User login
What is the diagnosis?
As the lesion was growing, getting more violaceous and indurated, a biopsy was performed. The biopsy showed multiple discrete lobules of dermal capillaries with slight extension into the superficial subcutis. Capillary lobules demonstrate the “cannonball-like” architecture often associated with tufted angioma, and some lobules showed bulging into adjacent thin-walled vessels. Spindled endothelial cells lining slit-like vessels were present in the mid dermis, although this comprises a minority of the lesion. The majority of the subcutis was uninvolved. The findings are overall most consistent with a tufted angioma.

Kaposiform hemangioendothelioma (KHE) has been considered given the presence of occasional slit-like vascular spaces; however, the lesion is predominantly superficial and therefore the lesion is best classified as tufted angioma. GLUT–1 staining was negative.
At the time of biopsy, blood work was ordered, which showed a normal complete blood count with normal number of platelets, slightly elevated D-dimer, and slightly low fibrinogen. Several repeat blood counts and coagulation tests once a week for a few weeks revealed no changes.
The patient was started on aspirin at a dose of 5 mg/kg per day. After a week on the medication the lesion was starting to get smaller and less red.
Tufted angiomas are a rare type of vascular tumor within the spectrum of kaposiform hemangioendotheliomas. Most cases present within the first year of life; some occur at birth. They usually present as papules, plaques, or erythematous, violaceous indurated nodules on the face, neck, trunk, and extremities. The lesions can also be present with hyperhidrosis and hypertrichosis. Clinically, the lesions will have to be differentiated from other vascular tumors such as infantile hemangiomas, congenital hemangiomas, and Kaposi’s sarcoma, as well as subcutaneous fat necrosis of the newborn, cellulitis, and nonaccidental trauma.
Pathogenesis of tufted angiomas is poorly understood. A recent case report found a somatic mutation on GNA14.This protein regulates Ras activity and modulates endothelial cell permeability and migration in response to FGF2 and VEGFA. The p.205L mutation causes activation of GNA14, which upregulates pERK-MAPK pathway, suggesting MAPK inhibition as a potential target for therapy. Clinically, tufted angioma can present in three patterns: uncomplicated tufted angioma (most common type); tufted angioma without thrombocytopenia but with chronic coagulopathy, as it was seen in our patient; and tufted angioma associated with Kasabach-Merritt phenomenon (KMP). KMP is characterized by thrombocytopenia in association with microangiopathic hemolytic anemia, consumptive coagulopathy, and enlarging vascular tumor. Treatment of uncomplicated tufted angioma will depend on symptomatology, size, and location of the lesion. Smaller lesions in noncosmetically sensitive areas can be treated with surgical excision. Cases that are not amenable to excision can be treated with aspirin. There are also reports of response to topical modalities including tacrolimus and timolol. For complicated cases associated with KMP, sirolimus, systemic corticosteroids, ticlopidine, interferon, or vincristine are recommended. Some lesions may demonstrate spontaneous regression.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Cohen S et al. Dermatol Online J. 2019 Sep 15;25(9):13030/qt6pv254mc.
Lim YH et al. Pediatr Dermatol. 2019 Nov;36(6):963-4.
Prasuna A, Rao PN. Indian Dermatol Online J. 2015;6:266-8.
As the lesion was growing, getting more violaceous and indurated, a biopsy was performed. The biopsy showed multiple discrete lobules of dermal capillaries with slight extension into the superficial subcutis. Capillary lobules demonstrate the “cannonball-like” architecture often associated with tufted angioma, and some lobules showed bulging into adjacent thin-walled vessels. Spindled endothelial cells lining slit-like vessels were present in the mid dermis, although this comprises a minority of the lesion. The majority of the subcutis was uninvolved. The findings are overall most consistent with a tufted angioma.
Kaposiform hemangioendothelioma (KHE) has been considered given the presence of occasional slit-like vascular spaces; however, the lesion is predominantly superficial and therefore the lesion is best classified as tufted angioma. GLUT–1 staining was negative.
At the time of biopsy, blood work was ordered, which showed a normal complete blood count with normal number of platelets, slightly elevated D-dimer, and slightly low fibrinogen. Several repeat blood counts and coagulation tests once a week for a few weeks revealed no changes.
The patient was started on aspirin at a dose of 5 mg/kg per day. After a week on the medication the lesion was starting to get smaller and less red.
Tufted angiomas are a rare type of vascular tumor within the spectrum of kaposiform hemangioendotheliomas. Most cases present within the first year of life; some occur at birth. They usually present as papules, plaques, or erythematous, violaceous indurated nodules on the face, neck, trunk, and extremities. The lesions can also be present with hyperhidrosis and hypertrichosis. Clinically, the lesions will have to be differentiated from other vascular tumors such as infantile hemangiomas, congenital hemangiomas, and Kaposi’s sarcoma, as well as subcutaneous fat necrosis of the newborn, cellulitis, and nonaccidental trauma.
Pathogenesis of tufted angiomas is poorly understood. A recent case report found a somatic mutation on GNA14.This protein regulates Ras activity and modulates endothelial cell permeability and migration in response to FGF2 and VEGFA. The p.205L mutation causes activation of GNA14, which upregulates pERK-MAPK pathway, suggesting MAPK inhibition as a potential target for therapy. Clinically, tufted angioma can present in three patterns: uncomplicated tufted angioma (most common type); tufted angioma without thrombocytopenia but with chronic coagulopathy, as it was seen in our patient; and tufted angioma associated with Kasabach-Merritt phenomenon (KMP). KMP is characterized by thrombocytopenia in association with microangiopathic hemolytic anemia, consumptive coagulopathy, and enlarging vascular tumor. Treatment of uncomplicated tufted angioma will depend on symptomatology, size, and location of the lesion. Smaller lesions in noncosmetically sensitive areas can be treated with surgical excision. Cases that are not amenable to excision can be treated with aspirin. There are also reports of response to topical modalities including tacrolimus and timolol. For complicated cases associated with KMP, sirolimus, systemic corticosteroids, ticlopidine, interferon, or vincristine are recommended. Some lesions may demonstrate spontaneous regression.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Cohen S et al. Dermatol Online J. 2019 Sep 15;25(9):13030/qt6pv254mc.
Lim YH et al. Pediatr Dermatol. 2019 Nov;36(6):963-4.
Prasuna A, Rao PN. Indian Dermatol Online J. 2015;6:266-8.
As the lesion was growing, getting more violaceous and indurated, a biopsy was performed. The biopsy showed multiple discrete lobules of dermal capillaries with slight extension into the superficial subcutis. Capillary lobules demonstrate the “cannonball-like” architecture often associated with tufted angioma, and some lobules showed bulging into adjacent thin-walled vessels. Spindled endothelial cells lining slit-like vessels were present in the mid dermis, although this comprises a minority of the lesion. The majority of the subcutis was uninvolved. The findings are overall most consistent with a tufted angioma.
Kaposiform hemangioendothelioma (KHE) has been considered given the presence of occasional slit-like vascular spaces; however, the lesion is predominantly superficial and therefore the lesion is best classified as tufted angioma. GLUT–1 staining was negative.
At the time of biopsy, blood work was ordered, which showed a normal complete blood count with normal number of platelets, slightly elevated D-dimer, and slightly low fibrinogen. Several repeat blood counts and coagulation tests once a week for a few weeks revealed no changes.
The patient was started on aspirin at a dose of 5 mg/kg per day. After a week on the medication the lesion was starting to get smaller and less red.
Tufted angiomas are a rare type of vascular tumor within the spectrum of kaposiform hemangioendotheliomas. Most cases present within the first year of life; some occur at birth. They usually present as papules, plaques, or erythematous, violaceous indurated nodules on the face, neck, trunk, and extremities. The lesions can also be present with hyperhidrosis and hypertrichosis. Clinically, the lesions will have to be differentiated from other vascular tumors such as infantile hemangiomas, congenital hemangiomas, and Kaposi’s sarcoma, as well as subcutaneous fat necrosis of the newborn, cellulitis, and nonaccidental trauma.
Pathogenesis of tufted angiomas is poorly understood. A recent case report found a somatic mutation on GNA14.This protein regulates Ras activity and modulates endothelial cell permeability and migration in response to FGF2 and VEGFA. The p.205L mutation causes activation of GNA14, which upregulates pERK-MAPK pathway, suggesting MAPK inhibition as a potential target for therapy. Clinically, tufted angioma can present in three patterns: uncomplicated tufted angioma (most common type); tufted angioma without thrombocytopenia but with chronic coagulopathy, as it was seen in our patient; and tufted angioma associated with Kasabach-Merritt phenomenon (KMP). KMP is characterized by thrombocytopenia in association with microangiopathic hemolytic anemia, consumptive coagulopathy, and enlarging vascular tumor. Treatment of uncomplicated tufted angioma will depend on symptomatology, size, and location of the lesion. Smaller lesions in noncosmetically sensitive areas can be treated with surgical excision. Cases that are not amenable to excision can be treated with aspirin. There are also reports of response to topical modalities including tacrolimus and timolol. For complicated cases associated with KMP, sirolimus, systemic corticosteroids, ticlopidine, interferon, or vincristine are recommended. Some lesions may demonstrate spontaneous regression.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Cohen S et al. Dermatol Online J. 2019 Sep 15;25(9):13030/qt6pv254mc.
Lim YH et al. Pediatr Dermatol. 2019 Nov;36(6):963-4.
Prasuna A, Rao PN. Indian Dermatol Online J. 2015;6:266-8.
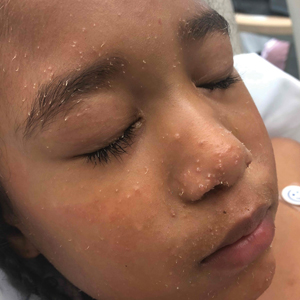
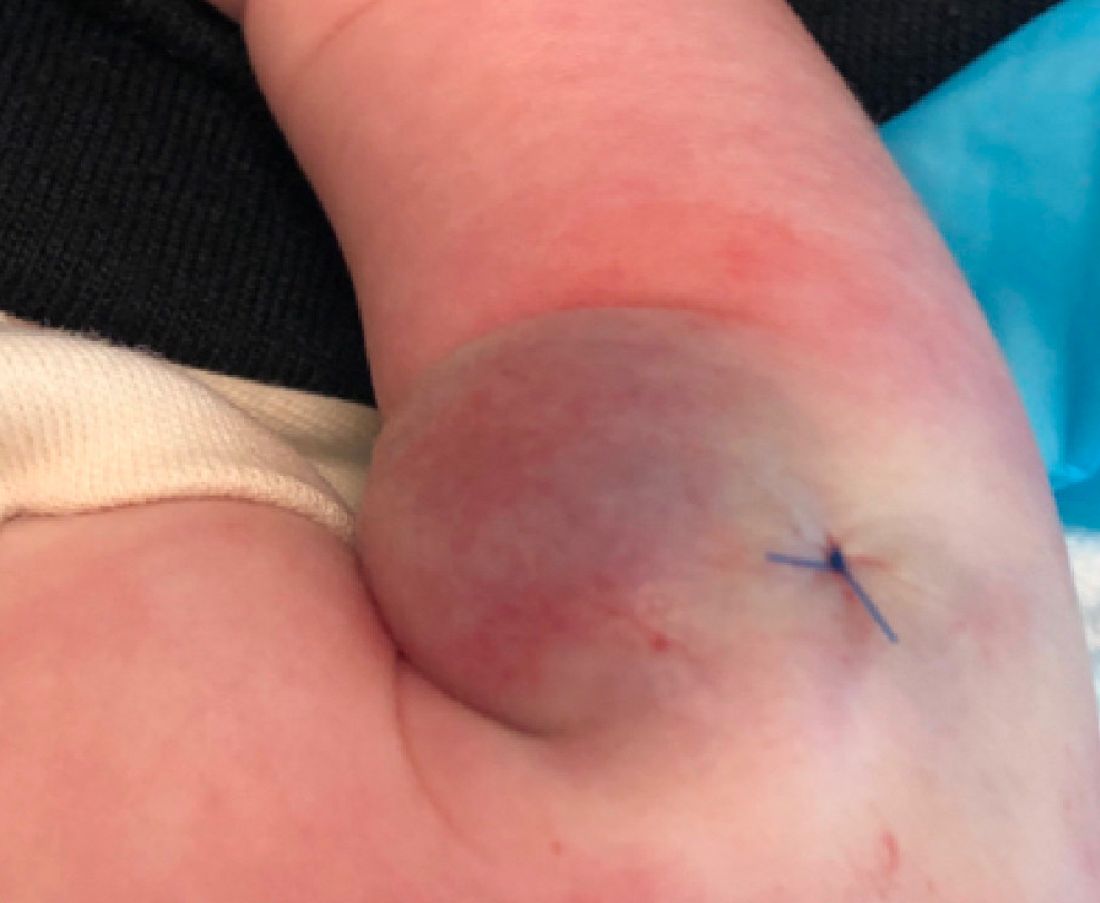
A 35-day-old female was referred to our pediatric dermatology clinic for evaluation of a red lesion on the right arm. The lesion presented at about 4 days of life as a red plaque (image 1 at 8 days of life).
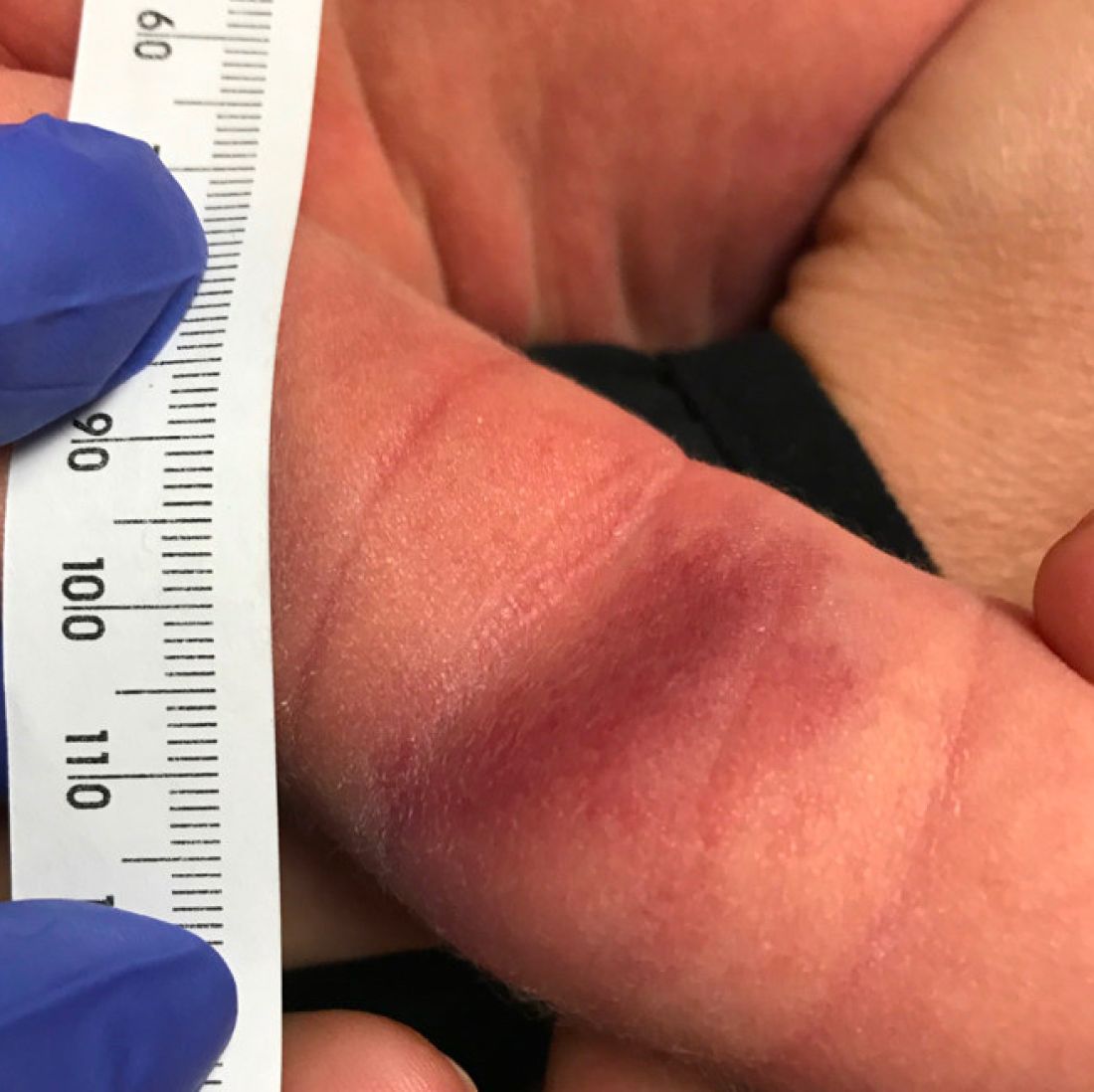
On the following days, the lesion started growing but it didn't seem to be tender or bothersome to the patient (image 2, at 35 days of life).
