User login
Rise in autism prevalence indicates earlier diagnosis
The prevalence of autism spectrum disorder in 4-year-olds rose from 2014 to 2016, indicating more early identification of ASD among the children born in 2012, compared with 2008, according to the Centers for Disease Control and Prevention.
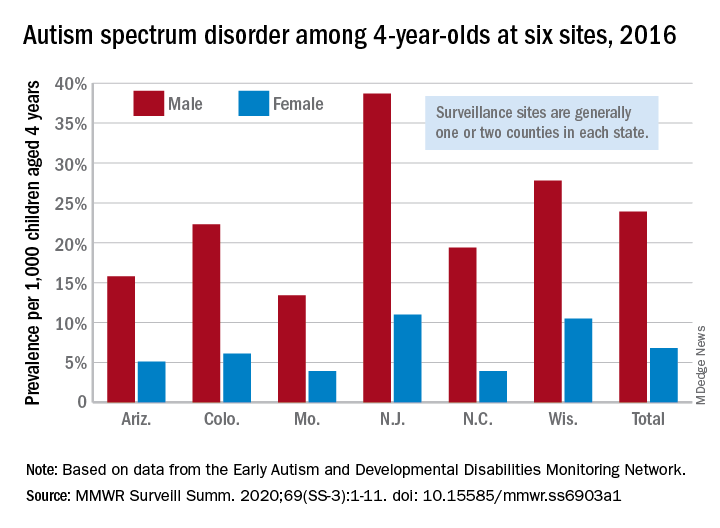
Data from individual surveillance sites in the CDC’s Early Autism and Developmental Disabilities Monitoring (Early ADDM) Network, however, show “wide variability in estimates [that] could reflect variable success in improving community identification,” Kelly A. Shaw, PhD, and associates wrote in MMWR Surveillance Summaries.
they reported.
“In addition, the cumulative incidence of ASD diagnoses at age 48 months was higher for children born in 2012 than for children born in 2008, which indicates a higher rate of diagnosis for the younger cohort,” wrote Dr. Shaw of the CDC’s National Center on Birth Defects and Developmental Disabilities, Atlanta, and associates.
A closer look at the six Early ADDM Network sites shows considerable variation in prevalence. The New Jersey site, consisting of one full county and part of another that includes metropolitan Newark, reported a rate of 25.3 per 1,000 – 38.7 for males and 11.0 for females – while the rates for Missouri – one county in metropolitan St. Louis – were 13.4 (male), 3.9 (female), and 8.8 (combined), the investigators wrote.
ASD prevalence across the six sites was 3.5 times higher among males (23.9 per 1,000) than females (6.8). “Cumulative incidence patterns also differed by sex, with a steady increase in diagnoses with age for boys but an apparent plateau for girls at approximately age 36 months,” they noted.
The median age at earliest diagnosis was 33 months for all sites, with North Carolina lowest at 29 months and Wisconsin highest at 36 months.
The overall median, Dr. Shaw and associates pointed out, is “well above the youngest age at which ASD can be identified, [so] work remains to improve early diagnosis so children can receive timely services.”
SOURCE: Shaw KA et al. MMWR Surveill Summ. 2020;69(SS-3):1-11. doi: 10.15585/mmwr.ss6903a1.
The prevalence of autism spectrum disorder in 4-year-olds rose from 2014 to 2016, indicating more early identification of ASD among the children born in 2012, compared with 2008, according to the Centers for Disease Control and Prevention.
Data from individual surveillance sites in the CDC’s Early Autism and Developmental Disabilities Monitoring (Early ADDM) Network, however, show “wide variability in estimates [that] could reflect variable success in improving community identification,” Kelly A. Shaw, PhD, and associates wrote in MMWR Surveillance Summaries.
they reported.
“In addition, the cumulative incidence of ASD diagnoses at age 48 months was higher for children born in 2012 than for children born in 2008, which indicates a higher rate of diagnosis for the younger cohort,” wrote Dr. Shaw of the CDC’s National Center on Birth Defects and Developmental Disabilities, Atlanta, and associates.
A closer look at the six Early ADDM Network sites shows considerable variation in prevalence. The New Jersey site, consisting of one full county and part of another that includes metropolitan Newark, reported a rate of 25.3 per 1,000 – 38.7 for males and 11.0 for females – while the rates for Missouri – one county in metropolitan St. Louis – were 13.4 (male), 3.9 (female), and 8.8 (combined), the investigators wrote.
ASD prevalence across the six sites was 3.5 times higher among males (23.9 per 1,000) than females (6.8). “Cumulative incidence patterns also differed by sex, with a steady increase in diagnoses with age for boys but an apparent plateau for girls at approximately age 36 months,” they noted.
The median age at earliest diagnosis was 33 months for all sites, with North Carolina lowest at 29 months and Wisconsin highest at 36 months.
The overall median, Dr. Shaw and associates pointed out, is “well above the youngest age at which ASD can be identified, [so] work remains to improve early diagnosis so children can receive timely services.”
SOURCE: Shaw KA et al. MMWR Surveill Summ. 2020;69(SS-3):1-11. doi: 10.15585/mmwr.ss6903a1.
The prevalence of autism spectrum disorder in 4-year-olds rose from 2014 to 2016, indicating more early identification of ASD among the children born in 2012, compared with 2008, according to the Centers for Disease Control and Prevention.
Data from individual surveillance sites in the CDC’s Early Autism and Developmental Disabilities Monitoring (Early ADDM) Network, however, show “wide variability in estimates [that] could reflect variable success in improving community identification,” Kelly A. Shaw, PhD, and associates wrote in MMWR Surveillance Summaries.
they reported.
“In addition, the cumulative incidence of ASD diagnoses at age 48 months was higher for children born in 2012 than for children born in 2008, which indicates a higher rate of diagnosis for the younger cohort,” wrote Dr. Shaw of the CDC’s National Center on Birth Defects and Developmental Disabilities, Atlanta, and associates.
A closer look at the six Early ADDM Network sites shows considerable variation in prevalence. The New Jersey site, consisting of one full county and part of another that includes metropolitan Newark, reported a rate of 25.3 per 1,000 – 38.7 for males and 11.0 for females – while the rates for Missouri – one county in metropolitan St. Louis – were 13.4 (male), 3.9 (female), and 8.8 (combined), the investigators wrote.
ASD prevalence across the six sites was 3.5 times higher among males (23.9 per 1,000) than females (6.8). “Cumulative incidence patterns also differed by sex, with a steady increase in diagnoses with age for boys but an apparent plateau for girls at approximately age 36 months,” they noted.
The median age at earliest diagnosis was 33 months for all sites, with North Carolina lowest at 29 months and Wisconsin highest at 36 months.
The overall median, Dr. Shaw and associates pointed out, is “well above the youngest age at which ASD can be identified, [so] work remains to improve early diagnosis so children can receive timely services.”
SOURCE: Shaw KA et al. MMWR Surveill Summ. 2020;69(SS-3):1-11. doi: 10.15585/mmwr.ss6903a1.
FROM MMWR SURVEILLANCE SUMMARIES
FDA issues EUA allowing hydroxychloroquine sulfate, chloroquine phosphate treatment in COVID-19
The Food and Drug Administration issued an Emergency Use Authorization on March 28, 2020, allowing for the usage of hydroxychloroquine sulfate and chloroquine phosphate products in certain hospitalized patients with COVID-19.

The products, currently stored by the Strategic National Stockpile, will be distributed by the SNS to states so that doctors may prescribe the drugs to adolescent and adult patients hospitalized with COVID-19 in the absence of appropriate or feasible clinical trials. The SNS will work with the Federal Emergency Management Agency to ship the products to states.
According to the Emergency Use Authorization, fact sheets will be provided to health care providers and patients with important information about hydroxychloroquine sulfate and chloroquine phosphate, including the risks of using them to treat COVID-19.
The Food and Drug Administration issued an Emergency Use Authorization on March 28, 2020, allowing for the usage of hydroxychloroquine sulfate and chloroquine phosphate products in certain hospitalized patients with COVID-19.
The products, currently stored by the Strategic National Stockpile, will be distributed by the SNS to states so that doctors may prescribe the drugs to adolescent and adult patients hospitalized with COVID-19 in the absence of appropriate or feasible clinical trials. The SNS will work with the Federal Emergency Management Agency to ship the products to states.
According to the Emergency Use Authorization, fact sheets will be provided to health care providers and patients with important information about hydroxychloroquine sulfate and chloroquine phosphate, including the risks of using them to treat COVID-19.
