User login
Black women at highest risk for asthma
Women are much more likely than men to have asthma, and , according to the Centers for Disease Control and Prevention.
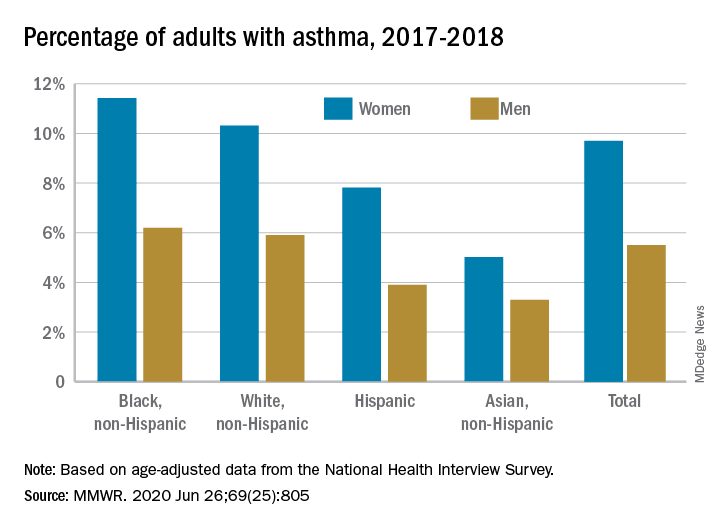
Among all women aged 18 years and older, 9.7% reported that they currently had asthma in 2017-2018, compared with 5.5% of men, based on age-adjusted data from the National Health Interview Survey.
The proportion of black, non-Hispanic women with asthma, however, was even higher, at 11.4%. White non-Hispanic women were next at 10.3%, followed by Hispanic (7.8%) and Asian (5.0%) women, the CDC reported June 26 in the Morbidity and Mortality Weekly Report.
The same pattern held for men: 6.2% of black men had asthma in 2017-2018, compared with 5.9% of whites, 3.9% of Hispanics, and 3.3% of Asian men, the CDC said.
SOURCE: MMWR. 2020 Jun 26;69(25):805.
Women are much more likely than men to have asthma, and , according to the Centers for Disease Control and Prevention.
Among all women aged 18 years and older, 9.7% reported that they currently had asthma in 2017-2018, compared with 5.5% of men, based on age-adjusted data from the National Health Interview Survey.
The proportion of black, non-Hispanic women with asthma, however, was even higher, at 11.4%. White non-Hispanic women were next at 10.3%, followed by Hispanic (7.8%) and Asian (5.0%) women, the CDC reported June 26 in the Morbidity and Mortality Weekly Report.
The same pattern held for men: 6.2% of black men had asthma in 2017-2018, compared with 5.9% of whites, 3.9% of Hispanics, and 3.3% of Asian men, the CDC said.
SOURCE: MMWR. 2020 Jun 26;69(25):805.
Women are much more likely than men to have asthma, and , according to the Centers for Disease Control and Prevention.
Among all women aged 18 years and older, 9.7% reported that they currently had asthma in 2017-2018, compared with 5.5% of men, based on age-adjusted data from the National Health Interview Survey.
The proportion of black, non-Hispanic women with asthma, however, was even higher, at 11.4%. White non-Hispanic women were next at 10.3%, followed by Hispanic (7.8%) and Asian (5.0%) women, the CDC reported June 26 in the Morbidity and Mortality Weekly Report.
The same pattern held for men: 6.2% of black men had asthma in 2017-2018, compared with 5.9% of whites, 3.9% of Hispanics, and 3.3% of Asian men, the CDC said.
SOURCE: MMWR. 2020 Jun 26;69(25):805.
FROM MMWR
ACIP approves flu vaccine recommendations for 2020-2021
– Fluzone high-dose quadrivalent, which replaces the trivalent Fluzone high-dose and Fluad quadrivalent (Seqirus), according to the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
![]()
At a virtual meeting on June 24, the committee voted unanimously to approve the vaccine recommendations for annual influenza immunization of all individuals aged 6 months and older. They also voted to accept some guidance and language changes to the recommendations.
The past flu season was unique in its overlap with the emergence of the COVID-19 coronavirus, which likely contributed to a third peak in reported cases of influenza-like illness at approximately week 14 of last season, said Lisa Grohskopf, MD, of the CDC’s influenza division, who presented data on last year’s activity and the updates for next season.
The CDC estimates that 39,000,000-56,000,000 flu illnesses occurred in the United States from Oct. 1, 2019, to April 4, 2020, said Dr. Grohskopf. Estimates also suggest as many as 740,000 hospitalizations and 62,000 deaths related to the seasonal flu.
Preliminary results of vaccine effectiveness showed 39% overall for the 2019-2020 season, with more substantial protection against influenza B and lower protection against A/H1N1pmd09.
Vaccine safety data from the Vaccine Adverse Event Reporting System and Vaccine Safety Datalink showed no new safety concerns for any flu vaccine types used last year, Dr. Grohskopf noted.
Based on this information, three components (A/H1N1pdm09, A/H3N2, and B/Victoria) have been updated for the 2020-2021 vaccines, said Dr. Grohskopf. The egg-based influenza vaccines will include hemagglutinin derived from an A/Guangdong-Maonan/SWL1536/2019(H1N1)pdm09–like virus, an A/Hong Kong/2671/2019(H3N2)–like virus and a B/Washington/02/2019 (Victoria lineage)–like virus, and (for quadrivalent vaccines) a B/Phuket/3073/2013 (Yamagata lineage)–like virus.
Nonegg vaccines will contain hemagglutinin derived from an A/Hawaii/70/2019 (H1N1)pdm09–like virus, an A/Hong Kong/45/2019 (H3N2)–like virus, a B/Washington/02/2019 (Victoria lineage)–like virus, and a B/Phuket/3073/2013 (Yamagata lineage)–like virus.
New guidance for next year’s flu season includes a change to the language in the contraindications and precautions table to simply read “Contraindications,” with more details in the text explaining package insert contraindications and ACIP recommendations, Dr. Grohskopf said. In addition, updated guidance clarifies that live-attenuated influenza vaccine quadravalents (LAIV4) should not be used in patients with cochlear implants, active cerebrospinal fluid leaks, and anatomical or functional asplenia, based on ACIP’s review of the latest evidence and the availability of alternative vaccines.
ACIP also updated guidance on the use of antivirals and LAIV4. Based on half-lives, language was added indicating that clinicians should assume interference if antivirals are given within certain intervals of LAIV4, Dr. Grohskopf explained. “Newer antivirals peramivir and baloxavir have longer half-lives than oseltamivir and zanamivir, and insufficient data are available on the use of LAIV4 in the setting of antiviral use.”
The ACIP members had no financial conflicts to disclose.
– Fluzone high-dose quadrivalent, which replaces the trivalent Fluzone high-dose and Fluad quadrivalent (Seqirus), according to the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
![]()
At a virtual meeting on June 24, the committee voted unanimously to approve the vaccine recommendations for annual influenza immunization of all individuals aged 6 months and older. They also voted to accept some guidance and language changes to the recommendations.
The past flu season was unique in its overlap with the emergence of the COVID-19 coronavirus, which likely contributed to a third peak in reported cases of influenza-like illness at approximately week 14 of last season, said Lisa Grohskopf, MD, of the CDC’s influenza division, who presented data on last year’s activity and the updates for next season.
The CDC estimates that 39,000,000-56,000,000 flu illnesses occurred in the United States from Oct. 1, 2019, to April 4, 2020, said Dr. Grohskopf. Estimates also suggest as many as 740,000 hospitalizations and 62,000 deaths related to the seasonal flu.
Preliminary results of vaccine effectiveness showed 39% overall for the 2019-2020 season, with more substantial protection against influenza B and lower protection against A/H1N1pmd09.
Vaccine safety data from the Vaccine Adverse Event Reporting System and Vaccine Safety Datalink showed no new safety concerns for any flu vaccine types used last year, Dr. Grohskopf noted.
Based on this information, three components (A/H1N1pdm09, A/H3N2, and B/Victoria) have been updated for the 2020-2021 vaccines, said Dr. Grohskopf. The egg-based influenza vaccines will include hemagglutinin derived from an A/Guangdong-Maonan/SWL1536/2019(H1N1)pdm09–like virus, an A/Hong Kong/2671/2019(H3N2)–like virus and a B/Washington/02/2019 (Victoria lineage)–like virus, and (for quadrivalent vaccines) a B/Phuket/3073/2013 (Yamagata lineage)–like virus.
Nonegg vaccines will contain hemagglutinin derived from an A/Hawaii/70/2019 (H1N1)pdm09–like virus, an A/Hong Kong/45/2019 (H3N2)–like virus, a B/Washington/02/2019 (Victoria lineage)–like virus, and a B/Phuket/3073/2013 (Yamagata lineage)–like virus.
New guidance for next year’s flu season includes a change to the language in the contraindications and precautions table to simply read “Contraindications,” with more details in the text explaining package insert contraindications and ACIP recommendations, Dr. Grohskopf said. In addition, updated guidance clarifies that live-attenuated influenza vaccine quadravalents (LAIV4) should not be used in patients with cochlear implants, active cerebrospinal fluid leaks, and anatomical or functional asplenia, based on ACIP’s review of the latest evidence and the availability of alternative vaccines.
