User login
FDA warns of possible temporary shortage of trach tube
manufactured by Smiths Medical caused by the closure of a large ethylene oxide sterilization facilities in Willowbrook, Ill., and the future planned closure of a similar facility.

The shortage may affect pediatric use because, although tubes are used for both adults and children, there are fewer alternative products on the market for pediatric patients. Parents and caregivers of children who use the Bivona tube are encouraged to check with Smiths Medical about available inventory and with their health care providers about alternative products.
Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in a press release, “I want to assure you that the FDA is working closely with the company to quickly resolve their sterilization challenges and bring these critical devices to the patients who need them as quickly as possible, which we anticipate will be made available again beginning the week of April 22.”
For patients currently using the Bivona tubes, Dr. Shuren noted, “The closure of the Willowbrook facility does not impact tubes already in use by patients at home or in health care settings. The company is communicating with patients about the tubes and how patients and caregivers can mitigate any potential impact, including reusing and cleaning tubes in accordance with the manufacturer’s instructions for use.”
Read the entire announcement at the FDA website.
manufactured by Smiths Medical caused by the closure of a large ethylene oxide sterilization facilities in Willowbrook, Ill., and the future planned closure of a similar facility.
The shortage may affect pediatric use because, although tubes are used for both adults and children, there are fewer alternative products on the market for pediatric patients. Parents and caregivers of children who use the Bivona tube are encouraged to check with Smiths Medical about available inventory and with their health care providers about alternative products.
Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in a press release, “I want to assure you that the FDA is working closely with the company to quickly resolve their sterilization challenges and bring these critical devices to the patients who need them as quickly as possible, which we anticipate will be made available again beginning the week of April 22.”
For patients currently using the Bivona tubes, Dr. Shuren noted, “The closure of the Willowbrook facility does not impact tubes already in use by patients at home or in health care settings. The company is communicating with patients about the tubes and how patients and caregivers can mitigate any potential impact, including reusing and cleaning tubes in accordance with the manufacturer’s instructions for use.”
Read the entire announcement at the FDA website.
manufactured by Smiths Medical caused by the closure of a large ethylene oxide sterilization facilities in Willowbrook, Ill., and the future planned closure of a similar facility.
The shortage may affect pediatric use because, although tubes are used for both adults and children, there are fewer alternative products on the market for pediatric patients. Parents and caregivers of children who use the Bivona tube are encouraged to check with Smiths Medical about available inventory and with their health care providers about alternative products.
Jeff Shuren, MD, director of the Center for Devices and Radiological Health, wrote in a press release, “I want to assure you that the FDA is working closely with the company to quickly resolve their sterilization challenges and bring these critical devices to the patients who need them as quickly as possible, which we anticipate will be made available again beginning the week of April 22.”
For patients currently using the Bivona tubes, Dr. Shuren noted, “The closure of the Willowbrook facility does not impact tubes already in use by patients at home or in health care settings. The company is communicating with patients about the tubes and how patients and caregivers can mitigate any potential impact, including reusing and cleaning tubes in accordance with the manufacturer’s instructions for use.”
Read the entire announcement at the FDA website.
FDA approves pembrolizumab for first-line stage III NSCLC
The who are not candidates for surgical resection or definitive chemoradiation, and for stage IV NSCLC.

Patients’ tumors must express programmed death-ligand 1 (PD-L1) as determined by an FDA-approved test (tumor proportion score ≥1%) and have no epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
The checkpoint inhibitor was previously approved as a single agent for the first-line treatment of patients with metastatic disease with PD-L1 expression at a higher level (TPS ≥50%), the FDA said in a press statement.
Approval was based on statistically significant overall survival improvement with pembrolizumab, compared with investigator’s choice of a carboplatin-containing regimen with either pemetrexed or paclitaxel in KEYNOTE‑042. The trial enrolled 1,274 patients with stage III or IV NSCLC who had not received prior systemic treatment for metastatic NSCLC and whose tumors expressed PD-L1 (TPS ≥1%).
Overall survival was improved in all three subgroups for pembrolizumab, compared with chemotherapy: in the TPS ≥50% subgroup, the TPS ≥20% subgroup, and the overall population (TPS ≥1%). The median overall survival in the TPS ≥1% population was 16.7 for pembrolizumab and 12.1 months for the chemotherapy arms (hazard ratio, 0.81; 95% confidence interval, 0.71-0.93; P = .0036). For the TPS ≥50% subgroup, the estimated median overall survival was 20 months for pembrolizumab and 12.2 months for the chemotherapy arm (HR, 0.69; 95% CI, 0.56-0.85; P = .0006).
The most common adverse reactions reported for patients who received pembrolizumab included fatigue, decreased appetite, dyspnea, cough, rash, constipation, diarrhea, nausea, hypothyroidism, pneumonia, pyrexia, and weight loss, the FDA said.
The recommended dose for NSCLC is 200 mg as an IV infusion over 30 minutes every 3 weeks.
The who are not candidates for surgical resection or definitive chemoradiation, and for stage IV NSCLC.
Patients’ tumors must express programmed death-ligand 1 (PD-L1) as determined by an FDA-approved test (tumor proportion score ≥1%) and have no epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
The checkpoint inhibitor was previously approved as a single agent for the first-line treatment of patients with metastatic disease with PD-L1 expression at a higher level (TPS ≥50%), the FDA said in a press statement.
Approval was based on statistically significant overall survival improvement with pembrolizumab, compared with investigator’s choice of a carboplatin-containing regimen with either pemetrexed or paclitaxel in KEYNOTE‑042. The trial enrolled 1,274 patients with stage III or IV NSCLC who had not received prior systemic treatment for metastatic NSCLC and whose tumors expressed PD-L1 (TPS ≥1%).
Overall survival was improved in all three subgroups for pembrolizumab, compared with chemotherapy: in the TPS ≥50% subgroup, the TPS ≥20% subgroup, and the overall population (TPS ≥1%). The median overall survival in the TPS ≥1% population was 16.7 for pembrolizumab and 12.1 months for the chemotherapy arms (hazard ratio, 0.81; 95% confidence interval, 0.71-0.93; P = .0036). For the TPS ≥50% subgroup, the estimated median overall survival was 20 months for pembrolizumab and 12.2 months for the chemotherapy arm (HR, 0.69; 95% CI, 0.56-0.85; P = .0006).
The most common adverse reactions reported for patients who received pembrolizumab included fatigue, decreased appetite, dyspnea, cough, rash, constipation, diarrhea, nausea, hypothyroidism, pneumonia, pyrexia, and weight loss, the FDA said.
The recommended dose for NSCLC is 200 mg as an IV infusion over 30 minutes every 3 weeks.
The who are not candidates for surgical resection or definitive chemoradiation, and for stage IV NSCLC.
Patients’ tumors must express programmed death-ligand 1 (PD-L1) as determined by an FDA-approved test (tumor proportion score ≥1%) and have no epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
The checkpoint inhibitor was previously approved as a single agent for the first-line treatment of patients with metastatic disease with PD-L1 expression at a higher level (TPS ≥50%), the FDA said in a press statement.
Approval was based on statistically significant overall survival improvement with pembrolizumab, compared with investigator’s choice of a carboplatin-containing regimen with either pemetrexed or paclitaxel in KEYNOTE‑042. The trial enrolled 1,274 patients with stage III or IV NSCLC who had not received prior systemic treatment for metastatic NSCLC and whose tumors expressed PD-L1 (TPS ≥1%).
