User login
Vascular Nodule on the Upper Chest
Vascular Nodule on the Upper Chest
THE DIAGNOSIS: Metastatic Renal Cell Carcinoma
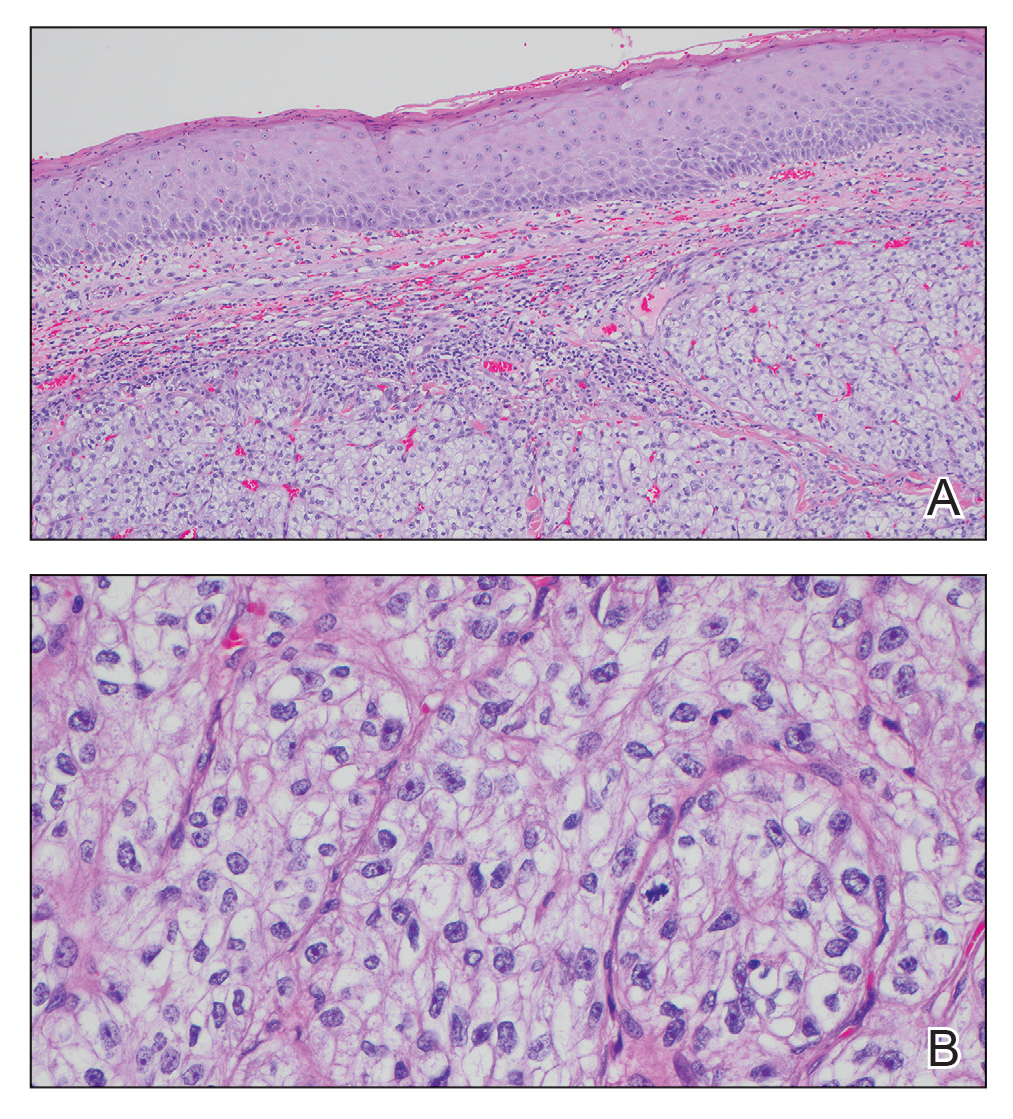
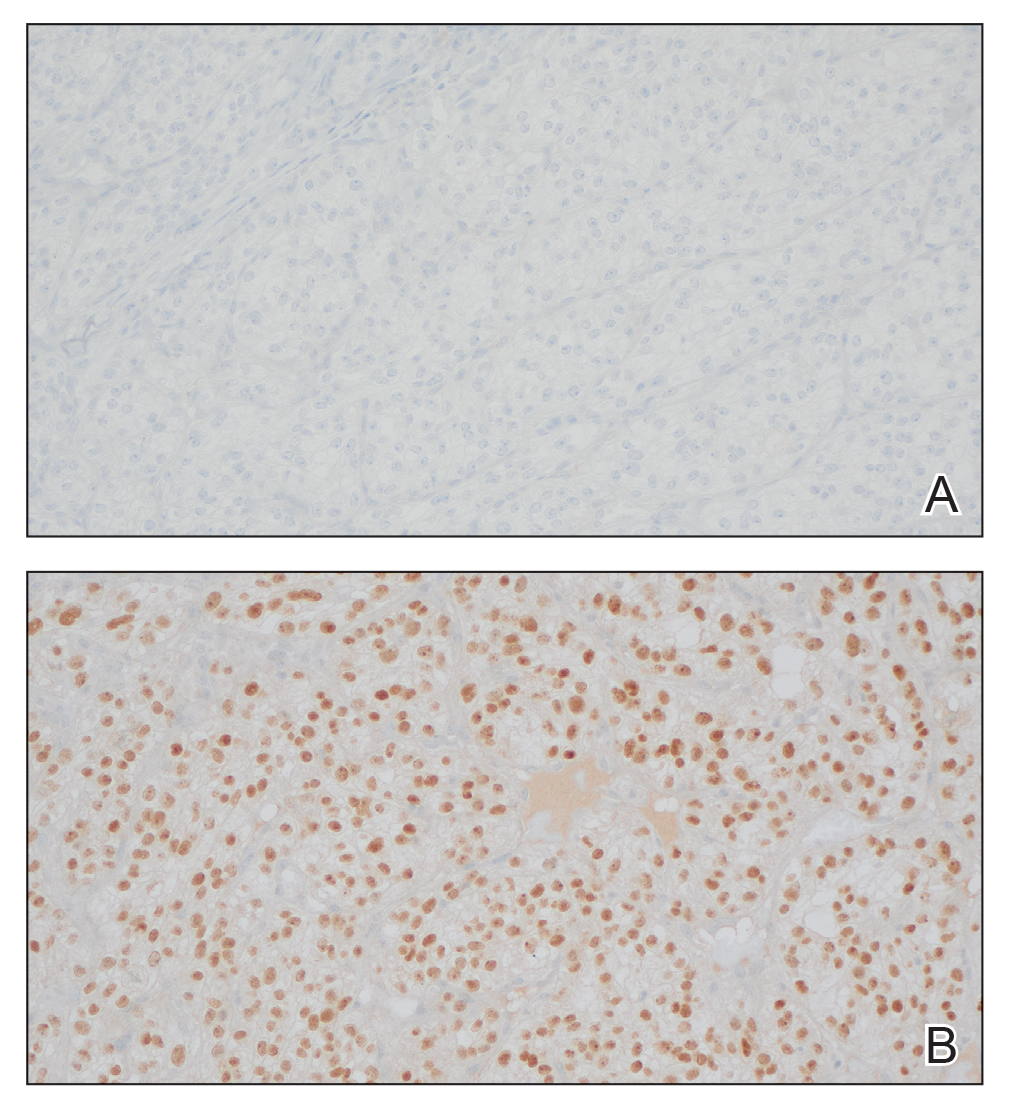
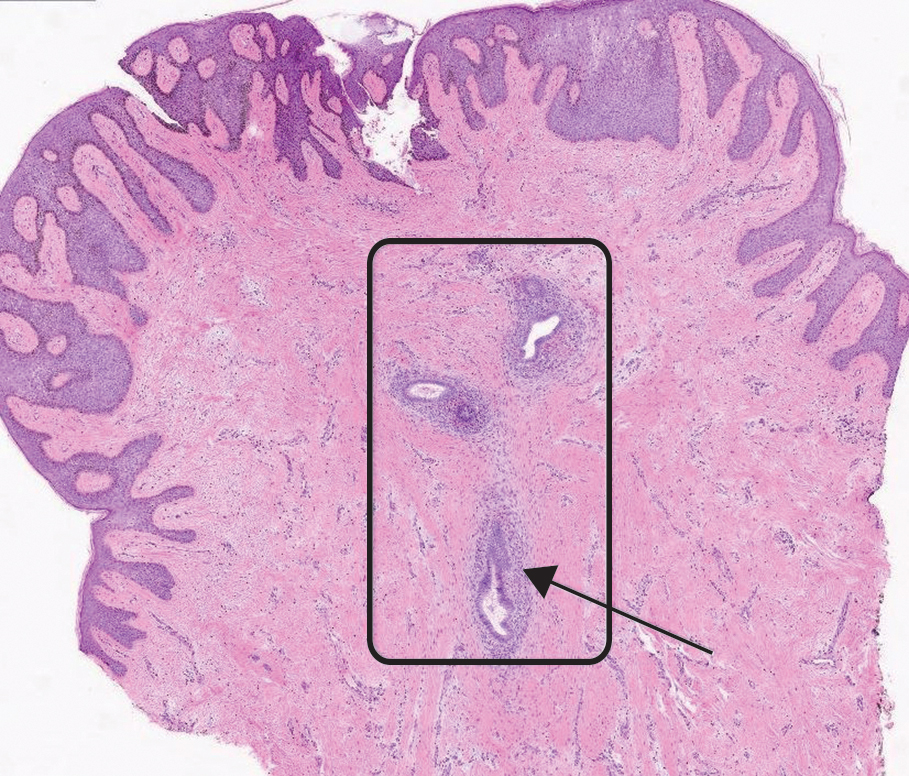
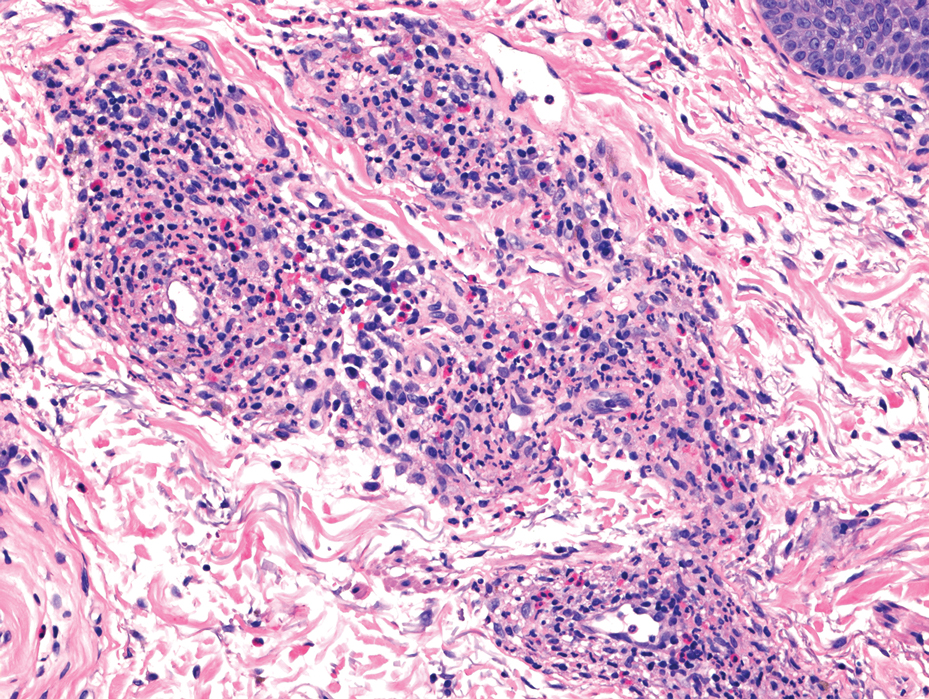
The shave biopsy revealed large cells with prominent nucleoli, clear cytoplasm, and thin cell borders in a nestlike arrangement (Figure 1). Immunohistochemical examination was negative for cytokeratin 5/6 and positive for PAX8 (Figure 2), which finalized the diagnosis of metastatic renal cell carcinoma (RCC). Later, our patient had a core biopsy-proven metastasis to the C6 spinous process, with concern for additional metastasis to the liver and lungs on positron emission tomography. Our patient’s treatment plan included pembrolizumab and axitinib to manage further cutaneous metastasis and radiation therapy for the C6 spinous process metastasis.
Renal cell carcinoma denotes cancer originating from the renal epithelium and is the most common kidney tumor in adults.1 Renal cell carcinoma accounts for more than 90% of kidney malignancies in the United States and has 3 main subtypes: clear cell RCC, papillary RCC, and chromophobe RCC.2 About 25% of cases metastasize, commonly to the lungs, liver, bones, lymph nodes, contralateral kidney, and adrenal glands.3
Cutaneous metastasis of RCC is rare, with an incidence of approximately 3.3%.4 Notably, 80% to 90% of patients with metastatic skin lesions had a prior diagnosis of RCC.2 Skin metastases associated with RCC predominantly are found on the face and scalp, appearing as nodular, swiftly expanding, circular, or oval-shaped growths. The robust vascular element of these lesions can lead to confusion with regard to the proper diagnosis, as they often resemble hemangiomas, pyogenic granulomas, or Kaposi sarcomas.4
Many cutaneous metastases linked to RCC exhibit a histomorphologic pattern consistent with clear cell adenocarcinoma.2 The malignant cells are large and possess transparent cytoplasm, round to oval nuclei, and prominent nucleoli. The cells can form glandular, acinar, or papillary arrangements; extravasated red blood cells frequently are found within the surrounding fibrovascular tissue.5 The presence of cytoplasmic glycogen can be revealed through periodic acid–Schiff staining. Other immunohistochemical markers commonly used to identify skin metastasis of RCC include epithelioid membrane antigen, carcinoembryonic antigen, and CD-10.1
Various mechanisms are involved in the cutaneous metastases of RCC. The most common pathway involves infiltration of the skin directly overlying the malignant renal mass; additional potential mechanisms include the introduction of abnormal cells into the skin during surgical or diagnostic interventions and their dissemination through the lymphatic system or bloodstream.1 Among urogenital malignancies other than RCC, skin metastases predominantly manifest in the abdominal region.2 Conversely, the head and neck region are more frequently impacted in RCC. The vascular composition of these tumors plays a role in facilitating the extension of cancer cells through the bloodstream, fostering the emergence of distant metastases.6
The development of cutaneous metastasis in RCC is associated with a poor prognosis, as most patients die within 6 months of detection.3 Treatment options thus are limited and palliative. Although local excision is an alternative treatment for localized cutaneous metastasis, it often provides little benefit in the presence of extensive metastasis; radiotherapy also has been shown to have a limited effect on primary RCC, though its devascularization of the lesion may be effective in metastatic cases.5 Immune checkpoint inhibitors such as nivolumab and ipilimumab have improved progression-free survival in patients with metastatic RCC, though uncertainty remains regarding their efficacy in attenuating cutaneous metastasis.5,6
- Kanwal R. Metastasis in renal cell carcinoma: biology and treatment. Adv Cancer Biol Metastasis. 2023;7:100094. doi:10.1016 /j.adcanc.2023.100094
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Bianchi M, Sun M, Jeldres C, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. 2012;23:973-980. doi:10.1093/annonc/mdr362
- Lorenzo-Rios D, Cruzval-O’Reilly E, Rabelo-Cartagena J. Facial cutaneous metastasis in renal cell carcinoma. Cureus. 2020;12:E12093. doi:10.7759/cureus.12093
- Iliescu CA, Beiu C, Racovit·a¢ A, et al. Atypical presentation of rapidly progressive cutaneous metastases of clear cell renal carcinoma: a case report. Medicina. 2024;60:1797. doi:10.3390/medicina60111797
- Joyce MJ. Management of skeletal metastases in renal cell carcinoma patients. In: Bukowski RM, Novick AC, eds. Clinical Management of Renal Tumors. Springer; 2008: 421-459.
THE DIAGNOSIS: Metastatic Renal Cell Carcinoma
The shave biopsy revealed large cells with prominent nucleoli, clear cytoplasm, and thin cell borders in a nestlike arrangement (Figure 1). Immunohistochemical examination was negative for cytokeratin 5/6 and positive for PAX8 (Figure 2), which finalized the diagnosis of metastatic renal cell carcinoma (RCC). Later, our patient had a core biopsy-proven metastasis to the C6 spinous process, with concern for additional metastasis to the liver and lungs on positron emission tomography. Our patient’s treatment plan included pembrolizumab and axitinib to manage further cutaneous metastasis and radiation therapy for the C6 spinous process metastasis.
Renal cell carcinoma denotes cancer originating from the renal epithelium and is the most common kidney tumor in adults.1 Renal cell carcinoma accounts for more than 90% of kidney malignancies in the United States and has 3 main subtypes: clear cell RCC, papillary RCC, and chromophobe RCC.2 About 25% of cases metastasize, commonly to the lungs, liver, bones, lymph nodes, contralateral kidney, and adrenal glands.3
Cutaneous metastasis of RCC is rare, with an incidence of approximately 3.3%.4 Notably, 80% to 90% of patients with metastatic skin lesions had a prior diagnosis of RCC.2 Skin metastases associated with RCC predominantly are found on the face and scalp, appearing as nodular, swiftly expanding, circular, or oval-shaped growths. The robust vascular element of these lesions can lead to confusion with regard to the proper diagnosis, as they often resemble hemangiomas, pyogenic granulomas, or Kaposi sarcomas.4
Many cutaneous metastases linked to RCC exhibit a histomorphologic pattern consistent with clear cell adenocarcinoma.2 The malignant cells are large and possess transparent cytoplasm, round to oval nuclei, and prominent nucleoli. The cells can form glandular, acinar, or papillary arrangements; extravasated red blood cells frequently are found within the surrounding fibrovascular tissue.5 The presence of cytoplasmic glycogen can be revealed through periodic acid–Schiff staining. Other immunohistochemical markers commonly used to identify skin metastasis of RCC include epithelioid membrane antigen, carcinoembryonic antigen, and CD-10.1
Various mechanisms are involved in the cutaneous metastases of RCC. The most common pathway involves infiltration of the skin directly overlying the malignant renal mass; additional potential mechanisms include the introduction of abnormal cells into the skin during surgical or diagnostic interventions and their dissemination through the lymphatic system or bloodstream.1 Among urogenital malignancies other than RCC, skin metastases predominantly manifest in the abdominal region.2 Conversely, the head and neck region are more frequently impacted in RCC. The vascular composition of these tumors plays a role in facilitating the extension of cancer cells through the bloodstream, fostering the emergence of distant metastases.6
The development of cutaneous metastasis in RCC is associated with a poor prognosis, as most patients die within 6 months of detection.3 Treatment options thus are limited and palliative. Although local excision is an alternative treatment for localized cutaneous metastasis, it often provides little benefit in the presence of extensive metastasis; radiotherapy also has been shown to have a limited effect on primary RCC, though its devascularization of the lesion may be effective in metastatic cases.5 Immune checkpoint inhibitors such as nivolumab and ipilimumab have improved progression-free survival in patients with metastatic RCC, though uncertainty remains regarding their efficacy in attenuating cutaneous metastasis.5,6
THE DIAGNOSIS: Metastatic Renal Cell Carcinoma
The shave biopsy revealed large cells with prominent nucleoli, clear cytoplasm, and thin cell borders in a nestlike arrangement (Figure 1). Immunohistochemical examination was negative for cytokeratin 5/6 and positive for PAX8 (Figure 2), which finalized the diagnosis of metastatic renal cell carcinoma (RCC). Later, our patient had a core biopsy-proven metastasis to the C6 spinous process, with concern for additional metastasis to the liver and lungs on positron emission tomography. Our patient’s treatment plan included pembrolizumab and axitinib to manage further cutaneous metastasis and radiation therapy for the C6 spinous process metastasis.