ACIP also updated guidance on the use of antivirals and LAIV4. Based on half-lives, language was added indicating that clinicians should assume interference if antivirals are given within certain intervals of LAIV4, Dr. Grohskopf explained. “Newer antivirals peramivir and baloxavir have longer half-lives than oseltamivir and zanamivir, and insufficient data are available on the use of LAIV4 in the setting of antiviral use.”
The ACIP members had no financial conflicts to disclose.
– Fluzone high-dose quadrivalent, which replaces the trivalent Fluzone high-dose and Fluad quadrivalent (Seqirus), according to the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
![]()
At a virtual meeting on June 24, the committee voted unanimously to approve the vaccine recommendations for annual influenza immunization of all individuals aged 6 months and older. They also voted to accept some guidance and language changes to the recommendations.
The past flu season was unique in its overlap with the emergence of the COVID-19 coronavirus, which likely contributed to a third peak in reported cases of influenza-like illness at approximately week 14 of last season, said Lisa Grohskopf, MD, of the CDC’s influenza division, who presented data on last year’s activity and the updates for next season.
The CDC estimates that 39,000,000-56,000,000 flu illnesses occurred in the United States from Oct. 1, 2019, to April 4, 2020, said Dr. Grohskopf. Estimates also suggest as many as 740,000 hospitalizations and 62,000 deaths related to the seasonal flu.
Preliminary results of vaccine effectiveness showed 39% overall for the 2019-2020 season, with more substantial protection against influenza B and lower protection against A/H1N1pmd09.
Vaccine safety data from the Vaccine Adverse Event Reporting System and Vaccine Safety Datalink showed no new safety concerns for any flu vaccine types used last year, Dr. Grohskopf noted.
Based on this information, three components (A/H1N1pdm09, A/H3N2, and B/Victoria) have been updated for the 2020-2021 vaccines, said Dr. Grohskopf. The egg-based influenza vaccines will include hemagglutinin derived from an A/Guangdong-Maonan/SWL1536/2019(H1N1)pdm09–like virus, an A/Hong Kong/2671/2019(H3N2)–like virus and a B/Washington/02/2019 (Victoria lineage)–like virus, and (for quadrivalent vaccines) a B/Phuket/3073/2013 (Yamagata lineage)–like virus.
Nonegg vaccines will contain hemagglutinin derived from an A/Hawaii/70/2019 (H1N1)pdm09–like virus, an A/Hong Kong/45/2019 (H3N2)–like virus, a B/Washington/02/2019 (Victoria lineage)–like virus, and a B/Phuket/3073/2013 (Yamagata lineage)–like virus.
New guidance for next year’s flu season includes a change to the language in the contraindications and precautions table to simply read “Contraindications,” with more details in the text explaining package insert contraindications and ACIP recommendations, Dr. Grohskopf said. In addition, updated guidance clarifies that live-attenuated influenza vaccine quadravalents (LAIV4) should not be used in patients with cochlear implants, active cerebrospinal fluid leaks, and anatomical or functional asplenia, based on ACIP’s review of the latest evidence and the availability of alternative vaccines.
ACIP also updated guidance on the use of antivirals and LAIV4. Based on half-lives, language was added indicating that clinicians should assume interference if antivirals are given within certain intervals of LAIV4, Dr. Grohskopf explained. “Newer antivirals peramivir and baloxavir have longer half-lives than oseltamivir and zanamivir, and insufficient data are available on the use of LAIV4 in the setting of antiviral use.”
The ACIP members had no financial conflicts to disclose.
COVID-19: Medicare data show long hospital stays, disparities
according to a new analysis by the Centers for Medicare & Medicaid Services.
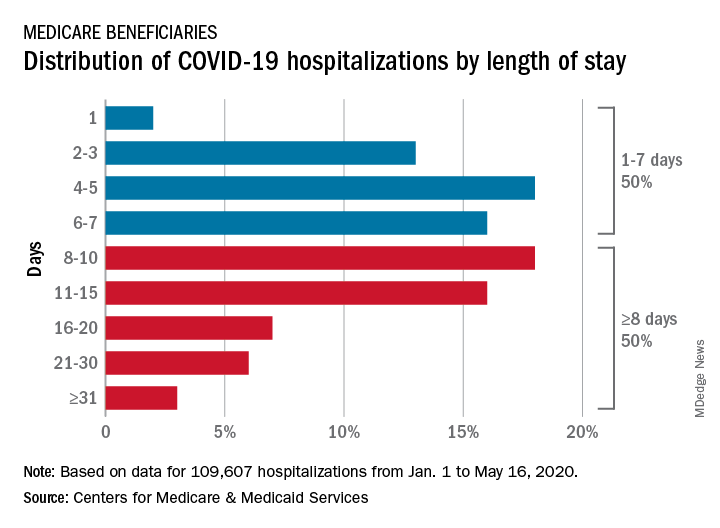
CMS encounter and claims data show almost 110,000 hospital stays for COVID-19 from Jan. 1 to May 16, 2020. Of the longer admissions, 18% were 8-10 days, 16% were 11-15 days, and another 16% were 16 days or longer, the CMS reported in a preliminary data snapshot released June 22.
The hospitalization rate for the Medicare population was 175 per 100,000 as of May 16, but the CMS data show a number of disparities involving race/ethnicity and other demographic characteristics were uncovered, such as the following:
- Black patients were hospitalized for COVID-19 at a much higher rate, at 465 per 100,000 beneficiaries, than were Hispanics (258), Asians (187), and whites (123).
- Residents of urban/suburban areas had a much higher hospitalization rate than did those living in rural areas: 205 versus 57 per 100,000.
- Beneficiaries enrolled in both Medicare and Medicaid had 473 hospitalizations per 100,000, but the rate for those with Medicare only was 112.
“The disparities in the data reflect longstanding challenges facing minority communities and low-income older adults, many of whom face structural challenges to their health that go far beyond what is traditionally considered ‘medical,’ ” CMS Administrator Seema Verma said in a separate statement.
according to a new analysis by the Centers for Medicare & Medicaid Services.
CMS encounter and claims data show almost 110,000 hospital stays for COVID-19 from Jan. 1 to May 16, 2020. Of the longer admissions, 18% were 8-10 days, 16% were 11-15 days, and another 16% were 16 days or longer, the CMS reported in a preliminary data snapshot released June 22.
The hospitalization rate for the Medicare population was 175 per 100,000 as of May 16, but the CMS data show a number of disparities involving race/ethnicity and other demographic characteristics were uncovered, such as the following:
- Black patients were hospitalized for COVID-19 at a much higher rate, at 465 per 100,000 beneficiaries, than were Hispanics (258), Asians (187), and whites (123).
- Residents of urban/suburban areas had a much higher hospitalization rate than did those living in rural areas: 205 versus 57 per 100,000.
- Beneficiaries enrolled in both Medicare and Medicaid had 473 hospitalizations per 100,000, but the rate for those with Medicare only was 112.
“The disparities in the data reflect longstanding challenges facing minority communities and low-income older adults, many of whom face structural challenges to their health that go far beyond what is traditionally considered ‘medical,’ ” CMS Administrator Seema Verma said in a separate statement.
according to a new analysis by the Centers for Medicare & Medicaid Services.
CMS encounter and claims data show almost 110,000 hospital stays for COVID-19 from Jan. 1 to May 16, 2020. Of the longer admissions, 18% were 8-10 days, 16% were 11-15 days, and another 16% were 16 days or longer, the CMS reported in a preliminary data snapshot released June 22.
The hospitalization rate for the Medicare population was 175 per 100,000 as of May 16, but the CMS data show a number of disparities involving race/ethnicity and other demographic characteristics were uncovered, such as the following:
- Black patients were hospitalized for COVID-19 at a much higher rate, at 465 per 100,000 beneficiaries, than were Hispanics (258), Asians (187), and whites (123).
- Residents of urban/suburban areas had a much higher hospitalization rate than did those living in rural areas: 205 versus 57 per 100,000.
- Beneficiaries enrolled in both Medicare and Medicaid had 473 hospitalizations per 100,000, but the rate for those with Medicare only was 112.
“The disparities in the data reflect longstanding challenges facing minority communities and low-income older adults, many of whom face structural challenges to their health that go far beyond what is traditionally considered ‘medical,’ ” CMS Administrator Seema Verma said in a separate statement.
FDA approves selinexor for relapsed/refractory DLBCL
The Food and Drug Administration has granted accelerated approval of selinexor, a nuclear transport inhibitor, for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
Selinexor (marketed as XPOVIO by Karyopharm Therapeutics) is intended for adult patients with relapsed/refractory DLBCL, not otherwise specified, including DLBCL arising from follicular lymphoma, after at least two lines of systemic therapy, according to the FDA’s announcement.
The FDA granted selinexor accelerated approval for this indication based on the response rate seen in the SADAL trial. Continued approval for this indication “may be contingent upon verification and description of clinical benefit in confirmatory trials,” according to the FDA.
The SADAL trial (NCT02227251) was a phase 2, single-arm, open-label study of patients with DLBCL who had previously received two to five systemic regimens. The patients received selinexor at 60 mg orally on days 1 and 3 of each week.
