User login
Pediatric Dermatology Consult - March 2018
Rosacea is a chronic inflammatory skin disorder characterized by flushing, telangiectasia, erythema, papules, and pustules, most classically of the central face. Fair-skinned individuals and women are more commonly affected than are men, with age of onset typically around 30 years and older.1 In children and adolescents, rosacea is rare, especially among prepubertal children, so other papulopustular disorders should be considered when a rosacealike picture is present.2 Recurrent chalazia are seen with ocular rosacea and may be a clue to the diagnosis of acne rosacea. An individual’s subtype of rosacea also may transform with time to a different or an additional subtype.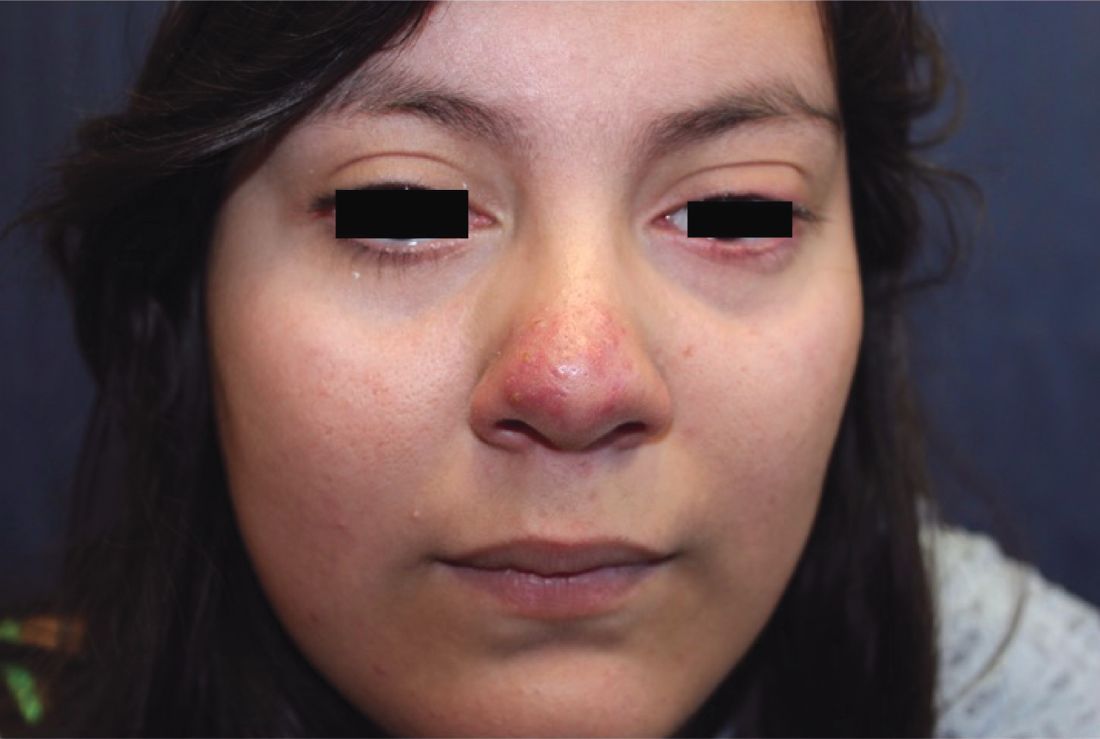
Papulopustular rosacea (subtype II) is characterized by the presence of erythematous, dome-shaped papules distributed in crops in the central facial region. Cheeks, nasolabial folds, and the chin are most commonly affected. Pustules may or may not be present, but comedones are notably absent in an exclusively rosacea disease process. If comedones are present, a diagnosis of acne vulgaris should be considered instead of, or in addition to, rosacea. Pediatric patients with rosacea frequently present with papules and/or pustules, following the development of flushing.2

Ocular rosacea (subtype IV) may range in severity from mild blepharitis to severe keratitis and corneal vascularization. Patients may complain of a foreign body sensation. On external exam, lid margin telangiectasias, blepharitis, conjunctivitis, conjunctival injection, and recurrent chalazia may be frequently seen.3 Ocular rosacea may present without any signs of cutaneous disease; it may be the only form of rosacea (15% of patients in one study of 20 patients had only ocular rosacea)4 or may herald the development of cutaneous involvement. In fact, in children, ocular rosacea is frequently the first sign of disease. (A total of 55% of patients in the same study had both ocular and cutaneous rosacea, with ocular symptoms manifesting before the cutaneous disease). Thus an index of suspicion for rosacea should be maintained when a child presents with ocular findings.2

Other dermatitides resembling rosacea include steroid rosacea, perioral dermatitis, and idiopathic facial aseptic granuloma. Steroid rosacea, also known as iatrosacea, describes an eruption of erythema, papules, and telangiectasias that is clinically indistinguishable from rosacea.6 It results from chronic use of topical steroids, generally high potency, or abrupt withdrawal of steroids. Steroid rosacea should be treated by discontinuation of the steroid via tapered withdrawal.7 Perioral dermatitis, also known as periorificial dermatitis, may also appear rosacea-form. It usually is located around the perioral and perinasal areas, but may extend to the periocular area.8 Idiopathic facial septic granuloma describes erythematous to violaceous nodules of the cheeks and eyelid in children, with chalazia frequently present; it is thought to be associated with rosacea.9
Although the exact pathophysiology of rosacea is unknown, it is clear that the dysregulation of the innate immune system plays a key role in the pathogenesis of rosacea. Studies have found that patients with rosacea have increased expression of cathelicidin, and its activating serine protease, kallikrein.5 Interestingly, UV light, a known trigger of rosacea, induces expression of cathelicidin and its inflammatory cascade.5 Neurovascular signaling is also aberrantly upregulated; vanilloid and ankyrin receptors have been shown to be active in rosacea, and are activated by rosacea-exacerbating stimuli, such as heat, inflammation, and spices. Higher levels of Demodex folliculorum and Staphylococcus epidermis also have been consistently found on the skin of rosacea patients, compared with healthy subjects, though it is unclear what role these pathogens play in the development of rosacea.
Treatment of rosacea is very important given its profound impact on quality of life; one study found that the odds ratio for depression in individuals with rosacea is 4.81.10 Patient education is essential, and patients should be encouraged to identify specific triggers so they can minimize exposure when feasible. Common triggers include hot and cold temperature, sunlight, wind, spicy foods, alcohol, exercise, emotional stress, and certain medications such as niacin. Topical steroids frequently are exacerbating, so patients should be encouraged to avoid them and use moisturizers often, given their skin’s increased transepidermal water loss and susceptibility to irritation. In addition, sunscreens are essential to reduce inflammation from reactive oxygen species, which aggravate rosacea.11 For pharmaceutical therapeutics, topical sodium sulfacetamide, metronidazole, and azelaic acid have been shown to be effective in rosacea. For persistent erythema, topical alpha-adrenergic receptor agonists including brimonidine tartrate and oxymetazoline have been shown to reduce erythema by vasoconstricting blood vessels, although some products are associated with a rebound erythema on treatment discontinuation. For moderate to severe rosacea, low-dose oral doxycycline at anti-inflammatory doses (less than 50 mg daily) is the mainstay of therapy. Other oral antibiotics and topical permethrin have been used, and topical ivermectin 1% cream has been approved for inflammatory rosacea.11 Oral beta-blockers also have been successfully used in some patients to reduce erythema and flushing, as well as isotretinoin for refractory, severe rosacea with improvement.
Allison Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Han or Dr. Eichenfield.
References
1. J Am Acad Dermatol. 2018 Jan;78(1):148-55.
2. Cutis. 2016 Jul;98(1):49-53.
3. J Eur Acad Dermatol Venereol. 2017 Oct;31(10):1732-8.
4. J Fr Ophtalmol. 2011 Dec;34(10):703-10.
5. J Am Acad Dermatol. 2015 May;72(5):749-58.
6. Indian J Dermatol. 2011 Jan;56(1):30-2.
7. Cutis, 2004. 74(2):99-103.
8. Pediatr Dermatol. 1992 Mar;9(1):22-6.
9. Pediatr Dermatol. 2015 Jul-Aug;32(4):e136-9.
10. Br J Dermatol. 2005 Dec;153(6):1176-81.
11. J Am Acad Dermatol. 2015 May;72(5):761-70.
Rosacea is a chronic inflammatory skin disorder characterized by flushing, telangiectasia, erythema, papules, and pustules, most classically of the central face. Fair-skinned individuals and women are more commonly affected than are men, with age of onset typically around 30 years and older.1 In children and adolescents, rosacea is rare, especially among prepubertal children, so other papulopustular disorders should be considered when a rosacealike picture is present.2 Recurrent chalazia are seen with ocular rosacea and may be a clue to the diagnosis of acne rosacea. An individual’s subtype of rosacea also may transform with time to a different or an additional subtype.
Papulopustular rosacea (subtype II) is characterized by the presence of erythematous, dome-shaped papules distributed in crops in the central facial region. Cheeks, nasolabial folds, and the chin are most commonly affected. Pustules may or may not be present, but comedones are notably absent in an exclusively rosacea disease process. If comedones are present, a diagnosis of acne vulgaris should be considered instead of, or in addition to, rosacea. Pediatric patients with rosacea frequently present with papules and/or pustules, following the development of flushing.2
Ocular rosacea (subtype IV) may range in severity from mild blepharitis to severe keratitis and corneal vascularization. Patients may complain of a foreign body sensation. On external exam, lid margin telangiectasias, blepharitis, conjunctivitis, conjunctival injection, and recurrent chalazia may be frequently seen.3 Ocular rosacea may present without any signs of cutaneous disease; it may be the only form of rosacea (15% of patients in one study of 20 patients had only ocular rosacea)4 or may herald the development of cutaneous involvement. In fact, in children, ocular rosacea is frequently the first sign of disease. (A total of 55% of patients in the same study had both ocular and cutaneous rosacea, with ocular symptoms manifesting before the cutaneous disease). Thus an index of suspicion for rosacea should be maintained when a child presents with ocular findings.2
Other dermatitides resembling rosacea include steroid rosacea, perioral dermatitis, and idiopathic facial aseptic granuloma. Steroid rosacea, also known as iatrosacea, describes an eruption of erythema, papules, and telangiectasias that is clinically indistinguishable from rosacea.6 It results from chronic use of topical steroids, generally high potency, or abrupt withdrawal of steroids. Steroid rosacea should be treated by discontinuation of the steroid via tapered withdrawal.7 Perioral dermatitis, also known as periorificial dermatitis, may also appear rosacea-form. It usually is located around the perioral and perinasal areas, but may extend to the periocular area.8 Idiopathic facial septic granuloma describes erythematous to violaceous nodules of the cheeks and eyelid in children, with chalazia frequently present; it is thought to be associated with rosacea.9
Although the exact pathophysiology of rosacea is unknown, it is clear that the dysregulation of the innate immune system plays a key role in the pathogenesis of rosacea. Studies have found that patients with rosacea have increased expression of cathelicidin, and its activating serine protease, kallikrein.5 Interestingly, UV light, a known trigger of rosacea, induces expression of cathelicidin and its inflammatory cascade.5 Neurovascular signaling is also aberrantly upregulated; vanilloid and ankyrin receptors have been shown to be active in rosacea, and are activated by rosacea-exacerbating stimuli, such as heat, inflammation, and spices. Higher levels of Demodex folliculorum and Staphylococcus epidermis also have been consistently found on the skin of rosacea patients, compared with healthy subjects, though it is unclear what role these pathogens play in the development of rosacea.
Treatment of rosacea is very important given its profound impact on quality of life; one study found that the odds ratio for depression in individuals with rosacea is 4.81.10 Patient education is essential, and patients should be encouraged to identify specific triggers so they can minimize exposure when feasible. Common triggers include hot and cold temperature, sunlight, wind, spicy foods, alcohol, exercise, emotional stress, and certain medications such as niacin. Topical steroids frequently are exacerbating, so patients should be encouraged to avoid them and use moisturizers often, given their skin’s increased transepidermal water loss and susceptibility to irritation. In addition, sunscreens are essential to reduce inflammation from reactive oxygen species, which aggravate rosacea.11 For pharmaceutical therapeutics, topical sodium sulfacetamide, metronidazole, and azelaic acid have been shown to be effective in rosacea. For persistent erythema, topical alpha-adrenergic receptor agonists including brimonidine tartrate and oxymetazoline have been shown to reduce erythema by vasoconstricting blood vessels, although some products are associated with a rebound erythema on treatment discontinuation. For moderate to severe rosacea, low-dose oral doxycycline at anti-inflammatory doses (less than 50 mg daily) is the mainstay of therapy. Other oral antibiotics and topical permethrin have been used, and topical ivermectin 1% cream has been approved for inflammatory rosacea.11 Oral beta-blockers also have been successfully used in some patients to reduce erythema and flushing, as well as isotretinoin for refractory, severe rosacea with improvement.
Allison Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Han or Dr. Eichenfield.
References
1. J Am Acad Dermatol. 2018 Jan;78(1):148-55.
2. Cutis. 2016 Jul;98(1):49-53.
3. J Eur Acad Dermatol Venereol. 2017 Oct;31(10):1732-8.
4. J Fr Ophtalmol. 2011 Dec;34(10):703-10.
5. J Am Acad Dermatol. 2015 May;72(5):749-58.
6. Indian J Dermatol. 2011 Jan;56(1):30-2.
7. Cutis, 2004. 74(2):99-103.
8. Pediatr Dermatol. 1992 Mar;9(1):22-6.
9. Pediatr Dermatol. 2015 Jul-Aug;32(4):e136-9.
10. Br J Dermatol. 2005 Dec;153(6):1176-81.
11. J Am Acad Dermatol. 2015 May;72(5):761-70.
Rosacea is a chronic inflammatory skin disorder characterized by flushing, telangiectasia, erythema, papules, and pustules, most classically of the central face. Fair-skinned individuals and women are more commonly affected than are men, with age of onset typically around 30 years and older.1 In children and adolescents, rosacea is rare, especially among prepubertal children, so other papulopustular disorders should be considered when a rosacealike picture is present.2 Recurrent chalazia are seen with ocular rosacea and may be a clue to the diagnosis of acne rosacea. An individual’s subtype of rosacea also may transform with time to a different or an additional subtype.
Papulopustular rosacea (subtype II) is characterized by the presence of erythematous, dome-shaped papules distributed in crops in the central facial region. Cheeks, nasolabial folds, and the chin are most commonly affected. Pustules may or may not be present, but comedones are notably absent in an exclusively rosacea disease process. If comedones are present, a diagnosis of acne vulgaris should be considered instead of, or in addition to, rosacea. Pediatric patients with rosacea frequently present with papules and/or pustules, following the development of flushing.2
Ocular rosacea (subtype IV) may range in severity from mild blepharitis to severe keratitis and corneal vascularization. Patients may complain of a foreign body sensation. On external exam, lid margin telangiectasias, blepharitis, conjunctivitis, conjunctival injection, and recurrent chalazia may be frequently seen.3 Ocular rosacea may present without any signs of cutaneous disease; it may be the only form of rosacea (15% of patients in one study of 20 patients had only ocular rosacea)4 or may herald the development of cutaneous involvement. In fact, in children, ocular rosacea is frequently the first sign of disease. (A total of 55% of patients in the same study had both ocular and cutaneous rosacea, with ocular symptoms manifesting before the cutaneous disease). Thus an index of suspicion for rosacea should be maintained when a child presents with ocular findings.2
Other dermatitides resembling rosacea include steroid rosacea, perioral dermatitis, and idiopathic facial aseptic granuloma. Steroid rosacea, also known as iatrosacea, describes an eruption of erythema, papules, and telangiectasias that is clinically indistinguishable from rosacea.6 It results from chronic use of topical steroids, generally high potency, or abrupt withdrawal of steroids. Steroid rosacea should be treated by discontinuation of the steroid via tapered withdrawal.7 Perioral dermatitis, also known as periorificial dermatitis, may also appear rosacea-form. It usually is located around the perioral and perinasal areas, but may extend to the periocular area.8 Idiopathic facial septic granuloma describes erythematous to violaceous nodules of the cheeks and eyelid in children, with chalazia frequently present; it is thought to be associated with rosacea.9
Although the exact pathophysiology of rosacea is unknown, it is clear that the dysregulation of the innate immune system plays a key role in the pathogenesis of rosacea. Studies have found that patients with rosacea have increased expression of cathelicidin, and its activating serine protease, kallikrein.5 Interestingly, UV light, a known trigger of rosacea, induces expression of cathelicidin and its inflammatory cascade.5 Neurovascular signaling is also aberrantly upregulated; vanilloid and ankyrin receptors have been shown to be active in rosacea, and are activated by rosacea-exacerbating stimuli, such as heat, inflammation, and spices. Higher levels of Demodex folliculorum and Staphylococcus epidermis also have been consistently found on the skin of rosacea patients, compared with healthy subjects, though it is unclear what role these pathogens play in the development of rosacea.
Treatment of rosacea is very important given its profound impact on quality of life; one study found that the odds ratio for depression in individuals with rosacea is 4.81.10 Patient education is essential, and patients should be encouraged to identify specific triggers so they can minimize exposure when feasible. Common triggers include hot and cold temperature, sunlight, wind, spicy foods, alcohol, exercise, emotional stress, and certain medications such as niacin. Topical steroids frequently are exacerbating, so patients should be encouraged to avoid them and use moisturizers often, given their skin’s increased transepidermal water loss and susceptibility to irritation. In addition, sunscreens are essential to reduce inflammation from reactive oxygen species, which aggravate rosacea.11 For pharmaceutical therapeutics, topical sodium sulfacetamide, metronidazole, and azelaic acid have been shown to be effective in rosacea. For persistent erythema, topical alpha-adrenergic receptor agonists including brimonidine tartrate and oxymetazoline have been shown to reduce erythema by vasoconstricting blood vessels, although some products are associated with a rebound erythema on treatment discontinuation. For moderate to severe rosacea, low-dose oral doxycycline at anti-inflammatory doses (less than 50 mg daily) is the mainstay of therapy. Other oral antibiotics and topical permethrin have been used, and topical ivermectin 1% cream has been approved for inflammatory rosacea.11 Oral beta-blockers also have been successfully used in some patients to reduce erythema and flushing, as well as isotretinoin for refractory, severe rosacea with improvement.
Allison Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Ms. Han or Dr. Eichenfield.
References
1. J Am Acad Dermatol. 2018 Jan;78(1):148-55.
2. Cutis. 2016 Jul;98(1):49-53.
3. J Eur Acad Dermatol Venereol. 2017 Oct;31(10):1732-8.
4. J Fr Ophtalmol. 2011 Dec;34(10):703-10.
5. J Am Acad Dermatol. 2015 May;72(5):749-58.
6. Indian J Dermatol. 2011 Jan;56(1):30-2.
7. Cutis, 2004. 74(2):99-103.
8. Pediatr Dermatol. 1992 Mar;9(1):22-6.
9. Pediatr Dermatol. 2015 Jul-Aug;32(4):e136-9.
10. Br J Dermatol. 2005 Dec;153(6):1176-81.
11. J Am Acad Dermatol. 2015 May;72(5):761-70.
A 16-year-old girl presented with a 6-month history of an erythematous eruption of small papules and pustules around the cheeks and nose. She states the erythema had started first, with periods of feeling flushed that became worse with sun exposure. She saw her primary care physician who prescribed topical steroids. After using the steroids, the rash became worse, and she developed papules and pustules.
Pediatric Dermatology Consult - February 2018
The patient was diagnosed with pityriasis rosea (PR) on the basis of the clinical findings; a biopsy was not performed. The patient’s pruritus was treated with oral hydroxyzine and topical 1% triamcinolone ointment. She experienced itch relief with these treatments. On follow-up at 3 months, the patient’s lesions had mostly resolved with some postinflammatory hyperpigmentation.
In some patients, flu-like symptoms precede the onset of skin lesions; this has led to speculation regarding a viral etiology for PR. This prodrome, which is present in as many as half of all cases,can include mild headache, low-grade fever, joint aches, or malaise.2 Pityriasis rosea is thought to occur secondary to a systemic activation of human herpesviruses (HHV) 6 and/or HHV-7. Three cases of PR have been reported in the setting of H1N1 influenza virus infection.3 In one small study, HHV-8 was detected by polymerase chain reaction in approximately 20% of biopsy samples of lesional skin in patients with PR.4 However, most research on a viral etiology for pityriasis rosea has focused on HHV-6 and to a lesser extent HHV-7. DNA from both viruses has been isolated from PR lesions, but at varying detection rates.5,6 Furthermore, HHV-7 DNA has been isolated in as many as 14% of normal individuals without pityriasis rosea, suggesting that the presence of this virus on the skin is fairly common.7
Pityriasis rosea occurs in males and females of all ethnicities, with a slight female predominance. It is rare in young children and older adults. Most cases occur in adolescents and in adults in their twenties and early thirties. Cases occur most frequently in fall and spring.8
The herald patch of pityriasis rosea is typically solitary, but cases with multiple herald patches have been described. The herald patch can range in size from 1-10 cm and usually contains the best example of trailing scale – scale seen on the inside edge of the annular lesion. The satellite lesions of pityriasis rosea are typically papules or plaques with a collarette of scale. These lesions usually are oriented along the Langer cleavage lines, giving them a “Christmas tree” configuration when they appear on the posterior trunk.