Renal cell carcinoma denotes cancer originating from the renal epithelium and is the most common kidney tumor in adults.1 Renal cell carcinoma accounts for more than 90% of kidney malignancies in the United States and has 3 main subtypes: clear cell RCC, papillary RCC, and chromophobe RCC.2 About 25% of cases metastasize, commonly to the lungs, liver, bones, lymph nodes, contralateral kidney, and adrenal glands.3
Cutaneous metastasis of RCC is rare, with an incidence of approximately 3.3%.4 Notably, 80% to 90% of patients with metastatic skin lesions had a prior diagnosis of RCC.2 Skin metastases associated with RCC predominantly are found on the face and scalp, appearing as nodular, swiftly expanding, circular, or oval-shaped growths. The robust vascular element of these lesions can lead to confusion with regard to the proper diagnosis, as they often resemble hemangiomas, pyogenic granulomas, or Kaposi sarcomas.4
Many cutaneous metastases linked to RCC exhibit a histomorphologic pattern consistent with clear cell adenocarcinoma.2 The malignant cells are large and possess transparent cytoplasm, round to oval nuclei, and prominent nucleoli. The cells can form glandular, acinar, or papillary arrangements; extravasated red blood cells frequently are found within the surrounding fibrovascular tissue.5 The presence of cytoplasmic glycogen can be revealed through periodic acid–Schiff staining. Other immunohistochemical markers commonly used to identify skin metastasis of RCC include epithelioid membrane antigen, carcinoembryonic antigen, and CD-10.1
Various mechanisms are involved in the cutaneous metastases of RCC. The most common pathway involves infiltration of the skin directly overlying the malignant renal mass; additional potential mechanisms include the introduction of abnormal cells into the skin during surgical or diagnostic interventions and their dissemination through the lymphatic system or bloodstream.1 Among urogenital malignancies other than RCC, skin metastases predominantly manifest in the abdominal region.2 Conversely, the head and neck region are more frequently impacted in RCC. The vascular composition of these tumors plays a role in facilitating the extension of cancer cells through the bloodstream, fostering the emergence of distant metastases.6
The development of cutaneous metastasis in RCC is associated with a poor prognosis, as most patients die within 6 months of detection.3 Treatment options thus are limited and palliative. Although local excision is an alternative treatment for localized cutaneous metastasis, it often provides little benefit in the presence of extensive metastasis; radiotherapy also has been shown to have a limited effect on primary RCC, though its devascularization of the lesion may be effective in metastatic cases.5 Immune checkpoint inhibitors such as nivolumab and ipilimumab have improved progression-free survival in patients with metastatic RCC, though uncertainty remains regarding their efficacy in attenuating cutaneous metastasis.5,6
- Kanwal R. Metastasis in renal cell carcinoma: biology and treatment. Adv Cancer Biol Metastasis. 2023;7:100094. doi:10.1016 /j.adcanc.2023.100094
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Bianchi M, Sun M, Jeldres C, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. 2012;23:973-980. doi:10.1093/annonc/mdr362
- Lorenzo-Rios D, Cruzval-O’Reilly E, Rabelo-Cartagena J. Facial cutaneous metastasis in renal cell carcinoma. Cureus. 2020;12:E12093. doi:10.7759/cureus.12093
- Iliescu CA, Beiu C, Racovit·a¢ A, et al. Atypical presentation of rapidly progressive cutaneous metastases of clear cell renal carcinoma: a case report. Medicina. 2024;60:1797. doi:10.3390/medicina60111797
- Joyce MJ. Management of skeletal metastases in renal cell carcinoma patients. In: Bukowski RM, Novick AC, eds. Clinical Management of Renal Tumors. Springer; 2008: 421-459.
- Kanwal R. Metastasis in renal cell carcinoma: biology and treatment. Adv Cancer Biol Metastasis. 2023;7:100094. doi:10.1016 /j.adcanc.2023.100094
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Bianchi M, Sun M, Jeldres C, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. 2012;23:973-980. doi:10.1093/annonc/mdr362
- Lorenzo-Rios D, Cruzval-O’Reilly E, Rabelo-Cartagena J. Facial cutaneous metastasis in renal cell carcinoma. Cureus. 2020;12:E12093. doi:10.7759/cureus.12093
- Iliescu CA, Beiu C, Racovit·a¢ A, et al. Atypical presentation of rapidly progressive cutaneous metastases of clear cell renal carcinoma: a case report. Medicina. 2024;60:1797. doi:10.3390/medicina60111797
- Joyce MJ. Management of skeletal metastases in renal cell carcinoma patients. In: Bukowski RM, Novick AC, eds. Clinical Management of Renal Tumors. Springer; 2008: 421-459.
Vascular Nodule on the Upper Chest
Vascular Nodule on the Upper Chest
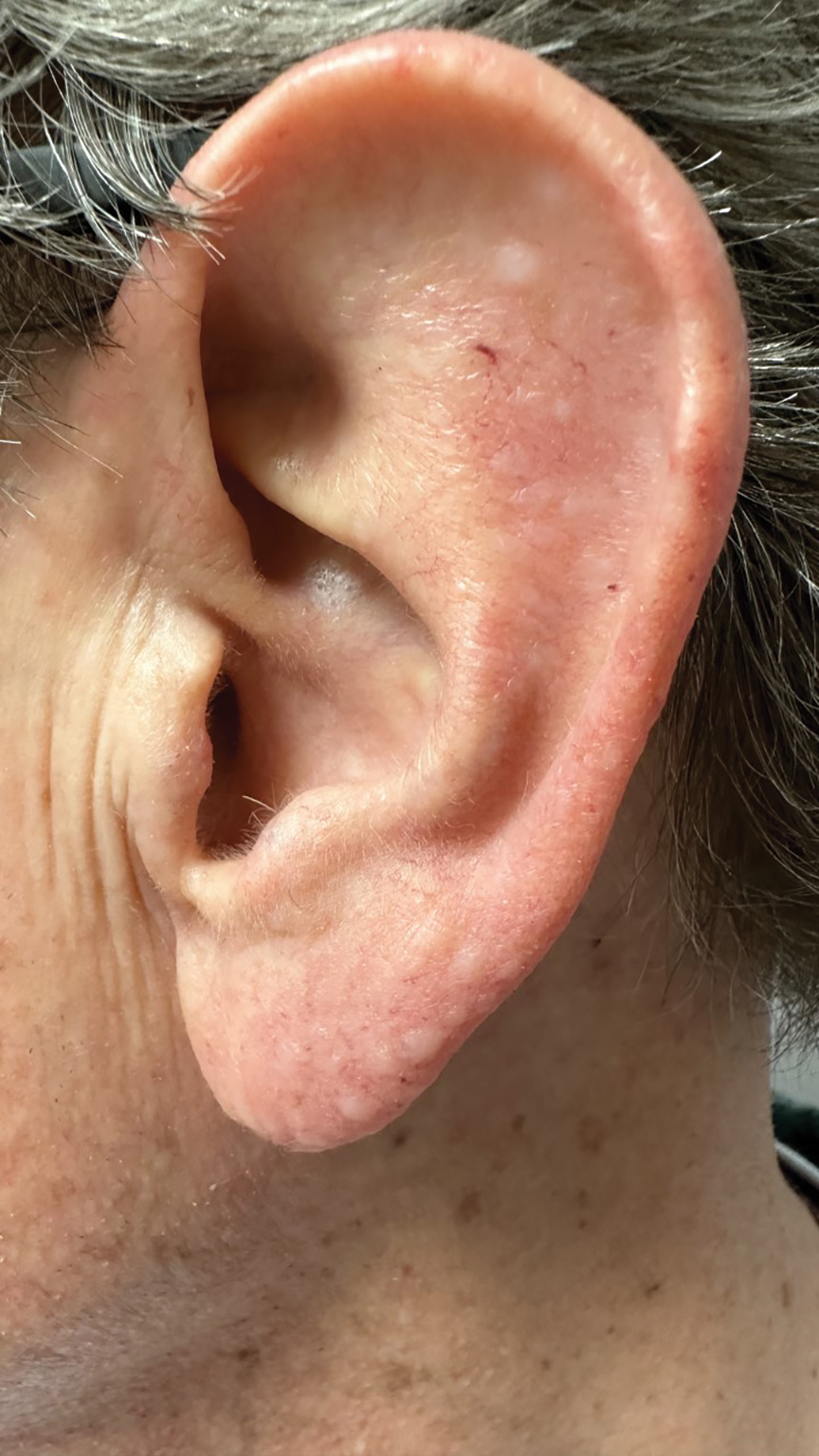
A 45-year-old man presented to the dermatology clinic with a bleeding nodule on the upper chest of 2 months’ duration. He had a history of a low-grade mucoepidermoid carcinoma of the left parotid gland that was diagnosed 14 years prior and was treated via parotidectomy with 1 positive lymph node removed. Two months prior to the current presentation, the patient presented to the emergency department with unintentional weight loss and fatigue and subsequently was diagnosed with clear cell renal cell carcinoma that was treated via radical nephrectomy.
At the current presentation, the patient denied any recent fatigue, fever, weight loss, shortness of breath, or abdominal pain but reported neck stiffness. Physical examination revealed a solitary, smooth, vascular, 1.5×1.5 cm nodule on the left upper chest with no overlying skin changes. The remainder of the skin examination was unremarkable. A shave biopsy of the nodule was performed.

A Painful Flesh-Colored Papule on the Shoulder
A Painful Flesh-Colored Papule on the Shoulder
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
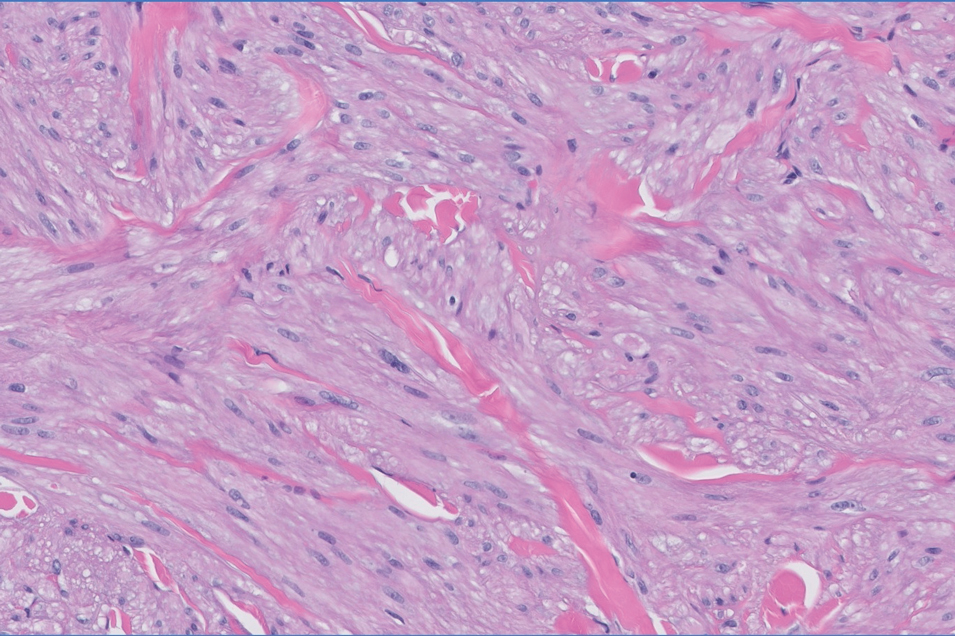
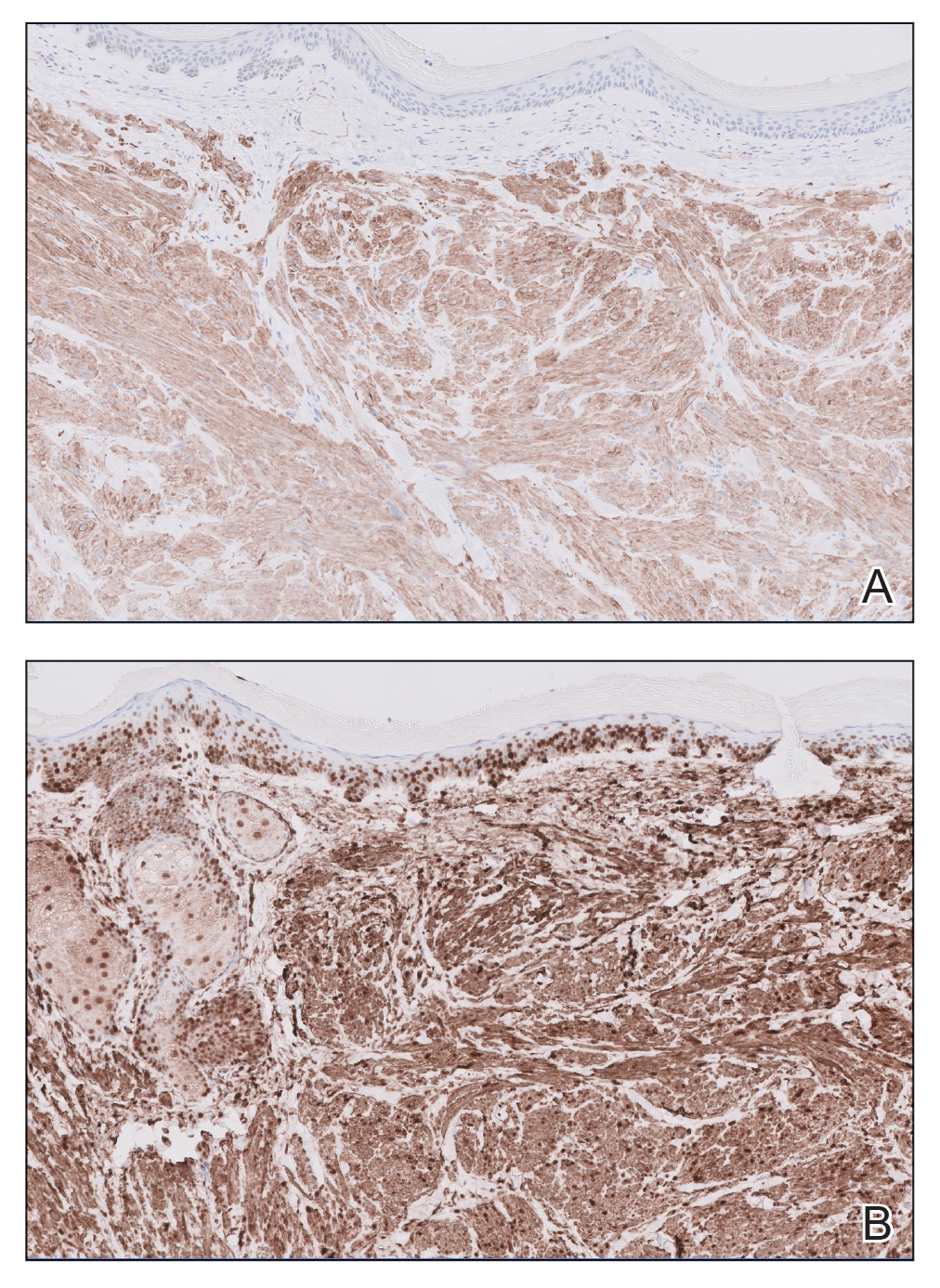

Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
A Painful Flesh-Colored Papule on the Shoulder
A Painful Flesh-Colored Papule on the Shoulder
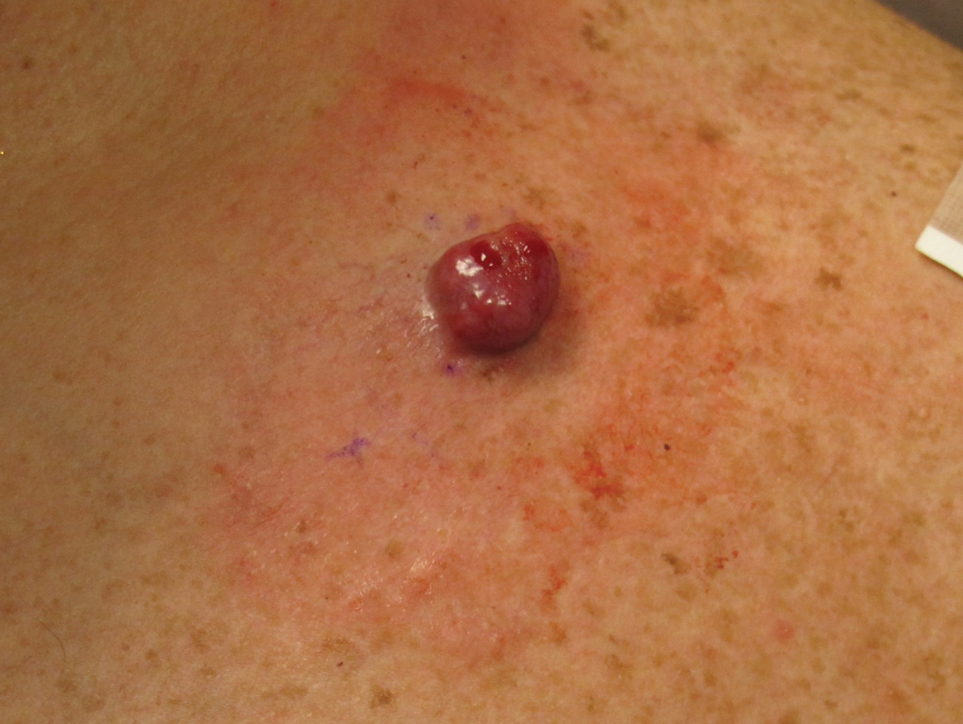
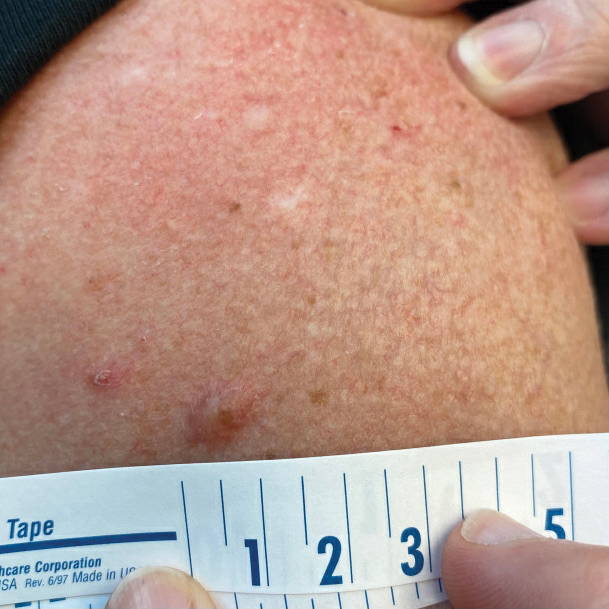
A 65-year-old woman with a history of metabolic syndrome presented to the family medicine clinic for evaluation of a papule on the right shoulder that had started small and increased in size over the past 3 years. Physical examination revealed a 1.0×0.8×0.1-cm, smooth, flesh-colored to light brown papule on the right shoulder that was notably tender to palpation. The patient reported that her paternal uncle had multiple skin lesions of similar morphology dispersed on the bilateral upper extremities. A shave biopsy of the lesion was performed.
Erythematous Annular Scaly Plaques on the Upper Chest
Erythematous Annular Scaly Plaques on the Upper Chest
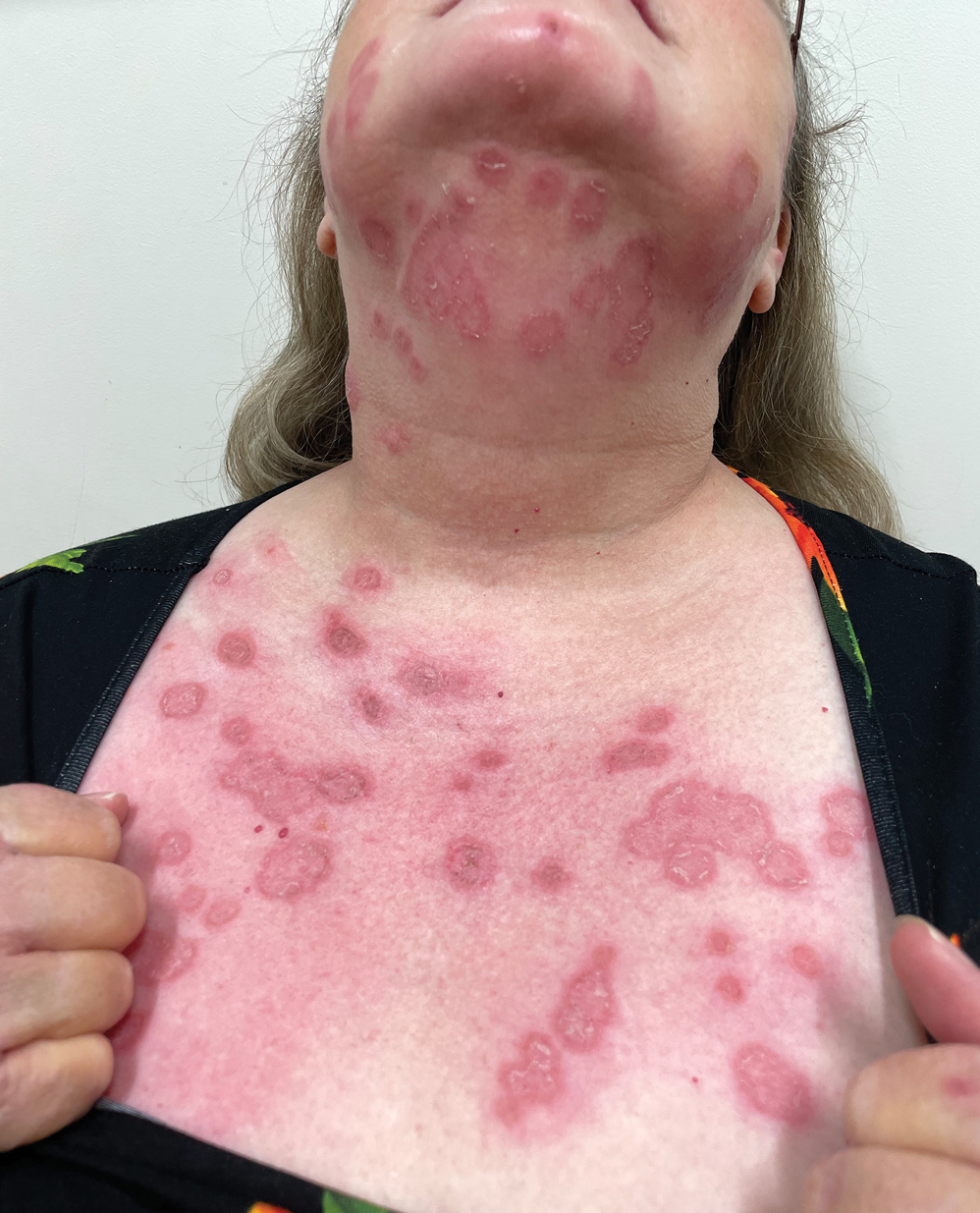
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.

Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.
Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.
Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
Erythematous Annular Scaly Plaques on the Upper Chest
Erythematous Annular Scaly Plaques on the Upper Chest
A 60-year-old woman with a history of keratinocyte carcinomas, hypertension, diabetes mellitus, and anxiety presented to the dermatology department with a widespread rash of more than 2 weeks’ duration. The patient had tried 1 to 2 days of self-treatment with triamcinolone cream that she had previously been prescribed for an unknown dermatitis and zinc oxide cream, which caused considerable inflammation of the rash and prompted her to discontinue use. She could not recall any recent use of new personal care products or medications or eating any new foods. She also denied any recent yard work, known arthropod bites, illnesses, prolonged sun exposure, or constitutional symptoms. Her medications included metformin, hydrochlorothiazide, losartan, and sertraline. She also reported taking daily supplements of vitamins D, K, and C as well as acetaminophen and ibuprofen as needed. Physical examination revealed several 2- to 4-cm, erythematous, annular, circinate, petaloid plaques with scale mostly on photodistributed areas of the central anterior chest, neck, lower cheeks, and chin as well as a few scattered lesions with similar morphology on the arms, lower abdomen, left buttock, and back.

Cyclically Bleeding Umbilical Papules
Cyclically Bleeding Umbilical Papules
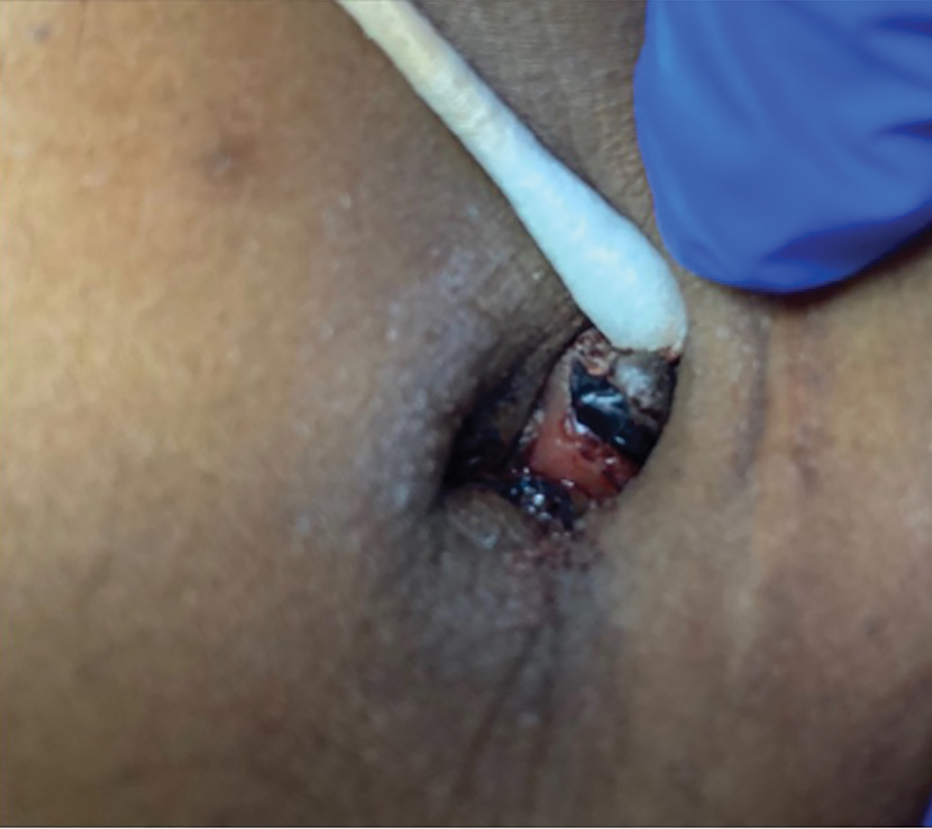
THE DIAGNOSIS: Cutaneous Endometriosis
On histopathology, a biopsy specimen of an umbilical papule showed a dermal lymphohistiocyticrich infiltrate, hemorrhage, and ectopic endometrial glands consistent with cutaneous endometriosis (CE)(Figure). Cutaneous endometriosis is a rare condition that typically affects females of reproductive potential and is characterized by endometrial glands and stroma within the dermis and hypodermis. Cutaneous endometriosis is classified as primary or secondary. There is no surgical history of the abdomen or pelvis in primary CE. In contrast, a history of abdominopelvic surgery is the defining characteristic of secondary CE, which is more common than primary CE and typically manifests as painful red, brown, or purple papules along preexisting surgical scars of the umbilicus, lower abdomen, or pelvic region.1 Our patient may have developed secondary CE related to the laparoscopic cholecystectomy performed 10 years prior. Surgical excision is considered the definitive treatment for CE, and hormonal therapy with danazol or leuprolide may help ameliorate symptoms.1 Our patient deferred any hormonal or surgical interventions to undergo fertility treatments for pregnancy.
Cyclical bleeding and pain that coincides with menstruation is consistent with CE; however, cyclical symptoms are not always present, which can lead to delayed or incorrect diagnosis. Biopsy and histopathologic analysis are required for definitive diagnosis and are critical for distinguishing CE from other conditions. The differential diagnosis in our patient included pyogenic granuloma, dermatofibrosarcoma protuberans, keloid, and cutaneous metastasis of a primary malignancy. Vascular lesions such as pyogenic granuloma can manifest with bleeding but have a characteristic histopathologic lobular capillary arrangement that was not present in our patient.
Dermatofibrosarcoma protuberans is a rare, slow-growing, malignant soft-tissue sarcoma that most commonly manifests on the trunk, arms, and legs.2 It is characterized by a slow-growing, indurated plaque that often is present for years and may suddenly progress into a smooth, red-brown, multinodular mass. Histopathology typically shows spindle cells infiltrating the dermis and subcutaneous tissue in storiform or whorled pattern with variations based on the tumor stage, as well as diffuse CD34 immunoreactivity.2
Keloids are dense, raised, hyperpigmented, fibrous nodules—sometimes with accompanying telangiectasias—that typically grow secondary to trauma and project past the boundaries of the initial trauma site.1 Keloids are more commonly seen in individuals with darker skin types and tend to grow larger in this population. Histopathology reveals thickened hyalinized collagen bundles, which were not seen in our patient.1
Metastatic skin lesions of the umbilicus are rare but can arise from internal malignancies including cancers of the lung, colon, and breast.3 We considered Sister Mary Joseph nodule, which is caused most commonly by metastasis of a primary gastrointestinal cancer and signifies poor prognosis. The histopathology of metastatic lesions would reveal the presence of atypical cells with cancer-specific markers. Histopathology along with the patient’s personal and family history, a comprehensive review of symptoms, and cancer screening may help with reaching the correct diagnosis.
The average duration between abdominopelvic surgery and onset of secondary CE symptoms is 3.7 to 5.3 years.4 Our patient presented 10 years post surgery and after cessation of oral contraception, which may suggest a potential role of hormonal contraception in delayed CE onset. Diagnosis of CE can be challenging due to atypical signs or symptoms, delayed onset, and lack of awareness among health care professionals. Patients with delayed diagnosis may endure multiple procedures, prolonged physical pain, and emotional distress. Furthermore, 30% to 50% of females with endometriosis experience infertility. Delayed diagnosis of CE compounded with associated age-related increase in oocyte atresia could potentially worsen fecundity as patients age.5 It is important to consider CE in the differential diagnosis of females of reproductive age who present with cyclical bleeding and abdominal or umbilical nodules.
- James WD, Elston D, Treat JR, et al. Andrews Diseases of the Skin: Clinical Dermatology. 13th ed. Elsevier; 2019. Accessed March 19, 2024. https://search.worldcat.org/title/1084979207
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Komurcugil I, Arslan Z, Bal ZI, et al. Cutaneous metastases different clinical presentations: case series and review of the literature. Dermatol Reports. 2022;15:9553.
- Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Published online September 16, 2019. Eur J Obstet Gynecol Reprod Biol X.
- Missmer SA, Hankinson SE, Spiegelman D, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784-796.
THE DIAGNOSIS: Cutaneous Endometriosis
On histopathology, a biopsy specimen of an umbilical papule showed a dermal lymphohistiocyticrich infiltrate, hemorrhage, and ectopic endometrial glands consistent with cutaneous endometriosis (CE)(Figure). Cutaneous endometriosis is a rare condition that typically affects females of reproductive potential and is characterized by endometrial glands and stroma within the dermis and hypodermis. Cutaneous endometriosis is classified as primary or secondary. There is no surgical history of the abdomen or pelvis in primary CE. In contrast, a history of abdominopelvic surgery is the defining characteristic of secondary CE, which is more common than primary CE and typically manifests as painful red, brown, or purple papules along preexisting surgical scars of the umbilicus, lower abdomen, or pelvic region.1 Our patient may have developed secondary CE related to the laparoscopic cholecystectomy performed 10 years prior. Surgical excision is considered the definitive treatment for CE, and hormonal therapy with danazol or leuprolide may help ameliorate symptoms.1 Our patient deferred any hormonal or surgical interventions to undergo fertility treatments for pregnancy.
Cyclical bleeding and pain that coincides with menstruation is consistent with CE; however, cyclical symptoms are not always present, which can lead to delayed or incorrect diagnosis. Biopsy and histopathologic analysis are required for definitive diagnosis and are critical for distinguishing CE from other conditions. The differential diagnosis in our patient included pyogenic granuloma, dermatofibrosarcoma protuberans, keloid, and cutaneous metastasis of a primary malignancy. Vascular lesions such as pyogenic granuloma can manifest with bleeding but have a characteristic histopathologic lobular capillary arrangement that was not present in our patient.
Dermatofibrosarcoma protuberans is a rare, slow-growing, malignant soft-tissue sarcoma that most commonly manifests on the trunk, arms, and legs.2 It is characterized by a slow-growing, indurated plaque that often is present for years and may suddenly progress into a smooth, red-brown, multinodular mass. Histopathology typically shows spindle cells infiltrating the dermis and subcutaneous tissue in storiform or whorled pattern with variations based on the tumor stage, as well as diffuse CD34 immunoreactivity.2
Keloids are dense, raised, hyperpigmented, fibrous nodules—sometimes with accompanying telangiectasias—that typically grow secondary to trauma and project past the boundaries of the initial trauma site.1 Keloids are more commonly seen in individuals with darker skin types and tend to grow larger in this population. Histopathology reveals thickened hyalinized collagen bundles, which were not seen in our patient.1
Metastatic skin lesions of the umbilicus are rare but can arise from internal malignancies including cancers of the lung, colon, and breast.3 We considered Sister Mary Joseph nodule, which is caused most commonly by metastasis of a primary gastrointestinal cancer and signifies poor prognosis. The histopathology of metastatic lesions would reveal the presence of atypical cells with cancer-specific markers. Histopathology along with the patient’s personal and family history, a comprehensive review of symptoms, and cancer screening may help with reaching the correct diagnosis.
The average duration between abdominopelvic surgery and onset of secondary CE symptoms is 3.7 to 5.3 years.4 Our patient presented 10 years post surgery and after cessation of oral contraception, which may suggest a potential role of hormonal contraception in delayed CE onset. Diagnosis of CE can be challenging due to atypical signs or symptoms, delayed onset, and lack of awareness among health care professionals. Patients with delayed diagnosis may endure multiple procedures, prolonged physical pain, and emotional distress. Furthermore, 30% to 50% of females with endometriosis experience infertility. Delayed diagnosis of CE compounded with associated age-related increase in oocyte atresia could potentially worsen fecundity as patients age.5 It is important to consider CE in the differential diagnosis of females of reproductive age who present with cyclical bleeding and abdominal or umbilical nodules.
THE DIAGNOSIS: Cutaneous Endometriosis
On histopathology, a biopsy specimen of an umbilical papule showed a dermal lymphohistiocyticrich infiltrate, hemorrhage, and ectopic endometrial glands consistent with cutaneous endometriosis (CE)(Figure). Cutaneous endometriosis is a rare condition that typically affects females of reproductive potential and is characterized by endometrial glands and stroma within the dermis and hypodermis. Cutaneous endometriosis is classified as primary or secondary. There is no surgical history of the abdomen or pelvis in primary CE. In contrast, a history of abdominopelvic surgery is the defining characteristic of secondary CE, which is more common than primary CE and typically manifests as painful red, brown, or purple papules along preexisting surgical scars of the umbilicus, lower abdomen, or pelvic region.1 Our patient may have developed secondary CE related to the laparoscopic cholecystectomy performed 10 years prior. Surgical excision is considered the definitive treatment for CE, and hormonal therapy with danazol or leuprolide may help ameliorate symptoms.1 Our patient deferred any hormonal or surgical interventions to undergo fertility treatments for pregnancy.
Cyclical bleeding and pain that coincides with menstruation is consistent with CE; however, cyclical symptoms are not always present, which can lead to delayed or incorrect diagnosis. Biopsy and histopathologic analysis are required for definitive diagnosis and are critical for distinguishing CE from other conditions. The differential diagnosis in our patient included pyogenic granuloma, dermatofibrosarcoma protuberans, keloid, and cutaneous metastasis of a primary malignancy. Vascular lesions such as pyogenic granuloma can manifest with bleeding but have a characteristic histopathologic lobular capillary arrangement that was not present in our patient.
Dermatofibrosarcoma protuberans is a rare, slow-growing, malignant soft-tissue sarcoma that most commonly manifests on the trunk, arms, and legs.2 It is characterized by a slow-growing, indurated plaque that often is present for years and may suddenly progress into a smooth, red-brown, multinodular mass. Histopathology typically shows spindle cells infiltrating the dermis and subcutaneous tissue in storiform or whorled pattern with variations based on the tumor stage, as well as diffuse CD34 immunoreactivity.2
Keloids are dense, raised, hyperpigmented, fibrous nodules—sometimes with accompanying telangiectasias—that typically grow secondary to trauma and project past the boundaries of the initial trauma site.1 Keloids are more commonly seen in individuals with darker skin types and tend to grow larger in this population. Histopathology reveals thickened hyalinized collagen bundles, which were not seen in our patient.1
Metastatic skin lesions of the umbilicus are rare but can arise from internal malignancies including cancers of the lung, colon, and breast.3 We considered Sister Mary Joseph nodule, which is caused most commonly by metastasis of a primary gastrointestinal cancer and signifies poor prognosis. The histopathology of metastatic lesions would reveal the presence of atypical cells with cancer-specific markers. Histopathology along with the patient’s personal and family history, a comprehensive review of symptoms, and cancer screening may help with reaching the correct diagnosis.
The average duration between abdominopelvic surgery and onset of secondary CE symptoms is 3.7 to 5.3 years.4 Our patient presented 10 years post surgery and after cessation of oral contraception, which may suggest a potential role of hormonal contraception in delayed CE onset. Diagnosis of CE can be challenging due to atypical signs or symptoms, delayed onset, and lack of awareness among health care professionals. Patients with delayed diagnosis may endure multiple procedures, prolonged physical pain, and emotional distress. Furthermore, 30% to 50% of females with endometriosis experience infertility. Delayed diagnosis of CE compounded with associated age-related increase in oocyte atresia could potentially worsen fecundity as patients age.5 It is important to consider CE in the differential diagnosis of females of reproductive age who present with cyclical bleeding and abdominal or umbilical nodules.
- James WD, Elston D, Treat JR, et al. Andrews Diseases of the Skin: Clinical Dermatology. 13th ed. Elsevier; 2019. Accessed March 19, 2024. https://search.worldcat.org/title/1084979207
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Komurcugil I, Arslan Z, Bal ZI, et al. Cutaneous metastases different clinical presentations: case series and review of the literature. Dermatol Reports. 2022;15:9553.
- Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Published online September 16, 2019. Eur J Obstet Gynecol Reprod Biol X.
- Missmer SA, Hankinson SE, Spiegelman D, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784-796.
- James WD, Elston D, Treat JR, et al. Andrews Diseases of the Skin: Clinical Dermatology. 13th ed. Elsevier; 2019. Accessed March 19, 2024. https://search.worldcat.org/title/1084979207
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Komurcugil I, Arslan Z, Bal ZI, et al. Cutaneous metastases different clinical presentations: case series and review of the literature. Dermatol Reports. 2022;15:9553.
- Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Published online September 16, 2019. Eur J Obstet Gynecol Reprod Biol X.
- Missmer SA, Hankinson SE, Spiegelman D, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784-796.
Cyclically Bleeding Umbilical Papules
Cyclically Bleeding Umbilical Papules
A 38-year-old nulligravid female with menorrhagia and dysmenorrhea presented with cyclical umbilical bleeding of 1 year’s duration. Shortly before the onset of symptoms, the patient had discontinued oral contraceptive therapy with the intent to become pregnant. She had an uncomplicated laparoscopic cholecystectomy 10 years prior, but her medical history was otherwise unremarkable. At the current presentation, physical examination revealed multilobular brown papules with serosanguineous crusting in the umbilicus.

Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
THE DIAGNOSIS: Granuloma Faciale
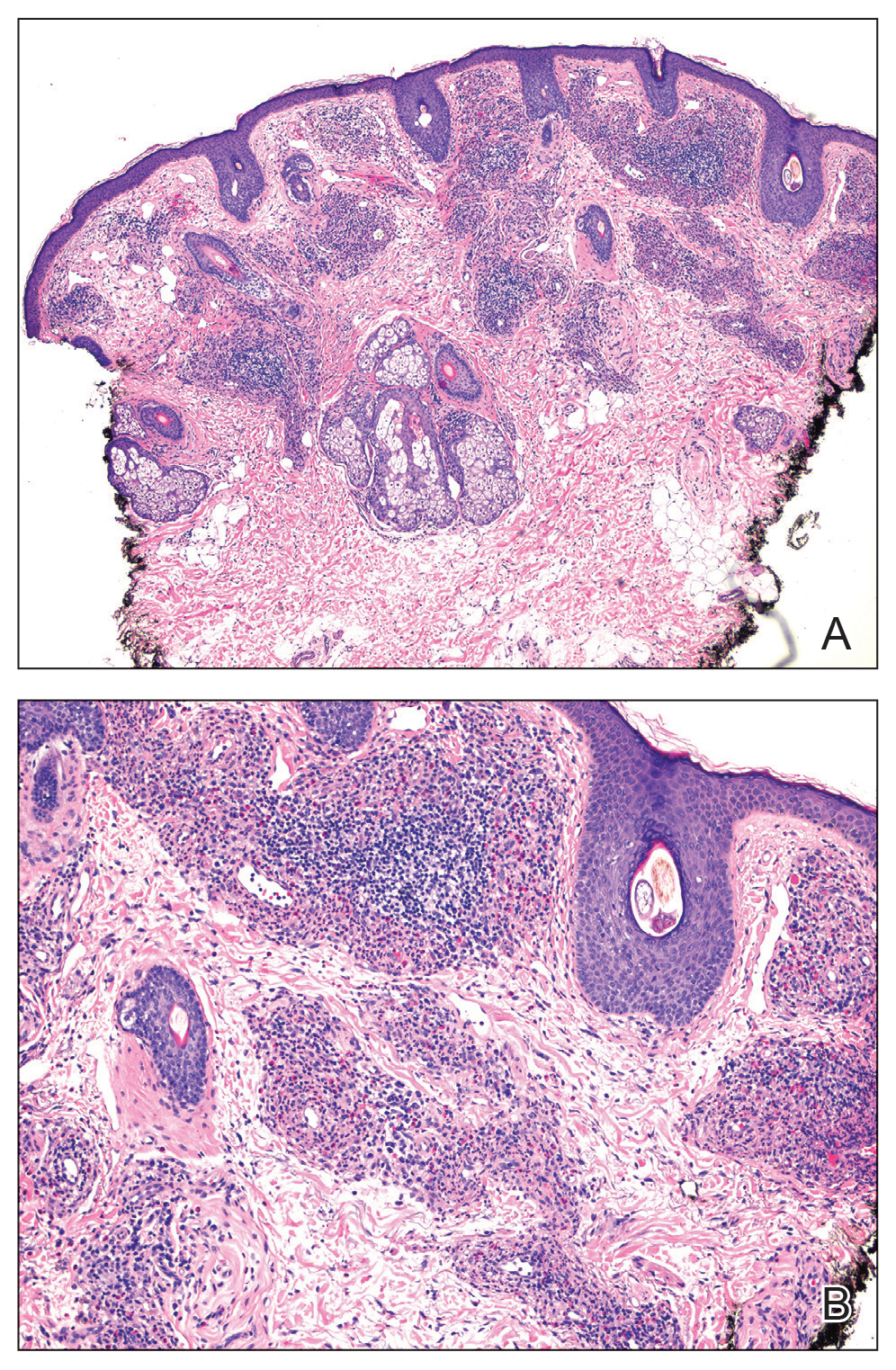
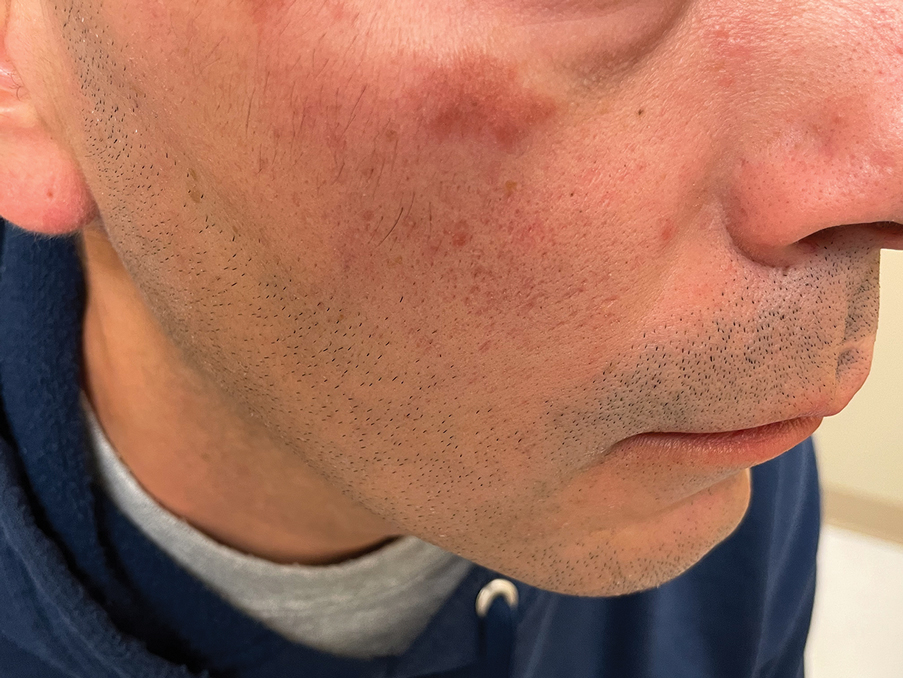
Histology revealed a dense mixed inflammatory cell infiltrate with conspicuous neutrophils and eosinophils in the upper to mid dermis with a narrow uninvolved grenz zone beneath the epidermis (Figures 1 and 2). These findings along with the clinical presentation (Figure 3) were consistent with a diagnosis of granuloma faciale (GF). Most often seen in middle-aged White men, GF is an uncommon localized inflammatory skin condition that often manifests as a single, well-defined, red-to-brown papule, nodule, or plaque on the face or other sun-exposed areas of the skin. Since numerous other skin diseases manifest similarly to GF, biopsy is necessary for definitive diagnosis.1 Histopathology of GF classically shows a mixed inflammatory infiltrate with a narrow band of uninvolved dermis separating it from the epidermis (grenz zone). Dilated follicular plugs and vascular changes frequently are appreciated. Despite its name, GF does not include granulomas and is thought to be similar to leukocytoclastic vasculitis.1 Reports of GF in the literature have shown immunohistochemical staining with the presence of CD4+ lymphocytes that secrete IL-5, a chemotactic agent responsible for attracting eosinophils that contributes to the eosinophilic infiltrate on histology.2
Topical corticosteroids and topical tacrolimus are the first-line treatments for GF. Intralesional corticosteroids also are a treatment option and can be used in combination with cryotherapy.1,3 Additionally, both topical and oral dapsone have been shown to be effective for GF.1 Oral dapsone is given at a dose of 50 mg to 150 mg once daily.1 Clofazimine, typically used as an antileprosy treatment, also has been efficacious in treating GF. Clofazimine has anti-inflammatory and antiproliferative effects on lymphocytes that may attenuate the inflammation underlying GF. It is prescribed at a dose of 300 mg once daily for 3 to 5 months.1
The differential diagnosis for GF is broad and includes tumid lupus erythematosus, Jessner lymphocytic infiltrate (JLI), cutaneous sarcoidosis, and mycosis fungoides. Tumid lupus erythematosus is a subtype of cutaneous lupus erythematosus that rarely is associated with systemic lupus manifestations. Tumid lupus erythematosus manifests as annular, indurated, erythematous plaques, whereas JLI manifests with erythematous papular to nodular lesions without scale on the upper back or face.4 Jessner lymphocytic infiltrate and tumid lupus erythematosus are histopathologically identical, with abundant dermal mucin deposition and a superficial and deep perivascular and periadnexal lymphocytic infiltrate. It is debatable whether JLI is a separate entity or a variant of tumid lupus erythematosus. Sarcoidosis is a granulomatous disease that manifests with a myriad of clinical features. The skin is the second most commonly involved organ.5 The most common morphology is numerous small, firm, nonscaly papules, typically on the face. Histology in cutaneous sarcoidosis will show lymphocyte-poor, noncaseating epithelioid cell granulomas with positive reticulin staining, which were not seen in our patient.6 Lastly, mycosis fungoides is the most common type of cutaneous T-cell lymphoma. It can manifest as patches, plaques, or tumors. The plaque stage may mimic GF as lesions are infiltrative, annular, and raised, with well-defined margins. Histopathology will show intraepidermal lymphocytes out of proportion with spongiosis.7
- Al Dhafiri M, Kaliyadan F. Granuloma faciale. StatPearls Publishing. Updated July 4, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539832/
- Chen A, Harview CL, Rand SE, et al. Refractory granuloma faciale successfully treated with adjunct topical JAK inhibitor. JAAD Case Rep. 2023;33:91-94. doi:10.1016/j.jdcr.2023.01.016
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551. doi:10.1046 /j.1365-4362.1997.00161.x
- Koritala T, Grubbs H, Crane J. Tumid lupus erythematosus. StatPearls Publishing. Updated June 28, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482515/
- Caplan A, Rosenbach M, Imadojemu S. Cutaneous sarcoidosis. Semin Respir Crit Care Med. 2020;41:689-699. doi:10.1055/s-0040-1713130
- Singh P, Jain E, Dhingra H, et al. Clinico-pathological spectrum of cutaneous sarcoidosis: an experience from a government institute in North India. Med Pharm Rep. 2020;93:241-245. doi:10.15386 /mpr-1384
- Vaidya T, Badri T. Mycosis fungoides. StatPearls Publishing. Updated July 31, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519572/
THE DIAGNOSIS: Granuloma Faciale
Histology revealed a dense mixed inflammatory cell infiltrate with conspicuous neutrophils and eosinophils in the upper to mid dermis with a narrow uninvolved grenz zone beneath the epidermis (Figures 1 and 2). These findings along with the clinical presentation (Figure 3) were consistent with a diagnosis of granuloma faciale (GF). Most often seen in middle-aged White men, GF is an uncommon localized inflammatory skin condition that often manifests as a single, well-defined, red-to-brown papule, nodule, or plaque on the face or other sun-exposed areas of the skin. Since numerous other skin diseases manifest similarly to GF, biopsy is necessary for definitive diagnosis.1 Histopathology of GF classically shows a mixed inflammatory infiltrate with a narrow band of uninvolved dermis separating it from the epidermis (grenz zone). Dilated follicular plugs and vascular changes frequently are appreciated. Despite its name, GF does not include granulomas and is thought to be similar to leukocytoclastic vasculitis.1 Reports of GF in the literature have shown immunohistochemical staining with the presence of CD4+ lymphocytes that secrete IL-5, a chemotactic agent responsible for attracting eosinophils that contributes to the eosinophilic infiltrate on histology.2
Topical corticosteroids and topical tacrolimus are the first-line treatments for GF. Intralesional corticosteroids also are a treatment option and can be used in combination with cryotherapy.1,3 Additionally, both topical and oral dapsone have been shown to be effective for GF.1 Oral dapsone is given at a dose of 50 mg to 150 mg once daily.1 Clofazimine, typically used as an antileprosy treatment, also has been efficacious in treating GF. Clofazimine has anti-inflammatory and antiproliferative effects on lymphocytes that may attenuate the inflammation underlying GF. It is prescribed at a dose of 300 mg once daily for 3 to 5 months.1
The differential diagnosis for GF is broad and includes tumid lupus erythematosus, Jessner lymphocytic infiltrate (JLI), cutaneous sarcoidosis, and mycosis fungoides. Tumid lupus erythematosus is a subtype of cutaneous lupus erythematosus that rarely is associated with systemic lupus manifestations. Tumid lupus erythematosus manifests as annular, indurated, erythematous plaques, whereas JLI manifests with erythematous papular to nodular lesions without scale on the upper back or face.4 Jessner lymphocytic infiltrate and tumid lupus erythematosus are histopathologically identical, with abundant dermal mucin deposition and a superficial and deep perivascular and periadnexal lymphocytic infiltrate. It is debatable whether JLI is a separate entity or a variant of tumid lupus erythematosus. Sarcoidosis is a granulomatous disease that manifests with a myriad of clinical features. The skin is the second most commonly involved organ.5 The most common morphology is numerous small, firm, nonscaly papules, typically on the face. Histology in cutaneous sarcoidosis will show lymphocyte-poor, noncaseating epithelioid cell granulomas with positive reticulin staining, which were not seen in our patient.6 Lastly, mycosis fungoides is the most common type of cutaneous T-cell lymphoma. It can manifest as patches, plaques, or tumors. The plaque stage may mimic GF as lesions are infiltrative, annular, and raised, with well-defined margins. Histopathology will show intraepidermal lymphocytes out of proportion with spongiosis.7
THE DIAGNOSIS: Granuloma Faciale
Histology revealed a dense mixed inflammatory cell infiltrate with conspicuous neutrophils and eosinophils in the upper to mid dermis with a narrow uninvolved grenz zone beneath the epidermis (Figures 1 and 2). These findings along with the clinical presentation (Figure 3) were consistent with a diagnosis of granuloma faciale (GF). Most often seen in middle-aged White men, GF is an uncommon localized inflammatory skin condition that often manifests as a single, well-defined, red-to-brown papule, nodule, or plaque on the face or other sun-exposed areas of the skin. Since numerous other skin diseases manifest similarly to GF, biopsy is necessary for definitive diagnosis.1 Histopathology of GF classically shows a mixed inflammatory infiltrate with a narrow band of uninvolved dermis separating it from the epidermis (grenz zone). Dilated follicular plugs and vascular changes frequently are appreciated. Despite its name, GF does not include granulomas and is thought to be similar to leukocytoclastic vasculitis.1 Reports of GF in the literature have shown immunohistochemical staining with the presence of CD4+ lymphocytes that secrete IL-5, a chemotactic agent responsible for attracting eosinophils that contributes to the eosinophilic infiltrate on histology.2
Topical corticosteroids and topical tacrolimus are the first-line treatments for GF. Intralesional corticosteroids also are a treatment option and can be used in combination with cryotherapy.1,3 Additionally, both topical and oral dapsone have been shown to be effective for GF.1 Oral dapsone is given at a dose of 50 mg to 150 mg once daily.1 Clofazimine, typically used as an antileprosy treatment, also has been efficacious in treating GF. Clofazimine has anti-inflammatory and antiproliferative effects on lymphocytes that may attenuate the inflammation underlying GF. It is prescribed at a dose of 300 mg once daily for 3 to 5 months.1
The differential diagnosis for GF is broad and includes tumid lupus erythematosus, Jessner lymphocytic infiltrate (JLI), cutaneous sarcoidosis, and mycosis fungoides. Tumid lupus erythematosus is a subtype of cutaneous lupus erythematosus that rarely is associated with systemic lupus manifestations. Tumid lupus erythematosus manifests as annular, indurated, erythematous plaques, whereas JLI manifests with erythematous papular to nodular lesions without scale on the upper back or face.4 Jessner lymphocytic infiltrate and tumid lupus erythematosus are histopathologically identical, with abundant dermal mucin deposition and a superficial and deep perivascular and periadnexal lymphocytic infiltrate. It is debatable whether JLI is a separate entity or a variant of tumid lupus erythematosus. Sarcoidosis is a granulomatous disease that manifests with a myriad of clinical features. The skin is the second most commonly involved organ.5 The most common morphology is numerous small, firm, nonscaly papules, typically on the face. Histology in cutaneous sarcoidosis will show lymphocyte-poor, noncaseating epithelioid cell granulomas with positive reticulin staining, which were not seen in our patient.6 Lastly, mycosis fungoides is the most common type of cutaneous T-cell lymphoma. It can manifest as patches, plaques, or tumors. The plaque stage may mimic GF as lesions are infiltrative, annular, and raised, with well-defined margins. Histopathology will show intraepidermal lymphocytes out of proportion with spongiosis.7
- Al Dhafiri M, Kaliyadan F. Granuloma faciale. StatPearls Publishing. Updated July 4, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539832/
- Chen A, Harview CL, Rand SE, et al. Refractory granuloma faciale successfully treated with adjunct topical JAK inhibitor. JAAD Case Rep. 2023;33:91-94. doi:10.1016/j.jdcr.2023.01.016
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551. doi:10.1046 /j.1365-4362.1997.00161.x
- Koritala T, Grubbs H, Crane J. Tumid lupus erythematosus. StatPearls Publishing. Updated June 28, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482515/
- Caplan A, Rosenbach M, Imadojemu S. Cutaneous sarcoidosis. Semin Respir Crit Care Med. 2020;41:689-699. doi:10.1055/s-0040-1713130
- Singh P, Jain E, Dhingra H, et al. Clinico-pathological spectrum of cutaneous sarcoidosis: an experience from a government institute in North India. Med Pharm Rep. 2020;93:241-245. doi:10.15386 /mpr-1384
- Vaidya T, Badri T. Mycosis fungoides. StatPearls Publishing. Updated July 31, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519572/
- Al Dhafiri M, Kaliyadan F. Granuloma faciale. StatPearls Publishing. Updated July 4, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539832/
- Chen A, Harview CL, Rand SE, et al. Refractory granuloma faciale successfully treated with adjunct topical JAK inhibitor. JAAD Case Rep. 2023;33:91-94. doi:10.1016/j.jdcr.2023.01.016
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551. doi:10.1046 /j.1365-4362.1997.00161.x
- Koritala T, Grubbs H, Crane J. Tumid lupus erythematosus. StatPearls Publishing. Updated June 28, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK482515/
- Caplan A, Rosenbach M, Imadojemu S. Cutaneous sarcoidosis. Semin Respir Crit Care Med. 2020;41:689-699. doi:10.1055/s-0040-1713130
- Singh P, Jain E, Dhingra H, et al. Clinico-pathological spectrum of cutaneous sarcoidosis: an experience from a government institute in North India. Med Pharm Rep. 2020;93:241-245. doi:10.15386 /mpr-1384
- Vaidya T, Badri T. Mycosis fungoides. StatPearls Publishing. Updated July 31, 2023. Accessed February 18, 2025. https://www.ncbi.nlm.nih.gov/books/NBK519572/
Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
Bilateral Brownish-Red Indurated Facial Plaques in an Adult Man
A 44-year-old man presented to the dermatology clinic with a facial rash of 2 years’ duration. The patient reported associated pruritus but no systemic symptoms. His medical history was relevant for childhood eczema. He had tried various over-the-counter treatments for the facial rash, including topical hydrocortisone, neomycin/bacitracin/polymyxin antibiotic ointment, moisturizers, and antihistamines, with no success. Physical examination demonstrated symmetric, well-circumscribed, circinate, brownish-red, indurated plaques without scaling on the cheeks. A 4-mm punch biopsy was obtained from a plaque on the left cheek.
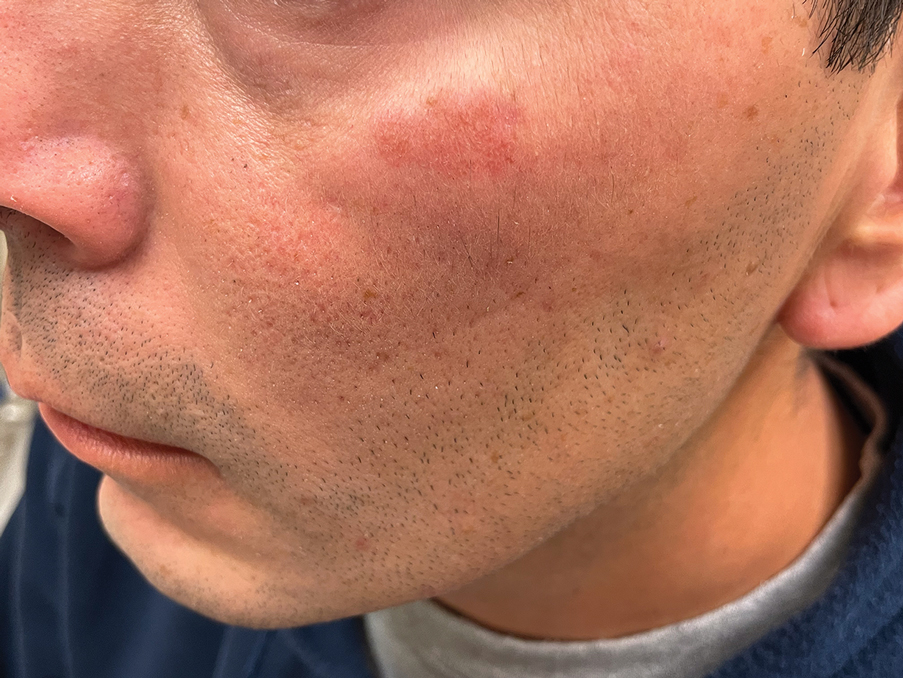

Fingernail Abnormalities in an Adolescent With a History of Toe Walking
Fingernail Abnormalities in an Adolescent With a History of Toe Walking
THE DIAGNOSIS: Nail-Patella Syndrome
Nail-patella syndrome (NPS) is an autosomaldominant disorder that is present in approximately 1 in 50,000 live births worldwide.1,2 It manifests with a spectrum of clinical findings affecting the nails, skeletal system, kidneys, and eyes.3 Most cases of NPS are caused by loss-of-function mutations in LMX1B,1 a gene encoding the LIM homeobox transcription factor.4 The LMX1B gene plays a critical role in the dorsoventral patterning of developing limbs.5 Mutations of this gene impair the development and function of podocytes and glomerular filtration slits6 and have been found to affect the development of the dopaminergic and mesencephalic serotoninergic neurons.2 Approximately 5% of patients with NPS have an unexplained genetic cause, suggesting an alternate mechanism for disease.1 Loss-of-function mutations also were observed in the Wnt inhibitory factor 1 gene (WIF1) in a family with an NPS-like presentation and could represent a novel cause of the condition.1 Regardless, NPS may be diagnosed clinically based on characteristic medical history, imaging, and physical examination findings.