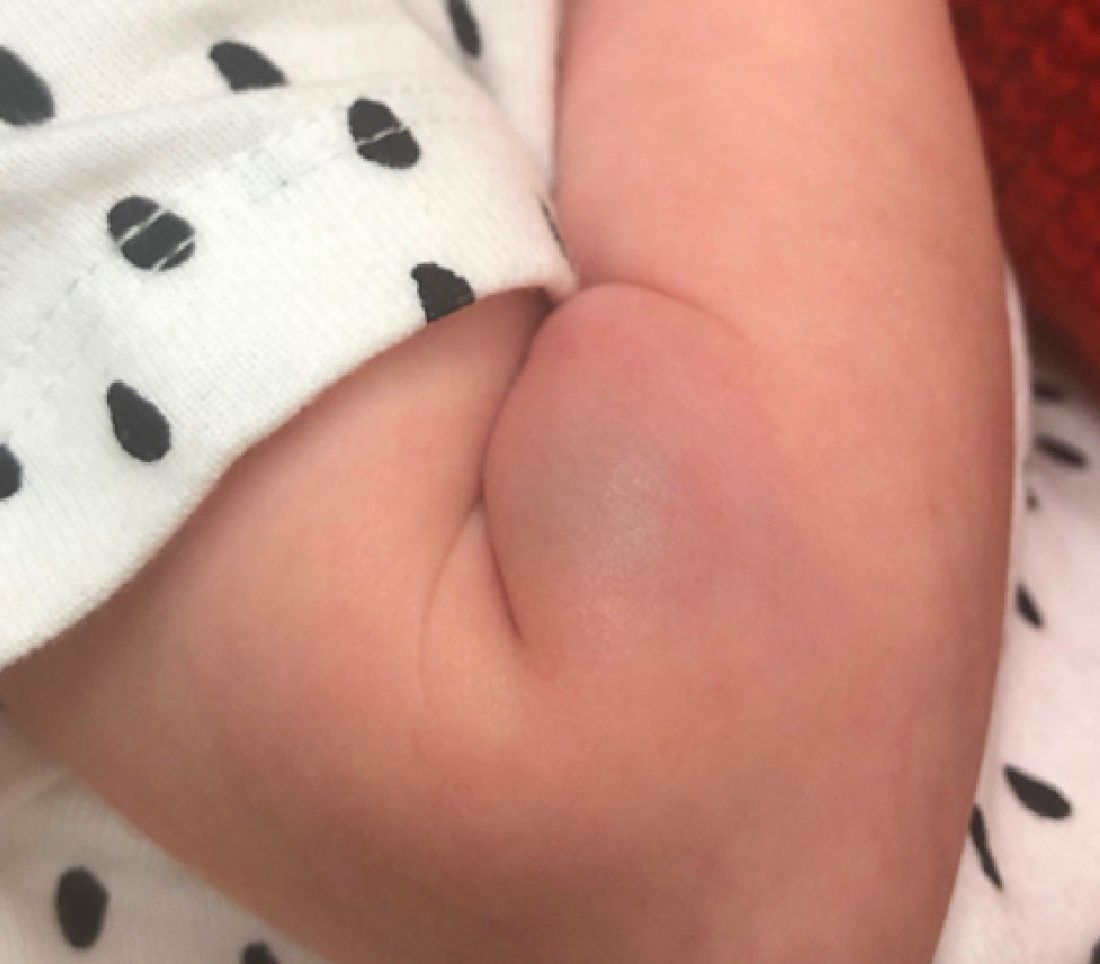
At a 2-week follow up the lesion was getting fuller and more violaceous. There was no history of fever and the lesion didn't appear tender to the touch.
She was born via normal spontaneous vaginal delivery. There were no complications and the mother received prenatal care.
On exam she had a red to violaceous nodule on the right arm (image 3 at 45 days of life).
What is the diagnosis?
Numerous morphologies of skin rashes have been described in the setting of COVID-19, including pernio, livedoid rash, exanthem, and vasculitis. This classic constellation of symptoms (palpable purpura on buttocks/legs, abdominal pain, arthralgia, hematuria) is highly consistent with Henoch-Schonlein purpura (HSP). There are now multiple case reports of COVID-19–associated HSP.

HSP is the most common type of childhood systemic vasculitis. It is mediated by immunoglobulin A (IgA) immune complex deposition and has been associated with respiratory tract infections, streptococcal species, parainfluenza virus, and human parvovirus B19, medications, vaccinations, and malignancies. HSP is usually a self-limiting disease, with a course over 4-6 weeks, and can affect multiple organs, including the skin, gastrointestinal tract, joints, and the kidneys. The diagnostic criteria include palpable purpura in the presence of one or more of the following: diffuse abdominal pain, arthritis or arthralgia, any biopsy showing predominant IgA deposition, and renal involvement in the form of hematuria or proteinuria. Renal disease is variable and is the most significant indicator of long-term prognosis. This teenager was treated with oral corticosteroids because of the severe periarticular edema and responded rapidly. His subsequent urine analyses normalized.
What is on the differential?
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal, complication of COVID-19 infection that causes inflammation of multiple organs, including the heart, lungs, kidneys, brain, skin, eyes, or the gastrointestinal tract. It commonly affects children around ages 8-9 years. Initial symptoms include fever, rash, red eyes, diarrhea, and vomiting that appear 2-6 weeks post COVID-19 infection. Like HSP, MIS-C can present with edema of the extremities, worsening hand/foot pain, and hematuria; however, the absence of both fever and the pattern of system involvement seen with MIS-C and classic findings in this patient are more consistent with HSP.

Reactive infectious mucocutaneous eruption (RIME) was recently coined to encompass both infection-associated Stevens-Johnson eruptions including Mycoplasma pneumoniae-induced rash and mucositis (MIRM) and mucocutaneous eruptions caused by nonmycoplasma pathogens (including Chlamydia pneumoniae, human parainfluenza virus 2, rhinovirus, adenovirus, enterovirus, human metapneumovirus, influenza B virus, and COVID-19). It is usually seen in male children and adolescents. Prodromal symptoms include cough, fever, and malaise and they precede the prominent feature of mucositis. Our patient’s lack of mucosal involvement is not consistent with RIME.
Perniosis (chilblains) is characterized by localized edematous patches of erythema or cyanosis on exposed extremities, that may be associated with cold exposure. Lesions are usually symmetric and self-limiting, and symptoms can include numbness, tingling, pruritus, burning, or pain. Pernio-like skin lesions have been seen during the COVID-19 pandemic, though many patients have negative testing for infection by PCR and serology. Pernio may also be seen with autoimmune diseases or malignancy.
Meningococcemia is a rare disease caused by infection with gram-negative diplococci bacteria Neisseria meningitidis and spreads through saliva or respiratory secretions. Its clinical presentation can vary widely, from transient fever to fulminant disease. It is characterized by upper respiratory tract infection, fever, and petechial lesions associated with thrombocytopenia and coagulopathy.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Laborada have no relevant financial disclosures.
References
AlGhoozi DA, AlKhayyat HM. BMJ Case Reports CP 2021;14:e239910.
Jacobi M et al. Pediatr Infect Dis J. 2021;40(2):e93-4.
Paller A, Mancini AJ. Hurwitz clinical pediatric dermatology: A textbook of skin disorders of childhood and adolescence. 4th ed. Philadelphia (PA): Elsevier Saunders; 2011.
Radia T et al. Paediatr Respir Rev. 2021;38:51-7.
Ramien ML. Clin Exp Dermatol. 2021;46(3):420-9.
Numerous morphologies of skin rashes have been described in the setting of COVID-19, including pernio, livedoid rash, exanthem, and vasculitis. This classic constellation of symptoms (palpable purpura on buttocks/legs, abdominal pain, arthralgia, hematuria) is highly consistent with Henoch-Schonlein purpura (HSP). There are now multiple case reports of COVID-19–associated HSP.
HSP is the most common type of childhood systemic vasculitis. It is mediated by immunoglobulin A (IgA) immune complex deposition and has been associated with respiratory tract infections, streptococcal species, parainfluenza virus, and human parvovirus B19, medications, vaccinations, and malignancies. HSP is usually a self-limiting disease, with a course over 4-6 weeks, and can affect multiple organs, including the skin, gastrointestinal tract, joints, and the kidneys. The diagnostic criteria include palpable purpura in the presence of one or more of the following: diffuse abdominal pain, arthritis or arthralgia, any biopsy showing predominant IgA deposition, and renal involvement in the form of hematuria or proteinuria. Renal disease is variable and is the most significant indicator of long-term prognosis. This teenager was treated with oral corticosteroids because of the severe periarticular edema and responded rapidly. His subsequent urine analyses normalized.
What is on the differential?
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal, complication of COVID-19 infection that causes inflammation of multiple organs, including the heart, lungs, kidneys, brain, skin, eyes, or the gastrointestinal tract. It commonly affects children around ages 8-9 years. Initial symptoms include fever, rash, red eyes, diarrhea, and vomiting that appear 2-6 weeks post COVID-19 infection. Like HSP, MIS-C can present with edema of the extremities, worsening hand/foot pain, and hematuria; however, the absence of both fever and the pattern of system involvement seen with MIS-C and classic findings in this patient are more consistent with HSP.
Reactive infectious mucocutaneous eruption (RIME) was recently coined to encompass both infection-associated Stevens-Johnson eruptions including Mycoplasma pneumoniae-induced rash and mucositis (MIRM) and mucocutaneous eruptions caused by nonmycoplasma pathogens (including Chlamydia pneumoniae, human parainfluenza virus 2, rhinovirus, adenovirus, enterovirus, human metapneumovirus, influenza B virus, and COVID-19). It is usually seen in male children and adolescents. Prodromal symptoms include cough, fever, and malaise and they precede the prominent feature of mucositis. Our patient’s lack of mucosal involvement is not consistent with RIME.
Perniosis (chilblains) is characterized by localized edematous patches of erythema or cyanosis on exposed extremities, that may be associated with cold exposure. Lesions are usually symmetric and self-limiting, and symptoms can include numbness, tingling, pruritus, burning, or pain. Pernio-like skin lesions have been seen during the COVID-19 pandemic, though many patients have negative testing for infection by PCR and serology. Pernio may also be seen with autoimmune diseases or malignancy.
Meningococcemia is a rare disease caused by infection with gram-negative diplococci bacteria Neisseria meningitidis and spreads through saliva or respiratory secretions. Its clinical presentation can vary widely, from transient fever to fulminant disease. It is characterized by upper respiratory tract infection, fever, and petechial lesions associated with thrombocytopenia and coagulopathy.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Laborada have no relevant financial disclosures.
References
AlGhoozi DA, AlKhayyat HM. BMJ Case Reports CP 2021;14:e239910.
Jacobi M et al. Pediatr Infect Dis J. 2021;40(2):e93-4.
Paller A, Mancini AJ. Hurwitz clinical pediatric dermatology: A textbook of skin disorders of childhood and adolescence. 4th ed. Philadelphia (PA): Elsevier Saunders; 2011.
Radia T et al. Paediatr Respir Rev. 2021;38:51-7.
Ramien ML. Clin Exp Dermatol. 2021;46(3):420-9.
Numerous morphologies of skin rashes have been described in the setting of COVID-19, including pernio, livedoid rash, exanthem, and vasculitis. This classic constellation of symptoms (palpable purpura on buttocks/legs, abdominal pain, arthralgia, hematuria) is highly consistent with Henoch-Schonlein purpura (HSP). There are now multiple case reports of COVID-19–associated HSP.
HSP is the most common type of childhood systemic vasculitis. It is mediated by immunoglobulin A (IgA) immune complex deposition and has been associated with respiratory tract infections, streptococcal species, parainfluenza virus, and human parvovirus B19, medications, vaccinations, and malignancies. HSP is usually a self-limiting disease, with a course over 4-6 weeks, and can affect multiple organs, including the skin, gastrointestinal tract, joints, and the kidneys. The diagnostic criteria include palpable purpura in the presence of one or more of the following: diffuse abdominal pain, arthritis or arthralgia, any biopsy showing predominant IgA deposition, and renal involvement in the form of hematuria or proteinuria. Renal disease is variable and is the most significant indicator of long-term prognosis. This teenager was treated with oral corticosteroids because of the severe periarticular edema and responded rapidly. His subsequent urine analyses normalized.
What is on the differential?
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal, complication of COVID-19 infection that causes inflammation of multiple organs, including the heart, lungs, kidneys, brain, skin, eyes, or the gastrointestinal tract. It commonly affects children around ages 8-9 years. Initial symptoms include fever, rash, red eyes, diarrhea, and vomiting that appear 2-6 weeks post COVID-19 infection. Like HSP, MIS-C can present with edema of the extremities, worsening hand/foot pain, and hematuria; however, the absence of both fever and the pattern of system involvement seen with MIS-C and classic findings in this patient are more consistent with HSP.
Reactive infectious mucocutaneous eruption (RIME) was recently coined to encompass both infection-associated Stevens-Johnson eruptions including Mycoplasma pneumoniae-induced rash and mucositis (MIRM) and mucocutaneous eruptions caused by nonmycoplasma pathogens (including Chlamydia pneumoniae, human parainfluenza virus 2, rhinovirus, adenovirus, enterovirus, human metapneumovirus, influenza B virus, and COVID-19). It is usually seen in male children and adolescents. Prodromal symptoms include cough, fever, and malaise and they precede the prominent feature of mucositis. Our patient’s lack of mucosal involvement is not consistent with RIME.
Perniosis (chilblains) is characterized by localized edematous patches of erythema or cyanosis on exposed extremities, that may be associated with cold exposure. Lesions are usually symmetric and self-limiting, and symptoms can include numbness, tingling, pruritus, burning, or pain. Pernio-like skin lesions have been seen during the COVID-19 pandemic, though many patients have negative testing for infection by PCR and serology. Pernio may also be seen with autoimmune diseases or malignancy.
Meningococcemia is a rare disease caused by infection with gram-negative diplococci bacteria Neisseria meningitidis and spreads through saliva or respiratory secretions. Its clinical presentation can vary widely, from transient fever to fulminant disease. It is characterized by upper respiratory tract infection, fever, and petechial lesions associated with thrombocytopenia and coagulopathy.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Laborada have no relevant financial disclosures.
