User login
A Practical Guide to Understanding and Treating Patellofemoral Pain
Take-Home Points
- Anterior knee pain is common, particularly in young females.
- For most patients, activity modification and rest will control the pain; continuing to engage in painful activity only prolongs symptoms.
- In physical therapy, core stability, weight loss, and hip strengthening are essential.
- Surgery is required only in a very small subset of patients with anterior knee pain.
- Traumatic and overload- related chondral defects that have resisted a reasonable amount of conservative (nonoperative) treatment may be arthroscopically assessed and treated when documented to cause persistent pain.
Anterior knee pain is common (AKP), particularly in young females. Understanding the biomechanics of a rapidly growing young female knee, whose pelvis is relatively wider than her male counterpart, helps greatly in understanding origins of AKP.1
Compared with males of similar weight and size, females often walk and run with increased valgus at the knee and internal rotation of the hip on heel strike. The patella contacts the lateral edge of the trochlea with more focal load on the distal lateral patella for a longer time in a female than in a male of similar stature because of the increased lateral force vector. Add the rigors of athletics, excessive body weight, use of high heels, or a predisposing structural anomaly, and painful focal overload can develop—resulting in pain on stairs, inability to run, and a visit to your office. Some male patients also develop AKP, often related to patellofemoral dysplasia or activity-related overload leading to a similar pattern and need for care. Fortunately, most young patients improve when they reduce physical activity, attain stable musculoskeletal maturity, or both.
In addition to focal articular overload occurring, retinacular structures about the anterior knee can be stressed by the structural imbalance resulting from the excessive and sudden internal rotation of the hip that occurs even during normal gait and often is related to female lower extremity function. Small nerve damage in the stressed retinaculum is an important cause of peripatellar pain2 and is best identified by clinical examination. Additionally, the infrapatellar fat pad may become pinched, causing synovial inflammation.
With these patients, reassurance can go a long way, and resting, taping, bracing, and anti-inflammatory medications are helpful. Dye3 has emphasized nonoperative treatmentand allowing patients to re-establish homeostatic balance of the patellofemoral joint. Establishing normal body weight plays a key role in the process, and focusing on lower extremity core stability, starting with increased strength in the hip external rotators, is important.4 In the majority of patients, these measures are all that is needed.
Traumatic Anterior Knee Pain
Direct trauma to the anterior knee causes an entirely different sort of pain. Knee pain may be retinacular, neuronal, synovial, bony, or articular. Nothing replaces careful, detailed clinical history taking and physical examination in determining the source of this pain. Much AKP, particularly in its early stages, is very focal. A specific injection of an anesthetic into a suspected retinacular pain location may solve the diagnostic dilemma. With many patients, paying attention to the specific degree of knee flexion in which the injury occurred helps in localizing the lesion. A flexed-knee impact injury (dashboard or fall directly onto anterior knee) is a common cause of articular damage on the mid or proximal patella and distal medial femoral condyle. Identifying this cause is particularly important in worker’s compensation cases, as the pattern is diagnostic of a direct blow to the knee and may confirm the patient’s history.
Treating painful patellofemoral lesions related to direct trauma can be difficult. Once they are identified and correlated with the physical examination and magnetic resonance imaging (MRI) findings, a treatment plan can be developed.
Examination, Testing, Imaging
Knowing how AKP started is important. Asking a patient to point to the origin of pain is essential. A pain diagram (having the patient draw a picture of the pain location) is also very helpful.5 Spontaneous onset suggests an underlying structural and/or functional problem rather than a traumatic event. Examination should include palpation of all structures and the retinaculum about the knee; careful appraisal of patella tracking, location of pain, and crepitus (angle of knee flexion), and evidence of possible pain referred from the back or hip; gait analysis for functional aberrations; assessment of patellar mobility; and standard radiographs, including a perfect lateral radiograph and a knee-flexion axial radiograph of no more than 30° to 45°. Computed tomography, radionuclide scintigraphy,3 and MRI can be very useful in select patients, but such imaging generally is not necessary in the management of routine AKP. However, these studies can be extremely helpful in patients with resistant pain.
Resistant Anterior Knee Pain
When nonoperative measures (rest, bracing, taping, physical therapy, activity modification) fail to relieve pain, more aggressive treatment may be warranted. The clinician must take extra time to listen to the patient, look for the precise source of the pain, and address it directly. Treatment depends on the specific source of pain. A chronically painful retinacular lesion or neuroma usually responds to release of the painful segment. After a retinacular source of pain has been identified and temporarily eliminated with injection of a local anesthetic, the pain source can be accurately resected and the patient quickly cured. When the chronically painful locus is an injured fat pad, resection provides complete relief.
For most orthopedic surgeons, the greatest dilemma is how to address a young person’s persistent pain in the setting of minimal objective evidence. In my experience with hundreds of arthroscopies, distinct distal lateral patella articular softening is common. In some cases, the degree of articular softening can be extreme, extending toward the central ridge or even across the center of the patella and involving 40% to 50% of the patella articular surface. This spongy, soft cartilage does not resist load normally, and in many cases pain is disabling. Most important is to acknowledge the problem, as many of these patients have been living with articular lesion pain for a year or more. As quality of life can be severely diminished by chronic patellofemoral pain, it behooves us to find answers and provide appropriate treatment. Although patients with this degree of articular softening and breakdown represent a small percentage of all patients with patellofemoral pain, identifying these cases is essential.
However benign-appearing, a resistant, painful patella articular lesion can be disabling. The key to treating a young person with a patella articular lesion objectively proved with imaging or arthroscopy is to inform the patient and family of the resistant nature of some lesions. In a referral patellofemoral practice, I see many patients who are disabled and depressed about the results of articular breakdown related to focal overload. Once the problem is identified, there is hope.
Prolonged rest and activity withdrawal usually help, but in some cases pain with stairs and daily activities continues. Running is usually impossible, which can be devastating for many young people.
My approach is to exhaust the nonoperative measures, which include focusing intensely on core stability training. The physical therapist must understand the importance of this treatment component; the patient must understand the importance of strengthening the hip external rotators and the vastus medialis oblique, modifying gait, avoiding pain-inducing activities, controlling weight, using proper footwear, and being patient. Applying heavy resistance to the quadriceps during rehabilitation will likely perpetuate or exacerbate the problem. The goals are to limit loading of the articular lesion and improve lower extremity function emphasizing reduction and balanced distribution of load.
Other Causes of Anterior Knee Pain
The possibility of an unusual source of pain should always be considered. Some causes (osteochondral lesion, bipartite patella, patella baja, radiographic evidence of focal overload) are apparent only on imaging. MRI may provide evidence of hypertrophic synovium, thickened fat pad, or patellar tendonitis. The physical examination is important in determining unusual sources of pain, such as those related to trauma or retinacular neuronal injury from direct impact. Pain referred from the hip or back can also cause AKP. As kinesiophobia may also play a role, it should be considered whenever an objective cause of the pain cannot be identified.
Surgery for Anterior Knee Pain
Surgery should be considered only after prolonged rest and healing have failed to resolve the pain caused by sustained direct trauma to the anterior knee. Physical therapy typically is not useful in direct trauma. If a painful traumatic articular lesion persists, then direct treatment—removing loose articular fragments and resurfacing or unloading a damaged articular surface—may be appropriate. In most cases, 6 to 12 months should be allowed before considering surgery. Meanwhile, rest, bracing, anti-inflammatory measures, reassurance, and work modification are the cornerstones of treatment.
After all conservative measures have failed in a patient with spontaneous-onset AKP related to repetitive focal overload, and disability caused by an objectively proven articular lesion related to mechanical dysfunction or dysplasia, diagnostic arthroscopy may be appropriate. Quantitation and characterization of the lesion with images and measurements are imperative in forming an optimal surgical plan. Remember that not all problems can be cured with surgery, and there is no patellofemoral problem that cannot potentially be made worse with improper surgery.
Am J Orthop. 2017;46(2):101-103. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Sanchis-Alfonso V, Dye SF. How to deal with anterior knee pain in the active young patient. Sports Health. 2016 Dec 5. [Epub ahead of print]
2. Fulkerson JP, Tennant R, Jaivin JS, Grunnet M. Histologic evidence of retinacular nerve injury associated with patellofemoral malalignment. Clin Orthop Relat Res. 1985;(197):196-205.
3. Dye SF. The pathophysiology of patellofemoral pain: a tissue homeostasis perspective. Clin Orthop Relat Res. 2005;(436):100-110.
4. Souza RB, Powers CM. Differences in hip kinematics, muscle strength, and muscle activation between subjects with and without patellofemoral pain. J Orthop Sports Phys Ther. 2009;39(1):12-19.
5. Post WR, Fulkerson J. Knee pain diagrams: correlation with physical examination findings in patients with anterior knee pain. Arthroscopy. 1994;10(6):618-623.
Take-Home Points
- Anterior knee pain is common, particularly in young females.
- For most patients, activity modification and rest will control the pain; continuing to engage in painful activity only prolongs symptoms.
- In physical therapy, core stability, weight loss, and hip strengthening are essential.
- Surgery is required only in a very small subset of patients with anterior knee pain.
- Traumatic and overload- related chondral defects that have resisted a reasonable amount of conservative (nonoperative) treatment may be arthroscopically assessed and treated when documented to cause persistent pain.
Anterior knee pain is common (AKP), particularly in young females. Understanding the biomechanics of a rapidly growing young female knee, whose pelvis is relatively wider than her male counterpart, helps greatly in understanding origins of AKP.1
Compared with males of similar weight and size, females often walk and run with increased valgus at the knee and internal rotation of the hip on heel strike. The patella contacts the lateral edge of the trochlea with more focal load on the distal lateral patella for a longer time in a female than in a male of similar stature because of the increased lateral force vector. Add the rigors of athletics, excessive body weight, use of high heels, or a predisposing structural anomaly, and painful focal overload can develop—resulting in pain on stairs, inability to run, and a visit to your office. Some male patients also develop AKP, often related to patellofemoral dysplasia or activity-related overload leading to a similar pattern and need for care. Fortunately, most young patients improve when they reduce physical activity, attain stable musculoskeletal maturity, or both.
In addition to focal articular overload occurring, retinacular structures about the anterior knee can be stressed by the structural imbalance resulting from the excessive and sudden internal rotation of the hip that occurs even during normal gait and often is related to female lower extremity function. Small nerve damage in the stressed retinaculum is an important cause of peripatellar pain2 and is best identified by clinical examination. Additionally, the infrapatellar fat pad may become pinched, causing synovial inflammation.
With these patients, reassurance can go a long way, and resting, taping, bracing, and anti-inflammatory medications are helpful. Dye3 has emphasized nonoperative treatmentand allowing patients to re-establish homeostatic balance of the patellofemoral joint. Establishing normal body weight plays a key role in the process, and focusing on lower extremity core stability, starting with increased strength in the hip external rotators, is important.4 In the majority of patients, these measures are all that is needed.
Traumatic Anterior Knee Pain
Direct trauma to the anterior knee causes an entirely different sort of pain. Knee pain may be retinacular, neuronal, synovial, bony, or articular. Nothing replaces careful, detailed clinical history taking and physical examination in determining the source of this pain. Much AKP, particularly in its early stages, is very focal. A specific injection of an anesthetic into a suspected retinacular pain location may solve the diagnostic dilemma. With many patients, paying attention to the specific degree of knee flexion in which the injury occurred helps in localizing the lesion. A flexed-knee impact injury (dashboard or fall directly onto anterior knee) is a common cause of articular damage on the mid or proximal patella and distal medial femoral condyle. Identifying this cause is particularly important in worker’s compensation cases, as the pattern is diagnostic of a direct blow to the knee and may confirm the patient’s history.
Treating painful patellofemoral lesions related to direct trauma can be difficult. Once they are identified and correlated with the physical examination and magnetic resonance imaging (MRI) findings, a treatment plan can be developed.
Examination, Testing, Imaging
Knowing how AKP started is important. Asking a patient to point to the origin of pain is essential. A pain diagram (having the patient draw a picture of the pain location) is also very helpful.5 Spontaneous onset suggests an underlying structural and/or functional problem rather than a traumatic event. Examination should include palpation of all structures and the retinaculum about the knee; careful appraisal of patella tracking, location of pain, and crepitus (angle of knee flexion), and evidence of possible pain referred from the back or hip; gait analysis for functional aberrations; assessment of patellar mobility; and standard radiographs, including a perfect lateral radiograph and a knee-flexion axial radiograph of no more than 30° to 45°. Computed tomography, radionuclide scintigraphy,3 and MRI can be very useful in select patients, but such imaging generally is not necessary in the management of routine AKP. However, these studies can be extremely helpful in patients with resistant pain.
Resistant Anterior Knee Pain
When nonoperative measures (rest, bracing, taping, physical therapy, activity modification) fail to relieve pain, more aggressive treatment may be warranted. The clinician must take extra time to listen to the patient, look for the precise source of the pain, and address it directly. Treatment depends on the specific source of pain. A chronically painful retinacular lesion or neuroma usually responds to release of the painful segment. After a retinacular source of pain has been identified and temporarily eliminated with injection of a local anesthetic, the pain source can be accurately resected and the patient quickly cured. When the chronically painful locus is an injured fat pad, resection provides complete relief.
For most orthopedic surgeons, the greatest dilemma is how to address a young person’s persistent pain in the setting of minimal objective evidence. In my experience with hundreds of arthroscopies, distinct distal lateral patella articular softening is common. In some cases, the degree of articular softening can be extreme, extending toward the central ridge or even across the center of the patella and involving 40% to 50% of the patella articular surface. This spongy, soft cartilage does not resist load normally, and in many cases pain is disabling. Most important is to acknowledge the problem, as many of these patients have been living with articular lesion pain for a year or more. As quality of life can be severely diminished by chronic patellofemoral pain, it behooves us to find answers and provide appropriate treatment. Although patients with this degree of articular softening and breakdown represent a small percentage of all patients with patellofemoral pain, identifying these cases is essential.
However benign-appearing, a resistant, painful patella articular lesion can be disabling. The key to treating a young person with a patella articular lesion objectively proved with imaging or arthroscopy is to inform the patient and family of the resistant nature of some lesions. In a referral patellofemoral practice, I see many patients who are disabled and depressed about the results of articular breakdown related to focal overload. Once the problem is identified, there is hope.
Prolonged rest and activity withdrawal usually help, but in some cases pain with stairs and daily activities continues. Running is usually impossible, which can be devastating for many young people.
My approach is to exhaust the nonoperative measures, which include focusing intensely on core stability training. The physical therapist must understand the importance of this treatment component; the patient must understand the importance of strengthening the hip external rotators and the vastus medialis oblique, modifying gait, avoiding pain-inducing activities, controlling weight, using proper footwear, and being patient. Applying heavy resistance to the quadriceps during rehabilitation will likely perpetuate or exacerbate the problem. The goals are to limit loading of the articular lesion and improve lower extremity function emphasizing reduction and balanced distribution of load.
Other Causes of Anterior Knee Pain
The possibility of an unusual source of pain should always be considered. Some causes (osteochondral lesion, bipartite patella, patella baja, radiographic evidence of focal overload) are apparent only on imaging. MRI may provide evidence of hypertrophic synovium, thickened fat pad, or patellar tendonitis. The physical examination is important in determining unusual sources of pain, such as those related to trauma or retinacular neuronal injury from direct impact. Pain referred from the hip or back can also cause AKP. As kinesiophobia may also play a role, it should be considered whenever an objective cause of the pain cannot be identified.
Surgery for Anterior Knee Pain
Surgery should be considered only after prolonged rest and healing have failed to resolve the pain caused by sustained direct trauma to the anterior knee. Physical therapy typically is not useful in direct trauma. If a painful traumatic articular lesion persists, then direct treatment—removing loose articular fragments and resurfacing or unloading a damaged articular surface—may be appropriate. In most cases, 6 to 12 months should be allowed before considering surgery. Meanwhile, rest, bracing, anti-inflammatory measures, reassurance, and work modification are the cornerstones of treatment.
After all conservative measures have failed in a patient with spontaneous-onset AKP related to repetitive focal overload, and disability caused by an objectively proven articular lesion related to mechanical dysfunction or dysplasia, diagnostic arthroscopy may be appropriate. Quantitation and characterization of the lesion with images and measurements are imperative in forming an optimal surgical plan. Remember that not all problems can be cured with surgery, and there is no patellofemoral problem that cannot potentially be made worse with improper surgery.
Am J Orthop. 2017;46(2):101-103. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Anterior knee pain is common, particularly in young females.
- For most patients, activity modification and rest will control the pain; continuing to engage in painful activity only prolongs symptoms.
- In physical therapy, core stability, weight loss, and hip strengthening are essential.
- Surgery is required only in a very small subset of patients with anterior knee pain.
- Traumatic and overload- related chondral defects that have resisted a reasonable amount of conservative (nonoperative) treatment may be arthroscopically assessed and treated when documented to cause persistent pain.
Anterior knee pain is common (AKP), particularly in young females. Understanding the biomechanics of a rapidly growing young female knee, whose pelvis is relatively wider than her male counterpart, helps greatly in understanding origins of AKP.1
Compared with males of similar weight and size, females often walk and run with increased valgus at the knee and internal rotation of the hip on heel strike. The patella contacts the lateral edge of the trochlea with more focal load on the distal lateral patella for a longer time in a female than in a male of similar stature because of the increased lateral force vector. Add the rigors of athletics, excessive body weight, use of high heels, or a predisposing structural anomaly, and painful focal overload can develop—resulting in pain on stairs, inability to run, and a visit to your office. Some male patients also develop AKP, often related to patellofemoral dysplasia or activity-related overload leading to a similar pattern and need for care. Fortunately, most young patients improve when they reduce physical activity, attain stable musculoskeletal maturity, or both.
In addition to focal articular overload occurring, retinacular structures about the anterior knee can be stressed by the structural imbalance resulting from the excessive and sudden internal rotation of the hip that occurs even during normal gait and often is related to female lower extremity function. Small nerve damage in the stressed retinaculum is an important cause of peripatellar pain2 and is best identified by clinical examination. Additionally, the infrapatellar fat pad may become pinched, causing synovial inflammation.
With these patients, reassurance can go a long way, and resting, taping, bracing, and anti-inflammatory medications are helpful. Dye3 has emphasized nonoperative treatmentand allowing patients to re-establish homeostatic balance of the patellofemoral joint. Establishing normal body weight plays a key role in the process, and focusing on lower extremity core stability, starting with increased strength in the hip external rotators, is important.4 In the majority of patients, these measures are all that is needed.
Traumatic Anterior Knee Pain
Direct trauma to the anterior knee causes an entirely different sort of pain. Knee pain may be retinacular, neuronal, synovial, bony, or articular. Nothing replaces careful, detailed clinical history taking and physical examination in determining the source of this pain. Much AKP, particularly in its early stages, is very focal. A specific injection of an anesthetic into a suspected retinacular pain location may solve the diagnostic dilemma. With many patients, paying attention to the specific degree of knee flexion in which the injury occurred helps in localizing the lesion. A flexed-knee impact injury (dashboard or fall directly onto anterior knee) is a common cause of articular damage on the mid or proximal patella and distal medial femoral condyle. Identifying this cause is particularly important in worker’s compensation cases, as the pattern is diagnostic of a direct blow to the knee and may confirm the patient’s history.
Treating painful patellofemoral lesions related to direct trauma can be difficult. Once they are identified and correlated with the physical examination and magnetic resonance imaging (MRI) findings, a treatment plan can be developed.
Examination, Testing, Imaging
Knowing how AKP started is important. Asking a patient to point to the origin of pain is essential. A pain diagram (having the patient draw a picture of the pain location) is also very helpful.5 Spontaneous onset suggests an underlying structural and/or functional problem rather than a traumatic event. Examination should include palpation of all structures and the retinaculum about the knee; careful appraisal of patella tracking, location of pain, and crepitus (angle of knee flexion), and evidence of possible pain referred from the back or hip; gait analysis for functional aberrations; assessment of patellar mobility; and standard radiographs, including a perfect lateral radiograph and a knee-flexion axial radiograph of no more than 30° to 45°. Computed tomography, radionuclide scintigraphy,3 and MRI can be very useful in select patients, but such imaging generally is not necessary in the management of routine AKP. However, these studies can be extremely helpful in patients with resistant pain.
Resistant Anterior Knee Pain
When nonoperative measures (rest, bracing, taping, physical therapy, activity modification) fail to relieve pain, more aggressive treatment may be warranted. The clinician must take extra time to listen to the patient, look for the precise source of the pain, and address it directly. Treatment depends on the specific source of pain. A chronically painful retinacular lesion or neuroma usually responds to release of the painful segment. After a retinacular source of pain has been identified and temporarily eliminated with injection of a local anesthetic, the pain source can be accurately resected and the patient quickly cured. When the chronically painful locus is an injured fat pad, resection provides complete relief.
For most orthopedic surgeons, the greatest dilemma is how to address a young person’s persistent pain in the setting of minimal objective evidence. In my experience with hundreds of arthroscopies, distinct distal lateral patella articular softening is common. In some cases, the degree of articular softening can be extreme, extending toward the central ridge or even across the center of the patella and involving 40% to 50% of the patella articular surface. This spongy, soft cartilage does not resist load normally, and in many cases pain is disabling. Most important is to acknowledge the problem, as many of these patients have been living with articular lesion pain for a year or more. As quality of life can be severely diminished by chronic patellofemoral pain, it behooves us to find answers and provide appropriate treatment. Although patients with this degree of articular softening and breakdown represent a small percentage of all patients with patellofemoral pain, identifying these cases is essential.
However benign-appearing, a resistant, painful patella articular lesion can be disabling. The key to treating a young person with a patella articular lesion objectively proved with imaging or arthroscopy is to inform the patient and family of the resistant nature of some lesions. In a referral patellofemoral practice, I see many patients who are disabled and depressed about the results of articular breakdown related to focal overload. Once the problem is identified, there is hope.
Prolonged rest and activity withdrawal usually help, but in some cases pain with stairs and daily activities continues. Running is usually impossible, which can be devastating for many young people.
My approach is to exhaust the nonoperative measures, which include focusing intensely on core stability training. The physical therapist must understand the importance of this treatment component; the patient must understand the importance of strengthening the hip external rotators and the vastus medialis oblique, modifying gait, avoiding pain-inducing activities, controlling weight, using proper footwear, and being patient. Applying heavy resistance to the quadriceps during rehabilitation will likely perpetuate or exacerbate the problem. The goals are to limit loading of the articular lesion and improve lower extremity function emphasizing reduction and balanced distribution of load.
Other Causes of Anterior Knee Pain
The possibility of an unusual source of pain should always be considered. Some causes (osteochondral lesion, bipartite patella, patella baja, radiographic evidence of focal overload) are apparent only on imaging. MRI may provide evidence of hypertrophic synovium, thickened fat pad, or patellar tendonitis. The physical examination is important in determining unusual sources of pain, such as those related to trauma or retinacular neuronal injury from direct impact. Pain referred from the hip or back can also cause AKP. As kinesiophobia may also play a role, it should be considered whenever an objective cause of the pain cannot be identified.
Surgery for Anterior Knee Pain
Surgery should be considered only after prolonged rest and healing have failed to resolve the pain caused by sustained direct trauma to the anterior knee. Physical therapy typically is not useful in direct trauma. If a painful traumatic articular lesion persists, then direct treatment—removing loose articular fragments and resurfacing or unloading a damaged articular surface—may be appropriate. In most cases, 6 to 12 months should be allowed before considering surgery. Meanwhile, rest, bracing, anti-inflammatory measures, reassurance, and work modification are the cornerstones of treatment.
After all conservative measures have failed in a patient with spontaneous-onset AKP related to repetitive focal overload, and disability caused by an objectively proven articular lesion related to mechanical dysfunction or dysplasia, diagnostic arthroscopy may be appropriate. Quantitation and characterization of the lesion with images and measurements are imperative in forming an optimal surgical plan. Remember that not all problems can be cured with surgery, and there is no patellofemoral problem that cannot potentially be made worse with improper surgery.
Am J Orthop. 2017;46(2):101-103. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Sanchis-Alfonso V, Dye SF. How to deal with anterior knee pain in the active young patient. Sports Health. 2016 Dec 5. [Epub ahead of print]
2. Fulkerson JP, Tennant R, Jaivin JS, Grunnet M. Histologic evidence of retinacular nerve injury associated with patellofemoral malalignment. Clin Orthop Relat Res. 1985;(197):196-205.
3. Dye SF. The pathophysiology of patellofemoral pain: a tissue homeostasis perspective. Clin Orthop Relat Res. 2005;(436):100-110.
4. Souza RB, Powers CM. Differences in hip kinematics, muscle strength, and muscle activation between subjects with and without patellofemoral pain. J Orthop Sports Phys Ther. 2009;39(1):12-19.
5. Post WR, Fulkerson J. Knee pain diagrams: correlation with physical examination findings in patients with anterior knee pain. Arthroscopy. 1994;10(6):618-623.
1. Sanchis-Alfonso V, Dye SF. How to deal with anterior knee pain in the active young patient. Sports Health. 2016 Dec 5. [Epub ahead of print]
2. Fulkerson JP, Tennant R, Jaivin JS, Grunnet M. Histologic evidence of retinacular nerve injury associated with patellofemoral malalignment. Clin Orthop Relat Res. 1985;(197):196-205.
3. Dye SF. The pathophysiology of patellofemoral pain: a tissue homeostasis perspective. Clin Orthop Relat Res. 2005;(436):100-110.
4. Souza RB, Powers CM. Differences in hip kinematics, muscle strength, and muscle activation between subjects with and without patellofemoral pain. J Orthop Sports Phys Ther. 2009;39(1):12-19.
5. Post WR, Fulkerson J. Knee pain diagrams: correlation with physical examination findings in patients with anterior knee pain. Arthroscopy. 1994;10(6):618-623.
Neoadjuvant and Adjuvant Therapy for Gastric Cancer
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
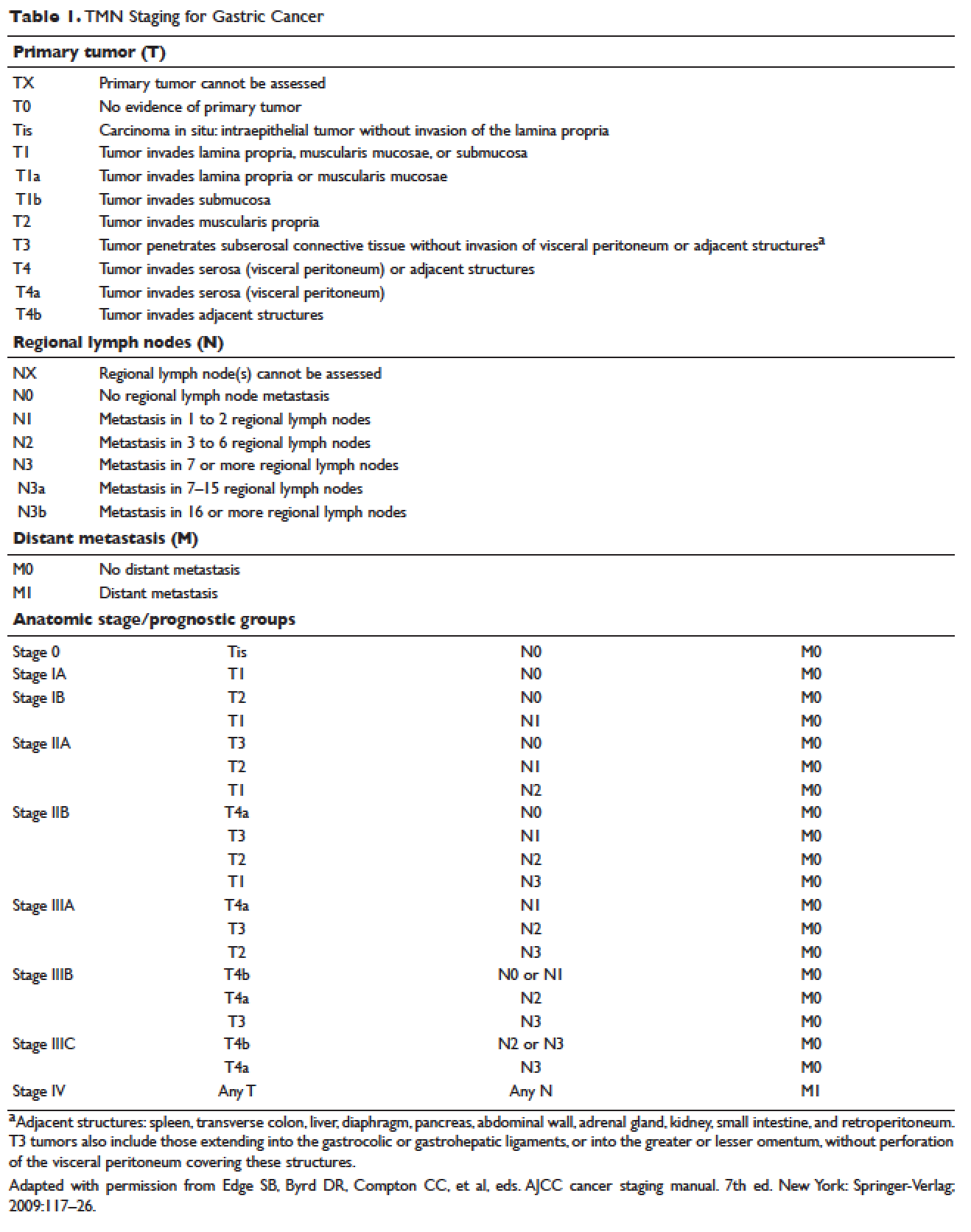
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
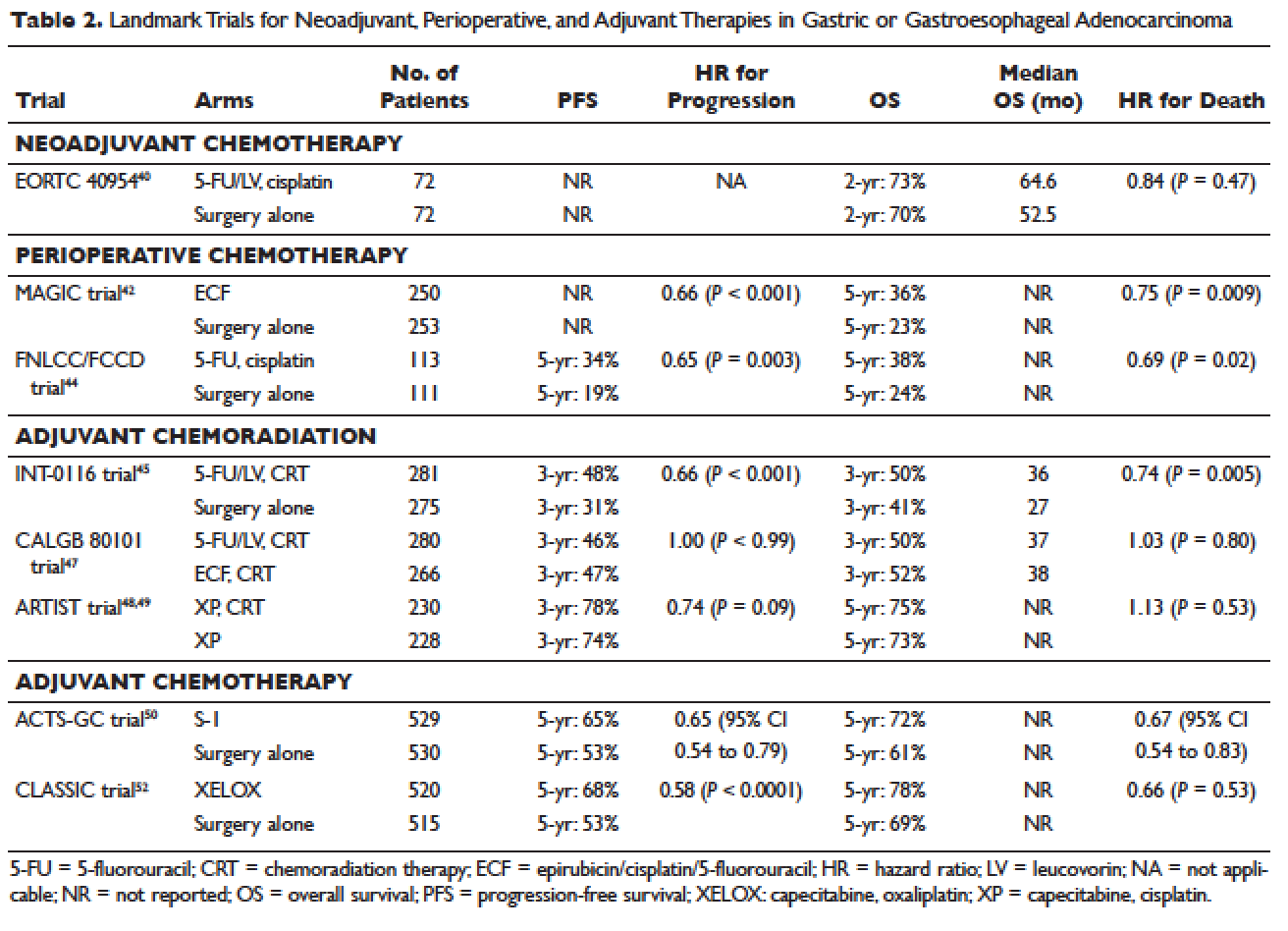
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
Hemophilia A and B: An Overview
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).

MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
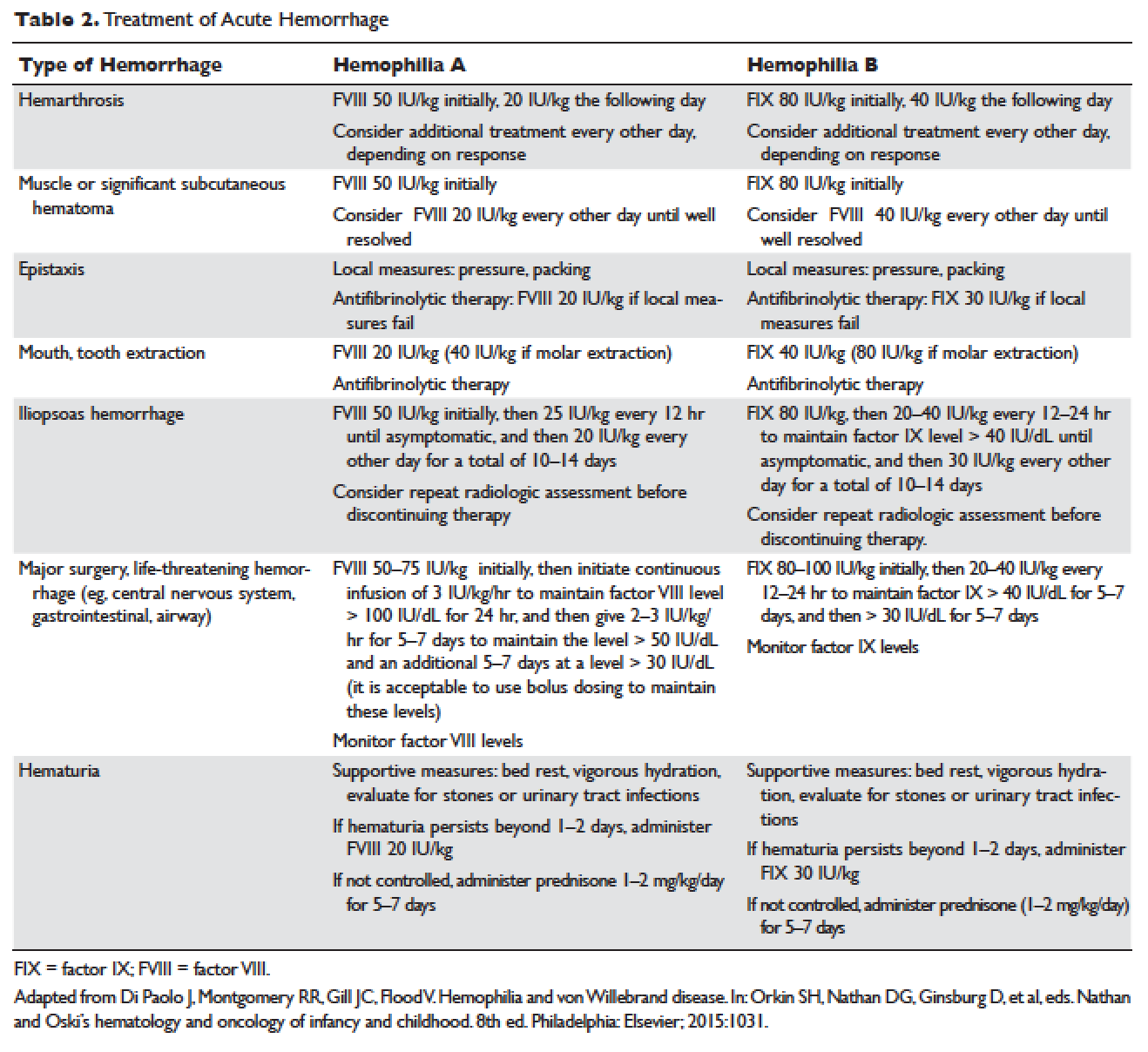
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
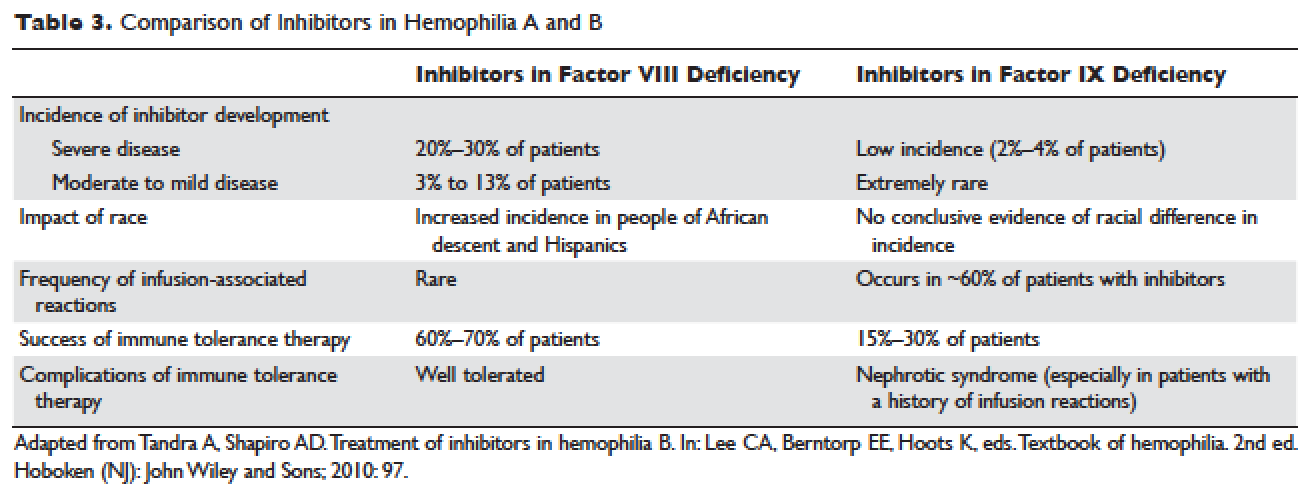
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.

Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82

ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
Mantle Cell Lymphoma
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
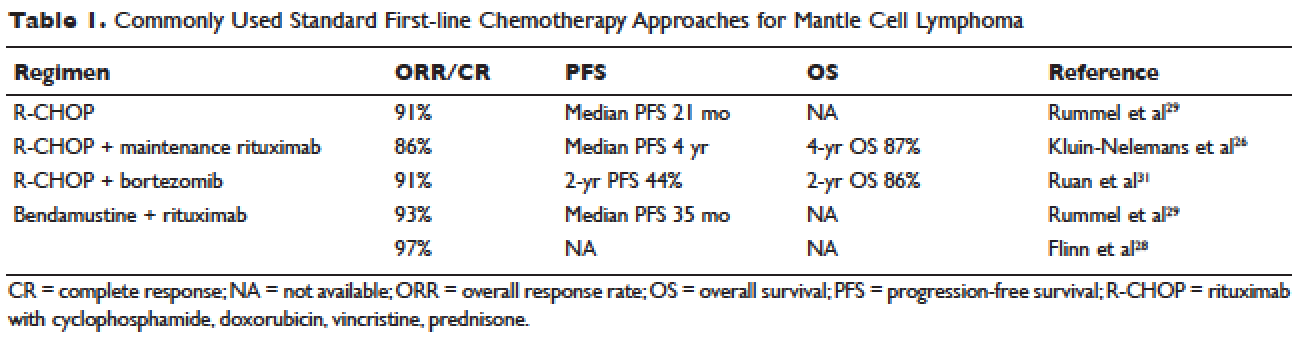
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
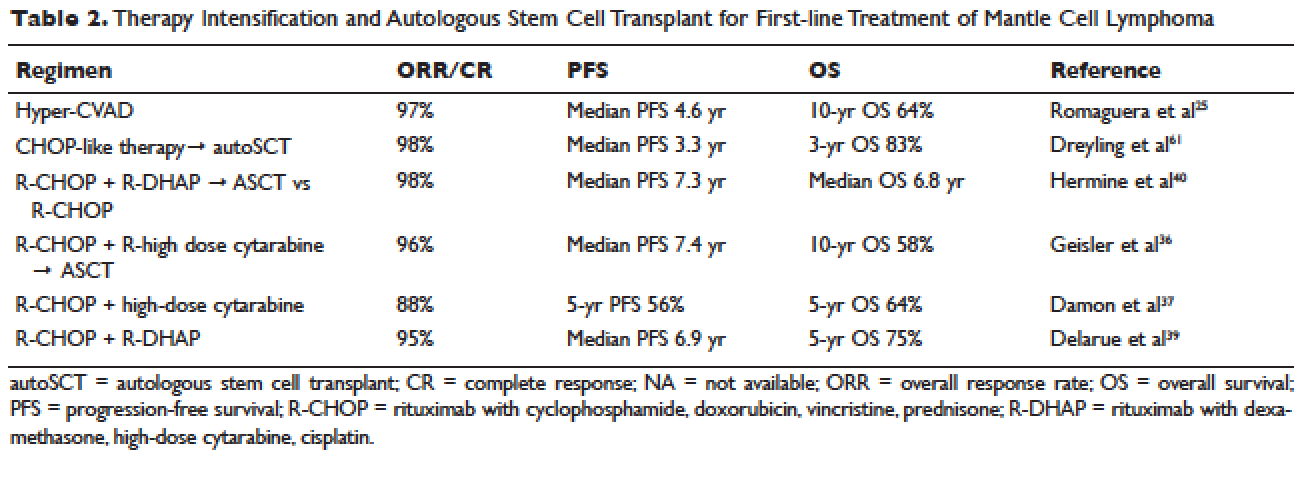
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
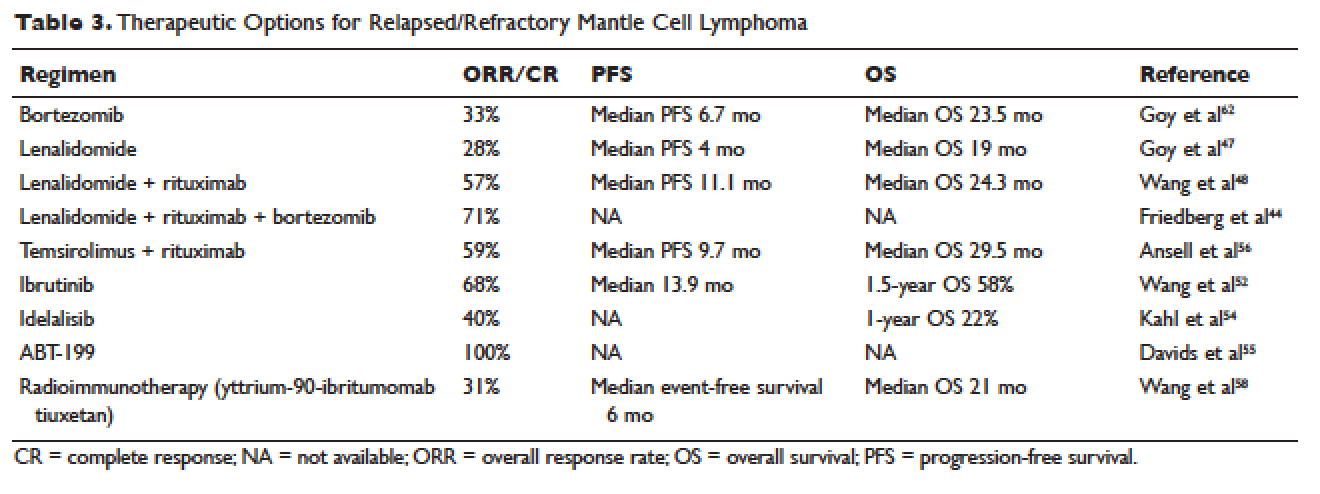
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
How and when umbilical cord gas analysis can justify your obstetric management
Umbilical cord blood (cord) gas values can aid both in understanding the cause of an infant’s acidosis and in providing reassurance that acute acidosis or asphyxia is not responsible for a compromised infant with a low Apgar score. Together with other clinical measurements (including fetal heart rate [FHR] tracings, Apgar scores, newborn nucleated red cell counts, and neonatal imaging), cord gas analysis can be remarkably helpful in determining the cause for a depressed newborn. It can help us determine, for example, if infant compromise was a result of an asphyxial event, and we often can differentiate whether the event was acute, prolonged, or occurred prior to presentation in labor. We further can use cord gas values to assess whether a decision for operative intervention for nonreassuring fetal well-being was appropriate (see “Brain injury at birth: Cord gas values presented as evidence at trial”). In addition, cord gas analysis can complement methods for determining fetal acidosis changes during labor, enabling improved assessment of FHR tracings.1−3
At 40 weeks' gestation, a woman presented to the hospital because of decreased fetal movement. On arrival, an external fetal heart-rate (FHR) monitor showed nonreassuring tracings, evidenced by absent to minimal variability and subtle decelerations occurring at 10- to 15-minute intervals. The on-call ObGyn requested induction of labor with oxytocin, and a low-dose infusion (1 mU/min) was initiated. An internal FHR monitor was then placed and late decelerations were observed with the first 2 induced contractions. The oxytocin infusion was discontinued and the ObGyn performed an emergency cesarean delivery. The infant's Apgar scores were 1, 2, and 2 at 1, 5, and 10 minutes, respectively. Cord samples were obtained and values from the umbilical artery were as follows: pH, 6.86; Pco2, 55 mm Hg; Po2, 6 mm Hg; and BDECF, 21.1 mmol/L. Values from the umbilical vein were: pH, 6.94; Pco2, 45 mm Hg; Po2, 17 mm Hg; and BDECF, 20.0 mmol/L. The infant was later diagnosed with a hypoxic brain injury resulting in cerebral palsy. At trial years later, the boy had cognitive and physical limitations and required 24-hour care.
The parents claimed that the ObGyn should have performed a cesarean delivery earlier when the external FHR monitor showed nonreassuring tracings.
The hospital and physician claimed that, while tracings were consistently nonreassuring, they were stable. They maintained that the child's brain damage was not due to a delivery delay, as the severe level of acidosis in both the umbilical artery and vein could not be a result of the few heart rate decelerations during the 2-hour period of monitoring prior to delivery. They argued that the clinical picture indicated a pre-hospital hypoxic event associated with decreased fetal movement.
A defense verdict was returned.
Case assessment
Cord gas results, together with other measures (eg, infant nucleated red blood cells, brain imaging) can aid the ObGyn in medicolegal cases. However, they are not always protective of adverse judgment.
I recommend checking umbilical cord blood gas values on all operative vaginal deliveries, cesarean deliveries for fetal concern, abnormal FHR patterns, clinical chorioamnionitis, multifetal gestations, premature deliveries, and all infants with low Apgar scores at 1 or 5 minutes. If you think you may need a cord gas analysis, go ahead and obtain it. Cord gas analysis often will aid in justifying your management or provide insight into the infant’s status.
Controversy remains as to the benefit of universal cord gas analysis. Assuming a variable cost of $15 for 2 (artery and vein) blood gas samples per neonate,4 the annual cost in the United States would be approximately $60 million. This would likely be cost effective as a result of medicolegal and educational benefits as well as potential improvements in perinatal outcome5 and reductions in special care nursery admissions.4
CASE 1: A newborn with unexpected acidosis
A 29-year-old woman (G2P1) at 38 weeks’ gestation was admitted to the hospital following an office visit during which oligohydramnios (amniotic fluid index, 3.5 cm) was found. The patient had a history of a prior cesarean delivery for failure to progress, and she desired a repeat cesarean delivery. Fetal monitoring revealed a heart rate of 140 beats per minute with moderate variability and uterine contractions every 3 to 5 minutes associated with moderate variable decelerations. A decision was made to proceed with the surgery. Blood samples were drawn for laboratory analysis, monitoring was discontinued, and the patient was taken to the operating room. An epidural anesthetic was placed and the cesarean delivery proceeded.
On uterine incision, there was no evidence of abruption or uterine rupture, but thick meconium-stained amniotic fluid was observed. A depressed infant was delivered, the umbilical cord clamped, and the infant handed to the pediatric team. Cord samples were obtained and values from the umbilical artery were as follows: pH, 6.80; P
What happened?
Read how to use cord gas values in practice
Using cord gas values in practice
Before analyzing the circumstances in Case 1,it is important to consider several key questions, including:
- What are the normal levels of cord pH, O2, CO2, and base deficit (BD)?
- How does cord gas indicate what happened during labor?
- What are the preventable errors in cord gas sampling or interpretation?
For a review of fetal cord gas physiology, see “Physiology of fetal cord gases: The basics”.
A review of basic fetal cord gas physiology will assist in understanding how values are interpreted.
Umbilical cord O2 and CO2
Fetal cord gas values result from the rapid transfer of gases and the slow clearance of acid across the placenta. Approximately 10% of maternal blood flow supplies the uteroplacental circulation, with the near-term placenta receiving approximately 70% of the uterine blood flow.1 Of the oxygen delivered, a surprising 50% provides for placental metabolism and 50% for the fetus. On the fetal side, 40% of fetal cardiac output supplies the umbilical circulation. Oxygen and carbon dioxide pass readily across the placental layers; exchange is limited by the amount of blood flow on both the maternal and the fetal side (flow limited). In the human placenta, maternal blood and fetal blood effectively travel in the same direction (concurrent exchange); thus, umbilical vein O2 and CO2 equilibrate with that in the maternal uterine vein.
Most of the O2 in fetal blood is carried by hemoglobin. Because of the markedly greater affinity of fetal hemoglobin for O2, the saturation curve is shifted to the left, resulting in increased hemoglobin saturation at the relatively low levels of fetal Po2. This greater affinity for oxygen results from the unique fetal hemoglobin gamma (γ) subunit, as compared with the adult beta (ß) subunit. Fetal hemoglobin has a reduced interaction with 2,3-bisphosphoglycerate, which itself decreases the affinity of adult hemoglobin for oxygen.
The majority of CO2 (85%) is carried as part of the bicarbonate buffer system. Fetal CO2 is converted into carbonic acid (H2CO3) in the red cell and dissociates into hydrogen (H+) and bicarbonate (HCO3−) ions, which diffuse out of the cell. When fetal blood reaches the placenta, this process is reversed and CO2 diffuses across the placenta to the maternal circulation. The production of H+ ions from CO2 explains the development of respiratory acidosis from high Pco2. In contrast, anaerobic metabolism, which produces lactic acid, results in metabolic acidosis.
Difference between pH and BD
The pH is calculated as the inverse log of the H+ ion concentration; thus, the pH falls as the H+ ion concentration exponentially increases, whether due to respiratory or metabolic acidosis. To quantify the more important metabolic acidosis, we use BD, which is a measure of how much of bicarbonate buffer base has been used by (lactic) acid. The BD and the base excess (BE) may be used interchangeably, with BE representing a negative number. Although BD represents the metabolic component of acidosis, a correction may be required to account for high levels of fetal Pco2 (see Case 1). In this situation, a more accurate measure is BD extracellular fluid (BDECF).
Why not just use pH? There are 2 major limitations to using pH as a measure of fetal or newborn acidosis. First, pH may be influenced by both respiratory and metabolic alterations, although only metabolic acidosis is associated with fetal neurologic injury.2 Furthermore, as pH is a log function, it does not change linearly with the amount of acid produced. In contrast to pH, BD is a measure of metabolic acidosis and changes in direct proportion to fetal acid production.
What about lactate? Measurements of lactate may also be included in blood gas analyses. Under hypoxic conditions, excess pyruvate is converted into lactate and released from the cell along with H+, resulting in acidosis. However, levels of umbilical cord lactate associated with neonatal hypoxic injury have not been established to the same degree as have pH or BD. Nevertheless, lactate has been measured in fetal scalp blood samples and offers the potential as a marker of fetal hypoxemia and acidosis.3
References
- Assali NS. Dynamics of the uteroplacental circulation in health and disease. Am J Perinatol. 1989;6(2):105-109.
- Low JA, Panagiotopoulos C, Derrick EJ. Newborn complications after intrapartum asphyxia with metabolic acidosis in the term fetus. Am J Obstet Gynecol. 1994;170(4):1081-1087.
- Mancho JP, Gamboa SM, Gimenez OR, Esteras RC, Solanilla BR, Mateo SC. Diagnostic accuracy of fetal scalp lactate for intrapartum acidosis compared with scalp pH [published online ahead of print October 8, 2016]. J Perinatal Med. doi: 10.1515/jpm-2016-004.
Normal values: The “20, 30, 40, 50 rule”
Among the values reported for umbilical blood gas, the pH, P
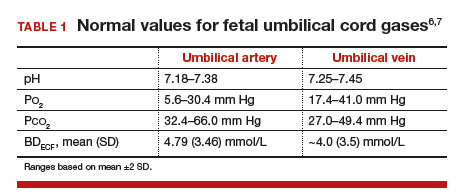
I recommend using the “20, 30, 40, 50 rule” as a simple tool for remembering normal umbilical artery and vein P
- P
o 2 values are lower than Pco 2 values; thus, the 20 and 30 represent Po 2 values - as fetal umbilical artery P
o 2 is lower than umbilical vein Po 2, 20 mm Hg represents the umbilical artery and 30 mm Hg represents the vein - P
co 2 values are higher in the umbilical artery than in the vein; thus, 50 mm Hg represents the umbilical artery and 40 mm Hg represents the umbilical vein.

Umbilical cord BD values change in relation to labor and FHR decelerations.8 Prior to labor, the normal fetus has a slight degree of acidosis (BD, 2 mmol/L). During the latent phase of labor, fetal BD typically does not change. With the increased frequency of contractions, BD may increase 1 mmol/L for every 3 to 6 hours during the active phase and up to 1 mmol/L per hour during the second stage, depending on FHR responses. Thus, following vaginal delivery the average umbilical artery BD is approximately 5 mmol/L and the umbilical vein BD is approximately 4 mmol/L. As lactate crosses the placenta slowly, BD values are typically only 1 mmol/L less in the umbilical vein than in the artery, unless there has been an obstruction to placental flow (see Case 1).
For pH, the umbilical artery value is always lower than that of the vein, a result of both the higher umbilical artery P
Possible causes of abnormal cord gas values
Because of the nearly fully saturated maternal hemoglobin under normal conditions, fetal arterial and venous P
In contrast, reduced fetal P
Effect of maternal oxygen administration on fetal oxygenation
Although maternal oxygen administration is commonly used during labor and delivery, controversy remains as to the benefit of oxygen supplementation.10 In a normal mother with oxygen saturation above 95%, the administration of oxygen will increase maternal arterial P
However, maternal oxygen supplementation may have marked benefit in cases in which maternal arterial P
How did the Case 1 circumstances lead to newborn acidosis?
Most noticeable in this case is the large difference in BD between the umbilical artery and vein and the high P
Whereas BD normally is only about 1 mmol/L greater in the umbilical artery versus in the vein, occasionally the arterial value is markedly greater than the vein value. This can occur when there is a cessation of blood flow through the placenta, as a result of complete umbilical cord obstruction, or when there is a uterine abruption. In these situations, the umbilical vein (which has not had blood flow) represents the fetal status prior to the occlusion event. In contrast, despite bradycardia, fetal heart pulsations mix blood within the umbilical artery and therefore the artery generally represents the fetal status at the time of birth.
In response to complete cord occlusion, fetal BD increases by approximately 1 mmol/L every 2 minutes. Consequently, an 8 mmol/L difference in BD between the umbilical artery and vein is consistent with a 16-minute period of umbilical occlusion or placental abruption. Also in response to complete umbilical cord occlusion, P
The umbilical vein BD is also elevated for early labor. This value suggests that repetitive, intermittent cord occlusions (evident on the initial fetal monitor tracing) likely resulted in this moderate acidosis prior to the complete cord occlusion in the final 16 minutes.
Thus, BD and P
Read more cases plus procedures, equipment for cord sampling
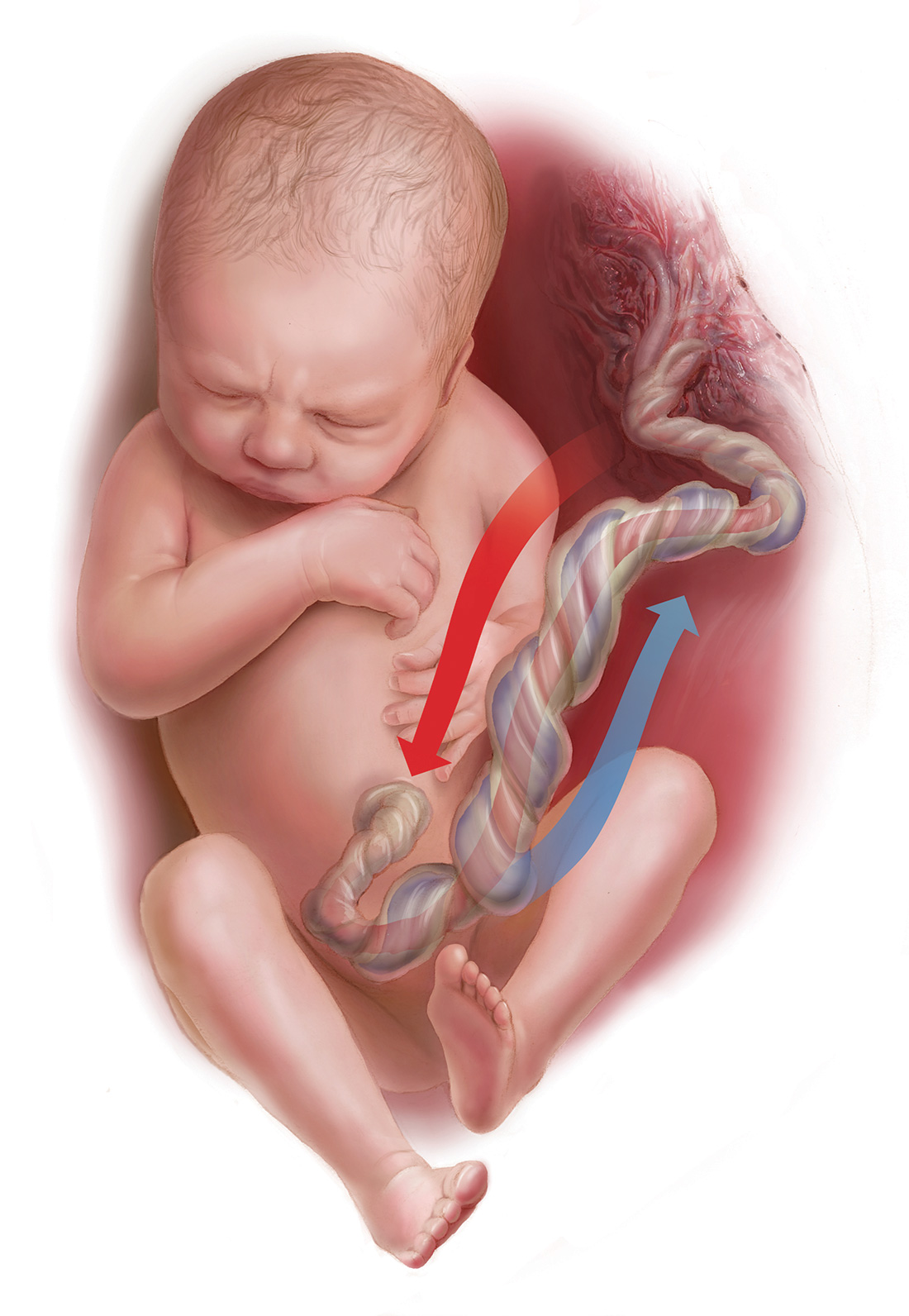
CASE 2: An infant with unusual umbilical artery values
An infant born via vacuum delivery for a prolonged second stage of labor had 1- and 5-minute Apgar scores of 8 and 9, respectively. Cord gas values were obtained, and analysis revealed that for the umbilical artery, the pH was 7.29; P
The resident asked, “How is the P
The curious Case 2 values suggest an air bubble
Although it is possible that the aberrant values in Case 2 could have resulted from switching the artery and vein samples, the pH is lower in the artery, and both the artery P
Related article:
Is neonatal injury more likely outside of a 30-minute decision-to-incision time interval for cesarean delivery?
CASE 3: A vigorous baby with significant acidosis
A baby with 1- and 5-minute Apgar scores of 9 and 9 was delivered by cesarean and remained vigorous. Umbilical cord analysis revealed an umbilical artery pH level of 7.15, with normal P
Was there a collection error in Case 3?
On occasion, a falsely low pH level and, thus, a falsely elevated BD may result from excessive heparin in the collection syringe. Heparin is acidotic and should be used only to coat the syringe. Although syringes in current use are often pre-heparinized, if one is drawing up heparin into the syringe, it should be coated and then fully expelled.
Umbilical cord sampling: Procedures and equipment
Many issues remain regarding the optimal storage of cord samples. Ideally, a doubly clamped section of the cord promptly should be sampled into glass syringes that can be placed on ice and rapidly measured for cord values.
Stability of umbilical cord samples within the cord is within 20 to 30 minutes. Delayed sampling of clamped cord sections generally has minimal effect on pH and P
Plastic syringes can introduce interference. Several studies have demonstrated that collection of samples in plastic may result in an increase in P
Use glass, and “ice” the sample if necessary. Although it has been suggested that placing samples on ice minimizes metabolism, the cooled plastic may in fact be more susceptible to oxygen diffusion. Thus, unless samples will be analyzed promptly, it is best to use glass syringes on ice.13,14
Related article:
Protecting the newborn brain—the final frontier in obstetric and neonatal care
What if the umbilical cord is torn?
Sometimes the umbilical cord is torn and discarded or cannot be accessed for other reasons. A sample can still be obtained, however, by aspirating the placental surface artery and vein vessels. Although there is some potential variance in pH, P
How do you obtain cord analysis when delaying cord clamping?
The American College of Obstetricians and Gynecologists (ACOG) now advises delayed cord clamping in term and preterm deliveries, which raises the question of how you obtain a blood sample in this setting. Importantly, ACOG recommends delayed cord clamping only in vigorous infants,16 whereas potentially compromised infants should be transferred rapidly for newborn care. Although several studies have demonstrated some variation in cord gas values with delayed cord clamping,17–21 clamping after pulsation has ceased or after the recommended 30 to 60 seconds following birth results in minimal change in BD values. Thus, do not hesitate to perform delayed cord clamping in vigorous infants.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187(1):1–9.
- Uccella S, Cromi A, Colombo G, et al. Prediction of fetal base excess values at birth using an algorithm to interpret fetal heart rate tracings: a retrospective validation. BJOG. 2012;119(13):1657–1664.
- Uccella S, Cromi A, Colombo GF, et al. Interobserver reliability to interpret intrapartum electronic fetal heart rate monitoring: does a standardized algorithm improve agreement among clinicians? J Obstet Gynaecol. 2015;35(3):241–245.
- White CR, Doherty DA, Cannon JW, Kohan R, Newnham JP, Pennell CE. Cost effectiveness of universal umbilical cord blood gas and lactate analysis in a tertiary level maternity unit. J Perinat Med. 2016;44(5):573–584.
- White CR, Doherty DA, Henderson JJ, Kohan R, Newnham JP, Pennell CE. Benefits of introducing universal umbilical cord blood gas and lactate analysis into an obstetric unit. Aust N Z J Obstet Gynaecol. 2010;50(4):318–328.
- Yeomans ER, Hauth JC, Gilstrap LC III, Strickland DM. Umbilical cord pH, Pco2, and bicarbonate following uncomplicated term vaginal deliveries. Am J Obstet Gynecol. 1985;151(6):798–800.
- Wiberg N, Källén K, Olofsson P. Base deficit estimation in umbilical cord blood is influenced by gestational age, choice of fetal fluid compartment, and algorithm for calculation. Am J Obstet Gynecol. 2006;195(6):1651–1656.
- Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187(1):1–9.
- Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123(4):896-901.
- Hamel MS, Anderson BL, Rouse DJ. Oxygen for intrauterine resuscitation: of unproved benefit and potentially harmful. Am J Obstet Gynecol. 2014;211(2):124–127.
- Owen P, Farrell TA, Steyn W. Umbilical cord blood gas analysis; a comparison of two simple methods of sample storage. Early Hum Dev. 1995;42(1):67–71.
- Armstrong L, Stenson B. Effect of delayed sampling on umbilical cord arterial and venous lactate and blood gases in clamped and unclamped vessels. Arch Dis Child Fetal Neonatal Ed. 2006;91(5):F342–F345.
- White CR, Mok T, Doherty DA, Henderson JJ, Newnham JP, Pennell CE. The effect of time, temperature and storage device on umbilical cord blood gas and lactate measurement: a randomized controlled trial. J Matern Fetal Neonatal Med. 2012;25(6):587–594.
- Knowles TP, Mullin RA, Hunter JA, Douce FH. Effects of syringe material, sample storage time, and temperature on blood gases and oxygen saturation in arterialized human blood samples. Respir Care. 2006;51(7):732–736.
- Nodwell A, Carmichael L, Ross M, Richardson B. Placental compared with umbilical cord blood to assess fetal blood gas and acid-base status. Obstet Gynecol. 2005;105(1):129–138.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 684. Delayed umbilical cord clamping after birth. Obstet Gynecol. 2017;129(1):e5–e10.
- De Paco C, Florido J, Garrido MC, Prados S, Navarrete L. Umbilical cord blood acid-base and gas analysis after early versus delayed cord clamping in neonates at term. Arch Gynecol Obstet. 2011;283(5):1011–1014.
- Valero J, Desantes D, Perales-Puchalt A, Rubio J, Diago Almela VJ, Perales A. Effect of delayed umbilical cord clamping on blood gas analysis. Eur J Obstet Gynecol Reprod Biol. 2012;162(1): 21–23.
- Andersson O, Hellström-Westas L, Andersson D, Clausen J, Domellöf M. Effects of delayed compared with early umbilical cord clamping on maternal postpartum hemorrhage and cord blood gas sampling: a randomized trial. Acta Obstet Gynecol Scand. 2013;92(5):567–574.
- Wiberg N, Källén K, Olofsson P. Delayed umbilical cord clamping at birth has effects on arterial and venous blood gases and lactate concentrations. BJOG. 2008;115(6):697–703.
- Mokarami P, Wiberg N, Olofsson P. Hidden acidosis: an explanation of acid-base and lactate changes occurring in umbilical cord blood after delayed sampling. BJOG. 2013;120(8):996–1002.

Dr. Ross is Distinguished Professor of Obstetrics and Gynecology and Public Health, Geffen School of Medicine at UCLA, Fielding School of Public Health at UCLA, Department of Obstetrics and Gynecology, Harbor/UCLA Medical Center, Torrance, California.
The author reports no financial relationships relevant to this article.
Dr. Ross is Distinguished Professor of Obstetrics and Gynecology and Public Health, Geffen School of Medicine at UCLA, Fielding School of Public Health at UCLA, Department of Obstetrics and Gynecology, Harbor/UCLA Medical Center, Torrance, California.
The author reports no financial relationships relevant to this article.
Dr. Ross is Distinguished Professor of Obstetrics and Gynecology and Public Health, Geffen School of Medicine at UCLA, Fielding School of Public Health at UCLA, Department of Obstetrics and Gynecology, Harbor/UCLA Medical Center, Torrance, California.
The author reports no financial relationships relevant to this article.
Umbilical cord blood (cord) gas values can aid both in understanding the cause of an infant’s acidosis and in providing reassurance that acute acidosis or asphyxia is not responsible for a compromised infant with a low Apgar score. Together with other clinical measurements (including fetal heart rate [FHR] tracings, Apgar scores, newborn nucleated red cell counts, and neonatal imaging), cord gas analysis can be remarkably helpful in determining the cause for a depressed newborn. It can help us determine, for example, if infant compromise was a result of an asphyxial event, and we often can differentiate whether the event was acute, prolonged, or occurred prior to presentation in labor. We further can use cord gas values to assess whether a decision for operative intervention for nonreassuring fetal well-being was appropriate (see “Brain injury at birth: Cord gas values presented as evidence at trial”). In addition, cord gas analysis can complement methods for determining fetal acidosis changes during labor, enabling improved assessment of FHR tracings.1−3
At 40 weeks' gestation, a woman presented to the hospital because of decreased fetal movement. On arrival, an external fetal heart-rate (FHR) monitor showed nonreassuring tracings, evidenced by absent to minimal variability and subtle decelerations occurring at 10- to 15-minute intervals. The on-call ObGyn requested induction of labor with oxytocin, and a low-dose infusion (1 mU/min) was initiated. An internal FHR monitor was then placed and late decelerations were observed with the first 2 induced contractions. The oxytocin infusion was discontinued and the ObGyn performed an emergency cesarean delivery. The infant's Apgar scores were 1, 2, and 2 at 1, 5, and 10 minutes, respectively. Cord samples were obtained and values from the umbilical artery were as follows: pH, 6.86; Pco2, 55 mm Hg; Po2, 6 mm Hg; and BDECF, 21.1 mmol/L. Values from the umbilical vein were: pH, 6.94; Pco2, 45 mm Hg; Po2, 17 mm Hg; and BDECF, 20.0 mmol/L. The infant was later diagnosed with a hypoxic brain injury resulting in cerebral palsy. At trial years later, the boy had cognitive and physical limitations and required 24-hour care.
The parents claimed that the ObGyn should have performed a cesarean delivery earlier when the external FHR monitor showed nonreassuring tracings.
The hospital and physician claimed that, while tracings were consistently nonreassuring, they were stable. They maintained that the child's brain damage was not due to a delivery delay, as the severe level of acidosis in both the umbilical artery and vein could not be a result of the few heart rate decelerations during the 2-hour period of monitoring prior to delivery. They argued that the clinical picture indicated a pre-hospital hypoxic event associated with decreased fetal movement.
A defense verdict was returned.
Case assessment
Cord gas results, together with other measures (eg, infant nucleated red blood cells, brain imaging) can aid the ObGyn in medicolegal cases. However, they are not always protective of adverse judgment.
I recommend checking umbilical cord blood gas values on all operative vaginal deliveries, cesarean deliveries for fetal concern, abnormal FHR patterns, clinical chorioamnionitis, multifetal gestations, premature deliveries, and all infants with low Apgar scores at 1 or 5 minutes. If you think you may need a cord gas analysis, go ahead and obtain it. Cord gas analysis often will aid in justifying your management or provide insight into the infant’s status.
Controversy remains as to the benefit of universal cord gas analysis. Assuming a variable cost of $15 for 2 (artery and vein) blood gas samples per neonate,4 the annual cost in the United States would be approximately $60 million. This would likely be cost effective as a result of medicolegal and educational benefits as well as potential improvements in perinatal outcome5 and reductions in special care nursery admissions.4
CASE 1: A newborn with unexpected acidosis
A 29-year-old woman (G2P1) at 38 weeks’ gestation was admitted to the hospital following an office visit during which oligohydramnios (amniotic fluid index, 3.5 cm) was found. The patient had a history of a prior cesarean delivery for failure to progress, and she desired a repeat cesarean delivery. Fetal monitoring revealed a heart rate of 140 beats per minute with moderate variability and uterine contractions every 3 to 5 minutes associated with moderate variable decelerations. A decision was made to proceed with the surgery. Blood samples were drawn for laboratory analysis, monitoring was discontinued, and the patient was taken to the operating room. An epidural anesthetic was placed and the cesarean delivery proceeded.
On uterine incision, there was no evidence of abruption or uterine rupture, but thick meconium-stained amniotic fluid was observed. A depressed infant was delivered, the umbilical cord clamped, and the infant handed to the pediatric team. Cord samples were obtained and values from the umbilical artery were as follows: pH, 6.80; P
What happened?
Read how to use cord gas values in practice
Using cord gas values in practice
Before analyzing the circumstances in Case 1,it is important to consider several key questions, including:
- What are the normal levels of cord pH, O2, CO2, and base deficit (BD)?
- How does cord gas indicate what happened during labor?
- What are the preventable errors in cord gas sampling or interpretation?
For a review of fetal cord gas physiology, see “Physiology of fetal cord gases: The basics”.
A review of basic fetal cord gas physiology will assist in understanding how values are interpreted.
Umbilical cord O2 and CO2
Fetal cord gas values result from the rapid transfer of gases and the slow clearance of acid across the placenta. Approximately 10% of maternal blood flow supplies the uteroplacental circulation, with the near-term placenta receiving approximately 70% of the uterine blood flow.1 Of the oxygen delivered, a surprising 50% provides for placental metabolism and 50% for the fetus. On the fetal side, 40% of fetal cardiac output supplies the umbilical circulation. Oxygen and carbon dioxide pass readily across the placental layers; exchange is limited by the amount of blood flow on both the maternal and the fetal side (flow limited). In the human placenta, maternal blood and fetal blood effectively travel in the same direction (concurrent exchange); thus, umbilical vein O2 and CO2 equilibrate with that in the maternal uterine vein.
Most of the O2 in fetal blood is carried by hemoglobin. Because of the markedly greater affinity of fetal hemoglobin for O2, the saturation curve is shifted to the left, resulting in increased hemoglobin saturation at the relatively low levels of fetal Po2. This greater affinity for oxygen results from the unique fetal hemoglobin gamma (γ) subunit, as compared with the adult beta (ß) subunit. Fetal hemoglobin has a reduced interaction with 2,3-bisphosphoglycerate, which itself decreases the affinity of adult hemoglobin for oxygen.
The majority of CO2 (85%) is carried as part of the bicarbonate buffer system. Fetal CO2 is converted into carbonic acid (H2CO3) in the red cell and dissociates into hydrogen (H+) and bicarbonate (HCO3−) ions, which diffuse out of the cell. When fetal blood reaches the placenta, this process is reversed and CO2 diffuses across the placenta to the maternal circulation. The production of H+ ions from CO2 explains the development of respiratory acidosis from high Pco2. In contrast, anaerobic metabolism, which produces lactic acid, results in metabolic acidosis.
Difference between pH and BD
The pH is calculated as the inverse log of the H+ ion concentration; thus, the pH falls as the H+ ion concentration exponentially increases, whether due to respiratory or metabolic acidosis. To quantify the more important metabolic acidosis, we use BD, which is a measure of how much of bicarbonate buffer base has been used by (lactic) acid. The BD and the base excess (BE) may be used interchangeably, with BE representing a negative number. Although BD represents the metabolic component of acidosis, a correction may be required to account for high levels of fetal Pco2 (see Case 1). In this situation, a more accurate measure is BD extracellular fluid (BDECF).
Why not just use pH? There are 2 major limitations to using pH as a measure of fetal or newborn acidosis. First, pH may be influenced by both respiratory and metabolic alterations, although only metabolic acidosis is associated with fetal neurologic injury.2 Furthermore, as pH is a log function, it does not change linearly with the amount of acid produced. In contrast to pH, BD is a measure of metabolic acidosis and changes in direct proportion to fetal acid production.
What about lactate? Measurements of lactate may also be included in blood gas analyses. Under hypoxic conditions, excess pyruvate is converted into lactate and released from the cell along with H+, resulting in acidosis. However, levels of umbilical cord lactate associated with neonatal hypoxic injury have not been established to the same degree as have pH or BD. Nevertheless, lactate has been measured in fetal scalp blood samples and offers the potential as a marker of fetal hypoxemia and acidosis.3
References
- Assali NS. Dynamics of the uteroplacental circulation in health and disease. Am J Perinatol. 1989;6(2):105-109.
- Low JA, Panagiotopoulos C, Derrick EJ. Newborn complications after intrapartum asphyxia with metabolic acidosis in the term fetus. Am J Obstet Gynecol. 1994;170(4):1081-1087.
- Mancho JP, Gamboa SM, Gimenez OR, Esteras RC, Solanilla BR, Mateo SC. Diagnostic accuracy of fetal scalp lactate for intrapartum acidosis compared with scalp pH [published online ahead of print October 8, 2016]. J Perinatal Med. doi: 10.1515/jpm-2016-004.
Normal values: The “20, 30, 40, 50 rule”
Among the values reported for umbilical blood gas, the pH, P
I recommend using the “20, 30, 40, 50 rule” as a simple tool for remembering normal umbilical artery and vein P
- P
o 2 values are lower than Pco 2 values; thus, the 20 and 30 represent Po 2 values - as fetal umbilical artery P
o 2 is lower than umbilical vein Po 2, 20 mm Hg represents the umbilical artery and 30 mm Hg represents the vein - P
co 2 values are higher in the umbilical artery than in the vein; thus, 50 mm Hg represents the umbilical artery and 40 mm Hg represents the umbilical vein.
Umbilical cord BD values change in relation to labor and FHR decelerations.8 Prior to labor, the normal fetus has a slight degree of acidosis (BD, 2 mmol/L). During the latent phase of labor, fetal BD typically does not change. With the increased frequency of contractions, BD may increase 1 mmol/L for every 3 to 6 hours during the active phase and up to 1 mmol/L per hour during the second stage, depending on FHR responses. Thus, following vaginal delivery the average umbilical artery BD is approximately 5 mmol/L and the umbilical vein BD is approximately 4 mmol/L. As lactate crosses the placenta slowly, BD values are typically only 1 mmol/L less in the umbilical vein than in the artery, unless there has been an obstruction to placental flow (see Case 1).
For pH, the umbilical artery value is always lower than that of the vein, a result of both the higher umbilical artery P
Possible causes of abnormal cord gas values
Because of the nearly fully saturated maternal hemoglobin under normal conditions, fetal arterial and venous P
In contrast, reduced fetal P
Effect of maternal oxygen administration on fetal oxygenation
Although maternal oxygen administration is commonly used during labor and delivery, controversy remains as to the benefit of oxygen supplementation.10 In a normal mother with oxygen saturation above 95%, the administration of oxygen will increase maternal arterial P
However, maternal oxygen supplementation may have marked benefit in cases in which maternal arterial P
How did the Case 1 circumstances lead to newborn acidosis?
Most noticeable in this case is the large difference in BD between the umbilical artery and vein and the high P
Whereas BD normally is only about 1 mmol/L greater in the umbilical artery versus in the vein, occasionally the arterial value is markedly greater than the vein value. This can occur when there is a cessation of blood flow through the placenta, as a result of complete umbilical cord obstruction, or when there is a uterine abruption. In these situations, the umbilical vein (which has not had blood flow) represents the fetal status prior to the occlusion event. In contrast, despite bradycardia, fetal heart pulsations mix blood within the umbilical artery and therefore the artery generally represents the fetal status at the time of birth.
In response to complete cord occlusion, fetal BD increases by approximately 1 mmol/L every 2 minutes. Consequently, an 8 mmol/L difference in BD between the umbilical artery and vein is consistent with a 16-minute period of umbilical occlusion or placental abruption. Also in response to complete umbilical cord occlusion, P
The umbilical vein BD is also elevated for early labor. This value suggests that repetitive, intermittent cord occlusions (evident on the initial fetal monitor tracing) likely resulted in this moderate acidosis prior to the complete cord occlusion in the final 16 minutes.
Thus, BD and P
Read more cases plus procedures, equipment for cord sampling
CASE 2: An infant with unusual umbilical artery values
An infant born via vacuum delivery for a prolonged second stage of labor had 1- and 5-minute Apgar scores of 8 and 9, respectively. Cord gas values were obtained, and analysis revealed that for the umbilical artery, the pH was 7.29; P
The resident asked, “How is the P
The curious Case 2 values suggest an air bubble
Although it is possible that the aberrant values in Case 2 could have resulted from switching the artery and vein samples, the pH is lower in the artery, and both the artery P
Related article:
Is neonatal injury more likely outside of a 30-minute decision-to-incision time interval for cesarean delivery?