Overall survival was improved in all three subgroups for pembrolizumab, compared with chemotherapy: in the TPS ≥50% subgroup, the TPS ≥20% subgroup, and the overall population (TPS ≥1%). The median overall survival in the TPS ≥1% population was 16.7 for pembrolizumab and 12.1 months for the chemotherapy arms (hazard ratio, 0.81; 95% confidence interval, 0.71-0.93; P = .0036). For the TPS ≥50% subgroup, the estimated median overall survival was 20 months for pembrolizumab and 12.2 months for the chemotherapy arm (HR, 0.69; 95% CI, 0.56-0.85; P = .0006).
The most common adverse reactions reported for patients who received pembrolizumab included fatigue, decreased appetite, dyspnea, cough, rash, constipation, diarrhea, nausea, hypothyroidism, pneumonia, pyrexia, and weight loss, the FDA said.
The recommended dose for NSCLC is 200 mg as an IV infusion over 30 minutes every 3 weeks.
FDA approves first two-drug tablet for HIV
The U.S. Food and Drug Administration has approved the first two-drug, fixed-dose, complete regimen for HIV-infected adults, according to an FDA press announcement.
Dovato (dolutegravir and lamivudine), a product of ViiV Healthcare, is intended to serve “as a complete regimen” for the treatment of HIV-1 infection in adults who have had no previous antiretroviral treatment and who have an infection with no known or suspected genetic substitutions associated with resistance to the individual components of Dovato.
“With this approval, patients who have never been treated have the option of taking a two-drug regimen in a single tablet while eliminating additional toxicity and potential drug interactions from a third drug,” said Debra Birnkrant, MD, director of the FDA’s Division of Antiviral Products.
The Dovato labeling includes a Boxed Warning that patients infected with both HIV and hepatitis B should add additional treatment for their HBV or consider a different drug regimen. The most common adverse reactions with Dovato were headache, diarrhea, nausea, insomnia, and fatigue. In addition, the FDA warned that, as there is a known risk for neural tube defects with dolutegravir, patients are advised to avoid use of Dovato at the time of conception through the first trimester of pregnancy.
[email protected]
The U.S. Food and Drug Administration has approved the first two-drug, fixed-dose, complete regimen for HIV-infected adults, according to an FDA press announcement.
Dovato (dolutegravir and lamivudine), a product of ViiV Healthcare, is intended to serve “as a complete regimen” for the treatment of HIV-1 infection in adults who have had no previous antiretroviral treatment and who have an infection with no known or suspected genetic substitutions associated with resistance to the individual components of Dovato.
“With this approval, patients who have never been treated have the option of taking a two-drug regimen in a single tablet while eliminating additional toxicity and potential drug interactions from a third drug,” said Debra Birnkrant, MD, director of the FDA’s Division of Antiviral Products.
The Dovato labeling includes a Boxed Warning that patients infected with both HIV and hepatitis B should add additional treatment for their HBV or consider a different drug regimen. The most common adverse reactions with Dovato were headache, diarrhea, nausea, insomnia, and fatigue. In addition, the FDA warned that, as there is a known risk for neural tube defects with dolutegravir, patients are advised to avoid use of Dovato at the time of conception through the first trimester of pregnancy.
[email protected]
The U.S. Food and Drug Administration has approved the first two-drug, fixed-dose, complete regimen for HIV-infected adults, according to an FDA press announcement.
Dovato (dolutegravir and lamivudine), a product of ViiV Healthcare, is intended to serve “as a complete regimen” for the treatment of HIV-1 infection in adults who have had no previous antiretroviral treatment and who have an infection with no known or suspected genetic substitutions associated with resistance to the individual components of Dovato.
“With this approval, patients who have never been treated have the option of taking a two-drug regimen in a single tablet while eliminating additional toxicity and potential drug interactions from a third drug,” said Debra Birnkrant, MD, director of the FDA’s Division of Antiviral Products.
The Dovato labeling includes a Boxed Warning that patients infected with both HIV and hepatitis B should add additional treatment for their HBV or consider a different drug regimen. The most common adverse reactions with Dovato were headache, diarrhea, nausea, insomnia, and fatigue. In addition, the FDA warned that, as there is a known risk for neural tube defects with dolutegravir, patients are advised to avoid use of Dovato at the time of conception through the first trimester of pregnancy.
[email protected]
Large measles outbreak reported in Michigan
A new measles outbreak in Michigan has already resulted in 39 cases, and four more states reported their first cases of 2019 during the week ending April 4, according to the Centers for Disease Control and Prevention
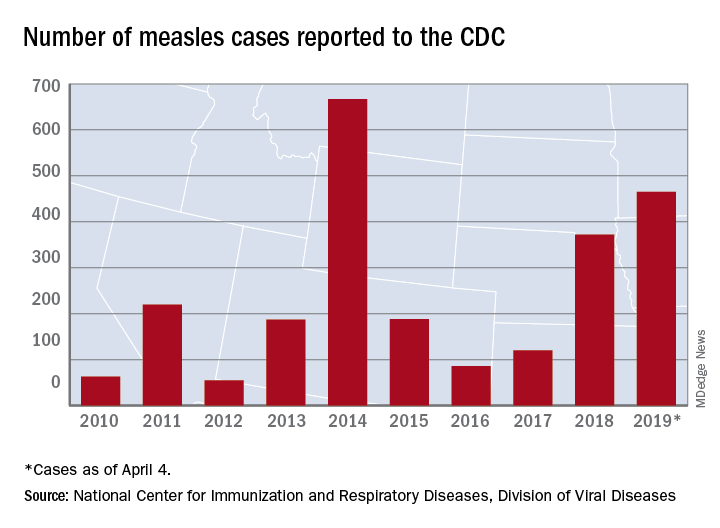
The measles virus has now infected individuals in Florida, Indiana, Massachusetts, and Nevada, which means that 19 states have now reported a total of 465 cases this year, and that is the second-highest total “reported in the U.S. since measles was eliminated in 2000,” the CDC said April 8.
The Michigan outbreak is mostly concentrated in Oakland County, where 38 cases have occurred. The county has posted an up-to-date list of exposure locations.
Not to be outdone, New York reported 45 new cases last week: 44 in Brooklyn and 1 in Queens. There have been 259 confirmed cases in the two boroughs since the outbreak began in October of last year.
Besides Michigan and New York City, there are five other outbreaks ongoing in the United States: Rockland County, N.Y.; Washington State (no new cases since March 22); Butte County, Calif.; Santa Cruz County, Calif.; and New Jersey, the CDC reported.
A judge in New York State temporarily blocked an order banning unimmunized children from public spaces in Rockland County and has set a hearing date of April 19, CNN reported. The ban, ordered by Rockland County Executive Ed Day, went into effect on March 27.
On April 2, the Maine Center for Disease Control & Prevention announced that an out-of-state resident with a confirmed case of measles had visited two health care offices – one in Falmouth and one in Westbrook – on March 27. No cases in Maine residents have been reported yet.
On a vaccine-related note, the Washington State Senate’s Health and Long Term Care Committee approved a proposal on April 1 that would “end the personal exemption for parents who don’t want their children vaccinated against measles,” the Spokane Spokesman-Review said. The bill, which would still allow medical and religious exemptions, has already passed the state’s House of Representatives and goes next to the full senate.
A new measles outbreak in Michigan has already resulted in 39 cases, and four more states reported their first cases of 2019 during the week ending April 4, according to the Centers for Disease Control and Prevention
The measles virus has now infected individuals in Florida, Indiana, Massachusetts, and Nevada, which means that 19 states have now reported a total of 465 cases this year, and that is the second-highest total “reported in the U.S. since measles was eliminated in 2000,” the CDC said April 8.
