia, which is more commonly seen as a presenting feature in stroke and is a rarely reported presentation of an SCSEH. However, neck pain is not a classical presenting feature in stroke, and this prompted us to order the CTA of the carotid arteries and CT scan of the head. This ultimately led to the correct diagnosis and prompt management, avoiding unnecessary and potentially dangerous anticoagulation.
Conclusion
Hemiplegia is an important, though rarely reported, presentation of an SCSEH and should not be misdiagnosed as an acute stroke. Neck pain in a patient presenting with unilateral weakness should be a red flag that prompts the emergency physician (EP) to search for alternative diagnoses to stroke. If a patient with an SCSEH presents to the ED, prompt and accurate recognition by an EP allows for early surgical intervention, which improves clinical outcome, aids neurological recovery, and minimizes long-term sequales.6
1. Nickel C, Nemec M, Bingisser R. Weakness as presenting symptom in emergency department. Swiss Med Wkly. 2009;139(17-18):271-272.
2. Asimos AW, Hockberger RS, Grayzel J. Evaluation of the adult with acute weakness in the emergency department. UpToDate web site. Available at: http://www.uptodate.com/contents/evaluation-of-the-adult-with-acute-weakness-in-the-emergency-department. Accessed April 12, 2016.
3. Pope JV, Edlow JA. Avoiding misdiagnosis in patients with neurological emergencies. Emerg Med Int. 2012;2012:949275.
4. Baek B, Hur J, Kwon Ki, Lee HK. Spontaneous spinal epidural hematoma. J Korean Neurosurg Soc. 2008; 44(1):40-42.
5. Fukui M, Swarnkar A, Williams R. Acute spontaneous spinal epidural hematomas. AJNR Am J Neuroradiol.1999;20(7):1365-1372.
6. Wang CC, Chang CH, Lin HJ, Lin KC, Kuo JR. Misdiagnosis of spontaneous cervical epidural haemorrhage. Eur Spine J. 2009;18(Suppl 2):210-212.
7. Hsieh CF, Lin HJ, Chen KT, Foo NP, Te AL. Acute spontaneous cervical spinal epidural hematoma with hemiparesis as the initial presentation. Eur J Emerg Med. 2006;13(1):36-38.
8. Dimou J, Jithoo R, Bush S. A patient with delayed traumatic cervical spinal epidural hematoma presenting with hemiparesis. J Clin Neurosci. 2010;17(3):404-405.
Unilateral weakness is a common ED presentation with a diverse etiology, including stroke.1,2 Studies have reported a misdiagnosis rate of stroke and transient ischemic attack of approximately 10%.3 This case presents an unusual stroke mimic where treatment with an anticoagulant could have led to adverse outcomes. It also highlights the importance of considering a spontaneous cervical spinal epidural hematoma (SCSEH) as a stroke mimic. This is especially pertinent when a patient’s symptoms are not fully consistent with an acute stroke, in order to avoid potentially dangerous anticoagulation and allow for prompt treatment of the hematoma.
Case
A 68-year-old woman presented to the ED via ambulance with the complaint of stroke-like symptoms. She had a 2-hour history of sudden-onset right arm and leg weakness with loss of sensation, and right-sided neck pain. She also complained of a mild right-sided headache, which had an insidious onset and had been present for the past 2 days. She had recently finished a course of antibiotics for a urinary tract infection. There was no history of trauma. The patient had no significant medical history, was taking no medications, and was a nonsmoker with a normal body mass index. Her family history was significant for cerebral vascular accidents.
On arrival at the ED, the patient had a blood pressure of 179/95 mm Hg; her other vital signs were normal. On examination, she had a right-sided hemiplegia, with a 0/5 power grading observed for motor strength for both her right arm and leg. She reported paresthesia in dermatomes C4 to C5 and L3 to L5. There was extreme tenderness when her right trapezius and upper paraspinal muscles were palpated, but she had no midline cervical spine tenderness and had full, though painful, range of movement of her neck. Her left side was unaffected. She had normal cranial nerves, no higher cortical dysfunction, a Glasgow Coma Scale (GCS) score of 15, and complete control of her bladder and bowel.
A computed tomography angiogram (CTA) of the carotid arteries and a CT scan of the head were ordered to rule out acute stroke and carotid artery dissection. The CT scan of the head showed no acute bleeding or evidence of infarction. However, the CTA raised suspicion of an SCSEH.
Subsequent magnetic resonance imaging showed a right posterior epidural lesion measuring 3 to 3.5 cm in length and 8 to 9 mm in maximal thickness, extending from the midpoint of C2 to the C3-C4 disc level. The lesion compressed the spinal cord, and this stenosis was made worse by posterior disc osteophytes. There was a single strongly enhancing vessel projecting within the hematoma, raising the suspicion of an active bleed (Figure).
Blood tests, including a coagulation screen, were normal and an electrocardiogram showed normal sinus rhythm at 57 beats/minute.
The patient was transferred to the neurosurgery team at the tertiary center, where she underwent an emergency C2-C3 laminectomy that day. She was discharged 6 days later and has made a full recovery with physiotherapy, regaining full function of her right arm and leg.
Discussion
Spontaneous cervical spinal epidural hematomas are rare, with an estimated annual incidence of 0.1 cases per 100,000 people.4 The etiology is largely unknown, but SCSEH has been attributed to a venous source.5 Multiple predisposing factors have been reported, including coagulopathies, anticoagulation, disc herniation, vascular malformations, neoplasms, and idiopathic causes.3,4
The most common presentation of an SCSEH is sudden onset of interscapular or cervical pain with paresthesia and even paralysis. Paraplegia and quadriplegia are common; however, hemiplegia as a presentation of an SCSEH is rarely reported in the literature.6-8
In a case report, Wang et al6 described a similar presentation and management of an SCSEH in the ED as our case. However, the patient in that report was initially treated for a stroke with heparin, and the authors commented that the unnecessary anticoagulation could have caused expansion of the hematoma and subsequent spinal cord compression.6
Both our patient and the patient described by Wang et al6 presented with hemiplegia, which is more commonly seen as a presenting feature in stroke and is a rarely reported presentation of an SCSEH. However, neck pain is not a classical presenting feature in stroke, and this prompted us to order the CTA of the carotid arteries and CT scan of the head. This ultimately led to the correct diagnosis and prompt management, avoiding unnecessary and potentially dangerous anticoagulation.
Conclusion
Hemiplegia is an important, though rarely reported, presentation of an SCSEH and should not be misdiagnosed as an acute stroke. Neck pain in a patient presenting with unilateral weakness should be a red flag that prompts the emergency physician (EP) to search for alternative diagnoses to stroke. If a patient with an SCSEH presents to the ED, prompt and accurate recognition by an EP allows for early surgical intervention, which improves clinical outcome, aids neurological recovery, and minimizes long-term sequales.6
Unilateral weakness is a common ED presentation with a diverse etiology, including stroke.1,2 Studies have reported a misdiagnosis rate of stroke and transient ischemic attack of approximately 10%.3 This case presents an unusual stroke mimic where treatment with an anticoagulant could have led to adverse outcomes. It also highlights the importance of considering a spontaneous cervical spinal epidural hematoma (SCSEH) as a stroke mimic. This is especially pertinent when a patient’s symptoms are not fully consistent with an acute stroke, in order to avoid potentially dangerous anticoagulation and allow for prompt treatment of the hematoma.
Case
A 68-year-old woman presented to the ED via ambulance with the complaint of stroke-like symptoms. She had a 2-hour history of sudden-onset right arm and leg weakness with loss of sensation, and right-sided neck pain. She also complained of a mild right-sided headache, which had an insidious onset and had been present for the past 2 days. She had recently finished a course of antibiotics for a urinary tract infection. There was no history of trauma. The patient had no significant medical history, was taking no medications, and was a nonsmoker with a normal body mass index. Her family history was significant for cerebral vascular accidents.
On arrival at the ED, the patient had a blood pressure of 179/95 mm Hg; her other vital signs were normal. On examination, she had a right-sided hemiplegia, with a 0/5 power grading observed for motor strength for both her right arm and leg. She reported paresthesia in dermatomes C4 to C5 and L3 to L5. There was extreme tenderness when her right trapezius and upper paraspinal muscles were palpated, but she had no midline cervical spine tenderness and had full, though painful, range of movement of her neck. Her left side was unaffected. She had normal cranial nerves, no higher cortical dysfunction, a Glasgow Coma Scale (GCS) score of 15, and complete control of her bladder and bowel.
A computed tomography angiogram (CTA) of the carotid arteries and a CT scan of the head were ordered to rule out acute stroke and carotid artery dissection. The CT scan of the head showed no acute bleeding or evidence of infarction. However, the CTA raised suspicion of an SCSEH.
Subsequent magnetic resonance imaging showed a right posterior epidural lesion measuring 3 to 3.5 cm in length and 8 to 9 mm in maximal thickness, extending from the midpoint of C2 to the C3-C4 disc level. The lesion compressed the spinal cord, and this stenosis was made worse by posterior disc osteophytes. There was a single strongly enhancing vessel projecting within the hematoma, raising the suspicion of an active bleed (Figure).
Blood tests, including a coagulation screen, were normal and an electrocardiogram showed normal sinus rhythm at 57 beats/minute.
The patient was transferred to the neurosurgery team at the tertiary center, where she underwent an emergency C2-C3 laminectomy that day. She was discharged 6 days later and has made a full recovery with physiotherapy, regaining full function of her right arm and leg.
