User login
Acute monocular vision loss: Don’t lose sight of the differential
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.

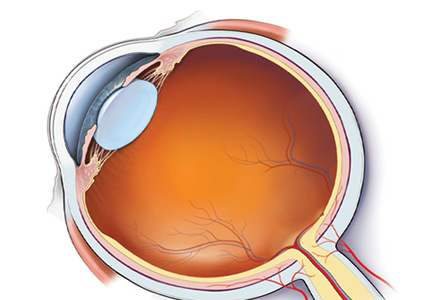
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history

A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.

An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations

A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).

For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
An 83-year-old man presented to the emergency department with acute, painless loss of vision in his left eye. His vision in that eye had been normal in the middle of the night when he woke to use the restroom, but on awakening 6 hours later he could perceive only light or darkness.
He denied headache, scalp tenderness, jaw claudication, fever, weight loss, myalgia, or other neurologic symptoms. He had not experienced any recent change in his vision before this presentation, including halos around lights, floaters, eye pain, or redness. However, 6 months ago he had undergone left cataract surgery (left phacoemulsification with intraocular implant) without complications. And he said that when he was 3 years old, he had sustained a serious injury to his right eye.
His medical history included ischemic heart disease and hypertension. His medications included losartan, furosemide, amlodipine, atorvastatin, and aspirin.
CAUSES OF ACUTE MONOCULAR VISION LOSS
1. Which of the following is the least likely cause of this patient’s acute monocular vision loss?
- Optic neuritis
- Retinal vein occlusion
- Retinal artery occlusion
- Pituitary apoplexy
- Retinal detachment
Acute vision loss is often so distressing to the patient that the emergency department may be the first step in evaluation. While its diagnosis and management often require an interdisciplinary effort, early evaluation and triage of this potential medical emergency is often done by clinicians without specialized training in ophthalmology.
The physiology of vision is complex and the list of possible causes of vision loss is long, but the differential diagnosis can be narrowed quickly by considering the time course of vision loss and the anatomic localization.1
The time course (including onset and tempo) of vision loss can classified as:
- Transient (ie, vision returned to normal by the time seen by clinician)
- Acute (instantaneous onset, ie, within seconds to minutes)
- Subacute (progression over days to weeks)
- Chronic (insidious progression over months to years).
Although acute vision loss is usually dramatic, insidious vision loss may occasionally be unnoticed for a surprisingly long time until the normal eye is inadvertently shielded.
Anatomic localization. Lesions anterior to the optic chiasm cause monocular vision loss, whereas lesions at or posterior to the chiasm lead to bilateral visual field defects. Problems leading to monocular blindness can be broadly divided into 3 anatomic categories (Figure 1):
- Ocular medial (including the cornea, anterior chamber, and lens)
- Retinal
- Neurologic (including the optic nerve and chiasm).
Clues from the history
A careful ophthalmic history is an essential initial step in the evaluation (Table 1). In addition, nonvisual symptoms can help narrow the differential diagnosis.
Nausea and vomiting often accompany acute elevation of intraocular pressure.
Focal neurologic deficits or other neurologic symptoms can point to a demyelinating disease such as multiple sclerosis.
Risk factors for vascular atherosclerotic disease such as diabetes, hypertension, and coronary artery disease raise concern for retinal, optic nerve, or cerebral ischemia.
Medications with anticholinergic and adrenergic properties can also precipitate monocular vision loss with acute angle-closure glaucoma.
Can we rule out anything yet?
Our patient presented with painless monocular vision loss. As discussed, causes of monocular vision loss can be localized to ocular abnormalities and prechiasmatic neurologic ones. Retinal detachment, occlusion of a retinal artery or vein, and optic neuritis are all important potential causes of acute monocular vision loss.
Pituitary apoplexy, on the other hand, is characterized by an acute increase in pituitary volume, often leading to compression of the optic chiasm resulting in a visual-field defect. It is most often characterized by binocular deficits (eg, bitemporal hemianopia) but is less likely to cause monocular vision loss.1
CASE CONTINUED: EXAMINATION
On examination, the patient appeared comfortable. His temperature was 97.6°F (36.4°C), pulse 59 beats per minute, respiratory rate 18 per minute, and blood pressure 153/56 mm Hg.
Heart and lung examinations were notable for a grade 3 of 6 midsystolic, low-pitched murmur in the aortic area radiating to the neck, bilateral carotid bruits, and clear lungs. The cardiac impulse was normal in location and character. There was no evidence of aortic insufficiency (including auscultation during exhalation phase while sitting upright).
Eye examination. Visual acuity in the right eye was 20/200 with correction (owing to his eye injury at age 3). With the left eye, he could see only light or darkness. The conjunctiva and sclera were normal.
The right pupil was irregular and measured 3 mm (baseline from his previous eye injury). The left pupil was 3.5 mm. The direct pupillary response was preserved, but a relative afferent pupillary defect was present: on the swinging flashlight test, the left pupil dilated when the flashlight was passed from the right to the left pupil. Extraocular movements were full and intact bilaterally. The rest of the neurologic examination was normal.
An ophthalmologist was urgently consulted. A dilated funduscopic examination of the left eye revealed peripapillary atrophy, tortuous vessels, a cherry red macular spot, and flame hemorrhages, but no disc edema or pallor (Figure 2).
FURTHER WORKUP
2. Which of the following investigations would be least useful and not indicated at this point for this patient?
- Carotid ultrasonography
- Electrocardiography and echocardiography
- Magnetic resonance angiography of the brain
- Computed tomographic (CT) angiography of the head and neck
- Testing for the factor V Leiden and prothrombin gene mutations
A systematic ocular physical examination can offer important diagnostic information (Table 2). Ophthalmoscopy directly examines the optic disc, macula, and retinal vasculature. To interpret the funduscopic examination, we need a basic understanding of the vascular supply to the eye (Figure 3).
For example, the cherry red spot within the macula in our patient is characteristic of central retinal artery occlusion and highlights the relationship between anatomy and pathophysiology. The retina’s blood supply is compromised, leading to an ischemic, white background (secondary to edema of the inner third of the retina), but the macula continues to be nourished by the posterior ciliary arteries. This contrast in color is accentuated by the underlying structures composing the fovea, which lacks the nerve fiber layer and ganglion cell layer, making the vascular bed more visible.2,3
Also in our patient, the marked reduction in visual acuity and relative afferent pupillary defect in the left eye point to unilateral optic nerve (or retinal ganglion cell) dysfunction. The findings on direct funduscopy were consistent with acute central retinal artery ischemia or occlusion. Central retinal artery occlusion can be either arteritic (due to inflammation, most often giant cell arteritis) or nonarteritic (due to atherosclerotic vascular disease).
Thus, carotid ultrasonography, electrocardiography, and transthoracic and transesophageal echocardiography are important components of the further workup. In addition, urgent brain imaging including either CT angiography or magnetic resonance angiography of the head and neck is indicated in all patients with central retinal artery occlusion.
Thrombophilia testing, including tests for the factor V Leiden and prothrombin gene mutations, is indicated in specific cases when a hypercoagulable state is suggested by components of the history, physical examination, and laboratory and radiologic testing. Thrombophilia testing would be low-yield and should not be part of the first-line testing in elderly patients with several atherosclerotic risk factors, such as our patient.
CASE CONTINUED: LABORATORY AND IMAGING EVIDENCE
Initial laboratory work showed:
- Mild microcytic anemia
- Erythrocyte sedimentation rate 77 mm/hour (reference range 1–10)
- C-reactive protein 4.0 mg/dL (reference range < 0.9).
The rest of the complete blood cell count and metabolic profile were unremarkable. His hemoglobin A1c value was 5.3% (reference range 4.8%–6.2%).
A neurologist was urgently consulted.
Magnetic resonance imaging of the brain without contrast revealed nonspecific white-matter disease with no evidence of ischemic stroke.
Magnetic resonance angiography of the head and neck with contrast demonstrated 20% to 40% stenosis in both carotid arteries with otherwise patent anterior and posterior circulation.
Continuous monitoring of the left carotid artery with transcranial Doppler ultrasonography was also ordered, and the study concluded there were no undetected microembolic events.
Transthoracic echocardiography showed aortic sclerosis with no other abnormalities.
Ophthalmic fluorescein angiography was performed and showed patchy choroidal hypoperfusion, severe delayed filling, and extensive pruning of the arterial circulation with no involvement of the posterior ciliary arteries.
Given the elevated inflammatory markers, pulse-dose intravenous methylprednisolone was started, and a temporal artery biopsy was planned.
CENTRAL RETINAL ARTERY OCCLUSION: NONARTERITIC VS ARTERITIC CAUSES
3. Which of the following is least useful to differentiate arteritic from nonarteritic causes of central retinal artery occlusion?
- Finding emboli in the retinal vasculature on funduscopy
- Temporal artery biopsy
- Measuring the C-reactive protein level and the erythrocyte sedimentation rate
- Echocardiography
- Positron-emission tomography (PET)
- Retinal fluorescein angiography
In patients diagnosed with central retinal artery occlusion, the next step is to differentiate between nonarteritic and arteritic causes, since separating them has therapeutic relevance.
The carotid artery is the main culprit for embolic disease affecting the central retinal artery, leading to the nonarteritic subtype. Thus, evaluation of acute retinal ischemia secondary to nonarteritic central retinal artery occlusion is similar to the evaluation of patients with an acute cerebral stroke.4 Studies have shown that 25% of patients diagnosed with central retinal artery occlusion have an additional ischemic insult in the cerebrovascular system, and these patients are at high risk of recurrent ocular or cerebral infarction. Workup includes diffusion-weighted MRI, angiography, echocardiography, and telemetry.5
Arteritic central retinal artery occlusion is most often caused by giant cell arteritis. The American College of Rheumatology classification criteria for giant cell arteritis include 3 of the following 5:
- Age 50 or older
- New onset of localized headache
- Temporal artery tenderness or decreased temporal artery pulse
- Erythrocyte sedimentation rate 50 mm/hour or greater
- Positive biopsy findings.6
Temporal artery biopsy is the gold standard for the diagnosis of giant cell arteritis and should be done whenever the disease is suspected.7,8 However, the test is invasive and imperfect, as a negative result does not completely rule out giant cell arteritis.9
Although a unilateral temporal artery biopsy can be falsely negative, several studies evaluating the efficacy of bilateral biopsies did not show significant improvement in the diagnostic yield.10,11
Ophthalmic fluorescein angiography is another helpful test for distinguishing nonarteritic from arteritic central retinal artery occlusion.12 Involvement of the posterior ciliary arteries usually occurs in giant cell arteritis, and this leads to choroidal malperfusion with or without retinal involvement. The optic nerve may also be infarcted by closure of the paraoptic vessels fed by the posterior ciliary vessels.12,13 Such involvement of multiple vessels would not be typical with nonarteritic central retinal artery occlusion. Thus, this finding is helpful in making the final diagnosis along with supplying possible prognostic information.13
PET-CT is emerging as a test for early inflammation in extracranial disease, but its utility for diagnosing intracranial disease is limited by high uptake of the tracer fluorodeoxyglucose by the brain and low resolution.14 Currently, it has no established role in the evaluation of patients with central retinal artery occlusion and would have no utility in differentiating arteritic vs nonarteritic causes of central retinal artery occlusion.
If giant cell arteritis is suspected, it is essential to start intravenous pulse-dose methylprednisolone early to prevent further vision loss in the contralateral eye. Treatment should not be delayed for invasive testing or temporal artery biopsy. Improvement in headache, jaw claudication, or scalp tenderness once steroids are initiated also helps support the diagnosis of giant cell arteritis.7
Unfortunately, visual symptoms may be irreversible despite treatment.
Our patient’s central retinal artery occlusion
This case highlights how difficult it is in practice to distinguish nonarteritic from arteritic central retinal artery occlusion.
Our patient had numerous cardiovascular risk factors, including known carotid and coronary artery disease, favoring a nonarteritic diagnosis.
On the other hand, his elevated inflammatory markers suggested an underlying inflammatory response. He lacked the characteristic headache and other systemic signs of giant cell arteritis, but this has been described in about 25% of patients.15 If emboli are seen on funduscopy, further workup for arteritic central retinal artery occlusion is not warranted, but emboli are not always present. Then again, absence of posterior ciliary artery involvement on fluorescein angiography pointed away from giant cell arteritis.
CASE CONTINUED: FINAL DIAGNOSIS
Biopsy of the left temporal artery showed intimal thickening with focal destruction of the internal elastic lamina by dystrophic calcification with no evidence of inflammatory infiltrates, giant cells, or granulomata in the adventitia, media, or intima. Based on the results of biopsy study and fluorescein angiography, we concluded that this was nonarteritic central retinal artery occlusion related to atherosclerotic disease.
Methylprednisolone was discontinued. The patient was discharged on aspirin, losartan, furosemide, amlodipine, and high-dose atorvastatin for standard stroke prevention. He was followed by the medical team and the ophthalmology department. At 6 weeks, there was only marginal improvement in the visual acuity of the left eye.
MANAGEMENT
4. Management of nonarteritic central retinal artery occlusion could include all of the following except which one?
- Ocular massage
- Intravenous thrombolysis
- Intra-arterial thrombolysis
- Risk-factor modification
- Intraocular steroid injection
In patients with acute vision loss from nonarteritic central retinal artery occlusion, acute strategies to restore retinal perfusion include noninvasive “standard” therapies and thrombolysis (intravenous or intra-arterial). Unfortunately, consensus and guidelines are lacking.
Traditional therapies include sublingual isosorbide dinitrate, systemic pentoxifylline, inhalation of a carbogen, hyperbaric oxygen, ocular massage, intravenous acetazolamide and mannitol, anterior chamber paracentesis, and systemic steroids. However, none of these have been shown to be more effective than placebo.16
Thrombolytic therapy, analogous to the treatment of patients with ischemic stroke or myocardial infarction, is more controversial in acute central retinal artery occlusion.13 Data from small case-series suggested that intra-arterial or intravenous thrombolysis might improve visual acuity with reasonable safety.17 On the other hand, a randomized study from the United Kingdom that compared intra-arterial thrombolysis within a 24-hour window and conservative measures concluded that thrombolysis should not be used.18
Thrombolysis is thus used only in selected patients on a case-specific basis with involvement of a multispecialty team including stroke neurologists, especially if patients present within hours of onset and have concomitant neurologic symptoms.
Treatment beyond the acute phase focuses on preventing complications of the eye ischemia and aggressively managing systemic atherosclerotic risk factors to decrease the incidence of further ischemic events. Other interventions include endarterectomy for significant carotid stenosis and anticoagulation to prevent cardioembolic embolization (such as atrial fibrillation). Most experts agree on the addition of an antiplatelet agent.13,19
Intraocular steroid injection can be used in the management of some retinal disorders but has no value in nonarteritic central retinal artery occlusion.
Vision recovery in nonarteritic central retinal artery occlusion is variable, but the prognosis is generally poor. The visual acuity on presentation, the onset of the symptoms, and collateral vessels are major factors influencing long-term recovery. Most of the recovery occurs within 7 days and involves peripheral vision rather than central vision. Several studies report some recovery in peripheral vision in approximately 30% to 35% of affected eyes.20–22
PROMPT ACTION MAY SAVE SIGHT
Vision loss is a common presenting symptom in the emergency setting. A meticulous history and systematic physical examination can narrow the differential diagnosis of this neuro-ophthalmologic emergency. Acute retinal ischemia from central retinal artery occlusion is the ocular equivalent of an ischemic stroke, and they share risk factors, diagnostic workup, and management approaches.
Both etiologic subtypes (ie, arteritic and nonarteritic) require prompt intervention by front-line physicians. If giant cell arteritis is suspected, corticosteroid therapy must be initiated to save the contralateral retina from ischemia. Suspicion of central retinal artery occlusion warrants immediate evaluation by a neurologist to consider thrombolysis. Prompt action and interdisciplinary care involving an ophthalmologist, neurologist, and emergency or internal medicine physician may save a patient from permanent visual disability.
KEY POINTS
- Monocular vision loss requires urgent evaluation with a multidisciplinary management approach.
- There are no consensus treatment guidelines for nonarteritic central retinal artery occlusion, but the workup includes a comprehensive stroke evaluation.
- Arteritic central retinal artery occlusion is most often due to giant cell arteritis, and when it is suspected, the patient should be empirically treated with steroids.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
- Glezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Arch Endocrinol Metab 2015; 59:259–264.
- Campbell WW. DeJong’s The Neurologic Examination. 7th ed. Philadelphia: Lippincott Williams & Wilkins, 2013.
- Biller J. Practical Neurology. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2012.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology 2009; 116:1928–1936.
- Biousse V. Acute retinal arterial ischemia: an emergency often ignored. Am J Ophthalmol 2014; 157:1119–1121.
- Hunder GG, Bloch DA, Michel BA, et al. American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33:1122–1128.
- Smith JH, Swanson JW. Giant cell arteritis. Headache 2014; 54:1273–1289.
- Hall S, Persellin S, Lie JT, O’Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet 1983; 2:1217–1220.
- Gabriel SE, O’Fallon WM, Achkar AA, Lie JT, Hunder GG. The use of clinical characteristics to predict the results of temporal artery biopsy among patients with suspected giant cell arteritis. J Rheumatol 1995; 22:93–96.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- Danesh-Meyer HV, Savino PJ, Eagle RC Jr, Kubis KC, Sergott RC. Low diagnostic yield with second biopsies in suspected giant cell arteritis. J Neuroophthalmol 2000; 20:213–215.
- Cavallerano AA. Ophthalmic fluorescein angiography. Optom Clin 1996; 5:1–23.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res 2011; 30:359–394.
- Khan A, Dasgupta B. Imaging in giant cell arteritis. Curr Rheumatol Rep 2015; 17:52.
- Biousse V, Newman N. Retinal and optic nerve ischemia. Continuum (Minneap Minn) 2014; 20:838–856.
- Fraser SG, Adams W. Interventions for acute non-arteritic central retinal artery occlusion. Cochrane Database Syst Rev 2009; 1:CD001989.
- Beatty S, Au Eong KG. Local intra-arterial fibrinolysis for acute occlusion of the central retinal artery: a meta-analysis of the published data. Br J Ophthalmol 2000; 84:914–916.
- Schumacher M, Schmidt D, Jurklies B, et al; EAGLE-Study Group. Central retinal artery occlusion: local intra-arterial fibrinolysis versus conservative treatment, a multicenter randomized trial. Ophthalmology 2010; 117:1367–1375.e1.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hayreh SS, Zimmerman MB. Central retinal artery occlusion: visual outcome. Am J Ophthalmol 2005; 140:376–391.
- Augsburger JJ, Magargal LE. Visual prognosis following treatment of acute central retinal artery obstruction. Br J Ophthalmol 1980; 64:913–917.
- Brown GC, Shields JA. Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 1979; 97:84–92.
Postoperative delirium in a 64-year-old woman
A 64-year-old woman undergoes elective T10-S1 nerve decompression with fusion for chronic idiopathic scoliosis. Soon afterward, she develops acute urinary retention attributed to an Escherichia coli urinary tract infection and narcotic medications. She is treated with antibiotics, an indwelling catheter is inserted, and her symptoms resolve. She is transferred to the inpatient physical rehabilitation unit.
On postoperative day 9, she develops an acute change in mental status, suddenly becoming extremely anxious and falsely believing she has a “terminal illness.” A psychiatrist suggests that these symptoms are a manifestation of delirium, given the patient’s recent surgery and exposure to benzodiazepine and narcotic medications. On postoperative day 10, she is awake but is now mute and uncooperative. An internist is consulted for an evaluation for encephalopathy and delirium.
MEDICAL HISTORY
Her medical history, obtained by chart review and interviewing her husband, includes well-controlled bipolar disorder over the last 4 years, with no episodes of frank psychosis or mania. She had a “bout of delirium” 4 years earlier attributed to a catastrophic life event, but the symptoms resolved after adjustment of her anxiolytic and mood-stabilizing drugs. She also has well-controlled hypertension, hypothyroidism, and gastroesophageal reflux. Her only surgery was her recent elective procedure.
She has a family history of dementia (Pick disease in her mother).
She is married, lives with her husband, and has an adult son. She is employed as a media specialist and also teaches English as a second language. Before this hospital admission, she was described as happy and content, though her primary psychiatrist had noted intermittent anxiety. Her husband does not suspect illicit drug use and denies significant alcohol or tobacco abuse.
A thorough review of systems is not possible, given her encephalopathy. But before her acute decline, she had complained of “choking on blood” and a subjective inability to swallow.
Her home medications include dextroamphetamine extended-release, alprazolam as needed for sleep, venlafaxine extended-release, lamotrigine, lisinopril, propranolol, amlodipine, atorvastatin, levothyroxine, omeprazole, iron, and vitamin B12. At the time of the evaluation, she is on her home medications with the addition of olanzapine, vitamin D, polyethylene glycol, and an intravenous infusion of dextrose 5% with 0.45% saline at a rate of 100 mL/hour. She has allergies to latex, penicillin, peanuts, and shellfish.
PHYSICAL EXAMINATION
On physical examination, the patient seems healthy and appears normal for her stated age. She is wearing a spinal brace and is in no apparent distress. She is afebrile, pulse 104 beats per minute, respirations 16 breaths per minute and unlabored, and oxygen saturation good on room air. The skin is normal. No thyromegaly, bruits, or lymphadenopathy is noted. Cardiovascular, respiratory, and abdominal examinations, though limited by the spinal brace, are unremarkable. She has no evidence of peripheral edema or vascular insufficiency. Muscle bulk and tone are adequate and symmetric.
She is awake and alert and able to follow simple commands with some prompting. She does not initiate movements spontaneously. She makes some eye contact but does not track or acknowledge the interviewer consistently and does not respond verbally to questions. Her sclera are nonicteric, the pupils are equally round and reactive to light, and the external ocular muscles are intact. There is no facial asymmetry, and the tongue protrudes at midline. She blinks appropriately to threat bilaterally. Strength is at least 3/5 in the upper extremities and 2/5 in the lower extremities, though the examination is limited by lack of patient cooperation. She shows minimal grimace on noxious stimulation but does not withdraw extremities. Reflexes are present and mildly depressed symmetrically. Plantar reflexes are downgoing bilaterally.
INITIAL LABORATORY EVALUATION
On initial laboratory testing, the serum sodium is 132 mmol/L (reference range 136–144), stable since admission. Point-of-care glucose is 98 mg/dL. Aspartate aminotransferase and alanine aminotransferase levels are mildly elevated at 59 U/L (13–35) and 51 U/L (7–38), respectively, but serum ammonia is undetectable. Vitamin B12, folate, thyroid-stimulating hormone, and free thyroxine are within the normal ranges. Leukocytosis is noted, with 14 × 109 cells/L (3.7–11.0), 86% neutrophils, and a mild left shift. Urinalysis is negative for leukocyte esterase, nitrites, and white blood cells.
APPROACH TO ALTERED MENTAL STATUS
1. Which of the following risk factors predisposes this patient to postoperative delirium?
- Hyponatremia
- Polypharmacy
- Family history of dementia
- Depression
Altered mental status, or encephalopathy, is one of the most common yet challenging conditions in medicine. When a consult is placed for altered mental status, it is important to determine the affected domain that has changed from the patient’s normal state. Changes can include alterations in consciousness, attention, behavior, cognition, language, speech, and praxis and can reflect varying degrees of cerebral dysfunction.