The Food and Drug Administration issued an Emergency Use Authorization on March 28, 2020, allowing for the usage of hydroxychloroquine sulfate and chloroquine phosphate products in certain hospitalized patients with COVID-19.
The products, currently stored by the Strategic National Stockpile, will be distributed by the SNS to states so that doctors may prescribe the drugs to adolescent and adult patients hospitalized with COVID-19 in the absence of appropriate or feasible clinical trials. The SNS will work with the Federal Emergency Management Agency to ship the products to states.
According to the Emergency Use Authorization, fact sheets will be provided to health care providers and patients with important information about hydroxychloroquine sulfate and chloroquine phosphate, including the risks of using them to treat COVID-19.
FDA OKs durvalumab combo for extensive-stage SCLC
The US Food and Drug Administration has approved the immunotherapy durvalumab (Imfinzi, AstraZeneca) in combination with etoposide and either carboplatin or cisplatin as first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Durvalumab plus chemotherapy “can be considered a new standard in ES-SCLC,” said Myung-Ju Ahn, MD, Sungkyunkwan University, Seoul, South Korea, last year at the European Society of Medical Oncology (ESMO) annual meeting, where he discussed results from the phase 3 trial known as CASPIAN.
The new approval is based on efficacy and safety data from that trial, conducted in patients with previously untreated ES-SCLC. In the experimental group (n = 268), durvalumab plus etoposide and a platinum agent (EP) was followed by maintenance durvalumab, and in the control group (n = 269) patients received the EP regimen alone.
Median overall survival (OS) was 13 months in the durvalumab plus chemotherapy group compared with 10.3 months in the chemotherapy alone group (hazard ratio 0.73; 95% confidence interval, 0.59-0.91; P = .0047).
Reporting these results, trial investigator Luis Paz-Ares, MD, Hospital Universitario 12 de Octubre, Madrid, put the new survival benefit in the context of standard treatments at the ESMO meeting last year.
“Initial response rates to etoposide plus a platinum are high, but responses are not durable and patients treated with EP typically relapse within 6 months of starting treatment with a median OS of approximately 10 months,” he said.
In addition to the primary endpoint of OS, additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Median PFS was not statistically significant with immunotherapy; it was 5.1 months (95% CI, 4.7-6.2) in the durvalumab plus chemotherapy group and 5.4 months (95% CI, 4.8-6.2) in the chemotherapy alone group (HR, 0.78; 95% CI, 0.65-0.94).
The investigator-assessed ORR was 68% in the durvalumab plus chemotherapy group and 58% in the chemotherapy alone group.
The most common adverse reactions (≥ 20%) in patients with ES-SCLC were nausea, fatigue/asthenia, and alopecia, according to the FDA.
At ESMO, Paz-Ares reported that rates of serious adverse events (AEs) were comparable at 30.9% and 36.1% for the durvalumab plus EP group vs. EP alone, respectively; rates of AEs leading to discontinuation were identical in both groups at 9.4%. Unsurprisingly, immune-mediated AEs were higher at 19.6% in the durvalumab combination group vs. 2.6% in the EP alone group.
In this setting, durvalumab is administered prior to chemotherapy on the same day. The recommended durvalumab dose (when administered with etoposide and carboplatin or cisplatin) is 1,500 mg every 3 weeks prior to chemotherapy and then every 4 weeks as a single-agent maintenance therapy.
Durvalumab is already approved for metastatic non–small cell lung cancer in patients whose tumors have only spread in the chest, and is also approved for use in urothelial cancer.
This article first appeared on Medscape.com.
The US Food and Drug Administration has approved the immunotherapy durvalumab (Imfinzi, AstraZeneca) in combination with etoposide and either carboplatin or cisplatin as first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Durvalumab plus chemotherapy “can be considered a new standard in ES-SCLC,” said Myung-Ju Ahn, MD, Sungkyunkwan University, Seoul, South Korea, last year at the European Society of Medical Oncology (ESMO) annual meeting, where he discussed results from the phase 3 trial known as CASPIAN.
The new approval is based on efficacy and safety data from that trial, conducted in patients with previously untreated ES-SCLC. In the experimental group (n = 268), durvalumab plus etoposide and a platinum agent (EP) was followed by maintenance durvalumab, and in the control group (n = 269) patients received the EP regimen alone.
Median overall survival (OS) was 13 months in the durvalumab plus chemotherapy group compared with 10.3 months in the chemotherapy alone group (hazard ratio 0.73; 95% confidence interval, 0.59-0.91; P = .0047).
Reporting these results, trial investigator Luis Paz-Ares, MD, Hospital Universitario 12 de Octubre, Madrid, put the new survival benefit in the context of standard treatments at the ESMO meeting last year.
“Initial response rates to etoposide plus a platinum are high, but responses are not durable and patients treated with EP typically relapse within 6 months of starting treatment with a median OS of approximately 10 months,” he said.
In addition to the primary endpoint of OS, additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Median PFS was not statistically significant with immunotherapy; it was 5.1 months (95% CI, 4.7-6.2) in the durvalumab plus chemotherapy group and 5.4 months (95% CI, 4.8-6.2) in the chemotherapy alone group (HR, 0.78; 95% CI, 0.65-0.94).
The investigator-assessed ORR was 68% in the durvalumab plus chemotherapy group and 58% in the chemotherapy alone group.
The most common adverse reactions (≥ 20%) in patients with ES-SCLC were nausea, fatigue/asthenia, and alopecia, according to the FDA.
At ESMO, Paz-Ares reported that rates of serious adverse events (AEs) were comparable at 30.9% and 36.1% for the durvalumab plus EP group vs. EP alone, respectively; rates of AEs leading to discontinuation were identical in both groups at 9.4%. Unsurprisingly, immune-mediated AEs were higher at 19.6% in the durvalumab combination group vs. 2.6% in the EP alone group.
In this setting, durvalumab is administered prior to chemotherapy on the same day. The recommended durvalumab dose (when administered with etoposide and carboplatin or cisplatin) is 1,500 mg every 3 weeks prior to chemotherapy and then every 4 weeks as a single-agent maintenance therapy.
Durvalumab is already approved for metastatic non–small cell lung cancer in patients whose tumors have only spread in the chest, and is also approved for use in urothelial cancer.
This article first appeared on Medscape.com.
The US Food and Drug Administration has approved the immunotherapy durvalumab (Imfinzi, AstraZeneca) in combination with etoposide and either carboplatin or cisplatin as first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Durvalumab plus chemotherapy “can be considered a new standard in ES-SCLC,” said Myung-Ju Ahn, MD, Sungkyunkwan University, Seoul, South Korea, last year at the European Society of Medical Oncology (ESMO) annual meeting, where he discussed results from the phase 3 trial known as CASPIAN.
The new approval is based on efficacy and safety data from that trial, conducted in patients with previously untreated ES-SCLC. In the experimental group (n = 268), durvalumab plus etoposide and a platinum agent (EP) was followed by maintenance durvalumab, and in the control group (n = 269) patients received the EP regimen alone.
Median overall survival (OS) was 13 months in the durvalumab plus chemotherapy group compared with 10.3 months in the chemotherapy alone group (hazard ratio 0.73; 95% confidence interval, 0.59-0.91; P = .0047).
Reporting these results, trial investigator Luis Paz-Ares, MD, Hospital Universitario 12 de Octubre, Madrid, put the new survival benefit in the context of standard treatments at the ESMO meeting last year.
“Initial response rates to etoposide plus a platinum are high, but responses are not durable and patients treated with EP typically relapse within 6 months of starting treatment with a median OS of approximately 10 months,” he said.
In addition to the primary endpoint of OS, additional efficacy outcome measures were investigator-assessed progression-free survival (PFS) and objective response rate (ORR).
Median PFS was not statistically significant with immunotherapy; it was 5.1 months (95% CI, 4.7-6.2) in the durvalumab plus chemotherapy group and 5.4 months (95% CI, 4.8-6.2) in the chemotherapy alone group (HR, 0.78; 95% CI, 0.65-0.94).
The investigator-assessed ORR was 68% in the durvalumab plus chemotherapy group and 58% in the chemotherapy alone group.
The most common adverse reactions (≥ 20%) in patients with ES-SCLC were nausea, fatigue/asthenia, and alopecia, according to the FDA.
At ESMO, Paz-Ares reported that rates of serious adverse events (AEs) were comparable at 30.9% and 36.1% for the durvalumab plus EP group vs. EP alone, respectively; rates of AEs leading to discontinuation were identical in both groups at 9.4%. Unsurprisingly, immune-mediated AEs were higher at 19.6% in the durvalumab combination group vs. 2.6% in the EP alone group.
