User login
FDA approves immunotherapy combo for liver cancer
Patients with advanced liver cancer have a new treatment option – the immunotherapy combination of nivolumab (Opdivo, Bristol-Myers Squibb) and ipilimumab (Yervoy, Bristol-Myers Squibb).
The US Food and Drug Administration (FDA) granted an accelerated approval of the combination for use in patients with advanced hepatocellular carcinoma (HCC) who have previously been treated with sorafenib (Nexavar, Bayer). Nivolumab is already approved as monotherapy for use in advanced HCC in patients who have previously been treated with sorafenib.
The immunotherapy combination has shown a response rate that is more than twice that seen with nivolumab alone. The combination was tested at three different dosage schedules in the single-arm phase 1/2 trial known as CheckMate-040, which was conducted in 148 patients with advanced HCC who had previously been treated with sorafenib.
The approval was based on one arm of this trial, a cohort of 49 patients who were treated with nivolumab 1 mg/kg IV and ipilimumab 3 mg/kg IV every 3 weeks for four doses, followed by nivolumab 240 mg every 2 weeks until disease progression or unacceptable toxicity.
After a minimum follow-up of 28 months, 33% (16/49) of these patients showed a response, with 8% (4/49) showing a complete response and 24% (12/49) a partial response.
In terms of duration of responses, 88% of the responses lasted at least 6 months, 56% at least 12 months, and 31% at least 24 months, according to the company.
The results that led to the 2017 approval of nivolumab monotherapy for advanced HCC, as previously reported by Medscape Medical News, come from a cohort of 154 patients who received nivolumab 3 mg/kg administered intravenously every 2 weeks.
The overall response rate was 14.3% (22 of 154 patients), with three patients (1.9%) showing a complete response and 19 patients (12.3%) a partial response. The duration of the responses ranged from 3.2 to 38.2+ months; 91% of those patients had responses of 6 months or longer, and 55% had responses of 12 months or longer.
Notably, patient responses in all arms were achieved regardless of baseline tumor PD-L1 status.
Aggressive disease, incidence is rising
“HCC is an aggressive disease in need of different treatment approaches,” said Anthony B. El-Khoueiry, MD, of the Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, in a company press statement.“The overall response rate observed in the Opdivo + Yervoy cohort of the CheckMate-040 trial underscores the potential of this dual immunotherapy as a possible treatment option for patients,” he commented. El-Khoueiry was lead investigator of the study and has received honoraria and consulting fees from Bristol-Myers Squibb.
“The incidence of liver cancer is rising in the United States, and HCC is the most common and aggressive form of the disease,” said Andrea Wilson, president and founder, Blue Faery: The Adrienne Wilson Liver Cancer Association.
“Today’s approval provides a new option for patients with HCC previously treated with sorafenib, giving the community more hope,” she said in the company press statement.
Safety profile
The nivolumab-ipilimumab combination had an “acceptable” safety profile overall in the CheckMate-040 trial, wrote lead study author Thomas Yao, MD, of the University at Hong Kong, China, and colleagues in their study abstract, which was presented at the 2019 annual meeting of the American Society of Clinical Oncology. Yao has received honoraria from Bristol-Myers Squibb and has served as a consultant to the company.
According to those data, 37% of patients had a grade 3-4 treatment-related adverse event (TRAE), the most common of which were pruritus and rash; 5% had grade 3–4 TRAEs that led to discontinuation.
Nivolumab is associated with pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, renal dysfunction, skin adverse reactions, encephalitis, other adverse reactions, and infusion-related reactions, as well as embryo-fetal toxicity. Ipilimumab has a boxed warning for immune-mediated adverse reactions.
Nivolumab alone is approved for use in the treatment of unresectable or metastatic melanoma, non–small cell lung cancer, small cell lung cancer, and classical Hodgkin lymphoma.
The combination of nivolumab with ipilimumab is also approved for use in the treatment of melanoma and renal cell carcinoma.
This article first appeared on Medscape.com.
Patients with advanced liver cancer have a new treatment option – the immunotherapy combination of nivolumab (Opdivo, Bristol-Myers Squibb) and ipilimumab (Yervoy, Bristol-Myers Squibb).
The US Food and Drug Administration (FDA) granted an accelerated approval of the combination for use in patients with advanced hepatocellular carcinoma (HCC) who have previously been treated with sorafenib (Nexavar, Bayer). Nivolumab is already approved as monotherapy for use in advanced HCC in patients who have previously been treated with sorafenib.
The immunotherapy combination has shown a response rate that is more than twice that seen with nivolumab alone. The combination was tested at three different dosage schedules in the single-arm phase 1/2 trial known as CheckMate-040, which was conducted in 148 patients with advanced HCC who had previously been treated with sorafenib.
The approval was based on one arm of this trial, a cohort of 49 patients who were treated with nivolumab 1 mg/kg IV and ipilimumab 3 mg/kg IV every 3 weeks for four doses, followed by nivolumab 240 mg every 2 weeks until disease progression or unacceptable toxicity.
After a minimum follow-up of 28 months, 33% (16/49) of these patients showed a response, with 8% (4/49) showing a complete response and 24% (12/49) a partial response.
In terms of duration of responses, 88% of the responses lasted at least 6 months, 56% at least 12 months, and 31% at least 24 months, according to the company.
The results that led to the 2017 approval of nivolumab monotherapy for advanced HCC, as previously reported by Medscape Medical News, come from a cohort of 154 patients who received nivolumab 3 mg/kg administered intravenously every 2 weeks.
The overall response rate was 14.3% (22 of 154 patients), with three patients (1.9%) showing a complete response and 19 patients (12.3%) a partial response. The duration of the responses ranged from 3.2 to 38.2+ months; 91% of those patients had responses of 6 months or longer, and 55% had responses of 12 months or longer.
Notably, patient responses in all arms were achieved regardless of baseline tumor PD-L1 status.
Aggressive disease, incidence is rising
“HCC is an aggressive disease in need of different treatment approaches,” said Anthony B. El-Khoueiry, MD, of the Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, in a company press statement.“The overall response rate observed in the Opdivo + Yervoy cohort of the CheckMate-040 trial underscores the potential of this dual immunotherapy as a possible treatment option for patients,” he commented. El-Khoueiry was lead investigator of the study and has received honoraria and consulting fees from Bristol-Myers Squibb.
“The incidence of liver cancer is rising in the United States, and HCC is the most common and aggressive form of the disease,” said Andrea Wilson, president and founder, Blue Faery: The Adrienne Wilson Liver Cancer Association.
“Today’s approval provides a new option for patients with HCC previously treated with sorafenib, giving the community more hope,” she said in the company press statement.
Safety profile
The nivolumab-ipilimumab combination had an “acceptable” safety profile overall in the CheckMate-040 trial, wrote lead study author Thomas Yao, MD, of the University at Hong Kong, China, and colleagues in their study abstract, which was presented at the 2019 annual meeting of the American Society of Clinical Oncology. Yao has received honoraria from Bristol-Myers Squibb and has served as a consultant to the company.
According to those data, 37% of patients had a grade 3-4 treatment-related adverse event (TRAE), the most common of which were pruritus and rash; 5% had grade 3–4 TRAEs that led to discontinuation.
Nivolumab is associated with pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, renal dysfunction, skin adverse reactions, encephalitis, other adverse reactions, and infusion-related reactions, as well as embryo-fetal toxicity. Ipilimumab has a boxed warning for immune-mediated adverse reactions.
Nivolumab alone is approved for use in the treatment of unresectable or metastatic melanoma, non–small cell lung cancer, small cell lung cancer, and classical Hodgkin lymphoma.
The combination of nivolumab with ipilimumab is also approved for use in the treatment of melanoma and renal cell carcinoma.
This article first appeared on Medscape.com.
Patients with advanced liver cancer have a new treatment option – the immunotherapy combination of nivolumab (Opdivo, Bristol-Myers Squibb) and ipilimumab (Yervoy, Bristol-Myers Squibb).
The US Food and Drug Administration (FDA) granted an accelerated approval of the combination for use in patients with advanced hepatocellular carcinoma (HCC) who have previously been treated with sorafenib (Nexavar, Bayer). Nivolumab is already approved as monotherapy for use in advanced HCC in patients who have previously been treated with sorafenib.
The immunotherapy combination has shown a response rate that is more than twice that seen with nivolumab alone. The combination was tested at three different dosage schedules in the single-arm phase 1/2 trial known as CheckMate-040, which was conducted in 148 patients with advanced HCC who had previously been treated with sorafenib.
The approval was based on one arm of this trial, a cohort of 49 patients who were treated with nivolumab 1 mg/kg IV and ipilimumab 3 mg/kg IV every 3 weeks for four doses, followed by nivolumab 240 mg every 2 weeks until disease progression or unacceptable toxicity.
After a minimum follow-up of 28 months, 33% (16/49) of these patients showed a response, with 8% (4/49) showing a complete response and 24% (12/49) a partial response.
In terms of duration of responses, 88% of the responses lasted at least 6 months, 56% at least 12 months, and 31% at least 24 months, according to the company.
The results that led to the 2017 approval of nivolumab monotherapy for advanced HCC, as previously reported by Medscape Medical News, come from a cohort of 154 patients who received nivolumab 3 mg/kg administered intravenously every 2 weeks.
The overall response rate was 14.3% (22 of 154 patients), with three patients (1.9%) showing a complete response and 19 patients (12.3%) a partial response. The duration of the responses ranged from 3.2 to 38.2+ months; 91% of those patients had responses of 6 months or longer, and 55% had responses of 12 months or longer.
Notably, patient responses in all arms were achieved regardless of baseline tumor PD-L1 status.
Aggressive disease, incidence is rising
“HCC is an aggressive disease in need of different treatment approaches,” said Anthony B. El-Khoueiry, MD, of the Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, in a company press statement.“The overall response rate observed in the Opdivo + Yervoy cohort of the CheckMate-040 trial underscores the potential of this dual immunotherapy as a possible treatment option for patients,” he commented. El-Khoueiry was lead investigator of the study and has received honoraria and consulting fees from Bristol-Myers Squibb.
