User login
Recurrent Nodule on the First Toe
Recurrent Nodule on the First Toe
THE DIAGNOSIS: Hidradenocarcinoma
Both the original and recurrent lesions were interpreted as a chondroid syringoma, a benign adnexal tumor; however, the third biopsy of the lesion revealed a low-grade adnexal neoplasm with irregular nests of variably sized epithelial cells demonstrating mild nuclear atypia and low mitotic activity. Given the multiple recurrences, accelerated growth, and more aggressive histologic findings, the patient was referred to our clinic for surgical management.
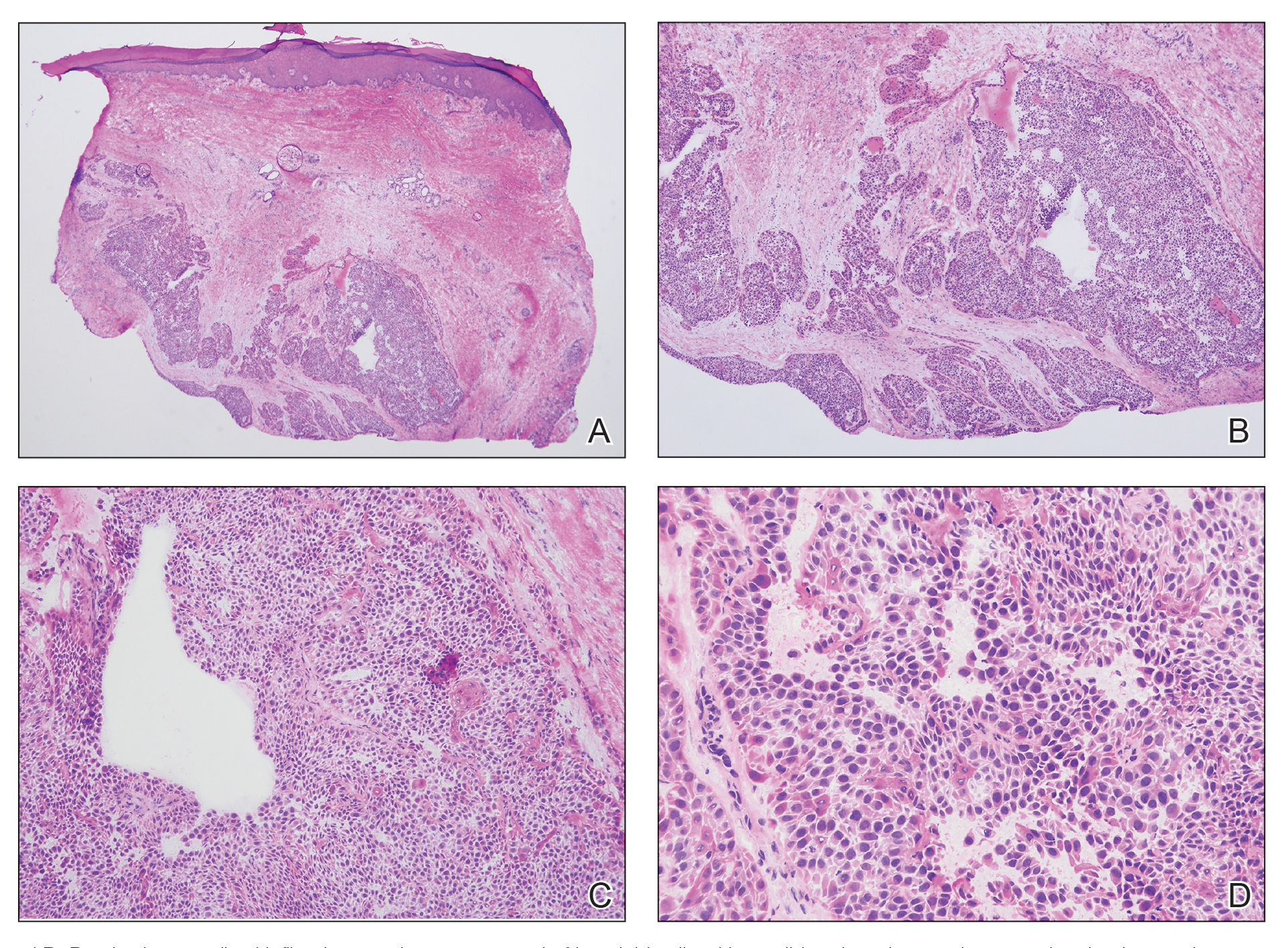
We elected to perform modified Mohs micrographic surgery (MMS) with permanent tissue sections to enable the application of immunohistochemical stains to fully characterize the tumor. Histopathology showed a poorly circumscribed infiltrative dermal neoplasm composed of basaloid cells with a solid and cystic growth pattern in a background of hyalinized, fibrotic stroma (Figure, A and B). There were focal clear cell and squamous features as well as focal ductal differentiation (Figure, C and D). No obvious papillary structures were noted. The tumor cells were positive for D2-40, and staining for CD31 failed to reveal lymphovascular invasion. Based on the infiltrative features in conjunction with the findings from the prior biopsies, a diagnosis of hidradenocarcinoma (HAC) was made. Deep and peripheral margins were cleared after 2 stages of MMS.
Initially described in 1954, HAC is an exceedingly rare adnexal tumor of apocrine and eccrine derivation.1 Historically, nomenclature for this entity has varied in the literature, including synonyms such as malignant nodular hidradenoma, malignant acrospiroma, solid-cystic adenocarcinoma, and malignant clear cell myoepithelioma.2,3 Approximately 6% of all malignant eccrine tumors worldwide are HACs, which account for only 1 in 13,000 dermatopathology specimens.1 These tumors may transform from clear cell hidradenomas (their benign counterparts) but more commonly arise de novo. Compared to benign hidradenomas, HACs are poorly circumscribed with infiltrative growth patterns on histopathology and may exhibit nuclear pleomorphism, prominent mitotic activity, necrosis, and perineural or vascular invasion.2
Clinically, HAC manifests as a 1- to 5-cm, solitary, firm, intradermal pink or violaceous nodule with possible ulceration.2,4 The nodule often is asymptomatic but may be tender, as in our patient. There seems to be no clear anatomic site of predilection, with approximately 42% of HACs localized to the head and neck and the remainder occurring on the trunk, arms, and legs.3,5-7 Females and males are affected equally, and lesions tend to arise in the seventh decade of life.7
Reports in the literature suggest that HAC is a very aggressive tumor with a generally poor prognosis.1 Several studies have found that up to half of tumors locally recur despite aggressive surgical management, and metastasis occurs in 20% to 60% of patients.3,8 However, a large study of US Surveillance, Epidemiology, and End Results data investigating the clinicopathologic characteristics of 289 patients with HAC revealed a more favorable prognosis.7 Mean overall survival and cancer-specific survival were greater than 13 years, and 10-year overall survival and cancer-specific survival rates were 60.2% and 90.5%, respectively.
Traditionally used to treat keratinocyte carcinomas, including basal cell carcinoma and squamous cell carcinoma, complete margin assessment with MMS is increasingly being utilized in the management of other cutaneous malignancies, including adnexal tumors.8 Due to its rarity, there remains no standard optimal treatment approach for HAC. One small retrospective study of 10 patients with HAC treated with MMS demonstrated favorable outcomes with no cases of recurrence, metastasis, or diseaserelated mortality in a mean 7-year follow-up period.9
Whole-body positron emission tomography/computed tomography performed in our patient approximately 1 month after MMS revealed mildly hypermetabolic left inguinal lymph nodes, which were thought to be reactive, and a question of small hypermetabolic foci in the liver. Follow-up computed tomography of the abdomen subsequently was performed and was negative for hepatic metastases. The patient will be monitored closely for local recurrence; however, the clearance of the tumor with MMS, which allowed complete margin assessment, is encouraging and supports MMS as superior to traditional surgical excision in the treatment of HAC. At his most recent examination 17 months after Mohs surgery, the patient remained tumor free.
Aggressive digital papillary adenocarcinoma (ADPA) is a rare malignant tumor originating in the sweat glands that can occur on the first toe but most commonly arises on the fingers. While both HAC and ADPA can manifest with an infiltrative growth pattern and cytologic atypia, ADPA classically reveals a well-circumscribed multinodular tumor in the dermis comprised of solid and cystic proliferation as well as papillary projections. In addition, ADPA has been described as having back-to-back glandular and ductal structures.10 Giant cell tumor of the tendon sheath is a benign fibrohistiocytic tumor that also typically manifests on the fingers but rarely can occur on the foot, including the first toe.11,12 This tumor is more common in women and most frequently affects individuals aged 30 to 50 years.12 Microscopically, giant cell tumor of the tendon sheath is characterized by a proliferation of osteoclastlike giant cells, epithelioid histiocytelike cells, mononuclear cells, and xanthomatous cells among collagenous bands.11
Osteosarcoma is an uncommon tumor of osteoidproducing cells that usually arises in the metaphysis of long bones and manifests as a tender subcutaneous mass. It has a bimodal age distribution, peaking in adolescents and adults older than 65 years.13 While very rare, osteosarcoma has been reported to occur in the bones of the feet, including the phalanges.14 Given the recurrent nature of our patient’s tumor, metastasis should always be considered; however, in his case, full-body imaging was negative for additional malignancy.
- Gauerke S, Driscoll JJ. Hidradenocarcinomas: a brief review and future directions. Arch Pathol Lab Med. 2010;134:781-785. doi:10.5858/134.5.781
- Ahn CS, Sangüeza OP. Malignant sweat gland tumors. Hematol Oncol Clin North Am. 2019;33:53-71. doi:10.1016/J.HOC.2018.09.002
- Ohta M, Hiramoto M, Fujii M, et al. Nodular hidradenocarcinoma on the scalp of a young woman: case report and review of literature. Dermatol Surg. 2004;30:1265-1268. doi:10.1111/J.1524-4725.2004.30390.X
- Souvatzidis P, Sbano P, Mandato F, et al. Malignant nodular hidradenoma of the skin: report of seven cases. J Eur Acad Dermatol Venereol. 2008;22:549-554. doi:10.1111/J.1468-3083.2007.02504.X
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach: case reports and review of the literature. Dermatolog Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Kazakov DV, Ivan D, Kutzner H, et al. Cutaneous hidradenocarcinoma: a clinicopathological, immunohistochemical, and molecular biologic study of 14 cases, including Her2/neu gene expression/ amplification, TP53 gene mutation analysis, and t(11;19) translocation. Am J Dermatopathol. 2009;31:236-247. doi:10.1097/DAD.0B013E3181984F10
- Gao T, Pan S, Li M, et al. Prognostic analysis of hidradenocarcinoma: a SEER-based observational study. Ann Med. 2022;54:454-463. doi:10 .1080/07853890.2022.2032313
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207. doi:10.1097/DSS.0000000000001167
- Tolkachjov SN, Hocker TL, Hochwalt PC, et al. Mohs micrographic surgery for the treatment of hidradenocarcinoma: the mayo clinic experience from 1993 to 2013. Dermatolog Surg. 2015;41:226-231. doi:10.1097/DSS.0000000000000242
- Weingertner N, Gressel A, Battistella M, et al. Aggressive digital papillary adenocarcinoma: a clinicopathological study of 19 cases. J Am Acad Dermatol. 2017;77:549-558.e1. doi:10.1016/J.JAAD.2017.02.028
- Paral KM, Petronic-Rosic V. Acral manifestations of soft tissue tumors. Clin Dermatol. 2017;35:85-98. doi:10.1016/J.CLINDER MATOL.2016.09.012
- Kondo RN, Crespigio J, Pavezzi PD, et al. Giant cell tumors of the tendon sheath in the left hallux. An Bras Dermatol. 2016;91:704-705. doi:10.1590/ABD1806-4841.20165769
- Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13. doi:10.1007/978-1-4419-0284-9_1
- Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462:109- 120. doi:10.1007/S00428-012-1339-3
THE DIAGNOSIS: Hidradenocarcinoma
Both the original and recurrent lesions were interpreted as a chondroid syringoma, a benign adnexal tumor; however, the third biopsy of the lesion revealed a low-grade adnexal neoplasm with irregular nests of variably sized epithelial cells demonstrating mild nuclear atypia and low mitotic activity. Given the multiple recurrences, accelerated growth, and more aggressive histologic findings, the patient was referred to our clinic for surgical management.
We elected to perform modified Mohs micrographic surgery (MMS) with permanent tissue sections to enable the application of immunohistochemical stains to fully characterize the tumor. Histopathology showed a poorly circumscribed infiltrative dermal neoplasm composed of basaloid cells with a solid and cystic growth pattern in a background of hyalinized, fibrotic stroma (Figure, A and B). There were focal clear cell and squamous features as well as focal ductal differentiation (Figure, C and D). No obvious papillary structures were noted. The tumor cells were positive for D2-40, and staining for CD31 failed to reveal lymphovascular invasion. Based on the infiltrative features in conjunction with the findings from the prior biopsies, a diagnosis of hidradenocarcinoma (HAC) was made. Deep and peripheral margins were cleared after 2 stages of MMS.
Initially described in 1954, HAC is an exceedingly rare adnexal tumor of apocrine and eccrine derivation.1 Historically, nomenclature for this entity has varied in the literature, including synonyms such as malignant nodular hidradenoma, malignant acrospiroma, solid-cystic adenocarcinoma, and malignant clear cell myoepithelioma.2,3 Approximately 6% of all malignant eccrine tumors worldwide are HACs, which account for only 1 in 13,000 dermatopathology specimens.1 These tumors may transform from clear cell hidradenomas (their benign counterparts) but more commonly arise de novo. Compared to benign hidradenomas, HACs are poorly circumscribed with infiltrative growth patterns on histopathology and may exhibit nuclear pleomorphism, prominent mitotic activity, necrosis, and perineural or vascular invasion.2
Clinically, HAC manifests as a 1- to 5-cm, solitary, firm, intradermal pink or violaceous nodule with possible ulceration.2,4 The nodule often is asymptomatic but may be tender, as in our patient. There seems to be no clear anatomic site of predilection, with approximately 42% of HACs localized to the head and neck and the remainder occurring on the trunk, arms, and legs.3,5-7 Females and males are affected equally, and lesions tend to arise in the seventh decade of life.7
Reports in the literature suggest that HAC is a very aggressive tumor with a generally poor prognosis.1 Several studies have found that up to half of tumors locally recur despite aggressive surgical management, and metastasis occurs in 20% to 60% of patients.3,8 However, a large study of US Surveillance, Epidemiology, and End Results data investigating the clinicopathologic characteristics of 289 patients with HAC revealed a more favorable prognosis.7 Mean overall survival and cancer-specific survival were greater than 13 years, and 10-year overall survival and cancer-specific survival rates were 60.2% and 90.5%, respectively.
Traditionally used to treat keratinocyte carcinomas, including basal cell carcinoma and squamous cell carcinoma, complete margin assessment with MMS is increasingly being utilized in the management of other cutaneous malignancies, including adnexal tumors.8 Due to its rarity, there remains no standard optimal treatment approach for HAC. One small retrospective study of 10 patients with HAC treated with MMS demonstrated favorable outcomes with no cases of recurrence, metastasis, or diseaserelated mortality in a mean 7-year follow-up period.9
Whole-body positron emission tomography/computed tomography performed in our patient approximately 1 month after MMS revealed mildly hypermetabolic left inguinal lymph nodes, which were thought to be reactive, and a question of small hypermetabolic foci in the liver. Follow-up computed tomography of the abdomen subsequently was performed and was negative for hepatic metastases. The patient will be monitored closely for local recurrence; however, the clearance of the tumor with MMS, which allowed complete margin assessment, is encouraging and supports MMS as superior to traditional surgical excision in the treatment of HAC. At his most recent examination 17 months after Mohs surgery, the patient remained tumor free.