Mimics
The herald patch of pityriasis rosea can resemble tinea corporis, and if there is any doubt as to the diagnosis, potassium hydroxide examination (also known as a KOH test) and/or fungal culture should be done to rule out a fungal etiology. However, certain features of this case, particularly the subsequent development of satellite lesions, are more consistent with pityriasis rosea.
Secondary syphilis should be considered in patients who are sexually active. The lesions of secondary syphilis are not typically pruritic, and involvement of the palms and soles is common (whereas such involvement is rare in pityriasis rosea).
Like pityriasis rosea, pityriasis lichenoides et varioliformis acuta (PLEVA) is characterized by papular lesions that resolve spontaneously; the lesions of PLEVA usually evolve to vesicular, necrotic, and purpuric papules that take longer to resolve than PR lesions. The lesions of PLEVA are more erythematous, pustular, and crusting than the lesions of pityriasis rosea.
Guttate psoriasis, which occurs following streptococcal pharyngitis in over 50% of patients, does not present with a herald lesion or distribution along Langer’s lines.10 If guttate psoriasis is suspected, rapid streptococcal testing of the throat or perianal area may be considered.
Nummular eczema presents as papules that enlarge to form erythematous, lichenified plaques that measure 1-2 cm in diameter. A relatively sudden eruption, such as this patient’s, would be unusual for nummular eczema. Also, nummular eczema typically occurs on xerotic skin, more often on the extremities than the trunk.
Diagnostic tests, treatment

Most patients do not require specific therapy for pityriasis rosea. Patients should be reassured that PR is typically a self-limited disease without long-term sequelae. Pregnant patients who develop pityriasis rosea in the first trimester may be at higher risk for spontaneous abortion,although data on the subject are sorely lacking.11 Oral antihistamines are useful in reducing pruritus associated with PR, and some patients experience relief by applying a low-potency topical corticosteroid.
In more severe cases, or in cases in which the patient is greatly distressed by the lesions, both broadband and narrowband UVB phototherapy effectively improve severity of lesions and reduces symptoms.12 These observations suggest that moderate sun exposure can help to reduce severity of PR lesions and hasten their resolution, but no studies assessing the effect of sun exposure on pityriasis rosea symptoms have been performed.
Furthermore, the possible role of the HHV-6 in PR has led some investigators to explore the utility of acyclovir in managing pityriasis rosea.13 One group recently found that 400 mg of acyclovir three times per day for 7 days decreased the number of lesions and pruritus associated with pityriasis rosea, compared those seen in controls, at 1-month follow-up.13
Timely recognition of the diagnosis, consideration of mimics, and ample reassurance are appropriate when approaching this disease.
Mr. Kusari is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. They have no relevant financial disclosures. Email them at [email protected].
References
1. Dermatology. 2015;231(1):9-14.
2. World J Clin Cases. 2017 Jun 16;5(6):203-11.
3. Pediatr Dermatol. 2011 May-Jun;28(3):341-2.
4. J Eur Acad Dermatol Venereol. 2006 Jul;20(6):667-71.
5. Dermatology. 1997;195(4):374-8.
6. J Invest Dermatol. 2005 Jun;124(6):1234-40.
7. Arch Dermatol. 1999 Sep;135(9):1070-2.
8. J Am Acad Dermatol. 1982 Jul;7(1):80-9.
9. Iran J Pediatr. 2010 Jun;20(2):237-41.
10. J Pediatr. 1988 Dec;113(6):1037-9.
11. J Am Acad Dermatol. 2008 May;58(5 Suppl 1):S78-83.
12. J Am Acad Dermatol. 1995 Dec;33(6):996-9.
13. Indian Dermatol Online J. 2015 May-Jun;6(3):181-4.
The patient was diagnosed with pityriasis rosea (PR) on the basis of the clinical findings; a biopsy was not performed. The patient’s pruritus was treated with oral hydroxyzine and topical 1% triamcinolone ointment. She experienced itch relief with these treatments. On follow-up at 3 months, the patient’s lesions had mostly resolved with some postinflammatory hyperpigmentation.
In some patients, flu-like symptoms precede the onset of skin lesions; this has led to speculation regarding a viral etiology for PR. This prodrome, which is present in as many as half of all cases,can include mild headache, low-grade fever, joint aches, or malaise.2 Pityriasis rosea is thought to occur secondary to a systemic activation of human herpesviruses (HHV) 6 and/or HHV-7. Three cases of PR have been reported in the setting of H1N1 influenza virus infection.3 In one small study, HHV-8 was detected by polymerase chain reaction in approximately 20% of biopsy samples of lesional skin in patients with PR.4 However, most research on a viral etiology for pityriasis rosea has focused on HHV-6 and to a lesser extent HHV-7. DNA from both viruses has been isolated from PR lesions, but at varying detection rates.5,6 Furthermore, HHV-7 DNA has been isolated in as many as 14% of normal individuals without pityriasis rosea, suggesting that the presence of this virus on the skin is fairly common.7
Pityriasis rosea occurs in males and females of all ethnicities, with a slight female predominance. It is rare in young children and older adults. Most cases occur in adolescents and in adults in their twenties and early thirties. Cases occur most frequently in fall and spring.8
The herald patch of pityriasis rosea is typically solitary, but cases with multiple herald patches have been described. The herald patch can range in size from 1-10 cm and usually contains the best example of trailing scale – scale seen on the inside edge of the annular lesion. The satellite lesions of pityriasis rosea are typically papules or plaques with a collarette of scale. These lesions usually are oriented along the Langer cleavage lines, giving them a “Christmas tree” configuration when they appear on the posterior trunk.
Mimics
The herald patch of pityriasis rosea can resemble tinea corporis, and if there is any doubt as to the diagnosis, potassium hydroxide examination (also known as a KOH test) and/or fungal culture should be done to rule out a fungal etiology. However, certain features of this case, particularly the subsequent development of satellite lesions, are more consistent with pityriasis rosea.
Secondary syphilis should be considered in patients who are sexually active. The lesions of secondary syphilis are not typically pruritic, and involvement of the palms and soles is common (whereas such involvement is rare in pityriasis rosea).
Like pityriasis rosea, pityriasis lichenoides et varioliformis acuta (PLEVA) is characterized by papular lesions that resolve spontaneously; the lesions of PLEVA usually evolve to vesicular, necrotic, and purpuric papules that take longer to resolve than PR lesions. The lesions of PLEVA are more erythematous, pustular, and crusting than the lesions of pityriasis rosea.
Guttate psoriasis, which occurs following streptococcal pharyngitis in over 50% of patients, does not present with a herald lesion or distribution along Langer’s lines.10 If guttate psoriasis is suspected, rapid streptococcal testing of the throat or perianal area may be considered.
Nummular eczema presents as papules that enlarge to form erythematous, lichenified plaques that measure 1-2 cm in diameter. A relatively sudden eruption, such as this patient’s, would be unusual for nummular eczema. Also, nummular eczema typically occurs on xerotic skin, more often on the extremities than the trunk.
Diagnostic tests, treatment
Most patients do not require specific therapy for pityriasis rosea. Patients should be reassured that PR is typically a self-limited disease without long-term sequelae. Pregnant patients who develop pityriasis rosea in the first trimester may be at higher risk for spontaneous abortion,although data on the subject are sorely lacking.11 Oral antihistamines are useful in reducing pruritus associated with PR, and some patients experience relief by applying a low-potency topical corticosteroid.
In more severe cases, or in cases in which the patient is greatly distressed by the lesions, both broadband and narrowband UVB phototherapy effectively improve severity of lesions and reduces symptoms.12 These observations suggest that moderate sun exposure can help to reduce severity of PR lesions and hasten their resolution, but no studies assessing the effect of sun exposure on pityriasis rosea symptoms have been performed.
Furthermore, the possible role of the HHV-6 in PR has led some investigators to explore the utility of acyclovir in managing pityriasis rosea.13 One group recently found that 400 mg of acyclovir three times per day for 7 days decreased the number of lesions and pruritus associated with pityriasis rosea, compared those seen in controls, at 1-month follow-up.13
Timely recognition of the diagnosis, consideration of mimics, and ample reassurance are appropriate when approaching this disease.
Mr. Kusari is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. They have no relevant financial disclosures. Email them at [email protected].
References
1. Dermatology. 2015;231(1):9-14.
2. World J Clin Cases. 2017 Jun 16;5(6):203-11.
3. Pediatr Dermatol. 2011 May-Jun;28(3):341-2.
4. J Eur Acad Dermatol Venereol. 2006 Jul;20(6):667-71.
5. Dermatology. 1997;195(4):374-8.
6. J Invest Dermatol. 2005 Jun;124(6):1234-40.
7. Arch Dermatol. 1999 Sep;135(9):1070-2.
8. J Am Acad Dermatol. 1982 Jul;7(1):80-9.
9. Iran J Pediatr. 2010 Jun;20(2):237-41.
10. J Pediatr. 1988 Dec;113(6):1037-9.
11. J Am Acad Dermatol. 2008 May;58(5 Suppl 1):S78-83.
12. J Am Acad Dermatol. 1995 Dec;33(6):996-9.
13. Indian Dermatol Online J. 2015 May-Jun;6(3):181-4.
The patient was diagnosed with pityriasis rosea (PR) on the basis of the clinical findings; a biopsy was not performed. The patient’s pruritus was treated with oral hydroxyzine and topical 1% triamcinolone ointment. She experienced itch relief with these treatments. On follow-up at 3 months, the patient’s lesions had mostly resolved with some postinflammatory hyperpigmentation.
In some patients, flu-like symptoms precede the onset of skin lesions; this has led to speculation regarding a viral etiology for PR. This prodrome, which is present in as many as half of all cases,can include mild headache, low-grade fever, joint aches, or malaise.2 Pityriasis rosea is thought to occur secondary to a systemic activation of human herpesviruses (HHV) 6 and/or HHV-7. Three cases of PR have been reported in the setting of H1N1 influenza virus infection.3 In one small study, HHV-8 was detected by polymerase chain reaction in approximately 20% of biopsy samples of lesional skin in patients with PR.4 However, most research on a viral etiology for pityriasis rosea has focused on HHV-6 and to a lesser extent HHV-7. DNA from both viruses has been isolated from PR lesions, but at varying detection rates.5,6 Furthermore, HHV-7 DNA has been isolated in as many as 14% of normal individuals without pityriasis rosea, suggesting that the presence of this virus on the skin is fairly common.7
Pityriasis rosea occurs in males and females of all ethnicities, with a slight female predominance. It is rare in young children and older adults. Most cases occur in adolescents and in adults in their twenties and early thirties. Cases occur most frequently in fall and spring.8
The herald patch of pityriasis rosea is typically solitary, but cases with multiple herald patches have been described. The herald patch can range in size from 1-10 cm and usually contains the best example of trailing scale – scale seen on the inside edge of the annular lesion. The satellite lesions of pityriasis rosea are typically papules or plaques with a collarette of scale. These lesions usually are oriented along the Langer cleavage lines, giving them a “Christmas tree” configuration when they appear on the posterior trunk.
Mimics
The herald patch of pityriasis rosea can resemble tinea corporis, and if there is any doubt as to the diagnosis, potassium hydroxide examination (also known as a KOH test) and/or fungal culture should be done to rule out a fungal etiology. However, certain features of this case, particularly the subsequent development of satellite lesions, are more consistent with pityriasis rosea.
Secondary syphilis should be considered in patients who are sexually active. The lesions of secondary syphilis are not typically pruritic, and involvement of the palms and soles is common (whereas such involvement is rare in pityriasis rosea).
Like pityriasis rosea, pityriasis lichenoides et varioliformis acuta (PLEVA) is characterized by papular lesions that resolve spontaneously; the lesions of PLEVA usually evolve to vesicular, necrotic, and purpuric papules that take longer to resolve than PR lesions. The lesions of PLEVA are more erythematous, pustular, and crusting than the lesions of pityriasis rosea.
Guttate psoriasis, which occurs following streptococcal pharyngitis in over 50% of patients, does not present with a herald lesion or distribution along Langer’s lines.10 If guttate psoriasis is suspected, rapid streptococcal testing of the throat or perianal area may be considered.
Nummular eczema presents as papules that enlarge to form erythematous, lichenified plaques that measure 1-2 cm in diameter. A relatively sudden eruption, such as this patient’s, would be unusual for nummular eczema. Also, nummular eczema typically occurs on xerotic skin, more often on the extremities than the trunk.
Diagnostic tests, treatment
Most patients do not require specific therapy for pityriasis rosea. Patients should be reassured that PR is typically a self-limited disease without long-term sequelae. Pregnant patients who develop pityriasis rosea in the first trimester may be at higher risk for spontaneous abortion,although data on the subject are sorely lacking.11 Oral antihistamines are useful in reducing pruritus associated with PR, and some patients experience relief by applying a low-potency topical corticosteroid.
In more severe cases, or in cases in which the patient is greatly distressed by the lesions, both broadband and narrowband UVB phototherapy effectively improve severity of lesions and reduces symptoms.12 These observations suggest that moderate sun exposure can help to reduce severity of PR lesions and hasten their resolution, but no studies assessing the effect of sun exposure on pityriasis rosea symptoms have been performed.
Furthermore, the possible role of the HHV-6 in PR has led some investigators to explore the utility of acyclovir in managing pityriasis rosea.13 One group recently found that 400 mg of acyclovir three times per day for 7 days decreased the number of lesions and pruritus associated with pityriasis rosea, compared those seen in controls, at 1-month follow-up.13
Timely recognition of the diagnosis, consideration of mimics, and ample reassurance are appropriate when approaching this disease.
Mr. Kusari is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. They have no relevant financial disclosures. Email them at [email protected].
References
1. Dermatology. 2015;231(1):9-14.
2. World J Clin Cases. 2017 Jun 16;5(6):203-11.
3. Pediatr Dermatol. 2011 May-Jun;28(3):341-2.
4. J Eur Acad Dermatol Venereol. 2006 Jul;20(6):667-71.
5. Dermatology. 1997;195(4):374-8.
6. J Invest Dermatol. 2005 Jun;124(6):1234-40.
7. Arch Dermatol. 1999 Sep;135(9):1070-2.
8. J Am Acad Dermatol. 1982 Jul;7(1):80-9.
9. Iran J Pediatr. 2010 Jun;20(2):237-41.
10. J Pediatr. 1988 Dec;113(6):1037-9.
11. J Am Acad Dermatol. 2008 May;58(5 Suppl 1):S78-83.
12. J Am Acad Dermatol. 1995 Dec;33(6):996-9.
13. Indian Dermatol Online J. 2015 May-Jun;6(3):181-4.
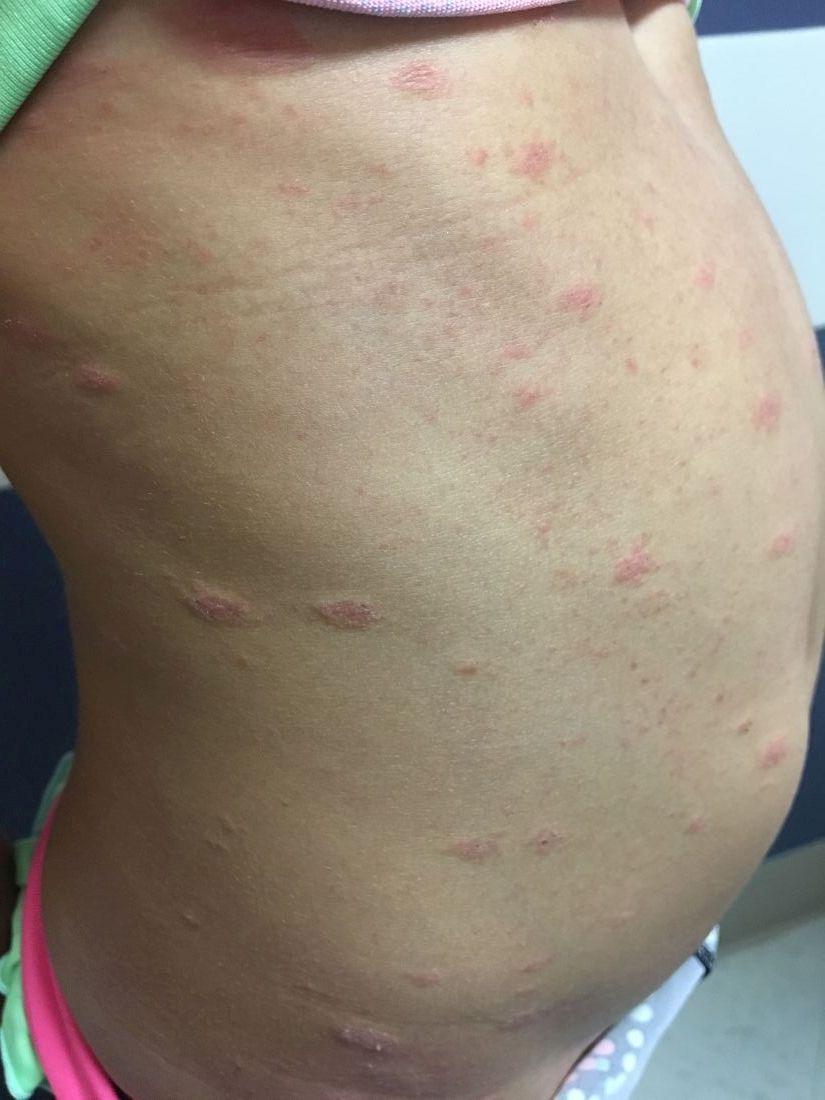
A 6-year-old female presents to the pediatric dermatology office with a 2-day history of a slightly itchy skin lesion on her back. Her birthday was a week prior, and her mother gave her a new kitten, and since then she has been playing with the kitten daily. She has tried some over-the-counter antifungal cream since the lesion first appeared, but there hasn’t been much improvement. The night prior to presenting to the office, the mother noticed more lesions developing on the child’s torso, and because of this, she became worried.
On physical exam, the patient is well appearing, and vital signs are normal. She has multiple scaly, pink, oval plaques and papules on her torso. There are no oral lesions, and her palms and soles are spared.
Pediatric Dermatology Consult - January 2018
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
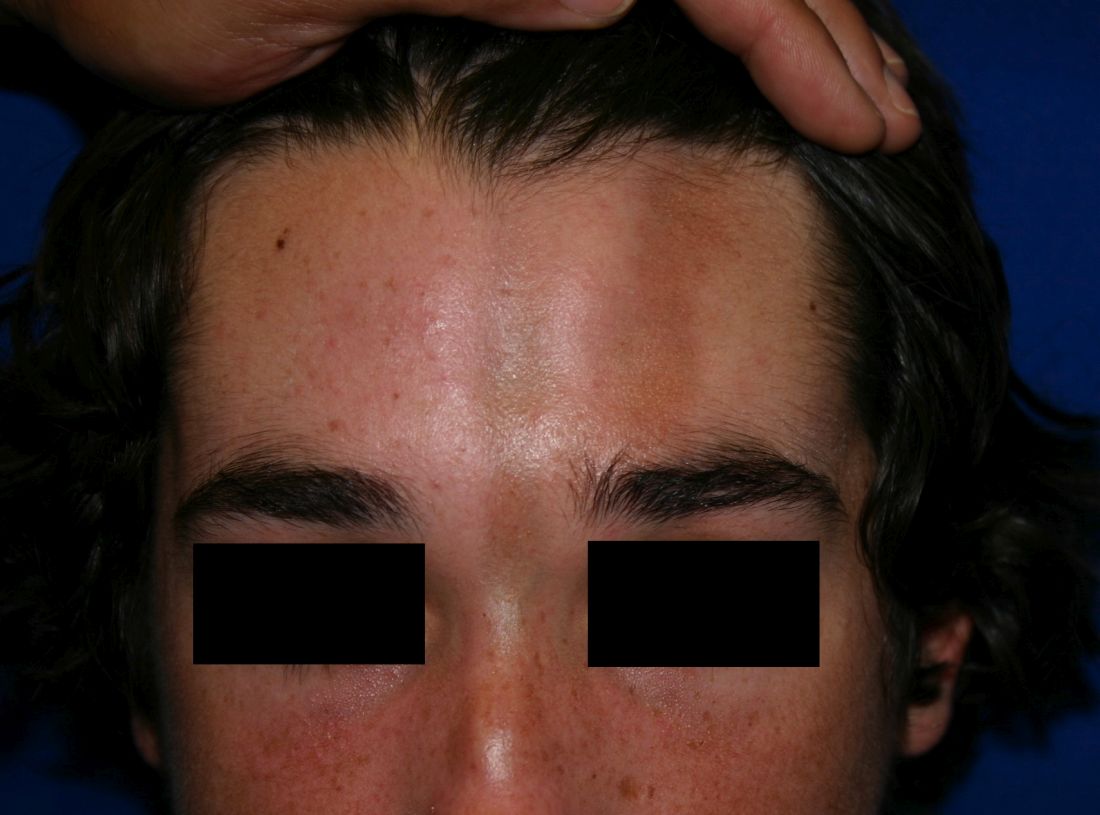
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
A 14-year-old patient presents to a dermatology clinic for a depression on his forehead, which has been there for about 2 years. A few years ago, he used to have a pruritic pink lesion on the forehead where the depression is now. He denies any symptoms.
