User login
FDA approves new interferon for polycythemia vera
, according to an agency press release.

Besremi has a longer half-life than do other pegylated interferon-alfas, allowing for dosing every 2 weeks instead of weekly. If red blood cell counts remain normal for a year, patients have the option of switching to once-monthly dosing. As with similar products, Besremi is self-administered as a subcutaneous injection.
It’s the first interferon approved in the United States specifically for polycythemia vera. Besremi is also approved for upfront therapy, unlike FDA’s first approval for the condition, the oral JAK inhibitor ruxolitinib (Jakafi), which is indicated only after hydroxyurea failure.
Taiwan-based maker PharmaEssentia said in another press release that it will roll Besremi out to the U.S. market in the coming weeks.
“As we begin working closely with the community to integrate this important treatment into clinical practice, we also continue to expand our scientific efforts to unlock the full potential of our pioneering molecule,” said Ko-Chung Lin, PhD, the company’s CEO.
As for unlocking the full potential, Besremi is under investigation for other interferon indications, including myelofibrosis, leukemia, and chronic hepatitis.
The FDA’s approval was based on results in 51 adults treated for an average of 5 years; 31 (61%) had a complete hematologic response, defined as a hematocrit below 45% with no phlebotomy for at least 2 months, plus normal platelet and white cell counts, normal spleen size, and no blood clots.
“Noninferiority to hydroxyurea regarding haematological response and normal spleen size was not shown at 12 months. However, response to ropeginterferon alfa-2b continued to increase over time with improved responses compared with hydroxyurea at 36 months,” investigators noted in an earlier report (Lancet Haematol. 2020 Mar;7[3]:e196-e208).
Besremi carries the same boxed warning as those of peginterferon alfa-2b (Pegintron) and peginterferon alfa-2a (Pegasys), which notes the risk of life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Related contraindications include severe depression and other psychiatric problems; liver impairment; serious or untreated autoimmune disease, and immunosuppression following organ transplant.
Influenza-like illness, arthralgia, fatigue, pruritis, nasopharyngitis, and musculoskeletal pain were the most common adverse events in studies, occurring in over 40% of subjects. Urinary tract infections, transient ischemic attacks, and depression were the most frequent serious complications, occurring in over 4%.
Labeling also notes the risk for fetal harm and the need for effective contraception.
Besremi was approved in Europe in 2019 and is approved in Taiwan and South Korea.
Polycythemia vera is a rare condition thought to be caused by acquired bone marrow stem cell mutations that trigger an overproduction of red blood cells. Patients are at increased risk of blood clots and emboli, and subsequent heart attacks, strokes, and other problems. There’s also the risk of transformation to secondary myelofibrosis or leukemia.
, according to an agency press release.
Besremi has a longer half-life than do other pegylated interferon-alfas, allowing for dosing every 2 weeks instead of weekly. If red blood cell counts remain normal for a year, patients have the option of switching to once-monthly dosing. As with similar products, Besremi is self-administered as a subcutaneous injection.
It’s the first interferon approved in the United States specifically for polycythemia vera. Besremi is also approved for upfront therapy, unlike FDA’s first approval for the condition, the oral JAK inhibitor ruxolitinib (Jakafi), which is indicated only after hydroxyurea failure.
Taiwan-based maker PharmaEssentia said in another press release that it will roll Besremi out to the U.S. market in the coming weeks.
“As we begin working closely with the community to integrate this important treatment into clinical practice, we also continue to expand our scientific efforts to unlock the full potential of our pioneering molecule,” said Ko-Chung Lin, PhD, the company’s CEO.
As for unlocking the full potential, Besremi is under investigation for other interferon indications, including myelofibrosis, leukemia, and chronic hepatitis.
The FDA’s approval was based on results in 51 adults treated for an average of 5 years; 31 (61%) had a complete hematologic response, defined as a hematocrit below 45% with no phlebotomy for at least 2 months, plus normal platelet and white cell counts, normal spleen size, and no blood clots.
“Noninferiority to hydroxyurea regarding haematological response and normal spleen size was not shown at 12 months. However, response to ropeginterferon alfa-2b continued to increase over time with improved responses compared with hydroxyurea at 36 months,” investigators noted in an earlier report (Lancet Haematol. 2020 Mar;7[3]:e196-e208).
Besremi carries the same boxed warning as those of peginterferon alfa-2b (Pegintron) and peginterferon alfa-2a (Pegasys), which notes the risk of life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Related contraindications include severe depression and other psychiatric problems; liver impairment; serious or untreated autoimmune disease, and immunosuppression following organ transplant.
Influenza-like illness, arthralgia, fatigue, pruritis, nasopharyngitis, and musculoskeletal pain were the most common adverse events in studies, occurring in over 40% of subjects. Urinary tract infections, transient ischemic attacks, and depression were the most frequent serious complications, occurring in over 4%.
Labeling also notes the risk for fetal harm and the need for effective contraception.
Besremi was approved in Europe in 2019 and is approved in Taiwan and South Korea.
Polycythemia vera is a rare condition thought to be caused by acquired bone marrow stem cell mutations that trigger an overproduction of red blood cells. Patients are at increased risk of blood clots and emboli, and subsequent heart attacks, strokes, and other problems. There’s also the risk of transformation to secondary myelofibrosis or leukemia.
, according to an agency press release.
Besremi has a longer half-life than do other pegylated interferon-alfas, allowing for dosing every 2 weeks instead of weekly. If red blood cell counts remain normal for a year, patients have the option of switching to once-monthly dosing. As with similar products, Besremi is self-administered as a subcutaneous injection.
It’s the first interferon approved in the United States specifically for polycythemia vera. Besremi is also approved for upfront therapy, unlike FDA’s first approval for the condition, the oral JAK inhibitor ruxolitinib (Jakafi), which is indicated only after hydroxyurea failure.
Taiwan-based maker PharmaEssentia said in another press release that it will roll Besremi out to the U.S. market in the coming weeks.
“As we begin working closely with the community to integrate this important treatment into clinical practice, we also continue to expand our scientific efforts to unlock the full potential of our pioneering molecule,” said Ko-Chung Lin, PhD, the company’s CEO.
As for unlocking the full potential, Besremi is under investigation for other interferon indications, including myelofibrosis, leukemia, and chronic hepatitis.
The FDA’s approval was based on results in 51 adults treated for an average of 5 years; 31 (61%) had a complete hematologic response, defined as a hematocrit below 45% with no phlebotomy for at least 2 months, plus normal platelet and white cell counts, normal spleen size, and no blood clots.
“Noninferiority to hydroxyurea regarding haematological response and normal spleen size was not shown at 12 months. However, response to ropeginterferon alfa-2b continued to increase over time with improved responses compared with hydroxyurea at 36 months,” investigators noted in an earlier report (Lancet Haematol. 2020 Mar;7[3]:e196-e208).
Besremi carries the same boxed warning as those of peginterferon alfa-2b (Pegintron) and peginterferon alfa-2a (Pegasys), which notes the risk of life-threatening neuropsychiatric, autoimmune, ischemic, and infectious disorders. Related contraindications include severe depression and other psychiatric problems; liver impairment; serious or untreated autoimmune disease, and immunosuppression following organ transplant.
Influenza-like illness, arthralgia, fatigue, pruritis, nasopharyngitis, and musculoskeletal pain were the most common adverse events in studies, occurring in over 40% of subjects. Urinary tract infections, transient ischemic attacks, and depression were the most frequent serious complications, occurring in over 4%.
Labeling also notes the risk for fetal harm and the need for effective contraception.
Besremi was approved in Europe in 2019 and is approved in Taiwan and South Korea.
Polycythemia vera is a rare condition thought to be caused by acquired bone marrow stem cell mutations that trigger an overproduction of red blood cells. Patients are at increased risk of blood clots and emboli, and subsequent heart attacks, strokes, and other problems. There’s also the risk of transformation to secondary myelofibrosis or leukemia.
FDA flags cardiac perforation risks during leadless pacemaker implantation
The Food and Drug Administration is reminding health care providers about the risk of major complications if cardiac perforation occurs during leadless pacemaker implantation.

Cardiac perforation is a rare complication and the overall risk associated with leadless pacemaker implantation appears similar to that with traditional transvenous pacemakers, the agency says. However, premarket clinical studies of the Micra leadless pacemaker (Medtronic) suggested major complications related to cardiac perforation appear to be more severe for those receiving a leadless pacemaker.
“Information from real-world use suggests that cardiac perforations associated with Micra leadless pacemakers are more likely to be associated with serious complications, such as cardiac tamponade or death, than with traditional pacemakers,” the FDA said Nov. 17 in a letter to health care professionals.
“The FDA is bringing this information to your attention as a reminder and to encourage you to report leadless pacemaker cardiac perforations and complications related to perforation to the manufacturer and the FDA,” it notes.
The Micra Transcatheter Pacing System in 2015 was the first leadless pacemaker approved in Europe, and was approved in the United States the following year with a mandated postapproval study to help assess continued safety and efficacy. The Micra device is currently the only approved leadless pacemaker in the United States.
The FDA continues to evaluate outcomes in patients who receive leadless pacing systems and recommends that health care providers discuss the risks and benefits of available pacing system options with patients as part of shared clinical decision-making.
Providers are advised to read and carefully follow the instructions for use and training for Medtronic’s Micra pacemaker.
Any adverse events or suspected adverse events related to the Micra Transcatheter Pacing System or any other pacemaker systems should be reported to the FDA through MedWatch, its adverse-event reporting program.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration is reminding health care providers about the risk of major complications if cardiac perforation occurs during leadless pacemaker implantation.
Cardiac perforation is a rare complication and the overall risk associated with leadless pacemaker implantation appears similar to that with traditional transvenous pacemakers, the agency says. However, premarket clinical studies of the Micra leadless pacemaker (Medtronic) suggested major complications related to cardiac perforation appear to be more severe for those receiving a leadless pacemaker.
“Information from real-world use suggests that cardiac perforations associated with Micra leadless pacemakers are more likely to be associated with serious complications, such as cardiac tamponade or death, than with traditional pacemakers,” the FDA said Nov. 17 in a letter to health care professionals.
“The FDA is bringing this information to your attention as a reminder and to encourage you to report leadless pacemaker cardiac perforations and complications related to perforation to the manufacturer and the FDA,” it notes.
The Micra Transcatheter Pacing System in 2015 was the first leadless pacemaker approved in Europe, and was approved in the United States the following year with a mandated postapproval study to help assess continued safety and efficacy. The Micra device is currently the only approved leadless pacemaker in the United States.