Discussion
Spontaneous cervical spinal epidural hematomas are rare, with an estimated annual incidence of 0.1 cases per 100,000 people.4 The etiology is largely unknown, but SCSEH has been attributed to a venous source.5 Multiple predisposing factors have been reported, including coagulopathies, anticoagulation, disc herniation, vascular malformations, neoplasms, and idiopathic causes.3,4
The most common presentation of an SCSEH is sudden onset of interscapular or cervical pain with paresthesia and even paralysis. Paraplegia and quadriplegia are common; however, hemiplegia as a presentation of an SCSEH is rarely reported in the literature.6-8
In a case report, Wang et al6 described a similar presentation and management of an SCSEH in the ED as our case. However, the patient in that report was initially treated for a stroke with heparin, and the authors commented that the unnecessary anticoagulation could have caused expansion of the hematoma and subsequent spinal cord compression.6
Both our patient and the patient described by Wang et al6 presented with hemiplegia, which is more commonly seen as a presenting feature in stroke and is a rarely reported presentation of an SCSEH. However, neck pain is not a classical presenting feature in stroke, and this prompted us to order the CTA of the carotid arteries and CT scan of the head. This ultimately led to the correct diagnosis and prompt management, avoiding unnecessary and potentially dangerous anticoagulation.
Conclusion
Hemiplegia is an important, though rarely reported, presentation of an SCSEH and should not be misdiagnosed as an acute stroke. Neck pain in a patient presenting with unilateral weakness should be a red flag that prompts the emergency physician (EP) to search for alternative diagnoses to stroke. If a patient with an SCSEH presents to the ED, prompt and accurate recognition by an EP allows for early surgical intervention, which improves clinical outcome, aids neurological recovery, and minimizes long-term sequales.6
1. Nickel C, Nemec M, Bingisser R. Weakness as presenting symptom in emergency department. Swiss Med Wkly. 2009;139(17-18):271-272.
2. Asimos AW, Hockberger RS, Grayzel J. Evaluation of the adult with acute weakness in the emergency department. UpToDate web site. Available at: http://www.uptodate.com/contents/evaluation-of-the-adult-with-acute-weakness-in-the-emergency-department. Accessed April 12, 2016.
3. Pope JV, Edlow JA. Avoiding misdiagnosis in patients with neurological emergencies. Emerg Med Int. 2012;2012:949275.
4. Baek B, Hur J, Kwon Ki, Lee HK. Spontaneous spinal epidural hematoma. J Korean Neurosurg Soc. 2008; 44(1):40-42.
5. Fukui M, Swarnkar A, Williams R. Acute spontaneous spinal epidural hematomas. AJNR Am J Neuroradiol.1999;20(7):1365-1372.
6. Wang CC, Chang CH, Lin HJ, Lin KC, Kuo JR. Misdiagnosis of spontaneous cervical epidural haemorrhage. Eur Spine J. 2009;18(Suppl 2):210-212.
7. Hsieh CF, Lin HJ, Chen KT, Foo NP, Te AL. Acute spontaneous cervical spinal epidural hematoma with hemiparesis as the initial presentation. Eur J Emerg Med. 2006;13(1):36-38.
8. Dimou J, Jithoo R, Bush S. A patient with delayed traumatic cervical spinal epidural hematoma presenting with hemiparesis. J Clin Neurosci. 2010;17(3):404-405.
1. Nickel C, Nemec M, Bingisser R. Weakness as presenting symptom in emergency department. Swiss Med Wkly. 2009;139(17-18):271-272.
2. Asimos AW, Hockberger RS, Grayzel J. Evaluation of the adult with acute weakness in the emergency department. UpToDate web site. Available at: http://www.uptodate.com/contents/evaluation-of-the-adult-with-acute-weakness-in-the-emergency-department. Accessed April 12, 2016.
3. Pope JV, Edlow JA. Avoiding misdiagnosis in patients with neurological emergencies. Emerg Med Int. 2012;2012:949275.
4. Baek B, Hur J, Kwon Ki, Lee HK. Spontaneous spinal epidural hematoma. J Korean Neurosurg Soc. 2008; 44(1):40-42.
5. Fukui M, Swarnkar A, Williams R. Acute spontaneous spinal epidural hematomas. AJNR Am J Neuroradiol.1999;20(7):1365-1372.
6. Wang CC, Chang CH, Lin HJ, Lin KC, Kuo JR. Misdiagnosis of spontaneous cervical epidural haemorrhage. Eur Spine J. 2009;18(Suppl 2):210-212.
7. Hsieh CF, Lin HJ, Chen KT, Foo NP, Te AL. Acute spontaneous cervical spinal epidural hematoma with hemiparesis as the initial presentation. Eur J Emerg Med. 2006;13(1):36-38.
8. Dimou J, Jithoo R, Bush S. A patient with delayed traumatic cervical spinal epidural hematoma presenting with hemiparesis. J Clin Neurosci. 2010;17(3):404-405.

Multiple Morphologically Distinct Cutaneous Granular Cell Tumors Occurring in a Single Patient
Case Report
A 27-year-old black man was admitted to the hospital with chills; night sweats; unintentional 25-lb weight loss; and multiple widespread, painful, progressively enlarging skin nodules of 3 months’ duration. The lesions had first developed on the back and later appeared on the face, trunk, arms, thighs, and genital region. He denied dysuria or urethral discharge. He had a remote history of adequately treated chlamydia infection but no other remarkable personal or family history.
| ||
| Figure 1. Firm subcutaneous nodules on the back with no epidermal change. | ||
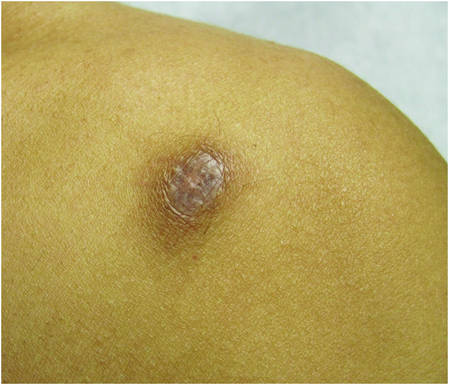
| ||
| Figure 2. Firm dermal papule on the anterior aspect of the left shoulder with violaceous hyperpigmentation (dermatofibromalike). |
Physical examination revealed a thin man with more than 20 lesions on the face, trunk, arms, thighs, and genital region ranging in size from 1 to 4 cm. Lesion morphologies varied greatly and included subcutaneous firm nodules with no epidermal change (Figure 1); dermatofibromalike nodules with overlying erythema and hyperpigmentation (Figure 2); condylomalike, verrucous, pink papulonodules (Figure 3); ulcerated angular plaques with rolled borders and palpable tumor extension deep (1–2 cm) to the subcutis (Figure 4); and a vegetative, eroded, exophytic tumor with palpable deep extension (Figure 4). A diffuse, erythematous, macular eruption also was noted on the trunk and bilateral arms and legs including the soles of both feet along with nontender cervical, axillary, and inguinal lymphadenopathy. The ocular, oral, and nasal mucosae were not affected.
The differential diagnosis for each lesion differed based on morphology. Infectious, inflammatory, and neoplastic processes were considered, including syphilis, dermatofibroma, dermatofibrosarcoma protuberans, metastatic disease, leukemia cutis, sarcoidosis, panniculitis, condyloma acuminatum, and vegetative herpes simplex virus infection (inguinal lesion).
Laboratory data revealed a reactive rapid plasma reagin with treponemal IgG titers of 1:64. Urine chlamydia RNA probe and lymphogranuloma venereum (LGV) serum antibodies also were positive. Human immunodeficiency virus screening was negative. Positron emission tomography–computerized tomography revealed enlarged and hypermetabolic lymphadenopathy above and below the diaphragm.
After therapy with intravenous penicillin G and oral doxycycline for concurrent secondary syphilis and LGV, the patient’s macular eruption and constitutional symptoms resolved within weeks of the initial presentation. His lymphadenopathy improved, his rapid plasma reagin titer decreased, and his chlamydia RNA became undetectable. However, the skin lesions remained unchanged.
Incisional biopsies of 4 clinically distinct skin lesions revealed well-delineated dermal proliferations of cells with eosinophilic granular cytoplasm and indistinct cell borders (Figure 5). Two specimens displayed marked epidermal hyperplasia (Figure 6).
No atypical mitotic figures were identified. Immunohistochemistry for S-100 protein was diffusely positive in the neoplastic cells. Immunohistochemistry for Treponema pallidum was negative.
No mycobacterial or fungal organisms were identified in acid-fast bacillus, periodic acid–Schiff, or Gomori methenamine-silver–stained sections. All 4 lesions had histopathologic findings characteristic of granular cell tumors (GCTs). A lesion in the left inguinal region (Figure 4 [medial lesion]), which initially was thought to be condyloma latum or a squamous cell carcinoma (SCC), also was later confirmed to be a GCT.
Repeat positron emission tomography–computerized tomography several weeks later confirmed resolution of the previously noted lymphadenopathy. Although 2 GCTs have not recurred after biopsy, the other 2, which the patient refused to have completely excised, continued to grow. Follow-up 2.5 years after hospitalization revealed persistence of the lesions with no remarkable morphological changes.