Confluent and reticulated papillomatosis
Confluent and reticulated papillomatosis of Gougerot and Carteaud, also known as Gougerot-Carteaud syndrome, is an uncommon skin disorder of young individuals characterized by hyperkeratotic or verrucous brown papules or plaques that coalesce centrally and by a reticulated pattern peripherally. It was first described by two French dermatologists, Gougerot and Carteaud, in 1927.1 Initially, the distinct entity of CARP was contested, with some dermatologists believing it to be a variant of acanthosis nigricans. However, CARP is now recognized as a distinct, though rare, dermatosis.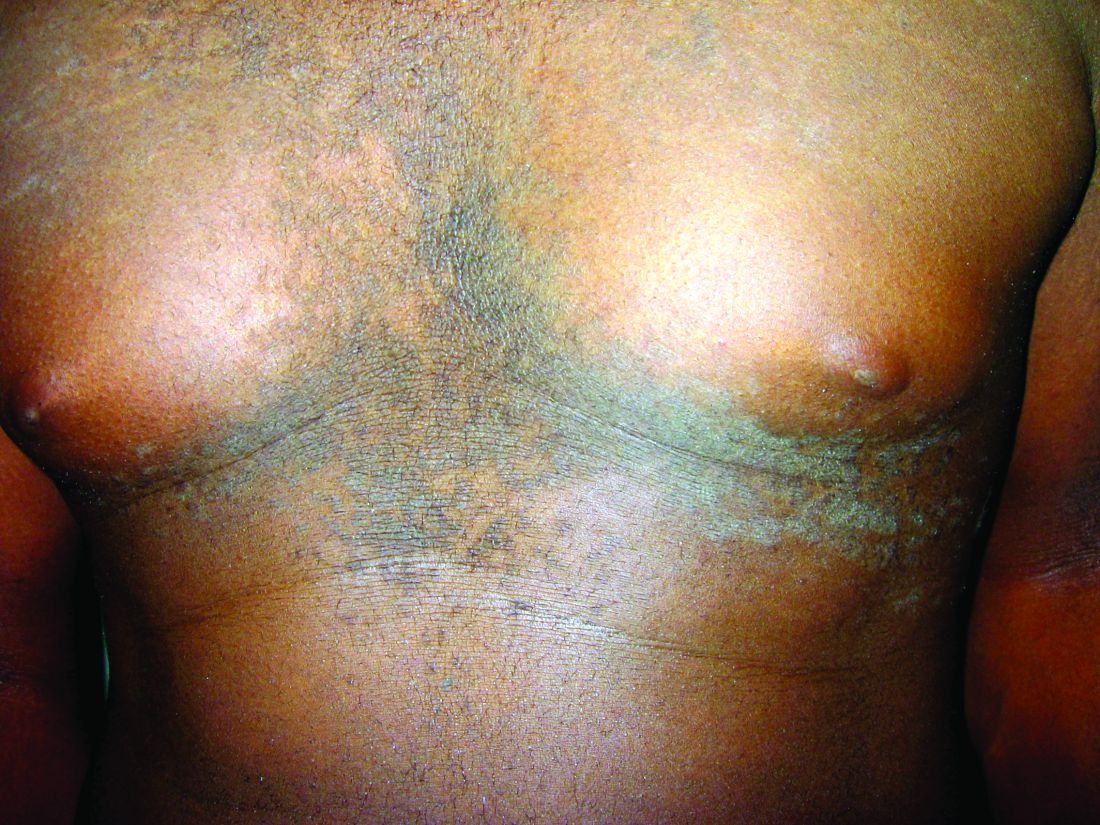
Histopathology reveals findings similar to those that may be found in acanthosis nigricans and epidermal nevi. Classic characteristics of CARP include hyperkeratosis, papillomatosis, increased basal melanin pigmentation, and mild acanthosis. Occasionally, there may be perivascular lymphocytic infiltrates in the superficial dermis.3,4
The etiology of CARP is unknown. CARP’s resolution in response to antibiotics and the isolation of two bacterial actinomycetes, Rhodococcus and Dietzia papillomatosis, from skin scrapings of CARP patients have led some to believe that its etiology is bacterial. However, no bacterial species have been consistently isolated from CARP patients. The prevailing theory of the past was that CARP was an abnormal host response to the fungus Malassezia furfur. Inconsistent detection of the fungus in skin scrapings, as well as persistence of the skin lesions after fungal clearance with antifungal therapy, has debunked this theory. An underlying disorder of keratinization resulting in hyperproliferation also has been suggested given reports of familial CARP and electron microscopy studies demonstrating focal-enhanced expression of keratin-16 in the stratum granulosom.5 Other theories include a cutaneous response to underlying endocrinopathies, ultraviolet light, and localized amyloidosis.1
Diagnosis and differential
CARP is poorly recognized by clinicians and frequently initially misdiagnosed due to its similar appearance to other disorders, most commonly tinea versicolor and acanthosis nigricans. Davis et al. proposed criteria for diagnosis of CARP requiring 1) presence of scaly, reticulated and papillomatous brown macules and patches; 2) distribution over the upper trunk and neck; 3) negative fungal staining of scales; 4) no improvement following antifungal treatment; and 5) improvement following minocycline.2
Tinea versicolor may appear similar to CARP, but unlike CARP, will respond to antifungal treatment and may demonstrate hyphae and yeast on KOH preparation. Acanthosis nigricans and CARP both may present with velvety, hyperpigmented plaques in individuals of obese habitus or with insulin resistance, but peripheral reticulation will be absent in acanthosis nigricans. However, acanthosis nigricans and CARP may coexist, and this coexistence is not uncommonly seen in individuals with obesity and/or insulin resistance. Darier’s disease may look similar to cases of CARP without pigmentary change, but it often will have accompanying nail changes. Macular or lichen amyloidosis may present with pruritic brown macules or papules, but skin biopsy will have positive amyloid staining. The use of 70% alcohol swabbing to diagnose terra firma-forme dermatosis, with lesions disappearing with swabbing, is classic and used to differentiate it from CARP. Other conditions to consider include seborrheic dermatitis, epidermal nevi, verruca plana, epidermodysplasia verruciformis, and acne vulgaris.1,2,4
Treatment
Minocycline is the first-line treatment for CARP: 80% of patients may have complete resolution with minocycline, while the remainder experience at least 50% clearance of skin lesions.2 However, recurrence after stopping minocycline treatment is not uncommon. The mechanism by which minocycline works is unknown. Second-line treatment for those who cannot tolerate minocycline are macrolide antibiotics.6 Other treatment options with reported success include oral isotretinoin and topical retinoids, including tretinoin gel and tazarotene cream.3,7 Appropriate strength topical corticosteroids may be used for pruritus.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, as well as the vice chair of the department of dermatology and a professor of dermatology and pediatrics at UC San Diego. They report having no conflicts of interest or financial disclosures. Email them at [email protected].
References
1. Clin Cosmet Investig Dermatol. 2016 Aug 25;9:217-23.
2. Br J Dermatol. 2006 Feb;154(2):287-93.
3. Arch Dermatol. 2012 Apr;148(4):505-8.
4. J Am Acad Dermatol. 2003 Dec;49(6):1182-4.
5. Arch Dermatol. 2002 Feb;138(2):276-7.
6. J Am Acad Dermatol. 2001;44(4):652-5.
7. Am J Clin Dermatol. 2006;7(5):305-13.
Confluent and reticulated papillomatosis of Gougerot and Carteaud, also known as Gougerot-Carteaud syndrome, is an uncommon skin disorder of young individuals characterized by hyperkeratotic or verrucous brown papules or plaques that coalesce centrally and by a reticulated pattern peripherally. It was first described by two French dermatologists, Gougerot and Carteaud, in 1927.1 Initially, the distinct entity of CARP was contested, with some dermatologists believing it to be a variant of acanthosis nigricans. However, CARP is now recognized as a distinct, though rare, dermatosis.
Histopathology reveals findings similar to those that may be found in acanthosis nigricans and epidermal nevi. Classic characteristics of CARP include hyperkeratosis, papillomatosis, increased basal melanin pigmentation, and mild acanthosis. Occasionally, there may be perivascular lymphocytic infiltrates in the superficial dermis.3,4
The etiology of CARP is unknown. CARP’s resolution in response to antibiotics and the isolation of two bacterial actinomycetes, Rhodococcus and Dietzia papillomatosis, from skin scrapings of CARP patients have led some to believe that its etiology is bacterial. However, no bacterial species have been consistently isolated from CARP patients. The prevailing theory of the past was that CARP was an abnormal host response to the fungus Malassezia furfur. Inconsistent detection of the fungus in skin scrapings, as well as persistence of the skin lesions after fungal clearance with antifungal therapy, has debunked this theory. An underlying disorder of keratinization resulting in hyperproliferation also has been suggested given reports of familial CARP and electron microscopy studies demonstrating focal-enhanced expression of keratin-16 in the stratum granulosom.5 Other theories include a cutaneous response to underlying endocrinopathies, ultraviolet light, and localized amyloidosis.1
Diagnosis and differential
CARP is poorly recognized by clinicians and frequently initially misdiagnosed due to its similar appearance to other disorders, most commonly tinea versicolor and acanthosis nigricans. Davis et al. proposed criteria for diagnosis of CARP requiring 1) presence of scaly, reticulated and papillomatous brown macules and patches; 2) distribution over the upper trunk and neck; 3) negative fungal staining of scales; 4) no improvement following antifungal treatment; and 5) improvement following minocycline.2
Tinea versicolor may appear similar to CARP, but unlike CARP, will respond to antifungal treatment and may demonstrate hyphae and yeast on KOH preparation. Acanthosis nigricans and CARP both may present with velvety, hyperpigmented plaques in individuals of obese habitus or with insulin resistance, but peripheral reticulation will be absent in acanthosis nigricans. However, acanthosis nigricans and CARP may coexist, and this coexistence is not uncommonly seen in individuals with obesity and/or insulin resistance. Darier’s disease may look similar to cases of CARP without pigmentary change, but it often will have accompanying nail changes. Macular or lichen amyloidosis may present with pruritic brown macules or papules, but skin biopsy will have positive amyloid staining. The use of 70% alcohol swabbing to diagnose terra firma-forme dermatosis, with lesions disappearing with swabbing, is classic and used to differentiate it from CARP. Other conditions to consider include seborrheic dermatitis, epidermal nevi, verruca plana, epidermodysplasia verruciformis, and acne vulgaris.1,2,4
Treatment
Minocycline is the first-line treatment for CARP: 80% of patients may have complete resolution with minocycline, while the remainder experience at least 50% clearance of skin lesions.2 However, recurrence after stopping minocycline treatment is not uncommon. The mechanism by which minocycline works is unknown. Second-line treatment for those who cannot tolerate minocycline are macrolide antibiotics.6 Other treatment options with reported success include oral isotretinoin and topical retinoids, including tretinoin gel and tazarotene cream.3,7 Appropriate strength topical corticosteroids may be used for pruritus.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, as well as the vice chair of the department of dermatology and a professor of dermatology and pediatrics at UC San Diego. They report having no conflicts of interest or financial disclosures. Email them at [email protected].
References
1. Clin Cosmet Investig Dermatol. 2016 Aug 25;9:217-23.
2. Br J Dermatol. 2006 Feb;154(2):287-93.
3. Arch Dermatol. 2012 Apr;148(4):505-8.
4. J Am Acad Dermatol. 2003 Dec;49(6):1182-4.
5. Arch Dermatol. 2002 Feb;138(2):276-7.
6. J Am Acad Dermatol. 2001;44(4):652-5.
7. Am J Clin Dermatol. 2006;7(5):305-13.
Confluent and reticulated papillomatosis of Gougerot and Carteaud, also known as Gougerot-Carteaud syndrome, is an uncommon skin disorder of young individuals characterized by hyperkeratotic or verrucous brown papules or plaques that coalesce centrally and by a reticulated pattern peripherally. It was first described by two French dermatologists, Gougerot and Carteaud, in 1927.1 Initially, the distinct entity of CARP was contested, with some dermatologists believing it to be a variant of acanthosis nigricans. However, CARP is now recognized as a distinct, though rare, dermatosis.
Histopathology reveals findings similar to those that may be found in acanthosis nigricans and epidermal nevi. Classic characteristics of CARP include hyperkeratosis, papillomatosis, increased basal melanin pigmentation, and mild acanthosis. Occasionally, there may be perivascular lymphocytic infiltrates in the superficial dermis.3,4
The etiology of CARP is unknown. CARP’s resolution in response to antibiotics and the isolation of two bacterial actinomycetes, Rhodococcus and Dietzia papillomatosis, from skin scrapings of CARP patients have led some to believe that its etiology is bacterial. However, no bacterial species have been consistently isolated from CARP patients. The prevailing theory of the past was that CARP was an abnormal host response to the fungus Malassezia furfur. Inconsistent detection of the fungus in skin scrapings, as well as persistence of the skin lesions after fungal clearance with antifungal therapy, has debunked this theory. An underlying disorder of keratinization resulting in hyperproliferation also has been suggested given reports of familial CARP and electron microscopy studies demonstrating focal-enhanced expression of keratin-16 in the stratum granulosom.5 Other theories include a cutaneous response to underlying endocrinopathies, ultraviolet light, and localized amyloidosis.1
Diagnosis and differential
CARP is poorly recognized by clinicians and frequently initially misdiagnosed due to its similar appearance to other disorders, most commonly tinea versicolor and acanthosis nigricans. Davis et al. proposed criteria for diagnosis of CARP requiring 1) presence of scaly, reticulated and papillomatous brown macules and patches; 2) distribution over the upper trunk and neck; 3) negative fungal staining of scales; 4) no improvement following antifungal treatment; and 5) improvement following minocycline.2
Tinea versicolor may appear similar to CARP, but unlike CARP, will respond to antifungal treatment and may demonstrate hyphae and yeast on KOH preparation. Acanthosis nigricans and CARP both may present with velvety, hyperpigmented plaques in individuals of obese habitus or with insulin resistance, but peripheral reticulation will be absent in acanthosis nigricans. However, acanthosis nigricans and CARP may coexist, and this coexistence is not uncommonly seen in individuals with obesity and/or insulin resistance. Darier’s disease may look similar to cases of CARP without pigmentary change, but it often will have accompanying nail changes. Macular or lichen amyloidosis may present with pruritic brown macules or papules, but skin biopsy will have positive amyloid staining. The use of 70% alcohol swabbing to diagnose terra firma-forme dermatosis, with lesions disappearing with swabbing, is classic and used to differentiate it from CARP. Other conditions to consider include seborrheic dermatitis, epidermal nevi, verruca plana, epidermodysplasia verruciformis, and acne vulgaris.1,2,4
Treatment
Minocycline is the first-line treatment for CARP: 80% of patients may have complete resolution with minocycline, while the remainder experience at least 50% clearance of skin lesions.2 However, recurrence after stopping minocycline treatment is not uncommon. The mechanism by which minocycline works is unknown. Second-line treatment for those who cannot tolerate minocycline are macrolide antibiotics.6 Other treatment options with reported success include oral isotretinoin and topical retinoids, including tretinoin gel and tazarotene cream.3,7 Appropriate strength topical corticosteroids may be used for pruritus.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego, as well as the vice chair of the department of dermatology and a professor of dermatology and pediatrics at UC San Diego. They report having no conflicts of interest or financial disclosures. Email them at [email protected].
References
1. Clin Cosmet Investig Dermatol. 2016 Aug 25;9:217-23.
2. Br J Dermatol. 2006 Feb;154(2):287-93.
3. Arch Dermatol. 2012 Apr;148(4):505-8.
4. J Am Acad Dermatol. 2003 Dec;49(6):1182-4.
5. Arch Dermatol. 2002 Feb;138(2):276-7.
6. J Am Acad Dermatol. 2001;44(4):652-5.
7. Am J Clin Dermatol. 2006;7(5):305-13.
A 17-year-old male presents to the dermatology clinic for brown lesions on his central chest and back that have been present for about a year. The brown areas gradually have become scaly over time. They are asymptomatic. His pediatrician had given him hydrocortisone ointment to apply to the lesions, but there was no improvement. Review of systems was otherwise negative.
Pediatric Dermatology Consult - November 2017
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at [email protected].
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.
15. Pediatrics. 2007 Nov;120(5):1079-87.
16. Arch Dis Child. 2010 Nov;95(11):877-82.
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at [email protected].
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.
15. Pediatrics. 2007 Nov;120(5):1079-87.
16. Arch Dis Child. 2010 Nov;95(11):877-82.
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at [email protected].
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.
15. Pediatrics. 2007 Nov;120(5):1079-87.
16. Arch Dis Child. 2010 Nov;95(11):877-82.
Clinical presentation
A healthy 9-year-old boy presents with 1 week history of a rash that began as “bruises” on both ankles that subsequently ascended over a few days to the proximal lower extremities and upper extremities. The rash has been painful and pruritic at times. The patient’s mother reports regular application of hydrocortisone cream for itch and pain relief, and this has been somewhat successful.
The patient has a history of longstanding constipation and abdominal pain, but over the past week has reported abdominal pain that is different and more severe than his usual abdominal pain. This abdominal pain has limited oral intake over the past 2 days. The patient and family also report bilateral pain of the wrists and elbows, which has limited his daily activities. The patient and mother deny fevers, chills, cough, coryza, and any sick contacts.
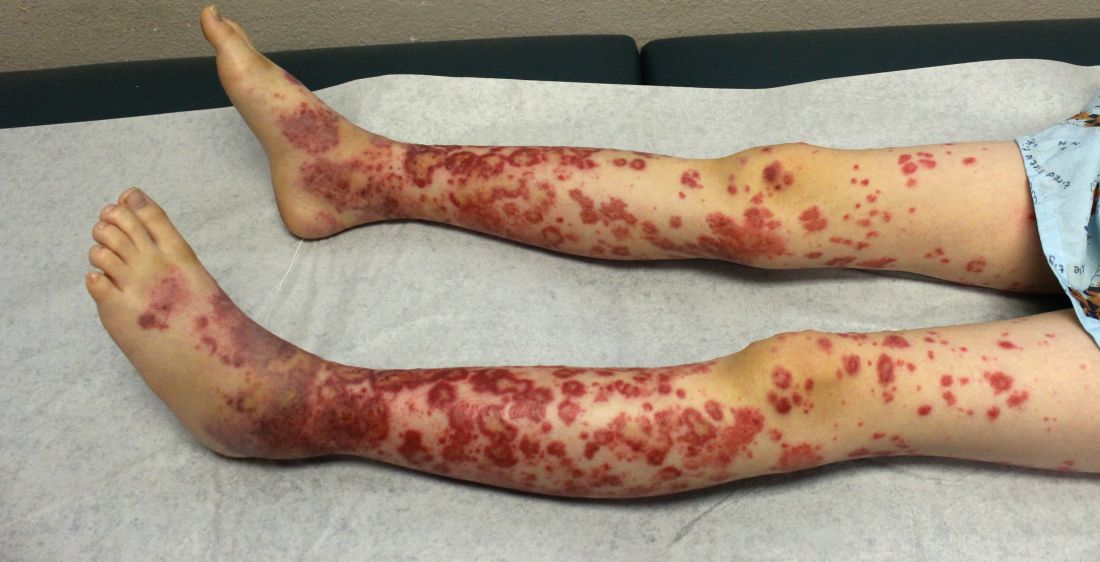
His vital signs are stable. On physical examination there is mild conjunctival injection, no intraoral lesions, and no lymphadenopathy or hepatosplenomegaly. The abdomen is not distended but it is tender to deep palpation. Bowel sounds are present. On skin examination, there are multiple purpuric annular plaques with central clearing, some with bullae and petechiae, on the bilateral buttocks and legs. There is bilateral pedal edema. On the arms, there are a few polymorphic pink and red annular to targetoid plaques.
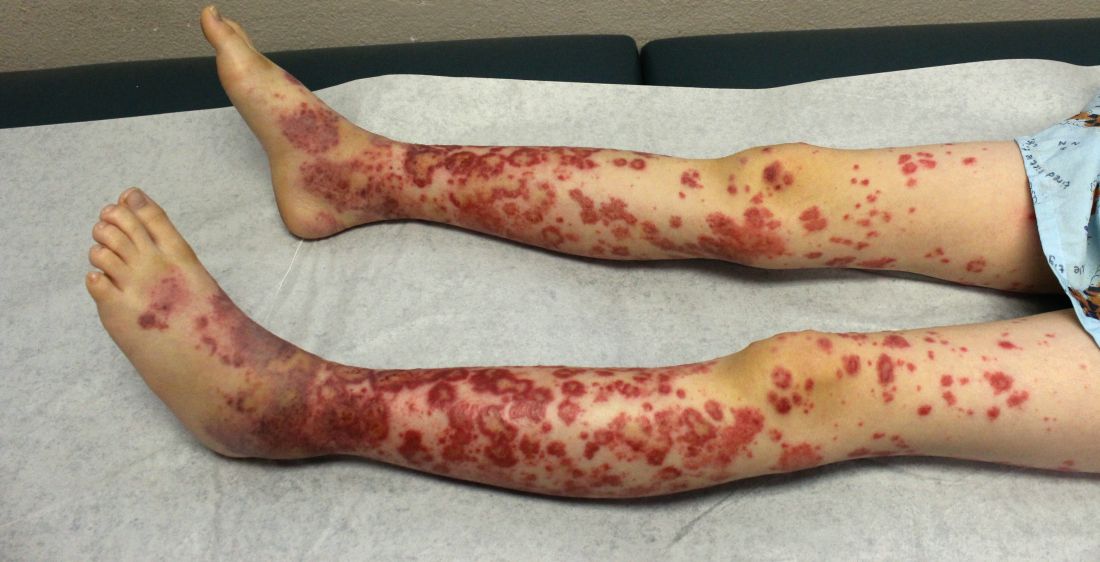
Pediatric Dermatology Consult - October 2017
Pyogenic granuloma
BY ALLISON HAN AND LAWRENCE F. EICHENFIELD, MD
Frontline Medical News
Pyogenic granuloma (PG), also known as lobular capillary hemangioma, is a benign, acquired vascular neoplasm of the skin and mucous membranes, and is fairly commonly found in children.1 Though most variants are superficial, PGs rarely may be discovered in subcutaneous, dermal, or satellite locations as well.2 Despite its name, no infectious or granulomatous disorder association with the vascular tumor has been discovered.