Aggressive digital papillary adenocarcinoma (ADPA) is a rare malignant tumor originating in the sweat glands that can occur on the first toe but most commonly arises on the fingers. While both HAC and ADPA can manifest with an infiltrative growth pattern and cytologic atypia, ADPA classically reveals a well-circumscribed multinodular tumor in the dermis comprised of solid and cystic proliferation as well as papillary projections. In addition, ADPA has been described as having back-to-back glandular and ductal structures.10 Giant cell tumor of the tendon sheath is a benign fibrohistiocytic tumor that also typically manifests on the fingers but rarely can occur on the foot, including the first toe.11,12 This tumor is more common in women and most frequently affects individuals aged 30 to 50 years.12 Microscopically, giant cell tumor of the tendon sheath is characterized by a proliferation of osteoclastlike giant cells, epithelioid histiocytelike cells, mononuclear cells, and xanthomatous cells among collagenous bands.11
Osteosarcoma is an uncommon tumor of osteoidproducing cells that usually arises in the metaphysis of long bones and manifests as a tender subcutaneous mass. It has a bimodal age distribution, peaking in adolescents and adults older than 65 years.13 While very rare, osteosarcoma has been reported to occur in the bones of the feet, including the phalanges.14 Given the recurrent nature of our patient’s tumor, metastasis should always be considered; however, in his case, full-body imaging was negative for additional malignancy.
THE DIAGNOSIS: Hidradenocarcinoma
Both the original and recurrent lesions were interpreted as a chondroid syringoma, a benign adnexal tumor; however, the third biopsy of the lesion revealed a low-grade adnexal neoplasm with irregular nests of variably sized epithelial cells demonstrating mild nuclear atypia and low mitotic activity. Given the multiple recurrences, accelerated growth, and more aggressive histologic findings, the patient was referred to our clinic for surgical management.
We elected to perform modified Mohs micrographic surgery (MMS) with permanent tissue sections to enable the application of immunohistochemical stains to fully characterize the tumor. Histopathology showed a poorly circumscribed infiltrative dermal neoplasm composed of basaloid cells with a solid and cystic growth pattern in a background of hyalinized, fibrotic stroma (Figure, A and B). There were focal clear cell and squamous features as well as focal ductal differentiation (Figure, C and D). No obvious papillary structures were noted. The tumor cells were positive for D2-40, and staining for CD31 failed to reveal lymphovascular invasion. Based on the infiltrative features in conjunction with the findings from the prior biopsies, a diagnosis of hidradenocarcinoma (HAC) was made. Deep and peripheral margins were cleared after 2 stages of MMS.
Initially described in 1954, HAC is an exceedingly rare adnexal tumor of apocrine and eccrine derivation.1 Historically, nomenclature for this entity has varied in the literature, including synonyms such as malignant nodular hidradenoma, malignant acrospiroma, solid-cystic adenocarcinoma, and malignant clear cell myoepithelioma.2,3 Approximately 6% of all malignant eccrine tumors worldwide are HACs, which account for only 1 in 13,000 dermatopathology specimens.1 These tumors may transform from clear cell hidradenomas (their benign counterparts) but more commonly arise de novo. Compared to benign hidradenomas, HACs are poorly circumscribed with infiltrative growth patterns on histopathology and may exhibit nuclear pleomorphism, prominent mitotic activity, necrosis, and perineural or vascular invasion.2
Clinically, HAC manifests as a 1- to 5-cm, solitary, firm, intradermal pink or violaceous nodule with possible ulceration.2,4 The nodule often is asymptomatic but may be tender, as in our patient. There seems to be no clear anatomic site of predilection, with approximately 42% of HACs localized to the head and neck and the remainder occurring on the trunk, arms, and legs.3,5-7 Females and males are affected equally, and lesions tend to arise in the seventh decade of life.7
Reports in the literature suggest that HAC is a very aggressive tumor with a generally poor prognosis.1 Several studies have found that up to half of tumors locally recur despite aggressive surgical management, and metastasis occurs in 20% to 60% of patients.3,8 However, a large study of US Surveillance, Epidemiology, and End Results data investigating the clinicopathologic characteristics of 289 patients with HAC revealed a more favorable prognosis.7 Mean overall survival and cancer-specific survival were greater than 13 years, and 10-year overall survival and cancer-specific survival rates were 60.2% and 90.5%, respectively.
Traditionally used to treat keratinocyte carcinomas, including basal cell carcinoma and squamous cell carcinoma, complete margin assessment with MMS is increasingly being utilized in the management of other cutaneous malignancies, including adnexal tumors.8 Due to its rarity, there remains no standard optimal treatment approach for HAC. One small retrospective study of 10 patients with HAC treated with MMS demonstrated favorable outcomes with no cases of recurrence, metastasis, or diseaserelated mortality in a mean 7-year follow-up period.9
Whole-body positron emission tomography/computed tomography performed in our patient approximately 1 month after MMS revealed mildly hypermetabolic left inguinal lymph nodes, which were thought to be reactive, and a question of small hypermetabolic foci in the liver. Follow-up computed tomography of the abdomen subsequently was performed and was negative for hepatic metastases. The patient will be monitored closely for local recurrence; however, the clearance of the tumor with MMS, which allowed complete margin assessment, is encouraging and supports MMS as superior to traditional surgical excision in the treatment of HAC. At his most recent examination 17 months after Mohs surgery, the patient remained tumor free.
Aggressive digital papillary adenocarcinoma (ADPA) is a rare malignant tumor originating in the sweat glands that can occur on the first toe but most commonly arises on the fingers. While both HAC and ADPA can manifest with an infiltrative growth pattern and cytologic atypia, ADPA classically reveals a well-circumscribed multinodular tumor in the dermis comprised of solid and cystic proliferation as well as papillary projections. In addition, ADPA has been described as having back-to-back glandular and ductal structures.10 Giant cell tumor of the tendon sheath is a benign fibrohistiocytic tumor that also typically manifests on the fingers but rarely can occur on the foot, including the first toe.11,12 This tumor is more common in women and most frequently affects individuals aged 30 to 50 years.12 Microscopically, giant cell tumor of the tendon sheath is characterized by a proliferation of osteoclastlike giant cells, epithelioid histiocytelike cells, mononuclear cells, and xanthomatous cells among collagenous bands.11
Osteosarcoma is an uncommon tumor of osteoidproducing cells that usually arises in the metaphysis of long bones and manifests as a tender subcutaneous mass. It has a bimodal age distribution, peaking in adolescents and adults older than 65 years.13 While very rare, osteosarcoma has been reported to occur in the bones of the feet, including the phalanges.14 Given the recurrent nature of our patient’s tumor, metastasis should always be considered; however, in his case, full-body imaging was negative for additional malignancy.
- Gauerke S, Driscoll JJ. Hidradenocarcinomas: a brief review and future directions. Arch Pathol Lab Med. 2010;134:781-785. doi:10.5858/134.5.781
- Ahn CS, Sangüeza OP. Malignant sweat gland tumors. Hematol Oncol Clin North Am. 2019;33:53-71. doi:10.1016/J.HOC.2018.09.002
- Ohta M, Hiramoto M, Fujii M, et al. Nodular hidradenocarcinoma on the scalp of a young woman: case report and review of literature. Dermatol Surg. 2004;30:1265-1268. doi:10.1111/J.1524-4725.2004.30390.X
- Souvatzidis P, Sbano P, Mandato F, et al. Malignant nodular hidradenoma of the skin: report of seven cases. J Eur Acad Dermatol Venereol. 2008;22:549-554. doi:10.1111/J.1468-3083.2007.02504.X
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach: case reports and review of the literature. Dermatolog Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Kazakov DV, Ivan D, Kutzner H, et al. Cutaneous hidradenocarcinoma: a clinicopathological, immunohistochemical, and molecular biologic study of 14 cases, including Her2/neu gene expression/ amplification, TP53 gene mutation analysis, and t(11;19) translocation. Am J Dermatopathol. 2009;31:236-247. doi:10.1097/DAD.0B013E3181984F10
- Gao T, Pan S, Li M, et al. Prognostic analysis of hidradenocarcinoma: a SEER-based observational study. Ann Med. 2022;54:454-463. doi:10 .1080/07853890.2022.2032313
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207. doi:10.1097/DSS.0000000000001167
- Tolkachjov SN, Hocker TL, Hochwalt PC, et al. Mohs micrographic surgery for the treatment of hidradenocarcinoma: the mayo clinic experience from 1993 to 2013. Dermatolog Surg. 2015;41:226-231. doi:10.1097/DSS.0000000000000242
- Weingertner N, Gressel A, Battistella M, et al. Aggressive digital papillary adenocarcinoma: a clinicopathological study of 19 cases. J Am Acad Dermatol. 2017;77:549-558.e1. doi:10.1016/J.JAAD.2017.02.028
- Paral KM, Petronic-Rosic V. Acral manifestations of soft tissue tumors. Clin Dermatol. 2017;35:85-98. doi:10.1016/J.CLINDER MATOL.2016.09.012
- Kondo RN, Crespigio J, Pavezzi PD, et al. Giant cell tumors of the tendon sheath in the left hallux. An Bras Dermatol. 2016;91:704-705. doi:10.1590/ABD1806-4841.20165769
- Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13. doi:10.1007/978-1-4419-0284-9_1
- Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462:109- 120. doi:10.1007/S00428-012-1339-3
- Gauerke S, Driscoll JJ. Hidradenocarcinomas: a brief review and future directions. Arch Pathol Lab Med. 2010;134:781-785. doi:10.5858/134.5.781
- Ahn CS, Sangüeza OP. Malignant sweat gland tumors. Hematol Oncol Clin North Am. 2019;33:53-71. doi:10.1016/J.HOC.2018.09.002
- Ohta M, Hiramoto M, Fujii M, et al. Nodular hidradenocarcinoma on the scalp of a young woman: case report and review of literature. Dermatol Surg. 2004;30:1265-1268. doi:10.1111/J.1524-4725.2004.30390.X
- Souvatzidis P, Sbano P, Mandato F, et al. Malignant nodular hidradenoma of the skin: report of seven cases. J Eur Acad Dermatol Venereol. 2008;22:549-554. doi:10.1111/J.1468-3083.2007.02504.X
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach: case reports and review of the literature. Dermatolog Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Kazakov DV, Ivan D, Kutzner H, et al. Cutaneous hidradenocarcinoma: a clinicopathological, immunohistochemical, and molecular biologic study of 14 cases, including Her2/neu gene expression/ amplification, TP53 gene mutation analysis, and t(11;19) translocation. Am J Dermatopathol. 2009;31:236-247. doi:10.1097/DAD.0B013E3181984F10
- Gao T, Pan S, Li M, et al. Prognostic analysis of hidradenocarcinoma: a SEER-based observational study. Ann Med. 2022;54:454-463. doi:10 .1080/07853890.2022.2032313
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207. doi:10.1097/DSS.0000000000001167
- Tolkachjov SN, Hocker TL, Hochwalt PC, et al. Mohs micrographic surgery for the treatment of hidradenocarcinoma: the mayo clinic experience from 1993 to 2013. Dermatolog Surg. 2015;41:226-231. doi:10.1097/DSS.0000000000000242
- Weingertner N, Gressel A, Battistella M, et al. Aggressive digital papillary adenocarcinoma: a clinicopathological study of 19 cases. J Am Acad Dermatol. 2017;77:549-558.e1. doi:10.1016/J.JAAD.2017.02.028
- Paral KM, Petronic-Rosic V. Acral manifestations of soft tissue tumors. Clin Dermatol. 2017;35:85-98. doi:10.1016/J.CLINDER MATOL.2016.09.012
- Kondo RN, Crespigio J, Pavezzi PD, et al. Giant cell tumors of the tendon sheath in the left hallux. An Bras Dermatol. 2016;91:704-705. doi:10.1590/ABD1806-4841.20165769
- Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13. doi:10.1007/978-1-4419-0284-9_1
- Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462:109- 120. doi:10.1007/S00428-012-1339-3
Recurrent Nodule on the First Toe
Recurrent Nodule on the First Toe
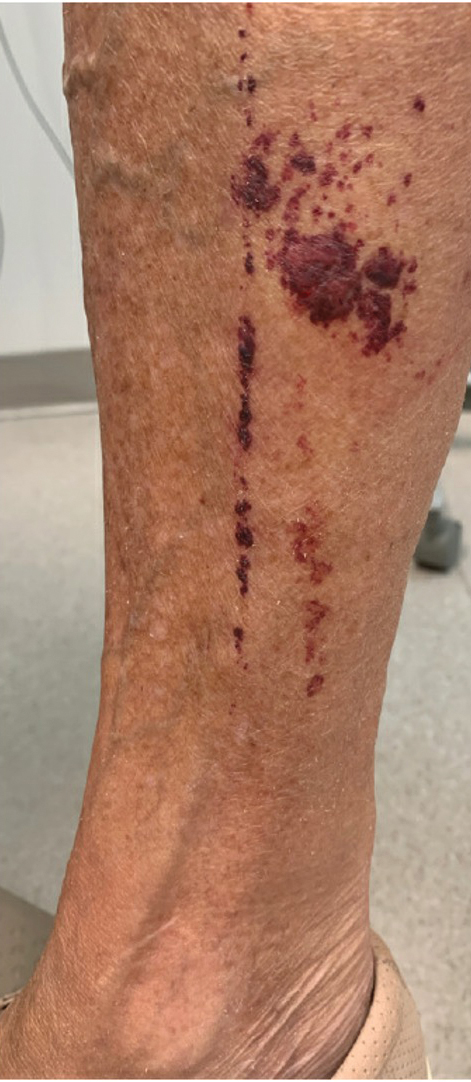
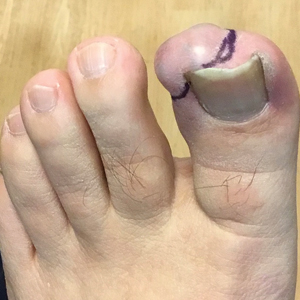
A 56-year-old man was referred to the dermatology clinic for treatment of a recurrent nodule on the left first toe. The lesion first appeared 12 years prior and was resected by an outside dermatologist, who diagnosed the lesion as benign based on biopsy results. Approximately 10 years later, the lesion began to grow back with a similar appearance to the original nodule; it again was diagnosed as benign based on another biopsy and excised by the outside dermatologist. Two years later, the patient had a second recurrence of the lesion, which was excised by his dermatologist. The biopsy report at that time identified the lesion as a low-grade adnexal neoplasm. The patient had a rapid recurrence of the tumor after 6 months and was referred to our clinic for Mohs micrographic surgery. Physical examination revealed a tender, 2.5×1.8-cm, firm, exophytic, subcutaneous nodule on the left first toe with no associated lymphadenopathy.