In this setting, durvalumab is administered prior to chemotherapy on the same day. The recommended durvalumab dose (when administered with etoposide and carboplatin or cisplatin) is 1,500 mg every 3 weeks prior to chemotherapy and then every 4 weeks as a single-agent maintenance therapy.
Durvalumab is already approved for metastatic non–small cell lung cancer in patients whose tumors have only spread in the chest, and is also approved for use in urothelial cancer.
This article first appeared on Medscape.com.
FDA approves ixekizumab for pediatric plaque psoriasis
The according to an announcement from Lilly.
Patients need to be candidates for systemic therapy or phototherapy and have no known hypersensitivity to the biologic.
The safety, tolerability, and efficacy of the interleukin-17a antagonist were demonstrated in a phase 3 study that included 171 patients aged 6-17 years with moderate to severe plaque psoriasis. At 12 weeks, 89% those on ixekizumab achieved a 75% improvement on Psoriasis Area and Severity Index score, compared with 25% of those on placebo, and 81% achieved a static Physician’s Global Assessment of clear or almost clear, compared with 11% of those on placebo, according to the Lilly statement.
The safety profile seen with ixekizumab (Taltz) among the pediatric patients with plaque psoriasis is consistent with what has been observed among adult patients, although there were higher rates of conjunctivitis, influenza, and urticaria among the pediatric patients, the statement noted. The biologic may increase the risk of infection, and patients should be evaluated for tuberculosis, hypersensitivity, and inflammatory bowel disease. It is also recommended that routine immunizations be completed before initiating treatment.
Ixekizumab was initially approved for treating adults with moderate to severe plaque psoriasis in 2016, followed by approvals for treatment of adults with active psoriatic arthritis in 2017, and for adults with ankylosing spondylitis in August 2019.
The biologic therapies – etanercept, a tumor necrosis factor blocker, and ustekinumab (Stelara), an IL-12/23 antagonist – were previously approved by the FDA for pediatric psoriasis, in children ages 4 years and older and 12 years and older, respectively.
Updated prescribing information for ixekizumab can be found on the Lilly website.
[email protected]
The according to an announcement from Lilly.
Patients need to be candidates for systemic therapy or phototherapy and have no known hypersensitivity to the biologic.
The safety, tolerability, and efficacy of the interleukin-17a antagonist were demonstrated in a phase 3 study that included 171 patients aged 6-17 years with moderate to severe plaque psoriasis. At 12 weeks, 89% those on ixekizumab achieved a 75% improvement on Psoriasis Area and Severity Index score, compared with 25% of those on placebo, and 81% achieved a static Physician’s Global Assessment of clear or almost clear, compared with 11% of those on placebo, according to the Lilly statement.
The safety profile seen with ixekizumab (Taltz) among the pediatric patients with plaque psoriasis is consistent with what has been observed among adult patients, although there were higher rates of conjunctivitis, influenza, and urticaria among the pediatric patients, the statement noted. The biologic may increase the risk of infection, and patients should be evaluated for tuberculosis, hypersensitivity, and inflammatory bowel disease. It is also recommended that routine immunizations be completed before initiating treatment.
Ixekizumab was initially approved for treating adults with moderate to severe plaque psoriasis in 2016, followed by approvals for treatment of adults with active psoriatic arthritis in 2017, and for adults with ankylosing spondylitis in August 2019.
The biologic therapies – etanercept, a tumor necrosis factor blocker, and ustekinumab (Stelara), an IL-12/23 antagonist – were previously approved by the FDA for pediatric psoriasis, in children ages 4 years and older and 12 years and older, respectively.
Updated prescribing information for ixekizumab can be found on the Lilly website.
[email protected]
The according to an announcement from Lilly.
Patients need to be candidates for systemic therapy or phototherapy and have no known hypersensitivity to the biologic.
The safety, tolerability, and efficacy of the interleukin-17a antagonist were demonstrated in a phase 3 study that included 171 patients aged 6-17 years with moderate to severe plaque psoriasis. At 12 weeks, 89% those on ixekizumab achieved a 75% improvement on Psoriasis Area and Severity Index score, compared with 25% of those on placebo, and 81% achieved a static Physician’s Global Assessment of clear or almost clear, compared with 11% of those on placebo, according to the Lilly statement.
The safety profile seen with ixekizumab (Taltz) among the pediatric patients with plaque psoriasis is consistent with what has been observed among adult patients, although there were higher rates of conjunctivitis, influenza, and urticaria among the pediatric patients, the statement noted. The biologic may increase the risk of infection, and patients should be evaluated for tuberculosis, hypersensitivity, and inflammatory bowel disease. It is also recommended that routine immunizations be completed before initiating treatment.
Ixekizumab was initially approved for treating adults with moderate to severe plaque psoriasis in 2016, followed by approvals for treatment of adults with active psoriatic arthritis in 2017, and for adults with ankylosing spondylitis in August 2019.
The biologic therapies – etanercept, a tumor necrosis factor blocker, and ustekinumab (Stelara), an IL-12/23 antagonist – were previously approved by the FDA for pediatric psoriasis, in children ages 4 years and older and 12 years and older, respectively.
Updated prescribing information for ixekizumab can be found on the Lilly website.
[email protected]
Flu activity measures continue COVID-19–related divergence
The 2019-2020 flu paradox continues in the United States: Fewer respiratory samples are testing positive for influenza, but more people are seeking care for respiratory symptoms because of COVID-19, according to the Centers for Disease Control and Prevention.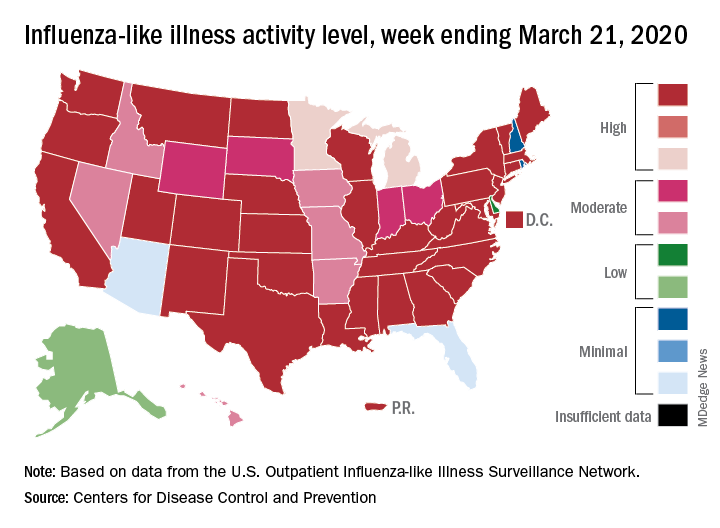
compared with 14.9% the week before, but outpatient visits for influenza-like illness (ILI) rose from 5.6% of all visits to 6.2% for third week of March, the CDC’s influenza division reported.
The CDC defines ILI as “fever (temperature of 100°F [37.8°C] or greater) and a cough and/or a sore throat without a known cause other than influenza.” The outpatient ILI visit rate needs to get below the national baseline of 2.4% for the CDC to call the end of the 2019-2020 flu season.
This week’s map shows that fewer states are at the highest level of ILI activity on the CDC’s 1-10 scale: 33 states plus Puerto Rico for the week ending March 21, compared with 35 and Puerto Rico the previous week. The number of states at level 10 had risen the two previous weeks, CDC data show.
“Influenza severity indicators remain moderate to low overall, but hospitalization rates differ by age group, with high rates among children and young adults,” the influenza division said.
Overall mortality also has not been high, but 155 children have died from the flu so far in 2019-2020, which is more than any season since the 2009 pandemic, the CDC noted.
The 2019-2020 flu paradox continues in the United States: Fewer respiratory samples are testing positive for influenza, but more people are seeking care for respiratory symptoms because of COVID-19, according to the Centers for Disease Control and Prevention.
compared with 14.9% the week before, but outpatient visits for influenza-like illness (ILI) rose from 5.6% of all visits to 6.2% for third week of March, the CDC’s influenza division reported.
The CDC defines ILI as “fever (temperature of 100°F [37.8°C] or greater) and a cough and/or a sore throat without a known cause other than influenza.” The outpatient ILI visit rate needs to get below the national baseline of 2.4% for the CDC to call the end of the 2019-2020 flu season.
This week’s map shows that fewer states are at the highest level of ILI activity on the CDC’s 1-10 scale: 33 states plus Puerto Rico for the week ending March 21, compared with 35 and Puerto Rico the previous week. The number of states at level 10 had risen the two previous weeks, CDC data show.
