User login
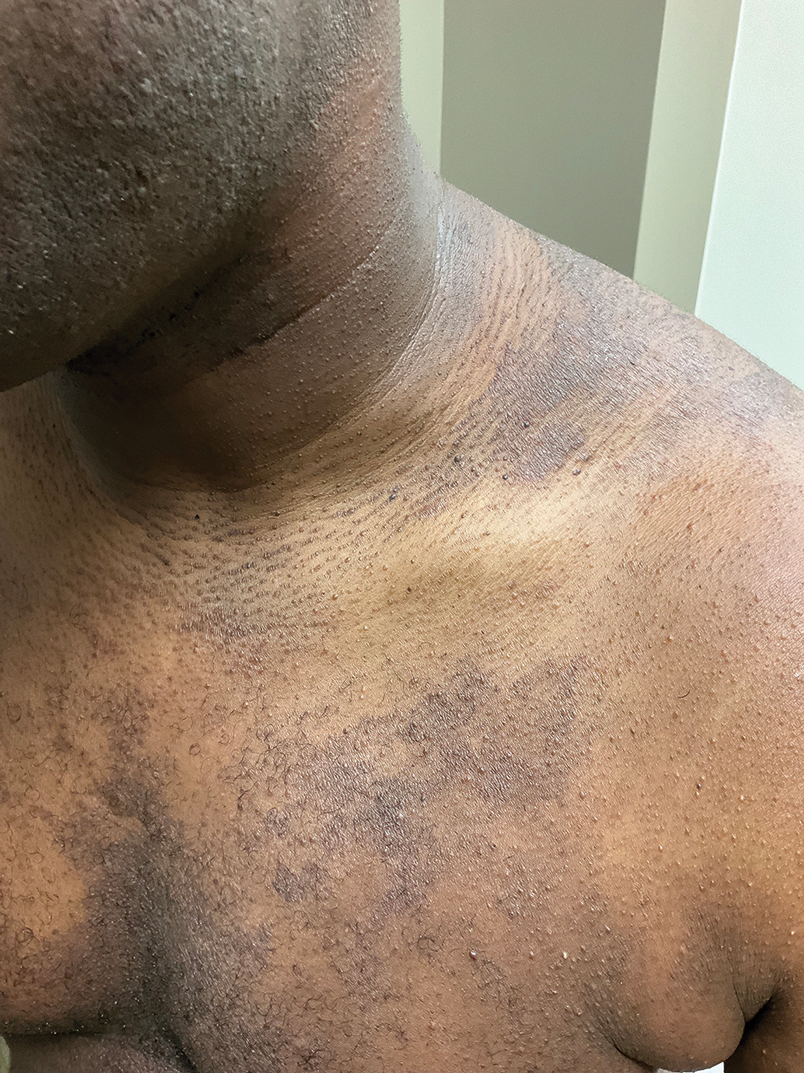
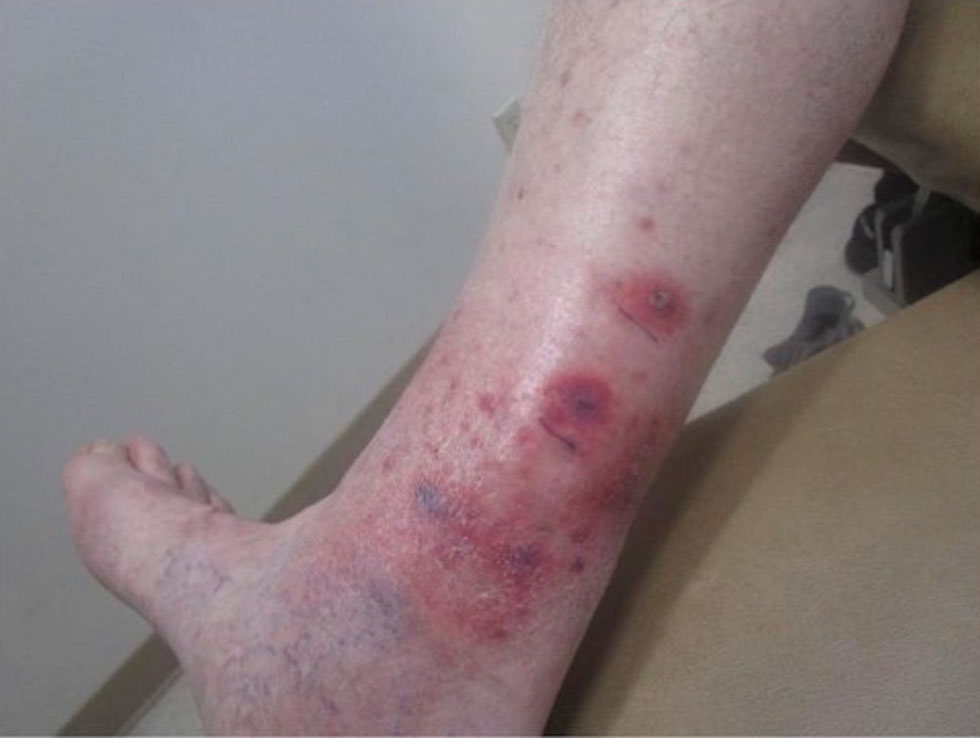
Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
A 78-year-old man was referred to our dermatology clinic for evaluation of nontender erythematous plaques and nodules with central ulceration on the right leg of 5 months’ duration. The patient’s medical history was remarkable for hyperlipidemia, gastroesophageal reflux disease, prostate cancer, and colon cancer status post resection. He denied any relevant travel history but noted that he was an avid hiker and suspected he may have obtained a puncture wound from a bush or a mosquito bite prior to the appearance of the lesions. Previous therapies prescribed by outside physicians and our practice included trimethoprim/sulfamethoxazole, ceftriaxone, levofloxacin, mupirocin, and topical corticosteroids, all with minimal benefit. Clinical examination on initial presentation revealed multiple ulcerations of the lower extremities present for more than 2 months. Punch biopsy of a sample lesion at the current presentation revealed granulomatous change, focal necrosis, and a mixed inflammatory cell infiltrate. Grocott-Gomori methenamine silver and periodic acid–Schiff stains were negative for fungal organisms. The initial acid-fast bacilli stain was negative for mycobacteria, and tissue culture showed no growth.

Exophytic Papule on the Hand
Exophytic Papule on the Hand
THE DIAGNOSIS: Kaposi Sarcoma
Histopathology revealed a serum crust on the surface of the specimen, and the dermis contained compact collections of spindled cells with interspersed erythrocytes (Figure 1). Human herpesvirus 8–stained sections highlighted many lesional cell nuclei (Figure 2). A diagnosis of Kaposi sarcoma (KS) was made based on these findings. The patient expressed interest in surgical excision; however, he was lost to follow-up.
Kaposi sarcoma is an indolent, multifocal, angioproliferative tumor that predominantly affects mucocutaneous sites with less frequent involvement of visceral organs. Kaposi sarcoma is categorized into 4 subtypes: epidemic, iatrogenic, endemic, and classic. Human herpesvirus 8, primarily transmitted through saliva or sexual contact, plays a central role in the pathogenesis of KS, as it drives disease development across all subtypes. The virus causes proliferation of endothelial cells and the formation of angioproliferative lesions characteristic of KS.1
Prevalence is highest in the epidemic subtype, in which patients with advanced HIV and low CD4 T-cell counts may develop KS lesions. Although KS is associated most commonly with HIV, it also has been observed in men who have sex with men regardless of their HIV status.2 Patients undergoing immunosuppressive therapy also may not maintain immune tolerance to previously or newly acquired human herpesvirus 8, leading to the development of iatrogenic KS. This subtype particularly manifests in patients receiving therapy for autoimmune conditions or organ transplants and often only regresses if immunosuppressive therapy is withdrawn.3,4
The endemic and classic subtypes of KS may occur in patients without any known immunocompromise. Endemic KS demonstrates a predilection for pediatric populations in Africa and exhibits less pronounced sex disparity.5 In Uganda and Zimbabwe, endemic KS is the leading cancer in men and the second most frequently occurring cancer in women.6 In contrast, classic KS generally affects older men of Eastern European and Mediterranean descent or Ashkenazi Jewish ancestry. Patients with classic KS generally exhibit a less aggressive disease trajectory relative to other subtypes; however, these patients have a substantial risk for a secondary hematologic malignancy, which may already coexist at the time of diagnosis or emerge subsequently.1,7
Our patient, a native of Eastern Europe, was negative for HIV and was in a monogamous relationship with his wife; therefore, he was likely to have had the classic subtype of KS. As KS is a multifocal disease, lesions may independently emerge at different times and locations on the body. Our patient presented with a new lesion on the hand several years after excision of a similar lesion on the face. Lesions suspicious for KS include slow-growing, painless, red or violaceous patches, nodules, plaques, or patches on the extremities, most commonly manifesting on the feet and ankles. Our differential diagnosis included pyogenic granuloma, amelanotic melanoma, squamous cell carcinoma, and angiosarcoma.
The prognosis in patients with classic KS is favorable, as it often is limited to cutaneous sites and less commonly manifests on visceral organs. Nonetheless, pulmonary and gastrointestinal involvement manifesting as hemoptysis and rectal bleeding, respectively, can occur. This underscores the potential for more serious complications in instances with visceral involvement. Treatment focuses on managing symptoms and preventing growth and progression of individual lesions. Additionally, treatment strategies aim to improve cosmetic outcomes and address any underlying immunosuppression that may exacerbate the condition.8
For most patients, local therapies such as surgical excision, cryotherapy, laser therapy, or intralesional chemotherapy will remove or reduce individual lesions. Patients with widespread cutaneous or extracutaneous disease may require immunomodulatory agents such as interferon α or chemotherapeutic agents such as anthracyclines or paclitaxel.8
Our case highlights the importance of considering risk factors beyond HIV status when including KS as part of the differential diagnosis in patients with atypical vascular lesions. Early recognition enables timely evaluation of potential associated conditions and informs subsequent management decisions.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Lanternier F, Lebbé C, Schartz N, et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS. 2008;22:1163-1168. doi:10.1097/QAD.0b013e3283031a8a
- Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation. 1997;64:669-673. doi:10.1097/00007890-199709150-00001
- Gallo Marin B, Maymone MBC, El Rayess F, et al. Kaposi sarcoma associated with tofacitinib use in a patient with rheumatoid arthritis. R I Med J (2013). 2023;106:18-20.
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. Updated June 5, 2023. Accessed May 15, 2026. https://www.ncbi.nlm .nih.gov/books/NBK534839/
- Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer. 2003;88:1-3. doi:10.1038 /sj.bjc.6600745
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi sarcoma. Mod Pathol. 2008;21:572-582. doi:10.1038/modpathol.2008.15
- Ceccarelli M, Facciolà A, Taibi R, et al. The treatment of Kaposi’s sarcoma: present and future options, a review of the literature. Eur Rev Med Pharmacol Sci. 2019;23:7488-7497. doi:10.26355 /eurrev_201909_18860
THE DIAGNOSIS: Kaposi Sarcoma
Histopathology revealed a serum crust on the surface of the specimen, and the dermis contained compact collections of spindled cells with interspersed erythrocytes (Figure 1). Human herpesvirus 8–stained sections highlighted many lesional cell nuclei (Figure 2). A diagnosis of Kaposi sarcoma (KS) was made based on these findings. The patient expressed interest in surgical excision; however, he was lost to follow-up.
Kaposi sarcoma is an indolent, multifocal, angioproliferative tumor that predominantly affects mucocutaneous sites with less frequent involvement of visceral organs. Kaposi sarcoma is categorized into 4 subtypes: epidemic, iatrogenic, endemic, and classic. Human herpesvirus 8, primarily transmitted through saliva or sexual contact, plays a central role in the pathogenesis of KS, as it drives disease development across all subtypes. The virus causes proliferation of endothelial cells and the formation of angioproliferative lesions characteristic of KS.1
Prevalence is highest in the epidemic subtype, in which patients with advanced HIV and low CD4 T-cell counts may develop KS lesions. Although KS is associated most commonly with HIV, it also has been observed in men who have sex with men regardless of their HIV status.2 Patients undergoing immunosuppressive therapy also may not maintain immune tolerance to previously or newly acquired human herpesvirus 8, leading to the development of iatrogenic KS. This subtype particularly manifests in patients receiving therapy for autoimmune conditions or organ transplants and often only regresses if immunosuppressive therapy is withdrawn.3,4
The endemic and classic subtypes of KS may occur in patients without any known immunocompromise. Endemic KS demonstrates a predilection for pediatric populations in Africa and exhibits less pronounced sex disparity.5 In Uganda and Zimbabwe, endemic KS is the leading cancer in men and the second most frequently occurring cancer in women.6 In contrast, classic KS generally affects older men of Eastern European and Mediterranean descent or Ashkenazi Jewish ancestry. Patients with classic KS generally exhibit a less aggressive disease trajectory relative to other subtypes; however, these patients have a substantial risk for a secondary hematologic malignancy, which may already coexist at the time of diagnosis or emerge subsequently.1,7
Our patient, a native of Eastern Europe, was negative for HIV and was in a monogamous relationship with his wife; therefore, he was likely to have had the classic subtype of KS. As KS is a multifocal disease, lesions may independently emerge at different times and locations on the body. Our patient presented with a new lesion on the hand several years after excision of a similar lesion on the face. Lesions suspicious for KS include slow-growing, painless, red or violaceous patches, nodules, plaques, or patches on the extremities, most commonly manifesting on the feet and ankles. Our differential diagnosis included pyogenic granuloma, amelanotic melanoma, squamous cell carcinoma, and angiosarcoma.
The prognosis in patients with classic KS is favorable, as it often is limited to cutaneous sites and less commonly manifests on visceral organs. Nonetheless, pulmonary and gastrointestinal involvement manifesting as hemoptysis and rectal bleeding, respectively, can occur. This underscores the potential for more serious complications in instances with visceral involvement. Treatment focuses on managing symptoms and preventing growth and progression of individual lesions. Additionally, treatment strategies aim to improve cosmetic outcomes and address any underlying immunosuppression that may exacerbate the condition.8
For most patients, local therapies such as surgical excision, cryotherapy, laser therapy, or intralesional chemotherapy will remove or reduce individual lesions. Patients with widespread cutaneous or extracutaneous disease may require immunomodulatory agents such as interferon α or chemotherapeutic agents such as anthracyclines or paclitaxel.8
Our case highlights the importance of considering risk factors beyond HIV status when including KS as part of the differential diagnosis in patients with atypical vascular lesions. Early recognition enables timely evaluation of potential associated conditions and informs subsequent management decisions.
THE DIAGNOSIS: Kaposi Sarcoma
Histopathology revealed a serum crust on the surface of the specimen, and the dermis contained compact collections of spindled cells with interspersed erythrocytes (Figure 1). Human herpesvirus 8–stained sections highlighted many lesional cell nuclei (Figure 2). A diagnosis of Kaposi sarcoma (KS) was made based on these findings. The patient expressed interest in surgical excision; however, he was lost to follow-up.
Kaposi sarcoma is an indolent, multifocal, angioproliferative tumor that predominantly affects mucocutaneous sites with less frequent involvement of visceral organs. Kaposi sarcoma is categorized into 4 subtypes: epidemic, iatrogenic, endemic, and classic. Human herpesvirus 8, primarily transmitted through saliva or sexual contact, plays a central role in the pathogenesis of KS, as it drives disease development across all subtypes. The virus causes proliferation of endothelial cells and the formation of angioproliferative lesions characteristic of KS.1
Prevalence is highest in the epidemic subtype, in which patients with advanced HIV and low CD4 T-cell counts may develop KS lesions. Although KS is associated most commonly with HIV, it also has been observed in men who have sex with men regardless of their HIV status.2 Patients undergoing immunosuppressive therapy also may not maintain immune tolerance to previously or newly acquired human herpesvirus 8, leading to the development of iatrogenic KS. This subtype particularly manifests in patients receiving therapy for autoimmune conditions or organ transplants and often only regresses if immunosuppressive therapy is withdrawn.3,4
The endemic and classic subtypes of KS may occur in patients without any known immunocompromise. Endemic KS demonstrates a predilection for pediatric populations in Africa and exhibits less pronounced sex disparity.5 In Uganda and Zimbabwe, endemic KS is the leading cancer in men and the second most frequently occurring cancer in women.6 In contrast, classic KS generally affects older men of Eastern European and Mediterranean descent or Ashkenazi Jewish ancestry. Patients with classic KS generally exhibit a less aggressive disease trajectory relative to other subtypes; however, these patients have a substantial risk for a secondary hematologic malignancy, which may already coexist at the time of diagnosis or emerge subsequently.1,7
Our patient, a native of Eastern Europe, was negative for HIV and was in a monogamous relationship with his wife; therefore, he was likely to have had the classic subtype of KS. As KS is a multifocal disease, lesions may independently emerge at different times and locations on the body. Our patient presented with a new lesion on the hand several years after excision of a similar lesion on the face. Lesions suspicious for KS include slow-growing, painless, red or violaceous patches, nodules, plaques, or patches on the extremities, most commonly manifesting on the feet and ankles. Our differential diagnosis included pyogenic granuloma, amelanotic melanoma, squamous cell carcinoma, and angiosarcoma.
The prognosis in patients with classic KS is favorable, as it often is limited to cutaneous sites and less commonly manifests on visceral organs. Nonetheless, pulmonary and gastrointestinal involvement manifesting as hemoptysis and rectal bleeding, respectively, can occur. This underscores the potential for more serious complications in instances with visceral involvement. Treatment focuses on managing symptoms and preventing growth and progression of individual lesions. Additionally, treatment strategies aim to improve cosmetic outcomes and address any underlying immunosuppression that may exacerbate the condition.8
For most patients, local therapies such as surgical excision, cryotherapy, laser therapy, or intralesional chemotherapy will remove or reduce individual lesions. Patients with widespread cutaneous or extracutaneous disease may require immunomodulatory agents such as interferon α or chemotherapeutic agents such as anthracyclines or paclitaxel.8
Our case highlights the importance of considering risk factors beyond HIV status when including KS as part of the differential diagnosis in patients with atypical vascular lesions. Early recognition enables timely evaluation of potential associated conditions and informs subsequent management decisions.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Lanternier F, Lebbé C, Schartz N, et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS. 2008;22:1163-1168. doi:10.1097/QAD.0b013e3283031a8a
- Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation. 1997;64:669-673. doi:10.1097/00007890-199709150-00001
- Gallo Marin B, Maymone MBC, El Rayess F, et al. Kaposi sarcoma associated with tofacitinib use in a patient with rheumatoid arthritis. R I Med J (2013). 2023;106:18-20.
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. Updated June 5, 2023. Accessed May 15, 2026. https://www.ncbi.nlm .nih.gov/books/NBK534839/
- Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer. 2003;88:1-3. doi:10.1038 /sj.bjc.6600745
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi sarcoma. Mod Pathol. 2008;21:572-582. doi:10.1038/modpathol.2008.15
- Ceccarelli M, Facciolà A, Taibi R, et al. The treatment of Kaposi’s sarcoma: present and future options, a review of the literature. Eur Rev Med Pharmacol Sci. 2019;23:7488-7497. doi:10.26355 /eurrev_201909_18860
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Lanternier F, Lebbé C, Schartz N, et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS. 2008;22:1163-1168. doi:10.1097/QAD.0b013e3283031a8a
- Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation. 1997;64:669-673. doi:10.1097/00007890-199709150-00001
- Gallo Marin B, Maymone MBC, El Rayess F, et al. Kaposi sarcoma associated with tofacitinib use in a patient with rheumatoid arthritis. R I Med J (2013). 2023;106:18-20.