Results in 134 patients showed an overall response rate of 29% (95% confidence interval: 22-38), with complete responses in 13% of patients. Of 39 patients who achieved a partial or complete response, 38% had a response duration of at least 6 months, and 15% had a response duration of at least 12 months, according to the FDA announcement.
The most common adverse reactions in this trial were fatigue, nausea, diarrhea, appetite decrease, weight decrease, constipation, vomiting, and pyrexia. Grade 3-4 laboratory abnormalities occurred in 15% or more of the patients, and comprised thrombocytopenia, lymphopenia, neutropenia, anemia, and hyponatremia.
Serious adverse reactions occurred in 46% of patients, most often from infection. Gastrointestinal toxicity occurred in 80% of patients, and any-grade hyponatremia occurred in 61%. Central neurological adverse reactions occurred in 25% of patients, including dizziness and mental status changes, according to the announcement.
Warnings and precautions for adverse events – including thrombocytopenia, neutropenia, gastrointestinal toxicity, hyponatremia, serious infection, neurological toxicity, and embryo-fetal toxicity – are provided in the prescribing information.
Selinexor acts through the inhibition of exportin-1 and leads to an accumulation of tumor suppressor proteins, a reduction in oncoproteins, and apoptosis of cancer cells. The drug was previously approved in 2019 for the treatment of relapsed/refractory multiple myeloma.
The SADAL trial was sponsored by Karyopharm Therapeutics.
SOURCE: U.S. Food and Drug Administration. 2020. Approval announcement.
The Food and Drug Administration has granted accelerated approval of selinexor, a nuclear transport inhibitor, for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
Selinexor (marketed as XPOVIO by Karyopharm Therapeutics) is intended for adult patients with relapsed/refractory DLBCL, not otherwise specified, including DLBCL arising from follicular lymphoma, after at least two lines of systemic therapy, according to the FDA’s announcement.
The FDA granted selinexor accelerated approval for this indication based on the response rate seen in the SADAL trial. Continued approval for this indication “may be contingent upon verification and description of clinical benefit in confirmatory trials,” according to the FDA.
The SADAL trial (NCT02227251) was a phase 2, single-arm, open-label study of patients with DLBCL who had previously received two to five systemic regimens. The patients received selinexor at 60 mg orally on days 1 and 3 of each week.
Results in 134 patients showed an overall response rate of 29% (95% confidence interval: 22-38), with complete responses in 13% of patients. Of 39 patients who achieved a partial or complete response, 38% had a response duration of at least 6 months, and 15% had a response duration of at least 12 months, according to the FDA announcement.
The most common adverse reactions in this trial were fatigue, nausea, diarrhea, appetite decrease, weight decrease, constipation, vomiting, and pyrexia. Grade 3-4 laboratory abnormalities occurred in 15% or more of the patients, and comprised thrombocytopenia, lymphopenia, neutropenia, anemia, and hyponatremia.
Serious adverse reactions occurred in 46% of patients, most often from infection. Gastrointestinal toxicity occurred in 80% of patients, and any-grade hyponatremia occurred in 61%. Central neurological adverse reactions occurred in 25% of patients, including dizziness and mental status changes, according to the announcement.
Warnings and precautions for adverse events – including thrombocytopenia, neutropenia, gastrointestinal toxicity, hyponatremia, serious infection, neurological toxicity, and embryo-fetal toxicity – are provided in the prescribing information.
Selinexor acts through the inhibition of exportin-1 and leads to an accumulation of tumor suppressor proteins, a reduction in oncoproteins, and apoptosis of cancer cells. The drug was previously approved in 2019 for the treatment of relapsed/refractory multiple myeloma.
The SADAL trial was sponsored by Karyopharm Therapeutics.
SOURCE: U.S. Food and Drug Administration. 2020. Approval announcement.
The Food and Drug Administration has granted accelerated approval of selinexor, a nuclear transport inhibitor, for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
Selinexor (marketed as XPOVIO by Karyopharm Therapeutics) is intended for adult patients with relapsed/refractory DLBCL, not otherwise specified, including DLBCL arising from follicular lymphoma, after at least two lines of systemic therapy, according to the FDA’s announcement.
The FDA granted selinexor accelerated approval for this indication based on the response rate seen in the SADAL trial. Continued approval for this indication “may be contingent upon verification and description of clinical benefit in confirmatory trials,” according to the FDA.
The SADAL trial (NCT02227251) was a phase 2, single-arm, open-label study of patients with DLBCL who had previously received two to five systemic regimens. The patients received selinexor at 60 mg orally on days 1 and 3 of each week.
Results in 134 patients showed an overall response rate of 29% (95% confidence interval: 22-38), with complete responses in 13% of patients. Of 39 patients who achieved a partial or complete response, 38% had a response duration of at least 6 months, and 15% had a response duration of at least 12 months, according to the FDA announcement.
The most common adverse reactions in this trial were fatigue, nausea, diarrhea, appetite decrease, weight decrease, constipation, vomiting, and pyrexia. Grade 3-4 laboratory abnormalities occurred in 15% or more of the patients, and comprised thrombocytopenia, lymphopenia, neutropenia, anemia, and hyponatremia.
Serious adverse reactions occurred in 46% of patients, most often from infection. Gastrointestinal toxicity occurred in 80% of patients, and any-grade hyponatremia occurred in 61%. Central neurological adverse reactions occurred in 25% of patients, including dizziness and mental status changes, according to the announcement.
Warnings and precautions for adverse events – including thrombocytopenia, neutropenia, gastrointestinal toxicity, hyponatremia, serious infection, neurological toxicity, and embryo-fetal toxicity – are provided in the prescribing information.
Selinexor acts through the inhibition of exportin-1 and leads to an accumulation of tumor suppressor proteins, a reduction in oncoproteins, and apoptosis of cancer cells. The drug was previously approved in 2019 for the treatment of relapsed/refractory multiple myeloma.
The SADAL trial was sponsored by Karyopharm Therapeutics.
SOURCE: U.S. Food and Drug Administration. 2020. Approval announcement.
FROM THE FDA
ED visits for life-threatening conditions declined early in COVID-19 pandemic
ED visits for myocardial infarction, stroke, and hyperglycemic crisis dropped substantially in the 10 weeks after COVID-19 was declared a national emergency on March 13, according to the Centers for Disease Control and Prevention.
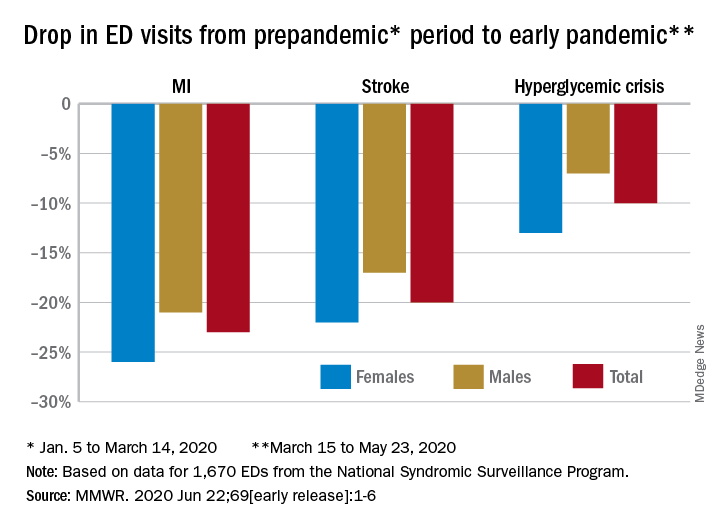
Compared with the 10-week period from Jan. 5 to March 14, ED visits were down by 23% for MI, 20% for stroke, and 10% for hyperglycemic crisis from March 15 to May 23, Samantha J. Lange, MPH, and associates at the CDC reported June 22 in the Morbidity and Mortality Weekly Report.
“A short-term decline of this magnitude … is biologically implausible for MI and stroke, especially for older adults, and unlikely for hyperglycemic crisis, and the finding suggests that patients with these conditions either could not access care or were delaying or avoiding seeking care during the early pandemic period,” they wrote.
The largest decreases in the actual number of visits for MI occurred among both men (down by 2,114, –24%) and women (down by 1,459, –25%) aged 65-74 years. For stroke, men aged 65-74 years had 1,406 (–19%) fewer visits to the ED and women 75-84 years had 1,642 (–23%) fewer visits, the CDC researchers said.
For hypoglycemic crisis, the largest declines during the early pandemic period occurred among younger adults: ED visits for men and women aged 18-44 years were down, respectively, by 419 (–8%) and 775 (–16%), they reported based on data from the National Syndromic Surveillance Program.
“Decreases in ED visits for hyperglycemic crisis might be less striking because patient recognition of this crisis is typically augmented by home glucose monitoring and not reliant upon symptoms alone, as is the case for MI and stroke,” Ms. Lange and her associates noted.
Charting weekly visit numbers showed that the drop for all three conditions actually started the week before the emergency was declared and reached its nadir the week after (March 22) for MI and 2 weeks later (March 29) for stroke and hypoglycemic crisis.