The Michigan outbreak is mostly concentrated in Oakland County, where 38 cases have occurred. The county has posted an up-to-date list of exposure locations.
Not to be outdone, New York reported 45 new cases last week: 44 in Brooklyn and 1 in Queens. There have been 259 confirmed cases in the two boroughs since the outbreak began in October of last year.
Besides Michigan and New York City, there are five other outbreaks ongoing in the United States: Rockland County, N.Y.; Washington State (no new cases since March 22); Butte County, Calif.; Santa Cruz County, Calif.; and New Jersey, the CDC reported.
A judge in New York State temporarily blocked an order banning unimmunized children from public spaces in Rockland County and has set a hearing date of April 19, CNN reported. The ban, ordered by Rockland County Executive Ed Day, went into effect on March 27.
On April 2, the Maine Center for Disease Control & Prevention announced that an out-of-state resident with a confirmed case of measles had visited two health care offices – one in Falmouth and one in Westbrook – on March 27. No cases in Maine residents have been reported yet.
On a vaccine-related note, the Washington State Senate’s Health and Long Term Care Committee approved a proposal on April 1 that would “end the personal exemption for parents who don’t want their children vaccinated against measles,” the Spokane Spokesman-Review said. The bill, which would still allow medical and religious exemptions, has already passed the state’s House of Representatives and goes next to the full senate.
A new measles outbreak in Michigan has already resulted in 39 cases, and four more states reported their first cases of 2019 during the week ending April 4, according to the Centers for Disease Control and Prevention
The measles virus has now infected individuals in Florida, Indiana, Massachusetts, and Nevada, which means that 19 states have now reported a total of 465 cases this year, and that is the second-highest total “reported in the U.S. since measles was eliminated in 2000,” the CDC said April 8.
The Michigan outbreak is mostly concentrated in Oakland County, where 38 cases have occurred. The county has posted an up-to-date list of exposure locations.
Not to be outdone, New York reported 45 new cases last week: 44 in Brooklyn and 1 in Queens. There have been 259 confirmed cases in the two boroughs since the outbreak began in October of last year.
Besides Michigan and New York City, there are five other outbreaks ongoing in the United States: Rockland County, N.Y.; Washington State (no new cases since March 22); Butte County, Calif.; Santa Cruz County, Calif.; and New Jersey, the CDC reported.
A judge in New York State temporarily blocked an order banning unimmunized children from public spaces in Rockland County and has set a hearing date of April 19, CNN reported. The ban, ordered by Rockland County Executive Ed Day, went into effect on March 27.
On April 2, the Maine Center for Disease Control & Prevention announced that an out-of-state resident with a confirmed case of measles had visited two health care offices – one in Falmouth and one in Westbrook – on March 27. No cases in Maine residents have been reported yet.
On a vaccine-related note, the Washington State Senate’s Health and Long Term Care Committee approved a proposal on April 1 that would “end the personal exemption for parents who don’t want their children vaccinated against measles,” the Spokane Spokesman-Review said. The bill, which would still allow medical and religious exemptions, has already passed the state’s House of Representatives and goes next to the full senate.
Flu activity falling but still elevated
Measures of influenza activity fell again as the flu season continues to make its later-than-usual departure this year, according to the Centers for Disease Control and Prevention.
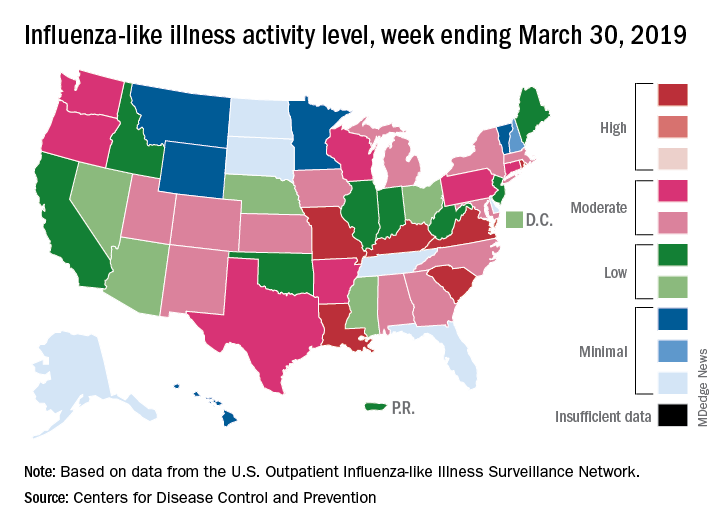
On the geographic front, the map of influenza-like illness (ILI) activity for the week ending March 30 shows that only 6 states are at level 10 on the CDC’s 1-10 scale, compared with 11 for the previous week, and that those same 6 states make up the entire membership of the high range of levels 8-10, which is down from 20 states a week ago, data from the CDC’s Outpatient ILI Surveillance Network show.
The proportion of outpatient visits for ILI, now at 3.2%, dropped for the sixth consecutive week after reaching its season high of 5.1% back in mid-February. The outpatient rate has now been at or above the national baseline of 2.2% for 19 weeks this season, the CDC’s influenza division said April 5, noting that the average for the past five seasons is 16 weeks.
Six flu-related pediatric deaths were reported in the week ending March 30, and the total is now 82 for the 2018-2019 season. Five of the six occurred during previous weeks of this season, and one occurred in the 2017-2018 season, the CDC said.
Measures of influenza activity fell again as the flu season continues to make its later-than-usual departure this year, according to the Centers for Disease Control and Prevention.
On the geographic front, the map of influenza-like illness (ILI) activity for the week ending March 30 shows that only 6 states are at level 10 on the CDC’s 1-10 scale, compared with 11 for the previous week, and that those same 6 states make up the entire membership of the high range of levels 8-10, which is down from 20 states a week ago, data from the CDC’s Outpatient ILI Surveillance Network show.
The proportion of outpatient visits for ILI, now at 3.2%, dropped for the sixth consecutive week after reaching its season high of 5.1% back in mid-February. The outpatient rate has now been at or above the national baseline of 2.2% for 19 weeks this season, the CDC’s influenza division said April 5, noting that the average for the past five seasons is 16 weeks.
Six flu-related pediatric deaths were reported in the week ending March 30, and the total is now 82 for the 2018-2019 season. Five of the six occurred during previous weeks of this season, and one occurred in the 2017-2018 season, the CDC said.
Measures of influenza activity fell again as the flu season continues to make its later-than-usual departure this year, according to the Centers for Disease Control and Prevention.
On the geographic front, the map of influenza-like illness (ILI) activity for the week ending March 30 shows that only 6 states are at level 10 on the CDC’s 1-10 scale, compared with 11 for the previous week, and that those same 6 states make up the entire membership of the high range of levels 8-10, which is down from 20 states a week ago, data from the CDC’s Outpatient ILI Surveillance Network show.
The proportion of outpatient visits for ILI, now at 3.2%, dropped for the sixth consecutive week after reaching its season high of 5.1% back in mid-February. The outpatient rate has now been at or above the national baseline of 2.2% for 19 weeks this season, the CDC’s influenza division said April 5, noting that the average for the past five seasons is 16 weeks.
Six flu-related pediatric deaths were reported in the week ending March 30, and the total is now 82 for the 2018-2019 season. Five of the six occurred during previous weeks of this season, and one occurred in the 2017-2018 season, the CDC said.