The FDA continues to evaluate outcomes in patients who receive leadless pacing systems and recommends that health care providers discuss the risks and benefits of available pacing system options with patients as part of shared clinical decision-making.
Providers are advised to read and carefully follow the instructions for use and training for Medtronic’s Micra pacemaker.
Any adverse events or suspected adverse events related to the Micra Transcatheter Pacing System or any other pacemaker systems should be reported to the FDA through MedWatch, its adverse-event reporting program.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration is reminding health care providers about the risk of major complications if cardiac perforation occurs during leadless pacemaker implantation.
Cardiac perforation is a rare complication and the overall risk associated with leadless pacemaker implantation appears similar to that with traditional transvenous pacemakers, the agency says. However, premarket clinical studies of the Micra leadless pacemaker (Medtronic) suggested major complications related to cardiac perforation appear to be more severe for those receiving a leadless pacemaker.
“Information from real-world use suggests that cardiac perforations associated with Micra leadless pacemakers are more likely to be associated with serious complications, such as cardiac tamponade or death, than with traditional pacemakers,” the FDA said Nov. 17 in a letter to health care professionals.
“The FDA is bringing this information to your attention as a reminder and to encourage you to report leadless pacemaker cardiac perforations and complications related to perforation to the manufacturer and the FDA,” it notes.
The Micra Transcatheter Pacing System in 2015 was the first leadless pacemaker approved in Europe, and was approved in the United States the following year with a mandated postapproval study to help assess continued safety and efficacy. The Micra device is currently the only approved leadless pacemaker in the United States.
The FDA continues to evaluate outcomes in patients who receive leadless pacing systems and recommends that health care providers discuss the risks and benefits of available pacing system options with patients as part of shared clinical decision-making.
Providers are advised to read and carefully follow the instructions for use and training for Medtronic’s Micra pacemaker.
Any adverse events or suspected adverse events related to the Micra Transcatheter Pacing System or any other pacemaker systems should be reported to the FDA through MedWatch, its adverse-event reporting program.
A version of this article first appeared on Medscape.com.
Biden seeks to return Califf as FDA chief
On Nov. 12, president Joe Biden said he will nominate Robert Califf, MD, to be commissioner of the U.S. Food and Drug Administration, the top U.S. regulator of drugs and medical devices.
Dr. Califf, a cardiologist, served as FDA chief in the Obama administration, leading the agency from Feb. 2016 to Jan. 2017.
The coming nomination ends nearly 11 months of speculation over Mr. Biden’s pick to the lead the agency during the ongoing pandemic. Janet Woodcock, MD, an FDA veteran, has been serving as acting commissioner. The White House faced a Tuesday deadline to make a nomination or see Dr. Woodcock’s tenure as acting chief expire under federal law.
The initial reaction to the idea of Dr. Califf’s return to the FDA drew mixed reactions.
The nonprofit watchdog Public Citizen issued a statement about its opposition to the potential nomination of Dr. Califf. Michael Carome, MD, director of Public Citizen’s Health Research Group, said the United States “desperately needs an FDA leader who will reverse the decades-long trend in which the agency’s relationship with the pharmaceutical and medical-device industries has grown dangerously cozier – resulting in regulatory capture of the agency by industry.”
But the idea of Dr. Califf returning to the FDA pleased Harlan Krumholz, MD, a cardiologist who has been a leader in outcomes research.
Dr. Krumholz tweeted that the Biden administration likely was testing the reaction to a possible Dr. Califf nomination before making it official. “I realize that this is being floated and not officially announced ... but the nomination of [Califf] just makes so much sense,” Dr. Krumholz tweeted. Dr. Califf’s “expertise as a researcher, policymaker, clinician are unparalleled. In a time of partisanship, he should be a slam-dunk confirmation.”
Dr. Califf’s 2016 Senate confirmation process was marked by dissent from several Democrats who questioned his ties to industry. But the chamber voted 89-4 to confirm him.
A version of this article first appeared on Medscape.com.
On Nov. 12, president Joe Biden said he will nominate Robert Califf, MD, to be commissioner of the U.S. Food and Drug Administration, the top U.S. regulator of drugs and medical devices.
Dr. Califf, a cardiologist, served as FDA chief in the Obama administration, leading the agency from Feb. 2016 to Jan. 2017.
The coming nomination ends nearly 11 months of speculation over Mr. Biden’s pick to the lead the agency during the ongoing pandemic. Janet Woodcock, MD, an FDA veteran, has been serving as acting commissioner. The White House faced a Tuesday deadline to make a nomination or see Dr. Woodcock’s tenure as acting chief expire under federal law.
The initial reaction to the idea of Dr. Califf’s return to the FDA drew mixed reactions.
The nonprofit watchdog Public Citizen issued a statement about its opposition to the potential nomination of Dr. Califf. Michael Carome, MD, director of Public Citizen’s Health Research Group, said the United States “desperately needs an FDA leader who will reverse the decades-long trend in which the agency’s relationship with the pharmaceutical and medical-device industries has grown dangerously cozier – resulting in regulatory capture of the agency by industry.”
But the idea of Dr. Califf returning to the FDA pleased Harlan Krumholz, MD, a cardiologist who has been a leader in outcomes research.
Dr. Krumholz tweeted that the Biden administration likely was testing the reaction to a possible Dr. Califf nomination before making it official. “I realize that this is being floated and not officially announced ... but the nomination of [Califf] just makes so much sense,” Dr. Krumholz tweeted. Dr. Califf’s “expertise as a researcher, policymaker, clinician are unparalleled. In a time of partisanship, he should be a slam-dunk confirmation.”
Dr. Califf’s 2016 Senate confirmation process was marked by dissent from several Democrats who questioned his ties to industry. But the chamber voted 89-4 to confirm him.
A version of this article first appeared on Medscape.com.
On Nov. 12, president Joe Biden said he will nominate Robert Califf, MD, to be commissioner of the U.S. Food and Drug Administration, the top U.S. regulator of drugs and medical devices.
Dr. Califf, a cardiologist, served as FDA chief in the Obama administration, leading the agency from Feb. 2016 to Jan. 2017.
The coming nomination ends nearly 11 months of speculation over Mr. Biden’s pick to the lead the agency during the ongoing pandemic. Janet Woodcock, MD, an FDA veteran, has been serving as acting commissioner. The White House faced a Tuesday deadline to make a nomination or see Dr. Woodcock’s tenure as acting chief expire under federal law.
The initial reaction to the idea of Dr. Califf’s return to the FDA drew mixed reactions.
The nonprofit watchdog Public Citizen issued a statement about its opposition to the potential nomination of Dr. Califf. Michael Carome, MD, director of Public Citizen’s Health Research Group, said the United States “desperately needs an FDA leader who will reverse the decades-long trend in which the agency’s relationship with the pharmaceutical and medical-device industries has grown dangerously cozier – resulting in regulatory capture of the agency by industry.”
But the idea of Dr. Califf returning to the FDA pleased Harlan Krumholz, MD, a cardiologist who has been a leader in outcomes research.
Dr. Krumholz tweeted that the Biden administration likely was testing the reaction to a possible Dr. Califf nomination before making it official. “I realize that this is being floated and not officially announced ... but the nomination of [Califf] just makes so much sense,” Dr. Krumholz tweeted. Dr. Califf’s “expertise as a researcher, policymaker, clinician are unparalleled. In a time of partisanship, he should be a slam-dunk confirmation.”
Dr. Califf’s 2016 Senate confirmation process was marked by dissent from several Democrats who questioned his ties to industry. But the chamber voted 89-4 to confirm him.
A version of this article first appeared on Medscape.com.
FDA stands firm on deadline for clozapine REMS recertification
Despite objections from the American Psychiatric Association and other national groups, federal regulators are sticking to the Nov. 15 deadline for the modified clozapine risk evaluation and mitigation strategy (REMS) program.
The modifications will require all those who prescribe and dispense clozapine to be recertified. Prescribers and pharmacies who fail to be certified by this date will no longer be able to prescribe/dispense the drug.
Clozapine is used to treat schizophrenia that is not well controlled with standard antipsychotics. It is also prescribed to patients with recurrent suicidal behavior associated with schizophrenia or schizoaffective disorder.
Although it is highly effective in some patients, it also carries serious risks. Specifically, it can decrease the neutrophil count, which can lead to severe neutropenia, serious infections, and death. As a result, those taking the drug must undergo regular absolute neutrophil count (ANC) monitoring. Clozapine REMS is intended to maximize the benefits of the drug and minimize risk.
The U.S. Food and Drug Administration first announced modifications to the program in July. Under the new rules, pharmacies will no longer be permitted to use telecommunication verification, also known as the switch system, to verify safe use conditions. Instead, the authorization to dispense will be obtained either through the contact center or online via the REMS website.
Furthermore, a new patient status form (PFS) will be used to document absolute ANC monitoring for all outpatients and the form must be submitted monthly.
In addition to the APA, opponents of the new recertification rules include the College of Psychiatric and Neurologic Pharmacists, the National Council for Mental Wellbeing, the National Association of State Mental Health Program Directors, the National Alliance on Mental Illness, the American Psychiatric Nurses Association, and the American Pharmacists Association.
No more access to patient data
Among other concerns, the coalition argues the new requirements will make it more difficult for clinicians to access data on patients’ previous clozapine history. Many clinicians depend on these data, which were maintained in the earlier REMS and in previous manufacturer-maintained clozapine monitoring systems.
The FDA told this news organization in a statement that the Clozapine Products Manufacturers’ Group “was not able to incorporate this data into the modified REMS database because of technical reasons. The new REMS contact center will have access to the historical data should a health care provider need the information,” the FDA said.
The FDA could not immediately provide this news organization with more detail on the nature of the “technical reasons” cited as a cause for the new hurdle that prevents clinicians from accessing these previously available data.
In a September letter to the FDA, the coalition pushed back, writing that it is “unreasonable to deny prescribers of this data that they entered. Furthermore, the lack of historical data could result in harm to patients in some circumstances.”
The coalition asked the FDA to intervene and ask the drug’s manufacturers to reinstate prescriber access to the information.
In a response to questions on this issue from this news organization, the FDA said in a written response the historical data had not been lost, but would be maintained “for record-keeping purposes.”
The FDA also said agency officials met on Oct. 10 with members of the professional organizations who authored the letter to hear their concerns about the clozapine REMS program. The CPMG, which has the responsibility for implementing the REMS, was also represented at the meeting, the FDA said.
“Based on concerns raised by stakeholders, the CPMG has recently begun to address some of the concerns raised in the letter including updating the prescriber/prescriber designee relationship and enrollment process to allow for a designee to be affiliated with multiple prescribers,” the FDA told this news organization.