“The incidence of liver cancer is rising in the United States, and HCC is the most common and aggressive form of the disease,” said Andrea Wilson, president and founder, Blue Faery: The Adrienne Wilson Liver Cancer Association.
“Today’s approval provides a new option for patients with HCC previously treated with sorafenib, giving the community more hope,” she said in the company press statement.
Safety profile
The nivolumab-ipilimumab combination had an “acceptable” safety profile overall in the CheckMate-040 trial, wrote lead study author Thomas Yao, MD, of the University at Hong Kong, China, and colleagues in their study abstract, which was presented at the 2019 annual meeting of the American Society of Clinical Oncology. Yao has received honoraria from Bristol-Myers Squibb and has served as a consultant to the company.
According to those data, 37% of patients had a grade 3-4 treatment-related adverse event (TRAE), the most common of which were pruritus and rash; 5% had grade 3–4 TRAEs that led to discontinuation.
Nivolumab is associated with pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, renal dysfunction, skin adverse reactions, encephalitis, other adverse reactions, and infusion-related reactions, as well as embryo-fetal toxicity. Ipilimumab has a boxed warning for immune-mediated adverse reactions.
Nivolumab alone is approved for use in the treatment of unresectable or metastatic melanoma, non–small cell lung cancer, small cell lung cancer, and classical Hodgkin lymphoma.
The combination of nivolumab with ipilimumab is also approved for use in the treatment of melanoma and renal cell carcinoma.
This article first appeared on Medscape.com.
FDA broadens nintedanib interstitial lung disease indication
A new indication for the tyrosine kinase inhibitor nintedanib approved by the Food and Drug Administration on March 9, 2020, broadened the drug’s targeted population to include patients with chronic fibrosing interstitial lung diseases with a progressive phenotype.

This new group of patients eligible for nintedanib treatment extends the drug’s labeling beyond patients with idiopathic pulmonary fibrosis (IPF) or interstitial lung disease (ILD) associated with systemic sclerosis or scleroderma, and may come close to doubling the total number of eligible patients.
The new, expanded indication “helps to fulfill an unmet treatment need, as patients with these life-threatening lung diseases have not had an approved medication until now,” said Banu Karimi-Shah, MD, acting deputy director of the division of pulmonary, allergy, and rheumatology products in the FDA’s Center for Drug Evaluation and Research, in a written agency statement that announced the new indication.
The FDA first approved nintedanib (Ofev) for treating IPF in October 2014, and then granted a second indication in September 2019 for ILD associated with systemic sclerosis or scleroderma.
A recent assessment of 1,285 Canadian patients diagnosed with fibrotic ILD and entered into a national registry (CARE-PF) showed that IPF was the associated diagnosis for 25% of patients, and that the majority of patients had other primary diagnoses such as connective tissue disease ILD in 33% of enrolled patients, unclassifiable ILD in 22%, chronic sensitivity pneumonitis in about 8%, sarcoidosis in 3%, as well as other types (BMC Pulm Med. 2019 Nov 27. doi: 10.1186/s12890-019-0986-4).
It remains unclear right now what percentage of patients with fibrotic ILD have the progressive form that would make them eligible for nintedanib treatment under the new indication, but it’s probably about another quarter of the entire ILD population, or roughly similar to the number of patients with an IPF etiology who are already eligible to get the drug, commented Martin Kolb, MD, a professor of respirology at McMaster University, Hamilton, Ont., and a coinvestigator on the CARE-PF registry. A goal of the registry, which has now enrolled nearly 3,700 ILD patients, is to track them serially to get a better handle on the prevalence of progressive disease. The percentage of patients with ILD associated with systemic sclerosis or scleroderma is “relatively small,” compared with these other two patients subgroups, Dr. Kolb said in an interview.
The evidence base for treating patients with progressive ILD is “really strong,” he noted, and comes primarily from a major trial reported last year – the INBUILD study – that randomized 663 patients to treatment with either nintedanib or placebo and showed that nintedanib treatment significantly cut the rate of decline in forced vital capacity during 1 year of treatment (New Engl J Med. 2019 Oct 31;381[18]:1718-27). The patients entered the study as referrals from routine practice with documented ILD with progressive fibrosis that was not responsive to treatment with steroids or other immunosuppressive drugs, and reflects real-world, community practice, Dr. Kolb said.
“Conceptually, it makes so much sense” to treat the patients enrolled in INBUILD, the same patients who fit the new indication, with an agent like nintedanib that slows fibrosis progression, and in some patients may bring progression to a virtual halt, said Dr. Kolb, a coinvestigator on the INBUILD study. Future treatment of these patients will likely involve coupling an antifibrotic drug like nintedanib with an anti-inflammatory agent, although combined treatment of this type needs more study, he noted. In the more than 5 years since nintedanib came onto the U.S. market, it has been used on more than 10,000 patients and has generated no new safety concerns beyond those first included in the drug’s label.
The INBUILD study was sponsored by Boehringer Ingelheim, the company that markets nintedanib. Dr. Kolb has been a consultant to, received honoraria from, and received research funding from Boehringer Ingelheim. He has also received consulting fees or honoraria from Genoa, Gilead, GlaxoSmithKline, Indalo, Prometic, Roche, and Third Pole, and he has received research funding from Actelion, Alkermes, Gilead, GlaxoSmithKline, Pharmaxis, Prometic, RespiVert, and Roche.
A new indication for the tyrosine kinase inhibitor nintedanib approved by the Food and Drug Administration on March 9, 2020, broadened the drug’s targeted population to include patients with chronic fibrosing interstitial lung diseases with a progressive phenotype.
This new group of patients eligible for nintedanib treatment extends the drug’s labeling beyond patients with idiopathic pulmonary fibrosis (IPF) or interstitial lung disease (ILD) associated with systemic sclerosis or scleroderma, and may come close to doubling the total number of eligible patients.
The new, expanded indication “helps to fulfill an unmet treatment need, as patients with these life-threatening lung diseases have not had an approved medication until now,” said Banu Karimi-Shah, MD, acting deputy director of the division of pulmonary, allergy, and rheumatology products in the FDA’s Center for Drug Evaluation and Research, in a written agency statement that announced the new indication.
The FDA first approved nintedanib (Ofev) for treating IPF in October 2014, and then granted a second indication in September 2019 for ILD associated with systemic sclerosis or scleroderma.
A recent assessment of 1,285 Canadian patients diagnosed with fibrotic ILD and entered into a national registry (CARE-PF) showed that IPF was the associated diagnosis for 25% of patients, and that the majority of patients had other primary diagnoses such as connective tissue disease ILD in 33% of enrolled patients, unclassifiable ILD in 22%, chronic sensitivity pneumonitis in about 8%, sarcoidosis in 3%, as well as other types (BMC Pulm Med. 2019 Nov 27. doi: 10.1186/s12890-019-0986-4).
It remains unclear right now what percentage of patients with fibrotic ILD have the progressive form that would make them eligible for nintedanib treatment under the new indication, but it’s probably about another quarter of the entire ILD population, or roughly similar to the number of patients with an IPF etiology who are already eligible to get the drug, commented Martin Kolb, MD, a professor of respirology at McMaster University, Hamilton, Ont., and a coinvestigator on the CARE-PF registry. A goal of the registry, which has now enrolled nearly 3,700 ILD patients, is to track them serially to get a better handle on the prevalence of progressive disease. The percentage of patients with ILD associated with systemic sclerosis or scleroderma is “relatively small,” compared with these other two patients subgroups, Dr. Kolb said in an interview.
The evidence base for treating patients with progressive ILD is “really strong,” he noted, and comes primarily from a major trial reported last year – the INBUILD study – that randomized 663 patients to treatment with either nintedanib or placebo and showed that nintedanib treatment significantly cut the rate of decline in forced vital capacity during 1 year of treatment (New Engl J Med. 2019 Oct 31;381[18]:1718-27). The patients entered the study as referrals from routine practice with documented ILD with progressive fibrosis that was not responsive to treatment with steroids or other immunosuppressive drugs, and reflects real-world, community practice, Dr. Kolb said.
“Conceptually, it makes so much sense” to treat the patients enrolled in INBUILD, the same patients who fit the new indication, with an agent like nintedanib that slows fibrosis progression, and in some patients may bring progression to a virtual halt, said Dr. Kolb, a coinvestigator on the INBUILD study. Future treatment of these patients will likely involve coupling an antifibrotic drug like nintedanib with an anti-inflammatory agent, although combined treatment of this type needs more study, he noted. In the more than 5 years since nintedanib came onto the U.S. market, it has been used on more than 10,000 patients and has generated no new safety concerns beyond those first included in the drug’s label.
The INBUILD study was sponsored by Boehringer Ingelheim, the company that markets nintedanib. Dr. Kolb has been a consultant to, received honoraria from, and received research funding from Boehringer Ingelheim. He has also received consulting fees or honoraria from Genoa, Gilead, GlaxoSmithKline, Indalo, Prometic, Roche, and Third Pole, and he has received research funding from Actelion, Alkermes, Gilead, GlaxoSmithKline, Pharmaxis, Prometic, RespiVert, and Roche.
A new indication for the tyrosine kinase inhibitor nintedanib approved by the Food and Drug Administration on March 9, 2020, broadened the drug’s targeted population to include patients with chronic fibrosing interstitial lung diseases with a progressive phenotype.
This new group of patients eligible for nintedanib treatment extends the drug’s labeling beyond patients with idiopathic pulmonary fibrosis (IPF) or interstitial lung disease (ILD) associated with systemic sclerosis or scleroderma, and may come close to doubling the total number of eligible patients.
The new, expanded indication “helps to fulfill an unmet treatment need, as patients with these life-threatening lung diseases have not had an approved medication until now,” said Banu Karimi-Shah, MD, acting deputy director of the division of pulmonary, allergy, and rheumatology products in the FDA’s Center for Drug Evaluation and Research, in a written agency statement that announced the new indication.
The FDA first approved nintedanib (Ofev) for treating IPF in October 2014, and then granted a second indication in September 2019 for ILD associated with systemic sclerosis or scleroderma.
