User login
Diagnosing Multiple Myeloma in Primary Care
IN THIS ARTICLE
- Presenting symptoms
- Diagnostic tests
- Differential diagnostic criteria
Multiple myeloma (MM) is a fatal, malignant neoplasm that originates in the plasma cells of bone marrow. A genetic mutation in the plasma cells creates myeloma cells, which replicate and produce monoclonal protein (M-protein). This accumulation of cells and abnormal protein can result in destruction and eventual marrow failure.1,2
MM’s insidious nature means it often goes undetected or misdiagnosed in its early stages; this delayed diagnosis can cause sequelae that limit quality of life. Furthermore, the five-year survival rate for myeloma varies by stage at which the disease is diagnosed: from 48% for distant (metastasized) myeloma to 71% for localized disease.3 It has also been noted that, in the past two decades, improvements in available treatment options and supportive care have contributed to a doubling of median survival time (from three years to six years).4 It is therefore paramount that providers be aware of MM and its signs to facilitate early diagnosis and treatment.
INCIDENCE AND EPIDEMIOLOGY
MM accounts for 1% of all cancers and about 10% of all hematologic malignancies.5 In 2017, the American Cancer Society estimated that more than 30,000 new cases of MM would be diagnosed in the United States.6 Additionally, MM was expected to cause more than 12,000 deaths last year.6
Median age at diagnosis is 69.3 In fact, 75% of men are older than 75 and 79% of women are older than 70 at diagnosis.1
Apart from age, other risk factors for MM have been identified but not fully explicated. For example, the disease is more common in men than in women (with men comprising two-thirds of new cases per year).3 MM is also two to three times more common in black than in white persons, making it the most common hematologic malignancy in this demographic group.3,7
The possibility of a genetic predisposition has also been studied. Several analyses have indicated an increased risk for MM in patients with a family history of the disease—as much as four times higher in those with an affected first-degree relative. This risk was further elevated in black compared with white patients (odds ratios, 17.4 and 1.5, respectively).7 However, many patients with MM have no relatives with this disorder.6,8
DISEASE PROGRESSION
Almost all patients who develop MM also experience an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance (MGUS). MGUS is present in 3% to 4% of the general population older than 50 and is often an incidental finding. This stage almost always precedes MM—but because it is asymptomatic, only 10% of individuals diagnosed with MM have a known history of MGUS.8
In some patients, an asymptomatic intermediate stage called smoldering multiple myeloma (SMM) can be identified. SMM progresses to MM at a rate of 10% per year for the first five years; the rate decreases to 3% per year over the following five years, and 1% per year after that.8
MM is not curable, but as noted, the survival rate is steadily increasing due to rapidly evolving treatment regimens. Discussion of treatment is outside the scope of this article, but early diagnosis can improve quality of life and clinical outcomes and prolong life expectancy.
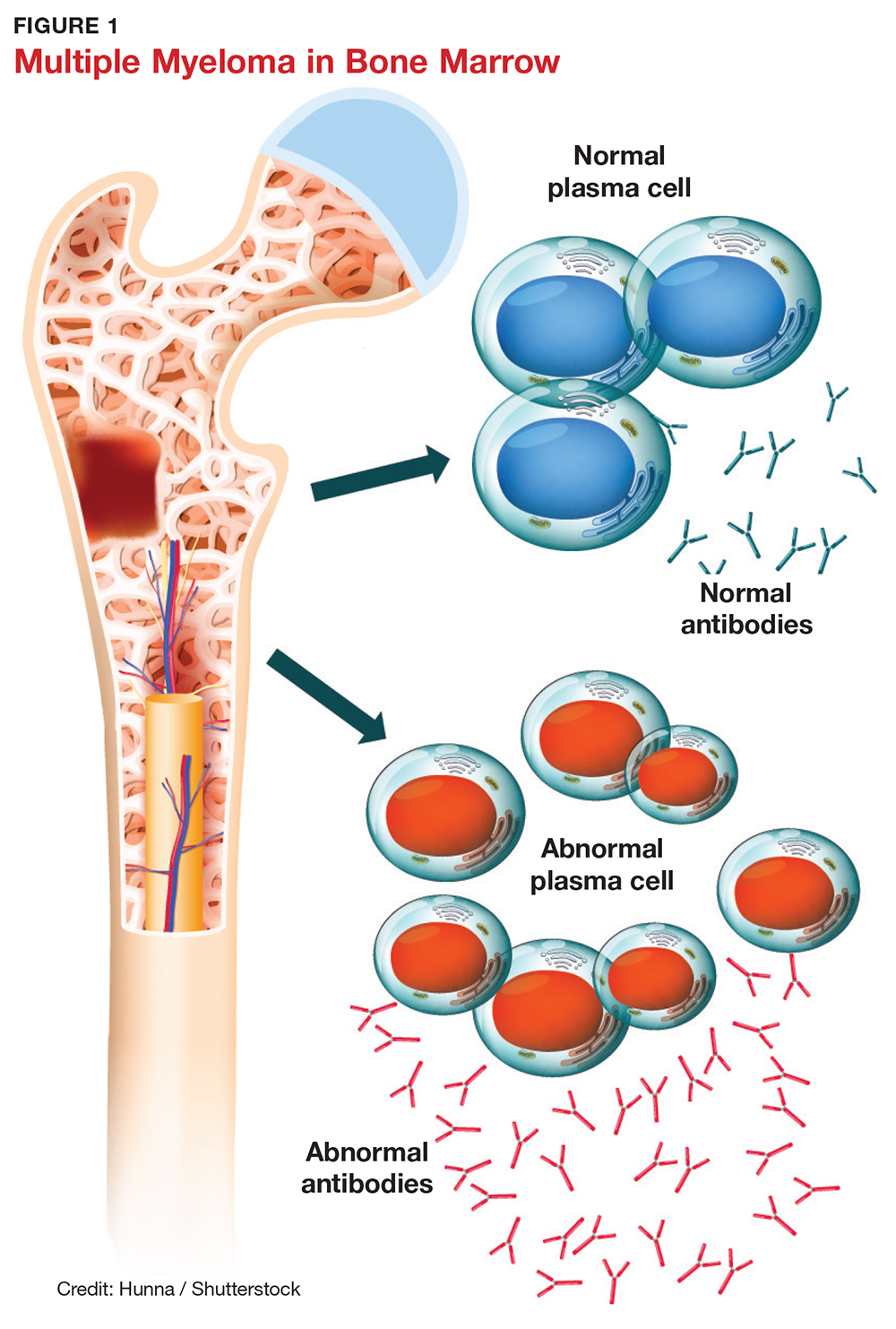
SYMPTOMS
The initial symptoms of MM can be nonspecific and may lead the provider to suspect a host of other conditions.2,6 (Those for advanced disease are also vague but tend to be more pronounced.) These may include fatigue, weakness, easy bruising or bleeding, and bone pain. Other common clinical manifestations of MM are anemia, chronic infection, bone disease, and/or renal failure.1,4 Patients may also experience loss of appetite, nausea, vomiting, increased thirst, and increased urination.9
Recent studies have shown that patients with SMM and/or MGUS also exhibit early signs of bone disease and increased risk for fracture.10 Eighty percent of patients who progress to MM have evidence of pathologic bone fractures.10 It is also possible for bones in the spine to weaken and collapse, pressing on the spinal nerves. This is known as spinal cord compression, which can manifest with sudden, severe back pain or numbness and/or muscle weakness (most often in the legs).6
MM must be included in the differential diagnosis, particularly when symptoms do not point to one specific disease process. Without early diagnosis, disease progression can result in complications such as bone fracture and osteoporosis, reduced kidney function, peripheral neuropathy, chronic anemia, and ultimately, death.2,6 The presence of bone fractures increases mortality risk by 20%.10
DIAGNOSTIC WORKUP
Evidence of MM may be discovered during routine bloodwork and screening tests, while presenting symptoms or subtle changes in lab results can raise suspicion for the disease. Initial bloodwork abnormalities include anemia, elevated calcium levels, renal insufficiency, and/or elevated protein levels.8
A combination of abnormalities in the complete blood count (CBC) and complete metabolic panel (CMP), along with symptoms, should alert the provider to the possibility of MM, prompting additional workup. Table 1 outlines suggested diagnostic tests; the possible findings are discussed below.
CBC. The CBC may reveal abnormalities including anemia (which occurs in 75% of patients with MM), thrombocytopenia, and leukopenia.1,8 These findings can contribute to fatigue, increased incidence of infection, and abnormal bruising of the skin.2,8
CMP. A CMP may show increases in serum calcium or protein. Hypercalcemia occurs in 15% of patients with MM, leading to symptoms such as loss of appetite, nausea, vomiting, increased urination, weakness, and confusion.8 An increase in protein may alter the albumin/globulin ratio, which should raise suspicion for MM. A decrease in albumin can signify disease severity. Also, the CMP may show worsening renal function and elevated serum creatinine, which occurs in 20% of patients with MM.8
Serum protein electrophoresis (SPEP). Suspicion of MM should prompt the clinician to evaluate proteins via SPEP. This test may be indicated for patients with anemia, hypercalcemia, bone pain, and unexplained neuropathy.9 The electrophoresis separates proteins based on their physical properties. This identifies the presence and amount of M-protein, which can determine the extent of the disease.1 M-protein is identified in approximately 82% of patients with MM using this test.8
Serum free light chain (FLC) assay. This diagnostic test can identify MM in individuals with high clinical suspicion for the disease but no discernible M-protein on SPEP; it increases sensitivity to 97%.8 The serum FLC assay evaluates for presence and ratio of free light chains—proteins produced by plasma cells. This test is also useful for monitoring treatment response and disease progression.1
Urine protein electrophoresis (UPEP). The UPEP separates proteins according to charge, which is helpful for classifying renal injury. Protein patterns are interpreted and may be reported as glomerular, tubular, or mixed. UPEP also tests for M-protein in the urine.1,11
24-hour urine. The 24-h urine test quantifies the amount and type of protein excreted in the urine and helps determine the extent of kidney disease.1
Skeletal survey. MM causes significant bone changes that can be identified with radiographic studies. The most common locations for fractures are the vertebral, pelvic, and clavicular areas.10 Currently, the skeletal survey is the gold standard for detecting fractures and osteolytic lesions associated with MM.10 Radiographic films ordered for other purposes may uncover abnormalities in bones.
Bone mineral density (BMD) test. Most often, BMD testing is used to evaluate treatment and progression of bone involvement. Because it can uncover osteopenia or osteoporosis, however, it can also be used to corroborate the diagnosis of MM.10
Once the presence of M-protein is identified, patients are referred for specialty care. At that time, further workup will include a bone marrow biopsy and imaging studies, such as additional radiographic films, CT scans (without contrast, as contrast dye can damage frail kidneys), and MRI.1,8 These diagnostic tests provide useful information for the classification of the disease and guide initiation of treatment.
CLASSIFICATION OF DISEASE
MM can be classified into three stages—MGUS, SMM, and MM—based on recommendations from the International Myeloma Working Group.12 Table 2 outlines the diagnostic criteria for each stage.
Individuals with MGUS and SMM are considered asymptomatic; guidelines do not recommend treatment for these patients. Those who are diagnosed with MM are referred to oncologists and treated based on current clinical practice guidelines.1
CONCLUSION
Multiple myeloma is a malignant neoplasm without a cure. Presenting symptoms may include anemia, bone pain, elevated creatinine or serum protein, fatigue, and hypercalcemia. Early diagnosis is key to early intervention and treatment, which can improve quality of life and clinical outcomes for those affected. Primary care providers play a major role in recognizing the subtle symptoms and ordering the appropriate diagnostic tests.
1. National Comprehensive Cancer Network. Multiple myeloma. NCCN clinical practice guidelines in oncology version 2.2015.
2. Rajkumar VS. Multiple myeloma symptoms, diagnosis, and staging. www.uptodate.com/contents/clinical-features-laboratory-manifestations-and-diagnosis-of-multiple-myeloma?source=machineLearning&search=multiple+myeloma&selectedTitle=1%7E150§ionRank=1&anchor=H25#H26. Accessed October 16, 2017.
3. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed October 26, 2017.
4. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208.
5. Moreau P, San Miguel J, Sonneveld M, et al. Multiple myeloma: ESMO clinical practice guidelines. Ann Oncol. 2017;28(4):iv52-iv61.
6. American Cancer Society. Multiple myeloma. www.cancer.org/cancer/multiplemyeloma/detailedguide. Accessed October 16, 2017.
7. Koura DT, Langston AA. Inherited predisposition to multiple myeloma. Ther Adv Hematol. 2013;4(4):291-297.
8. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91:101-119.
9. O’Connell T, Horita TJ, Kasravi B. Understanding and interpreting serum electrophoresis. Am Fam Physician. 2005; 71(1):105-112.
10. Kristinsson SY, Minter AR, Korde N, et al. Bone disease in multiple myeloma and precursor disease; novel diagnostic approaches and implications on clinical management. Expert Rev Mol Diagn. 2011;11(6):593-603.
11. Jacobs D, DeMott W, Oxley D. Laboratory Test Handbook: Concise With Disease Index. Hudson, OH: Lexi-Comp; 2004.
12. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9.
IN THIS ARTICLE
- Presenting symptoms
- Diagnostic tests
- Differential diagnostic criteria
Multiple myeloma (MM) is a fatal, malignant neoplasm that originates in the plasma cells of bone marrow. A genetic mutation in the plasma cells creates myeloma cells, which replicate and produce monoclonal protein (M-protein). This accumulation of cells and abnormal protein can result in destruction and eventual marrow failure.1,2
MM’s insidious nature means it often goes undetected or misdiagnosed in its early stages; this delayed diagnosis can cause sequelae that limit quality of life. Furthermore, the five-year survival rate for myeloma varies by stage at which the disease is diagnosed: from 48% for distant (metastasized) myeloma to 71% for localized disease.3 It has also been noted that, in the past two decades, improvements in available treatment options and supportive care have contributed to a doubling of median survival time (from three years to six years).4 It is therefore paramount that providers be aware of MM and its signs to facilitate early diagnosis and treatment.
INCIDENCE AND EPIDEMIOLOGY
MM accounts for 1% of all cancers and about 10% of all hematologic malignancies.5 In 2017, the American Cancer Society estimated that more than 30,000 new cases of MM would be diagnosed in the United States.6 Additionally, MM was expected to cause more than 12,000 deaths last year.6
Median age at diagnosis is 69.3 In fact, 75% of men are older than 75 and 79% of women are older than 70 at diagnosis.1
Apart from age, other risk factors for MM have been identified but not fully explicated. For example, the disease is more common in men than in women (with men comprising two-thirds of new cases per year).3 MM is also two to three times more common in black than in white persons, making it the most common hematologic malignancy in this demographic group.3,7
The possibility of a genetic predisposition has also been studied. Several analyses have indicated an increased risk for MM in patients with a family history of the disease—as much as four times higher in those with an affected first-degree relative. This risk was further elevated in black compared with white patients (odds ratios, 17.4 and 1.5, respectively).7 However, many patients with MM have no relatives with this disorder.6,8
DISEASE PROGRESSION
Almost all patients who develop MM also experience an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance (MGUS). MGUS is present in 3% to 4% of the general population older than 50 and is often an incidental finding. This stage almost always precedes MM—but because it is asymptomatic, only 10% of individuals diagnosed with MM have a known history of MGUS.8
In some patients, an asymptomatic intermediate stage called smoldering multiple myeloma (SMM) can be identified. SMM progresses to MM at a rate of 10% per year for the first five years; the rate decreases to 3% per year over the following five years, and 1% per year after that.8
MM is not curable, but as noted, the survival rate is steadily increasing due to rapidly evolving treatment regimens. Discussion of treatment is outside the scope of this article, but early diagnosis can improve quality of life and clinical outcomes and prolong life expectancy.
SYMPTOMS
The initial symptoms of MM can be nonspecific and may lead the provider to suspect a host of other conditions.2,6 (Those for advanced disease are also vague but tend to be more pronounced.) These may include fatigue, weakness, easy bruising or bleeding, and bone pain. Other common clinical manifestations of MM are anemia, chronic infection, bone disease, and/or renal failure.1,4 Patients may also experience loss of appetite, nausea, vomiting, increased thirst, and increased urination.9
Recent studies have shown that patients with SMM and/or MGUS also exhibit early signs of bone disease and increased risk for fracture.10 Eighty percent of patients who progress to MM have evidence of pathologic bone fractures.10 It is also possible for bones in the spine to weaken and collapse, pressing on the spinal nerves. This is known as spinal cord compression, which can manifest with sudden, severe back pain or numbness and/or muscle weakness (most often in the legs).6
MM must be included in the differential diagnosis, particularly when symptoms do not point to one specific disease process. Without early diagnosis, disease progression can result in complications such as bone fracture and osteoporosis, reduced kidney function, peripheral neuropathy, chronic anemia, and ultimately, death.2,6 The presence of bone fractures increases mortality risk by 20%.10
DIAGNOSTIC WORKUP
Evidence of MM may be discovered during routine bloodwork and screening tests, while presenting symptoms or subtle changes in lab results can raise suspicion for the disease. Initial bloodwork abnormalities include anemia, elevated calcium levels, renal insufficiency, and/or elevated protein levels.8
A combination of abnormalities in the complete blood count (CBC) and complete metabolic panel (CMP), along with symptoms, should alert the provider to the possibility of MM, prompting additional workup. Table 1 outlines suggested diagnostic tests; the possible findings are discussed below.
CBC. The CBC may reveal abnormalities including anemia (which occurs in 75% of patients with MM), thrombocytopenia, and leukopenia.1,8 These findings can contribute to fatigue, increased incidence of infection, and abnormal bruising of the skin.2,8
CMP. A CMP may show increases in serum calcium or protein. Hypercalcemia occurs in 15% of patients with MM, leading to symptoms such as loss of appetite, nausea, vomiting, increased urination, weakness, and confusion.8 An increase in protein may alter the albumin/globulin ratio, which should raise suspicion for MM. A decrease in albumin can signify disease severity. Also, the CMP may show worsening renal function and elevated serum creatinine, which occurs in 20% of patients with MM.8
Serum protein electrophoresis (SPEP). Suspicion of MM should prompt the clinician to evaluate proteins via SPEP. This test may be indicated for patients with anemia, hypercalcemia, bone pain, and unexplained neuropathy.9 The electrophoresis separates proteins based on their physical properties. This identifies the presence and amount of M-protein, which can determine the extent of the disease.1 M-protein is identified in approximately 82% of patients with MM using this test.8
Serum free light chain (FLC) assay. This diagnostic test can identify MM in individuals with high clinical suspicion for the disease but no discernible M-protein on SPEP; it increases sensitivity to 97%.8 The serum FLC assay evaluates for presence and ratio of free light chains—proteins produced by plasma cells. This test is also useful for monitoring treatment response and disease progression.1
Urine protein electrophoresis (UPEP). The UPEP separates proteins according to charge, which is helpful for classifying renal injury. Protein patterns are interpreted and may be reported as glomerular, tubular, or mixed. UPEP also tests for M-protein in the urine.1,11
24-hour urine. The 24-h urine test quantifies the amount and type of protein excreted in the urine and helps determine the extent of kidney disease.1
Skeletal survey. MM causes significant bone changes that can be identified with radiographic studies. The most common locations for fractures are the vertebral, pelvic, and clavicular areas.10 Currently, the skeletal survey is the gold standard for detecting fractures and osteolytic lesions associated with MM.10 Radiographic films ordered for other purposes may uncover abnormalities in bones.
Bone mineral density (BMD) test. Most often, BMD testing is used to evaluate treatment and progression of bone involvement. Because it can uncover osteopenia or osteoporosis, however, it can also be used to corroborate the diagnosis of MM.10
Once the presence of M-protein is identified, patients are referred for specialty care. At that time, further workup will include a bone marrow biopsy and imaging studies, such as additional radiographic films, CT scans (without contrast, as contrast dye can damage frail kidneys), and MRI.1,8 These diagnostic tests provide useful information for the classification of the disease and guide initiation of treatment.
CLASSIFICATION OF DISEASE
MM can be classified into three stages—MGUS, SMM, and MM—based on recommendations from the International Myeloma Working Group.12 Table 2 outlines the diagnostic criteria for each stage.
Individuals with MGUS and SMM are considered asymptomatic; guidelines do not recommend treatment for these patients. Those who are diagnosed with MM are referred to oncologists and treated based on current clinical practice guidelines.1
CONCLUSION
Multiple myeloma is a malignant neoplasm without a cure. Presenting symptoms may include anemia, bone pain, elevated creatinine or serum protein, fatigue, and hypercalcemia. Early diagnosis is key to early intervention and treatment, which can improve quality of life and clinical outcomes for those affected. Primary care providers play a major role in recognizing the subtle symptoms and ordering the appropriate diagnostic tests.
IN THIS ARTICLE
- Presenting symptoms
- Diagnostic tests
- Differential diagnostic criteria
Multiple myeloma (MM) is a fatal, malignant neoplasm that originates in the plasma cells of bone marrow. A genetic mutation in the plasma cells creates myeloma cells, which replicate and produce monoclonal protein (M-protein). This accumulation of cells and abnormal protein can result in destruction and eventual marrow failure.1,2
MM’s insidious nature means it often goes undetected or misdiagnosed in its early stages; this delayed diagnosis can cause sequelae that limit quality of life. Furthermore, the five-year survival rate for myeloma varies by stage at which the disease is diagnosed: from 48% for distant (metastasized) myeloma to 71% for localized disease.3 It has also been noted that, in the past two decades, improvements in available treatment options and supportive care have contributed to a doubling of median survival time (from three years to six years).4 It is therefore paramount that providers be aware of MM and its signs to facilitate early diagnosis and treatment.
INCIDENCE AND EPIDEMIOLOGY
MM accounts for 1% of all cancers and about 10% of all hematologic malignancies.5 In 2017, the American Cancer Society estimated that more than 30,000 new cases of MM would be diagnosed in the United States.6 Additionally, MM was expected to cause more than 12,000 deaths last year.6
Median age at diagnosis is 69.3 In fact, 75% of men are older than 75 and 79% of women are older than 70 at diagnosis.1
Apart from age, other risk factors for MM have been identified but not fully explicated. For example, the disease is more common in men than in women (with men comprising two-thirds of new cases per year).3 MM is also two to three times more common in black than in white persons, making it the most common hematologic malignancy in this demographic group.3,7
The possibility of a genetic predisposition has also been studied. Several analyses have indicated an increased risk for MM in patients with a family history of the disease—as much as four times higher in those with an affected first-degree relative. This risk was further elevated in black compared with white patients (odds ratios, 17.4 and 1.5, respectively).7 However, many patients with MM have no relatives with this disorder.6,8
DISEASE PROGRESSION
Almost all patients who develop MM also experience an asymptomatic premalignant stage called monoclonal gammopathy of undetermined significance (MGUS). MGUS is present in 3% to 4% of the general population older than 50 and is often an incidental finding. This stage almost always precedes MM—but because it is asymptomatic, only 10% of individuals diagnosed with MM have a known history of MGUS.8
In some patients, an asymptomatic intermediate stage called smoldering multiple myeloma (SMM) can be identified. SMM progresses to MM at a rate of 10% per year for the first five years; the rate decreases to 3% per year over the following five years, and 1% per year after that.8
MM is not curable, but as noted, the survival rate is steadily increasing due to rapidly evolving treatment regimens. Discussion of treatment is outside the scope of this article, but early diagnosis can improve quality of life and clinical outcomes and prolong life expectancy.
SYMPTOMS
The initial symptoms of MM can be nonspecific and may lead the provider to suspect a host of other conditions.2,6 (Those for advanced disease are also vague but tend to be more pronounced.) These may include fatigue, weakness, easy bruising or bleeding, and bone pain. Other common clinical manifestations of MM are anemia, chronic infection, bone disease, and/or renal failure.1,4 Patients may also experience loss of appetite, nausea, vomiting, increased thirst, and increased urination.9
Recent studies have shown that patients with SMM and/or MGUS also exhibit early signs of bone disease and increased risk for fracture.10 Eighty percent of patients who progress to MM have evidence of pathologic bone fractures.10 It is also possible for bones in the spine to weaken and collapse, pressing on the spinal nerves. This is known as spinal cord compression, which can manifest with sudden, severe back pain or numbness and/or muscle weakness (most often in the legs).6
MM must be included in the differential diagnosis, particularly when symptoms do not point to one specific disease process. Without early diagnosis, disease progression can result in complications such as bone fracture and osteoporosis, reduced kidney function, peripheral neuropathy, chronic anemia, and ultimately, death.2,6 The presence of bone fractures increases mortality risk by 20%.10
DIAGNOSTIC WORKUP
Evidence of MM may be discovered during routine bloodwork and screening tests, while presenting symptoms or subtle changes in lab results can raise suspicion for the disease. Initial bloodwork abnormalities include anemia, elevated calcium levels, renal insufficiency, and/or elevated protein levels.8
A combination of abnormalities in the complete blood count (CBC) and complete metabolic panel (CMP), along with symptoms, should alert the provider to the possibility of MM, prompting additional workup. Table 1 outlines suggested diagnostic tests; the possible findings are discussed below.
CBC. The CBC may reveal abnormalities including anemia (which occurs in 75% of patients with MM), thrombocytopenia, and leukopenia.1,8 These findings can contribute to fatigue, increased incidence of infection, and abnormal bruising of the skin.2,8
CMP. A CMP may show increases in serum calcium or protein. Hypercalcemia occurs in 15% of patients with MM, leading to symptoms such as loss of appetite, nausea, vomiting, increased urination, weakness, and confusion.8 An increase in protein may alter the albumin/globulin ratio, which should raise suspicion for MM. A decrease in albumin can signify disease severity. Also, the CMP may show worsening renal function and elevated serum creatinine, which occurs in 20% of patients with MM.8
Serum protein electrophoresis (SPEP). Suspicion of MM should prompt the clinician to evaluate proteins via SPEP. This test may be indicated for patients with anemia, hypercalcemia, bone pain, and unexplained neuropathy.9 The electrophoresis separates proteins based on their physical properties. This identifies the presence and amount of M-protein, which can determine the extent of the disease.1 M-protein is identified in approximately 82% of patients with MM using this test.8
Serum free light chain (FLC) assay. This diagnostic test can identify MM in individuals with high clinical suspicion for the disease but no discernible M-protein on SPEP; it increases sensitivity to 97%.8 The serum FLC assay evaluates for presence and ratio of free light chains—proteins produced by plasma cells. This test is also useful for monitoring treatment response and disease progression.1
Urine protein electrophoresis (UPEP). The UPEP separates proteins according to charge, which is helpful for classifying renal injury. Protein patterns are interpreted and may be reported as glomerular, tubular, or mixed. UPEP also tests for M-protein in the urine.1,11
24-hour urine. The 24-h urine test quantifies the amount and type of protein excreted in the urine and helps determine the extent of kidney disease.1
Skeletal survey. MM causes significant bone changes that can be identified with radiographic studies. The most common locations for fractures are the vertebral, pelvic, and clavicular areas.10 Currently, the skeletal survey is the gold standard for detecting fractures and osteolytic lesions associated with MM.10 Radiographic films ordered for other purposes may uncover abnormalities in bones.
Bone mineral density (BMD) test. Most often, BMD testing is used to evaluate treatment and progression of bone involvement. Because it can uncover osteopenia or osteoporosis, however, it can also be used to corroborate the diagnosis of MM.10
Once the presence of M-protein is identified, patients are referred for specialty care. At that time, further workup will include a bone marrow biopsy and imaging studies, such as additional radiographic films, CT scans (without contrast, as contrast dye can damage frail kidneys), and MRI.1,8 These diagnostic tests provide useful information for the classification of the disease and guide initiation of treatment.
CLASSIFICATION OF DISEASE
MM can be classified into three stages—MGUS, SMM, and MM—based on recommendations from the International Myeloma Working Group.12 Table 2 outlines the diagnostic criteria for each stage.
Individuals with MGUS and SMM are considered asymptomatic; guidelines do not recommend treatment for these patients. Those who are diagnosed with MM are referred to oncologists and treated based on current clinical practice guidelines.1
CONCLUSION
Multiple myeloma is a malignant neoplasm without a cure. Presenting symptoms may include anemia, bone pain, elevated creatinine or serum protein, fatigue, and hypercalcemia. Early diagnosis is key to early intervention and treatment, which can improve quality of life and clinical outcomes for those affected. Primary care providers play a major role in recognizing the subtle symptoms and ordering the appropriate diagnostic tests.
1. National Comprehensive Cancer Network. Multiple myeloma. NCCN clinical practice guidelines in oncology version 2.2015.
2. Rajkumar VS. Multiple myeloma symptoms, diagnosis, and staging. www.uptodate.com/contents/clinical-features-laboratory-manifestations-and-diagnosis-of-multiple-myeloma?source=machineLearning&search=multiple+myeloma&selectedTitle=1%7E150§ionRank=1&anchor=H25#H26. Accessed October 16, 2017.
3. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed October 26, 2017.
4. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208.
5. Moreau P, San Miguel J, Sonneveld M, et al. Multiple myeloma: ESMO clinical practice guidelines. Ann Oncol. 2017;28(4):iv52-iv61.
6. American Cancer Society. Multiple myeloma. www.cancer.org/cancer/multiplemyeloma/detailedguide. Accessed October 16, 2017.
7. Koura DT, Langston AA. Inherited predisposition to multiple myeloma. Ther Adv Hematol. 2013;4(4):291-297.
8. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91:101-119.
9. O’Connell T, Horita TJ, Kasravi B. Understanding and interpreting serum electrophoresis. Am Fam Physician. 2005; 71(1):105-112.
10. Kristinsson SY, Minter AR, Korde N, et al. Bone disease in multiple myeloma and precursor disease; novel diagnostic approaches and implications on clinical management. Expert Rev Mol Diagn. 2011;11(6):593-603.
11. Jacobs D, DeMott W, Oxley D. Laboratory Test Handbook: Concise With Disease Index. Hudson, OH: Lexi-Comp; 2004.
12. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9.
1. National Comprehensive Cancer Network. Multiple myeloma. NCCN clinical practice guidelines in oncology version 2.2015.
2. Rajkumar VS. Multiple myeloma symptoms, diagnosis, and staging. www.uptodate.com/contents/clinical-features-laboratory-manifestations-and-diagnosis-of-multiple-myeloma?source=machineLearning&search=multiple+myeloma&selectedTitle=1%7E150§ionRank=1&anchor=H25#H26. Accessed October 16, 2017.
3. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed October 26, 2017.
4. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385(9983):2197-2208.
5. Moreau P, San Miguel J, Sonneveld M, et al. Multiple myeloma: ESMO clinical practice guidelines. Ann Oncol. 2017;28(4):iv52-iv61.
6. American Cancer Society. Multiple myeloma. www.cancer.org/cancer/multiplemyeloma/detailedguide. Accessed October 16, 2017.
7. Koura DT, Langston AA. Inherited predisposition to multiple myeloma. Ther Adv Hematol. 2013;4(4):291-297.
8. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91:101-119.
9. O’Connell T, Horita TJ, Kasravi B. Understanding and interpreting serum electrophoresis. Am Fam Physician. 2005; 71(1):105-112.
10. Kristinsson SY, Minter AR, Korde N, et al. Bone disease in multiple myeloma and precursor disease; novel diagnostic approaches and implications on clinical management. Expert Rev Mol Diagn. 2011;11(6):593-603.
11. Jacobs D, DeMott W, Oxley D. Laboratory Test Handbook: Concise With Disease Index. Hudson, OH: Lexi-Comp; 2004.
12. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23(1):3-9.
Improving Strength and Balance for Long-Term Care Residents At Risk for Falling: Suggestions for Practice
From the Geriatric Education and Research in Aging Sciences Centre, McMaster University Hamilton, ON (Dr. McArthur) and the University of Waterloo and Research Institute for Aging, Waterloo, ON (Dr. Giangregorio), Canada
Abstract
- Objective: To synthesize the available literature on exercise and falls reduction interventions in long-term care (LTC) and provide practical information for clinicians and other decision makers.
- Methods: Review of positive trials included in systematic reviews.
- Results: Falls are a major concern for residents, families, clinicians, and decision-makers in LTC. Exercise is recommended as part of a multifactorial falls prevention program for residents in LTC. Strength and balance exercises should be incorporated into the multifactorial falls prevention program. They should be challenging and progressed as the residents’ abilities improve. Evidence suggests that exercises should be completed 2 to 3 times per week for a period longer than 6 months. Exercise programs in LTC should be resident-centered and should consider residents’ potential physical and cognitive impairments. Exercises in standing should be prioritized where appropriate.
- Conclusion: Appropriately challenging and progressive strength and balance exercises should be included in a multifactorial falls prevention program for residents in LTC.
Key words: long-term care; nursing homes; falls reduction; exercise.
Falls are common in long-term care (LTC) homes: the estimated falls rate is 1.5 falls per bed per year, which is 3 times greater than that for older adults living in the community [1]. Falls can have significant consequences for residents in LTC, including functional disability, fractures, pain, reduced quality of life, and death [1–6]. Indeed, 25% of residents who are hospitalized after a fall die within 1 year [3]. Consequently, falls prevention programs are important to help in reducing falls and averting the associated negative consequences.
Exercise may address the circumstances and physical deconditioning that often contribute to falls in LTC residents. Weight shifting [7], walking, and transferring [8–10], are common activities that precede falls, suggesting that balance, gait, and functional mobility training may be possible targets for prevention. Additionally, it is estimated that LTC residents spend three quarters of their waking time in sedentary activities [11,12] and have a high prevalence of sarcopenia [13–16]. Challenging balance training and resistance exercise are well-known intervention for reducing falls [17] and improving muscle strength for community-dwelling older adults [18]. However, evidence around balance and strength training for preventing falls in LTC is mixed [17,19,20], and careful planning and modification of exercises is necessary to meet the needs of LTC residents.
Residents in LTC are often medically complex, with multiple comorbidities [21] that can affect their ability to meaningfully participate in exercise. In Canada, 56.3% of residents have a diagnosis of Alzheimer’s or other dementias, 25.0% have diabetes, 14.4% have chronic obstructive pulmonary disease, and 21.2% have experienced a stroke [21]. Residents also often have significant functional impairments. For example, 97% of residents require assistance with basic activities of daily living [21]. Therefore, the lack of effect of exercise as a single falls prevention strategy observed in previous studies may be because the often complex, multimorbid LTC population likely requires a multifactorial approach to fall prevention [17]. Additionally, organizational aspects of LTC homes (eg, specific funds dedicated to employing exercise professionals and to support exercise programming) can affect residents’ engagement in exercise [22,23]. Subsequently, prescribing exercises in the LTC context must consider both resident characteristics and organizational features of the LTC home (eg, professionals available to support exercise programming).
A comprehensive exercise prescription describes the elements of an appropriate exercise program to facilitate implementation of that program. The exercise prescription should include a description of the type (eg, balance, strength) and intensity of exercises (eg, subjective or objective measurement of how hard the resident is working) included in the program [24]. The prescription should also include a description of the dose of exercise: frequency of exercise participation (eg, 2 days per week), duration of individual exercise sessions (eg, 30-minute sessions), and duration of exercise program (eg, 12-week program) [24]. Lastly, the prescription should describe the setting of the exercise program (eg, group or individual basis) and the professional delivering the program (eg, physiotherapist, fitness instructor) [24].
Therefore, the objectives of this article are to (1) synthesize studies demonstrating a positive effect of exercise on reducing falls for residents in LTC; (2) provide an overview of the principles of balance and strength training to guide clinicians in designing appropriate exercise prescription; and (3) make suggestions for clinical practice regarding an appropriate strength and balance exercise protocol by considering the influence of the LTC context.
Methods
To provide clinicians and other policy-makers with a description of which balance and strength exercises may be effective for preventing falls, we synthesized trials that demonstrated a positive effect on reducing falls or falls risk for residents in LTC. Studies were identified through a database search for systematic reviews in PubMed, Ovid, and Google Scholar using the keywords falls, long-term care, nursing homes, exercise, strength, balance, and systematic reviews. Our purpose was to provide practical information on what works to prevent falls through balance and strength training for residents in LTC rather than to evaluate the available evidence. Therefore, only positive trials from systematic reviews were discussed, as we wanted to present exercises that seem to have a positive effect on decreasing falls. Positive trials were defined as those included in identified systematic reviews with a risk or rate ratio and confidence intervals below 1.0.
We first provide an overview of the conclusions of the systematic reviews found in our search. Next, for each positive trial we describe the following elements of the exercise component of the intervention: frequency, time of sessions, length of program, intensity, type of exercise including a description of the specific exercises performed, whether the intervention was delivered in a group or on an individual basis, the professional delivering the intervention, and any other features of the intervention aside from the exercise component. We used the ProFaNE taxonomy definitions [25] to identify and describe each element of the exercise interventions. Frequency is the number of times per week that residents engage in sessions, time of sessions is the amount allocated to each exercise session, duration of program is how long the resident participates in the exercise program, and intensity is the subjective or objective report of how hard the resident is working [25]. The types of exercises described were those targeting balance defined as “...the efficient transfer of bodyweight from one part of the body to another or challenges specific aspects of the balance systems (eg, vestibular system)” [25], and strength defined as “...contracting the muscles against a resistance to ‘overload’ and bring about a training effect in the muscular system” [25]. Strength could be either an external resistance (eg, dumbbell) or using body weight against gravity (eg, squat) [25].
Results
We found 3 systematic reviews that include exercise programs to reduce falls in LTC homes [17,19,20]. Overall, evidence suggests that exercise should be included as part of a multifactorial falls prevention program for residents in LTC. There is limited evidence that exercise as a single intervention prevents falls, and some trials, albeit underpowered, even demonstrate an increased risk of falling in the exercise group compared to control [19]. With regards to specific exercise programs, the Cochrane review found that gait, balance, and functional training decrease the rate of falls but not the risk of falling [26–28], and the 2013 review by Silva et al [20] concluded that combined exercise programs (ie, multiple types of exercise) that include balance tasks, are completed frequently (2–3 times per week), and over a long term (greater than 6 months) were most effective at preventing falls [20].
A more recent systematic review and meta-analysis [17] also concluded that there was no evidence that exercise as a single intervention can prevent falls for residents in LTC. Table 1 provides a description of the exercise component of the seven positive trials [29–35] that were included in the 3 systematic reviews we identified in our search. 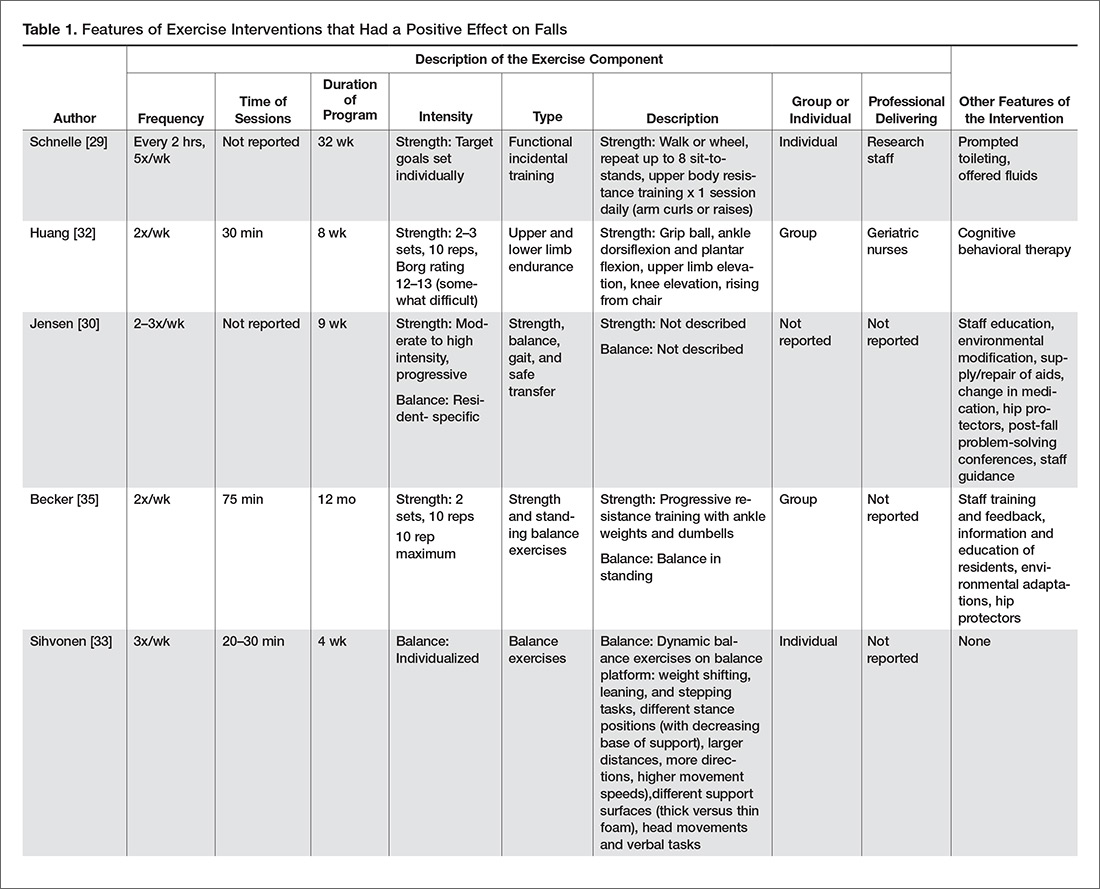
Type of Exercise
Balance Exercises
There were 4 positive trials that included balance exercises in their intervention [31,33–35]. Trials that had a positive effect on reducing falls and included balance training employed mostly dynamic balance exercises in standing (Table 1). However, only 2 of the 7 trials provided a detailed description of their balance exercises (Table 1) [26,34]. Jensen et al [30] and Dyer et al [31] did not include a description of the balance training performed but stated that balance was part of the multicomponent exercise program. Becker et al [36] stated that participants performed standing balance exercises, while Schnelle et al [39] and Huang et al [32] did not include balance training in their trial.
Strength Exercises
Of the 7 positive trials included in this review, 6 included strength exercises [29–32,34,35]. The strength activities used in trials where exercise had a positive effect on decreasing falls included functional activities [29,31] and progressive resistance training [31,36] (Table 1). Functional activities are those that replicate what a resident might be required to do in their everyday life, such as performing sit-to-stands out of a chair (Figure) 
Frequency, Time of Sessions, Duration of Program
In our description of positive trials, exercise was performed on 2 to 3 days per week for 20 to 75 minutes per session, for periods ranging from 4 to 52 weeks (Table 1).
Intensity
For the trials including balance exercises, one trial described the intensity as resident-specific [37] and another as individualized [33]. Two studies did not describe the intensity of their balance exercises [31,34]. The intensity of strength exercises included in the positive trials was individualized for one of the trial [29]. Two trials had participants complete 2 to 3 sets of 10 repetitions [32,35], with one indicating an intensity of 12–13 or “somewhat difficult” on the Borg Rating of Perceived Exertion Scale [32] and the other using a 10-rep max [35]. Two studies described their strength exercises as progressive [31,37], and one at a moderate to high intensity [30]. Lord et al prescribed 30 repetitions of each strength exercise [34].
Delivery of Intervention
Exercise was delivered in a group setting for 4 of the trials [31,32,34,36], individually for 2 of the trials [26,29], and the setting was not described for one of the trials (Table 1) [30]. Finally, only 3 of the 7 articles reported the professional delivering the intervention: one was research staff [29], one was geriatric nurses [32], and one was exercise assistants supported by a physiotherapist [31].
Discussion
There is limited evidence to support the use of strength and balance exercise as a single intervention to prevent falls in LTC. However, exercise should be included as part of a multifactorial falls prevention program. Trials that had a positive effect on decreasing falls training used dynamic balance exercises in standing, functional training, and progressive resistance training on 2 to 3 days per week, for 20 to 75 minutes per session, over 4 to 52 weeks. The intensity of balance exercises was individualized, and strength exercises were described as somewhat difficult or performed at a moderate to high intensity. Exercise was performed in a group or individually, and was delivered by research staff, geriatric nurses, exercise assistants supervised by physiotherapists, or more frequently, it was not reported who delivered the intervention.
Balance Training
Our work suggests that standing, dynamic balance exercises may be best to decrease falls. Example balance exercises include reducing the base of support (eg, standing with feet together instead of apart, or tandem with one foot in front), moving the center of gravity and control body position while standing (eg, reaching, weight shifting, stepping up or down), and standing without using arms for support or reducing reliance on the upper limbs for support (eg, use one hand on a handrail instead of two, or two fingers instead of the whole hand) [17]. It is well established that balance training programs, especially those including challenging exercises, can prevent falls in community-dwelling older adults [17]. However, the relationship is not as clear in LTC.
Strength Training
Reduced muscle strength has been identified as an important risk factor for falls [38]. There are also many psychological and metabolic benefits to strength training [39]. To induce change in muscular strength, resistance exercises need to be challenging and progressive. Our work suggests that strength training that is effective at decreasing falls is functional and progressive, and is completed at a moderate to high intensity. A resident should be able to do a strength exercise for one to two sets of 6 to 8 repetitions before being fatigued [40]. Once the resident can complete two sets of 13 to 15 repetitions easily the exercise should be progressed. Residents who are particularly deconditioned may need to begin with lower intensity strength exercises (eg, only do one set, with a lower resistance and progress to a higher resistance) [40]. Residents should perform resistance exercises for all major muscle groups [40]. Progression could include increasing the number of sets (eg, increase from one to two sets), the resistance (eg, holding dumbbells while squatting), or the intensity of the exercise (eg, squat lower or faster) [41].
Implementing Exercise Programs in LTC
Implementation of exercise programs into LTC homes should consider the dose of exercise (eg, time and frequency of sessions, duration of program), if they are delivered in a group or individual setting, and who is delivering them. First, trials included in this paper suggest that strength and balance exercises to prevent falls were delivered 2 to 3 times per week, for 20 to 75 minutes per session, over 4 to 52 weeks. Second, previous work has established that exercise programs delivered on 2 to 3 days per week over a period of more than 6 months are most effective at reducing falls in LTC [20]. Finally, a recent task force report from an international group of clinician researchers in LTC recommends twice weekly exercise sessions lasting 35 to 45 minutes each [40]. Therefore, strength and balance exercises to prevent falls in LTC should be delivered at least twice per week, for at least 20 minutes, for greater than 6 weeks’ duration.
Whether exercise should be performed in a group or individual setting remains unclear. Two of the 6 positive trials in this paper were completed individually, while 3 were in a group. The aforementioned task force also recommended that every resident who does not have contraindications to exercise must have an individualized exercise program as part of their health care plan [40]. However, whether the exercise program is provided on an individual basis or in a group setting was not delineated. Indeed, there are currently no recommendations concerning prioritizing group or individual exercise programs. Therefore, exercise programs being implemented into LTC homes should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings.
Finally, which professionals should deliver the exercise program is also uncertain. Only 3 of the positive trials in this paper described the professional delivering the intervention, with one being research staff, one geriatric nurses, and one exercise assistants supported by a physiotherapist. We suggest that professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC.
Modifications for Physical Impairments
Residents in LTC often have complex health needs, with multiple comorbidities (eg, stroke, Parkinson’s disease, multiple sclerosis) [21]. Modifications of strength and balance exercises may be required to accommodate for physical impairments (eg, hemiplegia, drop foot, freezing gait). For example, if a resident has hemiplegia and cannot fully activate the muscles of one arm, one can do resistance exercises with a dumbbell on the functioning side and active assisted range of motion (ie, the exercise provider assists the resident to achieve full range of motion against gravity) on the hemiparetic side. A resident with Parkinson’s disease who has freezing gait may need visual or rhythmical verbal cues to be able to accomplish standing balance tasks such as altered walking patterns (eg, wide or narrow stepping) [42].
Modifications for Cognitive Impairments
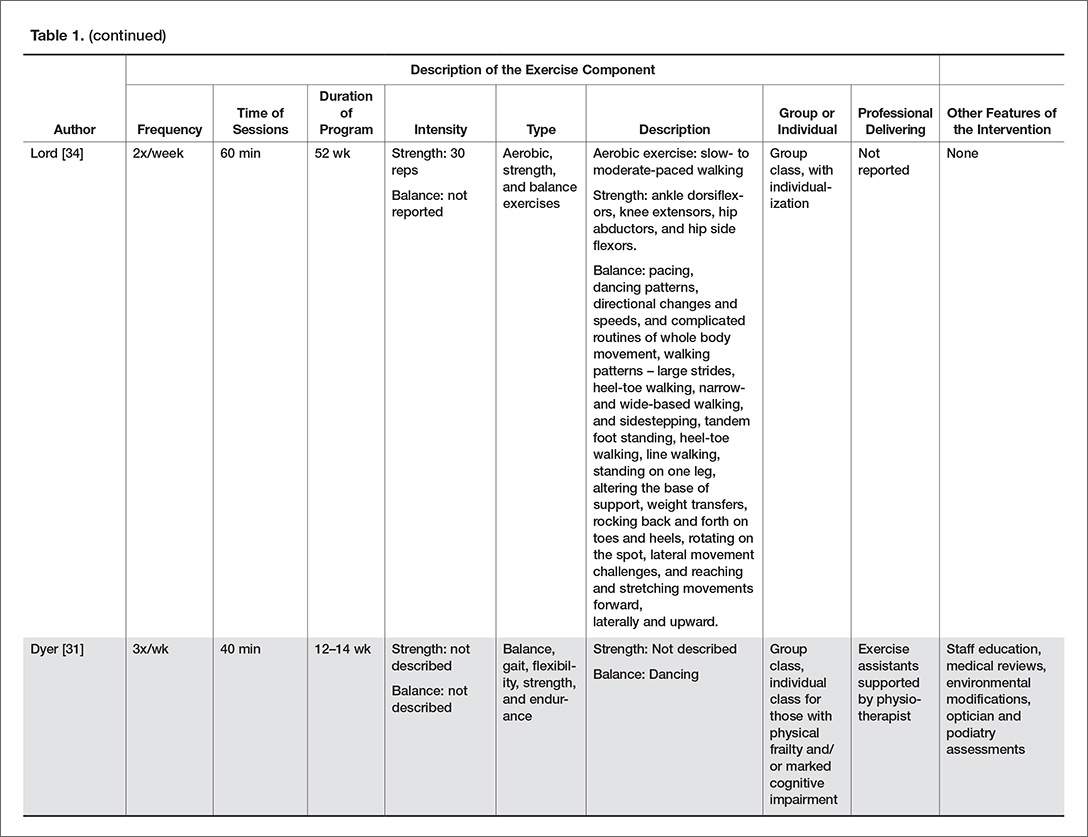
More than 80% of residents in LTC have some degree of cognitive impairment [21]. Cognitive impairment may be the result of stroke, depression, traumatic injuries, medications, and degenerative diseases such as Parkinson’s and Alzheimer’s disease [43]. A common misconception is that residents with cognitive impairment cannot benefit from exercise because they cannot learn new skills and have difficulty following directions. On the contrary, evidence suggests that exercise can improve functional mobility for residents with cognitive impairment [44,45].
Residents with cognitive impairment may require a different approach to facilitate participation in the desired exercises because of difficulty following multi-step directions, responsive behaviors, or increased distractibility [46]. Clear communication is key in improving the quality of interaction for residents with cognitive impairment. The Alzheimer Society of Ontario suggests 10 strategies for communicating with people with dementia [47], and we have provided suggestions of how to apply these communication strategies to the exercise context in LTC (Table 2). Other suggestions for engaging residents with cognitive impairment in strength and balance training include making the exercises functional (eg, ask them to pick something up of the floor to perform a squat, or reach a point on the wall to do calf raises) and playful (eg, toss a ball back and forth or sing a song about rowing to promote weight shifting) [48]. 
Standing versus Seated Exercises
Residents may not be able to participate in standing exercises for several reasons: perhaps the resident cannot stand or has severe balance impairments and a high falls risk; the resident may have poor insight into which exercises are safe to perform in standing versus sitting; or there may be limited supervision of a large group exercise class where the risk of falls is a concern. If balance impairments are a concern, where the risk of injury or falling while completing exercises in standing outweighs the benefit of doing the exercises, then seated exercises are appropriate. However, when residents are able, we recommend encouraging some or all exercises in standing, to facilitate carry over of strength gains into functional tasks such as being able to rise from a chair and walking. A recent study, comparing standing versus seated exercises for community dwelling older adults, saw greater functional gains for those who completed the standing exercises [49]. Therefore, strength and balance exercises should be performed in standing, where appropriate.
Resident-Centered Exercise for Falls Prevention
Putting the resident at the center of falls prevention is important. Previous work has found that older adults have expressed a strong preference for care that transcends traditional biomedical care and that values efficiency, consistency, and hierarchical decision making [50]. On the contrary, resident-centered care emphasizes well-being and quality of life as defined by the resident, values giving residents greater control over the nature of services they receive, and respects their rights to be involved in every day decision making [51,52]. Indeed, residents may choose to engage in risky behaviors that increase their risk of falls but also increases their quality of life. Previous work has found disconnects between residents’ perceived frailty and the potential ability of protective devices to prevent adverse events, such as falls and fractures [53]. Additionally, one study identified that older residents feared being labelled, so instead hid impairments and chose to refuse assistance and assistive devices [54]. For example, a resident with impaired balance and gait may choose to walk independently when they have been deemed as requiring a gait aid (eg, rollator walker). However, they may value walking without a gait aid and accept the increased risk of falling. Therefore, it is essential to find the delicate balance between respecting a resident’s right to make their own decisions and preventing adverse events, such as falls [52]. An example of this would be respecting a resident’s right to refuse to attend exercise programming even though the team may think they can benefit from strength and balance training.
There is limited evidence around falls prevention and resident-centered care. A recent systematic review [55] revealed that resident-centered care may increase falls rates [56,57]. However, the authors of the review attributed the increase in falls to differences in frailty between the control and intervention group [56], and to environmental factors (eg, slippery flooring material, lack of handrails) [57]. Additionally, these trials did not include an exercise program as part of the resident-centered care program. On the other hand, resident-centered care has been associated with reduction of boredom, helplessness, and depression [58,59]. Most studies included in the review were quasi-experimental, which significantly limits the evidence quality [55]. At this point in time, the evidence suggests that resident-centered care is important for mood and quality of life but may have a negative or no effect on reducing falls.
Multifactorial Falls Prevention Programs
While there are mixed results about the effect of exercise as a single intervention for reducing falls for residents in LTC, the literature clearly supports exercise as part of a multifactorial falls prevention program [17,20,60–62]. A 2015 umbrella review [62] of meta-analyses of randomized controlled trials of falls prevention interventions in LTC concluded that multifactorial interventions were the most effective at preventing falls in LTC. Additionally, recently developed recommendations for fracture prevention in LTC [61] suggest that balance, strength, and functional training should be included for residents who are not at high risk of fracture, while for those at high risk, exercise should be provided as part of a multifactorial falls prevention intervention. Clinicians must therefore incorporate elements aside from exercise into their falls prevention strategies. Interventions that have shown positive effects on reducing falls when delivered as part of multifactorial interventions include: staff and resident education [31,35,37], environmental modifications [31,35], supply/repair/provision of assistive devices [30], falls problem-solving conferences [30], urinary incontinence management [29], medication review [30], optician review [31], and cognitive behavioral therapy [32].
