User login
Flu now riding on COVID-19’s coattails
The viral tsunami that is COVID-19 has hit the United States, and influenza appears to be riding the crest of the wave.
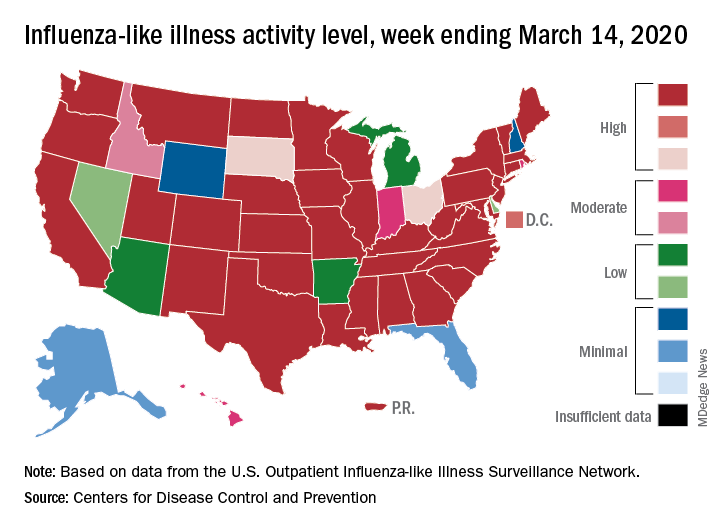
according to the Centers for Disease Control. Flu-related visits went from 5.2% of all outpatient visits the week before to 5.8% during the week ending March 14.
“The COVID-19 outbreak unfolding in the United States may affect healthcare seeking behavior which in turn would impact data from” the U.S. Outpatient Influenza-like Illness Surveillance Network, the CDC explained.
Data from clinical laboratories show that, despite the increased activity, fewer respiratory specimens tested positive for influenza: 15.3% for the week of March 8-14, compared with 21.1% the week before, the CDC’s influenza division said in its latest FluView report.
Influenza activity also increased slightly among the states, with 35 states and Puerto Rico at the highest level on the CDC’s 1-10 scale, versus 34 states and Puerto Rico the previous week. The count was down to 33 for the last week of February, CDC data show.
Severity measures remain mixed as overall hospitalization continues to be moderate but rates for children aged 0-4 years and adults aged 18-49 years are the highest on record and rates for children aged 5-17 years are the highest since the 2009 pandemic, the influenza division said.
Mortality data present a similar picture: The overall death rate is low, but the 149 flu-related deaths reported among children is the most for this point of the season since 2009, the CDC said.
The viral tsunami that is COVID-19 has hit the United States, and influenza appears to be riding the crest of the wave.
according to the Centers for Disease Control. Flu-related visits went from 5.2% of all outpatient visits the week before to 5.8% during the week ending March 14.
“The COVID-19 outbreak unfolding in the United States may affect healthcare seeking behavior which in turn would impact data from” the U.S. Outpatient Influenza-like Illness Surveillance Network, the CDC explained.
Data from clinical laboratories show that, despite the increased activity, fewer respiratory specimens tested positive for influenza: 15.3% for the week of March 8-14, compared with 21.1% the week before, the CDC’s influenza division said in its latest FluView report.
Influenza activity also increased slightly among the states, with 35 states and Puerto Rico at the highest level on the CDC’s 1-10 scale, versus 34 states and Puerto Rico the previous week. The count was down to 33 for the last week of February, CDC data show.
Severity measures remain mixed as overall hospitalization continues to be moderate but rates for children aged 0-4 years and adults aged 18-49 years are the highest on record and rates for children aged 5-17 years are the highest since the 2009 pandemic, the influenza division said.
Mortality data present a similar picture: The overall death rate is low, but the 149 flu-related deaths reported among children is the most for this point of the season since 2009, the CDC said.
The viral tsunami that is COVID-19 has hit the United States, and influenza appears to be riding the crest of the wave.
according to the Centers for Disease Control. Flu-related visits went from 5.2% of all outpatient visits the week before to 5.8% during the week ending March 14.
“The COVID-19 outbreak unfolding in the United States may affect healthcare seeking behavior which in turn would impact data from” the U.S. Outpatient Influenza-like Illness Surveillance Network, the CDC explained.
Data from clinical laboratories show that, despite the increased activity, fewer respiratory specimens tested positive for influenza: 15.3% for the week of March 8-14, compared with 21.1% the week before, the CDC’s influenza division said in its latest FluView report.
Influenza activity also increased slightly among the states, with 35 states and Puerto Rico at the highest level on the CDC’s 1-10 scale, versus 34 states and Puerto Rico the previous week. The count was down to 33 for the last week of February, CDC data show.
Severity measures remain mixed as overall hospitalization continues to be moderate but rates for children aged 0-4 years and adults aged 18-49 years are the highest on record and rates for children aged 5-17 years are the highest since the 2009 pandemic, the influenza division said.
Mortality data present a similar picture: The overall death rate is low, but the 149 flu-related deaths reported among children is the most for this point of the season since 2009, the CDC said.
20% of U.S. COVID-19 deaths were aged 20-64 years
*Correction, 3/20/2020: An earlier version of this story misstated the age range for COVID-19 deaths. The headline of this story was corrected to read "20% of COVID-19 deaths were aged 20-64 years" and the text was adjusted to reflect the correct age range.
A review of more than 4,000 U.S. patients who were diagnosed with novel coronavirus infection (COVID-19) shows that an unexpected 20% of deaths occurred among adults aged 20-64 years, and 20% of those hospitalized were aged 20-44 years.
The expectation has been that people over 65 are most vulnerable to COVID-19 infection, but this study indicates that, at least in the United States, a significant number of patients under 45 can land in the hospital and can even die of the disease.
To assess rates of hospitalization, admission to an ICU, and death among patients with COVID-19 by age group, the Centers for Disease Control and Prevention analyzed 4,226 COVID-19 cases in the United States that were reported between Feb. 12 and March 16.
Overall, older patients in this group were the most likely to be hospitalized, to be admitted to ICU, and to die of COVID-19. A total of 31% of the cases, 45% of hospitalizations, 53% of ICU admissions, and 80% of deaths occurred in patients aged 65 years and older. “Similar to reports from other countries, this finding suggests that the risk for serious disease and death from COVID-19 is higher in older age groups,” said the investigators. “In contrast, persons aged [19 years and younger] appear to have milder COVID-19 illness, with almost no hospitalizations or deaths reported to date in the United States in this age group.”
But compared with the under-19 group, patients aged 20-44 years appeared to be at higher risk for hospitalization and ICU admission, according to the data published March 18 in Morbidity and Mortality Weekly Report.