Visits for hypoglycemic crisis have largely returned to normal since those low points, but MI and stroke visits “remain below prepandemic levels” despite gradual increases through April and May, they said.
It has been reported that “deaths not associated with confirmed or probable COVID-19 might have been directly or indirectly attributed to the pandemic. The striking decline in ED visits for acute life-threatening conditions might partially explain observed excess mortality not associated with COVID-19,” the investigators wrote.
ED visits for myocardial infarction, stroke, and hyperglycemic crisis dropped substantially in the 10 weeks after COVID-19 was declared a national emergency on March 13, according to the Centers for Disease Control and Prevention.
Compared with the 10-week period from Jan. 5 to March 14, ED visits were down by 23% for MI, 20% for stroke, and 10% for hyperglycemic crisis from March 15 to May 23, Samantha J. Lange, MPH, and associates at the CDC reported June 22 in the Morbidity and Mortality Weekly Report.
“A short-term decline of this magnitude … is biologically implausible for MI and stroke, especially for older adults, and unlikely for hyperglycemic crisis, and the finding suggests that patients with these conditions either could not access care or were delaying or avoiding seeking care during the early pandemic period,” they wrote.
The largest decreases in the actual number of visits for MI occurred among both men (down by 2,114, –24%) and women (down by 1,459, –25%) aged 65-74 years. For stroke, men aged 65-74 years had 1,406 (–19%) fewer visits to the ED and women 75-84 years had 1,642 (–23%) fewer visits, the CDC researchers said.
For hypoglycemic crisis, the largest declines during the early pandemic period occurred among younger adults: ED visits for men and women aged 18-44 years were down, respectively, by 419 (–8%) and 775 (–16%), they reported based on data from the National Syndromic Surveillance Program.
“Decreases in ED visits for hyperglycemic crisis might be less striking because patient recognition of this crisis is typically augmented by home glucose monitoring and not reliant upon symptoms alone, as is the case for MI and stroke,” Ms. Lange and her associates noted.
Charting weekly visit numbers showed that the drop for all three conditions actually started the week before the emergency was declared and reached its nadir the week after (March 22) for MI and 2 weeks later (March 29) for stroke and hypoglycemic crisis.
Visits for hypoglycemic crisis have largely returned to normal since those low points, but MI and stroke visits “remain below prepandemic levels” despite gradual increases through April and May, they said.
It has been reported that “deaths not associated with confirmed or probable COVID-19 might have been directly or indirectly attributed to the pandemic. The striking decline in ED visits for acute life-threatening conditions might partially explain observed excess mortality not associated with COVID-19,” the investigators wrote.
ED visits for myocardial infarction, stroke, and hyperglycemic crisis dropped substantially in the 10 weeks after COVID-19 was declared a national emergency on March 13, according to the Centers for Disease Control and Prevention.
Compared with the 10-week period from Jan. 5 to March 14, ED visits were down by 23% for MI, 20% for stroke, and 10% for hyperglycemic crisis from March 15 to May 23, Samantha J. Lange, MPH, and associates at the CDC reported June 22 in the Morbidity and Mortality Weekly Report.
“A short-term decline of this magnitude … is biologically implausible for MI and stroke, especially for older adults, and unlikely for hyperglycemic crisis, and the finding suggests that patients with these conditions either could not access care or were delaying or avoiding seeking care during the early pandemic period,” they wrote.
The largest decreases in the actual number of visits for MI occurred among both men (down by 2,114, –24%) and women (down by 1,459, –25%) aged 65-74 years. For stroke, men aged 65-74 years had 1,406 (–19%) fewer visits to the ED and women 75-84 years had 1,642 (–23%) fewer visits, the CDC researchers said.
For hypoglycemic crisis, the largest declines during the early pandemic period occurred among younger adults: ED visits for men and women aged 18-44 years were down, respectively, by 419 (–8%) and 775 (–16%), they reported based on data from the National Syndromic Surveillance Program.
“Decreases in ED visits for hyperglycemic crisis might be less striking because patient recognition of this crisis is typically augmented by home glucose monitoring and not reliant upon symptoms alone, as is the case for MI and stroke,” Ms. Lange and her associates noted.
Charting weekly visit numbers showed that the drop for all three conditions actually started the week before the emergency was declared and reached its nadir the week after (March 22) for MI and 2 weeks later (March 29) for stroke and hypoglycemic crisis.
Visits for hypoglycemic crisis have largely returned to normal since those low points, but MI and stroke visits “remain below prepandemic levels” despite gradual increases through April and May, they said.
It has been reported that “deaths not associated with confirmed or probable COVID-19 might have been directly or indirectly attributed to the pandemic. The striking decline in ED visits for acute life-threatening conditions might partially explain observed excess mortality not associated with COVID-19,” the investigators wrote.
FROM MMWR
U.S. adults reach Healthy People 2020 cholesterol goal
Good news: High cholesterol is down in the United States. More good news: Low HDL cholesterol is down in the United States.
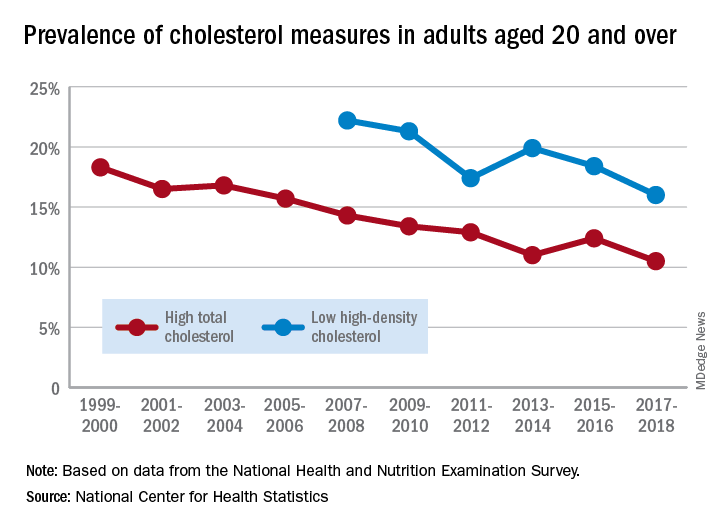
The prevalence of high total cholesterol in adults aged 20 years and older dropped from 18.3% in 1999-2000 to 10.5% in 2017-2018. And starting in 2007-2008, the prevalence of low HDL cholesterol declined from 22.2% to 16.0% in 2017-2018, the National Center for Health Statistics reported.
HDL cholesterol data before 2007 were not presented because of changes in laboratories and methods, but both trends are significant, and the decline in high total cholesterol means that the Healthy People 2020 goal of dropping prevalence to 13.5% has been met, said Margaret D. Carroll, MSPH, and Cheryl D. Fryar, MSPH, of the NCHS.
The demographic details, however, show some disparities hidden by the broader measures. The prevalence of low HDL cholesterol for women in 2015-2018 was 8.5%, but for men it was 26.6%, the NCHS investigators said.
And that Healthy People 2020 goal for total cholesterol? Age makes a difference: 7.5% of adults aged 20-39 years had high total cholesterol in 2015-2018, as did 11.4% of those aged 60 years and older, but those aged 40-59 years had a significantly higher prevalence of 15.7%, they reported.
Race/ethnicity was also a factor. Prevalence of low HDL was similar for white (16.6%) and Asian (15.8%) adults in 2015-2018, but black adults’ low HDL prevalence was significantly lower (11.9%) and Hispanics’ was significantly higher (21.9%), the researchers said.
The analysis was based on data from the National Health and Nutrition Examination Survey. The investigators defined high total cholesterol as a level of 240 mg/dL or more, and low HDL cholesterol as less than 40 mg/dL. LDL cholesterol was not included in the analysis.
Good news: High cholesterol is down in the United States. More good news: Low HDL cholesterol is down in the United States.
The prevalence of high total cholesterol in adults aged 20 years and older dropped from 18.3% in 1999-2000 to 10.5% in 2017-2018. And starting in 2007-2008, the prevalence of low HDL cholesterol declined from 22.2% to 16.0% in 2017-2018, the National Center for Health Statistics reported.
HDL cholesterol data before 2007 were not presented because of changes in laboratories and methods, but both trends are significant, and the decline in high total cholesterol means that the Healthy People 2020 goal of dropping prevalence to 13.5% has been met, said Margaret D. Carroll, MSPH, and Cheryl D. Fryar, MSPH, of the NCHS.
The demographic details, however, show some disparities hidden by the broader measures. The prevalence of low HDL cholesterol for women in 2015-2018 was 8.5%, but for men it was 26.6%, the NCHS investigators said.
And that Healthy People 2020 goal for total cholesterol? Age makes a difference: 7.5% of adults aged 20-39 years had high total cholesterol in 2015-2018, as did 11.4% of those aged 60 years and older, but those aged 40-59 years had a significantly higher prevalence of 15.7%, they reported.
Race/ethnicity was also a factor. Prevalence of low HDL was similar for white (16.6%) and Asian (15.8%) adults in 2015-2018, but black adults’ low HDL prevalence was significantly lower (11.9%) and Hispanics’ was significantly higher (21.9%), the researchers said.