PGs are characterized by small, erythematous to purple papules that rapidly grow to become pedunculated or sessile within weeks, and then stabilize. They may bleed profusely because of their friable vascular nature, ulcerate, and crust.3 They rarely resolve on their own, thus, removal frequently is sought.4 PGs most commonly develop on the head and neck, and slightly less commonly on the fingers, periungual areas, upper extremities, lips, gingiva, and conjunctiva. Most PGs grow to about 1 to 10 mm.5
Histologically, PGs are organized in a characteristic lobular pattern of hyperplastic capillary clusters after which they are named (lobular capillary hemangioma). Fibrous bands separate the lobules. Mitotic activity is often seen, likely reflecting the rapid growth of the tumor.2 Immunohistochemistry staining is notably negative for glucose transporter 1 (GLUT-1), unlike hemangiomas, which aids in differentiating the two lesions.6
The pathogenesis of PGs is poorly understood; some hypothesize that a traumatic event, such as an insect bite, may trigger aberrant overgrowth of granulation tissue, but this theory is contested as many PGs appear to arise de novo.5 PGs have a tendency to develop within vascular lesions, suggesting an angiogenic stimuli of development, and increased expression of angiogenic growth factors (vascular endothelial growth factor and mitogen-activated protein kinase pathways) have been found in PGs.7,8
PGs also arise frequently during pregnancy and resolve after childbirth, leading many to support a causal role of estrogen in PG development. A link between both steroid hormone and angiogenesis may exist, as a beta isoform of estrogen receptor has been discovered to have consistent expression in a variety of vascular proliferations, possibly underlying PG pathogenesis.2
Drug-induced cases of PGs have been reported as well, with use of systemic medications such as acitretin, human immunodeficiency virus protease inhibitors, and epidermal growth factor receptor inhibitors.9
Diagnosis and differential
The diagnosis of PGs is based on the characteristic history of a rapidly growing, red to purple nodule that frequently bleeds and often ulcerates, accompanied by the physical findings of a typical exophytic, friable, vascular lesion. Patients often present with the “band aid sign,” an irritated portion of skin from the adhesive of bandages worn to protect the bleeding lesion.
The classic history of profuse and recurrent bleeding with little agitation often enables differentiation of PG from other disorders. In young infants, PGs may look similar to infantile hemangiomas, however hemangiomas regress and involute spontaneously unlike PGs.10 Other disorders that may be differentiated from PGs by history include Spitz nevi, glomus tumors, and warts. Melanoma in children may be amelanotic and present as red papules or nodules as well.11 An association between Bartonella infection and PGs had been questioned in the past because of reported increased rates of Bartonella seropositivity in patients, but this has since been debunked.12
Treatment
Almost all PGs undergo treatment; 85%-90% of PGs are treated by localized numbing, followed by superficial excision and electrodessication of the base.13 This technique allows the PG to be removed in less than 5 minutes and avoids suture use. Alternative treatments include surgical excision, pulsed dye laser, cryotherapy with liquid nitrogen, timolol solution, imiquimod, or a combination of methods.3,4,5 Methods that produce tissue for histopathologic analysis are ideal to rule out malignancy or other lesions that may mimic PGs such as Spitz nevi. Pulsed dye laser offers low recurrence, but often requires multiple treatments, and young children may not tolerate the treatment well. Cryotherapy has few side effects, but has higher recurrence rates and requires multiple treatment sessions.14
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego, as well as vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Allison Han is a medical student at the university. Neither Dr. Eichenfield nor Ms. Han have any relevant financial disclosures. Email them at [email protected].
References
1. Br J Dermatol. 2014;171(3):466-73.
2. Am J Dermatopathol. 2007;29(4):408-11.
3. J Plast Reconstr Aesthet Surg. 2011;64(9):1216-20.
4. Br J Dermatol. 2014;171(6):1537-8.
5. Pediatr Dermatol. 2004;21(1):10-3.
6. Pediatr Dermatol. 2009;26(3):323-7.
7. J Am Acad Dermatol. 2000;42(2 Pt 1):275-9.
8. J Am Acad Dermatol. 2001;44(2):193-7.
9. Br J Dermatol. 2010;163(5):941-53.
10. Pediatrics. 1992;90(6):989-91.
11. J Am Acad Dermatol. 2013;68(6):913-25.
12. J Am Acad Dermatol. 2005;53(6):1065-6.
13. “Hurwitz Clinical Pediatric Dermatology,” 4th Edition: A Textbook of Skin Disorders of Childhood and Adolescence (New York: Elsevier Health Sciences, 2011).
14. Dermatol Surg. 2013;39(8):1137-46.
Pyogenic granuloma
BY ALLISON HAN AND LAWRENCE F. EICHENFIELD, MD
Frontline Medical News
Pyogenic granuloma (PG), also known as lobular capillary hemangioma, is a benign, acquired vascular neoplasm of the skin and mucous membranes, and is fairly commonly found in children.1 Though most variants are superficial, PGs rarely may be discovered in subcutaneous, dermal, or satellite locations as well.2 Despite its name, no infectious or granulomatous disorder association with the vascular tumor has been discovered.
PGs are characterized by small, erythematous to purple papules that rapidly grow to become pedunculated or sessile within weeks, and then stabilize. They may bleed profusely because of their friable vascular nature, ulcerate, and crust.3 They rarely resolve on their own, thus, removal frequently is sought.4 PGs most commonly develop on the head and neck, and slightly less commonly on the fingers, periungual areas, upper extremities, lips, gingiva, and conjunctiva. Most PGs grow to about 1 to 10 mm.5
Histologically, PGs are organized in a characteristic lobular pattern of hyperplastic capillary clusters after which they are named (lobular capillary hemangioma). Fibrous bands separate the lobules. Mitotic activity is often seen, likely reflecting the rapid growth of the tumor.2 Immunohistochemistry staining is notably negative for glucose transporter 1 (GLUT-1), unlike hemangiomas, which aids in differentiating the two lesions.6
The pathogenesis of PGs is poorly understood; some hypothesize that a traumatic event, such as an insect bite, may trigger aberrant overgrowth of granulation tissue, but this theory is contested as many PGs appear to arise de novo.5 PGs have a tendency to develop within vascular lesions, suggesting an angiogenic stimuli of development, and increased expression of angiogenic growth factors (vascular endothelial growth factor and mitogen-activated protein kinase pathways) have been found in PGs.7,8
PGs also arise frequently during pregnancy and resolve after childbirth, leading many to support a causal role of estrogen in PG development. A link between both steroid hormone and angiogenesis may exist, as a beta isoform of estrogen receptor has been discovered to have consistent expression in a variety of vascular proliferations, possibly underlying PG pathogenesis.2
Drug-induced cases of PGs have been reported as well, with use of systemic medications such as acitretin, human immunodeficiency virus protease inhibitors, and epidermal growth factor receptor inhibitors.9
Diagnosis and differential
The diagnosis of PGs is based on the characteristic history of a rapidly growing, red to purple nodule that frequently bleeds and often ulcerates, accompanied by the physical findings of a typical exophytic, friable, vascular lesion. Patients often present with the “band aid sign,” an irritated portion of skin from the adhesive of bandages worn to protect the bleeding lesion.
The classic history of profuse and recurrent bleeding with little agitation often enables differentiation of PG from other disorders. In young infants, PGs may look similar to infantile hemangiomas, however hemangiomas regress and involute spontaneously unlike PGs.10 Other disorders that may be differentiated from PGs by history include Spitz nevi, glomus tumors, and warts. Melanoma in children may be amelanotic and present as red papules or nodules as well.11 An association between Bartonella infection and PGs had been questioned in the past because of reported increased rates of Bartonella seropositivity in patients, but this has since been debunked.12
Treatment
Almost all PGs undergo treatment; 85%-90% of PGs are treated by localized numbing, followed by superficial excision and electrodessication of the base.13 This technique allows the PG to be removed in less than 5 minutes and avoids suture use. Alternative treatments include surgical excision, pulsed dye laser, cryotherapy with liquid nitrogen, timolol solution, imiquimod, or a combination of methods.3,4,5 Methods that produce tissue for histopathologic analysis are ideal to rule out malignancy or other lesions that may mimic PGs such as Spitz nevi. Pulsed dye laser offers low recurrence, but often requires multiple treatments, and young children may not tolerate the treatment well. Cryotherapy has few side effects, but has higher recurrence rates and requires multiple treatment sessions.14
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego, as well as vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Allison Han is a medical student at the university. Neither Dr. Eichenfield nor Ms. Han have any relevant financial disclosures. Email them at [email protected].
References
1. Br J Dermatol. 2014;171(3):466-73.
2. Am J Dermatopathol. 2007;29(4):408-11.
3. J Plast Reconstr Aesthet Surg. 2011;64(9):1216-20.
4. Br J Dermatol. 2014;171(6):1537-8.
5. Pediatr Dermatol. 2004;21(1):10-3.
6. Pediatr Dermatol. 2009;26(3):323-7.
7. J Am Acad Dermatol. 2000;42(2 Pt 1):275-9.
8. J Am Acad Dermatol. 2001;44(2):193-7.
9. Br J Dermatol. 2010;163(5):941-53.
10. Pediatrics. 1992;90(6):989-91.
11. J Am Acad Dermatol. 2013;68(6):913-25.
12. J Am Acad Dermatol. 2005;53(6):1065-6.
13. “Hurwitz Clinical Pediatric Dermatology,” 4th Edition: A Textbook of Skin Disorders of Childhood and Adolescence (New York: Elsevier Health Sciences, 2011).
14. Dermatol Surg. 2013;39(8):1137-46.
Pyogenic granuloma
BY ALLISON HAN AND LAWRENCE F. EICHENFIELD, MD
Frontline Medical News
Pyogenic granuloma (PG), also known as lobular capillary hemangioma, is a benign, acquired vascular neoplasm of the skin and mucous membranes, and is fairly commonly found in children.1 Though most variants are superficial, PGs rarely may be discovered in subcutaneous, dermal, or satellite locations as well.2 Despite its name, no infectious or granulomatous disorder association with the vascular tumor has been discovered.
PGs are characterized by small, erythematous to purple papules that rapidly grow to become pedunculated or sessile within weeks, and then stabilize. They may bleed profusely because of their friable vascular nature, ulcerate, and crust.3 They rarely resolve on their own, thus, removal frequently is sought.4 PGs most commonly develop on the head and neck, and slightly less commonly on the fingers, periungual areas, upper extremities, lips, gingiva, and conjunctiva. Most PGs grow to about 1 to 10 mm.5
Histologically, PGs are organized in a characteristic lobular pattern of hyperplastic capillary clusters after which they are named (lobular capillary hemangioma). Fibrous bands separate the lobules. Mitotic activity is often seen, likely reflecting the rapid growth of the tumor.2 Immunohistochemistry staining is notably negative for glucose transporter 1 (GLUT-1), unlike hemangiomas, which aids in differentiating the two lesions.6
The pathogenesis of PGs is poorly understood; some hypothesize that a traumatic event, such as an insect bite, may trigger aberrant overgrowth of granulation tissue, but this theory is contested as many PGs appear to arise de novo.5 PGs have a tendency to develop within vascular lesions, suggesting an angiogenic stimuli of development, and increased expression of angiogenic growth factors (vascular endothelial growth factor and mitogen-activated protein kinase pathways) have been found in PGs.7,8
PGs also arise frequently during pregnancy and resolve after childbirth, leading many to support a causal role of estrogen in PG development. A link between both steroid hormone and angiogenesis may exist, as a beta isoform of estrogen receptor has been discovered to have consistent expression in a variety of vascular proliferations, possibly underlying PG pathogenesis.2
Drug-induced cases of PGs have been reported as well, with use of systemic medications such as acitretin, human immunodeficiency virus protease inhibitors, and epidermal growth factor receptor inhibitors.9
Diagnosis and differential
The diagnosis of PGs is based on the characteristic history of a rapidly growing, red to purple nodule that frequently bleeds and often ulcerates, accompanied by the physical findings of a typical exophytic, friable, vascular lesion. Patients often present with the “band aid sign,” an irritated portion of skin from the adhesive of bandages worn to protect the bleeding lesion.
The classic history of profuse and recurrent bleeding with little agitation often enables differentiation of PG from other disorders. In young infants, PGs may look similar to infantile hemangiomas, however hemangiomas regress and involute spontaneously unlike PGs.10 Other disorders that may be differentiated from PGs by history include Spitz nevi, glomus tumors, and warts. Melanoma in children may be amelanotic and present as red papules or nodules as well.11 An association between Bartonella infection and PGs had been questioned in the past because of reported increased rates of Bartonella seropositivity in patients, but this has since been debunked.12
Treatment
Almost all PGs undergo treatment; 85%-90% of PGs are treated by localized numbing, followed by superficial excision and electrodessication of the base.13 This technique allows the PG to be removed in less than 5 minutes and avoids suture use. Alternative treatments include surgical excision, pulsed dye laser, cryotherapy with liquid nitrogen, timolol solution, imiquimod, or a combination of methods.3,4,5 Methods that produce tissue for histopathologic analysis are ideal to rule out malignancy or other lesions that may mimic PGs such as Spitz nevi. Pulsed dye laser offers low recurrence, but often requires multiple treatments, and young children may not tolerate the treatment well. Cryotherapy has few side effects, but has higher recurrence rates and requires multiple treatment sessions.14
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego, as well as vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Allison Han is a medical student at the university. Neither Dr. Eichenfield nor Ms. Han have any relevant financial disclosures. Email them at [email protected].
References
1. Br J Dermatol. 2014;171(3):466-73.
2. Am J Dermatopathol. 2007;29(4):408-11.
3. J Plast Reconstr Aesthet Surg. 2011;64(9):1216-20.
4. Br J Dermatol. 2014;171(6):1537-8.
5. Pediatr Dermatol. 2004;21(1):10-3.
6. Pediatr Dermatol. 2009;26(3):323-7.
7. J Am Acad Dermatol. 2000;42(2 Pt 1):275-9.
8. J Am Acad Dermatol. 2001;44(2):193-7.
9. Br J Dermatol. 2010;163(5):941-53.
10. Pediatrics. 1992;90(6):989-91.
11. J Am Acad Dermatol. 2013;68(6):913-25.
12. J Am Acad Dermatol. 2005;53(6):1065-6.
13. “Hurwitz Clinical Pediatric Dermatology,” 4th Edition: A Textbook of Skin Disorders of Childhood and Adolescence (New York: Elsevier Health Sciences, 2011).
14. Dermatol Surg. 2013;39(8):1137-46.
A 7-year-old healthy female presents to the dermatology clinic for evaluation of a bump on her lower lip that has been present for 3 months. It started as a small red papule and within a couple months rapidly increased in size, but now has stabilized. She denies pain and itching, but she notes that it frequently bleeds, and that the bleeding can be difficult to stop. She denies any trauma to the area. Review of systems was otherwise negative.
On examination, the patient is a well-developed, healthy appearing female. In the middle of her lower lip, there is a 1-cm pedunculated, raspberry-like exophytic nodule. It is reddish-purple with slight ulceration at the surface. There is no active bleeding. The remainder of the physical examination is normal.
Pediatric Dermatology Consult - August 2017
BY AYAN KUSARI AND CATALINA MATIZ, MD
The patient was diagnosed with eruptive vellus hair cysts (EVHC). Treatment with a keratolytic, such as 12% lactic acid cream, was recommended. Hydrocortisone 2.5% once daily as needed also was recommended to treat the patient’s itch.
EVHC are benign middermal cysts characterized by epidermoid keratinization of the cyst wall, as well as lamellar keratin and vellus hairs within the cyst.1 The term “eruptive vellus hair cysts” was first used to describe a longstanding hyperpigmented, monomorphous papular eruption in two children by Esterly, Fretzin, and Pinkus in 1977.2 Clinically, EVHC present as 1- to 3-mm follicular, dome-shaped papules that are often skin-colored but also have been described as being brown, gray, green or black colored.3,4 They appear suddenly and sometimes are associated with mild tenderness and pruritus.1,5 EVHC most commonly present on the anterior chest but also can present on the upper and lower extremities, face, neck, axillae, and buttocks.4
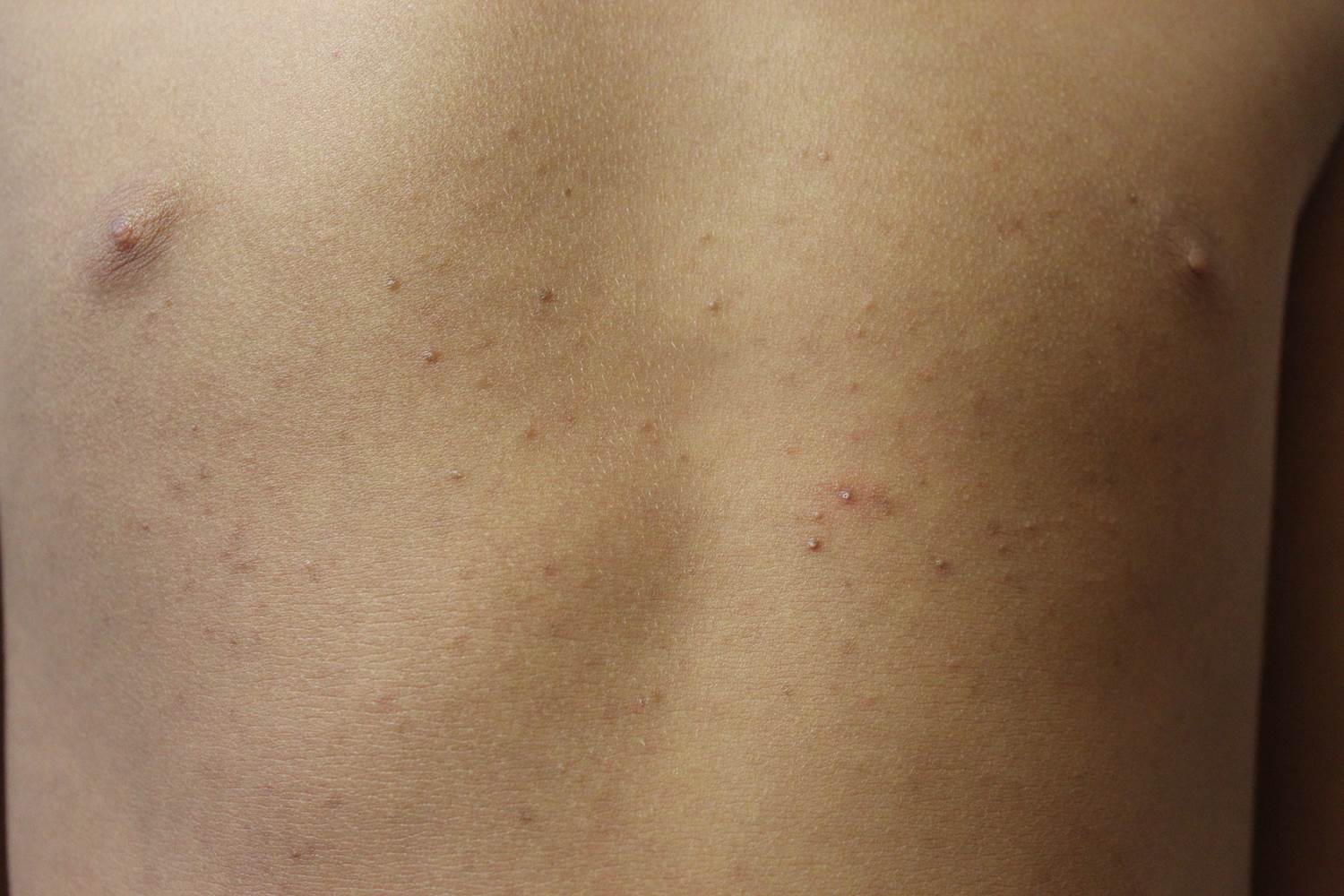
Furthermore, although spontaneous resolution is possible through transepidermal elimination of cyst products, cases may persist for years in the absence of treatment.1
Accurate diagnosis of eruptive vellus hair cysts is important to guide therapy.