Painful Oral, Groin, and Scalp Lesions in a Young Man
Painful Oral, Groin, and Scalp Lesions in a Young Man
THE DIAGNOSIS: Pemphigus Vegetans
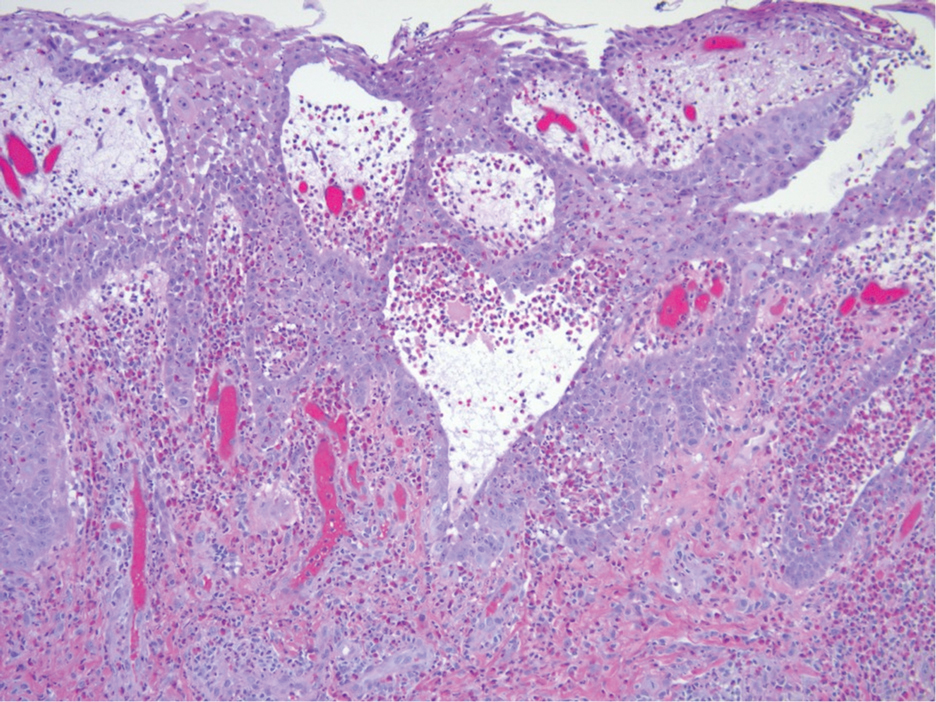
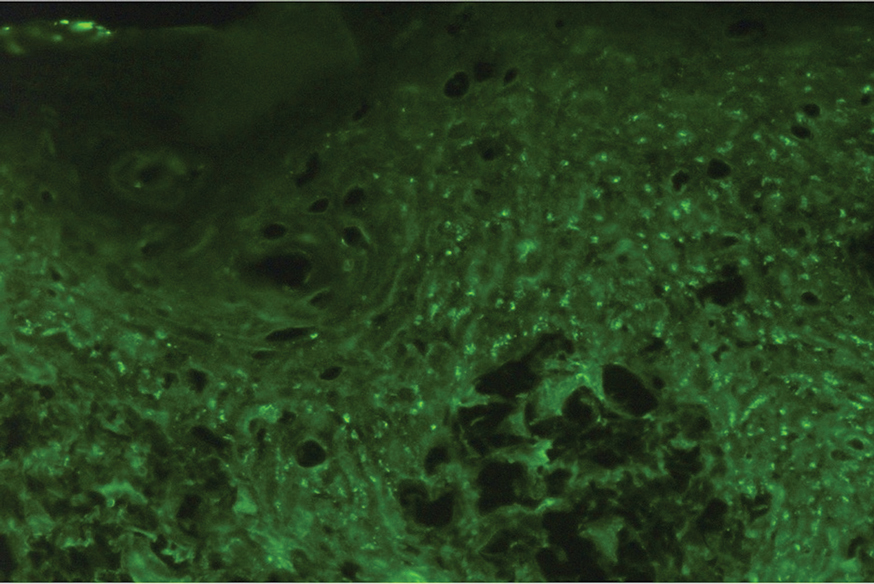
Histopathologic examination of the biopsies from the scalp and left anterior thigh revealed suprabasal clefting with acantholytic cells extending into the follicular infundibulum with eosinophilic pustules within the epidermis. The dermis contained perivascular lymphohistiocytic and eosinophilic inflammatory infiltrates without viral cytopathic effects (Figure 1). Direct immunofluorescence revealed strong IgG and moderate IgA pericellular deposition around keratinocyte cytoplasms (Figure 2). Serologic evaluation demonstrated anti–desmoglein 3 antibodies. Based on the clinical presentation and histopathologic correlation, a diagnosis of pemphigus vegetans was made.
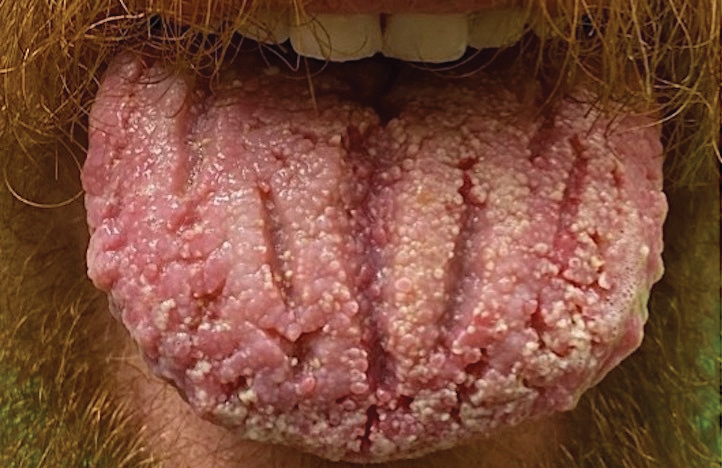
Pemphigus vegetans is a vesiculobullous autoimmune disease that is similar to pemphigus vulgaris but is characterized by the formation of vegetative plaques along the intertriginous areas and on the oral mucosa.1 It is the rarest variant of all pemphigus subtypes and was first described by Neumann in 1876.2 There are 2 subtypes of this variant: Hallopeau and Neumann, each with unique characteristics and physical manifestations. The Hallopeau type initially manifests with pustular lesions that rupture and evolve into erosions that commonly become infected. Gradually they merge and multiply to become more painful and vegetative.3 It has a more indolent course and typically responds well to treatment, and prolonged remission can be reached.4 The Neumann type is more severe and manifests with large vesiculobullous and erosive lesions that rupture and ulcerate, forming verrucous crusted vegetative plaques over the erosions.5 The erosions along the edge of the lesions induce new vegetation, becoming dry, hyperkeratotic, and fissured.3 The Neumann type often requires higher-dose steroids and typically is resistant to treatment.4 Patients can present with oral stomatitis and occasionally can develop a fissured or cerebriform appearance of the tongue, as seen in our patient (Figure 3).1,2 Nail changes include onychorrhexis, onychomadesis, subungual pustules, and ultimately nail atrophy.5
Pemphigus diseases are characterized by IgG autoantibodies against desmoglein 3 and/or desmoglein 1. These are components of desmosomes that are responsible for keratinocyte adhesion, disruption of which results in the blister formation seen in pemphigus subtypes. The unique physical manifestation of pemphigus vegetans is thought to be due not only to autoantibodies against desmogleins 1 and 3 but also to autoantibodies against desmocollin 1 and 2.1
Histopathologic examination reveals hyperkeratosis and pseudoepitheliomatous hyperplasia with acantholysis that creates a suprabasal cleft. Basal cells remain intact to the basement membrane by hemidesmosomes, resulting in a tombstone appearance. The Hallopeau type typically manifests with a large eosinophilic inflammatory response, leading to eosinophilic spongiosis and intraepidermal microabscesses. The Neumann type manifests with more of a neutrophilic and lymphocytic infiltrate, accompanied by the eosinophilic response.1 For evaluation, obtain histopathology as well as direct immunofluorescence or enzyme-linked immunosorbent assay to look for intracellular deposition of desmoglein autoantibodies.
First-line treatment for pemphigus vulgaris and its variants is rituximab, an anti-CD20 monoclonal antibody. It has also been shown to have therapeutic benefit with combination of corticosteroids and rituximab. Corticosteroids should be given at a dose of 1 mg/kg daily for 2 to 4 weeks. Other immunosuppressive agents (steroid sparing) include azathioprine, dapsone, mycophenolate mofetil, methotrexate, cyclophosphamide, cyclosporine, and intravenous immunoglobulin. Pulse therapy with intermittent intravenous corticosteroids and immunosuppressants is another second-line therapeutic option. Topical therapeutic options include steroids, tacrolimus, and nicotinamide with oral tetracycline at onset and relapse. The goal of therapy is to maintain remission for 1 year then slowly taper treatment over another year.1
Our patient initially was treated with prednisone, and subsequent courses of azathioprine and mycophenolate mofetil failed. He then was treated with 2 infusions of rituximab that were given 2 weeks apart. He was able to taper off the prednisone 1 month after the last infusion with complete remission of disease. He has been disease free for more than 9 months postinfusion.
Differential diagnoses for pemphigus vegetans can include bullous pemphigoid, bullous systemic lupus erythematosus, dermatitis herpetiformis, and pemphigus vulgaris. Lesion characteristics are key to differentiating pemphigus vegetans from other autoimmune blistering disorders. Bullous pemphigoid will manifest with tense blisters where pemphigus vulgaris will be flaccid; this is due to the difference in autoantibody targets between the conditions. Diagnosis depends on clinical presentation and histopathologic findings.
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed December 16, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Rebello MS, Ramesh BM, Sukumar D, et al. Cerebriform cutaneous lesions in pemphigus vegetans. Indian J Dermatol. 2016;61:206-208.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Ajbani AA, Mehta KS, Marfatia YS. Verrucous lesions over external genitalia as a presenting feature of pemphigus vegetans. Indian J Sex Transm Dis AIDS. 2019;40:176-179.
- Vinay K, De D, Handa S, et al. Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol. 2016;41:316-317.
THE DIAGNOSIS: Pemphigus Vegetans
Histopathologic examination of the biopsies from the scalp and left anterior thigh revealed suprabasal clefting with acantholytic cells extending into the follicular infundibulum with eosinophilic pustules within the epidermis. The dermis contained perivascular lymphohistiocytic and eosinophilic inflammatory infiltrates without viral cytopathic effects (Figure 1). Direct immunofluorescence revealed strong IgG and moderate IgA pericellular deposition around keratinocyte cytoplasms (Figure 2). Serologic evaluation demonstrated anti–desmoglein 3 antibodies. Based on the clinical presentation and histopathologic correlation, a diagnosis of pemphigus vegetans was made.
Pemphigus vegetans is a vesiculobullous autoimmune disease that is similar to pemphigus vulgaris but is characterized by the formation of vegetative plaques along the intertriginous areas and on the oral mucosa.1 It is the rarest variant of all pemphigus subtypes and was first described by Neumann in 1876.2 There are 2 subtypes of this variant: Hallopeau and Neumann, each with unique characteristics and physical manifestations. The Hallopeau type initially manifests with pustular lesions that rupture and evolve into erosions that commonly become infected. Gradually they merge and multiply to become more painful and vegetative.3 It has a more indolent course and typically responds well to treatment, and prolonged remission can be reached.4 The Neumann type is more severe and manifests with large vesiculobullous and erosive lesions that rupture and ulcerate, forming verrucous crusted vegetative plaques over the erosions.5 The erosions along the edge of the lesions induce new vegetation, becoming dry, hyperkeratotic, and fissured.3 The Neumann type often requires higher-dose steroids and typically is resistant to treatment.4 Patients can present with oral stomatitis and occasionally can develop a fissured or cerebriform appearance of the tongue, as seen in our patient (Figure 3).1,2 Nail changes include onychorrhexis, onychomadesis, subungual pustules, and ultimately nail atrophy.5
Pemphigus diseases are characterized by IgG autoantibodies against desmoglein 3 and/or desmoglein 1. These are components of desmosomes that are responsible for keratinocyte adhesion, disruption of which results in the blister formation seen in pemphigus subtypes. The unique physical manifestation of pemphigus vegetans is thought to be due not only to autoantibodies against desmogleins 1 and 3 but also to autoantibodies against desmocollin 1 and 2.1
Histopathologic examination reveals hyperkeratosis and pseudoepitheliomatous hyperplasia with acantholysis that creates a suprabasal cleft. Basal cells remain intact to the basement membrane by hemidesmosomes, resulting in a tombstone appearance. The Hallopeau type typically manifests with a large eosinophilic inflammatory response, leading to eosinophilic spongiosis and intraepidermal microabscesses. The Neumann type manifests with more of a neutrophilic and lymphocytic infiltrate, accompanied by the eosinophilic response.1 For evaluation, obtain histopathology as well as direct immunofluorescence or enzyme-linked immunosorbent assay to look for intracellular deposition of desmoglein autoantibodies.
First-line treatment for pemphigus vulgaris and its variants is rituximab, an anti-CD20 monoclonal antibody. It has also been shown to have therapeutic benefit with combination of corticosteroids and rituximab. Corticosteroids should be given at a dose of 1 mg/kg daily for 2 to 4 weeks. Other immunosuppressive agents (steroid sparing) include azathioprine, dapsone, mycophenolate mofetil, methotrexate, cyclophosphamide, cyclosporine, and intravenous immunoglobulin. Pulse therapy with intermittent intravenous corticosteroids and immunosuppressants is another second-line therapeutic option. Topical therapeutic options include steroids, tacrolimus, and nicotinamide with oral tetracycline at onset and relapse. The goal of therapy is to maintain remission for 1 year then slowly taper treatment over another year.1
Our patient initially was treated with prednisone, and subsequent courses of azathioprine and mycophenolate mofetil failed. He then was treated with 2 infusions of rituximab that were given 2 weeks apart. He was able to taper off the prednisone 1 month after the last infusion with complete remission of disease. He has been disease free for more than 9 months postinfusion.
Differential diagnoses for pemphigus vegetans can include bullous pemphigoid, bullous systemic lupus erythematosus, dermatitis herpetiformis, and pemphigus vulgaris. Lesion characteristics are key to differentiating pemphigus vegetans from other autoimmune blistering disorders. Bullous pemphigoid will manifest with tense blisters where pemphigus vulgaris will be flaccid; this is due to the difference in autoantibody targets between the conditions. Diagnosis depends on clinical presentation and histopathologic findings.
THE DIAGNOSIS: Pemphigus Vegetans
Histopathologic examination of the biopsies from the scalp and left anterior thigh revealed suprabasal clefting with acantholytic cells extending into the follicular infundibulum with eosinophilic pustules within the epidermis. The dermis contained perivascular lymphohistiocytic and eosinophilic inflammatory infiltrates without viral cytopathic effects (Figure 1). Direct immunofluorescence revealed strong IgG and moderate IgA pericellular deposition around keratinocyte cytoplasms (Figure 2). Serologic evaluation demonstrated anti–desmoglein 3 antibodies. Based on the clinical presentation and histopathologic correlation, a diagnosis of pemphigus vegetans was made.
Pemphigus vegetans is a vesiculobullous autoimmune disease that is similar to pemphigus vulgaris but is characterized by the formation of vegetative plaques along the intertriginous areas and on the oral mucosa.1 It is the rarest variant of all pemphigus subtypes and was first described by Neumann in 1876.2 There are 2 subtypes of this variant: Hallopeau and Neumann, each with unique characteristics and physical manifestations. The Hallopeau type initially manifests with pustular lesions that rupture and evolve into erosions that commonly become infected. Gradually they merge and multiply to become more painful and vegetative.3 It has a more indolent course and typically responds well to treatment, and prolonged remission can be reached.4 The Neumann type is more severe and manifests with large vesiculobullous and erosive lesions that rupture and ulcerate, forming verrucous crusted vegetative plaques over the erosions.5 The erosions along the edge of the lesions induce new vegetation, becoming dry, hyperkeratotic, and fissured.3 The Neumann type often requires higher-dose steroids and typically is resistant to treatment.4 Patients can present with oral stomatitis and occasionally can develop a fissured or cerebriform appearance of the tongue, as seen in our patient (Figure 3).1,2 Nail changes include onychorrhexis, onychomadesis, subungual pustules, and ultimately nail atrophy.5
Pemphigus diseases are characterized by IgG autoantibodies against desmoglein 3 and/or desmoglein 1. These are components of desmosomes that are responsible for keratinocyte adhesion, disruption of which results in the blister formation seen in pemphigus subtypes. The unique physical manifestation of pemphigus vegetans is thought to be due not only to autoantibodies against desmogleins 1 and 3 but also to autoantibodies against desmocollin 1 and 2.1
Histopathologic examination reveals hyperkeratosis and pseudoepitheliomatous hyperplasia with acantholysis that creates a suprabasal cleft. Basal cells remain intact to the basement membrane by hemidesmosomes, resulting in a tombstone appearance. The Hallopeau type typically manifests with a large eosinophilic inflammatory response, leading to eosinophilic spongiosis and intraepidermal microabscesses. The Neumann type manifests with more of a neutrophilic and lymphocytic infiltrate, accompanied by the eosinophilic response.1 For evaluation, obtain histopathology as well as direct immunofluorescence or enzyme-linked immunosorbent assay to look for intracellular deposition of desmoglein autoantibodies.
First-line treatment for pemphigus vulgaris and its variants is rituximab, an anti-CD20 monoclonal antibody. It has also been shown to have therapeutic benefit with combination of corticosteroids and rituximab. Corticosteroids should be given at a dose of 1 mg/kg daily for 2 to 4 weeks. Other immunosuppressive agents (steroid sparing) include azathioprine, dapsone, mycophenolate mofetil, methotrexate, cyclophosphamide, cyclosporine, and intravenous immunoglobulin. Pulse therapy with intermittent intravenous corticosteroids and immunosuppressants is another second-line therapeutic option. Topical therapeutic options include steroids, tacrolimus, and nicotinamide with oral tetracycline at onset and relapse. The goal of therapy is to maintain remission for 1 year then slowly taper treatment over another year.1
Our patient initially was treated with prednisone, and subsequent courses of azathioprine and mycophenolate mofetil failed. He then was treated with 2 infusions of rituximab that were given 2 weeks apart. He was able to taper off the prednisone 1 month after the last infusion with complete remission of disease. He has been disease free for more than 9 months postinfusion.