A recent assessment of 1,285 Canadian patients diagnosed with fibrotic ILD and entered into a national registry (CARE-PF) showed that IPF was the associated diagnosis for 25% of patients, and that the majority of patients had other primary diagnoses such as connective tissue disease ILD in 33% of enrolled patients, unclassifiable ILD in 22%, chronic sensitivity pneumonitis in about 8%, sarcoidosis in 3%, as well as other types (BMC Pulm Med. 2019 Nov 27. doi: 10.1186/s12890-019-0986-4).
It remains unclear right now what percentage of patients with fibrotic ILD have the progressive form that would make them eligible for nintedanib treatment under the new indication, but it’s probably about another quarter of the entire ILD population, or roughly similar to the number of patients with an IPF etiology who are already eligible to get the drug, commented Martin Kolb, MD, a professor of respirology at McMaster University, Hamilton, Ont., and a coinvestigator on the CARE-PF registry. A goal of the registry, which has now enrolled nearly 3,700 ILD patients, is to track them serially to get a better handle on the prevalence of progressive disease. The percentage of patients with ILD associated with systemic sclerosis or scleroderma is “relatively small,” compared with these other two patients subgroups, Dr. Kolb said in an interview.
The evidence base for treating patients with progressive ILD is “really strong,” he noted, and comes primarily from a major trial reported last year – the INBUILD study – that randomized 663 patients to treatment with either nintedanib or placebo and showed that nintedanib treatment significantly cut the rate of decline in forced vital capacity during 1 year of treatment (New Engl J Med. 2019 Oct 31;381[18]:1718-27). The patients entered the study as referrals from routine practice with documented ILD with progressive fibrosis that was not responsive to treatment with steroids or other immunosuppressive drugs, and reflects real-world, community practice, Dr. Kolb said.
“Conceptually, it makes so much sense” to treat the patients enrolled in INBUILD, the same patients who fit the new indication, with an agent like nintedanib that slows fibrosis progression, and in some patients may bring progression to a virtual halt, said Dr. Kolb, a coinvestigator on the INBUILD study. Future treatment of these patients will likely involve coupling an antifibrotic drug like nintedanib with an anti-inflammatory agent, although combined treatment of this type needs more study, he noted. In the more than 5 years since nintedanib came onto the U.S. market, it has been used on more than 10,000 patients and has generated no new safety concerns beyond those first included in the drug’s label.
The INBUILD study was sponsored by Boehringer Ingelheim, the company that markets nintedanib. Dr. Kolb has been a consultant to, received honoraria from, and received research funding from Boehringer Ingelheim. He has also received consulting fees or honoraria from Genoa, Gilead, GlaxoSmithKline, Indalo, Prometic, Roche, and Third Pole, and he has received research funding from Actelion, Alkermes, Gilead, GlaxoSmithKline, Pharmaxis, Prometic, RespiVert, and Roche.
FDA cancels or postpones meetings amid COVID-19 concerns
Officials at the Food and Drug Administration’s Center for Drug Evaluation and Research are taking the precautionary step of canceling or postponing advisory committee meetings and limiting staff travel in an effort to help curb the spread of the COVID-19.

“The outbreak of respiratory illness caused by a novel coronavirus, COVID-19, that started in China is spreading to other countries, including the United States,” CDER Director Janet Woodcock, MD, said in a memo to CDER staff. “As a precaution, FDA is canceling foreign official agency travel and limiting domestic travel to mission critical only, effective immediately and through April.”
Additionally, the memo notes that “CDER-organized external meetings, conferences, and workshops will be postponed or canceled from March 10 through April.”
“To mitigate the impact on our work, I encourage you to hold meetings with external stakeholders through teleconference, when possible,” she wrote.
Thus far, only a few CDER events on the FDA’s meeting webpage are listed as being canceled or postponed. Some of the affected meetings include a March 10 public meeting on patient-focused drug development for stimulant-use disorder, a March 11 meeting of the Nonprescription Drug Advisory Committee, and a March 30 public meeting on patient-focused drug development for vitiligo, all of which are postponed until further notice. The Center for Biologics Evaluation and Research also has postponed until further notice its U.S.–Japan Cellular and Gene Therapy Conference, originally scheduled for March 12.
Dr. Woodcock also noted in the memo that in relation to inspections, “we plan to use technology and established agreements with our foreign counterparts to minimize disruptions to the drug supply chain and to applications under review, so that Americans can continue to get their medications.”
Officials at the Food and Drug Administration’s Center for Drug Evaluation and Research are taking the precautionary step of canceling or postponing advisory committee meetings and limiting staff travel in an effort to help curb the spread of the COVID-19.
“The outbreak of respiratory illness caused by a novel coronavirus, COVID-19, that started in China is spreading to other countries, including the United States,” CDER Director Janet Woodcock, MD, said in a memo to CDER staff. “As a precaution, FDA is canceling foreign official agency travel and limiting domestic travel to mission critical only, effective immediately and through April.”
Additionally, the memo notes that “CDER-organized external meetings, conferences, and workshops will be postponed or canceled from March 10 through April.”
“To mitigate the impact on our work, I encourage you to hold meetings with external stakeholders through teleconference, when possible,” she wrote.
Thus far, only a few CDER events on the FDA’s meeting webpage are listed as being canceled or postponed. Some of the affected meetings include a March 10 public meeting on patient-focused drug development for stimulant-use disorder, a March 11 meeting of the Nonprescription Drug Advisory Committee, and a March 30 public meeting on patient-focused drug development for vitiligo, all of which are postponed until further notice. The Center for Biologics Evaluation and Research also has postponed until further notice its U.S.–Japan Cellular and Gene Therapy Conference, originally scheduled for March 12.
Dr. Woodcock also noted in the memo that in relation to inspections, “we plan to use technology and established agreements with our foreign counterparts to minimize disruptions to the drug supply chain and to applications under review, so that Americans can continue to get their medications.”
Officials at the Food and Drug Administration’s Center for Drug Evaluation and Research are taking the precautionary step of canceling or postponing advisory committee meetings and limiting staff travel in an effort to help curb the spread of the COVID-19.
“The outbreak of respiratory illness caused by a novel coronavirus, COVID-19, that started in China is spreading to other countries, including the United States,” CDER Director Janet Woodcock, MD, said in a memo to CDER staff. “As a precaution, FDA is canceling foreign official agency travel and limiting domestic travel to mission critical only, effective immediately and through April.”
Additionally, the memo notes that “CDER-organized external meetings, conferences, and workshops will be postponed or canceled from March 10 through April.”
“To mitigate the impact on our work, I encourage you to hold meetings with external stakeholders through teleconference, when possible,” she wrote.
Thus far, only a few CDER events on the FDA’s meeting webpage are listed as being canceled or postponed. Some of the affected meetings include a March 10 public meeting on patient-focused drug development for stimulant-use disorder, a March 11 meeting of the Nonprescription Drug Advisory Committee, and a March 30 public meeting on patient-focused drug development for vitiligo, all of which are postponed until further notice. The Center for Biologics Evaluation and Research also has postponed until further notice its U.S.–Japan Cellular and Gene Therapy Conference, originally scheduled for March 12.
Dr. Woodcock also noted in the memo that in relation to inspections, “we plan to use technology and established agreements with our foreign counterparts to minimize disruptions to the drug supply chain and to applications under review, so that Americans can continue to get their medications.”
TBI deaths from falls on the rise
A 17% surge in mortality from fall-related traumatic brain injuries from 2008 to 2017 was driven largely by increases among those aged 75 years and older, according to investigators from the Centers for Disease Control and Prevention.
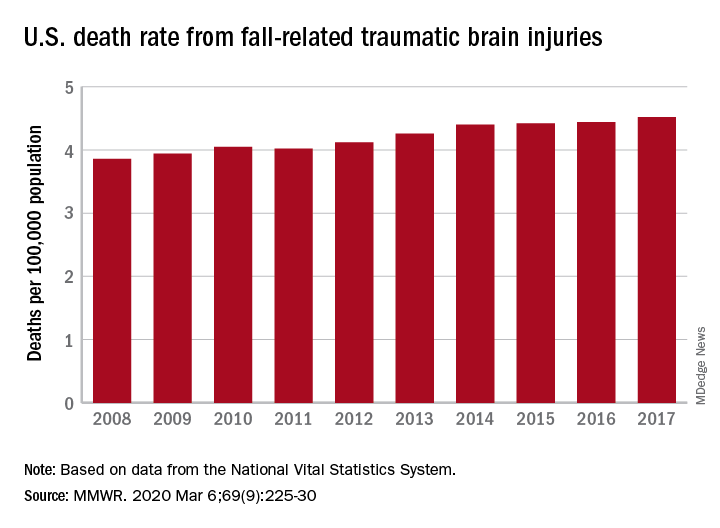
Nationally, the rate of deaths from traumatic brain injuries (TBIs) caused by unintentional falls rose from 3.86 per 100,000 population in 2008 to 4.52 per 100,000 in 2017, as the number of deaths went from 12,311 to 17,408, said Alexis B. Peterson, PhD, and Scott R. Kegler, PhD, of the CDC’s National Center for Injury Prevention and Control in Atlanta.
“This increase might be explained by longer survival following the onset of common diseases such as stroke, cancer, and heart disease or be attributable to the increasing population of older adults in the United States,” they suggested in the Mortality and Morbidity Weekly Report.
The rate of fall-related TBI among Americans aged 75 years and older increased by an average of 2.6% per year from 2008 to 2017, compared with 1.8% in those aged 55-74. Over that same time, death rates dropped for those aged 35-44 (–0.3%), 18-34 (–1.1%), and 0-17 (–4.3%), they said, based on data from the National Vital Statistics System’s multiple cause-of-death database.
The death rate increased fastest in residents of rural areas (2.9% per year), but deaths from fall-related TBI were up at all levels of urbanization. The largest central cities and fringe metro areas were up by 1.4% a year, with larger annual increases seen in medium-size cities (2.1%), small cities (2.2%), and small towns (2.1%), Dr. Peterson and Dr. Kegler said.
