User login
Gardasil-9 approved for prevention of head and neck cancers
The US Food and Drug Administration (FDA) has expanded the indication for the Gardasil-9 (Merck) vaccine to include prevention of oropharyngeal and other head and neck cancers caused by HPV types 16, 18, 31, 33, 45, 52, and 58.
This new indication is approved under the FDA’s accelerated approval program and is based on the vaccine’s effectiveness in preventing HPV-related anogenital disease. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory clinical trial, which is currently underway.
“At Merck, working to help prevent certain HPV-related cancers has been a priority for more than two decades,” Alain Luxembourg, MD, director, clinical research, Merck Research Laboratories, said in a statement. “Today’s approval for the prevention of HPV-related oropharyngeal and other head and neck cancers represents an important step in Merck’s mission to help reduce the number of men and women affected by certain HPV-related cancers.”
This new indication doesn’t affect the current recommendations that are already in place. In 2018, a supplemental application for Gardasil 9 was approved to include women and men aged 27 through 45 years for preventing a variety of cancers including cervical, vulvar, vaginal, and anal cancer as well as genital warts. But cancers of the head and neck were not included.
The original Gardasil vaccine came on the market in 2006, with an indication to prevent certain cancers and diseases caused by HPV types 6, 11, 16, and 18. It is no longer distributed in the United States.
In 2014, the FDA approved Gardasil 9, which extends the vaccine coverage for the initial four HPV types as five additional types (31, 33, 45, 52, and 58), and its initial indication was for use in both men and women between the ages of 9 through 26 years.
Head and neck cancers surpass cervical cancer
More than 2 decades ago, researchers first found a connection between HPV and a subset of head and neck cancers (Curr Opin Oncol. 1999;11(3):191-199). The cancers associated with HPV also appeared to have a different biology and disease pattern, as well as a better prognosis, compared with those that were unrelated. HPV is now responsible for the majority of oropharyngeal squamous cell cancers diagnosed in the United States.
A study published last year found that oral HPV infections were occurring with significantly less frequency among sexually active female adolescents who had received the quadrivalent vaccine, as compared with those who were unvaccinated.
These findings provided evidence that HPV vaccination was associated with a reduced frequency of HPV infection in the oral cavity, suggesting that vaccination could decrease the future risk of HPV-associated head and neck cancers.
The omission of head and neck cancers from the initial list of indications for the vaccine is notable because, according to data from the Centers for Disease Control and Prevention (CDC), oropharyngeal cancers are now the most common malignancy caused by HPV, surpassing cervical cancer.
Who will benefit?
An estimated 14 million new HPV infections occur every year in the United States, according to the CDC, and about 80% of individuals who are sexually active have been exposed at some point during their lifetime. In most people, however, the virus will clear on its own without causing any illness or symptoms.
In a Medscape videoblog, Sandra Adamson Fryhofer, MD, MACP, FRCP, helped clarify the adult population most likely to benefit from the vaccine. She pointed out that the HPV vaccine doesn’t treat HPV-related disease or help clear infections, and there are currently no clinical antibody tests or titers that can predict immunity.
“Many adults aged 27-45 have already been exposed to HPV early in life,” she said. Those in a long-term mutually monogamous relationship are not likely to get a new HPV infection. Those with multiple prior sex partners are more likely to have already been exposed to vaccine serotypes. For them, the vaccine will be less effective.”
Fryhofer added that individuals who are now at risk for exposure to a new HPV infection from a new sex partner are the ones most likely to benefit from HPV vaccination.
Confirmation needed
The FDA’s accelerated approval is contingent on confirmatory data, and Merck opened a clinical trial this past February to evaluate the efficacy, immunogenicity, and safety of the 9-valent HPV vaccine in men 20 to 45 years of age. The phase 3 multicenter randomized trial will have an estimated enrollment of 6000 men.
This article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has expanded the indication for the Gardasil-9 (Merck) vaccine to include prevention of oropharyngeal and other head and neck cancers caused by HPV types 16, 18, 31, 33, 45, 52, and 58.
This new indication is approved under the FDA’s accelerated approval program and is based on the vaccine’s effectiveness in preventing HPV-related anogenital disease. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory clinical trial, which is currently underway.
“At Merck, working to help prevent certain HPV-related cancers has been a priority for more than two decades,” Alain Luxembourg, MD, director, clinical research, Merck Research Laboratories, said in a statement. “Today’s approval for the prevention of HPV-related oropharyngeal and other head and neck cancers represents an important step in Merck’s mission to help reduce the number of men and women affected by certain HPV-related cancers.”
This new indication doesn’t affect the current recommendations that are already in place. In 2018, a supplemental application for Gardasil 9 was approved to include women and men aged 27 through 45 years for preventing a variety of cancers including cervical, vulvar, vaginal, and anal cancer as well as genital warts. But cancers of the head and neck were not included.
The original Gardasil vaccine came on the market in 2006, with an indication to prevent certain cancers and diseases caused by HPV types 6, 11, 16, and 18. It is no longer distributed in the United States.
In 2014, the FDA approved Gardasil 9, which extends the vaccine coverage for the initial four HPV types as five additional types (31, 33, 45, 52, and 58), and its initial indication was for use in both men and women between the ages of 9 through 26 years.
Head and neck cancers surpass cervical cancer
More than 2 decades ago, researchers first found a connection between HPV and a subset of head and neck cancers (Curr Opin Oncol. 1999;11(3):191-199). The cancers associated with HPV also appeared to have a different biology and disease pattern, as well as a better prognosis, compared with those that were unrelated. HPV is now responsible for the majority of oropharyngeal squamous cell cancers diagnosed in the United States.
A study published last year found that oral HPV infections were occurring with significantly less frequency among sexually active female adolescents who had received the quadrivalent vaccine, as compared with those who were unvaccinated.
These findings provided evidence that HPV vaccination was associated with a reduced frequency of HPV infection in the oral cavity, suggesting that vaccination could decrease the future risk of HPV-associated head and neck cancers.
The omission of head and neck cancers from the initial list of indications for the vaccine is notable because, according to data from the Centers for Disease Control and Prevention (CDC), oropharyngeal cancers are now the most common malignancy caused by HPV, surpassing cervical cancer.
Who will benefit?
An estimated 14 million new HPV infections occur every year in the United States, according to the CDC, and about 80% of individuals who are sexually active have been exposed at some point during their lifetime. In most people, however, the virus will clear on its own without causing any illness or symptoms.
In a Medscape videoblog, Sandra Adamson Fryhofer, MD, MACP, FRCP, helped clarify the adult population most likely to benefit from the vaccine. She pointed out that the HPV vaccine doesn’t treat HPV-related disease or help clear infections, and there are currently no clinical antibody tests or titers that can predict immunity.
“Many adults aged 27-45 have already been exposed to HPV early in life,” she said. Those in a long-term mutually monogamous relationship are not likely to get a new HPV infection. Those with multiple prior sex partners are more likely to have already been exposed to vaccine serotypes. For them, the vaccine will be less effective.”
Fryhofer added that individuals who are now at risk for exposure to a new HPV infection from a new sex partner are the ones most likely to benefit from HPV vaccination.
Confirmation needed
The FDA’s accelerated approval is contingent on confirmatory data, and Merck opened a clinical trial this past February to evaluate the efficacy, immunogenicity, and safety of the 9-valent HPV vaccine in men 20 to 45 years of age. The phase 3 multicenter randomized trial will have an estimated enrollment of 6000 men.