Nail changes are the most consistent feature of NPS. The nails may be absent, hypoplastic, dystrophic, ridged (horizontally or vertically), or pitted or may demonstrate characteristic triangular lacunae. Nail findings often are congenital, bilateral, and symmetrical. The first digits typically are most severely affected, with progressive improvement appreciated toward the fifth fingers, as seen in our patient. The nail changes can be subtle, sometimes manifesting only as a single triangular lacuna on both thumbnails. Toenail involvement is less common and, when present, tends to be even more discreet. In contrast to the fingernails, the fifth toenails are most commonly affected.7
There are many skeletal manifestations of NPS. Patellae may be absent, hypoplastic, or irregularly shaped on physical examination or imaging, and changes may involve one or both knees. The Figure shows plain radiographs of the knees with bilateral patellar subluxation. Elbow dysplasia or radial head subluxation may result in physical limitations in extension, pronation, or supination of the joint.7 In approximately 70% of patients seen with the disorder, imaging may reveal symmetric posterior and lateral bony projections from the iliac crests, known as iliac horns; when present, these are considered pathognomonic.8
Open-angle glaucoma is the most common ocular finding in NPS. Other less commonly associated eye abnormalities include hyperpigmentation of the pupillary margin (Lester iris).6 Renal involvement occurs in 30% to 50% of patients with NPS and is the main predictor of mortality, with percentages as high as 5% to 14%.7 Defects occur in the glomerular basement membrane and manifest clinically with hematuria and/or proteinuria. The course of proteinuria is unpredictable. Some cases remit spontaneously, while others remain asymptomatic, progress to nephrotic syndrome, or, although rare, advance to renal failure.7,9
Bowel symptoms, neurologic problems, vasomotor concerns, thin dental enamel, attention-deficit disorder or attention-deficit/hyperactivity disorder, and major depressive disorder all have been reported in association with NPS.2,7
Nail psoriasis typically exhibits nail pitting and onycholysis. Other manifestations include subungual hyperkeratosis, oil drop discoloration, and splinter hemorrhages. Topical and intralesional treatments are used to manage symptoms of the disease, as it can become debilitating if left untreated, unlike the nail disease seen in NPS.10 Onychomycosis can have a similar manifestation to psoriasis with sublingual hyperkeratosis of the nail, but it usually is caused by dermatophytes or yeasts such as Candida albicans. Onycholysis and thickening of the subungual region also can be seen. Diagnosis relies on direct microscopy and fungal culture, and a thorough patient history will help distinguish fungal vs nonfungal etiology. New-generation antifungals are used to eradicate the infection.11 Leukonychia manifests with white-appearing nails due to nail-plate or nail-bed abnormalities. Leukonychia can have multisystem involvement, but nails demonstrate a white discoloration rather than the other abnormalities discussed here.12 Hypohidrotic ectodermal dysplasia is a rare hereditary congenital disease that affects ectodermal structures and manifests with a triad of symptoms: hypotrichosis, hypohidrosis, and hypodontia. The condition often manifests in childhood with characteristic features such as light-pigmented sparse and fine hair. Physical growth as well as psychomotor development are within normal limits. Neither bone nor renal involvement is typical for hypohidrotic ectodermal dysplasia.13
Our case highlights the typical manifestation of NPS with multisystem involvement, demonstrating the complexity of the disease. For cases in which a clinical diagnosis of NPS is uncertain, gene-targeted or comprehensive genomic testing is recommended, as well as genetic counseling. Given the broad spectrum of clinical manifestations, it is imperative that patients undergo screening for musculoskeletal, renal, and ophthalmologic involvement. Treatment is targeted at symptom management and prevention of long-term complications, reliant on clinical presentation, and specific to each patient.
- Jones MC, Topol SE, Rueda M, et al. Mutation of WIF1: a potential novel cause of a nail-patella–like disorder. Genet Med. 2017;19:1179-1183.
- López-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in Nail-patella syndrome: potential association with LMX1B loss-of-function. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016; 30:1614-1617.
- Vollrath D, Jaramillo-Babb VL, Clough MV, et al. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet. 1998;7:1091-1098. Published correction appears in Hum Mol Genet. 1998;7:1333.
- Chen H, Lun Y, Ovchinnikov D, et al. Limb and kidney defects in LMX1B mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet. 1998;19:51-55.
- Witzgall R. Nail-patella syndrome. Pflugers Arch. 2017;469:927-936.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. University of Washington; 2003.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey. Orthop Traumatol Surg Res. 2015;101:959-962.
- Harita Y, Urae S, Akashio R, et al. Clinical and genetic characterization of nephropathy in patients with nail-patella syndrome. Eur J Hum Genet. 2020;28:1414-1421.
- Tan ES, Chong WS, Tey HL. Nail psoriasis. Am J Clin Dermatol. 2012; 13:375-388.
- Elewski BE. Onychomycosis: pathogenesis, diagnosis, and management. Clin Microbiol Rev. 1998;11:415-429.
- Iorizzo M, Starace M, Pasch MC. Leukonychia: what can white nails tell us? Am J Clin Dermatol. 2022;23:177-193.
- Wright JT, Grange DK, Fete M. Hypohidrotic ectodermal dysplasia. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1993-2024.
THE DIAGNOSIS: Nail-Patella Syndrome
Nail-patella syndrome (NPS) is an autosomaldominant disorder that is present in approximately 1 in 50,000 live births worldwide.1,2 It manifests with a spectrum of clinical findings affecting the nails, skeletal system, kidneys, and eyes.3 Most cases of NPS are caused by loss-of-function mutations in LMX1B,1 a gene encoding the LIM homeobox transcription factor.4 The LMX1B gene plays a critical role in the dorsoventral patterning of developing limbs.5 Mutations of this gene impair the development and function of podocytes and glomerular filtration slits6 and have been found to affect the development of the dopaminergic and mesencephalic serotoninergic neurons.2 Approximately 5% of patients with NPS have an unexplained genetic cause, suggesting an alternate mechanism for disease.1 Loss-of-function mutations also were observed in the Wnt inhibitory factor 1 gene (WIF1) in a family with an NPS-like presentation and could represent a novel cause of the condition.1 Regardless, NPS may be diagnosed clinically based on characteristic medical history, imaging, and physical examination findings.
Nail changes are the most consistent feature of NPS. The nails may be absent, hypoplastic, dystrophic, ridged (horizontally or vertically), or pitted or may demonstrate characteristic triangular lacunae. Nail findings often are congenital, bilateral, and symmetrical. The first digits typically are most severely affected, with progressive improvement appreciated toward the fifth fingers, as seen in our patient. The nail changes can be subtle, sometimes manifesting only as a single triangular lacuna on both thumbnails. Toenail involvement is less common and, when present, tends to be even more discreet. In contrast to the fingernails, the fifth toenails are most commonly affected.7
There are many skeletal manifestations of NPS. Patellae may be absent, hypoplastic, or irregularly shaped on physical examination or imaging, and changes may involve one or both knees. The Figure shows plain radiographs of the knees with bilateral patellar subluxation. Elbow dysplasia or radial head subluxation may result in physical limitations in extension, pronation, or supination of the joint.7 In approximately 70% of patients seen with the disorder, imaging may reveal symmetric posterior and lateral bony projections from the iliac crests, known as iliac horns; when present, these are considered pathognomonic.8
Open-angle glaucoma is the most common ocular finding in NPS. Other less commonly associated eye abnormalities include hyperpigmentation of the pupillary margin (Lester iris).6 Renal involvement occurs in 30% to 50% of patients with NPS and is the main predictor of mortality, with percentages as high as 5% to 14%.7 Defects occur in the glomerular basement membrane and manifest clinically with hematuria and/or proteinuria. The course of proteinuria is unpredictable. Some cases remit spontaneously, while others remain asymptomatic, progress to nephrotic syndrome, or, although rare, advance to renal failure.7,9
Bowel symptoms, neurologic problems, vasomotor concerns, thin dental enamel, attention-deficit disorder or attention-deficit/hyperactivity disorder, and major depressive disorder all have been reported in association with NPS.2,7
Nail psoriasis typically exhibits nail pitting and onycholysis. Other manifestations include subungual hyperkeratosis, oil drop discoloration, and splinter hemorrhages. Topical and intralesional treatments are used to manage symptoms of the disease, as it can become debilitating if left untreated, unlike the nail disease seen in NPS.10 Onychomycosis can have a similar manifestation to psoriasis with sublingual hyperkeratosis of the nail, but it usually is caused by dermatophytes or yeasts such as Candida albicans. Onycholysis and thickening of the subungual region also can be seen. Diagnosis relies on direct microscopy and fungal culture, and a thorough patient history will help distinguish fungal vs nonfungal etiology. New-generation antifungals are used to eradicate the infection.11 Leukonychia manifests with white-appearing nails due to nail-plate or nail-bed abnormalities. Leukonychia can have multisystem involvement, but nails demonstrate a white discoloration rather than the other abnormalities discussed here.12 Hypohidrotic ectodermal dysplasia is a rare hereditary congenital disease that affects ectodermal structures and manifests with a triad of symptoms: hypotrichosis, hypohidrosis, and hypodontia. The condition often manifests in childhood with characteristic features such as light-pigmented sparse and fine hair. Physical growth as well as psychomotor development are within normal limits. Neither bone nor renal involvement is typical for hypohidrotic ectodermal dysplasia.13
Our case highlights the typical manifestation of NPS with multisystem involvement, demonstrating the complexity of the disease. For cases in which a clinical diagnosis of NPS is uncertain, gene-targeted or comprehensive genomic testing is recommended, as well as genetic counseling. Given the broad spectrum of clinical manifestations, it is imperative that patients undergo screening for musculoskeletal, renal, and ophthalmologic involvement. Treatment is targeted at symptom management and prevention of long-term complications, reliant on clinical presentation, and specific to each patient.
THE DIAGNOSIS: Nail-Patella Syndrome
Nail-patella syndrome (NPS) is an autosomaldominant disorder that is present in approximately 1 in 50,000 live births worldwide.1,2 It manifests with a spectrum of clinical findings affecting the nails, skeletal system, kidneys, and eyes.3 Most cases of NPS are caused by loss-of-function mutations in LMX1B,1 a gene encoding the LIM homeobox transcription factor.4 The LMX1B gene plays a critical role in the dorsoventral patterning of developing limbs.5 Mutations of this gene impair the development and function of podocytes and glomerular filtration slits6 and have been found to affect the development of the dopaminergic and mesencephalic serotoninergic neurons.2 Approximately 5% of patients with NPS have an unexplained genetic cause, suggesting an alternate mechanism for disease.1 Loss-of-function mutations also were observed in the Wnt inhibitory factor 1 gene (WIF1) in a family with an NPS-like presentation and could represent a novel cause of the condition.1 Regardless, NPS may be diagnosed clinically based on characteristic medical history, imaging, and physical examination findings.
Nail changes are the most consistent feature of NPS. The nails may be absent, hypoplastic, dystrophic, ridged (horizontally or vertically), or pitted or may demonstrate characteristic triangular lacunae. Nail findings often are congenital, bilateral, and symmetrical. The first digits typically are most severely affected, with progressive improvement appreciated toward the fifth fingers, as seen in our patient. The nail changes can be subtle, sometimes manifesting only as a single triangular lacuna on both thumbnails. Toenail involvement is less common and, when present, tends to be even more discreet. In contrast to the fingernails, the fifth toenails are most commonly affected.7
There are many skeletal manifestations of NPS. Patellae may be absent, hypoplastic, or irregularly shaped on physical examination or imaging, and changes may involve one or both knees. The Figure shows plain radiographs of the knees with bilateral patellar subluxation. Elbow dysplasia or radial head subluxation may result in physical limitations in extension, pronation, or supination of the joint.7 In approximately 70% of patients seen with the disorder, imaging may reveal symmetric posterior and lateral bony projections from the iliac crests, known as iliac horns; when present, these are considered pathognomonic.8
Open-angle glaucoma is the most common ocular finding in NPS. Other less commonly associated eye abnormalities include hyperpigmentation of the pupillary margin (Lester iris).6 Renal involvement occurs in 30% to 50% of patients with NPS and is the main predictor of mortality, with percentages as high as 5% to 14%.7 Defects occur in the glomerular basement membrane and manifest clinically with hematuria and/or proteinuria. The course of proteinuria is unpredictable. Some cases remit spontaneously, while others remain asymptomatic, progress to nephrotic syndrome, or, although rare, advance to renal failure.7,9
Bowel symptoms, neurologic problems, vasomotor concerns, thin dental enamel, attention-deficit disorder or attention-deficit/hyperactivity disorder, and major depressive disorder all have been reported in association with NPS.2,7
Nail psoriasis typically exhibits nail pitting and onycholysis. Other manifestations include subungual hyperkeratosis, oil drop discoloration, and splinter hemorrhages. Topical and intralesional treatments are used to manage symptoms of the disease, as it can become debilitating if left untreated, unlike the nail disease seen in NPS.10 Onychomycosis can have a similar manifestation to psoriasis with sublingual hyperkeratosis of the nail, but it usually is caused by dermatophytes or yeasts such as Candida albicans. Onycholysis and thickening of the subungual region also can be seen. Diagnosis relies on direct microscopy and fungal culture, and a thorough patient history will help distinguish fungal vs nonfungal etiology. New-generation antifungals are used to eradicate the infection.11 Leukonychia manifests with white-appearing nails due to nail-plate or nail-bed abnormalities. Leukonychia can have multisystem involvement, but nails demonstrate a white discoloration rather than the other abnormalities discussed here.12 Hypohidrotic ectodermal dysplasia is a rare hereditary congenital disease that affects ectodermal structures and manifests with a triad of symptoms: hypotrichosis, hypohidrosis, and hypodontia. The condition often manifests in childhood with characteristic features such as light-pigmented sparse and fine hair. Physical growth as well as psychomotor development are within normal limits. Neither bone nor renal involvement is typical for hypohidrotic ectodermal dysplasia.13
Our case highlights the typical manifestation of NPS with multisystem involvement, demonstrating the complexity of the disease. For cases in which a clinical diagnosis of NPS is uncertain, gene-targeted or comprehensive genomic testing is recommended, as well as genetic counseling. Given the broad spectrum of clinical manifestations, it is imperative that patients undergo screening for musculoskeletal, renal, and ophthalmologic involvement. Treatment is targeted at symptom management and prevention of long-term complications, reliant on clinical presentation, and specific to each patient.
- Jones MC, Topol SE, Rueda M, et al. Mutation of WIF1: a potential novel cause of a nail-patella–like disorder. Genet Med. 2017;19:1179-1183.
- López-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in Nail-patella syndrome: potential association with LMX1B loss-of-function. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016; 30:1614-1617.
- Vollrath D, Jaramillo-Babb VL, Clough MV, et al. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet. 1998;7:1091-1098. Published correction appears in Hum Mol Genet. 1998;7:1333.
- Chen H, Lun Y, Ovchinnikov D, et al. Limb and kidney defects in LMX1B mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet. 1998;19:51-55.
- Witzgall R. Nail-patella syndrome. Pflugers Arch. 2017;469:927-936.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. University of Washington; 2003.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey. Orthop Traumatol Surg Res. 2015;101:959-962.
- Harita Y, Urae S, Akashio R, et al. Clinical and genetic characterization of nephropathy in patients with nail-patella syndrome. Eur J Hum Genet. 2020;28:1414-1421.
- Tan ES, Chong WS, Tey HL. Nail psoriasis. Am J Clin Dermatol. 2012; 13:375-388.
- Elewski BE. Onychomycosis: pathogenesis, diagnosis, and management. Clin Microbiol Rev. 1998;11:415-429.
- Iorizzo M, Starace M, Pasch MC. Leukonychia: what can white nails tell us? Am J Clin Dermatol. 2022;23:177-193.
- Wright JT, Grange DK, Fete M. Hypohidrotic ectodermal dysplasia. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1993-2024.
- Jones MC, Topol SE, Rueda M, et al. Mutation of WIF1: a potential novel cause of a nail-patella–like disorder. Genet Med. 2017;19:1179-1183.
- López-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in Nail-patella syndrome: potential association with LMX1B loss-of-function. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016; 30:1614-1617.
- Vollrath D, Jaramillo-Babb VL, Clough MV, et al. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet. 1998;7:1091-1098. Published correction appears in Hum Mol Genet. 1998;7:1333.
- Chen H, Lun Y, Ovchinnikov D, et al. Limb and kidney defects in LMX1B mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet. 1998;19:51-55.
- Witzgall R. Nail-patella syndrome. Pflugers Arch. 2017;469:927-936.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. University of Washington; 2003.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey. Orthop Traumatol Surg Res. 2015;101:959-962.
- Harita Y, Urae S, Akashio R, et al. Clinical and genetic characterization of nephropathy in patients with nail-patella syndrome. Eur J Hum Genet. 2020;28:1414-1421.
- Tan ES, Chong WS, Tey HL. Nail psoriasis. Am J Clin Dermatol. 2012; 13:375-388.
- Elewski BE. Onychomycosis: pathogenesis, diagnosis, and management. Clin Microbiol Rev. 1998;11:415-429.
- Iorizzo M, Starace M, Pasch MC. Leukonychia: what can white nails tell us? Am J Clin Dermatol. 2022;23:177-193.
- Wright JT, Grange DK, Fete M. Hypohidrotic ectodermal dysplasia. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1993-2024.
Fingernail Abnormalities in an Adolescent With a History of Toe Walking
Fingernail Abnormalities in an Adolescent With a History of Toe Walking
A 14-year-old boy with a history of toe walking, attention-deficit/hyperactivity disorder, and mixed receptive expressive language disorder presented to our pediatric dermatology clinic with fingernail abnormalities that had been present since birth. Physical examination revealed narrowing and longitudinal splitting of the nail plates with triangular lacunae and progressive improvement appreciated toward the fifth digits. The nail changes were most prominent on the first digits. A review of the patient’s medical record revealed incidental bilateral iliac horns of the pelvis on radiographs taken at age 18 months. The patient reported waxing and waning knee pain that worsened with prolonged activity and when climbing stairs. Urinalysis demonstrated mild hematuria without proteinuria. The patient was normotensive. There was no evidence of glaucoma, cataracts, or hyperpigmentation of the pupillary margin (Lester iris) on ophthalmologic examination. Genetic testing was performed.
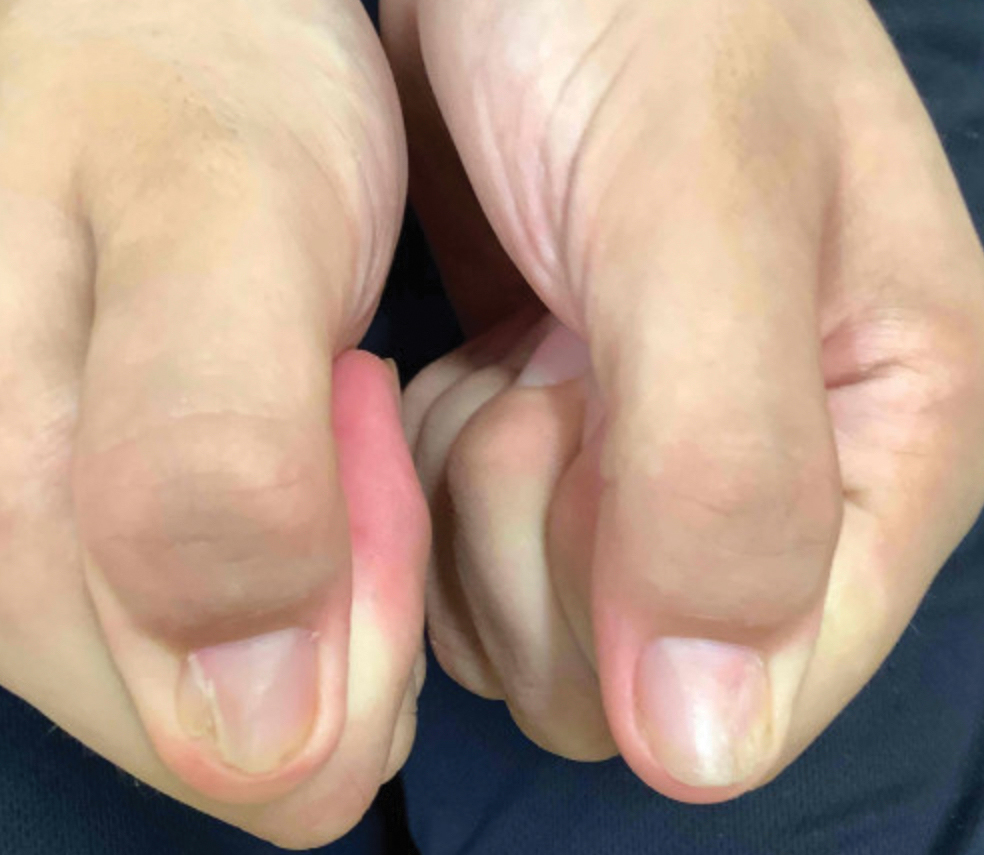

Exophytic Scaly Nodule on the Wrist
Exophytic Scaly Nodule on the Wrist
THE DIAGNOSIS: Atypical Spitz Tumor
The shave biopsy revealed extensive dermal proliferation with spitzoid cytomorphology containing large, spindled nuclei; prominent nucleoli; and abundant homogenous cytoplasm arranged in haphazard fascicles. The proliferation was associated with prominent pseudoepitheliomatous hyperplasia of the overlying epidermis, and anaplastic lymphoma kinase immunohistochemistry showed diffuse strong positivity. Fluorescence in situ hybridization confirmed fusion of the tropomyosin 3 (TPM3) and anaplastic lymphoma kinase (ALK) genes, which finalized the diagnosis of an ALK-mutated atypical spitz tumor. Due to the location and size of the lesion, Mohs micrographic surgery was performed to excise the tumor and clear the margins.