The analysis was based on data from the National Health and Nutrition Examination Survey. The investigators defined high total cholesterol as a level of 240 mg/dL or more, and low HDL cholesterol as less than 40 mg/dL. LDL cholesterol was not included in the analysis.
Good news: High cholesterol is down in the United States. More good news: Low HDL cholesterol is down in the United States.
The prevalence of high total cholesterol in adults aged 20 years and older dropped from 18.3% in 1999-2000 to 10.5% in 2017-2018. And starting in 2007-2008, the prevalence of low HDL cholesterol declined from 22.2% to 16.0% in 2017-2018, the National Center for Health Statistics reported.
HDL cholesterol data before 2007 were not presented because of changes in laboratories and methods, but both trends are significant, and the decline in high total cholesterol means that the Healthy People 2020 goal of dropping prevalence to 13.5% has been met, said Margaret D. Carroll, MSPH, and Cheryl D. Fryar, MSPH, of the NCHS.
The demographic details, however, show some disparities hidden by the broader measures. The prevalence of low HDL cholesterol for women in 2015-2018 was 8.5%, but for men it was 26.6%, the NCHS investigators said.
And that Healthy People 2020 goal for total cholesterol? Age makes a difference: 7.5% of adults aged 20-39 years had high total cholesterol in 2015-2018, as did 11.4% of those aged 60 years and older, but those aged 40-59 years had a significantly higher prevalence of 15.7%, they reported.
Race/ethnicity was also a factor. Prevalence of low HDL was similar for white (16.6%) and Asian (15.8%) adults in 2015-2018, but black adults’ low HDL prevalence was significantly lower (11.9%) and Hispanics’ was significantly higher (21.9%), the researchers said.
The analysis was based on data from the National Health and Nutrition Examination Survey. The investigators defined high total cholesterol as a level of 240 mg/dL or more, and low HDL cholesterol as less than 40 mg/dL. LDL cholesterol was not included in the analysis.
FDA gives thumbs up to tazemetostat for follicular lymphoma
The US Food and Drug Administration (FDA) has granted accelerated approval of the EZH2 inhibitor tazemetostat (Tazverik, Epizyme, Inc) for the treatment of relapsed or refractory follicular lymphoma in adult patients with tumors harboring an EZH2 mutation.
Eligible patients must have already received at least two prior systemic therapies and have tumors that are positive for an EZH2 mutation, as detected by an FDA-approved test. The FDA has also approved the cobas EZH2 Mutation Test (Roche Molecular Systems, Inc) as a companion diagnostic test for tazemetostat.
The new indication is also for adult patients with relapsed/refractory follicular lymphoma who have no other satisfactory alternative treatment options.
“In our view, there remains no clear standard of care in the relapsed and/or refractory [follicular lymphoma] population, as not all patients benefit from today’s available therapies,” said Shefali Agarwal, MD, chief medical officer of Epizyme, in a company press release. “Based on this label, physicians will have the ability to use their clinical discretion to prescribe tazemetostat for their relapsed or refractory patients regardless of EZH2 mutational status and without regard to a specific line of treatment where other options are not satisfactory.”
This accelerated approval is based on overall response rate and duration of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials, the FDA notes.
Tazemetostat acts as an inhibitor of EZH2 methyltransferase. Earlier this year, the drug was approved for the treatment of metastatic or locally advanced epithelioid sarcoma in cases in which complete resection is not possible. It is the first drug with this mechanism of action and is the first to be indicated for epithelioid sarcoma.
Promising Efficacy in Phase 2 Trial
The new approval for use in follicular lymphoma was based on results from an open-label, single-arm, multicenter phase 2 clinical trial involving patients who had experienced disease progression after being treated with at least two prior systemic regimens. The cohort was divided into two treatment groups: One group consisted of 45 patients with EZH2-activating mutations, the other included 54 patients with wild-type EZH2.
All patients received tazemetostat at 800 mg administered orally twice a day. The primary efficacy outcome measures were overall response rate and duration of response, in accordance with International Working Group Non-Hodgkin Lymphoma criteria.
The median duration of follow-up was 22 months for patients with EZH2-activating mutations and 36 months for those with wild-type tumors.
Among the 45 patients with an EZH2-activating mutation, the median number of lines of prior systemic therapy was 2.0 (range, 1 – 11). In 49% of patients, disease was refractory to rituximab, and in 49%, it was refractory to the patient’s last therapy.
The overall response rate was 69%; 12% of patients achieved a complete response, and 57% achieved a partial response. The median duration of response was 10.9 months and ongoing.
In the cohort of 54 patients with wild-type EZH2, the median number previous therapies was 3.0 (range, 1 – 8); in 59% of patients, disease was refractory to rituximab, and in 41%, it was refractory to the patient’s last therapy.
The overall response rate to tazemetostat treatment was 34%; 4% of patients achieved a complete response, and 30% achieved a partial response. The median duration of response was 13 months.
Serious adverse reactions occurred in 30% of patients. The most common were fatigue, upper respiratory tract infection, musculoskeletal pain, nausea, and abdominal pain. Eight patients (8%) discontinued treatment during the trial because of adverse events. There were no reported deaths. No black box warnings have been published, and there are no contraindications.
“The durable responses observed with this drug are notable in the context of the safety profile and route of oral, at-home administration, and will offer an important new option for physicians as we care for patients with relapsed/refractory follicular lymphoma,” said John Leonard, MD, in a company press release. He is associate dean for clinical research and Richard T. Silver Distinguished Professor of Hematology and Medical Oncology, Meyer Cancer Center, Weill Cornell Medicine and New York–Presbyterian Hospital, New York, and an investigator in the ongoing phase 1b/3 confirmatory trial for tazemetostat.
“Follicular lymphoma remains an incurable disease, and even with the availability of new drugs in recent years, there have remained important unmet needs in the treatment of follicular lymphoma,” he commented.
This article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has granted accelerated approval of the EZH2 inhibitor tazemetostat (Tazverik, Epizyme, Inc) for the treatment of relapsed or refractory follicular lymphoma in adult patients with tumors harboring an EZH2 mutation.
Eligible patients must have already received at least two prior systemic therapies and have tumors that are positive for an EZH2 mutation, as detected by an FDA-approved test. The FDA has also approved the cobas EZH2 Mutation Test (Roche Molecular Systems, Inc) as a companion diagnostic test for tazemetostat.
The new indication is also for adult patients with relapsed/refractory follicular lymphoma who have no other satisfactory alternative treatment options.
“In our view, there remains no clear standard of care in the relapsed and/or refractory [follicular lymphoma] population, as not all patients benefit from today’s available therapies,” said Shefali Agarwal, MD, chief medical officer of Epizyme, in a company press release. “Based on this label, physicians will have the ability to use their clinical discretion to prescribe tazemetostat for their relapsed or refractory patients regardless of EZH2 mutational status and without regard to a specific line of treatment where other options are not satisfactory.”
This accelerated approval is based on overall response rate and duration of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials, the FDA notes.
Tazemetostat acts as an inhibitor of EZH2 methyltransferase. Earlier this year, the drug was approved for the treatment of metastatic or locally advanced epithelioid sarcoma in cases in which complete resection is not possible. It is the first drug with this mechanism of action and is the first to be indicated for epithelioid sarcoma.
Promising Efficacy in Phase 2 Trial
The new approval for use in follicular lymphoma was based on results from an open-label, single-arm, multicenter phase 2 clinical trial involving patients who had experienced disease progression after being treated with at least two prior systemic regimens. The cohort was divided into two treatment groups: One group consisted of 45 patients with EZH2-activating mutations, the other included 54 patients with wild-type EZH2.
All patients received tazemetostat at 800 mg administered orally twice a day. The primary efficacy outcome measures were overall response rate and duration of response, in accordance with International Working Group Non-Hodgkin Lymphoma criteria.
The median duration of follow-up was 22 months for patients with EZH2-activating mutations and 36 months for those with wild-type tumors.
Among the 45 patients with an EZH2-activating mutation, the median number of lines of prior systemic therapy was 2.0 (range, 1 – 11). In 49% of patients, disease was refractory to rituximab, and in 49%, it was refractory to the patient’s last therapy.
The overall response rate was 69%; 12% of patients achieved a complete response, and 57% achieved a partial response. The median duration of response was 10.9 months and ongoing.
In the cohort of 54 patients with wild-type EZH2, the median number previous therapies was 3.0 (range, 1 – 8); in 59% of patients, disease was refractory to rituximab, and in 41%, it was refractory to the patient’s last therapy.
The overall response rate to tazemetostat treatment was 34%; 4% of patients achieved a complete response, and 30% achieved a partial response. The median duration of response was 13 months.
Serious adverse reactions occurred in 30% of patients. The most common were fatigue, upper respiratory tract infection, musculoskeletal pain, nausea, and abdominal pain. Eight patients (8%) discontinued treatment during the trial because of adverse events. There were no reported deaths. No black box warnings have been published, and there are no contraindications.
“The durable responses observed with this drug are notable in the context of the safety profile and route of oral, at-home administration, and will offer an important new option for physicians as we care for patients with relapsed/refractory follicular lymphoma,” said John Leonard, MD, in a company press release. He is associate dean for clinical research and Richard T. Silver Distinguished Professor of Hematology and Medical Oncology, Meyer Cancer Center, Weill Cornell Medicine and New York–Presbyterian Hospital, New York, and an investigator in the ongoing phase 1b/3 confirmatory trial for tazemetostat.