CASE 3: A vigorous baby with significant acidosis
A baby with 1- and 5-minute Apgar scores of 9 and 9 was delivered by cesarean and remained vigorous. Umbilical cord analysis revealed an umbilical artery pH level of 7.15, with normal P
Was there a collection error in Case 3?
On occasion, a falsely low pH level and, thus, a falsely elevated BD may result from excessive heparin in the collection syringe. Heparin is acidotic and should be used only to coat the syringe. Although syringes in current use are often pre-heparinized, if one is drawing up heparin into the syringe, it should be coated and then fully expelled.
Umbilical cord sampling: Procedures and equipment
Many issues remain regarding the optimal storage of cord samples. Ideally, a doubly clamped section of the cord promptly should be sampled into glass syringes that can be placed on ice and rapidly measured for cord values.
Stability of umbilical cord samples within the cord is within 20 to 30 minutes. Delayed sampling of clamped cord sections generally has minimal effect on pH and P
Plastic syringes can introduce interference. Several studies have demonstrated that collection of samples in plastic may result in an increase in P
Use glass, and “ice” the sample if necessary. Although it has been suggested that placing samples on ice minimizes metabolism, the cooled plastic may in fact be more susceptible to oxygen diffusion. Thus, unless samples will be analyzed promptly, it is best to use glass syringes on ice.13,14
Related article:
Protecting the newborn brain—the final frontier in obstetric and neonatal care
What if the umbilical cord is torn?
Sometimes the umbilical cord is torn and discarded or cannot be accessed for other reasons. A sample can still be obtained, however, by aspirating the placental surface artery and vein vessels. Although there is some potential variance in pH, P
How do you obtain cord analysis when delaying cord clamping?
The American College of Obstetricians and Gynecologists (ACOG) now advises delayed cord clamping in term and preterm deliveries, which raises the question of how you obtain a blood sample in this setting. Importantly, ACOG recommends delayed cord clamping only in vigorous infants,16 whereas potentially compromised infants should be transferred rapidly for newborn care. Although several studies have demonstrated some variation in cord gas values with delayed cord clamping,17–21 clamping after pulsation has ceased or after the recommended 30 to 60 seconds following birth results in minimal change in BD values. Thus, do not hesitate to perform delayed cord clamping in vigorous infants.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Umbilical cord blood (cord) gas values can aid both in understanding the cause of an infant’s acidosis and in providing reassurance that acute acidosis or asphyxia is not responsible for a compromised infant with a low Apgar score. Together with other clinical measurements (including fetal heart rate [FHR] tracings, Apgar scores, newborn nucleated red cell counts, and neonatal imaging), cord gas analysis can be remarkably helpful in determining the cause for a depressed newborn. It can help us determine, for example, if infant compromise was a result of an asphyxial event, and we often can differentiate whether the event was acute, prolonged, or occurred prior to presentation in labor. We further can use cord gas values to assess whether a decision for operative intervention for nonreassuring fetal well-being was appropriate (see “Brain injury at birth: Cord gas values presented as evidence at trial”). In addition, cord gas analysis can complement methods for determining fetal acidosis changes during labor, enabling improved assessment of FHR tracings.1−3
At 40 weeks' gestation, a woman presented to the hospital because of decreased fetal movement. On arrival, an external fetal heart-rate (FHR) monitor showed nonreassuring tracings, evidenced by absent to minimal variability and subtle decelerations occurring at 10- to 15-minute intervals. The on-call ObGyn requested induction of labor with oxytocin, and a low-dose infusion (1 mU/min) was initiated. An internal FHR monitor was then placed and late decelerations were observed with the first 2 induced contractions. The oxytocin infusion was discontinued and the ObGyn performed an emergency cesarean delivery. The infant's Apgar scores were 1, 2, and 2 at 1, 5, and 10 minutes, respectively. Cord samples were obtained and values from the umbilical artery were as follows: pH, 6.86; Pco2, 55 mm Hg; Po2, 6 mm Hg; and BDECF, 21.1 mmol/L. Values from the umbilical vein were: pH, 6.94; Pco2, 45 mm Hg; Po2, 17 mm Hg; and BDECF, 20.0 mmol/L. The infant was later diagnosed with a hypoxic brain injury resulting in cerebral palsy. At trial years later, the boy had cognitive and physical limitations and required 24-hour care.
The parents claimed that the ObGyn should have performed a cesarean delivery earlier when the external FHR monitor showed nonreassuring tracings.
The hospital and physician claimed that, while tracings were consistently nonreassuring, they were stable. They maintained that the child's brain damage was not due to a delivery delay, as the severe level of acidosis in both the umbilical artery and vein could not be a result of the few heart rate decelerations during the 2-hour period of monitoring prior to delivery. They argued that the clinical picture indicated a pre-hospital hypoxic event associated with decreased fetal movement.
A defense verdict was returned.
Case assessment
Cord gas results, together with other measures (eg, infant nucleated red blood cells, brain imaging) can aid the ObGyn in medicolegal cases. However, they are not always protective of adverse judgment.
I recommend checking umbilical cord blood gas values on all operative vaginal deliveries, cesarean deliveries for fetal concern, abnormal FHR patterns, clinical chorioamnionitis, multifetal gestations, premature deliveries, and all infants with low Apgar scores at 1 or 5 minutes. If you think you may need a cord gas analysis, go ahead and obtain it. Cord gas analysis often will aid in justifying your management or provide insight into the infant’s status.
Controversy remains as to the benefit of universal cord gas analysis. Assuming a variable cost of $15 for 2 (artery and vein) blood gas samples per neonate,4 the annual cost in the United States would be approximately $60 million. This would likely be cost effective as a result of medicolegal and educational benefits as well as potential improvements in perinatal outcome5 and reductions in special care nursery admissions.4
CASE 1: A newborn with unexpected acidosis
A 29-year-old woman (G2P1) at 38 weeks’ gestation was admitted to the hospital following an office visit during which oligohydramnios (amniotic fluid index, 3.5 cm) was found. The patient had a history of a prior cesarean delivery for failure to progress, and she desired a repeat cesarean delivery. Fetal monitoring revealed a heart rate of 140 beats per minute with moderate variability and uterine contractions every 3 to 5 minutes associated with moderate variable decelerations. A decision was made to proceed with the surgery. Blood samples were drawn for laboratory analysis, monitoring was discontinued, and the patient was taken to the operating room. An epidural anesthetic was placed and the cesarean delivery proceeded.
On uterine incision, there was no evidence of abruption or uterine rupture, but thick meconium-stained amniotic fluid was observed. A depressed infant was delivered, the umbilical cord clamped, and the infant handed to the pediatric team. Cord samples were obtained and values from the umbilical artery were as follows: pH, 6.80; P
What happened?
Read how to use cord gas values in practice
Using cord gas values in practice
Before analyzing the circumstances in Case 1,it is important to consider several key questions, including:
- What are the normal levels of cord pH, O2, CO2, and base deficit (BD)?
- How does cord gas indicate what happened during labor?
- What are the preventable errors in cord gas sampling or interpretation?
For a review of fetal cord gas physiology, see “Physiology of fetal cord gases: The basics”.
A review of basic fetal cord gas physiology will assist in understanding how values are interpreted.
Umbilical cord O2 and CO2
Fetal cord gas values result from the rapid transfer of gases and the slow clearance of acid across the placenta. Approximately 10% of maternal blood flow supplies the uteroplacental circulation, with the near-term placenta receiving approximately 70% of the uterine blood flow.1 Of the oxygen delivered, a surprising 50% provides for placental metabolism and 50% for the fetus. On the fetal side, 40% of fetal cardiac output supplies the umbilical circulation. Oxygen and carbon dioxide pass readily across the placental layers; exchange is limited by the amount of blood flow on both the maternal and the fetal side (flow limited). In the human placenta, maternal blood and fetal blood effectively travel in the same direction (concurrent exchange); thus, umbilical vein O2 and CO2 equilibrate with that in the maternal uterine vein.
Most of the O2 in fetal blood is carried by hemoglobin. Because of the markedly greater affinity of fetal hemoglobin for O2, the saturation curve is shifted to the left, resulting in increased hemoglobin saturation at the relatively low levels of fetal Po2. This greater affinity for oxygen results from the unique fetal hemoglobin gamma (γ) subunit, as compared with the adult beta (ß) subunit. Fetal hemoglobin has a reduced interaction with 2,3-bisphosphoglycerate, which itself decreases the affinity of adult hemoglobin for oxygen.
The majority of CO2 (85%) is carried as part of the bicarbonate buffer system. Fetal CO2 is converted into carbonic acid (H2CO3) in the red cell and dissociates into hydrogen (H+) and bicarbonate (HCO3−) ions, which diffuse out of the cell. When fetal blood reaches the placenta, this process is reversed and CO2 diffuses across the placenta to the maternal circulation. The production of H+ ions from CO2 explains the development of respiratory acidosis from high Pco2. In contrast, anaerobic metabolism, which produces lactic acid, results in metabolic acidosis.
Difference between pH and BD
The pH is calculated as the inverse log of the H+ ion concentration; thus, the pH falls as the H+ ion concentration exponentially increases, whether due to respiratory or metabolic acidosis. To quantify the more important metabolic acidosis, we use BD, which is a measure of how much of bicarbonate buffer base has been used by (lactic) acid. The BD and the base excess (BE) may be used interchangeably, with BE representing a negative number. Although BD represents the metabolic component of acidosis, a correction may be required to account for high levels of fetal Pco2 (see Case 1). In this situation, a more accurate measure is BD extracellular fluid (BDECF).
Why not just use pH? There are 2 major limitations to using pH as a measure of fetal or newborn acidosis. First, pH may be influenced by both respiratory and metabolic alterations, although only metabolic acidosis is associated with fetal neurologic injury.2 Furthermore, as pH is a log function, it does not change linearly with the amount of acid produced. In contrast to pH, BD is a measure of metabolic acidosis and changes in direct proportion to fetal acid production.
What about lactate? Measurements of lactate may also be included in blood gas analyses. Under hypoxic conditions, excess pyruvate is converted into lactate and released from the cell along with H+, resulting in acidosis. However, levels of umbilical cord lactate associated with neonatal hypoxic injury have not been established to the same degree as have pH or BD. Nevertheless, lactate has been measured in fetal scalp blood samples and offers the potential as a marker of fetal hypoxemia and acidosis.3
References
- Assali NS. Dynamics of the uteroplacental circulation in health and disease. Am J Perinatol. 1989;6(2):105-109.
- Low JA, Panagiotopoulos C, Derrick EJ. Newborn complications after intrapartum asphyxia with metabolic acidosis in the term fetus. Am J Obstet Gynecol. 1994;170(4):1081-1087.
- Mancho JP, Gamboa SM, Gimenez OR, Esteras RC, Solanilla BR, Mateo SC. Diagnostic accuracy of fetal scalp lactate for intrapartum acidosis compared with scalp pH [published online ahead of print October 8, 2016]. J Perinatal Med. doi: 10.1515/jpm-2016-004.
Normal values: The “20, 30, 40, 50 rule”
Among the values reported for umbilical blood gas, the pH, P
I recommend using the “20, 30, 40, 50 rule” as a simple tool for remembering normal umbilical artery and vein P
- P
o 2 values are lower than Pco 2 values; thus, the 20 and 30 represent Po 2 values - as fetal umbilical artery P
o 2 is lower than umbilical vein Po 2, 20 mm Hg represents the umbilical artery and 30 mm Hg represents the vein - P
co 2 values are higher in the umbilical artery than in the vein; thus, 50 mm Hg represents the umbilical artery and 40 mm Hg represents the umbilical vein.
Umbilical cord BD values change in relation to labor and FHR decelerations.8 Prior to labor, the normal fetus has a slight degree of acidosis (BD, 2 mmol/L). During the latent phase of labor, fetal BD typically does not change. With the increased frequency of contractions, BD may increase 1 mmol/L for every 3 to 6 hours during the active phase and up to 1 mmol/L per hour during the second stage, depending on FHR responses. Thus, following vaginal delivery the average umbilical artery BD is approximately 5 mmol/L and the umbilical vein BD is approximately 4 mmol/L. As lactate crosses the placenta slowly, BD values are typically only 1 mmol/L less in the umbilical vein than in the artery, unless there has been an obstruction to placental flow (see Case 1).
For pH, the umbilical artery value is always lower than that of the vein, a result of both the higher umbilical artery P
Possible causes of abnormal cord gas values
Because of the nearly fully saturated maternal hemoglobin under normal conditions, fetal arterial and venous P
In contrast, reduced fetal P
Effect of maternal oxygen administration on fetal oxygenation
Although maternal oxygen administration is commonly used during labor and delivery, controversy remains as to the benefit of oxygen supplementation.10 In a normal mother with oxygen saturation above 95%, the administration of oxygen will increase maternal arterial P
However, maternal oxygen supplementation may have marked benefit in cases in which maternal arterial P
How did the Case 1 circumstances lead to newborn acidosis?
Most noticeable in this case is the large difference in BD between the umbilical artery and vein and the high P
Whereas BD normally is only about 1 mmol/L greater in the umbilical artery versus in the vein, occasionally the arterial value is markedly greater than the vein value. This can occur when there is a cessation of blood flow through the placenta, as a result of complete umbilical cord obstruction, or when there is a uterine abruption. In these situations, the umbilical vein (which has not had blood flow) represents the fetal status prior to the occlusion event. In contrast, despite bradycardia, fetal heart pulsations mix blood within the umbilical artery and therefore the artery generally represents the fetal status at the time of birth.
In response to complete cord occlusion, fetal BD increases by approximately 1 mmol/L every 2 minutes. Consequently, an 8 mmol/L difference in BD between the umbilical artery and vein is consistent with a 16-minute period of umbilical occlusion or placental abruption. Also in response to complete umbilical cord occlusion, P
The umbilical vein BD is also elevated for early labor. This value suggests that repetitive, intermittent cord occlusions (evident on the initial fetal monitor tracing) likely resulted in this moderate acidosis prior to the complete cord occlusion in the final 16 minutes.
Thus, BD and P
Read more cases plus procedures, equipment for cord sampling
CASE 2: An infant with unusual umbilical artery values
An infant born via vacuum delivery for a prolonged second stage of labor had 1- and 5-minute Apgar scores of 8 and 9, respectively. Cord gas values were obtained, and analysis revealed that for the umbilical artery, the pH was 7.29; P
The resident asked, “How is the P
The curious Case 2 values suggest an air bubble
Although it is possible that the aberrant values in Case 2 could have resulted from switching the artery and vein samples, the pH is lower in the artery, and both the artery P
Related article:
Is neonatal injury more likely outside of a 30-minute decision-to-incision time interval for cesarean delivery?
CASE 3: A vigorous baby with significant acidosis
A baby with 1- and 5-minute Apgar scores of 9 and 9 was delivered by cesarean and remained vigorous. Umbilical cord analysis revealed an umbilical artery pH level of 7.15, with normal P
Was there a collection error in Case 3?
On occasion, a falsely low pH level and, thus, a falsely elevated BD may result from excessive heparin in the collection syringe. Heparin is acidotic and should be used only to coat the syringe. Although syringes in current use are often pre-heparinized, if one is drawing up heparin into the syringe, it should be coated and then fully expelled.
Umbilical cord sampling: Procedures and equipment
Many issues remain regarding the optimal storage of cord samples. Ideally, a doubly clamped section of the cord promptly should be sampled into glass syringes that can be placed on ice and rapidly measured for cord values.
Stability of umbilical cord samples within the cord is within 20 to 30 minutes. Delayed sampling of clamped cord sections generally has minimal effect on pH and P
Plastic syringes can introduce interference. Several studies have demonstrated that collection of samples in plastic may result in an increase in P
Use glass, and “ice” the sample if necessary. Although it has been suggested that placing samples on ice minimizes metabolism, the cooled plastic may in fact be more susceptible to oxygen diffusion. Thus, unless samples will be analyzed promptly, it is best to use glass syringes on ice.13,14
Related article:
Protecting the newborn brain—the final frontier in obstetric and neonatal care
What if the umbilical cord is torn?
Sometimes the umbilical cord is torn and discarded or cannot be accessed for other reasons. A sample can still be obtained, however, by aspirating the placental surface artery and vein vessels. Although there is some potential variance in pH, P
How do you obtain cord analysis when delaying cord clamping?
The American College of Obstetricians and Gynecologists (ACOG) now advises delayed cord clamping in term and preterm deliveries, which raises the question of how you obtain a blood sample in this setting. Importantly, ACOG recommends delayed cord clamping only in vigorous infants,16 whereas potentially compromised infants should be transferred rapidly for newborn care. Although several studies have demonstrated some variation in cord gas values with delayed cord clamping,17–21 clamping after pulsation has ceased or after the recommended 30 to 60 seconds following birth results in minimal change in BD values. Thus, do not hesitate to perform delayed cord clamping in vigorous infants.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187(1):1–9.
- Uccella S, Cromi A, Colombo G, et al. Prediction of fetal base excess values at birth using an algorithm to interpret fetal heart rate tracings: a retrospective validation. BJOG. 2012;119(13):1657–1664.
- Uccella S, Cromi A, Colombo GF, et al. Interobserver reliability to interpret intrapartum electronic fetal heart rate monitoring: does a standardized algorithm improve agreement among clinicians? J Obstet Gynaecol. 2015;35(3):241–245.
- White CR, Doherty DA, Cannon JW, Kohan R, Newnham JP, Pennell CE. Cost effectiveness of universal umbilical cord blood gas and lactate analysis in a tertiary level maternity unit. J Perinat Med. 2016;44(5):573–584.
- White CR, Doherty DA, Henderson JJ, Kohan R, Newnham JP, Pennell CE. Benefits of introducing universal umbilical cord blood gas and lactate analysis into an obstetric unit. Aust N Z J Obstet Gynaecol. 2010;50(4):318–328.
- Yeomans ER, Hauth JC, Gilstrap LC III, Strickland DM. Umbilical cord pH, Pco2, and bicarbonate following uncomplicated term vaginal deliveries. Am J Obstet Gynecol. 1985;151(6):798–800.
- Wiberg N, Källén K, Olofsson P. Base deficit estimation in umbilical cord blood is influenced by gestational age, choice of fetal fluid compartment, and algorithm for calculation. Am J Obstet Gynecol. 2006;195(6):1651–1656.
- Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187(1):1–9.
- Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123(4):896-901.
- Hamel MS, Anderson BL, Rouse DJ. Oxygen for intrauterine resuscitation: of unproved benefit and potentially harmful. Am J Obstet Gynecol. 2014;211(2):124–127.
- Owen P, Farrell TA, Steyn W. Umbilical cord blood gas analysis; a comparison of two simple methods of sample storage. Early Hum Dev. 1995;42(1):67–71.
- Armstrong L, Stenson B. Effect of delayed sampling on umbilical cord arterial and venous lactate and blood gases in clamped and unclamped vessels. Arch Dis Child Fetal Neonatal Ed. 2006;91(5):F342–F345.
- White CR, Mok T, Doherty DA, Henderson JJ, Newnham JP, Pennell CE. The effect of time, temperature and storage device on umbilical cord blood gas and lactate measurement: a randomized controlled trial. J Matern Fetal Neonatal Med. 2012;25(6):587–594.
- Knowles TP, Mullin RA, Hunter JA, Douce FH. Effects of syringe material, sample storage time, and temperature on blood gases and oxygen saturation in arterialized human blood samples. Respir Care. 2006;51(7):732–736.
- Nodwell A, Carmichael L, Ross M, Richardson B. Placental compared with umbilical cord blood to assess fetal blood gas and acid-base status. Obstet Gynecol. 2005;105(1):129–138.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 684. Delayed umbilical cord clamping after birth. Obstet Gynecol. 2017;129(1):e5–e10.
- De Paco C, Florido J, Garrido MC, Prados S, Navarrete L. Umbilical cord blood acid-base and gas analysis after early versus delayed cord clamping in neonates at term. Arch Gynecol Obstet. 2011;283(5):1011–1014.
- Valero J, Desantes D, Perales-Puchalt A, Rubio J, Diago Almela VJ, Perales A. Effect of delayed umbilical cord clamping on blood gas analysis. Eur J Obstet Gynecol Reprod Biol. 2012;162(1): 21–23.
- Andersson O, Hellström-Westas L, Andersson D, Clausen J, Domellöf M. Effects of delayed compared with early umbilical cord clamping on maternal postpartum hemorrhage and cord blood gas sampling: a randomized trial. Acta Obstet Gynecol Scand. 2013;92(5):567–574.
- Wiberg N, Källén K, Olofsson P. Delayed umbilical cord clamping at birth has effects on arterial and venous blood gases and lactate concentrations. BJOG. 2008;115(6):697–703.
- Mokarami P, Wiberg N, Olofsson P. Hidden acidosis: an explanation of acid-base and lactate changes occurring in umbilical cord blood after delayed sampling. BJOG. 2013;120(8):996–1002.
- Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187(1):1–9.
- Uccella S, Cromi A, Colombo G, et al. Prediction of fetal base excess values at birth using an algorithm to interpret fetal heart rate tracings: a retrospective validation. BJOG. 2012;119(13):1657–1664.
- Uccella S, Cromi A, Colombo GF, et al. Interobserver reliability to interpret intrapartum electronic fetal heart rate monitoring: does a standardized algorithm improve agreement among clinicians? J Obstet Gynaecol. 2015;35(3):241–245.
- White CR, Doherty DA, Cannon JW, Kohan R, Newnham JP, Pennell CE. Cost effectiveness of universal umbilical cord blood gas and lactate analysis in a tertiary level maternity unit. J Perinat Med. 2016;44(5):573–584.
- White CR, Doherty DA, Henderson JJ, Kohan R, Newnham JP, Pennell CE. Benefits of introducing universal umbilical cord blood gas and lactate analysis into an obstetric unit. Aust N Z J Obstet Gynaecol. 2010;50(4):318–328.
- Yeomans ER, Hauth JC, Gilstrap LC III, Strickland DM. Umbilical cord pH, Pco2, and bicarbonate following uncomplicated term vaginal deliveries. Am J Obstet Gynecol. 1985;151(6):798–800.
- Wiberg N, Källén K, Olofsson P. Base deficit estimation in umbilical cord blood is influenced by gestational age, choice of fetal fluid compartment, and algorithm for calculation. Am J Obstet Gynecol. 2006;195(6):1651–1656.
- Ross MG, Gala R. Use of umbilical artery base excess: algorithm for the timing of hypoxic injury. Am J Obstet Gynecol. 2002;187(1):1–9.
- Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123(4):896-901.
- Hamel MS, Anderson BL, Rouse DJ. Oxygen for intrauterine resuscitation: of unproved benefit and potentially harmful. Am J Obstet Gynecol. 2014;211(2):124–127.
- Owen P, Farrell TA, Steyn W. Umbilical cord blood gas analysis; a comparison of two simple methods of sample storage. Early Hum Dev. 1995;42(1):67–71.
- Armstrong L, Stenson B. Effect of delayed sampling on umbilical cord arterial and venous lactate and blood gases in clamped and unclamped vessels. Arch Dis Child Fetal Neonatal Ed. 2006;91(5):F342–F345.
- White CR, Mok T, Doherty DA, Henderson JJ, Newnham JP, Pennell CE. The effect of time, temperature and storage device on umbilical cord blood gas and lactate measurement: a randomized controlled trial. J Matern Fetal Neonatal Med. 2012;25(6):587–594.
- Knowles TP, Mullin RA, Hunter JA, Douce FH. Effects of syringe material, sample storage time, and temperature on blood gases and oxygen saturation in arterialized human blood samples. Respir Care. 2006;51(7):732–736.
- Nodwell A, Carmichael L, Ross M, Richardson B. Placental compared with umbilical cord blood to assess fetal blood gas and acid-base status. Obstet Gynecol. 2005;105(1):129–138.
- American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 684. Delayed umbilical cord clamping after birth. Obstet Gynecol. 2017;129(1):e5–e10.
- De Paco C, Florido J, Garrido MC, Prados S, Navarrete L. Umbilical cord blood acid-base and gas analysis after early versus delayed cord clamping in neonates at term. Arch Gynecol Obstet. 2011;283(5):1011–1014.
- Valero J, Desantes D, Perales-Puchalt A, Rubio J, Diago Almela VJ, Perales A. Effect of delayed umbilical cord clamping on blood gas analysis. Eur J Obstet Gynecol Reprod Biol. 2012;162(1): 21–23.
- Andersson O, Hellström-Westas L, Andersson D, Clausen J, Domellöf M. Effects of delayed compared with early umbilical cord clamping on maternal postpartum hemorrhage and cord blood gas sampling: a randomized trial. Acta Obstet Gynecol Scand. 2013;92(5):567–574.
- Wiberg N, Källén K, Olofsson P. Delayed umbilical cord clamping at birth has effects on arterial and venous blood gases and lactate concentrations. BJOG. 2008;115(6):697–703.
- Mokarami P, Wiberg N, Olofsson P. Hidden acidosis: an explanation of acid-base and lactate changes occurring in umbilical cord blood after delayed sampling. BJOG. 2013;120(8):996–1002.
2017 Update on ovarian cancer
In 2017, an estimated 22,240 women will be diagnosed with ovarian cancer, and 14,080 women will die of the disease.1 The high mortality associated with ovarian cancer is due largely to the inability to detect the disease early and the lack of effective therapeutics for women with recurrent disease. In this Update, we review important advances in the diagnosis and treatment of ovarian cancer.
Development of an effective screening tool for women at average risk has been an elusive challenge. The United Kingdom Collaborative Trial of Ovarian Cancer Screening (UKCTOCS) examined the efficacy of transvaginal ultrasound and cancer antigen 125 (CA 125) monitoring for ovarian cancer in a large cohort of women.
For women diagnosed with ovarian cancer, treatment paradigms for the initial management of the disease have shifted dramatically. Based on data from multiple randomized controlled trials, neoadjuvant chemotherapy (NACT) is being used more frequently. The American Society of Clinical Oncology and the Society of Gynecologic Oncology developed consensus recommendations for the appropriate use of NACT and primary cytoreductive surgery for women with ovarian cancer.
Finally, all of oncology has moved toward incorporating molecularly targeted therapeutics directed toward individual genetic abnormalities in tumors, so-called precision medicine. In ovarian cancer, poly(adenosine diphosphate [ADP]–ribose) polymerase (PARP) has emerged as an important target, particularly for women with BRCA gene pathway mutations. We describe a recently published randomized controlled trial of the PARP inhibitor niraparib.
Read about ovarian cancer screening tests
Is CA 125 or ultrasound screening appropriate for the general population?
Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945-956.
In the United States, the overall ovarian cancer 5-year survival rate is 46.2%, resulting in more than 14,000 deaths annually.2 The poor prognosis associated with this malignancy is largely attributable to the fact that almost 75% of women have stage III or stage IV disease at the time of diagnosis.2 Ovarian cancer is usually associated with vague, nonspecific symptoms as it progresses, which contributes to delayed diagnosis and increased mortality.
Multiple studies have examined pelvic ultrasonography and tumor markers, such as CA 125, as possible screening tools to increase early detection in asymptomatic women. However, neither modality alone or in combination has sufficient sensitivity or specificity to recommend it for use in the general population.3,4 Nevertheless, the search for an appropriate screening tool continues, and the UKCTOCS trial results have reinvigorated this discussion.5
The UKCTOCS findings
The UKCTOCS was a multicenter, randomized controlled trial in the United Kingdom in which researchers allocated 202,638 women aged 50 to 74 years to 1 of 3 groups: annual multimodal screening (MMS) with serum CA 125 interpreted with the use of the risk of ovarian cancer algorithm, annual transvaginal ultrasound screening (USS), or no screening. The median follow-up was more than 11 years.
The investigators found that equivalent rates of ovarian cancer were diagnosed in each group: 0.7% in the MMS group, 0.6% in the USS group, and 0.6% in the no-screening group. Overall, there was no significant reduction in the mortality rate from ovarian cancer in either of the 2 screening groups compared with the no-screening group.5
An important subset discovery
However, in a prespecified subset analysis excluding "prevalent cases" (women with ovarian cancer thought to be present prior to randomization and subsequent screening), ovarian cancer mortality was significantly lower in the MMS group compared with the no-screening group (P = .021). Compared with no screening, MMS was associated with a 20% reduction in mortality rate from ovarian cancer over time, with the most pronounced effects occurring at years 7 to 14 of follow-up, suggesting the possible increased effectiveness of screening over time.5
Related article:
Can CA 125 screening reduce mortality from ovarian cancer?
Concordance with other screening trials
While impressive in study magnitude and scope, the UKCTOCS results did not demonstrate a significant mortality benefit associated with MMS or USS when compared with no screening. Although the screening complications were low (<1% in both screening groups), the authors did note a false-positive surgery rate of 14 per 10,000 screens for the MMS group and 50 per 10,000 screens for the USS group. Based on the performance of screening in this trial, 641 women would need to be screened annually using MMS for 14 years to prevent 1 ovarian cancer death.
Like the UKCTOCS, the ovarian cancer-screening arm of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial in the United States was also unable to demonstrate a reduction in mortality rate with screening with CA 125 and transvaginal ultrasound. Importantly, more than one-third of women with a false-positive screen underwent surgery and 15% of them experienced a major complication.6 Based on these findings, the US Preventive Services Task Force grades screening for ovarian cancer as D, suggesting that the harms of screening may outweigh the benefits.7
While screening for ovarian cancer remains an important need, there is currently no evidence to suggest that serum tumor marker or ultrasound screening is appropriate in the general population. Studies using more specific screening tests or strategies targeted to higher-risk women are ongoing.
Read about patient selection for neoadjuvant chemotherapy
New clinical practice guideline advises neoadjuvant chemotherapy for certain women with ovarian cancer
Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(28):3460-3473.
It has long been held as a central dogma that primary cytoreductive surgery (PCS) is the preferred initial treatment for women with newly diagnosed ovarian cancer.8 However, PCS is associated with substantial morbidity, and the ability to achieve optimal cytoreduction (<1 cm of residual disease), an important prognostic factor, is often compromised in women with significant tumor burden.9,10
Neoadjuvant chemotherapy, in which chemotherapy is administered prior to surgical cytoreduction, challenges the traditional treatment paradigm for advanced-stage ovarian cancer. Several randomized controlled trials have reported equivalent survival for primary surgical cytoreduction and NACT. Importantly, women who received NACT had fewer complications and were more likely to have optimal cytoreduction at the time of surgery.11,12 These studies have limitations, however, and the role of NACT remains uncertain.
To help guide clinicians, the Society of Gynecologic Oncology and the American Society of Clinical Oncology convened an expert panel to provide recommendations and guidance on the evaluation of women for and the use of NACT in the setting of advanced ovarian cancer.13
Related article:
2015 Update on cancer
Recommendation: Clinical evaluation and patient selection
Strong clinical evidence supports that all women with suspected stage IIIC or stage IV ovarian cancer should be evaluated by a gynecologic oncologist prior to the initiation of therapy. The evaluation should include at least a computed tomography scan of the chest, abdomen, and pelvis to assess the extent of disease and resectability. A preoperative risk assessment should be performed to assess risk factors for increased morbidity and mortality.
Women who have a high perioperative risk profile or a low likelihood of achieving cytoreduction to 1 cm or less of residual tumor should receive NACT. Prior to the initiation of NACT, histologic confirmation of ovarian cancer should be obtained.13
Outcomes for neoadjuvant chemotherapy versus primary cytoreduction
Four phase 3 randomized controlled trials (EORTC 55971, CHORUS, JCOG0602, and SCORPION) suggest that NACT is noninferior to PCS with regard to progression-free survival and overall survival. NACT is associated with less perioperative and postoperative morbidity and mortality and is associated with shorter hospital stays.
To date, complete data are available only from the EORTC and CHORUS trials, which both demonstrated similar progression-free survival and overall survival for NACT and PCS. Critics have noted, however, that both trials have shorter median overall survival for the PCS groups than were previously reported in other phase 3 studies in the United States, suggesting the possibility of different patient populations or less aggressive "surgical effort." Thus, PCS remains the preferred management strategy for women with advanced-stage ovarian cancer in whom there is a high likelihood of optimal cytoreduction.13
Recommendation: Use of neoadjuvant chemotherapy
Patients who are appropriate candidates for NACT should be treated with a platinum and taxane doublet and should receive interval cytoreduction following 3 to 4 cycles of therapy if a favorable response is noted. Patients whose disease progresses despite NACT have a poor prognosis, and there is little role for surgical treatment with the exception of palliative purposes.13
Neoadjuvant chemotherapy is a noninferior and appropriate treatment option for women who are poor surgical candidates or who have a low likelihood of optimal cytoreduction. When optimal cytoreduction is possible, however, PCS is preferred (see FIGURE). The data on the efficacy of NACT for ovarian cancer have led to increased use of this treatment in the United States.
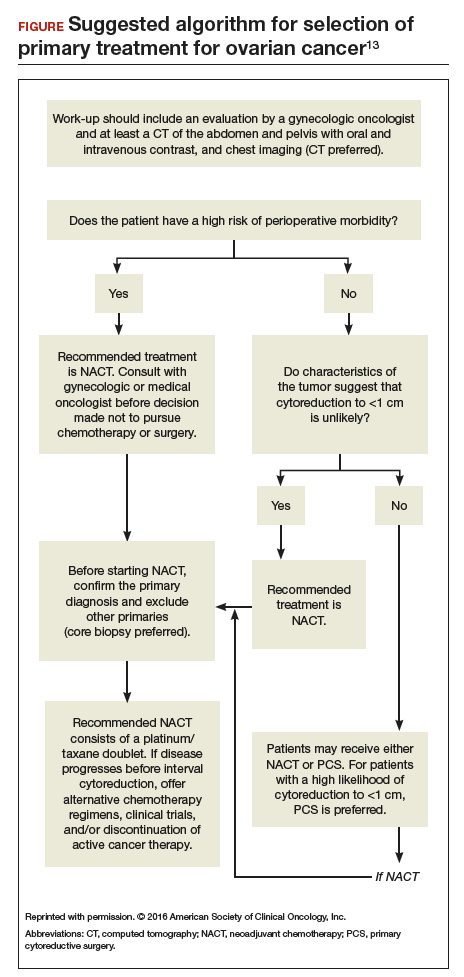
Read about a new PARP inhibitor for maintenance therapy
Niraparib is promising as maintenance therapy in ovarian cancer
Mirza MR, Monk BJ, Herrstedt J, et al; for the ENGOT-OV16/NOVA Investigators. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154-2164.
Approximately 85% of women with ovarian cancer will develop recurrent disease. Women with ovarian cancer are commonly treated with a range of antineoplastic agents over the course of their lifetime. As such, there is a great need for additional active therapeutic agents in this setting. Recently, substantial effort has been directed toward "precision" or "personalized medicine" in oncology.
Precision medicine, targeted therapies in oncology
Precision medicine refers to the customization of medical therapy based on the genetic characterization of the individual patient or the molecular profile of the patient's tumor. As a result of large-scale molecular profiling from projects such as the International Cancer Genome Consortium and The Cancer Genome Atlas, an abundance of molecular data has been generated through the characterization of multiple tumor types. This has led to the discovery of key cancer drivers, alterations, and specific molecular profiles that have distinct prognostic and treatment implications. These data, in combination with the commercial availability of molecular profiling tests, has made precision medicine a reality for women with ovarian cancer.
This wealth of new information has led to development of targeted therapeutics that block the growth and spread of cancer by acting on specific molecules or molecular pathways. Targeted therapies approved for cancer treatment include hormonal therapies, signal transduction inhibitors, gene expression modulators, apoptosis inducers, angiogenesis inhibitors, and immunotherapies.14
How PARP inhibitors work
PARP inhibitors are a class of agents that are emerging as important therapies for ovarian cancer. These agents block the nuclear protein PARP, which functions to detect and repair single-strand DNA breaks with the resulting accumulation of double-stranded DNA breaks.15 In the setting of DNA damage, the homologous recombination repair pathway is activated for repair. However, homologous recombination deficiencies (HRD) can arise as a result of BRCA1 or BRCA2 mutations or BRCA-independent pathways, which effectively disable this DNA repair pathway. As a result, when PARP inhibitors are used in patients with HRD, the cell cannot repair double-stranded DNA breaks and this leads to "synthetic lethality."16
Understanding this molecular mechanism of PARP inhibitors as well as the frequent abnormalities in the BRCA genes and HRD pathways in ovarian cancer has provided an important potential therapeutic target in ovarian cancer. A number of PARP inhibitors are now commercially available and are undergoing testing in ovarian cancer.
Related article:
Is a minimally invasive approach to hysterectomy for Gyn cancer utilized equally in all racial and income groups?
Niraparib for ovarian cancer
In a randomized, double-blind, phase 3 trial by Mizra and colleagues, 553 women with platinum-sensitive recurrent ovarian cancer who responded to therapy were divided according to the presence or absence of a germline BRCA (gBRCA) mutation and randomly assigned to niraparib 300 mg or placebo once daily. Women in the niraparib group had a significantly longer median duration of progression-free survival than did those in the placebo group. This was most pronounced in women in the gBRCA cohort (21.0 vs 5.5 months). Importantly, niraparib was associated with improved progression-free survival in HRD-positive patients without gBRCA mutations (12.9 vs 3.8 months) as well as in the HRD-negative subgroup (6.9 vs 3.8 months).17
Overall, niraparib was well tolerated. About 15% of women discontinued the drug due to toxicity. Significant (grade 3 or 4) adverse events were seen in three-quarters of women treated with niraparib, and they most commonly consisted of hematologic toxicities. Patient-reported outcomes were similar for both groups, indicating no significant effect from niraparib on quality of life.17
This study's results suggest that niraparib has clinical activity against ovarian cancer. Importantly, niraparib was active in women with gBRCA mutations, in those with HRD without a gBRCA mutation, and potentially in women without HRD. If approved by the US Food and Drug Administration, niraparib will join olaparib and rucaparib as a newly approved therapeutic agent for ovarian cancer. This study provides important evidence that suggests niraparib maintenance therapy may be an efficacious and important addition to the treatment armamentarium for platinum-sensitive ovarian cancer.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: ovarian cancer. https://seer.cancer.gov/statfacts/html/ovary.html. Accessed January 20, 2017.
- Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ. 1993;306(6884):1030–1034.
- van Nagell JR Jr, Pavlik EJ. Ovarian cancer screening. Clin Obstet Gynecol. 2012;55:43–51.
- Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945–956.
- Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening randomized controlled trial. JAMA. 2011;305(22):2295–2303.
- Moyer VA, US Preventive Services Task Force. Screening for ovarian cancer: US Preventive Services Task Force reaffirmation recommendation statement. Ann Intern Med. 2012;157(12):900–904.
- Schorge JO, McCann C, Del Carmen MG. Surgical debulking of ovarian cancer: what difference does it make? Rev Obstet Gynecol. 2010;3(3):111–117.
- Hoskins WJ, McGuire WP, Brady MF, et al. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am J Obstet Gynecol. 1994;170(4):974–979; discussion 979–980.
- Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol. 2002;20(5):1248–1259.
- Vergote I, Trope CG, Amant F, et al; European Organization for Research and Treatment of Cancer–Gynaecological Cancer Goup; NCIC Clinical Trials Group. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med. 2010;363(10):943–953.
- Kehoe S, Hook J, Nankivell M, et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet. 2015;386(9990):249–257.
- Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(28):3460–3473.
- National Cancer Institute. Targeted cancer therapies. https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet. Updated April 25, 2014. Accessed January 21, 2017.
- Drean A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85.
- Ledermann JA, El-Khouly F. PARP inhibitors in ovarian cancer: clinical evidence for informed treatment decisions. Br J Cancer. 2015;113(suppl 1):S10–S16.
- Mirza MR, Monk BJ, Herrstedt J, et al; ENGOT-OV16/NOVA Investigators. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164.

Dr. Wright is Sol Goldman Associate Professor, Chief of Division of Gynecologic Oncology, Vice Chair of Academic Affairs, Department of Obstetrics and Gynecology, Columbia University College of Physicians and Surgeons, New York, New York.

Dr. Buskwofie is Fellow in the Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, New York–Presbyterian/Weill Cornell Medical College, and the Columbia University Medical Center, New York, New York.
Dr. Wright reports that he is a consultant to Clovis Oncology and Tesaro, Inc. Dr. Buskwofie reports no financial relationships relevant to this article.
Dr. Wright is Sol Goldman Associate Professor, Chief of Division of Gynecologic Oncology, Vice Chair of Academic Affairs, Department of Obstetrics and Gynecology, Columbia University College of Physicians and Surgeons, New York, New York.