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. Updated June 5, 2023. Accessed May 15, 2026. https://www.ncbi.nlm .nih.gov/books/NBK534839/
- Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer. 2003;88:1-3. doi:10.1038 /sj.bjc.6600745
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi sarcoma. Mod Pathol. 2008;21:572-582. doi:10.1038/modpathol.2008.15
- Ceccarelli M, Facciolà A, Taibi R, et al. The treatment of Kaposi’s sarcoma: present and future options, a review of the literature. Eur Rev Med Pharmacol Sci. 2019;23:7488-7497. doi:10.26355 /eurrev_201909_18860
Exophytic Papule on the Hand
Exophytic Papule on the Hand
A man in his 70s with a history of hypertension was admitted to the hospital for symptomatic bradycardia. On the day of admission, he reported a growth on the left second digit of 1 month’s duration, for which dermatology was consulted. The patient said the growth was asymptomatic but occasionally would get caught on objects. He denied any recent fevers, weight loss, or fatigue. He also denied any trauma to the area or other inciting factors. The patient reported there were no lesions anywhere else on the body, but he did mention a similar mass had been excised from his face several years prior. He noted that he had immigrated to the United States from Eastern Europe within the past several years.
Results of laboratory testing at the current presentation, including a basic metabolic panel, complete blood count with differential, hepatic function panel, thyroid-stimulating hormone level, and HIV antigen/antibody testing, were unremarkable. Physical examination revealed a single, well-circumscribed, 6×6–mm, round, red, exophytic papule with a collarette of scale on the volar surface of the left second digit. The skin on both arms was otherwise unremarkable. There was no evidence of lymphadenopathy or mucosal involvement. A shave biopsy of the lesion was performed.
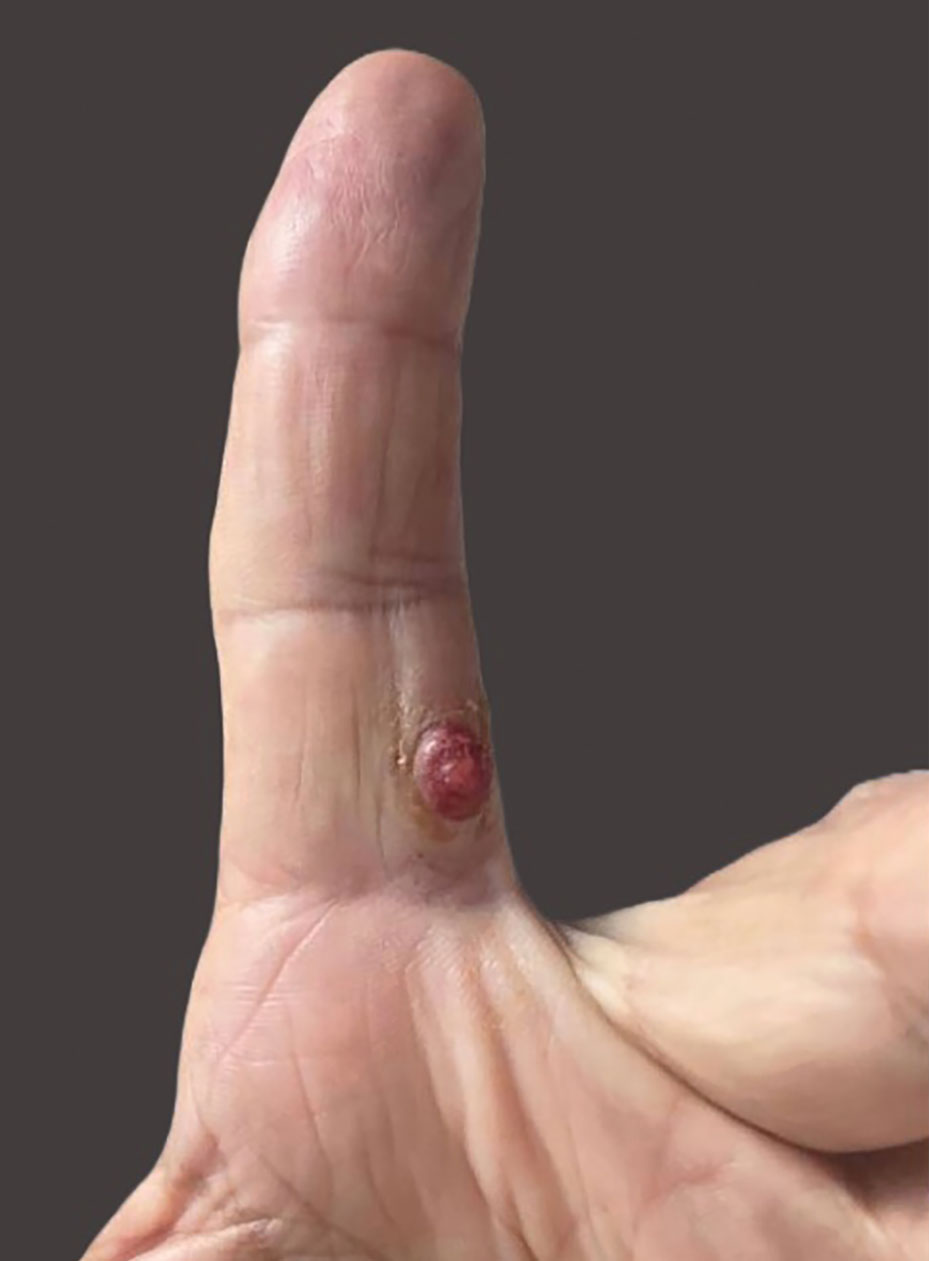

A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
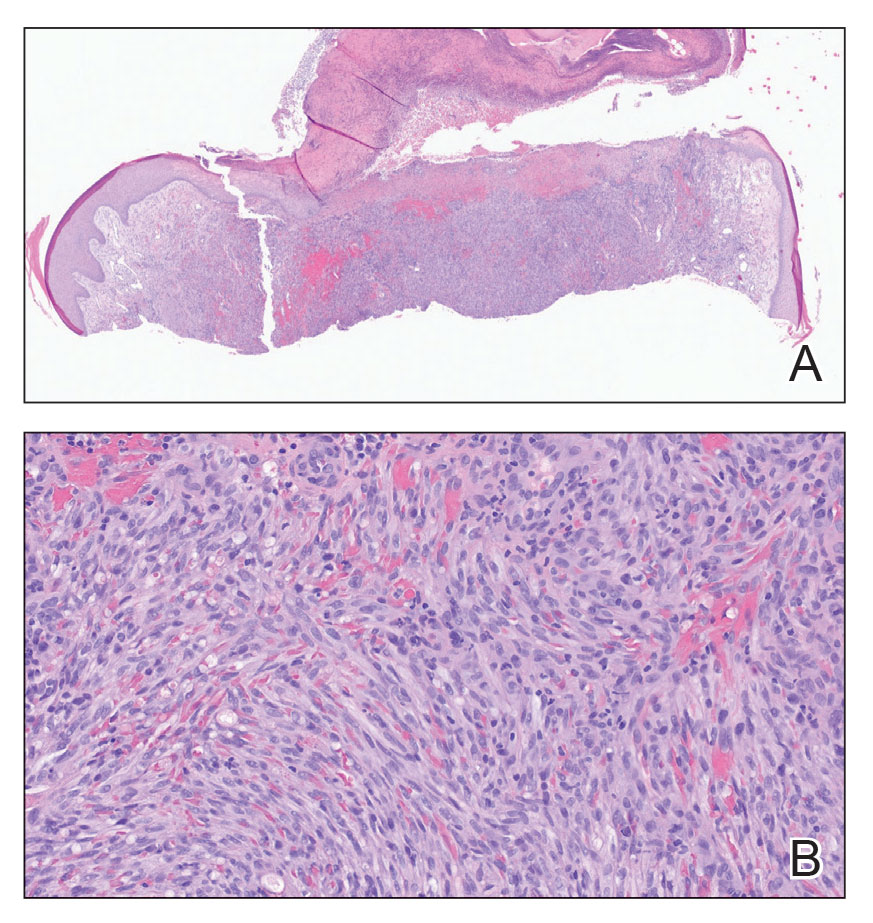
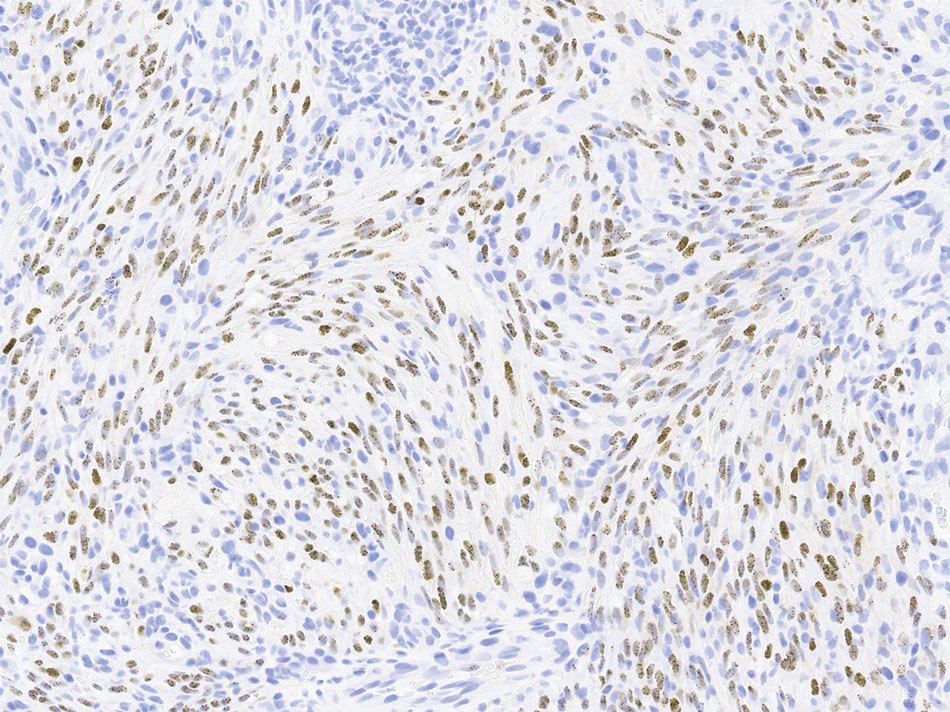
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
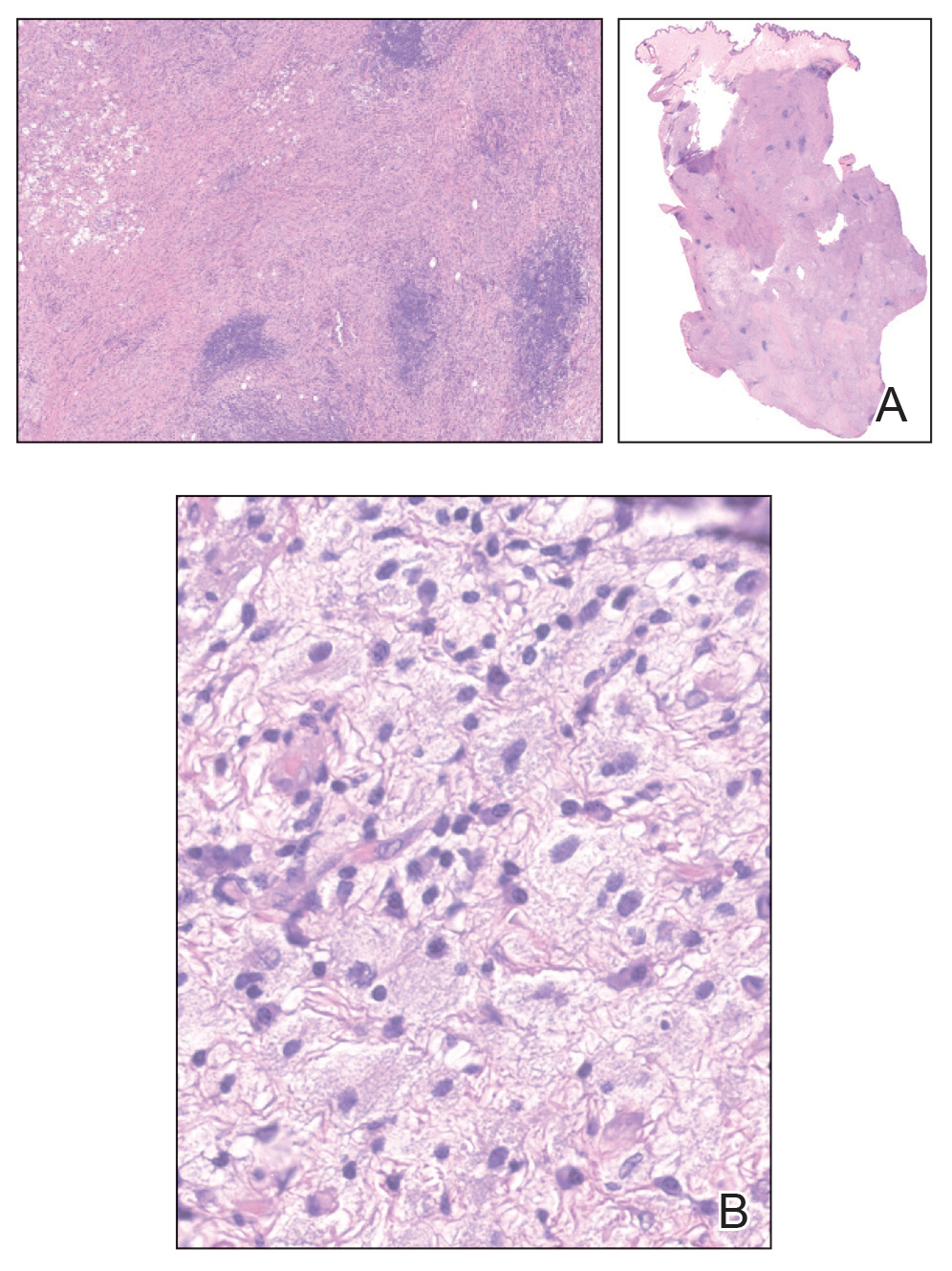
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
A 34-year-old man presented to our dermatology clinic for evaluation of a lesion in the right axilla of 1 year’s duration that had recently increased in size. The lesion was nontender and intermittently pruritic and was associated with focal hypohidrosis. The patient denied any fevers, chills, or recent weight change. His medical history was otherwise unremarkable. His only medications were daily ashwagandha and vitamin B and C supplements. On physical examination, a firm, 6-cm, subcutaneous nodule was noted in the right axilla with central alopecia and without a clear punctum. He had no palpable cervical, postauricular, or inguinal lymphadenopathy. The left axilla was clear, and there were no other relevant skin findings. Laboratory testing including a complete blood count, comprehensive metabolic panel, and sexually transmitted infections panel was unremarkable. Ultrasonography and subsequent magnetic resonance imaging of the right axilla showed a 4.9-cm nodule located in the subcutaneous fat with minimal deep infiltration and relatively smooth margins. An incisional biopsy of the lesion was performed.
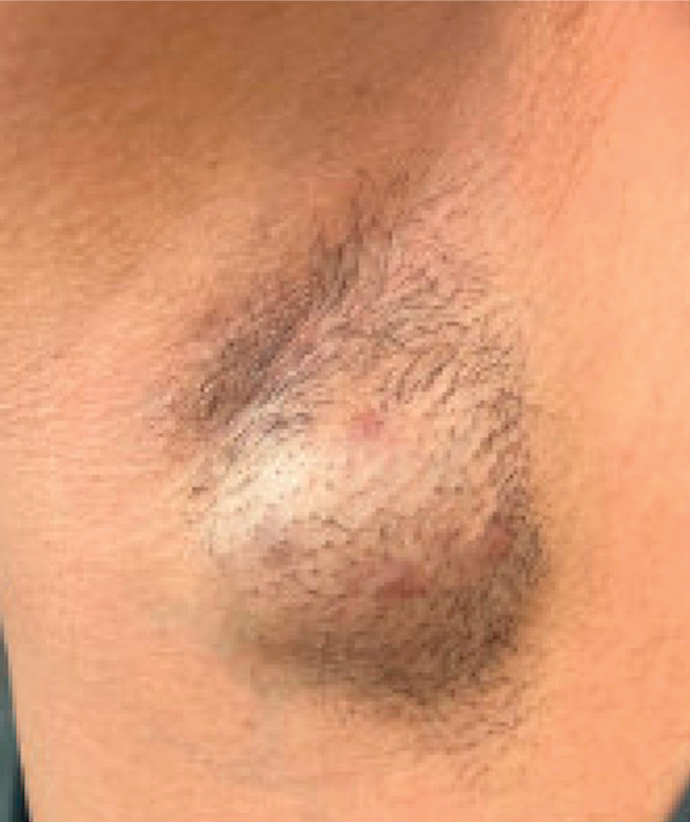

Multiple Papules and Pustules on the Face and Neck
Multiple Papules and Pustules on the Face and Neck
THE DIAGNOSIS: Demodicosis
Direct microscopic examination of the purulent fluid revealed a considerable number of actively motile Demodex mites (Figure). Based on the microscopy results and the patient’s history of prolonged topical immunosuppressive therapy, a known risk factor for Demodex overgrowth, a diagnosis of demodicosis was made. The patient was prescribed a single dose of oral metronidazole 2 g as well as metronidazole solution 0.5% to be applied 3 times daily. The folliculitis gradually improved and eventually resolved completely.
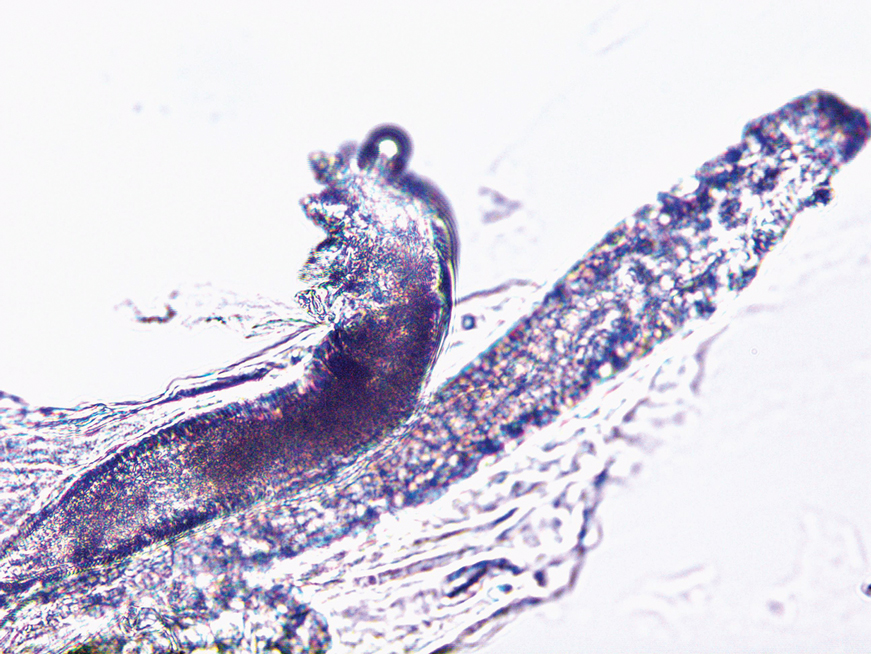
Demodex is a parasitic mite inhabiting the pilosebaceous units of human skin. Evidence suggests the vast majority of adults carry these mites. Demodex mites maintain a balance with the human immune system in appropriate microenvironments, with the immune system controlling their numbers without eliciting an inflammatory response; however, immunosuppression, as induced by topical corticosteroids and other immunomodulators, can lead to an increase in Demodex mite populations on facial skin. Clinical manifestations and severity of demodicosis are highly variable, ranging from nonspecific dry, sensitive skin and papules to nodules or granulomas, depending on mite density, the cutaneous microenvironment, and the host immune response.1 Consequently, demodicosis often is mistaken for other dermatologic conditions with similar skin lesions.