“Follicular lymphoma remains an incurable disease, and even with the availability of new drugs in recent years, there have remained important unmet needs in the treatment of follicular lymphoma,” he commented.
This article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has granted accelerated approval of the EZH2 inhibitor tazemetostat (Tazverik, Epizyme, Inc) for the treatment of relapsed or refractory follicular lymphoma in adult patients with tumors harboring an EZH2 mutation.
Eligible patients must have already received at least two prior systemic therapies and have tumors that are positive for an EZH2 mutation, as detected by an FDA-approved test. The FDA has also approved the cobas EZH2 Mutation Test (Roche Molecular Systems, Inc) as a companion diagnostic test for tazemetostat.
The new indication is also for adult patients with relapsed/refractory follicular lymphoma who have no other satisfactory alternative treatment options.
“In our view, there remains no clear standard of care in the relapsed and/or refractory [follicular lymphoma] population, as not all patients benefit from today’s available therapies,” said Shefali Agarwal, MD, chief medical officer of Epizyme, in a company press release. “Based on this label, physicians will have the ability to use their clinical discretion to prescribe tazemetostat for their relapsed or refractory patients regardless of EZH2 mutational status and without regard to a specific line of treatment where other options are not satisfactory.”
This accelerated approval is based on overall response rate and duration of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in confirmatory trials, the FDA notes.
Tazemetostat acts as an inhibitor of EZH2 methyltransferase. Earlier this year, the drug was approved for the treatment of metastatic or locally advanced epithelioid sarcoma in cases in which complete resection is not possible. It is the first drug with this mechanism of action and is the first to be indicated for epithelioid sarcoma.
Promising Efficacy in Phase 2 Trial
The new approval for use in follicular lymphoma was based on results from an open-label, single-arm, multicenter phase 2 clinical trial involving patients who had experienced disease progression after being treated with at least two prior systemic regimens. The cohort was divided into two treatment groups: One group consisted of 45 patients with EZH2-activating mutations, the other included 54 patients with wild-type EZH2.
All patients received tazemetostat at 800 mg administered orally twice a day. The primary efficacy outcome measures were overall response rate and duration of response, in accordance with International Working Group Non-Hodgkin Lymphoma criteria.
The median duration of follow-up was 22 months for patients with EZH2-activating mutations and 36 months for those with wild-type tumors.
Among the 45 patients with an EZH2-activating mutation, the median number of lines of prior systemic therapy was 2.0 (range, 1 – 11). In 49% of patients, disease was refractory to rituximab, and in 49%, it was refractory to the patient’s last therapy.
The overall response rate was 69%; 12% of patients achieved a complete response, and 57% achieved a partial response. The median duration of response was 10.9 months and ongoing.
In the cohort of 54 patients with wild-type EZH2, the median number previous therapies was 3.0 (range, 1 – 8); in 59% of patients, disease was refractory to rituximab, and in 41%, it was refractory to the patient’s last therapy.
The overall response rate to tazemetostat treatment was 34%; 4% of patients achieved a complete response, and 30% achieved a partial response. The median duration of response was 13 months.
Serious adverse reactions occurred in 30% of patients. The most common were fatigue, upper respiratory tract infection, musculoskeletal pain, nausea, and abdominal pain. Eight patients (8%) discontinued treatment during the trial because of adverse events. There were no reported deaths. No black box warnings have been published, and there are no contraindications.
“The durable responses observed with this drug are notable in the context of the safety profile and route of oral, at-home administration, and will offer an important new option for physicians as we care for patients with relapsed/refractory follicular lymphoma,” said John Leonard, MD, in a company press release. He is associate dean for clinical research and Richard T. Silver Distinguished Professor of Hematology and Medical Oncology, Meyer Cancer Center, Weill Cornell Medicine and New York–Presbyterian Hospital, New York, and an investigator in the ongoing phase 1b/3 confirmatory trial for tazemetostat.
“Follicular lymphoma remains an incurable disease, and even with the availability of new drugs in recent years, there have remained important unmet needs in the treatment of follicular lymphoma,” he commented.
This article first appeared on Medscape.com.
FDA makes Ilaris the first approved treatment for adult-onset Still’s disease
The Food and Drug Administration has expanded the indications for canakinumab (Ilaris) to include all patients with active Still’s disease older than 2 years, adding adult-onset Still’s disease (AOSD) to a previous approval for juvenile-onset Still’s disease, also known as systemic juvenile idiopathic arthritis (sJIA), making it the first approved treatment for AOSD, according to an FDA announcement.

The approval comes under a Priority Review designation that used “comparable pharmacokinetic exposure and extrapolation of established efficacy of canakinumab in patients with sJIA, as well as the safety of canakinumab in patients with AOSD and other diseases,” the FDA said.
The results from a randomized, double-blind, placebo-controlled study of 36 patients with AOSD aged 22-70 years showed that the efficacy and safety data in AOSD were generally consistent with the results of a pooled analysis of sJIA patients, according to Novartis, which markets canakinumab.
AOSD and sJIA share certain similarities, such as fever, arthritis, rash, and elevated markers of inflammation, which has led to suspicion that they are part of a continuum rather than wholly distinct, according to the agency. In addition, the role of interleukin-1 is well established in both diseases and is blocked by canakinumab.
The most common side effects (occurring in greater than 10% of patients) in sJIA studies included infections, abdominal pain, and injection-site reactions. Serious infections (e.g., pneumonia, varicella, gastroenteritis, measles, sepsis, otitis media, sinusitis, adenovirus, lymph node abscess, pharyngitis) were observed in approximately 4%-5%, according to the full prescribing information.
Canakinumab is also approved for the periodic fever syndromes of cryopyrin-associated periodic syndromes in adults and children aged 4 years and older (including familial cold auto-inflammatory syndrome and Muckle-Wells syndrome), tumor necrosis factor receptor associated periodic syndrome in adult and pediatric patients, hyperimmunoglobulin D syndrome/mevalonate kinase deficiency in adult and pediatric patients, and familial Mediterranean fever in adult and pediatric patients.
The Food and Drug Administration has expanded the indications for canakinumab (Ilaris) to include all patients with active Still’s disease older than 2 years, adding adult-onset Still’s disease (AOSD) to a previous approval for juvenile-onset Still’s disease, also known as systemic juvenile idiopathic arthritis (sJIA), making it the first approved treatment for AOSD, according to an FDA announcement.
The approval comes under a Priority Review designation that used “comparable pharmacokinetic exposure and extrapolation of established efficacy of canakinumab in patients with sJIA, as well as the safety of canakinumab in patients with AOSD and other diseases,” the FDA said.
The results from a randomized, double-blind, placebo-controlled study of 36 patients with AOSD aged 22-70 years showed that the efficacy and safety data in AOSD were generally consistent with the results of a pooled analysis of sJIA patients, according to Novartis, which markets canakinumab.
AOSD and sJIA share certain similarities, such as fever, arthritis, rash, and elevated markers of inflammation, which has led to suspicion that they are part of a continuum rather than wholly distinct, according to the agency. In addition, the role of interleukin-1 is well established in both diseases and is blocked by canakinumab.
The most common side effects (occurring in greater than 10% of patients) in sJIA studies included infections, abdominal pain, and injection-site reactions. Serious infections (e.g., pneumonia, varicella, gastroenteritis, measles, sepsis, otitis media, sinusitis, adenovirus, lymph node abscess, pharyngitis) were observed in approximately 4%-5%, according to the full prescribing information.
Canakinumab is also approved for the periodic fever syndromes of cryopyrin-associated periodic syndromes in adults and children aged 4 years and older (including familial cold auto-inflammatory syndrome and Muckle-Wells syndrome), tumor necrosis factor receptor associated periodic syndrome in adult and pediatric patients, hyperimmunoglobulin D syndrome/mevalonate kinase deficiency in adult and pediatric patients, and familial Mediterranean fever in adult and pediatric patients.
The Food and Drug Administration has expanded the indications for canakinumab (Ilaris) to include all patients with active Still’s disease older than 2 years, adding adult-onset Still’s disease (AOSD) to a previous approval for juvenile-onset Still’s disease, also known as systemic juvenile idiopathic arthritis (sJIA), making it the first approved treatment for AOSD, according to an FDA announcement.
The approval comes under a Priority Review designation that used “comparable pharmacokinetic exposure and extrapolation of established efficacy of canakinumab in patients with sJIA, as well as the safety of canakinumab in patients with AOSD and other diseases,” the FDA said.
The results from a randomized, double-blind, placebo-controlled study of 36 patients with AOSD aged 22-70 years showed that the efficacy and safety data in AOSD were generally consistent with the results of a pooled analysis of sJIA patients, according to Novartis, which markets canakinumab.
AOSD and sJIA share certain similarities, such as fever, arthritis, rash, and elevated markers of inflammation, which has led to suspicion that they are part of a continuum rather than wholly distinct, according to the agency. In addition, the role of interleukin-1 is well established in both diseases and is blocked by canakinumab.