Keratosis pilaris consists of follicular-based papules with variable erythema.4 It may be widespread – including over the anterior chest – but is most commonly seen on the cheeks, extensor surfaces of proximal upper extremities, and the anterior thighs.4 It is related to excessive keratinization, which leads to formation of horny plugs within hair-follicle orifices.1
Steatocystoma multiplex is typically characterized by firm, yellow-to-flesh–colored dermal cysts ranging from a few millimeters to 1 cm in size.1 They are sometimes clinically hard to distinguish from EVHC, and both are associated with keratin 17 gene mutations and type 2 pachyonychia congenita.1 Nonetheless, this patient’s lesions did not have any features – such as size or drainage – that would point toward steatocystoma multiplex or other skin findings suggestive of pachyonychia congenita.
Superficial folliculitis, also known as Bockhart’s impetigo, is an infection of the follicular ostium and typically presents with perifollicular pustules on an erythematous base that may be painful or pruritic and can occur throughout the corpus, including the anterior trunk.1
Acne vulgaris is a very common disease that involves the pilosebaceous unit and occurs most frequently on the face, back, upper arms, and chest. However, it is characterized by open and closed comedones, papules, and pustules and, in severe cases, nodules and cysts that may leave postinflammatory hyperpigmentation and scarring. Tiny, hyperpigmented, dome-shaped macules occurring exclusively on the chest would not be characteristic.
Patients with hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome, can present with EVHC. This condition is characterized by a triad of fair, sparse short hair; hyperthermia related to decreased sweating; and missing teeth.4 Although EVHC have been reported in association with hypohidrotic ectodermal dysplasia, this patient does not have any of the dysmorphic features associated with this syndrome.11
Patients with pachyonychia congenita (type 2) also may have EVHC as part of their presentation, but this patient does not have nail dystrophy, focal palmoplantar keratoderma, follicular keratoses, or multiple steatocysts which also are features of this condition.4
Treatment may be offered to patients who are distressed by the lesions or seek cosmesis. A 2012 review of 220 cases of EVHC found that topical retinoic acid, incision/excision, CO2 laser, erbium:yttrium-aluminum-garnet laser, needle evacuation, dermabrasion, and 10% urea cream were each associated with successful treatment in multiple cases.3
Forty years have passed since EVHC was identified as a distinct disease entity. Despite this, eruptive vellus hair cysts remains somewhat understudied, and further research is needed to determine an ideal treatment algorithm for patients with this condition. Our approach was to attempt noninvasive keratolytic therapy before considering retinoids or surgical options; we also recommended steroid treatment for symptom relief. Providers should keep EVHC in the differential for eruptions consisting of tiny papules so that appropriate treatment may be offered.
Dr. Matiz is a pediatric dermatologist at Rady Children’s Hospital, San Diego, and an assistant clinical professor in the department of pediatric and adolescent dermatology at the University of California, San Diego. Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures.
Email them at [email protected].
References
1. “Dermatology.” 3rd ed. (Philadelphia: Saunders, 2012).
2. Arch Dermatol. 1977 Apr;113(4):500-3.
3. Am J Clin Dermatol. 2012 Feb 1;13(1):19-28.
5. Indian Dermatol Online J. 2013 Jul;4(3):213-5.
6. J Am Acad Dermatol. 1980 Oct;3(4):425-9.
7. Am J Dermatopathol. 1997 Jun;19(3):250-3.
8. Hum Mol Genet. 1998 Jul;7(7):1143-8.
9. Dermatology. 1998;196(4):392-6.
BY AYAN KUSARI AND CATALINA MATIZ, MD
The patient was diagnosed with eruptive vellus hair cysts (EVHC). Treatment with a keratolytic, such as 12% lactic acid cream, was recommended. Hydrocortisone 2.5% once daily as needed also was recommended to treat the patient’s itch.
EVHC are benign middermal cysts characterized by epidermoid keratinization of the cyst wall, as well as lamellar keratin and vellus hairs within the cyst.1 The term “eruptive vellus hair cysts” was first used to describe a longstanding hyperpigmented, monomorphous papular eruption in two children by Esterly, Fretzin, and Pinkus in 1977.2 Clinically, EVHC present as 1- to 3-mm follicular, dome-shaped papules that are often skin-colored but also have been described as being brown, gray, green or black colored.3,4 They appear suddenly and sometimes are associated with mild tenderness and pruritus.1,5 EVHC most commonly present on the anterior chest but also can present on the upper and lower extremities, face, neck, axillae, and buttocks.4
Furthermore, although spontaneous resolution is possible through transepidermal elimination of cyst products, cases may persist for years in the absence of treatment.1
Accurate diagnosis of eruptive vellus hair cysts is important to guide therapy.
Keratosis pilaris consists of follicular-based papules with variable erythema.4 It may be widespread – including over the anterior chest – but is most commonly seen on the cheeks, extensor surfaces of proximal upper extremities, and the anterior thighs.4 It is related to excessive keratinization, which leads to formation of horny plugs within hair-follicle orifices.1
Steatocystoma multiplex is typically characterized by firm, yellow-to-flesh–colored dermal cysts ranging from a few millimeters to 1 cm in size.1 They are sometimes clinically hard to distinguish from EVHC, and both are associated with keratin 17 gene mutations and type 2 pachyonychia congenita.1 Nonetheless, this patient’s lesions did not have any features – such as size or drainage – that would point toward steatocystoma multiplex or other skin findings suggestive of pachyonychia congenita.
Superficial folliculitis, also known as Bockhart’s impetigo, is an infection of the follicular ostium and typically presents with perifollicular pustules on an erythematous base that may be painful or pruritic and can occur throughout the corpus, including the anterior trunk.1
Acne vulgaris is a very common disease that involves the pilosebaceous unit and occurs most frequently on the face, back, upper arms, and chest. However, it is characterized by open and closed comedones, papules, and pustules and, in severe cases, nodules and cysts that may leave postinflammatory hyperpigmentation and scarring. Tiny, hyperpigmented, dome-shaped macules occurring exclusively on the chest would not be characteristic.
Patients with hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome, can present with EVHC. This condition is characterized by a triad of fair, sparse short hair; hyperthermia related to decreased sweating; and missing teeth.4 Although EVHC have been reported in association with hypohidrotic ectodermal dysplasia, this patient does not have any of the dysmorphic features associated with this syndrome.11
Patients with pachyonychia congenita (type 2) also may have EVHC as part of their presentation, but this patient does not have nail dystrophy, focal palmoplantar keratoderma, follicular keratoses, or multiple steatocysts which also are features of this condition.4
Treatment may be offered to patients who are distressed by the lesions or seek cosmesis. A 2012 review of 220 cases of EVHC found that topical retinoic acid, incision/excision, CO2 laser, erbium:yttrium-aluminum-garnet laser, needle evacuation, dermabrasion, and 10% urea cream were each associated with successful treatment in multiple cases.3
Forty years have passed since EVHC was identified as a distinct disease entity. Despite this, eruptive vellus hair cysts remains somewhat understudied, and further research is needed to determine an ideal treatment algorithm for patients with this condition. Our approach was to attempt noninvasive keratolytic therapy before considering retinoids or surgical options; we also recommended steroid treatment for symptom relief. Providers should keep EVHC in the differential for eruptions consisting of tiny papules so that appropriate treatment may be offered.
Dr. Matiz is a pediatric dermatologist at Rady Children’s Hospital, San Diego, and an assistant clinical professor in the department of pediatric and adolescent dermatology at the University of California, San Diego. Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures.
Email them at [email protected].
References
1. “Dermatology.” 3rd ed. (Philadelphia: Saunders, 2012).
2. Arch Dermatol. 1977 Apr;113(4):500-3.
3. Am J Clin Dermatol. 2012 Feb 1;13(1):19-28.
5. Indian Dermatol Online J. 2013 Jul;4(3):213-5.
6. J Am Acad Dermatol. 1980 Oct;3(4):425-9.
7. Am J Dermatopathol. 1997 Jun;19(3):250-3.
8. Hum Mol Genet. 1998 Jul;7(7):1143-8.
9. Dermatology. 1998;196(4):392-6.
BY AYAN KUSARI AND CATALINA MATIZ, MD
The patient was diagnosed with eruptive vellus hair cysts (EVHC). Treatment with a keratolytic, such as 12% lactic acid cream, was recommended. Hydrocortisone 2.5% once daily as needed also was recommended to treat the patient’s itch.
EVHC are benign middermal cysts characterized by epidermoid keratinization of the cyst wall, as well as lamellar keratin and vellus hairs within the cyst.1 The term “eruptive vellus hair cysts” was first used to describe a longstanding hyperpigmented, monomorphous papular eruption in two children by Esterly, Fretzin, and Pinkus in 1977.2 Clinically, EVHC present as 1- to 3-mm follicular, dome-shaped papules that are often skin-colored but also have been described as being brown, gray, green or black colored.3,4 They appear suddenly and sometimes are associated with mild tenderness and pruritus.1,5 EVHC most commonly present on the anterior chest but also can present on the upper and lower extremities, face, neck, axillae, and buttocks.4
Furthermore, although spontaneous resolution is possible through transepidermal elimination of cyst products, cases may persist for years in the absence of treatment.1
Accurate diagnosis of eruptive vellus hair cysts is important to guide therapy.
Keratosis pilaris consists of follicular-based papules with variable erythema.4 It may be widespread – including over the anterior chest – but is most commonly seen on the cheeks, extensor surfaces of proximal upper extremities, and the anterior thighs.4 It is related to excessive keratinization, which leads to formation of horny plugs within hair-follicle orifices.1
Steatocystoma multiplex is typically characterized by firm, yellow-to-flesh–colored dermal cysts ranging from a few millimeters to 1 cm in size.1 They are sometimes clinically hard to distinguish from EVHC, and both are associated with keratin 17 gene mutations and type 2 pachyonychia congenita.1 Nonetheless, this patient’s lesions did not have any features – such as size or drainage – that would point toward steatocystoma multiplex or other skin findings suggestive of pachyonychia congenita.
Superficial folliculitis, also known as Bockhart’s impetigo, is an infection of the follicular ostium and typically presents with perifollicular pustules on an erythematous base that may be painful or pruritic and can occur throughout the corpus, including the anterior trunk.1
Acne vulgaris is a very common disease that involves the pilosebaceous unit and occurs most frequently on the face, back, upper arms, and chest. However, it is characterized by open and closed comedones, papules, and pustules and, in severe cases, nodules and cysts that may leave postinflammatory hyperpigmentation and scarring. Tiny, hyperpigmented, dome-shaped macules occurring exclusively on the chest would not be characteristic.
Patients with hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome, can present with EVHC. This condition is characterized by a triad of fair, sparse short hair; hyperthermia related to decreased sweating; and missing teeth.4 Although EVHC have been reported in association with hypohidrotic ectodermal dysplasia, this patient does not have any of the dysmorphic features associated with this syndrome.11
Patients with pachyonychia congenita (type 2) also may have EVHC as part of their presentation, but this patient does not have nail dystrophy, focal palmoplantar keratoderma, follicular keratoses, or multiple steatocysts which also are features of this condition.4
Treatment may be offered to patients who are distressed by the lesions or seek cosmesis. A 2012 review of 220 cases of EVHC found that topical retinoic acid, incision/excision, CO2 laser, erbium:yttrium-aluminum-garnet laser, needle evacuation, dermabrasion, and 10% urea cream were each associated with successful treatment in multiple cases.3
Forty years have passed since EVHC was identified as a distinct disease entity. Despite this, eruptive vellus hair cysts remains somewhat understudied, and further research is needed to determine an ideal treatment algorithm for patients with this condition. Our approach was to attempt noninvasive keratolytic therapy before considering retinoids or surgical options; we also recommended steroid treatment for symptom relief. Providers should keep EVHC in the differential for eruptions consisting of tiny papules so that appropriate treatment may be offered.
Dr. Matiz is a pediatric dermatologist at Rady Children’s Hospital, San Diego, and an assistant clinical professor in the department of pediatric and adolescent dermatology at the University of California, San Diego. Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures.
Email them at [email protected].
References
1. “Dermatology.” 3rd ed. (Philadelphia: Saunders, 2012).
2. Arch Dermatol. 1977 Apr;113(4):500-3.
3. Am J Clin Dermatol. 2012 Feb 1;13(1):19-28.
5. Indian Dermatol Online J. 2013 Jul;4(3):213-5.
6. J Am Acad Dermatol. 1980 Oct;3(4):425-9.
7. Am J Dermatopathol. 1997 Jun;19(3):250-3.
8. Hum Mol Genet. 1998 Jul;7(7):1143-8.
9. Dermatology. 1998;196(4):392-6.
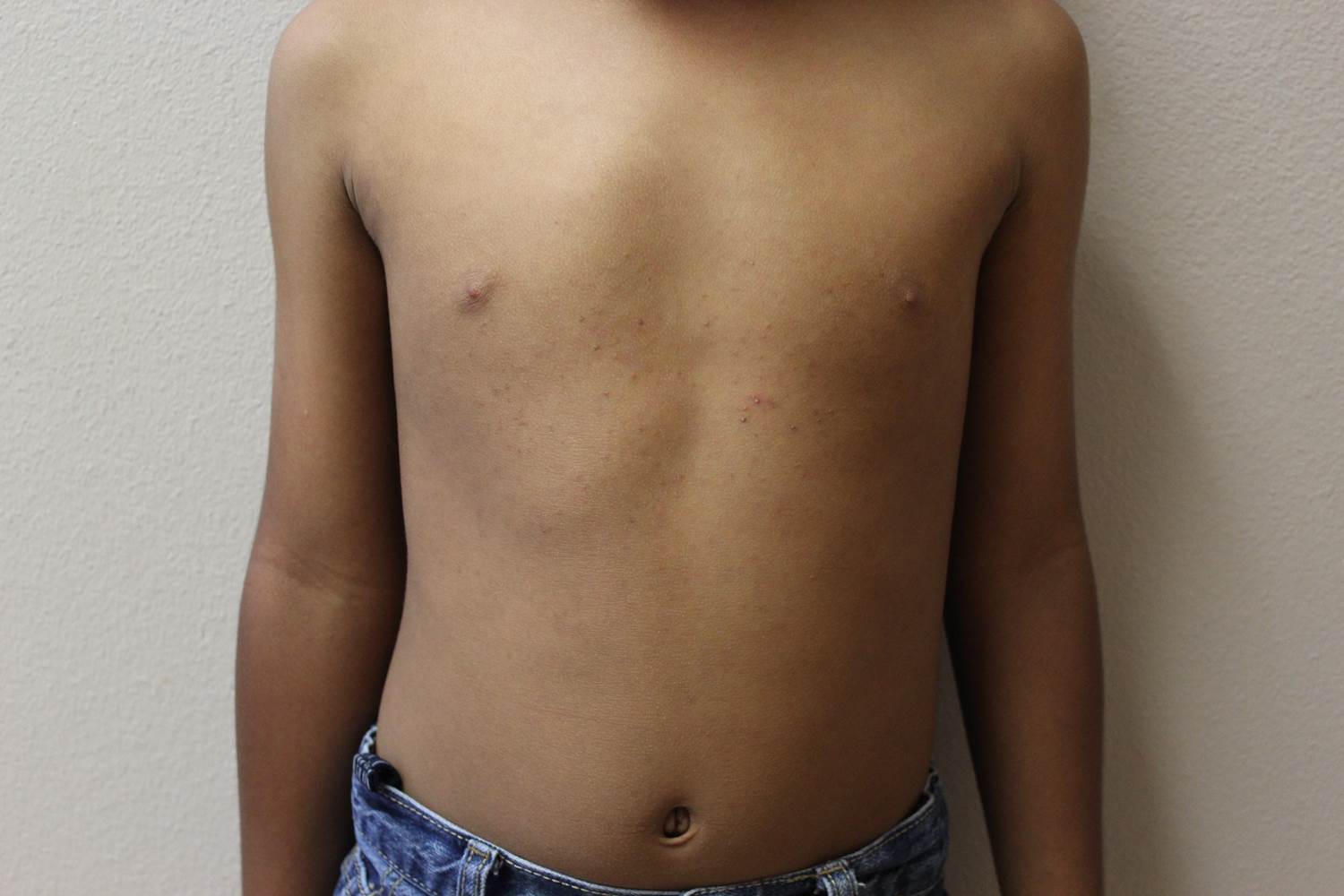
A 6-year-old boy presents with bumps on his chest and lower abdomen that have been present for 6 months. The patient’s mother states that the bumps are occasionally pruritic but not painful. She reports that the bumps first appeared on the chest and subsequently spread downward to involve the upper abdomen.
The patient is otherwise healthy. No similar lesions are present beyond the trunk. The patient’s past medical history and developmental history are unremarkable aside from bilateral amblyopia and high myopia. The patient’s mother denies any other family members with similar lesions. There is no history of teeth or nail abnormalities.
On exam, you find symmetrically distributed, firm, nontender, tiny 1- to 2-mm hyperpigmented dome-shaped papules on the anterior chest with no similar lesions elsewhere on the body. The remainder of the physical exam discloses no abnormalities.
50 years of pediatric dermatology
The world in pediatric dermatology has changed in incredible ways since 1967. In fact, pediatric dermatology was not an organized specialty until years later! This article will look back at some of the history of pediatric dermatology, exploring how different the field was 50 years ago, and how it has evolved into the vibrant field that it is. By looking at some disease states, and differences in practice in relation to the care of dermatologic conditions in children both by pediatricians and dermatologists, we can see the tremendous evolution in our understanding and management of pediatric skin conditions, and perhaps gain insight into the future.
Pediatric dermatology was fairly “neonatal” 50 years ago, with only a few practitioners in the field. Recognizing that up to 30% of pediatric primary care visits include a skin-related problem, and that there was limited training about skin diseases among primary care practitioners and inconsistent training amongst dermatologists, there was a clinical need for establishing the subspecialty of pediatric dermatology. The first international symposium was held in Mexico City in October 1972, and with this meeting the International Society of Pediatric Dermatology was founded. The Society for Pediatric Dermatology (SPD) began in 1973, with Alvin Jacobs, MD, Samuel Weinberg, MD, Nancy Esterly, MD, Sidney Hurwitz, MD, William Weston, MD, and Coleman Jacobson, MD, as some of the initial “founding mothers and fathers.” The journal Pediatric Dermatology released its first issue in 1982 (35 years ago), and the American Academy of Pediatrics did not have a section of dermatology until 1986.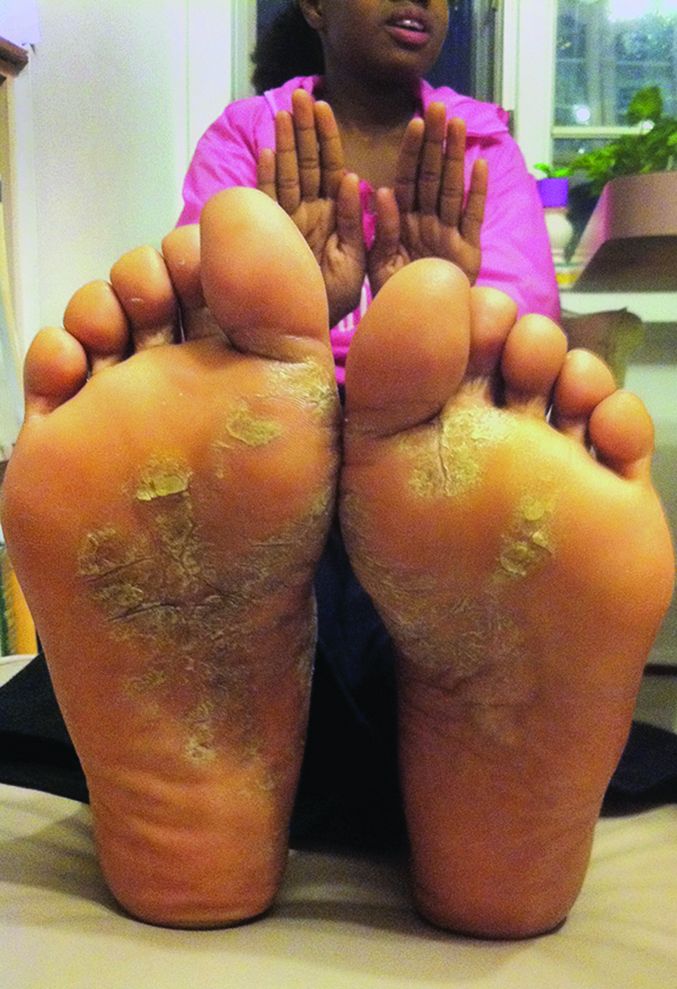
Pediatrics and dermatology: The interface
Many of the first generation of pediatric dermatologists trained as pediatricians prior to pursuing their dermatology work, with some being “assigned” dermatology as pediatric experts, while others did formal residencies in dermatology. This history is important, as pediatric dermatology was, and remains, integrated with pediatrics, even while training in dermatology residencies became standard practice. An important part of the development of the field has been the education of pediatricians and dermatologists by pediatric dermatologists, with a strong sensibility that improved training for both generalists and specialists about pediatric skin disease would yield better care for patients and families.
Initially, there were very few pediatric or dermatology programs in the United States that had pediatric dermatologists. Over the succeeding decades, this is now less common, although even now there are still dermatology and pediatric residency programs that do not have a pediatric dermatologist for either training or to serve their patients. The founding leaders of the SPD set a tone of collaboration nationally and internationally, reaching out to pediatric colleagues and dermatology associates from around the world, and establishing superb educational programs for the exchange of ideas, presentation of challenging cases, and promoting state of the art knowledge of the field. Through annual meetings of the SPD, conferences immediately preceding the American Academy of Dermatology annual meetings, the World Congress of Pediatric Dermatology, and other regional and international meetings, the field developed as the number of practitioners grew, and as the specialized published literature reflected new knowledge in diagnosis and therapy.