High Demodex mite density is considered a pathogenic factor in demodicosis; therefore, determining Demodex mite density is essential to the diagnosis of demodicosis. Standard skin surface biopsy and direct microscopic examination commonly are used methods for measuring Demodex mite density; however, the accuracy of these methods is subject to the technical proficiency of the investigator. Noninvasive examination tools like dermoscopy and confocal laser scanning also offer advantages in diagnosing demodicosis. Dermoscopy, by direct contact with skin lesions, typically reveals gelatinous filaments extending from the follicular openings.
Importantly, Demodex mite density alone does not determine the severity of clinical symptoms. In addition, mites may migrate to the skin surface or reside deep within follicles, rendering them difficult to detect with standard examination methods.1 Therefore, diagnostic criteria should extend beyond mite proliferation to include characteristic clinical lesions, response to acaricidal therapy, and normalization of mite density.
Rosacea was included in the differential diagnosis for our patient, but it typically manifests in the central facial area (eg, forehead, nose, chin). Patients may have a history of facial flushing associated with alcohol consumption, heat exposure, or emotional stress.2 Additionally, rosacea typically has an insidious onset and does not erupt suddenly within a short period of time; however, our patient presented with a sudden onset of widespread papules and pustules on the face without facial flushing, and there was no exacerbation upon exposure to heat or emotional stress. Furthermore, rosacea tends to be recurrent and challenging to cure, whereas our patient responded rapidly to treatment without recurrence. Therefore, the likelihood of rosacea was minimal. Histopathologic examination also can differentiate between rosacea and demodicosis. Histologically, the features of rosacea include dilated blood and lymphatic vessels and infiltration of T lymphocytes, macrophages, and mast cells around blood vessels, often with increased solar elastosis and dermal edema.3 Demodicosis can reveal Demodex mites within the infundibulum of hair follicles, with dense neutrophil and monocyte infiltration around and between the infundibula.4
Bacterial folliculitis is primarily characterized by perifollicular erythema, papules, and pustules, often accompanied by pain. Positive bacterial culture of purulent fluid is indicative.5 Our patient’s lesions shared certain similarities with bacterial folliculitis but lacked the characteristic pain, instead exhibiting pronounced pruritus. Remarkable therapeutic efficacy was observed following topical acaricidal treatment, thus rendering the diagnosis of bacterial folliculitis less probable.
Acne vulgaris is a noninfectious folliculitis caused by follicular occlusion. Abnormal keratinization leads to the obstruction of follicles by keratin, hindering the outflow of sebum from the follicles. Sebum accumulation within the follicles provides a rich substrate for Propionibacterium acnes, which metabolizes sebum into proinflammatory free fatty acids, resulting in the formation of comedones, papules, and pustules.5 Our patient did not exhibit comedonal lesions on the face and lacked a seborrheic complexion, hence diminishing the likelihood of acne vulgaris.
Tinea corporis is another intensely pruritic condition, especially in areas subjected to prolonged use of topical immunosuppressants. It is caused by dermatophyte fungi and typically manifests as erythematous pruritic patches, often presenting as ring-shaped lesions with active margins and sometimes accompanied by scaling.6 While long-term use of immunosuppressants may be a risk factor for fungal infections and increase the probability of tinea corporis, our patient’s presentation of papules and pustules without a ring-shaped configuration or scaling diminished the likelihood of tinea corporis.
Our patient represents an intriguing case of an eruptive form of demodicosis induced by long-term intermittent and inconsistent application of topical immunosuppressive agents. Demodicosis encompasses a spectrum of clinical presentations, including pityriasis folliculorum, rosacealike, folliculitislike, and perioral dermatitis–like forms.1 It is prone to misdiagnosis, as it is clinically similar to other conditions, such as acne, rosacea, or bacterial folliculitis, and it also is susceptible to missed diagnosis. Demodicosis tends to erupt in immunocompromised individuals, and the use of topical immunosuppressive and corticosteroid medications can exacerbate Demodex activity. Dermatologists should be aware that demodicosis is not a rare skin disorder, and timely identification and diagnosis can reduce the incidence of disease and improve quality of life for affected patients. Conversely, the consequences of misdiagnosis can be severe, with inappropriate treatment potentially exacerbating the condition.
- Paichitrojjana A. Demodex: the worst enemies are the ones that used to be friends. Dermatol Reports. 2022;14:9339. doi:10.4081 /dr.2022.9339
- Del RJ, Baldwin H, Bhatia N, et al. A review of the diagnostic and therapeutic gaps in rosacea management: consensus opinion. Dermatol Ther (Heidelb). 2024;14:271-284. doi:10.1007/s13555-023-01087-8
- Powell FC. The histopathology of rosacea: ‘where’s the beef?’ Dermatology. 2004;209:173-174. doi:10.1159/000079884
- Helou W, Avitan-Hersh E, Bergman R. Demodex folliculitis of the scalp: clinicopathological study of an uncommon entity. Am J Dermatopathol. 2016;38:658-663. doi:10.1097/DAD.0000000000000512
- Laureano AC, Schwartz RA, Cohen PJ. Facial bacterial infections: folliculitis. Clin Dermatol. 2014;32:711-714. doi:10.1016 /j.clindermatol.2014.02.009
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9. doi:10.7573/dic.2020-5-6
THE DIAGNOSIS: Demodicosis
Direct microscopic examination of the purulent fluid revealed a considerable number of actively motile Demodex mites (Figure). Based on the microscopy results and the patient’s history of prolonged topical immunosuppressive therapy, a known risk factor for Demodex overgrowth, a diagnosis of demodicosis was made. The patient was prescribed a single dose of oral metronidazole 2 g as well as metronidazole solution 0.5% to be applied 3 times daily. The folliculitis gradually improved and eventually resolved completely.
Demodex is a parasitic mite inhabiting the pilosebaceous units of human skin. Evidence suggests the vast majority of adults carry these mites. Demodex mites maintain a balance with the human immune system in appropriate microenvironments, with the immune system controlling their numbers without eliciting an inflammatory response; however, immunosuppression, as induced by topical corticosteroids and other immunomodulators, can lead to an increase in Demodex mite populations on facial skin. Clinical manifestations and severity of demodicosis are highly variable, ranging from nonspecific dry, sensitive skin and papules to nodules or granulomas, depending on mite density, the cutaneous microenvironment, and the host immune response.1 Consequently, demodicosis often is mistaken for other dermatologic conditions with similar skin lesions.
High Demodex mite density is considered a pathogenic factor in demodicosis; therefore, determining Demodex mite density is essential to the diagnosis of demodicosis. Standard skin surface biopsy and direct microscopic examination commonly are used methods for measuring Demodex mite density; however, the accuracy of these methods is subject to the technical proficiency of the investigator. Noninvasive examination tools like dermoscopy and confocal laser scanning also offer advantages in diagnosing demodicosis. Dermoscopy, by direct contact with skin lesions, typically reveals gelatinous filaments extending from the follicular openings.
Importantly, Demodex mite density alone does not determine the severity of clinical symptoms. In addition, mites may migrate to the skin surface or reside deep within follicles, rendering them difficult to detect with standard examination methods.1 Therefore, diagnostic criteria should extend beyond mite proliferation to include characteristic clinical lesions, response to acaricidal therapy, and normalization of mite density.
Rosacea was included in the differential diagnosis for our patient, but it typically manifests in the central facial area (eg, forehead, nose, chin). Patients may have a history of facial flushing associated with alcohol consumption, heat exposure, or emotional stress.2 Additionally, rosacea typically has an insidious onset and does not erupt suddenly within a short period of time; however, our patient presented with a sudden onset of widespread papules and pustules on the face without facial flushing, and there was no exacerbation upon exposure to heat or emotional stress. Furthermore, rosacea tends to be recurrent and challenging to cure, whereas our patient responded rapidly to treatment without recurrence. Therefore, the likelihood of rosacea was minimal. Histopathologic examination also can differentiate between rosacea and demodicosis. Histologically, the features of rosacea include dilated blood and lymphatic vessels and infiltration of T lymphocytes, macrophages, and mast cells around blood vessels, often with increased solar elastosis and dermal edema.3 Demodicosis can reveal Demodex mites within the infundibulum of hair follicles, with dense neutrophil and monocyte infiltration around and between the infundibula.4
Bacterial folliculitis is primarily characterized by perifollicular erythema, papules, and pustules, often accompanied by pain. Positive bacterial culture of purulent fluid is indicative.5 Our patient’s lesions shared certain similarities with bacterial folliculitis but lacked the characteristic pain, instead exhibiting pronounced pruritus. Remarkable therapeutic efficacy was observed following topical acaricidal treatment, thus rendering the diagnosis of bacterial folliculitis less probable.
Acne vulgaris is a noninfectious folliculitis caused by follicular occlusion. Abnormal keratinization leads to the obstruction of follicles by keratin, hindering the outflow of sebum from the follicles. Sebum accumulation within the follicles provides a rich substrate for Propionibacterium acnes, which metabolizes sebum into proinflammatory free fatty acids, resulting in the formation of comedones, papules, and pustules.5 Our patient did not exhibit comedonal lesions on the face and lacked a seborrheic complexion, hence diminishing the likelihood of acne vulgaris.
Tinea corporis is another intensely pruritic condition, especially in areas subjected to prolonged use of topical immunosuppressants. It is caused by dermatophyte fungi and typically manifests as erythematous pruritic patches, often presenting as ring-shaped lesions with active margins and sometimes accompanied by scaling.6 While long-term use of immunosuppressants may be a risk factor for fungal infections and increase the probability of tinea corporis, our patient’s presentation of papules and pustules without a ring-shaped configuration or scaling diminished the likelihood of tinea corporis.
Our patient represents an intriguing case of an eruptive form of demodicosis induced by long-term intermittent and inconsistent application of topical immunosuppressive agents. Demodicosis encompasses a spectrum of clinical presentations, including pityriasis folliculorum, rosacealike, folliculitislike, and perioral dermatitis–like forms.1 It is prone to misdiagnosis, as it is clinically similar to other conditions, such as acne, rosacea, or bacterial folliculitis, and it also is susceptible to missed diagnosis. Demodicosis tends to erupt in immunocompromised individuals, and the use of topical immunosuppressive and corticosteroid medications can exacerbate Demodex activity. Dermatologists should be aware that demodicosis is not a rare skin disorder, and timely identification and diagnosis can reduce the incidence of disease and improve quality of life for affected patients. Conversely, the consequences of misdiagnosis can be severe, with inappropriate treatment potentially exacerbating the condition.
THE DIAGNOSIS: Demodicosis
Direct microscopic examination of the purulent fluid revealed a considerable number of actively motile Demodex mites (Figure). Based on the microscopy results and the patient’s history of prolonged topical immunosuppressive therapy, a known risk factor for Demodex overgrowth, a diagnosis of demodicosis was made. The patient was prescribed a single dose of oral metronidazole 2 g as well as metronidazole solution 0.5% to be applied 3 times daily. The folliculitis gradually improved and eventually resolved completely.
Demodex is a parasitic mite inhabiting the pilosebaceous units of human skin. Evidence suggests the vast majority of adults carry these mites. Demodex mites maintain a balance with the human immune system in appropriate microenvironments, with the immune system controlling their numbers without eliciting an inflammatory response; however, immunosuppression, as induced by topical corticosteroids and other immunomodulators, can lead to an increase in Demodex mite populations on facial skin. Clinical manifestations and severity of demodicosis are highly variable, ranging from nonspecific dry, sensitive skin and papules to nodules or granulomas, depending on mite density, the cutaneous microenvironment, and the host immune response.1 Consequently, demodicosis often is mistaken for other dermatologic conditions with similar skin lesions.
High Demodex mite density is considered a pathogenic factor in demodicosis; therefore, determining Demodex mite density is essential to the diagnosis of demodicosis. Standard skin surface biopsy and direct microscopic examination commonly are used methods for measuring Demodex mite density; however, the accuracy of these methods is subject to the technical proficiency of the investigator. Noninvasive examination tools like dermoscopy and confocal laser scanning also offer advantages in diagnosing demodicosis. Dermoscopy, by direct contact with skin lesions, typically reveals gelatinous filaments extending from the follicular openings.
Importantly, Demodex mite density alone does not determine the severity of clinical symptoms. In addition, mites may migrate to the skin surface or reside deep within follicles, rendering them difficult to detect with standard examination methods.1 Therefore, diagnostic criteria should extend beyond mite proliferation to include characteristic clinical lesions, response to acaricidal therapy, and normalization of mite density.
Rosacea was included in the differential diagnosis for our patient, but it typically manifests in the central facial area (eg, forehead, nose, chin). Patients may have a history of facial flushing associated with alcohol consumption, heat exposure, or emotional stress.2 Additionally, rosacea typically has an insidious onset and does not erupt suddenly within a short period of time; however, our patient presented with a sudden onset of widespread papules and pustules on the face without facial flushing, and there was no exacerbation upon exposure to heat or emotional stress. Furthermore, rosacea tends to be recurrent and challenging to cure, whereas our patient responded rapidly to treatment without recurrence. Therefore, the likelihood of rosacea was minimal. Histopathologic examination also can differentiate between rosacea and demodicosis. Histologically, the features of rosacea include dilated blood and lymphatic vessels and infiltration of T lymphocytes, macrophages, and mast cells around blood vessels, often with increased solar elastosis and dermal edema.3 Demodicosis can reveal Demodex mites within the infundibulum of hair follicles, with dense neutrophil and monocyte infiltration around and between the infundibula.4
Bacterial folliculitis is primarily characterized by perifollicular erythema, papules, and pustules, often accompanied by pain. Positive bacterial culture of purulent fluid is indicative.5 Our patient’s lesions shared certain similarities with bacterial folliculitis but lacked the characteristic pain, instead exhibiting pronounced pruritus. Remarkable therapeutic efficacy was observed following topical acaricidal treatment, thus rendering the diagnosis of bacterial folliculitis less probable.
Acne vulgaris is a noninfectious folliculitis caused by follicular occlusion. Abnormal keratinization leads to the obstruction of follicles by keratin, hindering the outflow of sebum from the follicles. Sebum accumulation within the follicles provides a rich substrate for Propionibacterium acnes, which metabolizes sebum into proinflammatory free fatty acids, resulting in the formation of comedones, papules, and pustules.5 Our patient did not exhibit comedonal lesions on the face and lacked a seborrheic complexion, hence diminishing the likelihood of acne vulgaris.
Tinea corporis is another intensely pruritic condition, especially in areas subjected to prolonged use of topical immunosuppressants. It is caused by dermatophyte fungi and typically manifests as erythematous pruritic patches, often presenting as ring-shaped lesions with active margins and sometimes accompanied by scaling.6 While long-term use of immunosuppressants may be a risk factor for fungal infections and increase the probability of tinea corporis, our patient’s presentation of papules and pustules without a ring-shaped configuration or scaling diminished the likelihood of tinea corporis.
Our patient represents an intriguing case of an eruptive form of demodicosis induced by long-term intermittent and inconsistent application of topical immunosuppressive agents. Demodicosis encompasses a spectrum of clinical presentations, including pityriasis folliculorum, rosacealike, folliculitislike, and perioral dermatitis–like forms.1 It is prone to misdiagnosis, as it is clinically similar to other conditions, such as acne, rosacea, or bacterial folliculitis, and it also is susceptible to missed diagnosis. Demodicosis tends to erupt in immunocompromised individuals, and the use of topical immunosuppressive and corticosteroid medications can exacerbate Demodex activity. Dermatologists should be aware that demodicosis is not a rare skin disorder, and timely identification and diagnosis can reduce the incidence of disease and improve quality of life for affected patients. Conversely, the consequences of misdiagnosis can be severe, with inappropriate treatment potentially exacerbating the condition.
- Paichitrojjana A. Demodex: the worst enemies are the ones that used to be friends. Dermatol Reports. 2022;14:9339. doi:10.4081 /dr.2022.9339
- Del RJ, Baldwin H, Bhatia N, et al. A review of the diagnostic and therapeutic gaps in rosacea management: consensus opinion. Dermatol Ther (Heidelb). 2024;14:271-284. doi:10.1007/s13555-023-01087-8
- Powell FC. The histopathology of rosacea: ‘where’s the beef?’ Dermatology. 2004;209:173-174. doi:10.1159/000079884
- Helou W, Avitan-Hersh E, Bergman R. Demodex folliculitis of the scalp: clinicopathological study of an uncommon entity. Am J Dermatopathol. 2016;38:658-663. doi:10.1097/DAD.0000000000000512
- Laureano AC, Schwartz RA, Cohen PJ. Facial bacterial infections: folliculitis. Clin Dermatol. 2014;32:711-714. doi:10.1016 /j.clindermatol.2014.02.009
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9. doi:10.7573/dic.2020-5-6
- Paichitrojjana A. Demodex: the worst enemies are the ones that used to be friends. Dermatol Reports. 2022;14:9339. doi:10.4081 /dr.2022.9339
- Del RJ, Baldwin H, Bhatia N, et al. A review of the diagnostic and therapeutic gaps in rosacea management: consensus opinion. Dermatol Ther (Heidelb). 2024;14:271-284. doi:10.1007/s13555-023-01087-8
- Powell FC. The histopathology of rosacea: ‘where’s the beef?’ Dermatology. 2004;209:173-174. doi:10.1159/000079884
- Helou W, Avitan-Hersh E, Bergman R. Demodex folliculitis of the scalp: clinicopathological study of an uncommon entity. Am J Dermatopathol. 2016;38:658-663. doi:10.1097/DAD.0000000000000512
- Laureano AC, Schwartz RA, Cohen PJ. Facial bacterial infections: folliculitis. Clin Dermatol. 2014;32:711-714. doi:10.1016 /j.clindermatol.2014.02.009
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9. doi:10.7573/dic.2020-5-6
Multiple Papules and Pustules on the Face and Neck
Multiple Papules and Pustules on the Face and Neck
A 26-year-old woman presented to our clinic with multiple papules and pustules on the face and neck. One year prior, the patient had developed a pruritic rash on the face after using a new over-the-counter skin care product. An outside physician had diagnosed the rash as contact dermatitis and prescribed tacrolimus cream 0.1%. Initially, the patient noted improvement, but the rash recurred intermittently over the next year. She continued using the cream, but 2 months prior to the current presentation, the patient developed more papules and pustules on the face, prompting further evaluation.
Physical examination at the current presentation revealed widespread papules and pustules on the face and neck. Due to the patient’s aesthetic concerns, a more invasive biopsy was avoided, and purulent fluid from the lesions was collected for microscopic examination.