Conclusion and Suggestions for Clinical Practice
We suggest incorporating strength and balance exercises as part of a multifactorial falls prevention program for residents in LTC. Balance exercises should be challenging and dynamic (eg, weight shifting). Strength exercises should be of a moderate to high intensity (eg, can complete one to sets of 6 to 8 repetitions) and need to be progressed as the residents’ abilities improve. Residents should participate in strength and balance training on 2 to 3 days per week, for 30- to 45-minute sessions, for at least 6 months. Exercises in standing should be prioritized where appropriate. Exercise could be delivered in a group or individual format, but should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings. Professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC. Exercise programs in LTC should be resident-centered and consider residents’ potential physical and cognitive impairments.
Funding/support: Dr. Giangregorio was supported by grants from the Canadian Frailty Network and Canadian Institutes of Health Research.
1. Harris IA, Yong S, McEvoy L, Thorn L. A prospective study of the effect of nursing home residency on mortality following hip fracture. ANZ J Surg 2010;80:447–50.
2. Ooms ME, Vlasman P, Lips P, et al. The incidence of hip fractures in independent and institutionalized elderly people. Osteoporos Int 1994;4:6–10.
3. Ayoung-Chee P, McIntyre L, Ebel BE, et al. Long-term outcomes of ground-level falls in the elderly. J Trauma Acute Care Surg 2014;76:498–503.
4. Heinrich S, Rapp K, Rissmann U, et al. Cost of falls in old age: a systematic review. Osteoporos Int 2010;21: 891–902.
5. Rubenstein LZ, Josephson KR, Robbins AS. Falls in the nursing home. Ann Intern Med 1994;121:442–51.
6. Hartholt KA, van Beeck EF, Polinder S, et al. Societal consequences of falls in the older population: injuries, healthcare costs, and long-term reduced quality of life. J Trauma
2011;71:748–53.
7. Robinovitch SN, Feldman F, Yang Y, et al. Video capture of the circumstances of falls in elderly people residing in long-term care: an observational study. Lancet 2013;381:
47–54.
8. Rapp K, Becker C, Cameron ID, et al. Epidemiology of falls in residential aged care: analysis of more than 70,000 falls from residents of bavarian nursing homes. J Am Med Dir Assoc 2012;13:187.
9. Büchele G, Becker C, Cameron ID, et al. Predictors of serious consequences of falls in residential aged care: analysis of more than 70,000 falls from residents of Bavarian nursing homes. J Am Med Dir Assoc 2014;15:559–63.
10. McArthur C, Gonzalez DA, Roy E, Giangregorio L. What are the circumstances of falls and fractures in long-term care? Can J Aging / La Rev Can du Vieil 2016;35:491–8.
11. Chin A Paw MJM, van Poppel MNM, van Mechelen W. Effects of resistance and functional-skills training on habitual activity and constipation among older adults living in long-term care facilities: a randomized controlled trial. BMC Geriatr 2006;6:9.
12. Ikezoe T, Asakawa Y, Shima H, et al. Daytime physical activity patterns and physical fitness in institutionalized elderly women: an exploratory study. Arch Gerontol Geriatr 2013;57:221–5.
13. Senior HE, Henwood TR, Beller EM, et al. Prevalence and risk factors of sarcopenia among adults living in nursing homes. Maturitas 2015;82:418–23.
14. Smoliner C, Sieber CC, Wirth R. Prevalence of sarcopenia in geriatric hospitalized patients. J Am Med Dir Assoc 2014;15:267–72.
15. Landi F, Liperoti R, Fusco D, et al. Sarcopenia and mortality among older nursing home residents. J Am Med Dir Assoc 2012;13:121–6.
16. Yalcin A, Aras S, Atmis V, et al. Sarcopenia prevalence and factors associated with sarcopenia in older people living in a nursing home in Ankara Turkey. Geriatr Gerontol Int
2016;16:903–10.
17. Sherrington C, Michaleff ZA, Fairhall N, et al. Exercise to prevent falls in older adults: an updated systematic review and meta-analysis. Br J Sports Med October 2016.
18. Liu C, Latham NK. Progressive resistance strength training for improving physical function in older adults. In: Liu C, ed. Cochrane Database Syst Rev;2009:CD002759.
19. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev;2012:CD005465.
20. Silva RB, Eslick GD, Duque G. Exercise for falls and fracture prevention in long term care facilities: a systematic review and meta-analysis. J Am Med Dir Assoc 2013;14:685–9.
21. Hirdes JP, Mitchell L, Maxwell CJ, White N. Beyond the “iron lungs of gerontology”: Using evidence to shape the future of nursing homes in Canada. Can J Aging 2011;30: 371–90.
22. Benjamin K, Edwards N, Guitard P, et al. Factors that influence physical activity in long-term care: Perspectives of residents, staff, and significant others. Can J Aging 2011;30:247–58.
23. Benjamin K, Edwards N, Ploeg J, Legault F. Barriers to physical activity and restorative care for residents in long-term care: A review of the literature. J Aging Phys Act 2014;22:154–65.
24. American College of Sports Medicine. ACSM’s guidelines for exercise testing and prescription. 9th ed. American College of Sports Medicine; 2013.
25. Prevention of Falls Network Europe. Prevention of Falls Network Europe. Accessed 27 Nov 2017 at www.profane.eu.org/.
26. Sihvonen SE, Sipilä S, Era PA. Changes in postural balance in frail elderly women during a 4-week visual feedback training: a randomized controlled trial. Gerontology 2004;50:87–95.
27. Sakamoto K, Nakamura T, Hagino H, et al. Effects of unipedal standing balance exercise on the prevention of falls and hip fracture among clinically defined high-risk elderly individuals: a randomized controlled trial. J Orthop Sci 2006;11:467–72.
28. Shimada H, Obuchi S, Furuna T, Suzuki T. New intervention program for preventing falls among frail elderly people: the effects of perturbed walking exercise using a bilateral separated treadmill. Am J Phys Med Rehabil 2004;83:493–9.
29. Schnelle JF, Kapur K, Alessi C, et al. Does an exercise and incontinence intervention save healthcare costs in a nursing home population? J Am Geriatr Soc 2003;51:161–8.
30. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities: A cluster randomized trial. Ann Intern Med 2002;136:733–41.
31. Dyer CAE. Falls prevention in residential care homes: a randomised controlled trial. Age Ageing 2004;33:596–602.
32. Huang T-T, Chung M-L, Chen F-R, Chin Y-F, Wang B-H. Evaluation of a combined cognitive-behavioural and exercise intervention to manage fear of falling among elderly residents in nursing homes. Aging Ment Health 2016;20:2–12.
33. Sihvonen S, Sipilä S, Taskinen S, Era P. Fall incidence in frail older women after individualized visual feedback-based balance training. Gerontology 2004;50:411–6.
34. Lord SR, Castell S, Corcoran J, et al. The effect of group exercise on physical functioning and falls in frail older people living in retirement villages: a randomized, controlled trial. J Am Geriatr Soc 2003;51:1685–92.
35. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
36. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
37. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities. A cluster randomized trial. Ann Intern Med 2002;136:733–41.
38. Moreland JD, Richardson JA, Goldsmith CH, Clase CM. Muscle weakness and falls in older adults: a systematic review and meta-analysis. J Am Geriatr Soc 2004;52: 1121–9.
39. Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, et al. Exercise and physical activity for older adults. Med Sci Sport Exerc 2009;41:1510–30.
40. de Souto Barreto P, Morley JE, Chodzko-Zajko W, et al. Recommendations on physical activity and exercise for older adults living in long-term care facilities: a taskforce report. J Am Med Dir Assoc 2016;17:381–92.
41. American College of Sports Medicine. Progression models in resistance training for healthy adults. Med Sci Sport Exerc 2009;41:687–708.
42. Fietzek UM, Schroeteler FE, Ziegler K, et al. Randomized cross-over trial to investigate the efficacy of a two-week physiotherapy programme with repetitive exercises of cueing to reduce the severity of freezing of gait in patients with Parkinson’s disease. Clin Rehabil 2014;28:902–11.
43. Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: A systematic evidence review. Alzheimer Dement 2007;3:341–7.
44. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
45. Christofoletti G, Oliani MM, Gobbi S, et al. A controlled clinical trial on the effects of motor intervention on balance and cognition in institutionalized elderly patients with dementia. Clin Rehabil 2008;22:618–26.
46. van Alphen HJM, Hortobágyi T, van Heuvelen MJG. Barriers, motivators, and facilitators of physical activity in dementia patients: A systematic review. Arch Gerontol Geriatr 2016;66:109–18.
47. Alzheimer Society of Ontario. Rethink Dementia. Accessed 18 Sep 2017 at http://rethinkdementia.ca/.
48. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with Alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
49. Brach JS, Perera S, Gilmore S, et al. Effectiveness of a timing and coordination group exercise program to improve mobility in community-dwelling older adults. JAMA Intern Med August 2017.
50. Rosher RB, Robinson S. Impact of the Eden alternative on family satisfaction. J Am Med Dir Assoc 2005;6:189–93.
51. Crandall LG, White DL, Schuldheis S, Talerico KA. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47–56.
52. Sims-Gould J, McKay HA, Feldman F, et al. Autonomy, choice, patient-centered care, and hip protectors: the experience of residents and staff in long-term care. J Appl Gerontol 2014;33:690–709.
53. Robinovitch SN, Cronin T. Perception of postural limits in elderly nursing home and day care participants. J Gerontol A Biol Sci Med Sci 1999;54:B124-30.
54. Perkins MM, Ball MM, Whittington FJ, Hollingsworth C. Relational autonomy in assisted living: a focus on diverse care settings for older adults. J Aging Stud 2012;26:214–25.
55. Brownie S, Nancarrow S. Effects of person-centered care on residents and staff in aged-care facilities: a systematic review. Clin Interv Aging 2013;8:1–10.
56. Coleman MT, Looney S, O’Brien J, et al. The Eden Alternative: findings after 1 year of implementation. J Gerontol A Biol Sci Med Sci 2002;57:M422–7.
57. Chenoweth L, King MT, Jeon Y-H, et al. Caring for Aged Dementia Care Resident Study (CADRES) of personcentred care, dementia-care mapping, and usual care in dementia: a cluster-randomised trial. Lancet Neurol 2009;8: 317–25.
58. Bergman-Evans B. Beyond the basics. Effects of the Eden Alternative model on quality of life issues. J Gerontol Nurs 2004;30:27–34.
59. Robinson SB, Rosher RB. Tangling with the barriers to culture change: creating a resident-centered nursing home environment. J Gerontol Nurs 2006;32:19–25.
60. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev 2012;12.
61. Papaioannou A, Santesso N, Morin SN, et al. Recommendations for preventing fracture in long-term care. Can Med Assoc J 2015;187:1135–44.
62. Stubbs B, Denkinger MD, Brefka S, Dallmeier D. What works to prevent falls in older adults dwelling in long term care facilities and hospitals? An umbrella review of meta-analyses of randomised controlled trials. Maturitas 2015;81:335–42.
From the Geriatric Education and Research in Aging Sciences Centre, McMaster University Hamilton, ON (Dr. McArthur) and the University of Waterloo and Research Institute for Aging, Waterloo, ON (Dr. Giangregorio), Canada
Abstract
- Objective: To synthesize the available literature on exercise and falls reduction interventions in long-term care (LTC) and provide practical information for clinicians and other decision makers.
- Methods: Review of positive trials included in systematic reviews.
- Results: Falls are a major concern for residents, families, clinicians, and decision-makers in LTC. Exercise is recommended as part of a multifactorial falls prevention program for residents in LTC. Strength and balance exercises should be incorporated into the multifactorial falls prevention program. They should be challenging and progressed as the residents’ abilities improve. Evidence suggests that exercises should be completed 2 to 3 times per week for a period longer than 6 months. Exercise programs in LTC should be resident-centered and should consider residents’ potential physical and cognitive impairments. Exercises in standing should be prioritized where appropriate.
- Conclusion: Appropriately challenging and progressive strength and balance exercises should be included in a multifactorial falls prevention program for residents in LTC.
Key words: long-term care; nursing homes; falls reduction; exercise.
Falls are common in long-term care (LTC) homes: the estimated falls rate is 1.5 falls per bed per year, which is 3 times greater than that for older adults living in the community [1]. Falls can have significant consequences for residents in LTC, including functional disability, fractures, pain, reduced quality of life, and death [1–6]. Indeed, 25% of residents who are hospitalized after a fall die within 1 year [3]. Consequently, falls prevention programs are important to help in reducing falls and averting the associated negative consequences.
Exercise may address the circumstances and physical deconditioning that often contribute to falls in LTC residents. Weight shifting [7], walking, and transferring [8–10], are common activities that precede falls, suggesting that balance, gait, and functional mobility training may be possible targets for prevention. Additionally, it is estimated that LTC residents spend three quarters of their waking time in sedentary activities [11,12] and have a high prevalence of sarcopenia [13–16]. Challenging balance training and resistance exercise are well-known intervention for reducing falls [17] and improving muscle strength for community-dwelling older adults [18]. However, evidence around balance and strength training for preventing falls in LTC is mixed [17,19,20], and careful planning and modification of exercises is necessary to meet the needs of LTC residents.
Residents in LTC are often medically complex, with multiple comorbidities [21] that can affect their ability to meaningfully participate in exercise. In Canada, 56.3% of residents have a diagnosis of Alzheimer’s or other dementias, 25.0% have diabetes, 14.4% have chronic obstructive pulmonary disease, and 21.2% have experienced a stroke [21]. Residents also often have significant functional impairments. For example, 97% of residents require assistance with basic activities of daily living [21]. Therefore, the lack of effect of exercise as a single falls prevention strategy observed in previous studies may be because the often complex, multimorbid LTC population likely requires a multifactorial approach to fall prevention [17]. Additionally, organizational aspects of LTC homes (eg, specific funds dedicated to employing exercise professionals and to support exercise programming) can affect residents’ engagement in exercise [22,23]. Subsequently, prescribing exercises in the LTC context must consider both resident characteristics and organizational features of the LTC home (eg, professionals available to support exercise programming).
A comprehensive exercise prescription describes the elements of an appropriate exercise program to facilitate implementation of that program. The exercise prescription should include a description of the type (eg, balance, strength) and intensity of exercises (eg, subjective or objective measurement of how hard the resident is working) included in the program [24]. The prescription should also include a description of the dose of exercise: frequency of exercise participation (eg, 2 days per week), duration of individual exercise sessions (eg, 30-minute sessions), and duration of exercise program (eg, 12-week program) [24]. Lastly, the prescription should describe the setting of the exercise program (eg, group or individual basis) and the professional delivering the program (eg, physiotherapist, fitness instructor) [24].
Therefore, the objectives of this article are to (1) synthesize studies demonstrating a positive effect of exercise on reducing falls for residents in LTC; (2) provide an overview of the principles of balance and strength training to guide clinicians in designing appropriate exercise prescription; and (3) make suggestions for clinical practice regarding an appropriate strength and balance exercise protocol by considering the influence of the LTC context.
Methods
To provide clinicians and other policy-makers with a description of which balance and strength exercises may be effective for preventing falls, we synthesized trials that demonstrated a positive effect on reducing falls or falls risk for residents in LTC. Studies were identified through a database search for systematic reviews in PubMed, Ovid, and Google Scholar using the keywords falls, long-term care, nursing homes, exercise, strength, balance, and systematic reviews. Our purpose was to provide practical information on what works to prevent falls through balance and strength training for residents in LTC rather than to evaluate the available evidence. Therefore, only positive trials from systematic reviews were discussed, as we wanted to present exercises that seem to have a positive effect on decreasing falls. Positive trials were defined as those included in identified systematic reviews with a risk or rate ratio and confidence intervals below 1.0.
We first provide an overview of the conclusions of the systematic reviews found in our search. Next, for each positive trial we describe the following elements of the exercise component of the intervention: frequency, time of sessions, length of program, intensity, type of exercise including a description of the specific exercises performed, whether the intervention was delivered in a group or on an individual basis, the professional delivering the intervention, and any other features of the intervention aside from the exercise component. We used the ProFaNE taxonomy definitions [25] to identify and describe each element of the exercise interventions. Frequency is the number of times per week that residents engage in sessions, time of sessions is the amount allocated to each exercise session, duration of program is how long the resident participates in the exercise program, and intensity is the subjective or objective report of how hard the resident is working [25]. The types of exercises described were those targeting balance defined as “...the efficient transfer of bodyweight from one part of the body to another or challenges specific aspects of the balance systems (eg, vestibular system)” [25], and strength defined as “...contracting the muscles against a resistance to ‘overload’ and bring about a training effect in the muscular system” [25]. Strength could be either an external resistance (eg, dumbbell) or using body weight against gravity (eg, squat) [25].
Results
We found 3 systematic reviews that include exercise programs to reduce falls in LTC homes [17,19,20]. Overall, evidence suggests that exercise should be included as part of a multifactorial falls prevention program for residents in LTC. There is limited evidence that exercise as a single intervention prevents falls, and some trials, albeit underpowered, even demonstrate an increased risk of falling in the exercise group compared to control [19]. With regards to specific exercise programs, the Cochrane review found that gait, balance, and functional training decrease the rate of falls but not the risk of falling [26–28], and the 2013 review by Silva et al [20] concluded that combined exercise programs (ie, multiple types of exercise) that include balance tasks, are completed frequently (2–3 times per week), and over a long term (greater than 6 months) were most effective at preventing falls [20].
A more recent systematic review and meta-analysis [17] also concluded that there was no evidence that exercise as a single intervention can prevent falls for residents in LTC. Table 1 provides a description of the exercise component of the seven positive trials [29–35] that were included in the 3 systematic reviews we identified in our search.
Type of Exercise
Balance Exercises
There were 4 positive trials that included balance exercises in their intervention [31,33–35]. Trials that had a positive effect on reducing falls and included balance training employed mostly dynamic balance exercises in standing (Table 1). However, only 2 of the 7 trials provided a detailed description of their balance exercises (Table 1) [26,34]. Jensen et al [30] and Dyer et al [31] did not include a description of the balance training performed but stated that balance was part of the multicomponent exercise program. Becker et al [36] stated that participants performed standing balance exercises, while Schnelle et al [39] and Huang et al [32] did not include balance training in their trial.
Strength Exercises
Of the 7 positive trials included in this review, 6 included strength exercises [29–32,34,35]. The strength activities used in trials where exercise had a positive effect on decreasing falls included functional activities [29,31] and progressive resistance training [31,36] (Table 1). Functional activities are those that replicate what a resident might be required to do in their everyday life, such as performing sit-to-stands out of a chair (Figure)
Frequency, Time of Sessions, Duration of Program
In our description of positive trials, exercise was performed on 2 to 3 days per week for 20 to 75 minutes per session, for periods ranging from 4 to 52 weeks (Table 1).
Intensity
For the trials including balance exercises, one trial described the intensity as resident-specific [37] and another as individualized [33]. Two studies did not describe the intensity of their balance exercises [31,34]. The intensity of strength exercises included in the positive trials was individualized for one of the trial [29]. Two trials had participants complete 2 to 3 sets of 10 repetitions [32,35], with one indicating an intensity of 12–13 or “somewhat difficult” on the Borg Rating of Perceived Exertion Scale [32] and the other using a 10-rep max [35]. Two studies described their strength exercises as progressive [31,37], and one at a moderate to high intensity [30]. Lord et al prescribed 30 repetitions of each strength exercise [34].
Delivery of Intervention
Exercise was delivered in a group setting for 4 of the trials [31,32,34,36], individually for 2 of the trials [26,29], and the setting was not described for one of the trials (Table 1) [30]. Finally, only 3 of the 7 articles reported the professional delivering the intervention: one was research staff [29], one was geriatric nurses [32], and one was exercise assistants supported by a physiotherapist [31].
Discussion
There is limited evidence to support the use of strength and balance exercise as a single intervention to prevent falls in LTC. However, exercise should be included as part of a multifactorial falls prevention program. Trials that had a positive effect on decreasing falls training used dynamic balance exercises in standing, functional training, and progressive resistance training on 2 to 3 days per week, for 20 to 75 minutes per session, over 4 to 52 weeks. The intensity of balance exercises was individualized, and strength exercises were described as somewhat difficult or performed at a moderate to high intensity. Exercise was performed in a group or individually, and was delivered by research staff, geriatric nurses, exercise assistants supervised by physiotherapists, or more frequently, it was not reported who delivered the intervention.
Balance Training
Our work suggests that standing, dynamic balance exercises may be best to decrease falls. Example balance exercises include reducing the base of support (eg, standing with feet together instead of apart, or tandem with one foot in front), moving the center of gravity and control body position while standing (eg, reaching, weight shifting, stepping up or down), and standing without using arms for support or reducing reliance on the upper limbs for support (eg, use one hand on a handrail instead of two, or two fingers instead of the whole hand) [17]. It is well established that balance training programs, especially those including challenging exercises, can prevent falls in community-dwelling older adults [17]. However, the relationship is not as clear in LTC.
Strength Training
Reduced muscle strength has been identified as an important risk factor for falls [38]. There are also many psychological and metabolic benefits to strength training [39]. To induce change in muscular strength, resistance exercises need to be challenging and progressive. Our work suggests that strength training that is effective at decreasing falls is functional and progressive, and is completed at a moderate to high intensity. A resident should be able to do a strength exercise for one to two sets of 6 to 8 repetitions before being fatigued [40]. Once the resident can complete two sets of 13 to 15 repetitions easily the exercise should be progressed. Residents who are particularly deconditioned may need to begin with lower intensity strength exercises (eg, only do one set, with a lower resistance and progress to a higher resistance) [40]. Residents should perform resistance exercises for all major muscle groups [40]. Progression could include increasing the number of sets (eg, increase from one to two sets), the resistance (eg, holding dumbbells while squatting), or the intensity of the exercise (eg, squat lower or faster) [41].
Implementing Exercise Programs in LTC
Implementation of exercise programs into LTC homes should consider the dose of exercise (eg, time and frequency of sessions, duration of program), if they are delivered in a group or individual setting, and who is delivering them. First, trials included in this paper suggest that strength and balance exercises to prevent falls were delivered 2 to 3 times per week, for 20 to 75 minutes per session, over 4 to 52 weeks. Second, previous work has established that exercise programs delivered on 2 to 3 days per week over a period of more than 6 months are most effective at reducing falls in LTC [20]. Finally, a recent task force report from an international group of clinician researchers in LTC recommends twice weekly exercise sessions lasting 35 to 45 minutes each [40]. Therefore, strength and balance exercises to prevent falls in LTC should be delivered at least twice per week, for at least 20 minutes, for greater than 6 weeks’ duration.
Whether exercise should be performed in a group or individual setting remains unclear. Two of the 6 positive trials in this paper were completed individually, while 3 were in a group. The aforementioned task force also recommended that every resident who does not have contraindications to exercise must have an individualized exercise program as part of their health care plan [40]. However, whether the exercise program is provided on an individual basis or in a group setting was not delineated. Indeed, there are currently no recommendations concerning prioritizing group or individual exercise programs. Therefore, exercise programs being implemented into LTC homes should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings.
Finally, which professionals should deliver the exercise program is also uncertain. Only 3 of the positive trials in this paper described the professional delivering the intervention, with one being research staff, one geriatric nurses, and one exercise assistants supported by a physiotherapist. We suggest that professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC.
Modifications for Physical Impairments
Residents in LTC often have complex health needs, with multiple comorbidities (eg, stroke, Parkinson’s disease, multiple sclerosis) [21]. Modifications of strength and balance exercises may be required to accommodate for physical impairments (eg, hemiplegia, drop foot, freezing gait). For example, if a resident has hemiplegia and cannot fully activate the muscles of one arm, one can do resistance exercises with a dumbbell on the functioning side and active assisted range of motion (ie, the exercise provider assists the resident to achieve full range of motion against gravity) on the hemiparetic side. A resident with Parkinson’s disease who has freezing gait may need visual or rhythmical verbal cues to be able to accomplish standing balance tasks such as altered walking patterns (eg, wide or narrow stepping) [42].
Modifications for Cognitive Impairments
More than 80% of residents in LTC have some degree of cognitive impairment [21]. Cognitive impairment may be the result of stroke, depression, traumatic injuries, medications, and degenerative diseases such as Parkinson’s and Alzheimer’s disease [43]. A common misconception is that residents with cognitive impairment cannot benefit from exercise because they cannot learn new skills and have difficulty following directions. On the contrary, evidence suggests that exercise can improve functional mobility for residents with cognitive impairment [44,45].
Residents with cognitive impairment may require a different approach to facilitate participation in the desired exercises because of difficulty following multi-step directions, responsive behaviors, or increased distractibility [46]. Clear communication is key in improving the quality of interaction for residents with cognitive impairment. The Alzheimer Society of Ontario suggests 10 strategies for communicating with people with dementia [47], and we have provided suggestions of how to apply these communication strategies to the exercise context in LTC (Table 2). Other suggestions for engaging residents with cognitive impairment in strength and balance training include making the exercises functional (eg, ask them to pick something up of the floor to perform a squat, or reach a point on the wall to do calf raises) and playful (eg, toss a ball back and forth or sing a song about rowing to promote weight shifting) [48].
Standing versus Seated Exercises
Residents may not be able to participate in standing exercises for several reasons: perhaps the resident cannot stand or has severe balance impairments and a high falls risk; the resident may have poor insight into which exercises are safe to perform in standing versus sitting; or there may be limited supervision of a large group exercise class where the risk of falls is a concern. If balance impairments are a concern, where the risk of injury or falling while completing exercises in standing outweighs the benefit of doing the exercises, then seated exercises are appropriate. However, when residents are able, we recommend encouraging some or all exercises in standing, to facilitate carry over of strength gains into functional tasks such as being able to rise from a chair and walking. A recent study, comparing standing versus seated exercises for community dwelling older adults, saw greater functional gains for those who completed the standing exercises [49]. Therefore, strength and balance exercises should be performed in standing, where appropriate.
Resident-Centered Exercise for Falls Prevention
Putting the resident at the center of falls prevention is important. Previous work has found that older adults have expressed a strong preference for care that transcends traditional biomedical care and that values efficiency, consistency, and hierarchical decision making [50]. On the contrary, resident-centered care emphasizes well-being and quality of life as defined by the resident, values giving residents greater control over the nature of services they receive, and respects their rights to be involved in every day decision making [51,52]. Indeed, residents may choose to engage in risky behaviors that increase their risk of falls but also increases their quality of life. Previous work has found disconnects between residents’ perceived frailty and the potential ability of protective devices to prevent adverse events, such as falls and fractures [53]. Additionally, one study identified that older residents feared being labelled, so instead hid impairments and chose to refuse assistance and assistive devices [54]. For example, a resident with impaired balance and gait may choose to walk independently when they have been deemed as requiring a gait aid (eg, rollator walker). However, they may value walking without a gait aid and accept the increased risk of falling. Therefore, it is essential to find the delicate balance between respecting a resident’s right to make their own decisions and preventing adverse events, such as falls [52]. An example of this would be respecting a resident’s right to refuse to attend exercise programming even though the team may think they can benefit from strength and balance training.
There is limited evidence around falls prevention and resident-centered care. A recent systematic review [55] revealed that resident-centered care may increase falls rates [56,57]. However, the authors of the review attributed the increase in falls to differences in frailty between the control and intervention group [56], and to environmental factors (eg, slippery flooring material, lack of handrails) [57]. Additionally, these trials did not include an exercise program as part of the resident-centered care program. On the other hand, resident-centered care has been associated with reduction of boredom, helplessness, and depression [58,59]. Most studies included in the review were quasi-experimental, which significantly limits the evidence quality [55]. At this point in time, the evidence suggests that resident-centered care is important for mood and quality of life but may have a negative or no effect on reducing falls.
Multifactorial Falls Prevention Programs
While there are mixed results about the effect of exercise as a single intervention for reducing falls for residents in LTC, the literature clearly supports exercise as part of a multifactorial falls prevention program [17,20,60–62]. A 2015 umbrella review [62] of meta-analyses of randomized controlled trials of falls prevention interventions in LTC concluded that multifactorial interventions were the most effective at preventing falls in LTC. Additionally, recently developed recommendations for fracture prevention in LTC [61] suggest that balance, strength, and functional training should be included for residents who are not at high risk of fracture, while for those at high risk, exercise should be provided as part of a multifactorial falls prevention intervention. Clinicians must therefore incorporate elements aside from exercise into their falls prevention strategies. Interventions that have shown positive effects on reducing falls when delivered as part of multifactorial interventions include: staff and resident education [31,35,37], environmental modifications [31,35], supply/repair/provision of assistive devices [30], falls problem-solving conferences [30], urinary incontinence management [29], medication review [30], optician review [31], and cognitive behavioral therapy [32].
Conclusion and Suggestions for Clinical Practice
We suggest incorporating strength and balance exercises as part of a multifactorial falls prevention program for residents in LTC. Balance exercises should be challenging and dynamic (eg, weight shifting). Strength exercises should be of a moderate to high intensity (eg, can complete one to sets of 6 to 8 repetitions) and need to be progressed as the residents’ abilities improve. Residents should participate in strength and balance training on 2 to 3 days per week, for 30- to 45-minute sessions, for at least 6 months. Exercises in standing should be prioritized where appropriate. Exercise could be delivered in a group or individual format, but should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings. Professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC. Exercise programs in LTC should be resident-centered and consider residents’ potential physical and cognitive impairments.
Funding/support: Dr. Giangregorio was supported by grants from the Canadian Frailty Network and Canadian Institutes of Health Research.
From the Geriatric Education and Research in Aging Sciences Centre, McMaster University Hamilton, ON (Dr. McArthur) and the University of Waterloo and Research Institute for Aging, Waterloo, ON (Dr. Giangregorio), Canada
Abstract
- Objective: To synthesize the available literature on exercise and falls reduction interventions in long-term care (LTC) and provide practical information for clinicians and other decision makers.
- Methods: Review of positive trials included in systematic reviews.
- Results: Falls are a major concern for residents, families, clinicians, and decision-makers in LTC. Exercise is recommended as part of a multifactorial falls prevention program for residents in LTC. Strength and balance exercises should be incorporated into the multifactorial falls prevention program. They should be challenging and progressed as the residents’ abilities improve. Evidence suggests that exercises should be completed 2 to 3 times per week for a period longer than 6 months. Exercise programs in LTC should be resident-centered and should consider residents’ potential physical and cognitive impairments. Exercises in standing should be prioritized where appropriate.
- Conclusion: Appropriately challenging and progressive strength and balance exercises should be included in a multifactorial falls prevention program for residents in LTC.
Key words: long-term care; nursing homes; falls reduction; exercise.
Falls are common in long-term care (LTC) homes: the estimated falls rate is 1.5 falls per bed per year, which is 3 times greater than that for older adults living in the community [1]. Falls can have significant consequences for residents in LTC, including functional disability, fractures, pain, reduced quality of life, and death [1–6]. Indeed, 25% of residents who are hospitalized after a fall die within 1 year [3]. Consequently, falls prevention programs are important to help in reducing falls and averting the associated negative consequences.
Exercise may address the circumstances and physical deconditioning that often contribute to falls in LTC residents. Weight shifting [7], walking, and transferring [8–10], are common activities that precede falls, suggesting that balance, gait, and functional mobility training may be possible targets for prevention. Additionally, it is estimated that LTC residents spend three quarters of their waking time in sedentary activities [11,12] and have a high prevalence of sarcopenia [13–16]. Challenging balance training and resistance exercise are well-known intervention for reducing falls [17] and improving muscle strength for community-dwelling older adults [18]. However, evidence around balance and strength training for preventing falls in LTC is mixed [17,19,20], and careful planning and modification of exercises is necessary to meet the needs of LTC residents.
Residents in LTC are often medically complex, with multiple comorbidities [21] that can affect their ability to meaningfully participate in exercise. In Canada, 56.3% of residents have a diagnosis of Alzheimer’s or other dementias, 25.0% have diabetes, 14.4% have chronic obstructive pulmonary disease, and 21.2% have experienced a stroke [21]. Residents also often have significant functional impairments. For example, 97% of residents require assistance with basic activities of daily living [21]. Therefore, the lack of effect of exercise as a single falls prevention strategy observed in previous studies may be because the often complex, multimorbid LTC population likely requires a multifactorial approach to fall prevention [17]. Additionally, organizational aspects of LTC homes (eg, specific funds dedicated to employing exercise professionals and to support exercise programming) can affect residents’ engagement in exercise [22,23]. Subsequently, prescribing exercises in the LTC context must consider both resident characteristics and organizational features of the LTC home (eg, professionals available to support exercise programming).
A comprehensive exercise prescription describes the elements of an appropriate exercise program to facilitate implementation of that program. The exercise prescription should include a description of the type (eg, balance, strength) and intensity of exercises (eg, subjective or objective measurement of how hard the resident is working) included in the program [24]. The prescription should also include a description of the dose of exercise: frequency of exercise participation (eg, 2 days per week), duration of individual exercise sessions (eg, 30-minute sessions), and duration of exercise program (eg, 12-week program) [24]. Lastly, the prescription should describe the setting of the exercise program (eg, group or individual basis) and the professional delivering the program (eg, physiotherapist, fitness instructor) [24].
Therefore, the objectives of this article are to (1) synthesize studies demonstrating a positive effect of exercise on reducing falls for residents in LTC; (2) provide an overview of the principles of balance and strength training to guide clinicians in designing appropriate exercise prescription; and (3) make suggestions for clinical practice regarding an appropriate strength and balance exercise protocol by considering the influence of the LTC context.
Methods
To provide clinicians and other policy-makers with a description of which balance and strength exercises may be effective for preventing falls, we synthesized trials that demonstrated a positive effect on reducing falls or falls risk for residents in LTC. Studies were identified through a database search for systematic reviews in PubMed, Ovid, and Google Scholar using the keywords falls, long-term care, nursing homes, exercise, strength, balance, and systematic reviews. Our purpose was to provide practical information on what works to prevent falls through balance and strength training for residents in LTC rather than to evaluate the available evidence. Therefore, only positive trials from systematic reviews were discussed, as we wanted to present exercises that seem to have a positive effect on decreasing falls. Positive trials were defined as those included in identified systematic reviews with a risk or rate ratio and confidence intervals below 1.0.
We first provide an overview of the conclusions of the systematic reviews found in our search. Next, for each positive trial we describe the following elements of the exercise component of the intervention: frequency, time of sessions, length of program, intensity, type of exercise including a description of the specific exercises performed, whether the intervention was delivered in a group or on an individual basis, the professional delivering the intervention, and any other features of the intervention aside from the exercise component. We used the ProFaNE taxonomy definitions [25] to identify and describe each element of the exercise interventions. Frequency is the number of times per week that residents engage in sessions, time of sessions is the amount allocated to each exercise session, duration of program is how long the resident participates in the exercise program, and intensity is the subjective or objective report of how hard the resident is working [25]. The types of exercises described were those targeting balance defined as “...the efficient transfer of bodyweight from one part of the body to another or challenges specific aspects of the balance systems (eg, vestibular system)” [25], and strength defined as “...contracting the muscles against a resistance to ‘overload’ and bring about a training effect in the muscular system” [25]. Strength could be either an external resistance (eg, dumbbell) or using body weight against gravity (eg, squat) [25].
Results
We found 3 systematic reviews that include exercise programs to reduce falls in LTC homes [17,19,20]. Overall, evidence suggests that exercise should be included as part of a multifactorial falls prevention program for residents in LTC. There is limited evidence that exercise as a single intervention prevents falls, and some trials, albeit underpowered, even demonstrate an increased risk of falling in the exercise group compared to control [19]. With regards to specific exercise programs, the Cochrane review found that gait, balance, and functional training decrease the rate of falls but not the risk of falling [26–28], and the 2013 review by Silva et al [20] concluded that combined exercise programs (ie, multiple types of exercise) that include balance tasks, are completed frequently (2–3 times per week), and over a long term (greater than 6 months) were most effective at preventing falls [20].
A more recent systematic review and meta-analysis [17] also concluded that there was no evidence that exercise as a single intervention can prevent falls for residents in LTC. Table 1 provides a description of the exercise component of the seven positive trials [29–35] that were included in the 3 systematic reviews we identified in our search.
Type of Exercise
Balance Exercises
There were 4 positive trials that included balance exercises in their intervention [31,33–35]. Trials that had a positive effect on reducing falls and included balance training employed mostly dynamic balance exercises in standing (Table 1). However, only 2 of the 7 trials provided a detailed description of their balance exercises (Table 1) [26,34]. Jensen et al [30] and Dyer et al [31] did not include a description of the balance training performed but stated that balance was part of the multicomponent exercise program. Becker et al [36] stated that participants performed standing balance exercises, while Schnelle et al [39] and Huang et al [32] did not include balance training in their trial.
Strength Exercises
Of the 7 positive trials included in this review, 6 included strength exercises [29–32,34,35]. The strength activities used in trials where exercise had a positive effect on decreasing falls included functional activities [29,31] and progressive resistance training [31,36] (Table 1). Functional activities are those that replicate what a resident might be required to do in their everyday life, such as performing sit-to-stands out of a chair (Figure)
Frequency, Time of Sessions, Duration of Program
In our description of positive trials, exercise was performed on 2 to 3 days per week for 20 to 75 minutes per session, for periods ranging from 4 to 52 weeks (Table 1).
Intensity
For the trials including balance exercises, one trial described the intensity as resident-specific [37] and another as individualized [33]. Two studies did not describe the intensity of their balance exercises [31,34]. The intensity of strength exercises included in the positive trials was individualized for one of the trial [29]. Two trials had participants complete 2 to 3 sets of 10 repetitions [32,35], with one indicating an intensity of 12–13 or “somewhat difficult” on the Borg Rating of Perceived Exertion Scale [32] and the other using a 10-rep max [35]. Two studies described their strength exercises as progressive [31,37], and one at a moderate to high intensity [30]. Lord et al prescribed 30 repetitions of each strength exercise [34].
Delivery of Intervention
Exercise was delivered in a group setting for 4 of the trials [31,32,34,36], individually for 2 of the trials [26,29], and the setting was not described for one of the trials (Table 1) [30]. Finally, only 3 of the 7 articles reported the professional delivering the intervention: one was research staff [29], one was geriatric nurses [32], and one was exercise assistants supported by a physiotherapist [31].
Discussion
There is limited evidence to support the use of strength and balance exercise as a single intervention to prevent falls in LTC. However, exercise should be included as part of a multifactorial falls prevention program. Trials that had a positive effect on decreasing falls training used dynamic balance exercises in standing, functional training, and progressive resistance training on 2 to 3 days per week, for 20 to 75 minutes per session, over 4 to 52 weeks. The intensity of balance exercises was individualized, and strength exercises were described as somewhat difficult or performed at a moderate to high intensity. Exercise was performed in a group or individually, and was delivered by research staff, geriatric nurses, exercise assistants supervised by physiotherapists, or more frequently, it was not reported who delivered the intervention.
Balance Training
Our work suggests that standing, dynamic balance exercises may be best to decrease falls. Example balance exercises include reducing the base of support (eg, standing with feet together instead of apart, or tandem with one foot in front), moving the center of gravity and control body position while standing (eg, reaching, weight shifting, stepping up or down), and standing without using arms for support or reducing reliance on the upper limbs for support (eg, use one hand on a handrail instead of two, or two fingers instead of the whole hand) [17]. It is well established that balance training programs, especially those including challenging exercises, can prevent falls in community-dwelling older adults [17]. However, the relationship is not as clear in LTC.
Strength Training
Reduced muscle strength has been identified as an important risk factor for falls [38]. There are also many psychological and metabolic benefits to strength training [39]. To induce change in muscular strength, resistance exercises need to be challenging and progressive. Our work suggests that strength training that is effective at decreasing falls is functional and progressive, and is completed at a moderate to high intensity. A resident should be able to do a strength exercise for one to two sets of 6 to 8 repetitions before being fatigued [40]. Once the resident can complete two sets of 13 to 15 repetitions easily the exercise should be progressed. Residents who are particularly deconditioned may need to begin with lower intensity strength exercises (eg, only do one set, with a lower resistance and progress to a higher resistance) [40]. Residents should perform resistance exercises for all major muscle groups [40]. Progression could include increasing the number of sets (eg, increase from one to two sets), the resistance (eg, holding dumbbells while squatting), or the intensity of the exercise (eg, squat lower or faster) [41].
Implementing Exercise Programs in LTC
Implementation of exercise programs into LTC homes should consider the dose of exercise (eg, time and frequency of sessions, duration of program), if they are delivered in a group or individual setting, and who is delivering them. First, trials included in this paper suggest that strength and balance exercises to prevent falls were delivered 2 to 3 times per week, for 20 to 75 minutes per session, over 4 to 52 weeks. Second, previous work has established that exercise programs delivered on 2 to 3 days per week over a period of more than 6 months are most effective at reducing falls in LTC [20]. Finally, a recent task force report from an international group of clinician researchers in LTC recommends twice weekly exercise sessions lasting 35 to 45 minutes each [40]. Therefore, strength and balance exercises to prevent falls in LTC should be delivered at least twice per week, for at least 20 minutes, for greater than 6 weeks’ duration.
Whether exercise should be performed in a group or individual setting remains unclear. Two of the 6 positive trials in this paper were completed individually, while 3 were in a group. The aforementioned task force also recommended that every resident who does not have contraindications to exercise must have an individualized exercise program as part of their health care plan [40]. However, whether the exercise program is provided on an individual basis or in a group setting was not delineated. Indeed, there are currently no recommendations concerning prioritizing group or individual exercise programs. Therefore, exercise programs being implemented into LTC homes should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings.
Finally, which professionals should deliver the exercise program is also uncertain. Only 3 of the positive trials in this paper described the professional delivering the intervention, with one being research staff, one geriatric nurses, and one exercise assistants supported by a physiotherapist. We suggest that professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC.
Modifications for Physical Impairments
Residents in LTC often have complex health needs, with multiple comorbidities (eg, stroke, Parkinson’s disease, multiple sclerosis) [21]. Modifications of strength and balance exercises may be required to accommodate for physical impairments (eg, hemiplegia, drop foot, freezing gait). For example, if a resident has hemiplegia and cannot fully activate the muscles of one arm, one can do resistance exercises with a dumbbell on the functioning side and active assisted range of motion (ie, the exercise provider assists the resident to achieve full range of motion against gravity) on the hemiparetic side. A resident with Parkinson’s disease who has freezing gait may need visual or rhythmical verbal cues to be able to accomplish standing balance tasks such as altered walking patterns (eg, wide or narrow stepping) [42].
Modifications for Cognitive Impairments
More than 80% of residents in LTC have some degree of cognitive impairment [21]. Cognitive impairment may be the result of stroke, depression, traumatic injuries, medications, and degenerative diseases such as Parkinson’s and Alzheimer’s disease [43]. A common misconception is that residents with cognitive impairment cannot benefit from exercise because they cannot learn new skills and have difficulty following directions. On the contrary, evidence suggests that exercise can improve functional mobility for residents with cognitive impairment [44,45].
Residents with cognitive impairment may require a different approach to facilitate participation in the desired exercises because of difficulty following multi-step directions, responsive behaviors, or increased distractibility [46]. Clear communication is key in improving the quality of interaction for residents with cognitive impairment. The Alzheimer Society of Ontario suggests 10 strategies for communicating with people with dementia [47], and we have provided suggestions of how to apply these communication strategies to the exercise context in LTC (Table 2). Other suggestions for engaging residents with cognitive impairment in strength and balance training include making the exercises functional (eg, ask them to pick something up of the floor to perform a squat, or reach a point on the wall to do calf raises) and playful (eg, toss a ball back and forth or sing a song about rowing to promote weight shifting) [48].
Standing versus Seated Exercises
Residents may not be able to participate in standing exercises for several reasons: perhaps the resident cannot stand or has severe balance impairments and a high falls risk; the resident may have poor insight into which exercises are safe to perform in standing versus sitting; or there may be limited supervision of a large group exercise class where the risk of falls is a concern. If balance impairments are a concern, where the risk of injury or falling while completing exercises in standing outweighs the benefit of doing the exercises, then seated exercises are appropriate. However, when residents are able, we recommend encouraging some or all exercises in standing, to facilitate carry over of strength gains into functional tasks such as being able to rise from a chair and walking. A recent study, comparing standing versus seated exercises for community dwelling older adults, saw greater functional gains for those who completed the standing exercises [49]. Therefore, strength and balance exercises should be performed in standing, where appropriate.
Resident-Centered Exercise for Falls Prevention
Putting the resident at the center of falls prevention is important. Previous work has found that older adults have expressed a strong preference for care that transcends traditional biomedical care and that values efficiency, consistency, and hierarchical decision making [50]. On the contrary, resident-centered care emphasizes well-being and quality of life as defined by the resident, values giving residents greater control over the nature of services they receive, and respects their rights to be involved in every day decision making [51,52]. Indeed, residents may choose to engage in risky behaviors that increase their risk of falls but also increases their quality of life. Previous work has found disconnects between residents’ perceived frailty and the potential ability of protective devices to prevent adverse events, such as falls and fractures [53]. Additionally, one study identified that older residents feared being labelled, so instead hid impairments and chose to refuse assistance and assistive devices [54]. For example, a resident with impaired balance and gait may choose to walk independently when they have been deemed as requiring a gait aid (eg, rollator walker). However, they may value walking without a gait aid and accept the increased risk of falling. Therefore, it is essential to find the delicate balance between respecting a resident’s right to make their own decisions and preventing adverse events, such as falls [52]. An example of this would be respecting a resident’s right to refuse to attend exercise programming even though the team may think they can benefit from strength and balance training.
There is limited evidence around falls prevention and resident-centered care. A recent systematic review [55] revealed that resident-centered care may increase falls rates [56,57]. However, the authors of the review attributed the increase in falls to differences in frailty between the control and intervention group [56], and to environmental factors (eg, slippery flooring material, lack of handrails) [57]. Additionally, these trials did not include an exercise program as part of the resident-centered care program. On the other hand, resident-centered care has been associated with reduction of boredom, helplessness, and depression [58,59]. Most studies included in the review were quasi-experimental, which significantly limits the evidence quality [55]. At this point in time, the evidence suggests that resident-centered care is important for mood and quality of life but may have a negative or no effect on reducing falls.
Multifactorial Falls Prevention Programs
While there are mixed results about the effect of exercise as a single intervention for reducing falls for residents in LTC, the literature clearly supports exercise as part of a multifactorial falls prevention program [17,20,60–62]. A 2015 umbrella review [62] of meta-analyses of randomized controlled trials of falls prevention interventions in LTC concluded that multifactorial interventions were the most effective at preventing falls in LTC. Additionally, recently developed recommendations for fracture prevention in LTC [61] suggest that balance, strength, and functional training should be included for residents who are not at high risk of fracture, while for those at high risk, exercise should be provided as part of a multifactorial falls prevention intervention. Clinicians must therefore incorporate elements aside from exercise into their falls prevention strategies. Interventions that have shown positive effects on reducing falls when delivered as part of multifactorial interventions include: staff and resident education [31,35,37], environmental modifications [31,35], supply/repair/provision of assistive devices [30], falls problem-solving conferences [30], urinary incontinence management [29], medication review [30], optician review [31], and cognitive behavioral therapy [32].
Conclusion and Suggestions for Clinical Practice
We suggest incorporating strength and balance exercises as part of a multifactorial falls prevention program for residents in LTC. Balance exercises should be challenging and dynamic (eg, weight shifting). Strength exercises should be of a moderate to high intensity (eg, can complete one to sets of 6 to 8 repetitions) and need to be progressed as the residents’ abilities improve. Residents should participate in strength and balance training on 2 to 3 days per week, for 30- to 45-minute sessions, for at least 6 months. Exercises in standing should be prioritized where appropriate. Exercise could be delivered in a group or individual format, but should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings. Professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC. Exercise programs in LTC should be resident-centered and consider residents’ potential physical and cognitive impairments.
Funding/support: Dr. Giangregorio was supported by grants from the Canadian Frailty Network and Canadian Institutes of Health Research.
1. Harris IA, Yong S, McEvoy L, Thorn L. A prospective study of the effect of nursing home residency on mortality following hip fracture. ANZ J Surg 2010;80:447–50.
2. Ooms ME, Vlasman P, Lips P, et al. The incidence of hip fractures in independent and institutionalized elderly people. Osteoporos Int 1994;4:6–10.
3. Ayoung-Chee P, McIntyre L, Ebel BE, et al. Long-term outcomes of ground-level falls in the elderly. J Trauma Acute Care Surg 2014;76:498–503.
4. Heinrich S, Rapp K, Rissmann U, et al. Cost of falls in old age: a systematic review. Osteoporos Int 2010;21: 891–902.
5. Rubenstein LZ, Josephson KR, Robbins AS. Falls in the nursing home. Ann Intern Med 1994;121:442–51.
6. Hartholt KA, van Beeck EF, Polinder S, et al. Societal consequences of falls in the older population: injuries, healthcare costs, and long-term reduced quality of life. J Trauma
2011;71:748–53.
7. Robinovitch SN, Feldman F, Yang Y, et al. Video capture of the circumstances of falls in elderly people residing in long-term care: an observational study. Lancet 2013;381:
47–54.
8. Rapp K, Becker C, Cameron ID, et al. Epidemiology of falls in residential aged care: analysis of more than 70,000 falls from residents of bavarian nursing homes. J Am Med Dir Assoc 2012;13:187.
9. Büchele G, Becker C, Cameron ID, et al. Predictors of serious consequences of falls in residential aged care: analysis of more than 70,000 falls from residents of Bavarian nursing homes. J Am Med Dir Assoc 2014;15:559–63.
10. McArthur C, Gonzalez DA, Roy E, Giangregorio L. What are the circumstances of falls and fractures in long-term care? Can J Aging / La Rev Can du Vieil 2016;35:491–8.
11. Chin A Paw MJM, van Poppel MNM, van Mechelen W. Effects of resistance and functional-skills training on habitual activity and constipation among older adults living in long-term care facilities: a randomized controlled trial. BMC Geriatr 2006;6:9.
12. Ikezoe T, Asakawa Y, Shima H, et al. Daytime physical activity patterns and physical fitness in institutionalized elderly women: an exploratory study. Arch Gerontol Geriatr 2013;57:221–5.
13. Senior HE, Henwood TR, Beller EM, et al. Prevalence and risk factors of sarcopenia among adults living in nursing homes. Maturitas 2015;82:418–23.
14. Smoliner C, Sieber CC, Wirth R. Prevalence of sarcopenia in geriatric hospitalized patients. J Am Med Dir Assoc 2014;15:267–72.
15. Landi F, Liperoti R, Fusco D, et al. Sarcopenia and mortality among older nursing home residents. J Am Med Dir Assoc 2012;13:121–6.
16. Yalcin A, Aras S, Atmis V, et al. Sarcopenia prevalence and factors associated with sarcopenia in older people living in a nursing home in Ankara Turkey. Geriatr Gerontol Int
2016;16:903–10.
17. Sherrington C, Michaleff ZA, Fairhall N, et al. Exercise to prevent falls in older adults: an updated systematic review and meta-analysis. Br J Sports Med October 2016.
18. Liu C, Latham NK. Progressive resistance strength training for improving physical function in older adults. In: Liu C, ed. Cochrane Database Syst Rev;2009:CD002759.
19. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev;2012:CD005465.
20. Silva RB, Eslick GD, Duque G. Exercise for falls and fracture prevention in long term care facilities: a systematic review and meta-analysis. J Am Med Dir Assoc 2013;14:685–9.
21. Hirdes JP, Mitchell L, Maxwell CJ, White N. Beyond the “iron lungs of gerontology”: Using evidence to shape the future of nursing homes in Canada. Can J Aging 2011;30: 371–90.
22. Benjamin K, Edwards N, Guitard P, et al. Factors that influence physical activity in long-term care: Perspectives of residents, staff, and significant others. Can J Aging 2011;30:247–58.
23. Benjamin K, Edwards N, Ploeg J, Legault F. Barriers to physical activity and restorative care for residents in long-term care: A review of the literature. J Aging Phys Act 2014;22:154–65.
24. American College of Sports Medicine. ACSM’s guidelines for exercise testing and prescription. 9th ed. American College of Sports Medicine; 2013.
25. Prevention of Falls Network Europe. Prevention of Falls Network Europe. Accessed 27 Nov 2017 at www.profane.eu.org/.
26. Sihvonen SE, Sipilä S, Era PA. Changes in postural balance in frail elderly women during a 4-week visual feedback training: a randomized controlled trial. Gerontology 2004;50:87–95.
27. Sakamoto K, Nakamura T, Hagino H, et al. Effects of unipedal standing balance exercise on the prevention of falls and hip fracture among clinically defined high-risk elderly individuals: a randomized controlled trial. J Orthop Sci 2006;11:467–72.
28. Shimada H, Obuchi S, Furuna T, Suzuki T. New intervention program for preventing falls among frail elderly people: the effects of perturbed walking exercise using a bilateral separated treadmill. Am J Phys Med Rehabil 2004;83:493–9.
29. Schnelle JF, Kapur K, Alessi C, et al. Does an exercise and incontinence intervention save healthcare costs in a nursing home population? J Am Geriatr Soc 2003;51:161–8.
30. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities: A cluster randomized trial. Ann Intern Med 2002;136:733–41.
31. Dyer CAE. Falls prevention in residential care homes: a randomised controlled trial. Age Ageing 2004;33:596–602.
32. Huang T-T, Chung M-L, Chen F-R, Chin Y-F, Wang B-H. Evaluation of a combined cognitive-behavioural and exercise intervention to manage fear of falling among elderly residents in nursing homes. Aging Ment Health 2016;20:2–12.
33. Sihvonen S, Sipilä S, Taskinen S, Era P. Fall incidence in frail older women after individualized visual feedback-based balance training. Gerontology 2004;50:411–6.
34. Lord SR, Castell S, Corcoran J, et al. The effect of group exercise on physical functioning and falls in frail older people living in retirement villages: a randomized, controlled trial. J Am Geriatr Soc 2003;51:1685–92.
35. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
36. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
37. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities. A cluster randomized trial. Ann Intern Med 2002;136:733–41.
38. Moreland JD, Richardson JA, Goldsmith CH, Clase CM. Muscle weakness and falls in older adults: a systematic review and meta-analysis. J Am Geriatr Soc 2004;52: 1121–9.
39. Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, et al. Exercise and physical activity for older adults. Med Sci Sport Exerc 2009;41:1510–30.
40. de Souto Barreto P, Morley JE, Chodzko-Zajko W, et al. Recommendations on physical activity and exercise for older adults living in long-term care facilities: a taskforce report. J Am Med Dir Assoc 2016;17:381–92.
41. American College of Sports Medicine. Progression models in resistance training for healthy adults. Med Sci Sport Exerc 2009;41:687–708.
42. Fietzek UM, Schroeteler FE, Ziegler K, et al. Randomized cross-over trial to investigate the efficacy of a two-week physiotherapy programme with repetitive exercises of cueing to reduce the severity of freezing of gait in patients with Parkinson’s disease. Clin Rehabil 2014;28:902–11.
43. Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: A systematic evidence review. Alzheimer Dement 2007;3:341–7.
44. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
45. Christofoletti G, Oliani MM, Gobbi S, et al. A controlled clinical trial on the effects of motor intervention on balance and cognition in institutionalized elderly patients with dementia. Clin Rehabil 2008;22:618–26.
46. van Alphen HJM, Hortobágyi T, van Heuvelen MJG. Barriers, motivators, and facilitators of physical activity in dementia patients: A systematic review. Arch Gerontol Geriatr 2016;66:109–18.
47. Alzheimer Society of Ontario. Rethink Dementia. Accessed 18 Sep 2017 at http://rethinkdementia.ca/.
48. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with Alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
49. Brach JS, Perera S, Gilmore S, et al. Effectiveness of a timing and coordination group exercise program to improve mobility in community-dwelling older adults. JAMA Intern Med August 2017.
50. Rosher RB, Robinson S. Impact of the Eden alternative on family satisfaction. J Am Med Dir Assoc 2005;6:189–93.
51. Crandall LG, White DL, Schuldheis S, Talerico KA. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47–56.
52. Sims-Gould J, McKay HA, Feldman F, et al. Autonomy, choice, patient-centered care, and hip protectors: the experience of residents and staff in long-term care. J Appl Gerontol 2014;33:690–709.
53. Robinovitch SN, Cronin T. Perception of postural limits in elderly nursing home and day care participants. J Gerontol A Biol Sci Med Sci 1999;54:B124-30.
54. Perkins MM, Ball MM, Whittington FJ, Hollingsworth C. Relational autonomy in assisted living: a focus on diverse care settings for older adults. J Aging Stud 2012;26:214–25.
55. Brownie S, Nancarrow S. Effects of person-centered care on residents and staff in aged-care facilities: a systematic review. Clin Interv Aging 2013;8:1–10.
56. Coleman MT, Looney S, O’Brien J, et al. The Eden Alternative: findings after 1 year of implementation. J Gerontol A Biol Sci Med Sci 2002;57:M422–7.
57. Chenoweth L, King MT, Jeon Y-H, et al. Caring for Aged Dementia Care Resident Study (CADRES) of personcentred care, dementia-care mapping, and usual care in dementia: a cluster-randomised trial. Lancet Neurol 2009;8: 317–25.
58. Bergman-Evans B. Beyond the basics. Effects of the Eden Alternative model on quality of life issues. J Gerontol Nurs 2004;30:27–34.
59. Robinson SB, Rosher RB. Tangling with the barriers to culture change: creating a resident-centered nursing home environment. J Gerontol Nurs 2006;32:19–25.
60. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev 2012;12.
61. Papaioannou A, Santesso N, Morin SN, et al. Recommendations for preventing fracture in long-term care. Can Med Assoc J 2015;187:1135–44.
62. Stubbs B, Denkinger MD, Brefka S, Dallmeier D. What works to prevent falls in older adults dwelling in long term care facilities and hospitals? An umbrella review of meta-analyses of randomised controlled trials. Maturitas 2015;81:335–42.
1. Harris IA, Yong S, McEvoy L, Thorn L. A prospective study of the effect of nursing home residency on mortality following hip fracture. ANZ J Surg 2010;80:447–50.
2. Ooms ME, Vlasman P, Lips P, et al. The incidence of hip fractures in independent and institutionalized elderly people. Osteoporos Int 1994;4:6–10.
3. Ayoung-Chee P, McIntyre L, Ebel BE, et al. Long-term outcomes of ground-level falls in the elderly. J Trauma Acute Care Surg 2014;76:498–503.
4. Heinrich S, Rapp K, Rissmann U, et al. Cost of falls in old age: a systematic review. Osteoporos Int 2010;21: 891–902.
5. Rubenstein LZ, Josephson KR, Robbins AS. Falls in the nursing home. Ann Intern Med 1994;121:442–51.
6. Hartholt KA, van Beeck EF, Polinder S, et al. Societal consequences of falls in the older population: injuries, healthcare costs, and long-term reduced quality of life. J Trauma
2011;71:748–53.
7. Robinovitch SN, Feldman F, Yang Y, et al. Video capture of the circumstances of falls in elderly people residing in long-term care: an observational study. Lancet 2013;381:
47–54.
8. Rapp K, Becker C, Cameron ID, et al. Epidemiology of falls in residential aged care: analysis of more than 70,000 falls from residents of bavarian nursing homes. J Am Med Dir Assoc 2012;13:187.
9. Büchele G, Becker C, Cameron ID, et al. Predictors of serious consequences of falls in residential aged care: analysis of more than 70,000 falls from residents of Bavarian nursing homes. J Am Med Dir Assoc 2014;15:559–63.
10. McArthur C, Gonzalez DA, Roy E, Giangregorio L. What are the circumstances of falls and fractures in long-term care? Can J Aging / La Rev Can du Vieil 2016;35:491–8.
11. Chin A Paw MJM, van Poppel MNM, van Mechelen W. Effects of resistance and functional-skills training on habitual activity and constipation among older adults living in long-term care facilities: a randomized controlled trial. BMC Geriatr 2006;6:9.
12. Ikezoe T, Asakawa Y, Shima H, et al. Daytime physical activity patterns and physical fitness in institutionalized elderly women: an exploratory study. Arch Gerontol Geriatr 2013;57:221–5.
13. Senior HE, Henwood TR, Beller EM, et al. Prevalence and risk factors of sarcopenia among adults living in nursing homes. Maturitas 2015;82:418–23.
14. Smoliner C, Sieber CC, Wirth R. Prevalence of sarcopenia in geriatric hospitalized patients. J Am Med Dir Assoc 2014;15:267–72.
15. Landi F, Liperoti R, Fusco D, et al. Sarcopenia and mortality among older nursing home residents. J Am Med Dir Assoc 2012;13:121–6.
16. Yalcin A, Aras S, Atmis V, et al. Sarcopenia prevalence and factors associated with sarcopenia in older people living in a nursing home in Ankara Turkey. Geriatr Gerontol Int
2016;16:903–10.
17. Sherrington C, Michaleff ZA, Fairhall N, et al. Exercise to prevent falls in older adults: an updated systematic review and meta-analysis. Br J Sports Med October 2016.
18. Liu C, Latham NK. Progressive resistance strength training for improving physical function in older adults. In: Liu C, ed. Cochrane Database Syst Rev;2009:CD002759.
19. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev;2012:CD005465.
20. Silva RB, Eslick GD, Duque G. Exercise for falls and fracture prevention in long term care facilities: a systematic review and meta-analysis. J Am Med Dir Assoc 2013;14:685–9.
21. Hirdes JP, Mitchell L, Maxwell CJ, White N. Beyond the “iron lungs of gerontology”: Using evidence to shape the future of nursing homes in Canada. Can J Aging 2011;30: 371–90.
22. Benjamin K, Edwards N, Guitard P, et al. Factors that influence physical activity in long-term care: Perspectives of residents, staff, and significant others. Can J Aging 2011;30:247–58.
23. Benjamin K, Edwards N, Ploeg J, Legault F. Barriers to physical activity and restorative care for residents in long-term care: A review of the literature. J Aging Phys Act 2014;22:154–65.
24. American College of Sports Medicine. ACSM’s guidelines for exercise testing and prescription. 9th ed. American College of Sports Medicine; 2013.
25. Prevention of Falls Network Europe. Prevention of Falls Network Europe. Accessed 27 Nov 2017 at www.profane.eu.org/.
26. Sihvonen SE, Sipilä S, Era PA. Changes in postural balance in frail elderly women during a 4-week visual feedback training: a randomized controlled trial. Gerontology 2004;50:87–95.
27. Sakamoto K, Nakamura T, Hagino H, et al. Effects of unipedal standing balance exercise on the prevention of falls and hip fracture among clinically defined high-risk elderly individuals: a randomized controlled trial. J Orthop Sci 2006;11:467–72.
28. Shimada H, Obuchi S, Furuna T, Suzuki T. New intervention program for preventing falls among frail elderly people: the effects of perturbed walking exercise using a bilateral separated treadmill. Am J Phys Med Rehabil 2004;83:493–9.
29. Schnelle JF, Kapur K, Alessi C, et al. Does an exercise and incontinence intervention save healthcare costs in a nursing home population? J Am Geriatr Soc 2003;51:161–8.
30. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities: A cluster randomized trial. Ann Intern Med 2002;136:733–41.
31. Dyer CAE. Falls prevention in residential care homes: a randomised controlled trial. Age Ageing 2004;33:596–602.
32. Huang T-T, Chung M-L, Chen F-R, Chin Y-F, Wang B-H. Evaluation of a combined cognitive-behavioural and exercise intervention to manage fear of falling among elderly residents in nursing homes. Aging Ment Health 2016;20:2–12.
33. Sihvonen S, Sipilä S, Taskinen S, Era P. Fall incidence in frail older women after individualized visual feedback-based balance training. Gerontology 2004;50:411–6.
34. Lord SR, Castell S, Corcoran J, et al. The effect of group exercise on physical functioning and falls in frail older people living in retirement villages: a randomized, controlled trial. J Am Geriatr Soc 2003;51:1685–92.
35. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
36. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
37. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities. A cluster randomized trial. Ann Intern Med 2002;136:733–41.
38. Moreland JD, Richardson JA, Goldsmith CH, Clase CM. Muscle weakness and falls in older adults: a systematic review and meta-analysis. J Am Geriatr Soc 2004;52: 1121–9.
39. Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, et al. Exercise and physical activity for older adults. Med Sci Sport Exerc 2009;41:1510–30.
40. de Souto Barreto P, Morley JE, Chodzko-Zajko W, et al. Recommendations on physical activity and exercise for older adults living in long-term care facilities: a taskforce report. J Am Med Dir Assoc 2016;17:381–92.
41. American College of Sports Medicine. Progression models in resistance training for healthy adults. Med Sci Sport Exerc 2009;41:687–708.
42. Fietzek UM, Schroeteler FE, Ziegler K, et al. Randomized cross-over trial to investigate the efficacy of a two-week physiotherapy programme with repetitive exercises of cueing to reduce the severity of freezing of gait in patients with Parkinson’s disease. Clin Rehabil 2014;28:902–11.
43. Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: A systematic evidence review. Alzheimer Dement 2007;3:341–7.
44. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
45. Christofoletti G, Oliani MM, Gobbi S, et al. A controlled clinical trial on the effects of motor intervention on balance and cognition in institutionalized elderly patients with dementia. Clin Rehabil 2008;22:618–26.
46. van Alphen HJM, Hortobágyi T, van Heuvelen MJG. Barriers, motivators, and facilitators of physical activity in dementia patients: A systematic review. Arch Gerontol Geriatr 2016;66:109–18.
47. Alzheimer Society of Ontario. Rethink Dementia. Accessed 18 Sep 2017 at http://rethinkdementia.ca/.
48. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with Alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
49. Brach JS, Perera S, Gilmore S, et al. Effectiveness of a timing and coordination group exercise program to improve mobility in community-dwelling older adults. JAMA Intern Med August 2017.
50. Rosher RB, Robinson S. Impact of the Eden alternative on family satisfaction. J Am Med Dir Assoc 2005;6:189–93.
51. Crandall LG, White DL, Schuldheis S, Talerico KA. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47–56.
52. Sims-Gould J, McKay HA, Feldman F, et al. Autonomy, choice, patient-centered care, and hip protectors: the experience of residents and staff in long-term care. J Appl Gerontol 2014;33:690–709.
53. Robinovitch SN, Cronin T. Perception of postural limits in elderly nursing home and day care participants. J Gerontol A Biol Sci Med Sci 1999;54:B124-30.
54. Perkins MM, Ball MM, Whittington FJ, Hollingsworth C. Relational autonomy in assisted living: a focus on diverse care settings for older adults. J Aging Stud 2012;26:214–25.
55. Brownie S, Nancarrow S. Effects of person-centered care on residents and staff in aged-care facilities: a systematic review. Clin Interv Aging 2013;8:1–10.
56. Coleman MT, Looney S, O’Brien J, et al. The Eden Alternative: findings after 1 year of implementation. J Gerontol A Biol Sci Med Sci 2002;57:M422–7.
57. Chenoweth L, King MT, Jeon Y-H, et al. Caring for Aged Dementia Care Resident Study (CADRES) of personcentred care, dementia-care mapping, and usual care in dementia: a cluster-randomised trial. Lancet Neurol 2009;8: 317–25.
58. Bergman-Evans B. Beyond the basics. Effects of the Eden Alternative model on quality of life issues. J Gerontol Nurs 2004;30:27–34.
59. Robinson SB, Rosher RB. Tangling with the barriers to culture change: creating a resident-centered nursing home environment. J Gerontol Nurs 2006;32:19–25.
60. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev 2012;12.
61. Papaioannou A, Santesso N, Morin SN, et al. Recommendations for preventing fracture in long-term care. Can Med Assoc J 2015;187:1135–44.
62. Stubbs B, Denkinger MD, Brefka S, Dallmeier D. What works to prevent falls in older adults dwelling in long term care facilities and hospitals? An umbrella review of meta-analyses of randomised controlled trials. Maturitas 2015;81:335–42.
Screening for Metabolic Syndrome in People with Severe Mental Illness
From the University of California San Francisco, Department of Psychiatry, Weill Institute for Neurosciences, San Francisco, CA.
Abstract
- Objective: To review screening for metabolic syndrome in people with severe mental illness (SMI).
- Methods: Review of the literature.
- Results: Despite evidence-based metabolic screening guidelines, rates of metabolic screening remain low among people with SMI. Barriers to screening exist at the individual, organizational, and systems levels. Interventions to address these barriers range from point-of-care tools to systems-level reorganization towards population-based care.
- Conclusion: Greater systems-level interventions, particularly those that improve collaboration between mental health and primary care, are needed to improve metabolic monitoring and identify cardiovascular disease risk among people with SMI.
Key words: metabolic monitoring; severe mental illness; metabolic syndrome; integrated care.
People with severe mental illness (SMI) have a life expectancy 10 to 20 years shorter than the general population, and cardiometabolic risk factors contribute significantly to the increased morbidity and mortality seen in this population. To address this health disparity, metabolic monitoring guidelines have been proposed as a mechanism to identify metabolic risk factors. This paper aims to discuss metabolic syndrome and its risk factors, describe metabolic monitoring including current rates and barriers to screening, and identify interventions that may improve rates of screening for metabolic syndrome among people with SMI.
Metabolic syndrome has been conceptualized as a state of chronic low-grade inflammation and hypercoagulation associated with hypertension, dyslipidemia, glucose intolerance, insulin resistance, and visceral adiposity [1]. Per the modified National Cholesterol Education Program Adult Treatment Plan III (NCEP ATP III) guidelines, metabolic syndrome is defined as the presence of 3 of the following 5 parameters: (1) blood glucose > 100 mg/dL (or a person is taking a hypoglycemic medication), (2) high density lipoprotein (HDL) < 40 mg/dL in men or < 50 mg/dL in women, (3) triglycerides > 150 mg/dL (or taking a lipid lowering agent), (4) waist circumference > 40 inches in men or > 35 inches in women, and/or (5) blood pressure > 130/85 mm Hg (or taking an antihypertensive medication) [2,3] (Table 1). 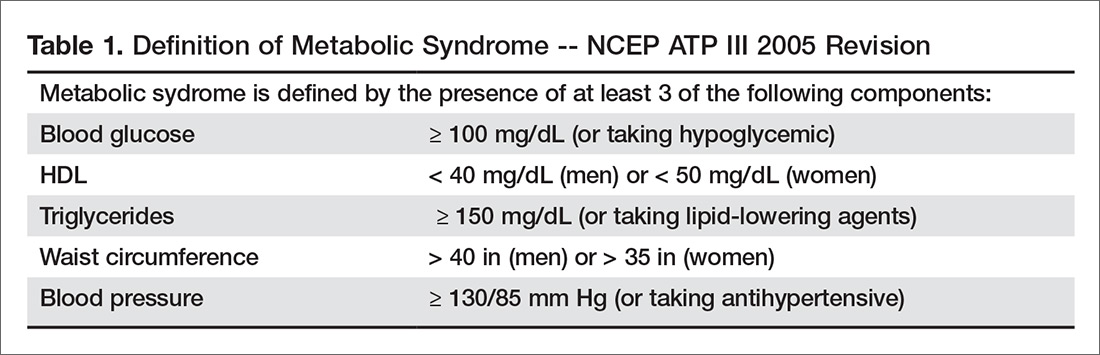
Metabolic syndrome is associated with an increased risk of diabetes mellitus, cardiovascular disease (including myocardial infarction and cerebrovascular accident), and all-cause mortality [3]. Other systemic effects related to metabolic syndrome include renal, hepatic, and skin manifestations such as chronic kidney disease, non-alcoholic steatohepatitis, and obstructive sleep apnea [1].
Epidemiology and Risk Factors
An estimated 34% of people in the United States meet criteria for metabolic syndrome, with worldwide estimates ranging widely from less than 10% to 84%. People with SMI (eg, bipolar disorder, schizoaffective disorder, schizophrenia) are at even greater risk of developing metabolic syndrome than the general population [4,5]. The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study demonstrated metabolic syndrome rates of 40.9% and 51.6% in men and women with a diagnosis of schizophrenia, respectively [6]. In a systematic review of bipolar disorder and metabolic syndrome, people with bipolar disorder showed higher rates of hypertriglyceridemia and hyperglycemia than controls [5].
People with SMI have been found to have significantly increased morbidity and mortality as compared to people without an SMI diagnosis, much of which has been attributed to increased cardiometabolic risk related to multiple factors [7]. Among adults with schizophrenia receiving Medicaid, Olfson et al found diabetes mellitus, ischemic heart disease, nonischemic heart disease, and cerebrovascular accident to be among the top 10 causes of death [7]. The mortality rate for people with SMI is estimated to be 2 to 3 times higher than the general population, and the life expectancy for people with SMI is estimated to be 10 to 20 years shorter than the general population [8–10]. Contributors to this disparity include modifiable health-related behaviors, social determinants of health, and iatrogenic sequelae of prescribed medications. Behavioral factors include poor nutrition, food insecurity, sedentary lifestyle, and smoking; side effects of commonly prescribed psychotropic medications, most notably atypical antipsychotics and mood stabilizers, also contribute to this disparity [7,11].
Both first- and second-generation antipsychotics have been shown to be associated with metabolic sequelae, including weight gain, elevated blood glucose, and insulin resistance [12–14]. Among psychotropic medications, the atypical or second-generation antipsychotics (SGAs) are a class of medications known to have significant metabolic side effects [15,16]. Studies comparing the metabolic consequences of individual SGAs have found significant variation within the class. Clozapine, olanzapine, quetiapine, and risperidone show significant likelihood of weight gain, hyperlipidemia, and hyperglycemia as well as other metabolic consequences [17]. Aripiprazole, lurasidone, and ziprasidone have shown little to no risk of metabolic sequelae [17].
Metabolic side effects of SGAs have been demonstrated in children, adolescents, and adults. There is evidence that adolescents may be particularly sensitive to these sequelae. Galling and colleagues found that adolescents treated with antipsychotics were at greater risk of developing type 2 diabetes mellitus as compared to both healthy controls and controls with psychiatric illness [18]. Kryzhanovskaya et al, looking at metabolic parameters associated with olanzapine use in adolescents and adults, found that both adolescents and adults showed metabolic sequelae and that adolescents had larger changes in weight gain and lipids compared with adults [19].
The mechanism of SGA impact on metabolic parameters remains incompletely understood, though is thought to be multifactorial, mediated primarily through weight gain with increased adiposity. SGA histamine (H1) receptor binding affinity is implicated in weight gain [20] and 5HT2C antagonism may also lead to an increase in appetite [21]. Other proposed mechanisms include changes in appetite through leptin resistance or decreased sensitivity to leptin, the hormone that mediates satiety. Zhang and colleagues found an increase in leptin levels in patients with schizophrenia prescribed antipsychotics, suggesting leptin dysregulation [21]. Additional studies suggest metabolic disturbances independent of weight gain including direct effects of SGAs on glucose and lipid metabolism [22].
If a person experiences a weight gain of 5% after starting an SGA, it is recommended that the dose be decreased or that they be switched to another psychotropic medication with lower likelihood of metabolic consequences [23]. The effectiveness of switching antipsychotic medications to one with lower metabolic risk to improve weight and lipids has been previously demonstrated [24]. If a patient develops diabetes in the context of an antipsychotic prescription, it is also recommended that the medication be switched to an antipsychotic with less risk of hyperglycemia, and if not possible, to target additional risk factors including weight, poor nutrition, and sedentary lifestyle [25]. The decision to switch medications or decrease dosage is often weighed against the psychiatric stability of the person and their overall response to the medication in the context of their treatment course [14].
Metabolic Monitoring
Given the increased risk of metabolic syndrome among people with SMI, and the association of metabolic syndrome with increased morbidity and all-cause mortality, there has been a growing awareness of the importance of screening for metabolic syndrome among people with SMI. Metabolic monitoring involves routine screening for metabolic parameters and assessment of metabolic risk factors among people with SMI who are prescribed antipsychotic medications. Various practice guidelines have been developed in the United States and internationally to assess for metabolic risk factors in people prescribed antipsychotic medications [26]. Current metabolic monitoring guidelines in the United States stem from 2004 consensus recommendations of the American Diabetes Association and American Psychiatric Association along with the American Association of Clinical Endocrinologists and the North American Association for the Study of Obesity for metabolic monitoring among people prescribed SGAs [23]. These recommendations include a time line for routine monitoring of weight/body mass index, waist circumference, blood pressure, fasting blood glucose or hemoglobin A1c, and fasting lipids (Table 2). Guidelines recommend screening at baseline, more frequently within the first 3 months, and then annually [23]. 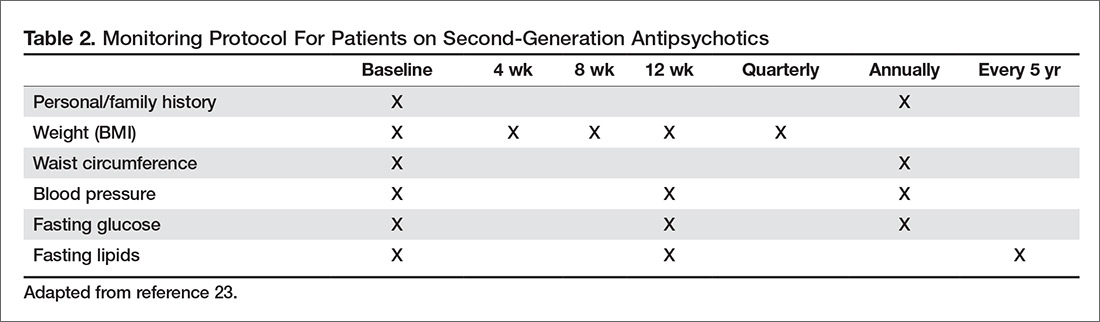
Though guidelines recommend measurement of waist circumference as a marker for metabolic health, body mass index is often used alone as a measure of obesity [27,28]. This may be due to the relative ease of obtaining weight over waist circumference. For example, weight is more likely to be part of clinic workflows and many providers may not be accustomed to measuring waist circumference. However, waist circumference does provide additional information regarding metabolic health [29], as central adiposity is a marker of cardiometabolic risk and related to insulin resistance [21]. Further modifications of the guidelines have included ethnicity-specific waist measurements [30].
There is evidence that non-fasting lipids may be substituted for fasting lipid panels, particularly for patients who may have difficulty adhering to fasting due to cognitive difficulties. Vanderlip and colleagues argue that fasting serum cholesterol panels are not necessary for screening for dyslipidemia given that non-HDL cholesterol is calculated based on total cholesterol and HDL, which do not substantially differ between fasting and non-fasting values [31]. Hemoglobin A1c is recommended as a screening test for blood glucose abnormalities given that it does not require a fasting state and can therefore be more easily obtained for many patients. The choice to obtain a fasting blood glucose versus hemoglobin A1c may depend on multiple factors, including that a person can adhere to fasting and the cost of the laboratory test.
Routine monitoring of metabolic parameters is an integral step in targeting interventions to treat metabolic syndrome. These interventions include lifestyle modifications and evidence-based treatment guidelines for management of associated dyslipidemia, hypertension, and type 2 diabetes mellitus.
Current Metabolic Screening Practices
Despite the presence of defined guidelines, estimates show persistently low rates of metabolic monitoring among adults prescribed SGAs [32]. One study of 3 state Medicaid programs showed little to no improvement in screening rates for glucose and lipids post dissemination of the 2004 APA/ADA guidelines [33]. They noted a nonsignificant change in rates of glucose testing from 27% to 30% and small change in lipid testing from 10% to 11% among patients prescribed SGAs between 2002–2005 [33]. Examining screening rates among Medicaid recipients in Missouri between 2010–2012, Morrato and colleagues found glucose testing rates of 80% with lipid testing remaining at 41% [34]. A retrospective study of adult Medicaid recipients prescribed first- and second-generation antipsychotics between 2008 and 2012 showed rates of screening for lipids and glucose to increase over time; glucose monitoring increased from 56.6% to 72.6% and lipids from 38.3% to 41.2% [35]. A review by Mangurian and colleagues suggested rates of glucose (fasting blood glucose or hemoglobin A1c) and lipid screening as low as 30% among people prescribed antipsychotic medications [14]. Furthermore, they underscore the impact of low screening rates, stating that if 20% of adults with SMI have diabetes and 70% remain unscreened, then approximately 2 million adults with SMI and diabetes in the United States would not be identified within our current system [14].
Higher rates of screening have been shown for Medicaid populations than commercially insured populations [36]. Haupt et al compared lipid and glucose testing pre- and post- ADA/APA guideline implementation among commercially insured patients. They found an increase from 8.4% to 10.5% post guideline implementation for baseline lipid testing and from 6.8% to 9.0% for lipid testing at 12 weeks post-antipsychotic initiation [36]. Baseline glucose testing increased from 17.3% to 21.8% and from 14.1% to 17.9 % at 12-week post antipsychotic initiation. In alignment with findings from other studies, testing rates were particularly low for children [36].
Low screening rates have been found among children and adolescents prescribed SGAs [37] despite evidence that youth may be at risk of developing more significant metabolic sequelae from SGAs [19]. Edelsohn and colleagues found an increase from 30% to 50% for glucose screening and from 19% to 28% for lipid screening among youth Medicaid recipients prescribed first- and second-generation antipsychotics between 2008 and 2012 [35]. Connolly and colleagues reported on metabolic screening rates for children and adolescents prescribed SGAs over the 8 years following announcement of the 2004 ADA/APA guidelines. Using insurance claims data, they found screening rates for fasting blood glucose and hemoglobin A1c temporarily increased following guideline dissemination, then dropped during the period 2004–2008, and again increased slightly [38].
Barriers to Screening
Barriers to screening exist at the level of the individual patient and provider as well as at the clinic and larger systems levels. Lack of provider awareness of evidence-based guidelines for metabolic monitoring despite the presence of the 2004 ADA/APA guidelines has been cited by researchers as an impediment to screening. In a survey of primary care clinicians in San Francisco, Mangurian et al found that 40% of primary care providers did not know about the ADA/APA consensus guidelines for metabolic monitoring. The same survey of primary care providers identified additional impediments to screening, including obstacles to collaboration with psychiatric providers and to scheduling patients for psychiatric follow-up [39]. Another clinician survey conducted by Parameswaran et al found that psychiatrists viewed psychiatric illness severity, lack of staff time, and lack of clinician time as significant barriers to metabolic screening. In addition, clinicians identified factors related to the complexity of coordinating care across systems as obstacles; these included barriers to coordinating follow-up with medical providers, long wait times for patients to see medical providers, and difficulty collaborating with medical providers [40].
Other systems-level barriers include lack of a population-based approach to screening (eg, registries) and lack of electronic record integration, which impedes the ability of primary care and psychiatry providers to share information related to the ordering of metabolic screening tests and prescribing of medications [41]. Mangurian calls for integration of electronic medical record systems between primary care and psychiatry, a population-based approach to metabolic monitoring utilizing registries and other elements of collaborative care models, and primary care consultation to aid in the treatment of metabolic abnormalities [41]. Amiel et al point to systems-level factors “including but not limited to … poor access to general medical services, inadequate medical record-keeping infrastructure, lack of in-system compliance incentives and lack of centralized oversight” [26].
Based on their experience implementing a computer-based intervention for metabolic monitoring, Lai et al propose that the following factors may influence providers’ engagement in metabolic monitoring: lack of apparent symptoms to suggest metabolic syndrome, patients’ lack of engagement in care, and poor access to care. They identify additional factors at the clinician level to include under-recognition of the need for metabolic monitoring, lack of familiarity with screening guidelines, lack of agreement with guidelines, and the potential for individual clinicians to forget to order tests [42]. At the systems-level, they identify the absence of ongoing training as a potential reason why sustained testing was not observed in their intervention [42].
In a 2011 survey of providers prescribing antipsychotic medication to Medicaid beneficiaries in Missouri, Morrato and colleagues found that factors limiting frequency of health care utilization were closely linked to lack of metabolic testing. They also noted disparities in screening guidelines may lead to lack of routine metabolic monitoring; providers may screen based on prescribed medication, diagnosis, or other risk factor based stratification depending on the guidelines followed [34].
Current Unmet Needs
Vulnerable Populations
Though rates of metabolic screening remain low for all groups prescribed antipsychotic medications, studies have consistently shown low rates of screening among children and adolescents [35,36]. Edelsohn and colleagues hypothesize that the cause of these low rates is multifactorial, including that guardians may be reluctant to have young people undergo blood draws [35]. Morrato and colleagues suggest that policymakers should focus initiatives on younger, healthier adults, who they found to have lower rates of screening [37].
Racial and ethnic minorities with SMI constitute another particularly vulnerable population, with some studies showing an increased risk of metabolic sequelae and lower likelihood of treatment for diabetes and other metabolic derangements among African American and Latino populations with SMI [14,43,44].
Integration of Care
Lack of widespread integration of care between mental health and primary care remains another unmet need [41]. Hasnain and colleagues recommend improved communication between mental health and primary care clinicians to coordinate care to improve rates of monitoring, facilitate early follow-up of metabolic abnormalities, and avoid duplication of monitoring efforts [45]. Morrato and colleagues recommend that efforts to increase rates of metabolic monitoring be targeted not only to providers practicing in community mental health centers, but also to other practice settings including primary care. They found that for 75% of people prescribed antipsychotic medications, the prescriptions were started by prescribing providers who practiced outside of a community mental health center [34] and recommend that educational initiatives and performance improvement interventions broaden to include primary care and other care settings [34].
Potential Interventions for Improvement
Early interventions to improve metabolic screening rates have included educational initiatives to teach providers about consensus guidelines. However, initiatives to educate clinicians on metabolic monitoring have shown to be inadequate to significantly improve rates of screening [33]. Therefore, subsequent initiatives have sought to influence screening rates by targeting behavior of individual clinicians with point-of-care tools, electronic reminders, or through systems-level reorganization towards population-based care [27,42,46].
A variety of clinical interventions focus on technologies that remind clinicians to order metabolic monitoring tests according to screening guidelines. One public mental health service in Queensland, Australia, created a standardized metabolic monitoring form to be uploaded to the electronic medical record. In their implementation study examining the efficacy of the metabolic monitoring form, they found that only 36% of the forms contained data. When data were recorded, there were significantly higher rates of documentation of measurements (weight, body mass index, blood pressure) rather than laboratory tests (including lipids and fasting blood glucose) [27].
Computerized reminder systems for metabolic monitoring have been studied in both outpatient and inpatient settings. Lai and colleagues studied the impact of a computerized reminder system on lab monitoring for metabolic parameters among outpatients with schizophrenia prescribed SGAs [42]. This intervention also included an educational component with discussion of metabolic monitoring for people prescribed SGAs at meetings with attending psychiatrists. Computer reminders were displayed when a provider failed to order fasting plasma glucose or lipids (cholesterol, triglyceride) for patients prescribed clozapine, olanzapine, quetiapine, or risperidone. The study found a statistically significant improvement in laboratory metabolic screening for patients prescribed SGAs after implementation, with the greatest impact 6-months post-intervention, though with subsequent decline in screening rates [42].
Psychiatric inpatient hospitalizations provide an opportunity to obtain testing at the time of treatment initiation and also for ongoing monitoring in a location where fasting laboratory tests may be more easily obtained given onsite phlebotomy. One intervention targeting psychiatric inpatients utilized a computerized physician order entry system with the goal to improve metabolic screening among patients prescribed SGAs. Set in a large academic medical setting, the study found inpatient metabolic monitoring rates did not change significantly after implementation of these pop-up computer alerts, comparing rates immediately and 4 years after implementation [46].
There has been increasing focus on integrating mental health and medical care in an effort to improve the health of people with mental illness [47]. Mangurian and colleagues found that the likelihood of diabetes mellitus screening doubled for people with severe mental illness who were seen for at least one primary care visit in addition to mental health treatment [48]. Haupt similarly found higher rates of metabolic screening among patients who had greater than one primary care visit [36]. Models of integration include both integration of medical services into mental health treatment as well as incorporation of mental health services into primary care. For people with SMI, integration efforts have largely focused on integrating primary care services into community mental health settings [49]. The Substance Abuse and Mental Health Service Administration’s (SAMHSA) Primary and Behavioral Health Care Integration (PBHCI) grants program and the Affordable Care Act’s Health Home Initiative are examples of federal incentive programs for improved integration between behavioral health and primary care [49]. In their evaluation of the PBHCI grant program, Scharf and colleagues presented findings that patients at 3 matched clinics with PCBHI grants showed improvement in some lipids, diastolic blood pressure, and fasting blood glucose, though not smoking or body mass index [50].
Conclusion
Several risk factors contribute to an increase in cardiometabolic risk for people with severe mental illness, including poor nutrition, sedentary lifestyle, social determinants of health, and prescribed antipsychotic medications. Metabolic monitoring aims to address these health disparities by screening for metabolic parameters and identifying abnormalities in order to target appropriate health interventions. Screening rates for metabolic parameters remain low for children, adolescents, and adults prescribed second-generation antipsychotics despite published guidelines and clinical interventions to improve screening. More system-wide interventions to improve collaboration between mental health and primary care are needed to enhance screening and prevent cardiovascular disease risk in this vulnerable population.
Corresponding author: Carrie Cunningham, MD, MPH, Zuckerberg San Francisco General Hospital, 1001 Potrero Ave, Suite 7M, San Francisco, CA 94110, [email protected].
Funding/support: Dr. Cunningham was supported by the UCSF-Zuckerberg San Francisco General Public Psychiatry Fellowship. Mr. Riano was supported by the NIH Center Grant from the National Institute of Diabetes and Digestive and Kidney Diseases for The Health Delivery Systems-Center for Diabetes Translational Research (CDTR) (P30DK092924) and by the UCSF-San Francisco General Hospital Public Psychiatry Fellowship. Dr. Mangurian received support from a grant from the NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R03 DK101857), as well as NIH Career Development Award (K23MH093689).
1. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014;2014.
2. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001;285:2486–97.
3. American Heart Association. What is metabolic syndrome? 2015.
4. Vancampfort D, Stubbs B, Mitchell AJ, et al. Risk of metabolic syndrome and its components in people with schizophrenia and related psychotic disorders, bipolar disorder and major depressive disorder: a systematic review and meta‐analysis. World Psychiatry 2015;14:339–47.
5. Czepielewski L, Daruy Filho L, Brietzke E, Grassi-Oliveira R. Bipolar disorder and metabolic syndrome: a systematic review. Rev Bras Psiquiatria 2013;35:88–93.
6. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005;80:19–32.
7. Olfson M, Gerhard T, Huang C, et al. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry 2015:1–10.
8. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry 2007;64:1123–31.
9. Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry 2015;72:334–41.
10. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chron Dis 2006;3:A42.
11. Williams J, Stubbs B, Gaughran F, Craig T. ‘Walk This Way’–a pilot of a health coaching intervention to reduce sedentary behaviour and increase low intensity exercise in people with serious mental illness: study protocol for a randomised controlled trial. Trials 2016;17:594.
12. Allison DB, Mentore JL, Heo M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry 1999;156:1686–96.
13. Chadda RK, Ramshankar P, Deb KS, Sood M. Metabolic syndrome in schizophrenia: differences between antipsychotic-naïve and treated patients. J Pharmacol Pharmacother 2013;4:176–86.
14. Mangurian C, Newcomer JW, Modlin C, Schillinger D. Diabetes and cardiovascular care among people with severe mental illness: a literature review. J Gen Intern Med 2016:1–9.
15. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005;19(Suppl 1):1–93.
16. Baptista T, De Mendoza S, Beaulieu S, et al. The metabolic syndrome during atypical antipsychotic drug treatment: mechanisms and management. Metab Syndr Relat Disord 2004;2:290–307.
17. Hert MDE, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry 2011;10:52–77.
18. Galling B, Roldan A, Nielsen RE, et al. Type 2 diabetes mellitus in youth exposed to antipsychotics: a systematic review and meta-analysis. JAMA Psychiatry 2016;73:247–59.
19. Kryzhanovskaya LA, Xu W, Millen BA, et al. Comparison of long-term (at least 24 weeks) weight gain and metabolic changes between adolescents and adults treated with olanzapine. J Child Adol Psychopharmacol 2012;22:157–65.
20. Nasrallah H. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiatry 2008;13:27–35.
21. Zhang Z-J, YAO Z-J, Liu W, et al. Effects of antipsychotics on fat deposition and changes in leptin and insulin levels. Br J Psychiatry 2004;184:58–62.
22. Kang SH, Lee JI. Metabolic disturbances independent of body mass in patients with schizophrenia taking atypical antipsychotics. Psychiatr Invest 2015;12:242–8.
23. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004; 596–601.
24. Weiden PJ, Newcomer JW, Loebel AD, et al. Long-term changes in weight and plasma lipids during maintenance treatment with ziprasidone. Neuropsychopharmacology 2008;33:985–94.
25. Henderson DC. Atypical antipsychotic-induced diabetes mellitus. CNS Drugs 2002;16:77–89.
26. Amiel JM, Mangurian CV, Ganguli R, Newcomer JW. Addressing cardiometabolic risk during treatment with antipsychotic medications. Curr Opin Psychiatry 2008;21:613–8.
27. Happell B, Platania-Phung C, Gaskin CJ, Stanton R. Use of an electronic metabolic monitoring form in a mental health service–a retrospective file audit. BMC Psychiatry 2016;16:109.
28. Rosenbaum S, Nijjar S, Watkins A, et al. Nurse‐assessed metabolic monitoring: A file audit of risk factor prevalence and impact of an intervention to enhance measurement of waist circumference. Int J Ment Health Nurs 2014;23:252–6.
29. Klein S, Allison DB, Heymsfield SB, et al. Waist circumference and cardiometabolic risk: a consensus statement from shaping America’s health: Association for Weight Management and Obesity Prevention; NAASO, the Obesity Society; the American Society for Nutrition; and the American Diabetes Association. Obesity 2007;15:1061–7.
30. Tan C-E, Ma S, Wai D, et al. Can we apply the National Cholesterol Education Program Adult Treatment Panel definition of the metabolic syndrome to Asians? Diabetes Care 2004;27:1182–6.
31. Vanderlip ER, Chwastiak LA, McCarron RM. Integrated care: nonfasting screening for cardiovascular risk among individuals taking second-generation antipsychotics. Psychiatr Serv 2014;65:573–6.
32. Mitchell A, Delaffon V, Vancampfort D, et al. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med 2012;42:125–47.
33. Morrato EH, Druss B, Hartung DM, et al. Metabolic testing rates in 3 state Medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch Gen Psychiatry 2010;67:17–24.
34. Morrato EH, Campagna EJ, Brewer SE, et al. Metabolic testing for adults in a state Medicaid program receiving antipsychotics: remaining barriers to achieving population health prevention goals. JAMA Psychiatry 2016;73:721–30.
35. Edelsohn GA, Parthasarathy M, Terhorst L, et al. Measurement of metabolic monitoring in youth and adult Medicaid recipients prescribed antipsychotics. J Manage Care Specialty Pharm 2015;21:769–77.
36. Haupt DW, Rosenblatt LC, Kim E, et al. Prevalence and predictors of lipid and glucose monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am J Psychiatry 2009;166:345–53.
37. Morrato EH, Nicol GE, Maahs D, et al. Metabolic screening in children receiving antipsychotic drug treatment. Arch Pediatr Adolesc Med 2010;164:344–51.
38. Connolly JG, Toomey TJ, Schneeweiss MC. Metabolic monitoring for youths initiating use of second-generation antipsychotics, 2003–2011. Psychiatr Serv 2015;66:604–9.
39. Mangurian C, Giwa F, Shumway M, et al. Primary care providers’ views on metabolic monitoring of outpatients taking antipsychotic medication. Psychiatr Serv 2013;64:597–9.
40. Parameswaran SG, Chang C, Swenson AK, et al. Roles in and barriers to metabolic screening for people taking antipsychotic medications: a survey of psychiatrists. Schizophren Res 2013;143:395–6.
41. Mangurian C. Patient-centered medical care in community mental health settings. Psychiatr Serv 2017;68:213-.
42. Lai C-L, Chan H-Y, Pan Y-J, Chen C-H. The effectiveness of a computer reminder system for laboratory monitoring of metabolic syndrome in schizophrenic outpatients using second-generation antipsychotics. Pharmacopsychiatry 2015;48:25–9.
43. Lambert BL, Chou C-H, Chang K-Y, et al. Antipsychotic exposure and type 2 diabetes among patients with schizophrenia: a matched case-control study of California Medicaid claims. Pharmacoepidemiol Drug Saf 2005;14:417–25.
44. Ramaswamy K, Kozma CM, Nasrallah H. Risk of diabetic ketoacidosis after exposure to risperidone or olanzapine. Drug Saf 2007;30:589–99.
45. Hasnain M, Vieweg WVR, Fredrickson SK, et al. Clinical monitoring and management of the metabolic syndrome in patients receiving atypical antipsychotic medications. Prim Care Diab 2009;3:5–15.
46. Lee J, Dalack G, Casher M, et al. Persistence of metabolic monitoring for psychiatry inpatients treated with second‐generation antipsychotics utilizing a computer‐based intervention. J Clin Pharm Therap 2016;41:209–13.
47. Katz MH. Improving the health of persons with serious mental illness. JAMA Intern Med 2015;175:1979–80.
48. Mangurian C, Newcomer JW, Vittinghoff E, et al. Diabetes screening among underserved adults with severe mental illness who take antipsychotic medications. JAMA Intern Med 2015;175:1977–9.
49. Gerrity M. Integrating primary care into behavioral health settings: What works. New York: Milbank Memorial Fund; 2014.
50. Scharf DM EN, Hackbarth NS, Horvitz-Lennon M, et al. Evaluation of the SAMHSA Primary and Behavioral Health Care Integration (PBHCI) Grant Program: Final Report (Task 13). 2014.
From the University of California San Francisco, Department of Psychiatry, Weill Institute for Neurosciences, San Francisco, CA.
Abstract
- Objective: To review screening for metabolic syndrome in people with severe mental illness (SMI).
- Methods: Review of the literature.
- Results: Despite evidence-based metabolic screening guidelines, rates of metabolic screening remain low among people with SMI. Barriers to screening exist at the individual, organizational, and systems levels. Interventions to address these barriers range from point-of-care tools to systems-level reorganization towards population-based care.
- Conclusion: Greater systems-level interventions, particularly those that improve collaboration between mental health and primary care, are needed to improve metabolic monitoring and identify cardiovascular disease risk among people with SMI.
Key words: metabolic monitoring; severe mental illness; metabolic syndrome; integrated care.
People with severe mental illness (SMI) have a life expectancy 10 to 20 years shorter than the general population, and cardiometabolic risk factors contribute significantly to the increased morbidity and mortality seen in this population. To address this health disparity, metabolic monitoring guidelines have been proposed as a mechanism to identify metabolic risk factors. This paper aims to discuss metabolic syndrome and its risk factors, describe metabolic monitoring including current rates and barriers to screening, and identify interventions that may improve rates of screening for metabolic syndrome among people with SMI.
Metabolic syndrome has been conceptualized as a state of chronic low-grade inflammation and hypercoagulation associated with hypertension, dyslipidemia, glucose intolerance, insulin resistance, and visceral adiposity [1]. Per the modified National Cholesterol Education Program Adult Treatment Plan III (NCEP ATP III) guidelines, metabolic syndrome is defined as the presence of 3 of the following 5 parameters: (1) blood glucose > 100 mg/dL (or a person is taking a hypoglycemic medication), (2) high density lipoprotein (HDL) < 40 mg/dL in men or < 50 mg/dL in women, (3) triglycerides > 150 mg/dL (or taking a lipid lowering agent), (4) waist circumference > 40 inches in men or > 35 inches in women, and/or (5) blood pressure > 130/85 mm Hg (or taking an antihypertensive medication) [2,3] (Table 1).
Metabolic syndrome is associated with an increased risk of diabetes mellitus, cardiovascular disease (including myocardial infarction and cerebrovascular accident), and all-cause mortality [3]. Other systemic effects related to metabolic syndrome include renal, hepatic, and skin manifestations such as chronic kidney disease, non-alcoholic steatohepatitis, and obstructive sleep apnea [1].
Epidemiology and Risk Factors
An estimated 34% of people in the United States meet criteria for metabolic syndrome, with worldwide estimates ranging widely from less than 10% to 84%. People with SMI (eg, bipolar disorder, schizoaffective disorder, schizophrenia) are at even greater risk of developing metabolic syndrome than the general population [4,5]. The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study demonstrated metabolic syndrome rates of 40.9% and 51.6% in men and women with a diagnosis of schizophrenia, respectively [6]. In a systematic review of bipolar disorder and metabolic syndrome, people with bipolar disorder showed higher rates of hypertriglyceridemia and hyperglycemia than controls [5].
People with SMI have been found to have significantly increased morbidity and mortality as compared to people without an SMI diagnosis, much of which has been attributed to increased cardiometabolic risk related to multiple factors [7]. Among adults with schizophrenia receiving Medicaid, Olfson et al found diabetes mellitus, ischemic heart disease, nonischemic heart disease, and cerebrovascular accident to be among the top 10 causes of death [7]. The mortality rate for people with SMI is estimated to be 2 to 3 times higher than the general population, and the life expectancy for people with SMI is estimated to be 10 to 20 years shorter than the general population [8–10]. Contributors to this disparity include modifiable health-related behaviors, social determinants of health, and iatrogenic sequelae of prescribed medications. Behavioral factors include poor nutrition, food insecurity, sedentary lifestyle, and smoking; side effects of commonly prescribed psychotropic medications, most notably atypical antipsychotics and mood stabilizers, also contribute to this disparity [7,11].
Both first- and second-generation antipsychotics have been shown to be associated with metabolic sequelae, including weight gain, elevated blood glucose, and insulin resistance [12–14]. Among psychotropic medications, the atypical or second-generation antipsychotics (SGAs) are a class of medications known to have significant metabolic side effects [15,16]. Studies comparing the metabolic consequences of individual SGAs have found significant variation within the class. Clozapine, olanzapine, quetiapine, and risperidone show significant likelihood of weight gain, hyperlipidemia, and hyperglycemia as well as other metabolic consequences [17]. Aripiprazole, lurasidone, and ziprasidone have shown little to no risk of metabolic sequelae [17].
Metabolic side effects of SGAs have been demonstrated in children, adolescents, and adults. There is evidence that adolescents may be particularly sensitive to these sequelae. Galling and colleagues found that adolescents treated with antipsychotics were at greater risk of developing type 2 diabetes mellitus as compared to both healthy controls and controls with psychiatric illness [18]. Kryzhanovskaya et al, looking at metabolic parameters associated with olanzapine use in adolescents and adults, found that both adolescents and adults showed metabolic sequelae and that adolescents had larger changes in weight gain and lipids compared with adults [19].
The mechanism of SGA impact on metabolic parameters remains incompletely understood, though is thought to be multifactorial, mediated primarily through weight gain with increased adiposity. SGA histamine (H1) receptor binding affinity is implicated in weight gain [20] and 5HT2C antagonism may also lead to an increase in appetite [21]. Other proposed mechanisms include changes in appetite through leptin resistance or decreased sensitivity to leptin, the hormone that mediates satiety. Zhang and colleagues found an increase in leptin levels in patients with schizophrenia prescribed antipsychotics, suggesting leptin dysregulation [21]. Additional studies suggest metabolic disturbances independent of weight gain including direct effects of SGAs on glucose and lipid metabolism [22].
If a person experiences a weight gain of 5% after starting an SGA, it is recommended that the dose be decreased or that they be switched to another psychotropic medication with lower likelihood of metabolic consequences [23]. The effectiveness of switching antipsychotic medications to one with lower metabolic risk to improve weight and lipids has been previously demonstrated [24]. If a patient develops diabetes in the context of an antipsychotic prescription, it is also recommended that the medication be switched to an antipsychotic with less risk of hyperglycemia, and if not possible, to target additional risk factors including weight, poor nutrition, and sedentary lifestyle [25]. The decision to switch medications or decrease dosage is often weighed against the psychiatric stability of the person and their overall response to the medication in the context of their treatment course [14].
Metabolic Monitoring
Given the increased risk of metabolic syndrome among people with SMI, and the association of metabolic syndrome with increased morbidity and all-cause mortality, there has been a growing awareness of the importance of screening for metabolic syndrome among people with SMI. Metabolic monitoring involves routine screening for metabolic parameters and assessment of metabolic risk factors among people with SMI who are prescribed antipsychotic medications. Various practice guidelines have been developed in the United States and internationally to assess for metabolic risk factors in people prescribed antipsychotic medications [26]. Current metabolic monitoring guidelines in the United States stem from 2004 consensus recommendations of the American Diabetes Association and American Psychiatric Association along with the American Association of Clinical Endocrinologists and the North American Association for the Study of Obesity for metabolic monitoring among people prescribed SGAs [23]. These recommendations include a time line for routine monitoring of weight/body mass index, waist circumference, blood pressure, fasting blood glucose or hemoglobin A1c, and fasting lipids (Table 2). Guidelines recommend screening at baseline, more frequently within the first 3 months, and then annually [23].
Though guidelines recommend measurement of waist circumference as a marker for metabolic health, body mass index is often used alone as a measure of obesity [27,28]. This may be due to the relative ease of obtaining weight over waist circumference. For example, weight is more likely to be part of clinic workflows and many providers may not be accustomed to measuring waist circumference. However, waist circumference does provide additional information regarding metabolic health [29], as central adiposity is a marker of cardiometabolic risk and related to insulin resistance [21]. Further modifications of the guidelines have included ethnicity-specific waist measurements [30].
There is evidence that non-fasting lipids may be substituted for fasting lipid panels, particularly for patients who may have difficulty adhering to fasting due to cognitive difficulties. Vanderlip and colleagues argue that fasting serum cholesterol panels are not necessary for screening for dyslipidemia given that non-HDL cholesterol is calculated based on total cholesterol and HDL, which do not substantially differ between fasting and non-fasting values [31]. Hemoglobin A1c is recommended as a screening test for blood glucose abnormalities given that it does not require a fasting state and can therefore be more easily obtained for many patients. The choice to obtain a fasting blood glucose versus hemoglobin A1c may depend on multiple factors, including that a person can adhere to fasting and the cost of the laboratory test.
Routine monitoring of metabolic parameters is an integral step in targeting interventions to treat metabolic syndrome. These interventions include lifestyle modifications and evidence-based treatment guidelines for management of associated dyslipidemia, hypertension, and type 2 diabetes mellitus.
Current Metabolic Screening Practices
Despite the presence of defined guidelines, estimates show persistently low rates of metabolic monitoring among adults prescribed SGAs [32]. One study of 3 state Medicaid programs showed little to no improvement in screening rates for glucose and lipids post dissemination of the 2004 APA/ADA guidelines [33]. They noted a nonsignificant change in rates of glucose testing from 27% to 30% and small change in lipid testing from 10% to 11% among patients prescribed SGAs between 2002–2005 [33]. Examining screening rates among Medicaid recipients in Missouri between 2010–2012, Morrato and colleagues found glucose testing rates of 80% with lipid testing remaining at 41% [34]. A retrospective study of adult Medicaid recipients prescribed first- and second-generation antipsychotics between 2008 and 2012 showed rates of screening for lipids and glucose to increase over time; glucose monitoring increased from 56.6% to 72.6% and lipids from 38.3% to 41.2% [35]. A review by Mangurian and colleagues suggested rates of glucose (fasting blood glucose or hemoglobin A1c) and lipid screening as low as 30% among people prescribed antipsychotic medications [14]. Furthermore, they underscore the impact of low screening rates, stating that if 20% of adults with SMI have diabetes and 70% remain unscreened, then approximately 2 million adults with SMI and diabetes in the United States would not be identified within our current system [14].
Higher rates of screening have been shown for Medicaid populations than commercially insured populations [36]. Haupt et al compared lipid and glucose testing pre- and post- ADA/APA guideline implementation among commercially insured patients. They found an increase from 8.4% to 10.5% post guideline implementation for baseline lipid testing and from 6.8% to 9.0% for lipid testing at 12 weeks post-antipsychotic initiation [36]. Baseline glucose testing increased from 17.3% to 21.8% and from 14.1% to 17.9 % at 12-week post antipsychotic initiation. In alignment with findings from other studies, testing rates were particularly low for children [36].
Low screening rates have been found among children and adolescents prescribed SGAs [37] despite evidence that youth may be at risk of developing more significant metabolic sequelae from SGAs [19]. Edelsohn and colleagues found an increase from 30% to 50% for glucose screening and from 19% to 28% for lipid screening among youth Medicaid recipients prescribed first- and second-generation antipsychotics between 2008 and 2012 [35]. Connolly and colleagues reported on metabolic screening rates for children and adolescents prescribed SGAs over the 8 years following announcement of the 2004 ADA/APA guidelines. Using insurance claims data, they found screening rates for fasting blood glucose and hemoglobin A1c temporarily increased following guideline dissemination, then dropped during the period 2004–2008, and again increased slightly [38].
Barriers to Screening
Barriers to screening exist at the level of the individual patient and provider as well as at the clinic and larger systems levels. Lack of provider awareness of evidence-based guidelines for metabolic monitoring despite the presence of the 2004 ADA/APA guidelines has been cited by researchers as an impediment to screening. In a survey of primary care clinicians in San Francisco, Mangurian et al found that 40% of primary care providers did not know about the ADA/APA consensus guidelines for metabolic monitoring. The same survey of primary care providers identified additional impediments to screening, including obstacles to collaboration with psychiatric providers and to scheduling patients for psychiatric follow-up [39]. Another clinician survey conducted by Parameswaran et al found that psychiatrists viewed psychiatric illness severity, lack of staff time, and lack of clinician time as significant barriers to metabolic screening. In addition, clinicians identified factors related to the complexity of coordinating care across systems as obstacles; these included barriers to coordinating follow-up with medical providers, long wait times for patients to see medical providers, and difficulty collaborating with medical providers [40].