The researchers excluded from their analysis patients who repatriated to the United States from Wuhan, China, and from Japan, including patients repatriated from cruise ships. Data on serious underlying health conditions were not available, and many cases were missing key data, they noted.
Among 508 patients known to have been hospitalized, 9% were aged 85 years or older, 36% were aged 65-84 years, 17% were aged 55-64 years, 18% were 45-54 years, and 20% were aged 20-44 years.
Among 121 patients admitted to an ICU, 7% were aged 85 years or older, 46% were aged 65-84 years, 36% were aged 45-64 years, and 12% were aged 20-44 years. Between 11% and 31% of patients with COVID-19 aged 75-84 years were admitted to an ICU.
Of 44 deaths, more than a third occurred among adults aged 85 years and older, and 46% occurred among adults aged 65-84 years, and 20% occurred among adults aged 20-64 years.
More follow-up time is needed to determine outcomes among active cases, the researchers said. These results also might overestimate the prevalence of severe disease because the initial approach to testing for COVID-19 focused on people with more severe disease. “These preliminary data also demonstrate that severe illness leading to hospitalization, including ICU admission and death, can occur in adults of any age with COVID-19,” according to the CDC.
SOURCE: CDC COVID-19 Response Team. MMWR Morb Mortal Wkly Rep. 2020 Mar 18. doi: 10.15585/mmwr.mm6912e2.
*Correction, 3/20/2020: An earlier version of this story misstated the age range for COVID-19 deaths. The headline of this story was corrected to read "20% of COVID-19 deaths were aged 20-64 years" and the text was adjusted to reflect the correct age range.
A review of more than 4,000 U.S. patients who were diagnosed with novel coronavirus infection (COVID-19) shows that an unexpected 20% of deaths occurred among adults aged 20-64 years, and 20% of those hospitalized were aged 20-44 years.
The expectation has been that people over 65 are most vulnerable to COVID-19 infection, but this study indicates that, at least in the United States, a significant number of patients under 45 can land in the hospital and can even die of the disease.
To assess rates of hospitalization, admission to an ICU, and death among patients with COVID-19 by age group, the Centers for Disease Control and Prevention analyzed 4,226 COVID-19 cases in the United States that were reported between Feb. 12 and March 16.
Overall, older patients in this group were the most likely to be hospitalized, to be admitted to ICU, and to die of COVID-19. A total of 31% of the cases, 45% of hospitalizations, 53% of ICU admissions, and 80% of deaths occurred in patients aged 65 years and older. “Similar to reports from other countries, this finding suggests that the risk for serious disease and death from COVID-19 is higher in older age groups,” said the investigators. “In contrast, persons aged [19 years and younger] appear to have milder COVID-19 illness, with almost no hospitalizations or deaths reported to date in the United States in this age group.”
But compared with the under-19 group, patients aged 20-44 years appeared to be at higher risk for hospitalization and ICU admission, according to the data published March 18 in Morbidity and Mortality Weekly Report.
The researchers excluded from their analysis patients who repatriated to the United States from Wuhan, China, and from Japan, including patients repatriated from cruise ships. Data on serious underlying health conditions were not available, and many cases were missing key data, they noted.
Among 508 patients known to have been hospitalized, 9% were aged 85 years or older, 36% were aged 65-84 years, 17% were aged 55-64 years, 18% were 45-54 years, and 20% were aged 20-44 years.
Among 121 patients admitted to an ICU, 7% were aged 85 years or older, 46% were aged 65-84 years, 36% were aged 45-64 years, and 12% were aged 20-44 years. Between 11% and 31% of patients with COVID-19 aged 75-84 years were admitted to an ICU.
Of 44 deaths, more than a third occurred among adults aged 85 years and older, and 46% occurred among adults aged 65-84 years, and 20% occurred among adults aged 20-64 years.
More follow-up time is needed to determine outcomes among active cases, the researchers said. These results also might overestimate the prevalence of severe disease because the initial approach to testing for COVID-19 focused on people with more severe disease. “These preliminary data also demonstrate that severe illness leading to hospitalization, including ICU admission and death, can occur in adults of any age with COVID-19,” according to the CDC.
SOURCE: CDC COVID-19 Response Team. MMWR Morb Mortal Wkly Rep. 2020 Mar 18. doi: 10.15585/mmwr.mm6912e2.
*Correction, 3/20/2020: An earlier version of this story misstated the age range for COVID-19 deaths. The headline of this story was corrected to read "20% of COVID-19 deaths were aged 20-64 years" and the text was adjusted to reflect the correct age range.
A review of more than 4,000 U.S. patients who were diagnosed with novel coronavirus infection (COVID-19) shows that an unexpected 20% of deaths occurred among adults aged 20-64 years, and 20% of those hospitalized were aged 20-44 years.
The expectation has been that people over 65 are most vulnerable to COVID-19 infection, but this study indicates that, at least in the United States, a significant number of patients under 45 can land in the hospital and can even die of the disease.
To assess rates of hospitalization, admission to an ICU, and death among patients with COVID-19 by age group, the Centers for Disease Control and Prevention analyzed 4,226 COVID-19 cases in the United States that were reported between Feb. 12 and March 16.
Overall, older patients in this group were the most likely to be hospitalized, to be admitted to ICU, and to die of COVID-19. A total of 31% of the cases, 45% of hospitalizations, 53% of ICU admissions, and 80% of deaths occurred in patients aged 65 years and older. “Similar to reports from other countries, this finding suggests that the risk for serious disease and death from COVID-19 is higher in older age groups,” said the investigators. “In contrast, persons aged [19 years and younger] appear to have milder COVID-19 illness, with almost no hospitalizations or deaths reported to date in the United States in this age group.”
But compared with the under-19 group, patients aged 20-44 years appeared to be at higher risk for hospitalization and ICU admission, according to the data published March 18 in Morbidity and Mortality Weekly Report.
The researchers excluded from their analysis patients who repatriated to the United States from Wuhan, China, and from Japan, including patients repatriated from cruise ships. Data on serious underlying health conditions were not available, and many cases were missing key data, they noted.
Among 508 patients known to have been hospitalized, 9% were aged 85 years or older, 36% were aged 65-84 years, 17% were aged 55-64 years, 18% were 45-54 years, and 20% were aged 20-44 years.
Among 121 patients admitted to an ICU, 7% were aged 85 years or older, 46% were aged 65-84 years, 36% were aged 45-64 years, and 12% were aged 20-44 years. Between 11% and 31% of patients with COVID-19 aged 75-84 years were admitted to an ICU.
Of 44 deaths, more than a third occurred among adults aged 85 years and older, and 46% occurred among adults aged 65-84 years, and 20% occurred among adults aged 20-64 years.