This article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) has expanded the indication for the Gardasil-9 (Merck) vaccine to include prevention of oropharyngeal and other head and neck cancers caused by HPV types 16, 18, 31, 33, 45, 52, and 58.
This new indication is approved under the FDA’s accelerated approval program and is based on the vaccine’s effectiveness in preventing HPV-related anogenital disease. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory clinical trial, which is currently underway.
“At Merck, working to help prevent certain HPV-related cancers has been a priority for more than two decades,” Alain Luxembourg, MD, director, clinical research, Merck Research Laboratories, said in a statement. “Today’s approval for the prevention of HPV-related oropharyngeal and other head and neck cancers represents an important step in Merck’s mission to help reduce the number of men and women affected by certain HPV-related cancers.”
This new indication doesn’t affect the current recommendations that are already in place. In 2018, a supplemental application for Gardasil 9 was approved to include women and men aged 27 through 45 years for preventing a variety of cancers including cervical, vulvar, vaginal, and anal cancer as well as genital warts. But cancers of the head and neck were not included.
The original Gardasil vaccine came on the market in 2006, with an indication to prevent certain cancers and diseases caused by HPV types 6, 11, 16, and 18. It is no longer distributed in the United States.
In 2014, the FDA approved Gardasil 9, which extends the vaccine coverage for the initial four HPV types as five additional types (31, 33, 45, 52, and 58), and its initial indication was for use in both men and women between the ages of 9 through 26 years.
Head and neck cancers surpass cervical cancer
More than 2 decades ago, researchers first found a connection between HPV and a subset of head and neck cancers (Curr Opin Oncol. 1999;11(3):191-199). The cancers associated with HPV also appeared to have a different biology and disease pattern, as well as a better prognosis, compared with those that were unrelated. HPV is now responsible for the majority of oropharyngeal squamous cell cancers diagnosed in the United States.
A study published last year found that oral HPV infections were occurring with significantly less frequency among sexually active female adolescents who had received the quadrivalent vaccine, as compared with those who were unvaccinated.
These findings provided evidence that HPV vaccination was associated with a reduced frequency of HPV infection in the oral cavity, suggesting that vaccination could decrease the future risk of HPV-associated head and neck cancers.
The omission of head and neck cancers from the initial list of indications for the vaccine is notable because, according to data from the Centers for Disease Control and Prevention (CDC), oropharyngeal cancers are now the most common malignancy caused by HPV, surpassing cervical cancer.
Who will benefit?
An estimated 14 million new HPV infections occur every year in the United States, according to the CDC, and about 80% of individuals who are sexually active have been exposed at some point during their lifetime. In most people, however, the virus will clear on its own without causing any illness or symptoms.
In a Medscape videoblog, Sandra Adamson Fryhofer, MD, MACP, FRCP, helped clarify the adult population most likely to benefit from the vaccine. She pointed out that the HPV vaccine doesn’t treat HPV-related disease or help clear infections, and there are currently no clinical antibody tests or titers that can predict immunity.
“Many adults aged 27-45 have already been exposed to HPV early in life,” she said. Those in a long-term mutually monogamous relationship are not likely to get a new HPV infection. Those with multiple prior sex partners are more likely to have already been exposed to vaccine serotypes. For them, the vaccine will be less effective.”
Fryhofer added that individuals who are now at risk for exposure to a new HPV infection from a new sex partner are the ones most likely to benefit from HPV vaccination.
Confirmation needed
The FDA’s accelerated approval is contingent on confirmatory data, and Merck opened a clinical trial this past February to evaluate the efficacy, immunogenicity, and safety of the 9-valent HPV vaccine in men 20 to 45 years of age. The phase 3 multicenter randomized trial will have an estimated enrollment of 6000 men.
This article first appeared on Medscape.com.
Nivolumab approved to treat esophageal squamous cell carcinoma
The Food and Drug Administration has approved nivolumab (Opdivo) for use in certain patients with esophageal squamous cell carcinoma (ESCC).
The checkpoint inhibitor is now approved to treat patients with unresectable advanced, recurrent, or metastatic ESCC who previously received fluoropyrimidine- and platinum-based chemotherapy.
Researchers tested nivolumab in this population in the ATTRACTION-3 trial (NCT02569242). The trial enrolled 419 patients.
The patients were randomized to receive nivolumab at 240 mg via intravenous infusion over 30 minutes every 2 weeks (n = 210) or investigator’s choice of taxane chemotherapy (n = 209), which consisted of docetaxel (75 mg/m2 intravenously every 3 weeks) or paclitaxel (100 mg/m2 intravenously once a week for 6 weeks followed by 1 week off).
Nivolumab significantly improved overall survival but not progression-free survival. The median progression-free survival was 1.7 months in the nivolumab arm and 3.4 months in the chemotherapy arm (hazard ratio, 1.1).
The median overall survival was 10.9 months in the nivolumab arm and 8.4 months in the chemotherapy arm (hazard ratio, 0.77; P = .0189). The overall survival benefit was observed regardless of tumor programmed death–ligand 1 expression.
Response rates were similar between the treatment arms, but responses were more durable with nivolumab. The overall responses rate was 19.3% in the nivolumab arm and 21.5% in the chemotherapy arm. The median duration of response was 6.9 months and 3.9 months, respectively.
Serious adverse events were reported in 38% of patients in the nivolumab arm. Serious adverse events occurring in at least 2% of patients were pneumonia, esophageal fistula, interstitial lung disease, and pyrexia.
Adverse events prompted 13% of patients to discontinue nivolumab and 27% to delay nivolumab treatment.
Fatal adverse events in patients on nivolumab included interstitial lung disease or pneumonitis (1.4%), pneumonia (1.0%), septic shock (0.5%), esophageal fistula (0.5%), gastrointestinal hemorrhage (0.5%), pulmonary embolism (0.5%), and sudden death (0.5%).
The recommended dose of nivolumab for ESCC is 240 mg every 2 weeks or 480 mg every 4 weeks. For more details, see the full prescribing information.
The Food and Drug Administration has approved nivolumab (Opdivo) for use in certain patients with esophageal squamous cell carcinoma (ESCC).
The checkpoint inhibitor is now approved to treat patients with unresectable advanced, recurrent, or metastatic ESCC who previously received fluoropyrimidine- and platinum-based chemotherapy.
Researchers tested nivolumab in this population in the ATTRACTION-3 trial (NCT02569242). The trial enrolled 419 patients.
The patients were randomized to receive nivolumab at 240 mg via intravenous infusion over 30 minutes every 2 weeks (n = 210) or investigator’s choice of taxane chemotherapy (n = 209), which consisted of docetaxel (75 mg/m2 intravenously every 3 weeks) or paclitaxel (100 mg/m2 intravenously once a week for 6 weeks followed by 1 week off).
Nivolumab significantly improved overall survival but not progression-free survival. The median progression-free survival was 1.7 months in the nivolumab arm and 3.4 months in the chemotherapy arm (hazard ratio, 1.1).
The median overall survival was 10.9 months in the nivolumab arm and 8.4 months in the chemotherapy arm (hazard ratio, 0.77; P = .0189). The overall survival benefit was observed regardless of tumor programmed death–ligand 1 expression.
Response rates were similar between the treatment arms, but responses were more durable with nivolumab. The overall responses rate was 19.3% in the nivolumab arm and 21.5% in the chemotherapy arm. The median duration of response was 6.9 months and 3.9 months, respectively.
Serious adverse events were reported in 38% of patients in the nivolumab arm. Serious adverse events occurring in at least 2% of patients were pneumonia, esophageal fistula, interstitial lung disease, and pyrexia.