Rates of TBI-related mortality in general are higher in rural areas, they noted, and “heterogeneity in the availability and accessibility of resources (e.g., access to high-level trauma centers and rehabilitative services) can result in disparities in postinjury outcomes.”
State-specific rates increased in 45 states, although Alaska was excluded from the analysis because of its small number of cases (less than 20). Increases were significant in 29 states, but none of the changes were significant in the 4 states with lower rates at the end of the study period, the investigators reported.
“In older adults, evidence-based fall prevention strategies can prevent falls and avert costly medical expenditures,” Dr. Peterson and Dr. Kegler said, suggesting that health care providers “consider prescribing exercises that incorporate balance, strength and gait activities, such as tai chi, and reviewing and managing medications linked to falls.”
SOURCE: Peterson AB, Kegler SR. MMWR. 2019 Mar 6;69(9):225-30.
A 17% surge in mortality from fall-related traumatic brain injuries from 2008 to 2017 was driven largely by increases among those aged 75 years and older, according to investigators from the Centers for Disease Control and Prevention.
Nationally, the rate of deaths from traumatic brain injuries (TBIs) caused by unintentional falls rose from 3.86 per 100,000 population in 2008 to 4.52 per 100,000 in 2017, as the number of deaths went from 12,311 to 17,408, said Alexis B. Peterson, PhD, and Scott R. Kegler, PhD, of the CDC’s National Center for Injury Prevention and Control in Atlanta.
“This increase might be explained by longer survival following the onset of common diseases such as stroke, cancer, and heart disease or be attributable to the increasing population of older adults in the United States,” they suggested in the Mortality and Morbidity Weekly Report.
The rate of fall-related TBI among Americans aged 75 years and older increased by an average of 2.6% per year from 2008 to 2017, compared with 1.8% in those aged 55-74. Over that same time, death rates dropped for those aged 35-44 (–0.3%), 18-34 (–1.1%), and 0-17 (–4.3%), they said, based on data from the National Vital Statistics System’s multiple cause-of-death database.
The death rate increased fastest in residents of rural areas (2.9% per year), but deaths from fall-related TBI were up at all levels of urbanization. The largest central cities and fringe metro areas were up by 1.4% a year, with larger annual increases seen in medium-size cities (2.1%), small cities (2.2%), and small towns (2.1%), Dr. Peterson and Dr. Kegler said.
Rates of TBI-related mortality in general are higher in rural areas, they noted, and “heterogeneity in the availability and accessibility of resources (e.g., access to high-level trauma centers and rehabilitative services) can result in disparities in postinjury outcomes.”
State-specific rates increased in 45 states, although Alaska was excluded from the analysis because of its small number of cases (less than 20). Increases were significant in 29 states, but none of the changes were significant in the 4 states with lower rates at the end of the study period, the investigators reported.
“In older adults, evidence-based fall prevention strategies can prevent falls and avert costly medical expenditures,” Dr. Peterson and Dr. Kegler said, suggesting that health care providers “consider prescribing exercises that incorporate balance, strength and gait activities, such as tai chi, and reviewing and managing medications linked to falls.”
SOURCE: Peterson AB, Kegler SR. MMWR. 2019 Mar 6;69(9):225-30.
A 17% surge in mortality from fall-related traumatic brain injuries from 2008 to 2017 was driven largely by increases among those aged 75 years and older, according to investigators from the Centers for Disease Control and Prevention.
Nationally, the rate of deaths from traumatic brain injuries (TBIs) caused by unintentional falls rose from 3.86 per 100,000 population in 2008 to 4.52 per 100,000 in 2017, as the number of deaths went from 12,311 to 17,408, said Alexis B. Peterson, PhD, and Scott R. Kegler, PhD, of the CDC’s National Center for Injury Prevention and Control in Atlanta.
“This increase might be explained by longer survival following the onset of common diseases such as stroke, cancer, and heart disease or be attributable to the increasing population of older adults in the United States,” they suggested in the Mortality and Morbidity Weekly Report.
The rate of fall-related TBI among Americans aged 75 years and older increased by an average of 2.6% per year from 2008 to 2017, compared with 1.8% in those aged 55-74. Over that same time, death rates dropped for those aged 35-44 (–0.3%), 18-34 (–1.1%), and 0-17 (–4.3%), they said, based on data from the National Vital Statistics System’s multiple cause-of-death database.
The death rate increased fastest in residents of rural areas (2.9% per year), but deaths from fall-related TBI were up at all levels of urbanization. The largest central cities and fringe metro areas were up by 1.4% a year, with larger annual increases seen in medium-size cities (2.1%), small cities (2.2%), and small towns (2.1%), Dr. Peterson and Dr. Kegler said.
Rates of TBI-related mortality in general are higher in rural areas, they noted, and “heterogeneity in the availability and accessibility of resources (e.g., access to high-level trauma centers and rehabilitative services) can result in disparities in postinjury outcomes.”
State-specific rates increased in 45 states, although Alaska was excluded from the analysis because of its small number of cases (less than 20). Increases were significant in 29 states, but none of the changes were significant in the 4 states with lower rates at the end of the study period, the investigators reported.
“In older adults, evidence-based fall prevention strategies can prevent falls and avert costly medical expenditures,” Dr. Peterson and Dr. Kegler said, suggesting that health care providers “consider prescribing exercises that incorporate balance, strength and gait activities, such as tai chi, and reviewing and managing medications linked to falls.”
SOURCE: Peterson AB, Kegler SR. MMWR. 2019 Mar 6;69(9):225-30.
FROM MMWR
Flu activity declines again but remains high
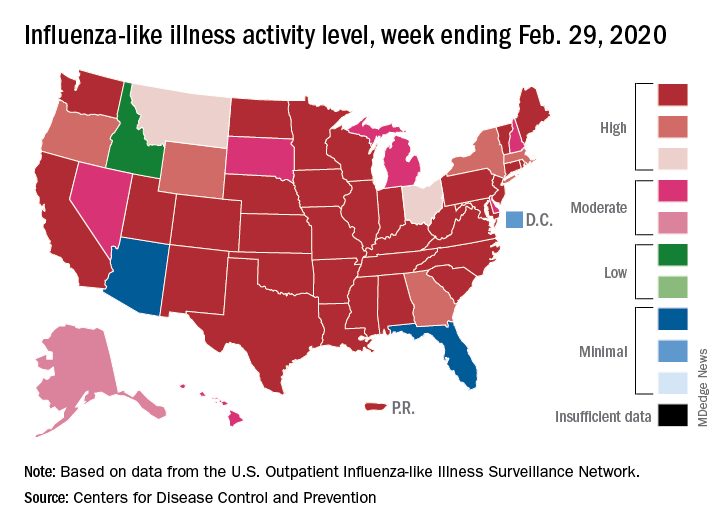
Outpatient visits to health care providers for influenza-like illness dropped from 5.5% the previous week to 5.3% of all visits for the week ending Feb. 29, the Centers for Disease Control and Prevention said on March 6.
The national baseline rate of 2.4% was first reached during the week of Nov. 9, 2019 – marking the start of flu season – and has remained at or above that level for 17 consecutive weeks. Last year’s season, which also was the longest in a decade, lasted 21 consecutive weeks but started 2 weeks later than the current season and had a lower outpatient-visit rate (4.5%) for the last week of February, CDC data show.
This season’s earlier start could mean that even a somewhat steep decline in visits to below the baseline rate – marking the end of the season – might take 5 or 6 weeks and would make 2019-2020 even longer than 2018-2019.
The activity situation on the state level reflects the small national decline. For the week ending Feb. 29, there were 33 states at level 10 on the CDC’s 1-10 activity scale, compared with 37 the week before, and a total of 40 in the “high” range of 8-10, compared with 43 the week before, the CDC’s influenza division reported.
The other main measure of influenza activity, percentage of respiratory specimens testing positive, also declined for the third week in a row and is now at 24.3% after reaching a high of 30.3% during the week of Feb. 2-8, the influenza division said.
The overall cumulative hospitalization rate continues to remain at a fairly typical 57.9 per 100,000 population, but rates for school-aged children (84.9 per 100,000) and young adults (31.2 per 100,000) are among the highest ever recorded at this point in the season. Mortality among children – now at 136 for 2019-2020 – is higher than for any season since reporting began in 2004, with the exception of the 2009 pandemic, the CDC said.
Outpatient visits to health care providers for influenza-like illness dropped from 5.5% the previous week to 5.3% of all visits for the week ending Feb. 29, the Centers for Disease Control and Prevention said on March 6.
The national baseline rate of 2.4% was first reached during the week of Nov. 9, 2019 – marking the start of flu season – and has remained at or above that level for 17 consecutive weeks. Last year’s season, which also was the longest in a decade, lasted 21 consecutive weeks but started 2 weeks later than the current season and had a lower outpatient-visit rate (4.5%) for the last week of February, CDC data show.
This season’s earlier start could mean that even a somewhat steep decline in visits to below the baseline rate – marking the end of the season – might take 5 or 6 weeks and would make 2019-2020 even longer than 2018-2019.
The activity situation on the state level reflects the small national decline. For the week ending Feb. 29, there were 33 states at level 10 on the CDC’s 1-10 activity scale, compared with 37 the week before, and a total of 40 in the “high” range of 8-10, compared with 43 the week before, the CDC’s influenza division reported.
The other main measure of influenza activity, percentage of respiratory specimens testing positive, also declined for the third week in a row and is now at 24.3% after reaching a high of 30.3% during the week of Feb. 2-8, the influenza division said.
The overall cumulative hospitalization rate continues to remain at a fairly typical 57.9 per 100,000 population, but rates for school-aged children (84.9 per 100,000) and young adults (31.2 per 100,000) are among the highest ever recorded at this point in the season. Mortality among children – now at 136 for 2019-2020 – is higher than for any season since reporting began in 2004, with the exception of the 2009 pandemic, the CDC said.
Outpatient visits to health care providers for influenza-like illness dropped from 5.5% the previous week to 5.3% of all visits for the week ending Feb. 29, the Centers for Disease Control and Prevention said on March 6.