“Influenza severity indicators remain moderate to low overall, but hospitalization rates differ by age group, with high rates among children and young adults,” the influenza division said.
Overall mortality also has not been high, but 155 children have died from the flu so far in 2019-2020, which is more than any season since the 2009 pandemic, the CDC noted.
The 2019-2020 flu paradox continues in the United States: Fewer respiratory samples are testing positive for influenza, but more people are seeking care for respiratory symptoms because of COVID-19, according to the Centers for Disease Control and Prevention.
compared with 14.9% the week before, but outpatient visits for influenza-like illness (ILI) rose from 5.6% of all visits to 6.2% for third week of March, the CDC’s influenza division reported.
The CDC defines ILI as “fever (temperature of 100°F [37.8°C] or greater) and a cough and/or a sore throat without a known cause other than influenza.” The outpatient ILI visit rate needs to get below the national baseline of 2.4% for the CDC to call the end of the 2019-2020 flu season.
This week’s map shows that fewer states are at the highest level of ILI activity on the CDC’s 1-10 scale: 33 states plus Puerto Rico for the week ending March 21, compared with 35 and Puerto Rico the previous week. The number of states at level 10 had risen the two previous weeks, CDC data show.
“Influenza severity indicators remain moderate to low overall, but hospitalization rates differ by age group, with high rates among children and young adults,” the influenza division said.
Overall mortality also has not been high, but 155 children have died from the flu so far in 2019-2020, which is more than any season since the 2009 pandemic, the CDC noted.
FDA okays emergency use of convalescent plasma for seriously ill COVID-19 patients
As the proportion of patients infected with COVID-19 continues to rise in the United States, the Food and Drug Administration is facilitating access to COVID-19 convalescent plasma for use in patients with serious or immediately life-threatening COVID-19 infections.

While clinical trials are underway to evaluate the safety and efficacy of administering convalescent plasma to patients with COVID-19, the FDA is granting clinicians permission for use of investigational convalescent plasma under single-patient emergency Investigational New Drug Applications (INDs), since no known cure exists and a vaccine is more than 1 year away from becoming available.
This allows the use of an investigational drug for the treatment of an individual patient by a licensed physician upon FDA authorization. This does not include the use of COVID-19 convalescent plasma for the prevention of infection, according to a statement issued by the agency on March 24.
“It is possible that convalescent plasma that contains antibodies to SARS-CoV-2 (the virus that causes COVID-19) might be effective against the infection,” the FDA statement reads. “Use of convalescent plasma has been studied in outbreaks of other respiratory infections, including the 2009-2010 H1N1 influenza virus pandemic, 2003 SARS-CoV-1 epidemic, and the 2012 MERS-CoV epidemic. Although promising, convalescent plasma has not been shown to be effective in every disease studied.”
“I think the FDA got caught initially a little flat-footed when it came to the development of COVID-19 tests, but they’re quickly catching up,” Peter J. Pitts, who was the FDA’s associate commissioner from 2002 to 2004, said in an interview. “I think that the attitude now is, ‘If it’s safe, let’s create a pathway to see how these things work in the real world.’ I think that’s going to be as true for treatments to lessen the symptoms and shorten the duration of the disease, as well as convalescent plasma as a potential alternative to a yet-to-be-developed vaccine.”
At the University of Washington School of Medicine, Seattle, Terry B. Gernsheimer, MD, and her colleagues are recruiting recovered COVID-19 patients to donate plasma for seriously ill patients affected with the virus. “The thought of using convalescent plasma makes total sense, because it’s immediately available, and it’s something that we can try to give people,” said Dr. Gernsheimer, a hematologist who is professor of medicine at the medical school. “It’s been used in China, and reports should be coming out shortly about their experience with this.”

In a case series that appeared in JAMA on March 27 (doi: 10.1001/jama.2020.4783), Chinese researchers led by Chenguang Shen, PhD, reported findings from five critically ill COVID-19 patients with acute respiratory distress syndrome who received a transfusion with convalescent plasma at Shenzhen Third People’s Hospital 10 and 22 days after hospital admission. The patients ranged in age from 36 to 73 years, three were men, and all were receiving mechanical ventilation at the time of treatment.
Dr. Shen and colleagues reported that viral loads decreased and became negative within 12 days following the transfusion. Three of the patients were discharged from the hospital after a length of stay that ranged from 51 to 55 days, and two remain in stable condition at 37 days after the transfusion. The researchers pointed out that all patients received antiviral agents, including interferon and lopinavir/ritonavir, during and following convalescent plasma treatment, “which also may have contributed to the viral clearance observed.”
Under the FDA policy on emergency IND use, COVID-19 convalescent plasma must only be collected from recovered individuals if they are eligible to donate blood, required testing must be performed, and the donation must be found suitable.
Potential donors “are going to be screened the way all blood donors are screened,” Dr. Gernsheimer said. “It’s not going to be any less safe than any unit of plasma that’s on the shelf that comes from our volunteer donors. There are always transfusion reactions that we have to worry about, [and] there are potentially unknown pathogens that we don’t yet know about that we are not yet testing for. It’s the regular risk we see with any unit of plasma.”
She added that COVID-19 survivors appear to start increasing their titer of the antibody around day 28. “We’ll be looking for recovered individuals who have had a documented infection, and whose symptoms started about 28 days before we collect,” she said.
The FDA advises clinicians to address several considerations for donor eligibility, including prior diagnosis of COVID-19 documented by a laboratory test; complete resolution of symptoms at least 14 days prior to donation; female donors negative for HLA antibodies or male donors, and negative results for COVID-19 either from one or more nasopharyngeal swab specimens or by a molecular diagnostic test from blood. [A partial list of available tests can be accessed on the FDA website.] The agency also advises that donors have defined SARS-CoV-2–neutralizing antibody titers, if testing can be conducted (optimally greater than 1:320).
Patients eligible to receive COVID-19 convalescent plasma must have a severe or immediately life-threatening infection with laboratory-confirmed COVID-19. The agency defines severe disease as dyspnea, respiratory frequency of 30 per minute or greater, blood oxygen saturation of 93% or less, partial pressure of arterial oxygen to fraction of inspired oxygen ratio of less than 300, and/or lung infiltrates of greater than 50% within 24-48 hours. Life-threatening disease is defined as respiratory failure, septic shock, and/or multiple organ dysfunction or failure. Patients must provide informed consent.
The potential risks of receiving COVID-19 convalescent plasma remain unknown, according to Dr. Gernsheimer. “What some people have thought about is, could there be such an inflammatory response with the virus that we would initially see these patients get worse?” she said. “My understanding is that has not occurred in China yet, but we don’t have all those data. But we always worry if we have something that’s going to cause inflammation around an infection, for example, that could initially make it more difficult to breathe if it’s a lung infection. So far, my understanding is that has not been seen.”
For COVID-19 convalescent plasma authorization requests that require a response within 4-8 hours, requesting clinicians may complete form 3296 and submit it by email to [email protected].
For COVID-19 convalescent plasma authorization requests that require a response in less than 4 hours, or if the clinician is unable to complete and submit form 3926 because of extenuating circumstances, verbal authorization can be sought by calling the FDA’s Office of Emergency Operations at 1-866-300-4374.
The FDA is working with the National Institutes of Health, the Centers for Disease Control and Prevention, and other government partners to develop protocols for use by multiple investigators in order to coordinate the collection and use of COVID-19 convalescent plasma.
“It’s crucial that data be captured for every patient so that we really understand what safety and effectiveness looks like on as close to a real-world level as we can, as quickly as we can,” said Mr. Pitts, who is president and cofounder of the Center for Medicine in the Public Interest, and who also does consulting work for the FDA. “I understand that health care professionals are overworked and overburdened right now. I applaud them for their heroic work. But that doesn’t mean that we can shirk off collecting the data. When I was at the FDA, I helped address the SARS epidemic. The agency attitude at that point was, ‘Let’s get things that just might work through the process, as long as the cure isn’t going to be worse than the disease.’ I think that’s the attitude that’s leading the charge today.”
As the proportion of patients infected with COVID-19 continues to rise in the United States, the Food and Drug Administration is facilitating access to COVID-19 convalescent plasma for use in patients with serious or immediately life-threatening COVID-19 infections.
While clinical trials are underway to evaluate the safety and efficacy of administering convalescent plasma to patients with COVID-19, the FDA is granting clinicians permission for use of investigational convalescent plasma under single-patient emergency Investigational New Drug Applications (INDs), since no known cure exists and a vaccine is more than 1 year away from becoming available.