FDA approves palbociclib for men with HR+/HER2- advanced breast cancer
The Food and Drug Administration has expanded the indication of palbociclib (Ibrance) in combination with specific endocrine therapies for hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer in men.
Approval was based on postmarketing reports and electronic health records showing that the safety profile for men is consistent with that of women, the FDA said in a statement.
Less than 1% of all cases of breast cancer occur in men, but in the majority of those cases the tumors do express hormone receptors. Men are more likely to be diagnosed at a more advanced stage of disease. “According to the current clinical practice standards, male patients with breast cancer are treated similarly to women with breast cancer,” the FDA said.
The kinase inhibitor palbociclib was initially approved in 2015, in combination with an aromatase inhibitor, for postmenopausal women as first-line treatment of advanced disease.
The most common side effects in patients taking palbociclib are infections, leukopenia, fatigue, nausea, stomatitis, anemia, hair loss, diarrhea, and thrombocytopenia.
Because of the potential for genotoxicity, the FDA advised health care providers to tell male patients with female partners of reproductive potential to use effective contraception during treatment with palbociclib and for 3 months after the last dose.
The Food and Drug Administration has expanded the indication of palbociclib (Ibrance) in combination with specific endocrine therapies for hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer in men.
Approval was based on postmarketing reports and electronic health records showing that the safety profile for men is consistent with that of women, the FDA said in a statement.
Less than 1% of all cases of breast cancer occur in men, but in the majority of those cases the tumors do express hormone receptors. Men are more likely to be diagnosed at a more advanced stage of disease. “According to the current clinical practice standards, male patients with breast cancer are treated similarly to women with breast cancer,” the FDA said.
The kinase inhibitor palbociclib was initially approved in 2015, in combination with an aromatase inhibitor, for postmenopausal women as first-line treatment of advanced disease.
The most common side effects in patients taking palbociclib are infections, leukopenia, fatigue, nausea, stomatitis, anemia, hair loss, diarrhea, and thrombocytopenia.
Because of the potential for genotoxicity, the FDA advised health care providers to tell male patients with female partners of reproductive potential to use effective contraception during treatment with palbociclib and for 3 months after the last dose.
The Food and Drug Administration has expanded the indication of palbociclib (Ibrance) in combination with specific endocrine therapies for hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer in men.
Approval was based on postmarketing reports and electronic health records showing that the safety profile for men is consistent with that of women, the FDA said in a statement.
Less than 1% of all cases of breast cancer occur in men, but in the majority of those cases the tumors do express hormone receptors. Men are more likely to be diagnosed at a more advanced stage of disease. “According to the current clinical practice standards, male patients with breast cancer are treated similarly to women with breast cancer,” the FDA said.
The kinase inhibitor palbociclib was initially approved in 2015, in combination with an aromatase inhibitor, for postmenopausal women as first-line treatment of advanced disease.
The most common side effects in patients taking palbociclib are infections, leukopenia, fatigue, nausea, stomatitis, anemia, hair loss, diarrhea, and thrombocytopenia.
Because of the potential for genotoxicity, the FDA advised health care providers to tell male patients with female partners of reproductive potential to use effective contraception during treatment with palbociclib and for 3 months after the last dose.
Powerful breast-implant testimony constrained by limited evidence
What’s the role of anecdotal medical histories in the era of evidence-based medicine?

But the anecdotal histories fell short of producing a clear committee consensus on dramatic, immediate changes in FDA policy, such as joining a renewed ban on certain types of breast implants linked with a rare lymphoma, a step recently taken by 38 other countries, including 33 European countries acting in concert through the European Union.
The disconnect between gripping testimony and limited panel recommendations was most stark for a complication that’s been named Breast Implant Illness (BII) by patients on the Internet. Many breast implant recipients have reported life-changing symptoms that appeared after implant placement, most often fatigue, joint and muscle pain, brain fog, neurologic symptoms, immune dysfunction, skin manifestations, and autoimmune disease or symptoms. By my count, 22 people spoke about their harrowing experiences with BII symptoms out of the 77 who stepped to the panel’s public-comment mic during 4 hours of public testimony over 2-days of hearings, often saying that they had experienced dramatic improvements after their implants came out. The meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee also heard presentations from two experts who ran some of the first reported studies on BII, or a BII-like syndrome called Autoimmune Syndrome Induced by Adjuvants (ASIA) described by Jan W.C. Tervaert, MD, professor of medicine and director of rheumatology at the University of Alberta in Edmonton. Dr. Tervaert and his associates published their findings about ASIA in the rheumatology literature last year (Clin Rheumatol. 2018 Feb;37[2]:483-93), and during his talk before the FDA panel, he said that silicone breast implants and the surgical mesh often used with them could be ASIA triggers.
Panel members seemed to mostly believe that the evidence they heard about BII did no more than hint at a possible association between breast implants and BII symptoms that required additional study. Many agreed on the need to include mention of the most common BII-linked patient complaints in informed consent material, but some were reluctant about even taking that step.

“I do not mention BII to patients. It’s not a disease; it’s a constellation of symptoms,” said panel member and plastic surgeon Pierre M. Chevray, MD, from Houston Methodist Hospital. The evidence for BII “is extremely anecdotal,” he said in an interview at the end of the 2-day session. Descriptions of BII “have been mainly published on social media. One reason why I don’t tell patients [about BII as part of informed consent] is because right now the evidence of a link is weak. We don’t yet even have a definition of this as an illness. A first step is to define it,” said Dr. Chevray, who has a very active implant practice. Other plastic surgeons were more accepting of BII as a real complication, although they agreed it needs much more study. During the testimony period, St. Louis plastic surgeon Patricia A. McGuire, MD, highlighted the challenge of teasing apart whether real symptoms are truly related to implants or are simply common ailments that accumulate during middle-age in many women. Dr. McGuire and some of her associates published an assessment of the challenges and possible solutions to studying BII that appeared shortly before the hearing (Plast Reconstr Surg. 2019 March;143[3S]:74S-81S),
Consensus recommendations from the panel to the FDA to address BII included having a single registry that would include all U.S. patients who receive breast implants (recently launched as the National Breast Implant Registry), inclusion of a control group, and collection of data at baseline and after regular follow-up intervals that includes a variety of measures relevant to autoimmune and rheumatologic disorders. Several panel members cited inadequate postmarketing safety surveillance by manufacturers in the years since breast implants returned to the U.S. market, and earlier in March, the FDA issued warning letters to two of the four companies that market U.S. breast implants over their inadequate long-term safety follow-up.
The panel’s decisions about the other major implant-associated health risk it considered, breast implant associated anaplastic large cell lymphoma (BIA-ALCL), faced a different sort of challenge. First described as linked to breast implants in 2011, today there is little doubt that BIA-ALCL is a consequence of breast implants, what several patients derisively called a “man-made cancer.” The key issue the committee grappled with was whether the calculated incidence of BIA-ALCL was at a frequency that warranted a ban on at least selected breast implant types. Mark W. Clemens, MD, a plastic surgeon at MD Anderson Cancer Center in Houston, told the panel that he calculated the Allergan Biocell group of implants, which have textured surfaces that allows for easier and more stable placement in patients, linked with an incidence of BIA-ALCL that was sevenfold to eightfold higher than that with smooth implants. That’s against a background of an overall incidence of about one case for every 20,000 U.S. implant recipients, Dr. Clemens said.