The national baseline rate of 2.4% was first reached during the week of Nov. 9, 2019 – marking the start of flu season – and has remained at or above that level for 17 consecutive weeks. Last year’s season, which also was the longest in a decade, lasted 21 consecutive weeks but started 2 weeks later than the current season and had a lower outpatient-visit rate (4.5%) for the last week of February, CDC data show.
This season’s earlier start could mean that even a somewhat steep decline in visits to below the baseline rate – marking the end of the season – might take 5 or 6 weeks and would make 2019-2020 even longer than 2018-2019.
The activity situation on the state level reflects the small national decline. For the week ending Feb. 29, there were 33 states at level 10 on the CDC’s 1-10 activity scale, compared with 37 the week before, and a total of 40 in the “high” range of 8-10, compared with 43 the week before, the CDC’s influenza division reported.
The other main measure of influenza activity, percentage of respiratory specimens testing positive, also declined for the third week in a row and is now at 24.3% after reaching a high of 30.3% during the week of Feb. 2-8, the influenza division said.
The overall cumulative hospitalization rate continues to remain at a fairly typical 57.9 per 100,000 population, but rates for school-aged children (84.9 per 100,000) and young adults (31.2 per 100,000) are among the highest ever recorded at this point in the season. Mortality among children – now at 136 for 2019-2020 – is higher than for any season since reporting began in 2004, with the exception of the 2009 pandemic, the CDC said.
FDA issues stronger warning on neuropsychiatric event risk linked to montelukast
The Food and Drug Administration has issued , a prescription drug for asthma and allergy.
The new boxed warning advises health care providers to avoid prescribing montelukast for patients with mild symptoms, particularly those with allergic rhinitis, the FDA said in a press release. The drug was first approved in 1998, and the product labeling was updated in 2008 to include information about neuropsychiatric adverse events reported with usage of montelukast.
While the Sentinel study, along with other observational studies, did not find an increased risk of mental health side effects with montelukast treatment, compared with inhaled corticosteroids, those studies had limitations that may have affected results, the FDA said in the Drug Safety Communication. However, the FDA has continued to receive reports of neuropsychiatric events – including agitation, depression, sleeping problems, and suicidal thoughts and actions – in patients receiving the medication.
“The incidence of neuropsychiatric events associated with montelukast is unknown, but some reports are serious, and many patients and health care professionals are not fully aware of these risks,” Sally Seymour, MD, director of the division of pulmonary, allergy and rheumatology products in the FDA’s Center for Drug Evaluation and Research, said in the press release. “There are many other safe and effective medications to treat allergies with extensive history of use and safety, such that many products are available over the counter without a prescription.”
In addition to the boxed warning, the FDA now requires a new medication guide to be given to patients with each montelukast prescription, the FDA said.
The Food and Drug Administration has issued , a prescription drug for asthma and allergy.
The new boxed warning advises health care providers to avoid prescribing montelukast for patients with mild symptoms, particularly those with allergic rhinitis, the FDA said in a press release. The drug was first approved in 1998, and the product labeling was updated in 2008 to include information about neuropsychiatric adverse events reported with usage of montelukast.
While the Sentinel study, along with other observational studies, did not find an increased risk of mental health side effects with montelukast treatment, compared with inhaled corticosteroids, those studies had limitations that may have affected results, the FDA said in the Drug Safety Communication. However, the FDA has continued to receive reports of neuropsychiatric events – including agitation, depression, sleeping problems, and suicidal thoughts and actions – in patients receiving the medication.
“The incidence of neuropsychiatric events associated with montelukast is unknown, but some reports are serious, and many patients and health care professionals are not fully aware of these risks,” Sally Seymour, MD, director of the division of pulmonary, allergy and rheumatology products in the FDA’s Center for Drug Evaluation and Research, said in the press release. “There are many other safe and effective medications to treat allergies with extensive history of use and safety, such that many products are available over the counter without a prescription.”
In addition to the boxed warning, the FDA now requires a new medication guide to be given to patients with each montelukast prescription, the FDA said.
The Food and Drug Administration has issued , a prescription drug for asthma and allergy.
The new boxed warning advises health care providers to avoid prescribing montelukast for patients with mild symptoms, particularly those with allergic rhinitis, the FDA said in a press release. The drug was first approved in 1998, and the product labeling was updated in 2008 to include information about neuropsychiatric adverse events reported with usage of montelukast.
While the Sentinel study, along with other observational studies, did not find an increased risk of mental health side effects with montelukast treatment, compared with inhaled corticosteroids, those studies had limitations that may have affected results, the FDA said in the Drug Safety Communication. However, the FDA has continued to receive reports of neuropsychiatric events – including agitation, depression, sleeping problems, and suicidal thoughts and actions – in patients receiving the medication.
“The incidence of neuropsychiatric events associated with montelukast is unknown, but some reports are serious, and many patients and health care professionals are not fully aware of these risks,” Sally Seymour, MD, director of the division of pulmonary, allergy and rheumatology products in the FDA’s Center for Drug Evaluation and Research, said in the press release. “There are many other safe and effective medications to treat allergies with extensive history of use and safety, such that many products are available over the counter without a prescription.”
In addition to the boxed warning, the FDA now requires a new medication guide to be given to patients with each montelukast prescription, the FDA said.
FDA moves to expand coronavirus testing capacity; CDC clarifies testing criteria
The White House Coronavirus Task Force appeared at a press briefing March 2 to provide updates about testing strategies and public health coordination to address the current outbreak of the coronavirus COVID-19. Speaking at the briefing, led by Vice President Mike Pence, Centers for Disease Control and Prevention (CDC) director Robert Redfield, MD, said, “Working with our public health partners we continue to be able to identify new community cases and use our public health efforts to aggressively confirm, isolate, and do contact tracking.” Calling state, local, tribal, and territorial public health departments “the backbone of the public health system in our country,” Dr. Redfield noted that he expected many more confirmed COVID-19 cases to emerge.

At least some of the expected increase in confirmed cases of COVID-19 will occur because of expanded testing capacity, noted several of the task force members. On Feb. 29, the Food and Drug Administration issued a the virus that is causing the current outbreak of COVID-19.
Highly qualified laboratories, including both those run by public agencies and private labs, are now authorized to begin using their own validated test for the virus as long as they submit an Emergency Use Authorization (EUA) to the Food and Drug Administration within 15 days of notifying the agency of validation.
“To effectively respond to the COVID-19 outbreak, rapid detection of cases and contacts, appropriate clinical management and infection control, and implementation of community mitigation efforts are critical. This can best be achieved with wide availability of testing capabilities in health care settings, reference and commercial laboratories, and at the point of care,” the agency wrote in a press announcement of the expedited test expansion.
On Feb. 4, the Secretary of the Department of Health & Human Services declared a coronavirus public health emergency. The FDA was then authorized to allow individual laboratories with validated coronavirus tests to begin testing samples immediately. The goal is a more rapid and expanded testing capacity in the United States.
“The global emergence of COVID-19 is concerning, and we appreciate the efforts of the FDA to help bring more testing capability to the U.S.,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD), said in the press release.
The new guidance that permits the immediate use of clinical tests after individual development and validation, said the FDA, only applies to labs already certified to perform high complexity testing under Clinical Laboratory Improvement Amendments. Many governmental, academic, and private laboratories fall into this category, however.
“Under this policy, we expect certain laboratories who develop validated tests for coronavirus would begin using them right away prior to FDA review,” said Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health. “We believe this action will support laboratories across the country working on this urgent public health situation,” he added in the press release.
“By the end of this week, close to a million tests will be available,” FDA Commissioner Stephen M. Hahn, MD, said during the March 2 briefing.*
Updated criteria
The CDC is maintaining updated criteria for the virus testing on its website. Testing criteria are based both on clinical features and epidemiologic risk.
Individuals with less severe clinical features – those who have either fever or signs and symptoms of lower respiratory disease such as cough or shortness of breath, but who don’t require hospitalization – should be tested if they have high epidemiologic risk. “High risk” is defined by the CDC as any individual, including health care workers, who has had close contact with a person with confirmed COVID-19 within the past 2 weeks. For health care workers, testing can be considered even if they have relatively mild respiratory symptoms or have had contact with a person who is suspected, but not yet confirmed, to have coronavirus.
In its testing guidance, the CDC recognizes that defining close contact is difficult. General guidelines are that individuals are considered to have been in close contact with a person who has COVID-19 if they were within about six feet of the person for a prolonged period, or cared for or have spent a prolonged amount of time in the same room or house as a person with confirmed COVID-19.
Individuals who have both fever and signs or symptoms of lower respiratory illness who require hospitalization should be tested if they have a history of travel from any affected geographic area within 14 days of the onset of their symptoms. The CDC now defines “affected geographic area” as any country or region that has at least a CDC Level 2 Travel Health Notice for COVID-19, so that the testing criteria themselves don’t need to be updated when new geographic areas are included in these alerts. As of March 3, China, Iran, Italy, Japan, and South Korea all have Level 2 or 3 travel alerts.
The CDC now recommends that any patient who has severe acute lower respiratory illness that requires hospitalization and doesn’t have an alternative diagnosis should be tested, even without any identified source of exposure.
“Despite seeing these new cases, the risk to the American people is low,” said the CDC’s Dr. Redfield. In response to a question from the press about how fast the coronavirus will spread across the United States, Dr. Redfield said, “From the beginning we’ve anticipated seeing community cases pop up.” He added that as these cases arise, testing and public health strategies will focus on unearthing linkages and contacts to learn how the virus is spreading. “We’ll use the public health strategies that we can to limit that transmission,” he said.
*An earlier version of this article misattributed this quote.
The White House Coronavirus Task Force appeared at a press briefing March 2 to provide updates about testing strategies and public health coordination to address the current outbreak of the coronavirus COVID-19. Speaking at the briefing, led by Vice President Mike Pence, Centers for Disease Control and Prevention (CDC) director Robert Redfield, MD, said, “Working with our public health partners we continue to be able to identify new community cases and use our public health efforts to aggressively confirm, isolate, and do contact tracking.” Calling state, local, tribal, and territorial public health departments “the backbone of the public health system in our country,” Dr. Redfield noted that he expected many more confirmed COVID-19 cases to emerge.