This allows the use of an investigational drug for the treatment of an individual patient by a licensed physician upon FDA authorization. This does not include the use of COVID-19 convalescent plasma for the prevention of infection, according to a statement issued by the agency on March 24.
“It is possible that convalescent plasma that contains antibodies to SARS-CoV-2 (the virus that causes COVID-19) might be effective against the infection,” the FDA statement reads. “Use of convalescent plasma has been studied in outbreaks of other respiratory infections, including the 2009-2010 H1N1 influenza virus pandemic, 2003 SARS-CoV-1 epidemic, and the 2012 MERS-CoV epidemic. Although promising, convalescent plasma has not been shown to be effective in every disease studied.”
“I think the FDA got caught initially a little flat-footed when it came to the development of COVID-19 tests, but they’re quickly catching up,” Peter J. Pitts, who was the FDA’s associate commissioner from 2002 to 2004, said in an interview. “I think that the attitude now is, ‘If it’s safe, let’s create a pathway to see how these things work in the real world.’ I think that’s going to be as true for treatments to lessen the symptoms and shorten the duration of the disease, as well as convalescent plasma as a potential alternative to a yet-to-be-developed vaccine.”
At the University of Washington School of Medicine, Seattle, Terry B. Gernsheimer, MD, and her colleagues are recruiting recovered COVID-19 patients to donate plasma for seriously ill patients affected with the virus. “The thought of using convalescent plasma makes total sense, because it’s immediately available, and it’s something that we can try to give people,” said Dr. Gernsheimer, a hematologist who is professor of medicine at the medical school. “It’s been used in China, and reports should be coming out shortly about their experience with this.”
In a case series that appeared in JAMA on March 27 (doi: 10.1001/jama.2020.4783), Chinese researchers led by Chenguang Shen, PhD, reported findings from five critically ill COVID-19 patients with acute respiratory distress syndrome who received a transfusion with convalescent plasma at Shenzhen Third People’s Hospital 10 and 22 days after hospital admission. The patients ranged in age from 36 to 73 years, three were men, and all were receiving mechanical ventilation at the time of treatment.
Dr. Shen and colleagues reported that viral loads decreased and became negative within 12 days following the transfusion. Three of the patients were discharged from the hospital after a length of stay that ranged from 51 to 55 days, and two remain in stable condition at 37 days after the transfusion. The researchers pointed out that all patients received antiviral agents, including interferon and lopinavir/ritonavir, during and following convalescent plasma treatment, “which also may have contributed to the viral clearance observed.”
Under the FDA policy on emergency IND use, COVID-19 convalescent plasma must only be collected from recovered individuals if they are eligible to donate blood, required testing must be performed, and the donation must be found suitable.
Potential donors “are going to be screened the way all blood donors are screened,” Dr. Gernsheimer said. “It’s not going to be any less safe than any unit of plasma that’s on the shelf that comes from our volunteer donors. There are always transfusion reactions that we have to worry about, [and] there are potentially unknown pathogens that we don’t yet know about that we are not yet testing for. It’s the regular risk we see with any unit of plasma.”
She added that COVID-19 survivors appear to start increasing their titer of the antibody around day 28. “We’ll be looking for recovered individuals who have had a documented infection, and whose symptoms started about 28 days before we collect,” she said.
The FDA advises clinicians to address several considerations for donor eligibility, including prior diagnosis of COVID-19 documented by a laboratory test; complete resolution of symptoms at least 14 days prior to donation; female donors negative for HLA antibodies or male donors, and negative results for COVID-19 either from one or more nasopharyngeal swab specimens or by a molecular diagnostic test from blood. [A partial list of available tests can be accessed on the FDA website.] The agency also advises that donors have defined SARS-CoV-2–neutralizing antibody titers, if testing can be conducted (optimally greater than 1:320).
Patients eligible to receive COVID-19 convalescent plasma must have a severe or immediately life-threatening infection with laboratory-confirmed COVID-19. The agency defines severe disease as dyspnea, respiratory frequency of 30 per minute or greater, blood oxygen saturation of 93% or less, partial pressure of arterial oxygen to fraction of inspired oxygen ratio of less than 300, and/or lung infiltrates of greater than 50% within 24-48 hours. Life-threatening disease is defined as respiratory failure, septic shock, and/or multiple organ dysfunction or failure. Patients must provide informed consent.
The potential risks of receiving COVID-19 convalescent plasma remain unknown, according to Dr. Gernsheimer. “What some people have thought about is, could there be such an inflammatory response with the virus that we would initially see these patients get worse?” she said. “My understanding is that has not occurred in China yet, but we don’t have all those data. But we always worry if we have something that’s going to cause inflammation around an infection, for example, that could initially make it more difficult to breathe if it’s a lung infection. So far, my understanding is that has not been seen.”
For COVID-19 convalescent plasma authorization requests that require a response within 4-8 hours, requesting clinicians may complete form 3296 and submit it by email to [email protected].
For COVID-19 convalescent plasma authorization requests that require a response in less than 4 hours, or if the clinician is unable to complete and submit form 3926 because of extenuating circumstances, verbal authorization can be sought by calling the FDA’s Office of Emergency Operations at 1-866-300-4374.
The FDA is working with the National Institutes of Health, the Centers for Disease Control and Prevention, and other government partners to develop protocols for use by multiple investigators in order to coordinate the collection and use of COVID-19 convalescent plasma.
“It’s crucial that data be captured for every patient so that we really understand what safety and effectiveness looks like on as close to a real-world level as we can, as quickly as we can,” said Mr. Pitts, who is president and cofounder of the Center for Medicine in the Public Interest, and who also does consulting work for the FDA. “I understand that health care professionals are overworked and overburdened right now. I applaud them for their heroic work. But that doesn’t mean that we can shirk off collecting the data. When I was at the FDA, I helped address the SARS epidemic. The agency attitude at that point was, ‘Let’s get things that just might work through the process, as long as the cure isn’t going to be worse than the disease.’ I think that’s the attitude that’s leading the charge today.”
As the proportion of patients infected with COVID-19 continues to rise in the United States, the Food and Drug Administration is facilitating access to COVID-19 convalescent plasma for use in patients with serious or immediately life-threatening COVID-19 infections.
While clinical trials are underway to evaluate the safety and efficacy of administering convalescent plasma to patients with COVID-19, the FDA is granting clinicians permission for use of investigational convalescent plasma under single-patient emergency Investigational New Drug Applications (INDs), since no known cure exists and a vaccine is more than 1 year away from becoming available.
This allows the use of an investigational drug for the treatment of an individual patient by a licensed physician upon FDA authorization. This does not include the use of COVID-19 convalescent plasma for the prevention of infection, according to a statement issued by the agency on March 24.
“It is possible that convalescent plasma that contains antibodies to SARS-CoV-2 (the virus that causes COVID-19) might be effective against the infection,” the FDA statement reads. “Use of convalescent plasma has been studied in outbreaks of other respiratory infections, including the 2009-2010 H1N1 influenza virus pandemic, 2003 SARS-CoV-1 epidemic, and the 2012 MERS-CoV epidemic. Although promising, convalescent plasma has not been shown to be effective in every disease studied.”
“I think the FDA got caught initially a little flat-footed when it came to the development of COVID-19 tests, but they’re quickly catching up,” Peter J. Pitts, who was the FDA’s associate commissioner from 2002 to 2004, said in an interview. “I think that the attitude now is, ‘If it’s safe, let’s create a pathway to see how these things work in the real world.’ I think that’s going to be as true for treatments to lessen the symptoms and shorten the duration of the disease, as well as convalescent plasma as a potential alternative to a yet-to-be-developed vaccine.”
At the University of Washington School of Medicine, Seattle, Terry B. Gernsheimer, MD, and her colleagues are recruiting recovered COVID-19 patients to donate plasma for seriously ill patients affected with the virus. “The thought of using convalescent plasma makes total sense, because it’s immediately available, and it’s something that we can try to give people,” said Dr. Gernsheimer, a hematologist who is professor of medicine at the medical school. “It’s been used in China, and reports should be coming out shortly about their experience with this.”
In a case series that appeared in JAMA on March 27 (doi: 10.1001/jama.2020.4783), Chinese researchers led by Chenguang Shen, PhD, reported findings from five critically ill COVID-19 patients with acute respiratory distress syndrome who received a transfusion with convalescent plasma at Shenzhen Third People’s Hospital 10 and 22 days after hospital admission. The patients ranged in age from 36 to 73 years, three were men, and all were receiving mechanical ventilation at the time of treatment.
