User login
Patient-Specific Implants in Severe Glenoid Bone Loss
ABSTRACT
Complex glenoid bone deformities present the treating surgeon with a complex reconstructive challenge. Although glenoid bone loss can be encountered in the primary setting (degenerative, congenital, post-traumatic), severe glenoid bone loss is encountered in most revision total shoulder arthroplasties. Severe glenoid bone loss is treated with various techniques including hemiarthroplasty, eccentric reaming, and glenoid reconstruction with bone autografts and allografts. Despite encouraging short- to mid-term results reported with these reconstruction techniques, the clinical and radiographic outcomes remain inconsistent and the high number of complications is a concern. To overcome this problem, more recently augmented components and patient specific implants were introduced. Using the computer-aided design and computer-aided manufacturing technology patient-specific implants have been created to reconstruct the glenoid vault in cases of severe glenoid bone loss.
In this article we describe a patient specific glenoid implant, its indication, technical aspects and surgical technique, based on the author's experience as well as a review of the current literature on custom glenoid implants.
Continue to: Total shoulder arthroplasty...
Total shoulder arthroplasty (TSA) is an effective operation for providing pain relief and improving function in patients with end-stage degenerative shoulder disease that is nonresponsive to nonoperative treatments.1-4 With the increasing number of arthroplasties performed, and the expanding indication for shoulder arthroplasty, the number of revision shoulder arthroplasties is also increasing.5-14 Complex glenoid bone deformities present the treating surgeon with a complex reconstructive challenge. Although glenoid bone loss can be seen in the primary setting (degenerative, congenital, and post-traumatic), severe glenoid bone loss is encountered mostly in revision TSAs.
Historically, patients with severe glenoid bone loss were treated with a hemiarthroplasty.15-17 However, due to inferior outcomes associated with the use of shoulder hemiarthroplasties compared with TSA in these cases,18-20 various techniques were developed with the aim of realigning the glenoid axis and securing the implants into the deficient glenoid vault.21-25 Options have included eccentric reaming, glenoid reconstruction with bone autografts and allografts, and more recently augmented components and patient-specific implants. Studies with eccentric reaming and reconstruction with bone graft during complex shoulder arthroplasty have reported encouraging short- to mid-term results, but the clinical and radiographic outcomes remain inconsistent, and the high number of complications is a concern.25-28
Complications with these techniques include component loosening, graft resorption, nonunion, failure of graft incorporation, infection, and instability.25-28
Computer-aided design and computer-aided manufacturing (CAD/CAM) of patient-specific implants have been used successfully by hip arthroplasty surgeons to deal with complex acetabular reconstructions in the setting of severe bone loss. More recently, the same technology has been used to reconstruct the glenoid vault in cases of severe glenoid bone loss.
In this article, we describe a patient-specific glenoid implant, its indication, and both technical aspects and the surgical technique, based on the authors’ experience as well as a review of the current literature on custom glenoid implants.
Continue to: PATIENT-SPECIFIC GLENOID COMPONENT
PATIENT-SPECIFIC GLENOID COMPONENT
The Vault Reconstruction System ([VRS], Zimmer Biomet) is a patient-specific glenoid vault reconstruction system developed with the use of CAD/CAM to address severe glenoid bone loss encountered during shoulder arthroplasty. For several years, the VRS was available only as a custom implant according to the US Food and Drug Administration rules, and therefore its use was limited to a few cases per year. Recently, a 510(k) envelope clearance was granted to use the VRS in reverse TSA to address significant glenoid bone defects.
The VRS is made of porous plasma spray titanium to provide high strength and flexibility, and allows for biologic fixation. This system can accommodate a restricted bone loss envelope of about 50 mm × 50 mm × 35 mm according to the previous experience of the manufacturer in the custom scenario, covering 96% of defects previously addressed. One 6.5-mm nonlocking central screw and a minimum of four 4.75-mm nonlocking or locking peripheral screws are required for optimal fixation of the implant in the native scapula. A custom boss can be added in to enhance fixation in the native scapula when the bone is sufficient. To facilitate the surgical procedure, a trial implant, a bone model of the scapula, and a custom boss reaming guide are 3-dimensional (3-D) and printed in sterilizable material. These are all provided as single-use disposable instruments and can be available for surgeons during both the initial plan review and surgery.

PREOPERATIVE PLANNING
Patients undergo a preoperative fine-cut 2-dimensional computed tomography scan of the scapula and adjacent humerus following a predefined protocol with a slice thickness of 2 mm to 3 mm. An accurate 3-D bone model of the scapula is obtained using a 3-D image processing software system (Figure 1). The 3-D scapular model is used to create a patient-specific glenoid implant proposal that is approved by the surgeon (Figure 2). Implant position, orientation, size, screw trajectory, and recommended bone removal, if necessary, are determined to create a more normal glenohumeral center of rotation and to secure a glenoid implant in severely deficient glenoid bone (Figure 3). Once the implant design is approved by the surgeon, the final patient-specific implant is manufactured.

SURGICAL TECHNIQUE
The exposure of the glenoid is a critical step for the successful implantation of the patient-specific glenoid implant. Soft tissue and scar tissue around the glenoid must be removed to allow for optimal fit of the custom-made reaming guide. Also, removal of the entire capsulolabral complex on the anteroinferior rim of the glenoid is essential to both enhance glenoid exposure and to allow a perfect fit of the guide to the pathologic bone stock. Attention should be paid during débridement and/or implant removal in case of revision, to make sure that no excessive bone is removed because the patient-specific guide is referenced to this anatomy. Excessive bone removal can change the orientation of the patient-specific guide and ultimately the fixation of the implant. Once the custom-made patient-specific guide is positioned, a 3.2-mm Steinmann pin is placed through the inserter for temporary fixation. The pin should engage or perforate the medial cortical wall to ensure that the subsequent reamer has a stable cannula over which to ream. After the glenoid is reamed, the final implant can be placed in the ideal position according to the preoperative planning. A central 6.5-mm nonlocking central screw and 4.75-mm nonlocking or locking peripheral screws are required to complete the fixation of the implant in the native scapula. Once the patient-specific glenoid component is positioned and strongly fixed to the bone, the glenosphere can be positioned according to the preoperative planning, and the reverse shoulder arthroplasty can be completed in the usual fashion.

CASE EXAMPLES
A 68-year-old woman underwent a TSA for end-stage osteoarthritis in 2000. The implant failed due to a cuff failure. The patient underwent several surgeries, including an open cuff repair, with no success. She had no active elevation preoperatively. Because of the significant glenoid bone loss, a patient-specific glenoid reconstruction was planned. Within 24 months after this surgery, the patient was able to get her hand to her head and elevate to 90º (Figures 4A-4F).

Continue to: In October 2013...
In October 2013, a 68-year-old man underwent a TSA for end-stage osteoarthritis. After 18 months, the implant failed due to active Propionibacterium acnes infection, which required excisional arthroplasty with insertion of an antibiotic spacer. Significant glenoid bone loss (Figure 5) and global soft-tissue deficiency caused substantial disability and led to an indication for a reverse TSA with a patient-specific glenoid vault reconstruction (Figures 6A-6D) after infection eradication. Within 20 months after this surgery, the patient had resumed a satisfactory range of motion (130º forward elevation, 20º external rotation) and outcome.

DISCUSSION
Although glenoid bone loss is often seen in the primary setting (degenerative, congenital, and post-traumatic), severe glenoid bone loss is encountered in most revision TSAs. The best treatment method for massive glenoid bone defects during complex shoulder arthroplasty remains uncertain. Options have included eccentric reaming, glenoid reconstruction with bone allograft and autograft, and more recently augmented components and patient-specific implants.21-25 The advent and availability of CAD/CAM technology have enabled shoulder surgeons to create patient-specific metal solutions to these challenging cases. Currently, only a few reports exist in the literature on patient-specific glenoid components in the setting of severe bone loss.29-32

Chammaa and colleagues29 reported the outcomes of 37 patients with a hip-inspired glenoid component (Total Shoulder Replacement, Stanmore Implants Worldwide). The 5-year results with this implant were promising, with a 16% revision rate and only 1 case of glenoid loosening.
Stoffelen and colleagues30 recently described the successful use of a patient-specific anatomic metal-backed glenoid component for the management of severe glenoid bone loss with excellent results at 2.5 years of follow-up. A different approach was pursued by Gunther and Lynch,31 who reported on 7 patients with a custom inset glenoid implant for deficient glenoid vaults. These circular anatomic, custom-made glenoid components were created with the intention of placing the implants partially inside the glenoid vault and relying partially on sclerotic cortical bone. Despite excellent results at 3 years of follow-up, their use is limited to specific defect geometries and cannot be used in cases of extreme bone loss.
CONCLUSION
We have described the use of a patient-specific glenoid component in 2 patients with severe glenoid bone loss. Despite the satisfactory clinical and short-term radiographic results, we acknowledge that longer-term follow-up is needed to confirm the efficacy of this type of reconstruction. We believe that patient-specific glenoid components represent a valuable addition to the armamentarium of shoulder surgeons who address complex glenoid bone deformities.
1. Chalmers PN, Gupta AK, Rahman Z, Bruce B, Romeo AA, Nicholson GP. Predictors of early complications of total shoulder arthroplasty. J Arthroplasty. 2014;29(4):856-860. doi:10.1016/j.arth.2013.07.002.
2. Deshmukh AV, Koris M, Zurakowski D, Thornhill TS. Total shoulder arthroplasty: long-term survivorship, functional outcome, and quality of life. J Shoulder Elbow Surg. 2005;14(5):471-479. doi:10.1016/j.jse.2005.02.009.
3. Montoya F, Magosch P, Scheiderer B, Lichtenberg S, Melean P, Habermeyer P. Midterm results of a total shoulder prosthesis fixed with a cementless glenoid component. J Shoulder Elbow Surg. 2013;22(5):628-635. doi:10.1016/j.jse.2012.07.005.
4. Torchia ME, Cofield RH, Settergren CR. Total shoulder arthroplasty with the Neer prosthesis: long-term results. J Shoulder Elbow Surg. 1997;6(6):495-505.
5. Antuna SA, Sperling JW, Cofield RH, Rowland CM. Glenoid revision surgery after total shoulder arthroplasty. J Shoulder Elbow Surg. 2001;10(3):217-224. doi:10.1067/mse.2001.113961.
6. Chalmers PN, Rahman Z, Romeo AA, Nicholson GP. Early dislocation after reverse total shoulder arthroplasty. J Shoulder Elbow Surg. 2014;23(5):737-744. doi:10.1016/j.jse.2013.08.015.
7. Farng E, Zingmond D, Krenek L, Soohoo NF. Factors predicting complication rates after primary shoulder arthroplasty. J Shoulder Elbow Surg. 2011;20(4):557-563. doi:10.1016/j.jse.2010.11.005.
8. Farshad M, Grogli M, Catanzaro S, Gerber C. Revision of reversed total shoulder arthroplasty. Indications and outcome. BMC Musculoskelet Disord. 2012;13(1):160. doi:10.1186/1471-2474-13-160.
9. Fevang BT, Lie SA, Havelin LI, Skredderstuen A, Furnes O. Risk factors for revision after shoulder arthroplasty: 1,825 shoulder arthroplasties from the Norwegian Arthroplasty Register. Acta Orthop. 2009;80(1):83-91.
10. Fox TJ, Cil A, Sperling JW, Sanchez-Sotelo J, Schleck CD, Cofield RH. Survival of the glenoid component in shoulder arthroplasty. J Shoulder Elbow Surg. 2009;18(6):859-863. doi:10.1016/j.jse.2008.11.020.
11. Rasmussen JV. Outcome and risk of revision following shoulder replacement in patients with glenohumeral osteoarthritis. Acta Orthop Suppl. 2014;85(355 suppl):1-23. doi:10.3109/17453674.2014.922007.
12. Rasmussen JV, Polk A, Brorson S, Sorensen AK, Olsen BS. Patient-reported outcome and risk of revision after shoulder replacement for osteoarthritis. 1,209 cases from the Danish Shoulder Arthroplasty Registry, 2006-2010. Acta Orthop. 2014;85(2):117-122. doi:10.3109/17453674.2014.893497.
13. Sajadi KR, Kwon YW, Zuckerman JD. Revision shoulder arthroplasty: an analysis of indications and outcomes. J Shoulder Elbow Surg. 2010;19(2):308-313. doi:10.1016/j.jse.2009.05.016.
14. Singh JA, Sperling JW, Cofield RH. Revision surgery following total shoulder arthroplasty: analysis of 2588 shoulders over three decades (1976 to 2008). J Bone Joint Surg Br. 2011;93(11):1513-1517. doi:10.1302/0301-620X.93B11.26938.
15. Levine WN, Djurasovic M, Glasson JM, Pollock RG, Flatow EL, Bigliani LU. Hemiarthroplasty for glenohumeral osteoarthritis: results correlated to degree of glenoid wear. J Shoulder Elbow Surg. 1997;6(5):449-454.
16. Levine WN, Fischer CR, Nguyen D, Flatow EL, Ahmad CS, Bigliani LU. Long-term follow-up of shoulder hemiarthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2012;94(22):e164. doi:10.2106/JBJS.K.00603.
17. Lynch JR, Franta AK, Montgomery WH, Lenters TR, Mounce D, Matsen FA. Self-assessed outcome at two to four years after shoulder hemiarthroplasty with concentric glenoid reaming. J Bone Joint Surg Am. 2007;89(6):1284-1292. doi:10.2106/JBJS.E.00942.
18. Iannotti JP, Norris TR. Influence of preoperative factors on outcome of shoulder arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2003;85-A(2):251-258.
19. Sperling JW, Cofield RH, Rowland CM. Neer hemiarthroplasty and Neer total shoulder arthroplasty in patients fifty years old or less. Long-term results. J Bone Joint Surg Am. 1998;80(4):464-473.
20. Strauss EJ, Roche C, Flurin PH, Wright T, Zuckerman JD. The glenoid in shoulder arthroplasty. J Shoulder Elbow Surg. 2009;18(5):819-833. doi:10.1016/j.jse.2009.05.008.
21. Cil A, Sperling JW, Cofield RH. Nonstandard glenoid components for bone deficiencies in shoulder arthroplasty. J Shoulder Elbow Surg. 2014;23(7):e149-e157. doi:10.1016/j.jse.2013.09.023.
22. Denard PJ, Walch G. Current concepts in the surgical management of primary glenohumeral arthritis with a biconcave glenoid. J Shoulder Elbow Surg. 2013;22(11):1589-1598. doi:10.1016/j.jse.2013.06.017.
23. Gunther SB, Lynch TL. Total shoulder replacement surgery with custom glenoid implants for severe bone deficiency. J Shoulder Elbow Surg. 2012;21(5):675-684. doi:10.1016/j.jse.2011.03.023.
24. Neer CS, Morrison DS. Glenoid bone-grafting in total shoulder arthroplasty. J Bone Joint Surg Am. 1988;70(8):1154-1162.
25. Steinmann SP, Cofield RH. Bone grafting for glenoid deficiency in total shoulder replacement. J Shoulder Elbow Surg. 2000;9(5):361-367. doi:10.1067/mse.2000.106921.
26. Iannotti JP, Frangiamore SJ. Fate of large structural allograft for treatment of severe uncontained glenoid bone deficiency. J Shoulder Elbow Surg. 2012:21(6):765-771. doi:10.1016/j.jse.2011.08.069.
27. Hill JM, Norris TR. Long-term results of total shoulder arthroplasty following bone-grafting of the glenoid. J Bone Joint Surg Am. 2001;83-A(6):877-883.
28. Hsu JE, Ricchetti ET, Huffman GR, Iannotti JP, Glaser DL. Addressing glenoid bone deficiency and asymptomatic posterior erosion in shoulder arthroplasty. J Shoulder Elbow Surg. 2013;22(9):1298-1308.
29. Chammaa R, Uri O, Lambert S. Primary shoulder arthroplasty using a custom-made hip-inspired implant for the treatment of advanced glenohumeral arthritis in the presence of severe glenoid bone loss. J Shoulder Elbow Surg. 2017;26(1):101-107. doi:10.1016/j.jse.2016.05.027.
30. Stoffelen DV, Eraly K, Debeer P. The use of 3D printing technology in reconstruction of a severe glenoid defect: a case report with 2.5 years of follow-up. J Shoulder Elbow Surg. 2015;24(8):e218-e222. doi:10.1016/j.jse.2015.04.006.
31. Gunther SB, Lynch TL. Total shoulder replacement surgery with custom glenoid implants for severe bone deficiency. J Shoulder Elbow Surg. 2012;21(5):675-684. doi:10.1016/j.jse.2011.03.023.
32. Dines DM, Gulotta L, Craig EV, Dines JS. Novel solution for massive glenoid defects in shoulder arthroplasty: a patient-specific glenoid vault reconstruction system. Am J Orthop. 2017;46(2):104-108.
ABSTRACT
Complex glenoid bone deformities present the treating surgeon with a complex reconstructive challenge. Although glenoid bone loss can be encountered in the primary setting (degenerative, congenital, post-traumatic), severe glenoid bone loss is encountered in most revision total shoulder arthroplasties. Severe glenoid bone loss is treated with various techniques including hemiarthroplasty, eccentric reaming, and glenoid reconstruction with bone autografts and allografts. Despite encouraging short- to mid-term results reported with these reconstruction techniques, the clinical and radiographic outcomes remain inconsistent and the high number of complications is a concern. To overcome this problem, more recently augmented components and patient specific implants were introduced. Using the computer-aided design and computer-aided manufacturing technology patient-specific implants have been created to reconstruct the glenoid vault in cases of severe glenoid bone loss.
In this article we describe a patient specific glenoid implant, its indication, technical aspects and surgical technique, based on the author's experience as well as a review of the current literature on custom glenoid implants.
Continue to: Total shoulder arthroplasty...
Total shoulder arthroplasty (TSA) is an effective operation for providing pain relief and improving function in patients with end-stage degenerative shoulder disease that is nonresponsive to nonoperative treatments.1-4 With the increasing number of arthroplasties performed, and the expanding indication for shoulder arthroplasty, the number of revision shoulder arthroplasties is also increasing.5-14 Complex glenoid bone deformities present the treating surgeon with a complex reconstructive challenge. Although glenoid bone loss can be seen in the primary setting (degenerative, congenital, and post-traumatic), severe glenoid bone loss is encountered mostly in revision TSAs.
Historically, patients with severe glenoid bone loss were treated with a hemiarthroplasty.15-17 However, due to inferior outcomes associated with the use of shoulder hemiarthroplasties compared with TSA in these cases,18-20 various techniques were developed with the aim of realigning the glenoid axis and securing the implants into the deficient glenoid vault.21-25 Options have included eccentric reaming, glenoid reconstruction with bone autografts and allografts, and more recently augmented components and patient-specific implants. Studies with eccentric reaming and reconstruction with bone graft during complex shoulder arthroplasty have reported encouraging short- to mid-term results, but the clinical and radiographic outcomes remain inconsistent, and the high number of complications is a concern.25-28
Complications with these techniques include component loosening, graft resorption, nonunion, failure of graft incorporation, infection, and instability.25-28
Computer-aided design and computer-aided manufacturing (CAD/CAM) of patient-specific implants have been used successfully by hip arthroplasty surgeons to deal with complex acetabular reconstructions in the setting of severe bone loss. More recently, the same technology has been used to reconstruct the glenoid vault in cases of severe glenoid bone loss.
In this article, we describe a patient-specific glenoid implant, its indication, and both technical aspects and the surgical technique, based on the authors’ experience as well as a review of the current literature on custom glenoid implants.
Continue to: PATIENT-SPECIFIC GLENOID COMPONENT
PATIENT-SPECIFIC GLENOID COMPONENT
The Vault Reconstruction System ([VRS], Zimmer Biomet) is a patient-specific glenoid vault reconstruction system developed with the use of CAD/CAM to address severe glenoid bone loss encountered during shoulder arthroplasty. For several years, the VRS was available only as a custom implant according to the US Food and Drug Administration rules, and therefore its use was limited to a few cases per year. Recently, a 510(k) envelope clearance was granted to use the VRS in reverse TSA to address significant glenoid bone defects.
The VRS is made of porous plasma spray titanium to provide high strength and flexibility, and allows for biologic fixation. This system can accommodate a restricted bone loss envelope of about 50 mm × 50 mm × 35 mm according to the previous experience of the manufacturer in the custom scenario, covering 96% of defects previously addressed. One 6.5-mm nonlocking central screw and a minimum of four 4.75-mm nonlocking or locking peripheral screws are required for optimal fixation of the implant in the native scapula. A custom boss can be added in to enhance fixation in the native scapula when the bone is sufficient. To facilitate the surgical procedure, a trial implant, a bone model of the scapula, and a custom boss reaming guide are 3-dimensional (3-D) and printed in sterilizable material. These are all provided as single-use disposable instruments and can be available for surgeons during both the initial plan review and surgery.
PREOPERATIVE PLANNING
Patients undergo a preoperative fine-cut 2-dimensional computed tomography scan of the scapula and adjacent humerus following a predefined protocol with a slice thickness of 2 mm to 3 mm. An accurate 3-D bone model of the scapula is obtained using a 3-D image processing software system (Figure 1). The 3-D scapular model is used to create a patient-specific glenoid implant proposal that is approved by the surgeon (Figure 2). Implant position, orientation, size, screw trajectory, and recommended bone removal, if necessary, are determined to create a more normal glenohumeral center of rotation and to secure a glenoid implant in severely deficient glenoid bone (Figure 3). Once the implant design is approved by the surgeon, the final patient-specific implant is manufactured.
SURGICAL TECHNIQUE
The exposure of the glenoid is a critical step for the successful implantation of the patient-specific glenoid implant. Soft tissue and scar tissue around the glenoid must be removed to allow for optimal fit of the custom-made reaming guide. Also, removal of the entire capsulolabral complex on the anteroinferior rim of the glenoid is essential to both enhance glenoid exposure and to allow a perfect fit of the guide to the pathologic bone stock. Attention should be paid during débridement and/or implant removal in case of revision, to make sure that no excessive bone is removed because the patient-specific guide is referenced to this anatomy. Excessive bone removal can change the orientation of the patient-specific guide and ultimately the fixation of the implant. Once the custom-made patient-specific guide is positioned, a 3.2-mm Steinmann pin is placed through the inserter for temporary fixation. The pin should engage or perforate the medial cortical wall to ensure that the subsequent reamer has a stable cannula over which to ream. After the glenoid is reamed, the final implant can be placed in the ideal position according to the preoperative planning. A central 6.5-mm nonlocking central screw and 4.75-mm nonlocking or locking peripheral screws are required to complete the fixation of the implant in the native scapula. Once the patient-specific glenoid component is positioned and strongly fixed to the bone, the glenosphere can be positioned according to the preoperative planning, and the reverse shoulder arthroplasty can be completed in the usual fashion.
CASE EXAMPLES
A 68-year-old woman underwent a TSA for end-stage osteoarthritis in 2000. The implant failed due to a cuff failure. The patient underwent several surgeries, including an open cuff repair, with no success. She had no active elevation preoperatively. Because of the significant glenoid bone loss, a patient-specific glenoid reconstruction was planned. Within 24 months after this surgery, the patient was able to get her hand to her head and elevate to 90º (Figures 4A-4F).
Continue to: In October 2013...
In October 2013, a 68-year-old man underwent a TSA for end-stage osteoarthritis. After 18 months, the implant failed due to active Propionibacterium acnes infection, which required excisional arthroplasty with insertion of an antibiotic spacer. Significant glenoid bone loss (Figure 5) and global soft-tissue deficiency caused substantial disability and led to an indication for a reverse TSA with a patient-specific glenoid vault reconstruction (Figures 6A-6D) after infection eradication. Within 20 months after this surgery, the patient had resumed a satisfactory range of motion (130º forward elevation, 20º external rotation) and outcome.
DISCUSSION
Although glenoid bone loss is often seen in the primary setting (degenerative, congenital, and post-traumatic), severe glenoid bone loss is encountered in most revision TSAs. The best treatment method for massive glenoid bone defects during complex shoulder arthroplasty remains uncertain. Options have included eccentric reaming, glenoid reconstruction with bone allograft and autograft, and more recently augmented components and patient-specific implants.21-25 The advent and availability of CAD/CAM technology have enabled shoulder surgeons to create patient-specific metal solutions to these challenging cases. Currently, only a few reports exist in the literature on patient-specific glenoid components in the setting of severe bone loss.29-32
Chammaa and colleagues29 reported the outcomes of 37 patients with a hip-inspired glenoid component (Total Shoulder Replacement, Stanmore Implants Worldwide). The 5-year results with this implant were promising, with a 16% revision rate and only 1 case of glenoid loosening.
Stoffelen and colleagues30 recently described the successful use of a patient-specific anatomic metal-backed glenoid component for the management of severe glenoid bone loss with excellent results at 2.5 years of follow-up. A different approach was pursued by Gunther and Lynch,31 who reported on 7 patients with a custom inset glenoid implant for deficient glenoid vaults. These circular anatomic, custom-made glenoid components were created with the intention of placing the implants partially inside the glenoid vault and relying partially on sclerotic cortical bone. Despite excellent results at 3 years of follow-up, their use is limited to specific defect geometries and cannot be used in cases of extreme bone loss.
CONCLUSION
We have described the use of a patient-specific glenoid component in 2 patients with severe glenoid bone loss. Despite the satisfactory clinical and short-term radiographic results, we acknowledge that longer-term follow-up is needed to confirm the efficacy of this type of reconstruction. We believe that patient-specific glenoid components represent a valuable addition to the armamentarium of shoulder surgeons who address complex glenoid bone deformities.
ABSTRACT
Complex glenoid bone deformities present the treating surgeon with a complex reconstructive challenge. Although glenoid bone loss can be encountered in the primary setting (degenerative, congenital, post-traumatic), severe glenoid bone loss is encountered in most revision total shoulder arthroplasties. Severe glenoid bone loss is treated with various techniques including hemiarthroplasty, eccentric reaming, and glenoid reconstruction with bone autografts and allografts. Despite encouraging short- to mid-term results reported with these reconstruction techniques, the clinical and radiographic outcomes remain inconsistent and the high number of complications is a concern. To overcome this problem, more recently augmented components and patient specific implants were introduced. Using the computer-aided design and computer-aided manufacturing technology patient-specific implants have been created to reconstruct the glenoid vault in cases of severe glenoid bone loss.
In this article we describe a patient specific glenoid implant, its indication, technical aspects and surgical technique, based on the author's experience as well as a review of the current literature on custom glenoid implants.
Continue to: Total shoulder arthroplasty...
Total shoulder arthroplasty (TSA) is an effective operation for providing pain relief and improving function in patients with end-stage degenerative shoulder disease that is nonresponsive to nonoperative treatments.1-4 With the increasing number of arthroplasties performed, and the expanding indication for shoulder arthroplasty, the number of revision shoulder arthroplasties is also increasing.5-14 Complex glenoid bone deformities present the treating surgeon with a complex reconstructive challenge. Although glenoid bone loss can be seen in the primary setting (degenerative, congenital, and post-traumatic), severe glenoid bone loss is encountered mostly in revision TSAs.
Historically, patients with severe glenoid bone loss were treated with a hemiarthroplasty.15-17 However, due to inferior outcomes associated with the use of shoulder hemiarthroplasties compared with TSA in these cases,18-20 various techniques were developed with the aim of realigning the glenoid axis and securing the implants into the deficient glenoid vault.21-25 Options have included eccentric reaming, glenoid reconstruction with bone autografts and allografts, and more recently augmented components and patient-specific implants. Studies with eccentric reaming and reconstruction with bone graft during complex shoulder arthroplasty have reported encouraging short- to mid-term results, but the clinical and radiographic outcomes remain inconsistent, and the high number of complications is a concern.25-28
Complications with these techniques include component loosening, graft resorption, nonunion, failure of graft incorporation, infection, and instability.25-28
Computer-aided design and computer-aided manufacturing (CAD/CAM) of patient-specific implants have been used successfully by hip arthroplasty surgeons to deal with complex acetabular reconstructions in the setting of severe bone loss. More recently, the same technology has been used to reconstruct the glenoid vault in cases of severe glenoid bone loss.
In this article, we describe a patient-specific glenoid implant, its indication, and both technical aspects and the surgical technique, based on the authors’ experience as well as a review of the current literature on custom glenoid implants.
Continue to: PATIENT-SPECIFIC GLENOID COMPONENT
PATIENT-SPECIFIC GLENOID COMPONENT
The Vault Reconstruction System ([VRS], Zimmer Biomet) is a patient-specific glenoid vault reconstruction system developed with the use of CAD/CAM to address severe glenoid bone loss encountered during shoulder arthroplasty. For several years, the VRS was available only as a custom implant according to the US Food and Drug Administration rules, and therefore its use was limited to a few cases per year. Recently, a 510(k) envelope clearance was granted to use the VRS in reverse TSA to address significant glenoid bone defects.
The VRS is made of porous plasma spray titanium to provide high strength and flexibility, and allows for biologic fixation. This system can accommodate a restricted bone loss envelope of about 50 mm × 50 mm × 35 mm according to the previous experience of the manufacturer in the custom scenario, covering 96% of defects previously addressed. One 6.5-mm nonlocking central screw and a minimum of four 4.75-mm nonlocking or locking peripheral screws are required for optimal fixation of the implant in the native scapula. A custom boss can be added in to enhance fixation in the native scapula when the bone is sufficient. To facilitate the surgical procedure, a trial implant, a bone model of the scapula, and a custom boss reaming guide are 3-dimensional (3-D) and printed in sterilizable material. These are all provided as single-use disposable instruments and can be available for surgeons during both the initial plan review and surgery.
PREOPERATIVE PLANNING
Patients undergo a preoperative fine-cut 2-dimensional computed tomography scan of the scapula and adjacent humerus following a predefined protocol with a slice thickness of 2 mm to 3 mm. An accurate 3-D bone model of the scapula is obtained using a 3-D image processing software system (Figure 1). The 3-D scapular model is used to create a patient-specific glenoid implant proposal that is approved by the surgeon (Figure 2). Implant position, orientation, size, screw trajectory, and recommended bone removal, if necessary, are determined to create a more normal glenohumeral center of rotation and to secure a glenoid implant in severely deficient glenoid bone (Figure 3). Once the implant design is approved by the surgeon, the final patient-specific implant is manufactured.
SURGICAL TECHNIQUE
The exposure of the glenoid is a critical step for the successful implantation of the patient-specific glenoid implant. Soft tissue and scar tissue around the glenoid must be removed to allow for optimal fit of the custom-made reaming guide. Also, removal of the entire capsulolabral complex on the anteroinferior rim of the glenoid is essential to both enhance glenoid exposure and to allow a perfect fit of the guide to the pathologic bone stock. Attention should be paid during débridement and/or implant removal in case of revision, to make sure that no excessive bone is removed because the patient-specific guide is referenced to this anatomy. Excessive bone removal can change the orientation of the patient-specific guide and ultimately the fixation of the implant. Once the custom-made patient-specific guide is positioned, a 3.2-mm Steinmann pin is placed through the inserter for temporary fixation. The pin should engage or perforate the medial cortical wall to ensure that the subsequent reamer has a stable cannula over which to ream. After the glenoid is reamed, the final implant can be placed in the ideal position according to the preoperative planning. A central 6.5-mm nonlocking central screw and 4.75-mm nonlocking or locking peripheral screws are required to complete the fixation of the implant in the native scapula. Once the patient-specific glenoid component is positioned and strongly fixed to the bone, the glenosphere can be positioned according to the preoperative planning, and the reverse shoulder arthroplasty can be completed in the usual fashion.
CASE EXAMPLES
A 68-year-old woman underwent a TSA for end-stage osteoarthritis in 2000. The implant failed due to a cuff failure. The patient underwent several surgeries, including an open cuff repair, with no success. She had no active elevation preoperatively. Because of the significant glenoid bone loss, a patient-specific glenoid reconstruction was planned. Within 24 months after this surgery, the patient was able to get her hand to her head and elevate to 90º (Figures 4A-4F).
Continue to: In October 2013...
In October 2013, a 68-year-old man underwent a TSA for end-stage osteoarthritis. After 18 months, the implant failed due to active Propionibacterium acnes infection, which required excisional arthroplasty with insertion of an antibiotic spacer. Significant glenoid bone loss (Figure 5) and global soft-tissue deficiency caused substantial disability and led to an indication for a reverse TSA with a patient-specific glenoid vault reconstruction (Figures 6A-6D) after infection eradication. Within 20 months after this surgery, the patient had resumed a satisfactory range of motion (130º forward elevation, 20º external rotation) and outcome.
DISCUSSION
Although glenoid bone loss is often seen in the primary setting (degenerative, congenital, and post-traumatic), severe glenoid bone loss is encountered in most revision TSAs. The best treatment method for massive glenoid bone defects during complex shoulder arthroplasty remains uncertain. Options have included eccentric reaming, glenoid reconstruction with bone allograft and autograft, and more recently augmented components and patient-specific implants.21-25 The advent and availability of CAD/CAM technology have enabled shoulder surgeons to create patient-specific metal solutions to these challenging cases. Currently, only a few reports exist in the literature on patient-specific glenoid components in the setting of severe bone loss.29-32
Chammaa and colleagues29 reported the outcomes of 37 patients with a hip-inspired glenoid component (Total Shoulder Replacement, Stanmore Implants Worldwide). The 5-year results with this implant were promising, with a 16% revision rate and only 1 case of glenoid loosening.
Stoffelen and colleagues30 recently described the successful use of a patient-specific anatomic metal-backed glenoid component for the management of severe glenoid bone loss with excellent results at 2.5 years of follow-up. A different approach was pursued by Gunther and Lynch,31 who reported on 7 patients with a custom inset glenoid implant for deficient glenoid vaults. These circular anatomic, custom-made glenoid components were created with the intention of placing the implants partially inside the glenoid vault and relying partially on sclerotic cortical bone. Despite excellent results at 3 years of follow-up, their use is limited to specific defect geometries and cannot be used in cases of extreme bone loss.
CONCLUSION
We have described the use of a patient-specific glenoid component in 2 patients with severe glenoid bone loss. Despite the satisfactory clinical and short-term radiographic results, we acknowledge that longer-term follow-up is needed to confirm the efficacy of this type of reconstruction. We believe that patient-specific glenoid components represent a valuable addition to the armamentarium of shoulder surgeons who address complex glenoid bone deformities.
1. Chalmers PN, Gupta AK, Rahman Z, Bruce B, Romeo AA, Nicholson GP. Predictors of early complications of total shoulder arthroplasty. J Arthroplasty. 2014;29(4):856-860. doi:10.1016/j.arth.2013.07.002.
2. Deshmukh AV, Koris M, Zurakowski D, Thornhill TS. Total shoulder arthroplasty: long-term survivorship, functional outcome, and quality of life. J Shoulder Elbow Surg. 2005;14(5):471-479. doi:10.1016/j.jse.2005.02.009.
3. Montoya F, Magosch P, Scheiderer B, Lichtenberg S, Melean P, Habermeyer P. Midterm results of a total shoulder prosthesis fixed with a cementless glenoid component. J Shoulder Elbow Surg. 2013;22(5):628-635. doi:10.1016/j.jse.2012.07.005.
4. Torchia ME, Cofield RH, Settergren CR. Total shoulder arthroplasty with the Neer prosthesis: long-term results. J Shoulder Elbow Surg. 1997;6(6):495-505.
5. Antuna SA, Sperling JW, Cofield RH, Rowland CM. Glenoid revision surgery after total shoulder arthroplasty. J Shoulder Elbow Surg. 2001;10(3):217-224. doi:10.1067/mse.2001.113961.
6. Chalmers PN, Rahman Z, Romeo AA, Nicholson GP. Early dislocation after reverse total shoulder arthroplasty. J Shoulder Elbow Surg. 2014;23(5):737-744. doi:10.1016/j.jse.2013.08.015.
7. Farng E, Zingmond D, Krenek L, Soohoo NF. Factors predicting complication rates after primary shoulder arthroplasty. J Shoulder Elbow Surg. 2011;20(4):557-563. doi:10.1016/j.jse.2010.11.005.
8. Farshad M, Grogli M, Catanzaro S, Gerber C. Revision of reversed total shoulder arthroplasty. Indications and outcome. BMC Musculoskelet Disord. 2012;13(1):160. doi:10.1186/1471-2474-13-160.
9. Fevang BT, Lie SA, Havelin LI, Skredderstuen A, Furnes O. Risk factors for revision after shoulder arthroplasty: 1,825 shoulder arthroplasties from the Norwegian Arthroplasty Register. Acta Orthop. 2009;80(1):83-91.
10. Fox TJ, Cil A, Sperling JW, Sanchez-Sotelo J, Schleck CD, Cofield RH. Survival of the glenoid component in shoulder arthroplasty. J Shoulder Elbow Surg. 2009;18(6):859-863. doi:10.1016/j.jse.2008.11.020.
11. Rasmussen JV. Outcome and risk of revision following shoulder replacement in patients with glenohumeral osteoarthritis. Acta Orthop Suppl. 2014;85(355 suppl):1-23. doi:10.3109/17453674.2014.922007.
12. Rasmussen JV, Polk A, Brorson S, Sorensen AK, Olsen BS. Patient-reported outcome and risk of revision after shoulder replacement for osteoarthritis. 1,209 cases from the Danish Shoulder Arthroplasty Registry, 2006-2010. Acta Orthop. 2014;85(2):117-122. doi:10.3109/17453674.2014.893497.
13. Sajadi KR, Kwon YW, Zuckerman JD. Revision shoulder arthroplasty: an analysis of indications and outcomes. J Shoulder Elbow Surg. 2010;19(2):308-313. doi:10.1016/j.jse.2009.05.016.
14. Singh JA, Sperling JW, Cofield RH. Revision surgery following total shoulder arthroplasty: analysis of 2588 shoulders over three decades (1976 to 2008). J Bone Joint Surg Br. 2011;93(11):1513-1517. doi:10.1302/0301-620X.93B11.26938.
15. Levine WN, Djurasovic M, Glasson JM, Pollock RG, Flatow EL, Bigliani LU. Hemiarthroplasty for glenohumeral osteoarthritis: results correlated to degree of glenoid wear. J Shoulder Elbow Surg. 1997;6(5):449-454.
16. Levine WN, Fischer CR, Nguyen D, Flatow EL, Ahmad CS, Bigliani LU. Long-term follow-up of shoulder hemiarthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2012;94(22):e164. doi:10.2106/JBJS.K.00603.
17. Lynch JR, Franta AK, Montgomery WH, Lenters TR, Mounce D, Matsen FA. Self-assessed outcome at two to four years after shoulder hemiarthroplasty with concentric glenoid reaming. J Bone Joint Surg Am. 2007;89(6):1284-1292. doi:10.2106/JBJS.E.00942.
18. Iannotti JP, Norris TR. Influence of preoperative factors on outcome of shoulder arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2003;85-A(2):251-258.
19. Sperling JW, Cofield RH, Rowland CM. Neer hemiarthroplasty and Neer total shoulder arthroplasty in patients fifty years old or less. Long-term results. J Bone Joint Surg Am. 1998;80(4):464-473.
20. Strauss EJ, Roche C, Flurin PH, Wright T, Zuckerman JD. The glenoid in shoulder arthroplasty. J Shoulder Elbow Surg. 2009;18(5):819-833. doi:10.1016/j.jse.2009.05.008.
21. Cil A, Sperling JW, Cofield RH. Nonstandard glenoid components for bone deficiencies in shoulder arthroplasty. J Shoulder Elbow Surg. 2014;23(7):e149-e157. doi:10.1016/j.jse.2013.09.023.
22. Denard PJ, Walch G. Current concepts in the surgical management of primary glenohumeral arthritis with a biconcave glenoid. J Shoulder Elbow Surg. 2013;22(11):1589-1598. doi:10.1016/j.jse.2013.06.017.
23. Gunther SB, Lynch TL. Total shoulder replacement surgery with custom glenoid implants for severe bone deficiency. J Shoulder Elbow Surg. 2012;21(5):675-684. doi:10.1016/j.jse.2011.03.023.
24. Neer CS, Morrison DS. Glenoid bone-grafting in total shoulder arthroplasty. J Bone Joint Surg Am. 1988;70(8):1154-1162.
25. Steinmann SP, Cofield RH. Bone grafting for glenoid deficiency in total shoulder replacement. J Shoulder Elbow Surg. 2000;9(5):361-367. doi:10.1067/mse.2000.106921.
26. Iannotti JP, Frangiamore SJ. Fate of large structural allograft for treatment of severe uncontained glenoid bone deficiency. J Shoulder Elbow Surg. 2012:21(6):765-771. doi:10.1016/j.jse.2011.08.069.
27. Hill JM, Norris TR. Long-term results of total shoulder arthroplasty following bone-grafting of the glenoid. J Bone Joint Surg Am. 2001;83-A(6):877-883.
28. Hsu JE, Ricchetti ET, Huffman GR, Iannotti JP, Glaser DL. Addressing glenoid bone deficiency and asymptomatic posterior erosion in shoulder arthroplasty. J Shoulder Elbow Surg. 2013;22(9):1298-1308.
29. Chammaa R, Uri O, Lambert S. Primary shoulder arthroplasty using a custom-made hip-inspired implant for the treatment of advanced glenohumeral arthritis in the presence of severe glenoid bone loss. J Shoulder Elbow Surg. 2017;26(1):101-107. doi:10.1016/j.jse.2016.05.027.
30. Stoffelen DV, Eraly K, Debeer P. The use of 3D printing technology in reconstruction of a severe glenoid defect: a case report with 2.5 years of follow-up. J Shoulder Elbow Surg. 2015;24(8):e218-e222. doi:10.1016/j.jse.2015.04.006.
31. Gunther SB, Lynch TL. Total shoulder replacement surgery with custom glenoid implants for severe bone deficiency. J Shoulder Elbow Surg. 2012;21(5):675-684. doi:10.1016/j.jse.2011.03.023.
32. Dines DM, Gulotta L, Craig EV, Dines JS. Novel solution for massive glenoid defects in shoulder arthroplasty: a patient-specific glenoid vault reconstruction system. Am J Orthop. 2017;46(2):104-108.
1. Chalmers PN, Gupta AK, Rahman Z, Bruce B, Romeo AA, Nicholson GP. Predictors of early complications of total shoulder arthroplasty. J Arthroplasty. 2014;29(4):856-860. doi:10.1016/j.arth.2013.07.002.
2. Deshmukh AV, Koris M, Zurakowski D, Thornhill TS. Total shoulder arthroplasty: long-term survivorship, functional outcome, and quality of life. J Shoulder Elbow Surg. 2005;14(5):471-479. doi:10.1016/j.jse.2005.02.009.
3. Montoya F, Magosch P, Scheiderer B, Lichtenberg S, Melean P, Habermeyer P. Midterm results of a total shoulder prosthesis fixed with a cementless glenoid component. J Shoulder Elbow Surg. 2013;22(5):628-635. doi:10.1016/j.jse.2012.07.005.
4. Torchia ME, Cofield RH, Settergren CR. Total shoulder arthroplasty with the Neer prosthesis: long-term results. J Shoulder Elbow Surg. 1997;6(6):495-505.
5. Antuna SA, Sperling JW, Cofield RH, Rowland CM. Glenoid revision surgery after total shoulder arthroplasty. J Shoulder Elbow Surg. 2001;10(3):217-224. doi:10.1067/mse.2001.113961.
6. Chalmers PN, Rahman Z, Romeo AA, Nicholson GP. Early dislocation after reverse total shoulder arthroplasty. J Shoulder Elbow Surg. 2014;23(5):737-744. doi:10.1016/j.jse.2013.08.015.
7. Farng E, Zingmond D, Krenek L, Soohoo NF. Factors predicting complication rates after primary shoulder arthroplasty. J Shoulder Elbow Surg. 2011;20(4):557-563. doi:10.1016/j.jse.2010.11.005.
8. Farshad M, Grogli M, Catanzaro S, Gerber C. Revision of reversed total shoulder arthroplasty. Indications and outcome. BMC Musculoskelet Disord. 2012;13(1):160. doi:10.1186/1471-2474-13-160.
9. Fevang BT, Lie SA, Havelin LI, Skredderstuen A, Furnes O. Risk factors for revision after shoulder arthroplasty: 1,825 shoulder arthroplasties from the Norwegian Arthroplasty Register. Acta Orthop. 2009;80(1):83-91.
10. Fox TJ, Cil A, Sperling JW, Sanchez-Sotelo J, Schleck CD, Cofield RH. Survival of the glenoid component in shoulder arthroplasty. J Shoulder Elbow Surg. 2009;18(6):859-863. doi:10.1016/j.jse.2008.11.020.
11. Rasmussen JV. Outcome and risk of revision following shoulder replacement in patients with glenohumeral osteoarthritis. Acta Orthop Suppl. 2014;85(355 suppl):1-23. doi:10.3109/17453674.2014.922007.
12. Rasmussen JV, Polk A, Brorson S, Sorensen AK, Olsen BS. Patient-reported outcome and risk of revision after shoulder replacement for osteoarthritis. 1,209 cases from the Danish Shoulder Arthroplasty Registry, 2006-2010. Acta Orthop. 2014;85(2):117-122. doi:10.3109/17453674.2014.893497.
13. Sajadi KR, Kwon YW, Zuckerman JD. Revision shoulder arthroplasty: an analysis of indications and outcomes. J Shoulder Elbow Surg. 2010;19(2):308-313. doi:10.1016/j.jse.2009.05.016.
14. Singh JA, Sperling JW, Cofield RH. Revision surgery following total shoulder arthroplasty: analysis of 2588 shoulders over three decades (1976 to 2008). J Bone Joint Surg Br. 2011;93(11):1513-1517. doi:10.1302/0301-620X.93B11.26938.
15. Levine WN, Djurasovic M, Glasson JM, Pollock RG, Flatow EL, Bigliani LU. Hemiarthroplasty for glenohumeral osteoarthritis: results correlated to degree of glenoid wear. J Shoulder Elbow Surg. 1997;6(5):449-454.
16. Levine WN, Fischer CR, Nguyen D, Flatow EL, Ahmad CS, Bigliani LU. Long-term follow-up of shoulder hemiarthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2012;94(22):e164. doi:10.2106/JBJS.K.00603.
17. Lynch JR, Franta AK, Montgomery WH, Lenters TR, Mounce D, Matsen FA. Self-assessed outcome at two to four years after shoulder hemiarthroplasty with concentric glenoid reaming. J Bone Joint Surg Am. 2007;89(6):1284-1292. doi:10.2106/JBJS.E.00942.
18. Iannotti JP, Norris TR. Influence of preoperative factors on outcome of shoulder arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2003;85-A(2):251-258.
19. Sperling JW, Cofield RH, Rowland CM. Neer hemiarthroplasty and Neer total shoulder arthroplasty in patients fifty years old or less. Long-term results. J Bone Joint Surg Am. 1998;80(4):464-473.
20. Strauss EJ, Roche C, Flurin PH, Wright T, Zuckerman JD. The glenoid in shoulder arthroplasty. J Shoulder Elbow Surg. 2009;18(5):819-833. doi:10.1016/j.jse.2009.05.008.
21. Cil A, Sperling JW, Cofield RH. Nonstandard glenoid components for bone deficiencies in shoulder arthroplasty. J Shoulder Elbow Surg. 2014;23(7):e149-e157. doi:10.1016/j.jse.2013.09.023.
22. Denard PJ, Walch G. Current concepts in the surgical management of primary glenohumeral arthritis with a biconcave glenoid. J Shoulder Elbow Surg. 2013;22(11):1589-1598. doi:10.1016/j.jse.2013.06.017.
23. Gunther SB, Lynch TL. Total shoulder replacement surgery with custom glenoid implants for severe bone deficiency. J Shoulder Elbow Surg. 2012;21(5):675-684. doi:10.1016/j.jse.2011.03.023.
24. Neer CS, Morrison DS. Glenoid bone-grafting in total shoulder arthroplasty. J Bone Joint Surg Am. 1988;70(8):1154-1162.
25. Steinmann SP, Cofield RH. Bone grafting for glenoid deficiency in total shoulder replacement. J Shoulder Elbow Surg. 2000;9(5):361-367. doi:10.1067/mse.2000.106921.
26. Iannotti JP, Frangiamore SJ. Fate of large structural allograft for treatment of severe uncontained glenoid bone deficiency. J Shoulder Elbow Surg. 2012:21(6):765-771. doi:10.1016/j.jse.2011.08.069.
27. Hill JM, Norris TR. Long-term results of total shoulder arthroplasty following bone-grafting of the glenoid. J Bone Joint Surg Am. 2001;83-A(6):877-883.
28. Hsu JE, Ricchetti ET, Huffman GR, Iannotti JP, Glaser DL. Addressing glenoid bone deficiency and asymptomatic posterior erosion in shoulder arthroplasty. J Shoulder Elbow Surg. 2013;22(9):1298-1308.
29. Chammaa R, Uri O, Lambert S. Primary shoulder arthroplasty using a custom-made hip-inspired implant for the treatment of advanced glenohumeral arthritis in the presence of severe glenoid bone loss. J Shoulder Elbow Surg. 2017;26(1):101-107. doi:10.1016/j.jse.2016.05.027.
30. Stoffelen DV, Eraly K, Debeer P. The use of 3D printing technology in reconstruction of a severe glenoid defect: a case report with 2.5 years of follow-up. J Shoulder Elbow Surg. 2015;24(8):e218-e222. doi:10.1016/j.jse.2015.04.006.
31. Gunther SB, Lynch TL. Total shoulder replacement surgery with custom glenoid implants for severe bone deficiency. J Shoulder Elbow Surg. 2012;21(5):675-684. doi:10.1016/j.jse.2011.03.023.
32. Dines DM, Gulotta L, Craig EV, Dines JS. Novel solution for massive glenoid defects in shoulder arthroplasty: a patient-specific glenoid vault reconstruction system. Am J Orthop. 2017;46(2):104-108.
TAKE-HOME POINTS
- With the increasing number of arthroplasties performed, and the expanding indication for shoulder arthroplasty, the number of revision shoulder arthroplasties is also increasing.
- Complex glenoid bone defects are sometimes encountered in revision shoulder arthroplasties.
- Glenoid reconstructions with bone graft have reported encouraging short- to mid-term results, but the high number of complications is a concern.
- Using the CAD/CAM technology patient-specific glenoid components have been created to reconstruct the glenoid vault in cases of severe glenoid bone loss.
- Short-term clinical and radiographic results of patient-specific glenoid components are encouraging, however longer-term follow-up are needed to confirm the efficacy of this type of reconstruction.