Differential diagnoses for pemphigus vegetans can include bullous pemphigoid, bullous systemic lupus erythematosus, dermatitis herpetiformis, and pemphigus vulgaris. Lesion characteristics are key to differentiating pemphigus vegetans from other autoimmune blistering disorders. Bullous pemphigoid will manifest with tense blisters where pemphigus vulgaris will be flaccid; this is due to the difference in autoantibody targets between the conditions. Diagnosis depends on clinical presentation and histopathologic findings.
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed December 16, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Rebello MS, Ramesh BM, Sukumar D, et al. Cerebriform cutaneous lesions in pemphigus vegetans. Indian J Dermatol. 2016;61:206-208.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Ajbani AA, Mehta KS, Marfatia YS. Verrucous lesions over external genitalia as a presenting feature of pemphigus vegetans. Indian J Sex Transm Dis AIDS. 2019;40:176-179.
- Vinay K, De D, Handa S, et al. Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol. 2016;41:316-317.
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls [Internet]. Updated June 26, 2023. Accessed December 16, 2024. https://www.ncbi.nlm.nih.gov/books/NBK545229/
- Rebello MS, Ramesh BM, Sukumar D, et al. Cerebriform cutaneous lesions in pemphigus vegetans. Indian J Dermatol. 2016;61:206-208.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Ajbani AA, Mehta KS, Marfatia YS. Verrucous lesions over external genitalia as a presenting feature of pemphigus vegetans. Indian J Sex Transm Dis AIDS. 2019;40:176-179.
- Vinay K, De D, Handa S, et al. Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol. 2016;41:316-317.
Painful Oral, Groin, and Scalp Lesions in a Young Man
Painful Oral, Groin, and Scalp Lesions in a Young Man
A 27-year-old man presented to the dermatology department with painful oral and groin lesions of 2 years’ duration as well as lip ulceration that had been present for 1 month. The patient also reported moderately tender scalp and face lesions that had been present for several weeks. The lip ulceration was previously treated by his primary care provider with valacyclovir (1 g daily for 2 weeks) without improvement. Six months prior to the current presentation, we treated the groin lesions as condyloma involving the perineum and genital region at our clinic with no response to cryotherapy, topical imiquimod, or extensive surgical excision with skin grafting. Pathology at the time showed condyloma but was negative for human papillomavirus. Physical examination at the current presentation revealed superficial erosions along the vermilion border. The oral mucosa exhibited cobblestoning, and fissures were present on the tongue. Eroded pink plaques studded with vesicles were present on the vertex scalp and left chin. The bilateral inguinal regions extending to anterior-lateral upper thighs and posterior buttocks revealed erythematous, arcuate, and annular erosive plaques with verrucous hyperkeratotic borders and fissuring on the leading edge. Pink erosive and verrucous erythematous plaques were noted on the penile shaft, scrotum, and perineum. Punch biopsies of the scalp and left anterior thigh as well as direct immunofluorescence were performed.

Demarcated Nonpruritic Lesions Following Antibiotic Therapy
Demarcated Nonpruritic Lesions Following Antibiotic Therapy
THE DIAGNOSIS: Fixed Drug Eruption
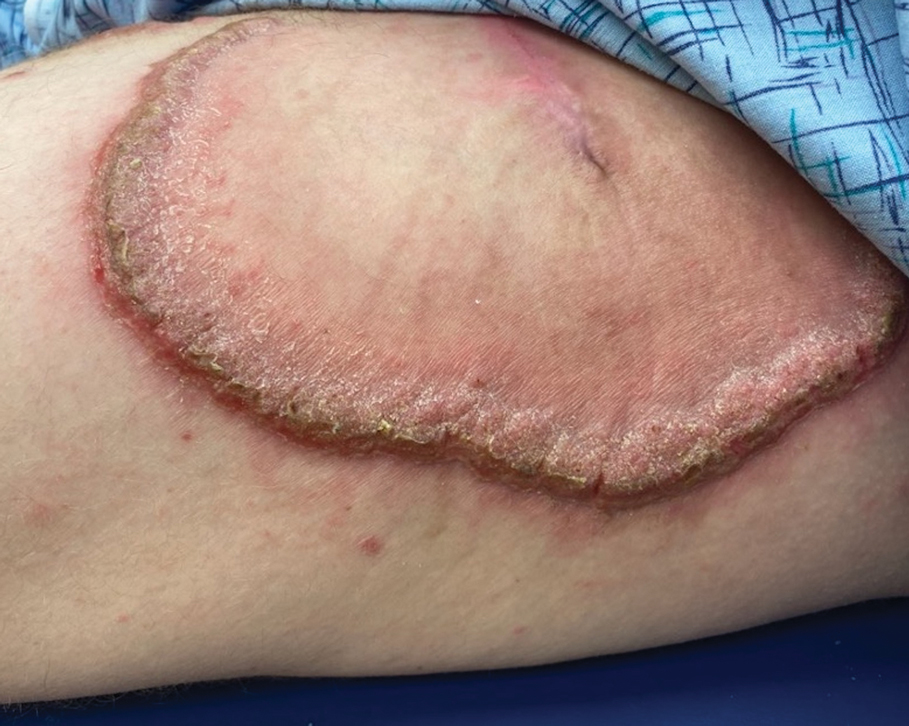
Based on the patient’s clinical presentation and history of similar eruptions, a diagnosis of levofloxacin-induced fixed drug eruption (FDE) was made. After cessation of the drug, the lesions resolved within 1 week without any residual postinflammatory hyperpigmentation.
Fixed drug eruption is an adverse cutaneous reaction characterized by the onset of a rash at a fixed location each time a specific medication is administered. Patients typically report a history of similar eruptions, often involving the upper and lower extremities, genital area, or mucous membranes. The most common causative agents vary, but retrospective analyses primarily implicate nonsteroidal anti-inflammatory drugs followed by antibiotics (eg, amoxicillin, levofloxacin, doxycycline) and antiepileptics.1,2
While FDE can be solitary or scattered, most patients have 5 or fewer lesions, with a mean interval of 48 hours from exposure to the causative agent to onset of the rash.1 The lesions can be differentiated by their typically solitary, well-demarcated, round or oval appearance; they also are erythematous to purple with a dusky center. The lesions may increase in size and number with each additional exposure to the offending medication.1,3 Postinflammatory hyperpigmentation may last for weeks to months after the acute inflammatory response has resolved.
The high risk for recurrence of FDE may be explained by the presence of tissue resident memory T (TRM) cells in the affected skin that evoke a characteristic clinical manifestation upon administration of a causative agent.2,3 Intraepidermal CD8+ TRM cells, which have an effectormemory phenotype, may contribute to the development of localized tissue damage; these cells demonstrate their effector function by the rapid increase in interferon gamma after challenge.2 Within 24 hours of administration of the offending medication, CD8+ TRM cells migrate upward in the epidermis, and their activity leads to the epidermal necrosis observed with FDE. The self-limiting nature of FDE can be explained by the action of CD4+ Foxp3+ regulatory T cells that migrate similarly and induce the production of IL-10, which limits the damage inflicted by the CD8+ T cells.1
Type I hypersensitivity reactions are IgE mediated; typically occur much more rapidly than FDE; and involve a raised urticarial rash, pruritus, and flushing. Urticaria is useful in identifying IgE-mediated reactions and mast cell degranulation. Previous exposure to the drug in question is required for diagnosis.4
Type IV delayed hypersensitivity reactions—including contact dermatitis and FDE—are mediated by T cells rather than IgE. These reactions occur at least 48 to 72 hours after drug exposure.4 Contact dermatitis follows exposure to an irritant but generally is limited to the site of contact and manifests with burning or stinging. Chronic contact dermatitis is characterized by erythema, scaling, and lichenification that may be associated with burning pain.
The target lesions of erythema multiforme are associated with the use of medications such as nonsteroidal anti-inflammatory drugs, antiepileptics, and antibiotics in fewer than 10% of cases. Infections are the predominant cause, with herpes simplex virus 1 being the most common etiology.5 Erythema multiforme lesions have 3 concentric segments: a dark red inflammatory zone surrounded by a pale ring of edema, both of which are surrounded by an erythematous halo. Lesions initially are distributed symmetrically on the extensor surfaces of the upper and lower extremities, but mucosal involvement may be present.5
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, involves fever and peripheral neutrophilia in addition to cutaneous erythematous eruptions and dermal neutrophilic infiltration on histopathology.6 Most cases are idiopathic but may occur in the setting of malignancy or drug administration. A major criterion for drug-induced Sweet syndrome is abrupt onset of painful erythematous plaques or nodules with pyrexia.6
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925. doi:10.3390/medicina57090925
- Tokura Y, Phadungsaksawasdi P, Kurihara K, et al. Pathophysiology of skin resident memory T cells. Front Immunol. 2021;11:618897. doi:10.3389/fimmu.2020.618897
- Mockenhaupt M. Bullous drug reactions. Acta Derm Venereol. 2020;100:adv00057. doi:10.2340/00015555-3408
- Böhm R, Proksch E, Schwarz T, et al. Drug hypersensitivity. Dtsch Arztebl Int. 2018;115:501-512. doi:10.3238/arztebl.2018.0501
- Trayes KP, Love G, Studdiford JS. Erythema multiforme: recognition and management. Am Fam Physician. 2019;100:82-88.
- Joshi TP, Friske SK, Hsiou DA, et al. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi:10.1007 /s40257-022-00673-4
THE DIAGNOSIS: Fixed Drug Eruption
Based on the patient’s clinical presentation and history of similar eruptions, a diagnosis of levofloxacin-induced fixed drug eruption (FDE) was made. After cessation of the drug, the lesions resolved within 1 week without any residual postinflammatory hyperpigmentation.
Fixed drug eruption is an adverse cutaneous reaction characterized by the onset of a rash at a fixed location each time a specific medication is administered. Patients typically report a history of similar eruptions, often involving the upper and lower extremities, genital area, or mucous membranes. The most common causative agents vary, but retrospective analyses primarily implicate nonsteroidal anti-inflammatory drugs followed by antibiotics (eg, amoxicillin, levofloxacin, doxycycline) and antiepileptics.1,2
While FDE can be solitary or scattered, most patients have 5 or fewer lesions, with a mean interval of 48 hours from exposure to the causative agent to onset of the rash.1 The lesions can be differentiated by their typically solitary, well-demarcated, round or oval appearance; they also are erythematous to purple with a dusky center. The lesions may increase in size and number with each additional exposure to the offending medication.1,3 Postinflammatory hyperpigmentation may last for weeks to months after the acute inflammatory response has resolved.
The high risk for recurrence of FDE may be explained by the presence of tissue resident memory T (TRM) cells in the affected skin that evoke a characteristic clinical manifestation upon administration of a causative agent.2,3 Intraepidermal CD8+ TRM cells, which have an effectormemory phenotype, may contribute to the development of localized tissue damage; these cells demonstrate their effector function by the rapid increase in interferon gamma after challenge.2 Within 24 hours of administration of the offending medication, CD8+ TRM cells migrate upward in the epidermis, and their activity leads to the epidermal necrosis observed with FDE. The self-limiting nature of FDE can be explained by the action of CD4+ Foxp3+ regulatory T cells that migrate similarly and induce the production of IL-10, which limits the damage inflicted by the CD8+ T cells.1
Type I hypersensitivity reactions are IgE mediated; typically occur much more rapidly than FDE; and involve a raised urticarial rash, pruritus, and flushing. Urticaria is useful in identifying IgE-mediated reactions and mast cell degranulation. Previous exposure to the drug in question is required for diagnosis.4
Type IV delayed hypersensitivity reactions—including contact dermatitis and FDE—are mediated by T cells rather than IgE. These reactions occur at least 48 to 72 hours after drug exposure.4 Contact dermatitis follows exposure to an irritant but generally is limited to the site of contact and manifests with burning or stinging. Chronic contact dermatitis is characterized by erythema, scaling, and lichenification that may be associated with burning pain.
The target lesions of erythema multiforme are associated with the use of medications such as nonsteroidal anti-inflammatory drugs, antiepileptics, and antibiotics in fewer than 10% of cases. Infections are the predominant cause, with herpes simplex virus 1 being the most common etiology.5 Erythema multiforme lesions have 3 concentric segments: a dark red inflammatory zone surrounded by a pale ring of edema, both of which are surrounded by an erythematous halo. Lesions initially are distributed symmetrically on the extensor surfaces of the upper and lower extremities, but mucosal involvement may be present.5
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, involves fever and peripheral neutrophilia in addition to cutaneous erythematous eruptions and dermal neutrophilic infiltration on histopathology.6 Most cases are idiopathic but may occur in the setting of malignancy or drug administration. A major criterion for drug-induced Sweet syndrome is abrupt onset of painful erythematous plaques or nodules with pyrexia.6
THE DIAGNOSIS: Fixed Drug Eruption
Based on the patient’s clinical presentation and history of similar eruptions, a diagnosis of levofloxacin-induced fixed drug eruption (FDE) was made. After cessation of the drug, the lesions resolved within 1 week without any residual postinflammatory hyperpigmentation.
Fixed drug eruption is an adverse cutaneous reaction characterized by the onset of a rash at a fixed location each time a specific medication is administered. Patients typically report a history of similar eruptions, often involving the upper and lower extremities, genital area, or mucous membranes. The most common causative agents vary, but retrospective analyses primarily implicate nonsteroidal anti-inflammatory drugs followed by antibiotics (eg, amoxicillin, levofloxacin, doxycycline) and antiepileptics.1,2
While FDE can be solitary or scattered, most patients have 5 or fewer lesions, with a mean interval of 48 hours from exposure to the causative agent to onset of the rash.1 The lesions can be differentiated by their typically solitary, well-demarcated, round or oval appearance; they also are erythematous to purple with a dusky center. The lesions may increase in size and number with each additional exposure to the offending medication.1,3 Postinflammatory hyperpigmentation may last for weeks to months after the acute inflammatory response has resolved.
The high risk for recurrence of FDE may be explained by the presence of tissue resident memory T (TRM) cells in the affected skin that evoke a characteristic clinical manifestation upon administration of a causative agent.2,3 Intraepidermal CD8+ TRM cells, which have an effectormemory phenotype, may contribute to the development of localized tissue damage; these cells demonstrate their effector function by the rapid increase in interferon gamma after challenge.2 Within 24 hours of administration of the offending medication, CD8+ TRM cells migrate upward in the epidermis, and their activity leads to the epidermal necrosis observed with FDE. The self-limiting nature of FDE can be explained by the action of CD4+ Foxp3+ regulatory T cells that migrate similarly and induce the production of IL-10, which limits the damage inflicted by the CD8+ T cells.1
Type I hypersensitivity reactions are IgE mediated; typically occur much more rapidly than FDE; and involve a raised urticarial rash, pruritus, and flushing. Urticaria is useful in identifying IgE-mediated reactions and mast cell degranulation. Previous exposure to the drug in question is required for diagnosis.4
Type IV delayed hypersensitivity reactions—including contact dermatitis and FDE—are mediated by T cells rather than IgE. These reactions occur at least 48 to 72 hours after drug exposure.4 Contact dermatitis follows exposure to an irritant but generally is limited to the site of contact and manifests with burning or stinging. Chronic contact dermatitis is characterized by erythema, scaling, and lichenification that may be associated with burning pain.