At least some of the expected increase in confirmed cases of COVID-19 will occur because of expanded testing capacity, noted several of the task force members. On Feb. 29, the Food and Drug Administration issued a the virus that is causing the current outbreak of COVID-19.
Highly qualified laboratories, including both those run by public agencies and private labs, are now authorized to begin using their own validated test for the virus as long as they submit an Emergency Use Authorization (EUA) to the Food and Drug Administration within 15 days of notifying the agency of validation.
“To effectively respond to the COVID-19 outbreak, rapid detection of cases and contacts, appropriate clinical management and infection control, and implementation of community mitigation efforts are critical. This can best be achieved with wide availability of testing capabilities in health care settings, reference and commercial laboratories, and at the point of care,” the agency wrote in a press announcement of the expedited test expansion.
On Feb. 4, the Secretary of the Department of Health & Human Services declared a coronavirus public health emergency. The FDA was then authorized to allow individual laboratories with validated coronavirus tests to begin testing samples immediately. The goal is a more rapid and expanded testing capacity in the United States.
“The global emergence of COVID-19 is concerning, and we appreciate the efforts of the FDA to help bring more testing capability to the U.S.,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD), said in the press release.
The new guidance that permits the immediate use of clinical tests after individual development and validation, said the FDA, only applies to labs already certified to perform high complexity testing under Clinical Laboratory Improvement Amendments. Many governmental, academic, and private laboratories fall into this category, however.
“Under this policy, we expect certain laboratories who develop validated tests for coronavirus would begin using them right away prior to FDA review,” said Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health. “We believe this action will support laboratories across the country working on this urgent public health situation,” he added in the press release.
“By the end of this week, close to a million tests will be available,” FDA Commissioner Stephen M. Hahn, MD, said during the March 2 briefing.*
Updated criteria
The CDC is maintaining updated criteria for the virus testing on its website. Testing criteria are based both on clinical features and epidemiologic risk.
Individuals with less severe clinical features – those who have either fever or signs and symptoms of lower respiratory disease such as cough or shortness of breath, but who don’t require hospitalization – should be tested if they have high epidemiologic risk. “High risk” is defined by the CDC as any individual, including health care workers, who has had close contact with a person with confirmed COVID-19 within the past 2 weeks. For health care workers, testing can be considered even if they have relatively mild respiratory symptoms or have had contact with a person who is suspected, but not yet confirmed, to have coronavirus.
In its testing guidance, the CDC recognizes that defining close contact is difficult. General guidelines are that individuals are considered to have been in close contact with a person who has COVID-19 if they were within about six feet of the person for a prolonged period, or cared for or have spent a prolonged amount of time in the same room or house as a person with confirmed COVID-19.
Individuals who have both fever and signs or symptoms of lower respiratory illness who require hospitalization should be tested if they have a history of travel from any affected geographic area within 14 days of the onset of their symptoms. The CDC now defines “affected geographic area” as any country or region that has at least a CDC Level 2 Travel Health Notice for COVID-19, so that the testing criteria themselves don’t need to be updated when new geographic areas are included in these alerts. As of March 3, China, Iran, Italy, Japan, and South Korea all have Level 2 or 3 travel alerts.
The CDC now recommends that any patient who has severe acute lower respiratory illness that requires hospitalization and doesn’t have an alternative diagnosis should be tested, even without any identified source of exposure.
“Despite seeing these new cases, the risk to the American people is low,” said the CDC’s Dr. Redfield. In response to a question from the press about how fast the coronavirus will spread across the United States, Dr. Redfield said, “From the beginning we’ve anticipated seeing community cases pop up.” He added that as these cases arise, testing and public health strategies will focus on unearthing linkages and contacts to learn how the virus is spreading. “We’ll use the public health strategies that we can to limit that transmission,” he said.
*An earlier version of this article misattributed this quote.
The White House Coronavirus Task Force appeared at a press briefing March 2 to provide updates about testing strategies and public health coordination to address the current outbreak of the coronavirus COVID-19. Speaking at the briefing, led by Vice President Mike Pence, Centers for Disease Control and Prevention (CDC) director Robert Redfield, MD, said, “Working with our public health partners we continue to be able to identify new community cases and use our public health efforts to aggressively confirm, isolate, and do contact tracking.” Calling state, local, tribal, and territorial public health departments “the backbone of the public health system in our country,” Dr. Redfield noted that he expected many more confirmed COVID-19 cases to emerge.
At least some of the expected increase in confirmed cases of COVID-19 will occur because of expanded testing capacity, noted several of the task force members. On Feb. 29, the Food and Drug Administration issued a the virus that is causing the current outbreak of COVID-19.
Highly qualified laboratories, including both those run by public agencies and private labs, are now authorized to begin using their own validated test for the virus as long as they submit an Emergency Use Authorization (EUA) to the Food and Drug Administration within 15 days of notifying the agency of validation.
“To effectively respond to the COVID-19 outbreak, rapid detection of cases and contacts, appropriate clinical management and infection control, and implementation of community mitigation efforts are critical. This can best be achieved with wide availability of testing capabilities in health care settings, reference and commercial laboratories, and at the point of care,” the agency wrote in a press announcement of the expedited test expansion.
On Feb. 4, the Secretary of the Department of Health & Human Services declared a coronavirus public health emergency. The FDA was then authorized to allow individual laboratories with validated coronavirus tests to begin testing samples immediately. The goal is a more rapid and expanded testing capacity in the United States.
“The global emergence of COVID-19 is concerning, and we appreciate the efforts of the FDA to help bring more testing capability to the U.S.,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD), said in the press release.
The new guidance that permits the immediate use of clinical tests after individual development and validation, said the FDA, only applies to labs already certified to perform high complexity testing under Clinical Laboratory Improvement Amendments. Many governmental, academic, and private laboratories fall into this category, however.
“Under this policy, we expect certain laboratories who develop validated tests for coronavirus would begin using them right away prior to FDA review,” said Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health. “We believe this action will support laboratories across the country working on this urgent public health situation,” he added in the press release.
“By the end of this week, close to a million tests will be available,” FDA Commissioner Stephen M. Hahn, MD, said during the March 2 briefing.*
Updated criteria
The CDC is maintaining updated criteria for the virus testing on its website. Testing criteria are based both on clinical features and epidemiologic risk.
Individuals with less severe clinical features – those who have either fever or signs and symptoms of lower respiratory disease such as cough or shortness of breath, but who don’t require hospitalization – should be tested if they have high epidemiologic risk. “High risk” is defined by the CDC as any individual, including health care workers, who has had close contact with a person with confirmed COVID-19 within the past 2 weeks. For health care workers, testing can be considered even if they have relatively mild respiratory symptoms or have had contact with a person who is suspected, but not yet confirmed, to have coronavirus.
In its testing guidance, the CDC recognizes that defining close contact is difficult. General guidelines are that individuals are considered to have been in close contact with a person who has COVID-19 if they were within about six feet of the person for a prolonged period, or cared for or have spent a prolonged amount of time in the same room or house as a person with confirmed COVID-19.
Individuals who have both fever and signs or symptoms of lower respiratory illness who require hospitalization should be tested if they have a history of travel from any affected geographic area within 14 days of the onset of their symptoms. The CDC now defines “affected geographic area” as any country or region that has at least a CDC Level 2 Travel Health Notice for COVID-19, so that the testing criteria themselves don’t need to be updated when new geographic areas are included in these alerts. As of March 3, China, Iran, Italy, Japan, and South Korea all have Level 2 or 3 travel alerts.
The CDC now recommends that any patient who has severe acute lower respiratory illness that requires hospitalization and doesn’t have an alternative diagnosis should be tested, even without any identified source of exposure.
“Despite seeing these new cases, the risk to the American people is low,” said the CDC’s Dr. Redfield. In response to a question from the press about how fast the coronavirus will spread across the United States, Dr. Redfield said, “From the beginning we’ve anticipated seeing community cases pop up.” He added that as these cases arise, testing and public health strategies will focus on unearthing linkages and contacts to learn how the virus is spreading. “We’ll use the public health strategies that we can to limit that transmission,” he said.
*An earlier version of this article misattributed this quote.
FROM A PRESS BRIEFING BY THE WHITE HOUSE CORONAVIRUS TASK FORCE
Upcoming vaccine may offset surge in polio subtypes
Although wild poliovirus type 3 has not been detected globally for 7 years, the number of wild type 1 cases increased from 33 in 2018 to 173 in 2019. In response, a modified oral vaccine is being developed, according to Stephen Cochi, MD, of the Centers for Disease Control and Prevention’s Center for Global Health.
![]()
Several factors, including a Taliban ban on house-to-house vaccination in Afghanistan and a delay of large-scale vaccinations in Pakistan contributed to the surge in polio infections, Dr. Cochi said in a presentation at the February meeting of the CDC’s Advisory Committee on Immunization Practices (ACIP).
In addition, circulating vaccine-derived polioviruses (cVDPV) outbreaks have occurred in multiple countries including sub-Saharan Africa, China, Pakistan, and the Philippines. These outbreaks threaten the success of the bivalent oral polio vaccine introduced in April 2016 in 155 countries, Dr. Cochi said.
Outbreaks tend to occur just outside targeted areas for campaigns, caused by decreasing population immunity, he said.
The novel OPV2 (nOPV2) is a genetic modification of the existing OPV2 vaccine designed to improve genetic stability, Dr. Cochi explained. The modifications would “decrease the risk of seeding new cVDPVs and the risk of vaccine-associated paralytic poliomyelitis (VAPP),” he said.
The Emergency Use Listing (EUL) was developed by the World Health Organization in response to the Ebola virus outbreak in 2014-2016 and is the fastest way to obtain regulatory review and approval of drug products, said Dr. Cochi.
A pilot plant has been established in Indonesia, and upon EUL approval, 4-8 million doses of the nOPV2 should be available for use in the second quarter of 2020, he concluded.