“The recent updates also enable prescriber designees to self-enroll/create credentials, send affiliation requests to prescribers, and attest/perform functions on behalf of prescribers. It is our understanding that the CPMG is disseminating this information to stakeholders,” the agency added.
Bigger burden, bad timing
In an interview, Robert Cotes, MD, who serves as a member of the clinical expert team for Serious Mental Illness Adviser, a joint initiative of the APA and the Substance Abuse and Mental Health Services Administration, said the changes to the clozapine REMS will likely increase the administrative burden on clinicians.
In the letter to the FDA, the APA and other groups in the coalition expressed concern about patient status forms, which are five pages long. The coalition has requested that the FDA develop a form where clinicians can submit monitoring results on multiple patients at one time, with PDF forms presented in a fillable format.
“The concern is that if the workflows become more laborious for people, it could result in treatment interruptions for individuals on clozapine or potentially it may steer prescribers away from using clozapine for people who may need it,” said Dr. Cotes.
Also commenting for this news organization, Raymond C. Love, PharmD, professor and vice chair at the University of Maryland School of Pharmacy, Baltimore, emphasized the poor timing of the switch to a new clozapine REMS.
“Pharmacies are overloaded right now due to COVID tests and influenza and COVID boosters and COVID vaccinations for kids,” making it a tough time to manage the demands of the clozapine REMS re-enrollment, he said.
A version of this article first appeared on Medscape.com.
Despite objections from the American Psychiatric Association and other national groups, federal regulators are sticking to the Nov. 15 deadline for the modified clozapine risk evaluation and mitigation strategy (REMS) program.
The modifications will require all those who prescribe and dispense clozapine to be recertified. Prescribers and pharmacies who fail to be certified by this date will no longer be able to prescribe/dispense the drug.
Clozapine is used to treat schizophrenia that is not well controlled with standard antipsychotics. It is also prescribed to patients with recurrent suicidal behavior associated with schizophrenia or schizoaffective disorder.
Although it is highly effective in some patients, it also carries serious risks. Specifically, it can decrease the neutrophil count, which can lead to severe neutropenia, serious infections, and death. As a result, those taking the drug must undergo regular absolute neutrophil count (ANC) monitoring. Clozapine REMS is intended to maximize the benefits of the drug and minimize risk.
The U.S. Food and Drug Administration first announced modifications to the program in July. Under the new rules, pharmacies will no longer be permitted to use telecommunication verification, also known as the switch system, to verify safe use conditions. Instead, the authorization to dispense will be obtained either through the contact center or online via the REMS website.
Furthermore, a new patient status form (PFS) will be used to document absolute ANC monitoring for all outpatients and the form must be submitted monthly.
In addition to the APA, opponents of the new recertification rules include the College of Psychiatric and Neurologic Pharmacists, the National Council for Mental Wellbeing, the National Association of State Mental Health Program Directors, the National Alliance on Mental Illness, the American Psychiatric Nurses Association, and the American Pharmacists Association.
No more access to patient data
Among other concerns, the coalition argues the new requirements will make it more difficult for clinicians to access data on patients’ previous clozapine history. Many clinicians depend on these data, which were maintained in the earlier REMS and in previous manufacturer-maintained clozapine monitoring systems.
The FDA told this news organization in a statement that the Clozapine Products Manufacturers’ Group “was not able to incorporate this data into the modified REMS database because of technical reasons. The new REMS contact center will have access to the historical data should a health care provider need the information,” the FDA said.
The FDA could not immediately provide this news organization with more detail on the nature of the “technical reasons” cited as a cause for the new hurdle that prevents clinicians from accessing these previously available data.
In a September letter to the FDA, the coalition pushed back, writing that it is “unreasonable to deny prescribers of this data that they entered. Furthermore, the lack of historical data could result in harm to patients in some circumstances.”
The coalition asked the FDA to intervene and ask the drug’s manufacturers to reinstate prescriber access to the information.
In a response to questions on this issue from this news organization, the FDA said in a written response the historical data had not been lost, but would be maintained “for record-keeping purposes.”
The FDA also said agency officials met on Oct. 10 with members of the professional organizations who authored the letter to hear their concerns about the clozapine REMS program. The CPMG, which has the responsibility for implementing the REMS, was also represented at the meeting, the FDA said.
“Based on concerns raised by stakeholders, the CPMG has recently begun to address some of the concerns raised in the letter including updating the prescriber/prescriber designee relationship and enrollment process to allow for a designee to be affiliated with multiple prescribers,” the FDA told this news organization.
“The recent updates also enable prescriber designees to self-enroll/create credentials, send affiliation requests to prescribers, and attest/perform functions on behalf of prescribers. It is our understanding that the CPMG is disseminating this information to stakeholders,” the agency added.
Bigger burden, bad timing
In an interview, Robert Cotes, MD, who serves as a member of the clinical expert team for Serious Mental Illness Adviser, a joint initiative of the APA and the Substance Abuse and Mental Health Services Administration, said the changes to the clozapine REMS will likely increase the administrative burden on clinicians.
In the letter to the FDA, the APA and other groups in the coalition expressed concern about patient status forms, which are five pages long. The coalition has requested that the FDA develop a form where clinicians can submit monitoring results on multiple patients at one time, with PDF forms presented in a fillable format.
“The concern is that if the workflows become more laborious for people, it could result in treatment interruptions for individuals on clozapine or potentially it may steer prescribers away from using clozapine for people who may need it,” said Dr. Cotes.
Also commenting for this news organization, Raymond C. Love, PharmD, professor and vice chair at the University of Maryland School of Pharmacy, Baltimore, emphasized the poor timing of the switch to a new clozapine REMS.
“Pharmacies are overloaded right now due to COVID tests and influenza and COVID boosters and COVID vaccinations for kids,” making it a tough time to manage the demands of the clozapine REMS re-enrollment, he said.
A version of this article first appeared on Medscape.com.
Despite objections from the American Psychiatric Association and other national groups, federal regulators are sticking to the Nov. 15 deadline for the modified clozapine risk evaluation and mitigation strategy (REMS) program.
The modifications will require all those who prescribe and dispense clozapine to be recertified. Prescribers and pharmacies who fail to be certified by this date will no longer be able to prescribe/dispense the drug.
Clozapine is used to treat schizophrenia that is not well controlled with standard antipsychotics. It is also prescribed to patients with recurrent suicidal behavior associated with schizophrenia or schizoaffective disorder.
Although it is highly effective in some patients, it also carries serious risks. Specifically, it can decrease the neutrophil count, which can lead to severe neutropenia, serious infections, and death. As a result, those taking the drug must undergo regular absolute neutrophil count (ANC) monitoring. Clozapine REMS is intended to maximize the benefits of the drug and minimize risk.
The U.S. Food and Drug Administration first announced modifications to the program in July. Under the new rules, pharmacies will no longer be permitted to use telecommunication verification, also known as the switch system, to verify safe use conditions. Instead, the authorization to dispense will be obtained either through the contact center or online via the REMS website.
Furthermore, a new patient status form (PFS) will be used to document absolute ANC monitoring for all outpatients and the form must be submitted monthly.
In addition to the APA, opponents of the new recertification rules include the College of Psychiatric and Neurologic Pharmacists, the National Council for Mental Wellbeing, the National Association of State Mental Health Program Directors, the National Alliance on Mental Illness, the American Psychiatric Nurses Association, and the American Pharmacists Association.
No more access to patient data
Among other concerns, the coalition argues the new requirements will make it more difficult for clinicians to access data on patients’ previous clozapine history. Many clinicians depend on these data, which were maintained in the earlier REMS and in previous manufacturer-maintained clozapine monitoring systems.
The FDA told this news organization in a statement that the Clozapine Products Manufacturers’ Group “was not able to incorporate this data into the modified REMS database because of technical reasons. The new REMS contact center will have access to the historical data should a health care provider need the information,” the FDA said.
The FDA could not immediately provide this news organization with more detail on the nature of the “technical reasons” cited as a cause for the new hurdle that prevents clinicians from accessing these previously available data.
In a September letter to the FDA, the coalition pushed back, writing that it is “unreasonable to deny prescribers of this data that they entered. Furthermore, the lack of historical data could result in harm to patients in some circumstances.”
The coalition asked the FDA to intervene and ask the drug’s manufacturers to reinstate prescriber access to the information.
In a response to questions on this issue from this news organization, the FDA said in a written response the historical data had not been lost, but would be maintained “for record-keeping purposes.”
The FDA also said agency officials met on Oct. 10 with members of the professional organizations who authored the letter to hear their concerns about the clozapine REMS program. The CPMG, which has the responsibility for implementing the REMS, was also represented at the meeting, the FDA said.
“Based on concerns raised by stakeholders, the CPMG has recently begun to address some of the concerns raised in the letter including updating the prescriber/prescriber designee relationship and enrollment process to allow for a designee to be affiliated with multiple prescribers,” the FDA told this news organization.
“The recent updates also enable prescriber designees to self-enroll/create credentials, send affiliation requests to prescribers, and attest/perform functions on behalf of prescribers. It is our understanding that the CPMG is disseminating this information to stakeholders,” the agency added.
Bigger burden, bad timing
In an interview, Robert Cotes, MD, who serves as a member of the clinical expert team for Serious Mental Illness Adviser, a joint initiative of the APA and the Substance Abuse and Mental Health Services Administration, said the changes to the clozapine REMS will likely increase the administrative burden on clinicians.
In the letter to the FDA, the APA and other groups in the coalition expressed concern about patient status forms, which are five pages long. The coalition has requested that the FDA develop a form where clinicians can submit monitoring results on multiple patients at one time, with PDF forms presented in a fillable format.
“The concern is that if the workflows become more laborious for people, it could result in treatment interruptions for individuals on clozapine or potentially it may steer prescribers away from using clozapine for people who may need it,” said Dr. Cotes.
Also commenting for this news organization, Raymond C. Love, PharmD, professor and vice chair at the University of Maryland School of Pharmacy, Baltimore, emphasized the poor timing of the switch to a new clozapine REMS.
“Pharmacies are overloaded right now due to COVID tests and influenza and COVID boosters and COVID vaccinations for kids,” making it a tough time to manage the demands of the clozapine REMS re-enrollment, he said.
A version of this article first appeared on Medscape.com.
FDA panel slams Endologix response to stent-graft safety issues
The Food and Drug Administration has long kept a watchful eye over successive iterations of endovascular stent graphs in the Endologix AFX line, designed for repair of abdominal aortic aneurysms (AAA). For years, the devices, first approved in 2011, have drawn safety alerts and recalls , stemming from what the agency says was a “higher than expected” risk for potentially injurious or fatal type III endoleaks.