References
AlGhoozi DA, AlKhayyat HM. BMJ Case Reports CP 2021;14:e239910.
Jacobi M et al. Pediatr Infect Dis J. 2021;40(2):e93-4.
Paller A, Mancini AJ. Hurwitz clinical pediatric dermatology: A textbook of skin disorders of childhood and adolescence. 4th ed. Philadelphia (PA): Elsevier Saunders; 2011.
Radia T et al. Paediatr Respir Rev. 2021;38:51-7.
Ramien ML. Clin Exp Dermatol. 2021;46(3):420-9.
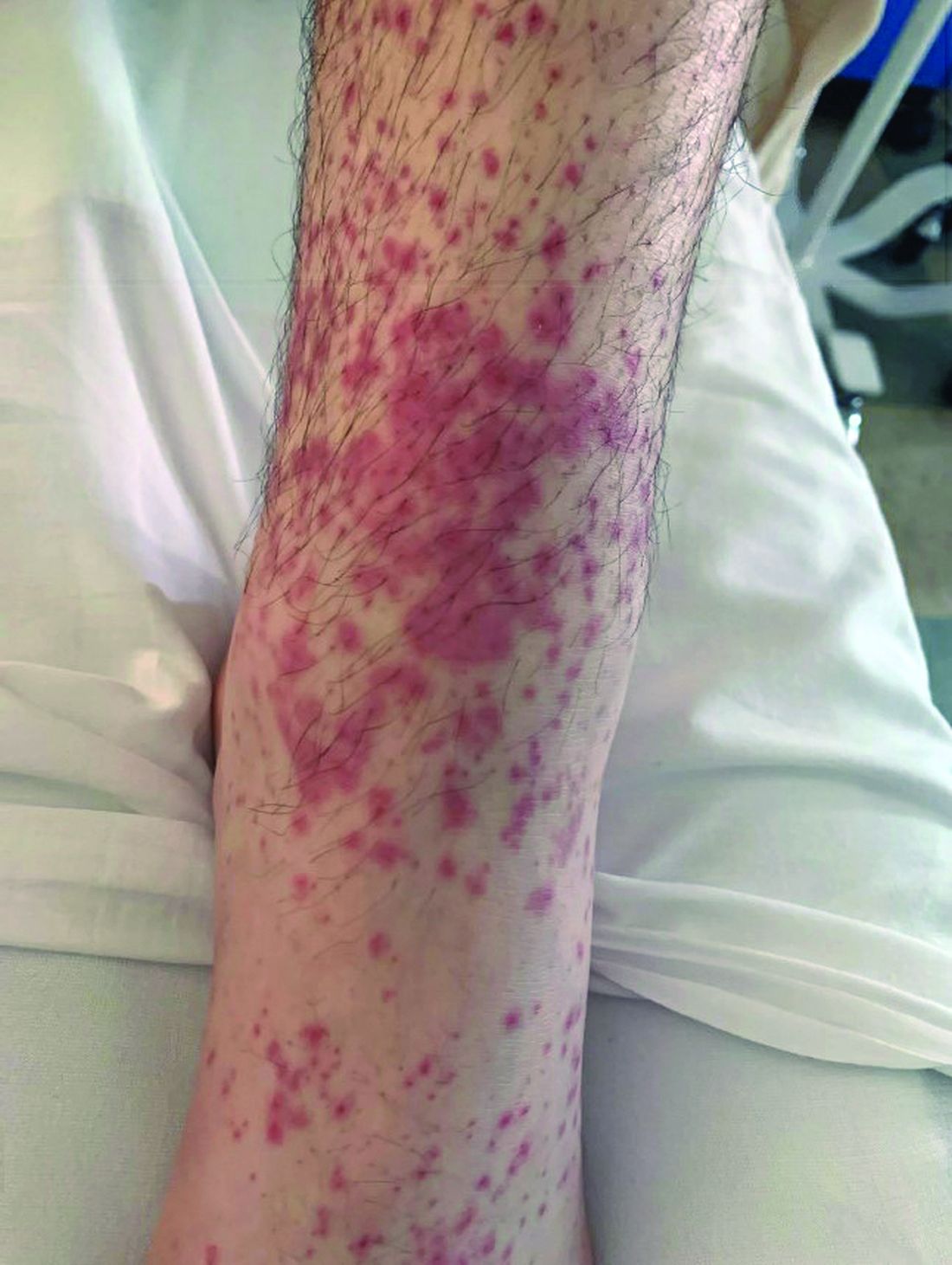

Linear Violaceous Papules in a Child
The Diagnosis: Linear Lichen Planus
The patient was clinically diagnosed with linear lichen planus and was started on betamethasone dipropionate ointment 0.05% applied once daily with improvement in both the pruritus and appearance at 4-month follow-up. A biopsy was deferred based on the parents’ wishes.
Lichen planus is an inflammatory disorder involving the skin and oral mucosa. Cutaneous lichen planus classically presents as flat-topped, violaceous, pruritic, polygonal papules with overlying fine white or grey lines known as Wickham striae.1 Postinflammatory hyperpigmentation is common, especially in patients with darker skin tones. Expected histologic findings include orthokeratosis, apoptotic keratinocytes, and bandlike lymphocytic infiltration at the dermoepidermal junction.1
An estimated 5% of cases of cutaneous lichen planus occur in children.2 A study of 316 children with lichen planus demonstrated that the classic morphology remained the most common presentation, while the linear variant was present in only 6.9% of pediatric cases.3 Linear lichen planus appears to be more common among children than adults. A study of 36 pediatric cases showed a greater representation of lichen planus in Black children (67% affected vs 21% cohort).2
Cutaneous lichen planus often clears spontaneously in approximately 1 year.4 Treatment in children primarily is focused on shortening the time to resolution and relieving pruritus, with topical corticosteroids as firstline therapy.3,4 Oral corticosteroids have a faster clinical response; greater efficacy; and more effectively prevent residual hyperpigmentation, which is especially relevant in individuals with darker skin.3 Nonetheless, oral corticosteroids are considered a second-line treatment due to their unfavorable side-effect profile. Additional treatment options include oral aromatic retinoids (acitretin) and phototherapy.3
Incontinentia pigmenti is characterized by a defect in the inhibitor of nuclear factor–κB kinase regulatory subunit gamma, IKBKG, gene on the X chromosome. Incontinentia pigmenti usually is lethal in males; in females, it leads to ectodermal dysplasia associated with skin findings in a blaschkoid distribution occurring in 4 stages.5 The verrucous stage is preceded by the vesicular stage and expected to occur within the first few months of life, making it unlikely in our 5-year-old patient. Inflammatory linear verrucous epidermal nevus usually occurs in children younger than 5 years and is characterized by psoriasiform papules coalescing into a plaque with substantial scale instead of Wickham striae, as seen in our patient.6 Lichen striatus consists of smaller, pink to flesh-colored papules that rarely are pruritic.7 It is more common among atopic individuals and is associated with postinflammatory hypopigmentation.8 Linear psoriasis presents similarly to inflammatory linear verrucous epidermal nevus, with greater erythema and scale compared to the fine lacy Wickham striae that were seen in our patient.8
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Walton KE, Bowers EV, Drolet BA, et al. Childhood lichen planus: demographics of a U.S. population. Pediatr Dermatol. 2010;27:34-38.
- Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31:59-67.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:E45-E52.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Payette MJ, Weston G, Humphrey S, et al. Lichen planus and other lichenoid dermatoses: kids are not just little people. Clin Dermatol. 2015;33:631-643.
- Moss C, Browne F. Mosaicism and linear lesions. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
The Diagnosis: Linear Lichen Planus
The patient was clinically diagnosed with linear lichen planus and was started on betamethasone dipropionate ointment 0.05% applied once daily with improvement in both the pruritus and appearance at 4-month follow-up. A biopsy was deferred based on the parents’ wishes.
Lichen planus is an inflammatory disorder involving the skin and oral mucosa. Cutaneous lichen planus classically presents as flat-topped, violaceous, pruritic, polygonal papules with overlying fine white or grey lines known as Wickham striae.1 Postinflammatory hyperpigmentation is common, especially in patients with darker skin tones. Expected histologic findings include orthokeratosis, apoptotic keratinocytes, and bandlike lymphocytic infiltration at the dermoepidermal junction.1
An estimated 5% of cases of cutaneous lichen planus occur in children.2 A study of 316 children with lichen planus demonstrated that the classic morphology remained the most common presentation, while the linear variant was present in only 6.9% of pediatric cases.3 Linear lichen planus appears to be more common among children than adults. A study of 36 pediatric cases showed a greater representation of lichen planus in Black children (67% affected vs 21% cohort).2
Cutaneous lichen planus often clears spontaneously in approximately 1 year.4 Treatment in children primarily is focused on shortening the time to resolution and relieving pruritus, with topical corticosteroids as firstline therapy.3,4 Oral corticosteroids have a faster clinical response; greater efficacy; and more effectively prevent residual hyperpigmentation, which is especially relevant in individuals with darker skin.3 Nonetheless, oral corticosteroids are considered a second-line treatment due to their unfavorable side-effect profile. Additional treatment options include oral aromatic retinoids (acitretin) and phototherapy.3
Incontinentia pigmenti is characterized by a defect in the inhibitor of nuclear factor–κB kinase regulatory subunit gamma, IKBKG, gene on the X chromosome. Incontinentia pigmenti usually is lethal in males; in females, it leads to ectodermal dysplasia associated with skin findings in a blaschkoid distribution occurring in 4 stages.5 The verrucous stage is preceded by the vesicular stage and expected to occur within the first few months of life, making it unlikely in our 5-year-old patient. Inflammatory linear verrucous epidermal nevus usually occurs in children younger than 5 years and is characterized by psoriasiform papules coalescing into a plaque with substantial scale instead of Wickham striae, as seen in our patient.6 Lichen striatus consists of smaller, pink to flesh-colored papules that rarely are pruritic.7 It is more common among atopic individuals and is associated with postinflammatory hypopigmentation.8 Linear psoriasis presents similarly to inflammatory linear verrucous epidermal nevus, with greater erythema and scale compared to the fine lacy Wickham striae that were seen in our patient.8
The Diagnosis: Linear Lichen Planus
The patient was clinically diagnosed with linear lichen planus and was started on betamethasone dipropionate ointment 0.05% applied once daily with improvement in both the pruritus and appearance at 4-month follow-up. A biopsy was deferred based on the parents’ wishes.
Lichen planus is an inflammatory disorder involving the skin and oral mucosa. Cutaneous lichen planus classically presents as flat-topped, violaceous, pruritic, polygonal papules with overlying fine white or grey lines known as Wickham striae.1 Postinflammatory hyperpigmentation is common, especially in patients with darker skin tones. Expected histologic findings include orthokeratosis, apoptotic keratinocytes, and bandlike lymphocytic infiltration at the dermoepidermal junction.1
An estimated 5% of cases of cutaneous lichen planus occur in children.2 A study of 316 children with lichen planus demonstrated that the classic morphology remained the most common presentation, while the linear variant was present in only 6.9% of pediatric cases.3 Linear lichen planus appears to be more common among children than adults. A study of 36 pediatric cases showed a greater representation of lichen planus in Black children (67% affected vs 21% cohort).2
Cutaneous lichen planus often clears spontaneously in approximately 1 year.4 Treatment in children primarily is focused on shortening the time to resolution and relieving pruritus, with topical corticosteroids as firstline therapy.3,4 Oral corticosteroids have a faster clinical response; greater efficacy; and more effectively prevent residual hyperpigmentation, which is especially relevant in individuals with darker skin.3 Nonetheless, oral corticosteroids are considered a second-line treatment due to their unfavorable side-effect profile. Additional treatment options include oral aromatic retinoids (acitretin) and phototherapy.3
Incontinentia pigmenti is characterized by a defect in the inhibitor of nuclear factor–κB kinase regulatory subunit gamma, IKBKG, gene on the X chromosome. Incontinentia pigmenti usually is lethal in males; in females, it leads to ectodermal dysplasia associated with skin findings in a blaschkoid distribution occurring in 4 stages.5 The verrucous stage is preceded by the vesicular stage and expected to occur within the first few months of life, making it unlikely in our 5-year-old patient. Inflammatory linear verrucous epidermal nevus usually occurs in children younger than 5 years and is characterized by psoriasiform papules coalescing into a plaque with substantial scale instead of Wickham striae, as seen in our patient.6 Lichen striatus consists of smaller, pink to flesh-colored papules that rarely are pruritic.7 It is more common among atopic individuals and is associated with postinflammatory hypopigmentation.8 Linear psoriasis presents similarly to inflammatory linear verrucous epidermal nevus, with greater erythema and scale compared to the fine lacy Wickham striae that were seen in our patient.8
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Walton KE, Bowers EV, Drolet BA, et al. Childhood lichen planus: demographics of a U.S. population. Pediatr Dermatol. 2010;27:34-38.
- Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31:59-67.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:E45-E52.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Payette MJ, Weston G, Humphrey S, et al. Lichen planus and other lichenoid dermatoses: kids are not just little people. Clin Dermatol. 2015;33:631-643.
- Moss C, Browne F. Mosaicism and linear lesions. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Tziotzios C, Lee JYW, Brier T, et al. Lichen planus and lichenoid dermatoses: clinical overview and molecular basis. J Am Acad Dermatol. 2018;79:789-804.
- Walton KE, Bowers EV, Drolet BA, et al. Childhood lichen planus: demographics of a U.S. population. Pediatr Dermatol. 2010;27:34-38.
- Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31:59-67.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:E45-E52.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
- Payette MJ, Weston G, Humphrey S, et al. Lichen planus and other lichenoid dermatoses: kids are not just little people. Clin Dermatol. 2015;33:631-643.
- Moss C, Browne F. Mosaicism and linear lesions. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1894-1916.
A 5-year-old Black girl presented to the dermatology clinic with a stable pruritic eruption on the right leg of 1 month’s duration. Over-the-counter hydrocortisone cream was applied for 3 days with no response. Physical examination revealed grouped, flat-topped, violaceous papules coalescing into plaques with overlying lacy white striae along the right lower leg, wrapping around to the right dorsal foot in a blaschkoid distribution. The patient was otherwise healthy and up-to-date on immunizations and had an unremarkable birth history.