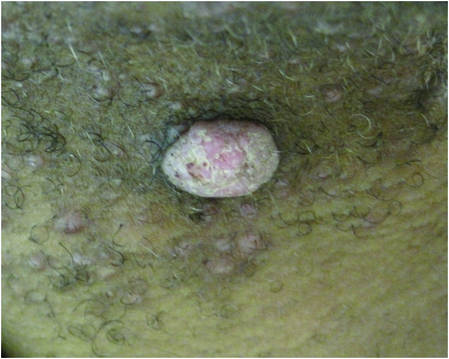
| 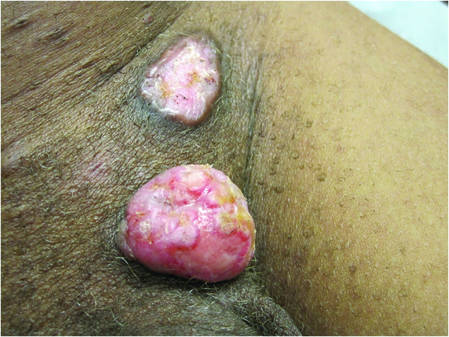
| |
| Figure 3. Verrucous pink papule on the right side of the neck. | Figure 4. Ulcerated angular plaque in the left inguinal/genital area with rolled borders and tumor extension deep to the subcutis adjacent to a vegetative, eroded, exophytic tumor with palpable deep extension. | |
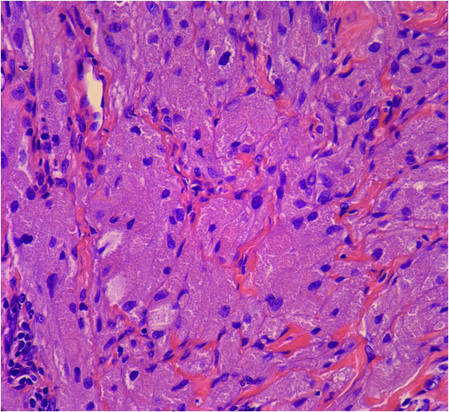
| 
| |
Figure 5. Large polygonal cells with eosinophilic granular cytoplasm, prominent bland nuclei, and indistinct cell borders (H&E, original magnification ×40). | Figure 6. Marked pseudoepitheliomatous hyperplasia (H&E, original magnification ×10). |
Comment
First described in 1854, GCTs are uncommon neoplasms of probable Schwann cell origin that can arise in almost any location of the body but most often appear on the skin and in the subcutaneous tissues and oral cavity.1,2 The commonly regarded rule of thirds describes its most favored locations: one-third on the tongue, one-third on the skin, and one-third in internal organs.3,4 Granular cell tumors occur with greater frequency in adults, females, and black individuals.1-5
Cutaneous GCTs usually present as solitary asymptomatic masses; however, multiple tumors have been noted in up to 25% of reported cases.4,6 In children, multiple cutaneous GCTs have been reported in the setting of neurofibromatosis type I as well as with other disorders.2,5,7-9
Cutaneous GCTs have been reported to range from sessile, pedunculated, or verrucous nodules to subcutaneous papules and nodules with no epidermal change. Our case not only illustrated the diverse clinical appearance of cutaneous GCTs but also demonstrated multiple morphologically distinct cutaneous GCTs occurring in a single patient. Of particular interest is our patient’s coexisting secondary syphilis and LGV infections, which can pose a diagnostic dilemma to the unsuspecting clinician. The manifold appearances of this patient’s GCTs resulted in a broad differential diagnosis. Syphilis (condyloma latum), condyloma acuminatum, LGV, metastatic disease, Kaposi sarcoma, lymphoma, dermatofibrosarcoma protuberans, leiomyoma, SCC, and deep fungal and atypical mycobacterial infection were all considerations. In 1981, Apisarnthanarax1 reviewed 88 cases of GCTs seen over a 15-year period and discovered that the preoperative clinical diagnoses were incorrect in all cases. Skin biopsy is necessary to diagnose GCT, and our patient’s case underscores the need for a thorough history, physical examination, and laboratory evaluation to rule out coexisting diseases.
Histopathology of cutaneous GCTs shows an unencapsulated dermal proliferation of large monotonous polygonal cells with blurred cell borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests. Nuclei are small, uniform, round, centrally located, and rarely contain mitoses.3 The presence of mitotic activity on histopathology does not necessarily portend malignant biological behavior.5 Overlying pseudoepitheliomatous hyperplasia has been reported in as many as 85% of GCTs and may mimic SCC.10 The neoplastic cells stain positively with S-100 protein, neuron-specific enolase, and peripheral nerve myelin proteins.3,4 The cytoplasmic granules are positive on periodic acid–Schiff staining and diastase resistant and will sometimes stain for CD68.1 Electron microscopy shows degraded myelinated axons intracellularly.4
Malignancy is rare and reportedly occurs in 1% to 3% of cases.4,5 Consideration of both clinical behavior and histopathology is important in distinguishing benign from malignant lesions. According to published reports, in GCTs that were regarded as malignant, size tended to be greater than 4 cm, growth was rapid, and metastases to regional lymph nodes were observed.4,5 Histologically, nuclear pleomorphism and atypia, cell spindling, vesicular nuclei with prominent nucleoli, necrosis, and high mitotic activity favor malignancy.1,3
Treatment is complete surgical excision. Observation is acceptable if tumors are asymptomatic and do not impede function. Regression of some GCTs has been induced with use of intralesional corticosteroids.5 Spontaneous regression is rare. Prior reports have emphasized the importance of long-term follow-up in patients with multiple GCTs to monitor for development of systemic lesions.4
1. Apisarnthanarax P. Granular cell tumor. an analysis of 16 cases and review of the literature. J Am Acad Dermatol. 1981;5:171-182.
2. Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol. 1991;24(2, pt 2):359-363.
3. Hazan C, Fangman W. Multiple cutaneous granular-cell tumors. Dermatol Online J. 2007;13:4.
4. Gross VL, Lynfield Y. Multiple cutaneous granular cell tumors: a case report and review of the literature. Cutis. 2002;69:343-346.
5. Martin RW 3rd, Neldner KH, Boyd AS, et al. Multiple cutaneous granular cell tumors and neurofibromatosis in childhood. a case report and review of the literature. Arch Dermatol. 1990;126:1051-1056.
6. Janousková G, Campr V, Konkol’ová R, et al. Multiple granular cell tumour. J Eur Acad Dermatol Venereol. 2004;18:347-349.
7. Gunson TH, Hashim N, Sharpe GR. Generalized lentiginosis, short stature, and multiple cutaneous nodules—quiz case. LEOPARD syndrome (LS) associated with multiple granular cell tumors (GCTs). Arch Dermatol. 2010;146:337-342.
8. De Raeve L, Roseeuw D, Otten J. Multiple cutaneous granular cell tumors in a child in remission for Hodgkin’s disease. J Am Acad Dermatol. 2002;47(2 suppl):S180-S182.
9. Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol. 2010;27:209-211.
10. Bangle R Jr. A morphological and histochemical study of the granular-cell myoblastoma. Cancer. 1952;5:950-965.
Case Report
A 27-year-old black man was admitted to the hospital with chills; night sweats; unintentional 25-lb weight loss; and multiple widespread, painful, progressively enlarging skin nodules of 3 months’ duration. The lesions had first developed on the back and later appeared on the face, trunk, arms, thighs, and genital region. He denied dysuria or urethral discharge. He had a remote history of adequately treated chlamydia infection but no other remarkable personal or family history.
|
| ||
| Figure 1. Firm subcutaneous nodules on the back with no epidermal change. | ||
|
| ||
| Figure 2. Firm dermal papule on the anterior aspect of the left shoulder with violaceous hyperpigmentation (dermatofibromalike). |
Physical examination revealed a thin man with more than 20 lesions on the face, trunk, arms, thighs, and genital region ranging in size from 1 to 4 cm. Lesion morphologies varied greatly and included subcutaneous firm nodules with no epidermal change (Figure 1); dermatofibromalike nodules with overlying erythema and hyperpigmentation (Figure 2); condylomalike, verrucous, pink papulonodules (Figure 3); ulcerated angular plaques with rolled borders and palpable tumor extension deep (1–2 cm) to the subcutis (Figure 4); and a vegetative, eroded, exophytic tumor with palpable deep extension (Figure 4). A diffuse, erythematous, macular eruption also was noted on the trunk and bilateral arms and legs including the soles of both feet along with nontender cervical, axillary, and inguinal lymphadenopathy. The ocular, oral, and nasal mucosae were not affected.
The differential diagnosis for each lesion differed based on morphology. Infectious, inflammatory, and neoplastic processes were considered, including syphilis, dermatofibroma, dermatofibrosarcoma protuberans, metastatic disease, leukemia cutis, sarcoidosis, panniculitis, condyloma acuminatum, and vegetative herpes simplex virus infection (inguinal lesion).
Laboratory data revealed a reactive rapid plasma reagin with treponemal IgG titers of 1:64. Urine chlamydia RNA probe and lymphogranuloma venereum (LGV) serum antibodies also were positive. Human immunodeficiency virus screening was negative. Positron emission tomography–computerized tomography revealed enlarged and hypermetabolic lymphadenopathy above and below the diaphragm.
After therapy with intravenous penicillin G and oral doxycycline for concurrent secondary syphilis and LGV, the patient’s macular eruption and constitutional symptoms resolved within weeks of the initial presentation. His lymphadenopathy improved, his rapid plasma reagin titer decreased, and his chlamydia RNA became undetectable. However, the skin lesions remained unchanged.
Incisional biopsies of 4 clinically distinct skin lesions revealed well-delineated dermal proliferations of cells with eosinophilic granular cytoplasm and indistinct cell borders (Figure 5). Two specimens displayed marked epidermal hyperplasia (Figure 6).
No atypical mitotic figures were identified. Immunohistochemistry for S-100 protein was diffusely positive in the neoplastic cells. Immunohistochemistry for Treponema pallidum was negative.
No mycobacterial or fungal organisms were identified in acid-fast bacillus, periodic acid–Schiff, or Gomori methenamine-silver–stained sections. All 4 lesions had histopathologic findings characteristic of granular cell tumors (GCTs). A lesion in the left inguinal region (Figure 4 [medial lesion]), which initially was thought to be condyloma latum or a squamous cell carcinoma (SCC), also was later confirmed to be a GCT.
