User login
Impact of Medicaid expansion on inpatient outcomes
The Affordable Care Act (ACA) has greatly increased the number of Medicaid enrollees since it expanded eligibility criteria on Jan. 1, 2014. Nearly 9 million additional U.S. adults now have Medicaid coverage, mostly in the 31 states and Washington, which have opted into Medicaid expansion.
The ACA has also had an important impact on hospital payer mix, mainly by decreasing the amount of uncompensated care in Medicaid-expansion states. Previous studies have shown disparities in the quality of inpatient care based on insurance type. Patients with Medicaid insurance often have longer hospitalizations and higher in-hospital mortality than commercially-insured patients and occasionally even than uninsured patients.

In our study published in the Journal of Hospital Medicine (2016 Dec. doi: 10.1002/jhm.2649), we evaluated the impact of state Medicaid expansion status on payer mix, length of stay, and in-hospital mortality for general medicine patients discharged from U.S. academic medical centers. We considered Jan. 1, 2014, to be the date of ACA implementation for all states except Michigan, New Hampshire, Pennsylvania, and Indiana, which had unique dates of Medicaid expansion.
We were able to identify 3,144,488 discharges from 156 hospitals in 24 Medicaid-expansion states and Washington, and 1,114,464 discharges from 55 hospitals in 14 nonexpansion states between October 2012 and September 2015.

Despite this difference in payer mix trends, state Medicaid expansion status was not associated with differences in overall length of stay or in-hospital mortality in our study. More precisely, we looked at the length of stay and mortality indices, or ratio of observed to expected values, to control for such potential confounders as disease severity and comorbid conditions.
One possible explanation for our findings is that the higher proportion of Medicare and commercially-insured patients overshadowed the contribution of Medicaid patients to the overall length of stay and mortality indices.
To our knowledge, our study is the first to look at the effect of ACA implementation on inpatient outcomes. Early evidence suggests that Medicaid expansion has improved outpatient outcomes. Low-income adults in Medicaid-expansion states have shown greater gains in access to primary care clinics and medications and in the diagnosis of certain chronic health conditions than those in non-expansion states. However, these changes would not necessarily lead to improvements in the length of stay or mortality indices for Medicaid-expansion hospitals, since the measures account for patient acuity on admission.
The take-home message from our study for health policy makers is that state Medicaid expansion status had a neutral effect on both length of stay and mortality indices. This should be reassuring for states considering expansion of their Medicaid programs in the future.
As a next step, it would be useful to see research on the impact of ACA implementation on other inpatient outcomes that may vary with insurance type, such as readmissions or hospital-acquired complications.
The take-home message for hospitalists is that there is more work to be done in reducing disparities in inpatient care based on payer status. Though not a primary focus of our study, we did see variation in the length of stay and mortality indices based on insurance type.
It is unclear whether these differences occurred because of variation in the expertise of inpatient providers, access to invasive procedures or medical therapies, the timeliness of discharge to post-acute care facilities, or other patient- or system-level factors. However, these disparities warrant our improvement efforts moving forward.
Mary Anderson, MD, and Christine Jones, MD, are hospitalists at the University of Colorado (Aurora) Hospital and assistant professors in the department of medicine at the University of Colorado.
The Affordable Care Act (ACA) has greatly increased the number of Medicaid enrollees since it expanded eligibility criteria on Jan. 1, 2014. Nearly 9 million additional U.S. adults now have Medicaid coverage, mostly in the 31 states and Washington, which have opted into Medicaid expansion.
The ACA has also had an important impact on hospital payer mix, mainly by decreasing the amount of uncompensated care in Medicaid-expansion states. Previous studies have shown disparities in the quality of inpatient care based on insurance type. Patients with Medicaid insurance often have longer hospitalizations and higher in-hospital mortality than commercially-insured patients and occasionally even than uninsured patients.
In our study published in the Journal of Hospital Medicine (2016 Dec. doi: 10.1002/jhm.2649), we evaluated the impact of state Medicaid expansion status on payer mix, length of stay, and in-hospital mortality for general medicine patients discharged from U.S. academic medical centers. We considered Jan. 1, 2014, to be the date of ACA implementation for all states except Michigan, New Hampshire, Pennsylvania, and Indiana, which had unique dates of Medicaid expansion.
We were able to identify 3,144,488 discharges from 156 hospitals in 24 Medicaid-expansion states and Washington, and 1,114,464 discharges from 55 hospitals in 14 nonexpansion states between October 2012 and September 2015.
Despite this difference in payer mix trends, state Medicaid expansion status was not associated with differences in overall length of stay or in-hospital mortality in our study. More precisely, we looked at the length of stay and mortality indices, or ratio of observed to expected values, to control for such potential confounders as disease severity and comorbid conditions.
One possible explanation for our findings is that the higher proportion of Medicare and commercially-insured patients overshadowed the contribution of Medicaid patients to the overall length of stay and mortality indices.
To our knowledge, our study is the first to look at the effect of ACA implementation on inpatient outcomes. Early evidence suggests that Medicaid expansion has improved outpatient outcomes. Low-income adults in Medicaid-expansion states have shown greater gains in access to primary care clinics and medications and in the diagnosis of certain chronic health conditions than those in non-expansion states. However, these changes would not necessarily lead to improvements in the length of stay or mortality indices for Medicaid-expansion hospitals, since the measures account for patient acuity on admission.
The take-home message from our study for health policy makers is that state Medicaid expansion status had a neutral effect on both length of stay and mortality indices. This should be reassuring for states considering expansion of their Medicaid programs in the future.
As a next step, it would be useful to see research on the impact of ACA implementation on other inpatient outcomes that may vary with insurance type, such as readmissions or hospital-acquired complications.
The take-home message for hospitalists is that there is more work to be done in reducing disparities in inpatient care based on payer status. Though not a primary focus of our study, we did see variation in the length of stay and mortality indices based on insurance type.
It is unclear whether these differences occurred because of variation in the expertise of inpatient providers, access to invasive procedures or medical therapies, the timeliness of discharge to post-acute care facilities, or other patient- or system-level factors. However, these disparities warrant our improvement efforts moving forward.
Mary Anderson, MD, and Christine Jones, MD, are hospitalists at the University of Colorado (Aurora) Hospital and assistant professors in the department of medicine at the University of Colorado.
The Affordable Care Act (ACA) has greatly increased the number of Medicaid enrollees since it expanded eligibility criteria on Jan. 1, 2014. Nearly 9 million additional U.S. adults now have Medicaid coverage, mostly in the 31 states and Washington, which have opted into Medicaid expansion.
The ACA has also had an important impact on hospital payer mix, mainly by decreasing the amount of uncompensated care in Medicaid-expansion states. Previous studies have shown disparities in the quality of inpatient care based on insurance type. Patients with Medicaid insurance often have longer hospitalizations and higher in-hospital mortality than commercially-insured patients and occasionally even than uninsured patients.
In our study published in the Journal of Hospital Medicine (2016 Dec. doi: 10.1002/jhm.2649), we evaluated the impact of state Medicaid expansion status on payer mix, length of stay, and in-hospital mortality for general medicine patients discharged from U.S. academic medical centers. We considered Jan. 1, 2014, to be the date of ACA implementation for all states except Michigan, New Hampshire, Pennsylvania, and Indiana, which had unique dates of Medicaid expansion.
We were able to identify 3,144,488 discharges from 156 hospitals in 24 Medicaid-expansion states and Washington, and 1,114,464 discharges from 55 hospitals in 14 nonexpansion states between October 2012 and September 2015.
Despite this difference in payer mix trends, state Medicaid expansion status was not associated with differences in overall length of stay or in-hospital mortality in our study. More precisely, we looked at the length of stay and mortality indices, or ratio of observed to expected values, to control for such potential confounders as disease severity and comorbid conditions.
One possible explanation for our findings is that the higher proportion of Medicare and commercially-insured patients overshadowed the contribution of Medicaid patients to the overall length of stay and mortality indices.
To our knowledge, our study is the first to look at the effect of ACA implementation on inpatient outcomes. Early evidence suggests that Medicaid expansion has improved outpatient outcomes. Low-income adults in Medicaid-expansion states have shown greater gains in access to primary care clinics and medications and in the diagnosis of certain chronic health conditions than those in non-expansion states. However, these changes would not necessarily lead to improvements in the length of stay or mortality indices for Medicaid-expansion hospitals, since the measures account for patient acuity on admission.
The take-home message from our study for health policy makers is that state Medicaid expansion status had a neutral effect on both length of stay and mortality indices. This should be reassuring for states considering expansion of their Medicaid programs in the future.
As a next step, it would be useful to see research on the impact of ACA implementation on other inpatient outcomes that may vary with insurance type, such as readmissions or hospital-acquired complications.
The take-home message for hospitalists is that there is more work to be done in reducing disparities in inpatient care based on payer status. Though not a primary focus of our study, we did see variation in the length of stay and mortality indices based on insurance type.
It is unclear whether these differences occurred because of variation in the expertise of inpatient providers, access to invasive procedures or medical therapies, the timeliness of discharge to post-acute care facilities, or other patient- or system-level factors. However, these disparities warrant our improvement efforts moving forward.
Mary Anderson, MD, and Christine Jones, MD, are hospitalists at the University of Colorado (Aurora) Hospital and assistant professors in the department of medicine at the University of Colorado.
OA drug development needs patient-focused approach to biomarkers and outcome measures
Advances in our understanding of the development and treatment of osteoarthritis have led to renewed interest in leveraging these insights to establish more ways to evaluate potential drug candidates in clinical trials, particularly through the use of qualified biomarkers as meaningful outcome measures.
It is with this goal in mind that the Arthritis Foundation and the Food and Drug Administration held the Accelerating Osteoarthritis Clinical Trials Workshop in Atlanta in February 2016,1 to discuss ways of enhancing the likelihood of successful OA trials, including possible “qualification” of biomarkers that can be used as endpoints in trials of OA treatments. Clinical trial endpoints would ideally be known to be clinically meaningful to the patient (such as pain, fatigue, functional status, etc.)2 and would reflect treatments that enable patients to feel and function better.

Patient engagement in different steps of the development process might be possible through patient registries, social media communities, smart phones, and wearable devices to be used as tools for capturing patient perceptions and mobility. These means for capturing dynamic data about domains of high interest to OA patients may lead to the development of better outcome measures that can be used as clinical trial endpoints.4 Researchers at the workshop presented new ways in which they are beginning to specialize in these techniques and methods that will be useful for turning the patient experience into usable data to aid in precision medicine.

Imaging outcomes and biomarkers are attractive for their potential to demonstrate an effect on the structural and pathophysiologic elements of OA in the time frame of a clinical trial, but rely on well-characterized relationships between those structural and pathophysiologic elements of OA and the clinical outcomes of OA. A recent advance is data showing that semiquantitative knee joint features, including cartilage thickness and surface area, meniscal morphology, and bone marrow lesions, were associated with clinically relevant OA progression and could be potentially useful as measures of efficacy in clinical trials of disease-modifying interventions.5 Being able to describe the clinical benefit to be expected from such changes is essential to use these outcomes in the benefit-risk assessment. How to include structural outcomes in OA trials will depend on the level of information available to characterize clinical benefit. With less information, structural outcomes may still be useful as adjunct or secondary endpoints. To be used as the primary endpoint to support approval, a high level of characterization would be needed about the relationship of the endpoint to anticipated clinical benefit. The FDA recommends that sponsors proposing such a primary endpoint should engage with the agency early in the development program.

Another option discussed was to study an accelerated OA population, that is, subjects (prior to joint replacement) with a history of trauma to a joint and other injuries predictive of early OA. In that setting, the study duration needed to demonstrate the relationship of structural and clinical outcomes may be more feasible.
A third option discussed was an end-stage OA trial that could enroll subjects who meet criteria for joint replacement. Such a study could possibly have outcomes that include delay in time to surgery, reduction in need for surgery, and clinical outcomes, such as need for concomitant analgesics, patient pain, and patient function. In any OA population, demonstrating a treatment’s effectiveness on clinical outcomes could be the sole basis for approval, in the context of an acceptable safety profile.

Dr. Niskar is national scientific director of the Arthritis Foundation. Dr. Yim is supervisory associate director of the division of pulmonary, allergy, and rheumatology products in the Office of New Drugs at the FDA’s Center for Drug Evaluation and Research. Dr. Kraus is professor of medicine, pathology, and orthopedic surgery at Duke University, Durham, N.C., and is a faculty member of the Duke Molecular Physiology Institute. Dr. Tuan is director of the Center for Cellular and Molecular Engineering and Distinguished Professor of Orthopedic Surgery at the University of Pittsburgh.
Dr. Niskar reports that the Arthritis Foundation received a donation from Samumed and a cosponsorship grant from the FDA to implement the Accelerating OA Clinical Trials Workshop. Dr. Kraus reports an Arthritis Foundation Delivering on Discovery Award that is outside of this editorial. Dr. Yim and Dr. Tuan disclosed no conflicts. Dr. Yim is relaying her personal views in this editorial, and they are not intended to convey official FDA policy, and no official support or endorsement by the FDA is provided or should be inferred. Samumed did not have a role in the writing of this editorial.
References
1. Bioworld. 2016;27:1-4.
2. Osteoarthritis Cartilage. 2015 May;23[5]:677-85.
3. Clinical Trials Transformation Initiative. PG engagement across the research & development continuum. 2015.
4. Sci Transl Med. 2016;8:336ps11.
5. Arthritis Rheumatol. 2016 Oct;68[10]:2422-31.
Advances in our understanding of the development and treatment of osteoarthritis have led to renewed interest in leveraging these insights to establish more ways to evaluate potential drug candidates in clinical trials, particularly through the use of qualified biomarkers as meaningful outcome measures.
It is with this goal in mind that the Arthritis Foundation and the Food and Drug Administration held the Accelerating Osteoarthritis Clinical Trials Workshop in Atlanta in February 2016,1 to discuss ways of enhancing the likelihood of successful OA trials, including possible “qualification” of biomarkers that can be used as endpoints in trials of OA treatments. Clinical trial endpoints would ideally be known to be clinically meaningful to the patient (such as pain, fatigue, functional status, etc.)2 and would reflect treatments that enable patients to feel and function better.
Patient engagement in different steps of the development process might be possible through patient registries, social media communities, smart phones, and wearable devices to be used as tools for capturing patient perceptions and mobility. These means for capturing dynamic data about domains of high interest to OA patients may lead to the development of better outcome measures that can be used as clinical trial endpoints.4 Researchers at the workshop presented new ways in which they are beginning to specialize in these techniques and methods that will be useful for turning the patient experience into usable data to aid in precision medicine.
Imaging outcomes and biomarkers are attractive for their potential to demonstrate an effect on the structural and pathophysiologic elements of OA in the time frame of a clinical trial, but rely on well-characterized relationships between those structural and pathophysiologic elements of OA and the clinical outcomes of OA. A recent advance is data showing that semiquantitative knee joint features, including cartilage thickness and surface area, meniscal morphology, and bone marrow lesions, were associated with clinically relevant OA progression and could be potentially useful as measures of efficacy in clinical trials of disease-modifying interventions.5 Being able to describe the clinical benefit to be expected from such changes is essential to use these outcomes in the benefit-risk assessment. How to include structural outcomes in OA trials will depend on the level of information available to characterize clinical benefit. With less information, structural outcomes may still be useful as adjunct or secondary endpoints. To be used as the primary endpoint to support approval, a high level of characterization would be needed about the relationship of the endpoint to anticipated clinical benefit. The FDA recommends that sponsors proposing such a primary endpoint should engage with the agency early in the development program.
Another option discussed was to study an accelerated OA population, that is, subjects (prior to joint replacement) with a history of trauma to a joint and other injuries predictive of early OA. In that setting, the study duration needed to demonstrate the relationship of structural and clinical outcomes may be more feasible.
A third option discussed was an end-stage OA trial that could enroll subjects who meet criteria for joint replacement. Such a study could possibly have outcomes that include delay in time to surgery, reduction in need for surgery, and clinical outcomes, such as need for concomitant analgesics, patient pain, and patient function. In any OA population, demonstrating a treatment’s effectiveness on clinical outcomes could be the sole basis for approval, in the context of an acceptable safety profile.
Dr. Niskar is national scientific director of the Arthritis Foundation. Dr. Yim is supervisory associate director of the division of pulmonary, allergy, and rheumatology products in the Office of New Drugs at the FDA’s Center for Drug Evaluation and Research. Dr. Kraus is professor of medicine, pathology, and orthopedic surgery at Duke University, Durham, N.C., and is a faculty member of the Duke Molecular Physiology Institute. Dr. Tuan is director of the Center for Cellular and Molecular Engineering and Distinguished Professor of Orthopedic Surgery at the University of Pittsburgh.
Dr. Niskar reports that the Arthritis Foundation received a donation from Samumed and a cosponsorship grant from the FDA to implement the Accelerating OA Clinical Trials Workshop. Dr. Kraus reports an Arthritis Foundation Delivering on Discovery Award that is outside of this editorial. Dr. Yim and Dr. Tuan disclosed no conflicts. Dr. Yim is relaying her personal views in this editorial, and they are not intended to convey official FDA policy, and no official support or endorsement by the FDA is provided or should be inferred. Samumed did not have a role in the writing of this editorial.
References
1. Bioworld. 2016;27:1-4.
2. Osteoarthritis Cartilage. 2015 May;23[5]:677-85.
3. Clinical Trials Transformation Initiative. PG engagement across the research & development continuum. 2015.
4. Sci Transl Med. 2016;8:336ps11.
5. Arthritis Rheumatol. 2016 Oct;68[10]:2422-31.
Advances in our understanding of the development and treatment of osteoarthritis have led to renewed interest in leveraging these insights to establish more ways to evaluate potential drug candidates in clinical trials, particularly through the use of qualified biomarkers as meaningful outcome measures.
It is with this goal in mind that the Arthritis Foundation and the Food and Drug Administration held the Accelerating Osteoarthritis Clinical Trials Workshop in Atlanta in February 2016,1 to discuss ways of enhancing the likelihood of successful OA trials, including possible “qualification” of biomarkers that can be used as endpoints in trials of OA treatments. Clinical trial endpoints would ideally be known to be clinically meaningful to the patient (such as pain, fatigue, functional status, etc.)2 and would reflect treatments that enable patients to feel and function better.
Patient engagement in different steps of the development process might be possible through patient registries, social media communities, smart phones, and wearable devices to be used as tools for capturing patient perceptions and mobility. These means for capturing dynamic data about domains of high interest to OA patients may lead to the development of better outcome measures that can be used as clinical trial endpoints.4 Researchers at the workshop presented new ways in which they are beginning to specialize in these techniques and methods that will be useful for turning the patient experience into usable data to aid in precision medicine.
Imaging outcomes and biomarkers are attractive for their potential to demonstrate an effect on the structural and pathophysiologic elements of OA in the time frame of a clinical trial, but rely on well-characterized relationships between those structural and pathophysiologic elements of OA and the clinical outcomes of OA. A recent advance is data showing that semiquantitative knee joint features, including cartilage thickness and surface area, meniscal morphology, and bone marrow lesions, were associated with clinically relevant OA progression and could be potentially useful as measures of efficacy in clinical trials of disease-modifying interventions.5 Being able to describe the clinical benefit to be expected from such changes is essential to use these outcomes in the benefit-risk assessment. How to include structural outcomes in OA trials will depend on the level of information available to characterize clinical benefit. With less information, structural outcomes may still be useful as adjunct or secondary endpoints. To be used as the primary endpoint to support approval, a high level of characterization would be needed about the relationship of the endpoint to anticipated clinical benefit. The FDA recommends that sponsors proposing such a primary endpoint should engage with the agency early in the development program.
Another option discussed was to study an accelerated OA population, that is, subjects (prior to joint replacement) with a history of trauma to a joint and other injuries predictive of early OA. In that setting, the study duration needed to demonstrate the relationship of structural and clinical outcomes may be more feasible.
A third option discussed was an end-stage OA trial that could enroll subjects who meet criteria for joint replacement. Such a study could possibly have outcomes that include delay in time to surgery, reduction in need for surgery, and clinical outcomes, such as need for concomitant analgesics, patient pain, and patient function. In any OA population, demonstrating a treatment’s effectiveness on clinical outcomes could be the sole basis for approval, in the context of an acceptable safety profile.
Dr. Niskar is national scientific director of the Arthritis Foundation. Dr. Yim is supervisory associate director of the division of pulmonary, allergy, and rheumatology products in the Office of New Drugs at the FDA’s Center for Drug Evaluation and Research. Dr. Kraus is professor of medicine, pathology, and orthopedic surgery at Duke University, Durham, N.C., and is a faculty member of the Duke Molecular Physiology Institute. Dr. Tuan is director of the Center for Cellular and Molecular Engineering and Distinguished Professor of Orthopedic Surgery at the University of Pittsburgh.
Dr. Niskar reports that the Arthritis Foundation received a donation from Samumed and a cosponsorship grant from the FDA to implement the Accelerating OA Clinical Trials Workshop. Dr. Kraus reports an Arthritis Foundation Delivering on Discovery Award that is outside of this editorial. Dr. Yim and Dr. Tuan disclosed no conflicts. Dr. Yim is relaying her personal views in this editorial, and they are not intended to convey official FDA policy, and no official support or endorsement by the FDA is provided or should be inferred. Samumed did not have a role in the writing of this editorial.
References
1. Bioworld. 2016;27:1-4.
2. Osteoarthritis Cartilage. 2015 May;23[5]:677-85.
3. Clinical Trials Transformation Initiative. PG engagement across the research & development continuum. 2015.
4. Sci Transl Med. 2016;8:336ps11.
5. Arthritis Rheumatol. 2016 Oct;68[10]:2422-31.
The Death of a Dream: Closing an NP Practice
For many nurse practitioners, having your own practice is the culmination of many years of planning and anticipation. I worked as an NP for 14 years in practices operated by others—hospitals and physicians—before I opened my own practice. During those years, I had observed which ways of doing things appeared productive and healing to me and which did not.
When the time came, having seen a need for more affordable health care that was not predicated on the assumption that every patient had health insurance, I opened a cash-only practice in the town where I resided. By eliminating the need for personnel and apparatus dedicated to insurance filing, I was able to charge about half of what other practices in the same location did for identical services. My chief goal was to be of service to the community, not to make the most money possible. I anticipated that volume would make up for the lower prices in the long run.
For about four years, our revenue grew slowly. I decided to risk all and stop teaching part-time in order to focus exclusively on my practice. This proved to be a good decision—for about one year. Then the recession hit my part of the country. Suddenly, the operation of a “cash-only” practice became an oxymoron, as many of the patients with already limited funds lost their jobs. These patients started to seek “free” care at area emergency departments, and the practice income plummeted. My revenue fell by one-half the first year and then one-half of that the next year.
In what would turn out to be my final year of practice ownership, I decided to accept a full-time position as faculty of a distance FNP program. I was practicing “on the side,” although in reality I was in my office full-time and teaching from there. I was already recognizing the difficulties of juggling my roles and responsibilities when a student in one of my classes asked what it would take for me to decide to close my practice, since I was (by this point) making no money from it.
As I pondered that question (my initial response was a quite honest “I don’t know”), I stepped onto the road toward closing my practice. From a business perspective, there was little point in keeping the practice open. However, from an emotional point of view, I had invested so much in building my dream—and, by extension, so had my family—that closing the practice seemed unthinkable.
THE DECISION
Opening a practice is a time of joy, pride, and a sense of accomplishment; closing that same practice induces a period of reflection, sadness, and even anger that circumstances did not allow continued operation. While financial considerations play a significant role in the decision to close a practice, they may not be the only, or the deciding, factor.
In my case, the financial shortfall of the practice led me to accept a teaching position in order to earn living expenses. The result of that decision was that my attention became divided: Sometimes I was in meetings or interacting with students—and for a few weeks per year, I was out of town—which meant less time devoted to seeing patients. Conversely, if I was with a patient, I of course could not be available to my students. Over time, I started to feel that I was not giving my all to either role as I shifted back and forth. Having given 100% to each of these roles at previous points in my career, I now felt that I was cheating my patients and my students.
The decision to close a practice may take months or even years before the actual process is started. I lived with my conundrum for about a year before I made the decision to close. I was exhausted—and while I was relieved to have the burden of deciding off my shoulders, it was now time to do the work of closing a practice.