Building upon the history of collaboration and reflecting the maturation of the field with a desire to influence the breadth and quantity of research in pediatric dermatology, the Pediatric Dermatology Research Alliance (PeDRA) was formed in 2012. This organization was formed to promote and facilitate high quality collaborative clinical, translational, educational, and basic science research in pediatric dermatology with a vision to create sustainable, collaborative networks to better understand, prevent, treat, and cure dermatologic diseases in children. This network is now composed of over 230 members representing over 68 institutions from the United States and Canada, but including involvement globally from Mexico, Europe, and the Middle East.
Examples of changing perspectives: hemangiomas
A good way to look at evolution of the field is take a look at some of the similarities and differences in clinical practice in relation to common and uncommon disease states.
A great example is hemangiomas. Some of the first natural history studies on hemangiomas were done in the early 1960s, establishing that many lesions had a typical clinical course of fairly rapid growth, plateau, and involution over time. Of course, the identification of hemangiomas of infancy (or “HOI” in the trade), was confused with vascular malformations, and no one had recognized variant tumors that were distinct, such as rapidly involuting and noninvoluting congenital hemangiomas (RICHs or NICHs), tufted angiomas, and hemangioendotheliomas. PHACE syndrome (posterior fossa brain malformations) had yet to be described (that was done in 1996 by Ilona Frieden and her colleagues). For a time period, hemangiomas were treated with X-rays, before the negative impact of such radiation was acknowledged. For many years after that, even deforming and functionally significant lesions were “followed clinically” for natural involution, presumably a backlash from the radiation therapy interventions.
This story also reflects how organized research efforts helped with the evolution of knowledge and clinical care. The Hemangioma of Infancy Group was formed to take a collaborative approach to characterize and study hemangiomas and related tumors. Beginning with energetic, insightful pediatric dermatologists, and little funding, they changed our knowledge base of how hemangiomas present, the risk factors for their development and the characteristics and multiple organ findings associated with PHACE and other syndromic hemangiomas.
Procedural pediatric dermatology: Tremendous revolution in surgery and laser
The first generation of pediatric dermatologists were considered medical dermatologist specialists. And how important this specialty work was! Acne, atopic dermatitis, psoriasis, diaper and seborrheic dermatitis, and rare genetic syndromes, these conditions were a major part of the work of early pediatric dermatologists (and remain so now). What was not common was for pediatric dermatologists to have procedural or surgical practices, while this now is routinely part of the work of specialists in the field. How did this shift occur?
The fundamental shift began to occur with the introduction of the pulsed dye laser in 1989 and the publication of a seminal article in the New England Journal of Medicine (1989 Feb 16;320[7]:416-21) on its utility in treating port-wine stains in children with minimal scarring. Vascular lesions including port-wine stains were common, and pediatric dermatologists managed these patients for both diagnosis and medical management. Also, dermatology residencies at this time offered training in cutaneous surgery, excisions (including Mohs surgery) and repairs, and trainees in pediatric dermatology were “trained up” to high levels of expertise. As lasers were incorporated into dermatology residency work and practices, pediatric dermatologists had the exposure and skill to do this work. An added advantage was having the pediatric knowledge of how to handle children and adolescents in an age appropriate manner, and consideration of methods to minimize the pain and anxiety of procedures. Within a few years, pediatric dermatologists were at the forefront of the use of topical anesthetics (EMLA and liposomal lidocaine) and had general anesthesia privileges for laser and excisional surgery.
So while pediatric dermatologists still do “small procedures” every hour in most practices (cryotherapy for warts, cantharidin for molluscum, shave and punch biopsies), a subset now have extensive procedural practices, which in recent years has extended to pigment lesion lasers (to treat nevus of Ota), hair lasers (to treat perineal areas to prevent pilonidal cyst recurrence or to treat hirsutism), and combinations of lasers to treat hypertrophic, constrictive, and/or deforming scars).
Inflammatory skin disorders: Bread and butter ... and peanut butter?
The care of pediatric inflammatory skin disorders has evolved, but more slowly for some diseases than others. Acne vulgaris now is recognized as much more common under age 12 years than previously, presumably reflecting earlier pubertal changes in our preteens. Over the past 30 years, therapy has evolved with the use of topical retinoids (still underused by pediatricians, considered a “practice gap”), hormonal therapy with combined oral contraceptives, and oral isotretinoin, a powerful but highly effective systemic agent for severe and refractory acne. Specific pediatric guidelines came much later. Pediatric acne expert recommendations were formulated by the American Acne and Rosacea Society and endorsed by the American Academy of Pediatrics in 2013 (Pediatrics. 2013;131:S163-86). Over the past few years, there is a push by experts for more judicious use of antibiotics for acne (oral and topical) to minimize the emergence of bacterial resistance.
Psoriasis has been a condition that has been “behind the revolution,” in that no biologic agent was approved for pediatric psoriasis in the United States until several months ago, lagging behind Europe and elsewhere in the world by almost a decade. Adult psoriasis has been recognized to be associated with a broad set of comorbidities, including obesity and early heart disease, and there is now research on how children are at risk as well, and new recommendations on how to screen children with psoriasis. Moderate to severe psoriasis in adults is now tremendously controllable with biologic agents targeting TNF-alpha, IL 12/23, and IL-17. Etanercept has been approved for children with psoriasis aged 4 years and older, and other biologic agents are under study.
Atopic dermatitis now is ready for its revolution! AD has increased in prevalence from around 5% of the pediatric population 30-plus years ago to 10%-15%. Treatment of most individuals has remained the same over the decades: Good skin care, frequent moisturizers, topical corticosteroids for flares, management of infection if noted. The topical calcineurin inhibitors (TCIs) broadened the therapeutic approach when introduced in 2000 and 2001, but the boxed warning resulted in some practitioners minimizing their utilization of these useful agents.

It has been recognized for years that children with AD have higher risk of developing food allergies than children without AD. A changing understanding of how early food exposure may induce tolerance is changing the world of allergy and influencing the care of children with AD. This is where the peanut butter (or other processed peanut, such as “Bamba”) may be life saving. New guidelines have come from the National Institute of Allergy and Infectious Diseases recommending that infants with severe eczema (or egg allergy, or both) have introduction of age-appropriate peanut-containing food as early as 4-6 months of age to reduce the risk of development of peanut allergy. It is recommended that these infants undergo early evaluation for possible sensitization to peanut protein, with referral to allergists for skin prick tests or serum IgE screens (though if positive, referral to allergists is appropriate), and assess the safety of going ahead with early feeding. It is hoped that following these new guidelines can minimize the development of peanut allergy.
The future
Where will pediatric skin disease, or more importantly, skin health over a lifetime be in 50 years? Can we cure or prevent the consequences of our lethal and life altering genetic diseases such as epidermolysis bullosa or our neurocutaneous disorders? Will our new insights into birthmarks (they are mostly somatic mutations) allow us to form specific, personalized therapies to minimize their impact? Will we be using computers equipped with imaging devices and algorithms to assess our patients’ moles, papules, and nodules? Will our vaccines have wiped out warts, molluscum, and perhaps, acne? Will we have cured our inflammatory skin disorders, or perhaps prevented them by interventions in the neonatal period? No predictions will be offered here, other than that we can look forward to incredible changes for our future generations of health care practitioners, patients, and families.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Eichenfield has served as a consultant for Anacor/Pfizer and Regeneron/Sanofi. Email him at [email protected].
The world in pediatric dermatology has changed in incredible ways since 1967. In fact, pediatric dermatology was not an organized specialty until years later! This article will look back at some of the history of pediatric dermatology, exploring how different the field was 50 years ago, and how it has evolved into the vibrant field that it is. By looking at some disease states, and differences in practice in relation to the care of dermatologic conditions in children both by pediatricians and dermatologists, we can see the tremendous evolution in our understanding and management of pediatric skin conditions, and perhaps gain insight into the future.
Pediatric dermatology was fairly “neonatal” 50 years ago, with only a few practitioners in the field. Recognizing that up to 30% of pediatric primary care visits include a skin-related problem, and that there was limited training about skin diseases among primary care practitioners and inconsistent training amongst dermatologists, there was a clinical need for establishing the subspecialty of pediatric dermatology. The first international symposium was held in Mexico City in October 1972, and with this meeting the International Society of Pediatric Dermatology was founded. The Society for Pediatric Dermatology (SPD) began in 1973, with Alvin Jacobs, MD, Samuel Weinberg, MD, Nancy Esterly, MD, Sidney Hurwitz, MD, William Weston, MD, and Coleman Jacobson, MD, as some of the initial “founding mothers and fathers.” The journal Pediatric Dermatology released its first issue in 1982 (35 years ago), and the American Academy of Pediatrics did not have a section of dermatology until 1986.
Pediatrics and dermatology: The interface
Many of the first generation of pediatric dermatologists trained as pediatricians prior to pursuing their dermatology work, with some being “assigned” dermatology as pediatric experts, while others did formal residencies in dermatology. This history is important, as pediatric dermatology was, and remains, integrated with pediatrics, even while training in dermatology residencies became standard practice. An important part of the development of the field has been the education of pediatricians and dermatologists by pediatric dermatologists, with a strong sensibility that improved training for both generalists and specialists about pediatric skin disease would yield better care for patients and families.
Initially, there were very few pediatric or dermatology programs in the United States that had pediatric dermatologists. Over the succeeding decades, this is now less common, although even now there are still dermatology and pediatric residency programs that do not have a pediatric dermatologist for either training or to serve their patients. The founding leaders of the SPD set a tone of collaboration nationally and internationally, reaching out to pediatric colleagues and dermatology associates from around the world, and establishing superb educational programs for the exchange of ideas, presentation of challenging cases, and promoting state of the art knowledge of the field. Through annual meetings of the SPD, conferences immediately preceding the American Academy of Dermatology annual meetings, the World Congress of Pediatric Dermatology, and other regional and international meetings, the field developed as the number of practitioners grew, and as the specialized published literature reflected new knowledge in diagnosis and therapy.
Building upon the history of collaboration and reflecting the maturation of the field with a desire to influence the breadth and quantity of research in pediatric dermatology, the Pediatric Dermatology Research Alliance (PeDRA) was formed in 2012. This organization was formed to promote and facilitate high quality collaborative clinical, translational, educational, and basic science research in pediatric dermatology with a vision to create sustainable, collaborative networks to better understand, prevent, treat, and cure dermatologic diseases in children. This network is now composed of over 230 members representing over 68 institutions from the United States and Canada, but including involvement globally from Mexico, Europe, and the Middle East.
Examples of changing perspectives: hemangiomas
A good way to look at evolution of the field is take a look at some of the similarities and differences in clinical practice in relation to common and uncommon disease states.
A great example is hemangiomas. Some of the first natural history studies on hemangiomas were done in the early 1960s, establishing that many lesions had a typical clinical course of fairly rapid growth, plateau, and involution over time. Of course, the identification of hemangiomas of infancy (or “HOI” in the trade), was confused with vascular malformations, and no one had recognized variant tumors that were distinct, such as rapidly involuting and noninvoluting congenital hemangiomas (RICHs or NICHs), tufted angiomas, and hemangioendotheliomas. PHACE syndrome (posterior fossa brain malformations) had yet to be described (that was done in 1996 by Ilona Frieden and her colleagues). For a time period, hemangiomas were treated with X-rays, before the negative impact of such radiation was acknowledged. For many years after that, even deforming and functionally significant lesions were “followed clinically” for natural involution, presumably a backlash from the radiation therapy interventions.
This story also reflects how organized research efforts helped with the evolution of knowledge and clinical care. The Hemangioma of Infancy Group was formed to take a collaborative approach to characterize and study hemangiomas and related tumors. Beginning with energetic, insightful pediatric dermatologists, and little funding, they changed our knowledge base of how hemangiomas present, the risk factors for their development and the characteristics and multiple organ findings associated with PHACE and other syndromic hemangiomas.
Procedural pediatric dermatology: Tremendous revolution in surgery and laser
The first generation of pediatric dermatologists were considered medical dermatologist specialists. And how important this specialty work was! Acne, atopic dermatitis, psoriasis, diaper and seborrheic dermatitis, and rare genetic syndromes, these conditions were a major part of the work of early pediatric dermatologists (and remain so now). What was not common was for pediatric dermatologists to have procedural or surgical practices, while this now is routinely part of the work of specialists in the field. How did this shift occur?
The fundamental shift began to occur with the introduction of the pulsed dye laser in 1989 and the publication of a seminal article in the New England Journal of Medicine (1989 Feb 16;320[7]:416-21) on its utility in treating port-wine stains in children with minimal scarring. Vascular lesions including port-wine stains were common, and pediatric dermatologists managed these patients for both diagnosis and medical management. Also, dermatology residencies at this time offered training in cutaneous surgery, excisions (including Mohs surgery) and repairs, and trainees in pediatric dermatology were “trained up” to high levels of expertise. As lasers were incorporated into dermatology residency work and practices, pediatric dermatologists had the exposure and skill to do this work. An added advantage was having the pediatric knowledge of how to handle children and adolescents in an age appropriate manner, and consideration of methods to minimize the pain and anxiety of procedures. Within a few years, pediatric dermatologists were at the forefront of the use of topical anesthetics (EMLA and liposomal lidocaine) and had general anesthesia privileges for laser and excisional surgery.
So while pediatric dermatologists still do “small procedures” every hour in most practices (cryotherapy for warts, cantharidin for molluscum, shave and punch biopsies), a subset now have extensive procedural practices, which in recent years has extended to pigment lesion lasers (to treat nevus of Ota), hair lasers (to treat perineal areas to prevent pilonidal cyst recurrence or to treat hirsutism), and combinations of lasers to treat hypertrophic, constrictive, and/or deforming scars).
Inflammatory skin disorders: Bread and butter ... and peanut butter?
The care of pediatric inflammatory skin disorders has evolved, but more slowly for some diseases than others. Acne vulgaris now is recognized as much more common under age 12 years than previously, presumably reflecting earlier pubertal changes in our preteens. Over the past 30 years, therapy has evolved with the use of topical retinoids (still underused by pediatricians, considered a “practice gap”), hormonal therapy with combined oral contraceptives, and oral isotretinoin, a powerful but highly effective systemic agent for severe and refractory acne. Specific pediatric guidelines came much later. Pediatric acne expert recommendations were formulated by the American Acne and Rosacea Society and endorsed by the American Academy of Pediatrics in 2013 (Pediatrics. 2013;131:S163-86). Over the past few years, there is a push by experts for more judicious use of antibiotics for acne (oral and topical) to minimize the emergence of bacterial resistance.
Psoriasis has been a condition that has been “behind the revolution,” in that no biologic agent was approved for pediatric psoriasis in the United States until several months ago, lagging behind Europe and elsewhere in the world by almost a decade. Adult psoriasis has been recognized to be associated with a broad set of comorbidities, including obesity and early heart disease, and there is now research on how children are at risk as well, and new recommendations on how to screen children with psoriasis. Moderate to severe psoriasis in adults is now tremendously controllable with biologic agents targeting TNF-alpha, IL 12/23, and IL-17. Etanercept has been approved for children with psoriasis aged 4 years and older, and other biologic agents are under study.
Atopic dermatitis now is ready for its revolution! AD has increased in prevalence from around 5% of the pediatric population 30-plus years ago to 10%-15%. Treatment of most individuals has remained the same over the decades: Good skin care, frequent moisturizers, topical corticosteroids for flares, management of infection if noted. The topical calcineurin inhibitors (TCIs) broadened the therapeutic approach when introduced in 2000 and 2001, but the boxed warning resulted in some practitioners minimizing their utilization of these useful agents.
It has been recognized for years that children with AD have higher risk of developing food allergies than children without AD. A changing understanding of how early food exposure may induce tolerance is changing the world of allergy and influencing the care of children with AD. This is where the peanut butter (or other processed peanut, such as “Bamba”) may be life saving. New guidelines have come from the National Institute of Allergy and Infectious Diseases recommending that infants with severe eczema (or egg allergy, or both) have introduction of age-appropriate peanut-containing food as early as 4-6 months of age to reduce the risk of development of peanut allergy. It is recommended that these infants undergo early evaluation for possible sensitization to peanut protein, with referral to allergists for skin prick tests or serum IgE screens (though if positive, referral to allergists is appropriate), and assess the safety of going ahead with early feeding. It is hoped that following these new guidelines can minimize the development of peanut allergy.
The future
Where will pediatric skin disease, or more importantly, skin health over a lifetime be in 50 years? Can we cure or prevent the consequences of our lethal and life altering genetic diseases such as epidermolysis bullosa or our neurocutaneous disorders? Will our new insights into birthmarks (they are mostly somatic mutations) allow us to form specific, personalized therapies to minimize their impact? Will we be using computers equipped with imaging devices and algorithms to assess our patients’ moles, papules, and nodules? Will our vaccines have wiped out warts, molluscum, and perhaps, acne? Will we have cured our inflammatory skin disorders, or perhaps prevented them by interventions in the neonatal period? No predictions will be offered here, other than that we can look forward to incredible changes for our future generations of health care practitioners, patients, and families.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Eichenfield has served as a consultant for Anacor/Pfizer and Regeneron/Sanofi. Email him at [email protected].
The world in pediatric dermatology has changed in incredible ways since 1967. In fact, pediatric dermatology was not an organized specialty until years later! This article will look back at some of the history of pediatric dermatology, exploring how different the field was 50 years ago, and how it has evolved into the vibrant field that it is. By looking at some disease states, and differences in practice in relation to the care of dermatologic conditions in children both by pediatricians and dermatologists, we can see the tremendous evolution in our understanding and management of pediatric skin conditions, and perhaps gain insight into the future.
Pediatric dermatology was fairly “neonatal” 50 years ago, with only a few practitioners in the field. Recognizing that up to 30% of pediatric primary care visits include a skin-related problem, and that there was limited training about skin diseases among primary care practitioners and inconsistent training amongst dermatologists, there was a clinical need for establishing the subspecialty of pediatric dermatology. The first international symposium was held in Mexico City in October 1972, and with this meeting the International Society of Pediatric Dermatology was founded. The Society for Pediatric Dermatology (SPD) began in 1973, with Alvin Jacobs, MD, Samuel Weinberg, MD, Nancy Esterly, MD, Sidney Hurwitz, MD, William Weston, MD, and Coleman Jacobson, MD, as some of the initial “founding mothers and fathers.” The journal Pediatric Dermatology released its first issue in 1982 (35 years ago), and the American Academy of Pediatrics did not have a section of dermatology until 1986.
Pediatrics and dermatology: The interface
Many of the first generation of pediatric dermatologists trained as pediatricians prior to pursuing their dermatology work, with some being “assigned” dermatology as pediatric experts, while others did formal residencies in dermatology. This history is important, as pediatric dermatology was, and remains, integrated with pediatrics, even while training in dermatology residencies became standard practice. An important part of the development of the field has been the education of pediatricians and dermatologists by pediatric dermatologists, with a strong sensibility that improved training for both generalists and specialists about pediatric skin disease would yield better care for patients and families.
Initially, there were very few pediatric or dermatology programs in the United States that had pediatric dermatologists. Over the succeeding decades, this is now less common, although even now there are still dermatology and pediatric residency programs that do not have a pediatric dermatologist for either training or to serve their patients. The founding leaders of the SPD set a tone of collaboration nationally and internationally, reaching out to pediatric colleagues and dermatology associates from around the world, and establishing superb educational programs for the exchange of ideas, presentation of challenging cases, and promoting state of the art knowledge of the field. Through annual meetings of the SPD, conferences immediately preceding the American Academy of Dermatology annual meetings, the World Congress of Pediatric Dermatology, and other regional and international meetings, the field developed as the number of practitioners grew, and as the specialized published literature reflected new knowledge in diagnosis and therapy.
Building upon the history of collaboration and reflecting the maturation of the field with a desire to influence the breadth and quantity of research in pediatric dermatology, the Pediatric Dermatology Research Alliance (PeDRA) was formed in 2012. This organization was formed to promote and facilitate high quality collaborative clinical, translational, educational, and basic science research in pediatric dermatology with a vision to create sustainable, collaborative networks to better understand, prevent, treat, and cure dermatologic diseases in children. This network is now composed of over 230 members representing over 68 institutions from the United States and Canada, but including involvement globally from Mexico, Europe, and the Middle East.
Examples of changing perspectives: hemangiomas
A good way to look at evolution of the field is take a look at some of the similarities and differences in clinical practice in relation to common and uncommon disease states.
A great example is hemangiomas. Some of the first natural history studies on hemangiomas were done in the early 1960s, establishing that many lesions had a typical clinical course of fairly rapid growth, plateau, and involution over time. Of course, the identification of hemangiomas of infancy (or “HOI” in the trade), was confused with vascular malformations, and no one had recognized variant tumors that were distinct, such as rapidly involuting and noninvoluting congenital hemangiomas (RICHs or NICHs), tufted angiomas, and hemangioendotheliomas. PHACE syndrome (posterior fossa brain malformations) had yet to be described (that was done in 1996 by Ilona Frieden and her colleagues). For a time period, hemangiomas were treated with X-rays, before the negative impact of such radiation was acknowledged. For many years after that, even deforming and functionally significant lesions were “followed clinically” for natural involution, presumably a backlash from the radiation therapy interventions.