The most common side effects (occurring in greater than 10% of patients) in sJIA studies included infections, abdominal pain, and injection-site reactions. Serious infections (e.g., pneumonia, varicella, gastroenteritis, measles, sepsis, otitis media, sinusitis, adenovirus, lymph node abscess, pharyngitis) were observed in approximately 4%-5%, according to the full prescribing information.
Canakinumab is also approved for the periodic fever syndromes of cryopyrin-associated periodic syndromes in adults and children aged 4 years and older (including familial cold auto-inflammatory syndrome and Muckle-Wells syndrome), tumor necrosis factor receptor associated periodic syndrome in adult and pediatric patients, hyperimmunoglobulin D syndrome/mevalonate kinase deficiency in adult and pediatric patients, and familial Mediterranean fever in adult and pediatric patients.
FDA approves Cosentyx for treatment of active nr-axSpA
The Food and Drug Administration has approved secukinumab (Cosentyx) for the treatment of active nonradiographic axial spondyloarthritis (nr-axSpA), according to an announcement from the drug’s manufacturer, Novartis.
FDA approval was based on results of the 2-year PREVENT trial, a randomized, double-blind, placebo-controlled, phase 3 study in 555 adults with active nr-axSpA who received a loading dose of 150 mg secukinumab subcutaneously weekly for 4 weeks, then maintenance dosing with 150 mg secukinumab monthly; 150 mg secukinumab monthly with no loading dose; or placebo. Patients were included if they were aged at least 18 years with 6 months or more of inflammatory back pain, had objective signs of inflammation (sacroiliitis on MRI and/or C-reactive protein at 5.0 mg/dL or higher), had active disease and spinal pain according to the Bath Ankylosing Spondylitis Disease Activity Index, had total back pain with a visual analog scale of 40 mm or greater, and had not received a tumor necrosis factor (TNF) inhibitor or had an inadequate response to no more than one TNF inhibitor. A total of 501 patients had not previously taken a biologic medication.
A significantly greater proportion of biologic-naive patients taking secukinumab in both active treatment arm met the trial’s primary endpoint of at least a 40% improvement in the Assessment of Spondyloarthritis International Society response criteria versus placebo after 52 weeks. Both loading and nonloading arms saw significant improvements in Ankylosing Spondylitis Quality of Life scores, compared with those in the placebo group.
The safety profile of secukinumab in PREVENT was shown to be consistent with previous clinical trials, with no new safety signals detected.
Secukinumab, a fully human monoclonal antibody that directly inhibits interleukin-17A, also received European Medicines Agency approval for the treatment of nr-axSpA in April 2020. It is already approved by the FDA for the treatment of moderate to severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis.
The Food and Drug Administration has approved secukinumab (Cosentyx) for the treatment of active nonradiographic axial spondyloarthritis (nr-axSpA), according to an announcement from the drug’s manufacturer, Novartis.
FDA approval was based on results of the 2-year PREVENT trial, a randomized, double-blind, placebo-controlled, phase 3 study in 555 adults with active nr-axSpA who received a loading dose of 150 mg secukinumab subcutaneously weekly for 4 weeks, then maintenance dosing with 150 mg secukinumab monthly; 150 mg secukinumab monthly with no loading dose; or placebo. Patients were included if they were aged at least 18 years with 6 months or more of inflammatory back pain, had objective signs of inflammation (sacroiliitis on MRI and/or C-reactive protein at 5.0 mg/dL or higher), had active disease and spinal pain according to the Bath Ankylosing Spondylitis Disease Activity Index, had total back pain with a visual analog scale of 40 mm or greater, and had not received a tumor necrosis factor (TNF) inhibitor or had an inadequate response to no more than one TNF inhibitor. A total of 501 patients had not previously taken a biologic medication.
A significantly greater proportion of biologic-naive patients taking secukinumab in both active treatment arm met the trial’s primary endpoint of at least a 40% improvement in the Assessment of Spondyloarthritis International Society response criteria versus placebo after 52 weeks. Both loading and nonloading arms saw significant improvements in Ankylosing Spondylitis Quality of Life scores, compared with those in the placebo group.
The safety profile of secukinumab in PREVENT was shown to be consistent with previous clinical trials, with no new safety signals detected.
Secukinumab, a fully human monoclonal antibody that directly inhibits interleukin-17A, also received European Medicines Agency approval for the treatment of nr-axSpA in April 2020. It is already approved by the FDA for the treatment of moderate to severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis.
The Food and Drug Administration has approved secukinumab (Cosentyx) for the treatment of active nonradiographic axial spondyloarthritis (nr-axSpA), according to an announcement from the drug’s manufacturer, Novartis.
FDA approval was based on results of the 2-year PREVENT trial, a randomized, double-blind, placebo-controlled, phase 3 study in 555 adults with active nr-axSpA who received a loading dose of 150 mg secukinumab subcutaneously weekly for 4 weeks, then maintenance dosing with 150 mg secukinumab monthly; 150 mg secukinumab monthly with no loading dose; or placebo. Patients were included if they were aged at least 18 years with 6 months or more of inflammatory back pain, had objective signs of inflammation (sacroiliitis on MRI and/or C-reactive protein at 5.0 mg/dL or higher), had active disease and spinal pain according to the Bath Ankylosing Spondylitis Disease Activity Index, had total back pain with a visual analog scale of 40 mm or greater, and had not received a tumor necrosis factor (TNF) inhibitor or had an inadequate response to no more than one TNF inhibitor. A total of 501 patients had not previously taken a biologic medication.
A significantly greater proportion of biologic-naive patients taking secukinumab in both active treatment arm met the trial’s primary endpoint of at least a 40% improvement in the Assessment of Spondyloarthritis International Society response criteria versus placebo after 52 weeks. Both loading and nonloading arms saw significant improvements in Ankylosing Spondylitis Quality of Life scores, compared with those in the placebo group.
The safety profile of secukinumab in PREVENT was shown to be consistent with previous clinical trials, with no new safety signals detected.
Secukinumab, a fully human monoclonal antibody that directly inhibits interleukin-17A, also received European Medicines Agency approval for the treatment of nr-axSpA in April 2020. It is already approved by the FDA for the treatment of moderate to severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis.
Lurbinectedin approved for metastatic SCLC
Patients with metastatic small-cell lung cancer (SCLC) whose disease has progressed after or during treatment with platinum-based chemotherapy now have a new option to try — lurbinectedin (Zepzelca, Jazz Pharma/PharmaMar).
The drug was granted accelerated approval by the US Food and Drug Administration (FDA) based on response data. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial, the FDA notes.
“Small-cell lung cancer is a disease with limited treatment options,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “While patients may initially respond to traditional chemotherapy, they often experience an aggressive recurrence that is historically resistant to treatment.”
“Seeing first-hand the aggressive nature of SCLC and knowing that the large majority of those diagnosed will experience relapse, I am excited to see an effective new treatment demonstrating durable responses,” Jeff Petty, MD, oncology specialist, Wake Forest Baptist Health, Winston-Salem, North Carolina, commented in the company press release. This new drug “is an important and much-needed addition to the treatment landscape for relapsing SCLC,” he added.
Approval based on monotherapy trial
The approval is based on a monotherapy clinical trial in 105 patients, which was published in May in Lancet Oncology, with first author José Trigo, MD, from the Hospital Universitario Virgen de la Victoria in Malaga, Spain.
These were adult patients with both platinum-sensitive and platinum-resistant SCLC who had disease progression after treatment with platinum-based chemotherapy. They were treated at 26 hospitals across six European countries and the US. All patients received lurbinectedin at 3.2 mg/m2 by intravenous infusion over 1 hour. Median follow-up was 17.1 months.
Overall response by investigator assessment was seen in 37 (35.2%) of the 105 patients. The response was greater (at 45%) among the patients with platinum-sensitive disease and smaller (22.2%) among those with platinum-resistant disease.
Lurbinectedin demonstrated a median duration of response of 5.3 months as measured by investigator assessment.
In a post-hoc analysis, among the 37 patients who had an initial objective response, the median overall survival was just over 1 year (12.6 months). It was even longer among patients who had platinum-sensitive disease (15.8 months), although it was shorter in patients with resistant disease (10.9 months).
These data are “particularly encouraging,” comment the authors of an accompanying editorial, led by Oscar Arrieta, MD, from the Thoracic Oncology Unit at the Instituto Nacional de Cancerología in Mexico City, Mexico. These response rates “outperform all previous results achieved with topotecan and other less established treatment schemes including cyclophosphamide, doxorubicin, and vincristine, or platinum re-challenge, in this setting.”
“Lurbinectedin represents an innovative approach to conventional anti-cancer drugs, with an elegant mechanism of action based on the inhibition of transcription-dependent replication stress and genome instability of tumor cells,” the editorialists comment. “The drug binds to specific DNA triplets commonly found in transcription sites and triggers cellular apoptosis.”
“At present, the only evidence-based second-line treatment approved for SCLC is topotecan, a topoisomerase 1 inhibitor with moderate activity in patients with sensitive disease, although its effect is much less evident in patients with resistant SCLC,” they continue.
“Overall, the study by Trigo and colleagues presents novel data for a very challenging disease for which few treatment options exist, and the data on response and survival do seem to outperform data from historical controls,” Arrieta and colleagues write.