Treatment of Biliary Tract Cancers
Introduction
Biliary tract carcinoma (BTC) is th
Epidemiology
In the United States, BTC is rare and accounts for approximately 4% of all gastrointestinal malignancies, with an estimated 6000 to 7000 cases of carcinoma of the gallbladder and 3000 to 4000 cases of carcinoma of the bile duct diagnosed annually.4 Among women, there is a 26-fold variation in BTC mortality worldwide, ranging from 0.8 deaths per 100,000 in South Africa to 21.2 per 100,000 in Chile.1,5 Interestingly, for American Indians in New Mexico, gallbladder cancer mortality rates (8.9 per 100,000) surpass those for breast and pancreatic cancers.6 The incidence of anatomical cholangiocarcinoma subtypes also varies regionally, reflecting disparities in genetic and environmental predisposing factors.2,7 In a large, single-center study in the United States, intrahepatic cholangiocarcinoma accounted for less than 10% of cases, perihilar accounted for 50%, and distal accounted for the remaining 40%.8 Importantly, intrahepatic cholangiocarcinoma is the second most common primary malignancy of the liver, and its incidence seems to be rising in many western countries. In the United States, there has been an estimated 128% rise over the past 40 years.4,9
BTC is associated with high mortality rates.10 Median overall survival (OS) for cholangiocarcinoma is 20 to 28 months and 5-year survival is around 25%.10 Most cholangiocarcinomas are diagnosed at advanced stages with unresectable tumors.10 Furthermore, outcomes following resection with curative intent are poor—median disease-free survival (DFS) of 12 to 36 months has been reported.11,12 Patients with intrahepatic disease have a better prognosis when compared with patients who have extrahepatic tumors.12 Gallbladder cancer, likewise, carries a poor overall prognosis; median OS is 32 months and 5-year survival is as low as 13%.6
Risk factors for BTC include intrinsic and extrinsic elements.6 Incidence of BTC increases with age, and diagnosis typically occurs in the sixth to eighth decade of life.5,6,13 In contrast to gallbladder cancer, the incidence of cholangiocarcinoma is slightly higher in men.9 Obesity, diabetes, and consumption of sweetened drinks also increase the risk for BTC.14–16 Cholelithiasis is the most prevalent risk factor for gallbladder cancer, and the risk is greater for larger stones.5 Around 1 in 5 patients with porcelain gallbladder will develop gallbladder carcinoma.17 Primary sclerosing cholangitis (PSC), chronic calculi of the bile duct, choledochal cysts, cirrhosis, hepatitis C, and liver fluke infections are well established risk factors for cholangiocarcinoma.7,12,18 PSC is one of the best described entities among these predisposing conditions. Lifetime prevalence of cholangiocarcinoma among patients with PSC ranges from 5% to 10%.18,19 These patients also present at a younger age; in one series, the median age at diagnosis for BTC arising from PSC was 39 years.18 It is important to recognize, however, that in most patients diagnosed with cholangiocarcinoma, no predisposing factors are identified.8
Diagnosis
Clinical Presentation
Clinical presentation of BTC depends upon anatomic location.20 Patients with early invasive gallbladder cancer are most often asymptomatic.21 When symptoms occur, they may be nonspecific and mimic cholelithiasis.21 The most common clinical presentations include jaundice, weight loss, and abdominal pain.21 Prior to widespread availability of imaging studies, the preoperative diagnosis rate for gallbladder cancer was as low as 10%.22 However, the accuracy of computed tomography (CT) has changed this scenario, with sensitivity ranging from 73% to 87% and specificity from 88% to 100%.21 As a result of its silent clinical character, cholangiocarcinoma is frequently difficult to diagnose.23 Perihilar and distal cholangiocarcinoma characteristically present with signs of biliary obstruction, and imaging and laboratory data can corroborate the presence of cholestasis.24 On examination, patients with extrahepatic cholangiocarcinoma may present with jaundice, hepatomegaly, and a palpable right upper quadrant mass.25 A palpable gallbladder (Courvoisier sign) can also be present.25 Intrahepatic cholangiocarcinoma presents differently, and patients are less likely to be jaundiced.23 Typical clinical features are nonspecific and include dull right upper quadrant pain, weight loss, and an elevated alkaline phosphatase level.23 Alternatively, asymptomatic patients can present with incidentally detected lesions, when imaging is obtained as part of the workup for other causes or during screening for hepatocellular carcinoma in patients with viral hepatitis or cirrhosis.23,26 Uncommonly, BTC patients present because of signs or symptoms related to metastatic disease or evidence of metastatic disease on imaging.
Pathology and Grading
The majority of BTCs are adenocarcinomas, corresponding to 90% of cholangiocarcinomas and 99% of gallbladder cancers.27,28 They are graded as well, moderately, or poorly differentiated.2 Adenosquamous and squamous cell carcinoma are responsible for most of the remaining cases.2,29 Cholangiocarcinomas are divided into 3 types, defined by the Liver Cancer Study Group of Japan: (1) mass-forming, (2) periductal-infiltrating, and (3) intraductal-growing.30,31 Mass-forming intrahepatic cholangiocarcinomas are characterized morphologically by a homogeneous gray-yellow mass with frequent satellite nodules and irregular but well-defined margins.17,30 Central necrosis and fibrosis are also common.30 In the periductal-infiltrating type, tumor typically grows along the bile duct wall without mass formation, resulting in concentric mural thickening and proximal biliary dilation.30 Intraductal-growing papillary cholangiocarcinoma is characterized by the presence of intraluminal papillary or tubular polypoid tumors of the intra- or extrahepatic bile ducts, with partial obstruction and proximal biliary dilation.30
Cholangiocarcinoma
Case Presentation
A previously healthy 59-year-old man presents to his primary care physician with a 3-month history of dull right upper quadrant pain associated with weight loss. The patient is markedly cachectic and abdominal examination reveals upper quadrant tenderness. Laboratory exams are significant for elevated alkaline phosphatase (500 U/L; reference range 45–115 U/L), cancer antigen 19-9 (CA 19-9, 73 U/mL; reference range ≤ 37 U/mL), and carcinoembryonic antigen (CEA , 20 ng/mL; reference range for nonsmokers ≤ 3.0 ng/mL). Aspartate aminotransferase, alanine aminotransferase, total bilirubin, and coagulation studies are within normal range. Ultrasound demonstrates a homogeneous mass with irregular borders in the right lobe of the liver. Triphasic contrast-enhanced CT scan demonstrates a tumor with ragged rim enhancement at the periphery, and portal venous phase shows gradual centripetal enhancement of the tumor with capsular retraction. No abdominal lymph nodes or extrahepatic tumors are noted (Figure 1, Image A).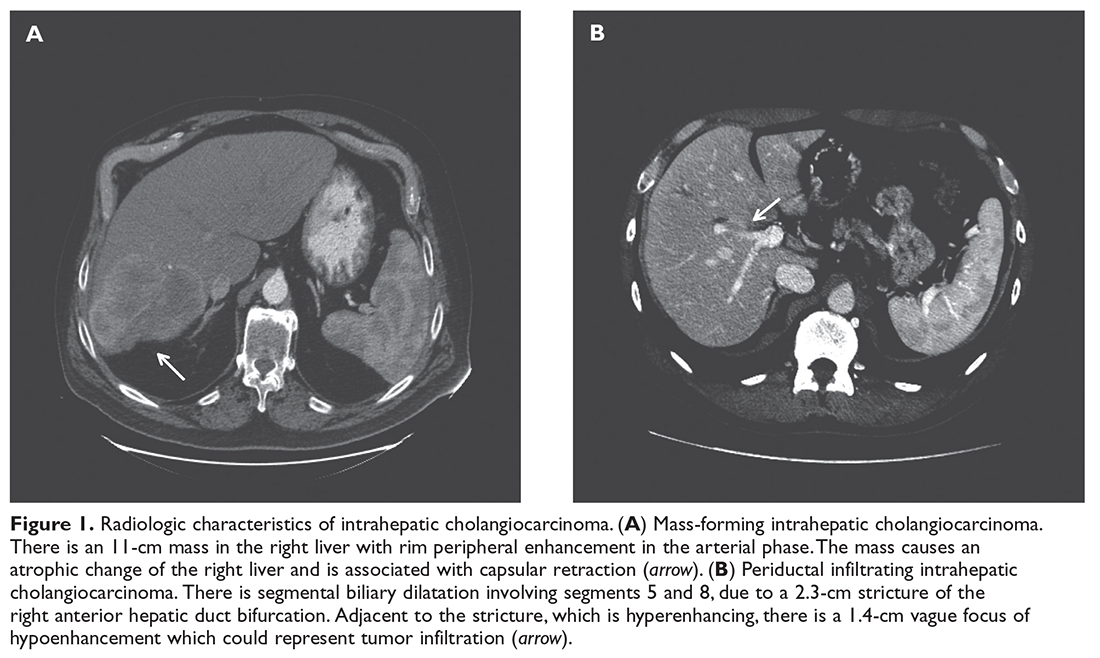
- What are the next diagnostic steps?
The most critical differential diagnosis of solid liver mass in patients without cirrhosis is cholangiocarcinoma and metastases from another primary site.32 Alternatively, when an intrahepatic lesion is noted on an imaging study in the setting of cirrhosis, the next diagnostic step is differentiation between cholangiocarcinoma and hepatocellular carcinoma (HCC).32 Triphasic contrast-enhanced CT and dynamic magnetic resonance imaging (MRI) are key diagnostic procedures.32,33 In the appropriate setting, classical imaging features in the arterial phase with washout in portal venous or delayed phase can be diagnostic of HCC and may obviate the need for a biopsy (Figure 2). 
Typical radiographic features of cholangiocarcinoma include a hypodense hepatic lesion that can be either well-defined or infiltrative and is frequently associated with biliary dilatation (Figure 1, Image A).33 The dense fibrotic nature of the tumor may cause capsular retraction, which is seen in up to 20% of cases.17 This finding is highly suggestive of cholangiocarcinoma and is rarely present in HCC.33 Following contrast administration, there is peripheral (rim) enhancement throughout both arterial and venous phases.32–34 However, these classic features were present in only 70% of cases in one study.35 Although intrahepatic cholangiocarcinomas are most commonly hypovascular, small mass-forming intrahepatic cholangiocarcinomas can often be arterially hyperenhancing and mimic HCC.33 Tumor enhancement on delayed CT imaging has been correlated with survival. Asayama et al demonstrated that tumors that exhibited delayed enhancement on CT in more than two-thirds of their volume were associated with a worse prognosis.36
Patients without cirrhosis who present with a localized lesion of the liver should undergo extensive evaluation for a primary cancer site.37 CT of the chest, abdomen, and pelvis with contrast should be obtained.37 Additionally, mammogram and endoscopic evaluation with esophagogastroduodenoscopy (EGD) and colonoscopy should be included in the work-up.37
Preoperative tumor markers are also included in the work-up. All patients with a solid liver lesion should have serum alpha-fetoprotein (AFP) levels checked. AFP is a serum glycoprotein recognized as a marker for HCC and is reported to detect preclinical HCC.38 However, serum concentrations are normal in up to 40% of small HCCs.38 Although no specific marker for cholangiocarcinoma has yet been identified, the presence of certain tumor markers in the serum of patients may be of diagnostic value, especially in patients with PSC. CA 19-9 and CEA are the best studied. Elevated levels of CA 19-9 prior to treatment are associated with a poorer prognosis, and CA 19-9 concentrations greater than 1000 U/mL are consistent with advanced disease.39,40 One large series evaluated the diagnostic value of serum CEA levels in 333 patients with PSC, 13% of whom were diagnosed with cholangiocarcinoma.34 A serum CEA level greater than 5.2 ng/mL had a sensitivity of 68.0% and specificity of 81.5%.38
If a biopsy is obtained, appropriate immunohistochemistry (IHC) can facilitate the diagnosis. BTC is strongly positive for CK-7 and CK-19.41 CK-7 positivity is not specific and is also common among metastatic cancers of the lung and breast; therefore, in some cases cholangiocarcinoma may be a diagnosis of exclusion. Immunostaining for monoclonal CEA is diffusely positive in up to 75% of cases.41 An IHC panel consisting of Hep Par-1, arginase-1, monoclonal CEA, CK-7, CK-20, TTF-1, MOC-31, and CDX-2 has been proposed to optimize the differential diagnosis of HCC, metastatic adenocarcinoma, and cholangiocarcinoma.41
Case Continued
CT of the chest, abdomen, and pelvis reveals no concerns for metastasis and no evidence of primary cancer elsewhere. EGD and colonoscopy are clear. AFP levels are within normal limits (2 ng/mL). Biopsy is performed and demonstrates adenocarcinoma. IHC studies demonstrate cells positive for monoclonal CEA, CK-7, CK-19, and MOC-31, and negative for Napsin A, TTF-1, and CK-20.
- How is cholangiocarcinoma staged and classified?
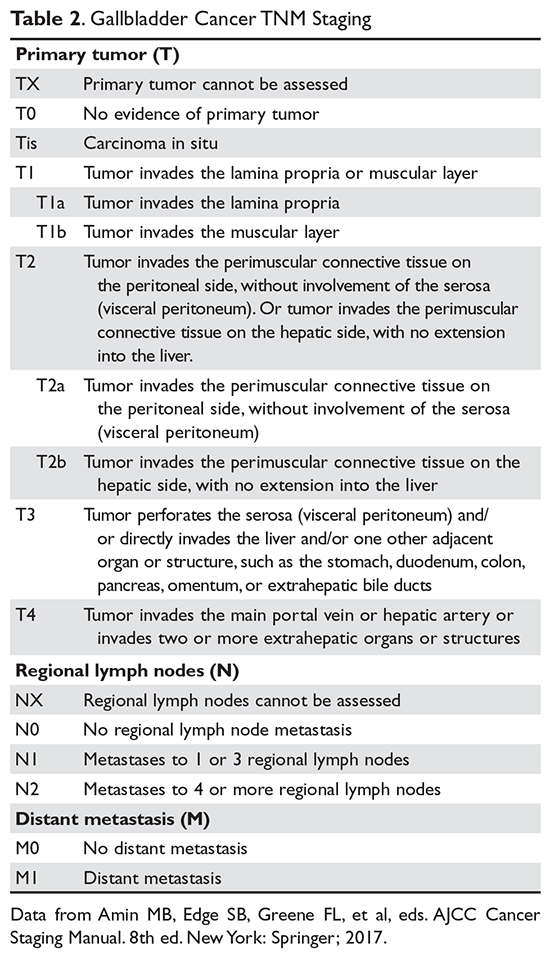
The purpose of the staging system is to provide information on prognosis and guidance for therapy. Prognostic factors and the therapeutic approaches for BTC differ depending upon their location in the biliary tree. Accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated (Table 1 and Table 2).23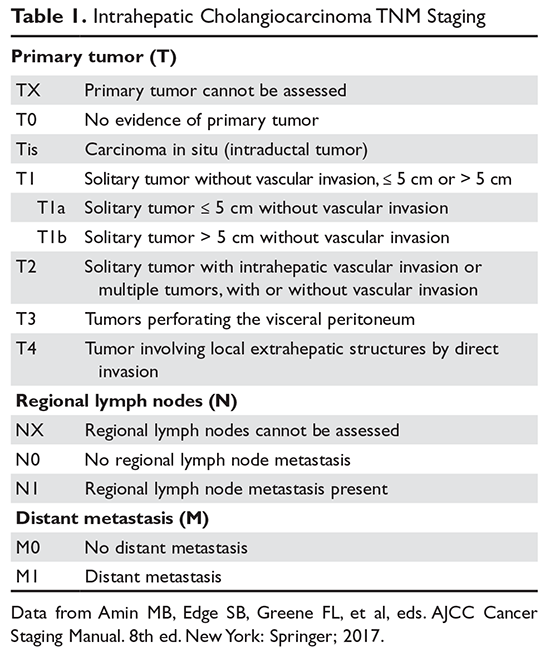
The Bismuth-Corlette classification is used to further classify perihilar cholangiocarcinoma according to patterns of hepatic duct involvement. Type I tumors are located below the confluence of the left and right hepatic ducts.42 Type II reach the confluence of the hepatic ducts.42 Type III occlude the common hepatic duct and either the right or left hepatic duct (IIIa and IIIb, respectively).42 Finally, type IV are multicentric, or involve the confluence and both the right and left hepatic ducts.42 Tumors that involve the common hepatic duct bifurcation are named Klatskin tumors.42
- What is the first-line treatment for localized cholangiocarcinomas?
Surgical resection is the only potentially curative treatment for localized cholangiocarcinoma, although fewer than 20% of patients are suitable for curative treatment, due to the presence of advanced disease at diagnosis.43,44 Available evidence supports the recommendation that resection with negative margins, regardless of extent, should be the goal of therapy for patients with potentially resectable disease.44 Extensive hepatic resections are often necessary to achieve clear margins since the majority of patients present with large masses. Substantial evidence corroborates that R0 resection is associated with better survival, whereas the benefit of wide compared to narrow (< 5–10 mm) margins is unclear.45 A recent analysis of 96 patients suggests that the proximal resection margin has more prognostic implications than distal margins.45
Surgical options and resectability criteria depend upon tumor location. Extent of tumor in the bile duct is one of the most important factors that determine resectability.17 Although multifocal liver tumors (including satellite lesions), lymph node metastases to the porta hepatis, and distant metastases are considered relative contraindications to surgery, surgical approaches can be considered in selected patients.43 Patient selection for surgery is facilitated by careful preoperative staging, which may include laparoscopy. Laparoscopic staging prior to resection may prevent unnecessary laparotomy in 30% to 45% of patients.42,46
- Is there a role for adjuvant treatment?
Recurrence following complete resection is a primary limitation for cure in BTC, which provides a rationale for the use of adjuvant therapy.47,48 In a sample of 79 patients with extrahepatic cholangiocarcinoma who underwent curative resection, the cumulative recurrence rate after 4 years was 56%.47 Initial recurrence at a distant site occurs in 40% to 50% of patients.48
Lymphovascular and perineural invasion, lymph node metastasis, and tumor size ≥ 5 cm have been reported as independent predictors of recurrence and mortality following resection.49 A 2017 meta-analysis which included 30 studies involving more than 22,499 patients reported a 41% reduction in the risk of death with adjuvant chemotherapy, which translated to a mean OS benefit of 4 months in an unselected population.49 Moreover, this study revealed inferior OS in patients given adjuvant radiation therapy (RT) in combination with chemotherapy.49 These results are in line with the previous meta-analysis by Horgan et al, which demonstrated that adjuvant RT seems to benefit only patients with R1 resections, with a possible detrimental effect in R0 disease.50 Therefore, adjuvant chemoradiation cannot be viewed as a standard practice following R0 resection, and should be reserved for those patients with positive margins (R1/ 2) to reduce local progression.
In the phase 3 BILCAP trial presented at ASCO 2017, 447 patients with completely resected cholangiocarcinoma or gallbladder cancer with adequate biliary drainage and Eastern Cooperative Oncology Group (ECOG) performance score ≤ 2 were randomly assigned to observation or capecitabine (1250 mg/m2 twice daily for days 1–14 every 21 days for 8 cycles).51 Surgical treatment achieved R0 resection in 62% of patients and 46% were node-negative. Median OS was 51 months for the capecitabine group and 36 months for the control arm (hazard ratio [HR] 0.80, 95% CI 0.63 to 1.04, P = 0.097). Analyses with adjustment for nodal status, grade of disease, and gender indicated a HR of 0.71 (P < 0.01). Median DFS was 25 months versus 18 months favoring the capecitabine group, and rates of grade 3 or 4 toxicity were less than anticipated. Following the results of this trial, adjuvant capecitabine should become the new standard of care.
- What is the treatment for locally advanced cholangiocarcinoma?
The optimal approach to patients with locally advanced unresectable cholangiocarcinoma has not been established. The prognosis for patients with either locally unresectable or locally recurrent disease is typically measured in months. Goals of palliative therapy are relief of symptoms and improvement in quality of life, and there is no role for surgical debulking.
Liver transplantation is a potentially curative option for selected patients with hilar or intrahepatic cholangiocarcinoma. Patients with lymph node-negative, non-disseminated, locally advanced hilar cholangiocarcinomas have 5-year survival rates ranging from 25% to 42% following transplantation.52 Retrospective data suggests that neoadjuvant chemoradiation followed by liver transplantation is highly effective for selected patients with hilar cholangiocarcinoma.52 However, these results require confirmation from prospective clinical evidence. It is important to recognize that liver transplantation plays no role in the management of distal cholangiocarcinoma or gallbladder cancer.
Rarely, patients with borderline resectable intrahepatic cholangiocarcinoma will have a sufficient response to chemotherapy to permit later resection, and, in such cases, starting with chemotherapy and then restaging to evaluate resectability is appropriate.54 A single-center, retrospective analysis including 186 patients by Le Roy et al evaluated survival in patients with locally advanced, unresectable intrahepatic cholangiocarcinoma who received primary chemotherapy, followed by surgery in those with secondary resectability.54 After a median of 6 cycles of chemotherapy, 53% of patients achieved resectability and underwent surgery with curative intent. These patients had similar short- and long-term results compared to patients with initially resectable intrahepatic cholangiocarcinoma who had surgery alone, with median OS reaching 24 months.54
Ablative radiotherapy is an additional option for localized inoperable intrahepatic cholangiocarcinoma. Tao and colleagues evaluated 79 consecutive patients with inoperable intrahepatic cholangiocarcinoma treated with definitive RT.55 Median tumor size was 7.9 cm and 89% of patients received chemotherapy before RT. Median OS was 30 months and 3-year OS was 44%. Radiation dose was the single most important prognostic factor, and higher doses correlated with improved local control and OS. A biologic equivalent dose (BED) greater than 80.5 Gy was identified as an ablative dose of RT for large intrahepatic cholangiocarcinomas. The 3-year OS for patients receiving BED greater than 80.5 Gy was 73% versus 38% for those receiving lower doses.
Case Continued
The patient is deemed to have resectable disease and undergoes surgical resection followed by adjuvant capecitabine for 8 cycles. Unfortunately, after 1 year, follow-up imaging identifies bilateral enlarging lung nodules. Biopsy is performed and confirms metastatic cholangiocarcinoma.
- What is the treatment for metastatic BTC?
The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. A benefit of chemotherapy over best supportive care for cholangiocarcinoma was demonstrated in an early phase 3 trial that randomly assigned 90 patients with advanced pancreatic or biliary cancer (37 with bile duct cancer) to receive either fluorouracil (FU) -based systemic chemotherapy or best supportive care. Results showed that chemotherapy significantly improved OS (6 months versus 2.5 months).56 Chemotherapy is also beneficial for patients with unresectable gallbladder cancer. In a single-center randomized study including 81 patients with unresectable gallbladder cancer, gemcitabine and oxaliplatin (GEMOX) improved progression-free survival (PFS) and OS compared to best supportive care.57 Treatment for metastatic cholangiocarcinoma and gallbladder cancer follows the same algorithm.
In 2010, cisplatin plus gemcitabine was established as a reference regimen for first-line therapy by the ABC-02 study, in which 410 patients with locally advanced or metastatic bile duct, gallbladder, or ampullary cancer were randomly assigned to 6 courses of cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2 on days 1 and 8, every 21 days) or gemcitabine alone (1000 mg/m2 days 1, 8, 15, every 28 days).58 OS was significantly greater with combination therapy (11.7 versus 8.1 months), and PFS also favored the combination arm (8 versus 5 months). Toxicity was comparable in both groups, with the exception of significantly higher rates of grade 3 or 4 neutropenia with gemcitabine plus cisplatin (25% versus 17%), and higher rates of grade 3 or 4 abnormal liver function with gemcitabine alone (27% versus 17%). Most quality-of-life scales showed a trend favoring combined therapy.58 A smaller, identically designed Japanese phase 3 randomized trial achieved similar results, demonstrating greater OS with cisplatin plus gemcitabine compared to gemcitabine alone (11.2 versus 7.7 months).59
The gemcitabine plus cisplatin combination has not been directly compared with other gemcitabine combinations in phase 3 trials. A pooled analysis of 104 trials of a variety of chemotherapy regimens in advanced biliary cancer concluded that the gemcitabine plus cisplatin regimen offered the highest rates of objective response and tumor control compared with either gemcitabine-free or cisplatin-free regimens.60 However, this did not translate into significant benefit in terms of either time to tumor progression or median OS. It is important to note that this analysis did not include results of the subsequent ABC-02 trial.
There is no standard treatment for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. There are no completed prospective phase 3 trials supporting the use of second-line chemotherapy after failure of first-line chemotherapy in BTC, and the selection of candidates for second-line therapy as well as the optimal regimen are not established.61 The ongoing phase 2 multicenter ABC-06 trial is evaluating oxaliplatin plus short-term infusional FU and leucovorin (FOLFOX) versus best supportive care for second-line therapy. In a systematic review including 23 studies (14 phase 2 clinical trials and 9 retrospective studies) with 761 patients with BTC, the median OS was 7.2 months.
The optimal selection of candidates for second-line chemotherapy is not established. Two independent studies suggest that patients who have a good performance status (0 or 1), disease control with the first-line chemotherapy, low CA 19-9 level, and possibly previous surgery on their primary tumor, have the longest survival with second-line chemotherapy. However, whether these characteristics predict for chemotherapy responsiveness or more favorable biologic behavior is not clear.62,63 No particular regimen has proved superior to any other, and the choice of second-line regimen remains empiric.
For patients with adequate performance status, examples of other conventional chemotherapy regimens with demonstrated activity that could be considered for second-line therapy include: FOLFOX or capecitabine, gemcitabine plus capecitabine, capecitabine plus cisplatin, or irinotecan plus short-term infusional FU and leucovorin (FOLFIRI) with or without bevacizumab.64 For selected patients, second-line molecularly targeted therapy using erlotinib plus bevacizumab may be considered. However, this regimen is very costly.64 Examples of other regimens with demonstrated activity in phase 2 trials include GEMOX, gemcitabine plus fluoropyrimidine, and fluoropyrimidine plus oxaliplatin or cisplatin.64
There is promising data from studies of targeted therapy for specific molecular subgroups. A recent phase 2 trial evaluated the activity of BGJ398, an orally bioavailable, selective, ATP-competitive pan inhibitor of human fibroblast growth factor receptor (FGFR) kinase, in patients with FGFR-altered advanced cholangiocarcinoma.65 The overall response rate was 14.8% (18.8% FGFR2 fusions only) and disease control rate was 75.4% (83.3% FGFR2 fusions only). All responsive tumors contained FGFR2 fusions. Adverse events were manageable, and grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%). Those included hyperphosphatemia, stomatitis, and palmar-plantar erythrodysesthesia. Javle and colleagues also identified HER2/neu blockade as a promising treatment strategy for gallbladder cancer patients with this gene amplification.66 This retrospective analysis included 9 patients with gallbladder cancer and 5 patients with cholangiocarcinoma who received HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab). In the gallbladder cancer group, HER2/neu gene amplification or overexpression was detected in 8 cases. These patients experienced disease stability (n = 3), partial response (n = 4), or complete response (n = 1) with HER2/neu–directed therapy. Median duration of response was 40 weeks. The cholangiocarcinoma cases treated in this series had no radiological responses despite HER2/neu mutations or amplification.
Gallbladder Cancer
Case Presentation
A 57-year-old woman from Chile presents with a 3-week history of progressive right upper quadrant abdominal pain. She denies nausea, vomiting, dysphagia, odynophagia, alterations in bowel habits, fever, or jaundice. Her past medical history is significant for obesity and hypertension. She has no history of smoking, alcohol, or illicit drug use. Laboratory studies show marked leukocytosis (23,800/µL) with neutrophilia (91%). Liver function test results are within normal limits. Ultrasound of the abdomen reveals gallbladder wall thickening and cholelithiasis.
The patient undergoes an uneventful laparoscopic cholecystectomy and is discharged from the hospital after 48 hours. Pathology report reveals a moderately differentiated adenocarcinoma of the gallbladder invading the perimuscular connective tissue (T2). No lymph nodes are identified in the specimen.
- What is the appropriate surgical management of gallbladder cancer?
Gallbladder cancer can be diagnosed preoperatively or can be found incidentally by intraoperative or pathological findings. In one large series, gallbladder cancer was incidentally found during 0.25% of laparoscopic cholecystectomies.67
For patients who are diagnosed with previously unsuspected gallbladder cancer by pathology findings, the extent of tumor invasion (T stage) indicates the need for re-resection (Figure 3).64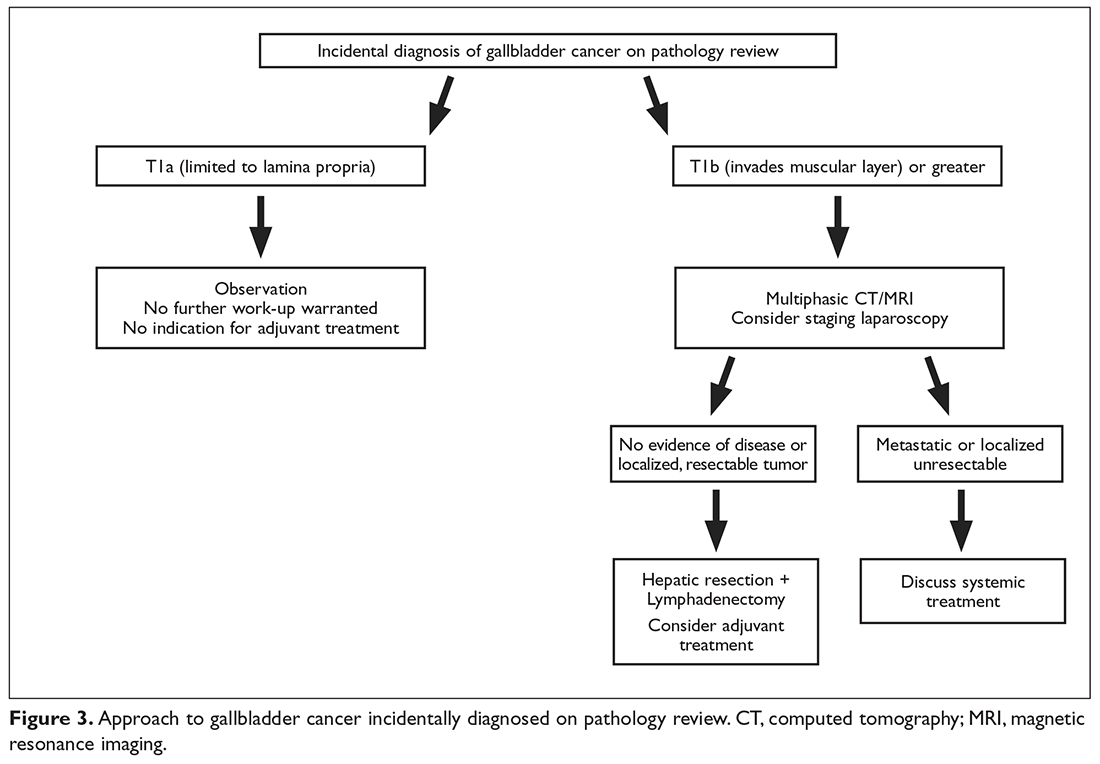
Alternatively, when gallbladder cancer is documented or suspected preoperatively, adequate imaging is important to identify patients with absolute contraindications to resection. Contraindications to surgery include metastasis, extensive involvement of the hepatoduodenal ligament, encasement of major vessels, and involvement of celiac, peripancreatic, periduodenal, or superior mesenteric nodes.72 Notwithstanding, retrospective series suggest individual patients may benefit, and surgical indications in advanced disease should be determined on an individual basis.73 Staging imaging should be obtained using multiphasic contrast-enhanced CT or MRI of the chest, abdomen, and pelvis. PET-scan can be used in selected cases where metastatic disease is suspected.64 Laparoscopic diagnostic staging should be considered prior to resection.64 This procedure can identify previously unknown contraindications to tumor resection in as much as 23% of patients, and the yield is significantly higher in locally advanced tumors.73
Patients with a diagnosis of potentially resectable, localized gallbladder cancer should be offered definitive surgery. Extended cholecystectomy is recommended for patients stage T2 or above. This procedure involves wedge resection of the gallbladder bed or a segmentectomy IVb/V and lymph node dissection, which should include the cystic duct, common bile duct, posterior superior pancreaticoduodenal lymph nodes, and those around the hepatoduodenal ligament.72 Bile duct excision should be performed if there is malignant involvement.64
Conclusion
BTCs are anatomically and clinically heterogeneous tumors. Prognostic factors and therapeutic approaches for BTCs differ depending upon their location in the biliary tree and, accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated. Surgical resection is the only potentially curative treatment for localized BTC. However, recurrence following complete resection is a primary limitation for cure, which provides a rationale for the use of adjuvant therapy. The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. Multiple randomized clinical trials have demonstrated a benefit of chemotherapy for metastatic disease. For patients with adequate performance status, second-line therapy can be considered, and data from studies that evaluated targeted therapy for specific molecular subgroups is promising.
1. Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2(Suppl 1):e000152.
2. Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2017 Oct 10. doi: 10.1038/nrclinonc.2017.157.
3. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist 2008;13:415–23.
4. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2014 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2017.
5. Torre LA, Siegel RL, Islami F, et al. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol 2017 Aug 18. doi: 10.1016/j.cgh.2017.08.017.
6. Lau CSM, Zywot A, Mahendraraj K, Chamberlain CS. Gallbladder carcinoma in the United States: a population based clinical outcomes study involving 22,343 patients from the Surveillance, Epidemiology, and End Result Database (1973–2013). HPB Surg 2017;2017:1532835. doi:10.1155/2017/1532835.
7. Hughes T, O’Connor T, Techasen A, et al. Opisthorchiasis and cholangiocarcinoma in Southeast Asia: an unresolved problem. Int J Gen Med 2017;10:227–37.
8. DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–62.
9. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9.
10. Yao KJ, Jabbour S, Parekh N, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117.
11. Choi SB, Kim KS, Choi JY, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol 2009;16:3048–56.
12. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96.
13. Duffy A, Capanu M, Abou-Alfa GK, et al. Gallbladder cancer (GBC): 10-year experience at Memorial Sloan-Kettering Cancer Centre (MSKCC). J Surg Oncol 2008;98:485–9.
14. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer — viewpoint of the IARC Working Group. N Engl J Med 2016;375:794–8.
15. Chen J, Han Y, Xu C, et al. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases. Eur J Cancer Prev 2015;24:89–99.
16. Larsson SC, Giovannucci EL, Wolk A. Sweetened beverage consumption and risk of biliary tract and gallbladder cancer in a prospective study. J Natl Cancer Inst 2016;108: doi: 10.1093/jnci/djw125.
17. Gore RM. Biliary tract neoplasms: diagnosis and staging. Cancer Imaging 2007;7(Special Issue A):S15–23.
18. Broome U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610–5.
19. Burak K, Angulo P, Pasha T, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523–6.
20. Rodrigues J, Diehl DL. Cholangiocarcinoma: clinical manifestations and diagnosis. Tech Gastrointest Endosc 2016;18:75–82.
21. Mitchell CH, Johnson PT, Fishman EK, et al. Features suggestive of gallbladder malignancy. J Comput Assist Tomogr 2014;38:235–41.
22. Beltz WR, Condon RE. Primary carcinoma of the gallbladder. Ann Surg 1974;180:180–4.
23. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512–22.
24. Patel T. Cholangiocarcinoma—controversies and challenges. Nat Rev Gastroenterol Hepatol 2011;8:189–200.
25. Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 1996;224:463–73.
26. Bartella I, Dufour JF. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J Gastrointestin Liver Dis 2015;24:481-9.
27. Yamaguchi K, Enjoji M. Carcinoma of the gallbladder: a clinicopathology of 103 patients and a newly proposed staging. Cancer 1988;62:1425–32.
28. Esposito I, Schirmacher P. Pathological aspects of cholangiocarcinoma. HPB. 2008;10:83–6.
29. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol 2016;5:62.
30. Chung YE, Kim MJ, Park YN, et al. Varying appearances of cholangiocarcinoma: radiologic-pathologic correlation. Radiographics 2009;29:683–700.
31. Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg 2003;10:288–91.
32. Rao PN. Nodule in liver: investigations, differential diagnosis and follow-up. J Clin Exp Hepatol 2014;4(Suppl 3):S57–62.
33. Kim TK, Lee E, Jang HJ. Imaging findings of mimickers of hepatocellular carcinoma. Clin Mol Hepatol 2015;21:326–43.
34. Hennedige TP, Neo WT, Venkatesh SK. Imaging of malignancies of the biliary tract- an update. Cancer Imaging 2014;14:14.
35. Kim SH, Lee CH, Kim BH, et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J Comput Assist Tomogr 2012;36:704–9.
36. Asayama Y, Yoshimitsu K, Irie H, et al. Delayed-phase dynamic CT enhancement as a prognostic factor for mass-forming intrahepatic cholangiocarcinoma. Radiology 2006;238:150–5.
37. National Comprehensive Cancer Network. Cancer of unknown primary. www.nccn.org/professionals/physician_gls/pdf/bone.pdf. Accessed 1 Dec 2017.
38. Kefeli A, Basyigit S, Yeniova AO. Diagnosis of hepatocellular carcinoma. In: Abdeldayem HM, ed. Updates in liver cancer. London: InTech; 2017.
39. Bergquist JR, Ivanics T, Storlie CB, et al. Implications of CA19-9 elevation for survival, staging, and treatment sequencing in intrahepatic cholangiocarcinoma: A national cohort analysis. J Surg Oncol 2016;114:475–82.
40. Chung YJ, Choi DW, Choi SH, et al. Prognostic factors following surgical resection of distal bile duct cancer. J Korean Surg Soc 2013;85:212–8.
41. Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol 2002;33:1175–81.
42. Paul A, Kaiser GM, Molmenti EP, et al. Klatskin tumors and the accuracy of the Bismuth-Corlette classification. Am Surg 2011;77:1695–9.
43. Cannavale A, Santoni M, Gazzetti M, et al. Updated management of malignant biliary tract tumors: an illustrative review. J Vasc Interv Radiol 2016;27:1056–69.
44. Matsuo K, Rocha FG, Ito K, et al. The Blumgart preoperative staging system for hilar cholangiocarcinoma: analysis of resectability and outcomes in 380 patients. J Am Coll Surg 2012;215:343–55.
45. Yoo T, Park SJ, Han SS, et al. Proximal resection margins: more prognostic than distal resection margins in patients undergoing hilar cholangiocarcinoma resection. Cancer Res Treat 2017 Nov 16; doi.org/10.4143/crt.2017.320.
46. Joseph S, Connor S, Garden OJ. Staging laparoscopy for cholangiocarcinoma. HPB 2008;10:116–9.
47. Jarnagin WR, Ruo L, Little SA, et al. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: implications for adjuvant therapeutic strategies. Cancer 2003;98:1689–700.
48. Kobayashi A, Miwa S, Nakata T, Miyagawa S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br J Surg 2010;97:56–64.
49. Ghidini M, Tomasello G, Botticelli A, et al. Adjuvant chemotherapy for resected biliary tract cancers: a systematic review and meta-analysis. HPB 2017;19:741–8.
50. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934–40.
51. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study [abstract]. J Clin Oncol 2017 35:15_suppl:4006-4006.
52. Darwish Murad S, Kim WR, Darnois DM, et al. Efficacy of neoadjuvant chemoradiation followed by liver transplantation for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88–98.
53. Sapisochin G, Facciuto M, Rubbia-Brandt L, et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016;64:1178–88.
54. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg 2017 Aug 31. doi: 10.1002/bjs.10641.
55. Tao R, Krishnan S, Bhosale PR, et al. Ablative radiotherapy doses lead to a substantial prolongation of survival in patients with inoperable intrahepatic cholangiocarcinoma: a retrospective dose response analysis. J Clin Oncol 2016;34:219–26.
56. Glimelius B, Hoffman K, SjÓdén PO, et al. 555 Palliative chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Eur J Cancer 1995;31:S118.
57. Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol 2010;28:4581–6.
58. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
59. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469–74.
60. Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
61. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38.
62. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290–7.
63. Fornaro L, Cereda S, Aprile G, et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br J Cancer 2014;110:2165–9.
64. National Comprehensive Cancer Network. Hepatobiliary cancer. www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. Accessed 12 Nov 2017.
65. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2017 Nov 28;JCO2017755009.
66. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58.
67. Konstantinidis IT, Deshpande V, Genevay M, et al. Trends in presentation and survival for gallbladder cancer during a period of more than 4 decades: a single-institution experience. Arch Surg 2009;144:441–47.
68. Singh S, Agarwal AK. Gallbladder cancer: the role of laparoscopy and radical resection. Ann Surg 2009;250:494–5.
69. Kapoor VK, Haribhakti SP. Extended cholecystectomy for carcinoma of the gall bladder. Trop Gastroenterol 1995;16:74–5.
70. Ethun CG, Postlewait LM, Le N, et al. Association of optimal time Interval to re-resection for incidental gallbladder cancer with overall survival: a multi-Institution analysis from the US extrahepatic biliary malignancy consortium. JAMA Surg 2017;152:143–9.
71. Goetze TO, Paolucci V. Benefits of reoperation of T2 and more advanced incidental gallbladder carcinoma: analysis of the German registry. Ann Surg 2008;247:104–8.
72. Nishio H, Nagino M, Ebata T, et al. Aggressive surgery for stage IV gallbladder carcinoma; what are the contraindications? J Hepatobiliary Pancreat Surg 2007;14:351–7.
73. Agarwal AK, Kalayarasan R, Javed A, et al. The role of staging laparoscopy in primary gallbladder cancer--an analysis of 409 patients: a prospective study to evaluate the role of staging laparoscopy in the management of gallbladder cancer. Ann Surg 2013;258:318–23.
Introduction
Biliary tract carcinoma (BTC) is th
Epidemiology
In the United States, BTC is rare and accounts for approximately 4% of all gastrointestinal malignancies, with an estimated 6000 to 7000 cases of carcinoma of the gallbladder and 3000 to 4000 cases of carcinoma of the bile duct diagnosed annually.4 Among women, there is a 26-fold variation in BTC mortality worldwide, ranging from 0.8 deaths per 100,000 in South Africa to 21.2 per 100,000 in Chile.1,5 Interestingly, for American Indians in New Mexico, gallbladder cancer mortality rates (8.9 per 100,000) surpass those for breast and pancreatic cancers.6 The incidence of anatomical cholangiocarcinoma subtypes also varies regionally, reflecting disparities in genetic and environmental predisposing factors.2,7 In a large, single-center study in the United States, intrahepatic cholangiocarcinoma accounted for less than 10% of cases, perihilar accounted for 50%, and distal accounted for the remaining 40%.8 Importantly, intrahepatic cholangiocarcinoma is the second most common primary malignancy of the liver, and its incidence seems to be rising in many western countries. In the United States, there has been an estimated 128% rise over the past 40 years.4,9
BTC is associated with high mortality rates.10 Median overall survival (OS) for cholangiocarcinoma is 20 to 28 months and 5-year survival is around 25%.10 Most cholangiocarcinomas are diagnosed at advanced stages with unresectable tumors.10 Furthermore, outcomes following resection with curative intent are poor—median disease-free survival (DFS) of 12 to 36 months has been reported.11,12 Patients with intrahepatic disease have a better prognosis when compared with patients who have extrahepatic tumors.12 Gallbladder cancer, likewise, carries a poor overall prognosis; median OS is 32 months and 5-year survival is as low as 13%.6
Risk factors for BTC include intrinsic and extrinsic elements.6 Incidence of BTC increases with age, and diagnosis typically occurs in the sixth to eighth decade of life.5,6,13 In contrast to gallbladder cancer, the incidence of cholangiocarcinoma is slightly higher in men.9 Obesity, diabetes, and consumption of sweetened drinks also increase the risk for BTC.14–16 Cholelithiasis is the most prevalent risk factor for gallbladder cancer, and the risk is greater for larger stones.5 Around 1 in 5 patients with porcelain gallbladder will develop gallbladder carcinoma.17 Primary sclerosing cholangitis (PSC), chronic calculi of the bile duct, choledochal cysts, cirrhosis, hepatitis C, and liver fluke infections are well established risk factors for cholangiocarcinoma.7,12,18 PSC is one of the best described entities among these predisposing conditions. Lifetime prevalence of cholangiocarcinoma among patients with PSC ranges from 5% to 10%.18,19 These patients also present at a younger age; in one series, the median age at diagnosis for BTC arising from PSC was 39 years.18 It is important to recognize, however, that in most patients diagnosed with cholangiocarcinoma, no predisposing factors are identified.8
Diagnosis
Clinical Presentation
Clinical presentation of BTC depends upon anatomic location.20 Patients with early invasive gallbladder cancer are most often asymptomatic.21 When symptoms occur, they may be nonspecific and mimic cholelithiasis.21 The most common clinical presentations include jaundice, weight loss, and abdominal pain.21 Prior to widespread availability of imaging studies, the preoperative diagnosis rate for gallbladder cancer was as low as 10%.22 However, the accuracy of computed tomography (CT) has changed this scenario, with sensitivity ranging from 73% to 87% and specificity from 88% to 100%.21 As a result of its silent clinical character, cholangiocarcinoma is frequently difficult to diagnose.23 Perihilar and distal cholangiocarcinoma characteristically present with signs of biliary obstruction, and imaging and laboratory data can corroborate the presence of cholestasis.24 On examination, patients with extrahepatic cholangiocarcinoma may present with jaundice, hepatomegaly, and a palpable right upper quadrant mass.25 A palpable gallbladder (Courvoisier sign) can also be present.25 Intrahepatic cholangiocarcinoma presents differently, and patients are less likely to be jaundiced.23 Typical clinical features are nonspecific and include dull right upper quadrant pain, weight loss, and an elevated alkaline phosphatase level.23 Alternatively, asymptomatic patients can present with incidentally detected lesions, when imaging is obtained as part of the workup for other causes or during screening for hepatocellular carcinoma in patients with viral hepatitis or cirrhosis.23,26 Uncommonly, BTC patients present because of signs or symptoms related to metastatic disease or evidence of metastatic disease on imaging.
Pathology and Grading
The majority of BTCs are adenocarcinomas, corresponding to 90% of cholangiocarcinomas and 99% of gallbladder cancers.27,28 They are graded as well, moderately, or poorly differentiated.2 Adenosquamous and squamous cell carcinoma are responsible for most of the remaining cases.2,29 Cholangiocarcinomas are divided into 3 types, defined by the Liver Cancer Study Group of Japan: (1) mass-forming, (2) periductal-infiltrating, and (3) intraductal-growing.30,31 Mass-forming intrahepatic cholangiocarcinomas are characterized morphologically by a homogeneous gray-yellow mass with frequent satellite nodules and irregular but well-defined margins.17,30 Central necrosis and fibrosis are also common.30 In the periductal-infiltrating type, tumor typically grows along the bile duct wall without mass formation, resulting in concentric mural thickening and proximal biliary dilation.30 Intraductal-growing papillary cholangiocarcinoma is characterized by the presence of intraluminal papillary or tubular polypoid tumors of the intra- or extrahepatic bile ducts, with partial obstruction and proximal biliary dilation.30
Cholangiocarcinoma
Case Presentation
A previously healthy 59-year-old man presents to his primary care physician with a 3-month history of dull right upper quadrant pain associated with weight loss. The patient is markedly cachectic and abdominal examination reveals upper quadrant tenderness. Laboratory exams are significant for elevated alkaline phosphatase (500 U/L; reference range 45–115 U/L), cancer antigen 19-9 (CA 19-9, 73 U/mL; reference range ≤ 37 U/mL), and carcinoembryonic antigen (CEA , 20 ng/mL; reference range for nonsmokers ≤ 3.0 ng/mL). Aspartate aminotransferase, alanine aminotransferase, total bilirubin, and coagulation studies are within normal range. Ultrasound demonstrates a homogeneous mass with irregular borders in the right lobe of the liver. Triphasic contrast-enhanced CT scan demonstrates a tumor with ragged rim enhancement at the periphery, and portal venous phase shows gradual centripetal enhancement of the tumor with capsular retraction. No abdominal lymph nodes or extrahepatic tumors are noted (Figure 1, Image A).
- What are the next diagnostic steps?
The most critical differential diagnosis of solid liver mass in patients without cirrhosis is cholangiocarcinoma and metastases from another primary site.32 Alternatively, when an intrahepatic lesion is noted on an imaging study in the setting of cirrhosis, the next diagnostic step is differentiation between cholangiocarcinoma and hepatocellular carcinoma (HCC).32 Triphasic contrast-enhanced CT and dynamic magnetic resonance imaging (MRI) are key diagnostic procedures.32,33 In the appropriate setting, classical imaging features in the arterial phase with washout in portal venous or delayed phase can be diagnostic of HCC and may obviate the need for a biopsy (Figure 2).
Typical radiographic features of cholangiocarcinoma include a hypodense hepatic lesion that can be either well-defined or infiltrative and is frequently associated with biliary dilatation (Figure 1, Image A).33 The dense fibrotic nature of the tumor may cause capsular retraction, which is seen in up to 20% of cases.17 This finding is highly suggestive of cholangiocarcinoma and is rarely present in HCC.33 Following contrast administration, there is peripheral (rim) enhancement throughout both arterial and venous phases.32–34 However, these classic features were present in only 70% of cases in one study.35 Although intrahepatic cholangiocarcinomas are most commonly hypovascular, small mass-forming intrahepatic cholangiocarcinomas can often be arterially hyperenhancing and mimic HCC.33 Tumor enhancement on delayed CT imaging has been correlated with survival. Asayama et al demonstrated that tumors that exhibited delayed enhancement on CT in more than two-thirds of their volume were associated with a worse prognosis.36
Patients without cirrhosis who present with a localized lesion of the liver should undergo extensive evaluation for a primary cancer site.37 CT of the chest, abdomen, and pelvis with contrast should be obtained.37 Additionally, mammogram and endoscopic evaluation with esophagogastroduodenoscopy (EGD) and colonoscopy should be included in the work-up.37
Preoperative tumor markers are also included in the work-up. All patients with a solid liver lesion should have serum alpha-fetoprotein (AFP) levels checked. AFP is a serum glycoprotein recognized as a marker for HCC and is reported to detect preclinical HCC.38 However, serum concentrations are normal in up to 40% of small HCCs.38 Although no specific marker for cholangiocarcinoma has yet been identified, the presence of certain tumor markers in the serum of patients may be of diagnostic value, especially in patients with PSC. CA 19-9 and CEA are the best studied. Elevated levels of CA 19-9 prior to treatment are associated with a poorer prognosis, and CA 19-9 concentrations greater than 1000 U/mL are consistent with advanced disease.39,40 One large series evaluated the diagnostic value of serum CEA levels in 333 patients with PSC, 13% of whom were diagnosed with cholangiocarcinoma.34 A serum CEA level greater than 5.2 ng/mL had a sensitivity of 68.0% and specificity of 81.5%.38
If a biopsy is obtained, appropriate immunohistochemistry (IHC) can facilitate the diagnosis. BTC is strongly positive for CK-7 and CK-19.41 CK-7 positivity is not specific and is also common among metastatic cancers of the lung and breast; therefore, in some cases cholangiocarcinoma may be a diagnosis of exclusion. Immunostaining for monoclonal CEA is diffusely positive in up to 75% of cases.41 An IHC panel consisting of Hep Par-1, arginase-1, monoclonal CEA, CK-7, CK-20, TTF-1, MOC-31, and CDX-2 has been proposed to optimize the differential diagnosis of HCC, metastatic adenocarcinoma, and cholangiocarcinoma.41
Case Continued
CT of the chest, abdomen, and pelvis reveals no concerns for metastasis and no evidence of primary cancer elsewhere. EGD and colonoscopy are clear. AFP levels are within normal limits (2 ng/mL). Biopsy is performed and demonstrates adenocarcinoma. IHC studies demonstrate cells positive for monoclonal CEA, CK-7, CK-19, and MOC-31, and negative for Napsin A, TTF-1, and CK-20.
- How is cholangiocarcinoma staged and classified?
The purpose of the staging system is to provide information on prognosis and guidance for therapy. Prognostic factors and the therapeutic approaches for BTC differ depending upon their location in the biliary tree. Accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated (Table 1 and Table 2).23
The Bismuth-Corlette classification is used to further classify perihilar cholangiocarcinoma according to patterns of hepatic duct involvement. Type I tumors are located below the confluence of the left and right hepatic ducts.42 Type II reach the confluence of the hepatic ducts.42 Type III occlude the common hepatic duct and either the right or left hepatic duct (IIIa and IIIb, respectively).42 Finally, type IV are multicentric, or involve the confluence and both the right and left hepatic ducts.42 Tumors that involve the common hepatic duct bifurcation are named Klatskin tumors.42
- What is the first-line treatment for localized cholangiocarcinomas?
Surgical resection is the only potentially curative treatment for localized cholangiocarcinoma, although fewer than 20% of patients are suitable for curative treatment, due to the presence of advanced disease at diagnosis.43,44 Available evidence supports the recommendation that resection with negative margins, regardless of extent, should be the goal of therapy for patients with potentially resectable disease.44 Extensive hepatic resections are often necessary to achieve clear margins since the majority of patients present with large masses. Substantial evidence corroborates that R0 resection is associated with better survival, whereas the benefit of wide compared to narrow (< 5–10 mm) margins is unclear.45 A recent analysis of 96 patients suggests that the proximal resection margin has more prognostic implications than distal margins.45
Surgical options and resectability criteria depend upon tumor location. Extent of tumor in the bile duct is one of the most important factors that determine resectability.17 Although multifocal liver tumors (including satellite lesions), lymph node metastases to the porta hepatis, and distant metastases are considered relative contraindications to surgery, surgical approaches can be considered in selected patients.43 Patient selection for surgery is facilitated by careful preoperative staging, which may include laparoscopy. Laparoscopic staging prior to resection may prevent unnecessary laparotomy in 30% to 45% of patients.42,46
- Is there a role for adjuvant treatment?
Recurrence following complete resection is a primary limitation for cure in BTC, which provides a rationale for the use of adjuvant therapy.47,48 In a sample of 79 patients with extrahepatic cholangiocarcinoma who underwent curative resection, the cumulative recurrence rate after 4 years was 56%.47 Initial recurrence at a distant site occurs in 40% to 50% of patients.48
Lymphovascular and perineural invasion, lymph node metastasis, and tumor size ≥ 5 cm have been reported as independent predictors of recurrence and mortality following resection.49 A 2017 meta-analysis which included 30 studies involving more than 22,499 patients reported a 41% reduction in the risk of death with adjuvant chemotherapy, which translated to a mean OS benefit of 4 months in an unselected population.49 Moreover, this study revealed inferior OS in patients given adjuvant radiation therapy (RT) in combination with chemotherapy.49 These results are in line with the previous meta-analysis by Horgan et al, which demonstrated that adjuvant RT seems to benefit only patients with R1 resections, with a possible detrimental effect in R0 disease.50 Therefore, adjuvant chemoradiation cannot be viewed as a standard practice following R0 resection, and should be reserved for those patients with positive margins (R1/ 2) to reduce local progression.
In the phase 3 BILCAP trial presented at ASCO 2017, 447 patients with completely resected cholangiocarcinoma or gallbladder cancer with adequate biliary drainage and Eastern Cooperative Oncology Group (ECOG) performance score ≤ 2 were randomly assigned to observation or capecitabine (1250 mg/m2 twice daily for days 1–14 every 21 days for 8 cycles).51 Surgical treatment achieved R0 resection in 62% of patients and 46% were node-negative. Median OS was 51 months for the capecitabine group and 36 months for the control arm (hazard ratio [HR] 0.80, 95% CI 0.63 to 1.04, P = 0.097). Analyses with adjustment for nodal status, grade of disease, and gender indicated a HR of 0.71 (P < 0.01). Median DFS was 25 months versus 18 months favoring the capecitabine group, and rates of grade 3 or 4 toxicity were less than anticipated. Following the results of this trial, adjuvant capecitabine should become the new standard of care.
- What is the treatment for locally advanced cholangiocarcinoma?
The optimal approach to patients with locally advanced unresectable cholangiocarcinoma has not been established. The prognosis for patients with either locally unresectable or locally recurrent disease is typically measured in months. Goals of palliative therapy are relief of symptoms and improvement in quality of life, and there is no role for surgical debulking.
Liver transplantation is a potentially curative option for selected patients with hilar or intrahepatic cholangiocarcinoma. Patients with lymph node-negative, non-disseminated, locally advanced hilar cholangiocarcinomas have 5-year survival rates ranging from 25% to 42% following transplantation.52 Retrospective data suggests that neoadjuvant chemoradiation followed by liver transplantation is highly effective for selected patients with hilar cholangiocarcinoma.52 However, these results require confirmation from prospective clinical evidence. It is important to recognize that liver transplantation plays no role in the management of distal cholangiocarcinoma or gallbladder cancer.
Rarely, patients with borderline resectable intrahepatic cholangiocarcinoma will have a sufficient response to chemotherapy to permit later resection, and, in such cases, starting with chemotherapy and then restaging to evaluate resectability is appropriate.54 A single-center, retrospective analysis including 186 patients by Le Roy et al evaluated survival in patients with locally advanced, unresectable intrahepatic cholangiocarcinoma who received primary chemotherapy, followed by surgery in those with secondary resectability.54 After a median of 6 cycles of chemotherapy, 53% of patients achieved resectability and underwent surgery with curative intent. These patients had similar short- and long-term results compared to patients with initially resectable intrahepatic cholangiocarcinoma who had surgery alone, with median OS reaching 24 months.54
Ablative radiotherapy is an additional option for localized inoperable intrahepatic cholangiocarcinoma. Tao and colleagues evaluated 79 consecutive patients with inoperable intrahepatic cholangiocarcinoma treated with definitive RT.55 Median tumor size was 7.9 cm and 89% of patients received chemotherapy before RT. Median OS was 30 months and 3-year OS was 44%. Radiation dose was the single most important prognostic factor, and higher doses correlated with improved local control and OS. A biologic equivalent dose (BED) greater than 80.5 Gy was identified as an ablative dose of RT for large intrahepatic cholangiocarcinomas. The 3-year OS for patients receiving BED greater than 80.5 Gy was 73% versus 38% for those receiving lower doses.
Case Continued
The patient is deemed to have resectable disease and undergoes surgical resection followed by adjuvant capecitabine for 8 cycles. Unfortunately, after 1 year, follow-up imaging identifies bilateral enlarging lung nodules. Biopsy is performed and confirms metastatic cholangiocarcinoma.
- What is the treatment for metastatic BTC?
The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. A benefit of chemotherapy over best supportive care for cholangiocarcinoma was demonstrated in an early phase 3 trial that randomly assigned 90 patients with advanced pancreatic or biliary cancer (37 with bile duct cancer) to receive either fluorouracil (FU) -based systemic chemotherapy or best supportive care. Results showed that chemotherapy significantly improved OS (6 months versus 2.5 months).56 Chemotherapy is also beneficial for patients with unresectable gallbladder cancer. In a single-center randomized study including 81 patients with unresectable gallbladder cancer, gemcitabine and oxaliplatin (GEMOX) improved progression-free survival (PFS) and OS compared to best supportive care.57 Treatment for metastatic cholangiocarcinoma and gallbladder cancer follows the same algorithm.
In 2010, cisplatin plus gemcitabine was established as a reference regimen for first-line therapy by the ABC-02 study, in which 410 patients with locally advanced or metastatic bile duct, gallbladder, or ampullary cancer were randomly assigned to 6 courses of cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2 on days 1 and 8, every 21 days) or gemcitabine alone (1000 mg/m2 days 1, 8, 15, every 28 days).58 OS was significantly greater with combination therapy (11.7 versus 8.1 months), and PFS also favored the combination arm (8 versus 5 months). Toxicity was comparable in both groups, with the exception of significantly higher rates of grade 3 or 4 neutropenia with gemcitabine plus cisplatin (25% versus 17%), and higher rates of grade 3 or 4 abnormal liver function with gemcitabine alone (27% versus 17%). Most quality-of-life scales showed a trend favoring combined therapy.58 A smaller, identically designed Japanese phase 3 randomized trial achieved similar results, demonstrating greater OS with cisplatin plus gemcitabine compared to gemcitabine alone (11.2 versus 7.7 months).59
The gemcitabine plus cisplatin combination has not been directly compared with other gemcitabine combinations in phase 3 trials. A pooled analysis of 104 trials of a variety of chemotherapy regimens in advanced biliary cancer concluded that the gemcitabine plus cisplatin regimen offered the highest rates of objective response and tumor control compared with either gemcitabine-free or cisplatin-free regimens.60 However, this did not translate into significant benefit in terms of either time to tumor progression or median OS. It is important to note that this analysis did not include results of the subsequent ABC-02 trial.
There is no standard treatment for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. There are no completed prospective phase 3 trials supporting the use of second-line chemotherapy after failure of first-line chemotherapy in BTC, and the selection of candidates for second-line therapy as well as the optimal regimen are not established.61 The ongoing phase 2 multicenter ABC-06 trial is evaluating oxaliplatin plus short-term infusional FU and leucovorin (FOLFOX) versus best supportive care for second-line therapy. In a systematic review including 23 studies (14 phase 2 clinical trials and 9 retrospective studies) with 761 patients with BTC, the median OS was 7.2 months.
The optimal selection of candidates for second-line chemotherapy is not established. Two independent studies suggest that patients who have a good performance status (0 or 1), disease control with the first-line chemotherapy, low CA 19-9 level, and possibly previous surgery on their primary tumor, have the longest survival with second-line chemotherapy. However, whether these characteristics predict for chemotherapy responsiveness or more favorable biologic behavior is not clear.62,63 No particular regimen has proved superior to any other, and the choice of second-line regimen remains empiric.
For patients with adequate performance status, examples of other conventional chemotherapy regimens with demonstrated activity that could be considered for second-line therapy include: FOLFOX or capecitabine, gemcitabine plus capecitabine, capecitabine plus cisplatin, or irinotecan plus short-term infusional FU and leucovorin (FOLFIRI) with or without bevacizumab.64 For selected patients, second-line molecularly targeted therapy using erlotinib plus bevacizumab may be considered. However, this regimen is very costly.64 Examples of other regimens with demonstrated activity in phase 2 trials include GEMOX, gemcitabine plus fluoropyrimidine, and fluoropyrimidine plus oxaliplatin or cisplatin.64
There is promising data from studies of targeted therapy for specific molecular subgroups. A recent phase 2 trial evaluated the activity of BGJ398, an orally bioavailable, selective, ATP-competitive pan inhibitor of human fibroblast growth factor receptor (FGFR) kinase, in patients with FGFR-altered advanced cholangiocarcinoma.65 The overall response rate was 14.8% (18.8% FGFR2 fusions only) and disease control rate was 75.4% (83.3% FGFR2 fusions only). All responsive tumors contained FGFR2 fusions. Adverse events were manageable, and grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%). Those included hyperphosphatemia, stomatitis, and palmar-plantar erythrodysesthesia. Javle and colleagues also identified HER2/neu blockade as a promising treatment strategy for gallbladder cancer patients with this gene amplification.66 This retrospective analysis included 9 patients with gallbladder cancer and 5 patients with cholangiocarcinoma who received HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab). In the gallbladder cancer group, HER2/neu gene amplification or overexpression was detected in 8 cases. These patients experienced disease stability (n = 3), partial response (n = 4), or complete response (n = 1) with HER2/neu–directed therapy. Median duration of response was 40 weeks. The cholangiocarcinoma cases treated in this series had no radiological responses despite HER2/neu mutations or amplification.
Gallbladder Cancer
Case Presentation
A 57-year-old woman from Chile presents with a 3-week history of progressive right upper quadrant abdominal pain. She denies nausea, vomiting, dysphagia, odynophagia, alterations in bowel habits, fever, or jaundice. Her past medical history is significant for obesity and hypertension. She has no history of smoking, alcohol, or illicit drug use. Laboratory studies show marked leukocytosis (23,800/µL) with neutrophilia (91%). Liver function test results are within normal limits. Ultrasound of the abdomen reveals gallbladder wall thickening and cholelithiasis.
The patient undergoes an uneventful laparoscopic cholecystectomy and is discharged from the hospital after 48 hours. Pathology report reveals a moderately differentiated adenocarcinoma of the gallbladder invading the perimuscular connective tissue (T2). No lymph nodes are identified in the specimen.
- What is the appropriate surgical management of gallbladder cancer?
Gallbladder cancer can be diagnosed preoperatively or can be found incidentally by intraoperative or pathological findings. In one large series, gallbladder cancer was incidentally found during 0.25% of laparoscopic cholecystectomies.67
For patients who are diagnosed with previously unsuspected gallbladder cancer by pathology findings, the extent of tumor invasion (T stage) indicates the need for re-resection (Figure 3).64
Alternatively, when gallbladder cancer is documented or suspected preoperatively, adequate imaging is important to identify patients with absolute contraindications to resection. Contraindications to surgery include metastasis, extensive involvement of the hepatoduodenal ligament, encasement of major vessels, and involvement of celiac, peripancreatic, periduodenal, or superior mesenteric nodes.72 Notwithstanding, retrospective series suggest individual patients may benefit, and surgical indications in advanced disease should be determined on an individual basis.73 Staging imaging should be obtained using multiphasic contrast-enhanced CT or MRI of the chest, abdomen, and pelvis. PET-scan can be used in selected cases where metastatic disease is suspected.64 Laparoscopic diagnostic staging should be considered prior to resection.64 This procedure can identify previously unknown contraindications to tumor resection in as much as 23% of patients, and the yield is significantly higher in locally advanced tumors.73
Patients with a diagnosis of potentially resectable, localized gallbladder cancer should be offered definitive surgery. Extended cholecystectomy is recommended for patients stage T2 or above. This procedure involves wedge resection of the gallbladder bed or a segmentectomy IVb/V and lymph node dissection, which should include the cystic duct, common bile duct, posterior superior pancreaticoduodenal lymph nodes, and those around the hepatoduodenal ligament.72 Bile duct excision should be performed if there is malignant involvement.64
Conclusion
BTCs are anatomically and clinically heterogeneous tumors. Prognostic factors and therapeutic approaches for BTCs differ depending upon their location in the biliary tree and, accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated. Surgical resection is the only potentially curative treatment for localized BTC. However, recurrence following complete resection is a primary limitation for cure, which provides a rationale for the use of adjuvant therapy. The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. Multiple randomized clinical trials have demonstrated a benefit of chemotherapy for metastatic disease. For patients with adequate performance status, second-line therapy can be considered, and data from studies that evaluated targeted therapy for specific molecular subgroups is promising.
Introduction
Biliary tract carcinoma (BTC) is th
Epidemiology
In the United States, BTC is rare and accounts for approximately 4% of all gastrointestinal malignancies, with an estimated 6000 to 7000 cases of carcinoma of the gallbladder and 3000 to 4000 cases of carcinoma of the bile duct diagnosed annually.4 Among women, there is a 26-fold variation in BTC mortality worldwide, ranging from 0.8 deaths per 100,000 in South Africa to 21.2 per 100,000 in Chile.1,5 Interestingly, for American Indians in New Mexico, gallbladder cancer mortality rates (8.9 per 100,000) surpass those for breast and pancreatic cancers.6 The incidence of anatomical cholangiocarcinoma subtypes also varies regionally, reflecting disparities in genetic and environmental predisposing factors.2,7 In a large, single-center study in the United States, intrahepatic cholangiocarcinoma accounted for less than 10% of cases, perihilar accounted for 50%, and distal accounted for the remaining 40%.8 Importantly, intrahepatic cholangiocarcinoma is the second most common primary malignancy of the liver, and its incidence seems to be rising in many western countries. In the United States, there has been an estimated 128% rise over the past 40 years.4,9
BTC is associated with high mortality rates.10 Median overall survival (OS) for cholangiocarcinoma is 20 to 28 months and 5-year survival is around 25%.10 Most cholangiocarcinomas are diagnosed at advanced stages with unresectable tumors.10 Furthermore, outcomes following resection with curative intent are poor—median disease-free survival (DFS) of 12 to 36 months has been reported.11,12 Patients with intrahepatic disease have a better prognosis when compared with patients who have extrahepatic tumors.12 Gallbladder cancer, likewise, carries a poor overall prognosis; median OS is 32 months and 5-year survival is as low as 13%.6
Risk factors for BTC include intrinsic and extrinsic elements.6 Incidence of BTC increases with age, and diagnosis typically occurs in the sixth to eighth decade of life.5,6,13 In contrast to gallbladder cancer, the incidence of cholangiocarcinoma is slightly higher in men.9 Obesity, diabetes, and consumption of sweetened drinks also increase the risk for BTC.14–16 Cholelithiasis is the most prevalent risk factor for gallbladder cancer, and the risk is greater for larger stones.5 Around 1 in 5 patients with porcelain gallbladder will develop gallbladder carcinoma.17 Primary sclerosing cholangitis (PSC), chronic calculi of the bile duct, choledochal cysts, cirrhosis, hepatitis C, and liver fluke infections are well established risk factors for cholangiocarcinoma.7,12,18 PSC is one of the best described entities among these predisposing conditions. Lifetime prevalence of cholangiocarcinoma among patients with PSC ranges from 5% to 10%.18,19 These patients also present at a younger age; in one series, the median age at diagnosis for BTC arising from PSC was 39 years.18 It is important to recognize, however, that in most patients diagnosed with cholangiocarcinoma, no predisposing factors are identified.8
Diagnosis
Clinical Presentation
Clinical presentation of BTC depends upon anatomic location.20 Patients with early invasive gallbladder cancer are most often asymptomatic.21 When symptoms occur, they may be nonspecific and mimic cholelithiasis.21 The most common clinical presentations include jaundice, weight loss, and abdominal pain.21 Prior to widespread availability of imaging studies, the preoperative diagnosis rate for gallbladder cancer was as low as 10%.22 However, the accuracy of computed tomography (CT) has changed this scenario, with sensitivity ranging from 73% to 87% and specificity from 88% to 100%.21 As a result of its silent clinical character, cholangiocarcinoma is frequently difficult to diagnose.23 Perihilar and distal cholangiocarcinoma characteristically present with signs of biliary obstruction, and imaging and laboratory data can corroborate the presence of cholestasis.24 On examination, patients with extrahepatic cholangiocarcinoma may present with jaundice, hepatomegaly, and a palpable right upper quadrant mass.25 A palpable gallbladder (Courvoisier sign) can also be present.25 Intrahepatic cholangiocarcinoma presents differently, and patients are less likely to be jaundiced.23 Typical clinical features are nonspecific and include dull right upper quadrant pain, weight loss, and an elevated alkaline phosphatase level.23 Alternatively, asymptomatic patients can present with incidentally detected lesions, when imaging is obtained as part of the workup for other causes or during screening for hepatocellular carcinoma in patients with viral hepatitis or cirrhosis.23,26 Uncommonly, BTC patients present because of signs or symptoms related to metastatic disease or evidence of metastatic disease on imaging.
Pathology and Grading
The majority of BTCs are adenocarcinomas, corresponding to 90% of cholangiocarcinomas and 99% of gallbladder cancers.27,28 They are graded as well, moderately, or poorly differentiated.2 Adenosquamous and squamous cell carcinoma are responsible for most of the remaining cases.2,29 Cholangiocarcinomas are divided into 3 types, defined by the Liver Cancer Study Group of Japan: (1) mass-forming, (2) periductal-infiltrating, and (3) intraductal-growing.30,31 Mass-forming intrahepatic cholangiocarcinomas are characterized morphologically by a homogeneous gray-yellow mass with frequent satellite nodules and irregular but well-defined margins.17,30 Central necrosis and fibrosis are also common.30 In the periductal-infiltrating type, tumor typically grows along the bile duct wall without mass formation, resulting in concentric mural thickening and proximal biliary dilation.30 Intraductal-growing papillary cholangiocarcinoma is characterized by the presence of intraluminal papillary or tubular polypoid tumors of the intra- or extrahepatic bile ducts, with partial obstruction and proximal biliary dilation.30