Repeat positron emission tomography–computerized tomography several weeks later confirmed resolution of the previously noted lymphadenopathy. Although 2 GCTs have not recurred after biopsy, the other 2, which the patient refused to have completely excised, continued to grow. Follow-up 2.5 years after hospitalization revealed persistence of the lesions with no remarkable morphological changes.
|
|
| |
| Figure 3. Verrucous pink papule on the right side of the neck. | Figure 4. Ulcerated angular plaque in the left inguinal/genital area with rolled borders and tumor extension deep to the subcutis adjacent to a vegetative, eroded, exophytic tumor with palpable deep extension. | |
|
|
| |
Figure 5. Large polygonal cells with eosinophilic granular cytoplasm, prominent bland nuclei, and indistinct cell borders (H&E, original magnification ×40). | Figure 6. Marked pseudoepitheliomatous hyperplasia (H&E, original magnification ×10). |
Comment
First described in 1854, GCTs are uncommon neoplasms of probable Schwann cell origin that can arise in almost any location of the body but most often appear on the skin and in the subcutaneous tissues and oral cavity.1,2 The commonly regarded rule of thirds describes its most favored locations: one-third on the tongue, one-third on the skin, and one-third in internal organs.3,4 Granular cell tumors occur with greater frequency in adults, females, and black individuals.1-5
Cutaneous GCTs usually present as solitary asymptomatic masses; however, multiple tumors have been noted in up to 25% of reported cases.4,6 In children, multiple cutaneous GCTs have been reported in the setting of neurofibromatosis type I as well as with other disorders.2,5,7-9
Cutaneous GCTs have been reported to range from sessile, pedunculated, or verrucous nodules to subcutaneous papules and nodules with no epidermal change. Our case not only illustrated the diverse clinical appearance of cutaneous GCTs but also demonstrated multiple morphologically distinct cutaneous GCTs occurring in a single patient. Of particular interest is our patient’s coexisting secondary syphilis and LGV infections, which can pose a diagnostic dilemma to the unsuspecting clinician. The manifold appearances of this patient’s GCTs resulted in a broad differential diagnosis. Syphilis (condyloma latum), condyloma acuminatum, LGV, metastatic disease, Kaposi sarcoma, lymphoma, dermatofibrosarcoma protuberans, leiomyoma, SCC, and deep fungal and atypical mycobacterial infection were all considerations. In 1981, Apisarnthanarax1 reviewed 88 cases of GCTs seen over a 15-year period and discovered that the preoperative clinical diagnoses were incorrect in all cases. Skin biopsy is necessary to diagnose GCT, and our patient’s case underscores the need for a thorough history, physical examination, and laboratory evaluation to rule out coexisting diseases.
Histopathology of cutaneous GCTs shows an unencapsulated dermal proliferation of large monotonous polygonal cells with blurred cell borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests. Nuclei are small, uniform, round, centrally located, and rarely contain mitoses.3 The presence of mitotic activity on histopathology does not necessarily portend malignant biological behavior.5 Overlying pseudoepitheliomatous hyperplasia has been reported in as many as 85% of GCTs and may mimic SCC.10 The neoplastic cells stain positively with S-100 protein, neuron-specific enolase, and peripheral nerve myelin proteins.3,4 The cytoplasmic granules are positive on periodic acid–Schiff staining and diastase resistant and will sometimes stain for CD68.1 Electron microscopy shows degraded myelinated axons intracellularly.4
Malignancy is rare and reportedly occurs in 1% to 3% of cases.4,5 Consideration of both clinical behavior and histopathology is important in distinguishing benign from malignant lesions. According to published reports, in GCTs that were regarded as malignant, size tended to be greater than 4 cm, growth was rapid, and metastases to regional lymph nodes were observed.4,5 Histologically, nuclear pleomorphism and atypia, cell spindling, vesicular nuclei with prominent nucleoli, necrosis, and high mitotic activity favor malignancy.1,3
Treatment is complete surgical excision. Observation is acceptable if tumors are asymptomatic and do not impede function. Regression of some GCTs has been induced with use of intralesional corticosteroids.5 Spontaneous regression is rare. Prior reports have emphasized the importance of long-term follow-up in patients with multiple GCTs to monitor for development of systemic lesions.4
Case Report
A 27-year-old black man was admitted to the hospital with chills; night sweats; unintentional 25-lb weight loss; and multiple widespread, painful, progressively enlarging skin nodules of 3 months’ duration. The lesions had first developed on the back and later appeared on the face, trunk, arms, thighs, and genital region. He denied dysuria or urethral discharge. He had a remote history of adequately treated chlamydia infection but no other remarkable personal or family history.
|
| ||
| Figure 1. Firm subcutaneous nodules on the back with no epidermal change. | ||
|
| ||
| Figure 2. Firm dermal papule on the anterior aspect of the left shoulder with violaceous hyperpigmentation (dermatofibromalike). |
Physical examination revealed a thin man with more than 20 lesions on the face, trunk, arms, thighs, and genital region ranging in size from 1 to 4 cm. Lesion morphologies varied greatly and included subcutaneous firm nodules with no epidermal change (Figure 1); dermatofibromalike nodules with overlying erythema and hyperpigmentation (Figure 2); condylomalike, verrucous, pink papulonodules (Figure 3); ulcerated angular plaques with rolled borders and palpable tumor extension deep (1–2 cm) to the subcutis (Figure 4); and a vegetative, eroded, exophytic tumor with palpable deep extension (Figure 4). A diffuse, erythematous, macular eruption also was noted on the trunk and bilateral arms and legs including the soles of both feet along with nontender cervical, axillary, and inguinal lymphadenopathy. The ocular, oral, and nasal mucosae were not affected.
The differential diagnosis for each lesion differed based on morphology. Infectious, inflammatory, and neoplastic processes were considered, including syphilis, dermatofibroma, dermatofibrosarcoma protuberans, metastatic disease, leukemia cutis, sarcoidosis, panniculitis, condyloma acuminatum, and vegetative herpes simplex virus infection (inguinal lesion).
Laboratory data revealed a reactive rapid plasma reagin with treponemal IgG titers of 1:64. Urine chlamydia RNA probe and lymphogranuloma venereum (LGV) serum antibodies also were positive. Human immunodeficiency virus screening was negative. Positron emission tomography–computerized tomography revealed enlarged and hypermetabolic lymphadenopathy above and below the diaphragm.
After therapy with intravenous penicillin G and oral doxycycline for concurrent secondary syphilis and LGV, the patient’s macular eruption and constitutional symptoms resolved within weeks of the initial presentation. His lymphadenopathy improved, his rapid plasma reagin titer decreased, and his chlamydia RNA became undetectable. However, the skin lesions remained unchanged.
Incisional biopsies of 4 clinically distinct skin lesions revealed well-delineated dermal proliferations of cells with eosinophilic granular cytoplasm and indistinct cell borders (Figure 5). Two specimens displayed marked epidermal hyperplasia (Figure 6).
No atypical mitotic figures were identified. Immunohistochemistry for S-100 protein was diffusely positive in the neoplastic cells. Immunohistochemistry for Treponema pallidum was negative.
No mycobacterial or fungal organisms were identified in acid-fast bacillus, periodic acid–Schiff, or Gomori methenamine-silver–stained sections. All 4 lesions had histopathologic findings characteristic of granular cell tumors (GCTs). A lesion in the left inguinal region (Figure 4 [medial lesion]), which initially was thought to be condyloma latum or a squamous cell carcinoma (SCC), also was later confirmed to be a GCT.
Repeat positron emission tomography–computerized tomography several weeks later confirmed resolution of the previously noted lymphadenopathy. Although 2 GCTs have not recurred after biopsy, the other 2, which the patient refused to have completely excised, continued to grow. Follow-up 2.5 years after hospitalization revealed persistence of the lesions with no remarkable morphological changes.
|
|
| |
| Figure 3. Verrucous pink papule on the right side of the neck. | Figure 4. Ulcerated angular plaque in the left inguinal/genital area with rolled borders and tumor extension deep to the subcutis adjacent to a vegetative, eroded, exophytic tumor with palpable deep extension. | |
|
|
| |
Figure 5. Large polygonal cells with eosinophilic granular cytoplasm, prominent bland nuclei, and indistinct cell borders (H&E, original magnification ×40). | Figure 6. Marked pseudoepitheliomatous hyperplasia (H&E, original magnification ×10). |
Comment
First described in 1854, GCTs are uncommon neoplasms of probable Schwann cell origin that can arise in almost any location of the body but most often appear on the skin and in the subcutaneous tissues and oral cavity.1,2 The commonly regarded rule of thirds describes its most favored locations: one-third on the tongue, one-third on the skin, and one-third in internal organs.3,4 Granular cell tumors occur with greater frequency in adults, females, and black individuals.1-5
Cutaneous GCTs usually present as solitary asymptomatic masses; however, multiple tumors have been noted in up to 25% of reported cases.4,6 In children, multiple cutaneous GCTs have been reported in the setting of neurofibromatosis type I as well as with other disorders.2,5,7-9
Cutaneous GCTs have been reported to range from sessile, pedunculated, or verrucous nodules to subcutaneous papules and nodules with no epidermal change. Our case not only illustrated the diverse clinical appearance of cutaneous GCTs but also demonstrated multiple morphologically distinct cutaneous GCTs occurring in a single patient. Of particular interest is our patient’s coexisting secondary syphilis and LGV infections, which can pose a diagnostic dilemma to the unsuspecting clinician. The manifold appearances of this patient’s GCTs resulted in a broad differential diagnosis. Syphilis (condyloma latum), condyloma acuminatum, LGV, metastatic disease, Kaposi sarcoma, lymphoma, dermatofibrosarcoma protuberans, leiomyoma, SCC, and deep fungal and atypical mycobacterial infection were all considerations. In 1981, Apisarnthanarax1 reviewed 88 cases of GCTs seen over a 15-year period and discovered that the preoperative clinical diagnoses were incorrect in all cases. Skin biopsy is necessary to diagnose GCT, and our patient’s case underscores the need for a thorough history, physical examination, and laboratory evaluation to rule out coexisting diseases.