More follow-up time is needed to determine outcomes among active cases, the researchers said. These results also might overestimate the prevalence of severe disease because the initial approach to testing for COVID-19 focused on people with more severe disease. “These preliminary data also demonstrate that severe illness leading to hospitalization, including ICU admission and death, can occur in adults of any age with COVID-19,” according to the CDC.
SOURCE: CDC COVID-19 Response Team. MMWR Morb Mortal Wkly Rep. 2020 Mar 18. doi: 10.15585/mmwr.mm6912e2.
FDA advises stopping SGLT2 inhibitor treatment prior to surgery
The new changes affect canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin, and were made because surgery may put patients being treated with SGLT2 inhibitors at a higher risk of ketoacidosis. Canagliflozin, dapagliflozin, and empagliflozin should be discontinued 3 days before scheduled surgery, and ertugliflozin should be stopped at least 4 days before, the agency noted in a press release. Blood glucose should be monitored after drug discontinuation and appropriately managed before surgery.
“The SGLT2 inhibitor may be restarted once the patient’s oral intake is back to baseline and any other risk factors for ketoacidosis are resolved,” the agency added.
SGLT2 inhibitors lower blood sugar by causing the kidney to remove sugar from the body through urine. Side effects for the drugs vary, but include urinary tract infections and genital mycotic infection. Patients with severe renal impairment or end-stage renal disease, who are on dialysis treatment, or who have a known hypersensitivity to the medication should not take SGLT2 inhibitors, the FDA said.
The new changes affect canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin, and were made because surgery may put patients being treated with SGLT2 inhibitors at a higher risk of ketoacidosis. Canagliflozin, dapagliflozin, and empagliflozin should be discontinued 3 days before scheduled surgery, and ertugliflozin should be stopped at least 4 days before, the agency noted in a press release. Blood glucose should be monitored after drug discontinuation and appropriately managed before surgery.
“The SGLT2 inhibitor may be restarted once the patient’s oral intake is back to baseline and any other risk factors for ketoacidosis are resolved,” the agency added.
SGLT2 inhibitors lower blood sugar by causing the kidney to remove sugar from the body through urine. Side effects for the drugs vary, but include urinary tract infections and genital mycotic infection. Patients with severe renal impairment or end-stage renal disease, who are on dialysis treatment, or who have a known hypersensitivity to the medication should not take SGLT2 inhibitors, the FDA said.
The new changes affect canagliflozin, dapagliflozin, empagliflozin, and ertugliflozin, and were made because surgery may put patients being treated with SGLT2 inhibitors at a higher risk of ketoacidosis. Canagliflozin, dapagliflozin, and empagliflozin should be discontinued 3 days before scheduled surgery, and ertugliflozin should be stopped at least 4 days before, the agency noted in a press release. Blood glucose should be monitored after drug discontinuation and appropriately managed before surgery.
“The SGLT2 inhibitor may be restarted once the patient’s oral intake is back to baseline and any other risk factors for ketoacidosis are resolved,” the agency added.
SGLT2 inhibitors lower blood sugar by causing the kidney to remove sugar from the body through urine. Side effects for the drugs vary, but include urinary tract infections and genital mycotic infection. Patients with severe renal impairment or end-stage renal disease, who are on dialysis treatment, or who have a known hypersensitivity to the medication should not take SGLT2 inhibitors, the FDA said.
FDA provides flexibility to improve COVID-19 test availability

First, the FDA is giving states more flexibility to approve and implement testing for COVID-19.
“States can set up a system in which they take responsibility for authorizing such tests and the laboratories will not engage with the FDA,” agency Commissioner Stephen Hahn, MD, said in a March 16 statement announcing the policy updates. “Laboratories developing tests in these states can engage directly with the appropriate state authorities, instead of with the FDA.”
A copy of the updated guidance document can be found here.
Dr. Hahn added that laboratories working within this authority granted to states will not have to pursue an emergency use authorization (EUA). New York state was previously granted a waiver to allow for more state oversight over the introduction of diagnostic testing.
Second, the FDA is expanding guidance issued on Feb. 29 on who can develop diagnostic tests. Originally, the Feb. 29 guidance was aimed at labs certified to perform high-complexity testing consistent with requirements outlined in the Clinical Laboratory Improvement Amendments.
“Under the update published today, the agency does not intend to object to commercial manufacturers distributing and labs using new commercially developed tests prior to the FDA granting an EUA, under certain circumstances,” Commissioner Hahn said, adding that a number of commercial manufacturers are developing tests for the coronavirus with the intent of submitting an EUA request.
“During this public health emergency, the FDA does not intend to object to the distribution and use of these tests for specimen testing for a reasonable period of time after the manufacturer’s validation of the test while the manufacturer is preparing its EUA request,” he added.
The updated guidance also provides recommendations for test developers working on serologic tests for COVID-19.
During a March 16 conference call with reporters, Commissioner Hahn said the flexibility would add a “significant number of tests and we believe this will be a surge to meet the demand that we expect to see, although it is somewhat difficult” to quantify the number of tests this new flexibility will bring to the market.
First, the FDA is giving states more flexibility to approve and implement testing for COVID-19.
“States can set up a system in which they take responsibility for authorizing such tests and the laboratories will not engage with the FDA,” agency Commissioner Stephen Hahn, MD, said in a March 16 statement announcing the policy updates. “Laboratories developing tests in these states can engage directly with the appropriate state authorities, instead of with the FDA.”
A copy of the updated guidance document can be found here.
Dr. Hahn added that laboratories working within this authority granted to states will not have to pursue an emergency use authorization (EUA). New York state was previously granted a waiver to allow for more state oversight over the introduction of diagnostic testing.
Second, the FDA is expanding guidance issued on Feb. 29 on who can develop diagnostic tests. Originally, the Feb. 29 guidance was aimed at labs certified to perform high-complexity testing consistent with requirements outlined in the Clinical Laboratory Improvement Amendments.
“Under the update published today, the agency does not intend to object to commercial manufacturers distributing and labs using new commercially developed tests prior to the FDA granting an EUA, under certain circumstances,” Commissioner Hahn said, adding that a number of commercial manufacturers are developing tests for the coronavirus with the intent of submitting an EUA request.
“During this public health emergency, the FDA does not intend to object to the distribution and use of these tests for specimen testing for a reasonable period of time after the manufacturer’s validation of the test while the manufacturer is preparing its EUA request,” he added.
The updated guidance also provides recommendations for test developers working on serologic tests for COVID-19.
During a March 16 conference call with reporters, Commissioner Hahn said the flexibility would add a “significant number of tests and we believe this will be a surge to meet the demand that we expect to see, although it is somewhat difficult” to quantify the number of tests this new flexibility will bring to the market.