Cholangiocarcinoma
Case Presentation
A previously healthy 59-year-old man presents to his primary care physician with a 3-month history of dull right upper quadrant pain associated with weight loss. The patient is markedly cachectic and abdominal examination reveals upper quadrant tenderness. Laboratory exams are significant for elevated alkaline phosphatase (500 U/L; reference range 45–115 U/L), cancer antigen 19-9 (CA 19-9, 73 U/mL; reference range ≤ 37 U/mL), and carcinoembryonic antigen (CEA , 20 ng/mL; reference range for nonsmokers ≤ 3.0 ng/mL). Aspartate aminotransferase, alanine aminotransferase, total bilirubin, and coagulation studies are within normal range. Ultrasound demonstrates a homogeneous mass with irregular borders in the right lobe of the liver. Triphasic contrast-enhanced CT scan demonstrates a tumor with ragged rim enhancement at the periphery, and portal venous phase shows gradual centripetal enhancement of the tumor with capsular retraction. No abdominal lymph nodes or extrahepatic tumors are noted (Figure 1, Image A).
- What are the next diagnostic steps?
The most critical differential diagnosis of solid liver mass in patients without cirrhosis is cholangiocarcinoma and metastases from another primary site.32 Alternatively, when an intrahepatic lesion is noted on an imaging study in the setting of cirrhosis, the next diagnostic step is differentiation between cholangiocarcinoma and hepatocellular carcinoma (HCC).32 Triphasic contrast-enhanced CT and dynamic magnetic resonance imaging (MRI) are key diagnostic procedures.32,33 In the appropriate setting, classical imaging features in the arterial phase with washout in portal venous or delayed phase can be diagnostic of HCC and may obviate the need for a biopsy (Figure 2).
Typical radiographic features of cholangiocarcinoma include a hypodense hepatic lesion that can be either well-defined or infiltrative and is frequently associated with biliary dilatation (Figure 1, Image A).33 The dense fibrotic nature of the tumor may cause capsular retraction, which is seen in up to 20% of cases.17 This finding is highly suggestive of cholangiocarcinoma and is rarely present in HCC.33 Following contrast administration, there is peripheral (rim) enhancement throughout both arterial and venous phases.32–34 However, these classic features were present in only 70% of cases in one study.35 Although intrahepatic cholangiocarcinomas are most commonly hypovascular, small mass-forming intrahepatic cholangiocarcinomas can often be arterially hyperenhancing and mimic HCC.33 Tumor enhancement on delayed CT imaging has been correlated with survival. Asayama et al demonstrated that tumors that exhibited delayed enhancement on CT in more than two-thirds of their volume were associated with a worse prognosis.36
Patients without cirrhosis who present with a localized lesion of the liver should undergo extensive evaluation for a primary cancer site.37 CT of the chest, abdomen, and pelvis with contrast should be obtained.37 Additionally, mammogram and endoscopic evaluation with esophagogastroduodenoscopy (EGD) and colonoscopy should be included in the work-up.37
Preoperative tumor markers are also included in the work-up. All patients with a solid liver lesion should have serum alpha-fetoprotein (AFP) levels checked. AFP is a serum glycoprotein recognized as a marker for HCC and is reported to detect preclinical HCC.38 However, serum concentrations are normal in up to 40% of small HCCs.38 Although no specific marker for cholangiocarcinoma has yet been identified, the presence of certain tumor markers in the serum of patients may be of diagnostic value, especially in patients with PSC. CA 19-9 and CEA are the best studied. Elevated levels of CA 19-9 prior to treatment are associated with a poorer prognosis, and CA 19-9 concentrations greater than 1000 U/mL are consistent with advanced disease.39,40 One large series evaluated the diagnostic value of serum CEA levels in 333 patients with PSC, 13% of whom were diagnosed with cholangiocarcinoma.34 A serum CEA level greater than 5.2 ng/mL had a sensitivity of 68.0% and specificity of 81.5%.38
If a biopsy is obtained, appropriate immunohistochemistry (IHC) can facilitate the diagnosis. BTC is strongly positive for CK-7 and CK-19.41 CK-7 positivity is not specific and is also common among metastatic cancers of the lung and breast; therefore, in some cases cholangiocarcinoma may be a diagnosis of exclusion. Immunostaining for monoclonal CEA is diffusely positive in up to 75% of cases.41 An IHC panel consisting of Hep Par-1, arginase-1, monoclonal CEA, CK-7, CK-20, TTF-1, MOC-31, and CDX-2 has been proposed to optimize the differential diagnosis of HCC, metastatic adenocarcinoma, and cholangiocarcinoma.41
Case Continued
CT of the chest, abdomen, and pelvis reveals no concerns for metastasis and no evidence of primary cancer elsewhere. EGD and colonoscopy are clear. AFP levels are within normal limits (2 ng/mL). Biopsy is performed and demonstrates adenocarcinoma. IHC studies demonstrate cells positive for monoclonal CEA, CK-7, CK-19, and MOC-31, and negative for Napsin A, TTF-1, and CK-20.
- How is cholangiocarcinoma staged and classified?
The purpose of the staging system is to provide information on prognosis and guidance for therapy. Prognostic factors and the therapeutic approaches for BTC differ depending upon their location in the biliary tree. Accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated (Table 1 and Table 2).23
The Bismuth-Corlette classification is used to further classify perihilar cholangiocarcinoma according to patterns of hepatic duct involvement. Type I tumors are located below the confluence of the left and right hepatic ducts.42 Type II reach the confluence of the hepatic ducts.42 Type III occlude the common hepatic duct and either the right or left hepatic duct (IIIa and IIIb, respectively).42 Finally, type IV are multicentric, or involve the confluence and both the right and left hepatic ducts.42 Tumors that involve the common hepatic duct bifurcation are named Klatskin tumors.42
- What is the first-line treatment for localized cholangiocarcinomas?
Surgical resection is the only potentially curative treatment for localized cholangiocarcinoma, although fewer than 20% of patients are suitable for curative treatment, due to the presence of advanced disease at diagnosis.43,44 Available evidence supports the recommendation that resection with negative margins, regardless of extent, should be the goal of therapy for patients with potentially resectable disease.44 Extensive hepatic resections are often necessary to achieve clear margins since the majority of patients present with large masses. Substantial evidence corroborates that R0 resection is associated with better survival, whereas the benefit of wide compared to narrow (< 5–10 mm) margins is unclear.45 A recent analysis of 96 patients suggests that the proximal resection margin has more prognostic implications than distal margins.45
Surgical options and resectability criteria depend upon tumor location. Extent of tumor in the bile duct is one of the most important factors that determine resectability.17 Although multifocal liver tumors (including satellite lesions), lymph node metastases to the porta hepatis, and distant metastases are considered relative contraindications to surgery, surgical approaches can be considered in selected patients.43 Patient selection for surgery is facilitated by careful preoperative staging, which may include laparoscopy. Laparoscopic staging prior to resection may prevent unnecessary laparotomy in 30% to 45% of patients.42,46
- Is there a role for adjuvant treatment?
Recurrence following complete resection is a primary limitation for cure in BTC, which provides a rationale for the use of adjuvant therapy.47,48 In a sample of 79 patients with extrahepatic cholangiocarcinoma who underwent curative resection, the cumulative recurrence rate after 4 years was 56%.47 Initial recurrence at a distant site occurs in 40% to 50% of patients.48
Lymphovascular and perineural invasion, lymph node metastasis, and tumor size ≥ 5 cm have been reported as independent predictors of recurrence and mortality following resection.49 A 2017 meta-analysis which included 30 studies involving more than 22,499 patients reported a 41% reduction in the risk of death with adjuvant chemotherapy, which translated to a mean OS benefit of 4 months in an unselected population.49 Moreover, this study revealed inferior OS in patients given adjuvant radiation therapy (RT) in combination with chemotherapy.49 These results are in line with the previous meta-analysis by Horgan et al, which demonstrated that adjuvant RT seems to benefit only patients with R1 resections, with a possible detrimental effect in R0 disease.50 Therefore, adjuvant chemoradiation cannot be viewed as a standard practice following R0 resection, and should be reserved for those patients with positive margins (R1/ 2) to reduce local progression.
In the phase 3 BILCAP trial presented at ASCO 2017, 447 patients with completely resected cholangiocarcinoma or gallbladder cancer with adequate biliary drainage and Eastern Cooperative Oncology Group (ECOG) performance score ≤ 2 were randomly assigned to observation or capecitabine (1250 mg/m2 twice daily for days 1–14 every 21 days for 8 cycles).51 Surgical treatment achieved R0 resection in 62% of patients and 46% were node-negative. Median OS was 51 months for the capecitabine group and 36 months for the control arm (hazard ratio [HR] 0.80, 95% CI 0.63 to 1.04, P = 0.097). Analyses with adjustment for nodal status, grade of disease, and gender indicated a HR of 0.71 (P < 0.01). Median DFS was 25 months versus 18 months favoring the capecitabine group, and rates of grade 3 or 4 toxicity were less than anticipated. Following the results of this trial, adjuvant capecitabine should become the new standard of care.
- What is the treatment for locally advanced cholangiocarcinoma?
The optimal approach to patients with locally advanced unresectable cholangiocarcinoma has not been established. The prognosis for patients with either locally unresectable or locally recurrent disease is typically measured in months. Goals of palliative therapy are relief of symptoms and improvement in quality of life, and there is no role for surgical debulking.
Liver transplantation is a potentially curative option for selected patients with hilar or intrahepatic cholangiocarcinoma. Patients with lymph node-negative, non-disseminated, locally advanced hilar cholangiocarcinomas have 5-year survival rates ranging from 25% to 42% following transplantation.52 Retrospective data suggests that neoadjuvant chemoradiation followed by liver transplantation is highly effective for selected patients with hilar cholangiocarcinoma.52 However, these results require confirmation from prospective clinical evidence. It is important to recognize that liver transplantation plays no role in the management of distal cholangiocarcinoma or gallbladder cancer.
Rarely, patients with borderline resectable intrahepatic cholangiocarcinoma will have a sufficient response to chemotherapy to permit later resection, and, in such cases, starting with chemotherapy and then restaging to evaluate resectability is appropriate.54 A single-center, retrospective analysis including 186 patients by Le Roy et al evaluated survival in patients with locally advanced, unresectable intrahepatic cholangiocarcinoma who received primary chemotherapy, followed by surgery in those with secondary resectability.54 After a median of 6 cycles of chemotherapy, 53% of patients achieved resectability and underwent surgery with curative intent. These patients had similar short- and long-term results compared to patients with initially resectable intrahepatic cholangiocarcinoma who had surgery alone, with median OS reaching 24 months.54
Ablative radiotherapy is an additional option for localized inoperable intrahepatic cholangiocarcinoma. Tao and colleagues evaluated 79 consecutive patients with inoperable intrahepatic cholangiocarcinoma treated with definitive RT.55 Median tumor size was 7.9 cm and 89% of patients received chemotherapy before RT. Median OS was 30 months and 3-year OS was 44%. Radiation dose was the single most important prognostic factor, and higher doses correlated with improved local control and OS. A biologic equivalent dose (BED) greater than 80.5 Gy was identified as an ablative dose of RT for large intrahepatic cholangiocarcinomas. The 3-year OS for patients receiving BED greater than 80.5 Gy was 73% versus 38% for those receiving lower doses.
Case Continued
The patient is deemed to have resectable disease and undergoes surgical resection followed by adjuvant capecitabine for 8 cycles. Unfortunately, after 1 year, follow-up imaging identifies bilateral enlarging lung nodules. Biopsy is performed and confirms metastatic cholangiocarcinoma.
- What is the treatment for metastatic BTC?
The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. A benefit of chemotherapy over best supportive care for cholangiocarcinoma was demonstrated in an early phase 3 trial that randomly assigned 90 patients with advanced pancreatic or biliary cancer (37 with bile duct cancer) to receive either fluorouracil (FU) -based systemic chemotherapy or best supportive care. Results showed that chemotherapy significantly improved OS (6 months versus 2.5 months).56 Chemotherapy is also beneficial for patients with unresectable gallbladder cancer. In a single-center randomized study including 81 patients with unresectable gallbladder cancer, gemcitabine and oxaliplatin (GEMOX) improved progression-free survival (PFS) and OS compared to best supportive care.57 Treatment for metastatic cholangiocarcinoma and gallbladder cancer follows the same algorithm.
In 2010, cisplatin plus gemcitabine was established as a reference regimen for first-line therapy by the ABC-02 study, in which 410 patients with locally advanced or metastatic bile duct, gallbladder, or ampullary cancer were randomly assigned to 6 courses of cisplatin (25 mg/m2) plus gemcitabine (1000 mg/m2 on days 1 and 8, every 21 days) or gemcitabine alone (1000 mg/m2 days 1, 8, 15, every 28 days).58 OS was significantly greater with combination therapy (11.7 versus 8.1 months), and PFS also favored the combination arm (8 versus 5 months). Toxicity was comparable in both groups, with the exception of significantly higher rates of grade 3 or 4 neutropenia with gemcitabine plus cisplatin (25% versus 17%), and higher rates of grade 3 or 4 abnormal liver function with gemcitabine alone (27% versus 17%). Most quality-of-life scales showed a trend favoring combined therapy.58 A smaller, identically designed Japanese phase 3 randomized trial achieved similar results, demonstrating greater OS with cisplatin plus gemcitabine compared to gemcitabine alone (11.2 versus 7.7 months).59
The gemcitabine plus cisplatin combination has not been directly compared with other gemcitabine combinations in phase 3 trials. A pooled analysis of 104 trials of a variety of chemotherapy regimens in advanced biliary cancer concluded that the gemcitabine plus cisplatin regimen offered the highest rates of objective response and tumor control compared with either gemcitabine-free or cisplatin-free regimens.60 However, this did not translate into significant benefit in terms of either time to tumor progression or median OS. It is important to note that this analysis did not include results of the subsequent ABC-02 trial.
There is no standard treatment for patients with cholangiocarcinoma for whom first-line gemcitabine-based therapy fails. There are no completed prospective phase 3 trials supporting the use of second-line chemotherapy after failure of first-line chemotherapy in BTC, and the selection of candidates for second-line therapy as well as the optimal regimen are not established.61 The ongoing phase 2 multicenter ABC-06 trial is evaluating oxaliplatin plus short-term infusional FU and leucovorin (FOLFOX) versus best supportive care for second-line therapy. In a systematic review including 23 studies (14 phase 2 clinical trials and 9 retrospective studies) with 761 patients with BTC, the median OS was 7.2 months.
The optimal selection of candidates for second-line chemotherapy is not established. Two independent studies suggest that patients who have a good performance status (0 or 1), disease control with the first-line chemotherapy, low CA 19-9 level, and possibly previous surgery on their primary tumor, have the longest survival with second-line chemotherapy. However, whether these characteristics predict for chemotherapy responsiveness or more favorable biologic behavior is not clear.62,63 No particular regimen has proved superior to any other, and the choice of second-line regimen remains empiric.
For patients with adequate performance status, examples of other conventional chemotherapy regimens with demonstrated activity that could be considered for second-line therapy include: FOLFOX or capecitabine, gemcitabine plus capecitabine, capecitabine plus cisplatin, or irinotecan plus short-term infusional FU and leucovorin (FOLFIRI) with or without bevacizumab.64 For selected patients, second-line molecularly targeted therapy using erlotinib plus bevacizumab may be considered. However, this regimen is very costly.64 Examples of other regimens with demonstrated activity in phase 2 trials include GEMOX, gemcitabine plus fluoropyrimidine, and fluoropyrimidine plus oxaliplatin or cisplatin.64
There is promising data from studies of targeted therapy for specific molecular subgroups. A recent phase 2 trial evaluated the activity of BGJ398, an orally bioavailable, selective, ATP-competitive pan inhibitor of human fibroblast growth factor receptor (FGFR) kinase, in patients with FGFR-altered advanced cholangiocarcinoma.65 The overall response rate was 14.8% (18.8% FGFR2 fusions only) and disease control rate was 75.4% (83.3% FGFR2 fusions only). All responsive tumors contained FGFR2 fusions. Adverse events were manageable, and grade 3 or 4 treatment-related adverse events occurred in 25 patients (41%). Those included hyperphosphatemia, stomatitis, and palmar-plantar erythrodysesthesia. Javle and colleagues also identified HER2/neu blockade as a promising treatment strategy for gallbladder cancer patients with this gene amplification.66 This retrospective analysis included 9 patients with gallbladder cancer and 5 patients with cholangiocarcinoma who received HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab). In the gallbladder cancer group, HER2/neu gene amplification or overexpression was detected in 8 cases. These patients experienced disease stability (n = 3), partial response (n = 4), or complete response (n = 1) with HER2/neu–directed therapy. Median duration of response was 40 weeks. The cholangiocarcinoma cases treated in this series had no radiological responses despite HER2/neu mutations or amplification.
Gallbladder Cancer
Case Presentation
A 57-year-old woman from Chile presents with a 3-week history of progressive right upper quadrant abdominal pain. She denies nausea, vomiting, dysphagia, odynophagia, alterations in bowel habits, fever, or jaundice. Her past medical history is significant for obesity and hypertension. She has no history of smoking, alcohol, or illicit drug use. Laboratory studies show marked leukocytosis (23,800/µL) with neutrophilia (91%). Liver function test results are within normal limits. Ultrasound of the abdomen reveals gallbladder wall thickening and cholelithiasis.
The patient undergoes an uneventful laparoscopic cholecystectomy and is discharged from the hospital after 48 hours. Pathology report reveals a moderately differentiated adenocarcinoma of the gallbladder invading the perimuscular connective tissue (T2). No lymph nodes are identified in the specimen.
- What is the appropriate surgical management of gallbladder cancer?
Gallbladder cancer can be diagnosed preoperatively or can be found incidentally by intraoperative or pathological findings. In one large series, gallbladder cancer was incidentally found during 0.25% of laparoscopic cholecystectomies.67
For patients who are diagnosed with previously unsuspected gallbladder cancer by pathology findings, the extent of tumor invasion (T stage) indicates the need for re-resection (Figure 3).64
Alternatively, when gallbladder cancer is documented or suspected preoperatively, adequate imaging is important to identify patients with absolute contraindications to resection. Contraindications to surgery include metastasis, extensive involvement of the hepatoduodenal ligament, encasement of major vessels, and involvement of celiac, peripancreatic, periduodenal, or superior mesenteric nodes.72 Notwithstanding, retrospective series suggest individual patients may benefit, and surgical indications in advanced disease should be determined on an individual basis.73 Staging imaging should be obtained using multiphasic contrast-enhanced CT or MRI of the chest, abdomen, and pelvis. PET-scan can be used in selected cases where metastatic disease is suspected.64 Laparoscopic diagnostic staging should be considered prior to resection.64 This procedure can identify previously unknown contraindications to tumor resection in as much as 23% of patients, and the yield is significantly higher in locally advanced tumors.73
Patients with a diagnosis of potentially resectable, localized gallbladder cancer should be offered definitive surgery. Extended cholecystectomy is recommended for patients stage T2 or above. This procedure involves wedge resection of the gallbladder bed or a segmentectomy IVb/V and lymph node dissection, which should include the cystic duct, common bile duct, posterior superior pancreaticoduodenal lymph nodes, and those around the hepatoduodenal ligament.72 Bile duct excision should be performed if there is malignant involvement.64
Conclusion
BTCs are anatomically and clinically heterogeneous tumors. Prognostic factors and therapeutic approaches for BTCs differ depending upon their location in the biliary tree and, accordingly, TNM classification systems for intrahepatic, hilar, and distal cholangiocarcinoma and gallbladder cancer have been separated. Surgical resection is the only potentially curative treatment for localized BTC. However, recurrence following complete resection is a primary limitation for cure, which provides a rationale for the use of adjuvant therapy. The prognosis of patients with advanced BTC is poor and OS for those undergoing supportive care alone is short. Multiple randomized clinical trials have demonstrated a benefit of chemotherapy for metastatic disease. For patients with adequate performance status, second-line therapy can be considered, and data from studies that evaluated targeted therapy for specific molecular subgroups is promising.
1. Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2(Suppl 1):e000152.
2. Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2017 Oct 10. doi: 10.1038/nrclinonc.2017.157.
3. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist 2008;13:415–23.
4. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2014 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2017.
5. Torre LA, Siegel RL, Islami F, et al. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol 2017 Aug 18. doi: 10.1016/j.cgh.2017.08.017.
6. Lau CSM, Zywot A, Mahendraraj K, Chamberlain CS. Gallbladder carcinoma in the United States: a population based clinical outcomes study involving 22,343 patients from the Surveillance, Epidemiology, and End Result Database (1973–2013). HPB Surg 2017;2017:1532835. doi:10.1155/2017/1532835.
7. Hughes T, O’Connor T, Techasen A, et al. Opisthorchiasis and cholangiocarcinoma in Southeast Asia: an unresolved problem. Int J Gen Med 2017;10:227–37.
8. DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–62.
9. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9.
10. Yao KJ, Jabbour S, Parekh N, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117.
11. Choi SB, Kim KS, Choi JY, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol 2009;16:3048–56.
12. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96.
13. Duffy A, Capanu M, Abou-Alfa GK, et al. Gallbladder cancer (GBC): 10-year experience at Memorial Sloan-Kettering Cancer Centre (MSKCC). J Surg Oncol 2008;98:485–9.
14. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer — viewpoint of the IARC Working Group. N Engl J Med 2016;375:794–8.
15. Chen J, Han Y, Xu C, et al. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases. Eur J Cancer Prev 2015;24:89–99.
16. Larsson SC, Giovannucci EL, Wolk A. Sweetened beverage consumption and risk of biliary tract and gallbladder cancer in a prospective study. J Natl Cancer Inst 2016;108: doi: 10.1093/jnci/djw125.
17. Gore RM. Biliary tract neoplasms: diagnosis and staging. Cancer Imaging 2007;7(Special Issue A):S15–23.
18. Broome U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610–5.
19. Burak K, Angulo P, Pasha T, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523–6.
20. Rodrigues J, Diehl DL. Cholangiocarcinoma: clinical manifestations and diagnosis. Tech Gastrointest Endosc 2016;18:75–82.
21. Mitchell CH, Johnson PT, Fishman EK, et al. Features suggestive of gallbladder malignancy. J Comput Assist Tomogr 2014;38:235–41.
22. Beltz WR, Condon RE. Primary carcinoma of the gallbladder. Ann Surg 1974;180:180–4.
23. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512–22.
24. Patel T. Cholangiocarcinoma—controversies and challenges. Nat Rev Gastroenterol Hepatol 2011;8:189–200.
25. Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 1996;224:463–73.
26. Bartella I, Dufour JF. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J Gastrointestin Liver Dis 2015;24:481-9.
27. Yamaguchi K, Enjoji M. Carcinoma of the gallbladder: a clinicopathology of 103 patients and a newly proposed staging. Cancer 1988;62:1425–32.
28. Esposito I, Schirmacher P. Pathological aspects of cholangiocarcinoma. HPB. 2008;10:83–6.
29. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol 2016;5:62.
30. Chung YE, Kim MJ, Park YN, et al. Varying appearances of cholangiocarcinoma: radiologic-pathologic correlation. Radiographics 2009;29:683–700.
31. Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg 2003;10:288–91.
32. Rao PN. Nodule in liver: investigations, differential diagnosis and follow-up. J Clin Exp Hepatol 2014;4(Suppl 3):S57–62.
33. Kim TK, Lee E, Jang HJ. Imaging findings of mimickers of hepatocellular carcinoma. Clin Mol Hepatol 2015;21:326–43.
34. Hennedige TP, Neo WT, Venkatesh SK. Imaging of malignancies of the biliary tract- an update. Cancer Imaging 2014;14:14.
35. Kim SH, Lee CH, Kim BH, et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J Comput Assist Tomogr 2012;36:704–9.
36. Asayama Y, Yoshimitsu K, Irie H, et al. Delayed-phase dynamic CT enhancement as a prognostic factor for mass-forming intrahepatic cholangiocarcinoma. Radiology 2006;238:150–5.
37. National Comprehensive Cancer Network. Cancer of unknown primary. www.nccn.org/professionals/physician_gls/pdf/bone.pdf. Accessed 1 Dec 2017.
38. Kefeli A, Basyigit S, Yeniova AO. Diagnosis of hepatocellular carcinoma. In: Abdeldayem HM, ed. Updates in liver cancer. London: InTech; 2017.
39. Bergquist JR, Ivanics T, Storlie CB, et al. Implications of CA19-9 elevation for survival, staging, and treatment sequencing in intrahepatic cholangiocarcinoma: A national cohort analysis. J Surg Oncol 2016;114:475–82.
40. Chung YJ, Choi DW, Choi SH, et al. Prognostic factors following surgical resection of distal bile duct cancer. J Korean Surg Soc 2013;85:212–8.
41. Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol 2002;33:1175–81.
42. Paul A, Kaiser GM, Molmenti EP, et al. Klatskin tumors and the accuracy of the Bismuth-Corlette classification. Am Surg 2011;77:1695–9.
43. Cannavale A, Santoni M, Gazzetti M, et al. Updated management of malignant biliary tract tumors: an illustrative review. J Vasc Interv Radiol 2016;27:1056–69.
44. Matsuo K, Rocha FG, Ito K, et al. The Blumgart preoperative staging system for hilar cholangiocarcinoma: analysis of resectability and outcomes in 380 patients. J Am Coll Surg 2012;215:343–55.
45. Yoo T, Park SJ, Han SS, et al. Proximal resection margins: more prognostic than distal resection margins in patients undergoing hilar cholangiocarcinoma resection. Cancer Res Treat 2017 Nov 16; doi.org/10.4143/crt.2017.320.
46. Joseph S, Connor S, Garden OJ. Staging laparoscopy for cholangiocarcinoma. HPB 2008;10:116–9.
47. Jarnagin WR, Ruo L, Little SA, et al. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: implications for adjuvant therapeutic strategies. Cancer 2003;98:1689–700.
48. Kobayashi A, Miwa S, Nakata T, Miyagawa S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br J Surg 2010;97:56–64.
49. Ghidini M, Tomasello G, Botticelli A, et al. Adjuvant chemotherapy for resected biliary tract cancers: a systematic review and meta-analysis. HPB 2017;19:741–8.
50. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934–40.
51. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study [abstract]. J Clin Oncol 2017 35:15_suppl:4006-4006.
52. Darwish Murad S, Kim WR, Darnois DM, et al. Efficacy of neoadjuvant chemoradiation followed by liver transplantation for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88–98.
53. Sapisochin G, Facciuto M, Rubbia-Brandt L, et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016;64:1178–88.
54. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg 2017 Aug 31. doi: 10.1002/bjs.10641.
55. Tao R, Krishnan S, Bhosale PR, et al. Ablative radiotherapy doses lead to a substantial prolongation of survival in patients with inoperable intrahepatic cholangiocarcinoma: a retrospective dose response analysis. J Clin Oncol 2016;34:219–26.
56. Glimelius B, Hoffman K, SjÓdén PO, et al. 555 Palliative chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Eur J Cancer 1995;31:S118.
57. Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol 2010;28:4581–6.
58. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
59. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469–74.
60. Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
61. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38.
62. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290–7.
63. Fornaro L, Cereda S, Aprile G, et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br J Cancer 2014;110:2165–9.
64. National Comprehensive Cancer Network. Hepatobiliary cancer. www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. Accessed 12 Nov 2017.
65. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2017 Nov 28;JCO2017755009.
66. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58.
67. Konstantinidis IT, Deshpande V, Genevay M, et al. Trends in presentation and survival for gallbladder cancer during a period of more than 4 decades: a single-institution experience. Arch Surg 2009;144:441–47.
68. Singh S, Agarwal AK. Gallbladder cancer: the role of laparoscopy and radical resection. Ann Surg 2009;250:494–5.
69. Kapoor VK, Haribhakti SP. Extended cholecystectomy for carcinoma of the gall bladder. Trop Gastroenterol 1995;16:74–5.
70. Ethun CG, Postlewait LM, Le N, et al. Association of optimal time Interval to re-resection for incidental gallbladder cancer with overall survival: a multi-Institution analysis from the US extrahepatic biliary malignancy consortium. JAMA Surg 2017;152:143–9.
71. Goetze TO, Paolucci V. Benefits of reoperation of T2 and more advanced incidental gallbladder carcinoma: analysis of the German registry. Ann Surg 2008;247:104–8.
72. Nishio H, Nagino M, Ebata T, et al. Aggressive surgery for stage IV gallbladder carcinoma; what are the contraindications? J Hepatobiliary Pancreat Surg 2007;14:351–7.
73. Agarwal AK, Kalayarasan R, Javed A, et al. The role of staging laparoscopy in primary gallbladder cancer--an analysis of 409 patients: a prospective study to evaluate the role of staging laparoscopy in the management of gallbladder cancer. Ann Surg 2013;258:318–23.
1. Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017;2(Suppl 1):e000152.
2. Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2017 Oct 10. doi: 10.1038/nrclinonc.2017.157.
3. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist 2008;13:415–23.
4. U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999-2014 Incidence and Mortality Web-based Report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; 2017.
5. Torre LA, Siegel RL, Islami F, et al. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol 2017 Aug 18. doi: 10.1016/j.cgh.2017.08.017.
6. Lau CSM, Zywot A, Mahendraraj K, Chamberlain CS. Gallbladder carcinoma in the United States: a population based clinical outcomes study involving 22,343 patients from the Surveillance, Epidemiology, and End Result Database (1973–2013). HPB Surg 2017;2017:1532835. doi:10.1155/2017/1532835.
7. Hughes T, O’Connor T, Techasen A, et al. Opisthorchiasis and cholangiocarcinoma in Southeast Asia: an unresolved problem. Int J Gen Med 2017;10:227–37.
8. DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg 2007;245:755–62.
9. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 2016;21:594–9.
10. Yao KJ, Jabbour S, Parekh N, et al. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol 2016;16:117.
11. Choi SB, Kim KS, Choi JY, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol 2009;16:3048–56.
12. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84–96.
13. Duffy A, Capanu M, Abou-Alfa GK, et al. Gallbladder cancer (GBC): 10-year experience at Memorial Sloan-Kettering Cancer Centre (MSKCC). J Surg Oncol 2008;98:485–9.
14. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer — viewpoint of the IARC Working Group. N Engl J Med 2016;375:794–8.
15. Chen J, Han Y, Xu C, et al. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases. Eur J Cancer Prev 2015;24:89–99.
16. Larsson SC, Giovannucci EL, Wolk A. Sweetened beverage consumption and risk of biliary tract and gallbladder cancer in a prospective study. J Natl Cancer Inst 2016;108: doi: 10.1093/jnci/djw125.
17. Gore RM. Biliary tract neoplasms: diagnosis and staging. Cancer Imaging 2007;7(Special Issue A):S15–23.
18. Broome U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610–5.
19. Burak K, Angulo P, Pasha T, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004;99:523–6.
20. Rodrigues J, Diehl DL. Cholangiocarcinoma: clinical manifestations and diagnosis. Tech Gastrointest Endosc 2016;18:75–82.
21. Mitchell CH, Johnson PT, Fishman EK, et al. Features suggestive of gallbladder malignancy. J Comput Assist Tomogr 2014;38:235–41.
22. Beltz WR, Condon RE. Primary carcinoma of the gallbladder. Ann Surg 1974;180:180–4.
23. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512–22.
24. Patel T. Cholangiocarcinoma—controversies and challenges. Nat Rev Gastroenterol Hepatol 2011;8:189–200.
25. Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 1996;224:463–73.
26. Bartella I, Dufour JF. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J Gastrointestin Liver Dis 2015;24:481-9.
27. Yamaguchi K, Enjoji M. Carcinoma of the gallbladder: a clinicopathology of 103 patients and a newly proposed staging. Cancer 1988;62:1425–32.
28. Esposito I, Schirmacher P. Pathological aspects of cholangiocarcinoma. HPB. 2008;10:83–6.
29. Silva VWK, Askan G, Daniel TD, et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol 2016;5:62.
30. Chung YE, Kim MJ, Park YN, et al. Varying appearances of cholangiocarcinoma: radiologic-pathologic correlation. Radiographics 2009;29:683–700.
31. Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg 2003;10:288–91.
32. Rao PN. Nodule in liver: investigations, differential diagnosis and follow-up. J Clin Exp Hepatol 2014;4(Suppl 3):S57–62.
33. Kim TK, Lee E, Jang HJ. Imaging findings of mimickers of hepatocellular carcinoma. Clin Mol Hepatol 2015;21:326–43.
34. Hennedige TP, Neo WT, Venkatesh SK. Imaging of malignancies of the biliary tract- an update. Cancer Imaging 2014;14:14.
35. Kim SH, Lee CH, Kim BH, et al. Typical and atypical imaging findings of intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging. J Comput Assist Tomogr 2012;36:704–9.
36. Asayama Y, Yoshimitsu K, Irie H, et al. Delayed-phase dynamic CT enhancement as a prognostic factor for mass-forming intrahepatic cholangiocarcinoma. Radiology 2006;238:150–5.
37. National Comprehensive Cancer Network. Cancer of unknown primary. www.nccn.org/professionals/physician_gls/pdf/bone.pdf. Accessed 1 Dec 2017.
38. Kefeli A, Basyigit S, Yeniova AO. Diagnosis of hepatocellular carcinoma. In: Abdeldayem HM, ed. Updates in liver cancer. London: InTech; 2017.
39. Bergquist JR, Ivanics T, Storlie CB, et al. Implications of CA19-9 elevation for survival, staging, and treatment sequencing in intrahepatic cholangiocarcinoma: A national cohort analysis. J Surg Oncol 2016;114:475–82.
40. Chung YJ, Choi DW, Choi SH, et al. Prognostic factors following surgical resection of distal bile duct cancer. J Korean Surg Soc 2013;85:212–8.
41. Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol 2002;33:1175–81.
42. Paul A, Kaiser GM, Molmenti EP, et al. Klatskin tumors and the accuracy of the Bismuth-Corlette classification. Am Surg 2011;77:1695–9.
43. Cannavale A, Santoni M, Gazzetti M, et al. Updated management of malignant biliary tract tumors: an illustrative review. J Vasc Interv Radiol 2016;27:1056–69.
44. Matsuo K, Rocha FG, Ito K, et al. The Blumgart preoperative staging system for hilar cholangiocarcinoma: analysis of resectability and outcomes in 380 patients. J Am Coll Surg 2012;215:343–55.
45. Yoo T, Park SJ, Han SS, et al. Proximal resection margins: more prognostic than distal resection margins in patients undergoing hilar cholangiocarcinoma resection. Cancer Res Treat 2017 Nov 16; doi.org/10.4143/crt.2017.320.
46. Joseph S, Connor S, Garden OJ. Staging laparoscopy for cholangiocarcinoma. HPB 2008;10:116–9.
47. Jarnagin WR, Ruo L, Little SA, et al. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: implications for adjuvant therapeutic strategies. Cancer 2003;98:1689–700.
48. Kobayashi A, Miwa S, Nakata T, Miyagawa S. Disease recurrence patterns after R0 resection of hilar cholangiocarcinoma. Br J Surg 2010;97:56–64.
49. Ghidini M, Tomasello G, Botticelli A, et al. Adjuvant chemotherapy for resected biliary tract cancers: a systematic review and meta-analysis. HPB 2017;19:741–8.
50. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol 2012;30:1934–40.
51. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study [abstract]. J Clin Oncol 2017 35:15_suppl:4006-4006.
52. Darwish Murad S, Kim WR, Darnois DM, et al. Efficacy of neoadjuvant chemoradiation followed by liver transplantation for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88–98.
53. Sapisochin G, Facciuto M, Rubbia-Brandt L, et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: International retrospective study supporting a prospective assessment. Hepatology 2016;64:1178–88.
54. Le Roy B, Gelli M, Pittau G, et al. Neoadjuvant chemotherapy for initially unresectable intrahepatic cholangiocarcinoma. Br J Surg 2017 Aug 31. doi: 10.1002/bjs.10641.
55. Tao R, Krishnan S, Bhosale PR, et al. Ablative radiotherapy doses lead to a substantial prolongation of survival in patients with inoperable intrahepatic cholangiocarcinoma: a retrospective dose response analysis. J Clin Oncol 2016;34:219–26.
56. Glimelius B, Hoffman K, SjÓdén PO, et al. 555 Palliative chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Eur J Cancer 1995;31:S118.
57. Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol 2010;28:4581–6.
58. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81.
59. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469–74.
60. Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer 2007;96:896–902.
61. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328–38.
62. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: A large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290–7.
63. Fornaro L, Cereda S, Aprile G, et al. Multivariate prognostic factors analysis for second-line chemotherapy in advanced biliary tract cancer. Br J Cancer 2014;110:2165–9.
64. National Comprehensive Cancer Network. Hepatobiliary cancer. www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf. Accessed 12 Nov 2017.
65. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2017 Nov 28;JCO2017755009.
66. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol 2015;8:58.
67. Konstantinidis IT, Deshpande V, Genevay M, et al. Trends in presentation and survival for gallbladder cancer during a period of more than 4 decades: a single-institution experience. Arch Surg 2009;144:441–47.
68. Singh S, Agarwal AK. Gallbladder cancer: the role of laparoscopy and radical resection. Ann Surg 2009;250:494–5.
69. Kapoor VK, Haribhakti SP. Extended cholecystectomy for carcinoma of the gall bladder. Trop Gastroenterol 1995;16:74–5.
70. Ethun CG, Postlewait LM, Le N, et al. Association of optimal time Interval to re-resection for incidental gallbladder cancer with overall survival: a multi-Institution analysis from the US extrahepatic biliary malignancy consortium. JAMA Surg 2017;152:143–9.
71. Goetze TO, Paolucci V. Benefits of reoperation of T2 and more advanced incidental gallbladder carcinoma: analysis of the German registry. Ann Surg 2008;247:104–8.
72. Nishio H, Nagino M, Ebata T, et al. Aggressive surgery for stage IV gallbladder carcinoma; what are the contraindications? J Hepatobiliary Pancreat Surg 2007;14:351–7.
73. Agarwal AK, Kalayarasan R, Javed A, et al. The role of staging laparoscopy in primary gallbladder cancer--an analysis of 409 patients: a prospective study to evaluate the role of staging laparoscopy in the management of gallbladder cancer. Ann Surg 2013;258:318–23.
Polycythemia Vera and Essential Thrombocythemia: Current Management
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30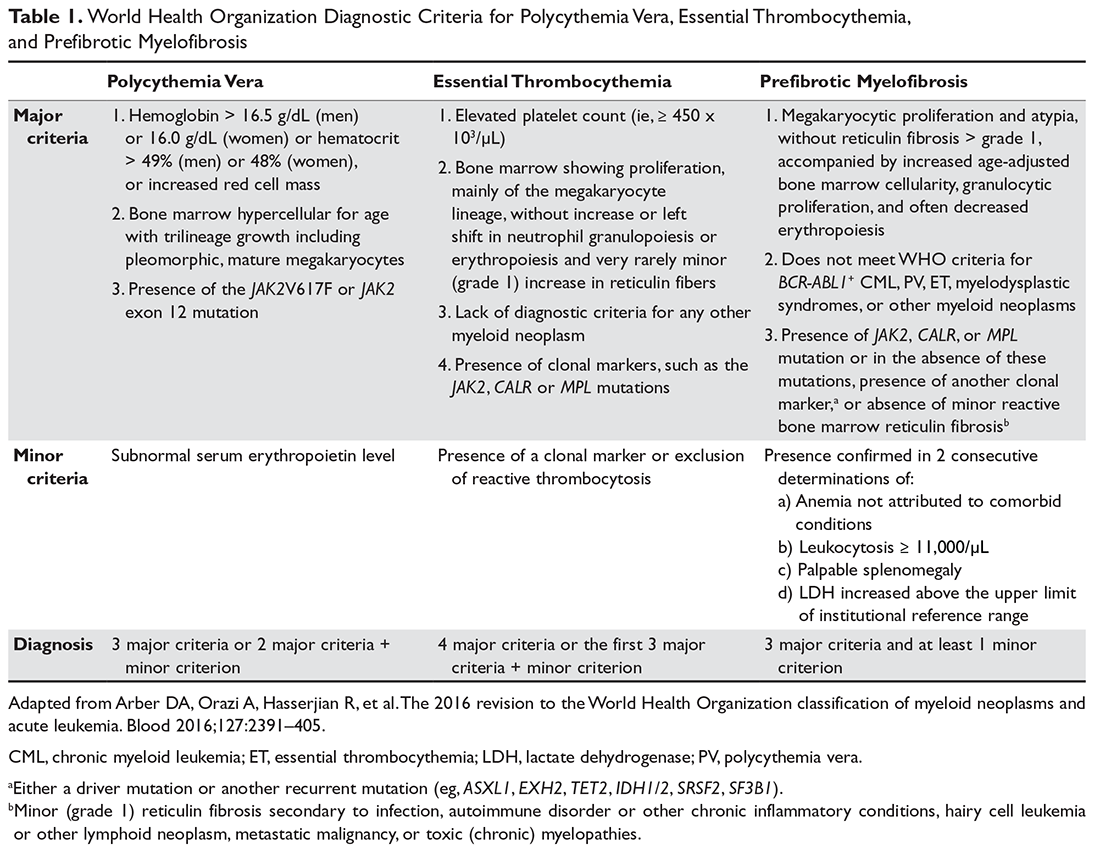
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55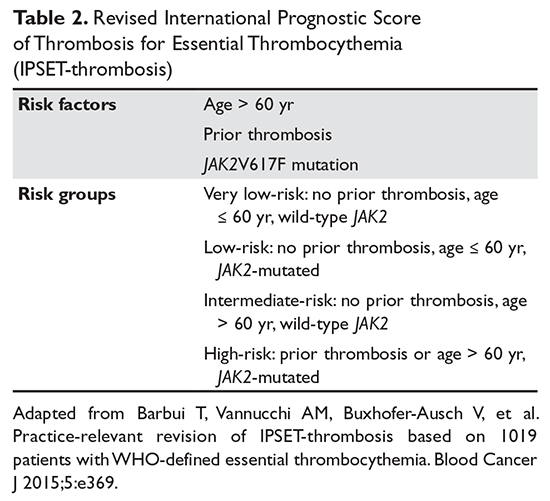
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1. 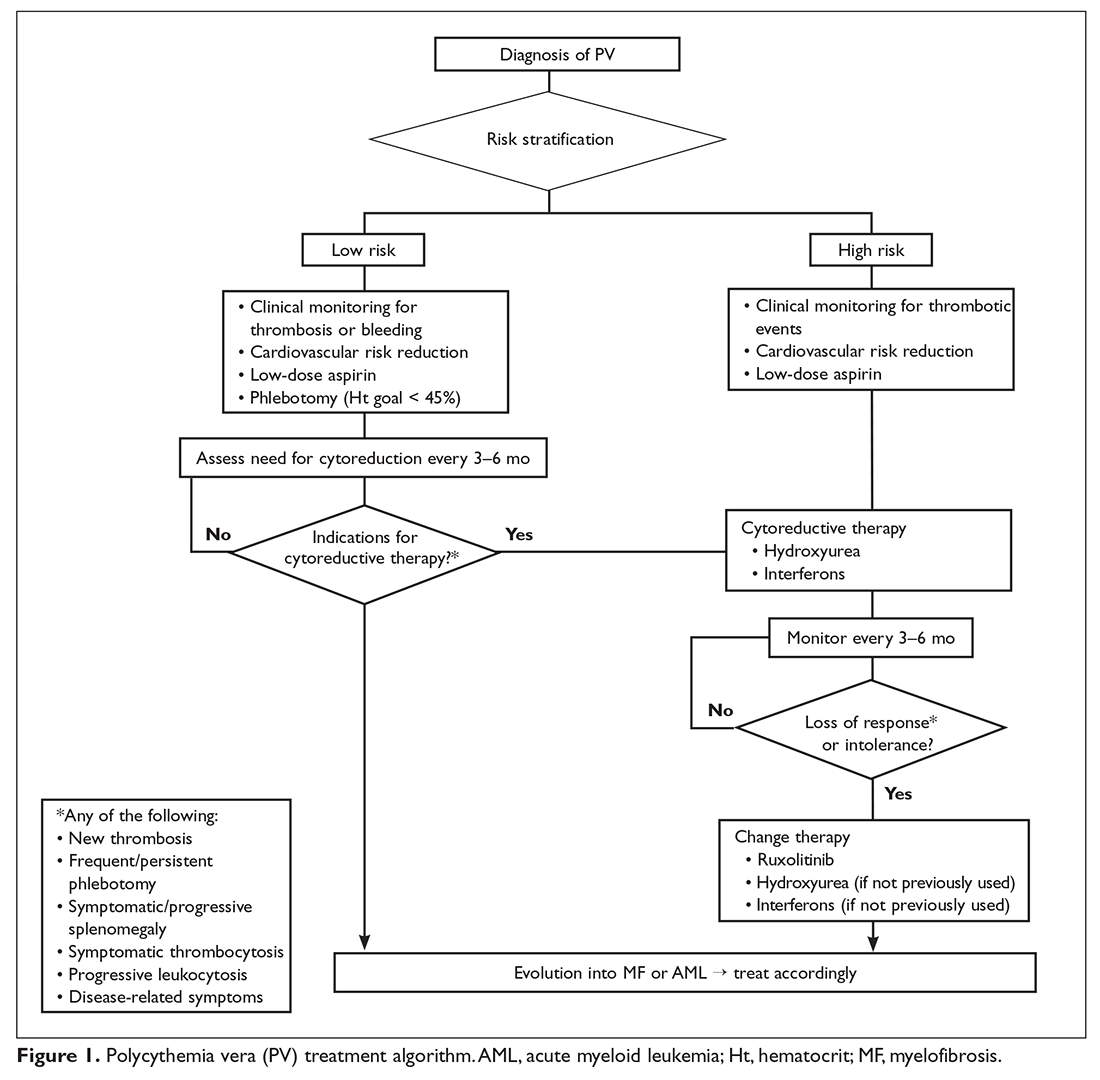
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.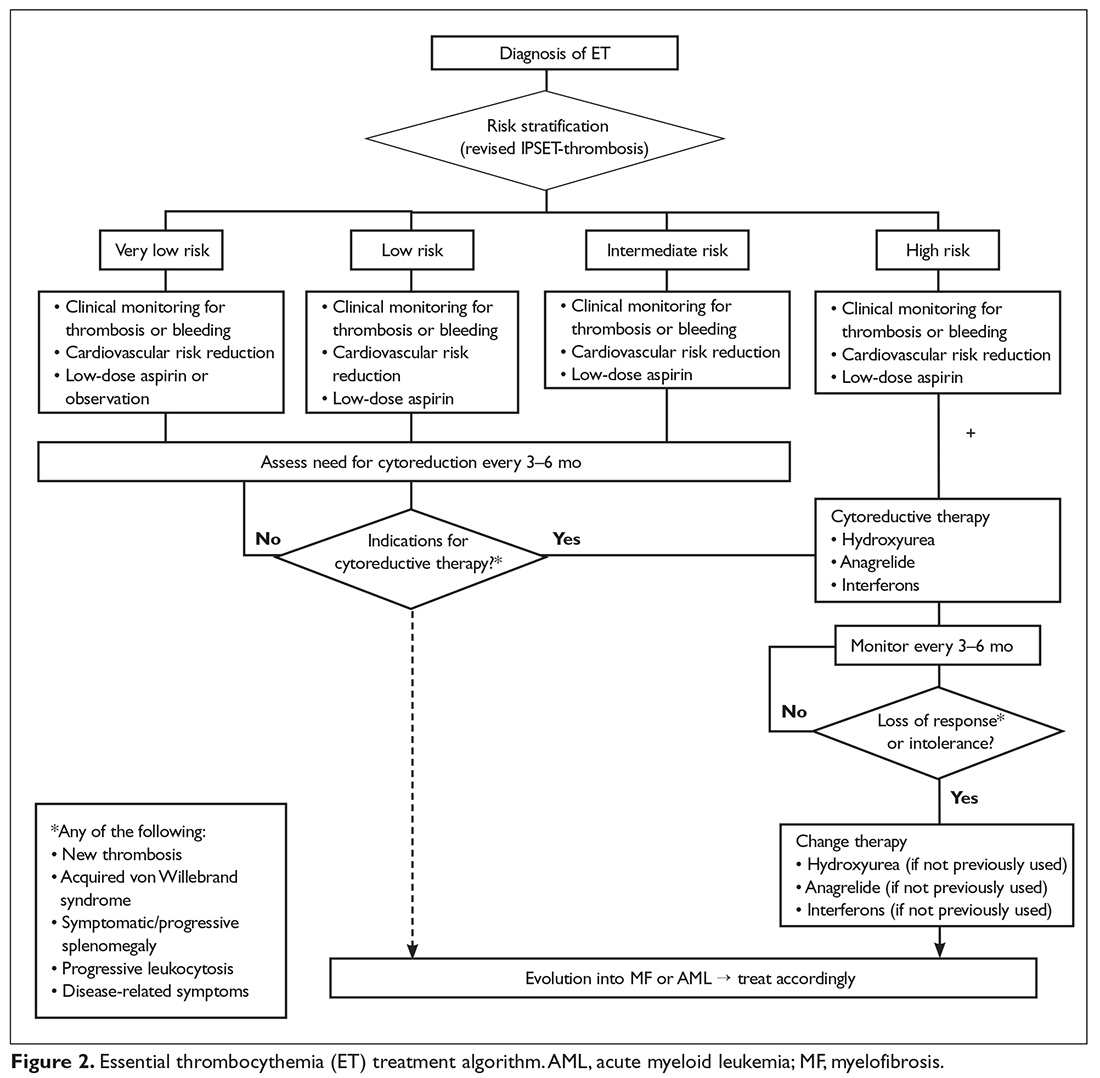
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94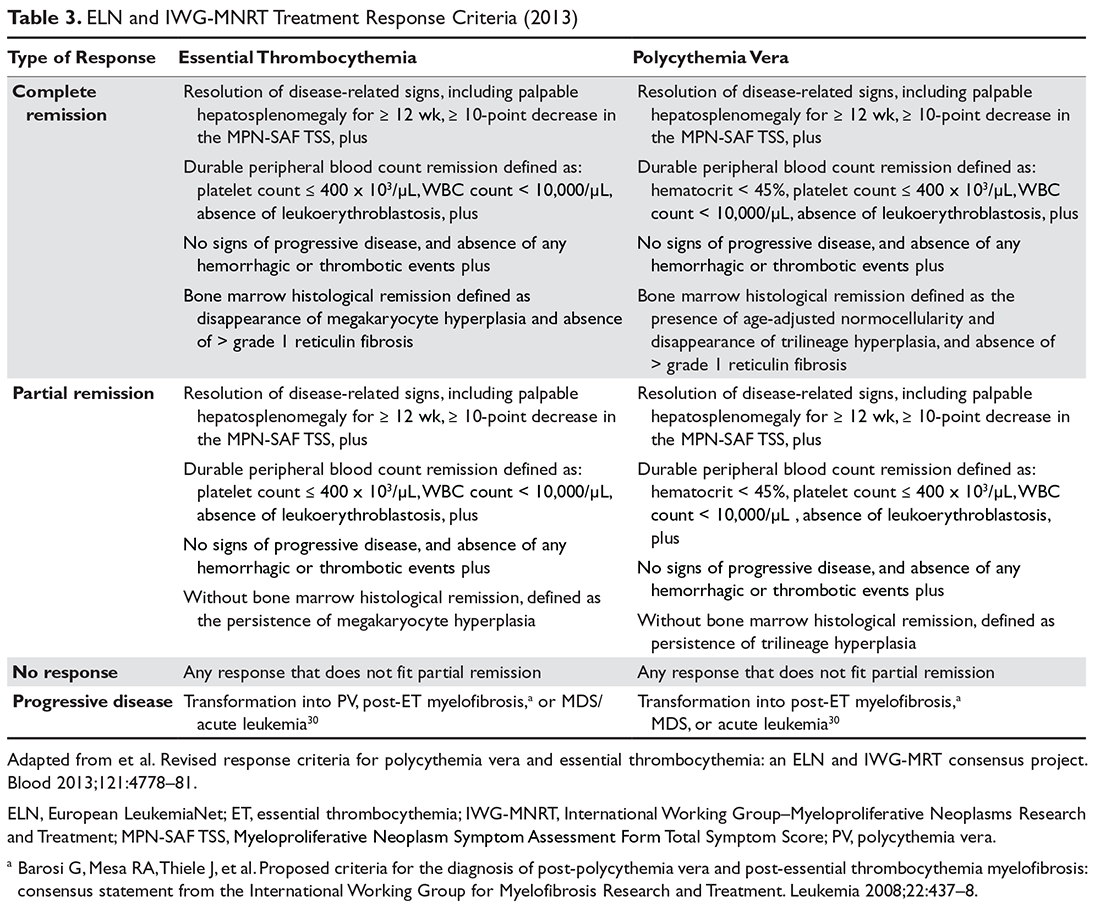
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4. 
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1.
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4.
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET), along with primary myelofibrosis (PMF), belong to the group of Philadelphia-negative myeloproliferative neoplasms (MPN). All these malignancies arise from the clonal proliferation of an aberrant hematopoietic stem cell, but are characterized by distinct clinical phenotypes.1,2 Although the clinical course of PV and ET is indolent, it can be complicated by thrombohemorrhagic episodes and/or evolution into myelofibrosis and/or acute myeloid leukemia (AML).3 Since vascular events are the most frequent life-threatening complications of PV and ET, therapeutic strategies are aimed at reducing this risk. Treatment may also help control other disease-associated symptoms.4 No therapy has been shown to prevent evolution of PV or ET into myelofibrosis or AML. The discovery of the Janus kinase 2 (JAK2)/V617F mutation in most patients with PV and over half of those with ET (and PMF)5,6 has opened new avenues of research and led to the development of targeted therapies, such as the JAK1/2 inhibitor ruxolitinib, for patients with MPN.7,8
Epidemiology
PV and ET are typically diagnosed in the fifth to seventh decade of life.9 Although these disorders are generally associated with a long clinical course, survival of patients with PV or ET may be shorter than that of the general population.10–13 Estimating the incidence and prevalence of MPN is a challenge because most patients remain asymptomatic for long periods of time and do not seek medical attention.13 The annual incidence rates of PV and ET are estimated at 0.01 to 2.61 and 0.21 to 2.53 per 100,000, respectively. PV occurs slightly more frequently in males, whereas ET has a predilection for females.14 Given the long course and low mortality associated with these disorders, the prevalence of PV and ET are significantly higher than the respective incidence: up to 47 and 57 per 100,000, respectively.15–17
Molecular Pathogenesis
In 2005 researchers discovered a gain-of-function mutation of the JAK2 gene in nearly all patients with PV and more than half of those with ET and PMF.5,6,18,19 JAK2 is a non-receptor tyrosine kinase that plays a central role in normal hematopoiesis. Substitution of a valine for a phenylalanine at codon 617 (ie, V617F) leads to its constitutive activation and signaling through the JAK-STAT pathway.5,6,18,19 More rarely (and exclusively in patients with PV), JAK2 mutations involve exon 12.20–22 The vast majority of JAK2-negative ET patients harbor mutations in either the myeloproliferative leukemia (MPL) gene, which encodes the thrombopoietin receptor,23–25 or the calreticulin (CALR) gene,26,27 which encodes for a chaperone protein that plays a role in cellular proliferation, differentiation, and apoptosis.28 Both the MPL and CALR mutations ultimately result in the constitutive activation of the JAK-STAT pathway. Thus, JAK2, MPL, and CALR alterations are collectively referred to as driver mutations. Moreover, because these mutations affect the same oncogenic pathway (ie, JAK-STAT), they are almost always mutually exclusive in a given patient. Patients with ET (or myelofibrosis) who are wild-type for JAK2, MPL, and CALR are referred to as having “triple-negative” disease. Many recurrent non-driver mutations are also found in patients with MPN that are not exclusive of each other (ie, patients may have many at the same time), and involve for example ten-eleven translocation-2 (TET2), additional sex combs like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), isocitrate dehydrogenase 1 and isocitrate dehydrogenase 2 (IDH1/2), and DNA methyltransferase 3A (DNMT3A) genes, among others.29 The biologic and prognostic significance of these non-driver alterations remain to be fully defined in ET and PV.
Diagnosis and Risk Assessment
Case Presentations
Patient A is a 68-year-old man with a history of gouty arthritis who presents with a 6-month history of recurrent headaches and itching that increases after a hot shower. Over the past 2 months, he has also noticed worsening fatigue and redness of his face. He is a nonsmoker. Physical exam reveals erythromelalgia (ie, erythema, edema, and warmth) of the upper and lower extremities, scattered scratch marks, and splenomegaly 4 cm below the costal margin. Complete blood count (CBC) shows a white blood cell (WBC) count of 8100/µL, hemoglobin 194 g/L, and platelets 582 × 103/µL. Serum erythropoietin level is decreased at 2 mU/mL. Peripheral blood testing reveals a JAK2V617F mutation.
Patient B is a 51-year-old woman with a history of severe depression treated with sertraline and hypertension controlled with lisinopril and amlodipine who presents to her primary care physician for her “50-year-old physical.” She denies symptoms and is a nonsmoker. Physical exam is unrevealing. CBC shows a WBC count of 7400/µL (normal differential), hemoglobin 135 g/L, and platelets 1282 × 103/µL. A bone marrow biopsy shows normal cellularity with clusters of large, hyperlobulated megakaryocytes. Reverse transcriptase-polymerase chain reaction fails to reveal a BCR-ABL fusion product. The patient is diagnosed with ET.
Diagnostic Criteria
Diagnostic criteria for PV and ET according to the World Health Organization (WHO) classification30 are summarized in Table 1. Criteria for the diagnosis of prefibrotic myelofibrosis are included as well since this entity was formally recognized as separate from ET and part of the PMF spectrum in the 2016 WHO classification of myeloid tumors.30
Risk Stratification
Thrombohemorrhagic events, evolution into myelofibrosis, and leukemic transformation are the most serious complications in the course of PV or ET. Only thrombohemorrhagic events are, at least partially, preventable. Arterial or venous thrombotic complications are observed at rates of 1.8 to 10.9 per 100 patient-years in PV (arterial thrombosis being more common than venous) and 0.74 to 7.7 per 100 patient-years in ET, depending on the risk group35 and the presence of other factors (see below).
Thrombosis Risk Stratification in PV
The risk stratification of patients with PV is based on 2 factors: age ≥ 60 years and prior history of thrombosis. If either is present the patient is assigned to the high-risk category, whereas if none is present the patient is considered at low risk.36 In addition, high hematocrit37 and high WBC,38 but not thrombocytosis, have been associated with the development of vascular complications. In one study, the risk of new arterial thrombosis was increased by the presence of leukoerythroblastosis, hypertension, and prior arterial thrombosis, while karyotypic abnormalities and prior venous thrombosis were predictors of new venous thrombosis.39 Another emerging risk factor for thrombosis in patients with PV is high JAK2 allele burden (ie, the normal-to-mutated gene product ratio), although the evidence supporting this conclusion is equivocal.40
Thrombosis Risk Stratification in ET
Traditionally, in ET patients, thrombotic risk was assessed using the same 2 factors (age ≥ 60 years and prior history of thrombosis), separating patients into low- and high-risk groups. However, the prognostication of ET patients has been refined recently with the identification of new relevant factors. In particular, the impact of JAK2 mutations on thrombotic risk has been thoroughly studied. Clinically, the presence of JAK2V617F is associated with older age, higher hemoglobin and hematocrit, lower platelet counts, more frequent need for cytoreductive treatment, and greater tendency to evolve into PV (a rare event).41,42 Many,41,43–46 but not all,47–51 studies suggested a correlation between JAK2 mutation and risk of both arterial and venous thrombosis. Although infrequent, a JAK2V617F homozygous state (ie, the mutation is present in both alleles) might confer an even higher thrombotic risk.52 Moreover, the impact of the JAK2 mutation on vascular events persists over time,53 particularly in patients with high or unstable mutation burden.54 Based on JAK2V617F’s influence on the thrombotic risk of ET patients, a new prognostic score was proposed, the International Prognostic Score for ET (IPSET)-thrombosis (Table 2). The revised version of this model is currently endorsed by the National Comprehensive Cancer Network and divides patients into 4 risk groups: high, intermediate, low, and very low. Treatment recommendations vary according to the risk group (as described below).55
Other thrombotic risk factors have been identified, but deemed not significant enough to be included in the model. Cardiovascular risk factors (hypercholesterolemia, hypertension, smoking, diabetes mellitus) can increase the risk of vascular events,56–59 as can splenomegaly60 and baseline or persistent leukocytosis.61–63 Thrombocytosis has been correlated with thrombotic risk in some studies,64–68 whereas others did not support this conclusion and/or suggested a lower rate of thrombosis and, in some cases, increased risk of bleeding in ET patients with platelet counts greater than 1000 × 103/µL (due to acquired von Willebrand syndrome).56,61,63,68,69
CALR mutations tend to occur in younger males with lower hemoglobin and WBC count, higher platelet count, and greater marrow megakaryocytic predominance as compared to JAK2 mutations.26,27,70–72 The associated incidence of thrombosis was less than 10% at 15 years in patients with CALR mutations, lower than the incidence reported for ET patients with JAK2V617F mutations.73 The presence of the mutation per se does not appear to affect the thrombotic risk.74–76 Information on the thrombotic risk associated with MPL mutations or a triple-negative state is scarce. In both instances, however, the risk appears to be lower than with the JAK2 mutation.73,77–79
Venous thromboembolism in patients with PV or ET may occur at unusual sites, such as the splanchnic or cerebral venous systems.80 Risk factors for unusual venous thromboembolism include younger age,81 female gender (especially with concomitant use of oral contraceptive pills),82 and splenomegaly/splenectomy.83JAK2 mutation has also been associated with thrombosis at unusual sites. However, the prevalence of MPN or JAK2V617F in patients presenting with splanchnic venous thromboembolism has varied.80 In addition, MPN may be occult (ie, no clinical or laboratory abnormalities) in around 15% of patients.84 Screening for JAK2V617F and underlying MPN is recommended in patients presenting with isolated unexplained splanchnic venous thromboembolism. Treatment entails long-term anticoagulation therapy. JAK2V617F screening in patients with nonsplanchnic venous thromboembolism is not recommended, as its prevalence in this group is low (< 3%).85,86
Treatment
Cases Continued
Patient A is diagnosed with PV based on the presence of 2 major criteria (elevated hemoglobin and presence of the JAK2V617F mutation) and 1 minor criterion (low erythropoietin level). Given his age, he belongs to the high-risk disease category. He is now seeking advice regarding the management of his newly diagnosed PV.
Patient B presents to the emergency department with right lower extremity swelling and is found to have deep femoral thrombosis extending to the iliac vein. Five days after being discharged from the emergency department, she presents for follow-up. She is taking warfarin compliantly and her INR is within therapeutic range. The patient now has high-risk ET and would like to know more about thrombosis in her condition and how to best manage her risk.
Risk-Adapted Therapy
Low-Risk PV
All patients with PV should receive counseling to mitigate cardiovascular risk factors, including smoking cessation, lifestyle modifications, and lipid-lowering therapy, as indicated. Furthermore, all PV patients should receive acetylsalicylic acid (ASA) to decrease their risk for thrombosis and control vasomotor symptoms.55,87 Aspirin 81 to 100 mg daily is the preferred regimen because it provides adequate antithrombotic effect without the associated bleeding risk of higher-dose aspirin.88 Low-risk PV patients should also receive periodic phlebotomies to reduce and maintain their hematocrit below 45%. This recommendation is based on the results of the Cytoreductive Therapy in Polycythemia Vera (CYTO PV) randomized controlled trial. In the CYTO PV study, patients receiving more intense therapy to maintain the hematocrit below 45% had a lower incidence of cardiovascular-related deaths or major thrombotic events than those with hematocrit goals of 45% to 50% (2.7% versus 9.8%).89 Cytoreduction is an option for low-risk patients who do not tolerate phlebotomy or require frequent phlebotomy, or who have disease-related bleeding, severe symptoms, symptomatic splenomegaly, or progressive leukocytosis.38
High-Risk PV
Patients older than 60 years and/or with a history of thrombosis should be considered for cytoreductive therapy in addition to the above measures. Front-line cytoreductive therapies include hydroxyurea or interferon (IFN)- alfa.87 Hydroxyurea is a potent ribonucleotide reductase inhibitor that interferes with DNA repair and is the treatment of choice for most high-risk patients with PV.90 In a small trial hydroxyurea reduced the risk of thrombosis compared with historical controls treated with phlebotomy alone.91 Hydroxyurea is generally well tolerated; common side effects include cytopenias, nail changes, and mucosal and/or skin ulcers. Although never formally proven to be leukemogenic, this agent should be used with caution in younger patients.87 Indeed, in the original study, the rates of transformation were 5.9% and 1.5% for patients receiving hydroxyurea and phlebotomy alone,92 respectively, although an independent role for hydroxyurea in leukemic transformation was not supported in the much larger European Collaboration on Low-dose Aspirin in Polycythemia Vera (ECLAP) study.93 About 70% of patients will have a sustained response to hydroxyurea,94 while the remaining patients become resistant to or intolerant of the drug. Resistant individuals have a higher risk of progression to acute leukemia and death.95
IFN alfa is a pleiotropic antitumor agent that has found application in many types of malignancies96 and is sometimes employed as treatment for patients with newly diagnosed high-risk PV. Early studies showed responses in up to 100% of cases,97,98 albeit at the expense of a high discontinuation rate due to adverse events, such as flu-like symptoms, fatigue, and neuropsychiatric manifestations.99 A newer formulation of the drug obtained by adding a polyethylene glycol (PEG) moiety to the native IFN alfa molecule (PEG-IFN alfa) was shown to have a longer half-life, greater stability, less immunogenicity, and, potentially, better tolerability.100 Pilot phase 2 trials of PEG-IFN alfa-2a demonstrated its remarkable activity, with symptomatic and hematologic responses seen in the majority of patients (which, in some cases, persisted beyond discontinuation), and reasonable tolerability, with long-term discontinuation rates of around 20% to 30%.101–103 In some patients JAK2V617F became undetectable over time.104 Results of 2 ongoing trials, MDP-RC111 (single-arm study, PEG-IFN alfa-2a in high-risk PV or ET [NCT01259817]) and MPD-RC112 (randomized controlled trial, PEG-IFN alfa-2a versus hydroxyurea in the same population [NCT01258856]), will shed light on the role of PEG-IFN alfa in the management of patients with high-risk PV or ET. In 2 phase 2 studies of PEG-IFN alfa-2b, complete responses were seen in 70% to 100% of patients and discontinuation occurred in around a third of cases.105,106 A new, longer-acting formulation of PEG-IFN alfa-2a (peg-proline INF alfa-2b, AOP2014) is also undergoing clinical development.107,108
The approach to treatment of PV based on thrombotic risk level is illustrated in Figure 1.
Very Low- and Low-Risk ET
Like patients with PV, individuals with ET should undergo rigorous cardiovascular risk management and generally receive ASA to decrease their thrombotic risk and improve symptom control. Antiplatelet therapy may not be warranted in patients with documented
Intermediate-Risk ET
This category includes patients older than 60 years but without thrombosis or JAK2 mutations. These individuals would have been considered high risk (and thus candidates for cytoreductive therapy) according to the traditional risk stratification. Guidelines currently recommend ASA as the sole therapy for these patients, while reserving cytoreduction for those who experience thrombosis (ie, become high-risk) or have uncontrolled vasomotor or general symptoms, symptomatic splenomegaly, symptomatic thrombocytosis, or progressive leukocytosis.
High-Risk ET
For patients with ET in need of cytoreductive therapy (ie, those with prior thrombosis or older than 60 years with a JAK2V617F mutation), first-line options include hydroxyurea, IFN, and anagrelide. Hydroxyurea remains the treatment of choice in the majority of patients.110 In a seminal study, 114 patients with ET were randomly assigned to either observation or hydroxyurea treatment with the goal of maintaining the platelet count below 600 × 103/µL. At a median follow-up of 27 months, patients in the hydroxyurea group had a lower thrombosis rate (3.6% versus 24%, P = 0.003) and longer thrombosis-free survival, regardless of the use of antiplatelet drugs.64
Anagrelide, a selective inhibitor of megakaryocytic differentiation and proliferation, was compared with hydroxyurea in patients with ET in 2 randomized trials. In the first (N = 809), the group receiving anagrelide had a higher risk of arterial thrombosis, major bleeding, and fibrotic evolution, but lower incidence of venous thrombosis. Hydroxyurea was better tolerated, mainly due to anagrelide-related cardiovascular adverse events.111 As a result of this study, hydroxyurea is often preferred to anagrelide as front-line therapy for patients with newly diagnosed high-risk ET. In the second, more recent study (N = 259), however, the 2 agents proved equivalent in terms of major or minor arterial or venous thrombosis, as well as discontinuation rate.112 The discrepancy between the 2 trials may be partly explained by the different ET diagnostic criteria used, with the latter only enrolling patients with WHO-defined true ET, while the former utilized Polycythemia Vera Study Group-ET diagnostic criteria that included patients with increases in other blood counts or varying degrees of marrow fibrosis.
Interferons were studied in ET in parallel with PV. PEG-IFN alfa-2a proved effective in patients with ET, with responses observed in 80% of patients.103 PEG-IFN alfa-2b produced similar results, with responses in 70% to 90% of patients in small studies and discontinuation observed in 20% to 38% of cases.105,106,113 Because the very long-term leukemogenic potential of hydroxyurea has remained somewhat uncertain, anagrelide or IFN might be preferable choices in younger patients.
The approach to treatment of ET based on thrombotic risk level is illustrated in Figure 2.
Assessing Response to Therapy
For both patients with PV and ET the endpoint of treatment set forth for clinical trials has been the achievement of a clinicohematologic response. However, studies have failed to show a correlation between response and reduction of the thrombohemorrhagic risk.114 Therefore, proposed clinical trial response criteria were revised to include absence of hemorrhagic or thrombotic events as part of the definition of response (Table 3).94
Cases Continued
Patient A was initially treated with phlebotomies and his blood counts were subsequently controlled with hydroxyurea, which he took uninterruptedly at an average dose of 2.5 g daily. He also took ASA daily throughout. Now, 18 months after the start of therapy, he presents with a complaint of fatigue for the past 3 months, which more recently has been associated with recurrent itching. A repeat CBC shows a WBC count of 17,200/µL, hemoglobin 181 g/L, and platelets 940 × 103/µL.
Patient B presents for scheduled follow-up. She has had no further thrombotic episodes. However, she spontaneously discontinued hydroxyurea 1 month ago because of worsening mouth ulcers that impaired her ability to eat even small meals. She seeks recommendations for further treatment options.
Approach to Patients Refractory to or Intolerant of First-Line Therapy
According to the European LeukemiaNet recommendations, an inadequate response to hydroxyurea in patients with PV (or myelofibrosis) is defined as a need for phlebotomy to maintain hematocrit below < 45%, platelet count > 400 × 103/µL, and a WBC count > 10,000/µL, or failure to reduce splenomegaly > 10 cm by > 50% at a dose of ≥ 2 g/day or maximum tolerated dose. Historically, treatment options for patients with PV or ET who failed first-line therapy (most commonly hydroxyurea) have included alkylating agents, such as busulfan, chlorambucil, or pipobroman, and phosphorus (P)-32. However, the use of these drugs is limited by the associated risk of leukemic transformation.93,115,116 The use of IFN (or anagrelide for ET) is often considered in patients previously treated with hydroxyurea, and vice versa.
Ruxolitinib is a JAK1 and JAK2 inhibitor currently approved for the treatment of PV patients refractory to or intolerant of hydroxyurea.7 Following promising results of a phase 2 trial,117 ruxolitinib 10 mg twice daily was compared with best available therapy in the pivotal RESPONSE trial (N = 222). Ruxolitinib proved superior in achieving hematocrit control, reduction of spleen volume, and improvement of symptoms. Grade 3-4 hematologic toxicity was infrequent and similar in the 2 arms.118 In addition, longer follow-up of that study suggested a lower rate of thrombotic events in patients receiving ruxolitinib (1.8 versus 8.2 per 100 patient-years).119 In a similarly designed randomized phase 3 study in PV patients without splenomegaly (RESPONSE-2), more patients in the ruxolitinib arm had hematocrit reduction without an increase in toxicity. Based on the results of the above studies, ruxolitinib can be considered a standard of care for second-line therapy in this post-hydroxyurea patient population.120
Ruxolitinib is also being tested in patients with high-risk ET who have become resistant to, or were intolerant of hydroxyurea, but currently has no approved indication in this setting.121,122 Common side effects of ruxolitinib include cytopenias (especially anemia), increased risk of infections, hyperlipidemia, and increased risk of non-melanoma skin cancer.
Novel Agents
Novel agents that have been studied in patients with PV and ET are histone deacetylase inhibitors, murine double minute 2 (MDM2, or HDM2 for their human counterpart) inhibitors (which restore the function of p53), Bcl-2 homology domain 3 mimetics such as navitoclax and venetoclax, and, for patients with ET, the telomerase inhibitor imetelstat.123
Disease Evolution
Cases Continued
Patient A’s PV has been well controlled with PEG-IFN alfa-2a 90 μg subcutaneously weekly. However, he now presents with a complaint of worsening fatigue and early satiety. On exam the patient appears ill and splenomegaly is appreciated 12 cm below the costal margin. CBC shows a WBC count of 2600/µL, hemoglobin 73 g/L, and platelets 122 × 103/µL. Peripheral blood smear reveals leukoerythroblastosis and dacrocytosis. CBC 6 months ago was normal. A bone marrow biopsy is consistent with myelofibrosis.
After discontinuing hydroxyurea, patient B’s ET has been well controlled with anagrelide. However, for the past 4 weeks she has complained of severe fatigue and easy bruising. Physical exam reveals a pale, ill-appearing woman with scattered bruises. CBC shows a WBC count of 14,600/µL with 44% myeloblasts, hemoglobin 73 g/L, and platelets 22 × 103/µL. CBC 6 months ago was normal. A bone marrow biopsy is consistent with leukemic transformation of ET.
Post-PV/Post-ET Myelofibrosis
Diagnostic criteria for post-PV and post-ET myelofibrosis are outlined in Table 4.
Leukemic Transformation
The presence of more than 20% blasts in peripheral blood or bone marrow in a patient with MPN defines leukemic transformation. This occurs in up to 5% to 10% of patients and may or may not be preceded by a myelofibrosis phase.126 In cases of extramedullary transformation, a lower percentage of blasts can be seen in the bone marrow compared to the peripheral blood. The pathogenesis of leukemic transformation has remained elusive, but it is believed to be associated with genetic instability, which facilitates the acquisition of additional mutations, including those of TET2, ASXL1, EZH2 and DNMT3, IDH1/2, and TP53.127
Clinical risk factors for leukemic transformation include advanced age, karyotypic abnormalities, prior therapy with alkylating agents or P-32, splenectomy, increased peripheral blood or bone marrow blasts, leukocytosis, anemia, thrombocytopenia, and cytogenetic abnormalities. Hydroxyurea, interferon, and ruxolitinib have not been shown to have leukemogenic potential thus far. Prognosis of leukemic transformation is uniformly poor and patient survival rarely exceeds 6 months.
There is no standard of care for leukemic transformation of MPN (MPN-LT). Treatment options range from low-intensity regimens to more aggressive AML-type induction chemotherapy. No strategy appears clearly superior to others.128 Hematopoietic stem cell transplantation is the only therapy that provides clinically meaningful benefit to patients,129 but it is applicable only to a minority of patients with chemosensitive disease and good performance status.130 Notable experimental approaches to MPN–LT include hypomethylating agents, such as decitabine131 or azacitidine,132 with or without ruxolitinib.133-135
Conclusion
PV and ET are rare, chronic myeloid disorders. Patients typically experience a long clinical course and enjoy near-normal quality of life if properly managed. The 2 most important life-limiting complications of PV and ET are thrombohemorrhagic events and myelofibrosis/AML transformation. Vascular events are at least in part preventable with counseling on risk factors, phlebotomy (for patients with PV), antiplatelet therapy, and cytoreduction with hydroxyurea, IFNs, or anagrelide (for patients with ET). In addition, ruxolitinib was recently approved for PV patients after hydroxyurea failure. PV/ET transformation in myelofibrosis or AML is part of the natural history of the disease and no therapy has been shown to prevent it. Treatment follows recommendations set forth for PMF and AML, but results are generally poorer and novel strategies are needed to improve patients’ outcomes.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
1. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
2. Alvarez-Larran A, Pereira A, Arellano-Rodrigo E, et al. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol 2013;161:865–71.
3. Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013;88:507–16.
4. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets 2006;17:528–44.
5. James C, Ugo V, Couedic J-PL, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–8.
6. Kralovics R, Passamonti F, Buser A, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders.N Engl J Med 2005;352:1779–90.
7. Verstovsek S, Mesa R, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
8. Harrison C, Kiladjian J, Al-Ali H, et al. JAK Inhibition with ruxolitinib versus best available therapy for myelofibrosis.N Engl J Med 2012;366:787–98.
9. Berglund S, Zettervall O. Incidence of polycythemia vera in a defined population. Eur J Haematol 1992;48:20–6.
10. Rozman C, Giralt M, Feliu E, et al. Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 1991;67:2658–63.
11. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004;117:755–61.
12. Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 2012;30:2995–3001.
13. Wolanskyj AP, Schwager SM, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc 2006;81:159–66.
14. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol 2016;174:382–96.
15. Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 2014;89:581–7.
16. Moulard O, Mehta J, Fryzek J, et al. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol 2014;92:289–97.
17. Stein BL, Gotlib J, Arcasoy M, et al. Historical views, conventional approaches, and evolving management strategies for myeloproliferative neoplasms. J Natl Compr Canc Netw 2015;13:424–34.
18. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97.
19. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–61.
20. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356:459–68.
21. Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007;21:1960–3.
22. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011;117:2813–6.
23. Abe M, Suzuki K, Inagaki O, et al. A novel MPL point mutation resulting in thrombopoietin-independent activation. Leukemia 2002;16:1500–6.
24. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
25. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–6.
26. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405.
27. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90.
28. Wang WA, Groenendyk J, Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 2012;44:842–6.
29. Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit Rev Oncol Hematol 2016;98:375–89.
30. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
31. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
32. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005;23:2224–32.
33. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–8.
34. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103.
35. Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost 2013;11:1215–27.
36. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood 2013;122:2176-–84.
37. Pearson TC, Wetherley-Mein G. Vascular occlusive episodes and venous haematocrit in primary myeloproliferative polychytemia. Lancet 1978;2:1219–22.
38. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007;109:2446–52.
39. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013;27:1874–81.