Dr. Shen and colleagues reported that viral loads decreased and became negative within 12 days following the transfusion. Three of the patients were discharged from the hospital after a length of stay that ranged from 51 to 55 days, and two remain in stable condition at 37 days after the transfusion. The researchers pointed out that all patients received antiviral agents, including interferon and lopinavir/ritonavir, during and following convalescent plasma treatment, “which also may have contributed to the viral clearance observed.”
Under the FDA policy on emergency IND use, COVID-19 convalescent plasma must only be collected from recovered individuals if they are eligible to donate blood, required testing must be performed, and the donation must be found suitable.
Potential donors “are going to be screened the way all blood donors are screened,” Dr. Gernsheimer said. “It’s not going to be any less safe than any unit of plasma that’s on the shelf that comes from our volunteer donors. There are always transfusion reactions that we have to worry about, [and] there are potentially unknown pathogens that we don’t yet know about that we are not yet testing for. It’s the regular risk we see with any unit of plasma.”
She added that COVID-19 survivors appear to start increasing their titer of the antibody around day 28. “We’ll be looking for recovered individuals who have had a documented infection, and whose symptoms started about 28 days before we collect,” she said.
The FDA advises clinicians to address several considerations for donor eligibility, including prior diagnosis of COVID-19 documented by a laboratory test; complete resolution of symptoms at least 14 days prior to donation; female donors negative for HLA antibodies or male donors, and negative results for COVID-19 either from one or more nasopharyngeal swab specimens or by a molecular diagnostic test from blood. [A partial list of available tests can be accessed on the FDA website.] The agency also advises that donors have defined SARS-CoV-2–neutralizing antibody titers, if testing can be conducted (optimally greater than 1:320).
Patients eligible to receive COVID-19 convalescent plasma must have a severe or immediately life-threatening infection with laboratory-confirmed COVID-19. The agency defines severe disease as dyspnea, respiratory frequency of 30 per minute or greater, blood oxygen saturation of 93% or less, partial pressure of arterial oxygen to fraction of inspired oxygen ratio of less than 300, and/or lung infiltrates of greater than 50% within 24-48 hours. Life-threatening disease is defined as respiratory failure, septic shock, and/or multiple organ dysfunction or failure. Patients must provide informed consent.
The potential risks of receiving COVID-19 convalescent plasma remain unknown, according to Dr. Gernsheimer. “What some people have thought about is, could there be such an inflammatory response with the virus that we would initially see these patients get worse?” she said. “My understanding is that has not occurred in China yet, but we don’t have all those data. But we always worry if we have something that’s going to cause inflammation around an infection, for example, that could initially make it more difficult to breathe if it’s a lung infection. So far, my understanding is that has not been seen.”
For COVID-19 convalescent plasma authorization requests that require a response within 4-8 hours, requesting clinicians may complete form 3296 and submit it by email to [email protected].
For COVID-19 convalescent plasma authorization requests that require a response in less than 4 hours, or if the clinician is unable to complete and submit form 3926 because of extenuating circumstances, verbal authorization can be sought by calling the FDA’s Office of Emergency Operations at 1-866-300-4374.
The FDA is working with the National Institutes of Health, the Centers for Disease Control and Prevention, and other government partners to develop protocols for use by multiple investigators in order to coordinate the collection and use of COVID-19 convalescent plasma.
“It’s crucial that data be captured for every patient so that we really understand what safety and effectiveness looks like on as close to a real-world level as we can, as quickly as we can,” said Mr. Pitts, who is president and cofounder of the Center for Medicine in the Public Interest, and who also does consulting work for the FDA. “I understand that health care professionals are overworked and overburdened right now. I applaud them for their heroic work. But that doesn’t mean that we can shirk off collecting the data. When I was at the FDA, I helped address the SARS epidemic. The agency attitude at that point was, ‘Let’s get things that just might work through the process, as long as the cure isn’t going to be worse than the disease.’ I think that’s the attitude that’s leading the charge today.”
FDA approves ozanimod for relapsing and secondary progressive forms of MS
The Food and Drug Administration has approved the oral medication ozanimod (Zeposia) for relapsing forms of multiple sclerosis (MS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, according to a release from Bristol-Myers Squibb.
Ozanimod is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. It blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. Although its therapeutic mechanism of action in MS is unknown, it may involve the reduction of lymphocyte migration into the central nervous system. A genetic test is not required to start the drug, and no patient observation is required for the first dose, although up-titration of initial doses are required to reach the maintenance dose because a transient decrease in heart rate and atrioventricular conduction delays may occur, according to the company.
The approval is based on a pair of head-to-head studies that compared it with interferon beta-1a (Avonex) and together included more than 2,600 patients. It delivered better efficacy in terms of relative reduction in annualized relapse rate (48% at 1 year and 38% at 2 years). It also demonstrated better relative reduction of the number of T1-weighted gadolinium-enhanced brain lesions (63% fewer at 1 year and 53% fewer at 2 years) and number of new or enlarging T2 lesions (48% fewer at 1 year and 42% at 2 years).
Ozanimod is contraindicated in patients who, in the past 6 months, experienced a myocardial infarction, unstable angina, stroke, or other conditions. It is associated with other health risks, including infections, liver injury, additive immunosuppressive effects from prior immune-modulating therapies, and increased blood pressure. Certain assessments, such as recent complete blood count, ECG, liver function test, and current and prior medications and vaccinations, are required before initiation of treatment.
In its announcement, Bristol-Myers Squibb said that it has decided to delay the commercial launch of ozanimod during the COVID-19 pandemic until a later date.
The drug is also in development for additional immune-inflammatory indications, including ulcerative colitis and Crohn’s disease.
The full prescribing information can be found on the company’s website.
The Food and Drug Administration has approved the oral medication ozanimod (Zeposia) for relapsing forms of multiple sclerosis (MS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, according to a release from Bristol-Myers Squibb.
Ozanimod is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. It blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. Although its therapeutic mechanism of action in MS is unknown, it may involve the reduction of lymphocyte migration into the central nervous system. A genetic test is not required to start the drug, and no patient observation is required for the first dose, although up-titration of initial doses are required to reach the maintenance dose because a transient decrease in heart rate and atrioventricular conduction delays may occur, according to the company.
The approval is based on a pair of head-to-head studies that compared it with interferon beta-1a (Avonex) and together included more than 2,600 patients. It delivered better efficacy in terms of relative reduction in annualized relapse rate (48% at 1 year and 38% at 2 years). It also demonstrated better relative reduction of the number of T1-weighted gadolinium-enhanced brain lesions (63% fewer at 1 year and 53% fewer at 2 years) and number of new or enlarging T2 lesions (48% fewer at 1 year and 42% at 2 years).
Ozanimod is contraindicated in patients who, in the past 6 months, experienced a myocardial infarction, unstable angina, stroke, or other conditions. It is associated with other health risks, including infections, liver injury, additive immunosuppressive effects from prior immune-modulating therapies, and increased blood pressure. Certain assessments, such as recent complete blood count, ECG, liver function test, and current and prior medications and vaccinations, are required before initiation of treatment.
In its announcement, Bristol-Myers Squibb said that it has decided to delay the commercial launch of ozanimod during the COVID-19 pandemic until a later date.
The drug is also in development for additional immune-inflammatory indications, including ulcerative colitis and Crohn’s disease.
The full prescribing information can be found on the company’s website.
The Food and Drug Administration has approved the oral medication ozanimod (Zeposia) for relapsing forms of multiple sclerosis (MS), including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, according to a release from Bristol-Myers Squibb.
Ozanimod is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. It blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. Although its therapeutic mechanism of action in MS is unknown, it may involve the reduction of lymphocyte migration into the central nervous system. A genetic test is not required to start the drug, and no patient observation is required for the first dose, although up-titration of initial doses are required to reach the maintenance dose because a transient decrease in heart rate and atrioventricular conduction delays may occur, according to the company.
The approval is based on a pair of head-to-head studies that compared it with interferon beta-1a (Avonex) and together included more than 2,600 patients. It delivered better efficacy in terms of relative reduction in annualized relapse rate (48% at 1 year and 38% at 2 years). It also demonstrated better relative reduction of the number of T1-weighted gadolinium-enhanced brain lesions (63% fewer at 1 year and 53% fewer at 2 years) and number of new or enlarging T2 lesions (48% fewer at 1 year and 42% at 2 years).