The target lesions of erythema multiforme are associated with the use of medications such as nonsteroidal anti-inflammatory drugs, antiepileptics, and antibiotics in fewer than 10% of cases. Infections are the predominant cause, with herpes simplex virus 1 being the most common etiology.5 Erythema multiforme lesions have 3 concentric segments: a dark red inflammatory zone surrounded by a pale ring of edema, both of which are surrounded by an erythematous halo. Lesions initially are distributed symmetrically on the extensor surfaces of the upper and lower extremities, but mucosal involvement may be present.5
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, involves fever and peripheral neutrophilia in addition to cutaneous erythematous eruptions and dermal neutrophilic infiltration on histopathology.6 Most cases are idiopathic but may occur in the setting of malignancy or drug administration. A major criterion for drug-induced Sweet syndrome is abrupt onset of painful erythematous plaques or nodules with pyrexia.6
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925. doi:10.3390/medicina57090925
- Tokura Y, Phadungsaksawasdi P, Kurihara K, et al. Pathophysiology of skin resident memory T cells. Front Immunol. 2021;11:618897. doi:10.3389/fimmu.2020.618897
- Mockenhaupt M. Bullous drug reactions. Acta Derm Venereol. 2020;100:adv00057. doi:10.2340/00015555-3408
- Böhm R, Proksch E, Schwarz T, et al. Drug hypersensitivity. Dtsch Arztebl Int. 2018;115:501-512. doi:10.3238/arztebl.2018.0501
- Trayes KP, Love G, Studdiford JS. Erythema multiforme: recognition and management. Am Fam Physician. 2019;100:82-88.
- Joshi TP, Friske SK, Hsiou DA, et al. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi:10.1007 /s40257-022-00673-4
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925. doi:10.3390/medicina57090925
- Tokura Y, Phadungsaksawasdi P, Kurihara K, et al. Pathophysiology of skin resident memory T cells. Front Immunol. 2021;11:618897. doi:10.3389/fimmu.2020.618897
- Mockenhaupt M. Bullous drug reactions. Acta Derm Venereol. 2020;100:adv00057. doi:10.2340/00015555-3408
- Böhm R, Proksch E, Schwarz T, et al. Drug hypersensitivity. Dtsch Arztebl Int. 2018;115:501-512. doi:10.3238/arztebl.2018.0501
- Trayes KP, Love G, Studdiford JS. Erythema multiforme: recognition and management. Am Fam Physician. 2019;100:82-88.
- Joshi TP, Friske SK, Hsiou DA, et al. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi:10.1007 /s40257-022-00673-4
Demarcated Nonpruritic Lesions Following Antibiotic Therapy
Demarcated Nonpruritic Lesions Following Antibiotic Therapy
A 35-year-old man was admitted to the hospital for treatment of cellulitis that required antibiotic therapy. Two days after administration of a single dose of intravenous levofloxacin, he developed demarcated nonpruritic and painless lesions on the abdomen (top) and right upper extremity (bottom). He was afebrile through the entire 1-week hospital course and denied use of any topical products prior to hospitalization. The patient reported a history of similar rashes associated with the use of levofloxacin.


Fluctuant Subcutaneous Nodule in the Axilla of an Adolescent Female
Fluctuant Subcutaneous Nodule in the Axilla of an Adolescent Female
The Diagnosis: Accessory Breast
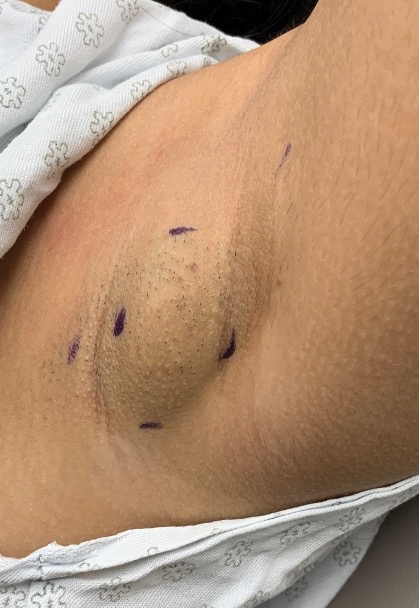
A diagnosis of accessory breast was confirmed on histopathology, which demonstrated a slightly hyperplastic and hyperpigmented epidermis. The dermis contained an increased number of smooth muscle bundles with the presence of apocrine glands and mammary lobules (Figure). Tenderness of the mass fluctuated according to the patient’s menstrual cycle, which supported a diagnosis of accessory breast over lipoma. The patient had no signs of infection or other systemic symptoms that were suggestive of lymphadenopathy. Unlike an epidermoid inclusion cyst, our patient’s mass presented as poorly defined and boggy in texture. Biopsy results were not consistent with malignancy, ruling out soft tissue sarcoma.
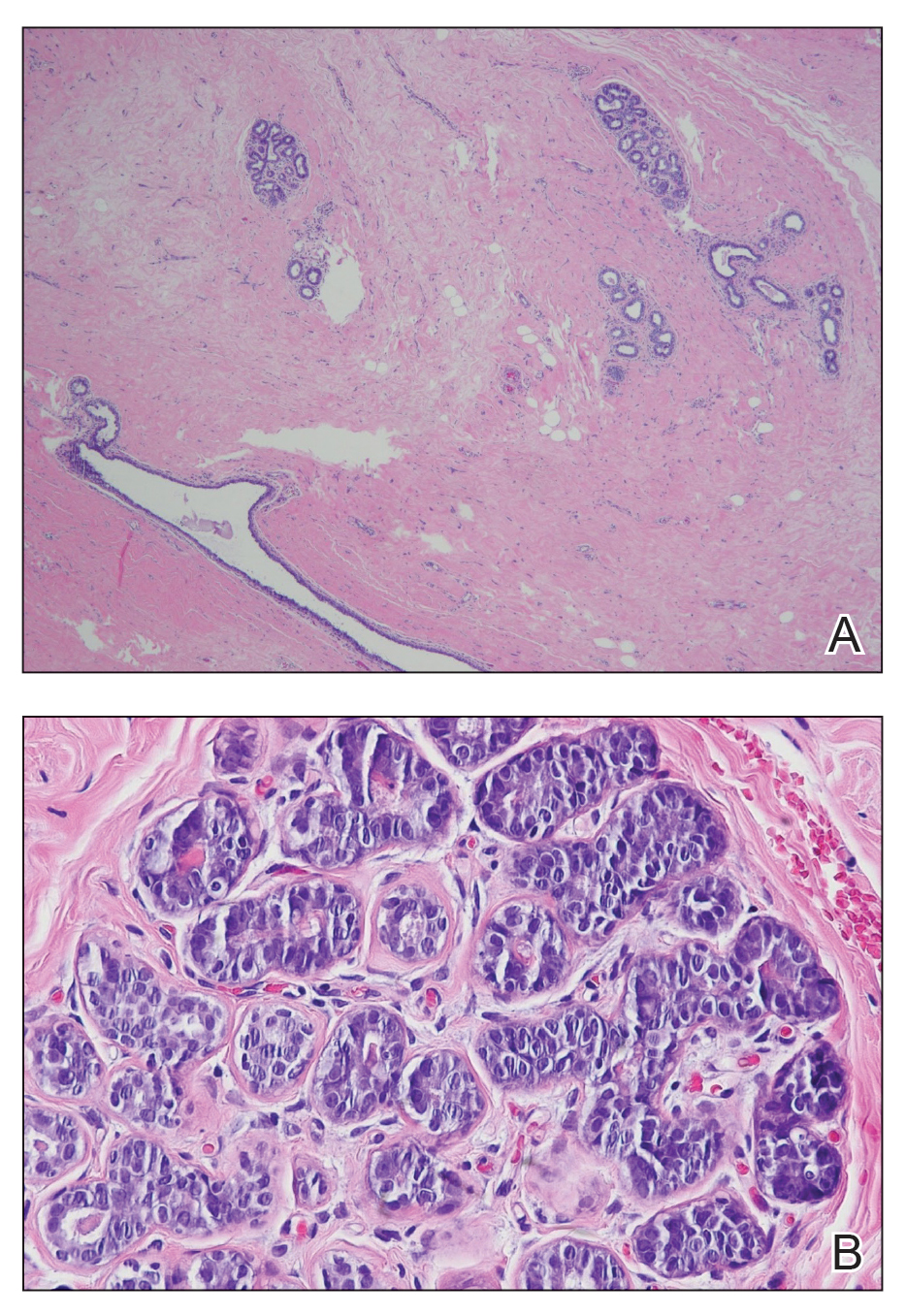
B, Myoepithelial cells lined a stratified columnar epithelium, characteristic of breast tissue (H&E, original magnification x40).
Accessory breasts are characterized by the presence of breast tissue outside the breast and can be found anywhere along the milk line from the axillae to the vulva.1 The prevalence of accessory breasts is 2% to 6% of women, with an average age of presentation for treatment of 42 years.2 Ninety percent of accessory breasts are found in the thorax, 5% are found in the abdomen, and 5% are found in the axillae.3 Incidence is uncommon in adolescents; however, in addition to our patient, there are several cases in the literature of adolescents with accessory breasts in the axillae.4,5
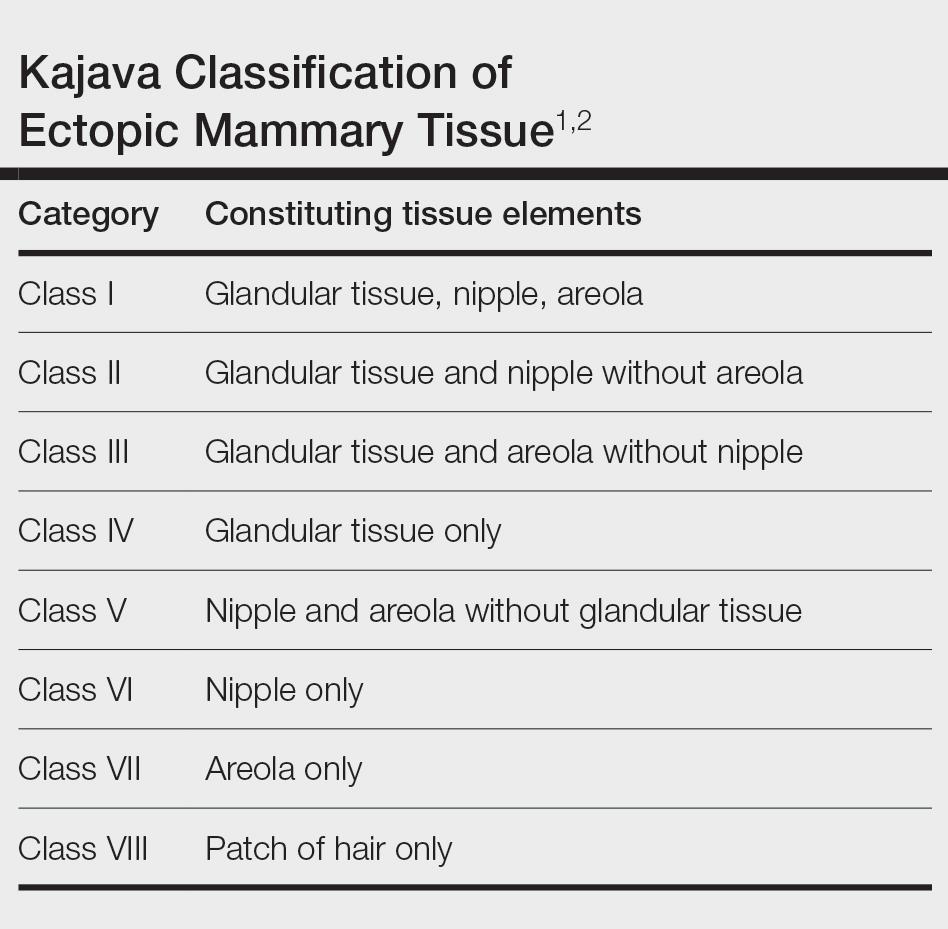
Ectopic mammary tissue is divided into 8 classes based on the Kajava classification system (Table). In a retrospective study of adolescent females with accessory breasts, 91% (10/11) of patients were classified as class IV, and 1 was class II.6 Similarly, our patient was classified as class IV since her accessory breast was composed entirely of glandular tissue and did not include an areola and nipple.
Supernumerary breast structures such as areolas and nipples typically are diagnosed at birth, whereas supernumerary breast tissue is not diagnosed until after hormonal stimulation typically seen during puberty, pregnancy, or breastfeeding. Common symptoms include cyclic pain with menstruation, fluctuation in the size of the mass, and tenderness of the ectopic tissue. There also can be restricted range of motion and increased irritation from clothing. Ultrasonography generally shows a hypoechoic septate indicative of mammary tissue.6 Diagnosis is confirmed by histopathologic studies that show mammary lobules in the dermis with smooth muscle, mammary ducts connected to the nipple, and connective stroma.6
If bothersome, ectopic breast tissue can be surgically removed, either by direct excision or suction lipectomy depending on the size of the mass.2 Postoperative complications are low but can include seroma, bleeding, infection, remnant tissue, or undesired cosmetic results. As with normal breast tissue, ectopic breast tissue can manifest with benign and malignant pathologies.
In conclusion, accessory breast is a benign condition that can cause cyclical pain with menstruation, restricted range of motion, discomfort, anxiety, and cosmetic problems. It is important to keep this diagnosis on the differential when evaluating a soft tissue mass that appears in the axillary region.
- Loukas M, Clarke P, Tubbs RS. Accessory breasts: a historical and current perspective. Am Surg. 2007;73:525-528.
- Bartsich SA, Ofodile FA. Accessory breast tissue in the axilla: classification and treatment. Plast Reconstr Surg. 2011;128:35E-36E. doi:10.1097/PRS.0b013e3182173f95
- Mazine K, Bouassria A, Elbouhaddouti H. Bilateral supernumerary axillary breasts: a case report. Pan Afr Med J. 2020;36:282. doi:10.11604 /pamj.2020.36.282.20445
- Patel RV, Govani D, Patel R, et al. Adolescent right axillary accessory breast with galactorrhoea. BMJ Case Rep. 2014;2014:bcr2014204215. doi:10.1136/bcr-2014-204215
- Surd A, Mironescu A, Gocan H. Fibroadenoma in axillary supernumerary breast in a 17-year-old girl: case report. J Pediatr Adolesc Gynecol. 2016;29:E79-E81. doi:10.1016/j.jpag.2016.04.008
- De la Torre M, Lorca-García C, de Tomás E, et al. Axillary ectopic breast tissue in the adolescent. Pediatr Surg Int. 2022;38:1445-1451. doi:10.1007/s00383-022-05184-1
The Diagnosis: Accessory Breast
A diagnosis of accessory breast was confirmed on histopathology, which demonstrated a slightly hyperplastic and hyperpigmented epidermis. The dermis contained an increased number of smooth muscle bundles with the presence of apocrine glands and mammary lobules (Figure). Tenderness of the mass fluctuated according to the patient’s menstrual cycle, which supported a diagnosis of accessory breast over lipoma. The patient had no signs of infection or other systemic symptoms that were suggestive of lymphadenopathy. Unlike an epidermoid inclusion cyst, our patient’s mass presented as poorly defined and boggy in texture. Biopsy results were not consistent with malignancy, ruling out soft tissue sarcoma.
B, Myoepithelial cells lined a stratified columnar epithelium, characteristic of breast tissue (H&E, original magnification x40).
Accessory breasts are characterized by the presence of breast tissue outside the breast and can be found anywhere along the milk line from the axillae to the vulva.1 The prevalence of accessory breasts is 2% to 6% of women, with an average age of presentation for treatment of 42 years.2 Ninety percent of accessory breasts are found in the thorax, 5% are found in the abdomen, and 5% are found in the axillae.3 Incidence is uncommon in adolescents; however, in addition to our patient, there are several cases in the literature of adolescents with accessory breasts in the axillae.4,5
Ectopic mammary tissue is divided into 8 classes based on the Kajava classification system (Table). In a retrospective study of adolescent females with accessory breasts, 91% (10/11) of patients were classified as class IV, and 1 was class II.6 Similarly, our patient was classified as class IV since her accessory breast was composed entirely of glandular tissue and did not include an areola and nipple.
Supernumerary breast structures such as areolas and nipples typically are diagnosed at birth, whereas supernumerary breast tissue is not diagnosed until after hormonal stimulation typically seen during puberty, pregnancy, or breastfeeding. Common symptoms include cyclic pain with menstruation, fluctuation in the size of the mass, and tenderness of the ectopic tissue. There also can be restricted range of motion and increased irritation from clothing. Ultrasonography generally shows a hypoechoic septate indicative of mammary tissue.6 Diagnosis is confirmed by histopathologic studies that show mammary lobules in the dermis with smooth muscle, mammary ducts connected to the nipple, and connective stroma.6
If bothersome, ectopic breast tissue can be surgically removed, either by direct excision or suction lipectomy depending on the size of the mass.2 Postoperative complications are low but can include seroma, bleeding, infection, remnant tissue, or undesired cosmetic results. As with normal breast tissue, ectopic breast tissue can manifest with benign and malignant pathologies.