Histopathology of cutaneous GCTs shows an unencapsulated dermal proliferation of large monotonous polygonal cells with blurred cell borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests. Nuclei are small, uniform, round, centrally located, and rarely contain mitoses.3 The presence of mitotic activity on histopathology does not necessarily portend malignant biological behavior.5 Overlying pseudoepitheliomatous hyperplasia has been reported in as many as 85% of GCTs and may mimic SCC.10 The neoplastic cells stain positively with S-100 protein, neuron-specific enolase, and peripheral nerve myelin proteins.3,4 The cytoplasmic granules are positive on periodic acid–Schiff staining and diastase resistant and will sometimes stain for CD68.1 Electron microscopy shows degraded myelinated axons intracellularly.4
Malignancy is rare and reportedly occurs in 1% to 3% of cases.4,5 Consideration of both clinical behavior and histopathology is important in distinguishing benign from malignant lesions. According to published reports, in GCTs that were regarded as malignant, size tended to be greater than 4 cm, growth was rapid, and metastases to regional lymph nodes were observed.4,5 Histologically, nuclear pleomorphism and atypia, cell spindling, vesicular nuclei with prominent nucleoli, necrosis, and high mitotic activity favor malignancy.1,3
Treatment is complete surgical excision. Observation is acceptable if tumors are asymptomatic and do not impede function. Regression of some GCTs has been induced with use of intralesional corticosteroids.5 Spontaneous regression is rare. Prior reports have emphasized the importance of long-term follow-up in patients with multiple GCTs to monitor for development of systemic lesions.4
1. Apisarnthanarax P. Granular cell tumor. an analysis of 16 cases and review of the literature. J Am Acad Dermatol. 1981;5:171-182.
2. Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol. 1991;24(2, pt 2):359-363.
3. Hazan C, Fangman W. Multiple cutaneous granular-cell tumors. Dermatol Online J. 2007;13:4.
4. Gross VL, Lynfield Y. Multiple cutaneous granular cell tumors: a case report and review of the literature. Cutis. 2002;69:343-346.
5. Martin RW 3rd, Neldner KH, Boyd AS, et al. Multiple cutaneous granular cell tumors and neurofibromatosis in childhood. a case report and review of the literature. Arch Dermatol. 1990;126:1051-1056.
6. Janousková G, Campr V, Konkol’ová R, et al. Multiple granular cell tumour. J Eur Acad Dermatol Venereol. 2004;18:347-349.
7. Gunson TH, Hashim N, Sharpe GR. Generalized lentiginosis, short stature, and multiple cutaneous nodules—quiz case. LEOPARD syndrome (LS) associated with multiple granular cell tumors (GCTs). Arch Dermatol. 2010;146:337-342.
8. De Raeve L, Roseeuw D, Otten J. Multiple cutaneous granular cell tumors in a child in remission for Hodgkin’s disease. J Am Acad Dermatol. 2002;47(2 suppl):S180-S182.
9. Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol. 2010;27:209-211.
10. Bangle R Jr. A morphological and histochemical study of the granular-cell myoblastoma. Cancer. 1952;5:950-965.
1. Apisarnthanarax P. Granular cell tumor. an analysis of 16 cases and review of the literature. J Am Acad Dermatol. 1981;5:171-182.
2. Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol. 1991;24(2, pt 2):359-363.
3. Hazan C, Fangman W. Multiple cutaneous granular-cell tumors. Dermatol Online J. 2007;13:4.
4. Gross VL, Lynfield Y. Multiple cutaneous granular cell tumors: a case report and review of the literature. Cutis. 2002;69:343-346.
5. Martin RW 3rd, Neldner KH, Boyd AS, et al. Multiple cutaneous granular cell tumors and neurofibromatosis in childhood. a case report and review of the literature. Arch Dermatol. 1990;126:1051-1056.
6. Janousková G, Campr V, Konkol’ová R, et al. Multiple granular cell tumour. J Eur Acad Dermatol Venereol. 2004;18:347-349.
7. Gunson TH, Hashim N, Sharpe GR. Generalized lentiginosis, short stature, and multiple cutaneous nodules—quiz case. LEOPARD syndrome (LS) associated with multiple granular cell tumors (GCTs). Arch Dermatol. 2010;146:337-342.
8. De Raeve L, Roseeuw D, Otten J. Multiple cutaneous granular cell tumors in a child in remission for Hodgkin’s disease. J Am Acad Dermatol. 2002;47(2 suppl):S180-S182.
9. Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol. 2010;27:209-211.
10. Bangle R Jr. A morphological and histochemical study of the granular-cell myoblastoma. Cancer. 1952;5:950-965.
Practice Points
- Granular cell tumors (GCTs) typically present as solitary lesions; however, multiple lesions occur in approximately 25% of cases.
- Granular cell tumors have a variable clinical appearance and may mimic malignant neoplasms (eg, squamous cell carcinoma) as well as infectious diseases (eg, condyloma, syphilis).
- The histological features of GCTs are distinctive, including an unencapsulated dermal proliferation of monotonous polygonal cells with indistinct borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests.
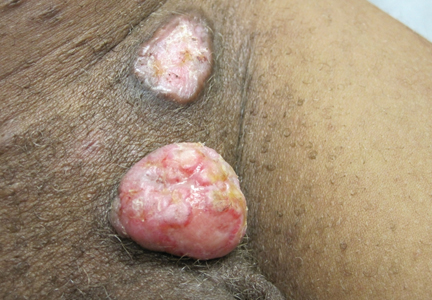
Pityriasis Lichenoides Chronica Presenting With Bilateral Palmoplantar Involvement
Pityriasis lichenoides is an uncommon, acquired, idiopathic, self-limiting skin disease that poses a challenge to patients and clinicians to diagnose and treat. Several variants exist including pityriasis lichenoides et varioliformis acuta (PLEVA), pityriasis lichenoides chronica (PLC), and febrile ulceronecrotic Mucha-Habermann disease. Precise classification can be difficult due to an overlap of clinical and histologic features. The spectrum of this inflammatory skin disorder is characterized by recurrent crops of spontaneously regressing papulosquamous, polymorphic, and ulceronecrotic papules affecting the trunk and extremities. Pityriasis lichenoides is a monoclonal T-cell disorder that needs careful follow-up because it can progress, though rarely, to cutaneous T-cell lymphoma. In this case report we describe a patient with a rare presentation of PLC exhibiting bilateral palmoplantar involvement and mimicking psoriasis. We review the literature and discuss the clinical course, pathogenesis, and current treatment modalities of PLC.
Case Report
A 61-year-old woman presented with a recurrent itchy rash on the legs, feet, hands, and trunk of several months’ duration. Her medical history included Helicobacter pylori–associated peptic ulcer disease and hypertension. She was not taking any prescription medications. She reported no alcohol or tobacco use or any personal or family history of skin disease. For many years she had lived part-time in Hong Kong, and she was concerned that her skin condition might be infectious or allergic in nature because she had observed similar skin lesions in Hong Kong natives who attributed the outbreaks of rash to “bad water.”
Physical examination revealed reddish brown crusted papules and plaques scattered bilaterally over the legs and feet (Figure 1); serpiginous scaly patches on the hips, thighs, and back; and thick hyperkeratotic psoriasiform plaques with yellow scale and crust on the palms and soles (Figure 2). The nails and oral mucosa were unaffected. Histopathologic evaluation of the lesions obtained from the superior aspect of the thigh showed parakeratotic scale and a lichenoid lymphocytic infiltrate in the papillary dermis consistent with PLC (Figure 3).
The patient was started on tetracycline 500 mg twice daily for 10 days and on narrowband UVB (NB-UVB) therapy at 350 J/cm2 with incremental increases of 60 J/cm2 at each treatment for a maximum dose of 770 J/cm2. She received 9 treatments in total over 1 month and noted some improvement in overall appearance of the lesions, mostly over the trunk and extremities. Palmoplantar lesions were resistant to treatment. Therapy with NB-UVB was discontinued, as the patient had to return to Hong Kong. Given the brief course of NB-UVB therapy, it was hard to assess why the palmoplantar lesions failed to respond to treatment.
Comment
Subtypes
Pityriasis lichenoides is a unique inflammatory disorder that usually presents with guttate papules in various stages of evolution ranging from acute hemorrhagic, vesicular, or ulcerated lesions to chronic pink papules with adherent micalike scale. Two ends of the spectrum are PLEVA and PLC. Papule distribution often is diffuse, affecting both the trunk and extremities, but involvement can be confined to the trunk producing a central distribution or restricted to the extremities giving a peripheral pattern. A purely acral localization is uncommon and rarely has been documented in the literature.1
Pityriasis lichenoides et varioliformis acuta typically presents with an acute polymorphous eruption of 2- to 3-mm erythematous macules that evolve into papules with a fine, micaceous, centrally attached scale. The center of the papule then undergoes hemorrhagic necrosis, becomes ulcerated with reddish brown crust, and may heal with a varioliform scar. Symptoms may include a burning sensation and pruritus. Successive crops may persist for weeks, months, and sometimes years.2
Febrile ulceronecrotic Mucha-Habermann disease is an acute and severe generalized eruption of ulceronecrotic plaques. Extensive painful necrosis of the skin may follow and there is an increased risk for secondary infection.2 Systemic symptoms may include fever, sore throat, diarrhea, and abdominal pain. Febrile ulceronecrotic Mucha-Habermann disease has a mortality rate of 25% and should be treated as a dermatologic emergency.2
Pityriasis lichenoides chronica has a more gradual presentation and indolent course than PLEVA. It most commonly presents as small asymptomatic polymorphous red-brown maculopapules with micaceous scale.3 Papules spontaneously flatten over a few weeks. Postinflammatory hypopigmentation or hyperpigmentation may persist once the lesions resolve. Similar to PLEVA, PLC has a relapsing course but with longer periods of remission. Pityriasis lichenoides chronica usually involves the trunk and proximal extremities, but acral distributions, as in our case, have been described. This rare variant of pityriasis lichenoides may be underrecognized and underdiagnosed due to its resemblance to psoriasis.1
The prevalence and incidence of PLC in the general population is unknown. There appears to be no predominance based on gender, ethnicity, or geographical location, and it occurs in both children and adults. One study showed the average age to be 29 years.2
Etiology
The cause of pityriasis lichenoides is unknown, but there are 3 popular theories regarding its pathogenesis: a hypersensitivity response due to an infectious agent, an inflammatory response to a T-cell dyscrasia, or an immune complex–mediated hypersensitivity vasculitis.2 The theory of an infectious cause has been proposed due to reports of disease clustering in families and communities.2,3 Elevated titers of certain pathogens and clearing of the disease after pathogen-specific treatment also have been reported. Possible triggers cited in the literature include the Epstein-Barr virus, Toxoplasma gondii, parvovirus B19, adenovirus, human immunodeficiency virus, freeze-dried live attenuated measles vaccine, Staphylococcus aureus, and group A β-hemolytic streptococci.2,3
Some reported cases of pityriasis lichenoides have demonstrated T-cell clonality. Weinberg et al4 found a significantly higher number of clonal T cells in PLEVA than in PLC (P=.008) and hypothesized that PLEVA is actually a benign clonal T-cell disorder arising from a specific subset of T cells in PLC. Malignant transformation of pityriasis lichenoides has been reported but is rare.3
Differential Diagnosis
Historically, pityriasis lichenoides has been confused with many other dermatoses. With palmoplantar involvement, consider other papulosquamous disorders such as palmoplantar psoriasis, lichen planus, cutaneous T-cell lymphoma, lymphomatoid papulosis, vasculitis, and secondary syphilis. Rule out alternative diagnoses with histologic examination; assessments of nails, oral mucosa, joints, and constitutional symptoms; and laboratory testing.