Other systems-level barriers include lack of a population-based approach to screening (eg, registries) and lack of electronic record integration, which impedes the ability of primary care and psychiatry providers to share information related to the ordering of metabolic screening tests and prescribing of medications [41]. Mangurian calls for integration of electronic medical record systems between primary care and psychiatry, a population-based approach to metabolic monitoring utilizing registries and other elements of collaborative care models, and primary care consultation to aid in the treatment of metabolic abnormalities [41]. Amiel et al point to systems-level factors “including but not limited to … poor access to general medical services, inadequate medical record-keeping infrastructure, lack of in-system compliance incentives and lack of centralized oversight” [26].
Based on their experience implementing a computer-based intervention for metabolic monitoring, Lai et al propose that the following factors may influence providers’ engagement in metabolic monitoring: lack of apparent symptoms to suggest metabolic syndrome, patients’ lack of engagement in care, and poor access to care. They identify additional factors at the clinician level to include under-recognition of the need for metabolic monitoring, lack of familiarity with screening guidelines, lack of agreement with guidelines, and the potential for individual clinicians to forget to order tests [42]. At the systems-level, they identify the absence of ongoing training as a potential reason why sustained testing was not observed in their intervention [42].
In a 2011 survey of providers prescribing antipsychotic medication to Medicaid beneficiaries in Missouri, Morrato and colleagues found that factors limiting frequency of health care utilization were closely linked to lack of metabolic testing. They also noted disparities in screening guidelines may lead to lack of routine metabolic monitoring; providers may screen based on prescribed medication, diagnosis, or other risk factor based stratification depending on the guidelines followed [34].
Current Unmet Needs
Vulnerable Populations
Though rates of metabolic screening remain low for all groups prescribed antipsychotic medications, studies have consistently shown low rates of screening among children and adolescents [35,36]. Edelsohn and colleagues hypothesize that the cause of these low rates is multifactorial, including that guardians may be reluctant to have young people undergo blood draws [35]. Morrato and colleagues suggest that policymakers should focus initiatives on younger, healthier adults, who they found to have lower rates of screening [37].
Racial and ethnic minorities with SMI constitute another particularly vulnerable population, with some studies showing an increased risk of metabolic sequelae and lower likelihood of treatment for diabetes and other metabolic derangements among African American and Latino populations with SMI [14,43,44].
Integration of Care
Lack of widespread integration of care between mental health and primary care remains another unmet need [41]. Hasnain and colleagues recommend improved communication between mental health and primary care clinicians to coordinate care to improve rates of monitoring, facilitate early follow-up of metabolic abnormalities, and avoid duplication of monitoring efforts [45]. Morrato and colleagues recommend that efforts to increase rates of metabolic monitoring be targeted not only to providers practicing in community mental health centers, but also to other practice settings including primary care. They found that for 75% of people prescribed antipsychotic medications, the prescriptions were started by prescribing providers who practiced outside of a community mental health center [34] and recommend that educational initiatives and performance improvement interventions broaden to include primary care and other care settings [34].
Potential Interventions for Improvement
Early interventions to improve metabolic screening rates have included educational initiatives to teach providers about consensus guidelines. However, initiatives to educate clinicians on metabolic monitoring have shown to be inadequate to significantly improve rates of screening [33]. Therefore, subsequent initiatives have sought to influence screening rates by targeting behavior of individual clinicians with point-of-care tools, electronic reminders, or through systems-level reorganization towards population-based care [27,42,46].
A variety of clinical interventions focus on technologies that remind clinicians to order metabolic monitoring tests according to screening guidelines. One public mental health service in Queensland, Australia, created a standardized metabolic monitoring form to be uploaded to the electronic medical record. In their implementation study examining the efficacy of the metabolic monitoring form, they found that only 36% of the forms contained data. When data were recorded, there were significantly higher rates of documentation of measurements (weight, body mass index, blood pressure) rather than laboratory tests (including lipids and fasting blood glucose) [27].
Computerized reminder systems for metabolic monitoring have been studied in both outpatient and inpatient settings. Lai and colleagues studied the impact of a computerized reminder system on lab monitoring for metabolic parameters among outpatients with schizophrenia prescribed SGAs [42]. This intervention also included an educational component with discussion of metabolic monitoring for people prescribed SGAs at meetings with attending psychiatrists. Computer reminders were displayed when a provider failed to order fasting plasma glucose or lipids (cholesterol, triglyceride) for patients prescribed clozapine, olanzapine, quetiapine, or risperidone. The study found a statistically significant improvement in laboratory metabolic screening for patients prescribed SGAs after implementation, with the greatest impact 6-months post-intervention, though with subsequent decline in screening rates [42].
Psychiatric inpatient hospitalizations provide an opportunity to obtain testing at the time of treatment initiation and also for ongoing monitoring in a location where fasting laboratory tests may be more easily obtained given onsite phlebotomy. One intervention targeting psychiatric inpatients utilized a computerized physician order entry system with the goal to improve metabolic screening among patients prescribed SGAs. Set in a large academic medical setting, the study found inpatient metabolic monitoring rates did not change significantly after implementation of these pop-up computer alerts, comparing rates immediately and 4 years after implementation [46].
There has been increasing focus on integrating mental health and medical care in an effort to improve the health of people with mental illness [47]. Mangurian and colleagues found that the likelihood of diabetes mellitus screening doubled for people with severe mental illness who were seen for at least one primary care visit in addition to mental health treatment [48]. Haupt similarly found higher rates of metabolic screening among patients who had greater than one primary care visit [36]. Models of integration include both integration of medical services into mental health treatment as well as incorporation of mental health services into primary care. For people with SMI, integration efforts have largely focused on integrating primary care services into community mental health settings [49]. The Substance Abuse and Mental Health Service Administration’s (SAMHSA) Primary and Behavioral Health Care Integration (PBHCI) grants program and the Affordable Care Act’s Health Home Initiative are examples of federal incentive programs for improved integration between behavioral health and primary care [49]. In their evaluation of the PBHCI grant program, Scharf and colleagues presented findings that patients at 3 matched clinics with PCBHI grants showed improvement in some lipids, diastolic blood pressure, and fasting blood glucose, though not smoking or body mass index [50].
Conclusion
Several risk factors contribute to an increase in cardiometabolic risk for people with severe mental illness, including poor nutrition, sedentary lifestyle, social determinants of health, and prescribed antipsychotic medications. Metabolic monitoring aims to address these health disparities by screening for metabolic parameters and identifying abnormalities in order to target appropriate health interventions. Screening rates for metabolic parameters remain low for children, adolescents, and adults prescribed second-generation antipsychotics despite published guidelines and clinical interventions to improve screening. More system-wide interventions to improve collaboration between mental health and primary care are needed to enhance screening and prevent cardiovascular disease risk in this vulnerable population.
Corresponding author: Carrie Cunningham, MD, MPH, Zuckerberg San Francisco General Hospital, 1001 Potrero Ave, Suite 7M, San Francisco, CA 94110, [email protected].
Funding/support: Dr. Cunningham was supported by the UCSF-Zuckerberg San Francisco General Public Psychiatry Fellowship. Mr. Riano was supported by the NIH Center Grant from the National Institute of Diabetes and Digestive and Kidney Diseases for The Health Delivery Systems-Center for Diabetes Translational Research (CDTR) (P30DK092924) and by the UCSF-San Francisco General Hospital Public Psychiatry Fellowship. Dr. Mangurian received support from a grant from the NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R03 DK101857), as well as NIH Career Development Award (K23MH093689).
From the University of California San Francisco, Department of Psychiatry, Weill Institute for Neurosciences, San Francisco, CA.
Abstract
- Objective: To review screening for metabolic syndrome in people with severe mental illness (SMI).
- Methods: Review of the literature.
- Results: Despite evidence-based metabolic screening guidelines, rates of metabolic screening remain low among people with SMI. Barriers to screening exist at the individual, organizational, and systems levels. Interventions to address these barriers range from point-of-care tools to systems-level reorganization towards population-based care.
- Conclusion: Greater systems-level interventions, particularly those that improve collaboration between mental health and primary care, are needed to improve metabolic monitoring and identify cardiovascular disease risk among people with SMI.
Key words: metabolic monitoring; severe mental illness; metabolic syndrome; integrated care.
People with severe mental illness (SMI) have a life expectancy 10 to 20 years shorter than the general population, and cardiometabolic risk factors contribute significantly to the increased morbidity and mortality seen in this population. To address this health disparity, metabolic monitoring guidelines have been proposed as a mechanism to identify metabolic risk factors. This paper aims to discuss metabolic syndrome and its risk factors, describe metabolic monitoring including current rates and barriers to screening, and identify interventions that may improve rates of screening for metabolic syndrome among people with SMI.
Metabolic syndrome has been conceptualized as a state of chronic low-grade inflammation and hypercoagulation associated with hypertension, dyslipidemia, glucose intolerance, insulin resistance, and visceral adiposity [1]. Per the modified National Cholesterol Education Program Adult Treatment Plan III (NCEP ATP III) guidelines, metabolic syndrome is defined as the presence of 3 of the following 5 parameters: (1) blood glucose > 100 mg/dL (or a person is taking a hypoglycemic medication), (2) high density lipoprotein (HDL) < 40 mg/dL in men or < 50 mg/dL in women, (3) triglycerides > 150 mg/dL (or taking a lipid lowering agent), (4) waist circumference > 40 inches in men or > 35 inches in women, and/or (5) blood pressure > 130/85 mm Hg (or taking an antihypertensive medication) [2,3] (Table 1).
Metabolic syndrome is associated with an increased risk of diabetes mellitus, cardiovascular disease (including myocardial infarction and cerebrovascular accident), and all-cause mortality [3]. Other systemic effects related to metabolic syndrome include renal, hepatic, and skin manifestations such as chronic kidney disease, non-alcoholic steatohepatitis, and obstructive sleep apnea [1].
Epidemiology and Risk Factors
An estimated 34% of people in the United States meet criteria for metabolic syndrome, with worldwide estimates ranging widely from less than 10% to 84%. People with SMI (eg, bipolar disorder, schizoaffective disorder, schizophrenia) are at even greater risk of developing metabolic syndrome than the general population [4,5]. The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study demonstrated metabolic syndrome rates of 40.9% and 51.6% in men and women with a diagnosis of schizophrenia, respectively [6]. In a systematic review of bipolar disorder and metabolic syndrome, people with bipolar disorder showed higher rates of hypertriglyceridemia and hyperglycemia than controls [5].
People with SMI have been found to have significantly increased morbidity and mortality as compared to people without an SMI diagnosis, much of which has been attributed to increased cardiometabolic risk related to multiple factors [7]. Among adults with schizophrenia receiving Medicaid, Olfson et al found diabetes mellitus, ischemic heart disease, nonischemic heart disease, and cerebrovascular accident to be among the top 10 causes of death [7]. The mortality rate for people with SMI is estimated to be 2 to 3 times higher than the general population, and the life expectancy for people with SMI is estimated to be 10 to 20 years shorter than the general population [8–10]. Contributors to this disparity include modifiable health-related behaviors, social determinants of health, and iatrogenic sequelae of prescribed medications. Behavioral factors include poor nutrition, food insecurity, sedentary lifestyle, and smoking; side effects of commonly prescribed psychotropic medications, most notably atypical antipsychotics and mood stabilizers, also contribute to this disparity [7,11].
Both first- and second-generation antipsychotics have been shown to be associated with metabolic sequelae, including weight gain, elevated blood glucose, and insulin resistance [12–14]. Among psychotropic medications, the atypical or second-generation antipsychotics (SGAs) are a class of medications known to have significant metabolic side effects [15,16]. Studies comparing the metabolic consequences of individual SGAs have found significant variation within the class. Clozapine, olanzapine, quetiapine, and risperidone show significant likelihood of weight gain, hyperlipidemia, and hyperglycemia as well as other metabolic consequences [17]. Aripiprazole, lurasidone, and ziprasidone have shown little to no risk of metabolic sequelae [17].
Metabolic side effects of SGAs have been demonstrated in children, adolescents, and adults. There is evidence that adolescents may be particularly sensitive to these sequelae. Galling and colleagues found that adolescents treated with antipsychotics were at greater risk of developing type 2 diabetes mellitus as compared to both healthy controls and controls with psychiatric illness [18]. Kryzhanovskaya et al, looking at metabolic parameters associated with olanzapine use in adolescents and adults, found that both adolescents and adults showed metabolic sequelae and that adolescents had larger changes in weight gain and lipids compared with adults [19].
The mechanism of SGA impact on metabolic parameters remains incompletely understood, though is thought to be multifactorial, mediated primarily through weight gain with increased adiposity. SGA histamine (H1) receptor binding affinity is implicated in weight gain [20] and 5HT2C antagonism may also lead to an increase in appetite [21]. Other proposed mechanisms include changes in appetite through leptin resistance or decreased sensitivity to leptin, the hormone that mediates satiety. Zhang and colleagues found an increase in leptin levels in patients with schizophrenia prescribed antipsychotics, suggesting leptin dysregulation [21]. Additional studies suggest metabolic disturbances independent of weight gain including direct effects of SGAs on glucose and lipid metabolism [22].
If a person experiences a weight gain of 5% after starting an SGA, it is recommended that the dose be decreased or that they be switched to another psychotropic medication with lower likelihood of metabolic consequences [23]. The effectiveness of switching antipsychotic medications to one with lower metabolic risk to improve weight and lipids has been previously demonstrated [24]. If a patient develops diabetes in the context of an antipsychotic prescription, it is also recommended that the medication be switched to an antipsychotic with less risk of hyperglycemia, and if not possible, to target additional risk factors including weight, poor nutrition, and sedentary lifestyle [25]. The decision to switch medications or decrease dosage is often weighed against the psychiatric stability of the person and their overall response to the medication in the context of their treatment course [14].
Metabolic Monitoring
Given the increased risk of metabolic syndrome among people with SMI, and the association of metabolic syndrome with increased morbidity and all-cause mortality, there has been a growing awareness of the importance of screening for metabolic syndrome among people with SMI. Metabolic monitoring involves routine screening for metabolic parameters and assessment of metabolic risk factors among people with SMI who are prescribed antipsychotic medications. Various practice guidelines have been developed in the United States and internationally to assess for metabolic risk factors in people prescribed antipsychotic medications [26]. Current metabolic monitoring guidelines in the United States stem from 2004 consensus recommendations of the American Diabetes Association and American Psychiatric Association along with the American Association of Clinical Endocrinologists and the North American Association for the Study of Obesity for metabolic monitoring among people prescribed SGAs [23]. These recommendations include a time line for routine monitoring of weight/body mass index, waist circumference, blood pressure, fasting blood glucose or hemoglobin A1c, and fasting lipids (Table 2). Guidelines recommend screening at baseline, more frequently within the first 3 months, and then annually [23].
Though guidelines recommend measurement of waist circumference as a marker for metabolic health, body mass index is often used alone as a measure of obesity [27,28]. This may be due to the relative ease of obtaining weight over waist circumference. For example, weight is more likely to be part of clinic workflows and many providers may not be accustomed to measuring waist circumference. However, waist circumference does provide additional information regarding metabolic health [29], as central adiposity is a marker of cardiometabolic risk and related to insulin resistance [21]. Further modifications of the guidelines have included ethnicity-specific waist measurements [30].
There is evidence that non-fasting lipids may be substituted for fasting lipid panels, particularly for patients who may have difficulty adhering to fasting due to cognitive difficulties. Vanderlip and colleagues argue that fasting serum cholesterol panels are not necessary for screening for dyslipidemia given that non-HDL cholesterol is calculated based on total cholesterol and HDL, which do not substantially differ between fasting and non-fasting values [31]. Hemoglobin A1c is recommended as a screening test for blood glucose abnormalities given that it does not require a fasting state and can therefore be more easily obtained for many patients. The choice to obtain a fasting blood glucose versus hemoglobin A1c may depend on multiple factors, including that a person can adhere to fasting and the cost of the laboratory test.
Routine monitoring of metabolic parameters is an integral step in targeting interventions to treat metabolic syndrome. These interventions include lifestyle modifications and evidence-based treatment guidelines for management of associated dyslipidemia, hypertension, and type 2 diabetes mellitus.
Current Metabolic Screening Practices
Despite the presence of defined guidelines, estimates show persistently low rates of metabolic monitoring among adults prescribed SGAs [32]. One study of 3 state Medicaid programs showed little to no improvement in screening rates for glucose and lipids post dissemination of the 2004 APA/ADA guidelines [33]. They noted a nonsignificant change in rates of glucose testing from 27% to 30% and small change in lipid testing from 10% to 11% among patients prescribed SGAs between 2002–2005 [33]. Examining screening rates among Medicaid recipients in Missouri between 2010–2012, Morrato and colleagues found glucose testing rates of 80% with lipid testing remaining at 41% [34]. A retrospective study of adult Medicaid recipients prescribed first- and second-generation antipsychotics between 2008 and 2012 showed rates of screening for lipids and glucose to increase over time; glucose monitoring increased from 56.6% to 72.6% and lipids from 38.3% to 41.2% [35]. A review by Mangurian and colleagues suggested rates of glucose (fasting blood glucose or hemoglobin A1c) and lipid screening as low as 30% among people prescribed antipsychotic medications [14]. Furthermore, they underscore the impact of low screening rates, stating that if 20% of adults with SMI have diabetes and 70% remain unscreened, then approximately 2 million adults with SMI and diabetes in the United States would not be identified within our current system [14].
Higher rates of screening have been shown for Medicaid populations than commercially insured populations [36]. Haupt et al compared lipid and glucose testing pre- and post- ADA/APA guideline implementation among commercially insured patients. They found an increase from 8.4% to 10.5% post guideline implementation for baseline lipid testing and from 6.8% to 9.0% for lipid testing at 12 weeks post-antipsychotic initiation [36]. Baseline glucose testing increased from 17.3% to 21.8% and from 14.1% to 17.9 % at 12-week post antipsychotic initiation. In alignment with findings from other studies, testing rates were particularly low for children [36].
Low screening rates have been found among children and adolescents prescribed SGAs [37] despite evidence that youth may be at risk of developing more significant metabolic sequelae from SGAs [19]. Edelsohn and colleagues found an increase from 30% to 50% for glucose screening and from 19% to 28% for lipid screening among youth Medicaid recipients prescribed first- and second-generation antipsychotics between 2008 and 2012 [35]. Connolly and colleagues reported on metabolic screening rates for children and adolescents prescribed SGAs over the 8 years following announcement of the 2004 ADA/APA guidelines. Using insurance claims data, they found screening rates for fasting blood glucose and hemoglobin A1c temporarily increased following guideline dissemination, then dropped during the period 2004–2008, and again increased slightly [38].
Barriers to Screening
Barriers to screening exist at the level of the individual patient and provider as well as at the clinic and larger systems levels. Lack of provider awareness of evidence-based guidelines for metabolic monitoring despite the presence of the 2004 ADA/APA guidelines has been cited by researchers as an impediment to screening. In a survey of primary care clinicians in San Francisco, Mangurian et al found that 40% of primary care providers did not know about the ADA/APA consensus guidelines for metabolic monitoring. The same survey of primary care providers identified additional impediments to screening, including obstacles to collaboration with psychiatric providers and to scheduling patients for psychiatric follow-up [39]. Another clinician survey conducted by Parameswaran et al found that psychiatrists viewed psychiatric illness severity, lack of staff time, and lack of clinician time as significant barriers to metabolic screening. In addition, clinicians identified factors related to the complexity of coordinating care across systems as obstacles; these included barriers to coordinating follow-up with medical providers, long wait times for patients to see medical providers, and difficulty collaborating with medical providers [40].
Other systems-level barriers include lack of a population-based approach to screening (eg, registries) and lack of electronic record integration, which impedes the ability of primary care and psychiatry providers to share information related to the ordering of metabolic screening tests and prescribing of medications [41]. Mangurian calls for integration of electronic medical record systems between primary care and psychiatry, a population-based approach to metabolic monitoring utilizing registries and other elements of collaborative care models, and primary care consultation to aid in the treatment of metabolic abnormalities [41]. Amiel et al point to systems-level factors “including but not limited to … poor access to general medical services, inadequate medical record-keeping infrastructure, lack of in-system compliance incentives and lack of centralized oversight” [26].
Based on their experience implementing a computer-based intervention for metabolic monitoring, Lai et al propose that the following factors may influence providers’ engagement in metabolic monitoring: lack of apparent symptoms to suggest metabolic syndrome, patients’ lack of engagement in care, and poor access to care. They identify additional factors at the clinician level to include under-recognition of the need for metabolic monitoring, lack of familiarity with screening guidelines, lack of agreement with guidelines, and the potential for individual clinicians to forget to order tests [42]. At the systems-level, they identify the absence of ongoing training as a potential reason why sustained testing was not observed in their intervention [42].
In a 2011 survey of providers prescribing antipsychotic medication to Medicaid beneficiaries in Missouri, Morrato and colleagues found that factors limiting frequency of health care utilization were closely linked to lack of metabolic testing. They also noted disparities in screening guidelines may lead to lack of routine metabolic monitoring; providers may screen based on prescribed medication, diagnosis, or other risk factor based stratification depending on the guidelines followed [34].
Current Unmet Needs
Vulnerable Populations
Though rates of metabolic screening remain low for all groups prescribed antipsychotic medications, studies have consistently shown low rates of screening among children and adolescents [35,36]. Edelsohn and colleagues hypothesize that the cause of these low rates is multifactorial, including that guardians may be reluctant to have young people undergo blood draws [35]. Morrato and colleagues suggest that policymakers should focus initiatives on younger, healthier adults, who they found to have lower rates of screening [37].
Racial and ethnic minorities with SMI constitute another particularly vulnerable population, with some studies showing an increased risk of metabolic sequelae and lower likelihood of treatment for diabetes and other metabolic derangements among African American and Latino populations with SMI [14,43,44].
Integration of Care
Lack of widespread integration of care between mental health and primary care remains another unmet need [41]. Hasnain and colleagues recommend improved communication between mental health and primary care clinicians to coordinate care to improve rates of monitoring, facilitate early follow-up of metabolic abnormalities, and avoid duplication of monitoring efforts [45]. Morrato and colleagues recommend that efforts to increase rates of metabolic monitoring be targeted not only to providers practicing in community mental health centers, but also to other practice settings including primary care. They found that for 75% of people prescribed antipsychotic medications, the prescriptions were started by prescribing providers who practiced outside of a community mental health center [34] and recommend that educational initiatives and performance improvement interventions broaden to include primary care and other care settings [34].
Potential Interventions for Improvement
Early interventions to improve metabolic screening rates have included educational initiatives to teach providers about consensus guidelines. However, initiatives to educate clinicians on metabolic monitoring have shown to be inadequate to significantly improve rates of screening [33]. Therefore, subsequent initiatives have sought to influence screening rates by targeting behavior of individual clinicians with point-of-care tools, electronic reminders, or through systems-level reorganization towards population-based care [27,42,46].
A variety of clinical interventions focus on technologies that remind clinicians to order metabolic monitoring tests according to screening guidelines. One public mental health service in Queensland, Australia, created a standardized metabolic monitoring form to be uploaded to the electronic medical record. In their implementation study examining the efficacy of the metabolic monitoring form, they found that only 36% of the forms contained data. When data were recorded, there were significantly higher rates of documentation of measurements (weight, body mass index, blood pressure) rather than laboratory tests (including lipids and fasting blood glucose) [27].
Computerized reminder systems for metabolic monitoring have been studied in both outpatient and inpatient settings. Lai and colleagues studied the impact of a computerized reminder system on lab monitoring for metabolic parameters among outpatients with schizophrenia prescribed SGAs [42]. This intervention also included an educational component with discussion of metabolic monitoring for people prescribed SGAs at meetings with attending psychiatrists. Computer reminders were displayed when a provider failed to order fasting plasma glucose or lipids (cholesterol, triglyceride) for patients prescribed clozapine, olanzapine, quetiapine, or risperidone. The study found a statistically significant improvement in laboratory metabolic screening for patients prescribed SGAs after implementation, with the greatest impact 6-months post-intervention, though with subsequent decline in screening rates [42].
Psychiatric inpatient hospitalizations provide an opportunity to obtain testing at the time of treatment initiation and also for ongoing monitoring in a location where fasting laboratory tests may be more easily obtained given onsite phlebotomy. One intervention targeting psychiatric inpatients utilized a computerized physician order entry system with the goal to improve metabolic screening among patients prescribed SGAs. Set in a large academic medical setting, the study found inpatient metabolic monitoring rates did not change significantly after implementation of these pop-up computer alerts, comparing rates immediately and 4 years after implementation [46].
There has been increasing focus on integrating mental health and medical care in an effort to improve the health of people with mental illness [47]. Mangurian and colleagues found that the likelihood of diabetes mellitus screening doubled for people with severe mental illness who were seen for at least one primary care visit in addition to mental health treatment [48]. Haupt similarly found higher rates of metabolic screening among patients who had greater than one primary care visit [36]. Models of integration include both integration of medical services into mental health treatment as well as incorporation of mental health services into primary care. For people with SMI, integration efforts have largely focused on integrating primary care services into community mental health settings [49]. The Substance Abuse and Mental Health Service Administration’s (SAMHSA) Primary and Behavioral Health Care Integration (PBHCI) grants program and the Affordable Care Act’s Health Home Initiative are examples of federal incentive programs for improved integration between behavioral health and primary care [49]. In their evaluation of the PBHCI grant program, Scharf and colleagues presented findings that patients at 3 matched clinics with PCBHI grants showed improvement in some lipids, diastolic blood pressure, and fasting blood glucose, though not smoking or body mass index [50].
Conclusion
Several risk factors contribute to an increase in cardiometabolic risk for people with severe mental illness, including poor nutrition, sedentary lifestyle, social determinants of health, and prescribed antipsychotic medications. Metabolic monitoring aims to address these health disparities by screening for metabolic parameters and identifying abnormalities in order to target appropriate health interventions. Screening rates for metabolic parameters remain low for children, adolescents, and adults prescribed second-generation antipsychotics despite published guidelines and clinical interventions to improve screening. More system-wide interventions to improve collaboration between mental health and primary care are needed to enhance screening and prevent cardiovascular disease risk in this vulnerable population.
Corresponding author: Carrie Cunningham, MD, MPH, Zuckerberg San Francisco General Hospital, 1001 Potrero Ave, Suite 7M, San Francisco, CA 94110, [email protected].
Funding/support: Dr. Cunningham was supported by the UCSF-Zuckerberg San Francisco General Public Psychiatry Fellowship. Mr. Riano was supported by the NIH Center Grant from the National Institute of Diabetes and Digestive and Kidney Diseases for The Health Delivery Systems-Center for Diabetes Translational Research (CDTR) (P30DK092924) and by the UCSF-San Francisco General Hospital Public Psychiatry Fellowship. Dr. Mangurian received support from a grant from the NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R03 DK101857), as well as NIH Career Development Award (K23MH093689).
1. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014;2014.
2. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001;285:2486–97.
3. American Heart Association. What is metabolic syndrome? 2015.
4. Vancampfort D, Stubbs B, Mitchell AJ, et al. Risk of metabolic syndrome and its components in people with schizophrenia and related psychotic disorders, bipolar disorder and major depressive disorder: a systematic review and meta‐analysis. World Psychiatry 2015;14:339–47.
5. Czepielewski L, Daruy Filho L, Brietzke E, Grassi-Oliveira R. Bipolar disorder and metabolic syndrome: a systematic review. Rev Bras Psiquiatria 2013;35:88–93.
6. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005;80:19–32.
7. Olfson M, Gerhard T, Huang C, et al. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry 2015:1–10.
8. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry 2007;64:1123–31.
9. Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry 2015;72:334–41.
10. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chron Dis 2006;3:A42.
11. Williams J, Stubbs B, Gaughran F, Craig T. ‘Walk This Way’–a pilot of a health coaching intervention to reduce sedentary behaviour and increase low intensity exercise in people with serious mental illness: study protocol for a randomised controlled trial. Trials 2016;17:594.
12. Allison DB, Mentore JL, Heo M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry 1999;156:1686–96.
13. Chadda RK, Ramshankar P, Deb KS, Sood M. Metabolic syndrome in schizophrenia: differences between antipsychotic-naïve and treated patients. J Pharmacol Pharmacother 2013;4:176–86.
14. Mangurian C, Newcomer JW, Modlin C, Schillinger D. Diabetes and cardiovascular care among people with severe mental illness: a literature review. J Gen Intern Med 2016:1–9.
15. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005;19(Suppl 1):1–93.
16. Baptista T, De Mendoza S, Beaulieu S, et al. The metabolic syndrome during atypical antipsychotic drug treatment: mechanisms and management. Metab Syndr Relat Disord 2004;2:290–307.
17. Hert MDE, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry 2011;10:52–77.
18. Galling B, Roldan A, Nielsen RE, et al. Type 2 diabetes mellitus in youth exposed to antipsychotics: a systematic review and meta-analysis. JAMA Psychiatry 2016;73:247–59.
19. Kryzhanovskaya LA, Xu W, Millen BA, et al. Comparison of long-term (at least 24 weeks) weight gain and metabolic changes between adolescents and adults treated with olanzapine. J Child Adol Psychopharmacol 2012;22:157–65.
20. Nasrallah H. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiatry 2008;13:27–35.
21. Zhang Z-J, YAO Z-J, Liu W, et al. Effects of antipsychotics on fat deposition and changes in leptin and insulin levels. Br J Psychiatry 2004;184:58–62.
22. Kang SH, Lee JI. Metabolic disturbances independent of body mass in patients with schizophrenia taking atypical antipsychotics. Psychiatr Invest 2015;12:242–8.
23. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004; 596–601.
24. Weiden PJ, Newcomer JW, Loebel AD, et al. Long-term changes in weight and plasma lipids during maintenance treatment with ziprasidone. Neuropsychopharmacology 2008;33:985–94.
25. Henderson DC. Atypical antipsychotic-induced diabetes mellitus. CNS Drugs 2002;16:77–89.
26. Amiel JM, Mangurian CV, Ganguli R, Newcomer JW. Addressing cardiometabolic risk during treatment with antipsychotic medications. Curr Opin Psychiatry 2008;21:613–8.
27. Happell B, Platania-Phung C, Gaskin CJ, Stanton R. Use of an electronic metabolic monitoring form in a mental health service–a retrospective file audit. BMC Psychiatry 2016;16:109.
28. Rosenbaum S, Nijjar S, Watkins A, et al. Nurse‐assessed metabolic monitoring: A file audit of risk factor prevalence and impact of an intervention to enhance measurement of waist circumference. Int J Ment Health Nurs 2014;23:252–6.
29. Klein S, Allison DB, Heymsfield SB, et al. Waist circumference and cardiometabolic risk: a consensus statement from shaping America’s health: Association for Weight Management and Obesity Prevention; NAASO, the Obesity Society; the American Society for Nutrition; and the American Diabetes Association. Obesity 2007;15:1061–7.
30. Tan C-E, Ma S, Wai D, et al. Can we apply the National Cholesterol Education Program Adult Treatment Panel definition of the metabolic syndrome to Asians? Diabetes Care 2004;27:1182–6.
31. Vanderlip ER, Chwastiak LA, McCarron RM. Integrated care: nonfasting screening for cardiovascular risk among individuals taking second-generation antipsychotics. Psychiatr Serv 2014;65:573–6.
32. Mitchell A, Delaffon V, Vancampfort D, et al. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med 2012;42:125–47.
33. Morrato EH, Druss B, Hartung DM, et al. Metabolic testing rates in 3 state Medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch Gen Psychiatry 2010;67:17–24.
34. Morrato EH, Campagna EJ, Brewer SE, et al. Metabolic testing for adults in a state Medicaid program receiving antipsychotics: remaining barriers to achieving population health prevention goals. JAMA Psychiatry 2016;73:721–30.
35. Edelsohn GA, Parthasarathy M, Terhorst L, et al. Measurement of metabolic monitoring in youth and adult Medicaid recipients prescribed antipsychotics. J Manage Care Specialty Pharm 2015;21:769–77.
36. Haupt DW, Rosenblatt LC, Kim E, et al. Prevalence and predictors of lipid and glucose monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am J Psychiatry 2009;166:345–53.
37. Morrato EH, Nicol GE, Maahs D, et al. Metabolic screening in children receiving antipsychotic drug treatment. Arch Pediatr Adolesc Med 2010;164:344–51.
38. Connolly JG, Toomey TJ, Schneeweiss MC. Metabolic monitoring for youths initiating use of second-generation antipsychotics, 2003–2011. Psychiatr Serv 2015;66:604–9.
39. Mangurian C, Giwa F, Shumway M, et al. Primary care providers’ views on metabolic monitoring of outpatients taking antipsychotic medication. Psychiatr Serv 2013;64:597–9.
40. Parameswaran SG, Chang C, Swenson AK, et al. Roles in and barriers to metabolic screening for people taking antipsychotic medications: a survey of psychiatrists. Schizophren Res 2013;143:395–6.
41. Mangurian C. Patient-centered medical care in community mental health settings. Psychiatr Serv 2017;68:213-.
42. Lai C-L, Chan H-Y, Pan Y-J, Chen C-H. The effectiveness of a computer reminder system for laboratory monitoring of metabolic syndrome in schizophrenic outpatients using second-generation antipsychotics. Pharmacopsychiatry 2015;48:25–9.
43. Lambert BL, Chou C-H, Chang K-Y, et al. Antipsychotic exposure and type 2 diabetes among patients with schizophrenia: a matched case-control study of California Medicaid claims. Pharmacoepidemiol Drug Saf 2005;14:417–25.
44. Ramaswamy K, Kozma CM, Nasrallah H. Risk of diabetic ketoacidosis after exposure to risperidone or olanzapine. Drug Saf 2007;30:589–99.
45. Hasnain M, Vieweg WVR, Fredrickson SK, et al. Clinical monitoring and management of the metabolic syndrome in patients receiving atypical antipsychotic medications. Prim Care Diab 2009;3:5–15.
46. Lee J, Dalack G, Casher M, et al. Persistence of metabolic monitoring for psychiatry inpatients treated with second‐generation antipsychotics utilizing a computer‐based intervention. J Clin Pharm Therap 2016;41:209–13.
47. Katz MH. Improving the health of persons with serious mental illness. JAMA Intern Med 2015;175:1979–80.
48. Mangurian C, Newcomer JW, Vittinghoff E, et al. Diabetes screening among underserved adults with severe mental illness who take antipsychotic medications. JAMA Intern Med 2015;175:1977–9.
49. Gerrity M. Integrating primary care into behavioral health settings: What works. New York: Milbank Memorial Fund; 2014.
50. Scharf DM EN, Hackbarth NS, Horvitz-Lennon M, et al. Evaluation of the SAMHSA Primary and Behavioral Health Care Integration (PBHCI) Grant Program: Final Report (Task 13). 2014.
1. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract 2014;2014.
2. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001;285:2486–97.
3. American Heart Association. What is metabolic syndrome? 2015.
4. Vancampfort D, Stubbs B, Mitchell AJ, et al. Risk of metabolic syndrome and its components in people with schizophrenia and related psychotic disorders, bipolar disorder and major depressive disorder: a systematic review and meta‐analysis. World Psychiatry 2015;14:339–47.
5. Czepielewski L, Daruy Filho L, Brietzke E, Grassi-Oliveira R. Bipolar disorder and metabolic syndrome: a systematic review. Rev Bras Psiquiatria 2013;35:88–93.
6. McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res 2005;80:19–32.
7. Olfson M, Gerhard T, Huang C, et al. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry 2015:1–10.
8. Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry 2007;64:1123–31.
9. Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry 2015;72:334–41.
10. Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev Chron Dis 2006;3:A42.
11. Williams J, Stubbs B, Gaughran F, Craig T. ‘Walk This Way’–a pilot of a health coaching intervention to reduce sedentary behaviour and increase low intensity exercise in people with serious mental illness: study protocol for a randomised controlled trial. Trials 2016;17:594.
12. Allison DB, Mentore JL, Heo M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry 1999;156:1686–96.
13. Chadda RK, Ramshankar P, Deb KS, Sood M. Metabolic syndrome in schizophrenia: differences between antipsychotic-naïve and treated patients. J Pharmacol Pharmacother 2013;4:176–86.
14. Mangurian C, Newcomer JW, Modlin C, Schillinger D. Diabetes and cardiovascular care among people with severe mental illness: a literature review. J Gen Intern Med 2016:1–9.
15. Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005;19(Suppl 1):1–93.
16. Baptista T, De Mendoza S, Beaulieu S, et al. The metabolic syndrome during atypical antipsychotic drug treatment: mechanisms and management. Metab Syndr Relat Disord 2004;2:290–307.
17. Hert MDE, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry 2011;10:52–77.
18. Galling B, Roldan A, Nielsen RE, et al. Type 2 diabetes mellitus in youth exposed to antipsychotics: a systematic review and meta-analysis. JAMA Psychiatry 2016;73:247–59.
19. Kryzhanovskaya LA, Xu W, Millen BA, et al. Comparison of long-term (at least 24 weeks) weight gain and metabolic changes between adolescents and adults treated with olanzapine. J Child Adol Psychopharmacol 2012;22:157–65.
20. Nasrallah H. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiatry 2008;13:27–35.
21. Zhang Z-J, YAO Z-J, Liu W, et al. Effects of antipsychotics on fat deposition and changes in leptin and insulin levels. Br J Psychiatry 2004;184:58–62.
22. Kang SH, Lee JI. Metabolic disturbances independent of body mass in patients with schizophrenia taking atypical antipsychotics. Psychiatr Invest 2015;12:242–8.
23. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004; 596–601.
24. Weiden PJ, Newcomer JW, Loebel AD, et al. Long-term changes in weight and plasma lipids during maintenance treatment with ziprasidone. Neuropsychopharmacology 2008;33:985–94.
25. Henderson DC. Atypical antipsychotic-induced diabetes mellitus. CNS Drugs 2002;16:77–89.
26. Amiel JM, Mangurian CV, Ganguli R, Newcomer JW. Addressing cardiometabolic risk during treatment with antipsychotic medications. Curr Opin Psychiatry 2008;21:613–8.
27. Happell B, Platania-Phung C, Gaskin CJ, Stanton R. Use of an electronic metabolic monitoring form in a mental health service–a retrospective file audit. BMC Psychiatry 2016;16:109.
28. Rosenbaum S, Nijjar S, Watkins A, et al. Nurse‐assessed metabolic monitoring: A file audit of risk factor prevalence and impact of an intervention to enhance measurement of waist circumference. Int J Ment Health Nurs 2014;23:252–6.
29. Klein S, Allison DB, Heymsfield SB, et al. Waist circumference and cardiometabolic risk: a consensus statement from shaping America’s health: Association for Weight Management and Obesity Prevention; NAASO, the Obesity Society; the American Society for Nutrition; and the American Diabetes Association. Obesity 2007;15:1061–7.
30. Tan C-E, Ma S, Wai D, et al. Can we apply the National Cholesterol Education Program Adult Treatment Panel definition of the metabolic syndrome to Asians? Diabetes Care 2004;27:1182–6.
31. Vanderlip ER, Chwastiak LA, McCarron RM. Integrated care: nonfasting screening for cardiovascular risk among individuals taking second-generation antipsychotics. Psychiatr Serv 2014;65:573–6.
32. Mitchell A, Delaffon V, Vancampfort D, et al. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med 2012;42:125–47.
33. Morrato EH, Druss B, Hartung DM, et al. Metabolic testing rates in 3 state Medicaid programs after FDA warnings and ADA/APA recommendations for second-generation antipsychotic drugs. Arch Gen Psychiatry 2010;67:17–24.
34. Morrato EH, Campagna EJ, Brewer SE, et al. Metabolic testing for adults in a state Medicaid program receiving antipsychotics: remaining barriers to achieving population health prevention goals. JAMA Psychiatry 2016;73:721–30.
35. Edelsohn GA, Parthasarathy M, Terhorst L, et al. Measurement of metabolic monitoring in youth and adult Medicaid recipients prescribed antipsychotics. J Manage Care Specialty Pharm 2015;21:769–77.
36. Haupt DW, Rosenblatt LC, Kim E, et al. Prevalence and predictors of lipid and glucose monitoring in commercially insured patients treated with second-generation antipsychotic agents. Am J Psychiatry 2009;166:345–53.
37. Morrato EH, Nicol GE, Maahs D, et al. Metabolic screening in children receiving antipsychotic drug treatment. Arch Pediatr Adolesc Med 2010;164:344–51.
38. Connolly JG, Toomey TJ, Schneeweiss MC. Metabolic monitoring for youths initiating use of second-generation antipsychotics, 2003–2011. Psychiatr Serv 2015;66:604–9.
39. Mangurian C, Giwa F, Shumway M, et al. Primary care providers’ views on metabolic monitoring of outpatients taking antipsychotic medication. Psychiatr Serv 2013;64:597–9.
40. Parameswaran SG, Chang C, Swenson AK, et al. Roles in and barriers to metabolic screening for people taking antipsychotic medications: a survey of psychiatrists. Schizophren Res 2013;143:395–6.
41. Mangurian C. Patient-centered medical care in community mental health settings. Psychiatr Serv 2017;68:213-.
42. Lai C-L, Chan H-Y, Pan Y-J, Chen C-H. The effectiveness of a computer reminder system for laboratory monitoring of metabolic syndrome in schizophrenic outpatients using second-generation antipsychotics. Pharmacopsychiatry 2015;48:25–9.
43. Lambert BL, Chou C-H, Chang K-Y, et al. Antipsychotic exposure and type 2 diabetes among patients with schizophrenia: a matched case-control study of California Medicaid claims. Pharmacoepidemiol Drug Saf 2005;14:417–25.
44. Ramaswamy K, Kozma CM, Nasrallah H. Risk of diabetic ketoacidosis after exposure to risperidone or olanzapine. Drug Saf 2007;30:589–99.
45. Hasnain M, Vieweg WVR, Fredrickson SK, et al. Clinical monitoring and management of the metabolic syndrome in patients receiving atypical antipsychotic medications. Prim Care Diab 2009;3:5–15.
46. Lee J, Dalack G, Casher M, et al. Persistence of metabolic monitoring for psychiatry inpatients treated with second‐generation antipsychotics utilizing a computer‐based intervention. J Clin Pharm Therap 2016;41:209–13.
47. Katz MH. Improving the health of persons with serious mental illness. JAMA Intern Med 2015;175:1979–80.
48. Mangurian C, Newcomer JW, Vittinghoff E, et al. Diabetes screening among underserved adults with severe mental illness who take antipsychotic medications. JAMA Intern Med 2015;175:1977–9.
49. Gerrity M. Integrating primary care into behavioral health settings: What works. New York: Milbank Memorial Fund; 2014.
50. Scharf DM EN, Hackbarth NS, Horvitz-Lennon M, et al. Evaluation of the SAMHSA Primary and Behavioral Health Care Integration (PBHCI) Grant Program: Final Report (Task 13). 2014.
2018 Update on obstetrics
The past year brought new information and guidance from the American College of Obstetricians and Gynecologists (ACOG) on many relevant obstetric topics, making it difficult to choose just a few for this Update. Opioid use in pregnancy was an obvious choice given the national media attention and the potential opportunity for intervention in pregnancy for both the mother and the fetus/newborn. Postpartum hemorrhage, an “oldie but goodie,” was chosen for several reasons: It got a new definition, a new focus on multidisciplinary care, and an exciting novel tool for the treatment toolbox. Finally, given the rapidly changing technology, new screening recommendations, and the complexity of counseling, carrier screening was chosen as a genetic hot topic for this year.
Opioids, obstetrics, and opportunities
Reddy UM, Davis JM, Ren Z, Greene MF; Opioid Use in Pregnancy, Neonatal Abstinence Syndrome, and Childhood Outcomes Workshop Invited Speakers. Opioid use in pregnancy, neonatal abstinence syndrome, and childhood outcomes: Executive summary of a joint workshop. Obstet Gynecol. 2017;130(1):10-28.
ACOG Committee on Obstetric Practice. ACOG committee opinion No. 711: Opioid use and opioid use disorder in pregnancy. Obstet Gynecol. 2017;130(2):e81-e94.
The term "opioid epidemic" is omnipresent in both the lay media and the medical literature. In the past decade, the United States has had a huge increase in the number of opioid prescriptions, the rate of admissions and deaths due to prescription opioid misuse and abuse, and an increased rate of heroin use attributed to prior prescription opioid use.
Obstetrics is unique in that opioid use and abuse disorders affect 2 patients simultaneously (the mother and fetus), and the treatment options are somewhat at odds in that they need to balance a stable maternal status and intrauterine environment with the risk of neonatal abstinence syndrome (NAS). Additionally, pregnancy is an opportunity for a woman with opioid use disorder to have access to medical care (possibly for the first time) leading to the diagnosis and treatment of her disease. As the clinicians on the front line, obstetricians therefore require education and guidance on best practice for management of opioid use in pregnancy.
In 2017, Reddy and colleagues, as part of a joint workshop on opioid use in pregnancy, and a committee opinion from ACOG provided the following recommendations.
Screening
Universally screen for substance use, starting at the first prenatal visit; this is recommended over risk factor-based screening.
Use a validated screening tool. A tool such as a questionnaire is recommended as the first-line screening test (for example, the 4Ps screen, the National Institute on Drug Abuse Quick Screen, and the CRAFFT Screening Interview).
Do not universally screen urine and hair for drugs. This type of screening has many limitations, such as the limited number of substances tested, false-positive results, and inaccurate determination of the frequency or timing of drug use. Information regarding the consequences of the test must be provided, and patient consent must be obtained prior to performing the test.
Treatment
Use medication-assisted treatment with buprenorphine or methadone, which is preferred to medically supervised withdrawal. Medication-assisted treatment prevents withdrawal symptoms and cravings, decreases the risk of relapse, improves compliance with prenatal care and addiction treatment programs, and leads to better obstetric outcomes (higher birth weight, lower rate of preterm birth, lower perinatal mortality).
Know that buprenorphine has several advantages over methadone, including the convenience of an outpatient prescription, a lower risk of overdose, and improved neonatal outcomes (higher birth weight, lower doses of morphine to treat NAS, shorter treatment duration).
Prioritize methadone as the preferred option for pregnant women who are already receiving methadone treatment (changing to buprenorphine may precipitate withdrawal), those with a long-standing history of or multi-substance abuse, and those who have failed other treatment programs.
Prenatal care
Screen for comorbid conditions such as sexually transmitted infections, other medications or substance use, social conditions, and mental health disorders.
Perform ultrasonography serially to monitor fetal growth because of the increased risk of fetal growth restriction.
Consult with anesthesiology for pain control recommendations for labor and delivery and with neonatalogy/pediatrics for NAS counseling.
Intrapartum/postpartum care
Recognize heightened pain. Women with opioid use disorder have increased sensitivity to painful stimuli.
Continue the maintenance dose of methadone or buprenorphine throughout hospitalization, with short-acting opioids added for a brief period for postoperative pain.
Prioritize regional anesthesia for pain control in labor or for cesarean delivery.
Consider alternative therapies such as regional blocks, nonopioid medications (nonsteroidal anti-inflammatory drugs, acetaminophen), or relaxation/mindfulness training.
Avoid mixed antagonist and agonist narcotics (butorphanol, nalbuphine, pentazocine) as they may cause acute withdrawal.
Encourage breastfeeding to decrease the severity of NAS and maternal stress and increase maternal-child bonding and maternal confidence.
Offer contraceptive counseling and services immediately postpartum in the hospital, with strong consideration for long-acting reversible contraception.
Opioid prescribing practices
Opioids are prescribed in excess post–cesarean delivery. Several recent studies have demonstrated that most women are prescribed opioids post–cesarean delivery in excess of the amount they use (median 30–40 tablets prescribed, median 20 tablets used).1,2 The leftover opioid medication usually is not discarded and therefore is at risk for diversion or misuse. A small subset of patients will use all the opioids prescribed and feel as though they have not received enough medication.
Prescribe post–cesarean delivery opioids more appropriately by considering individual inpatient opioid requirements or a shared decision-making model.3
Prioritize acetaminophen and ibuprofen during breastfeeding. In a recent editorial in OBG Management, Robert L. Barbieri, MD, recommended that whenever possible, acetaminophen and ibuprofen should be the first-line treatment for breastfeeding women, and narcotics that are metabolized by CYP2D6 should be avoided to reduce the risk to the newborn.4
Universal screening for substance use should be performed in all pregnant women, and clinicians should offer medication-assisted treatment in conjunction with prenatal care and other supportive services as the standard therapy for opioid use disorder. More selective, patient-specific opioid prescribing practices should be applied in the obstetric population.
Read about new strategies for postpartum hemorrhage.
Postpartum hemorrhage: New definitions and new strategies for stemming the flow
ACOG Committee on Practice Bulletins—Obstetrics. ACOG practice bulletin No. 183: Postpartum hemorrhage. Obstet Gynecol. 2017;130(4):e168-e186.
From the very first sentence of the new ACOG practice bulletin, postpartum hemorrhage (PPH) is redefined as "cumulative blood loss greater than or equal to 1,000 mL or blood loss accompanied by signs or symptoms of hypovolemia within 24 hours after the birth process (includes intrapartum loss) regardless of route of delivery." Although this does not seem to be a huge change from the traditional teaching of a 500-mL blood loss at vaginal delivery and a 1,000-mL loss at cesarean delivery, it reflects a shift in focus from simply responding to a certain amount of bleeding to using a multidisciplinary action plan for treating this leading cause of maternal mortality worldwide.
Focus on developing a PPH action plan
As part of the shift toward a multidisciplinary action plan for PPH, all obstetric team members should be aware of the following:
- For most postpartum women, by the time they begin to show signs of hemodynamic compromise, the amount of blood loss approaches 25% of their total blood volume (1,500 mL). Lactic acidosis, systemic inflammation, and a consumptive coagulopathy result.
- Risk stratification prior to delivery, recognition and identification of the source of bleeding, and aggressive early resuscitation to prevent hypovolemia are paramount. Experience gleaned from trauma massive transfusion protocols suggests that judicious transfusion of packed red blood cells, fresh frozen plasma, and platelets in a 1:1:1 ratio is appropriate for obstetric patients. Additionally, patients with low fibrinogen levels should be treated with cryoprecipitate.
- The use of fixed transfusion ratios and standardized protocols for recognition and management of PPH has been demonstrated to increase earlier intervention and resolution of hemorrhage at an earlier stage, although the maternal outcomes results have been mixed.
- Multidisciplinary team drills and simulation exercises also should be considered to help solidify training of an institution's teams responsible for PPH response.
Novel management option: Tranexamic acid
In addition to these strategies, there is a new recommendation for managing refractory PPH: tranexamic acid, which works by binding to lysine receptors on plasminogen and plasmin, inhibiting plasmin-mediated fibrin degradation.5 Previously, tranexamic acid was known to be effective in trauma, heart surgery, and in patients with thrombophilias. Pacheco and colleagues recently demonstrated reduced mortality from obstetric bleeding if tranexamic acid was given within 3 hours of delivery, without increased thrombotic complications.5 ACOG recommends its use if initial medical therapy fails, while the World Health Organization strongly recommends that tranexamic acid be part of a standard PPH package for all cases of PPH (TABLE).6

Postpartum hemorrhage requires early, aggressive, and multidisciplinary coordination to ensure that 1) patients at risk for hemorrhage are identified for preventive measures; 2) existing hemorrhage is recognized and quickly treated, first with noninvasive methods and then with more definitive surgical treatments; and 3) blood product replacement follows an evidence-based standardized protocol. Tranexamic acid is recommended as an adjunct treatment for PPH (of any cause) and should be used within 3 hours of delivery.
Read about new ACOG guidance on prepregnancy and prenatal screening.
Carrier screening—choose something
ACOG Committee on Genetics. Committee opinion No. 690: Carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35-e40.
ACOG Committee on Genetics. Committee opinion No. 691: Carrier screening for genetic conditions. Obstet Gynecol. 2017;129(3):e41-e55.
Ideally, carrier screening should be offered prior to pregnancy to fully inform couples of their reproductive risks and options for pregnancy. If not performed in the preconception period, carrier screening should be offered to all pregnant women. If a patient chooses screening and screens positive for a particular disorder, her reproductive partner should then be offered screening so that the risk of having an affected child can be determined.
New ACOG guidance on prepregnancy and prenatal screening
Carrier screening recommendations have evolved as the technology available has expanded. All 3 of the following strategies now are considered "acceptable" according to 2 recently published ACOG committee opinions.
Traditional ethnic-specific carrier screening, previously ACOG's sole recommendation, involves offering specific genetic screening to patients from populations with a high prevalence for certain conditions. One such example is Tay-Sachs disease screening in Ashkenazi Jewish patients.
Panethnic screening, which takes into account mixed or uncertain backgrounds, involves screening for a certain panel of disorders and is available to all patients regardless of their background (for example, cystic fibrosis screening offered to all pregnant patients).
Expanded carrier screening is when a large number of disorders can be screened for simultaneously for a lower cost than previous testing strategies. Expanded carrier screening panels vary in number and which conditions are tested by the laboratory. An ideal expanded carrier screening panel has been debated in the literature but not agreed on.7
ObGyns and practices therefore are encouraged to develop a standard counseling and screening protocol to offer to all their patients while being flexible to make available any patient-requested screening that is outside their protocol. Pretest and posttest counseling, including a thorough family history, is essential (as with any genetic testing) and should include residual risk after testing, potential need for specific familial mutation testing instead of general carrier screening, and issues with consanguinity.
Three essential screens
Regardless of the screening strategy chosen from the above options, 3 screening tests should be offered to all pregnant women or couples considering pregnancy (either individually or in the context of an expanded screening panel):
- Cystic fibrosis. At the least, a panel of the 23 most common mutations should be used. More expanded panels, which include hundreds of mutations, increase detection in non-Caucasian populations and for milder forms of the disease or infertility-related mutations.
- Hemoglobinopathies (sickle cell, α- and β-thalassemia). Complete blood count and red blood indices are recommended for all, with hemoglobin electrophoresis recommended for patients of African, Middle Eastern, Mediterranean, or West Indian descent or if mean corpuscular volume is low.
- Spinal muscular atrophy (SMA). The most recent addition to ACOG's recommendations for general carrier screening due to the relatively high carrier frequency (1-in-40 to 1-in-60) and the severity of the disease, SMA causes degeneration of the spinal cord neurons, skeletal muscular atrophy, and overall weakness. Screening is via polymerase chain reaction for SMN1 copy number: 2 copies are normal, and 1 copy indicates a carrier of the SMN1 deletion. About 3% to 4% of patients will screen negative but still will be "carriers" due to having 2 copies of the SMN1 gene on 1 chromosome and no copies on the other chromosome.
All pregnant patients or patients considering pregnancy should be offered carrier screening as standard reproductive care, including screening for cystic fibrosis, hemoglobinopathies, and spinal muscular atrophy. Ethnic, panethnic, or expanded carrier screening (and patient-requested specific screening) all are acceptable options, and a standard screening and counseling protocol should be determined by the ObGyn or practice.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Bateman BT, Cole NM, Maeda A, et al. Patterns of opioid prescription and use after cesarean delivery. Obstet Gynecol. 2017;130(1):29–35.
- Osmundson SS, Schornack LA, Grasch JL, Zuckerwise LC, Young JL, Richardson MD. Postdischarge opioid use after cesarean delivery. Obstet Gynecol. 2017;130(1):36–41.