Adverse events prompted 13% of patients to discontinue nivolumab and 27% to delay nivolumab treatment.
Fatal adverse events in patients on nivolumab included interstitial lung disease or pneumonitis (1.4%), pneumonia (1.0%), septic shock (0.5%), esophageal fistula (0.5%), gastrointestinal hemorrhage (0.5%), pulmonary embolism (0.5%), and sudden death (0.5%).
The recommended dose of nivolumab for ESCC is 240 mg every 2 weeks or 480 mg every 4 weeks. For more details, see the full prescribing information.
The Food and Drug Administration has approved nivolumab (Opdivo) for use in certain patients with esophageal squamous cell carcinoma (ESCC).
The checkpoint inhibitor is now approved to treat patients with unresectable advanced, recurrent, or metastatic ESCC who previously received fluoropyrimidine- and platinum-based chemotherapy.
Researchers tested nivolumab in this population in the ATTRACTION-3 trial (NCT02569242). The trial enrolled 419 patients.
The patients were randomized to receive nivolumab at 240 mg via intravenous infusion over 30 minutes every 2 weeks (n = 210) or investigator’s choice of taxane chemotherapy (n = 209), which consisted of docetaxel (75 mg/m2 intravenously every 3 weeks) or paclitaxel (100 mg/m2 intravenously once a week for 6 weeks followed by 1 week off).
Nivolumab significantly improved overall survival but not progression-free survival. The median progression-free survival was 1.7 months in the nivolumab arm and 3.4 months in the chemotherapy arm (hazard ratio, 1.1).
The median overall survival was 10.9 months in the nivolumab arm and 8.4 months in the chemotherapy arm (hazard ratio, 0.77; P = .0189). The overall survival benefit was observed regardless of tumor programmed death–ligand 1 expression.
Response rates were similar between the treatment arms, but responses were more durable with nivolumab. The overall responses rate was 19.3% in the nivolumab arm and 21.5% in the chemotherapy arm. The median duration of response was 6.9 months and 3.9 months, respectively.
Serious adverse events were reported in 38% of patients in the nivolumab arm. Serious adverse events occurring in at least 2% of patients were pneumonia, esophageal fistula, interstitial lung disease, and pyrexia.
Adverse events prompted 13% of patients to discontinue nivolumab and 27% to delay nivolumab treatment.
Fatal adverse events in patients on nivolumab included interstitial lung disease or pneumonitis (1.4%), pneumonia (1.0%), septic shock (0.5%), esophageal fistula (0.5%), gastrointestinal hemorrhage (0.5%), pulmonary embolism (0.5%), and sudden death (0.5%).
The recommended dose of nivolumab for ESCC is 240 mg every 2 weeks or 480 mg every 4 weeks. For more details, see the full prescribing information.
Mental health visits account for 19% of ED costs
Emergency department visits for mental and substance use disorders (MUSDs) cost $14.6 billion in 2017, representing 19% of the total for all ED visits that year, according to the Agency for Healthcare Quality and Research.
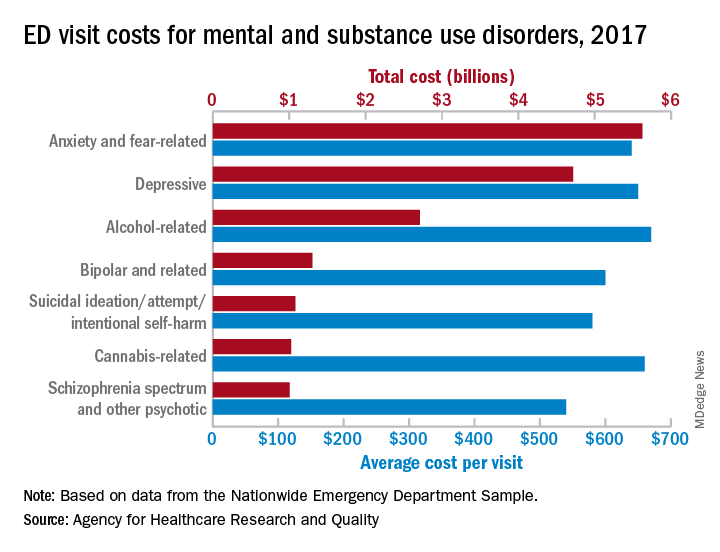
In terms of the total number of visits for MUSDs, 23.1 million, the proportion was slightly lower: 16% of all ED visits for the year, Zeynal Karaca, PhD, a senior economist with AHRQ, and Brian J. Moore, PhD, a senior research leader at IBM Watson Health, said in a recent statistical brief.
Put those figures together and the average visit for an MUSD diagnosis cost $630 and that is 19% higher than the average of $530 for all 145 million ED visits, they reported based on data from the Nationwide Emergency Department Sample.
The most costly MUSD diagnosis in 2017 was anxiety and fear-related disorders, with a total of $5.6 billion for ED visits, followed by depressive disorders at $4.7 billion and alcohol-related disorders at $2.7 billion. Some ED visits may involve more than one MUSD diagnosis, so the sum of all the individual diagnoses does not agree with the total for the entire MUSD category, the researchers noted.
On a per-visit basis, in 2017. [It was not included in the graph because it was 13th.] Other disorders with high per-visit costs were alcohol-related ($670), cannabis-related ($660), and depressive and stimulant-related (both with $650), Dr. Karaca and Dr. Moore said.
Patients with MUSDs who were routinely discharged after an ED visit in 2017 represented a much lower share of the total MUSD cost (68.0%), compared with the overall group of ED visitors (81.4%), but MUSD visits resulting in an inpatient admission made up a larger proportion of costs (19.0%), compared with all visits (9.5%), they said.
Costs between MUSD visits and all ED visits also differed by patient age. Visits by patients aged 0-9 years represented only 0.7% of MUSD-related ED costs but 5.6% of the overall cost, but the respective figures for those aged 45-64 were 36.2% for MUSD costs and 28.5% for the total ED cost, they reported.
SOURCE: Karaca Z and Moore BJ. HCUP Statistical Brief #257. May 12, 2020.
Emergency department visits for mental and substance use disorders (MUSDs) cost $14.6 billion in 2017, representing 19% of the total for all ED visits that year, according to the Agency for Healthcare Quality and Research.
In terms of the total number of visits for MUSDs, 23.1 million, the proportion was slightly lower: 16% of all ED visits for the year, Zeynal Karaca, PhD, a senior economist with AHRQ, and Brian J. Moore, PhD, a senior research leader at IBM Watson Health, said in a recent statistical brief.
Put those figures together and the average visit for an MUSD diagnosis cost $630 and that is 19% higher than the average of $530 for all 145 million ED visits, they reported based on data from the Nationwide Emergency Department Sample.
The most costly MUSD diagnosis in 2017 was anxiety and fear-related disorders, with a total of $5.6 billion for ED visits, followed by depressive disorders at $4.7 billion and alcohol-related disorders at $2.7 billion. Some ED visits may involve more than one MUSD diagnosis, so the sum of all the individual diagnoses does not agree with the total for the entire MUSD category, the researchers noted.
On a per-visit basis, in 2017. [It was not included in the graph because it was 13th.] Other disorders with high per-visit costs were alcohol-related ($670), cannabis-related ($660), and depressive and stimulant-related (both with $650), Dr. Karaca and Dr. Moore said.
Patients with MUSDs who were routinely discharged after an ED visit in 2017 represented a much lower share of the total MUSD cost (68.0%), compared with the overall group of ED visitors (81.4%), but MUSD visits resulting in an inpatient admission made up a larger proportion of costs (19.0%), compared with all visits (9.5%), they said.