In conclusion, accessory breast is a benign condition that can cause cyclical pain with menstruation, restricted range of motion, discomfort, anxiety, and cosmetic problems. It is important to keep this diagnosis on the differential when evaluating a soft tissue mass that appears in the axillary region.
The Diagnosis: Accessory Breast
A diagnosis of accessory breast was confirmed on histopathology, which demonstrated a slightly hyperplastic and hyperpigmented epidermis. The dermis contained an increased number of smooth muscle bundles with the presence of apocrine glands and mammary lobules (Figure). Tenderness of the mass fluctuated according to the patient’s menstrual cycle, which supported a diagnosis of accessory breast over lipoma. The patient had no signs of infection or other systemic symptoms that were suggestive of lymphadenopathy. Unlike an epidermoid inclusion cyst, our patient’s mass presented as poorly defined and boggy in texture. Biopsy results were not consistent with malignancy, ruling out soft tissue sarcoma.
B, Myoepithelial cells lined a stratified columnar epithelium, characteristic of breast tissue (H&E, original magnification x40).
Accessory breasts are characterized by the presence of breast tissue outside the breast and can be found anywhere along the milk line from the axillae to the vulva.1 The prevalence of accessory breasts is 2% to 6% of women, with an average age of presentation for treatment of 42 years.2 Ninety percent of accessory breasts are found in the thorax, 5% are found in the abdomen, and 5% are found in the axillae.3 Incidence is uncommon in adolescents; however, in addition to our patient, there are several cases in the literature of adolescents with accessory breasts in the axillae.4,5
Ectopic mammary tissue is divided into 8 classes based on the Kajava classification system (Table). In a retrospective study of adolescent females with accessory breasts, 91% (10/11) of patients were classified as class IV, and 1 was class II.6 Similarly, our patient was classified as class IV since her accessory breast was composed entirely of glandular tissue and did not include an areola and nipple.
Supernumerary breast structures such as areolas and nipples typically are diagnosed at birth, whereas supernumerary breast tissue is not diagnosed until after hormonal stimulation typically seen during puberty, pregnancy, or breastfeeding. Common symptoms include cyclic pain with menstruation, fluctuation in the size of the mass, and tenderness of the ectopic tissue. There also can be restricted range of motion and increased irritation from clothing. Ultrasonography generally shows a hypoechoic septate indicative of mammary tissue.6 Diagnosis is confirmed by histopathologic studies that show mammary lobules in the dermis with smooth muscle, mammary ducts connected to the nipple, and connective stroma.6
If bothersome, ectopic breast tissue can be surgically removed, either by direct excision or suction lipectomy depending on the size of the mass.2 Postoperative complications are low but can include seroma, bleeding, infection, remnant tissue, or undesired cosmetic results. As with normal breast tissue, ectopic breast tissue can manifest with benign and malignant pathologies.
In conclusion, accessory breast is a benign condition that can cause cyclical pain with menstruation, restricted range of motion, discomfort, anxiety, and cosmetic problems. It is important to keep this diagnosis on the differential when evaluating a soft tissue mass that appears in the axillary region.
- Loukas M, Clarke P, Tubbs RS. Accessory breasts: a historical and current perspective. Am Surg. 2007;73:525-528.
- Bartsich SA, Ofodile FA. Accessory breast tissue in the axilla: classification and treatment. Plast Reconstr Surg. 2011;128:35E-36E. doi:10.1097/PRS.0b013e3182173f95
- Mazine K, Bouassria A, Elbouhaddouti H. Bilateral supernumerary axillary breasts: a case report. Pan Afr Med J. 2020;36:282. doi:10.11604 /pamj.2020.36.282.20445
- Patel RV, Govani D, Patel R, et al. Adolescent right axillary accessory breast with galactorrhoea. BMJ Case Rep. 2014;2014:bcr2014204215. doi:10.1136/bcr-2014-204215
- Surd A, Mironescu A, Gocan H. Fibroadenoma in axillary supernumerary breast in a 17-year-old girl: case report. J Pediatr Adolesc Gynecol. 2016;29:E79-E81. doi:10.1016/j.jpag.2016.04.008
- De la Torre M, Lorca-García C, de Tomás E, et al. Axillary ectopic breast tissue in the adolescent. Pediatr Surg Int. 2022;38:1445-1451. doi:10.1007/s00383-022-05184-1
- Loukas M, Clarke P, Tubbs RS. Accessory breasts: a historical and current perspective. Am Surg. 2007;73:525-528.
- Bartsich SA, Ofodile FA. Accessory breast tissue in the axilla: classification and treatment. Plast Reconstr Surg. 2011;128:35E-36E. doi:10.1097/PRS.0b013e3182173f95
- Mazine K, Bouassria A, Elbouhaddouti H. Bilateral supernumerary axillary breasts: a case report. Pan Afr Med J. 2020;36:282. doi:10.11604 /pamj.2020.36.282.20445
- Patel RV, Govani D, Patel R, et al. Adolescent right axillary accessory breast with galactorrhoea. BMJ Case Rep. 2014;2014:bcr2014204215. doi:10.1136/bcr-2014-204215
- Surd A, Mironescu A, Gocan H. Fibroadenoma in axillary supernumerary breast in a 17-year-old girl: case report. J Pediatr Adolesc Gynecol. 2016;29:E79-E81. doi:10.1016/j.jpag.2016.04.008
- De la Torre M, Lorca-García C, de Tomás E, et al. Axillary ectopic breast tissue in the adolescent. Pediatr Surg Int. 2022;38:1445-1451. doi:10.1007/s00383-022-05184-1
Fluctuant Subcutaneous Nodule in the Axilla of an Adolescent Female
Fluctuant Subcutaneous Nodule in the Axilla of an Adolescent Female
A 15-year-old adolescent female with an unremarkable medical history presented to the dermatology clinic with a mass in the left axilla of 2 years’ duration. The patient reported that there was no drainage of the lesion nor did she have any other similar lesions. She reported tenderness of the lesion during menstruation that resolved after this phase ended. Dermatologic examination revealed a solitary 4.4-cm, flesh-colored, poorly defined, boggy, fluctuant subcutaneous nodule with no central punctum or surface changes. Ultrasonography of the axilla showed a 6.4-cm hypoechoic heterogenous mass. A biopsy of the lesion was performed.
Longitudinal Depression on the Right Thumbnail
THE DIAGNOSIS: Habit-Tic Deformity
Habit-tic deformity is a cause of nail dystrophy that commonly arises in children and adults due to subconscious repetitive and self-injurious manipulation of the nail bed or cuticle, which ultimately damages the nail matrix.1,2 It can be considered a variant of onychotillomania.1
Characteristic features of habit-tic deformity include a longitudinal depression on the central nail plate with transverse ridges,1 which can be more prominent on the dominant hand.3 Patients typically note a long duration of nail deformity, often without insight into its etiology.2 Diagnosis relies on careful assessment of the clinical presentation and the patient’s history to rule out other differential diagnoses. Based on our patient’s clinical presentation and history, we excluded wart, squamous cell carcinoma, eczema, psoriasis, lichen planus, autoimmune connective tissue disease, onychomycosis, paronychia, pincer nail deformity, and Beau line as potential diagnoses. Biopsy also can be performed to exclude these diagnoses from the differential if the cause is unclear following clinical examination.
Treatment for habit-tic deformity involves identifying and addressing the underlying habit. Barrier methods such as bandages and cyanoacrylate adhesives that prevent further manipulation of the nail matrix are effective treatments for habit-tic deformity.2 A multidisciplinary approach with psychiatry may be optimal to identify underlying psychological comorbidities and break the habit through behavior interventions and medications.4 Nail dystrophy generally improves once the habit is disrupted; however, a younger age of onset may carry a worse prognosis.3 Patients should be counseled that the affected nail may never grow normally.
Our patient was advised to use fluocinonide ointment 0.05% to reduce inflammation of the proximal nail fold and to cover the thumbnail with a bandage to prevent picking. He also was counseled that the nail may show ongoing abnormal growth. Minimal improvement was noted after 6 months.
- Rieder EA, Tosti A. Onychotillomania: an underrecognized disorder. J Am Acad Dermatol. 2016;75:1245-1250.doi:10.1016/j.jaad.2016
- Ring DS. Inexpensive solution for habit-tic deformity. Arch Dermatol. 2010;146:1222-1223. doi:10.1001/archdermatol.2010.287
- Horne MI, Utzig JB, Rieder EA, et al. Alopecia areata and habit tic deformities. Skin Appendage Disord. 2018;4:323-325. doi:10.1159/000486540
- Sonthalia S, Sharma P, Kapoor J, et al. Habit tic deformity: need fora comprehensive approach. Skin Appendage Disord. 2019;5:117-118.doi:10.1159/000489320 .05.036
THE DIAGNOSIS: Habit-Tic Deformity
Habit-tic deformity is a cause of nail dystrophy that commonly arises in children and adults due to subconscious repetitive and self-injurious manipulation of the nail bed or cuticle, which ultimately damages the nail matrix.1,2 It can be considered a variant of onychotillomania.1
Characteristic features of habit-tic deformity include a longitudinal depression on the central nail plate with transverse ridges,1 which can be more prominent on the dominant hand.3 Patients typically note a long duration of nail deformity, often without insight into its etiology.2 Diagnosis relies on careful assessment of the clinical presentation and the patient’s history to rule out other differential diagnoses. Based on our patient’s clinical presentation and history, we excluded wart, squamous cell carcinoma, eczema, psoriasis, lichen planus, autoimmune connective tissue disease, onychomycosis, paronychia, pincer nail deformity, and Beau line as potential diagnoses. Biopsy also can be performed to exclude these diagnoses from the differential if the cause is unclear following clinical examination.
Treatment for habit-tic deformity involves identifying and addressing the underlying habit. Barrier methods such as bandages and cyanoacrylate adhesives that prevent further manipulation of the nail matrix are effective treatments for habit-tic deformity.2 A multidisciplinary approach with psychiatry may be optimal to identify underlying psychological comorbidities and break the habit through behavior interventions and medications.4 Nail dystrophy generally improves once the habit is disrupted; however, a younger age of onset may carry a worse prognosis.3 Patients should be counseled that the affected nail may never grow normally.
Our patient was advised to use fluocinonide ointment 0.05% to reduce inflammation of the proximal nail fold and to cover the thumbnail with a bandage to prevent picking. He also was counseled that the nail may show ongoing abnormal growth. Minimal improvement was noted after 6 months.
THE DIAGNOSIS: Habit-Tic Deformity
Habit-tic deformity is a cause of nail dystrophy that commonly arises in children and adults due to subconscious repetitive and self-injurious manipulation of the nail bed or cuticle, which ultimately damages the nail matrix.1,2 It can be considered a variant of onychotillomania.1
Characteristic features of habit-tic deformity include a longitudinal depression on the central nail plate with transverse ridges,1 which can be more prominent on the dominant hand.3 Patients typically note a long duration of nail deformity, often without insight into its etiology.2 Diagnosis relies on careful assessment of the clinical presentation and the patient’s history to rule out other differential diagnoses. Based on our patient’s clinical presentation and history, we excluded wart, squamous cell carcinoma, eczema, psoriasis, lichen planus, autoimmune connective tissue disease, onychomycosis, paronychia, pincer nail deformity, and Beau line as potential diagnoses. Biopsy also can be performed to exclude these diagnoses from the differential if the cause is unclear following clinical examination.
Treatment for habit-tic deformity involves identifying and addressing the underlying habit. Barrier methods such as bandages and cyanoacrylate adhesives that prevent further manipulation of the nail matrix are effective treatments for habit-tic deformity.2 A multidisciplinary approach with psychiatry may be optimal to identify underlying psychological comorbidities and break the habit through behavior interventions and medications.4 Nail dystrophy generally improves once the habit is disrupted; however, a younger age of onset may carry a worse prognosis.3 Patients should be counseled that the affected nail may never grow normally.
Our patient was advised to use fluocinonide ointment 0.05% to reduce inflammation of the proximal nail fold and to cover the thumbnail with a bandage to prevent picking. He also was counseled that the nail may show ongoing abnormal growth. Minimal improvement was noted after 6 months.
- Rieder EA, Tosti A. Onychotillomania: an underrecognized disorder. J Am Acad Dermatol. 2016;75:1245-1250.doi:10.1016/j.jaad.2016
- Ring DS. Inexpensive solution for habit-tic deformity. Arch Dermatol. 2010;146:1222-1223. doi:10.1001/archdermatol.2010.287
- Horne MI, Utzig JB, Rieder EA, et al. Alopecia areata and habit tic deformities. Skin Appendage Disord. 2018;4:323-325. doi:10.1159/000486540
- Sonthalia S, Sharma P, Kapoor J, et al. Habit tic deformity: need fora comprehensive approach. Skin Appendage Disord. 2019;5:117-118.doi:10.1159/000489320 .05.036
- Rieder EA, Tosti A. Onychotillomania: an underrecognized disorder. J Am Acad Dermatol. 2016;75:1245-1250.doi:10.1016/j.jaad.2016
- Ring DS. Inexpensive solution for habit-tic deformity. Arch Dermatol. 2010;146:1222-1223. doi:10.1001/archdermatol.2010.287
- Horne MI, Utzig JB, Rieder EA, et al. Alopecia areata and habit tic deformities. Skin Appendage Disord. 2018;4:323-325. doi:10.1159/000486540
- Sonthalia S, Sharma P, Kapoor J, et al. Habit tic deformity: need fora comprehensive approach. Skin Appendage Disord. 2019;5:117-118.doi:10.1159/000489320 .05.036
A healthy 13-year-old boy presented to the dermatology department with dystrophy of the right thumbnail of 3 to 4 years’ duration. A 5-mm-wide, depressed median longitudinal groove with a fir tree pattern was noted on the central nail plate. The patient noted that the groove had been gradually deepening. There was erythema, edema, and lichenification of the proximal nailfold without vascular changes, and the lunula was enlarged. No hyperkeratosis, subungual debris, erythematous nail folds, or inward curvature of the lateral aspects of the nail were noted. The patient denied any pruritus, pain, discomfort, or bleeding; he also denied any recent illness or trauma to the nail. None of the other nails were affected, and no other lesions or rashes were observed elsewhere on the body. The patient was unsure if he picked at the nail but acknowledged that he may have done so subconsciously. He had no history of eczema, psoriasis, or autoimmune connective tissue disorders.
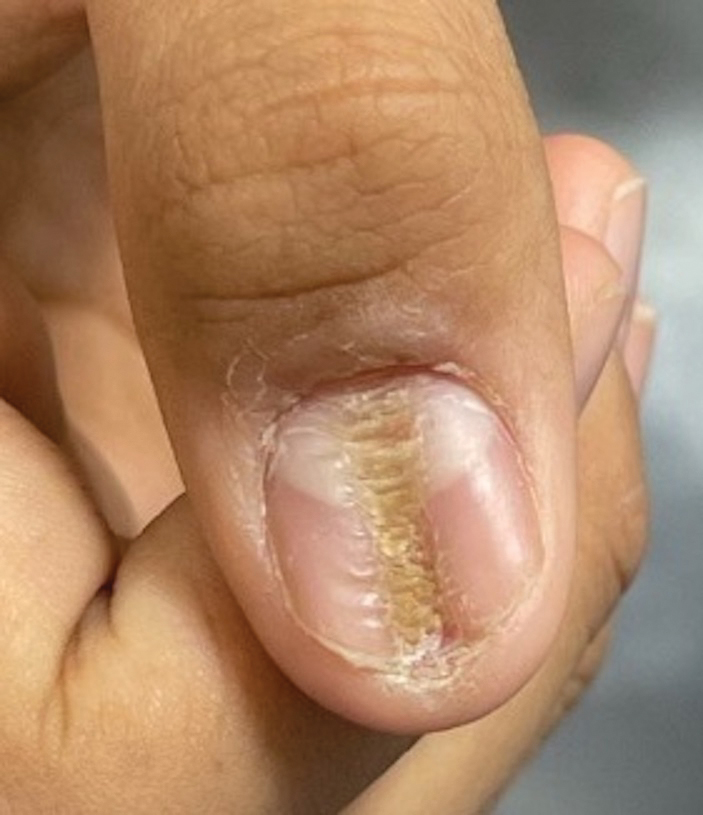
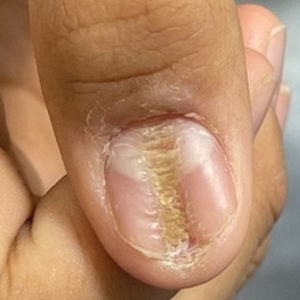
Hyperkeratotic Papules and Black Macules on the Hands
THE DIAGNOSIS: Acral Hemorrhagic Darier Disease
Darier disease (DD), also known as keratosis follicularis, is a rare autosomal-dominant genodermatosis caused by mutations in the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene (ATP2A2). This gene encodes the enzyme sarcoplasmic/endoplasmic reticulum calcium ATPase 2, which results in abnormal calcium signaling in keratinocytes and leads to dyskeratosis.1 Darier disease commonly manifests in the second decade of life with hyperkeratotic papules coalescing into plaques, often accompanied by erosions and fissures that cause discomfort and pruritus. Darier disease also is associated with characteristic nail findings such as the classic candy cane nails and V-shaped nicking.