NOTIFYING YOUR STAKEHOLDERS
Once the decision to close a practice is reached, the provider/owner ceases to exist in a vacuum. There are stakeholders who need to be notified—some obvious, some less so.
I started by breaking the news to my family, the people who had supported me in opening my practice (and even helped me find and refurbish furniture for my waiting room!). Although they had been aware of my internal debate, they had not lived with the decision process as I had. Having resolved at least some of my own emotions, I now had to watch others experience many of those same feelings.
Next, I had to tell my employees of the decision. Through attrition, my staff had already shrunk to two: a receptionist and a part-time licensed vocational nurse (LVN). Like my family, they had to process their own emotions about the closure. I had anticipated that the people who worked for me, concerned about their future, might choose to accept another job before we officially closed. My LVN—who had observed the practice dwindling in the preceding two years—seemed prepared for my decision. She stuck it out with me until the end and was a huge help with the influx of patients requesting records. (My MD—required by Texas law to delegate prescriptive authority to me—had already relocated his practice and was ill, so he was content with my decision.)
Of course, the biggest stakeholders in a practice are the patients. Notifying them of the impending closure is the most important action you will take (aside from making the decision to close). Although you can place notices in the local media (newspapers, TV, radio) to announce the closure of your practice to the community, you should send a notification letter directly to your patients. It should be sent at least 60 to 90 days before the closure date—and certainly not less than 30 days in any case—giving patients adequate time to find new providers and arrange for their records to be transferred.1,2 The letter should include
- A statement of gratitude for the patient’s business
- The dates of the transition period
- What is expected of the patient (eg, does he/she need to come and pick up his/her records?)
- An explanation for the closure3
I composed a letter to be sent to all patients who had been seen within the past 18 months. In it, I thanked them for being a part of the practice and gave them 60 days’ notice of the intent to close. For many patients, this was an emotional time; many understandably worried how their health care needs would be met in the future. Some responded with sadness that I had not been able to make the practice a success.
AVOIDING “ABANDONMENT”
Ideally, a provider who wants to get out of the business should seek to sell the practice—but this is not always feasible.3,4 When closure is the best (or only) option, it is important to avoid even the appearance of abandonment.
Besides giving adequate notice of practice closure, providers must have a plan for the dispersal of patients.1 Be prepared to give recommendations for new providers. Depending on the practice location (rural or urban), options may vary.
I made a concerted effort to refer patients to new providers, with the caveat that if the patient did not feel a particular provider was a good match, he/she should seek another provider of his/her choosing. Unlike in a purchased practice, where patients “go with” the practice, patients from a closed practice may be referred to one, several, or even many other providers.5
Provisions must be made to store patient records so that they are retrievable for a specified period of time. The requirements vary by state, so consultation of the state board’s rules and regulations—and/or an attorney—is in order.3 In general, the proscribed time period is seven to 10 years for adults and seven to 10 years after the patient turns 18 for pediatric patients.2 In some states, the retention time may be as short as three years for adults.1

OTHER PRACTICAL CONSIDERATIONS
While people will be your priority as you work through the process of closing, you will have “stuff” to deal with. What will you do with the furnishings and equipment? Obviously, anything that was borrowed can be returned. Beyond that, your options are to sell (to another provider or even a patient), donate, or repurpose items.
The orthopedic exam table from my practice went to a private school for their athletic training facility. Screens went to my neighbor, a chiropractor. My preschool-aged grandsons were thrilled to be given the children’s art supplies and books that had once graced my waiting area. One of my patients bought some decorative vases and a bookcase. The painting that had been carefully chosen to pull together my waiting room now hangs in my library at home.
As the closure date approaches, the practice environment may begin to look bare as furnishings are sold or moved. One item you will want to buy, however, is a fresh ink cartridge for your copier/printer. As patients request documents, you’ll use it!
RESPONSE AND AFTERMATH
The practice may be very busy immediately following the receipt of notification letters—but don’t be fooled into thinking you have made the wrong decision. The first month after the letters went out advising of the closure, my practice was busier than it had ever been! This tapered off in the second month, though.
Most patients, once they’ve heard the news, will want prescription refills and/or their records. Some may just want to know what happened to result in the closure. Remember that to the patient, this seems like a sudden decision—no matter how long you have deliberated about it.
What surprised me most, however, was that new patients continued to present to the practice, seeking care for acute issues. While I did provide this, I made them aware from the beginning that the practice was in the process of closing and that I could not assume the responsibility of being their primary provider. I made sure to provide these patients with recommendations for other providers.
Slowly the rush will settle down, as patients start to move on to other providers. A few may drop in to see you socially. On the day I closed my practice, several patients came in just to say goodbye and wish me well.
The last things I did in my practice were turn off the lights and leave a sign on the door stating that the practice was now closed.
RECOVERY
The time needed to recover from the closure of a practice will differ. Factors include how long the practice was open and how the clinician normally deals with a setback.6 For some, relief that the pressures of ownership are over may be the predominant emotion. Having a steady, stable salary in a new position goes a long way toward making the transition easier! Although if possible, take some time between closing the practice and starting a new job.
Do not be surprised if negative emotions manifest at odd times, as feelings of sadness, regret, and even a sense of failure are worked through. Life does go on—and nurse practitioners are resilient. Find a way to use the knowledge gained from your practice in your new endeavors, whatever they may be.
For me, the healing process would have started sooner if I had acknowledged how difficult giving up the dream of having my own practice was. If I had sought out others with similar experience or even talked with a counselor, my journey through this process could have been expedited. When I started to share my story, one frequently asked question was “How did you get through this?” This showed me that others could learn from my experience.
1. Tatooles JJ, Brunell A. What you need to know before leaving a medical practice: a primer for moving on. AAOS NOW. 2015;22-23.
2. Kern SI. Take these steps if selling or closing a practice. Med Econ. 2010;87(20):66.
3. Weiss GG. How to close a practice. Med Econ. 2004;81:69.
4. Zaumeyer C. How to Start an Independent Practice: The Nurse Practitioner’s Guide to Success. Philadelphia: F. A. Davis, Publishers; 2003.
5. Barrett W. The legal corner: Eleven essential steps to purchasing or selling a medical practice. J Med Pract Manage. 2014;29(5):275-277.
6. McBride JL. Personal issues to consider before leaving independent practice. Fam Pract Manag. 2013;20(4):9-12.
For many nurse practitioners, having your own practice is the culmination of many years of planning and anticipation. I worked as an NP for 14 years in practices operated by others—hospitals and physicians—before I opened my own practice. During those years, I had observed which ways of doing things appeared productive and healing to me and which did not.
When the time came, having seen a need for more affordable health care that was not predicated on the assumption that every patient had health insurance, I opened a cash-only practice in the town where I resided. By eliminating the need for personnel and apparatus dedicated to insurance filing, I was able to charge about half of what other practices in the same location did for identical services. My chief goal was to be of service to the community, not to make the most money possible. I anticipated that volume would make up for the lower prices in the long run.
For about four years, our revenue grew slowly. I decided to risk all and stop teaching part-time in order to focus exclusively on my practice. This proved to be a good decision—for about one year. Then the recession hit my part of the country. Suddenly, the operation of a “cash-only” practice became an oxymoron, as many of the patients with already limited funds lost their jobs. These patients started to seek “free” care at area emergency departments, and the practice income plummeted. My revenue fell by one-half the first year and then one-half of that the next year.
In what would turn out to be my final year of practice ownership, I decided to accept a full-time position as faculty of a distance FNP program. I was practicing “on the side,” although in reality I was in my office full-time and teaching from there. I was already recognizing the difficulties of juggling my roles and responsibilities when a student in one of my classes asked what it would take for me to decide to close my practice, since I was (by this point) making no money from it.
As I pondered that question (my initial response was a quite honest “I don’t know”), I stepped onto the road toward closing my practice. From a business perspective, there was little point in keeping the practice open. However, from an emotional point of view, I had invested so much in building my dream—and, by extension, so had my family—that closing the practice seemed unthinkable.
THE DECISION
Opening a practice is a time of joy, pride, and a sense of accomplishment; closing that same practice induces a period of reflection, sadness, and even anger that circumstances did not allow continued operation. While financial considerations play a significant role in the decision to close a practice, they may not be the only, or the deciding, factor.
In my case, the financial shortfall of the practice led me to accept a teaching position in order to earn living expenses. The result of that decision was that my attention became divided: Sometimes I was in meetings or interacting with students—and for a few weeks per year, I was out of town—which meant less time devoted to seeing patients. Conversely, if I was with a patient, I of course could not be available to my students. Over time, I started to feel that I was not giving my all to either role as I shifted back and forth. Having given 100% to each of these roles at previous points in my career, I now felt that I was cheating my patients and my students.
The decision to close a practice may take months or even years before the actual process is started. I lived with my conundrum for about a year before I made the decision to close. I was exhausted—and while I was relieved to have the burden of deciding off my shoulders, it was now time to do the work of closing a practice.
NOTIFYING YOUR STAKEHOLDERS
Once the decision to close a practice is reached, the provider/owner ceases to exist in a vacuum. There are stakeholders who need to be notified—some obvious, some less so.
I started by breaking the news to my family, the people who had supported me in opening my practice (and even helped me find and refurbish furniture for my waiting room!). Although they had been aware of my internal debate, they had not lived with the decision process as I had. Having resolved at least some of my own emotions, I now had to watch others experience many of those same feelings.
Next, I had to tell my employees of the decision. Through attrition, my staff had already shrunk to two: a receptionist and a part-time licensed vocational nurse (LVN). Like my family, they had to process their own emotions about the closure. I had anticipated that the people who worked for me, concerned about their future, might choose to accept another job before we officially closed. My LVN—who had observed the practice dwindling in the preceding two years—seemed prepared for my decision. She stuck it out with me until the end and was a huge help with the influx of patients requesting records. (My MD—required by Texas law to delegate prescriptive authority to me—had already relocated his practice and was ill, so he was content with my decision.)
Of course, the biggest stakeholders in a practice are the patients. Notifying them of the impending closure is the most important action you will take (aside from making the decision to close). Although you can place notices in the local media (newspapers, TV, radio) to announce the closure of your practice to the community, you should send a notification letter directly to your patients. It should be sent at least 60 to 90 days before the closure date—and certainly not less than 30 days in any case—giving patients adequate time to find new providers and arrange for their records to be transferred.1,2 The letter should include
- A statement of gratitude for the patient’s business
- The dates of the transition period
- What is expected of the patient (eg, does he/she need to come and pick up his/her records?)
- An explanation for the closure3
I composed a letter to be sent to all patients who had been seen within the past 18 months. In it, I thanked them for being a part of the practice and gave them 60 days’ notice of the intent to close. For many patients, this was an emotional time; many understandably worried how their health care needs would be met in the future. Some responded with sadness that I had not been able to make the practice a success.
AVOIDING “ABANDONMENT”
Ideally, a provider who wants to get out of the business should seek to sell the practice—but this is not always feasible.3,4 When closure is the best (or only) option, it is important to avoid even the appearance of abandonment.
Besides giving adequate notice of practice closure, providers must have a plan for the dispersal of patients.1 Be prepared to give recommendations for new providers. Depending on the practice location (rural or urban), options may vary.
I made a concerted effort to refer patients to new providers, with the caveat that if the patient did not feel a particular provider was a good match, he/she should seek another provider of his/her choosing. Unlike in a purchased practice, where patients “go with” the practice, patients from a closed practice may be referred to one, several, or even many other providers.5
Provisions must be made to store patient records so that they are retrievable for a specified period of time. The requirements vary by state, so consultation of the state board’s rules and regulations—and/or an attorney—is in order.3 In general, the proscribed time period is seven to 10 years for adults and seven to 10 years after the patient turns 18 for pediatric patients.2 In some states, the retention time may be as short as three years for adults.1
OTHER PRACTICAL CONSIDERATIONS
While people will be your priority as you work through the process of closing, you will have “stuff” to deal with. What will you do with the furnishings and equipment? Obviously, anything that was borrowed can be returned. Beyond that, your options are to sell (to another provider or even a patient), donate, or repurpose items.
The orthopedic exam table from my practice went to a private school for their athletic training facility. Screens went to my neighbor, a chiropractor. My preschool-aged grandsons were thrilled to be given the children’s art supplies and books that had once graced my waiting area. One of my patients bought some decorative vases and a bookcase. The painting that had been carefully chosen to pull together my waiting room now hangs in my library at home.
As the closure date approaches, the practice environment may begin to look bare as furnishings are sold or moved. One item you will want to buy, however, is a fresh ink cartridge for your copier/printer. As patients request documents, you’ll use it!
RESPONSE AND AFTERMATH
The practice may be very busy immediately following the receipt of notification letters—but don’t be fooled into thinking you have made the wrong decision. The first month after the letters went out advising of the closure, my practice was busier than it had ever been! This tapered off in the second month, though.
Most patients, once they’ve heard the news, will want prescription refills and/or their records. Some may just want to know what happened to result in the closure. Remember that to the patient, this seems like a sudden decision—no matter how long you have deliberated about it.
What surprised me most, however, was that new patients continued to present to the practice, seeking care for acute issues. While I did provide this, I made them aware from the beginning that the practice was in the process of closing and that I could not assume the responsibility of being their primary provider. I made sure to provide these patients with recommendations for other providers.
Slowly the rush will settle down, as patients start to move on to other providers. A few may drop in to see you socially. On the day I closed my practice, several patients came in just to say goodbye and wish me well.
The last things I did in my practice were turn off the lights and leave a sign on the door stating that the practice was now closed.
RECOVERY
The time needed to recover from the closure of a practice will differ. Factors include how long the practice was open and how the clinician normally deals with a setback.6 For some, relief that the pressures of ownership are over may be the predominant emotion. Having a steady, stable salary in a new position goes a long way toward making the transition easier! Although if possible, take some time between closing the practice and starting a new job.
Do not be surprised if negative emotions manifest at odd times, as feelings of sadness, regret, and even a sense of failure are worked through. Life does go on—and nurse practitioners are resilient. Find a way to use the knowledge gained from your practice in your new endeavors, whatever they may be.
For me, the healing process would have started sooner if I had acknowledged how difficult giving up the dream of having my own practice was. If I had sought out others with similar experience or even talked with a counselor, my journey through this process could have been expedited. When I started to share my story, one frequently asked question was “How did you get through this?” This showed me that others could learn from my experience.
For many nurse practitioners, having your own practice is the culmination of many years of planning and anticipation. I worked as an NP for 14 years in practices operated by others—hospitals and physicians—before I opened my own practice. During those years, I had observed which ways of doing things appeared productive and healing to me and which did not.
When the time came, having seen a need for more affordable health care that was not predicated on the assumption that every patient had health insurance, I opened a cash-only practice in the town where I resided. By eliminating the need for personnel and apparatus dedicated to insurance filing, I was able to charge about half of what other practices in the same location did for identical services. My chief goal was to be of service to the community, not to make the most money possible. I anticipated that volume would make up for the lower prices in the long run.
For about four years, our revenue grew slowly. I decided to risk all and stop teaching part-time in order to focus exclusively on my practice. This proved to be a good decision—for about one year. Then the recession hit my part of the country. Suddenly, the operation of a “cash-only” practice became an oxymoron, as many of the patients with already limited funds lost their jobs. These patients started to seek “free” care at area emergency departments, and the practice income plummeted. My revenue fell by one-half the first year and then one-half of that the next year.
In what would turn out to be my final year of practice ownership, I decided to accept a full-time position as faculty of a distance FNP program. I was practicing “on the side,” although in reality I was in my office full-time and teaching from there. I was already recognizing the difficulties of juggling my roles and responsibilities when a student in one of my classes asked what it would take for me to decide to close my practice, since I was (by this point) making no money from it.
As I pondered that question (my initial response was a quite honest “I don’t know”), I stepped onto the road toward closing my practice. From a business perspective, there was little point in keeping the practice open. However, from an emotional point of view, I had invested so much in building my dream—and, by extension, so had my family—that closing the practice seemed unthinkable.
THE DECISION
Opening a practice is a time of joy, pride, and a sense of accomplishment; closing that same practice induces a period of reflection, sadness, and even anger that circumstances did not allow continued operation. While financial considerations play a significant role in the decision to close a practice, they may not be the only, or the deciding, factor.
In my case, the financial shortfall of the practice led me to accept a teaching position in order to earn living expenses. The result of that decision was that my attention became divided: Sometimes I was in meetings or interacting with students—and for a few weeks per year, I was out of town—which meant less time devoted to seeing patients. Conversely, if I was with a patient, I of course could not be available to my students. Over time, I started to feel that I was not giving my all to either role as I shifted back and forth. Having given 100% to each of these roles at previous points in my career, I now felt that I was cheating my patients and my students.
The decision to close a practice may take months or even years before the actual process is started. I lived with my conundrum for about a year before I made the decision to close. I was exhausted—and while I was relieved to have the burden of deciding off my shoulders, it was now time to do the work of closing a practice.
NOTIFYING YOUR STAKEHOLDERS
Once the decision to close a practice is reached, the provider/owner ceases to exist in a vacuum. There are stakeholders who need to be notified—some obvious, some less so.
I started by breaking the news to my family, the people who had supported me in opening my practice (and even helped me find and refurbish furniture for my waiting room!). Although they had been aware of my internal debate, they had not lived with the decision process as I had. Having resolved at least some of my own emotions, I now had to watch others experience many of those same feelings.
Next, I had to tell my employees of the decision. Through attrition, my staff had already shrunk to two: a receptionist and a part-time licensed vocational nurse (LVN). Like my family, they had to process their own emotions about the closure. I had anticipated that the people who worked for me, concerned about their future, might choose to accept another job before we officially closed. My LVN—who had observed the practice dwindling in the preceding two years—seemed prepared for my decision. She stuck it out with me until the end and was a huge help with the influx of patients requesting records. (My MD—required by Texas law to delegate prescriptive authority to me—had already relocated his practice and was ill, so he was content with my decision.)
Of course, the biggest stakeholders in a practice are the patients. Notifying them of the impending closure is the most important action you will take (aside from making the decision to close). Although you can place notices in the local media (newspapers, TV, radio) to announce the closure of your practice to the community, you should send a notification letter directly to your patients. It should be sent at least 60 to 90 days before the closure date—and certainly not less than 30 days in any case—giving patients adequate time to find new providers and arrange for their records to be transferred.1,2 The letter should include
- A statement of gratitude for the patient’s business
- The dates of the transition period
- What is expected of the patient (eg, does he/she need to come and pick up his/her records?)
- An explanation for the closure3
I composed a letter to be sent to all patients who had been seen within the past 18 months. In it, I thanked them for being a part of the practice and gave them 60 days’ notice of the intent to close. For many patients, this was an emotional time; many understandably worried how their health care needs would be met in the future. Some responded with sadness that I had not been able to make the practice a success.
AVOIDING “ABANDONMENT”
Ideally, a provider who wants to get out of the business should seek to sell the practice—but this is not always feasible.3,4 When closure is the best (or only) option, it is important to avoid even the appearance of abandonment.
Besides giving adequate notice of practice closure, providers must have a plan for the dispersal of patients.1 Be prepared to give recommendations for new providers. Depending on the practice location (rural or urban), options may vary.
I made a concerted effort to refer patients to new providers, with the caveat that if the patient did not feel a particular provider was a good match, he/she should seek another provider of his/her choosing. Unlike in a purchased practice, where patients “go with” the practice, patients from a closed practice may be referred to one, several, or even many other providers.5
Provisions must be made to store patient records so that they are retrievable for a specified period of time. The requirements vary by state, so consultation of the state board’s rules and regulations—and/or an attorney—is in order.3 In general, the proscribed time period is seven to 10 years for adults and seven to 10 years after the patient turns 18 for pediatric patients.2 In some states, the retention time may be as short as three years for adults.1
OTHER PRACTICAL CONSIDERATIONS
While people will be your priority as you work through the process of closing, you will have “stuff” to deal with. What will you do with the furnishings and equipment? Obviously, anything that was borrowed can be returned. Beyond that, your options are to sell (to another provider or even a patient), donate, or repurpose items.
The orthopedic exam table from my practice went to a private school for their athletic training facility. Screens went to my neighbor, a chiropractor. My preschool-aged grandsons were thrilled to be given the children’s art supplies and books that had once graced my waiting area. One of my patients bought some decorative vases and a bookcase. The painting that had been carefully chosen to pull together my waiting room now hangs in my library at home.
As the closure date approaches, the practice environment may begin to look bare as furnishings are sold or moved. One item you will want to buy, however, is a fresh ink cartridge for your copier/printer. As patients request documents, you’ll use it!
RESPONSE AND AFTERMATH
The practice may be very busy immediately following the receipt of notification letters—but don’t be fooled into thinking you have made the wrong decision. The first month after the letters went out advising of the closure, my practice was busier than it had ever been! This tapered off in the second month, though.
Most patients, once they’ve heard the news, will want prescription refills and/or their records. Some may just want to know what happened to result in the closure. Remember that to the patient, this seems like a sudden decision—no matter how long you have deliberated about it.
What surprised me most, however, was that new patients continued to present to the practice, seeking care for acute issues. While I did provide this, I made them aware from the beginning that the practice was in the process of closing and that I could not assume the responsibility of being their primary provider. I made sure to provide these patients with recommendations for other providers.
Slowly the rush will settle down, as patients start to move on to other providers. A few may drop in to see you socially. On the day I closed my practice, several patients came in just to say goodbye and wish me well.
The last things I did in my practice were turn off the lights and leave a sign on the door stating that the practice was now closed.
RECOVERY
The time needed to recover from the closure of a practice will differ. Factors include how long the practice was open and how the clinician normally deals with a setback.6 For some, relief that the pressures of ownership are over may be the predominant emotion. Having a steady, stable salary in a new position goes a long way toward making the transition easier! Although if possible, take some time between closing the practice and starting a new job.
Do not be surprised if negative emotions manifest at odd times, as feelings of sadness, regret, and even a sense of failure are worked through. Life does go on—and nurse practitioners are resilient. Find a way to use the knowledge gained from your practice in your new endeavors, whatever they may be.
For me, the healing process would have started sooner if I had acknowledged how difficult giving up the dream of having my own practice was. If I had sought out others with similar experience or even talked with a counselor, my journey through this process could have been expedited. When I started to share my story, one frequently asked question was “How did you get through this?” This showed me that others could learn from my experience.
1. Tatooles JJ, Brunell A. What you need to know before leaving a medical practice: a primer for moving on. AAOS NOW. 2015;22-23.
2. Kern SI. Take these steps if selling or closing a practice. Med Econ. 2010;87(20):66.
3. Weiss GG. How to close a practice. Med Econ. 2004;81:69.
4. Zaumeyer C. How to Start an Independent Practice: The Nurse Practitioner’s Guide to Success. Philadelphia: F. A. Davis, Publishers; 2003.
5. Barrett W. The legal corner: Eleven essential steps to purchasing or selling a medical practice. J Med Pract Manage. 2014;29(5):275-277.
6. McBride JL. Personal issues to consider before leaving independent practice. Fam Pract Manag. 2013;20(4):9-12.
1. Tatooles JJ, Brunell A. What you need to know before leaving a medical practice: a primer for moving on. AAOS NOW. 2015;22-23.
2. Kern SI. Take these steps if selling or closing a practice. Med Econ. 2010;87(20):66.
3. Weiss GG. How to close a practice. Med Econ. 2004;81:69.
4. Zaumeyer C. How to Start an Independent Practice: The Nurse Practitioner’s Guide to Success. Philadelphia: F. A. Davis, Publishers; 2003.
5. Barrett W. The legal corner: Eleven essential steps to purchasing or selling a medical practice. J Med Pract Manage. 2014;29(5):275-277.
6. McBride JL. Personal issues to consider before leaving independent practice. Fam Pract Manag. 2013;20(4):9-12.
The war on pain
When your peer group is dominated by folks in their early 70s, conversations at dinner parties and lobster bakes invariably morph into storytelling competitions between the survivors of recent hospitalizations and medical procedures. I try to redirect this tedious and repetitive chatter with a topic from my standard collection of conversation re-starters that includes “How about those Red Sox?” and “How’s your granddaughter’s soccer season going?” But sadly I am not always successful.
Often embedded in these tales of medical misadventure are stories of unfortunate experiences with pain medications. Sometimes the story includes a description of how prescribed pain medication created symptoms that were far worse than the pain it was intended to treat. Vomiting, constipation, and “feeling goofy” are high on the list of complaints.