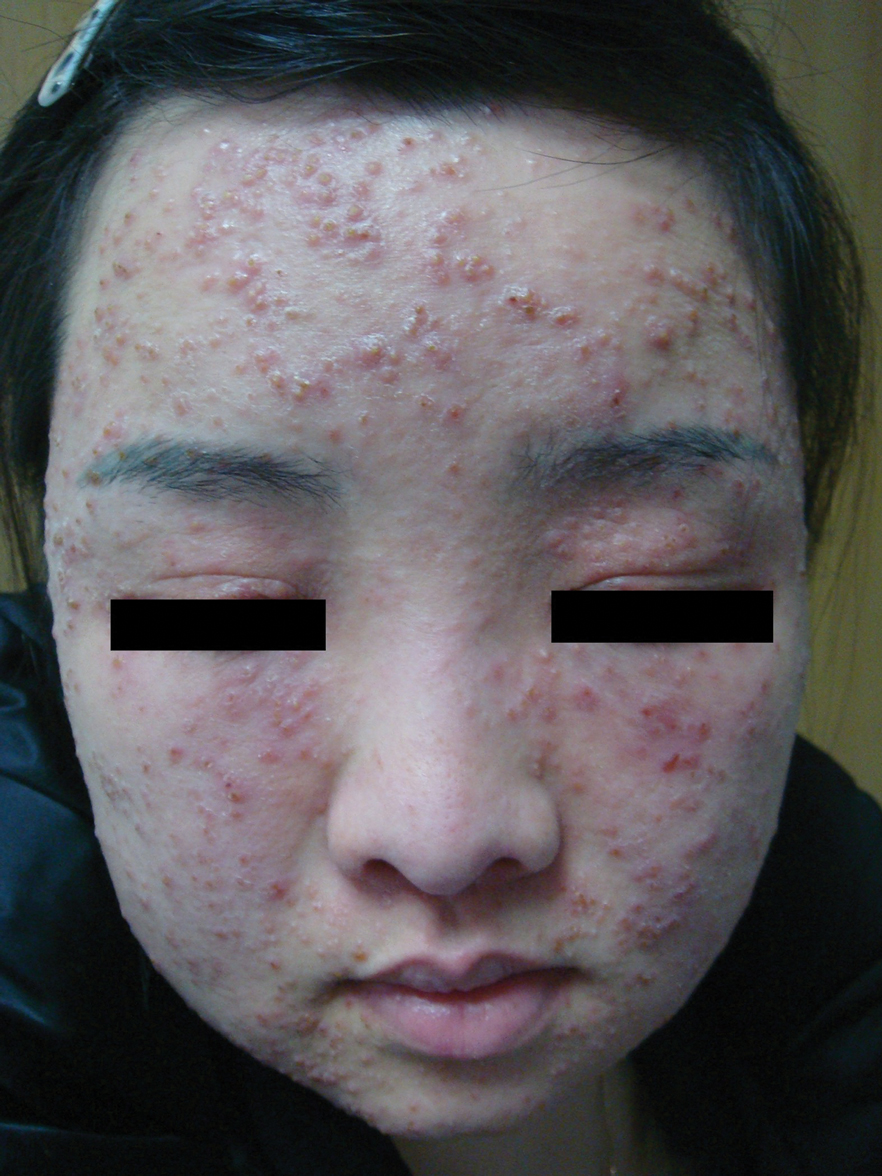

Enlarging Mass on the Scalp
Enlarging Mass on the Scalp
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
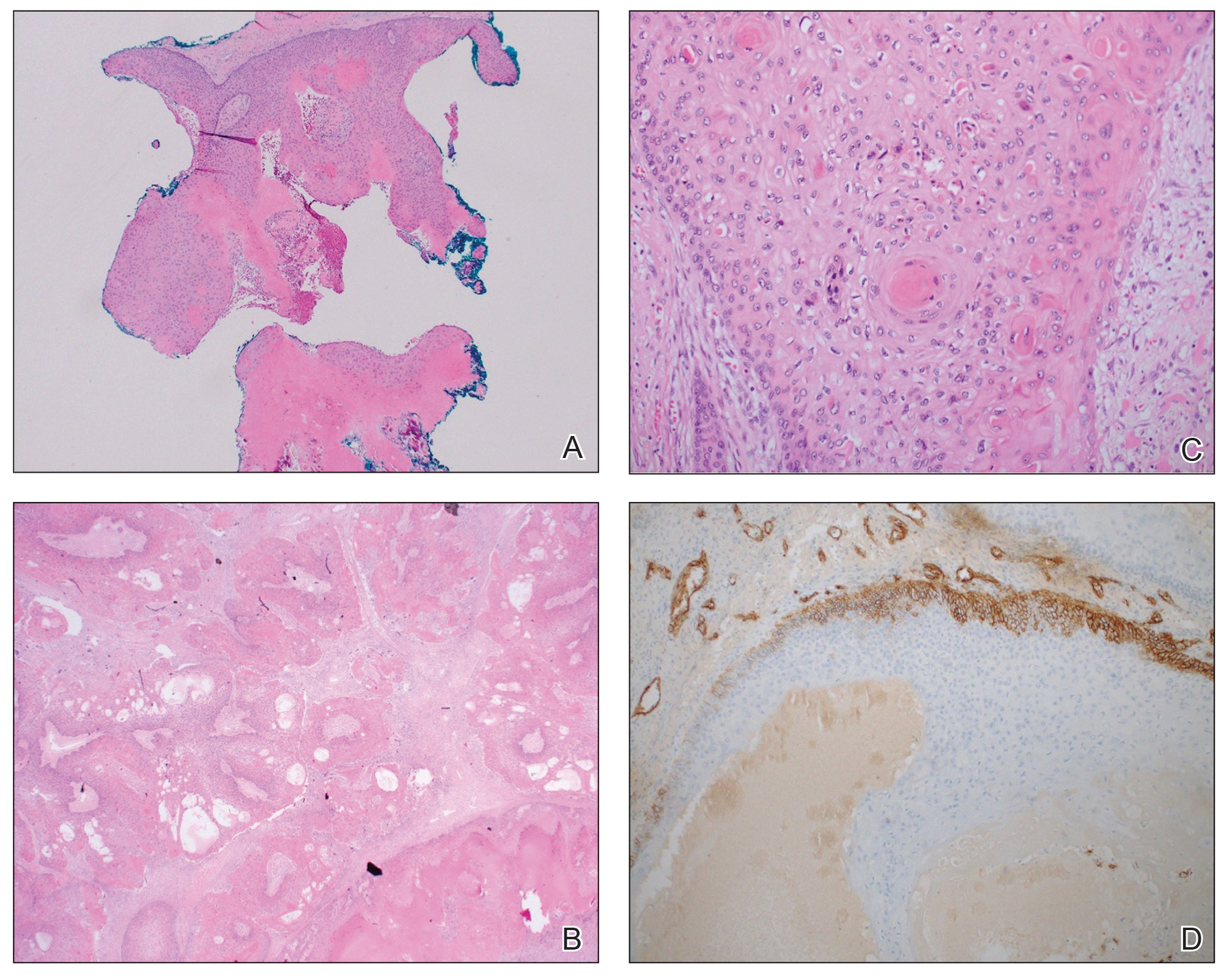
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
Enlarging Mass on the Scalp
Enlarging Mass on the Scalp
A 61-year-old woman presented to the emergency department with worsening pain and bleeding from a scalp tumor of 16 years’ duration. Initially noted as a small nodule on the left parietal scalp on computed tomography of the head, the mass had grown rapidly in recent years and currently measured 22×10×15 cm. At prior consultations with plastic and general surgery, the patient had declined surgical intervention. At the current presentation, biopsies were performed by plastic surgery, and a dermatopathology consultation was ordered. Histopathology revealed atypical keratinocytes, nuclear pleomorphism, lobulating epithelial masses with trichilemmal keratinization, and CD34 positivity. Subsequent computed tomography and positron emission tomography of the head showed occipital skull erosion and bilateral cervical lymphadenopathy, suggesting metastasis.
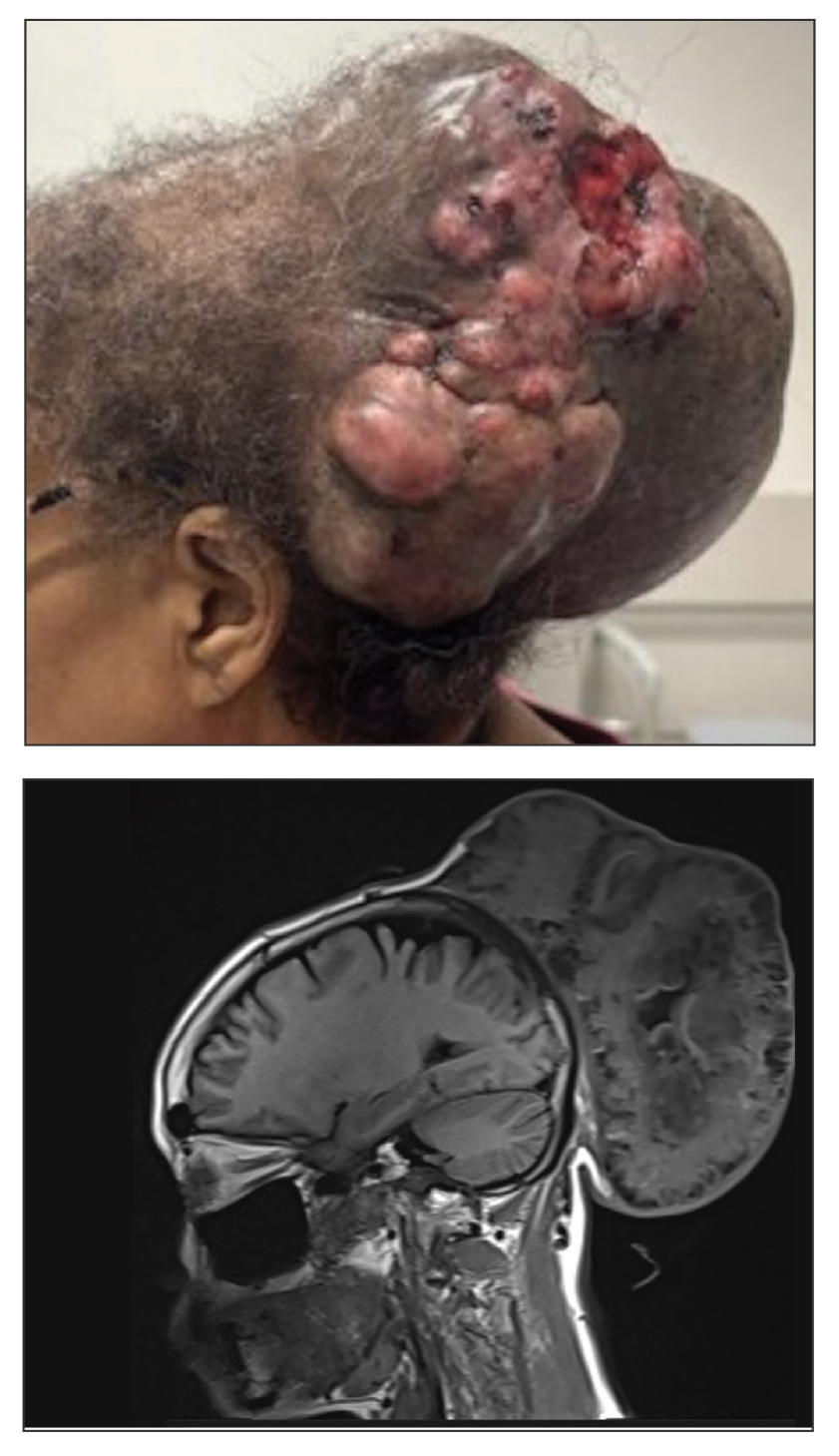

Painful, Purpuric, Nodular Lesion With an Irregular Surface on the Shoulder
Painful, Purpuric, Nodular Lesion With an Irregular Surface on the Shoulder
THE DIAGNOSIS: Cutaneous Leiomyosarcoma
Based on the clinical and histopathologic findings, our patient was diagnosed with primary cutaneous leiomyosarcoma (LMS), a rare soft-tissue neoplasm that arises from smooth muscle and typically manifests as a firm pink nodule.1 The neoplasm may occur in the area of a prior traumatic injury or develop spontaneously without an identifiable cause.1-3 Cutaneous LMS represents 2% to 3% of all soft-tissue sarcomas worldwide, with an estimated incidence of 1 in 500,000 annually.1,4 Men who are in their fifth to seventh decades of life are at the highest risk for LMS.1
Histologically, cutaneous LMS can be subclassified as dermal, which has a low metastatic risk and excellent prognosis, or subcutaneous, which is associated with poorer outcomes and vascular muscle origin.1 In our case, hematoxylin and eosin staining revealed fascicles of smooth muscle fibers with hypercellularity, atypia, and mitotic figures (Figure). The neoplasm stained positive for desmin, vimentin, and smooth muscle actin and negative for SOX10, Melan-A, PRAME (preferentially expressed antigen in melanoma), CD34, and Factor XIIIa.1
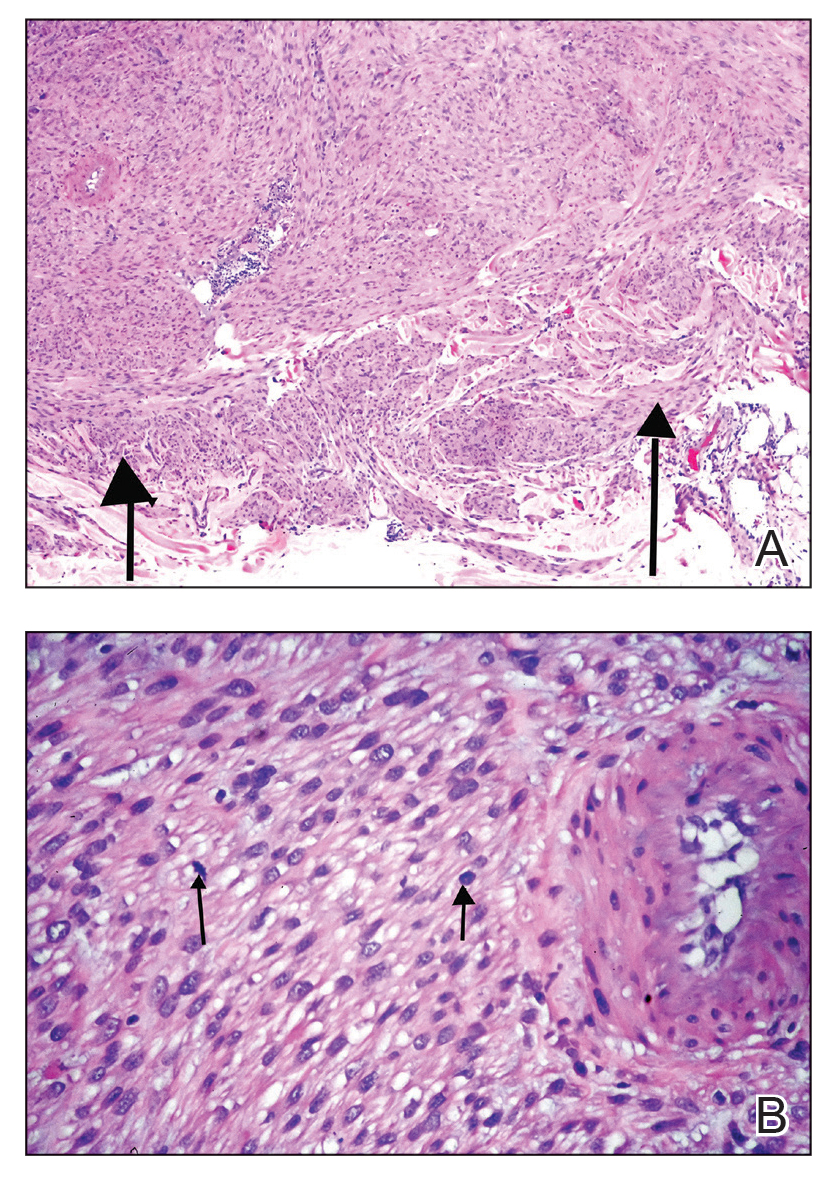
Standard treatment for LMS is surgical excision.5 Poor prognostic factors include lesions with a diameter of 5 cm or larger, deep subcutaneous tumor invasion, and distant metastases.2,5
The differential diagnosis may include dermatofibrosarcoma protuberans, which can have a similar pink nodular appearance and also may manifest after injury6; however, this lesion would stain positive for CD34 on histopathology.1 Nodular melanoma also can manifest as a solitary red, raised lesion, but it would stain positive for SOX10, PRAME, and Melan-A on histopathology.7 Basal cell carcinoma, which also may have a similar clinical appearance, is associated with nests of basaloid cells and palisading nuclei histologically.8 Lastly, atypical fibroxanthoma also manifests as a red nodule or plaque and is associated with atypical mitotic figures on histology; however, it notably stains negative for desmin.9
In summary, cutaneous LMS should be included in the differential diagnosis for raised, pink nodules. Given its nonspecific clinical presentation, this rare and malignant neoplasm requires biopsy and immunohistochemical staining for accurate diagnosis.
- Soares Queirós C, Filipe P, Soares de Almeida L. Cutaneous leiomyosarcoma: a 20-year retrospective study and review of the literature. Anais Brasileiros de Dermatologia. 2021;96:278-283. doi:10.1016/j.abd.2020.10.003
- Kim NG, Kim JO, Park YJ, et al. Cutaneous leiomyosarcoma of the face. Arch Craniofac Surg. 2017;18:145-148. doi:10.7181/acfs.2017.18.2.145
- Litaiem N, Tabka M, Nefiss M, et al. Cutaneous leiomyosarcoma mimicking arteriovenous malformation. Clin Case Rep. 2020;8:2538-2540. doi:10.1002/ccr3.3222
- Hmida L, Letaief F, Doghri R, et al. Cutaneous leiomyosarcoma on the trunk: an unusual presentation with an aggressive course - case report and review of literature. Pan Afr Med J. 2018;31:190. doi:10.11604/pamj.2018.31.190.16682
- Cazzato G, Sergi MC, Sablone S, et al. Advanced cutaneous leiomyosarcoma of the forearm. Dermatopathology (Basel). 2021;8:40-44. doi:10.3390/dermatopathology8010008
- Li Y, Wang C, Xiang B, et al. Clinical features, pathological findings and treatment of recurrent dermatofibrosarcoma protuberans. J Cancer. 2017;8:1319-1323. doi:10.7150/jca.17988
- Hernandez LE, Frech FS, Mohsin N, et al. Nodular melanoma: a review of pathogenesis, presentation, diagnosis and treatment. J Dermatol & Skin Sci. 2021;3:25-30. doi:10.29245/2767-5092/2021/3.1144
- Steele RB, Puckette Y. Basal cell carcinoma. StatPearls [Internet]. StatPearls Publishing; 2026. Updated November 7, 2025. Accessed March 3, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482439/
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls [Internet]. StatPearls Publishing; 2026. Updated September 18, 2022. Accessed March 3, 2026. http://www.ncbi.nlm.nih.gov/books/NBK459342/
THE DIAGNOSIS: Cutaneous Leiomyosarcoma
Based on the clinical and histopathologic findings, our patient was diagnosed with primary cutaneous leiomyosarcoma (LMS), a rare soft-tissue neoplasm that arises from smooth muscle and typically manifests as a firm pink nodule.1 The neoplasm may occur in the area of a prior traumatic injury or develop spontaneously without an identifiable cause.1-3 Cutaneous LMS represents 2% to 3% of all soft-tissue sarcomas worldwide, with an estimated incidence of 1 in 500,000 annually.1,4 Men who are in their fifth to seventh decades of life are at the highest risk for LMS.1
Histologically, cutaneous LMS can be subclassified as dermal, which has a low metastatic risk and excellent prognosis, or subcutaneous, which is associated with poorer outcomes and vascular muscle origin.1 In our case, hematoxylin and eosin staining revealed fascicles of smooth muscle fibers with hypercellularity, atypia, and mitotic figures (Figure). The neoplasm stained positive for desmin, vimentin, and smooth muscle actin and negative for SOX10, Melan-A, PRAME (preferentially expressed antigen in melanoma), CD34, and Factor XIIIa.1
Standard treatment for LMS is surgical excision.5 Poor prognostic factors include lesions with a diameter of 5 cm or larger, deep subcutaneous tumor invasion, and distant metastases.2,5
The differential diagnosis may include dermatofibrosarcoma protuberans, which can have a similar pink nodular appearance and also may manifest after injury6; however, this lesion would stain positive for CD34 on histopathology.1 Nodular melanoma also can manifest as a solitary red, raised lesion, but it would stain positive for SOX10, PRAME, and Melan-A on histopathology.7 Basal cell carcinoma, which also may have a similar clinical appearance, is associated with nests of basaloid cells and palisading nuclei histologically.8 Lastly, atypical fibroxanthoma also manifests as a red nodule or plaque and is associated with atypical mitotic figures on histology; however, it notably stains negative for desmin.9
In summary, cutaneous LMS should be included in the differential diagnosis for raised, pink nodules. Given its nonspecific clinical presentation, this rare and malignant neoplasm requires biopsy and immunohistochemical staining for accurate diagnosis.