Costs between MUSD visits and all ED visits also differed by patient age. Visits by patients aged 0-9 years represented only 0.7% of MUSD-related ED costs but 5.6% of the overall cost, but the respective figures for those aged 45-64 were 36.2% for MUSD costs and 28.5% for the total ED cost, they reported.
SOURCE: Karaca Z and Moore BJ. HCUP Statistical Brief #257. May 12, 2020.
Emergency department visits for mental and substance use disorders (MUSDs) cost $14.6 billion in 2017, representing 19% of the total for all ED visits that year, according to the Agency for Healthcare Quality and Research.
In terms of the total number of visits for MUSDs, 23.1 million, the proportion was slightly lower: 16% of all ED visits for the year, Zeynal Karaca, PhD, a senior economist with AHRQ, and Brian J. Moore, PhD, a senior research leader at IBM Watson Health, said in a recent statistical brief.
Put those figures together and the average visit for an MUSD diagnosis cost $630 and that is 19% higher than the average of $530 for all 145 million ED visits, they reported based on data from the Nationwide Emergency Department Sample.
The most costly MUSD diagnosis in 2017 was anxiety and fear-related disorders, with a total of $5.6 billion for ED visits, followed by depressive disorders at $4.7 billion and alcohol-related disorders at $2.7 billion. Some ED visits may involve more than one MUSD diagnosis, so the sum of all the individual diagnoses does not agree with the total for the entire MUSD category, the researchers noted.
On a per-visit basis, in 2017. [It was not included in the graph because it was 13th.] Other disorders with high per-visit costs were alcohol-related ($670), cannabis-related ($660), and depressive and stimulant-related (both with $650), Dr. Karaca and Dr. Moore said.
Patients with MUSDs who were routinely discharged after an ED visit in 2017 represented a much lower share of the total MUSD cost (68.0%), compared with the overall group of ED visitors (81.4%), but MUSD visits resulting in an inpatient admission made up a larger proportion of costs (19.0%), compared with all visits (9.5%), they said.
Costs between MUSD visits and all ED visits also differed by patient age. Visits by patients aged 0-9 years represented only 0.7% of MUSD-related ED costs but 5.6% of the overall cost, but the respective figures for those aged 45-64 were 36.2% for MUSD costs and 28.5% for the total ED cost, they reported.
SOURCE: Karaca Z and Moore BJ. HCUP Statistical Brief #257. May 12, 2020.
By the numbers: Asthma-COPD overlap deaths
Death rates for combined asthma and chronic obstructive pulmonary disease declined during 1999-2016, but the risk remains higher among women, compared with men, and in certain occupations, according to a recent report from the Centers for Disease Control and Prevention.
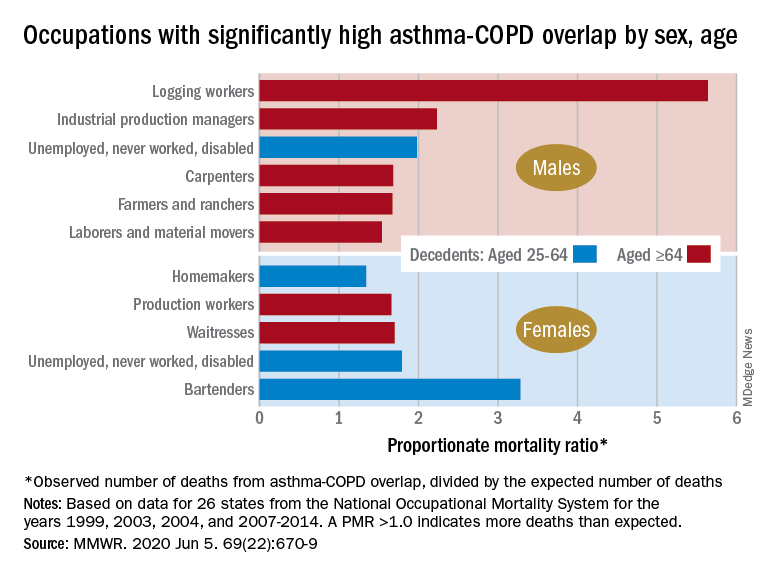
There is also an association between mortality and nonworking status among adults aged 25-64 years, which “suggests that asthma-COPD overlap might be associated with substantial morbidity,” Katelynn E. Dodd, MPH, and associates at the CDC’s National Institute for Occupational Safety and Health said in the Morbidity and Mortality Weekly Report. “These patients have been reported to have worse health outcomes than do those with asthma or COPD alone.”
For females with asthma-COPD overlap, the age-adjusted death rate among adults aged 25 years and older dropped from 7.71 per million in 1999 to 4.01 in 2016, with corresponding rates of 6.70 and 3.01 per million for males, they reported.
In 1999-2016, a total of 18,766 U.S. decedents aged ≥25 years had both asthma and COPD assigned as the underlying or contributing cause of death (12,028 women and 6,738 men), for an overall death rate of 5.03 per million persons (women, 5.59; men, 4.30), data from the National Vital Statistics System show.
Additional analysis, based on the calculation of proportionate mortality ratios (PMRs), also showed that mortality varied by occupational status and age for both males and females, the investigators said, noting that workplace exposures, such as dusts and secondhand smoke, are known to cause both asthma and COPD.
The PMR represents the observed number of deaths from asthma-COPD overlap in a specified industry or occupation, divided by the expected number of deaths, so a value over 1.0 indicates that there were more deaths associated with the condition than expected, Ms. Dodd and her associates explained.
Among female decedents, the occupation with the highest PMR that was statistically significant was bartending at 3.28. For men, the highest significant PMR, 5.64, occurred in logging workers. Those rates, however, only applied to one of the two age groups: 25-64 years in women and ≥65 in men, based on data from the National Occupational Mortality Surveillance, which included information from 26 states for the years 1999, 2003, 2004, and 2007-2014.
Occupationally speaking, the one area of common ground between males and females was lack of occupation. PMRs for those aged 25-64 years “were significantly elevated among men (1.98) and women (1.79) who were unemployed, never worked, or were disabled workers,” they said. PMRs were elevated for nonworking older males and females but were not significant.
The elevated PMRs suggest “that asthma-COPD overlap might be associated with substantial morbidity resulting in loss of employment [because] retired and unemployed persons might have left the workforce because of severe asthma or COPD,” the investigators wrote.
SOURCE: Dodd KE et al. MMWR. 2020 Jun 5. 69(22):670-9.
Death rates for combined asthma and chronic obstructive pulmonary disease declined during 1999-2016, but the risk remains higher among women, compared with men, and in certain occupations, according to a recent report from the Centers for Disease Control and Prevention.
There is also an association between mortality and nonworking status among adults aged 25-64 years, which “suggests that asthma-COPD overlap might be associated with substantial morbidity,” Katelynn E. Dodd, MPH, and associates at the CDC’s National Institute for Occupational Safety and Health said in the Morbidity and Mortality Weekly Report. “These patients have been reported to have worse health outcomes than do those with asthma or COPD alone.”
For females with asthma-COPD overlap, the age-adjusted death rate among adults aged 25 years and older dropped from 7.71 per million in 1999 to 4.01 in 2016, with corresponding rates of 6.70 and 3.01 per million for males, they reported.
In 1999-2016, a total of 18,766 U.S. decedents aged ≥25 years had both asthma and COPD assigned as the underlying or contributing cause of death (12,028 women and 6,738 men), for an overall death rate of 5.03 per million persons (women, 5.59; men, 4.30), data from the National Vital Statistics System show.