Dr. Buskwofie is Fellow in the Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, New York–Presbyterian/Weill Cornell Medical College, and the Columbia University Medical Center, New York, New York.
Dr. Wright reports that he is a consultant to Clovis Oncology and Tesaro, Inc. Dr. Buskwofie reports no financial relationships relevant to this article.
Dr. Wright is Sol Goldman Associate Professor, Chief of Division of Gynecologic Oncology, Vice Chair of Academic Affairs, Department of Obstetrics and Gynecology, Columbia University College of Physicians and Surgeons, New York, New York.
Dr. Buskwofie is Fellow in the Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, New York–Presbyterian/Weill Cornell Medical College, and the Columbia University Medical Center, New York, New York.
Dr. Wright reports that he is a consultant to Clovis Oncology and Tesaro, Inc. Dr. Buskwofie reports no financial relationships relevant to this article.
In 2017, an estimated 22,240 women will be diagnosed with ovarian cancer, and 14,080 women will die of the disease.1 The high mortality associated with ovarian cancer is due largely to the inability to detect the disease early and the lack of effective therapeutics for women with recurrent disease. In this Update, we review important advances in the diagnosis and treatment of ovarian cancer.
Development of an effective screening tool for women at average risk has been an elusive challenge. The United Kingdom Collaborative Trial of Ovarian Cancer Screening (UKCTOCS) examined the efficacy of transvaginal ultrasound and cancer antigen 125 (CA 125) monitoring for ovarian cancer in a large cohort of women.
For women diagnosed with ovarian cancer, treatment paradigms for the initial management of the disease have shifted dramatically. Based on data from multiple randomized controlled trials, neoadjuvant chemotherapy (NACT) is being used more frequently. The American Society of Clinical Oncology and the Society of Gynecologic Oncology developed consensus recommendations for the appropriate use of NACT and primary cytoreductive surgery for women with ovarian cancer.
Finally, all of oncology has moved toward incorporating molecularly targeted therapeutics directed toward individual genetic abnormalities in tumors, so-called precision medicine. In ovarian cancer, poly(adenosine diphosphate [ADP]–ribose) polymerase (PARP) has emerged as an important target, particularly for women with BRCA gene pathway mutations. We describe a recently published randomized controlled trial of the PARP inhibitor niraparib.
Read about ovarian cancer screening tests
Is CA 125 or ultrasound screening appropriate for the general population?
Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945-956.
In the United States, the overall ovarian cancer 5-year survival rate is 46.2%, resulting in more than 14,000 deaths annually.2 The poor prognosis associated with this malignancy is largely attributable to the fact that almost 75% of women have stage III or stage IV disease at the time of diagnosis.2 Ovarian cancer is usually associated with vague, nonspecific symptoms as it progresses, which contributes to delayed diagnosis and increased mortality.
Multiple studies have examined pelvic ultrasonography and tumor markers, such as CA 125, as possible screening tools to increase early detection in asymptomatic women. However, neither modality alone or in combination has sufficient sensitivity or specificity to recommend it for use in the general population.3,4 Nevertheless, the search for an appropriate screening tool continues, and the UKCTOCS trial results have reinvigorated this discussion.5
The UKCTOCS findings
The UKCTOCS was a multicenter, randomized controlled trial in the United Kingdom in which researchers allocated 202,638 women aged 50 to 74 years to 1 of 3 groups: annual multimodal screening (MMS) with serum CA 125 interpreted with the use of the risk of ovarian cancer algorithm, annual transvaginal ultrasound screening (USS), or no screening. The median follow-up was more than 11 years.
The investigators found that equivalent rates of ovarian cancer were diagnosed in each group: 0.7% in the MMS group, 0.6% in the USS group, and 0.6% in the no-screening group. Overall, there was no significant reduction in the mortality rate from ovarian cancer in either of the 2 screening groups compared with the no-screening group.5
An important subset discovery
However, in a prespecified subset analysis excluding "prevalent cases" (women with ovarian cancer thought to be present prior to randomization and subsequent screening), ovarian cancer mortality was significantly lower in the MMS group compared with the no-screening group (P = .021). Compared with no screening, MMS was associated with a 20% reduction in mortality rate from ovarian cancer over time, with the most pronounced effects occurring at years 7 to 14 of follow-up, suggesting the possible increased effectiveness of screening over time.5
Related article:
Can CA 125 screening reduce mortality from ovarian cancer?
Concordance with other screening trials
While impressive in study magnitude and scope, the UKCTOCS results did not demonstrate a significant mortality benefit associated with MMS or USS when compared with no screening. Although the screening complications were low (<1% in both screening groups), the authors did note a false-positive surgery rate of 14 per 10,000 screens for the MMS group and 50 per 10,000 screens for the USS group. Based on the performance of screening in this trial, 641 women would need to be screened annually using MMS for 14 years to prevent 1 ovarian cancer death.
Like the UKCTOCS, the ovarian cancer-screening arm of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial in the United States was also unable to demonstrate a reduction in mortality rate with screening with CA 125 and transvaginal ultrasound. Importantly, more than one-third of women with a false-positive screen underwent surgery and 15% of them experienced a major complication.6 Based on these findings, the US Preventive Services Task Force grades screening for ovarian cancer as D, suggesting that the harms of screening may outweigh the benefits.7
While screening for ovarian cancer remains an important need, there is currently no evidence to suggest that serum tumor marker or ultrasound screening is appropriate in the general population. Studies using more specific screening tests or strategies targeted to higher-risk women are ongoing.
Read about patient selection for neoadjuvant chemotherapy
New clinical practice guideline advises neoadjuvant chemotherapy for certain women with ovarian cancer
Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(28):3460-3473.
It has long been held as a central dogma that primary cytoreductive surgery (PCS) is the preferred initial treatment for women with newly diagnosed ovarian cancer.8 However, PCS is associated with substantial morbidity, and the ability to achieve optimal cytoreduction (<1 cm of residual disease), an important prognostic factor, is often compromised in women with significant tumor burden.9,10
Neoadjuvant chemotherapy, in which chemotherapy is administered prior to surgical cytoreduction, challenges the traditional treatment paradigm for advanced-stage ovarian cancer. Several randomized controlled trials have reported equivalent survival for primary surgical cytoreduction and NACT. Importantly, women who received NACT had fewer complications and were more likely to have optimal cytoreduction at the time of surgery.11,12 These studies have limitations, however, and the role of NACT remains uncertain.
To help guide clinicians, the Society of Gynecologic Oncology and the American Society of Clinical Oncology convened an expert panel to provide recommendations and guidance on the evaluation of women for and the use of NACT in the setting of advanced ovarian cancer.13
Related article:
2015 Update on cancer
Recommendation: Clinical evaluation and patient selection
Strong clinical evidence supports that all women with suspected stage IIIC or stage IV ovarian cancer should be evaluated by a gynecologic oncologist prior to the initiation of therapy. The evaluation should include at least a computed tomography scan of the chest, abdomen, and pelvis to assess the extent of disease and resectability. A preoperative risk assessment should be performed to assess risk factors for increased morbidity and mortality.
Women who have a high perioperative risk profile or a low likelihood of achieving cytoreduction to 1 cm or less of residual tumor should receive NACT. Prior to the initiation of NACT, histologic confirmation of ovarian cancer should be obtained.13
Outcomes for neoadjuvant chemotherapy versus primary cytoreduction
Four phase 3 randomized controlled trials (EORTC 55971, CHORUS, JCOG0602, and SCORPION) suggest that NACT is noninferior to PCS with regard to progression-free survival and overall survival. NACT is associated with less perioperative and postoperative morbidity and mortality and is associated with shorter hospital stays.
To date, complete data are available only from the EORTC and CHORUS trials, which both demonstrated similar progression-free survival and overall survival for NACT and PCS. Critics have noted, however, that both trials have shorter median overall survival for the PCS groups than were previously reported in other phase 3 studies in the United States, suggesting the possibility of different patient populations or less aggressive "surgical effort." Thus, PCS remains the preferred management strategy for women with advanced-stage ovarian cancer in whom there is a high likelihood of optimal cytoreduction.13
Recommendation: Use of neoadjuvant chemotherapy
Patients who are appropriate candidates for NACT should be treated with a platinum and taxane doublet and should receive interval cytoreduction following 3 to 4 cycles of therapy if a favorable response is noted. Patients whose disease progresses despite NACT have a poor prognosis, and there is little role for surgical treatment with the exception of palliative purposes.13
Neoadjuvant chemotherapy is a noninferior and appropriate treatment option for women who are poor surgical candidates or who have a low likelihood of optimal cytoreduction. When optimal cytoreduction is possible, however, PCS is preferred (see FIGURE). The data on the efficacy of NACT for ovarian cancer have led to increased use of this treatment in the United States.
Read about a new PARP inhibitor for maintenance therapy
Niraparib is promising as maintenance therapy in ovarian cancer
Mirza MR, Monk BJ, Herrstedt J, et al; for the ENGOT-OV16/NOVA Investigators. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154-2164.
Approximately 85% of women with ovarian cancer will develop recurrent disease. Women with ovarian cancer are commonly treated with a range of antineoplastic agents over the course of their lifetime. As such, there is a great need for additional active therapeutic agents in this setting. Recently, substantial effort has been directed toward "precision" or "personalized medicine" in oncology.
Precision medicine, targeted therapies in oncology
Precision medicine refers to the customization of medical therapy based on the genetic characterization of the individual patient or the molecular profile of the patient's tumor. As a result of large-scale molecular profiling from projects such as the International Cancer Genome Consortium and The Cancer Genome Atlas, an abundance of molecular data has been generated through the characterization of multiple tumor types. This has led to the discovery of key cancer drivers, alterations, and specific molecular profiles that have distinct prognostic and treatment implications. These data, in combination with the commercial availability of molecular profiling tests, has made precision medicine a reality for women with ovarian cancer.
This wealth of new information has led to development of targeted therapeutics that block the growth and spread of cancer by acting on specific molecules or molecular pathways. Targeted therapies approved for cancer treatment include hormonal therapies, signal transduction inhibitors, gene expression modulators, apoptosis inducers, angiogenesis inhibitors, and immunotherapies.14
How PARP inhibitors work
PARP inhibitors are a class of agents that are emerging as important therapies for ovarian cancer. These agents block the nuclear protein PARP, which functions to detect and repair single-strand DNA breaks with the resulting accumulation of double-stranded DNA breaks.15 In the setting of DNA damage, the homologous recombination repair pathway is activated for repair. However, homologous recombination deficiencies (HRD) can arise as a result of BRCA1 or BRCA2 mutations or BRCA-independent pathways, which effectively disable this DNA repair pathway. As a result, when PARP inhibitors are used in patients with HRD, the cell cannot repair double-stranded DNA breaks and this leads to "synthetic lethality."16
Understanding this molecular mechanism of PARP inhibitors as well as the frequent abnormalities in the BRCA genes and HRD pathways in ovarian cancer has provided an important potential therapeutic target in ovarian cancer. A number of PARP inhibitors are now commercially available and are undergoing testing in ovarian cancer.
Related article:
Is a minimally invasive approach to hysterectomy for Gyn cancer utilized equally in all racial and income groups?
Niraparib for ovarian cancer
In a randomized, double-blind, phase 3 trial by Mizra and colleagues, 553 women with platinum-sensitive recurrent ovarian cancer who responded to therapy were divided according to the presence or absence of a germline BRCA (gBRCA) mutation and randomly assigned to niraparib 300 mg or placebo once daily. Women in the niraparib group had a significantly longer median duration of progression-free survival than did those in the placebo group. This was most pronounced in women in the gBRCA cohort (21.0 vs 5.5 months). Importantly, niraparib was associated with improved progression-free survival in HRD-positive patients without gBRCA mutations (12.9 vs 3.8 months) as well as in the HRD-negative subgroup (6.9 vs 3.8 months).17
Overall, niraparib was well tolerated. About 15% of women discontinued the drug due to toxicity. Significant (grade 3 or 4) adverse events were seen in three-quarters of women treated with niraparib, and they most commonly consisted of hematologic toxicities. Patient-reported outcomes were similar for both groups, indicating no significant effect from niraparib on quality of life.17
This study's results suggest that niraparib has clinical activity against ovarian cancer. Importantly, niraparib was active in women with gBRCA mutations, in those with HRD without a gBRCA mutation, and potentially in women without HRD. If approved by the US Food and Drug Administration, niraparib will join olaparib and rucaparib as a newly approved therapeutic agent for ovarian cancer. This study provides important evidence that suggests niraparib maintenance therapy may be an efficacious and important addition to the treatment armamentarium for platinum-sensitive ovarian cancer.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In 2017, an estimated 22,240 women will be diagnosed with ovarian cancer, and 14,080 women will die of the disease.1 The high mortality associated with ovarian cancer is due largely to the inability to detect the disease early and the lack of effective therapeutics for women with recurrent disease. In this Update, we review important advances in the diagnosis and treatment of ovarian cancer.
Development of an effective screening tool for women at average risk has been an elusive challenge. The United Kingdom Collaborative Trial of Ovarian Cancer Screening (UKCTOCS) examined the efficacy of transvaginal ultrasound and cancer antigen 125 (CA 125) monitoring for ovarian cancer in a large cohort of women.
For women diagnosed with ovarian cancer, treatment paradigms for the initial management of the disease have shifted dramatically. Based on data from multiple randomized controlled trials, neoadjuvant chemotherapy (NACT) is being used more frequently. The American Society of Clinical Oncology and the Society of Gynecologic Oncology developed consensus recommendations for the appropriate use of NACT and primary cytoreductive surgery for women with ovarian cancer.
Finally, all of oncology has moved toward incorporating molecularly targeted therapeutics directed toward individual genetic abnormalities in tumors, so-called precision medicine. In ovarian cancer, poly(adenosine diphosphate [ADP]–ribose) polymerase (PARP) has emerged as an important target, particularly for women with BRCA gene pathway mutations. We describe a recently published randomized controlled trial of the PARP inhibitor niraparib.
Read about ovarian cancer screening tests
Is CA 125 or ultrasound screening appropriate for the general population?
Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945-956.
In the United States, the overall ovarian cancer 5-year survival rate is 46.2%, resulting in more than 14,000 deaths annually.2 The poor prognosis associated with this malignancy is largely attributable to the fact that almost 75% of women have stage III or stage IV disease at the time of diagnosis.2 Ovarian cancer is usually associated with vague, nonspecific symptoms as it progresses, which contributes to delayed diagnosis and increased mortality.
Multiple studies have examined pelvic ultrasonography and tumor markers, such as CA 125, as possible screening tools to increase early detection in asymptomatic women. However, neither modality alone or in combination has sufficient sensitivity or specificity to recommend it for use in the general population.3,4 Nevertheless, the search for an appropriate screening tool continues, and the UKCTOCS trial results have reinvigorated this discussion.5
The UKCTOCS findings
The UKCTOCS was a multicenter, randomized controlled trial in the United Kingdom in which researchers allocated 202,638 women aged 50 to 74 years to 1 of 3 groups: annual multimodal screening (MMS) with serum CA 125 interpreted with the use of the risk of ovarian cancer algorithm, annual transvaginal ultrasound screening (USS), or no screening. The median follow-up was more than 11 years.
The investigators found that equivalent rates of ovarian cancer were diagnosed in each group: 0.7% in the MMS group, 0.6% in the USS group, and 0.6% in the no-screening group. Overall, there was no significant reduction in the mortality rate from ovarian cancer in either of the 2 screening groups compared with the no-screening group.5
An important subset discovery
However, in a prespecified subset analysis excluding "prevalent cases" (women with ovarian cancer thought to be present prior to randomization and subsequent screening), ovarian cancer mortality was significantly lower in the MMS group compared with the no-screening group (P = .021). Compared with no screening, MMS was associated with a 20% reduction in mortality rate from ovarian cancer over time, with the most pronounced effects occurring at years 7 to 14 of follow-up, suggesting the possible increased effectiveness of screening over time.5
Related article:
Can CA 125 screening reduce mortality from ovarian cancer?
Concordance with other screening trials
While impressive in study magnitude and scope, the UKCTOCS results did not demonstrate a significant mortality benefit associated with MMS or USS when compared with no screening. Although the screening complications were low (<1% in both screening groups), the authors did note a false-positive surgery rate of 14 per 10,000 screens for the MMS group and 50 per 10,000 screens for the USS group. Based on the performance of screening in this trial, 641 women would need to be screened annually using MMS for 14 years to prevent 1 ovarian cancer death.
Like the UKCTOCS, the ovarian cancer-screening arm of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial in the United States was also unable to demonstrate a reduction in mortality rate with screening with CA 125 and transvaginal ultrasound. Importantly, more than one-third of women with a false-positive screen underwent surgery and 15% of them experienced a major complication.6 Based on these findings, the US Preventive Services Task Force grades screening for ovarian cancer as D, suggesting that the harms of screening may outweigh the benefits.7
While screening for ovarian cancer remains an important need, there is currently no evidence to suggest that serum tumor marker or ultrasound screening is appropriate in the general population. Studies using more specific screening tests or strategies targeted to higher-risk women are ongoing.
Read about patient selection for neoadjuvant chemotherapy
New clinical practice guideline advises neoadjuvant chemotherapy for certain women with ovarian cancer
Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(28):3460-3473.
It has long been held as a central dogma that primary cytoreductive surgery (PCS) is the preferred initial treatment for women with newly diagnosed ovarian cancer.8 However, PCS is associated with substantial morbidity, and the ability to achieve optimal cytoreduction (<1 cm of residual disease), an important prognostic factor, is often compromised in women with significant tumor burden.9,10
Neoadjuvant chemotherapy, in which chemotherapy is administered prior to surgical cytoreduction, challenges the traditional treatment paradigm for advanced-stage ovarian cancer. Several randomized controlled trials have reported equivalent survival for primary surgical cytoreduction and NACT. Importantly, women who received NACT had fewer complications and were more likely to have optimal cytoreduction at the time of surgery.11,12 These studies have limitations, however, and the role of NACT remains uncertain.
To help guide clinicians, the Society of Gynecologic Oncology and the American Society of Clinical Oncology convened an expert panel to provide recommendations and guidance on the evaluation of women for and the use of NACT in the setting of advanced ovarian cancer.13
Related article:
2015 Update on cancer
Recommendation: Clinical evaluation and patient selection
Strong clinical evidence supports that all women with suspected stage IIIC or stage IV ovarian cancer should be evaluated by a gynecologic oncologist prior to the initiation of therapy. The evaluation should include at least a computed tomography scan of the chest, abdomen, and pelvis to assess the extent of disease and resectability. A preoperative risk assessment should be performed to assess risk factors for increased morbidity and mortality.
Women who have a high perioperative risk profile or a low likelihood of achieving cytoreduction to 1 cm or less of residual tumor should receive NACT. Prior to the initiation of NACT, histologic confirmation of ovarian cancer should be obtained.13
Outcomes for neoadjuvant chemotherapy versus primary cytoreduction
Four phase 3 randomized controlled trials (EORTC 55971, CHORUS, JCOG0602, and SCORPION) suggest that NACT is noninferior to PCS with regard to progression-free survival and overall survival. NACT is associated with less perioperative and postoperative morbidity and mortality and is associated with shorter hospital stays.
To date, complete data are available only from the EORTC and CHORUS trials, which both demonstrated similar progression-free survival and overall survival for NACT and PCS. Critics have noted, however, that both trials have shorter median overall survival for the PCS groups than were previously reported in other phase 3 studies in the United States, suggesting the possibility of different patient populations or less aggressive "surgical effort." Thus, PCS remains the preferred management strategy for women with advanced-stage ovarian cancer in whom there is a high likelihood of optimal cytoreduction.13
Recommendation: Use of neoadjuvant chemotherapy
Patients who are appropriate candidates for NACT should be treated with a platinum and taxane doublet and should receive interval cytoreduction following 3 to 4 cycles of therapy if a favorable response is noted. Patients whose disease progresses despite NACT have a poor prognosis, and there is little role for surgical treatment with the exception of palliative purposes.13
Neoadjuvant chemotherapy is a noninferior and appropriate treatment option for women who are poor surgical candidates or who have a low likelihood of optimal cytoreduction. When optimal cytoreduction is possible, however, PCS is preferred (see FIGURE). The data on the efficacy of NACT for ovarian cancer have led to increased use of this treatment in the United States.
Read about a new PARP inhibitor for maintenance therapy
Niraparib is promising as maintenance therapy in ovarian cancer
Mirza MR, Monk BJ, Herrstedt J, et al; for the ENGOT-OV16/NOVA Investigators. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154-2164.
Approximately 85% of women with ovarian cancer will develop recurrent disease. Women with ovarian cancer are commonly treated with a range of antineoplastic agents over the course of their lifetime. As such, there is a great need for additional active therapeutic agents in this setting. Recently, substantial effort has been directed toward "precision" or "personalized medicine" in oncology.
Precision medicine, targeted therapies in oncology
Precision medicine refers to the customization of medical therapy based on the genetic characterization of the individual patient or the molecular profile of the patient's tumor. As a result of large-scale molecular profiling from projects such as the International Cancer Genome Consortium and The Cancer Genome Atlas, an abundance of molecular data has been generated through the characterization of multiple tumor types. This has led to the discovery of key cancer drivers, alterations, and specific molecular profiles that have distinct prognostic and treatment implications. These data, in combination with the commercial availability of molecular profiling tests, has made precision medicine a reality for women with ovarian cancer.
This wealth of new information has led to development of targeted therapeutics that block the growth and spread of cancer by acting on specific molecules or molecular pathways. Targeted therapies approved for cancer treatment include hormonal therapies, signal transduction inhibitors, gene expression modulators, apoptosis inducers, angiogenesis inhibitors, and immunotherapies.14
How PARP inhibitors work
PARP inhibitors are a class of agents that are emerging as important therapies for ovarian cancer. These agents block the nuclear protein PARP, which functions to detect and repair single-strand DNA breaks with the resulting accumulation of double-stranded DNA breaks.15 In the setting of DNA damage, the homologous recombination repair pathway is activated for repair. However, homologous recombination deficiencies (HRD) can arise as a result of BRCA1 or BRCA2 mutations or BRCA-independent pathways, which effectively disable this DNA repair pathway. As a result, when PARP inhibitors are used in patients with HRD, the cell cannot repair double-stranded DNA breaks and this leads to "synthetic lethality."16
Understanding this molecular mechanism of PARP inhibitors as well as the frequent abnormalities in the BRCA genes and HRD pathways in ovarian cancer has provided an important potential therapeutic target in ovarian cancer. A number of PARP inhibitors are now commercially available and are undergoing testing in ovarian cancer.
Related article:
Is a minimally invasive approach to hysterectomy for Gyn cancer utilized equally in all racial and income groups?
Niraparib for ovarian cancer
In a randomized, double-blind, phase 3 trial by Mizra and colleagues, 553 women with platinum-sensitive recurrent ovarian cancer who responded to therapy were divided according to the presence or absence of a germline BRCA (gBRCA) mutation and randomly assigned to niraparib 300 mg or placebo once daily. Women in the niraparib group had a significantly longer median duration of progression-free survival than did those in the placebo group. This was most pronounced in women in the gBRCA cohort (21.0 vs 5.5 months). Importantly, niraparib was associated with improved progression-free survival in HRD-positive patients without gBRCA mutations (12.9 vs 3.8 months) as well as in the HRD-negative subgroup (6.9 vs 3.8 months).17
Overall, niraparib was well tolerated. About 15% of women discontinued the drug due to toxicity. Significant (grade 3 or 4) adverse events were seen in three-quarters of women treated with niraparib, and they most commonly consisted of hematologic toxicities. Patient-reported outcomes were similar for both groups, indicating no significant effect from niraparib on quality of life.17
This study's results suggest that niraparib has clinical activity against ovarian cancer. Importantly, niraparib was active in women with gBRCA mutations, in those with HRD without a gBRCA mutation, and potentially in women without HRD. If approved by the US Food and Drug Administration, niraparib will join olaparib and rucaparib as a newly approved therapeutic agent for ovarian cancer. This study provides important evidence that suggests niraparib maintenance therapy may be an efficacious and important addition to the treatment armamentarium for platinum-sensitive ovarian cancer.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: ovarian cancer. https://seer.cancer.gov/statfacts/html/ovary.html. Accessed January 20, 2017.
- Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ. 1993;306(6884):1030–1034.
- van Nagell JR Jr, Pavlik EJ. Ovarian cancer screening. Clin Obstet Gynecol. 2012;55:43–51.
- Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945–956.
- Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening randomized controlled trial. JAMA. 2011;305(22):2295–2303.
- Moyer VA, US Preventive Services Task Force. Screening for ovarian cancer: US Preventive Services Task Force reaffirmation recommendation statement. Ann Intern Med. 2012;157(12):900–904.
- Schorge JO, McCann C, Del Carmen MG. Surgical debulking of ovarian cancer: what difference does it make? Rev Obstet Gynecol. 2010;3(3):111–117.
- Hoskins WJ, McGuire WP, Brady MF, et al. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am J Obstet Gynecol. 1994;170(4):974–979; discussion 979–980.
- Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol. 2002;20(5):1248–1259.
- Vergote I, Trope CG, Amant F, et al; European Organization for Research and Treatment of Cancer–Gynaecological Cancer Goup; NCIC Clinical Trials Group. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med. 2010;363(10):943–953.
- Kehoe S, Hook J, Nankivell M, et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet. 2015;386(9990):249–257.
- Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(28):3460–3473.
- National Cancer Institute. Targeted cancer therapies. https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet. Updated April 25, 2014. Accessed January 21, 2017.
- Drean A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85.
- Ledermann JA, El-Khouly F. PARP inhibitors in ovarian cancer: clinical evidence for informed treatment decisions. Br J Cancer. 2015;113(suppl 1):S10–S16.
- Mirza MR, Monk BJ, Herrstedt J, et al; ENGOT-OV16/NOVA Investigators. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: ovarian cancer. https://seer.cancer.gov/statfacts/html/ovary.html. Accessed January 20, 2017.
- Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ. 1993;306(6884):1030–1034.
- van Nagell JR Jr, Pavlik EJ. Ovarian cancer screening. Clin Obstet Gynecol. 2012;55:43–51.
- Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet. 2016;387(10022):945–956.
- Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening randomized controlled trial. JAMA. 2011;305(22):2295–2303.
- Moyer VA, US Preventive Services Task Force. Screening for ovarian cancer: US Preventive Services Task Force reaffirmation recommendation statement. Ann Intern Med. 2012;157(12):900–904.
- Schorge JO, McCann C, Del Carmen MG. Surgical debulking of ovarian cancer: what difference does it make? Rev Obstet Gynecol. 2010;3(3):111–117.
- Hoskins WJ, McGuire WP, Brady MF, et al. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am J Obstet Gynecol. 1994;170(4):974–979; discussion 979–980.
- Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol. 2002;20(5):1248–1259.
- Vergote I, Trope CG, Amant F, et al; European Organization for Research and Treatment of Cancer–Gynaecological Cancer Goup; NCIC Clinical Trials Group. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med. 2010;363(10):943–953.
- Kehoe S, Hook J, Nankivell M, et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet. 2015;386(9990):249–257.
- Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(28):3460–3473.
- National Cancer Institute. Targeted cancer therapies. https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet. Updated April 25, 2014. Accessed January 21, 2017.
- Drean A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85.
- Ledermann JA, El-Khouly F. PARP inhibitors in ovarian cancer: clinical evidence for informed treatment decisions. Br J Cancer. 2015;113(suppl 1):S10–S16.
- Mirza MR, Monk BJ, Herrstedt J, et al; ENGOT-OV16/NOVA Investigators. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164.
Anticoagulation Management Outcomes in Veterans: Office vs Telephone Visits
Oral anticoagulation with warfarin is used for the treatment and prevention of a variety of thrombotic disorders, including deep venous thrombosis (DVT), pulmonary embolism (PE), stroke prevention in atrial fibrillation (AF) and atrial flutter, and other hypercoagulable conditions. Although a mainstay in the treatment for these conditions, warfarin requires close monitoring due to its narrow therapeutic range, extensive drug and dietary interactions, and dosage variability among patients.1 Patients outside the therapeutic range are at risk of having a thrombotic or bleeding event that could lead to hospitalization or fatality.1 To reduce the risk of these events, patients on warfarin are managed by dose adjustment based on the international normalized ratio (INR). Research has shown that patients on warfarin in pharmacist-managed specialty anticoagulation clinics have more consistent monitoring and lower rates of adverse events (AEs) compared with traditional physician or nurse clinics.2-6 Management through these clinics can be achieved through office visits or telephone visits.
There are advantages and disadvantages to each model of anticoagulation management for patients.Telephone clinics provide time and cost savings, increased access to care, and convenience. However, disadvantages include missed phone calls or inability to contact the patient, difficulty for the patient to hear the provider’s instructions over the phone, and patient unavailability when a critical INR is of concern. Office visits are beneficial in that providers can provide both written and verbal instruction to patients, perform visual or physical patient assessments, and provide timely care if needed. Disadvantages of office visits may include long wait times and inconvenience for patients who live far away.
Telephone anticoagulation clinics have been evaluated for their efficacy and cost-effectiveness in several studies.5,7,8 However, few studies are available that compare patient outcomes between office visits and telephone visits. Two prior studies comparing groups of anticoagulation patients managed by telephone or by office visit concluded that there is no difference in outcomes between the 2 management models.9,10 However, a retrospective study by Stoudenmire and colleagues examined extreme INR values (≤ 1.5 or ≥ 4.5) in each management model and found that telephone clinic patients have a significant increase in extreme INR values but no difference in AEs between the 2 management models.11
The VA North Texas Health Care System (VANTHCS) includes a major medical center, 3 outlying medical facilities, and 5 community-based outpatient clinics (CBOCs). A centralized pharmacist-managed anticoagulation clinic is used to manage more than 2,500 VANTHCS anticoagulation patients. To meet the National Patient Safety Goal measures and provide consistent management across the system, all anticoagulation patients from CBOCs and medical facilities are enrolled in the clinic.12 To facilitate access to care, many patients transitioned from office visits to telephone visits. It was essential to evaluate the transition of patients from office to telephone visits to ensure continued stability and continuity of care across both models. The objective of this study was to determine whether a difference in anticoagulation outcomes exists when patients are transitioned from office to telephone visits.
Methods
The VANTHCS anticoagulation clinic policy for office visits requires that patients arrive at the Dallas VAMC 2 hours before their appointment for INR lab draw. During the office visit, the anticoagulation pharmacist evaluates the INR and pertinent changes since the previous visit. The patient is provided verbal instructions and a written dosage adjustment card. Telephone clinic protocol is similar to office visits with a few exceptions. Any patient, regardless of INR stability, may be enrolled in the telephone clinic as long as the patient provides consent and has a working telephone with voice mail. Patients enrolled in the telephone clinic access blood draws at the nearest VA facility and are given a questionnaire that includes pertinent questions asked during an office visit. Anticoagulation pharmacists evaluate the questionnaire and INR then contact the patient within 1 business day to provide the patient with instructions. If a patient fails to answer the telephone, the anticoagulation pharmacist leaves a voicemail message.
Study Design
This retrospective study was conducted by chart review using Computerized Patient Record System (CPRS) at VANTHCS on patients who met inclusion criteria between January 1, 2011 and May 31, 2014, and it was approved by the institutional review board and research and development committee. The study included patients aged ≥ 18 years on warfarin therapy managed by the VANTHCS anticoagulation clinic who were previously managed in office visits for ≥ 180 days before the telephone transition, then in telephone visits for another ≥ 180 days. Only INR values obtained through the VANTHCS anticoagulation clinic were assessed.
Patients were excluded from the study if they were not managed by the VANTHCS anticoagulation clinic or received direct oral anticoagulants (DOACs). The INR values were excluded if they were nonclinic related INR values (ie, results reported that do not reflect management by the anticoagulation clinic), the first INR after hospitalization, or INRs obtained during the first month of initial warfarin treatment for a patient.
For all patients included in the study, demographic information, goal INR range (2 to 3 or 2.5 to 3.5), indication for warfarin therapy, and duration of warfarin therapy (defined as the first prescription filled for warfarin at the VA) were obtained. Individual INR values were obtained for each patient during the period of investigation and type of visit (office or telephone) for each INR drawn was specified. Any major bleeding or thrombotic events (bleed requiring an emergency department [ED] visit, hospitalization, vitamin K administration, blood transfusion, and/or warfarin therapy hold/discontinuation) were documented. Procedures and number of hospitalizations also during the investigation were recorded.
The primary outcomes measures evaluated INRs for time in therapeutic range (TTR) using the Rosendaal method and percentage of INRs within range.13 The therapeutic range was either 2 to 3 or 2.5 to 3.5 (the “strict range” for INR management). Because many patients fluctuate around the strict range and it is common to avoid therapy adjustment based on slightly elevated or lower values, a “nonstrict” range (1.8 to 3.2 or 2.3 to 3.7) also was evaluated.14 The secondary outcomes examined differences between the 2 management models in rates of major AEs, including thrombosis and major bleeding events as defined earlier.Frequencies, percentages, and other descriptive statistics were used to describe nominal data. A paired t test was used to compare TTR of patients transitioned from office to telephone visits. A P value of < .05 was used for statistical significance.
Results
A total of 111 patients met inclusion criteria (Table 1). Most patients were elderly males with AF or atrial flutter as their primary indication for warfarin therapy. No statistically significant difference was found for percentage INRs in strict range (56.8% in office vs 56.9% in telephone, P = .98) or TTR (65.9% in office vs 62.72% in telephone, P = .23) for patients who transitioned from office to telephone visits (Table 2). Similar results were found within the nonstrict range. 
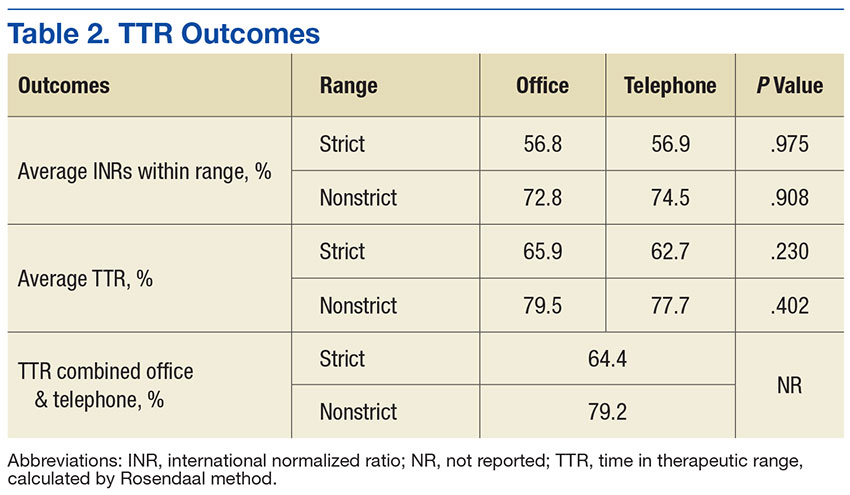
In examining safety, 5 major AEs occurred. One patient had 2 thrombotic pulmonary embolism events. This patient had a history of nonadherence with warfarin therapy. Three major bleeding events occurred (2 in the telephone group and 1 in the office group). Two bleeding events led to ED visits, and 1 event led to hospitalization. Although 43% of patients had a procedure during the study period, only a portion of patients received bridging with low-molecular-weight heparin (LMWH). None of the 3 reported bleeding events discovered during the study were associated with recent LMWH use. No events were fatal (Table 3).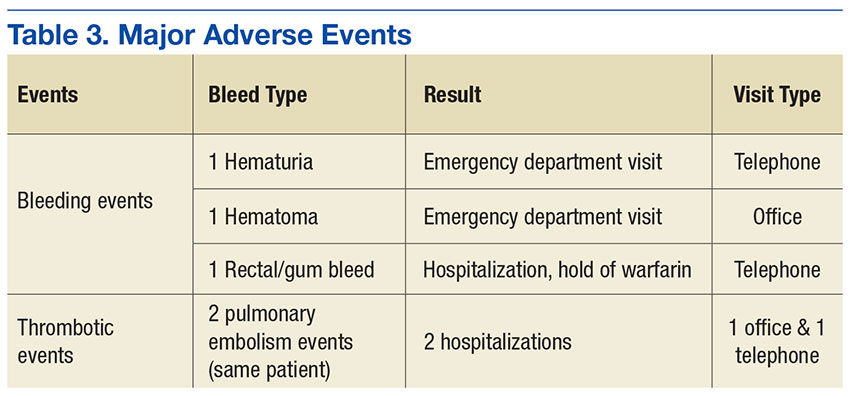
Discussion
This study demonstrates that patients transitioned from office to telephone visits for warfarin management will have no significant change in their TTR. Additionally, patients had similar rates of major AEs before and after transition, although there were few events overall.
Previous research comparing anticoagulation outcomes in telephone vs office visits also has described outcomes to be similar between these 2 management models. Wittkowsky and colleagues examined 2 university-affiliated clinics to evaluate warfarin outcomes and AEs in patients in each management model (office vs telephone) and found no difference in outcomes between the 2 management models.9
Staresinic and colleagues designed a prospective study of 192 patients to evaluate TTR and AEs of the 2 management models at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin.10 This study found no difference between the 2 groups in percentage of time maintained within INR range or AEs and concluded that the telephone model was effective for anticoagulant management.
A retrospective study by Stoudenmire and colleagues evaluated office vs telephone management effects on extreme INR values (≤ 1.5 or ≥ 4.5), TTR, and AEs.11 This study found overall TTR and AEs to be similar between groups, but the telephone clinic had a 2-fold increase in extreme INR values compared with the office clinic.11
The current study differs from the previously discussed studies in that it evaluated outcomes for the same patients before and after the transition to telephone. This study did not exclude specific patients from telephone clinic. In the Wittkowsky study, patients were enrolled in the telephone clinic based on criteria such as patient disability or living long distances from the clinic.9 Additionally, in the current study, patients transitioned to telephone visits did not have scheduled office visits for anticoagulation management. In contrast, patients in the Staresinic study had routine anticoagulation office visits every 3 months, thus it was not a true telephone-only clinic.10
This study’s findings support prior studies’ findings that telephone clinics are acceptable for anticoagulation management. Furthermore, safety does not seem to be affected when transitioning patients, although there were few AEs to review. Providers can use telephone clinics to potentially decrease cost and facilitate access to care for patients.
Limitations
Patients were required to be in office and telephone for a sequential 6 months, and this may have produced selection biases toward patients who adhered to appointments and who were on long-term warfarin therapy. Many patients that were excluded from the study transitioned back and forth between the 2 management models. Due to the retrospective nature of this study, the authors were unable to control for all confounding variables. Patients also were not randomly assigned to be transitioned from office to telephone. Although a strength of this study was the limited telephone clinic selection criteria, there may be a few individual situations in which the pharmacist’s clinical judgment influenced the transition to the telephone clinic, creating selection bias.
There may be time bias present as clinical guidelines, providers, and clinic population size differed over the study period and might have influenced management. The population of VA patients was mainly elderly males; therefore, the study results may not be applicable to other populations. Last, the results of the study are reflective of the VANTHCS clinic structure and may not be applicable to other clinic designs.
Conclusion
Veterans in a pharmacist-managed anticoagulation clinic experienced the same outcomes in terms of TTR and major AEs when transitioned from the traditional face-to-face office visits to telephone visits. The study supports the safety and efficacy of transitioning patients from a pharmacist-managed anticoagulation office clinic to telephone clinic.
1. Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(suppl 6):160S-198S.
2. Rudd KM, Dier JG. Comparison of two different models of anticoagulation management services with usual medical care. Pharmacotherapy. 2010;30(4):330-338.
3. Bungard TJ, Gardner L, Archer SL, et al. Evaluation of a pharmacist-managed anticoagulation clinic: improving patient care. Open Med. 2009;3(1):e16-e21.
4. Chiquette E, Amato MG, Bussey HI. Comparison of an anticoagulation clinic with usual medical care: anticoagulation control, patient outcomes, and health care costs. Arch Intern Med. 1998;158(15):1641-1647.
5. Waterman AD, Banet G, Milligan PE, et al. Patient and physician satisfaction with a telephone-based anticoagulation service. J Gen Intern Med. 2001;16(7):460-463.
6. Hasan SS, Shamala R, Syed IA, et al. Factors affecting warfarin-related knowledge and INR control of patients attending physician- and pharmacist-managed anticoagulation clinics. J Pharm Pract. 2011;24(5):485-493.
7. Hassan S, Naboush A, Radbel J, et al. Telephone-based anticoagulation management in the homebound setting: a retrospective observational study. Int J Gen Med. 2013;6:869-875.