Electrolyte abnormalities
Disorders of sodium homeostasis are common in hospitalized patients and may contribute to the onset of delirium. Hyponatremia is especially frequent and often iatrogenic, with a prevalence significantly higher in women (2.1% vs 1.3%, P = .0044) and in the elderly.2
Neurologic manifestations are often the result of cerebral edema due to osmolar volume shifts.3–6 Acute hyponatremic encephalopathy is most likely to occur when sodium shifts are rapid, usually within 24 hours, and is often seen in postoperative patients requiring significant volume resuscitation with hypotonic fluids.6 Young premenopausal women appear to be at especially high risk of permanent brain damage secondary to hyponatremic encephalopathy,7 a finding that may reflect the limited compliance within the intracranial vault and lack of significant involutional parenchymal changes that occur with aging.8–11
Aging also has important effects on fluid balance, as restoration of body fluid homeostasis is slower in older patients.12
Hormonal effects of estrogen appear to play a synergistic role in the expression of arginine vasopressin in postmenopausal women, further contributing to hyponatremia.
Although our patient has mild hyponatremia, there has been no acute change in her sodium balance since admission to the hospital, and so it is unlikely to be the cause of her acute delirium. Her mild hyponatremia may in part be from hypo-osmolar maintenance fluids with dextrose 5% and 0.45% normal saline.
Mild chronic hyponatremia may affect balance and has been associated with increased mortality risk in certain chronic disease states, but this is unlikely to be the main cause of acute delirium.
Polypharmacy
Patients admitted to the hospital with polypharmacy are at high risk of drug-induced delirium. In approaching delirium, a patient’s medications should be evaluated for interactions, as well as for possible effects of newly prescribed drugs. New medications that affect cytochrome P450 enzymes warrant investigation, as do drugs with narrow therapeutic windows that the patient has been using long-term.
Consultation with a clinical pharmacist is often helpful. Macrolides, protease inhibitors, and nondihydropyridine calcium channel blockers are common P450 inhibitors, while many anticonvulsants are known inducers of the P450 system. Selective serotonin reuptake inhibitors and diuretics can lead to electrolyte imbalances such as hyponatremia, which may further predispose to bouts of delirium, as described above.
The patient’s extensive list of psychoactive medications makes polypharmacy a significant risk factor for delirium. Quetiapine and venlafaxine both cause sedation and increase the risk of serotonin syndrome. However, in this case, the patient does not have marked fever, rigidity, or hyperreflexia to corroborate that diagnosis.
Dementia
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), defines dementia as a disorder involving cognitive impairment in at least 1 cognitive domain, with a significant decline from a previous level of functioning.1 These impairments need not necessarily occur separately from bouts of delirium, but the time course for most forms of dementia tends to be progressive over a subacute to chronic duration.
Dementia increases the risk for acute confusion and delirium in hospitalized patients.13 This is partly reflected by pathophysiologic changes that leave elderly patients susceptible to the effects of anticholinergic drugs.14 Structural changes due to small-vessel ischemia may also predispose patients to seizures in the setting of metabolic derangement or critical illness. Diagnosing dementia thus remains a challenge, as dementia must be clearly distinguished from other disorders such as delirium and depression.
The acute change in this patient’s case makes the isolated diagnosis of dementia much less likely than other causes of altered mental status. Also, her previous level of function does not suggest a clinically significant personal history of impairment.
Mental illness
Several studies have examined the link between preoperative mental health disorders and postoperative delirium.15–17 Depression appears to be a risk factor for postoperative delirium in patients undergoing elective orthopedic surgery,15 and this includes elderly patients.16 While a clear etiologic link has yet to be determined, disruption of circadian rhythm and abnormal cerebral response to stress may play a role. Studies have also suggested an association between schizophrenia and delirium, though this may be related to perioperative suspension of medications.17
Bipolar disorder has not been well studied with regard to postoperative complications. However, this patient has had a previous episode of decompensated mania, therefore making bipolar disorder a plausible condition in the differential diagnosis.
CASE CONTINUED: ACUTE DETERIORATION
Without a clearly identifiable cause for our patient’s acute confusional state, neurology and medical consultants recommend neuroimaging.
Computed tomography (CT) and magnetic resonance imaging (MRI) without contrast are ordered and performed on postoperative day 11 and demonstrate chronic small-vessel ischemic disease, consistent with our patient’s age, as well as frontotemporal atrophy. There is no evidence of mass effect, bleeding, or acute ischemia.
Overnight, she becomes obtunded, and the rapid response team is called. Her vital signs appear stable, and she is afebrile. Basic laboratory studies, imaging, and electrocardiography are repeated, and the results are unchanged from recent tests. She is transferred to the intensive care unit (ICU) for closer monitoring.
2. What is most likely cause of the patient’s declining mental status, and what is the next appropriate step?
- Acute stroke: repeat MRI with contrast
- Urinary tract infection: order blood and urine cultures, and start empiric antibiotics
- Neuroleptic malignant syndrome: start dantrolene
- Seizures: order electroencephalography (EEG)
Acute stroke
Acute stroke can affect mental status and consciousness through several pathways. Stroke syndromes can vary in presentation depending on the level of cortical and subcortical involvement, with clinical manifestations including confusion, aphasia, neglect, and inattention. Wakefulness and the ability to maintain consciousness is impaired, with disruption of the ascending reticular activating system, often seen in injuries to the brainstem. Large territorial or hemispheric infarcts, with subsequent cerebral edema, can also disrupt this system and lead to cerebral herniation and coma.
MRI without contrast is extremely sensitive for ischemia and can typically detect ischemia in acute stroke within 3 to 30 minutes.18–20 Repeating the study with contrast is unlikely to provide additional benefit.
In our patient’s case, the lack of localizing neurologic symptoms, in addition to her recent negative neuroimaging workup, makes the diagnosis of acute stroke unlikely.
Infection
The role of severe infection in patients with altered mental status is well documented and likely relates to diffuse cerebral dysfunction caused by an inflammatory cascade. Less well understood is the role of occult infection, especially urinary tract infection, in otherwise immunocompetent patients. Urinary tract infection has long been thought to cause delirium in otherwise asymptomatic elderly patients, but few studies have examined this relationship, and those studies have been shown to have significant methodologic errors.21 In the absence of better data, urinary tract infection as the cause of frank delirium in an otherwise well patient should be viewed with skepticism, and alternative causes should be sought.
Although the patient has a nonspecific leukocytosis, her benign urinalysis and lack of corroborating evidence makes urinary tract infection an unlikely cause of her frank delirium.
Neuroleptic malignant syndrome
Neuroleptic malignant syndrome is defined as fever, rigidity, mental status changes, and autonomic instability after exposure to antidopaminergic drugs. It is classically seen after administration of typical antipsychotics, though atypical antipsychotics and antiemetic drugs may be implicated as well.
Patients often exhibit agitation and confusion, which when severe may progress to mutism and catatonia. Likewise, psychotropic drugs such as quetiapine and venlafaxine, used in combination, have the additional risk of serotonin syndrome.
Additional symptoms include hyperreflexia, ataxia, and myoclonus. Withdrawal of the causative agent and supportive care are the mainstays of therapy. Targeted therapies with agents such as dantrolene, bromocriptine, and amantadine have also been reported anecdotally, but their efficacy is unclear, with variable results.22
As noted earlier, the addition of quetiapine to the patient’s already lengthy medication list could conceivably cause neuroleptic malignant syndrome or serotonin syndrome and should be considered. However, additional neurologic findings to confirm this diagnosis are lacking.
Seizures
Nonconvulsive seizure, particularly nonconvulsive status epilepticus (NCSE), is not well recognized and is particularly challenging to diagnose without EEG. In several case series of patients presenting to the emergency room with altered mental status, NCSE was found in 16% to 28% of patients in whom EEG was performed after an initial evaluation failed to show an obvious cause for the delirium.23,24 Historical features are unreliable for ruling out NCSE as a cause of delirium, as up to 41% of patients in whom the condition is ultimately diagnosed have only confusion as the presenting clinical symptom.25
Likewise, alternating ictal and postictal periods may mimic the typical waxing and waning course classically associated with delirium of other causes. Physical findings such as nystagmus, anisocoria, and hippus may be helpful but are often overlooked or absent. EEG is thus an essential requirement for the diagnosis.26
Given the lack of a clear diagnosis, a workup with EEG should be considered in this patient.
CASE CONTINUED: ADDITIONAL SIGNS
In the ICU, our patient is evaluated by the intensivist team. Her vital signs are stable, and while she is now awakening, she is unable to follow commands and remains mute. She does not initiate movement spontaneously but offers slight resistance to passive movements, holding and maintaining postures her extremities are placed in. She keeps her eyes closed, but when opened by the examining physician, dysconjugate gaze and anisocoria are noted.
3. What clinical entity is most consistent with these physical findings, and what is the next step in management?
- Catatonia secondary to bipolar disorder type I: challenge with intravenous lorazepam 2 mg
- Oculomotor nerve palsy due to enlarging intracranial aneurysm: aggressive blood pressure lowering, elevation of the head of the bed
- Toxic leukoencephalopathy: supportive care and withdrawal of the causative agent
- NCSE: challenge with intravenous lorazepam 2 mg and order EEG
Catatonia
The DSM-5 defines catatonia as a behavioral syndrome complicating an underlying psychiatric or medical condition, as opposed to a distinct diagnosis. It is most commonly encountered in psychiatric illnesses including bipolar disorder, major depression, and schizophrenia. Akinesis, stupor, mutism, and “waxy” flexibility often dominate the clinical picture.
The pathophysiology is poorly defined, but likely involves neurotransmitter imbalances particularly with an increase in N-methyl-d-aspartate (NMDA) activity and suppression of gamma-aminobutyric acid (GABA) activity. This hypothesis is supported by the finding that benzodiazepines, electroconvulsive therapy, and NMDA antagonists such as amantadine are all effective in treating catatonia.27,28 Findings of focal neurologic abnormalities warrant further investigation. EEG may be necessary to differentiate catatonia from NCSE, as both may respond to a benzodiazepine challenge.
As pure catatonia is a diagnosis of exclusion, further workup, including EEG, is necessary to confirm the diagnosis.
Oculomotor nerve palsy
Anisocoria together with dysconjugate gaze should prompt consideration of a lesion involving the oculomotor nerve. Loss of tonic muscle activity from the lateral rectus and superior oblique cause a downward and outward gaze. Furthermore, loss of parasympathetic tone occurs with compressive palsies of the oculomotor nerve, clinically manifesting as a mydriatic and unreactive pupil with ptosis. Given its anatomic course and proximity to other vascular and parenchymal structures, the oculomotor nerve is vulnerable to compression from many sources, including aneurysmal dilation (especially of the posterior cerebral artery), uncal herniation, and inflammation of the cavernous sinus.
Noncontrast CT and lumbar puncture are very sensitive for making the diagnosis of sentinel bleeding within the first 24 hours,29 whereas computed tomographic angiography and magnetic resonance angiography can reliably detect unruptured aneurysms as small as 3 mm.30
Conditions that can lead to oculomotor palsy are unlikely to cause an acute gain in appendicular muscle tone, as noted by the catatonia this patient is demonstrating. Also, mass lesions or bleeding associated with oculomotor palsy is likely to cause acute loss of tone. Chronic upper-motor neuron lesions lead to spasticity rather than the waxy flexibility seen in this patient. In our patient, the findings of isolated anisocoria without further clinical evidence of oculomotor nerve compression make this diagnosis unlikely.
Toxic leukoencephalopathy
Toxic leukoencephalopathy—widespread destruction of myelin, particularly in the white matter tracts that support higher cortical functions—can be caused by antineoplastic agents, immunosuppressant agents, and industrial solvents, as well as by abuse of vaporized drugs such as heroin (“chasing the dragon”). In its mild forms it may cause behavioral disturbances or inattention. In severe forms, a neurobehavioral syndrome of akinetic mutism may be present and can mimic catatonia.31
The diagnosis is often based on the clinical history and neuroimaging, particularly MRI, which demonstrates hyperintensity of the white matter tracts in T2-weighted images.32
This patient does not have a clear history of exposure to an agent typically associated with toxic leukoencephalopathy and does not have the corroborating MRI findings to support this diagnosis.
CASE CONTINUED
Because recent neuroimaging revealed no structural brain lesions and no cause for brain herniation, the patient receives a challenge of 2 mg of intravenous lorazepam to treat potential NCSE. Subsequent improvement is noted in her anisocoria, gaze deviation, and encephalopathy. EEG reveals frequent focal seizures arising from mesial frontal regions with bilateral hemisphere propagation, consistent with bifrontal focal NCSE.
As our patient is being transferred to a room for continuous EEG monitoring, her condition begins to deteriorate, and she again becomes more encephalopathic, with anisocoria and dysconjugate gaze. Additional doses of lorazepam are given (to complete a 0.1-mg/kg load), and additional therapy with intravenous fosphenytoin (20-mg/kg load) is given. Intubation is done for airway protection.
Continuous EEG monitoring reveals multiple frequent electrographic seizures arising from the bifrontal territories, concerning for persistent focal NCSE. A midazolam drip is initiated for EEG burst suppression of cerebral activity. Over 24 hours, EEG shows resolution of seizure activity. As the patient is weaned from sedation, she awakens and follows commands consistently, tolerating extubation without complications. Her neurologic status remains stable over the next 48 hours, having returned to her neurologic baseline level of functioning. She is able to be transferred out of the ICU in stable condition while continuing on scheduled antiepileptic therapy with phenytoin.
ALTERED MENTAL STATUS IN INPATIENTS
Altered mental status is one of the most frequently encountered reasons for medical consultation from nonmedical services. The workup and management of metabolic, toxic, psychiatric, and neurologic causes requires a deep appreciation for the broad differential diagnosis and a multidisciplinary approach. Physicians caring for these patients should avoid prematurely drawing conclusions when the patient’s clinical condition fails to respond to typical measures.
Delirium is a challenging adverse event in older patients during hospitalization, with a significant national financial burden of $164 billion per year.33 The prevalence of delirium in adults on hospital admission is estimated as 14% to 24%, with an inpatient hospitalization incidence ranging from 6% to 56% in general hospital patients.34 In addition, postoperative delirium has been reported in 15% to 53% of older patients.35
While delirium is preventable in 30% to 40% of cases,36,37 it remains an important independent prognostic determinant of hospital outcomes.38–40
Delirium in hospitalized patients requires a thorough, individualized workup. In our patient’s case, the clinical findings of hypoactive delirium were found to be manifestations of NCSE, a rare life-threatening and potentially reversible neurologic disease.
While establishing seizures as a diagnosis, careful attention must first be directed towards investigating environmental or metabolic triggers that may be inciting the disease. This often involves a similar workup for metabolic derangements, as seen in the approach to delirium.
 without contrast reveals significant frontotemporal atrophy (blue arrows) and deep sulci within the frontal lobe, features not as prevalent in occipital and cerebellar territories (red arrows).")
The diagnosis of NCSE, while made in this patient’s case, remains challenging. Careful physical examination should assess for automatisms, “negative” symptoms (staring, aphasia, weakness), and “positive” symptoms (hallucinations, psychosis). Cataplexy, mutism, and other acute psychiatric features have been associated with NCSE,44 highlighting the importance of EEG. A trial of a benzodiazepine in conjunction with clinical and EEG monitoring may help guide clinical decision- making.
As there is no current universally accepted definition for NCSE nor an accepted agreement on required EEG diagnostic features at this time,41 accurate diagnosis is most likely to be obtained in facilities with both subspecialty neurologic consultation and EEG capabilities.
Our patient’s family history of Pick disease is interesting, as this is a progressive form of frontotemporal dementia with both sporadic and genetically linked cases. Recent studies have shown evidence that patients with neurodegenerative disease have increased seizure frequency early in the disease course,31 and efforts are under way to establish the incidence of first unprovoked seizure in patients with frontotemporal dementia. In our patient’s case, resolution of seizure activity yielded a return to her baseline level of neurologic function.
Early use of selective serotonin reuptake inhibitors has been shown to help with the behavioral symptoms of frontotemporal dementia,45 but increasing requirements over time may indicate progression of neurodegeneration and should warrant further appropriate investigation.
In our patient’s case, escalating dose requirements may have reflected worsening frontotemporal atrophy. However, the diagnosis of a neurodegenerative disease such as frontotemporal dementia in a patient such as ours is not definitively established at this time and is being investigated on an outpatient basis.
Given the frequency of delirium and its many risk factors in the inpatient setting, verifying a causative diagnosis can be difficult. Detailed consideration of the patient’s individual clinical circumstances, often in concert with appropriate subspecialty consultations, is essential to the evaluation. Although it is time-intensive, multidisciplinary intervention can lead to safer outcomes and shorter hospital stays.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013. http://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596. Accessed July 7, 2017.
- Mohan S, Gu S, Parikh A, Radhakrishnan J. Prevalence of hyponatremia and association with mortality: results from NHANES. Am J Med 2013; 126:1127–1137.e1.
- Sterns RH. Disorders of plasma sodium—causes, consequences, and correction. N Engl J Med 2015; 372:55–65.
- Rose B, Post T. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001.
- McManus ML, Churchwell KB, Strange K. Regulation of cell volume in health and disease. N Engl J Med 1995; 333:1260–1266.
- Strange K. Regulation of solute and water balance and cell volume in the central nervous system. J Am Soc Nephrol 1992; 3:12–27.
- Ayus JC, Wheeler JM, Arieff AI. Postoperative hyponatremic encephalopathy in menstruant women. Ann Intern Med 1992; 117:891–897.
- Gur RC, Mozley PD, Resnick SM, et al. Gender differences in age effect on brain atrophy measured by magnetic resonance imaging. Proc Natl Acad Sci USA 1991; 88:2845–2849.
- Rosomoff HL, Zugibe FT. Distribution of intracranial contents in experimental edema. Arch Neurol 1963; 9:26–34.
- Melton JE, Nattie EE. Brain and CSF water and ions during dilutional and isosmotic hyponatremia in the rat. Am J Physiol 1983; 244:R724–R732.
- Nattie EE, Edwards WH. Brain and CSF water and ions in newborn puppies during acute hypo- and hypernatremia. J Appl Physiol Respir Environ Exerc Physiol 1981; 51:1086–1091.
- Stachenfeld NS, DiPietro L, Palter SF, Nadel ER. Estrogen influences osmotic secretion of AVP and body water balance in postmenopausal women. Am J Physiol 1998; 274:R187–R195.
- Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc 2002; 50:1723–1732.
- de Smet Y, Ruberg M, Serdaru M, Dubois B, Lhermitte F, Agid Y. Confusion, dementia and anticholinergics in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1982; 45:1161–1164.
- Mollon B, Mahure SA, Ding DY, Zuckerman JD, Kwon YW. The influence of a history of clinical depression on peri-operative outcomes in elective total shoulder arthroplasty: a ten-year national analysis. Bone Joint J 2016; 98-B:818–824.
- Kosar CM, Tabloski PA, Travison TG, et al. Effect of preoperative pain and depressive symptoms on the development of postoperative delirium. Lancet Psychiatry 2014; 1:431–436.
- Copeland LA, Zeber JE, Pugh MJ, Mortensen EM, Restrepo MI, Lawrence VA. Postoperative complications in the seriously mentally ill: a systematic review of the literature. Ann Surg 2008; 248:31–38.
- Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann Neurol 1995; 37:231–241.
- Sorensen AG, Buonanno FS, Gonzalez RG, et al. Hyperacute stroke: evaluation with combined multisection diffusion-weighted and hemodynamically weighted echo-planar MR imaging. Radiology 1996; 199:391–401.
- Li F, Han S, Tatlisumak T, et al. A new method to improve in-bore middle cerebral artery occlusion in rats: demonstration with diffusion—and perfusion—weighted imaging. Stroke 1998; 29:1715–1720.
- Balogun SA, Philbrick JT. Delirium, a symptom of UTI in the elderly: fact or fable? A systematic review. Can Geriatr J 2013; 17:22–26.
- Reulbach U, Dütsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007; 11:R4.
- Naeije G, Depondt C, Meeus C, Korpak K, Pepersack T, Legros B. EEG patterns compatible with nonconvulsive status epilepticus are common in elderly patients with delirium: a prospective study with continuous EEG monitoring. Epilepsy Behav 2014; 36:18–21.
- Veran O, Kahane P, Thomas P, Hamelin S, Sabourdy C, Vercueil L. De novo epileptic confusion in the elderly: a 1-year prospective study. Epilepsia 2010; 51:1030–1035.
- Sutter R, Rüegg S, Kaplan PW. Epidemiology, diagnosis, and management of nonconvulsive status epilepticus. Opening Pandora’s box. Neurol Clin Pract 2012; 2:275–286.
- Husain AM, Horn GJ, Jacobson MP. Non-convulsive status epilepticus: usefulness of clinical features in selecting patients for urgent EEG. J Neurol Neurosurg Psychiatry 2003; 74:189–191.
- Ungvari GS, Chiu HF, Chow LY, Lau BS, Tang WK. Lorazepam for chronic catatonia: a randomized, double-blind, placebo-controlled cross-over study. Psychopharmacology (Berl) 1999; 142:393–398.
- Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci 2007; 19:406– 412.
- Perry JJ, Spacek A, Forbes M, et al. Is the combination of negative computed tomography result and negative lumbar puncture result sufficient to rule out subarachnoid hemorrhage? Ann Emerg Med 2008; 51:707–713.
- Li MH, Cheng YS, Li YD, et al. Large-cohort comparison between three-dimensional time-of-flight magnetic resonance and rotational digital subtraction angiographies in intracranial aneurysm detection. Stroke 2009; 40:3127–3129.
- Filley CM, Kleinschmidt-DeMasters BK. Toxic leukoencephalopathy. N Engl J Med 2001; 345:425–432.
- Magnetic resonance imaging of the central nervous system. Council on Scientific Affairs. Report of the Panel on Magnetic Resonance Imaging. JAMA 1988; 259:1211–1222.
- Leslie DL, Marcantonio ER, Zhang Y, Leo-Summers L, Inouye SK. One-year health care costs associated with delirium in the elderly population. Arch Intern Med 2008; 168:27–32.
- Inouye SK. Delirium in hospitalized older patients. Clin Geriatr Med 1998; 14:745–764.
- Agostini JV, Inouye SK, Hazzard W, Blass J. Delirium. In: Principles of Geriatric Medicine and Gerontology. 5th ed. New York, NY: McGraw-Hill; 2003:1503–1515.
- Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med 1999; 340:669–676.
- Marcantonio ER, Flacker JM, Wright RJ, Resnick NM. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc 2001; 49:516–522.
- Inouye SK, Rushing JT, Foreman MD, Palmer RM, Pompei P. Does delirium contribute to poor hospital outcomes? A three-site epidemiologic study. J Gen Intern Med 1998; 13:234–242.
- Rothschild JM, Bates DW, Leape LL. Preventable medical injuries in older patients. Arch Intern Med 2000; 160:2717–2728.
- Gillick MR, Serrell NA, Gillick LS. Adverse consequences of hospitalization in the elderly. Soc Sci Med 1982; 16:1033–1038.
- Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav 2000; 1:301-314.
- Rosenow F, Hamer HM, Knake S. The epidemiology of convulsive and nonconvulsive status epilepticus. Epilepsia 2007; 48(suppl 8):82–84.
- Woodford HJ, George J, Jackson M. Non-convulsive status epilepticus: a practical approach to diagnosis in confused older people. Postgrad Med J 2015; 91:655–661.
- Kaplan PW. Nonconvulsive status epilepticus in the emergency room. Epilepsia 1996; 37:643–650.
- Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry 1997; 58:212–216.
A 64-year-old woman undergoes elective T10-S1 nerve decompression with fusion for chronic idiopathic scoliosis. Soon afterward, she develops acute urinary retention attributed to an Escherichia coli urinary tract infection and narcotic medications. She is treated with antibiotics, an indwelling catheter is inserted, and her symptoms resolve. She is transferred to the inpatient physical rehabilitation unit.
On postoperative day 9, she develops an acute change in mental status, suddenly becoming extremely anxious and falsely believing she has a “terminal illness.” A psychiatrist suggests that these symptoms are a manifestation of delirium, given the patient’s recent surgery and exposure to benzodiazepine and narcotic medications. On postoperative day 10, she is awake but is now mute and uncooperative. An internist is consulted for an evaluation for encephalopathy and delirium.
MEDICAL HISTORY
Her medical history, obtained by chart review and interviewing her husband, includes well-controlled bipolar disorder over the last 4 years, with no episodes of frank psychosis or mania. She had a “bout of delirium” 4 years earlier attributed to a catastrophic life event, but the symptoms resolved after adjustment of her anxiolytic and mood-stabilizing drugs. She also has well-controlled hypertension, hypothyroidism, and gastroesophageal reflux. Her only surgery was her recent elective procedure.
She has a family history of dementia (Pick disease in her mother).
She is married, lives with her husband, and has an adult son. She is employed as a media specialist and also teaches English as a second language. Before this hospital admission, she was described as happy and content, though her primary psychiatrist had noted intermittent anxiety. Her husband does not suspect illicit drug use and denies significant alcohol or tobacco abuse.
A thorough review of systems is not possible, given her encephalopathy. But before her acute decline, she had complained of “choking on blood” and a subjective inability to swallow.
Her home medications include dextroamphetamine extended-release, alprazolam as needed for sleep, venlafaxine extended-release, lamotrigine, lisinopril, propranolol, amlodipine, atorvastatin, levothyroxine, omeprazole, iron, and vitamin B12. At the time of the evaluation, she is on her home medications with the addition of olanzapine, vitamin D, polyethylene glycol, and an intravenous infusion of dextrose 5% with 0.45% saline at a rate of 100 mL/hour. She has allergies to latex, penicillin, peanuts, and shellfish.
PHYSICAL EXAMINATION
On physical examination, the patient seems healthy and appears normal for her stated age. She is wearing a spinal brace and is in no apparent distress. She is afebrile, pulse 104 beats per minute, respirations 16 breaths per minute and unlabored, and oxygen saturation good on room air. The skin is normal. No thyromegaly, bruits, or lymphadenopathy is noted. Cardiovascular, respiratory, and abdominal examinations, though limited by the spinal brace, are unremarkable. She has no evidence of peripheral edema or vascular insufficiency. Muscle bulk and tone are adequate and symmetric.
She is awake and alert and able to follow simple commands with some prompting. She does not initiate movements spontaneously. She makes some eye contact but does not track or acknowledge the interviewer consistently and does not respond verbally to questions. Her sclera are nonicteric, the pupils are equally round and reactive to light, and the external ocular muscles are intact. There is no facial asymmetry, and the tongue protrudes at midline. She blinks appropriately to threat bilaterally. Strength is at least 3/5 in the upper extremities and 2/5 in the lower extremities, though the examination is limited by lack of patient cooperation. She shows minimal grimace on noxious stimulation but does not withdraw extremities. Reflexes are present and mildly depressed symmetrically. Plantar reflexes are downgoing bilaterally.
INITIAL LABORATORY EVALUATION
On initial laboratory testing, the serum sodium is 132 mmol/L (reference range 136–144), stable since admission. Point-of-care glucose is 98 mg/dL. Aspartate aminotransferase and alanine aminotransferase levels are mildly elevated at 59 U/L (13–35) and 51 U/L (7–38), respectively, but serum ammonia is undetectable. Vitamin B12, folate, thyroid-stimulating hormone, and free thyroxine are within the normal ranges. Leukocytosis is noted, with 14 × 109 cells/L (3.7–11.0), 86% neutrophils, and a mild left shift. Urinalysis is negative for leukocyte esterase, nitrites, and white blood cells.
APPROACH TO ALTERED MENTAL STATUS
1. Which of the following risk factors predisposes this patient to postoperative delirium?
- Hyponatremia
- Polypharmacy
- Family history of dementia
- Depression
Altered mental status, or encephalopathy, is one of the most common yet challenging conditions in medicine. When a consult is placed for altered mental status, it is important to determine the affected domain that has changed from the patient’s normal state. Changes can include alterations in consciousness, attention, behavior, cognition, language, speech, and praxis and can reflect varying degrees of cerebral dysfunction.
Electrolyte abnormalities
Disorders of sodium homeostasis are common in hospitalized patients and may contribute to the onset of delirium. Hyponatremia is especially frequent and often iatrogenic, with a prevalence significantly higher in women (2.1% vs 1.3%, P = .0044) and in the elderly.2
Neurologic manifestations are often the result of cerebral edema due to osmolar volume shifts.3–6 Acute hyponatremic encephalopathy is most likely to occur when sodium shifts are rapid, usually within 24 hours, and is often seen in postoperative patients requiring significant volume resuscitation with hypotonic fluids.6 Young premenopausal women appear to be at especially high risk of permanent brain damage secondary to hyponatremic encephalopathy,7 a finding that may reflect the limited compliance within the intracranial vault and lack of significant involutional parenchymal changes that occur with aging.8–11
Aging also has important effects on fluid balance, as restoration of body fluid homeostasis is slower in older patients.12
Hormonal effects of estrogen appear to play a synergistic role in the expression of arginine vasopressin in postmenopausal women, further contributing to hyponatremia.
Although our patient has mild hyponatremia, there has been no acute change in her sodium balance since admission to the hospital, and so it is unlikely to be the cause of her acute delirium. Her mild hyponatremia may in part be from hypo-osmolar maintenance fluids with dextrose 5% and 0.45% normal saline.
Mild chronic hyponatremia may affect balance and has been associated with increased mortality risk in certain chronic disease states, but this is unlikely to be the main cause of acute delirium.
Polypharmacy
Patients admitted to the hospital with polypharmacy are at high risk of drug-induced delirium. In approaching delirium, a patient’s medications should be evaluated for interactions, as well as for possible effects of newly prescribed drugs. New medications that affect cytochrome P450 enzymes warrant investigation, as do drugs with narrow therapeutic windows that the patient has been using long-term.
Consultation with a clinical pharmacist is often helpful. Macrolides, protease inhibitors, and nondihydropyridine calcium channel blockers are common P450 inhibitors, while many anticonvulsants are known inducers of the P450 system. Selective serotonin reuptake inhibitors and diuretics can lead to electrolyte imbalances such as hyponatremia, which may further predispose to bouts of delirium, as described above.
The patient’s extensive list of psychoactive medications makes polypharmacy a significant risk factor for delirium. Quetiapine and venlafaxine both cause sedation and increase the risk of serotonin syndrome. However, in this case, the patient does not have marked fever, rigidity, or hyperreflexia to corroborate that diagnosis.
Dementia
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), defines dementia as a disorder involving cognitive impairment in at least 1 cognitive domain, with a significant decline from a previous level of functioning.1 These impairments need not necessarily occur separately from bouts of delirium, but the time course for most forms of dementia tends to be progressive over a subacute to chronic duration.
Dementia increases the risk for acute confusion and delirium in hospitalized patients.13 This is partly reflected by pathophysiologic changes that leave elderly patients susceptible to the effects of anticholinergic drugs.14 Structural changes due to small-vessel ischemia may also predispose patients to seizures in the setting of metabolic derangement or critical illness. Diagnosing dementia thus remains a challenge, as dementia must be clearly distinguished from other disorders such as delirium and depression.
The acute change in this patient’s case makes the isolated diagnosis of dementia much less likely than other causes of altered mental status. Also, her previous level of function does not suggest a clinically significant personal history of impairment.
Mental illness
Several studies have examined the link between preoperative mental health disorders and postoperative delirium.15–17 Depression appears to be a risk factor for postoperative delirium in patients undergoing elective orthopedic surgery,15 and this includes elderly patients.16 While a clear etiologic link has yet to be determined, disruption of circadian rhythm and abnormal cerebral response to stress may play a role. Studies have also suggested an association between schizophrenia and delirium, though this may be related to perioperative suspension of medications.17
Bipolar disorder has not been well studied with regard to postoperative complications. However, this patient has had a previous episode of decompensated mania, therefore making bipolar disorder a plausible condition in the differential diagnosis.
CASE CONTINUED: ACUTE DETERIORATION
Without a clearly identifiable cause for our patient’s acute confusional state, neurology and medical consultants recommend neuroimaging.
Computed tomography (CT) and magnetic resonance imaging (MRI) without contrast are ordered and performed on postoperative day 11 and demonstrate chronic small-vessel ischemic disease, consistent with our patient’s age, as well as frontotemporal atrophy. There is no evidence of mass effect, bleeding, or acute ischemia.
Overnight, she becomes obtunded, and the rapid response team is called. Her vital signs appear stable, and she is afebrile. Basic laboratory studies, imaging, and electrocardiography are repeated, and the results are unchanged from recent tests. She is transferred to the intensive care unit (ICU) for closer monitoring.
2. What is most likely cause of the patient’s declining mental status, and what is the next appropriate step?
- Acute stroke: repeat MRI with contrast
- Urinary tract infection: order blood and urine cultures, and start empiric antibiotics
- Neuroleptic malignant syndrome: start dantrolene
- Seizures: order electroencephalography (EEG)
Acute stroke
Acute stroke can affect mental status and consciousness through several pathways. Stroke syndromes can vary in presentation depending on the level of cortical and subcortical involvement, with clinical manifestations including confusion, aphasia, neglect, and inattention. Wakefulness and the ability to maintain consciousness is impaired, with disruption of the ascending reticular activating system, often seen in injuries to the brainstem. Large territorial or hemispheric infarcts, with subsequent cerebral edema, can also disrupt this system and lead to cerebral herniation and coma.
MRI without contrast is extremely sensitive for ischemia and can typically detect ischemia in acute stroke within 3 to 30 minutes.18–20 Repeating the study with contrast is unlikely to provide additional benefit.
In our patient’s case, the lack of localizing neurologic symptoms, in addition to her recent negative neuroimaging workup, makes the diagnosis of acute stroke unlikely.
Infection
The role of severe infection in patients with altered mental status is well documented and likely relates to diffuse cerebral dysfunction caused by an inflammatory cascade. Less well understood is the role of occult infection, especially urinary tract infection, in otherwise immunocompetent patients. Urinary tract infection has long been thought to cause delirium in otherwise asymptomatic elderly patients, but few studies have examined this relationship, and those studies have been shown to have significant methodologic errors.21 In the absence of better data, urinary tract infection as the cause of frank delirium in an otherwise well patient should be viewed with skepticism, and alternative causes should be sought.
Although the patient has a nonspecific leukocytosis, her benign urinalysis and lack of corroborating evidence makes urinary tract infection an unlikely cause of her frank delirium.
Neuroleptic malignant syndrome
Neuroleptic malignant syndrome is defined as fever, rigidity, mental status changes, and autonomic instability after exposure to antidopaminergic drugs. It is classically seen after administration of typical antipsychotics, though atypical antipsychotics and antiemetic drugs may be implicated as well.
Patients often exhibit agitation and confusion, which when severe may progress to mutism and catatonia. Likewise, psychotropic drugs such as quetiapine and venlafaxine, used in combination, have the additional risk of serotonin syndrome.
Additional symptoms include hyperreflexia, ataxia, and myoclonus. Withdrawal of the causative agent and supportive care are the mainstays of therapy. Targeted therapies with agents such as dantrolene, bromocriptine, and amantadine have also been reported anecdotally, but their efficacy is unclear, with variable results.22
As noted earlier, the addition of quetiapine to the patient’s already lengthy medication list could conceivably cause neuroleptic malignant syndrome or serotonin syndrome and should be considered. However, additional neurologic findings to confirm this diagnosis are lacking.
Seizures
Nonconvulsive seizure, particularly nonconvulsive status epilepticus (NCSE), is not well recognized and is particularly challenging to diagnose without EEG. In several case series of patients presenting to the emergency room with altered mental status, NCSE was found in 16% to 28% of patients in whom EEG was performed after an initial evaluation failed to show an obvious cause for the delirium.23,24 Historical features are unreliable for ruling out NCSE as a cause of delirium, as up to 41% of patients in whom the condition is ultimately diagnosed have only confusion as the presenting clinical symptom.25
Likewise, alternating ictal and postictal periods may mimic the typical waxing and waning course classically associated with delirium of other causes. Physical findings such as nystagmus, anisocoria, and hippus may be helpful but are often overlooked or absent. EEG is thus an essential requirement for the diagnosis.26
Given the lack of a clear diagnosis, a workup with EEG should be considered in this patient.
CASE CONTINUED: ADDITIONAL SIGNS
In the ICU, our patient is evaluated by the intensivist team. Her vital signs are stable, and while she is now awakening, she is unable to follow commands and remains mute. She does not initiate movement spontaneously but offers slight resistance to passive movements, holding and maintaining postures her extremities are placed in. She keeps her eyes closed, but when opened by the examining physician, dysconjugate gaze and anisocoria are noted.
3. What clinical entity is most consistent with these physical findings, and what is the next step in management?
- Catatonia secondary to bipolar disorder type I: challenge with intravenous lorazepam 2 mg
- Oculomotor nerve palsy due to enlarging intracranial aneurysm: aggressive blood pressure lowering, elevation of the head of the bed
- Toxic leukoencephalopathy: supportive care and withdrawal of the causative agent
- NCSE: challenge with intravenous lorazepam 2 mg and order EEG
Catatonia
The DSM-5 defines catatonia as a behavioral syndrome complicating an underlying psychiatric or medical condition, as opposed to a distinct diagnosis. It is most commonly encountered in psychiatric illnesses including bipolar disorder, major depression, and schizophrenia. Akinesis, stupor, mutism, and “waxy” flexibility often dominate the clinical picture.
The pathophysiology is poorly defined, but likely involves neurotransmitter imbalances particularly with an increase in N-methyl-d-aspartate (NMDA) activity and suppression of gamma-aminobutyric acid (GABA) activity. This hypothesis is supported by the finding that benzodiazepines, electroconvulsive therapy, and NMDA antagonists such as amantadine are all effective in treating catatonia.27,28 Findings of focal neurologic abnormalities warrant further investigation. EEG may be necessary to differentiate catatonia from NCSE, as both may respond to a benzodiazepine challenge.
As pure catatonia is a diagnosis of exclusion, further workup, including EEG, is necessary to confirm the diagnosis.
Oculomotor nerve palsy
Anisocoria together with dysconjugate gaze should prompt consideration of a lesion involving the oculomotor nerve. Loss of tonic muscle activity from the lateral rectus and superior oblique cause a downward and outward gaze. Furthermore, loss of parasympathetic tone occurs with compressive palsies of the oculomotor nerve, clinically manifesting as a mydriatic and unreactive pupil with ptosis. Given its anatomic course and proximity to other vascular and parenchymal structures, the oculomotor nerve is vulnerable to compression from many sources, including aneurysmal dilation (especially of the posterior cerebral artery), uncal herniation, and inflammation of the cavernous sinus.
Noncontrast CT and lumbar puncture are very sensitive for making the diagnosis of sentinel bleeding within the first 24 hours,29 whereas computed tomographic angiography and magnetic resonance angiography can reliably detect unruptured aneurysms as small as 3 mm.30
Conditions that can lead to oculomotor palsy are unlikely to cause an acute gain in appendicular muscle tone, as noted by the catatonia this patient is demonstrating. Also, mass lesions or bleeding associated with oculomotor palsy is likely to cause acute loss of tone. Chronic upper-motor neuron lesions lead to spasticity rather than the waxy flexibility seen in this patient. In our patient, the findings of isolated anisocoria without further clinical evidence of oculomotor nerve compression make this diagnosis unlikely.
Toxic leukoencephalopathy
Toxic leukoencephalopathy—widespread destruction of myelin, particularly in the white matter tracts that support higher cortical functions—can be caused by antineoplastic agents, immunosuppressant agents, and industrial solvents, as well as by abuse of vaporized drugs such as heroin (“chasing the dragon”). In its mild forms it may cause behavioral disturbances or inattention. In severe forms, a neurobehavioral syndrome of akinetic mutism may be present and can mimic catatonia.31
The diagnosis is often based on the clinical history and neuroimaging, particularly MRI, which demonstrates hyperintensity of the white matter tracts in T2-weighted images.32
This patient does not have a clear history of exposure to an agent typically associated with toxic leukoencephalopathy and does not have the corroborating MRI findings to support this diagnosis.
CASE CONTINUED
Because recent neuroimaging revealed no structural brain lesions and no cause for brain herniation, the patient receives a challenge of 2 mg of intravenous lorazepam to treat potential NCSE. Subsequent improvement is noted in her anisocoria, gaze deviation, and encephalopathy. EEG reveals frequent focal seizures arising from mesial frontal regions with bilateral hemisphere propagation, consistent with bifrontal focal NCSE.
As our patient is being transferred to a room for continuous EEG monitoring, her condition begins to deteriorate, and she again becomes more encephalopathic, with anisocoria and dysconjugate gaze. Additional doses of lorazepam are given (to complete a 0.1-mg/kg load), and additional therapy with intravenous fosphenytoin (20-mg/kg load) is given. Intubation is done for airway protection.
Continuous EEG monitoring reveals multiple frequent electrographic seizures arising from the bifrontal territories, concerning for persistent focal NCSE. A midazolam drip is initiated for EEG burst suppression of cerebral activity. Over 24 hours, EEG shows resolution of seizure activity. As the patient is weaned from sedation, she awakens and follows commands consistently, tolerating extubation without complications. Her neurologic status remains stable over the next 48 hours, having returned to her neurologic baseline level of functioning. She is able to be transferred out of the ICU in stable condition while continuing on scheduled antiepileptic therapy with phenytoin.
ALTERED MENTAL STATUS IN INPATIENTS
Altered mental status is one of the most frequently encountered reasons for medical consultation from nonmedical services. The workup and management of metabolic, toxic, psychiatric, and neurologic causes requires a deep appreciation for the broad differential diagnosis and a multidisciplinary approach. Physicians caring for these patients should avoid prematurely drawing conclusions when the patient’s clinical condition fails to respond to typical measures.
Delirium is a challenging adverse event in older patients during hospitalization, with a significant national financial burden of $164 billion per year.33 The prevalence of delirium in adults on hospital admission is estimated as 14% to 24%, with an inpatient hospitalization incidence ranging from 6% to 56% in general hospital patients.34 In addition, postoperative delirium has been reported in 15% to 53% of older patients.35
While delirium is preventable in 30% to 40% of cases,36,37 it remains an important independent prognostic determinant of hospital outcomes.38–40
Delirium in hospitalized patients requires a thorough, individualized workup. In our patient’s case, the clinical findings of hypoactive delirium were found to be manifestations of NCSE, a rare life-threatening and potentially reversible neurologic disease.
While establishing seizures as a diagnosis, careful attention must first be directed towards investigating environmental or metabolic triggers that may be inciting the disease. This often involves a similar workup for metabolic derangements, as seen in the approach to delirium.
The diagnosis of NCSE, while made in this patient’s case, remains challenging. Careful physical examination should assess for automatisms, “negative” symptoms (staring, aphasia, weakness), and “positive” symptoms (hallucinations, psychosis). Cataplexy, mutism, and other acute psychiatric features have been associated with NCSE,44 highlighting the importance of EEG. A trial of a benzodiazepine in conjunction with clinical and EEG monitoring may help guide clinical decision- making.
As there is no current universally accepted definition for NCSE nor an accepted agreement on required EEG diagnostic features at this time,41 accurate diagnosis is most likely to be obtained in facilities with both subspecialty neurologic consultation and EEG capabilities.
Our patient’s family history of Pick disease is interesting, as this is a progressive form of frontotemporal dementia with both sporadic and genetically linked cases. Recent studies have shown evidence that patients with neurodegenerative disease have increased seizure frequency early in the disease course,31 and efforts are under way to establish the incidence of first unprovoked seizure in patients with frontotemporal dementia. In our patient’s case, resolution of seizure activity yielded a return to her baseline level of neurologic function.
Early use of selective serotonin reuptake inhibitors has been shown to help with the behavioral symptoms of frontotemporal dementia,45 but increasing requirements over time may indicate progression of neurodegeneration and should warrant further appropriate investigation.
In our patient’s case, escalating dose requirements may have reflected worsening frontotemporal atrophy. However, the diagnosis of a neurodegenerative disease such as frontotemporal dementia in a patient such as ours is not definitively established at this time and is being investigated on an outpatient basis.
Given the frequency of delirium and its many risk factors in the inpatient setting, verifying a causative diagnosis can be difficult. Detailed consideration of the patient’s individual clinical circumstances, often in concert with appropriate subspecialty consultations, is essential to the evaluation. Although it is time-intensive, multidisciplinary intervention can lead to safer outcomes and shorter hospital stays.
A 64-year-old woman undergoes elective T10-S1 nerve decompression with fusion for chronic idiopathic scoliosis. Soon afterward, she develops acute urinary retention attributed to an Escherichia coli urinary tract infection and narcotic medications. She is treated with antibiotics, an indwelling catheter is inserted, and her symptoms resolve. She is transferred to the inpatient physical rehabilitation unit.
On postoperative day 9, she develops an acute change in mental status, suddenly becoming extremely anxious and falsely believing she has a “terminal illness.” A psychiatrist suggests that these symptoms are a manifestation of delirium, given the patient’s recent surgery and exposure to benzodiazepine and narcotic medications. On postoperative day 10, she is awake but is now mute and uncooperative. An internist is consulted for an evaluation for encephalopathy and delirium.
MEDICAL HISTORY
Her medical history, obtained by chart review and interviewing her husband, includes well-controlled bipolar disorder over the last 4 years, with no episodes of frank psychosis or mania. She had a “bout of delirium” 4 years earlier attributed to a catastrophic life event, but the symptoms resolved after adjustment of her anxiolytic and mood-stabilizing drugs. She also has well-controlled hypertension, hypothyroidism, and gastroesophageal reflux. Her only surgery was her recent elective procedure.
She has a family history of dementia (Pick disease in her mother).
She is married, lives with her husband, and has an adult son. She is employed as a media specialist and also teaches English as a second language. Before this hospital admission, she was described as happy and content, though her primary psychiatrist had noted intermittent anxiety. Her husband does not suspect illicit drug use and denies significant alcohol or tobacco abuse.
A thorough review of systems is not possible, given her encephalopathy. But before her acute decline, she had complained of “choking on blood” and a subjective inability to swallow.
Her home medications include dextroamphetamine extended-release, alprazolam as needed for sleep, venlafaxine extended-release, lamotrigine, lisinopril, propranolol, amlodipine, atorvastatin, levothyroxine, omeprazole, iron, and vitamin B12. At the time of the evaluation, she is on her home medications with the addition of olanzapine, vitamin D, polyethylene glycol, and an intravenous infusion of dextrose 5% with 0.45% saline at a rate of 100 mL/hour. She has allergies to latex, penicillin, peanuts, and shellfish.
PHYSICAL EXAMINATION
On physical examination, the patient seems healthy and appears normal for her stated age. She is wearing a spinal brace and is in no apparent distress. She is afebrile, pulse 104 beats per minute, respirations 16 breaths per minute and unlabored, and oxygen saturation good on room air. The skin is normal. No thyromegaly, bruits, or lymphadenopathy is noted. Cardiovascular, respiratory, and abdominal examinations, though limited by the spinal brace, are unremarkable. She has no evidence of peripheral edema or vascular insufficiency. Muscle bulk and tone are adequate and symmetric.
She is awake and alert and able to follow simple commands with some prompting. She does not initiate movements spontaneously. She makes some eye contact but does not track or acknowledge the interviewer consistently and does not respond verbally to questions. Her sclera are nonicteric, the pupils are equally round and reactive to light, and the external ocular muscles are intact. There is no facial asymmetry, and the tongue protrudes at midline. She blinks appropriately to threat bilaterally. Strength is at least 3/5 in the upper extremities and 2/5 in the lower extremities, though the examination is limited by lack of patient cooperation. She shows minimal grimace on noxious stimulation but does not withdraw extremities. Reflexes are present and mildly depressed symmetrically. Plantar reflexes are downgoing bilaterally.
INITIAL LABORATORY EVALUATION
On initial laboratory testing, the serum sodium is 132 mmol/L (reference range 136–144), stable since admission. Point-of-care glucose is 98 mg/dL. Aspartate aminotransferase and alanine aminotransferase levels are mildly elevated at 59 U/L (13–35) and 51 U/L (7–38), respectively, but serum ammonia is undetectable. Vitamin B12, folate, thyroid-stimulating hormone, and free thyroxine are within the normal ranges. Leukocytosis is noted, with 14 × 109 cells/L (3.7–11.0), 86% neutrophils, and a mild left shift. Urinalysis is negative for leukocyte esterase, nitrites, and white blood cells.
APPROACH TO ALTERED MENTAL STATUS
1. Which of the following risk factors predisposes this patient to postoperative delirium?
- Hyponatremia
- Polypharmacy
- Family history of dementia
- Depression
Altered mental status, or encephalopathy, is one of the most common yet challenging conditions in medicine. When a consult is placed for altered mental status, it is important to determine the affected domain that has changed from the patient’s normal state. Changes can include alterations in consciousness, attention, behavior, cognition, language, speech, and praxis and can reflect varying degrees of cerebral dysfunction.
Electrolyte abnormalities
Disorders of sodium homeostasis are common in hospitalized patients and may contribute to the onset of delirium. Hyponatremia is especially frequent and often iatrogenic, with a prevalence significantly higher in women (2.1% vs 1.3%, P = .0044) and in the elderly.2
Neurologic manifestations are often the result of cerebral edema due to osmolar volume shifts.3–6 Acute hyponatremic encephalopathy is most likely to occur when sodium shifts are rapid, usually within 24 hours, and is often seen in postoperative patients requiring significant volume resuscitation with hypotonic fluids.6 Young premenopausal women appear to be at especially high risk of permanent brain damage secondary to hyponatremic encephalopathy,7 a finding that may reflect the limited compliance within the intracranial vault and lack of significant involutional parenchymal changes that occur with aging.8–11
Aging also has important effects on fluid balance, as restoration of body fluid homeostasis is slower in older patients.12
Hormonal effects of estrogen appear to play a synergistic role in the expression of arginine vasopressin in postmenopausal women, further contributing to hyponatremia.
Although our patient has mild hyponatremia, there has been no acute change in her sodium balance since admission to the hospital, and so it is unlikely to be the cause of her acute delirium. Her mild hyponatremia may in part be from hypo-osmolar maintenance fluids with dextrose 5% and 0.45% normal saline.
Mild chronic hyponatremia may affect balance and has been associated with increased mortality risk in certain chronic disease states, but this is unlikely to be the main cause of acute delirium.
Polypharmacy
Patients admitted to the hospital with polypharmacy are at high risk of drug-induced delirium. In approaching delirium, a patient’s medications should be evaluated for interactions, as well as for possible effects of newly prescribed drugs. New medications that affect cytochrome P450 enzymes warrant investigation, as do drugs with narrow therapeutic windows that the patient has been using long-term.
Consultation with a clinical pharmacist is often helpful. Macrolides, protease inhibitors, and nondihydropyridine calcium channel blockers are common P450 inhibitors, while many anticonvulsants are known inducers of the P450 system. Selective serotonin reuptake inhibitors and diuretics can lead to electrolyte imbalances such as hyponatremia, which may further predispose to bouts of delirium, as described above.
The patient’s extensive list of psychoactive medications makes polypharmacy a significant risk factor for delirium. Quetiapine and venlafaxine both cause sedation and increase the risk of serotonin syndrome. However, in this case, the patient does not have marked fever, rigidity, or hyperreflexia to corroborate that diagnosis.
Dementia
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), defines dementia as a disorder involving cognitive impairment in at least 1 cognitive domain, with a significant decline from a previous level of functioning.1 These impairments need not necessarily occur separately from bouts of delirium, but the time course for most forms of dementia tends to be progressive over a subacute to chronic duration.
Dementia increases the risk for acute confusion and delirium in hospitalized patients.13 This is partly reflected by pathophysiologic changes that leave elderly patients susceptible to the effects of anticholinergic drugs.14 Structural changes due to small-vessel ischemia may also predispose patients to seizures in the setting of metabolic derangement or critical illness. Diagnosing dementia thus remains a challenge, as dementia must be clearly distinguished from other disorders such as delirium and depression.
The acute change in this patient’s case makes the isolated diagnosis of dementia much less likely than other causes of altered mental status. Also, her previous level of function does not suggest a clinically significant personal history of impairment.
Mental illness
Several studies have examined the link between preoperative mental health disorders and postoperative delirium.15–17 Depression appears to be a risk factor for postoperative delirium in patients undergoing elective orthopedic surgery,15 and this includes elderly patients.16 While a clear etiologic link has yet to be determined, disruption of circadian rhythm and abnormal cerebral response to stress may play a role. Studies have also suggested an association between schizophrenia and delirium, though this may be related to perioperative suspension of medications.17
Bipolar disorder has not been well studied with regard to postoperative complications. However, this patient has had a previous episode of decompensated mania, therefore making bipolar disorder a plausible condition in the differential diagnosis.
CASE CONTINUED: ACUTE DETERIORATION
Without a clearly identifiable cause for our patient’s acute confusional state, neurology and medical consultants recommend neuroimaging.
Computed tomography (CT) and magnetic resonance imaging (MRI) without contrast are ordered and performed on postoperative day 11 and demonstrate chronic small-vessel ischemic disease, consistent with our patient’s age, as well as frontotemporal atrophy. There is no evidence of mass effect, bleeding, or acute ischemia.
Overnight, she becomes obtunded, and the rapid response team is called. Her vital signs appear stable, and she is afebrile. Basic laboratory studies, imaging, and electrocardiography are repeated, and the results are unchanged from recent tests. She is transferred to the intensive care unit (ICU) for closer monitoring.
2. What is most likely cause of the patient’s declining mental status, and what is the next appropriate step?
- Acute stroke: repeat MRI with contrast
- Urinary tract infection: order blood and urine cultures, and start empiric antibiotics
- Neuroleptic malignant syndrome: start dantrolene
- Seizures: order electroencephalography (EEG)
Acute stroke
Acute stroke can affect mental status and consciousness through several pathways. Stroke syndromes can vary in presentation depending on the level of cortical and subcortical involvement, with clinical manifestations including confusion, aphasia, neglect, and inattention. Wakefulness and the ability to maintain consciousness is impaired, with disruption of the ascending reticular activating system, often seen in injuries to the brainstem. Large territorial or hemispheric infarcts, with subsequent cerebral edema, can also disrupt this system and lead to cerebral herniation and coma.
MRI without contrast is extremely sensitive for ischemia and can typically detect ischemia in acute stroke within 3 to 30 minutes.18–20 Repeating the study with contrast is unlikely to provide additional benefit.
In our patient’s case, the lack of localizing neurologic symptoms, in addition to her recent negative neuroimaging workup, makes the diagnosis of acute stroke unlikely.
Infection
The role of severe infection in patients with altered mental status is well documented and likely relates to diffuse cerebral dysfunction caused by an inflammatory cascade. Less well understood is the role of occult infection, especially urinary tract infection, in otherwise immunocompetent patients. Urinary tract infection has long been thought to cause delirium in otherwise asymptomatic elderly patients, but few studies have examined this relationship, and those studies have been shown to have significant methodologic errors.21 In the absence of better data, urinary tract infection as the cause of frank delirium in an otherwise well patient should be viewed with skepticism, and alternative causes should be sought.
Although the patient has a nonspecific leukocytosis, her benign urinalysis and lack of corroborating evidence makes urinary tract infection an unlikely cause of her frank delirium.
Neuroleptic malignant syndrome
Neuroleptic malignant syndrome is defined as fever, rigidity, mental status changes, and autonomic instability after exposure to antidopaminergic drugs. It is classically seen after administration of typical antipsychotics, though atypical antipsychotics and antiemetic drugs may be implicated as well.
Patients often exhibit agitation and confusion, which when severe may progress to mutism and catatonia. Likewise, psychotropic drugs such as quetiapine and venlafaxine, used in combination, have the additional risk of serotonin syndrome.
Additional symptoms include hyperreflexia, ataxia, and myoclonus. Withdrawal of the causative agent and supportive care are the mainstays of therapy. Targeted therapies with agents such as dantrolene, bromocriptine, and amantadine have also been reported anecdotally, but their efficacy is unclear, with variable results.22
As noted earlier, the addition of quetiapine to the patient’s already lengthy medication list could conceivably cause neuroleptic malignant syndrome or serotonin syndrome and should be considered. However, additional neurologic findings to confirm this diagnosis are lacking.
Seizures
Nonconvulsive seizure, particularly nonconvulsive status epilepticus (NCSE), is not well recognized and is particularly challenging to diagnose without EEG. In several case series of patients presenting to the emergency room with altered mental status, NCSE was found in 16% to 28% of patients in whom EEG was performed after an initial evaluation failed to show an obvious cause for the delirium.23,24 Historical features are unreliable for ruling out NCSE as a cause of delirium, as up to 41% of patients in whom the condition is ultimately diagnosed have only confusion as the presenting clinical symptom.25
Likewise, alternating ictal and postictal periods may mimic the typical waxing and waning course classically associated with delirium of other causes. Physical findings such as nystagmus, anisocoria, and hippus may be helpful but are often overlooked or absent. EEG is thus an essential requirement for the diagnosis.26
Given the lack of a clear diagnosis, a workup with EEG should be considered in this patient.
CASE CONTINUED: ADDITIONAL SIGNS
In the ICU, our patient is evaluated by the intensivist team. Her vital signs are stable, and while she is now awakening, she is unable to follow commands and remains mute. She does not initiate movement spontaneously but offers slight resistance to passive movements, holding and maintaining postures her extremities are placed in. She keeps her eyes closed, but when opened by the examining physician, dysconjugate gaze and anisocoria are noted.
3. What clinical entity is most consistent with these physical findings, and what is the next step in management?
- Catatonia secondary to bipolar disorder type I: challenge with intravenous lorazepam 2 mg
- Oculomotor nerve palsy due to enlarging intracranial aneurysm: aggressive blood pressure lowering, elevation of the head of the bed
- Toxic leukoencephalopathy: supportive care and withdrawal of the causative agent
- NCSE: challenge with intravenous lorazepam 2 mg and order EEG
Catatonia
The DSM-5 defines catatonia as a behavioral syndrome complicating an underlying psychiatric or medical condition, as opposed to a distinct diagnosis. It is most commonly encountered in psychiatric illnesses including bipolar disorder, major depression, and schizophrenia. Akinesis, stupor, mutism, and “waxy” flexibility often dominate the clinical picture.
The pathophysiology is poorly defined, but likely involves neurotransmitter imbalances particularly with an increase in N-methyl-d-aspartate (NMDA) activity and suppression of gamma-aminobutyric acid (GABA) activity. This hypothesis is supported by the finding that benzodiazepines, electroconvulsive therapy, and NMDA antagonists such as amantadine are all effective in treating catatonia.27,28 Findings of focal neurologic abnormalities warrant further investigation. EEG may be necessary to differentiate catatonia from NCSE, as both may respond to a benzodiazepine challenge.
As pure catatonia is a diagnosis of exclusion, further workup, including EEG, is necessary to confirm the diagnosis.
Oculomotor nerve palsy
Anisocoria together with dysconjugate gaze should prompt consideration of a lesion involving the oculomotor nerve. Loss of tonic muscle activity from the lateral rectus and superior oblique cause a downward and outward gaze. Furthermore, loss of parasympathetic tone occurs with compressive palsies of the oculomotor nerve, clinically manifesting as a mydriatic and unreactive pupil with ptosis. Given its anatomic course and proximity to other vascular and parenchymal structures, the oculomotor nerve is vulnerable to compression from many sources, including aneurysmal dilation (especially of the posterior cerebral artery), uncal herniation, and inflammation of the cavernous sinus.
Noncontrast CT and lumbar puncture are very sensitive for making the diagnosis of sentinel bleeding within the first 24 hours,29 whereas computed tomographic angiography and magnetic resonance angiography can reliably detect unruptured aneurysms as small as 3 mm.30
Conditions that can lead to oculomotor palsy are unlikely to cause an acute gain in appendicular muscle tone, as noted by the catatonia this patient is demonstrating. Also, mass lesions or bleeding associated with oculomotor palsy is likely to cause acute loss of tone. Chronic upper-motor neuron lesions lead to spasticity rather than the waxy flexibility seen in this patient. In our patient, the findings of isolated anisocoria without further clinical evidence of oculomotor nerve compression make this diagnosis unlikely.
Toxic leukoencephalopathy
Toxic leukoencephalopathy—widespread destruction of myelin, particularly in the white matter tracts that support higher cortical functions—can be caused by antineoplastic agents, immunosuppressant agents, and industrial solvents, as well as by abuse of vaporized drugs such as heroin (“chasing the dragon”). In its mild forms it may cause behavioral disturbances or inattention. In severe forms, a neurobehavioral syndrome of akinetic mutism may be present and can mimic catatonia.31
The diagnosis is often based on the clinical history and neuroimaging, particularly MRI, which demonstrates hyperintensity of the white matter tracts in T2-weighted images.32
This patient does not have a clear history of exposure to an agent typically associated with toxic leukoencephalopathy and does not have the corroborating MRI findings to support this diagnosis.
CASE CONTINUED
Because recent neuroimaging revealed no structural brain lesions and no cause for brain herniation, the patient receives a challenge of 2 mg of intravenous lorazepam to treat potential NCSE. Subsequent improvement is noted in her anisocoria, gaze deviation, and encephalopathy. EEG reveals frequent focal seizures arising from mesial frontal regions with bilateral hemisphere propagation, consistent with bifrontal focal NCSE.
As our patient is being transferred to a room for continuous EEG monitoring, her condition begins to deteriorate, and she again becomes more encephalopathic, with anisocoria and dysconjugate gaze. Additional doses of lorazepam are given (to complete a 0.1-mg/kg load), and additional therapy with intravenous fosphenytoin (20-mg/kg load) is given. Intubation is done for airway protection.
Continuous EEG monitoring reveals multiple frequent electrographic seizures arising from the bifrontal territories, concerning for persistent focal NCSE. A midazolam drip is initiated for EEG burst suppression of cerebral activity. Over 24 hours, EEG shows resolution of seizure activity. As the patient is weaned from sedation, she awakens and follows commands consistently, tolerating extubation without complications. Her neurologic status remains stable over the next 48 hours, having returned to her neurologic baseline level of functioning. She is able to be transferred out of the ICU in stable condition while continuing on scheduled antiepileptic therapy with phenytoin.
ALTERED MENTAL STATUS IN INPATIENTS
Altered mental status is one of the most frequently encountered reasons for medical consultation from nonmedical services. The workup and management of metabolic, toxic, psychiatric, and neurologic causes requires a deep appreciation for the broad differential diagnosis and a multidisciplinary approach. Physicians caring for these patients should avoid prematurely drawing conclusions when the patient’s clinical condition fails to respond to typical measures.
Delirium is a challenging adverse event in older patients during hospitalization, with a significant national financial burden of $164 billion per year.33 The prevalence of delirium in adults on hospital admission is estimated as 14% to 24%, with an inpatient hospitalization incidence ranging from 6% to 56% in general hospital patients.34 In addition, postoperative delirium has been reported in 15% to 53% of older patients.35
While delirium is preventable in 30% to 40% of cases,36,37 it remains an important independent prognostic determinant of hospital outcomes.38–40
Delirium in hospitalized patients requires a thorough, individualized workup. In our patient’s case, the clinical findings of hypoactive delirium were found to be manifestations of NCSE, a rare life-threatening and potentially reversible neurologic disease.
While establishing seizures as a diagnosis, careful attention must first be directed towards investigating environmental or metabolic triggers that may be inciting the disease. This often involves a similar workup for metabolic derangements, as seen in the approach to delirium.
The diagnosis of NCSE, while made in this patient’s case, remains challenging. Careful physical examination should assess for automatisms, “negative” symptoms (staring, aphasia, weakness), and “positive” symptoms (hallucinations, psychosis). Cataplexy, mutism, and other acute psychiatric features have been associated with NCSE,44 highlighting the importance of EEG. A trial of a benzodiazepine in conjunction with clinical and EEG monitoring may help guide clinical decision- making.
As there is no current universally accepted definition for NCSE nor an accepted agreement on required EEG diagnostic features at this time,41 accurate diagnosis is most likely to be obtained in facilities with both subspecialty neurologic consultation and EEG capabilities.
Our patient’s family history of Pick disease is interesting, as this is a progressive form of frontotemporal dementia with both sporadic and genetically linked cases. Recent studies have shown evidence that patients with neurodegenerative disease have increased seizure frequency early in the disease course,31 and efforts are under way to establish the incidence of first unprovoked seizure in patients with frontotemporal dementia. In our patient’s case, resolution of seizure activity yielded a return to her baseline level of neurologic function.
Early use of selective serotonin reuptake inhibitors has been shown to help with the behavioral symptoms of frontotemporal dementia,45 but increasing requirements over time may indicate progression of neurodegeneration and should warrant further appropriate investigation.
In our patient’s case, escalating dose requirements may have reflected worsening frontotemporal atrophy. However, the diagnosis of a neurodegenerative disease such as frontotemporal dementia in a patient such as ours is not definitively established at this time and is being investigated on an outpatient basis.
Given the frequency of delirium and its many risk factors in the inpatient setting, verifying a causative diagnosis can be difficult. Detailed consideration of the patient’s individual clinical circumstances, often in concert with appropriate subspecialty consultations, is essential to the evaluation. Although it is time-intensive, multidisciplinary intervention can lead to safer outcomes and shorter hospital stays.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013. http://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596. Accessed July 7, 2017.
- Mohan S, Gu S, Parikh A, Radhakrishnan J. Prevalence of hyponatremia and association with mortality: results from NHANES. Am J Med 2013; 126:1127–1137.e1.
- Sterns RH. Disorders of plasma sodium—causes, consequences, and correction. N Engl J Med 2015; 372:55–65.
- Rose B, Post T. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001.
- McManus ML, Churchwell KB, Strange K. Regulation of cell volume in health and disease. N Engl J Med 1995; 333:1260–1266.
- Strange K. Regulation of solute and water balance and cell volume in the central nervous system. J Am Soc Nephrol 1992; 3:12–27.
- Ayus JC, Wheeler JM, Arieff AI. Postoperative hyponatremic encephalopathy in menstruant women. Ann Intern Med 1992; 117:891–897.
- Gur RC, Mozley PD, Resnick SM, et al. Gender differences in age effect on brain atrophy measured by magnetic resonance imaging. Proc Natl Acad Sci USA 1991; 88:2845–2849.
- Rosomoff HL, Zugibe FT. Distribution of intracranial contents in experimental edema. Arch Neurol 1963; 9:26–34.
- Melton JE, Nattie EE. Brain and CSF water and ions during dilutional and isosmotic hyponatremia in the rat. Am J Physiol 1983; 244:R724–R732.
- Nattie EE, Edwards WH. Brain and CSF water and ions in newborn puppies during acute hypo- and hypernatremia. J Appl Physiol Respir Environ Exerc Physiol 1981; 51:1086–1091.
- Stachenfeld NS, DiPietro L, Palter SF, Nadel ER. Estrogen influences osmotic secretion of AVP and body water balance in postmenopausal women. Am J Physiol 1998; 274:R187–R195.
- Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc 2002; 50:1723–1732.
- de Smet Y, Ruberg M, Serdaru M, Dubois B, Lhermitte F, Agid Y. Confusion, dementia and anticholinergics in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1982; 45:1161–1164.
- Mollon B, Mahure SA, Ding DY, Zuckerman JD, Kwon YW. The influence of a history of clinical depression on peri-operative outcomes in elective total shoulder arthroplasty: a ten-year national analysis. Bone Joint J 2016; 98-B:818–824.
- Kosar CM, Tabloski PA, Travison TG, et al. Effect of preoperative pain and depressive symptoms on the development of postoperative delirium. Lancet Psychiatry 2014; 1:431–436.
- Copeland LA, Zeber JE, Pugh MJ, Mortensen EM, Restrepo MI, Lawrence VA. Postoperative complications in the seriously mentally ill: a systematic review of the literature. Ann Surg 2008; 248:31–38.
- Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann Neurol 1995; 37:231–241.
- Sorensen AG, Buonanno FS, Gonzalez RG, et al. Hyperacute stroke: evaluation with combined multisection diffusion-weighted and hemodynamically weighted echo-planar MR imaging. Radiology 1996; 199:391–401.
- Li F, Han S, Tatlisumak T, et al. A new method to improve in-bore middle cerebral artery occlusion in rats: demonstration with diffusion—and perfusion—weighted imaging. Stroke 1998; 29:1715–1720.
- Balogun SA, Philbrick JT. Delirium, a symptom of UTI in the elderly: fact or fable? A systematic review. Can Geriatr J 2013; 17:22–26.
- Reulbach U, Dütsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007; 11:R4.
- Naeije G, Depondt C, Meeus C, Korpak K, Pepersack T, Legros B. EEG patterns compatible with nonconvulsive status epilepticus are common in elderly patients with delirium: a prospective study with continuous EEG monitoring. Epilepsy Behav 2014; 36:18–21.
- Veran O, Kahane P, Thomas P, Hamelin S, Sabourdy C, Vercueil L. De novo epileptic confusion in the elderly: a 1-year prospective study. Epilepsia 2010; 51:1030–1035.
- Sutter R, Rüegg S, Kaplan PW. Epidemiology, diagnosis, and management of nonconvulsive status epilepticus. Opening Pandora’s box. Neurol Clin Pract 2012; 2:275–286.
- Husain AM, Horn GJ, Jacobson MP. Non-convulsive status epilepticus: usefulness of clinical features in selecting patients for urgent EEG. J Neurol Neurosurg Psychiatry 2003; 74:189–191.
- Ungvari GS, Chiu HF, Chow LY, Lau BS, Tang WK. Lorazepam for chronic catatonia: a randomized, double-blind, placebo-controlled cross-over study. Psychopharmacology (Berl) 1999; 142:393–398.
- Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci 2007; 19:406– 412.
- Perry JJ, Spacek A, Forbes M, et al. Is the combination of negative computed tomography result and negative lumbar puncture result sufficient to rule out subarachnoid hemorrhage? Ann Emerg Med 2008; 51:707–713.
- Li MH, Cheng YS, Li YD, et al. Large-cohort comparison between three-dimensional time-of-flight magnetic resonance and rotational digital subtraction angiographies in intracranial aneurysm detection. Stroke 2009; 40:3127–3129.
- Filley CM, Kleinschmidt-DeMasters BK. Toxic leukoencephalopathy. N Engl J Med 2001; 345:425–432.
- Magnetic resonance imaging of the central nervous system. Council on Scientific Affairs. Report of the Panel on Magnetic Resonance Imaging. JAMA 1988; 259:1211–1222.
- Leslie DL, Marcantonio ER, Zhang Y, Leo-Summers L, Inouye SK. One-year health care costs associated with delirium in the elderly population. Arch Intern Med 2008; 168:27–32.
- Inouye SK. Delirium in hospitalized older patients. Clin Geriatr Med 1998; 14:745–764.
- Agostini JV, Inouye SK, Hazzard W, Blass J. Delirium. In: Principles of Geriatric Medicine and Gerontology. 5th ed. New York, NY: McGraw-Hill; 2003:1503–1515.
- Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med 1999; 340:669–676.
- Marcantonio ER, Flacker JM, Wright RJ, Resnick NM. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc 2001; 49:516–522.
- Inouye SK, Rushing JT, Foreman MD, Palmer RM, Pompei P. Does delirium contribute to poor hospital outcomes? A three-site epidemiologic study. J Gen Intern Med 1998; 13:234–242.
- Rothschild JM, Bates DW, Leape LL. Preventable medical injuries in older patients. Arch Intern Med 2000; 160:2717–2728.
- Gillick MR, Serrell NA, Gillick LS. Adverse consequences of hospitalization in the elderly. Soc Sci Med 1982; 16:1033–1038.
- Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav 2000; 1:301-314.
- Rosenow F, Hamer HM, Knake S. The epidemiology of convulsive and nonconvulsive status epilepticus. Epilepsia 2007; 48(suppl 8):82–84.
- Woodford HJ, George J, Jackson M. Non-convulsive status epilepticus: a practical approach to diagnosis in confused older people. Postgrad Med J 2015; 91:655–661.
- Kaplan PW. Nonconvulsive status epilepticus in the emergency room. Epilepsia 1996; 37:643–650.
- Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry 1997; 58:212–216.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013. http://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596. Accessed July 7, 2017.
- Mohan S, Gu S, Parikh A, Radhakrishnan J. Prevalence of hyponatremia and association with mortality: results from NHANES. Am J Med 2013; 126:1127–1137.e1.
- Sterns RH. Disorders of plasma sodium—causes, consequences, and correction. N Engl J Med 2015; 372:55–65.
- Rose B, Post T. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001.
- McManus ML, Churchwell KB, Strange K. Regulation of cell volume in health and disease. N Engl J Med 1995; 333:1260–1266.
- Strange K. Regulation of solute and water balance and cell volume in the central nervous system. J Am Soc Nephrol 1992; 3:12–27.
- Ayus JC, Wheeler JM, Arieff AI. Postoperative hyponatremic encephalopathy in menstruant women. Ann Intern Med 1992; 117:891–897.
- Gur RC, Mozley PD, Resnick SM, et al. Gender differences in age effect on brain atrophy measured by magnetic resonance imaging. Proc Natl Acad Sci USA 1991; 88:2845–2849.
- Rosomoff HL, Zugibe FT. Distribution of intracranial contents in experimental edema. Arch Neurol 1963; 9:26–34.
- Melton JE, Nattie EE. Brain and CSF water and ions during dilutional and isosmotic hyponatremia in the rat. Am J Physiol 1983; 244:R724–R732.
- Nattie EE, Edwards WH. Brain and CSF water and ions in newborn puppies during acute hypo- and hypernatremia. J Appl Physiol Respir Environ Exerc Physiol 1981; 51:1086–1091.
- Stachenfeld NS, DiPietro L, Palter SF, Nadel ER. Estrogen influences osmotic secretion of AVP and body water balance in postmenopausal women. Am J Physiol 1998; 274:R187–R195.
- Fick DM, Agostini JV, Inouye SK. Delirium superimposed on dementia: a systematic review. J Am Geriatr Soc 2002; 50:1723–1732.
- de Smet Y, Ruberg M, Serdaru M, Dubois B, Lhermitte F, Agid Y. Confusion, dementia and anticholinergics in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1982; 45:1161–1164.
- Mollon B, Mahure SA, Ding DY, Zuckerman JD, Kwon YW. The influence of a history of clinical depression on peri-operative outcomes in elective total shoulder arthroplasty: a ten-year national analysis. Bone Joint J 2016; 98-B:818–824.
- Kosar CM, Tabloski PA, Travison TG, et al. Effect of preoperative pain and depressive symptoms on the development of postoperative delirium. Lancet Psychiatry 2014; 1:431–436.
- Copeland LA, Zeber JE, Pugh MJ, Mortensen EM, Restrepo MI, Lawrence VA. Postoperative complications in the seriously mentally ill: a systematic review of the literature. Ann Surg 2008; 248:31–38.
- Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann Neurol 1995; 37:231–241.
- Sorensen AG, Buonanno FS, Gonzalez RG, et al. Hyperacute stroke: evaluation with combined multisection diffusion-weighted and hemodynamically weighted echo-planar MR imaging. Radiology 1996; 199:391–401.
- Li F, Han S, Tatlisumak T, et al. A new method to improve in-bore middle cerebral artery occlusion in rats: demonstration with diffusion—and perfusion—weighted imaging. Stroke 1998; 29:1715–1720.
- Balogun SA, Philbrick JT. Delirium, a symptom of UTI in the elderly: fact or fable? A systematic review. Can Geriatr J 2013; 17:22–26.
- Reulbach U, Dütsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007; 11:R4.
- Naeije G, Depondt C, Meeus C, Korpak K, Pepersack T, Legros B. EEG patterns compatible with nonconvulsive status epilepticus are common in elderly patients with delirium: a prospective study with continuous EEG monitoring. Epilepsy Behav 2014; 36:18–21.
- Veran O, Kahane P, Thomas P, Hamelin S, Sabourdy C, Vercueil L. De novo epileptic confusion in the elderly: a 1-year prospective study. Epilepsia 2010; 51:1030–1035.
- Sutter R, Rüegg S, Kaplan PW. Epidemiology, diagnosis, and management of nonconvulsive status epilepticus. Opening Pandora’s box. Neurol Clin Pract 2012; 2:275–286.
- Husain AM, Horn GJ, Jacobson MP. Non-convulsive status epilepticus: usefulness of clinical features in selecting patients for urgent EEG. J Neurol Neurosurg Psychiatry 2003; 74:189–191.
- Ungvari GS, Chiu HF, Chow LY, Lau BS, Tang WK. Lorazepam for chronic catatonia: a randomized, double-blind, placebo-controlled cross-over study. Psychopharmacology (Berl) 1999; 142:393–398.
- Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci 2007; 19:406– 412.
- Perry JJ, Spacek A, Forbes M, et al. Is the combination of negative computed tomography result and negative lumbar puncture result sufficient to rule out subarachnoid hemorrhage? Ann Emerg Med 2008; 51:707–713.
- Li MH, Cheng YS, Li YD, et al. Large-cohort comparison between three-dimensional time-of-flight magnetic resonance and rotational digital subtraction angiographies in intracranial aneurysm detection. Stroke 2009; 40:3127–3129.
- Filley CM, Kleinschmidt-DeMasters BK. Toxic leukoencephalopathy. N Engl J Med 2001; 345:425–432.
- Magnetic resonance imaging of the central nervous system. Council on Scientific Affairs. Report of the Panel on Magnetic Resonance Imaging. JAMA 1988; 259:1211–1222.
- Leslie DL, Marcantonio ER, Zhang Y, Leo-Summers L, Inouye SK. One-year health care costs associated with delirium in the elderly population. Arch Intern Med 2008; 168:27–32.
- Inouye SK. Delirium in hospitalized older patients. Clin Geriatr Med 1998; 14:745–764.
- Agostini JV, Inouye SK, Hazzard W, Blass J. Delirium. In: Principles of Geriatric Medicine and Gerontology. 5th ed. New York, NY: McGraw-Hill; 2003:1503–1515.
- Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med 1999; 340:669–676.
- Marcantonio ER, Flacker JM, Wright RJ, Resnick NM. Reducing delirium after hip fracture: a randomized trial. J Am Geriatr Soc 2001; 49:516–522.
- Inouye SK, Rushing JT, Foreman MD, Palmer RM, Pompei P. Does delirium contribute to poor hospital outcomes? A three-site epidemiologic study. J Gen Intern Med 1998; 13:234–242.
- Rothschild JM, Bates DW, Leape LL. Preventable medical injuries in older patients. Arch Intern Med 2000; 160:2717–2728.
- Gillick MR, Serrell NA, Gillick LS. Adverse consequences of hospitalization in the elderly. Soc Sci Med 1982; 16:1033–1038.
- Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav 2000; 1:301-314.
- Rosenow F, Hamer HM, Knake S. The epidemiology of convulsive and nonconvulsive status epilepticus. Epilepsia 2007; 48(suppl 8):82–84.
- Woodford HJ, George J, Jackson M. Non-convulsive status epilepticus: a practical approach to diagnosis in confused older people. Postgrad Med J 2015; 91:655–661.
- Kaplan PW. Nonconvulsive status epilepticus in the emergency room. Epilepsia 1996; 37:643–650.
- Swartz JR, Miller BL, Lesser IM, Darby AL. Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry 1997; 58:212–216.
Another complication of cirrhosis
A 53-year-old Native American woman with a history of liver cirrhosis secondary to alcohol abuse presents to the emergency department after 2 days of diffuse abdominal pain and weakness. The pain was sudden in onset and has progressed relentlessly over the last day, reaching 9 on a scale of 10 in severity. Family members say that her oral intake has been decreased for the last 2 days, but she has had no fever, vomiting, change in bowel habit, blood in stool, or black stool. She has never undergone surgery, and has had one uncomplicated pregnancy.
Physical examination
Vital signs:
- Blood pressure 82/57 mm Hg
- Heart rate 96 beats per minute
- Temperature 37.3°C (99.1°F)
- Respiratory rate 16 per minute
- Oxygen saturation 92% while receiving oxygen at 2 L/minute.
The patient is somnolent and has scleral icterus. Her cardiopulmonary examination is normal. Her abdomen is tense, distended, and diffusely tender. She has bilateral +2 pitting edema in her lower extremities. She is oriented to person only and is noted to have asterixis. Her baseline Model for End-stage Liver Disease score is 18 points on a scale of 6 (less ill) to 40 (gravely ill).
Laboratory studies:
- Hemoglobin 9.8 g/dL (reference range 11.5–15.5)
- Platelet count 100 × 109/L (150–400)
- White blood cell count 9.9 × 109/L (3.7–11.0)
- Serum creatinine 1.06 mg/dL (0.58–0.96)
- Bilirubin 6.3 mg/dL (0.2–1.3)
- International normalized ratio of the prothrombin time 2.15 (0.8–1.2)
- Blood urea nitrogen 13 mg/dL (7–21)
- Serum albumin 2.7 g/dL (3.9–4.9).
Intravenous fluid resuscitation is initiated but the patient remains hypotensive, and on repeat laboratory testing 4 hours later her hemoglobin level has dropped to 7.3 mg/dL.
DIFFERENTIAL DIAGNOSIS
1. Which of the following are likely causes of this patient’s presentation?
- Splenic arterial aneurysm rupture
- Spontaneous bacterial peritonitis
- Variceal hemorrhage
- Portal vein thrombosis
- Abdominal aortic aneurysm rupture
Ruptured splenic artery aneurysm
Splenic artery aneurysms are the third most common intra-abdominal aneurysm, after those of the abdominal aorta and iliac artery.1 They are often asymptomatic and are being detected more frequently because of increased use of computed tomography (CT).2 Symptomatic splenic artery aneurysms may present with abdominal pain and have the potential to rupture, which can be life-threatening.3,4
This patient may have a ruptured splenic artery aneurysm, given her hemodynamic shock.
Spontaneous bacterial peritonitis
Ten percent to 20% of hospitalized patients with cirrhosis and ascites develop spontaneous bacterial peritonitis. Patients may present with ascites and abdominal pain, tenderness to palpation, fever, encephalopathy, or worsening liver and renal function.
Diagnostic paracentesis is paramount to delineate the cause of ascites; one should calculate the serum-ascites albumin gradient and obtain a cell count and culture of the ascitic fluid. The diagnosis of spontaneous bacterial peritonitis can be made if the ascitic fluid polymorphonuclear cell count is 0.25 × 109/L or higher, even if the ascitic fluid culture is negative.5,6 Simultaneous blood cultures should also be collected, as 50% of cases are associated with bacteremia.
The in-hospital mortality rate of an episode of spontaneous bacterial peritonitis has been reduced to 10% to 20% thanks to prompt diagnosis and empiric treatment with third-generation cephalosporins.7
Five percent of cases of infected ascites fluid are due to secondary bacterial peritonitis from a perforated viscus or a loculated abscess, which cannot be differentiated clinically from spontaneous bacterial peritonitis but can be diagnosed with CT.8
This patient may be presenting with septic shock secondary to either of these causes.
Variceal hemorrhage
Half of patients with cirrhosis have gastroesophageal varices due to portal hypertension. Endoscopic surveillance is warranted, as the risk of hemorrhage is 12% to 15% per year, and the mortality rate approaches 15% to 20% with each episode. Prompt resuscitation, diagnosis, and control of bleeding is paramount.
Esophagogastroduodenoscopy is used for both diagnosis and intervention. Short-term prophylactic use of antibiotics improves survival by preventing infections in the event bleeding recurs.9–11
Our patient may be presenting with hemodynamic shock from bleeding esophageal varices.
Portal vein thrombosis
Portal vein thrombosis is a common complication of cirrhosis, occurring in 5% to 28% of patients. The risk increases with the severity of liver disease and in association with hepatocellular carcinoma.12 Forty-three percent of cases are discovered incidentally in asymptomatic patients during ultrasonography, 39% present with upper gastrointestinal bleeding, and 18% present with abdominal pain.13,14
Portal vein thrombosis is the complete or partial obstruction of blood flow due to a thrombus in the lumen of the portal vein. Contrast ultrasonography and CT can be used to establish the diagnosis.15
Anticoagulation is recommended in cases of complete thrombosis in candidates for living-donor liver transplant and for those at risk of mesenteric ischemia because of the thrombus extending into the mesenteric veins. In symptomatic patients, the decision to initiate anticoagulation should be made on a case-by-case basis after appropriate screening and management of varices.16–18
Our patient’s thrombocytopenia reflects the severity of portal hypertension and increases her risk of portal vein thrombosis, but this is unlikely to be the sole cause of the hemodynamic compromise in this patient.
Ruptured abdominal aortic aneurysm
Rupture of an abdominal aortic aneurysm is a medical emergency, with a mortality rate approaching 90%. Risk factors for abdominal aortic aneurysms are smoking, male sex, age over 65, history of cardiovascular disease, hypertension, and a family history of abdominal aortic aneurysm, especially if a first-degree relative is affected.19 Endovascular repair is associated with lower rates of death and complications compared with open repair.20
The patient does not have any of those risk factors, making this diagnosis less likely.
CASE CONTINUED: RUPTURED SPLENIC ARTERY ANEURYSM