Additional analysis, based on the calculation of proportionate mortality ratios (PMRs), also showed that mortality varied by occupational status and age for both males and females, the investigators said, noting that workplace exposures, such as dusts and secondhand smoke, are known to cause both asthma and COPD.
The PMR represents the observed number of deaths from asthma-COPD overlap in a specified industry or occupation, divided by the expected number of deaths, so a value over 1.0 indicates that there were more deaths associated with the condition than expected, Ms. Dodd and her associates explained.
Among female decedents, the occupation with the highest PMR that was statistically significant was bartending at 3.28. For men, the highest significant PMR, 5.64, occurred in logging workers. Those rates, however, only applied to one of the two age groups: 25-64 years in women and ≥65 in men, based on data from the National Occupational Mortality Surveillance, which included information from 26 states for the years 1999, 2003, 2004, and 2007-2014.
Occupationally speaking, the one area of common ground between males and females was lack of occupation. PMRs for those aged 25-64 years “were significantly elevated among men (1.98) and women (1.79) who were unemployed, never worked, or were disabled workers,” they said. PMRs were elevated for nonworking older males and females but were not significant.
The elevated PMRs suggest “that asthma-COPD overlap might be associated with substantial morbidity resulting in loss of employment [because] retired and unemployed persons might have left the workforce because of severe asthma or COPD,” the investigators wrote.
SOURCE: Dodd KE et al. MMWR. 2020 Jun 5. 69(22):670-9.
Death rates for combined asthma and chronic obstructive pulmonary disease declined during 1999-2016, but the risk remains higher among women, compared with men, and in certain occupations, according to a recent report from the Centers for Disease Control and Prevention.
There is also an association between mortality and nonworking status among adults aged 25-64 years, which “suggests that asthma-COPD overlap might be associated with substantial morbidity,” Katelynn E. Dodd, MPH, and associates at the CDC’s National Institute for Occupational Safety and Health said in the Morbidity and Mortality Weekly Report. “These patients have been reported to have worse health outcomes than do those with asthma or COPD alone.”
For females with asthma-COPD overlap, the age-adjusted death rate among adults aged 25 years and older dropped from 7.71 per million in 1999 to 4.01 in 2016, with corresponding rates of 6.70 and 3.01 per million for males, they reported.
In 1999-2016, a total of 18,766 U.S. decedents aged ≥25 years had both asthma and COPD assigned as the underlying or contributing cause of death (12,028 women and 6,738 men), for an overall death rate of 5.03 per million persons (women, 5.59; men, 4.30), data from the National Vital Statistics System show.
Additional analysis, based on the calculation of proportionate mortality ratios (PMRs), also showed that mortality varied by occupational status and age for both males and females, the investigators said, noting that workplace exposures, such as dusts and secondhand smoke, are known to cause both asthma and COPD.
The PMR represents the observed number of deaths from asthma-COPD overlap in a specified industry or occupation, divided by the expected number of deaths, so a value over 1.0 indicates that there were more deaths associated with the condition than expected, Ms. Dodd and her associates explained.
Among female decedents, the occupation with the highest PMR that was statistically significant was bartending at 3.28. For men, the highest significant PMR, 5.64, occurred in logging workers. Those rates, however, only applied to one of the two age groups: 25-64 years in women and ≥65 in men, based on data from the National Occupational Mortality Surveillance, which included information from 26 states for the years 1999, 2003, 2004, and 2007-2014.
Occupationally speaking, the one area of common ground between males and females was lack of occupation. PMRs for those aged 25-64 years “were significantly elevated among men (1.98) and women (1.79) who were unemployed, never worked, or were disabled workers,” they said. PMRs were elevated for nonworking older males and females but were not significant.
The elevated PMRs suggest “that asthma-COPD overlap might be associated with substantial morbidity resulting in loss of employment [because] retired and unemployed persons might have left the workforce because of severe asthma or COPD,” the investigators wrote.
SOURCE: Dodd KE et al. MMWR. 2020 Jun 5. 69(22):670-9.
FROM MMWR
FDA approves new antibiotic for HABP/VABP treatment
in people aged 18 years and older.

Approval for Recarbrio was based on results of a randomized, controlled clinical trial of 535 hospitalized adults with hospital-acquired and ventilator-associated bacterial pneumonia who received either Recarbrio or piperacillin-tazobactam. After 28 days, 16% of patients who received Recarbrio and 21% of patients who received piperacillin-tazobactam had died.
The most common adverse events associated with Recarbrio are increased alanine aminotransferase/ aspartate aminotransferase, anemia, diarrhea, hypokalemia, and hyponatremia. Recarbrio was previously approved by the FDA to treat patients with complicated urinary tract infections and complicated intra-abdominal infections who have limited or no alternative treatment options, according to an FDA press release.
“As a public health agency, the FDA addresses the threat of antimicrobial-resistant infections by facilitating the development of safe and effective new treatments. These efforts provide more options to fight serious bacterial infections and get new, safe and effective therapies to patients as soon as possible,” said Sumathi Nambiar, MD, MPH, director of the division of anti-infectives within the office of infectious disease at the Center for Drug Evaluation and Research.
in people aged 18 years and older.
Approval for Recarbrio was based on results of a randomized, controlled clinical trial of 535 hospitalized adults with hospital-acquired and ventilator-associated bacterial pneumonia who received either Recarbrio or piperacillin-tazobactam. After 28 days, 16% of patients who received Recarbrio and 21% of patients who received piperacillin-tazobactam had died.
The most common adverse events associated with Recarbrio are increased alanine aminotransferase/ aspartate aminotransferase, anemia, diarrhea, hypokalemia, and hyponatremia. Recarbrio was previously approved by the FDA to treat patients with complicated urinary tract infections and complicated intra-abdominal infections who have limited or no alternative treatment options, according to an FDA press release.
“As a public health agency, the FDA addresses the threat of antimicrobial-resistant infections by facilitating the development of safe and effective new treatments. These efforts provide more options to fight serious bacterial infections and get new, safe and effective therapies to patients as soon as possible,” said Sumathi Nambiar, MD, MPH, director of the division of anti-infectives within the office of infectious disease at the Center for Drug Evaluation and Research.
in people aged 18 years and older.
Approval for Recarbrio was based on results of a randomized, controlled clinical trial of 535 hospitalized adults with hospital-acquired and ventilator-associated bacterial pneumonia who received either Recarbrio or piperacillin-tazobactam. After 28 days, 16% of patients who received Recarbrio and 21% of patients who received piperacillin-tazobactam had died.
The most common adverse events associated with Recarbrio are increased alanine aminotransferase/ aspartate aminotransferase, anemia, diarrhea, hypokalemia, and hyponatremia. Recarbrio was previously approved by the FDA to treat patients with complicated urinary tract infections and complicated intra-abdominal infections who have limited or no alternative treatment options, according to an FDA press release.
“As a public health agency, the FDA addresses the threat of antimicrobial-resistant infections by facilitating the development of safe and effective new treatments. These efforts provide more options to fight serious bacterial infections and get new, safe and effective therapies to patients as soon as possible,” said Sumathi Nambiar, MD, MPH, director of the division of anti-infectives within the office of infectious disease at the Center for Drug Evaluation and Research.
Americans avoided emergency departments early in the pandemic
compared with the corresponding period in 2019, according to a report from the Centers for Disease Control and Prevention.