Spitz nevi are uncommon benign melanocytic neoplasms that typically occur in pediatric populations.1 Atypical spitz nevi comprised fewer than 17% of all childhood melanocytic nevi in the United States and can be considered in the broader category of spitzoid tumors. Spitz nevi are divided into 3 classes: Spitz nevus, atypical Spitz nevus, and spitzoid melanoma. Atypical Spitz nevi have typical Spitz nevus and spitzoid melanoma features and often can be difficult to distinguish on dermoscopy. Malignant Spitz tumors typically occur in the fifth decade of life, though the age distribution can vary widely.1
Black patients are less likely to be diagnosed with Spitz nevi, potentially due to a lower prevalence in this population, thus limiting the clinician’s clinical exposure and leading to increased rates of misdiagnoses.2 Spitz nevi usually manifest as well-circumscribed, dome-shaped papules and frequently are described as pink to red due to increased vascularity and limited melanin content1; however, these lesions may appear more violaceous, dusky, or dark brown in darker skin types. Additionally, approximately 71% of patients in a clinical review of Spitz nevi had a pigmented lesion, ranging from light brown to black.3 It is important for dermatologists to understand that the contrast in color between the nevus and the surrounding skin may not be as striking, prominent, or clinically concerning, particularly in darker skin types, such as in our patient.
Spitz nevi frequently manifest as rapidly growing solitary lesions most frequently developing in the lower legs (shown in 41% of lesions in one report).4 However, a recent retrospective review indicated that Spitz nevi in Black patients most commonly were found on the upper extremities, as was seen in our patient.2 Compared to typical and common Spitz nevi, atypical Spitz nevi often are greater than 10 mm in diameter and have features of ulceration.
Diagnosing atypical spitzoid melanocytic lesions requires adequate clinical suspicion and confirmation via biopsy. Under dermoscopy, typical Spitz nevi often display a starburst or globular pattern with pinpoint vessels, though it can have variable manifestations of both patterns. Atypical Spitz nevi can be challenging to distinguish from melanoma on dermoscopy since both conditions can have atypical pigment networks or structureless homogenous areas.1 Consequently, there often is a lower threshold for biopsy and possible follow-up excision for atypical Spitz nevi. Histopathology of atypical Spitz nevi includes epithelioid and spindle melanocytes but can share features of melanomas, including areas of prominent pagetoid spread, asymmetry, and poor circumscription.5 Furthermore, atypical Spitz nevi with ALK gene fusion, as seen in our patient, have been shown in the literature to demonstrate distinct histopathologic features, such as wedge-shaped extension into the dermis or a bulbous lower border that can resemble pseudoepitheliomatous hyperplasia.6
The differential diagnosis for this rapidly growing scaly nodule also should include pyogenic granuloma, bacillary angiomatosis, Kaposi sarcoma, and amelanotic melanoma. Pyogenic granuloma is a rapidly growing, benign, vascular tumor that often becomes ulcerated and can occur in any age group.7 Pyogenic granuloma frequently appears at sites of trauma as a solitary, bright pink to red, friable, pedunculated papule and often manifests on the arms, hands, and face, similar to atypical Spitz nevi, though they can appear anywhere on the body. Histology shows a lobular capillary network with a central feeder vessel.7
Bacillary angiomatosis is an uncommon cutaneous infection associated with vascular proliferation and neovascularization due to the gram-negative organism Bartonella henselae.8 Bacillary nodules typically are reddish to purple and appear on the arms, sometimes with central ulceration and bleeding. Patients may present with multiple papules and nodules of varying sizes, as the lesions can arise in crops and follow a sporotrichoid pattern. Most patients with bacillary angiomatosis are immunosuppressed, though it rarely can affect immunocompetent patients. Histologically, bacillary angiomatosis is similar to pyogenic granuloma, though Gram or Warthin-Starry stains can help differentiate B henselae.8
Kaposi sarcoma is a malignant vascular neoplasm that often manifests in immunocompromised patients as violaceous, purple, or red patches, plaques, and nodules on the skin or oral mucosa. Histopathology shows spindle cell proliferation of irregular complex vascular channels dissecting through the dermis. Human herpesvirus 8 immunohistochemistry can be used to confirm diagnosis on histopathology.9 In contrast, amelanotic melanoma consists of lack of pigmentation, asymmetry with polymorphous vascular pattern, and high mitotic rate and is commonly found in sun-exposed areas. Dermoscopic features include irregular globules with blue-whitish veil.10
Treatment of atypical Spitz nevi depends mainly on the age of the patient and the histologic features of the nevus. Adults with atypical Spitz nevi frequently require excision, while the preferred choice for treatment in children with common Spitz nevi is regular clinical monitoring when there are no concerning clinical, dermoscopic, or histologic features.8 Compared to common Spitz nevi, atypical Spitz nevi have more melanoma-like features, resulting in a stronger recommendation for excision. Excision allows for a more thorough histologic evaluation and minimizes the likelihood of a recurrent atypical lesion.11 In all cases, close clinical follow-up is recommended to monitor for reoccurrence.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084. doi:10.1016/j.jaad.2011.04.040
- Farid YI, Honda KS. Spitz nevi in African Americans: a retrospective chart review of 11 patients. J Cutan Pathol. 2021;48:511-518. doi:10.1111 /cup.13903
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20-25. doi: 10.1159/000246051
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, et al. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267-275. doi: 10.1097 /DAD.0b013e3181c52b99
- Brown A, Sawyer JD, Neumeister MW. Spitz nevus: review and update. Clin Plast Surg 2021;48:677-686. doi: 10.1016/j.cps.2021.06.002 [published Online First: 20210818]
- Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. doi: 10.1097/PAS.0000000000000387
- Sarwal P, Lapumnuaypol K. Pyogenic granuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556077/
- Akram SM, Anwar MY, Thandra KC, et al. Bacillary angiomatosis. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 4, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448092/
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Pizzichetta MA, Talamini R, Stanganelli I, et al. Amelanotic/ hypomelanotic melanoma: clinical and dermoscopic features. Br J Dermatol 2004;150(6):1117-1124. doi: 10.1111/j.1365-2133.2004.05928.x
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part II. natural history and management. J Am Acad Dermatol 2011;65:1087-1092. doi:10.1016/j.jaad.2011.06.045
THE DIAGNOSIS: Atypical Spitz Tumor
The shave biopsy revealed extensive dermal proliferation with spitzoid cytomorphology containing large, spindled nuclei; prominent nucleoli; and abundant homogenous cytoplasm arranged in haphazard fascicles. The proliferation was associated with prominent pseudoepitheliomatous hyperplasia of the overlying epidermis, and anaplastic lymphoma kinase immunohistochemistry showed diffuse strong positivity. Fluorescence in situ hybridization confirmed fusion of the tropomyosin 3 (TPM3) and anaplastic lymphoma kinase (ALK) genes, which finalized the diagnosis of an ALK-mutated atypical spitz tumor. Due to the location and size of the lesion, Mohs micrographic surgery was performed to excise the tumor and clear the margins.
Spitz nevi are uncommon benign melanocytic neoplasms that typically occur in pediatric populations.1 Atypical spitz nevi comprised fewer than 17% of all childhood melanocytic nevi in the United States and can be considered in the broader category of spitzoid tumors. Spitz nevi are divided into 3 classes: Spitz nevus, atypical Spitz nevus, and spitzoid melanoma. Atypical Spitz nevi have typical Spitz nevus and spitzoid melanoma features and often can be difficult to distinguish on dermoscopy. Malignant Spitz tumors typically occur in the fifth decade of life, though the age distribution can vary widely.1
Black patients are less likely to be diagnosed with Spitz nevi, potentially due to a lower prevalence in this population, thus limiting the clinician’s clinical exposure and leading to increased rates of misdiagnoses.2 Spitz nevi usually manifest as well-circumscribed, dome-shaped papules and frequently are described as pink to red due to increased vascularity and limited melanin content1; however, these lesions may appear more violaceous, dusky, or dark brown in darker skin types. Additionally, approximately 71% of patients in a clinical review of Spitz nevi had a pigmented lesion, ranging from light brown to black.3 It is important for dermatologists to understand that the contrast in color between the nevus and the surrounding skin may not be as striking, prominent, or clinically concerning, particularly in darker skin types, such as in our patient.
Spitz nevi frequently manifest as rapidly growing solitary lesions most frequently developing in the lower legs (shown in 41% of lesions in one report).4 However, a recent retrospective review indicated that Spitz nevi in Black patients most commonly were found on the upper extremities, as was seen in our patient.2 Compared to typical and common Spitz nevi, atypical Spitz nevi often are greater than 10 mm in diameter and have features of ulceration.
Diagnosing atypical spitzoid melanocytic lesions requires adequate clinical suspicion and confirmation via biopsy. Under dermoscopy, typical Spitz nevi often display a starburst or globular pattern with pinpoint vessels, though it can have variable manifestations of both patterns. Atypical Spitz nevi can be challenging to distinguish from melanoma on dermoscopy since both conditions can have atypical pigment networks or structureless homogenous areas.1 Consequently, there often is a lower threshold for biopsy and possible follow-up excision for atypical Spitz nevi. Histopathology of atypical Spitz nevi includes epithelioid and spindle melanocytes but can share features of melanomas, including areas of prominent pagetoid spread, asymmetry, and poor circumscription.5 Furthermore, atypical Spitz nevi with ALK gene fusion, as seen in our patient, have been shown in the literature to demonstrate distinct histopathologic features, such as wedge-shaped extension into the dermis or a bulbous lower border that can resemble pseudoepitheliomatous hyperplasia.6
The differential diagnosis for this rapidly growing scaly nodule also should include pyogenic granuloma, bacillary angiomatosis, Kaposi sarcoma, and amelanotic melanoma. Pyogenic granuloma is a rapidly growing, benign, vascular tumor that often becomes ulcerated and can occur in any age group.7 Pyogenic granuloma frequently appears at sites of trauma as a solitary, bright pink to red, friable, pedunculated papule and often manifests on the arms, hands, and face, similar to atypical Spitz nevi, though they can appear anywhere on the body. Histology shows a lobular capillary network with a central feeder vessel.7
Bacillary angiomatosis is an uncommon cutaneous infection associated with vascular proliferation and neovascularization due to the gram-negative organism Bartonella henselae.8 Bacillary nodules typically are reddish to purple and appear on the arms, sometimes with central ulceration and bleeding. Patients may present with multiple papules and nodules of varying sizes, as the lesions can arise in crops and follow a sporotrichoid pattern. Most patients with bacillary angiomatosis are immunosuppressed, though it rarely can affect immunocompetent patients. Histologically, bacillary angiomatosis is similar to pyogenic granuloma, though Gram or Warthin-Starry stains can help differentiate B henselae.8
Kaposi sarcoma is a malignant vascular neoplasm that often manifests in immunocompromised patients as violaceous, purple, or red patches, plaques, and nodules on the skin or oral mucosa. Histopathology shows spindle cell proliferation of irregular complex vascular channels dissecting through the dermis. Human herpesvirus 8 immunohistochemistry can be used to confirm diagnosis on histopathology.9 In contrast, amelanotic melanoma consists of lack of pigmentation, asymmetry with polymorphous vascular pattern, and high mitotic rate and is commonly found in sun-exposed areas. Dermoscopic features include irregular globules with blue-whitish veil.10
Treatment of atypical Spitz nevi depends mainly on the age of the patient and the histologic features of the nevus. Adults with atypical Spitz nevi frequently require excision, while the preferred choice for treatment in children with common Spitz nevi is regular clinical monitoring when there are no concerning clinical, dermoscopic, or histologic features.8 Compared to common Spitz nevi, atypical Spitz nevi have more melanoma-like features, resulting in a stronger recommendation for excision. Excision allows for a more thorough histologic evaluation and minimizes the likelihood of a recurrent atypical lesion.11 In all cases, close clinical follow-up is recommended to monitor for reoccurrence.
THE DIAGNOSIS: Atypical Spitz Tumor
The shave biopsy revealed extensive dermal proliferation with spitzoid cytomorphology containing large, spindled nuclei; prominent nucleoli; and abundant homogenous cytoplasm arranged in haphazard fascicles. The proliferation was associated with prominent pseudoepitheliomatous hyperplasia of the overlying epidermis, and anaplastic lymphoma kinase immunohistochemistry showed diffuse strong positivity. Fluorescence in situ hybridization confirmed fusion of the tropomyosin 3 (TPM3) and anaplastic lymphoma kinase (ALK) genes, which finalized the diagnosis of an ALK-mutated atypical spitz tumor. Due to the location and size of the lesion, Mohs micrographic surgery was performed to excise the tumor and clear the margins.
Spitz nevi are uncommon benign melanocytic neoplasms that typically occur in pediatric populations.1 Atypical spitz nevi comprised fewer than 17% of all childhood melanocytic nevi in the United States and can be considered in the broader category of spitzoid tumors. Spitz nevi are divided into 3 classes: Spitz nevus, atypical Spitz nevus, and spitzoid melanoma. Atypical Spitz nevi have typical Spitz nevus and spitzoid melanoma features and often can be difficult to distinguish on dermoscopy. Malignant Spitz tumors typically occur in the fifth decade of life, though the age distribution can vary widely.1
Black patients are less likely to be diagnosed with Spitz nevi, potentially due to a lower prevalence in this population, thus limiting the clinician’s clinical exposure and leading to increased rates of misdiagnoses.2 Spitz nevi usually manifest as well-circumscribed, dome-shaped papules and frequently are described as pink to red due to increased vascularity and limited melanin content1; however, these lesions may appear more violaceous, dusky, or dark brown in darker skin types. Additionally, approximately 71% of patients in a clinical review of Spitz nevi had a pigmented lesion, ranging from light brown to black.3 It is important for dermatologists to understand that the contrast in color between the nevus and the surrounding skin may not be as striking, prominent, or clinically concerning, particularly in darker skin types, such as in our patient.
Spitz nevi frequently manifest as rapidly growing solitary lesions most frequently developing in the lower legs (shown in 41% of lesions in one report).4 However, a recent retrospective review indicated that Spitz nevi in Black patients most commonly were found on the upper extremities, as was seen in our patient.2 Compared to typical and common Spitz nevi, atypical Spitz nevi often are greater than 10 mm in diameter and have features of ulceration.
Diagnosing atypical spitzoid melanocytic lesions requires adequate clinical suspicion and confirmation via biopsy. Under dermoscopy, typical Spitz nevi often display a starburst or globular pattern with pinpoint vessels, though it can have variable manifestations of both patterns. Atypical Spitz nevi can be challenging to distinguish from melanoma on dermoscopy since both conditions can have atypical pigment networks or structureless homogenous areas.1 Consequently, there often is a lower threshold for biopsy and possible follow-up excision for atypical Spitz nevi. Histopathology of atypical Spitz nevi includes epithelioid and spindle melanocytes but can share features of melanomas, including areas of prominent pagetoid spread, asymmetry, and poor circumscription.5 Furthermore, atypical Spitz nevi with ALK gene fusion, as seen in our patient, have been shown in the literature to demonstrate distinct histopathologic features, such as wedge-shaped extension into the dermis or a bulbous lower border that can resemble pseudoepitheliomatous hyperplasia.6
The differential diagnosis for this rapidly growing scaly nodule also should include pyogenic granuloma, bacillary angiomatosis, Kaposi sarcoma, and amelanotic melanoma. Pyogenic granuloma is a rapidly growing, benign, vascular tumor that often becomes ulcerated and can occur in any age group.7 Pyogenic granuloma frequently appears at sites of trauma as a solitary, bright pink to red, friable, pedunculated papule and often manifests on the arms, hands, and face, similar to atypical Spitz nevi, though they can appear anywhere on the body. Histology shows a lobular capillary network with a central feeder vessel.7
Bacillary angiomatosis is an uncommon cutaneous infection associated with vascular proliferation and neovascularization due to the gram-negative organism Bartonella henselae.8 Bacillary nodules typically are reddish to purple and appear on the arms, sometimes with central ulceration and bleeding. Patients may present with multiple papules and nodules of varying sizes, as the lesions can arise in crops and follow a sporotrichoid pattern. Most patients with bacillary angiomatosis are immunosuppressed, though it rarely can affect immunocompetent patients. Histologically, bacillary angiomatosis is similar to pyogenic granuloma, though Gram or Warthin-Starry stains can help differentiate B henselae.8
Kaposi sarcoma is a malignant vascular neoplasm that often manifests in immunocompromised patients as violaceous, purple, or red patches, plaques, and nodules on the skin or oral mucosa. Histopathology shows spindle cell proliferation of irregular complex vascular channels dissecting through the dermis. Human herpesvirus 8 immunohistochemistry can be used to confirm diagnosis on histopathology.9 In contrast, amelanotic melanoma consists of lack of pigmentation, asymmetry with polymorphous vascular pattern, and high mitotic rate and is commonly found in sun-exposed areas. Dermoscopic features include irregular globules with blue-whitish veil.10
Treatment of atypical Spitz nevi depends mainly on the age of the patient and the histologic features of the nevus. Adults with atypical Spitz nevi frequently require excision, while the preferred choice for treatment in children with common Spitz nevi is regular clinical monitoring when there are no concerning clinical, dermoscopic, or histologic features.8 Compared to common Spitz nevi, atypical Spitz nevi have more melanoma-like features, resulting in a stronger recommendation for excision. Excision allows for a more thorough histologic evaluation and minimizes the likelihood of a recurrent atypical lesion.11 In all cases, close clinical follow-up is recommended to monitor for reoccurrence.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084. doi:10.1016/j.jaad.2011.04.040
- Farid YI, Honda KS. Spitz nevi in African Americans: a retrospective chart review of 11 patients. J Cutan Pathol. 2021;48:511-518. doi:10.1111 /cup.13903
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20-25. doi: 10.1159/000246051
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, et al. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267-275. doi: 10.1097 /DAD.0b013e3181c52b99
- Brown A, Sawyer JD, Neumeister MW. Spitz nevus: review and update. Clin Plast Surg 2021;48:677-686. doi: 10.1016/j.cps.2021.06.002 [published Online First: 20210818]
- Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. doi: 10.1097/PAS.0000000000000387
- Sarwal P, Lapumnuaypol K. Pyogenic granuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556077/
- Akram SM, Anwar MY, Thandra KC, et al. Bacillary angiomatosis. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 4, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448092/
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Pizzichetta MA, Talamini R, Stanganelli I, et al. Amelanotic/ hypomelanotic melanoma: clinical and dermoscopic features. Br J Dermatol 2004;150(6):1117-1124. doi: 10.1111/j.1365-2133.2004.05928.x
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part II. natural history and management. J Am Acad Dermatol 2011;65:1087-1092. doi:10.1016/j.jaad.2011.06.045
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084. doi:10.1016/j.jaad.2011.04.040
- Farid YI, Honda KS. Spitz nevi in African Americans: a retrospective chart review of 11 patients. J Cutan Pathol. 2021;48:511-518. doi:10.1111 /cup.13903
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20-25. doi: 10.1159/000246051
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, et al. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267-275. doi: 10.1097 /DAD.0b013e3181c52b99
- Brown A, Sawyer JD, Neumeister MW. Spitz nevus: review and update. Clin Plast Surg 2021;48:677-686. doi: 10.1016/j.cps.2021.06.002 [published Online First: 20210818]
- Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. doi: 10.1097/PAS.0000000000000387
- Sarwal P, Lapumnuaypol K. Pyogenic granuloma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556077/
- Akram SM, Anwar MY, Thandra KC, et al. Bacillary angiomatosis. StatPearls [Internet]. StatPearls Publishing; 2024. Updated July 4, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448092/
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. StatPearls Publishing; 2024. Updated June 5, 2023. Accessed December 4, 2024. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Pizzichetta MA, Talamini R, Stanganelli I, et al. Amelanotic/ hypomelanotic melanoma: clinical and dermoscopic features. Br J Dermatol 2004;150(6):1117-1124. doi: 10.1111/j.1365-2133.2004.05928.x
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part II. natural history and management. J Am Acad Dermatol 2011;65:1087-1092. doi:10.1016/j.jaad.2011.06.045
Exophytic Scaly Nodule on the Wrist
Exophytic Scaly Nodule on the Wrist
A 30-year-old Black man presented to the dermatology clinic with a rapidly growing, exophytic, scaly nodule on the right volar wrist of 2 months’ duration. The patient’s medical history was otherwise unremarkable. Physical examination revealed an irregularly bordered, red to violaceous, scaly, eroded, exophytic nodule on the wrist that was 2 cm in diameter with a surrounding adherent white-yellow crust. The patient had presumed the nodule was a wart and had been self-treating with over-the-counter salicylic acid and cryotherapy with no relief. He denied any bleeding or pruritus. The rest of the skin examination was unremarkable. A shave biopsy was performed for further evaluation.