As part of the latest review process, Endologix recently showed regulators data from a rare randomized trial of the AAA endovascular aneurysm repair (EVAR) technique. The company said the recent postmarket study LEOPARD suggested the type III endoleaks – blood seeping around or through the device into the aneurysm – are no more common with the current AFX2 system than with other available AAA stent-grafts.
Technical upgrades to its AFX line of EVAR devices in recent years have largely resolved the safety issues identified in previous models, the company argued.
But the company’s case was unconvincing for a majority of the FDA Circulatory System Devices Advisory Panel that assembled virtually on Nov. 2. A number of panelists questioned the earnestness with which Endologix worked to rectify the safety alert and recall issues. Many also decried the real-world relevance of the randomized trial presented as evidence, with its follow-up time of only a few years.
The panel that included more than a dozen clinicians – mostly surgeons or interventional cardiologists or radiologists – were not instructed to formally vote on the issues. But it ultimately advised the FDA that more exacting studies with longer follow-ups appear needed to show that the device’s benefits in routine use outweigh its risks, especially for type III endoleaks.
“There isn’t a tremendous amount of confidence” that Endologix had enacted sufficient risk-mitigation measures in the wake of the safety alerts and recalls, chair Richard A. Lange, MD, MBA, Foster School of Medicine and Texas Tech University Health Sciences Center, El Paso, said when summarizing the panel’s take on the day’s proceedings.
Although the stent-graft’s safety seemed improved with recent design changes, the panel wasn’t convinced the upgrades could take the credit, or even that they were aimed specifically at preventing endoleaks, Dr. Lange said. “Nobody feels assurance that the problem has been solved.”
“I believe that the type-three endoleaks pose a challenge to patients, and I have not seen enough data to assure me with a degree of certainty that that problem no longer persists,” said panelist Joaquin E. Cigarroa, MD, a cardiologist at Oregon Health & Science University, Portland. His take on the LEOPARD trial, moreover, is that it “does not refute that there is an issue, given the duration of follow-up.”
On the other hand, a majority of the panel agreed that, currently, the AFX2’s benefits would likely outweigh risks for patients in narrowly defined high-risk anatomic or clinical scenarios and those with no other endovascular or surgical option.
“I do believe that there are patient subsets where the Endologix graft can play an important and vital role,” surgeon Keith B. Allen, MD, St. Luke’s Mid America Heart & Vascular Institute, Kansas City, Missouri, offered from the panel.
“In patients that don’t have aneurysmal disease but have distal bifurcation proximal iliac disease, it can be a very nice graft to use and solves a problem,” he said. “To remove that graft completely from the market, I believe, would deny a subset of patients.”
But for aortic aneurysms in routine practice, Dr. Allen said, “I think there are some red flags with it.”
Joining the day’s proceedings as a public commenter, surgeon Mark Conrad, MD, St. Elizabeth’s Hospital, Boston, agreed that “there’s not one commercial device out there that is able to handle every anatomy.”
Having options for patients is important, he said, because “the biggest problems we run into are when somebody only uses one graft, and they try to make that fit everything.”
Another public commenter offered a similar take. “I think we haven’t done a great job in the vascular surgery community really honing in on the detailed nuances that separate one device from another,” said Naiem Nassiri, MD, Yale New Haven Hospital Heart & Vascular Center, Connecticut.
The Endologix device, he said, “serves a very specific role under certain anatomic configurations and limitations, and really, truly fills a gap” left by other available grafts. It suits a very specific niche, “and I think it needs to be explored further for that.”
Endologix representatives who advise clinicians could play a better role in familiarizing operators with the EVAR system’s strengths and limitations, proposed several panelists, including Minhaj S. Khaja, MD, MBA, interventional radiologist at UVA Health and the University of Virginia, Charlottesville.
“There definitely needs to be more education of the clinical reps as well as the physicians implanting these devices,” he said, regarding the type III leaks, patient selection issues, appropriate imaging follow-up, “and the potential for increased reintervention.”
All public commenters, Dr. Lange observed, had been invited to disclose potential conflicts of interest, but it was not mandatory and none did so during the public forum. Disclosures of potential conflicts for the panelists are available on the FDA site.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has long kept a watchful eye over successive iterations of endovascular stent graphs in the Endologix AFX line, designed for repair of abdominal aortic aneurysms (AAA). For years, the devices, first approved in 2011, have drawn safety alerts and recalls , stemming from what the agency says was a “higher than expected” risk for potentially injurious or fatal type III endoleaks.
As part of the latest review process, Endologix recently showed regulators data from a rare randomized trial of the AAA endovascular aneurysm repair (EVAR) technique. The company said the recent postmarket study LEOPARD suggested the type III endoleaks – blood seeping around or through the device into the aneurysm – are no more common with the current AFX2 system than with other available AAA stent-grafts.
Technical upgrades to its AFX line of EVAR devices in recent years have largely resolved the safety issues identified in previous models, the company argued.
But the company’s case was unconvincing for a majority of the FDA Circulatory System Devices Advisory Panel that assembled virtually on Nov. 2. A number of panelists questioned the earnestness with which Endologix worked to rectify the safety alert and recall issues. Many also decried the real-world relevance of the randomized trial presented as evidence, with its follow-up time of only a few years.
The panel that included more than a dozen clinicians – mostly surgeons or interventional cardiologists or radiologists – were not instructed to formally vote on the issues. But it ultimately advised the FDA that more exacting studies with longer follow-ups appear needed to show that the device’s benefits in routine use outweigh its risks, especially for type III endoleaks.
“There isn’t a tremendous amount of confidence” that Endologix had enacted sufficient risk-mitigation measures in the wake of the safety alerts and recalls, chair Richard A. Lange, MD, MBA, Foster School of Medicine and Texas Tech University Health Sciences Center, El Paso, said when summarizing the panel’s take on the day’s proceedings.
Although the stent-graft’s safety seemed improved with recent design changes, the panel wasn’t convinced the upgrades could take the credit, or even that they were aimed specifically at preventing endoleaks, Dr. Lange said. “Nobody feels assurance that the problem has been solved.”
“I believe that the type-three endoleaks pose a challenge to patients, and I have not seen enough data to assure me with a degree of certainty that that problem no longer persists,” said panelist Joaquin E. Cigarroa, MD, a cardiologist at Oregon Health & Science University, Portland. His take on the LEOPARD trial, moreover, is that it “does not refute that there is an issue, given the duration of follow-up.”
On the other hand, a majority of the panel agreed that, currently, the AFX2’s benefits would likely outweigh risks for patients in narrowly defined high-risk anatomic or clinical scenarios and those with no other endovascular or surgical option.
“I do believe that there are patient subsets where the Endologix graft can play an important and vital role,” surgeon Keith B. Allen, MD, St. Luke’s Mid America Heart & Vascular Institute, Kansas City, Missouri, offered from the panel.
“In patients that don’t have aneurysmal disease but have distal bifurcation proximal iliac disease, it can be a very nice graft to use and solves a problem,” he said. “To remove that graft completely from the market, I believe, would deny a subset of patients.”
But for aortic aneurysms in routine practice, Dr. Allen said, “I think there are some red flags with it.”
Joining the day’s proceedings as a public commenter, surgeon Mark Conrad, MD, St. Elizabeth’s Hospital, Boston, agreed that “there’s not one commercial device out there that is able to handle every anatomy.”
Having options for patients is important, he said, because “the biggest problems we run into are when somebody only uses one graft, and they try to make that fit everything.”
Another public commenter offered a similar take. “I think we haven’t done a great job in the vascular surgery community really honing in on the detailed nuances that separate one device from another,” said Naiem Nassiri, MD, Yale New Haven Hospital Heart & Vascular Center, Connecticut.
The Endologix device, he said, “serves a very specific role under certain anatomic configurations and limitations, and really, truly fills a gap” left by other available grafts. It suits a very specific niche, “and I think it needs to be explored further for that.”
Endologix representatives who advise clinicians could play a better role in familiarizing operators with the EVAR system’s strengths and limitations, proposed several panelists, including Minhaj S. Khaja, MD, MBA, interventional radiologist at UVA Health and the University of Virginia, Charlottesville.
“There definitely needs to be more education of the clinical reps as well as the physicians implanting these devices,” he said, regarding the type III leaks, patient selection issues, appropriate imaging follow-up, “and the potential for increased reintervention.”
All public commenters, Dr. Lange observed, had been invited to disclose potential conflicts of interest, but it was not mandatory and none did so during the public forum. Disclosures of potential conflicts for the panelists are available on the FDA site.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has long kept a watchful eye over successive iterations of endovascular stent graphs in the Endologix AFX line, designed for repair of abdominal aortic aneurysms (AAA). For years, the devices, first approved in 2011, have drawn safety alerts and recalls , stemming from what the agency says was a “higher than expected” risk for potentially injurious or fatal type III endoleaks.
As part of the latest review process, Endologix recently showed regulators data from a rare randomized trial of the AAA endovascular aneurysm repair (EVAR) technique. The company said the recent postmarket study LEOPARD suggested the type III endoleaks – blood seeping around or through the device into the aneurysm – are no more common with the current AFX2 system than with other available AAA stent-grafts.
Technical upgrades to its AFX line of EVAR devices in recent years have largely resolved the safety issues identified in previous models, the company argued.
But the company’s case was unconvincing for a majority of the FDA Circulatory System Devices Advisory Panel that assembled virtually on Nov. 2. A number of panelists questioned the earnestness with which Endologix worked to rectify the safety alert and recall issues. Many also decried the real-world relevance of the randomized trial presented as evidence, with its follow-up time of only a few years.
The panel that included more than a dozen clinicians – mostly surgeons or interventional cardiologists or radiologists – were not instructed to formally vote on the issues. But it ultimately advised the FDA that more exacting studies with longer follow-ups appear needed to show that the device’s benefits in routine use outweigh its risks, especially for type III endoleaks.
“There isn’t a tremendous amount of confidence” that Endologix had enacted sufficient risk-mitigation measures in the wake of the safety alerts and recalls, chair Richard A. Lange, MD, MBA, Foster School of Medicine and Texas Tech University Health Sciences Center, El Paso, said when summarizing the panel’s take on the day’s proceedings.
Although the stent-graft’s safety seemed improved with recent design changes, the panel wasn’t convinced the upgrades could take the credit, or even that they were aimed specifically at preventing endoleaks, Dr. Lange said. “Nobody feels assurance that the problem has been solved.”