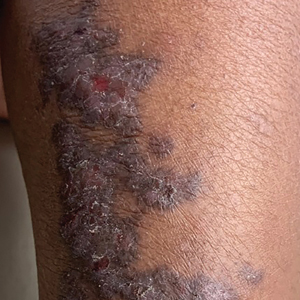
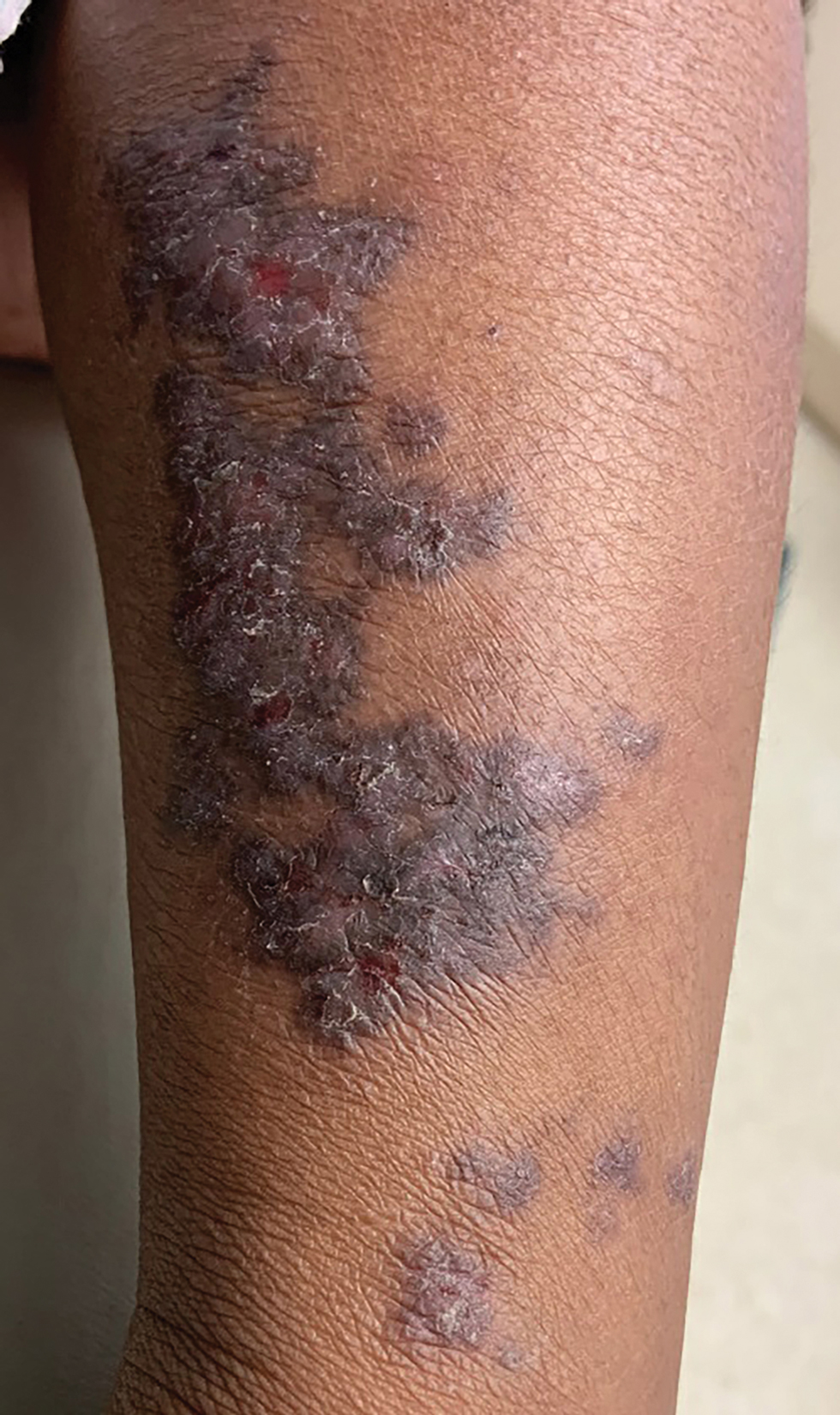
Flesh-Colored Papule in the Nose of a Child
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
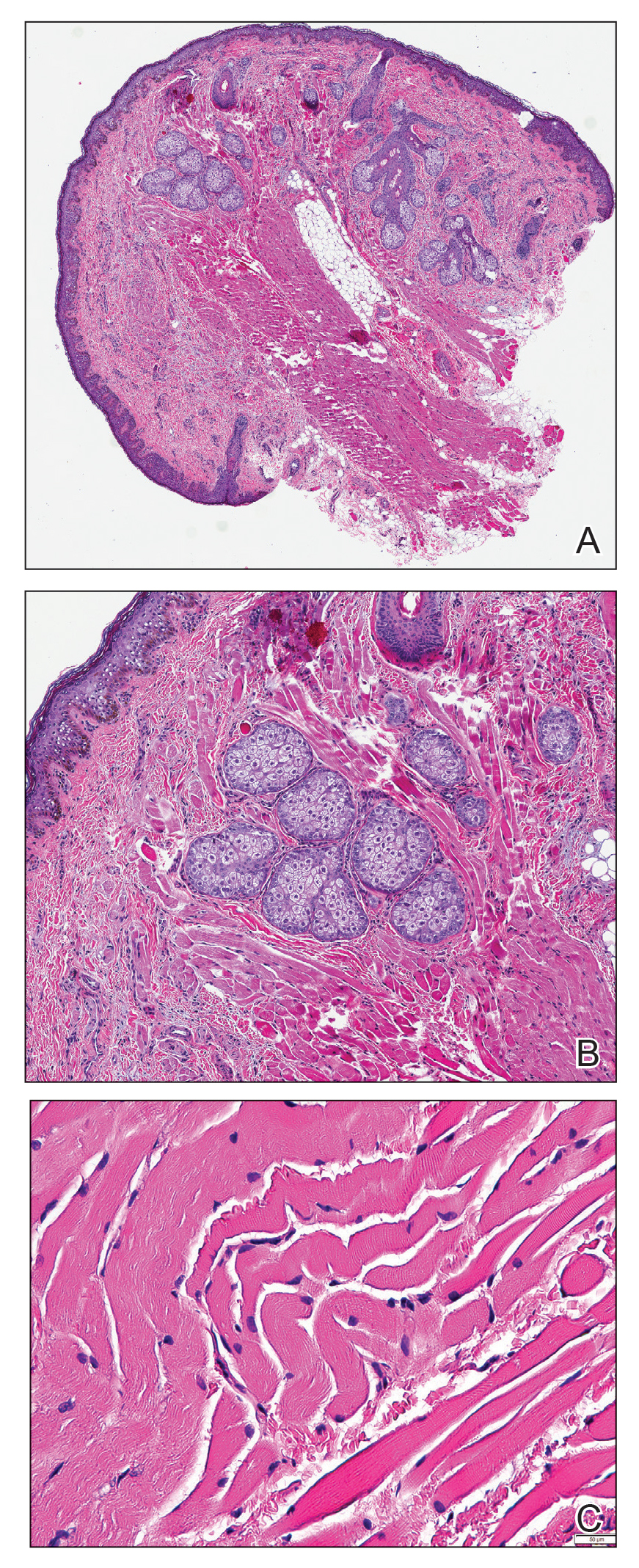
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
The Diagnosis: Striated Muscle Hamartoma
Histopathologic evaluation revealed a dome-shaped papule with a center composed of mature striated muscle bundles, vellus hairs, sebaceous lobules, and nerve twigs (Figure) consistent with a diagnosis of striated muscle hamartoma (SMH).
Striated muscle hamartoma was first described in 1986 by Hendrick et al1 with 2 cases in neonates. Biopsies of the lesions taken from the upper lip and sternum showed a characteristic histology consisting of dermal striated muscle fibers and nerve bundles in the central core of the papules associated with a marked number of adnexa. In 1989, the diagnosis of rhabdomyomatous mesenchymal hamartoma was described, which showed similar findings.2 Cases reported since these entities were discovered have used the terms striated muscle hamartoma and rhabdomyomatous mesenchymal hamartoma interchangeably.3
Most commonly found on the head and neck, SMH has now been observed in diverse locations including the sternum, hallux, vagina, and oral cavity.1-15 Many reported cases describe lesions around or in the nose.4,7,8 Multiple congenital anomalies have been described alongside SMH and may be associated with this entity including amniotic bands, cleft lip and palate, coloboma, and Delleman syndrome.1,3,4 Almost all of the lesions present as a sessile or pedunculated papule, polyp, nodule, or plaque measuring from 0.3 cm up to 4.9 cm and typically are present since birth.3,5,15 However, there are a few cases of lesions presenting in adults with no prior history.5,6,15
Microscopically, SMH is defined by a dermal lesion with a core comprised of mature skeletal muscle admixed with adipose tissue, adnexa, nerve bundles, and fibrovascular tissue.1 There are other entities that should be considered before making the diagnosis of SMH. Other hamartomas such as accessory tragus, connective tissue nevus, fibrous hamartoma of infancy, and nevus lipomatosis may present similarly; however, these lesions classically lack skeletal muscle. Benign triton tumors, or neuromuscular hamartomas, are rare lesions composed of skeletal muscle and abundant, intimately associated neural tissue. Neuromuscular hamartomas frequently involve large nerves.16 Rhabdomyomas also should be considered. Adult rhabdomyomas are composed of eosinophilic polygonal cells with granular cytoplasm and occasional cross-striations. Fetal rhabdomyomas have multiple histologic types and are defined by a variable myxoid stroma, eosinophilic spindled cells, and rhabdomyocytes in various stages of maturity. Genital rhabdomyomas histopathologically appear similar to fetal rhabdomyomas but are confined to the genital region. The skeletal muscle present in rhabdomyomas typically is less differentiated.17 TMature skeletal bundles should be a dominant component of the lesion before diagnosing SMH.
Typically presenting as congenital lesions in the head and neck region, papules with a dermal core of mature skeletal muscle associated with adnexa and nerve twigs should prompt consideration of a diagnosis of SMH or rhabdomyomatous mesenchymal hamartoma. These lesions are benign and usually are cured with complete excision.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol. 1986;3:153-157.
- Mills AE. Rhabdomyomatous mesenchymal hamartoma of skin. Am J Dermatopathol. 1989;1:58-63.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: an unusual dermal entity with a report of two cases and a review of the literature. J Cutan Pathol. 2002;29:238-243.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman’s syndrome. J Cutan Pathol. 1994;21:40-46.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005;21:185-188.
- Harris MA, Dutton JJ, Proia AD. Striated muscle hamartoma of the eyelid in an adult woman. Ophthalmic Plast Reconstr Surg. 2008;24:492-494.
- Nakanishi H, Hashimoto I, Takiwaki H, et al. Striated muscle hamartoma of the nostril. J Dermatol. 1995;22:504-507.
- Farris PE, Manning S, Veatch F. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol. 1994;16:73-75.
- Grilli R, Escalonilla P, Soriano ML, et al. The so-called striated muscle hamartoma is a hamartoma of cutaneous adnexa and mesenchyme, but not of striated muscle. Acta Derm Venereol. 1998;78:390.
- Sampat K, Cheesman E, Siminas S. Perianal rhabdomyomatous mesenchymal hamartoma. Ann R Coll Surg Engl. 2017;99:E193-E195.
- Brinster NK, Farmer ER. Rhabdomyomatous mesenchymal hamartoma presenting on a digit. J Cutan Pathol. 2009;36:61-63.
- Han SH, Song HJ, Hong WK, et al. Rhabdomyomatous mesenchymal hamartoma of the vagina. Pediatr Dermatol. 2009;26:753-755.
- De la Sotta P, Salomone C, González S. Rhabdomyomatous (mesenchymal) hamartoma of the tongue: report of a case. J Oral Pathol Med. 2007;36:58-59.
- Magro G, Di Benedetto A, Sanges G, et al. Rhabdomyomatous mesenchymal hamartoma of oral cavity: an unusual location for such a rare lesion. Virchows Arch. 2005;446:346-347.
- Wang Y, Zhao H, Yue X, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a big subcutaneous mass on the neck: a case report. J Med Case Rep. 2014;8:410.
- Amita K, Shankar SV, Nischal KC, et al. Benign triton tumor: a rare entity in head and neck region. Korean J Pathol. 2013;47:74-76.
- Walsh S, Hurt M. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32:485-491.
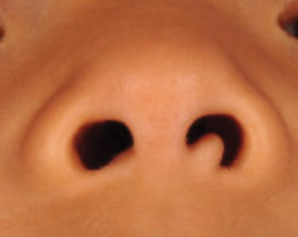
A 4-year-old girl presented to our clinic with an asymptomatic flesh-colored papule in the left nostril. The lesion had been present since birth and grew in relation to the patient with no rapid changes. There had been no pigmentation changes and no bleeding, pain, or itching. The patient’s birth and developmental history were normal. Physical examination revealed a singular, 10×5-mm, flesh-colored, pedunculated mass on the left nasal sill. There were no additional lesions present. An excisional biopsy was performed and submitted for pathologic diagnosis.

Teen boy’s knee lesion has changed
A biopsy of the lesion was performed which showed an increased number of eccrine glands and blood vessels within the dermis. Some areas showed an increase in adipocytes and smooth muscle bundles. The changes were consistent with eccrine angiomatous hamartoma (EAH).

The boy was referred to vascular laser therapy for treatment of the lesion.
EAH is a rare benign vascular growth characterized by an increased number of mature eccrine glands and blood vessels in the dermis and subcutis. The lesions are mostly present on the extremities, but cases of diffuse congenital lesions and lesions on the face and trunk have also been described. The lesions can be seen at birth or during the first years of life in about half of the cases, and the others tend to occur later in puberty and rarely in adulthood.1
Clinically, EAH lesions present as red, yellow to brown papules and plaques. Different dermoscopic patterns have been described which include the popcorn pattern that presents as yellow, confluent nodules with popcornlike shapes over a background of erythema, and linear arborizing vessels. The spitzoid pattern are brown globules on a background of erythema and pseudoreticular pigmentation around the globules. The verrucous hemangiomalike pattern has a bluish-white hue, reddish-blue or bluish lacunae, as seen in our patient.2-4
Most of the lesions are asymptomatic, but in some patients, they can be associated with pain, hyperhidrosis, and sometimes bleeding. Hyperhidrosis has been reported early in the presentation or during puberty or pregnancy. Our patient had started on amphetamines when hyperhidrosis occurred. Hyperhidrosis is a knowns side effect of this type of medication and may have had a role in the increased sweating noted on the hamartoma.
EAH can clinically look like verrucous hemangiomas, angiokeratomas, and vascular malformations, and histopathology may be needed to differentiate between them. Eccrine nevi and EAH can be similar. Hyperhidrosis is an early and predominant component of eccrine nevi, compared with one-third of EAH.
The exact etiology of this lesion is not known. It is thought to be caused by an abnormal differentiation of the epithelium, adnexal structure, and the mesenchyme during organogenesis.3 No other associated conditions have been described with EAH.
EAH are benign lesions that rarely require treatment. If the lesions are symptomatic or because of cosmetic reasons, they can be removed surgically. There are some reports of successful treatment with pulse dual-wavelength sequential 595- and 1064-nm lasers.5 Botulinum toxin has also been used in cases of symptomatic hyperhidrosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no conflicts. Email her at [email protected].
References
1. Smith SD et al. Pediatr Dermatol. 2019 Nov;36(6):909-12.
2. Patterson AT et al. Am J Dermatopathol. 2016;38:413-7.
3. Garcıa-Garcıa SC et al. JAAD Case Rep. 2018;4(2):165-7.
4. Awatef Kelati et al. JAAD Case Rep. 2018;4(8)835-6.
5. Felgueiras J et al. Dermatol Surg. 2015 Mar;41(3):428-30.
A biopsy of the lesion was performed which showed an increased number of eccrine glands and blood vessels within the dermis. Some areas showed an increase in adipocytes and smooth muscle bundles. The changes were consistent with eccrine angiomatous hamartoma (EAH).
The boy was referred to vascular laser therapy for treatment of the lesion.
EAH is a rare benign vascular growth characterized by an increased number of mature eccrine glands and blood vessels in the dermis and subcutis. The lesions are mostly present on the extremities, but cases of diffuse congenital lesions and lesions on the face and trunk have also been described. The lesions can be seen at birth or during the first years of life in about half of the cases, and the others tend to occur later in puberty and rarely in adulthood.1
Clinically, EAH lesions present as red, yellow to brown papules and plaques. Different dermoscopic patterns have been described which include the popcorn pattern that presents as yellow, confluent nodules with popcornlike shapes over a background of erythema, and linear arborizing vessels. The spitzoid pattern are brown globules on a background of erythema and pseudoreticular pigmentation around the globules. The verrucous hemangiomalike pattern has a bluish-white hue, reddish-blue or bluish lacunae, as seen in our patient.2-4
Most of the lesions are asymptomatic, but in some patients, they can be associated with pain, hyperhidrosis, and sometimes bleeding. Hyperhidrosis has been reported early in the presentation or during puberty or pregnancy. Our patient had started on amphetamines when hyperhidrosis occurred. Hyperhidrosis is a knowns side effect of this type of medication and may have had a role in the increased sweating noted on the hamartoma.