Emergency CT of the abdomen and pelvis with contrast enhancement shows a large left intraperitoneal hematoma with active extravasation from a ruptured splenic artery aneurysm (Figure 1). The patient receives packed red blood cells and fresh-frozen plasma before being transferred to our hospital.
2. Which of the following is false regarding splenic artery aneurysms?
- They are the most common type of splanchnic arterial aneurysm
- True aneurysms are more common than pseudoaneurysms
- Asymptomatic aneurysms are discovered incidentally during assessment for other radiographic indications
- Splenic artery aneurysm in portal hypertension is the result of athero-sclerotic changes to the vascular intima
Splenic artery aneurysm in portal hypertension is not the result of atherosclerotic change to the vascular intima.
Splenic artery aneurysms are the most common type of splanchnic artery aneurysm.1 True aneurysms involve all 3 layers of the arterial wall, ie, intima, media, and adventitia. Cirrhosis and portal hypertension are associated with true aneurysm formation. The proposed mechanism of aneurysm formation is increased splenic blood flow in response to portal congestion with resultant hemodynamic stress that disrupts arterial wall structure, leading to aneurysmal dilation.21
In earlier reports, the incidence of true splenic artery aneurysm in portal hypertension varied from 2.9% to 50%, the latter representing autopsy findings of small aneurysms that were found in the splenic hilum of patients with cirrhosis.22–25 The incidence of clinically significant aneurysms in cirrhosis is unknown but incidental asymptomatic aneurysm is being detected more frequently on imaging studies pursued for screening purposes.26
The risk of rupture is low, only 2% to 10% in older studies and likely even lower now due to increased incidental detection in asymptomatic patients.27 However, emergent management of rupture at a tertiary care facility is paramount, as the mortality rate of ruptured splenic artery aneurysm is 29% to 36%.1,26,28
Splenic artery pseudoaneurysm is rarer and has a different pathophysiologic process than true aneurysm. It usually arises in the setting of trauma, pancreatitis, or postsurgery.29,30 Pseudoaneurysm is more likely to rupture, owing to compromise in the vascular wall integrity.4,21,28 As a result, treatment is indicated for every pseudoaneurysm regardless of size.
RISK FACTORS FOR SPLENIC ARTERY ANEURYSM
3. Which of the following is true regarding our patient’s risk of splenic artery aneurysm?
- Liver cirrhosis and portal hypertension are her greatest risk factors for it
- Female sex and prior pregnancy are her greatest risk factors for it
- Being Native American makes it more likely that the patient has splenic artery aneurysm secondary to collagen vascular disease
- Her risk of rupture would diminish after receiving a liver transplant
Liver cirrhosis and portal hypertension are her greatest risk factors for splenic artery aneurysm.
Risk factors for true aneurysm include hypertension, atherosclerosis, portal hypertension with or without liver cirrhosis, liver transplant, third trimester of pregnancy, and multiparity.1,4,26,28,31 Splenic artery aneurysm is usually diagnosed in the sixth decade. It may be 4 times as common in women, given a hormonal influence.32 Cirrhosis is also associated with massive splenic artery aneurysm (≥ 5 cm). Although rare, massive splenic artery aneurysm is more frequent in men (the male-to-female ratio is 1.78:1) and has a heightened risk of rupture.28 The incidence of rupture increases to around 3% to 4% after liver transplant.33 Rare causes of true aneurysm include fibrodysplasia, collagen vascular disease (eg, Loeys-Dietz and type IV Ehler-Danlos syndromes), vasculitis (eg, polyarteritis nodosa due to amphetamine abuse), and mycotic aneurysms.24,25,28,29
This patient’s age, sex, and history of cirrhosis puts her at increased risk of splenic artery aneurysm. The risk of rupture is highest in the peripartum period and in patients with cirrhosis who become pregnant. Although being Native American portends an increased risk for collagen vascular disease, the latter is unlikely to be a contributing factor.
TREATMENT OF SPLENIC ARTERY ANEURYSM
4. Which of the following is false regarding treatment of splenic artery aneurysms?
- Aneurysms larger than 2 cm and those that are expanding require repair
- Treatment should be offered if the patient has symptoms attributable to the aneurysm
- Asymptomatic aneurysms in pregnant women can be followed with watchful waiting
- Minimally invasive therapies such as percutaneous embolization may be a good option in poor operative candidates
Asymptomatic aneurysms in pregnant women should not be followed with watchful waiting—they should be repaired, as rupture carries a maternal mortality rate of 75% and a fetal mortality rate of 95%.34
Complications of splenic artery aneurysm depend on the type of aneurysm and its predisposing factors. Indications for treatment of true aneurysms include:
- Symptoms attributable to the aneurysm (hence, the second answer choice above is true)
- Diameter 2 cm or greater or enlarging diameter (hence, the first answer choice is true)
- Women of childbearing age in anticipation of pregnancy
- Need for surgical intervention such as portocaval shunt and liver transplant.
Conservative management is associated with a late mortality risk of 4.9%.2 Interventional options include percutaneous embolization or stenting; or laparotomy with splenic artery ligation or excision with or without splenectomy.1,28,35–37
Endovascular and open surgical repair have both been used to treat splenic artery aneurysms. The method used depends on the patient’s surgical history and aneurysm anatomy such as splenic artery tortuosity hindering passage of a catheter. Open surgery is associated with longer intraoperative time and length of hospital stay and higher rates of 30-day mortality and perioperative morbidity.38–41 With endovascular repair, the complication of persistent or recurrent flow occurs in 3% to 5% of cases by 30 days; hence, postprocedural surveillance is recommended.42–44 Endovascular repair has a higher reintervention rate but may still be more cost-effective than open surgical repair.
Because patients with cirrhosis have a higher risk of surgical complications,45 elective endovascular treatment may be an option for patients with aneurysms at high risk of rupturing. Endovascular treatment of visceral aneurysms is associated with complications such as postembolization syndrome (fever, abdominal pain, pleural effusion, and pancreatitis), access site hematoma, splenic infarction, and persistent abdominal pain.42
Patients with cirrhosis as the cause of splenic artery aneurysm tend to need longer hospitalization after endovascular treatment, but there is insufficient evidence to suggest that they are at higher risk of other complications.37
CASE CONTINUED: SPLENIC ARTERY EMBOLIZATION

The patient undergoes emergency splenic artery embolization, performed by an interventional radiology team (Figure 2 and Figure 3). Over the next few days, her mental status improves and her abdominal pain resolves. Her hemoglobin level remains stable after the procedure.