- Prabhu M, McQuaid-Hanson E, Hopp S, et al. A shared decision-making intervention to guide opioid prescribing after cesarean delivery. Obstet Gynecol. 2017;130(1):42–46.
- Barbieri RL. Stop using codeine, oxycodone, hydrocodone, tramadol, and aspirin in women who are breastfeeding. OBG Manag. 2017;29(10):8–12.
- Pacheco LD, Hankins GD, Saad AF, Costantine MM, Chiossi G, Saade GR. Tranexamic acid for the management of obstetric hemorrhage. Obstet Gynecol. 2017;130(4);765–769.
- WHO recommendation on tranexamic acid for the treatment of postpartum haemorrhage. Geneva, Switzerland: World Health Organization; 2017.
- Stevens B, Krstic N, Jones M, Murphy L, Hoskovec J. Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol. 2017;130(2):279–284.

Dr. Pauli is Associate Professor and Attending Perinatologist, Division of Maternal-Fetal
Medicine, Department of Obstetrics and Gynecology, Penn State Health, Milton S.
Hershey Medical Center, Hershey, Pennsylvania.
The author reports no financial relationships relevant to this article
Dr. Pauli is Associate Professor and Attending Perinatologist, Division of Maternal-Fetal
Medicine, Department of Obstetrics and Gynecology, Penn State Health, Milton S.
Hershey Medical Center, Hershey, Pennsylvania.
The author reports no financial relationships relevant to this article
Dr. Pauli is Associate Professor and Attending Perinatologist, Division of Maternal-Fetal
Medicine, Department of Obstetrics and Gynecology, Penn State Health, Milton S.
Hershey Medical Center, Hershey, Pennsylvania.
The author reports no financial relationships relevant to this article
The past year brought new information and guidance from the American College of Obstetricians and Gynecologists (ACOG) on many relevant obstetric topics, making it difficult to choose just a few for this Update. Opioid use in pregnancy was an obvious choice given the national media attention and the potential opportunity for intervention in pregnancy for both the mother and the fetus/newborn. Postpartum hemorrhage, an “oldie but goodie,” was chosen for several reasons: It got a new definition, a new focus on multidisciplinary care, and an exciting novel tool for the treatment toolbox. Finally, given the rapidly changing technology, new screening recommendations, and the complexity of counseling, carrier screening was chosen as a genetic hot topic for this year.
Opioids, obstetrics, and opportunities
Reddy UM, Davis JM, Ren Z, Greene MF; Opioid Use in Pregnancy, Neonatal Abstinence Syndrome, and Childhood Outcomes Workshop Invited Speakers. Opioid use in pregnancy, neonatal abstinence syndrome, and childhood outcomes: Executive summary of a joint workshop. Obstet Gynecol. 2017;130(1):10-28.
ACOG Committee on Obstetric Practice. ACOG committee opinion No. 711: Opioid use and opioid use disorder in pregnancy. Obstet Gynecol. 2017;130(2):e81-e94.
The term "opioid epidemic" is omnipresent in both the lay media and the medical literature. In the past decade, the United States has had a huge increase in the number of opioid prescriptions, the rate of admissions and deaths due to prescription opioid misuse and abuse, and an increased rate of heroin use attributed to prior prescription opioid use.
Obstetrics is unique in that opioid use and abuse disorders affect 2 patients simultaneously (the mother and fetus), and the treatment options are somewhat at odds in that they need to balance a stable maternal status and intrauterine environment with the risk of neonatal abstinence syndrome (NAS). Additionally, pregnancy is an opportunity for a woman with opioid use disorder to have access to medical care (possibly for the first time) leading to the diagnosis and treatment of her disease. As the clinicians on the front line, obstetricians therefore require education and guidance on best practice for management of opioid use in pregnancy.
In 2017, Reddy and colleagues, as part of a joint workshop on opioid use in pregnancy, and a committee opinion from ACOG provided the following recommendations.
Screening
Universally screen for substance use, starting at the first prenatal visit; this is recommended over risk factor-based screening.
Use a validated screening tool. A tool such as a questionnaire is recommended as the first-line screening test (for example, the 4Ps screen, the National Institute on Drug Abuse Quick Screen, and the CRAFFT Screening Interview).
Do not universally screen urine and hair for drugs. This type of screening has many limitations, such as the limited number of substances tested, false-positive results, and inaccurate determination of the frequency or timing of drug use. Information regarding the consequences of the test must be provided, and patient consent must be obtained prior to performing the test.
Treatment
Use medication-assisted treatment with buprenorphine or methadone, which is preferred to medically supervised withdrawal. Medication-assisted treatment prevents withdrawal symptoms and cravings, decreases the risk of relapse, improves compliance with prenatal care and addiction treatment programs, and leads to better obstetric outcomes (higher birth weight, lower rate of preterm birth, lower perinatal mortality).
Know that buprenorphine has several advantages over methadone, including the convenience of an outpatient prescription, a lower risk of overdose, and improved neonatal outcomes (higher birth weight, lower doses of morphine to treat NAS, shorter treatment duration).
Prioritize methadone as the preferred option for pregnant women who are already receiving methadone treatment (changing to buprenorphine may precipitate withdrawal), those with a long-standing history of or multi-substance abuse, and those who have failed other treatment programs.
Prenatal care
Screen for comorbid conditions such as sexually transmitted infections, other medications or substance use, social conditions, and mental health disorders.
Perform ultrasonography serially to monitor fetal growth because of the increased risk of fetal growth restriction.
Consult with anesthesiology for pain control recommendations for labor and delivery and with neonatalogy/pediatrics for NAS counseling.
Intrapartum/postpartum care
Recognize heightened pain. Women with opioid use disorder have increased sensitivity to painful stimuli.
Continue the maintenance dose of methadone or buprenorphine throughout hospitalization, with short-acting opioids added for a brief period for postoperative pain.
Prioritize regional anesthesia for pain control in labor or for cesarean delivery.
Consider alternative therapies such as regional blocks, nonopioid medications (nonsteroidal anti-inflammatory drugs, acetaminophen), or relaxation/mindfulness training.
Avoid mixed antagonist and agonist narcotics (butorphanol, nalbuphine, pentazocine) as they may cause acute withdrawal.
Encourage breastfeeding to decrease the severity of NAS and maternal stress and increase maternal-child bonding and maternal confidence.
Offer contraceptive counseling and services immediately postpartum in the hospital, with strong consideration for long-acting reversible contraception.
Opioid prescribing practices
Opioids are prescribed in excess post–cesarean delivery. Several recent studies have demonstrated that most women are prescribed opioids post–cesarean delivery in excess of the amount they use (median 30–40 tablets prescribed, median 20 tablets used).1,2 The leftover opioid medication usually is not discarded and therefore is at risk for diversion or misuse. A small subset of patients will use all the opioids prescribed and feel as though they have not received enough medication.
Prescribe post–cesarean delivery opioids more appropriately by considering individual inpatient opioid requirements or a shared decision-making model.3
Prioritize acetaminophen and ibuprofen during breastfeeding. In a recent editorial in OBG Management, Robert L. Barbieri, MD, recommended that whenever possible, acetaminophen and ibuprofen should be the first-line treatment for breastfeeding women, and narcotics that are metabolized by CYP2D6 should be avoided to reduce the risk to the newborn.4
Universal screening for substance use should be performed in all pregnant women, and clinicians should offer medication-assisted treatment in conjunction with prenatal care and other supportive services as the standard therapy for opioid use disorder. More selective, patient-specific opioid prescribing practices should be applied in the obstetric population.
Read about new strategies for postpartum hemorrhage.
Postpartum hemorrhage: New definitions and new strategies for stemming the flow
ACOG Committee on Practice Bulletins—Obstetrics. ACOG practice bulletin No. 183: Postpartum hemorrhage. Obstet Gynecol. 2017;130(4):e168-e186.
From the very first sentence of the new ACOG practice bulletin, postpartum hemorrhage (PPH) is redefined as "cumulative blood loss greater than or equal to 1,000 mL or blood loss accompanied by signs or symptoms of hypovolemia within 24 hours after the birth process (includes intrapartum loss) regardless of route of delivery." Although this does not seem to be a huge change from the traditional teaching of a 500-mL blood loss at vaginal delivery and a 1,000-mL loss at cesarean delivery, it reflects a shift in focus from simply responding to a certain amount of bleeding to using a multidisciplinary action plan for treating this leading cause of maternal mortality worldwide.
Focus on developing a PPH action plan
As part of the shift toward a multidisciplinary action plan for PPH, all obstetric team members should be aware of the following:
- For most postpartum women, by the time they begin to show signs of hemodynamic compromise, the amount of blood loss approaches 25% of their total blood volume (1,500 mL). Lactic acidosis, systemic inflammation, and a consumptive coagulopathy result.
- Risk stratification prior to delivery, recognition and identification of the source of bleeding, and aggressive early resuscitation to prevent hypovolemia are paramount. Experience gleaned from trauma massive transfusion protocols suggests that judicious transfusion of packed red blood cells, fresh frozen plasma, and platelets in a 1:1:1 ratio is appropriate for obstetric patients. Additionally, patients with low fibrinogen levels should be treated with cryoprecipitate.
- The use of fixed transfusion ratios and standardized protocols for recognition and management of PPH has been demonstrated to increase earlier intervention and resolution of hemorrhage at an earlier stage, although the maternal outcomes results have been mixed.
- Multidisciplinary team drills and simulation exercises also should be considered to help solidify training of an institution's teams responsible for PPH response.
Novel management option: Tranexamic acid
In addition to these strategies, there is a new recommendation for managing refractory PPH: tranexamic acid, which works by binding to lysine receptors on plasminogen and plasmin, inhibiting plasmin-mediated fibrin degradation.5 Previously, tranexamic acid was known to be effective in trauma, heart surgery, and in patients with thrombophilias. Pacheco and colleagues recently demonstrated reduced mortality from obstetric bleeding if tranexamic acid was given within 3 hours of delivery, without increased thrombotic complications.5 ACOG recommends its use if initial medical therapy fails, while the World Health Organization strongly recommends that tranexamic acid be part of a standard PPH package for all cases of PPH (TABLE).6
Postpartum hemorrhage requires early, aggressive, and multidisciplinary coordination to ensure that 1) patients at risk for hemorrhage are identified for preventive measures; 2) existing hemorrhage is recognized and quickly treated, first with noninvasive methods and then with more definitive surgical treatments; and 3) blood product replacement follows an evidence-based standardized protocol. Tranexamic acid is recommended as an adjunct treatment for PPH (of any cause) and should be used within 3 hours of delivery.
Read about new ACOG guidance on prepregnancy and prenatal screening.
Carrier screening—choose something
ACOG Committee on Genetics. Committee opinion No. 690: Carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35-e40.
ACOG Committee on Genetics. Committee opinion No. 691: Carrier screening for genetic conditions. Obstet Gynecol. 2017;129(3):e41-e55.
Ideally, carrier screening should be offered prior to pregnancy to fully inform couples of their reproductive risks and options for pregnancy. If not performed in the preconception period, carrier screening should be offered to all pregnant women. If a patient chooses screening and screens positive for a particular disorder, her reproductive partner should then be offered screening so that the risk of having an affected child can be determined.
New ACOG guidance on prepregnancy and prenatal screening
Carrier screening recommendations have evolved as the technology available has expanded. All 3 of the following strategies now are considered "acceptable" according to 2 recently published ACOG committee opinions.
Traditional ethnic-specific carrier screening, previously ACOG's sole recommendation, involves offering specific genetic screening to patients from populations with a high prevalence for certain conditions. One such example is Tay-Sachs disease screening in Ashkenazi Jewish patients.
Panethnic screening, which takes into account mixed or uncertain backgrounds, involves screening for a certain panel of disorders and is available to all patients regardless of their background (for example, cystic fibrosis screening offered to all pregnant patients).
Expanded carrier screening is when a large number of disorders can be screened for simultaneously for a lower cost than previous testing strategies. Expanded carrier screening panels vary in number and which conditions are tested by the laboratory. An ideal expanded carrier screening panel has been debated in the literature but not agreed on.7
ObGyns and practices therefore are encouraged to develop a standard counseling and screening protocol to offer to all their patients while being flexible to make available any patient-requested screening that is outside their protocol. Pretest and posttest counseling, including a thorough family history, is essential (as with any genetic testing) and should include residual risk after testing, potential need for specific familial mutation testing instead of general carrier screening, and issues with consanguinity.
Three essential screens
Regardless of the screening strategy chosen from the above options, 3 screening tests should be offered to all pregnant women or couples considering pregnancy (either individually or in the context of an expanded screening panel):
- Cystic fibrosis. At the least, a panel of the 23 most common mutations should be used. More expanded panels, which include hundreds of mutations, increase detection in non-Caucasian populations and for milder forms of the disease or infertility-related mutations.
- Hemoglobinopathies (sickle cell, α- and β-thalassemia). Complete blood count and red blood indices are recommended for all, with hemoglobin electrophoresis recommended for patients of African, Middle Eastern, Mediterranean, or West Indian descent or if mean corpuscular volume is low.
- Spinal muscular atrophy (SMA). The most recent addition to ACOG's recommendations for general carrier screening due to the relatively high carrier frequency (1-in-40 to 1-in-60) and the severity of the disease, SMA causes degeneration of the spinal cord neurons, skeletal muscular atrophy, and overall weakness. Screening is via polymerase chain reaction for SMN1 copy number: 2 copies are normal, and 1 copy indicates a carrier of the SMN1 deletion. About 3% to 4% of patients will screen negative but still will be "carriers" due to having 2 copies of the SMN1 gene on 1 chromosome and no copies on the other chromosome.
All pregnant patients or patients considering pregnancy should be offered carrier screening as standard reproductive care, including screening for cystic fibrosis, hemoglobinopathies, and spinal muscular atrophy. Ethnic, panethnic, or expanded carrier screening (and patient-requested specific screening) all are acceptable options, and a standard screening and counseling protocol should be determined by the ObGyn or practice.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The past year brought new information and guidance from the American College of Obstetricians and Gynecologists (ACOG) on many relevant obstetric topics, making it difficult to choose just a few for this Update. Opioid use in pregnancy was an obvious choice given the national media attention and the potential opportunity for intervention in pregnancy for both the mother and the fetus/newborn. Postpartum hemorrhage, an “oldie but goodie,” was chosen for several reasons: It got a new definition, a new focus on multidisciplinary care, and an exciting novel tool for the treatment toolbox. Finally, given the rapidly changing technology, new screening recommendations, and the complexity of counseling, carrier screening was chosen as a genetic hot topic for this year.
Opioids, obstetrics, and opportunities
Reddy UM, Davis JM, Ren Z, Greene MF; Opioid Use in Pregnancy, Neonatal Abstinence Syndrome, and Childhood Outcomes Workshop Invited Speakers. Opioid use in pregnancy, neonatal abstinence syndrome, and childhood outcomes: Executive summary of a joint workshop. Obstet Gynecol. 2017;130(1):10-28.
ACOG Committee on Obstetric Practice. ACOG committee opinion No. 711: Opioid use and opioid use disorder in pregnancy. Obstet Gynecol. 2017;130(2):e81-e94.
The term "opioid epidemic" is omnipresent in both the lay media and the medical literature. In the past decade, the United States has had a huge increase in the number of opioid prescriptions, the rate of admissions and deaths due to prescription opioid misuse and abuse, and an increased rate of heroin use attributed to prior prescription opioid use.
Obstetrics is unique in that opioid use and abuse disorders affect 2 patients simultaneously (the mother and fetus), and the treatment options are somewhat at odds in that they need to balance a stable maternal status and intrauterine environment with the risk of neonatal abstinence syndrome (NAS). Additionally, pregnancy is an opportunity for a woman with opioid use disorder to have access to medical care (possibly for the first time) leading to the diagnosis and treatment of her disease. As the clinicians on the front line, obstetricians therefore require education and guidance on best practice for management of opioid use in pregnancy.
In 2017, Reddy and colleagues, as part of a joint workshop on opioid use in pregnancy, and a committee opinion from ACOG provided the following recommendations.
Screening
Universally screen for substance use, starting at the first prenatal visit; this is recommended over risk factor-based screening.
Use a validated screening tool. A tool such as a questionnaire is recommended as the first-line screening test (for example, the 4Ps screen, the National Institute on Drug Abuse Quick Screen, and the CRAFFT Screening Interview).
Do not universally screen urine and hair for drugs. This type of screening has many limitations, such as the limited number of substances tested, false-positive results, and inaccurate determination of the frequency or timing of drug use. Information regarding the consequences of the test must be provided, and patient consent must be obtained prior to performing the test.
Treatment
Use medication-assisted treatment with buprenorphine or methadone, which is preferred to medically supervised withdrawal. Medication-assisted treatment prevents withdrawal symptoms and cravings, decreases the risk of relapse, improves compliance with prenatal care and addiction treatment programs, and leads to better obstetric outcomes (higher birth weight, lower rate of preterm birth, lower perinatal mortality).
Know that buprenorphine has several advantages over methadone, including the convenience of an outpatient prescription, a lower risk of overdose, and improved neonatal outcomes (higher birth weight, lower doses of morphine to treat NAS, shorter treatment duration).
Prioritize methadone as the preferred option for pregnant women who are already receiving methadone treatment (changing to buprenorphine may precipitate withdrawal), those with a long-standing history of or multi-substance abuse, and those who have failed other treatment programs.
Prenatal care
Screen for comorbid conditions such as sexually transmitted infections, other medications or substance use, social conditions, and mental health disorders.
Perform ultrasonography serially to monitor fetal growth because of the increased risk of fetal growth restriction.
Consult with anesthesiology for pain control recommendations for labor and delivery and with neonatalogy/pediatrics for NAS counseling.
Intrapartum/postpartum care
Recognize heightened pain. Women with opioid use disorder have increased sensitivity to painful stimuli.
Continue the maintenance dose of methadone or buprenorphine throughout hospitalization, with short-acting opioids added for a brief period for postoperative pain.
Prioritize regional anesthesia for pain control in labor or for cesarean delivery.
Consider alternative therapies such as regional blocks, nonopioid medications (nonsteroidal anti-inflammatory drugs, acetaminophen), or relaxation/mindfulness training.
Avoid mixed antagonist and agonist narcotics (butorphanol, nalbuphine, pentazocine) as they may cause acute withdrawal.
Encourage breastfeeding to decrease the severity of NAS and maternal stress and increase maternal-child bonding and maternal confidence.
Offer contraceptive counseling and services immediately postpartum in the hospital, with strong consideration for long-acting reversible contraception.
Opioid prescribing practices
Opioids are prescribed in excess post–cesarean delivery. Several recent studies have demonstrated that most women are prescribed opioids post–cesarean delivery in excess of the amount they use (median 30–40 tablets prescribed, median 20 tablets used).1,2 The leftover opioid medication usually is not discarded and therefore is at risk for diversion or misuse. A small subset of patients will use all the opioids prescribed and feel as though they have not received enough medication.
Prescribe post–cesarean delivery opioids more appropriately by considering individual inpatient opioid requirements or a shared decision-making model.3
Prioritize acetaminophen and ibuprofen during breastfeeding. In a recent editorial in OBG Management, Robert L. Barbieri, MD, recommended that whenever possible, acetaminophen and ibuprofen should be the first-line treatment for breastfeeding women, and narcotics that are metabolized by CYP2D6 should be avoided to reduce the risk to the newborn.4
Universal screening for substance use should be performed in all pregnant women, and clinicians should offer medication-assisted treatment in conjunction with prenatal care and other supportive services as the standard therapy for opioid use disorder. More selective, patient-specific opioid prescribing practices should be applied in the obstetric population.
Read about new strategies for postpartum hemorrhage.
Postpartum hemorrhage: New definitions and new strategies for stemming the flow
ACOG Committee on Practice Bulletins—Obstetrics. ACOG practice bulletin No. 183: Postpartum hemorrhage. Obstet Gynecol. 2017;130(4):e168-e186.
From the very first sentence of the new ACOG practice bulletin, postpartum hemorrhage (PPH) is redefined as "cumulative blood loss greater than or equal to 1,000 mL or blood loss accompanied by signs or symptoms of hypovolemia within 24 hours after the birth process (includes intrapartum loss) regardless of route of delivery." Although this does not seem to be a huge change from the traditional teaching of a 500-mL blood loss at vaginal delivery and a 1,000-mL loss at cesarean delivery, it reflects a shift in focus from simply responding to a certain amount of bleeding to using a multidisciplinary action plan for treating this leading cause of maternal mortality worldwide.
Focus on developing a PPH action plan
As part of the shift toward a multidisciplinary action plan for PPH, all obstetric team members should be aware of the following:
- For most postpartum women, by the time they begin to show signs of hemodynamic compromise, the amount of blood loss approaches 25% of their total blood volume (1,500 mL). Lactic acidosis, systemic inflammation, and a consumptive coagulopathy result.
- Risk stratification prior to delivery, recognition and identification of the source of bleeding, and aggressive early resuscitation to prevent hypovolemia are paramount. Experience gleaned from trauma massive transfusion protocols suggests that judicious transfusion of packed red blood cells, fresh frozen plasma, and platelets in a 1:1:1 ratio is appropriate for obstetric patients. Additionally, patients with low fibrinogen levels should be treated with cryoprecipitate.
- The use of fixed transfusion ratios and standardized protocols for recognition and management of PPH has been demonstrated to increase earlier intervention and resolution of hemorrhage at an earlier stage, although the maternal outcomes results have been mixed.
- Multidisciplinary team drills and simulation exercises also should be considered to help solidify training of an institution's teams responsible for PPH response.
Novel management option: Tranexamic acid
In addition to these strategies, there is a new recommendation for managing refractory PPH: tranexamic acid, which works by binding to lysine receptors on plasminogen and plasmin, inhibiting plasmin-mediated fibrin degradation.5 Previously, tranexamic acid was known to be effective in trauma, heart surgery, and in patients with thrombophilias. Pacheco and colleagues recently demonstrated reduced mortality from obstetric bleeding if tranexamic acid was given within 3 hours of delivery, without increased thrombotic complications.5 ACOG recommends its use if initial medical therapy fails, while the World Health Organization strongly recommends that tranexamic acid be part of a standard PPH package for all cases of PPH (TABLE).6
Postpartum hemorrhage requires early, aggressive, and multidisciplinary coordination to ensure that 1) patients at risk for hemorrhage are identified for preventive measures; 2) existing hemorrhage is recognized and quickly treated, first with noninvasive methods and then with more definitive surgical treatments; and 3) blood product replacement follows an evidence-based standardized protocol. Tranexamic acid is recommended as an adjunct treatment for PPH (of any cause) and should be used within 3 hours of delivery.
Read about new ACOG guidance on prepregnancy and prenatal screening.
Carrier screening—choose something
ACOG Committee on Genetics. Committee opinion No. 690: Carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35-e40.
ACOG Committee on Genetics. Committee opinion No. 691: Carrier screening for genetic conditions. Obstet Gynecol. 2017;129(3):e41-e55.
Ideally, carrier screening should be offered prior to pregnancy to fully inform couples of their reproductive risks and options for pregnancy. If not performed in the preconception period, carrier screening should be offered to all pregnant women. If a patient chooses screening and screens positive for a particular disorder, her reproductive partner should then be offered screening so that the risk of having an affected child can be determined.
New ACOG guidance on prepregnancy and prenatal screening
Carrier screening recommendations have evolved as the technology available has expanded. All 3 of the following strategies now are considered "acceptable" according to 2 recently published ACOG committee opinions.
Traditional ethnic-specific carrier screening, previously ACOG's sole recommendation, involves offering specific genetic screening to patients from populations with a high prevalence for certain conditions. One such example is Tay-Sachs disease screening in Ashkenazi Jewish patients.
Panethnic screening, which takes into account mixed or uncertain backgrounds, involves screening for a certain panel of disorders and is available to all patients regardless of their background (for example, cystic fibrosis screening offered to all pregnant patients).
Expanded carrier screening is when a large number of disorders can be screened for simultaneously for a lower cost than previous testing strategies. Expanded carrier screening panels vary in number and which conditions are tested by the laboratory. An ideal expanded carrier screening panel has been debated in the literature but not agreed on.7
ObGyns and practices therefore are encouraged to develop a standard counseling and screening protocol to offer to all their patients while being flexible to make available any patient-requested screening that is outside their protocol. Pretest and posttest counseling, including a thorough family history, is essential (as with any genetic testing) and should include residual risk after testing, potential need for specific familial mutation testing instead of general carrier screening, and issues with consanguinity.
Three essential screens
Regardless of the screening strategy chosen from the above options, 3 screening tests should be offered to all pregnant women or couples considering pregnancy (either individually or in the context of an expanded screening panel):
- Cystic fibrosis. At the least, a panel of the 23 most common mutations should be used. More expanded panels, which include hundreds of mutations, increase detection in non-Caucasian populations and for milder forms of the disease or infertility-related mutations.
- Hemoglobinopathies (sickle cell, α- and β-thalassemia). Complete blood count and red blood indices are recommended for all, with hemoglobin electrophoresis recommended for patients of African, Middle Eastern, Mediterranean, or West Indian descent or if mean corpuscular volume is low.
- Spinal muscular atrophy (SMA). The most recent addition to ACOG's recommendations for general carrier screening due to the relatively high carrier frequency (1-in-40 to 1-in-60) and the severity of the disease, SMA causes degeneration of the spinal cord neurons, skeletal muscular atrophy, and overall weakness. Screening is via polymerase chain reaction for SMN1 copy number: 2 copies are normal, and 1 copy indicates a carrier of the SMN1 deletion. About 3% to 4% of patients will screen negative but still will be "carriers" due to having 2 copies of the SMN1 gene on 1 chromosome and no copies on the other chromosome.
All pregnant patients or patients considering pregnancy should be offered carrier screening as standard reproductive care, including screening for cystic fibrosis, hemoglobinopathies, and spinal muscular atrophy. Ethnic, panethnic, or expanded carrier screening (and patient-requested specific screening) all are acceptable options, and a standard screening and counseling protocol should be determined by the ObGyn or practice.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Bateman BT, Cole NM, Maeda A, et al. Patterns of opioid prescription and use after cesarean delivery. Obstet Gynecol. 2017;130(1):29–35.
- Osmundson SS, Schornack LA, Grasch JL, Zuckerwise LC, Young JL, Richardson MD. Postdischarge opioid use after cesarean delivery. Obstet Gynecol. 2017;130(1):36–41.
- Prabhu M, McQuaid-Hanson E, Hopp S, et al. A shared decision-making intervention to guide opioid prescribing after cesarean delivery. Obstet Gynecol. 2017;130(1):42–46.
- Barbieri RL. Stop using codeine, oxycodone, hydrocodone, tramadol, and aspirin in women who are breastfeeding. OBG Manag. 2017;29(10):8–12.
- Pacheco LD, Hankins GD, Saad AF, Costantine MM, Chiossi G, Saade GR. Tranexamic acid for the management of obstetric hemorrhage. Obstet Gynecol. 2017;130(4);765–769.
- WHO recommendation on tranexamic acid for the treatment of postpartum haemorrhage. Geneva, Switzerland: World Health Organization; 2017.
- Stevens B, Krstic N, Jones M, Murphy L, Hoskovec J. Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol. 2017;130(2):279–284.
- Bateman BT, Cole NM, Maeda A, et al. Patterns of opioid prescription and use after cesarean delivery. Obstet Gynecol. 2017;130(1):29–35.
- Osmundson SS, Schornack LA, Grasch JL, Zuckerwise LC, Young JL, Richardson MD. Postdischarge opioid use after cesarean delivery. Obstet Gynecol. 2017;130(1):36–41.
- Prabhu M, McQuaid-Hanson E, Hopp S, et al. A shared decision-making intervention to guide opioid prescribing after cesarean delivery. Obstet Gynecol. 2017;130(1):42–46.
- Barbieri RL. Stop using codeine, oxycodone, hydrocodone, tramadol, and aspirin in women who are breastfeeding. OBG Manag. 2017;29(10):8–12.
- Pacheco LD, Hankins GD, Saad AF, Costantine MM, Chiossi G, Saade GR. Tranexamic acid for the management of obstetric hemorrhage. Obstet Gynecol. 2017;130(4);765–769.
- WHO recommendation on tranexamic acid for the treatment of postpartum haemorrhage. Geneva, Switzerland: World Health Organization; 2017.
- Stevens B, Krstic N, Jones M, Murphy L, Hoskovec J. Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol. 2017;130(2):279–284.

8 common questions about newborn circumcision
In the United States, circumcision is the fourth most common surgical procedure—behind cataract removal, cesarean delivery, and joint replacement.1 This operation, which dates to ancient times, is chosen for medical, personal, or religious reasons. It is performed on 77% of males born in the United States and on 42% of those born elsewhere who are living in this country.2 Whether it is performed depends not only on the parents’ race, ethnic background, and religion but also on region: US circumcision rates range from 74% in the Midwest to 30% in the West, and in between are the Northeast (67%) and the South (61%).3
Circumcision is not without controversy. Some claim that it is unnecessary cosmetic surgery, that it is genital mutilation, that the patient cannot choose it or object to it, or that it decreases sexual satisfaction.
In this article, I review 8 common questions about circumcision and provide data-based answers to them.
1. Should a newborn be circumcised?
For many years, the medical benefits of circumcision were scientifically ambiguous. With no clear answers, some thought that parents should base their decision for or against circumcision not on any potential medical benefit but rather on their family or religious tradition, or on a social standard, that is, what the majority of families in their community do.
Over the past 20 years, a growing body of evidence has demonstrated real medical benefits of circumcision. In 2012, the American Academy of Pediatrics (AAP), which previously had been neutral on the subject, issued a task force report concluding that the health benefits of circumcision outweigh its risks and justify access to the procedure.3,4 However, the report stopped short of recommending circumcision.
Opponents have expressed several concerns about circumcision. First, they say, it is painful and unnecessary, and performing it when life has just begun takes the decision away from the adult-to-be, who may want to be uncircumcised as an adult but will have no recourse. Second, they say circumcision will diminish the adult’s sexual pleasure. However, there is no proof this occurs, and it is unclear how the claim could be adequately verified.5
Health benefits of circumcision3
- Prevention of phimosis and balanoposthitis (inflammation of glans and foreskin), penile retraction disorders, and penile cancer
- Fewer infant urinary tract infections
- Decreased spread of human papillomavirus–related disease, including cervical cancer and its precursors, to sexual partners
- Lower risk of acquiring, harboring, and spreading human immunodeficiency virus infection, herpes virus infection, and other sexually transmitted diseases
- Easier genital hygiene
- No need for circumcision later in life, when the procedure is more involved
2. What is the best analgesia for circumcision?
Although in decades past circumcision was often performed without any analgesia, in the United States analgesia is now standard of care. The AAP Task Force on Circumcision formalized this standard in a 2012 policy statement.4 For newborn circumcision, analgesia can be given in the form of analgesic cream, penile ring block, or dorsal nerve block.
Analgesic EMLA cream (a mixture of local anesthetics such as lidocaine 2.5%/prilocaine 2.5%) is easy to use but is minimally effective in relieving circumcision pain,6 although some investigators have reported it is efficacious compared with placebo.7 When used, the analgesic cream is applied 30 to 60 minutes before circumcision.
Both penile ring block and dorsal nerve block with 1% lidocaine are easy to administer and are very effective.8,9 They are best used with buffered lidocaine, which partially relieves the burning that occurs with injection. With both methods, the smaller the needle used (preferably 30 gauge), the better.
These 2 block methods have different injection sites. For the ring block, small amounts of lidocaine (1 to 1.5 mL) are given in a series of injections around the entire circumference of the base of the penis. The dorsal block targets the 2 dorsal nerves located at 10 o’clock and 2 o’clock at the base of the penis. Epinephrine, given its vasoconstrictive properties and the potential for necrosis, should never be used with local analgesia for penile infiltration.
Analgesia can be supplemented with comfort measures, such as a pacifier, sugar water, gentle rubbing on the forehead, and soothing speech.10
Related article:
Circumcision impedes viral disease. Will opposition fade?
3. What conditions are required for safe circumcision?
As circumcision is not medically required and need not occur in the days immediately after birth, it should be performed only when conditions are optimal:
- A pediatrician or other practitioner must first examine the newborn.
- The newborn must be full-term, healthy, and stable.
- The best time to circumcise a baby born prematurely is right before discharge from the intensive care nursery.
- The penis must be of normal size and without anatomical defect—no micropenis, hypospadias, or penoscrotal webbing.
- The lower abdominal fat pad must not be so large that it will cause the shaft’s skin to cover the exposed penile head.
- If there is a family history of a bleeding disorder, the newborn must be evaluated for the disorder before the circumcision.
- The newborn must have received his vitamin K shot.
4. What is the best circumcision method?
Circumcision can be performed with the Gomco circumcision clamp, the Mogen circumcision clamp, or the PlastiBell circumcision device. Each device works well, provides excellent results, and has its pluses and minuses. Practitioners should use the device with which they are most familiar and comfortable, which likely will be the device they used in training.
In the United States, the Gomco clamp is perhaps the most commonly used device. It provides good cosmetic results, and its metal “bell” protects the entire head of the penis. Of the 3 methods, however, it is the most difficult—the partially cut foreskin must be threaded between the bell and the clamp frame before the clamp is tightened. In many cases, too, there is bleeding at the penile frenulum.
The Mogen clamp, another commonly used device, also is used in traditional Jewish circumcisions. Of the 3 methods, it is the quickest, produces the best hemostasis, and is associated with the least discomfort.10 To those unfamiliar with the method, there may seem to be a potential for amputation of the head of the penis, but actually there virtually is no risk, as an indentation on the penile side of the clamp protects the penile head.
The PlastiBell device is very easy to use but must stay on until the foreskin becomes necrotic and the bell and foreskin fall off on their own—a process that takes 7 to 10 days. Many parents dislike this method because its final result is not immediate and they have to contend with a medical implement during their newborn’s first week home.
Electrocautery is not recommended. Some clinicians, especially urologists, use electrocautery as the cutting mechanism for circumcision. A review of the literature, however, reveals that electrocautery has not been studied head-to-head against traditional techniques, and that various significant complications—transected penile head, severe burns, meatal stenosis—have been reported.11,12 It is certainly not a mainstream procedure for neonatal circumcision.
Evaluate penile anatomy for abnormalities
Before performing any circumcision, the head of the penis should be examined to rule out hypospadias or other penile abnormalities. This is because the foreskin is utilized in certain penile repair procedures. The pediatrician should perform an initial examination of the penis at the formal newborn physical within 24 hours of delivery. The clinician performing the circumcision should re-examine the penis just before the procedure is begun—by pushing back the foreskin as much as possible—as well as during the procedure, once the foreskin is lifted off the penile head but before the foreskin is excised.
Read about how to ensure the best outcome of circumcision.
5. When is the best time to perform a circumcision?
The medical literature provides no firm answer to this question. The younger the baby, the easier it is to perform a circumcision as a simple procedure with local anesthesia. The older the baby, the larger the penis and the more aware the baby will be of his surroundings. Both these factors will make the procedure more difficult.
Most clinicians would be reluctant to perform a circumcision in the office or clinic after the baby is 6 to 8 weeks old. If a family desires their son to be circumcised after that time—or a medical condition precludes earlier circumcision—the procedure is best performed by a pediatric urologist in the operating room.
Related article:
Circumcision accident: $1.3M verdict
6. What are the potential complications of circumcision?
The rate of circumcision complications is very low: 0.2%.13 That being said, the 3 most common types of complications are postoperative bleeding, infection, and damage to the penis.
Far and away the most common complication is postoperative bleeding , usually at the frenulum of the head of the penis (the 6 o’clock position). In most cases, the bleeding is light to moderate. It is controlled with direct pressure applied for several minutes, the use of processed gelatin (Gelfoam) or cellulose (Surgicel), sparing use of silver nitrate, or placement of a polyglycolic acid (Vicryl) 5-0 suture.
Infection, an unusual occurrence, is seen within 24 to 72 hours after circumcision. It is marked by swelling, redness, and a foul-smelling mucus discharge. This discharge must be differentiated from dried fibrin, which is commonly seen on the head of the penis in the days after circumcision but has no odor or association with erythema, fever, or infant fussiness. True infection should be treated, in collaboration with the child’s pediatrician, with a staphylococcal-sensitive penicillin (such as dicloxacillin).
More serious is damage to the penis, which ranges from accidental dilation of the meatus to partial amputation of the penile glans. Any such injury should immediately prompt a consultation with a pediatric urologist.
More of a nuisance than a complication is the sliding of the penile shaft’s skin up and over the glans. This is a relatively frequent occurrence after normal, successful circumcisions. Parents of an affected newborn should be instructed to gently slide the skin back until the head of the penis is completely exposed again. After several days, the skin will adhere to its proper position on the shaft.
- Just before the procedure, have a face-to-face discussion with the parents. Confirm that they want the circumcision done, explain exactly what it entails, and let them know they will receive complete aftercare instructions.
- Make sure one of the parents signs the consent form.
- Circumcise the right baby! Check the identification bracelet and confirm that the newborn’s hospital and chart numbers match.
- Prevent excessive hip movement by securing the baby's legs. The usual solution is a specially designed plastic restraint board with Velcro straps for the legs.
- Examine the infant’s penile anatomy prior to the procedure to make certain it is normal.
- For pain relief, administer enough analgesia, as either dorsal nerve block or penile ring block (the best methods). Before injection, draw the plunger of the syringe back to make certain that the needle is not in a blood vessel.
- During the procedure, make sure the entire membranous layer of foreskin covering the head of the penis is separated from the glans.
- Watch the penis for several minutes after the circumcision to make sure there is no bleeding.
7. What is a Jewish ritual circumcision?
For their newborn’s circumcision, Jewish parents may choose a bris ceremony, formally called a brit milah, in fulfillment of religious tradition. The ceremony involves a brief religious service, circumcision with the traditional Mogen clamp, a special blessing, and an official religious naming rite. The bris traditionally is performed by a mohel, a rabbi or other religious official trained in circumcision. Many parents have the bris done by a mohel who is a medical doctor. In the United States, the availability of both types of mohels varies.
8. Who should perform circumcisions—obstetricians or pediatricians?
The answer to this question depends on where you practice. In some communities or hospitals, the obstetrician performs newborn circumcision, while in other places the pediatrician does. In addition, depending on local circumstances or the specific population involved, circumcisions may be performed by a pediatric urologist, nurse practitioner, or even out of hospital by a trained religiously affiliated practitioner.
Obstetricians began doing circumcisions for 2 reasons. First, obstetricians are surgically trained whereas pediatricians are not. It was therefore thought to be more appropriate for obstetricians to do this minor surgical procedure. Second, circumcisions used to be done right in the delivery room shortly after delivery. It was thought that the crying induced by performing the circumcision helped clear the baby’s lungs and invigorated sluggish babies. Now, however, in-hospital circumcisions are usually done in the days following delivery, after the baby has had the opportunity to undergo his first physical examination to make sure that all is well and that the penile anatomy is normal.
Clinician experience, proper protocol contribute to a safe procedure
In the United States, a large percentage of male infants are circumcised. Although circumcision has known medical benefits, the procedure generally is performed for family, religious, or cultural reasons. Circumcision is a safe and straightforward procedure but has its risks and potential complications. As with most surgeries, the best outcomes are achieved by practitioners who are well trained, who perform the procedure under supervision until their experience is sufficient, and who follow correct protocol during the entire operation.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Dallas ME. The 10 most common surgeries in the US. Healthgrades website. https://www.healthgrades.com/explore/the-10-most-common-surgeries-in-the-us. Reviewed August 15, 2017. Accessed October 2, 2017.
- Laumann EO, Masi CM, Zuckerman EW. Circumcision in the United States: prevalence, prophylactic effects, and sexual practice. JAMA. 1997;277(13):1052–1057.
- American Academy of Pediatrics Task Force on Circumcision. Male circumcision. Pediatrics. 2012;130(3):e756–e785.
- American Academy of Pediatrics Task Force on Circumcision. Circumcision policy statement. Pediatrics. 2012;130(3):585–586.
- Morris BJ, Krieger JN. Does male circumcision affect sexual function, sensitivity, or satisfaction? A systematic review. J Sex Med. 2013;10(11):2644–2657.
- Howard FM, Howard CR, Fortune K, Generelli P, Zolnoun D, tenHoopen C. A randomized, placebo-controlled comparison of EMLA and dorsal penile nerve block for pain relief during neonatal circumcision. Prim Care Update Ob Gyns. 1998;5(4):196.
- Taddio A, Stevens B, Craig K, et al. Efficacy and safety of lidocaine-prilocaine cream for pain during circumcision. N Engl J Med. 1997;336(17):1197–1201.
- Lander J, Brady-Fryer B, Metcalfe JB, Nazarali S, Muttitt S. Comparison of ring block, dorsal penile nerve block, and topical anesthesia for neonatal circumcision: a randomized controlled trial. JAMA. 1997;278(24):2157–2162.
- Hardwick-Smith S, Mastrobattista JM, Wallace PA, Ritchey ML. Ring block for neonatal circumcision. Obstet Gynecol. 1998;91(6):930–934.
- Kaufman GE, Cimo S, Miller LW, Blass EM. An evaluation of the effects of sucrose on neonatal pain with 2 commonly used circumcision methods. Am J Obstet Gynecol. 2002;186(3):564–568.
- Tucker SC, Cerqueiro J, Sterne GD, Bracka A. Circumcision: a refined technique and 5 year review. Ann R Coll Surg Engl. 2001;83(2):121–125.
- Fraser ID, Tjoe J. Circumcision using bipolar scissors can be a safe and simple operation. Ann R Coll Surg Engl. 2000;82(3):190–191.
- Wiswell TE, Geschke DW. Risks from circumcision during the first month of life compared with those for uncircumcised boys. Pediatrics. 1989;83(6):1011–1015.

Dr. Lerner is Assistant Clinical Professor, Department of Obstetrics and Gynecology, Harvard Medical School, Boston, Massachusetts.
The author reports no financial relationships relevant to this article.
Dr. Lerner is Assistant Clinical Professor, Department of Obstetrics and Gynecology, Harvard Medical School, Boston, Massachusetts.
The author reports no financial relationships relevant to this article.
Dr. Lerner is Assistant Clinical Professor, Department of Obstetrics and Gynecology, Harvard Medical School, Boston, Massachusetts.
The author reports no financial relationships relevant to this article.
In the United States, circumcision is the fourth most common surgical procedure—behind cataract removal, cesarean delivery, and joint replacement.1 This operation, which dates to ancient times, is chosen for medical, personal, or religious reasons. It is performed on 77% of males born in the United States and on 42% of those born elsewhere who are living in this country.2 Whether it is performed depends not only on the parents’ race, ethnic background, and religion but also on region: US circumcision rates range from 74% in the Midwest to 30% in the West, and in between are the Northeast (67%) and the South (61%).3
Circumcision is not without controversy. Some claim that it is unnecessary cosmetic surgery, that it is genital mutilation, that the patient cannot choose it or object to it, or that it decreases sexual satisfaction.
In this article, I review 8 common questions about circumcision and provide data-based answers to them.
1. Should a newborn be circumcised?
For many years, the medical benefits of circumcision were scientifically ambiguous. With no clear answers, some thought that parents should base their decision for or against circumcision not on any potential medical benefit but rather on their family or religious tradition, or on a social standard, that is, what the majority of families in their community do.
Over the past 20 years, a growing body of evidence has demonstrated real medical benefits of circumcision. In 2012, the American Academy of Pediatrics (AAP), which previously had been neutral on the subject, issued a task force report concluding that the health benefits of circumcision outweigh its risks and justify access to the procedure.3,4 However, the report stopped short of recommending circumcision.
Opponents have expressed several concerns about circumcision. First, they say, it is painful and unnecessary, and performing it when life has just begun takes the decision away from the adult-to-be, who may want to be uncircumcised as an adult but will have no recourse. Second, they say circumcision will diminish the adult’s sexual pleasure. However, there is no proof this occurs, and it is unclear how the claim could be adequately verified.5
Health benefits of circumcision3
- Prevention of phimosis and balanoposthitis (inflammation of glans and foreskin), penile retraction disorders, and penile cancer
- Fewer infant urinary tract infections
- Decreased spread of human papillomavirus–related disease, including cervical cancer and its precursors, to sexual partners
- Lower risk of acquiring, harboring, and spreading human immunodeficiency virus infection, herpes virus infection, and other sexually transmitted diseases
- Easier genital hygiene
- No need for circumcision later in life, when the procedure is more involved
2. What is the best analgesia for circumcision?
Although in decades past circumcision was often performed without any analgesia, in the United States analgesia is now standard of care. The AAP Task Force on Circumcision formalized this standard in a 2012 policy statement.4 For newborn circumcision, analgesia can be given in the form of analgesic cream, penile ring block, or dorsal nerve block.
Analgesic EMLA cream (a mixture of local anesthetics such as lidocaine 2.5%/prilocaine 2.5%) is easy to use but is minimally effective in relieving circumcision pain,6 although some investigators have reported it is efficacious compared with placebo.7 When used, the analgesic cream is applied 30 to 60 minutes before circumcision.
Both penile ring block and dorsal nerve block with 1% lidocaine are easy to administer and are very effective.8,9 They are best used with buffered lidocaine, which partially relieves the burning that occurs with injection. With both methods, the smaller the needle used (preferably 30 gauge), the better.
These 2 block methods have different injection sites. For the ring block, small amounts of lidocaine (1 to 1.5 mL) are given in a series of injections around the entire circumference of the base of the penis. The dorsal block targets the 2 dorsal nerves located at 10 o’clock and 2 o’clock at the base of the penis. Epinephrine, given its vasoconstrictive properties and the potential for necrosis, should never be used with local analgesia for penile infiltration.
Analgesia can be supplemented with comfort measures, such as a pacifier, sugar water, gentle rubbing on the forehead, and soothing speech.10
Related article:
Circumcision impedes viral disease. Will opposition fade?
3. What conditions are required for safe circumcision?
As circumcision is not medically required and need not occur in the days immediately after birth, it should be performed only when conditions are optimal:
- A pediatrician or other practitioner must first examine the newborn.
- The newborn must be full-term, healthy, and stable.
- The best time to circumcise a baby born prematurely is right before discharge from the intensive care nursery.
- The penis must be of normal size and without anatomical defect—no micropenis, hypospadias, or penoscrotal webbing.
- The lower abdominal fat pad must not be so large that it will cause the shaft’s skin to cover the exposed penile head.
- If there is a family history of a bleeding disorder, the newborn must be evaluated for the disorder before the circumcision.
- The newborn must have received his vitamin K shot.
4. What is the best circumcision method?
Circumcision can be performed with the Gomco circumcision clamp, the Mogen circumcision clamp, or the PlastiBell circumcision device. Each device works well, provides excellent results, and has its pluses and minuses. Practitioners should use the device with which they are most familiar and comfortable, which likely will be the device they used in training.
In the United States, the Gomco clamp is perhaps the most commonly used device. It provides good cosmetic results, and its metal “bell” protects the entire head of the penis. Of the 3 methods, however, it is the most difficult—the partially cut foreskin must be threaded between the bell and the clamp frame before the clamp is tightened. In many cases, too, there is bleeding at the penile frenulum.
The Mogen clamp, another commonly used device, also is used in traditional Jewish circumcisions. Of the 3 methods, it is the quickest, produces the best hemostasis, and is associated with the least discomfort.10 To those unfamiliar with the method, there may seem to be a potential for amputation of the head of the penis, but actually there virtually is no risk, as an indentation on the penile side of the clamp protects the penile head.
The PlastiBell device is very easy to use but must stay on until the foreskin becomes necrotic and the bell and foreskin fall off on their own—a process that takes 7 to 10 days. Many parents dislike this method because its final result is not immediate and they have to contend with a medical implement during their newborn’s first week home.
Electrocautery is not recommended. Some clinicians, especially urologists, use electrocautery as the cutting mechanism for circumcision. A review of the literature, however, reveals that electrocautery has not been studied head-to-head against traditional techniques, and that various significant complications—transected penile head, severe burns, meatal stenosis—have been reported.11,12 It is certainly not a mainstream procedure for neonatal circumcision.
Evaluate penile anatomy for abnormalities
Before performing any circumcision, the head of the penis should be examined to rule out hypospadias or other penile abnormalities. This is because the foreskin is utilized in certain penile repair procedures. The pediatrician should perform an initial examination of the penis at the formal newborn physical within 24 hours of delivery. The clinician performing the circumcision should re-examine the penis just before the procedure is begun—by pushing back the foreskin as much as possible—as well as during the procedure, once the foreskin is lifted off the penile head but before the foreskin is excised.
Read about how to ensure the best outcome of circumcision.
5. When is the best time to perform a circumcision?
The medical literature provides no firm answer to this question. The younger the baby, the easier it is to perform a circumcision as a simple procedure with local anesthesia. The older the baby, the larger the penis and the more aware the baby will be of his surroundings. Both these factors will make the procedure more difficult.
Most clinicians would be reluctant to perform a circumcision in the office or clinic after the baby is 6 to 8 weeks old. If a family desires their son to be circumcised after that time—or a medical condition precludes earlier circumcision—the procedure is best performed by a pediatric urologist in the operating room.
Related article:
Circumcision accident: $1.3M verdict
6. What are the potential complications of circumcision?
The rate of circumcision complications is very low: 0.2%.13 That being said, the 3 most common types of complications are postoperative bleeding, infection, and damage to the penis.
Far and away the most common complication is postoperative bleeding , usually at the frenulum of the head of the penis (the 6 o’clock position). In most cases, the bleeding is light to moderate. It is controlled with direct pressure applied for several minutes, the use of processed gelatin (Gelfoam) or cellulose (Surgicel), sparing use of silver nitrate, or placement of a polyglycolic acid (Vicryl) 5-0 suture.
Infection, an unusual occurrence, is seen within 24 to 72 hours after circumcision. It is marked by swelling, redness, and a foul-smelling mucus discharge. This discharge must be differentiated from dried fibrin, which is commonly seen on the head of the penis in the days after circumcision but has no odor or association with erythema, fever, or infant fussiness. True infection should be treated, in collaboration with the child’s pediatrician, with a staphylococcal-sensitive penicillin (such as dicloxacillin).
More serious is damage to the penis, which ranges from accidental dilation of the meatus to partial amputation of the penile glans. Any such injury should immediately prompt a consultation with a pediatric urologist.
More of a nuisance than a complication is the sliding of the penile shaft’s skin up and over the glans. This is a relatively frequent occurrence after normal, successful circumcisions. Parents of an affected newborn should be instructed to gently slide the skin back until the head of the penis is completely exposed again. After several days, the skin will adhere to its proper position on the shaft.
- Just before the procedure, have a face-to-face discussion with the parents. Confirm that they want the circumcision done, explain exactly what it entails, and let them know they will receive complete aftercare instructions.
- Make sure one of the parents signs the consent form.
- Circumcise the right baby! Check the identification bracelet and confirm that the newborn’s hospital and chart numbers match.
- Prevent excessive hip movement by securing the baby's legs. The usual solution is a specially designed plastic restraint board with Velcro straps for the legs.
- Examine the infant’s penile anatomy prior to the procedure to make certain it is normal.
- For pain relief, administer enough analgesia, as either dorsal nerve block or penile ring block (the best methods). Before injection, draw the plunger of the syringe back to make certain that the needle is not in a blood vessel.
- During the procedure, make sure the entire membranous layer of foreskin covering the head of the penis is separated from the glans.
- Watch the penis for several minutes after the circumcision to make sure there is no bleeding.
7. What is a Jewish ritual circumcision?
For their newborn’s circumcision, Jewish parents may choose a bris ceremony, formally called a brit milah, in fulfillment of religious tradition. The ceremony involves a brief religious service, circumcision with the traditional Mogen clamp, a special blessing, and an official religious naming rite. The bris traditionally is performed by a mohel, a rabbi or other religious official trained in circumcision. Many parents have the bris done by a mohel who is a medical doctor. In the United States, the availability of both types of mohels varies.
8. Who should perform circumcisions—obstetricians or pediatricians?
The answer to this question depends on where you practice. In some communities or hospitals, the obstetrician performs newborn circumcision, while in other places the pediatrician does. In addition, depending on local circumstances or the specific population involved, circumcisions may be performed by a pediatric urologist, nurse practitioner, or even out of hospital by a trained religiously affiliated practitioner.
Obstetricians began doing circumcisions for 2 reasons. First, obstetricians are surgically trained whereas pediatricians are not. It was therefore thought to be more appropriate for obstetricians to do this minor surgical procedure. Second, circumcisions used to be done right in the delivery room shortly after delivery. It was thought that the crying induced by performing the circumcision helped clear the baby’s lungs and invigorated sluggish babies. Now, however, in-hospital circumcisions are usually done in the days following delivery, after the baby has had the opportunity to undergo his first physical examination to make sure that all is well and that the penile anatomy is normal.
Clinician experience, proper protocol contribute to a safe procedure
In the United States, a large percentage of male infants are circumcised. Although circumcision has known medical benefits, the procedure generally is performed for family, religious, or cultural reasons. Circumcision is a safe and straightforward procedure but has its risks and potential complications. As with most surgeries, the best outcomes are achieved by practitioners who are well trained, who perform the procedure under supervision until their experience is sufficient, and who follow correct protocol during the entire operation.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In the United States, circumcision is the fourth most common surgical procedure—behind cataract removal, cesarean delivery, and joint replacement.1 This operation, which dates to ancient times, is chosen for medical, personal, or religious reasons. It is performed on 77% of males born in the United States and on 42% of those born elsewhere who are living in this country.2 Whether it is performed depends not only on the parents’ race, ethnic background, and religion but also on region: US circumcision rates range from 74% in the Midwest to 30% in the West, and in between are the Northeast (67%) and the South (61%).3
Circumcision is not without controversy. Some claim that it is unnecessary cosmetic surgery, that it is genital mutilation, that the patient cannot choose it or object to it, or that it decreases sexual satisfaction.
In this article, I review 8 common questions about circumcision and provide data-based answers to them.
1. Should a newborn be circumcised?
For many years, the medical benefits of circumcision were scientifically ambiguous. With no clear answers, some thought that parents should base their decision for or against circumcision not on any potential medical benefit but rather on their family or religious tradition, or on a social standard, that is, what the majority of families in their community do.
Over the past 20 years, a growing body of evidence has demonstrated real medical benefits of circumcision. In 2012, the American Academy of Pediatrics (AAP), which previously had been neutral on the subject, issued a task force report concluding that the health benefits of circumcision outweigh its risks and justify access to the procedure.3,4 However, the report stopped short of recommending circumcision.
Opponents have expressed several concerns about circumcision. First, they say, it is painful and unnecessary, and performing it when life has just begun takes the decision away from the adult-to-be, who may want to be uncircumcised as an adult but will have no recourse. Second, they say circumcision will diminish the adult’s sexual pleasure. However, there is no proof this occurs, and it is unclear how the claim could be adequately verified.5
Health benefits of circumcision3
- Prevention of phimosis and balanoposthitis (inflammation of glans and foreskin), penile retraction disorders, and penile cancer
- Fewer infant urinary tract infections
- Decreased spread of human papillomavirus–related disease, including cervical cancer and its precursors, to sexual partners
- Lower risk of acquiring, harboring, and spreading human immunodeficiency virus infection, herpes virus infection, and other sexually transmitted diseases
- Easier genital hygiene
- No need for circumcision later in life, when the procedure is more involved
2. What is the best analgesia for circumcision?
Although in decades past circumcision was often performed without any analgesia, in the United States analgesia is now standard of care. The AAP Task Force on Circumcision formalized this standard in a 2012 policy statement.4 For newborn circumcision, analgesia can be given in the form of analgesic cream, penile ring block, or dorsal nerve block.