THE DIAGNOSIS: Cutaneous Leiomyosarcoma
Based on the clinical and histopathologic findings, our patient was diagnosed with primary cutaneous leiomyosarcoma (LMS), a rare soft-tissue neoplasm that arises from smooth muscle and typically manifests as a firm pink nodule.1 The neoplasm may occur in the area of a prior traumatic injury or develop spontaneously without an identifiable cause.1-3 Cutaneous LMS represents 2% to 3% of all soft-tissue sarcomas worldwide, with an estimated incidence of 1 in 500,000 annually.1,4 Men who are in their fifth to seventh decades of life are at the highest risk for LMS.1
Histologically, cutaneous LMS can be subclassified as dermal, which has a low metastatic risk and excellent prognosis, or subcutaneous, which is associated with poorer outcomes and vascular muscle origin.1 In our case, hematoxylin and eosin staining revealed fascicles of smooth muscle fibers with hypercellularity, atypia, and mitotic figures (Figure). The neoplasm stained positive for desmin, vimentin, and smooth muscle actin and negative for SOX10, Melan-A, PRAME (preferentially expressed antigen in melanoma), CD34, and Factor XIIIa.1
Standard treatment for LMS is surgical excision.5 Poor prognostic factors include lesions with a diameter of 5 cm or larger, deep subcutaneous tumor invasion, and distant metastases.2,5
The differential diagnosis may include dermatofibrosarcoma protuberans, which can have a similar pink nodular appearance and also may manifest after injury6; however, this lesion would stain positive for CD34 on histopathology.1 Nodular melanoma also can manifest as a solitary red, raised lesion, but it would stain positive for SOX10, PRAME, and Melan-A on histopathology.7 Basal cell carcinoma, which also may have a similar clinical appearance, is associated with nests of basaloid cells and palisading nuclei histologically.8 Lastly, atypical fibroxanthoma also manifests as a red nodule or plaque and is associated with atypical mitotic figures on histology; however, it notably stains negative for desmin.9
In summary, cutaneous LMS should be included in the differential diagnosis for raised, pink nodules. Given its nonspecific clinical presentation, this rare and malignant neoplasm requires biopsy and immunohistochemical staining for accurate diagnosis.
- Soares Queirós C, Filipe P, Soares de Almeida L. Cutaneous leiomyosarcoma: a 20-year retrospective study and review of the literature. Anais Brasileiros de Dermatologia. 2021;96:278-283. doi:10.1016/j.abd.2020.10.003
- Kim NG, Kim JO, Park YJ, et al. Cutaneous leiomyosarcoma of the face. Arch Craniofac Surg. 2017;18:145-148. doi:10.7181/acfs.2017.18.2.145
- Litaiem N, Tabka M, Nefiss M, et al. Cutaneous leiomyosarcoma mimicking arteriovenous malformation. Clin Case Rep. 2020;8:2538-2540. doi:10.1002/ccr3.3222
- Hmida L, Letaief F, Doghri R, et al. Cutaneous leiomyosarcoma on the trunk: an unusual presentation with an aggressive course - case report and review of literature. Pan Afr Med J. 2018;31:190. doi:10.11604/pamj.2018.31.190.16682
- Cazzato G, Sergi MC, Sablone S, et al. Advanced cutaneous leiomyosarcoma of the forearm. Dermatopathology (Basel). 2021;8:40-44. doi:10.3390/dermatopathology8010008
- Li Y, Wang C, Xiang B, et al. Clinical features, pathological findings and treatment of recurrent dermatofibrosarcoma protuberans. J Cancer. 2017;8:1319-1323. doi:10.7150/jca.17988
- Hernandez LE, Frech FS, Mohsin N, et al. Nodular melanoma: a review of pathogenesis, presentation, diagnosis and treatment. J Dermatol & Skin Sci. 2021;3:25-30. doi:10.29245/2767-5092/2021/3.1144
- Steele RB, Puckette Y. Basal cell carcinoma. StatPearls [Internet]. StatPearls Publishing; 2026. Updated November 7, 2025. Accessed March 3, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482439/
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls [Internet]. StatPearls Publishing; 2026. Updated September 18, 2022. Accessed March 3, 2026. http://www.ncbi.nlm.nih.gov/books/NBK459342/
- Soares Queirós C, Filipe P, Soares de Almeida L. Cutaneous leiomyosarcoma: a 20-year retrospective study and review of the literature. Anais Brasileiros de Dermatologia. 2021;96:278-283. doi:10.1016/j.abd.2020.10.003
- Kim NG, Kim JO, Park YJ, et al. Cutaneous leiomyosarcoma of the face. Arch Craniofac Surg. 2017;18:145-148. doi:10.7181/acfs.2017.18.2.145
- Litaiem N, Tabka M, Nefiss M, et al. Cutaneous leiomyosarcoma mimicking arteriovenous malformation. Clin Case Rep. 2020;8:2538-2540. doi:10.1002/ccr3.3222
- Hmida L, Letaief F, Doghri R, et al. Cutaneous leiomyosarcoma on the trunk: an unusual presentation with an aggressive course - case report and review of literature. Pan Afr Med J. 2018;31:190. doi:10.11604/pamj.2018.31.190.16682
- Cazzato G, Sergi MC, Sablone S, et al. Advanced cutaneous leiomyosarcoma of the forearm. Dermatopathology (Basel). 2021;8:40-44. doi:10.3390/dermatopathology8010008
- Li Y, Wang C, Xiang B, et al. Clinical features, pathological findings and treatment of recurrent dermatofibrosarcoma protuberans. J Cancer. 2017;8:1319-1323. doi:10.7150/jca.17988
- Hernandez LE, Frech FS, Mohsin N, et al. Nodular melanoma: a review of pathogenesis, presentation, diagnosis and treatment. J Dermatol & Skin Sci. 2021;3:25-30. doi:10.29245/2767-5092/2021/3.1144
- Steele RB, Puckette Y. Basal cell carcinoma. StatPearls [Internet]. StatPearls Publishing; 2026. Updated November 7, 2025. Accessed March 3, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482439/
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls [Internet]. StatPearls Publishing; 2026. Updated September 18, 2022. Accessed March 3, 2026. http://www.ncbi.nlm.nih.gov/books/NBK459342/
Painful, Purpuric, Nodular Lesion With an Irregular Surface on the Shoulder
Painful, Purpuric, Nodular Lesion With an Irregular Surface on the Shoulder
A 53-year-old man presented to the dermatology clinic for evaluation of a painful, purpuric, nodular lesion on the left shoulder of 3 months’ duration. The lesion had an irregular surface that was surrounded by an erythematous ring. Biopsy revealed fascicles of eosinophilic cells within the dermis. The nuclei were heterogeneous in size and shape and had blunted ends. Frequent atypia and mitotic figures were observed, and the lesion extended into the subcutis. Immunostaining was positive for desmin and smooth muscle actin and negative for SOX10, Melan-A, PRAME (preferentially expressed antigen in melanoma), CD34, and Factor XIIIa.
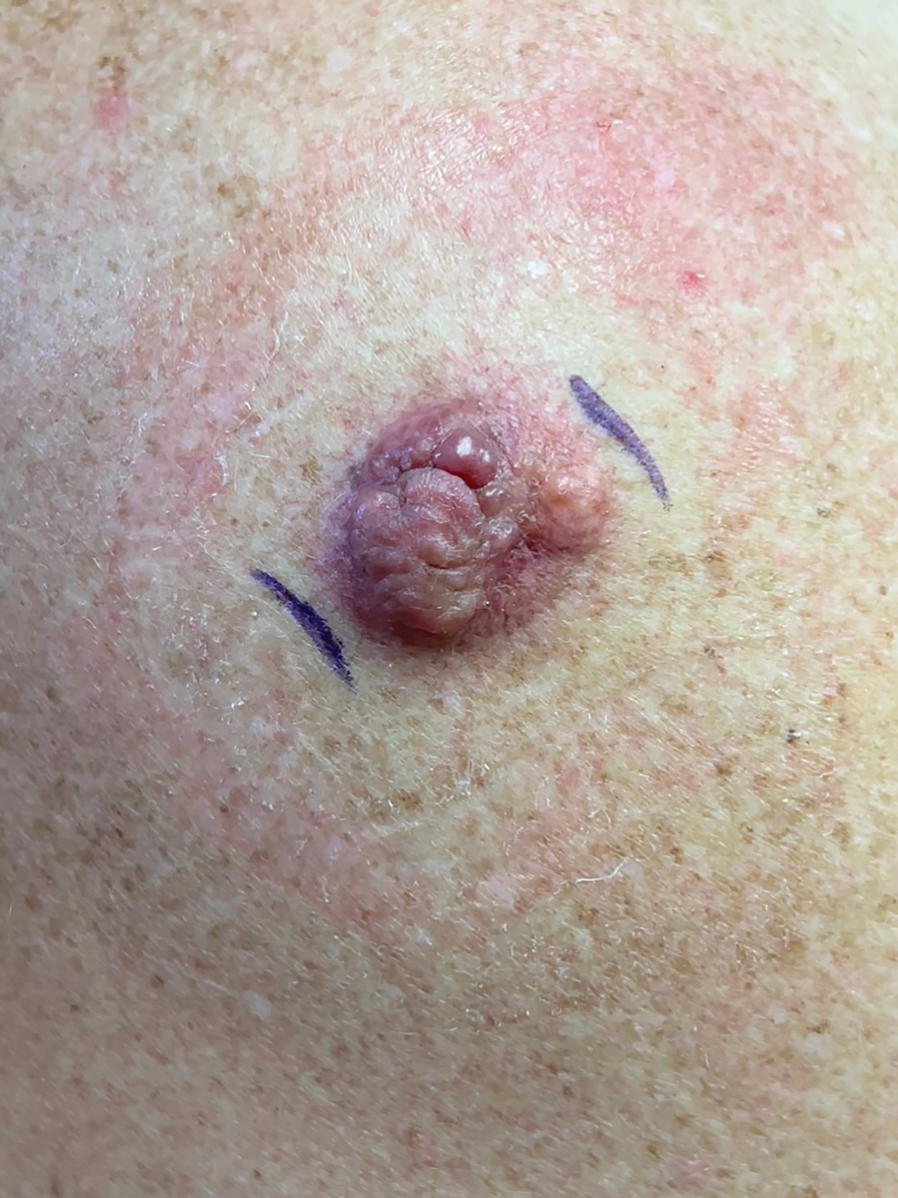

Asymptomatic Plaque and Nodule on the Nose
Asymptomatic Plaque and Nodule on the Nose
THE DIAGNOSIS: Coexisting Squamous Cell Carcinoma and Basal Cell Carcinoma
Dermoscopy of the plaque showed central ulceration with blood spots surrounded by branched linear vessels, which was suggestive of squamous cell carcinoma (SCC)(Figure 1A). The nodule showed shiny, white-red, structureless areas with small gray spots, bright white crystalline streaks, and short fine telangiectasias suggestive of basal cell carcinoma (BCC)(Figure 1B). Histopathology showed that the plaque had irregular nests, cords, and sheets of neoplastic keratinocytes invading the dermis (Figure 2A) and the nodule had discrete nests of basaloid cells with peripheral palisading in the dermis (Figure 2B), which confirmed the diagnosis of coexisting SCC and BCC. The patient underwent surgical excision of the lesions, which achieved clear margins. At the 2-year follow-up, there was no sign of recurrence.
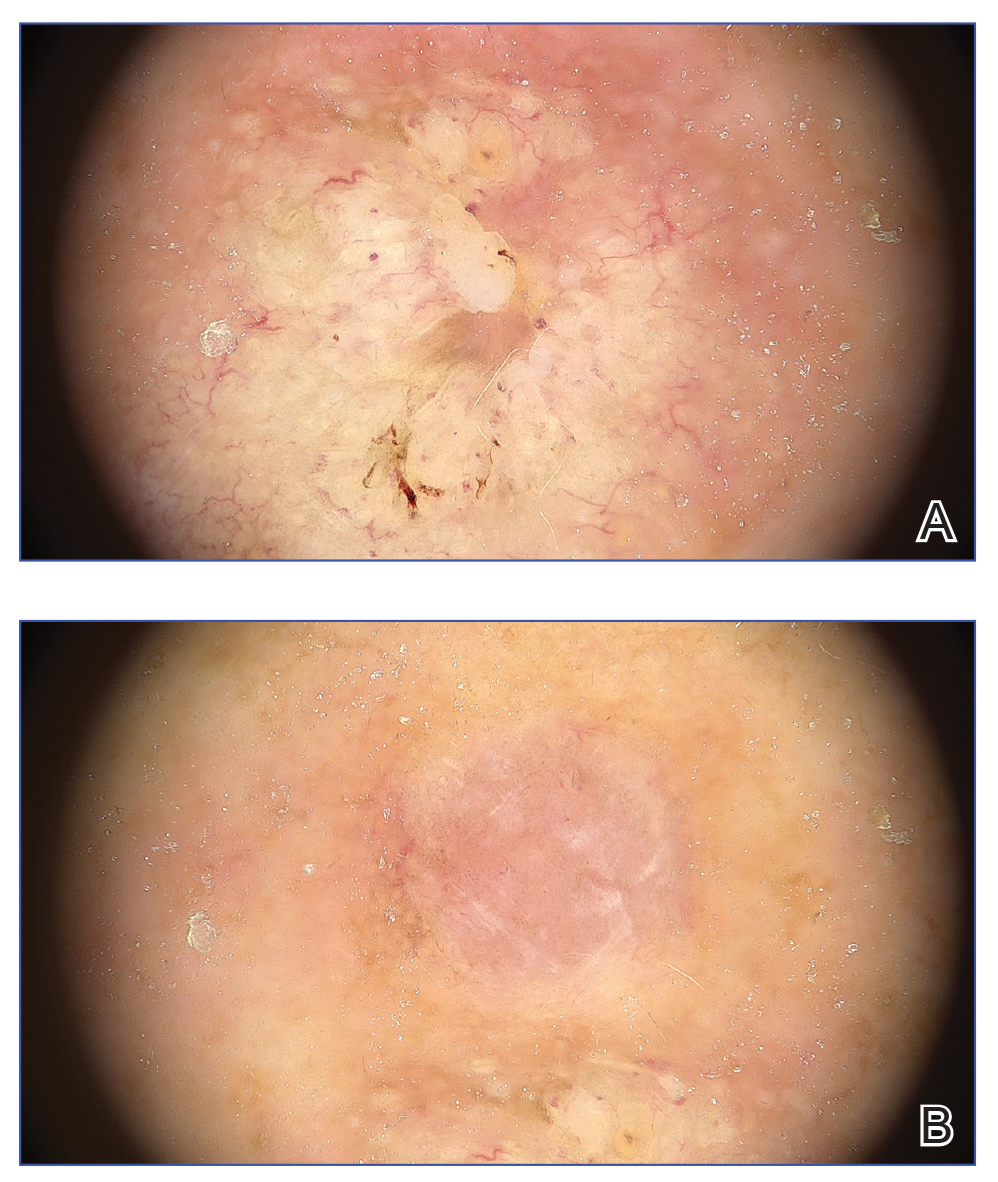
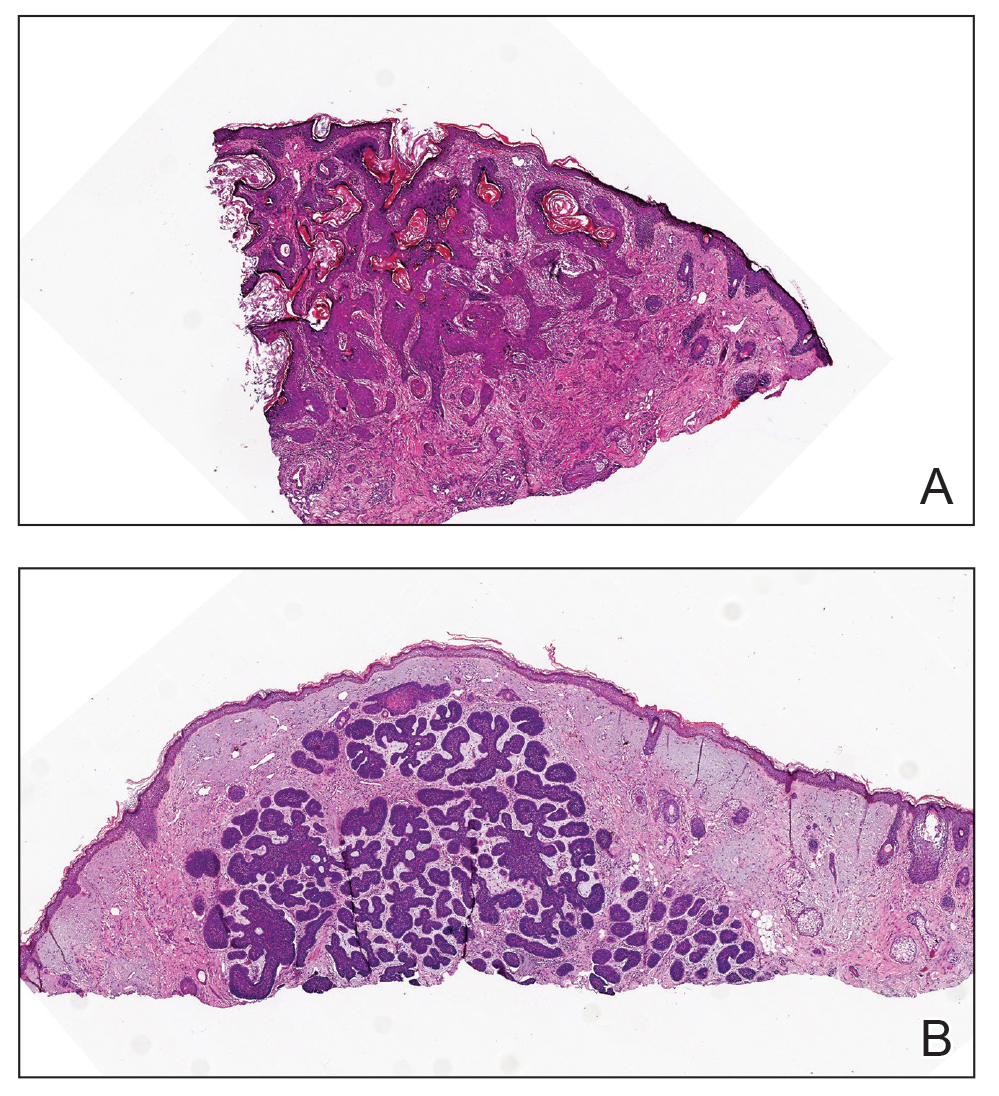
Squamous cell carcinoma is the second most frequent cancer in humans. Older patients are more susceptible due to chronic UV exposure.1 Basal cell carcinoma is the most common human cancer worldwide.2 These skin cancers have different clinical manifestations, pathologic features, treatment methods, and prognoses. The coexistence of 2 types of skin cancer presents a diagnostic challenge. Possible causes of this phenomenon are not clear. It may simply be a coincidence since the lesions typically occur in sun-exposed areas such as the nose, which may be affected by photodamage.3 According to the field cancerization theory, chronically sun-exposed areas are at higher risk for development of coexisting skin cancers.4 A more interesting explanation is the interaction theory, which suggests that one tumor produces epidermal or stromal changes that induce the formation of a second independent tumor via the paracrine effect (ie, growth mediators from nearby cells).4
Dermoscopy is an important noninvasive diagnostic tool for the evaluation of skin cancer, particularly early detection. Dermoscopic findings of blood vessels, ulcers, the fiber sign, blood spots, white structureless areas, keratin, and centered vessels indicate a diagnosis of SCC.5 In contrast, common dermoscopic findings for BCC include arborizing vessels, ulceration, shiny white structures, and blue-gray ovoid nests or globules.6
Irritated seborrheic keratosis is an inflammatory variant of seborrheic keratosis, which often is challenging to identify clinically due to its similar features with SCC; however, SCC is more likely to demonstrate dotted or branched vessels, white structureless areas, white circles around follicles, irregular or peripheral vessel patterns, and central scales on dermoscopy. In contrast, irritated seborrheic keratosis is more likely to have hairpin vessels, regular vessel patterns, and white halos around vessels, which may aid in the differentiation between the two entities.7
Due to the higher sensitivity of dermoscopy for detecting pigmented BCC compared to nonpigmented BCC, it holds substantial diagnostic value in Asian populations, in whom pigmented BCC is the most common subtype.6,8 However, the lack of pigmentation in the nodule in our case posed a diagnostic challenge, as the diagnosis of BCC had to rely on subtle vascular and shiny white structures rather than more obvious pigment clues. This absence of pigment, however, also helped rule out pigmented BCC as a diagnosis for the nodule. Short fine telangiectasias is the second most common vascular pattern in BCC, and bright white structures are highly suggestive of nonpigmented BCC.6 Therefore, dermoscopic findings of bright-white structures with fine telangiectasias should be alerted to the possibility of nonpigmented BCC.