Histopathology
Pityriasis lichenoides et varioliformis acuta and PLC are similar with subtle and gradually evolving differences, supporting the notion that these disorders are polar ends of the same disease spectrum.2 Pityriasis lichenoides et varioliformis acuta typically produces a dense wedge-shaped dermal infiltrate composed of CD8+ T cells and histiocytes most concentrated along the basal layer with lymphocytic exocytosis into the epidermis and perivascular inflammation. The epidermis also demonstrates spongiosis, necrosis and apoptosis of keratinocytes, neutrophilic inclusions, vacuolar degeneration, intraepidermal vesicles and ulceration, and focal parakeratosis with scale and crust. In contrast, PLC is less exaggerated than PLEVA with a superficial bandlike lymphocytic infiltrate in which CD4+ T cells predominate with minimal perivascular involvement. Immunohistochemical studies reveal that CD8+ cells predominate in PLEVA, while CD4+ cells predominate in PLC. Staining for HLA-DR–positive keratinocytes yields stronger and more diffuse findings in PLEVA than in PLC and is considered a marker for the former.2
Treatment
There is no standard treatment of pityriasis lichenoides. However, combination therapy is considered the best approach. To date, phototherapy has been the most effective modality and is considered a first-line treatment of PLC. Variants of phototherapy include UVB, NB-UVB, psoralen plus UVA, and UVA1.5 One study showed UVA1 (340–400 nm) treatment to be effective and well tolerated at a medium dose of 60 J/cm2.6 Narrowband UVB has become a well-used phototherapy for a variety of skin conditions including pityriasis lichenoides. In a study by Aydogan et al,5 NB-UVB was safe and effective for the management of PLEVA and PLC. The authors also argue that it has added advantages over other phototherapies, including a more immunosuppressive effect on lymphoproliferation that causes a greater depletion of T cells in skin lesions, possibly due to its deeper dermal penetration compared with broadband UVB. Narrowband UVB also is safe in children.5 Tapering of phototherapy has been recommended to prevent relapses.3
If infection is a suspected contributor to the problem, treat as needed. The antibiotics tetracycline, erythromycin, and dapsone have been used with success, as well as the antiviral acyclovir. Tetracycline and erythromycin also may confer anti-inflammatory benefits. A gradual taper of these agents is advised to prevent recurrences. Topical corticosteroids and coal tar may help alleviate pruritus and inflammation; however, they do not affect the course of the disease.3 In one report, the topical immunomodulator tacrolimus markedly reduced lesions, most likely due to its anti-inflammatory effect. After discontinuation of the medication, lesions recurred but were less severe.7
Clinical Recommendations
Early diagnosis and management of pityriasis lichenoides is essential. At this time, screening for pathogens is not advised unless the patient has specific symptoms of infection. Due to the history of recurrence with this disease, combination therapy is recommended with a gradual taper of all modalities. Because of the rare but possible transformation to malignancy, careful follow-up and repeated biopsies have been advised in chronic intermittent disease.3
- Kossard S. Acral pityriasis lichenoides. Australas J Dermatol. 2002;43:68-71.
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-568; quiz 573-576.
- Khachemoune A, Blyumin ML. Pityriasis lichenoides: pathophysiology, classification, and treatment. Am J Clin Dermatol. 2007;8:29-36.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dematol. 2002;138:1063-1067.
- Aydogan K, Saricaoglu H, Turan H. Narrowband UVB (311nm, TL01) phototherapy for pityriasis lichenoides. Photodermatol Photoimmunol Photomed. 2008;24:128-133.
- Pinton P, Capezzera R, Zane C, et al. Medium-dose ultraviolet A1 therapy for pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronic. J Am Acad Dermatol. 2002;47:401-414.
- Simon D, Boudny C, Nievergelt H, et al. Successful treatment of pityriasis lichenoides with topical tacrolimus. Br J Dermatol. 2004;150:1033-1035.
Pityriasis lichenoides is an uncommon, acquired, idiopathic, self-limiting skin disease that poses a challenge to patients and clinicians to diagnose and treat. Several variants exist including pityriasis lichenoides et varioliformis acuta (PLEVA), pityriasis lichenoides chronica (PLC), and febrile ulceronecrotic Mucha-Habermann disease. Precise classification can be difficult due to an overlap of clinical and histologic features. The spectrum of this inflammatory skin disorder is characterized by recurrent crops of spontaneously regressing papulosquamous, polymorphic, and ulceronecrotic papules affecting the trunk and extremities. Pityriasis lichenoides is a monoclonal T-cell disorder that needs careful follow-up because it can progress, though rarely, to cutaneous T-cell lymphoma. In this case report we describe a patient with a rare presentation of PLC exhibiting bilateral palmoplantar involvement and mimicking psoriasis. We review the literature and discuss the clinical course, pathogenesis, and current treatment modalities of PLC.
Case Report
A 61-year-old woman presented with a recurrent itchy rash on the legs, feet, hands, and trunk of several months’ duration. Her medical history included Helicobacter pylori–associated peptic ulcer disease and hypertension. She was not taking any prescription medications. She reported no alcohol or tobacco use or any personal or family history of skin disease. For many years she had lived part-time in Hong Kong, and she was concerned that her skin condition might be infectious or allergic in nature because she had observed similar skin lesions in Hong Kong natives who attributed the outbreaks of rash to “bad water.”
Physical examination revealed reddish brown crusted papules and plaques scattered bilaterally over the legs and feet (Figure 1); serpiginous scaly patches on the hips, thighs, and back; and thick hyperkeratotic psoriasiform plaques with yellow scale and crust on the palms and soles (Figure 2). The nails and oral mucosa were unaffected. Histopathologic evaluation of the lesions obtained from the superior aspect of the thigh showed parakeratotic scale and a lichenoid lymphocytic infiltrate in the papillary dermis consistent with PLC (Figure 3).
The patient was started on tetracycline 500 mg twice daily for 10 days and on narrowband UVB (NB-UVB) therapy at 350 J/cm2 with incremental increases of 60 J/cm2 at each treatment for a maximum dose of 770 J/cm2. She received 9 treatments in total over 1 month and noted some improvement in overall appearance of the lesions, mostly over the trunk and extremities. Palmoplantar lesions were resistant to treatment. Therapy with NB-UVB was discontinued, as the patient had to return to Hong Kong. Given the brief course of NB-UVB therapy, it was hard to assess why the palmoplantar lesions failed to respond to treatment.