EAH can clinically look like verrucous hemangiomas, angiokeratomas, and vascular malformations, and histopathology may be needed to differentiate between them. Eccrine nevi and EAH can be similar. Hyperhidrosis is an early and predominant component of eccrine nevi, compared with one-third of EAH.
The exact etiology of this lesion is not known. It is thought to be caused by an abnormal differentiation of the epithelium, adnexal structure, and the mesenchyme during organogenesis.3 No other associated conditions have been described with EAH.
EAH are benign lesions that rarely require treatment. If the lesions are symptomatic or because of cosmetic reasons, they can be removed surgically. There are some reports of successful treatment with pulse dual-wavelength sequential 595- and 1064-nm lasers.5 Botulinum toxin has also been used in cases of symptomatic hyperhidrosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no conflicts. Email her at [email protected].
References
1. Smith SD et al. Pediatr Dermatol. 2019 Nov;36(6):909-12.
2. Patterson AT et al. Am J Dermatopathol. 2016;38:413-7.
3. Garcıa-Garcıa SC et al. JAAD Case Rep. 2018;4(2):165-7.
4. Awatef Kelati et al. JAAD Case Rep. 2018;4(8)835-6.
5. Felgueiras J et al. Dermatol Surg. 2015 Mar;41(3):428-30.
A biopsy of the lesion was performed which showed an increased number of eccrine glands and blood vessels within the dermis. Some areas showed an increase in adipocytes and smooth muscle bundles. The changes were consistent with eccrine angiomatous hamartoma (EAH).
The boy was referred to vascular laser therapy for treatment of the lesion.
EAH is a rare benign vascular growth characterized by an increased number of mature eccrine glands and blood vessels in the dermis and subcutis. The lesions are mostly present on the extremities, but cases of diffuse congenital lesions and lesions on the face and trunk have also been described. The lesions can be seen at birth or during the first years of life in about half of the cases, and the others tend to occur later in puberty and rarely in adulthood.1
Clinically, EAH lesions present as red, yellow to brown papules and plaques. Different dermoscopic patterns have been described which include the popcorn pattern that presents as yellow, confluent nodules with popcornlike shapes over a background of erythema, and linear arborizing vessels. The spitzoid pattern are brown globules on a background of erythema and pseudoreticular pigmentation around the globules. The verrucous hemangiomalike pattern has a bluish-white hue, reddish-blue or bluish lacunae, as seen in our patient.2-4
Most of the lesions are asymptomatic, but in some patients, they can be associated with pain, hyperhidrosis, and sometimes bleeding. Hyperhidrosis has been reported early in the presentation or during puberty or pregnancy. Our patient had started on amphetamines when hyperhidrosis occurred. Hyperhidrosis is a knowns side effect of this type of medication and may have had a role in the increased sweating noted on the hamartoma.
EAH can clinically look like verrucous hemangiomas, angiokeratomas, and vascular malformations, and histopathology may be needed to differentiate between them. Eccrine nevi and EAH can be similar. Hyperhidrosis is an early and predominant component of eccrine nevi, compared with one-third of EAH.
The exact etiology of this lesion is not known. It is thought to be caused by an abnormal differentiation of the epithelium, adnexal structure, and the mesenchyme during organogenesis.3 No other associated conditions have been described with EAH.
EAH are benign lesions that rarely require treatment. If the lesions are symptomatic or because of cosmetic reasons, they can be removed surgically. There are some reports of successful treatment with pulse dual-wavelength sequential 595- and 1064-nm lasers.5 Botulinum toxin has also been used in cases of symptomatic hyperhidrosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no conflicts. Email her at [email protected].
References
1. Smith SD et al. Pediatr Dermatol. 2019 Nov;36(6):909-12.
2. Patterson AT et al. Am J Dermatopathol. 2016;38:413-7.
3. Garcıa-Garcıa SC et al. JAAD Case Rep. 2018;4(2):165-7.
4. Awatef Kelati et al. JAAD Case Rep. 2018;4(8)835-6.
5. Felgueiras J et al. Dermatol Surg. 2015 Mar;41(3):428-30.
A 14-year-old male was referred to our pediatric dermatology clinic for evaluation of a lesion on the left knee that appeared at 1 year of age. The lesion has been growing with him and was not symptomatic until 6 months prior to the consultation, when it started bleeding and feeling wet.
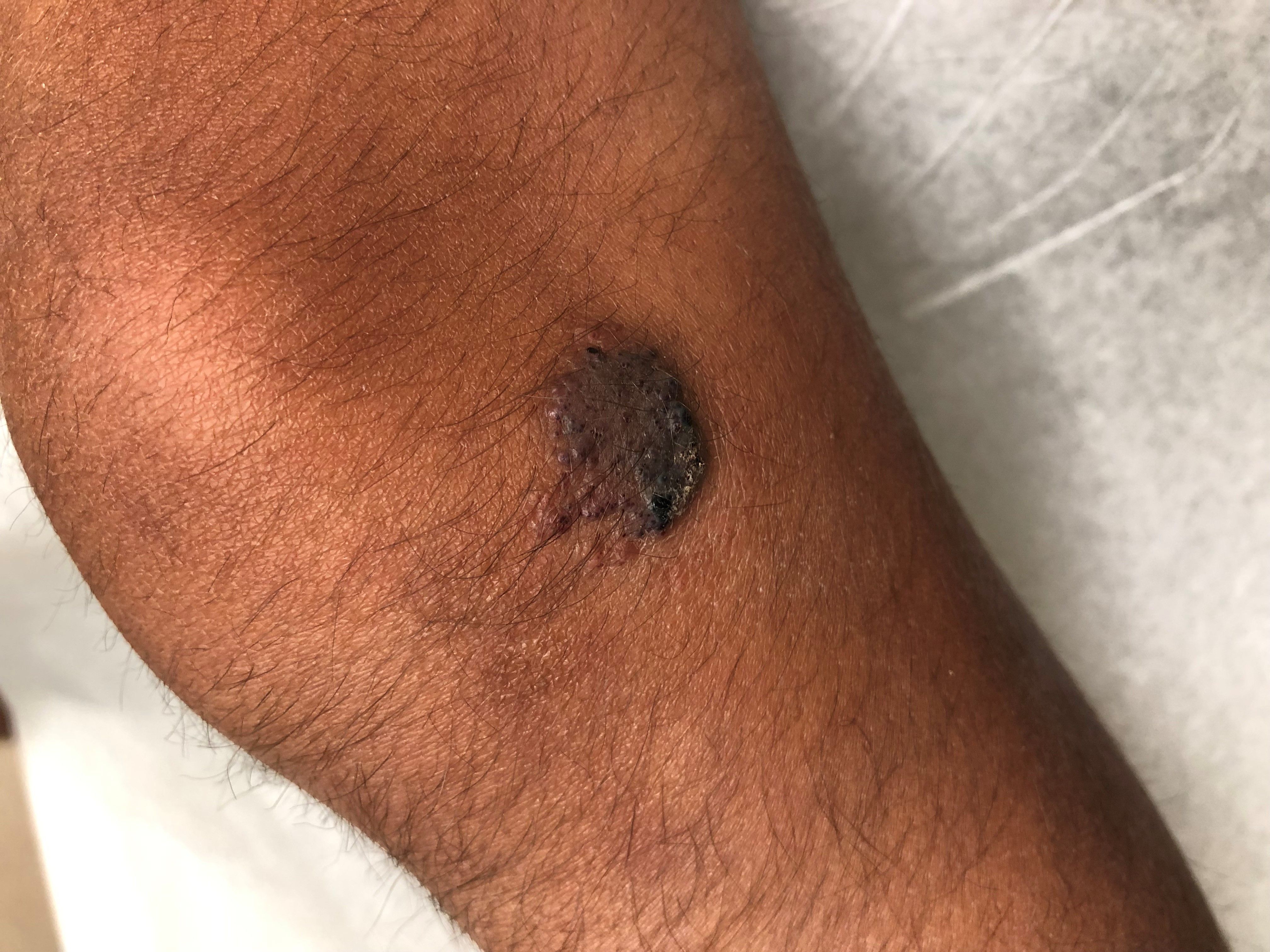
He has a history of attention-deficit/hyperactivity disorder managed with dextroamphetamine-amphetamine. The changes noted on the knee lesion seem to occur at the same time that his ADHD medication was started.
On physical exam he had a violaceous circular plaque on the left knee.
On dermoscopy the lesion showed multiple dilated red and violaceous lacunae and whitish blue hue.
A female toddler presents with an itchy yellow nodule
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
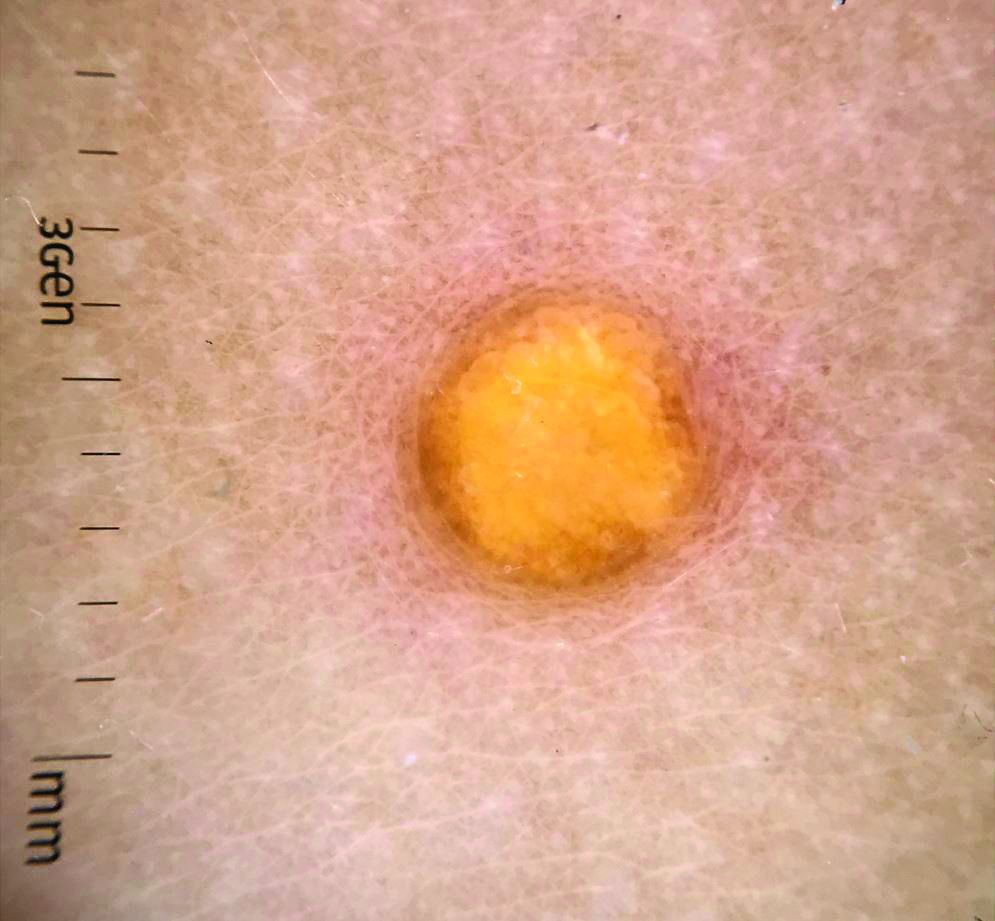
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.

Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
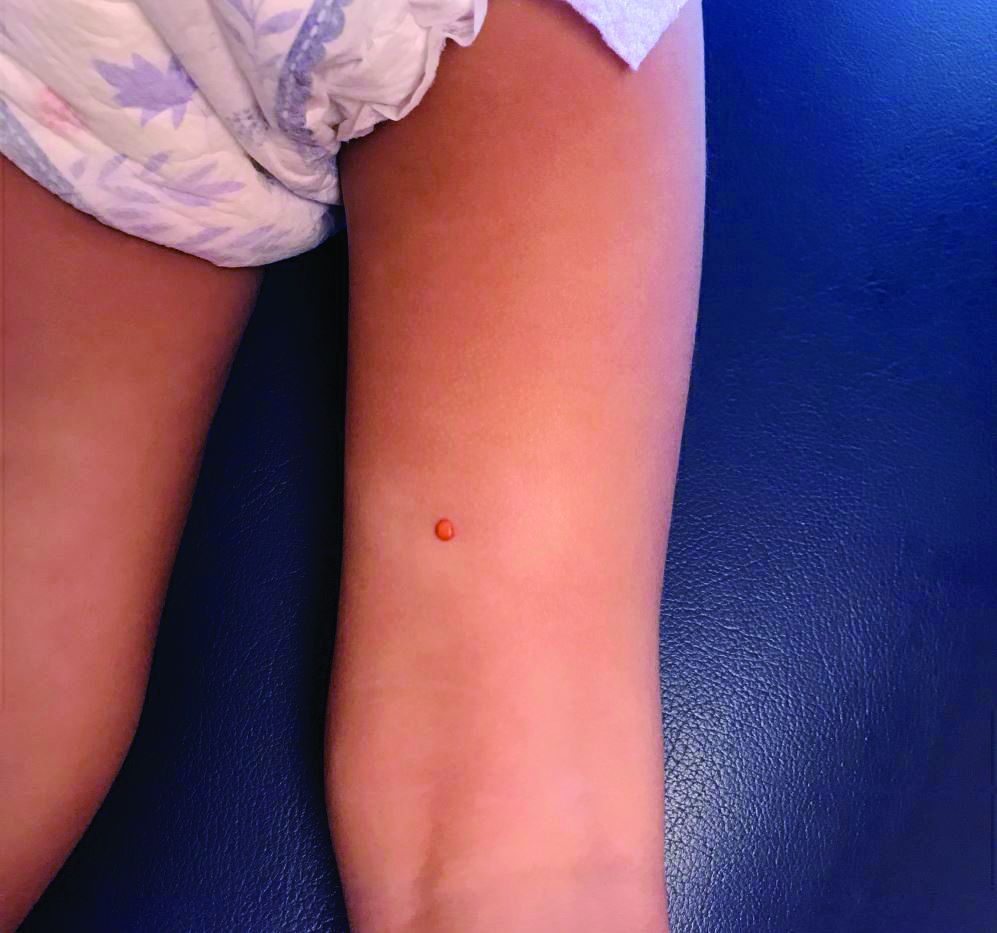
Lesions on the Thigh After an Organ Transplant
The Diagnosis: Microcystic Lymphatic Malformation
The shave biopsy demonstrated numerous thin-walled vascular spaces filled with lymphatic fluid within the dermis (Figure), consistent with a diagnosis of microcystic lymphatic malformation (LM). Lymphatic malformations represent a class of benign vascular lesions consisting of anomalous or dilated lymphatic vessels, which can be broadly categorized as macrocystic (formerly cavernous lymphangioma or cystic hygroma), microcystic (formerly lymphangioma circumscriptum), or mixed.1 Patients often will present with pruritus, crusting, secondary infection, edema, or oozing.2 The superficial blebs of microcystic LMs resemble frog spawn and range in color from clear to pink, brawny, or deep maroon.3 Although the lymphatic vessels involved in microcystic LMs appear disconnected from the major lymphatic circulation,3 systemic fluid overload could plausibly promote lesional swelling and tenderness; we attributed our patient's worsening symptoms to the cumulative 7.8 L of intravenous fluid he received intraoperatively during his cardiac transplant. The excess fluid allowed communication between lymphatic cisterns and thin-walled vesicles on the skin surface through dilated channels. Overall, LMs represent roughly 26% of pediatric benign vascular tumors and approximately 4% of all vascular tumors.4
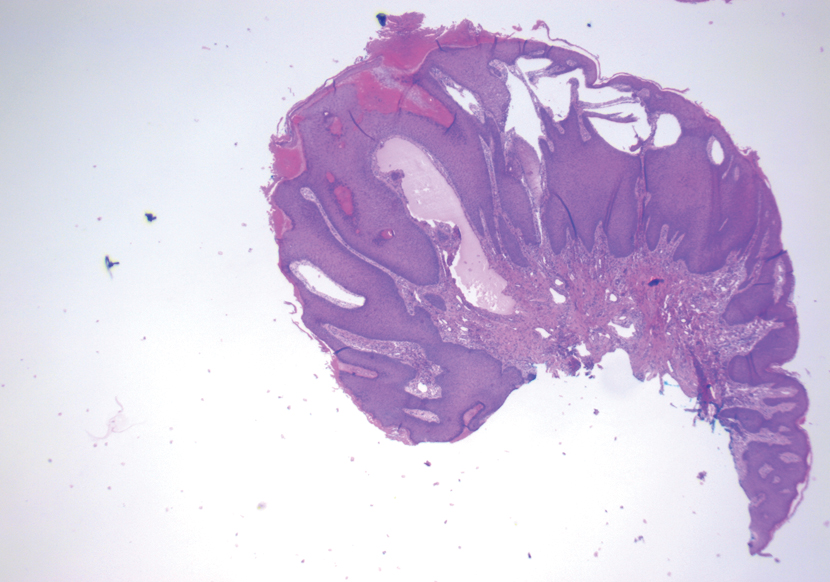
Although microcystic LMs may appear especially vascular or verrucous, the differential diagnosis for our patient's LM included condyloma acuminatum,5,6 condyloma lata,7 epidermal nevus, and lymphangiosarcoma. Epidermal nevi are congenital lesions, varying in appearance from velvety to verrucous patches and plaques that often evolve during puberty and become thicker, more verrucous, and hyperpigmented. Keratinocytic epidermal nevus syndromes and other entities such as nevus sebaceous have been associated with somatic mutations affecting proteins in the fibroblast growth factor receptor signaling pathway (eg, FGFR3, HRAS).8 Although the clinical appearance alone may be similar, lymphangiosarcoma can be distinguished from LM via biopsy.