8. Moherman LJ, Kolar MM. Complication rates for a telephone-based anticoagulation service. Am J Health Syst Pharm. 1999;56(15):1540-1542.
9. Wittkowsky AK, Nutescu EA, Blackburn J, et al. Outcomes of oral anticoagulant therapy managed by telephone vs in-office visits in an anticoagulation clinic setting. Chest. 2006;130(5):1385-1389.
10. Staresinic AG, Sorkness CA, Goodman BM, Pigarelli DW. Comparison of outcomes using 2 delivery models of anticoagulation care. Arch Intern Med. 2006;166(9):997-1002.
11. Stoudenmire LG, DeRemer CE, Elewa H. Telephone versus office-based management of warfarin: impact on international normalized ratios and outcomes. Int J Hematol. 2014;100(2):119-124.
12. The Joint Commission. National Patient Safety Goals Effective January 1, 2015. http://www.jointcommission.org/assets/1/6/2015_NPSG_AHC1.PDF. Published 2014. Accessed November 23, 2016.
13. Rosendaal FR, Cannegieter SC, van der Meer FJ, Briët E. A method to determine the optimal intensity of oral anticoagulant therapy. Thromb Haemost. 1993;69(3):236-239.
14. Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(suppl 2):7S-47S.
Oral anticoagulation with warfarin is used for the treatment and prevention of a variety of thrombotic disorders, including deep venous thrombosis (DVT), pulmonary embolism (PE), stroke prevention in atrial fibrillation (AF) and atrial flutter, and other hypercoagulable conditions. Although a mainstay in the treatment for these conditions, warfarin requires close monitoring due to its narrow therapeutic range, extensive drug and dietary interactions, and dosage variability among patients.1 Patients outside the therapeutic range are at risk of having a thrombotic or bleeding event that could lead to hospitalization or fatality.1 To reduce the risk of these events, patients on warfarin are managed by dose adjustment based on the international normalized ratio (INR). Research has shown that patients on warfarin in pharmacist-managed specialty anticoagulation clinics have more consistent monitoring and lower rates of adverse events (AEs) compared with traditional physician or nurse clinics.2-6 Management through these clinics can be achieved through office visits or telephone visits.
There are advantages and disadvantages to each model of anticoagulation management for patients.Telephone clinics provide time and cost savings, increased access to care, and convenience. However, disadvantages include missed phone calls or inability to contact the patient, difficulty for the patient to hear the provider’s instructions over the phone, and patient unavailability when a critical INR is of concern. Office visits are beneficial in that providers can provide both written and verbal instruction to patients, perform visual or physical patient assessments, and provide timely care if needed. Disadvantages of office visits may include long wait times and inconvenience for patients who live far away.
Telephone anticoagulation clinics have been evaluated for their efficacy and cost-effectiveness in several studies.5,7,8 However, few studies are available that compare patient outcomes between office visits and telephone visits. Two prior studies comparing groups of anticoagulation patients managed by telephone or by office visit concluded that there is no difference in outcomes between the 2 management models.9,10 However, a retrospective study by Stoudenmire and colleagues examined extreme INR values (≤ 1.5 or ≥ 4.5) in each management model and found that telephone clinic patients have a significant increase in extreme INR values but no difference in AEs between the 2 management models.11
The VA North Texas Health Care System (VANTHCS) includes a major medical center, 3 outlying medical facilities, and 5 community-based outpatient clinics (CBOCs). A centralized pharmacist-managed anticoagulation clinic is used to manage more than 2,500 VANTHCS anticoagulation patients. To meet the National Patient Safety Goal measures and provide consistent management across the system, all anticoagulation patients from CBOCs and medical facilities are enrolled in the clinic.12 To facilitate access to care, many patients transitioned from office visits to telephone visits. It was essential to evaluate the transition of patients from office to telephone visits to ensure continued stability and continuity of care across both models. The objective of this study was to determine whether a difference in anticoagulation outcomes exists when patients are transitioned from office to telephone visits.
Methods
The VANTHCS anticoagulation clinic policy for office visits requires that patients arrive at the Dallas VAMC 2 hours before their appointment for INR lab draw. During the office visit, the anticoagulation pharmacist evaluates the INR and pertinent changes since the previous visit. The patient is provided verbal instructions and a written dosage adjustment card. Telephone clinic protocol is similar to office visits with a few exceptions. Any patient, regardless of INR stability, may be enrolled in the telephone clinic as long as the patient provides consent and has a working telephone with voice mail. Patients enrolled in the telephone clinic access blood draws at the nearest VA facility and are given a questionnaire that includes pertinent questions asked during an office visit. Anticoagulation pharmacists evaluate the questionnaire and INR then contact the patient within 1 business day to provide the patient with instructions. If a patient fails to answer the telephone, the anticoagulation pharmacist leaves a voicemail message.
Study Design
This retrospective study was conducted by chart review using Computerized Patient Record System (CPRS) at VANTHCS on patients who met inclusion criteria between January 1, 2011 and May 31, 2014, and it was approved by the institutional review board and research and development committee. The study included patients aged ≥ 18 years on warfarin therapy managed by the VANTHCS anticoagulation clinic who were previously managed in office visits for ≥ 180 days before the telephone transition, then in telephone visits for another ≥ 180 days. Only INR values obtained through the VANTHCS anticoagulation clinic were assessed.
Patients were excluded from the study if they were not managed by the VANTHCS anticoagulation clinic or received direct oral anticoagulants (DOACs). The INR values were excluded if they were nonclinic related INR values (ie, results reported that do not reflect management by the anticoagulation clinic), the first INR after hospitalization, or INRs obtained during the first month of initial warfarin treatment for a patient.
For all patients included in the study, demographic information, goal INR range (2 to 3 or 2.5 to 3.5), indication for warfarin therapy, and duration of warfarin therapy (defined as the first prescription filled for warfarin at the VA) were obtained. Individual INR values were obtained for each patient during the period of investigation and type of visit (office or telephone) for each INR drawn was specified. Any major bleeding or thrombotic events (bleed requiring an emergency department [ED] visit, hospitalization, vitamin K administration, blood transfusion, and/or warfarin therapy hold/discontinuation) were documented. Procedures and number of hospitalizations also during the investigation were recorded.
The primary outcomes measures evaluated INRs for time in therapeutic range (TTR) using the Rosendaal method and percentage of INRs within range.13 The therapeutic range was either 2 to 3 or 2.5 to 3.5 (the “strict range” for INR management). Because many patients fluctuate around the strict range and it is common to avoid therapy adjustment based on slightly elevated or lower values, a “nonstrict” range (1.8 to 3.2 or 2.3 to 3.7) also was evaluated.14 The secondary outcomes examined differences between the 2 management models in rates of major AEs, including thrombosis and major bleeding events as defined earlier.Frequencies, percentages, and other descriptive statistics were used to describe nominal data. A paired t test was used to compare TTR of patients transitioned from office to telephone visits. A P value of < .05 was used for statistical significance.
Results
A total of 111 patients met inclusion criteria (Table 1). Most patients were elderly males with AF or atrial flutter as their primary indication for warfarin therapy. No statistically significant difference was found for percentage INRs in strict range (56.8% in office vs 56.9% in telephone, P = .98) or TTR (65.9% in office vs 62.72% in telephone, P = .23) for patients who transitioned from office to telephone visits (Table 2). Similar results were found within the nonstrict range.
In examining safety, 5 major AEs occurred. One patient had 2 thrombotic pulmonary embolism events. This patient had a history of nonadherence with warfarin therapy. Three major bleeding events occurred (2 in the telephone group and 1 in the office group). Two bleeding events led to ED visits, and 1 event led to hospitalization. Although 43% of patients had a procedure during the study period, only a portion of patients received bridging with low-molecular-weight heparin (LMWH). None of the 3 reported bleeding events discovered during the study were associated with recent LMWH use. No events were fatal (Table 3).
Discussion
This study demonstrates that patients transitioned from office to telephone visits for warfarin management will have no significant change in their TTR. Additionally, patients had similar rates of major AEs before and after transition, although there were few events overall.
Previous research comparing anticoagulation outcomes in telephone vs office visits also has described outcomes to be similar between these 2 management models. Wittkowsky and colleagues examined 2 university-affiliated clinics to evaluate warfarin outcomes and AEs in patients in each management model (office vs telephone) and found no difference in outcomes between the 2 management models.9
Staresinic and colleagues designed a prospective study of 192 patients to evaluate TTR and AEs of the 2 management models at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin.10 This study found no difference between the 2 groups in percentage of time maintained within INR range or AEs and concluded that the telephone model was effective for anticoagulant management.
A retrospective study by Stoudenmire and colleagues evaluated office vs telephone management effects on extreme INR values (≤ 1.5 or ≥ 4.5), TTR, and AEs.11 This study found overall TTR and AEs to be similar between groups, but the telephone clinic had a 2-fold increase in extreme INR values compared with the office clinic.11
The current study differs from the previously discussed studies in that it evaluated outcomes for the same patients before and after the transition to telephone. This study did not exclude specific patients from telephone clinic. In the Wittkowsky study, patients were enrolled in the telephone clinic based on criteria such as patient disability or living long distances from the clinic.9 Additionally, in the current study, patients transitioned to telephone visits did not have scheduled office visits for anticoagulation management. In contrast, patients in the Staresinic study had routine anticoagulation office visits every 3 months, thus it was not a true telephone-only clinic.10
This study’s findings support prior studies’ findings that telephone clinics are acceptable for anticoagulation management. Furthermore, safety does not seem to be affected when transitioning patients, although there were few AEs to review. Providers can use telephone clinics to potentially decrease cost and facilitate access to care for patients.
Limitations
Patients were required to be in office and telephone for a sequential 6 months, and this may have produced selection biases toward patients who adhered to appointments and who were on long-term warfarin therapy. Many patients that were excluded from the study transitioned back and forth between the 2 management models. Due to the retrospective nature of this study, the authors were unable to control for all confounding variables. Patients also were not randomly assigned to be transitioned from office to telephone. Although a strength of this study was the limited telephone clinic selection criteria, there may be a few individual situations in which the pharmacist’s clinical judgment influenced the transition to the telephone clinic, creating selection bias.
There may be time bias present as clinical guidelines, providers, and clinic population size differed over the study period and might have influenced management. The population of VA patients was mainly elderly males; therefore, the study results may not be applicable to other populations. Last, the results of the study are reflective of the VANTHCS clinic structure and may not be applicable to other clinic designs.
Conclusion
Veterans in a pharmacist-managed anticoagulation clinic experienced the same outcomes in terms of TTR and major AEs when transitioned from the traditional face-to-face office visits to telephone visits. The study supports the safety and efficacy of transitioning patients from a pharmacist-managed anticoagulation office clinic to telephone clinic.
Oral anticoagulation with warfarin is used for the treatment and prevention of a variety of thrombotic disorders, including deep venous thrombosis (DVT), pulmonary embolism (PE), stroke prevention in atrial fibrillation (AF) and atrial flutter, and other hypercoagulable conditions. Although a mainstay in the treatment for these conditions, warfarin requires close monitoring due to its narrow therapeutic range, extensive drug and dietary interactions, and dosage variability among patients.1 Patients outside the therapeutic range are at risk of having a thrombotic or bleeding event that could lead to hospitalization or fatality.1 To reduce the risk of these events, patients on warfarin are managed by dose adjustment based on the international normalized ratio (INR). Research has shown that patients on warfarin in pharmacist-managed specialty anticoagulation clinics have more consistent monitoring and lower rates of adverse events (AEs) compared with traditional physician or nurse clinics.2-6 Management through these clinics can be achieved through office visits or telephone visits.
There are advantages and disadvantages to each model of anticoagulation management for patients.Telephone clinics provide time and cost savings, increased access to care, and convenience. However, disadvantages include missed phone calls or inability to contact the patient, difficulty for the patient to hear the provider’s instructions over the phone, and patient unavailability when a critical INR is of concern. Office visits are beneficial in that providers can provide both written and verbal instruction to patients, perform visual or physical patient assessments, and provide timely care if needed. Disadvantages of office visits may include long wait times and inconvenience for patients who live far away.
Telephone anticoagulation clinics have been evaluated for their efficacy and cost-effectiveness in several studies.5,7,8 However, few studies are available that compare patient outcomes between office visits and telephone visits. Two prior studies comparing groups of anticoagulation patients managed by telephone or by office visit concluded that there is no difference in outcomes between the 2 management models.9,10 However, a retrospective study by Stoudenmire and colleagues examined extreme INR values (≤ 1.5 or ≥ 4.5) in each management model and found that telephone clinic patients have a significant increase in extreme INR values but no difference in AEs between the 2 management models.11
The VA North Texas Health Care System (VANTHCS) includes a major medical center, 3 outlying medical facilities, and 5 community-based outpatient clinics (CBOCs). A centralized pharmacist-managed anticoagulation clinic is used to manage more than 2,500 VANTHCS anticoagulation patients. To meet the National Patient Safety Goal measures and provide consistent management across the system, all anticoagulation patients from CBOCs and medical facilities are enrolled in the clinic.12 To facilitate access to care, many patients transitioned from office visits to telephone visits. It was essential to evaluate the transition of patients from office to telephone visits to ensure continued stability and continuity of care across both models. The objective of this study was to determine whether a difference in anticoagulation outcomes exists when patients are transitioned from office to telephone visits.
Methods
The VANTHCS anticoagulation clinic policy for office visits requires that patients arrive at the Dallas VAMC 2 hours before their appointment for INR lab draw. During the office visit, the anticoagulation pharmacist evaluates the INR and pertinent changes since the previous visit. The patient is provided verbal instructions and a written dosage adjustment card. Telephone clinic protocol is similar to office visits with a few exceptions. Any patient, regardless of INR stability, may be enrolled in the telephone clinic as long as the patient provides consent and has a working telephone with voice mail. Patients enrolled in the telephone clinic access blood draws at the nearest VA facility and are given a questionnaire that includes pertinent questions asked during an office visit. Anticoagulation pharmacists evaluate the questionnaire and INR then contact the patient within 1 business day to provide the patient with instructions. If a patient fails to answer the telephone, the anticoagulation pharmacist leaves a voicemail message.
Study Design
This retrospective study was conducted by chart review using Computerized Patient Record System (CPRS) at VANTHCS on patients who met inclusion criteria between January 1, 2011 and May 31, 2014, and it was approved by the institutional review board and research and development committee. The study included patients aged ≥ 18 years on warfarin therapy managed by the VANTHCS anticoagulation clinic who were previously managed in office visits for ≥ 180 days before the telephone transition, then in telephone visits for another ≥ 180 days. Only INR values obtained through the VANTHCS anticoagulation clinic were assessed.
Patients were excluded from the study if they were not managed by the VANTHCS anticoagulation clinic or received direct oral anticoagulants (DOACs). The INR values were excluded if they were nonclinic related INR values (ie, results reported that do not reflect management by the anticoagulation clinic), the first INR after hospitalization, or INRs obtained during the first month of initial warfarin treatment for a patient.
For all patients included in the study, demographic information, goal INR range (2 to 3 or 2.5 to 3.5), indication for warfarin therapy, and duration of warfarin therapy (defined as the first prescription filled for warfarin at the VA) were obtained. Individual INR values were obtained for each patient during the period of investigation and type of visit (office or telephone) for each INR drawn was specified. Any major bleeding or thrombotic events (bleed requiring an emergency department [ED] visit, hospitalization, vitamin K administration, blood transfusion, and/or warfarin therapy hold/discontinuation) were documented. Procedures and number of hospitalizations also during the investigation were recorded.
The primary outcomes measures evaluated INRs for time in therapeutic range (TTR) using the Rosendaal method and percentage of INRs within range.13 The therapeutic range was either 2 to 3 or 2.5 to 3.5 (the “strict range” for INR management). Because many patients fluctuate around the strict range and it is common to avoid therapy adjustment based on slightly elevated or lower values, a “nonstrict” range (1.8 to 3.2 or 2.3 to 3.7) also was evaluated.14 The secondary outcomes examined differences between the 2 management models in rates of major AEs, including thrombosis and major bleeding events as defined earlier.Frequencies, percentages, and other descriptive statistics were used to describe nominal data. A paired t test was used to compare TTR of patients transitioned from office to telephone visits. A P value of < .05 was used for statistical significance.
Results
A total of 111 patients met inclusion criteria (Table 1). Most patients were elderly males with AF or atrial flutter as their primary indication for warfarin therapy. No statistically significant difference was found for percentage INRs in strict range (56.8% in office vs 56.9% in telephone, P = .98) or TTR (65.9% in office vs 62.72% in telephone, P = .23) for patients who transitioned from office to telephone visits (Table 2). Similar results were found within the nonstrict range.
In examining safety, 5 major AEs occurred. One patient had 2 thrombotic pulmonary embolism events. This patient had a history of nonadherence with warfarin therapy. Three major bleeding events occurred (2 in the telephone group and 1 in the office group). Two bleeding events led to ED visits, and 1 event led to hospitalization. Although 43% of patients had a procedure during the study period, only a portion of patients received bridging with low-molecular-weight heparin (LMWH). None of the 3 reported bleeding events discovered during the study were associated with recent LMWH use. No events were fatal (Table 3).
Discussion
This study demonstrates that patients transitioned from office to telephone visits for warfarin management will have no significant change in their TTR. Additionally, patients had similar rates of major AEs before and after transition, although there were few events overall.
Previous research comparing anticoagulation outcomes in telephone vs office visits also has described outcomes to be similar between these 2 management models. Wittkowsky and colleagues examined 2 university-affiliated clinics to evaluate warfarin outcomes and AEs in patients in each management model (office vs telephone) and found no difference in outcomes between the 2 management models.9
Staresinic and colleagues designed a prospective study of 192 patients to evaluate TTR and AEs of the 2 management models at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin.10 This study found no difference between the 2 groups in percentage of time maintained within INR range or AEs and concluded that the telephone model was effective for anticoagulant management.
A retrospective study by Stoudenmire and colleagues evaluated office vs telephone management effects on extreme INR values (≤ 1.5 or ≥ 4.5), TTR, and AEs.11 This study found overall TTR and AEs to be similar between groups, but the telephone clinic had a 2-fold increase in extreme INR values compared with the office clinic.11
The current study differs from the previously discussed studies in that it evaluated outcomes for the same patients before and after the transition to telephone. This study did not exclude specific patients from telephone clinic. In the Wittkowsky study, patients were enrolled in the telephone clinic based on criteria such as patient disability or living long distances from the clinic.9 Additionally, in the current study, patients transitioned to telephone visits did not have scheduled office visits for anticoagulation management. In contrast, patients in the Staresinic study had routine anticoagulation office visits every 3 months, thus it was not a true telephone-only clinic.10
This study’s findings support prior studies’ findings that telephone clinics are acceptable for anticoagulation management. Furthermore, safety does not seem to be affected when transitioning patients, although there were few AEs to review. Providers can use telephone clinics to potentially decrease cost and facilitate access to care for patients.
Limitations
Patients were required to be in office and telephone for a sequential 6 months, and this may have produced selection biases toward patients who adhered to appointments and who were on long-term warfarin therapy. Many patients that were excluded from the study transitioned back and forth between the 2 management models. Due to the retrospective nature of this study, the authors were unable to control for all confounding variables. Patients also were not randomly assigned to be transitioned from office to telephone. Although a strength of this study was the limited telephone clinic selection criteria, there may be a few individual situations in which the pharmacist’s clinical judgment influenced the transition to the telephone clinic, creating selection bias.
There may be time bias present as clinical guidelines, providers, and clinic population size differed over the study period and might have influenced management. The population of VA patients was mainly elderly males; therefore, the study results may not be applicable to other populations. Last, the results of the study are reflective of the VANTHCS clinic structure and may not be applicable to other clinic designs.
Conclusion
Veterans in a pharmacist-managed anticoagulation clinic experienced the same outcomes in terms of TTR and major AEs when transitioned from the traditional face-to-face office visits to telephone visits. The study supports the safety and efficacy of transitioning patients from a pharmacist-managed anticoagulation office clinic to telephone clinic.
1. Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(suppl 6):160S-198S.
2. Rudd KM, Dier JG. Comparison of two different models of anticoagulation management services with usual medical care. Pharmacotherapy. 2010;30(4):330-338.
3. Bungard TJ, Gardner L, Archer SL, et al. Evaluation of a pharmacist-managed anticoagulation clinic: improving patient care. Open Med. 2009;3(1):e16-e21.
4. Chiquette E, Amato MG, Bussey HI. Comparison of an anticoagulation clinic with usual medical care: anticoagulation control, patient outcomes, and health care costs. Arch Intern Med. 1998;158(15):1641-1647.
5. Waterman AD, Banet G, Milligan PE, et al. Patient and physician satisfaction with a telephone-based anticoagulation service. J Gen Intern Med. 2001;16(7):460-463.
6. Hasan SS, Shamala R, Syed IA, et al. Factors affecting warfarin-related knowledge and INR control of patients attending physician- and pharmacist-managed anticoagulation clinics. J Pharm Pract. 2011;24(5):485-493.
7. Hassan S, Naboush A, Radbel J, et al. Telephone-based anticoagulation management in the homebound setting: a retrospective observational study. Int J Gen Med. 2013;6:869-875.
8. Moherman LJ, Kolar MM. Complication rates for a telephone-based anticoagulation service. Am J Health Syst Pharm. 1999;56(15):1540-1542.
9. Wittkowsky AK, Nutescu EA, Blackburn J, et al. Outcomes of oral anticoagulant therapy managed by telephone vs in-office visits in an anticoagulation clinic setting. Chest. 2006;130(5):1385-1389.
10. Staresinic AG, Sorkness CA, Goodman BM, Pigarelli DW. Comparison of outcomes using 2 delivery models of anticoagulation care. Arch Intern Med. 2006;166(9):997-1002.
11. Stoudenmire LG, DeRemer CE, Elewa H. Telephone versus office-based management of warfarin: impact on international normalized ratios and outcomes. Int J Hematol. 2014;100(2):119-124.
12. The Joint Commission. National Patient Safety Goals Effective January 1, 2015. http://www.jointcommission.org/assets/1/6/2015_NPSG_AHC1.PDF. Published 2014. Accessed November 23, 2016.
13. Rosendaal FR, Cannegieter SC, van der Meer FJ, Briët E. A method to determine the optimal intensity of oral anticoagulant therapy. Thromb Haemost. 1993;69(3):236-239.
14. Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(suppl 2):7S-47S.
1. Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(suppl 6):160S-198S.
2. Rudd KM, Dier JG. Comparison of two different models of anticoagulation management services with usual medical care. Pharmacotherapy. 2010;30(4):330-338.
3. Bungard TJ, Gardner L, Archer SL, et al. Evaluation of a pharmacist-managed anticoagulation clinic: improving patient care. Open Med. 2009;3(1):e16-e21.
4. Chiquette E, Amato MG, Bussey HI. Comparison of an anticoagulation clinic with usual medical care: anticoagulation control, patient outcomes, and health care costs. Arch Intern Med. 1998;158(15):1641-1647.
5. Waterman AD, Banet G, Milligan PE, et al. Patient and physician satisfaction with a telephone-based anticoagulation service. J Gen Intern Med. 2001;16(7):460-463.
6. Hasan SS, Shamala R, Syed IA, et al. Factors affecting warfarin-related knowledge and INR control of patients attending physician- and pharmacist-managed anticoagulation clinics. J Pharm Pract. 2011;24(5):485-493.
7. Hassan S, Naboush A, Radbel J, et al. Telephone-based anticoagulation management in the homebound setting: a retrospective observational study. Int J Gen Med. 2013;6:869-875.
8. Moherman LJ, Kolar MM. Complication rates for a telephone-based anticoagulation service. Am J Health Syst Pharm. 1999;56(15):1540-1542.
9. Wittkowsky AK, Nutescu EA, Blackburn J, et al. Outcomes of oral anticoagulant therapy managed by telephone vs in-office visits in an anticoagulation clinic setting. Chest. 2006;130(5):1385-1389.
10. Staresinic AG, Sorkness CA, Goodman BM, Pigarelli DW. Comparison of outcomes using 2 delivery models of anticoagulation care. Arch Intern Med. 2006;166(9):997-1002.
11. Stoudenmire LG, DeRemer CE, Elewa H. Telephone versus office-based management of warfarin: impact on international normalized ratios and outcomes. Int J Hematol. 2014;100(2):119-124.
12. The Joint Commission. National Patient Safety Goals Effective January 1, 2015. http://www.jointcommission.org/assets/1/6/2015_NPSG_AHC1.PDF. Published 2014. Accessed November 23, 2016.
13. Rosendaal FR, Cannegieter SC, van der Meer FJ, Briët E. A method to determine the optimal intensity of oral anticoagulant therapy. Thromb Haemost. 1993;69(3):236-239.
14. Guyatt GH, Akl EA, Crowther M, Gutterman DD, Schuünemann HJ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(suppl 2):7S-47S.
Management of Proximal Biceps Pathology in Overhead Athletes: What Is the Role of Biceps Tenodesis?
Take Home Points
- Outcomes after SLAP repair remain guarded.
- Physical examination is key in determining proper management of biceps pathology.
- When performing SLAP repair, knotless technology may prevent future cartilage or rotator cuff injury.
- Revision of SLAP repair is best handled with biceps tenodesis.
- Subpectoral biceps tenodesis avoids residual groove pain.
In recent decades, the long head of the biceps (LHB) tendon has been recognized as a pain generator in the shoulder of throwing athletes. The LHB muscle and its role in glenohumeral kinematics remains largely in question. The LHB tendon varies in size but most commonly is 5 mm to 6mm in diameter and about 9 cm in length, inserting on the superior labrum and supraglenoid tubercle after traveling through the bicipital groove.1 The many conditions that can develop along the course of the biceps tendon include overall biceps tendonitis, biceps tendon subluxation or instability, and injuries to the superior anterior to posterior area of the labrum.
These injuries can occur in young overhead athletes as well as manual laborers and older overhead recreational athletes. Pitching is the most common activity that leads to proximal biceps tendon disorders. The 6 phases of the pitch are linked in a kinetic chain that generates energy that is then translated to high velocity. The amount of force that is exerted on the shoulder during pitching and especially after ball release is impressive, and the athlete’s shoulder changes in many ways as it adapts to the motion.2-5 The late-cocking and deceleration phases are most commonly associated with proximal biceps pathology and the “peel-back” phenomenon. Other common activities that lead to biceps tendon issues in a young population are volleyball, baseball, tennis, softball, swimming, and cricket. Shoulder arthroscopies performed in older patients show degenerative biceps and labrum tears, which should be treated appropriately but perhaps different from how they are treated in overhead athletes.6-8 Further, many professional athletes have asymptomatic superior labrum anterior-posterior (SLAP) tears.9
Mechanism of Injury
Overhead throwing is commonly thought to be the mechanism by which lesions are created in the biceps–labrum complex (BLC). Pitching in particular generates incredible force and torque within the shoulder. In professional pitchers, the resulting throwing speed creates forces regularly in excess of 1000 N.3 These forces effect internal compensatory changes and internal derangement of the BLC. These changes often involve internal rotation deficits and alterations in the rotator cuff, which may contribute to glenohumeral instability and altered joint kinematics.10
Repetitive overhead activity is largely considered the mechanism of injury in this population, though more specific mechanisms have been described, including the peel-back mechanism11 and the posterior superior glenoid impingement. There is little evidence that preventive programs have any effect on decreasing the incidence of SLAP tears in overhead athletes.
Preoperative Evaluation
Preoperative evaluation is arguably the most important step in treating a patient with persistent or recurrent symptoms consistent with a SLAP tear. Evaluation includes thorough history, physical examination, and review of any prior injuries or surgical procedures. The physical examination should focus on maneuvers that define where the problem is occurring. Although SLAP tears are most common in this population, disorders of the biceps tendon within the groove, including inflammation and instability, should be ruled out with physical examination and advanced imaging. Palpation for groove tenderness, impingement-type complaints, internal rotation loss, and SLAP provocative testing are crucial in the diagnosis.12,13 The cause of symptoms may be multifactorial and include the often encountered concomitant pathology of rotator cuff tears, internal impingement, and instability.
Standard radiographs (Grashey anteroposterior, scapular/lateral, axillary lateral) and magnetic resonance imaging (MRI) with or without arthrography can be helpful in identifying and characterizing most SLAP tears as well as failed SLAP tear repairs. However, MRI is often positive for SLAP tears in asymptomatic patients, and diagnosing SLAP tears with MRI is often a challenge.14 MRI can help in determining concomitant pathology, including rotator cuff injury and cysts causing nerve compression. Correlation with clinical examination and patient history is most crucial. Conservative treatment (rest, activity modification, use of oral anti-inflammatory medications) typically is attempted and coordinated with respect to the athlete’s season of play.15,16
Classification
In overhead throwing athletes, SLAP tears typically are associated with anterior shoulder pain. Associated shoulder instability and significant glenohumeral dysfunction are not uncommon in athletes with lesions of the BLC. In 1985, Andrews and colleagues17 were the first to describe SLAP tears in overhead athletes (73 patients). Later, Snyder and colleagues18,19 further classified these lesions into 4 types based on tear stability and location, and they coined the acronym SLAP (Figure 1). 
Type I lesions typically are described as fraying at the inner margin of the labrum and are common in throwers, even asymptomatic throwers. Type II lesions, separations of the biceps and labrum from the superior glenoid (≥5 mm of excursion), are the most commonly occurring and treated variant in throwing athletes.20-22 Intraoperative evaluation for a peel-back lesion (placing the arm in abduction with external rotation), rather than for a sulcus of 1 mm to 2 mm, may confirm a type II SLAP tear.20,23,24 It is often important to consider the direction of tear propagation as well. Type III lesions include those with an intact BLC (but with a bucket-handle tear of the superior labral complex and an intact biceps tendon), whereas type IV lesions involve additional extension of the tear into the biceps tendon.18,19The classification systems are well defined. Nevertheless, management of SLAP lesions remains controversial.
Options for Surgical Treatment
SLAP Tear Repair—Outcomes
The incidence of SLAP tear repairs has increased dramatically in recent years.6,25 There are various SLAP tear repair methods, but the most common consists of repairing the labrum and biceps anchor. Management of type II SLAP lesions remains controversial. Several prospective studies have found overall improvement after SLAP tear repair.26-31 Other series have reported less encouraging outcomes, including dissatisfaction with persistent pain and inability to return to throwing.28,32 A 2010 systematic review found that the percentage of patients who returned to their preinjury level of play was only 64%, and outcomes for overhead throwing athletes were even worse—only 22% to 60% of these patients returned to their previous level.33 The right surgery for SLAP tears in this population continues to be an area of uncertainty for many surgeons.
Failed SLAP tear repairs (poor outcomes) have become common in overhead throwing athletes. The reasons for these failed repairs are unclear, but several possible explanations have been offered. One is that labral repair may result in permanent alterations in pitching biomechanics, which may lead to an inability to regain velocity and command.3 Another is that the athlete’s shoulder may remain unstable even after repair.10Hardware complications are a significant concern in this high-level population. Suture anchor pullout or iatrogenic cartilage damage may occur during instrumentation or as a result of suture anchor reactive changes. In addition, there are several reports of glenoid osteochondrolysis (Figure 2) caused by prominent hardware or prominent knots.34-39 

Stiffness after SLAP tear repair is a significant problem, with most patients taking up to 6 months to regain full motion.26,48 Overtensioning of the labrum and the glenohumeral ligaments may be the cause, and the solution may be to place anchors posterior (vs anterior) to the biceps insertion. In a large prospective military study, mean forward flexion and external rotation were reduced at final follow-up.31 These outcomes are less acceptable to overhead throwing athletes, who rely on motion for high-end throwing activities.
Primary Biceps Tenodesis—Outcomes
A 2015 database study found a 1.7-fold increase in biceps tenodesis over the preceding 5 years.49 However, relatively few procedures included in the study were performed in patients age younger than 30 years. For many older non-overhead throwers with type II tears, SLAP tear repair has become less popular as a treatment option.32 There is a dearth of knowledge about the outcomes of subpectoral biceps tenodesis as a primary treatment for biceps tendonitis and an associated SLAP tear. Although type I tears historically have been treated with débridement, débridement is seldom used for concomitant biceps tendonitis. It should be coupled with careful clinical examination.
In recent years, biceps tenodesis has been proposed as an alternative to repair for SLAP tears, particularly in older patients.24,44 For obvious reasons, however, there has been some trepidation about performing biceps tenodesis in throwing athletes. Some authors have proposed biceps tenodesis as primary treatment for isolated SLAP tears. Boileau and colleagues44 compared the outcomes of treatment of isolated type II SLAP lesions in 25 consecutive patients. For 10 patients, repair involved suture anchors; for the other 15, arthroscopic biceps tenodesis was performed with an absorbable interference screw. Six of the 10 suture anchor patients were disappointed with their outcome (persistent pain or inability to return to sport), whereas 14 of the 15 biceps tenodesis patients were satisfied. The authors concluded that arthroscopic biceps tenodesis is an effective alternative to repair for type II SLAP lesions, though their study was not isolated to overhead athletes (tenodesis group mean age, 52 years).
In a 2014 series of cases, Ek and colleagues50 reported good outcomes of SLAP tear repair and biceps tenodesis. Again, though, tenodesis was used in older patients, and repair in younger, more active patients, with no high-level athletes in either group. There was no difference in return to sport between groups. In a study of patients who underwent primary biceps tenodesis, Gupta and colleagues51 found 80% excellent outcomes (improved shoulder outcome scores) in select SLAP tear patients, including 8 athletes, 88% of whom were overhead athletes. Gottschalk and colleagues52 reported on differences in prospectively collected outcome data (age, sex, SLAP lesion type II or IV) for primary biceps tenodesis in a series of 33 patients. Twenty-six of the 29 patients who completed follow-up returned to their previous level of activity. These studies suggest that primary biceps tenodesis may be an alternative with lower failure rates in the treatment of SLAP tears in middle-aged patients, and in overhead athletes, though additional specific studies are needed to focus on overhead athletes on a larger scale.
Revision SLAP Tear Repair Versus Biceps Tenodesis
Failed arthroscopic SLAP tear repairs, which are increasingly common, present a unique treatment challenge. In a 2013 prospective cohort series, Gupta and colleagues46 found excellent clinical outcomes of subpectoral biceps tenodesis for failed type II SLAP tears. The authors reported a postoperative SANE (Single Assessment Numeric Evaluation) score of 70.4%, an SST (Simple Shoulder Test) score of 9.33, and an ASES (American Shoulder and Elbow Surgeons) score of 77.96, along with reasonable health-related quality-of-life scores. Werner and colleagues53 evaluated 2-year outcomes of biceps tenodesis performed after SLAP tear repair in 24 patients and found a return to almost normal range of motion as well as good clinical outcome scores. Significantly worse outcomes were found for patients with open worker’s compensation claims.
McCormick and colleagues26 prospectively evaluated the efficacy of biceps tenodesis for failed type II SLAP tear repair in 46 patients. Improvement was noted across all outcome assessments during follow-up (mean, 3.6 years). From these findings, we might conclude that biceps tenodesis is a more predictable option for failed SLAP tear repair, and that it has a relatively low complication rate. However, most investigators have used a heterogeneous patient population, as opposed to overhead athletes specifically. To our knowledge, no one has evaluated the specific population of overhead throwers with failed SLAP tear repairs. In addition, no one has conducted randomized controlled trials comparing débridement, biceps tenodesis, and repair for failed SLAP tear repairs.
Postoperative Considerations
When overhead athletes and their surgeons are considering surgical options, they must take rehabilitation and return to play into account. Many surgeons think the possible marginal clinical benefit of SLAP tear repair may not be worth the protracted rehabilitation. In most practices, rehabilitation after biceps tenodesis is less involved. Discussing the advantages and disadvantages of these 2 procedures can be helpful in decision making.
Dein and colleagues54 reported the case of a middle-aged pitcher who sustained a fracture after biceps tenodesis with an interference screw. Cases like this are concerning. Surgeons should consider altering the rehabilitation regimen when planning postoperative care in cases of biceps tenodesis in throwers. Other reported complications of open tenodesis are deep infection, thrombosis, postoperative stiffness, and nerve injury.55-58
Consequences for Overhead Throwers
The unknown role of the BLC leaves surgeons wary when considering biceps tenodesis for elite athletes. Some have postulated that removing the intra-articular portion of the LHB may cause microinstability and alter joint kinematics.10,59-61 Others have suggested the biceps is desynchronized from the other musculature and is not functionally important.62 Disruption of one portion of the superior labrum may result in instability on the opposite side of the glenoid.10,61 Biomechanical studies, both cadaveric and in vivo, have tried to create proper loads to the LHB and evaluate the kinematics of the shoulder before and after biceps tenodesis and SLAP tear repair.59,60 Using a cadaveric model, Strauss and colleagues63 found that type II SLAP lesions resulted in increased glenohumeral translation compared with baseline. Biceps tenodesis did not restore normal translation, but this did not negatively affect stability in the presence of a SLAP lesion. The consensus is that the role of the biceps is controversial at best.
Several studies have used electromyography (EMG) to evaluate LHB functioning. In 2014, Chalmers and colleagues59 used surface EMG and motion analysis to evaluate 18 pitchers: 6 underwent SLAP tear repair, 5 underwent biceps tenodesis, and 7 were uninjured controls. There were no significant differences in the activity of the LHB muscle, the short head of the biceps muscle, the deltoid, the infraspinatus, or the latissimus among the 3 groups. Motion analysis showed that the normal pattern of muscular activation within the LHB muscle was more closely restored by biceps tenodesis than by SLAP tear repair. In addition, thoracic rotation patterns were significantly more altered in the SLAP tear repair patients than in the uninjured controls. As the authors noted, given the low frequency with which biceps tenodesis is performed in overhead athletes, it is unlikely that larger scale studies will be conducted without a multicenter effort.
Recommendations and Our Preferred Technique
Which surgical option is best for treating symptomatic SLAP lesions in overhead athletes remains unclear. Many athletes struggle to return to high-level play after SLAP tear repair. Whether the same is true after biceps tenodesis is yet to be determined because of the low frequency with which biceps tenodesis is performed in high-level overhead athletes. The options for fixation, technique, and fixation location are equally broad. In this section, we outline our general line of thinking for cases of proximal biceps pathology.
In each case, we perform glenohumeral arthroscopy to evaluate the BLC and identify any other pathology. For overhead athletes who are younger than 30 years and lack bicipital groove pain or signs of gross tendinopathy, we favor arthroscopic SLAP tear repair. Repair is usually performed through an anterior working portal for suture passage and a Wilmington portal for anchor placement. We use knotless technology to achieve stable fixation and stay posterior to the biceps anchor insertion.
For the prevention of any potential pain from the bicipital groove in carefully selected patients—recreational overhead athletes and patients who want a less involved surgical recovery—we favor open subpectoral biceps tenodesis rather than arthroscopic tenodesis. The outcomes of biceps tenodesis are consistent, according to the literature.47,57,64 Moreover, the open approach is favored for the incidence of postoperative stiffness in the arthroscopic population.65 Tendons can be fixed with multiple procedures, including soft-tissue tenodesis, interference screw fixation, and surface anchors. We favor using a tenodesis screw in the subpectoral location, as outlined by Mazzocca and colleagues.64Our algorithm for SLAP lesions is evolving with our understanding of this complex disease process. For young overhead throwers with type II SLAP lesions, we favor arthroscopic SLAP tear repair with knotless technology. For older recreational overhead athletes, we favor biceps tenodesis in the subpectoral region after diagnostic arthroscopy plus biceps tenotomy with or without additional SLAP tear fixation, depending on the stability of the biceps anchor (Figures 4A, 4B). 
Conclusion
Overhead athletes who present with symptomatic SLAP lesions often provide a treatment dilemma. Although SLAP tear repair historically has been standard treatment, biceps tenodesis represents a consistent surgical option with low complication rates and low revision rates. It is likely that, as additional data on glenohumeral kinematics and outcomes in young athletes become available, improved decision-making algorithms will follow.
Am J Orthop. 2017;46(1):E71-E78. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Elser F, Braun S, Dewing CB, Giphart JE, Millett PJ. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27(4):581-592.
2. Fedoriw WW, Ramkumar P, McCulloch PC, Lintner DM. Return to play after treatment of superior labral tears in professional baseball players. Am J Sports Med. 2014;42(5):1155-1160.
3. Fleisig GS, Andrews JR, Dillman CJ, Escamilla RF. Kinetics of baseball pitching with implications about injury mechanisms. Am J Sports Med. 1995;23(2):233-239.