Many testifying patients, including several of the eight who described a personal history of BIA-ALCL, called for a ban on the sale of at least some breast implants because of their role in causing lymphoma. That sentiment was shared by Dr. Chevray, who endorsed a ban on “salt-loss” implants (the method that makes Biocell implants) during his closing comments to his fellow panel members. But earlier during panel discussions, others on the committee pushed back against implant bans, leaving the FDA’s eventual decision on this issue unclear. Evidence presented during the hearings suggests that implants cause ALCL by triggering a local “inflammatory milieu” and that different types of implants can have varying levels of potency for producing this milieu.

Perhaps the closest congruence between what patients called for and what the committee recommended was on informed consent. “No doubt, patients feel that informed consent failed them,” concluded panel member Karen E. Burke, MD, a New York dermatologist who was one of two panel discussants for the topic.
In addition to many suggestions on how to improve informed consent and public awareness lobbed at FDA staffers during the session by panel members, the final public comment of the 2 days came from Laurie A. Casas, MD, a Chicago plastic surgeon affiliated with the University of Chicago and a member of the board of directors of the American Society of Aesthetic Plastic Surgery (also know as the Aesthetic Society). During her testimony, Dr. Casas said “Over the past 2 days, we heard that patients need a structured educational checklist for informed consent. The Aesthetic Society hears you,” and promised that the website of the Society’s publication, the Aesthetic Surgery Journal, will soon feature a safety checklist for people receiving breast implants that will get updated as new information becomes available. She also highlighted the need for a comprehensive registry and long-term follow-up of implant recipients by the plastic surgeons who treated them.
In addition to better informed consent, patients who came to the hearing clearly also hoped to raise awareness in the general American public about the potential dangers from breast implants and the need to follow patients who receive implants. The 2 days of hearing accomplished that in part just by taking place. The New York Times and The Washington Post ran at least a couple of articles apiece on implant safety just before or during the hearings, while a more regional paper, the Philadelphia Inquirer, ran one article, as presumably did many other newspapers, broadcast outlets, and websites across America. Much of the coverage focused on compelling and moving personal stories from patients.
Women who have been having adverse effects from breast implants “have felt dismissed,” noted panel member Natalie C. Portis, PhD, a clinical psychologist from Oakland, Calif., and the patient representative on the advisory committee. “We need to listen to women that something real is happening.”
Dr. Tervaert, Dr. Chevray, Dr. McGuire, Dr. Clemens, Dr. Burke, Dr. Casas, and Dr. Portis had no relevant commercial disclosures.
What’s the role of anecdotal medical histories in the era of evidence-based medicine?
But the anecdotal histories fell short of producing a clear committee consensus on dramatic, immediate changes in FDA policy, such as joining a renewed ban on certain types of breast implants linked with a rare lymphoma, a step recently taken by 38 other countries, including 33 European countries acting in concert through the European Union.
The disconnect between gripping testimony and limited panel recommendations was most stark for a complication that’s been named Breast Implant Illness (BII) by patients on the Internet. Many breast implant recipients have reported life-changing symptoms that appeared after implant placement, most often fatigue, joint and muscle pain, brain fog, neurologic symptoms, immune dysfunction, skin manifestations, and autoimmune disease or symptoms. By my count, 22 people spoke about their harrowing experiences with BII symptoms out of the 77 who stepped to the panel’s public-comment mic during 4 hours of public testimony over 2-days of hearings, often saying that they had experienced dramatic improvements after their implants came out. The meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee also heard presentations from two experts who ran some of the first reported studies on BII, or a BII-like syndrome called Autoimmune Syndrome Induced by Adjuvants (ASIA) described by Jan W.C. Tervaert, MD, professor of medicine and director of rheumatology at the University of Alberta in Edmonton. Dr. Tervaert and his associates published their findings about ASIA in the rheumatology literature last year (Clin Rheumatol. 2018 Feb;37[2]:483-93), and during his talk before the FDA panel, he said that silicone breast implants and the surgical mesh often used with them could be ASIA triggers.
Panel members seemed to mostly believe that the evidence they heard about BII did no more than hint at a possible association between breast implants and BII symptoms that required additional study. Many agreed on the need to include mention of the most common BII-linked patient complaints in informed consent material, but some were reluctant about even taking that step.
“I do not mention BII to patients. It’s not a disease; it’s a constellation of symptoms,” said panel member and plastic surgeon Pierre M. Chevray, MD, from Houston Methodist Hospital. The evidence for BII “is extremely anecdotal,” he said in an interview at the end of the 2-day session. Descriptions of BII “have been mainly published on social media. One reason why I don’t tell patients [about BII as part of informed consent] is because right now the evidence of a link is weak. We don’t yet even have a definition of this as an illness. A first step is to define it,” said Dr. Chevray, who has a very active implant practice. Other plastic surgeons were more accepting of BII as a real complication, although they agreed it needs much more study. During the testimony period, St. Louis plastic surgeon Patricia A. McGuire, MD, highlighted the challenge of teasing apart whether real symptoms are truly related to implants or are simply common ailments that accumulate during middle-age in many women. Dr. McGuire and some of her associates published an assessment of the challenges and possible solutions to studying BII that appeared shortly before the hearing (Plast Reconstr Surg. 2019 March;143[3S]:74S-81S),
Consensus recommendations from the panel to the FDA to address BII included having a single registry that would include all U.S. patients who receive breast implants (recently launched as the National Breast Implant Registry), inclusion of a control group, and collection of data at baseline and after regular follow-up intervals that includes a variety of measures relevant to autoimmune and rheumatologic disorders. Several panel members cited inadequate postmarketing safety surveillance by manufacturers in the years since breast implants returned to the U.S. market, and earlier in March, the FDA issued warning letters to two of the four companies that market U.S. breast implants over their inadequate long-term safety follow-up.
The panel’s decisions about the other major implant-associated health risk it considered, breast implant associated anaplastic large cell lymphoma (BIA-ALCL), faced a different sort of challenge. First described as linked to breast implants in 2011, today there is little doubt that BIA-ALCL is a consequence of breast implants, what several patients derisively called a “man-made cancer.” The key issue the committee grappled with was whether the calculated incidence of BIA-ALCL was at a frequency that warranted a ban on at least selected breast implant types. Mark W. Clemens, MD, a plastic surgeon at MD Anderson Cancer Center in Houston, told the panel that he calculated the Allergan Biocell group of implants, which have textured surfaces that allows for easier and more stable placement in patients, linked with an incidence of BIA-ALCL that was sevenfold to eightfold higher than that with smooth implants. That’s against a background of an overall incidence of about one case for every 20,000 U.S. implant recipients, Dr. Clemens said.
Many testifying patients, including several of the eight who described a personal history of BIA-ALCL, called for a ban on the sale of at least some breast implants because of their role in causing lymphoma. That sentiment was shared by Dr. Chevray, who endorsed a ban on “salt-loss” implants (the method that makes Biocell implants) during his closing comments to his fellow panel members. But earlier during panel discussions, others on the committee pushed back against implant bans, leaving the FDA’s eventual decision on this issue unclear. Evidence presented during the hearings suggests that implants cause ALCL by triggering a local “inflammatory milieu” and that different types of implants can have varying levels of potency for producing this milieu.
Perhaps the closest congruence between what patients called for and what the committee recommended was on informed consent. “No doubt, patients feel that informed consent failed them,” concluded panel member Karen E. Burke, MD, a New York dermatologist who was one of two panel discussants for the topic.