There are several methods to diagnose LM. Duplex sonography is possibly the best noninvasive method to identify the flow between venous valves. Magnetic resonance imaging can detect larger occurrences of LM, and lymphangiography can be utilized to confirm a normal or abnormal lymphatic network.4 Treatment options are broad, including surgical excision, laser ablation, and topical sirolimus. Hypertonic saline sclerotherapy can be injected into the afflicted lymphatic channels to decrease inflammation, erythema, and hyperpigmentation without further treatment or major side effects.4
However, the benefits of sclerotherapy alone in the treatment of LM often come gradually, and radiofrequency ablation may need to be utilized to achieve more immediate results.2 Overall, outcomes are highly variable, but favorable outcomes often can be difficult to obtain due to a high recurrence rate.2,8 Our patient's symptoms improved during his postoperative recovery, and he declined further intervention.
- Elluru RG, Balakrishnan K, Padua HM. Lymphatic malformations: diagnosis and management. Semin Pediatr Surg. 2014;23:178-185. doi:10.1053/j.sempedsurg.2014.07.002
- Niti K, Manish P. Microcystic lymphatic malformation (lymphangioma circumscriptum) treated using a minimally invasive technique of radiofrequency ablation and sclerotherapy. Dermatol Surg. 2010;36:1711-1717. doi:10.1111/j.1524-4725.2010.01723.x
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111 /j.1365-4632.2009.04226.x
- Bikowski JB, Dumont AM. Lymphangioma circumscriptum: treatment with hypertonic saline sclerotherapy. J Am Acad Dermatol. 2005;53:442-444. doi:10.1016/j.jaad.2005.04.086
- Costa-Silva M, Fernandes I, Rodrigues AG, et al. Anogenital warts in pediatric population. An Bras Dermatol. 2017;92:675-681. doi:10.1590 /abd1806-4841.201756411
- Darmstadt GL. Perianal lymphangioma circumscriptum mistaken for genital warts. Pediatrics 1996;98;461.
- Bruins FG, van Deudekom FJA, de Vries HJC. Syphilitic condylomata lata mimicking anogenital warts. BMJ. 2015;350:h1259. doi:10.1136 /bmj.h1259
- Asch S, Sugarman JL. Epidermal nevus syndromes: new insights into whorls and swirls. Pediatr Dermatol. 2018;35:21-29. doi:10.1111 /pde.13273
The Diagnosis: Microcystic Lymphatic Malformation
The shave biopsy demonstrated numerous thin-walled vascular spaces filled with lymphatic fluid within the dermis (Figure), consistent with a diagnosis of microcystic lymphatic malformation (LM). Lymphatic malformations represent a class of benign vascular lesions consisting of anomalous or dilated lymphatic vessels, which can be broadly categorized as macrocystic (formerly cavernous lymphangioma or cystic hygroma), microcystic (formerly lymphangioma circumscriptum), or mixed.1 Patients often will present with pruritus, crusting, secondary infection, edema, or oozing.2 The superficial blebs of microcystic LMs resemble frog spawn and range in color from clear to pink, brawny, or deep maroon.3 Although the lymphatic vessels involved in microcystic LMs appear disconnected from the major lymphatic circulation,3 systemic fluid overload could plausibly promote lesional swelling and tenderness; we attributed our patient's worsening symptoms to the cumulative 7.8 L of intravenous fluid he received intraoperatively during his cardiac transplant. The excess fluid allowed communication between lymphatic cisterns and thin-walled vesicles on the skin surface through dilated channels. Overall, LMs represent roughly 26% of pediatric benign vascular tumors and approximately 4% of all vascular tumors.4
Although microcystic LMs may appear especially vascular or verrucous, the differential diagnosis for our patient's LM included condyloma acuminatum,5,6 condyloma lata,7 epidermal nevus, and lymphangiosarcoma. Epidermal nevi are congenital lesions, varying in appearance from velvety to verrucous patches and plaques that often evolve during puberty and become thicker, more verrucous, and hyperpigmented. Keratinocytic epidermal nevus syndromes and other entities such as nevus sebaceous have been associated with somatic mutations affecting proteins in the fibroblast growth factor receptor signaling pathway (eg, FGFR3, HRAS).8 Although the clinical appearance alone may be similar, lymphangiosarcoma can be distinguished from LM via biopsy.
There are several methods to diagnose LM. Duplex sonography is possibly the best noninvasive method to identify the flow between venous valves. Magnetic resonance imaging can detect larger occurrences of LM, and lymphangiography can be utilized to confirm a normal or abnormal lymphatic network.4 Treatment options are broad, including surgical excision, laser ablation, and topical sirolimus. Hypertonic saline sclerotherapy can be injected into the afflicted lymphatic channels to decrease inflammation, erythema, and hyperpigmentation without further treatment or major side effects.4
However, the benefits of sclerotherapy alone in the treatment of LM often come gradually, and radiofrequency ablation may need to be utilized to achieve more immediate results.2 Overall, outcomes are highly variable, but favorable outcomes often can be difficult to obtain due to a high recurrence rate.2,8 Our patient's symptoms improved during his postoperative recovery, and he declined further intervention.
The Diagnosis: Microcystic Lymphatic Malformation
The shave biopsy demonstrated numerous thin-walled vascular spaces filled with lymphatic fluid within the dermis (Figure), consistent with a diagnosis of microcystic lymphatic malformation (LM). Lymphatic malformations represent a class of benign vascular lesions consisting of anomalous or dilated lymphatic vessels, which can be broadly categorized as macrocystic (formerly cavernous lymphangioma or cystic hygroma), microcystic (formerly lymphangioma circumscriptum), or mixed.1 Patients often will present with pruritus, crusting, secondary infection, edema, or oozing.2 The superficial blebs of microcystic LMs resemble frog spawn and range in color from clear to pink, brawny, or deep maroon.3 Although the lymphatic vessels involved in microcystic LMs appear disconnected from the major lymphatic circulation,3 systemic fluid overload could plausibly promote lesional swelling and tenderness; we attributed our patient's worsening symptoms to the cumulative 7.8 L of intravenous fluid he received intraoperatively during his cardiac transplant. The excess fluid allowed communication between lymphatic cisterns and thin-walled vesicles on the skin surface through dilated channels. Overall, LMs represent roughly 26% of pediatric benign vascular tumors and approximately 4% of all vascular tumors.4
Although microcystic LMs may appear especially vascular or verrucous, the differential diagnosis for our patient's LM included condyloma acuminatum,5,6 condyloma lata,7 epidermal nevus, and lymphangiosarcoma. Epidermal nevi are congenital lesions, varying in appearance from velvety to verrucous patches and plaques that often evolve during puberty and become thicker, more verrucous, and hyperpigmented. Keratinocytic epidermal nevus syndromes and other entities such as nevus sebaceous have been associated with somatic mutations affecting proteins in the fibroblast growth factor receptor signaling pathway (eg, FGFR3, HRAS).8 Although the clinical appearance alone may be similar, lymphangiosarcoma can be distinguished from LM via biopsy.
There are several methods to diagnose LM. Duplex sonography is possibly the best noninvasive method to identify the flow between venous valves. Magnetic resonance imaging can detect larger occurrences of LM, and lymphangiography can be utilized to confirm a normal or abnormal lymphatic network.4 Treatment options are broad, including surgical excision, laser ablation, and topical sirolimus. Hypertonic saline sclerotherapy can be injected into the afflicted lymphatic channels to decrease inflammation, erythema, and hyperpigmentation without further treatment or major side effects.4
However, the benefits of sclerotherapy alone in the treatment of LM often come gradually, and radiofrequency ablation may need to be utilized to achieve more immediate results.2 Overall, outcomes are highly variable, but favorable outcomes often can be difficult to obtain due to a high recurrence rate.2,8 Our patient's symptoms improved during his postoperative recovery, and he declined further intervention.
- Elluru RG, Balakrishnan K, Padua HM. Lymphatic malformations: diagnosis and management. Semin Pediatr Surg. 2014;23:178-185. doi:10.1053/j.sempedsurg.2014.07.002
- Niti K, Manish P. Microcystic lymphatic malformation (lymphangioma circumscriptum) treated using a minimally invasive technique of radiofrequency ablation and sclerotherapy. Dermatol Surg. 2010;36:1711-1717. doi:10.1111/j.1524-4725.2010.01723.x
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111 /j.1365-4632.2009.04226.x
- Bikowski JB, Dumont AM. Lymphangioma circumscriptum: treatment with hypertonic saline sclerotherapy. J Am Acad Dermatol. 2005;53:442-444. doi:10.1016/j.jaad.2005.04.086
- Costa-Silva M, Fernandes I, Rodrigues AG, et al. Anogenital warts in pediatric population. An Bras Dermatol. 2017;92:675-681. doi:10.1590 /abd1806-4841.201756411
- Darmstadt GL. Perianal lymphangioma circumscriptum mistaken for genital warts. Pediatrics 1996;98;461.
- Bruins FG, van Deudekom FJA, de Vries HJC. Syphilitic condylomata lata mimicking anogenital warts. BMJ. 2015;350:h1259. doi:10.1136 /bmj.h1259
- Asch S, Sugarman JL. Epidermal nevus syndromes: new insights into whorls and swirls. Pediatr Dermatol. 2018;35:21-29. doi:10.1111 /pde.13273
- Elluru RG, Balakrishnan K, Padua HM. Lymphatic malformations: diagnosis and management. Semin Pediatr Surg. 2014;23:178-185. doi:10.1053/j.sempedsurg.2014.07.002
- Niti K, Manish P. Microcystic lymphatic malformation (lymphangioma circumscriptum) treated using a minimally invasive technique of radiofrequency ablation and sclerotherapy. Dermatol Surg. 2010;36:1711-1717. doi:10.1111/j.1524-4725.2010.01723.x
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295. doi:10.1111 /j.1365-4632.2009.04226.x
- Bikowski JB, Dumont AM. Lymphangioma circumscriptum: treatment with hypertonic saline sclerotherapy. J Am Acad Dermatol. 2005;53:442-444. doi:10.1016/j.jaad.2005.04.086
- Costa-Silva M, Fernandes I, Rodrigues AG, et al. Anogenital warts in pediatric population. An Bras Dermatol. 2017;92:675-681. doi:10.1590 /abd1806-4841.201756411
- Darmstadt GL. Perianal lymphangioma circumscriptum mistaken for genital warts. Pediatrics 1996;98;461.
- Bruins FG, van Deudekom FJA, de Vries HJC. Syphilitic condylomata lata mimicking anogenital warts. BMJ. 2015;350:h1259. doi:10.1136 /bmj.h1259
- Asch S, Sugarman JL. Epidermal nevus syndromes: new insights into whorls and swirls. Pediatr Dermatol. 2018;35:21-29. doi:10.1111 /pde.13273
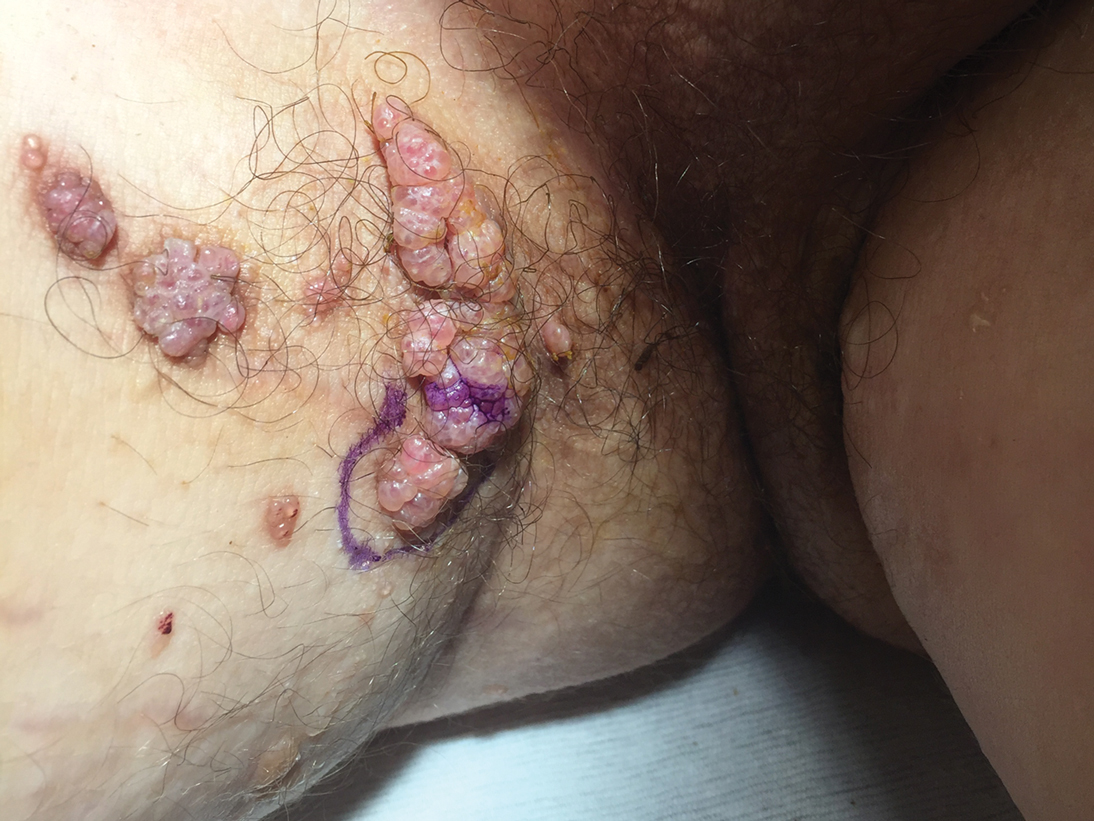
A 17-year-old adolescent boy presented with increasingly painful genital warts on the right thigh, groin, and scrotum that had been present since birth. The patient had a medical history of cardiac transplantation in the months prior to presentation and was on immunosuppressive therapy. The lesions had become more swollen and bothersome in the weeks following the transplantation and now prevented him from ambulating due to discomfort. He denied any history of sexual contact or oral lesions. Physical examination revealed numerous translucent and hemorrhagic vesicles clustered and linearly distributed on the right medial thigh. A shave biopsy of a vesicle was performed.