These caches of unused opioids, many of which were never needed in the first place, are evidence of why our health care has become so expensive, and also represent the seeds from which the addiction epidemic has grown. Ironically, they also are collateral damage from an unsuccessful and sometimes misguided war on pain.
It isn’t clear exactly when or where the war on pain began, but I’m sure those who fired the first shots were understandably concerned that many patients with incurable and terminal conditions were suffering needlessly because their pain was being under-treated. Coincidently came the realization that the sooner we could get postoperative patients on their feet and taking deep breaths, the fewer complications we would see. And the more adequately we treated their pain, the sooner we could get those patients moving and breathing optimally.
In a good faith effort to be more “scientific” about pain management, patients were asked to rate their pain and smiley face charts appeared. Unfortunately, somewhere along the line came the mantra that not only should no patient’s pain go unmeasured, but no patient’s pain should go unmedicated.
The federal government entered the war when the Centers for Medicare & Medicaid Services issued the directive that hospitals ask patients who were being discharged if their pain had been well controlled and how often did the hospital staff do what they could to ease their pain? The answers to these questions, along with others, was collected and used in assessing a hospital’s quality of care and determining its level of reimbursement.
So far, there is insufficient data to determine how frequently this directive on pain management induced hospitals to over-prescribe medication, but it certainly hasn’t been associated with a decline in opioid abuse. It is reasonable to suspect that this salvo by the government has resulted in some collateral damage as it encouraged a steady flow of unused and unnecessary prescription narcotics out of the hospital and on to the streets.
The good news is that there has been enough concern voiced about the unintended effect of these pain management questions that the CMS has decided to eliminate financial pressure clinicians might feel to over-prescribe medications by withdrawing the questions from the patient discharge questionnaire.
The bad news is that we continue to fight the war on pain with a limited arsenal. As long as clinicians simply believe that no pain should go unmedicated, they will continue to miss opportunities to use other modalities such as counseling, physical therapy, and education that can be effective without the risk of collateral damage. Instead of asking the patient (who may not know the answer), we should be asking ourselves if we have been doing everything we could to help the patient deal with his pain. The answer is often not written on prescription pads.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
When your peer group is dominated by folks in their early 70s, conversations at dinner parties and lobster bakes invariably morph into storytelling competitions between the survivors of recent hospitalizations and medical procedures. I try to redirect this tedious and repetitive chatter with a topic from my standard collection of conversation re-starters that includes “How about those Red Sox?” and “How’s your granddaughter’s soccer season going?” But sadly I am not always successful.
Often embedded in these tales of medical misadventure are stories of unfortunate experiences with pain medications. Sometimes the story includes a description of how prescribed pain medication created symptoms that were far worse than the pain it was intended to treat. Vomiting, constipation, and “feeling goofy” are high on the list of complaints.
These caches of unused opioids, many of which were never needed in the first place, are evidence of why our health care has become so expensive, and also represent the seeds from which the addiction epidemic has grown. Ironically, they also are collateral damage from an unsuccessful and sometimes misguided war on pain.
It isn’t clear exactly when or where the war on pain began, but I’m sure those who fired the first shots were understandably concerned that many patients with incurable and terminal conditions were suffering needlessly because their pain was being under-treated. Coincidently came the realization that the sooner we could get postoperative patients on their feet and taking deep breaths, the fewer complications we would see. And the more adequately we treated their pain, the sooner we could get those patients moving and breathing optimally.
In a good faith effort to be more “scientific” about pain management, patients were asked to rate their pain and smiley face charts appeared. Unfortunately, somewhere along the line came the mantra that not only should no patient’s pain go unmeasured, but no patient’s pain should go unmedicated.
The federal government entered the war when the Centers for Medicare & Medicaid Services issued the directive that hospitals ask patients who were being discharged if their pain had been well controlled and how often did the hospital staff do what they could to ease their pain? The answers to these questions, along with others, was collected and used in assessing a hospital’s quality of care and determining its level of reimbursement.
So far, there is insufficient data to determine how frequently this directive on pain management induced hospitals to over-prescribe medication, but it certainly hasn’t been associated with a decline in opioid abuse. It is reasonable to suspect that this salvo by the government has resulted in some collateral damage as it encouraged a steady flow of unused and unnecessary prescription narcotics out of the hospital and on to the streets.
The good news is that there has been enough concern voiced about the unintended effect of these pain management questions that the CMS has decided to eliminate financial pressure clinicians might feel to over-prescribe medications by withdrawing the questions from the patient discharge questionnaire.
The bad news is that we continue to fight the war on pain with a limited arsenal. As long as clinicians simply believe that no pain should go unmedicated, they will continue to miss opportunities to use other modalities such as counseling, physical therapy, and education that can be effective without the risk of collateral damage. Instead of asking the patient (who may not know the answer), we should be asking ourselves if we have been doing everything we could to help the patient deal with his pain. The answer is often not written on prescription pads.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
When your peer group is dominated by folks in their early 70s, conversations at dinner parties and lobster bakes invariably morph into storytelling competitions between the survivors of recent hospitalizations and medical procedures. I try to redirect this tedious and repetitive chatter with a topic from my standard collection of conversation re-starters that includes “How about those Red Sox?” and “How’s your granddaughter’s soccer season going?” But sadly I am not always successful.
Often embedded in these tales of medical misadventure are stories of unfortunate experiences with pain medications. Sometimes the story includes a description of how prescribed pain medication created symptoms that were far worse than the pain it was intended to treat. Vomiting, constipation, and “feeling goofy” are high on the list of complaints.
These caches of unused opioids, many of which were never needed in the first place, are evidence of why our health care has become so expensive, and also represent the seeds from which the addiction epidemic has grown. Ironically, they also are collateral damage from an unsuccessful and sometimes misguided war on pain.
It isn’t clear exactly when or where the war on pain began, but I’m sure those who fired the first shots were understandably concerned that many patients with incurable and terminal conditions were suffering needlessly because their pain was being under-treated. Coincidently came the realization that the sooner we could get postoperative patients on their feet and taking deep breaths, the fewer complications we would see. And the more adequately we treated their pain, the sooner we could get those patients moving and breathing optimally.
In a good faith effort to be more “scientific” about pain management, patients were asked to rate their pain and smiley face charts appeared. Unfortunately, somewhere along the line came the mantra that not only should no patient’s pain go unmeasured, but no patient’s pain should go unmedicated.
The federal government entered the war when the Centers for Medicare & Medicaid Services issued the directive that hospitals ask patients who were being discharged if their pain had been well controlled and how often did the hospital staff do what they could to ease their pain? The answers to these questions, along with others, was collected and used in assessing a hospital’s quality of care and determining its level of reimbursement.
So far, there is insufficient data to determine how frequently this directive on pain management induced hospitals to over-prescribe medication, but it certainly hasn’t been associated with a decline in opioid abuse. It is reasonable to suspect that this salvo by the government has resulted in some collateral damage as it encouraged a steady flow of unused and unnecessary prescription narcotics out of the hospital and on to the streets.
The good news is that there has been enough concern voiced about the unintended effect of these pain management questions that the CMS has decided to eliminate financial pressure clinicians might feel to over-prescribe medications by withdrawing the questions from the patient discharge questionnaire.
The bad news is that we continue to fight the war on pain with a limited arsenal. As long as clinicians simply believe that no pain should go unmedicated, they will continue to miss opportunities to use other modalities such as counseling, physical therapy, and education that can be effective without the risk of collateral damage. Instead of asking the patient (who may not know the answer), we should be asking ourselves if we have been doing everything we could to help the patient deal with his pain. The answer is often not written on prescription pads.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
EMTALA – statutory law
This is the first of a two-part series.
Question: Which of the following statements regarding the Emergency Medical Treatment & Labor Act (EMTALA) is correct?
A. Deals with the standard of care in emergency medicine.
B. Provides safeguards for uninsured and nonpaying patients with an emergency medical condition.
C. Mandates uniform screening and treatment stabilization prior to transfer, irrespective of the hospital’s capability.
D. Is mostly directed at hospitals and emergency department staff doctors, but excludes on-call physicians.
E. Violations can result in fines, loss of Medicare provider participation, or even imprisonment.
Answer: B. In 1985, the CBS investigative news show “60 Minutes” ran an exposé on abuses in the emergency departments of U.S. hospitals, featuring the case of Eugene Barnes, a 32-year-old man brought to the Brookside Hospital emergency department (ED) in San Pablo, Calif., with a penetrating stab wound.