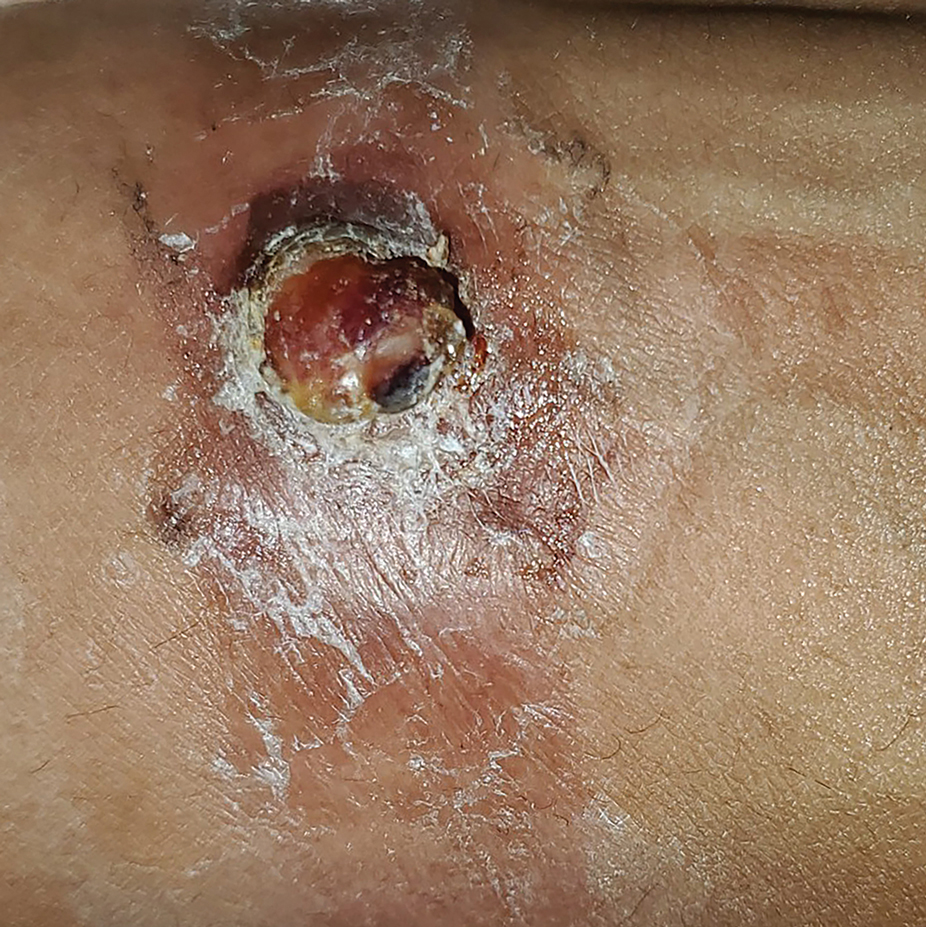

Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
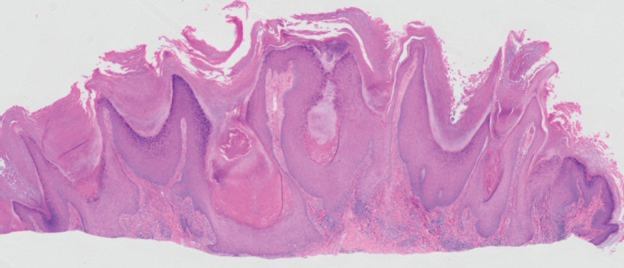
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
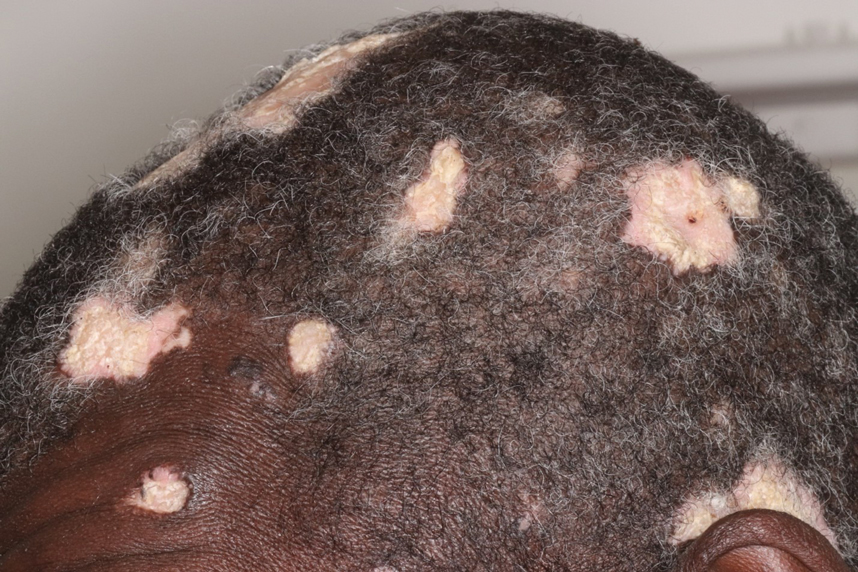
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
THE DIAGNOSIS: Hypertrophic Lupus Erythematosus
The biopsy of the face collected at the initial appointment revealed interface dermatitis with epidermal hyperplasia with no parakeratosis or eosinophils (Figure 1). Microscopic findings were suggestive of hypertrophic lupus erythematosus (HLE) or hypertrophic lichen planus. The rapid plasma reagin and HIV labs collected at the initial appointment were negative, and a review of systems was negative for systemic symptoms. Considering these results and the clinical distribution of the lesions primarily affecting sun-exposed areas of the upper body, a final diagnosis of HLE was made. The patient was counseled on the importance of photoprotection and was started on hydroxychloroquine.
Hypertrophic lupus erythematosus, a rare variant of chronic cutaneous lupus erythematosus (CCLE), typically manifests as verrucous plaques or nodules commonly found on sun-exposed areas of the body, as was observed in our patient on the face, scalp (Figures 2 and 3), chest, and upper extremities.1 Lesions can have a variable appearance, from hyperkeratotic ulcers to depigmented plaques and keratoacanthomalike lesions.2 On histopathology, HLE falls into the category of lichenoid interface dermatitis and commonly demonstrates hyperkeratosis, acanthosis, follicular plugging, superficial and deep infiltrate, and increased mucin deposition in the dermis.3
Although rare, it is critical to remain vigilant for the development of squamous cell carcinoma in patients with chronic untreated CCLE. Hypertrophic lupus erythematosus, specifically, is the most likely variant to give rise to invasive squamous cell carcinoma and can be more aggressive as a result of this malignant transformation.3,4 Ruling out squamous cell carcinoma in the setting of HLE can be achieved by staining for CD123, as HLE commonly is associated with many CD123+ plasmacytoid dendritic cells adjacent to the epithelium, unlike squamous cell carcinoma.3 Fortunately no evidence of invasive squamous cell carcinoma, including cellular atypia or increased mitotic figures, was seen on histology in our patient.
A thorough history and physical examination are essential for screening for HLE, as positive antinuclear antibodies are observed only in half of the patients diagnosed with CCLE.5 Furthermore, antinuclear antibodies sometimes can be negative in patients with HLE who have end-stage organ involvement.
Hypertrophic lupus erythematosus can be challenging to treat. First-line therapies include antimalarials, topical steroids, and sun-protective measures. Intralesional triamcinolone injection also can be used as an adjunctive therapy to expedite the treatment response.6 Evidence supports good response following treatment with acitretin or a combination of isotretinoin and hydroxychloroquine.2 Another therapeutic strategy is implementing immunosuppressants such as methotrexate, mycophenolate mofetil, and azathioprine for persistent disease. Immunomodulators such as thalidomide historically have been shown to treat severe recalcitrant cases of HLE but typically are reserved for extreme cases due to adverse effects. Biologic agents such as intravenous immunoglobulins and rituximab have been shown to treat CCLE successfully, but routine use is limited due to high cost and lack of strong clinical trials.7
There have been reports of experimental therapies such as monoclonal antibodies (eg, anifrolumab and tocilizumab therapy) providing remission for patients with refractory CCLE, but information on their efficacy—specifically in patients with HLE—is lacking.8 Chronic cutaneous lupus erythematosus and its variants require further investigation regarding which treatment options provide the greatest benefit while minimizing adverse effects.
It is important to distinguish HLE from other potential diagnoses. Features of HLE can mimic hypertrophic lichen planus; however, the latter typically appears on the legs while HLE appears more commonly on the upper extremities and face in a photodistributed pattern.9 Since HLE has a lichenoid appearance histologically, it may appear clinically similar to hypertrophic lichen planus. Although not performed in our patient due to cost, direct immunofluorescence can aid in distinguishing HLE from hypertrophic lichen planus. Chronic cutaneous lupus erythematosus shows a granular pattern of deposition of IgM (primarily), IgG, IgA, and C3. In contrast, hypertrophic lichen planus exhibits cytoid bodies that stain positive for IgM as well as linear deposition of fibrinogen along the basement membrane.3,10
Blastomycosis also can lead to development of verrucous plaques in sun-exposed areas, but the lesions typically originate as pustules that ulcerate over time. Lesions also can manifest with central scarring and a heaped edge.3 Unlike HLE, pseudoepitheliomatous hyperplasia with mixed infiltrate and intradermal pustules are seen in blastomycosis.3 Fungal organisms often are seen on pathology and are relatively large and uniform in size and shape, are found within giant cells, and have a thick refractile asymmetrical wall.11 In rupioid psoriasis, skin lesions mostly are widespread and are not limited to sun-exposed areas. Additionally, biopsies from active rupioid lesions typically show psoriasiform epidermal hyperplasia with parakeratosis with no interface inflammation—a key differentiator.12 In secondary syphilis, chancres often are missed and are not reported by patients. Clinically, secondary syphilis often manifests as scaly patches and plaques with palmar involvement and positive rapid plasma reagin, which was negative in our patient.13 Histologically, secondary syphilis can exhibit a vacuolar or lichenoid interface dermatitis; however, it typically exhibits slender acanthosis with long rete ridges and neutrophils in the stratum corneum.3 Furthermore, plasma cells are present in about two-thirds of cases in the United States, with obliteration of the lumen of small vessels and perivascular histiocytes and lymphocytes with apparent cytoplasm commonly seen on pathology. Silver staining or immunostaining for Treponema pallidum may reveal the spirochetes that cause this condition.3
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophic lupus erythematosus: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892. doi:10.1111/j.1600-0560.2011.01779.x
- Narang T, Sharma M, Gulati N, et al. Extensive hypertrophic lupus erythematosus: atypical presentation. Indian J Dermatol. 2012;57:504. doi:10.4103/0019-5154.103085
- Elston D, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Saunders/ Elsevier; 2018.
- Melikoglu MA, Melikoglu M, Demirci E, et al. Discoid lupus erythematosus- associated cutaneous squamous cell carcinoma in systemic lupus erythematosus. Eurasian J Med. 2022;54:204-205. doi:10.5152 /eurasianjmed. 2022.21062
- Patsinakidis N, Gambichler T, Lahner N, et al. Cutaneous characteristics and association with antinuclear antibodies in 402 patients with different subtypes of lupus erythematosus. J Eur Acad Dermatol Venereol. 2016;30:2097-2104. doi:10.1111/jdv.13769
- Kulkarni S, Kar S, Madke B, et al. A rare presentation of verrucous/ hypertrophic lupus erythematosus: a variant of cutaneous LE. Indian Dermatol Online J. 2014;5:87. doi:10.4103/2229-5178.126048
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-Based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Blum FR, Sampath AJ, Foulke GT. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin Exp Dermatol. 2022;47:1998- 2001. doi:10.1111/ced.15335
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:E3555. doi:10.7759/cureus.3555
- Demirci GT, Altunay IK, Sarýkaya S, et al. Lupus erythematosus and lichen planus overlap syndrome: a case report with a rapid response to topical corticosteroid therapy. Dermatol Reports. 2011 25;3:E48. doi:10.4081/dr.2011.e48
- Caldito EG, Antia C, Petronic-Rosic V. Cutaneous blastomycosis. JAMA Dermatol. 2022;158:1064. doi:10.1001/jamadermatol.2022.3151
- Ip KHK, Cheng HS, Oliver FG. Rupioid psoriasis. JAMA Dermatol. 2021;157:859. doi:10.1001/jamadermatol.2021.0451
- Trawinski H. Secondary syphilis. Dtsch Arztebl Int. 2021;118:249. doi:10.3238/arztebl.m2021.0107
Verrucous Plaques on Sun-Exposed Areas
Verrucous Plaques on Sun-Exposed Areas
A 54-year-old man with no notable medical history presented to an outpatient dermatology clinic with multiple skin lesions on sun-exposed areas including the face, chest, scalp, and bilateral upper extremities. The patient reported that he had not seen a doctor for 26 years. He noted that the lesions had been present for many years but was unsure of the exact timeframe. Physical examination revealed verrucous plaques with a violaceous rim and central hypopigmentation on the chest, scalp, face, and arms. Scarring alopecia also was noted on the scalp with no associated pain or pruritus. Antinuclear antibody and extractable nuclear antigen tests were negative, and urine analysis was normal. A shave biopsy of the chest was performed for histopathologic evaluation. Rapid plasma reagin tests and HIV antibody tests also were performed.
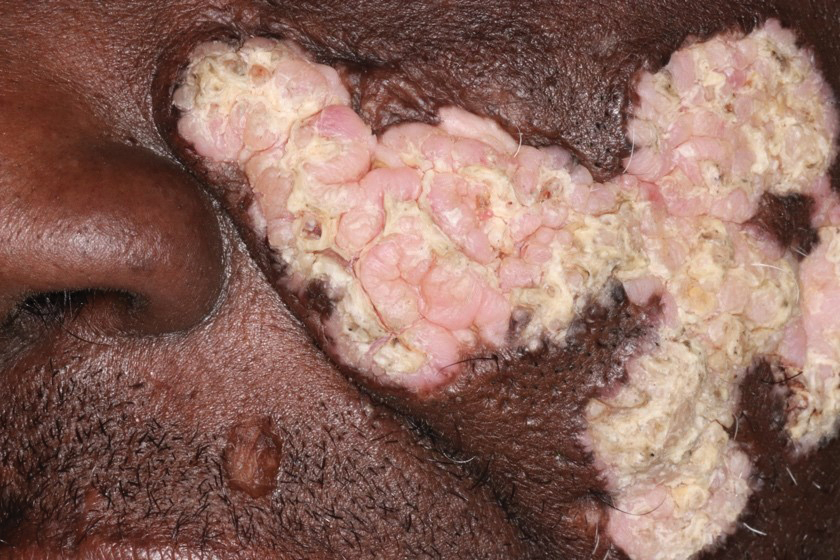
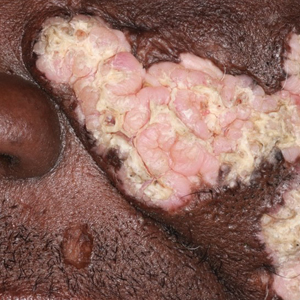
Bilateral Ankle Ulcerations and Gangrene of the Toes
Bilateral Ankle Ulcerations and Gangrene of the Toes
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
Bilateral Ankle Ulcerations and Gangrene of the Toes
Bilateral Ankle Ulcerations and Gangrene of the Toes
A 74-year-old woman presented to the hospital with large tender ulcerations on both ankles as well as gangrene of the toes of 6 to 8 weeks’ duration. The patient had a history of hypertension as well as seropositive nonerosive rheumatoid arthritis that had been diagnosed 8 years prior and was well controlled with leflunomide and prednisone as needed for flares. She denied any history of similar ulcers as well as any recent illnesses, medication changes, or joint pain or swelling. She was evaluated by vascular surgery 1 week prior to the current presentation, at which time her ankle-brachial index score was normal. Skin examination revealed noninflammatory retiform purpura surrounding ulcerations on both ankles (top) and necrosis of all toes (bottom) with peripheral retiform purpura. Joint examination revealed swan neck deformities of multiple fingers with normal range of motion, and there was no effusion or tenderness of the joints of the fingers on palpation. No rheumatoid nodules were present. Laboratory testing revealed elevated rheumatoid factor, anti–cyclic citrullinated peptide, C-reactive protein, and anti–Sjögren syndrome–related antigen A levels and low C4 levels. Cryoglobulins, antineutrophil cytoplasmic antibodies, and serum protein electrophoresis were negative. Biopsy of an ulcer on the right ankle showed medium-sized vessel vasculitis with fibrinoid necrosis, including endothelium necrosis and a perivascular lymphocytic infiltrate. Direct immunofluorescence demonstrated dense, granular, intraperivascular deposition of IgM and IgG with slightly weaker deposition of IgA, C3, and C5b-9 in the dermis and subcutis with a greater effect on medium-sized vessels.
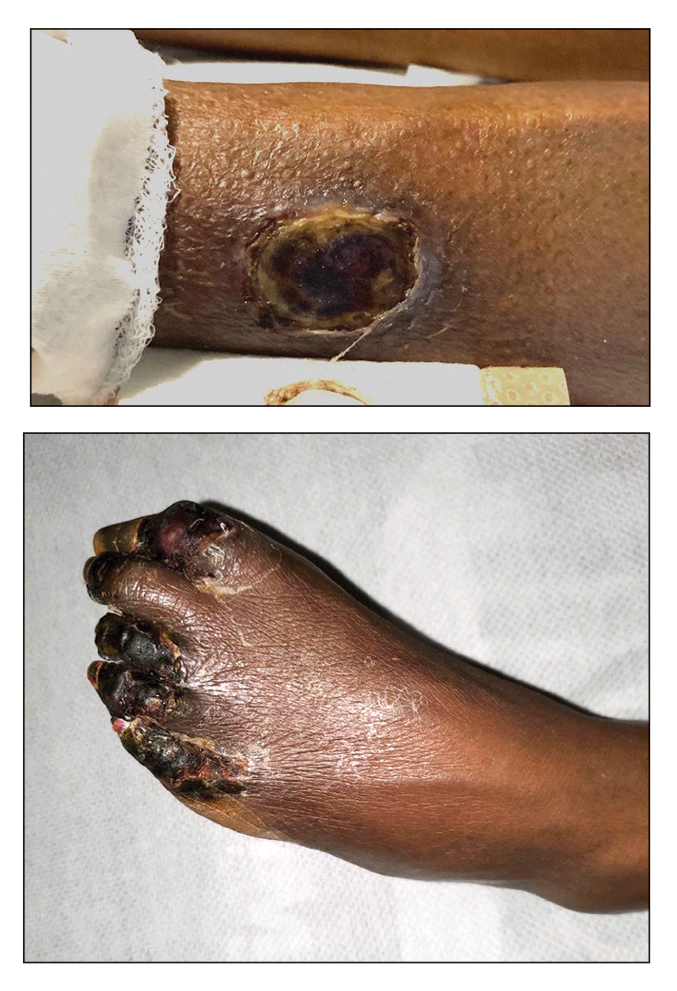
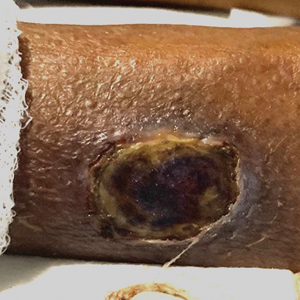
Dome-Shaped White Papules on the Earlobe
Dome-Shaped White Papules on the Earlobe
THE DIAGNOSIS: Trichodiscoma
Histologic evaluation revealed an unremarkable epidermal surface and a subjacent well-demarcated superficial dermal nodule showing a proliferation, sometimes fascicular, of wavy and spindled fibroblasts with some stellate forms within a variably loose fibrous stroma. Some angioplasia and vascular ectasia also were seen (Figure). A diagnosis of trichodiscoma was made based on these histologic findings.