“I believe that the type-three endoleaks pose a challenge to patients, and I have not seen enough data to assure me with a degree of certainty that that problem no longer persists,” said panelist Joaquin E. Cigarroa, MD, a cardiologist at Oregon Health & Science University, Portland. His take on the LEOPARD trial, moreover, is that it “does not refute that there is an issue, given the duration of follow-up.”
On the other hand, a majority of the panel agreed that, currently, the AFX2’s benefits would likely outweigh risks for patients in narrowly defined high-risk anatomic or clinical scenarios and those with no other endovascular or surgical option.
“I do believe that there are patient subsets where the Endologix graft can play an important and vital role,” surgeon Keith B. Allen, MD, St. Luke’s Mid America Heart & Vascular Institute, Kansas City, Missouri, offered from the panel.
“In patients that don’t have aneurysmal disease but have distal bifurcation proximal iliac disease, it can be a very nice graft to use and solves a problem,” he said. “To remove that graft completely from the market, I believe, would deny a subset of patients.”
But for aortic aneurysms in routine practice, Dr. Allen said, “I think there are some red flags with it.”
Joining the day’s proceedings as a public commenter, surgeon Mark Conrad, MD, St. Elizabeth’s Hospital, Boston, agreed that “there’s not one commercial device out there that is able to handle every anatomy.”
Having options for patients is important, he said, because “the biggest problems we run into are when somebody only uses one graft, and they try to make that fit everything.”
Another public commenter offered a similar take. “I think we haven’t done a great job in the vascular surgery community really honing in on the detailed nuances that separate one device from another,” said Naiem Nassiri, MD, Yale New Haven Hospital Heart & Vascular Center, Connecticut.
The Endologix device, he said, “serves a very specific role under certain anatomic configurations and limitations, and really, truly fills a gap” left by other available grafts. It suits a very specific niche, “and I think it needs to be explored further for that.”
Endologix representatives who advise clinicians could play a better role in familiarizing operators with the EVAR system’s strengths and limitations, proposed several panelists, including Minhaj S. Khaja, MD, MBA, interventional radiologist at UVA Health and the University of Virginia, Charlottesville.
“There definitely needs to be more education of the clinical reps as well as the physicians implanting these devices,” he said, regarding the type III leaks, patient selection issues, appropriate imaging follow-up, “and the potential for increased reintervention.”
All public commenters, Dr. Lange observed, had been invited to disclose potential conflicts of interest, but it was not mandatory and none did so during the public forum. Disclosures of potential conflicts for the panelists are available on the FDA site.
A version of this article first appeared on Medscape.com.
Expected spike in acute flaccid myelitis did not occur in 2020
suggested researchers at the Centers for Disease Control and Prevention.
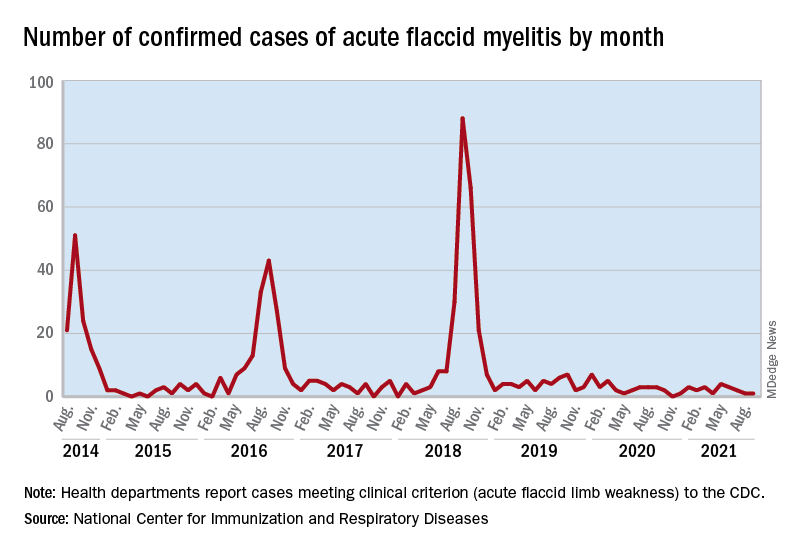
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
suggested researchers at the Centers for Disease Control and Prevention.
Acute flaccid myelitis (AFM) is an uncommon but serious complication of some viral infections, including West Nile virus and nonpolio enteroviruses. It is “characterized by sudden onset of limb weakness and lesions in the gray matter of the spinal cord,” they said, and more than 90% of cases occur in young children.
Cases of AFM, which can lead to respiratory insufficiency and permanent paralysis, spiked during the late summer and early fall in 2014, 2016, and 2018 and were expected to do so again in 2020, Sarah Kidd, MD, and associates at the division of viral diseases at the CDC’s National Center for Immunization and Respiratory Diseases, Atlanta, said in the Morbidity and Mortality Weekly Report.
Monthly peaks in those previous years – each occurring in September – reached 51 cases in 2014, 43 cases in 2016, and 88 cases in 2018, but in 2020 there was only 1 case reported in September, with a high of 4 coming in May, CDC data show. The total number of cases for 2020 (32) was, in fact, lower than in 2019, when 47 were reported.
The investigators’ main objective was to see if there were any differences between the 2018 and 2019-2020 cases. Reports from state health departments to the CDC showed that, in 2019-2020, “patients were older; more likely to have lower limb involvement; and less likely to have upper limb involvement, prodromal illness, [cerebrospinal fluid] pleocytosis, or specimens that tested positive for EV [enterovirus]-D68” than patients from 2018, Dr. Kidd and associates said.
Mask wearing and reduced in-school attendance may have decreased circulation of EV-D68 – the enterovirus type most often detected in the stool and respiratory specimens of AFM patients – as was seen with other respiratory viruses, such as influenza and respiratory syncytial virus, in 2020. Previous studies have suggested that EV-D68 drives the increases in cases during peak years, the researchers noted.
The absence of such an increase “in 2020 reflects a deviation from the previously observed biennial pattern, and it is unclear when the next increase in AFM should be expected. Clinicians should continue to maintain vigilance and suspect AFM in any child with acute flaccid limb weakness, particularly in the setting of recent febrile or respiratory illness,” they wrote.
FROM MMWR
New single-button blood glucose monitor available in U.S.
The POGO Automatic Blood Glucose Monitoring System (Intuity Medical) has been cleared by the U.S. Food and Drug Administration for people with diabetes aged 13 years and older.
It contains a 10-test cartridge, and once loaded and the monitor is turned on, the user only has to press their finger on a button to activate POGO Automatic, which then does all the work of lancing and blood collection, followed by a 4-second countdown and a result. Users only need to carry the monitor and not separate lancets or strips.
An app called Patterns is available for iOS and Android that allows the results from the device to automatically sync via Bluetooth. It visually presents glucose trends and enables data sharing with health care providers.
“We know that people with diabetes are more effective at managing their diabetes when they regularly check their blood glucose and use the information to take action,” said Daniel Einhorn, MD, medical director of Scripps Whittier Diabetes Institute, president of Diabetes and Endocrine Associates, and chairperson of the Intuity Medical Scientific Advisory Board, in a company statement.
“My patients and millions of others with diabetes have struggled for decades with the burden of checking their glucose because it’s complicated, there’s a lot to carry around, and it’s intrusive,” he added. “What they’ve needed is a simple, quick, and truly discreet way to check their blood glucose, so they’ll actually do it.”
How does POGO compare with CGM?
Continuous glucose monitors (CGMs), such as the Abbott FreeStyle Libre, Dexcom G6, and Eversense implant, are increasingly employed by people with type 1 diabetes, and some with type 2 diabetes, to keep a close eye on their blood glucose levels.
Asked how the POGO device compares with CGM systems, Intuity Chief Commercial Officer Dean Zikria said: “While [CGM] is certainly an important option for a subset of people with diabetes, CGM is a very different technology, requiring a user to wear a sensor and transmitter on their body.”
“Patients also need to obtain a prescription in order to use CGM.”
“Conversely, POGO Automatic is available with or without a prescription. POGO Automatic also gives people who do not want to wear a device on their body a new choice other than traditional blood glucose monitoring,” Mr. Zikria added.
The POGO system is available at U.S. pharmacies, including CVS and Walgreens, and can also be purchased online.
The device costs $68 from the company website and a pack of 5 cartridges (each containing 10 tests, with an aim of people performing 1-2 tests per day) costs a further $32 as a one-off, or $32 per month as a subscription.
The product is also eligible for purchase using Flexible Spending Accounts and Health Savings Accounts.
A version of this article first appeared on Medscape.com.
The POGO Automatic Blood Glucose Monitoring System (Intuity Medical) has been cleared by the U.S. Food and Drug Administration for people with diabetes aged 13 years and older.
It contains a 10-test cartridge, and once loaded and the monitor is turned on, the user only has to press their finger on a button to activate POGO Automatic, which then does all the work of lancing and blood collection, followed by a 4-second countdown and a result. Users only need to carry the monitor and not separate lancets or strips.
An app called Patterns is available for iOS and Android that allows the results from the device to automatically sync via Bluetooth. It visually presents glucose trends and enables data sharing with health care providers.
“We know that people with diabetes are more effective at managing their diabetes when they regularly check their blood glucose and use the information to take action,” said Daniel Einhorn, MD, medical director of Scripps Whittier Diabetes Institute, president of Diabetes and Endocrine Associates, and chairperson of the Intuity Medical Scientific Advisory Board, in a company statement.
“My patients and millions of others with diabetes have struggled for decades with the burden of checking their glucose because it’s complicated, there’s a lot to carry around, and it’s intrusive,” he added. “What they’ve needed is a simple, quick, and truly discreet way to check their blood glucose, so they’ll actually do it.”
How does POGO compare with CGM?
Continuous glucose monitors (CGMs), such as the Abbott FreeStyle Libre, Dexcom G6, and Eversense implant, are increasingly employed by people with type 1 diabetes, and some with type 2 diabetes, to keep a close eye on their blood glucose levels.
Asked how the POGO device compares with CGM systems, Intuity Chief Commercial Officer Dean Zikria said: “While [CGM] is certainly an important option for a subset of people with diabetes, CGM is a very different technology, requiring a user to wear a sensor and transmitter on their body.”
“Patients also need to obtain a prescription in order to use CGM.”
“Conversely, POGO Automatic is available with or without a prescription. POGO Automatic also gives people who do not want to wear a device on their body a new choice other than traditional blood glucose monitoring,” Mr. Zikria added.
The POGO system is available at U.S. pharmacies, including CVS and Walgreens, and can also be purchased online.
The device costs $68 from the company website and a pack of 5 cartridges (each containing 10 tests, with an aim of people performing 1-2 tests per day) costs a further $32 as a one-off, or $32 per month as a subscription.