In another case, William Jenness, injured in an auto accident, died after a delayed transfer to a county hospital, because the original hospital required a $1,000 deposit in advance before initiating treatment.
In response to the widespread perception that uninsured patients were being denied treatment in the nation’s emergency departments, Congress enacted the Emergency Medical Treatment & Labor Act.1
Originally referred to as the “antidumping law,” EMTALA was designed to prevent hospitals from transferring financially undesirable patients to public hospitals without providing a medical screening examination and stabilizing treatment prior to transfer.
The purpose and intent of the law is to ensure that all patients who come to the ED have access to emergency services, although the statute itself is silent on payment ability.
EMTALA is not meant to replace or override state tort law, and does not deal with quality of care issues that may arise in the emergency department. Over the 30-year period since its enactment, EMTALA has received mixed reviews, with one scholar complaining that the statute is sloppily drafted and the premise of the statute, silly at best.2
EMTALA defines an emergency medical condition as:
1. A medical condition manifesting itself by acute symptoms of sufficient severity (including severe pain, psychiatric disturbances, and/or symptoms of substance abuse) such that the absence of immediate medical attention could reasonably be expected to result in placing the health of the individual (or, with respect to a pregnant woman, the health of the woman or her unborn child) in serious jeopardy, cause serious impairment to bodily functions, or result in serious dysfunction of any bodily organ or part.
2. With respect to a pregnant woman who is having contractions, there is inadequate time to effect a safe transfer to another hospital before delivery, or that transfer may pose a threat to the health or safety of the woman or the unborn child.3
Whether an emergency medical condition exists is determined by a medical screening exam (MSE). EMTALA is about a process directed at the well-being and safety of all patients with a medical emergency who come to the ED, defined as being licensed by the state or held out to the public as a place that provides care for emergency medical conditions. Hospital-based outpatient clinics that handle less than one-third of emergency visits and physician offices are exempt.
All patients who present to the ED seeking treatment are entitled to an MSE, and EDs are required to post such notification on their premises. A triage nurse may not be qualified to conduct the MSE unless he or she possesses special competencies, and has approval from the medical staff and the hospital’s governing body.
It is important that the MSE be documented soon after the patient’s arrival to determine if the medical condition warrants immediate treatment. It is definitely not acceptable to delay performing an MSE while awaiting information on insurance coverage, and one cannot “hold” the patient and delay stabilizing treatment because of the carrier’s insistence on using only certain approved facilities.
EMTALA requires that the screening exam be “appropriate,” but the statute does not define the term except to note that it is to be “within the capability of the hospital’s emergency department.” However, it is generally recognized that triage alone is insufficient, and the screening exam should be based on the patient’s symptoms and performed by a qualified person.
The important point is that it is uniformly applied, without discrimination, to all who seek treatment in the ED. The hospital itself is expected to have in place policies addressing the broad aspects of the screening process in a nondisparate manner.
The second key issue under the EMTALA statute concerns treatment and transfer.4 If an emergency medical condition exists, treatment must be provided until the emergency is resolved or stabilized.
Under the law, a patient is considered stable for transfer (or discharge) if the treating physician determines that no material deterioration is reasonably likely to occur during or as a result of the transfer between facilities. A receiving hospital is obligated to report any individual who has been transferred in an unstable condition in violation of EMTALA.
However, in the event the hospital does not have the capability to stabilize the emergency medical condition, EMTALA mandates an appropriate transfer, under prescribed conditions, to another hospital whose specialized capabilities obligate it to cooperate. The ED physician in the sending hospital will directly request acceptance of such a transfer. If the patient is unstable, the physician must certify that the medical benefits expected from the transfer outweigh the risks, unless the patient insists on a transfer in writing after being informed of the hospital’s obligations under EMTALA and the risks of transfer.
Furthermore, the transferring hospital must: 1. provide ongoing care within its capability until transfer to minimize transfer risks, 2. provide copies of medical records, 3. confirm that the receiving facility has space and qualified personnel to treat the condition and has agreed to accept the transfer, and 4. ensure that the transfer is made with qualified personnel and appropriate medical equipment.
On-call physicians at both transferring and accepting facilities are also subject to EMTALA. The U.S. Department of Health & Human Services’ Office of Inspector General (OIG) has promulgated rules regarding on-call physicians, even touching on reimbursement.
The American College of Emergency Physicians subscribes to the view that hospitals, medical staff, and payers share an ethical responsibility for the provision of emergency care, acknowledging that EDs require a reliable on-call system that provides for the availability of medical staff members for consultation and participation in the evaluation and treatment of emergency patients.5
Penalties for EMTALA violations include fines up to $50,000 per violation, and/or nullification of Medicare provider agreements. There is a 2-year statute of limitations for civil enforcement of any violation,6 carried out by the OIG and the Centers for Medicare & Medicaid Services (CMS).
A receiving facility, having suffered financial loss as a result of another hospital’s violation of EMTALA, can bring suit to recover damages, and patients may bring private lawsuits against hospitals, though not against physicians. EMTALA, being a civil rather than a criminal statute, does not impose any prison terms.
Investigations and citations by the OIG/CMS are common, with about half of all hospitals subjected to EMTALA investigations and a quarter receiving a violation citation over a recent 10-year period.
However, a recently published study covering 2002-2015 found that, despite 40% of investigations ending up with EMTALA violations, only 3% of investigations triggered fines – and none resulted in suspension of Medicare provider participation.7
There were a total of 192 settlements, or an average of 14 per year for the 4,000 hospitals in the United States. Most were for failing to provide screening (75%) and stabilization (42%). The vast majority of violations affected hospitals, and only eight physicians were involved.
Fines against hospitals and physicians totaled $6,357,000 (averages, $33,435 and $25,625, respectively). Patient dumping attributable to insurance or financial discrimination accounted for 15.6% of settlements.
References
1. 42 USC §1395dd et seq.
2. Chest. 2015 Jun;147(6):1691-6.
3. 42 USC §1395dd(a).
4. 42 USC §1395dd(b)(c).
5. “EMTALA and On-call Responsibility for Emergency Department Patients,” American College of Emergency Physicians.
6. 42 USC §1395dd(d).
7. West J Emerg Med. 2016 May;17(3):245-51.
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
This is the first of a two-part series.
Question: Which of the following statements regarding the Emergency Medical Treatment & Labor Act (EMTALA) is correct?
A. Deals with the standard of care in emergency medicine.
B. Provides safeguards for uninsured and nonpaying patients with an emergency medical condition.
C. Mandates uniform screening and treatment stabilization prior to transfer, irrespective of the hospital’s capability.
D. Is mostly directed at hospitals and emergency department staff doctors, but excludes on-call physicians.
E. Violations can result in fines, loss of Medicare provider participation, or even imprisonment.
Answer: B. In 1985, the CBS investigative news show “60 Minutes” ran an exposé on abuses in the emergency departments of U.S. hospitals, featuring the case of Eugene Barnes, a 32-year-old man brought to the Brookside Hospital emergency department (ED) in San Pablo, Calif., with a penetrating stab wound.
In another case, William Jenness, injured in an auto accident, died after a delayed transfer to a county hospital, because the original hospital required a $1,000 deposit in advance before initiating treatment.
In response to the widespread perception that uninsured patients were being denied treatment in the nation’s emergency departments, Congress enacted the Emergency Medical Treatment & Labor Act.1
Originally referred to as the “antidumping law,” EMTALA was designed to prevent hospitals from transferring financially undesirable patients to public hospitals without providing a medical screening examination and stabilizing treatment prior to transfer.
The purpose and intent of the law is to ensure that all patients who come to the ED have access to emergency services, although the statute itself is silent on payment ability.
EMTALA is not meant to replace or override state tort law, and does not deal with quality of care issues that may arise in the emergency department. Over the 30-year period since its enactment, EMTALA has received mixed reviews, with one scholar complaining that the statute is sloppily drafted and the premise of the statute, silly at best.2
EMTALA defines an emergency medical condition as:
1. A medical condition manifesting itself by acute symptoms of sufficient severity (including severe pain, psychiatric disturbances, and/or symptoms of substance abuse) such that the absence of immediate medical attention could reasonably be expected to result in placing the health of the individual (or, with respect to a pregnant woman, the health of the woman or her unborn child) in serious jeopardy, cause serious impairment to bodily functions, or result in serious dysfunction of any bodily organ or part.
2. With respect to a pregnant woman who is having contractions, there is inadequate time to effect a safe transfer to another hospital before delivery, or that transfer may pose a threat to the health or safety of the woman or the unborn child.3
Whether an emergency medical condition exists is determined by a medical screening exam (MSE). EMTALA is about a process directed at the well-being and safety of all patients with a medical emergency who come to the ED, defined as being licensed by the state or held out to the public as a place that provides care for emergency medical conditions. Hospital-based outpatient clinics that handle less than one-third of emergency visits and physician offices are exempt.
All patients who present to the ED seeking treatment are entitled to an MSE, and EDs are required to post such notification on their premises. A triage nurse may not be qualified to conduct the MSE unless he or she possesses special competencies, and has approval from the medical staff and the hospital’s governing body.
It is important that the MSE be documented soon after the patient’s arrival to determine if the medical condition warrants immediate treatment. It is definitely not acceptable to delay performing an MSE while awaiting information on insurance coverage, and one cannot “hold” the patient and delay stabilizing treatment because of the carrier’s insistence on using only certain approved facilities.
EMTALA requires that the screening exam be “appropriate,” but the statute does not define the term except to note that it is to be “within the capability of the hospital’s emergency department.” However, it is generally recognized that triage alone is insufficient, and the screening exam should be based on the patient’s symptoms and performed by a qualified person.
The important point is that it is uniformly applied, without discrimination, to all who seek treatment in the ED. The hospital itself is expected to have in place policies addressing the broad aspects of the screening process in a nondisparate manner.
The second key issue under the EMTALA statute concerns treatment and transfer.4 If an emergency medical condition exists, treatment must be provided until the emergency is resolved or stabilized.
Under the law, a patient is considered stable for transfer (or discharge) if the treating physician determines that no material deterioration is reasonably likely to occur during or as a result of the transfer between facilities. A receiving hospital is obligated to report any individual who has been transferred in an unstable condition in violation of EMTALA.
However, in the event the hospital does not have the capability to stabilize the emergency medical condition, EMTALA mandates an appropriate transfer, under prescribed conditions, to another hospital whose specialized capabilities obligate it to cooperate. The ED physician in the sending hospital will directly request acceptance of such a transfer. If the patient is unstable, the physician must certify that the medical benefits expected from the transfer outweigh the risks, unless the patient insists on a transfer in writing after being informed of the hospital’s obligations under EMTALA and the risks of transfer.
Furthermore, the transferring hospital must: 1. provide ongoing care within its capability until transfer to minimize transfer risks, 2. provide copies of medical records, 3. confirm that the receiving facility has space and qualified personnel to treat the condition and has agreed to accept the transfer, and 4. ensure that the transfer is made with qualified personnel and appropriate medical equipment.
On-call physicians at both transferring and accepting facilities are also subject to EMTALA. The U.S. Department of Health & Human Services’ Office of Inspector General (OIG) has promulgated rules regarding on-call physicians, even touching on reimbursement.
The American College of Emergency Physicians subscribes to the view that hospitals, medical staff, and payers share an ethical responsibility for the provision of emergency care, acknowledging that EDs require a reliable on-call system that provides for the availability of medical staff members for consultation and participation in the evaluation and treatment of emergency patients.5
Penalties for EMTALA violations include fines up to $50,000 per violation, and/or nullification of Medicare provider agreements. There is a 2-year statute of limitations for civil enforcement of any violation,6 carried out by the OIG and the Centers for Medicare & Medicaid Services (CMS).
A receiving facility, having suffered financial loss as a result of another hospital’s violation of EMTALA, can bring suit to recover damages, and patients may bring private lawsuits against hospitals, though not against physicians. EMTALA, being a civil rather than a criminal statute, does not impose any prison terms.
Investigations and citations by the OIG/CMS are common, with about half of all hospitals subjected to EMTALA investigations and a quarter receiving a violation citation over a recent 10-year period.
However, a recently published study covering 2002-2015 found that, despite 40% of investigations ending up with EMTALA violations, only 3% of investigations triggered fines – and none resulted in suspension of Medicare provider participation.7
There were a total of 192 settlements, or an average of 14 per year for the 4,000 hospitals in the United States. Most were for failing to provide screening (75%) and stabilization (42%). The vast majority of violations affected hospitals, and only eight physicians were involved.
Fines against hospitals and physicians totaled $6,357,000 (averages, $33,435 and $25,625, respectively). Patient dumping attributable to insurance or financial discrimination accounted for 15.6% of settlements.
References
1. 42 USC §1395dd et seq.
2. Chest. 2015 Jun;147(6):1691-6.
3. 42 USC §1395dd(a).
4. 42 USC §1395dd(b)(c).
5. “EMTALA and On-call Responsibility for Emergency Department Patients,” American College of Emergency Physicians.
6. 42 USC §1395dd(d).
7. West J Emerg Med. 2016 May;17(3):245-51.
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
This is the first of a two-part series.
Question: Which of the following statements regarding the Emergency Medical Treatment & Labor Act (EMTALA) is correct?
A. Deals with the standard of care in emergency medicine.
B. Provides safeguards for uninsured and nonpaying patients with an emergency medical condition.
C. Mandates uniform screening and treatment stabilization prior to transfer, irrespective of the hospital’s capability.
D. Is mostly directed at hospitals and emergency department staff doctors, but excludes on-call physicians.
E. Violations can result in fines, loss of Medicare provider participation, or even imprisonment.
Answer: B. In 1985, the CBS investigative news show “60 Minutes” ran an exposé on abuses in the emergency departments of U.S. hospitals, featuring the case of Eugene Barnes, a 32-year-old man brought to the Brookside Hospital emergency department (ED) in San Pablo, Calif., with a penetrating stab wound.
In another case, William Jenness, injured in an auto accident, died after a delayed transfer to a county hospital, because the original hospital required a $1,000 deposit in advance before initiating treatment.
In response to the widespread perception that uninsured patients were being denied treatment in the nation’s emergency departments, Congress enacted the Emergency Medical Treatment & Labor Act.1
Originally referred to as the “antidumping law,” EMTALA was designed to prevent hospitals from transferring financially undesirable patients to public hospitals without providing a medical screening examination and stabilizing treatment prior to transfer.
The purpose and intent of the law is to ensure that all patients who come to the ED have access to emergency services, although the statute itself is silent on payment ability.
EMTALA is not meant to replace or override state tort law, and does not deal with quality of care issues that may arise in the emergency department. Over the 30-year period since its enactment, EMTALA has received mixed reviews, with one scholar complaining that the statute is sloppily drafted and the premise of the statute, silly at best.2
EMTALA defines an emergency medical condition as:
1. A medical condition manifesting itself by acute symptoms of sufficient severity (including severe pain, psychiatric disturbances, and/or symptoms of substance abuse) such that the absence of immediate medical attention could reasonably be expected to result in placing the health of the individual (or, with respect to a pregnant woman, the health of the woman or her unborn child) in serious jeopardy, cause serious impairment to bodily functions, or result in serious dysfunction of any bodily organ or part.
2. With respect to a pregnant woman who is having contractions, there is inadequate time to effect a safe transfer to another hospital before delivery, or that transfer may pose a threat to the health or safety of the woman or the unborn child.3
Whether an emergency medical condition exists is determined by a medical screening exam (MSE). EMTALA is about a process directed at the well-being and safety of all patients with a medical emergency who come to the ED, defined as being licensed by the state or held out to the public as a place that provides care for emergency medical conditions. Hospital-based outpatient clinics that handle less than one-third of emergency visits and physician offices are exempt.
All patients who present to the ED seeking treatment are entitled to an MSE, and EDs are required to post such notification on their premises. A triage nurse may not be qualified to conduct the MSE unless he or she possesses special competencies, and has approval from the medical staff and the hospital’s governing body.
It is important that the MSE be documented soon after the patient’s arrival to determine if the medical condition warrants immediate treatment. It is definitely not acceptable to delay performing an MSE while awaiting information on insurance coverage, and one cannot “hold” the patient and delay stabilizing treatment because of the carrier’s insistence on using only certain approved facilities.
EMTALA requires that the screening exam be “appropriate,” but the statute does not define the term except to note that it is to be “within the capability of the hospital’s emergency department.” However, it is generally recognized that triage alone is insufficient, and the screening exam should be based on the patient’s symptoms and performed by a qualified person.
The important point is that it is uniformly applied, without discrimination, to all who seek treatment in the ED. The hospital itself is expected to have in place policies addressing the broad aspects of the screening process in a nondisparate manner.
The second key issue under the EMTALA statute concerns treatment and transfer.4 If an emergency medical condition exists, treatment must be provided until the emergency is resolved or stabilized.
Under the law, a patient is considered stable for transfer (or discharge) if the treating physician determines that no material deterioration is reasonably likely to occur during or as a result of the transfer between facilities. A receiving hospital is obligated to report any individual who has been transferred in an unstable condition in violation of EMTALA.
However, in the event the hospital does not have the capability to stabilize the emergency medical condition, EMTALA mandates an appropriate transfer, under prescribed conditions, to another hospital whose specialized capabilities obligate it to cooperate. The ED physician in the sending hospital will directly request acceptance of such a transfer. If the patient is unstable, the physician must certify that the medical benefits expected from the transfer outweigh the risks, unless the patient insists on a transfer in writing after being informed of the hospital’s obligations under EMTALA and the risks of transfer.
Furthermore, the transferring hospital must: 1. provide ongoing care within its capability until transfer to minimize transfer risks, 2. provide copies of medical records, 3. confirm that the receiving facility has space and qualified personnel to treat the condition and has agreed to accept the transfer, and 4. ensure that the transfer is made with qualified personnel and appropriate medical equipment.
On-call physicians at both transferring and accepting facilities are also subject to EMTALA. The U.S. Department of Health & Human Services’ Office of Inspector General (OIG) has promulgated rules regarding on-call physicians, even touching on reimbursement.
The American College of Emergency Physicians subscribes to the view that hospitals, medical staff, and payers share an ethical responsibility for the provision of emergency care, acknowledging that EDs require a reliable on-call system that provides for the availability of medical staff members for consultation and participation in the evaluation and treatment of emergency patients.5
Penalties for EMTALA violations include fines up to $50,000 per violation, and/or nullification of Medicare provider agreements. There is a 2-year statute of limitations for civil enforcement of any violation,6 carried out by the OIG and the Centers for Medicare & Medicaid Services (CMS).
A receiving facility, having suffered financial loss as a result of another hospital’s violation of EMTALA, can bring suit to recover damages, and patients may bring private lawsuits against hospitals, though not against physicians. EMTALA, being a civil rather than a criminal statute, does not impose any prison terms.
Investigations and citations by the OIG/CMS are common, with about half of all hospitals subjected to EMTALA investigations and a quarter receiving a violation citation over a recent 10-year period.
However, a recently published study covering 2002-2015 found that, despite 40% of investigations ending up with EMTALA violations, only 3% of investigations triggered fines – and none resulted in suspension of Medicare provider participation.7
There were a total of 192 settlements, or an average of 14 per year for the 4,000 hospitals in the United States. Most were for failing to provide screening (75%) and stabilization (42%). The vast majority of violations affected hospitals, and only eight physicians were involved.
Fines against hospitals and physicians totaled $6,357,000 (averages, $33,435 and $25,625, respectively). Patient dumping attributable to insurance or financial discrimination accounted for 15.6% of settlements.
References
1. 42 USC §1395dd et seq.
2. Chest. 2015 Jun;147(6):1691-6.
3. 42 USC §1395dd(a).
4. 42 USC §1395dd(b)(c).
5. “EMTALA and On-call Responsibility for Emergency Department Patients,” American College of Emergency Physicians.
6. 42 USC §1395dd(d).
7. West J Emerg Med. 2016 May;17(3):245-51.
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
Tips for avoiding nerve injuries in gynecologic surgery
Upper- and lower-extremity injuries can occur during gynecologic surgery. The incidence of lower-extremity injury is 1.1%-1.9% and upper-extremity injuries can occur in 0.16% of cases.1-5 Fortunately, most of the injuries are transient, sensory injuries that resolve spontaneously. However, a small percentage of injuries result in long-term sequelae.
The pathophysiology of the nerve injuries can be mechanistically separated into three categories: neuropraxia, axonotmesis, and neurotmesis. Neuropraxia results from nerve demyelination at the site of injury because of compression and typically resolves within weeks to months as the nerve is remyelinated. Axonotmesis results from severe compression with axon damage. This may take up to a year to resolve as axonal regeneration proceeds at the rate of 1 mm per day. This can be separated into second and third degree and refers to the severity of damage and the resultant persistent deficit. Neurotmesis results from complete transection and is associated with a poor prognosis without reparative surgery.

Brachial plexus
Stretch injury is the most common reason for a brachial plexus injury. This can occur if the arm board is extended to greater than 90 degrees from the patient’s torso or if the patient’s arm falls off of the arm board. Careful positioning and securing the patient’s arm on the arm board before draping can avoid this injury. A brachial plexus injury can also occur if shoulder braces are placed too laterally during minimally invasive surgery. Radial nerve injuries can occur if there is too much pressure on the humerus during positioning. Ulnar injuries arise from pressure placed on the medial aspect of the elbow.
Tip #1: When tucking a patient’s arm for minimally invasive surgery, appropriate padding should be placed around the elbow and wrist, and the arm should be in the “thumbs-up” position.
Tip #2: Shoulder blocks should be placed over the acromioclavicular (AC) joint.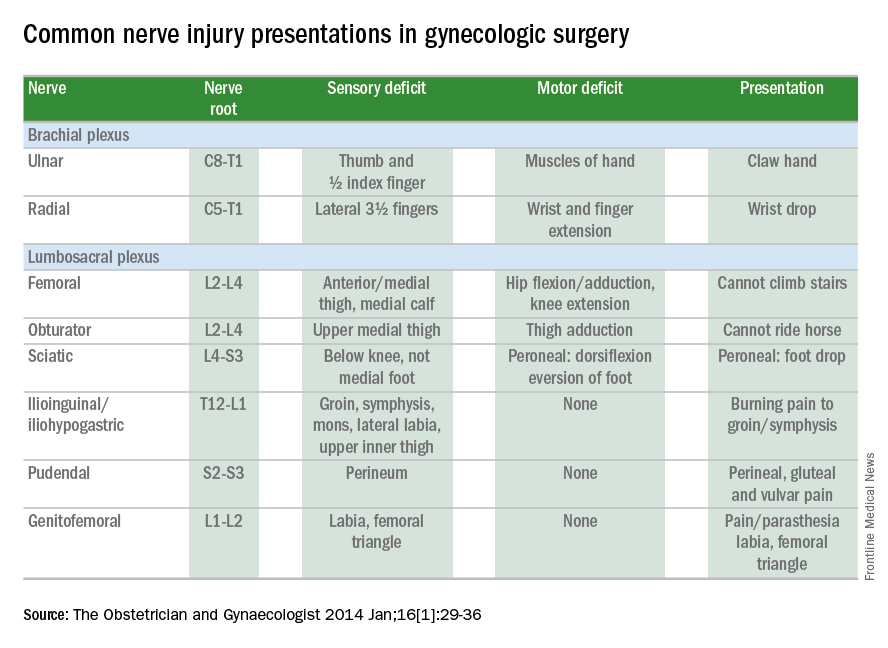
Lumbosacral plexus
The femoral nerve is the nerve most commonly injured during gynecologic surgery and this usually occurs because of compression of the nerve from the lateral blades of self-retaining retractors. One study showed an 8% incidence of injury from self-retaining retractors, compared with less than 1% when the retractors were not used.6 The femoral nerve can also be stretched when patients are placed in the lithotomy position and the hip is hyperflexed.
As with brachial injury prevention, patients should be positioned prior to draping and care must be taken to not hyperflex or externally rotate the hip during minimally invasive surgical procedures. With the introduction of robot-assisted surgery, care must be taken when docking the robot and surgeons must resist excessive movement of the stirrups.
Tip #3: During laparotomy, surgeons should use the shortest blades that allow for adequate visualization and check the blades during the procedure to ensure that excessive pressure is not placed on the psoas muscle. Consider intermittently releasing the pressure on the lateral blades during other portions of the procedure.
Tip #4: Make sure the stirrups are at the same height and that the leg is in line with the patient’s contralateral shoulder.
Obturator nerve injuries can occur during retroperitoneal dissection for pelvic lymphadenectomy (obturator nodes) and can be either a transection or a cautery injury. It can also be injured during urogynecologic procedures including paravaginal defect repairs and during the placement of transobturator tapes.
The sciatic nerve and its branch, the common peroneal, are generally injured because of excessive stretch or pressure. Both nerves can be injured from hyperflexion of the thigh and the common peroneal can suffer a pressure injury as it courses around the lateral head of the fibula. Therefore, care during lithotomy positioning with both candy cane and Allen stirrups is critical during vaginal surgery.
Tip #5: Ensure that the lateral fibula is not touching the stirrup or that padding is placed between the fibular head and the stirrup.
The ilioinguinal and iliohypogastric nerves are typically injured via suture entrapment from low transverse skin incisions, though laparoscopic injury has also been reported. The incidence after a Pfannenstiel incision is about 3.7%.7
Tip #6: Avoid extending the low transverse incision beyond the lateral margin of the rectus muscle, and do not extend the fascial closure suture more than 1.5 cm from the lateral edge of the fascial incision to avoid catching the nerve with the suture.
The pudendal nerve is most commonly injured during vaginal procedures such as sacrospinous fixation. Pain is typically worse when seated.
The genitofemoral nerve is typically injured during retroperitoneal lymph node dissection, particularly the external iliac nodes. The nerve is small and runs lateral to the external iliac artery. It can suffer cautery and transection injuries. Usually, the paresthesias over the mons pubis, labia majora, and medial inner thigh are temporary.
Tip #7: Care should be taken to identify and spare the nerve during retroperitoneal dissection or external iliac node removal.
Nerve injuries during gynecologic surgery are common and are a significant cause of potential morbidity. While occasionally unavoidable and inherent to the surgical procedure, many times the injury could be prevented with proper attention and care to patient positioning and retractor use. Gynecologists should be aware of the risks and have a through understanding of the anatomy. However, should an injury occur, the patient can be reassured that most are self-limited and full recovery is generally expected. In a prospective study, the median time to resolution of symptoms was 31.5 days (range, 1 day to 6 months).5
References
1. The Obstetrician & Gynaecologist 2014;16:29-36.
2. Gynecol Oncol. 1988 Nov;31(3):462-6.
3. Fertil Steril. 1993 Oct;60(4):729-32.
4. J Minim Invasive Gynecol. 2007 Sep-Oct;14(5):664-72.
5. Am J Obstet Gynecol. 2009 Nov;201(5):531.e1-7.
6. Eur J Obstet Gynecol Reprod Biol. 1985 Dec;20(6):385-92.
7. Obstet Gynecol. 2008 Apr;111(4):839-46.
Dr. Gehrig is professor and director of gynecologic oncology at the University of North Carolina at Chapel Hill. She reported having no financial disclosures relevant to this column. Email her at [email protected].
Upper- and lower-extremity injuries can occur during gynecologic surgery. The incidence of lower-extremity injury is 1.1%-1.9% and upper-extremity injuries can occur in 0.16% of cases.1-5 Fortunately, most of the injuries are transient, sensory injuries that resolve spontaneously. However, a small percentage of injuries result in long-term sequelae.
The pathophysiology of the nerve injuries can be mechanistically separated into three categories: neuropraxia, axonotmesis, and neurotmesis. Neuropraxia results from nerve demyelination at the site of injury because of compression and typically resolves within weeks to months as the nerve is remyelinated. Axonotmesis results from severe compression with axon damage. This may take up to a year to resolve as axonal regeneration proceeds at the rate of 1 mm per day. This can be separated into second and third degree and refers to the severity of damage and the resultant persistent deficit. Neurotmesis results from complete transection and is associated with a poor prognosis without reparative surgery.
Brachial plexus
Stretch injury is the most common reason for a brachial plexus injury. This can occur if the arm board is extended to greater than 90 degrees from the patient’s torso or if the patient’s arm falls off of the arm board. Careful positioning and securing the patient’s arm on the arm board before draping can avoid this injury. A brachial plexus injury can also occur if shoulder braces are placed too laterally during minimally invasive surgery. Radial nerve injuries can occur if there is too much pressure on the humerus during positioning. Ulnar injuries arise from pressure placed on the medial aspect of the elbow.
Tip #1: When tucking a patient’s arm for minimally invasive surgery, appropriate padding should be placed around the elbow and wrist, and the arm should be in the “thumbs-up” position.
Tip #2: Shoulder blocks should be placed over the acromioclavicular (AC) joint.
Lumbosacral plexus
The femoral nerve is the nerve most commonly injured during gynecologic surgery and this usually occurs because of compression of the nerve from the lateral blades of self-retaining retractors. One study showed an 8% incidence of injury from self-retaining retractors, compared with less than 1% when the retractors were not used.6 The femoral nerve can also be stretched when patients are placed in the lithotomy position and the hip is hyperflexed.
As with brachial injury prevention, patients should be positioned prior to draping and care must be taken to not hyperflex or externally rotate the hip during minimally invasive surgical procedures. With the introduction of robot-assisted surgery, care must be taken when docking the robot and surgeons must resist excessive movement of the stirrups.
Tip #3: During laparotomy, surgeons should use the shortest blades that allow for adequate visualization and check the blades during the procedure to ensure that excessive pressure is not placed on the psoas muscle. Consider intermittently releasing the pressure on the lateral blades during other portions of the procedure.
Tip #4: Make sure the stirrups are at the same height and that the leg is in line with the patient’s contralateral shoulder.
Obturator nerve injuries can occur during retroperitoneal dissection for pelvic lymphadenectomy (obturator nodes) and can be either a transection or a cautery injury. It can also be injured during urogynecologic procedures including paravaginal defect repairs and during the placement of transobturator tapes.
The sciatic nerve and its branch, the common peroneal, are generally injured because of excessive stretch or pressure. Both nerves can be injured from hyperflexion of the thigh and the common peroneal can suffer a pressure injury as it courses around the lateral head of the fibula. Therefore, care during lithotomy positioning with both candy cane and Allen stirrups is critical during vaginal surgery.
Tip #5: Ensure that the lateral fibula is not touching the stirrup or that padding is placed between the fibular head and the stirrup.
The ilioinguinal and iliohypogastric nerves are typically injured via suture entrapment from low transverse skin incisions, though laparoscopic injury has also been reported. The incidence after a Pfannenstiel incision is about 3.7%.7
Tip #6: Avoid extending the low transverse incision beyond the lateral margin of the rectus muscle, and do not extend the fascial closure suture more than 1.5 cm from the lateral edge of the fascial incision to avoid catching the nerve with the suture.
The pudendal nerve is most commonly injured during vaginal procedures such as sacrospinous fixation. Pain is typically worse when seated.
The genitofemoral nerve is typically injured during retroperitoneal lymph node dissection, particularly the external iliac nodes. The nerve is small and runs lateral to the external iliac artery. It can suffer cautery and transection injuries. Usually, the paresthesias over the mons pubis, labia majora, and medial inner thigh are temporary.
Tip #7: Care should be taken to identify and spare the nerve during retroperitoneal dissection or external iliac node removal.
Nerve injuries during gynecologic surgery are common and are a significant cause of potential morbidity. While occasionally unavoidable and inherent to the surgical procedure, many times the injury could be prevented with proper attention and care to patient positioning and retractor use. Gynecologists should be aware of the risks and have a through understanding of the anatomy. However, should an injury occur, the patient can be reassured that most are self-limited and full recovery is generally expected. In a prospective study, the median time to resolution of symptoms was 31.5 days (range, 1 day to 6 months).5
References
1. The Obstetrician & Gynaecologist 2014;16:29-36.
2. Gynecol Oncol. 1988 Nov;31(3):462-6.
3. Fertil Steril. 1993 Oct;60(4):729-32.
4. J Minim Invasive Gynecol. 2007 Sep-Oct;14(5):664-72.
5. Am J Obstet Gynecol. 2009 Nov;201(5):531.e1-7.
6. Eur J Obstet Gynecol Reprod Biol. 1985 Dec;20(6):385-92.
7. Obstet Gynecol. 2008 Apr;111(4):839-46.
Dr. Gehrig is professor and director of gynecologic oncology at the University of North Carolina at Chapel Hill. She reported having no financial disclosures relevant to this column. Email her at [email protected].
Upper- and lower-extremity injuries can occur during gynecologic surgery. The incidence of lower-extremity injury is 1.1%-1.9% and upper-extremity injuries can occur in 0.16% of cases.1-5 Fortunately, most of the injuries are transient, sensory injuries that resolve spontaneously. However, a small percentage of injuries result in long-term sequelae.
The pathophysiology of the nerve injuries can be mechanistically separated into three categories: neuropraxia, axonotmesis, and neurotmesis. Neuropraxia results from nerve demyelination at the site of injury because of compression and typically resolves within weeks to months as the nerve is remyelinated. Axonotmesis results from severe compression with axon damage. This may take up to a year to resolve as axonal regeneration proceeds at the rate of 1 mm per day. This can be separated into second and third degree and refers to the severity of damage and the resultant persistent deficit. Neurotmesis results from complete transection and is associated with a poor prognosis without reparative surgery.
Brachial plexus
Stretch injury is the most common reason for a brachial plexus injury. This can occur if the arm board is extended to greater than 90 degrees from the patient’s torso or if the patient’s arm falls off of the arm board. Careful positioning and securing the patient’s arm on the arm board before draping can avoid this injury. A brachial plexus injury can also occur if shoulder braces are placed too laterally during minimally invasive surgery. Radial nerve injuries can occur if there is too much pressure on the humerus during positioning. Ulnar injuries arise from pressure placed on the medial aspect of the elbow.
Tip #1: When tucking a patient’s arm for minimally invasive surgery, appropriate padding should be placed around the elbow and wrist, and the arm should be in the “thumbs-up” position.
Tip #2: Shoulder blocks should be placed over the acromioclavicular (AC) joint.
Lumbosacral plexus
The femoral nerve is the nerve most commonly injured during gynecologic surgery and this usually occurs because of compression of the nerve from the lateral blades of self-retaining retractors. One study showed an 8% incidence of injury from self-retaining retractors, compared with less than 1% when the retractors were not used.6 The femoral nerve can also be stretched when patients are placed in the lithotomy position and the hip is hyperflexed.
As with brachial injury prevention, patients should be positioned prior to draping and care must be taken to not hyperflex or externally rotate the hip during minimally invasive surgical procedures. With the introduction of robot-assisted surgery, care must be taken when docking the robot and surgeons must resist excessive movement of the stirrups.
Tip #3: During laparotomy, surgeons should use the shortest blades that allow for adequate visualization and check the blades during the procedure to ensure that excessive pressure is not placed on the psoas muscle. Consider intermittently releasing the pressure on the lateral blades during other portions of the procedure.
Tip #4: Make sure the stirrups are at the same height and that the leg is in line with the patient’s contralateral shoulder.
Obturator nerve injuries can occur during retroperitoneal dissection for pelvic lymphadenectomy (obturator nodes) and can be either a transection or a cautery injury. It can also be injured during urogynecologic procedures including paravaginal defect repairs and during the placement of transobturator tapes.
The sciatic nerve and its branch, the common peroneal, are generally injured because of excessive stretch or pressure. Both nerves can be injured from hyperflexion of the thigh and the common peroneal can suffer a pressure injury as it courses around the lateral head of the fibula. Therefore, care during lithotomy positioning with both candy cane and Allen stirrups is critical during vaginal surgery.
Tip #5: Ensure that the lateral fibula is not touching the stirrup or that padding is placed between the fibular head and the stirrup.
The ilioinguinal and iliohypogastric nerves are typically injured via suture entrapment from low transverse skin incisions, though laparoscopic injury has also been reported. The incidence after a Pfannenstiel incision is about 3.7%.7
Tip #6: Avoid extending the low transverse incision beyond the lateral margin of the rectus muscle, and do not extend the fascial closure suture more than 1.5 cm from the lateral edge of the fascial incision to avoid catching the nerve with the suture.
The pudendal nerve is most commonly injured during vaginal procedures such as sacrospinous fixation. Pain is typically worse when seated.
The genitofemoral nerve is typically injured during retroperitoneal lymph node dissection, particularly the external iliac nodes. The nerve is small and runs lateral to the external iliac artery. It can suffer cautery and transection injuries. Usually, the paresthesias over the mons pubis, labia majora, and medial inner thigh are temporary.
Tip #7: Care should be taken to identify and spare the nerve during retroperitoneal dissection or external iliac node removal.
Nerve injuries during gynecologic surgery are common and are a significant cause of potential morbidity. While occasionally unavoidable and inherent to the surgical procedure, many times the injury could be prevented with proper attention and care to patient positioning and retractor use. Gynecologists should be aware of the risks and have a through understanding of the anatomy. However, should an injury occur, the patient can be reassured that most are self-limited and full recovery is generally expected. In a prospective study, the median time to resolution of symptoms was 31.5 days (range, 1 day to 6 months).5
References
1. The Obstetrician & Gynaecologist 2014;16:29-36.
2. Gynecol Oncol. 1988 Nov;31(3):462-6.
3. Fertil Steril. 1993 Oct;60(4):729-32.
4. J Minim Invasive Gynecol. 2007 Sep-Oct;14(5):664-72.
5. Am J Obstet Gynecol. 2009 Nov;201(5):531.e1-7.
6. Eur J Obstet Gynecol Reprod Biol. 1985 Dec;20(6):385-92.
7. Obstet Gynecol. 2008 Apr;111(4):839-46.
Dr. Gehrig is professor and director of gynecologic oncology at the University of North Carolina at Chapel Hill. She reported having no financial disclosures relevant to this column. Email her at [email protected].
Examining the safety of lipid-lowering drugs in pregnancy
Lipid-lowering medications are some of the most commonly prescribed drugs in the United States. But while much is known about their general safety, the data are limited when it comes to pregnancy and breastfeeding.
Antilipemic agents are a pharmacologic class that contains 18 drugs. The class is divided into eight subclasses: bile acid sequestrants; fibric acid derivatives, HMG-CoA inhibitors; immunoglobulins; monoclonal antibodies; oligonucleotide inhibitors; vitamins; as well as two miscellaneous drugs, ezetimibe (Zetia) and lomitapide (Juxtapid). Another antilipemic – dextrothyroxine – has been removed from the market by the manufacturer.
Bile acid sequestrants
Bile acid sequestrants include cholestyramine (Prevalite, Questran), colesevelam (Welchol), and colestipol (Colestid). These drugs have the potential to cause fetal toxicity. This assessment is based on their mechanism of action. These agents are not absorbed systemically, or absorption is very poor and they bind bile acids into a nonabsorbable complex. This action can reduce intestinal absorption of fat-soluble vitamins A, D, E, and K.