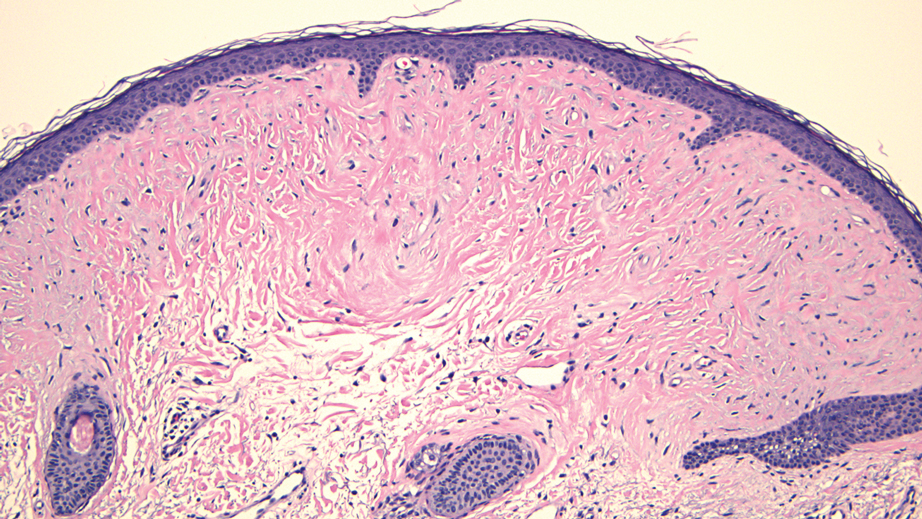
While the patient’s personal and family history of pneumothorax originally had been attributed to other causes, the diagnosis of trichodiscoma raised suspicion for Birt-Hogg-Dubé syndrome due to the classic association of skin lesions (often trichodiscomas), renal cell carcinoma, and spontaneous pneumothorax in this condition. The patient was sent for genetic testing for the associated folliculin (FLCN) gene, which was positive and thereby confirmed the diagnosis of Birt-Hogg-Dubé syndrome. At the most recent follow-up almost 2 years after initial presentation, the lesions on the earlobe were stable. The patient has since undergone screening for abdominal and renal neoplasia with negative results, and he has had no other occurrences of pneumothorax.
Our case highlights the association between trichodiscomas and Birt-Hogg-Dubé syndrome, which necessitates screening for renal cell carcinoma, pneumothorax, and lung cysts.1 Birt-Hogg-Dubé syndrome is an autosomal- dominant disorder of the skin and lungs that is characterized by a predisposition for renal carcinoma, pneumothorax, and colon polyps as well as cutaneous markers that include fibrofolliculomas, acrochordons, and trichodiscomas; the trichodiscomas tend to manifest as numerous smooth, flesh-colored or grayish-white papules on the face, ears, neck, and/or upper trunk.1
Trichodiscomas are benign lesions and do not require treatment2; however, if they are cosmetically bothersome to the patient, surgical excision is an option for single lesions. For more widespread cutaneous disease, combination therapy with a CO2 laser and erbium-doped yttrium aluminum garnet laser may be utilized.3 The differential diagnosis for trichodiscoma includes basal cell carcinoma, fibrous papule, dermal nevus, and trichofolliculoma.
Basal cell carcinoma is the most common type of skin cancer.4 Clinically, it typically manifests as pink or flesh-colored papules on the head or neck, often with overlying ulceration or telangiectasia. Due to its association with chronic sun exposure, the median age of diagnosis for basal cell carcinoma is 68 years. Histopathologically, basal cell carcinoma is characterized by islands or nests of atypical basaloid cells with palisading cells at the periphery.4 Treatment depends on the location and size of the lesion, but Mohs micrographic surgery is the most common intervention on the face and ears.5
In contrast, fibrous papules are benign lesions that manifest clinically as small, firm, flesh-colored papules that most commonly are found on the nose.6,7 On dermatopathology, classic findings include fibrovascular proliferation and scattered multinucleated triangular or stellate cells in the upper dermis.7 Due to the benign nature of the lesion, treatment is not required6; however, shave excision, electrodessication, and laser therapies can be attempted if the patient chooses to pursue treatment.8
Dermal nevus is a type of benign acquired melanocytic nevus that manifests clinically as a light-brown to flesh-colored, dome-shaped or papillomatous papule.9 It typically develops in areas that are exposed to the sun, including the face.10 There also have been cases of dermal nevi on the ear.11 Histopathology shows melanocytic nevus cells that have completely detached from the epidermis and are located entirely in the dermis.12 While dermal nevi are benign and treatment is not necessary, surgical excision is an option for patients who request removal.13
Trichofolliculoma is a benign tumor of the adnexa that shows follicular differentiation on histopathology.14 On physical examination, it manifests as an isolated flesh-colored papule or nodule with a central pore from which tufted hairs protrude. These lesions usually appear on the face or scalp and occur more commonly in women than in men. While these may be clinically indistinguishable from trichodiscomas, the absence of protruding hair in our patient’s case makes trichofolliculoma less likely. When biopsied, histopathology classically shows a cystically dilated hair follicle with keratinous material and several mature and immature branched follicular structures. Preferred treatment for trichofolliculomas is surgical excision, and recurrence is rare.14
- Toro JR, Glenn G, Duray P, et al. Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135:1195-202. doi:10.1001/archderm.135.10.1195
- Tong Y, Coda AB, Schneider JA, et al. Familial multiple trichodiscomas: case report and concise review. Cureus. 2017;9:E1596. doi:10.7759/cureus.1596
- Riley J, Athalye L, Tran D, et al. Concomitant fibrofolliculoma and trichodiscoma on the abdomen. Cutis. 2018;102:E30-E32.
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. StatPearls [Internet]. Updated March 13, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482439/
- Bittner GC, Kubo EM, Fantini BC, et al. Auricular reconstruction after Mohs micrographic surgery: analysis of 101 cases. An Bras Dermatol. 2021;96:408-415. doi:10.1016/j.abd.2020.12.008
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560. doi:10.1097/DAD.0000000000001083
- Jacyk WK, Rütten A, Requena L. Fibrous papule of the face with granular cells. Dermatology. 2008;216:56-59. doi:10.1159/000109359
- Macri A, Kwan E, Tanner LS. Cutaneous angiofibroma. StatPearls [Internet]. Updated July 19, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482470/
- Sardana K, Chakravarty P, Goel K. Optimal management of common acquired melanocytic nevi (moles): current perspectives. Clin Cosmet Investig Dermatol. 2014;7:89-103. doi:10.2147/CCID.S57782
- Conforti C, Giuffrida R, Agozzino M, et al. Basal cell carcinoma and dermal nevi of the face: comparison of localization and dermatoscopic features. Int J Dermatol. 2021;60:996-1002. doi:10.1111/ijd.15554
- Alves RV, Brandão FH, Aquino JE, et al. Intradermal melanocytic nevus of the external auditory canal. Braz J Otorhinolaryngol. 2005;71:104-106. doi: 10.1016/s1808-8694(15)31295-7
- Muradia I, Khunger N, Yadav AK. A clinical, dermoscopic, and histopathological analysis of common acquired melanocytic nevi in skin of color. J Clin Aesthet Dermatol. 2022;15:41-51.
- Sardana K, Chakravarty P, Goel K. Optimal management of common acquired melanocytic nevi (moles): current perspectives. Clin Cosmet Investig Dermatol. 2014;7:89-103. doi:10.2147/CCID.S57782
- Massara B, Sellami K, Graja S, et al. Trichofolliculoma: a case series. J Clin Aesthet Dermatol. 2023;16:41-43.
THE DIAGNOSIS: Trichodiscoma
Histologic evaluation revealed an unremarkable epidermal surface and a subjacent well-demarcated superficial dermal nodule showing a proliferation, sometimes fascicular, of wavy and spindled fibroblasts with some stellate forms within a variably loose fibrous stroma. Some angioplasia and vascular ectasia also were seen (Figure). A diagnosis of trichodiscoma was made based on these histologic findings.
While the patient’s personal and family history of pneumothorax originally had been attributed to other causes, the diagnosis of trichodiscoma raised suspicion for Birt-Hogg-Dubé syndrome due to the classic association of skin lesions (often trichodiscomas), renal cell carcinoma, and spontaneous pneumothorax in this condition. The patient was sent for genetic testing for the associated folliculin (FLCN) gene, which was positive and thereby confirmed the diagnosis of Birt-Hogg-Dubé syndrome. At the most recent follow-up almost 2 years after initial presentation, the lesions on the earlobe were stable. The patient has since undergone screening for abdominal and renal neoplasia with negative results, and he has had no other occurrences of pneumothorax.
Our case highlights the association between trichodiscomas and Birt-Hogg-Dubé syndrome, which necessitates screening for renal cell carcinoma, pneumothorax, and lung cysts.1 Birt-Hogg-Dubé syndrome is an autosomal- dominant disorder of the skin and lungs that is characterized by a predisposition for renal carcinoma, pneumothorax, and colon polyps as well as cutaneous markers that include fibrofolliculomas, acrochordons, and trichodiscomas; the trichodiscomas tend to manifest as numerous smooth, flesh-colored or grayish-white papules on the face, ears, neck, and/or upper trunk.1
Trichodiscomas are benign lesions and do not require treatment2; however, if they are cosmetically bothersome to the patient, surgical excision is an option for single lesions. For more widespread cutaneous disease, combination therapy with a CO2 laser and erbium-doped yttrium aluminum garnet laser may be utilized.3 The differential diagnosis for trichodiscoma includes basal cell carcinoma, fibrous papule, dermal nevus, and trichofolliculoma.
Basal cell carcinoma is the most common type of skin cancer.4 Clinically, it typically manifests as pink or flesh-colored papules on the head or neck, often with overlying ulceration or telangiectasia. Due to its association with chronic sun exposure, the median age of diagnosis for basal cell carcinoma is 68 years. Histopathologically, basal cell carcinoma is characterized by islands or nests of atypical basaloid cells with palisading cells at the periphery.4 Treatment depends on the location and size of the lesion, but Mohs micrographic surgery is the most common intervention on the face and ears.5
In contrast, fibrous papules are benign lesions that manifest clinically as small, firm, flesh-colored papules that most commonly are found on the nose.6,7 On dermatopathology, classic findings include fibrovascular proliferation and scattered multinucleated triangular or stellate cells in the upper dermis.7 Due to the benign nature of the lesion, treatment is not required6; however, shave excision, electrodessication, and laser therapies can be attempted if the patient chooses to pursue treatment.8
Dermal nevus is a type of benign acquired melanocytic nevus that manifests clinically as a light-brown to flesh-colored, dome-shaped or papillomatous papule.9 It typically develops in areas that are exposed to the sun, including the face.10 There also have been cases of dermal nevi on the ear.11 Histopathology shows melanocytic nevus cells that have completely detached from the epidermis and are located entirely in the dermis.12 While dermal nevi are benign and treatment is not necessary, surgical excision is an option for patients who request removal.13
Trichofolliculoma is a benign tumor of the adnexa that shows follicular differentiation on histopathology.14 On physical examination, it manifests as an isolated flesh-colored papule or nodule with a central pore from which tufted hairs protrude. These lesions usually appear on the face or scalp and occur more commonly in women than in men. While these may be clinically indistinguishable from trichodiscomas, the absence of protruding hair in our patient’s case makes trichofolliculoma less likely. When biopsied, histopathology classically shows a cystically dilated hair follicle with keratinous material and several mature and immature branched follicular structures. Preferred treatment for trichofolliculomas is surgical excision, and recurrence is rare.14
THE DIAGNOSIS: Trichodiscoma
Histologic evaluation revealed an unremarkable epidermal surface and a subjacent well-demarcated superficial dermal nodule showing a proliferation, sometimes fascicular, of wavy and spindled fibroblasts with some stellate forms within a variably loose fibrous stroma. Some angioplasia and vascular ectasia also were seen (Figure). A diagnosis of trichodiscoma was made based on these histologic findings.
While the patient’s personal and family history of pneumothorax originally had been attributed to other causes, the diagnosis of trichodiscoma raised suspicion for Birt-Hogg-Dubé syndrome due to the classic association of skin lesions (often trichodiscomas), renal cell carcinoma, and spontaneous pneumothorax in this condition. The patient was sent for genetic testing for the associated folliculin (FLCN) gene, which was positive and thereby confirmed the diagnosis of Birt-Hogg-Dubé syndrome. At the most recent follow-up almost 2 years after initial presentation, the lesions on the earlobe were stable. The patient has since undergone screening for abdominal and renal neoplasia with negative results, and he has had no other occurrences of pneumothorax.
Our case highlights the association between trichodiscomas and Birt-Hogg-Dubé syndrome, which necessitates screening for renal cell carcinoma, pneumothorax, and lung cysts.1 Birt-Hogg-Dubé syndrome is an autosomal- dominant disorder of the skin and lungs that is characterized by a predisposition for renal carcinoma, pneumothorax, and colon polyps as well as cutaneous markers that include fibrofolliculomas, acrochordons, and trichodiscomas; the trichodiscomas tend to manifest as numerous smooth, flesh-colored or grayish-white papules on the face, ears, neck, and/or upper trunk.1
Trichodiscomas are benign lesions and do not require treatment2; however, if they are cosmetically bothersome to the patient, surgical excision is an option for single lesions. For more widespread cutaneous disease, combination therapy with a CO2 laser and erbium-doped yttrium aluminum garnet laser may be utilized.3 The differential diagnosis for trichodiscoma includes basal cell carcinoma, fibrous papule, dermal nevus, and trichofolliculoma.
Basal cell carcinoma is the most common type of skin cancer.4 Clinically, it typically manifests as pink or flesh-colored papules on the head or neck, often with overlying ulceration or telangiectasia. Due to its association with chronic sun exposure, the median age of diagnosis for basal cell carcinoma is 68 years. Histopathologically, basal cell carcinoma is characterized by islands or nests of atypical basaloid cells with palisading cells at the periphery.4 Treatment depends on the location and size of the lesion, but Mohs micrographic surgery is the most common intervention on the face and ears.5
In contrast, fibrous papules are benign lesions that manifest clinically as small, firm, flesh-colored papules that most commonly are found on the nose.6,7 On dermatopathology, classic findings include fibrovascular proliferation and scattered multinucleated triangular or stellate cells in the upper dermis.7 Due to the benign nature of the lesion, treatment is not required6; however, shave excision, electrodessication, and laser therapies can be attempted if the patient chooses to pursue treatment.8
Dermal nevus is a type of benign acquired melanocytic nevus that manifests clinically as a light-brown to flesh-colored, dome-shaped or papillomatous papule.9 It typically develops in areas that are exposed to the sun, including the face.10 There also have been cases of dermal nevi on the ear.11 Histopathology shows melanocytic nevus cells that have completely detached from the epidermis and are located entirely in the dermis.12 While dermal nevi are benign and treatment is not necessary, surgical excision is an option for patients who request removal.13
Trichofolliculoma is a benign tumor of the adnexa that shows follicular differentiation on histopathology.14 On physical examination, it manifests as an isolated flesh-colored papule or nodule with a central pore from which tufted hairs protrude. These lesions usually appear on the face or scalp and occur more commonly in women than in men. While these may be clinically indistinguishable from trichodiscomas, the absence of protruding hair in our patient’s case makes trichofolliculoma less likely. When biopsied, histopathology classically shows a cystically dilated hair follicle with keratinous material and several mature and immature branched follicular structures. Preferred treatment for trichofolliculomas is surgical excision, and recurrence is rare.14
- Toro JR, Glenn G, Duray P, et al. Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135:1195-202. doi:10.1001/archderm.135.10.1195
- Tong Y, Coda AB, Schneider JA, et al. Familial multiple trichodiscomas: case report and concise review. Cureus. 2017;9:E1596. doi:10.7759/cureus.1596
- Riley J, Athalye L, Tran D, et al. Concomitant fibrofolliculoma and trichodiscoma on the abdomen. Cutis. 2018;102:E30-E32.
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. StatPearls [Internet]. Updated March 13, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482439/
- Bittner GC, Kubo EM, Fantini BC, et al. Auricular reconstruction after Mohs micrographic surgery: analysis of 101 cases. An Bras Dermatol. 2021;96:408-415. doi:10.1016/j.abd.2020.12.008
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560. doi:10.1097/DAD.0000000000001083
- Jacyk WK, Rütten A, Requena L. Fibrous papule of the face with granular cells. Dermatology. 2008;216:56-59. doi:10.1159/000109359
- Macri A, Kwan E, Tanner LS. Cutaneous angiofibroma. StatPearls [Internet]. Updated July 19, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482470/
- Sardana K, Chakravarty P, Goel K. Optimal management of common acquired melanocytic nevi (moles): current perspectives. Clin Cosmet Investig Dermatol. 2014;7:89-103. doi:10.2147/CCID.S57782
- Conforti C, Giuffrida R, Agozzino M, et al. Basal cell carcinoma and dermal nevi of the face: comparison of localization and dermatoscopic features. Int J Dermatol. 2021;60:996-1002. doi:10.1111/ijd.15554
- Alves RV, Brandão FH, Aquino JE, et al. Intradermal melanocytic nevus of the external auditory canal. Braz J Otorhinolaryngol. 2005;71:104-106. doi: 10.1016/s1808-8694(15)31295-7
- Muradia I, Khunger N, Yadav AK. A clinical, dermoscopic, and histopathological analysis of common acquired melanocytic nevi in skin of color. J Clin Aesthet Dermatol. 2022;15:41-51.
- Sardana K, Chakravarty P, Goel K. Optimal management of common acquired melanocytic nevi (moles): current perspectives. Clin Cosmet Investig Dermatol. 2014;7:89-103. doi:10.2147/CCID.S57782
- Massara B, Sellami K, Graja S, et al. Trichofolliculoma: a case series. J Clin Aesthet Dermatol. 2023;16:41-43.
- Toro JR, Glenn G, Duray P, et al. Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135:1195-202. doi:10.1001/archderm.135.10.1195
- Tong Y, Coda AB, Schneider JA, et al. Familial multiple trichodiscomas: case report and concise review. Cureus. 2017;9:E1596. doi:10.7759/cureus.1596
- Riley J, Athalye L, Tran D, et al. Concomitant fibrofolliculoma and trichodiscoma on the abdomen. Cutis. 2018;102:E30-E32.
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. StatPearls [Internet]. Updated March 13, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482439/
- Bittner GC, Kubo EM, Fantini BC, et al. Auricular reconstruction after Mohs micrographic surgery: analysis of 101 cases. An Bras Dermatol. 2021;96:408-415. doi:10.1016/j.abd.2020.12.008
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560. doi:10.1097/DAD.0000000000001083
- Jacyk WK, Rütten A, Requena L. Fibrous papule of the face with granular cells. Dermatology. 2008;216:56-59. doi:10.1159/000109359
- Macri A, Kwan E, Tanner LS. Cutaneous angiofibroma. StatPearls [Internet]. Updated July 19, 2024. Accessed December 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482470/
- Sardana K, Chakravarty P, Goel K. Optimal management of common acquired melanocytic nevi (moles): current perspectives. Clin Cosmet Investig Dermatol. 2014;7:89-103. doi:10.2147/CCID.S57782
- Conforti C, Giuffrida R, Agozzino M, et al. Basal cell carcinoma and dermal nevi of the face: comparison of localization and dermatoscopic features. Int J Dermatol. 2021;60:996-1002. doi:10.1111/ijd.15554
- Alves RV, Brandão FH, Aquino JE, et al. Intradermal melanocytic nevus of the external auditory canal. Braz J Otorhinolaryngol. 2005;71:104-106. doi: 10.1016/s1808-8694(15)31295-7
- Muradia I, Khunger N, Yadav AK. A clinical, dermoscopic, and histopathological analysis of common acquired melanocytic nevi in skin of color. J Clin Aesthet Dermatol. 2022;15:41-51.
- Sardana K, Chakravarty P, Goel K. Optimal management of common acquired melanocytic nevi (moles): current perspectives. Clin Cosmet Investig Dermatol. 2014;7:89-103. doi:10.2147/CCID.S57782
- Massara B, Sellami K, Graja S, et al. Trichofolliculoma: a case series. J Clin Aesthet Dermatol. 2023;16:41-43.
Dome-Shaped White Papules on the Earlobe
Dome-Shaped White Papules on the Earlobe
A 70-year-old man presented to the dermatology clinic for a routine full-body skin examination that revealed multiple asymptomatic, dome-shaped, white papules on the left posterior earlobe. The patient had a personal and family history of spontaneous pneumothorax and no history of cancer. A shave biopsy of one of the papules was performed.