The product is also eligible for purchase using Flexible Spending Accounts and Health Savings Accounts.
A version of this article first appeared on Medscape.com.
The POGO Automatic Blood Glucose Monitoring System (Intuity Medical) has been cleared by the U.S. Food and Drug Administration for people with diabetes aged 13 years and older.
It contains a 10-test cartridge, and once loaded and the monitor is turned on, the user only has to press their finger on a button to activate POGO Automatic, which then does all the work of lancing and blood collection, followed by a 4-second countdown and a result. Users only need to carry the monitor and not separate lancets or strips.
An app called Patterns is available for iOS and Android that allows the results from the device to automatically sync via Bluetooth. It visually presents glucose trends and enables data sharing with health care providers.
“We know that people with diabetes are more effective at managing their diabetes when they regularly check their blood glucose and use the information to take action,” said Daniel Einhorn, MD, medical director of Scripps Whittier Diabetes Institute, president of Diabetes and Endocrine Associates, and chairperson of the Intuity Medical Scientific Advisory Board, in a company statement.
“My patients and millions of others with diabetes have struggled for decades with the burden of checking their glucose because it’s complicated, there’s a lot to carry around, and it’s intrusive,” he added. “What they’ve needed is a simple, quick, and truly discreet way to check their blood glucose, so they’ll actually do it.”
How does POGO compare with CGM?
Continuous glucose monitors (CGMs), such as the Abbott FreeStyle Libre, Dexcom G6, and Eversense implant, are increasingly employed by people with type 1 diabetes, and some with type 2 diabetes, to keep a close eye on their blood glucose levels.
Asked how the POGO device compares with CGM systems, Intuity Chief Commercial Officer Dean Zikria said: “While [CGM] is certainly an important option for a subset of people with diabetes, CGM is a very different technology, requiring a user to wear a sensor and transmitter on their body.”
“Patients also need to obtain a prescription in order to use CGM.”
“Conversely, POGO Automatic is available with or without a prescription. POGO Automatic also gives people who do not want to wear a device on their body a new choice other than traditional blood glucose monitoring,” Mr. Zikria added.
The POGO system is available at U.S. pharmacies, including CVS and Walgreens, and can also be purchased online.
The device costs $68 from the company website and a pack of 5 cartridges (each containing 10 tests, with an aim of people performing 1-2 tests per day) costs a further $32 as a one-off, or $32 per month as a subscription.
The product is also eligible for purchase using Flexible Spending Accounts and Health Savings Accounts.
A version of this article first appeared on Medscape.com.
CDC endorses Pfizer’s COVID-19 vaccine for young kids
– meaning the shots are now available for immediate use.
The Nov. 2 decision came mere hours after experts that advise the CDC on vaccinations strongly recommended the vaccine for this age group.
“Together, with science leading the charge, we have taken another important step forward in our nation’s fight against the virus that causes COVID-19. We know millions of parents are eager to get their children vaccinated and with this decision, we now have recommended that about 28 million children receive a COVID-19 vaccine. As a mom, I encourage parents with questions to talk to their pediatrician, school nurse, or local pharmacist to learn more about the vaccine and the importance of getting their children vaccinated,” Dr. Walensky said in a prepared statement.
President Joe Biden applauded Dr. Walensky’s endorsement: “Today, we have reached a turning point in our battle against COVID-19: authorization of a safe, effective vaccine for children age 5 to 11. It will allow parents to end months of anxious worrying about their kids, and reduce the extent to which children spread the virus to others. It is a major step forward for our nation in our fight to defeat the virus,” he said in a statement.
The 14 members of the Advisory Committee on Immunization Practices (ACIP) voted unanimously earlier in the day to recommend the vaccine for kids.
“I feel like I have a responsibility to make this vaccine available to children and their parents,” said committee member Beth Bell, MD, MPH, a clinical professor at the University of Washington in Seattle. Bell noted that all evidence the committee had reviewed pointed to a vaccine that was safe and effective for younger children.
“If I had a grandchild, I would certainly get that grandchild vaccinated as soon as possible,” she said.
Their recommendations follow the U.S. Food and Drug Administration’s emergency authorization of Pfizer-BioNTech’s vaccine for this same age group last week.
“I’m voting for this because I think it could have a huge positive impact on [kids’] health and their social and emotional wellbeing,” said Grace Lee, MD, a professor of pediatrics at Stanford University School of Medicine, who chairs the CDC’s ACIP.
She noted that, though masks are available to reduce the risk for kids, they aren’t perfect and transmission still occurs.
“Vaccines are really the only consistent and reliable way to provide that protection,” Lee said.
The vaccine for children is two doses given 3 weeks apart. Each dose is 10 micrograms, which is one-third of the dose used in adults and teens.
To avoid confusion, the smaller dose for kids will come in bottles with orange labels and orange tops. The vaccine for adults is packaged in purple.
The CDC also addressed the question of kids who are close to age 12 when they get their first dose.
In general, pediatricians allow for a 4-day grace period around birthdays to determine which dose is needed. That will be the same with the COVID-19 vaccine.
For kids who are 11 when they start the series, they should get another 10-microgram dose after they turn 12 a few weeks later.
COVID-19 cases in this age group have climbed sharply over the summer and into the fall as schools have fully reopened, sometimes without the benefit of masks.
In the first week of October, roughly 10% of all COVID-19 cases recorded in the United States were among children ages 5 through 11. Since the start of pandemic, about 1.9 million children in this age group have been infected, though that’s almost certainly an undercount. More than 8,300 have been hospitalized, and 94 children have died.
Children of color have been disproportionately impacted. More than two-thirds of hospitalized children have been black or Hispanic.
Weighing benefits and risks
In clinical trials that included more than 4,600 children, the most common adverse events were pain and swelling at the injection site. They could also have side effects like fevers, fatigue, headache, chills, and sometimes swollen lymph nodes.
These kinds of side effects appear to be less common in children ages 5 to 11 than they have been in teens and adults, and they were temporary.
No cases of myocarditis or pericarditis were seen in the studies, but myocarditis is a very rare side effect, and the studies were too small to pick up these cases.
Still, doctors say they’re watching for it. In general, the greatest risk for myocarditis after vaccination has been seen in younger males between the ages of 12 and 30.
Even without COVID-19 or vaccines in the mix, doctors expect to see as many as two cases of myocarditis for every million people over the course of a week. The risk for myocarditis jumps up to about 11 cases for every million doses of mRNA vaccine given to men ages 25 to 30. It’s between 37 and 69 cases per million doses in boys between the ages of 12 and 24.
Still, experts say the possibility of this rare risk shouldn’t deter parents from vaccinating younger children.
Here’s why: The risk for myocarditis is higher after COVID-19 infection than after vaccination. Younger children have a lower risk for myocarditis than teens and young adults, suggesting that this side effect may be less frequent in this age group, although that remains to be seen.
Additionally, the smaller dose authorized for children is expected to minimize the risk for myocarditis even further.
The CDC says parents should call their doctor if a child develops pain in their chest, has trouble breathing, or feels like they have a beating or fluttering heart after vaccination.
What about benefits?
Models looking at the impact of vaccines in this age group predict that, nationally, cases would drop by about 8% if children are vaccinated.
The models also suggested that vaccination of kids this age would slow — but not stop — the emergence of new variants.
For every million doses, the CDC’s modeling predicts that more than 56,000 COVID-19 infections would be prevented in this age group, along with dozens of hospitalizations, and post-COVID conditions like multisystem inflammatory syndrome in children.
CDC experts estimate that just 10 kids would need to be vaccinated over 6 months to prevent a single case of COVID-19.
The CDC pointed out that vaccinating kids may help slow transmission of the virus and would give parents and other caregivers greater confidence in participating in school and extracurricular activities.
CDC experts said they would use a variety of systems, including hospital networks, the open Vaccines and Adverse Events Reporting System (VAERS) database, the cell-phone based V-SAFE app, and insurance claims databases to keep an eye out for any rare adverse events related to the vaccines in children.
This article, a version of which first appeared on Medscape.com, was updated on Nov. 3, 2021.
– meaning the shots are now available for immediate use.
The Nov. 2 decision came mere hours after experts that advise the CDC on vaccinations strongly recommended the vaccine for this age group.
“Together, with science leading the charge, we have taken another important step forward in our nation’s fight against the virus that causes COVID-19. We know millions of parents are eager to get their children vaccinated and with this decision, we now have recommended that about 28 million children receive a COVID-19 vaccine. As a mom, I encourage parents with questions to talk to their pediatrician, school nurse, or local pharmacist to learn more about the vaccine and the importance of getting their children vaccinated,” Dr. Walensky said in a prepared statement.
President Joe Biden applauded Dr. Walensky’s endorsement: “Today, we have reached a turning point in our battle against COVID-19: authorization of a safe, effective vaccine for children age 5 to 11. It will allow parents to end months of anxious worrying about their kids, and reduce the extent to which children spread the virus to others. It is a major step forward for our nation in our fight to defeat the virus,” he said in a statement.
The 14 members of the Advisory Committee on Immunization Practices (ACIP) voted unanimously earlier in the day to recommend the vaccine for kids.
“I feel like I have a responsibility to make this vaccine available to children and their parents,” said committee member Beth Bell, MD, MPH, a clinical professor at the University of Washington in Seattle. Bell noted that all evidence the committee had reviewed pointed to a vaccine that was safe and effective for younger children.
“If I had a grandchild, I would certainly get that grandchild vaccinated as soon as possible,” she said.
Their recommendations follow the U.S. Food and Drug Administration’s emergency authorization of Pfizer-BioNTech’s vaccine for this same age group last week.
“I’m voting for this because I think it could have a huge positive impact on [kids’] health and their social and emotional wellbeing,” said Grace Lee, MD, a professor of pediatrics at Stanford University School of Medicine, who chairs the CDC’s ACIP.
She noted that, though masks are available to reduce the risk for kids, they aren’t perfect and transmission still occurs.
“Vaccines are really the only consistent and reliable way to provide that protection,” Lee said.
The vaccine for children is two doses given 3 weeks apart. Each dose is 10 micrograms, which is one-third of the dose used in adults and teens.
To avoid confusion, the smaller dose for kids will come in bottles with orange labels and orange tops. The vaccine for adults is packaged in purple.
The CDC also addressed the question of kids who are close to age 12 when they get their first dose.
In general, pediatricians allow for a 4-day grace period around birthdays to determine which dose is needed. That will be the same with the COVID-19 vaccine.
For kids who are 11 when they start the series, they should get another 10-microgram dose after they turn 12 a few weeks later.