Basosquamous carcinoma has clinical and dermoscopic features between SCC and BCC, and the presence of dermatoscopic features from both BCC and SCC should raise suspicion, but the diagnosis is particularly challenging because its presentation is nonspecific.9 We need to be vigilant about the possibility of coexistence of 2 types of skin cancer, and that regular physical examination and dermatoscopy are very important for early detection and diagnosis.
- Corchado-Cobos R, García-Sancha N, González-Sarmiento R, et al. Cutaneous squamous cell carcinoma: from biology to therapy. Int J Mol Sci. 2020;21:2956. doi:10.3390/ijms21082956
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241-250. doi:10.1001/archderm.123.2.241
- Cornejo KM, Deng AC. Malignant melanoma within squamous cell carcinoma and basal cell carcinoma: is it a combined or collision tumor? a case report and review of the literature. Am J Dermatopathol. 2013;35:226-34. doi:10.1097/DAD.0b013e3182545e27
- Ertop Dog˘an P, Akay BN, Okçu Heper A, et al. Dermatoscopic findings and dermatopathological correlates in clinical variants of actinic keratosis, Bowen’s disease, keratoacanthoma, and squamous cell carcinoma. Dermatol Ther. 2021;34:E14877. doi:10.1111/dth.14877.
- Álvarez-Salafranca M, Ara M, Zaballos P. Dermoscopy in basal cell carcinoma: an updated review. Actas Dermosifiliogr (Engl Ed). 2021;112:330-338. doi:10.1016/j.ad.2020.11.011
- Papageorgiou C, Spyridis I, Manoli SM, et al. Accuracy of dermoscopic criteria for the differential diagnosis between irritated seborrheic keratosis and squamous cell carcinoma. J Am Acad Dermatol. 2021;85:1143-1150. doi:10.1016/j.jaad.2020.02.019
- Cheng SY, Luk NM, Chong LY. Special features of non-melanoma skin cancer in Hong Kong Chinese patients: 10-year retrospective study. Hong Kong Med J. 2001;7:22-28.
- Murgia G, Denaro N, Boggio F, et al. Basosquamous carcinoma: comprehensive clinical and histopathological aspects, novel imaging tools, and therapeutic approaches. Cells. 2023;23:2737. doi:10.3390/cells12232737
THE DIAGNOSIS: Coexisting Squamous Cell Carcinoma and Basal Cell Carcinoma
Dermoscopy of the plaque showed central ulceration with blood spots surrounded by branched linear vessels, which was suggestive of squamous cell carcinoma (SCC)(Figure 1A). The nodule showed shiny, white-red, structureless areas with small gray spots, bright white crystalline streaks, and short fine telangiectasias suggestive of basal cell carcinoma (BCC)(Figure 1B). Histopathology showed that the plaque had irregular nests, cords, and sheets of neoplastic keratinocytes invading the dermis (Figure 2A) and the nodule had discrete nests of basaloid cells with peripheral palisading in the dermis (Figure 2B), which confirmed the diagnosis of coexisting SCC and BCC. The patient underwent surgical excision of the lesions, which achieved clear margins. At the 2-year follow-up, there was no sign of recurrence.
Squamous cell carcinoma is the second most frequent cancer in humans. Older patients are more susceptible due to chronic UV exposure.1 Basal cell carcinoma is the most common human cancer worldwide.2 These skin cancers have different clinical manifestations, pathologic features, treatment methods, and prognoses. The coexistence of 2 types of skin cancer presents a diagnostic challenge. Possible causes of this phenomenon are not clear. It may simply be a coincidence since the lesions typically occur in sun-exposed areas such as the nose, which may be affected by photodamage.3 According to the field cancerization theory, chronically sun-exposed areas are at higher risk for development of coexisting skin cancers.4 A more interesting explanation is the interaction theory, which suggests that one tumor produces epidermal or stromal changes that induce the formation of a second independent tumor via the paracrine effect (ie, growth mediators from nearby cells).4
Dermoscopy is an important noninvasive diagnostic tool for the evaluation of skin cancer, particularly early detection. Dermoscopic findings of blood vessels, ulcers, the fiber sign, blood spots, white structureless areas, keratin, and centered vessels indicate a diagnosis of SCC.5 In contrast, common dermoscopic findings for BCC include arborizing vessels, ulceration, shiny white structures, and blue-gray ovoid nests or globules.6
Irritated seborrheic keratosis is an inflammatory variant of seborrheic keratosis, which often is challenging to identify clinically due to its similar features with SCC; however, SCC is more likely to demonstrate dotted or branched vessels, white structureless areas, white circles around follicles, irregular or peripheral vessel patterns, and central scales on dermoscopy. In contrast, irritated seborrheic keratosis is more likely to have hairpin vessels, regular vessel patterns, and white halos around vessels, which may aid in the differentiation between the two entities.7
Due to the higher sensitivity of dermoscopy for detecting pigmented BCC compared to nonpigmented BCC, it holds substantial diagnostic value in Asian populations, in whom pigmented BCC is the most common subtype.6,8 However, the lack of pigmentation in the nodule in our case posed a diagnostic challenge, as the diagnosis of BCC had to rely on subtle vascular and shiny white structures rather than more obvious pigment clues. This absence of pigment, however, also helped rule out pigmented BCC as a diagnosis for the nodule. Short fine telangiectasias is the second most common vascular pattern in BCC, and bright white structures are highly suggestive of nonpigmented BCC.6 Therefore, dermoscopic findings of bright-white structures with fine telangiectasias should be alerted to the possibility of nonpigmented BCC.
Basosquamous carcinoma has clinical and dermoscopic features between SCC and BCC, and the presence of dermatoscopic features from both BCC and SCC should raise suspicion, but the diagnosis is particularly challenging because its presentation is nonspecific.9 We need to be vigilant about the possibility of coexistence of 2 types of skin cancer, and that regular physical examination and dermatoscopy are very important for early detection and diagnosis.
THE DIAGNOSIS: Coexisting Squamous Cell Carcinoma and Basal Cell Carcinoma
Dermoscopy of the plaque showed central ulceration with blood spots surrounded by branched linear vessels, which was suggestive of squamous cell carcinoma (SCC)(Figure 1A). The nodule showed shiny, white-red, structureless areas with small gray spots, bright white crystalline streaks, and short fine telangiectasias suggestive of basal cell carcinoma (BCC)(Figure 1B). Histopathology showed that the plaque had irregular nests, cords, and sheets of neoplastic keratinocytes invading the dermis (Figure 2A) and the nodule had discrete nests of basaloid cells with peripheral palisading in the dermis (Figure 2B), which confirmed the diagnosis of coexisting SCC and BCC. The patient underwent surgical excision of the lesions, which achieved clear margins. At the 2-year follow-up, there was no sign of recurrence.
Squamous cell carcinoma is the second most frequent cancer in humans. Older patients are more susceptible due to chronic UV exposure.1 Basal cell carcinoma is the most common human cancer worldwide.2 These skin cancers have different clinical manifestations, pathologic features, treatment methods, and prognoses. The coexistence of 2 types of skin cancer presents a diagnostic challenge. Possible causes of this phenomenon are not clear. It may simply be a coincidence since the lesions typically occur in sun-exposed areas such as the nose, which may be affected by photodamage.3 According to the field cancerization theory, chronically sun-exposed areas are at higher risk for development of coexisting skin cancers.4 A more interesting explanation is the interaction theory, which suggests that one tumor produces epidermal or stromal changes that induce the formation of a second independent tumor via the paracrine effect (ie, growth mediators from nearby cells).4
Dermoscopy is an important noninvasive diagnostic tool for the evaluation of skin cancer, particularly early detection. Dermoscopic findings of blood vessels, ulcers, the fiber sign, blood spots, white structureless areas, keratin, and centered vessels indicate a diagnosis of SCC.5 In contrast, common dermoscopic findings for BCC include arborizing vessels, ulceration, shiny white structures, and blue-gray ovoid nests or globules.6
Irritated seborrheic keratosis is an inflammatory variant of seborrheic keratosis, which often is challenging to identify clinically due to its similar features with SCC; however, SCC is more likely to demonstrate dotted or branched vessels, white structureless areas, white circles around follicles, irregular or peripheral vessel patterns, and central scales on dermoscopy. In contrast, irritated seborrheic keratosis is more likely to have hairpin vessels, regular vessel patterns, and white halos around vessels, which may aid in the differentiation between the two entities.7
Due to the higher sensitivity of dermoscopy for detecting pigmented BCC compared to nonpigmented BCC, it holds substantial diagnostic value in Asian populations, in whom pigmented BCC is the most common subtype.6,8 However, the lack of pigmentation in the nodule in our case posed a diagnostic challenge, as the diagnosis of BCC had to rely on subtle vascular and shiny white structures rather than more obvious pigment clues. This absence of pigment, however, also helped rule out pigmented BCC as a diagnosis for the nodule. Short fine telangiectasias is the second most common vascular pattern in BCC, and bright white structures are highly suggestive of nonpigmented BCC.6 Therefore, dermoscopic findings of bright-white structures with fine telangiectasias should be alerted to the possibility of nonpigmented BCC.
Basosquamous carcinoma has clinical and dermoscopic features between SCC and BCC, and the presence of dermatoscopic features from both BCC and SCC should raise suspicion, but the diagnosis is particularly challenging because its presentation is nonspecific.9 We need to be vigilant about the possibility of coexistence of 2 types of skin cancer, and that regular physical examination and dermatoscopy are very important for early detection and diagnosis.
- Corchado-Cobos R, García-Sancha N, González-Sarmiento R, et al. Cutaneous squamous cell carcinoma: from biology to therapy. Int J Mol Sci. 2020;21:2956. doi:10.3390/ijms21082956
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241-250. doi:10.1001/archderm.123.2.241
- Cornejo KM, Deng AC. Malignant melanoma within squamous cell carcinoma and basal cell carcinoma: is it a combined or collision tumor? a case report and review of the literature. Am J Dermatopathol. 2013;35:226-34. doi:10.1097/DAD.0b013e3182545e27
- Ertop Dog˘an P, Akay BN, Okçu Heper A, et al. Dermatoscopic findings and dermatopathological correlates in clinical variants of actinic keratosis, Bowen’s disease, keratoacanthoma, and squamous cell carcinoma. Dermatol Ther. 2021;34:E14877. doi:10.1111/dth.14877.
- Álvarez-Salafranca M, Ara M, Zaballos P. Dermoscopy in basal cell carcinoma: an updated review. Actas Dermosifiliogr (Engl Ed). 2021;112:330-338. doi:10.1016/j.ad.2020.11.011
- Papageorgiou C, Spyridis I, Manoli SM, et al. Accuracy of dermoscopic criteria for the differential diagnosis between irritated seborrheic keratosis and squamous cell carcinoma. J Am Acad Dermatol. 2021;85:1143-1150. doi:10.1016/j.jaad.2020.02.019
- Cheng SY, Luk NM, Chong LY. Special features of non-melanoma skin cancer in Hong Kong Chinese patients: 10-year retrospective study. Hong Kong Med J. 2001;7:22-28.
- Murgia G, Denaro N, Boggio F, et al. Basosquamous carcinoma: comprehensive clinical and histopathological aspects, novel imaging tools, and therapeutic approaches. Cells. 2023;23:2737. doi:10.3390/cells12232737
- Corchado-Cobos R, García-Sancha N, González-Sarmiento R, et al. Cutaneous squamous cell carcinoma: from biology to therapy. Int J Mol Sci. 2020;21:2956. doi:10.3390/ijms21082956
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241-250. doi:10.1001/archderm.123.2.241
- Cornejo KM, Deng AC. Malignant melanoma within squamous cell carcinoma and basal cell carcinoma: is it a combined or collision tumor? a case report and review of the literature. Am J Dermatopathol. 2013;35:226-34. doi:10.1097/DAD.0b013e3182545e27
- Ertop Dog˘an P, Akay BN, Okçu Heper A, et al. Dermatoscopic findings and dermatopathological correlates in clinical variants of actinic keratosis, Bowen’s disease, keratoacanthoma, and squamous cell carcinoma. Dermatol Ther. 2021;34:E14877. doi:10.1111/dth.14877.
- Álvarez-Salafranca M, Ara M, Zaballos P. Dermoscopy in basal cell carcinoma: an updated review. Actas Dermosifiliogr (Engl Ed). 2021;112:330-338. doi:10.1016/j.ad.2020.11.011
- Papageorgiou C, Spyridis I, Manoli SM, et al. Accuracy of dermoscopic criteria for the differential diagnosis between irritated seborrheic keratosis and squamous cell carcinoma. J Am Acad Dermatol. 2021;85:1143-1150. doi:10.1016/j.jaad.2020.02.019
- Cheng SY, Luk NM, Chong LY. Special features of non-melanoma skin cancer in Hong Kong Chinese patients: 10-year retrospective study. Hong Kong Med J. 2001;7:22-28.
- Murgia G, Denaro N, Boggio F, et al. Basosquamous carcinoma: comprehensive clinical and histopathological aspects, novel imaging tools, and therapeutic approaches. Cells. 2023;23:2737. doi:10.3390/cells12232737
Asymptomatic Plaque and Nodule on the Nose
Asymptomatic Plaque and Nodule on the Nose
An 80-year-old Asian woman presented to the hospital dermatology clinic for evaluation of 2 lesions on the nose of 2 years’ duration. The patient reported that the lesions had initially manifested as an asymptomatic red rash, but within the past month, the affected area had grown in diameter with a rough surface and occasional bleeding. Additionally, a smaller new rash appeared around the original plaque. She had no personal or family history of skin cancer. Physical examination revealed a 1.5-cm reddish plaque on the tip of the nose with a central ulcer filled with viscous exudate. Another 0.5-cm reddish nodule with a smooth surface also was noted adjacent to the plaque. Dermoscopy and a skin biopsy of both lesions were performed.

Pseudoleukonychia of the Distal Fingernails
Pseudoleukonychia of the Distal Fingernails
THE DIAGNOSIS: Pseudopsoriatic Nails With Pterygium Inversum Unguis
Based on the clinical findings and the patient’s history of gel manicures, a diagnosis of pseudopsoriatic nails with pterygium inversum unguis (PIU) was made. The patient was advised to avoid gel manicures and any other chemical or mechanical trauma to the nails. No other treatment was administered. Improvements including healthy nail growth and disappearing color and structure changes within the nail plates were noted at 2 months’ follow-up.
The durability and availability of gel manicures has been increasingly popular due to their ideal cosmetic results. A gel manicure involves applying a gel nail polish (GNP) containing acrylate or methacrylate monomers that harden after exposure to UV light through a photopolymerization reaction. Acrylate polymers including ethylene glycol dimethacrylate, 2-hydroxyethyl acrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, methyl methacrylate, and tetrahydrofurfuryl methacrylate are known to cause allergic contact dermatitis in patients who wear acrylate-based GNP.1Hydroxyethyl methacrylate is the most common sensitizer among these acrylates. Fingertip dry dermatitis, fissured painful pulpitis of the fingers, and periungual erythema are the most common manifestations of methacrylate allergy; however, there also are reports of onycholysis and onychodystrophy in patients with severe allergic contact dermatitis caused by acrylates.2,3
In contrast to common public misconception that GNP may strengthen the nails, scientific evidence has shown otherwise. Besides allergic contact dermatitis, mechanical damage and UV-induced skin manifestations have been reported in association with GNP.1,3,4 Pseudopsoriatic nails are characterized by onycholysis accompanied by subungual hyperkeratosis, closely resembling the nail findings seen in psoriasis. This condition may occur due to mechanical damage and acrylate sensitization.2,4 Pterygium inversum unguis, also known as ventral pterygium, occurs as a result of hyponychium trauma due to either application or removal processes of GNP and/ or exposure to chemical ingredients and is one of the most striking clinical manifestations of GNP use.5 In our patient, all fingernails were affected by PIU.
Patients presenting with pseudopsoriatic nail changes and/or PIU should be questioned about potential exposure to GNP and/or sculpted nails, also known as custom artificial nails or nail prostheses. Diagnosis primarily is made clinically, but microbial cultures or skin biopsy may be required to exclude psoriasis and fungal infections in some patients. Patch testing with acrylate series in particular also is necessary in patients presenting with hand dermatitis. As it is the most common contact sensitizer in the acrylic material of the GNPs, screening for 2-hydroxyethyl methacrylate allergy is recommended in these patients.1 Almost all adverse effects related to use of GNP may be reversible upon discontinuation of exposure.
- Litaiem N, Baklouti M, Zeglaoui F. Side effects of gel nail polish: a systematic review. Clin Dermatol. 2022;40:706-715. doi:10.1016 /j.clindermatol.2022.07.008
- Engelina S, Shim TN. Atypical cases of pseudo-psoriatic nails associated with acrylate contact allergy. Contact Dermatitis. 2021; 84:342-344. doi:10.1111/cod.13741
- Draelos ZD. Nail cosmetics and adornment. Dermatol Clinics. 2021;39:351-359. doi:10.1016/j.det.2021.01.001
- Rieder EA, Tosti A. Cosmetically induced disorders of the nail with update on contemporary nail manicures. J Clin Aesthet Dermatol. 2016;9:39-44.
- Cervantes J, Sanchez M, Eber AE, et al. Pterygium inversum unguis secondary to gel polish. J Eur Acad Dermatol Venereol. 2018;32:160-163. doi:10.1111/jdv.14603
THE DIAGNOSIS: Pseudopsoriatic Nails With Pterygium Inversum Unguis
Based on the clinical findings and the patient’s history of gel manicures, a diagnosis of pseudopsoriatic nails with pterygium inversum unguis (PIU) was made. The patient was advised to avoid gel manicures and any other chemical or mechanical trauma to the nails. No other treatment was administered. Improvements including healthy nail growth and disappearing color and structure changes within the nail plates were noted at 2 months’ follow-up.