This story also reflects how organized research efforts helped with the evolution of knowledge and clinical care. The Hemangioma of Infancy Group was formed to take a collaborative approach to characterize and study hemangiomas and related tumors. Beginning with energetic, insightful pediatric dermatologists, and little funding, they changed our knowledge base of how hemangiomas present, the risk factors for their development and the characteristics and multiple organ findings associated with PHACE and other syndromic hemangiomas.
Procedural pediatric dermatology: Tremendous revolution in surgery and laser
The first generation of pediatric dermatologists were considered medical dermatologist specialists. And how important this specialty work was! Acne, atopic dermatitis, psoriasis, diaper and seborrheic dermatitis, and rare genetic syndromes, these conditions were a major part of the work of early pediatric dermatologists (and remain so now). What was not common was for pediatric dermatologists to have procedural or surgical practices, while this now is routinely part of the work of specialists in the field. How did this shift occur?
The fundamental shift began to occur with the introduction of the pulsed dye laser in 1989 and the publication of a seminal article in the New England Journal of Medicine (1989 Feb 16;320[7]:416-21) on its utility in treating port-wine stains in children with minimal scarring. Vascular lesions including port-wine stains were common, and pediatric dermatologists managed these patients for both diagnosis and medical management. Also, dermatology residencies at this time offered training in cutaneous surgery, excisions (including Mohs surgery) and repairs, and trainees in pediatric dermatology were “trained up” to high levels of expertise. As lasers were incorporated into dermatology residency work and practices, pediatric dermatologists had the exposure and skill to do this work. An added advantage was having the pediatric knowledge of how to handle children and adolescents in an age appropriate manner, and consideration of methods to minimize the pain and anxiety of procedures. Within a few years, pediatric dermatologists were at the forefront of the use of topical anesthetics (EMLA and liposomal lidocaine) and had general anesthesia privileges for laser and excisional surgery.
So while pediatric dermatologists still do “small procedures” every hour in most practices (cryotherapy for warts, cantharidin for molluscum, shave and punch biopsies), a subset now have extensive procedural practices, which in recent years has extended to pigment lesion lasers (to treat nevus of Ota), hair lasers (to treat perineal areas to prevent pilonidal cyst recurrence or to treat hirsutism), and combinations of lasers to treat hypertrophic, constrictive, and/or deforming scars).
Inflammatory skin disorders: Bread and butter ... and peanut butter?
The care of pediatric inflammatory skin disorders has evolved, but more slowly for some diseases than others. Acne vulgaris now is recognized as much more common under age 12 years than previously, presumably reflecting earlier pubertal changes in our preteens. Over the past 30 years, therapy has evolved with the use of topical retinoids (still underused by pediatricians, considered a “practice gap”), hormonal therapy with combined oral contraceptives, and oral isotretinoin, a powerful but highly effective systemic agent for severe and refractory acne. Specific pediatric guidelines came much later. Pediatric acne expert recommendations were formulated by the American Acne and Rosacea Society and endorsed by the American Academy of Pediatrics in 2013 (Pediatrics. 2013;131:S163-86). Over the past few years, there is a push by experts for more judicious use of antibiotics for acne (oral and topical) to minimize the emergence of bacterial resistance.
Psoriasis has been a condition that has been “behind the revolution,” in that no biologic agent was approved for pediatric psoriasis in the United States until several months ago, lagging behind Europe and elsewhere in the world by almost a decade. Adult psoriasis has been recognized to be associated with a broad set of comorbidities, including obesity and early heart disease, and there is now research on how children are at risk as well, and new recommendations on how to screen children with psoriasis. Moderate to severe psoriasis in adults is now tremendously controllable with biologic agents targeting TNF-alpha, IL 12/23, and IL-17. Etanercept has been approved for children with psoriasis aged 4 years and older, and other biologic agents are under study.
Atopic dermatitis now is ready for its revolution! AD has increased in prevalence from around 5% of the pediatric population 30-plus years ago to 10%-15%. Treatment of most individuals has remained the same over the decades: Good skin care, frequent moisturizers, topical corticosteroids for flares, management of infection if noted. The topical calcineurin inhibitors (TCIs) broadened the therapeutic approach when introduced in 2000 and 2001, but the boxed warning resulted in some practitioners minimizing their utilization of these useful agents.
It has been recognized for years that children with AD have higher risk of developing food allergies than children without AD. A changing understanding of how early food exposure may induce tolerance is changing the world of allergy and influencing the care of children with AD. This is where the peanut butter (or other processed peanut, such as “Bamba”) may be life saving. New guidelines have come from the National Institute of Allergy and Infectious Diseases recommending that infants with severe eczema (or egg allergy, or both) have introduction of age-appropriate peanut-containing food as early as 4-6 months of age to reduce the risk of development of peanut allergy. It is recommended that these infants undergo early evaluation for possible sensitization to peanut protein, with referral to allergists for skin prick tests or serum IgE screens (though if positive, referral to allergists is appropriate), and assess the safety of going ahead with early feeding. It is hoped that following these new guidelines can minimize the development of peanut allergy.
The future
Where will pediatric skin disease, or more importantly, skin health over a lifetime be in 50 years? Can we cure or prevent the consequences of our lethal and life altering genetic diseases such as epidermolysis bullosa or our neurocutaneous disorders? Will our new insights into birthmarks (they are mostly somatic mutations) allow us to form specific, personalized therapies to minimize their impact? Will we be using computers equipped with imaging devices and algorithms to assess our patients’ moles, papules, and nodules? Will our vaccines have wiped out warts, molluscum, and perhaps, acne? Will we have cured our inflammatory skin disorders, or perhaps prevented them by interventions in the neonatal period? No predictions will be offered here, other than that we can look forward to incredible changes for our future generations of health care practitioners, patients, and families.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Eichenfield has served as a consultant for Anacor/Pfizer and Regeneron/Sanofi. Email him at [email protected].
Pediatric Dermatology Consult - July 2017
BY JUSLEEN AHLUWALIA, MD, AND LAWRENCE F. EICHENFIELD, MD
Consider immunologic blistering diseases in the differential diagnosis of unusual vesicles and bullae in infants and children.
The patient was diagnosed with infantile bullous pemphigoid. Treatment was initiated with clobetasol 0.05% ointment twice daily to the affected areas. Despite treatment with topical corticosteroids, the patient continued to manifest bullae and urticariforme plaques on his legs and trunk. He was then started on oral prednisolone 1 mg/kg per day with gastrointestinal ulcer prophylaxis. He improved within 3 weeks of treatment.
Overall, childhood BP carries a good prognosis and responds well to topical or oral steroids.1 Most patients achieve remission within a few weeks to months of treatments; however, disease course can vary with several relapses over a few years. The longest duration of disease that has been reported is 3 years.1 Second-line treatments – such as dapsone, sulfapyridine, mycophenolate mofetil, administered alone or concurrently with steroids – may be considered for those who fail to respond to conventional treatment.1,5 Intravenous immunoglobulin has reported to be efficacious and well-tolerated in a few cases of treatment-resistant childhood BP.1,5
Differentiation of infantile bullous pemphigoid from other common and uncommon vesiculobullous diseases is necessary. Bullous impetigo (A), caused by Streptococcus pyogenes or Staphylococcus aureus can present with vesicles and bullae due to staphylococcal exfoliative toxins (exfoliatin A-D) which cause separation at the superficial epidermal level. Hand, foot, and mouth disease (HFMD) (B) is an acute Coxsackie viral infection, classically involving the oral mucosa, palms, and soles, although broad variations in presentations include “eczema coxsackium,” Gianotti-Crosti–type lesions, and petechiae.6 Linear IgA dermatosis (C) is an immunobullous condition occurring most frequently in perioral and perineal regions. The disease is characterized by a linear deposition of IgA along the dermal-epidermal junction. Dermatitis herpetiformis (E) is an autoimmune, papulovesicular eruption most commonly involving extensor surfaces of extremities and is characterized by granular deposition of IgA in the papillary dermis.
This patient’s systemic steroids were successfully tapered over several months without disease recurrence. It is unknown why this immunobullous disease is usually self-limited in children, unlike BP in adults, which has significant morbidity or mortality. While uncommon, it is important to consider immunologic blistering diseases in the differential diagnosis of unusual vesicles and bullae in infants and children.
Dr. Ahluwalia is with the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Neither of the physicians has relevant financial disclosures. Email them at [email protected].
References
1. Pediatr Dermatol. 2016 Mar-Apr;33(2):e77-81.
2. Orphanet J Rare Dis. 2014 Dec 10;9:185.
3. J Am Acad Dermatol. 2008 Jan;58(1):41-8.
4. Arch Dermatol. 1991 Mar;127(3):378-86.
BY JUSLEEN AHLUWALIA, MD, AND LAWRENCE F. EICHENFIELD, MD
Consider immunologic blistering diseases in the differential diagnosis of unusual vesicles and bullae in infants and children.
The patient was diagnosed with infantile bullous pemphigoid. Treatment was initiated with clobetasol 0.05% ointment twice daily to the affected areas. Despite treatment with topical corticosteroids, the patient continued to manifest bullae and urticariforme plaques on his legs and trunk. He was then started on oral prednisolone 1 mg/kg per day with gastrointestinal ulcer prophylaxis. He improved within 3 weeks of treatment.
Overall, childhood BP carries a good prognosis and responds well to topical or oral steroids.1 Most patients achieve remission within a few weeks to months of treatments; however, disease course can vary with several relapses over a few years. The longest duration of disease that has been reported is 3 years.1 Second-line treatments – such as dapsone, sulfapyridine, mycophenolate mofetil, administered alone or concurrently with steroids – may be considered for those who fail to respond to conventional treatment.1,5 Intravenous immunoglobulin has reported to be efficacious and well-tolerated in a few cases of treatment-resistant childhood BP.1,5
Differentiation of infantile bullous pemphigoid from other common and uncommon vesiculobullous diseases is necessary. Bullous impetigo (A), caused by Streptococcus pyogenes or Staphylococcus aureus can present with vesicles and bullae due to staphylococcal exfoliative toxins (exfoliatin A-D) which cause separation at the superficial epidermal level. Hand, foot, and mouth disease (HFMD) (B) is an acute Coxsackie viral infection, classically involving the oral mucosa, palms, and soles, although broad variations in presentations include “eczema coxsackium,” Gianotti-Crosti–type lesions, and petechiae.6 Linear IgA dermatosis (C) is an immunobullous condition occurring most frequently in perioral and perineal regions. The disease is characterized by a linear deposition of IgA along the dermal-epidermal junction. Dermatitis herpetiformis (E) is an autoimmune, papulovesicular eruption most commonly involving extensor surfaces of extremities and is characterized by granular deposition of IgA in the papillary dermis.
This patient’s systemic steroids were successfully tapered over several months without disease recurrence. It is unknown why this immunobullous disease is usually self-limited in children, unlike BP in adults, which has significant morbidity or mortality. While uncommon, it is important to consider immunologic blistering diseases in the differential diagnosis of unusual vesicles and bullae in infants and children.
Dr. Ahluwalia is with the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Neither of the physicians has relevant financial disclosures. Email them at [email protected].
References
1. Pediatr Dermatol. 2016 Mar-Apr;33(2):e77-81.
2. Orphanet J Rare Dis. 2014 Dec 10;9:185.
3. J Am Acad Dermatol. 2008 Jan;58(1):41-8.
4. Arch Dermatol. 1991 Mar;127(3):378-86.
BY JUSLEEN AHLUWALIA, MD, AND LAWRENCE F. EICHENFIELD, MD
Consider immunologic blistering diseases in the differential diagnosis of unusual vesicles and bullae in infants and children.
The patient was diagnosed with infantile bullous pemphigoid. Treatment was initiated with clobetasol 0.05% ointment twice daily to the affected areas. Despite treatment with topical corticosteroids, the patient continued to manifest bullae and urticariforme plaques on his legs and trunk. He was then started on oral prednisolone 1 mg/kg per day with gastrointestinal ulcer prophylaxis. He improved within 3 weeks of treatment.
Overall, childhood BP carries a good prognosis and responds well to topical or oral steroids.1 Most patients achieve remission within a few weeks to months of treatments; however, disease course can vary with several relapses over a few years. The longest duration of disease that has been reported is 3 years.1 Second-line treatments – such as dapsone, sulfapyridine, mycophenolate mofetil, administered alone or concurrently with steroids – may be considered for those who fail to respond to conventional treatment.1,5 Intravenous immunoglobulin has reported to be efficacious and well-tolerated in a few cases of treatment-resistant childhood BP.1,5
Differentiation of infantile bullous pemphigoid from other common and uncommon vesiculobullous diseases is necessary. Bullous impetigo (A), caused by Streptococcus pyogenes or Staphylococcus aureus can present with vesicles and bullae due to staphylococcal exfoliative toxins (exfoliatin A-D) which cause separation at the superficial epidermal level. Hand, foot, and mouth disease (HFMD) (B) is an acute Coxsackie viral infection, classically involving the oral mucosa, palms, and soles, although broad variations in presentations include “eczema coxsackium,” Gianotti-Crosti–type lesions, and petechiae.6 Linear IgA dermatosis (C) is an immunobullous condition occurring most frequently in perioral and perineal regions. The disease is characterized by a linear deposition of IgA along the dermal-epidermal junction. Dermatitis herpetiformis (E) is an autoimmune, papulovesicular eruption most commonly involving extensor surfaces of extremities and is characterized by granular deposition of IgA in the papillary dermis.
This patient’s systemic steroids were successfully tapered over several months without disease recurrence. It is unknown why this immunobullous disease is usually self-limited in children, unlike BP in adults, which has significant morbidity or mortality. While uncommon, it is important to consider immunologic blistering diseases in the differential diagnosis of unusual vesicles and bullae in infants and children.
Dr. Ahluwalia is with the division of pediatric and adolescent dermatology, Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego and professor of dermatology and pediatrics at the University of California, San Diego. Neither of the physicians has relevant financial disclosures. Email them at [email protected].
References
1. Pediatr Dermatol. 2016 Mar-Apr;33(2):e77-81.
2. Orphanet J Rare Dis. 2014 Dec 10;9:185.
3. J Am Acad Dermatol. 2008 Jan;58(1):41-8.
4. Arch Dermatol. 1991 Mar;127(3):378-86.
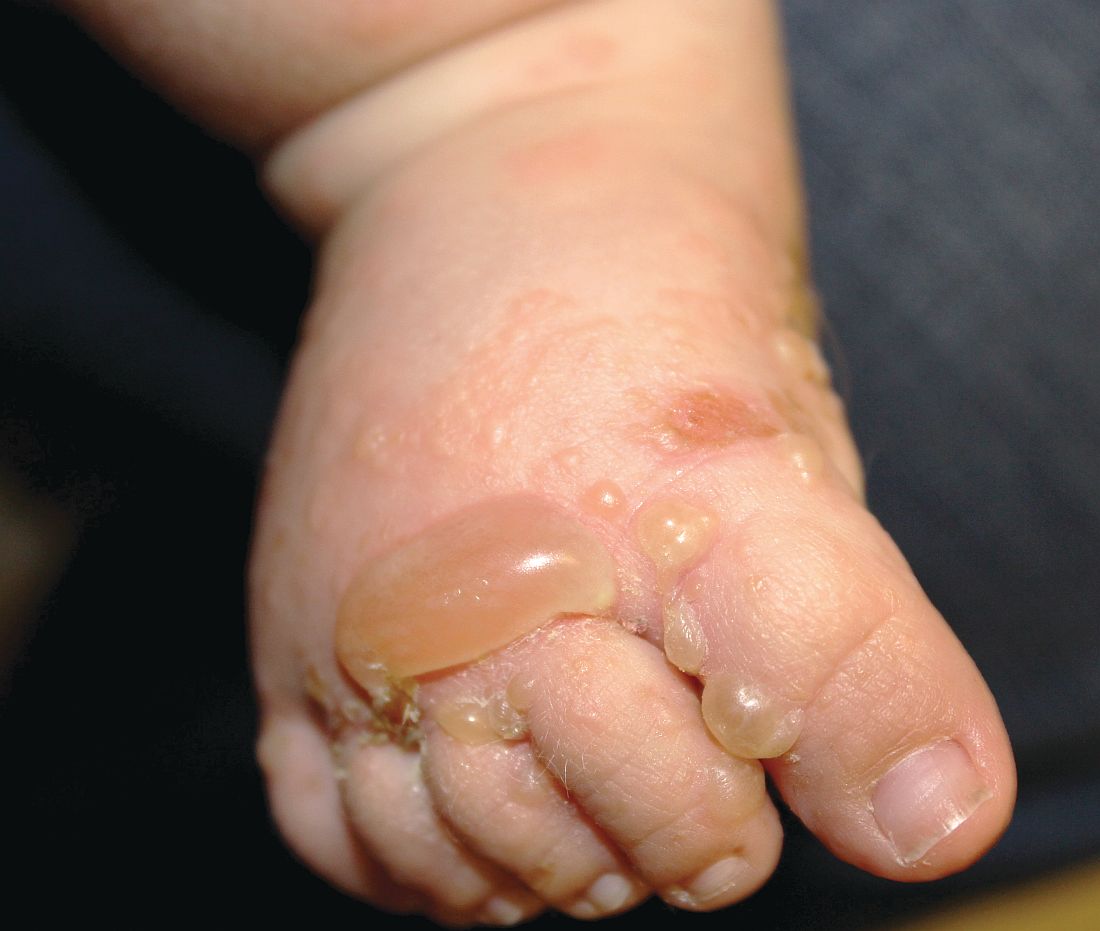
A 3-month-old otherwise healthy boy was referred for a 2-week history of a nonpruritic, urticariforme eruption that progressed to blisters on the trunk, hands, and feet. The patient had a 4-day history of a nonproductive cough preceding the rash. He was otherwise active with normal feeding and bowel habits. His family history was negative for dermatologic or autoimmune disease.
On examination, the patient is a well-developed, active male with widespread erythematous infiltrated papules and arcuate plaques, tense vesiculobullous lesions, and multiple erosions on the trunk and extremities. Mucous membranes and perineum were spared.
Bacterial culture of a lesion was negative. Consult from dermatology was obtained and a biopsy performed.
Histologic examination showed a subepidermal unilocular blister with serous fluid containing predominately eosinophils and lymphocytes. Direct immunofluorescence revealed linear immunoglobulin G and C3 deposition along the basement membrane.
Pediatric Dermatology Consult - June 2017
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
Serum sickness
How does serum sickness–like reaction present?
At the time of initial evaluation, the patient presented with a recent history of fever, edema of the hands and feet, limited gait, and a diffuse, persistent serpiginous and annular dermatitis for 48 hours. Of note, the patient was prescribed a 10-day course of amoxicillin for otitis media 8 days prior to initial presentation.
The distribution and morphology of our patient’s cutaneous eruption, in combination with the systemic symptoms, facial and acral edema, lymphadenopathy, and arthralgia, were highly suggestive of serum sickness–like reaction (SSLR). SSLR is an allergic reaction characterized by a cutaneous eruption, arthralgias, fever, lymphadenopathy, facial edema, and malaise. True serum sickness was originally distinguished in 1905 as a self-limited illness that occurred in several patients after administration of equine diphtheria antitoxin.1 Today this reaction is rarely noted in the pediatric population.
Pathogenically, true serum sickness represents a type III arthus hypersensitivity reaction to proteins in toxins or drugs, mediated by circulating antigen-antibody complexes. In contrast, SSLR is not associated with circulating immune complexes or hypocomplementemia.2 Histopathology frequently resembles the findings of urticaria, including a perivascular and dermal inflammatory infiltrate with associated neutrophils, eosinophils, and lymphocytes.2 Several hypotheses concerning the etiology of SSLR have been suggested in the literature, most commonly including an inflammatory response to defective drug metabolism. However, definitive pathology remains unknown.3
Initially, an urticarial rash and low-grade fever typically develop 7-21 days after exposure to the offending agent or sooner in individuals previously sensitized to the drug. The lesions quickly evolve to the classic purpuric lesions of SSLR over the following 24-48 hours. These dermatologic findings include pink to red oval and/or polycyclic lesions with large central areas of purple discoloration, classically involving the trunk, extremities, and/or face. The lesions may be discrete, scattered, or confluent.2
Several medications are associated with SSLR, but cefaclor is most frequently implicated, occurring in 0.2% of treated children.4,5 Other common culprits include penicillins, tetracycline, sulfonamides, macrolides, ciprofloxacin, rifampin, griseofulvin, bupropion, and fluoxetine.6-10 More recently, biologic agents have been implicated in SSLR, including rituximab, efalizumab, and infliximab.11,12 SSLR also has been described in association with immunizations (hepatitis B, tetanus, and rabies) and active infections with hepatitis B or C.13,14
The diagnosis of SSLR is primarily clinical, formulated based on the constellation of characteristic lesions, fever, adenopathy, facial edema, and/or arthralgia in conjunction with recent history (within 7-21 days) of offending medications or agents. Supporting laboratory findings include normal or mildly low complement C3 and C4 levels and mild proteinuria. Abnormalities in liver and renal function test results are rare, in contrast to true serum sickness. Biopsy and histopathology may be utilized to exclude other diagnoses.2
An extensive variety of cutaneous conditions bearing a resemblance to SSLR were considered in the differential, including urticaria multiforme, Kawasaki disease, erythema multiforme, and urticarial vasculitis. A common challenge for practitioners is distinguishing SSLR from similar dermatoses, particularly urticarial multiforme (UM), which some experts consider clinically related to SSLR. Despite the cutaneous similarities of UM and SSLR, including urticarial plaques with an associated central duskiness, SSLR tends to have a more delayed onset following offending agent exposure and more extracutaneous symptoms, especially arthralgias and/or arthritis, lymphadenopathy, and higher fevers.15 The rash of UM classically presents 1-3 days following agent exposure or illness, whereas SSLR presentation is delayed 1-3 weeks. Further distinguishing UM is the transient nature of the individual lesions, each lasting less than 24 hours.