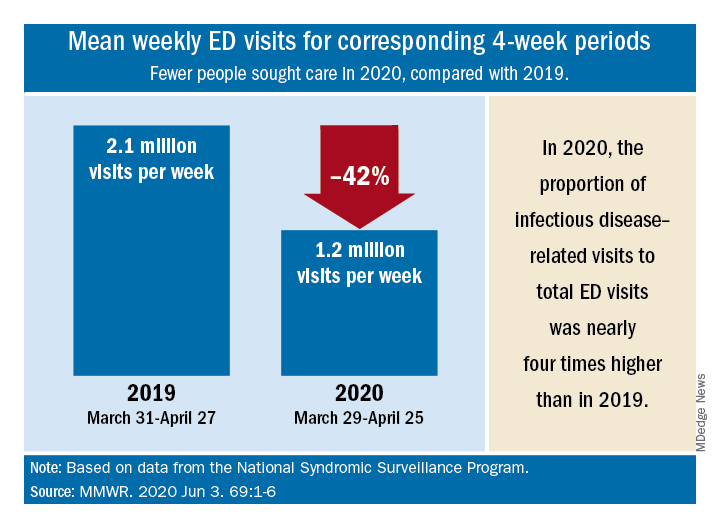
“The striking decline in ED visits nationwide … suggests that the pandemic has altered the use of the ED by the public,” Kathleen P. Hartnett, PhD, and associates at the CDC said June 3 in the Mortality and Morbidity Weekly Report.
The weekly mean was just over 1.2 million ED visits for the 4 weeks from March 29 to April 25, 2020, compared with the nearly 2.2 million visits per week recorded from March 31 to April 27, 2019 – a drop of 42%, based on an analysis of data from the National Syndromic Surveillance Program.
Despite that drop, ED visits for infectious disease–related reasons, taken as a proportion of all 1.2 ED visits during the early pandemic period, were 3.8 times higher than the comparison period in 2019, the investigators reported.
ED visits also were higher in 2020 for specified and unspecified lower respiratory disease not including influenza, pneumonia, asthma, or bronchitis (prevalence ratio of 1.99, compared with 2019), cardiac arrest and ventricular fibrillation (PR, 1.98), and pneumonia not caused by tuberculosis (PR, 1.91), Dr. Hartnett and associates said.
Prevalence ratios for the early pandemic period were down for most other conditions, with some of the largest decreases seen for influenza (PR, 0.16), otitis media (PR, 0.35), and neoplasm-related encounters (PR, 0.40), they said.
Visits have increased each week since reaching their lowest point during April 12-18, but the number for the most recent full week, May 24-30, which was not included in the analysis, was still 26% lower than the corresponding week in 2019, the CDC team pointed out.
“Some persons could be delaying care for conditions that might result in additional mortality if left untreated,” the investigators noted, and those “who use the ED as a safety net because they lack access to primary care and telemedicine might be disproportionately affected if they avoid seeking care because of concerns about the infection risk in the ED.”
SOURCE: Hartnett KP et al. MMWR. 2020 Jun 3. 69:1-6.
compared with the corresponding period in 2019, according to a report from the Centers for Disease Control and Prevention.
“The striking decline in ED visits nationwide … suggests that the pandemic has altered the use of the ED by the public,” Kathleen P. Hartnett, PhD, and associates at the CDC said June 3 in the Mortality and Morbidity Weekly Report.
The weekly mean was just over 1.2 million ED visits for the 4 weeks from March 29 to April 25, 2020, compared with the nearly 2.2 million visits per week recorded from March 31 to April 27, 2019 – a drop of 42%, based on an analysis of data from the National Syndromic Surveillance Program.
Despite that drop, ED visits for infectious disease–related reasons, taken as a proportion of all 1.2 ED visits during the early pandemic period, were 3.8 times higher than the comparison period in 2019, the investigators reported.
ED visits also were higher in 2020 for specified and unspecified lower respiratory disease not including influenza, pneumonia, asthma, or bronchitis (prevalence ratio of 1.99, compared with 2019), cardiac arrest and ventricular fibrillation (PR, 1.98), and pneumonia not caused by tuberculosis (PR, 1.91), Dr. Hartnett and associates said.
Prevalence ratios for the early pandemic period were down for most other conditions, with some of the largest decreases seen for influenza (PR, 0.16), otitis media (PR, 0.35), and neoplasm-related encounters (PR, 0.40), they said.
Visits have increased each week since reaching their lowest point during April 12-18, but the number for the most recent full week, May 24-30, which was not included in the analysis, was still 26% lower than the corresponding week in 2019, the CDC team pointed out.
“Some persons could be delaying care for conditions that might result in additional mortality if left untreated,” the investigators noted, and those “who use the ED as a safety net because they lack access to primary care and telemedicine might be disproportionately affected if they avoid seeking care because of concerns about the infection risk in the ED.”
SOURCE: Hartnett KP et al. MMWR. 2020 Jun 3. 69:1-6.
compared with the corresponding period in 2019, according to a report from the Centers for Disease Control and Prevention.
“The striking decline in ED visits nationwide … suggests that the pandemic has altered the use of the ED by the public,” Kathleen P. Hartnett, PhD, and associates at the CDC said June 3 in the Mortality and Morbidity Weekly Report.
The weekly mean was just over 1.2 million ED visits for the 4 weeks from March 29 to April 25, 2020, compared with the nearly 2.2 million visits per week recorded from March 31 to April 27, 2019 – a drop of 42%, based on an analysis of data from the National Syndromic Surveillance Program.
Despite that drop, ED visits for infectious disease–related reasons, taken as a proportion of all 1.2 ED visits during the early pandemic period, were 3.8 times higher than the comparison period in 2019, the investigators reported.
ED visits also were higher in 2020 for specified and unspecified lower respiratory disease not including influenza, pneumonia, asthma, or bronchitis (prevalence ratio of 1.99, compared with 2019), cardiac arrest and ventricular fibrillation (PR, 1.98), and pneumonia not caused by tuberculosis (PR, 1.91), Dr. Hartnett and associates said.
Prevalence ratios for the early pandemic period were down for most other conditions, with some of the largest decreases seen for influenza (PR, 0.16), otitis media (PR, 0.35), and neoplasm-related encounters (PR, 0.40), they said.
Visits have increased each week since reaching their lowest point during April 12-18, but the number for the most recent full week, May 24-30, which was not included in the analysis, was still 26% lower than the corresponding week in 2019, the CDC team pointed out.
“Some persons could be delaying care for conditions that might result in additional mortality if left untreated,” the investigators noted, and those “who use the ED as a safety net because they lack access to primary care and telemedicine might be disproportionately affected if they avoid seeking care because of concerns about the infection risk in the ED.”
SOURCE: Hartnett KP et al. MMWR. 2020 Jun 3. 69:1-6.
FROM MMWR
FDA approves ramucirumab-erlotinib combo for metastatic NSCLC
The approval was supported by results from the phase 3 RELAY trial (Lancet Oncol. 2019 Dec;20[12]:1655-69). The trial enrolled 449 patients with previously untreated, EGFR-mutated, metastatic NSCLC.
Patients received either ramucirumab at 10 mg/kg or placebo every 2 weeks as an intravenous infusion in combination with erlotinib at 150 mg orally once daily. Patients continued treatment until they progressed or developed unacceptable toxicity. The median progression-free survival was 19.4 months in the ramucirumab-erlotinib arm, compared with 12.4 months in the placebo-erlotinib arm (hazard ratio, 0.59; 95% confidence interval, 0.46-0.76; P < .0001). The overall response rate was 76% in the ramucirumab arm and 75% in the placebo arm. The median duration of response was 18.0 months and 11.1 months, respectively. Overall survival data were not mature at the final analysis.