The surgical and interventional radiology teams discuss the risk of repeat intervention with the patient and her family, who prefer a nonoperative approach. She is managed supportively in the intensive care unit and is finally discharged home in stable condition and is scheduled for outpatient follow-up.
SUSPECT THIS FATAL CONDITION
The low prevalence of ruptured splenic artery aneurysm may lead physicians to attribute septic shock to spontaneous bacterial peritonitis or hemorrhagic shock from gastroesophageal varices in patients with cirrhosis, but a high index of suspicion and early recognition of this rare disease can lead to timely diagnosis and treatment of this highly fatal complication.
KEY POINTS
- Splenic artery aneurysm is a common complication of cirrhosis, often diagnosed incidentally.
- Elective embolization should be considered for asymptomatic splenic artery aneurysms larger than 2 cm in diameter, clinically symptomatic aneurysms, women of childbearing age, and patients who are candidates for liver transplant.
- Although splenic artery aneurysm rupture is rare, it has a high mortality rate and warrants a high index of suspicion to institute prompt specialized intervention.
- We recommend that physicians consider splenic artery aneurysm rupture in their differential diagnoses in patients with liver cirrhosis presenting with abdominal pain, altered mental status, and hemodynamic shock.
- Bakhos CT, McIntosh BC, Nukta FA, et al. Staged arterial embolization and surgical resection of a giant splenic artery aneurysm. Ann Vasc Surg 2007; 21:208–210.
- Hogendoorn W, Lavida A, Hunink MG, et al. Open repair, endovascular repair, and conservative management of true splenic artery aneurysms. J Vasc Surg 2014; 60:1667–1676.e1.
- Algudkar A. Unruptured splenic artery aneurysm presenting as epigastric pain. JRSM Short Rep 2010; 1:24.
- Abbas MA, Stone WM, Fowl RJ, et al. Splenic artery aneurysms: two decades experience at Mayo Clinic. Ann Vasc Surg 2002; 16:442–449.
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2:399–407.
- Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology 1984; 4:1209–1211.
- Garcia-Tsao G. Spontaneous bacterial peritonitis: a historical perspective. J Hepatol 2004; 41:522–527.
- Soriano G, Castellote J, Alvarez C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol 2010; 52:39–44.
- D’Amico G, De Franchis R; Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010; 31:366–374.
- Kobori L, van der Kolk MJ, de Jong KP, et al. Splenic artery aneurysms in liver transplant patients. Liver Transplant Group. J Hepatol 1997; 27:890–893.
- Manzano-Robleda Mdel C, Barranco-Fragoso B, Uribe M, Mendez-Sanchez N. Portal vein thrombosis: what is new? Ann Hepatol 2015; 14:20–27.
- Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016; 151:574–577.e3.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Manzanet G, Sanjuan F, Orbis P, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl 2001; 7:125–131.
- John BV, Konjeti R, Aggarwal A, et al. Impact of untreated portal vein thrombosis on pre and post liver transplant outcomes in cirrhosis. Ann Hepatol 2013; 12:952–958.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery/Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—summary of recommendations. J Vasc Interv Radiol 2006; 17:1383–1397.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med 2008; 358:464–474.
- Ohta M, Hashizume M, Ueno K, Tanoue K, Sugimachi K, Hasuo K. Hemodynamic study of splenic artery aneurysm in portal hypertension. Hepatogastroenterology 1994; 41:181–184.
- Sunagozaka H, Tsuji H, Mizukoshi E, et al. The development and clinical features of splenic aneurysm associated with liver cirrhosis. Liver Int 2006; 26:291–297.
- Manenti F, Williams R. Injection studies of the splenic vasculature in portal hypertension. Gut 1966; 7:175–180.
- Stanley JC, Fry WJ. Pathogenesis and clinical significance of splenic artery aneurysms. Surgery 1974; 76:898–909.
- Lee PC, Rhee RY, Gordon RY, Fung JJ, Webster MW. Management of splenic artery aneurysms: the significance of portal and essential hypertension. J Am Coll Surg 1999; 189:483–490.
- Al-Habbal Y, Christophi C, Muralidharan V. Aneurysms of the splenic artery—a review. Surgeon 2010; 8:223–231.
- Mattar SG, Lumsden AB. The management of splenic artery aneurysms: experience with 23 cases. Am J Surg 1995; 169:580–584.
- Akbulut S, Otan E. Management of giant splenic artery aneurysm: comprehensive literature review. Medicine (Baltimore) 2015; 94:e1016.
- Agrawal GA, Johnson PT, Fishman EK. Splenic artery aneurysms and pseudoaneurysms: clinical distinctions and CT appearances. AJR Am J Roentgenol 2007; 188:992–999.
- Tessier DJ, Stone WM, Fowl RJ, et al. Clinical features and management of splenic artery pseudoaneurysm: case series and cumulative review of literature. J Vasc Surg 2003; 38:969–974.
- Dave SP, Reis ED, Hossain A, Taub PJ, Kerstein MD, Hollier LH. Splenic artery aneurysm in the 1990s. Ann Vasc Surg 2000; 14:223–229.
- Parrish J, Maxwell C, Beecroft JR. Splenic artery aneurysm in pregnancy. J Obstet Gynaecol Can 2015; 37:816–818.
- Moon DB, Lee SG, Hwang S, et al. Characteristics and management of splenic artery aneurysms in adult living donor liver transplant recipients. Liver Transpl 2009; 15:1535–1541.
- Sadat U, Dar O, Walsh S, Varty K. Splenic artery aneurysms in pregnancy—a systematic review. Int J Surg 2008; 6:261–265.
- Geoghegan T, McAuley G, Snow A, Torreggiani WC. Emergency embolization of multiple splenic artery pseudoaneurysms associated with portal hypertension complicating cystic fibrosis. Australas Radiol 2007; 51(suppl):B337–B339.
- Jiang R, Ding X, Jian W, Jiang J, Hu S, Zhang Z. Combined endovascular embolization and open surgery for splenic artery aneurysm with arteriovenous fistula. Ann Vasc Surg 2016; 30:311.e1–311.e4.
- Naganuma M, Matsui H, Koizumi J, Fushimi K, Yasunaga H. Short-term outcomes following elective transcatheter arterial embolization for splenic artery aneurysms: data from a nationwide administrative database. Acta Radiol Open 2015; 4:2047981615574354.
- Batagini NC, El-Arousy H, Clair DG, Kirksey L. Open versus endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Ann Vasc Surg 2016; 35:1–8.
- Marone EM, Mascia D, Kahlberg A, Brioschi C, Tshomba Y, Chiesa R. Is open repair still the gold standard in visceral artery aneurysm management? Ann Vasc Surg 2011; 25:936–946.
- Sticco A, Aggarwal A, Shapiro M, Pratt A, Rissuci D, D'Ayala M. A comparison of open and endovascular treatment strategies for the management of splenic artery aneurysms. Vascular 2016; 24:487–491.
- Hogendoorn W, Lavida A, Hunink MG, et al. Cost-effectiveness of endovascular repair, open repair, and conservative management of splenic artery aneurysms. J Vasc Surg 2015; 61:1432–1440.
- Fankhauser GT, Stone WM, Naidu SG, et al; Mayo Vascular Research Center Consortium. The minimally invasive management of visceral artery aneurysms and pseudoaneurysms. J Vasc Surg 2011; 53:966–970.
- Lagana D, Carrafiello G, Mangini M, et al. Multimodal approach to endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Eur J Radiol 2006; 59:104–111.
- Guillon R, Garcier JM, Abergel A, et al. Management of splenic artery aneurysms and false aneurysms with endovascular treatment in 12 patients. Cardiovasc Intervent Radiol 2003; 26:256–260.
- Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for end-stage liver disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg 2005; 242:244–251.
A 53-year-old Native American woman with a history of liver cirrhosis secondary to alcohol abuse presents to the emergency department after 2 days of diffuse abdominal pain and weakness. The pain was sudden in onset and has progressed relentlessly over the last day, reaching 9 on a scale of 10 in severity. Family members say that her oral intake has been decreased for the last 2 days, but she has had no fever, vomiting, change in bowel habit, blood in stool, or black stool. She has never undergone surgery, and has had one uncomplicated pregnancy.
Physical examination
Vital signs:
- Blood pressure 82/57 mm Hg
- Heart rate 96 beats per minute
- Temperature 37.3°C (99.1°F)
- Respiratory rate 16 per minute
- Oxygen saturation 92% while receiving oxygen at 2 L/minute.
The patient is somnolent and has scleral icterus. Her cardiopulmonary examination is normal. Her abdomen is tense, distended, and diffusely tender. She has bilateral +2 pitting edema in her lower extremities. She is oriented to person only and is noted to have asterixis. Her baseline Model for End-stage Liver Disease score is 18 points on a scale of 6 (less ill) to 40 (gravely ill).
Laboratory studies:
- Hemoglobin 9.8 g/dL (reference range 11.5–15.5)
- Platelet count 100 × 109/L (150–400)
- White blood cell count 9.9 × 109/L (3.7–11.0)
- Serum creatinine 1.06 mg/dL (0.58–0.96)
- Bilirubin 6.3 mg/dL (0.2–1.3)
- International normalized ratio of the prothrombin time 2.15 (0.8–1.2)
- Blood urea nitrogen 13 mg/dL (7–21)
- Serum albumin 2.7 g/dL (3.9–4.9).
Intravenous fluid resuscitation is initiated but the patient remains hypotensive, and on repeat laboratory testing 4 hours later her hemoglobin level has dropped to 7.3 mg/dL.
DIFFERENTIAL DIAGNOSIS
1. Which of the following are likely causes of this patient’s presentation?
- Splenic arterial aneurysm rupture
- Spontaneous bacterial peritonitis
- Variceal hemorrhage
- Portal vein thrombosis
- Abdominal aortic aneurysm rupture
Ruptured splenic artery aneurysm
Splenic artery aneurysms are the third most common intra-abdominal aneurysm, after those of the abdominal aorta and iliac artery.1 They are often asymptomatic and are being detected more frequently because of increased use of computed tomography (CT).2 Symptomatic splenic artery aneurysms may present with abdominal pain and have the potential to rupture, which can be life-threatening.3,4
This patient may have a ruptured splenic artery aneurysm, given her hemodynamic shock.
Spontaneous bacterial peritonitis
Ten percent to 20% of hospitalized patients with cirrhosis and ascites develop spontaneous bacterial peritonitis. Patients may present with ascites and abdominal pain, tenderness to palpation, fever, encephalopathy, or worsening liver and renal function.
Diagnostic paracentesis is paramount to delineate the cause of ascites; one should calculate the serum-ascites albumin gradient and obtain a cell count and culture of the ascitic fluid. The diagnosis of spontaneous bacterial peritonitis can be made if the ascitic fluid polymorphonuclear cell count is 0.25 × 109/L or higher, even if the ascitic fluid culture is negative.5,6 Simultaneous blood cultures should also be collected, as 50% of cases are associated with bacteremia.
The in-hospital mortality rate of an episode of spontaneous bacterial peritonitis has been reduced to 10% to 20% thanks to prompt diagnosis and empiric treatment with third-generation cephalosporins.7
Five percent of cases of infected ascites fluid are due to secondary bacterial peritonitis from a perforated viscus or a loculated abscess, which cannot be differentiated clinically from spontaneous bacterial peritonitis but can be diagnosed with CT.8
This patient may be presenting with septic shock secondary to either of these causes.
Variceal hemorrhage
Half of patients with cirrhosis have gastroesophageal varices due to portal hypertension. Endoscopic surveillance is warranted, as the risk of hemorrhage is 12% to 15% per year, and the mortality rate approaches 15% to 20% with each episode. Prompt resuscitation, diagnosis, and control of bleeding is paramount.
Esophagogastroduodenoscopy is used for both diagnosis and intervention. Short-term prophylactic use of antibiotics improves survival by preventing infections in the event bleeding recurs.9–11
Our patient may be presenting with hemodynamic shock from bleeding esophageal varices.
Portal vein thrombosis
Portal vein thrombosis is a common complication of cirrhosis, occurring in 5% to 28% of patients. The risk increases with the severity of liver disease and in association with hepatocellular carcinoma.12 Forty-three percent of cases are discovered incidentally in asymptomatic patients during ultrasonography, 39% present with upper gastrointestinal bleeding, and 18% present with abdominal pain.13,14
Portal vein thrombosis is the complete or partial obstruction of blood flow due to a thrombus in the lumen of the portal vein. Contrast ultrasonography and CT can be used to establish the diagnosis.15
Anticoagulation is recommended in cases of complete thrombosis in candidates for living-donor liver transplant and for those at risk of mesenteric ischemia because of the thrombus extending into the mesenteric veins. In symptomatic patients, the decision to initiate anticoagulation should be made on a case-by-case basis after appropriate screening and management of varices.16–18
Our patient’s thrombocytopenia reflects the severity of portal hypertension and increases her risk of portal vein thrombosis, but this is unlikely to be the sole cause of the hemodynamic compromise in this patient.
Ruptured abdominal aortic aneurysm
Rupture of an abdominal aortic aneurysm is a medical emergency, with a mortality rate approaching 90%. Risk factors for abdominal aortic aneurysms are smoking, male sex, age over 65, history of cardiovascular disease, hypertension, and a family history of abdominal aortic aneurysm, especially if a first-degree relative is affected.19 Endovascular repair is associated with lower rates of death and complications compared with open repair.20
The patient does not have any of those risk factors, making this diagnosis less likely.
CASE CONTINUED: RUPTURED SPLENIC ARTERY ANEURYSM
Emergency CT of the abdomen and pelvis with contrast enhancement shows a large left intraperitoneal hematoma with active extravasation from a ruptured splenic artery aneurysm (Figure 1). The patient receives packed red blood cells and fresh-frozen plasma before being transferred to our hospital.
2. Which of the following is false regarding splenic artery aneurysms?
- They are the most common type of splanchnic arterial aneurysm
- True aneurysms are more common than pseudoaneurysms
- Asymptomatic aneurysms are discovered incidentally during assessment for other radiographic indications
- Splenic artery aneurysm in portal hypertension is the result of athero-sclerotic changes to the vascular intima
Splenic artery aneurysm in portal hypertension is not the result of atherosclerotic change to the vascular intima.
Splenic artery aneurysms are the most common type of splanchnic artery aneurysm.1 True aneurysms involve all 3 layers of the arterial wall, ie, intima, media, and adventitia. Cirrhosis and portal hypertension are associated with true aneurysm formation. The proposed mechanism of aneurysm formation is increased splenic blood flow in response to portal congestion with resultant hemodynamic stress that disrupts arterial wall structure, leading to aneurysmal dilation.21
In earlier reports, the incidence of true splenic artery aneurysm in portal hypertension varied from 2.9% to 50%, the latter representing autopsy findings of small aneurysms that were found in the splenic hilum of patients with cirrhosis.22–25 The incidence of clinically significant aneurysms in cirrhosis is unknown but incidental asymptomatic aneurysm is being detected more frequently on imaging studies pursued for screening purposes.26
The risk of rupture is low, only 2% to 10% in older studies and likely even lower now due to increased incidental detection in asymptomatic patients.27 However, emergent management of rupture at a tertiary care facility is paramount, as the mortality rate of ruptured splenic artery aneurysm is 29% to 36%.1,26,28
Splenic artery pseudoaneurysm is rarer and has a different pathophysiologic process than true aneurysm. It usually arises in the setting of trauma, pancreatitis, or postsurgery.29,30 Pseudoaneurysm is more likely to rupture, owing to compromise in the vascular wall integrity.4,21,28 As a result, treatment is indicated for every pseudoaneurysm regardless of size.
RISK FACTORS FOR SPLENIC ARTERY ANEURYSM
3. Which of the following is true regarding our patient’s risk of splenic artery aneurysm?
- Liver cirrhosis and portal hypertension are her greatest risk factors for it
- Female sex and prior pregnancy are her greatest risk factors for it
- Being Native American makes it more likely that the patient has splenic artery aneurysm secondary to collagen vascular disease
- Her risk of rupture would diminish after receiving a liver transplant
Liver cirrhosis and portal hypertension are her greatest risk factors for splenic artery aneurysm.
Risk factors for true aneurysm include hypertension, atherosclerosis, portal hypertension with or without liver cirrhosis, liver transplant, third trimester of pregnancy, and multiparity.1,4,26,28,31 Splenic artery aneurysm is usually diagnosed in the sixth decade. It may be 4 times as common in women, given a hormonal influence.32 Cirrhosis is also associated with massive splenic artery aneurysm (≥ 5 cm). Although rare, massive splenic artery aneurysm is more frequent in men (the male-to-female ratio is 1.78:1) and has a heightened risk of rupture.28 The incidence of rupture increases to around 3% to 4% after liver transplant.33 Rare causes of true aneurysm include fibrodysplasia, collagen vascular disease (eg, Loeys-Dietz and type IV Ehler-Danlos syndromes), vasculitis (eg, polyarteritis nodosa due to amphetamine abuse), and mycotic aneurysms.24,25,28,29
This patient’s age, sex, and history of cirrhosis puts her at increased risk of splenic artery aneurysm. The risk of rupture is highest in the peripartum period and in patients with cirrhosis who become pregnant. Although being Native American portends an increased risk for collagen vascular disease, the latter is unlikely to be a contributing factor.
TREATMENT OF SPLENIC ARTERY ANEURYSM
4. Which of the following is false regarding treatment of splenic artery aneurysms?
- Aneurysms larger than 2 cm and those that are expanding require repair
- Treatment should be offered if the patient has symptoms attributable to the aneurysm
- Asymptomatic aneurysms in pregnant women can be followed with watchful waiting
- Minimally invasive therapies such as percutaneous embolization may be a good option in poor operative candidates
Asymptomatic aneurysms in pregnant women should not be followed with watchful waiting—they should be repaired, as rupture carries a maternal mortality rate of 75% and a fetal mortality rate of 95%.34
Complications of splenic artery aneurysm depend on the type of aneurysm and its predisposing factors. Indications for treatment of true aneurysms include:
- Symptoms attributable to the aneurysm (hence, the second answer choice above is true)
- Diameter 2 cm or greater or enlarging diameter (hence, the first answer choice is true)
- Women of childbearing age in anticipation of pregnancy
- Need for surgical intervention such as portocaval shunt and liver transplant.
Conservative management is associated with a late mortality risk of 4.9%.2 Interventional options include percutaneous embolization or stenting; or laparotomy with splenic artery ligation or excision with or without splenectomy.1,28,35–37
Endovascular and open surgical repair have both been used to treat splenic artery aneurysms. The method used depends on the patient’s surgical history and aneurysm anatomy such as splenic artery tortuosity hindering passage of a catheter. Open surgery is associated with longer intraoperative time and length of hospital stay and higher rates of 30-day mortality and perioperative morbidity.38–41 With endovascular repair, the complication of persistent or recurrent flow occurs in 3% to 5% of cases by 30 days; hence, postprocedural surveillance is recommended.42–44 Endovascular repair has a higher reintervention rate but may still be more cost-effective than open surgical repair.
Because patients with cirrhosis have a higher risk of surgical complications,45 elective endovascular treatment may be an option for patients with aneurysms at high risk of rupturing. Endovascular treatment of visceral aneurysms is associated with complications such as postembolization syndrome (fever, abdominal pain, pleural effusion, and pancreatitis), access site hematoma, splenic infarction, and persistent abdominal pain.42
Patients with cirrhosis as the cause of splenic artery aneurysm tend to need longer hospitalization after endovascular treatment, but there is insufficient evidence to suggest that they are at higher risk of other complications.37
CASE CONTINUED: SPLENIC ARTERY EMBOLIZATION
The patient undergoes emergency splenic artery embolization, performed by an interventional radiology team (Figure 2 and Figure 3). Over the next few days, her mental status improves and her abdominal pain resolves. Her hemoglobin level remains stable after the procedure.
The surgical and interventional radiology teams discuss the risk of repeat intervention with the patient and her family, who prefer a nonoperative approach. She is managed supportively in the intensive care unit and is finally discharged home in stable condition and is scheduled for outpatient follow-up.
SUSPECT THIS FATAL CONDITION
The low prevalence of ruptured splenic artery aneurysm may lead physicians to attribute septic shock to spontaneous bacterial peritonitis or hemorrhagic shock from gastroesophageal varices in patients with cirrhosis, but a high index of suspicion and early recognition of this rare disease can lead to timely diagnosis and treatment of this highly fatal complication.
KEY POINTS
- Splenic artery aneurysm is a common complication of cirrhosis, often diagnosed incidentally.
- Elective embolization should be considered for asymptomatic splenic artery aneurysms larger than 2 cm in diameter, clinically symptomatic aneurysms, women of childbearing age, and patients who are candidates for liver transplant.
- Although splenic artery aneurysm rupture is rare, it has a high mortality rate and warrants a high index of suspicion to institute prompt specialized intervention.
- We recommend that physicians consider splenic artery aneurysm rupture in their differential diagnoses in patients with liver cirrhosis presenting with abdominal pain, altered mental status, and hemodynamic shock.
A 53-year-old Native American woman with a history of liver cirrhosis secondary to alcohol abuse presents to the emergency department after 2 days of diffuse abdominal pain and weakness. The pain was sudden in onset and has progressed relentlessly over the last day, reaching 9 on a scale of 10 in severity. Family members say that her oral intake has been decreased for the last 2 days, but she has had no fever, vomiting, change in bowel habit, blood in stool, or black stool. She has never undergone surgery, and has had one uncomplicated pregnancy.
Physical examination
Vital signs:
- Blood pressure 82/57 mm Hg
- Heart rate 96 beats per minute
- Temperature 37.3°C (99.1°F)
- Respiratory rate 16 per minute
- Oxygen saturation 92% while receiving oxygen at 2 L/minute.
The patient is somnolent and has scleral icterus. Her cardiopulmonary examination is normal. Her abdomen is tense, distended, and diffusely tender. She has bilateral +2 pitting edema in her lower extremities. She is oriented to person only and is noted to have asterixis. Her baseline Model for End-stage Liver Disease score is 18 points on a scale of 6 (less ill) to 40 (gravely ill).
Laboratory studies:
- Hemoglobin 9.8 g/dL (reference range 11.5–15.5)
- Platelet count 100 × 109/L (150–400)
- White blood cell count 9.9 × 109/L (3.7–11.0)
- Serum creatinine 1.06 mg/dL (0.58–0.96)
- Bilirubin 6.3 mg/dL (0.2–1.3)
- International normalized ratio of the prothrombin time 2.15 (0.8–1.2)
- Blood urea nitrogen 13 mg/dL (7–21)
- Serum albumin 2.7 g/dL (3.9–4.9).
Intravenous fluid resuscitation is initiated but the patient remains hypotensive, and on repeat laboratory testing 4 hours later her hemoglobin level has dropped to 7.3 mg/dL.
DIFFERENTIAL DIAGNOSIS
1. Which of the following are likely causes of this patient’s presentation?
- Splenic arterial aneurysm rupture
- Spontaneous bacterial peritonitis
- Variceal hemorrhage
- Portal vein thrombosis
- Abdominal aortic aneurysm rupture
Ruptured splenic artery aneurysm
Splenic artery aneurysms are the third most common intra-abdominal aneurysm, after those of the abdominal aorta and iliac artery.1 They are often asymptomatic and are being detected more frequently because of increased use of computed tomography (CT).2 Symptomatic splenic artery aneurysms may present with abdominal pain and have the potential to rupture, which can be life-threatening.3,4
This patient may have a ruptured splenic artery aneurysm, given her hemodynamic shock.
Spontaneous bacterial peritonitis
Ten percent to 20% of hospitalized patients with cirrhosis and ascites develop spontaneous bacterial peritonitis. Patients may present with ascites and abdominal pain, tenderness to palpation, fever, encephalopathy, or worsening liver and renal function.
Diagnostic paracentesis is paramount to delineate the cause of ascites; one should calculate the serum-ascites albumin gradient and obtain a cell count and culture of the ascitic fluid. The diagnosis of spontaneous bacterial peritonitis can be made if the ascitic fluid polymorphonuclear cell count is 0.25 × 109/L or higher, even if the ascitic fluid culture is negative.5,6 Simultaneous blood cultures should also be collected, as 50% of cases are associated with bacteremia.
The in-hospital mortality rate of an episode of spontaneous bacterial peritonitis has been reduced to 10% to 20% thanks to prompt diagnosis and empiric treatment with third-generation cephalosporins.7
Five percent of cases of infected ascites fluid are due to secondary bacterial peritonitis from a perforated viscus or a loculated abscess, which cannot be differentiated clinically from spontaneous bacterial peritonitis but can be diagnosed with CT.8
This patient may be presenting with septic shock secondary to either of these causes.
Variceal hemorrhage
Half of patients with cirrhosis have gastroesophageal varices due to portal hypertension. Endoscopic surveillance is warranted, as the risk of hemorrhage is 12% to 15% per year, and the mortality rate approaches 15% to 20% with each episode. Prompt resuscitation, diagnosis, and control of bleeding is paramount.
Esophagogastroduodenoscopy is used for both diagnosis and intervention. Short-term prophylactic use of antibiotics improves survival by preventing infections in the event bleeding recurs.9–11
Our patient may be presenting with hemodynamic shock from bleeding esophageal varices.
Portal vein thrombosis
Portal vein thrombosis is a common complication of cirrhosis, occurring in 5% to 28% of patients. The risk increases with the severity of liver disease and in association with hepatocellular carcinoma.12 Forty-three percent of cases are discovered incidentally in asymptomatic patients during ultrasonography, 39% present with upper gastrointestinal bleeding, and 18% present with abdominal pain.13,14
Portal vein thrombosis is the complete or partial obstruction of blood flow due to a thrombus in the lumen of the portal vein. Contrast ultrasonography and CT can be used to establish the diagnosis.15
Anticoagulation is recommended in cases of complete thrombosis in candidates for living-donor liver transplant and for those at risk of mesenteric ischemia because of the thrombus extending into the mesenteric veins. In symptomatic patients, the decision to initiate anticoagulation should be made on a case-by-case basis after appropriate screening and management of varices.16–18
Our patient’s thrombocytopenia reflects the severity of portal hypertension and increases her risk of portal vein thrombosis, but this is unlikely to be the sole cause of the hemodynamic compromise in this patient.
Ruptured abdominal aortic aneurysm
Rupture of an abdominal aortic aneurysm is a medical emergency, with a mortality rate approaching 90%. Risk factors for abdominal aortic aneurysms are smoking, male sex, age over 65, history of cardiovascular disease, hypertension, and a family history of abdominal aortic aneurysm, especially if a first-degree relative is affected.19 Endovascular repair is associated with lower rates of death and complications compared with open repair.20
The patient does not have any of those risk factors, making this diagnosis less likely.
CASE CONTINUED: RUPTURED SPLENIC ARTERY ANEURYSM
Emergency CT of the abdomen and pelvis with contrast enhancement shows a large left intraperitoneal hematoma with active extravasation from a ruptured splenic artery aneurysm (Figure 1). The patient receives packed red blood cells and fresh-frozen plasma before being transferred to our hospital.
2. Which of the following is false regarding splenic artery aneurysms?
- They are the most common type of splanchnic arterial aneurysm
- True aneurysms are more common than pseudoaneurysms
- Asymptomatic aneurysms are discovered incidentally during assessment for other radiographic indications
- Splenic artery aneurysm in portal hypertension is the result of athero-sclerotic changes to the vascular intima
Splenic artery aneurysm in portal hypertension is not the result of atherosclerotic change to the vascular intima.
Splenic artery aneurysms are the most common type of splanchnic artery aneurysm.1 True aneurysms involve all 3 layers of the arterial wall, ie, intima, media, and adventitia. Cirrhosis and portal hypertension are associated with true aneurysm formation. The proposed mechanism of aneurysm formation is increased splenic blood flow in response to portal congestion with resultant hemodynamic stress that disrupts arterial wall structure, leading to aneurysmal dilation.21
In earlier reports, the incidence of true splenic artery aneurysm in portal hypertension varied from 2.9% to 50%, the latter representing autopsy findings of small aneurysms that were found in the splenic hilum of patients with cirrhosis.22–25 The incidence of clinically significant aneurysms in cirrhosis is unknown but incidental asymptomatic aneurysm is being detected more frequently on imaging studies pursued for screening purposes.26
The risk of rupture is low, only 2% to 10% in older studies and likely even lower now due to increased incidental detection in asymptomatic patients.27 However, emergent management of rupture at a tertiary care facility is paramount, as the mortality rate of ruptured splenic artery aneurysm is 29% to 36%.1,26,28
Splenic artery pseudoaneurysm is rarer and has a different pathophysiologic process than true aneurysm. It usually arises in the setting of trauma, pancreatitis, or postsurgery.29,30 Pseudoaneurysm is more likely to rupture, owing to compromise in the vascular wall integrity.4,21,28 As a result, treatment is indicated for every pseudoaneurysm regardless of size.
RISK FACTORS FOR SPLENIC ARTERY ANEURYSM
3. Which of the following is true regarding our patient’s risk of splenic artery aneurysm?
- Liver cirrhosis and portal hypertension are her greatest risk factors for it
- Female sex and prior pregnancy are her greatest risk factors for it
- Being Native American makes it more likely that the patient has splenic artery aneurysm secondary to collagen vascular disease
- Her risk of rupture would diminish after receiving a liver transplant
Liver cirrhosis and portal hypertension are her greatest risk factors for splenic artery aneurysm.
Risk factors for true aneurysm include hypertension, atherosclerosis, portal hypertension with or without liver cirrhosis, liver transplant, third trimester of pregnancy, and multiparity.1,4,26,28,31 Splenic artery aneurysm is usually diagnosed in the sixth decade. It may be 4 times as common in women, given a hormonal influence.32 Cirrhosis is also associated with massive splenic artery aneurysm (≥ 5 cm). Although rare, massive splenic artery aneurysm is more frequent in men (the male-to-female ratio is 1.78:1) and has a heightened risk of rupture.28 The incidence of rupture increases to around 3% to 4% after liver transplant.33 Rare causes of true aneurysm include fibrodysplasia, collagen vascular disease (eg, Loeys-Dietz and type IV Ehler-Danlos syndromes), vasculitis (eg, polyarteritis nodosa due to amphetamine abuse), and mycotic aneurysms.24,25,28,29
This patient’s age, sex, and history of cirrhosis puts her at increased risk of splenic artery aneurysm. The risk of rupture is highest in the peripartum period and in patients with cirrhosis who become pregnant. Although being Native American portends an increased risk for collagen vascular disease, the latter is unlikely to be a contributing factor.
TREATMENT OF SPLENIC ARTERY ANEURYSM
4. Which of the following is false regarding treatment of splenic artery aneurysms?
- Aneurysms larger than 2 cm and those that are expanding require repair
- Treatment should be offered if the patient has symptoms attributable to the aneurysm
- Asymptomatic aneurysms in pregnant women can be followed with watchful waiting
- Minimally invasive therapies such as percutaneous embolization may be a good option in poor operative candidates
Asymptomatic aneurysms in pregnant women should not be followed with watchful waiting—they should be repaired, as rupture carries a maternal mortality rate of 75% and a fetal mortality rate of 95%.34
Complications of splenic artery aneurysm depend on the type of aneurysm and its predisposing factors. Indications for treatment of true aneurysms include:
- Symptoms attributable to the aneurysm (hence, the second answer choice above is true)
- Diameter 2 cm or greater or enlarging diameter (hence, the first answer choice is true)
- Women of childbearing age in anticipation of pregnancy
- Need for surgical intervention such as portocaval shunt and liver transplant.
Conservative management is associated with a late mortality risk of 4.9%.2 Interventional options include percutaneous embolization or stenting; or laparotomy with splenic artery ligation or excision with or without splenectomy.1,28,35–37
Endovascular and open surgical repair have both been used to treat splenic artery aneurysms. The method used depends on the patient’s surgical history and aneurysm anatomy such as splenic artery tortuosity hindering passage of a catheter. Open surgery is associated with longer intraoperative time and length of hospital stay and higher rates of 30-day mortality and perioperative morbidity.38–41 With endovascular repair, the complication of persistent or recurrent flow occurs in 3% to 5% of cases by 30 days; hence, postprocedural surveillance is recommended.42–44 Endovascular repair has a higher reintervention rate but may still be more cost-effective than open surgical repair.
Because patients with cirrhosis have a higher risk of surgical complications,45 elective endovascular treatment may be an option for patients with aneurysms at high risk of rupturing. Endovascular treatment of visceral aneurysms is associated with complications such as postembolization syndrome (fever, abdominal pain, pleural effusion, and pancreatitis), access site hematoma, splenic infarction, and persistent abdominal pain.42
Patients with cirrhosis as the cause of splenic artery aneurysm tend to need longer hospitalization after endovascular treatment, but there is insufficient evidence to suggest that they are at higher risk of other complications.37
CASE CONTINUED: SPLENIC ARTERY EMBOLIZATION
The patient undergoes emergency splenic artery embolization, performed by an interventional radiology team (Figure 2 and Figure 3). Over the next few days, her mental status improves and her abdominal pain resolves. Her hemoglobin level remains stable after the procedure.
The surgical and interventional radiology teams discuss the risk of repeat intervention with the patient and her family, who prefer a nonoperative approach. She is managed supportively in the intensive care unit and is finally discharged home in stable condition and is scheduled for outpatient follow-up.
SUSPECT THIS FATAL CONDITION
The low prevalence of ruptured splenic artery aneurysm may lead physicians to attribute septic shock to spontaneous bacterial peritonitis or hemorrhagic shock from gastroesophageal varices in patients with cirrhosis, but a high index of suspicion and early recognition of this rare disease can lead to timely diagnosis and treatment of this highly fatal complication.
KEY POINTS
- Splenic artery aneurysm is a common complication of cirrhosis, often diagnosed incidentally.
- Elective embolization should be considered for asymptomatic splenic artery aneurysms larger than 2 cm in diameter, clinically symptomatic aneurysms, women of childbearing age, and patients who are candidates for liver transplant.
- Although splenic artery aneurysm rupture is rare, it has a high mortality rate and warrants a high index of suspicion to institute prompt specialized intervention.
- We recommend that physicians consider splenic artery aneurysm rupture in their differential diagnoses in patients with liver cirrhosis presenting with abdominal pain, altered mental status, and hemodynamic shock.
- Bakhos CT, McIntosh BC, Nukta FA, et al. Staged arterial embolization and surgical resection of a giant splenic artery aneurysm. Ann Vasc Surg 2007; 21:208–210.
- Hogendoorn W, Lavida A, Hunink MG, et al. Open repair, endovascular repair, and conservative management of true splenic artery aneurysms. J Vasc Surg 2014; 60:1667–1676.e1.
- Algudkar A. Unruptured splenic artery aneurysm presenting as epigastric pain. JRSM Short Rep 2010; 1:24.
- Abbas MA, Stone WM, Fowl RJ, et al. Splenic artery aneurysms: two decades experience at Mayo Clinic. Ann Vasc Surg 2002; 16:442–449.
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2:399–407.
- Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology 1984; 4:1209–1211.
- Garcia-Tsao G. Spontaneous bacterial peritonitis: a historical perspective. J Hepatol 2004; 41:522–527.
- Soriano G, Castellote J, Alvarez C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol 2010; 52:39–44.
- D’Amico G, De Franchis R; Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010; 31:366–374.
- Kobori L, van der Kolk MJ, de Jong KP, et al. Splenic artery aneurysms in liver transplant patients. Liver Transplant Group. J Hepatol 1997; 27:890–893.
- Manzano-Robleda Mdel C, Barranco-Fragoso B, Uribe M, Mendez-Sanchez N. Portal vein thrombosis: what is new? Ann Hepatol 2015; 14:20–27.
- Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016; 151:574–577.e3.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Manzanet G, Sanjuan F, Orbis P, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl 2001; 7:125–131.
- John BV, Konjeti R, Aggarwal A, et al. Impact of untreated portal vein thrombosis on pre and post liver transplant outcomes in cirrhosis. Ann Hepatol 2013; 12:952–958.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery/Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—summary of recommendations. J Vasc Interv Radiol 2006; 17:1383–1397.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med 2008; 358:464–474.
- Ohta M, Hashizume M, Ueno K, Tanoue K, Sugimachi K, Hasuo K. Hemodynamic study of splenic artery aneurysm in portal hypertension. Hepatogastroenterology 1994; 41:181–184.
- Sunagozaka H, Tsuji H, Mizukoshi E, et al. The development and clinical features of splenic aneurysm associated with liver cirrhosis. Liver Int 2006; 26:291–297.
- Manenti F, Williams R. Injection studies of the splenic vasculature in portal hypertension. Gut 1966; 7:175–180.
- Stanley JC, Fry WJ. Pathogenesis and clinical significance of splenic artery aneurysms. Surgery 1974; 76:898–909.
- Lee PC, Rhee RY, Gordon RY, Fung JJ, Webster MW. Management of splenic artery aneurysms: the significance of portal and essential hypertension. J Am Coll Surg 1999; 189:483–490.
- Al-Habbal Y, Christophi C, Muralidharan V. Aneurysms of the splenic artery—a review. Surgeon 2010; 8:223–231.
- Mattar SG, Lumsden AB. The management of splenic artery aneurysms: experience with 23 cases. Am J Surg 1995; 169:580–584.
- Akbulut S, Otan E. Management of giant splenic artery aneurysm: comprehensive literature review. Medicine (Baltimore) 2015; 94:e1016.
- Agrawal GA, Johnson PT, Fishman EK. Splenic artery aneurysms and pseudoaneurysms: clinical distinctions and CT appearances. AJR Am J Roentgenol 2007; 188:992–999.
- Tessier DJ, Stone WM, Fowl RJ, et al. Clinical features and management of splenic artery pseudoaneurysm: case series and cumulative review of literature. J Vasc Surg 2003; 38:969–974.
- Dave SP, Reis ED, Hossain A, Taub PJ, Kerstein MD, Hollier LH. Splenic artery aneurysm in the 1990s. Ann Vasc Surg 2000; 14:223–229.
- Parrish J, Maxwell C, Beecroft JR. Splenic artery aneurysm in pregnancy. J Obstet Gynaecol Can 2015; 37:816–818.
- Moon DB, Lee SG, Hwang S, et al. Characteristics and management of splenic artery aneurysms in adult living donor liver transplant recipients. Liver Transpl 2009; 15:1535–1541.
- Sadat U, Dar O, Walsh S, Varty K. Splenic artery aneurysms in pregnancy—a systematic review. Int J Surg 2008; 6:261–265.
- Geoghegan T, McAuley G, Snow A, Torreggiani WC. Emergency embolization of multiple splenic artery pseudoaneurysms associated with portal hypertension complicating cystic fibrosis. Australas Radiol 2007; 51(suppl):B337–B339.
- Jiang R, Ding X, Jian W, Jiang J, Hu S, Zhang Z. Combined endovascular embolization and open surgery for splenic artery aneurysm with arteriovenous fistula. Ann Vasc Surg 2016; 30:311.e1–311.e4.
- Naganuma M, Matsui H, Koizumi J, Fushimi K, Yasunaga H. Short-term outcomes following elective transcatheter arterial embolization for splenic artery aneurysms: data from a nationwide administrative database. Acta Radiol Open 2015; 4:2047981615574354.
- Batagini NC, El-Arousy H, Clair DG, Kirksey L. Open versus endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Ann Vasc Surg 2016; 35:1–8.
- Marone EM, Mascia D, Kahlberg A, Brioschi C, Tshomba Y, Chiesa R. Is open repair still the gold standard in visceral artery aneurysm management? Ann Vasc Surg 2011; 25:936–946.
- Sticco A, Aggarwal A, Shapiro M, Pratt A, Rissuci D, D'Ayala M. A comparison of open and endovascular treatment strategies for the management of splenic artery aneurysms. Vascular 2016; 24:487–491.
- Hogendoorn W, Lavida A, Hunink MG, et al. Cost-effectiveness of endovascular repair, open repair, and conservative management of splenic artery aneurysms. J Vasc Surg 2015; 61:1432–1440.
- Fankhauser GT, Stone WM, Naidu SG, et al; Mayo Vascular Research Center Consortium. The minimally invasive management of visceral artery aneurysms and pseudoaneurysms. J Vasc Surg 2011; 53:966–970.
- Lagana D, Carrafiello G, Mangini M, et al. Multimodal approach to endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Eur J Radiol 2006; 59:104–111.
- Guillon R, Garcier JM, Abergel A, et al. Management of splenic artery aneurysms and false aneurysms with endovascular treatment in 12 patients. Cardiovasc Intervent Radiol 2003; 26:256–260.
- Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for end-stage liver disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg 2005; 242:244–251.
- Bakhos CT, McIntosh BC, Nukta FA, et al. Staged arterial embolization and surgical resection of a giant splenic artery aneurysm. Ann Vasc Surg 2007; 21:208–210.
- Hogendoorn W, Lavida A, Hunink MG, et al. Open repair, endovascular repair, and conservative management of true splenic artery aneurysms. J Vasc Surg 2014; 60:1667–1676.e1.
- Algudkar A. Unruptured splenic artery aneurysm presenting as epigastric pain. JRSM Short Rep 2010; 1:24.
- Abbas MA, Stone WM, Fowl RJ, et al. Splenic artery aneurysms: two decades experience at Mayo Clinic. Ann Vasc Surg 2002; 16:442–449.
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2:399–407.
- Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology 1984; 4:1209–1211.
- Garcia-Tsao G. Spontaneous bacterial peritonitis: a historical perspective. J Hepatol 2004; 41:522–527.
- Soriano G, Castellote J, Alvarez C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol 2010; 52:39–44.
- D’Amico G, De Franchis R; Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010; 31:366–374.
- Kobori L, van der Kolk MJ, de Jong KP, et al. Splenic artery aneurysms in liver transplant patients. Liver Transplant Group. J Hepatol 1997; 27:890–893.
- Manzano-Robleda Mdel C, Barranco-Fragoso B, Uribe M, Mendez-Sanchez N. Portal vein thrombosis: what is new? Ann Hepatol 2015; 14:20–27.
- Sarin SK, Philips CA, Kamath PS, et al. Toward a comprehensive new classification of portal vein thrombosis in patients with cirrhosis. Gastroenterology 2016; 151:574–577.e3.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Manzanet G, Sanjuan F, Orbis P, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl 2001; 7:125–131.
- John BV, Konjeti R, Aggarwal A, et al. Impact of untreated portal vein thrombosis on pre and post liver transplant outcomes in cirrhosis. Ann Hepatol 2013; 12:952–958.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery/Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—summary of recommendations. J Vasc Interv Radiol 2006; 17:1383–1397.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med 2008; 358:464–474.
- Ohta M, Hashizume M, Ueno K, Tanoue K, Sugimachi K, Hasuo K. Hemodynamic study of splenic artery aneurysm in portal hypertension. Hepatogastroenterology 1994; 41:181–184.
- Sunagozaka H, Tsuji H, Mizukoshi E, et al. The development and clinical features of splenic aneurysm associated with liver cirrhosis. Liver Int 2006; 26:291–297.
- Manenti F, Williams R. Injection studies of the splenic vasculature in portal hypertension. Gut 1966; 7:175–180.
- Stanley JC, Fry WJ. Pathogenesis and clinical significance of splenic artery aneurysms. Surgery 1974; 76:898–909.
- Lee PC, Rhee RY, Gordon RY, Fung JJ, Webster MW. Management of splenic artery aneurysms: the significance of portal and essential hypertension. J Am Coll Surg 1999; 189:483–490.
- Al-Habbal Y, Christophi C, Muralidharan V. Aneurysms of the splenic artery—a review. Surgeon 2010; 8:223–231.
- Mattar SG, Lumsden AB. The management of splenic artery aneurysms: experience with 23 cases. Am J Surg 1995; 169:580–584.
- Akbulut S, Otan E. Management of giant splenic artery aneurysm: comprehensive literature review. Medicine (Baltimore) 2015; 94:e1016.
- Agrawal GA, Johnson PT, Fishman EK. Splenic artery aneurysms and pseudoaneurysms: clinical distinctions and CT appearances. AJR Am J Roentgenol 2007; 188:992–999.
- Tessier DJ, Stone WM, Fowl RJ, et al. Clinical features and management of splenic artery pseudoaneurysm: case series and cumulative review of literature. J Vasc Surg 2003; 38:969–974.
- Dave SP, Reis ED, Hossain A, Taub PJ, Kerstein MD, Hollier LH. Splenic artery aneurysm in the 1990s. Ann Vasc Surg 2000; 14:223–229.
- Parrish J, Maxwell C, Beecroft JR. Splenic artery aneurysm in pregnancy. J Obstet Gynaecol Can 2015; 37:816–818.
- Moon DB, Lee SG, Hwang S, et al. Characteristics and management of splenic artery aneurysms in adult living donor liver transplant recipients. Liver Transpl 2009; 15:1535–1541.
- Sadat U, Dar O, Walsh S, Varty K. Splenic artery aneurysms in pregnancy—a systematic review. Int J Surg 2008; 6:261–265.
- Geoghegan T, McAuley G, Snow A, Torreggiani WC. Emergency embolization of multiple splenic artery pseudoaneurysms associated with portal hypertension complicating cystic fibrosis. Australas Radiol 2007; 51(suppl):B337–B339.
- Jiang R, Ding X, Jian W, Jiang J, Hu S, Zhang Z. Combined endovascular embolization and open surgery for splenic artery aneurysm with arteriovenous fistula. Ann Vasc Surg 2016; 30:311.e1–311.e4.
- Naganuma M, Matsui H, Koizumi J, Fushimi K, Yasunaga H. Short-term outcomes following elective transcatheter arterial embolization for splenic artery aneurysms: data from a nationwide administrative database. Acta Radiol Open 2015; 4:2047981615574354.
- Batagini NC, El-Arousy H, Clair DG, Kirksey L. Open versus endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Ann Vasc Surg 2016; 35:1–8.
- Marone EM, Mascia D, Kahlberg A, Brioschi C, Tshomba Y, Chiesa R. Is open repair still the gold standard in visceral artery aneurysm management? Ann Vasc Surg 2011; 25:936–946.
- Sticco A, Aggarwal A, Shapiro M, Pratt A, Rissuci D, D'Ayala M. A comparison of open and endovascular treatment strategies for the management of splenic artery aneurysms. Vascular 2016; 24:487–491.
- Hogendoorn W, Lavida A, Hunink MG, et al. Cost-effectiveness of endovascular repair, open repair, and conservative management of splenic artery aneurysms. J Vasc Surg 2015; 61:1432–1440.
- Fankhauser GT, Stone WM, Naidu SG, et al; Mayo Vascular Research Center Consortium. The minimally invasive management of visceral artery aneurysms and pseudoaneurysms. J Vasc Surg 2011; 53:966–970.
- Lagana D, Carrafiello G, Mangini M, et al. Multimodal approach to endovascular treatment of visceral artery aneurysms and pseudoaneurysms. Eur J Radiol 2006; 59:104–111.
- Guillon R, Garcier JM, Abergel A, et al. Management of splenic artery aneurysms and false aneurysms with endovascular treatment in 12 patients. Cardiovasc Intervent Radiol 2003; 26:256–260.
- Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for end-stage liver disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg 2005; 242:244–251.
Weight loss, fatigue, and renal failure
A black 37-year-old man has gradually lost 100 lb (45 kg) over the past 2 years, and reports progressive fatigue and malaise as well. He has not noted swollen lymph nodes, fever, or night sweats. He denies dyspnea, cough, or chest pain. He has no skin rashes, and no dry or red eyes or visual changes. He reports no flank pain, dysuria, frank hematuria, foamy urine, decline in urine output, or difficulty voiding.
He has no history of significant medical conditions. He does not drink, smoke, or use recreational drugs. He is not taking any prescription medications and has not been using nonsteroidal anti-inflammatory drugs (NSAIDs) or combination analgesics. He does not have a family history of kidney disease.
Physical examination. He appears relaxed and comfortable. He does not have nasal polyps or signs of pharyngeal inflammation. He has no apparent lymphadenopathy. His breath sounds are normal without rales or wheezes. Cardiac examination reveals a regular rhythm, with no rub or murmurs. The abdomen is soft and nontender with no flank pain or groin tenderness. The skin is intact with no rash or nodules.
- Temperature 98.4ºF (36.9ºC)
- Blood pressure 125/70 mm Hg
- Heart rate 102 beats per minute
- Respiratory rate 19 per minute
- Oxygen saturation 99% while breathing room air
- Weight 194 lb (88 kg)
- Body mass index 28 kg/m2.

Laboratory testing (Table 1) reveals severe renal insufficiency with anemia:
- Serum creatinine 9 mg/dL (reference range 0.5–1.2)
- Estimated glomerular filtration rate (eGFR) 8 mL/min/1.73m2 (using the Modification of Diet in Renal Disease Study equation).
His serum calcium level is normal, but his serum phosphorus is 5.3 mg/dL (reference range 2.5–4.6), and his parathyroid hormone level is 317 pg/mL (12–88), consistent with hyperparathyroidism secondary to chronic kidney disease. His 25-hydroxyvitamin D level is less than 13 ng/mL (30–80), and angiotensin-converting enzyme (ACE) is 129 U/L (9–67 U/L). His urinary calcium level is less than 3.0 mg/dL.
Urinalysis:
- Urine protein 100 mg/dL (0–20)
- No urine crystals
- 3 to 5 coarse granular urine casts per high-power field
- No hematuria or pyuria.