Ozanimod is contraindicated in patients who, in the past 6 months, experienced a myocardial infarction, unstable angina, stroke, or other conditions. It is associated with other health risks, including infections, liver injury, additive immunosuppressive effects from prior immune-modulating therapies, and increased blood pressure. Certain assessments, such as recent complete blood count, ECG, liver function test, and current and prior medications and vaccinations, are required before initiation of treatment.
In its announcement, Bristol-Myers Squibb said that it has decided to delay the commercial launch of ozanimod during the COVID-19 pandemic until a later date.
The drug is also in development for additional immune-inflammatory indications, including ulcerative colitis and Crohn’s disease.
The full prescribing information can be found on the company’s website.
FDA to allow alternative respiratory devices to treat COVID-19
“Whenever possible, health care facilities should use FDA-cleared conventional/standard full-featured ventilators when necessary to support patients with respiratory failure, or a device subject to an Emergency Use Authorization (EUA), if any,” FDA stated in a guidance document issued March 22.
“However, to help ensure the availability of the greatest possible number of devices for this purpose, ... FDA does not intend to object to limited modifications to indications, claims, functionality, or to the hardware, software, or materials of FDA-cleared devices used to support patients with respiratory failure or respiratory insufficiency, without prior submission of a premarket notification” for the duration of the declared national emergency related to the COVID-19 pandemic.
FDA Commissioner Stephen Hahn, MD, said in a statement that the agency is doing everything it can to support patients, health care professionals, and others during this pandemic.
“One of the most impactful steps we can take is to help with access and availability to life-saving medical treatments,” he said. “Our policy issued today demonstrates our ability to react and adapt quickly during this pandemic and help very ill patients access the lifesaving ventilator support they need. To do that, we are providing maximum regulatory flexibility to facilitate an increase in ventilator inventory, while still providing crucial FDA oversight. We believe this action will immediately increase ventilator availability.”
The document identified examples of where modifications would not create undue risk, including the use of powered emergency ventilators and anesthesia gas machines for patients needing mechanical ventilation; the use of ventilators outside of their cleared environment; the use of devices used to treat patients with sleep apnea, such as CPAPs and BiPAPs, to treat respiratory insufficiency when appropriate design mitigations are in place to minimize aerosolization; and the use of oxygen concentrators for primary supply when medically necessary and clinically appropriate.
The agency also is allowing for changes to the hardware, software, and materials to FDA-cleared ventilators and anesthesia gas machines, such as modifications to motors, batteries, or other electrical components; material changes to components in the gas pathways or with other patient tissue contact; the introduction of filtration to minimize aerosolization; and other hardware and software modifications.
FDA is also allowing for products to be used past their indicated shelf life.
“Whenever possible, health care facilities should use FDA-cleared conventional/standard full-featured ventilators when necessary to support patients with respiratory failure, or a device subject to an Emergency Use Authorization (EUA), if any,” FDA stated in a guidance document issued March 22.
“However, to help ensure the availability of the greatest possible number of devices for this purpose, ... FDA does not intend to object to limited modifications to indications, claims, functionality, or to the hardware, software, or materials of FDA-cleared devices used to support patients with respiratory failure or respiratory insufficiency, without prior submission of a premarket notification” for the duration of the declared national emergency related to the COVID-19 pandemic.
FDA Commissioner Stephen Hahn, MD, said in a statement that the agency is doing everything it can to support patients, health care professionals, and others during this pandemic.
“One of the most impactful steps we can take is to help with access and availability to life-saving medical treatments,” he said. “Our policy issued today demonstrates our ability to react and adapt quickly during this pandemic and help very ill patients access the lifesaving ventilator support they need. To do that, we are providing maximum regulatory flexibility to facilitate an increase in ventilator inventory, while still providing crucial FDA oversight. We believe this action will immediately increase ventilator availability.”
The document identified examples of where modifications would not create undue risk, including the use of powered emergency ventilators and anesthesia gas machines for patients needing mechanical ventilation; the use of ventilators outside of their cleared environment; the use of devices used to treat patients with sleep apnea, such as CPAPs and BiPAPs, to treat respiratory insufficiency when appropriate design mitigations are in place to minimize aerosolization; and the use of oxygen concentrators for primary supply when medically necessary and clinically appropriate.
The agency also is allowing for changes to the hardware, software, and materials to FDA-cleared ventilators and anesthesia gas machines, such as modifications to motors, batteries, or other electrical components; material changes to components in the gas pathways or with other patient tissue contact; the introduction of filtration to minimize aerosolization; and other hardware and software modifications.
FDA is also allowing for products to be used past their indicated shelf life.
“Whenever possible, health care facilities should use FDA-cleared conventional/standard full-featured ventilators when necessary to support patients with respiratory failure, or a device subject to an Emergency Use Authorization (EUA), if any,” FDA stated in a guidance document issued March 22.
“However, to help ensure the availability of the greatest possible number of devices for this purpose, ... FDA does not intend to object to limited modifications to indications, claims, functionality, or to the hardware, software, or materials of FDA-cleared devices used to support patients with respiratory failure or respiratory insufficiency, without prior submission of a premarket notification” for the duration of the declared national emergency related to the COVID-19 pandemic.
FDA Commissioner Stephen Hahn, MD, said in a statement that the agency is doing everything it can to support patients, health care professionals, and others during this pandemic.
“One of the most impactful steps we can take is to help with access and availability to life-saving medical treatments,” he said. “Our policy issued today demonstrates our ability to react and adapt quickly during this pandemic and help very ill patients access the lifesaving ventilator support they need. To do that, we are providing maximum regulatory flexibility to facilitate an increase in ventilator inventory, while still providing crucial FDA oversight. We believe this action will immediately increase ventilator availability.”
The document identified examples of where modifications would not create undue risk, including the use of powered emergency ventilators and anesthesia gas machines for patients needing mechanical ventilation; the use of ventilators outside of their cleared environment; the use of devices used to treat patients with sleep apnea, such as CPAPs and BiPAPs, to treat respiratory insufficiency when appropriate design mitigations are in place to minimize aerosolization; and the use of oxygen concentrators for primary supply when medically necessary and clinically appropriate.
The agency also is allowing for changes to the hardware, software, and materials to FDA-cleared ventilators and anesthesia gas machines, such as modifications to motors, batteries, or other electrical components; material changes to components in the gas pathways or with other patient tissue contact; the introduction of filtration to minimize aerosolization; and other hardware and software modifications.
FDA is also allowing for products to be used past their indicated shelf life.
FDA issues warning on fecal transplant transmission of SARS-CoV-2
and that additional safety procedures may be required.

The risk of SARS-CoV-2 transmission through fecal microbiota transplant is unknown, but “several recent studies have documented the presence of SARS-CoV-2 ribonucleic acid (RNA) and/or SARS-CoV-2 virus in stool of infected individuals,” the FDA said in the press release. The testing of nasopharyngeal specimens from stool donors may not be available, and the availability and sensitivity of direct testing of stool for SARS-CoV-2 is currently unknown.
Because of the risk of serious adverse events, the FDA has issued several recommendations for any medically necessary usage of fecal microbiota transplantation involving stool samples donated after Dec. 1, 2019.
- Donor screening with questions directed at identifying those currently or recently infected with SARS-CoV-2.
- Testing donors and/or donor stool for SARS-CoV-2, as feasible.
- Development of criteria for exclusion of donors and donor stool based on screening and testing.
- Informed consent that includes information about the potential for transmission of SARS-CoV-2 via fecal microbiota transplantation, including transplantation prepared from stool from donors who are asymptomatic for COVID-19.
“As the scientific community learns more about SARS-CoV-2 and COVID-19, FDA will provide further information as warranted,” the agency said.
and that additional safety procedures may be required.
The risk of SARS-CoV-2 transmission through fecal microbiota transplant is unknown, but “several recent studies have documented the presence of SARS-CoV-2 ribonucleic acid (RNA) and/or SARS-CoV-2 virus in stool of infected individuals,” the FDA said in the press release. The testing of nasopharyngeal specimens from stool donors may not be available, and the availability and sensitivity of direct testing of stool for SARS-CoV-2 is currently unknown.
Because of the risk of serious adverse events, the FDA has issued several recommendations for any medically necessary usage of fecal microbiota transplantation involving stool samples donated after Dec. 1, 2019.
- Donor screening with questions directed at identifying those currently or recently infected with SARS-CoV-2.
- Testing donors and/or donor stool for SARS-CoV-2, as feasible.
- Development of criteria for exclusion of donors and donor stool based on screening and testing.
- Informed consent that includes information about the potential for transmission of SARS-CoV-2 via fecal microbiota transplantation, including transplantation prepared from stool from donors who are asymptomatic for COVID-19.