Reports of fetal harm have not been located for the other two agents in this class, but there is only one case report involving five women for colesevelam and no reports for colestipol. Nevertheless, both of these drugs have the potential to cause fetal hemorrhage if they are taken for prolonged periods in pregnancy.
Fibric acid derivatives
The fibric acid derivatives subclass includes fenofibrate (Tricor, Lofibra) and gemfibrozil (Lopid).
Six reports, involving 13 pregnancies, have described the use of gemfibrozil during all phases of pregnancy. No teratogenic effects were observed in these cases. In one woman, similar concentrations of gemfibrozil and its active metabolite were found in the umbilical vein and artery at levels within the normal reference for adults.
Statins
There are seven HMG-CoA inhibitors, known as statins: atorvastatin (Lipitor), fluvastatin (Lescol), lovastatin (Mevacor), pitavastatin (Livalo), pravastatin (Pravachol), rosuvastatin (Crestor), and simvastatin (Zocor).
The interruption of cholesterol-lowering therapy during pregnancy should have no effect on the long-term treatment of hyperlipidemia. Moreover, cholesterol and products synthesized by cholesterol are important during fetal development as shown by the rise in maternal cholesterol levels during pregnancy. Although the potential for embryo-fetal harm has not been clearly documented, and that potential may eventually be confirmed as low, the use of these agents in the first trimester are best classified as contraindicated.
One consideration in estimating the embryo-fetal risk of statins is their classification as either lipophilic or hydrophilic. Three of the seven statins are hydrophilic (fluvastatin, pravastatin, and rosuvastatin); the remaining four agents are lipophilic. In a 2004 review of 70 reports, all adverse birth outcomes were reported following exposure to lipophilic statins (atorvastatin, lovastatin, or simvastatin) and none with the hydrophilic pravastatin. The authors stated that the findings were due to the fact that lipophilic agents equilibrate between maternal and embryonic compartments, whereas pravastatin is minimally present in the embryo.3 If this is indeed the case, and a statin must be used during pregnancy, fluvastatin, pravastatin, or rosuvastatin appears to be best.
Pravastatin also has been used for the prevention and treatment of preeclampsia.5,6 Although the teratogenic potential of these agents has not been fully determined, the risk for birth defects, if any, appears to be low even when exposure occurs during organogenesis.7,8,9 Nevertheless, avoiding these products during the first trimester appears to be best.
Immunoglobulins
The only immunoglobulin in the antilipemic class is evolocumab (Repatha), which has no human pregnancy data. It is an immunoglobulin G2 that is indicated as an adjunct to diet and maximally tolerated statin therapy. It is also indicated as an adjunct to diet and other low-density lipoprotein–lowering therapies in patients with homozygous familial hypercholesterolemia who require additional lowering. No adverse embryo-fetal effects were observed in monkeys. Because statins are contraindicated in the first trimester, the drug, if combined with a statin, can also be classified as contraindicated. However, if the drug is used alone, the embryo-fetal risk appears to be low based on the animal data.
Monoclonal antibodies
The protein alirocumab (Praluent) is a human monoclonal antibody. It is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease. There are no human pregnancy data. The animal data in rats and monkeys suggest low embryo-fetal risk. However, suppression of the humoral immune response to keyhole hemocyanin antigen was observed in infant monkeys at 4-6 months of age. The significance of this in human infants is apparently unknown. Because statins are contraindicated in the first trimester, the drug should not be used with these agents during that period.
Oligonucleotide inhibitors
No reports describing the use of mipomersen (Kynamro), an oligonucleotide inhibitor of apolipoprotein B-100 synthesis, in human pregnancy have been located. The drug is indicated as an adjunct to lipid-lowering medications and diet to reduce low-density lipoprotein cholesterol, apolipoprotein B, total cholesterol, and non–high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. It has a very long (1-2 months) elimination half-life. The drug caused fetal toxicity in rats, but not in mice or rabbits.
Vitamins
Niacin is a water-soluble B complex vitamin that is converted in vivo to niacinamide. Niacin has no known embryo-fetal risk.
Miscellaneous agents
The two agents in the miscellaneous category are ezetimibe and lomitapide. Ezetimibe is indicated, either alone or in combination with a statin, as adjunctive therapy to diet for the reduction of cholesterol and triglycerides. Statins are contraindicated in the first trimester, but ezetimibe alone could be used during that period if treatment of the mother was mandated. The drug caused no problems in rabbits, but in rats, a dose 10 times the human exposure increased the incidence of skeletal abnormalities. In one report, a woman with homozygous familial hypercholesterolemia was treated with direct adsorption of lipoprotein apheresis, ezetimibe, and rosuvastatin. When pregnancy was discovered (gestational age not specified), the two drugs were stopped but biweekly apheresis was continued. At 37 weeks’ gestation, the patient gave birth to a healthy 2,400-g male infant.10
There are no human pregnancy data with lomitapide. It is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including low-density lipoprotein apheresis where available, to reduce LDL cholesterol, total cholesterol, apolipoprotein B, and non–high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. At doses less than 10 times the human dose, the drug caused congenital malformations and embryo-fetal death in rats, rabbits, and ferrets. The manufacturer classifies the drug as contraindicated in pregnancy because of the animal data.
Breastfeeding
Only niacin, pravastatin, and rosuvastatin have data regarding human milk concentrations. Niacin and its active form – niacinamide – are excreted into breast milk.
The average peak milk level in 11 lactating women given pravastatin 20 mg twice daily for 2.5 days was 3.9 mcg/L, whereas the level for the active metabolite was 2.1 mcg/L. Based on these data, a fully breastfed infant would receive daily about 1.4% of the mother’s weight-adjusted dose.11
A 31-year-old woman was treated with rosuvastatin for familial hypercholesterolemia while breastfeeding her infant. The drug was stopped during breastfeeding but was restarted at 33 days post partum. Breast milk concentrations of the drug were 1.2 times serum levels (about 22 ng/mL vs. 18 ng/mL). Unfortunately, no information was provided on the status of the nursing infant.12
Three of the above agents have high molecular weights - alirocumab, evolocumab, and mipomersen - and are probably not excreted into mature breast milk. Moreover, colesevelam is not absorbed, and very small amounts of colestipol are absorbed by mothers. Several antilipemic agents have characteristics (for example, low molecular weight or long elimination half-life) that suggest they will be excreted into breast milk: ezetimibe, fenofibric acid (active metabolite of fenofibrate), gemfibrozil, lomitapide, and all the statins.
Taken in sum, all of the antilipemics, with the exception of niacin, have the potential to cause a deficiency of fat-soluble vitamins (A, D, E, K) in mother’s milk and in the nursing infant. Deficiency is a concern for all of these vitamins, but especially for vitamin K, because it could cause bruising, petechiae, hematomas, and bleeding in the nursing infant. In addition, antilipemics could cause low levels in milk of cholesterol and lipids, which are required by a nursing infant. Consequently, they should not be used by mothers who are breastfeeding an infant.
References
1. Br J Obstet Gynaecol. 1995 Feb;102(2):169-70.
2. J Matern Fetal Neonatal Med. 2015 May;28(8):954-8.
3. Am J Med Genet A. 2004 Dec 15;131(3):287-98.
4. J Clin Invest. 2016 Aug 1;126(8):2933-40.
5. Hypertension. 2015 Sep;66(3):687-97.
6. Am J Obstet Gynecol. 2016 Jun;214(6):720.e1-720.e17.
7. Birth Defects Res A Clin Mol Teratol. 2005 Nov;73(11):888-96.
8. Reprod Toxicol. 2008 Oct;26(2):175-7.
9. Ann Pharmacother. 2012 Oct;46(10):1419-24.
10. Open Cardiovasc Med J. 2015 Dec 29;9:114-7.
11. J Clin Pharmacol. 1988;28:942.
12. Am J Med. 2013 Sep;126(9):e7-e8.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, and Washington State University, Spokane. He is coauthor of “Drugs in Pregnancy and Lactation,” and coeditor of “Diseases, Complications, and Drug Therapy in Obstetrics.” He has no relevant financial disclosures.
Lipid-lowering medications are some of the most commonly prescribed drugs in the United States. But while much is known about their general safety, the data are limited when it comes to pregnancy and breastfeeding.
Antilipemic agents are a pharmacologic class that contains 18 drugs. The class is divided into eight subclasses: bile acid sequestrants; fibric acid derivatives, HMG-CoA inhibitors; immunoglobulins; monoclonal antibodies; oligonucleotide inhibitors; vitamins; as well as two miscellaneous drugs, ezetimibe (Zetia) and lomitapide (Juxtapid). Another antilipemic – dextrothyroxine – has been removed from the market by the manufacturer.
Bile acid sequestrants
Bile acid sequestrants include cholestyramine (Prevalite, Questran), colesevelam (Welchol), and colestipol (Colestid). These drugs have the potential to cause fetal toxicity. This assessment is based on their mechanism of action. These agents are not absorbed systemically, or absorption is very poor and they bind bile acids into a nonabsorbable complex. This action can reduce intestinal absorption of fat-soluble vitamins A, D, E, and K.
Reports of fetal harm have not been located for the other two agents in this class, but there is only one case report involving five women for colesevelam and no reports for colestipol. Nevertheless, both of these drugs have the potential to cause fetal hemorrhage if they are taken for prolonged periods in pregnancy.
Fibric acid derivatives
The fibric acid derivatives subclass includes fenofibrate (Tricor, Lofibra) and gemfibrozil (Lopid).
Six reports, involving 13 pregnancies, have described the use of gemfibrozil during all phases of pregnancy. No teratogenic effects were observed in these cases. In one woman, similar concentrations of gemfibrozil and its active metabolite were found in the umbilical vein and artery at levels within the normal reference for adults.
Statins
There are seven HMG-CoA inhibitors, known as statins: atorvastatin (Lipitor), fluvastatin (Lescol), lovastatin (Mevacor), pitavastatin (Livalo), pravastatin (Pravachol), rosuvastatin (Crestor), and simvastatin (Zocor).
The interruption of cholesterol-lowering therapy during pregnancy should have no effect on the long-term treatment of hyperlipidemia. Moreover, cholesterol and products synthesized by cholesterol are important during fetal development as shown by the rise in maternal cholesterol levels during pregnancy. Although the potential for embryo-fetal harm has not been clearly documented, and that potential may eventually be confirmed as low, the use of these agents in the first trimester are best classified as contraindicated.
One consideration in estimating the embryo-fetal risk of statins is their classification as either lipophilic or hydrophilic. Three of the seven statins are hydrophilic (fluvastatin, pravastatin, and rosuvastatin); the remaining four agents are lipophilic. In a 2004 review of 70 reports, all adverse birth outcomes were reported following exposure to lipophilic statins (atorvastatin, lovastatin, or simvastatin) and none with the hydrophilic pravastatin. The authors stated that the findings were due to the fact that lipophilic agents equilibrate between maternal and embryonic compartments, whereas pravastatin is minimally present in the embryo.3 If this is indeed the case, and a statin must be used during pregnancy, fluvastatin, pravastatin, or rosuvastatin appears to be best.
Pravastatin also has been used for the prevention and treatment of preeclampsia.5,6 Although the teratogenic potential of these agents has not been fully determined, the risk for birth defects, if any, appears to be low even when exposure occurs during organogenesis.7,8,9 Nevertheless, avoiding these products during the first trimester appears to be best.
Immunoglobulins
The only immunoglobulin in the antilipemic class is evolocumab (Repatha), which has no human pregnancy data. It is an immunoglobulin G2 that is indicated as an adjunct to diet and maximally tolerated statin therapy. It is also indicated as an adjunct to diet and other low-density lipoprotein–lowering therapies in patients with homozygous familial hypercholesterolemia who require additional lowering. No adverse embryo-fetal effects were observed in monkeys. Because statins are contraindicated in the first trimester, the drug, if combined with a statin, can also be classified as contraindicated. However, if the drug is used alone, the embryo-fetal risk appears to be low based on the animal data.
Monoclonal antibodies
The protein alirocumab (Praluent) is a human monoclonal antibody. It is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease. There are no human pregnancy data. The animal data in rats and monkeys suggest low embryo-fetal risk. However, suppression of the humoral immune response to keyhole hemocyanin antigen was observed in infant monkeys at 4-6 months of age. The significance of this in human infants is apparently unknown. Because statins are contraindicated in the first trimester, the drug should not be used with these agents during that period.
Oligonucleotide inhibitors
No reports describing the use of mipomersen (Kynamro), an oligonucleotide inhibitor of apolipoprotein B-100 synthesis, in human pregnancy have been located. The drug is indicated as an adjunct to lipid-lowering medications and diet to reduce low-density lipoprotein cholesterol, apolipoprotein B, total cholesterol, and non–high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. It has a very long (1-2 months) elimination half-life. The drug caused fetal toxicity in rats, but not in mice or rabbits.
Vitamins
Niacin is a water-soluble B complex vitamin that is converted in vivo to niacinamide. Niacin has no known embryo-fetal risk.
Miscellaneous agents
The two agents in the miscellaneous category are ezetimibe and lomitapide. Ezetimibe is indicated, either alone or in combination with a statin, as adjunctive therapy to diet for the reduction of cholesterol and triglycerides. Statins are contraindicated in the first trimester, but ezetimibe alone could be used during that period if treatment of the mother was mandated. The drug caused no problems in rabbits, but in rats, a dose 10 times the human exposure increased the incidence of skeletal abnormalities. In one report, a woman with homozygous familial hypercholesterolemia was treated with direct adsorption of lipoprotein apheresis, ezetimibe, and rosuvastatin. When pregnancy was discovered (gestational age not specified), the two drugs were stopped but biweekly apheresis was continued. At 37 weeks’ gestation, the patient gave birth to a healthy 2,400-g male infant.10
There are no human pregnancy data with lomitapide. It is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including low-density lipoprotein apheresis where available, to reduce LDL cholesterol, total cholesterol, apolipoprotein B, and non–high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. At doses less than 10 times the human dose, the drug caused congenital malformations and embryo-fetal death in rats, rabbits, and ferrets. The manufacturer classifies the drug as contraindicated in pregnancy because of the animal data.
Breastfeeding
Only niacin, pravastatin, and rosuvastatin have data regarding human milk concentrations. Niacin and its active form – niacinamide – are excreted into breast milk.
The average peak milk level in 11 lactating women given pravastatin 20 mg twice daily for 2.5 days was 3.9 mcg/L, whereas the level for the active metabolite was 2.1 mcg/L. Based on these data, a fully breastfed infant would receive daily about 1.4% of the mother’s weight-adjusted dose.11
A 31-year-old woman was treated with rosuvastatin for familial hypercholesterolemia while breastfeeding her infant. The drug was stopped during breastfeeding but was restarted at 33 days post partum. Breast milk concentrations of the drug were 1.2 times serum levels (about 22 ng/mL vs. 18 ng/mL). Unfortunately, no information was provided on the status of the nursing infant.12
Three of the above agents have high molecular weights - alirocumab, evolocumab, and mipomersen - and are probably not excreted into mature breast milk. Moreover, colesevelam is not absorbed, and very small amounts of colestipol are absorbed by mothers. Several antilipemic agents have characteristics (for example, low molecular weight or long elimination half-life) that suggest they will be excreted into breast milk: ezetimibe, fenofibric acid (active metabolite of fenofibrate), gemfibrozil, lomitapide, and all the statins.
Taken in sum, all of the antilipemics, with the exception of niacin, have the potential to cause a deficiency of fat-soluble vitamins (A, D, E, K) in mother’s milk and in the nursing infant. Deficiency is a concern for all of these vitamins, but especially for vitamin K, because it could cause bruising, petechiae, hematomas, and bleeding in the nursing infant. In addition, antilipemics could cause low levels in milk of cholesterol and lipids, which are required by a nursing infant. Consequently, they should not be used by mothers who are breastfeeding an infant.
References
1. Br J Obstet Gynaecol. 1995 Feb;102(2):169-70.
2. J Matern Fetal Neonatal Med. 2015 May;28(8):954-8.
3. Am J Med Genet A. 2004 Dec 15;131(3):287-98.
4. J Clin Invest. 2016 Aug 1;126(8):2933-40.
5. Hypertension. 2015 Sep;66(3):687-97.
6. Am J Obstet Gynecol. 2016 Jun;214(6):720.e1-720.e17.
7. Birth Defects Res A Clin Mol Teratol. 2005 Nov;73(11):888-96.
8. Reprod Toxicol. 2008 Oct;26(2):175-7.
9. Ann Pharmacother. 2012 Oct;46(10):1419-24.
10. Open Cardiovasc Med J. 2015 Dec 29;9:114-7.
11. J Clin Pharmacol. 1988;28:942.
12. Am J Med. 2013 Sep;126(9):e7-e8.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, and Washington State University, Spokane. He is coauthor of “Drugs in Pregnancy and Lactation,” and coeditor of “Diseases, Complications, and Drug Therapy in Obstetrics.” He has no relevant financial disclosures.
Lipid-lowering medications are some of the most commonly prescribed drugs in the United States. But while much is known about their general safety, the data are limited when it comes to pregnancy and breastfeeding.
Antilipemic agents are a pharmacologic class that contains 18 drugs. The class is divided into eight subclasses: bile acid sequestrants; fibric acid derivatives, HMG-CoA inhibitors; immunoglobulins; monoclonal antibodies; oligonucleotide inhibitors; vitamins; as well as two miscellaneous drugs, ezetimibe (Zetia) and lomitapide (Juxtapid). Another antilipemic – dextrothyroxine – has been removed from the market by the manufacturer.
Bile acid sequestrants
Bile acid sequestrants include cholestyramine (Prevalite, Questran), colesevelam (Welchol), and colestipol (Colestid). These drugs have the potential to cause fetal toxicity. This assessment is based on their mechanism of action. These agents are not absorbed systemically, or absorption is very poor and they bind bile acids into a nonabsorbable complex. This action can reduce intestinal absorption of fat-soluble vitamins A, D, E, and K.
Reports of fetal harm have not been located for the other two agents in this class, but there is only one case report involving five women for colesevelam and no reports for colestipol. Nevertheless, both of these drugs have the potential to cause fetal hemorrhage if they are taken for prolonged periods in pregnancy.
Fibric acid derivatives
The fibric acid derivatives subclass includes fenofibrate (Tricor, Lofibra) and gemfibrozil (Lopid).
Six reports, involving 13 pregnancies, have described the use of gemfibrozil during all phases of pregnancy. No teratogenic effects were observed in these cases. In one woman, similar concentrations of gemfibrozil and its active metabolite were found in the umbilical vein and artery at levels within the normal reference for adults.
Statins
There are seven HMG-CoA inhibitors, known as statins: atorvastatin (Lipitor), fluvastatin (Lescol), lovastatin (Mevacor), pitavastatin (Livalo), pravastatin (Pravachol), rosuvastatin (Crestor), and simvastatin (Zocor).
The interruption of cholesterol-lowering therapy during pregnancy should have no effect on the long-term treatment of hyperlipidemia. Moreover, cholesterol and products synthesized by cholesterol are important during fetal development as shown by the rise in maternal cholesterol levels during pregnancy. Although the potential for embryo-fetal harm has not been clearly documented, and that potential may eventually be confirmed as low, the use of these agents in the first trimester are best classified as contraindicated.
One consideration in estimating the embryo-fetal risk of statins is their classification as either lipophilic or hydrophilic. Three of the seven statins are hydrophilic (fluvastatin, pravastatin, and rosuvastatin); the remaining four agents are lipophilic. In a 2004 review of 70 reports, all adverse birth outcomes were reported following exposure to lipophilic statins (atorvastatin, lovastatin, or simvastatin) and none with the hydrophilic pravastatin. The authors stated that the findings were due to the fact that lipophilic agents equilibrate between maternal and embryonic compartments, whereas pravastatin is minimally present in the embryo.3 If this is indeed the case, and a statin must be used during pregnancy, fluvastatin, pravastatin, or rosuvastatin appears to be best.
Pravastatin also has been used for the prevention and treatment of preeclampsia.5,6 Although the teratogenic potential of these agents has not been fully determined, the risk for birth defects, if any, appears to be low even when exposure occurs during organogenesis.7,8,9 Nevertheless, avoiding these products during the first trimester appears to be best.
Immunoglobulins
The only immunoglobulin in the antilipemic class is evolocumab (Repatha), which has no human pregnancy data. It is an immunoglobulin G2 that is indicated as an adjunct to diet and maximally tolerated statin therapy. It is also indicated as an adjunct to diet and other low-density lipoprotein–lowering therapies in patients with homozygous familial hypercholesterolemia who require additional lowering. No adverse embryo-fetal effects were observed in monkeys. Because statins are contraindicated in the first trimester, the drug, if combined with a statin, can also be classified as contraindicated. However, if the drug is used alone, the embryo-fetal risk appears to be low based on the animal data.
Monoclonal antibodies
The protein alirocumab (Praluent) is a human monoclonal antibody. It is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease. There are no human pregnancy data. The animal data in rats and monkeys suggest low embryo-fetal risk. However, suppression of the humoral immune response to keyhole hemocyanin antigen was observed in infant monkeys at 4-6 months of age. The significance of this in human infants is apparently unknown. Because statins are contraindicated in the first trimester, the drug should not be used with these agents during that period.
Oligonucleotide inhibitors
No reports describing the use of mipomersen (Kynamro), an oligonucleotide inhibitor of apolipoprotein B-100 synthesis, in human pregnancy have been located. The drug is indicated as an adjunct to lipid-lowering medications and diet to reduce low-density lipoprotein cholesterol, apolipoprotein B, total cholesterol, and non–high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. It has a very long (1-2 months) elimination half-life. The drug caused fetal toxicity in rats, but not in mice or rabbits.
Vitamins
Niacin is a water-soluble B complex vitamin that is converted in vivo to niacinamide. Niacin has no known embryo-fetal risk.
Miscellaneous agents
The two agents in the miscellaneous category are ezetimibe and lomitapide. Ezetimibe is indicated, either alone or in combination with a statin, as adjunctive therapy to diet for the reduction of cholesterol and triglycerides. Statins are contraindicated in the first trimester, but ezetimibe alone could be used during that period if treatment of the mother was mandated. The drug caused no problems in rabbits, but in rats, a dose 10 times the human exposure increased the incidence of skeletal abnormalities. In one report, a woman with homozygous familial hypercholesterolemia was treated with direct adsorption of lipoprotein apheresis, ezetimibe, and rosuvastatin. When pregnancy was discovered (gestational age not specified), the two drugs were stopped but biweekly apheresis was continued. At 37 weeks’ gestation, the patient gave birth to a healthy 2,400-g male infant.10
There are no human pregnancy data with lomitapide. It is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including low-density lipoprotein apheresis where available, to reduce LDL cholesterol, total cholesterol, apolipoprotein B, and non–high-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. At doses less than 10 times the human dose, the drug caused congenital malformations and embryo-fetal death in rats, rabbits, and ferrets. The manufacturer classifies the drug as contraindicated in pregnancy because of the animal data.
Breastfeeding
Only niacin, pravastatin, and rosuvastatin have data regarding human milk concentrations. Niacin and its active form – niacinamide – are excreted into breast milk.
The average peak milk level in 11 lactating women given pravastatin 20 mg twice daily for 2.5 days was 3.9 mcg/L, whereas the level for the active metabolite was 2.1 mcg/L. Based on these data, a fully breastfed infant would receive daily about 1.4% of the mother’s weight-adjusted dose.11
A 31-year-old woman was treated with rosuvastatin for familial hypercholesterolemia while breastfeeding her infant. The drug was stopped during breastfeeding but was restarted at 33 days post partum. Breast milk concentrations of the drug were 1.2 times serum levels (about 22 ng/mL vs. 18 ng/mL). Unfortunately, no information was provided on the status of the nursing infant.12
Three of the above agents have high molecular weights - alirocumab, evolocumab, and mipomersen - and are probably not excreted into mature breast milk. Moreover, colesevelam is not absorbed, and very small amounts of colestipol are absorbed by mothers. Several antilipemic agents have characteristics (for example, low molecular weight or long elimination half-life) that suggest they will be excreted into breast milk: ezetimibe, fenofibric acid (active metabolite of fenofibrate), gemfibrozil, lomitapide, and all the statins.
Taken in sum, all of the antilipemics, with the exception of niacin, have the potential to cause a deficiency of fat-soluble vitamins (A, D, E, K) in mother’s milk and in the nursing infant. Deficiency is a concern for all of these vitamins, but especially for vitamin K, because it could cause bruising, petechiae, hematomas, and bleeding in the nursing infant. In addition, antilipemics could cause low levels in milk of cholesterol and lipids, which are required by a nursing infant. Consequently, they should not be used by mothers who are breastfeeding an infant.
References
1. Br J Obstet Gynaecol. 1995 Feb;102(2):169-70.
2. J Matern Fetal Neonatal Med. 2015 May;28(8):954-8.
3. Am J Med Genet A. 2004 Dec 15;131(3):287-98.
4. J Clin Invest. 2016 Aug 1;126(8):2933-40.
5. Hypertension. 2015 Sep;66(3):687-97.
6. Am J Obstet Gynecol. 2016 Jun;214(6):720.e1-720.e17.
7. Birth Defects Res A Clin Mol Teratol. 2005 Nov;73(11):888-96.
8. Reprod Toxicol. 2008 Oct;26(2):175-7.
9. Ann Pharmacother. 2012 Oct;46(10):1419-24.
10. Open Cardiovasc Med J. 2015 Dec 29;9:114-7.
11. J Clin Pharmacol. 1988;28:942.
12. Am J Med. 2013 Sep;126(9):e7-e8.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, and Washington State University, Spokane. He is coauthor of “Drugs in Pregnancy and Lactation,” and coeditor of “Diseases, Complications, and Drug Therapy in Obstetrics.” He has no relevant financial disclosures.
Alarming gaps in gestational diabetes care
BY E. ALBERT REECE, MD, PhD, MBA
Much attention has been given in the media to the incidence of prediabetes in the general population. The Centers for Disease Control and Prevention estimates that approximately 86 million adults have prediabetes, and that the incidence of this condition is similar across racial and ethnic groups. Indeed, the seriousness of this public health concern prompted the Centers for Medicare & Medicaid Services to expand Medicare coverage for interventions for people with prediabetes, a move that was finalized in November 2016.
Despite a widespread focus on the need to prevent prediabetes from becoming type 2 diabetes, women diagnosed with gestational diabetes mellitus (GDM), which accounts for about 9% of women in the United States, may not be receiving critical advice and care.