Acral hemorrhagic lesions are a rare manifestation of DD. Clinically, these lesions can manifest as hemorrhagic macules, papules, and/or vesicles, most commonly occurring following local trauma or retinoid use. Patients with these lesions are believed to have either specific mutations in the ATP2A2 gene that impair sarcoplasmic/endoplasmic reticulum calcium ATPase 2 function in the vascular endothelium or a mutation in the sarcoplasmic/endoplasmic reticulum calcium ATPase protein itself, leading to dysregulation of mitochondrial homeostasis from within the cell, provoking oxidative stress and causing detrimental effects on blood vessels.2 Patients with this variant can present with all the features of classic DD concomitantly, with varying symptom severity or distinct clinical features during separate episodic flares, or as the sole manifestation. Other nonclassical lesions of DD include acral keratoderma, giant comedones, keloidlike vegetations, and leucodermic macules (Figure).3
Acral hemorrhagic DD may appear either in isolation or in tandem with more traditional symptoms, necessitating consideration of other possible differential diagnoses such as acrokeratosis verruciformis of Hopf (AKV), porphyria cutanea tarda, bullous lichen planus (BLP), and hemorrhagic lichen sclerosus.
Sometimes regarded as a variant of DD, AKV is an autosomal- dominant genodermatosis characterized by flat or verrucous hyperkeratotic papules on the hands and feet. In AKV, the nails also may be affected, with changes including striations, subungual hyperkeratosis, and V-shaped nicking of the distal nails. Although our patient displayed features of AKV, it has not been associated with acral hemorrhagic macules, making this diagnosis less likely than DD.4
Porphyria cutanea tarda, a condition caused by decreased levels of uroporphyrinogen decarboxylase, also can cause skin manifestations such as blistering as well as increased skin fragility, predominantly in sun-exposed areas.5 Our patient’s lack of photosensitivity and absence of other common symptoms of this disorder, such as hypertrichosis and hyperpigmentation, made porphyria cutanea tarda less likely.
Bullous lichen planus is a rare subtype of lichen planus characterized by tense bullae arising from preexisting lichen planus lesions or appearing de novo, most commonly manifesting on the oral mucosa or the legs.6 The bullae associated with BLP can rupture and form ulcers—a symptom that could potentially be mistaken for hemorrhagic macules like the ones observed in our patient. However, BLP typically is characterized by erythematous, violaceous, polygonal papules commonly appearing on the oral mucosa and the legs with blisters developing near or on pre-existing lichen planus lesions. These are different from the hyperkeratotic papules and leucodermic macules seen in our patient, which aligned more closely with the clinical presentation of DD.
Hemorrhagic lichen sclerosus presents with white atrophic patches and plaques and hemorrhagic bullae, which may resemble the leucodermic macules and hemorrhagic macules of DD. However, hemorrhagic lichen sclerosus most commonly involves the genital area in postmenopausal women. Extragenital manifestations of lichen sclerosus, although less common, can occur and typically manifest on the thighs, buttocks, breasts, back, chest, axillae, shoulders, and wrists.7 Notably, these hemorrhagic lesions typically are surrounded by hypopigmented skin and display an atrophic appearance.
Management of DD can be challenging. General measures include sun protection, heat avoidance, and friction reduction. Retinoids are considered the first-line therapy for severe DD, as they help normalize keratinocyte differentiation and reduce keratotic scaling.8 Topical corticosteroids can help manage inflammation and reduce the risk for secondary infections. Our patient responded well to this treatment approach, with a notable reduction in the number and severity of the hyperkeratotic plaques and resolution of the acral hemorrhagic lesions.
- Savignac M, Edir A, Simon M, et al. Darier disease: a disease model of impaired calcium homeostasis in the skin. Biochim Biophys Acta. 2011;1813:1111-1117. doi:10.1016/j.bbamcr.2010.12.006
- Hong E, Hu R, Posligua A, et al. Acral hemorrhagic Darier disease: a case report of a rare presentation and literature review. JAAD Case Rep. 2023;31:93-96. doi:10.1016/j.jdcr.2022.05.030
- Yeshurun A, Ziv M, Cohen-Barak E, et al. An update on the cutaneous manifestations of Darier disease. J Cutan Med Surg. 2021;25:498-503. doi:10.1177/1203475421999331
- Williams GM, Lincoln M. Acrokeratosis verruciformis of Hopf. In: StatPearls. StatPearls Publishing; May 1, 2023.
- Shah A, Bhatt H. Cutanea tarda porphyria. In: StatPearls. StatPearls Publishing; April 17, 2023.
- Liakopoulou A, Rallis E. Bullous lichen planus—a review. J Dermatol Case Rep. 2017;11:1-4. doi:10.3315/jdcr.2017.1239
- Arnold N, Manway M, Stephenson S, et al. Extragenital bullous lichen sclerosus on the anterior lower extremities: report of a case and literature review. Dermatol Online J. 2017;23:13030
- Haber RN, Dib NG. Management of Darier disease: a review of the literature and update. Indian J Dermatol Venereol Leprol. 2021;87:14-21. doi:10.25259/IJDVL_963_19 /qt8dn3p7kv.
THE DIAGNOSIS: Acral Hemorrhagic Darier Disease
Darier disease (DD), also known as keratosis follicularis, is a rare autosomal-dominant genodermatosis caused by mutations in the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene (ATP2A2). This gene encodes the enzyme sarcoplasmic/endoplasmic reticulum calcium ATPase 2, which results in abnormal calcium signaling in keratinocytes and leads to dyskeratosis.1 Darier disease commonly manifests in the second decade of life with hyperkeratotic papules coalescing into plaques, often accompanied by erosions and fissures that cause discomfort and pruritus. Darier disease also is associated with characteristic nail findings such as the classic candy cane nails and V-shaped nicking.
Acral hemorrhagic lesions are a rare manifestation of DD. Clinically, these lesions can manifest as hemorrhagic macules, papules, and/or vesicles, most commonly occurring following local trauma or retinoid use. Patients with these lesions are believed to have either specific mutations in the ATP2A2 gene that impair sarcoplasmic/endoplasmic reticulum calcium ATPase 2 function in the vascular endothelium or a mutation in the sarcoplasmic/endoplasmic reticulum calcium ATPase protein itself, leading to dysregulation of mitochondrial homeostasis from within the cell, provoking oxidative stress and causing detrimental effects on blood vessels.2 Patients with this variant can present with all the features of classic DD concomitantly, with varying symptom severity or distinct clinical features during separate episodic flares, or as the sole manifestation. Other nonclassical lesions of DD include acral keratoderma, giant comedones, keloidlike vegetations, and leucodermic macules (Figure).3
Acral hemorrhagic DD may appear either in isolation or in tandem with more traditional symptoms, necessitating consideration of other possible differential diagnoses such as acrokeratosis verruciformis of Hopf (AKV), porphyria cutanea tarda, bullous lichen planus (BLP), and hemorrhagic lichen sclerosus.
Sometimes regarded as a variant of DD, AKV is an autosomal- dominant genodermatosis characterized by flat or verrucous hyperkeratotic papules on the hands and feet. In AKV, the nails also may be affected, with changes including striations, subungual hyperkeratosis, and V-shaped nicking of the distal nails. Although our patient displayed features of AKV, it has not been associated with acral hemorrhagic macules, making this diagnosis less likely than DD.4
Porphyria cutanea tarda, a condition caused by decreased levels of uroporphyrinogen decarboxylase, also can cause skin manifestations such as blistering as well as increased skin fragility, predominantly in sun-exposed areas.5 Our patient’s lack of photosensitivity and absence of other common symptoms of this disorder, such as hypertrichosis and hyperpigmentation, made porphyria cutanea tarda less likely.
Bullous lichen planus is a rare subtype of lichen planus characterized by tense bullae arising from preexisting lichen planus lesions or appearing de novo, most commonly manifesting on the oral mucosa or the legs.6 The bullae associated with BLP can rupture and form ulcers—a symptom that could potentially be mistaken for hemorrhagic macules like the ones observed in our patient. However, BLP typically is characterized by erythematous, violaceous, polygonal papules commonly appearing on the oral mucosa and the legs with blisters developing near or on pre-existing lichen planus lesions. These are different from the hyperkeratotic papules and leucodermic macules seen in our patient, which aligned more closely with the clinical presentation of DD.
Hemorrhagic lichen sclerosus presents with white atrophic patches and plaques and hemorrhagic bullae, which may resemble the leucodermic macules and hemorrhagic macules of DD. However, hemorrhagic lichen sclerosus most commonly involves the genital area in postmenopausal women. Extragenital manifestations of lichen sclerosus, although less common, can occur and typically manifest on the thighs, buttocks, breasts, back, chest, axillae, shoulders, and wrists.7 Notably, these hemorrhagic lesions typically are surrounded by hypopigmented skin and display an atrophic appearance.
Management of DD can be challenging. General measures include sun protection, heat avoidance, and friction reduction. Retinoids are considered the first-line therapy for severe DD, as they help normalize keratinocyte differentiation and reduce keratotic scaling.8 Topical corticosteroids can help manage inflammation and reduce the risk for secondary infections. Our patient responded well to this treatment approach, with a notable reduction in the number and severity of the hyperkeratotic plaques and resolution of the acral hemorrhagic lesions.
THE DIAGNOSIS: Acral Hemorrhagic Darier Disease
Darier disease (DD), also known as keratosis follicularis, is a rare autosomal-dominant genodermatosis caused by mutations in the ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 gene (ATP2A2). This gene encodes the enzyme sarcoplasmic/endoplasmic reticulum calcium ATPase 2, which results in abnormal calcium signaling in keratinocytes and leads to dyskeratosis.1 Darier disease commonly manifests in the second decade of life with hyperkeratotic papules coalescing into plaques, often accompanied by erosions and fissures that cause discomfort and pruritus. Darier disease also is associated with characteristic nail findings such as the classic candy cane nails and V-shaped nicking.
Acral hemorrhagic lesions are a rare manifestation of DD. Clinically, these lesions can manifest as hemorrhagic macules, papules, and/or vesicles, most commonly occurring following local trauma or retinoid use. Patients with these lesions are believed to have either specific mutations in the ATP2A2 gene that impair sarcoplasmic/endoplasmic reticulum calcium ATPase 2 function in the vascular endothelium or a mutation in the sarcoplasmic/endoplasmic reticulum calcium ATPase protein itself, leading to dysregulation of mitochondrial homeostasis from within the cell, provoking oxidative stress and causing detrimental effects on blood vessels.2 Patients with this variant can present with all the features of classic DD concomitantly, with varying symptom severity or distinct clinical features during separate episodic flares, or as the sole manifestation. Other nonclassical lesions of DD include acral keratoderma, giant comedones, keloidlike vegetations, and leucodermic macules (Figure).3
Acral hemorrhagic DD may appear either in isolation or in tandem with more traditional symptoms, necessitating consideration of other possible differential diagnoses such as acrokeratosis verruciformis of Hopf (AKV), porphyria cutanea tarda, bullous lichen planus (BLP), and hemorrhagic lichen sclerosus.
Sometimes regarded as a variant of DD, AKV is an autosomal- dominant genodermatosis characterized by flat or verrucous hyperkeratotic papules on the hands and feet. In AKV, the nails also may be affected, with changes including striations, subungual hyperkeratosis, and V-shaped nicking of the distal nails. Although our patient displayed features of AKV, it has not been associated with acral hemorrhagic macules, making this diagnosis less likely than DD.4
Porphyria cutanea tarda, a condition caused by decreased levels of uroporphyrinogen decarboxylase, also can cause skin manifestations such as blistering as well as increased skin fragility, predominantly in sun-exposed areas.5 Our patient’s lack of photosensitivity and absence of other common symptoms of this disorder, such as hypertrichosis and hyperpigmentation, made porphyria cutanea tarda less likely.
Bullous lichen planus is a rare subtype of lichen planus characterized by tense bullae arising from preexisting lichen planus lesions or appearing de novo, most commonly manifesting on the oral mucosa or the legs.6 The bullae associated with BLP can rupture and form ulcers—a symptom that could potentially be mistaken for hemorrhagic macules like the ones observed in our patient. However, BLP typically is characterized by erythematous, violaceous, polygonal papules commonly appearing on the oral mucosa and the legs with blisters developing near or on pre-existing lichen planus lesions. These are different from the hyperkeratotic papules and leucodermic macules seen in our patient, which aligned more closely with the clinical presentation of DD.
Hemorrhagic lichen sclerosus presents with white atrophic patches and plaques and hemorrhagic bullae, which may resemble the leucodermic macules and hemorrhagic macules of DD. However, hemorrhagic lichen sclerosus most commonly involves the genital area in postmenopausal women. Extragenital manifestations of lichen sclerosus, although less common, can occur and typically manifest on the thighs, buttocks, breasts, back, chest, axillae, shoulders, and wrists.7 Notably, these hemorrhagic lesions typically are surrounded by hypopigmented skin and display an atrophic appearance.
Management of DD can be challenging. General measures include sun protection, heat avoidance, and friction reduction. Retinoids are considered the first-line therapy for severe DD, as they help normalize keratinocyte differentiation and reduce keratotic scaling.8 Topical corticosteroids can help manage inflammation and reduce the risk for secondary infections. Our patient responded well to this treatment approach, with a notable reduction in the number and severity of the hyperkeratotic plaques and resolution of the acral hemorrhagic lesions.
- Savignac M, Edir A, Simon M, et al. Darier disease: a disease model of impaired calcium homeostasis in the skin. Biochim Biophys Acta. 2011;1813:1111-1117. doi:10.1016/j.bbamcr.2010.12.006
- Hong E, Hu R, Posligua A, et al. Acral hemorrhagic Darier disease: a case report of a rare presentation and literature review. JAAD Case Rep. 2023;31:93-96. doi:10.1016/j.jdcr.2022.05.030
- Yeshurun A, Ziv M, Cohen-Barak E, et al. An update on the cutaneous manifestations of Darier disease. J Cutan Med Surg. 2021;25:498-503. doi:10.1177/1203475421999331
- Williams GM, Lincoln M. Acrokeratosis verruciformis of Hopf. In: StatPearls. StatPearls Publishing; May 1, 2023.
- Shah A, Bhatt H. Cutanea tarda porphyria. In: StatPearls. StatPearls Publishing; April 17, 2023.
- Liakopoulou A, Rallis E. Bullous lichen planus—a review. J Dermatol Case Rep. 2017;11:1-4. doi:10.3315/jdcr.2017.1239
- Arnold N, Manway M, Stephenson S, et al. Extragenital bullous lichen sclerosus on the anterior lower extremities: report of a case and literature review. Dermatol Online J. 2017;23:13030
- Haber RN, Dib NG. Management of Darier disease: a review of the literature and update. Indian J Dermatol Venereol Leprol. 2021;87:14-21. doi:10.25259/IJDVL_963_19 /qt8dn3p7kv.