40. Barbui T, Falanga A. Molecular biomarkers of thrombosis in myeloproliferative neoplasms. Thromb Res 2016;140 Suppl 1:S71–75.
41. Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005;366:1945–53.
42. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014;124:2507–13.
43. Qin Y, Wang X, Zhao C, et al. The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis. Int J Hematol 2015;102:170–80.
44. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica 2008;93:1412–4.
45. Lussana F, Dentali F, Abbate R, et al. Screening for thrombophilia and antithrombotic prophylaxis in pregnancy: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 2009;124:e19–25.
46. Dahabreh IJ, Zoi K, Giannouli S, et al. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res 2009;33:67–73.
47. Cho YU, Chi HS, Lee EH, et al. Comparison of clinicopathologic findings according to JAK2 V617F mutation in patients with essential thrombocythemia. Int J Hematol 2009;89:39–44.
48. Wolanskyj AP, Lasho TL, Schwager SM, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005;131:208–13.
49. Antonioli E, Guglielmelli P, Pancrazzi A, et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005;19:1847–9.
50. Palandri F, Catani L, Testoni N, et al. Long-term follow-up of 386 consecutive patients with essential thrombocythemia: safety of cytoreductive therapy. Am J Hematol 2009;84:215–20.
51. Chim CS, Sim JP, Chan CC, et al. Impact of JAK2V617F mutation on thrombosis and myeloid transformation in essential thrombocythemia: a multivariate analysis by Cox regression in 141 patients. Hematology 2010;15:187–92.
52. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007;110:840–6.
53. Carobbio A, Finazzi G, Antonioli E, et al. JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009;37:1016–21.
54. Alvarez-Larran A, Bellosillo B, Pereira A, et al. JAK2V617F monitoring in polycythemia vera and essential thrombocythemia: clinical usefulness for predicting myelofibrotic transformation and thrombotic events. Am J Hematol 2014;89:517–23.
55. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J 2015;5:e369.
56. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011;117:5857–9.
57. Alvarez-Larran A, Cervantes F, Bellosillo B, et al. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007;21:1218–23.
58. Jantunen R, Juvonen E, Ikkala E, et al. The predictive value of vascular risk factors and gender for the development of thrombotic complications in essential thrombocythemia. Ann Hematol 2001;80:74–8.
59. Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia 1999;13:150–4.
60. Haider M, Gangat N, Hanson C, Tefferi A. Splenomegaly and thrombosis risk in essential thrombocythemia: the mayo clinic experience. Am J Hematol 2016;91:E296–297.
61. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood 2008;112:3135–7.
62. Palandri F, Polverelli N, Catani L, et al. Impact of leukocytosis on thrombotic risk and survival in 532 patients with essential thrombocythemia: a retrospective study. Ann Hematol 2011;90:933–8.
63. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012;120:1409–11.
64. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–6.
65. van Genderen PJ, Mulder PG, Waleboer M, et al. Prevention and treatment of thrombotic complications in essential thrombocythaemia: efficacy and safety of aspirin. Br J Haematol 1997;97:179–84.
66. Storen EC, Tefferi A. Long-term use of anagrelide in young patients with essential thrombocythemia. Blood 2001;97:863–6.
67. De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica 2008;93:372–80.
68. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010;116:1205–10.
69. Palandri F, Polverelli N, Catani L, et al. Bleeding in essential thrombocythaemia: a retrospective analysis on 565 patients. Br J Haematol 2012;156:281–4.
70. Rotunno G, Mannarelli C, Guglielmelli P, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 2014;123:1552–5.
71. Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014;28:2300–3.
72. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014;123:1544–51.
73. Palandri F, Latagliata R, Polverelli N, et al. Mutations and long-term outcome of 217 young patients with essential thrombocythemia or early primary myelofibrosis. Leukemia 2015;29:1344–9.
74. Fu R, Xuan M, Zhou Y, et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: clinical implications in diagnosis, prognosis and treatment. Leukemia 2014;28:1912–4.
75. Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol 2014;89:E121–4.
76. Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8.
77. Rumi E, Pietra D, Guglielmelli P, et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013;121:4388–95.
78. Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008;112:141–9.
79. Gangat N, Wassie EA, Lasho TL, et al. Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol 2015;94:31–6.
80. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 2013;162:730–47.
81. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymph 2013;54:1989–95.
82. Landolfi R, Di Gennaro L, Nicolazzi MA, et al. Polycythemia vera: gender-related phenotypic differences. Intern Emerg Med 2012;7:509–15.
83. Winslow ER, Brunt LM, Drebin JA, et al. Portal vein thrombosis after splenectomy. Am J Surg 2002;184:631–6.
84. Smalberg JH, Arends LR, Valla DC, et al. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 2012;120:4921–8.
85. Dentali F, Squizzato A, Brivio L, et al. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood 2009;113:5617–23.
86. Pardanani A, Lasho TL, Hussein K, et al. JAK2V617F mutation screening as part of the hypercoagulable work-up in the absence of splanchnic venous thrombosis or overt myeloproliferative neoplasm: assessment of value in a series of 664 consecutive patients. Mayo Clin Proc 2008;83:457–9.
87. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011;29:761–70.
88. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004;350:114–24.
89. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013;368:22–33.
90. Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 2011;29:3907–13.
91. Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 1986;23:167–71.
92. Fruchtman SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997;34:17–23.
93. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 2005;105:2664–70.
94. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013;121:4778–81.
95. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012;119:1363–9.
96. Stein BL, Tiu RV. Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 2013;33:145–53.
97. Ludwig H, Cortelezzi A, Van Camp BG, et al. Treatment with recombinant interferon-alpha-2C: multiple myeloma and thrombocythaemia in myeloproliferative diseases. Oncology 1985;42 Suppl 1:19–25.
98. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer 2006;107:451–8.
99. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–15.
100. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs 2008;22:315–29.
101. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008;112:3065–72.
102. Turlure P, Cambier N, Roussel M, et al. Complete hematological, molecular and histological remissions without cytoreductive treatment lasting after pegylated-interferon {alpha}-2a (peg-IFN{alpha}-2a) therapy in polycythemia vera (PV): long term results of a phase 2 trial [abstract]. Blood 2011;118(21). Abstract 280.
103. Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 2009;27:5418–24.
104. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon a-2a. Blood 2013;122:893–901.
105. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon alpha-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer 2006;106:2397–405.
106. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer 2007;110:2012–18.
107. Them NC, Bagienski K, Berg T, et al. Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 2015;90:288–94.
108. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients [abstract]. Blood 2016;128(suppl 22). Abstract 475.
109. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol 1997;75:215–20.
110. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995;332:1132–7.
111. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005;353:33–45.
112. Gisslinger H, Gotic M, Holowiecki J, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013;121:1720–8.
113. Alvarado Y, Cortes J, Verstovsek S, et al. Pilot study of pegylated interferon-alpha 2b in patients with essential thrombocythemia. Cancer Chemother Pharmacol 2003;51:81–6.
114. Barosi G, Tefferi A, Barbui T, ad hoc committee ‘Definition of clinically relevant outcomes for contemporarily clinical trials in Ph-neg M. Do current response criteria in classical Ph-negative myeloproliferative neoplasms capture benefit for patients? Leukemia 2012;26:1148–9.
115. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 2011;29:2410–5.
116. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 2014;93:2037–43.
117. Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20.
118. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib in polycythemia vera resistant to or intolerant of hydroxyurea. N Engl J Med 2015; 372:426–35.
119. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016;101:821–9.
120. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017;18:88–99.
121. Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term results from a phase II open-label study of ruxolitinib in patients with essential thrombocythemia refractory to or intolerant of hydroxyurea [abstract]. Blood 2014;124. Abstract 1847.
122. Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib versus best available therapy for ET intolerant or resistant to hydroxycarbamide in a randomized trial. Blood 2017 Aug 9. pii: blood-2017-05-785790 .
123. Bose P, Verstovsek S. Drug development pipeline for myeloproliferative neoplasms: potential future impact on guidelines and management. J Natl Compr Canc Netw 2016;14:1613–24.
124. Cerquozzi S, Teffieri A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 2015;Nov 13;5:e366.
125. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 2008;111:3383–7.
126. Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007;31:737–40.
127. Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 2014;21:65–71.
128. Chihara D, Kantarjian HM, Newberry KJ, et al. Survival outcome of patients with acute myeloid leukemia transformed from myeloproliferative neoplasms [abstract]. Blood 2016;128. Abstract 1940.
129. Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood 2008;112:1628–37.
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work? Curr Hematol Malig Rep 2012;7:78–86.
131. Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res 2015;39:950–6.
132. Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 2010;116:3735–42.
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015;15:171–6.
134. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in patients with blast-phase MPN and post-MPN AML: results of a phase I study (Myeloproliferative Disorders Research Consortium 109 trial) [abstract]. Blood 2016;128. Abstract 1124.
135. Bose P, Verstovsek S, Gasior Y, et al. Phase I/II study of ruxolitinib (RUX) with decitabine (DAC) in patients with post-myeloproliferative neoplasm acute myeloid leukemia (post-MPN AML): phase I results [abstract]. Blood 2016;128. Abstract 4262.
A Year 3 Progress Report on Graduate Medical Education Expansion in the Veterans Choice Act
The VHA is the largest healthcare delivery system in the U.S. It includes 146 medical centers (VAMCs), 1,063 community-based outpatient centers (CBOCs) and various other sites of care. General Omar Bradley, the first VA Secretary, established education as one of VA’s 4 statutory missions in Policy Memorandum No.2.1 In addition to training physicians to care for active-duty service members and veterans, 38 USC §7302 directs the VA to assist in providing an adequate supply of health personnel. The 4 statutory missions of the VA are inclusive of not only developing, operating, and maintaining a health care system for veterans, but also including contingency support services as part of emergency preparedness, conducting research, and offering a program of education for health professions.
Background
Today, with few exceptions, the VHA does not act as a graduate medical education (GME) sponsoring institution. Through its Office of Academic Affiliations (OAA), the VHA develops partnerships with Liaison Committee for Medical Education (LCME)/American Osteopathic Association (AOA)-approved medical colleges/universities and with institutions that sponsor Accreditation Council for Graduate Medical Education (ACGME)/AOA-accredited residency program-sponsoring institutions. These collaborations include 144 out of 149 allopathic medical schools and all 34 osteopathic medical schools. The VHA provided training to 43,565 medical residents and 24,683 medical students through these partnerships in 2017.2 Since funding of the GME positions is not provided through the Centers for Medicare & Medicaid Services (CMS), program sponsors may use these partnerships to expand GME positions beyond their funding (but not ACGME) cap.
The gap between supply and demand of physicians continues to grow nationally.3,4 This gap is particularly significant in rural and other underserved areas. U.S. Census Bureau data show that about 5 million veterans (24%) live in rural areas.5 Compared with the urban veteran population, the rural veteran experiences higher disease prevalence and lower physical and mental quality-of-life scores.6 Addressing the problem of physician shortages is a mission-critical priority for the VHA.7
With an eye toward enhancing 2 of the 4 statutory missions of the VA and to mitigate the shortage of physicians and improve the access of veterans to VHA medical services, on August 7, 2014, the Veterans Access, Choice, and Accountability Act of 2014 (Public Law [PL] 113-146), known as the Choice Act was enacted.8 Title III, §301(b) of the Choice Act requires VHA to increase GME residency positions by:
Establishing new medical residency programs, or ensuring that already established medical residency programs have a sufficient number of residency positions, at any VHA medical facility that is: (a) experiencing a shortage of physicians and (b) located in a community that is designated as a health professional shortage area.
The legislation specifies that priority must be placed on medical occupations that experience the largest staffing shortages throughout the VHA and “programs in primary care, mental health, and any other specialty that the Secretary of the VA determines appropriate.” The Choice Act authorized the VHA to increase the number of GME residency positions by up to 1,500 over a 5-year period. In December 2016, as amended by PL 114–315, Title VI, §617(a), this authorization was extended by another 5 years for a total of 10 years and will run through 2024.9
GME Development/Distribution
To distribute these newly created GME positions as mandated by Congress, the OAA is using a system with 3 types of request for proposal (RFP) applications. These include planning, infrastructure, and position grants. This phased approach was taken with the understanding that the development of new training sites requires a properly staffed education office and dedicated faculty time. Planning and infrastructure grants provide start-up funds for smaller VAMCs, allowing them to keep facility resources focused on their clinical mission.
Planning grants (of up to $250,000 over 2 years) primarily were designed for VA facilities with no or low numbers of physician residents at the desired teaching location. Priority was given to facilities in rural and/or underserved areas as well as those developing new affiliations. Applications were reviewed by OAA staff along with peer-selected Designated Education Officers (DEOs) from VA facilities across the nation that were not applying for the grants. Awards were based on the priorities mentioned earlier, with additional credit for programs focused on 2 VHA fundamental services areas—primary care and/or mental health training. Facilities receiving planning grants were mentored by an OAA physician staff member, anticipating a 2- to 3-year time line to request positions and begin GME training.
Infrastructure grants (of up to $520,000 used over 2-3 years) were designed as bridge funds after approval of Veterans Access, Choice, and Accountability Act (VACAA) GME positions. Infrastructure grants are appropriate to sustain a local education office, develop VA faculty, purchase equipment, and make minor modifications to the clinical space in the VAMCs or CBOCs to enhance the learning environment during the period before VA supportive funds from the Veterans Equitable Resource Allocation (VERA) (similar to indirect GME funds from CMS) become available. Applications were managed the same as planning grant submissions.
Position RFPs, unlike planning and infrastructure RFPs, are available to all VAMCs. The primary purpose of the VACAA Position RFP is to fund new positions in primary care and psychiatry. Graduate medical education positions in subspecialty programs also are considered when there is documentation of critical need to improve access to these services. Applications were reviewed by OAA staff along with selected DEOs from VA facilities around the U.S. Award criteria prioritized primary care (family medicine, internal medicine, geriatrics), and mental health (psychiatry and psychiatry subspecialties). Priority also was given to positions in areas with a documented shortage of physicians and areas with high concentrations of veterans.
Current Progress
To date the OAA has offered 3 RFP cycles consisting of planning/infrastructure grants, and 4 RFP cycles for salary/benefit support for additional resident full-time equivalent (FTE) positions. Resident positions were defined as residency or fellowship FTEs that were part of an ACGME or AOA-accredited training program. Figure 1 illustrates the geographic distribution of awarded GME positions. 
In primary care specialties (family medicine, internal medicine, and geriatrics, a total of 349.4 FTE positions have been approved (Table 1). Due to a low number of applications, only 6.3 of these positions were awarded in geriatrics. In mental health, 167.6 FTE positions have been approved, whereas in critical needs specialties (needed to support rural/underserved healthcare and improve specialty access) 256.5 FTE positions have been added. 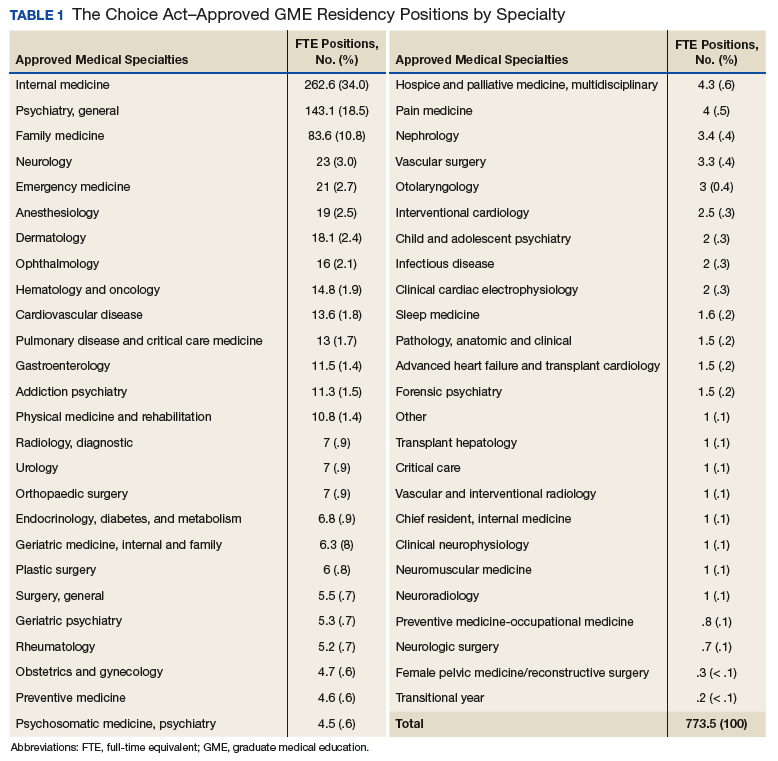
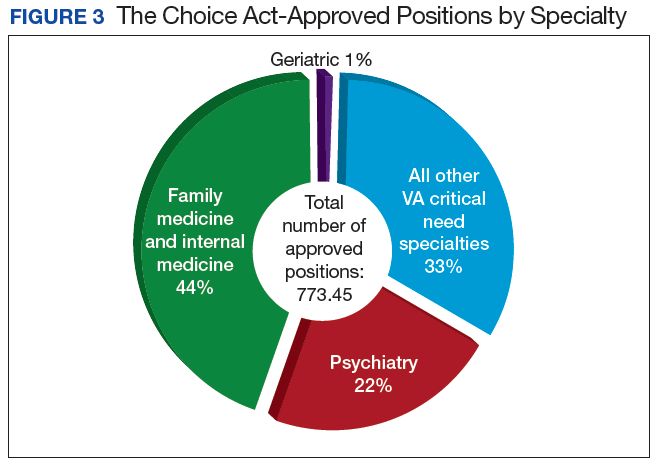
Discussion
There are several important desired short-term outcomes from VACAA. The first is improved access to high-quality care for both rural and urban veterans. There is an emphasis on primary care and mental health because shortages in these areas across the U.S. are well established.3,4,10 Likewise, rural areas have been prioritized because often there is a disparity of care. 
One area of concern is the small number of applicants in geriatrics. Even with VACAA specifically targeting geriatrics as a primary care specialty, we have only received enough applications to approve 6.3 positions over the first 3 years of the program. As the veteran and overall population in the U.S. ages, it is important to develop a medical workforce that is willing and able to address their needs.
The VACAA statute is not intended to alter medical students’ career choice but rather to provide funded positions for those choosing primary care, geriatrics, psychiatry (including psychiatric subspecialties), and experience in the VA clinical settings. The hope is that this experience will encourage practitioners to competently care for veterans after training in the VA and/or other civilian settings.
By enabling smaller VA facilities to become training sites through planning and infrastructure grants, residents have the opportunity to gain experience in more rural settings. Physicians who choose to train in rural areas are likely to spend time practicing in those areas after they complete training.15 The process of developing facilities with no GME into training sitestakes time and resources. Establishing an education office and choosing site directors and core faculty are all important steps that must be done before resident rotations begin. Resources provided through VACAA have enabled the VHA to reduce the number of VAMCs with no GME activity to just 3.
Another benefit of VACAA GME expansion is the opportunity to engage new LCME/AOA-accredited medical schools and ACGME/AOA-accredited residency-sponsoring institutions.16,17 Representatives of these institutions may have perceived a reluctance of their local VAs to develop GME affiliations in the past. This statute has enabled many VAMCs to use nontraditional training sites and modalities to overcome barriers and create new academic affiliations.
However, VACAA only provides funds for training that occurs in established VA sites of care. This can hinder the development of partnerships where other funding sources are required for non-VA rotations. Another VACAA limitation is that it does not fund undergraduate medical education as does the Armed Forces Health Professional Scholarship Program (HPSP). In addition, the primary financial relationship is between the VA and the sponsoring institution, thus VHA cannot send residents to underserved locations.
Conclusion
The VHA has a rich tradition of educating physician and other health care providers in the U.S. More than 60% of U.S. trained physicians received a portion of their training through VHA.2 Through VACAA GME expansion initiative, the 113th Congress has asked VHA to continue its important training mission “to bind up the Nations wounds” and “to care for him who shall have borne the battle.”18
Acknowledgments
In memoriam – Robert Louis Jesse MD, PhD. Dr. Jesse, the Chief of the Office of Academic Affiliations passed away on September 2, 2017, at age 64. He had an illustrious medical career as a cardiologist and served in many leadership roles including Principal Deputy Under Secretary for Health in the U.S. Department of Veterans Affairs. His expertise, visionary leadership, and friendship will be missed by all involved in the VA’s academic training mission but particularly by those of us who worked for and with him at OAA.
1. U.S. Department of Veteran Affairs. Policy Memorandum No. 2. Policy in association of veterans’ hospitals with medical schools. https://www.va.gov/oaa/Archive/PolicyMemo2.pdf. Published January 30, 1947. Accessed December 13, 2017.
2. U.S. Department of Veteran Affairs, Office of Academic Affiliations. 2017 statistics: health professions trainees. https://www .va.gov/OAA/docs/OAA_Statistics.pdf. Accessed January 8, 2018.
3. IHS, Inc. The complexities of physician supply and demand 2016 update: projections from 2014 to 2025, final report. https://www.aamc.org/download/458082/data/2016_complexities_of_supply_and_demand_projections.pdf. Published April 5, 2016. Accessed December 13, 2017.
4. Petterson SM, Liaw WR, Tran C, Bazemore AW. Estimating the residency expansion required to avoid projected primary care physician shortages by 2035. Ann Fam Med. 2015;13(2):107-114.
5. Holder KA. Veterans in rural America 2011-2015. https://www.census.gov/content/dam/Census/library/publica tions/2017/acs/acs-36.pdf. Published January 2017. Accessed January 18, 2018.
6. Weeks WB, Wallace AE, Wang S, Lee A, Kazis LE. Rural-urban disparities in health-related quality of life within disease categories of veterans. J Rural Health. 2006;22(3):204-211.
7. U.S. Government Accountability Office. GAO-18-124. VHA Physician Staffing and Recruitment. https://www.gao.gov/assets/690/687853.pdf. Published October 19, 2017. Accessed January 23, 2018.
8. Veterans Access, Choice, and Accountability Act, section 301 (b): Increase of graduate medical education residency positions, 38 USC § 74 (2014) .
9. Jeff Miller and Richard Blumenthal Veterans Health Care and Benefits Improvement Act of 2016, 38 USC §101 (2016).
10. Thomas KC, Ellis AR, Konrad TR, Holzer CE, Morrissey JP. County-level estimates of mental health professional shortage in the United States. Psychiatr Serv. 2009;60(10):1323-1328.
11. Garibaldi RA, Popkave C, Bylsma W. Career plans for trainees in internal medicine residency programs. Acad Med. 2005;80(5):507-512.
12. West CP, Dupras DM. General medicine vs subspecialty career plans among internal medicine residents. JAMA. 2012;308(21):2241-2247.
13. Stimmel B, Haddow S, Smith L. The practice of general internal medicine by subspecialists. J Urban Health. 1998;75(1):184-190.
14. Shea JA, Kleetke PR, Wozniak GD, Polsky D, Escarce JJ. Self-reported physician specialties and the primary care content of medical practice: a study of the AMA physician masterfile. American Medical Association. Med Care. 1999;37(4):333-338.
15. Rabinowitz HK, Diamond JJ, Markham FW, Paynter NP. Critical factors for designing
16. Accredited MD programs in the United States. http://lcme.org /directory/accredited-u-s-programs/. Updated December 12, 2017. Accessed January 8, 2018.
17. Osteopathic medical schools. http://www.osteopathic.org/in side-aoa/about/affiliates/Pages/osteopathic-medical-schools.aspx Published 2017. Accessed January 8, 2018.
18. Lincoln A. Second inaugural address. https://www.va.gov/opa/publications/celebrate/vamotto.pdf. Accessed January 8. 2018.
The VHA is the largest healthcare delivery system in the U.S. It includes 146 medical centers (VAMCs), 1,063 community-based outpatient centers (CBOCs) and various other sites of care. General Omar Bradley, the first VA Secretary, established education as one of VA’s 4 statutory missions in Policy Memorandum No.2.1 In addition to training physicians to care for active-duty service members and veterans, 38 USC §7302 directs the VA to assist in providing an adequate supply of health personnel. The 4 statutory missions of the VA are inclusive of not only developing, operating, and maintaining a health care system for veterans, but also including contingency support services as part of emergency preparedness, conducting research, and offering a program of education for health professions.
Background
Today, with few exceptions, the VHA does not act as a graduate medical education (GME) sponsoring institution. Through its Office of Academic Affiliations (OAA), the VHA develops partnerships with Liaison Committee for Medical Education (LCME)/American Osteopathic Association (AOA)-approved medical colleges/universities and with institutions that sponsor Accreditation Council for Graduate Medical Education (ACGME)/AOA-accredited residency program-sponsoring institutions. These collaborations include 144 out of 149 allopathic medical schools and all 34 osteopathic medical schools. The VHA provided training to 43,565 medical residents and 24,683 medical students through these partnerships in 2017.2 Since funding of the GME positions is not provided through the Centers for Medicare & Medicaid Services (CMS), program sponsors may use these partnerships to expand GME positions beyond their funding (but not ACGME) cap.
The gap between supply and demand of physicians continues to grow nationally.3,4 This gap is particularly significant in rural and other underserved areas. U.S. Census Bureau data show that about 5 million veterans (24%) live in rural areas.5 Compared with the urban veteran population, the rural veteran experiences higher disease prevalence and lower physical and mental quality-of-life scores.6 Addressing the problem of physician shortages is a mission-critical priority for the VHA.7
With an eye toward enhancing 2 of the 4 statutory missions of the VA and to mitigate the shortage of physicians and improve the access of veterans to VHA medical services, on August 7, 2014, the Veterans Access, Choice, and Accountability Act of 2014 (Public Law [PL] 113-146), known as the Choice Act was enacted.8 Title III, §301(b) of the Choice Act requires VHA to increase GME residency positions by:
Establishing new medical residency programs, or ensuring that already established medical residency programs have a sufficient number of residency positions, at any VHA medical facility that is: (a) experiencing a shortage of physicians and (b) located in a community that is designated as a health professional shortage area.
The legislation specifies that priority must be placed on medical occupations that experience the largest staffing shortages throughout the VHA and “programs in primary care, mental health, and any other specialty that the Secretary of the VA determines appropriate.” The Choice Act authorized the VHA to increase the number of GME residency positions by up to 1,500 over a 5-year period. In December 2016, as amended by PL 114–315, Title VI, §617(a), this authorization was extended by another 5 years for a total of 10 years and will run through 2024.9
GME Development/Distribution
To distribute these newly created GME positions as mandated by Congress, the OAA is using a system with 3 types of request for proposal (RFP) applications. These include planning, infrastructure, and position grants. This phased approach was taken with the understanding that the development of new training sites requires a properly staffed education office and dedicated faculty time. Planning and infrastructure grants provide start-up funds for smaller VAMCs, allowing them to keep facility resources focused on their clinical mission.
Planning grants (of up to $250,000 over 2 years) primarily were designed for VA facilities with no or low numbers of physician residents at the desired teaching location. Priority was given to facilities in rural and/or underserved areas as well as those developing new affiliations. Applications were reviewed by OAA staff along with peer-selected Designated Education Officers (DEOs) from VA facilities across the nation that were not applying for the grants. Awards were based on the priorities mentioned earlier, with additional credit for programs focused on 2 VHA fundamental services areas—primary care and/or mental health training. Facilities receiving planning grants were mentored by an OAA physician staff member, anticipating a 2- to 3-year time line to request positions and begin GME training.
Infrastructure grants (of up to $520,000 used over 2-3 years) were designed as bridge funds after approval of Veterans Access, Choice, and Accountability Act (VACAA) GME positions. Infrastructure grants are appropriate to sustain a local education office, develop VA faculty, purchase equipment, and make minor modifications to the clinical space in the VAMCs or CBOCs to enhance the learning environment during the period before VA supportive funds from the Veterans Equitable Resource Allocation (VERA) (similar to indirect GME funds from CMS) become available. Applications were managed the same as planning grant submissions.
Position RFPs, unlike planning and infrastructure RFPs, are available to all VAMCs. The primary purpose of the VACAA Position RFP is to fund new positions in primary care and psychiatry. Graduate medical education positions in subspecialty programs also are considered when there is documentation of critical need to improve access to these services. Applications were reviewed by OAA staff along with selected DEOs from VA facilities around the U.S. Award criteria prioritized primary care (family medicine, internal medicine, geriatrics), and mental health (psychiatry and psychiatry subspecialties). Priority also was given to positions in areas with a documented shortage of physicians and areas with high concentrations of veterans.
Current Progress
To date the OAA has offered 3 RFP cycles consisting of planning/infrastructure grants, and 4 RFP cycles for salary/benefit support for additional resident full-time equivalent (FTE) positions. Resident positions were defined as residency or fellowship FTEs that were part of an ACGME or AOA-accredited training program. Figure 1 illustrates the geographic distribution of awarded GME positions.
In primary care specialties (family medicine, internal medicine, and geriatrics, a total of 349.4 FTE positions have been approved (Table 1). Due to a low number of applications, only 6.3 of these positions were awarded in geriatrics. In mental health, 167.6 FTE positions have been approved, whereas in critical needs specialties (needed to support rural/underserved healthcare and improve specialty access) 256.5 FTE positions have been added.
Discussion
There are several important desired short-term outcomes from VACAA. The first is improved access to high-quality care for both rural and urban veterans. There is an emphasis on primary care and mental health because shortages in these areas across the U.S. are well established.3,4,10 Likewise, rural areas have been prioritized because often there is a disparity of care.
One area of concern is the small number of applicants in geriatrics. Even with VACAA specifically targeting geriatrics as a primary care specialty, we have only received enough applications to approve 6.3 positions over the first 3 years of the program. As the veteran and overall population in the U.S. ages, it is important to develop a medical workforce that is willing and able to address their needs.
The VACAA statute is not intended to alter medical students’ career choice but rather to provide funded positions for those choosing primary care, geriatrics, psychiatry (including psychiatric subspecialties), and experience in the VA clinical settings. The hope is that this experience will encourage practitioners to competently care for veterans after training in the VA and/or other civilian settings.
By enabling smaller VA facilities to become training sites through planning and infrastructure grants, residents have the opportunity to gain experience in more rural settings. Physicians who choose to train in rural areas are likely to spend time practicing in those areas after they complete training.15 The process of developing facilities with no GME into training sitestakes time and resources. Establishing an education office and choosing site directors and core faculty are all important steps that must be done before resident rotations begin. Resources provided through VACAA have enabled the VHA to reduce the number of VAMCs with no GME activity to just 3.
Another benefit of VACAA GME expansion is the opportunity to engage new LCME/AOA-accredited medical schools and ACGME/AOA-accredited residency-sponsoring institutions.16,17 Representatives of these institutions may have perceived a reluctance of their local VAs to develop GME affiliations in the past. This statute has enabled many VAMCs to use nontraditional training sites and modalities to overcome barriers and create new academic affiliations.
However, VACAA only provides funds for training that occurs in established VA sites of care. This can hinder the development of partnerships where other funding sources are required for non-VA rotations. Another VACAA limitation is that it does not fund undergraduate medical education as does the Armed Forces Health Professional Scholarship Program (HPSP). In addition, the primary financial relationship is between the VA and the sponsoring institution, thus VHA cannot send residents to underserved locations.
Conclusion
The VHA has a rich tradition of educating physician and other health care providers in the U.S. More than 60% of U.S. trained physicians received a portion of their training through VHA.2 Through VACAA GME expansion initiative, the 113th Congress has asked VHA to continue its important training mission “to bind up the Nations wounds” and “to care for him who shall have borne the battle.”18
Acknowledgments
In memoriam – Robert Louis Jesse MD, PhD. Dr. Jesse, the Chief of the Office of Academic Affiliations passed away on September 2, 2017, at age 64. He had an illustrious medical career as a cardiologist and served in many leadership roles including Principal Deputy Under Secretary for Health in the U.S. Department of Veterans Affairs. His expertise, visionary leadership, and friendship will be missed by all involved in the VA’s academic training mission but particularly by those of us who worked for and with him at OAA.
The VHA is the largest healthcare delivery system in the U.S. It includes 146 medical centers (VAMCs), 1,063 community-based outpatient centers (CBOCs) and various other sites of care. General Omar Bradley, the first VA Secretary, established education as one of VA’s 4 statutory missions in Policy Memorandum No.2.1 In addition to training physicians to care for active-duty service members and veterans, 38 USC §7302 directs the VA to assist in providing an adequate supply of health personnel. The 4 statutory missions of the VA are inclusive of not only developing, operating, and maintaining a health care system for veterans, but also including contingency support services as part of emergency preparedness, conducting research, and offering a program of education for health professions.
Background
Today, with few exceptions, the VHA does not act as a graduate medical education (GME) sponsoring institution. Through its Office of Academic Affiliations (OAA), the VHA develops partnerships with Liaison Committee for Medical Education (LCME)/American Osteopathic Association (AOA)-approved medical colleges/universities and with institutions that sponsor Accreditation Council for Graduate Medical Education (ACGME)/AOA-accredited residency program-sponsoring institutions. These collaborations include 144 out of 149 allopathic medical schools and all 34 osteopathic medical schools. The VHA provided training to 43,565 medical residents and 24,683 medical students through these partnerships in 2017.2 Since funding of the GME positions is not provided through the Centers for Medicare & Medicaid Services (CMS), program sponsors may use these partnerships to expand GME positions beyond their funding (but not ACGME) cap.
The gap between supply and demand of physicians continues to grow nationally.3,4 This gap is particularly significant in rural and other underserved areas. U.S. Census Bureau data show that about 5 million veterans (24%) live in rural areas.5 Compared with the urban veteran population, the rural veteran experiences higher disease prevalence and lower physical and mental quality-of-life scores.6 Addressing the problem of physician shortages is a mission-critical priority for the VHA.7
With an eye toward enhancing 2 of the 4 statutory missions of the VA and to mitigate the shortage of physicians and improve the access of veterans to VHA medical services, on August 7, 2014, the Veterans Access, Choice, and Accountability Act of 2014 (Public Law [PL] 113-146), known as the Choice Act was enacted.8 Title III, §301(b) of the Choice Act requires VHA to increase GME residency positions by:
Establishing new medical residency programs, or ensuring that already established medical residency programs have a sufficient number of residency positions, at any VHA medical facility that is: (a) experiencing a shortage of physicians and (b) located in a community that is designated as a health professional shortage area.
The legislation specifies that priority must be placed on medical occupations that experience the largest staffing shortages throughout the VHA and “programs in primary care, mental health, and any other specialty that the Secretary of the VA determines appropriate.” The Choice Act authorized the VHA to increase the number of GME residency positions by up to 1,500 over a 5-year period. In December 2016, as amended by PL 114–315, Title VI, §617(a), this authorization was extended by another 5 years for a total of 10 years and will run through 2024.9
GME Development/Distribution
To distribute these newly created GME positions as mandated by Congress, the OAA is using a system with 3 types of request for proposal (RFP) applications. These include planning, infrastructure, and position grants. This phased approach was taken with the understanding that the development of new training sites requires a properly staffed education office and dedicated faculty time. Planning and infrastructure grants provide start-up funds for smaller VAMCs, allowing them to keep facility resources focused on their clinical mission.
Planning grants (of up to $250,000 over 2 years) primarily were designed for VA facilities with no or low numbers of physician residents at the desired teaching location. Priority was given to facilities in rural and/or underserved areas as well as those developing new affiliations. Applications were reviewed by OAA staff along with peer-selected Designated Education Officers (DEOs) from VA facilities across the nation that were not applying for the grants. Awards were based on the priorities mentioned earlier, with additional credit for programs focused on 2 VHA fundamental services areas—primary care and/or mental health training. Facilities receiving planning grants were mentored by an OAA physician staff member, anticipating a 2- to 3-year time line to request positions and begin GME training.
Infrastructure grants (of up to $520,000 used over 2-3 years) were designed as bridge funds after approval of Veterans Access, Choice, and Accountability Act (VACAA) GME positions. Infrastructure grants are appropriate to sustain a local education office, develop VA faculty, purchase equipment, and make minor modifications to the clinical space in the VAMCs or CBOCs to enhance the learning environment during the period before VA supportive funds from the Veterans Equitable Resource Allocation (VERA) (similar to indirect GME funds from CMS) become available. Applications were managed the same as planning grant submissions.
Position RFPs, unlike planning and infrastructure RFPs, are available to all VAMCs. The primary purpose of the VACAA Position RFP is to fund new positions in primary care and psychiatry. Graduate medical education positions in subspecialty programs also are considered when there is documentation of critical need to improve access to these services. Applications were reviewed by OAA staff along with selected DEOs from VA facilities around the U.S. Award criteria prioritized primary care (family medicine, internal medicine, geriatrics), and mental health (psychiatry and psychiatry subspecialties). Priority also was given to positions in areas with a documented shortage of physicians and areas with high concentrations of veterans.
Current Progress
To date the OAA has offered 3 RFP cycles consisting of planning/infrastructure grants, and 4 RFP cycles for salary/benefit support for additional resident full-time equivalent (FTE) positions. Resident positions were defined as residency or fellowship FTEs that were part of an ACGME or AOA-accredited training program. Figure 1 illustrates the geographic distribution of awarded GME positions.
In primary care specialties (family medicine, internal medicine, and geriatrics, a total of 349.4 FTE positions have been approved (Table 1). Due to a low number of applications, only 6.3 of these positions were awarded in geriatrics. In mental health, 167.6 FTE positions have been approved, whereas in critical needs specialties (needed to support rural/underserved healthcare and improve specialty access) 256.5 FTE positions have been added.
Discussion
There are several important desired short-term outcomes from VACAA. The first is improved access to high-quality care for both rural and urban veterans. There is an emphasis on primary care and mental health because shortages in these areas across the U.S. are well established.3,4,10 Likewise, rural areas have been prioritized because often there is a disparity of care.
One area of concern is the small number of applicants in geriatrics. Even with VACAA specifically targeting geriatrics as a primary care specialty, we have only received enough applications to approve 6.3 positions over the first 3 years of the program. As the veteran and overall population in the U.S. ages, it is important to develop a medical workforce that is willing and able to address their needs.
The VACAA statute is not intended to alter medical students’ career choice but rather to provide funded positions for those choosing primary care, geriatrics, psychiatry (including psychiatric subspecialties), and experience in the VA clinical settings. The hope is that this experience will encourage practitioners to competently care for veterans after training in the VA and/or other civilian settings.
By enabling smaller VA facilities to become training sites through planning and infrastructure grants, residents have the opportunity to gain experience in more rural settings. Physicians who choose to train in rural areas are likely to spend time practicing in those areas after they complete training.15 The process of developing facilities with no GME into training sitestakes time and resources. Establishing an education office and choosing site directors and core faculty are all important steps that must be done before resident rotations begin. Resources provided through VACAA have enabled the VHA to reduce the number of VAMCs with no GME activity to just 3.
Another benefit of VACAA GME expansion is the opportunity to engage new LCME/AOA-accredited medical schools and ACGME/AOA-accredited residency-sponsoring institutions.16,17 Representatives of these institutions may have perceived a reluctance of their local VAs to develop GME affiliations in the past. This statute has enabled many VAMCs to use nontraditional training sites and modalities to overcome barriers and create new academic affiliations.
However, VACAA only provides funds for training that occurs in established VA sites of care. This can hinder the development of partnerships where other funding sources are required for non-VA rotations. Another VACAA limitation is that it does not fund undergraduate medical education as does the Armed Forces Health Professional Scholarship Program (HPSP). In addition, the primary financial relationship is between the VA and the sponsoring institution, thus VHA cannot send residents to underserved locations.
Conclusion
The VHA has a rich tradition of educating physician and other health care providers in the U.S. More than 60% of U.S. trained physicians received a portion of their training through VHA.2 Through VACAA GME expansion initiative, the 113th Congress has asked VHA to continue its important training mission “to bind up the Nations wounds” and “to care for him who shall have borne the battle.”18
Acknowledgments
In memoriam – Robert Louis Jesse MD, PhD. Dr. Jesse, the Chief of the Office of Academic Affiliations passed away on September 2, 2017, at age 64. He had an illustrious medical career as a cardiologist and served in many leadership roles including Principal Deputy Under Secretary for Health in the U.S. Department of Veterans Affairs. His expertise, visionary leadership, and friendship will be missed by all involved in the VA’s academic training mission but particularly by those of us who worked for and with him at OAA.
1. U.S. Department of Veteran Affairs. Policy Memorandum No. 2. Policy in association of veterans’ hospitals with medical schools. https://www.va.gov/oaa/Archive/PolicyMemo2.pdf. Published January 30, 1947. Accessed December 13, 2017.
2. U.S. Department of Veteran Affairs, Office of Academic Affiliations. 2017 statistics: health professions trainees. https://www .va.gov/OAA/docs/OAA_Statistics.pdf. Accessed January 8, 2018.
3. IHS, Inc. The complexities of physician supply and demand 2016 update: projections from 2014 to 2025, final report. https://www.aamc.org/download/458082/data/2016_complexities_of_supply_and_demand_projections.pdf. Published April 5, 2016. Accessed December 13, 2017.
4. Petterson SM, Liaw WR, Tran C, Bazemore AW. Estimating the residency expansion required to avoid projected primary care physician shortages by 2035. Ann Fam Med. 2015;13(2):107-114.
5. Holder KA. Veterans in rural America 2011-2015. https://www.census.gov/content/dam/Census/library/publica tions/2017/acs/acs-36.pdf. Published January 2017. Accessed January 18, 2018.
6. Weeks WB, Wallace AE, Wang S, Lee A, Kazis LE. Rural-urban disparities in health-related quality of life within disease categories of veterans. J Rural Health. 2006;22(3):204-211.
7. U.S. Government Accountability Office. GAO-18-124. VHA Physician Staffing and Recruitment. https://www.gao.gov/assets/690/687853.pdf. Published October 19, 2017. Accessed January 23, 2018.
8. Veterans Access, Choice, and Accountability Act, section 301 (b): Increase of graduate medical education residency positions, 38 USC § 74 (2014) .
9. Jeff Miller and Richard Blumenthal Veterans Health Care and Benefits Improvement Act of 2016, 38 USC §101 (2016).
10. Thomas KC, Ellis AR, Konrad TR, Holzer CE, Morrissey JP. County-level estimates of mental health professional shortage in the United States. Psychiatr Serv. 2009;60(10):1323-1328.
11. Garibaldi RA, Popkave C, Bylsma W. Career plans for trainees in internal medicine residency programs. Acad Med. 2005;80(5):507-512.
12. West CP, Dupras DM. General medicine vs subspecialty career plans among internal medicine residents. JAMA. 2012;308(21):2241-2247.
13. Stimmel B, Haddow S, Smith L. The practice of general internal medicine by subspecialists. J Urban Health. 1998;75(1):184-190.
14. Shea JA, Kleetke PR, Wozniak GD, Polsky D, Escarce JJ. Self-reported physician specialties and the primary care content of medical practice: a study of the AMA physician masterfile. American Medical Association. Med Care. 1999;37(4):333-338.
15. Rabinowitz HK, Diamond JJ, Markham FW, Paynter NP. Critical factors for designing
16. Accredited MD programs in the United States. http://lcme.org /directory/accredited-u-s-programs/. Updated December 12, 2017. Accessed January 8, 2018.
17. Osteopathic medical schools. http://www.osteopathic.org/in side-aoa/about/affiliates/Pages/osteopathic-medical-schools.aspx Published 2017. Accessed January 8, 2018.
18. Lincoln A. Second inaugural address. https://www.va.gov/opa/publications/celebrate/vamotto.pdf. Accessed January 8. 2018.
1. U.S. Department of Veteran Affairs. Policy Memorandum No. 2. Policy in association of veterans’ hospitals with medical schools. https://www.va.gov/oaa/Archive/PolicyMemo2.pdf. Published January 30, 1947. Accessed December 13, 2017.
2. U.S. Department of Veteran Affairs, Office of Academic Affiliations. 2017 statistics: health professions trainees. https://www .va.gov/OAA/docs/OAA_Statistics.pdf. Accessed January 8, 2018.
3. IHS, Inc. The complexities of physician supply and demand 2016 update: projections from 2014 to 2025, final report. https://www.aamc.org/download/458082/data/2016_complexities_of_supply_and_demand_projections.pdf. Published April 5, 2016. Accessed December 13, 2017.
4. Petterson SM, Liaw WR, Tran C, Bazemore AW. Estimating the residency expansion required to avoid projected primary care physician shortages by 2035. Ann Fam Med. 2015;13(2):107-114.
5. Holder KA. Veterans in rural America 2011-2015. https://www.census.gov/content/dam/Census/library/publica tions/2017/acs/acs-36.pdf. Published January 2017. Accessed January 18, 2018.
6. Weeks WB, Wallace AE, Wang S, Lee A, Kazis LE. Rural-urban disparities in health-related quality of life within disease categories of veterans. J Rural Health. 2006;22(3):204-211.
7. U.S. Government Accountability Office. GAO-18-124. VHA Physician Staffing and Recruitment. https://www.gao.gov/assets/690/687853.pdf. Published October 19, 2017. Accessed January 23, 2018.
8. Veterans Access, Choice, and Accountability Act, section 301 (b): Increase of graduate medical education residency positions, 38 USC § 74 (2014) .
9. Jeff Miller and Richard Blumenthal Veterans Health Care and Benefits Improvement Act of 2016, 38 USC §101 (2016).
10. Thomas KC, Ellis AR, Konrad TR, Holzer CE, Morrissey JP. County-level estimates of mental health professional shortage in the United States. Psychiatr Serv. 2009;60(10):1323-1328.
11. Garibaldi RA, Popkave C, Bylsma W. Career plans for trainees in internal medicine residency programs. Acad Med. 2005;80(5):507-512.
12. West CP, Dupras DM. General medicine vs subspecialty career plans among internal medicine residents. JAMA. 2012;308(21):2241-2247.
13. Stimmel B, Haddow S, Smith L. The practice of general internal medicine by subspecialists. J Urban Health. 1998;75(1):184-190.
14. Shea JA, Kleetke PR, Wozniak GD, Polsky D, Escarce JJ. Self-reported physician specialties and the primary care content of medical practice: a study of the AMA physician masterfile. American Medical Association. Med Care. 1999;37(4):333-338.
15. Rabinowitz HK, Diamond JJ, Markham FW, Paynter NP. Critical factors for designing
16. Accredited MD programs in the United States. http://lcme.org /directory/accredited-u-s-programs/. Updated December 12, 2017. Accessed January 8, 2018.
17. Osteopathic medical schools. http://www.osteopathic.org/in side-aoa/about/affiliates/Pages/osteopathic-medical-schools.aspx Published 2017. Accessed January 8, 2018.
18. Lincoln A. Second inaugural address. https://www.va.gov/opa/publications/celebrate/vamotto.pdf. Accessed January 8. 2018.
Postsurgical pain: Optimizing relief while minimizing use of opioids
CASE Managing pain associated with prolapse and SUI surgery
A 46-year-old woman (G4P4) described 3 years of worsening symptoms related to recurrent stage-3 palpable uterine prolapse. She had associated symptomatic stress urinary incontinence. She had been treated for uterine prolapse 5 years ago with vaginal hysterectomy, bilateral salpingectomy, and high uterosacral-ligament suspension.
After consultation, the patient elected to undergo laparoscopic sacral colpopexy, a mid-urethral sling, and possible anterior and posterior colporrhaphy. Appropriate discussion about the risks and benefits of mesh was provided preoperatively. The surgical team judged her to be highly motivated; she wanted same-day outpatient surgery so that she could go home and then return to work. She had excellent support at home.
How would you counsel this patient about expected postoperative pain? Which medications would you administer to her preoperatively and perioperatively? Which ones would you prescribe for her to manage pain postoperatively?
Adverse impact of prescription opioids in the United States
Although fewer than 5% of the world’s population live in the United States, nearly 80% of the world’s opioids are written for them.1 In 2012, 259 million prescriptions were written for opioids in the United States—more than enough to give every American adult their own bottle of pills.2 Sadly, drug overdose is now a leading cause of accidental death in the United States, with 52,404 lethal drug overdoses in 2015. A startling statistic is that prescription opioid abuse is driving this epidemic, with 20,101 overdose deaths related to prescription pain relievers and 12,990 overdose deaths related to heroin in 2015.3
It is likely that there are multiple reasons prescribing of opioids is epidemic. Surgical pain is a common indication for opioid prescriptions; fewer than half of patients who undergo surgery report adequate postoperative pain relief.4 Recognition of these deficits in pain management has inspired national campaigns to improve patients’ experience with pain and aggressively address pain with drugs such as opioids.5
At the same time, marketing efforts by the pharmaceutical industry sought to reassure the medical community that patients would not become addicted to prescription opioid pain relievers if physical pain was the indication for such prescriptions. In response, health care providers began to prescribe opioids at a greater rate. As providers were encouraged to increase prescriptions, opioid medications began to be misused—and only then did it become clear that these medications are, in fact, highly addictive.6 Opioid abuse and overdose rates began to increase; in 2015, more than 33,000 Americans died because of an opioid overdose, including prescription opioids and heroin7 (FIGURE). In fact, although most people recognize the threat posed by illegal heroin, most of the 2 million who abused opioids in 2015 in the United States suffered from prescription abuse; only about a quarter, or about 600,000, abused heroin.8 In addition, more than 80% of people who abuse heroin initially abused prescription opioids.9
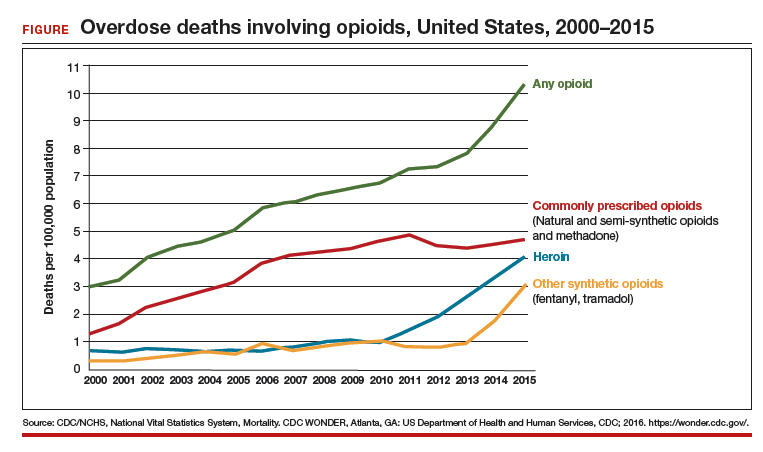
Read about medications and strategies for multimodal pain management.
Multimodal approach to pain management
The goals of postsurgical pain treatment are to relieve suffering, optimize bodily functioning after surgery, limit length of the stay, and optimize patient satisfaction. Pain-control regimens should consider the specific surgical procedure and the patient’s medical, psychological, and physical conditions; age; level of fear or anxiety; personal preference; and response to previous treatments.10
Optimally, postsurgical pain management starts well before the day of surgery. Employing such strategies as Enhanced Recovery after Surgery (ERAS) protocols does not necessarily mean providing the same care for every patient, every time. Rather, ERAS serves as a checklist to ensure that all applicable categories of pain medication and pain-control strategies are considered, selected, and dosed according to individual needs.11 (See “Preoperative management of pain expectations.”)
Ideally, before surgery, provide the patient with an opportunity to learn that:
- Her expectations about postsurgical pain should be realistic, and that freedom from pain is not realistic.
- Pain-reduction options should optimize her bodily function and mobility, reduce the degree to which pain interferes with activities, and relieve associated psychological stressors.
- Inherent in the pain management plan should be a goal of minimizing the risks of opioid misuse, abuse, and addiction—for the patient and for her family members and friends.
Opioids
Opioids have been employed to treat pain for 700 years.12 They are powerful pain relievers because they target central mechanisms involved in the perception of pain. Regrettably, because of their central action, opioids have many adverse effects in addition to being highly addictive.
Nonopioid alternatives
Expert consensus, including recommendations of the World Health Organization,11 favors using nonopioids as first-line medications to address surgical pain. Nonopioid analgesic options are acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and adjuvant medications. In addition, nonanalgesic medications such as sedatives, sleep aids, and muscle relaxants can relieve postsurgical pain. Optimal use of these nonopioid medications can significantly reduce or eliminate the need for opioid medications to treat pain. Goals are to 1) reserve opioids for the most severe pain and 2) minimize the number of doses/pills of opioids required to control postsurgical pain.
Acetaminophen. At dosages of 325 to 1,000 mg orally every 4 to 6 hours, to a maximum dosage of 4,000 mg/d, acetaminophen can be used to treat mild pain and, in combination with other medications, moderate-to-severe pain. The drug also can be administered intravenously (IV), although use of the IV route is limited in many hospitals because of its significantly higher expense compared to the oral form.
The mechanism of action of acetaminophen is unique among pain relievers; it can therefore be used in combination with other pain relievers to more effectively treat pain with fewer concerns about medication-induced adverse effects or opioid overdose. However, keep in mind when considering combining analgesics, that acetaminophen is an active ingredient in hundreds of over-the-counter (OTC) and prescription formulations, and that a combination of more than one acetaminophen-containing product can create the risk of overdose.
Acetaminophen should be used with caution in patients with liver disease. That being said, multiple trials have documented safe use in normal body weight adults who do not have hepatic disease, at dosages as high as 4,000 mg over a 24-hour period.13
NSAIDs. A combination of an NSAID and acetaminophen has been documented to reduce the amount of opioid medications required to treat postsurgical pain. In most circumstances, especially for minor surgery, acetaminophen and NSAIDS can be administered just before surgery starts. This preoperative treatment, called “preventive analgesia” or “preemptive analgesia,” has been demonstrated in multiple clinical trials to reduce postoperative pain.14
Adjuvant pain medications. Antidepressants, antiepileptic agents, and muscle relaxants—agents that have a primary indication for a condition (or conditions) other than pain and do not directly provide analgesia—have been used as adjuvant pain medications. When employed with traditional analgesics, they have been demonstrated to reduce postsurgical pain scores and the amount of opioids required. These medications need to be used cautiously because some are associated with serious sedation and vertigo (TABLE). Take caution when using adjuvant pain medications in patients older than 65 years; guidance on their use in older patients has been outlined by the American Geriatrics Society and other professional organizations.15
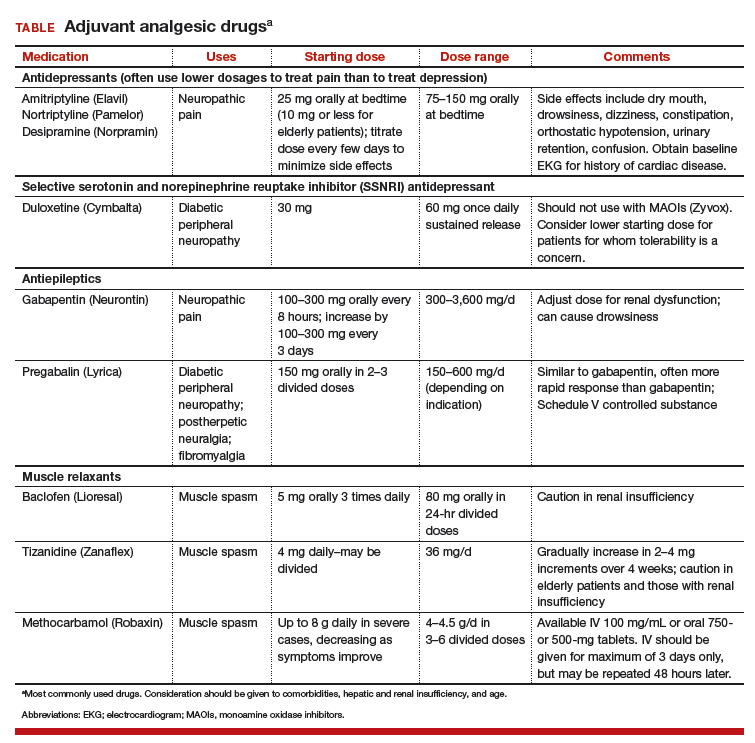
Case Continued
The patient was given the expectation that the 11-mm left lower-quadrant port site would likely be the most bothersome site of pain—a rating of 4 or 5 on a visual analogue scale of 1 to 10, on postoperative day 1, while standing. The other 3 (5-mm) laparoscopic ports, she was told, would, typically, be less bothersome. The patient was educated regarding the role of analgesics and adjuvant medications and cautioned not to exceed 4,000 mg of acetaminophen in any 24-hour period. She was told that gabapentin may make her feel sedated or dizzy, or both; she was encouraged to hold this medication if she found these adverse effects bothersome or limiting.
The following multimodal pain management was established.
Preoperatively, the patient was given:
- Acetaminophen 1.5 g orally (as a liquid, 45 mL of a suspension of 500 mg/15 mL liquid), 2 to 3 hours preoperatively; the surgical suite did not stock IV acetaminophen.
- Gabapentin 600 mg orally, with a sip of water, the morning of surgery.
- Celecoxib 100 mg orally, with a sip of water, the morning of surgery.
Prescriptions for home postoperative pain management were provided preoperatively:
- OTC acetaminophen 1,000 mg (as 2 500-mgtablets) taken as a scheduled dose every 8 hours for the first 48 hours postoperatively.
- Meloxicam 15 mg daily as the NSAID, taken as a scheduled dose once per day for the first 48 hours postoperatively, then as needed.
- Gabapentin 300 mg (in addition to the preoperative dose, above), taken as a scheduled dose every 8 hours for the first 48 hours postoperatively, then as needed.
- Oxycodone 5 mg (without acetaminophen) for breakthrough pain.
Intraoperatively:
- Meticulous attention was paid to patient positioning, to reduce the possibility of back and upper- and lower-extremity injury postoperatively.
- A corticosteroid (dexamethasone 8 mg IV) was administered to minimize postoperative nausea and vomiting and as an adjuvant medication for postoperative pain control.
- Careful attention was paid to limit residual CO2 gas and intraoperative intra-abdominal pressures.
- All laparoscopic port sites were injected with 30 mL of 0.25% bupivacaine with epinephrine, extending to subcutaneous, fascial, and peritoneal layers.
Read about why a multimodal approach is best for postsurgical pain.
Why a multimodal plan to treat pain?
Pain following laparoscopy has been associated with many variables, including patient positioning, port size and placement, amount of port manipulation, and gas retention. After a laparoscopic surgical procedure, patients report pain in the abdomen, back, and shoulders.
Postsurgical pain has 3 components:
- Shoulder pain, thought to result from phrenic nerve irritation caused by lingering CO2 in the abdominal cavity.
- Visceral pain, occurring secondary to stretching of the abdominal cavity.
- Somatic pain, caused by the surgical incision; of the 3 components to pain, somatic pain can have the least impact because laparoscopic incisions are small.
For our patient, prior to the incisions being made, she received local anesthesia intraoperatively to the laparoscopic port sites to include the subcutaneous, fascial, and peritoneal layers. Involving these layers allows for more of a block. An ultrasonography-guided transversus abdominis plane (TAP) block, if available, is highly effective at decreasing postoperative pain, but its efficacy is dependent on the anatomy and the skill of the physician (whether anesthesiologist, gynecologist, or surgeon) who is placing it.16
We used dexamethasone 8 mg IV, intraoperatively because this single dose has been shown to decrease the perception of pain postoperatively. Dexamethasone also has been shown to decrease consumption of oxycodone during the 24 hours after laparoscopic gynecologic surgery.17
CO2 used to insufflate the patient’s abdomen can take as long as 2 days to fully resorb, resulting in increased pain. This discomfort has been described as delayed; the patient might not notice it until she goes home. In a study, 70% of patients had shoulder discomfort following laparoscopy 24 hours after their procedure.18 For this reason, we employed several techniques to reduce this effect:
- We reduced the intra-abdominal pressure limit to 10 mm Hg (from 15 mm Hg) once dissection was complete.
- At the end of the procedure, careful attention was paid to removing as much intra-abdominal gas as possible, including placing the patient in the Trendelenburg position and having the anesthesiologist induce a Valsalva maneuver. This action has been shown to significantly improve pain control compared to placebo intervention.19
- We used humidified CO2, which has been demonstrated to reduce pain in laparoscopic surgery.20
Preemptively, we provided this patient with acetaminophen, celecoxib, and gabapentin, which have been demonstrated to be effective in gynecologic patients to decrease the need for postoperative opioids.21 Also, our patient received counseling, with specific expectations for what to expect following the surgical procedure.
CASE Resolved
Our patient did exceptionally well following surgery. She used only one of the oxycodone pills and did not require unplanned interventions. She took gabapentin, acetaminophen, and meloxicam at their scheduled doses for 2 days. She continued to use meloxicam for 4 more days for mild abdominal pain, then discontinued all medications.She flushed her 9 unused oxycodone pills down the toilet. (See “A word about disposal of ‘excess’ opioids”22) The patient returned to her administrative duties at work 2 weeks after the procedure and reported that she was “very satisfied” with her surgical experience.
The US Food and Drug Administration (FDA) recommends disposing of certain drugs through a take-back program or, if such a program is not readily available, by flushing them down a toilet or sink. In a recent study, investigators concluded that opioids on the FDA's so-called flush list include most opioids in clinical use--even if the entire supply prescribed is to be flushed down the drain. Conservative estimates of environmental degradation were employed in the study; the investigators' conclusion was that these drugs pose a "negligible" eco-toxicologic risk.1
Reference
- Khan U, Bloom RA, Nicell JA, Laurenson JP. Risks associated with the environmental release of pharmaceuticals on the U.S. Food and Drug Administration "flush list". Sci Total Environ. 2017;609:1023-1040.
In conclusion
Postoperative pain is a complex entity that must be considered to require individualized strategies and, possibly, multiple interventions. Optimally, thorough education, including pain management options, is provided to the patient prior to surgery. Given the current state of opioid abuse in the United States, all gynecologic surgeons should be familiar with multimodal pain therapy and how to employ nonmedical techniques to reduce postsurgical pain without relying solely on opioids. (See “Online resources for pain management”.)
- Drug Disposal Information
(US Department of Justice Drug Enforcement Administration)
https://www.deadiversion.usdoj.gov/drug_disposal/index.html - Surgical Pain Consortium
http://surgicalpainconsortium.org/
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- United Nations International Narcotics Control Board. Narcotic drugs: Report 2016: Estimated world requirements for 2017-statistics for 2015. New York, NY. https://www.incb.org/documents/Narcotic-Drugs/Technical-Publications/2016/Narcotic_Drugs_Publication_2016.pdf. Published 2017. Accessed January 7, 2018.
- Centers for Disease Control and Prevention. Opioid painkiller prescribing: Where you live makes a difference. Atlanta, GA. https://www.cdc.gov/vitalsigns/pdf/2014-07-vitalsigns.pdf. Published July 2014. Accessed January 5, 2018.
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445–1452.
- Gan TJ, Habib AS, Miller TE, et al. Incidence, patient satisfaction, and perceptions of post-surgical pain: results from a US national survey. Curr Med Res Opin. 2014;30(1):149–160.
- Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625.
- Van Zee A. The promotion and marketing of OxyContin: commercial triumph, public health tragedy. Am J Public Health. 2009;99(2):221–227.
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445–1452.
- US Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. Results from the 2015 national survey on drug use and health: Detailed tables. Rockville, MD. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.htm. Published 2016. Accessed January 5, 2018.
- Muhuri PK, Gfroerer JC, Davies MC. Associations of nonmedical pain reliever use and initiation of heroin use in the United States. CBHSQ Data Rev. http://www.samhsa.gov/data/sites/default/files/DR006/DR006/nonmedical-pain-reliever-use-2013.htm. Published August 2013. Accessed January 5, 2018.
- Joshi GP. Multimodal analgesia techniques and postoperative rehabilitation. Anesthesiol Clin North America. 2005;23(1):185–202.
- Oderda G. Challenges in the management of acute postsurgical pain. Pharmacotherapy. 2012;32(9 suppl):6S–11S.
- Brownstein, MJ. A brief history of opiates, opioid peptides, and opioid receptors. Proc Natl Acad Sci USA. 1993;90(12):5391–5393.
- US Food and Drug Administration. Acetaminophen. https://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm165107.htm. Published November 2017. Accessed January 7, 2018.
- Ong CK, Seymour RA, Lirk P, Merry AF. Combining paracetamol (acetaminophen) with nonsteroidal anti-inflammatory drugs: a qualitative systematic review of analgesic efficacy for acute postoperative pain. Anesth Analg. 2010;110(4):1170–1179.
- Hanlon JT, Semla TP, Schmader KE. Alternative medications for medications included in the use of high‐risk medications in the elderly and potentially harmful drug–disease interactions in the elderly quality measures. J Am Geriatr Soc. 2015;63(12):e8–e18.
- Joshi GP, Janis JE, Haas EM, et al. Surgical site infiltration for abdominal surgery: A novel neuroanatomical-based approach. Plast Reconstr Surg Glob Open. 2016;4(12):e1181. https://insights.ovid.com/crossref?an=01720096-201612000-00021. Accessed January 5, 2018.
- Jokela RM, Ahonen JV, Tallgren MK, et al. The effective analgesic dose of dexamethasone after laparoscopic hysterectomy. Anesth Analg. 2009;109(2):607–615.
- Hohlrieder M, Brimacombe J, Eschertzhuber S, et al. A study of airway management using the ProSeal LMA laryngeal mask airway compared with the tracheal tube on postoperative analgesia requirements following gynaecological laparoscopic surgery. Anaesthesia. 2007;62(9):913–918.
- Phelps P, Cakmakkaya OS, Apfel CC, Radke OC. A simple clinical maneuver to reduce laparoscopy-induced shoulder pain: a randomized controlled trial. Obstet Gynecol. 2008;111(5):1155–1160.
- Sammour T, Kahokehr A, Hill AG. Meta‐analysis of the effect of warm humidified insufflation on pain after laparoscopy. Br J Surg. 2008;95(8):950–956.
- Reagan KM, O’Sullivan DM, Gannon R, Steinberg AC. Decreasing postoperative narcotics in reconstructive pelvic surgery; A randomized controlled trial. Am J Obstet Gynecol. 2017;217(3):325.e1–e10.
- Khan U, Bloom RA, Nicell JA, Laurenson JP. Risks associated with the environmental release of pharmaceuticals on the U.S. Food and Drug Administration “flush list”. Sci Total Environ. 2017;609:1023–1040.