The investigators analyzed data collected via the National Health and Nutrition Examination Survey from 2007-2012, and identified 284 women with a history of GDM. Only 67% of these women received diabetes screening, and approximately one-third of women included in the study had undiagnosed prediabetes and diabetes. The authors concluded that prediabetes in women who have had GDM may be underdiagnosed. They argued that women with GDM should be encouraged to have additional health visits and screenings to prevent the development of prediabetes or diabetes. Considering the fact that a number of studies have shown that GDM predisposes a woman to developing type 2 diabetes, the University of Illinois findings are alarming.
As ob.gyns., we have increasingly become a woman’s only health care practitioner. Although individuals may skip annual exams with a primary care physician, during which blood work is typically drawn, many women will see their ob.gyn. for regular check-ups. Therefore, we have a unique role to play in our patients’ lifelong health. This is especially important during pregnancy, when it may be easy to focus only on the mother’s health as it pertains to the health of the baby, rather than her health in pregnancy as it may affect her long-term well-being.
We have invited Robert Ratner, MD, the chief scientific and medical officer at the American Diabetes Association, to discuss the need to carefully follow up with patients who have had GDM and to educate them about their risk for developing type 2 diabetes later in life.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
Why postpartum GDM follow-up is so important
BY ROBERT E. RATNER, MD
Much of the attention paid to diagnosing gestational diabetes has focused on the fetus and on babies being born very large. However, it is important to appreciate that the original definitions of the condition were based entirely on the long-term outcomes of the mother.
John O’Sullivan, MD, and statistician Claire Mahan published diagnostic criteria in 1964 after performing 3-hour oral glucose tolerance tests (OGTTs) in more than 500 unselected women during their pregnancies, and then following these women and babies out as far as 23 years. Retrospectively, Dr. O’Sullivan and Ms. Mahan defined gestational diabetes mellitus (GDM) as glucose values exceeding two standard deviations above the mean on two out of four OGTT values.