First, the FDA is giving states more flexibility to approve and implement testing for COVID-19.
“States can set up a system in which they take responsibility for authorizing such tests and the laboratories will not engage with the FDA,” agency Commissioner Stephen Hahn, MD, said in a March 16 statement announcing the policy updates. “Laboratories developing tests in these states can engage directly with the appropriate state authorities, instead of with the FDA.”
A copy of the updated guidance document can be found here.
Dr. Hahn added that laboratories working within this authority granted to states will not have to pursue an emergency use authorization (EUA). New York state was previously granted a waiver to allow for more state oversight over the introduction of diagnostic testing.
Second, the FDA is expanding guidance issued on Feb. 29 on who can develop diagnostic tests. Originally, the Feb. 29 guidance was aimed at labs certified to perform high-complexity testing consistent with requirements outlined in the Clinical Laboratory Improvement Amendments.
“Under the update published today, the agency does not intend to object to commercial manufacturers distributing and labs using new commercially developed tests prior to the FDA granting an EUA, under certain circumstances,” Commissioner Hahn said, adding that a number of commercial manufacturers are developing tests for the coronavirus with the intent of submitting an EUA request.
“During this public health emergency, the FDA does not intend to object to the distribution and use of these tests for specimen testing for a reasonable period of time after the manufacturer’s validation of the test while the manufacturer is preparing its EUA request,” he added.
The updated guidance also provides recommendations for test developers working on serologic tests for COVID-19.
During a March 16 conference call with reporters, Commissioner Hahn said the flexibility would add a “significant number of tests and we believe this will be a surge to meet the demand that we expect to see, although it is somewhat difficult” to quantify the number of tests this new flexibility will bring to the market.
Trump to governors: Don’t wait for feds on medical supplies
President Donald Trump has advised state governors not to wait on the federal government when it comes to ensuring readiness for a surge in patients from the COVID-19 outbreak.
“If they are able to get ventilators, respirators, if they are able to get certain things without having to go through the longer process of federal government,” they should order on their own and bypass the federal government ordering system, the president stated during a March 16 press briefing.
That being said, he noted that the federal government is “ordering tremendous numbers of ventilators, respirators, [and] masks,” although he could not give a specific number on how much has been ordered or how many has already been stockpiled.
“It is always going to be faster if they can get them directly, if they need them, and I have given them authorization to order directly,” President Trump said.
The comments came as the White House revised recommendations on gatherings. The new guidelines now limit gatherings to no more than 10 people. Officials are further advising Americans to self-quarantine for 2 weeks if they are sick, if someone in their house is sick, or if someone in their house has tested positive for COVID-19.
Additionally, the White House called on Americans to limit discretionary travel and to avoid eating and drinking in restaurants, bars, and food courts during the next 15 days, even if they are feeling healthy and are asymptomatic.
“With several weeks of focused action, we can turn the corner and turn it quickly,” the president said.
In terms of testing, the Food and Drug Administration has granted emergency use authorization to two commercial diagnostic tests: Thermo Fisher for its TaqPath COVID-19 Combo Kit and Roche for its cobas SARS-CoV-2 test. White House officials said up to 1 million tests will be available this week, with 2 million next week.
The president also announced that phase 1 testing of a vaccine has begun. The test involves more than 40 healthy volunteers in the Seattle area who will receive three shots over the trial period. Phase 1 testing is generally conducted to determine safety of a new therapeutic.
President Donald Trump has advised state governors not to wait on the federal government when it comes to ensuring readiness for a surge in patients from the COVID-19 outbreak.
“If they are able to get ventilators, respirators, if they are able to get certain things without having to go through the longer process of federal government,” they should order on their own and bypass the federal government ordering system, the president stated during a March 16 press briefing.
That being said, he noted that the federal government is “ordering tremendous numbers of ventilators, respirators, [and] masks,” although he could not give a specific number on how much has been ordered or how many has already been stockpiled.
“It is always going to be faster if they can get them directly, if they need them, and I have given them authorization to order directly,” President Trump said.
The comments came as the White House revised recommendations on gatherings. The new guidelines now limit gatherings to no more than 10 people. Officials are further advising Americans to self-quarantine for 2 weeks if they are sick, if someone in their house is sick, or if someone in their house has tested positive for COVID-19.
Additionally, the White House called on Americans to limit discretionary travel and to avoid eating and drinking in restaurants, bars, and food courts during the next 15 days, even if they are feeling healthy and are asymptomatic.
“With several weeks of focused action, we can turn the corner and turn it quickly,” the president said.
In terms of testing, the Food and Drug Administration has granted emergency use authorization to two commercial diagnostic tests: Thermo Fisher for its TaqPath COVID-19 Combo Kit and Roche for its cobas SARS-CoV-2 test. White House officials said up to 1 million tests will be available this week, with 2 million next week.
The president also announced that phase 1 testing of a vaccine has begun. The test involves more than 40 healthy volunteers in the Seattle area who will receive three shots over the trial period. Phase 1 testing is generally conducted to determine safety of a new therapeutic.
President Donald Trump has advised state governors not to wait on the federal government when it comes to ensuring readiness for a surge in patients from the COVID-19 outbreak.
“If they are able to get ventilators, respirators, if they are able to get certain things without having to go through the longer process of federal government,” they should order on their own and bypass the federal government ordering system, the president stated during a March 16 press briefing.
That being said, he noted that the federal government is “ordering tremendous numbers of ventilators, respirators, [and] masks,” although he could not give a specific number on how much has been ordered or how many has already been stockpiled.
“It is always going to be faster if they can get them directly, if they need them, and I have given them authorization to order directly,” President Trump said.
The comments came as the White House revised recommendations on gatherings. The new guidelines now limit gatherings to no more than 10 people. Officials are further advising Americans to self-quarantine for 2 weeks if they are sick, if someone in their house is sick, or if someone in their house has tested positive for COVID-19.
Additionally, the White House called on Americans to limit discretionary travel and to avoid eating and drinking in restaurants, bars, and food courts during the next 15 days, even if they are feeling healthy and are asymptomatic.
“With several weeks of focused action, we can turn the corner and turn it quickly,” the president said.
In terms of testing, the Food and Drug Administration has granted emergency use authorization to two commercial diagnostic tests: Thermo Fisher for its TaqPath COVID-19 Combo Kit and Roche for its cobas SARS-CoV-2 test. White House officials said up to 1 million tests will be available this week, with 2 million next week.
The president also announced that phase 1 testing of a vaccine has begun. The test involves more than 40 healthy volunteers in the Seattle area who will receive three shots over the trial period. Phase 1 testing is generally conducted to determine safety of a new therapeutic.