In addition to many suggestions on how to improve informed consent and public awareness lobbed at FDA staffers during the session by panel members, the final public comment of the 2 days came from Laurie A. Casas, MD, a Chicago plastic surgeon affiliated with the University of Chicago and a member of the board of directors of the American Society of Aesthetic Plastic Surgery (also know as the Aesthetic Society). During her testimony, Dr. Casas said “Over the past 2 days, we heard that patients need a structured educational checklist for informed consent. The Aesthetic Society hears you,” and promised that the website of the Society’s publication, the Aesthetic Surgery Journal, will soon feature a safety checklist for people receiving breast implants that will get updated as new information becomes available. She also highlighted the need for a comprehensive registry and long-term follow-up of implant recipients by the plastic surgeons who treated them.
In addition to better informed consent, patients who came to the hearing clearly also hoped to raise awareness in the general American public about the potential dangers from breast implants and the need to follow patients who receive implants. The 2 days of hearing accomplished that in part just by taking place. The New York Times and The Washington Post ran at least a couple of articles apiece on implant safety just before or during the hearings, while a more regional paper, the Philadelphia Inquirer, ran one article, as presumably did many other newspapers, broadcast outlets, and websites across America. Much of the coverage focused on compelling and moving personal stories from patients.
Women who have been having adverse effects from breast implants “have felt dismissed,” noted panel member Natalie C. Portis, PhD, a clinical psychologist from Oakland, Calif., and the patient representative on the advisory committee. “We need to listen to women that something real is happening.”
Dr. Tervaert, Dr. Chevray, Dr. McGuire, Dr. Clemens, Dr. Burke, Dr. Casas, and Dr. Portis had no relevant commercial disclosures.
What’s the role of anecdotal medical histories in the era of evidence-based medicine?
But the anecdotal histories fell short of producing a clear committee consensus on dramatic, immediate changes in FDA policy, such as joining a renewed ban on certain types of breast implants linked with a rare lymphoma, a step recently taken by 38 other countries, including 33 European countries acting in concert through the European Union.
The disconnect between gripping testimony and limited panel recommendations was most stark for a complication that’s been named Breast Implant Illness (BII) by patients on the Internet. Many breast implant recipients have reported life-changing symptoms that appeared after implant placement, most often fatigue, joint and muscle pain, brain fog, neurologic symptoms, immune dysfunction, skin manifestations, and autoimmune disease or symptoms. By my count, 22 people spoke about their harrowing experiences with BII symptoms out of the 77 who stepped to the panel’s public-comment mic during 4 hours of public testimony over 2-days of hearings, often saying that they had experienced dramatic improvements after their implants came out. The meeting of the General and Plastic Surgery Devices Panel of the Medical Devices Advisory Committee also heard presentations from two experts who ran some of the first reported studies on BII, or a BII-like syndrome called Autoimmune Syndrome Induced by Adjuvants (ASIA) described by Jan W.C. Tervaert, MD, professor of medicine and director of rheumatology at the University of Alberta in Edmonton. Dr. Tervaert and his associates published their findings about ASIA in the rheumatology literature last year (Clin Rheumatol. 2018 Feb;37[2]:483-93), and during his talk before the FDA panel, he said that silicone breast implants and the surgical mesh often used with them could be ASIA triggers.
Panel members seemed to mostly believe that the evidence they heard about BII did no more than hint at a possible association between breast implants and BII symptoms that required additional study. Many agreed on the need to include mention of the most common BII-linked patient complaints in informed consent material, but some were reluctant about even taking that step.
“I do not mention BII to patients. It’s not a disease; it’s a constellation of symptoms,” said panel member and plastic surgeon Pierre M. Chevray, MD, from Houston Methodist Hospital. The evidence for BII “is extremely anecdotal,” he said in an interview at the end of the 2-day session. Descriptions of BII “have been mainly published on social media. One reason why I don’t tell patients [about BII as part of informed consent] is because right now the evidence of a link is weak. We don’t yet even have a definition of this as an illness. A first step is to define it,” said Dr. Chevray, who has a very active implant practice. Other plastic surgeons were more accepting of BII as a real complication, although they agreed it needs much more study. During the testimony period, St. Louis plastic surgeon Patricia A. McGuire, MD, highlighted the challenge of teasing apart whether real symptoms are truly related to implants or are simply common ailments that accumulate during middle-age in many women. Dr. McGuire and some of her associates published an assessment of the challenges and possible solutions to studying BII that appeared shortly before the hearing (Plast Reconstr Surg. 2019 March;143[3S]:74S-81S),
Consensus recommendations from the panel to the FDA to address BII included having a single registry that would include all U.S. patients who receive breast implants (recently launched as the National Breast Implant Registry), inclusion of a control group, and collection of data at baseline and after regular follow-up intervals that includes a variety of measures relevant to autoimmune and rheumatologic disorders. Several panel members cited inadequate postmarketing safety surveillance by manufacturers in the years since breast implants returned to the U.S. market, and earlier in March, the FDA issued warning letters to two of the four companies that market U.S. breast implants over their inadequate long-term safety follow-up.
The panel’s decisions about the other major implant-associated health risk it considered, breast implant associated anaplastic large cell lymphoma (BIA-ALCL), faced a different sort of challenge. First described as linked to breast implants in 2011, today there is little doubt that BIA-ALCL is a consequence of breast implants, what several patients derisively called a “man-made cancer.” The key issue the committee grappled with was whether the calculated incidence of BIA-ALCL was at a frequency that warranted a ban on at least selected breast implant types. Mark W. Clemens, MD, a plastic surgeon at MD Anderson Cancer Center in Houston, told the panel that he calculated the Allergan Biocell group of implants, which have textured surfaces that allows for easier and more stable placement in patients, linked with an incidence of BIA-ALCL that was sevenfold to eightfold higher than that with smooth implants. That’s against a background of an overall incidence of about one case for every 20,000 U.S. implant recipients, Dr. Clemens said.
Many testifying patients, including several of the eight who described a personal history of BIA-ALCL, called for a ban on the sale of at least some breast implants because of their role in causing lymphoma. That sentiment was shared by Dr. Chevray, who endorsed a ban on “salt-loss” implants (the method that makes Biocell implants) during his closing comments to his fellow panel members. But earlier during panel discussions, others on the committee pushed back against implant bans, leaving the FDA’s eventual decision on this issue unclear. Evidence presented during the hearings suggests that implants cause ALCL by triggering a local “inflammatory milieu” and that different types of implants can have varying levels of potency for producing this milieu.
Perhaps the closest congruence between what patients called for and what the committee recommended was on informed consent. “No doubt, patients feel that informed consent failed them,” concluded panel member Karen E. Burke, MD, a New York dermatologist who was one of two panel discussants for the topic.
In addition to many suggestions on how to improve informed consent and public awareness lobbed at FDA staffers during the session by panel members, the final public comment of the 2 days came from Laurie A. Casas, MD, a Chicago plastic surgeon affiliated with the University of Chicago and a member of the board of directors of the American Society of Aesthetic Plastic Surgery (also know as the Aesthetic Society). During her testimony, Dr. Casas said “Over the past 2 days, we heard that patients need a structured educational checklist for informed consent. The Aesthetic Society hears you,” and promised that the website of the Society’s publication, the Aesthetic Surgery Journal, will soon feature a safety checklist for people receiving breast implants that will get updated as new information becomes available. She also highlighted the need for a comprehensive registry and long-term follow-up of implant recipients by the plastic surgeons who treated them.
In addition to better informed consent, patients who came to the hearing clearly also hoped to raise awareness in the general American public about the potential dangers from breast implants and the need to follow patients who receive implants. The 2 days of hearing accomplished that in part just by taking place. The New York Times and The Washington Post ran at least a couple of articles apiece on implant safety just before or during the hearings, while a more regional paper, the Philadelphia Inquirer, ran one article, as presumably did many other newspapers, broadcast outlets, and websites across America. Much of the coverage focused on compelling and moving personal stories from patients.