They came to their conclusions after tracking the later development of diabetes outside of pregnancy. More than 20 years later, 70% of women with the higher OGTT values had developed type 2 diabetes, compared with approximately 10% of women who did not have higher values during pregnancy. The O’Sullivan criteria were established, essentially, based on their association with the development of diabetes after pregnancy. In addition to being a significant predictor of subsequent diabetes, a history of GDM also conferred a three- to fourfold increase in maternal mortality.
Fifty-some years later, these findings have been affirmed through additional research and are the crux of what drives the current recommendations for postpartum follow-up of women with a history of GDM.
Long-term maternal risks
Postpartum, the current recommendation from both the American Diabetes Association and the American College of Obstetricians and Gynecologists is that women with GDM be tested at 6-12 weeks after delivery to ensure that the diabetes has resolved.
This recommendation for initial postpartum testing carries with it a stipulation that’s different from subsequent postpartum testing. It says that postpartum testing at 6-12 weeks should be performed with either a fasting glucose test or a 2-hour OGTT. Since hemoglobin A1c may still be impacted by the rapid red blood cell turnover in pregnancy or blood loss at delivery, A1c testing lacks sensitivity for identifying diabetes during this window of time.
Initial postpartum testing also serves as a way to identify whether the diabetes during pregnancy was preexisting or purely secondary to the hormonal changes associated with the pregnancy.
If this first postpartum test shows diabetes, the patient most likely had preexisting diabetes, and therapy must be initiated immediately. In the case of a normal result, the patient remains at higher risk for the development of type 2 diabetes essentially for the rest of her life and should be tested at least every 3 years for the occurrence of the disease.
Much of the increased risk for different ethnic groups occurs within 5 years of the index pregnancy. This was shown in a systematic review led by Catherine Kim, MD; the review examined more than two dozen studies with follow-up of up to 28 years postpartum. The cumulative incidence of type 2 diabetes increased markedly in the first 5 years and then appeared to plateau after 10 years (Diabetes Care. 2002 Oct;25[10]:1862-8).
The best data on late-occurring diabetes following GDM comes from the multicenter National Institutes of Health–sponsored Diabetes Prevention Program (DPP) trial, which randomized more than 3,000 individuals with baseline impaired glucose tolerance – or prediabetes – to one of two interventions: metformin therapy or intensive lifestyle intervention, or to placebo.
Within this population, there were more than 1,700 women who had a previous live birth. Of these women, 350 reported a history of GDM at a mean of 12 years since the delivery of their first GDM pregnancy. The DPP gave us the opportunity, therefore, to look at a large group of women about 12 years away from their GDM pregnancy who had abnormal glucose levels but had not reached the level of type 2 diabetes, and compare them with women with similarly impaired glucose tolerance who did not have a history of GDM.
There were interesting similarities and differences. Women with a GDM history were on average 8 years younger than women without a GDM history, but they had comparable BMIs. In addition, within the placebo arm, we could observe the natural history of glucose intolerance in women with and without a history of GDM. Despite both groups entering the study with equivalent degrees of impaired glucose tolerance and similar BMI, women with a history of GDM had a 71% higher risk of developing diabetes during the 3-year intervention period than that of parous women without a history of GDM (J Clin Endocrinol Metab. 2008 Dec;93[12]:4774-9).
Clearly, there was something about the history of GDM that puts these women at greater risk for diabetes than women who had the same impaired glucose tolerance, but no GDM. The study demonstrated that GDM is an exceptionally strong predictor of the development of type 2 diabetes, even for those who manage to escape diabetes for the first 10 years.
Postpartum prevention
The DPP demonstrated, moreover, that intensive lifestyle therapy and metformin not only were both effective, but that they were equally effective, in delaying or preventing diabetes in women with impaired glucose tolerance and a history of GDM. Both reduced the risk by about 50% at 3 years. This was striking because in parous women without GDM, the reductions were 49% and 14%, respectively. Metformin thus appeared to be more effective in women with a history of GDM.
The effects of the interventions persisted over a 10-year follow up of the DPP population. In women with a history of GDM, the intensive lifestyle intervention and metformin reduced progression to diabetes by 35% and 40%, respectively, over 10 years (J Clin Endocrinol Metab. 2015 Apr;100[4]:1646-53).
Pregnancy presents a stress test for beta cell function, and gestational diabetes clearly is a harbinger of further deterioration in beta-cell function and metabolic abnormalities in the mother. Because of these risks and because early intervention makes a difference, surveillance is critically important. Most women see their ob.gyn. as their primary care physician in the 10 years following a pregnancy – the time when more than 50% of all cases of subsequent diabetes will occur – and many continue to see their ob.gyns. in the longer term, as their risk continues to linger.
Immediately after a pregnancy with GDM, ob.gyns. can counsel women not only about their risks of developing type 2 diabetes and the importance of screening, but also about the beneficial impact of lifestyle modification, caloric restriction and weight loss if necessary, and increased exercise. Mothers should also know that GDM is a family affair, and that lifestyle changes that are beneficial for the mother will be equally beneficial for the baby.
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study taught us that there are continuous linear relationships between maternal glucose and adverse fetal outcomes like birth weight and percent body fat greater than the 90th percentile. Longitudinal studies of the Pima Indians showed us that offspring of women who had diabetes during pregnancy were more likely to be obese and more likely to develop diabetes than offspring of women who did not have diabetes during pregnancy. Even when GDM has been well treated and controlled, we should have heightened awareness to the potential risks in the fetus and the growing child and adolescent.
Patients who are found to have subsequent type 2 diabetes should know that aggressive therapy early on in the natural history of the disease reduces the risk of microvascular and macrovascular complications. And as the DPP has demonstrated, lifestyle interventions and metformin may also keep women who are found to have prediabetes outside of pregnancy from progressing on to diabetes.
Dr. Ratner is the chief scientific and medical officer for the American Diabetes Association. He reported having no financial disclosures relevant to this Master Class.
BY E. ALBERT REECE, MD, PhD, MBA
Much attention has been given in the media to the incidence of prediabetes in the general population. The Centers for Disease Control and Prevention estimates that approximately 86 million adults have prediabetes, and that the incidence of this condition is similar across racial and ethnic groups. Indeed, the seriousness of this public health concern prompted the Centers for Medicare & Medicaid Services to expand Medicare coverage for interventions for people with prediabetes, a move that was finalized in November 2016.
Despite a widespread focus on the need to prevent prediabetes from becoming type 2 diabetes, women diagnosed with gestational diabetes mellitus (GDM), which accounts for about 9% of women in the United States, may not be receiving critical advice and care.
The investigators analyzed data collected via the National Health and Nutrition Examination Survey from 2007-2012, and identified 284 women with a history of GDM. Only 67% of these women received diabetes screening, and approximately one-third of women included in the study had undiagnosed prediabetes and diabetes. The authors concluded that prediabetes in women who have had GDM may be underdiagnosed. They argued that women with GDM should be encouraged to have additional health visits and screenings to prevent the development of prediabetes or diabetes. Considering the fact that a number of studies have shown that GDM predisposes a woman to developing type 2 diabetes, the University of Illinois findings are alarming.
As ob.gyns., we have increasingly become a woman’s only health care practitioner. Although individuals may skip annual exams with a primary care physician, during which blood work is typically drawn, many women will see their ob.gyn. for regular check-ups. Therefore, we have a unique role to play in our patients’ lifelong health. This is especially important during pregnancy, when it may be easy to focus only on the mother’s health as it pertains to the health of the baby, rather than her health in pregnancy as it may affect her long-term well-being.
We have invited Robert Ratner, MD, the chief scientific and medical officer at the American Diabetes Association, to discuss the need to carefully follow up with patients who have had GDM and to educate them about their risk for developing type 2 diabetes later in life.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
Why postpartum GDM follow-up is so important
BY ROBERT E. RATNER, MD
Much of the attention paid to diagnosing gestational diabetes has focused on the fetus and on babies being born very large. However, it is important to appreciate that the original definitions of the condition were based entirely on the long-term outcomes of the mother.
John O’Sullivan, MD, and statistician Claire Mahan published diagnostic criteria in 1964 after performing 3-hour oral glucose tolerance tests (OGTTs) in more than 500 unselected women during their pregnancies, and then following these women and babies out as far as 23 years. Retrospectively, Dr. O’Sullivan and Ms. Mahan defined gestational diabetes mellitus (GDM) as glucose values exceeding two standard deviations above the mean on two out of four OGTT values.
They came to their conclusions after tracking the later development of diabetes outside of pregnancy. More than 20 years later, 70% of women with the higher OGTT values had developed type 2 diabetes, compared with approximately 10% of women who did not have higher values during pregnancy. The O’Sullivan criteria were established, essentially, based on their association with the development of diabetes after pregnancy. In addition to being a significant predictor of subsequent diabetes, a history of GDM also conferred a three- to fourfold increase in maternal mortality.
Fifty-some years later, these findings have been affirmed through additional research and are the crux of what drives the current recommendations for postpartum follow-up of women with a history of GDM.
Long-term maternal risks
Postpartum, the current recommendation from both the American Diabetes Association and the American College of Obstetricians and Gynecologists is that women with GDM be tested at 6-12 weeks after delivery to ensure that the diabetes has resolved.
This recommendation for initial postpartum testing carries with it a stipulation that’s different from subsequent postpartum testing. It says that postpartum testing at 6-12 weeks should be performed with either a fasting glucose test or a 2-hour OGTT. Since hemoglobin A1c may still be impacted by the rapid red blood cell turnover in pregnancy or blood loss at delivery, A1c testing lacks sensitivity for identifying diabetes during this window of time.
Initial postpartum testing also serves as a way to identify whether the diabetes during pregnancy was preexisting or purely secondary to the hormonal changes associated with the pregnancy.
If this first postpartum test shows diabetes, the patient most likely had preexisting diabetes, and therapy must be initiated immediately. In the case of a normal result, the patient remains at higher risk for the development of type 2 diabetes essentially for the rest of her life and should be tested at least every 3 years for the occurrence of the disease.
Much of the increased risk for different ethnic groups occurs within 5 years of the index pregnancy. This was shown in a systematic review led by Catherine Kim, MD; the review examined more than two dozen studies with follow-up of up to 28 years postpartum. The cumulative incidence of type 2 diabetes increased markedly in the first 5 years and then appeared to plateau after 10 years (Diabetes Care. 2002 Oct;25[10]:1862-8).
The best data on late-occurring diabetes following GDM comes from the multicenter National Institutes of Health–sponsored Diabetes Prevention Program (DPP) trial, which randomized more than 3,000 individuals with baseline impaired glucose tolerance – or prediabetes – to one of two interventions: metformin therapy or intensive lifestyle intervention, or to placebo.
Within this population, there were more than 1,700 women who had a previous live birth. Of these women, 350 reported a history of GDM at a mean of 12 years since the delivery of their first GDM pregnancy. The DPP gave us the opportunity, therefore, to look at a large group of women about 12 years away from their GDM pregnancy who had abnormal glucose levels but had not reached the level of type 2 diabetes, and compare them with women with similarly impaired glucose tolerance who did not have a history of GDM.
There were interesting similarities and differences. Women with a GDM history were on average 8 years younger than women without a GDM history, but they had comparable BMIs. In addition, within the placebo arm, we could observe the natural history of glucose intolerance in women with and without a history of GDM. Despite both groups entering the study with equivalent degrees of impaired glucose tolerance and similar BMI, women with a history of GDM had a 71% higher risk of developing diabetes during the 3-year intervention period than that of parous women without a history of GDM (J Clin Endocrinol Metab. 2008 Dec;93[12]:4774-9).
Clearly, there was something about the history of GDM that puts these women at greater risk for diabetes than women who had the same impaired glucose tolerance, but no GDM. The study demonstrated that GDM is an exceptionally strong predictor of the development of type 2 diabetes, even for those who manage to escape diabetes for the first 10 years.
Postpartum prevention
The DPP demonstrated, moreover, that intensive lifestyle therapy and metformin not only were both effective, but that they were equally effective, in delaying or preventing diabetes in women with impaired glucose tolerance and a history of GDM. Both reduced the risk by about 50% at 3 years. This was striking because in parous women without GDM, the reductions were 49% and 14%, respectively. Metformin thus appeared to be more effective in women with a history of GDM.
The effects of the interventions persisted over a 10-year follow up of the DPP population. In women with a history of GDM, the intensive lifestyle intervention and metformin reduced progression to diabetes by 35% and 40%, respectively, over 10 years (J Clin Endocrinol Metab. 2015 Apr;100[4]:1646-53).
Pregnancy presents a stress test for beta cell function, and gestational diabetes clearly is a harbinger of further deterioration in beta-cell function and metabolic abnormalities in the mother. Because of these risks and because early intervention makes a difference, surveillance is critically important. Most women see their ob.gyn. as their primary care physician in the 10 years following a pregnancy – the time when more than 50% of all cases of subsequent diabetes will occur – and many continue to see their ob.gyns. in the longer term, as their risk continues to linger.
Immediately after a pregnancy with GDM, ob.gyns. can counsel women not only about their risks of developing type 2 diabetes and the importance of screening, but also about the beneficial impact of lifestyle modification, caloric restriction and weight loss if necessary, and increased exercise. Mothers should also know that GDM is a family affair, and that lifestyle changes that are beneficial for the mother will be equally beneficial for the baby.
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study taught us that there are continuous linear relationships between maternal glucose and adverse fetal outcomes like birth weight and percent body fat greater than the 90th percentile. Longitudinal studies of the Pima Indians showed us that offspring of women who had diabetes during pregnancy were more likely to be obese and more likely to develop diabetes than offspring of women who did not have diabetes during pregnancy. Even when GDM has been well treated and controlled, we should have heightened awareness to the potential risks in the fetus and the growing child and adolescent.
Patients who are found to have subsequent type 2 diabetes should know that aggressive therapy early on in the natural history of the disease reduces the risk of microvascular and macrovascular complications. And as the DPP has demonstrated, lifestyle interventions and metformin may also keep women who are found to have prediabetes outside of pregnancy from progressing on to diabetes.
Dr. Ratner is the chief scientific and medical officer for the American Diabetes Association. He reported having no financial disclosures relevant to this Master Class.
BY E. ALBERT REECE, MD, PhD, MBA
Much attention has been given in the media to the incidence of prediabetes in the general population. The Centers for Disease Control and Prevention estimates that approximately 86 million adults have prediabetes, and that the incidence of this condition is similar across racial and ethnic groups. Indeed, the seriousness of this public health concern prompted the Centers for Medicare & Medicaid Services to expand Medicare coverage for interventions for people with prediabetes, a move that was finalized in November 2016.
Despite a widespread focus on the need to prevent prediabetes from becoming type 2 diabetes, women diagnosed with gestational diabetes mellitus (GDM), which accounts for about 9% of women in the United States, may not be receiving critical advice and care.
The investigators analyzed data collected via the National Health and Nutrition Examination Survey from 2007-2012, and identified 284 women with a history of GDM. Only 67% of these women received diabetes screening, and approximately one-third of women included in the study had undiagnosed prediabetes and diabetes. The authors concluded that prediabetes in women who have had GDM may be underdiagnosed. They argued that women with GDM should be encouraged to have additional health visits and screenings to prevent the development of prediabetes or diabetes. Considering the fact that a number of studies have shown that GDM predisposes a woman to developing type 2 diabetes, the University of Illinois findings are alarming.
As ob.gyns., we have increasingly become a woman’s only health care practitioner. Although individuals may skip annual exams with a primary care physician, during which blood work is typically drawn, many women will see their ob.gyn. for regular check-ups. Therefore, we have a unique role to play in our patients’ lifelong health. This is especially important during pregnancy, when it may be easy to focus only on the mother’s health as it pertains to the health of the baby, rather than her health in pregnancy as it may affect her long-term well-being.
We have invited Robert Ratner, MD, the chief scientific and medical officer at the American Diabetes Association, to discuss the need to carefully follow up with patients who have had GDM and to educate them about their risk for developing type 2 diabetes later in life.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
Why postpartum GDM follow-up is so important
BY ROBERT E. RATNER, MD
Much of the attention paid to diagnosing gestational diabetes has focused on the fetus and on babies being born very large. However, it is important to appreciate that the original definitions of the condition were based entirely on the long-term outcomes of the mother.
John O’Sullivan, MD, and statistician Claire Mahan published diagnostic criteria in 1964 after performing 3-hour oral glucose tolerance tests (OGTTs) in more than 500 unselected women during their pregnancies, and then following these women and babies out as far as 23 years. Retrospectively, Dr. O’Sullivan and Ms. Mahan defined gestational diabetes mellitus (GDM) as glucose values exceeding two standard deviations above the mean on two out of four OGTT values.
They came to their conclusions after tracking the later development of diabetes outside of pregnancy. More than 20 years later, 70% of women with the higher OGTT values had developed type 2 diabetes, compared with approximately 10% of women who did not have higher values during pregnancy. The O’Sullivan criteria were established, essentially, based on their association with the development of diabetes after pregnancy. In addition to being a significant predictor of subsequent diabetes, a history of GDM also conferred a three- to fourfold increase in maternal mortality.
Fifty-some years later, these findings have been affirmed through additional research and are the crux of what drives the current recommendations for postpartum follow-up of women with a history of GDM.
Long-term maternal risks
Postpartum, the current recommendation from both the American Diabetes Association and the American College of Obstetricians and Gynecologists is that women with GDM be tested at 6-12 weeks after delivery to ensure that the diabetes has resolved.
This recommendation for initial postpartum testing carries with it a stipulation that’s different from subsequent postpartum testing. It says that postpartum testing at 6-12 weeks should be performed with either a fasting glucose test or a 2-hour OGTT. Since hemoglobin A1c may still be impacted by the rapid red blood cell turnover in pregnancy or blood loss at delivery, A1c testing lacks sensitivity for identifying diabetes during this window of time.
Initial postpartum testing also serves as a way to identify whether the diabetes during pregnancy was preexisting or purely secondary to the hormonal changes associated with the pregnancy.
If this first postpartum test shows diabetes, the patient most likely had preexisting diabetes, and therapy must be initiated immediately. In the case of a normal result, the patient remains at higher risk for the development of type 2 diabetes essentially for the rest of her life and should be tested at least every 3 years for the occurrence of the disease.
Much of the increased risk for different ethnic groups occurs within 5 years of the index pregnancy. This was shown in a systematic review led by Catherine Kim, MD; the review examined more than two dozen studies with follow-up of up to 28 years postpartum. The cumulative incidence of type 2 diabetes increased markedly in the first 5 years and then appeared to plateau after 10 years (Diabetes Care. 2002 Oct;25[10]:1862-8).
The best data on late-occurring diabetes following GDM comes from the multicenter National Institutes of Health–sponsored Diabetes Prevention Program (DPP) trial, which randomized more than 3,000 individuals with baseline impaired glucose tolerance – or prediabetes – to one of two interventions: metformin therapy or intensive lifestyle intervention, or to placebo.
Within this population, there were more than 1,700 women who had a previous live birth. Of these women, 350 reported a history of GDM at a mean of 12 years since the delivery of their first GDM pregnancy. The DPP gave us the opportunity, therefore, to look at a large group of women about 12 years away from their GDM pregnancy who had abnormal glucose levels but had not reached the level of type 2 diabetes, and compare them with women with similarly impaired glucose tolerance who did not have a history of GDM.
There were interesting similarities and differences. Women with a GDM history were on average 8 years younger than women without a GDM history, but they had comparable BMIs. In addition, within the placebo arm, we could observe the natural history of glucose intolerance in women with and without a history of GDM. Despite both groups entering the study with equivalent degrees of impaired glucose tolerance and similar BMI, women with a history of GDM had a 71% higher risk of developing diabetes during the 3-year intervention period than that of parous women without a history of GDM (J Clin Endocrinol Metab. 2008 Dec;93[12]:4774-9).
Clearly, there was something about the history of GDM that puts these women at greater risk for diabetes than women who had the same impaired glucose tolerance, but no GDM. The study demonstrated that GDM is an exceptionally strong predictor of the development of type 2 diabetes, even for those who manage to escape diabetes for the first 10 years.
Postpartum prevention
The DPP demonstrated, moreover, that intensive lifestyle therapy and metformin not only were both effective, but that they were equally effective, in delaying or preventing diabetes in women with impaired glucose tolerance and a history of GDM. Both reduced the risk by about 50% at 3 years. This was striking because in parous women without GDM, the reductions were 49% and 14%, respectively. Metformin thus appeared to be more effective in women with a history of GDM.
The effects of the interventions persisted over a 10-year follow up of the DPP population. In women with a history of GDM, the intensive lifestyle intervention and metformin reduced progression to diabetes by 35% and 40%, respectively, over 10 years (J Clin Endocrinol Metab. 2015 Apr;100[4]:1646-53).
Pregnancy presents a stress test for beta cell function, and gestational diabetes clearly is a harbinger of further deterioration in beta-cell function and metabolic abnormalities in the mother. Because of these risks and because early intervention makes a difference, surveillance is critically important. Most women see their ob.gyn. as their primary care physician in the 10 years following a pregnancy – the time when more than 50% of all cases of subsequent diabetes will occur – and many continue to see their ob.gyns. in the longer term, as their risk continues to linger.
Immediately after a pregnancy with GDM, ob.gyns. can counsel women not only about their risks of developing type 2 diabetes and the importance of screening, but also about the beneficial impact of lifestyle modification, caloric restriction and weight loss if necessary, and increased exercise. Mothers should also know that GDM is a family affair, and that lifestyle changes that are beneficial for the mother will be equally beneficial for the baby.
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study taught us that there are continuous linear relationships between maternal glucose and adverse fetal outcomes like birth weight and percent body fat greater than the 90th percentile. Longitudinal studies of the Pima Indians showed us that offspring of women who had diabetes during pregnancy were more likely to be obese and more likely to develop diabetes than offspring of women who did not have diabetes during pregnancy. Even when GDM has been well treated and controlled, we should have heightened awareness to the potential risks in the fetus and the growing child and adolescent.
Patients who are found to have subsequent type 2 diabetes should know that aggressive therapy early on in the natural history of the disease reduces the risk of microvascular and macrovascular complications. And as the DPP has demonstrated, lifestyle interventions and metformin may also keep women who are found to have prediabetes outside of pregnancy from progressing on to diabetes.
Dr. Ratner is the chief scientific and medical officer for the American Diabetes Association. He reported having no financial disclosures relevant to this Master Class.
Critical Care Commentary
According to IBM, over 2 quintillion bytes of data are generated every day (that’s a 2 with 18 zeros!), with over 90% of the data in the world today generated in the past 2 years alone.
In our private lives, much of this information is generated through online shopping, web surfing, and popular websites such as Facebook and Twitter. Companies are making incredible efforts to collect these data and to use it to improve how they relate to customers and, ultimately, to make more money. For example, companies like Google, Amazon, Facebook, and Netflix collect enormous amounts of data and then use algorithms to provide real-time suggestions for what their customers might want to rent, buy, or click on. These algorithms, which companies use for anything from predicting customer behavior to facial recognition, were developed in the field of machine learning, a branch of computer science that focuses on how to learn from data.