Urticarial vasculitis (UV), morphologically similar to SSLR and UM, causes persistent urticarial-like plaques that last longer than 24-48 hours and resolve with bruising. Biopsy results revealing leukocytoclastic vasculitis distinguish UV, but case reports exist reporting leukocytoclastic vasculitis in association with SSLR.
Erythema multiforme (EM), a hypersensitivity reaction usually triggered by infections (most commonly herpes simplex virus), also may appear morphologically similar to SSLR. These lesions also persist for longer than 24-48 hours and often have a target appearance with central duskiness that sometimes can blister. Patients may have mucosal involvement with vesicles and erosions, compared with patients with SSLR in whom mucosal lesions are rarely seen. Medications are an uncommon trigger for EM, occurring in less than 10% of cases, and alternative drug eruptions including SSLR should be excluded prior to diagnosis.
Other disorders commonly presenting with similar extracutaneous manifestations to SSLR and concomitant dermatitis were further excluded based on the morphologic appearance of the lesions and other clinical dissimilarities. Henoch-Schönlein purpura, which also may present with arthralgia, fever, GI symptoms, and extremity edema, was unlikely given the urticarial appearance of the lesions, rather than palpable purpura. Furthermore, the arthritis/arthralgia of Henoch-Schönlein purpura is typically oligoarticular, affecting the large joints of the lower extremities most frequently.
If the patient presents with persistent fever for more than 5 days, Kawasaki disease may be considered in the differential and ruled out with clinical and laboratory assessment. The presence of fever, lymphadenopathy, and acute rash similarly should prompt consideration of a clinical and laboratory work-up for drug reaction with eosinophilia and systemic symptoms. Morphologically, this drug reaction is characterized by a maculopapular eruption more than 3 weeks after exposure to an offending agent. Patients with SSLR respond favorably to cessation of the causative drug and supportive care with antipyretics, NSAIDs, and antihistamines. The cutaneous and extracutaneous manifestations of this drug reaction typically disappear within 2-3 weeks after discontinuation of the offending agent. If symptoms are severe, a short systemic corticosteroid course may be warranted. Our patient’s symptoms completely resolved within 2-3 weeks of discontinuing amoxicillin therapy.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital–San Diego, University of California, San Diego. Dr. Waldman is a clinical research fellow at the hospital. Email them at [email protected].
References
1. von Pirquet, C. Frh, and Bela Schick. “Serum Sickness.” Translated by B. Schick. (London: Bailliere, Tindall and Cox, 1951).
2. Cutis. 2002 May;69(5):395-7.
3. J Pediatr. 1994 Nov;125(5 Pt 1):805-11.
4. J Am Acad Dermatol. 1991 Nov;25(5 Pt 1):805-8.
5. J Paediatr Child Health. 2003 Dec;39(9):677-81.
6. Ann Pharmacother. 1996 May;30(5):481-3.
7. J Hosp Med. 2011 Apr;6(4):231-2.
8. Allergy Asthma Immunol Res. 2014 Mar;6(2):183-5.
9. Ann Pharmacother. 2004 Apr;38(4):609-11.
10. Ann Pharmacother. 2000 Apr;34(4):471-3.
11. J Clin Rheumatol. 2013 Sep;19(6):360.
12. Int J Rheum Dis. 2012 Feb;15(1):e6-7.
13. Am J Med Sci. 2013 May;345(5):412-3.
14. Semin Cutan Med Surg. 2007 Sep;26(3):179-87.
15. J Clin Aesthet Dermatol. 2013 Mar;6(3):34-9.
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
Serum sickness
How does serum sickness–like reaction present?
At the time of initial evaluation, the patient presented with a recent history of fever, edema of the hands and feet, limited gait, and a diffuse, persistent serpiginous and annular dermatitis for 48 hours. Of note, the patient was prescribed a 10-day course of amoxicillin for otitis media 8 days prior to initial presentation.
The distribution and morphology of our patient’s cutaneous eruption, in combination with the systemic symptoms, facial and acral edema, lymphadenopathy, and arthralgia, were highly suggestive of serum sickness–like reaction (SSLR). SSLR is an allergic reaction characterized by a cutaneous eruption, arthralgias, fever, lymphadenopathy, facial edema, and malaise. True serum sickness was originally distinguished in 1905 as a self-limited illness that occurred in several patients after administration of equine diphtheria antitoxin.1 Today this reaction is rarely noted in the pediatric population.
Pathogenically, true serum sickness represents a type III arthus hypersensitivity reaction to proteins in toxins or drugs, mediated by circulating antigen-antibody complexes. In contrast, SSLR is not associated with circulating immune complexes or hypocomplementemia.2 Histopathology frequently resembles the findings of urticaria, including a perivascular and dermal inflammatory infiltrate with associated neutrophils, eosinophils, and lymphocytes.2 Several hypotheses concerning the etiology of SSLR have been suggested in the literature, most commonly including an inflammatory response to defective drug metabolism. However, definitive pathology remains unknown.3
Initially, an urticarial rash and low-grade fever typically develop 7-21 days after exposure to the offending agent or sooner in individuals previously sensitized to the drug. The lesions quickly evolve to the classic purpuric lesions of SSLR over the following 24-48 hours. These dermatologic findings include pink to red oval and/or polycyclic lesions with large central areas of purple discoloration, classically involving the trunk, extremities, and/or face. The lesions may be discrete, scattered, or confluent.2
Several medications are associated with SSLR, but cefaclor is most frequently implicated, occurring in 0.2% of treated children.4,5 Other common culprits include penicillins, tetracycline, sulfonamides, macrolides, ciprofloxacin, rifampin, griseofulvin, bupropion, and fluoxetine.6-10 More recently, biologic agents have been implicated in SSLR, including rituximab, efalizumab, and infliximab.11,12 SSLR also has been described in association with immunizations (hepatitis B, tetanus, and rabies) and active infections with hepatitis B or C.13,14
The diagnosis of SSLR is primarily clinical, formulated based on the constellation of characteristic lesions, fever, adenopathy, facial edema, and/or arthralgia in conjunction with recent history (within 7-21 days) of offending medications or agents. Supporting laboratory findings include normal or mildly low complement C3 and C4 levels and mild proteinuria. Abnormalities in liver and renal function test results are rare, in contrast to true serum sickness. Biopsy and histopathology may be utilized to exclude other diagnoses.2
An extensive variety of cutaneous conditions bearing a resemblance to SSLR were considered in the differential, including urticaria multiforme, Kawasaki disease, erythema multiforme, and urticarial vasculitis. A common challenge for practitioners is distinguishing SSLR from similar dermatoses, particularly urticarial multiforme (UM), which some experts consider clinically related to SSLR. Despite the cutaneous similarities of UM and SSLR, including urticarial plaques with an associated central duskiness, SSLR tends to have a more delayed onset following offending agent exposure and more extracutaneous symptoms, especially arthralgias and/or arthritis, lymphadenopathy, and higher fevers.15 The rash of UM classically presents 1-3 days following agent exposure or illness, whereas SSLR presentation is delayed 1-3 weeks. Further distinguishing UM is the transient nature of the individual lesions, each lasting less than 24 hours.
Urticarial vasculitis (UV), morphologically similar to SSLR and UM, causes persistent urticarial-like plaques that last longer than 24-48 hours and resolve with bruising. Biopsy results revealing leukocytoclastic vasculitis distinguish UV, but case reports exist reporting leukocytoclastic vasculitis in association with SSLR.
Erythema multiforme (EM), a hypersensitivity reaction usually triggered by infections (most commonly herpes simplex virus), also may appear morphologically similar to SSLR. These lesions also persist for longer than 24-48 hours and often have a target appearance with central duskiness that sometimes can blister. Patients may have mucosal involvement with vesicles and erosions, compared with patients with SSLR in whom mucosal lesions are rarely seen. Medications are an uncommon trigger for EM, occurring in less than 10% of cases, and alternative drug eruptions including SSLR should be excluded prior to diagnosis.
Other disorders commonly presenting with similar extracutaneous manifestations to SSLR and concomitant dermatitis were further excluded based on the morphologic appearance of the lesions and other clinical dissimilarities. Henoch-Schönlein purpura, which also may present with arthralgia, fever, GI symptoms, and extremity edema, was unlikely given the urticarial appearance of the lesions, rather than palpable purpura. Furthermore, the arthritis/arthralgia of Henoch-Schönlein purpura is typically oligoarticular, affecting the large joints of the lower extremities most frequently.
If the patient presents with persistent fever for more than 5 days, Kawasaki disease may be considered in the differential and ruled out with clinical and laboratory assessment. The presence of fever, lymphadenopathy, and acute rash similarly should prompt consideration of a clinical and laboratory work-up for drug reaction with eosinophilia and systemic symptoms. Morphologically, this drug reaction is characterized by a maculopapular eruption more than 3 weeks after exposure to an offending agent. Patients with SSLR respond favorably to cessation of the causative drug and supportive care with antipyretics, NSAIDs, and antihistamines. The cutaneous and extracutaneous manifestations of this drug reaction typically disappear within 2-3 weeks after discontinuation of the offending agent. If symptoms are severe, a short systemic corticosteroid course may be warranted. Our patient’s symptoms completely resolved within 2-3 weeks of discontinuing amoxicillin therapy.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital–San Diego, University of California, San Diego. Dr. Waldman is a clinical research fellow at the hospital. Email them at [email protected].
References
1. von Pirquet, C. Frh, and Bela Schick. “Serum Sickness.” Translated by B. Schick. (London: Bailliere, Tindall and Cox, 1951).
2. Cutis. 2002 May;69(5):395-7.
3. J Pediatr. 1994 Nov;125(5 Pt 1):805-11.
4. J Am Acad Dermatol. 1991 Nov;25(5 Pt 1):805-8.
5. J Paediatr Child Health. 2003 Dec;39(9):677-81.
6. Ann Pharmacother. 1996 May;30(5):481-3.
7. J Hosp Med. 2011 Apr;6(4):231-2.
8. Allergy Asthma Immunol Res. 2014 Mar;6(2):183-5.
9. Ann Pharmacother. 2004 Apr;38(4):609-11.
10. Ann Pharmacother. 2000 Apr;34(4):471-3.
11. J Clin Rheumatol. 2013 Sep;19(6):360.
12. Int J Rheum Dis. 2012 Feb;15(1):e6-7.
13. Am J Med Sci. 2013 May;345(5):412-3.
14. Semin Cutan Med Surg. 2007 Sep;26(3):179-87.
15. J Clin Aesthet Dermatol. 2013 Mar;6(3):34-9.
BY CATALINA MATIZ, MD, AND ANDREA WALDMAN, MD
Serum sickness
How does serum sickness–like reaction present?
At the time of initial evaluation, the patient presented with a recent history of fever, edema of the hands and feet, limited gait, and a diffuse, persistent serpiginous and annular dermatitis for 48 hours. Of note, the patient was prescribed a 10-day course of amoxicillin for otitis media 8 days prior to initial presentation.
The distribution and morphology of our patient’s cutaneous eruption, in combination with the systemic symptoms, facial and acral edema, lymphadenopathy, and arthralgia, were highly suggestive of serum sickness–like reaction (SSLR). SSLR is an allergic reaction characterized by a cutaneous eruption, arthralgias, fever, lymphadenopathy, facial edema, and malaise. True serum sickness was originally distinguished in 1905 as a self-limited illness that occurred in several patients after administration of equine diphtheria antitoxin.1 Today this reaction is rarely noted in the pediatric population.
Pathogenically, true serum sickness represents a type III arthus hypersensitivity reaction to proteins in toxins or drugs, mediated by circulating antigen-antibody complexes. In contrast, SSLR is not associated with circulating immune complexes or hypocomplementemia.2 Histopathology frequently resembles the findings of urticaria, including a perivascular and dermal inflammatory infiltrate with associated neutrophils, eosinophils, and lymphocytes.2 Several hypotheses concerning the etiology of SSLR have been suggested in the literature, most commonly including an inflammatory response to defective drug metabolism. However, definitive pathology remains unknown.3
Initially, an urticarial rash and low-grade fever typically develop 7-21 days after exposure to the offending agent or sooner in individuals previously sensitized to the drug. The lesions quickly evolve to the classic purpuric lesions of SSLR over the following 24-48 hours. These dermatologic findings include pink to red oval and/or polycyclic lesions with large central areas of purple discoloration, classically involving the trunk, extremities, and/or face. The lesions may be discrete, scattered, or confluent.2
Several medications are associated with SSLR, but cefaclor is most frequently implicated, occurring in 0.2% of treated children.4,5 Other common culprits include penicillins, tetracycline, sulfonamides, macrolides, ciprofloxacin, rifampin, griseofulvin, bupropion, and fluoxetine.6-10 More recently, biologic agents have been implicated in SSLR, including rituximab, efalizumab, and infliximab.11,12 SSLR also has been described in association with immunizations (hepatitis B, tetanus, and rabies) and active infections with hepatitis B or C.13,14
The diagnosis of SSLR is primarily clinical, formulated based on the constellation of characteristic lesions, fever, adenopathy, facial edema, and/or arthralgia in conjunction with recent history (within 7-21 days) of offending medications or agents. Supporting laboratory findings include normal or mildly low complement C3 and C4 levels and mild proteinuria. Abnormalities in liver and renal function test results are rare, in contrast to true serum sickness. Biopsy and histopathology may be utilized to exclude other diagnoses.2
An extensive variety of cutaneous conditions bearing a resemblance to SSLR were considered in the differential, including urticaria multiforme, Kawasaki disease, erythema multiforme, and urticarial vasculitis. A common challenge for practitioners is distinguishing SSLR from similar dermatoses, particularly urticarial multiforme (UM), which some experts consider clinically related to SSLR. Despite the cutaneous similarities of UM and SSLR, including urticarial plaques with an associated central duskiness, SSLR tends to have a more delayed onset following offending agent exposure and more extracutaneous symptoms, especially arthralgias and/or arthritis, lymphadenopathy, and higher fevers.15 The rash of UM classically presents 1-3 days following agent exposure or illness, whereas SSLR presentation is delayed 1-3 weeks. Further distinguishing UM is the transient nature of the individual lesions, each lasting less than 24 hours.
Urticarial vasculitis (UV), morphologically similar to SSLR and UM, causes persistent urticarial-like plaques that last longer than 24-48 hours and resolve with bruising. Biopsy results revealing leukocytoclastic vasculitis distinguish UV, but case reports exist reporting leukocytoclastic vasculitis in association with SSLR.
Erythema multiforme (EM), a hypersensitivity reaction usually triggered by infections (most commonly herpes simplex virus), also may appear morphologically similar to SSLR. These lesions also persist for longer than 24-48 hours and often have a target appearance with central duskiness that sometimes can blister. Patients may have mucosal involvement with vesicles and erosions, compared with patients with SSLR in whom mucosal lesions are rarely seen. Medications are an uncommon trigger for EM, occurring in less than 10% of cases, and alternative drug eruptions including SSLR should be excluded prior to diagnosis.
Other disorders commonly presenting with similar extracutaneous manifestations to SSLR and concomitant dermatitis were further excluded based on the morphologic appearance of the lesions and other clinical dissimilarities. Henoch-Schönlein purpura, which also may present with arthralgia, fever, GI symptoms, and extremity edema, was unlikely given the urticarial appearance of the lesions, rather than palpable purpura. Furthermore, the arthritis/arthralgia of Henoch-Schönlein purpura is typically oligoarticular, affecting the large joints of the lower extremities most frequently.
If the patient presents with persistent fever for more than 5 days, Kawasaki disease may be considered in the differential and ruled out with clinical and laboratory assessment. The presence of fever, lymphadenopathy, and acute rash similarly should prompt consideration of a clinical and laboratory work-up for drug reaction with eosinophilia and systemic symptoms. Morphologically, this drug reaction is characterized by a maculopapular eruption more than 3 weeks after exposure to an offending agent. Patients with SSLR respond favorably to cessation of the causative drug and supportive care with antipyretics, NSAIDs, and antihistamines. The cutaneous and extracutaneous manifestations of this drug reaction typically disappear within 2-3 weeks after discontinuation of the offending agent. If symptoms are severe, a short systemic corticosteroid course may be warranted. Our patient’s symptoms completely resolved within 2-3 weeks of discontinuing amoxicillin therapy.
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital–San Diego, University of California, San Diego. Dr. Waldman is a clinical research fellow at the hospital. Email them at [email protected].
References
1. von Pirquet, C. Frh, and Bela Schick. “Serum Sickness.” Translated by B. Schick. (London: Bailliere, Tindall and Cox, 1951).
2. Cutis. 2002 May;69(5):395-7.
3. J Pediatr. 1994 Nov;125(5 Pt 1):805-11.
4. J Am Acad Dermatol. 1991 Nov;25(5 Pt 1):805-8.
5. J Paediatr Child Health. 2003 Dec;39(9):677-81.
6. Ann Pharmacother. 1996 May;30(5):481-3.
7. J Hosp Med. 2011 Apr;6(4):231-2.
8. Allergy Asthma Immunol Res. 2014 Mar;6(2):183-5.
9. Ann Pharmacother. 2004 Apr;38(4):609-11.
10. Ann Pharmacother. 2000 Apr;34(4):471-3.
11. J Clin Rheumatol. 2013 Sep;19(6):360.
12. Int J Rheum Dis. 2012 Feb;15(1):e6-7.
13. Am J Med Sci. 2013 May;345(5):412-3.
14. Semin Cutan Med Surg. 2007 Sep;26(3):179-87.
15. J Clin Aesthet Dermatol. 2013 Mar;6(3):34-9.
Make the diagnosis
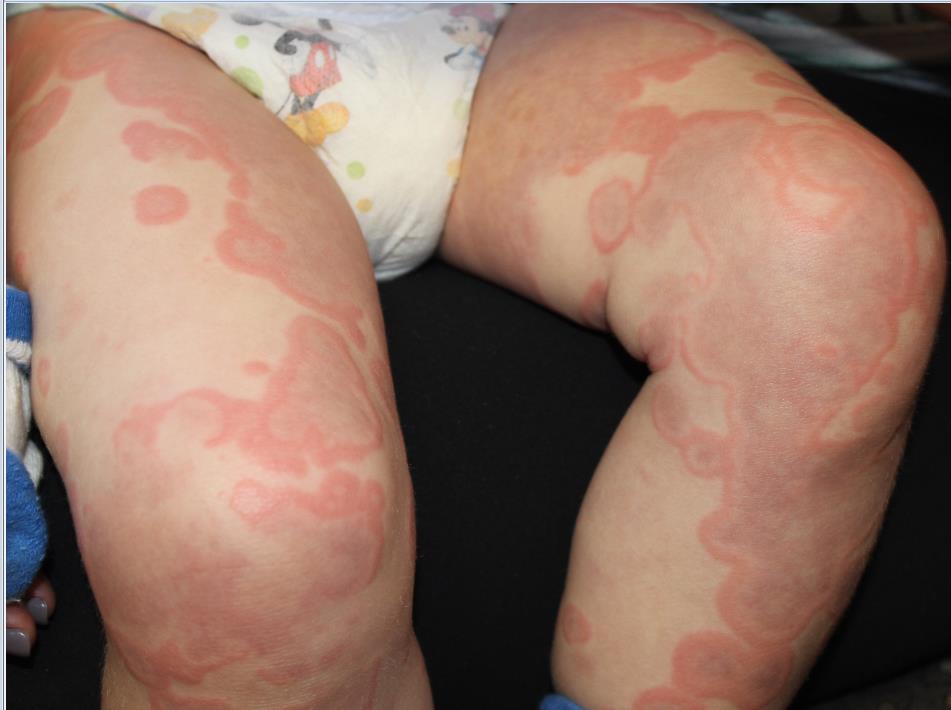
The day prior to presentation, the family took the patient to the ED for evaluation of the lesions and concomitant swelling of the face, eyelids, hands, and feet, as well as fever and irritability. The ED discharged the patient after comprehensive examination and arranged follow-up in the dermatology clinic the following day. Review of systems further revealed limited gait. Parents denied any recent travel or pets residing in the home. Family history is noncontributory.
Physical exam
The patient is a well-appearing toddler, who is in no acute distress but is irritable. Patient is afebrile, with a temperature of 99.5°F, and vital signs are within normal limits. On skin examination, there are serpiginous and annular confluent erythematous urticarial plaques with a central ecchymotic discoloration over the eyes, peripheral malar distribution, neck, chest, abdomen, back, buttock, extremities, hands, and feet. There is notable eyelid, hand, and feet edema. There was no mucosal involvement. The patient has posterior cervical lymphadenopathy but no hepatomegaly or splenomegaly. The patient refused to walk.