Adverse events that were more common in the ramucirumab arm were infections, hypertension, stomatitis, proteinuria, alopecia, epistaxis, and peripheral edema. Full prescribing information is available on the FDA website.
The approval was supported by results from the phase 3 RELAY trial (Lancet Oncol. 2019 Dec;20[12]:1655-69). The trial enrolled 449 patients with previously untreated, EGFR-mutated, metastatic NSCLC.
Patients received either ramucirumab at 10 mg/kg or placebo every 2 weeks as an intravenous infusion in combination with erlotinib at 150 mg orally once daily. Patients continued treatment until they progressed or developed unacceptable toxicity. The median progression-free survival was 19.4 months in the ramucirumab-erlotinib arm, compared with 12.4 months in the placebo-erlotinib arm (hazard ratio, 0.59; 95% confidence interval, 0.46-0.76; P < .0001). The overall response rate was 76% in the ramucirumab arm and 75% in the placebo arm. The median duration of response was 18.0 months and 11.1 months, respectively. Overall survival data were not mature at the final analysis.
Adverse events that were more common in the ramucirumab arm were infections, hypertension, stomatitis, proteinuria, alopecia, epistaxis, and peripheral edema. Full prescribing information is available on the FDA website.
The approval was supported by results from the phase 3 RELAY trial (Lancet Oncol. 2019 Dec;20[12]:1655-69). The trial enrolled 449 patients with previously untreated, EGFR-mutated, metastatic NSCLC.
Patients received either ramucirumab at 10 mg/kg or placebo every 2 weeks as an intravenous infusion in combination with erlotinib at 150 mg orally once daily. Patients continued treatment until they progressed or developed unacceptable toxicity. The median progression-free survival was 19.4 months in the ramucirumab-erlotinib arm, compared with 12.4 months in the placebo-erlotinib arm (hazard ratio, 0.59; 95% confidence interval, 0.46-0.76; P < .0001). The overall response rate was 76% in the ramucirumab arm and 75% in the placebo arm. The median duration of response was 18.0 months and 11.1 months, respectively. Overall survival data were not mature at the final analysis.
Adverse events that were more common in the ramucirumab arm were infections, hypertension, stomatitis, proteinuria, alopecia, epistaxis, and peripheral edema. Full prescribing information is available on the FDA website.
FDA approves mAb combo for hepatocellular carcinoma
The approval was supported by results from the IMbrave150 trial (N Engl J Med. 2020;382:1894-1905). This phase 3 trial enrolled 501 patients with hepatocellular carcinoma who were randomized to receive either sorafenib or atezolizumab plus bevacizumab.
The median overall survival was not reached in patients who received atezolizumab plus bevacizumab, but it was 13.2 months in patients who received sorafenib (hazard ratio, 0.58; 95% confidence interval, 0.42-0.79; P = .0006). The median progression-free survival was 6.8 months in patients who received atezolizumab plus bevacizumab and 4.3 months for those who received sorafenib.
The most common adverse events seen in the atezolizumab-bevacizumab arm were hypertension, fatigue, and proteinuria.
The recommended atezolizumab dose is 1,200 mg, followed by 15 mg/kg bevacizumab on the same day every 3 weeks.
The FDA collaborated with regulatory agencies from Canada, Australia, and Singapore on the review of the atezolizumab application, as part of Project Orbis. The FDA approved the application ahead of schedule. It is still under review for the other agencies.
The approval was supported by results from the IMbrave150 trial (N Engl J Med. 2020;382:1894-1905). This phase 3 trial enrolled 501 patients with hepatocellular carcinoma who were randomized to receive either sorafenib or atezolizumab plus bevacizumab.
The median overall survival was not reached in patients who received atezolizumab plus bevacizumab, but it was 13.2 months in patients who received sorafenib (hazard ratio, 0.58; 95% confidence interval, 0.42-0.79; P = .0006). The median progression-free survival was 6.8 months in patients who received atezolizumab plus bevacizumab and 4.3 months for those who received sorafenib.
The most common adverse events seen in the atezolizumab-bevacizumab arm were hypertension, fatigue, and proteinuria.
The recommended atezolizumab dose is 1,200 mg, followed by 15 mg/kg bevacizumab on the same day every 3 weeks.
The FDA collaborated with regulatory agencies from Canada, Australia, and Singapore on the review of the atezolizumab application, as part of Project Orbis. The FDA approved the application ahead of schedule. It is still under review for the other agencies.
The approval was supported by results from the IMbrave150 trial (N Engl J Med. 2020;382:1894-1905). This phase 3 trial enrolled 501 patients with hepatocellular carcinoma who were randomized to receive either sorafenib or atezolizumab plus bevacizumab.
The median overall survival was not reached in patients who received atezolizumab plus bevacizumab, but it was 13.2 months in patients who received sorafenib (hazard ratio, 0.58; 95% confidence interval, 0.42-0.79; P = .0006). The median progression-free survival was 6.8 months in patients who received atezolizumab plus bevacizumab and 4.3 months for those who received sorafenib.
The most common adverse events seen in the atezolizumab-bevacizumab arm were hypertension, fatigue, and proteinuria.
The recommended atezolizumab dose is 1,200 mg, followed by 15 mg/kg bevacizumab on the same day every 3 weeks.
The FDA collaborated with regulatory agencies from Canada, Australia, and Singapore on the review of the atezolizumab application, as part of Project Orbis. The FDA approved the application ahead of schedule. It is still under review for the other agencies.
FDA approves ixekizumab for nonradiographic axSpA
The Food and Drug Administration has extended approval of ixekizumab (Taltz) to the treatment of nonradiographic axial spondyloarthritis (nr-axSpA), according to a press release from its manufacturer, Eli Lilly. Specifically, this supplemental biologics license application refers to nr-axSpA with objective signs of inflammation.
The monoclonal interleukin-17A antagonist has three other indications, including ankylosing spondylitis in adults, psoriatic arthritis in adults, and plaque psoriasis in adults and children aged 6 years and older. It is the first IL-17A antagonist to receive FDA approval for nr-axSpA.
Approval for this indication was based on the phase 3, randomized, double-blind COAST-X trial, which put 96 nr-axSpA patients on 80-mg injections of ixekizumab every 4 weeks and 105 on placebo. After 52 weeks, ixekizumab was superior on the trial’s primary endpoint: 30% of patients had achieved a 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS 40), compared with 13% of patients on placebo (P = .0045).
Warnings and precautions for ixekizumab include considering potentially increased risk of infection and inflammatory bowel disease, as well as evaluating patients for tuberculosis before treatment. The most common adverse reactions (≥1%) are injection-site reactions, upper respiratory tract infections, nausea, and tinea infections. The safety profile for ixekizumab among nr-axSpA patients is mostly consistent with that seen among patients receiving it for other indications, according to Lilly. The full prescribing information is available on Lilly’s website.
The Food and Drug Administration has extended approval of ixekizumab (Taltz) to the treatment of nonradiographic axial spondyloarthritis (nr-axSpA), according to a press release from its manufacturer, Eli Lilly. Specifically, this supplemental biologics license application refers to nr-axSpA with objective signs of inflammation.
The monoclonal interleukin-17A antagonist has three other indications, including ankylosing spondylitis in adults, psoriatic arthritis in adults, and plaque psoriasis in adults and children aged 6 years and older. It is the first IL-17A antagonist to receive FDA approval for nr-axSpA.