4. Aydin N, Sirin E, Arya A. Superior labrum anterior to posterior lesions of the shoulder: diagnosis and arthroscopic management. World J Orthop. 2014;5(3):344-350.
5. Barber A, Field LD, Ryu R. Biceps tendon and superior labrum injuries: decision-marking. J Bone Joint Surg Am. 2007;89(8):1844-1855.
6. Onyekwelu I, Khatib O, Zuckerman JD, Rokito AS, Kwon YW. The rising incidence of arthroscopic superior labrum anterior and posterior (SLAP) repairs. J Shoulder Elbow Surg. 2012;21(6):728-731.
7. Patterson BM, Creighton RA, Spang JT, Roberson JR, Kamath GV. Surgical trends in the treatment of superior labrum anterior and posterior lesions of the shoulder: analysis of data from the American Board of Orthopaedic Surgery Certification Examination Database. Am J Sports Med. 2014;42(8):1904-1910.
8. Walton DM, Sadi J. Identifying SLAP lesions: a meta-analysis of clinical tests and exercise in clinical reasoning. Phys Ther Sport. 2008;9(4):167-176.
9. Lesniak BP, Baraga MG, Jose J, Smith MK, Cunningham S, Kaplan LD. Glenohumeral findings on magnetic resonance imaging correlate with innings pitched in asymptomatic pitchers. Am J Sports Med. 2013;41(9):2022-2027.
10. Mihata T, McGarry MH, Tibone JE, Fitzpatrick MJ, Kinoshita M, Lee TQ. Biomechanical assessment of type II superior labral anterior-posterior (SLAP) lesions associated with anterior shoulder capsular laxity as seen in throwers: a cadaveric study. Am J Sports Med. 2008;36(8):1604-1610.
11. Burkhart SS, Morgan CD, Kibler WB. The disabled throwing shoulder: spectrum of pathology. Part II: evaluation and treatment of SLAP lesions in throwers. Arthroscopy. 2003;19(5):531-539.
12. Meserve BB, Cleland JA, Boucher TR. A meta-analysis examining clinical test utility for assessing superior labral anterior posterior lesions. Am J Sports Med. 2009;37(11):2252-2258.
13. Pandya NK, Colton A, Webner D, Sennett B, Huffman GR. Physical examination and magnetic resonance imaging in the diagnosis of superior labrum anterior-posterior lesions of the shoulder: a sensitivity analysis. Arthroscopy. 2008;24(3):311-317.
14. Amin MF, Youssef AO. The diagnostic value of magnetic resonance arthrography of the shoulder in detection and grading of SLAP lesions: comparison with arthroscopic findings. Eur J Radiol. 2012;81(9):2343-2347.
15. Cook C, Beaty S, Kissenberth MJ, Siffri P, Pill SG, Hawkins RJ. Diagnostic accuracy of five orthopedic clinical tests for diagnosis of superior labrum anterior posterior (SLAP) lesions. J Shoulder Elbow Surg. 2012;21(1):13-22.
16. Edwards SL, Lee JA, Bell JE, et al. Nonoperative treatment of superior labrum anterior posterior tears: improvements in pain, function, and quality of life. Am J Sports Med. 2010;38(7):1456-1461.
17. Andrews JR, Carson WG Jr, McLeod WD. Glenoid labrum tears related to the long head of the biceps. Am J Sports Med. 1985;13(5):337-341.
18. Snyder SJ, Karzel RP, Del Pizzo W, Ferkel RD, Friedman MJ. SLAP lesions of the shoulder. Arthroscopy. 1990;6(4):274-279.
19. Snyder SJ, Banas MP, Karzel RP. An analysis of 140 injuries to the superior glenoid labrum. J Shoulder Elbow Surg. 1995;4(4):243-248.
20. Morgan CD, Burkhart SS, Palmeri M, Gillespie M. Type II SLAP lesions: three subtypes and their relationships to superior instability and rotator cuff tears. Arthroscopy. 1998;14(6):553-565.
21. Weber SC, Martin DF, Seiler JG 3rd, Harrast JJ. Superior labrum anterior and posterior lesions of the shoulder: incidence rates, complications, and outcomes as reported by American Board of Orthopedic Surgery. Part II candidates. Am J Sports Med. 2012;40(7):1538-1543.
22. Keener JD, Brophy RH. Superior labral tears of the shoulder: pathogenesis, evaluation, and treatment. J Am Acad Orthop Surg. 2009;17(10):627-637.
23. Chen CH, Hsu KY, Chen WJ, Shih CH. Incidence and severity of biceps long head tendon lesion in patients with complete rotator cuff tears. J Trauma. 2005;58(6):1189-1193.
24. Nho SJ, Strauss EJ, Lenart BA, et al. Long head of the biceps tendinopathy: diagnosis and management. J Am Acad Orthop Surg. 2010;18(11):645-656.
25. Zhang AL, Kreulen C, Ngo SS, Hame SL, Wang JC, Gamradt SC. Demographic trends in arthroscopic SLAP repair in the United States. Am J Sports Med. 2012;40(5):1144-1147.
26. McCormick F, Bhatia S, Chalmers P, Gupta A, Verma N, Romeo AA. The management of type II superior labral anterior to posterior injuries. Orthop Clin North Am. 2014;45(1):121-128.
27. Brockmeier SF, Voos JE, Williams RJ 3rd, Altchek DW, Cordasco FA, Allen AA; Hospital for Special Surgery Sports Medicine and Shoulder Service. Outcomes after arthroscopic repair of type-II SLAP lesions. J Bone Joint Surg Am. 2009;91(7):1595-1603.
28. Boileau P, Parratte S, Chuinard C, Roussanne Y, Shia D, Bicknell R. Arthroscopic treatment of isolated type II SLAP lesions: biceps tenodesis as an alternative to reinsertion. Am J Sports Med. 2009;37(5):929-936.
29. Denard PJ, Lädermann A, Burkhart SS. Long-term outcome after arthroscopic repair of type II SLAP lesions: results according to age and workers’ compensation status. Arthroscopy. 2012;28(4):451-457.
30. Friel NA, Karas V, Slabaugh MA, Cole BJ. Outcomes of type II superior labrum, anterior to posterior (SLAP) repair: prospective evaluation at a minimum two-year follow-up. J Shoulder Elbow Surg. 2010;19(6):859-867.
31. Provencher MT, McCormick F, Dewing C, McIntire S, Solomon D. A prospective analysis of 179 type 2 superior labrum anterior and posterior repairs: outcomes and factors associated with success and failure. Am J Sports Med. 2013;41(4):880-886.
32. Gupta AK, Chalmers PN, Klosterman EL, et al. Subpectoral biceps tenodesis for bicipital tendonitis with SLAP tear. Orthopedics. 2015;38(1):e48-e53.
33. Gorantla K, Gill C, Wright RW. The outcome of type II SLAP repair: a systematic review. Arthroscopy. 2010;26(4):537-545.
34. Katz LM, Hsu S, Miller SL, et al. Poor outcomes after SLAP repair: descriptive analysis and prognosis. Arthroscopy. 2009;25(8):849-855.
35. Park MJ, Hsu JE, Harper C, Sennett BJ, Huffman GR. Poly-L/D-lactic acid anchors are associated with reoperation and failure of SLAP repairs. Arthroscopy. 2011;27(10):1335-1340.
36. Sassmannshausen G, Sukay M, Mair SD. Broken or dislodged poly-L-lactic acid bioabsorbable tacks in patients after SLAP lesion surgery. Arthroscopy. 2006;22(6):615-619.
37. Uggen C, Wei A, Glousman RE, et al. Biomechanical comparison of knotless anchor repair versus simple suture repair for type II SLAP lesions. Arthroscopy. 2009;25(10):1085-1092.
38. Weber SC. Surgical management of the failed SLAP repair. Sports Med Arthrosc. 2010;18(3):162-166.
39. Wilkerson JP, Zvijac JE, Uribe JW, Schürhoff MR, Green JB. Failure of polymerized lactic acid tacks in shoulder surgery. J Shoulder Elbow Surg. 2003;12(2):117-121.
40. Weber S. Surgical management of the failed SLAP lesion. Arthroscopy. 2008;24(suppl):e8-e9.
41. Schrøder CP, Skare O, Gjengedal E, Uppheim G, Reikerås O, Brox JI. Long-term results after SLAP repair: a 5-year follow-up study of 107 patients with comparison of patients aged over and under 40 years. Arthroscopy. 2012;28(11):1601-1607.
42. Mazzocca AD, Cote MP, Arciero CL, Romeo AA, Arciero RA. Clinical outcomes after subpectoral biceps tenodesis with an interference screw. Am J Sports Med. 2008;36(10):1922-1929.
43. Mazzocca AD, McCarthy MB, Ledgard FA, et al. Histomorphologic changes of the long head of the biceps tendon in common shoulder pathologies. Arthroscopy. 2013;29(6):972-981.
44. Boileau P, Parratte S, Chuinard C, Roussanne Y, Shia D, Bicknell R. Arthroscopic treatment of isolated type II SLAP lesions: biceps tenodesis as an alternative to reinsertion. Am J Sports Med. 2009;37(5):929-936.
45. Boileau P, Krishnan SG, Coste JS, Walch G. Arthroscopic biceps tenodesis: a new technique using bioabsorbable interference screw fixation. Arthroscopy. 2002;18(9):1002-1012.
46. Gupta AK, Bruce B, Klosterman EL, McCormick F, Harris J, Romeo AA. Subpectoral biceps tenodesis for failed type II SLAP repair. Orthopedics. 2013;36(6):e723-e728.
47. Provencher MT, LeClere LE, Romeo AA. Subpectoral biceps tenodesis. Sports Med Arthrosc. 2008;16(3):170-176.
48. McCarty LP 3rd, Buss DD, Datta MW, Freehill MQ, Giveans MR. Complications observed following labral or rotator cuff repair with use of poly-L-lactic acid implants. J Bone Joint Surg Am. 2013;95(6):507-511.
49. Werner BC, Brockmeier SF, Gwathmey FW. Trends in long head biceps tenodesis. Am J Sports Med. 2015;43(3):570-578.
50. Ek ET, Shi LL, Tompson JD, Freehill MT, Warner JJ. Surgical treatment of isolated type II superior labrum anterior-posterior (SLAP) lesions: repair versus biceps tenodesis. J Shoulder Elbow Surg. 2014;23(7):1059-1065.
51. Gupta AK, Chalmers PN, Klosterman EL, et al. Subpectoral biceps tenodesis for bicipital tendonitis with SLAP tear. Orthopedics. 2015;38(1):e48-e53.
52. Gottschalk MB, Karas SG, Ghattas TN, Burdette R. Subpectoral biceps tenodesis for the treatment of type II and IV superior labral anterior and posterior lesions. Am J Sports Med. 2014;42(9):2128-2135.
53. Werner BC, Pehlivan HC, Hart JM, et al. Biceps tenodesis is a viable option for salvage of failed SLAP repair. J Shoulder Elbow Surg. 2014;23(8):e179-e184.
54. Dein EJ, Huri G, Gordon JC, McFarland EG. A humerus fracture in a baseball pitcher after biceps tenodesis [published correction appears in Am J Sports Med. 2014;42(6):NP39]. Am J Sports Med. 2014;42(4):877-879.
55. Nho SJ, Reiff SN, Verma NN, Slabaugh MA, Mazzocca AD, Romeo AA. Complications associated with subpectoral biceps tenodesis: low rates of incidence following surgery. J Shoulder Elbow Surg. 2010;19(5):764-768.
56. Osbahr DC, Diamond AB, Speer KP. The cosmetic appearance of the biceps muscle after long-head tenotomy versus tenodesis. Arthroscopy. 2002;18(5):483-487.
57. Romeo AA, Mazzocca AD, Tauro JC. Arthroscopic biceps tenodesis. Arthroscopy. 2004;20(2):206-213.
58. Ma H, Van Heest A, Glisson C, Patel S. Musculocutaneous nerve entrapment: an unusual complication after biceps tenodesis. Am J Sports Med. 2009;37(12):2467-2469.
59. Chalmers PN, Trombley R, Cip J, et al. Postoperative restoration of upper extremity motion and neuromuscular control during the overhand pitch: evaluation of tenodesis and repair for superior labral anterior-posterior tears. Am J Sports Med. 2014;42(12):2825-2836.
60. Giphart JE, Elser F, Dewing CB, Torry MR, Millett PJ. The long head of the biceps tendon has minimal effect on in vivo glenohumeral kinematics: a biplane fluoroscopy study. Am J Sports Med. 2012;40(1):202-212.
61. Grossman MG, Tibone JE, McGarry MH, Schneider DJ, Veneziani S, Lee TQ. A cadaveric model of the throwing shoulder: a possible etiology of superior labrum anterior-to-posterior lesions. J Bone Joint Surg Am. 2005;87(4):824-831.
62. Hawkes DH, Alizadehkhaiyat O, Fisher AC, Kemp GJ, Roebuck MM, Frostick SP. Normal shoulder muscular activation and co-ordination during a shoulder elevation task based on activities of daily living: an electromyographic study. J Orthop Res. 2012;30(1):53-60.
63. Strauss EJ, Salata MJ, Sershon RA, et al. Role of the superior labrum after biceps tenodesis in glenohumeral stability. J Shoulder Elbow Surg. 2014;23(4):485-491.
64. Mazzocca AD, Bicos J, Santangelo S, Romeo AA, Arciero RA. The biomechanical evaluation of four fixation techniques for proximal biceps tenodesis. Arthroscopy. 2005;21(11):1296-1306.
65. Werner BC, Pehlivan HC, Hart JM, et al. Increased incidence of postoperative stiffness after arthroscopic compared with open biceps tenodesis. Arthroscopy. 2014;30(9):1075-1084.
66. Denard PJ, Dai X, Hanypsiak BT, Burkhart SS. Anatomy of the biceps tendon: implications for restoring physiological length–tension relation during biceps tenodesis with interference screw fixation. Arthroscopy. 2012;28(10):1352-1358.
67. Mazzocca AD, Rios CG, Romeo AA, Arciero RA. Subpectoral biceps tenodesis with interference screw fixation. Arthroscopy. 2005;21(7):896.
Take Home Points
- Outcomes after SLAP repair remain guarded.
- Physical examination is key in determining proper management of biceps pathology.
- When performing SLAP repair, knotless technology may prevent future cartilage or rotator cuff injury.
- Revision of SLAP repair is best handled with biceps tenodesis.
- Subpectoral biceps tenodesis avoids residual groove pain.
In recent decades, the long head of the biceps (LHB) tendon has been recognized as a pain generator in the shoulder of throwing athletes. The LHB muscle and its role in glenohumeral kinematics remains largely in question. The LHB tendon varies in size but most commonly is 5 mm to 6mm in diameter and about 9 cm in length, inserting on the superior labrum and supraglenoid tubercle after traveling through the bicipital groove.1 The many conditions that can develop along the course of the biceps tendon include overall biceps tendonitis, biceps tendon subluxation or instability, and injuries to the superior anterior to posterior area of the labrum.
These injuries can occur in young overhead athletes as well as manual laborers and older overhead recreational athletes. Pitching is the most common activity that leads to proximal biceps tendon disorders. The 6 phases of the pitch are linked in a kinetic chain that generates energy that is then translated to high velocity. The amount of force that is exerted on the shoulder during pitching and especially after ball release is impressive, and the athlete’s shoulder changes in many ways as it adapts to the motion.2-5 The late-cocking and deceleration phases are most commonly associated with proximal biceps pathology and the “peel-back” phenomenon. Other common activities that lead to biceps tendon issues in a young population are volleyball, baseball, tennis, softball, swimming, and cricket. Shoulder arthroscopies performed in older patients show degenerative biceps and labrum tears, which should be treated appropriately but perhaps different from how they are treated in overhead athletes.6-8 Further, many professional athletes have asymptomatic superior labrum anterior-posterior (SLAP) tears.9
Mechanism of Injury
Overhead throwing is commonly thought to be the mechanism by which lesions are created in the biceps–labrum complex (BLC). Pitching in particular generates incredible force and torque within the shoulder. In professional pitchers, the resulting throwing speed creates forces regularly in excess of 1000 N.3 These forces effect internal compensatory changes and internal derangement of the BLC. These changes often involve internal rotation deficits and alterations in the rotator cuff, which may contribute to glenohumeral instability and altered joint kinematics.10
Repetitive overhead activity is largely considered the mechanism of injury in this population, though more specific mechanisms have been described, including the peel-back mechanism11 and the posterior superior glenoid impingement. There is little evidence that preventive programs have any effect on decreasing the incidence of SLAP tears in overhead athletes.
Preoperative Evaluation
Preoperative evaluation is arguably the most important step in treating a patient with persistent or recurrent symptoms consistent with a SLAP tear. Evaluation includes thorough history, physical examination, and review of any prior injuries or surgical procedures. The physical examination should focus on maneuvers that define where the problem is occurring. Although SLAP tears are most common in this population, disorders of the biceps tendon within the groove, including inflammation and instability, should be ruled out with physical examination and advanced imaging. Palpation for groove tenderness, impingement-type complaints, internal rotation loss, and SLAP provocative testing are crucial in the diagnosis.12,13 The cause of symptoms may be multifactorial and include the often encountered concomitant pathology of rotator cuff tears, internal impingement, and instability.
Standard radiographs (Grashey anteroposterior, scapular/lateral, axillary lateral) and magnetic resonance imaging (MRI) with or without arthrography can be helpful in identifying and characterizing most SLAP tears as well as failed SLAP tear repairs. However, MRI is often positive for SLAP tears in asymptomatic patients, and diagnosing SLAP tears with MRI is often a challenge.14 MRI can help in determining concomitant pathology, including rotator cuff injury and cysts causing nerve compression. Correlation with clinical examination and patient history is most crucial. Conservative treatment (rest, activity modification, use of oral anti-inflammatory medications) typically is attempted and coordinated with respect to the athlete’s season of play.15,16
Classification
In overhead throwing athletes, SLAP tears typically are associated with anterior shoulder pain. Associated shoulder instability and significant glenohumeral dysfunction are not uncommon in athletes with lesions of the BLC. In 1985, Andrews and colleagues17 were the first to describe SLAP tears in overhead athletes (73 patients). Later, Snyder and colleagues18,19 further classified these lesions into 4 types based on tear stability and location, and they coined the acronym SLAP (Figure 1).
Type I lesions typically are described as fraying at the inner margin of the labrum and are common in throwers, even asymptomatic throwers. Type II lesions, separations of the biceps and labrum from the superior glenoid (≥5 mm of excursion), are the most commonly occurring and treated variant in throwing athletes.20-22 Intraoperative evaluation for a peel-back lesion (placing the arm in abduction with external rotation), rather than for a sulcus of 1 mm to 2 mm, may confirm a type II SLAP tear.20,23,24 It is often important to consider the direction of tear propagation as well. Type III lesions include those with an intact BLC (but with a bucket-handle tear of the superior labral complex and an intact biceps tendon), whereas type IV lesions involve additional extension of the tear into the biceps tendon.18,19The classification systems are well defined. Nevertheless, management of SLAP lesions remains controversial.
Options for Surgical Treatment
SLAP Tear Repair—Outcomes
The incidence of SLAP tear repairs has increased dramatically in recent years.6,25 There are various SLAP tear repair methods, but the most common consists of repairing the labrum and biceps anchor. Management of type II SLAP lesions remains controversial. Several prospective studies have found overall improvement after SLAP tear repair.26-31 Other series have reported less encouraging outcomes, including dissatisfaction with persistent pain and inability to return to throwing.28,32 A 2010 systematic review found that the percentage of patients who returned to their preinjury level of play was only 64%, and outcomes for overhead throwing athletes were even worse—only 22% to 60% of these patients returned to their previous level.33 The right surgery for SLAP tears in this population continues to be an area of uncertainty for many surgeons.
Failed SLAP tear repairs (poor outcomes) have become common in overhead throwing athletes. The reasons for these failed repairs are unclear, but several possible explanations have been offered. One is that labral repair may result in permanent alterations in pitching biomechanics, which may lead to an inability to regain velocity and command.3 Another is that the athlete’s shoulder may remain unstable even after repair.10Hardware complications are a significant concern in this high-level population. Suture anchor pullout or iatrogenic cartilage damage may occur during instrumentation or as a result of suture anchor reactive changes. In addition, there are several reports of glenoid osteochondrolysis (Figure 2) caused by prominent hardware or prominent knots.34-39
Stiffness after SLAP tear repair is a significant problem, with most patients taking up to 6 months to regain full motion.26,48 Overtensioning of the labrum and the glenohumeral ligaments may be the cause, and the solution may be to place anchors posterior (vs anterior) to the biceps insertion. In a large prospective military study, mean forward flexion and external rotation were reduced at final follow-up.31 These outcomes are less acceptable to overhead throwing athletes, who rely on motion for high-end throwing activities.
Primary Biceps Tenodesis—Outcomes
A 2015 database study found a 1.7-fold increase in biceps tenodesis over the preceding 5 years.49 However, relatively few procedures included in the study were performed in patients age younger than 30 years. For many older non-overhead throwers with type II tears, SLAP tear repair has become less popular as a treatment option.32 There is a dearth of knowledge about the outcomes of subpectoral biceps tenodesis as a primary treatment for biceps tendonitis and an associated SLAP tear. Although type I tears historically have been treated with débridement, débridement is seldom used for concomitant biceps tendonitis. It should be coupled with careful clinical examination.
In recent years, biceps tenodesis has been proposed as an alternative to repair for SLAP tears, particularly in older patients.24,44 For obvious reasons, however, there has been some trepidation about performing biceps tenodesis in throwing athletes. Some authors have proposed biceps tenodesis as primary treatment for isolated SLAP tears. Boileau and colleagues44 compared the outcomes of treatment of isolated type II SLAP lesions in 25 consecutive patients. For 10 patients, repair involved suture anchors; for the other 15, arthroscopic biceps tenodesis was performed with an absorbable interference screw. Six of the 10 suture anchor patients were disappointed with their outcome (persistent pain or inability to return to sport), whereas 14 of the 15 biceps tenodesis patients were satisfied. The authors concluded that arthroscopic biceps tenodesis is an effective alternative to repair for type II SLAP lesions, though their study was not isolated to overhead athletes (tenodesis group mean age, 52 years).
In a 2014 series of cases, Ek and colleagues50 reported good outcomes of SLAP tear repair and biceps tenodesis. Again, though, tenodesis was used in older patients, and repair in younger, more active patients, with no high-level athletes in either group. There was no difference in return to sport between groups. In a study of patients who underwent primary biceps tenodesis, Gupta and colleagues51 found 80% excellent outcomes (improved shoulder outcome scores) in select SLAP tear patients, including 8 athletes, 88% of whom were overhead athletes. Gottschalk and colleagues52 reported on differences in prospectively collected outcome data (age, sex, SLAP lesion type II or IV) for primary biceps tenodesis in a series of 33 patients. Twenty-six of the 29 patients who completed follow-up returned to their previous level of activity. These studies suggest that primary biceps tenodesis may be an alternative with lower failure rates in the treatment of SLAP tears in middle-aged patients, and in overhead athletes, though additional specific studies are needed to focus on overhead athletes on a larger scale.
Revision SLAP Tear Repair Versus Biceps Tenodesis
Failed arthroscopic SLAP tear repairs, which are increasingly common, present a unique treatment challenge. In a 2013 prospective cohort series, Gupta and colleagues46 found excellent clinical outcomes of subpectoral biceps tenodesis for failed type II SLAP tears. The authors reported a postoperative SANE (Single Assessment Numeric Evaluation) score of 70.4%, an SST (Simple Shoulder Test) score of 9.33, and an ASES (American Shoulder and Elbow Surgeons) score of 77.96, along with reasonable health-related quality-of-life scores. Werner and colleagues53 evaluated 2-year outcomes of biceps tenodesis performed after SLAP tear repair in 24 patients and found a return to almost normal range of motion as well as good clinical outcome scores. Significantly worse outcomes were found for patients with open worker’s compensation claims.
McCormick and colleagues26 prospectively evaluated the efficacy of biceps tenodesis for failed type II SLAP tear repair in 46 patients. Improvement was noted across all outcome assessments during follow-up (mean, 3.6 years). From these findings, we might conclude that biceps tenodesis is a more predictable option for failed SLAP tear repair, and that it has a relatively low complication rate. However, most investigators have used a heterogeneous patient population, as opposed to overhead athletes specifically. To our knowledge, no one has evaluated the specific population of overhead throwers with failed SLAP tear repairs. In addition, no one has conducted randomized controlled trials comparing débridement, biceps tenodesis, and repair for failed SLAP tear repairs.
Postoperative Considerations
When overhead athletes and their surgeons are considering surgical options, they must take rehabilitation and return to play into account. Many surgeons think the possible marginal clinical benefit of SLAP tear repair may not be worth the protracted rehabilitation. In most practices, rehabilitation after biceps tenodesis is less involved. Discussing the advantages and disadvantages of these 2 procedures can be helpful in decision making.
Dein and colleagues54 reported the case of a middle-aged pitcher who sustained a fracture after biceps tenodesis with an interference screw. Cases like this are concerning. Surgeons should consider altering the rehabilitation regimen when planning postoperative care in cases of biceps tenodesis in throwers. Other reported complications of open tenodesis are deep infection, thrombosis, postoperative stiffness, and nerve injury.55-58
Consequences for Overhead Throwers
The unknown role of the BLC leaves surgeons wary when considering biceps tenodesis for elite athletes. Some have postulated that removing the intra-articular portion of the LHB may cause microinstability and alter joint kinematics.10,59-61 Others have suggested the biceps is desynchronized from the other musculature and is not functionally important.62 Disruption of one portion of the superior labrum may result in instability on the opposite side of the glenoid.10,61 Biomechanical studies, both cadaveric and in vivo, have tried to create proper loads to the LHB and evaluate the kinematics of the shoulder before and after biceps tenodesis and SLAP tear repair.59,60 Using a cadaveric model, Strauss and colleagues63 found that type II SLAP lesions resulted in increased glenohumeral translation compared with baseline. Biceps tenodesis did not restore normal translation, but this did not negatively affect stability in the presence of a SLAP lesion. The consensus is that the role of the biceps is controversial at best.
Several studies have used electromyography (EMG) to evaluate LHB functioning. In 2014, Chalmers and colleagues59 used surface EMG and motion analysis to evaluate 18 pitchers: 6 underwent SLAP tear repair, 5 underwent biceps tenodesis, and 7 were uninjured controls. There were no significant differences in the activity of the LHB muscle, the short head of the biceps muscle, the deltoid, the infraspinatus, or the latissimus among the 3 groups. Motion analysis showed that the normal pattern of muscular activation within the LHB muscle was more closely restored by biceps tenodesis than by SLAP tear repair. In addition, thoracic rotation patterns were significantly more altered in the SLAP tear repair patients than in the uninjured controls. As the authors noted, given the low frequency with which biceps tenodesis is performed in overhead athletes, it is unlikely that larger scale studies will be conducted without a multicenter effort.
Recommendations and Our Preferred Technique
Which surgical option is best for treating symptomatic SLAP lesions in overhead athletes remains unclear. Many athletes struggle to return to high-level play after SLAP tear repair. Whether the same is true after biceps tenodesis is yet to be determined because of the low frequency with which biceps tenodesis is performed in high-level overhead athletes. The options for fixation, technique, and fixation location are equally broad. In this section, we outline our general line of thinking for cases of proximal biceps pathology.
In each case, we perform glenohumeral arthroscopy to evaluate the BLC and identify any other pathology. For overhead athletes who are younger than 30 years and lack bicipital groove pain or signs of gross tendinopathy, we favor arthroscopic SLAP tear repair. Repair is usually performed through an anterior working portal for suture passage and a Wilmington portal for anchor placement. We use knotless technology to achieve stable fixation and stay posterior to the biceps anchor insertion.
For the prevention of any potential pain from the bicipital groove in carefully selected patients—recreational overhead athletes and patients who want a less involved surgical recovery—we favor open subpectoral biceps tenodesis rather than arthroscopic tenodesis. The outcomes of biceps tenodesis are consistent, according to the literature.47,57,64 Moreover, the open approach is favored for the incidence of postoperative stiffness in the arthroscopic population.65 Tendons can be fixed with multiple procedures, including soft-tissue tenodesis, interference screw fixation, and surface anchors. We favor using a tenodesis screw in the subpectoral location, as outlined by Mazzocca and colleagues.64Our algorithm for SLAP lesions is evolving with our understanding of this complex disease process. For young overhead throwers with type II SLAP lesions, we favor arthroscopic SLAP tear repair with knotless technology. For older recreational overhead athletes, we favor biceps tenodesis in the subpectoral region after diagnostic arthroscopy plus biceps tenotomy with or without additional SLAP tear fixation, depending on the stability of the biceps anchor (Figures 4A, 4B).
Conclusion
Overhead athletes who present with symptomatic SLAP lesions often provide a treatment dilemma. Although SLAP tear repair historically has been standard treatment, biceps tenodesis represents a consistent surgical option with low complication rates and low revision rates. It is likely that, as additional data on glenohumeral kinematics and outcomes in young athletes become available, improved decision-making algorithms will follow.
Am J Orthop. 2017;46(1):E71-E78. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Take Home Points
- Outcomes after SLAP repair remain guarded.
- Physical examination is key in determining proper management of biceps pathology.
- When performing SLAP repair, knotless technology may prevent future cartilage or rotator cuff injury.
- Revision of SLAP repair is best handled with biceps tenodesis.
- Subpectoral biceps tenodesis avoids residual groove pain.
In recent decades, the long head of the biceps (LHB) tendon has been recognized as a pain generator in the shoulder of throwing athletes. The LHB muscle and its role in glenohumeral kinematics remains largely in question. The LHB tendon varies in size but most commonly is 5 mm to 6mm in diameter and about 9 cm in length, inserting on the superior labrum and supraglenoid tubercle after traveling through the bicipital groove.1 The many conditions that can develop along the course of the biceps tendon include overall biceps tendonitis, biceps tendon subluxation or instability, and injuries to the superior anterior to posterior area of the labrum.
These injuries can occur in young overhead athletes as well as manual laborers and older overhead recreational athletes. Pitching is the most common activity that leads to proximal biceps tendon disorders. The 6 phases of the pitch are linked in a kinetic chain that generates energy that is then translated to high velocity. The amount of force that is exerted on the shoulder during pitching and especially after ball release is impressive, and the athlete’s shoulder changes in many ways as it adapts to the motion.2-5 The late-cocking and deceleration phases are most commonly associated with proximal biceps pathology and the “peel-back” phenomenon. Other common activities that lead to biceps tendon issues in a young population are volleyball, baseball, tennis, softball, swimming, and cricket. Shoulder arthroscopies performed in older patients show degenerative biceps and labrum tears, which should be treated appropriately but perhaps different from how they are treated in overhead athletes.6-8 Further, many professional athletes have asymptomatic superior labrum anterior-posterior (SLAP) tears.9
Mechanism of Injury
Overhead throwing is commonly thought to be the mechanism by which lesions are created in the biceps–labrum complex (BLC). Pitching in particular generates incredible force and torque within the shoulder. In professional pitchers, the resulting throwing speed creates forces regularly in excess of 1000 N.3 These forces effect internal compensatory changes and internal derangement of the BLC. These changes often involve internal rotation deficits and alterations in the rotator cuff, which may contribute to glenohumeral instability and altered joint kinematics.10
Repetitive overhead activity is largely considered the mechanism of injury in this population, though more specific mechanisms have been described, including the peel-back mechanism11 and the posterior superior glenoid impingement. There is little evidence that preventive programs have any effect on decreasing the incidence of SLAP tears in overhead athletes.
Preoperative Evaluation
Preoperative evaluation is arguably the most important step in treating a patient with persistent or recurrent symptoms consistent with a SLAP tear. Evaluation includes thorough history, physical examination, and review of any prior injuries or surgical procedures. The physical examination should focus on maneuvers that define where the problem is occurring. Although SLAP tears are most common in this population, disorders of the biceps tendon within the groove, including inflammation and instability, should be ruled out with physical examination and advanced imaging. Palpation for groove tenderness, impingement-type complaints, internal rotation loss, and SLAP provocative testing are crucial in the diagnosis.12,13 The cause of symptoms may be multifactorial and include the often encountered concomitant pathology of rotator cuff tears, internal impingement, and instability.
Standard radiographs (Grashey anteroposterior, scapular/lateral, axillary lateral) and magnetic resonance imaging (MRI) with or without arthrography can be helpful in identifying and characterizing most SLAP tears as well as failed SLAP tear repairs. However, MRI is often positive for SLAP tears in asymptomatic patients, and diagnosing SLAP tears with MRI is often a challenge.14 MRI can help in determining concomitant pathology, including rotator cuff injury and cysts causing nerve compression. Correlation with clinical examination and patient history is most crucial. Conservative treatment (rest, activity modification, use of oral anti-inflammatory medications) typically is attempted and coordinated with respect to the athlete’s season of play.15,16
Classification
In overhead throwing athletes, SLAP tears typically are associated with anterior shoulder pain. Associated shoulder instability and significant glenohumeral dysfunction are not uncommon in athletes with lesions of the BLC. In 1985, Andrews and colleagues17 were the first to describe SLAP tears in overhead athletes (73 patients). Later, Snyder and colleagues18,19 further classified these lesions into 4 types based on tear stability and location, and they coined the acronym SLAP (Figure 1).
Type I lesions typically are described as fraying at the inner margin of the labrum and are common in throwers, even asymptomatic throwers. Type II lesions, separations of the biceps and labrum from the superior glenoid (≥5 mm of excursion), are the most commonly occurring and treated variant in throwing athletes.20-22 Intraoperative evaluation for a peel-back lesion (placing the arm in abduction with external rotation), rather than for a sulcus of 1 mm to 2 mm, may confirm a type II SLAP tear.20,23,24 It is often important to consider the direction of tear propagation as well. Type III lesions include those with an intact BLC (but with a bucket-handle tear of the superior labral complex and an intact biceps tendon), whereas type IV lesions involve additional extension of the tear into the biceps tendon.18,19The classification systems are well defined. Nevertheless, management of SLAP lesions remains controversial.
Options for Surgical Treatment
SLAP Tear Repair—Outcomes
The incidence of SLAP tear repairs has increased dramatically in recent years.6,25 There are various SLAP tear repair methods, but the most common consists of repairing the labrum and biceps anchor. Management of type II SLAP lesions remains controversial. Several prospective studies have found overall improvement after SLAP tear repair.26-31 Other series have reported less encouraging outcomes, including dissatisfaction with persistent pain and inability to return to throwing.28,32 A 2010 systematic review found that the percentage of patients who returned to their preinjury level of play was only 64%, and outcomes for overhead throwing athletes were even worse—only 22% to 60% of these patients returned to their previous level.33 The right surgery for SLAP tears in this population continues to be an area of uncertainty for many surgeons.
Failed SLAP tear repairs (poor outcomes) have become common in overhead throwing athletes. The reasons for these failed repairs are unclear, but several possible explanations have been offered. One is that labral repair may result in permanent alterations in pitching biomechanics, which may lead to an inability to regain velocity and command.3 Another is that the athlete’s shoulder may remain unstable even after repair.10Hardware complications are a significant concern in this high-level population. Suture anchor pullout or iatrogenic cartilage damage may occur during instrumentation or as a result of suture anchor reactive changes. In addition, there are several reports of glenoid osteochondrolysis (Figure 2) caused by prominent hardware or prominent knots.34-39
Stiffness after SLAP tear repair is a significant problem, with most patients taking up to 6 months to regain full motion.26,48 Overtensioning of the labrum and the glenohumeral ligaments may be the cause, and the solution may be to place anchors posterior (vs anterior) to the biceps insertion. In a large prospective military study, mean forward flexion and external rotation were reduced at final follow-up.31 These outcomes are less acceptable to overhead throwing athletes, who rely on motion for high-end throwing activities.
Primary Biceps Tenodesis—Outcomes
A 2015 database study found a 1.7-fold increase in biceps tenodesis over the preceding 5 years.49 However, relatively few procedures included in the study were performed in patients age younger than 30 years. For many older non-overhead throwers with type II tears, SLAP tear repair has become less popular as a treatment option.32 There is a dearth of knowledge about the outcomes of subpectoral biceps tenodesis as a primary treatment for biceps tendonitis and an associated SLAP tear. Although type I tears historically have been treated with débridement, débridement is seldom used for concomitant biceps tendonitis. It should be coupled with careful clinical examination.
In recent years, biceps tenodesis has been proposed as an alternative to repair for SLAP tears, particularly in older patients.24,44 For obvious reasons, however, there has been some trepidation about performing biceps tenodesis in throwing athletes. Some authors have proposed biceps tenodesis as primary treatment for isolated SLAP tears. Boileau and colleagues44 compared the outcomes of treatment of isolated type II SLAP lesions in 25 consecutive patients. For 10 patients, repair involved suture anchors; for the other 15, arthroscopic biceps tenodesis was performed with an absorbable interference screw. Six of the 10 suture anchor patients were disappointed with their outcome (persistent pain or inability to return to sport), whereas 14 of the 15 biceps tenodesis patients were satisfied. The authors concluded that arthroscopic biceps tenodesis is an effective alternative to repair for type II SLAP lesions, though their study was not isolated to overhead athletes (tenodesis group mean age, 52 years).
In a 2014 series of cases, Ek and colleagues50 reported good outcomes of SLAP tear repair and biceps tenodesis. Again, though, tenodesis was used in older patients, and repair in younger, more active patients, with no high-level athletes in either group. There was no difference in return to sport between groups. In a study of patients who underwent primary biceps tenodesis, Gupta and colleagues51 found 80% excellent outcomes (improved shoulder outcome scores) in select SLAP tear patients, including 8 athletes, 88% of whom were overhead athletes. Gottschalk and colleagues52 reported on differences in prospectively collected outcome data (age, sex, SLAP lesion type II or IV) for primary biceps tenodesis in a series of 33 patients. Twenty-six of the 29 patients who completed follow-up returned to their previous level of activity. These studies suggest that primary biceps tenodesis may be an alternative with lower failure rates in the treatment of SLAP tears in middle-aged patients, and in overhead athletes, though additional specific studies are needed to focus on overhead athletes on a larger scale.
Revision SLAP Tear Repair Versus Biceps Tenodesis
Failed arthroscopic SLAP tear repairs, which are increasingly common, present a unique treatment challenge. In a 2013 prospective cohort series, Gupta and colleagues46 found excellent clinical outcomes of subpectoral biceps tenodesis for failed type II SLAP tears. The authors reported a postoperative SANE (Single Assessment Numeric Evaluation) score of 70.4%, an SST (Simple Shoulder Test) score of 9.33, and an ASES (American Shoulder and Elbow Surgeons) score of 77.96, along with reasonable health-related quality-of-life scores. Werner and colleagues53 evaluated 2-year outcomes of biceps tenodesis performed after SLAP tear repair in 24 patients and found a return to almost normal range of motion as well as good clinical outcome scores. Significantly worse outcomes were found for patients with open worker’s compensation claims.
McCormick and colleagues26 prospectively evaluated the efficacy of biceps tenodesis for failed type II SLAP tear repair in 46 patients. Improvement was noted across all outcome assessments during follow-up (mean, 3.6 years). From these findings, we might conclude that biceps tenodesis is a more predictable option for failed SLAP tear repair, and that it has a relatively low complication rate. However, most investigators have used a heterogeneous patient population, as opposed to overhead athletes specifically. To our knowledge, no one has evaluated the specific population of overhead throwers with failed SLAP tear repairs. In addition, no one has conducted randomized controlled trials comparing débridement, biceps tenodesis, and repair for failed SLAP tear repairs.
Postoperative Considerations
When overhead athletes and their surgeons are considering surgical options, they must take rehabilitation and return to play into account. Many surgeons think the possible marginal clinical benefit of SLAP tear repair may not be worth the protracted rehabilitation. In most practices, rehabilitation after biceps tenodesis is less involved. Discussing the advantages and disadvantages of these 2 procedures can be helpful in decision making.