President declares national emergency for COVID-19, ramps up testing capability
President Donald Trump has declared a national emergency to allow for additional resources to combat the COVID-19 pandemic and announced increased testing capacity in partnership with private industry.
During a March 13 press conference, the president said the declaration would “open up access to up to $50 billion” for states and territories in combating the spread of the disease.
He also called on all states to “set up emergency operation centers, effective immediately” and for every hospital “to activate its emergency preparedness plan so that they can meet the needs of Americans everywhere.”
Additionally, he said the declaration will confer broad new authority on the Department of Health & Human Services Secretary Alex Azar that will allow him to “immediately waive provisions of applicable laws and regulations to give doctors, all hospitals, and health care providers maximum flexibility to respond to the virus and care for patients.”
Some of the powers he highlighted included the ability to waive laws to enable telehealth; to waive certain federal license requirements to allow doctors licensed in one state to offer services in other states; the ability to waive limits on beds in critical access hospitals; and to waive rules that hinder hospitals from hiring additional physicians.
The president also announced that more testing capacity will be made available within the next week, in partnership with private industry.
“We want to make sure that those who need a test can get a test very safely, quickly, and conveniently, but we don’t want people to take a test if we feel that they shouldn’t be doing it,” he said.
To help make that determination, a website, developed with Google, is expected to be launched the weekend of March 13 to will allow individuals to input their symptoms and risk factors to help determine if they should be tested. If certain criteria are met, the website will provide locations for drive-through testing facilities. Individuals will be tested using a nasal swab and will receive results within 24-36 hours.
The testing is being done in partnership with retailers, including Target and Walmart (who are providing parking lot space for the pop-up testing facilities) and testing companies LabCorp and Quest Diagnostics.
The new test was developed by Roche and just received emergency use authorization from the Food and Drug Administration.
“We therefore expect up to a half-million additional tests will be available early next week,” President Trump said, adding that testing locations will “probably” be announced on Sunday, March 15.
A second application for a new test, submitted by Thermo Fisher, is currently under review at the FDA and is expected to be approved within the next 24 hours, he said. This would add an additional 1.4 million tests in the next week and 5 million within a month, according to the president.
President Donald Trump has declared a national emergency to allow for additional resources to combat the COVID-19 pandemic and announced increased testing capacity in partnership with private industry.
During a March 13 press conference, the president said the declaration would “open up access to up to $50 billion” for states and territories in combating the spread of the disease.
He also called on all states to “set up emergency operation centers, effective immediately” and for every hospital “to activate its emergency preparedness plan so that they can meet the needs of Americans everywhere.”
Additionally, he said the declaration will confer broad new authority on the Department of Health & Human Services Secretary Alex Azar that will allow him to “immediately waive provisions of applicable laws and regulations to give doctors, all hospitals, and health care providers maximum flexibility to respond to the virus and care for patients.”
Some of the powers he highlighted included the ability to waive laws to enable telehealth; to waive certain federal license requirements to allow doctors licensed in one state to offer services in other states; the ability to waive limits on beds in critical access hospitals; and to waive rules that hinder hospitals from hiring additional physicians.
The president also announced that more testing capacity will be made available within the next week, in partnership with private industry.
“We want to make sure that those who need a test can get a test very safely, quickly, and conveniently, but we don’t want people to take a test if we feel that they shouldn’t be doing it,” he said.
To help make that determination, a website, developed with Google, is expected to be launched the weekend of March 13 to will allow individuals to input their symptoms and risk factors to help determine if they should be tested. If certain criteria are met, the website will provide locations for drive-through testing facilities. Individuals will be tested using a nasal swab and will receive results within 24-36 hours.
The testing is being done in partnership with retailers, including Target and Walmart (who are providing parking lot space for the pop-up testing facilities) and testing companies LabCorp and Quest Diagnostics.
The new test was developed by Roche and just received emergency use authorization from the Food and Drug Administration.
“We therefore expect up to a half-million additional tests will be available early next week,” President Trump said, adding that testing locations will “probably” be announced on Sunday, March 15.
A second application for a new test, submitted by Thermo Fisher, is currently under review at the FDA and is expected to be approved within the next 24 hours, he said. This would add an additional 1.4 million tests in the next week and 5 million within a month, according to the president.
President Donald Trump has declared a national emergency to allow for additional resources to combat the COVID-19 pandemic and announced increased testing capacity in partnership with private industry.
During a March 13 press conference, the president said the declaration would “open up access to up to $50 billion” for states and territories in combating the spread of the disease.
He also called on all states to “set up emergency operation centers, effective immediately” and for every hospital “to activate its emergency preparedness plan so that they can meet the needs of Americans everywhere.”
Additionally, he said the declaration will confer broad new authority on the Department of Health & Human Services Secretary Alex Azar that will allow him to “immediately waive provisions of applicable laws and regulations to give doctors, all hospitals, and health care providers maximum flexibility to respond to the virus and care for patients.”
Some of the powers he highlighted included the ability to waive laws to enable telehealth; to waive certain federal license requirements to allow doctors licensed in one state to offer services in other states; the ability to waive limits on beds in critical access hospitals; and to waive rules that hinder hospitals from hiring additional physicians.
The president also announced that more testing capacity will be made available within the next week, in partnership with private industry.
“We want to make sure that those who need a test can get a test very safely, quickly, and conveniently, but we don’t want people to take a test if we feel that they shouldn’t be doing it,” he said.
To help make that determination, a website, developed with Google, is expected to be launched the weekend of March 13 to will allow individuals to input their symptoms and risk factors to help determine if they should be tested. If certain criteria are met, the website will provide locations for drive-through testing facilities. Individuals will be tested using a nasal swab and will receive results within 24-36 hours.
The testing is being done in partnership with retailers, including Target and Walmart (who are providing parking lot space for the pop-up testing facilities) and testing companies LabCorp and Quest Diagnostics.
The new test was developed by Roche and just received emergency use authorization from the Food and Drug Administration.
“We therefore expect up to a half-million additional tests will be available early next week,” President Trump said, adding that testing locations will “probably” be announced on Sunday, March 15.
A second application for a new test, submitted by Thermo Fisher, is currently under review at the FDA and is expected to be approved within the next 24 hours, he said. This would add an additional 1.4 million tests in the next week and 5 million within a month, according to the president.