Renal ultrasonography shows normal kidneys with no hydronephrosis.
Renal biopsy study demonstrates noncaseating granulomatous interstitial nephritis (Figure 1).
GRANULOMATOUS INTERSTITIAL NEPHRITIS
1. Based on the information above, what is the most likely cause of this patient’s kidney disease?
- Medication
- Granulomatosis with polyangiitis
- Sarcoidosis
- Infection
Granulomatous interstitial nephritis is a histologic diagnosis that is present in up to 1% of renal biopsies. It has been associated with medications, infections, sarcoidosis, crystal deposits, paraproteinemia, and granulomatosis with polyangiitis and also is seen in an idiopathic form.
Medicines implicated include anticonvulsants, antibiotics, NSAIDs, allopurinol, and diuretics.
Mycobacteria and fungi are the main infective causes and seem to be the main causative factor in cases of renal transplant.1 Granulomas are usually not found on kidney biopsy in granulomatosis with polyangiitis, and that diagnosis is usually made by the presence of antiproteinase 3 antibodies.2
Sarcoidosis is the most likely diagnosis in this patient after excluding implicated medications, infection, and vasculitis and confirming the presence of granulomatous interstitial nephritis on renal biopsy.
SARCOIDOSIS: A MULTISYSTEM DISEASE
Sarcoidosis is a multisystem inflammatory disease of unknown cause, characterized by noncaseating epithelioid granulomas. It can involve any organ but most commonly the thoracic and peripheral lymph nodes.3,4 Involvement of the eyes and skin is also relatively common.
Extrapulmonary involvement occurs in more than 30% of cases of sarcoidosis, almost always with concomitant thoracic involvement.5,6 Isolated extrathoracic sarcoidosis is unusual, found in only 2% of patients in a sarcoidosis case-control study.5
Current theory suggests that sarcoidosis develops from a cell-mediated immune response triggered by one or more unidentified antigens in people with a genetic predisposition.7
Sarcoidosis affects men and women of all ages, most often adults ages 20 to 40; but more recently, it has increased in US adults over age 55.8 The condition is more prevalent in Northern Europe and Japan, and in blacks in the United States.7
HOW COMMON IS RENAL INVOLVEMENT IN SARCOIDOSIS?
2. What is the likelihood of finding clinically apparent renal involvement in a patient with sarcoidosis?
- Greater than 70%
- Greater than 50%
- Up to 50%
- Less than 10%
The prevalence of renal involvement in sarcoidosis is hard to determine due to differences in study design and patient populations included in the available reports, and because renal involvement may be silent for many years. Recent studies have reported impaired renal function in 0.7% to 9.7% of cases: eg, a case-control study of 736 patients reported clinically apparent renal involvement in 0.7% of patients,5 and in a series of 818 patients, the incidence was 1%.9 In earlier studies, depending on the diagnostic criteria, the incidence ranged from 1.1% to 9.7%.10
The prevalence of renal involvement may also be underestimated because it can be asymptomatic, and the number of granulomas may be so few that they are absent in a small biopsy specimen. A higher prevalence of renal involvement in sarcoidosis is reported from autopsy studies, although many cases remained clinically silent. These studies have reported renal noncaseating granulomas in 7% to 23% of sarcoidosis patients.11–13
PRESENTATION OF RENAL SARCOIDOSIS
3. What is the most common presentation in isolated renal sarcoidosis?
- Sterile pyuria
- Elevated urine eosinophils
- Renal insufficiency
- Painless hematuria
Renal manifestations of sarcoidosis include hypercalcemia, hypercalciuria, nephrocalcinosis, nephrolithiasis, and impaired renal function.14 Renal involvement can occur during the course of existing sarcoidosis, at the time of first presentation, or even as the sole presentation of the disease.1,11,15 In patients with isolated renal sarcoidosis, the most common presentation is renal insufficiency.15,16
Two main pathways for nephron insult that have been validated are granulomatous infiltration of the renal interstitium and disordered calcium homeostasis.11,17 Though extremely rare, various types of glomerular disease, renal tubular defects, and renal vascular involvement such as renal artery granulomatous angiitis have been documented.18
Hypercalcemia in sarcoidosis
Sarcoidosis is known to cause hypercalcemia by increasing calcium absorption secondary to 1,25-dihydroxyvitamin D production from granulomas. Our patient’s case is unusual, as renal failure was the sole extrapulmonary manifestation of sarcoidosis without hypercalcemia.
In sarcoidosis, extrarenal production of 1-alpha-hydroxylase by activated macrophages inappropriately increases levels of 1,25-dihydroxyvitamin D (calcitriol). Subsequently, serum calcium levels are increased. Unlike its renal equivalent, granulomatous 1-alpha-hydroxylase evades the normal negative feedback of hypercalcemia, so that increased calcitriol levels are sustained, leading to hypercalcemia, often accompanied by hypercalciuria.19
Disruption of calcium homeostasis affects renal function through several mechanisms. Hypercalcemia promotes vasoconstriction of the afferent arteriole, leading to a reduction in the GFR. Intracellular calcium overload can contribute to acute tubular necrosis and intratubular precipitation of calcium, leading to tubular obstruction. Hypercalciuria predisposes to nephrolithiasis and obstructive uropathy. Chronic hypercalcemia and hypercalciuria, if untreated, cause progressive interstitial inflammation and deposition of calcium in the kidney parenchyma and tubules, resulting in nephrocalcinosis. In some cases, nephrocalcinosis leads to chronic kidney injury and renal dysfunction.
HISTOLOGIC FEATURES
4. What is the characteristic histologic feature of renal sarcoidosis?
- Membranous glomerulonephritis
- Mesangioproliferative glomerulonephritis
- Minimal change disease
- Granulomatous interstitial nephritis
- Immunoglobulin (Ig) A nephropathy
Granulomatous interstitial nephritis is the most typical histologic feature of renal sarcoidosis.4,20–22 However, interstitial nephritis without granulomas is found in up to one-third of patients with sarcoid interstitial nephritis.15,23
Patients with sarcoid granulomatous interstitial nephritis usually present with elevated serum creatinine with or without mild proteinuria (< 1 g/24 hours).1,15,16 Advanced renal failure (stage 4 or 5 chronic kidney disease) is relatively common at the time of presentation.1,15,16 In the 2 largest case series of renal sarcoidosis to date, the mean presenting serum creatinine levels were 3.0 and 4.8 mg/dL.11,15 The most common clinical syndrome associated with sarcoidosis and granulomatous interstitial nephritis is chronic kidney disease with a decline in renal function, which if untreated can occur over weeks to months.21 Acute renal failure as an initial presentation is also well documented.15,24
Even though glomerular involvement in sarcoidosis is rare, different kinds of glomerulonephritis have been reported, including membranous glomerulonephritis, mesangioproliferative glomerulonephritis, IgA nephropathy, minimal change disease, focal segmental sclerosis, and crescentic glomerulonephritis.25
DIAGNOSIS OF RENAL SARCOIDOSIS
5. How is renal sarcoidosis diagnosed?
- By exclusion
- Complete urine analysis and renal function assessment
- Renal biopsy
- Computed tomography
- Renal ultrasonography
The diagnosis of renal sarcoidosis is one of exclusion. Sarcoidosis must be considered in the differential diagnosis of renal failure of unknown origin, especially if disordered calcium homeostasis is also present. If clinically suspected, diagnosis usually requires pathohistologic demonstration of typical granulomatous lesions in the kidneys or in one or more organ systems.26
In cases of sarcoidosis with granulomatous interstitial nephritis with isolated renal failure as a presenting feature, other causes of granulomatous interstitial nephritis must be ruled out. A number of drug reactions are associated with interstitial nephritis, most commonly with antibiotics, NSAIDs, and diuretics. Although granulomatous interstitial nephritis may develop as a reaction to some drugs, most cases of drug-induced interstitial nephritis do not involve granulomatous interstitial nephritis.
Other causes of granulomatous interstitial infiltrates include granulomatous infection by mycobacteria, fungi, or Brucella; foreign-body reaction such as cholesterol atheroemboli; heroin; lymphoma; or autoimmune disease such as tubulointerstitial nephritis with uveitis syndrome, granulomatosis with polyangiitis, or Crohn disease.27,28 The absence of characteristic kidney biopsy findings does not exclude the diagnosis because renal sarcoidosis can be focal and easily missed on biopsy.29
Urinary manifestations of renal sarcoidosis are usually not specific. In renal sarcoidosis with interstitial nephritis with or without granulomas, proteinuria is mild or absent, usually less than 1.0 g/day.11,15,16 Urine studies may show a “bland” sediment (ie, without red or white blood cells) or may show sterile pyuria or microscopic hematuria. In glomerular disease, more overt proteinuria or the presence of red blood cell casts is more typical.
Hypercalciuria, nephrocalcinosis, and nephrolithiasis are nonspecific abnormalities that may be present in patients with sarcoidosis. In this regard, an elevated urine calcium level may support the diagnosis of renal sarcoidosis.
Computed tomography and renal ultrasonography may aid in diagnosis by detecting nephrocalcinosis or nephrolithiasis.
The serum ACE level is elevated in 55% to 60% of patients with sarcoidosis, but it may also be elevated in other granulomatous diseases or in chronic kidney disease from various causes.5 Therefore, considering its nonspecificity, the serum ACE level has a limited role in the diagnosis of sarcoidosis.30 Using the ACE level as a marker for disease activity and response to treatment remains controversial because levels do not correlate with disease activity.5,11
TREATMENT OF RENAL SARCOIDOSIS
6. Which is a first-line therapy for renal sarcoidosis?
- Corticosteroids
- Azathioprine
- Mycophenolate mofetil
- Infliximab
- Adalimumab
Treatment of impaired calcium homeostasis in sarcoidosis includes hydration; reducing intake of calcium, vitamin D, and oxalate; and limiting sun exposure.11,31 For more significant hypercalcemia (eg, serum calcium levels > 11 mg/dL) or nephrolithiasis, corticosteroid therapy is the first choice and should be implemented at the first sign of renal involvement. Corticosteroids inhibit the activity of 1-alpha-hydroxylase in macrophages, thereby reducing the production of 1,25-dihydroxyvitamin D.
Chloroquine and hydroxychloroquine have been mentioned in the literature as alternatives to corticosteroids.32 But the effect of these agents is less predictable and is slower than treatment with corticosteroids. Ketoconazole has no effect on granuloma formation but corrects hypercalcemia by inhibiting calcitriol production, and can be used as an adjunct for treating hypercalcemia and hypercalciuria.
Corticosteroids are the mainstay of treatment for renal sarcoidosis, including granulomatous interstitial nephritis and interstitial nephritis without granulomas. Most patients experience significant improvement in renal function. However, full recovery is rare, likely as a result of long-standing disease with some degree of already established irreversible renal injury.16
Corticosteroid dosage
There is no standard dosing protocol, but patients with impaired renal function due to biopsy-proven renal sarcoidosis should receive prednisone 0.5 to 1 mg/kg/day, depending on the severity of the disease, in a single dose every morning.
The optimal dosing and duration of maintenance therapy are unknown. Based on studies to date, the initial dosing should be maintained for 4 weeks, after which it can be tapered by 5 mg each week down to a maintenance dosage of 5 to 10 mg/day.4
Patients with a poor response after 4 weeks tend to have a worse renal outcome and are more susceptible to relapse.15 Fortunately, relapse often responds to increased corticosteroid doses.11,15 In the case of relapse, the dose should be increased to the lowest effective dose and continued for 4 weeks, then tapered more gradually.
A total of 24 months of treatment seems necessary to be effective and to prevent relapse.15 Some authors have proposed a lifelong maintenance dose for patients with frequent relapses, and some propose it for all patients.4
Other agents
Tumor necrosis factor (TNF)-blocking agents. Considering the critical role TNF plays in granuloma formation, anti-TNF-alpha agents are useful in steroid-resistant sarcoidosis.33 A thorough workup is necessary before starting these agents because of the increased risk of serious infection, including reactivation of latent tuberculosis. Of the current TNF-blocking agents, infliximab is most often used in renal sarcoidosis.34 Experience with adalimumab is more limited, though promising results indicate it could be an alternative for patients who do not tolerate infliximab.35
Azathioprine, mycophenolate mofetil, or methotrexate may also be used as a second-line agent if treatment with corticosteroids is not tolerated or does not control the disease. The evidence in support of these agents is limited. In small series, they have allowed sustainable control of renal function while reducing the steroid dose. Currently, these agents are used for patients resistant to corticosteroid therapy, who would otherwise need prolonged high-dose corticosteroid treatment, or who have corticosteroid intolerance; they allow a more effective steroid taper and maintenance of stable renal function.15,36
The data supporting a standardized treatment of renal sarcoidosis are limited. For steroid intolerance or resistance, cytotoxic drugs and selected anti-TNF-alpha agents, as mentioned above, have shown promise in improving or stabilizing serum creatinine levels. Further exploration is required as to which agent or combination is better at limiting the disease process with fewer adverse effects.
Our patient was initially treated with corticosteroids and was ultimately weaned to a maintenance dose of 5 mg/day. He was followed as an outpatient and was started on mycophenolate mofetil in place of higher steroid doses. His renal function stabilized, but he was lost to follow-up after 2 years.
KEY POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly involves the lungs, skin, and reticuloendothelial system.
- Renal involvement in sarcoidosis is likely underestimated due to its often clinically silent nature and the possibility of missing typical granulomatous lesions in a small or less-than-optimal biopsy sample.
- Manifestations of renal sarcoidosis include disrupted calcium homeostasis, nephrocalcinosis, nephrolithiasis, and renal failure.
- Because the clinical and histopathologic manifestations of renal sarcoidosis are nonspecific, the diagnosis is one of exclusion. In patients with renal failure or with hypercalcemia or hypercalciuria of unknown cause, renal sarcoidosis should be included in the differential diagnosis. Patients with chronic sarcoidosis should also be screened for renal impairment.
- Granulomatous interstitial nephritis is the classic histologic finding of renal sarcoidosis. Nonetheless, up to one-third of patients have interstitial nephritis without granulomas.
- Corticosteroids are the mainstay of treatment for renal sarcoidosis. An initial dose of oral prednisone 0.5 to 1 mg/kg/day should be maintained for 4 weeks and then gradually tapered to 5 to 10 mg/day for a total of 24 months. Some patients require lifelong therapy.
- Several immunosuppressive and cytotoxic agents may be used in cases of corticosteroid intolerance or to aid in effective taper of corticosteroids.
- Joss N, Morris S, Young B, Geddes C. Granulomatous interstitial nephritis. Clin J Am Soc Nephrol 2007; 2:222–230.
- Lutalo PM, D'Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener's granulomatosis). J Autoimmun 2014; 48–49:94–98.
- Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med 1997; 336:1224–1234.
- Rajakariar R, Sharples EJ, Raftery MJ, Sheaff M, Yaqoob MM. Sarcoid tubulo-interstitial nephritis: long-term outcome and response to corticosteroid therapy. Kidney Int 2006; 70:165–169.
- Baughman RP, Teirstein AS, Judson MA, et al; Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001; 164:1885–1889.
- Rizzato G, Palmieri G, Agrati AM, Zanussi C. The organ-specific extrapulmonary presentation of sarcoidosis: a frequent occurrence but a challenge to an early diagnosis. A 3-year-long prospective observational study. Sarcoidosis Vasc Diffuse Lung Dis 2004; 21:119–126.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007; 357:2153–2165.
- Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016; 13:1244–1252.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med 1983; 52:525–533.
- Mayock RL, Bertrand P, Morrison CE, Scott JH. Manifestations of sarcoidosis. Analysis of 145 patients, with a review of nine series selected from the literature. Am J Med 1963; 35:67–89.
- Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist's perspective. Am J Kidney Dis 2006; 48:856–870.
- Longcope WT, Freiman DG. A study of sarcoidosis; based on a combined investigation of 160 cases including 30 autopsies from The Johns Hopkins Hospital and Massachusetts General Hospital. Medicine (Baltimore) 1952; 31:1–132.
- Branson JH, Park JH. Sarcoidosis hepatic involvement: presentation of a case with fatal liver involvement; including autopsy findings and review of the evidence for sarcoid involvement of the liver as found in the literature. Ann Intern Med 1954; 40:111–145.
- Muther RS, McCarron DA, Bennett WM. Renal manifestations of sarcoidosis. Arch Intern Med 1981; 141:643–645.
- Mahevas M, Lescure FX, Boffa JJ, et al. Renal sarcoidosis: clinical, laboratory, and histologic presentation and outcome in 47 patients. Medicine (Baltimore) 2009; 88:98–106.
- Robson MG, Banerjee D, Hopster D, Cairns HS. Seven cases of granulomatous interstitial nephritis in the absence of extrarenal sarcoid. Nephrol Dial Transplant 2003; 18:280–284.
- Casella FJ, Allon M. The kidney in sarcoidosis. J Am Soc Nephrol 1993; 3:1555–1562.
- Rafat C, Bobrie G, Chedid A, Nochy D, Hernigou A, Plouin PF. Sarcoidosis presenting as severe renin-dependent hypertension due to kidney vascular injury. Clin Kidney J 2014; 7:383–386.
- Reichel H, Koeffler HP, Barbers R, Norman AW. Regulation of 1,25-dihydroxyvitamin D3 production by cultured alveolar macrophages from normal human donors and from patients with pulmonary sarcoidosis. J Clin Endocrinol Metab 1987; 65:1201–1209.
- Brause M, Magnusson K, Degenhardt S, Helmchen U, Grabensee B. Renal involvement in sarcoidosis—a report of 6 cases. Clin Nephrol 2002; 57:142–148.
- Hannedouche T, Grateau G, Noel LH, et al. Renal granulomatous sarcoidosis: report of six cases. Nephrol Dial Transplant 1990; 5:18–24.
- Kettritz R, Goebel U, Fiebeler A, Schneider W, Luft F. The protean face of sarcoidosis revisited. Nephrol Dial Transplant 2006; 21:2690–2694.
- Bergner R, Hoffmann M, Waldherr R, Uppenkamp M. Frequency of kidney disease in chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2003; 20:126–132.
- O’Riordan E, Willert RP, Reeve R, et al. Isolated sarcoid granulomatous interstitial nephritis: review of five cases at one center. Clin Nephrol 2001; 55:297–302.
- Gobel U, Kettritz R, Schneider W, Luft F. The protean face of renal sarcoidosis. J Am Soc Nephrol 2001; 12:616–623.
- Statement on sarcoidosis. Joint statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999; 160:736–755.
- Bijol V, Mendez GP, Nose V, Rennke HG. Granulomatous interstitial nephritis: a clinicopathologic study of 46 cases from a single institution. Int J Surg Pathol 2006; 14:57–63.
- Mignon F, Mery JP, Mougenot B, Ronco P, Roland J, Morel-Maroger L. Granulomatous interstitial nephritis. Adv Nephrol Necker Hosp 1984; 13:219–245.
- Shah R, Shidham G, Agarwal A, Albawardi A, Nadasdy T. Diagnostic utility of kidney biopsy in patients with sarcoidosis and acute kidney injury. Int J Nephrol Renovasc Dis 2011; 4:131–136.
- Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis—its value in present clinical practice. Ann Clin Biochem 1989; 26:13–18.
- Demetriou ET, Pietras SM, Holick MF. Hypercalcemia and soft tissue calcification owing to sarcoidosis: the sunlight-cola connection. J Bone Miner Res 2010; 25:1695–1699.
- Beegle SH, Barba K, Gobunsuy R, Judson MA. Current and emerging pharmacological treatments for sarcoidosis: a review. Drug Des Devel Ther 2013; 7:325–338.
- Roberts SD, Wilkes DS, Burgett RA, Knox KS. Refractory sarcoidosis responding to infliximab. Chest 2003; 124:2028–2031.
- Ahmed MM, Mubashir E, Dossabhoy NR. Isolated renal sarcoidosis: a rare presentation of a rare disease treated with infliximab. Clin Rheumatol 2007; 26:1346–1349.
- Gupta R, Beaudet L, Moore J, Mehta T. Treatment of sarcoid granulomatous interstitial nephritis with adalimumab. NDT Plus 2009; 2:139–142.
- Moudgil A, Przygodzki RM, Kher KK. Successful steroid-sparing treatment of renal limited sarcoidosis with mycophenolate mofetil. Pediatr Nephrol 2006; 21:281–285.
A black 37-year-old man has gradually lost 100 lb (45 kg) over the past 2 years, and reports progressive fatigue and malaise as well. He has not noted swollen lymph nodes, fever, or night sweats. He denies dyspnea, cough, or chest pain. He has no skin rashes, and no dry or red eyes or visual changes. He reports no flank pain, dysuria, frank hematuria, foamy urine, decline in urine output, or difficulty voiding.
He has no history of significant medical conditions. He does not drink, smoke, or use recreational drugs. He is not taking any prescription medications and has not been using nonsteroidal anti-inflammatory drugs (NSAIDs) or combination analgesics. He does not have a family history of kidney disease.
Physical examination. He appears relaxed and comfortable. He does not have nasal polyps or signs of pharyngeal inflammation. He has no apparent lymphadenopathy. His breath sounds are normal without rales or wheezes. Cardiac examination reveals a regular rhythm, with no rub or murmurs. The abdomen is soft and nontender with no flank pain or groin tenderness. The skin is intact with no rash or nodules.
- Temperature 98.4ºF (36.9ºC)
- Blood pressure 125/70 mm Hg
- Heart rate 102 beats per minute
- Respiratory rate 19 per minute
- Oxygen saturation 99% while breathing room air
- Weight 194 lb (88 kg)
- Body mass index 28 kg/m2.
Laboratory testing (Table 1) reveals severe renal insufficiency with anemia:
- Serum creatinine 9 mg/dL (reference range 0.5–1.2)
- Estimated glomerular filtration rate (eGFR) 8 mL/min/1.73m2 (using the Modification of Diet in Renal Disease Study equation).
His serum calcium level is normal, but his serum phosphorus is 5.3 mg/dL (reference range 2.5–4.6), and his parathyroid hormone level is 317 pg/mL (12–88), consistent with hyperparathyroidism secondary to chronic kidney disease. His 25-hydroxyvitamin D level is less than 13 ng/mL (30–80), and angiotensin-converting enzyme (ACE) is 129 U/L (9–67 U/L). His urinary calcium level is less than 3.0 mg/dL.
Urinalysis:
- Urine protein 100 mg/dL (0–20)
- No urine crystals
- 3 to 5 coarse granular urine casts per high-power field
- No hematuria or pyuria.
Renal ultrasonography shows normal kidneys with no hydronephrosis.
Renal biopsy study demonstrates noncaseating granulomatous interstitial nephritis (Figure 1).
GRANULOMATOUS INTERSTITIAL NEPHRITIS
1. Based on the information above, what is the most likely cause of this patient’s kidney disease?
- Medication
- Granulomatosis with polyangiitis
- Sarcoidosis
- Infection
Granulomatous interstitial nephritis is a histologic diagnosis that is present in up to 1% of renal biopsies. It has been associated with medications, infections, sarcoidosis, crystal deposits, paraproteinemia, and granulomatosis with polyangiitis and also is seen in an idiopathic form.
Medicines implicated include anticonvulsants, antibiotics, NSAIDs, allopurinol, and diuretics.
Mycobacteria and fungi are the main infective causes and seem to be the main causative factor in cases of renal transplant.1 Granulomas are usually not found on kidney biopsy in granulomatosis with polyangiitis, and that diagnosis is usually made by the presence of antiproteinase 3 antibodies.2
Sarcoidosis is the most likely diagnosis in this patient after excluding implicated medications, infection, and vasculitis and confirming the presence of granulomatous interstitial nephritis on renal biopsy.
SARCOIDOSIS: A MULTISYSTEM DISEASE
Sarcoidosis is a multisystem inflammatory disease of unknown cause, characterized by noncaseating epithelioid granulomas. It can involve any organ but most commonly the thoracic and peripheral lymph nodes.3,4 Involvement of the eyes and skin is also relatively common.
Extrapulmonary involvement occurs in more than 30% of cases of sarcoidosis, almost always with concomitant thoracic involvement.5,6 Isolated extrathoracic sarcoidosis is unusual, found in only 2% of patients in a sarcoidosis case-control study.5
Current theory suggests that sarcoidosis develops from a cell-mediated immune response triggered by one or more unidentified antigens in people with a genetic predisposition.7
Sarcoidosis affects men and women of all ages, most often adults ages 20 to 40; but more recently, it has increased in US adults over age 55.8 The condition is more prevalent in Northern Europe and Japan, and in blacks in the United States.7
HOW COMMON IS RENAL INVOLVEMENT IN SARCOIDOSIS?
2. What is the likelihood of finding clinically apparent renal involvement in a patient with sarcoidosis?
- Greater than 70%
- Greater than 50%
- Up to 50%
- Less than 10%
The prevalence of renal involvement in sarcoidosis is hard to determine due to differences in study design and patient populations included in the available reports, and because renal involvement may be silent for many years. Recent studies have reported impaired renal function in 0.7% to 9.7% of cases: eg, a case-control study of 736 patients reported clinically apparent renal involvement in 0.7% of patients,5 and in a series of 818 patients, the incidence was 1%.9 In earlier studies, depending on the diagnostic criteria, the incidence ranged from 1.1% to 9.7%.10
The prevalence of renal involvement may also be underestimated because it can be asymptomatic, and the number of granulomas may be so few that they are absent in a small biopsy specimen. A higher prevalence of renal involvement in sarcoidosis is reported from autopsy studies, although many cases remained clinically silent. These studies have reported renal noncaseating granulomas in 7% to 23% of sarcoidosis patients.11–13
PRESENTATION OF RENAL SARCOIDOSIS
3. What is the most common presentation in isolated renal sarcoidosis?
- Sterile pyuria
- Elevated urine eosinophils
- Renal insufficiency
- Painless hematuria
Renal manifestations of sarcoidosis include hypercalcemia, hypercalciuria, nephrocalcinosis, nephrolithiasis, and impaired renal function.14 Renal involvement can occur during the course of existing sarcoidosis, at the time of first presentation, or even as the sole presentation of the disease.1,11,15 In patients with isolated renal sarcoidosis, the most common presentation is renal insufficiency.15,16
Two main pathways for nephron insult that have been validated are granulomatous infiltration of the renal interstitium and disordered calcium homeostasis.11,17 Though extremely rare, various types of glomerular disease, renal tubular defects, and renal vascular involvement such as renal artery granulomatous angiitis have been documented.18
Hypercalcemia in sarcoidosis
Sarcoidosis is known to cause hypercalcemia by increasing calcium absorption secondary to 1,25-dihydroxyvitamin D production from granulomas. Our patient’s case is unusual, as renal failure was the sole extrapulmonary manifestation of sarcoidosis without hypercalcemia.
In sarcoidosis, extrarenal production of 1-alpha-hydroxylase by activated macrophages inappropriately increases levels of 1,25-dihydroxyvitamin D (calcitriol). Subsequently, serum calcium levels are increased. Unlike its renal equivalent, granulomatous 1-alpha-hydroxylase evades the normal negative feedback of hypercalcemia, so that increased calcitriol levels are sustained, leading to hypercalcemia, often accompanied by hypercalciuria.19
Disruption of calcium homeostasis affects renal function through several mechanisms. Hypercalcemia promotes vasoconstriction of the afferent arteriole, leading to a reduction in the GFR. Intracellular calcium overload can contribute to acute tubular necrosis and intratubular precipitation of calcium, leading to tubular obstruction. Hypercalciuria predisposes to nephrolithiasis and obstructive uropathy. Chronic hypercalcemia and hypercalciuria, if untreated, cause progressive interstitial inflammation and deposition of calcium in the kidney parenchyma and tubules, resulting in nephrocalcinosis. In some cases, nephrocalcinosis leads to chronic kidney injury and renal dysfunction.
HISTOLOGIC FEATURES
4. What is the characteristic histologic feature of renal sarcoidosis?
- Membranous glomerulonephritis
- Mesangioproliferative glomerulonephritis
- Minimal change disease
- Granulomatous interstitial nephritis
- Immunoglobulin (Ig) A nephropathy
Granulomatous interstitial nephritis is the most typical histologic feature of renal sarcoidosis.4,20–22 However, interstitial nephritis without granulomas is found in up to one-third of patients with sarcoid interstitial nephritis.15,23
Patients with sarcoid granulomatous interstitial nephritis usually present with elevated serum creatinine with or without mild proteinuria (< 1 g/24 hours).1,15,16 Advanced renal failure (stage 4 or 5 chronic kidney disease) is relatively common at the time of presentation.1,15,16 In the 2 largest case series of renal sarcoidosis to date, the mean presenting serum creatinine levels were 3.0 and 4.8 mg/dL.11,15 The most common clinical syndrome associated with sarcoidosis and granulomatous interstitial nephritis is chronic kidney disease with a decline in renal function, which if untreated can occur over weeks to months.21 Acute renal failure as an initial presentation is also well documented.15,24
Even though glomerular involvement in sarcoidosis is rare, different kinds of glomerulonephritis have been reported, including membranous glomerulonephritis, mesangioproliferative glomerulonephritis, IgA nephropathy, minimal change disease, focal segmental sclerosis, and crescentic glomerulonephritis.25
DIAGNOSIS OF RENAL SARCOIDOSIS
5. How is renal sarcoidosis diagnosed?
- By exclusion
- Complete urine analysis and renal function assessment
- Renal biopsy
- Computed tomography
- Renal ultrasonography
The diagnosis of renal sarcoidosis is one of exclusion. Sarcoidosis must be considered in the differential diagnosis of renal failure of unknown origin, especially if disordered calcium homeostasis is also present. If clinically suspected, diagnosis usually requires pathohistologic demonstration of typical granulomatous lesions in the kidneys or in one or more organ systems.26
In cases of sarcoidosis with granulomatous interstitial nephritis with isolated renal failure as a presenting feature, other causes of granulomatous interstitial nephritis must be ruled out. A number of drug reactions are associated with interstitial nephritis, most commonly with antibiotics, NSAIDs, and diuretics. Although granulomatous interstitial nephritis may develop as a reaction to some drugs, most cases of drug-induced interstitial nephritis do not involve granulomatous interstitial nephritis.
Other causes of granulomatous interstitial infiltrates include granulomatous infection by mycobacteria, fungi, or Brucella; foreign-body reaction such as cholesterol atheroemboli; heroin; lymphoma; or autoimmune disease such as tubulointerstitial nephritis with uveitis syndrome, granulomatosis with polyangiitis, or Crohn disease.27,28 The absence of characteristic kidney biopsy findings does not exclude the diagnosis because renal sarcoidosis can be focal and easily missed on biopsy.29
Urinary manifestations of renal sarcoidosis are usually not specific. In renal sarcoidosis with interstitial nephritis with or without granulomas, proteinuria is mild or absent, usually less than 1.0 g/day.11,15,16 Urine studies may show a “bland” sediment (ie, without red or white blood cells) or may show sterile pyuria or microscopic hematuria. In glomerular disease, more overt proteinuria or the presence of red blood cell casts is more typical.
Hypercalciuria, nephrocalcinosis, and nephrolithiasis are nonspecific abnormalities that may be present in patients with sarcoidosis. In this regard, an elevated urine calcium level may support the diagnosis of renal sarcoidosis.
Computed tomography and renal ultrasonography may aid in diagnosis by detecting nephrocalcinosis or nephrolithiasis.
The serum ACE level is elevated in 55% to 60% of patients with sarcoidosis, but it may also be elevated in other granulomatous diseases or in chronic kidney disease from various causes.5 Therefore, considering its nonspecificity, the serum ACE level has a limited role in the diagnosis of sarcoidosis.30 Using the ACE level as a marker for disease activity and response to treatment remains controversial because levels do not correlate with disease activity.5,11
TREATMENT OF RENAL SARCOIDOSIS
6. Which is a first-line therapy for renal sarcoidosis?
- Corticosteroids
- Azathioprine
- Mycophenolate mofetil
- Infliximab
- Adalimumab
Treatment of impaired calcium homeostasis in sarcoidosis includes hydration; reducing intake of calcium, vitamin D, and oxalate; and limiting sun exposure.11,31 For more significant hypercalcemia (eg, serum calcium levels > 11 mg/dL) or nephrolithiasis, corticosteroid therapy is the first choice and should be implemented at the first sign of renal involvement. Corticosteroids inhibit the activity of 1-alpha-hydroxylase in macrophages, thereby reducing the production of 1,25-dihydroxyvitamin D.
Chloroquine and hydroxychloroquine have been mentioned in the literature as alternatives to corticosteroids.32 But the effect of these agents is less predictable and is slower than treatment with corticosteroids. Ketoconazole has no effect on granuloma formation but corrects hypercalcemia by inhibiting calcitriol production, and can be used as an adjunct for treating hypercalcemia and hypercalciuria.
Corticosteroids are the mainstay of treatment for renal sarcoidosis, including granulomatous interstitial nephritis and interstitial nephritis without granulomas. Most patients experience significant improvement in renal function. However, full recovery is rare, likely as a result of long-standing disease with some degree of already established irreversible renal injury.16
Corticosteroid dosage
There is no standard dosing protocol, but patients with impaired renal function due to biopsy-proven renal sarcoidosis should receive prednisone 0.5 to 1 mg/kg/day, depending on the severity of the disease, in a single dose every morning.
The optimal dosing and duration of maintenance therapy are unknown. Based on studies to date, the initial dosing should be maintained for 4 weeks, after which it can be tapered by 5 mg each week down to a maintenance dosage of 5 to 10 mg/day.4
Patients with a poor response after 4 weeks tend to have a worse renal outcome and are more susceptible to relapse.15 Fortunately, relapse often responds to increased corticosteroid doses.11,15 In the case of relapse, the dose should be increased to the lowest effective dose and continued for 4 weeks, then tapered more gradually.
A total of 24 months of treatment seems necessary to be effective and to prevent relapse.15 Some authors have proposed a lifelong maintenance dose for patients with frequent relapses, and some propose it for all patients.4
Other agents
Tumor necrosis factor (TNF)-blocking agents. Considering the critical role TNF plays in granuloma formation, anti-TNF-alpha agents are useful in steroid-resistant sarcoidosis.33 A thorough workup is necessary before starting these agents because of the increased risk of serious infection, including reactivation of latent tuberculosis. Of the current TNF-blocking agents, infliximab is most often used in renal sarcoidosis.34 Experience with adalimumab is more limited, though promising results indicate it could be an alternative for patients who do not tolerate infliximab.35
Azathioprine, mycophenolate mofetil, or methotrexate may also be used as a second-line agent if treatment with corticosteroids is not tolerated or does not control the disease. The evidence in support of these agents is limited. In small series, they have allowed sustainable control of renal function while reducing the steroid dose. Currently, these agents are used for patients resistant to corticosteroid therapy, who would otherwise need prolonged high-dose corticosteroid treatment, or who have corticosteroid intolerance; they allow a more effective steroid taper and maintenance of stable renal function.15,36
The data supporting a standardized treatment of renal sarcoidosis are limited. For steroid intolerance or resistance, cytotoxic drugs and selected anti-TNF-alpha agents, as mentioned above, have shown promise in improving or stabilizing serum creatinine levels. Further exploration is required as to which agent or combination is better at limiting the disease process with fewer adverse effects.
Our patient was initially treated with corticosteroids and was ultimately weaned to a maintenance dose of 5 mg/day. He was followed as an outpatient and was started on mycophenolate mofetil in place of higher steroid doses. His renal function stabilized, but he was lost to follow-up after 2 years.
KEY POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly involves the lungs, skin, and reticuloendothelial system.
- Renal involvement in sarcoidosis is likely underestimated due to its often clinically silent nature and the possibility of missing typical granulomatous lesions in a small or less-than-optimal biopsy sample.
- Manifestations of renal sarcoidosis include disrupted calcium homeostasis, nephrocalcinosis, nephrolithiasis, and renal failure.
- Because the clinical and histopathologic manifestations of renal sarcoidosis are nonspecific, the diagnosis is one of exclusion. In patients with renal failure or with hypercalcemia or hypercalciuria of unknown cause, renal sarcoidosis should be included in the differential diagnosis. Patients with chronic sarcoidosis should also be screened for renal impairment.
- Granulomatous interstitial nephritis is the classic histologic finding of renal sarcoidosis. Nonetheless, up to one-third of patients have interstitial nephritis without granulomas.
- Corticosteroids are the mainstay of treatment for renal sarcoidosis. An initial dose of oral prednisone 0.5 to 1 mg/kg/day should be maintained for 4 weeks and then gradually tapered to 5 to 10 mg/day for a total of 24 months. Some patients require lifelong therapy.
- Several immunosuppressive and cytotoxic agents may be used in cases of corticosteroid intolerance or to aid in effective taper of corticosteroids.
A black 37-year-old man has gradually lost 100 lb (45 kg) over the past 2 years, and reports progressive fatigue and malaise as well. He has not noted swollen lymph nodes, fever, or night sweats. He denies dyspnea, cough, or chest pain. He has no skin rashes, and no dry or red eyes or visual changes. He reports no flank pain, dysuria, frank hematuria, foamy urine, decline in urine output, or difficulty voiding.
He has no history of significant medical conditions. He does not drink, smoke, or use recreational drugs. He is not taking any prescription medications and has not been using nonsteroidal anti-inflammatory drugs (NSAIDs) or combination analgesics. He does not have a family history of kidney disease.
Physical examination. He appears relaxed and comfortable. He does not have nasal polyps or signs of pharyngeal inflammation. He has no apparent lymphadenopathy. His breath sounds are normal without rales or wheezes. Cardiac examination reveals a regular rhythm, with no rub or murmurs. The abdomen is soft and nontender with no flank pain or groin tenderness. The skin is intact with no rash or nodules.
- Temperature 98.4ºF (36.9ºC)
- Blood pressure 125/70 mm Hg
- Heart rate 102 beats per minute
- Respiratory rate 19 per minute
- Oxygen saturation 99% while breathing room air
- Weight 194 lb (88 kg)
- Body mass index 28 kg/m2.
Laboratory testing (Table 1) reveals severe renal insufficiency with anemia:
- Serum creatinine 9 mg/dL (reference range 0.5–1.2)
- Estimated glomerular filtration rate (eGFR) 8 mL/min/1.73m2 (using the Modification of Diet in Renal Disease Study equation).
His serum calcium level is normal, but his serum phosphorus is 5.3 mg/dL (reference range 2.5–4.6), and his parathyroid hormone level is 317 pg/mL (12–88), consistent with hyperparathyroidism secondary to chronic kidney disease. His 25-hydroxyvitamin D level is less than 13 ng/mL (30–80), and angiotensin-converting enzyme (ACE) is 129 U/L (9–67 U/L). His urinary calcium level is less than 3.0 mg/dL.
Urinalysis:
- Urine protein 100 mg/dL (0–20)
- No urine crystals
- 3 to 5 coarse granular urine casts per high-power field
- No hematuria or pyuria.
Renal ultrasonography shows normal kidneys with no hydronephrosis.
Renal biopsy study demonstrates noncaseating granulomatous interstitial nephritis (Figure 1).
GRANULOMATOUS INTERSTITIAL NEPHRITIS
1. Based on the information above, what is the most likely cause of this patient’s kidney disease?
- Medication
- Granulomatosis with polyangiitis
- Sarcoidosis
- Infection
Granulomatous interstitial nephritis is a histologic diagnosis that is present in up to 1% of renal biopsies. It has been associated with medications, infections, sarcoidosis, crystal deposits, paraproteinemia, and granulomatosis with polyangiitis and also is seen in an idiopathic form.
Medicines implicated include anticonvulsants, antibiotics, NSAIDs, allopurinol, and diuretics.
Mycobacteria and fungi are the main infective causes and seem to be the main causative factor in cases of renal transplant.1 Granulomas are usually not found on kidney biopsy in granulomatosis with polyangiitis, and that diagnosis is usually made by the presence of antiproteinase 3 antibodies.2
Sarcoidosis is the most likely diagnosis in this patient after excluding implicated medications, infection, and vasculitis and confirming the presence of granulomatous interstitial nephritis on renal biopsy.
SARCOIDOSIS: A MULTISYSTEM DISEASE
Sarcoidosis is a multisystem inflammatory disease of unknown cause, characterized by noncaseating epithelioid granulomas. It can involve any organ but most commonly the thoracic and peripheral lymph nodes.3,4 Involvement of the eyes and skin is also relatively common.
Extrapulmonary involvement occurs in more than 30% of cases of sarcoidosis, almost always with concomitant thoracic involvement.5,6 Isolated extrathoracic sarcoidosis is unusual, found in only 2% of patients in a sarcoidosis case-control study.5
Current theory suggests that sarcoidosis develops from a cell-mediated immune response triggered by one or more unidentified antigens in people with a genetic predisposition.7
Sarcoidosis affects men and women of all ages, most often adults ages 20 to 40; but more recently, it has increased in US adults over age 55.8 The condition is more prevalent in Northern Europe and Japan, and in blacks in the United States.7
HOW COMMON IS RENAL INVOLVEMENT IN SARCOIDOSIS?
2. What is the likelihood of finding clinically apparent renal involvement in a patient with sarcoidosis?
- Greater than 70%
- Greater than 50%
- Up to 50%
- Less than 10%
The prevalence of renal involvement in sarcoidosis is hard to determine due to differences in study design and patient populations included in the available reports, and because renal involvement may be silent for many years. Recent studies have reported impaired renal function in 0.7% to 9.7% of cases: eg, a case-control study of 736 patients reported clinically apparent renal involvement in 0.7% of patients,5 and in a series of 818 patients, the incidence was 1%.9 In earlier studies, depending on the diagnostic criteria, the incidence ranged from 1.1% to 9.7%.10
The prevalence of renal involvement may also be underestimated because it can be asymptomatic, and the number of granulomas may be so few that they are absent in a small biopsy specimen. A higher prevalence of renal involvement in sarcoidosis is reported from autopsy studies, although many cases remained clinically silent. These studies have reported renal noncaseating granulomas in 7% to 23% of sarcoidosis patients.11–13
PRESENTATION OF RENAL SARCOIDOSIS
3. What is the most common presentation in isolated renal sarcoidosis?
- Sterile pyuria
- Elevated urine eosinophils
- Renal insufficiency
- Painless hematuria
Renal manifestations of sarcoidosis include hypercalcemia, hypercalciuria, nephrocalcinosis, nephrolithiasis, and impaired renal function.14 Renal involvement can occur during the course of existing sarcoidosis, at the time of first presentation, or even as the sole presentation of the disease.1,11,15 In patients with isolated renal sarcoidosis, the most common presentation is renal insufficiency.15,16
Two main pathways for nephron insult that have been validated are granulomatous infiltration of the renal interstitium and disordered calcium homeostasis.11,17 Though extremely rare, various types of glomerular disease, renal tubular defects, and renal vascular involvement such as renal artery granulomatous angiitis have been documented.18
Hypercalcemia in sarcoidosis
Sarcoidosis is known to cause hypercalcemia by increasing calcium absorption secondary to 1,25-dihydroxyvitamin D production from granulomas. Our patient’s case is unusual, as renal failure was the sole extrapulmonary manifestation of sarcoidosis without hypercalcemia.
In sarcoidosis, extrarenal production of 1-alpha-hydroxylase by activated macrophages inappropriately increases levels of 1,25-dihydroxyvitamin D (calcitriol). Subsequently, serum calcium levels are increased. Unlike its renal equivalent, granulomatous 1-alpha-hydroxylase evades the normal negative feedback of hypercalcemia, so that increased calcitriol levels are sustained, leading to hypercalcemia, often accompanied by hypercalciuria.19
Disruption of calcium homeostasis affects renal function through several mechanisms. Hypercalcemia promotes vasoconstriction of the afferent arteriole, leading to a reduction in the GFR. Intracellular calcium overload can contribute to acute tubular necrosis and intratubular precipitation of calcium, leading to tubular obstruction. Hypercalciuria predisposes to nephrolithiasis and obstructive uropathy. Chronic hypercalcemia and hypercalciuria, if untreated, cause progressive interstitial inflammation and deposition of calcium in the kidney parenchyma and tubules, resulting in nephrocalcinosis. In some cases, nephrocalcinosis leads to chronic kidney injury and renal dysfunction.
HISTOLOGIC FEATURES
4. What is the characteristic histologic feature of renal sarcoidosis?
- Membranous glomerulonephritis
- Mesangioproliferative glomerulonephritis
- Minimal change disease
- Granulomatous interstitial nephritis
- Immunoglobulin (Ig) A nephropathy
Granulomatous interstitial nephritis is the most typical histologic feature of renal sarcoidosis.4,20–22 However, interstitial nephritis without granulomas is found in up to one-third of patients with sarcoid interstitial nephritis.15,23
Patients with sarcoid granulomatous interstitial nephritis usually present with elevated serum creatinine with or without mild proteinuria (< 1 g/24 hours).1,15,16 Advanced renal failure (stage 4 or 5 chronic kidney disease) is relatively common at the time of presentation.1,15,16 In the 2 largest case series of renal sarcoidosis to date, the mean presenting serum creatinine levels were 3.0 and 4.8 mg/dL.11,15 The most common clinical syndrome associated with sarcoidosis and granulomatous interstitial nephritis is chronic kidney disease with a decline in renal function, which if untreated can occur over weeks to months.21 Acute renal failure as an initial presentation is also well documented.15,24
Even though glomerular involvement in sarcoidosis is rare, different kinds of glomerulonephritis have been reported, including membranous glomerulonephritis, mesangioproliferative glomerulonephritis, IgA nephropathy, minimal change disease, focal segmental sclerosis, and crescentic glomerulonephritis.25
DIAGNOSIS OF RENAL SARCOIDOSIS
5. How is renal sarcoidosis diagnosed?
- By exclusion
- Complete urine analysis and renal function assessment
- Renal biopsy
- Computed tomography
- Renal ultrasonography
The diagnosis of renal sarcoidosis is one of exclusion. Sarcoidosis must be considered in the differential diagnosis of renal failure of unknown origin, especially if disordered calcium homeostasis is also present. If clinically suspected, diagnosis usually requires pathohistologic demonstration of typical granulomatous lesions in the kidneys or in one or more organ systems.26
In cases of sarcoidosis with granulomatous interstitial nephritis with isolated renal failure as a presenting feature, other causes of granulomatous interstitial nephritis must be ruled out. A number of drug reactions are associated with interstitial nephritis, most commonly with antibiotics, NSAIDs, and diuretics. Although granulomatous interstitial nephritis may develop as a reaction to some drugs, most cases of drug-induced interstitial nephritis do not involve granulomatous interstitial nephritis.
Other causes of granulomatous interstitial infiltrates include granulomatous infection by mycobacteria, fungi, or Brucella; foreign-body reaction such as cholesterol atheroemboli; heroin; lymphoma; or autoimmune disease such as tubulointerstitial nephritis with uveitis syndrome, granulomatosis with polyangiitis, or Crohn disease.27,28 The absence of characteristic kidney biopsy findings does not exclude the diagnosis because renal sarcoidosis can be focal and easily missed on biopsy.29
Urinary manifestations of renal sarcoidosis are usually not specific. In renal sarcoidosis with interstitial nephritis with or without granulomas, proteinuria is mild or absent, usually less than 1.0 g/day.11,15,16 Urine studies may show a “bland” sediment (ie, without red or white blood cells) or may show sterile pyuria or microscopic hematuria. In glomerular disease, more overt proteinuria or the presence of red blood cell casts is more typical.
Hypercalciuria, nephrocalcinosis, and nephrolithiasis are nonspecific abnormalities that may be present in patients with sarcoidosis. In this regard, an elevated urine calcium level may support the diagnosis of renal sarcoidosis.
Computed tomography and renal ultrasonography may aid in diagnosis by detecting nephrocalcinosis or nephrolithiasis.
The serum ACE level is elevated in 55% to 60% of patients with sarcoidosis, but it may also be elevated in other granulomatous diseases or in chronic kidney disease from various causes.5 Therefore, considering its nonspecificity, the serum ACE level has a limited role in the diagnosis of sarcoidosis.30 Using the ACE level as a marker for disease activity and response to treatment remains controversial because levels do not correlate with disease activity.5,11
TREATMENT OF RENAL SARCOIDOSIS
6. Which is a first-line therapy for renal sarcoidosis?
- Corticosteroids
- Azathioprine
- Mycophenolate mofetil
- Infliximab
- Adalimumab
Treatment of impaired calcium homeostasis in sarcoidosis includes hydration; reducing intake of calcium, vitamin D, and oxalate; and limiting sun exposure.11,31 For more significant hypercalcemia (eg, serum calcium levels > 11 mg/dL) or nephrolithiasis, corticosteroid therapy is the first choice and should be implemented at the first sign of renal involvement. Corticosteroids inhibit the activity of 1-alpha-hydroxylase in macrophages, thereby reducing the production of 1,25-dihydroxyvitamin D.
Chloroquine and hydroxychloroquine have been mentioned in the literature as alternatives to corticosteroids.32 But the effect of these agents is less predictable and is slower than treatment with corticosteroids. Ketoconazole has no effect on granuloma formation but corrects hypercalcemia by inhibiting calcitriol production, and can be used as an adjunct for treating hypercalcemia and hypercalciuria.
Corticosteroids are the mainstay of treatment for renal sarcoidosis, including granulomatous interstitial nephritis and interstitial nephritis without granulomas. Most patients experience significant improvement in renal function. However, full recovery is rare, likely as a result of long-standing disease with some degree of already established irreversible renal injury.16
Corticosteroid dosage
There is no standard dosing protocol, but patients with impaired renal function due to biopsy-proven renal sarcoidosis should receive prednisone 0.5 to 1 mg/kg/day, depending on the severity of the disease, in a single dose every morning.
The optimal dosing and duration of maintenance therapy are unknown. Based on studies to date, the initial dosing should be maintained for 4 weeks, after which it can be tapered by 5 mg each week down to a maintenance dosage of 5 to 10 mg/day.4
Patients with a poor response after 4 weeks tend to have a worse renal outcome and are more susceptible to relapse.15 Fortunately, relapse often responds to increased corticosteroid doses.11,15 In the case of relapse, the dose should be increased to the lowest effective dose and continued for 4 weeks, then tapered more gradually.
A total of 24 months of treatment seems necessary to be effective and to prevent relapse.15 Some authors have proposed a lifelong maintenance dose for patients with frequent relapses, and some propose it for all patients.4
Other agents
Tumor necrosis factor (TNF)-blocking agents. Considering the critical role TNF plays in granuloma formation, anti-TNF-alpha agents are useful in steroid-resistant sarcoidosis.33 A thorough workup is necessary before starting these agents because of the increased risk of serious infection, including reactivation of latent tuberculosis. Of the current TNF-blocking agents, infliximab is most often used in renal sarcoidosis.34 Experience with adalimumab is more limited, though promising results indicate it could be an alternative for patients who do not tolerate infliximab.35
Azathioprine, mycophenolate mofetil, or methotrexate may also be used as a second-line agent if treatment with corticosteroids is not tolerated or does not control the disease. The evidence in support of these agents is limited. In small series, they have allowed sustainable control of renal function while reducing the steroid dose. Currently, these agents are used for patients resistant to corticosteroid therapy, who would otherwise need prolonged high-dose corticosteroid treatment, or who have corticosteroid intolerance; they allow a more effective steroid taper and maintenance of stable renal function.15,36
The data supporting a standardized treatment of renal sarcoidosis are limited. For steroid intolerance or resistance, cytotoxic drugs and selected anti-TNF-alpha agents, as mentioned above, have shown promise in improving or stabilizing serum creatinine levels. Further exploration is required as to which agent or combination is better at limiting the disease process with fewer adverse effects.
Our patient was initially treated with corticosteroids and was ultimately weaned to a maintenance dose of 5 mg/day. He was followed as an outpatient and was started on mycophenolate mofetil in place of higher steroid doses. His renal function stabilized, but he was lost to follow-up after 2 years.
KEY POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly involves the lungs, skin, and reticuloendothelial system.
- Renal involvement in sarcoidosis is likely underestimated due to its often clinically silent nature and the possibility of missing typical granulomatous lesions in a small or less-than-optimal biopsy sample.
- Manifestations of renal sarcoidosis include disrupted calcium homeostasis, nephrocalcinosis, nephrolithiasis, and renal failure.
- Because the clinical and histopathologic manifestations of renal sarcoidosis are nonspecific, the diagnosis is one of exclusion. In patients with renal failure or with hypercalcemia or hypercalciuria of unknown cause, renal sarcoidosis should be included in the differential diagnosis. Patients with chronic sarcoidosis should also be screened for renal impairment.
- Granulomatous interstitial nephritis is the classic histologic finding of renal sarcoidosis. Nonetheless, up to one-third of patients have interstitial nephritis without granulomas.
- Corticosteroids are the mainstay of treatment for renal sarcoidosis. An initial dose of oral prednisone 0.5 to 1 mg/kg/day should be maintained for 4 weeks and then gradually tapered to 5 to 10 mg/day for a total of 24 months. Some patients require lifelong therapy.
- Several immunosuppressive and cytotoxic agents may be used in cases of corticosteroid intolerance or to aid in effective taper of corticosteroids.
- Joss N, Morris S, Young B, Geddes C. Granulomatous interstitial nephritis. Clin J Am Soc Nephrol 2007; 2:222–230.
- Lutalo PM, D'Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener's granulomatosis). J Autoimmun 2014; 48–49:94–98.
- Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med 1997; 336:1224–1234.
- Rajakariar R, Sharples EJ, Raftery MJ, Sheaff M, Yaqoob MM. Sarcoid tubulo-interstitial nephritis: long-term outcome and response to corticosteroid therapy. Kidney Int 2006; 70:165–169.
- Baughman RP, Teirstein AS, Judson MA, et al; Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001; 164:1885–1889.
- Rizzato G, Palmieri G, Agrati AM, Zanussi C. The organ-specific extrapulmonary presentation of sarcoidosis: a frequent occurrence but a challenge to an early diagnosis. A 3-year-long prospective observational study. Sarcoidosis Vasc Diffuse Lung Dis 2004; 21:119–126.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007; 357:2153–2165.
- Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016; 13:1244–1252.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med 1983; 52:525–533.
- Mayock RL, Bertrand P, Morrison CE, Scott JH. Manifestations of sarcoidosis. Analysis of 145 patients, with a review of nine series selected from the literature. Am J Med 1963; 35:67–89.
- Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist's perspective. Am J Kidney Dis 2006; 48:856–870.
- Longcope WT, Freiman DG. A study of sarcoidosis; based on a combined investigation of 160 cases including 30 autopsies from The Johns Hopkins Hospital and Massachusetts General Hospital. Medicine (Baltimore) 1952; 31:1–132.
- Branson JH, Park JH. Sarcoidosis hepatic involvement: presentation of a case with fatal liver involvement; including autopsy findings and review of the evidence for sarcoid involvement of the liver as found in the literature. Ann Intern Med 1954; 40:111–145.
- Muther RS, McCarron DA, Bennett WM. Renal manifestations of sarcoidosis. Arch Intern Med 1981; 141:643–645.
- Mahevas M, Lescure FX, Boffa JJ, et al. Renal sarcoidosis: clinical, laboratory, and histologic presentation and outcome in 47 patients. Medicine (Baltimore) 2009; 88:98–106.
- Robson MG, Banerjee D, Hopster D, Cairns HS. Seven cases of granulomatous interstitial nephritis in the absence of extrarenal sarcoid. Nephrol Dial Transplant 2003; 18:280–284.
- Casella FJ, Allon M. The kidney in sarcoidosis. J Am Soc Nephrol 1993; 3:1555–1562.
- Rafat C, Bobrie G, Chedid A, Nochy D, Hernigou A, Plouin PF. Sarcoidosis presenting as severe renin-dependent hypertension due to kidney vascular injury. Clin Kidney J 2014; 7:383–386.
- Reichel H, Koeffler HP, Barbers R, Norman AW. Regulation of 1,25-dihydroxyvitamin D3 production by cultured alveolar macrophages from normal human donors and from patients with pulmonary sarcoidosis. J Clin Endocrinol Metab 1987; 65:1201–1209.
- Brause M, Magnusson K, Degenhardt S, Helmchen U, Grabensee B. Renal involvement in sarcoidosis—a report of 6 cases. Clin Nephrol 2002; 57:142–148.
- Hannedouche T, Grateau G, Noel LH, et al. Renal granulomatous sarcoidosis: report of six cases. Nephrol Dial Transplant 1990; 5:18–24.
- Kettritz R, Goebel U, Fiebeler A, Schneider W, Luft F. The protean face of sarcoidosis revisited. Nephrol Dial Transplant 2006; 21:2690–2694.
- Bergner R, Hoffmann M, Waldherr R, Uppenkamp M. Frequency of kidney disease in chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2003; 20:126–132.
- O’Riordan E, Willert RP, Reeve R, et al. Isolated sarcoid granulomatous interstitial nephritis: review of five cases at one center. Clin Nephrol 2001; 55:297–302.
- Gobel U, Kettritz R, Schneider W, Luft F. The protean face of renal sarcoidosis. J Am Soc Nephrol 2001; 12:616–623.
- Statement on sarcoidosis. Joint statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999; 160:736–755.
- Bijol V, Mendez GP, Nose V, Rennke HG. Granulomatous interstitial nephritis: a clinicopathologic study of 46 cases from a single institution. Int J Surg Pathol 2006; 14:57–63.
- Mignon F, Mery JP, Mougenot B, Ronco P, Roland J, Morel-Maroger L. Granulomatous interstitial nephritis. Adv Nephrol Necker Hosp 1984; 13:219–245.
- Shah R, Shidham G, Agarwal A, Albawardi A, Nadasdy T. Diagnostic utility of kidney biopsy in patients with sarcoidosis and acute kidney injury. Int J Nephrol Renovasc Dis 2011; 4:131–136.
- Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis—its value in present clinical practice. Ann Clin Biochem 1989; 26:13–18.
- Demetriou ET, Pietras SM, Holick MF. Hypercalcemia and soft tissue calcification owing to sarcoidosis: the sunlight-cola connection. J Bone Miner Res 2010; 25:1695–1699.
- Beegle SH, Barba K, Gobunsuy R, Judson MA. Current and emerging pharmacological treatments for sarcoidosis: a review. Drug Des Devel Ther 2013; 7:325–338.
- Roberts SD, Wilkes DS, Burgett RA, Knox KS. Refractory sarcoidosis responding to infliximab. Chest 2003; 124:2028–2031.
- Ahmed MM, Mubashir E, Dossabhoy NR. Isolated renal sarcoidosis: a rare presentation of a rare disease treated with infliximab. Clin Rheumatol 2007; 26:1346–1349.
- Gupta R, Beaudet L, Moore J, Mehta T. Treatment of sarcoid granulomatous interstitial nephritis with adalimumab. NDT Plus 2009; 2:139–142.
- Moudgil A, Przygodzki RM, Kher KK. Successful steroid-sparing treatment of renal limited sarcoidosis with mycophenolate mofetil. Pediatr Nephrol 2006; 21:281–285.
- Joss N, Morris S, Young B, Geddes C. Granulomatous interstitial nephritis. Clin J Am Soc Nephrol 2007; 2:222–230.
- Lutalo PM, D'Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener's granulomatosis). J Autoimmun 2014; 48–49:94–98.
- Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med 1997; 336:1224–1234.
- Rajakariar R, Sharples EJ, Raftery MJ, Sheaff M, Yaqoob MM. Sarcoid tubulo-interstitial nephritis: long-term outcome and response to corticosteroid therapy. Kidney Int 2006; 70:165–169.
- Baughman RP, Teirstein AS, Judson MA, et al; Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001; 164:1885–1889.
- Rizzato G, Palmieri G, Agrati AM, Zanussi C. The organ-specific extrapulmonary presentation of sarcoidosis: a frequent occurrence but a challenge to an early diagnosis. A 3-year-long prospective observational study. Sarcoidosis Vasc Diffuse Lung Dis 2004; 21:119–126.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007; 357:2153–2165.
- Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016; 13:1244–1252.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med 1983; 52:525–533.
- Mayock RL, Bertrand P, Morrison CE, Scott JH. Manifestations of sarcoidosis. Analysis of 145 patients, with a review of nine series selected from the literature. Am J Med 1963; 35:67–89.
- Berliner AR, Haas M, Choi MJ. Sarcoidosis: the nephrologist's perspective. Am J Kidney Dis 2006; 48:856–870.
- Longcope WT, Freiman DG. A study of sarcoidosis; based on a combined investigation of 160 cases including 30 autopsies from The Johns Hopkins Hospital and Massachusetts General Hospital. Medicine (Baltimore) 1952; 31:1–132.
- Branson JH, Park JH. Sarcoidosis hepatic involvement: presentation of a case with fatal liver involvement; including autopsy findings and review of the evidence for sarcoid involvement of the liver as found in the literature. Ann Intern Med 1954; 40:111–145.
- Muther RS, McCarron DA, Bennett WM. Renal manifestations of sarcoidosis. Arch Intern Med 1981; 141:643–645.
- Mahevas M, Lescure FX, Boffa JJ, et al. Renal sarcoidosis: clinical, laboratory, and histologic presentation and outcome in 47 patients. Medicine (Baltimore) 2009; 88:98–106.
- Robson MG, Banerjee D, Hopster D, Cairns HS. Seven cases of granulomatous interstitial nephritis in the absence of extrarenal sarcoid. Nephrol Dial Transplant 2003; 18:280–284.
- Casella FJ, Allon M. The kidney in sarcoidosis. J Am Soc Nephrol 1993; 3:1555–1562.
- Rafat C, Bobrie G, Chedid A, Nochy D, Hernigou A, Plouin PF. Sarcoidosis presenting as severe renin-dependent hypertension due to kidney vascular injury. Clin Kidney J 2014; 7:383–386.
- Reichel H, Koeffler HP, Barbers R, Norman AW. Regulation of 1,25-dihydroxyvitamin D3 production by cultured alveolar macrophages from normal human donors and from patients with pulmonary sarcoidosis. J Clin Endocrinol Metab 1987; 65:1201–1209.
- Brause M, Magnusson K, Degenhardt S, Helmchen U, Grabensee B. Renal involvement in sarcoidosis—a report of 6 cases. Clin Nephrol 2002; 57:142–148.
- Hannedouche T, Grateau G, Noel LH, et al. Renal granulomatous sarcoidosis: report of six cases. Nephrol Dial Transplant 1990; 5:18–24.
- Kettritz R, Goebel U, Fiebeler A, Schneider W, Luft F. The protean face of sarcoidosis revisited. Nephrol Dial Transplant 2006; 21:2690–2694.
- Bergner R, Hoffmann M, Waldherr R, Uppenkamp M. Frequency of kidney disease in chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2003; 20:126–132.
- O’Riordan E, Willert RP, Reeve R, et al. Isolated sarcoid granulomatous interstitial nephritis: review of five cases at one center. Clin Nephrol 2001; 55:297–302.
- Gobel U, Kettritz R, Schneider W, Luft F. The protean face of renal sarcoidosis. J Am Soc Nephrol 2001; 12:616–623.
- Statement on sarcoidosis. Joint statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999; 160:736–755.
- Bijol V, Mendez GP, Nose V, Rennke HG. Granulomatous interstitial nephritis: a clinicopathologic study of 46 cases from a single institution. Int J Surg Pathol 2006; 14:57–63.
- Mignon F, Mery JP, Mougenot B, Ronco P, Roland J, Morel-Maroger L. Granulomatous interstitial nephritis. Adv Nephrol Necker Hosp 1984; 13:219–245.
- Shah R, Shidham G, Agarwal A, Albawardi A, Nadasdy T. Diagnostic utility of kidney biopsy in patients with sarcoidosis and acute kidney injury. Int J Nephrol Renovasc Dis 2011; 4:131–136.
- Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis—its value in present clinical practice. Ann Clin Biochem 1989; 26:13–18.
- Demetriou ET, Pietras SM, Holick MF. Hypercalcemia and soft tissue calcification owing to sarcoidosis: the sunlight-cola connection. J Bone Miner Res 2010; 25:1695–1699.
- Beegle SH, Barba K, Gobunsuy R, Judson MA. Current and emerging pharmacological treatments for sarcoidosis: a review. Drug Des Devel Ther 2013; 7:325–338.
- Roberts SD, Wilkes DS, Burgett RA, Knox KS. Refractory sarcoidosis responding to infliximab. Chest 2003; 124:2028–2031.
- Ahmed MM, Mubashir E, Dossabhoy NR. Isolated renal sarcoidosis: a rare presentation of a rare disease treated with infliximab. Clin Rheumatol 2007; 26:1346–1349.
- Gupta R, Beaudet L, Moore J, Mehta T. Treatment of sarcoid granulomatous interstitial nephritis with adalimumab. NDT Plus 2009; 2:139–142.
- Moudgil A, Przygodzki RM, Kher KK. Successful steroid-sparing treatment of renal limited sarcoidosis with mycophenolate mofetil. Pediatr Nephrol 2006; 21:281–285.
A man with progressive dysphagia
A 71-year-old man was referred to the gastroenterology department for evaluation of 9 months of progressive swallowing difficulties associated with epigastric and chest discomfort.
He was a previous smoker (17 pack-years), with a history of coronary artery disease, hypertension, and cervical spinal stenosis requiring decompressive laminectomy with a postoperative course complicated by episodes of aspiration.
DYSPHAGIA: OROPHARYNGEAL OR ESOPHAGEAL
Difficulty swallowing (dysphagia) can be caused by problems in the oropharynx or in the esophagus. Difficulty initiating a swallow can be thought of as oropharyngeal dysphagia, whereas the intermittent sensation of food stuck in the neck or chest is considered esophageal dysphagia.
Focused questioning can help differentiate oropharyngeal symptoms from esophageal symptoms. For example, difficulty clearing secretions or passing the food bolus beyond the mouth or frequent coughing spells while eating is consistent with oropharyngeal dysphagia and suggests a neurologic cause. Our patient, however, presented with a constellation of symptoms more suggestive of esophageal dysphagia.
When eliciting a history of esophageal symptoms, it is crucial to determine the progression of swallowing difficulty, as well as how it directly relates to eating solids or liquids, or both. Difficulty swallowing solid foods that has progressed over time to include liquids would raise concern for an obstruction such as a stricture, ring, or malignancy. On the other hand, abrupt onset of intermittent dysphagia to both solids and liquids would raise concern for a motility disorder of the esophagus. This patient presented with an abrupt onset of intermittent symptoms to both solids and liquids that was associated with substernal chest pain.
Once coronary disease was ruled out by cardiac biomarker testing, electrocardiography, and a pharmacologic stress test, our patient underwent upper endoscopy, which showed a normal esophageal mucosa without masses or obstruction and no evidence of peptic ulcer disease.
WHAT IS THE NEXT STEP?
When upper endoscopy is negative and cardiac causes and gastroesophageal reflux disease have been ruled out, an esophageal motility disorder should be considered.
1. After obstruction has been ruled out with upper endoscopy, which should be the next step in the investigation of esophageal dysphagia?
- A 24-hour pH recording
- Barium esophagography
- Modified barium swallow
- Computed tomography of the chest
Barium esophagography is the optimal fluoroscopic study to evaluate the esophageal phase of the swallow. This study requires the patient to swallow a thick barium solution and a 13-mm barium pill under video analysis. It is useful early in the investigation of esophageal dysphagia because it can potentially reveal areas of esophageal luminal narrowing not detected endoscopically, as well as detail the rate of esophageal emptying.1
The modified barium swallow, which is performed with the assistance of a speech pathologist, is similar but only shows the oropharynx as far as the cervical esophagus. Therefore, it would be the best fluoroscopic test to assess patients with possible aspiration or oropharyngeal dysphagia, whereas barium esophagography would be the test of choice in evaluating esophageal dysmotility or mechanical obstruction.
pH testing may be helpful in diagnosing gastroesophageal reflux disease but is less helpful in the evaluation of dysphagia.
Computed tomography of the chest may be useful to evaluate for extrinsic compression of the esophagus, but it is not the best next step in the evaluation of dysphagia.