Approval for this indication was based on the phase 3, randomized, double-blind COAST-X trial, which put 96 nr-axSpA patients on 80-mg injections of ixekizumab every 4 weeks and 105 on placebo. After 52 weeks, ixekizumab was superior on the trial’s primary endpoint: 30% of patients had achieved a 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS 40), compared with 13% of patients on placebo (P = .0045).
Warnings and precautions for ixekizumab include considering potentially increased risk of infection and inflammatory bowel disease, as well as evaluating patients for tuberculosis before treatment. The most common adverse reactions (≥1%) are injection-site reactions, upper respiratory tract infections, nausea, and tinea infections. The safety profile for ixekizumab among nr-axSpA patients is mostly consistent with that seen among patients receiving it for other indications, according to Lilly. The full prescribing information is available on Lilly’s website.
The Food and Drug Administration has extended approval of ixekizumab (Taltz) to the treatment of nonradiographic axial spondyloarthritis (nr-axSpA), according to a press release from its manufacturer, Eli Lilly. Specifically, this supplemental biologics license application refers to nr-axSpA with objective signs of inflammation.
The monoclonal interleukin-17A antagonist has three other indications, including ankylosing spondylitis in adults, psoriatic arthritis in adults, and plaque psoriasis in adults and children aged 6 years and older. It is the first IL-17A antagonist to receive FDA approval for nr-axSpA.
Approval for this indication was based on the phase 3, randomized, double-blind COAST-X trial, which put 96 nr-axSpA patients on 80-mg injections of ixekizumab every 4 weeks and 105 on placebo. After 52 weeks, ixekizumab was superior on the trial’s primary endpoint: 30% of patients had achieved a 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS 40), compared with 13% of patients on placebo (P = .0045).
Warnings and precautions for ixekizumab include considering potentially increased risk of infection and inflammatory bowel disease, as well as evaluating patients for tuberculosis before treatment. The most common adverse reactions (≥1%) are injection-site reactions, upper respiratory tract infections, nausea, and tinea infections. The safety profile for ixekizumab among nr-axSpA patients is mostly consistent with that seen among patients receiving it for other indications, according to Lilly. The full prescribing information is available on Lilly’s website.
Frontline nivo-ipi plus chemo approved for metastatic NSCLC
The Food and Drug Administration has approved the combination of nivolumab (Opdivo), ipilimumab (Yervoy), and two cycles of platinum-doublet chemotherapy as frontline treatment for patients with metastatic or recurrent non–small cell lung cancer (NSCLC) who have no EGFR or ALK genomic tumor aberrations.
The FDA collaborated with the Australian Therapeutic Goods Administration, Health Canada, and Singapore’s Health Sciences Authority on the review that led to this approval, as part of Project Orbis. The FDA approved the application 2 months ahead of schedule.
The combination chemotherapy was investigated in the CHECKMATE-9LA trial (NCT03215706), which enrolled patients with metastatic or recurrent NSCLC.
Patients were randomized to receive nivolumab plus ipilimumab and two cycles of platinum-doublet chemotherapy (n = 361) or platinum-doublet chemotherapy for four cycles (n = 358).
There was a significant overall survival benefit in the nivolumab-ipilimumab arm, compared with the chemotherapy-only arm. The median overall survival was 14.1 months and 10.7 months, respectively (hazard ratio, 0.69; P = .0006).
The median progression-free survival was 6.8 months in the nivolumab-ipilimumab arm and 5 months in the chemotherapy-only arm (HR, 0.70; P = .0001). The overall response rate was 38% and 25%, respectively (P = .0003).
The most common adverse events in the nivolumab-ipilimumab arm, which occurred in at least 20% of patients, were fatigue, musculoskeletal pain, nausea, diarrhea, rash, decreased appetite, constipation, and pruritus.
Serious adverse events occurred in 57% of patients in the nivolumab-ipilimumab arm. Fatal adverse events occurred in seven patients (2%) in that arm. Fatal events were hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
For more details, see the full prescribing information for nivolumab or ipilimumab. Nivolumab and ipilimumab are both products of Bristol-Myers Squibb.
The Food and Drug Administration has approved the combination of nivolumab (Opdivo), ipilimumab (Yervoy), and two cycles of platinum-doublet chemotherapy as frontline treatment for patients with metastatic or recurrent non–small cell lung cancer (NSCLC) who have no EGFR or ALK genomic tumor aberrations.
The FDA collaborated with the Australian Therapeutic Goods Administration, Health Canada, and Singapore’s Health Sciences Authority on the review that led to this approval, as part of Project Orbis. The FDA approved the application 2 months ahead of schedule.
The combination chemotherapy was investigated in the CHECKMATE-9LA trial (NCT03215706), which enrolled patients with metastatic or recurrent NSCLC.
Patients were randomized to receive nivolumab plus ipilimumab and two cycles of platinum-doublet chemotherapy (n = 361) or platinum-doublet chemotherapy for four cycles (n = 358).
There was a significant overall survival benefit in the nivolumab-ipilimumab arm, compared with the chemotherapy-only arm. The median overall survival was 14.1 months and 10.7 months, respectively (hazard ratio, 0.69; P = .0006).
The median progression-free survival was 6.8 months in the nivolumab-ipilimumab arm and 5 months in the chemotherapy-only arm (HR, 0.70; P = .0001). The overall response rate was 38% and 25%, respectively (P = .0003).
The most common adverse events in the nivolumab-ipilimumab arm, which occurred in at least 20% of patients, were fatigue, musculoskeletal pain, nausea, diarrhea, rash, decreased appetite, constipation, and pruritus.
Serious adverse events occurred in 57% of patients in the nivolumab-ipilimumab arm. Fatal adverse events occurred in seven patients (2%) in that arm. Fatal events were hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
For more details, see the full prescribing information for nivolumab or ipilimumab. Nivolumab and ipilimumab are both products of Bristol-Myers Squibb.
The Food and Drug Administration has approved the combination of nivolumab (Opdivo), ipilimumab (Yervoy), and two cycles of platinum-doublet chemotherapy as frontline treatment for patients with metastatic or recurrent non–small cell lung cancer (NSCLC) who have no EGFR or ALK genomic tumor aberrations.
The FDA collaborated with the Australian Therapeutic Goods Administration, Health Canada, and Singapore’s Health Sciences Authority on the review that led to this approval, as part of Project Orbis. The FDA approved the application 2 months ahead of schedule.
The combination chemotherapy was investigated in the CHECKMATE-9LA trial (NCT03215706), which enrolled patients with metastatic or recurrent NSCLC.
Patients were randomized to receive nivolumab plus ipilimumab and two cycles of platinum-doublet chemotherapy (n = 361) or platinum-doublet chemotherapy for four cycles (n = 358).
There was a significant overall survival benefit in the nivolumab-ipilimumab arm, compared with the chemotherapy-only arm. The median overall survival was 14.1 months and 10.7 months, respectively (hazard ratio, 0.69; P = .0006).
The median progression-free survival was 6.8 months in the nivolumab-ipilimumab arm and 5 months in the chemotherapy-only arm (HR, 0.70; P = .0001). The overall response rate was 38% and 25%, respectively (P = .0003).
The most common adverse events in the nivolumab-ipilimumab arm, which occurred in at least 20% of patients, were fatigue, musculoskeletal pain, nausea, diarrhea, rash, decreased appetite, constipation, and pruritus.
Serious adverse events occurred in 57% of patients in the nivolumab-ipilimumab arm. Fatal adverse events occurred in seven patients (2%) in that arm. Fatal events were hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
For more details, see the full prescribing information for nivolumab or ipilimumab. Nivolumab and ipilimumab are both products of Bristol-Myers Squibb.