Analgesic EMLA cream (a mixture of local anesthetics such as lidocaine 2.5%/prilocaine 2.5%) is easy to use but is minimally effective in relieving circumcision pain,6 although some investigators have reported it is efficacious compared with placebo.7 When used, the analgesic cream is applied 30 to 60 minutes before circumcision.
Both penile ring block and dorsal nerve block with 1% lidocaine are easy to administer and are very effective.8,9 They are best used with buffered lidocaine, which partially relieves the burning that occurs with injection. With both methods, the smaller the needle used (preferably 30 gauge), the better.
These 2 block methods have different injection sites. For the ring block, small amounts of lidocaine (1 to 1.5 mL) are given in a series of injections around the entire circumference of the base of the penis. The dorsal block targets the 2 dorsal nerves located at 10 o’clock and 2 o’clock at the base of the penis. Epinephrine, given its vasoconstrictive properties and the potential for necrosis, should never be used with local analgesia for penile infiltration.
Analgesia can be supplemented with comfort measures, such as a pacifier, sugar water, gentle rubbing on the forehead, and soothing speech.10
Related article:
Circumcision impedes viral disease. Will opposition fade?
3. What conditions are required for safe circumcision?
As circumcision is not medically required and need not occur in the days immediately after birth, it should be performed only when conditions are optimal:
- A pediatrician or other practitioner must first examine the newborn.
- The newborn must be full-term, healthy, and stable.
- The best time to circumcise a baby born prematurely is right before discharge from the intensive care nursery.
- The penis must be of normal size and without anatomical defect—no micropenis, hypospadias, or penoscrotal webbing.
- The lower abdominal fat pad must not be so large that it will cause the shaft’s skin to cover the exposed penile head.
- If there is a family history of a bleeding disorder, the newborn must be evaluated for the disorder before the circumcision.
- The newborn must have received his vitamin K shot.
4. What is the best circumcision method?
Circumcision can be performed with the Gomco circumcision clamp, the Mogen circumcision clamp, or the PlastiBell circumcision device. Each device works well, provides excellent results, and has its pluses and minuses. Practitioners should use the device with which they are most familiar and comfortable, which likely will be the device they used in training.
In the United States, the Gomco clamp is perhaps the most commonly used device. It provides good cosmetic results, and its metal “bell” protects the entire head of the penis. Of the 3 methods, however, it is the most difficult—the partially cut foreskin must be threaded between the bell and the clamp frame before the clamp is tightened. In many cases, too, there is bleeding at the penile frenulum.
The Mogen clamp, another commonly used device, also is used in traditional Jewish circumcisions. Of the 3 methods, it is the quickest, produces the best hemostasis, and is associated with the least discomfort.10 To those unfamiliar with the method, there may seem to be a potential for amputation of the head of the penis, but actually there virtually is no risk, as an indentation on the penile side of the clamp protects the penile head.
The PlastiBell device is very easy to use but must stay on until the foreskin becomes necrotic and the bell and foreskin fall off on their own—a process that takes 7 to 10 days. Many parents dislike this method because its final result is not immediate and they have to contend with a medical implement during their newborn’s first week home.
Electrocautery is not recommended. Some clinicians, especially urologists, use electrocautery as the cutting mechanism for circumcision. A review of the literature, however, reveals that electrocautery has not been studied head-to-head against traditional techniques, and that various significant complications—transected penile head, severe burns, meatal stenosis—have been reported.11,12 It is certainly not a mainstream procedure for neonatal circumcision.
Evaluate penile anatomy for abnormalities
Before performing any circumcision, the head of the penis should be examined to rule out hypospadias or other penile abnormalities. This is because the foreskin is utilized in certain penile repair procedures. The pediatrician should perform an initial examination of the penis at the formal newborn physical within 24 hours of delivery. The clinician performing the circumcision should re-examine the penis just before the procedure is begun—by pushing back the foreskin as much as possible—as well as during the procedure, once the foreskin is lifted off the penile head but before the foreskin is excised.
Read about how to ensure the best outcome of circumcision.
5. When is the best time to perform a circumcision?
The medical literature provides no firm answer to this question. The younger the baby, the easier it is to perform a circumcision as a simple procedure with local anesthesia. The older the baby, the larger the penis and the more aware the baby will be of his surroundings. Both these factors will make the procedure more difficult.
Most clinicians would be reluctant to perform a circumcision in the office or clinic after the baby is 6 to 8 weeks old. If a family desires their son to be circumcised after that time—or a medical condition precludes earlier circumcision—the procedure is best performed by a pediatric urologist in the operating room.
Related article:
Circumcision accident: $1.3M verdict
6. What are the potential complications of circumcision?
The rate of circumcision complications is very low: 0.2%.13 That being said, the 3 most common types of complications are postoperative bleeding, infection, and damage to the penis.
Far and away the most common complication is postoperative bleeding , usually at the frenulum of the head of the penis (the 6 o’clock position). In most cases, the bleeding is light to moderate. It is controlled with direct pressure applied for several minutes, the use of processed gelatin (Gelfoam) or cellulose (Surgicel), sparing use of silver nitrate, or placement of a polyglycolic acid (Vicryl) 5-0 suture.
Infection, an unusual occurrence, is seen within 24 to 72 hours after circumcision. It is marked by swelling, redness, and a foul-smelling mucus discharge. This discharge must be differentiated from dried fibrin, which is commonly seen on the head of the penis in the days after circumcision but has no odor or association with erythema, fever, or infant fussiness. True infection should be treated, in collaboration with the child’s pediatrician, with a staphylococcal-sensitive penicillin (such as dicloxacillin).
More serious is damage to the penis, which ranges from accidental dilation of the meatus to partial amputation of the penile glans. Any such injury should immediately prompt a consultation with a pediatric urologist.
More of a nuisance than a complication is the sliding of the penile shaft’s skin up and over the glans. This is a relatively frequent occurrence after normal, successful circumcisions. Parents of an affected newborn should be instructed to gently slide the skin back until the head of the penis is completely exposed again. After several days, the skin will adhere to its proper position on the shaft.
- Just before the procedure, have a face-to-face discussion with the parents. Confirm that they want the circumcision done, explain exactly what it entails, and let them know they will receive complete aftercare instructions.
- Make sure one of the parents signs the consent form.
- Circumcise the right baby! Check the identification bracelet and confirm that the newborn’s hospital and chart numbers match.
- Prevent excessive hip movement by securing the baby's legs. The usual solution is a specially designed plastic restraint board with Velcro straps for the legs.
- Examine the infant’s penile anatomy prior to the procedure to make certain it is normal.
- For pain relief, administer enough analgesia, as either dorsal nerve block or penile ring block (the best methods). Before injection, draw the plunger of the syringe back to make certain that the needle is not in a blood vessel.
- During the procedure, make sure the entire membranous layer of foreskin covering the head of the penis is separated from the glans.
- Watch the penis for several minutes after the circumcision to make sure there is no bleeding.
7. What is a Jewish ritual circumcision?
For their newborn’s circumcision, Jewish parents may choose a bris ceremony, formally called a brit milah, in fulfillment of religious tradition. The ceremony involves a brief religious service, circumcision with the traditional Mogen clamp, a special blessing, and an official religious naming rite. The bris traditionally is performed by a mohel, a rabbi or other religious official trained in circumcision. Many parents have the bris done by a mohel who is a medical doctor. In the United States, the availability of both types of mohels varies.
8. Who should perform circumcisions—obstetricians or pediatricians?
The answer to this question depends on where you practice. In some communities or hospitals, the obstetrician performs newborn circumcision, while in other places the pediatrician does. In addition, depending on local circumstances or the specific population involved, circumcisions may be performed by a pediatric urologist, nurse practitioner, or even out of hospital by a trained religiously affiliated practitioner.
Obstetricians began doing circumcisions for 2 reasons. First, obstetricians are surgically trained whereas pediatricians are not. It was therefore thought to be more appropriate for obstetricians to do this minor surgical procedure. Second, circumcisions used to be done right in the delivery room shortly after delivery. It was thought that the crying induced by performing the circumcision helped clear the baby’s lungs and invigorated sluggish babies. Now, however, in-hospital circumcisions are usually done in the days following delivery, after the baby has had the opportunity to undergo his first physical examination to make sure that all is well and that the penile anatomy is normal.
Clinician experience, proper protocol contribute to a safe procedure
In the United States, a large percentage of male infants are circumcised. Although circumcision has known medical benefits, the procedure generally is performed for family, religious, or cultural reasons. Circumcision is a safe and straightforward procedure but has its risks and potential complications. As with most surgeries, the best outcomes are achieved by practitioners who are well trained, who perform the procedure under supervision until their experience is sufficient, and who follow correct protocol during the entire operation.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Dallas ME. The 10 most common surgeries in the US. Healthgrades website. https://www.healthgrades.com/explore/the-10-most-common-surgeries-in-the-us. Reviewed August 15, 2017. Accessed October 2, 2017.
- Laumann EO, Masi CM, Zuckerman EW. Circumcision in the United States: prevalence, prophylactic effects, and sexual practice. JAMA. 1997;277(13):1052–1057.
- American Academy of Pediatrics Task Force on Circumcision. Male circumcision. Pediatrics. 2012;130(3):e756–e785.
- American Academy of Pediatrics Task Force on Circumcision. Circumcision policy statement. Pediatrics. 2012;130(3):585–586.
- Morris BJ, Krieger JN. Does male circumcision affect sexual function, sensitivity, or satisfaction? A systematic review. J Sex Med. 2013;10(11):2644–2657.
- Howard FM, Howard CR, Fortune K, Generelli P, Zolnoun D, tenHoopen C. A randomized, placebo-controlled comparison of EMLA and dorsal penile nerve block for pain relief during neonatal circumcision. Prim Care Update Ob Gyns. 1998;5(4):196.
- Taddio A, Stevens B, Craig K, et al. Efficacy and safety of lidocaine-prilocaine cream for pain during circumcision. N Engl J Med. 1997;336(17):1197–1201.
- Lander J, Brady-Fryer B, Metcalfe JB, Nazarali S, Muttitt S. Comparison of ring block, dorsal penile nerve block, and topical anesthesia for neonatal circumcision: a randomized controlled trial. JAMA. 1997;278(24):2157–2162.
- Hardwick-Smith S, Mastrobattista JM, Wallace PA, Ritchey ML. Ring block for neonatal circumcision. Obstet Gynecol. 1998;91(6):930–934.
- Kaufman GE, Cimo S, Miller LW, Blass EM. An evaluation of the effects of sucrose on neonatal pain with 2 commonly used circumcision methods. Am J Obstet Gynecol. 2002;186(3):564–568.
- Tucker SC, Cerqueiro J, Sterne GD, Bracka A. Circumcision: a refined technique and 5 year review. Ann R Coll Surg Engl. 2001;83(2):121–125.
- Fraser ID, Tjoe J. Circumcision using bipolar scissors can be a safe and simple operation. Ann R Coll Surg Engl. 2000;82(3):190–191.
- Wiswell TE, Geschke DW. Risks from circumcision during the first month of life compared with those for uncircumcised boys. Pediatrics. 1989;83(6):1011–1015.
- Dallas ME. The 10 most common surgeries in the US. Healthgrades website. https://www.healthgrades.com/explore/the-10-most-common-surgeries-in-the-us. Reviewed August 15, 2017. Accessed October 2, 2017.
- Laumann EO, Masi CM, Zuckerman EW. Circumcision in the United States: prevalence, prophylactic effects, and sexual practice. JAMA. 1997;277(13):1052–1057.
- American Academy of Pediatrics Task Force on Circumcision. Male circumcision. Pediatrics. 2012;130(3):e756–e785.
- American Academy of Pediatrics Task Force on Circumcision. Circumcision policy statement. Pediatrics. 2012;130(3):585–586.
- Morris BJ, Krieger JN. Does male circumcision affect sexual function, sensitivity, or satisfaction? A systematic review. J Sex Med. 2013;10(11):2644–2657.
- Howard FM, Howard CR, Fortune K, Generelli P, Zolnoun D, tenHoopen C. A randomized, placebo-controlled comparison of EMLA and dorsal penile nerve block for pain relief during neonatal circumcision. Prim Care Update Ob Gyns. 1998;5(4):196.
- Taddio A, Stevens B, Craig K, et al. Efficacy and safety of lidocaine-prilocaine cream for pain during circumcision. N Engl J Med. 1997;336(17):1197–1201.
- Lander J, Brady-Fryer B, Metcalfe JB, Nazarali S, Muttitt S. Comparison of ring block, dorsal penile nerve block, and topical anesthesia for neonatal circumcision: a randomized controlled trial. JAMA. 1997;278(24):2157–2162.
- Hardwick-Smith S, Mastrobattista JM, Wallace PA, Ritchey ML. Ring block for neonatal circumcision. Obstet Gynecol. 1998;91(6):930–934.
- Kaufman GE, Cimo S, Miller LW, Blass EM. An evaluation of the effects of sucrose on neonatal pain with 2 commonly used circumcision methods. Am J Obstet Gynecol. 2002;186(3):564–568.
- Tucker SC, Cerqueiro J, Sterne GD, Bracka A. Circumcision: a refined technique and 5 year review. Ann R Coll Surg Engl. 2001;83(2):121–125.
- Fraser ID, Tjoe J. Circumcision using bipolar scissors can be a safe and simple operation. Ann R Coll Surg Engl. 2000;82(3):190–191.
- Wiswell TE, Geschke DW. Risks from circumcision during the first month of life compared with those for uncircumcised boys. Pediatrics. 1989;83(6):1011–1015.

Innovative Therapies for Severe Asthma
More than 39.5 million people in the U.S. have been diagnosed with asthma, and about 3,400 deaths occur annually due to asthma complications.1 Although the prevalence of atopy and asthma have increased over the past few decades in western countries, control and outcomes are improving.2 Use of asthma protocols and early recognition by the primary care provider (PCP) are among the main reasons for trends toward decreased hospitalization and fewer asthma-related deaths.3,4
In addition to the mainstay of treatments, including trigger avoidance, inhaled corticosteroids (ICS), and rescue bronchodilators, new therapies have been developed to supplement the treatment of severe persistent asthma, which constitutes about 5% to 10% of asthma cases. Severe asthma is defined as asthma that is unresponsive to baseline therapy.5
Three sets of guidelines and recommendations exist to provide structure to asthma treatment decision making. The Expert Panel Report-3 (EPR-3) was created by the National Education and Prevention Program (NAEPP) and was last published in 2007. The NAEPP favors a stepwise approach, based on asthma severity and age group.3 The International European Respiratory Society (ERS) and American Thoracic Society (ATS) task force report was updated in 2014.5 The Global Initiative for Asthma (GINA) report, updated in 2016, now includes several of the advances in asthma care for those patients refractory to standard treatments.
Asthma Therapies
In this review, the authors cover therapies for severe asthma that are becoming more important for PCPs to consider, including exhaled nitric oxide (NO) levels, the use of tiotropium for asthma, the applicability of biologic agents, the use of allergen immunotherapy, and the usefulness of roflumilast. This review also covers antileukotriene therapy, bronchial thermoplasty, and a discussion of long-acting beta-agonist (LABA) therapy.
Fractional Exhaled Nitric Oxide
Nitric oxide is present in the exhaled breath and is elevated in those with eosinophilic asthma.6 The role of NO in asthma pathology is complex, involving proinflammatory qualities that contribute to airway hyperresponsiveness (AHR) and as a weak mediator of smooth muscle relaxation. In exhaled air, NO correlates with up-regulation of NO synthase (NOS), which occurs with inflammation, therefore, quantifying airway inflammation.6-8
There has been some variability in the evidence supporting the use of fractional exhaled NO (FeNO) levels as a diagnostic tool. Some studies have suggested that FeNO is also elevated in other nonasthma conditions, such as eosinophilic bronchitis, atopy, and allergic rhinitis. Also, FeNO levels have been shown to be variably influenced by smoking, bronchoconstriction, and viral respiratory infections.9 However, FeNO levels > 50 ppb correlated most strongly with eosinophilic asthma and steroid responsiveness.9
Fractional exhaled NO tests now can be performed in the PCP office with NIOX VERO (Chicago, IL), a small, relatively inexpensive device. Although the 2016 GINA guidelines and the 2015 ERS/ATS guidelines do not offer specific recommendations for use and do not support withholding ICS based on FeNO test results, guidelines for FeNO use do exist. In 2011, ATS published a specific set of FeNO interpretive guidelines for office-based use.9 When performed in conjunction with standard testing, FeNO levels can provide valuable clinically relevant information, such as (1) detection of eosinophilic airway inflammation; (2) determining the likelihood of corticosteroid responsiveness; (3) monitoring of airway inflammation to determine the need for steroids; and (4) unmasking of otherwise unsuspected nonadherence to corticosteroid therapy (Table 1). 
Tiotropium as an Adjunct Treatment
Tiotropium is a long-acting inhaled anticholinergic. A sentinel 1984 study by Gross and Skorodin demonstrated that parasympathetic activity is the dominant reversible component in patients with chronic obstructive pulmonary disease (COPD), including emphysema.10 In addition, all achievable bronchodilation was obtained with an inhaled anticholinergic compared with that of separate or simultaneous administration of adrenergics. Sympathetic neural pathways are sparse in human lungs and have their endings on the cells of the cholinergic postganglionic fibers, because sympathetic terminals on airway smooth muscle cells are rare or nonexistent.11 Therefore, sympathetic modulation or activation of beta cells could change the parasympathetic tone.11
The FDA approved the addition of tiotropium for treating asthma in September 2015 for patients aged ≥ 12 years. The use of tiotropium is supported by both the ERS/ASTS and GINA 2016 guidelines. The recommended and approved dose of tiotropium for asthma is 2.5 µg daily (the recommended dose for COPD treatment is 5 µg).12 A recent phase 3 study compared 2.5 µg vs 5 µg dosing with ICS but no LABA in adolescents, noting significant improvement with the 2.5 µg dose.13 Adding tiotropium to ICS + LABA in patients with severe symptomatic asthma has been associated with positive results in initial studies by Kersjens and colleagues.14 Even as early as 2010, the use of tiotropium was shown to produce statistically significant improvement in morning peak expiratory flow (PEF), with a mean difference of 25.8 L/min (n = 210, P < .001).15
Tiotropium also has been shown to provide a sustained reduction in lung hyperinflation for those with COPD, thus providing an improvement in exertional dyspnea and exercise tolerance. On day 42 of a randomized, double-blinded, placebo-controlled, parallel-group study of 187 patients, vital capacity and inspiratory capacity were noted to be increased with decreases in residual volume and functional residual capacity. Exercise endurance times increased by 105 ± 40 sec (21%).16 This effect has not been studied yet in a population of patients with asthma; however, the same principles may hold true.
Biologic Agents
Recent asthma research has been focused on disrupting the inflammatory cascade. Both GINA and ERS/ATS divide asthma into allergic vs nonallergic endotypes. Allergic asthma usually is manifested by sputum eosinophilia and high serum eosinophil counts, whereas other endotypes include aspirin-sensitive and exercise-induced asthmas that present with a neutrophilic predominance. Nonallergic asthma is more severe typically and has been linked to steroid resistance.17 Many differentphenotypes have been identified, but the main categories include eosinophilic, neutrophilic, mixed, and paucigranulocytic.18
Mast cells, bronchial epithelium, and macrophages are involved in asthma progression. Targeting the cytokines produced by these pathways can be achieved through direct and indirect modulation. Interleukin (IL)-13 is central to development of AHR, and its effect is mediated through binding to its receptor and IL-4 receptor α complexes.19 Patients with severe asthma with an eosinophilic phenotype can benefit with the use of the new biologics, which decrease the amount of eosinophilia in lung tissue by blocking specific receptors for IL-5.
Omalizumab
Omalizumab, an anti-immunoglobulin E (IgE) antibody, has been shown to be helpful in treating patients with allergic asthma. Omalizumab is a 95% humanized IgE monoclonal antibody (MAB) that binds to the IgE molecule at the fc region and prevents IgE from binding to cell-surface receptors. In a humanized MAB, only the hypervariable regions are from mouse origin vs the newer completely human MABs. Omalizumab forms small, biologically inert IgE+ anti-IgE complexes that cannot activate the complement cascade. The serum free IgE level is decreased.20 Approved in 2003 for those aged ≤ 12 years, its use is restricted to patients with severe asthma, allergic sensitization (positive allergen skin testing), and an elevated serum IgE level (30-700 IU/mL). It is administered subcutaneously every 2 to 4 weeks, based on body weight and serum IgE levels.
For those with baseline eosinophil counts > 300 µL, addition of omalizumab most likely has been shown to improve quality of life (QOL) and reduce exacerbations, the use of rescue medications, ICS dosages, and ED visits.21-26 The most dangerous adverse effect (AE) was found to be an anaphylaxis rate of 0.09%, most frequently occurring in the first 2 hours after the first dose. Therefore, the patient must be monitored for 2 hours after the first dose and 30 minutes after subsequent doses. Epinephrine injection also should be prescribed. Although a 5-year prospective cohort study and retrospective pooled analysis of more than 10,000 patients did not support any relationship with malignancy.27,28 A higher incidence of cardiovascular and cerebrovascular AEs has been observed, and the FDA issued a safety announcement regarding this finding.29
Both ERS/ATS and GINA 2016 recommended that a therapeutic trial of omalizumab should be performed in all patients with severe confirmed IgE dependent allergic asthma.4,5 If there is no response in 4 months, it is unlikely that further administration would be beneficial.
Mepolizumab
Interleukin-5 is a key cytokine in the eosinophil life pathway. There are receptors for IL-5 on eosinophils, basophils, and β cells.30 Mepolizumab is an anti-IL-5 antibody for those with refractory eosinophilic asthma and a history of continued exacerbations. It has beneficial effects in the management of persistent airways eosinophilia among corticosteroid-resistant subjects. In a 2009 study, rates of exacerbations at 50 weeks were significantly lower than with placebo (2.0 vs 3.4 mean exacerbations per subject, 95% confidence interval [CI], 0.32-0.92; P = .002) as were eosinophil counts in blood and sputum (P < .001 and P = .002 respectively.31 A 2014 randomized, double-blind trial by Ortega and colleagues demonstrated reduction in rate of asthma exacerbations (primary outcome) to 47% (95% CI, 29-61) among patients receiving IV dosing and 53% (95% CI, 37-65) in the oral mepolizumab group (P < .001 for both groups, n = 576).32
In addition, there is significant data to show that even if the patient did not respond to omalizumab, he or she might still respond to mepolizumab. Data were collected from 2 randomized, double-blind, placebo-controlled studies with rate of exacerbation and percentage reduction in oral corticosteroid dose as the primary outcomes. In one of the studies (n = 576), the subjects were noted to have prior omalizumab use but still decreased exacerbation rate by 57%.33
Reslizumab
Reslizumab also is an FDA-approved anti-IL-5 antibody. It binds directly to IL-5 and prevents it from binding to eosinophils.34 For adults with severe eosinophilic asthma and refractory exacerbations, the goal of reslizumab therapy is to reduce eosinophil maturation, recruitment, and activation. Reslizumab is delivered in a weight-based IV dose (3 mg/kg) every 4 weeks. The FDA has issued a boxed warning for a 0.3% anaphylaxis rate.35 The most common AEs are elevated creatinine kinase, musculoskeletal pain, and oropharyngeal pain. Use of reslizumab resulted in greater reduction in sputum eosinophils, improvements in airway function, and a trend toward greater asthma control compared with that of placebo.34
Other Biologic Therapies
Many biologics are being developed as medical researchers continue to understand more of the mechanisms and pathways that contribute to allergic disease (Table 2). Dupilumab is an IL-4 inhibitor designated as a “breakthrough therapy” in 2014 by the FDA. This biologic blocks the downstream signaling events induced by IL-4 and IL-13 by binding to a subunit of the IL-4 receptor in the complexes. It has been found beneficial for those with high blood eosinophil counts and moderate-to-severe asthma and decreased asthma exacerbations when LABA and ICS were withdrawn.36,37 
Fevipiprant is a prostaglandin D2 inhibitor that blocks T-helper type 2 (Th2) cell migration and subsequent bronchoconstriction and cytokine effects with decreased IL-4, IL-5, and IL-13. Although sputum eosinophil percentage was noted to be decreased in a study involving 61 patients randomized to treatment for 12 weeks, asthma QOL questionnaires and prebronchodilator spirometry did not change.38,39
Benralizumab is an anti-IL-5 receptor antibody that has been more effective in reduction of airway and blood eosinophils levels compared with that of mepolizumab (undetectable vs 52% reduction), within 24 hours of IV dosing. In contrast, the anti-IL-5 antibodies take about 4 weeks to decrease eosinophil levels in blood and sputum.34 There have been no documented AEs aside from nasopharyngitis and injection site reactions and no safety concerns to date. It is currently undergoing phase 3 trials.40
Immunotherapies
Allergen immunotherapy is recommended for mild-to-moderate asthma. A 2010 Cochrane Review found that subcutaneous immunotherapy compared with placebo demonstrated improvements in bronchial hyperresponsiveness and decreased medication use.41 Expert Panel Report-3 guidelines recommend consideration of immunotherapy for mild-to-moderate asthma.5 While ERS/ATS guidelines for severe asthma do not address allergen immunotherapy, GINA guidelines incorporate it as Evidence A for treating modifiable risk factors to reduce exacerbations, although the efficacy is limited.6
Roflumilast
Roflumilast is a selective PDE4 inhibitor that has shown an anti-inflammatory effect in COPD. Studies evaluating the reversibility and prevention of airway remodeling showed good promise in mouse models.42 Data from 8 placebo-controlled, double-blind, phase 1, 2, and 3 studies conducted at 14 sites in Europe, North America, and South Africa from 1997 to 2005 showed reduced sputum eosinophil and neutrophil counts, consistent with findings during COPD treatment. However, forced expiratory volume in one second (FEV1) and PEF values were unchanged, suggesting that there was no acute bronchodilatory effect with roflumilast therapy.43 Roflumilast is not addressed in the 2016 GINA guidelines and at this time does not have a role in the treatment of severe asthma.
Antileukotrienes
After the activation of mast cells and eosinophils, leukotrienes are generated by 5-lipoxygenase from arachidonic acid and create bronchoconstriction, vasodilation, increased mucus production, increased recruitment of eosinophils, and decreased ciliary motility. Some studies have encouraged addingleukotriene receptor blockers (both montelukast and zafirlukast) to ICS therapy44,45 and to therapy for patients with aspirin-intolerant asthma or allergic asthma.46,47 However, other studies have shown them to be of limited benefit.48,49 A recent Cochrane Reviewof 18 randomized-controlled trials with 7,208 adults and children compared ICS + leukotriene receptor antagonist (LTRA) vs ICS + LABA.50 The ICS + LABA resulted in greater improvements in lung function, symptoms score, and rates of exacerbations.50
Most recommendations recognize the limitations of antileukotriene medications and agree that they are an adjunct rather than primary therapy. The GINA 2016 guidelines support the use of LTRAs in mild asthma, stating that although LTRAs are less effective than ICS (Evidence A), they may be appropriate for initial controller treatment for some patients who are unable or unwilling to use ICS or for patients with concurrent allergic rhinitis (Evidence B).51,52
Zileuton is a different type of antileukotriene. It inhibits leukotrienes B4, C4, D4, and E4 by inhibition of 5-lipoxygenase, interfering with leukotriene formation. It is approved for patients aged ≥ 12 years and is more expensive than montelukast or zafirlukast. Most studies supporting its use were conducted in patients with mild-to-moderate asthma on β-agonist therapy only. A 1988 study showed that zileuton therapy improved FEV1, reduced nasal symptoms, and decreased bronchial responsiveness to inhaled aspirin and histamine.53 All but 1 study patient were on ICS or oral corticosteroids. Zileuton was noted to be effective for patients with aspirin-intolerant asthma.
Some earlier studies reported that a small number of subjects had an increase in transaminases that resolved when they discontinued the medication. Therefore, it is recommended to check baseline laboratory results every 2 to 3 months.54,55 Neither GINA nor ERS/ATS guidelines address the use of zileuton.
Bronchial Thermoplasty
With asthma there is marked hypertrophy and hyperplasia that occurs in the airway smooth muscle. The airway of the patient with asthma also is lined with cells that promote inflammation. Thermal energy is used to perform controlled destruction of the inflammatory lining and pathologic hyperplasia. Three sequential bronchoscopies are performed 3 weeks apart to treat the right lower lobe, left lower lobe, and bilateral upper lobes. The right middle lobe is not treated due to its smaller diameter. Each bronchoscopy takes about 30 to 60 minutes. Patients are given perioperative steroids.56
Three large phase 3 clinical trials have evaluated the efficacy of bronchial thermoplasty (BT). The AIR (Asthma Intervention Research) trial in 2007 was a randomized controlled study of 112 patients with moderate or severe asthma that showed improved exacerbation rates, symptom-free days and QOL scores (1.3 ± 1.0 vs 0.6 ± 1.1; P = .003), but no difference in prebronchodilator FEV1 or AHR.57 There was a significant reduction in the rate of mild exacerbations and increase in morning PEF rates.57 Findings at 5 years showed improved AHR but no difference in frequency of need for oral corticosteroids and frequency of hospital or emergency department (ED) visits.58
The Research in Severe Asthma (RISA) clinical trial was a randomized controlled study (n = 32, 15 randomly assigned to BT) that showed improved prebronchodilator FEV1 in patients with severe, symptomatic asthma and baseline FEV1 of 62% to 66% with half the patients requiring oral corticosteroids (percentage predicted; 14.9 ± 17.4 vs -0.9 ± 22.3; P =.04).57 Quality of life scores were also significantly improved. At 5 years (14 BT patients were followed), the frequency of hospitalizations and ED visits decreased.59
The 2010 AIR2 study was a randomized, double blind, sham-controlled study (n = 288) developed to address the limitations of the 2 previous studies. It excluded the severest asthma cases, and its blinded nature was created with sham bronchoscopy to eliminate possible placebo effect. The study questionnaires showed improved QOL overall (79% vs 64%); however, there was a definite placebo effect noted.60 Decreased frequency of severe exacerbations as well as ED visits and days lost from work or school also were documented as secondary endpoints. At 5 years, decreased frequency of severe exacerbations and ED visits continued in the control group (85% consented to follow-up).61 Importantly, despite the placebo effect in QOL scores, there were no improvements in exacerbation rates or hospitalizations in those receiving sham bronchoscopy at the 1-year mark.61
Although more longitudinal studies need to be planned, including evaluation of those with the most severe asthma, there seems to be a sustained improvement in patients. Those who have received BT generally are found to have reduced airway smooth muscle with lower concentration of key inflammatory cytokines on follow-up bronchoscopy. However, variability in response has been documented.56 There has been no documented deterioration in pulmonary function with BT, and no significant structural abnormalities have been seen on high-resolution computed tomography.56,58 Both GINA 2016 and ERS/ATS support the use of BT in the context of adults with severe asthma, calling for more long-term studies to address delayed benefits and safety.
LABA Inhalers
A multicenter, double-blind, 26-week study of 11,693 patients randomized to ICS + LABA (budesonide/formoterol) vs ICS (budesonide) alone has shown no increased AEs in either arm. The study found that treatment with budesonide/formoterol was associated with lower risk of asthma exacerbations than using budesonide alone (16.5%; P = .002).62
The safety of adding a LABA to fluticasone also has been evaluated recently. A 2016 study of almost 12,000 patients (aged > 12 years) compared fluticasone proprionate alone vs fluticasone with salmeterol.63 There were no asthma-related deaths, but 2 patients in the fluticasone-only group were intubated with asthma complications. The risk of a severe asthma exacerbation seemed to be lower in the combination group (8% vs 10%; P < .001).63
A 2014 Cochrane Review supported the view that LABAs in adults seem to be safe when used concurrently with an ICS with a A-level recommendation, based on consistent good-quality, patient-oriented evidence.64 Multiple organizations have issued guidelines to this effect in the past, but previous results of studies showed that asthma deaths and a small increase in nonfatal serious AEs were noted in those using LABA monotherapy alone.64
NAEPP (EPR-3) and ERS/ATS recommend stepwise increases in the dose of ICS in combination with a LABA. The GINA guidelines recommend controller therapy to include combination IHS and LABA but with the consideration of higher doses of ICS than are routinely recommended for general use.
Inhaler and Inhaler Combinations
Many different inhalers of ICS alone and ICS/LABA combinations exist on the market today. There are differences in delivery that affect patient preference but these differences have not been found to improve delivery. Small particle ICS therapy could possibly correlate with improved delivery to the small airways.65 There are 3 preparations of inhaled steroids that fit in to this group, including beclomethasone, ciclesonide, and flunisolide. Other inhaled steroid formulations include budesonide, fluticasone propionate, fluticasone furoate, and mometasone.
Combination therapy (ICS + LABA) inhalers also are widely available. They include budesonide/formoterol, fluticasone proprionate/salmeterol, mometasone/formoterol, and the newer fluticasone furoate/vilanterol, a once-daily combination approved for those aged ≥ 18 years.
Conclusion
The treatment of severe asthma has progressed from simple manipulation of avoidance, bronchodilators, and corticosteroids to include many other treatments that have improved QOL for patients with refractory asthma. Although many of these options are delivered in coordination with an allergy and pulmonary specialist, it is important for the PCP to have a good knowledge base and awareness of additional treatments that are currently available.
1. Centers for Disease Control and Prevention. Asthma facts: CDC’s national asthma control program grantees. https://www.cdc.gov/asthma/pdfs/asthma_facts_program_grantees.pdf. Published July 2013. Accessed November 9, 2017.
2. Wilson DH, Adams RJ, Tucker G, Appleton S, Taylor AW, Ruffin RE. Trends in asthma prevalence and population changes in South Australia, 1990-2003. Med J Aust. 2006;184(5):226-229.
3. National Asthma Education Prevention Program. Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120(suppl 5):S94-S138.
4. Global Initiative for Asthma. Global strategy for asthma management and prevention: 2016 update. http://ginasthma.org/wp-content/up loads/2016/04/wms-GINA-2016-main-report-final.pdf. Accessed November 9, 2017.
5. Chung KF, Wenzel SE, Brozek JL, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343-373.
6. Reid DW, Johns DP, Feltis B, Ward C, Walters EH. Exhaled nitric oxide continues to reflect airway hyperresponsiveness and disease activity in inhaled corticosteroid-treated adult asthmatic patients. Respirology. 2003;8(4):479-486.
7. De Sanctis GT, MacLean JA, Hamada K, et al. Contribution of nitric oxide synthases 1, 2, and 3 to airway hyperresponsiveness and inflammation in a murine model of asthma. J Exp Med. 1999;189(10):1621-1630.
8. Ricciardolo FL. Multiple roles of nitric oxide in the airways. Thorax. 2003;58(2):175-182.
9. Dweik RA, Boggs PB, Erzurum SC, et al; American Thoracic Society Committee on Interpretation of Exhaled Nitric Oxide Levels (FENO) for Clinical Applications. An official ATS clinical practice guideline: interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am J Respir Crit Care Med. 2011;184(5):602-615.
10. Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med. 1984;311(7):421-425.
11. Gelb AF, Nadel JA. Affirmation of the adoration of the vagi and role of tiotropium in asthmatic patients. J Allergy Clin Immunol. 2016;138(4):1011-1013.
12. Chin SJ, Durmowicz AG, Chowdhury BA. Tiotropium respimat is effective for the treatment of asthma at a dose lower than that for chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13(2):173-179.
13. Hamelmann E, Bateman ED, Vogelberg C, et al. Tiotropium add-on therapy in adolescents with moderate asthma: a 1-year randomized controlled trial. J Allergy Clin Immunol. 2016;138(2):441-450.e8.
14. Kerstjens HA, Casale TB, Bleeker ER, et al. Tiotropium or salmeterol as add-on therapy to inhaled corticosteroids for patients with moderate symptomatic asthma: two replicate, double-blind, placebo-controlled, parallel-group, active-comparator, randomised trials. Lancet Respir Med. 2015;3(5):367-376.
15. Peters SP, Kunselman SJ, Icitovic N, et al; National Heart, Lung, and Blood Institute Asthma Clinical Research Network. Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med. 2010;363(18):1715-1726.
16. O’Donnell DE, Flüge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnea, and exercise tolerance in COPD. Eur Respir J. 2004;23(6):832-840.
17. Lötvall J, Akdis CA, Bacharier LB, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355-360.
18. Wenzel SE. Phenotypes in asthma: useful guides for therapy, distinct biological processes, or both? Am J Respir Crit Care Med. 2004;170(6):579-580.
19. Wang E, Hoyte FC. Traditional therapies for severe asthma. Immunol Allergy Clin North Am. 2016;36(3):581-608.
20. Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med. 2006;354(25):2689-2695.
21. Bousquet J, Wenzel S, Holgate S, Lumry W, Freeman P, Fox H. Predicting response to omalizumab, an anti-IgE antibody, in patients with allergic asthma. Chest. 2004;125(4):1378-1386.
22. Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. 2005;60(3):309-316.
23. Hanania NA, Alpan O, Hamilos DL, et al. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011;154(9):573-582.
24. Holgate ST, Djukanovic´ R, Casale T, Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: an update on anti-inflammatory activity and clinical efficacy. Clin Exp Allergy. 2005;35(4):408-416.
25. Finn A, Gross G, van Bavel J, et al. Omalizumab improves asthma-related quality of life in patients with severe allergic asthma. J Allergy Clin Immunol. 2003;111(2):278-284.
26. Busse W, Corren J, Lanier BQ, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108(2):184-190.
27. Long A, Rahmaoui A, Rothman KJ, et al. Incidence of malignancy in patients with moderate-to-severe asthma treated with or without omalizumab. J Allergy Clin Immunol. 2014;134(3):560-567.e4.
28. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6.
29. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA approves label changes for asthma drug Xolair (omalizumab), including describing slightly higher risk of heart and brain adverse events. http://www.fda.gov/Drugs /DrugSafety/ucm414911.htm. Updated February 10, 2016. Accessed November 9, 2017.
30. Tan HT, Sugita K, Akdis CA. Novel biologicals for the treatment of allergic diseases and asthma. Curr Allergy Asthma Rep. 2016;16(10):70.
31. Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360(10):973-984.
32. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198-1207.
33. Magnan A, Bourdin A, Prazma CM, et al. Treatment response with mepolizumab in severe eosinophilic asthma patients with previous omalizumab treatment. Allergy. 2016;71(9):1335-1344.
34. Castro M, Mathur S, Hargreave F, et al; Res-5-0010 Study Group. Resilizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184(10):1125-1132.
35. Cinqair [package insert]. Frazier, PA: Teva Respiratory; 2016.
36. Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455-2466.
37. Chung KF. Dupilumab: a potential new treatment for severe asthma. Lancet. 2016;388(10039):3-4.
38. Gonem S, Berair R, Singapuri A, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2016;4(9):699-707.
39. Erpenbeck VJ, Popov TA, Miller D, et al. The oral CRTh2 antagonist QAWO39 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther. 2016;39:54-63.
40. Tan LD, Bratt JM, Godor D, Louie S, Kenyon NJ. Benralizumab: a unique IL-5 inhibitor for severe asthma. J Asthma Allergy. 2016;9:71-81.
41. Abramson MJ, Puy RM, Weiner JM. Injection allergen immunotherapy for asthma. Cochrane Database Syst Rev. 2010;(8):CD001186.
42. Kim SW, Kim JH, Park CK, et al. Effect of roflumilast on airway remodeling in a murine model of chronic asthma. Clin Exp Allergy. 2016;46(5):754-763.
43. Bardin P, Kanniess F, Gauvreau G, Bredenbröker D, Rabe KF. Roflumilast for asthma: efficacy findings in mechanism of action studies. Pulm Pharmacol Ther. 2015;(suppl 35):S4-S10.
44. Virchow JC Jr, Prasse A, Naya I, Summerton L, Harris A. Zafirlukast improves asthma control in patients receiving high-dose inhaled corticosteroids. Am J Resp Crit Care Med. 2000;162(2, pt 1):558-585.
45. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
46. Dahlén SE, Malmström K, Nizankowska E, et al. Improvement of aspirin intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(1):9-14.
47. Price DB, Swern A, Tozzi CA, Philip G, Polos P. Effect of montelukast on lung function in asthma patients with allergic rhinitis: analysis from the COMPACT trial. Allergy. 2006; 61(6):737-742.
48. Robinson DS, Campbell D, Barnes PJ. Addition of leukotriene antagonists to therapy in chronic persistent asthma: a randomised double-blind placebo-controlled trial. Lancet. 2001;357(9273):2007-2011.
49. Chauhan BF, Ducharme FM. Addition to inhaled corticosteroids of long-acting beta 2 agonists versus anti-leukotrienes for chronic asthma. Cochrane Database Syst Rev. 2014;(1):CD003137.
50. Chauhan BF, Ducharme FM. Anti-leukotriene agents compared to inhaled corticosteroids in the management of recurrent and/or chronic asthma in adults and children. Cochrane Database Syst Rev. 2012;(5):CD002314.
51. Philip G, Nayak AS, Berger WE, et al. The effect of montelukast on rhinitis symptoms in patients with asthma and seasonal allergic rhinitis. Curr Med Res Opin. 2004;20(10):1549-1558.
52. Wilson AM, Dempsey OJ, Sims EJ, Lipworth BJ. A comparison of topical budesonide and oral montelukast in seasonal allergic rhinitis and asthma. Clin Exp Allergy. 2001;31(4):616-624.
53. Dahlén B, Nizankowska E, Szczeklik A, et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am J Respir Crit Care Med. 1998;157(4, pt 1):1187-1194.
54. Israel E, Cohn J, Dubé L, Drazen JM. Effect of treatment with zileuton, a 5-lipoxygenase inhibitor, in patients with asthma. A randomized controlled trial. Zilueton Clinical Trial Group. JAMA. 1996;275(12):931-936.
55. Nelson H, Kemp J, Berger W, et al. Efficacy of zileuton controlled-release tablets administered twice daily in the treatment of moderate persistent asthma: a 3-month randomized controlled study. Ann Allergy Asthma Immunol. 2007;99(2):178-184.
56. Laxmanan B, Egressy K, Murgu SD, White SR, Hogarth DK. Advances in bronchial thermoplasty. Chest. 2016;150(3):694-704.
57. Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med. 2007;356(13):1327-1337.
58. Thomson NC, Rubin AS, Niven RM, et al; AIR Trial Study Group. Long-term (5 year) safety of bronchial thermoplasty: Asthma Intervention Research (AIR) trial. BMC Pulm Med. 2011:11:8.
59. Pavord, ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med. 2007;176(12):1185-1191.
60. Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med. 2010;181(2):116-124.
61. Wechsler ME, Laviolette M, Rubin AS, et al; Asthma Intervention Research 2 Trial Study Group. Bronchial thermoplasty: long-term safety and effectiveness in patients with severe persistent asthma. J Allergy Clin Immunol. 2013;132(6):1295-1302.
62. Peters SP, Bleecker ER, Canonica GW, et al. Serious asthma events with budesonide plus formoterol vs budesonide alone. N Engl J Med. 2016;375(9):850-860.
63. Stempel DA, Raphiou IH, Kral KM, et al; AUSTRI Investigators. Serious asthma events with fluticasone plus salmeterol versus fluticasone alone. N Engl J Med. 2016;374(19):1822-1830.
64. Kew KM, Dias S, Cates CJ. Long-acting inhaled therapy (beta-agonists, anticholinergics and steroids) for COPD: a network meta-analysis. Cochrane Database Syst Rev. 2014;(3):CD010844.
65. Finkas LK, Martin R. Role of small airways in asthma. Immunol Allergy Clin North Am. 2016;36(3):473-482.
More than 39.5 million people in the U.S. have been diagnosed with asthma, and about 3,400 deaths occur annually due to asthma complications.1 Although the prevalence of atopy and asthma have increased over the past few decades in western countries, control and outcomes are improving.2 Use of asthma protocols and early recognition by the primary care provider (PCP) are among the main reasons for trends toward decreased hospitalization and fewer asthma-related deaths.3,4
In addition to the mainstay of treatments, including trigger avoidance, inhaled corticosteroids (ICS), and rescue bronchodilators, new therapies have been developed to supplement the treatment of severe persistent asthma, which constitutes about 5% to 10% of asthma cases. Severe asthma is defined as asthma that is unresponsive to baseline therapy.5
Three sets of guidelines and recommendations exist to provide structure to asthma treatment decision making. The Expert Panel Report-3 (EPR-3) was created by the National Education and Prevention Program (NAEPP) and was last published in 2007. The NAEPP favors a stepwise approach, based on asthma severity and age group.3 The International European Respiratory Society (ERS) and American Thoracic Society (ATS) task force report was updated in 2014.5 The Global Initiative for Asthma (GINA) report, updated in 2016, now includes several of the advances in asthma care for those patients refractory to standard treatments.
Asthma Therapies
In this review, the authors cover therapies for severe asthma that are becoming more important for PCPs to consider, including exhaled nitric oxide (NO) levels, the use of tiotropium for asthma, the applicability of biologic agents, the use of allergen immunotherapy, and the usefulness of roflumilast. This review also covers antileukotriene therapy, bronchial thermoplasty, and a discussion of long-acting beta-agonist (LABA) therapy.
Fractional Exhaled Nitric Oxide
Nitric oxide is present in the exhaled breath and is elevated in those with eosinophilic asthma.6 The role of NO in asthma pathology is complex, involving proinflammatory qualities that contribute to airway hyperresponsiveness (AHR) and as a weak mediator of smooth muscle relaxation. In exhaled air, NO correlates with up-regulation of NO synthase (NOS), which occurs with inflammation, therefore, quantifying airway inflammation.6-8
There has been some variability in the evidence supporting the use of fractional exhaled NO (FeNO) levels as a diagnostic tool. Some studies have suggested that FeNO is also elevated in other nonasthma conditions, such as eosinophilic bronchitis, atopy, and allergic rhinitis. Also, FeNO levels have been shown to be variably influenced by smoking, bronchoconstriction, and viral respiratory infections.9 However, FeNO levels > 50 ppb correlated most strongly with eosinophilic asthma and steroid responsiveness.9
Fractional exhaled NO tests now can be performed in the PCP office with NIOX VERO (Chicago, IL), a small, relatively inexpensive device. Although the 2016 GINA guidelines and the 2015 ERS/ATS guidelines do not offer specific recommendations for use and do not support withholding ICS based on FeNO test results, guidelines for FeNO use do exist. In 2011, ATS published a specific set of FeNO interpretive guidelines for office-based use.9 When performed in conjunction with standard testing, FeNO levels can provide valuable clinically relevant information, such as (1) detection of eosinophilic airway inflammation; (2) determining the likelihood of corticosteroid responsiveness; (3) monitoring of airway inflammation to determine the need for steroids; and (4) unmasking of otherwise unsuspected nonadherence to corticosteroid therapy (Table 1).
Tiotropium as an Adjunct Treatment
Tiotropium is a long-acting inhaled anticholinergic. A sentinel 1984 study by Gross and Skorodin demonstrated that parasympathetic activity is the dominant reversible component in patients with chronic obstructive pulmonary disease (COPD), including emphysema.10 In addition, all achievable bronchodilation was obtained with an inhaled anticholinergic compared with that of separate or simultaneous administration of adrenergics. Sympathetic neural pathways are sparse in human lungs and have their endings on the cells of the cholinergic postganglionic fibers, because sympathetic terminals on airway smooth muscle cells are rare or nonexistent.11 Therefore, sympathetic modulation or activation of beta cells could change the parasympathetic tone.11
The FDA approved the addition of tiotropium for treating asthma in September 2015 for patients aged ≥ 12 years. The use of tiotropium is supported by both the ERS/ASTS and GINA 2016 guidelines. The recommended and approved dose of tiotropium for asthma is 2.5 µg daily (the recommended dose for COPD treatment is 5 µg).12 A recent phase 3 study compared 2.5 µg vs 5 µg dosing with ICS but no LABA in adolescents, noting significant improvement with the 2.5 µg dose.13 Adding tiotropium to ICS + LABA in patients with severe symptomatic asthma has been associated with positive results in initial studies by Kersjens and colleagues.14 Even as early as 2010, the use of tiotropium was shown to produce statistically significant improvement in morning peak expiratory flow (PEF), with a mean difference of 25.8 L/min (n = 210, P < .001).15
Tiotropium also has been shown to provide a sustained reduction in lung hyperinflation for those with COPD, thus providing an improvement in exertional dyspnea and exercise tolerance. On day 42 of a randomized, double-blinded, placebo-controlled, parallel-group study of 187 patients, vital capacity and inspiratory capacity were noted to be increased with decreases in residual volume and functional residual capacity. Exercise endurance times increased by 105 ± 40 sec (21%).16 This effect has not been studied yet in a population of patients with asthma; however, the same principles may hold true.
Biologic Agents
Recent asthma research has been focused on disrupting the inflammatory cascade. Both GINA and ERS/ATS divide asthma into allergic vs nonallergic endotypes. Allergic asthma usually is manifested by sputum eosinophilia and high serum eosinophil counts, whereas other endotypes include aspirin-sensitive and exercise-induced asthmas that present with a neutrophilic predominance. Nonallergic asthma is more severe typically and has been linked to steroid resistance.17 Many differentphenotypes have been identified, but the main categories include eosinophilic, neutrophilic, mixed, and paucigranulocytic.18
Mast cells, bronchial epithelium, and macrophages are involved in asthma progression. Targeting the cytokines produced by these pathways can be achieved through direct and indirect modulation. Interleukin (IL)-13 is central to development of AHR, and its effect is mediated through binding to its receptor and IL-4 receptor α complexes.19 Patients with severe asthma with an eosinophilic phenotype can benefit with the use of the new biologics, which decrease the amount of eosinophilia in lung tissue by blocking specific receptors for IL-5.
Omalizumab
Omalizumab, an anti-immunoglobulin E (IgE) antibody, has been shown to be helpful in treating patients with allergic asthma. Omalizumab is a 95% humanized IgE monoclonal antibody (MAB) that binds to the IgE molecule at the fc region and prevents IgE from binding to cell-surface receptors. In a humanized MAB, only the hypervariable regions are from mouse origin vs the newer completely human MABs. Omalizumab forms small, biologically inert IgE+ anti-IgE complexes that cannot activate the complement cascade. The serum free IgE level is decreased.20 Approved in 2003 for those aged ≤ 12 years, its use is restricted to patients with severe asthma, allergic sensitization (positive allergen skin testing), and an elevated serum IgE level (30-700 IU/mL). It is administered subcutaneously every 2 to 4 weeks, based on body weight and serum IgE levels.
For those with baseline eosinophil counts > 300 µL, addition of omalizumab most likely has been shown to improve quality of life (QOL) and reduce exacerbations, the use of rescue medications, ICS dosages, and ED visits.21-26 The most dangerous adverse effect (AE) was found to be an anaphylaxis rate of 0.09%, most frequently occurring in the first 2 hours after the first dose. Therefore, the patient must be monitored for 2 hours after the first dose and 30 minutes after subsequent doses. Epinephrine injection also should be prescribed. Although a 5-year prospective cohort study and retrospective pooled analysis of more than 10,000 patients did not support any relationship with malignancy.27,28 A higher incidence of cardiovascular and cerebrovascular AEs has been observed, and the FDA issued a safety announcement regarding this finding.29
Both ERS/ATS and GINA 2016 recommended that a therapeutic trial of omalizumab should be performed in all patients with severe confirmed IgE dependent allergic asthma.4,5 If there is no response in 4 months, it is unlikely that further administration would be beneficial.
Mepolizumab
Interleukin-5 is a key cytokine in the eosinophil life pathway. There are receptors for IL-5 on eosinophils, basophils, and β cells.30 Mepolizumab is an anti-IL-5 antibody for those with refractory eosinophilic asthma and a history of continued exacerbations. It has beneficial effects in the management of persistent airways eosinophilia among corticosteroid-resistant subjects. In a 2009 study, rates of exacerbations at 50 weeks were significantly lower than with placebo (2.0 vs 3.4 mean exacerbations per subject, 95% confidence interval [CI], 0.32-0.92; P = .002) as were eosinophil counts in blood and sputum (P < .001 and P = .002 respectively.31 A 2014 randomized, double-blind trial by Ortega and colleagues demonstrated reduction in rate of asthma exacerbations (primary outcome) to 47% (95% CI, 29-61) among patients receiving IV dosing and 53% (95% CI, 37-65) in the oral mepolizumab group (P < .001 for both groups, n = 576).32
In addition, there is significant data to show that even if the patient did not respond to omalizumab, he or she might still respond to mepolizumab. Data were collected from 2 randomized, double-blind, placebo-controlled studies with rate of exacerbation and percentage reduction in oral corticosteroid dose as the primary outcomes. In one of the studies (n = 576), the subjects were noted to have prior omalizumab use but still decreased exacerbation rate by 57%.33
Reslizumab
Reslizumab also is an FDA-approved anti-IL-5 antibody. It binds directly to IL-5 and prevents it from binding to eosinophils.34 For adults with severe eosinophilic asthma and refractory exacerbations, the goal of reslizumab therapy is to reduce eosinophil maturation, recruitment, and activation. Reslizumab is delivered in a weight-based IV dose (3 mg/kg) every 4 weeks. The FDA has issued a boxed warning for a 0.3% anaphylaxis rate.35 The most common AEs are elevated creatinine kinase, musculoskeletal pain, and oropharyngeal pain. Use of reslizumab resulted in greater reduction in sputum eosinophils, improvements in airway function, and a trend toward greater asthma control compared with that of placebo.34
Other Biologic Therapies
Many biologics are being developed as medical researchers continue to understand more of the mechanisms and pathways that contribute to allergic disease (Table 2). Dupilumab is an IL-4 inhibitor designated as a “breakthrough therapy” in 2014 by the FDA. This biologic blocks the downstream signaling events induced by IL-4 and IL-13 by binding to a subunit of the IL-4 receptor in the complexes. It has been found beneficial for those with high blood eosinophil counts and moderate-to-severe asthma and decreased asthma exacerbations when LABA and ICS were withdrawn.36,37
Fevipiprant is a prostaglandin D2 inhibitor that blocks T-helper type 2 (Th2) cell migration and subsequent bronchoconstriction and cytokine effects with decreased IL-4, IL-5, and IL-13. Although sputum eosinophil percentage was noted to be decreased in a study involving 61 patients randomized to treatment for 12 weeks, asthma QOL questionnaires and prebronchodilator spirometry did not change.38,39
Benralizumab is an anti-IL-5 receptor antibody that has been more effective in reduction of airway and blood eosinophils levels compared with that of mepolizumab (undetectable vs 52% reduction), within 24 hours of IV dosing. In contrast, the anti-IL-5 antibodies take about 4 weeks to decrease eosinophil levels in blood and sputum.34 There have been no documented AEs aside from nasopharyngitis and injection site reactions and no safety concerns to date. It is currently undergoing phase 3 trials.40
Immunotherapies
Allergen immunotherapy is recommended for mild-to-moderate asthma. A 2010 Cochrane Review found that subcutaneous immunotherapy compared with placebo demonstrated improvements in bronchial hyperresponsiveness and decreased medication use.41 Expert Panel Report-3 guidelines recommend consideration of immunotherapy for mild-to-moderate asthma.5 While ERS/ATS guidelines for severe asthma do not address allergen immunotherapy, GINA guidelines incorporate it as Evidence A for treating modifiable risk factors to reduce exacerbations, although the efficacy is limited.6
Roflumilast
Roflumilast is a selective PDE4 inhibitor that has shown an anti-inflammatory effect in COPD. Studies evaluating the reversibility and prevention of airway remodeling showed good promise in mouse models.42 Data from 8 placebo-controlled, double-blind, phase 1, 2, and 3 studies conducted at 14 sites in Europe, North America, and South Africa from 1997 to 2005 showed reduced sputum eosinophil and neutrophil counts, consistent with findings during COPD treatment. However, forced expiratory volume in one second (FEV1) and PEF values were unchanged, suggesting that there was no acute bronchodilatory effect with roflumilast therapy.43 Roflumilast is not addressed in the 2016 GINA guidelines and at this time does not have a role in the treatment of severe asthma.