Dr. Nihira is Clinical Professor, Obstetrics and Gynecology, UC Riverside School of Medicine, Riverside, California.

Dr. Steinberg is Associate Chief and FPMRS Fellowship Director, Department of Obstetrics and Gynecology, Hartford Hospital, Hartford, Connecticut.
Dr. Nihira reports that he is a consultant to Pacira. Dr. Steinberg reports no financial relationships relevant to this article.
Dr. Nihira is Clinical Professor, Obstetrics and Gynecology, UC Riverside School of Medicine, Riverside, California.
Dr. Steinberg is Associate Chief and FPMRS Fellowship Director, Department of Obstetrics and Gynecology, Hartford Hospital, Hartford, Connecticut.
Dr. Nihira reports that he is a consultant to Pacira. Dr. Steinberg reports no financial relationships relevant to this article.
Dr. Nihira is Clinical Professor, Obstetrics and Gynecology, UC Riverside School of Medicine, Riverside, California.
Dr. Steinberg is Associate Chief and FPMRS Fellowship Director, Department of Obstetrics and Gynecology, Hartford Hospital, Hartford, Connecticut.
Dr. Nihira reports that he is a consultant to Pacira. Dr. Steinberg reports no financial relationships relevant to this article.
CASE Managing pain associated with prolapse and SUI surgery
A 46-year-old woman (G4P4) described 3 years of worsening symptoms related to recurrent stage-3 palpable uterine prolapse. She had associated symptomatic stress urinary incontinence. She had been treated for uterine prolapse 5 years ago with vaginal hysterectomy, bilateral salpingectomy, and high uterosacral-ligament suspension.
After consultation, the patient elected to undergo laparoscopic sacral colpopexy, a mid-urethral sling, and possible anterior and posterior colporrhaphy. Appropriate discussion about the risks and benefits of mesh was provided preoperatively. The surgical team judged her to be highly motivated; she wanted same-day outpatient surgery so that she could go home and then return to work. She had excellent support at home.
How would you counsel this patient about expected postoperative pain? Which medications would you administer to her preoperatively and perioperatively? Which ones would you prescribe for her to manage pain postoperatively?
Adverse impact of prescription opioids in the United States
Although fewer than 5% of the world’s population live in the United States, nearly 80% of the world’s opioids are written for them.1 In 2012, 259 million prescriptions were written for opioids in the United States—more than enough to give every American adult their own bottle of pills.2 Sadly, drug overdose is now a leading cause of accidental death in the United States, with 52,404 lethal drug overdoses in 2015. A startling statistic is that prescription opioid abuse is driving this epidemic, with 20,101 overdose deaths related to prescription pain relievers and 12,990 overdose deaths related to heroin in 2015.3
It is likely that there are multiple reasons prescribing of opioids is epidemic. Surgical pain is a common indication for opioid prescriptions; fewer than half of patients who undergo surgery report adequate postoperative pain relief.4 Recognition of these deficits in pain management has inspired national campaigns to improve patients’ experience with pain and aggressively address pain with drugs such as opioids.5
At the same time, marketing efforts by the pharmaceutical industry sought to reassure the medical community that patients would not become addicted to prescription opioid pain relievers if physical pain was the indication for such prescriptions. In response, health care providers began to prescribe opioids at a greater rate. As providers were encouraged to increase prescriptions, opioid medications began to be misused—and only then did it become clear that these medications are, in fact, highly addictive.6 Opioid abuse and overdose rates began to increase; in 2015, more than 33,000 Americans died because of an opioid overdose, including prescription opioids and heroin7 (FIGURE). In fact, although most people recognize the threat posed by illegal heroin, most of the 2 million who abused opioids in 2015 in the United States suffered from prescription abuse; only about a quarter, or about 600,000, abused heroin.8 In addition, more than 80% of people who abuse heroin initially abused prescription opioids.9
Read about medications and strategies for multimodal pain management.
Multimodal approach to pain management
The goals of postsurgical pain treatment are to relieve suffering, optimize bodily functioning after surgery, limit length of the stay, and optimize patient satisfaction. Pain-control regimens should consider the specific surgical procedure and the patient’s medical, psychological, and physical conditions; age; level of fear or anxiety; personal preference; and response to previous treatments.10
Optimally, postsurgical pain management starts well before the day of surgery. Employing such strategies as Enhanced Recovery after Surgery (ERAS) protocols does not necessarily mean providing the same care for every patient, every time. Rather, ERAS serves as a checklist to ensure that all applicable categories of pain medication and pain-control strategies are considered, selected, and dosed according to individual needs.11 (See “Preoperative management of pain expectations.”)
Ideally, before surgery, provide the patient with an opportunity to learn that:
- Her expectations about postsurgical pain should be realistic, and that freedom from pain is not realistic.
- Pain-reduction options should optimize her bodily function and mobility, reduce the degree to which pain interferes with activities, and relieve associated psychological stressors.
- Inherent in the pain management plan should be a goal of minimizing the risks of opioid misuse, abuse, and addiction—for the patient and for her family members and friends.
Opioids
Opioids have been employed to treat pain for 700 years.12 They are powerful pain relievers because they target central mechanisms involved in the perception of pain. Regrettably, because of their central action, opioids have many adverse effects in addition to being highly addictive.
Nonopioid alternatives
Expert consensus, including recommendations of the World Health Organization,11 favors using nonopioids as first-line medications to address surgical pain. Nonopioid analgesic options are acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and adjuvant medications. In addition, nonanalgesic medications such as sedatives, sleep aids, and muscle relaxants can relieve postsurgical pain. Optimal use of these nonopioid medications can significantly reduce or eliminate the need for opioid medications to treat pain. Goals are to 1) reserve opioids for the most severe pain and 2) minimize the number of doses/pills of opioids required to control postsurgical pain.
Acetaminophen. At dosages of 325 to 1,000 mg orally every 4 to 6 hours, to a maximum dosage of 4,000 mg/d, acetaminophen can be used to treat mild pain and, in combination with other medications, moderate-to-severe pain. The drug also can be administered intravenously (IV), although use of the IV route is limited in many hospitals because of its significantly higher expense compared to the oral form.
The mechanism of action of acetaminophen is unique among pain relievers; it can therefore be used in combination with other pain relievers to more effectively treat pain with fewer concerns about medication-induced adverse effects or opioid overdose. However, keep in mind when considering combining analgesics, that acetaminophen is an active ingredient in hundreds of over-the-counter (OTC) and prescription formulations, and that a combination of more than one acetaminophen-containing product can create the risk of overdose.
Acetaminophen should be used with caution in patients with liver disease. That being said, multiple trials have documented safe use in normal body weight adults who do not have hepatic disease, at dosages as high as 4,000 mg over a 24-hour period.13
NSAIDs. A combination of an NSAID and acetaminophen has been documented to reduce the amount of opioid medications required to treat postsurgical pain. In most circumstances, especially for minor surgery, acetaminophen and NSAIDS can be administered just before surgery starts. This preoperative treatment, called “preventive analgesia” or “preemptive analgesia,” has been demonstrated in multiple clinical trials to reduce postoperative pain.14
Adjuvant pain medications. Antidepressants, antiepileptic agents, and muscle relaxants—agents that have a primary indication for a condition (or conditions) other than pain and do not directly provide analgesia—have been used as adjuvant pain medications. When employed with traditional analgesics, they have been demonstrated to reduce postsurgical pain scores and the amount of opioids required. These medications need to be used cautiously because some are associated with serious sedation and vertigo (TABLE). Take caution when using adjuvant pain medications in patients older than 65 years; guidance on their use in older patients has been outlined by the American Geriatrics Society and other professional organizations.15
Case Continued
The patient was given the expectation that the 11-mm left lower-quadrant port site would likely be the most bothersome site of pain—a rating of 4 or 5 on a visual analogue scale of 1 to 10, on postoperative day 1, while standing. The other 3 (5-mm) laparoscopic ports, she was told, would, typically, be less bothersome. The patient was educated regarding the role of analgesics and adjuvant medications and cautioned not to exceed 4,000 mg of acetaminophen in any 24-hour period. She was told that gabapentin may make her feel sedated or dizzy, or both; she was encouraged to hold this medication if she found these adverse effects bothersome or limiting.
The following multimodal pain management was established.
Preoperatively, the patient was given:
- Acetaminophen 1.5 g orally (as a liquid, 45 mL of a suspension of 500 mg/15 mL liquid), 2 to 3 hours preoperatively; the surgical suite did not stock IV acetaminophen.
- Gabapentin 600 mg orally, with a sip of water, the morning of surgery.
- Celecoxib 100 mg orally, with a sip of water, the morning of surgery.
Prescriptions for home postoperative pain management were provided preoperatively:
- OTC acetaminophen 1,000 mg (as 2 500-mgtablets) taken as a scheduled dose every 8 hours for the first 48 hours postoperatively.
- Meloxicam 15 mg daily as the NSAID, taken as a scheduled dose once per day for the first 48 hours postoperatively, then as needed.
- Gabapentin 300 mg (in addition to the preoperative dose, above), taken as a scheduled dose every 8 hours for the first 48 hours postoperatively, then as needed.
- Oxycodone 5 mg (without acetaminophen) for breakthrough pain.
Intraoperatively:
- Meticulous attention was paid to patient positioning, to reduce the possibility of back and upper- and lower-extremity injury postoperatively.
- A corticosteroid (dexamethasone 8 mg IV) was administered to minimize postoperative nausea and vomiting and as an adjuvant medication for postoperative pain control.
- Careful attention was paid to limit residual CO2 gas and intraoperative intra-abdominal pressures.
- All laparoscopic port sites were injected with 30 mL of 0.25% bupivacaine with epinephrine, extending to subcutaneous, fascial, and peritoneal layers.
Read about why a multimodal approach is best for postsurgical pain.
Why a multimodal plan to treat pain?
Pain following laparoscopy has been associated with many variables, including patient positioning, port size and placement, amount of port manipulation, and gas retention. After a laparoscopic surgical procedure, patients report pain in the abdomen, back, and shoulders.
Postsurgical pain has 3 components:
- Shoulder pain, thought to result from phrenic nerve irritation caused by lingering CO2 in the abdominal cavity.
- Visceral pain, occurring secondary to stretching of the abdominal cavity.
- Somatic pain, caused by the surgical incision; of the 3 components to pain, somatic pain can have the least impact because laparoscopic incisions are small.
For our patient, prior to the incisions being made, she received local anesthesia intraoperatively to the laparoscopic port sites to include the subcutaneous, fascial, and peritoneal layers. Involving these layers allows for more of a block. An ultrasonography-guided transversus abdominis plane (TAP) block, if available, is highly effective at decreasing postoperative pain, but its efficacy is dependent on the anatomy and the skill of the physician (whether anesthesiologist, gynecologist, or surgeon) who is placing it.16
We used dexamethasone 8 mg IV, intraoperatively because this single dose has been shown to decrease the perception of pain postoperatively. Dexamethasone also has been shown to decrease consumption of oxycodone during the 24 hours after laparoscopic gynecologic surgery.17
CO2 used to insufflate the patient’s abdomen can take as long as 2 days to fully resorb, resulting in increased pain. This discomfort has been described as delayed; the patient might not notice it until she goes home. In a study, 70% of patients had shoulder discomfort following laparoscopy 24 hours after their procedure.18 For this reason, we employed several techniques to reduce this effect:
- We reduced the intra-abdominal pressure limit to 10 mm Hg (from 15 mm Hg) once dissection was complete.
- At the end of the procedure, careful attention was paid to removing as much intra-abdominal gas as possible, including placing the patient in the Trendelenburg position and having the anesthesiologist induce a Valsalva maneuver. This action has been shown to significantly improve pain control compared to placebo intervention.19
- We used humidified CO2, which has been demonstrated to reduce pain in laparoscopic surgery.20
Preemptively, we provided this patient with acetaminophen, celecoxib, and gabapentin, which have been demonstrated to be effective in gynecologic patients to decrease the need for postoperative opioids.21 Also, our patient received counseling, with specific expectations for what to expect following the surgical procedure.
CASE Resolved
Our patient did exceptionally well following surgery. She used only one of the oxycodone pills and did not require unplanned interventions. She took gabapentin, acetaminophen, and meloxicam at their scheduled doses for 2 days. She continued to use meloxicam for 4 more days for mild abdominal pain, then discontinued all medications.She flushed her 9 unused oxycodone pills down the toilet. (See “A word about disposal of ‘excess’ opioids”22) The patient returned to her administrative duties at work 2 weeks after the procedure and reported that she was “very satisfied” with her surgical experience.
The US Food and Drug Administration (FDA) recommends disposing of certain drugs through a take-back program or, if such a program is not readily available, by flushing them down a toilet or sink. In a recent study, investigators concluded that opioids on the FDA's so-called flush list include most opioids in clinical use--even if the entire supply prescribed is to be flushed down the drain. Conservative estimates of environmental degradation were employed in the study; the investigators' conclusion was that these drugs pose a "negligible" eco-toxicologic risk.1
Reference
- Khan U, Bloom RA, Nicell JA, Laurenson JP. Risks associated with the environmental release of pharmaceuticals on the U.S. Food and Drug Administration "flush list". Sci Total Environ. 2017;609:1023-1040.
In conclusion
Postoperative pain is a complex entity that must be considered to require individualized strategies and, possibly, multiple interventions. Optimally, thorough education, including pain management options, is provided to the patient prior to surgery. Given the current state of opioid abuse in the United States, all gynecologic surgeons should be familiar with multimodal pain therapy and how to employ nonmedical techniques to reduce postsurgical pain without relying solely on opioids. (See “Online resources for pain management”.)
- Drug Disposal Information
(US Department of Justice Drug Enforcement Administration)
https://www.deadiversion.usdoj.gov/drug_disposal/index.html - Surgical Pain Consortium
http://surgicalpainconsortium.org/
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
CASE Managing pain associated with prolapse and SUI surgery
A 46-year-old woman (G4P4) described 3 years of worsening symptoms related to recurrent stage-3 palpable uterine prolapse. She had associated symptomatic stress urinary incontinence. She had been treated for uterine prolapse 5 years ago with vaginal hysterectomy, bilateral salpingectomy, and high uterosacral-ligament suspension.
After consultation, the patient elected to undergo laparoscopic sacral colpopexy, a mid-urethral sling, and possible anterior and posterior colporrhaphy. Appropriate discussion about the risks and benefits of mesh was provided preoperatively. The surgical team judged her to be highly motivated; she wanted same-day outpatient surgery so that she could go home and then return to work. She had excellent support at home.
How would you counsel this patient about expected postoperative pain? Which medications would you administer to her preoperatively and perioperatively? Which ones would you prescribe for her to manage pain postoperatively?
Adverse impact of prescription opioids in the United States
Although fewer than 5% of the world’s population live in the United States, nearly 80% of the world’s opioids are written for them.1 In 2012, 259 million prescriptions were written for opioids in the United States—more than enough to give every American adult their own bottle of pills.2 Sadly, drug overdose is now a leading cause of accidental death in the United States, with 52,404 lethal drug overdoses in 2015. A startling statistic is that prescription opioid abuse is driving this epidemic, with 20,101 overdose deaths related to prescription pain relievers and 12,990 overdose deaths related to heroin in 2015.3
It is likely that there are multiple reasons prescribing of opioids is epidemic. Surgical pain is a common indication for opioid prescriptions; fewer than half of patients who undergo surgery report adequate postoperative pain relief.4 Recognition of these deficits in pain management has inspired national campaigns to improve patients’ experience with pain and aggressively address pain with drugs such as opioids.5
At the same time, marketing efforts by the pharmaceutical industry sought to reassure the medical community that patients would not become addicted to prescription opioid pain relievers if physical pain was the indication for such prescriptions. In response, health care providers began to prescribe opioids at a greater rate. As providers were encouraged to increase prescriptions, opioid medications began to be misused—and only then did it become clear that these medications are, in fact, highly addictive.6 Opioid abuse and overdose rates began to increase; in 2015, more than 33,000 Americans died because of an opioid overdose, including prescription opioids and heroin7 (FIGURE). In fact, although most people recognize the threat posed by illegal heroin, most of the 2 million who abused opioids in 2015 in the United States suffered from prescription abuse; only about a quarter, or about 600,000, abused heroin.8 In addition, more than 80% of people who abuse heroin initially abused prescription opioids.9
Read about medications and strategies for multimodal pain management.
Multimodal approach to pain management
The goals of postsurgical pain treatment are to relieve suffering, optimize bodily functioning after surgery, limit length of the stay, and optimize patient satisfaction. Pain-control regimens should consider the specific surgical procedure and the patient’s medical, psychological, and physical conditions; age; level of fear or anxiety; personal preference; and response to previous treatments.10
Optimally, postsurgical pain management starts well before the day of surgery. Employing such strategies as Enhanced Recovery after Surgery (ERAS) protocols does not necessarily mean providing the same care for every patient, every time. Rather, ERAS serves as a checklist to ensure that all applicable categories of pain medication and pain-control strategies are considered, selected, and dosed according to individual needs.11 (See “Preoperative management of pain expectations.”)
Ideally, before surgery, provide the patient with an opportunity to learn that:
- Her expectations about postsurgical pain should be realistic, and that freedom from pain is not realistic.
- Pain-reduction options should optimize her bodily function and mobility, reduce the degree to which pain interferes with activities, and relieve associated psychological stressors.
- Inherent in the pain management plan should be a goal of minimizing the risks of opioid misuse, abuse, and addiction—for the patient and for her family members and friends.
Opioids
Opioids have been employed to treat pain for 700 years.12 They are powerful pain relievers because they target central mechanisms involved in the perception of pain. Regrettably, because of their central action, opioids have many adverse effects in addition to being highly addictive.
Nonopioid alternatives
Expert consensus, including recommendations of the World Health Organization,11 favors using nonopioids as first-line medications to address surgical pain. Nonopioid analgesic options are acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and adjuvant medications. In addition, nonanalgesic medications such as sedatives, sleep aids, and muscle relaxants can relieve postsurgical pain. Optimal use of these nonopioid medications can significantly reduce or eliminate the need for opioid medications to treat pain. Goals are to 1) reserve opioids for the most severe pain and 2) minimize the number of doses/pills of opioids required to control postsurgical pain.
Acetaminophen. At dosages of 325 to 1,000 mg orally every 4 to 6 hours, to a maximum dosage of 4,000 mg/d, acetaminophen can be used to treat mild pain and, in combination with other medications, moderate-to-severe pain. The drug also can be administered intravenously (IV), although use of the IV route is limited in many hospitals because of its significantly higher expense compared to the oral form.
The mechanism of action of acetaminophen is unique among pain relievers; it can therefore be used in combination with other pain relievers to more effectively treat pain with fewer concerns about medication-induced adverse effects or opioid overdose. However, keep in mind when considering combining analgesics, that acetaminophen is an active ingredient in hundreds of over-the-counter (OTC) and prescription formulations, and that a combination of more than one acetaminophen-containing product can create the risk of overdose.
Acetaminophen should be used with caution in patients with liver disease. That being said, multiple trials have documented safe use in normal body weight adults who do not have hepatic disease, at dosages as high as 4,000 mg over a 24-hour period.13
NSAIDs. A combination of an NSAID and acetaminophen has been documented to reduce the amount of opioid medications required to treat postsurgical pain. In most circumstances, especially for minor surgery, acetaminophen and NSAIDS can be administered just before surgery starts. This preoperative treatment, called “preventive analgesia” or “preemptive analgesia,” has been demonstrated in multiple clinical trials to reduce postoperative pain.14
Adjuvant pain medications. Antidepressants, antiepileptic agents, and muscle relaxants—agents that have a primary indication for a condition (or conditions) other than pain and do not directly provide analgesia—have been used as adjuvant pain medications. When employed with traditional analgesics, they have been demonstrated to reduce postsurgical pain scores and the amount of opioids required. These medications need to be used cautiously because some are associated with serious sedation and vertigo (TABLE). Take caution when using adjuvant pain medications in patients older than 65 years; guidance on their use in older patients has been outlined by the American Geriatrics Society and other professional organizations.15
Case Continued
The patient was given the expectation that the 11-mm left lower-quadrant port site would likely be the most bothersome site of pain—a rating of 4 or 5 on a visual analogue scale of 1 to 10, on postoperative day 1, while standing. The other 3 (5-mm) laparoscopic ports, she was told, would, typically, be less bothersome. The patient was educated regarding the role of analgesics and adjuvant medications and cautioned not to exceed 4,000 mg of acetaminophen in any 24-hour period. She was told that gabapentin may make her feel sedated or dizzy, or both; she was encouraged to hold this medication if she found these adverse effects bothersome or limiting.
The following multimodal pain management was established.
Preoperatively, the patient was given:
- Acetaminophen 1.5 g orally (as a liquid, 45 mL of a suspension of 500 mg/15 mL liquid), 2 to 3 hours preoperatively; the surgical suite did not stock IV acetaminophen.
- Gabapentin 600 mg orally, with a sip of water, the morning of surgery.
- Celecoxib 100 mg orally, with a sip of water, the morning of surgery.
Prescriptions for home postoperative pain management were provided preoperatively:
- OTC acetaminophen 1,000 mg (as 2 500-mgtablets) taken as a scheduled dose every 8 hours for the first 48 hours postoperatively.
- Meloxicam 15 mg daily as the NSAID, taken as a scheduled dose once per day for the first 48 hours postoperatively, then as needed.
- Gabapentin 300 mg (in addition to the preoperative dose, above), taken as a scheduled dose every 8 hours for the first 48 hours postoperatively, then as needed.
- Oxycodone 5 mg (without acetaminophen) for breakthrough pain.
Intraoperatively:
- Meticulous attention was paid to patient positioning, to reduce the possibility of back and upper- and lower-extremity injury postoperatively.
- A corticosteroid (dexamethasone 8 mg IV) was administered to minimize postoperative nausea and vomiting and as an adjuvant medication for postoperative pain control.
- Careful attention was paid to limit residual CO2 gas and intraoperative intra-abdominal pressures.
- All laparoscopic port sites were injected with 30 mL of 0.25% bupivacaine with epinephrine, extending to subcutaneous, fascial, and peritoneal layers.
Read about why a multimodal approach is best for postsurgical pain.
Why a multimodal plan to treat pain?
Pain following laparoscopy has been associated with many variables, including patient positioning, port size and placement, amount of port manipulation, and gas retention. After a laparoscopic surgical procedure, patients report pain in the abdomen, back, and shoulders.
Postsurgical pain has 3 components:
- Shoulder pain, thought to result from phrenic nerve irritation caused by lingering CO2 in the abdominal cavity.
- Visceral pain, occurring secondary to stretching of the abdominal cavity.
- Somatic pain, caused by the surgical incision; of the 3 components to pain, somatic pain can have the least impact because laparoscopic incisions are small.
For our patient, prior to the incisions being made, she received local anesthesia intraoperatively to the laparoscopic port sites to include the subcutaneous, fascial, and peritoneal layers. Involving these layers allows for more of a block. An ultrasonography-guided transversus abdominis plane (TAP) block, if available, is highly effective at decreasing postoperative pain, but its efficacy is dependent on the anatomy and the skill of the physician (whether anesthesiologist, gynecologist, or surgeon) who is placing it.16
We used dexamethasone 8 mg IV, intraoperatively because this single dose has been shown to decrease the perception of pain postoperatively. Dexamethasone also has been shown to decrease consumption of oxycodone during the 24 hours after laparoscopic gynecologic surgery.17
CO2 used to insufflate the patient’s abdomen can take as long as 2 days to fully resorb, resulting in increased pain. This discomfort has been described as delayed; the patient might not notice it until she goes home. In a study, 70% of patients had shoulder discomfort following laparoscopy 24 hours after their procedure.18 For this reason, we employed several techniques to reduce this effect:
- We reduced the intra-abdominal pressure limit to 10 mm Hg (from 15 mm Hg) once dissection was complete.
- At the end of the procedure, careful attention was paid to removing as much intra-abdominal gas as possible, including placing the patient in the Trendelenburg position and having the anesthesiologist induce a Valsalva maneuver. This action has been shown to significantly improve pain control compared to placebo intervention.19
- We used humidified CO2, which has been demonstrated to reduce pain in laparoscopic surgery.20
Preemptively, we provided this patient with acetaminophen, celecoxib, and gabapentin, which have been demonstrated to be effective in gynecologic patients to decrease the need for postoperative opioids.21 Also, our patient received counseling, with specific expectations for what to expect following the surgical procedure.
CASE Resolved
Our patient did exceptionally well following surgery. She used only one of the oxycodone pills and did not require unplanned interventions. She took gabapentin, acetaminophen, and meloxicam at their scheduled doses for 2 days. She continued to use meloxicam for 4 more days for mild abdominal pain, then discontinued all medications.She flushed her 9 unused oxycodone pills down the toilet. (See “A word about disposal of ‘excess’ opioids”22) The patient returned to her administrative duties at work 2 weeks after the procedure and reported that she was “very satisfied” with her surgical experience.
The US Food and Drug Administration (FDA) recommends disposing of certain drugs through a take-back program or, if such a program is not readily available, by flushing them down a toilet or sink. In a recent study, investigators concluded that opioids on the FDA's so-called flush list include most opioids in clinical use--even if the entire supply prescribed is to be flushed down the drain. Conservative estimates of environmental degradation were employed in the study; the investigators' conclusion was that these drugs pose a "negligible" eco-toxicologic risk.1
Reference
- Khan U, Bloom RA, Nicell JA, Laurenson JP. Risks associated with the environmental release of pharmaceuticals on the U.S. Food and Drug Administration "flush list". Sci Total Environ. 2017;609:1023-1040.
In conclusion
Postoperative pain is a complex entity that must be considered to require individualized strategies and, possibly, multiple interventions. Optimally, thorough education, including pain management options, is provided to the patient prior to surgery. Given the current state of opioid abuse in the United States, all gynecologic surgeons should be familiar with multimodal pain therapy and how to employ nonmedical techniques to reduce postsurgical pain without relying solely on opioids. (See “Online resources for pain management”.)
- Drug Disposal Information
(US Department of Justice Drug Enforcement Administration)
https://www.deadiversion.usdoj.gov/drug_disposal/index.html - Surgical Pain Consortium
http://surgicalpainconsortium.org/
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- United Nations International Narcotics Control Board. Narcotic drugs: Report 2016: Estimated world requirements for 2017-statistics for 2015. New York, NY. https://www.incb.org/documents/Narcotic-Drugs/Technical-Publications/2016/Narcotic_Drugs_Publication_2016.pdf. Published 2017. Accessed January 7, 2018.
- Centers for Disease Control and Prevention. Opioid painkiller prescribing: Where you live makes a difference. Atlanta, GA. https://www.cdc.gov/vitalsigns/pdf/2014-07-vitalsigns.pdf. Published July 2014. Accessed January 5, 2018.
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445–1452.
- Gan TJ, Habib AS, Miller TE, et al. Incidence, patient satisfaction, and perceptions of post-surgical pain: results from a US national survey. Curr Med Res Opin. 2014;30(1):149–160.
- Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625.
- Van Zee A. The promotion and marketing of OxyContin: commercial triumph, public health tragedy. Am J Public Health. 2009;99(2):221–227.
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445–1452.
- US Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. Results from the 2015 national survey on drug use and health: Detailed tables. Rockville, MD. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.htm. Published 2016. Accessed January 5, 2018.
- Muhuri PK, Gfroerer JC, Davies MC. Associations of nonmedical pain reliever use and initiation of heroin use in the United States. CBHSQ Data Rev. http://www.samhsa.gov/data/sites/default/files/DR006/DR006/nonmedical-pain-reliever-use-2013.htm. Published August 2013. Accessed January 5, 2018.
- Joshi GP. Multimodal analgesia techniques and postoperative rehabilitation. Anesthesiol Clin North America. 2005;23(1):185–202.
- Oderda G. Challenges in the management of acute postsurgical pain. Pharmacotherapy. 2012;32(9 suppl):6S–11S.
- Brownstein, MJ. A brief history of opiates, opioid peptides, and opioid receptors. Proc Natl Acad Sci USA. 1993;90(12):5391–5393.
- US Food and Drug Administration. Acetaminophen. https://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm165107.htm. Published November 2017. Accessed January 7, 2018.
- Ong CK, Seymour RA, Lirk P, Merry AF. Combining paracetamol (acetaminophen) with nonsteroidal anti-inflammatory drugs: a qualitative systematic review of analgesic efficacy for acute postoperative pain. Anesth Analg. 2010;110(4):1170–1179.
- Hanlon JT, Semla TP, Schmader KE. Alternative medications for medications included in the use of high‐risk medications in the elderly and potentially harmful drug–disease interactions in the elderly quality measures. J Am Geriatr Soc. 2015;63(12):e8–e18.
- Joshi GP, Janis JE, Haas EM, et al. Surgical site infiltration for abdominal surgery: A novel neuroanatomical-based approach. Plast Reconstr Surg Glob Open. 2016;4(12):e1181. https://insights.ovid.com/crossref?an=01720096-201612000-00021. Accessed January 5, 2018.
- Jokela RM, Ahonen JV, Tallgren MK, et al. The effective analgesic dose of dexamethasone after laparoscopic hysterectomy. Anesth Analg. 2009;109(2):607–615.
- Hohlrieder M, Brimacombe J, Eschertzhuber S, et al. A study of airway management using the ProSeal LMA laryngeal mask airway compared with the tracheal tube on postoperative analgesia requirements following gynaecological laparoscopic surgery. Anaesthesia. 2007;62(9):913–918.
- Phelps P, Cakmakkaya OS, Apfel CC, Radke OC. A simple clinical maneuver to reduce laparoscopy-induced shoulder pain: a randomized controlled trial. Obstet Gynecol. 2008;111(5):1155–1160.
- Sammour T, Kahokehr A, Hill AG. Meta‐analysis of the effect of warm humidified insufflation on pain after laparoscopy. Br J Surg. 2008;95(8):950–956.
- Reagan KM, O’Sullivan DM, Gannon R, Steinberg AC. Decreasing postoperative narcotics in reconstructive pelvic surgery; A randomized controlled trial. Am J Obstet Gynecol. 2017;217(3):325.e1–e10.
- Khan U, Bloom RA, Nicell JA, Laurenson JP. Risks associated with the environmental release of pharmaceuticals on the U.S. Food and Drug Administration “flush list”. Sci Total Environ. 2017;609:1023–1040.
- United Nations International Narcotics Control Board. Narcotic drugs: Report 2016: Estimated world requirements for 2017-statistics for 2015. New York, NY. https://www.incb.org/documents/Narcotic-Drugs/Technical-Publications/2016/Narcotic_Drugs_Publication_2016.pdf. Published 2017. Accessed January 7, 2018.
- Centers for Disease Control and Prevention. Opioid painkiller prescribing: Where you live makes a difference. Atlanta, GA. https://www.cdc.gov/vitalsigns/pdf/2014-07-vitalsigns.pdf. Published July 2014. Accessed January 5, 2018.
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445–1452.
- Gan TJ, Habib AS, Miller TE, et al. Incidence, patient satisfaction, and perceptions of post-surgical pain: results from a US national survey. Curr Med Res Opin. 2014;30(1):149–160.
- Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625.
- Van Zee A. The promotion and marketing of OxyContin: commercial triumph, public health tragedy. Am J Public Health. 2009;99(2):221–227.
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445–1452.
- US Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. Results from the 2015 national survey on drug use and health: Detailed tables. Rockville, MD. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015/NSDUH-DetTabs-2015.htm. Published 2016. Accessed January 5, 2018.
- Muhuri PK, Gfroerer JC, Davies MC. Associations of nonmedical pain reliever use and initiation of heroin use in the United States. CBHSQ Data Rev. http://www.samhsa.gov/data/sites/default/files/DR006/DR006/nonmedical-pain-reliever-use-2013.htm. Published August 2013. Accessed January 5, 2018.
- Joshi GP. Multimodal analgesia techniques and postoperative rehabilitation. Anesthesiol Clin North America. 2005;23(1):185–202.
- Oderda G. Challenges in the management of acute postsurgical pain. Pharmacotherapy. 2012;32(9 suppl):6S–11S.
- Brownstein, MJ. A brief history of opiates, opioid peptides, and opioid receptors. Proc Natl Acad Sci USA. 1993;90(12):5391–5393.
- US Food and Drug Administration. Acetaminophen. https://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm165107.htm. Published November 2017. Accessed January 7, 2018.
- Ong CK, Seymour RA, Lirk P, Merry AF. Combining paracetamol (acetaminophen) with nonsteroidal anti-inflammatory drugs: a qualitative systematic review of analgesic efficacy for acute postoperative pain. Anesth Analg. 2010;110(4):1170–1179.
- Hanlon JT, Semla TP, Schmader KE. Alternative medications for medications included in the use of high‐risk medications in the elderly and potentially harmful drug–disease interactions in the elderly quality measures. J Am Geriatr Soc. 2015;63(12):e8–e18.
- Joshi GP, Janis JE, Haas EM, et al. Surgical site infiltration for abdominal surgery: A novel neuroanatomical-based approach. Plast Reconstr Surg Glob Open. 2016;4(12):e1181. https://insights.ovid.com/crossref?an=01720096-201612000-00021. Accessed January 5, 2018.
- Jokela RM, Ahonen JV, Tallgren MK, et al. The effective analgesic dose of dexamethasone after laparoscopic hysterectomy. Anesth Analg. 2009;109(2):607–615.
- Hohlrieder M, Brimacombe J, Eschertzhuber S, et al. A study of airway management using the ProSeal LMA laryngeal mask airway compared with the tracheal tube on postoperative analgesia requirements following gynaecological laparoscopic surgery. Anaesthesia. 2007;62(9):913–918.
- Phelps P, Cakmakkaya OS, Apfel CC, Radke OC. A simple clinical maneuver to reduce laparoscopy-induced shoulder pain: a randomized controlled trial. Obstet Gynecol. 2008;111(5):1155–1160.
- Sammour T, Kahokehr A, Hill AG. Meta‐analysis of the effect of warm humidified insufflation on pain after laparoscopy. Br J Surg. 2008;95(8):950–956.
- Reagan KM, O’Sullivan DM, Gannon R, Steinberg AC. Decreasing postoperative narcotics in reconstructive pelvic surgery; A randomized controlled trial. Am J Obstet Gynecol. 2017;217(3):325.e1–e10.
- Khan U, Bloom RA, Nicell JA, Laurenson JP. Risks associated with the environmental release of pharmaceuticals on the U.S. Food and Drug Administration “flush list”. Sci Total Environ. 2017;609:1023–1040.
2018 Update on fertility
Clinicians always should consider endometriosis in the diagnostic work-up of an infertility patient. But the diagnosis of endometriosis is often difficult, and management is complex. In this Update, we summarize international consensus documents on endometriosis with the aim of enhancing clinicians’ ability to make evidence-based decisions. In addition, we explore the interesting results of a large hysterosalpingography trial in which 2 different contrast mediums were used. Finally, we urge all clinicians to adapt the new standardized lexicon of infertility and fertility care terms that comprise the recently revised international glossary.
Endometriosis and infertility: The knowns and unknowns
Johnson NP, Hummelshoj L, Adamson GD, et al; World Endometriosis Society Sao Paulo Consortium. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. 2017;32(2):315-324.
Johnson NP, Hummelshoj L; World Endometriosis Society Montpellier Consortium. Consensus on current management of endometriosis. Hum Reprod. 2013;28(6):1552-1568.
Rogers PA, Adamson GD, Al-Jefout M, et al; WES/WERF Consortium for Research Priorities in Endometriosis. Research priorities for endometriosis. Reprod Sci. 2017;24(2):202-226.
Endometriosis is defined as "a disease characterized by the presence of endometrium-like epithelium and stroma outside the endometrium and myometrium. Intrapelvic endometriosis can be located superficially on the peritoneum (peritoneal endometriosis), can extend 5 mm or more beneath the peritoneum (deep endometriosis) or can be present as an ovarian endometriotic cyst (endometrioma)."1 Always consider endometriosis in the infertile patient.
Although many professional societies and numerous Cochrane Database Systematic Reviews have provided guidelines on endometriosis, controversy and uncertainty remain. The World Endometriosis Society (WES) and the World Endometriosis Research Foundation (WERF), however, have now published several consensus documents that assess the global literature and professional organization guidelines in a structured, consensus-driven process.2-4 These WES and WERF documents consolidate known information and can be used to inform the clinician in making evidence-linked diagnostic and treatment decisions. Recommendations offered in this discussion are based on those documents.
Establishing the diagnosis can be difficult
Diagnosis of endometriosis is often difficult and is delayed an average of 7 years from onset of symptoms. These include severe dysmenorrhea, deep dyspareunia, chronic pelvic pain, ovulation pain, cyclical or perimenstrual symptoms (bowel or bladder associated) with or without abnormal bleeding, chronic fatigue, and infertility. A major difficulty is that the predictive value of any one symptom or set of symptoms remains uncertain, as each of these symptoms can have other causes, and a significant proportion of affected women are asymptomatic.
For a definitive diagnosis of endometriosis, visual inspection of the pelvis at laparoscopy is the gold standard investigation, unless disease is visible in the vagina or elsewhere. Positive histology confirms the diagnosis of endometriosis; negative histology does not exclude it. Whether histology should be obtained if peritoneal disease alone is present is controversial: visual inspection usually is adequate, but histologic confirmation of at least one lesion is ideal. In cases of ovarian endometrioma (>4 cm in diameter) and in deeply infiltrating disease, histology should be obtained to identify endometriosis and to exclude rare instances of malignancy.
Compared with laparoscopy, transvaginal ultrasonography (TVUS) has no value in diagnosing peritoneal endometriosis, but it is a useful tool for both making and excluding the diagnosis of an ovarian endometrioma. TVUS may have a role in the diagnosis of disease involving the bladder or rectum.
At present, evidence is insufficient to indicate that magnetic resonance imaging (MRI) is useful for diagnosing or excluding endometriosis compared with laparoscopy. MRI should be reserved for when ultrasound results are equivocal in cases of rectovaginal or bladder endometriosis.
Serum cancer antigen 125 (CA 125) levels may be elevated in endometriosis. However, measuring serum CA 125 levels has no value as a diagnostic tool.
No fertility benefit with ovarian suppression
More than 2 dozen randomized controlled trials (RCTs) provide strong evidence that there is no fertility benefit from ovarian suppression. The drug costs and delayed time to pregnancy mean that ovarian suppression with oral contraceptives, other progestational agents, or gonadotropin-releasing hormone (GnRH) agonists before fertility treatment is not indicated, with the possible exception of using it prior to in vitro fertilization (IVF).
Ovarian suppression also has been suggested as beneficial in conjunction with surgery. However, at least 16 RCTs have failed to show fertility improvement when ovarian suppression is given either preoperatively or postoperatively. Again, the delay in attempting pregnancy, drug costs, and adverse effects render ovarian suppression not appropriate.
While ovarian suppression has not been shown to increase pregnancy rates, ovarian stimulation (OS) likely does, especially when combined with intrauterine insemination (IUI).5
Laparoscopy: Appropriate for selected patients
A major decision for clinicians and patients dealing with infertility is whether to perform a laparoscopy, both for diagnostic and for treatment reasons. Currently, data are insufficient to recommend laparoscopic surgery prior to OS/IUI unless there is a history of evidence of anatomic disease and/or the patient has sufficient pain to justify the physical, emotional, financial, and time costs of laparoscopy. Laparoscopy therefore can be considered as possibly appropriate in younger women (<37 years of age) with short duration of infertility (<4 years), normal male factor, normal or treatable uterus, normal or treatable ovulation disorder, and limited prior treatment.
It is important to consider what disease might be found and how much of an increase in fertility can be obtained by treatment, so that the number needed to treat (NNT) can be used as an estimate of the potential value of laparoscopy in a given patient. A patient also should have no contraindications to laparoscopy and accept 9 to 15 months of attempting pregnancy before undergoing IVF treatment.
When laparoscopy is performed for minimal to mild disease, the odds ratio for pregnancy is 1.66 with treatment. It is important to remove all visible disease without injuring healthy tissue. When disease is moderate to severe, there is often severe anatomic distortion and a very low background pregnancy rate. Numerous uncontrolled trials show benefit of operative laparoscopy, especially for invasive, adhesive, and cystic endometriosis. However, repeat surgery is rarely indicated. After surgery, the Endometriosis Fertility Index (EFI) can be used to determine prognosis and plan management (FIGURE 1).6 An easy-to-use electronic EFI calculator is available online at www.endometriosisefi.com.
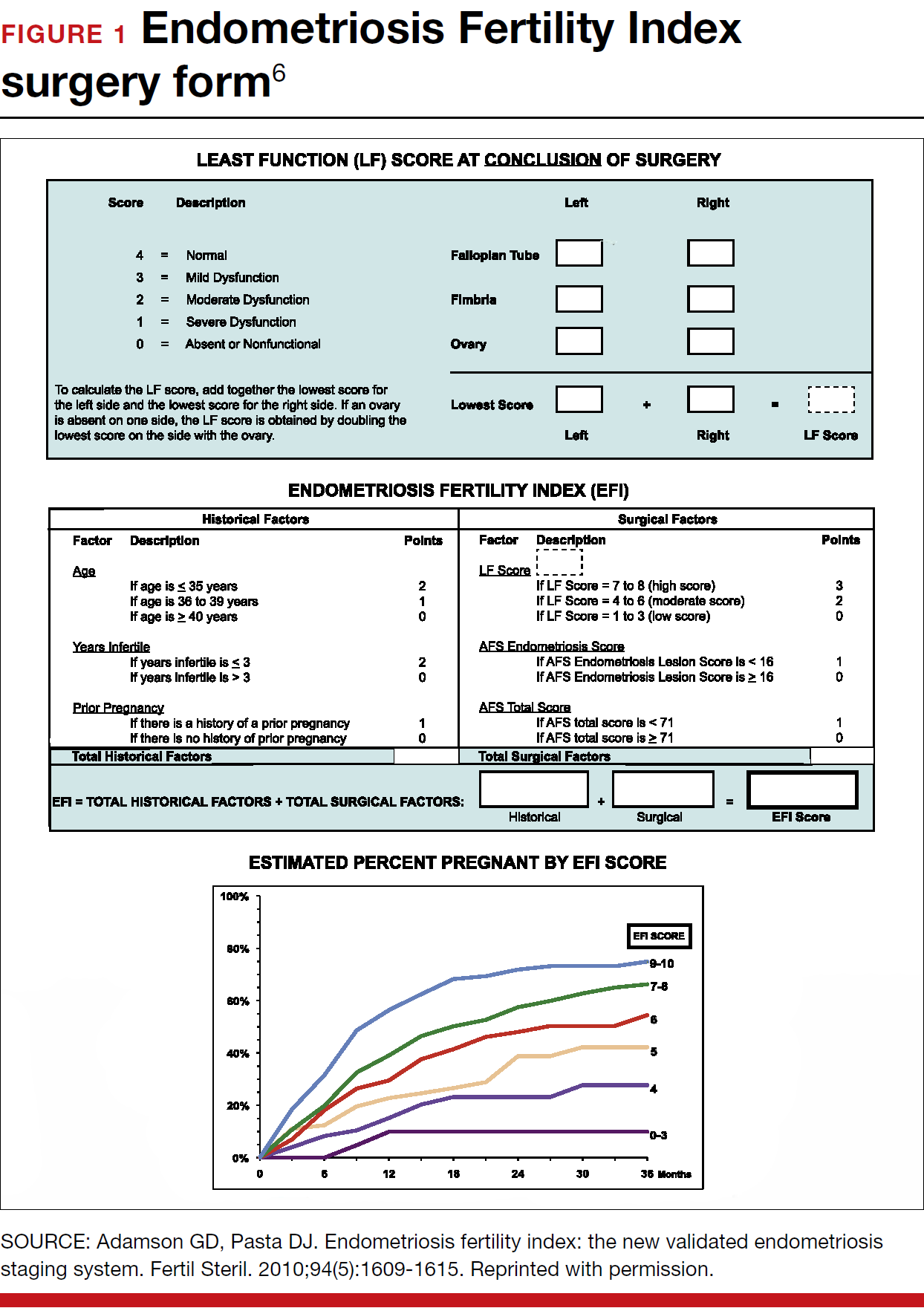
Management of endometriomas
Endometriomas are often operated on because of pain. Initial pain relief occurs in 60% to 100% of patients, but cysts recur following stripping about 10% of the time, and drainage without stripping, about 20%. With recurrence, pain is present about 75% of the time.
Pregnancy rates following endometrioma treatment depend on patient age and the status of the pelvis following operative intervention. This can be determined from the EFI. Often, the dilemma with endometriomas is how aggressive to be in removing them. The principles involved are to remove all the cyst wall if possible, but absolutely to minimize ovarian tissue damage, because reduced ovarian reserve is a possible major negative consequence of ovarian surgery.
Recommendations
While endometriosis is often a cause of infertility, often infertile patients do not have endometriosis. A careful history, physical examination, and ultrasonography, and possibly other imaging studies, are prerequisites to careful clinical judgment in diagnosing and treating infertile patients who might or do have endometriosis.
When pelvic pain is present, initially nonsteroidal anti-inflammatory drugs (NSAIDs), oral contraceptives (OCs), progestational agents, or an intrauterine device can be helpful. These ovarian suppression medications do not increase fertility, however, and should be stopped in any patient who desires to get pregnant.
When pelvic and male fertility factors appear reasonably normal (even if minimal or mild endometriosis is suspected), treatment with clomiphene 100 mg on cycle days 3 through 7 and IUI for 3 to 6 cycles is an effective first step. However, if the patient has persistent pain and/or infertility without other significant infertility factors, then diagnostic laparoscopy with intraoperative treatment of disease is indicated.
Surgery well performed is effective treatment for all stages of endometriosis and endometriomas, both for infertility and for pain. Repeat surgery, however, is rarely indicated because of limited results, so it is important to obtain the best possible result on the first surgery. Surgery is indicated for large endometriomas (>4 cm). Endometriosis has almost no effect on the IVF live birth rate unless ovarian reserve has been reduced by endometriomas or surgery, so endometriosis surgery should be performed by skilled and experienced surgeons.
Endometriosis is a complex disease that can cause infertility. Its diagnosis and management are frequently difficult, requiring knowledge, experience, and good medical judgment and surgical skills. However, if evidence-linked principles are followed, effective treatment plans and good outcomes can be obtained for most patients.
Read about why oil-based contrast may be better than water-based contrast with HSG.
Oil-based contrast medium use in hysterosalpingography is associated with higher pregnancy rates compared with water-based contrast
Dreyer K, van Rijswijk J, Mijatovic V, et al. Oil-based or water-based contrast for hysterosalpingography in infertile women. N Engl J Med. 2017;376(21):2043-2052.
Hysterosalpingography (HSG) to assess tubal patency has been a mainstay of infertility diagnosis for decades. Some, but not all, studies also have suggested that pregnancy rates are higher after this tubal flushing procedure, especially if performed with oil contrast.7,8 A recent multicenter, randomized, controlled trial by Dreyer and colleagues that compared ongoing pregnancy rates and other outcomes among women who had HSG with oil contrast versus with water contrast provides additional valuable information.9
Trial details
In this study, 1,294 infertile women in 27 academic, teaching and nonteaching hospitals were screened for trial eligibility; 1,119 women provided written informed consent. Of these, 557 women were randomly assigned to HSG with oil contrast and 562 to water contrast. The women had spontaneous menstrual cycles, had been attempting pregnancy for at least 1 year, and had indications for HSG.
Exclusion criteria were known endocrine disorders, fewer than 8 menstrual cycles per year, a high risk of tubal disease, iodine allergy, and a total motile sperm count after sperm wash of less than 3 million/mL in the male partner (or a total motile sperm count of less than 1 million/mL when an analysis after sperm wash was not performed).
Just prior to undergoing HSG, the women were randomly assigned to receive either oil contrast or water contrast medium. (The trial was not blinded to participants or caregivers.) HSG was performed according to local protocols using cervical vacuum cup, metal cannula (hysterophore), or balloon catheter and approximately 5 to 10 mL of contrast medium.
After HSG, couples received expectant management when the predicted likelihood of pregnancy within 12 months, based on the prognostic model of Hunault, was 30% or greater.10 IUI was offered for pregnancy likelihood less than 30%, mild male infertility, or failure after a period of expectant management. IUI with or without mild ovarian stimulation (2-3 follicles) with clomiphene or gonadotropins was initiated after a minimum of 2 months of expectant management after HSG.
The primary outcome measure was ongoing pregnancy, defined as a positive fetal heartbeat on ultrasonographic examination after 12 weeks of gestation, with the first day of the last menstrual cycle for the pregnancy within 6 months after randomization. Secondary outcome measures were clinical pregnancy, live birth, miscarriage, ectopic pregnancy, time to pregnancy, and pain scores after HSG. All data were analyzed according to intention-to-treat.
Pregnancy rates increased with oil-contrast HSG
The baseline characteristics of the 2 groups were similar. HSG showed bilateral tubal patency in 477 of 554 women (86.1%) in the oil contrast group and in 491 of 554 women (88.6%) who received the water contrast (rate ratio, 0.97; 95% confidence interval [CI], 0.93-1.02). Bilateral tubal occlusion occurred in 9 women in the oil group (1.6%) and in 13 in the water group (2.3%) (relative risk, 0.69; 95% CI, 0.30-1.61).
A total of 58.3% of the women assigned to oil contrast and 57.2% of those assigned to water contrast received expectant management. Similar percentages of women in the oil group and in the water group underwent IUI (39.7% and 41.0%, respectively), IVF or intracytoplasmic sperm injection (ICSI) (2.3% and 2.2%), laparoscopy (6.2% in each group), and hysteroscopy (4.4% and 4.2%).
Ongoing pregnancy occurred in 220 of 554 women (39.7%) in the oil contrast group and in 161 of 554 women (29.1%) in the water contrast group (rate ratio, 1.37; 95% CI, 1.16-1.61; P<.001). The median time to the onset of pregnancy in the oil group was 2.7 months (interquartile range, 1.5-4.7) (FIGURE 2), while in the water group it was 3.1 months (interquartile range, 1.6-4.8) (P = .44).
While the proportion of women getting pregnant with or without the different interventions was similar in both groups, the live birth rate was 38.8% in the oil group versus 28.1% in the water group (rate ratio, 1.38; 95% CI, 1.17-1.64; P<.001). Three of 554 women (0.5%) assigned to oil contrast and 4 of 554 women (0.7%) in the water contrast group had an adverse event during the trial period. Three women (1.4%), all in the oil group, delivered a child with a congenital anomaly.
Why this study is important
This is the largest and best methodologic study on this clinical issue. It showed higher pregnancy and live birth rates within 6 months of HSG performed with oil compared with water. Although the study was not blinded, the group similarities and objective outcomes support minimal bias. Importantly, these results can be generalized only to women with similar inclusion characteristics.
It is unclear why oil HSG might enhance fertility. Suggested mechanisms include flushing of debris and/or mucous plugs or an effect on peritoneal macrophages or endometrial receptivity. Since HSG is minimally invasive and inexpensive, and the 10% increase in pregnancy rates corresponds to an NNT of 10, it is reasonable to consider, although formal cost-effectiveness data are lacking.
Concerns include the rare theoretical risk of intravasation with subsequent allergic reaction or fat embolism. Three infants in the oil group and none in the water group had congenital anomalies. This is likely due to chance, since this rate is not higher than that in the general population and no other data suggest an increased risk. Comparison of these results with other new techniques, such as sonohysterography (saline infusion sonogram), awaits further studies.
Recommendation
HSG with oil contrast should be considered a potential therapeutic as well as diagnostic intervention in selected patients.
HSG is an important diagnostic test for most infertility patients. The fact that a therapeutic benefit probably also is associated with oil-based HSG increases the clinical indications for this test.
Read about new definitions of infertility terminology you should know.
Infertility glossary is newly updated
Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393-406.
Terms and definitions used in infertility and fertility care frequently have had different meanings for different stakeholders, especially on a global basis. This can result in misunderstandings and inappropriate interpretation and comparison of published information and research. To help address these issues, international fertility organizations recently developed an updated glossary on infertilityterminology.
The consensus process for updating the glossary
The International Glossary on Infertility and Fertility Care, 2017, was recently published simultaneously in Fertility and Sterility and Human Reproduction. This is the second revision; the first glossary was published in 2006 and revised in 2009. This revision's 25 lead experts began work in 2014. Their teams of professionals interacted by electronic mail, at international and regional society meetings, and at 2 consultations held in Geneva, Switzerland. This glossary represents consensus agreement reached on 283 evidence-driven terms and definitions.
The work was led by the International Committee for Monitoring Assisted Reproductive Technologies in partnership with the American Society for Reproductive Medicine, European Society of Human Reproduction and Embryology, International Federation of Fertility Societies, March of Dimes, African Fertility Society, Groupe Inter-africain d'Etude de Recherche et d'Application sur la Fertilité, Asian Pacific Initiative on Reproduction, Middle East Fertility Society, Red Latinoamericana de Reproducción Asistida, and the International Federation of Gynecology and Obstetrics.
All together, 108 international professional experts (clinicians, basic scientists, epidemiologists, and social scientists), along with national and regional representatives of infertile persons, participated in the development of this evidence-base driven glossary. As such, these definitions now set the standard for international communication among clinicians, scientists, and policymakers.
Definition of infertility is broadened
The definitions take account of ethics, human rights, cultural sensitivities, ethnic minorities, and gender equality. For example, the first modification included broadening the concept of infertility to be an "impairment of individuals" in their capacity to reproduce, irrespective of whether the individual has a partner. (See “Broadened definition of infertility” below). Reproductive rights are individual human rights and do not depend on a relationship with another individual. The revised definition also reinforces the concept of infertility as a disease that can generate an impairment of function.
Infertility: A disease characterized by the failure to establish a clinical pregnancy after 12 months of regular, unprotected sexual intercourse or due to an impairment of a person’s capacity to reproduce either as an individual or with his/her partner. Fertility interventions may be initiated in less than 1 year based on medical, sexual and reproductive history, age, physical findings and diagnostic testing. Infertility is a disease, which generates disability as an impairment of function.
Reference
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406
New--and changed--definitions
Certain terms need to be consistent with those used currently internationally, for example, at which gestational age a miscarriage/abortion becomes a stillbirth.
Some terms are confusing, such as subfertility, which does not define a different or less severe fertility status than infertility, does not exist before infertility is diagnosed, and should not be confused with sterility, which is a permanent state of infertility. The term subfertility therefore is redundant and has been removed and replaced by infertility (See “Some terms with an important new definition” below).
- Clinical pregnancy
- Conception (removed from glossary)
- Diminished ovarian reserve
- Fertility care
- Hypospermia (replaces oligospermia)
- Ovarian reserve
- Pregnancy
- Preimplantation genetic testing
- Spontaneous abortion/miscarriage
- Subfertility (should be used interchangeably with infertility)
Reference
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406.
In a different context, the term conception, and its derivatives such as conceiving or conceived, was removed because it cannot be described biologically during the process of reproduction. Instead, terms such as fertilization, implantation, pregnancy, and live birth should be used.
Important male terms also changed: oligospermia is a term for low semen volume that is now replaced by hypospermia to avoid confusion with oligozoospermia, which is low concentration of spermatozoa in the ejaculate below the lower reference limit. When reporting results, the reference criteria should be specified.
Lastly, owing to the lack of standardization in determining the burden of infertility, and to better ensure comparability of prevalence data published globally, this glossary includes definitions for terms frequently used in epidemiology and public health. Examples include voluntary and involuntary childlessness, primary and secondary infertility, fertility care, fecundity, and fecundability, among others.
Getting the word out
The glossary has been approved by all of the participating organizations who are assisting in its distribution. It is being presented at national and international meetings and is used in The FIGO Fertility Toolbox (www.fertilitytool.com). It is hoped that all professionals and other stakeholders will begin to use its terminology globally to provide quality care and ensure consistency in registering specific fertility care interventions and more accurate reporting of their outcomes.
The language we use determines our individual and collective understanding of the scientific and clinical care of our patients. This glossary provides an essential and comprehensive standardization of terms and definitions essential to quality reproductive health care.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406.
- Johnson NP, Hummelshoj L; World Endometriosis Society Montpellier Consortium. Consensus on current management of endometriosis. Hum Reprod. 2013;28(6):1552–1568.
- Rogers PA, Adamson GD, Al-Jefout M, et al; WES/WERF Consortium for Research Priorities in Endometriosis. Research priorities for endometriosis. Reprod Sci. 2017;24(2):202–226.
- Johnson NP, Hummelshoj L, Adamson GD, et al; World Endometriosis Society Sao Paulo Consortium. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. 2017;32(2):315–324.
- Practice Committee of the American Society for Reproductive Medicine. Endometriosis and infertility: a committee opinion. Fertil Steril. 2012;98(3):591–598.
- Adamson GD, Pasta DJ. Endometriosis fertility index: the new, validated endometriosis staging system. Fertil Steril. 2010;94(5):1609–1615.
- Weir WC, Weir DR. Therapeutic value of salpingograms in infertility. Fertil Steril. 1951;2(6);514–522.
- Johnson NP, Farquhar CM, Hadden WE, Suckling J, Yu Y, Sadler L. The FLUSH trial—flushing with lipiodol for unexplained (and endometriosis-related) subfertility by hysterosalpingography: a randomized trial. Hum Reprod. 2004;19(9):2043–2051.
- Dreyer K, van Rijswijk J, Mijatovic V, et al. Oil-based or water-based contrast for hysterosalpingography in infertile women. N Engl J Med. 2017;376(21):2043–2052.
- Van der Steeg JW, Steures P, Eijkemans MJ, et al; Collaborative Effort for Clinical Evaluation in Reproductive Medicine. Pregnancy is predictable: a large-scale prospective external validation of the prediction of spontaneous pregnancy in sub-fertile couples. Hum Reprod. 2007;22(2):536–542.