Our patient underwent barium esophagography, which revealed tertiary contractions in the mid and distal esophagus with slight narrowing of the lower cervical esophagus (Figure 1). (Primary contractions are elicited when initiating a swallow that propels the food bolus through the esophagus, while secondary contractions follow in response to esophageal distention to move all remaining esophageal contents from the thoracic esophagus. Tertiary contractions are abnormal, nonpropulsive, spontaneous contractions of the esophageal body that are initiated without swallowing.2)
EOSINOPHILIC ESOPHAGITIS
Histologic study of biopsies of the mid and distal esophagus from our patient’s upper endoscopy revealed 5 eosinophils per high-power field.
2. Does this patient meet the criteria for the diagnosis of eosinophilic esophagitis?
- Yes
- No
No. Having eosinophils in the esophagus is not enough to diagnose eosinophilic esophagitis, as eosinophils are also common in patients with gastroesophageal reflux disease.
Eosinophilic esophagitis is defined as a chronic immune-mediated esophageal disease with histologically eosinophil-predominant inflammation (with more than 15 eosinophils per high-power field). The diagnosis is additionally based on symptoms and endoscopic appearance.3 When investigating possible eosinophilic esophagitis, it is recommended that 2 to 4 samples be obtained from at least 2 different locations in the esophagus (eg, proximal and distal), because the inflammatory changes can be patchy.
WHAT DOES THE PATIENT HAVE?
3. What is the likely cause of this patient’s dysphagia?
- Eosinophilic esophagitis
- Achalasia
- Esophageal spasm
- Extrinsic compression
- Esophageal malignancy
Eosinophilic esophagitis causes characteristic symptoms that include difficulty swallowing, chest pain that does not respond to antisecretory therapy, and regurgitation of undigested food. As we discussed above, this patient has only 5 eosinophils per high-power field and does not meet the histologic criteria for eosinophilic esophagitis.
Achalasia has a characteristic “bird’s beak” appearance on esophagography that results from distal tapering of the esophagus to the gastroesophageal junction,1 and this is not apparent on our patient’s study.
Review of this patient’s esophagogram also does not reveal any extrinsic compression, esophageal malignancy, or distal tapering suggesting achalasia. In light of the abrupt onset of symptoms related to both solids and liquids associated with atypical chest pain, the primary concern should be for esophageal spasm.
ONE MORE TEST
4. What study would you order next to better elucidate the cause of this patient’s esophageal disorder?
- High-resolution esophageal manometry
- Esophagogastroduodenoscopy (EGD) with endoscopic ultrasonography
- 24-hour pH and impedance testing
- Wireless motility capsule