Dein and colleagues54 reported the case of a middle-aged pitcher who sustained a fracture after biceps tenodesis with an interference screw. Cases like this are concerning. Surgeons should consider altering the rehabilitation regimen when planning postoperative care in cases of biceps tenodesis in throwers. Other reported complications of open tenodesis are deep infection, thrombosis, postoperative stiffness, and nerve injury.55-58
Consequences for Overhead Throwers
The unknown role of the BLC leaves surgeons wary when considering biceps tenodesis for elite athletes. Some have postulated that removing the intra-articular portion of the LHB may cause microinstability and alter joint kinematics.10,59-61 Others have suggested the biceps is desynchronized from the other musculature and is not functionally important.62 Disruption of one portion of the superior labrum may result in instability on the opposite side of the glenoid.10,61 Biomechanical studies, both cadaveric and in vivo, have tried to create proper loads to the LHB and evaluate the kinematics of the shoulder before and after biceps tenodesis and SLAP tear repair.59,60 Using a cadaveric model, Strauss and colleagues63 found that type II SLAP lesions resulted in increased glenohumeral translation compared with baseline. Biceps tenodesis did not restore normal translation, but this did not negatively affect stability in the presence of a SLAP lesion. The consensus is that the role of the biceps is controversial at best.
Several studies have used electromyography (EMG) to evaluate LHB functioning. In 2014, Chalmers and colleagues59 used surface EMG and motion analysis to evaluate 18 pitchers: 6 underwent SLAP tear repair, 5 underwent biceps tenodesis, and 7 were uninjured controls. There were no significant differences in the activity of the LHB muscle, the short head of the biceps muscle, the deltoid, the infraspinatus, or the latissimus among the 3 groups. Motion analysis showed that the normal pattern of muscular activation within the LHB muscle was more closely restored by biceps tenodesis than by SLAP tear repair. In addition, thoracic rotation patterns were significantly more altered in the SLAP tear repair patients than in the uninjured controls. As the authors noted, given the low frequency with which biceps tenodesis is performed in overhead athletes, it is unlikely that larger scale studies will be conducted without a multicenter effort.
Recommendations and Our Preferred Technique
Which surgical option is best for treating symptomatic SLAP lesions in overhead athletes remains unclear. Many athletes struggle to return to high-level play after SLAP tear repair. Whether the same is true after biceps tenodesis is yet to be determined because of the low frequency with which biceps tenodesis is performed in high-level overhead athletes. The options for fixation, technique, and fixation location are equally broad. In this section, we outline our general line of thinking for cases of proximal biceps pathology.
In each case, we perform glenohumeral arthroscopy to evaluate the BLC and identify any other pathology. For overhead athletes who are younger than 30 years and lack bicipital groove pain or signs of gross tendinopathy, we favor arthroscopic SLAP tear repair. Repair is usually performed through an anterior working portal for suture passage and a Wilmington portal for anchor placement. We use knotless technology to achieve stable fixation and stay posterior to the biceps anchor insertion.
For the prevention of any potential pain from the bicipital groove in carefully selected patients—recreational overhead athletes and patients who want a less involved surgical recovery—we favor open subpectoral biceps tenodesis rather than arthroscopic tenodesis. The outcomes of biceps tenodesis are consistent, according to the literature.47,57,64 Moreover, the open approach is favored for the incidence of postoperative stiffness in the arthroscopic population.65 Tendons can be fixed with multiple procedures, including soft-tissue tenodesis, interference screw fixation, and surface anchors. We favor using a tenodesis screw in the subpectoral location, as outlined by Mazzocca and colleagues.64Our algorithm for SLAP lesions is evolving with our understanding of this complex disease process. For young overhead throwers with type II SLAP lesions, we favor arthroscopic SLAP tear repair with knotless technology. For older recreational overhead athletes, we favor biceps tenodesis in the subpectoral region after diagnostic arthroscopy plus biceps tenotomy with or without additional SLAP tear fixation, depending on the stability of the biceps anchor (Figures 4A, 4B).
Conclusion
Overhead athletes who present with symptomatic SLAP lesions often provide a treatment dilemma. Although SLAP tear repair historically has been standard treatment, biceps tenodesis represents a consistent surgical option with low complication rates and low revision rates. It is likely that, as additional data on glenohumeral kinematics and outcomes in young athletes become available, improved decision-making algorithms will follow.
Am J Orthop. 2017;46(1):E71-E78. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Elser F, Braun S, Dewing CB, Giphart JE, Millett PJ. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27(4):581-592.
2. Fedoriw WW, Ramkumar P, McCulloch PC, Lintner DM. Return to play after treatment of superior labral tears in professional baseball players. Am J Sports Med. 2014;42(5):1155-1160.
3. Fleisig GS, Andrews JR, Dillman CJ, Escamilla RF. Kinetics of baseball pitching with implications about injury mechanisms. Am J Sports Med. 1995;23(2):233-239.
4. Aydin N, Sirin E, Arya A. Superior labrum anterior to posterior lesions of the shoulder: diagnosis and arthroscopic management. World J Orthop. 2014;5(3):344-350.
5. Barber A, Field LD, Ryu R. Biceps tendon and superior labrum injuries: decision-marking. J Bone Joint Surg Am. 2007;89(8):1844-1855.
6. Onyekwelu I, Khatib O, Zuckerman JD, Rokito AS, Kwon YW. The rising incidence of arthroscopic superior labrum anterior and posterior (SLAP) repairs. J Shoulder Elbow Surg. 2012;21(6):728-731.
7. Patterson BM, Creighton RA, Spang JT, Roberson JR, Kamath GV. Surgical trends in the treatment of superior labrum anterior and posterior lesions of the shoulder: analysis of data from the American Board of Orthopaedic Surgery Certification Examination Database. Am J Sports Med. 2014;42(8):1904-1910.
8. Walton DM, Sadi J. Identifying SLAP lesions: a meta-analysis of clinical tests and exercise in clinical reasoning. Phys Ther Sport. 2008;9(4):167-176.
9. Lesniak BP, Baraga MG, Jose J, Smith MK, Cunningham S, Kaplan LD. Glenohumeral findings on magnetic resonance imaging correlate with innings pitched in asymptomatic pitchers. Am J Sports Med. 2013;41(9):2022-2027.
10. Mihata T, McGarry MH, Tibone JE, Fitzpatrick MJ, Kinoshita M, Lee TQ. Biomechanical assessment of type II superior labral anterior-posterior (SLAP) lesions associated with anterior shoulder capsular laxity as seen in throwers: a cadaveric study. Am J Sports Med. 2008;36(8):1604-1610.
11. Burkhart SS, Morgan CD, Kibler WB. The disabled throwing shoulder: spectrum of pathology. Part II: evaluation and treatment of SLAP lesions in throwers. Arthroscopy. 2003;19(5):531-539.
12. Meserve BB, Cleland JA, Boucher TR. A meta-analysis examining clinical test utility for assessing superior labral anterior posterior lesions. Am J Sports Med. 2009;37(11):2252-2258.
13. Pandya NK, Colton A, Webner D, Sennett B, Huffman GR. Physical examination and magnetic resonance imaging in the diagnosis of superior labrum anterior-posterior lesions of the shoulder: a sensitivity analysis. Arthroscopy. 2008;24(3):311-317.
14. Amin MF, Youssef AO. The diagnostic value of magnetic resonance arthrography of the shoulder in detection and grading of SLAP lesions: comparison with arthroscopic findings. Eur J Radiol. 2012;81(9):2343-2347.
15. Cook C, Beaty S, Kissenberth MJ, Siffri P, Pill SG, Hawkins RJ. Diagnostic accuracy of five orthopedic clinical tests for diagnosis of superior labrum anterior posterior (SLAP) lesions. J Shoulder Elbow Surg. 2012;21(1):13-22.
16. Edwards SL, Lee JA, Bell JE, et al. Nonoperative treatment of superior labrum anterior posterior tears: improvements in pain, function, and quality of life. Am J Sports Med. 2010;38(7):1456-1461.
17. Andrews JR, Carson WG Jr, McLeod WD. Glenoid labrum tears related to the long head of the biceps. Am J Sports Med. 1985;13(5):337-341.
18. Snyder SJ, Karzel RP, Del Pizzo W, Ferkel RD, Friedman MJ. SLAP lesions of the shoulder. Arthroscopy. 1990;6(4):274-279.
19. Snyder SJ, Banas MP, Karzel RP. An analysis of 140 injuries to the superior glenoid labrum. J Shoulder Elbow Surg. 1995;4(4):243-248.
20. Morgan CD, Burkhart SS, Palmeri M, Gillespie M. Type II SLAP lesions: three subtypes and their relationships to superior instability and rotator cuff tears. Arthroscopy. 1998;14(6):553-565.
21. Weber SC, Martin DF, Seiler JG 3rd, Harrast JJ. Superior labrum anterior and posterior lesions of the shoulder: incidence rates, complications, and outcomes as reported by American Board of Orthopedic Surgery. Part II candidates. Am J Sports Med. 2012;40(7):1538-1543.
22. Keener JD, Brophy RH. Superior labral tears of the shoulder: pathogenesis, evaluation, and treatment. J Am Acad Orthop Surg. 2009;17(10):627-637.
23. Chen CH, Hsu KY, Chen WJ, Shih CH. Incidence and severity of biceps long head tendon lesion in patients with complete rotator cuff tears. J Trauma. 2005;58(6):1189-1193.
24. Nho SJ, Strauss EJ, Lenart BA, et al. Long head of the biceps tendinopathy: diagnosis and management. J Am Acad Orthop Surg. 2010;18(11):645-656.
25. Zhang AL, Kreulen C, Ngo SS, Hame SL, Wang JC, Gamradt SC. Demographic trends in arthroscopic SLAP repair in the United States. Am J Sports Med. 2012;40(5):1144-1147.
26. McCormick F, Bhatia S, Chalmers P, Gupta A, Verma N, Romeo AA. The management of type II superior labral anterior to posterior injuries. Orthop Clin North Am. 2014;45(1):121-128.
27. Brockmeier SF, Voos JE, Williams RJ 3rd, Altchek DW, Cordasco FA, Allen AA; Hospital for Special Surgery Sports Medicine and Shoulder Service. Outcomes after arthroscopic repair of type-II SLAP lesions. J Bone Joint Surg Am. 2009;91(7):1595-1603.
28. Boileau P, Parratte S, Chuinard C, Roussanne Y, Shia D, Bicknell R. Arthroscopic treatment of isolated type II SLAP lesions: biceps tenodesis as an alternative to reinsertion. Am J Sports Med. 2009;37(5):929-936.
29. Denard PJ, Lädermann A, Burkhart SS. Long-term outcome after arthroscopic repair of type II SLAP lesions: results according to age and workers’ compensation status. Arthroscopy. 2012;28(4):451-457.
30. Friel NA, Karas V, Slabaugh MA, Cole BJ. Outcomes of type II superior labrum, anterior to posterior (SLAP) repair: prospective evaluation at a minimum two-year follow-up. J Shoulder Elbow Surg. 2010;19(6):859-867.
31. Provencher MT, McCormick F, Dewing C, McIntire S, Solomon D. A prospective analysis of 179 type 2 superior labrum anterior and posterior repairs: outcomes and factors associated with success and failure. Am J Sports Med. 2013;41(4):880-886.
32. Gupta AK, Chalmers PN, Klosterman EL, et al. Subpectoral biceps tenodesis for bicipital tendonitis with SLAP tear. Orthopedics. 2015;38(1):e48-e53.
33. Gorantla K, Gill C, Wright RW. The outcome of type II SLAP repair: a systematic review. Arthroscopy. 2010;26(4):537-545.
34. Katz LM, Hsu S, Miller SL, et al. Poor outcomes after SLAP repair: descriptive analysis and prognosis. Arthroscopy. 2009;25(8):849-855.
35. Park MJ, Hsu JE, Harper C, Sennett BJ, Huffman GR. Poly-L/D-lactic acid anchors are associated with reoperation and failure of SLAP repairs. Arthroscopy. 2011;27(10):1335-1340.
36. Sassmannshausen G, Sukay M, Mair SD. Broken or dislodged poly-L-lactic acid bioabsorbable tacks in patients after SLAP lesion surgery. Arthroscopy. 2006;22(6):615-619.
37. Uggen C, Wei A, Glousman RE, et al. Biomechanical comparison of knotless anchor repair versus simple suture repair for type II SLAP lesions. Arthroscopy. 2009;25(10):1085-1092.
38. Weber SC. Surgical management of the failed SLAP repair. Sports Med Arthrosc. 2010;18(3):162-166.
39. Wilkerson JP, Zvijac JE, Uribe JW, Schürhoff MR, Green JB. Failure of polymerized lactic acid tacks in shoulder surgery. J Shoulder Elbow Surg. 2003;12(2):117-121.
40. Weber S. Surgical management of the failed SLAP lesion. Arthroscopy. 2008;24(suppl):e8-e9.
41. Schrøder CP, Skare O, Gjengedal E, Uppheim G, Reikerås O, Brox JI. Long-term results after SLAP repair: a 5-year follow-up study of 107 patients with comparison of patients aged over and under 40 years. Arthroscopy. 2012;28(11):1601-1607.
42. Mazzocca AD, Cote MP, Arciero CL, Romeo AA, Arciero RA. Clinical outcomes after subpectoral biceps tenodesis with an interference screw. Am J Sports Med. 2008;36(10):1922-1929.
43. Mazzocca AD, McCarthy MB, Ledgard FA, et al. Histomorphologic changes of the long head of the biceps tendon in common shoulder pathologies. Arthroscopy. 2013;29(6):972-981.
44. Boileau P, Parratte S, Chuinard C, Roussanne Y, Shia D, Bicknell R. Arthroscopic treatment of isolated type II SLAP lesions: biceps tenodesis as an alternative to reinsertion. Am J Sports Med. 2009;37(5):929-936.
45. Boileau P, Krishnan SG, Coste JS, Walch G. Arthroscopic biceps tenodesis: a new technique using bioabsorbable interference screw fixation. Arthroscopy. 2002;18(9):1002-1012.
46. Gupta AK, Bruce B, Klosterman EL, McCormick F, Harris J, Romeo AA. Subpectoral biceps tenodesis for failed type II SLAP repair. Orthopedics. 2013;36(6):e723-e728.
47. Provencher MT, LeClere LE, Romeo AA. Subpectoral biceps tenodesis. Sports Med Arthrosc. 2008;16(3):170-176.
48. McCarty LP 3rd, Buss DD, Datta MW, Freehill MQ, Giveans MR. Complications observed following labral or rotator cuff repair with use of poly-L-lactic acid implants. J Bone Joint Surg Am. 2013;95(6):507-511.
49. Werner BC, Brockmeier SF, Gwathmey FW. Trends in long head biceps tenodesis. Am J Sports Med. 2015;43(3):570-578.
50. Ek ET, Shi LL, Tompson JD, Freehill MT, Warner JJ. Surgical treatment of isolated type II superior labrum anterior-posterior (SLAP) lesions: repair versus biceps tenodesis. J Shoulder Elbow Surg. 2014;23(7):1059-1065.
51. Gupta AK, Chalmers PN, Klosterman EL, et al. Subpectoral biceps tenodesis for bicipital tendonitis with SLAP tear. Orthopedics. 2015;38(1):e48-e53.
52. Gottschalk MB, Karas SG, Ghattas TN, Burdette R. Subpectoral biceps tenodesis for the treatment of type II and IV superior labral anterior and posterior lesions. Am J Sports Med. 2014;42(9):2128-2135.
53. Werner BC, Pehlivan HC, Hart JM, et al. Biceps tenodesis is a viable option for salvage of failed SLAP repair. J Shoulder Elbow Surg. 2014;23(8):e179-e184.
54. Dein EJ, Huri G, Gordon JC, McFarland EG. A humerus fracture in a baseball pitcher after biceps tenodesis [published correction appears in Am J Sports Med. 2014;42(6):NP39]. Am J Sports Med. 2014;42(4):877-879.
55. Nho SJ, Reiff SN, Verma NN, Slabaugh MA, Mazzocca AD, Romeo AA. Complications associated with subpectoral biceps tenodesis: low rates of incidence following surgery. J Shoulder Elbow Surg. 2010;19(5):764-768.
56. Osbahr DC, Diamond AB, Speer KP. The cosmetic appearance of the biceps muscle after long-head tenotomy versus tenodesis. Arthroscopy. 2002;18(5):483-487.
57. Romeo AA, Mazzocca AD, Tauro JC. Arthroscopic biceps tenodesis. Arthroscopy. 2004;20(2):206-213.
58. Ma H, Van Heest A, Glisson C, Patel S. Musculocutaneous nerve entrapment: an unusual complication after biceps tenodesis. Am J Sports Med. 2009;37(12):2467-2469.
59. Chalmers PN, Trombley R, Cip J, et al. Postoperative restoration of upper extremity motion and neuromuscular control during the overhand pitch: evaluation of tenodesis and repair for superior labral anterior-posterior tears. Am J Sports Med. 2014;42(12):2825-2836.
60. Giphart JE, Elser F, Dewing CB, Torry MR, Millett PJ. The long head of the biceps tendon has minimal effect on in vivo glenohumeral kinematics: a biplane fluoroscopy study. Am J Sports Med. 2012;40(1):202-212.
61. Grossman MG, Tibone JE, McGarry MH, Schneider DJ, Veneziani S, Lee TQ. A cadaveric model of the throwing shoulder: a possible etiology of superior labrum anterior-to-posterior lesions. J Bone Joint Surg Am. 2005;87(4):824-831.
62. Hawkes DH, Alizadehkhaiyat O, Fisher AC, Kemp GJ, Roebuck MM, Frostick SP. Normal shoulder muscular activation and co-ordination during a shoulder elevation task based on activities of daily living: an electromyographic study. J Orthop Res. 2012;30(1):53-60.
63. Strauss EJ, Salata MJ, Sershon RA, et al. Role of the superior labrum after biceps tenodesis in glenohumeral stability. J Shoulder Elbow Surg. 2014;23(4):485-491.
64. Mazzocca AD, Bicos J, Santangelo S, Romeo AA, Arciero RA. The biomechanical evaluation of four fixation techniques for proximal biceps tenodesis. Arthroscopy. 2005;21(11):1296-1306.
65. Werner BC, Pehlivan HC, Hart JM, et al. Increased incidence of postoperative stiffness after arthroscopic compared with open biceps tenodesis. Arthroscopy. 2014;30(9):1075-1084.
66. Denard PJ, Dai X, Hanypsiak BT, Burkhart SS. Anatomy of the biceps tendon: implications for restoring physiological length–tension relation during biceps tenodesis with interference screw fixation. Arthroscopy. 2012;28(10):1352-1358.
67. Mazzocca AD, Rios CG, Romeo AA, Arciero RA. Subpectoral biceps tenodesis with interference screw fixation. Arthroscopy. 2005;21(7):896.
1. Elser F, Braun S, Dewing CB, Giphart JE, Millett PJ. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27(4):581-592.
2. Fedoriw WW, Ramkumar P, McCulloch PC, Lintner DM. Return to play after treatment of superior labral tears in professional baseball players. Am J Sports Med. 2014;42(5):1155-1160.
3. Fleisig GS, Andrews JR, Dillman CJ, Escamilla RF. Kinetics of baseball pitching with implications about injury mechanisms. Am J Sports Med. 1995;23(2):233-239.
4. Aydin N, Sirin E, Arya A. Superior labrum anterior to posterior lesions of the shoulder: diagnosis and arthroscopic management. World J Orthop. 2014;5(3):344-350.
5. Barber A, Field LD, Ryu R. Biceps tendon and superior labrum injuries: decision-marking. J Bone Joint Surg Am. 2007;89(8):1844-1855.
6. Onyekwelu I, Khatib O, Zuckerman JD, Rokito AS, Kwon YW. The rising incidence of arthroscopic superior labrum anterior and posterior (SLAP) repairs. J Shoulder Elbow Surg. 2012;21(6):728-731.
7. Patterson BM, Creighton RA, Spang JT, Roberson JR, Kamath GV. Surgical trends in the treatment of superior labrum anterior and posterior lesions of the shoulder: analysis of data from the American Board of Orthopaedic Surgery Certification Examination Database. Am J Sports Med. 2014;42(8):1904-1910.
8. Walton DM, Sadi J. Identifying SLAP lesions: a meta-analysis of clinical tests and exercise in clinical reasoning. Phys Ther Sport. 2008;9(4):167-176.
9. Lesniak BP, Baraga MG, Jose J, Smith MK, Cunningham S, Kaplan LD. Glenohumeral findings on magnetic resonance imaging correlate with innings pitched in asymptomatic pitchers. Am J Sports Med. 2013;41(9):2022-2027.
10. Mihata T, McGarry MH, Tibone JE, Fitzpatrick MJ, Kinoshita M, Lee TQ. Biomechanical assessment of type II superior labral anterior-posterior (SLAP) lesions associated with anterior shoulder capsular laxity as seen in throwers: a cadaveric study. Am J Sports Med. 2008;36(8):1604-1610.
11. Burkhart SS, Morgan CD, Kibler WB. The disabled throwing shoulder: spectrum of pathology. Part II: evaluation and treatment of SLAP lesions in throwers. Arthroscopy. 2003;19(5):531-539.
12. Meserve BB, Cleland JA, Boucher TR. A meta-analysis examining clinical test utility for assessing superior labral anterior posterior lesions. Am J Sports Med. 2009;37(11):2252-2258.
13. Pandya NK, Colton A, Webner D, Sennett B, Huffman GR. Physical examination and magnetic resonance imaging in the diagnosis of superior labrum anterior-posterior lesions of the shoulder: a sensitivity analysis. Arthroscopy. 2008;24(3):311-317.
14. Amin MF, Youssef AO. The diagnostic value of magnetic resonance arthrography of the shoulder in detection and grading of SLAP lesions: comparison with arthroscopic findings. Eur J Radiol. 2012;81(9):2343-2347.
15. Cook C, Beaty S, Kissenberth MJ, Siffri P, Pill SG, Hawkins RJ. Diagnostic accuracy of five orthopedic clinical tests for diagnosis of superior labrum anterior posterior (SLAP) lesions. J Shoulder Elbow Surg. 2012;21(1):13-22.
16. Edwards SL, Lee JA, Bell JE, et al. Nonoperative treatment of superior labrum anterior posterior tears: improvements in pain, function, and quality of life. Am J Sports Med. 2010;38(7):1456-1461.
17. Andrews JR, Carson WG Jr, McLeod WD. Glenoid labrum tears related to the long head of the biceps. Am J Sports Med. 1985;13(5):337-341.
18. Snyder SJ, Karzel RP, Del Pizzo W, Ferkel RD, Friedman MJ. SLAP lesions of the shoulder. Arthroscopy. 1990;6(4):274-279.
19. Snyder SJ, Banas MP, Karzel RP. An analysis of 140 injuries to the superior glenoid labrum. J Shoulder Elbow Surg. 1995;4(4):243-248.
20. Morgan CD, Burkhart SS, Palmeri M, Gillespie M. Type II SLAP lesions: three subtypes and their relationships to superior instability and rotator cuff tears. Arthroscopy. 1998;14(6):553-565.
21. Weber SC, Martin DF, Seiler JG 3rd, Harrast JJ. Superior labrum anterior and posterior lesions of the shoulder: incidence rates, complications, and outcomes as reported by American Board of Orthopedic Surgery. Part II candidates. Am J Sports Med. 2012;40(7):1538-1543.
22. Keener JD, Brophy RH. Superior labral tears of the shoulder: pathogenesis, evaluation, and treatment. J Am Acad Orthop Surg. 2009;17(10):627-637.
23. Chen CH, Hsu KY, Chen WJ, Shih CH. Incidence and severity of biceps long head tendon lesion in patients with complete rotator cuff tears. J Trauma. 2005;58(6):1189-1193.
24. Nho SJ, Strauss EJ, Lenart BA, et al. Long head of the biceps tendinopathy: diagnosis and management. J Am Acad Orthop Surg. 2010;18(11):645-656.
25. Zhang AL, Kreulen C, Ngo SS, Hame SL, Wang JC, Gamradt SC. Demographic trends in arthroscopic SLAP repair in the United States. Am J Sports Med. 2012;40(5):1144-1147.
26. McCormick F, Bhatia S, Chalmers P, Gupta A, Verma N, Romeo AA. The management of type II superior labral anterior to posterior injuries. Orthop Clin North Am. 2014;45(1):121-128.
27. Brockmeier SF, Voos JE, Williams RJ 3rd, Altchek DW, Cordasco FA, Allen AA; Hospital for Special Surgery Sports Medicine and Shoulder Service. Outcomes after arthroscopic repair of type-II SLAP lesions. J Bone Joint Surg Am. 2009;91(7):1595-1603.
28. Boileau P, Parratte S, Chuinard C, Roussanne Y, Shia D, Bicknell R. Arthroscopic treatment of isolated type II SLAP lesions: biceps tenodesis as an alternative to reinsertion. Am J Sports Med. 2009;37(5):929-936.
29. Denard PJ, Lädermann A, Burkhart SS. Long-term outcome after arthroscopic repair of type II SLAP lesions: results according to age and workers’ compensation status. Arthroscopy. 2012;28(4):451-457.
30. Friel NA, Karas V, Slabaugh MA, Cole BJ. Outcomes of type II superior labrum, anterior to posterior (SLAP) repair: prospective evaluation at a minimum two-year follow-up. J Shoulder Elbow Surg. 2010;19(6):859-867.
31. Provencher MT, McCormick F, Dewing C, McIntire S, Solomon D. A prospective analysis of 179 type 2 superior labrum anterior and posterior repairs: outcomes and factors associated with success and failure. Am J Sports Med. 2013;41(4):880-886.
32. Gupta AK, Chalmers PN, Klosterman EL, et al. Subpectoral biceps tenodesis for bicipital tendonitis with SLAP tear. Orthopedics. 2015;38(1):e48-e53.
33. Gorantla K, Gill C, Wright RW. The outcome of type II SLAP repair: a systematic review. Arthroscopy. 2010;26(4):537-545.
34. Katz LM, Hsu S, Miller SL, et al. Poor outcomes after SLAP repair: descriptive analysis and prognosis. Arthroscopy. 2009;25(8):849-855.
35. Park MJ, Hsu JE, Harper C, Sennett BJ, Huffman GR. Poly-L/D-lactic acid anchors are associated with reoperation and failure of SLAP repairs. Arthroscopy. 2011;27(10):1335-1340.
36. Sassmannshausen G, Sukay M, Mair SD. Broken or dislodged poly-L-lactic acid bioabsorbable tacks in patients after SLAP lesion surgery. Arthroscopy. 2006;22(6):615-619.
37. Uggen C, Wei A, Glousman RE, et al. Biomechanical comparison of knotless anchor repair versus simple suture repair for type II SLAP lesions. Arthroscopy. 2009;25(10):1085-1092.
38. Weber SC. Surgical management of the failed SLAP repair. Sports Med Arthrosc. 2010;18(3):162-166.
39. Wilkerson JP, Zvijac JE, Uribe JW, Schürhoff MR, Green JB. Failure of polymerized lactic acid tacks in shoulder surgery. J Shoulder Elbow Surg. 2003;12(2):117-121.
40. Weber S. Surgical management of the failed SLAP lesion. Arthroscopy. 2008;24(suppl):e8-e9.
41. Schrøder CP, Skare O, Gjengedal E, Uppheim G, Reikerås O, Brox JI. Long-term results after SLAP repair: a 5-year follow-up study of 107 patients with comparison of patients aged over and under 40 years. Arthroscopy. 2012;28(11):1601-1607.
42. Mazzocca AD, Cote MP, Arciero CL, Romeo AA, Arciero RA. Clinical outcomes after subpectoral biceps tenodesis with an interference screw. Am J Sports Med. 2008;36(10):1922-1929.
43. Mazzocca AD, McCarthy MB, Ledgard FA, et al. Histomorphologic changes of the long head of the biceps tendon in common shoulder pathologies. Arthroscopy. 2013;29(6):972-981.
44. Boileau P, Parratte S, Chuinard C, Roussanne Y, Shia D, Bicknell R. Arthroscopic treatment of isolated type II SLAP lesions: biceps tenodesis as an alternative to reinsertion. Am J Sports Med. 2009;37(5):929-936.
45. Boileau P, Krishnan SG, Coste JS, Walch G. Arthroscopic biceps tenodesis: a new technique using bioabsorbable interference screw fixation. Arthroscopy. 2002;18(9):1002-1012.
46. Gupta AK, Bruce B, Klosterman EL, McCormick F, Harris J, Romeo AA. Subpectoral biceps tenodesis for failed type II SLAP repair. Orthopedics. 2013;36(6):e723-e728.
47. Provencher MT, LeClere LE, Romeo AA. Subpectoral biceps tenodesis. Sports Med Arthrosc. 2008;16(3):170-176.
48. McCarty LP 3rd, Buss DD, Datta MW, Freehill MQ, Giveans MR. Complications observed following labral or rotator cuff repair with use of poly-L-lactic acid implants. J Bone Joint Surg Am. 2013;95(6):507-511.
49. Werner BC, Brockmeier SF, Gwathmey FW. Trends in long head biceps tenodesis. Am J Sports Med. 2015;43(3):570-578.
50. Ek ET, Shi LL, Tompson JD, Freehill MT, Warner JJ. Surgical treatment of isolated type II superior labrum anterior-posterior (SLAP) lesions: repair versus biceps tenodesis. J Shoulder Elbow Surg. 2014;23(7):1059-1065.
51. Gupta AK, Chalmers PN, Klosterman EL, et al. Subpectoral biceps tenodesis for bicipital tendonitis with SLAP tear. Orthopedics. 2015;38(1):e48-e53.
52. Gottschalk MB, Karas SG, Ghattas TN, Burdette R. Subpectoral biceps tenodesis for the treatment of type II and IV superior labral anterior and posterior lesions. Am J Sports Med. 2014;42(9):2128-2135.
53. Werner BC, Pehlivan HC, Hart JM, et al. Biceps tenodesis is a viable option for salvage of failed SLAP repair. J Shoulder Elbow Surg. 2014;23(8):e179-e184.
54. Dein EJ, Huri G, Gordon JC, McFarland EG. A humerus fracture in a baseball pitcher after biceps tenodesis [published correction appears in Am J Sports Med. 2014;42(6):NP39]. Am J Sports Med. 2014;42(4):877-879.
55. Nho SJ, Reiff SN, Verma NN, Slabaugh MA, Mazzocca AD, Romeo AA. Complications associated with subpectoral biceps tenodesis: low rates of incidence following surgery. J Shoulder Elbow Surg. 2010;19(5):764-768.
56. Osbahr DC, Diamond AB, Speer KP. The cosmetic appearance of the biceps muscle after long-head tenotomy versus tenodesis. Arthroscopy. 2002;18(5):483-487.
57. Romeo AA, Mazzocca AD, Tauro JC. Arthroscopic biceps tenodesis. Arthroscopy. 2004;20(2):206-213.
58. Ma H, Van Heest A, Glisson C, Patel S. Musculocutaneous nerve entrapment: an unusual complication after biceps tenodesis. Am J Sports Med. 2009;37(12):2467-2469.
59. Chalmers PN, Trombley R, Cip J, et al. Postoperative restoration of upper extremity motion and neuromuscular control during the overhand pitch: evaluation of tenodesis and repair for superior labral anterior-posterior tears. Am J Sports Med. 2014;42(12):2825-2836.
60. Giphart JE, Elser F, Dewing CB, Torry MR, Millett PJ. The long head of the biceps tendon has minimal effect on in vivo glenohumeral kinematics: a biplane fluoroscopy study. Am J Sports Med. 2012;40(1):202-212.
61. Grossman MG, Tibone JE, McGarry MH, Schneider DJ, Veneziani S, Lee TQ. A cadaveric model of the throwing shoulder: a possible etiology of superior labrum anterior-to-posterior lesions. J Bone Joint Surg Am. 2005;87(4):824-831.
62. Hawkes DH, Alizadehkhaiyat O, Fisher AC, Kemp GJ, Roebuck MM, Frostick SP. Normal shoulder muscular activation and co-ordination during a shoulder elevation task based on activities of daily living: an electromyographic study. J Orthop Res. 2012;30(1):53-60.
63. Strauss EJ, Salata MJ, Sershon RA, et al. Role of the superior labrum after biceps tenodesis in glenohumeral stability. J Shoulder Elbow Surg. 2014;23(4):485-491.
64. Mazzocca AD, Bicos J, Santangelo S, Romeo AA, Arciero RA. The biomechanical evaluation of four fixation techniques for proximal biceps tenodesis. Arthroscopy. 2005;21(11):1296-1306.
65. Werner BC, Pehlivan HC, Hart JM, et al. Increased incidence of postoperative stiffness after arthroscopic compared with open biceps tenodesis. Arthroscopy. 2014;30(9):1075-1084.
66. Denard PJ, Dai X, Hanypsiak BT, Burkhart SS. Anatomy of the biceps tendon: implications for restoring physiological length–tension relation during biceps tenodesis with interference screw fixation. Arthroscopy. 2012;28(10):1352-1358.
67. Mazzocca AD, Rios CG, Romeo AA, Arciero RA. Subpectoral biceps tenodesis with interference screw fixation. Arthroscopy. 2005;21(7):896.
Actinomycetoma: An Update on Diagnosis and Treatment
Mycetoma is a subcutaneous disease that can be caused by aerobic bacteria (actinomycetoma) or fungi (eumycetoma). Diagnosis is based on clinical manifestations, including swelling and deformity of affected areas, as well as the presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate that contains the microorganisms.
The worldwide proportion of mycetomas is 60% actinomycetomas and 40% eumycetomas.1 The disease is endemic in tropical, subtropical, and temperate regions, predominating between latitudes 30°N and 15°S. Most cases occur in Africa, especially Sudan, Mauritania, and Senegal; India; Yemen; and Pakistan. In the Americas, the countries with the most reported cases are Mexico and Venezuela.1
Although mycetoma is rare in developed countries, migration of patients from endemic areas makes knowledge of this condition crucial for dermatologists worldwide. We present a review of the current concepts in the epidemiology, clinical presentation, diagnosis, and treatment of actinomycetoma.
Epidemiology
Actinomycetoma is more common in Latin America, with Mexico having the highest incidence. At last count, there were 2631 cases reported in Mexico.2 The majority of cases of mycetoma in Mexico are actinomycetoma (98%), including Nocardia (86%) and Actinomadura madurae (10%). Eumycetoma is rare in Mexico, constituting only 2% of cases.2 Worldwide, men are affected more commonly than women, which is thought to be related to a higher occupational risk during agricultural labor.
Clinical Features
Mycetoma can affect the skin, subcutaneous tissue, bones, and occasionally the internal organs. It is characterized by swelling, deformation of the affected area, and fistulae that drain serosanguineous or purulent exudates.
In Mexico, 60% of cases of mycetoma affect the lower extremities; the feet are the most commonly affected area, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.1 Other sites include the hands, shoulders, and abdominal wall. The head and neck area are seldom affected.3 Mycetoma lesions grow and disseminate locally. Bone lesions are possible depending on the osteophilic affinity of the etiological agent and on the interactions between the fungus and the host’s immune system. In severe advanced cases of mycetoma, the lesions may involve tendons and nerves. Dissemination via blood or lymphatics is extremely rare.4
Diagnosis
Diagnosis of actinomycetoma is suspected based on clinical features and confirmed by direct examination of exudates with Lugol iodine or saline solution. On direct microscopy, actinomycetes are recognized by the production of filaments with a width of 0.5 to 1 μm. On hematoxylin and eosin stain, the small grains of Nocardia appear eosinophilic with a blue center and pink filaments. On Gram stain, actinomycetoma grains show positive branching filaments. Culture of grains recovered from aspirated material or biopsy specimens provides specific etiologic diagnosis. Cultures should be held for at least 4 weeks. Additionally, there are some enzymatic, molecular, and serologic tests available for diagnosis.5-7 Serologic diagnosis is available in a few centers in Mexico and can be helpful in some cases for diagnosis or follow-up during treatment. Antibodies can be determined via enzyme-linked immunosorbent assay, Western blot analysis, immunodiffusion, or counterimmunoelectrophoresis.8
The causative agents of actinomycetoma can be isolated in Sabouraud dextrose agar. Deep wedge biopsies (or puncture aspiration) are useful in observing the diagnostic grains, which can be identified adequately with Gram stain. Grains usually are surrounded and/or infiltrated by neutrophils. The size, form, and color of grains can identify the causative agent.1 The granules of Nocardia are small (80–130 mm) and reniform or wormlike, with club structures in their periphery (Figure 1). Actinomadura madurae is characterized by large, white-yellow granules that can be seen with the naked eye (1–3 mm). On microscopic examination with hematoxylin and eosin stain, these grains are purple and exhibit peripheral pink pseudofilaments (Figure 2).2 The grains of Actinomadura pelletieri are large (1–3 mm) and red or violaceous. They fragment or break easily, giving the appearance of a broken dish (Figure 3). Streptomyces somaliensis forms round grains approximately 0.5 to 1 cm in diameter. These grains stain poorly and are extremely hard. Cutting the grains during processing results in striation, giving them the appearance of a potato chip (Figure 4).2

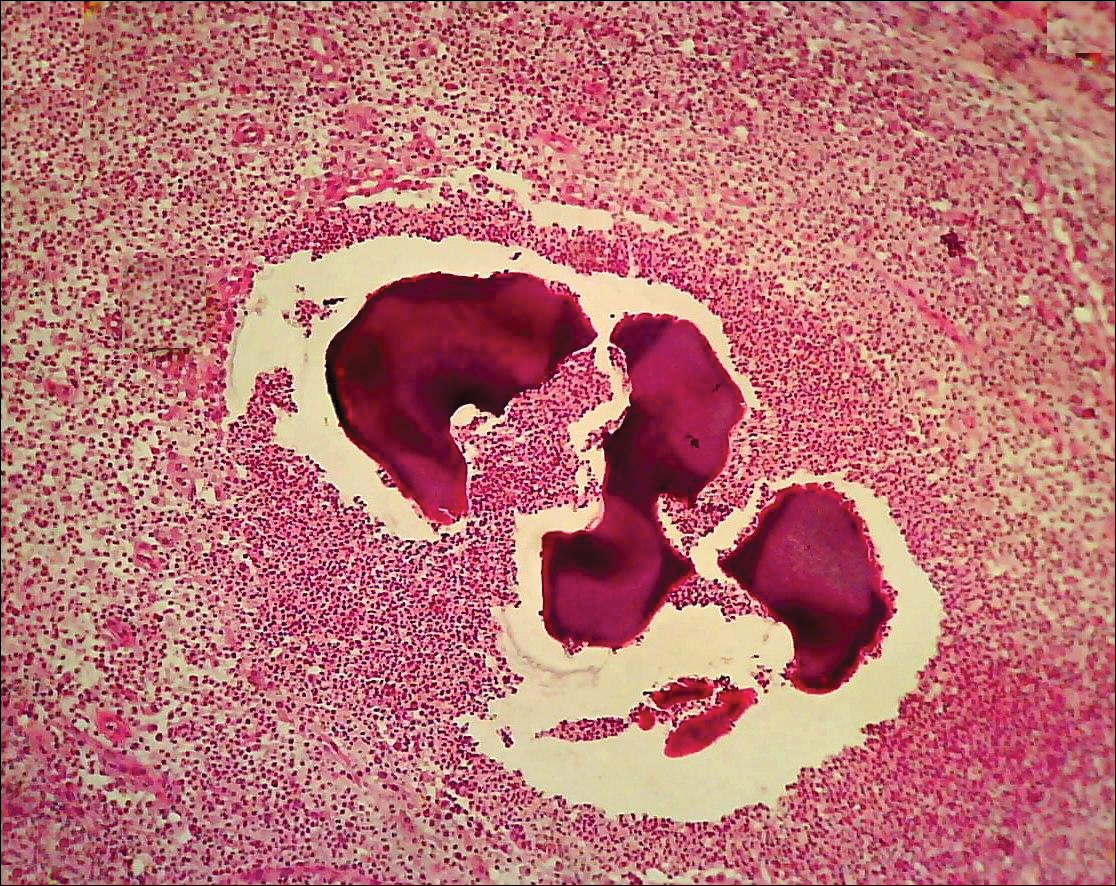
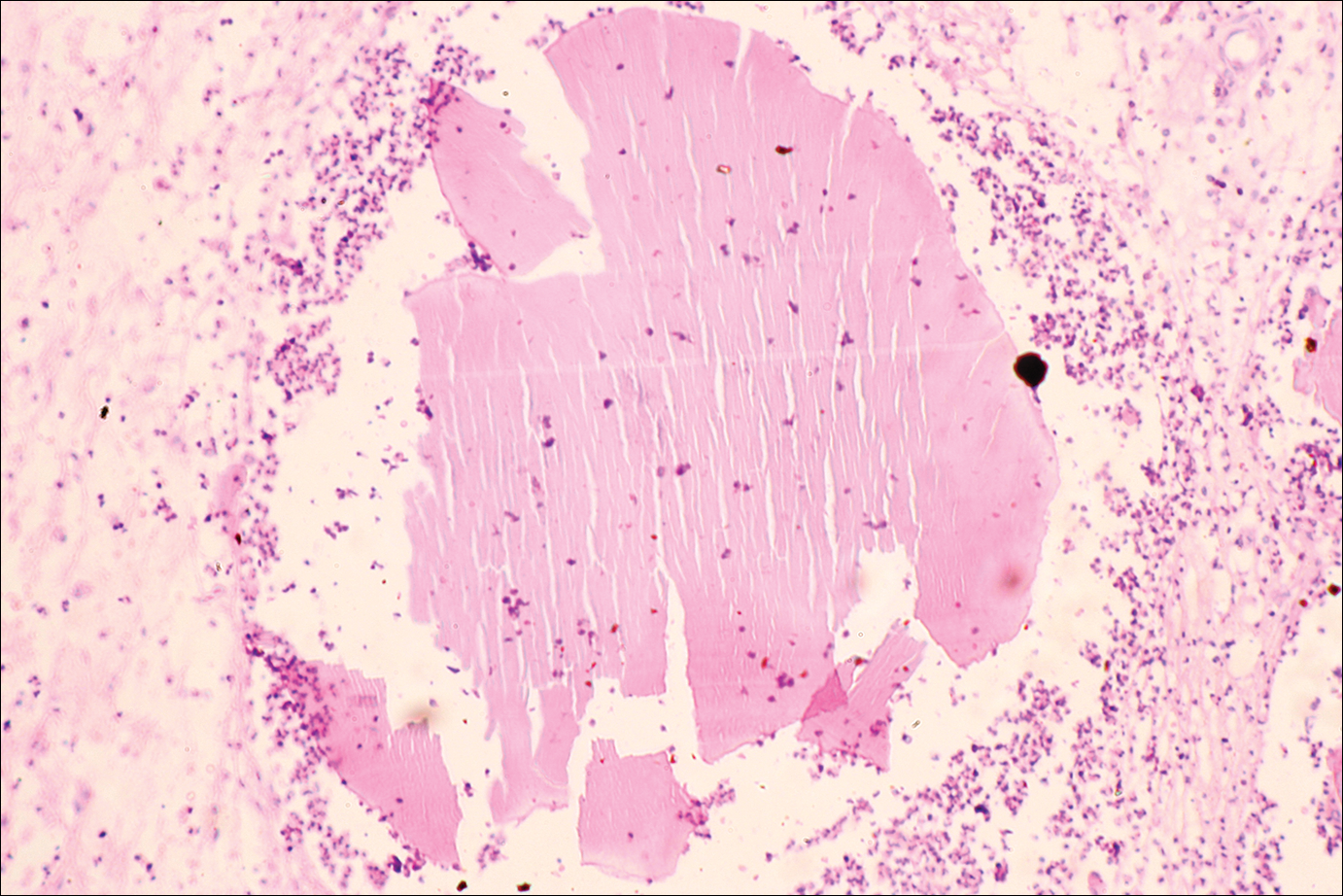
Treatment of Actinomycetoma
Precise identification of the etiologic agent is essential to provide effective treatment of actinomycetoma. Without treatment, or in resistant cases, progressive osseous and visceral involvement is inevitable.9 Actinomycetoma without osseous involvement usually responds well to medical treatment.