Women who have been having adverse effects from breast implants “have felt dismissed,” noted panel member Natalie C. Portis, PhD, a clinical psychologist from Oakland, Calif., and the patient representative on the advisory committee. “We need to listen to women that something real is happening.”
Dr. Tervaert, Dr. Chevray, Dr. McGuire, Dr. Clemens, Dr. Burke, Dr. Casas, and Dr. Portis had no relevant commercial disclosures.
FDA concerned about e-cigs/seizures in youth
the agency announced April 3.

Between 2010 and early 2019, the FDA and poison control centers received 35 reports of seizures that mentioned the use of e-cigarettes. Most reports involved youth or young adults, and the reports have increased slightly since June 2018, the announcement says.
“We want to be clear that we don’t yet know if there’s a direct relationship between the use of e-cigarettes and a risk of seizure,” said FDA Commissioner Scott Gottlieb, MD, and Principal Deputy Commissioner Amy Abernethy, MD, PhD, in a statement. “We believe these 35 cases warrant scientific investigation into whether there is in fact a connection.”
In addition, the FDA is trying to determine whether any e-cigarette product-specific factors may be associated with the risk of seizures.
Seizures have been reported after a few puffs or up to 1 day after e-cigarette use and among first-time and experienced users. A few patients had a prior history of seizures or also used other substances, such as marijuana or amphetamines.
“While 35 cases may not seem like much compared to the total number of people using e-cigarettes, we are nonetheless concerned by these reported cases. We also recognized that not all of the cases may be reported,” Dr. Gottlieb and Dr. Abernethy said.
Although seizures are known side effects of nicotine toxicity and have been reported in the context of intentional or accidental swallowing of e-cigarette liquid, the voluntary reports of seizures occurring with vaping could represent a new safety issue, the FDA said.
The agency encouraged people to report cases via an online safety reporting portal. It also provided redacted case reports that involve vaping and seizures.
the agency announced April 3.
Between 2010 and early 2019, the FDA and poison control centers received 35 reports of seizures that mentioned the use of e-cigarettes. Most reports involved youth or young adults, and the reports have increased slightly since June 2018, the announcement says.
“We want to be clear that we don’t yet know if there’s a direct relationship between the use of e-cigarettes and a risk of seizure,” said FDA Commissioner Scott Gottlieb, MD, and Principal Deputy Commissioner Amy Abernethy, MD, PhD, in a statement. “We believe these 35 cases warrant scientific investigation into whether there is in fact a connection.”
In addition, the FDA is trying to determine whether any e-cigarette product-specific factors may be associated with the risk of seizures.
Seizures have been reported after a few puffs or up to 1 day after e-cigarette use and among first-time and experienced users. A few patients had a prior history of seizures or also used other substances, such as marijuana or amphetamines.
“While 35 cases may not seem like much compared to the total number of people using e-cigarettes, we are nonetheless concerned by these reported cases. We also recognized that not all of the cases may be reported,” Dr. Gottlieb and Dr. Abernethy said.
Although seizures are known side effects of nicotine toxicity and have been reported in the context of intentional or accidental swallowing of e-cigarette liquid, the voluntary reports of seizures occurring with vaping could represent a new safety issue, the FDA said.
The agency encouraged people to report cases via an online safety reporting portal. It also provided redacted case reports that involve vaping and seizures.
the agency announced April 3.
Between 2010 and early 2019, the FDA and poison control centers received 35 reports of seizures that mentioned the use of e-cigarettes. Most reports involved youth or young adults, and the reports have increased slightly since June 2018, the announcement says.
“We want to be clear that we don’t yet know if there’s a direct relationship between the use of e-cigarettes and a risk of seizure,” said FDA Commissioner Scott Gottlieb, MD, and Principal Deputy Commissioner Amy Abernethy, MD, PhD, in a statement. “We believe these 35 cases warrant scientific investigation into whether there is in fact a connection.”
In addition, the FDA is trying to determine whether any e-cigarette product-specific factors may be associated with the risk of seizures.
Seizures have been reported after a few puffs or up to 1 day after e-cigarette use and among first-time and experienced users. A few patients had a prior history of seizures or also used other substances, such as marijuana or amphetamines.
“While 35 cases may not seem like much compared to the total number of people using e-cigarettes, we are nonetheless concerned by these reported cases. We also recognized that not all of the cases may be reported,” Dr. Gottlieb and Dr. Abernethy said.
Although seizures are known side effects of nicotine toxicity and have been reported in the context of intentional or accidental swallowing of e-cigarette liquid, the voluntary reports of seizures occurring with vaping could represent a new safety issue, the FDA said.
The agency encouraged people to report cases via an online safety reporting portal. It also provided redacted case reports that involve vaping and seizures.
Measles: Latest weekly count is the highest of the year
according to the Centers for Disease Control and Prevention.
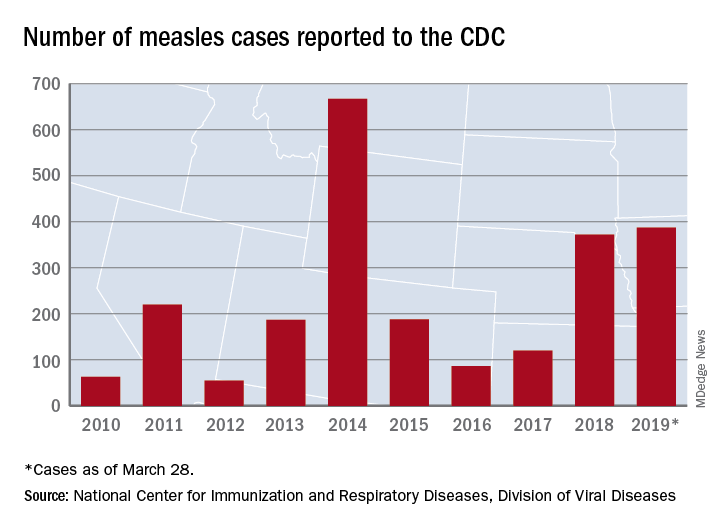
The 73 new cases of measles reported to the CDC during the week ending March 28 – more than any other single week so far in 2019 – brings the total number of cases for the year to 387, the CDC reported April 1. That surpasses the 372 reported in 2018 and is now the highest annual count since 667 cases were reported in 2014.
The ongoing outbreak in Rockland County, N.Y., which resulted in 6 new cases there last week and 52 for the year, prompted County Executive Ed Day to declare a state of emergency effective March 27 that bars unvaccinated individuals under age 18 years from public places for the next 30 days unless they receive an MMR vaccination.
“As this outbreak has continued our inspectors have begun to meet resistance from those they are trying to protect. They have been hung up on or told not to call again. They’ve been told ‘we’re not discussing this, do not come back,’ when visiting the homes of infected individuals as part of their investigations. This type of response is unacceptable and irresponsible. It endangers the health and well-being of others and displays a shocking lack of responsibility and concern for others in our community,” Mr. Day said in a written statement.
In addition to Rockland County, the CDC is currently tracking five other outbreaks: New York City, mainly Brooklyn (33 new cases last week); Washington state (74 cases for the year, but no new cases in the last week); New Jersey (10 total cases, with 8 related to an outbreak in Ocean and Monmouth Counties); and two in California (16 total cases, with 11 related to the outbreaks). One of the California outbreaks and the New Jersey outbreak are new, but the CDC is no longer reporting outbreaks in Texas and Illinois, so the total stays at six nationwide.