Esophageal manometry (Figure 2) is used to evaluate the function and coordination of the muscles of the esophagus, as in disorders of esophageal motility.
High-resolution manometry is the gold standard for evaluation of esophageal motility. It is appropriate in evaluating dysphagia or noncardiac chest pain without evidence of mechanical obstruction, ulceration, or inflammation.4,5
High-resolution manometry differs from conventional manometry in that the catheter has more sensors to measure intraluminal pressure (36 rather than the usual 7 to 12). The data are translated into pressure topography plots (Figure 3).6,7
 to red (high). This study shows a normal distal latency (black arrow) of 6.7 seconds")
Updated guidelines on how to interpret the findings of high-resolution manometry are known as the Chicago 3.0 criteria.4 According to this system, esophageal motility disorders are grouped on the basis of lower esophageal sphincter relaxation and then further subdivided based on the character of peristalsis.
EGD with endoscopic ultrasonography would not be appropriate at this time because there is little suspicion of an extraluminal mass that needs to be investigated.
A 24-hour pH and impedance study is helpful in determining the presence of esophageal acid exposure in patients presenting with gastroesophageal reflux disease. This patient does not have symptoms of heartburn or regurgitation; therefore, this investigation would not be of value.
A wireless motility capsule would help in investigating gastric and small-bowel motility and may be useful in the future for this patient, but at this point it would provide little additional utility.
ESOPHAGEAL SPASM
 less than 4.5 seconds")
Our patient underwent high-resolution esophageal manometry. The results (Figure 4) revealed a normal resting pressure in the lower esophageal sphincter and complete relaxation in all swallows. The body of the esophagus demonstrated premature contractions in 90% of swallows. Overall, these findings were consistent with the diagnosis of distal esophageal spasm.
TREATMENTS FOR ESOPHAGEAL SPASM
In addition to incorporating data obtained from endoscopy, esophagography, and manometry, it is crucial to identify the patient’s predominant symptom when planning treatment. For example, is the prevailing symptom dysphagia or chest pain? Additional consideration must be given to medical, surgical, and psychiatric comorbidities.
5. Which of the following is appropriate medical therapy for esophageal spasm?
- Calcium channel blockers
- Nitrates
- Hydralazine
- Phosphodiesterase 5 (PDE5) inhibitors
- All of the above
All of these have been used to treat distal esophageal spasm as well as other hypercontractile esophageal motility disorders.8–20
Calcium channel blockers have proven to be effective in randomized controlled trials. Diltiazem has been shown to be beneficial at doses ranging from 60 to 90 mg, as has nifedipine 10 to 20 mg 3 times daily. Although different drugs of this class tend to relax the lower esophageal sphincter to different degrees, when choosing among them in patients with hypercontractile disorders there is little concern for potentially precipitating reflux.8–13
Nitrates, hydralazine, and PDE5 inhibitors have been effective in uncontrolled studies but have not been studied in randomized trials.14–17
Other treatments. Patients may also benefit from neuromodulators such as trazodone and imipramine for chest pain and optimization of antisecretory therapy if they have concomitant gastroesophageal reflux disease.18–20
Patients who have documented esophageal hypercontractility along with reflux disease confirmed by an abnormal pH study show significant improvement in their chest pain symptoms with high doses of a proton pump inhibitor (PPI). As our patient presented with chest pain and dysphagia, a dedicated pH study was not needed, and we could progress straight to manometry and a trial of a PPI.
Our patient was started on a PPI and nifedipine but developed a pruritic rash. As rash does not preclude using another medication in the same class, his treatment was changed to diltiazem 30 mg by mouth 3 times a day, and his dysphagia improved. However, he continued to experience intermittent chest pain with swallowing. After discussion of neuromodulator therapy, he declined additional pharmacologic therapy.
A NONPHARMACOLOGIC TREATMENT?
6. Which of the following would you offer this patient as a nonpharmacologic alternative for his esophageal pain?
- St. John’s wort
- Ginkgo biloba
- Ginseng
- Peppermint extract
- Eucalyptus oil
In a small, open-label study in patients with esophageal spasm, the use of 5 drops of commercially available 11% peppermint extract in 10 mL of water significantly decreased simultaneous contractions and resolved chest pain.21 Esophageal manometry was performed 10 minutes after the peppermint solution was consumed, and the results showed improvement in esophageal spasm. While the authors of this study did not make any formal recommendations, the findings suggest that peppermint extract should be given 10 minutes before meals.
There is no evidence for or against the use of the other nonpharmacologic treatments mentioned here.
PAIN RELIEF
7. If a pharmacologic approach were chosen, which would be the best option for pain relief in this patient?
- Oxycodone 5 mg every 8 hours
- Acetaminophen 650 mg every 8 hours
- Ibuprofen 400 mg every evening at bedtime
- Trazodone 100 mg every evening at bedtime
- Imipramine 50 mg every evening at bedtime
- Aripiprazole 5 mg by mouth every day
Trazodone would be the most appropriate of these options. Doses of 100 mg to 150 mg every evening at bedtime have been shown to significantly improve global assessment scores of pain at 6 weeks.18
Imipramine 50 mg every evening at bedtime would be another option and also has been shown to reduce chest pain.19
Even though these were the doses that were investigated, in clinical practice it is common to start at lower doses (trazodone 50 mg or imipramine 10 mg) and to then titrate every 4 weeks based on the patient’s response.
Opiates (eg, oxycodone) should be avoided, as they can cause esophageal motility disorders such as spasm or achalasia.22
Acetaminophen and aripiprazole have not been studied exclusively for their effect on chest pain related to esophageal spasm.
RECURRENT SYMPTOMS
The patient’s dysphagia initially decreased while he was taking diltiazem 30 mg 3 times a day, but it recurred after 6 months. The dosage was increased to 60 mg 3 times a day over the course of the next year, with minimal response. (The maximum dose is 90 mg 4 times a day, but because of side effects of lightheadedness and dizziness, out patient could not tolerate more than 60 mg 3 times a day).
ENDOSCOPIC THERAPY
8. What endoscopic therapies are appropriate for patients with esophageal spasm that does not respond to medication?
- Bougie dilation
- Balloon dilation
- Onabotulinum toxin injection
- Expandable mesh stent placement
- Mucosal sclerotherapy
Onabotulinum toxin injections have been shown to improve dysphagia when given in a linear pattern.23
Endoscopic dilation has not been shown to be beneficial in this setting, as a study found no difference in efficacy between therapeutic (54-French) and sham (24-French) bougie dilation.24
Our patient received 100 units of onabotulinum toxin (10 units every centimeter in the distal 10 cm of the esophagus). Afterward, he experienced resolution of dysphagia, with only mild intermittent chest pain, which was controlled by taking peppermint extract as needed. The symptoms returned approximately 1 year later but responded to repeat endoscopy with onabotulinum toxin injections.23,25
Peroral endoscopic myotomy
 and advanced to the distal esophagus (B or C) or the lower esophageal sphincter (D), where the m")
Another relatively new endoscopic treatment for esophageal motility disorders is peroral endoscopic myotomy (Figure 5). During this procedure a tiny incision is made in the esophageal mucosa, permitting the endoscope to tunnel within the lining. The smooth muscle of the distal esophagus and lower esophageal sphincter is then cut, thereby freeing either the spastic muscle (in distal esophageal spasm) or the hyperactive lower esophageal sphincter (in achalasia).26,27
In an open trial, after undergoing peroral endoscopic myotomy for esophageal spasm and hypercontractile esophagus, 89% of patients had complete relief of dysphagia, and 92% had palliation of chest pain.28 Of note, the rate of relief of dysphagia was higher for patients with achalasia (98%) than for nonachalasia patients (71%).
- Vaezi MF, Pandolfino JE, Vela MF. ACG clinical guideline: diagnosis and management of achalasia. Am J Gastroenterol 2013; 108:1238–1249;
- Hellemans J, Vantrappen G. Physiology. In: Vantrappen G, Hellemans J, eds. Diseases of the esophagus. New York, NY: Springer-Verlag Berlin, Heidelberg; 1974:40–102.
- Dellon ES, Gonsalves N, Hirano I, Furuta GT, Liacouras CA, Katzka DA; American College of Gastroenterology. ACG clinical guideline: evidenced based approach to the diagnosis and management of esophageal eosinophilia and eosinophilic esophagitis (EoE). Am J Gastroenterol 2013; 108:679–692.
- Kahrilas PJ, Bredenoord AJ, Fox M, et al; International High Resolution Manometry Working Group. The Chicago classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil 2015; 27:160–174.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Ghosh SK, Pandolfino JE, Zhang Q, Jarosz A, Shah N, Kahrilas PJ. Quantifying esophageal peristalsis with high-resolution manometry: a study of 75 asymptomatic volunteers. Am J Physiol Gastrointest Liver Physiol 2006; 290:G988–G997.
- Kahrilas PJ, Sifrim D. High-resolution manometry and impedance-pH/manometry: valuable tools in clinical and investigational esophagology. Gastroenterology 2008; 135:756–769.
- Cattau EL Jr, Castell DO, Johnson DA, et al. Diltiazem therapy for symptoms associated with nutcracker esophagus. Am J Gastroenterol 1991; 86:272–276.
- Richter JE, Dalton CB, Bradley LA, Castell DO. Oral nifedipine in the treatment of noncardiac chest pain in patients with the nutcracker esophagus. Gastroenterology 1987; 93:21–28.
- Drenth JP, Bos LP, Engels LG. Efficacy of diltiazem in the treatment of diffuse oesophageal spasm. Aliment Pharmacol Ther 1990; 4:411–416.
- Thomas E, Witt P, Willis M, Morse J. Nifedipine therapy for diffuse esophageal spasm. South Med J 1986; 79:847–849.
- Davies HA, Lewis MJ, Rhodes J, Henderson AH. Trial of nifedipine for prevention of oesophageal spasm. Digestion 1987; 36:81–83.
- Richter JE, Dalton CB, Bradley LA, Castell DO. Oral nifedipine in the treatment of noncardiac chest pain in patients with the nutcracker esophagus. Gastroenterology 1987; 93:21–28.
- Tursi A, Brandimarte G, Gasbarrini G. Transdermal slow-release long-acting isosorbide dinitrate for ‘nutcracker’ oesophagus: an open study. Eur J Gastroenterol Hepatol 2000; 12:1061–1062.
- Mellow MH. Effect of isosorbide and hydralazine in painful primary esophageal motility disorders. Gastroenterology 1982; 83:364–370.
- Fox M, Sweis R, Wong T, Anggiansah A. Sildenafil relieves symptoms and normalizes motility in patients with oesophageal spasm: a report of two cases. Neurogastroenterol Motil 2007; 19:798–803.
- Orlando RC, Bozymski EM. Clinical and manometric effects of nitroglycerin in diffuse esophageal spasm. N Engl J Med 1973; 289:23–25.
- Clouse RE, Lustman PJ, Eckert TC, Ferney DM, Griffith LS. Low-dose trazodone for symptomatic patients with esophageal contraction abnormalities. A double-blind, placebo-controlled trial. Gastroenterology 1987; 92:1027–1036.
- Cannon RO 3rd, Quyyumi AA, Mincemoyer R, et al. Imipramine in patients with chest pain despite normal coronary angiograms. N Engl J Med 1994; 330:1411–1417.
- Achem SR, Kolts BE, Wears R, Burton L, Richter JE. Chest pain associated with nutcracker esophagus: a preliminary study of the role of gastroesophageal reflux. Am J Gastroenterol 1993; 88:187–192.
- Pimentel M, Bonorris GG, Chow EJ, Lin HC. Peppermint oil improves the manometric findings in diffuse esophageal spasm. J Clin Gastroenterol 2001; 33:27–31.
- Kraichely RE, Arora AS, Murray JA. Opiate-induced oesophageal dysmotility. Aliment Pharmacol Ther 2010; 31:601–606.
- Storr M, Allescher HD, Rösch T, Born P, Weigert N, Classen M. Treatment of symptomatic diffuse esophageal spasm by endoscopic injections of botulinum toxin: a prospective study with long-term follow-up. Gastrointest Endosc 2001; 54:754–759.
- Winters C, Artnak EJ, Benjamin SB, Castell DO. Esophageal bougienage in symptomatic patients with the nutcracker esophagus. A primary esophageal motility disorder. JAMA 1984; 252:363–366.
- Vanuytsel T, Bisschops R, Farré R, et al. Botulinum toxin reduces dysphagia in patients with nonachalasia primary esophageal motility disorders. Clin Gastroenterol Hepatol 2013; 11:1115–1121.e2.
- Khashab MA, Messallam AA, Onimaru M, et al. International multicenter experience with peroral endoscopic myotomy for the treatment of spastic esophageal disorders refractory to medical therapy (with video). Gastrointest Endosc 2015; 81:1170–1177.
- Leconte M, Douard R, Gaudric M, Dumontier I, Chaussade S, Dousset B. Functional results after extended myotomy for diffuse oesophageal spasm. Br J Surg 2007; 94:1113–1118.
- Sharata AM, Dunst CM, Pescarus R, et al. Peroral endoscopic myotomy (POEM) for esophageal primary motility disorders: analysis of 100 consecutive patients. J Gastrointest Surg 2015; 19:161–170.
A 71-year-old man was referred to the gastroenterology department for evaluation of 9 months of progressive swallowing difficulties associated with epigastric and chest discomfort.
He was a previous smoker (17 pack-years), with a history of coronary artery disease, hypertension, and cervical spinal stenosis requiring decompressive laminectomy with a postoperative course complicated by episodes of aspiration.
DYSPHAGIA: OROPHARYNGEAL OR ESOPHAGEAL
Difficulty swallowing (dysphagia) can be caused by problems in the oropharynx or in the esophagus. Difficulty initiating a swallow can be thought of as oropharyngeal dysphagia, whereas the intermittent sensation of food stuck in the neck or chest is considered esophageal dysphagia.
Focused questioning can help differentiate oropharyngeal symptoms from esophageal symptoms. For example, difficulty clearing secretions or passing the food bolus beyond the mouth or frequent coughing spells while eating is consistent with oropharyngeal dysphagia and suggests a neurologic cause. Our patient, however, presented with a constellation of symptoms more suggestive of esophageal dysphagia.
When eliciting a history of esophageal symptoms, it is crucial to determine the progression of swallowing difficulty, as well as how it directly relates to eating solids or liquids, or both. Difficulty swallowing solid foods that has progressed over time to include liquids would raise concern for an obstruction such as a stricture, ring, or malignancy. On the other hand, abrupt onset of intermittent dysphagia to both solids and liquids would raise concern for a motility disorder of the esophagus. This patient presented with an abrupt onset of intermittent symptoms to both solids and liquids that was associated with substernal chest pain.
Once coronary disease was ruled out by cardiac biomarker testing, electrocardiography, and a pharmacologic stress test, our patient underwent upper endoscopy, which showed a normal esophageal mucosa without masses or obstruction and no evidence of peptic ulcer disease.
WHAT IS THE NEXT STEP?
When upper endoscopy is negative and cardiac causes and gastroesophageal reflux disease have been ruled out, an esophageal motility disorder should be considered.
1. After obstruction has been ruled out with upper endoscopy, which should be the next step in the investigation of esophageal dysphagia?
- A 24-hour pH recording
- Barium esophagography
- Modified barium swallow
- Computed tomography of the chest
Barium esophagography is the optimal fluoroscopic study to evaluate the esophageal phase of the swallow. This study requires the patient to swallow a thick barium solution and a 13-mm barium pill under video analysis. It is useful early in the investigation of esophageal dysphagia because it can potentially reveal areas of esophageal luminal narrowing not detected endoscopically, as well as detail the rate of esophageal emptying.1
The modified barium swallow, which is performed with the assistance of a speech pathologist, is similar but only shows the oropharynx as far as the cervical esophagus. Therefore, it would be the best fluoroscopic test to assess patients with possible aspiration or oropharyngeal dysphagia, whereas barium esophagography would be the test of choice in evaluating esophageal dysmotility or mechanical obstruction.
pH testing may be helpful in diagnosing gastroesophageal reflux disease but is less helpful in the evaluation of dysphagia.
Computed tomography of the chest may be useful to evaluate for extrinsic compression of the esophagus, but it is not the best next step in the evaluation of dysphagia.
Our patient underwent barium esophagography, which revealed tertiary contractions in the mid and distal esophagus with slight narrowing of the lower cervical esophagus (Figure 1). (Primary contractions are elicited when initiating a swallow that propels the food bolus through the esophagus, while secondary contractions follow in response to esophageal distention to move all remaining esophageal contents from the thoracic esophagus. Tertiary contractions are abnormal, nonpropulsive, spontaneous contractions of the esophageal body that are initiated without swallowing.2)
EOSINOPHILIC ESOPHAGITIS
Histologic study of biopsies of the mid and distal esophagus from our patient’s upper endoscopy revealed 5 eosinophils per high-power field.
2. Does this patient meet the criteria for the diagnosis of eosinophilic esophagitis?
- Yes
- No
No. Having eosinophils in the esophagus is not enough to diagnose eosinophilic esophagitis, as eosinophils are also common in patients with gastroesophageal reflux disease.
Eosinophilic esophagitis is defined as a chronic immune-mediated esophageal disease with histologically eosinophil-predominant inflammation (with more than 15 eosinophils per high-power field). The diagnosis is additionally based on symptoms and endoscopic appearance.3 When investigating possible eosinophilic esophagitis, it is recommended that 2 to 4 samples be obtained from at least 2 different locations in the esophagus (eg, proximal and distal), because the inflammatory changes can be patchy.
WHAT DOES THE PATIENT HAVE?
3. What is the likely cause of this patient’s dysphagia?
- Eosinophilic esophagitis
- Achalasia
- Esophageal spasm
- Extrinsic compression
- Esophageal malignancy
Eosinophilic esophagitis causes characteristic symptoms that include difficulty swallowing, chest pain that does not respond to antisecretory therapy, and regurgitation of undigested food. As we discussed above, this patient has only 5 eosinophils per high-power field and does not meet the histologic criteria for eosinophilic esophagitis.
Achalasia has a characteristic “bird’s beak” appearance on esophagography that results from distal tapering of the esophagus to the gastroesophageal junction,1 and this is not apparent on our patient’s study.
Review of this patient’s esophagogram also does not reveal any extrinsic compression, esophageal malignancy, or distal tapering suggesting achalasia. In light of the abrupt onset of symptoms related to both solids and liquids associated with atypical chest pain, the primary concern should be for esophageal spasm.
ONE MORE TEST
4. What study would you order next to better elucidate the cause of this patient’s esophageal disorder?
- High-resolution esophageal manometry
- Esophagogastroduodenoscopy (EGD) with endoscopic ultrasonography
- 24-hour pH and impedance testing
- Wireless motility capsule
Esophageal manometry (Figure 2) is used to evaluate the function and coordination of the muscles of the esophagus, as in disorders of esophageal motility.
High-resolution manometry is the gold standard for evaluation of esophageal motility. It is appropriate in evaluating dysphagia or noncardiac chest pain without evidence of mechanical obstruction, ulceration, or inflammation.4,5
High-resolution manometry differs from conventional manometry in that the catheter has more sensors to measure intraluminal pressure (36 rather than the usual 7 to 12). The data are translated into pressure topography plots (Figure 3).6,7
Updated guidelines on how to interpret the findings of high-resolution manometry are known as the Chicago 3.0 criteria.4 According to this system, esophageal motility disorders are grouped on the basis of lower esophageal sphincter relaxation and then further subdivided based on the character of peristalsis.
EGD with endoscopic ultrasonography would not be appropriate at this time because there is little suspicion of an extraluminal mass that needs to be investigated.
A 24-hour pH and impedance study is helpful in determining the presence of esophageal acid exposure in patients presenting with gastroesophageal reflux disease. This patient does not have symptoms of heartburn or regurgitation; therefore, this investigation would not be of value.
A wireless motility capsule would help in investigating gastric and small-bowel motility and may be useful in the future for this patient, but at this point it would provide little additional utility.
ESOPHAGEAL SPASM
Our patient underwent high-resolution esophageal manometry. The results (Figure 4) revealed a normal resting pressure in the lower esophageal sphincter and complete relaxation in all swallows. The body of the esophagus demonstrated premature contractions in 90% of swallows. Overall, these findings were consistent with the diagnosis of distal esophageal spasm.
TREATMENTS FOR ESOPHAGEAL SPASM
In addition to incorporating data obtained from endoscopy, esophagography, and manometry, it is crucial to identify the patient’s predominant symptom when planning treatment. For example, is the prevailing symptom dysphagia or chest pain? Additional consideration must be given to medical, surgical, and psychiatric comorbidities.
5. Which of the following is appropriate medical therapy for esophageal spasm?
- Calcium channel blockers
- Nitrates
- Hydralazine
- Phosphodiesterase 5 (PDE5) inhibitors
- All of the above
All of these have been used to treat distal esophageal spasm as well as other hypercontractile esophageal motility disorders.8–20
Calcium channel blockers have proven to be effective in randomized controlled trials. Diltiazem has been shown to be beneficial at doses ranging from 60 to 90 mg, as has nifedipine 10 to 20 mg 3 times daily. Although different drugs of this class tend to relax the lower esophageal sphincter to different degrees, when choosing among them in patients with hypercontractile disorders there is little concern for potentially precipitating reflux.8–13
Nitrates, hydralazine, and PDE5 inhibitors have been effective in uncontrolled studies but have not been studied in randomized trials.14–17
Other treatments. Patients may also benefit from neuromodulators such as trazodone and imipramine for chest pain and optimization of antisecretory therapy if they have concomitant gastroesophageal reflux disease.18–20
Patients who have documented esophageal hypercontractility along with reflux disease confirmed by an abnormal pH study show significant improvement in their chest pain symptoms with high doses of a proton pump inhibitor (PPI). As our patient presented with chest pain and dysphagia, a dedicated pH study was not needed, and we could progress straight to manometry and a trial of a PPI.
Our patient was started on a PPI and nifedipine but developed a pruritic rash. As rash does not preclude using another medication in the same class, his treatment was changed to diltiazem 30 mg by mouth 3 times a day, and his dysphagia improved. However, he continued to experience intermittent chest pain with swallowing. After discussion of neuromodulator therapy, he declined additional pharmacologic therapy.
A NONPHARMACOLOGIC TREATMENT?
6. Which of the following would you offer this patient as a nonpharmacologic alternative for his esophageal pain?
- St. John’s wort
- Ginkgo biloba
- Ginseng
- Peppermint extract
- Eucalyptus oil
In a small, open-label study in patients with esophageal spasm, the use of 5 drops of commercially available 11% peppermint extract in 10 mL of water significantly decreased simultaneous contractions and resolved chest pain.21 Esophageal manometry was performed 10 minutes after the peppermint solution was consumed, and the results showed improvement in esophageal spasm. While the authors of this study did not make any formal recommendations, the findings suggest that peppermint extract should be given 10 minutes before meals.
There is no evidence for or against the use of the other nonpharmacologic treatments mentioned here.
PAIN RELIEF
7. If a pharmacologic approach were chosen, which would be the best option for pain relief in this patient?
- Oxycodone 5 mg every 8 hours
- Acetaminophen 650 mg every 8 hours
- Ibuprofen 400 mg every evening at bedtime
- Trazodone 100 mg every evening at bedtime
- Imipramine 50 mg every evening at bedtime
- Aripiprazole 5 mg by mouth every day
Trazodone would be the most appropriate of these options. Doses of 100 mg to 150 mg every evening at bedtime have been shown to significantly improve global assessment scores of pain at 6 weeks.18
Imipramine 50 mg every evening at bedtime would be another option and also has been shown to reduce chest pain.19
Even though these were the doses that were investigated, in clinical practice it is common to start at lower doses (trazodone 50 mg or imipramine 10 mg) and to then titrate every 4 weeks based on the patient’s response.
Opiates (eg, oxycodone) should be avoided, as they can cause esophageal motility disorders such as spasm or achalasia.22
Acetaminophen and aripiprazole have not been studied exclusively for their effect on chest pain related to esophageal spasm.
RECURRENT SYMPTOMS
The patient’s dysphagia initially decreased while he was taking diltiazem 30 mg 3 times a day, but it recurred after 6 months. The dosage was increased to 60 mg 3 times a day over the course of the next year, with minimal response. (The maximum dose is 90 mg 4 times a day, but because of side effects of lightheadedness and dizziness, out patient could not tolerate more than 60 mg 3 times a day).
ENDOSCOPIC THERAPY
8. What endoscopic therapies are appropriate for patients with esophageal spasm that does not respond to medication?
- Bougie dilation
- Balloon dilation
- Onabotulinum toxin injection
- Expandable mesh stent placement
- Mucosal sclerotherapy
Onabotulinum toxin injections have been shown to improve dysphagia when given in a linear pattern.23
Endoscopic dilation has not been shown to be beneficial in this setting, as a study found no difference in efficacy between therapeutic (54-French) and sham (24-French) bougie dilation.24
Our patient received 100 units of onabotulinum toxin (10 units every centimeter in the distal 10 cm of the esophagus). Afterward, he experienced resolution of dysphagia, with only mild intermittent chest pain, which was controlled by taking peppermint extract as needed. The symptoms returned approximately 1 year later but responded to repeat endoscopy with onabotulinum toxin injections.23,25
Peroral endoscopic myotomy
Another relatively new endoscopic treatment for esophageal motility disorders is peroral endoscopic myotomy (Figure 5). During this procedure a tiny incision is made in the esophageal mucosa, permitting the endoscope to tunnel within the lining. The smooth muscle of the distal esophagus and lower esophageal sphincter is then cut, thereby freeing either the spastic muscle (in distal esophageal spasm) or the hyperactive lower esophageal sphincter (in achalasia).26,27
In an open trial, after undergoing peroral endoscopic myotomy for esophageal spasm and hypercontractile esophagus, 89% of patients had complete relief of dysphagia, and 92% had palliation of chest pain.28 Of note, the rate of relief of dysphagia was higher for patients with achalasia (98%) than for nonachalasia patients (71%).
A 71-year-old man was referred to the gastroenterology department for evaluation of 9 months of progressive swallowing difficulties associated with epigastric and chest discomfort.
He was a previous smoker (17 pack-years), with a history of coronary artery disease, hypertension, and cervical spinal stenosis requiring decompressive laminectomy with a postoperative course complicated by episodes of aspiration.
DYSPHAGIA: OROPHARYNGEAL OR ESOPHAGEAL
Difficulty swallowing (dysphagia) can be caused by problems in the oropharynx or in the esophagus. Difficulty initiating a swallow can be thought of as oropharyngeal dysphagia, whereas the intermittent sensation of food stuck in the neck or chest is considered esophageal dysphagia.
Focused questioning can help differentiate oropharyngeal symptoms from esophageal symptoms. For example, difficulty clearing secretions or passing the food bolus beyond the mouth or frequent coughing spells while eating is consistent with oropharyngeal dysphagia and suggests a neurologic cause. Our patient, however, presented with a constellation of symptoms more suggestive of esophageal dysphagia.
When eliciting a history of esophageal symptoms, it is crucial to determine the progression of swallowing difficulty, as well as how it directly relates to eating solids or liquids, or both. Difficulty swallowing solid foods that has progressed over time to include liquids would raise concern for an obstruction such as a stricture, ring, or malignancy. On the other hand, abrupt onset of intermittent dysphagia to both solids and liquids would raise concern for a motility disorder of the esophagus. This patient presented with an abrupt onset of intermittent symptoms to both solids and liquids that was associated with substernal chest pain.
Once coronary disease was ruled out by cardiac biomarker testing, electrocardiography, and a pharmacologic stress test, our patient underwent upper endoscopy, which showed a normal esophageal mucosa without masses or obstruction and no evidence of peptic ulcer disease.
WHAT IS THE NEXT STEP?
When upper endoscopy is negative and cardiac causes and gastroesophageal reflux disease have been ruled out, an esophageal motility disorder should be considered.
1. After obstruction has been ruled out with upper endoscopy, which should be the next step in the investigation of esophageal dysphagia?
- A 24-hour pH recording
- Barium esophagography
- Modified barium swallow
- Computed tomography of the chest
Barium esophagography is the optimal fluoroscopic study to evaluate the esophageal phase of the swallow. This study requires the patient to swallow a thick barium solution and a 13-mm barium pill under video analysis. It is useful early in the investigation of esophageal dysphagia because it can potentially reveal areas of esophageal luminal narrowing not detected endoscopically, as well as detail the rate of esophageal emptying.1
The modified barium swallow, which is performed with the assistance of a speech pathologist, is similar but only shows the oropharynx as far as the cervical esophagus. Therefore, it would be the best fluoroscopic test to assess patients with possible aspiration or oropharyngeal dysphagia, whereas barium esophagography would be the test of choice in evaluating esophageal dysmotility or mechanical obstruction.
pH testing may be helpful in diagnosing gastroesophageal reflux disease but is less helpful in the evaluation of dysphagia.
Computed tomography of the chest may be useful to evaluate for extrinsic compression of the esophagus, but it is not the best next step in the evaluation of dysphagia.
Our patient underwent barium esophagography, which revealed tertiary contractions in the mid and distal esophagus with slight narrowing of the lower cervical esophagus (Figure 1). (Primary contractions are elicited when initiating a swallow that propels the food bolus through the esophagus, while secondary contractions follow in response to esophageal distention to move all remaining esophageal contents from the thoracic esophagus. Tertiary contractions are abnormal, nonpropulsive, spontaneous contractions of the esophageal body that are initiated without swallowing.2)
EOSINOPHILIC ESOPHAGITIS
Histologic study of biopsies of the mid and distal esophagus from our patient’s upper endoscopy revealed 5 eosinophils per high-power field.
2. Does this patient meet the criteria for the diagnosis of eosinophilic esophagitis?
- Yes
- No
No. Having eosinophils in the esophagus is not enough to diagnose eosinophilic esophagitis, as eosinophils are also common in patients with gastroesophageal reflux disease.
Eosinophilic esophagitis is defined as a chronic immune-mediated esophageal disease with histologically eosinophil-predominant inflammation (with more than 15 eosinophils per high-power field). The diagnosis is additionally based on symptoms and endoscopic appearance.3 When investigating possible eosinophilic esophagitis, it is recommended that 2 to 4 samples be obtained from at least 2 different locations in the esophagus (eg, proximal and distal), because the inflammatory changes can be patchy.
WHAT DOES THE PATIENT HAVE?
3. What is the likely cause of this patient’s dysphagia?
- Eosinophilic esophagitis
- Achalasia
- Esophageal spasm
- Extrinsic compression
- Esophageal malignancy
Eosinophilic esophagitis causes characteristic symptoms that include difficulty swallowing, chest pain that does not respond to antisecretory therapy, and regurgitation of undigested food. As we discussed above, this patient has only 5 eosinophils per high-power field and does not meet the histologic criteria for eosinophilic esophagitis.
Achalasia has a characteristic “bird’s beak” appearance on esophagography that results from distal tapering of the esophagus to the gastroesophageal junction,1 and this is not apparent on our patient’s study.
Review of this patient’s esophagogram also does not reveal any extrinsic compression, esophageal malignancy, or distal tapering suggesting achalasia. In light of the abrupt onset of symptoms related to both solids and liquids associated with atypical chest pain, the primary concern should be for esophageal spasm.
ONE MORE TEST
4. What study would you order next to better elucidate the cause of this patient’s esophageal disorder?
- High-resolution esophageal manometry
- Esophagogastroduodenoscopy (EGD) with endoscopic ultrasonography
- 24-hour pH and impedance testing
- Wireless motility capsule
Esophageal manometry (Figure 2) is used to evaluate the function and coordination of the muscles of the esophagus, as in disorders of esophageal motility.
High-resolution manometry is the gold standard for evaluation of esophageal motility. It is appropriate in evaluating dysphagia or noncardiac chest pain without evidence of mechanical obstruction, ulceration, or inflammation.4,5
High-resolution manometry differs from conventional manometry in that the catheter has more sensors to measure intraluminal pressure (36 rather than the usual 7 to 12). The data are translated into pressure topography plots (Figure 3).6,7
Updated guidelines on how to interpret the findings of high-resolution manometry are known as the Chicago 3.0 criteria.4 According to this system, esophageal motility disorders are grouped on the basis of lower esophageal sphincter relaxation and then further subdivided based on the character of peristalsis.
EGD with endoscopic ultrasonography would not be appropriate at this time because there is little suspicion of an extraluminal mass that needs to be investigated.
A 24-hour pH and impedance study is helpful in determining the presence of esophageal acid exposure in patients presenting with gastroesophageal reflux disease. This patient does not have symptoms of heartburn or regurgitation; therefore, this investigation would not be of value.
A wireless motility capsule would help in investigating gastric and small-bowel motility and may be useful in the future for this patient, but at this point it would provide little additional utility.
ESOPHAGEAL SPASM
Our patient underwent high-resolution esophageal manometry. The results (Figure 4) revealed a normal resting pressure in the lower esophageal sphincter and complete relaxation in all swallows. The body of the esophagus demonstrated premature contractions in 90% of swallows. Overall, these findings were consistent with the diagnosis of distal esophageal spasm.
TREATMENTS FOR ESOPHAGEAL SPASM
In addition to incorporating data obtained from endoscopy, esophagography, and manometry, it is crucial to identify the patient’s predominant symptom when planning treatment. For example, is the prevailing symptom dysphagia or chest pain? Additional consideration must be given to medical, surgical, and psychiatric comorbidities.
5. Which of the following is appropriate medical therapy for esophageal spasm?
- Calcium channel blockers
- Nitrates
- Hydralazine
- Phosphodiesterase 5 (PDE5) inhibitors
- All of the above
All of these have been used to treat distal esophageal spasm as well as other hypercontractile esophageal motility disorders.8–20
Calcium channel blockers have proven to be effective in randomized controlled trials. Diltiazem has been shown to be beneficial at doses ranging from 60 to 90 mg, as has nifedipine 10 to 20 mg 3 times daily. Although different drugs of this class tend to relax the lower esophageal sphincter to different degrees, when choosing among them in patients with hypercontractile disorders there is little concern for potentially precipitating reflux.8–13
Nitrates, hydralazine, and PDE5 inhibitors have been effective in uncontrolled studies but have not been studied in randomized trials.14–17
Other treatments. Patients may also benefit from neuromodulators such as trazodone and imipramine for chest pain and optimization of antisecretory therapy if they have concomitant gastroesophageal reflux disease.18–20
Patients who have documented esophageal hypercontractility along with reflux disease confirmed by an abnormal pH study show significant improvement in their chest pain symptoms with high doses of a proton pump inhibitor (PPI). As our patient presented with chest pain and dysphagia, a dedicated pH study was not needed, and we could progress straight to manometry and a trial of a PPI.
Our patient was started on a PPI and nifedipine but developed a pruritic rash. As rash does not preclude using another medication in the same class, his treatment was changed to diltiazem 30 mg by mouth 3 times a day, and his dysphagia improved. However, he continued to experience intermittent chest pain with swallowing. After discussion of neuromodulator therapy, he declined additional pharmacologic therapy.
A NONPHARMACOLOGIC TREATMENT?
6. Which of the following would you offer this patient as a nonpharmacologic alternative for his esophageal pain?
- St. John’s wort
- Ginkgo biloba
- Ginseng
- Peppermint extract
- Eucalyptus oil
In a small, open-label study in patients with esophageal spasm, the use of 5 drops of commercially available 11% peppermint extract in 10 mL of water significantly decreased simultaneous contractions and resolved chest pain.21 Esophageal manometry was performed 10 minutes after the peppermint solution was consumed, and the results showed improvement in esophageal spasm. While the authors of this study did not make any formal recommendations, the findings suggest that peppermint extract should be given 10 minutes before meals.
There is no evidence for or against the use of the other nonpharmacologic treatments mentioned here.
PAIN RELIEF
7. If a pharmacologic approach were chosen, which would be the best option for pain relief in this patient?
- Oxycodone 5 mg every 8 hours
- Acetaminophen 650 mg every 8 hours
- Ibuprofen 400 mg every evening at bedtime
- Trazodone 100 mg every evening at bedtime
- Imipramine 50 mg every evening at bedtime
- Aripiprazole 5 mg by mouth every day
Trazodone would be the most appropriate of these options. Doses of 100 mg to 150 mg every evening at bedtime have been shown to significantly improve global assessment scores of pain at 6 weeks.18
Imipramine 50 mg every evening at bedtime would be another option and also has been shown to reduce chest pain.19
Even though these were the doses that were investigated, in clinical practice it is common to start at lower doses (trazodone 50 mg or imipramine 10 mg) and to then titrate every 4 weeks based on the patient’s response.
Opiates (eg, oxycodone) should be avoided, as they can cause esophageal motility disorders such as spasm or achalasia.22
Acetaminophen and aripiprazole have not been studied exclusively for their effect on chest pain related to esophageal spasm.
RECURRENT SYMPTOMS
The patient’s dysphagia initially decreased while he was taking diltiazem 30 mg 3 times a day, but it recurred after 6 months. The dosage was increased to 60 mg 3 times a day over the course of the next year, with minimal response. (The maximum dose is 90 mg 4 times a day, but because of side effects of lightheadedness and dizziness, out patient could not tolerate more than 60 mg 3 times a day).
ENDOSCOPIC THERAPY
8. What endoscopic therapies are appropriate for patients with esophageal spasm that does not respond to medication?
- Bougie dilation
- Balloon dilation
- Onabotulinum toxin injection
- Expandable mesh stent placement
- Mucosal sclerotherapy
Onabotulinum toxin injections have been shown to improve dysphagia when given in a linear pattern.23
Endoscopic dilation has not been shown to be beneficial in this setting, as a study found no difference in efficacy between therapeutic (54-French) and sham (24-French) bougie dilation.24
Our patient received 100 units of onabotulinum toxin (10 units every centimeter in the distal 10 cm of the esophagus). Afterward, he experienced resolution of dysphagia, with only mild intermittent chest pain, which was controlled by taking peppermint extract as needed. The symptoms returned approximately 1 year later but responded to repeat endoscopy with onabotulinum toxin injections.23,25
Peroral endoscopic myotomy
Another relatively new endoscopic treatment for esophageal motility disorders is peroral endoscopic myotomy (Figure 5). During this procedure a tiny incision is made in the esophageal mucosa, permitting the endoscope to tunnel within the lining. The smooth muscle of the distal esophagus and lower esophageal sphincter is then cut, thereby freeing either the spastic muscle (in distal esophageal spasm) or the hyperactive lower esophageal sphincter (in achalasia).26,27
In an open trial, after undergoing peroral endoscopic myotomy for esophageal spasm and hypercontractile esophagus, 89% of patients had complete relief of dysphagia, and 92% had palliation of chest pain.28 Of note, the rate of relief of dysphagia was higher for patients with achalasia (98%) than for nonachalasia patients (71%).
- Vaezi MF, Pandolfino JE, Vela MF. ACG clinical guideline: diagnosis and management of achalasia. Am J Gastroenterol 2013; 108:1238–1249;
- Hellemans J, Vantrappen G. Physiology. In: Vantrappen G, Hellemans J, eds. Diseases of the esophagus. New York, NY: Springer-Verlag Berlin, Heidelberg; 1974:40–102.
- Dellon ES, Gonsalves N, Hirano I, Furuta GT, Liacouras CA, Katzka DA; American College of Gastroenterology. ACG clinical guideline: evidenced based approach to the diagnosis and management of esophageal eosinophilia and eosinophilic esophagitis (EoE). Am J Gastroenterol 2013; 108:679–692.
- Kahrilas PJ, Bredenoord AJ, Fox M, et al; International High Resolution Manometry Working Group. The Chicago classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil 2015; 27:160–174.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Ghosh SK, Pandolfino JE, Zhang Q, Jarosz A, Shah N, Kahrilas PJ. Quantifying esophageal peristalsis with high-resolution manometry: a study of 75 asymptomatic volunteers. Am J Physiol Gastrointest Liver Physiol 2006; 290:G988–G997.
- Kahrilas PJ, Sifrim D. High-resolution manometry and impedance-pH/manometry: valuable tools in clinical and investigational esophagology. Gastroenterology 2008; 135:756–769.
- Cattau EL Jr, Castell DO, Johnson DA, et al. Diltiazem therapy for symptoms associated with nutcracker esophagus. Am J Gastroenterol 1991; 86:272–276.
- Richter JE, Dalton CB, Bradley LA, Castell DO. Oral nifedipine in the treatment of noncardiac chest pain in patients with the nutcracker esophagus. Gastroenterology 1987; 93:21–28.
- Drenth JP, Bos LP, Engels LG. Efficacy of diltiazem in the treatment of diffuse oesophageal spasm. Aliment Pharmacol Ther 1990; 4:411–416.
- Thomas E, Witt P, Willis M, Morse J. Nifedipine therapy for diffuse esophageal spasm. South Med J 1986; 79:847–849.
- Davies HA, Lewis MJ, Rhodes J, Henderson AH. Trial of nifedipine for prevention of oesophageal spasm. Digestion 1987; 36:81–83.
- Richter JE, Dalton CB, Bradley LA, Castell DO. Oral nifedipine in the treatment of noncardiac chest pain in patients with the nutcracker esophagus. Gastroenterology 1987; 93:21–28.
- Tursi A, Brandimarte G, Gasbarrini G. Transdermal slow-release long-acting isosorbide dinitrate for ‘nutcracker’ oesophagus: an open study. Eur J Gastroenterol Hepatol 2000; 12:1061–1062.
- Mellow MH. Effect of isosorbide and hydralazine in painful primary esophageal motility disorders. Gastroenterology 1982; 83:364–370.
- Fox M, Sweis R, Wong T, Anggiansah A. Sildenafil relieves symptoms and normalizes motility in patients with oesophageal spasm: a report of two cases. Neurogastroenterol Motil 2007; 19:798–803.
- Orlando RC, Bozymski EM. Clinical and manometric effects of nitroglycerin in diffuse esophageal spasm. N Engl J Med 1973; 289:23–25.
- Clouse RE, Lustman PJ, Eckert TC, Ferney DM, Griffith LS. Low-dose trazodone for symptomatic patients with esophageal contraction abnormalities. A double-blind, placebo-controlled trial. Gastroenterology 1987; 92:1027–1036.
- Cannon RO 3rd, Quyyumi AA, Mincemoyer R, et al. Imipramine in patients with chest pain despite normal coronary angiograms. N Engl J Med 1994; 330:1411–1417.
- Achem SR, Kolts BE, Wears R, Burton L, Richter JE. Chest pain associated with nutcracker esophagus: a preliminary study of the role of gastroesophageal reflux. Am J Gastroenterol 1993; 88:187–192.
- Pimentel M, Bonorris GG, Chow EJ, Lin HC. Peppermint oil improves the manometric findings in diffuse esophageal spasm. J Clin Gastroenterol 2001; 33:27–31.
- Kraichely RE, Arora AS, Murray JA. Opiate-induced oesophageal dysmotility. Aliment Pharmacol Ther 2010; 31:601–606.
- Storr M, Allescher HD, Rösch T, Born P, Weigert N, Classen M. Treatment of symptomatic diffuse esophageal spasm by endoscopic injections of botulinum toxin: a prospective study with long-term follow-up. Gastrointest Endosc 2001; 54:754–759.
- Winters C, Artnak EJ, Benjamin SB, Castell DO. Esophageal bougienage in symptomatic patients with the nutcracker esophagus. A primary esophageal motility disorder. JAMA 1984; 252:363–366.
- Vanuytsel T, Bisschops R, Farré R, et al. Botulinum toxin reduces dysphagia in patients with nonachalasia primary esophageal motility disorders. Clin Gastroenterol Hepatol 2013; 11:1115–1121.e2.
- Khashab MA, Messallam AA, Onimaru M, et al. International multicenter experience with peroral endoscopic myotomy for the treatment of spastic esophageal disorders refractory to medical therapy (with video). Gastrointest Endosc 2015; 81:1170–1177.
- Leconte M, Douard R, Gaudric M, Dumontier I, Chaussade S, Dousset B. Functional results after extended myotomy for diffuse oesophageal spasm. Br J Surg 2007; 94:1113–1118.
- Sharata AM, Dunst CM, Pescarus R, et al. Peroral endoscopic myotomy (POEM) for esophageal primary motility disorders: analysis of 100 consecutive patients. J Gastrointest Surg 2015; 19:161–170.
- Vaezi MF, Pandolfino JE, Vela MF. ACG clinical guideline: diagnosis and management of achalasia. Am J Gastroenterol 2013; 108:1238–1249;
- Hellemans J, Vantrappen G. Physiology. In: Vantrappen G, Hellemans J, eds. Diseases of the esophagus. New York, NY: Springer-Verlag Berlin, Heidelberg; 1974:40–102.
- Dellon ES, Gonsalves N, Hirano I, Furuta GT, Liacouras CA, Katzka DA; American College of Gastroenterology. ACG clinical guideline: evidenced based approach to the diagnosis and management of esophageal eosinophilia and eosinophilic esophagitis (EoE). Am J Gastroenterol 2013; 108:679–692.
- Kahrilas PJ, Bredenoord AJ, Fox M, et al; International High Resolution Manometry Working Group. The Chicago classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil 2015; 27:160–174.
- Pandolfino JE, Kahrilas PJ; American Gastroenterological Association. AGA technical review on the clinical use of esophageal manometry. Gastroenterology 2005; 128:209–224.
- Ghosh SK, Pandolfino JE, Zhang Q, Jarosz A, Shah N, Kahrilas PJ. Quantifying esophageal peristalsis with high-resolution manometry: a study of 75 asymptomatic volunteers. Am J Physiol Gastrointest Liver Physiol 2006; 290:G988–G997.
- Kahrilas PJ, Sifrim D. High-resolution manometry and impedance-pH/manometry: valuable tools in clinical and investigational esophagology. Gastroenterology 2008; 135:756–769.
- Cattau EL Jr, Castell DO, Johnson DA, et al. Diltiazem therapy for symptoms associated with nutcracker esophagus. Am J Gastroenterol 1991; 86:272–276.
- Richter JE, Dalton CB, Bradley LA, Castell DO. Oral nifedipine in the treatment of noncardiac chest pain in patients with the nutcracker esophagus. Gastroenterology 1987; 93:21–28.
- Drenth JP, Bos LP, Engels LG. Efficacy of diltiazem in the treatment of diffuse oesophageal spasm. Aliment Pharmacol Ther 1990; 4:411–416.
- Thomas E, Witt P, Willis M, Morse J. Nifedipine therapy for diffuse esophageal spasm. South Med J 1986; 79:847–849.
- Davies HA, Lewis MJ, Rhodes J, Henderson AH. Trial of nifedipine for prevention of oesophageal spasm. Digestion 1987; 36:81–83.
- Richter JE, Dalton CB, Bradley LA, Castell DO. Oral nifedipine in the treatment of noncardiac chest pain in patients with the nutcracker esophagus. Gastroenterology 1987; 93:21–28.
- Tursi A, Brandimarte G, Gasbarrini G. Transdermal slow-release long-acting isosorbide dinitrate for ‘nutcracker’ oesophagus: an open study. Eur J Gastroenterol Hepatol 2000; 12:1061–1062.
- Mellow MH. Effect of isosorbide and hydralazine in painful primary esophageal motility disorders. Gastroenterology 1982; 83:364–370.
- Fox M, Sweis R, Wong T, Anggiansah A. Sildenafil relieves symptoms and normalizes motility in patients with oesophageal spasm: a report of two cases. Neurogastroenterol Motil 2007; 19:798–803.
- Orlando RC, Bozymski EM. Clinical and manometric effects of nitroglycerin in diffuse esophageal spasm. N Engl J Med 1973; 289:23–25.
- Clouse RE, Lustman PJ, Eckert TC, Ferney DM, Griffith LS. Low-dose trazodone for symptomatic patients with esophageal contraction abnormalities. A double-blind, placebo-controlled trial. Gastroenterology 1987; 92:1027–1036.
- Cannon RO 3rd, Quyyumi AA, Mincemoyer R, et al. Imipramine in patients with chest pain despite normal coronary angiograms. N Engl J Med 1994; 330:1411–1417.
- Achem SR, Kolts BE, Wears R, Burton L, Richter JE. Chest pain associated with nutcracker esophagus: a preliminary study of the role of gastroesophageal reflux. Am J Gastroenterol 1993; 88:187–192.
- Pimentel M, Bonorris GG, Chow EJ, Lin HC. Peppermint oil improves the manometric findings in diffuse esophageal spasm. J Clin Gastroenterol 2001; 33:27–31.
- Kraichely RE, Arora AS, Murray JA. Opiate-induced oesophageal dysmotility. Aliment Pharmacol Ther 2010; 31:601–606.
- Storr M, Allescher HD, Rösch T, Born P, Weigert N, Classen M. Treatment of symptomatic diffuse esophageal spasm by endoscopic injections of botulinum toxin: a prospective study with long-term follow-up. Gastrointest Endosc 2001; 54:754–759.
- Winters C, Artnak EJ, Benjamin SB, Castell DO. Esophageal bougienage in symptomatic patients with the nutcracker esophagus. A primary esophageal motility disorder. JAMA 1984; 252:363–366.
- Vanuytsel T, Bisschops R, Farré R, et al. Botulinum toxin reduces dysphagia in patients with nonachalasia primary esophageal motility disorders. Clin Gastroenterol Hepatol 2013; 11:1115–1121.e2.
- Khashab MA, Messallam AA, Onimaru M, et al. International multicenter experience with peroral endoscopic myotomy for the treatment of spastic esophageal disorders refractory to medical therapy (with video). Gastrointest Endosc 2015; 81:1170–1177.
- Leconte M, Douard R, Gaudric M, Dumontier I, Chaussade S, Dousset B. Functional results after extended myotomy for diffuse oesophageal spasm. Br J Surg 2007; 94:1113–1118.
- Sharata AM, Dunst CM, Pescarus R, et al. Peroral endoscopic myotomy (POEM) for esophageal primary motility disorders: analysis of 100 consecutive patients. J Gastrointest Surg 2015; 19:161–170.
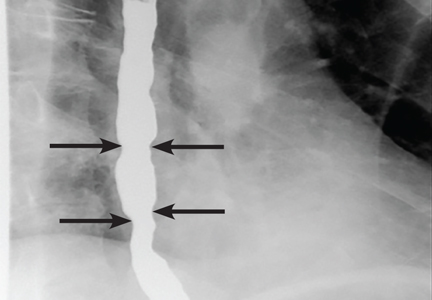
Confusion and hypercalcemia in an 80-year-old man
A retired 80-year-old man presented to the emergency department after 10 days of increasing polydipsia, polyuria, dry mouth, confusion, and slurred speech. He also reported that he had gradually and unintentionally lost 20 pounds and had loss of appetite, constipation, and chronic itching. He denied fevers, chills, night sweats, nausea, vomiting, and abdominal pain.
Medical history. He had type 2 diabetes mellitus that was well controlled by oral hypoglycemics, hypothyroidism treated with levothyroxine in stable doses, and chronic hepatitis C complicated by liver cirrhosis without focal hepatic lesions. He also had hypertension, well controlled with hydrochlorothiazide and losartan. For his long-standing pruritus he had tried prescription drugs including gabapentin and pregabalin without improvement. He had also seen a naturopathic practitioner, who had prescribed supplements that relieved the symptoms.
Examination. The patient was in no acute distress. He appeared thin, with a weight of 140 lb and a body mass index of 21 kg/m2. His temperature was 36.8°C (98.2°F), blood pressure 198/82 mm Hg, heart rate 72 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97%. His skin was without jaundice or rashes. The mucous membranes in the oropharynx were dry.
Neurologic examination revealed mild confusion, dysarthria, and ataxic gait. Sensation to light touch, pinprick, and vibration was intact. Generalized weakness was noted. Cranial nerves II through XII were intact. Deep tendon reflexes were symmetrically globally suppressed. Asterixis was absent. The remainder of the physical examination was unremarkable.
Laboratory values in the emergency department. We initially suspected he had symptomatic hyperglycemia, but a bedside blood glucose value of 113 mg/dL ruled this out. Other initial laboratory values:
- Blood urea nitrogen 31 mg/dL (reference range 9–24)
- Serum creatinine 1.7 mg/dL (0.73–1.22; an earlier value had been 1.0 mg/dL)
- Total serum calcium 14.4 mg/dL (8.6–10.0)
Complete blood cell counts were unremarkable. Computed tomography of the head was negative for acute pathology.

In view of the patient’s hypercalcemia, he was given aggressive intravenous fluid resuscitation (2 L of normal saline over 2 hours) and was admitted to the hospital. His laboratory values on admission are shown in Table 1. Fluid resuscitation was continued while the laboratory results were pending.
CAUSES OF HYPERCALCEMIA
1. Based on this information, which is the most likely cause of this patient’s hypercalcemia?
- Primary hyperparathyroidism
- Malignancy
- Hyperthyroidism
- Hypervitaminosis D
- Sarcoidosis
Traditionally, the workup for hypercalcemia in an outpatient starts with measuring the serum parathyroid hormone (PTH) level. Based on the results, a further evaluation of PTH-mediated vs PTH-independent causes of hypercalcemia would be initiated.
Primary hyperparathyroidism and malignancy account for 90% of all cases of hypercalcemia. The serum PTH concentration is usually high in primary hyperparathyroidism but low in malignancy, which helps distinguish the conditions from each other.1
Primary hyperparathyroidism
In primary hyperparathyroidism, there is overproduction of PTH, most commonly from a parathyroid adenoma, though parathyroid hyperplasia or, more rarely, parathyroid carcinoma can also overproduce the hormone.
PTH increases serum calcium levels through 3 primary mechanisms: increasing bone resorption, increasing intestinal absorption of calcium, and decreasing renal excretion of calcium. It also induces renal phosphorus excretion.
Typically, in primary hyperparathyroidism, the increases in serum calcium are small (with serum levels of total calcium rising to no higher than 11 mg/dL) and often intermittent.2 Our patient had extremely high serum calcium, low PTH, and high phosphorus levels—all of which are inconsistent with primary hyperparathyroidism.
Malignancy
In some solid tumors, the major mechanism of hypercalcemia is secretion of PTH-related peptide (PTHrP) through promotion of osteoclast function and also increased renal absorption of calcium.3 Hematologic malignancies (eg, multiple myeloma) produce osteoclast-activating factors such as RANK ligand, lymphotoxin, and interleukin 6. Direct tumor invasion of bone can cause osteolysis and subsequent hypercalcemia.4 These mechanisms are usually associated with a fall in PTH.
Less commonly, tumors can also increase levels of 1,25-dihydroxyvitamin D or produce PTH independently of the parathyroid gland.5 There have also been reports of severe hypercalcemia from hepatocellular carcinoma due to PTHrP production.6
Our patient is certainly at risk for malignancy, given his long-standing history of hepatitis C and cirrhosis. He also had a mildly elevated alpha fetoprotein level and suppressed PTH. However, his PTHrP level was normal, and ultrasonography done recently to screen for hepatocellular carcinoma (recommended every 6 months by the American Association for the Study of Liver Diseases in high-risk patients) was negative.7
Multiple myeloma screening involves testing with serum protein electrophoresis with immunofixation in combination with either a serum free light chain assay or 24-hour urine protein electrophoresis with immunofixation. This provides a 97% sensitivity.8 In this patient, these tests for multiple myeloma were negative.
Hyperthyroidism
As many as half of all patients with hyperthyroidism have elevated levels of ionized serum calcium.9 Increased osteoclastic activity is the likely mechanism. Hyperthyroid patients have increased levels of serum interleukin 6 and increased sensitivity of bone to this factor. This cytokine induces differentiation of monocytic cells into osteoclast precursors.10 These patients also have normal or low PTH levels.9
Our patient was receiving levothyroxine for hypothyroidism, but there was no evidence that the dosage was too high, as his thyroid-stimulating hormone level was within an acceptable range.
Hypervitaminosis D