The treatment of choice for actinomycetoma involving Nocardia brasiliensis is a combination of dapsone 100 to 200 mg once daily and trimethoprim-sulfamethoxazole (TMP-SMX) 80/400 to 160/800 mg once daily for 2 to 3 years.10 Other treatments have included the following: (1) amikacin 15 mg/kg or 500 mg intramuscularly twice daily for 3 weeks plus dapsone 100 to 200 mg once daily plus TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years (amikacin, however, is expensive and potentially toxic [nephrotoxicity and ototoxicity] and therefore is used only in resistant cases); (2) dapsone 100 to 200 mg once daily or TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years plus intramuscular kanamycin 15 mg/kg once daily for 2 weeks at the beginning of treatment, alternating with rest periods to reduce the risk for nephrotoxicity and ototoxicity10; (3) dapsone 1.5 mg/kg orally twice daily plus phosphomycin 500 mg once daily; (4) dapsone 1.5 mg/kg orally twice daily plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month, then the same dose every other day for 1 to 2 months monitoring for ototoxicity; and (5) TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years or rifampicin (15–20 mg/kg/d) plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month at the beginning of treatment, then the same dose every other day for 2 to 3 months until a total dose of 60 g is administered, monitoring for ototoxicity.11 Audiometric tests and creatinine levels must be performed every 5 weeks during the treatment to monitor toxicity.10
The best results for infections with A pelletieri, A madurae, and S somaliensis have been with streptomycin (1 g once daily in adults; 20 mg/kg once daily in children) until a total dose of 50 g is reached in combination with TMP-SMX or dapsone12 (Figure 5). Alternatives for A madurae infections include streptomycin plus oral clofazimine (100 mg once daily), oral rifampicin (300 mg twice daily), oral tetracycline (1 g once daily), oral isoniazid (300–600 mg once daily), or oral minocycline (100 mg twice daily; also effective for A pelletieri).

More recently, other drugs have been used such as carbapenems (eg, imipenem, meropenem), which have wide-spectrum efficacy and are resistant to β-lactamases. Patients should be hospitalized to receive intravenous therapy with imipenem.2 Carbapenems are effective against gram-positive and gram-negative as well as Nocardia species.13,14 Mycetoma that is resistant, severe, or has visceral involvement can be treated with a combination of amikacin and imipenem.15,16 Meropenem is a similar drug that is available as an oral formulation. Both imipenem and meropenem are recommended in cases with bone involvement.17,18 Alternatives for resistant cases include amoxicillin–clavulanic acid 500/125 mg orally 3 times daily for 3 to 6 months or intravenous cefotaxime 1 g every 8 hours plus intramuscular amikacin 500 mg twice daily plus oral levamisole 300 mg once weekly for 4 weeks.19-23
For resistant cases associated with Nocardia species, clindamycin plus quinolones (eg, ciprofloxacin, moxifloxacin, garenoxacin) at a dose of 25 mg/kg once daily for at least 3 months has been suggested in in vivo studies.23
Overall, the cure rate for actinomycetoma treated with any of the prior therapies ranges from 60% to 90%. Treatment must be modified or stopped if there is clinical or laboratory evidence of drug toxicity.13,24 Surgical treatment of actinomycetoma is contraindicated, as it may cause hematogenous dissemination.
Prognosis
Actinomycetomas of a few months’ duration and without bone involvement respond well to therapy. If no therapy is provided or if there is resistance, the functional and cosmetic prognosis is poor, mainly for the feet. There is a risk for spine involvement with mycetoma on the back and posterior head. Thoracic lesions may penetrate into the lungs. The muscular fascia impedes the penetration of abdominal lesions, but the inguinal canals can offer a path for intra-abdominal dissemination.4 Advanced cases lead to a poor general condition of patients, difficulty in using affected extremities, and in extreme cases even death.
The criteria used to guide the discontinuation of initial therapy for any mycetoma include a decrease in the volume of the lesion, closure of fistulae, 3 consecutive negative monthly cultures, imaging studies showing bone regeneration, lack of echoes and cavities on echography, and absence of grains on examination of fine-needle aspirates.11 After the initial treatment protocol is finished, most experts recommend continuing treatment with dapsone 100 to 300 mg once daily for several years to prevent recurrence.12
Prevention
Mycetoma is a disease associated with poverty. It could be prevented by improving living conditions and by regular use of shoes in rural populations.2
Conclusion
Mycetoma is a chronic infection that develops after traumatic inoculation of the skin with either true fungi or aerobic actinomycetes. The resultant infections are known as eumycetoma or actinomycetoma, respectively. The etiologic agents can be found in the so-called grains. Black grains suggest a fungal infection, minute white grains suggest Nocardia, and red grains are due to A pelletieri. Larger white grains or yellow-white grains may be fungal or actinomycotic in origin.
Specific diagnosis requires direct examination, culture, and biopsy. The treatment of choice for actinomycetoma by N brasiliensis is a combination of dapsone 100 to 200 mg once daily and TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years. Other effective treatments include aminoglycosides (eg, amikacine, streptomycin) and quinolones. More recently, some other agents have been used such as carbapenems and natural products of Streptomyces cattleya (imipenem), which have wide-spectrum efficacy and are resistant to β-lactamases.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R. Micología Medica Ilustrada. 4th ed. Mexico City, Mexico: McGraw-Hill Interamericana; 2011:125-146.
- McGinnis MR. Mycetoma. Dermatol Clin. 1996;14:97-104.
- Fahal AH. Mycetoma: Clinico-pathological Monograph. Khartoum, Sudan: University of Khartoum Press; 2006:20-23, 81-82.
- Estrada-Chavez GE, Vega-Memije ME, Arenas R, et al. Eumycotic mycetoma caused by Madurella mycetomatis successfully treated with antifungals, surgery, and topical negative pressure therapy. Int J Dermatol. 2009;48:401-403.
- Chávez G, Arenas R, Pérez-Polito A, et al. Eumycetic mycetoma due to Madurella mycetomatis. report of six cases. Rev Iberoam Micol. 1998;15:90-93.
- Vasquez del Mercado E, Arenas R, Moreno G. Sequelae and long-term consequences of systemic and subcutaneous mycoses. In: Fratamico PM, Smith JL, Brogden KA, eds. Sequelae and Long-term Consequences of Infectious Diseases. Washington, DC: ASM Press; 2009:415-420.
- Mancini N, Ossi CM, Perotti M, et al. Molecular mycological diagnosis and correct antimycotic treatments. J Clin Microbiol. 2005;43:3584-3585.
- Arenas R, Lavalle P. Micetoma (madura foot). In: Arenas R, Estrada R, eds. Tropical Dermatology. Austin, TX: Landes Bioscience; 2001:51-61.
- Welsh O, Sauceda E, González J, et al. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Fahal AH. Mycetoma: clinico-pathological monograph. In: Fahal AH. Evidence Based Guidelines for the Management of Mycetoma Patients. Khartoum, Sudan: University of Khartoum Press; 2002:5-15.
- Welsh O, Salinas MC, Rodríguez MA. Treatment of eumycetoma and actinomycetoma. Curr Top Med Mycol. 1995;6:47-71.
- Valle ACF, Welsh O, Vera-Cabrera L. Subcutaneous mycoses—mycetoma. In: Tyring SK, Lupi O, Hengge UR, eds. Tropical Dermatology. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:197-200.
- Fuentes A, Arenas R, Reyes M, et al. Actinomicetoma por Nocardia sp. Informe de cinco casos tratados con imipenem solo o combinado con amikacina. Gac Med Mex. 2006;142:247-252.
- Gombert ME, Aulicino TM, DuBouchet L, et al. Therapy of experimental cerebral nocardiosis with imipenem, amikacin, trimethoprim-sulfamethoxazole, and minocylina. Antimicrob Agents Chemother. 1986;30:270-273.
- Calandra GB, Ricci FM, Wang C, et al. Safety and tolerance comparison of imipenem-cilastatin to cephalotin and cefazolin. J Antimicrob Chemother. 1983;12:125-131.
- Ameen M, Arenas R, Vasquez del Mercado E, et al. Efficacy of imipenem therapy for Nocardia actinomycetomas refractory to sulfonamides. J Am Acad Dermatol. 2010;62:239-246.
- Ameen M, Vargas F, Vasquez del Mercado E, et al. Successful treatment of Nocardia actinomycetoma with meropenem and amikacin combination therapy. Int J Dermatol. 2011;50:443-445.
- Ameen M, Arenas R. Emerging therapeutic regimes for the management of mycetomas. Expert Opin Pharmacother. 2008;9:2077-2085.
- Vera-Cabrera L, Daw-Garza A, Said-Fernández S, et al. Therapeutic effect of a novel oxazolidinone, DA-7867 in BALB/c mice infected with Nocardia brasiliensis. PloS Negl Trop Dis. 2008;2:e289.
- Gómez A, Saúl A, Bonifaz A. Amoxicillin and clavulanic acid in the treatment of actinomicetoma. Int J Dermatol. 1993;32:218-220.
- Méndez-Tovar L, Serrano-Jaen L, Almeida-Arvizu VM. Cefotaxima mas amikacina asociadas a inmunomodulación en el tratamiento de actinomicetoma resistente a tratamiento convencional. Gac Med Mex. 1999;135:517-521.
- Chacon-Moreno BE, Welsh O, Cavazos-Rocha N, et al. Efficacy of ciprofloxacin and moxifloxacin against Nocardia brasiliensis in vitro in an experimental model of actinomycetoma in BALB/c mice. Antimicrob Agents Chemother. 2009;53:295-297.
- Welsh O. Treatment of actinomycetoma. Arch Med Res. 1993;24:413-415.
Mycetoma is a subcutaneous disease that can be caused by aerobic bacteria (actinomycetoma) or fungi (eumycetoma). Diagnosis is based on clinical manifestations, including swelling and deformity of affected areas, as well as the presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate that contains the microorganisms.
The worldwide proportion of mycetomas is 60% actinomycetomas and 40% eumycetomas.1 The disease is endemic in tropical, subtropical, and temperate regions, predominating between latitudes 30°N and 15°S. Most cases occur in Africa, especially Sudan, Mauritania, and Senegal; India; Yemen; and Pakistan. In the Americas, the countries with the most reported cases are Mexico and Venezuela.1
Although mycetoma is rare in developed countries, migration of patients from endemic areas makes knowledge of this condition crucial for dermatologists worldwide. We present a review of the current concepts in the epidemiology, clinical presentation, diagnosis, and treatment of actinomycetoma.
Epidemiology
Actinomycetoma is more common in Latin America, with Mexico having the highest incidence. At last count, there were 2631 cases reported in Mexico.2 The majority of cases of mycetoma in Mexico are actinomycetoma (98%), including Nocardia (86%) and Actinomadura madurae (10%). Eumycetoma is rare in Mexico, constituting only 2% of cases.2 Worldwide, men are affected more commonly than women, which is thought to be related to a higher occupational risk during agricultural labor.
Clinical Features
Mycetoma can affect the skin, subcutaneous tissue, bones, and occasionally the internal organs. It is characterized by swelling, deformation of the affected area, and fistulae that drain serosanguineous or purulent exudates.
In Mexico, 60% of cases of mycetoma affect the lower extremities; the feet are the most commonly affected area, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.1 Other sites include the hands, shoulders, and abdominal wall. The head and neck area are seldom affected.3 Mycetoma lesions grow and disseminate locally. Bone lesions are possible depending on the osteophilic affinity of the etiological agent and on the interactions between the fungus and the host’s immune system. In severe advanced cases of mycetoma, the lesions may involve tendons and nerves. Dissemination via blood or lymphatics is extremely rare.4
Diagnosis
Diagnosis of actinomycetoma is suspected based on clinical features and confirmed by direct examination of exudates with Lugol iodine or saline solution. On direct microscopy, actinomycetes are recognized by the production of filaments with a width of 0.5 to 1 μm. On hematoxylin and eosin stain, the small grains of Nocardia appear eosinophilic with a blue center and pink filaments. On Gram stain, actinomycetoma grains show positive branching filaments. Culture of grains recovered from aspirated material or biopsy specimens provides specific etiologic diagnosis. Cultures should be held for at least 4 weeks. Additionally, there are some enzymatic, molecular, and serologic tests available for diagnosis.5-7 Serologic diagnosis is available in a few centers in Mexico and can be helpful in some cases for diagnosis or follow-up during treatment. Antibodies can be determined via enzyme-linked immunosorbent assay, Western blot analysis, immunodiffusion, or counterimmunoelectrophoresis.8
The causative agents of actinomycetoma can be isolated in Sabouraud dextrose agar. Deep wedge biopsies (or puncture aspiration) are useful in observing the diagnostic grains, which can be identified adequately with Gram stain. Grains usually are surrounded and/or infiltrated by neutrophils. The size, form, and color of grains can identify the causative agent.1 The granules of Nocardia are small (80–130 mm) and reniform or wormlike, with club structures in their periphery (Figure 1). Actinomadura madurae is characterized by large, white-yellow granules that can be seen with the naked eye (1–3 mm). On microscopic examination with hematoxylin and eosin stain, these grains are purple and exhibit peripheral pink pseudofilaments (Figure 2).2 The grains of Actinomadura pelletieri are large (1–3 mm) and red or violaceous. They fragment or break easily, giving the appearance of a broken dish (Figure 3). Streptomyces somaliensis forms round grains approximately 0.5 to 1 cm in diameter. These grains stain poorly and are extremely hard. Cutting the grains during processing results in striation, giving them the appearance of a potato chip (Figure 4).2
Treatment of Actinomycetoma
Precise identification of the etiologic agent is essential to provide effective treatment of actinomycetoma. Without treatment, or in resistant cases, progressive osseous and visceral involvement is inevitable.9 Actinomycetoma without osseous involvement usually responds well to medical treatment.
The treatment of choice for actinomycetoma involving Nocardia brasiliensis is a combination of dapsone 100 to 200 mg once daily and trimethoprim-sulfamethoxazole (TMP-SMX) 80/400 to 160/800 mg once daily for 2 to 3 years.10 Other treatments have included the following: (1) amikacin 15 mg/kg or 500 mg intramuscularly twice daily for 3 weeks plus dapsone 100 to 200 mg once daily plus TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years (amikacin, however, is expensive and potentially toxic [nephrotoxicity and ototoxicity] and therefore is used only in resistant cases); (2) dapsone 100 to 200 mg once daily or TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years plus intramuscular kanamycin 15 mg/kg once daily for 2 weeks at the beginning of treatment, alternating with rest periods to reduce the risk for nephrotoxicity and ototoxicity10; (3) dapsone 1.5 mg/kg orally twice daily plus phosphomycin 500 mg once daily; (4) dapsone 1.5 mg/kg orally twice daily plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month, then the same dose every other day for 1 to 2 months monitoring for ototoxicity; and (5) TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years or rifampicin (15–20 mg/kg/d) plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month at the beginning of treatment, then the same dose every other day for 2 to 3 months until a total dose of 60 g is administered, monitoring for ototoxicity.11 Audiometric tests and creatinine levels must be performed every 5 weeks during the treatment to monitor toxicity.10
The best results for infections with A pelletieri, A madurae, and S somaliensis have been with streptomycin (1 g once daily in adults; 20 mg/kg once daily in children) until a total dose of 50 g is reached in combination with TMP-SMX or dapsone12 (Figure 5). Alternatives for A madurae infections include streptomycin plus oral clofazimine (100 mg once daily), oral rifampicin (300 mg twice daily), oral tetracycline (1 g once daily), oral isoniazid (300–600 mg once daily), or oral minocycline (100 mg twice daily; also effective for A pelletieri).
More recently, other drugs have been used such as carbapenems (eg, imipenem, meropenem), which have wide-spectrum efficacy and are resistant to β-lactamases. Patients should be hospitalized to receive intravenous therapy with imipenem.2 Carbapenems are effective against gram-positive and gram-negative as well as Nocardia species.13,14 Mycetoma that is resistant, severe, or has visceral involvement can be treated with a combination of amikacin and imipenem.15,16 Meropenem is a similar drug that is available as an oral formulation. Both imipenem and meropenem are recommended in cases with bone involvement.17,18 Alternatives for resistant cases include amoxicillin–clavulanic acid 500/125 mg orally 3 times daily for 3 to 6 months or intravenous cefotaxime 1 g every 8 hours plus intramuscular amikacin 500 mg twice daily plus oral levamisole 300 mg once weekly for 4 weeks.19-23
For resistant cases associated with Nocardia species, clindamycin plus quinolones (eg, ciprofloxacin, moxifloxacin, garenoxacin) at a dose of 25 mg/kg once daily for at least 3 months has been suggested in in vivo studies.23
Overall, the cure rate for actinomycetoma treated with any of the prior therapies ranges from 60% to 90%. Treatment must be modified or stopped if there is clinical or laboratory evidence of drug toxicity.13,24 Surgical treatment of actinomycetoma is contraindicated, as it may cause hematogenous dissemination.
Prognosis
Actinomycetomas of a few months’ duration and without bone involvement respond well to therapy. If no therapy is provided or if there is resistance, the functional and cosmetic prognosis is poor, mainly for the feet. There is a risk for spine involvement with mycetoma on the back and posterior head. Thoracic lesions may penetrate into the lungs. The muscular fascia impedes the penetration of abdominal lesions, but the inguinal canals can offer a path for intra-abdominal dissemination.4 Advanced cases lead to a poor general condition of patients, difficulty in using affected extremities, and in extreme cases even death.
The criteria used to guide the discontinuation of initial therapy for any mycetoma include a decrease in the volume of the lesion, closure of fistulae, 3 consecutive negative monthly cultures, imaging studies showing bone regeneration, lack of echoes and cavities on echography, and absence of grains on examination of fine-needle aspirates.11 After the initial treatment protocol is finished, most experts recommend continuing treatment with dapsone 100 to 300 mg once daily for several years to prevent recurrence.12
Prevention
Mycetoma is a disease associated with poverty. It could be prevented by improving living conditions and by regular use of shoes in rural populations.2
Conclusion
Mycetoma is a chronic infection that develops after traumatic inoculation of the skin with either true fungi or aerobic actinomycetes. The resultant infections are known as eumycetoma or actinomycetoma, respectively. The etiologic agents can be found in the so-called grains. Black grains suggest a fungal infection, minute white grains suggest Nocardia, and red grains are due to A pelletieri. Larger white grains or yellow-white grains may be fungal or actinomycotic in origin.
Specific diagnosis requires direct examination, culture, and biopsy. The treatment of choice for actinomycetoma by N brasiliensis is a combination of dapsone 100 to 200 mg once daily and TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years. Other effective treatments include aminoglycosides (eg, amikacine, streptomycin) and quinolones. More recently, some other agents have been used such as carbapenems and natural products of Streptomyces cattleya (imipenem), which have wide-spectrum efficacy and are resistant to β-lactamases.
Mycetoma is a subcutaneous disease that can be caused by aerobic bacteria (actinomycetoma) or fungi (eumycetoma). Diagnosis is based on clinical manifestations, including swelling and deformity of affected areas, as well as the presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate that contains the microorganisms.
The worldwide proportion of mycetomas is 60% actinomycetomas and 40% eumycetomas.1 The disease is endemic in tropical, subtropical, and temperate regions, predominating between latitudes 30°N and 15°S. Most cases occur in Africa, especially Sudan, Mauritania, and Senegal; India; Yemen; and Pakistan. In the Americas, the countries with the most reported cases are Mexico and Venezuela.1
Although mycetoma is rare in developed countries, migration of patients from endemic areas makes knowledge of this condition crucial for dermatologists worldwide. We present a review of the current concepts in the epidemiology, clinical presentation, diagnosis, and treatment of actinomycetoma.
Epidemiology
Actinomycetoma is more common in Latin America, with Mexico having the highest incidence. At last count, there were 2631 cases reported in Mexico.2 The majority of cases of mycetoma in Mexico are actinomycetoma (98%), including Nocardia (86%) and Actinomadura madurae (10%). Eumycetoma is rare in Mexico, constituting only 2% of cases.2 Worldwide, men are affected more commonly than women, which is thought to be related to a higher occupational risk during agricultural labor.
Clinical Features
Mycetoma can affect the skin, subcutaneous tissue, bones, and occasionally the internal organs. It is characterized by swelling, deformation of the affected area, and fistulae that drain serosanguineous or purulent exudates.
In Mexico, 60% of cases of mycetoma affect the lower extremities; the feet are the most commonly affected area, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.1 Other sites include the hands, shoulders, and abdominal wall. The head and neck area are seldom affected.3 Mycetoma lesions grow and disseminate locally. Bone lesions are possible depending on the osteophilic affinity of the etiological agent and on the interactions between the fungus and the host’s immune system. In severe advanced cases of mycetoma, the lesions may involve tendons and nerves. Dissemination via blood or lymphatics is extremely rare.4
Diagnosis
Diagnosis of actinomycetoma is suspected based on clinical features and confirmed by direct examination of exudates with Lugol iodine or saline solution. On direct microscopy, actinomycetes are recognized by the production of filaments with a width of 0.5 to 1 μm. On hematoxylin and eosin stain, the small grains of Nocardia appear eosinophilic with a blue center and pink filaments. On Gram stain, actinomycetoma grains show positive branching filaments. Culture of grains recovered from aspirated material or biopsy specimens provides specific etiologic diagnosis. Cultures should be held for at least 4 weeks. Additionally, there are some enzymatic, molecular, and serologic tests available for diagnosis.5-7 Serologic diagnosis is available in a few centers in Mexico and can be helpful in some cases for diagnosis or follow-up during treatment. Antibodies can be determined via enzyme-linked immunosorbent assay, Western blot analysis, immunodiffusion, or counterimmunoelectrophoresis.8
The causative agents of actinomycetoma can be isolated in Sabouraud dextrose agar. Deep wedge biopsies (or puncture aspiration) are useful in observing the diagnostic grains, which can be identified adequately with Gram stain. Grains usually are surrounded and/or infiltrated by neutrophils. The size, form, and color of grains can identify the causative agent.1 The granules of Nocardia are small (80–130 mm) and reniform or wormlike, with club structures in their periphery (Figure 1). Actinomadura madurae is characterized by large, white-yellow granules that can be seen with the naked eye (1–3 mm). On microscopic examination with hematoxylin and eosin stain, these grains are purple and exhibit peripheral pink pseudofilaments (Figure 2).2 The grains of Actinomadura pelletieri are large (1–3 mm) and red or violaceous. They fragment or break easily, giving the appearance of a broken dish (Figure 3). Streptomyces somaliensis forms round grains approximately 0.5 to 1 cm in diameter. These grains stain poorly and are extremely hard. Cutting the grains during processing results in striation, giving them the appearance of a potato chip (Figure 4).2
Treatment of Actinomycetoma
Precise identification of the etiologic agent is essential to provide effective treatment of actinomycetoma. Without treatment, or in resistant cases, progressive osseous and visceral involvement is inevitable.9 Actinomycetoma without osseous involvement usually responds well to medical treatment.
The treatment of choice for actinomycetoma involving Nocardia brasiliensis is a combination of dapsone 100 to 200 mg once daily and trimethoprim-sulfamethoxazole (TMP-SMX) 80/400 to 160/800 mg once daily for 2 to 3 years.10 Other treatments have included the following: (1) amikacin 15 mg/kg or 500 mg intramuscularly twice daily for 3 weeks plus dapsone 100 to 200 mg once daily plus TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years (amikacin, however, is expensive and potentially toxic [nephrotoxicity and ototoxicity] and therefore is used only in resistant cases); (2) dapsone 100 to 200 mg once daily or TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years plus intramuscular kanamycin 15 mg/kg once daily for 2 weeks at the beginning of treatment, alternating with rest periods to reduce the risk for nephrotoxicity and ototoxicity10; (3) dapsone 1.5 mg/kg orally twice daily plus phosphomycin 500 mg once daily; (4) dapsone 1.5 mg/kg orally twice daily plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month, then the same dose every other day for 1 to 2 months monitoring for ototoxicity; and (5) TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years or rifampicin (15–20 mg/kg/d) plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month at the beginning of treatment, then the same dose every other day for 2 to 3 months until a total dose of 60 g is administered, monitoring for ototoxicity.11 Audiometric tests and creatinine levels must be performed every 5 weeks during the treatment to monitor toxicity.10
The best results for infections with A pelletieri, A madurae, and S somaliensis have been with streptomycin (1 g once daily in adults; 20 mg/kg once daily in children) until a total dose of 50 g is reached in combination with TMP-SMX or dapsone12 (Figure 5). Alternatives for A madurae infections include streptomycin plus oral clofazimine (100 mg once daily), oral rifampicin (300 mg twice daily), oral tetracycline (1 g once daily), oral isoniazid (300–600 mg once daily), or oral minocycline (100 mg twice daily; also effective for A pelletieri).
More recently, other drugs have been used such as carbapenems (eg, imipenem, meropenem), which have wide-spectrum efficacy and are resistant to β-lactamases. Patients should be hospitalized to receive intravenous therapy with imipenem.2 Carbapenems are effective against gram-positive and gram-negative as well as Nocardia species.13,14 Mycetoma that is resistant, severe, or has visceral involvement can be treated with a combination of amikacin and imipenem.15,16 Meropenem is a similar drug that is available as an oral formulation. Both imipenem and meropenem are recommended in cases with bone involvement.17,18 Alternatives for resistant cases include amoxicillin–clavulanic acid 500/125 mg orally 3 times daily for 3 to 6 months or intravenous cefotaxime 1 g every 8 hours plus intramuscular amikacin 500 mg twice daily plus oral levamisole 300 mg once weekly for 4 weeks.19-23
For resistant cases associated with Nocardia species, clindamycin plus quinolones (eg, ciprofloxacin, moxifloxacin, garenoxacin) at a dose of 25 mg/kg once daily for at least 3 months has been suggested in in vivo studies.23
Overall, the cure rate for actinomycetoma treated with any of the prior therapies ranges from 60% to 90%. Treatment must be modified or stopped if there is clinical or laboratory evidence of drug toxicity.13,24 Surgical treatment of actinomycetoma is contraindicated, as it may cause hematogenous dissemination.
Prognosis
Actinomycetomas of a few months’ duration and without bone involvement respond well to therapy. If no therapy is provided or if there is resistance, the functional and cosmetic prognosis is poor, mainly for the feet. There is a risk for spine involvement with mycetoma on the back and posterior head. Thoracic lesions may penetrate into the lungs. The muscular fascia impedes the penetration of abdominal lesions, but the inguinal canals can offer a path for intra-abdominal dissemination.4 Advanced cases lead to a poor general condition of patients, difficulty in using affected extremities, and in extreme cases even death.
The criteria used to guide the discontinuation of initial therapy for any mycetoma include a decrease in the volume of the lesion, closure of fistulae, 3 consecutive negative monthly cultures, imaging studies showing bone regeneration, lack of echoes and cavities on echography, and absence of grains on examination of fine-needle aspirates.11 After the initial treatment protocol is finished, most experts recommend continuing treatment with dapsone 100 to 300 mg once daily for several years to prevent recurrence.12
Prevention
Mycetoma is a disease associated with poverty. It could be prevented by improving living conditions and by regular use of shoes in rural populations.2
Conclusion
Mycetoma is a chronic infection that develops after traumatic inoculation of the skin with either true fungi or aerobic actinomycetes. The resultant infections are known as eumycetoma or actinomycetoma, respectively. The etiologic agents can be found in the so-called grains. Black grains suggest a fungal infection, minute white grains suggest Nocardia, and red grains are due to A pelletieri. Larger white grains or yellow-white grains may be fungal or actinomycotic in origin.
Specific diagnosis requires direct examination, culture, and biopsy. The treatment of choice for actinomycetoma by N brasiliensis is a combination of dapsone 100 to 200 mg once daily and TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years. Other effective treatments include aminoglycosides (eg, amikacine, streptomycin) and quinolones. More recently, some other agents have been used such as carbapenems and natural products of Streptomyces cattleya (imipenem), which have wide-spectrum efficacy and are resistant to β-lactamases.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R. Micología Medica Ilustrada. 4th ed. Mexico City, Mexico: McGraw-Hill Interamericana; 2011:125-146.
- McGinnis MR. Mycetoma. Dermatol Clin. 1996;14:97-104.
- Fahal AH. Mycetoma: Clinico-pathological Monograph. Khartoum, Sudan: University of Khartoum Press; 2006:20-23, 81-82.
- Estrada-Chavez GE, Vega-Memije ME, Arenas R, et al. Eumycotic mycetoma caused by Madurella mycetomatis successfully treated with antifungals, surgery, and topical negative pressure therapy. Int J Dermatol. 2009;48:401-403.
- Chávez G, Arenas R, Pérez-Polito A, et al. Eumycetic mycetoma due to Madurella mycetomatis. report of six cases. Rev Iberoam Micol. 1998;15:90-93.
- Vasquez del Mercado E, Arenas R, Moreno G. Sequelae and long-term consequences of systemic and subcutaneous mycoses. In: Fratamico PM, Smith JL, Brogden KA, eds. Sequelae and Long-term Consequences of Infectious Diseases. Washington, DC: ASM Press; 2009:415-420.
- Mancini N, Ossi CM, Perotti M, et al. Molecular mycological diagnosis and correct antimycotic treatments. J Clin Microbiol. 2005;43:3584-3585.
- Arenas R, Lavalle P. Micetoma (madura foot). In: Arenas R, Estrada R, eds. Tropical Dermatology. Austin, TX: Landes Bioscience; 2001:51-61.
- Welsh O, Sauceda E, González J, et al. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Fahal AH. Mycetoma: clinico-pathological monograph. In: Fahal AH. Evidence Based Guidelines for the Management of Mycetoma Patients. Khartoum, Sudan: University of Khartoum Press; 2002:5-15.
- Welsh O, Salinas MC, Rodríguez MA. Treatment of eumycetoma and actinomycetoma. Curr Top Med Mycol. 1995;6:47-71.
- Valle ACF, Welsh O, Vera-Cabrera L. Subcutaneous mycoses—mycetoma. In: Tyring SK, Lupi O, Hengge UR, eds. Tropical Dermatology. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:197-200.
- Fuentes A, Arenas R, Reyes M, et al. Actinomicetoma por Nocardia sp. Informe de cinco casos tratados con imipenem solo o combinado con amikacina. Gac Med Mex. 2006;142:247-252.
- Gombert ME, Aulicino TM, DuBouchet L, et al. Therapy of experimental cerebral nocardiosis with imipenem, amikacin, trimethoprim-sulfamethoxazole, and minocylina. Antimicrob Agents Chemother. 1986;30:270-273.
- Calandra GB, Ricci FM, Wang C, et al. Safety and tolerance comparison of imipenem-cilastatin to cephalotin and cefazolin. J Antimicrob Chemother. 1983;12:125-131.
- Ameen M, Arenas R, Vasquez del Mercado E, et al. Efficacy of imipenem therapy for Nocardia actinomycetomas refractory to sulfonamides. J Am Acad Dermatol. 2010;62:239-246.
- Ameen M, Vargas F, Vasquez del Mercado E, et al. Successful treatment of Nocardia actinomycetoma with meropenem and amikacin combination therapy. Int J Dermatol. 2011;50:443-445.
- Ameen M, Arenas R. Emerging therapeutic regimes for the management of mycetomas. Expert Opin Pharmacother. 2008;9:2077-2085.
- Vera-Cabrera L, Daw-Garza A, Said-Fernández S, et al. Therapeutic effect of a novel oxazolidinone, DA-7867 in BALB/c mice infected with Nocardia brasiliensis. PloS Negl Trop Dis. 2008;2:e289.
- Gómez A, Saúl A, Bonifaz A. Amoxicillin and clavulanic acid in the treatment of actinomicetoma. Int J Dermatol. 1993;32:218-220.
- Méndez-Tovar L, Serrano-Jaen L, Almeida-Arvizu VM. Cefotaxima mas amikacina asociadas a inmunomodulación en el tratamiento de actinomicetoma resistente a tratamiento convencional. Gac Med Mex. 1999;135:517-521.
- Chacon-Moreno BE, Welsh O, Cavazos-Rocha N, et al. Efficacy of ciprofloxacin and moxifloxacin against Nocardia brasiliensis in vitro in an experimental model of actinomycetoma in BALB/c mice. Antimicrob Agents Chemother. 2009;53:295-297.
- Welsh O. Treatment of actinomycetoma. Arch Med Res. 1993;24:413-415.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R. Micología Medica Ilustrada. 4th ed. Mexico City, Mexico: McGraw-Hill Interamericana; 2011:125-146.
- McGinnis MR. Mycetoma. Dermatol Clin. 1996;14:97-104.
- Fahal AH. Mycetoma: Clinico-pathological Monograph. Khartoum, Sudan: University of Khartoum Press; 2006:20-23, 81-82.
- Estrada-Chavez GE, Vega-Memije ME, Arenas R, et al. Eumycotic mycetoma caused by Madurella mycetomatis successfully treated with antifungals, surgery, and topical negative pressure therapy. Int J Dermatol. 2009;48:401-403.
- Chávez G, Arenas R, Pérez-Polito A, et al. Eumycetic mycetoma due to Madurella mycetomatis. report of six cases. Rev Iberoam Micol. 1998;15:90-93.
- Vasquez del Mercado E, Arenas R, Moreno G. Sequelae and long-term consequences of systemic and subcutaneous mycoses. In: Fratamico PM, Smith JL, Brogden KA, eds. Sequelae and Long-term Consequences of Infectious Diseases. Washington, DC: ASM Press; 2009:415-420.
- Mancini N, Ossi CM, Perotti M, et al. Molecular mycological diagnosis and correct antimycotic treatments. J Clin Microbiol. 2005;43:3584-3585.
- Arenas R, Lavalle P. Micetoma (madura foot). In: Arenas R, Estrada R, eds. Tropical Dermatology. Austin, TX: Landes Bioscience; 2001:51-61.
- Welsh O, Sauceda E, González J, et al. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Fahal AH. Mycetoma: clinico-pathological monograph. In: Fahal AH. Evidence Based Guidelines for the Management of Mycetoma Patients. Khartoum, Sudan: University of Khartoum Press; 2002:5-15.
- Welsh O, Salinas MC, Rodríguez MA. Treatment of eumycetoma and actinomycetoma. Curr Top Med Mycol. 1995;6:47-71.
- Valle ACF, Welsh O, Vera-Cabrera L. Subcutaneous mycoses—mycetoma. In: Tyring SK, Lupi O, Hengge UR, eds. Tropical Dermatology. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:197-200.
- Fuentes A, Arenas R, Reyes M, et al. Actinomicetoma por Nocardia sp. Informe de cinco casos tratados con imipenem solo o combinado con amikacina. Gac Med Mex. 2006;142:247-252.
- Gombert ME, Aulicino TM, DuBouchet L, et al. Therapy of experimental cerebral nocardiosis with imipenem, amikacin, trimethoprim-sulfamethoxazole, and minocylina. Antimicrob Agents Chemother. 1986;30:270-273.
- Calandra GB, Ricci FM, Wang C, et al. Safety and tolerance comparison of imipenem-cilastatin to cephalotin and cefazolin. J Antimicrob Chemother. 1983;12:125-131.
- Ameen M, Arenas R, Vasquez del Mercado E, et al. Efficacy of imipenem therapy for Nocardia actinomycetomas refractory to sulfonamides. J Am Acad Dermatol. 2010;62:239-246.
- Ameen M, Vargas F, Vasquez del Mercado E, et al. Successful treatment of Nocardia actinomycetoma with meropenem and amikacin combination therapy. Int J Dermatol. 2011;50:443-445.
- Ameen M, Arenas R. Emerging therapeutic regimes for the management of mycetomas. Expert Opin Pharmacother. 2008;9:2077-2085.
- Vera-Cabrera L, Daw-Garza A, Said-Fernández S, et al. Therapeutic effect of a novel oxazolidinone, DA-7867 in BALB/c mice infected with Nocardia brasiliensis. PloS Negl Trop Dis. 2008;2:e289.
- Gómez A, Saúl A, Bonifaz A. Amoxicillin and clavulanic acid in the treatment of actinomicetoma. Int J Dermatol. 1993;32:218-220.
- Méndez-Tovar L, Serrano-Jaen L, Almeida-Arvizu VM. Cefotaxima mas amikacina asociadas a inmunomodulación en el tratamiento de actinomicetoma resistente a tratamiento convencional. Gac Med Mex. 1999;135:517-521.
- Chacon-Moreno BE, Welsh O, Cavazos-Rocha N, et al. Efficacy of ciprofloxacin and moxifloxacin against Nocardia brasiliensis in vitro in an experimental model of actinomycetoma in BALB/c mice. Antimicrob Agents Chemother. 2009;53:295-297.
- Welsh O. Treatment of actinomycetoma. Arch Med Res. 1993;24:413-415.
Practice Points
- Diagnosis of actinomycetoma is based on clinical manifestations including increased swelling and deformity of affected areas, presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate containing microorganisms.
- The feet are the most commonly affected location, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.
- Specific diagnosis of actinomycetoma requires clinical examination as well as direct examination of culture and biopsy results.
- Overall, the cure rate for actinomycetoma ranges from 60% to 90%.