Antileukotrienes
After the activation of mast cells and eosinophils, leukotrienes are generated by 5-lipoxygenase from arachidonic acid and create bronchoconstriction, vasodilation, increased mucus production, increased recruitment of eosinophils, and decreased ciliary motility. Some studies have encouraged addingleukotriene receptor blockers (both montelukast and zafirlukast) to ICS therapy44,45 and to therapy for patients with aspirin-intolerant asthma or allergic asthma.46,47 However, other studies have shown them to be of limited benefit.48,49 A recent Cochrane Reviewof 18 randomized-controlled trials with 7,208 adults and children compared ICS + leukotriene receptor antagonist (LTRA) vs ICS + LABA.50 The ICS + LABA resulted in greater improvements in lung function, symptoms score, and rates of exacerbations.50
Most recommendations recognize the limitations of antileukotriene medications and agree that they are an adjunct rather than primary therapy. The GINA 2016 guidelines support the use of LTRAs in mild asthma, stating that although LTRAs are less effective than ICS (Evidence A), they may be appropriate for initial controller treatment for some patients who are unable or unwilling to use ICS or for patients with concurrent allergic rhinitis (Evidence B).51,52
Zileuton is a different type of antileukotriene. It inhibits leukotrienes B4, C4, D4, and E4 by inhibition of 5-lipoxygenase, interfering with leukotriene formation. It is approved for patients aged ≥ 12 years and is more expensive than montelukast or zafirlukast. Most studies supporting its use were conducted in patients with mild-to-moderate asthma on β-agonist therapy only. A 1988 study showed that zileuton therapy improved FEV1, reduced nasal symptoms, and decreased bronchial responsiveness to inhaled aspirin and histamine.53 All but 1 study patient were on ICS or oral corticosteroids. Zileuton was noted to be effective for patients with aspirin-intolerant asthma.
Some earlier studies reported that a small number of subjects had an increase in transaminases that resolved when they discontinued the medication. Therefore, it is recommended to check baseline laboratory results every 2 to 3 months.54,55 Neither GINA nor ERS/ATS guidelines address the use of zileuton.
Bronchial Thermoplasty
With asthma there is marked hypertrophy and hyperplasia that occurs in the airway smooth muscle. The airway of the patient with asthma also is lined with cells that promote inflammation. Thermal energy is used to perform controlled destruction of the inflammatory lining and pathologic hyperplasia. Three sequential bronchoscopies are performed 3 weeks apart to treat the right lower lobe, left lower lobe, and bilateral upper lobes. The right middle lobe is not treated due to its smaller diameter. Each bronchoscopy takes about 30 to 60 minutes. Patients are given perioperative steroids.56
Three large phase 3 clinical trials have evaluated the efficacy of bronchial thermoplasty (BT). The AIR (Asthma Intervention Research) trial in 2007 was a randomized controlled study of 112 patients with moderate or severe asthma that showed improved exacerbation rates, symptom-free days and QOL scores (1.3 ± 1.0 vs 0.6 ± 1.1; P = .003), but no difference in prebronchodilator FEV1 or AHR.57 There was a significant reduction in the rate of mild exacerbations and increase in morning PEF rates.57 Findings at 5 years showed improved AHR but no difference in frequency of need for oral corticosteroids and frequency of hospital or emergency department (ED) visits.58
The Research in Severe Asthma (RISA) clinical trial was a randomized controlled study (n = 32, 15 randomly assigned to BT) that showed improved prebronchodilator FEV1 in patients with severe, symptomatic asthma and baseline FEV1 of 62% to 66% with half the patients requiring oral corticosteroids (percentage predicted; 14.9 ± 17.4 vs -0.9 ± 22.3; P =.04).57 Quality of life scores were also significantly improved. At 5 years (14 BT patients were followed), the frequency of hospitalizations and ED visits decreased.59
The 2010 AIR2 study was a randomized, double blind, sham-controlled study (n = 288) developed to address the limitations of the 2 previous studies. It excluded the severest asthma cases, and its blinded nature was created with sham bronchoscopy to eliminate possible placebo effect. The study questionnaires showed improved QOL overall (79% vs 64%); however, there was a definite placebo effect noted.60 Decreased frequency of severe exacerbations as well as ED visits and days lost from work or school also were documented as secondary endpoints. At 5 years, decreased frequency of severe exacerbations and ED visits continued in the control group (85% consented to follow-up).61 Importantly, despite the placebo effect in QOL scores, there were no improvements in exacerbation rates or hospitalizations in those receiving sham bronchoscopy at the 1-year mark.61
Although more longitudinal studies need to be planned, including evaluation of those with the most severe asthma, there seems to be a sustained improvement in patients. Those who have received BT generally are found to have reduced airway smooth muscle with lower concentration of key inflammatory cytokines on follow-up bronchoscopy. However, variability in response has been documented.56 There has been no documented deterioration in pulmonary function with BT, and no significant structural abnormalities have been seen on high-resolution computed tomography.56,58 Both GINA 2016 and ERS/ATS support the use of BT in the context of adults with severe asthma, calling for more long-term studies to address delayed benefits and safety.
LABA Inhalers
A multicenter, double-blind, 26-week study of 11,693 patients randomized to ICS + LABA (budesonide/formoterol) vs ICS (budesonide) alone has shown no increased AEs in either arm. The study found that treatment with budesonide/formoterol was associated with lower risk of asthma exacerbations than using budesonide alone (16.5%; P = .002).62
The safety of adding a LABA to fluticasone also has been evaluated recently. A 2016 study of almost 12,000 patients (aged > 12 years) compared fluticasone proprionate alone vs fluticasone with salmeterol.63 There were no asthma-related deaths, but 2 patients in the fluticasone-only group were intubated with asthma complications. The risk of a severe asthma exacerbation seemed to be lower in the combination group (8% vs 10%; P < .001).63
A 2014 Cochrane Review supported the view that LABAs in adults seem to be safe when used concurrently with an ICS with a A-level recommendation, based on consistent good-quality, patient-oriented evidence.64 Multiple organizations have issued guidelines to this effect in the past, but previous results of studies showed that asthma deaths and a small increase in nonfatal serious AEs were noted in those using LABA monotherapy alone.64
NAEPP (EPR-3) and ERS/ATS recommend stepwise increases in the dose of ICS in combination with a LABA. The GINA guidelines recommend controller therapy to include combination IHS and LABA but with the consideration of higher doses of ICS than are routinely recommended for general use.
Inhaler and Inhaler Combinations
Many different inhalers of ICS alone and ICS/LABA combinations exist on the market today. There are differences in delivery that affect patient preference but these differences have not been found to improve delivery. Small particle ICS therapy could possibly correlate with improved delivery to the small airways.65 There are 3 preparations of inhaled steroids that fit in to this group, including beclomethasone, ciclesonide, and flunisolide. Other inhaled steroid formulations include budesonide, fluticasone propionate, fluticasone furoate, and mometasone.
Combination therapy (ICS + LABA) inhalers also are widely available. They include budesonide/formoterol, fluticasone proprionate/salmeterol, mometasone/formoterol, and the newer fluticasone furoate/vilanterol, a once-daily combination approved for those aged ≥ 18 years.
Conclusion
The treatment of severe asthma has progressed from simple manipulation of avoidance, bronchodilators, and corticosteroids to include many other treatments that have improved QOL for patients with refractory asthma. Although many of these options are delivered in coordination with an allergy and pulmonary specialist, it is important for the PCP to have a good knowledge base and awareness of additional treatments that are currently available.
More than 39.5 million people in the U.S. have been diagnosed with asthma, and about 3,400 deaths occur annually due to asthma complications.1 Although the prevalence of atopy and asthma have increased over the past few decades in western countries, control and outcomes are improving.2 Use of asthma protocols and early recognition by the primary care provider (PCP) are among the main reasons for trends toward decreased hospitalization and fewer asthma-related deaths.3,4
In addition to the mainstay of treatments, including trigger avoidance, inhaled corticosteroids (ICS), and rescue bronchodilators, new therapies have been developed to supplement the treatment of severe persistent asthma, which constitutes about 5% to 10% of asthma cases. Severe asthma is defined as asthma that is unresponsive to baseline therapy.5
Three sets of guidelines and recommendations exist to provide structure to asthma treatment decision making. The Expert Panel Report-3 (EPR-3) was created by the National Education and Prevention Program (NAEPP) and was last published in 2007. The NAEPP favors a stepwise approach, based on asthma severity and age group.3 The International European Respiratory Society (ERS) and American Thoracic Society (ATS) task force report was updated in 2014.5 The Global Initiative for Asthma (GINA) report, updated in 2016, now includes several of the advances in asthma care for those patients refractory to standard treatments.
Asthma Therapies
In this review, the authors cover therapies for severe asthma that are becoming more important for PCPs to consider, including exhaled nitric oxide (NO) levels, the use of tiotropium for asthma, the applicability of biologic agents, the use of allergen immunotherapy, and the usefulness of roflumilast. This review also covers antileukotriene therapy, bronchial thermoplasty, and a discussion of long-acting beta-agonist (LABA) therapy.
Fractional Exhaled Nitric Oxide
Nitric oxide is present in the exhaled breath and is elevated in those with eosinophilic asthma.6 The role of NO in asthma pathology is complex, involving proinflammatory qualities that contribute to airway hyperresponsiveness (AHR) and as a weak mediator of smooth muscle relaxation. In exhaled air, NO correlates with up-regulation of NO synthase (NOS), which occurs with inflammation, therefore, quantifying airway inflammation.6-8
There has been some variability in the evidence supporting the use of fractional exhaled NO (FeNO) levels as a diagnostic tool. Some studies have suggested that FeNO is also elevated in other nonasthma conditions, such as eosinophilic bronchitis, atopy, and allergic rhinitis. Also, FeNO levels have been shown to be variably influenced by smoking, bronchoconstriction, and viral respiratory infections.9 However, FeNO levels > 50 ppb correlated most strongly with eosinophilic asthma and steroid responsiveness.9
Fractional exhaled NO tests now can be performed in the PCP office with NIOX VERO (Chicago, IL), a small, relatively inexpensive device. Although the 2016 GINA guidelines and the 2015 ERS/ATS guidelines do not offer specific recommendations for use and do not support withholding ICS based on FeNO test results, guidelines for FeNO use do exist. In 2011, ATS published a specific set of FeNO interpretive guidelines for office-based use.9 When performed in conjunction with standard testing, FeNO levels can provide valuable clinically relevant information, such as (1) detection of eosinophilic airway inflammation; (2) determining the likelihood of corticosteroid responsiveness; (3) monitoring of airway inflammation to determine the need for steroids; and (4) unmasking of otherwise unsuspected nonadherence to corticosteroid therapy (Table 1).
Tiotropium as an Adjunct Treatment
Tiotropium is a long-acting inhaled anticholinergic. A sentinel 1984 study by Gross and Skorodin demonstrated that parasympathetic activity is the dominant reversible component in patients with chronic obstructive pulmonary disease (COPD), including emphysema.10 In addition, all achievable bronchodilation was obtained with an inhaled anticholinergic compared with that of separate or simultaneous administration of adrenergics. Sympathetic neural pathways are sparse in human lungs and have their endings on the cells of the cholinergic postganglionic fibers, because sympathetic terminals on airway smooth muscle cells are rare or nonexistent.11 Therefore, sympathetic modulation or activation of beta cells could change the parasympathetic tone.11
The FDA approved the addition of tiotropium for treating asthma in September 2015 for patients aged ≥ 12 years. The use of tiotropium is supported by both the ERS/ASTS and GINA 2016 guidelines. The recommended and approved dose of tiotropium for asthma is 2.5 µg daily (the recommended dose for COPD treatment is 5 µg).12 A recent phase 3 study compared 2.5 µg vs 5 µg dosing with ICS but no LABA in adolescents, noting significant improvement with the 2.5 µg dose.13 Adding tiotropium to ICS + LABA in patients with severe symptomatic asthma has been associated with positive results in initial studies by Kersjens and colleagues.14 Even as early as 2010, the use of tiotropium was shown to produce statistically significant improvement in morning peak expiratory flow (PEF), with a mean difference of 25.8 L/min (n = 210, P < .001).15
Tiotropium also has been shown to provide a sustained reduction in lung hyperinflation for those with COPD, thus providing an improvement in exertional dyspnea and exercise tolerance. On day 42 of a randomized, double-blinded, placebo-controlled, parallel-group study of 187 patients, vital capacity and inspiratory capacity were noted to be increased with decreases in residual volume and functional residual capacity. Exercise endurance times increased by 105 ± 40 sec (21%).16 This effect has not been studied yet in a population of patients with asthma; however, the same principles may hold true.
Biologic Agents
Recent asthma research has been focused on disrupting the inflammatory cascade. Both GINA and ERS/ATS divide asthma into allergic vs nonallergic endotypes. Allergic asthma usually is manifested by sputum eosinophilia and high serum eosinophil counts, whereas other endotypes include aspirin-sensitive and exercise-induced asthmas that present with a neutrophilic predominance. Nonallergic asthma is more severe typically and has been linked to steroid resistance.17 Many differentphenotypes have been identified, but the main categories include eosinophilic, neutrophilic, mixed, and paucigranulocytic.18
Mast cells, bronchial epithelium, and macrophages are involved in asthma progression. Targeting the cytokines produced by these pathways can be achieved through direct and indirect modulation. Interleukin (IL)-13 is central to development of AHR, and its effect is mediated through binding to its receptor and IL-4 receptor α complexes.19 Patients with severe asthma with an eosinophilic phenotype can benefit with the use of the new biologics, which decrease the amount of eosinophilia in lung tissue by blocking specific receptors for IL-5.
Omalizumab
Omalizumab, an anti-immunoglobulin E (IgE) antibody, has been shown to be helpful in treating patients with allergic asthma. Omalizumab is a 95% humanized IgE monoclonal antibody (MAB) that binds to the IgE molecule at the fc region and prevents IgE from binding to cell-surface receptors. In a humanized MAB, only the hypervariable regions are from mouse origin vs the newer completely human MABs. Omalizumab forms small, biologically inert IgE+ anti-IgE complexes that cannot activate the complement cascade. The serum free IgE level is decreased.20 Approved in 2003 for those aged ≤ 12 years, its use is restricted to patients with severe asthma, allergic sensitization (positive allergen skin testing), and an elevated serum IgE level (30-700 IU/mL). It is administered subcutaneously every 2 to 4 weeks, based on body weight and serum IgE levels.
For those with baseline eosinophil counts > 300 µL, addition of omalizumab most likely has been shown to improve quality of life (QOL) and reduce exacerbations, the use of rescue medications, ICS dosages, and ED visits.21-26 The most dangerous adverse effect (AE) was found to be an anaphylaxis rate of 0.09%, most frequently occurring in the first 2 hours after the first dose. Therefore, the patient must be monitored for 2 hours after the first dose and 30 minutes after subsequent doses. Epinephrine injection also should be prescribed. Although a 5-year prospective cohort study and retrospective pooled analysis of more than 10,000 patients did not support any relationship with malignancy.27,28 A higher incidence of cardiovascular and cerebrovascular AEs has been observed, and the FDA issued a safety announcement regarding this finding.29
Both ERS/ATS and GINA 2016 recommended that a therapeutic trial of omalizumab should be performed in all patients with severe confirmed IgE dependent allergic asthma.4,5 If there is no response in 4 months, it is unlikely that further administration would be beneficial.
Mepolizumab
Interleukin-5 is a key cytokine in the eosinophil life pathway. There are receptors for IL-5 on eosinophils, basophils, and β cells.30 Mepolizumab is an anti-IL-5 antibody for those with refractory eosinophilic asthma and a history of continued exacerbations. It has beneficial effects in the management of persistent airways eosinophilia among corticosteroid-resistant subjects. In a 2009 study, rates of exacerbations at 50 weeks were significantly lower than with placebo (2.0 vs 3.4 mean exacerbations per subject, 95% confidence interval [CI], 0.32-0.92; P = .002) as were eosinophil counts in blood and sputum (P < .001 and P = .002 respectively.31 A 2014 randomized, double-blind trial by Ortega and colleagues demonstrated reduction in rate of asthma exacerbations (primary outcome) to 47% (95% CI, 29-61) among patients receiving IV dosing and 53% (95% CI, 37-65) in the oral mepolizumab group (P < .001 for both groups, n = 576).32
In addition, there is significant data to show that even if the patient did not respond to omalizumab, he or she might still respond to mepolizumab. Data were collected from 2 randomized, double-blind, placebo-controlled studies with rate of exacerbation and percentage reduction in oral corticosteroid dose as the primary outcomes. In one of the studies (n = 576), the subjects were noted to have prior omalizumab use but still decreased exacerbation rate by 57%.33
Reslizumab
Reslizumab also is an FDA-approved anti-IL-5 antibody. It binds directly to IL-5 and prevents it from binding to eosinophils.34 For adults with severe eosinophilic asthma and refractory exacerbations, the goal of reslizumab therapy is to reduce eosinophil maturation, recruitment, and activation. Reslizumab is delivered in a weight-based IV dose (3 mg/kg) every 4 weeks. The FDA has issued a boxed warning for a 0.3% anaphylaxis rate.35 The most common AEs are elevated creatinine kinase, musculoskeletal pain, and oropharyngeal pain. Use of reslizumab resulted in greater reduction in sputum eosinophils, improvements in airway function, and a trend toward greater asthma control compared with that of placebo.34
Other Biologic Therapies
Many biologics are being developed as medical researchers continue to understand more of the mechanisms and pathways that contribute to allergic disease (Table 2). Dupilumab is an IL-4 inhibitor designated as a “breakthrough therapy” in 2014 by the FDA. This biologic blocks the downstream signaling events induced by IL-4 and IL-13 by binding to a subunit of the IL-4 receptor in the complexes. It has been found beneficial for those with high blood eosinophil counts and moderate-to-severe asthma and decreased asthma exacerbations when LABA and ICS were withdrawn.36,37
Fevipiprant is a prostaglandin D2 inhibitor that blocks T-helper type 2 (Th2) cell migration and subsequent bronchoconstriction and cytokine effects with decreased IL-4, IL-5, and IL-13. Although sputum eosinophil percentage was noted to be decreased in a study involving 61 patients randomized to treatment for 12 weeks, asthma QOL questionnaires and prebronchodilator spirometry did not change.38,39
Benralizumab is an anti-IL-5 receptor antibody that has been more effective in reduction of airway and blood eosinophils levels compared with that of mepolizumab (undetectable vs 52% reduction), within 24 hours of IV dosing. In contrast, the anti-IL-5 antibodies take about 4 weeks to decrease eosinophil levels in blood and sputum.34 There have been no documented AEs aside from nasopharyngitis and injection site reactions and no safety concerns to date. It is currently undergoing phase 3 trials.40
Immunotherapies
Allergen immunotherapy is recommended for mild-to-moderate asthma. A 2010 Cochrane Review found that subcutaneous immunotherapy compared with placebo demonstrated improvements in bronchial hyperresponsiveness and decreased medication use.41 Expert Panel Report-3 guidelines recommend consideration of immunotherapy for mild-to-moderate asthma.5 While ERS/ATS guidelines for severe asthma do not address allergen immunotherapy, GINA guidelines incorporate it as Evidence A for treating modifiable risk factors to reduce exacerbations, although the efficacy is limited.6
Roflumilast
Roflumilast is a selective PDE4 inhibitor that has shown an anti-inflammatory effect in COPD. Studies evaluating the reversibility and prevention of airway remodeling showed good promise in mouse models.42 Data from 8 placebo-controlled, double-blind, phase 1, 2, and 3 studies conducted at 14 sites in Europe, North America, and South Africa from 1997 to 2005 showed reduced sputum eosinophil and neutrophil counts, consistent with findings during COPD treatment. However, forced expiratory volume in one second (FEV1) and PEF values were unchanged, suggesting that there was no acute bronchodilatory effect with roflumilast therapy.43 Roflumilast is not addressed in the 2016 GINA guidelines and at this time does not have a role in the treatment of severe asthma.
Antileukotrienes
After the activation of mast cells and eosinophils, leukotrienes are generated by 5-lipoxygenase from arachidonic acid and create bronchoconstriction, vasodilation, increased mucus production, increased recruitment of eosinophils, and decreased ciliary motility. Some studies have encouraged addingleukotriene receptor blockers (both montelukast and zafirlukast) to ICS therapy44,45 and to therapy for patients with aspirin-intolerant asthma or allergic asthma.46,47 However, other studies have shown them to be of limited benefit.48,49 A recent Cochrane Reviewof 18 randomized-controlled trials with 7,208 adults and children compared ICS + leukotriene receptor antagonist (LTRA) vs ICS + LABA.50 The ICS + LABA resulted in greater improvements in lung function, symptoms score, and rates of exacerbations.50
Most recommendations recognize the limitations of antileukotriene medications and agree that they are an adjunct rather than primary therapy. The GINA 2016 guidelines support the use of LTRAs in mild asthma, stating that although LTRAs are less effective than ICS (Evidence A), they may be appropriate for initial controller treatment for some patients who are unable or unwilling to use ICS or for patients with concurrent allergic rhinitis (Evidence B).51,52
Zileuton is a different type of antileukotriene. It inhibits leukotrienes B4, C4, D4, and E4 by inhibition of 5-lipoxygenase, interfering with leukotriene formation. It is approved for patients aged ≥ 12 years and is more expensive than montelukast or zafirlukast. Most studies supporting its use were conducted in patients with mild-to-moderate asthma on β-agonist therapy only. A 1988 study showed that zileuton therapy improved FEV1, reduced nasal symptoms, and decreased bronchial responsiveness to inhaled aspirin and histamine.53 All but 1 study patient were on ICS or oral corticosteroids. Zileuton was noted to be effective for patients with aspirin-intolerant asthma.
Some earlier studies reported that a small number of subjects had an increase in transaminases that resolved when they discontinued the medication. Therefore, it is recommended to check baseline laboratory results every 2 to 3 months.54,55 Neither GINA nor ERS/ATS guidelines address the use of zileuton.
Bronchial Thermoplasty
With asthma there is marked hypertrophy and hyperplasia that occurs in the airway smooth muscle. The airway of the patient with asthma also is lined with cells that promote inflammation. Thermal energy is used to perform controlled destruction of the inflammatory lining and pathologic hyperplasia. Three sequential bronchoscopies are performed 3 weeks apart to treat the right lower lobe, left lower lobe, and bilateral upper lobes. The right middle lobe is not treated due to its smaller diameter. Each bronchoscopy takes about 30 to 60 minutes. Patients are given perioperative steroids.56
Three large phase 3 clinical trials have evaluated the efficacy of bronchial thermoplasty (BT). The AIR (Asthma Intervention Research) trial in 2007 was a randomized controlled study of 112 patients with moderate or severe asthma that showed improved exacerbation rates, symptom-free days and QOL scores (1.3 ± 1.0 vs 0.6 ± 1.1; P = .003), but no difference in prebronchodilator FEV1 or AHR.57 There was a significant reduction in the rate of mild exacerbations and increase in morning PEF rates.57 Findings at 5 years showed improved AHR but no difference in frequency of need for oral corticosteroids and frequency of hospital or emergency department (ED) visits.58
The Research in Severe Asthma (RISA) clinical trial was a randomized controlled study (n = 32, 15 randomly assigned to BT) that showed improved prebronchodilator FEV1 in patients with severe, symptomatic asthma and baseline FEV1 of 62% to 66% with half the patients requiring oral corticosteroids (percentage predicted; 14.9 ± 17.4 vs -0.9 ± 22.3; P =.04).57 Quality of life scores were also significantly improved. At 5 years (14 BT patients were followed), the frequency of hospitalizations and ED visits decreased.59
The 2010 AIR2 study was a randomized, double blind, sham-controlled study (n = 288) developed to address the limitations of the 2 previous studies. It excluded the severest asthma cases, and its blinded nature was created with sham bronchoscopy to eliminate possible placebo effect. The study questionnaires showed improved QOL overall (79% vs 64%); however, there was a definite placebo effect noted.60 Decreased frequency of severe exacerbations as well as ED visits and days lost from work or school also were documented as secondary endpoints. At 5 years, decreased frequency of severe exacerbations and ED visits continued in the control group (85% consented to follow-up).61 Importantly, despite the placebo effect in QOL scores, there were no improvements in exacerbation rates or hospitalizations in those receiving sham bronchoscopy at the 1-year mark.61
Although more longitudinal studies need to be planned, including evaluation of those with the most severe asthma, there seems to be a sustained improvement in patients. Those who have received BT generally are found to have reduced airway smooth muscle with lower concentration of key inflammatory cytokines on follow-up bronchoscopy. However, variability in response has been documented.56 There has been no documented deterioration in pulmonary function with BT, and no significant structural abnormalities have been seen on high-resolution computed tomography.56,58 Both GINA 2016 and ERS/ATS support the use of BT in the context of adults with severe asthma, calling for more long-term studies to address delayed benefits and safety.
LABA Inhalers
A multicenter, double-blind, 26-week study of 11,693 patients randomized to ICS + LABA (budesonide/formoterol) vs ICS (budesonide) alone has shown no increased AEs in either arm. The study found that treatment with budesonide/formoterol was associated with lower risk of asthma exacerbations than using budesonide alone (16.5%; P = .002).62
The safety of adding a LABA to fluticasone also has been evaluated recently. A 2016 study of almost 12,000 patients (aged > 12 years) compared fluticasone proprionate alone vs fluticasone with salmeterol.63 There were no asthma-related deaths, but 2 patients in the fluticasone-only group were intubated with asthma complications. The risk of a severe asthma exacerbation seemed to be lower in the combination group (8% vs 10%; P < .001).63
A 2014 Cochrane Review supported the view that LABAs in adults seem to be safe when used concurrently with an ICS with a A-level recommendation, based on consistent good-quality, patient-oriented evidence.64 Multiple organizations have issued guidelines to this effect in the past, but previous results of studies showed that asthma deaths and a small increase in nonfatal serious AEs were noted in those using LABA monotherapy alone.64
NAEPP (EPR-3) and ERS/ATS recommend stepwise increases in the dose of ICS in combination with a LABA. The GINA guidelines recommend controller therapy to include combination IHS and LABA but with the consideration of higher doses of ICS than are routinely recommended for general use.
Inhaler and Inhaler Combinations
Many different inhalers of ICS alone and ICS/LABA combinations exist on the market today. There are differences in delivery that affect patient preference but these differences have not been found to improve delivery. Small particle ICS therapy could possibly correlate with improved delivery to the small airways.65 There are 3 preparations of inhaled steroids that fit in to this group, including beclomethasone, ciclesonide, and flunisolide. Other inhaled steroid formulations include budesonide, fluticasone propionate, fluticasone furoate, and mometasone.
Combination therapy (ICS + LABA) inhalers also are widely available. They include budesonide/formoterol, fluticasone proprionate/salmeterol, mometasone/formoterol, and the newer fluticasone furoate/vilanterol, a once-daily combination approved for those aged ≥ 18 years.
Conclusion
The treatment of severe asthma has progressed from simple manipulation of avoidance, bronchodilators, and corticosteroids to include many other treatments that have improved QOL for patients with refractory asthma. Although many of these options are delivered in coordination with an allergy and pulmonary specialist, it is important for the PCP to have a good knowledge base and awareness of additional treatments that are currently available.
1. Centers for Disease Control and Prevention. Asthma facts: CDC’s national asthma control program grantees. https://www.cdc.gov/asthma/pdfs/asthma_facts_program_grantees.pdf. Published July 2013. Accessed November 9, 2017.
2. Wilson DH, Adams RJ, Tucker G, Appleton S, Taylor AW, Ruffin RE. Trends in asthma prevalence and population changes in South Australia, 1990-2003. Med J Aust. 2006;184(5):226-229.
3. National Asthma Education Prevention Program. Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120(suppl 5):S94-S138.
4. Global Initiative for Asthma. Global strategy for asthma management and prevention: 2016 update. http://ginasthma.org/wp-content/up loads/2016/04/wms-GINA-2016-main-report-final.pdf. Accessed November 9, 2017.
5. Chung KF, Wenzel SE, Brozek JL, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343-373.
6. Reid DW, Johns DP, Feltis B, Ward C, Walters EH. Exhaled nitric oxide continues to reflect airway hyperresponsiveness and disease activity in inhaled corticosteroid-treated adult asthmatic patients. Respirology. 2003;8(4):479-486.
7. De Sanctis GT, MacLean JA, Hamada K, et al. Contribution of nitric oxide synthases 1, 2, and 3 to airway hyperresponsiveness and inflammation in a murine model of asthma. J Exp Med. 1999;189(10):1621-1630.
8. Ricciardolo FL. Multiple roles of nitric oxide in the airways. Thorax. 2003;58(2):175-182.
9. Dweik RA, Boggs PB, Erzurum SC, et al; American Thoracic Society Committee on Interpretation of Exhaled Nitric Oxide Levels (FENO) for Clinical Applications. An official ATS clinical practice guideline: interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am J Respir Crit Care Med. 2011;184(5):602-615.
10. Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med. 1984;311(7):421-425.
11. Gelb AF, Nadel JA. Affirmation of the adoration of the vagi and role of tiotropium in asthmatic patients. J Allergy Clin Immunol. 2016;138(4):1011-1013.
12. Chin SJ, Durmowicz AG, Chowdhury BA. Tiotropium respimat is effective for the treatment of asthma at a dose lower than that for chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13(2):173-179.
13. Hamelmann E, Bateman ED, Vogelberg C, et al. Tiotropium add-on therapy in adolescents with moderate asthma: a 1-year randomized controlled trial. J Allergy Clin Immunol. 2016;138(2):441-450.e8.
14. Kerstjens HA, Casale TB, Bleeker ER, et al. Tiotropium or salmeterol as add-on therapy to inhaled corticosteroids for patients with moderate symptomatic asthma: two replicate, double-blind, placebo-controlled, parallel-group, active-comparator, randomised trials. Lancet Respir Med. 2015;3(5):367-376.
15. Peters SP, Kunselman SJ, Icitovic N, et al; National Heart, Lung, and Blood Institute Asthma Clinical Research Network. Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med. 2010;363(18):1715-1726.
16. O’Donnell DE, Flüge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnea, and exercise tolerance in COPD. Eur Respir J. 2004;23(6):832-840.
17. Lötvall J, Akdis CA, Bacharier LB, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355-360.
18. Wenzel SE. Phenotypes in asthma: useful guides for therapy, distinct biological processes, or both? Am J Respir Crit Care Med. 2004;170(6):579-580.
19. Wang E, Hoyte FC. Traditional therapies for severe asthma. Immunol Allergy Clin North Am. 2016;36(3):581-608.
20. Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med. 2006;354(25):2689-2695.
21. Bousquet J, Wenzel S, Holgate S, Lumry W, Freeman P, Fox H. Predicting response to omalizumab, an anti-IgE antibody, in patients with allergic asthma. Chest. 2004;125(4):1378-1386.
22. Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. 2005;60(3):309-316.
23. Hanania NA, Alpan O, Hamilos DL, et al. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011;154(9):573-582.
24. Holgate ST, Djukanovic´ R, Casale T, Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: an update on anti-inflammatory activity and clinical efficacy. Clin Exp Allergy. 2005;35(4):408-416.
25. Finn A, Gross G, van Bavel J, et al. Omalizumab improves asthma-related quality of life in patients with severe allergic asthma. J Allergy Clin Immunol. 2003;111(2):278-284.
26. Busse W, Corren J, Lanier BQ, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108(2):184-190.
27. Long A, Rahmaoui A, Rothman KJ, et al. Incidence of malignancy in patients with moderate-to-severe asthma treated with or without omalizumab. J Allergy Clin Immunol. 2014;134(3):560-567.e4.
28. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6.
29. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA approves label changes for asthma drug Xolair (omalizumab), including describing slightly higher risk of heart and brain adverse events. http://www.fda.gov/Drugs /DrugSafety/ucm414911.htm. Updated February 10, 2016. Accessed November 9, 2017.
30. Tan HT, Sugita K, Akdis CA. Novel biologicals for the treatment of allergic diseases and asthma. Curr Allergy Asthma Rep. 2016;16(10):70.
31. Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360(10):973-984.
32. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198-1207.
33. Magnan A, Bourdin A, Prazma CM, et al. Treatment response with mepolizumab in severe eosinophilic asthma patients with previous omalizumab treatment. Allergy. 2016;71(9):1335-1344.
34. Castro M, Mathur S, Hargreave F, et al; Res-5-0010 Study Group. Resilizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184(10):1125-1132.
35. Cinqair [package insert]. Frazier, PA: Teva Respiratory; 2016.
36. Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455-2466.
37. Chung KF. Dupilumab: a potential new treatment for severe asthma. Lancet. 2016;388(10039):3-4.
38. Gonem S, Berair R, Singapuri A, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2016;4(9):699-707.
39. Erpenbeck VJ, Popov TA, Miller D, et al. The oral CRTh2 antagonist QAWO39 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther. 2016;39:54-63.
40. Tan LD, Bratt JM, Godor D, Louie S, Kenyon NJ. Benralizumab: a unique IL-5 inhibitor for severe asthma. J Asthma Allergy. 2016;9:71-81.
41. Abramson MJ, Puy RM, Weiner JM. Injection allergen immunotherapy for asthma. Cochrane Database Syst Rev. 2010;(8):CD001186.
42. Kim SW, Kim JH, Park CK, et al. Effect of roflumilast on airway remodeling in a murine model of chronic asthma. Clin Exp Allergy. 2016;46(5):754-763.
43. Bardin P, Kanniess F, Gauvreau G, Bredenbröker D, Rabe KF. Roflumilast for asthma: efficacy findings in mechanism of action studies. Pulm Pharmacol Ther. 2015;(suppl 35):S4-S10.
44. Virchow JC Jr, Prasse A, Naya I, Summerton L, Harris A. Zafirlukast improves asthma control in patients receiving high-dose inhaled corticosteroids. Am J Resp Crit Care Med. 2000;162(2, pt 1):558-585.
45. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
46. Dahlén SE, Malmström K, Nizankowska E, et al. Improvement of aspirin intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(1):9-14.
47. Price DB, Swern A, Tozzi CA, Philip G, Polos P. Effect of montelukast on lung function in asthma patients with allergic rhinitis: analysis from the COMPACT trial. Allergy. 2006; 61(6):737-742.
48. Robinson DS, Campbell D, Barnes PJ. Addition of leukotriene antagonists to therapy in chronic persistent asthma: a randomised double-blind placebo-controlled trial. Lancet. 2001;357(9273):2007-2011.
49. Chauhan BF, Ducharme FM. Addition to inhaled corticosteroids of long-acting beta 2 agonists versus anti-leukotrienes for chronic asthma. Cochrane Database Syst Rev. 2014;(1):CD003137.
50. Chauhan BF, Ducharme FM. Anti-leukotriene agents compared to inhaled corticosteroids in the management of recurrent and/or chronic asthma in adults and children. Cochrane Database Syst Rev. 2012;(5):CD002314.
51. Philip G, Nayak AS, Berger WE, et al. The effect of montelukast on rhinitis symptoms in patients with asthma and seasonal allergic rhinitis. Curr Med Res Opin. 2004;20(10):1549-1558.
52. Wilson AM, Dempsey OJ, Sims EJ, Lipworth BJ. A comparison of topical budesonide and oral montelukast in seasonal allergic rhinitis and asthma. Clin Exp Allergy. 2001;31(4):616-624.
53. Dahlén B, Nizankowska E, Szczeklik A, et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am J Respir Crit Care Med. 1998;157(4, pt 1):1187-1194.
54. Israel E, Cohn J, Dubé L, Drazen JM. Effect of treatment with zileuton, a 5-lipoxygenase inhibitor, in patients with asthma. A randomized controlled trial. Zilueton Clinical Trial Group. JAMA. 1996;275(12):931-936.
55. Nelson H, Kemp J, Berger W, et al. Efficacy of zileuton controlled-release tablets administered twice daily in the treatment of moderate persistent asthma: a 3-month randomized controlled study. Ann Allergy Asthma Immunol. 2007;99(2):178-184.
56. Laxmanan B, Egressy K, Murgu SD, White SR, Hogarth DK. Advances in bronchial thermoplasty. Chest. 2016;150(3):694-704.
57. Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med. 2007;356(13):1327-1337.
58. Thomson NC, Rubin AS, Niven RM, et al; AIR Trial Study Group. Long-term (5 year) safety of bronchial thermoplasty: Asthma Intervention Research (AIR) trial. BMC Pulm Med. 2011:11:8.
59. Pavord, ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med. 2007;176(12):1185-1191.
60. Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med. 2010;181(2):116-124.
61. Wechsler ME, Laviolette M, Rubin AS, et al; Asthma Intervention Research 2 Trial Study Group. Bronchial thermoplasty: long-term safety and effectiveness in patients with severe persistent asthma. J Allergy Clin Immunol. 2013;132(6):1295-1302.
62. Peters SP, Bleecker ER, Canonica GW, et al. Serious asthma events with budesonide plus formoterol vs budesonide alone. N Engl J Med. 2016;375(9):850-860.
63. Stempel DA, Raphiou IH, Kral KM, et al; AUSTRI Investigators. Serious asthma events with fluticasone plus salmeterol versus fluticasone alone. N Engl J Med. 2016;374(19):1822-1830.
64. Kew KM, Dias S, Cates CJ. Long-acting inhaled therapy (beta-agonists, anticholinergics and steroids) for COPD: a network meta-analysis. Cochrane Database Syst Rev. 2014;(3):CD010844.
65. Finkas LK, Martin R. Role of small airways in asthma. Immunol Allergy Clin North Am. 2016;36(3):473-482.
1. Centers for Disease Control and Prevention. Asthma facts: CDC’s national asthma control program grantees. https://www.cdc.gov/asthma/pdfs/asthma_facts_program_grantees.pdf. Published July 2013. Accessed November 9, 2017.
2. Wilson DH, Adams RJ, Tucker G, Appleton S, Taylor AW, Ruffin RE. Trends in asthma prevalence and population changes in South Australia, 1990-2003. Med J Aust. 2006;184(5):226-229.
3. National Asthma Education Prevention Program. Expert Panel Report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. J Allergy Clin Immunol. 2007;120(suppl 5):S94-S138.
4. Global Initiative for Asthma. Global strategy for asthma management and prevention: 2016 update. http://ginasthma.org/wp-content/up loads/2016/04/wms-GINA-2016-main-report-final.pdf. Accessed November 9, 2017.
5. Chung KF, Wenzel SE, Brozek JL, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343-373.
6. Reid DW, Johns DP, Feltis B, Ward C, Walters EH. Exhaled nitric oxide continues to reflect airway hyperresponsiveness and disease activity in inhaled corticosteroid-treated adult asthmatic patients. Respirology. 2003;8(4):479-486.
7. De Sanctis GT, MacLean JA, Hamada K, et al. Contribution of nitric oxide synthases 1, 2, and 3 to airway hyperresponsiveness and inflammation in a murine model of asthma. J Exp Med. 1999;189(10):1621-1630.
8. Ricciardolo FL. Multiple roles of nitric oxide in the airways. Thorax. 2003;58(2):175-182.
9. Dweik RA, Boggs PB, Erzurum SC, et al; American Thoracic Society Committee on Interpretation of Exhaled Nitric Oxide Levels (FENO) for Clinical Applications. An official ATS clinical practice guideline: interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am J Respir Crit Care Med. 2011;184(5):602-615.
10. Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med. 1984;311(7):421-425.
11. Gelb AF, Nadel JA. Affirmation of the adoration of the vagi and role of tiotropium in asthmatic patients. J Allergy Clin Immunol. 2016;138(4):1011-1013.
12. Chin SJ, Durmowicz AG, Chowdhury BA. Tiotropium respimat is effective for the treatment of asthma at a dose lower than that for chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13(2):173-179.
13. Hamelmann E, Bateman ED, Vogelberg C, et al. Tiotropium add-on therapy in adolescents with moderate asthma: a 1-year randomized controlled trial. J Allergy Clin Immunol. 2016;138(2):441-450.e8.
14. Kerstjens HA, Casale TB, Bleeker ER, et al. Tiotropium or salmeterol as add-on therapy to inhaled corticosteroids for patients with moderate symptomatic asthma: two replicate, double-blind, placebo-controlled, parallel-group, active-comparator, randomised trials. Lancet Respir Med. 2015;3(5):367-376.
15. Peters SP, Kunselman SJ, Icitovic N, et al; National Heart, Lung, and Blood Institute Asthma Clinical Research Network. Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med. 2010;363(18):1715-1726.
16. O’Donnell DE, Flüge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnea, and exercise tolerance in COPD. Eur Respir J. 2004;23(6):832-840.
17. Lötvall J, Akdis CA, Bacharier LB, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355-360.
18. Wenzel SE. Phenotypes in asthma: useful guides for therapy, distinct biological processes, or both? Am J Respir Crit Care Med. 2004;170(6):579-580.
19. Wang E, Hoyte FC. Traditional therapies for severe asthma. Immunol Allergy Clin North Am. 2016;36(3):581-608.
20. Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med. 2006;354(25):2689-2695.
21. Bousquet J, Wenzel S, Holgate S, Lumry W, Freeman P, Fox H. Predicting response to omalizumab, an anti-IgE antibody, in patients with allergic asthma. Chest. 2004;125(4):1378-1386.
22. Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. 2005;60(3):309-316.
23. Hanania NA, Alpan O, Hamilos DL, et al. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011;154(9):573-582.
24. Holgate ST, Djukanovic´ R, Casale T, Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: an update on anti-inflammatory activity and clinical efficacy. Clin Exp Allergy. 2005;35(4):408-416.
25. Finn A, Gross G, van Bavel J, et al. Omalizumab improves asthma-related quality of life in patients with severe allergic asthma. J Allergy Clin Immunol. 2003;111(2):278-284.
26. Busse W, Corren J, Lanier BQ, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108(2):184-190.
27. Long A, Rahmaoui A, Rothman KJ, et al. Incidence of malignancy in patients with moderate-to-severe asthma treated with or without omalizumab. J Allergy Clin Immunol. 2014;134(3):560-567.e4.
28. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6.
29. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA approves label changes for asthma drug Xolair (omalizumab), including describing slightly higher risk of heart and brain adverse events. http://www.fda.gov/Drugs /DrugSafety/ucm414911.htm. Updated February 10, 2016. Accessed November 9, 2017.
30. Tan HT, Sugita K, Akdis CA. Novel biologicals for the treatment of allergic diseases and asthma. Curr Allergy Asthma Rep. 2016;16(10):70.
31. Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360(10):973-984.
32. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198-1207.
33. Magnan A, Bourdin A, Prazma CM, et al. Treatment response with mepolizumab in severe eosinophilic asthma patients with previous omalizumab treatment. Allergy. 2016;71(9):1335-1344.
34. Castro M, Mathur S, Hargreave F, et al; Res-5-0010 Study Group. Resilizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184(10):1125-1132.
35. Cinqair [package insert]. Frazier, PA: Teva Respiratory; 2016.
36. Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455-2466.
37. Chung KF. Dupilumab: a potential new treatment for severe asthma. Lancet. 2016;388(10039):3-4.
38. Gonem S, Berair R, Singapuri A, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2016;4(9):699-707.
39. Erpenbeck VJ, Popov TA, Miller D, et al. The oral CRTh2 antagonist QAWO39 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther. 2016;39:54-63.
40. Tan LD, Bratt JM, Godor D, Louie S, Kenyon NJ. Benralizumab: a unique IL-5 inhibitor for severe asthma. J Asthma Allergy. 2016;9:71-81.
41. Abramson MJ, Puy RM, Weiner JM. Injection allergen immunotherapy for asthma. Cochrane Database Syst Rev. 2010;(8):CD001186.
42. Kim SW, Kim JH, Park CK, et al. Effect of roflumilast on airway remodeling in a murine model of chronic asthma. Clin Exp Allergy. 2016;46(5):754-763.
43. Bardin P, Kanniess F, Gauvreau G, Bredenbröker D, Rabe KF. Roflumilast for asthma: efficacy findings in mechanism of action studies. Pulm Pharmacol Ther. 2015;(suppl 35):S4-S10.
44. Virchow JC Jr, Prasse A, Naya I, Summerton L, Harris A. Zafirlukast improves asthma control in patients receiving high-dose inhaled corticosteroids. Am J Resp Crit Care Med. 2000;162(2, pt 1):558-585.
45. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
46. Dahlén SE, Malmström K, Nizankowska E, et al. Improvement of aspirin intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(1):9-14.
47. Price DB, Swern A, Tozzi CA, Philip G, Polos P. Effect of montelukast on lung function in asthma patients with allergic rhinitis: analysis from the COMPACT trial. Allergy. 2006; 61(6):737-742.
48. Robinson DS, Campbell D, Barnes PJ. Addition of leukotriene antagonists to therapy in chronic persistent asthma: a randomised double-blind placebo-controlled trial. Lancet. 2001;357(9273):2007-2011.
49. Chauhan BF, Ducharme FM. Addition to inhaled corticosteroids of long-acting beta 2 agonists versus anti-leukotrienes for chronic asthma. Cochrane Database Syst Rev. 2014;(1):CD003137.
50. Chauhan BF, Ducharme FM. Anti-leukotriene agents compared to inhaled corticosteroids in the management of recurrent and/or chronic asthma in adults and children. Cochrane Database Syst Rev. 2012;(5):CD002314.
51. Philip G, Nayak AS, Berger WE, et al. The effect of montelukast on rhinitis symptoms in patients with asthma and seasonal allergic rhinitis. Curr Med Res Opin. 2004;20(10):1549-1558.
52. Wilson AM, Dempsey OJ, Sims EJ, Lipworth BJ. A comparison of topical budesonide and oral montelukast in seasonal allergic rhinitis and asthma. Clin Exp Allergy. 2001;31(4):616-624.
53. Dahlén B, Nizankowska E, Szczeklik A, et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am J Respir Crit Care Med. 1998;157(4, pt 1):1187-1194.
54. Israel E, Cohn J, Dubé L, Drazen JM. Effect of treatment with zileuton, a 5-lipoxygenase inhibitor, in patients with asthma. A randomized controlled trial. Zilueton Clinical Trial Group. JAMA. 1996;275(12):931-936.
55. Nelson H, Kemp J, Berger W, et al. Efficacy of zileuton controlled-release tablets administered twice daily in the treatment of moderate persistent asthma: a 3-month randomized controlled study. Ann Allergy Asthma Immunol. 2007;99(2):178-184.
56. Laxmanan B, Egressy K, Murgu SD, White SR, Hogarth DK. Advances in bronchial thermoplasty. Chest. 2016;150(3):694-704.
57. Cox G, Thomson NC, Rubin AS, et al; AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med. 2007;356(13):1327-1337.
58. Thomson NC, Rubin AS, Niven RM, et al; AIR Trial Study Group. Long-term (5 year) safety of bronchial thermoplasty: Asthma Intervention Research (AIR) trial. BMC Pulm Med. 2011:11:8.
59. Pavord, ID, Cox G, Thomson NC, et al; RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med. 2007;176(12):1185-1191.
60. Castro M, Rubin AS, Laviolette M, et al; AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med. 2010;181(2):116-124.
61. Wechsler ME, Laviolette M, Rubin AS, et al; Asthma Intervention Research 2 Trial Study Group. Bronchial thermoplasty: long-term safety and effectiveness in patients with severe persistent asthma. J Allergy Clin Immunol. 2013;132(6):1295-1302.
62. Peters SP, Bleecker ER, Canonica GW, et al. Serious asthma events with budesonide plus formoterol vs budesonide alone. N Engl J Med. 2016;375(9):850-860.
63. Stempel DA, Raphiou IH, Kral KM, et al; AUSTRI Investigators. Serious asthma events with fluticasone plus salmeterol versus fluticasone alone. N Engl J Med. 2016;374(19):1822-1830.
64. Kew KM, Dias S, Cates CJ. Long-acting inhaled therapy (beta-agonists, anticholinergics and steroids) for COPD: a network meta-analysis. Cochrane Database Syst Rev. 2014;(3):CD010844.
65. Finkas LK, Martin R. Role of small airways in asthma. Immunol Allergy Clin North Am. 2016;36(3):473-482.
2017 Update on bone health
Bone health remains one of the most important health care concerns in the United States today. In 2004, the Surgeon General released a report on bone health and osteoporosis. According to the report’s introduction:
This first-ever Surgeon General’s Report on bone health and osteoporosis illustrates the large burden that bone disease places on our Nation and its citizens. Like other chronic diseases that disproportionately affect the elderly, the prevalence of bone disease and fractures is projected to increase markedly as the population ages. If these predictions come true, bone disease and fractures will have a tremendous negative impact on the future well-being of Americans. But as this report makes clear, they need not come true: by working together we can change the picture of aging in America. Osteoporosis and fractures…no longer should be thought of as an inevitable part of growing old. By focusing on prevention and lifestyle changes, including physical activity and nutrition, as well as early diagnosis and appropriate treatment, Americans can avoid much of the damaging impact of bone disease.1
Related article:
2016 Update on bone health
Although men also experience osteoporosis as they age, in women the rapid loss of bone at menopause makes their disease burden much greater. As women’s health care providers, we stand at the front line for preventing, diagnosing, and treating osteoporosis to reduce the impact of this disease. In this Update I focus on important information that has emerged in the past year.
Read about new ACP guidelines to assess fracture risk
Guidelines for therapy: How to assess fracture risk and when to treat
American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. ACOG Practice Bulletin No. 129: Osteoporosis. Obstet Gynecol. 2012;120(3):718-734.
Qaseem A, Forciea MA, McLean RM, Denberg TD; Clinical Guidelines Committee of the American College of Physicians. Treatment of low bone density or osteoporosis to prevent fractures in men and women: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166(11):818-839.
A crucial component for good bone health maintenance and osteoporotic fracture prevention is understanding the current guidelines for therapy. The most recent practice bulletin of the American College of Obstetricians and Gynecologists (ACOG) on osteoporosis was published in 2012. ACOG states that treatment be recommended for women who have a bone mineral density (BMD) T-score of -2.5 or lower.
For women in the low bone mass category (T-score between -1 and -2.5), use of the Fracture Risk Assessment Tool (FRAX) calculator can assist in making an informed treatment decision.2 Based on the FRAX calculator, women who have a 10-year risk of major osteoporotic fracture of 20% or greater, or a risk of hip fracture of 3% or greater, are candidates for pharmacologic therapy.
Women who have experienced a low-trauma fracture (especially of the vertebra or hip) also are candidates for treatment, even in the absence of osteoporosis on a dual-energy x-ray absorptiometry (DXA) report.
Related article:
Women’s Preventive Services Initiative Guidelines provide consensus for practicing ObGyns
Updated recommendations from the ACP
The 2017 guideline published by the American College of Physicians (ACP), whose target audience is "all clinicians," recommends that, for women who have known osteoporosis, clinicians offer pharmacologic treatment with alendronate, risedronate, zoledronic acid, or denosumab to reduce the risk for hip and vertebral fractures.
In addition, the ACP recommends that clinicians make the decision whether or not to treat osteopenic women 65 years of age or older who are at a high risk for fracture based on a discussion of patient preferences, fracture risk profile, and benefits, harms, and costs of medications. This may seem somewhat contradictory to ACOG's guidance vis-a-vis women younger than 65 years of age.
The ACP further states that given the limited evidence supporting the benefit of treatment, the balance of benefits and harms in treating osteopenic women is most favorable when the risk for fracture is high. Women younger than 65 years with osteopenia and women older than 65 years with mild osteopenia (T-score between -1.0 and -1.5) will benefit less than women who are 65 years of age or older with severe osteopenia (T-score <-2.0).
Risk factors and risk assessment tools
Clinicians can use their own judgment based on risk factors for fracture (lower body weight, smoking, weight loss, family history of fractures, decreased physical activity, alcohol or caffeine use, low calcium and vitamin D intake, corticosteroid use), or they can use a risk assessment tool. Several risk assessment tools, such as the FRAX calculator mentioned earlier, are available to predict fracture risk among untreated people with low bone density. Although the FRAX calculator is widely used, there is no evidence from randomized controlled trials demonstrating a benefit of fracture reduction when FRAX scores are used in treatment decision making.
Duration of therapy. The ACP recommends that clinicians treat osteoporotic women with pharmacologic therapy for 5 years. Bone density monitoring is not recommended during the 5-year treatment period for osteoporosis in women; current evidence does not show any benefit for bone density monitoring during treatment.
Moderate-quality evidence demonstrated that women treated with antiresorptive therapies (including bisphosphonates, raloxifene, and teriparatide) benefited from reduced fractures, even if no increase in BMD occurred or if BMD decreased.
As before, all women with osteoporosis or a previous low-trauma fracture should be treated. Use of the FRAX calculator should involve clinician judgment, and other risk factors should be taken into account. For most women, treatment should be continued for 5 years. There is no benefit in continued bone mass assessment (DXA testing) while a patient is on pharmacologic therapy.
Read about fracture risk after stopping HT
Another WHI update: No increase in fractures after stopping HT
Watts NB, Cauley JA, Jackson RD, et al; Women's Health Initiative Investigators. No increase in fractures after stopping hormone therapy: results from the Women's Health Initiative. J Clin Endocrinol Metab. 2017;102(1):302-308.
The analysis and reanalysis of the Women's Health Initiative (WHI) trial data seems never-ending, yet the article by Watts and colleagues is important. Although the WHI hormone therapy (HT) trials showed that treatment protects against hip and total fractures, a later observational report suggested loss of benefit and rebound increased risk after HT was discontinued.3 The purpose of the Watts' study was to examine fractures after stopping HT.
Related article:
Did long-term follow-up of WHI participants reveal any mortality increase among women who received HT?
Details of the study
Two placebo-controlled randomized trials served as the study setting. The study included WHI participants (n = 15,187) who continued to take active HT or placebo through the intervention period and who did not take HT in the postintervention period. The trial interventions included conjugated equine estrogen (CEE) plus medroxyprogesterone acetate (MPA) for women with natural menopause and CEE alone for women with prior hysterectomy. The investigators recorded total fractures and hip fractures through 5 years after HT discontinuation.