Dr. Adamson is Founder and CEO of Advanced Reproductive Care, Inc (ARC Fertility); Clinical Professor, ACF, at Stanford University School of Medicine; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose.

Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility and Chair, Department of Reproductive Endocrinology and Infertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson reports being a consultant to AbbVie, Bayer, Ferring, Guerbet, Hernest, and Merck, and that he has equity in ARC Fertility. Dr. Abusief reports no financial relationships relevant to this article.
Dr. Adamson is Founder and CEO of Advanced Reproductive Care, Inc (ARC Fertility); Clinical Professor, ACF, at Stanford University School of Medicine; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose.
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility and Chair, Department of Reproductive Endocrinology and Infertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson reports being a consultant to AbbVie, Bayer, Ferring, Guerbet, Hernest, and Merck, and that he has equity in ARC Fertility. Dr. Abusief reports no financial relationships relevant to this article.
Dr. Adamson is Founder and CEO of Advanced Reproductive Care, Inc (ARC Fertility); Clinical Professor, ACF, at Stanford University School of Medicine; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose.
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility and Chair, Department of Reproductive Endocrinology and Infertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson reports being a consultant to AbbVie, Bayer, Ferring, Guerbet, Hernest, and Merck, and that he has equity in ARC Fertility. Dr. Abusief reports no financial relationships relevant to this article.
Clinicians always should consider endometriosis in the diagnostic work-up of an infertility patient. But the diagnosis of endometriosis is often difficult, and management is complex. In this Update, we summarize international consensus documents on endometriosis with the aim of enhancing clinicians’ ability to make evidence-based decisions. In addition, we explore the interesting results of a large hysterosalpingography trial in which 2 different contrast mediums were used. Finally, we urge all clinicians to adapt the new standardized lexicon of infertility and fertility care terms that comprise the recently revised international glossary.
Endometriosis and infertility: The knowns and unknowns
Johnson NP, Hummelshoj L, Adamson GD, et al; World Endometriosis Society Sao Paulo Consortium. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. 2017;32(2):315-324.
Johnson NP, Hummelshoj L; World Endometriosis Society Montpellier Consortium. Consensus on current management of endometriosis. Hum Reprod. 2013;28(6):1552-1568.
Rogers PA, Adamson GD, Al-Jefout M, et al; WES/WERF Consortium for Research Priorities in Endometriosis. Research priorities for endometriosis. Reprod Sci. 2017;24(2):202-226.
Endometriosis is defined as "a disease characterized by the presence of endometrium-like epithelium and stroma outside the endometrium and myometrium. Intrapelvic endometriosis can be located superficially on the peritoneum (peritoneal endometriosis), can extend 5 mm or more beneath the peritoneum (deep endometriosis) or can be present as an ovarian endometriotic cyst (endometrioma)."1 Always consider endometriosis in the infertile patient.
Although many professional societies and numerous Cochrane Database Systematic Reviews have provided guidelines on endometriosis, controversy and uncertainty remain. The World Endometriosis Society (WES) and the World Endometriosis Research Foundation (WERF), however, have now published several consensus documents that assess the global literature and professional organization guidelines in a structured, consensus-driven process.2-4 These WES and WERF documents consolidate known information and can be used to inform the clinician in making evidence-linked diagnostic and treatment decisions. Recommendations offered in this discussion are based on those documents.
Establishing the diagnosis can be difficult
Diagnosis of endometriosis is often difficult and is delayed an average of 7 years from onset of symptoms. These include severe dysmenorrhea, deep dyspareunia, chronic pelvic pain, ovulation pain, cyclical or perimenstrual symptoms (bowel or bladder associated) with or without abnormal bleeding, chronic fatigue, and infertility. A major difficulty is that the predictive value of any one symptom or set of symptoms remains uncertain, as each of these symptoms can have other causes, and a significant proportion of affected women are asymptomatic.
For a definitive diagnosis of endometriosis, visual inspection of the pelvis at laparoscopy is the gold standard investigation, unless disease is visible in the vagina or elsewhere. Positive histology confirms the diagnosis of endometriosis; negative histology does not exclude it. Whether histology should be obtained if peritoneal disease alone is present is controversial: visual inspection usually is adequate, but histologic confirmation of at least one lesion is ideal. In cases of ovarian endometrioma (>4 cm in diameter) and in deeply infiltrating disease, histology should be obtained to identify endometriosis and to exclude rare instances of malignancy.
Compared with laparoscopy, transvaginal ultrasonography (TVUS) has no value in diagnosing peritoneal endometriosis, but it is a useful tool for both making and excluding the diagnosis of an ovarian endometrioma. TVUS may have a role in the diagnosis of disease involving the bladder or rectum.
At present, evidence is insufficient to indicate that magnetic resonance imaging (MRI) is useful for diagnosing or excluding endometriosis compared with laparoscopy. MRI should be reserved for when ultrasound results are equivocal in cases of rectovaginal or bladder endometriosis.
Serum cancer antigen 125 (CA 125) levels may be elevated in endometriosis. However, measuring serum CA 125 levels has no value as a diagnostic tool.
No fertility benefit with ovarian suppression
More than 2 dozen randomized controlled trials (RCTs) provide strong evidence that there is no fertility benefit from ovarian suppression. The drug costs and delayed time to pregnancy mean that ovarian suppression with oral contraceptives, other progestational agents, or gonadotropin-releasing hormone (GnRH) agonists before fertility treatment is not indicated, with the possible exception of using it prior to in vitro fertilization (IVF).
Ovarian suppression also has been suggested as beneficial in conjunction with surgery. However, at least 16 RCTs have failed to show fertility improvement when ovarian suppression is given either preoperatively or postoperatively. Again, the delay in attempting pregnancy, drug costs, and adverse effects render ovarian suppression not appropriate.
While ovarian suppression has not been shown to increase pregnancy rates, ovarian stimulation (OS) likely does, especially when combined with intrauterine insemination (IUI).5
Laparoscopy: Appropriate for selected patients
A major decision for clinicians and patients dealing with infertility is whether to perform a laparoscopy, both for diagnostic and for treatment reasons. Currently, data are insufficient to recommend laparoscopic surgery prior to OS/IUI unless there is a history of evidence of anatomic disease and/or the patient has sufficient pain to justify the physical, emotional, financial, and time costs of laparoscopy. Laparoscopy therefore can be considered as possibly appropriate in younger women (<37 years of age) with short duration of infertility (<4 years), normal male factor, normal or treatable uterus, normal or treatable ovulation disorder, and limited prior treatment.
It is important to consider what disease might be found and how much of an increase in fertility can be obtained by treatment, so that the number needed to treat (NNT) can be used as an estimate of the potential value of laparoscopy in a given patient. A patient also should have no contraindications to laparoscopy and accept 9 to 15 months of attempting pregnancy before undergoing IVF treatment.
When laparoscopy is performed for minimal to mild disease, the odds ratio for pregnancy is 1.66 with treatment. It is important to remove all visible disease without injuring healthy tissue. When disease is moderate to severe, there is often severe anatomic distortion and a very low background pregnancy rate. Numerous uncontrolled trials show benefit of operative laparoscopy, especially for invasive, adhesive, and cystic endometriosis. However, repeat surgery is rarely indicated. After surgery, the Endometriosis Fertility Index (EFI) can be used to determine prognosis and plan management (FIGURE 1).6 An easy-to-use electronic EFI calculator is available online at www.endometriosisefi.com.
Management of endometriomas
Endometriomas are often operated on because of pain. Initial pain relief occurs in 60% to 100% of patients, but cysts recur following stripping about 10% of the time, and drainage without stripping, about 20%. With recurrence, pain is present about 75% of the time.
Pregnancy rates following endometrioma treatment depend on patient age and the status of the pelvis following operative intervention. This can be determined from the EFI. Often, the dilemma with endometriomas is how aggressive to be in removing them. The principles involved are to remove all the cyst wall if possible, but absolutely to minimize ovarian tissue damage, because reduced ovarian reserve is a possible major negative consequence of ovarian surgery.
Recommendations
While endometriosis is often a cause of infertility, often infertile patients do not have endometriosis. A careful history, physical examination, and ultrasonography, and possibly other imaging studies, are prerequisites to careful clinical judgment in diagnosing and treating infertile patients who might or do have endometriosis.
When pelvic pain is present, initially nonsteroidal anti-inflammatory drugs (NSAIDs), oral contraceptives (OCs), progestational agents, or an intrauterine device can be helpful. These ovarian suppression medications do not increase fertility, however, and should be stopped in any patient who desires to get pregnant.
When pelvic and male fertility factors appear reasonably normal (even if minimal or mild endometriosis is suspected), treatment with clomiphene 100 mg on cycle days 3 through 7 and IUI for 3 to 6 cycles is an effective first step. However, if the patient has persistent pain and/or infertility without other significant infertility factors, then diagnostic laparoscopy with intraoperative treatment of disease is indicated.
Surgery well performed is effective treatment for all stages of endometriosis and endometriomas, both for infertility and for pain. Repeat surgery, however, is rarely indicated because of limited results, so it is important to obtain the best possible result on the first surgery. Surgery is indicated for large endometriomas (>4 cm). Endometriosis has almost no effect on the IVF live birth rate unless ovarian reserve has been reduced by endometriomas or surgery, so endometriosis surgery should be performed by skilled and experienced surgeons.
Endometriosis is a complex disease that can cause infertility. Its diagnosis and management are frequently difficult, requiring knowledge, experience, and good medical judgment and surgical skills. However, if evidence-linked principles are followed, effective treatment plans and good outcomes can be obtained for most patients.
Read about why oil-based contrast may be better than water-based contrast with HSG.
Oil-based contrast medium use in hysterosalpingography is associated with higher pregnancy rates compared with water-based contrast
Dreyer K, van Rijswijk J, Mijatovic V, et al. Oil-based or water-based contrast for hysterosalpingography in infertile women. N Engl J Med. 2017;376(21):2043-2052.
Hysterosalpingography (HSG) to assess tubal patency has been a mainstay of infertility diagnosis for decades. Some, but not all, studies also have suggested that pregnancy rates are higher after this tubal flushing procedure, especially if performed with oil contrast.7,8 A recent multicenter, randomized, controlled trial by Dreyer and colleagues that compared ongoing pregnancy rates and other outcomes among women who had HSG with oil contrast versus with water contrast provides additional valuable information.9
Trial details
In this study, 1,294 infertile women in 27 academic, teaching and nonteaching hospitals were screened for trial eligibility; 1,119 women provided written informed consent. Of these, 557 women were randomly assigned to HSG with oil contrast and 562 to water contrast. The women had spontaneous menstrual cycles, had been attempting pregnancy for at least 1 year, and had indications for HSG.
Exclusion criteria were known endocrine disorders, fewer than 8 menstrual cycles per year, a high risk of tubal disease, iodine allergy, and a total motile sperm count after sperm wash of less than 3 million/mL in the male partner (or a total motile sperm count of less than 1 million/mL when an analysis after sperm wash was not performed).
Just prior to undergoing HSG, the women were randomly assigned to receive either oil contrast or water contrast medium. (The trial was not blinded to participants or caregivers.) HSG was performed according to local protocols using cervical vacuum cup, metal cannula (hysterophore), or balloon catheter and approximately 5 to 10 mL of contrast medium.
After HSG, couples received expectant management when the predicted likelihood of pregnancy within 12 months, based on the prognostic model of Hunault, was 30% or greater.10 IUI was offered for pregnancy likelihood less than 30%, mild male infertility, or failure after a period of expectant management. IUI with or without mild ovarian stimulation (2-3 follicles) with clomiphene or gonadotropins was initiated after a minimum of 2 months of expectant management after HSG.
The primary outcome measure was ongoing pregnancy, defined as a positive fetal heartbeat on ultrasonographic examination after 12 weeks of gestation, with the first day of the last menstrual cycle for the pregnancy within 6 months after randomization. Secondary outcome measures were clinical pregnancy, live birth, miscarriage, ectopic pregnancy, time to pregnancy, and pain scores after HSG. All data were analyzed according to intention-to-treat.
Pregnancy rates increased with oil-contrast HSG
The baseline characteristics of the 2 groups were similar. HSG showed bilateral tubal patency in 477 of 554 women (86.1%) in the oil contrast group and in 491 of 554 women (88.6%) who received the water contrast (rate ratio, 0.97; 95% confidence interval [CI], 0.93-1.02). Bilateral tubal occlusion occurred in 9 women in the oil group (1.6%) and in 13 in the water group (2.3%) (relative risk, 0.69; 95% CI, 0.30-1.61).
A total of 58.3% of the women assigned to oil contrast and 57.2% of those assigned to water contrast received expectant management. Similar percentages of women in the oil group and in the water group underwent IUI (39.7% and 41.0%, respectively), IVF or intracytoplasmic sperm injection (ICSI) (2.3% and 2.2%), laparoscopy (6.2% in each group), and hysteroscopy (4.4% and 4.2%).
Ongoing pregnancy occurred in 220 of 554 women (39.7%) in the oil contrast group and in 161 of 554 women (29.1%) in the water contrast group (rate ratio, 1.37; 95% CI, 1.16-1.61; P<.001). The median time to the onset of pregnancy in the oil group was 2.7 months (interquartile range, 1.5-4.7) (FIGURE 2), while in the water group it was 3.1 months (interquartile range, 1.6-4.8) (P = .44).
While the proportion of women getting pregnant with or without the different interventions was similar in both groups, the live birth rate was 38.8% in the oil group versus 28.1% in the water group (rate ratio, 1.38; 95% CI, 1.17-1.64; P<.001). Three of 554 women (0.5%) assigned to oil contrast and 4 of 554 women (0.7%) in the water contrast group had an adverse event during the trial period. Three women (1.4%), all in the oil group, delivered a child with a congenital anomaly.
Why this study is important
This is the largest and best methodologic study on this clinical issue. It showed higher pregnancy and live birth rates within 6 months of HSG performed with oil compared with water. Although the study was not blinded, the group similarities and objective outcomes support minimal bias. Importantly, these results can be generalized only to women with similar inclusion characteristics.
It is unclear why oil HSG might enhance fertility. Suggested mechanisms include flushing of debris and/or mucous plugs or an effect on peritoneal macrophages or endometrial receptivity. Since HSG is minimally invasive and inexpensive, and the 10% increase in pregnancy rates corresponds to an NNT of 10, it is reasonable to consider, although formal cost-effectiveness data are lacking.
Concerns include the rare theoretical risk of intravasation with subsequent allergic reaction or fat embolism. Three infants in the oil group and none in the water group had congenital anomalies. This is likely due to chance, since this rate is not higher than that in the general population and no other data suggest an increased risk. Comparison of these results with other new techniques, such as sonohysterography (saline infusion sonogram), awaits further studies.
Recommendation
HSG with oil contrast should be considered a potential therapeutic as well as diagnostic intervention in selected patients.
HSG is an important diagnostic test for most infertility patients. The fact that a therapeutic benefit probably also is associated with oil-based HSG increases the clinical indications for this test.
Read about new definitions of infertility terminology you should know.
Infertility glossary is newly updated
Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393-406.
Terms and definitions used in infertility and fertility care frequently have had different meanings for different stakeholders, especially on a global basis. This can result in misunderstandings and inappropriate interpretation and comparison of published information and research. To help address these issues, international fertility organizations recently developed an updated glossary on infertilityterminology.
The consensus process for updating the glossary
The International Glossary on Infertility and Fertility Care, 2017, was recently published simultaneously in Fertility and Sterility and Human Reproduction. This is the second revision; the first glossary was published in 2006 and revised in 2009. This revision's 25 lead experts began work in 2014. Their teams of professionals interacted by electronic mail, at international and regional society meetings, and at 2 consultations held in Geneva, Switzerland. This glossary represents consensus agreement reached on 283 evidence-driven terms and definitions.
The work was led by the International Committee for Monitoring Assisted Reproductive Technologies in partnership with the American Society for Reproductive Medicine, European Society of Human Reproduction and Embryology, International Federation of Fertility Societies, March of Dimes, African Fertility Society, Groupe Inter-africain d'Etude de Recherche et d'Application sur la Fertilité, Asian Pacific Initiative on Reproduction, Middle East Fertility Society, Red Latinoamericana de Reproducción Asistida, and the International Federation of Gynecology and Obstetrics.
All together, 108 international professional experts (clinicians, basic scientists, epidemiologists, and social scientists), along with national and regional representatives of infertile persons, participated in the development of this evidence-base driven glossary. As such, these definitions now set the standard for international communication among clinicians, scientists, and policymakers.
Definition of infertility is broadened
The definitions take account of ethics, human rights, cultural sensitivities, ethnic minorities, and gender equality. For example, the first modification included broadening the concept of infertility to be an "impairment of individuals" in their capacity to reproduce, irrespective of whether the individual has a partner. (See “Broadened definition of infertility” below). Reproductive rights are individual human rights and do not depend on a relationship with another individual. The revised definition also reinforces the concept of infertility as a disease that can generate an impairment of function.
Infertility: A disease characterized by the failure to establish a clinical pregnancy after 12 months of regular, unprotected sexual intercourse or due to an impairment of a person’s capacity to reproduce either as an individual or with his/her partner. Fertility interventions may be initiated in less than 1 year based on medical, sexual and reproductive history, age, physical findings and diagnostic testing. Infertility is a disease, which generates disability as an impairment of function.
Reference
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406
New--and changed--definitions
Certain terms need to be consistent with those used currently internationally, for example, at which gestational age a miscarriage/abortion becomes a stillbirth.
Some terms are confusing, such as subfertility, which does not define a different or less severe fertility status than infertility, does not exist before infertility is diagnosed, and should not be confused with sterility, which is a permanent state of infertility. The term subfertility therefore is redundant and has been removed and replaced by infertility (See “Some terms with an important new definition” below).
- Clinical pregnancy
- Conception (removed from glossary)
- Diminished ovarian reserve
- Fertility care
- Hypospermia (replaces oligospermia)
- Ovarian reserve
- Pregnancy
- Preimplantation genetic testing
- Spontaneous abortion/miscarriage
- Subfertility (should be used interchangeably with infertility)
Reference
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406.
In a different context, the term conception, and its derivatives such as conceiving or conceived, was removed because it cannot be described biologically during the process of reproduction. Instead, terms such as fertilization, implantation, pregnancy, and live birth should be used.
Important male terms also changed: oligospermia is a term for low semen volume that is now replaced by hypospermia to avoid confusion with oligozoospermia, which is low concentration of spermatozoa in the ejaculate below the lower reference limit. When reporting results, the reference criteria should be specified.
Lastly, owing to the lack of standardization in determining the burden of infertility, and to better ensure comparability of prevalence data published globally, this glossary includes definitions for terms frequently used in epidemiology and public health. Examples include voluntary and involuntary childlessness, primary and secondary infertility, fertility care, fecundity, and fecundability, among others.
Getting the word out
The glossary has been approved by all of the participating organizations who are assisting in its distribution. It is being presented at national and international meetings and is used in The FIGO Fertility Toolbox (www.fertilitytool.com). It is hoped that all professionals and other stakeholders will begin to use its terminology globally to provide quality care and ensure consistency in registering specific fertility care interventions and more accurate reporting of their outcomes.
The language we use determines our individual and collective understanding of the scientific and clinical care of our patients. This glossary provides an essential and comprehensive standardization of terms and definitions essential to quality reproductive health care.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Clinicians always should consider endometriosis in the diagnostic work-up of an infertility patient. But the diagnosis of endometriosis is often difficult, and management is complex. In this Update, we summarize international consensus documents on endometriosis with the aim of enhancing clinicians’ ability to make evidence-based decisions. In addition, we explore the interesting results of a large hysterosalpingography trial in which 2 different contrast mediums were used. Finally, we urge all clinicians to adapt the new standardized lexicon of infertility and fertility care terms that comprise the recently revised international glossary.
Endometriosis and infertility: The knowns and unknowns
Johnson NP, Hummelshoj L, Adamson GD, et al; World Endometriosis Society Sao Paulo Consortium. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. 2017;32(2):315-324.
Johnson NP, Hummelshoj L; World Endometriosis Society Montpellier Consortium. Consensus on current management of endometriosis. Hum Reprod. 2013;28(6):1552-1568.
Rogers PA, Adamson GD, Al-Jefout M, et al; WES/WERF Consortium for Research Priorities in Endometriosis. Research priorities for endometriosis. Reprod Sci. 2017;24(2):202-226.
Endometriosis is defined as "a disease characterized by the presence of endometrium-like epithelium and stroma outside the endometrium and myometrium. Intrapelvic endometriosis can be located superficially on the peritoneum (peritoneal endometriosis), can extend 5 mm or more beneath the peritoneum (deep endometriosis) or can be present as an ovarian endometriotic cyst (endometrioma)."1 Always consider endometriosis in the infertile patient.
Although many professional societies and numerous Cochrane Database Systematic Reviews have provided guidelines on endometriosis, controversy and uncertainty remain. The World Endometriosis Society (WES) and the World Endometriosis Research Foundation (WERF), however, have now published several consensus documents that assess the global literature and professional organization guidelines in a structured, consensus-driven process.2-4 These WES and WERF documents consolidate known information and can be used to inform the clinician in making evidence-linked diagnostic and treatment decisions. Recommendations offered in this discussion are based on those documents.
Establishing the diagnosis can be difficult
Diagnosis of endometriosis is often difficult and is delayed an average of 7 years from onset of symptoms. These include severe dysmenorrhea, deep dyspareunia, chronic pelvic pain, ovulation pain, cyclical or perimenstrual symptoms (bowel or bladder associated) with or without abnormal bleeding, chronic fatigue, and infertility. A major difficulty is that the predictive value of any one symptom or set of symptoms remains uncertain, as each of these symptoms can have other causes, and a significant proportion of affected women are asymptomatic.
For a definitive diagnosis of endometriosis, visual inspection of the pelvis at laparoscopy is the gold standard investigation, unless disease is visible in the vagina or elsewhere. Positive histology confirms the diagnosis of endometriosis; negative histology does not exclude it. Whether histology should be obtained if peritoneal disease alone is present is controversial: visual inspection usually is adequate, but histologic confirmation of at least one lesion is ideal. In cases of ovarian endometrioma (>4 cm in diameter) and in deeply infiltrating disease, histology should be obtained to identify endometriosis and to exclude rare instances of malignancy.
Compared with laparoscopy, transvaginal ultrasonography (TVUS) has no value in diagnosing peritoneal endometriosis, but it is a useful tool for both making and excluding the diagnosis of an ovarian endometrioma. TVUS may have a role in the diagnosis of disease involving the bladder or rectum.
At present, evidence is insufficient to indicate that magnetic resonance imaging (MRI) is useful for diagnosing or excluding endometriosis compared with laparoscopy. MRI should be reserved for when ultrasound results are equivocal in cases of rectovaginal or bladder endometriosis.
Serum cancer antigen 125 (CA 125) levels may be elevated in endometriosis. However, measuring serum CA 125 levels has no value as a diagnostic tool.
No fertility benefit with ovarian suppression
More than 2 dozen randomized controlled trials (RCTs) provide strong evidence that there is no fertility benefit from ovarian suppression. The drug costs and delayed time to pregnancy mean that ovarian suppression with oral contraceptives, other progestational agents, or gonadotropin-releasing hormone (GnRH) agonists before fertility treatment is not indicated, with the possible exception of using it prior to in vitro fertilization (IVF).
Ovarian suppression also has been suggested as beneficial in conjunction with surgery. However, at least 16 RCTs have failed to show fertility improvement when ovarian suppression is given either preoperatively or postoperatively. Again, the delay in attempting pregnancy, drug costs, and adverse effects render ovarian suppression not appropriate.
While ovarian suppression has not been shown to increase pregnancy rates, ovarian stimulation (OS) likely does, especially when combined with intrauterine insemination (IUI).5
Laparoscopy: Appropriate for selected patients
A major decision for clinicians and patients dealing with infertility is whether to perform a laparoscopy, both for diagnostic and for treatment reasons. Currently, data are insufficient to recommend laparoscopic surgery prior to OS/IUI unless there is a history of evidence of anatomic disease and/or the patient has sufficient pain to justify the physical, emotional, financial, and time costs of laparoscopy. Laparoscopy therefore can be considered as possibly appropriate in younger women (<37 years of age) with short duration of infertility (<4 years), normal male factor, normal or treatable uterus, normal or treatable ovulation disorder, and limited prior treatment.
It is important to consider what disease might be found and how much of an increase in fertility can be obtained by treatment, so that the number needed to treat (NNT) can be used as an estimate of the potential value of laparoscopy in a given patient. A patient also should have no contraindications to laparoscopy and accept 9 to 15 months of attempting pregnancy before undergoing IVF treatment.
When laparoscopy is performed for minimal to mild disease, the odds ratio for pregnancy is 1.66 with treatment. It is important to remove all visible disease without injuring healthy tissue. When disease is moderate to severe, there is often severe anatomic distortion and a very low background pregnancy rate. Numerous uncontrolled trials show benefit of operative laparoscopy, especially for invasive, adhesive, and cystic endometriosis. However, repeat surgery is rarely indicated. After surgery, the Endometriosis Fertility Index (EFI) can be used to determine prognosis and plan management (FIGURE 1).6 An easy-to-use electronic EFI calculator is available online at www.endometriosisefi.com.
Management of endometriomas
Endometriomas are often operated on because of pain. Initial pain relief occurs in 60% to 100% of patients, but cysts recur following stripping about 10% of the time, and drainage without stripping, about 20%. With recurrence, pain is present about 75% of the time.
Pregnancy rates following endometrioma treatment depend on patient age and the status of the pelvis following operative intervention. This can be determined from the EFI. Often, the dilemma with endometriomas is how aggressive to be in removing them. The principles involved are to remove all the cyst wall if possible, but absolutely to minimize ovarian tissue damage, because reduced ovarian reserve is a possible major negative consequence of ovarian surgery.
Recommendations
While endometriosis is often a cause of infertility, often infertile patients do not have endometriosis. A careful history, physical examination, and ultrasonography, and possibly other imaging studies, are prerequisites to careful clinical judgment in diagnosing and treating infertile patients who might or do have endometriosis.
When pelvic pain is present, initially nonsteroidal anti-inflammatory drugs (NSAIDs), oral contraceptives (OCs), progestational agents, or an intrauterine device can be helpful. These ovarian suppression medications do not increase fertility, however, and should be stopped in any patient who desires to get pregnant.
When pelvic and male fertility factors appear reasonably normal (even if minimal or mild endometriosis is suspected), treatment with clomiphene 100 mg on cycle days 3 through 7 and IUI for 3 to 6 cycles is an effective first step. However, if the patient has persistent pain and/or infertility without other significant infertility factors, then diagnostic laparoscopy with intraoperative treatment of disease is indicated.
Surgery well performed is effective treatment for all stages of endometriosis and endometriomas, both for infertility and for pain. Repeat surgery, however, is rarely indicated because of limited results, so it is important to obtain the best possible result on the first surgery. Surgery is indicated for large endometriomas (>4 cm). Endometriosis has almost no effect on the IVF live birth rate unless ovarian reserve has been reduced by endometriomas or surgery, so endometriosis surgery should be performed by skilled and experienced surgeons.
Endometriosis is a complex disease that can cause infertility. Its diagnosis and management are frequently difficult, requiring knowledge, experience, and good medical judgment and surgical skills. However, if evidence-linked principles are followed, effective treatment plans and good outcomes can be obtained for most patients.
Read about why oil-based contrast may be better than water-based contrast with HSG.
Oil-based contrast medium use in hysterosalpingography is associated with higher pregnancy rates compared with water-based contrast
Dreyer K, van Rijswijk J, Mijatovic V, et al. Oil-based or water-based contrast for hysterosalpingography in infertile women. N Engl J Med. 2017;376(21):2043-2052.
Hysterosalpingography (HSG) to assess tubal patency has been a mainstay of infertility diagnosis for decades. Some, but not all, studies also have suggested that pregnancy rates are higher after this tubal flushing procedure, especially if performed with oil contrast.7,8 A recent multicenter, randomized, controlled trial by Dreyer and colleagues that compared ongoing pregnancy rates and other outcomes among women who had HSG with oil contrast versus with water contrast provides additional valuable information.9
Trial details
In this study, 1,294 infertile women in 27 academic, teaching and nonteaching hospitals were screened for trial eligibility; 1,119 women provided written informed consent. Of these, 557 women were randomly assigned to HSG with oil contrast and 562 to water contrast. The women had spontaneous menstrual cycles, had been attempting pregnancy for at least 1 year, and had indications for HSG.
Exclusion criteria were known endocrine disorders, fewer than 8 menstrual cycles per year, a high risk of tubal disease, iodine allergy, and a total motile sperm count after sperm wash of less than 3 million/mL in the male partner (or a total motile sperm count of less than 1 million/mL when an analysis after sperm wash was not performed).
Just prior to undergoing HSG, the women were randomly assigned to receive either oil contrast or water contrast medium. (The trial was not blinded to participants or caregivers.) HSG was performed according to local protocols using cervical vacuum cup, metal cannula (hysterophore), or balloon catheter and approximately 5 to 10 mL of contrast medium.
After HSG, couples received expectant management when the predicted likelihood of pregnancy within 12 months, based on the prognostic model of Hunault, was 30% or greater.10 IUI was offered for pregnancy likelihood less than 30%, mild male infertility, or failure after a period of expectant management. IUI with or without mild ovarian stimulation (2-3 follicles) with clomiphene or gonadotropins was initiated after a minimum of 2 months of expectant management after HSG.
The primary outcome measure was ongoing pregnancy, defined as a positive fetal heartbeat on ultrasonographic examination after 12 weeks of gestation, with the first day of the last menstrual cycle for the pregnancy within 6 months after randomization. Secondary outcome measures were clinical pregnancy, live birth, miscarriage, ectopic pregnancy, time to pregnancy, and pain scores after HSG. All data were analyzed according to intention-to-treat.
Pregnancy rates increased with oil-contrast HSG
The baseline characteristics of the 2 groups were similar. HSG showed bilateral tubal patency in 477 of 554 women (86.1%) in the oil contrast group and in 491 of 554 women (88.6%) who received the water contrast (rate ratio, 0.97; 95% confidence interval [CI], 0.93-1.02). Bilateral tubal occlusion occurred in 9 women in the oil group (1.6%) and in 13 in the water group (2.3%) (relative risk, 0.69; 95% CI, 0.30-1.61).
A total of 58.3% of the women assigned to oil contrast and 57.2% of those assigned to water contrast received expectant management. Similar percentages of women in the oil group and in the water group underwent IUI (39.7% and 41.0%, respectively), IVF or intracytoplasmic sperm injection (ICSI) (2.3% and 2.2%), laparoscopy (6.2% in each group), and hysteroscopy (4.4% and 4.2%).
Ongoing pregnancy occurred in 220 of 554 women (39.7%) in the oil contrast group and in 161 of 554 women (29.1%) in the water contrast group (rate ratio, 1.37; 95% CI, 1.16-1.61; P<.001). The median time to the onset of pregnancy in the oil group was 2.7 months (interquartile range, 1.5-4.7) (FIGURE 2), while in the water group it was 3.1 months (interquartile range, 1.6-4.8) (P = .44).
While the proportion of women getting pregnant with or without the different interventions was similar in both groups, the live birth rate was 38.8% in the oil group versus 28.1% in the water group (rate ratio, 1.38; 95% CI, 1.17-1.64; P<.001). Three of 554 women (0.5%) assigned to oil contrast and 4 of 554 women (0.7%) in the water contrast group had an adverse event during the trial period. Three women (1.4%), all in the oil group, delivered a child with a congenital anomaly.
Why this study is important
This is the largest and best methodologic study on this clinical issue. It showed higher pregnancy and live birth rates within 6 months of HSG performed with oil compared with water. Although the study was not blinded, the group similarities and objective outcomes support minimal bias. Importantly, these results can be generalized only to women with similar inclusion characteristics.
It is unclear why oil HSG might enhance fertility. Suggested mechanisms include flushing of debris and/or mucous plugs or an effect on peritoneal macrophages or endometrial receptivity. Since HSG is minimally invasive and inexpensive, and the 10% increase in pregnancy rates corresponds to an NNT of 10, it is reasonable to consider, although formal cost-effectiveness data are lacking.
Concerns include the rare theoretical risk of intravasation with subsequent allergic reaction or fat embolism. Three infants in the oil group and none in the water group had congenital anomalies. This is likely due to chance, since this rate is not higher than that in the general population and no other data suggest an increased risk. Comparison of these results with other new techniques, such as sonohysterography (saline infusion sonogram), awaits further studies.
Recommendation
HSG with oil contrast should be considered a potential therapeutic as well as diagnostic intervention in selected patients.
HSG is an important diagnostic test for most infertility patients. The fact that a therapeutic benefit probably also is associated with oil-based HSG increases the clinical indications for this test.
Read about new definitions of infertility terminology you should know.
Infertility glossary is newly updated
Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393-406.
Terms and definitions used in infertility and fertility care frequently have had different meanings for different stakeholders, especially on a global basis. This can result in misunderstandings and inappropriate interpretation and comparison of published information and research. To help address these issues, international fertility organizations recently developed an updated glossary on infertilityterminology.
The consensus process for updating the glossary
The International Glossary on Infertility and Fertility Care, 2017, was recently published simultaneously in Fertility and Sterility and Human Reproduction. This is the second revision; the first glossary was published in 2006 and revised in 2009. This revision's 25 lead experts began work in 2014. Their teams of professionals interacted by electronic mail, at international and regional society meetings, and at 2 consultations held in Geneva, Switzerland. This glossary represents consensus agreement reached on 283 evidence-driven terms and definitions.
The work was led by the International Committee for Monitoring Assisted Reproductive Technologies in partnership with the American Society for Reproductive Medicine, European Society of Human Reproduction and Embryology, International Federation of Fertility Societies, March of Dimes, African Fertility Society, Groupe Inter-africain d'Etude de Recherche et d'Application sur la Fertilité, Asian Pacific Initiative on Reproduction, Middle East Fertility Society, Red Latinoamericana de Reproducción Asistida, and the International Federation of Gynecology and Obstetrics.
All together, 108 international professional experts (clinicians, basic scientists, epidemiologists, and social scientists), along with national and regional representatives of infertile persons, participated in the development of this evidence-base driven glossary. As such, these definitions now set the standard for international communication among clinicians, scientists, and policymakers.
Definition of infertility is broadened
The definitions take account of ethics, human rights, cultural sensitivities, ethnic minorities, and gender equality. For example, the first modification included broadening the concept of infertility to be an "impairment of individuals" in their capacity to reproduce, irrespective of whether the individual has a partner. (See “Broadened definition of infertility” below). Reproductive rights are individual human rights and do not depend on a relationship with another individual. The revised definition also reinforces the concept of infertility as a disease that can generate an impairment of function.
Infertility: A disease characterized by the failure to establish a clinical pregnancy after 12 months of regular, unprotected sexual intercourse or due to an impairment of a person’s capacity to reproduce either as an individual or with his/her partner. Fertility interventions may be initiated in less than 1 year based on medical, sexual and reproductive history, age, physical findings and diagnostic testing. Infertility is a disease, which generates disability as an impairment of function.
Reference
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406
New--and changed--definitions
Certain terms need to be consistent with those used currently internationally, for example, at which gestational age a miscarriage/abortion becomes a stillbirth.
Some terms are confusing, such as subfertility, which does not define a different or less severe fertility status than infertility, does not exist before infertility is diagnosed, and should not be confused with sterility, which is a permanent state of infertility. The term subfertility therefore is redundant and has been removed and replaced by infertility (See “Some terms with an important new definition” below).
- Clinical pregnancy
- Conception (removed from glossary)
- Diminished ovarian reserve
- Fertility care
- Hypospermia (replaces oligospermia)
- Ovarian reserve
- Pregnancy
- Preimplantation genetic testing
- Spontaneous abortion/miscarriage
- Subfertility (should be used interchangeably with infertility)
Reference
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406.
In a different context, the term conception, and its derivatives such as conceiving or conceived, was removed because it cannot be described biologically during the process of reproduction. Instead, terms such as fertilization, implantation, pregnancy, and live birth should be used.
Important male terms also changed: oligospermia is a term for low semen volume that is now replaced by hypospermia to avoid confusion with oligozoospermia, which is low concentration of spermatozoa in the ejaculate below the lower reference limit. When reporting results, the reference criteria should be specified.
Lastly, owing to the lack of standardization in determining the burden of infertility, and to better ensure comparability of prevalence data published globally, this glossary includes definitions for terms frequently used in epidemiology and public health. Examples include voluntary and involuntary childlessness, primary and secondary infertility, fertility care, fecundity, and fecundability, among others.
Getting the word out
The glossary has been approved by all of the participating organizations who are assisting in its distribution. It is being presented at national and international meetings and is used in The FIGO Fertility Toolbox (www.fertilitytool.com). It is hoped that all professionals and other stakeholders will begin to use its terminology globally to provide quality care and ensure consistency in registering specific fertility care interventions and more accurate reporting of their outcomes.
The language we use determines our individual and collective understanding of the scientific and clinical care of our patients. This glossary provides an essential and comprehensive standardization of terms and definitions essential to quality reproductive health care.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406.
- Johnson NP, Hummelshoj L; World Endometriosis Society Montpellier Consortium. Consensus on current management of endometriosis. Hum Reprod. 2013;28(6):1552–1568.
- Rogers PA, Adamson GD, Al-Jefout M, et al; WES/WERF Consortium for Research Priorities in Endometriosis. Research priorities for endometriosis. Reprod Sci. 2017;24(2):202–226.
- Johnson NP, Hummelshoj L, Adamson GD, et al; World Endometriosis Society Sao Paulo Consortium. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. 2017;32(2):315–324.
- Practice Committee of the American Society for Reproductive Medicine. Endometriosis and infertility: a committee opinion. Fertil Steril. 2012;98(3):591–598.
- Adamson GD, Pasta DJ. Endometriosis fertility index: the new, validated endometriosis staging system. Fertil Steril. 2010;94(5):1609–1615.
- Weir WC, Weir DR. Therapeutic value of salpingograms in infertility. Fertil Steril. 1951;2(6);514–522.
- Johnson NP, Farquhar CM, Hadden WE, Suckling J, Yu Y, Sadler L. The FLUSH trial—flushing with lipiodol for unexplained (and endometriosis-related) subfertility by hysterosalpingography: a randomized trial. Hum Reprod. 2004;19(9):2043–2051.
- Dreyer K, van Rijswijk J, Mijatovic V, et al. Oil-based or water-based contrast for hysterosalpingography in infertile women. N Engl J Med. 2017;376(21):2043–2052.
- Van der Steeg JW, Steures P, Eijkemans MJ, et al; Collaborative Effort for Clinical Evaluation in Reproductive Medicine. Pregnancy is predictable: a large-scale prospective external validation of the prediction of spontaneous pregnancy in sub-fertile couples. Hum Reprod. 2007;22(2):536–542.
- Zegers-Hochchild F, Adamson GD, Dyer S, et al. The International Glossary on Infertility and Fertility Care, 2017. Fertil Steril. 2017;108(3):393–406.
- Johnson NP, Hummelshoj L; World Endometriosis Society Montpellier Consortium. Consensus on current management of endometriosis. Hum Reprod. 2013;28(6):1552–1568.
- Rogers PA, Adamson GD, Al-Jefout M, et al; WES/WERF Consortium for Research Priorities in Endometriosis. Research priorities for endometriosis. Reprod Sci. 2017;24(2):202–226.
- Johnson NP, Hummelshoj L, Adamson GD, et al; World Endometriosis Society Sao Paulo Consortium. World Endometriosis Society consensus on the classification of endometriosis. Hum Reprod. 2017;32(2):315–324.
- Practice Committee of the American Society for Reproductive Medicine. Endometriosis and infertility: a committee opinion. Fertil Steril. 2012;98(3):591–598.
- Adamson GD, Pasta DJ. Endometriosis fertility index: the new, validated endometriosis staging system. Fertil Steril. 2010;94(5):1609–1615.
- Weir WC, Weir DR. Therapeutic value of salpingograms in infertility. Fertil Steril. 1951;2(6);514–522.
- Johnson NP, Farquhar CM, Hadden WE, Suckling J, Yu Y, Sadler L. The FLUSH trial—flushing with lipiodol for unexplained (and endometriosis-related) subfertility by hysterosalpingography: a randomized trial. Hum Reprod. 2004;19(9):2043–2051.
- Dreyer K, van Rijswijk J, Mijatovic V, et al. Oil-based or water-based contrast for hysterosalpingography in infertile women. N Engl J Med. 2017;376(21):2043–2052.
- Van der Steeg JW, Steures P, Eijkemans MJ, et al; Collaborative Effort for Clinical Evaluation in Reproductive Medicine. Pregnancy is predictable: a large-scale prospective external validation of the prediction of spontaneous pregnancy in sub-fertile couples. Hum Reprod. 2007;22(2):536–542.
3 cases of chronic pelvic pain managed with nonsurgical, nonopioid therapies
Chronic pelvic pain (CPP) is defined as noncyclic pain in the pelvis, anterior abdominal wall, back, or buttocks that has been present for at least 6 months and is severe enough to cause functional disability or require medical care.1 CPP is very common, with an estimated prevalence of 15% to 20%. It accounts for 20% of gynecology visits and 15% of hysterectomies in the United States, and it is believed to account for $2.8 billion in direct health care spending annually.2–5
Caring for patients with CPP can be very challenging. They often arrive at your office frustrated, having seen multiple providers or having undergone multiple surgeries. They may come to you whether you are a general ObGyn or subspecialize in maternal-fetal medicine, oncology, reproductive endocrinology, urogynecology, or adolescent gynecology. From interactions with other providers or their own family members, these patients may have received the message—either subtly or overtly—that their pain is “all in their head.” As such, some patients may resist any implication that their pain does not have an anatomic source. It is therefore critical to have appropriate tools for evaluating and managing the complex problem of CPP.
Perform a thorough and thoughtful assessment
Chronic pelvic pain often presents as a constellation of symptoms with contributions from multiple sources, as opposed to a single disease entity. Occasionally there is a single cause of pain, such as a large endometrioma or degenerating fibroid, where surgery can be curative. But more commonly the pain arises from multiple organ systems. In such cases, surgery may be unnecessary and, often, can worsen pain.
Thoughtful evaluation is critical in the CPP population. Take a thorough patient history to determine the characteristics of pain (cyclic or constant, widespread or localized), exacerbating factors, sleep disturbances, fatigue, and current coping strategies. Focus a comprehensive physical examination on identifying the maneuvers that reproduce the patient’s pain, and include an examination of the pelvic floor muscles.6 In most cases, pelvic ultrasonography provides adequate evaluation for anatomic sources of pain.
Chronic pain does not behave like acute injury or postsurgical pain. Continuous peripheral pain signals for a prolonged period can lead to changes in how the brain processes pain; specifically, the brain can begin to amplify pain signals. This “central pain amplification” is characterized clinically by widespread pain, fatigue, sleep disturbances, memory difficulties, and somatic symptoms. Central pain amplification occurs in many chronic pain conditions, including fibromyalgia, interstitial cystitis, irritable bowel syndrome, low back pain, chronic headaches, and temporomandibular joint disorder.7,8 Recent clinical and functional magnetic resonance imaging (MRI) studies demonstrate central pain amplification in many patients with CPP.9–12 Notably, these findings are independent of the presence or severity of endometriosis.
In this article we discuss many therapies that have not been specifically studied in patients with CPP, and treatment efficacy is extrapolated from other conditions with chronic pain amplification, such as fibromyalgia or interstitial cystitis. Additionally, many treatments for conditions associated with central pain amplification are used off-label, that is, the US Food and Drug Administration (FDA) has not approved the medication for treatment of these specific conditions. This should be disclosed to patients during counseling.
Discuss treatment expectations with patients
Educating patients regarding the pathophysiology of chronic pain and setting reasonable expectations is the cornerstone of providing patient-centered care for this complex condition. We start most of our discussions about treatment options by telling patients that while we may not cure their pain, we will provide them with medical, surgical, and behavioral strategies that will reduce their pain, improve their function, and enhance their quality of life.
Surprisingly, most patients say that a cure is not their goal. They just want to feel better so they can return to work or activities, fully participate in family life, or not feel exhausted all the time. As such, a multimodal treatment plan is generally the best strategy for achieving a satisfactory improvement in symptoms.
Read about treating a case of continued pain after endometriosis treatment.
CASE 1 Patient’s pain continues after endometriosis excision
A 32-year-old woman (G1P1) reports having CPP for 8 years. She underwent excision of stage 1 endometriosis last year, which resulted in a modest improvement in pain for 6 months. Her pain is worse during menses, at the end of the day, and with vaginal intercourse (both during and lasting for 1 to 2 days after). On examination, you find diffuse pelvic floor tenderness but no adnexal masses or rectovaginal nodularity on palpation.
What treatment options would you consider for this patient?
Multimodal treatment often needed to manage CPP symptoms
The patient described in Case 1 may benefit from a combination of therapies that include analgesics, hormone suppression agents, and physical therapy (PT) (TABLE).
Analgesics
Nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen and naproxen, work by inhibiting cyclooxygenase enzyme, which decreases assembly of peripheral prostaglandins and thromboxane. In a large Cochrane review, NSAIDs were associated with moderate or excellent pain relief for approximately 50% of patients with dysmenorrhea, and they have been shown to reduce menstrual flow due to decreased production of uterine prostaglandins.13 There is little evidence for use of NSAIDs in chronic pain conditions.
Acetaminophen’s mechanism of action is unclear, but the drug likely inhibits central prostaglandin synthesis, and it works synergistically with other analgesics.
Opioids act on μ and δ opioid receptors in the central and peripheral nervous systems as well as in the gastrointestinal system. No evidence supports opioid use in CPP or other chronic pain conditions. Long-term opioid use is associated with a multitude of adverse effects, risk for dependence, and the induction of opioid-induced hyperalgesia (in which patients develop greater sensitivity to pain stimuli).
Analgesics, specifically NSAIDs, can be considered for use in patients with dysmenorrhea, cyclic pain exacerbation, or a suspected inflammatory component of pain. Best practices include scheduling NSAID use before the onset of menses and continuing the drugs on a scheduled basis throughout. NSAIDs should be used for a brief period, and regular use on an empty stomach should be avoided.
Hormone suppression
Many types of hormone suppression therapy are available, including combined estrogen-progestin medications, progestin-only medications, and gonadotropin-releasing hormone (GnRH) agonists and antagonists.
Combined estrogen-progestin medications include oral contraceptive pills (OCPs), vaginal rings, and transdermal patches. Combined estrogen-progestin methods cause atrophy of eutopic and ectopic endometrium and suppress GnRH.
Progestin-only methods include oral formulations, the levonorgestrel intrauterine device, intramuscular and subcuticular injections, and subdermal implants. Progestin-only methods lead to atrophy of eutopic and ectopic endometrium.
A GnRH agonist, leuprolide depot works by downregulating luteinizing hormone and follicle stimulating hormone release from the pituitary, causing suppression of ovarian follicular development and ovulation, leading to a hypoestrogenic state.
Combined estrogen-progestin formulations and progestin-only options are often considered first-line therapy for dysmenorrhea and endometriosis.13 Continuous administration, with the goal of inducing amenorrhea, is effective in the treatment of dysmenorrhea. Several randomized controlled trials have shown that different types of hormone suppression agents are, essentially, equally effective.13–15 Treatment recommendations therefore should focus on adverse effects, cost, and patient preference. GnRH agonists and norethindrone are not FDA approved for the treatment of endometriosis.
It may be appropriate to consider use of hormone suppression therapy in patients with menstrual exacerbation of pain symptoms, including those with a history of endometriosis. We generally advise patients that the goal is amenorrhea and that achieving it often involves a process of trying different formulations to find the best fit. Remember that GnRH agonists are dependent on a functional hypothalamic-pituitary-ovarian axis, and they are unlikely to be effective in women with suspected residual endometriosis who have had a bilateral oophorectomy.
Physical therapy
For CPP, PT typically targets musculoskeletal dysfunction in the pelvic floor, abdominal wall, hips, and back. Interventions include muscle control, mobilization, and biofeedback. Pelvic PT has been shown to improve pain and dyspareunia in patients with CPP, coccydynia, and vestibulodynia.16–18 One large study found a significant, patient-directed decrease in pain medication use after pelvic floor PT.19 Pelvic PT for patients with interstitial cystitis and pelvic floor tenderness resulted in improved pain and bladder symptoms.20
Pelvic PT can be considered for patients with pain reproducible with palpation of the pelvic floor, abdominal wall, paraspinal-lumbar muscles, or sacroiliac joints. Best practices include referral to a therapist who has specialized training in CPP, including pelvic floor therapy. It is important to clearly list the indication for referral, as many of these therapists also treat stress urinary incontinence. The wrong exercises can result in increased hypercontractility of pelvic floor muscles, which can worsen pelvic pain.
It is also critical to clarify expectations with your patient at the time of PT referral. Specifically, advise patients that when beginning therapy, it is common to experience a temporary increase in discomfort of the pelvic muscles. Inform patients also to expect that their therapist will perform internal manipulation of the pelvic floor muscles through the vagina, as this can be surprising for some patients. Finally, counsel patients that their adherence to daily home exercises improves their chance of a durable, long-term successful response.21
CASE 1 Treatment recommendations
For treatment of this patient’s CPP, consider scheduled naproxen therapy during menses, continuous OCPs, and referral for pelvic floor PT.
Read about treating a case of pain, sleep disturbance, and depression.
CASE 2 Patient with long-standing CPP, multiple diagnoses, and sleep problems
A 30-year-old woman (G2P2) reports having had CPP for 17 years. She is amenorrheic with continuous OCP treatment. She had experienced some improvement with pelvic PT. The patient reports that she has daily pain with intermittent pain flares and that she is exhausted and has poor sleep quality, which she attributes to pain. She has been diagnosed with interstitial cystitis, irritable bowel syndrome, and temporomandibular joint disorder. She has a history of depression, which she feels is well controlled with bupropion. Physical examination reveals that the patient has diffuse but mild pain in the pelvic floor and abdominal wall muscles.
What further pain management options can you offer for this patient?
Managing pain, sleep disturbance, and depression
This patient has been living with CPP for many years, and she has sleep difficulties that might be exacerbating pain or result from pain (or both). She is already on continuous OCPs and has had some relief with pelvic PT. Other options that may help with her multiple issues include antidepressants, cyclobenzaprine, and calcium channel blockers.
Antidepressants
Several classes of antidepressants have been used in the treatment of chronic pain conditions, specifically, tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). Commonly used TCAs include amitriptyline, nortriptyline, desipramine, and doxepin. Commonly used SNRIs are duloxetine and milnacipran. Both TCAs and SNRIs increase the availability of norepinephrine and serotonin, which are thought to act on the descending pain inhibitory systems to decrease pain sensitivity. Of note, most selective serotonin reuptake inhibitors (SSRIs) at typical doses do not exert a significant enough impact on norepinephrine to be useful for chronic pain.22
Evidence is limited on the use of antidepressants for treating CPP. Amitriptyline is the most extensively studied antidepressant. Amitriptyline treatment resulted in modest pain improvement in patients with CPP and fibromyalgia.23,24 Bothersome anticholinergic effects, including fatigue, dry mouth, and constipation, often are reported with TCAs. Adverse effects tend to be less with nortriptyline or desipramine compared with amitriptyline, but possibly at the expense of efficacy.
While SNRIs have not yet been studied in CPP, several investigations have shown that they improve pain and quality of life in fibromyalgia patients.22,25
Antidepressant therapy may be appropriate for patients with suspected central pain amplification, widespread pain, and sleep disturbances. Best practices include patient education and careful discussion of this option with your patient. We suggest that clinicians explain that antidepressant medications alter the function of neurotransmitters, which modulate pain signals. While neurotransmitters also are involved in mood modulation, this is not the therapeutic goal in this circumstance. In addition, the doses used for the effective treatment of chronic pain are significantly lower than those needed to treat depression effectively.
Patients often need to hear that you believe that their pain is real and is not a manifestation of depression or another mood disorder. If you suspect that the patient also has untreated depression, address this as its own issue and use medications that have greater efficacy for mood symptoms.
Because many antidepressants can cause sedation, they are best taken before bedtime. Also, slow dose titration over several weeks will reduce the chance of bothersome adverse effects. Counsel patients that efficacy is not generally seen until at goal dose for several weeks. Be aware of interactions with other medications that can cause serotonin syndrome.
Cyclobenzaprine
Cyclobenzaprine is a muscle relaxant that also has activity in the central nervous system. The drug’s precise mechanism of action is not known, but it appears to potentiate norepinephrine and bind to serotonin receptors. Thus, it also likely has some TCA-like activity.
Cyclobenzaprine has not been studied in patients with CPP. In fibromyalgia patients, however, it produced significant improvements in pain, sleep, fatigue, and tenderness.26,27 In our anecdotal experience with CPP patients, cyclobenzaprine has been one of the most impactful therapies. It hits the “chronic pain triad,” meaning that it helps with myofascial pain, neuropathic pain, and sleep disturbances.
Cyclobenzaprine treatment may be considered for patients with myofascial pain, sleep disturbances, and clinical symptoms of central pain amplification. Best practices include starting with low (5 mg) scheduled doses at bedtime and slowly titrating the dose. Drowsiness is a very common side effect, so we try to use that to the patient’s advantage to help with sleep quality.
Notably, sleep disturbances are highly prevalent in patients with chronic pain.28 The relationship appears to be bidirectional, meaning that chronic pain negatively impacts sleep quality, and poor sleep quality causes amplified perception of pain.28–30 Interventions that improve sleep quality have been associated with improvements in pain, coping, mood, and functional status.31 Helping a patient to improve her sleep generally requires a multifaceted approach. It always involves “sleep hygiene” or a behavioral component, and pharmacologic assistance may be considered when improved sleep hygiene does not provide adequately improved sleep quality.
Calcium channel blockers
Gabapentin and pregabalin are calcium channel blockers that inhibit the reuptake of glutamate, norepinephrine, and substance P, which helps to decrease pain sensitivity. They also act as membrane stabilizers, reducing hyperexcitability of peripheral and central nerves. Studies have shown that in patients with CPP, gabapentin resulted in improved pain and mood symptoms with few adverse effects.23,32 Patients with fibromyalgia had improvements in pain, sleep, quality of life, fatigue, and anxiety with both gabapentin and pregabalin.33
It is appropriate to consider use of gabapentin or pregabalin in patients with central pain amplification and sleep disturbances. Best practices include starting with a low dose at bedtime. Traditionally, gabapentin is given in 3 equal doses throughout the day. In our experience, patients report less daytime drowsiness and better sleep quality if two-thirds of the daily dose is given at night, with the remaining daily dose broken up into 2 smaller daytime doses. Slow titration over several weeks will reduce risk of bothersome adverse effects. Patients should be counseled that efficacy is not generally seen until treatment is at goal dose for several weeks.
CASE 2 Treatment recommendations
For this patient with daily pelvic pain, multiple diagnoses that have a pain component, and poor sleep quality, consider a treatment plan that includes scheduled cyclobenzaprine, improved sleep hygiene, and, if needed, gabapentin.
Read about treating a case of focal pain.
CASE 3 Cesarean delivery, hysterectomy, and continued pelvic pain
A 38-year-old woman (G2P2) has had CPP for the past 10 years. She developed persistent left lower-quadrant pain after cesarean delivery of her son. She had a hysterectomy 2 years ago for CPP, after which her pain worsened. She describes daily pain with intermittent flares. On examination, the patient has focal left lower-quadrant pain lateral to the left apex of her Pfannenstiel incision.
What treatment approach would be appropriate for this patient?
Focal pain requires a precisely targeted treatment
This patient with focal left lower-quadrant pain is a candidate for anesthetic trigger point injections in the affected area near her Pfannenstiel incision.
Anesthetic injections
Consider the presence of trigger points and peripheral neuropathy in patients with focal abdominal wall pain. Trigger points are focal, palpable nodules within muscles. They are markedly painful to palpation and are associated with referred pain, motor dysfunction, and occasionally autonomic symptoms. They frequently are seen in abdominal wall or pelvic floor muscles in patients with CPP and are caused by abnormal neuromuscular depolarization.
The ilioinguinal, iliohypogastric, and genitofemoral nerves are in close proximity to Pfannenstiel and laparoscopic port site incisions. These nerves may be injured directly during surgery, but they also may be compressed by postoperative scarring.
Anesthetics, such as lidocaine and bupivacaine, which act as sodium channel blockers, can be injected into this area, and improvement often substantially outlasts the anesthetic’s duration of action. While these drugs’ mechanism of action is not clear, theories include altered function of sodium channels on sensory nerves with repeated anesthetic exposure, dry needling that occurs during injection, hydrodissection of tight connective tissue bands surrounding neuromuscular bundles, or depletion of substance P and neuropeptides as a result of injection.34,35
In several studies, patients with CPP reported decreased pain with lidocaine injections in pelvic floor or abdominal wall trigger points.36–38 Patients with fibromyalgia reported improvement in pain and a decreased need for NSAIDs with bupivacaine trigger point injections.39 While abdominal wall nerve blocks have not been extensively studied in patients with chronic neuropathic pain following gynecologic surgery, they have been shown to substantially improve chronic neuropathic pain following inguinal hernia repair.40
Anesthetic injections appropriately may be considered in patients with focal pain in a muscle or in the distribution of abdominal wall nerves, palpation of which reproduces pain symptoms. Patients with diffuse pain are less likely to benefit from anesthetic injections. Best practices include careful examination with attention to areas of prior abdominal incisions.
Our practice is to inject each affected area with a mix of 9 mL of 1% lidocaine and 1 mL of sodium bicarbonate. If a patient reports at least 24 hours of improvement, we repeat the injection in 2 to 4 weeks. The goal is for the patient to experience a progressively longer duration of benefit with subsequent injections. We perform repeat injections shortly after pain begins to recur at that site. The patient should eventually graduate from receiving regular injections and may return for a remedial injection if pain recurs.
CASE 3 Treatment recommendations
For this patient with persistent focal left-lower quadrant pain and a defined trigger point near her Pfannenstiel incision, consider anesthetic injection in the left lower quadrant.
Work toward realistic symptom improvement
Remember that living with chronic pain is exhausting, and empathy with a patient-centered approach is the most important ingredient for patient improvement and satisfaction. Discuss realistic expectations with patients. Remind them that there is no magic bullet for the complex problem of CPP, and that chronic conditions generally do not improve overnight. Focus on improving the patient’s function and quality of life, and applaud symptom improvement rather than focusing on complete pain resolution.
As these visits often require a good deal of patient education, budget more appointment time if feasible. We find that scheduling frequent return visits (approximately every 3 to 4 months) allows timely treatment follow-up so that changes may be made if needed. If you have maximized your available treatment options, referring the patient to a specialist with additional training in CPP is a sensible next step.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Howard FM. Chronic pelvic pain. Obstet Gynecol. 2003;101(3):594–611.
- Mathias SD, Kuppermann M, Liberman RF, Lipschutz RC, Steege JF. Chronic pelvic pain: prevalence, health-related uality of life, and economic correlates. Obstet Gynecol. 1996;87(3):321–327.
- Gelbaya TA, El-Halwagy HE. Focus on primary care: chronic pelvic pain in women. Obstet Gynecol Surv. 2001;56(12):757–764.
- Broder MS, Kanouse DE, Mittman BS, Bernstein SJ. The appropriateness of recommendations for hysterectomy. Obstet Gynecol. 2000;95(2):199–205.
- Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am J Obstet Gynecol. 2008;198(1):34.e1–34.e7.
- Steege JF, Siedhoff MT. Chronic pelvic pain. Obstet Gynecol. 2014;124(3):616–629.
- Williams DA, Clauw DJ. Understanding fibromyalgia: lessons from the broader pain research community. J Pain. 2009;10(8):777–791.
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 suppl):S2–S15.
- Brawn J, Morotti M, Zondervan KT, Becker CM, Vincent K. Central changes associated with chronic pelvic pain and endometriosis. Hum Reprod Update. 2014;20(5):737–747.
- As-Sanie S, Harris RE, Harte SE, Tu FF, Neshewat G, Clauw DJ. Increased pressure pain sensitivity in women with chronic pelvic pain. Obstet Gynecol. 2013;122(5):1047–1055.
- As-Sanie S, Kim J, Schmidt-Wilcke T, et al. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J Pain. 2016;17(1):1–13.
- As-Sanie S, Harris RE, Napadow V, et al. Changes in regional gray matter volume in women with chronic pelvic pain: a voxel-based morphometry study. Pain. 2012;153(5):1006–1014.
- Marjoribanks J, Ayeleke RO, Farquhar C, Proctor M. Nonsteroidal anti-inflammatory drugs for dysmenorrhoea. Cochrane Database Syst Rev. 2015;(7):CD001751.
- Falcone T, Lebovic DI. Clinical management of endometriosis. Obstet Gynecol. 2011;118(3):691–705.
- Brown J, Pan A, Hart RJ. Gonadotrophin-releasing hormone analogues for pain associated with endometriosis. Cochrane Database Syst Rev. 2010;(12):CD008475.
- Zoorob D, South M, Karram M, et al. A pilot randomized trial of levator injections versus physical therapy for treatment of pelvic floor myalgia and sexual pain. Int Urogynecol J. 2015;26(6):845–852.
- Scott KM, Fisher LW, Bernstein IH, Bradley MH. The treatment of chronic coccydynia and postcoccygectomy pain with pelvic floor physical therapy. PM R. 2017;9(4):367–376.
- Goldfinger C, Pukall CF, Thibault-Gagnon S, McLean L, Chamberlain S. Effectiveness of cognitive-behavioral therapy and physical therapy for provoked vestibulodynia: a randomized pilot study. J Sex Med. 2016;13(1):88–94.
- Anderson RU, Harvey RH, Wise D, Nevin Smith J, Nathanson BH, Sawyer T. Chronic pelvic pain syndrome: reduction of medication use after pelvic floor physical therapy with an internal myofascial trigger point wand. Appl Psychophysiol Biofeedback. 2015;40(1):45–52.
- FitzGerald MP, Payne CK, Lukacz ES, et al. Randomized multicenter clinical trial of myofascial physical therapy in women with interstitial cystitis/painful bladder syndrome and pelvic floor tenderness. J Urol. 2012;187(6):2113–2118.
- FitzGerald MP, Kotarinos R. Rehabilitation of the short pelvic floor. II: Treatment of the patient with the short pelvic floor. Int Urogynecol J Pelvic Floor Dysfunct. 2003;14(4):269–275.
- Arnold LM. Duloxetine and other antidepressants in the treatment of patients with fibromyalgia. Pain Med. 2007;8(suppl 2):S63–S74.
- Sator-Katzenschlager SM, Scharbert G, Kress HG, et al. Chronic pelvic pain treated with gabapentin and amitriptyline: a randomized controlled pilot study. Wien Klin Wochenschr. 2005;117(21–22):761–78.
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;12:CD008242.
- Gendreau RM, Thorn MD, Gendreau JF, et al. Efficacy of milnacipran in patients with fibromyalgia. J Rheumatol. 2005;32(10):1975–1985.
- Tofferi JK, Jackson JL, O’Malley PG. Treatment of fibromyalgia with cyclobenzaprine: a meta-analysis. Arthritis Rheum. 2004;51(1):9–13.
- Moldofsky H, Harris HW, Archambault WT, Kwong T, Lederman S. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: a double-blind randomized placebo-controlled study. J Rheumatol. 2011;38(12):2653–2463.
- Cheatle MD, Foster S, Pinkett A, Lesneski M, Qu D, Dhingra L. Assessing and managing sleep disturbance in patients with chronic pain. Sleep Med Clin. 2016;11(4):531–541.
- Larson RA, Carter JR. Total sleep deprivation and pain perception during cold noxious stimuli in humans. Scand J Pain. 2016;13:12–16.
- Generaal E, Vogelzangs N, Penninx BW, Dekker J. Insomnia, sleep duration, depressive symptoms, and the onset of chronic multisite musculoskeletal pain [published online ahead of print January 1, 2017]. Sleep. doi:10.1093/sleep/zsw030.
- Gerhart JI, Burns JW, Post KM, et al. Relationships between sleep quality and pain-related factors for people with chronic low back pain: tests of reciprocal and time of day effects. Ann Behav Med. 2017;51(3):365–375.
- Lewis SC, Bhattacharya S, Wu O, et al. Gabapentin for the management of chronic pelvic pain in women (GaPP1): a pilot randomised controlled trial. PLoS One. 2016;11(4):e0153037.
- Häuser W, Bernardy K, Uçeyler N, Sommer C. Treatment of fibromyalgia syndrome with gabapentin and pregabalin—a meta-analysis of randomized controlled trials. Pain. 2009;145(1–2):69–81.
- Scott NA, Guo B, Barton PM, Gerwin RD. Trigger point injections for chronic non-malignant musculoskeletal pain: a systematic review. Pain Med. 2009;10(1):54–69.
- Hameroff SR, Crago BR, Blitt CD, Womble J, Kanel J. Comparison of bupivacaine, etidocaine, and saline for trigger-point therapy. Anesth Analg. 1981;60(10):752–755.
- Montenegro ML, Braz CA, Rosa-e-Silva JC, Candido-dos-Reis FJ, Nogueira AA, Poli-Neto OB. Anaesthetic injection versus ischemic compression for the pain relief of abdominal wall trigger points in women with chronic pelvic pain. BMC Anesthesiol. 2015;15:175.
- Kim DS, Jeong TY, Kim YK, Chang WH, Yoon JG, Lee SC. Usefulness of a myofascial trigger point injection for groin pain in patients with chronic prostatitis/chronic pelvic pain syndrome: a pilot study. Arch Phys Med Rehabil. 2013;94(5):930–936.
- Huang QM, Liu L. Wet needling of myofascial trigger points in abdominal muscles for treatment of primary dysmenorrhoea. Acupunct Med. 2014;32(4):346–349.
- Affaitati G, Fabrizio A, Savini A, et al. A randomized, controlled study comparing a lidocaine patch, a placebo patch, and anesthetic injection for treatment of trigger points in patients with myofascial pain syndrome: evaluation of pain and somatic pain thresholds. Clin Ther. 2009;31(4):705–720.
- Thomassen I, van Suijlekom JA, van de Gaag A, Ponten JE, Nienhuijs SW. Ultrasound-guided ilioinguinal/iliohypogastric nerve blocks for chronic pain after inguinal hernia repair. Hernia. 2013;17(3):329–332.