Comment
Subtypes
Pityriasis lichenoides is a unique inflammatory disorder that usually presents with guttate papules in various stages of evolution ranging from acute hemorrhagic, vesicular, or ulcerated lesions to chronic pink papules with adherent micalike scale. Two ends of the spectrum are PLEVA and PLC. Papule distribution often is diffuse, affecting both the trunk and extremities, but involvement can be confined to the trunk producing a central distribution or restricted to the extremities giving a peripheral pattern. A purely acral localization is uncommon and rarely has been documented in the literature.1
Pityriasis lichenoides et varioliformis acuta typically presents with an acute polymorphous eruption of 2- to 3-mm erythematous macules that evolve into papules with a fine, micaceous, centrally attached scale. The center of the papule then undergoes hemorrhagic necrosis, becomes ulcerated with reddish brown crust, and may heal with a varioliform scar. Symptoms may include a burning sensation and pruritus. Successive crops may persist for weeks, months, and sometimes years.2
Febrile ulceronecrotic Mucha-Habermann disease is an acute and severe generalized eruption of ulceronecrotic plaques. Extensive painful necrosis of the skin may follow and there is an increased risk for secondary infection.2 Systemic symptoms may include fever, sore throat, diarrhea, and abdominal pain. Febrile ulceronecrotic Mucha-Habermann disease has a mortality rate of 25% and should be treated as a dermatologic emergency.2
Pityriasis lichenoides chronica has a more gradual presentation and indolent course than PLEVA. It most commonly presents as small asymptomatic polymorphous red-brown maculopapules with micaceous scale.3 Papules spontaneously flatten over a few weeks. Postinflammatory hypopigmentation or hyperpigmentation may persist once the lesions resolve. Similar to PLEVA, PLC has a relapsing course but with longer periods of remission. Pityriasis lichenoides chronica usually involves the trunk and proximal extremities, but acral distributions, as in our case, have been described. This rare variant of pityriasis lichenoides may be underrecognized and underdiagnosed due to its resemblance to psoriasis.1
The prevalence and incidence of PLC in the general population is unknown. There appears to be no predominance based on gender, ethnicity, or geographical location, and it occurs in both children and adults. One study showed the average age to be 29 years.2
Etiology
The cause of pityriasis lichenoides is unknown, but there are 3 popular theories regarding its pathogenesis: a hypersensitivity response due to an infectious agent, an inflammatory response to a T-cell dyscrasia, or an immune complex–mediated hypersensitivity vasculitis.2 The theory of an infectious cause has been proposed due to reports of disease clustering in families and communities.2,3 Elevated titers of certain pathogens and clearing of the disease after pathogen-specific treatment also have been reported. Possible triggers cited in the literature include the Epstein-Barr virus, Toxoplasma gondii, parvovirus B19, adenovirus, human immunodeficiency virus, freeze-dried live attenuated measles vaccine, Staphylococcus aureus, and group A β-hemolytic streptococci.2,3
Some reported cases of pityriasis lichenoides have demonstrated T-cell clonality. Weinberg et al4 found a significantly higher number of clonal T cells in PLEVA than in PLC (P=.008) and hypothesized that PLEVA is actually a benign clonal T-cell disorder arising from a specific subset of T cells in PLC. Malignant transformation of pityriasis lichenoides has been reported but is rare.3
Differential Diagnosis
Historically, pityriasis lichenoides has been confused with many other dermatoses. With palmoplantar involvement, consider other papulosquamous disorders such as palmoplantar psoriasis, lichen planus, cutaneous T-cell lymphoma, lymphomatoid papulosis, vasculitis, and secondary syphilis. Rule out alternative diagnoses with histologic examination; assessments of nails, oral mucosa, joints, and constitutional symptoms; and laboratory testing.
Histopathology
Pityriasis lichenoides et varioliformis acuta and PLC are similar with subtle and gradually evolving differences, supporting the notion that these disorders are polar ends of the same disease spectrum.2 Pityriasis lichenoides et varioliformis acuta typically produces a dense wedge-shaped dermal infiltrate composed of CD8+ T cells and histiocytes most concentrated along the basal layer with lymphocytic exocytosis into the epidermis and perivascular inflammation. The epidermis also demonstrates spongiosis, necrosis and apoptosis of keratinocytes, neutrophilic inclusions, vacuolar degeneration, intraepidermal vesicles and ulceration, and focal parakeratosis with scale and crust. In contrast, PLC is less exaggerated than PLEVA with a superficial bandlike lymphocytic infiltrate in which CD4+ T cells predominate with minimal perivascular involvement. Immunohistochemical studies reveal that CD8+ cells predominate in PLEVA, while CD4+ cells predominate in PLC. Staining for HLA-DR–positive keratinocytes yields stronger and more diffuse findings in PLEVA than in PLC and is considered a marker for the former.2
Treatment
There is no standard treatment of pityriasis lichenoides. However, combination therapy is considered the best approach. To date, phototherapy has been the most effective modality and is considered a first-line treatment of PLC. Variants of phototherapy include UVB, NB-UVB, psoralen plus UVA, and UVA1.5 One study showed UVA1 (340–400 nm) treatment to be effective and well tolerated at a medium dose of 60 J/cm2.6 Narrowband UVB has become a well-used phototherapy for a variety of skin conditions including pityriasis lichenoides. In a study by Aydogan et al,5 NB-UVB was safe and effective for the management of PLEVA and PLC. The authors also argue that it has added advantages over other phototherapies, including a more immunosuppressive effect on lymphoproliferation that causes a greater depletion of T cells in skin lesions, possibly due to its deeper dermal penetration compared with broadband UVB. Narrowband UVB also is safe in children.5 Tapering of phototherapy has been recommended to prevent relapses.3
If infection is a suspected contributor to the problem, treat as needed. The antibiotics tetracycline, erythromycin, and dapsone have been used with success, as well as the antiviral acyclovir. Tetracycline and erythromycin also may confer anti-inflammatory benefits. A gradual taper of these agents is advised to prevent recurrences. Topical corticosteroids and coal tar may help alleviate pruritus and inflammation; however, they do not affect the course of the disease.3 In one report, the topical immunomodulator tacrolimus markedly reduced lesions, most likely due to its anti-inflammatory effect. After discontinuation of the medication, lesions recurred but were less severe.7
Clinical Recommendations
Early diagnosis and management of pityriasis lichenoides is essential. At this time, screening for pathogens is not advised unless the patient has specific symptoms of infection. Due to the history of recurrence with this disease, combination therapy is recommended with a gradual taper of all modalities. Because of the rare but possible transformation to malignancy, careful follow-up and repeated biopsies have been advised in chronic intermittent disease.3
Pityriasis lichenoides is an uncommon, acquired, idiopathic, self-limiting skin disease that poses a challenge to patients and clinicians to diagnose and treat. Several variants exist including pityriasis lichenoides et varioliformis acuta (PLEVA), pityriasis lichenoides chronica (PLC), and febrile ulceronecrotic Mucha-Habermann disease. Precise classification can be difficult due to an overlap of clinical and histologic features. The spectrum of this inflammatory skin disorder is characterized by recurrent crops of spontaneously regressing papulosquamous, polymorphic, and ulceronecrotic papules affecting the trunk and extremities. Pityriasis lichenoides is a monoclonal T-cell disorder that needs careful follow-up because it can progress, though rarely, to cutaneous T-cell lymphoma. In this case report we describe a patient with a rare presentation of PLC exhibiting bilateral palmoplantar involvement and mimicking psoriasis. We review the literature and discuss the clinical course, pathogenesis, and current treatment modalities of PLC.
Case Report
A 61-year-old woman presented with a recurrent itchy rash on the legs, feet, hands, and trunk of several months’ duration. Her medical history included Helicobacter pylori–associated peptic ulcer disease and hypertension. She was not taking any prescription medications. She reported no alcohol or tobacco use or any personal or family history of skin disease. For many years she had lived part-time in Hong Kong, and she was concerned that her skin condition might be infectious or allergic in nature because she had observed similar skin lesions in Hong Kong natives who attributed the outbreaks of rash to “bad water.”
Physical examination revealed reddish brown crusted papules and plaques scattered bilaterally over the legs and feet (Figure 1); serpiginous scaly patches on the hips, thighs, and back; and thick hyperkeratotic psoriasiform plaques with yellow scale and crust on the palms and soles (Figure 2). The nails and oral mucosa were unaffected. Histopathologic evaluation of the lesions obtained from the superior aspect of the thigh showed parakeratotic scale and a lichenoid lymphocytic infiltrate in the papillary dermis consistent with PLC (Figure 3).
The patient was started on tetracycline 500 mg twice daily for 10 days and on narrowband UVB (NB-UVB) therapy at 350 J/cm2 with incremental increases of 60 J/cm2 at each treatment for a maximum dose of 770 J/cm2. She received 9 treatments in total over 1 month and noted some improvement in overall appearance of the lesions, mostly over the trunk and extremities. Palmoplantar lesions were resistant to treatment. Therapy with NB-UVB was discontinued, as the patient had to return to Hong Kong. Given the brief course of NB-UVB therapy, it was hard to assess why the palmoplantar lesions failed to respond to treatment.
Comment
Subtypes
Pityriasis lichenoides is a unique inflammatory disorder that usually presents with guttate papules in various stages of evolution ranging from acute hemorrhagic, vesicular, or ulcerated lesions to chronic pink papules with adherent micalike scale. Two ends of the spectrum are PLEVA and PLC. Papule distribution often is diffuse, affecting both the trunk and extremities, but involvement can be confined to the trunk producing a central distribution or restricted to the extremities giving a peripheral pattern. A purely acral localization is uncommon and rarely has been documented in the literature.1
Pityriasis lichenoides et varioliformis acuta typically presents with an acute polymorphous eruption of 2- to 3-mm erythematous macules that evolve into papules with a fine, micaceous, centrally attached scale. The center of the papule then undergoes hemorrhagic necrosis, becomes ulcerated with reddish brown crust, and may heal with a varioliform scar. Symptoms may include a burning sensation and pruritus. Successive crops may persist for weeks, months, and sometimes years.2
Febrile ulceronecrotic Mucha-Habermann disease is an acute and severe generalized eruption of ulceronecrotic plaques. Extensive painful necrosis of the skin may follow and there is an increased risk for secondary infection.2 Systemic symptoms may include fever, sore throat, diarrhea, and abdominal pain. Febrile ulceronecrotic Mucha-Habermann disease has a mortality rate of 25% and should be treated as a dermatologic emergency.2
Pityriasis lichenoides chronica has a more gradual presentation and indolent course than PLEVA. It most commonly presents as small asymptomatic polymorphous red-brown maculopapules with micaceous scale.3 Papules spontaneously flatten over a few weeks. Postinflammatory hypopigmentation or hyperpigmentation may persist once the lesions resolve. Similar to PLEVA, PLC has a relapsing course but with longer periods of remission. Pityriasis lichenoides chronica usually involves the trunk and proximal extremities, but acral distributions, as in our case, have been described. This rare variant of pityriasis lichenoides may be underrecognized and underdiagnosed due to its resemblance to psoriasis.1
The prevalence and incidence of PLC in the general population is unknown. There appears to be no predominance based on gender, ethnicity, or geographical location, and it occurs in both children and adults. One study showed the average age to be 29 years.2
Etiology
The cause of pityriasis lichenoides is unknown, but there are 3 popular theories regarding its pathogenesis: a hypersensitivity response due to an infectious agent, an inflammatory response to a T-cell dyscrasia, or an immune complex–mediated hypersensitivity vasculitis.2 The theory of an infectious cause has been proposed due to reports of disease clustering in families and communities.2,3 Elevated titers of certain pathogens and clearing of the disease after pathogen-specific treatment also have been reported. Possible triggers cited in the literature include the Epstein-Barr virus, Toxoplasma gondii, parvovirus B19, adenovirus, human immunodeficiency virus, freeze-dried live attenuated measles vaccine, Staphylococcus aureus, and group A β-hemolytic streptococci.2,3
Some reported cases of pityriasis lichenoides have demonstrated T-cell clonality. Weinberg et al4 found a significantly higher number of clonal T cells in PLEVA than in PLC (P=.008) and hypothesized that PLEVA is actually a benign clonal T-cell disorder arising from a specific subset of T cells in PLC. Malignant transformation of pityriasis lichenoides has been reported but is rare.3
Differential Diagnosis
Historically, pityriasis lichenoides has been confused with many other dermatoses. With palmoplantar involvement, consider other papulosquamous disorders such as palmoplantar psoriasis, lichen planus, cutaneous T-cell lymphoma, lymphomatoid papulosis, vasculitis, and secondary syphilis. Rule out alternative diagnoses with histologic examination; assessments of nails, oral mucosa, joints, and constitutional symptoms; and laboratory testing.