COVID-19 cases in this age group have climbed sharply over the summer and into the fall as schools have fully reopened, sometimes without the benefit of masks.
In the first week of October, roughly 10% of all COVID-19 cases recorded in the United States were among children ages 5 through 11. Since the start of pandemic, about 1.9 million children in this age group have been infected, though that’s almost certainly an undercount. More than 8,300 have been hospitalized, and 94 children have died.
Children of color have been disproportionately impacted. More than two-thirds of hospitalized children have been black or Hispanic.
Weighing benefits and risks
In clinical trials that included more than 4,600 children, the most common adverse events were pain and swelling at the injection site. They could also have side effects like fevers, fatigue, headache, chills, and sometimes swollen lymph nodes.
These kinds of side effects appear to be less common in children ages 5 to 11 than they have been in teens and adults, and they were temporary.
No cases of myocarditis or pericarditis were seen in the studies, but myocarditis is a very rare side effect, and the studies were too small to pick up these cases.
Still, doctors say they’re watching for it. In general, the greatest risk for myocarditis after vaccination has been seen in younger males between the ages of 12 and 30.
Even without COVID-19 or vaccines in the mix, doctors expect to see as many as two cases of myocarditis for every million people over the course of a week. The risk for myocarditis jumps up to about 11 cases for every million doses of mRNA vaccine given to men ages 25 to 30. It’s between 37 and 69 cases per million doses in boys between the ages of 12 and 24.
Still, experts say the possibility of this rare risk shouldn’t deter parents from vaccinating younger children.
Here’s why: The risk for myocarditis is higher after COVID-19 infection than after vaccination. Younger children have a lower risk for myocarditis than teens and young adults, suggesting that this side effect may be less frequent in this age group, although that remains to be seen.
Additionally, the smaller dose authorized for children is expected to minimize the risk for myocarditis even further.
The CDC says parents should call their doctor if a child develops pain in their chest, has trouble breathing, or feels like they have a beating or fluttering heart after vaccination.
What about benefits?
Models looking at the impact of vaccines in this age group predict that, nationally, cases would drop by about 8% if children are vaccinated.
The models also suggested that vaccination of kids this age would slow — but not stop — the emergence of new variants.
For every million doses, the CDC’s modeling predicts that more than 56,000 COVID-19 infections would be prevented in this age group, along with dozens of hospitalizations, and post-COVID conditions like multisystem inflammatory syndrome in children.
CDC experts estimate that just 10 kids would need to be vaccinated over 6 months to prevent a single case of COVID-19.
The CDC pointed out that vaccinating kids may help slow transmission of the virus and would give parents and other caregivers greater confidence in participating in school and extracurricular activities.
CDC experts said they would use a variety of systems, including hospital networks, the open Vaccines and Adverse Events Reporting System (VAERS) database, the cell-phone based V-SAFE app, and insurance claims databases to keep an eye out for any rare adverse events related to the vaccines in children.
This article, a version of which first appeared on Medscape.com, was updated on Nov. 3, 2021.
– meaning the shots are now available for immediate use.
The Nov. 2 decision came mere hours after experts that advise the CDC on vaccinations strongly recommended the vaccine for this age group.
“Together, with science leading the charge, we have taken another important step forward in our nation’s fight against the virus that causes COVID-19. We know millions of parents are eager to get their children vaccinated and with this decision, we now have recommended that about 28 million children receive a COVID-19 vaccine. As a mom, I encourage parents with questions to talk to their pediatrician, school nurse, or local pharmacist to learn more about the vaccine and the importance of getting their children vaccinated,” Dr. Walensky said in a prepared statement.
President Joe Biden applauded Dr. Walensky’s endorsement: “Today, we have reached a turning point in our battle against COVID-19: authorization of a safe, effective vaccine for children age 5 to 11. It will allow parents to end months of anxious worrying about their kids, and reduce the extent to which children spread the virus to others. It is a major step forward for our nation in our fight to defeat the virus,” he said in a statement.
The 14 members of the Advisory Committee on Immunization Practices (ACIP) voted unanimously earlier in the day to recommend the vaccine for kids.
“I feel like I have a responsibility to make this vaccine available to children and their parents,” said committee member Beth Bell, MD, MPH, a clinical professor at the University of Washington in Seattle. Bell noted that all evidence the committee had reviewed pointed to a vaccine that was safe and effective for younger children.
“If I had a grandchild, I would certainly get that grandchild vaccinated as soon as possible,” she said.
Their recommendations follow the U.S. Food and Drug Administration’s emergency authorization of Pfizer-BioNTech’s vaccine for this same age group last week.
“I’m voting for this because I think it could have a huge positive impact on [kids’] health and their social and emotional wellbeing,” said Grace Lee, MD, a professor of pediatrics at Stanford University School of Medicine, who chairs the CDC’s ACIP.
She noted that, though masks are available to reduce the risk for kids, they aren’t perfect and transmission still occurs.
“Vaccines are really the only consistent and reliable way to provide that protection,” Lee said.
The vaccine for children is two doses given 3 weeks apart. Each dose is 10 micrograms, which is one-third of the dose used in adults and teens.
To avoid confusion, the smaller dose for kids will come in bottles with orange labels and orange tops. The vaccine for adults is packaged in purple.
The CDC also addressed the question of kids who are close to age 12 when they get their first dose.
In general, pediatricians allow for a 4-day grace period around birthdays to determine which dose is needed. That will be the same with the COVID-19 vaccine.
For kids who are 11 when they start the series, they should get another 10-microgram dose after they turn 12 a few weeks later.
COVID-19 cases in this age group have climbed sharply over the summer and into the fall as schools have fully reopened, sometimes without the benefit of masks.
In the first week of October, roughly 10% of all COVID-19 cases recorded in the United States were among children ages 5 through 11. Since the start of pandemic, about 1.9 million children in this age group have been infected, though that’s almost certainly an undercount. More than 8,300 have been hospitalized, and 94 children have died.
Children of color have been disproportionately impacted. More than two-thirds of hospitalized children have been black or Hispanic.
Weighing benefits and risks
In clinical trials that included more than 4,600 children, the most common adverse events were pain and swelling at the injection site. They could also have side effects like fevers, fatigue, headache, chills, and sometimes swollen lymph nodes.
These kinds of side effects appear to be less common in children ages 5 to 11 than they have been in teens and adults, and they were temporary.
No cases of myocarditis or pericarditis were seen in the studies, but myocarditis is a very rare side effect, and the studies were too small to pick up these cases.
Still, doctors say they’re watching for it. In general, the greatest risk for myocarditis after vaccination has been seen in younger males between the ages of 12 and 30.
Even without COVID-19 or vaccines in the mix, doctors expect to see as many as two cases of myocarditis for every million people over the course of a week. The risk for myocarditis jumps up to about 11 cases for every million doses of mRNA vaccine given to men ages 25 to 30. It’s between 37 and 69 cases per million doses in boys between the ages of 12 and 24.
Still, experts say the possibility of this rare risk shouldn’t deter parents from vaccinating younger children.
Here’s why: The risk for myocarditis is higher after COVID-19 infection than after vaccination. Younger children have a lower risk for myocarditis than teens and young adults, suggesting that this side effect may be less frequent in this age group, although that remains to be seen.
Additionally, the smaller dose authorized for children is expected to minimize the risk for myocarditis even further.
The CDC says parents should call their doctor if a child develops pain in their chest, has trouble breathing, or feels like they have a beating or fluttering heart after vaccination.
What about benefits?
Models looking at the impact of vaccines in this age group predict that, nationally, cases would drop by about 8% if children are vaccinated.
The models also suggested that vaccination of kids this age would slow — but not stop — the emergence of new variants.
For every million doses, the CDC’s modeling predicts that more than 56,000 COVID-19 infections would be prevented in this age group, along with dozens of hospitalizations, and post-COVID conditions like multisystem inflammatory syndrome in children.
CDC experts estimate that just 10 kids would need to be vaccinated over 6 months to prevent a single case of COVID-19.
The CDC pointed out that vaccinating kids may help slow transmission of the virus and would give parents and other caregivers greater confidence in participating in school and extracurricular activities.
CDC experts said they would use a variety of systems, including hospital networks, the open Vaccines and Adverse Events Reporting System (VAERS) database, the cell-phone based V-SAFE app, and insurance claims databases to keep an eye out for any rare adverse events related to the vaccines in children.
This article, a version of which first appeared on Medscape.com, was updated on Nov. 3, 2021.
FDA class I recall of CardioSave hybrid/rescue IABPs
Datascope/Getinge/Maquet is recalling CardioSave Hybrid and Rescue intra-aortic balloon pumps (IABPs) because some battery packs may have a shortened run time and fail unexpectedly, according to a medical device recall notice posted on the U.S. Food and Drug Administration website.
The FDA has identified this as a class I recall, the most serious type of recall, because of the risk for serious injury or death.
The recalled IABPs have substandard batteries that do not meet performance specifications and were mistakenly released to a limited number of customers.
If a patient requires life-supporting therapy with an IABP and the device does not work or stops working during use because of battery failure, the patient will be at risk for serious injury, including death, the FDA cautions.
Both IABP monitors display battery life and have low battery alarms when alternative power sources are needed.
Datascope/Getting/Maquet has received six complaints but no reports of injury or death related to this issue.
“However, there is a potential for underreporting since the end user reporting a failed battery or short battery run time cannot be aware that they originally received a substandard battery,” the FDA said.
The recall involves 137 battery packs distributed in the United States between Sept. 23, 2017, and Aug. 17, 2021. Product codes and lot numbers are available in the recall notice.
The company sent an urgent medical device removal letter to customers requesting that they check inventory to determine if there are any CardioSave LiIon battery packs with part number/reference number 0146-00-0097 and with serial numbers listed in the letter.
Customers are asked to replace any affected battery with an unaffected battery and remove the affected product from areas of use.
The company will issue credit or a replacement battery at no cost to the facility upon receipt of the response form attached to the letter.
Distributors who shipped any affected product to customers are asked to forward the device removal letter to customers.
All customers, regardless of whether or not they have defective batteries, are asked to complete and sign the response form to acknowledge that they received the notification and disposed of the affected batteries.
Completed forms can be scanned and emailed to Datascope/Getinge/Maquet at [email protected] or by FAX to 1-877-446-3360.
Customers who have questions about this recall should contact their Datascope/Getinge/Maquet sales representative or, for technical questions, customer service (1-888-943-8872, option 2), Monday through Friday, 8:00 a.m. to 6:00 p.m. ET.
Any adverse events or suspected adverse events related to the recalled CardioSave Hybrid/Rescue IABPs should be reported to the FDA through MedWatch, its adverse event reporting program.