In related news from California, state Sen. Richard Pan (D), a pediatrician, and Assemblywoman Lorena Gonzalez (D) introduced a bill to monitor vaccine exemptions “by requiring the state health department to vet each medical exemption form written by physicians [and to] maintain a database of exemptions that would allow officials to monitor which doctors are granting the exemptions,” the Los Angeles Times reported.
according to the Centers for Disease Control and Prevention.
The 73 new cases of measles reported to the CDC during the week ending March 28 – more than any other single week so far in 2019 – brings the total number of cases for the year to 387, the CDC reported April 1. That surpasses the 372 reported in 2018 and is now the highest annual count since 667 cases were reported in 2014.
The ongoing outbreak in Rockland County, N.Y., which resulted in 6 new cases there last week and 52 for the year, prompted County Executive Ed Day to declare a state of emergency effective March 27 that bars unvaccinated individuals under age 18 years from public places for the next 30 days unless they receive an MMR vaccination.
“As this outbreak has continued our inspectors have begun to meet resistance from those they are trying to protect. They have been hung up on or told not to call again. They’ve been told ‘we’re not discussing this, do not come back,’ when visiting the homes of infected individuals as part of their investigations. This type of response is unacceptable and irresponsible. It endangers the health and well-being of others and displays a shocking lack of responsibility and concern for others in our community,” Mr. Day said in a written statement.
In addition to Rockland County, the CDC is currently tracking five other outbreaks: New York City, mainly Brooklyn (33 new cases last week); Washington state (74 cases for the year, but no new cases in the last week); New Jersey (10 total cases, with 8 related to an outbreak in Ocean and Monmouth Counties); and two in California (16 total cases, with 11 related to the outbreaks). One of the California outbreaks and the New Jersey outbreak are new, but the CDC is no longer reporting outbreaks in Texas and Illinois, so the total stays at six nationwide.
In related news from California, state Sen. Richard Pan (D), a pediatrician, and Assemblywoman Lorena Gonzalez (D) introduced a bill to monitor vaccine exemptions “by requiring the state health department to vet each medical exemption form written by physicians [and to] maintain a database of exemptions that would allow officials to monitor which doctors are granting the exemptions,” the Los Angeles Times reported.
according to the Centers for Disease Control and Prevention.
The 73 new cases of measles reported to the CDC during the week ending March 28 – more than any other single week so far in 2019 – brings the total number of cases for the year to 387, the CDC reported April 1. That surpasses the 372 reported in 2018 and is now the highest annual count since 667 cases were reported in 2014.
The ongoing outbreak in Rockland County, N.Y., which resulted in 6 new cases there last week and 52 for the year, prompted County Executive Ed Day to declare a state of emergency effective March 27 that bars unvaccinated individuals under age 18 years from public places for the next 30 days unless they receive an MMR vaccination.
“As this outbreak has continued our inspectors have begun to meet resistance from those they are trying to protect. They have been hung up on or told not to call again. They’ve been told ‘we’re not discussing this, do not come back,’ when visiting the homes of infected individuals as part of their investigations. This type of response is unacceptable and irresponsible. It endangers the health and well-being of others and displays a shocking lack of responsibility and concern for others in our community,” Mr. Day said in a written statement.
In addition to Rockland County, the CDC is currently tracking five other outbreaks: New York City, mainly Brooklyn (33 new cases last week); Washington state (74 cases for the year, but no new cases in the last week); New Jersey (10 total cases, with 8 related to an outbreak in Ocean and Monmouth Counties); and two in California (16 total cases, with 11 related to the outbreaks). One of the California outbreaks and the New Jersey outbreak are new, but the CDC is no longer reporting outbreaks in Texas and Illinois, so the total stays at six nationwide.
In related news from California, state Sen. Richard Pan (D), a pediatrician, and Assemblywoman Lorena Gonzalez (D) introduced a bill to monitor vaccine exemptions “by requiring the state health department to vet each medical exemption form written by physicians [and to] maintain a database of exemptions that would allow officials to monitor which doctors are granting the exemptions,” the Los Angeles Times reported.
FDA approves multiple ambrisentan generics for patients with PAH
The Food and Drug Administration has approved multiple generics for ambrisentan (Letairis) tablets, as well as their associated risk evaluation and mitigation strategies (REMS), for patients with pulmonary arterial hypertension.
A total of four generics were approved, licensed to Mylan Pharmaceuticals, Sun Pharma Global, Watson Laboratories, and Zydus Pharmaceuticals. All four were approved at 5-mg and 10-mg doses. According to the label, the most common adverse events associated with ambrisentan include peripheral edema, nasal congestion, sinusitis, and flushing.
In addition to the generics, the FDA also approved two shared system REMS programs for ambrisentan. The first, Ambrisentan REMS, includes the brand sponsor and three abbreviated new drug applications. The FDA also approved two shared system REMS programs for ambrisentan. The first, Ambrisentan REMS, is composed of the brand sponsor and three abbreviated new drug applications. The second, PS-Ambrisentan REMS, is a parallel system and currently is constituted by one abbreviated new drug application.
“With the approval of these first generics ... patients will now have access to additional products (brand-name and generic) and additional types of pharmacies to fill their prescriptions,” the FDA said.
Find the full press release on the FDA website.
The Food and Drug Administration has approved multiple generics for ambrisentan (Letairis) tablets, as well as their associated risk evaluation and mitigation strategies (REMS), for patients with pulmonary arterial hypertension.
A total of four generics were approved, licensed to Mylan Pharmaceuticals, Sun Pharma Global, Watson Laboratories, and Zydus Pharmaceuticals. All four were approved at 5-mg and 10-mg doses. According to the label, the most common adverse events associated with ambrisentan include peripheral edema, nasal congestion, sinusitis, and flushing.
In addition to the generics, the FDA also approved two shared system REMS programs for ambrisentan. The first, Ambrisentan REMS, includes the brand sponsor and three abbreviated new drug applications. The FDA also approved two shared system REMS programs for ambrisentan. The first, Ambrisentan REMS, is composed of the brand sponsor and three abbreviated new drug applications. The second, PS-Ambrisentan REMS, is a parallel system and currently is constituted by one abbreviated new drug application.
“With the approval of these first generics ... patients will now have access to additional products (brand-name and generic) and additional types of pharmacies to fill their prescriptions,” the FDA said.
Find the full press release on the FDA website.
The Food and Drug Administration has approved multiple generics for ambrisentan (Letairis) tablets, as well as their associated risk evaluation and mitigation strategies (REMS), for patients with pulmonary arterial hypertension.
A total of four generics were approved, licensed to Mylan Pharmaceuticals, Sun Pharma Global, Watson Laboratories, and Zydus Pharmaceuticals. All four were approved at 5-mg and 10-mg doses. According to the label, the most common adverse events associated with ambrisentan include peripheral edema, nasal congestion, sinusitis, and flushing.
In addition to the generics, the FDA also approved two shared system REMS programs for ambrisentan. The first, Ambrisentan REMS, includes the brand sponsor and three abbreviated new drug applications. The FDA also approved two shared system REMS programs for ambrisentan. The first, Ambrisentan REMS, is composed of the brand sponsor and three abbreviated new drug applications. The second, PS-Ambrisentan REMS, is a parallel system and currently is constituted by one abbreviated new drug application.
“With the approval of these first generics ... patients will now have access to additional products (brand-name and generic) and additional types of pharmacies to fill their prescriptions,” the FDA said.
Find the full press release on the FDA website.