Dr. Cochi had no relevant financial conflicts to disclose.
Although wild poliovirus type 3 has not been detected globally for 7 years, the number of wild type 1 cases increased from 33 in 2018 to 173 in 2019. In response, a modified oral vaccine is being developed, according to Stephen Cochi, MD, of the Centers for Disease Control and Prevention’s Center for Global Health.
![]()
Several factors, including a Taliban ban on house-to-house vaccination in Afghanistan and a delay of large-scale vaccinations in Pakistan contributed to the surge in polio infections, Dr. Cochi said in a presentation at the February meeting of the CDC’s Advisory Committee on Immunization Practices (ACIP).
In addition, circulating vaccine-derived polioviruses (cVDPV) outbreaks have occurred in multiple countries including sub-Saharan Africa, China, Pakistan, and the Philippines. These outbreaks threaten the success of the bivalent oral polio vaccine introduced in April 2016 in 155 countries, Dr. Cochi said.
Outbreaks tend to occur just outside targeted areas for campaigns, caused by decreasing population immunity, he said.
The novel OPV2 (nOPV2) is a genetic modification of the existing OPV2 vaccine designed to improve genetic stability, Dr. Cochi explained. The modifications would “decrease the risk of seeding new cVDPVs and the risk of vaccine-associated paralytic poliomyelitis (VAPP),” he said.
The Emergency Use Listing (EUL) was developed by the World Health Organization in response to the Ebola virus outbreak in 2014-2016 and is the fastest way to obtain regulatory review and approval of drug products, said Dr. Cochi.
A pilot plant has been established in Indonesia, and upon EUL approval, 4-8 million doses of the nOPV2 should be available for use in the second quarter of 2020, he concluded.
Dr. Cochi had no relevant financial conflicts to disclose.
Although wild poliovirus type 3 has not been detected globally for 7 years, the number of wild type 1 cases increased from 33 in 2018 to 173 in 2019. In response, a modified oral vaccine is being developed, according to Stephen Cochi, MD, of the Centers for Disease Control and Prevention’s Center for Global Health.
![]()
Several factors, including a Taliban ban on house-to-house vaccination in Afghanistan and a delay of large-scale vaccinations in Pakistan contributed to the surge in polio infections, Dr. Cochi said in a presentation at the February meeting of the CDC’s Advisory Committee on Immunization Practices (ACIP).
In addition, circulating vaccine-derived polioviruses (cVDPV) outbreaks have occurred in multiple countries including sub-Saharan Africa, China, Pakistan, and the Philippines. These outbreaks threaten the success of the bivalent oral polio vaccine introduced in April 2016 in 155 countries, Dr. Cochi said.
Outbreaks tend to occur just outside targeted areas for campaigns, caused by decreasing population immunity, he said.
The novel OPV2 (nOPV2) is a genetic modification of the existing OPV2 vaccine designed to improve genetic stability, Dr. Cochi explained. The modifications would “decrease the risk of seeding new cVDPVs and the risk of vaccine-associated paralytic poliomyelitis (VAPP),” he said.
The Emergency Use Listing (EUL) was developed by the World Health Organization in response to the Ebola virus outbreak in 2014-2016 and is the fastest way to obtain regulatory review and approval of drug products, said Dr. Cochi.
A pilot plant has been established in Indonesia, and upon EUL approval, 4-8 million doses of the nOPV2 should be available for use in the second quarter of 2020, he concluded.
Dr. Cochi had no relevant financial conflicts to disclose.
FROM AN ACIP MEETING
FDA approves new drug for relapsed/refractory multiple myeloma
The U.S. Food and Drug Administration today approved isatuximab (Sarclisa, Sanofi) in combination with pomalidomide (Revlimid, Celgene) and dexamethasone for the treatment of adult patients with multiple myeloma who have received two or more prior therapies including lenalidomide and a proteasome inhibitor.
Isatuximab is an anti-CD38 monoclonal antibody administered by intravenous infusion that works by helping the immune system attack multiple myeloma cancer cells.
“While there is no cure for multiple myeloma, Sarclisa is now another CD38-directed treatment option added to the list of FDA-approved treatments of patients with multiple myeloma who have progressive disease after previous therapies,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
“In the clinical trial, there was a 40% reduction in the risk of disease progression or death with this therapy,” he added.
The new approval is based on results from ICARIA-MM, an open-label, randomized phase 3 clinical trial of isatuximab among 307 patients in this setting.
In the trial, at a median follow-up of 11.6 months, median progression-free survival was 11.5 months in the isatuximab-pomalidomide-dexamethasone group versus 6.5 months in the pomalidomide-dexamethasone group (hazard ratio, 0.60; P = .001), as reported last year. Overall response rates were 60.4% for the triplet-treated group versus 35.3% for the doublet-treated group.
The most common side effects for isatuximab included neutropenia, infusion-related reactions, pneumonia, upper respiratory tract infection, diarrhea, anemia, lymphopenia, and thrombocytopenia.
Deaths because of treatment-related adverse events were reported for one patient (less than 1%) in the isatuximab-pomalidomide-dexamethasone group (sepsis) and two patients (1%) in the pomalidomide-dexamethasone group (pneumonia and urinary tract infection).
The drug can also cause serious side effects, including IV infusion-related reactions. In the case of a grade 3 or higher reaction, the drug should be permanently discontinued and health care professionals should institute appropriate medical management.
The FDA notes there have been higher incidences of second primary malignancies observed in a controlled clinical trial of patients with multiple myeloma receiving the drug.
The FDA also highlighted that laboratory test interference may be caused by isatuximab and that blood banks should be informed that patients are receiving the drug. Isatuximab may interfere with, for example, antibody screening for patients who need a blood transfusion. Isatuximab may also interfere with the assays used to monitor M-protein, which may impact the determination of complete response.
This article originally appeared on Medscape.com.
The U.S. Food and Drug Administration today approved isatuximab (Sarclisa, Sanofi) in combination with pomalidomide (Revlimid, Celgene) and dexamethasone for the treatment of adult patients with multiple myeloma who have received two or more prior therapies including lenalidomide and a proteasome inhibitor.
Isatuximab is an anti-CD38 monoclonal antibody administered by intravenous infusion that works by helping the immune system attack multiple myeloma cancer cells.
“While there is no cure for multiple myeloma, Sarclisa is now another CD38-directed treatment option added to the list of FDA-approved treatments of patients with multiple myeloma who have progressive disease after previous therapies,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
“In the clinical trial, there was a 40% reduction in the risk of disease progression or death with this therapy,” he added.
The new approval is based on results from ICARIA-MM, an open-label, randomized phase 3 clinical trial of isatuximab among 307 patients in this setting.
In the trial, at a median follow-up of 11.6 months, median progression-free survival was 11.5 months in the isatuximab-pomalidomide-dexamethasone group versus 6.5 months in the pomalidomide-dexamethasone group (hazard ratio, 0.60; P = .001), as reported last year. Overall response rates were 60.4% for the triplet-treated group versus 35.3% for the doublet-treated group.
The most common side effects for isatuximab included neutropenia, infusion-related reactions, pneumonia, upper respiratory tract infection, diarrhea, anemia, lymphopenia, and thrombocytopenia.
Deaths because of treatment-related adverse events were reported for one patient (less than 1%) in the isatuximab-pomalidomide-dexamethasone group (sepsis) and two patients (1%) in the pomalidomide-dexamethasone group (pneumonia and urinary tract infection).
The drug can also cause serious side effects, including IV infusion-related reactions. In the case of a grade 3 or higher reaction, the drug should be permanently discontinued and health care professionals should institute appropriate medical management.
The FDA notes there have been higher incidences of second primary malignancies observed in a controlled clinical trial of patients with multiple myeloma receiving the drug.
The FDA also highlighted that laboratory test interference may be caused by isatuximab and that blood banks should be informed that patients are receiving the drug. Isatuximab may interfere with, for example, antibody screening for patients who need a blood transfusion. Isatuximab may also interfere with the assays used to monitor M-protein, which may impact the determination of complete response.
This article originally appeared on Medscape.com.
The U.S. Food and Drug Administration today approved isatuximab (Sarclisa, Sanofi) in combination with pomalidomide (Revlimid, Celgene) and dexamethasone for the treatment of adult patients with multiple myeloma who have received two or more prior therapies including lenalidomide and a proteasome inhibitor.
Isatuximab is an anti-CD38 monoclonal antibody administered by intravenous infusion that works by helping the immune system attack multiple myeloma cancer cells.
“While there is no cure for multiple myeloma, Sarclisa is now another CD38-directed treatment option added to the list of FDA-approved treatments of patients with multiple myeloma who have progressive disease after previous therapies,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
“In the clinical trial, there was a 40% reduction in the risk of disease progression or death with this therapy,” he added.
The new approval is based on results from ICARIA-MM, an open-label, randomized phase 3 clinical trial of isatuximab among 307 patients in this setting.
In the trial, at a median follow-up of 11.6 months, median progression-free survival was 11.5 months in the isatuximab-pomalidomide-dexamethasone group versus 6.5 months in the pomalidomide-dexamethasone group (hazard ratio, 0.60; P = .001), as reported last year. Overall response rates were 60.4% for the triplet-treated group versus 35.3% for the doublet-treated group.
The most common side effects for isatuximab included neutropenia, infusion-related reactions, pneumonia, upper respiratory tract infection, diarrhea, anemia, lymphopenia, and thrombocytopenia.
Deaths because of treatment-related adverse events were reported for one patient (less than 1%) in the isatuximab-pomalidomide-dexamethasone group (sepsis) and two patients (1%) in the pomalidomide-dexamethasone group (pneumonia and urinary tract infection).
The drug can also cause serious side effects, including IV infusion-related reactions. In the case of a grade 3 or higher reaction, the drug should be permanently discontinued and health care professionals should institute appropriate medical management.
The FDA notes there have been higher incidences of second primary malignancies observed in a controlled clinical trial of patients with multiple myeloma receiving the drug.