Histopathology
Pityriasis lichenoides et varioliformis acuta and PLC are similar with subtle and gradually evolving differences, supporting the notion that these disorders are polar ends of the same disease spectrum.2 Pityriasis lichenoides et varioliformis acuta typically produces a dense wedge-shaped dermal infiltrate composed of CD8+ T cells and histiocytes most concentrated along the basal layer with lymphocytic exocytosis into the epidermis and perivascular inflammation. The epidermis also demonstrates spongiosis, necrosis and apoptosis of keratinocytes, neutrophilic inclusions, vacuolar degeneration, intraepidermal vesicles and ulceration, and focal parakeratosis with scale and crust. In contrast, PLC is less exaggerated than PLEVA with a superficial bandlike lymphocytic infiltrate in which CD4+ T cells predominate with minimal perivascular involvement. Immunohistochemical studies reveal that CD8+ cells predominate in PLEVA, while CD4+ cells predominate in PLC. Staining for HLA-DR–positive keratinocytes yields stronger and more diffuse findings in PLEVA than in PLC and is considered a marker for the former.2
Treatment
There is no standard treatment of pityriasis lichenoides. However, combination therapy is considered the best approach. To date, phototherapy has been the most effective modality and is considered a first-line treatment of PLC. Variants of phototherapy include UVB, NB-UVB, psoralen plus UVA, and UVA1.5 One study showed UVA1 (340–400 nm) treatment to be effective and well tolerated at a medium dose of 60 J/cm2.6 Narrowband UVB has become a well-used phototherapy for a variety of skin conditions including pityriasis lichenoides. In a study by Aydogan et al,5 NB-UVB was safe and effective for the management of PLEVA and PLC. The authors also argue that it has added advantages over other phototherapies, including a more immunosuppressive effect on lymphoproliferation that causes a greater depletion of T cells in skin lesions, possibly due to its deeper dermal penetration compared with broadband UVB. Narrowband UVB also is safe in children.5 Tapering of phototherapy has been recommended to prevent relapses.3
If infection is a suspected contributor to the problem, treat as needed. The antibiotics tetracycline, erythromycin, and dapsone have been used with success, as well as the antiviral acyclovir. Tetracycline and erythromycin also may confer anti-inflammatory benefits. A gradual taper of these agents is advised to prevent recurrences. Topical corticosteroids and coal tar may help alleviate pruritus and inflammation; however, they do not affect the course of the disease.3 In one report, the topical immunomodulator tacrolimus markedly reduced lesions, most likely due to its anti-inflammatory effect. After discontinuation of the medication, lesions recurred but were less severe.7
Clinical Recommendations
Early diagnosis and management of pityriasis lichenoides is essential. At this time, screening for pathogens is not advised unless the patient has specific symptoms of infection. Due to the history of recurrence with this disease, combination therapy is recommended with a gradual taper of all modalities. Because of the rare but possible transformation to malignancy, careful follow-up and repeated biopsies have been advised in chronic intermittent disease.3
- Kossard S. Acral pityriasis lichenoides. Australas J Dermatol. 2002;43:68-71.
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-568; quiz 573-576.
- Khachemoune A, Blyumin ML. Pityriasis lichenoides: pathophysiology, classification, and treatment. Am J Clin Dermatol. 2007;8:29-36.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dematol. 2002;138:1063-1067.
- Aydogan K, Saricaoglu H, Turan H. Narrowband UVB (311nm, TL01) phototherapy for pityriasis lichenoides. Photodermatol Photoimmunol Photomed. 2008;24:128-133.
- Pinton P, Capezzera R, Zane C, et al. Medium-dose ultraviolet A1 therapy for pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronic. J Am Acad Dermatol. 2002;47:401-414.
- Simon D, Boudny C, Nievergelt H, et al. Successful treatment of pityriasis lichenoides with topical tacrolimus. Br J Dermatol. 2004;150:1033-1035.
- Kossard S. Acral pityriasis lichenoides. Australas J Dermatol. 2002;43:68-71.
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-568; quiz 573-576.
- Khachemoune A, Blyumin ML. Pityriasis lichenoides: pathophysiology, classification, and treatment. Am J Clin Dermatol. 2007;8:29-36.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dematol. 2002;138:1063-1067.
- Aydogan K, Saricaoglu H, Turan H. Narrowband UVB (311nm, TL01) phototherapy for pityriasis lichenoides. Photodermatol Photoimmunol Photomed. 2008;24:128-133.
- Pinton P, Capezzera R, Zane C, et al. Medium-dose ultraviolet A1 therapy for pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronic. J Am Acad Dermatol. 2002;47:401-414.
- Simon D, Boudny C, Nievergelt H, et al. Successful treatment of pityriasis lichenoides with topical tacrolimus. Br J Dermatol. 2004;150:1033-1035.
Practice Points
- Diagnosis of pityriasis lichenoides may be difficult due to a wide spectrum of clinical presentations.
- Pityriasis lichenoides chronica (PLC) with palmoplantar involvement may mimic psoriasis.
- Screening for infections is not recommended in patients with PLC unless the patient has other symptoms pointing to a specific infection.
- Phototherapy currently is the most effective treatment modality for PLC.
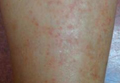
Acute benzodiazepine toxicity exacerbated by concomitant oral olanzapine in a patient with pancreatic cancer
Severe eosinophilia associated with cholangiocarcinoma
It is widely recognized that eosinophils are found in tumor infiltrates and that their mechanism of action is associated with particular symptoms and prognosis. However, the causes of and reasons for this process remain unclear, as does the exact mechanism by which it occurs. We report on the case of a 71-year-old woman with cholangiocellar carcinoma (CCC) with a marked eosinophilia. When the patient presented at the hospital, she said she was suffering from fatigue, depression, and pain.
Click on the PDF icon at the top of this introduction to read the full article.
It is widely recognized that eosinophils are found in tumor infiltrates and that their mechanism of action is associated with particular symptoms and prognosis. However, the causes of and reasons for this process remain unclear, as does the exact mechanism by which it occurs. We report on the case of a 71-year-old woman with cholangiocellar carcinoma (CCC) with a marked eosinophilia. When the patient presented at the hospital, she said she was suffering from fatigue, depression, and pain.
Click on the PDF icon at the top of this introduction to read the full article.
It is widely recognized that eosinophils are found in tumor infiltrates and that their mechanism of action is associated with particular symptoms and prognosis. However, the causes of and reasons for this process remain unclear, as does the exact mechanism by which it occurs. We report on the case of a 71-year-old woman with cholangiocellar carcinoma (CCC) with a marked eosinophilia. When the patient presented at the hospital, she said she was suffering from fatigue, depression, and pain.
Click on the PDF icon at the top of this introduction to read the full article.
An unusual case of non-small-cell lung cancer presenting as spontaneous cardiac tamponade
Hemorrhagic pericardial effusion with associated cardiac tamponade as a de novo sign of malignancy is seen in about 2% of patients.1 Consequently, cardiac tamponade is an oncologic emergency and considered a unique presentation of a malignancy.2 Cancer emergency is defined as an acute condition that is caused directly by the cancer itself or its treatment and requires intervention to avoid death or significant morbidity.3 The mechanism by which cardiac tamponade is classified as a life-threatening emergency stems from its impairment of right ventricular filling, resulting in ventricular diastolic collapse and decreased cardiac output, which can ultimately lead to death.4
Click on the PDF icon at the top of this introduction to read the full article.
Hemorrhagic pericardial effusion with associated cardiac tamponade as a de novo sign of malignancy is seen in about 2% of patients.1 Consequently, cardiac tamponade is an oncologic emergency and considered a unique presentation of a malignancy.2 Cancer emergency is defined as an acute condition that is caused directly by the cancer itself or its treatment and requires intervention to avoid death or significant morbidity.3 The mechanism by which cardiac tamponade is classified as a life-threatening emergency stems from its impairment of right ventricular filling, resulting in ventricular diastolic collapse and decreased cardiac output, which can ultimately lead to death.4
Click on the PDF icon at the top of this introduction to read the full article.
Hemorrhagic pericardial effusion with associated cardiac tamponade as a de novo sign of malignancy is seen in about 2% of patients.1 Consequently, cardiac tamponade is an oncologic emergency and considered a unique presentation of a malignancy.2 Cancer emergency is defined as an acute condition that is caused directly by the cancer itself or its treatment and requires intervention to avoid death or significant morbidity.3 The mechanism by which cardiac tamponade is classified as a life-threatening emergency stems from its impairment of right ventricular filling, resulting in ventricular diastolic collapse and decreased cardiac output, which can ultimately lead to death.4
Click on the PDF icon at the top of this introduction to read the full article.