After weeks of decline, influenza activity increases slightly
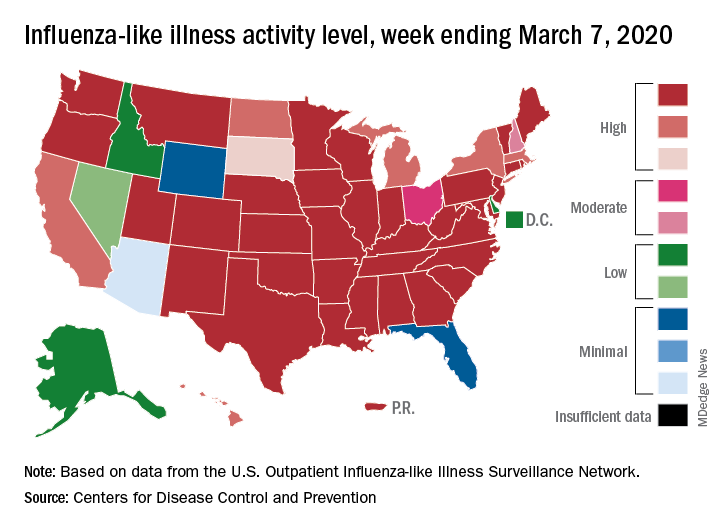
The two leading measures of influenza activity – the percentage of respiratory specimens testing positive for influenza and the proportion of visits to health care providers for influenza-like illness (ILI) – had been following a similar downward path since mid-February. But during the week ending March 7, their paths diverged, according to the Centers for Disease Control and Prevention.
The percentage of respiratory specimens testing positive for influenza dropped for the fourth consecutive week, falling from 26.1% to 21.5%, while the proportion of visits to health care providers for ILI increased from 5.1% to 5.2%, the CDC’s influenza division reported.
One possible explanation for that rise: “The largest increases in ILI activity occurred in areas of the country where COVID-19 is most prevalent. More people may be seeking care for respiratory illness than usual at this time,” the influenza division said March 13 in its weekly Fluview report.
This week’s map puts 34 states and Puerto Rico at level 10 on the CDC’s 1-10 scale of ILI activity, one more state than the week before, and 43 jurisdictions in the “high” range of 8-10, compared with 42 the previous week, the CDC said.
Rates of hospitalizations associated with influenza “remain moderate compared to recent seasons, but rates for children 0-4 years and adults 18-49 years are now the highest CDC has on record for these age groups, surpassing rates reported during the 2009 H1N1 pandemic,” the Fluview report said. Rates for children aged 5-17 years “are higher than any recent regular season but remain lower than rates experienced by this age group during the pandemic.”
The number of pediatric deaths this season is now up to 144, equaling the total for all of the 2018-2019 season. This year’s count led the CDC to invoke 2009 again, since it “is higher for the same time period than in every season since reporting began in 2004-2005, except for the 2009 pandemic.”
For the 2019-2020 season so far there have been 36 million flu illnesses, 370,000 hospitalizations, and 22,000 deaths from flu and pneumonia, the CDC estimated.
The two leading measures of influenza activity – the percentage of respiratory specimens testing positive for influenza and the proportion of visits to health care providers for influenza-like illness (ILI) – had been following a similar downward path since mid-February. But during the week ending March 7, their paths diverged, according to the Centers for Disease Control and Prevention.
The percentage of respiratory specimens testing positive for influenza dropped for the fourth consecutive week, falling from 26.1% to 21.5%, while the proportion of visits to health care providers for ILI increased from 5.1% to 5.2%, the CDC’s influenza division reported.
One possible explanation for that rise: “The largest increases in ILI activity occurred in areas of the country where COVID-19 is most prevalent. More people may be seeking care for respiratory illness than usual at this time,” the influenza division said March 13 in its weekly Fluview report.
This week’s map puts 34 states and Puerto Rico at level 10 on the CDC’s 1-10 scale of ILI activity, one more state than the week before, and 43 jurisdictions in the “high” range of 8-10, compared with 42 the previous week, the CDC said.
Rates of hospitalizations associated with influenza “remain moderate compared to recent seasons, but rates for children 0-4 years and adults 18-49 years are now the highest CDC has on record for these age groups, surpassing rates reported during the 2009 H1N1 pandemic,” the Fluview report said. Rates for children aged 5-17 years “are higher than any recent regular season but remain lower than rates experienced by this age group during the pandemic.”
The number of pediatric deaths this season is now up to 144, equaling the total for all of the 2018-2019 season. This year’s count led the CDC to invoke 2009 again, since it “is higher for the same time period than in every season since reporting began in 2004-2005, except for the 2009 pandemic.”
For the 2019-2020 season so far there have been 36 million flu illnesses, 370,000 hospitalizations, and 22,000 deaths from flu and pneumonia, the CDC estimated.
The two leading measures of influenza activity – the percentage of respiratory specimens testing positive for influenza and the proportion of visits to health care providers for influenza-like illness (ILI) – had been following a similar downward path since mid-February. But during the week ending March 7, their paths diverged, according to the Centers for Disease Control and Prevention.
The percentage of respiratory specimens testing positive for influenza dropped for the fourth consecutive week, falling from 26.1% to 21.5%, while the proportion of visits to health care providers for ILI increased from 5.1% to 5.2%, the CDC’s influenza division reported.
One possible explanation for that rise: “The largest increases in ILI activity occurred in areas of the country where COVID-19 is most prevalent. More people may be seeking care for respiratory illness than usual at this time,” the influenza division said March 13 in its weekly Fluview report.
This week’s map puts 34 states and Puerto Rico at level 10 on the CDC’s 1-10 scale of ILI activity, one more state than the week before, and 43 jurisdictions in the “high” range of 8-10, compared with 42 the previous week, the CDC said.
Rates of hospitalizations associated with influenza “remain moderate compared to recent seasons, but rates for children 0-4 years and adults 18-49 years are now the highest CDC has on record for these age groups, surpassing rates reported during the 2009 H1N1 pandemic,” the Fluview report said. Rates for children aged 5-17 years “are higher than any recent regular season but remain lower than rates experienced by this age group during the pandemic.”
The number of pediatric deaths this season is now up to 144, equaling the total for all of the 2018-2019 season. This year’s count led the CDC to invoke 2009 again, since it “is higher for the same time period than in every season since reporting began in 2004-2005, except for the 2009 pandemic.”
For the 2019-2020 season so far there have been 36 million flu illnesses, 370,000 hospitalizations, and 22,000 deaths from flu and pneumonia, the CDC estimated.
Age divide seen in colorectal cancer screening use
Roughly 79% of adults aged 65-75 years were up to date with their colorectal cancer (CRC) screening in 2018, compared with a significantly lower 63% of those aged 50-64, according to the Centers for Disease Control and Prevention.