What’s under my toenail?
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.

Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.
Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.
Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
A 13-year-old female was seen by her pediatrician for a lesion that had been on her right toe for about 6 months. She is unaware of any trauma to the area. The lesion has been growing slowly and recently it started lifting up the nail, became tender, and was bleeding, which is the reason why she sought care.
At the pediatrician's office, he noted a pink crusted papule under the nail. The nail was lifting up and was tender to the touch. She is a healthy girl who is not taking any medications and has no allergies. There is no family history of similar lesions.
The pediatrician took a picture of the lesion and he send it to our pediatric teledermatology service for consultation.
An otherwise healthy 1-month-old female presents with lesions on the face, scalp, and chest
A potassium hydroxide preparation (KOH) from skin scrapings from the scalp lesions demonstrated no fungal elements. Further laboratory work up revealed a normal blood cell count, normal liver enzymes, an antinuclear antibody (ANA) titer of less than 1:80, a positive anti–Sjögren’s syndrome type B (SSB) antibody but negative anti–Sjögren’s syndrome type A (SSA) antibody and anti-U1RNP antibody. An electrocardiogram revealed no abnormalities. Liver function tests were normal. The complete blood count showed mild thrombocytopenia. Given the typical skin lesions and the positive SSB test and associated thrombocytopenia, the baby was diagnosed with neonatal lupus erythematosus.
Because of the diagnosis of neonatal lupus the mother was also tested and was found to have an elevated ANA of 1:640, positive SSB and antiphospholipid antibodies. The mother was healthy and her review of systems was negative for any collagen vascular disease–related symptoms.
Discussion
Neonatal lupus erythematosus (NLE) is a rare form of systemic lupus erythematosus (SLE) believed to be caused by transplacental transfer of anti-Ro (Sjögren’s syndrome antigen A, SSA), or, less commonly, anti-La (Sjögren’s syndrome antigen B, SSB) from mothers who are positive for these antibodies. Approximately 95% of NLE is associated with maternal anti-SSA; of these cases, 40% are also associated with maternal anti-SSB.1 Only about 2% of children of mothers who have anti-SSA or anti-SSB develop NLE, a finding that has led some researchers to postulate that maternal factors, fetal genetic factors, and environmental factors determine which children of anti-SSA or SSB positive mothers develop NLE.
A recent review found no association between the development of NLE and fetal birth weight, prematurity, or age.3 Over half of mothers of children who develop NLE are asymptomatic at the time of diagnosis of the neonate,3 though many become symptomatic in following years. Of mothers who are symptomatic, SLE and undifferentiated autoimmune syndrome are the most common diagnoses, though NLE has been rarely reported in the offspring of mothers with Sjögren’s syndrome, rheumatoid arthritis, and psoriasis.4,5
Fetal genetics are not an absolute determinant of development of NLE, as discordance in the development of NLE in twins has been reported. However, certain genetic relationships have been established. Fetal mutations in tumor necrosis factor–alpha appear to increase the likelihood of cutaneous manifestations. Mutations in transforming growth factor beta appear to increase the likelihood of cardiac manifestations, and experiments in cultured mouse cardiocytes have shown anti-SSB antibodies to impair macrophage phagocytosis of apoptotic cells in the developing fetal heart. These observations taken together suggest a fibroblast-mediated response to unphagocytosed cardiocyte debris may account for conduction abnormalities in neonates with NLE-induced heart block.6
Cutaneous disease in NLE is possible at birth, but more skin findings develop upon exposure to the sun. Nearly 80% of neonates affected by NLE develop cutaneous manifestations in the first few months of life. The head, neck, and extensor surfaces of the arms are most commonly affected, presumably because they are most likely to be exposed to the sun. Erythematous, annular, or discoid lesions are most common, and periorbital erythema with or without scale (“raccoon eyes”) should prompt consideration of NLE. However, annular, or discoid lesions are sometimes not present in NLE; telangiectasias, bullae, atrophic divots (“ice-pick scars”) or ulcerations may be seen instead. Lesions in the genital area have been described in fewer than 5% of patients with NLE.
The differential diagnosis of annular, scaly lesions in neonates includes annular erythema of infancy, tinea corporis, and seborrheic dermatitis. Annular erythema of infancy is a rare skin condition characterized by a cyclical eruption of erythematous annular lesions with minimal scaling which resolve spontaneously within a few weeks to months without leaving scaring or pigment changes. There is no treatment needed as the lesions self-resolve.7 Acute urticaria can sometimes appear similar to NLE but these are not scaly and also the lesions will disappear within 24-36 hours, compared with NLE lesions, which may take weeks to months to go away. Seborrheic dermatitis is a common skin condition seen in newborns with in the first few weeks of life and can present as scaly annular erythematous plaques on the face, scalp, torso, and the diaper area. Seborrheic dermatitis usually responds well to a combination of an antiyeast cream and a low-potency topical corticosteroid medication.

When NLE is suspected, diagnostic testing for lupus antibodies (anti-SSA, anti-SSB, and anti-U1RNP) in both maternal and neonatal serum should be undertaken. The presence of a characteristic rash plus maternal or neonatal antibodies is sufficient to make the diagnosis. If the rash is less characteristic, a biopsy showing an interface dermatitis can help solidify the diagnosis. Neonates with cutaneous manifestations of lupus may also have systemic disease. The most common and serious complication is heart block, whose pathophysiology is described above. Neonates with evidence of first-, second-, or third-degree heart block should be referred to a pediatric cardiologist for careful monitoring and management. Hepatic involvement has been reported, but is usually mild. Hematologic abnormalities have also been described that include anemia, neutropenia, and thrombocytopenia, which resolve by 9 months of age. Central nervous system involvement may rarely occur. The mainstay of treatment for the rash in NLE is diligent sun avoidance and sun protection. Topical corticosteroids may be used, but are not needed as the rash typically resolves by 9 months to 1 year without treatment. Mothers who have one child with NLE should be advised that they are more likely to have another with NLE – the risk is as high as 30%-40% in the second child. Hydroxychloroquine taken during subsequent pregnancies can reduce the incidence of cardiac complications,8 as can the so-called “triple therapy” of plasmapheresis, steroids, and IVIg.9
The cutaneous manifestations of NLE are usually self-limiting. However, they can serve as important clues that can prompt diagnosis of SLE in the mother, investigation of cardiac complications in the infant, and appropriate preventative care in future pregnancies.
Dr. Matiz is with the department of dermatology, Southern California Permanente Medical Group, San Diego. Mr. Kusari is with the department of dermatology, University of California, San Francisco.
References
1. Moretti D et al. Int J Dermatol. 2014;53(12):1508-12.
2. Buyon JP et al. Nature Clin Prac Rheum. 2009;5(3):139-48.
3. Li Y-Q et al. Int J Rheum Dis. 2015;18(7):761-7.
4. Rivera TL et al. Annals Rheum Dis. 2009;68(6):828-35.
5. Li L et al. Zhonghua er ke za zhi 2011;49(2):146-50.
6. Izmirly PM et al. Clin Rheumatol. 2011;30(12):1641-5.
7. Toledo-Alberola F and Betlloch-Mas I. Actas Dermosifiliogr. 2010 Jul;101(6):473-84.
8. Izmirly PM et al. Circulation. 2012;126(1):76-82.
9. Martinez-Sanchez N et al. Autoimmun Rev. 2015;14(5):423-8.
A potassium hydroxide preparation (KOH) from skin scrapings from the scalp lesions demonstrated no fungal elements. Further laboratory work up revealed a normal blood cell count, normal liver enzymes, an antinuclear antibody (ANA) titer of less than 1:80, a positive anti–Sjögren’s syndrome type B (SSB) antibody but negative anti–Sjögren’s syndrome type A (SSA) antibody and anti-U1RNP antibody. An electrocardiogram revealed no abnormalities. Liver function tests were normal. The complete blood count showed mild thrombocytopenia. Given the typical skin lesions and the positive SSB test and associated thrombocytopenia, the baby was diagnosed with neonatal lupus erythematosus.
Because of the diagnosis of neonatal lupus the mother was also tested and was found to have an elevated ANA of 1:640, positive SSB and antiphospholipid antibodies. The mother was healthy and her review of systems was negative for any collagen vascular disease–related symptoms.
Discussion
Neonatal lupus erythematosus (NLE) is a rare form of systemic lupus erythematosus (SLE) believed to be caused by transplacental transfer of anti-Ro (Sjögren’s syndrome antigen A, SSA), or, less commonly, anti-La (Sjögren’s syndrome antigen B, SSB) from mothers who are positive for these antibodies. Approximately 95% of NLE is associated with maternal anti-SSA; of these cases, 40% are also associated with maternal anti-SSB.1 Only about 2% of children of mothers who have anti-SSA or anti-SSB develop NLE, a finding that has led some researchers to postulate that maternal factors, fetal genetic factors, and environmental factors determine which children of anti-SSA or SSB positive mothers develop NLE.
A recent review found no association between the development of NLE and fetal birth weight, prematurity, or age.3 Over half of mothers of children who develop NLE are asymptomatic at the time of diagnosis of the neonate,3 though many become symptomatic in following years. Of mothers who are symptomatic, SLE and undifferentiated autoimmune syndrome are the most common diagnoses, though NLE has been rarely reported in the offspring of mothers with Sjögren’s syndrome, rheumatoid arthritis, and psoriasis.4,5
Fetal genetics are not an absolute determinant of development of NLE, as discordance in the development of NLE in twins has been reported. However, certain genetic relationships have been established. Fetal mutations in tumor necrosis factor–alpha appear to increase the likelihood of cutaneous manifestations. Mutations in transforming growth factor beta appear to increase the likelihood of cardiac manifestations, and experiments in cultured mouse cardiocytes have shown anti-SSB antibodies to impair macrophage phagocytosis of apoptotic cells in the developing fetal heart. These observations taken together suggest a fibroblast-mediated response to unphagocytosed cardiocyte debris may account for conduction abnormalities in neonates with NLE-induced heart block.6
Cutaneous disease in NLE is possible at birth, but more skin findings develop upon exposure to the sun. Nearly 80% of neonates affected by NLE develop cutaneous manifestations in the first few months of life. The head, neck, and extensor surfaces of the arms are most commonly affected, presumably because they are most likely to be exposed to the sun. Erythematous, annular, or discoid lesions are most common, and periorbital erythema with or without scale (“raccoon eyes”) should prompt consideration of NLE. However, annular, or discoid lesions are sometimes not present in NLE; telangiectasias, bullae, atrophic divots (“ice-pick scars”) or ulcerations may be seen instead. Lesions in the genital area have been described in fewer than 5% of patients with NLE.
The differential diagnosis of annular, scaly lesions in neonates includes annular erythema of infancy, tinea corporis, and seborrheic dermatitis. Annular erythema of infancy is a rare skin condition characterized by a cyclical eruption of erythematous annular lesions with minimal scaling which resolve spontaneously within a few weeks to months without leaving scaring or pigment changes. There is no treatment needed as the lesions self-resolve.7 Acute urticaria can sometimes appear similar to NLE but these are not scaly and also the lesions will disappear within 24-36 hours, compared with NLE lesions, which may take weeks to months to go away. Seborrheic dermatitis is a common skin condition seen in newborns with in the first few weeks of life and can present as scaly annular erythematous plaques on the face, scalp, torso, and the diaper area. Seborrheic dermatitis usually responds well to a combination of an antiyeast cream and a low-potency topical corticosteroid medication.
When NLE is suspected, diagnostic testing for lupus antibodies (anti-SSA, anti-SSB, and anti-U1RNP) in both maternal and neonatal serum should be undertaken. The presence of a characteristic rash plus maternal or neonatal antibodies is sufficient to make the diagnosis. If the rash is less characteristic, a biopsy showing an interface dermatitis can help solidify the diagnosis. Neonates with cutaneous manifestations of lupus may also have systemic disease. The most common and serious complication is heart block, whose pathophysiology is described above. Neonates with evidence of first-, second-, or third-degree heart block should be referred to a pediatric cardiologist for careful monitoring and management. Hepatic involvement has been reported, but is usually mild. Hematologic abnormalities have also been described that include anemia, neutropenia, and thrombocytopenia, which resolve by 9 months of age. Central nervous system involvement may rarely occur. The mainstay of treatment for the rash in NLE is diligent sun avoidance and sun protection. Topical corticosteroids may be used, but are not needed as the rash typically resolves by 9 months to 1 year without treatment. Mothers who have one child with NLE should be advised that they are more likely to have another with NLE – the risk is as high as 30%-40% in the second child. Hydroxychloroquine taken during subsequent pregnancies can reduce the incidence of cardiac complications,8 as can the so-called “triple therapy” of plasmapheresis, steroids, and IVIg.9
The cutaneous manifestations of NLE are usually self-limiting. However, they can serve as important clues that can prompt diagnosis of SLE in the mother, investigation of cardiac complications in the infant, and appropriate preventative care in future pregnancies.
Dr. Matiz is with the department of dermatology, Southern California Permanente Medical Group, San Diego. Mr. Kusari is with the department of dermatology, University of California, San Francisco.
References
1. Moretti D et al. Int J Dermatol. 2014;53(12):1508-12.
2. Buyon JP et al. Nature Clin Prac Rheum. 2009;5(3):139-48.
3. Li Y-Q et al. Int J Rheum Dis. 2015;18(7):761-7.
4. Rivera TL et al. Annals Rheum Dis. 2009;68(6):828-35.
5. Li L et al. Zhonghua er ke za zhi 2011;49(2):146-50.
6. Izmirly PM et al. Clin Rheumatol. 2011;30(12):1641-5.
7. Toledo-Alberola F and Betlloch-Mas I. Actas Dermosifiliogr. 2010 Jul;101(6):473-84.
8. Izmirly PM et al. Circulation. 2012;126(1):76-82.
9. Martinez-Sanchez N et al. Autoimmun Rev. 2015;14(5):423-8.
A potassium hydroxide preparation (KOH) from skin scrapings from the scalp lesions demonstrated no fungal elements. Further laboratory work up revealed a normal blood cell count, normal liver enzymes, an antinuclear antibody (ANA) titer of less than 1:80, a positive anti–Sjögren’s syndrome type B (SSB) antibody but negative anti–Sjögren’s syndrome type A (SSA) antibody and anti-U1RNP antibody. An electrocardiogram revealed no abnormalities. Liver function tests were normal. The complete blood count showed mild thrombocytopenia. Given the typical skin lesions and the positive SSB test and associated thrombocytopenia, the baby was diagnosed with neonatal lupus erythematosus.
Because of the diagnosis of neonatal lupus the mother was also tested and was found to have an elevated ANA of 1:640, positive SSB and antiphospholipid antibodies. The mother was healthy and her review of systems was negative for any collagen vascular disease–related symptoms.