Vitamin D precursors arise from the skin and from the diet. These precursors are hydroxylated in the liver and then the kidneys to biologically active 1,25-dihydroxyvitamin D (Figure 1).11 Vitamin D’s primary actions are in the intestines to increase absorption of calcium and in bone to induce osteoclast action. These actions raise the serum calcium level, which in turn lowers the PTH level through negative feedback on the parathyroid gland.
Most vitamin D supplements consist of the inactive precursor cholecalciferol (vitamin D3). To assess the degree of supplementation, 25-hydroxyvitamin D levels, which indicate the size of the body’s vitamin D reservoir, are measured.11,12
Our patient’s 25-hydroxyvitamin D level is extremely elevated, well beyond the 250-ng/mL upper limit that is considered safe.13 His low PTH level, lack of other likely causes, and history of supplement use point toward the diagnosis of hypervitaminosis D.
Sarcoidosis
Up to 10% of patients with sarcoidosis have hypercalcemia that is not mediated by PTH. Hypercalcemia in sarcoidosis has several potential mechanisms, including increased activity of the enzyme 1-alpha hydroxylase with a subsequent increase in physiologically active 1,25-dihydroxyvitamin D3 production.14
Our patient had elevated levels of 25-hydroxyvitamin D, but his biologically active 1,25-dihydroxyvitamin D level remained within the laboratory’s reference range.
LESS LIKELY CAUSES OF HYPERCALCEMIA
2. Which of the following would be least likely to cause hypercalcemia?
- Thiazide diuretics
- Over-the-counter antacid tablets
- Lithium
- Vitamin A supplementation
- Proton pump inhibitors
Thiazide diuretics
This class of drugs is well known to cause hypercalcemia. The most familiar of the mechanisms is a reduction in urinary calcium excretion. There is also an increase in intestinal absorption of dietary calcium. Evidence is increasing that most patients (as many as two-thirds) who develop hypercalcemia while using a thiazide diuretic have subclinical primary hyperparathyroidism that is uncovered with use of the diuretic.
Of importance, the hypercalcemia that thiazide diuretics cause is mild. In a series of 72 patients with thiazide-induced hypercalcemia, the average serum calcium level was 10.7 mg/dL.15
Our patient was receiving a thiazide diuretic but presented with severe hypercalcemia, which is inconsistent with thiazide-induced hypercalcemia.
Over-the-counter antacid tablets
Calcium carbonate, a popular over-the-counter antacid, can cause a milk-alkali syndrome that is defined by ingestion of excessive calcium and alkalotic substances, leading to metabolic alkalosis, hypercalcemia, and renal insufficiency. To induce this syndrome generally requires up to 4 g of calcium intake daily, but even lower levels (1.0 to 1.5 g) are known to cause it.16
Lithium
Lithium is known to cause hypercalcemia. Multiple mechanisms have been proposed, including direct action on renal tubules and the intestines leading to calcium reabsorption and stimulation of PTH release. Interestingly, parathyroid gland hyperplasia has been noted in long-term users of lithium. An often-proposed mechanism is that lithium increases the threshold at which the parathyroid glands slow their production of PTH, making them less sensitive to serum calcium levels.17
Vitamin A supplementation
Multiple case reports have linked hypercalcemia to ingestion of large doses of vitamin A. The mechanism is thought to be increased bone resorption.18.19
Although our patient reported supplement use, he denied taking vitamin A in any form.
Proton pump inhibitors
Proton pump inhibitors are not known to cause hypercalcemia. On the contrary, case reports suggest that prolonged use of proton pump inhibitors is associated with hypocalcemia and hypomagnesemia, although the mechanism is still not fully understood. A low magnesium level is known to reduce PTH secretion and also skeletal responsiveness to PTH, which can lead to profound hypocalcemia.20
CASE CONTINUED
On further questioning, the patient revealed that the supplement prescribed by his naturopathic practitioner contained vitamin D. Although he had been instructed to take 1 tablet weekly, he had begun taking it daily with his other routine medications, resulting in a daily dose in excess of 60,000 IU of cholecalciferol (vitamin D3). The recommended dose is no more than 4,000 IU/day.
The supplement was immediately discontinued. His hydrochlorothiazide was also held due to its known effect of reducing urinary calcium excretion.
INITIAL TREATMENT OF HYPERCALCEMIA
3. Which of the following treatments is not recommended as part of this patient’s initial treatment?
- Bisphosphonates
- Calcitonin
- Intravenous fluids
- Furosemide
Our patient met the criteria for the diagnosis of hypercalcemic crisis, usually defined as an albumin-corrected serum calcium level higher than 14 mg/dL associated with multiorgan dysfunction resulting from the hypercalcemia.21 The mnemonic “stones, bones, abdominal moans, and psychic groans” captures the renal, skeletal, gastrointestinal, and neurologic manifestations.1
Bisphosphonates
Bisphosphonates are analogues of pyrophosphonates, which are normally incorporated into bone. Unlike pyrophosphonates, bisphosphonates inhibit osteoclast function. They are often used to treat hypercalcemia of any cause, although they are currently approved by the US Food and Drug Administration for treating hypercalcemia of malignancy only. As intravenous monotherapy, they are superior to other forms of treatment and are among the first-line agents in management.
Two bisphosphonates shown to be effective in hypercalcemia are zoledronate and pamidronate. Pamidronate begins to lower serum calcium levels within 2 days, with a peak effect at around 6 days.22 However, in studies comparing the 2 drugs, zoledronate has been shown to be more effective in normalizing serum calcium, with the additional benefit of having a much more rapid infusion time.23 Zoledronate is contraindicated in patients with creatinine clearance less than 30 mL/min; however, pamidronate may continue to be used.24
Calcitonin
This hormone inhibits bone resorption and increases excretion of calcium in the kidneys. It is not recommended for use alone because of its short duration of action and tachyphylaxis, but it can be used in combination with other agents, particularly in hypercalcemic crisis.22 It has the most rapid onset (within 2 hours) of the available medications, and when used in combination with bisphosphonates it produces a more substantial and rapid reduction in serum calcium.25,26
In a patient such as ours, with severe hypercalcemia and evidence of neurologic consequences, calcitonin should be used for its rapid and effective action in lowering serum calcium as other interventions take effect.
Intravenous fluids
Like our patient, many patients with significant hypercalcemia have volume depletion as a result of calciuresis-induced polyuria. Many also have nephrogenic diabetes insipidus from the cytotoxic effect of calcium on renal cells, leading to further volume depletion.27
All management approaches call for fluid repletion as an initial step in hypercalcemia. However, for severe hypercalcemia, volume resuscitation alone is unlikely to completely correct the imbalance. In addition to correcting dehydration, giving fluids increases glomerular filtration, allowing for increased secretion of calcium at the distal tubule.28 The recommendation is 2.5 to 4 L of normal saline over the first 24 hours, with continued aggressive hydration until good urine output is established.21
Our patient, in addition to having acute kidney injury thought to be due to prerenal azotemia, appeared to be volume-depleted and was given aggressive intravenous hydration.
Furosemide
Furosemide inhibits calcium reabsorption at the thick ascending loop of Henle, but this effect depends on the glomerular filtration rate. While our patient would likely eventually benefit from furosemide, it should not be considered the first-line therapy, as diuretic use in the setting of volume depletion can cause circulatory collapse.29 A relative contraindication was his presentation with acute kidney injury.
LONG-TERM TREATMENT
4. In the continued management of a patient with vitamin D toxicity with severe hypercalcemia, which of the following provides prolonged benefit?
- Intravenous hydrocortisone
- Fluid repletion
- Pamidronate
- Calcium-restricted diet
Much has been postulated concerning the mechanism of vitamin D intoxication and subsequent hypercalcemia. Studies have shown it is not an increase in dietary calcium absorption that drives the hypercalcemia but rather an increase in bone resorption. As such, bisphosphonates such as pamidronate have been shown to have a dramatic and rapid effect on severe hypercalcemia from vitamin D toxicity. The duration of action varies but is typically between 1 and 2 weeks.22,30
Corticosteroids such as hydrocortisone are also indicated in situations of severe toxicity. They block the action of 1-alpha-hydroxylase, which converts inactive 25-hydroxyvitamin D to the active 1,25-dihydroxyvitamin D. Corticosteroids have also been shown to more directly reduce calcium resorption from bone and intestine in addition to increasing calciuresis.31 A small study in the United Kingdom noted that while bisphosphonates and steroids were equally effective in reducing serum calcium levels, bisphosphonates accomplished this reduction more rapidly, with a time to therapeutic effect of 9 days as opposed to 22 days.
Fluid hydration, though necessary, is unlikely to produce complete correction on its own, as previously discussed.
THE PATIENT RECOVERS
The patient was treated with intravenous fluids over 3 days and received 1 dose of pamidronate. Calcitonin was provided over the first 48 hours after presentation to more rapidly reduce his calcium levels. He was advised to avoid taking the supplements prescribed by his naturopathic practitioner.
On follow-up with an endocrinologist 1 week later, his symptoms had entirely resolved, and his calcium level was 10.5 mg/dL.
TAKE-AWAY POINTS
- A good medication history includes over-the-counter products such as vitamin D supplements, as more and more people are taking them.
- The level of 25-hydroxyvitamin D should be monitored within 3 to 4 months after initiating treatment for vitamin D deficiency.11
- Vitamin D toxicity can have profound consequences, which are usually seen when levels of 25-hydroxyvitamin D rise above 250 ng/mL.13
- The Institute of Medicine recommends that the dosage of vitamin D supplements be no more than 4,000 IU/day and that doses may need to be lowered to account for concurrent use of hypercalcemia-inducing drugs and other vitamin D-containing supplements.32
- Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician 2003; 67:1959–1966.
- al Zahrani A, Levine MA. Primary hyperparathyroidism. Lancet 1997; 349:1233–1238.
- Mundy GR, Edwards JR. PTH-related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol 2008; 19:672–675.
- Ratcliffe WA, Hutchesson AC, Bundred NJ, Ratcliffe JG. Role of assays for parathyroid-hormone-related protein in investigation of hypercalcaemia. Lancet 1992; 339:164–167.
- Hewison M, Kantorovich V, Liker HR, et al. Vitamin D-mediated hypercalcemia in lymphoma: evidence for hormone production by tumor-adjacent macrophages. J Bone Miner Res 2003; 18:579–582.
- Ghobrial MW, George J, Mannam S, Henien SR. Severe hypercalcemia as an initial presenting manifestation of hepatocellular carcinoma. Can J Gastroenterol 2002; 16:607–609.
- Zhao C, Nguyen MH. Hepatocellular carcinoma screening and surveillance: practice guidelines and real-life practice. J Clin Gastroenterol 2016; 50:120–133.
- Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 2016; 91:101–119.
- Burman KD, Monchik JM, Earll JM, Wartofsky L. Ionized and total serum calcium and parathyroid hormone in hyperthyroidism. Ann Intern Med 1976; 84:668–671.
- Iqbal AA, Burgess EH, Gallina DL, Nanes MS, Cook CB. Hypercalcemia in hyperthyroidism: patterns of serum calcium, parathyroid hormone, and 1,25-dihydroxyvitamin D3 levels during management of thyrotoxicosis. Endocr Pract 2003; 9:517–521.
- Holick MF. Vitamin D deficiency. N Engl J Med 2007; 357:266–281.
- Wolpowitz D, Gilchrest BA. The vitamin D questions: how much do you need and how should you get it? J Am Acad Dermatol 2006; 54:301–317.
- Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr 2008; 88:582S–586S.
- Inui N, Murayama A, Sasaki S, et al. Correlation between 25-hydroxyvitamin D3 1 alpha-hydroxylase gene expression in alveolar macrophages and the activity of sarcoidosis. Am J Med 2001; 110:687–693.
- Wermers RA, Kearns AE, Jenkins GD, Melton LJ 3rd. Incidence and clinical spectrum of thiazide-associated hypercalcemia. Am J Med 2007; 120:911.e9–e15.
- Patel AM, Goldfarb S. Got calcium? Welcome to the calcium-alkali syndrome. J Am Soc Nephrol 2010; 21:1440–1443.
- Shapiro HI, Davis KA. Hypercalcemia and “primary” hyperparathyroidism during lithium therapy. Am J Psychiatry 2015; 172:12–15.
- Farrington K, Miller P, Varghese Z, Baillod RA, Moorhead JF. Vitamin A toxicity and hypercalcaemia in chronic renal failure. Br Med J (Clin Res Ed) 1981; 282:1999–2002.
- Frame B, Jackson CE, Reynolds WA, Umphrey JE. Hypercalcemia and skeletal effects in chronic hypervitaminosis A. Ann Intern Med 1974; 80:44–48.
- Florentin M, Elisaf MS. Proton pump inhibitor-induced hypomagnesemia: a new challenge. World J Nephrol 2012; 1:151–154.
- Ahmad S, Kuraganti G, Steenkamp D. Hypercalcemic crisis: a clinical review. Am J Med 2015; 128:239–245.
- Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med 1993; 95:297–304.
- Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol 2001; 19:558–567.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Bilezikian JP. Management of acute hypercalcemia. N Engl J Med 1992; 326:1196–1203.
- Ralston SH. Medical management of hypercalcaemia. Br J Clin Pharmacol 1992; 34:11–20.
- Garofeanu CG, Weir M, Rosas-Arellano MP, Henson G, Garg AX, Clark WF. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis 2005; 45:626–637.
- Hosking DJ, Cowley A, Bucknall CA. Rehydration in the treatment of severe hypercalcaemia. Q J Med 1981; 50:473–481.
- Suki WN, Yium JJ, Von Minden M, Saller-Hebert C, Eknoyan G, Martinez-Maldonado M. Acute treatment of hypercalcemia with furosemide. N Engl J Med 1970; 283:836–840.
- Selby PL, Davies M, Marks JS, Mawer EB. Vitamin D intoxication causes hypercalcaemia by increased bone resorption which responds to pamidronate. Clin Endocrinol 1995; 43:531–536.
- Davies M, Mawer EB, Freemont AJ. The osteodystrophy of hypervitaminosis D: a metabolic study. Q J Med 1986; 61:911–919.
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, et al, eds. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press (US); 2011.
A retired 80-year-old man presented to the emergency department after 10 days of increasing polydipsia, polyuria, dry mouth, confusion, and slurred speech. He also reported that he had gradually and unintentionally lost 20 pounds and had loss of appetite, constipation, and chronic itching. He denied fevers, chills, night sweats, nausea, vomiting, and abdominal pain.
Medical history. He had type 2 diabetes mellitus that was well controlled by oral hypoglycemics, hypothyroidism treated with levothyroxine in stable doses, and chronic hepatitis C complicated by liver cirrhosis without focal hepatic lesions. He also had hypertension, well controlled with hydrochlorothiazide and losartan. For his long-standing pruritus he had tried prescription drugs including gabapentin and pregabalin without improvement. He had also seen a naturopathic practitioner, who had prescribed supplements that relieved the symptoms.
Examination. The patient was in no acute distress. He appeared thin, with a weight of 140 lb and a body mass index of 21 kg/m2. His temperature was 36.8°C (98.2°F), blood pressure 198/82 mm Hg, heart rate 72 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97%. His skin was without jaundice or rashes. The mucous membranes in the oropharynx were dry.
Neurologic examination revealed mild confusion, dysarthria, and ataxic gait. Sensation to light touch, pinprick, and vibration was intact. Generalized weakness was noted. Cranial nerves II through XII were intact. Deep tendon reflexes were symmetrically globally suppressed. Asterixis was absent. The remainder of the physical examination was unremarkable.
Laboratory values in the emergency department. We initially suspected he had symptomatic hyperglycemia, but a bedside blood glucose value of 113 mg/dL ruled this out. Other initial laboratory values:
- Blood urea nitrogen 31 mg/dL (reference range 9–24)
- Serum creatinine 1.7 mg/dL (0.73–1.22; an earlier value had been 1.0 mg/dL)
- Total serum calcium 14.4 mg/dL (8.6–10.0)
Complete blood cell counts were unremarkable. Computed tomography of the head was negative for acute pathology.
In view of the patient’s hypercalcemia, he was given aggressive intravenous fluid resuscitation (2 L of normal saline over 2 hours) and was admitted to the hospital. His laboratory values on admission are shown in Table 1. Fluid resuscitation was continued while the laboratory results were pending.
CAUSES OF HYPERCALCEMIA
1. Based on this information, which is the most likely cause of this patient’s hypercalcemia?
- Primary hyperparathyroidism
- Malignancy
- Hyperthyroidism
- Hypervitaminosis D
- Sarcoidosis
Traditionally, the workup for hypercalcemia in an outpatient starts with measuring the serum parathyroid hormone (PTH) level. Based on the results, a further evaluation of PTH-mediated vs PTH-independent causes of hypercalcemia would be initiated.
Primary hyperparathyroidism and malignancy account for 90% of all cases of hypercalcemia. The serum PTH concentration is usually high in primary hyperparathyroidism but low in malignancy, which helps distinguish the conditions from each other.1
Primary hyperparathyroidism
In primary hyperparathyroidism, there is overproduction of PTH, most commonly from a parathyroid adenoma, though parathyroid hyperplasia or, more rarely, parathyroid carcinoma can also overproduce the hormone.
PTH increases serum calcium levels through 3 primary mechanisms: increasing bone resorption, increasing intestinal absorption of calcium, and decreasing renal excretion of calcium. It also induces renal phosphorus excretion.
Typically, in primary hyperparathyroidism, the increases in serum calcium are small (with serum levels of total calcium rising to no higher than 11 mg/dL) and often intermittent.2 Our patient had extremely high serum calcium, low PTH, and high phosphorus levels—all of which are inconsistent with primary hyperparathyroidism.
Malignancy
In some solid tumors, the major mechanism of hypercalcemia is secretion of PTH-related peptide (PTHrP) through promotion of osteoclast function and also increased renal absorption of calcium.3 Hematologic malignancies (eg, multiple myeloma) produce osteoclast-activating factors such as RANK ligand, lymphotoxin, and interleukin 6. Direct tumor invasion of bone can cause osteolysis and subsequent hypercalcemia.4 These mechanisms are usually associated with a fall in PTH.
Less commonly, tumors can also increase levels of 1,25-dihydroxyvitamin D or produce PTH independently of the parathyroid gland.5 There have also been reports of severe hypercalcemia from hepatocellular carcinoma due to PTHrP production.6
Our patient is certainly at risk for malignancy, given his long-standing history of hepatitis C and cirrhosis. He also had a mildly elevated alpha fetoprotein level and suppressed PTH. However, his PTHrP level was normal, and ultrasonography done recently to screen for hepatocellular carcinoma (recommended every 6 months by the American Association for the Study of Liver Diseases in high-risk patients) was negative.7
Multiple myeloma screening involves testing with serum protein electrophoresis with immunofixation in combination with either a serum free light chain assay or 24-hour urine protein electrophoresis with immunofixation. This provides a 97% sensitivity.8 In this patient, these tests for multiple myeloma were negative.
Hyperthyroidism
As many as half of all patients with hyperthyroidism have elevated levels of ionized serum calcium.9 Increased osteoclastic activity is the likely mechanism. Hyperthyroid patients have increased levels of serum interleukin 6 and increased sensitivity of bone to this factor. This cytokine induces differentiation of monocytic cells into osteoclast precursors.10 These patients also have normal or low PTH levels.9
Our patient was receiving levothyroxine for hypothyroidism, but there was no evidence that the dosage was too high, as his thyroid-stimulating hormone level was within an acceptable range.
Hypervitaminosis D
Vitamin D precursors arise from the skin and from the diet. These precursors are hydroxylated in the liver and then the kidneys to biologically active 1,25-dihydroxyvitamin D (Figure 1).11 Vitamin D’s primary actions are in the intestines to increase absorption of calcium and in bone to induce osteoclast action. These actions raise the serum calcium level, which in turn lowers the PTH level through negative feedback on the parathyroid gland.
Most vitamin D supplements consist of the inactive precursor cholecalciferol (vitamin D3). To assess the degree of supplementation, 25-hydroxyvitamin D levels, which indicate the size of the body’s vitamin D reservoir, are measured.11,12
Our patient’s 25-hydroxyvitamin D level is extremely elevated, well beyond the 250-ng/mL upper limit that is considered safe.13 His low PTH level, lack of other likely causes, and history of supplement use point toward the diagnosis of hypervitaminosis D.
Sarcoidosis
Up to 10% of patients with sarcoidosis have hypercalcemia that is not mediated by PTH. Hypercalcemia in sarcoidosis has several potential mechanisms, including increased activity of the enzyme 1-alpha hydroxylase with a subsequent increase in physiologically active 1,25-dihydroxyvitamin D3 production.14
Our patient had elevated levels of 25-hydroxyvitamin D, but his biologically active 1,25-dihydroxyvitamin D level remained within the laboratory’s reference range.
LESS LIKELY CAUSES OF HYPERCALCEMIA
2. Which of the following would be least likely to cause hypercalcemia?
- Thiazide diuretics
- Over-the-counter antacid tablets
- Lithium
- Vitamin A supplementation
- Proton pump inhibitors
Thiazide diuretics
This class of drugs is well known to cause hypercalcemia. The most familiar of the mechanisms is a reduction in urinary calcium excretion. There is also an increase in intestinal absorption of dietary calcium. Evidence is increasing that most patients (as many as two-thirds) who develop hypercalcemia while using a thiazide diuretic have subclinical primary hyperparathyroidism that is uncovered with use of the diuretic.
Of importance, the hypercalcemia that thiazide diuretics cause is mild. In a series of 72 patients with thiazide-induced hypercalcemia, the average serum calcium level was 10.7 mg/dL.15
Our patient was receiving a thiazide diuretic but presented with severe hypercalcemia, which is inconsistent with thiazide-induced hypercalcemia.
Over-the-counter antacid tablets
Calcium carbonate, a popular over-the-counter antacid, can cause a milk-alkali syndrome that is defined by ingestion of excessive calcium and alkalotic substances, leading to metabolic alkalosis, hypercalcemia, and renal insufficiency. To induce this syndrome generally requires up to 4 g of calcium intake daily, but even lower levels (1.0 to 1.5 g) are known to cause it.16
Lithium
Lithium is known to cause hypercalcemia. Multiple mechanisms have been proposed, including direct action on renal tubules and the intestines leading to calcium reabsorption and stimulation of PTH release. Interestingly, parathyroid gland hyperplasia has been noted in long-term users of lithium. An often-proposed mechanism is that lithium increases the threshold at which the parathyroid glands slow their production of PTH, making them less sensitive to serum calcium levels.17
Vitamin A supplementation
Multiple case reports have linked hypercalcemia to ingestion of large doses of vitamin A. The mechanism is thought to be increased bone resorption.18.19
Although our patient reported supplement use, he denied taking vitamin A in any form.
Proton pump inhibitors
Proton pump inhibitors are not known to cause hypercalcemia. On the contrary, case reports suggest that prolonged use of proton pump inhibitors is associated with hypocalcemia and hypomagnesemia, although the mechanism is still not fully understood. A low magnesium level is known to reduce PTH secretion and also skeletal responsiveness to PTH, which can lead to profound hypocalcemia.20
CASE CONTINUED
On further questioning, the patient revealed that the supplement prescribed by his naturopathic practitioner contained vitamin D. Although he had been instructed to take 1 tablet weekly, he had begun taking it daily with his other routine medications, resulting in a daily dose in excess of 60,000 IU of cholecalciferol (vitamin D3). The recommended dose is no more than 4,000 IU/day.
The supplement was immediately discontinued. His hydrochlorothiazide was also held due to its known effect of reducing urinary calcium excretion.
INITIAL TREATMENT OF HYPERCALCEMIA
3. Which of the following treatments is not recommended as part of this patient’s initial treatment?
- Bisphosphonates
- Calcitonin
- Intravenous fluids
- Furosemide
Our patient met the criteria for the diagnosis of hypercalcemic crisis, usually defined as an albumin-corrected serum calcium level higher than 14 mg/dL associated with multiorgan dysfunction resulting from the hypercalcemia.21 The mnemonic “stones, bones, abdominal moans, and psychic groans” captures the renal, skeletal, gastrointestinal, and neurologic manifestations.1
Bisphosphonates
Bisphosphonates are analogues of pyrophosphonates, which are normally incorporated into bone. Unlike pyrophosphonates, bisphosphonates inhibit osteoclast function. They are often used to treat hypercalcemia of any cause, although they are currently approved by the US Food and Drug Administration for treating hypercalcemia of malignancy only. As intravenous monotherapy, they are superior to other forms of treatment and are among the first-line agents in management.
Two bisphosphonates shown to be effective in hypercalcemia are zoledronate and pamidronate. Pamidronate begins to lower serum calcium levels within 2 days, with a peak effect at around 6 days.22 However, in studies comparing the 2 drugs, zoledronate has been shown to be more effective in normalizing serum calcium, with the additional benefit of having a much more rapid infusion time.23 Zoledronate is contraindicated in patients with creatinine clearance less than 30 mL/min; however, pamidronate may continue to be used.24
Calcitonin
This hormone inhibits bone resorption and increases excretion of calcium in the kidneys. It is not recommended for use alone because of its short duration of action and tachyphylaxis, but it can be used in combination with other agents, particularly in hypercalcemic crisis.22 It has the most rapid onset (within 2 hours) of the available medications, and when used in combination with bisphosphonates it produces a more substantial and rapid reduction in serum calcium.25,26
In a patient such as ours, with severe hypercalcemia and evidence of neurologic consequences, calcitonin should be used for its rapid and effective action in lowering serum calcium as other interventions take effect.
Intravenous fluids
Like our patient, many patients with significant hypercalcemia have volume depletion as a result of calciuresis-induced polyuria. Many also have nephrogenic diabetes insipidus from the cytotoxic effect of calcium on renal cells, leading to further volume depletion.27
All management approaches call for fluid repletion as an initial step in hypercalcemia. However, for severe hypercalcemia, volume resuscitation alone is unlikely to completely correct the imbalance. In addition to correcting dehydration, giving fluids increases glomerular filtration, allowing for increased secretion of calcium at the distal tubule.28 The recommendation is 2.5 to 4 L of normal saline over the first 24 hours, with continued aggressive hydration until good urine output is established.21
Our patient, in addition to having acute kidney injury thought to be due to prerenal azotemia, appeared to be volume-depleted and was given aggressive intravenous hydration.
Furosemide
Furosemide inhibits calcium reabsorption at the thick ascending loop of Henle, but this effect depends on the glomerular filtration rate. While our patient would likely eventually benefit from furosemide, it should not be considered the first-line therapy, as diuretic use in the setting of volume depletion can cause circulatory collapse.29 A relative contraindication was his presentation with acute kidney injury.
LONG-TERM TREATMENT
4. In the continued management of a patient with vitamin D toxicity with severe hypercalcemia, which of the following provides prolonged benefit?
- Intravenous hydrocortisone
- Fluid repletion
- Pamidronate
- Calcium-restricted diet
Much has been postulated concerning the mechanism of vitamin D intoxication and subsequent hypercalcemia. Studies have shown it is not an increase in dietary calcium absorption that drives the hypercalcemia but rather an increase in bone resorption. As such, bisphosphonates such as pamidronate have been shown to have a dramatic and rapid effect on severe hypercalcemia from vitamin D toxicity. The duration of action varies but is typically between 1 and 2 weeks.22,30
Corticosteroids such as hydrocortisone are also indicated in situations of severe toxicity. They block the action of 1-alpha-hydroxylase, which converts inactive 25-hydroxyvitamin D to the active 1,25-dihydroxyvitamin D. Corticosteroids have also been shown to more directly reduce calcium resorption from bone and intestine in addition to increasing calciuresis.31 A small study in the United Kingdom noted that while bisphosphonates and steroids were equally effective in reducing serum calcium levels, bisphosphonates accomplished this reduction more rapidly, with a time to therapeutic effect of 9 days as opposed to 22 days.
Fluid hydration, though necessary, is unlikely to produce complete correction on its own, as previously discussed.
THE PATIENT RECOVERS
The patient was treated with intravenous fluids over 3 days and received 1 dose of pamidronate. Calcitonin was provided over the first 48 hours after presentation to more rapidly reduce his calcium levels. He was advised to avoid taking the supplements prescribed by his naturopathic practitioner.
On follow-up with an endocrinologist 1 week later, his symptoms had entirely resolved, and his calcium level was 10.5 mg/dL.
TAKE-AWAY POINTS
- A good medication history includes over-the-counter products such as vitamin D supplements, as more and more people are taking them.
- The level of 25-hydroxyvitamin D should be monitored within 3 to 4 months after initiating treatment for vitamin D deficiency.11
- Vitamin D toxicity can have profound consequences, which are usually seen when levels of 25-hydroxyvitamin D rise above 250 ng/mL.13
- The Institute of Medicine recommends that the dosage of vitamin D supplements be no more than 4,000 IU/day and that doses may need to be lowered to account for concurrent use of hypercalcemia-inducing drugs and other vitamin D-containing supplements.32
A retired 80-year-old man presented to the emergency department after 10 days of increasing polydipsia, polyuria, dry mouth, confusion, and slurred speech. He also reported that he had gradually and unintentionally lost 20 pounds and had loss of appetite, constipation, and chronic itching. He denied fevers, chills, night sweats, nausea, vomiting, and abdominal pain.
Medical history. He had type 2 diabetes mellitus that was well controlled by oral hypoglycemics, hypothyroidism treated with levothyroxine in stable doses, and chronic hepatitis C complicated by liver cirrhosis without focal hepatic lesions. He also had hypertension, well controlled with hydrochlorothiazide and losartan. For his long-standing pruritus he had tried prescription drugs including gabapentin and pregabalin without improvement. He had also seen a naturopathic practitioner, who had prescribed supplements that relieved the symptoms.
Examination. The patient was in no acute distress. He appeared thin, with a weight of 140 lb and a body mass index of 21 kg/m2. His temperature was 36.8°C (98.2°F), blood pressure 198/82 mm Hg, heart rate 72 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 97%. His skin was without jaundice or rashes. The mucous membranes in the oropharynx were dry.
Neurologic examination revealed mild confusion, dysarthria, and ataxic gait. Sensation to light touch, pinprick, and vibration was intact. Generalized weakness was noted. Cranial nerves II through XII were intact. Deep tendon reflexes were symmetrically globally suppressed. Asterixis was absent. The remainder of the physical examination was unremarkable.
Laboratory values in the emergency department. We initially suspected he had symptomatic hyperglycemia, but a bedside blood glucose value of 113 mg/dL ruled this out. Other initial laboratory values:
- Blood urea nitrogen 31 mg/dL (reference range 9–24)
- Serum creatinine 1.7 mg/dL (0.73–1.22; an earlier value had been 1.0 mg/dL)
- Total serum calcium 14.4 mg/dL (8.6–10.0)
Complete blood cell counts were unremarkable. Computed tomography of the head was negative for acute pathology.
In view of the patient’s hypercalcemia, he was given aggressive intravenous fluid resuscitation (2 L of normal saline over 2 hours) and was admitted to the hospital. His laboratory values on admission are shown in Table 1. Fluid resuscitation was continued while the laboratory results were pending.
CAUSES OF HYPERCALCEMIA
1. Based on this information, which is the most likely cause of this patient’s hypercalcemia?
- Primary hyperparathyroidism
- Malignancy
- Hyperthyroidism
- Hypervitaminosis D
- Sarcoidosis
Traditionally, the workup for hypercalcemia in an outpatient starts with measuring the serum parathyroid hormone (PTH) level. Based on the results, a further evaluation of PTH-mediated vs PTH-independent causes of hypercalcemia would be initiated.
Primary hyperparathyroidism and malignancy account for 90% of all cases of hypercalcemia. The serum PTH concentration is usually high in primary hyperparathyroidism but low in malignancy, which helps distinguish the conditions from each other.1
Primary hyperparathyroidism
In primary hyperparathyroidism, there is overproduction of PTH, most commonly from a parathyroid adenoma, though parathyroid hyperplasia or, more rarely, parathyroid carcinoma can also overproduce the hormone.
PTH increases serum calcium levels through 3 primary mechanisms: increasing bone resorption, increasing intestinal absorption of calcium, and decreasing renal excretion of calcium. It also induces renal phosphorus excretion.
Typically, in primary hyperparathyroidism, the increases in serum calcium are small (with serum levels of total calcium rising to no higher than 11 mg/dL) and often intermittent.2 Our patient had extremely high serum calcium, low PTH, and high phosphorus levels—all of which are inconsistent with primary hyperparathyroidism.
Malignancy
In some solid tumors, the major mechanism of hypercalcemia is secretion of PTH-related peptide (PTHrP) through promotion of osteoclast function and also increased renal absorption of calcium.3 Hematologic malignancies (eg, multiple myeloma) produce osteoclast-activating factors such as RANK ligand, lymphotoxin, and interleukin 6. Direct tumor invasion of bone can cause osteolysis and subsequent hypercalcemia.4 These mechanisms are usually associated with a fall in PTH.
Less commonly, tumors can also increase levels of 1,25-dihydroxyvitamin D or produce PTH independently of the parathyroid gland.5 There have also been reports of severe hypercalcemia from hepatocellular carcinoma due to PTHrP production.6
Our patient is certainly at risk for malignancy, given his long-standing history of hepatitis C and cirrhosis. He also had a mildly elevated alpha fetoprotein level and suppressed PTH. However, his PTHrP level was normal, and ultrasonography done recently to screen for hepatocellular carcinoma (recommended every 6 months by the American Association for the Study of Liver Diseases in high-risk patients) was negative.7
Multiple myeloma screening involves testing with serum protein electrophoresis with immunofixation in combination with either a serum free light chain assay or 24-hour urine protein electrophoresis with immunofixation. This provides a 97% sensitivity.8 In this patient, these tests for multiple myeloma were negative.
Hyperthyroidism
As many as half of all patients with hyperthyroidism have elevated levels of ionized serum calcium.9 Increased osteoclastic activity is the likely mechanism. Hyperthyroid patients have increased levels of serum interleukin 6 and increased sensitivity of bone to this factor. This cytokine induces differentiation of monocytic cells into osteoclast precursors.10 These patients also have normal or low PTH levels.9
Our patient was receiving levothyroxine for hypothyroidism, but there was no evidence that the dosage was too high, as his thyroid-stimulating hormone level was within an acceptable range.
Hypervitaminosis D
Vitamin D precursors arise from the skin and from the diet. These precursors are hydroxylated in the liver and then the kidneys to biologically active 1,25-dihydroxyvitamin D (Figure 1).11 Vitamin D’s primary actions are in the intestines to increase absorption of calcium and in bone to induce osteoclast action. These actions raise the serum calcium level, which in turn lowers the PTH level through negative feedback on the parathyroid gland.
Most vitamin D supplements consist of the inactive precursor cholecalciferol (vitamin D3). To assess the degree of supplementation, 25-hydroxyvitamin D levels, which indicate the size of the body’s vitamin D reservoir, are measured.11,12
Our patient’s 25-hydroxyvitamin D level is extremely elevated, well beyond the 250-ng/mL upper limit that is considered safe.13 His low PTH level, lack of other likely causes, and history of supplement use point toward the diagnosis of hypervitaminosis D.
Sarcoidosis
Up to 10% of patients with sarcoidosis have hypercalcemia that is not mediated by PTH. Hypercalcemia in sarcoidosis has several potential mechanisms, including increased activity of the enzyme 1-alpha hydroxylase with a subsequent increase in physiologically active 1,25-dihydroxyvitamin D3 production.14
Our patient had elevated levels of 25-hydroxyvitamin D, but his biologically active 1,25-dihydroxyvitamin D level remained within the laboratory’s reference range.
LESS LIKELY CAUSES OF HYPERCALCEMIA
2. Which of the following would be least likely to cause hypercalcemia?
- Thiazide diuretics
- Over-the-counter antacid tablets
- Lithium
- Vitamin A supplementation
- Proton pump inhibitors
Thiazide diuretics
This class of drugs is well known to cause hypercalcemia. The most familiar of the mechanisms is a reduction in urinary calcium excretion. There is also an increase in intestinal absorption of dietary calcium. Evidence is increasing that most patients (as many as two-thirds) who develop hypercalcemia while using a thiazide diuretic have subclinical primary hyperparathyroidism that is uncovered with use of the diuretic.
Of importance, the hypercalcemia that thiazide diuretics cause is mild. In a series of 72 patients with thiazide-induced hypercalcemia, the average serum calcium level was 10.7 mg/dL.15
Our patient was receiving a thiazide diuretic but presented with severe hypercalcemia, which is inconsistent with thiazide-induced hypercalcemia.
Over-the-counter antacid tablets
Calcium carbonate, a popular over-the-counter antacid, can cause a milk-alkali syndrome that is defined by ingestion of excessive calcium and alkalotic substances, leading to metabolic alkalosis, hypercalcemia, and renal insufficiency. To induce this syndrome generally requires up to 4 g of calcium intake daily, but even lower levels (1.0 to 1.5 g) are known to cause it.16
Lithium
Lithium is known to cause hypercalcemia. Multiple mechanisms have been proposed, including direct action on renal tubules and the intestines leading to calcium reabsorption and stimulation of PTH release. Interestingly, parathyroid gland hyperplasia has been noted in long-term users of lithium. An often-proposed mechanism is that lithium increases the threshold at which the parathyroid glands slow their production of PTH, making them less sensitive to serum calcium levels.17
Vitamin A supplementation
Multiple case reports have linked hypercalcemia to ingestion of large doses of vitamin A. The mechanism is thought to be increased bone resorption.18.19
Although our patient reported supplement use, he denied taking vitamin A in any form.
Proton pump inhibitors
Proton pump inhibitors are not known to cause hypercalcemia. On the contrary, case reports suggest that prolonged use of proton pump inhibitors is associated with hypocalcemia and hypomagnesemia, although the mechanism is still not fully understood. A low magnesium level is known to reduce PTH secretion and also skeletal responsiveness to PTH, which can lead to profound hypocalcemia.20
CASE CONTINUED
On further questioning, the patient revealed that the supplement prescribed by his naturopathic practitioner contained vitamin D. Although he had been instructed to take 1 tablet weekly, he had begun taking it daily with his other routine medications, resulting in a daily dose in excess of 60,000 IU of cholecalciferol (vitamin D3). The recommended dose is no more than 4,000 IU/day.
The supplement was immediately discontinued. His hydrochlorothiazide was also held due to its known effect of reducing urinary calcium excretion.
INITIAL TREATMENT OF HYPERCALCEMIA
3. Which of the following treatments is not recommended as part of this patient’s initial treatment?
- Bisphosphonates
- Calcitonin
- Intravenous fluids
- Furosemide
Our patient met the criteria for the diagnosis of hypercalcemic crisis, usually defined as an albumin-corrected serum calcium level higher than 14 mg/dL associated with multiorgan dysfunction resulting from the hypercalcemia.21 The mnemonic “stones, bones, abdominal moans, and psychic groans” captures the renal, skeletal, gastrointestinal, and neurologic manifestations.1
Bisphosphonates
Bisphosphonates are analogues of pyrophosphonates, which are normally incorporated into bone. Unlike pyrophosphonates, bisphosphonates inhibit osteoclast function. They are often used to treat hypercalcemia of any cause, although they are currently approved by the US Food and Drug Administration for treating hypercalcemia of malignancy only. As intravenous monotherapy, they are superior to other forms of treatment and are among the first-line agents in management.
Two bisphosphonates shown to be effective in hypercalcemia are zoledronate and pamidronate. Pamidronate begins to lower serum calcium levels within 2 days, with a peak effect at around 6 days.22 However, in studies comparing the 2 drugs, zoledronate has been shown to be more effective in normalizing serum calcium, with the additional benefit of having a much more rapid infusion time.23 Zoledronate is contraindicated in patients with creatinine clearance less than 30 mL/min; however, pamidronate may continue to be used.24
Calcitonin
This hormone inhibits bone resorption and increases excretion of calcium in the kidneys. It is not recommended for use alone because of its short duration of action and tachyphylaxis, but it can be used in combination with other agents, particularly in hypercalcemic crisis.22 It has the most rapid onset (within 2 hours) of the available medications, and when used in combination with bisphosphonates it produces a more substantial and rapid reduction in serum calcium.25,26
In a patient such as ours, with severe hypercalcemia and evidence of neurologic consequences, calcitonin should be used for its rapid and effective action in lowering serum calcium as other interventions take effect.
Intravenous fluids
Like our patient, many patients with significant hypercalcemia have volume depletion as a result of calciuresis-induced polyuria. Many also have nephrogenic diabetes insipidus from the cytotoxic effect of calcium on renal cells, leading to further volume depletion.27
All management approaches call for fluid repletion as an initial step in hypercalcemia. However, for severe hypercalcemia, volume resuscitation alone is unlikely to completely correct the imbalance. In addition to correcting dehydration, giving fluids increases glomerular filtration, allowing for increased secretion of calcium at the distal tubule.28 The recommendation is 2.5 to 4 L of normal saline over the first 24 hours, with continued aggressive hydration until good urine output is established.21
Our patient, in addition to having acute kidney injury thought to be due to prerenal azotemia, appeared to be volume-depleted and was given aggressive intravenous hydration.
Furosemide
Furosemide inhibits calcium reabsorption at the thick ascending loop of Henle, but this effect depends on the glomerular filtration rate. While our patient would likely eventually benefit from furosemide, it should not be considered the first-line therapy, as diuretic use in the setting of volume depletion can cause circulatory collapse.29 A relative contraindication was his presentation with acute kidney injury.
LONG-TERM TREATMENT
4. In the continued management of a patient with vitamin D toxicity with severe hypercalcemia, which of the following provides prolonged benefit?
- Intravenous hydrocortisone
- Fluid repletion
- Pamidronate
- Calcium-restricted diet
Much has been postulated concerning the mechanism of vitamin D intoxication and subsequent hypercalcemia. Studies have shown it is not an increase in dietary calcium absorption that drives the hypercalcemia but rather an increase in bone resorption. As such, bisphosphonates such as pamidronate have been shown to have a dramatic and rapid effect on severe hypercalcemia from vitamin D toxicity. The duration of action varies but is typically between 1 and 2 weeks.22,30
Corticosteroids such as hydrocortisone are also indicated in situations of severe toxicity. They block the action of 1-alpha-hydroxylase, which converts inactive 25-hydroxyvitamin D to the active 1,25-dihydroxyvitamin D. Corticosteroids have also been shown to more directly reduce calcium resorption from bone and intestine in addition to increasing calciuresis.31 A small study in the United Kingdom noted that while bisphosphonates and steroids were equally effective in reducing serum calcium levels, bisphosphonates accomplished this reduction more rapidly, with a time to therapeutic effect of 9 days as opposed to 22 days.
Fluid hydration, though necessary, is unlikely to produce complete correction on its own, as previously discussed.
THE PATIENT RECOVERS
The patient was treated with intravenous fluids over 3 days and received 1 dose of pamidronate. Calcitonin was provided over the first 48 hours after presentation to more rapidly reduce his calcium levels. He was advised to avoid taking the supplements prescribed by his naturopathic practitioner.
On follow-up with an endocrinologist 1 week later, his symptoms had entirely resolved, and his calcium level was 10.5 mg/dL.
TAKE-AWAY POINTS
- A good medication history includes over-the-counter products such as vitamin D supplements, as more and more people are taking them.
- The level of 25-hydroxyvitamin D should be monitored within 3 to 4 months after initiating treatment for vitamin D deficiency.11
- Vitamin D toxicity can have profound consequences, which are usually seen when levels of 25-hydroxyvitamin D rise above 250 ng/mL.13
- The Institute of Medicine recommends that the dosage of vitamin D supplements be no more than 4,000 IU/day and that doses may need to be lowered to account for concurrent use of hypercalcemia-inducing drugs and other vitamin D-containing supplements.32
- Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician 2003; 67:1959–1966.
- al Zahrani A, Levine MA. Primary hyperparathyroidism. Lancet 1997; 349:1233–1238.
- Mundy GR, Edwards JR. PTH-related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol 2008; 19:672–675.
- Ratcliffe WA, Hutchesson AC, Bundred NJ, Ratcliffe JG. Role of assays for parathyroid-hormone-related protein in investigation of hypercalcaemia. Lancet 1992; 339:164–167.
- Hewison M, Kantorovich V, Liker HR, et al. Vitamin D-mediated hypercalcemia in lymphoma: evidence for hormone production by tumor-adjacent macrophages. J Bone Miner Res 2003; 18:579–582.
- Ghobrial MW, George J, Mannam S, Henien SR. Severe hypercalcemia as an initial presenting manifestation of hepatocellular carcinoma. Can J Gastroenterol 2002; 16:607–609.
- Zhao C, Nguyen MH. Hepatocellular carcinoma screening and surveillance: practice guidelines and real-life practice. J Clin Gastroenterol 2016; 50:120–133.
- Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 2016; 91:101–119.
- Burman KD, Monchik JM, Earll JM, Wartofsky L. Ionized and total serum calcium and parathyroid hormone in hyperthyroidism. Ann Intern Med 1976; 84:668–671.
- Iqbal AA, Burgess EH, Gallina DL, Nanes MS, Cook CB. Hypercalcemia in hyperthyroidism: patterns of serum calcium, parathyroid hormone, and 1,25-dihydroxyvitamin D3 levels during management of thyrotoxicosis. Endocr Pract 2003; 9:517–521.
- Holick MF. Vitamin D deficiency. N Engl J Med 2007; 357:266–281.
- Wolpowitz D, Gilchrest BA. The vitamin D questions: how much do you need and how should you get it? J Am Acad Dermatol 2006; 54:301–317.
- Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr 2008; 88:582S–586S.
- Inui N, Murayama A, Sasaki S, et al. Correlation between 25-hydroxyvitamin D3 1 alpha-hydroxylase gene expression in alveolar macrophages and the activity of sarcoidosis. Am J Med 2001; 110:687–693.
- Wermers RA, Kearns AE, Jenkins GD, Melton LJ 3rd. Incidence and clinical spectrum of thiazide-associated hypercalcemia. Am J Med 2007; 120:911.e9–e15.
- Patel AM, Goldfarb S. Got calcium? Welcome to the calcium-alkali syndrome. J Am Soc Nephrol 2010; 21:1440–1443.
- Shapiro HI, Davis KA. Hypercalcemia and “primary” hyperparathyroidism during lithium therapy. Am J Psychiatry 2015; 172:12–15.
- Farrington K, Miller P, Varghese Z, Baillod RA, Moorhead JF. Vitamin A toxicity and hypercalcaemia in chronic renal failure. Br Med J (Clin Res Ed) 1981; 282:1999–2002.
- Frame B, Jackson CE, Reynolds WA, Umphrey JE. Hypercalcemia and skeletal effects in chronic hypervitaminosis A. Ann Intern Med 1974; 80:44–48.
- Florentin M, Elisaf MS. Proton pump inhibitor-induced hypomagnesemia: a new challenge. World J Nephrol 2012; 1:151–154.
- Ahmad S, Kuraganti G, Steenkamp D. Hypercalcemic crisis: a clinical review. Am J Med 2015; 128:239–245.
- Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med 1993; 95:297–304.
- Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol 2001; 19:558–567.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Bilezikian JP. Management of acute hypercalcemia. N Engl J Med 1992; 326:1196–1203.
- Ralston SH. Medical management of hypercalcaemia. Br J Clin Pharmacol 1992; 34:11–20.
- Garofeanu CG, Weir M, Rosas-Arellano MP, Henson G, Garg AX, Clark WF. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis 2005; 45:626–637.
- Hosking DJ, Cowley A, Bucknall CA. Rehydration in the treatment of severe hypercalcaemia. Q J Med 1981; 50:473–481.
- Suki WN, Yium JJ, Von Minden M, Saller-Hebert C, Eknoyan G, Martinez-Maldonado M. Acute treatment of hypercalcemia with furosemide. N Engl J Med 1970; 283:836–840.
- Selby PL, Davies M, Marks JS, Mawer EB. Vitamin D intoxication causes hypercalcaemia by increased bone resorption which responds to pamidronate. Clin Endocrinol 1995; 43:531–536.
- Davies M, Mawer EB, Freemont AJ. The osteodystrophy of hypervitaminosis D: a metabolic study. Q J Med 1986; 61:911–919.
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, et al, eds. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press (US); 2011.
- Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician 2003; 67:1959–1966.
- al Zahrani A, Levine MA. Primary hyperparathyroidism. Lancet 1997; 349:1233–1238.
- Mundy GR, Edwards JR. PTH-related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol 2008; 19:672–675.
- Ratcliffe WA, Hutchesson AC, Bundred NJ, Ratcliffe JG. Role of assays for parathyroid-hormone-related protein in investigation of hypercalcaemia. Lancet 1992; 339:164–167.
- Hewison M, Kantorovich V, Liker HR, et al. Vitamin D-mediated hypercalcemia in lymphoma: evidence for hormone production by tumor-adjacent macrophages. J Bone Miner Res 2003; 18:579–582.
- Ghobrial MW, George J, Mannam S, Henien SR. Severe hypercalcemia as an initial presenting manifestation of hepatocellular carcinoma. Can J Gastroenterol 2002; 16:607–609.
- Zhao C, Nguyen MH. Hepatocellular carcinoma screening and surveillance: practice guidelines and real-life practice. J Clin Gastroenterol 2016; 50:120–133.
- Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 2016; 91:101–119.
- Burman KD, Monchik JM, Earll JM, Wartofsky L. Ionized and total serum calcium and parathyroid hormone in hyperthyroidism. Ann Intern Med 1976; 84:668–671.
- Iqbal AA, Burgess EH, Gallina DL, Nanes MS, Cook CB. Hypercalcemia in hyperthyroidism: patterns of serum calcium, parathyroid hormone, and 1,25-dihydroxyvitamin D3 levels during management of thyrotoxicosis. Endocr Pract 2003; 9:517–521.
- Holick MF. Vitamin D deficiency. N Engl J Med 2007; 357:266–281.
- Wolpowitz D, Gilchrest BA. The vitamin D questions: how much do you need and how should you get it? J Am Acad Dermatol 2006; 54:301–317.
- Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr 2008; 88:582S–586S.
- Inui N, Murayama A, Sasaki S, et al. Correlation between 25-hydroxyvitamin D3 1 alpha-hydroxylase gene expression in alveolar macrophages and the activity of sarcoidosis. Am J Med 2001; 110:687–693.
- Wermers RA, Kearns AE, Jenkins GD, Melton LJ 3rd. Incidence and clinical spectrum of thiazide-associated hypercalcemia. Am J Med 2007; 120:911.e9–e15.
- Patel AM, Goldfarb S. Got calcium? Welcome to the calcium-alkali syndrome. J Am Soc Nephrol 2010; 21:1440–1443.
- Shapiro HI, Davis KA. Hypercalcemia and “primary” hyperparathyroidism during lithium therapy. Am J Psychiatry 2015; 172:12–15.
- Farrington K, Miller P, Varghese Z, Baillod RA, Moorhead JF. Vitamin A toxicity and hypercalcaemia in chronic renal failure. Br Med J (Clin Res Ed) 1981; 282:1999–2002.
- Frame B, Jackson CE, Reynolds WA, Umphrey JE. Hypercalcemia and skeletal effects in chronic hypervitaminosis A. Ann Intern Med 1974; 80:44–48.
- Florentin M, Elisaf MS. Proton pump inhibitor-induced hypomagnesemia: a new challenge. World J Nephrol 2012; 1:151–154.
- Ahmad S, Kuraganti G, Steenkamp D. Hypercalcemic crisis: a clinical review. Am J Med 2015; 128:239–245.
- Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med 1993; 95:297–304.
- Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol 2001; 19:558–567.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Bilezikian JP. Management of acute hypercalcemia. N Engl J Med 1992; 326:1196–1203.
- Ralston SH. Medical management of hypercalcaemia. Br J Clin Pharmacol 1992; 34:11–20.
- Garofeanu CG, Weir M, Rosas-Arellano MP, Henson G, Garg AX, Clark WF. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis 2005; 45:626–637.
- Hosking DJ, Cowley A, Bucknall CA. Rehydration in the treatment of severe hypercalcaemia. Q J Med 1981; 50:473–481.
- Suki WN, Yium JJ, Von Minden M, Saller-Hebert C, Eknoyan G, Martinez-Maldonado M. Acute treatment of hypercalcemia with furosemide. N Engl J Med 1970; 283:836–840.
- Selby PL, Davies M, Marks JS, Mawer EB. Vitamin D intoxication causes hypercalcaemia by increased bone resorption which responds to pamidronate. Clin Endocrinol 1995; 43:531–536.
- Davies M, Mawer EB, Freemont AJ. The osteodystrophy of hypervitaminosis D: a metabolic study. Q J Med 1986; 61:911–919.
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, et al, eds. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press (US); 2011.