The editorialists also note that, in this trial, a few patients had received immunotherapy as part of their first-line treatment, and some of these patients (5 of 8 patients, 68%) had “an outstanding rate of durable response to lurbinectedin.” This raises the possibility of a synergistic effect between immunotherapy and lurbinectedin, as the combination seems to enhance immune memory and impair subsequent tumor growth, they add. Further trials will need to explore sequencing of therapy, they suggest.
A large phase 3 study known as ATLANTIS is currently underway.
The most common grade 3-4 adverse events in the present trial were hematologic abnormalities: anemia (9% of patients), leukopenia (29%), neutropenia (46%), and thrombocytopenia (7%). Serious treatment-related adverse events occurred in 10% of patients, of which neutropenia and febrile neutropenia were the most common (5% each). No treatment-related deaths were reported.
The study was funded by PharmaMar. Trigo and coauthors, and Arrieta and fellow editorialists, all report relationships with pharmaceutical companies, as detailed in the published articles.
This article first appeared on Medscape.com.
Patients with metastatic small-cell lung cancer (SCLC) whose disease has progressed after or during treatment with platinum-based chemotherapy now have a new option to try — lurbinectedin (Zepzelca, Jazz Pharma/PharmaMar).
The drug was granted accelerated approval by the US Food and Drug Administration (FDA) based on response data. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial, the FDA notes.
“Small-cell lung cancer is a disease with limited treatment options,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “While patients may initially respond to traditional chemotherapy, they often experience an aggressive recurrence that is historically resistant to treatment.”
“Seeing first-hand the aggressive nature of SCLC and knowing that the large majority of those diagnosed will experience relapse, I am excited to see an effective new treatment demonstrating durable responses,” Jeff Petty, MD, oncology specialist, Wake Forest Baptist Health, Winston-Salem, North Carolina, commented in the company press release. This new drug “is an important and much-needed addition to the treatment landscape for relapsing SCLC,” he added.
Approval based on monotherapy trial
The approval is based on a monotherapy clinical trial in 105 patients, which was published in May in Lancet Oncology, with first author José Trigo, MD, from the Hospital Universitario Virgen de la Victoria in Malaga, Spain.
These were adult patients with both platinum-sensitive and platinum-resistant SCLC who had disease progression after treatment with platinum-based chemotherapy. They were treated at 26 hospitals across six European countries and the US. All patients received lurbinectedin at 3.2 mg/m2 by intravenous infusion over 1 hour. Median follow-up was 17.1 months.
Overall response by investigator assessment was seen in 37 (35.2%) of the 105 patients. The response was greater (at 45%) among the patients with platinum-sensitive disease and smaller (22.2%) among those with platinum-resistant disease.
Lurbinectedin demonstrated a median duration of response of 5.3 months as measured by investigator assessment.
In a post-hoc analysis, among the 37 patients who had an initial objective response, the median overall survival was just over 1 year (12.6 months). It was even longer among patients who had platinum-sensitive disease (15.8 months), although it was shorter in patients with resistant disease (10.9 months).
These data are “particularly encouraging,” comment the authors of an accompanying editorial, led by Oscar Arrieta, MD, from the Thoracic Oncology Unit at the Instituto Nacional de Cancerología in Mexico City, Mexico. These response rates “outperform all previous results achieved with topotecan and other less established treatment schemes including cyclophosphamide, doxorubicin, and vincristine, or platinum re-challenge, in this setting.”
“Lurbinectedin represents an innovative approach to conventional anti-cancer drugs, with an elegant mechanism of action based on the inhibition of transcription-dependent replication stress and genome instability of tumor cells,” the editorialists comment. “The drug binds to specific DNA triplets commonly found in transcription sites and triggers cellular apoptosis.”
“At present, the only evidence-based second-line treatment approved for SCLC is topotecan, a topoisomerase 1 inhibitor with moderate activity in patients with sensitive disease, although its effect is much less evident in patients with resistant SCLC,” they continue.
“Overall, the study by Trigo and colleagues presents novel data for a very challenging disease for which few treatment options exist, and the data on response and survival do seem to outperform data from historical controls,” Arrieta and colleagues write.
The editorialists also note that, in this trial, a few patients had received immunotherapy as part of their first-line treatment, and some of these patients (5 of 8 patients, 68%) had “an outstanding rate of durable response to lurbinectedin.” This raises the possibility of a synergistic effect between immunotherapy and lurbinectedin, as the combination seems to enhance immune memory and impair subsequent tumor growth, they add. Further trials will need to explore sequencing of therapy, they suggest.
A large phase 3 study known as ATLANTIS is currently underway.
The most common grade 3-4 adverse events in the present trial were hematologic abnormalities: anemia (9% of patients), leukopenia (29%), neutropenia (46%), and thrombocytopenia (7%). Serious treatment-related adverse events occurred in 10% of patients, of which neutropenia and febrile neutropenia were the most common (5% each). No treatment-related deaths were reported.
The study was funded by PharmaMar. Trigo and coauthors, and Arrieta and fellow editorialists, all report relationships with pharmaceutical companies, as detailed in the published articles.
This article first appeared on Medscape.com.
Patients with metastatic small-cell lung cancer (SCLC) whose disease has progressed after or during treatment with platinum-based chemotherapy now have a new option to try — lurbinectedin (Zepzelca, Jazz Pharma/PharmaMar).
The drug was granted accelerated approval by the US Food and Drug Administration (FDA) based on response data. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial, the FDA notes.
“Small-cell lung cancer is a disease with limited treatment options,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “While patients may initially respond to traditional chemotherapy, they often experience an aggressive recurrence that is historically resistant to treatment.”
“Seeing first-hand the aggressive nature of SCLC and knowing that the large majority of those diagnosed will experience relapse, I am excited to see an effective new treatment demonstrating durable responses,” Jeff Petty, MD, oncology specialist, Wake Forest Baptist Health, Winston-Salem, North Carolina, commented in the company press release. This new drug “is an important and much-needed addition to the treatment landscape for relapsing SCLC,” he added.
Approval based on monotherapy trial
The approval is based on a monotherapy clinical trial in 105 patients, which was published in May in Lancet Oncology, with first author José Trigo, MD, from the Hospital Universitario Virgen de la Victoria in Malaga, Spain.
These were adult patients with both platinum-sensitive and platinum-resistant SCLC who had disease progression after treatment with platinum-based chemotherapy. They were treated at 26 hospitals across six European countries and the US. All patients received lurbinectedin at 3.2 mg/m2 by intravenous infusion over 1 hour. Median follow-up was 17.1 months.
Overall response by investigator assessment was seen in 37 (35.2%) of the 105 patients. The response was greater (at 45%) among the patients with platinum-sensitive disease and smaller (22.2%) among those with platinum-resistant disease.
Lurbinectedin demonstrated a median duration of response of 5.3 months as measured by investigator assessment.
In a post-hoc analysis, among the 37 patients who had an initial objective response, the median overall survival was just over 1 year (12.6 months). It was even longer among patients who had platinum-sensitive disease (15.8 months), although it was shorter in patients with resistant disease (10.9 months).
These data are “particularly encouraging,” comment the authors of an accompanying editorial, led by Oscar Arrieta, MD, from the Thoracic Oncology Unit at the Instituto Nacional de Cancerología in Mexico City, Mexico. These response rates “outperform all previous results achieved with topotecan and other less established treatment schemes including cyclophosphamide, doxorubicin, and vincristine, or platinum re-challenge, in this setting.”
“Lurbinectedin represents an innovative approach to conventional anti-cancer drugs, with an elegant mechanism of action based on the inhibition of transcription-dependent replication stress and genome instability of tumor cells,” the editorialists comment. “The drug binds to specific DNA triplets commonly found in transcription sites and triggers cellular apoptosis.”
“At present, the only evidence-based second-line treatment approved for SCLC is topotecan, a topoisomerase 1 inhibitor with moderate activity in patients with sensitive disease, although its effect is much less evident in patients with resistant SCLC,” they continue.
“Overall, the study by Trigo and colleagues presents novel data for a very challenging disease for which few treatment options exist, and the data on response and survival do seem to outperform data from historical controls,” Arrieta and colleagues write.
The editorialists also note that, in this trial, a few patients had received immunotherapy as part of their first-line treatment, and some of these patients (5 of 8 patients, 68%) had “an outstanding rate of durable response to lurbinectedin.” This raises the possibility of a synergistic effect between immunotherapy and lurbinectedin, as the combination seems to enhance immune memory and impair subsequent tumor growth, they add. Further trials will need to explore sequencing of therapy, they suggest.
A large phase 3 study known as ATLANTIS is currently underway.
The most common grade 3-4 adverse events in the present trial were hematologic abnormalities: anemia (9% of patients), leukopenia (29%), neutropenia (46%), and thrombocytopenia (7%). Serious treatment-related adverse events occurred in 10% of patients, of which neutropenia and febrile neutropenia were the most common (5% each). No treatment-related deaths were reported.
The study was funded by PharmaMar. Trigo and coauthors, and Arrieta and fellow editorialists, all report relationships with pharmaceutical companies, as detailed in the published articles.
This article first appeared on Medscape.com.