“As the scientific community learns more about SARS-CoV-2 and COVID-19, FDA will provide further information as warranted,” the agency said.
and that additional safety procedures may be required.
The risk of SARS-CoV-2 transmission through fecal microbiota transplant is unknown, but “several recent studies have documented the presence of SARS-CoV-2 ribonucleic acid (RNA) and/or SARS-CoV-2 virus in stool of infected individuals,” the FDA said in the press release. The testing of nasopharyngeal specimens from stool donors may not be available, and the availability and sensitivity of direct testing of stool for SARS-CoV-2 is currently unknown.
Because of the risk of serious adverse events, the FDA has issued several recommendations for any medically necessary usage of fecal microbiota transplantation involving stool samples donated after Dec. 1, 2019.
- Donor screening with questions directed at identifying those currently or recently infected with SARS-CoV-2.
- Testing donors and/or donor stool for SARS-CoV-2, as feasible.
- Development of criteria for exclusion of donors and donor stool based on screening and testing.
- Informed consent that includes information about the potential for transmission of SARS-CoV-2 via fecal microbiota transplantation, including transplantation prepared from stool from donors who are asymptomatic for COVID-19.
“As the scientific community learns more about SARS-CoV-2 and COVID-19, FDA will provide further information as warranted,” the agency said.
High rate of asymptomatic COVID-19 seen in cruise ship passengers
The high rate of asymptomatic COVID-19 infections aboard the cruise ship Diamond Princess “could partially explain the high attack rate among” the passengers and crew, according to the Centers for Disease Control and Prevention.

Testing of the 3,711 passengers and crew aboard the ship – the source of the largest outbreak outside of China during the initial stages of the pandemic – revealed that 19.2% were positive for COVID-19, Leah F. Moriarty, MPH, and associates reported in the Morbidity and Mortality Weekly Report.
At the time of testing, 46.5% (331) of the 712 infected individuals were asymptomatic, and “statistical models of the Diamond Princess outbreak suggest that 17.9% of infected persons never developed symptoms,” wrote Ms. Moriarty of the CDC COVID-19 response team, and associates.
RNA from the SARS-CoV-2 virus was found on surfaces in cabins up to 17 days after they had been vacated by passengers but before the cabins had been disinfected, the investigators noted.
The Diamond Princess departed from Yokohama, Japan, on Jan. 20, 2020, and returned on Feb. 3 after making six stops in three countries. The ship was quarantined upon its return because a symptomatic passenger who had departed Jan. 25 in Hong Kong tested positive for SARS-CoV-2, Ms. Moriarty and associates explained.
Of the 381 people from the ship who were symptomatic and tested positive, 37 (9.7%) needed intensive care and 9 (1.3%) died. There were 428 Americans on the ship, of whom 107 (25.0%) tested positive and 11 remained hospitalized in Japan as of March 13, they said.
“Many other cruise ships have since been implicated in SARS-CoV-2 transmission,” the investigators said, including the Grand Princess, which sailed out of San Francisco with 3,571 people on Feb. 21 and returned to Oakland on March 8.
That ship had been the site of virus transmission during its previous voyage from Feb. 11 to Feb. 21, from which more than 20 cases have been identified. During the latter trip, 21 of 45 passengers and crew tested positive before the ship docked. During the subsequent land-based quarantine, there have been 78 positive tests among the 469 people tested as of March 21, a rate of 16.6%, the research team reported.
“Public health responses to cruise ship outbreaks require extensive resources,” they wrote. “These responses required the coordination of stakeholders across multiple sectors, including U.S. government departments and agencies, foreign ministries of health, foreign embassies, state and local public health departments, hospitals, laboratories, and cruise ship companies.”
SOURCE: Moriarty LF et al. MMWR. 2020 Mar 23;69[early release]:1-6.
The high rate of asymptomatic COVID-19 infections aboard the cruise ship Diamond Princess “could partially explain the high attack rate among” the passengers and crew, according to the Centers for Disease Control and Prevention.
Testing of the 3,711 passengers and crew aboard the ship – the source of the largest outbreak outside of China during the initial stages of the pandemic – revealed that 19.2% were positive for COVID-19, Leah F. Moriarty, MPH, and associates reported in the Morbidity and Mortality Weekly Report.
At the time of testing, 46.5% (331) of the 712 infected individuals were asymptomatic, and “statistical models of the Diamond Princess outbreak suggest that 17.9% of infected persons never developed symptoms,” wrote Ms. Moriarty of the CDC COVID-19 response team, and associates.
RNA from the SARS-CoV-2 virus was found on surfaces in cabins up to 17 days after they had been vacated by passengers but before the cabins had been disinfected, the investigators noted.
The Diamond Princess departed from Yokohama, Japan, on Jan. 20, 2020, and returned on Feb. 3 after making six stops in three countries. The ship was quarantined upon its return because a symptomatic passenger who had departed Jan. 25 in Hong Kong tested positive for SARS-CoV-2, Ms. Moriarty and associates explained.
Of the 381 people from the ship who were symptomatic and tested positive, 37 (9.7%) needed intensive care and 9 (1.3%) died. There were 428 Americans on the ship, of whom 107 (25.0%) tested positive and 11 remained hospitalized in Japan as of March 13, they said.
“Many other cruise ships have since been implicated in SARS-CoV-2 transmission,” the investigators said, including the Grand Princess, which sailed out of San Francisco with 3,571 people on Feb. 21 and returned to Oakland on March 8.
That ship had been the site of virus transmission during its previous voyage from Feb. 11 to Feb. 21, from which more than 20 cases have been identified. During the latter trip, 21 of 45 passengers and crew tested positive before the ship docked. During the subsequent land-based quarantine, there have been 78 positive tests among the 469 people tested as of March 21, a rate of 16.6%, the research team reported.
“Public health responses to cruise ship outbreaks require extensive resources,” they wrote. “These responses required the coordination of stakeholders across multiple sectors, including U.S. government departments and agencies, foreign ministries of health, foreign embassies, state and local public health departments, hospitals, laboratories, and cruise ship companies.”
SOURCE: Moriarty LF et al. MMWR. 2020 Mar 23;69[early release]:1-6.
The high rate of asymptomatic COVID-19 infections aboard the cruise ship Diamond Princess “could partially explain the high attack rate among” the passengers and crew, according to the Centers for Disease Control and Prevention.
Testing of the 3,711 passengers and crew aboard the ship – the source of the largest outbreak outside of China during the initial stages of the pandemic – revealed that 19.2% were positive for COVID-19, Leah F. Moriarty, MPH, and associates reported in the Morbidity and Mortality Weekly Report.
At the time of testing, 46.5% (331) of the 712 infected individuals were asymptomatic, and “statistical models of the Diamond Princess outbreak suggest that 17.9% of infected persons never developed symptoms,” wrote Ms. Moriarty of the CDC COVID-19 response team, and associates.
RNA from the SARS-CoV-2 virus was found on surfaces in cabins up to 17 days after they had been vacated by passengers but before the cabins had been disinfected, the investigators noted.
The Diamond Princess departed from Yokohama, Japan, on Jan. 20, 2020, and returned on Feb. 3 after making six stops in three countries. The ship was quarantined upon its return because a symptomatic passenger who had departed Jan. 25 in Hong Kong tested positive for SARS-CoV-2, Ms. Moriarty and associates explained.
Of the 381 people from the ship who were symptomatic and tested positive, 37 (9.7%) needed intensive care and 9 (1.3%) died. There were 428 Americans on the ship, of whom 107 (25.0%) tested positive and 11 remained hospitalized in Japan as of March 13, they said.
“Many other cruise ships have since been implicated in SARS-CoV-2 transmission,” the investigators said, including the Grand Princess, which sailed out of San Francisco with 3,571 people on Feb. 21 and returned to Oakland on March 8.
That ship had been the site of virus transmission during its previous voyage from Feb. 11 to Feb. 21, from which more than 20 cases have been identified. During the latter trip, 21 of 45 passengers and crew tested positive before the ship docked. During the subsequent land-based quarantine, there have been 78 positive tests among the 469 people tested as of March 21, a rate of 16.6%, the research team reported.
“Public health responses to cruise ship outbreaks require extensive resources,” they wrote. “These responses required the coordination of stakeholders across multiple sectors, including U.S. government departments and agencies, foreign ministries of health, foreign embassies, state and local public health departments, hospitals, laboratories, and cruise ship companies.”
SOURCE: Moriarty LF et al. MMWR. 2020 Mar 23;69[early release]:1-6.
FROM MMWR