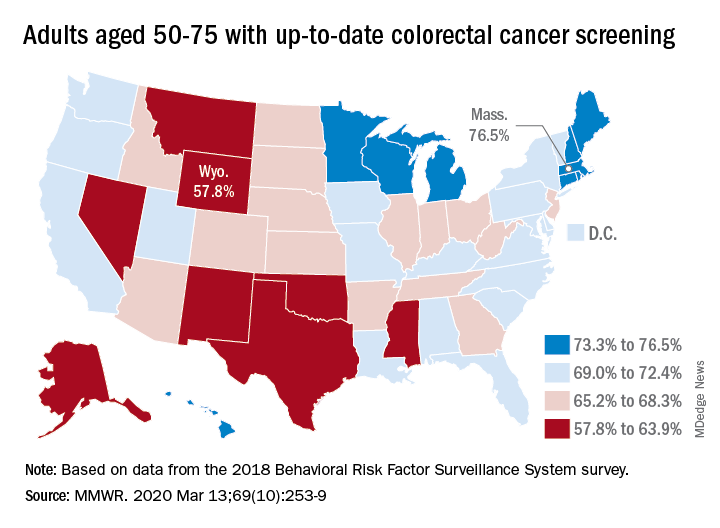
That works out to almost 69% of Americans aged 50-75 years with up-to-date CRC screening, which was defined as a blood stool test in the past year, sigmoidoscopy in the past 5 years, and/or colonoscopy in the past 10 years, Djenaba A. Joseph, MD, and associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion wrote in the Morbidity and Mortality Weekly Report.
“CRC screening has increased steadily among adults over the past 20 years,” the authors noted. However, they observed a lower rate of screening in the 50-64 age group (vs. the 65-75 age group) that was consistent and significant across all demographic groups studied: sex, race/ethnicity, education, annual income, residence location, health insurance status, and regular health care provider status.
The range of screening rates in those groups went from a low of 32.6% (in 50- to 64-year-olds who had no health insurance) to a high of 87.1% (in 65- to 75-year-olds who had annual household incomes of $75,000 and over).
Up-to-date CRC screening rates were higher for the 65-75 age group in every state. The highest screening rate was observed in Rhode Island, at 84.9% in the 65-75 age group. Massachusetts had the highest rate for 50- to 64-year-olds, at 72.1%, and the highest rate for all ages studied (50-75 years), at 76.5%. Wyoming was lowest in all three categories: 51.5% in the 50-64 age group, 68.5% in the 65-75 age group, and 57.8% in the 50-75 age group.
“To achieve further increases in CRC screening to maximize benefit, specific efforts to increase screening in persons aged 50-64 years are needed,” Dr. Joseph and colleagues wrote. They added that efforts might include “providing education about insurance coverage for preventive services, providing clear communication about test options, and conducting research to identify and understand barriers and facilitators to CRC screening specific to this younger age group to inform effective interventions to increase screening.”
SOURCE: Joseph DA et al. MMWR. 2020 Mar 13;69(10):253-9.
Roughly 79% of adults aged 65-75 years were up to date with their colorectal cancer (CRC) screening in 2018, compared with a significantly lower 63% of those aged 50-64, according to the Centers for Disease Control and Prevention.
That works out to almost 69% of Americans aged 50-75 years with up-to-date CRC screening, which was defined as a blood stool test in the past year, sigmoidoscopy in the past 5 years, and/or colonoscopy in the past 10 years, Djenaba A. Joseph, MD, and associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion wrote in the Morbidity and Mortality Weekly Report.
“CRC screening has increased steadily among adults over the past 20 years,” the authors noted. However, they observed a lower rate of screening in the 50-64 age group (vs. the 65-75 age group) that was consistent and significant across all demographic groups studied: sex, race/ethnicity, education, annual income, residence location, health insurance status, and regular health care provider status.
The range of screening rates in those groups went from a low of 32.6% (in 50- to 64-year-olds who had no health insurance) to a high of 87.1% (in 65- to 75-year-olds who had annual household incomes of $75,000 and over).
Up-to-date CRC screening rates were higher for the 65-75 age group in every state. The highest screening rate was observed in Rhode Island, at 84.9% in the 65-75 age group. Massachusetts had the highest rate for 50- to 64-year-olds, at 72.1%, and the highest rate for all ages studied (50-75 years), at 76.5%. Wyoming was lowest in all three categories: 51.5% in the 50-64 age group, 68.5% in the 65-75 age group, and 57.8% in the 50-75 age group.
“To achieve further increases in CRC screening to maximize benefit, specific efforts to increase screening in persons aged 50-64 years are needed,” Dr. Joseph and colleagues wrote. They added that efforts might include “providing education about insurance coverage for preventive services, providing clear communication about test options, and conducting research to identify and understand barriers and facilitators to CRC screening specific to this younger age group to inform effective interventions to increase screening.”
SOURCE: Joseph DA et al. MMWR. 2020 Mar 13;69(10):253-9.
Roughly 79% of adults aged 65-75 years were up to date with their colorectal cancer (CRC) screening in 2018, compared with a significantly lower 63% of those aged 50-64, according to the Centers for Disease Control and Prevention.
That works out to almost 69% of Americans aged 50-75 years with up-to-date CRC screening, which was defined as a blood stool test in the past year, sigmoidoscopy in the past 5 years, and/or colonoscopy in the past 10 years, Djenaba A. Joseph, MD, and associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion wrote in the Morbidity and Mortality Weekly Report.
“CRC screening has increased steadily among adults over the past 20 years,” the authors noted. However, they observed a lower rate of screening in the 50-64 age group (vs. the 65-75 age group) that was consistent and significant across all demographic groups studied: sex, race/ethnicity, education, annual income, residence location, health insurance status, and regular health care provider status.
The range of screening rates in those groups went from a low of 32.6% (in 50- to 64-year-olds who had no health insurance) to a high of 87.1% (in 65- to 75-year-olds who had annual household incomes of $75,000 and over).
Up-to-date CRC screening rates were higher for the 65-75 age group in every state. The highest screening rate was observed in Rhode Island, at 84.9% in the 65-75 age group. Massachusetts had the highest rate for 50- to 64-year-olds, at 72.1%, and the highest rate for all ages studied (50-75 years), at 76.5%. Wyoming was lowest in all three categories: 51.5% in the 50-64 age group, 68.5% in the 65-75 age group, and 57.8% in the 50-75 age group.
“To achieve further increases in CRC screening to maximize benefit, specific efforts to increase screening in persons aged 50-64 years are needed,” Dr. Joseph and colleagues wrote. They added that efforts might include “providing education about insurance coverage for preventive services, providing clear communication about test options, and conducting research to identify and understand barriers and facilitators to CRC screening specific to this younger age group to inform effective interventions to increase screening.”
SOURCE: Joseph DA et al. MMWR. 2020 Mar 13;69(10):253-9.