Discussion
Neonatal lupus erythematosus (NLE) is a rare form of systemic lupus erythematosus (SLE) believed to be caused by transplacental transfer of anti-Ro (Sjögren’s syndrome antigen A, SSA), or, less commonly, anti-La (Sjögren’s syndrome antigen B, SSB) from mothers who are positive for these antibodies. Approximately 95% of NLE is associated with maternal anti-SSA; of these cases, 40% are also associated with maternal anti-SSB.1 Only about 2% of children of mothers who have anti-SSA or anti-SSB develop NLE, a finding that has led some researchers to postulate that maternal factors, fetal genetic factors, and environmental factors determine which children of anti-SSA or SSB positive mothers develop NLE.
A recent review found no association between the development of NLE and fetal birth weight, prematurity, or age.3 Over half of mothers of children who develop NLE are asymptomatic at the time of diagnosis of the neonate,3 though many become symptomatic in following years. Of mothers who are symptomatic, SLE and undifferentiated autoimmune syndrome are the most common diagnoses, though NLE has been rarely reported in the offspring of mothers with Sjögren’s syndrome, rheumatoid arthritis, and psoriasis.4,5
Fetal genetics are not an absolute determinant of development of NLE, as discordance in the development of NLE in twins has been reported. However, certain genetic relationships have been established. Fetal mutations in tumor necrosis factor–alpha appear to increase the likelihood of cutaneous manifestations. Mutations in transforming growth factor beta appear to increase the likelihood of cardiac manifestations, and experiments in cultured mouse cardiocytes have shown anti-SSB antibodies to impair macrophage phagocytosis of apoptotic cells in the developing fetal heart. These observations taken together suggest a fibroblast-mediated response to unphagocytosed cardiocyte debris may account for conduction abnormalities in neonates with NLE-induced heart block.6
Cutaneous disease in NLE is possible at birth, but more skin findings develop upon exposure to the sun. Nearly 80% of neonates affected by NLE develop cutaneous manifestations in the first few months of life. The head, neck, and extensor surfaces of the arms are most commonly affected, presumably because they are most likely to be exposed to the sun. Erythematous, annular, or discoid lesions are most common, and periorbital erythema with or without scale (“raccoon eyes”) should prompt consideration of NLE. However, annular, or discoid lesions are sometimes not present in NLE; telangiectasias, bullae, atrophic divots (“ice-pick scars”) or ulcerations may be seen instead. Lesions in the genital area have been described in fewer than 5% of patients with NLE.
The differential diagnosis of annular, scaly lesions in neonates includes annular erythema of infancy, tinea corporis, and seborrheic dermatitis. Annular erythema of infancy is a rare skin condition characterized by a cyclical eruption of erythematous annular lesions with minimal scaling which resolve spontaneously within a few weeks to months without leaving scaring or pigment changes. There is no treatment needed as the lesions self-resolve.7 Acute urticaria can sometimes appear similar to NLE but these are not scaly and also the lesions will disappear within 24-36 hours, compared with NLE lesions, which may take weeks to months to go away. Seborrheic dermatitis is a common skin condition seen in newborns with in the first few weeks of life and can present as scaly annular erythematous plaques on the face, scalp, torso, and the diaper area. Seborrheic dermatitis usually responds well to a combination of an antiyeast cream and a low-potency topical corticosteroid medication.
When NLE is suspected, diagnostic testing for lupus antibodies (anti-SSA, anti-SSB, and anti-U1RNP) in both maternal and neonatal serum should be undertaken. The presence of a characteristic rash plus maternal or neonatal antibodies is sufficient to make the diagnosis. If the rash is less characteristic, a biopsy showing an interface dermatitis can help solidify the diagnosis. Neonates with cutaneous manifestations of lupus may also have systemic disease. The most common and serious complication is heart block, whose pathophysiology is described above. Neonates with evidence of first-, second-, or third-degree heart block should be referred to a pediatric cardiologist for careful monitoring and management. Hepatic involvement has been reported, but is usually mild. Hematologic abnormalities have also been described that include anemia, neutropenia, and thrombocytopenia, which resolve by 9 months of age. Central nervous system involvement may rarely occur. The mainstay of treatment for the rash in NLE is diligent sun avoidance and sun protection. Topical corticosteroids may be used, but are not needed as the rash typically resolves by 9 months to 1 year without treatment. Mothers who have one child with NLE should be advised that they are more likely to have another with NLE – the risk is as high as 30%-40% in the second child. Hydroxychloroquine taken during subsequent pregnancies can reduce the incidence of cardiac complications,8 as can the so-called “triple therapy” of plasmapheresis, steroids, and IVIg.9
The cutaneous manifestations of NLE are usually self-limiting. However, they can serve as important clues that can prompt diagnosis of SLE in the mother, investigation of cardiac complications in the infant, and appropriate preventative care in future pregnancies.
Dr. Matiz is with the department of dermatology, Southern California Permanente Medical Group, San Diego. Mr. Kusari is with the department of dermatology, University of California, San Francisco.
References
1. Moretti D et al. Int J Dermatol. 2014;53(12):1508-12.
2. Buyon JP et al. Nature Clin Prac Rheum. 2009;5(3):139-48.
3. Li Y-Q et al. Int J Rheum Dis. 2015;18(7):761-7.
4. Rivera TL et al. Annals Rheum Dis. 2009;68(6):828-35.
5. Li L et al. Zhonghua er ke za zhi 2011;49(2):146-50.
6. Izmirly PM et al. Clin Rheumatol. 2011;30(12):1641-5.
7. Toledo-Alberola F and Betlloch-Mas I. Actas Dermosifiliogr. 2010 Jul;101(6):473-84.
8. Izmirly PM et al. Circulation. 2012;126(1):76-82.
9. Martinez-Sanchez N et al. Autoimmun Rev. 2015;14(5):423-8.
A 1-month-old, full-term female, born via normal vaginal delivery, presented to the dermatology clinic with a 3-week history of recurrent skin lesions on the scalp, face, and chest. The mother has been treating the lesions with breast milk and most recently with clotrimazole cream without resolution. 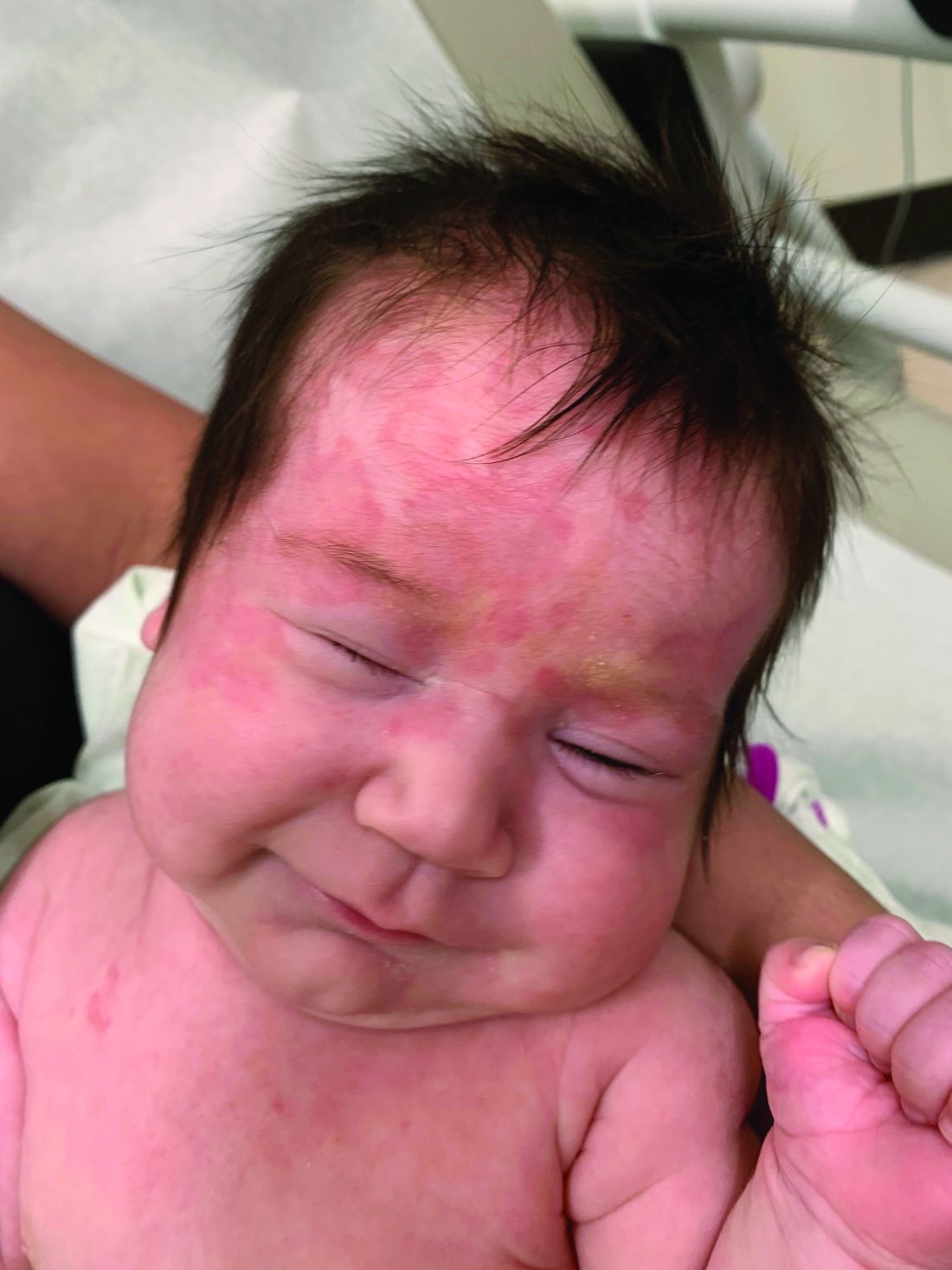
The mother of the baby is a healthy 32-year-old female with no past medical history. She had adequate prenatal care, and all the prenatal infectious and genetic tests were normal. The baby has been healthy and growing well. There is no history of associated fevers, chills, or any other symptoms. The family took no recent trips, and the parents are not affected. There are no other children at home and they have a cat and a dog. The family history is noncontributory.
On physical examination the baby was not in acute distress and her vital signs were normal. On skin examination she had several erythematous annular plaques and patches on the face, scalp, and upper chest (Fig. 1). There was no liver or spleen enlargement and no lymphadenopathy was palpated on exam.
Widespread Hyperkeratotic Papules in a Transplant Recipient
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
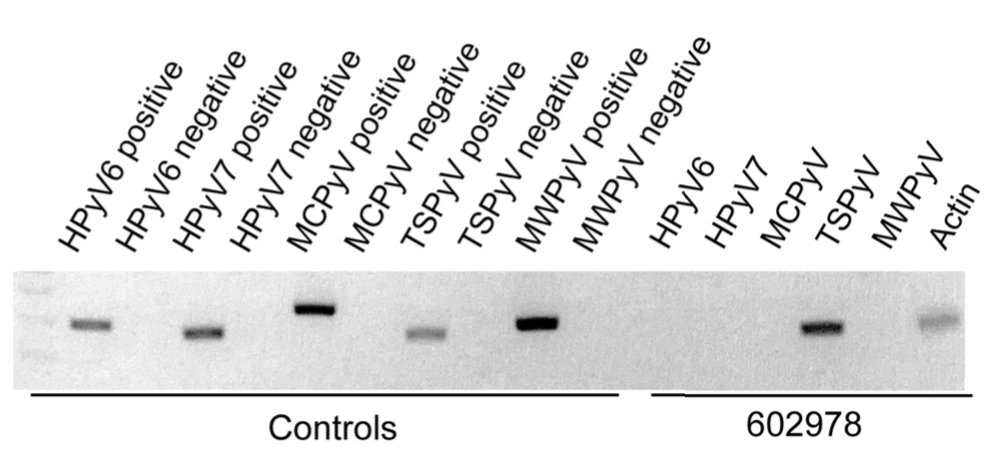
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
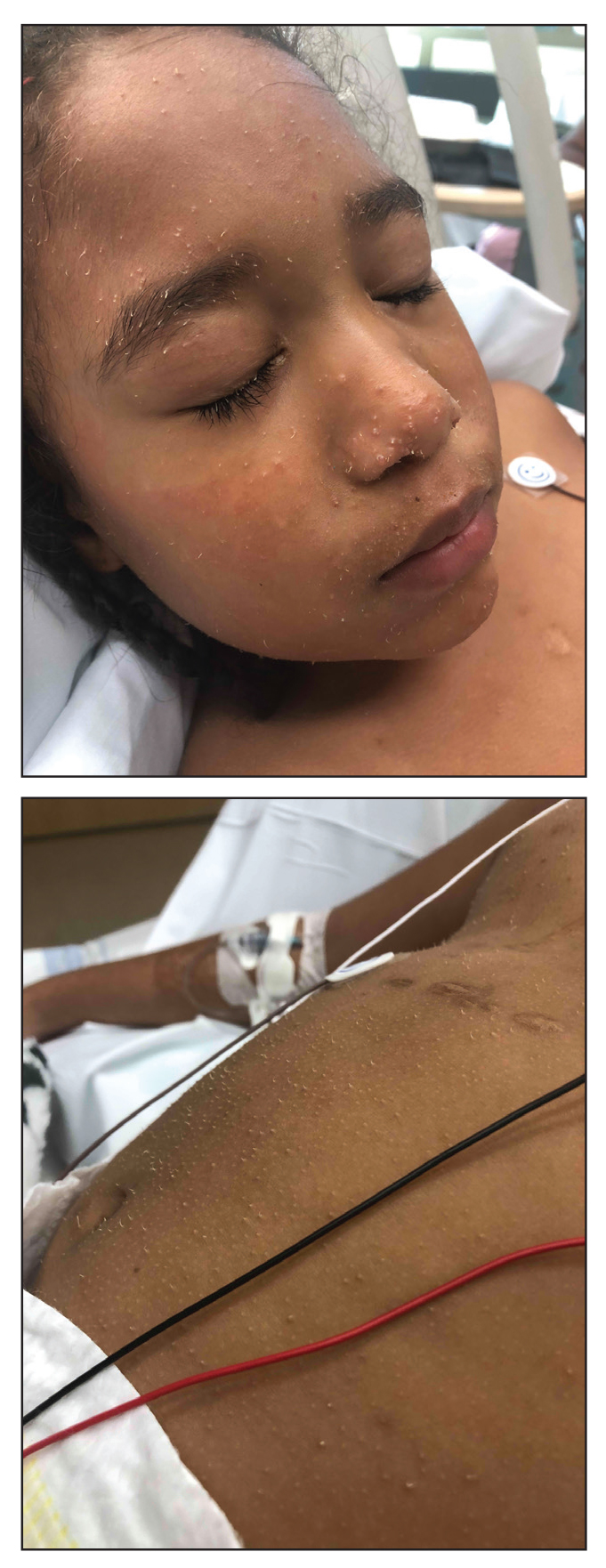
A 4-year-old girl with a history of cardiac transplantation 1 year prior for dilated cardiomyopathy presented to the dermatology consultation service with widespread hyperkeratotic papules of 2 months’ duration. The eruption initially had appeared on the face with subsequent involvement of the trunk and extremities. Her immunosuppressive medications included oral tacrolimus and mycophenolate mofetil. No over-the-counter or prescription treatments had been used for the eruption; the patient’s mother had been manually extracting the spicules from the nose, cheeks, and forehead with tweezers. The lesions were asymptomatic with only mild follicular erythema. Physical examination revealed multiple folliculocentric keratinous spicules on the nose, cheeks, forehead (top), trunk (bottom), arms, and legs.