A version of this article first appeared on Medscape.com.
Datascope/Getinge/Maquet is recalling CardioSave Hybrid and Rescue intra-aortic balloon pumps (IABPs) because some battery packs may have a shortened run time and fail unexpectedly, according to a medical device recall notice posted on the U.S. Food and Drug Administration website.
The FDA has identified this as a class I recall, the most serious type of recall, because of the risk for serious injury or death.
The recalled IABPs have substandard batteries that do not meet performance specifications and were mistakenly released to a limited number of customers.
If a patient requires life-supporting therapy with an IABP and the device does not work or stops working during use because of battery failure, the patient will be at risk for serious injury, including death, the FDA cautions.
Both IABP monitors display battery life and have low battery alarms when alternative power sources are needed.
Datascope/Getting/Maquet has received six complaints but no reports of injury or death related to this issue.
“However, there is a potential for underreporting since the end user reporting a failed battery or short battery run time cannot be aware that they originally received a substandard battery,” the FDA said.
The recall involves 137 battery packs distributed in the United States between Sept. 23, 2017, and Aug. 17, 2021. Product codes and lot numbers are available in the recall notice.
The company sent an urgent medical device removal letter to customers requesting that they check inventory to determine if there are any CardioSave LiIon battery packs with part number/reference number 0146-00-0097 and with serial numbers listed in the letter.
Customers are asked to replace any affected battery with an unaffected battery and remove the affected product from areas of use.
The company will issue credit or a replacement battery at no cost to the facility upon receipt of the response form attached to the letter.
Distributors who shipped any affected product to customers are asked to forward the device removal letter to customers.
All customers, regardless of whether or not they have defective batteries, are asked to complete and sign the response form to acknowledge that they received the notification and disposed of the affected batteries.
Completed forms can be scanned and emailed to Datascope/Getinge/Maquet at [email protected] or by FAX to 1-877-446-3360.
Customers who have questions about this recall should contact their Datascope/Getinge/Maquet sales representative or, for technical questions, customer service (1-888-943-8872, option 2), Monday through Friday, 8:00 a.m. to 6:00 p.m. ET.
Any adverse events or suspected adverse events related to the recalled CardioSave Hybrid/Rescue IABPs should be reported to the FDA through MedWatch, its adverse event reporting program.
A version of this article first appeared on Medscape.com.
Datascope/Getinge/Maquet is recalling CardioSave Hybrid and Rescue intra-aortic balloon pumps (IABPs) because some battery packs may have a shortened run time and fail unexpectedly, according to a medical device recall notice posted on the U.S. Food and Drug Administration website.
The FDA has identified this as a class I recall, the most serious type of recall, because of the risk for serious injury or death.
The recalled IABPs have substandard batteries that do not meet performance specifications and were mistakenly released to a limited number of customers.
If a patient requires life-supporting therapy with an IABP and the device does not work or stops working during use because of battery failure, the patient will be at risk for serious injury, including death, the FDA cautions.
Both IABP monitors display battery life and have low battery alarms when alternative power sources are needed.
Datascope/Getting/Maquet has received six complaints but no reports of injury or death related to this issue.
“However, there is a potential for underreporting since the end user reporting a failed battery or short battery run time cannot be aware that they originally received a substandard battery,” the FDA said.
The recall involves 137 battery packs distributed in the United States between Sept. 23, 2017, and Aug. 17, 2021. Product codes and lot numbers are available in the recall notice.
The company sent an urgent medical device removal letter to customers requesting that they check inventory to determine if there are any CardioSave LiIon battery packs with part number/reference number 0146-00-0097 and with serial numbers listed in the letter.
Customers are asked to replace any affected battery with an unaffected battery and remove the affected product from areas of use.
The company will issue credit or a replacement battery at no cost to the facility upon receipt of the response form attached to the letter.
Distributors who shipped any affected product to customers are asked to forward the device removal letter to customers.
All customers, regardless of whether or not they have defective batteries, are asked to complete and sign the response form to acknowledge that they received the notification and disposed of the affected batteries.
Completed forms can be scanned and emailed to Datascope/Getinge/Maquet at [email protected] or by FAX to 1-877-446-3360.
Customers who have questions about this recall should contact their Datascope/Getinge/Maquet sales representative or, for technical questions, customer service (1-888-943-8872, option 2), Monday through Friday, 8:00 a.m. to 6:00 p.m. ET.
Any adverse events or suspected adverse events related to the recalled CardioSave Hybrid/Rescue IABPs should be reported to the FDA through MedWatch, its adverse event reporting program.
A version of this article first appeared on Medscape.com.
COVID-19 vaccines provide 5 times the protection of natural immunity, CDC study says
, according to a new study published recently in the CDC’s Morbidity and Mortality Weekly Report.
The research team concluded that vaccination can provide a higher, stronger, and more consistent level of immunity against COVID-19 hospitalization than infection alone for at least six months.
“We now have additional evidence that reaffirms the importance of COVID-19 vaccines, even if you have had prior infection,” Rochelle Walensky, MD, director of the CDC, said in a statement.
“This study adds more to the body of knowledge demonstrating the protection of vaccines against severe disease from COVID-19,” she said. “The best way to stop COVID-19, including the emergence of variants, is with widespread COVID-19 vaccination and with disease prevention actions such as mask wearing, washing hands often, physical distancing and staying home when sick.”
Researchers looked at data from the VISION Network, which included more than 201,000 hospitalizations for COVID-like illness at 187 hospitals across nine states between Jan. 1 to Sept. 2. Among those, more than 94,000 had rapid testing for the coronavirus, and 7,300 had a lab-confirmed test for COVID-19.
The research team found that unvaccinated people with a prior infection within 3 to 6 months were about 5-1/2 times more likely to have laboratory-confirmed COVID-19 than those who were fully vaccinated within 3 to 6 months with the Pfizer or Moderna shots. They found similar results when looking at the months that the Delta variant was the dominant strain of the coronavirus.
Protection from the Moderna vaccine “appeared to be higher” than for the Pfizer vaccine, the study authors wrote. The boost in protection also “trended higher” among older adults, as compared to those under age 65.
Importantly, the research team noted, these estimates may change over time as immunity wanes. Future studies should consider infection-induced and vaccine-induced immunity as time passes during the pandemic, they wrote.
Additional research is also needed for the Johnson & Johnson vaccine, they wrote. Those who have received the Johnson & Johnson vaccine are currently recommended to receive a booster shot at least two months after the first shot.
Overall, “all eligible persons should be vaccinated against COVID-19 as soon as possible, including unvaccinated persons previously infected,” the research team concluded.
A version of this article first appeared on WebMD.com.
, according to a new study published recently in the CDC’s Morbidity and Mortality Weekly Report.
The research team concluded that vaccination can provide a higher, stronger, and more consistent level of immunity against COVID-19 hospitalization than infection alone for at least six months.
“We now have additional evidence that reaffirms the importance of COVID-19 vaccines, even if you have had prior infection,” Rochelle Walensky, MD, director of the CDC, said in a statement.
“This study adds more to the body of knowledge demonstrating the protection of vaccines against severe disease from COVID-19,” she said. “The best way to stop COVID-19, including the emergence of variants, is with widespread COVID-19 vaccination and with disease prevention actions such as mask wearing, washing hands often, physical distancing and staying home when sick.”
Researchers looked at data from the VISION Network, which included more than 201,000 hospitalizations for COVID-like illness at 187 hospitals across nine states between Jan. 1 to Sept. 2. Among those, more than 94,000 had rapid testing for the coronavirus, and 7,300 had a lab-confirmed test for COVID-19.
The research team found that unvaccinated people with a prior infection within 3 to 6 months were about 5-1/2 times more likely to have laboratory-confirmed COVID-19 than those who were fully vaccinated within 3 to 6 months with the Pfizer or Moderna shots. They found similar results when looking at the months that the Delta variant was the dominant strain of the coronavirus.
Protection from the Moderna vaccine “appeared to be higher” than for the Pfizer vaccine, the study authors wrote. The boost in protection also “trended higher” among older adults, as compared to those under age 65.
Importantly, the research team noted, these estimates may change over time as immunity wanes. Future studies should consider infection-induced and vaccine-induced immunity as time passes during the pandemic, they wrote.
Additional research is also needed for the Johnson & Johnson vaccine, they wrote. Those who have received the Johnson & Johnson vaccine are currently recommended to receive a booster shot at least two months after the first shot.
Overall, “all eligible persons should be vaccinated against COVID-19 as soon as possible, including unvaccinated persons previously infected,” the research team concluded.
A version of this article first appeared on WebMD.com.
, according to a new study published recently in the CDC’s Morbidity and Mortality Weekly Report.
The research team concluded that vaccination can provide a higher, stronger, and more consistent level of immunity against COVID-19 hospitalization than infection alone for at least six months.
“We now have additional evidence that reaffirms the importance of COVID-19 vaccines, even if you have had prior infection,” Rochelle Walensky, MD, director of the CDC, said in a statement.
“This study adds more to the body of knowledge demonstrating the protection of vaccines against severe disease from COVID-19,” she said. “The best way to stop COVID-19, including the emergence of variants, is with widespread COVID-19 vaccination and with disease prevention actions such as mask wearing, washing hands often, physical distancing and staying home when sick.”
Researchers looked at data from the VISION Network, which included more than 201,000 hospitalizations for COVID-like illness at 187 hospitals across nine states between Jan. 1 to Sept. 2. Among those, more than 94,000 had rapid testing for the coronavirus, and 7,300 had a lab-confirmed test for COVID-19.
The research team found that unvaccinated people with a prior infection within 3 to 6 months were about 5-1/2 times more likely to have laboratory-confirmed COVID-19 than those who were fully vaccinated within 3 to 6 months with the Pfizer or Moderna shots. They found similar results when looking at the months that the Delta variant was the dominant strain of the coronavirus.
Protection from the Moderna vaccine “appeared to be higher” than for the Pfizer vaccine, the study authors wrote. The boost in protection also “trended higher” among older adults, as compared to those under age 65.
Importantly, the research team noted, these estimates may change over time as immunity wanes. Future studies should consider infection-induced and vaccine-induced immunity as time passes during the pandemic, they wrote.
Additional research is also needed for the Johnson & Johnson vaccine, they wrote. Those who have received the Johnson & Johnson vaccine are currently recommended to receive a booster shot at least two months after the first shot.
Overall, “all eligible persons should be vaccinated against COVID-19 as soon as possible, including unvaccinated persons previously infected,” the research team concluded.
A version of this article first appeared on WebMD.com.