Big data and critical care
Although the “big data” revolution has proliferated across the private sector, medicine has been slow to utilize the data we painstakingly collect in hospitals every day in order to improve patient care.
Clinicians typically rely on their intuition and the few clinical trials that their patients would have been included in to make decisions, and evidence-based clinical decision support tools are often not available or not used. The tools and scores we have at our disposal are often oversimplified so that they can be calculated by hand and usually rely on the clinician to manually gather information from the electronic health record (EHR) to calculate the score. However, this is starting to change. From partnerships between IBM Watson and hospitals, to groups developing and implementing clinical decision support tools in the EHR, it is clear that hospitals are becoming increasingly interested in learning from and using the enormous amount of data that are just sitting in the hospital records.
Although there are many areas in medicine that stand to benefit from harnessing the data available in the EHR to improve patient care, critical care should be one of the specialties that benefits the most. With the variety and frequency of monitoring that critically ill patients receive, there are large swaths of data available to collect, analyze, and harness to improve patient care. The current glut of information results in data overload and alarm fatigue for today’s clinicians, but intelligent use of these data holds promise for making care safer and more efficient and effective.
Groups have already begun using these data to develop tools to identify patients with ARDS (Herasevich V, et al. Intensive Care Med. 2009;35[6]:1018-23), patients at risk of adverse drug reactions (Harinstein LM, et al. J Crit Care. 2012;27[3]:242-9), and those with sepsis (Tafelski S, et al. J Int Med Res. 2010;38:1605-16).
Furthermore, groups have begun “crowdsourcing” critical care problems by making large datasets publicly available, such as the Multi-parameter Intelligent Monitoring in Intensive Care (MIMIC) database, which now holds clinical data from over 40,000 ICU stays from Beth Israel Deaconess Medical Center. Continued efforts to utilize data from patients in the ICU have the potential to revolutionize the care in hospitals today.
An important area of critical care that has seen a rapid rise in the use of EHR data to create decision support tools is in the early detection of critical illness. Given that many in-hospital cardiac arrests occur outside the ICU and delays in transferring critically ill patients to the ICU increase morbidity and mortality (Churpek MM, et al. J Hosp Med. 2016;11[11]:757-62), detecting critical illness early is incredibly important.
For millennia, clinicians have relied on their intuition and experience to determine which patients have a poor prognosis or need increased levels of care. In the 1990s, rapid response teams (RRTs) were developed, with the goal of identifying and treating critical illness earlier. Along with them came early warning scores, which are objective tools that typically use vital sign abnormalities to detect patients at high risk of clinical deterioration. RRTs and the early warning scores used to activate them have proliferated around the world, including in the United States, and scores like the Modified Early Warning Score (MEWS) are available for automatic calculation in the EHR.
However, taking a tool such as the MEWS that can easily be calculated by hand and making our expensive EHRs calculate it is a lot like buying a Ferrari just to drive it around the parking lot. There is no reason to limit our decision support tools to simple algorithms with only a few variables, especially when patients’ lives are at stake.
Several groups around the country have, therefore, begun to utilize other variables in the EHR, such as laboratory values, to create integrated decision support tools for the early identification of critical illness. For example, Kollef and colleagues developed a statistical model to identify critical illness and implemented it on the wards to activate their RRT, which resulted in decreased lengths of stay in the intervention group (Kollef MH, et al. J Hosp Med. 2014;9[7]:424-9).
Escobar et al. developed a model to predict ICU transfer or non-DNR deaths in the Kaiser system and found it to be more accurate than the MEWS in a validation cohort (Escobar GJ, et al. J Hosp Med. 2012;7[5]:388-95). A clinical trial of their system is ongoing.
Finally, our group developed a model called eCART in a multicenter study of over 250,000 patients and has since implemented it in our hospital. An early “black-box” study found that eCART detected more patients who went on to experience a cardiac arrest or ICU transfer than our usual care RRT and it did so 24 hours earlier (Kang MA, et al. Crit Care Med. 2016;44[8]:1468-73). These scores and many more will likely become commonplace in hospitals to provide an objective and accurate way to identify critically ill patients earlier, which may result in decreased preventable morbidity and mortality.
Future directions
There are several important future directions at the intersection of big data and critical care.
First, efforts to collect, store, and share the highly granular data in the ICU are paramount for successful and generalizable research collaborations. Although there are often institutional barriers to data sharing to surmount, efforts such as the MIMIC database provide a roadmap for how ICU data can be shared and problems “crowdsourced” in order to allow researchers access to these data for high quality research.
Second, efforts to fuse randomized controlled trials with big data, such as randomized, embedded, multifactorial, adaptive platform (REMAP) trials, have the potential to greatly enhance the way trials are done in the future. REMAP trials would be embedded in the EHR, provide the ability to study multiple therapies at once, and adapt the randomization scheme to ensure that patients are not harmed by interventions that are clearly detrimental while the study is ongoing (Angus DC. JAMA. 2015;314[8]:767-8).
Finally, it is important that we move beyond the classic statistical methods that are commonly used to develop decision support tools and increase our use of more modern machine learning techniques that companies in the private sector use every day. For example, our group found that classic regression methods were the least accurate of all the methods we studied for detecting clinical deterioration on the wards (Churpek MM, et al. Crit Care Med. 2016;44[2]:368-74). In the future, methods such as the random forest and neural network should become commonplace in the critical care literature.
The big data revolution is here, both in our private lives and in the hospital. The future will bring continued efforts to use data to identify critical illness earlier, improve the care of patients in the ICU, and implement smarter and more efficient clinical trials. This should rapidly increase the generation and utilization of new knowledge and will have a profound impact on the way we care for critically ill patients.
Dr. Churpek is assistant professor, section of pulmonary and critical care medicine, department of medicine at University of Chicago.
Editor’s comment
Why should busy ICU clinicians bother with big data? Isn’t this simply a “flash in the pan” phenomenon that has sprung up in the aftermath of the electronic medical records (EMRs) mandated by the Affordable Care Act? Are concerns valid that clinical data–based algorithms will lead to an endless stream of alerts akin to the ubiquitous pop-up ads for mortgage refinancing, herbal Viagra, and online gambling that has resulted from commercial data mining?
In this Critical Care Commentary, Dr. Matthew Churpek convincingly outlines the potential inherent in the big data generated by our collective ICUs. These benefits are manifesting themselves not just in the data populated within the EMR – but also in the novel ways we can now design and execute studies. And for those who aren’t yet convinced, recall that payers already use the treasure trove of information within our EMRs against us in the forms of self-serving quality metrics, punitive reimbursement, and unvalidated hospital comparison sites.
Lee E. Morrow, MD, FCCP, is the editor of the Critical Care Commentary section of CHEST Physician.
According to IBM, over 2 quintillion bytes of data are generated every day (that’s a 2 with 18 zeros!), with over 90% of the data in the world today generated in the past 2 years alone.
In our private lives, much of this information is generated through online shopping, web surfing, and popular websites such as Facebook and Twitter. Companies are making incredible efforts to collect these data and to use it to improve how they relate to customers and, ultimately, to make more money. For example, companies like Google, Amazon, Facebook, and Netflix collect enormous amounts of data and then use algorithms to provide real-time suggestions for what their customers might want to rent, buy, or click on. These algorithms, which companies use for anything from predicting customer behavior to facial recognition, were developed in the field of machine learning, a branch of computer science that focuses on how to learn from data.
Big data and critical care
Although the “big data” revolution has proliferated across the private sector, medicine has been slow to utilize the data we painstakingly collect in hospitals every day in order to improve patient care.
Clinicians typically rely on their intuition and the few clinical trials that their patients would have been included in to make decisions, and evidence-based clinical decision support tools are often not available or not used. The tools and scores we have at our disposal are often oversimplified so that they can be calculated by hand and usually rely on the clinician to manually gather information from the electronic health record (EHR) to calculate the score. However, this is starting to change. From partnerships between IBM Watson and hospitals, to groups developing and implementing clinical decision support tools in the EHR, it is clear that hospitals are becoming increasingly interested in learning from and using the enormous amount of data that are just sitting in the hospital records.
Although there are many areas in medicine that stand to benefit from harnessing the data available in the EHR to improve patient care, critical care should be one of the specialties that benefits the most. With the variety and frequency of monitoring that critically ill patients receive, there are large swaths of data available to collect, analyze, and harness to improve patient care. The current glut of information results in data overload and alarm fatigue for today’s clinicians, but intelligent use of these data holds promise for making care safer and more efficient and effective.
Groups have already begun using these data to develop tools to identify patients with ARDS (Herasevich V, et al. Intensive Care Med. 2009;35[6]:1018-23), patients at risk of adverse drug reactions (Harinstein LM, et al. J Crit Care. 2012;27[3]:242-9), and those with sepsis (Tafelski S, et al. J Int Med Res. 2010;38:1605-16).
Furthermore, groups have begun “crowdsourcing” critical care problems by making large datasets publicly available, such as the Multi-parameter Intelligent Monitoring in Intensive Care (MIMIC) database, which now holds clinical data from over 40,000 ICU stays from Beth Israel Deaconess Medical Center. Continued efforts to utilize data from patients in the ICU have the potential to revolutionize the care in hospitals today.
An important area of critical care that has seen a rapid rise in the use of EHR data to create decision support tools is in the early detection of critical illness. Given that many in-hospital cardiac arrests occur outside the ICU and delays in transferring critically ill patients to the ICU increase morbidity and mortality (Churpek MM, et al. J Hosp Med. 2016;11[11]:757-62), detecting critical illness early is incredibly important.
For millennia, clinicians have relied on their intuition and experience to determine which patients have a poor prognosis or need increased levels of care. In the 1990s, rapid response teams (RRTs) were developed, with the goal of identifying and treating critical illness earlier. Along with them came early warning scores, which are objective tools that typically use vital sign abnormalities to detect patients at high risk of clinical deterioration. RRTs and the early warning scores used to activate them have proliferated around the world, including in the United States, and scores like the Modified Early Warning Score (MEWS) are available for automatic calculation in the EHR.
However, taking a tool such as the MEWS that can easily be calculated by hand and making our expensive EHRs calculate it is a lot like buying a Ferrari just to drive it around the parking lot. There is no reason to limit our decision support tools to simple algorithms with only a few variables, especially when patients’ lives are at stake.
Several groups around the country have, therefore, begun to utilize other variables in the EHR, such as laboratory values, to create integrated decision support tools for the early identification of critical illness. For example, Kollef and colleagues developed a statistical model to identify critical illness and implemented it on the wards to activate their RRT, which resulted in decreased lengths of stay in the intervention group (Kollef MH, et al. J Hosp Med. 2014;9[7]:424-9).
Escobar et al. developed a model to predict ICU transfer or non-DNR deaths in the Kaiser system and found it to be more accurate than the MEWS in a validation cohort (Escobar GJ, et al. J Hosp Med. 2012;7[5]:388-95). A clinical trial of their system is ongoing.
Finally, our group developed a model called eCART in a multicenter study of over 250,000 patients and has since implemented it in our hospital. An early “black-box” study found that eCART detected more patients who went on to experience a cardiac arrest or ICU transfer than our usual care RRT and it did so 24 hours earlier (Kang MA, et al. Crit Care Med. 2016;44[8]:1468-73). These scores and many more will likely become commonplace in hospitals to provide an objective and accurate way to identify critically ill patients earlier, which may result in decreased preventable morbidity and mortality.
Future directions
There are several important future directions at the intersection of big data and critical care.
First, efforts to collect, store, and share the highly granular data in the ICU are paramount for successful and generalizable research collaborations. Although there are often institutional barriers to data sharing to surmount, efforts such as the MIMIC database provide a roadmap for how ICU data can be shared and problems “crowdsourced” in order to allow researchers access to these data for high quality research.
Second, efforts to fuse randomized controlled trials with big data, such as randomized, embedded, multifactorial, adaptive platform (REMAP) trials, have the potential to greatly enhance the way trials are done in the future. REMAP trials would be embedded in the EHR, provide the ability to study multiple therapies at once, and adapt the randomization scheme to ensure that patients are not harmed by interventions that are clearly detrimental while the study is ongoing (Angus DC. JAMA. 2015;314[8]:767-8).
Finally, it is important that we move beyond the classic statistical methods that are commonly used to develop decision support tools and increase our use of more modern machine learning techniques that companies in the private sector use every day. For example, our group found that classic regression methods were the least accurate of all the methods we studied for detecting clinical deterioration on the wards (Churpek MM, et al. Crit Care Med. 2016;44[2]:368-74). In the future, methods such as the random forest and neural network should become commonplace in the critical care literature.
The big data revolution is here, both in our private lives and in the hospital. The future will bring continued efforts to use data to identify critical illness earlier, improve the care of patients in the ICU, and implement smarter and more efficient clinical trials. This should rapidly increase the generation and utilization of new knowledge and will have a profound impact on the way we care for critically ill patients.
Dr. Churpek is assistant professor, section of pulmonary and critical care medicine, department of medicine at University of Chicago.
Editor’s comment
Why should busy ICU clinicians bother with big data? Isn’t this simply a “flash in the pan” phenomenon that has sprung up in the aftermath of the electronic medical records (EMRs) mandated by the Affordable Care Act? Are concerns valid that clinical data–based algorithms will lead to an endless stream of alerts akin to the ubiquitous pop-up ads for mortgage refinancing, herbal Viagra, and online gambling that has resulted from commercial data mining?
In this Critical Care Commentary, Dr. Matthew Churpek convincingly outlines the potential inherent in the big data generated by our collective ICUs. These benefits are manifesting themselves not just in the data populated within the EMR – but also in the novel ways we can now design and execute studies. And for those who aren’t yet convinced, recall that payers already use the treasure trove of information within our EMRs against us in the forms of self-serving quality metrics, punitive reimbursement, and unvalidated hospital comparison sites.
Lee E. Morrow, MD, FCCP, is the editor of the Critical Care Commentary section of CHEST Physician.
According to IBM, over 2 quintillion bytes of data are generated every day (that’s a 2 with 18 zeros!), with over 90% of the data in the world today generated in the past 2 years alone.
In our private lives, much of this information is generated through online shopping, web surfing, and popular websites such as Facebook and Twitter. Companies are making incredible efforts to collect these data and to use it to improve how they relate to customers and, ultimately, to make more money. For example, companies like Google, Amazon, Facebook, and Netflix collect enormous amounts of data and then use algorithms to provide real-time suggestions for what their customers might want to rent, buy, or click on. These algorithms, which companies use for anything from predicting customer behavior to facial recognition, were developed in the field of machine learning, a branch of computer science that focuses on how to learn from data.
Big data and critical care
Although the “big data” revolution has proliferated across the private sector, medicine has been slow to utilize the data we painstakingly collect in hospitals every day in order to improve patient care.
Clinicians typically rely on their intuition and the few clinical trials that their patients would have been included in to make decisions, and evidence-based clinical decision support tools are often not available or not used. The tools and scores we have at our disposal are often oversimplified so that they can be calculated by hand and usually rely on the clinician to manually gather information from the electronic health record (EHR) to calculate the score. However, this is starting to change. From partnerships between IBM Watson and hospitals, to groups developing and implementing clinical decision support tools in the EHR, it is clear that hospitals are becoming increasingly interested in learning from and using the enormous amount of data that are just sitting in the hospital records.
Although there are many areas in medicine that stand to benefit from harnessing the data available in the EHR to improve patient care, critical care should be one of the specialties that benefits the most. With the variety and frequency of monitoring that critically ill patients receive, there are large swaths of data available to collect, analyze, and harness to improve patient care. The current glut of information results in data overload and alarm fatigue for today’s clinicians, but intelligent use of these data holds promise for making care safer and more efficient and effective.
Groups have already begun using these data to develop tools to identify patients with ARDS (Herasevich V, et al. Intensive Care Med. 2009;35[6]:1018-23), patients at risk of adverse drug reactions (Harinstein LM, et al. J Crit Care. 2012;27[3]:242-9), and those with sepsis (Tafelski S, et al. J Int Med Res. 2010;38:1605-16).
Furthermore, groups have begun “crowdsourcing” critical care problems by making large datasets publicly available, such as the Multi-parameter Intelligent Monitoring in Intensive Care (MIMIC) database, which now holds clinical data from over 40,000 ICU stays from Beth Israel Deaconess Medical Center. Continued efforts to utilize data from patients in the ICU have the potential to revolutionize the care in hospitals today.
An important area of critical care that has seen a rapid rise in the use of EHR data to create decision support tools is in the early detection of critical illness. Given that many in-hospital cardiac arrests occur outside the ICU and delays in transferring critically ill patients to the ICU increase morbidity and mortality (Churpek MM, et al. J Hosp Med. 2016;11[11]:757-62), detecting critical illness early is incredibly important.
For millennia, clinicians have relied on their intuition and experience to determine which patients have a poor prognosis or need increased levels of care. In the 1990s, rapid response teams (RRTs) were developed, with the goal of identifying and treating critical illness earlier. Along with them came early warning scores, which are objective tools that typically use vital sign abnormalities to detect patients at high risk of clinical deterioration. RRTs and the early warning scores used to activate them have proliferated around the world, including in the United States, and scores like the Modified Early Warning Score (MEWS) are available for automatic calculation in the EHR.
However, taking a tool such as the MEWS that can easily be calculated by hand and making our expensive EHRs calculate it is a lot like buying a Ferrari just to drive it around the parking lot. There is no reason to limit our decision support tools to simple algorithms with only a few variables, especially when patients’ lives are at stake.
Several groups around the country have, therefore, begun to utilize other variables in the EHR, such as laboratory values, to create integrated decision support tools for the early identification of critical illness. For example, Kollef and colleagues developed a statistical model to identify critical illness and implemented it on the wards to activate their RRT, which resulted in decreased lengths of stay in the intervention group (Kollef MH, et al. J Hosp Med. 2014;9[7]:424-9).
Escobar et al. developed a model to predict ICU transfer or non-DNR deaths in the Kaiser system and found it to be more accurate than the MEWS in a validation cohort (Escobar GJ, et al. J Hosp Med. 2012;7[5]:388-95). A clinical trial of their system is ongoing.
Finally, our group developed a model called eCART in a multicenter study of over 250,000 patients and has since implemented it in our hospital. An early “black-box” study found that eCART detected more patients who went on to experience a cardiac arrest or ICU transfer than our usual care RRT and it did so 24 hours earlier (Kang MA, et al. Crit Care Med. 2016;44[8]:1468-73). These scores and many more will likely become commonplace in hospitals to provide an objective and accurate way to identify critically ill patients earlier, which may result in decreased preventable morbidity and mortality.
Future directions
There are several important future directions at the intersection of big data and critical care.
First, efforts to collect, store, and share the highly granular data in the ICU are paramount for successful and generalizable research collaborations. Although there are often institutional barriers to data sharing to surmount, efforts such as the MIMIC database provide a roadmap for how ICU data can be shared and problems “crowdsourced” in order to allow researchers access to these data for high quality research.
Second, efforts to fuse randomized controlled trials with big data, such as randomized, embedded, multifactorial, adaptive platform (REMAP) trials, have the potential to greatly enhance the way trials are done in the future. REMAP trials would be embedded in the EHR, provide the ability to study multiple therapies at once, and adapt the randomization scheme to ensure that patients are not harmed by interventions that are clearly detrimental while the study is ongoing (Angus DC. JAMA. 2015;314[8]:767-8).
Finally, it is important that we move beyond the classic statistical methods that are commonly used to develop decision support tools and increase our use of more modern machine learning techniques that companies in the private sector use every day. For example, our group found that classic regression methods were the least accurate of all the methods we studied for detecting clinical deterioration on the wards (Churpek MM, et al. Crit Care Med. 2016;44[2]:368-74). In the future, methods such as the random forest and neural network should become commonplace in the critical care literature.
The big data revolution is here, both in our private lives and in the hospital. The future will bring continued efforts to use data to identify critical illness earlier, improve the care of patients in the ICU, and implement smarter and more efficient clinical trials. This should rapidly increase the generation and utilization of new knowledge and will have a profound impact on the way we care for critically ill patients.
Dr. Churpek is assistant professor, section of pulmonary and critical care medicine, department of medicine at University of Chicago.
Editor’s comment
Why should busy ICU clinicians bother with big data? Isn’t this simply a “flash in the pan” phenomenon that has sprung up in the aftermath of the electronic medical records (EMRs) mandated by the Affordable Care Act? Are concerns valid that clinical data–based algorithms will lead to an endless stream of alerts akin to the ubiquitous pop-up ads for mortgage refinancing, herbal Viagra, and online gambling that has resulted from commercial data mining?
In this Critical Care Commentary, Dr. Matthew Churpek convincingly outlines the potential inherent in the big data generated by our collective ICUs. These benefits are manifesting themselves not just in the data populated within the EMR – but also in the novel ways we can now design and execute studies. And for those who aren’t yet convinced, recall that payers already use the treasure trove of information within our EMRs against us in the forms of self-serving quality metrics, punitive reimbursement, and unvalidated hospital comparison sites.
Lee E. Morrow, MD, FCCP, is the editor of the Critical Care Commentary section of CHEST Physician.

Thank You to Our 2016 Peer Reviewers
The editors of Emergency Medicine acknowledge the help of the journal’s editorial board members, other emergency physicians, and colleagues in other specialties who reviewed manuscripts in 2016. On behalf of our readers, who are the beneficiaries of your efforts, we thank you.
Alfred Z. Abuhamad, MD
Department of Obstetrics and Gynecology
Eastern Virginia Medical School
John E. Arbo, MD
Division of Emergency Medicine and Pulmonary Critical Care Medicine
Weill Cornell Medical College, Cornell University
David P. Calfee, MD
Division of Infectious Diseases
Weill Cornell Medical College, Cornell University
Richard M. Cantor, MD, FAAP, FACEP
Emergency Department, Pediatrics
Upstate Medical University
Wallace A. Carter, MD, FACEP
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Sunday Clark, ScD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Theodore R. Delbridge, MD
Department of Emergency
Medicine East Carolina University, Brody School of Medicine
Joseph J. Fins, MD
Division of Medical Ethics Internal Medicine
Weill Cornell Medical College, Cornell University
Ron W. Flenner, MD
Department of Internal Medicine
Eastern Virginia Medical School
E. John Gallagher, MD
Department of Emergency Medicine
Albert Einstein College of Medicine
Marianne Gausche-Hill, MD
Department of Emergency Medicine
Harbor-UCLA Medical Center
Keith D. Hentel, MD
Department of Radiology
Weill Cornell Medical College, Cornell University
Barry J. Knapp, MD
Department of Emergency Medicine
Eastern Virginia Medical School
Richard I. Lapin, MD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Anthony C. Mustalish, MD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Lewis S. Nelson, MD
Department of Emergency Medicine
Rutgers New Jersey Medical School
Debra Perina, MD
Department of Emergency Medicine
University of Virginia, Charlottesville
Constance Peterson, MA
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Shari L. Platt, MD, FAAP
Division of Pediatric Emergency Medicine
Weill Cornell Medical College, Cornell University
Rama B. Rao, MD
Division of Toxicology
Weill Cornell Medical College, Cornell University
Earl J. Reisdorff, MD
Executive Director
American Board of Emergency Medicine
Thomas M. Scalea, MD, FACS, FCCM
Program in Trauma
University of Maryland School of Medicine
Edward J. Schenk, MD
Pulmonary Critical Care Medicine
Weill Cornell Medical College, Cornell University
Christopher K. Schott, MD, MS
Department of Emergency Medicine and Critical Care Medicine
University of Pittsburgh
Adam J Singer, MD
Department of Emergency Medicine
Stony Brook University and Medical Center
Sarah A. Stahmer, MD, FACEP
Division of Emergency Medicine
Duke University Medical Center
Michael E. Stern, MD
Division of Geriatric Emergency Medicine
Weill Cornell Medical College, Cornell University
Susan Stone, MD
Emergency Medicine/Palliative Care
University of California, Los Angeles
Todd Taylor, MD
Department of Emergency Medicine
Emory University School of Medicine
The editors of Emergency Medicine acknowledge the help of the journal’s editorial board members, other emergency physicians, and colleagues in other specialties who reviewed manuscripts in 2016. On behalf of our readers, who are the beneficiaries of your efforts, we thank you.
Alfred Z. Abuhamad, MD
Department of Obstetrics and Gynecology
Eastern Virginia Medical School
John E. Arbo, MD
Division of Emergency Medicine and Pulmonary Critical Care Medicine
Weill Cornell Medical College, Cornell University
David P. Calfee, MD
Division of Infectious Diseases
Weill Cornell Medical College, Cornell University
Richard M. Cantor, MD, FAAP, FACEP
Emergency Department, Pediatrics
Upstate Medical University
Wallace A. Carter, MD, FACEP
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Sunday Clark, ScD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Theodore R. Delbridge, MD
Department of Emergency
Medicine East Carolina University, Brody School of Medicine
Joseph J. Fins, MD
Division of Medical Ethics Internal Medicine
Weill Cornell Medical College, Cornell University
Ron W. Flenner, MD
Department of Internal Medicine
Eastern Virginia Medical School
E. John Gallagher, MD
Department of Emergency Medicine
Albert Einstein College of Medicine
Marianne Gausche-Hill, MD
Department of Emergency Medicine
Harbor-UCLA Medical Center
Keith D. Hentel, MD
Department of Radiology
Weill Cornell Medical College, Cornell University
Barry J. Knapp, MD
Department of Emergency Medicine
Eastern Virginia Medical School
Richard I. Lapin, MD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Anthony C. Mustalish, MD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Lewis S. Nelson, MD
Department of Emergency Medicine
Rutgers New Jersey Medical School
Debra Perina, MD
Department of Emergency Medicine
University of Virginia, Charlottesville
Constance Peterson, MA
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Shari L. Platt, MD, FAAP
Division of Pediatric Emergency Medicine
Weill Cornell Medical College, Cornell University
Rama B. Rao, MD
Division of Toxicology
Weill Cornell Medical College, Cornell University
Earl J. Reisdorff, MD
Executive Director
American Board of Emergency Medicine
Thomas M. Scalea, MD, FACS, FCCM
Program in Trauma
University of Maryland School of Medicine
Edward J. Schenk, MD
Pulmonary Critical Care Medicine
Weill Cornell Medical College, Cornell University
Christopher K. Schott, MD, MS
Department of Emergency Medicine and Critical Care Medicine
University of Pittsburgh
Adam J Singer, MD
Department of Emergency Medicine
Stony Brook University and Medical Center
Sarah A. Stahmer, MD, FACEP
Division of Emergency Medicine
Duke University Medical Center
Michael E. Stern, MD
Division of Geriatric Emergency Medicine
Weill Cornell Medical College, Cornell University
Susan Stone, MD
Emergency Medicine/Palliative Care
University of California, Los Angeles
Todd Taylor, MD
Department of Emergency Medicine
Emory University School of Medicine
The editors of Emergency Medicine acknowledge the help of the journal’s editorial board members, other emergency physicians, and colleagues in other specialties who reviewed manuscripts in 2016. On behalf of our readers, who are the beneficiaries of your efforts, we thank you.
Alfred Z. Abuhamad, MD
Department of Obstetrics and Gynecology
Eastern Virginia Medical School
John E. Arbo, MD
Division of Emergency Medicine and Pulmonary Critical Care Medicine
Weill Cornell Medical College, Cornell University
David P. Calfee, MD
Division of Infectious Diseases
Weill Cornell Medical College, Cornell University
Richard M. Cantor, MD, FAAP, FACEP
Emergency Department, Pediatrics
Upstate Medical University
Wallace A. Carter, MD, FACEP
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Sunday Clark, ScD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Theodore R. Delbridge, MD
Department of Emergency
Medicine East Carolina University, Brody School of Medicine
Joseph J. Fins, MD
Division of Medical Ethics Internal Medicine
Weill Cornell Medical College, Cornell University
Ron W. Flenner, MD
Department of Internal Medicine
Eastern Virginia Medical School
E. John Gallagher, MD
Department of Emergency Medicine
Albert Einstein College of Medicine
Marianne Gausche-Hill, MD
Department of Emergency Medicine
Harbor-UCLA Medical Center
Keith D. Hentel, MD
Department of Radiology
Weill Cornell Medical College, Cornell University
Barry J. Knapp, MD
Department of Emergency Medicine
Eastern Virginia Medical School
Richard I. Lapin, MD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Anthony C. Mustalish, MD
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Lewis S. Nelson, MD
Department of Emergency Medicine
Rutgers New Jersey Medical School
Debra Perina, MD
Department of Emergency Medicine
University of Virginia, Charlottesville
Constance Peterson, MA
Department of Emergency Medicine
Weill Cornell Medical College, Cornell University
Shari L. Platt, MD, FAAP
Division of Pediatric Emergency Medicine
Weill Cornell Medical College, Cornell University
Rama B. Rao, MD
Division of Toxicology
Weill Cornell Medical College, Cornell University
Earl J. Reisdorff, MD
Executive Director
American Board of Emergency Medicine
Thomas M. Scalea, MD, FACS, FCCM
Program in Trauma
University of Maryland School of Medicine
Edward J. Schenk, MD
Pulmonary Critical Care Medicine
Weill Cornell Medical College, Cornell University
Christopher K. Schott, MD, MS
Department of Emergency Medicine and Critical Care Medicine
University of Pittsburgh
Adam J Singer, MD
Department of Emergency Medicine
Stony Brook University and Medical Center
Sarah A. Stahmer, MD, FACEP
Division of Emergency Medicine
Duke University Medical Center
Michael E. Stern, MD
Division of Geriatric Emergency Medicine
Weill Cornell Medical College, Cornell University
Susan Stone, MD
Emergency Medicine/Palliative Care
University of California, Los Angeles
Todd Taylor, MD
Department of Emergency Medicine
Emory University School of Medicine