The FDA also highlighted that laboratory test interference may be caused by isatuximab and that blood banks should be informed that patients are receiving the drug. Isatuximab may interfere with, for example, antibody screening for patients who need a blood transfusion. Isatuximab may also interfere with the assays used to monitor M-protein, which may impact the determination of complete response.
This article originally appeared on Medscape.com.
CDC revises COVID-19 test kits, broadens ‘person under investigation’ definition
In a telebriefing on the COVID-19 outbreak, Nancy Messonnier, MD, director of the National Center for Immunization and Respiratory Diseases at .

The definition has been revised “to meet the needs of this rapidly evolving situation,” she said. The new PUI definition includes travel to more geographic areas to reflect this past week’s marked uptick in coronavirus activity in Italy and Iran. In addition to these countries and China, recent travel to Japan or South Korea also constitutes an epidemiologic risk factor which, in conjunction with clinical features, warrant an individual being classified as a PUI. These five countries each now have widespread person-to-person transmission of the virus.
Dr. Messonnier left open the possibility that the PUI definition would continue to evolve if such transmission within communities becomes more common. Asked whether the small number of U.S. cases thus might be an artifact of low test volumes, she said, “We aggressively controlled our borders to slow the spread. This was an intentional U.S. strategy. The CDC has always had the capacity to test rapidly from the time the sequence was available. ...We have been testing aggressively.”
The original PUI definition, she explained, emphasized individuals with fever, cough, or trouble breathing who had traveled recently from areas with COVID-19 activity, in particular China’s Hubei province. “We have been most focused on symptomatic people who are closely linked to, or who had, travel history, but our criteria also allow for clinical discretion,” she said. “There is no substitute for an astute clinician on the front lines of patient care.”
The first COVID-19 case from person-to-person spread was reported on Feb. 27. “At this time, we don’t know how or where this person became infected,” said Dr. Messonnier, although investigations are still underway. She responded to a question about whether the CDC delayed allowing COVID-19 testing for the patient for several days, as was reported in some media accounts. “According to CDC records, the first call we got was Feb. 23,” when public health officials in California reported a severely ill person with no travel abroad and no known contacts with individuals that would trigger suspicions for coronavirus. The CDC recommended COVID-19 testing on that day, she said.
Dr. Messonnier declined to answer questions about a whistleblower report alleging improper training and inadequate protective measures for Department of Health & Human Services workers at the quarantine center at Travis Air Force Base, Calif.
Dr. Messonnier said that the CDC has been working closely with the Food and Drug Administration to address problems with the COVID-19 test kits that were unusable because of a large number of indeterminate results. The two agencies together have determined that of the three reactions that were initially deemed necessary for a definitive COVID-19 diagnosis, just two are sufficient, so new kits that omit the problematic chemical are being manufactured and distributed.
These new kits are rapidly being made available; the goal, said Dr. Messonnier, is to have to state and local public health departments equipped with test kits by about March 7.
As local tests become available, the most updated information will be coming from state and local public health departments, she stressed, adding that the CDC would continue to update case counts on Monday, Wednesday, and Friday of each week. Procedures are being developed for the management of patients presumed to have COVID-19, where local health departments see positive tests but the mandatory CDC confirmatory test hasn’t been completed.
While new cases emerge across Europe and Asia, China’s earlier COVID-19 explosion seems to be slowing. “It’s really good news that the case counts in China are decreasing,” both for the well-being of that country’s citizens, and as a sign of the disease’s potential global effects, said Dr. Messonnier. She added that epidemiologists and mathematical modelers are parsing case fatality rates as well.
She advised health care providers and public health officials to keep abreast of changes in CDC guidance by checking frequently at https://www.cdc.gov/coronavirus/2019-ncov/index.html.
In a telebriefing on the COVID-19 outbreak, Nancy Messonnier, MD, director of the National Center for Immunization and Respiratory Diseases at .
The definition has been revised “to meet the needs of this rapidly evolving situation,” she said. The new PUI definition includes travel to more geographic areas to reflect this past week’s marked uptick in coronavirus activity in Italy and Iran. In addition to these countries and China, recent travel to Japan or South Korea also constitutes an epidemiologic risk factor which, in conjunction with clinical features, warrant an individual being classified as a PUI. These five countries each now have widespread person-to-person transmission of the virus.
Dr. Messonnier left open the possibility that the PUI definition would continue to evolve if such transmission within communities becomes more common. Asked whether the small number of U.S. cases thus might be an artifact of low test volumes, she said, “We aggressively controlled our borders to slow the spread. This was an intentional U.S. strategy. The CDC has always had the capacity to test rapidly from the time the sequence was available. ...We have been testing aggressively.”
The original PUI definition, she explained, emphasized individuals with fever, cough, or trouble breathing who had traveled recently from areas with COVID-19 activity, in particular China’s Hubei province. “We have been most focused on symptomatic people who are closely linked to, or who had, travel history, but our criteria also allow for clinical discretion,” she said. “There is no substitute for an astute clinician on the front lines of patient care.”
The first COVID-19 case from person-to-person spread was reported on Feb. 27. “At this time, we don’t know how or where this person became infected,” said Dr. Messonnier, although investigations are still underway. She responded to a question about whether the CDC delayed allowing COVID-19 testing for the patient for several days, as was reported in some media accounts. “According to CDC records, the first call we got was Feb. 23,” when public health officials in California reported a severely ill person with no travel abroad and no known contacts with individuals that would trigger suspicions for coronavirus. The CDC recommended COVID-19 testing on that day, she said.
Dr. Messonnier declined to answer questions about a whistleblower report alleging improper training and inadequate protective measures for Department of Health & Human Services workers at the quarantine center at Travis Air Force Base, Calif.
Dr. Messonnier said that the CDC has been working closely with the Food and Drug Administration to address problems with the COVID-19 test kits that were unusable because of a large number of indeterminate results. The two agencies together have determined that of the three reactions that were initially deemed necessary for a definitive COVID-19 diagnosis, just two are sufficient, so new kits that omit the problematic chemical are being manufactured and distributed.
These new kits are rapidly being made available; the goal, said Dr. Messonnier, is to have to state and local public health departments equipped with test kits by about March 7.
As local tests become available, the most updated information will be coming from state and local public health departments, she stressed, adding that the CDC would continue to update case counts on Monday, Wednesday, and Friday of each week. Procedures are being developed for the management of patients presumed to have COVID-19, where local health departments see positive tests but the mandatory CDC confirmatory test hasn’t been completed.
While new cases emerge across Europe and Asia, China’s earlier COVID-19 explosion seems to be slowing. “It’s really good news that the case counts in China are decreasing,” both for the well-being of that country’s citizens, and as a sign of the disease’s potential global effects, said Dr. Messonnier. She added that epidemiologists and mathematical modelers are parsing case fatality rates as well.
She advised health care providers and public health officials to keep abreast of changes in CDC guidance by checking frequently at https://www.cdc.gov/coronavirus/2019-ncov/index.html.
In a telebriefing on the COVID-19 outbreak, Nancy Messonnier, MD, director of the National Center for Immunization and Respiratory Diseases at .
The definition has been revised “to meet the needs of this rapidly evolving situation,” she said. The new PUI definition includes travel to more geographic areas to reflect this past week’s marked uptick in coronavirus activity in Italy and Iran. In addition to these countries and China, recent travel to Japan or South Korea also constitutes an epidemiologic risk factor which, in conjunction with clinical features, warrant an individual being classified as a PUI. These five countries each now have widespread person-to-person transmission of the virus.
Dr. Messonnier left open the possibility that the PUI definition would continue to evolve if such transmission within communities becomes more common. Asked whether the small number of U.S. cases thus might be an artifact of low test volumes, she said, “We aggressively controlled our borders to slow the spread. This was an intentional U.S. strategy. The CDC has always had the capacity to test rapidly from the time the sequence was available. ...We have been testing aggressively.”
The original PUI definition, she explained, emphasized individuals with fever, cough, or trouble breathing who had traveled recently from areas with COVID-19 activity, in particular China’s Hubei province. “We have been most focused on symptomatic people who are closely linked to, or who had, travel history, but our criteria also allow for clinical discretion,” she said. “There is no substitute for an astute clinician on the front lines of patient care.”
The first COVID-19 case from person-to-person spread was reported on Feb. 27. “At this time, we don’t know how or where this person became infected,” said Dr. Messonnier, although investigations are still underway. She responded to a question about whether the CDC delayed allowing COVID-19 testing for the patient for several days, as was reported in some media accounts. “According to CDC records, the first call we got was Feb. 23,” when public health officials in California reported a severely ill person with no travel abroad and no known contacts with individuals that would trigger suspicions for coronavirus. The CDC recommended COVID-19 testing on that day, she said.
Dr. Messonnier declined to answer questions about a whistleblower report alleging improper training and inadequate protective measures for Department of Health & Human Services workers at the quarantine center at Travis Air Force Base, Calif.
Dr. Messonnier said that the CDC has been working closely with the Food and Drug Administration to address problems with the COVID-19 test kits that were unusable because of a large number of indeterminate results. The two agencies together have determined that of the three reactions that were initially deemed necessary for a definitive COVID-19 diagnosis, just two are sufficient, so new kits that omit the problematic chemical are being manufactured and distributed.
These new kits are rapidly being made available; the goal, said Dr. Messonnier, is to have to state and local public health departments equipped with test kits by about March 7.
As local tests become available, the most updated information will be coming from state and local public health departments, she stressed, adding that the CDC would continue to update case counts on Monday, Wednesday, and Friday of each week. Procedures are being developed for the management of patients presumed to have COVID-19, where local health departments see positive tests but the mandatory CDC confirmatory test hasn’t been completed.
While new cases emerge across Europe and Asia, China’s earlier COVID-19 explosion seems to be slowing. “It’s really good news that the case counts in China are decreasing,” both for the well-being of that country’s citizens, and as a sign of the disease’s potential global effects, said Dr. Messonnier. She added that epidemiologists and mathematical modelers are parsing case fatality rates as well.
She advised health care providers and public health officials to keep abreast of changes in CDC guidance by checking frequently at https://www.cdc.gov/coronavirus/2019-ncov/index.html.
REPORTING FROM A CDC BRIEFING