The durability and availability of gel manicures has been increasingly popular due to their ideal cosmetic results. A gel manicure involves applying a gel nail polish (GNP) containing acrylate or methacrylate monomers that harden after exposure to UV light through a photopolymerization reaction. Acrylate polymers including ethylene glycol dimethacrylate, 2-hydroxyethyl acrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, methyl methacrylate, and tetrahydrofurfuryl methacrylate are known to cause allergic contact dermatitis in patients who wear acrylate-based GNP.1Hydroxyethyl methacrylate is the most common sensitizer among these acrylates. Fingertip dry dermatitis, fissured painful pulpitis of the fingers, and periungual erythema are the most common manifestations of methacrylate allergy; however, there also are reports of onycholysis and onychodystrophy in patients with severe allergic contact dermatitis caused by acrylates.2,3
In contrast to common public misconception that GNP may strengthen the nails, scientific evidence has shown otherwise. Besides allergic contact dermatitis, mechanical damage and UV-induced skin manifestations have been reported in association with GNP.1,3,4 Pseudopsoriatic nails are characterized by onycholysis accompanied by subungual hyperkeratosis, closely resembling the nail findings seen in psoriasis. This condition may occur due to mechanical damage and acrylate sensitization.2,4 Pterygium inversum unguis, also known as ventral pterygium, occurs as a result of hyponychium trauma due to either application or removal processes of GNP and/ or exposure to chemical ingredients and is one of the most striking clinical manifestations of GNP use.5 In our patient, all fingernails were affected by PIU.
Patients presenting with pseudopsoriatic nail changes and/or PIU should be questioned about potential exposure to GNP and/or sculpted nails, also known as custom artificial nails or nail prostheses. Diagnosis primarily is made clinically, but microbial cultures or skin biopsy may be required to exclude psoriasis and fungal infections in some patients. Patch testing with acrylate series in particular also is necessary in patients presenting with hand dermatitis. As it is the most common contact sensitizer in the acrylic material of the GNPs, screening for 2-hydroxyethyl methacrylate allergy is recommended in these patients.1 Almost all adverse effects related to use of GNP may be reversible upon discontinuation of exposure.
THE DIAGNOSIS: Pseudopsoriatic Nails With Pterygium Inversum Unguis
Based on the clinical findings and the patient’s history of gel manicures, a diagnosis of pseudopsoriatic nails with pterygium inversum unguis (PIU) was made. The patient was advised to avoid gel manicures and any other chemical or mechanical trauma to the nails. No other treatment was administered. Improvements including healthy nail growth and disappearing color and structure changes within the nail plates were noted at 2 months’ follow-up.
The durability and availability of gel manicures has been increasingly popular due to their ideal cosmetic results. A gel manicure involves applying a gel nail polish (GNP) containing acrylate or methacrylate monomers that harden after exposure to UV light through a photopolymerization reaction. Acrylate polymers including ethylene glycol dimethacrylate, 2-hydroxyethyl acrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, methyl methacrylate, and tetrahydrofurfuryl methacrylate are known to cause allergic contact dermatitis in patients who wear acrylate-based GNP.1Hydroxyethyl methacrylate is the most common sensitizer among these acrylates. Fingertip dry dermatitis, fissured painful pulpitis of the fingers, and periungual erythema are the most common manifestations of methacrylate allergy; however, there also are reports of onycholysis and onychodystrophy in patients with severe allergic contact dermatitis caused by acrylates.2,3
In contrast to common public misconception that GNP may strengthen the nails, scientific evidence has shown otherwise. Besides allergic contact dermatitis, mechanical damage and UV-induced skin manifestations have been reported in association with GNP.1,3,4 Pseudopsoriatic nails are characterized by onycholysis accompanied by subungual hyperkeratosis, closely resembling the nail findings seen in psoriasis. This condition may occur due to mechanical damage and acrylate sensitization.2,4 Pterygium inversum unguis, also known as ventral pterygium, occurs as a result of hyponychium trauma due to either application or removal processes of GNP and/ or exposure to chemical ingredients and is one of the most striking clinical manifestations of GNP use.5 In our patient, all fingernails were affected by PIU.
Patients presenting with pseudopsoriatic nail changes and/or PIU should be questioned about potential exposure to GNP and/or sculpted nails, also known as custom artificial nails or nail prostheses. Diagnosis primarily is made clinically, but microbial cultures or skin biopsy may be required to exclude psoriasis and fungal infections in some patients. Patch testing with acrylate series in particular also is necessary in patients presenting with hand dermatitis. As it is the most common contact sensitizer in the acrylic material of the GNPs, screening for 2-hydroxyethyl methacrylate allergy is recommended in these patients.1 Almost all adverse effects related to use of GNP may be reversible upon discontinuation of exposure.
- Litaiem N, Baklouti M, Zeglaoui F. Side effects of gel nail polish: a systematic review. Clin Dermatol. 2022;40:706-715. doi:10.1016 /j.clindermatol.2022.07.008
- Engelina S, Shim TN. Atypical cases of pseudo-psoriatic nails associated with acrylate contact allergy. Contact Dermatitis. 2021; 84:342-344. doi:10.1111/cod.13741
- Draelos ZD. Nail cosmetics and adornment. Dermatol Clinics. 2021;39:351-359. doi:10.1016/j.det.2021.01.001
- Rieder EA, Tosti A. Cosmetically induced disorders of the nail with update on contemporary nail manicures. J Clin Aesthet Dermatol. 2016;9:39-44.
- Cervantes J, Sanchez M, Eber AE, et al. Pterygium inversum unguis secondary to gel polish. J Eur Acad Dermatol Venereol. 2018;32:160-163. doi:10.1111/jdv.14603
- Litaiem N, Baklouti M, Zeglaoui F. Side effects of gel nail polish: a systematic review. Clin Dermatol. 2022;40:706-715. doi:10.1016 /j.clindermatol.2022.07.008
- Engelina S, Shim TN. Atypical cases of pseudo-psoriatic nails associated with acrylate contact allergy. Contact Dermatitis. 2021; 84:342-344. doi:10.1111/cod.13741
- Draelos ZD. Nail cosmetics and adornment. Dermatol Clinics. 2021;39:351-359. doi:10.1016/j.det.2021.01.001
- Rieder EA, Tosti A. Cosmetically induced disorders of the nail with update on contemporary nail manicures. J Clin Aesthet Dermatol. 2016;9:39-44.
- Cervantes J, Sanchez M, Eber AE, et al. Pterygium inversum unguis secondary to gel polish. J Eur Acad Dermatol Venereol. 2018;32:160-163. doi:10.1111/jdv.14603
Pseudoleukonychia of the Distal Fingernails
Pseudoleukonychia of the Distal Fingernails
An otherwise healthy 36-year-old woman presented to the dermatology department for evaluation of disfiguring nail changes and subungual verrucous skin lesions of 3 weeks’ duration. A review of systems and the patient’s personal and family history were unremarkable. She denied any recent trauma or chemical exposure but noted that she had regularly been patronizing a beauty salon for gel manicures over the past year; her most recent visit was 6 weeks prior to the current presentation. She previously was treated at another dermatology clinic with local corticosteroid creams without any improvement. Dermatologic examination revealed pseudoleukonychia of the distal fingernails surrounded by an erythematous and/or haemorrhagic border. Overgrowth and adherence of the hyponychium to the nail plate also was noted in almost all the fingernails. A prior complete blood cell count and biochemistry panel were within reference range.
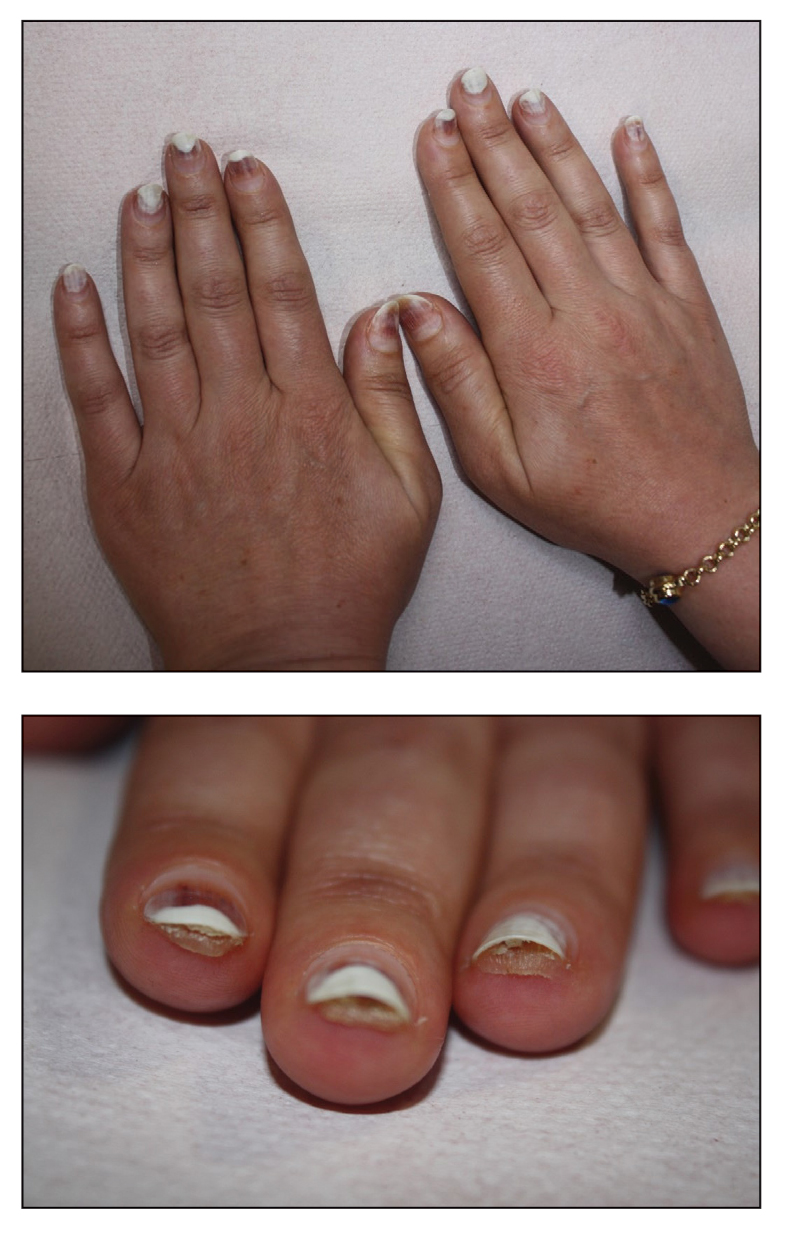

Black Patches on the Angles of the Mandible
Black Patches on the Angles of the Mandible
THE DIAGNOSIS: Black Dermographism
Black dermographism is characterized by asymptomatic black discoloration on the skin caused by contact with various metals, most commonly gold but also silver, nickel, zinc, lead, and aluminum.1 These metallic particles have a black appearance as they do not reflect light.2 Our patient was wearing gold hoop earrings at presentation, which were near the black patches. Certain topical products (eg, makeup, sunscreens [especially those containing zinc oxide or titanium oxide], toothpaste) can abrade metal, causing it to deposit on the skin and absorb light.3 The black discoloration is not permanent and can be prevented by avoiding contact between inciting products and metals.2 No further diagnostic testing is necessary, and the patches will self-resolve if contact with the product is avoided.
Our patient noted that she wore a physical sunscreen daily, but the black patches were present only when she wore the gold hoop earrings. Given this history and physical examination findings in the office, it was suspected she had black dermographism due to her gold earrings and topical sunscreen. The patient was advised to avoid wearing the gold earrings.
Black dermographism is a misnomer because it is not a true urticarial reaction but rather a false dermographism; therefore, patients will not experience pruritus or erythema.1 True dermographism is an inducible urticarial eruption from pressure or trauma to the skin. The clinical appearance is notable for erythematous wheals in the shape of the external force applied.4 Two other types of false dermographism include white dermographism, which occurs secondary to allergic contact dermatitis, and yellow dermographism, which is caused by bile deposits on the skin.4
Additional diagnoses were able to be ruled out for the following reasons: cutaneous mastocytosis can manifest with red-brown maculopapular lesions often accompanied by the Darier sign, which includes swelling, pruritus, and erythema but was not present in our patient.4 Allergic contact dermatitis manifests as a delayed eczematous reaction around 48 to 72 hours after exposure to an allergen. Our patient’s lesions formed while wearing gold earrings but did not manifest with a hypersensitivity reaction. Of note, symptomatic dermographism has been reported to mimic latex allergy.5 Ecchymosis may appear as erythematous, violaceous, or yellow-green patches depending on the stage but develops due to leakage from broken blood vessels secondary to trauma, which was not reported in our patient. Type I hypersensitivity reactions can occur minutes to hours after exposure to an allergen but typically manifest with a wheal-and-flare presentation.
Black dermographism from gold earrings can mimic concerning skin disorders or poor hygiene, causing unnecessary anxiety. Understanding that it is a harmless reaction between gold and certain topical products can reassure patients and prevent unnecessary testing or treatments.
- Zawar V, Kumavat S, Pawar M. Black dermographism: an uncommon cause of skin discoloration. Indian Dermatol Online J. 2018;9:216-217. doi:10.4103/idoj.IDOJ_228_17
- Lowe E, Lim S. Black dermographism. JAMA Dermatol. 2017; 153:352-353.
- Fisher AA. Black dermographism: mechanism for formation of black color. Cutis. 1993;52(1):17-19.
- Nobles T, Muse ME, Schmieder GJ. Dermatographism. In: StatPearls [Internet]. StatPearls Publishing; February 20, 2023.
- Golberg O, Johnston GA, Wilkinson M. Symptomatic dermographism mimicking latex allergy. Dermatitis. 2014;25:101-103. doi:10.1097 /DER.0000000000000016
THE DIAGNOSIS: Black Dermographism
Black dermographism is characterized by asymptomatic black discoloration on the skin caused by contact with various metals, most commonly gold but also silver, nickel, zinc, lead, and aluminum.1 These metallic particles have a black appearance as they do not reflect light.2 Our patient was wearing gold hoop earrings at presentation, which were near the black patches. Certain topical products (eg, makeup, sunscreens [especially those containing zinc oxide or titanium oxide], toothpaste) can abrade metal, causing it to deposit on the skin and absorb light.3 The black discoloration is not permanent and can be prevented by avoiding contact between inciting products and metals.2 No further diagnostic testing is necessary, and the patches will self-resolve if contact with the product is avoided.
Our patient noted that she wore a physical sunscreen daily, but the black patches were present only when she wore the gold hoop earrings. Given this history and physical examination findings in the office, it was suspected she had black dermographism due to her gold earrings and topical sunscreen. The patient was advised to avoid wearing the gold earrings.
Black dermographism is a misnomer because it is not a true urticarial reaction but rather a false dermographism; therefore, patients will not experience pruritus or erythema.1 True dermographism is an inducible urticarial eruption from pressure or trauma to the skin. The clinical appearance is notable for erythematous wheals in the shape of the external force applied.4 Two other types of false dermographism include white dermographism, which occurs secondary to allergic contact dermatitis, and yellow dermographism, which is caused by bile deposits on the skin.4
Additional diagnoses were able to be ruled out for the following reasons: cutaneous mastocytosis can manifest with red-brown maculopapular lesions often accompanied by the Darier sign, which includes swelling, pruritus, and erythema but was not present in our patient.4 Allergic contact dermatitis manifests as a delayed eczematous reaction around 48 to 72 hours after exposure to an allergen. Our patient’s lesions formed while wearing gold earrings but did not manifest with a hypersensitivity reaction. Of note, symptomatic dermographism has been reported to mimic latex allergy.5 Ecchymosis may appear as erythematous, violaceous, or yellow-green patches depending on the stage but develops due to leakage from broken blood vessels secondary to trauma, which was not reported in our patient. Type I hypersensitivity reactions can occur minutes to hours after exposure to an allergen but typically manifest with a wheal-and-flare presentation.
Black dermographism from gold earrings can mimic concerning skin disorders or poor hygiene, causing unnecessary anxiety. Understanding that it is a harmless reaction between gold and certain topical products can reassure patients and prevent unnecessary testing or treatments.
THE DIAGNOSIS: Black Dermographism
Black dermographism is characterized by asymptomatic black discoloration on the skin caused by contact with various metals, most commonly gold but also silver, nickel, zinc, lead, and aluminum.1 These metallic particles have a black appearance as they do not reflect light.2 Our patient was wearing gold hoop earrings at presentation, which were near the black patches. Certain topical products (eg, makeup, sunscreens [especially those containing zinc oxide or titanium oxide], toothpaste) can abrade metal, causing it to deposit on the skin and absorb light.3 The black discoloration is not permanent and can be prevented by avoiding contact between inciting products and metals.2 No further diagnostic testing is necessary, and the patches will self-resolve if contact with the product is avoided.
Our patient noted that she wore a physical sunscreen daily, but the black patches were present only when she wore the gold hoop earrings. Given this history and physical examination findings in the office, it was suspected she had black dermographism due to her gold earrings and topical sunscreen. The patient was advised to avoid wearing the gold earrings.
Black dermographism is a misnomer because it is not a true urticarial reaction but rather a false dermographism; therefore, patients will not experience pruritus or erythema.1 True dermographism is an inducible urticarial eruption from pressure or trauma to the skin. The clinical appearance is notable for erythematous wheals in the shape of the external force applied.4 Two other types of false dermographism include white dermographism, which occurs secondary to allergic contact dermatitis, and yellow dermographism, which is caused by bile deposits on the skin.4
Additional diagnoses were able to be ruled out for the following reasons: cutaneous mastocytosis can manifest with red-brown maculopapular lesions often accompanied by the Darier sign, which includes swelling, pruritus, and erythema but was not present in our patient.4 Allergic contact dermatitis manifests as a delayed eczematous reaction around 48 to 72 hours after exposure to an allergen. Our patient’s lesions formed while wearing gold earrings but did not manifest with a hypersensitivity reaction. Of note, symptomatic dermographism has been reported to mimic latex allergy.5 Ecchymosis may appear as erythematous, violaceous, or yellow-green patches depending on the stage but develops due to leakage from broken blood vessels secondary to trauma, which was not reported in our patient. Type I hypersensitivity reactions can occur minutes to hours after exposure to an allergen but typically manifest with a wheal-and-flare presentation.
Black dermographism from gold earrings can mimic concerning skin disorders or poor hygiene, causing unnecessary anxiety. Understanding that it is a harmless reaction between gold and certain topical products can reassure patients and prevent unnecessary testing or treatments.
- Zawar V, Kumavat S, Pawar M. Black dermographism: an uncommon cause of skin discoloration. Indian Dermatol Online J. 2018;9:216-217. doi:10.4103/idoj.IDOJ_228_17
- Lowe E, Lim S. Black dermographism. JAMA Dermatol. 2017; 153:352-353.
- Fisher AA. Black dermographism: mechanism for formation of black color. Cutis. 1993;52(1):17-19.
- Nobles T, Muse ME, Schmieder GJ. Dermatographism. In: StatPearls [Internet]. StatPearls Publishing; February 20, 2023.
- Golberg O, Johnston GA, Wilkinson M. Symptomatic dermographism mimicking latex allergy. Dermatitis. 2014;25:101-103. doi:10.1097 /DER.0000000000000016
- Zawar V, Kumavat S, Pawar M. Black dermographism: an uncommon cause of skin discoloration. Indian Dermatol Online J. 2018;9:216-217. doi:10.4103/idoj.IDOJ_228_17
- Lowe E, Lim S. Black dermographism. JAMA Dermatol. 2017; 153:352-353.
- Fisher AA. Black dermographism: mechanism for formation of black color. Cutis. 1993;52(1):17-19.
- Nobles T, Muse ME, Schmieder GJ. Dermatographism. In: StatPearls [Internet]. StatPearls Publishing; February 20, 2023.