FROM MMWR
FDA, FTC uniting to promote biosimilars
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also sponsored a public workshop on March 9 to discuss competition for biologics.
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also sponsored a public workshop on March 9 to discuss competition for biologics.
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also sponsored a public workshop on March 9 to discuss competition for biologics.
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
Cancer mortality continues to decline while cancer incidence rises in women
according to the Annual Report to the Nation on the Status of Cancer.
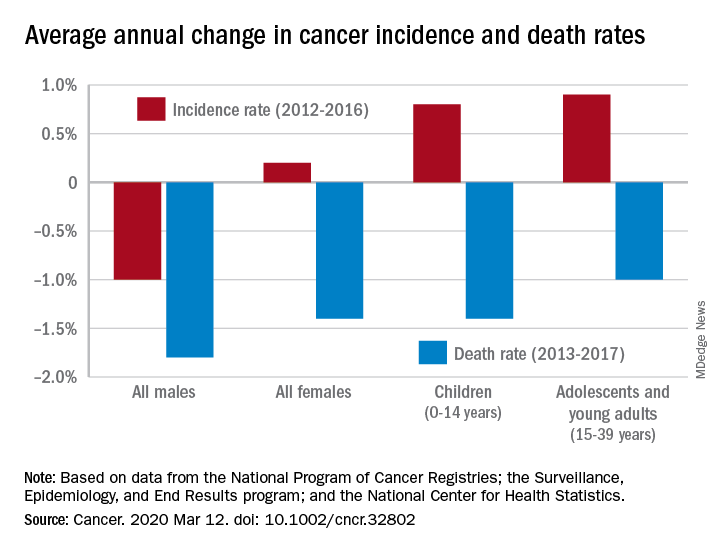
During 2013-2017, the overall age-standardized death rate for all cancers was 158.2 per 100,000 population, and the average decline over that period was 1.5% per year. The average annual change was greater for men (–1.8%) than women (–1.4%) for 2013-2017, but the death rate was higher for men (189.3 per 100,000 vs. 135.5 per 100,000) for those years, S. Jane Henley of the Centers for Disease Control and Prevention and associates reported in Cancer.
“The drops in mortality we’re seeing are real, sustained, and a strong indication of what we can do when we work to prevent and treat cancer,” William G. Cance, MD, chief medical and scientific officer of the America Cancer Society, said in a written statement accompanying the report.
Overall cancer incidence for the most recent 5-year period (2012-2016) was 447.9 per 100,000, with rates of 487.9 for men and 421.4 for women, the investigators said.
Incidence dropped by 0.6% per year overall, but that hides a major difference between men, who saw a decrease of 1.0% a year, and women, who experienced an annual increase of 0.2%.
Over those 5 years, cancer incidence also increased by 0.8% annually among children aged 0-14 years and by 0.9% in adolescents and young adults aged 15-39 years, Ms. Henley and associates said in the report, which is a collaborative effort between the CDC, the National Cancer Institute, the American Cancer Society, and the North American Association of Central Cancer Registries.
“[W]e must not be complacent. The cancer incidence data – especially the increase in cancer among women – is a clear reminder that there is more work ahead,” Norman E. Sharpless, MD, director of the National Cancer Institute, said in the accompanying statement.
SOURCE: Henley SJ et al. Cancer. 2020 Mar 12. doi: 10.1002/cncr.32802.
according to the Annual Report to the Nation on the Status of Cancer.
During 2013-2017, the overall age-standardized death rate for all cancers was 158.2 per 100,000 population, and the average decline over that period was 1.5% per year. The average annual change was greater for men (–1.8%) than women (–1.4%) for 2013-2017, but the death rate was higher for men (189.3 per 100,000 vs. 135.5 per 100,000) for those years, S. Jane Henley of the Centers for Disease Control and Prevention and associates reported in Cancer.
“The drops in mortality we’re seeing are real, sustained, and a strong indication of what we can do when we work to prevent and treat cancer,” William G. Cance, MD, chief medical and scientific officer of the America Cancer Society, said in a written statement accompanying the report.
Overall cancer incidence for the most recent 5-year period (2012-2016) was 447.9 per 100,000, with rates of 487.9 for men and 421.4 for women, the investigators said.
Incidence dropped by 0.6% per year overall, but that hides a major difference between men, who saw a decrease of 1.0% a year, and women, who experienced an annual increase of 0.2%.
Over those 5 years, cancer incidence also increased by 0.8% annually among children aged 0-14 years and by 0.9% in adolescents and young adults aged 15-39 years, Ms. Henley and associates said in the report, which is a collaborative effort between the CDC, the National Cancer Institute, the American Cancer Society, and the North American Association of Central Cancer Registries.
“[W]e must not be complacent. The cancer incidence data – especially the increase in cancer among women – is a clear reminder that there is more work ahead,” Norman E. Sharpless, MD, director of the National Cancer Institute, said in the accompanying statement.
SOURCE: Henley SJ et al. Cancer. 2020 Mar 12. doi: 10.1002/cncr.32802.
according to the Annual Report to the Nation on the Status of Cancer.
During 2013-2017, the overall age-standardized death rate for all cancers was 158.2 per 100,000 population, and the average decline over that period was 1.5% per year. The average annual change was greater for men (–1.8%) than women (–1.4%) for 2013-2017, but the death rate was higher for men (189.3 per 100,000 vs. 135.5 per 100,000) for those years, S. Jane Henley of the Centers for Disease Control and Prevention and associates reported in Cancer.
“The drops in mortality we’re seeing are real, sustained, and a strong indication of what we can do when we work to prevent and treat cancer,” William G. Cance, MD, chief medical and scientific officer of the America Cancer Society, said in a written statement accompanying the report.
Overall cancer incidence for the most recent 5-year period (2012-2016) was 447.9 per 100,000, with rates of 487.9 for men and 421.4 for women, the investigators said.
Incidence dropped by 0.6% per year overall, but that hides a major difference between men, who saw a decrease of 1.0% a year, and women, who experienced an annual increase of 0.2%.
Over those 5 years, cancer incidence also increased by 0.8% annually among children aged 0-14 years and by 0.9% in adolescents and young adults aged 15-39 years, Ms. Henley and associates said in the report, which is a collaborative effort between the CDC, the National Cancer Institute, the American Cancer Society, and the North American Association of Central Cancer Registries.
“[W]e must not be complacent. The cancer incidence data – especially the increase in cancer among women – is a clear reminder that there is more work ahead,” Norman E. Sharpless, MD, director of the National Cancer Institute, said in the accompanying statement.
SOURCE: Henley SJ et al. Cancer. 2020 Mar 12. doi: 10.1002/cncr.32802.
FROM CANCER