Dr. Till is Assistant Professor, Minimally Invasive Gynecologic Surgery, Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor.

Dr. As-Sanie is Associate Professor and Director, Minimally Invasive Gynecologic Surgery, Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor.
Dr. Till reports no financial relationships relevant to this article. Dr. As-Sanie reports that she is a consultant to AbbVie.
Dr. Till is Assistant Professor, Minimally Invasive Gynecologic Surgery, Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor.
Dr. As-Sanie is Associate Professor and Director, Minimally Invasive Gynecologic Surgery, Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor.
Dr. Till reports no financial relationships relevant to this article. Dr. As-Sanie reports that she is a consultant to AbbVie.
Dr. Till is Assistant Professor, Minimally Invasive Gynecologic Surgery, Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor.
Dr. As-Sanie is Associate Professor and Director, Minimally Invasive Gynecologic Surgery, Department of Obstetrics and Gynecology, University of Michigan, Ann Arbor.
Dr. Till reports no financial relationships relevant to this article. Dr. As-Sanie reports that she is a consultant to AbbVie.
Chronic pelvic pain (CPP) is defined as noncyclic pain in the pelvis, anterior abdominal wall, back, or buttocks that has been present for at least 6 months and is severe enough to cause functional disability or require medical care.1 CPP is very common, with an estimated prevalence of 15% to 20%. It accounts for 20% of gynecology visits and 15% of hysterectomies in the United States, and it is believed to account for $2.8 billion in direct health care spending annually.2–5
Caring for patients with CPP can be very challenging. They often arrive at your office frustrated, having seen multiple providers or having undergone multiple surgeries. They may come to you whether you are a general ObGyn or subspecialize in maternal-fetal medicine, oncology, reproductive endocrinology, urogynecology, or adolescent gynecology. From interactions with other providers or their own family members, these patients may have received the message—either subtly or overtly—that their pain is “all in their head.” As such, some patients may resist any implication that their pain does not have an anatomic source. It is therefore critical to have appropriate tools for evaluating and managing the complex problem of CPP.
Perform a thorough and thoughtful assessment
Chronic pelvic pain often presents as a constellation of symptoms with contributions from multiple sources, as opposed to a single disease entity. Occasionally there is a single cause of pain, such as a large endometrioma or degenerating fibroid, where surgery can be curative. But more commonly the pain arises from multiple organ systems. In such cases, surgery may be unnecessary and, often, can worsen pain.
Thoughtful evaluation is critical in the CPP population. Take a thorough patient history to determine the characteristics of pain (cyclic or constant, widespread or localized), exacerbating factors, sleep disturbances, fatigue, and current coping strategies. Focus a comprehensive physical examination on identifying the maneuvers that reproduce the patient’s pain, and include an examination of the pelvic floor muscles.6 In most cases, pelvic ultrasonography provides adequate evaluation for anatomic sources of pain.
Chronic pain does not behave like acute injury or postsurgical pain. Continuous peripheral pain signals for a prolonged period can lead to changes in how the brain processes pain; specifically, the brain can begin to amplify pain signals. This “central pain amplification” is characterized clinically by widespread pain, fatigue, sleep disturbances, memory difficulties, and somatic symptoms. Central pain amplification occurs in many chronic pain conditions, including fibromyalgia, interstitial cystitis, irritable bowel syndrome, low back pain, chronic headaches, and temporomandibular joint disorder.7,8 Recent clinical and functional magnetic resonance imaging (MRI) studies demonstrate central pain amplification in many patients with CPP.9–12 Notably, these findings are independent of the presence or severity of endometriosis.
In this article we discuss many therapies that have not been specifically studied in patients with CPP, and treatment efficacy is extrapolated from other conditions with chronic pain amplification, such as fibromyalgia or interstitial cystitis. Additionally, many treatments for conditions associated with central pain amplification are used off-label, that is, the US Food and Drug Administration (FDA) has not approved the medication for treatment of these specific conditions. This should be disclosed to patients during counseling.
Discuss treatment expectations with patients
Educating patients regarding the pathophysiology of chronic pain and setting reasonable expectations is the cornerstone of providing patient-centered care for this complex condition. We start most of our discussions about treatment options by telling patients that while we may not cure their pain, we will provide them with medical, surgical, and behavioral strategies that will reduce their pain, improve their function, and enhance their quality of life.
Surprisingly, most patients say that a cure is not their goal. They just want to feel better so they can return to work or activities, fully participate in family life, or not feel exhausted all the time. As such, a multimodal treatment plan is generally the best strategy for achieving a satisfactory improvement in symptoms.
Read about treating a case of continued pain after endometriosis treatment.
CASE 1 Patient’s pain continues after endometriosis excision
A 32-year-old woman (G1P1) reports having CPP for 8 years. She underwent excision of stage 1 endometriosis last year, which resulted in a modest improvement in pain for 6 months. Her pain is worse during menses, at the end of the day, and with vaginal intercourse (both during and lasting for 1 to 2 days after). On examination, you find diffuse pelvic floor tenderness but no adnexal masses or rectovaginal nodularity on palpation.
What treatment options would you consider for this patient?
Multimodal treatment often needed to manage CPP symptoms
The patient described in Case 1 may benefit from a combination of therapies that include analgesics, hormone suppression agents, and physical therapy (PT) (TABLE).
Analgesics
Nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen and naproxen, work by inhibiting cyclooxygenase enzyme, which decreases assembly of peripheral prostaglandins and thromboxane. In a large Cochrane review, NSAIDs were associated with moderate or excellent pain relief for approximately 50% of patients with dysmenorrhea, and they have been shown to reduce menstrual flow due to decreased production of uterine prostaglandins.13 There is little evidence for use of NSAIDs in chronic pain conditions.
Acetaminophen’s mechanism of action is unclear, but the drug likely inhibits central prostaglandin synthesis, and it works synergistically with other analgesics.
Opioids act on μ and δ opioid receptors in the central and peripheral nervous systems as well as in the gastrointestinal system. No evidence supports opioid use in CPP or other chronic pain conditions. Long-term opioid use is associated with a multitude of adverse effects, risk for dependence, and the induction of opioid-induced hyperalgesia (in which patients develop greater sensitivity to pain stimuli).
Analgesics, specifically NSAIDs, can be considered for use in patients with dysmenorrhea, cyclic pain exacerbation, or a suspected inflammatory component of pain. Best practices include scheduling NSAID use before the onset of menses and continuing the drugs on a scheduled basis throughout. NSAIDs should be used for a brief period, and regular use on an empty stomach should be avoided.
Hormone suppression
Many types of hormone suppression therapy are available, including combined estrogen-progestin medications, progestin-only medications, and gonadotropin-releasing hormone (GnRH) agonists and antagonists.
Combined estrogen-progestin medications include oral contraceptive pills (OCPs), vaginal rings, and transdermal patches. Combined estrogen-progestin methods cause atrophy of eutopic and ectopic endometrium and suppress GnRH.
Progestin-only methods include oral formulations, the levonorgestrel intrauterine device, intramuscular and subcuticular injections, and subdermal implants. Progestin-only methods lead to atrophy of eutopic and ectopic endometrium.
A GnRH agonist, leuprolide depot works by downregulating luteinizing hormone and follicle stimulating hormone release from the pituitary, causing suppression of ovarian follicular development and ovulation, leading to a hypoestrogenic state.
Combined estrogen-progestin formulations and progestin-only options are often considered first-line therapy for dysmenorrhea and endometriosis.13 Continuous administration, with the goal of inducing amenorrhea, is effective in the treatment of dysmenorrhea. Several randomized controlled trials have shown that different types of hormone suppression agents are, essentially, equally effective.13–15 Treatment recommendations therefore should focus on adverse effects, cost, and patient preference. GnRH agonists and norethindrone are not FDA approved for the treatment of endometriosis.
It may be appropriate to consider use of hormone suppression therapy in patients with menstrual exacerbation of pain symptoms, including those with a history of endometriosis. We generally advise patients that the goal is amenorrhea and that achieving it often involves a process of trying different formulations to find the best fit. Remember that GnRH agonists are dependent on a functional hypothalamic-pituitary-ovarian axis, and they are unlikely to be effective in women with suspected residual endometriosis who have had a bilateral oophorectomy.
Physical therapy
For CPP, PT typically targets musculoskeletal dysfunction in the pelvic floor, abdominal wall, hips, and back. Interventions include muscle control, mobilization, and biofeedback. Pelvic PT has been shown to improve pain and dyspareunia in patients with CPP, coccydynia, and vestibulodynia.16–18 One large study found a significant, patient-directed decrease in pain medication use after pelvic floor PT.19 Pelvic PT for patients with interstitial cystitis and pelvic floor tenderness resulted in improved pain and bladder symptoms.20
Pelvic PT can be considered for patients with pain reproducible with palpation of the pelvic floor, abdominal wall, paraspinal-lumbar muscles, or sacroiliac joints. Best practices include referral to a therapist who has specialized training in CPP, including pelvic floor therapy. It is important to clearly list the indication for referral, as many of these therapists also treat stress urinary incontinence. The wrong exercises can result in increased hypercontractility of pelvic floor muscles, which can worsen pelvic pain.
It is also critical to clarify expectations with your patient at the time of PT referral. Specifically, advise patients that when beginning therapy, it is common to experience a temporary increase in discomfort of the pelvic muscles. Inform patients also to expect that their therapist will perform internal manipulation of the pelvic floor muscles through the vagina, as this can be surprising for some patients. Finally, counsel patients that their adherence to daily home exercises improves their chance of a durable, long-term successful response.21
CASE 1 Treatment recommendations
For treatment of this patient’s CPP, consider scheduled naproxen therapy during menses, continuous OCPs, and referral for pelvic floor PT.
Read about treating a case of pain, sleep disturbance, and depression.
CASE 2 Patient with long-standing CPP, multiple diagnoses, and sleep problems
A 30-year-old woman (G2P2) reports having had CPP for 17 years. She is amenorrheic with continuous OCP treatment. She had experienced some improvement with pelvic PT. The patient reports that she has daily pain with intermittent pain flares and that she is exhausted and has poor sleep quality, which she attributes to pain. She has been diagnosed with interstitial cystitis, irritable bowel syndrome, and temporomandibular joint disorder. She has a history of depression, which she feels is well controlled with bupropion. Physical examination reveals that the patient has diffuse but mild pain in the pelvic floor and abdominal wall muscles.
What further pain management options can you offer for this patient?
Managing pain, sleep disturbance, and depression
This patient has been living with CPP for many years, and she has sleep difficulties that might be exacerbating pain or result from pain (or both). She is already on continuous OCPs and has had some relief with pelvic PT. Other options that may help with her multiple issues include antidepressants, cyclobenzaprine, and calcium channel blockers.
Antidepressants
Several classes of antidepressants have been used in the treatment of chronic pain conditions, specifically, tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). Commonly used TCAs include amitriptyline, nortriptyline, desipramine, and doxepin. Commonly used SNRIs are duloxetine and milnacipran. Both TCAs and SNRIs increase the availability of norepinephrine and serotonin, which are thought to act on the descending pain inhibitory systems to decrease pain sensitivity. Of note, most selective serotonin reuptake inhibitors (SSRIs) at typical doses do not exert a significant enough impact on norepinephrine to be useful for chronic pain.22
Evidence is limited on the use of antidepressants for treating CPP. Amitriptyline is the most extensively studied antidepressant. Amitriptyline treatment resulted in modest pain improvement in patients with CPP and fibromyalgia.23,24 Bothersome anticholinergic effects, including fatigue, dry mouth, and constipation, often are reported with TCAs. Adverse effects tend to be less with nortriptyline or desipramine compared with amitriptyline, but possibly at the expense of efficacy.
While SNRIs have not yet been studied in CPP, several investigations have shown that they improve pain and quality of life in fibromyalgia patients.22,25
Antidepressant therapy may be appropriate for patients with suspected central pain amplification, widespread pain, and sleep disturbances. Best practices include patient education and careful discussion of this option with your patient. We suggest that clinicians explain that antidepressant medications alter the function of neurotransmitters, which modulate pain signals. While neurotransmitters also are involved in mood modulation, this is not the therapeutic goal in this circumstance. In addition, the doses used for the effective treatment of chronic pain are significantly lower than those needed to treat depression effectively.
Patients often need to hear that you believe that their pain is real and is not a manifestation of depression or another mood disorder. If you suspect that the patient also has untreated depression, address this as its own issue and use medications that have greater efficacy for mood symptoms.
Because many antidepressants can cause sedation, they are best taken before bedtime. Also, slow dose titration over several weeks will reduce the chance of bothersome adverse effects. Counsel patients that efficacy is not generally seen until at goal dose for several weeks. Be aware of interactions with other medications that can cause serotonin syndrome.
Cyclobenzaprine
Cyclobenzaprine is a muscle relaxant that also has activity in the central nervous system. The drug’s precise mechanism of action is not known, but it appears to potentiate norepinephrine and bind to serotonin receptors. Thus, it also likely has some TCA-like activity.
Cyclobenzaprine has not been studied in patients with CPP. In fibromyalgia patients, however, it produced significant improvements in pain, sleep, fatigue, and tenderness.26,27 In our anecdotal experience with CPP patients, cyclobenzaprine has been one of the most impactful therapies. It hits the “chronic pain triad,” meaning that it helps with myofascial pain, neuropathic pain, and sleep disturbances.
Cyclobenzaprine treatment may be considered for patients with myofascial pain, sleep disturbances, and clinical symptoms of central pain amplification. Best practices include starting with low (5 mg) scheduled doses at bedtime and slowly titrating the dose. Drowsiness is a very common side effect, so we try to use that to the patient’s advantage to help with sleep quality.
Notably, sleep disturbances are highly prevalent in patients with chronic pain.28 The relationship appears to be bidirectional, meaning that chronic pain negatively impacts sleep quality, and poor sleep quality causes amplified perception of pain.28–30 Interventions that improve sleep quality have been associated with improvements in pain, coping, mood, and functional status.31 Helping a patient to improve her sleep generally requires a multifaceted approach. It always involves “sleep hygiene” or a behavioral component, and pharmacologic assistance may be considered when improved sleep hygiene does not provide adequately improved sleep quality.
Calcium channel blockers
Gabapentin and pregabalin are calcium channel blockers that inhibit the reuptake of glutamate, norepinephrine, and substance P, which helps to decrease pain sensitivity. They also act as membrane stabilizers, reducing hyperexcitability of peripheral and central nerves. Studies have shown that in patients with CPP, gabapentin resulted in improved pain and mood symptoms with few adverse effects.23,32 Patients with fibromyalgia had improvements in pain, sleep, quality of life, fatigue, and anxiety with both gabapentin and pregabalin.33
It is appropriate to consider use of gabapentin or pregabalin in patients with central pain amplification and sleep disturbances. Best practices include starting with a low dose at bedtime. Traditionally, gabapentin is given in 3 equal doses throughout the day. In our experience, patients report less daytime drowsiness and better sleep quality if two-thirds of the daily dose is given at night, with the remaining daily dose broken up into 2 smaller daytime doses. Slow titration over several weeks will reduce risk of bothersome adverse effects. Patients should be counseled that efficacy is not generally seen until treatment is at goal dose for several weeks.
CASE 2 Treatment recommendations
For this patient with daily pelvic pain, multiple diagnoses that have a pain component, and poor sleep quality, consider a treatment plan that includes scheduled cyclobenzaprine, improved sleep hygiene, and, if needed, gabapentin.
Read about treating a case of focal pain.
CASE 3 Cesarean delivery, hysterectomy, and continued pelvic pain
A 38-year-old woman (G2P2) has had CPP for the past 10 years. She developed persistent left lower-quadrant pain after cesarean delivery of her son. She had a hysterectomy 2 years ago for CPP, after which her pain worsened. She describes daily pain with intermittent flares. On examination, the patient has focal left lower-quadrant pain lateral to the left apex of her Pfannenstiel incision.
What treatment approach would be appropriate for this patient?
Focal pain requires a precisely targeted treatment
This patient with focal left lower-quadrant pain is a candidate for anesthetic trigger point injections in the affected area near her Pfannenstiel incision.
Anesthetic injections
Consider the presence of trigger points and peripheral neuropathy in patients with focal abdominal wall pain. Trigger points are focal, palpable nodules within muscles. They are markedly painful to palpation and are associated with referred pain, motor dysfunction, and occasionally autonomic symptoms. They frequently are seen in abdominal wall or pelvic floor muscles in patients with CPP and are caused by abnormal neuromuscular depolarization.
The ilioinguinal, iliohypogastric, and genitofemoral nerves are in close proximity to Pfannenstiel and laparoscopic port site incisions. These nerves may be injured directly during surgery, but they also may be compressed by postoperative scarring.
Anesthetics, such as lidocaine and bupivacaine, which act as sodium channel blockers, can be injected into this area, and improvement often substantially outlasts the anesthetic’s duration of action. While these drugs’ mechanism of action is not clear, theories include altered function of sodium channels on sensory nerves with repeated anesthetic exposure, dry needling that occurs during injection, hydrodissection of tight connective tissue bands surrounding neuromuscular bundles, or depletion of substance P and neuropeptides as a result of injection.34,35
In several studies, patients with CPP reported decreased pain with lidocaine injections in pelvic floor or abdominal wall trigger points.36–38 Patients with fibromyalgia reported improvement in pain and a decreased need for NSAIDs with bupivacaine trigger point injections.39 While abdominal wall nerve blocks have not been extensively studied in patients with chronic neuropathic pain following gynecologic surgery, they have been shown to substantially improve chronic neuropathic pain following inguinal hernia repair.40
Anesthetic injections appropriately may be considered in patients with focal pain in a muscle or in the distribution of abdominal wall nerves, palpation of which reproduces pain symptoms. Patients with diffuse pain are less likely to benefit from anesthetic injections. Best practices include careful examination with attention to areas of prior abdominal incisions.
Our practice is to inject each affected area with a mix of 9 mL of 1% lidocaine and 1 mL of sodium bicarbonate. If a patient reports at least 24 hours of improvement, we repeat the injection in 2 to 4 weeks. The goal is for the patient to experience a progressively longer duration of benefit with subsequent injections. We perform repeat injections shortly after pain begins to recur at that site. The patient should eventually graduate from receiving regular injections and may return for a remedial injection if pain recurs.
CASE 3 Treatment recommendations
For this patient with persistent focal left-lower quadrant pain and a defined trigger point near her Pfannenstiel incision, consider anesthetic injection in the left lower quadrant.
Work toward realistic symptom improvement
Remember that living with chronic pain is exhausting, and empathy with a patient-centered approach is the most important ingredient for patient improvement and satisfaction. Discuss realistic expectations with patients. Remind them that there is no magic bullet for the complex problem of CPP, and that chronic conditions generally do not improve overnight. Focus on improving the patient’s function and quality of life, and applaud symptom improvement rather than focusing on complete pain resolution.
As these visits often require a good deal of patient education, budget more appointment time if feasible. We find that scheduling frequent return visits (approximately every 3 to 4 months) allows timely treatment follow-up so that changes may be made if needed. If you have maximized your available treatment options, referring the patient to a specialist with additional training in CPP is a sensible next step.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Chronic pelvic pain (CPP) is defined as noncyclic pain in the pelvis, anterior abdominal wall, back, or buttocks that has been present for at least 6 months and is severe enough to cause functional disability or require medical care.1 CPP is very common, with an estimated prevalence of 15% to 20%. It accounts for 20% of gynecology visits and 15% of hysterectomies in the United States, and it is believed to account for $2.8 billion in direct health care spending annually.2–5
Caring for patients with CPP can be very challenging. They often arrive at your office frustrated, having seen multiple providers or having undergone multiple surgeries. They may come to you whether you are a general ObGyn or subspecialize in maternal-fetal medicine, oncology, reproductive endocrinology, urogynecology, or adolescent gynecology. From interactions with other providers or their own family members, these patients may have received the message—either subtly or overtly—that their pain is “all in their head.” As such, some patients may resist any implication that their pain does not have an anatomic source. It is therefore critical to have appropriate tools for evaluating and managing the complex problem of CPP.
Perform a thorough and thoughtful assessment
Chronic pelvic pain often presents as a constellation of symptoms with contributions from multiple sources, as opposed to a single disease entity. Occasionally there is a single cause of pain, such as a large endometrioma or degenerating fibroid, where surgery can be curative. But more commonly the pain arises from multiple organ systems. In such cases, surgery may be unnecessary and, often, can worsen pain.
Thoughtful evaluation is critical in the CPP population. Take a thorough patient history to determine the characteristics of pain (cyclic or constant, widespread or localized), exacerbating factors, sleep disturbances, fatigue, and current coping strategies. Focus a comprehensive physical examination on identifying the maneuvers that reproduce the patient’s pain, and include an examination of the pelvic floor muscles.6 In most cases, pelvic ultrasonography provides adequate evaluation for anatomic sources of pain.
Chronic pain does not behave like acute injury or postsurgical pain. Continuous peripheral pain signals for a prolonged period can lead to changes in how the brain processes pain; specifically, the brain can begin to amplify pain signals. This “central pain amplification” is characterized clinically by widespread pain, fatigue, sleep disturbances, memory difficulties, and somatic symptoms. Central pain amplification occurs in many chronic pain conditions, including fibromyalgia, interstitial cystitis, irritable bowel syndrome, low back pain, chronic headaches, and temporomandibular joint disorder.7,8 Recent clinical and functional magnetic resonance imaging (MRI) studies demonstrate central pain amplification in many patients with CPP.9–12 Notably, these findings are independent of the presence or severity of endometriosis.
In this article we discuss many therapies that have not been specifically studied in patients with CPP, and treatment efficacy is extrapolated from other conditions with chronic pain amplification, such as fibromyalgia or interstitial cystitis. Additionally, many treatments for conditions associated with central pain amplification are used off-label, that is, the US Food and Drug Administration (FDA) has not approved the medication for treatment of these specific conditions. This should be disclosed to patients during counseling.
Discuss treatment expectations with patients
Educating patients regarding the pathophysiology of chronic pain and setting reasonable expectations is the cornerstone of providing patient-centered care for this complex condition. We start most of our discussions about treatment options by telling patients that while we may not cure their pain, we will provide them with medical, surgical, and behavioral strategies that will reduce their pain, improve their function, and enhance their quality of life.
Surprisingly, most patients say that a cure is not their goal. They just want to feel better so they can return to work or activities, fully participate in family life, or not feel exhausted all the time. As such, a multimodal treatment plan is generally the best strategy for achieving a satisfactory improvement in symptoms.
Read about treating a case of continued pain after endometriosis treatment.
CASE 1 Patient’s pain continues after endometriosis excision
A 32-year-old woman (G1P1) reports having CPP for 8 years. She underwent excision of stage 1 endometriosis last year, which resulted in a modest improvement in pain for 6 months. Her pain is worse during menses, at the end of the day, and with vaginal intercourse (both during and lasting for 1 to 2 days after). On examination, you find diffuse pelvic floor tenderness but no adnexal masses or rectovaginal nodularity on palpation.
What treatment options would you consider for this patient?
Multimodal treatment often needed to manage CPP symptoms
The patient described in Case 1 may benefit from a combination of therapies that include analgesics, hormone suppression agents, and physical therapy (PT) (TABLE).
Analgesics
Nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen and naproxen, work by inhibiting cyclooxygenase enzyme, which decreases assembly of peripheral prostaglandins and thromboxane. In a large Cochrane review, NSAIDs were associated with moderate or excellent pain relief for approximately 50% of patients with dysmenorrhea, and they have been shown to reduce menstrual flow due to decreased production of uterine prostaglandins.13 There is little evidence for use of NSAIDs in chronic pain conditions.
Acetaminophen’s mechanism of action is unclear, but the drug likely inhibits central prostaglandin synthesis, and it works synergistically with other analgesics.
Opioids act on μ and δ opioid receptors in the central and peripheral nervous systems as well as in the gastrointestinal system. No evidence supports opioid use in CPP or other chronic pain conditions. Long-term opioid use is associated with a multitude of adverse effects, risk for dependence, and the induction of opioid-induced hyperalgesia (in which patients develop greater sensitivity to pain stimuli).
Analgesics, specifically NSAIDs, can be considered for use in patients with dysmenorrhea, cyclic pain exacerbation, or a suspected inflammatory component of pain. Best practices include scheduling NSAID use before the onset of menses and continuing the drugs on a scheduled basis throughout. NSAIDs should be used for a brief period, and regular use on an empty stomach should be avoided.
Hormone suppression
Many types of hormone suppression therapy are available, including combined estrogen-progestin medications, progestin-only medications, and gonadotropin-releasing hormone (GnRH) agonists and antagonists.
Combined estrogen-progestin medications include oral contraceptive pills (OCPs), vaginal rings, and transdermal patches. Combined estrogen-progestin methods cause atrophy of eutopic and ectopic endometrium and suppress GnRH.
Progestin-only methods include oral formulations, the levonorgestrel intrauterine device, intramuscular and subcuticular injections, and subdermal implants. Progestin-only methods lead to atrophy of eutopic and ectopic endometrium.
A GnRH agonist, leuprolide depot works by downregulating luteinizing hormone and follicle stimulating hormone release from the pituitary, causing suppression of ovarian follicular development and ovulation, leading to a hypoestrogenic state.
Combined estrogen-progestin formulations and progestin-only options are often considered first-line therapy for dysmenorrhea and endometriosis.13 Continuous administration, with the goal of inducing amenorrhea, is effective in the treatment of dysmenorrhea. Several randomized controlled trials have shown that different types of hormone suppression agents are, essentially, equally effective.13–15 Treatment recommendations therefore should focus on adverse effects, cost, and patient preference. GnRH agonists and norethindrone are not FDA approved for the treatment of endometriosis.
It may be appropriate to consider use of hormone suppression therapy in patients with menstrual exacerbation of pain symptoms, including those with a history of endometriosis. We generally advise patients that the goal is amenorrhea and that achieving it often involves a process of trying different formulations to find the best fit. Remember that GnRH agonists are dependent on a functional hypothalamic-pituitary-ovarian axis, and they are unlikely to be effective in women with suspected residual endometriosis who have had a bilateral oophorectomy.
Physical therapy
For CPP, PT typically targets musculoskeletal dysfunction in the pelvic floor, abdominal wall, hips, and back. Interventions include muscle control, mobilization, and biofeedback. Pelvic PT has been shown to improve pain and dyspareunia in patients with CPP, coccydynia, and vestibulodynia.16–18 One large study found a significant, patient-directed decrease in pain medication use after pelvic floor PT.19 Pelvic PT for patients with interstitial cystitis and pelvic floor tenderness resulted in improved pain and bladder symptoms.20
Pelvic PT can be considered for patients with pain reproducible with palpation of the pelvic floor, abdominal wall, paraspinal-lumbar muscles, or sacroiliac joints. Best practices include referral to a therapist who has specialized training in CPP, including pelvic floor therapy. It is important to clearly list the indication for referral, as many of these therapists also treat stress urinary incontinence. The wrong exercises can result in increased hypercontractility of pelvic floor muscles, which can worsen pelvic pain.
It is also critical to clarify expectations with your patient at the time of PT referral. Specifically, advise patients that when beginning therapy, it is common to experience a temporary increase in discomfort of the pelvic muscles. Inform patients also to expect that their therapist will perform internal manipulation of the pelvic floor muscles through the vagina, as this can be surprising for some patients. Finally, counsel patients that their adherence to daily home exercises improves their chance of a durable, long-term successful response.21
CASE 1 Treatment recommendations
For treatment of this patient’s CPP, consider scheduled naproxen therapy during menses, continuous OCPs, and referral for pelvic floor PT.
Read about treating a case of pain, sleep disturbance, and depression.
CASE 2 Patient with long-standing CPP, multiple diagnoses, and sleep problems
A 30-year-old woman (G2P2) reports having had CPP for 17 years. She is amenorrheic with continuous OCP treatment. She had experienced some improvement with pelvic PT. The patient reports that she has daily pain with intermittent pain flares and that she is exhausted and has poor sleep quality, which she attributes to pain. She has been diagnosed with interstitial cystitis, irritable bowel syndrome, and temporomandibular joint disorder. She has a history of depression, which she feels is well controlled with bupropion. Physical examination reveals that the patient has diffuse but mild pain in the pelvic floor and abdominal wall muscles.
What further pain management options can you offer for this patient?
Managing pain, sleep disturbance, and depression
This patient has been living with CPP for many years, and she has sleep difficulties that might be exacerbating pain or result from pain (or both). She is already on continuous OCPs and has had some relief with pelvic PT. Other options that may help with her multiple issues include antidepressants, cyclobenzaprine, and calcium channel blockers.
Antidepressants
Several classes of antidepressants have been used in the treatment of chronic pain conditions, specifically, tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). Commonly used TCAs include amitriptyline, nortriptyline, desipramine, and doxepin. Commonly used SNRIs are duloxetine and milnacipran. Both TCAs and SNRIs increase the availability of norepinephrine and serotonin, which are thought to act on the descending pain inhibitory systems to decrease pain sensitivity. Of note, most selective serotonin reuptake inhibitors (SSRIs) at typical doses do not exert a significant enough impact on norepinephrine to be useful for chronic pain.22
Evidence is limited on the use of antidepressants for treating CPP. Amitriptyline is the most extensively studied antidepressant. Amitriptyline treatment resulted in modest pain improvement in patients with CPP and fibromyalgia.23,24 Bothersome anticholinergic effects, including fatigue, dry mouth, and constipation, often are reported with TCAs. Adverse effects tend to be less with nortriptyline or desipramine compared with amitriptyline, but possibly at the expense of efficacy.
While SNRIs have not yet been studied in CPP, several investigations have shown that they improve pain and quality of life in fibromyalgia patients.22,25
Antidepressant therapy may be appropriate for patients with suspected central pain amplification, widespread pain, and sleep disturbances. Best practices include patient education and careful discussion of this option with your patient. We suggest that clinicians explain that antidepressant medications alter the function of neurotransmitters, which modulate pain signals. While neurotransmitters also are involved in mood modulation, this is not the therapeutic goal in this circumstance. In addition, the doses used for the effective treatment of chronic pain are significantly lower than those needed to treat depression effectively.
Patients often need to hear that you believe that their pain is real and is not a manifestation of depression or another mood disorder. If you suspect that the patient also has untreated depression, address this as its own issue and use medications that have greater efficacy for mood symptoms.
Because many antidepressants can cause sedation, they are best taken before bedtime. Also, slow dose titration over several weeks will reduce the chance of bothersome adverse effects. Counsel patients that efficacy is not generally seen until at goal dose for several weeks. Be aware of interactions with other medications that can cause serotonin syndrome.
Cyclobenzaprine
Cyclobenzaprine is a muscle relaxant that also has activity in the central nervous system. The drug’s precise mechanism of action is not known, but it appears to potentiate norepinephrine and bind to serotonin receptors. Thus, it also likely has some TCA-like activity.
Cyclobenzaprine has not been studied in patients with CPP. In fibromyalgia patients, however, it produced significant improvements in pain, sleep, fatigue, and tenderness.26,27 In our anecdotal experience with CPP patients, cyclobenzaprine has been one of the most impactful therapies. It hits the “chronic pain triad,” meaning that it helps with myofascial pain, neuropathic pain, and sleep disturbances.
Cyclobenzaprine treatment may be considered for patients with myofascial pain, sleep disturbances, and clinical symptoms of central pain amplification. Best practices include starting with low (5 mg) scheduled doses at bedtime and slowly titrating the dose. Drowsiness is a very common side effect, so we try to use that to the patient’s advantage to help with sleep quality.
Notably, sleep disturbances are highly prevalent in patients with chronic pain.28 The relationship appears to be bidirectional, meaning that chronic pain negatively impacts sleep quality, and poor sleep quality causes amplified perception of pain.28–30 Interventions that improve sleep quality have been associated with improvements in pain, coping, mood, and functional status.31 Helping a patient to improve her sleep generally requires a multifaceted approach. It always involves “sleep hygiene” or a behavioral component, and pharmacologic assistance may be considered when improved sleep hygiene does not provide adequately improved sleep quality.
Calcium channel blockers
Gabapentin and pregabalin are calcium channel blockers that inhibit the reuptake of glutamate, norepinephrine, and substance P, which helps to decrease pain sensitivity. They also act as membrane stabilizers, reducing hyperexcitability of peripheral and central nerves. Studies have shown that in patients with CPP, gabapentin resulted in improved pain and mood symptoms with few adverse effects.23,32 Patients with fibromyalgia had improvements in pain, sleep, quality of life, fatigue, and anxiety with both gabapentin and pregabalin.33
It is appropriate to consider use of gabapentin or pregabalin in patients with central pain amplification and sleep disturbances. Best practices include starting with a low dose at bedtime. Traditionally, gabapentin is given in 3 equal doses throughout the day. In our experience, patients report less daytime drowsiness and better sleep quality if two-thirds of the daily dose is given at night, with the remaining daily dose broken up into 2 smaller daytime doses. Slow titration over several weeks will reduce risk of bothersome adverse effects. Patients should be counseled that efficacy is not generally seen until treatment is at goal dose for several weeks.
CASE 2 Treatment recommendations
For this patient with daily pelvic pain, multiple diagnoses that have a pain component, and poor sleep quality, consider a treatment plan that includes scheduled cyclobenzaprine, improved sleep hygiene, and, if needed, gabapentin.
Read about treating a case of focal pain.
CASE 3 Cesarean delivery, hysterectomy, and continued pelvic pain
A 38-year-old woman (G2P2) has had CPP for the past 10 years. She developed persistent left lower-quadrant pain after cesarean delivery of her son. She had a hysterectomy 2 years ago for CPP, after which her pain worsened. She describes daily pain with intermittent flares. On examination, the patient has focal left lower-quadrant pain lateral to the left apex of her Pfannenstiel incision.
What treatment approach would be appropriate for this patient?
Focal pain requires a precisely targeted treatment
This patient with focal left lower-quadrant pain is a candidate for anesthetic trigger point injections in the affected area near her Pfannenstiel incision.
Anesthetic injections
Consider the presence of trigger points and peripheral neuropathy in patients with focal abdominal wall pain. Trigger points are focal, palpable nodules within muscles. They are markedly painful to palpation and are associated with referred pain, motor dysfunction, and occasionally autonomic symptoms. They frequently are seen in abdominal wall or pelvic floor muscles in patients with CPP and are caused by abnormal neuromuscular depolarization.
The ilioinguinal, iliohypogastric, and genitofemoral nerves are in close proximity to Pfannenstiel and laparoscopic port site incisions. These nerves may be injured directly during surgery, but they also may be compressed by postoperative scarring.
Anesthetics, such as lidocaine and bupivacaine, which act as sodium channel blockers, can be injected into this area, and improvement often substantially outlasts the anesthetic’s duration of action. While these drugs’ mechanism of action is not clear, theories include altered function of sodium channels on sensory nerves with repeated anesthetic exposure, dry needling that occurs during injection, hydrodissection of tight connective tissue bands surrounding neuromuscular bundles, or depletion of substance P and neuropeptides as a result of injection.34,35
In several studies, patients with CPP reported decreased pain with lidocaine injections in pelvic floor or abdominal wall trigger points.36–38 Patients with fibromyalgia reported improvement in pain and a decreased need for NSAIDs with bupivacaine trigger point injections.39 While abdominal wall nerve blocks have not been extensively studied in patients with chronic neuropathic pain following gynecologic surgery, they have been shown to substantially improve chronic neuropathic pain following inguinal hernia repair.40
Anesthetic injections appropriately may be considered in patients with focal pain in a muscle or in the distribution of abdominal wall nerves, palpation of which reproduces pain symptoms. Patients with diffuse pain are less likely to benefit from anesthetic injections. Best practices include careful examination with attention to areas of prior abdominal incisions.
Our practice is to inject each affected area with a mix of 9 mL of 1% lidocaine and 1 mL of sodium bicarbonate. If a patient reports at least 24 hours of improvement, we repeat the injection in 2 to 4 weeks. The goal is for the patient to experience a progressively longer duration of benefit with subsequent injections. We perform repeat injections shortly after pain begins to recur at that site. The patient should eventually graduate from receiving regular injections and may return for a remedial injection if pain recurs.
CASE 3 Treatment recommendations
For this patient with persistent focal left-lower quadrant pain and a defined trigger point near her Pfannenstiel incision, consider anesthetic injection in the left lower quadrant.
Work toward realistic symptom improvement
Remember that living with chronic pain is exhausting, and empathy with a patient-centered approach is the most important ingredient for patient improvement and satisfaction. Discuss realistic expectations with patients. Remind them that there is no magic bullet for the complex problem of CPP, and that chronic conditions generally do not improve overnight. Focus on improving the patient’s function and quality of life, and applaud symptom improvement rather than focusing on complete pain resolution.
As these visits often require a good deal of patient education, budget more appointment time if feasible. We find that scheduling frequent return visits (approximately every 3 to 4 months) allows timely treatment follow-up so that changes may be made if needed. If you have maximized your available treatment options, referring the patient to a specialist with additional training in CPP is a sensible next step.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Howard FM. Chronic pelvic pain. Obstet Gynecol. 2003;101(3):594–611.
- Mathias SD, Kuppermann M, Liberman RF, Lipschutz RC, Steege JF. Chronic pelvic pain: prevalence, health-related uality of life, and economic correlates. Obstet Gynecol. 1996;87(3):321–327.
- Gelbaya TA, El-Halwagy HE. Focus on primary care: chronic pelvic pain in women. Obstet Gynecol Surv. 2001;56(12):757–764.
- Broder MS, Kanouse DE, Mittman BS, Bernstein SJ. The appropriateness of recommendations for hysterectomy. Obstet Gynecol. 2000;95(2):199–205.
- Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am J Obstet Gynecol. 2008;198(1):34.e1–34.e7.
- Steege JF, Siedhoff MT. Chronic pelvic pain. Obstet Gynecol. 2014;124(3):616–629.
- Williams DA, Clauw DJ. Understanding fibromyalgia: lessons from the broader pain research community. J Pain. 2009;10(8):777–791.
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 suppl):S2–S15.
- Brawn J, Morotti M, Zondervan KT, Becker CM, Vincent K. Central changes associated with chronic pelvic pain and endometriosis. Hum Reprod Update. 2014;20(5):737–747.
- As-Sanie S, Harris RE, Harte SE, Tu FF, Neshewat G, Clauw DJ. Increased pressure pain sensitivity in women with chronic pelvic pain. Obstet Gynecol. 2013;122(5):1047–1055.
- As-Sanie S, Kim J, Schmidt-Wilcke T, et al. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J Pain. 2016;17(1):1–13.
- As-Sanie S, Harris RE, Napadow V, et al. Changes in regional gray matter volume in women with chronic pelvic pain: a voxel-based morphometry study. Pain. 2012;153(5):1006–1014.
- Marjoribanks J, Ayeleke RO, Farquhar C, Proctor M. Nonsteroidal anti-inflammatory drugs for dysmenorrhoea. Cochrane Database Syst Rev. 2015;(7):CD001751.
- Falcone T, Lebovic DI. Clinical management of endometriosis. Obstet Gynecol. 2011;118(3):691–705.
- Brown J, Pan A, Hart RJ. Gonadotrophin-releasing hormone analogues for pain associated with endometriosis. Cochrane Database Syst Rev. 2010;(12):CD008475.
- Zoorob D, South M, Karram M, et al. A pilot randomized trial of levator injections versus physical therapy for treatment of pelvic floor myalgia and sexual pain. Int Urogynecol J. 2015;26(6):845–852.
- Scott KM, Fisher LW, Bernstein IH, Bradley MH. The treatment of chronic coccydynia and postcoccygectomy pain with pelvic floor physical therapy. PM R. 2017;9(4):367–376.
- Goldfinger C, Pukall CF, Thibault-Gagnon S, McLean L, Chamberlain S. Effectiveness of cognitive-behavioral therapy and physical therapy for provoked vestibulodynia: a randomized pilot study. J Sex Med. 2016;13(1):88–94.
- Anderson RU, Harvey RH, Wise D, Nevin Smith J, Nathanson BH, Sawyer T. Chronic pelvic pain syndrome: reduction of medication use after pelvic floor physical therapy with an internal myofascial trigger point wand. Appl Psychophysiol Biofeedback. 2015;40(1):45–52.
- FitzGerald MP, Payne CK, Lukacz ES, et al. Randomized multicenter clinical trial of myofascial physical therapy in women with interstitial cystitis/painful bladder syndrome and pelvic floor tenderness. J Urol. 2012;187(6):2113–2118.
- FitzGerald MP, Kotarinos R. Rehabilitation of the short pelvic floor. II: Treatment of the patient with the short pelvic floor. Int Urogynecol J Pelvic Floor Dysfunct. 2003;14(4):269–275.
- Arnold LM. Duloxetine and other antidepressants in the treatment of patients with fibromyalgia. Pain Med. 2007;8(suppl 2):S63–S74.
- Sator-Katzenschlager SM, Scharbert G, Kress HG, et al. Chronic pelvic pain treated with gabapentin and amitriptyline: a randomized controlled pilot study. Wien Klin Wochenschr. 2005;117(21–22):761–78.
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;12:CD008242.
- Gendreau RM, Thorn MD, Gendreau JF, et al. Efficacy of milnacipran in patients with fibromyalgia. J Rheumatol. 2005;32(10):1975–1985.
- Tofferi JK, Jackson JL, O’Malley PG. Treatment of fibromyalgia with cyclobenzaprine: a meta-analysis. Arthritis Rheum. 2004;51(1):9–13.
- Moldofsky H, Harris HW, Archambault WT, Kwong T, Lederman S. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: a double-blind randomized placebo-controlled study. J Rheumatol. 2011;38(12):2653–2463.
- Cheatle MD, Foster S, Pinkett A, Lesneski M, Qu D, Dhingra L. Assessing and managing sleep disturbance in patients with chronic pain. Sleep Med Clin. 2016;11(4):531–541.
- Larson RA, Carter JR. Total sleep deprivation and pain perception during cold noxious stimuli in humans. Scand J Pain. 2016;13:12–16.
- Generaal E, Vogelzangs N, Penninx BW, Dekker J. Insomnia, sleep duration, depressive symptoms, and the onset of chronic multisite musculoskeletal pain [published online ahead of print January 1, 2017]. Sleep. doi:10.1093/sleep/zsw030.
- Gerhart JI, Burns JW, Post KM, et al. Relationships between sleep quality and pain-related factors for people with chronic low back pain: tests of reciprocal and time of day effects. Ann Behav Med. 2017;51(3):365–375.
- Lewis SC, Bhattacharya S, Wu O, et al. Gabapentin for the management of chronic pelvic pain in women (GaPP1): a pilot randomised controlled trial. PLoS One. 2016;11(4):e0153037.
- Häuser W, Bernardy K, Uçeyler N, Sommer C. Treatment of fibromyalgia syndrome with gabapentin and pregabalin—a meta-analysis of randomized controlled trials. Pain. 2009;145(1–2):69–81.
- Scott NA, Guo B, Barton PM, Gerwin RD. Trigger point injections for chronic non-malignant musculoskeletal pain: a systematic review. Pain Med. 2009;10(1):54–69.
- Hameroff SR, Crago BR, Blitt CD, Womble J, Kanel J. Comparison of bupivacaine, etidocaine, and saline for trigger-point therapy. Anesth Analg. 1981;60(10):752–755.
- Montenegro ML, Braz CA, Rosa-e-Silva JC, Candido-dos-Reis FJ, Nogueira AA, Poli-Neto OB. Anaesthetic injection versus ischemic compression for the pain relief of abdominal wall trigger points in women with chronic pelvic pain. BMC Anesthesiol. 2015;15:175.
- Kim DS, Jeong TY, Kim YK, Chang WH, Yoon JG, Lee SC. Usefulness of a myofascial trigger point injection for groin pain in patients with chronic prostatitis/chronic pelvic pain syndrome: a pilot study. Arch Phys Med Rehabil. 2013;94(5):930–936.
- Huang QM, Liu L. Wet needling of myofascial trigger points in abdominal muscles for treatment of primary dysmenorrhoea. Acupunct Med. 2014;32(4):346–349.
- Affaitati G, Fabrizio A, Savini A, et al. A randomized, controlled study comparing a lidocaine patch, a placebo patch, and anesthetic injection for treatment of trigger points in patients with myofascial pain syndrome: evaluation of pain and somatic pain thresholds. Clin Ther. 2009;31(4):705–720.
- Thomassen I, van Suijlekom JA, van de Gaag A, Ponten JE, Nienhuijs SW. Ultrasound-guided ilioinguinal/iliohypogastric nerve blocks for chronic pain after inguinal hernia repair. Hernia. 2013;17(3):329–332.
- Howard FM. Chronic pelvic pain. Obstet Gynecol. 2003;101(3):594–611.
- Mathias SD, Kuppermann M, Liberman RF, Lipschutz RC, Steege JF. Chronic pelvic pain: prevalence, health-related uality of life, and economic correlates. Obstet Gynecol. 1996;87(3):321–327.
- Gelbaya TA, El-Halwagy HE. Focus on primary care: chronic pelvic pain in women. Obstet Gynecol Surv. 2001;56(12):757–764.
- Broder MS, Kanouse DE, Mittman BS, Bernstein SJ. The appropriateness of recommendations for hysterectomy. Obstet Gynecol. 2000;95(2):199–205.
- Whiteman MK, Hillis SD, Jamieson DJ, et al. Inpatient hysterectomy surveillance in the United States, 2000–2004. Am J Obstet Gynecol. 2008;198(1):34.e1–34.e7.
- Steege JF, Siedhoff MT. Chronic pelvic pain. Obstet Gynecol. 2014;124(3):616–629.
- Williams DA, Clauw DJ. Understanding fibromyalgia: lessons from the broader pain research community. J Pain. 2009;10(8):777–791.
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 suppl):S2–S15.
- Brawn J, Morotti M, Zondervan KT, Becker CM, Vincent K. Central changes associated with chronic pelvic pain and endometriosis. Hum Reprod Update. 2014;20(5):737–747.
- As-Sanie S, Harris RE, Harte SE, Tu FF, Neshewat G, Clauw DJ. Increased pressure pain sensitivity in women with chronic pelvic pain. Obstet Gynecol. 2013;122(5):1047–1055.
- As-Sanie S, Kim J, Schmidt-Wilcke T, et al. Functional connectivity is associated with altered brain chemistry in women with endometriosis-associated chronic pelvic pain. J Pain. 2016;17(1):1–13.
- As-Sanie S, Harris RE, Napadow V, et al. Changes in regional gray matter volume in women with chronic pelvic pain: a voxel-based morphometry study. Pain. 2012;153(5):1006–1014.
- Marjoribanks J, Ayeleke RO, Farquhar C, Proctor M. Nonsteroidal anti-inflammatory drugs for dysmenorrhoea. Cochrane Database Syst Rev. 2015;(7):CD001751.
- Falcone T, Lebovic DI. Clinical management of endometriosis. Obstet Gynecol. 2011;118(3):691–705.
- Brown J, Pan A, Hart RJ. Gonadotrophin-releasing hormone analogues for pain associated with endometriosis. Cochrane Database Syst Rev. 2010;(12):CD008475.
- Zoorob D, South M, Karram M, et al. A pilot randomized trial of levator injections versus physical therapy for treatment of pelvic floor myalgia and sexual pain. Int Urogynecol J. 2015;26(6):845–852.
- Scott KM, Fisher LW, Bernstein IH, Bradley MH. The treatment of chronic coccydynia and postcoccygectomy pain with pelvic floor physical therapy. PM R. 2017;9(4):367–376.
- Goldfinger C, Pukall CF, Thibault-Gagnon S, McLean L, Chamberlain S. Effectiveness of cognitive-behavioral therapy and physical therapy for provoked vestibulodynia: a randomized pilot study. J Sex Med. 2016;13(1):88–94.
- Anderson RU, Harvey RH, Wise D, Nevin Smith J, Nathanson BH, Sawyer T. Chronic pelvic pain syndrome: reduction of medication use after pelvic floor physical therapy with an internal myofascial trigger point wand. Appl Psychophysiol Biofeedback. 2015;40(1):45–52.
- FitzGerald MP, Payne CK, Lukacz ES, et al. Randomized multicenter clinical trial of myofascial physical therapy in women with interstitial cystitis/painful bladder syndrome and pelvic floor tenderness. J Urol. 2012;187(6):2113–2118.
- FitzGerald MP, Kotarinos R. Rehabilitation of the short pelvic floor. II: Treatment of the patient with the short pelvic floor. Int Urogynecol J Pelvic Floor Dysfunct. 2003;14(4):269–275.
- Arnold LM. Duloxetine and other antidepressants in the treatment of patients with fibromyalgia. Pain Med. 2007;8(suppl 2):S63–S74.
- Sator-Katzenschlager SM, Scharbert G, Kress HG, et al. Chronic pelvic pain treated with gabapentin and amitriptyline: a randomized controlled pilot study. Wien Klin Wochenschr. 2005;117(21–22):761–78.
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;12:CD008242.
- Gendreau RM, Thorn MD, Gendreau JF, et al. Efficacy of milnacipran in patients with fibromyalgia. J Rheumatol. 2005;32(10):1975–1985.
- Tofferi JK, Jackson JL, O’Malley PG. Treatment of fibromyalgia with cyclobenzaprine: a meta-analysis. Arthritis Rheum. 2004;51(1):9–13.
- Moldofsky H, Harris HW, Archambault WT, Kwong T, Lederman S. Effects of bedtime very low dose cyclobenzaprine on symptoms and sleep physiology in patients with fibromyalgia syndrome: a double-blind randomized placebo-controlled study. J Rheumatol. 2011;38(12):2653–2463.
- Cheatle MD, Foster S, Pinkett A, Lesneski M, Qu D, Dhingra L. Assessing and managing sleep disturbance in patients with chronic pain. Sleep Med Clin. 2016;11(4):531–541.
- Larson RA, Carter JR. Total sleep deprivation and pain perception during cold noxious stimuli in humans. Scand J Pain. 2016;13:12–16.
- Generaal E, Vogelzangs N, Penninx BW, Dekker J. Insomnia, sleep duration, depressive symptoms, and the onset of chronic multisite musculoskeletal pain [published online ahead of print January 1, 2017]. Sleep. doi:10.1093/sleep/zsw030.
- Gerhart JI, Burns JW, Post KM, et al. Relationships between sleep quality and pain-related factors for people with chronic low back pain: tests of reciprocal and time of day effects. Ann Behav Med. 2017;51(3):365–375.
- Lewis SC, Bhattacharya S, Wu O, et al. Gabapentin for the management of chronic pelvic pain in women (GaPP1): a pilot randomised controlled trial. PLoS One. 2016;11(4):e0153037.
- Häuser W, Bernardy K, Uçeyler N, Sommer C. Treatment of fibromyalgia syndrome with gabapentin and pregabalin—a meta-analysis of randomized controlled trials. Pain. 2009;145(1–2):69–81.
- Scott NA, Guo B, Barton PM, Gerwin RD. Trigger point injections for chronic non-malignant musculoskeletal pain: a systematic review. Pain Med. 2009;10(1):54–69.
- Hameroff SR, Crago BR, Blitt CD, Womble J, Kanel J. Comparison of bupivacaine, etidocaine, and saline for trigger-point therapy. Anesth Analg. 1981;60(10):752–755.
- Montenegro ML, Braz CA, Rosa-e-Silva JC, Candido-dos-Reis FJ, Nogueira AA, Poli-Neto OB. Anaesthetic injection versus ischemic compression for the pain relief of abdominal wall trigger points in women with chronic pelvic pain. BMC Anesthesiol. 2015;15:175.
- Kim DS, Jeong TY, Kim YK, Chang WH, Yoon JG, Lee SC. Usefulness of a myofascial trigger point injection for groin pain in patients with chronic prostatitis/chronic pelvic pain syndrome: a pilot study. Arch Phys Med Rehabil. 2013;94(5):930–936.
- Huang QM, Liu L. Wet needling of myofascial trigger points in abdominal muscles for treatment of primary dysmenorrhoea. Acupunct Med. 2014;32(4):346–349.
- Affaitati G, Fabrizio A, Savini A, et al. A randomized, controlled study comparing a lidocaine patch, a placebo patch, and anesthetic injection for treatment of trigger points in patients with myofascial pain syndrome: evaluation of pain and somatic pain thresholds. Clin Ther. 2009;31(4):705–720.
- Thomassen I, van Suijlekom JA, van de Gaag A, Ponten JE, Nienhuijs SW. Ultrasound-guided ilioinguinal/iliohypogastric nerve blocks for chronic pain after inguinal hernia repair. Hernia. 2013;17(3):329–332.