- Golberg O, Johnston GA, Wilkinson M. Symptomatic dermographism mimicking latex allergy. Dermatitis. 2014;25:101-103. doi:10.1097 /DER.0000000000000016
Black Patches on the Angles of the Mandible
Black Patches on the Angles of the Mandible
A 30-year-old woman presented for evaluation of intermittent pigmented patches on the face of several months’ duration. The patches would form during the day and disappear when the patient woke up the next morning. She denied any associated pruritus, pain, redness, or recent trauma to the area. Her medical history was otherwise unremarkable. Physical examination revealed ill-defined black patches on both mandibular angles (top). The following day, the patient sent a photograph from home, and the patch was absent (bottom).

Diffusely Scattered Linear Folliculopapular Eruption
Diffusely Scattered Linear Folliculopapular Eruption
THE DIAGNOSIS: Disseminate and Recurrent Infundibulofolliculitis
Histopathology demonstrated a lymphocyte-predominant infundibular infiltrate with mild spongiosis and lymphocytic exocytosis; a mild, superficial perivascular infiltrate also was present. The surrounding skin was largely normal with no notable papillomatosis, acanthosis, or hyperkeratosis (Figure 1). The clinical presentation and histopathologic findings led to the diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF). The patient was started on a 2-week course of once-daily ammonium lactate lotion 12% and urea cream 40% and twice-daily triamcinolone ointment 0.1%. The patient was instructed to take a 1-week break before this regimen was repeated. Isotretinoin 0.5 mg/kg/d for 2 to 4 months was considered and will be an option if there is no improvement at follow-up.
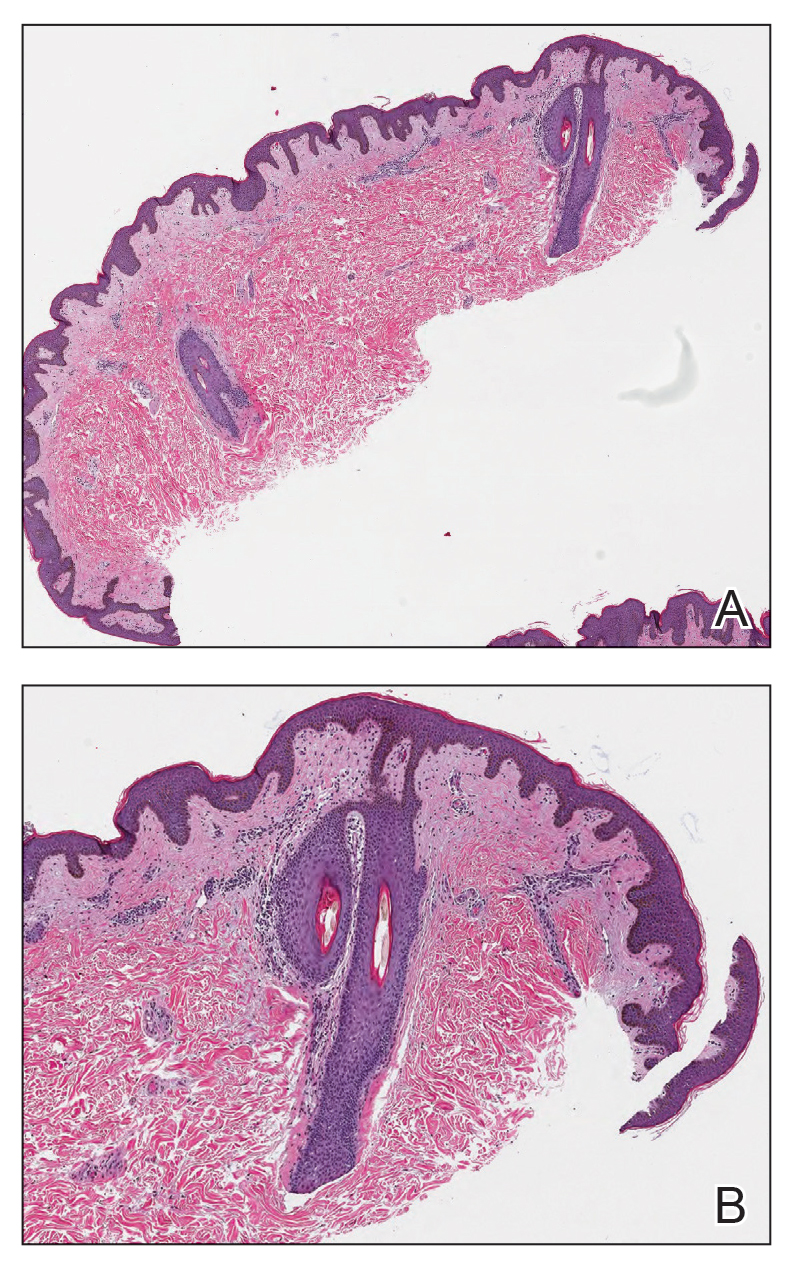
Disseminate and recurrent infundibulofolliculitis is a rare noninfectious folliculitis that initially was described by Hitch and Lund1 in 1968. Males of African descent are most commonly affected by DRIF, but the condition is not limited to this population.2,3 It manifests as asymptomatic, flesh-colored, monomorphic, follicular papules distributed on the trunk and proximal extremities. Pustules can be present, and hair may be seen protruding from them. As the name suggests, DRIF is associated with histopathologic changes that are prominent at the infundibulum of hair follicles.3,4 Disseminate and recurrent infundibulofolliculitis can persist for months to years because it often is resistant to treatment. Treatments include topical monotherapies such as corticosteroids, calcineurin inhibitors, or retinoids; combination topical treatments; antibiotics; and isotretinoin.2 Recurrent remission and exacerbation occurs in many patients.3
The classic manifestations of DRIF, including follicular, monomorphic, flesh-colored papules distributed on the neck, trunk, and proximal upper extremities, were seen in our patient (Figure 2). These findings along with the skin biopsy identifying a lymphocytic infundibular infiltrate led to the diagnosis of DRIF. The papules associated with DRIF can be recurrent or chronic. The lesions in this patient were chronic and persistent.
Despite limited evidence, it has been suggested that DRIF may be a manifestation of atopic dermatitis in patients with darker skin tones. In our case, the patient had a history of childhood eczema. Other hypotheses have proposed that DRIF could be a nonspecific reaction to a currently unknown antigen. A causative infectious agent has not been identified, although the search continues. There is speculation that DRIF could be an overt expression of normal follicular prominence, but the presence of occasional pustules and lymphocyte- predominant infundibular infiltrate negates that.3
Confluent and reticulated papillomatosis was included in the differential for our patient and manifests as asymptomatic hyperpigmented papules and plaques frequently occurring on the upper trunk, neck, and axilla; however, these lesions have a peripheral netlike configuration, as the name suggests. Additionally, this condition is thought to have an infectious component (Dietzia papillomatosis) and responds to antibiotic treatment.5 Follicular eczema also was high in the differential diagnosis but usually is seasonal and pruritic, and histopathology typically shows the features of spongiotic dermatitis. It also would respond well to topical steroids.6 Another condition high on the differential was juxtaclavicular beaded lines, which also manifests as flesh-colored follicular papules distributed on the upper trunk; however, histopathology usually shows features of hyperplastic pilosebaceous units along with spongiosis and exocytosis.7 Pityrosporum folliculitis initially was considered, but the patient only endorsed occasional pruritus. Additionally, no fungal elements were observed.
Currently, there are no definitive treatments for DRIF. The topical treatments available include midpotency corticosteroids, tretinoin, calcineurin inhibitors, 12% lactic acid, and 20% to 40% urea. The systemic therapies are high-dose oral vitamin A (100,000 IU/d), isotretinoin, and psoralen plus UVA.8-10
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Ma BC, Sahni VN, Sahni DR, et al. Disseminate and recurrent infundibulofolliculitis: an under-recognized yet treatable entity. J Drugs Dermatol. 2021;20:1353-1354. doi:10.36849/jdd.6173
- Nair SP, Gomathy M, Kumar GN. Disseminate and recurrent infundibulo- folliculitis in an Indian patient: a case report with review of literature. Indian Dermatol Online J. 2017;8:39-41. doi:10.4103/2229- 5178.198775
- Rekha S, Kumar V, Rao P, et al. Disseminate and recurrent infundibulofolliculitis. Indian J Dermatol. 2019;64:404-406. doi:10.4103/ijd.IJD_77_18
- Jones AL, Koerner RJ, Natarajan S, et al. Dietzia papillomatosis sp. nov., a novel actinomycete isolated from the skin of an immunocompetent patient with confluent and reticulated papillomatosis. Int J Syst Evol Microbiol. 2008;58(pt 1):68-72. doi:10.1099/ijs.0.65178-0
- Cohen PR. Follicular contact dermatitis revisited: a review emphasizing neomycin-associated follicular contact dermatitis. World J Clin Cases. 2014;2:815-821. doi:10.12998/wjcc.v2.i12.815
- Butterworth T, Johnson WC. Justa-clavicular beaded lines. Arch Dermatol. 1974;110:891-893.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulo-folliculitis responsive to treatment with isotretinoin. J Dermatol. 2002;29:431-434.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-76.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
THE DIAGNOSIS: Disseminate and Recurrent Infundibulofolliculitis
Histopathology demonstrated a lymphocyte-predominant infundibular infiltrate with mild spongiosis and lymphocytic exocytosis; a mild, superficial perivascular infiltrate also was present. The surrounding skin was largely normal with no notable papillomatosis, acanthosis, or hyperkeratosis (Figure 1). The clinical presentation and histopathologic findings led to the diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF). The patient was started on a 2-week course of once-daily ammonium lactate lotion 12% and urea cream 40% and twice-daily triamcinolone ointment 0.1%. The patient was instructed to take a 1-week break before this regimen was repeated. Isotretinoin 0.5 mg/kg/d for 2 to 4 months was considered and will be an option if there is no improvement at follow-up.
Disseminate and recurrent infundibulofolliculitis is a rare noninfectious folliculitis that initially was described by Hitch and Lund1 in 1968. Males of African descent are most commonly affected by DRIF, but the condition is not limited to this population.2,3 It manifests as asymptomatic, flesh-colored, monomorphic, follicular papules distributed on the trunk and proximal extremities. Pustules can be present, and hair may be seen protruding from them. As the name suggests, DRIF is associated with histopathologic changes that are prominent at the infundibulum of hair follicles.3,4 Disseminate and recurrent infundibulofolliculitis can persist for months to years because it often is resistant to treatment. Treatments include topical monotherapies such as corticosteroids, calcineurin inhibitors, or retinoids; combination topical treatments; antibiotics; and isotretinoin.2 Recurrent remission and exacerbation occurs in many patients.3
The classic manifestations of DRIF, including follicular, monomorphic, flesh-colored papules distributed on the neck, trunk, and proximal upper extremities, were seen in our patient (Figure 2). These findings along with the skin biopsy identifying a lymphocytic infundibular infiltrate led to the diagnosis of DRIF. The papules associated with DRIF can be recurrent or chronic. The lesions in this patient were chronic and persistent.
Despite limited evidence, it has been suggested that DRIF may be a manifestation of atopic dermatitis in patients with darker skin tones. In our case, the patient had a history of childhood eczema. Other hypotheses have proposed that DRIF could be a nonspecific reaction to a currently unknown antigen. A causative infectious agent has not been identified, although the search continues. There is speculation that DRIF could be an overt expression of normal follicular prominence, but the presence of occasional pustules and lymphocyte- predominant infundibular infiltrate negates that.3
Confluent and reticulated papillomatosis was included in the differential for our patient and manifests as asymptomatic hyperpigmented papules and plaques frequently occurring on the upper trunk, neck, and axilla; however, these lesions have a peripheral netlike configuration, as the name suggests. Additionally, this condition is thought to have an infectious component (Dietzia papillomatosis) and responds to antibiotic treatment.5 Follicular eczema also was high in the differential diagnosis but usually is seasonal and pruritic, and histopathology typically shows the features of spongiotic dermatitis. It also would respond well to topical steroids.6 Another condition high on the differential was juxtaclavicular beaded lines, which also manifests as flesh-colored follicular papules distributed on the upper trunk; however, histopathology usually shows features of hyperplastic pilosebaceous units along with spongiosis and exocytosis.7 Pityrosporum folliculitis initially was considered, but the patient only endorsed occasional pruritus. Additionally, no fungal elements were observed.
Currently, there are no definitive treatments for DRIF. The topical treatments available include midpotency corticosteroids, tretinoin, calcineurin inhibitors, 12% lactic acid, and 20% to 40% urea. The systemic therapies are high-dose oral vitamin A (100,000 IU/d), isotretinoin, and psoralen plus UVA.8-10
THE DIAGNOSIS: Disseminate and Recurrent Infundibulofolliculitis
Histopathology demonstrated a lymphocyte-predominant infundibular infiltrate with mild spongiosis and lymphocytic exocytosis; a mild, superficial perivascular infiltrate also was present. The surrounding skin was largely normal with no notable papillomatosis, acanthosis, or hyperkeratosis (Figure 1). The clinical presentation and histopathologic findings led to the diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF). The patient was started on a 2-week course of once-daily ammonium lactate lotion 12% and urea cream 40% and twice-daily triamcinolone ointment 0.1%. The patient was instructed to take a 1-week break before this regimen was repeated. Isotretinoin 0.5 mg/kg/d for 2 to 4 months was considered and will be an option if there is no improvement at follow-up.
Disseminate and recurrent infundibulofolliculitis is a rare noninfectious folliculitis that initially was described by Hitch and Lund1 in 1968. Males of African descent are most commonly affected by DRIF, but the condition is not limited to this population.2,3 It manifests as asymptomatic, flesh-colored, monomorphic, follicular papules distributed on the trunk and proximal extremities. Pustules can be present, and hair may be seen protruding from them. As the name suggests, DRIF is associated with histopathologic changes that are prominent at the infundibulum of hair follicles.3,4 Disseminate and recurrent infundibulofolliculitis can persist for months to years because it often is resistant to treatment. Treatments include topical monotherapies such as corticosteroids, calcineurin inhibitors, or retinoids; combination topical treatments; antibiotics; and isotretinoin.2 Recurrent remission and exacerbation occurs in many patients.3
The classic manifestations of DRIF, including follicular, monomorphic, flesh-colored papules distributed on the neck, trunk, and proximal upper extremities, were seen in our patient (Figure 2). These findings along with the skin biopsy identifying a lymphocytic infundibular infiltrate led to the diagnosis of DRIF. The papules associated with DRIF can be recurrent or chronic. The lesions in this patient were chronic and persistent.
Despite limited evidence, it has been suggested that DRIF may be a manifestation of atopic dermatitis in patients with darker skin tones. In our case, the patient had a history of childhood eczema. Other hypotheses have proposed that DRIF could be a nonspecific reaction to a currently unknown antigen. A causative infectious agent has not been identified, although the search continues. There is speculation that DRIF could be an overt expression of normal follicular prominence, but the presence of occasional pustules and lymphocyte- predominant infundibular infiltrate negates that.3
Confluent and reticulated papillomatosis was included in the differential for our patient and manifests as asymptomatic hyperpigmented papules and plaques frequently occurring on the upper trunk, neck, and axilla; however, these lesions have a peripheral netlike configuration, as the name suggests. Additionally, this condition is thought to have an infectious component (Dietzia papillomatosis) and responds to antibiotic treatment.5 Follicular eczema also was high in the differential diagnosis but usually is seasonal and pruritic, and histopathology typically shows the features of spongiotic dermatitis. It also would respond well to topical steroids.6 Another condition high on the differential was juxtaclavicular beaded lines, which also manifests as flesh-colored follicular papules distributed on the upper trunk; however, histopathology usually shows features of hyperplastic pilosebaceous units along with spongiosis and exocytosis.7 Pityrosporum folliculitis initially was considered, but the patient only endorsed occasional pruritus. Additionally, no fungal elements were observed.
Currently, there are no definitive treatments for DRIF. The topical treatments available include midpotency corticosteroids, tretinoin, calcineurin inhibitors, 12% lactic acid, and 20% to 40% urea. The systemic therapies are high-dose oral vitamin A (100,000 IU/d), isotretinoin, and psoralen plus UVA.8-10
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Ma BC, Sahni VN, Sahni DR, et al. Disseminate and recurrent infundibulofolliculitis: an under-recognized yet treatable entity. J Drugs Dermatol. 2021;20:1353-1354. doi:10.36849/jdd.6173
- Nair SP, Gomathy M, Kumar GN. Disseminate and recurrent infundibulo- folliculitis in an Indian patient: a case report with review of literature. Indian Dermatol Online J. 2017;8:39-41. doi:10.4103/2229- 5178.198775
- Rekha S, Kumar V, Rao P, et al. Disseminate and recurrent infundibulofolliculitis. Indian J Dermatol. 2019;64:404-406. doi:10.4103/ijd.IJD_77_18
- Jones AL, Koerner RJ, Natarajan S, et al. Dietzia papillomatosis sp. nov., a novel actinomycete isolated from the skin of an immunocompetent patient with confluent and reticulated papillomatosis. Int J Syst Evol Microbiol. 2008;58(pt 1):68-72. doi:10.1099/ijs.0.65178-0
- Cohen PR. Follicular contact dermatitis revisited: a review emphasizing neomycin-associated follicular contact dermatitis. World J Clin Cases. 2014;2:815-821. doi:10.12998/wjcc.v2.i12.815
- Butterworth T, Johnson WC. Justa-clavicular beaded lines. Arch Dermatol. 1974;110:891-893.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulo-folliculitis responsive to treatment with isotretinoin. J Dermatol. 2002;29:431-434.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-76.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Ma BC, Sahni VN, Sahni DR, et al. Disseminate and recurrent infundibulofolliculitis: an under-recognized yet treatable entity. J Drugs Dermatol. 2021;20:1353-1354. doi:10.36849/jdd.6173
- Nair SP, Gomathy M, Kumar GN. Disseminate and recurrent infundibulo- folliculitis in an Indian patient: a case report with review of literature. Indian Dermatol Online J. 2017;8:39-41. doi:10.4103/2229- 5178.198775
- Rekha S, Kumar V, Rao P, et al. Disseminate and recurrent infundibulofolliculitis. Indian J Dermatol. 2019;64:404-406. doi:10.4103/ijd.IJD_77_18
- Jones AL, Koerner RJ, Natarajan S, et al. Dietzia papillomatosis sp. nov., a novel actinomycete isolated from the skin of an immunocompetent patient with confluent and reticulated papillomatosis. Int J Syst Evol Microbiol. 2008;58(pt 1):68-72. doi:10.1099/ijs.0.65178-0
- Cohen PR. Follicular contact dermatitis revisited: a review emphasizing neomycin-associated follicular contact dermatitis. World J Clin Cases. 2014;2:815-821. doi:10.12998/wjcc.v2.i12.815
- Butterworth T, Johnson WC. Justa-clavicular beaded lines. Arch Dermatol. 1974;110:891-893.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulo-folliculitis responsive to treatment with isotretinoin. J Dermatol. 2002;29:431-434.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-76.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
Diffusely Scattered Linear Folliculopapular Eruption
Diffusely Scattered Linear Folliculopapular Eruption
A 31-year-old man with a darker skin tone and a history of childhood eczema presented with papules on the trunk and upper arms of several years’ duration. The papules were persistent and were generally asymptomatic but occasionally pruritic. The patient previously had self-treated with over-the counter lotions and topical hydrocortisone with no appreciable changes. On physical examination, a hyperpigmented patch with follicular monomorphic papules was noted across the upper back along with confluent papules and plaques predominantly on the trunk and upper arms. Additionally, the patient had several monomorphic papules in a linear distribution on the neck. Review of systems and examination of the remaining skin were unremarkable. A biopsy from a representative papule on the left upper back was performed.