Findings on fractures. Hip fractures occurred infrequently, with approximately 2.5 per 1,000 person-years. This finding was similar between trials and in former HT users and placebo groups.
No difference was found in total fractures in the CEE plus MPA trial for former HT users compared with former placebo users (28.9 per 1,000 person-years and 29.9 per 1,000 person-years, respectively; hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.87-1.09; P = .63). In the CEE-alone trial, however, total fractures were higher in former placebo users (36.9 per 1,000 person-years) compared with the former active-treatment group (31.1 per 1,000 person-years). This finding suggests a residual benefit of CEE in reducing total fractures (HR, 0.85; 95% CI, 0.73-0.98; P = .03).
Investigators' takeaway. The authors concluded that, after discontinuing HT, there was no evidence of increased fracture risk (sustained or transient) in former HT users compared with former placebo users. In the CEE-alone trial, there was a residual benefit for total fracture reduction in former HT users compared with placebo users.
Gynecologists have long believed that on stopping HT, the loss of bone mass will follow at the same rate as it would at natural menopause. These WHI trials demonstrate, however, that through 5 years, women who stopped HT had no increase in hip or total fractures, and hysterectomized women who stopped estrogen therapy actually had fewer fractures than the placebo group. Keep in mind that this large cohort was not chosen based on risk of osteoporotic fractures. In fact, baseline bone mass was not even measured in these women, making the results even more "real world."
Read about reassessing FRAX scores
A new look at fracture risk assessment scores
Gourlay ML, Overman RA, Fine JP, et al; Women's Health Initiative Investigators. Time to clinically relevant fracture risk scores in postmenopausal women. Am J Med. 2017;130:862.e15-e23.
Jiang X, Gruner M, Trémollieres F, et al. Diagnostic accuracy of FRAX in predicting the 10-year risk of osteoporotic fractures using the USA treatment thresholds: a systematic review and meta-analysis. Bone. 2017;99:20-25.
The FRAX score has become a popular form of triage for women who do not yet meet the bone mass criteria of osteoporosis. Current practice guidelines recommend use of fracture risk scores for screening and pharmacologic therapeutic decision making. Some newer data, however, may give rise to questions about its utility, especially in younger women.
Fracture risk analysis in a large postmenopausal population
Gourlay and colleagues conducted a retrospective competing risk analysis of new occurrence of treatment-level and screening-level fracture risk scores. Study participants were postmenopausal women aged 50 years and older who had not previously received pharmacologic treatment and had not had a first hip or clinical vertebral facture.
Details of the study
In 54,280 postmenopausal women aged 50 to 64 years who did not have a bone mineral density test, the time for 10% to develop a treatment-level FRAX score could not be estimated accurately because the incidence of treatment-level scores was rare.
A total of 6,096 women had FRAX scores calculated with bone mineral density testing. In this group, the estimated unadjusted time to treatment-level FRAX scores was 7.6 years (95% CI, 6.6-8.7) for those aged 65 to 69, and 5.1 years (95% CI, 3.5-7.5) for women aged 75 to 79 at baseline.
Of 17,967 women aged 50 to 64 who had a screening-level FRAX at baseline, 100 (0.6%) experienced a hip or clinical vertebral fracture by age 65 years.
Age is key factor. Gourlay and colleagues concluded that postmenopausal women who had subthreshold fracture risk scores at baseline would be unlikely to develop a treatment-level FRAX score between ages 50 and 64. The increased incidence of treatment-level fracture risk scores, osteoporosis, and major osteoporotic fracture after age 65, however, supports more frequent consideration of FRAX assessment and bone mineral density testing.
Related article:
2015 Update on osteoporosis
Meta-analysis of FRAX tool accuracy
In another study, Jiang and colleagues conducted a systematic review and meta-analysis to determine how the FRAX score performed in predicting the 10-year risk of major osteoporotic fractures and hip fractures. The investigators used the US treatment thresholds.
Details of the study
Seven studies (n = 57,027) were analyzed to assess the diagnostic accuracy of FRAX in predicting major osteoporotic fractures; 20% was used as the 10-year fracture risk threshold for intervention. The mean sensitivity and specificity, along with their 95% CIs, were 10.25% (3.76%-25.06%) and 97.02% (91.17%-99.03%), respectively.
For hip fracture prediction, 6 studies (n = 50,944) were analyzed, and 3% was used as the 10-year fracture risk threshold. The mean sensitivity and specificity, along with their 95% CIs, were 45.70% (24.88%-68.13%) and 84.70% (76.41%-90.44%), respectively.
Predictive value of FRAX. The authors concluded that, using the 10-year intervention thresholds of 20% for major osteoporotic fracture and 3% for hip fracture, FRAX performed better in identifying individuals who will not have a major osteoporotic fracture or hip fracture within 10 years than in identifying those who will experience a fracture. A substantial number of those who developed fractures, especially major osteoporotic fracture within 10 years of follow up, were missed by the baseline FRAX assessment.
Increasing age is still arguably among the most important factors for decreasing bone health. Older women are more likely to develop treatment-level FRAX scores more quickly than younger women. In addition, the FRAX tool is better in predicting which women will not develop a fracture in the next 10 years than in predicting those who will experience a fracture.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- United States Office of the Surgeon General. Bone health and osteoporosis: a report of the Surgeon General. Rockville, Maryland: Office of the Surgeon General (US); 2004. https://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed November 6, 2017.
- Centre for Metabolic Bone Diseases, University of Sheffield, United Kingdom. FRAX Fracture Risk Assessment Tool website. www.sheffield.ac.uk/FRAX. Accessed November 6, 2017.
- Yates J, Barrett-Connor E, Barlas S, Chen YT, Miller PD, Siris ES. Rapid loss of hip fracture protection after estrogen cessation: evidence from the National Osteoporosis Risk Assessment. Obstet Gynecol. 2004;103(3):440–446.

Dr. Goldstein is Professor in the Department of Obstetrics and Gynecology, New York
University School of Medicine, and Director of Gynecologic Ultrasound and Co-
Director of Bone Densitometry and Body Composition at New York University Medical
Center in New York, New York. He serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article
Dr. Goldstein is Professor in the Department of Obstetrics and Gynecology, New York
University School of Medicine, and Director of Gynecologic Ultrasound and Co-
Director of Bone Densitometry and Body Composition at New York University Medical
Center in New York, New York. He serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article
Dr. Goldstein is Professor in the Department of Obstetrics and Gynecology, New York
University School of Medicine, and Director of Gynecologic Ultrasound and Co-
Director of Bone Densitometry and Body Composition at New York University Medical
Center in New York, New York. He serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article
Bone health remains one of the most important health care concerns in the United States today. In 2004, the Surgeon General released a report on bone health and osteoporosis. According to the report’s introduction:
This first-ever Surgeon General’s Report on bone health and osteoporosis illustrates the large burden that bone disease places on our Nation and its citizens. Like other chronic diseases that disproportionately affect the elderly, the prevalence of bone disease and fractures is projected to increase markedly as the population ages. If these predictions come true, bone disease and fractures will have a tremendous negative impact on the future well-being of Americans. But as this report makes clear, they need not come true: by working together we can change the picture of aging in America. Osteoporosis and fractures…no longer should be thought of as an inevitable part of growing old. By focusing on prevention and lifestyle changes, including physical activity and nutrition, as well as early diagnosis and appropriate treatment, Americans can avoid much of the damaging impact of bone disease.1
Related article:
2016 Update on bone health
Although men also experience osteoporosis as they age, in women the rapid loss of bone at menopause makes their disease burden much greater. As women’s health care providers, we stand at the front line for preventing, diagnosing, and treating osteoporosis to reduce the impact of this disease. In this Update I focus on important information that has emerged in the past year.
Read about new ACP guidelines to assess fracture risk
Guidelines for therapy: How to assess fracture risk and when to treat
American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. ACOG Practice Bulletin No. 129: Osteoporosis. Obstet Gynecol. 2012;120(3):718-734.
Qaseem A, Forciea MA, McLean RM, Denberg TD; Clinical Guidelines Committee of the American College of Physicians. Treatment of low bone density or osteoporosis to prevent fractures in men and women: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166(11):818-839.
A crucial component for good bone health maintenance and osteoporotic fracture prevention is understanding the current guidelines for therapy. The most recent practice bulletin of the American College of Obstetricians and Gynecologists (ACOG) on osteoporosis was published in 2012. ACOG states that treatment be recommended for women who have a bone mineral density (BMD) T-score of -2.5 or lower.
For women in the low bone mass category (T-score between -1 and -2.5), use of the Fracture Risk Assessment Tool (FRAX) calculator can assist in making an informed treatment decision.2 Based on the FRAX calculator, women who have a 10-year risk of major osteoporotic fracture of 20% or greater, or a risk of hip fracture of 3% or greater, are candidates for pharmacologic therapy.
Women who have experienced a low-trauma fracture (especially of the vertebra or hip) also are candidates for treatment, even in the absence of osteoporosis on a dual-energy x-ray absorptiometry (DXA) report.
Related article:
Women’s Preventive Services Initiative Guidelines provide consensus for practicing ObGyns
Updated recommendations from the ACP
The 2017 guideline published by the American College of Physicians (ACP), whose target audience is "all clinicians," recommends that, for women who have known osteoporosis, clinicians offer pharmacologic treatment with alendronate, risedronate, zoledronic acid, or denosumab to reduce the risk for hip and vertebral fractures.
In addition, the ACP recommends that clinicians make the decision whether or not to treat osteopenic women 65 years of age or older who are at a high risk for fracture based on a discussion of patient preferences, fracture risk profile, and benefits, harms, and costs of medications. This may seem somewhat contradictory to ACOG's guidance vis-a-vis women younger than 65 years of age.
The ACP further states that given the limited evidence supporting the benefit of treatment, the balance of benefits and harms in treating osteopenic women is most favorable when the risk for fracture is high. Women younger than 65 years with osteopenia and women older than 65 years with mild osteopenia (T-score between -1.0 and -1.5) will benefit less than women who are 65 years of age or older with severe osteopenia (T-score <-2.0).
Risk factors and risk assessment tools
Clinicians can use their own judgment based on risk factors for fracture (lower body weight, smoking, weight loss, family history of fractures, decreased physical activity, alcohol or caffeine use, low calcium and vitamin D intake, corticosteroid use), or they can use a risk assessment tool. Several risk assessment tools, such as the FRAX calculator mentioned earlier, are available to predict fracture risk among untreated people with low bone density. Although the FRAX calculator is widely used, there is no evidence from randomized controlled trials demonstrating a benefit of fracture reduction when FRAX scores are used in treatment decision making.
Duration of therapy. The ACP recommends that clinicians treat osteoporotic women with pharmacologic therapy for 5 years. Bone density monitoring is not recommended during the 5-year treatment period for osteoporosis in women; current evidence does not show any benefit for bone density monitoring during treatment.
Moderate-quality evidence demonstrated that women treated with antiresorptive therapies (including bisphosphonates, raloxifene, and teriparatide) benefited from reduced fractures, even if no increase in BMD occurred or if BMD decreased.
As before, all women with osteoporosis or a previous low-trauma fracture should be treated. Use of the FRAX calculator should involve clinician judgment, and other risk factors should be taken into account. For most women, treatment should be continued for 5 years. There is no benefit in continued bone mass assessment (DXA testing) while a patient is on pharmacologic therapy.
Read about fracture risk after stopping HT
Another WHI update: No increase in fractures after stopping HT
Watts NB, Cauley JA, Jackson RD, et al; Women's Health Initiative Investigators. No increase in fractures after stopping hormone therapy: results from the Women's Health Initiative. J Clin Endocrinol Metab. 2017;102(1):302-308.
The analysis and reanalysis of the Women's Health Initiative (WHI) trial data seems never-ending, yet the article by Watts and colleagues is important. Although the WHI hormone therapy (HT) trials showed that treatment protects against hip and total fractures, a later observational report suggested loss of benefit and rebound increased risk after HT was discontinued.3 The purpose of the Watts' study was to examine fractures after stopping HT.
Related article:
Did long-term follow-up of WHI participants reveal any mortality increase among women who received HT?
Details of the study
Two placebo-controlled randomized trials served as the study setting. The study included WHI participants (n = 15,187) who continued to take active HT or placebo through the intervention period and who did not take HT in the postintervention period. The trial interventions included conjugated equine estrogen (CEE) plus medroxyprogesterone acetate (MPA) for women with natural menopause and CEE alone for women with prior hysterectomy. The investigators recorded total fractures and hip fractures through 5 years after HT discontinuation.
Findings on fractures. Hip fractures occurred infrequently, with approximately 2.5 per 1,000 person-years. This finding was similar between trials and in former HT users and placebo groups.
No difference was found in total fractures in the CEE plus MPA trial for former HT users compared with former placebo users (28.9 per 1,000 person-years and 29.9 per 1,000 person-years, respectively; hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.87-1.09; P = .63). In the CEE-alone trial, however, total fractures were higher in former placebo users (36.9 per 1,000 person-years) compared with the former active-treatment group (31.1 per 1,000 person-years). This finding suggests a residual benefit of CEE in reducing total fractures (HR, 0.85; 95% CI, 0.73-0.98; P = .03).
Investigators' takeaway. The authors concluded that, after discontinuing HT, there was no evidence of increased fracture risk (sustained or transient) in former HT users compared with former placebo users. In the CEE-alone trial, there was a residual benefit for total fracture reduction in former HT users compared with placebo users.
Gynecologists have long believed that on stopping HT, the loss of bone mass will follow at the same rate as it would at natural menopause. These WHI trials demonstrate, however, that through 5 years, women who stopped HT had no increase in hip or total fractures, and hysterectomized women who stopped estrogen therapy actually had fewer fractures than the placebo group. Keep in mind that this large cohort was not chosen based on risk of osteoporotic fractures. In fact, baseline bone mass was not even measured in these women, making the results even more "real world."
Read about reassessing FRAX scores
A new look at fracture risk assessment scores
Gourlay ML, Overman RA, Fine JP, et al; Women's Health Initiative Investigators. Time to clinically relevant fracture risk scores in postmenopausal women. Am J Med. 2017;130:862.e15-e23.
Jiang X, Gruner M, Trémollieres F, et al. Diagnostic accuracy of FRAX in predicting the 10-year risk of osteoporotic fractures using the USA treatment thresholds: a systematic review and meta-analysis. Bone. 2017;99:20-25.
The FRAX score has become a popular form of triage for women who do not yet meet the bone mass criteria of osteoporosis. Current practice guidelines recommend use of fracture risk scores for screening and pharmacologic therapeutic decision making. Some newer data, however, may give rise to questions about its utility, especially in younger women.
Fracture risk analysis in a large postmenopausal population
Gourlay and colleagues conducted a retrospective competing risk analysis of new occurrence of treatment-level and screening-level fracture risk scores. Study participants were postmenopausal women aged 50 years and older who had not previously received pharmacologic treatment and had not had a first hip or clinical vertebral facture.
Details of the study
In 54,280 postmenopausal women aged 50 to 64 years who did not have a bone mineral density test, the time for 10% to develop a treatment-level FRAX score could not be estimated accurately because the incidence of treatment-level scores was rare.
A total of 6,096 women had FRAX scores calculated with bone mineral density testing. In this group, the estimated unadjusted time to treatment-level FRAX scores was 7.6 years (95% CI, 6.6-8.7) for those aged 65 to 69, and 5.1 years (95% CI, 3.5-7.5) for women aged 75 to 79 at baseline.
Of 17,967 women aged 50 to 64 who had a screening-level FRAX at baseline, 100 (0.6%) experienced a hip or clinical vertebral fracture by age 65 years.
Age is key factor. Gourlay and colleagues concluded that postmenopausal women who had subthreshold fracture risk scores at baseline would be unlikely to develop a treatment-level FRAX score between ages 50 and 64. The increased incidence of treatment-level fracture risk scores, osteoporosis, and major osteoporotic fracture after age 65, however, supports more frequent consideration of FRAX assessment and bone mineral density testing.
Related article:
2015 Update on osteoporosis
Meta-analysis of FRAX tool accuracy
In another study, Jiang and colleagues conducted a systematic review and meta-analysis to determine how the FRAX score performed in predicting the 10-year risk of major osteoporotic fractures and hip fractures. The investigators used the US treatment thresholds.
Details of the study
Seven studies (n = 57,027) were analyzed to assess the diagnostic accuracy of FRAX in predicting major osteoporotic fractures; 20% was used as the 10-year fracture risk threshold for intervention. The mean sensitivity and specificity, along with their 95% CIs, were 10.25% (3.76%-25.06%) and 97.02% (91.17%-99.03%), respectively.
For hip fracture prediction, 6 studies (n = 50,944) were analyzed, and 3% was used as the 10-year fracture risk threshold. The mean sensitivity and specificity, along with their 95% CIs, were 45.70% (24.88%-68.13%) and 84.70% (76.41%-90.44%), respectively.
Predictive value of FRAX. The authors concluded that, using the 10-year intervention thresholds of 20% for major osteoporotic fracture and 3% for hip fracture, FRAX performed better in identifying individuals who will not have a major osteoporotic fracture or hip fracture within 10 years than in identifying those who will experience a fracture. A substantial number of those who developed fractures, especially major osteoporotic fracture within 10 years of follow up, were missed by the baseline FRAX assessment.
Increasing age is still arguably among the most important factors for decreasing bone health. Older women are more likely to develop treatment-level FRAX scores more quickly than younger women. In addition, the FRAX tool is better in predicting which women will not develop a fracture in the next 10 years than in predicting those who will experience a fracture.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Bone health remains one of the most important health care concerns in the United States today. In 2004, the Surgeon General released a report on bone health and osteoporosis. According to the report’s introduction:
This first-ever Surgeon General’s Report on bone health and osteoporosis illustrates the large burden that bone disease places on our Nation and its citizens. Like other chronic diseases that disproportionately affect the elderly, the prevalence of bone disease and fractures is projected to increase markedly as the population ages. If these predictions come true, bone disease and fractures will have a tremendous negative impact on the future well-being of Americans. But as this report makes clear, they need not come true: by working together we can change the picture of aging in America. Osteoporosis and fractures…no longer should be thought of as an inevitable part of growing old. By focusing on prevention and lifestyle changes, including physical activity and nutrition, as well as early diagnosis and appropriate treatment, Americans can avoid much of the damaging impact of bone disease.1
Related article:
2016 Update on bone health
Although men also experience osteoporosis as they age, in women the rapid loss of bone at menopause makes their disease burden much greater. As women’s health care providers, we stand at the front line for preventing, diagnosing, and treating osteoporosis to reduce the impact of this disease. In this Update I focus on important information that has emerged in the past year.
Read about new ACP guidelines to assess fracture risk
Guidelines for therapy: How to assess fracture risk and when to treat
American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. ACOG Practice Bulletin No. 129: Osteoporosis. Obstet Gynecol. 2012;120(3):718-734.
Qaseem A, Forciea MA, McLean RM, Denberg TD; Clinical Guidelines Committee of the American College of Physicians. Treatment of low bone density or osteoporosis to prevent fractures in men and women: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166(11):818-839.
A crucial component for good bone health maintenance and osteoporotic fracture prevention is understanding the current guidelines for therapy. The most recent practice bulletin of the American College of Obstetricians and Gynecologists (ACOG) on osteoporosis was published in 2012. ACOG states that treatment be recommended for women who have a bone mineral density (BMD) T-score of -2.5 or lower.
For women in the low bone mass category (T-score between -1 and -2.5), use of the Fracture Risk Assessment Tool (FRAX) calculator can assist in making an informed treatment decision.2 Based on the FRAX calculator, women who have a 10-year risk of major osteoporotic fracture of 20% or greater, or a risk of hip fracture of 3% or greater, are candidates for pharmacologic therapy.
Women who have experienced a low-trauma fracture (especially of the vertebra or hip) also are candidates for treatment, even in the absence of osteoporosis on a dual-energy x-ray absorptiometry (DXA) report.
Related article:
Women’s Preventive Services Initiative Guidelines provide consensus for practicing ObGyns
Updated recommendations from the ACP
The 2017 guideline published by the American College of Physicians (ACP), whose target audience is "all clinicians," recommends that, for women who have known osteoporosis, clinicians offer pharmacologic treatment with alendronate, risedronate, zoledronic acid, or denosumab to reduce the risk for hip and vertebral fractures.
In addition, the ACP recommends that clinicians make the decision whether or not to treat osteopenic women 65 years of age or older who are at a high risk for fracture based on a discussion of patient preferences, fracture risk profile, and benefits, harms, and costs of medications. This may seem somewhat contradictory to ACOG's guidance vis-a-vis women younger than 65 years of age.
The ACP further states that given the limited evidence supporting the benefit of treatment, the balance of benefits and harms in treating osteopenic women is most favorable when the risk for fracture is high. Women younger than 65 years with osteopenia and women older than 65 years with mild osteopenia (T-score between -1.0 and -1.5) will benefit less than women who are 65 years of age or older with severe osteopenia (T-score <-2.0).
Risk factors and risk assessment tools
Clinicians can use their own judgment based on risk factors for fracture (lower body weight, smoking, weight loss, family history of fractures, decreased physical activity, alcohol or caffeine use, low calcium and vitamin D intake, corticosteroid use), or they can use a risk assessment tool. Several risk assessment tools, such as the FRAX calculator mentioned earlier, are available to predict fracture risk among untreated people with low bone density. Although the FRAX calculator is widely used, there is no evidence from randomized controlled trials demonstrating a benefit of fracture reduction when FRAX scores are used in treatment decision making.
Duration of therapy. The ACP recommends that clinicians treat osteoporotic women with pharmacologic therapy for 5 years. Bone density monitoring is not recommended during the 5-year treatment period for osteoporosis in women; current evidence does not show any benefit for bone density monitoring during treatment.
Moderate-quality evidence demonstrated that women treated with antiresorptive therapies (including bisphosphonates, raloxifene, and teriparatide) benefited from reduced fractures, even if no increase in BMD occurred or if BMD decreased.
As before, all women with osteoporosis or a previous low-trauma fracture should be treated. Use of the FRAX calculator should involve clinician judgment, and other risk factors should be taken into account. For most women, treatment should be continued for 5 years. There is no benefit in continued bone mass assessment (DXA testing) while a patient is on pharmacologic therapy.
Read about fracture risk after stopping HT
Another WHI update: No increase in fractures after stopping HT
Watts NB, Cauley JA, Jackson RD, et al; Women's Health Initiative Investigators. No increase in fractures after stopping hormone therapy: results from the Women's Health Initiative. J Clin Endocrinol Metab. 2017;102(1):302-308.
The analysis and reanalysis of the Women's Health Initiative (WHI) trial data seems never-ending, yet the article by Watts and colleagues is important. Although the WHI hormone therapy (HT) trials showed that treatment protects against hip and total fractures, a later observational report suggested loss of benefit and rebound increased risk after HT was discontinued.3 The purpose of the Watts' study was to examine fractures after stopping HT.
Related article:
Did long-term follow-up of WHI participants reveal any mortality increase among women who received HT?
Details of the study
Two placebo-controlled randomized trials served as the study setting. The study included WHI participants (n = 15,187) who continued to take active HT or placebo through the intervention period and who did not take HT in the postintervention period. The trial interventions included conjugated equine estrogen (CEE) plus medroxyprogesterone acetate (MPA) for women with natural menopause and CEE alone for women with prior hysterectomy. The investigators recorded total fractures and hip fractures through 5 years after HT discontinuation.
Findings on fractures. Hip fractures occurred infrequently, with approximately 2.5 per 1,000 person-years. This finding was similar between trials and in former HT users and placebo groups.
No difference was found in total fractures in the CEE plus MPA trial for former HT users compared with former placebo users (28.9 per 1,000 person-years and 29.9 per 1,000 person-years, respectively; hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.87-1.09; P = .63). In the CEE-alone trial, however, total fractures were higher in former placebo users (36.9 per 1,000 person-years) compared with the former active-treatment group (31.1 per 1,000 person-years). This finding suggests a residual benefit of CEE in reducing total fractures (HR, 0.85; 95% CI, 0.73-0.98; P = .03).
Investigators' takeaway. The authors concluded that, after discontinuing HT, there was no evidence of increased fracture risk (sustained or transient) in former HT users compared with former placebo users. In the CEE-alone trial, there was a residual benefit for total fracture reduction in former HT users compared with placebo users.
Gynecologists have long believed that on stopping HT, the loss of bone mass will follow at the same rate as it would at natural menopause. These WHI trials demonstrate, however, that through 5 years, women who stopped HT had no increase in hip or total fractures, and hysterectomized women who stopped estrogen therapy actually had fewer fractures than the placebo group. Keep in mind that this large cohort was not chosen based on risk of osteoporotic fractures. In fact, baseline bone mass was not even measured in these women, making the results even more "real world."
Read about reassessing FRAX scores
A new look at fracture risk assessment scores
Gourlay ML, Overman RA, Fine JP, et al; Women's Health Initiative Investigators. Time to clinically relevant fracture risk scores in postmenopausal women. Am J Med. 2017;130:862.e15-e23.
Jiang X, Gruner M, Trémollieres F, et al. Diagnostic accuracy of FRAX in predicting the 10-year risk of osteoporotic fractures using the USA treatment thresholds: a systematic review and meta-analysis. Bone. 2017;99:20-25.
The FRAX score has become a popular form of triage for women who do not yet meet the bone mass criteria of osteoporosis. Current practice guidelines recommend use of fracture risk scores for screening and pharmacologic therapeutic decision making. Some newer data, however, may give rise to questions about its utility, especially in younger women.
Fracture risk analysis in a large postmenopausal population
Gourlay and colleagues conducted a retrospective competing risk analysis of new occurrence of treatment-level and screening-level fracture risk scores. Study participants were postmenopausal women aged 50 years and older who had not previously received pharmacologic treatment and had not had a first hip or clinical vertebral facture.
Details of the study
In 54,280 postmenopausal women aged 50 to 64 years who did not have a bone mineral density test, the time for 10% to develop a treatment-level FRAX score could not be estimated accurately because the incidence of treatment-level scores was rare.
A total of 6,096 women had FRAX scores calculated with bone mineral density testing. In this group, the estimated unadjusted time to treatment-level FRAX scores was 7.6 years (95% CI, 6.6-8.7) for those aged 65 to 69, and 5.1 years (95% CI, 3.5-7.5) for women aged 75 to 79 at baseline.
Of 17,967 women aged 50 to 64 who had a screening-level FRAX at baseline, 100 (0.6%) experienced a hip or clinical vertebral fracture by age 65 years.
Age is key factor. Gourlay and colleagues concluded that postmenopausal women who had subthreshold fracture risk scores at baseline would be unlikely to develop a treatment-level FRAX score between ages 50 and 64. The increased incidence of treatment-level fracture risk scores, osteoporosis, and major osteoporotic fracture after age 65, however, supports more frequent consideration of FRAX assessment and bone mineral density testing.
Related article:
2015 Update on osteoporosis
Meta-analysis of FRAX tool accuracy
In another study, Jiang and colleagues conducted a systematic review and meta-analysis to determine how the FRAX score performed in predicting the 10-year risk of major osteoporotic fractures and hip fractures. The investigators used the US treatment thresholds.
Details of the study
Seven studies (n = 57,027) were analyzed to assess the diagnostic accuracy of FRAX in predicting major osteoporotic fractures; 20% was used as the 10-year fracture risk threshold for intervention. The mean sensitivity and specificity, along with their 95% CIs, were 10.25% (3.76%-25.06%) and 97.02% (91.17%-99.03%), respectively.
For hip fracture prediction, 6 studies (n = 50,944) were analyzed, and 3% was used as the 10-year fracture risk threshold. The mean sensitivity and specificity, along with their 95% CIs, were 45.70% (24.88%-68.13%) and 84.70% (76.41%-90.44%), respectively.
Predictive value of FRAX. The authors concluded that, using the 10-year intervention thresholds of 20% for major osteoporotic fracture and 3% for hip fracture, FRAX performed better in identifying individuals who will not have a major osteoporotic fracture or hip fracture within 10 years than in identifying those who will experience a fracture. A substantial number of those who developed fractures, especially major osteoporotic fracture within 10 years of follow up, were missed by the baseline FRAX assessment.
Increasing age is still arguably among the most important factors for decreasing bone health. Older women are more likely to develop treatment-level FRAX scores more quickly than younger women. In addition, the FRAX tool is better in predicting which women will not develop a fracture in the next 10 years than in predicting those who will experience a fracture.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- United States Office of the Surgeon General. Bone health and osteoporosis: a report of the Surgeon General. Rockville, Maryland: Office of the Surgeon General (US); 2004. https://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed November 6, 2017.
- Centre for Metabolic Bone Diseases, University of Sheffield, United Kingdom. FRAX Fracture Risk Assessment Tool website. www.sheffield.ac.uk/FRAX. Accessed November 6, 2017.
- Yates J, Barrett-Connor E, Barlas S, Chen YT, Miller PD, Siris ES. Rapid loss of hip fracture protection after estrogen cessation: evidence from the National Osteoporosis Risk Assessment. Obstet Gynecol. 2004;103(3):440–446.
- United States Office of the Surgeon General. Bone health and osteoporosis: a report of the Surgeon General. Rockville, Maryland: Office of the Surgeon General (US); 2004. https://www.ncbi.nlm.nih.gov/books/NBK45513/. Accessed November 6, 2017.
- Centre for Metabolic Bone Diseases, University of Sheffield, United Kingdom. FRAX Fracture Risk Assessment Tool website. www.sheffield.ac.uk/FRAX. Accessed November 6, 2017.
- Yates J, Barrett-Connor E, Barlas S, Chen YT, Miller PD, Siris ES. Rapid loss of hip fracture protection after estrogen cessation: evidence from the National Osteoporosis Risk Assessment. Obstet Gynecol. 2004;103(3):440–446.
The importance of weight management and exercise: Practical advice for your patients
Over the past 3 decades, the prevalence of overweight and obesity has increased dramatically in the United States. A study published in 2016 showed the age-adjusted prevalence of obesity in 2013–2014 was 35% among men and 40.4% among women.1 It comes as no surprise that increased reliance on inexpensive fast foods coupled with progressively more sedentary lifestyles have been implicated as causative factors.2
With the rise in obesity also has come an attendant rise in related chronic diseases, such as type 2 diabetes mellitus and cardiovascular disease. Women who are obese are also at risk for certain women’s health conditions, such as polycystic ovary syndrome, breast cancer, and endometrial cancer.
It is clear that curbing this public health crisis will require concerted efforts from individuals, clinicians, and policy makers, as well as changes in societal norms.
Linda D. Bradley, MD: I think it is important for us not to lecture our patients. I could list all of the things that patients should or could do to prevent or even reverse disease states, in terms of eating right and exercising, but I think motivational interviewing is a more productive approach to elicit and evoke change (see “Principles and practice of motivational interviewing”). I used to preach to my patients. I would say, “You know, if you stay at this weight, you’re going to get diabetes, you’re going to increase your breast cancer risk, you’re going to have abnormal bleeding, you’re not going to be able to get pregnant,” and so on. It is easy to slip into that in the 7 minutes that you have with your patient, but to me, that is not the right way.
With motivational interviewing, our interactions with patients are shaped by:
- asking
- advising
- assisting
- arranging.
We begin by asking permission: “Do you mind if we talk about your weight?” or “Can we talk about your level of exercise?” Once the patient has granted permission, we ask open-ended questions and use reflective listening: “What I hear you saying is that you are concerned you will not be able to lose the weight,” or “It sounds like you don’t like to exercise, but you are worried about the health consequences of that.”
Utilizing motivational interviewing to help patients identify thoughts and feelings that contribute to unhealthy behaviors--and replacing those thoughts and feelings with new thought patterns that aid in behavior change--has been shown to be an effective and efficient facilitator for change. By incorporating the following principles of motivational interviewing into practice, clinicians can have an important impact on the prevention or management of serious diseases in women1:
- Express empathy and avoid arguments. "I know it has been difficult for you to take the first step to losing weight. That is something that is difficult for a lot of my patients. How can I help you take that first step?"
- Develop discrepancies to help the patient understand the difference between her behavior and her goals. "You have said that you would like to lose some weight. I think you know that exercise would help with that. Why do you think it has been hard for you to start exercising more?"
- Roll with resistance and provide personalized feedback to help the patient find ways to succeed. "What I hear you saying is your work schedule does not allow you time to work out at the gym. What about walking during lunch breaks or taking the stairs instead of the elevator--is that something you think you can commit to doing?"
- Support self-efficacy and elicit self-motivation. "What would you like to see differently about your health? What makes you think you need to change? What happens if you don't change?"
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243-246.
I find these skills useful for addressing anything from smoking to drinking to weight management to excessive shopping—any extreme behavior that is affecting a patient negatively. When a patient is not ready to talk about her clinical problems or make changes, I let her know my door is always open to her and that I have many resources available to help her when she is ready (TABLE).4 In those cases, I might say something like, “I have many patients who really don’t want to talk about this when I first ask them, but I just want you to know, Mrs. Jones, that I want you to succeed and I want you to be healthy. We have a team approach to taking care of all of you, and when you are ready, we are here to help.”

Related article:
2017 Update on fertility: Effects of obesity on reproduction
It is important to provide practical advice to patients—including how much to exercise, the importance of keeping a food journal, and determining a goal for slow, safe weight loss—and provide resources as necessary (such as for Weight Watchers, nutrition, and dieticians). Each day we have more than 30 opportunities to select foods to eat, drink, or purchase. Have a plan and advise your patients do the same. Recommend patients cook their own meals. Suggest weight loss apps. Counsel them to celebrate successes, find a buddy (for social support), practice positive self-talk (positive language), and plan for challenges (travel, parties, working late) and setbacks, which do not need to become a fall. Find an activity or exercise that the patient enjoys and tell them to seek professional help if needed.
Read about how to educate your patients on wellness.
Dr. Bradley: About 86% of the health care dollars spent in the United States are due to chronic diseases, and chronic diseases are the leading cause of death and disability in the country.5 The most common chronic diseases—cardiovascular disease, hypertension, type 2 diabetes, colon cancer, depression, dementia, cognitive problems, higher rates of fractures—all have been associated, at least in part, with unhealthy food choices and lack of exercise. That applies to breast cancer, too.
The good news is, we can prevent and even reverse disease. As Hippocrates said, let food be thy medicine and medicine be thy food. We have all seen success stories where consistent exercise and dietary changes definitely change the paradigm for what the disease state represents. A multiplicity of factors affect poor health—noncompliance, obesity, smoking—but when we begin to make consistent, healthy changes with diet and exercise, this creates a sort of domino effect.
In the book Us! Our Life. Our Health. Our Legacy,1 co-authored by Dr. Bradley and her colleague, Margaret L. McKenzie, MD, the authors highlight the 10 healthiest behaviors to bring about youthfulness and robust health:
- Walk at least 30-45 minutes per day most days of the week.
- Engage in resistance training 2-3 days per week.
- Eat a primarily plant-based diet made up of a variety of whole foods.
- Do not smoke.
- Maintain a waist line that measures less than half your height.
- Drink alcohol only in moderation.
- Get 7-8 hours of sleep most nights.
- Forgive.
- Have gratitude.
- Believe in something greater than yourself.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
Dr. Bradley: I think we need to get to the root cause of these clinical problems and provide the resources and support that patients need to reverse or even prevent these diseases. Clinicians need to become more aware—be an example and a role model. Our patients are watching us as much as we are watching them. Together, we can form good partnerships in order to promote better health.
Dr. Bradley: I think when you are about to be a change agent for your body and become what I call the best version of yourself, you can have these great ideas, but you need to turn those ideas into actions and make them consistent. And we know that is difficult to do, so I do try to have patients write down specific goals, their plan for achieving them, and list the reasons why it is important for them to reach their goals. That gives them something tangible to look at when the going gets tough. It is also important to work into the contract ways to reward positive behaviors when goals are met, and to plan for challenges and setbacks and how to get back on track.
I also encourage patients to document their progress and learn how to make quick adjustments when necessary to get back on track. Another important element involves setting milestones—by what date are you going to reach this goal? Like any other contract, I have my patients date and sign their wellness contracts. I also encourage them to visualize what their new self is going to look like, how they will feel when they reach their goal, what they will wear, and what activities they will engage in.
Related article:
Obesity medicine: How to incorporate it into your practice
Dr. Bradley: I do, but the amount of nutrition education that most of us get in medical school is minimal to nonexistent and not practical. As physicians, we know that food is health, exercise is fitness, and that our patients need both of them. We also know that we did not get this information in school and that our education was more about treating disease than preventing disease. Many of us were not trained in robot surgery either, because it did not exist. So what did we do? We took classes, attended lectures, read books, and learned. We can do the same with wellness. There are many courses around the country. We have to begin to relearn and reteach ourselves about health, nutrition, and exercise and then pass that information on to our patients—be a resource and a guide. We should be able to write a prescription for health as quickly as we can write a prescription for insulin or a statin.
I also bring up portion distortion with my patients. The National Institutes of Health has resources on their website (https://www.nhlbi.nih.gov/health/educational /wecan/eat-right/portion-distortion.html) that include great visuals that show portion sizes 20 years ago and what they are now. For instance, 20 years ago a bagel was 3 inches and 140 calories; today’s bagel is 6 inches and 350 calories (plus whatever toppings are added). I tell that to my patients and then explain how much more exercise is needed to burn off just that 1 bagel.
Related article:
How to help your patients control gestational weight gain
Dr. Bradley: They may not know that term directly, but I think people understand that you have the potential to pass on poor lifestyle and/or health issues related to how things are when you are in utero and later in life. It gets back to letting people know to be healthy in pregnancy and even pre-pregnancy, and that includes one’s emotional state, physical state, and spiritual state. We are what we are in our mother’s womb. Getting the best start in life starts with a healthy mom, healthy dad, and a healthy environment.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA.2016;315(21):2284Arial–2291.
- Sturm R, An R. Obesity and economic environments. CA Cancer J Clin. 2014;64(5):337Arial–350.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243–246.
- Centers for Disease Control and Prevention. Chronic disease overview. https://www.cdc.gov/chronicdisease/overview/index.htm. Updated June 28, 2017. Accessed November 3, 2017.

Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Over the past 3 decades, the prevalence of overweight and obesity has increased dramatically in the United States. A study published in 2016 showed the age-adjusted prevalence of obesity in 2013–2014 was 35% among men and 40.4% among women.1 It comes as no surprise that increased reliance on inexpensive fast foods coupled with progressively more sedentary lifestyles have been implicated as causative factors.2
With the rise in obesity also has come an attendant rise in related chronic diseases, such as type 2 diabetes mellitus and cardiovascular disease. Women who are obese are also at risk for certain women’s health conditions, such as polycystic ovary syndrome, breast cancer, and endometrial cancer.
It is clear that curbing this public health crisis will require concerted efforts from individuals, clinicians, and policy makers, as well as changes in societal norms.
Linda D. Bradley, MD: I think it is important for us not to lecture our patients. I could list all of the things that patients should or could do to prevent or even reverse disease states, in terms of eating right and exercising, but I think motivational interviewing is a more productive approach to elicit and evoke change (see “Principles and practice of motivational interviewing”). I used to preach to my patients. I would say, “You know, if you stay at this weight, you’re going to get diabetes, you’re going to increase your breast cancer risk, you’re going to have abnormal bleeding, you’re not going to be able to get pregnant,” and so on. It is easy to slip into that in the 7 minutes that you have with your patient, but to me, that is not the right way.
With motivational interviewing, our interactions with patients are shaped by:
- asking
- advising
- assisting
- arranging.
We begin by asking permission: “Do you mind if we talk about your weight?” or “Can we talk about your level of exercise?” Once the patient has granted permission, we ask open-ended questions and use reflective listening: “What I hear you saying is that you are concerned you will not be able to lose the weight,” or “It sounds like you don’t like to exercise, but you are worried about the health consequences of that.”
Utilizing motivational interviewing to help patients identify thoughts and feelings that contribute to unhealthy behaviors--and replacing those thoughts and feelings with new thought patterns that aid in behavior change--has been shown to be an effective and efficient facilitator for change. By incorporating the following principles of motivational interviewing into practice, clinicians can have an important impact on the prevention or management of serious diseases in women1:
- Express empathy and avoid arguments. "I know it has been difficult for you to take the first step to losing weight. That is something that is difficult for a lot of my patients. How can I help you take that first step?"
- Develop discrepancies to help the patient understand the difference between her behavior and her goals. "You have said that you would like to lose some weight. I think you know that exercise would help with that. Why do you think it has been hard for you to start exercising more?"
- Roll with resistance and provide personalized feedback to help the patient find ways to succeed. "What I hear you saying is your work schedule does not allow you time to work out at the gym. What about walking during lunch breaks or taking the stairs instead of the elevator--is that something you think you can commit to doing?"
- Support self-efficacy and elicit self-motivation. "What would you like to see differently about your health? What makes you think you need to change? What happens if you don't change?"
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243-246.
I find these skills useful for addressing anything from smoking to drinking to weight management to excessive shopping—any extreme behavior that is affecting a patient negatively. When a patient is not ready to talk about her clinical problems or make changes, I let her know my door is always open to her and that I have many resources available to help her when she is ready (TABLE).4 In those cases, I might say something like, “I have many patients who really don’t want to talk about this when I first ask them, but I just want you to know, Mrs. Jones, that I want you to succeed and I want you to be healthy. We have a team approach to taking care of all of you, and when you are ready, we are here to help.”
Related article:
2017 Update on fertility: Effects of obesity on reproduction
It is important to provide practical advice to patients—including how much to exercise, the importance of keeping a food journal, and determining a goal for slow, safe weight loss—and provide resources as necessary (such as for Weight Watchers, nutrition, and dieticians). Each day we have more than 30 opportunities to select foods to eat, drink, or purchase. Have a plan and advise your patients do the same. Recommend patients cook their own meals. Suggest weight loss apps. Counsel them to celebrate successes, find a buddy (for social support), practice positive self-talk (positive language), and plan for challenges (travel, parties, working late) and setbacks, which do not need to become a fall. Find an activity or exercise that the patient enjoys and tell them to seek professional help if needed.
Read about how to educate your patients on wellness.
Dr. Bradley: About 86% of the health care dollars spent in the United States are due to chronic diseases, and chronic diseases are the leading cause of death and disability in the country.5 The most common chronic diseases—cardiovascular disease, hypertension, type 2 diabetes, colon cancer, depression, dementia, cognitive problems, higher rates of fractures—all have been associated, at least in part, with unhealthy food choices and lack of exercise. That applies to breast cancer, too.
The good news is, we can prevent and even reverse disease. As Hippocrates said, let food be thy medicine and medicine be thy food. We have all seen success stories where consistent exercise and dietary changes definitely change the paradigm for what the disease state represents. A multiplicity of factors affect poor health—noncompliance, obesity, smoking—but when we begin to make consistent, healthy changes with diet and exercise, this creates a sort of domino effect.
In the book Us! Our Life. Our Health. Our Legacy,1 co-authored by Dr. Bradley and her colleague, Margaret L. McKenzie, MD, the authors highlight the 10 healthiest behaviors to bring about youthfulness and robust health:
- Walk at least 30-45 minutes per day most days of the week.
- Engage in resistance training 2-3 days per week.
- Eat a primarily plant-based diet made up of a variety of whole foods.
- Do not smoke.
- Maintain a waist line that measures less than half your height.
- Drink alcohol only in moderation.
- Get 7-8 hours of sleep most nights.
- Forgive.
- Have gratitude.
- Believe in something greater than yourself.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
Dr. Bradley: I think we need to get to the root cause of these clinical problems and provide the resources and support that patients need to reverse or even prevent these diseases. Clinicians need to become more aware—be an example and a role model. Our patients are watching us as much as we are watching them. Together, we can form good partnerships in order to promote better health.
Dr. Bradley: I think when you are about to be a change agent for your body and become what I call the best version of yourself, you can have these great ideas, but you need to turn those ideas into actions and make them consistent. And we know that is difficult to do, so I do try to have patients write down specific goals, their plan for achieving them, and list the reasons why it is important for them to reach their goals. That gives them something tangible to look at when the going gets tough. It is also important to work into the contract ways to reward positive behaviors when goals are met, and to plan for challenges and setbacks and how to get back on track.
I also encourage patients to document their progress and learn how to make quick adjustments when necessary to get back on track. Another important element involves setting milestones—by what date are you going to reach this goal? Like any other contract, I have my patients date and sign their wellness contracts. I also encourage them to visualize what their new self is going to look like, how they will feel when they reach their goal, what they will wear, and what activities they will engage in.
Related article:
Obesity medicine: How to incorporate it into your practice
Dr. Bradley: I do, but the amount of nutrition education that most of us get in medical school is minimal to nonexistent and not practical. As physicians, we know that food is health, exercise is fitness, and that our patients need both of them. We also know that we did not get this information in school and that our education was more about treating disease than preventing disease. Many of us were not trained in robot surgery either, because it did not exist. So what did we do? We took classes, attended lectures, read books, and learned. We can do the same with wellness. There are many courses around the country. We have to begin to relearn and reteach ourselves about health, nutrition, and exercise and then pass that information on to our patients—be a resource and a guide. We should be able to write a prescription for health as quickly as we can write a prescription for insulin or a statin.
I also bring up portion distortion with my patients. The National Institutes of Health has resources on their website (https://www.nhlbi.nih.gov/health/educational /wecan/eat-right/portion-distortion.html) that include great visuals that show portion sizes 20 years ago and what they are now. For instance, 20 years ago a bagel was 3 inches and 140 calories; today’s bagel is 6 inches and 350 calories (plus whatever toppings are added). I tell that to my patients and then explain how much more exercise is needed to burn off just that 1 bagel.
Related article:
How to help your patients control gestational weight gain
Dr. Bradley: They may not know that term directly, but I think people understand that you have the potential to pass on poor lifestyle and/or health issues related to how things are when you are in utero and later in life. It gets back to letting people know to be healthy in pregnancy and even pre-pregnancy, and that includes one’s emotional state, physical state, and spiritual state. We are what we are in our mother’s womb. Getting the best start in life starts with a healthy mom, healthy dad, and a healthy environment.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Over the past 3 decades, the prevalence of overweight and obesity has increased dramatically in the United States. A study published in 2016 showed the age-adjusted prevalence of obesity in 2013–2014 was 35% among men and 40.4% among women.1 It comes as no surprise that increased reliance on inexpensive fast foods coupled with progressively more sedentary lifestyles have been implicated as causative factors.2
With the rise in obesity also has come an attendant rise in related chronic diseases, such as type 2 diabetes mellitus and cardiovascular disease. Women who are obese are also at risk for certain women’s health conditions, such as polycystic ovary syndrome, breast cancer, and endometrial cancer.
It is clear that curbing this public health crisis will require concerted efforts from individuals, clinicians, and policy makers, as well as changes in societal norms.
Linda D. Bradley, MD: I think it is important for us not to lecture our patients. I could list all of the things that patients should or could do to prevent or even reverse disease states, in terms of eating right and exercising, but I think motivational interviewing is a more productive approach to elicit and evoke change (see “Principles and practice of motivational interviewing”). I used to preach to my patients. I would say, “You know, if you stay at this weight, you’re going to get diabetes, you’re going to increase your breast cancer risk, you’re going to have abnormal bleeding, you’re not going to be able to get pregnant,” and so on. It is easy to slip into that in the 7 minutes that you have with your patient, but to me, that is not the right way.
With motivational interviewing, our interactions with patients are shaped by:
- asking
- advising
- assisting
- arranging.
We begin by asking permission: “Do you mind if we talk about your weight?” or “Can we talk about your level of exercise?” Once the patient has granted permission, we ask open-ended questions and use reflective listening: “What I hear you saying is that you are concerned you will not be able to lose the weight,” or “It sounds like you don’t like to exercise, but you are worried about the health consequences of that.”
Utilizing motivational interviewing to help patients identify thoughts and feelings that contribute to unhealthy behaviors--and replacing those thoughts and feelings with new thought patterns that aid in behavior change--has been shown to be an effective and efficient facilitator for change. By incorporating the following principles of motivational interviewing into practice, clinicians can have an important impact on the prevention or management of serious diseases in women1:
- Express empathy and avoid arguments. "I know it has been difficult for you to take the first step to losing weight. That is something that is difficult for a lot of my patients. How can I help you take that first step?"
- Develop discrepancies to help the patient understand the difference between her behavior and her goals. "You have said that you would like to lose some weight. I think you know that exercise would help with that. Why do you think it has been hard for you to start exercising more?"
- Roll with resistance and provide personalized feedback to help the patient find ways to succeed. "What I hear you saying is your work schedule does not allow you time to work out at the gym. What about walking during lunch breaks or taking the stairs instead of the elevator--is that something you think you can commit to doing?"
- Support self-efficacy and elicit self-motivation. "What would you like to see differently about your health? What makes you think you need to change? What happens if you don't change?"
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243-246.
I find these skills useful for addressing anything from smoking to drinking to weight management to excessive shopping—any extreme behavior that is affecting a patient negatively. When a patient is not ready to talk about her clinical problems or make changes, I let her know my door is always open to her and that I have many resources available to help her when she is ready (TABLE).4 In those cases, I might say something like, “I have many patients who really don’t want to talk about this when I first ask them, but I just want you to know, Mrs. Jones, that I want you to succeed and I want you to be healthy. We have a team approach to taking care of all of you, and when you are ready, we are here to help.”
Related article:
2017 Update on fertility: Effects of obesity on reproduction
It is important to provide practical advice to patients—including how much to exercise, the importance of keeping a food journal, and determining a goal for slow, safe weight loss—and provide resources as necessary (such as for Weight Watchers, nutrition, and dieticians). Each day we have more than 30 opportunities to select foods to eat, drink, or purchase. Have a plan and advise your patients do the same. Recommend patients cook their own meals. Suggest weight loss apps. Counsel them to celebrate successes, find a buddy (for social support), practice positive self-talk (positive language), and plan for challenges (travel, parties, working late) and setbacks, which do not need to become a fall. Find an activity or exercise that the patient enjoys and tell them to seek professional help if needed.
Read about how to educate your patients on wellness.
Dr. Bradley: About 86% of the health care dollars spent in the United States are due to chronic diseases, and chronic diseases are the leading cause of death and disability in the country.5 The most common chronic diseases—cardiovascular disease, hypertension, type 2 diabetes, colon cancer, depression, dementia, cognitive problems, higher rates of fractures—all have been associated, at least in part, with unhealthy food choices and lack of exercise. That applies to breast cancer, too.
The good news is, we can prevent and even reverse disease. As Hippocrates said, let food be thy medicine and medicine be thy food. We have all seen success stories where consistent exercise and dietary changes definitely change the paradigm for what the disease state represents. A multiplicity of factors affect poor health—noncompliance, obesity, smoking—but when we begin to make consistent, healthy changes with diet and exercise, this creates a sort of domino effect.
In the book Us! Our Life. Our Health. Our Legacy,1 co-authored by Dr. Bradley and her colleague, Margaret L. McKenzie, MD, the authors highlight the 10 healthiest behaviors to bring about youthfulness and robust health:
- Walk at least 30-45 minutes per day most days of the week.
- Engage in resistance training 2-3 days per week.
- Eat a primarily plant-based diet made up of a variety of whole foods.
- Do not smoke.
- Maintain a waist line that measures less than half your height.
- Drink alcohol only in moderation.
- Get 7-8 hours of sleep most nights.
- Forgive.
- Have gratitude.
- Believe in something greater than yourself.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
Dr. Bradley: I think we need to get to the root cause of these clinical problems and provide the resources and support that patients need to reverse or even prevent these diseases. Clinicians need to become more aware—be an example and a role model. Our patients are watching us as much as we are watching them. Together, we can form good partnerships in order to promote better health.
Dr. Bradley: I think when you are about to be a change agent for your body and become what I call the best version of yourself, you can have these great ideas, but you need to turn those ideas into actions and make them consistent. And we know that is difficult to do, so I do try to have patients write down specific goals, their plan for achieving them, and list the reasons why it is important for them to reach their goals. That gives them something tangible to look at when the going gets tough. It is also important to work into the contract ways to reward positive behaviors when goals are met, and to plan for challenges and setbacks and how to get back on track.
I also encourage patients to document their progress and learn how to make quick adjustments when necessary to get back on track. Another important element involves setting milestones—by what date are you going to reach this goal? Like any other contract, I have my patients date and sign their wellness contracts. I also encourage them to visualize what their new self is going to look like, how they will feel when they reach their goal, what they will wear, and what activities they will engage in.
Related article:
Obesity medicine: How to incorporate it into your practice
Dr. Bradley: I do, but the amount of nutrition education that most of us get in medical school is minimal to nonexistent and not practical. As physicians, we know that food is health, exercise is fitness, and that our patients need both of them. We also know that we did not get this information in school and that our education was more about treating disease than preventing disease. Many of us were not trained in robot surgery either, because it did not exist. So what did we do? We took classes, attended lectures, read books, and learned. We can do the same with wellness. There are many courses around the country. We have to begin to relearn and reteach ourselves about health, nutrition, and exercise and then pass that information on to our patients—be a resource and a guide. We should be able to write a prescription for health as quickly as we can write a prescription for insulin or a statin.
I also bring up portion distortion with my patients. The National Institutes of Health has resources on their website (https://www.nhlbi.nih.gov/health/educational /wecan/eat-right/portion-distortion.html) that include great visuals that show portion sizes 20 years ago and what they are now. For instance, 20 years ago a bagel was 3 inches and 140 calories; today’s bagel is 6 inches and 350 calories (plus whatever toppings are added). I tell that to my patients and then explain how much more exercise is needed to burn off just that 1 bagel.
Related article:
How to help your patients control gestational weight gain
Dr. Bradley: They may not know that term directly, but I think people understand that you have the potential to pass on poor lifestyle and/or health issues related to how things are when you are in utero and later in life. It gets back to letting people know to be healthy in pregnancy and even pre-pregnancy, and that includes one’s emotional state, physical state, and spiritual state. We are what we are in our mother’s womb. Getting the best start in life starts with a healthy mom, healthy dad, and a healthy environment.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA.2016;315(21):2284Arial–2291.
- Sturm R, An R. Obesity and economic environments. CA Cancer J Clin. 2014;64(5):337Arial–350.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243–246.
- Centers for Disease Control and Prevention. Chronic disease overview. https://www.cdc.gov/chronicdisease/overview/index.htm. Updated June 28, 2017. Accessed November 3, 2017.
- Flegal KM, Kruszon-Moran D, Carroll MD, et al. Trends in obesity among adults in the United States, 2005 to 2014. JAMA.2016;315(21):2284Arial–2291.
- Sturm R, An R. Obesity and economic environments. CA Cancer J Clin. 2014;64(5):337Arial–350.
- Bradley LD, McKenzie ML. Us! Our life. Our health. Our legacy. Las Vegas, Nevada: The Literary Front Publishing Co., LLC; 2016.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 423: Motivational interviewing: a tool for behavioral change. Obstet Gynecol. 2009;113(1):243–246.
- Centers for Disease Control and Prevention. Chronic disease overview. https://www.cdc.gov/chronicdisease/overview/index.htm. Updated June 28, 2017. Accessed November 3, 2017.