Psoriasis Treatment in HIV-Positive Patients: A Systematic Review of Systemic Immunosuppressive Therapies
The prevalence of psoriasis among human immunodeficiency virus (HIV)–positive patients in the United States is reported to be approximately 1% to 3%, which is similar to the rates reported for the general population.1 Recalcitrant cases of psoriasis in patients with no history of the condition can be the initial manifestation of HIV infection. In patients with preexisting psoriasis, a flare of their disease can be seen following infection, and progression of HIV correlates with worsening psoriasis.2 Psoriatic arthropathy also affects 23% to 50% of HIV-positive patients with psoriasis worldwide, which may be higher than the general population,1 with more severe joint disease.
The management of psoriatic disease in the HIV-positive population is challenging. The current first-line recommendations for treatment include topical therapies, phototherapy, and highly active antiretroviral therapy (HAART), followed by oral retinoids as second-line agents.3 However, the clinical course of psoriasis in HIV-positive patients often is progressive and refractory2; therefore, these therapies often are inadequate to control both skin and joint manifestations. Most other currently available systemic therapies for psoriatic disease are immunosuppressive, which poses a distinct clinical challenge because HIV-positive patients are already immunocompromised.
There currently are many systemic immunosuppressive agents used for the treatment of psoriatic disease, including oral agents (eg, methotrexate, hydroxyurea, cyclosporine), as well as newer biologic medications, including tumor necrosis factor (TNF) α inhibitors etanercept, adalimumab, infliximab, golimumab, and certolizumab pegol. Golimumab and certolizumab pegol currently are indicated for psoriatic arthritis only. Other newer biologic therapies include ustekinumab, which inhibits IL-12 and IL-23, and secukinumab, which inhibits IL-17A. The purpose of this systematic review is to evaluate the most current literature to explore the efficacy and safety data as they pertain to systemic immunosuppressive therapies for the treatment of psoriatic disease in HIV-positive individuals.
Methods
To investigate the efficacy and safety of systemic immunosuppressive therapies for psoriatic disease in HIV-positive individuals, a PubMed search of articles indexed for MEDLINE (1985-2015) was conducted using the terms psoriasis and HIV and psoriatic arthritis and HIV combined with each of the following systemic immunosuppressive agents: methotrexate, hydroxyurea, cyclosporine, etanercept, adalimumab, infliximab, golimumab, certolizumab pegol, ustekinumab, and secukinumab. Pediatric cases and articles that were not available in the English language were excluded.
For each case, patient demographic information (ie, age, sex), prior failed psoriasis treatments, and history of HAART were documented. The dosing regimen of the systemic agent was noted when different from the US Food and Drug administration–approved dosage for psoriasis or psoriatic arthritis. The duration of immunosuppressive therapy as well as pretreatment and posttreatment CD4 and viral counts (when available) were collected. The response to treatment and adverse effects were summarized.
Results
Our review of the literature yielded a total of 25 reported cases of systemic immunosuppressive therapies used to treat psoriatic disease in HIV-positive patients, including methotrexate, cyclosporine, etanercept, adalimumab, in-fliximab, and ustekinumab (Table). There were no reports of the use of hydroxyurea, golimumab, certolizumab pegol, or secukinumab to treat psoriatic disease in this patient population.
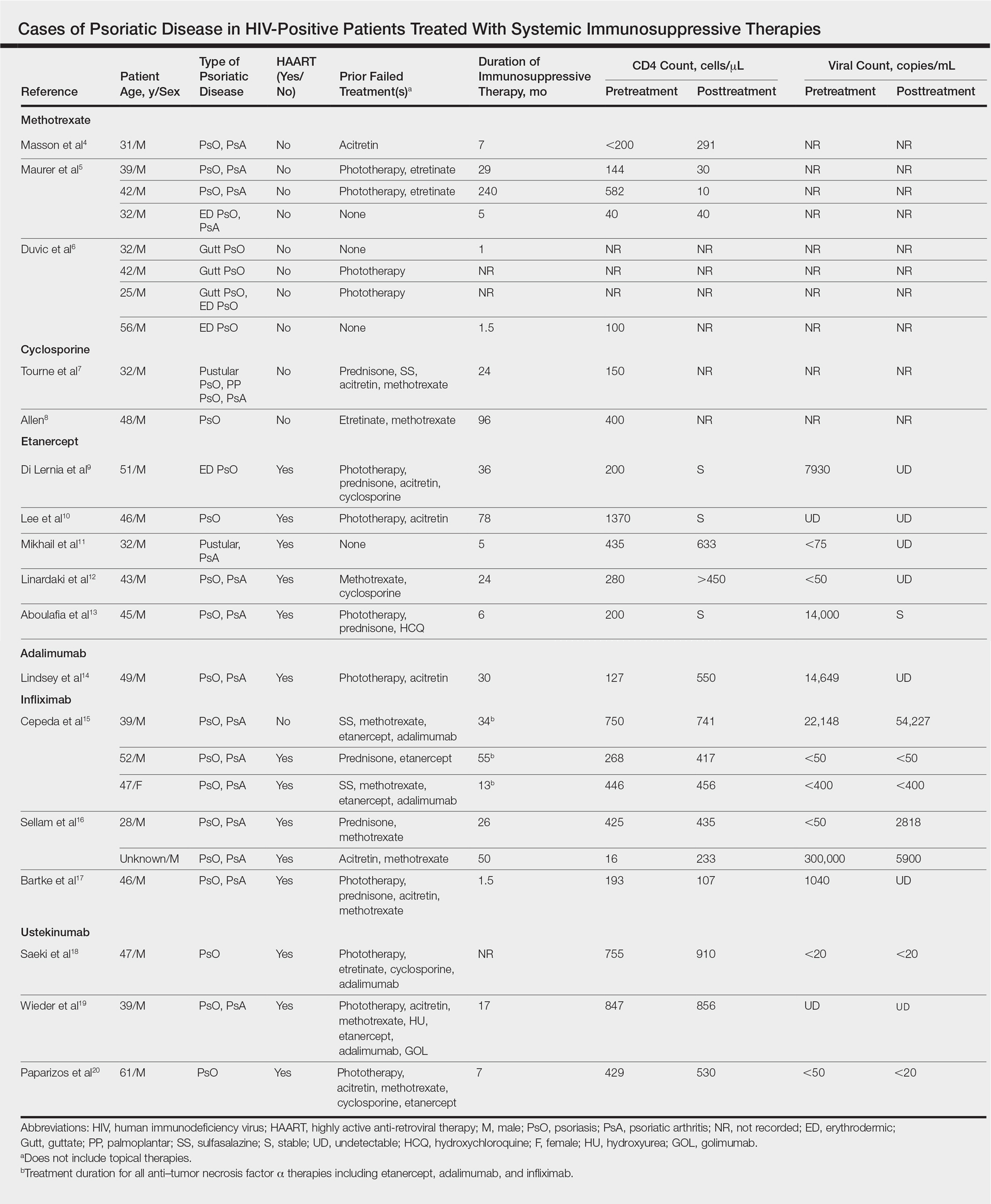
Methotrexate
Eight individual cases of methotrexate used to treat psoriasis and/or psoriatic arthritis in HIV-positive patients were reported.4-6 Duvic et al6 described 4 patients with psoriatic disease that was treated with methotrexate with varying efficacy. One patient developed toxic encephalopathy, which improved after discontinuation of methotrexate; however, he died 5 months later from pneumocystis pneumonia. In this early study, none of the 4 patients were on antiretroviral therapy for HIV.6
In the cases reported by Masson et al4 and Maurer et al,5 4 patients were treated with a single antiretroviral agent and received appropriate prophylaxis against opportunistic infections. In 1 case, methotrexate was given at a chemotherapeutic dose of 525 mg once weekly for Kaposi sarcoma.4 In 2 of 4 cases, the patients developed pneumocystis pneumonia.4,5
Cyclosporine
There were 2 case reports of successful treatment of psoriatic disease with cyclosporine in HIV-positive patients.7,8 Skin and joint manifestations improved rapidly without reports of infection for 27 and 8 years.8 Both patients were treated with one antiretroviral agent.7,8
Etanercept
There were 5 case reports of successful treatment of psoriatic disease with etanercept. In all 5 cases the patients were on HAART, and the CD4 count increased or remained stable and viral count became undetectable or remained stable following treatment.9-13 In 2 cases, the patient also had hepatitis C virus, which remained stable throughout the treatment period.9,12 The maximum duration of treatment was 6 years, with only 1 reported adverse event.13 In this case reported by Aboulafia et al,13 the patient experienced recurrent polymicrobial infections, including enterococcal cellulitis, cystitis, and bacteremia, as well as pseudomonas pneumonia and septic arthritis. Therapy was discontinued at 6 months. Four months after discontinuation of etanercept, the patient died from infectious causes.13
Adalimumab
There was 1 case of successful treatment of psoriatic disease with adalimumab in an HIV-positive patient. In this case, the patient was on HAART, and CD4 and viral counts improved substantially after 30 months of treatment.14
Infliximab
Six individual cases of successful treatment of psoriatic disease with infliximab were reported.15-17 In a report by Cepeda et al,15 HIV-positive patients with various rheumatologic diseases were chosen to receive etanercept followed by adalimumab and/or infliximab if clinical improvement was not observed on etanercept. In 3 patients with psoriasis and psoriatic arthritis, inadequate response was observed on etanercept. Two of these 3 patients received adalimumab with only partial response. All 3 were treated with infliximab in the end and showed excellent response. One of the patients experienced facial abscess responsive to antibiotics and was continued on infliximab therapy without further complications. In all 6 cases of infliximab therapy, the patients were on HAART, and CD4 and viral counts improved or remained stable.15
Ustekinumab
There were 3 case reports of successful treatment of psoriatic disease with ustekinumab in HIV-positive patients on HAART. CD4 and viral counts improved or remained stable.18-20
Comment
Currently, all of the systemic immunosuppressive therapies approved for psoriatic disease have a warning by the US Food and Drug Administration for increased risk of serious infection. Given such labels, these therapies are not routinely prescribed for HIV-positive patients who are already immunocompromised; however, many HIV-positive patients have severe psoriatic disease that cannot be adequately treated with first- and second-line therapies including topical agents, phototherapy, or oral retinoids.
Our comprehensive review yielded a total of 25 reported cases of systemic immunosuppressive therapies used to treat psoriatic disease in HIV-positive patients including methotrexate, cyclosporine, etanercept, adalimumab, in-fliximab, and ustekinumab. Although data are limited to case reports and case series, some trends were observed.
Efficacy
In most of the cases reviewed, the patients had inadequate improvement of psoriatic disease with first- and second-line therapies, which included antiretrovirals alone, topical agents, phototherapy, and oral retinoids. Some cases reported poor response to methotrexate and cyclosporine.4-8 Biologic agents were effective in many such cases.
Safety
Overall, there were 11 cases in which the patient was not on adequate HAART while being treated with systemic immunosuppressive therapy for psoriatic disease.4-8,15 Of them, 3 were associated with serious infection while on methotrexate.5,6 There was only 1 report of serious infection13 of 14 cases in which the patient was on concomitant HAART. In this case, which reported polymicrobial infections and subsequent death of the patient, the infections continued after discontinuing etanercept; thus, the association is unclear. Interestingly, despite multiple infections, the CD4 and viral counts were stable throughout treatment with etanercept.13
From reviewing the 4 total cases5,6,13 of serious infection, HAART appears to be a valuable concomitant treatment during systemic immunosuppressive therapy for HIV-positive patients; however, it does not necessarily prevent serious infections from occurring, and thus the clinician’s diligence in monitoring for signs and symptoms of infection remains important.
CD4 and Viral Counts
Although reports of CD4 and viral counts were not available in earlier studies,4-8 there were 15 cases that reported consistent pretreatment and posttreatment CD4 and viral counts during treatment with etanercept, adalimumab, infliximab, and ustekinumab.9-20 In all cases, the CD4 count was stable or increased. Similarly, the viral count was stable or decreased. All patients, except 1 by Cepeda et al,15 were on concomitant HAART.9-14,16-20
Although data are limited, treatment of psoriatic disease with biologic agents when used in combination with HAART may have beneficial effects on CD4 and viral counts. Tumor necrosis factor has a role in HIV expression through the action of nuclear factor κβ.21 An increase in TNF levels is shown to be associated with increased viral count, decreased CD4 count, and increased symptoms of HIV progression, such as fever, fatigue, cachexia, and dementia.22 Although more studies are necessary, TNF-α inhibitors may have a positive effect on HIV while simultaneously treating psoriatic disease. Other cytokines (eg, IL-12, IL-23, IL-17) involved in the mechanism of action of other biologic agents (ustekinumab and secukinumab) have not been shown to be directly associated with HIV activity; however, studies have shown that IL-10 has a role in inhibiting HIV-1 replication and inhibits secretion of proinflammatory cytokines such as IL-12 and TNF-α.21 It may be speculated that the inhibition of IL-12 and TNF-α may create a positive feedback effect to increase IL-10, which in turn inhibits HIV replication.
Conclusion
Although there are limited data on the efficacy and safety of systemic immunosuppressive therapies for the treatment of psoriatic disease in HIV-positive patients, a review of 25 individual cases suggest that these treatments are not only required but also are sufficient to treat some of the most resistant cases. It is possible that with adequate concomitant HAART and monitoring for signs and symptoms of infection, the likelihood of serious infection may be low. Furthermore, biologic agents may have a positive effect over other systemic immunosuppressive agents, such as methotrexate and cyclosporine, in improving CD4 and viral counts when used in combination with HAART. Although randomized controlled trials are necessary, current biologic therapies such as etanercept, adalimumab, infliximab, and ustekinumab may be safe viable options as third-line treatment of severe psoriasis in the HIV-positive population.
- Mallon
E, Bunker CB. HIV-associated psoriasis. AIDS Patient Care STDS. 2000;14:239-246. - Montaz
eri A, Kanitakis J, Bazex J. Psoriasis and HIV infection. Int J Dermatol. 1996;35:475-479. - Menon
K, Van Vorhees AS, Bebo BF, et al; National Psoriasis Foundation. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010;62:291-299. - Masso
n C, Chennebault JM, Leclech C. Is HIV infection contraindication to the use of methotrexate in psoriatic arthritis? J Rheumatol. 1995;22:2191. - Maurer
TA, Zackheim HS, Tuffanelli L, et al. The use of methotrexate for treatment of psoriasis in patients with HIV infection. J Am Acad Dermatol. 1994;31:372-375. - Duvic
M, Johnson TM, Rapini RP, et al. Acquired immunodeficiency syndrome-associated psoriasis and Reiter’s syndrome. Arch Dermatol. 1987;123:1622-1632. - Tourne
L, Durez P, Van Vooren JP, et al. Alleviation of HIV-associated psoriasis and psoriatic arthritis with cyclosporine. J Am Acad Dermatol. 1997;37:501-502. - Allen
BR. Use of cyclosporine for psoriasis in HIV-positive patient. Lancet. 1992;339:686. - Di Ler
nia V, Zoboli G, Ficarelli E. Long-term management of HIV/hepatitis C virus associated psoriasis with etanercept. Indian J Dermatol Venereol Leprol. 2013;79:444. - Lee E
S, Heller MM, Kamangar F, et al. Long-term etanercept use for severe generalized psoriasis in an HIV-infected individual: a case study. J Drugs Dermatol. 2012;11:413-414. - Mikha
il M, Weinberg JM, Smith BL. Successful treatment with etanercept of von Zumbusch pustular psoriasis in a patient with human immunodeficiency virus. Arch Dermatol. 2008;144:453-456. - Linar
daki G, Katsarou O, Ioannidou P, et al. Effective etanercept treatment for psoriatic arthritis complicating concomitant human immunodeficiency virus and hepatitis C virus infection. J Rheumatol. 2007;34:1353-1355. - Aboul
afia DM, Bundow D, Wilske K, et al. Etanercept for the treatment of human immunodeficiency virus-associated psoriatic arthritis. Mayo Clin Proc. 2000;75:1093-1098. - Linds
ey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871. - Ceped
a EJ, Williams FM, Ishimori ML, et al. The use of anti-tumor necrosis factor therapy in HIV-positive individuals with rheumatic disease. Ann Rheum Dis. 2008;67:710-712. - Sella
m J, Bouvard B, Masson C, et al. Use of infliximab to treat psoriatic arthritis in HIV-positive patients. Joint Bone Spine. 2007;74:197-200. - Bartk
e U, Venten I, Kreuter A, et al. Human immunodeficiency virus-associated psoriasis and psoriatic arthritis treated with infliximab. Br J Dermatol. 2004;150:784-786. - Saeki
H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655. - Wiede
r S, Routt E, Levitt J, et al. Treatment of refractory psoriasis with ustekinumab in an HIV-positive patient: a case presentation and review of the biologic literature. Psoriasis Forum. 2014;20:96-102. - Papar
izos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatol Treat. 2012;23:398-399. - Kedzierska K, Crowe SM, Turville S, et al. The influence of cytokines, chemokines, and their receptors on HIV-1 replication in monocytes and macrophages. Rev Med Virol. 2003;13:39-56.
- Emer JJ. Is there a potential role for anti-tumor necrosis factor therapy in patients with human immunodeficiency virus? J Clin Aesthet Dermatol. 2009;2:29-35.
The prevalence of psoriasis among human immunodeficiency virus (HIV)–positive patients in the United States is reported to be approximately 1% to 3%, which is similar to the rates reported for the general population.1 Recalcitrant cases of psoriasis in patients with no history of the condition can be the initial manifestation of HIV infection. In patients with preexisting psoriasis, a flare of their disease can be seen following infection, and progression of HIV correlates with worsening psoriasis.2 Psoriatic arthropathy also affects 23% to 50% of HIV-positive patients with psoriasis worldwide, which may be higher than the general population,1 with more severe joint disease.
The management of psoriatic disease in the HIV-positive population is challenging. The current first-line recommendations for treatment include topical therapies, phototherapy, and highly active antiretroviral therapy (HAART), followed by oral retinoids as second-line agents.3 However, the clinical course of psoriasis in HIV-positive patients often is progressive and refractory2; therefore, these therapies often are inadequate to control both skin and joint manifestations. Most other currently available systemic therapies for psoriatic disease are immunosuppressive, which poses a distinct clinical challenge because HIV-positive patients are already immunocompromised.
There currently are many systemic immunosuppressive agents used for the treatment of psoriatic disease, including oral agents (eg, methotrexate, hydroxyurea, cyclosporine), as well as newer biologic medications, including tumor necrosis factor (TNF) α inhibitors etanercept, adalimumab, infliximab, golimumab, and certolizumab pegol. Golimumab and certolizumab pegol currently are indicated for psoriatic arthritis only. Other newer biologic therapies include ustekinumab, which inhibits IL-12 and IL-23, and secukinumab, which inhibits IL-17A. The purpose of this systematic review is to evaluate the most current literature to explore the efficacy and safety data as they pertain to systemic immunosuppressive therapies for the treatment of psoriatic disease in HIV-positive individuals.
Methods
To investigate the efficacy and safety of systemic immunosuppressive therapies for psoriatic disease in HIV-positive individuals, a PubMed search of articles indexed for MEDLINE (1985-2015) was conducted using the terms psoriasis and HIV and psoriatic arthritis and HIV combined with each of the following systemic immunosuppressive agents: methotrexate, hydroxyurea, cyclosporine, etanercept, adalimumab, infliximab, golimumab, certolizumab pegol, ustekinumab, and secukinumab. Pediatric cases and articles that were not available in the English language were excluded.
For each case, patient demographic information (ie, age, sex), prior failed psoriasis treatments, and history of HAART were documented. The dosing regimen of the systemic agent was noted when different from the US Food and Drug administration–approved dosage for psoriasis or psoriatic arthritis. The duration of immunosuppressive therapy as well as pretreatment and posttreatment CD4 and viral counts (when available) were collected. The response to treatment and adverse effects were summarized.
Results
Our review of the literature yielded a total of 25 reported cases of systemic immunosuppressive therapies used to treat psoriatic disease in HIV-positive patients, including methotrexate, cyclosporine, etanercept, adalimumab, in-fliximab, and ustekinumab (Table). There were no reports of the use of hydroxyurea, golimumab, certolizumab pegol, or secukinumab to treat psoriatic disease in this patient population.
Methotrexate
Eight individual cases of methotrexate used to treat psoriasis and/or psoriatic arthritis in HIV-positive patients were reported.4-6 Duvic et al6 described 4 patients with psoriatic disease that was treated with methotrexate with varying efficacy. One patient developed toxic encephalopathy, which improved after discontinuation of methotrexate; however, he died 5 months later from pneumocystis pneumonia. In this early study, none of the 4 patients were on antiretroviral therapy for HIV.6
In the cases reported by Masson et al4 and Maurer et al,5 4 patients were treated with a single antiretroviral agent and received appropriate prophylaxis against opportunistic infections. In 1 case, methotrexate was given at a chemotherapeutic dose of 525 mg once weekly for Kaposi sarcoma.4 In 2 of 4 cases, the patients developed pneumocystis pneumonia.4,5
Cyclosporine
There were 2 case reports of successful treatment of psoriatic disease with cyclosporine in HIV-positive patients.7,8 Skin and joint manifestations improved rapidly without reports of infection for 27 and 8 years.8 Both patients were treated with one antiretroviral agent.7,8
Etanercept
There were 5 case reports of successful treatment of psoriatic disease with etanercept. In all 5 cases the patients were on HAART, and the CD4 count increased or remained stable and viral count became undetectable or remained stable following treatment.9-13 In 2 cases, the patient also had hepatitis C virus, which remained stable throughout the treatment period.9,12 The maximum duration of treatment was 6 years, with only 1 reported adverse event.13 In this case reported by Aboulafia et al,13 the patient experienced recurrent polymicrobial infections, including enterococcal cellulitis, cystitis, and bacteremia, as well as pseudomonas pneumonia and septic arthritis. Therapy was discontinued at 6 months. Four months after discontinuation of etanercept, the patient died from infectious causes.13
Adalimumab
There was 1 case of successful treatment of psoriatic disease with adalimumab in an HIV-positive patient. In this case, the patient was on HAART, and CD4 and viral counts improved substantially after 30 months of treatment.14
Infliximab
Six individual cases of successful treatment of psoriatic disease with infliximab were reported.15-17 In a report by Cepeda et al,15 HIV-positive patients with various rheumatologic diseases were chosen to receive etanercept followed by adalimumab and/or infliximab if clinical improvement was not observed on etanercept. In 3 patients with psoriasis and psoriatic arthritis, inadequate response was observed on etanercept. Two of these 3 patients received adalimumab with only partial response. All 3 were treated with infliximab in the end and showed excellent response. One of the patients experienced facial abscess responsive to antibiotics and was continued on infliximab therapy without further complications. In all 6 cases of infliximab therapy, the patients were on HAART, and CD4 and viral counts improved or remained stable.15
Ustekinumab
There were 3 case reports of successful treatment of psoriatic disease with ustekinumab in HIV-positive patients on HAART. CD4 and viral counts improved or remained stable.18-20
Comment
Currently, all of the systemic immunosuppressive therapies approved for psoriatic disease have a warning by the US Food and Drug Administration for increased risk of serious infection. Given such labels, these therapies are not routinely prescribed for HIV-positive patients who are already immunocompromised; however, many HIV-positive patients have severe psoriatic disease that cannot be adequately treated with first- and second-line therapies including topical agents, phototherapy, or oral retinoids.
Our comprehensive review yielded a total of 25 reported cases of systemic immunosuppressive therapies used to treat psoriatic disease in HIV-positive patients including methotrexate, cyclosporine, etanercept, adalimumab, in-fliximab, and ustekinumab. Although data are limited to case reports and case series, some trends were observed.
Efficacy
In most of the cases reviewed, the patients had inadequate improvement of psoriatic disease with first- and second-line therapies, which included antiretrovirals alone, topical agents, phototherapy, and oral retinoids. Some cases reported poor response to methotrexate and cyclosporine.4-8 Biologic agents were effective in many such cases.
Safety
Overall, there were 11 cases in which the patient was not on adequate HAART while being treated with systemic immunosuppressive therapy for psoriatic disease.4-8,15 Of them, 3 were associated with serious infection while on methotrexate.5,6 There was only 1 report of serious infection13 of 14 cases in which the patient was on concomitant HAART. In this case, which reported polymicrobial infections and subsequent death of the patient, the infections continued after discontinuing etanercept; thus, the association is unclear. Interestingly, despite multiple infections, the CD4 and viral counts were stable throughout treatment with etanercept.13
From reviewing the 4 total cases5,6,13 of serious infection, HAART appears to be a valuable concomitant treatment during systemic immunosuppressive therapy for HIV-positive patients; however, it does not necessarily prevent serious infections from occurring, and thus the clinician’s diligence in monitoring for signs and symptoms of infection remains important.
CD4 and Viral Counts
Although reports of CD4 and viral counts were not available in earlier studies,4-8 there were 15 cases that reported consistent pretreatment and posttreatment CD4 and viral counts during treatment with etanercept, adalimumab, infliximab, and ustekinumab.9-20 In all cases, the CD4 count was stable or increased. Similarly, the viral count was stable or decreased. All patients, except 1 by Cepeda et al,15 were on concomitant HAART.9-14,16-20
Although data are limited, treatment of psoriatic disease with biologic agents when used in combination with HAART may have beneficial effects on CD4 and viral counts. Tumor necrosis factor has a role in HIV expression through the action of nuclear factor κβ.21 An increase in TNF levels is shown to be associated with increased viral count, decreased CD4 count, and increased symptoms of HIV progression, such as fever, fatigue, cachexia, and dementia.22 Although more studies are necessary, TNF-α inhibitors may have a positive effect on HIV while simultaneously treating psoriatic disease. Other cytokines (eg, IL-12, IL-23, IL-17) involved in the mechanism of action of other biologic agents (ustekinumab and secukinumab) have not been shown to be directly associated with HIV activity; however, studies have shown that IL-10 has a role in inhibiting HIV-1 replication and inhibits secretion of proinflammatory cytokines such as IL-12 and TNF-α.21 It may be speculated that the inhibition of IL-12 and TNF-α may create a positive feedback effect to increase IL-10, which in turn inhibits HIV replication.
Conclusion
Although there are limited data on the efficacy and safety of systemic immunosuppressive therapies for the treatment of psoriatic disease in HIV-positive patients, a review of 25 individual cases suggest that these treatments are not only required but also are sufficient to treat some of the most resistant cases. It is possible that with adequate concomitant HAART and monitoring for signs and symptoms of infection, the likelihood of serious infection may be low. Furthermore, biologic agents may have a positive effect over other systemic immunosuppressive agents, such as methotrexate and cyclosporine, in improving CD4 and viral counts when used in combination with HAART. Although randomized controlled trials are necessary, current biologic therapies such as etanercept, adalimumab, infliximab, and ustekinumab may be safe viable options as third-line treatment of severe psoriasis in the HIV-positive population.
The prevalence of psoriasis among human immunodeficiency virus (HIV)–positive patients in the United States is reported to be approximately 1% to 3%, which is similar to the rates reported for the general population.1 Recalcitrant cases of psoriasis in patients with no history of the condition can be the initial manifestation of HIV infection. In patients with preexisting psoriasis, a flare of their disease can be seen following infection, and progression of HIV correlates with worsening psoriasis.2 Psoriatic arthropathy also affects 23% to 50% of HIV-positive patients with psoriasis worldwide, which may be higher than the general population,1 with more severe joint disease.
The management of psoriatic disease in the HIV-positive population is challenging. The current first-line recommendations for treatment include topical therapies, phototherapy, and highly active antiretroviral therapy (HAART), followed by oral retinoids as second-line agents.3 However, the clinical course of psoriasis in HIV-positive patients often is progressive and refractory2; therefore, these therapies often are inadequate to control both skin and joint manifestations. Most other currently available systemic therapies for psoriatic disease are immunosuppressive, which poses a distinct clinical challenge because HIV-positive patients are already immunocompromised.
There currently are many systemic immunosuppressive agents used for the treatment of psoriatic disease, including oral agents (eg, methotrexate, hydroxyurea, cyclosporine), as well as newer biologic medications, including tumor necrosis factor (TNF) α inhibitors etanercept, adalimumab, infliximab, golimumab, and certolizumab pegol. Golimumab and certolizumab pegol currently are indicated for psoriatic arthritis only. Other newer biologic therapies include ustekinumab, which inhibits IL-12 and IL-23, and secukinumab, which inhibits IL-17A. The purpose of this systematic review is to evaluate the most current literature to explore the efficacy and safety data as they pertain to systemic immunosuppressive therapies for the treatment of psoriatic disease in HIV-positive individuals.
Methods
To investigate the efficacy and safety of systemic immunosuppressive therapies for psoriatic disease in HIV-positive individuals, a PubMed search of articles indexed for MEDLINE (1985-2015) was conducted using the terms psoriasis and HIV and psoriatic arthritis and HIV combined with each of the following systemic immunosuppressive agents: methotrexate, hydroxyurea, cyclosporine, etanercept, adalimumab, infliximab, golimumab, certolizumab pegol, ustekinumab, and secukinumab. Pediatric cases and articles that were not available in the English language were excluded.
For each case, patient demographic information (ie, age, sex), prior failed psoriasis treatments, and history of HAART were documented. The dosing regimen of the systemic agent was noted when different from the US Food and Drug administration–approved dosage for psoriasis or psoriatic arthritis. The duration of immunosuppressive therapy as well as pretreatment and posttreatment CD4 and viral counts (when available) were collected. The response to treatment and adverse effects were summarized.
Results
Our review of the literature yielded a total of 25 reported cases of systemic immunosuppressive therapies used to treat psoriatic disease in HIV-positive patients, including methotrexate, cyclosporine, etanercept, adalimumab, in-fliximab, and ustekinumab (Table). There were no reports of the use of hydroxyurea, golimumab, certolizumab pegol, or secukinumab to treat psoriatic disease in this patient population.
Methotrexate
Eight individual cases of methotrexate used to treat psoriasis and/or psoriatic arthritis in HIV-positive patients were reported.4-6 Duvic et al6 described 4 patients with psoriatic disease that was treated with methotrexate with varying efficacy. One patient developed toxic encephalopathy, which improved after discontinuation of methotrexate; however, he died 5 months later from pneumocystis pneumonia. In this early study, none of the 4 patients were on antiretroviral therapy for HIV.6
In the cases reported by Masson et al4 and Maurer et al,5 4 patients were treated with a single antiretroviral agent and received appropriate prophylaxis against opportunistic infections. In 1 case, methotrexate was given at a chemotherapeutic dose of 525 mg once weekly for Kaposi sarcoma.4 In 2 of 4 cases, the patients developed pneumocystis pneumonia.4,5
Cyclosporine
There were 2 case reports of successful treatment of psoriatic disease with cyclosporine in HIV-positive patients.7,8 Skin and joint manifestations improved rapidly without reports of infection for 27 and 8 years.8 Both patients were treated with one antiretroviral agent.7,8
Etanercept
There were 5 case reports of successful treatment of psoriatic disease with etanercept. In all 5 cases the patients were on HAART, and the CD4 count increased or remained stable and viral count became undetectable or remained stable following treatment.9-13 In 2 cases, the patient also had hepatitis C virus, which remained stable throughout the treatment period.9,12 The maximum duration of treatment was 6 years, with only 1 reported adverse event.13 In this case reported by Aboulafia et al,13 the patient experienced recurrent polymicrobial infections, including enterococcal cellulitis, cystitis, and bacteremia, as well as pseudomonas pneumonia and septic arthritis. Therapy was discontinued at 6 months. Four months after discontinuation of etanercept, the patient died from infectious causes.13
Adalimumab
There was 1 case of successful treatment of psoriatic disease with adalimumab in an HIV-positive patient. In this case, the patient was on HAART, and CD4 and viral counts improved substantially after 30 months of treatment.14
Infliximab
Six individual cases of successful treatment of psoriatic disease with infliximab were reported.15-17 In a report by Cepeda et al,15 HIV-positive patients with various rheumatologic diseases were chosen to receive etanercept followed by adalimumab and/or infliximab if clinical improvement was not observed on etanercept. In 3 patients with psoriasis and psoriatic arthritis, inadequate response was observed on etanercept. Two of these 3 patients received adalimumab with only partial response. All 3 were treated with infliximab in the end and showed excellent response. One of the patients experienced facial abscess responsive to antibiotics and was continued on infliximab therapy without further complications. In all 6 cases of infliximab therapy, the patients were on HAART, and CD4 and viral counts improved or remained stable.15
Ustekinumab
There were 3 case reports of successful treatment of psoriatic disease with ustekinumab in HIV-positive patients on HAART. CD4 and viral counts improved or remained stable.18-20
Comment
Currently, all of the systemic immunosuppressive therapies approved for psoriatic disease have a warning by the US Food and Drug Administration for increased risk of serious infection. Given such labels, these therapies are not routinely prescribed for HIV-positive patients who are already immunocompromised; however, many HIV-positive patients have severe psoriatic disease that cannot be adequately treated with first- and second-line therapies including topical agents, phototherapy, or oral retinoids.
Our comprehensive review yielded a total of 25 reported cases of systemic immunosuppressive therapies used to treat psoriatic disease in HIV-positive patients including methotrexate, cyclosporine, etanercept, adalimumab, in-fliximab, and ustekinumab. Although data are limited to case reports and case series, some trends were observed.
Efficacy
In most of the cases reviewed, the patients had inadequate improvement of psoriatic disease with first- and second-line therapies, which included antiretrovirals alone, topical agents, phototherapy, and oral retinoids. Some cases reported poor response to methotrexate and cyclosporine.4-8 Biologic agents were effective in many such cases.
Safety
Overall, there were 11 cases in which the patient was not on adequate HAART while being treated with systemic immunosuppressive therapy for psoriatic disease.4-8,15 Of them, 3 were associated with serious infection while on methotrexate.5,6 There was only 1 report of serious infection13 of 14 cases in which the patient was on concomitant HAART. In this case, which reported polymicrobial infections and subsequent death of the patient, the infections continued after discontinuing etanercept; thus, the association is unclear. Interestingly, despite multiple infections, the CD4 and viral counts were stable throughout treatment with etanercept.13
From reviewing the 4 total cases5,6,13 of serious infection, HAART appears to be a valuable concomitant treatment during systemic immunosuppressive therapy for HIV-positive patients; however, it does not necessarily prevent serious infections from occurring, and thus the clinician’s diligence in monitoring for signs and symptoms of infection remains important.
CD4 and Viral Counts
Although reports of CD4 and viral counts were not available in earlier studies,4-8 there were 15 cases that reported consistent pretreatment and posttreatment CD4 and viral counts during treatment with etanercept, adalimumab, infliximab, and ustekinumab.9-20 In all cases, the CD4 count was stable or increased. Similarly, the viral count was stable or decreased. All patients, except 1 by Cepeda et al,15 were on concomitant HAART.9-14,16-20
Although data are limited, treatment of psoriatic disease with biologic agents when used in combination with HAART may have beneficial effects on CD4 and viral counts. Tumor necrosis factor has a role in HIV expression through the action of nuclear factor κβ.21 An increase in TNF levels is shown to be associated with increased viral count, decreased CD4 count, and increased symptoms of HIV progression, such as fever, fatigue, cachexia, and dementia.22 Although more studies are necessary, TNF-α inhibitors may have a positive effect on HIV while simultaneously treating psoriatic disease. Other cytokines (eg, IL-12, IL-23, IL-17) involved in the mechanism of action of other biologic agents (ustekinumab and secukinumab) have not been shown to be directly associated with HIV activity; however, studies have shown that IL-10 has a role in inhibiting HIV-1 replication and inhibits secretion of proinflammatory cytokines such as IL-12 and TNF-α.21 It may be speculated that the inhibition of IL-12 and TNF-α may create a positive feedback effect to increase IL-10, which in turn inhibits HIV replication.
Conclusion
Although there are limited data on the efficacy and safety of systemic immunosuppressive therapies for the treatment of psoriatic disease in HIV-positive patients, a review of 25 individual cases suggest that these treatments are not only required but also are sufficient to treat some of the most resistant cases. It is possible that with adequate concomitant HAART and monitoring for signs and symptoms of infection, the likelihood of serious infection may be low. Furthermore, biologic agents may have a positive effect over other systemic immunosuppressive agents, such as methotrexate and cyclosporine, in improving CD4 and viral counts when used in combination with HAART. Although randomized controlled trials are necessary, current biologic therapies such as etanercept, adalimumab, infliximab, and ustekinumab may be safe viable options as third-line treatment of severe psoriasis in the HIV-positive population.
- Mallon
E, Bunker CB. HIV-associated psoriasis. AIDS Patient Care STDS. 2000;14:239-246. - Montaz
eri A, Kanitakis J, Bazex J. Psoriasis and HIV infection. Int J Dermatol. 1996;35:475-479. - Menon
K, Van Vorhees AS, Bebo BF, et al; National Psoriasis Foundation. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010;62:291-299. - Masso
n C, Chennebault JM, Leclech C. Is HIV infection contraindication to the use of methotrexate in psoriatic arthritis? J Rheumatol. 1995;22:2191. - Maurer
TA, Zackheim HS, Tuffanelli L, et al. The use of methotrexate for treatment of psoriasis in patients with HIV infection. J Am Acad Dermatol. 1994;31:372-375. - Duvic
M, Johnson TM, Rapini RP, et al. Acquired immunodeficiency syndrome-associated psoriasis and Reiter’s syndrome. Arch Dermatol. 1987;123:1622-1632. - Tourne
L, Durez P, Van Vooren JP, et al. Alleviation of HIV-associated psoriasis and psoriatic arthritis with cyclosporine. J Am Acad Dermatol. 1997;37:501-502. - Allen
BR. Use of cyclosporine for psoriasis in HIV-positive patient. Lancet. 1992;339:686. - Di Ler
nia V, Zoboli G, Ficarelli E. Long-term management of HIV/hepatitis C virus associated psoriasis with etanercept. Indian J Dermatol Venereol Leprol. 2013;79:444. - Lee E
S, Heller MM, Kamangar F, et al. Long-term etanercept use for severe generalized psoriasis in an HIV-infected individual: a case study. J Drugs Dermatol. 2012;11:413-414. - Mikha
il M, Weinberg JM, Smith BL. Successful treatment with etanercept of von Zumbusch pustular psoriasis in a patient with human immunodeficiency virus. Arch Dermatol. 2008;144:453-456. - Linar
daki G, Katsarou O, Ioannidou P, et al. Effective etanercept treatment for psoriatic arthritis complicating concomitant human immunodeficiency virus and hepatitis C virus infection. J Rheumatol. 2007;34:1353-1355. - Aboul
afia DM, Bundow D, Wilske K, et al. Etanercept for the treatment of human immunodeficiency virus-associated psoriatic arthritis. Mayo Clin Proc. 2000;75:1093-1098. - Linds
ey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871. - Ceped
a EJ, Williams FM, Ishimori ML, et al. The use of anti-tumor necrosis factor therapy in HIV-positive individuals with rheumatic disease. Ann Rheum Dis. 2008;67:710-712. - Sella
m J, Bouvard B, Masson C, et al. Use of infliximab to treat psoriatic arthritis in HIV-positive patients. Joint Bone Spine. 2007;74:197-200. - Bartk
e U, Venten I, Kreuter A, et al. Human immunodeficiency virus-associated psoriasis and psoriatic arthritis treated with infliximab. Br J Dermatol. 2004;150:784-786. - Saeki
H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655. - Wiede
r S, Routt E, Levitt J, et al. Treatment of refractory psoriasis with ustekinumab in an HIV-positive patient: a case presentation and review of the biologic literature. Psoriasis Forum. 2014;20:96-102. - Papar
izos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatol Treat. 2012;23:398-399. - Kedzierska K, Crowe SM, Turville S, et al. The influence of cytokines, chemokines, and their receptors on HIV-1 replication in monocytes and macrophages. Rev Med Virol. 2003;13:39-56.
- Emer JJ. Is there a potential role for anti-tumor necrosis factor therapy in patients with human immunodeficiency virus? J Clin Aesthet Dermatol. 2009;2:29-35.
- Mallon
E, Bunker CB. HIV-associated psoriasis. AIDS Patient Care STDS. 2000;14:239-246. - Montaz
eri A, Kanitakis J, Bazex J. Psoriasis and HIV infection. Int J Dermatol. 1996;35:475-479. - Menon
K, Van Vorhees AS, Bebo BF, et al; National Psoriasis Foundation. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation. J Am Acad Dermatol. 2010;62:291-299. - Masso
n C, Chennebault JM, Leclech C. Is HIV infection contraindication to the use of methotrexate in psoriatic arthritis? J Rheumatol. 1995;22:2191. - Maurer
TA, Zackheim HS, Tuffanelli L, et al. The use of methotrexate for treatment of psoriasis in patients with HIV infection. J Am Acad Dermatol. 1994;31:372-375. - Duvic
M, Johnson TM, Rapini RP, et al. Acquired immunodeficiency syndrome-associated psoriasis and Reiter’s syndrome. Arch Dermatol. 1987;123:1622-1632. - Tourne
L, Durez P, Van Vooren JP, et al. Alleviation of HIV-associated psoriasis and psoriatic arthritis with cyclosporine. J Am Acad Dermatol. 1997;37:501-502. - Allen
BR. Use of cyclosporine for psoriasis in HIV-positive patient. Lancet. 1992;339:686. - Di Ler
nia V, Zoboli G, Ficarelli E. Long-term management of HIV/hepatitis C virus associated psoriasis with etanercept. Indian J Dermatol Venereol Leprol. 2013;79:444. - Lee E
S, Heller MM, Kamangar F, et al. Long-term etanercept use for severe generalized psoriasis in an HIV-infected individual: a case study. J Drugs Dermatol. 2012;11:413-414. - Mikha
il M, Weinberg JM, Smith BL. Successful treatment with etanercept of von Zumbusch pustular psoriasis in a patient with human immunodeficiency virus. Arch Dermatol. 2008;144:453-456. - Linar
daki G, Katsarou O, Ioannidou P, et al. Effective etanercept treatment for psoriatic arthritis complicating concomitant human immunodeficiency virus and hepatitis C virus infection. J Rheumatol. 2007;34:1353-1355. - Aboul
afia DM, Bundow D, Wilske K, et al. Etanercept for the treatment of human immunodeficiency virus-associated psoriatic arthritis. Mayo Clin Proc. 2000;75:1093-1098. - Linds
ey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871. - Ceped
a EJ, Williams FM, Ishimori ML, et al. The use of anti-tumor necrosis factor therapy in HIV-positive individuals with rheumatic disease. Ann Rheum Dis. 2008;67:710-712. - Sella
m J, Bouvard B, Masson C, et al. Use of infliximab to treat psoriatic arthritis in HIV-positive patients. Joint Bone Spine. 2007;74:197-200. - Bartk
e U, Venten I, Kreuter A, et al. Human immunodeficiency virus-associated psoriasis and psoriatic arthritis treated with infliximab. Br J Dermatol. 2004;150:784-786. - Saeki
H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655. - Wiede
r S, Routt E, Levitt J, et al. Treatment of refractory psoriasis with ustekinumab in an HIV-positive patient: a case presentation and review of the biologic literature. Psoriasis Forum. 2014;20:96-102. - Papar
izos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatol Treat. 2012;23:398-399. - Kedzierska K, Crowe SM, Turville S, et al. The influence of cytokines, chemokines, and their receptors on HIV-1 replication in monocytes and macrophages. Rev Med Virol. 2003;13:39-56.
- Emer JJ. Is there a potential role for anti-tumor necrosis factor therapy in patients with human immunodeficiency virus? J Clin Aesthet Dermatol. 2009;2:29-35.
Practice Points
- There are limited data on the use of systemic immunosuppressive therapies for the treatment of psoriatic disease in human immunodeficiency virus–positive patients.
- The limited data suggest that biologic therapies may be effective for cases of psoriasis recalcitrant to other systemic agents and may have a positive effect on CD4 and viral counts when used in combination with highly active antiretroviral therapy.
- Further research is needed.
A Review of Neurologic Complications of Biologic Therapy in Plaque Psoriasis
Biologic agents have provided patients with moderate to severe psoriasis with treatment alternatives that have improved systemic safety profiles and disease control1; however, case reports of associated neurologic complications have been emerging. Tumor necrosis factor α (TNF-α) inhibitors have been associated with central and peripheral demyelinating disorders. Notably, efalizumab was withdrawn from the market for its association with fatal cases of progressive multifocal leukoencephalopathy (PML).2,3 It is imperative for dermatologists to be familiar with the clinical presentation, evaluation, and diagnostic criteria of neurologic complications of biologic agents used in the treatment of psoriasis.
Leukoencephalopathy
Progressive multifocal leukoencephalopathy is a fatal demyelinating neurodegenerative disease caused by reactivation of the ubiquitous John Cunningham virus. Primary asymptomatic infection is thought to occur during childhood, then the virus remains latent. Reactivation usually occurs during severe immunosuppression and is classically described in human immunodeficiency virus infection, lymphoproliferative disorders, and other forms of cancer.4 A summary of PML and its association with biologics is found in Table 1.5-13 Few case reports of TNF-α inhibitor–associated PML exist, mostly in the presence of confounding factors such as immunosuppression or underlying autoimmune disease.10-13 Presenting symptoms of PML often are subacute, rapidly progressive, and can be focal or multifocal and include motor, cognitive, and visual deficits. Of note, there are 2 reported cases of ustekinumab associated with reversible posterior leukoencephalopathy syndrome, which is a hypertensive encephalopathy characterized by headache, altered mental status, vision abnormalities, and seizures.14,15 Fortunately, this disease is reversible with blood pressure control and removal of the immunosuppressive agent.16

Demyelinating Disorders
Clinical presentation of demyelinating events associated with biologic agents are varied but include optic neuritis, multiple sclerosis, transverse myelitis, and Guillain-Barré syndrome, among others.17-28 These demyelinating disorders with their salient features and associated biologics are summarized in Table 2.17-20,22-28 Patients on biologic agents, especially TNF-α inhibitors, with new-onset visual, motor, or sensory changes warrant closer inspection. Currently, there are no data on any neurologic side effects occurring with the new biologic secukinumab.29
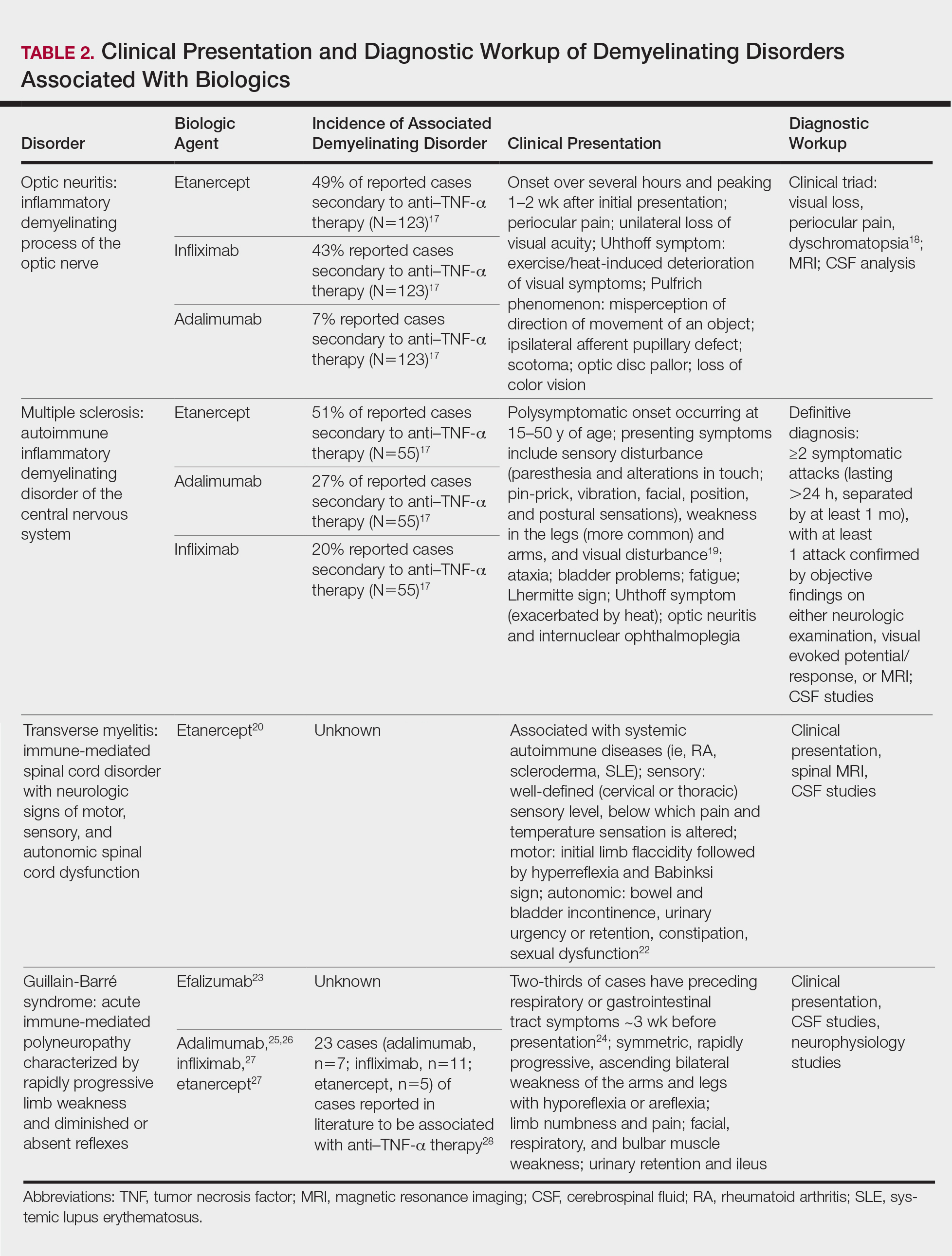
Conclusion
Biologic agents are effective in treating moderate to severe plaque psoriasis, but awareness of associated neurological adverse effects, though rare, is important to consider. Physicians need to be able to counsel patients concerning these risks and promote informed decision-making prior to initiating biologics. Patients with a personal or strong family history of demyelinating disease should be considered for alternative treatment options before initiating anti–TNF-α therapy. Since the withdrawal of efalizumab, no new cases of PML have been reported in patients who were previously on a long-term course. Dermatologists should be vigilant in detecting signs of neurological complications so that an expedited evaluation and neurology referral may prevent progression of disease.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- FDA Statement on the Voluntary Withdrawal of Raptiva From the U.S. Market. US Food and Drug Administration website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm143347.htm. Published April 8, 2009. Accessed December 21, 2017.
- Kothary N, Diak IL, Brinker A, et al. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol. 2011;65:546-551.
- Tavazzi E, Ferrante P, Khalili K. Progressive multifocal leukoencephalopathy: an unexpected complication of modern therapeutic monoclonal antibody therapies. Clin Microbiol Infect. 2011;17:1776-1780.
- Korman BD, Tyler KL, Korman NJ. Progressive multifocal leukoencephalopathy, efalizumab, and immunosuppression: a cautionary tale for dermatologists. Arch Dermatol. 2009;145:937-942.
- Sudhakar P, Bachman DM, Mark AS, et al. Progressive multifocal leukoencephalopathy: recent advances and a neuro-ophthalmological review. J Neuroophthalmol. 2015;35:296-305.
- Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology. 2013;80:1430-1438.
- Koralnik IJ, Boden D, Mai VX, et al. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology. 1999;52:253-260.
- Clifford DB, Ances B, Costello C, et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol. 2011;68:1156-1164.
- Babi MA, Pendlebury W, Braff S, et al. JC virus PCR detection is not infallible: a fulminant case of progressive multifocal leukoencephalopathy with false-negative cerebrospinal fluid studies despite progressive clinical course and radiological findings [published online March 12, 2015]. Case Rep Neurol Med. 2015;2015:643216.
- Ray M, Curtis JR, Baddley JW. A case report of progressive multifocal leucoencephalopathy (PML) associated with adalimumab. Ann Rheum Dis. 2014;73:1429-1430.
- Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leukoencephalopathy in a patient treated with infliximab. Arthritis Rheum. 2010;62:3191-3195.
- Graff-Radford J, Robinson MT, Warsame RM, et al. Progressive multifocal leukoencephalopathy in a patient treated with etanercept. Neurologist. 2012;18:85-87.
- Dickson L, Menter A. Reversible posterior leukoencephalopathy syndrome (RPLS) in a psoriasis patient treated with ustekinumab. J Drugs Dermatol. 2017;16:177-179.
- Gratton D, Szapary P, Goyal K, et al. Reversible posterior leukoencephalopathy syndrome in a patient treated with ustekinumab: case report and review of the literature. Arch Dermatol. 2011;147:1197-1202.
- Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334:494-500.
- Ramos-Casals M, Roberto-Perez A, Diaz-Lagares C, et al. Autoimmune diseases induced by biological agents: a double-edged sword? Autoimmun Rev. 2010;9:188-193.
- Hoorbakht H, Bagherkashi F. Optic neuritis, its differential diagnosis and management. Open Ophthalmol J. 2012;6:65-72.
- Richards RG, Sampson FC, Beard SM, et al. A review of the natural history and epidemiology of multiple sclerosis: implications for resource allocation and health economic models. Health Technol Assess. 2002;6:1-73.
- Caracseghi F, Izquierdo-Blasco J, Sanchez-Montanez A, et al. Etanercept-induced myelopathy in a pediatric case of blau syndrome [published online January 15, 2012]. Case Rep Rheumatol. 2011;2011:134106.
- Fromont A, De Seze J, Fleury MC, et al. Inflammatory demyelinating events following treatment with anti-tumor necrosis factor. Cytokine. 2009;45:55-57.
- Sellner J, Lüthi N, Schüpbach WM, et al. Diagnostic workup of patients with acute transverse myelitis: spectrum of clinical presentation, neuroimaging and laboratory findings. Spinal Cord. 2009;47:312-317.
- Turatti M, Tamburin S, Idone D, et al. Guillain-Barré syndrome after short-course efalizumab treatment. J Neurol. 2010;257:1404-1405.
- Koga M, Yuki N, Hirata K. Antecedent symptoms in Guillain-Barré syndrome: an important indicator for clinical and serological subgroups. Acta Neurol Scand. 2001;103:278-287.
- Cesarini M, Angelucci E, Foglietta T, et al. Guillain-Barré syndrome after treatment with human anti-tumor necrosis factor alpha (adalimumab) in a Crohn’s disease patient: case report and literature review [published online July 28, 2011]. J Crohns Colitis. 2011;5:619-622.
- Soto-Cabrera E, Hernández-Martínez A, Yañez H, et al. Guillain-Barré syndrome. Its association with alpha tumor necrosis factor [in Spanish]. Rev Med Inst Mex Seguro Soc. 2012;50:565-567.
- Shin IS, Baer AN, Kwon HJ, et al. Guillain-Barré and Miller Fisher syndromes occurring with tumor necrosis factor alpha antagonist therapy. Arthritis Rheum. 2006;54:1429-1434.
- Alvarez-Lario B, Prieto-Tejedo R, Colazo-Burlato M, et al. Severe Guillain-Barré syndrome in a patient receiving anti-TNF therapy. consequence or coincidence. a case-based review. Clin Rheumatol. 2013;32:1407-1412.
- Garnock-Jones KP. Secukinumab: a review in moderate to severe plaque psoriasis. Am J Clin Dermatol. 2015;16:323-330.
Biologic agents have provided patients with moderate to severe psoriasis with treatment alternatives that have improved systemic safety profiles and disease control1; however, case reports of associated neurologic complications have been emerging. Tumor necrosis factor α (TNF-α) inhibitors have been associated with central and peripheral demyelinating disorders. Notably, efalizumab was withdrawn from the market for its association with fatal cases of progressive multifocal leukoencephalopathy (PML).2,3 It is imperative for dermatologists to be familiar with the clinical presentation, evaluation, and diagnostic criteria of neurologic complications of biologic agents used in the treatment of psoriasis.
Leukoencephalopathy
Progressive multifocal leukoencephalopathy is a fatal demyelinating neurodegenerative disease caused by reactivation of the ubiquitous John Cunningham virus. Primary asymptomatic infection is thought to occur during childhood, then the virus remains latent. Reactivation usually occurs during severe immunosuppression and is classically described in human immunodeficiency virus infection, lymphoproliferative disorders, and other forms of cancer.4 A summary of PML and its association with biologics is found in Table 1.5-13 Few case reports of TNF-α inhibitor–associated PML exist, mostly in the presence of confounding factors such as immunosuppression or underlying autoimmune disease.10-13 Presenting symptoms of PML often are subacute, rapidly progressive, and can be focal or multifocal and include motor, cognitive, and visual deficits. Of note, there are 2 reported cases of ustekinumab associated with reversible posterior leukoencephalopathy syndrome, which is a hypertensive encephalopathy characterized by headache, altered mental status, vision abnormalities, and seizures.14,15 Fortunately, this disease is reversible with blood pressure control and removal of the immunosuppressive agent.16
Demyelinating Disorders
Clinical presentation of demyelinating events associated with biologic agents are varied but include optic neuritis, multiple sclerosis, transverse myelitis, and Guillain-Barré syndrome, among others.17-28 These demyelinating disorders with their salient features and associated biologics are summarized in Table 2.17-20,22-28 Patients on biologic agents, especially TNF-α inhibitors, with new-onset visual, motor, or sensory changes warrant closer inspection. Currently, there are no data on any neurologic side effects occurring with the new biologic secukinumab.29
Conclusion
Biologic agents are effective in treating moderate to severe plaque psoriasis, but awareness of associated neurological adverse effects, though rare, is important to consider. Physicians need to be able to counsel patients concerning these risks and promote informed decision-making prior to initiating biologics. Patients with a personal or strong family history of demyelinating disease should be considered for alternative treatment options before initiating anti–TNF-α therapy. Since the withdrawal of efalizumab, no new cases of PML have been reported in patients who were previously on a long-term course. Dermatologists should be vigilant in detecting signs of neurological complications so that an expedited evaluation and neurology referral may prevent progression of disease.
Biologic agents have provided patients with moderate to severe psoriasis with treatment alternatives that have improved systemic safety profiles and disease control1; however, case reports of associated neurologic complications have been emerging. Tumor necrosis factor α (TNF-α) inhibitors have been associated with central and peripheral demyelinating disorders. Notably, efalizumab was withdrawn from the market for its association with fatal cases of progressive multifocal leukoencephalopathy (PML).2,3 It is imperative for dermatologists to be familiar with the clinical presentation, evaluation, and diagnostic criteria of neurologic complications of biologic agents used in the treatment of psoriasis.
Leukoencephalopathy
Progressive multifocal leukoencephalopathy is a fatal demyelinating neurodegenerative disease caused by reactivation of the ubiquitous John Cunningham virus. Primary asymptomatic infection is thought to occur during childhood, then the virus remains latent. Reactivation usually occurs during severe immunosuppression and is classically described in human immunodeficiency virus infection, lymphoproliferative disorders, and other forms of cancer.4 A summary of PML and its association with biologics is found in Table 1.5-13 Few case reports of TNF-α inhibitor–associated PML exist, mostly in the presence of confounding factors such as immunosuppression or underlying autoimmune disease.10-13 Presenting symptoms of PML often are subacute, rapidly progressive, and can be focal or multifocal and include motor, cognitive, and visual deficits. Of note, there are 2 reported cases of ustekinumab associated with reversible posterior leukoencephalopathy syndrome, which is a hypertensive encephalopathy characterized by headache, altered mental status, vision abnormalities, and seizures.14,15 Fortunately, this disease is reversible with blood pressure control and removal of the immunosuppressive agent.16
Demyelinating Disorders
Clinical presentation of demyelinating events associated with biologic agents are varied but include optic neuritis, multiple sclerosis, transverse myelitis, and Guillain-Barré syndrome, among others.17-28 These demyelinating disorders with their salient features and associated biologics are summarized in Table 2.17-20,22-28 Patients on biologic agents, especially TNF-α inhibitors, with new-onset visual, motor, or sensory changes warrant closer inspection. Currently, there are no data on any neurologic side effects occurring with the new biologic secukinumab.29
Conclusion
Biologic agents are effective in treating moderate to severe plaque psoriasis, but awareness of associated neurological adverse effects, though rare, is important to consider. Physicians need to be able to counsel patients concerning these risks and promote informed decision-making prior to initiating biologics. Patients with a personal or strong family history of demyelinating disease should be considered for alternative treatment options before initiating anti–TNF-α therapy. Since the withdrawal of efalizumab, no new cases of PML have been reported in patients who were previously on a long-term course. Dermatologists should be vigilant in detecting signs of neurological complications so that an expedited evaluation and neurology referral may prevent progression of disease.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- FDA Statement on the Voluntary Withdrawal of Raptiva From the U.S. Market. US Food and Drug Administration website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm143347.htm. Published April 8, 2009. Accessed December 21, 2017.
- Kothary N, Diak IL, Brinker A, et al. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol. 2011;65:546-551.
- Tavazzi E, Ferrante P, Khalili K. Progressive multifocal leukoencephalopathy: an unexpected complication of modern therapeutic monoclonal antibody therapies. Clin Microbiol Infect. 2011;17:1776-1780.
- Korman BD, Tyler KL, Korman NJ. Progressive multifocal leukoencephalopathy, efalizumab, and immunosuppression: a cautionary tale for dermatologists. Arch Dermatol. 2009;145:937-942.
- Sudhakar P, Bachman DM, Mark AS, et al. Progressive multifocal leukoencephalopathy: recent advances and a neuro-ophthalmological review. J Neuroophthalmol. 2015;35:296-305.
- Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology. 2013;80:1430-1438.
- Koralnik IJ, Boden D, Mai VX, et al. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology. 1999;52:253-260.
- Clifford DB, Ances B, Costello C, et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol. 2011;68:1156-1164.
- Babi MA, Pendlebury W, Braff S, et al. JC virus PCR detection is not infallible: a fulminant case of progressive multifocal leukoencephalopathy with false-negative cerebrospinal fluid studies despite progressive clinical course and radiological findings [published online March 12, 2015]. Case Rep Neurol Med. 2015;2015:643216.
- Ray M, Curtis JR, Baddley JW. A case report of progressive multifocal leucoencephalopathy (PML) associated with adalimumab. Ann Rheum Dis. 2014;73:1429-1430.
- Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leukoencephalopathy in a patient treated with infliximab. Arthritis Rheum. 2010;62:3191-3195.
- Graff-Radford J, Robinson MT, Warsame RM, et al. Progressive multifocal leukoencephalopathy in a patient treated with etanercept. Neurologist. 2012;18:85-87.
- Dickson L, Menter A. Reversible posterior leukoencephalopathy syndrome (RPLS) in a psoriasis patient treated with ustekinumab. J Drugs Dermatol. 2017;16:177-179.
- Gratton D, Szapary P, Goyal K, et al. Reversible posterior leukoencephalopathy syndrome in a patient treated with ustekinumab: case report and review of the literature. Arch Dermatol. 2011;147:1197-1202.
- Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334:494-500.
- Ramos-Casals M, Roberto-Perez A, Diaz-Lagares C, et al. Autoimmune diseases induced by biological agents: a double-edged sword? Autoimmun Rev. 2010;9:188-193.
- Hoorbakht H, Bagherkashi F. Optic neuritis, its differential diagnosis and management. Open Ophthalmol J. 2012;6:65-72.
- Richards RG, Sampson FC, Beard SM, et al. A review of the natural history and epidemiology of multiple sclerosis: implications for resource allocation and health economic models. Health Technol Assess. 2002;6:1-73.
- Caracseghi F, Izquierdo-Blasco J, Sanchez-Montanez A, et al. Etanercept-induced myelopathy in a pediatric case of blau syndrome [published online January 15, 2012]. Case Rep Rheumatol. 2011;2011:134106.
- Fromont A, De Seze J, Fleury MC, et al. Inflammatory demyelinating events following treatment with anti-tumor necrosis factor. Cytokine. 2009;45:55-57.
- Sellner J, Lüthi N, Schüpbach WM, et al. Diagnostic workup of patients with acute transverse myelitis: spectrum of clinical presentation, neuroimaging and laboratory findings. Spinal Cord. 2009;47:312-317.
- Turatti M, Tamburin S, Idone D, et al. Guillain-Barré syndrome after short-course efalizumab treatment. J Neurol. 2010;257:1404-1405.
- Koga M, Yuki N, Hirata K. Antecedent symptoms in Guillain-Barré syndrome: an important indicator for clinical and serological subgroups. Acta Neurol Scand. 2001;103:278-287.
- Cesarini M, Angelucci E, Foglietta T, et al. Guillain-Barré syndrome after treatment with human anti-tumor necrosis factor alpha (adalimumab) in a Crohn’s disease patient: case report and literature review [published online July 28, 2011]. J Crohns Colitis. 2011;5:619-622.
- Soto-Cabrera E, Hernández-Martínez A, Yañez H, et al. Guillain-Barré syndrome. Its association with alpha tumor necrosis factor [in Spanish]. Rev Med Inst Mex Seguro Soc. 2012;50:565-567.
- Shin IS, Baer AN, Kwon HJ, et al. Guillain-Barré and Miller Fisher syndromes occurring with tumor necrosis factor alpha antagonist therapy. Arthritis Rheum. 2006;54:1429-1434.
- Alvarez-Lario B, Prieto-Tejedo R, Colazo-Burlato M, et al. Severe Guillain-Barré syndrome in a patient receiving anti-TNF therapy. consequence or coincidence. a case-based review. Clin Rheumatol. 2013;32:1407-1412.
- Garnock-Jones KP. Secukinumab: a review in moderate to severe plaque psoriasis. Am J Clin Dermatol. 2015;16:323-330.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- FDA Statement on the Voluntary Withdrawal of Raptiva From the U.S. Market. US Food and Drug Administration website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm143347.htm. Published April 8, 2009. Accessed December 21, 2017.
- Kothary N, Diak IL, Brinker A, et al. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol. 2011;65:546-551.
- Tavazzi E, Ferrante P, Khalili K. Progressive multifocal leukoencephalopathy: an unexpected complication of modern therapeutic monoclonal antibody therapies. Clin Microbiol Infect. 2011;17:1776-1780.
- Korman BD, Tyler KL, Korman NJ. Progressive multifocal leukoencephalopathy, efalizumab, and immunosuppression: a cautionary tale for dermatologists. Arch Dermatol. 2009;145:937-942.
- Sudhakar P, Bachman DM, Mark AS, et al. Progressive multifocal leukoencephalopathy: recent advances and a neuro-ophthalmological review. J Neuroophthalmol. 2015;35:296-305.
- Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology. 2013;80:1430-1438.
- Koralnik IJ, Boden D, Mai VX, et al. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology. 1999;52:253-260.
- Clifford DB, Ances B, Costello C, et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol. 2011;68:1156-1164.
- Babi MA, Pendlebury W, Braff S, et al. JC virus PCR detection is not infallible: a fulminant case of progressive multifocal leukoencephalopathy with false-negative cerebrospinal fluid studies despite progressive clinical course and radiological findings [published online March 12, 2015]. Case Rep Neurol Med. 2015;2015:643216.
- Ray M, Curtis JR, Baddley JW. A case report of progressive multifocal leucoencephalopathy (PML) associated with adalimumab. Ann Rheum Dis. 2014;73:1429-1430.
- Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leukoencephalopathy in a patient treated with infliximab. Arthritis Rheum. 2010;62:3191-3195.
- Graff-Radford J, Robinson MT, Warsame RM, et al. Progressive multifocal leukoencephalopathy in a patient treated with etanercept. Neurologist. 2012;18:85-87.
- Dickson L, Menter A. Reversible posterior leukoencephalopathy syndrome (RPLS) in a psoriasis patient treated with ustekinumab. J Drugs Dermatol. 2017;16:177-179.
- Gratton D, Szapary P, Goyal K, et al. Reversible posterior leukoencephalopathy syndrome in a patient treated with ustekinumab: case report and review of the literature. Arch Dermatol. 2011;147:1197-1202.
- Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334:494-500.
- Ramos-Casals M, Roberto-Perez A, Diaz-Lagares C, et al. Autoimmune diseases induced by biological agents: a double-edged sword? Autoimmun Rev. 2010;9:188-193.
- Hoorbakht H, Bagherkashi F. Optic neuritis, its differential diagnosis and management. Open Ophthalmol J. 2012;6:65-72.
- Richards RG, Sampson FC, Beard SM, et al. A review of the natural history and epidemiology of multiple sclerosis: implications for resource allocation and health economic models. Health Technol Assess. 2002;6:1-73.
- Caracseghi F, Izquierdo-Blasco J, Sanchez-Montanez A, et al. Etanercept-induced myelopathy in a pediatric case of blau syndrome [published online January 15, 2012]. Case Rep Rheumatol. 2011;2011:134106.
- Fromont A, De Seze J, Fleury MC, et al. Inflammatory demyelinating events following treatment with anti-tumor necrosis factor. Cytokine. 2009;45:55-57.
- Sellner J, Lüthi N, Schüpbach WM, et al. Diagnostic workup of patients with acute transverse myelitis: spectrum of clinical presentation, neuroimaging and laboratory findings. Spinal Cord. 2009;47:312-317.
- Turatti M, Tamburin S, Idone D, et al. Guillain-Barré syndrome after short-course efalizumab treatment. J Neurol. 2010;257:1404-1405.
- Koga M, Yuki N, Hirata K. Antecedent symptoms in Guillain-Barré syndrome: an important indicator for clinical and serological subgroups. Acta Neurol Scand. 2001;103:278-287.
- Cesarini M, Angelucci E, Foglietta T, et al. Guillain-Barré syndrome after treatment with human anti-tumor necrosis factor alpha (adalimumab) in a Crohn’s disease patient: case report and literature review [published online July 28, 2011]. J Crohns Colitis. 2011;5:619-622.
- Soto-Cabrera E, Hernández-Martínez A, Yañez H, et al. Guillain-Barré syndrome. Its association with alpha tumor necrosis factor [in Spanish]. Rev Med Inst Mex Seguro Soc. 2012;50:565-567.
- Shin IS, Baer AN, Kwon HJ, et al. Guillain-Barré and Miller Fisher syndromes occurring with tumor necrosis factor alpha antagonist therapy. Arthritis Rheum. 2006;54:1429-1434.
- Alvarez-Lario B, Prieto-Tejedo R, Colazo-Burlato M, et al. Severe Guillain-Barré syndrome in a patient receiving anti-TNF therapy. consequence or coincidence. a case-based review. Clin Rheumatol. 2013;32:1407-1412.
- Garnock-Jones KP. Secukinumab: a review in moderate to severe plaque psoriasis. Am J Clin Dermatol. 2015;16:323-330.
Practice Points
- Patients with a personal or strong family history of demyelinating disease should be considered for alternative treatment options before initiating anti–tumor necrosis factor (TNF) α therapy.
- Patients on biologic agents, especially TNF-α inhibitors, with subacute or rapidly progressive visual, motor, or sensory changes or a single neurologic deficit may warrant referral to neurology and/or neuroimaging.