- Savignac M, Edir A, Simon M, et al. Darier disease: a disease model of impaired calcium homeostasis in the skin. Biochim Biophys Acta. 2011;1813:1111-1117. doi:10.1016/j.bbamcr.2010.12.006
- Hong E, Hu R, Posligua A, et al. Acral hemorrhagic Darier disease: a case report of a rare presentation and literature review. JAAD Case Rep. 2023;31:93-96. doi:10.1016/j.jdcr.2022.05.030
- Yeshurun A, Ziv M, Cohen-Barak E, et al. An update on the cutaneous manifestations of Darier disease. J Cutan Med Surg. 2021;25:498-503. doi:10.1177/1203475421999331
- Williams GM, Lincoln M. Acrokeratosis verruciformis of Hopf. In: StatPearls. StatPearls Publishing; May 1, 2023.
- Shah A, Bhatt H. Cutanea tarda porphyria. In: StatPearls. StatPearls Publishing; April 17, 2023.
- Liakopoulou A, Rallis E. Bullous lichen planus—a review. J Dermatol Case Rep. 2017;11:1-4. doi:10.3315/jdcr.2017.1239
- Arnold N, Manway M, Stephenson S, et al. Extragenital bullous lichen sclerosus on the anterior lower extremities: report of a case and literature review. Dermatol Online J. 2017;23:13030
- Haber RN, Dib NG. Management of Darier disease: a review of the literature and update. Indian J Dermatol Venereol Leprol. 2021;87:14-21. doi:10.25259/IJDVL_963_19 /qt8dn3p7kv.
An elderly woman with a long history of hyperkeratotic papules on the abdomen, forearms, dorsal hands, and skinfolds presented with new lesions on the dorsal hands that had developed over the preceding few months after a lapse in treatment with her previous dermatologist. Her medical history was otherwise unremarkable. Physical examination revealed hyperkeratotic papules, black hemorrhagic macules with jagged borders, and a thin hemorrhagic plaque on the dorsal hands. Nail findings were notable for alternating white and red longitudinal bands with nicking of the distal nail plates. She also had scattered leucodermic macules over the trunk, feet, arms, and legs, as well as numerous hyperkeratotic papules coalescing into plaques over the mons pubis and in the inguinal folds.
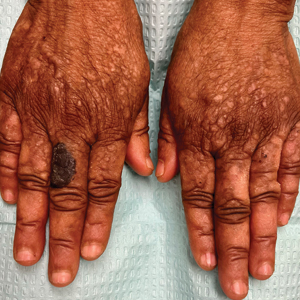
Eruption of Multiple Linear Hyperpigmented Plaques
THE DIAGNOSIS: Chemotherapy-Induced Flagellate Dermatitis
Based on the clinical presentation and temporal relation with chemotherapy, a diagnosis of bleomycininduced flagellate dermatitis (FD) was made, as bleomycin is the only chemotherapeutic agent from this regimen that has been linked with FD.1,2 Laboratory findings revealed eosinophilia, further supporting a druginduced dermatitis. The patient was treated with oral steroids and diphenhydramine to alleviate itching and discomfort. The chemotherapy was temporarily discontinued until symptomatic improvement was observed within 2 to 3 days.
Flagellate dermatitis is characterized by unique erythematous, linear, intermingled streaks of adjoining firm papules—often preceded by a prodrome of global pruritus—that eventually become hyperpigmented as the erythema subsides. The clinical manifestation of FD can be idiopathic; true/mechanical (dermatitis artefacta, abuse, sadomasochism); chemotherapy induced (peplomycin, trastuzumab, cisplatin, docetaxel, bendamustine); toxin induced (shiitake mushroom, cnidarian stings, Paederus insects); related to rheumatologic diseases (dermatomyositis, adult-onset Still disease), dermatographism, phytophotodermatitis, or poison ivy dermatitis; or induced by chikungunya fever.1
The term flagellate originates from the Latin word flagellum, which pertains to the distinctive whiplike pattern. It was first described by Moulin et al3 in 1970 in reference to bleomycin-induced linear hyperpigmentation. Bleomycin, a glycopeptide antibiotic derived from Streptomyces verticillus, is used to treat Hodgkin lymphoma, squamous cell carcinoma, and germ cell tumors. The worldwide incidence of bleomycin-induced FD is 8% to 22% and commonly is associated with a cumulative dose greater than 100 U.2 Clinical presentation is variable in terms of onset, distribution, and morphology of the eruption and could be independent of dose, route of administration, or type of malignancy being treated. The flagellate rash commonly involves the trunk, arms, and legs; can develop within hours to 6 months of starting bleomycin therapy; often is preceded by generalized itching; and eventually heals with hyperpigmentation.
Possible mechanisms of bleomycin-induced FD include localized melanogenesis, inflammatory pigmentary incontinence, alterations to normal pigmentation patterns, cytotoxic effects of the drug itself, minor trauma/ scratching leading to increased blood flow and causing local accumulation of bleomycin, heat recall, and reduced epidermal turnover leading to extended interaction between keratinocytes and melanocytes.2 Heat exposure can act as a trigger for bleomycin-induced skin rash recall even months after the treatment is stopped.
Apart from discontinuing the drug, there is no specific treatment available for bleomycin-induced FD. The primary objective of treatment is to alleviate pruritus, which often involves the use of topical or systemic corticosteroids and oral antihistamines. The duration of treatment depends on the patient’s clinical response. Once treatment is discontinued, FD typically resolves within 6 to 8 months. However, there can be a permanent postinflammatory hyperpigmentation in the affected area.4 Although there is a concern for increased mortality after postponement of chemotherapy,5 the decision to proceed with or discontinue the chemotherapy regimen necessitates a comprehensive interdisciplinary discussion and a meticulous assessment of the risks and benefits that is customized to each individual patient. Flagellate dermatitis can reoccur with bleomycin re-exposure; a combined approach of proactive topical and systemic steroid treatment seems to diminish the likelihood of FD recurrence.5
Our case underscores the importance of recognizing, detecting, and managing FD promptly in individuals undergoing bleomycin-based chemotherapy. Medical professionals should familiarize themselves with this distinct adverse effect linked to bleomycin, enabling prompt discontinuation if necessary, and educate patients about the condition’s typically temporary nature, thereby alleviating their concerns.
- Bhushan P, Manjul P, Baliyan V. Flagellate dermatoses. Indian J Dermatol Venereol Leprol. 2014;80:149-152.
- Ziemer M, Goetze S, Juhasz K, et al. Flagellate dermatitis as a bleomycinspecific adverse effect of cytostatic therapy: a clinical-histopathologic correlation. Am J Clin Dermatol. 2011;12:68-76. doi:10.2165/11537080-000000000-00000
- Moulin G, Fière B, Beyvin A. Cutaneous pigmentation caused by bleomycin. Article in French. Bull Soc Fr Dermatol Syphiligr. 1970;77:293-296.
- Biswas A, Chaudhari PB, Sharma P, et al. Bleomycin induced flagellate erythema: revisiting a unique complication. J Cancer Res Ther. 2013;9:500-503. doi:10.4103/0973-1482.119358
- Hanna TP, King WD, Thibodeau S, et al. Mortality due to cancer treatment delay: systematic review and meta-analysis. BMJ. 2020;371:m4087. doi:10.1136/bmj.m4087
THE DIAGNOSIS: Chemotherapy-Induced Flagellate Dermatitis
Based on the clinical presentation and temporal relation with chemotherapy, a diagnosis of bleomycininduced flagellate dermatitis (FD) was made, as bleomycin is the only chemotherapeutic agent from this regimen that has been linked with FD.1,2 Laboratory findings revealed eosinophilia, further supporting a druginduced dermatitis. The patient was treated with oral steroids and diphenhydramine to alleviate itching and discomfort. The chemotherapy was temporarily discontinued until symptomatic improvement was observed within 2 to 3 days.
Flagellate dermatitis is characterized by unique erythematous, linear, intermingled streaks of adjoining firm papules—often preceded by a prodrome of global pruritus—that eventually become hyperpigmented as the erythema subsides. The clinical manifestation of FD can be idiopathic; true/mechanical (dermatitis artefacta, abuse, sadomasochism); chemotherapy induced (peplomycin, trastuzumab, cisplatin, docetaxel, bendamustine); toxin induced (shiitake mushroom, cnidarian stings, Paederus insects); related to rheumatologic diseases (dermatomyositis, adult-onset Still disease), dermatographism, phytophotodermatitis, or poison ivy dermatitis; or induced by chikungunya fever.1
The term flagellate originates from the Latin word flagellum, which pertains to the distinctive whiplike pattern. It was first described by Moulin et al3 in 1970 in reference to bleomycin-induced linear hyperpigmentation. Bleomycin, a glycopeptide antibiotic derived from Streptomyces verticillus, is used to treat Hodgkin lymphoma, squamous cell carcinoma, and germ cell tumors. The worldwide incidence of bleomycin-induced FD is 8% to 22% and commonly is associated with a cumulative dose greater than 100 U.2 Clinical presentation is variable in terms of onset, distribution, and morphology of the eruption and could be independent of dose, route of administration, or type of malignancy being treated. The flagellate rash commonly involves the trunk, arms, and legs; can develop within hours to 6 months of starting bleomycin therapy; often is preceded by generalized itching; and eventually heals with hyperpigmentation.
Possible mechanisms of bleomycin-induced FD include localized melanogenesis, inflammatory pigmentary incontinence, alterations to normal pigmentation patterns, cytotoxic effects of the drug itself, minor trauma/ scratching leading to increased blood flow and causing local accumulation of bleomycin, heat recall, and reduced epidermal turnover leading to extended interaction between keratinocytes and melanocytes.2 Heat exposure can act as a trigger for bleomycin-induced skin rash recall even months after the treatment is stopped.
Apart from discontinuing the drug, there is no specific treatment available for bleomycin-induced FD. The primary objective of treatment is to alleviate pruritus, which often involves the use of topical or systemic corticosteroids and oral antihistamines. The duration of treatment depends on the patient’s clinical response. Once treatment is discontinued, FD typically resolves within 6 to 8 months. However, there can be a permanent postinflammatory hyperpigmentation in the affected area.4 Although there is a concern for increased mortality after postponement of chemotherapy,5 the decision to proceed with or discontinue the chemotherapy regimen necessitates a comprehensive interdisciplinary discussion and a meticulous assessment of the risks and benefits that is customized to each individual patient. Flagellate dermatitis can reoccur with bleomycin re-exposure; a combined approach of proactive topical and systemic steroid treatment seems to diminish the likelihood of FD recurrence.5
Our case underscores the importance of recognizing, detecting, and managing FD promptly in individuals undergoing bleomycin-based chemotherapy. Medical professionals should familiarize themselves with this distinct adverse effect linked to bleomycin, enabling prompt discontinuation if necessary, and educate patients about the condition’s typically temporary nature, thereby alleviating their concerns.
THE DIAGNOSIS: Chemotherapy-Induced Flagellate Dermatitis
Based on the clinical presentation and temporal relation with chemotherapy, a diagnosis of bleomycininduced flagellate dermatitis (FD) was made, as bleomycin is the only chemotherapeutic agent from this regimen that has been linked with FD.1,2 Laboratory findings revealed eosinophilia, further supporting a druginduced dermatitis. The patient was treated with oral steroids and diphenhydramine to alleviate itching and discomfort. The chemotherapy was temporarily discontinued until symptomatic improvement was observed within 2 to 3 days.
Flagellate dermatitis is characterized by unique erythematous, linear, intermingled streaks of adjoining firm papules—often preceded by a prodrome of global pruritus—that eventually become hyperpigmented as the erythema subsides. The clinical manifestation of FD can be idiopathic; true/mechanical (dermatitis artefacta, abuse, sadomasochism); chemotherapy induced (peplomycin, trastuzumab, cisplatin, docetaxel, bendamustine); toxin induced (shiitake mushroom, cnidarian stings, Paederus insects); related to rheumatologic diseases (dermatomyositis, adult-onset Still disease), dermatographism, phytophotodermatitis, or poison ivy dermatitis; or induced by chikungunya fever.1
The term flagellate originates from the Latin word flagellum, which pertains to the distinctive whiplike pattern. It was first described by Moulin et al3 in 1970 in reference to bleomycin-induced linear hyperpigmentation. Bleomycin, a glycopeptide antibiotic derived from Streptomyces verticillus, is used to treat Hodgkin lymphoma, squamous cell carcinoma, and germ cell tumors. The worldwide incidence of bleomycin-induced FD is 8% to 22% and commonly is associated with a cumulative dose greater than 100 U.2 Clinical presentation is variable in terms of onset, distribution, and morphology of the eruption and could be independent of dose, route of administration, or type of malignancy being treated. The flagellate rash commonly involves the trunk, arms, and legs; can develop within hours to 6 months of starting bleomycin therapy; often is preceded by generalized itching; and eventually heals with hyperpigmentation.
Possible mechanisms of bleomycin-induced FD include localized melanogenesis, inflammatory pigmentary incontinence, alterations to normal pigmentation patterns, cytotoxic effects of the drug itself, minor trauma/ scratching leading to increased blood flow and causing local accumulation of bleomycin, heat recall, and reduced epidermal turnover leading to extended interaction between keratinocytes and melanocytes.2 Heat exposure can act as a trigger for bleomycin-induced skin rash recall even months after the treatment is stopped.
Apart from discontinuing the drug, there is no specific treatment available for bleomycin-induced FD. The primary objective of treatment is to alleviate pruritus, which often involves the use of topical or systemic corticosteroids and oral antihistamines. The duration of treatment depends on the patient’s clinical response. Once treatment is discontinued, FD typically resolves within 6 to 8 months. However, there can be a permanent postinflammatory hyperpigmentation in the affected area.4 Although there is a concern for increased mortality after postponement of chemotherapy,5 the decision to proceed with or discontinue the chemotherapy regimen necessitates a comprehensive interdisciplinary discussion and a meticulous assessment of the risks and benefits that is customized to each individual patient. Flagellate dermatitis can reoccur with bleomycin re-exposure; a combined approach of proactive topical and systemic steroid treatment seems to diminish the likelihood of FD recurrence.5
Our case underscores the importance of recognizing, detecting, and managing FD promptly in individuals undergoing bleomycin-based chemotherapy. Medical professionals should familiarize themselves with this distinct adverse effect linked to bleomycin, enabling prompt discontinuation if necessary, and educate patients about the condition’s typically temporary nature, thereby alleviating their concerns.
- Bhushan P, Manjul P, Baliyan V. Flagellate dermatoses. Indian J Dermatol Venereol Leprol. 2014;80:149-152.
- Ziemer M, Goetze S, Juhasz K, et al. Flagellate dermatitis as a bleomycinspecific adverse effect of cytostatic therapy: a clinical-histopathologic correlation. Am J Clin Dermatol. 2011;12:68-76. doi:10.2165/11537080-000000000-00000
- Moulin G, Fière B, Beyvin A. Cutaneous pigmentation caused by bleomycin. Article in French. Bull Soc Fr Dermatol Syphiligr. 1970;77:293-296.
- Biswas A, Chaudhari PB, Sharma P, et al. Bleomycin induced flagellate erythema: revisiting a unique complication. J Cancer Res Ther. 2013;9:500-503. doi:10.4103/0973-1482.119358
- Hanna TP, King WD, Thibodeau S, et al. Mortality due to cancer treatment delay: systematic review and meta-analysis. BMJ. 2020;371:m4087. doi:10.1136/bmj.m4087
- Bhushan P, Manjul P, Baliyan V. Flagellate dermatoses. Indian J Dermatol Venereol Leprol. 2014;80:149-152.
- Ziemer M, Goetze S, Juhasz K, et al. Flagellate dermatitis as a bleomycinspecific adverse effect of cytostatic therapy: a clinical-histopathologic correlation. Am J Clin Dermatol. 2011;12:68-76. doi:10.2165/11537080-000000000-00000
- Moulin G, Fière B, Beyvin A. Cutaneous pigmentation caused by bleomycin. Article in French. Bull Soc Fr Dermatol Syphiligr. 1970;77:293-296.
- Biswas A, Chaudhari PB, Sharma P, et al. Bleomycin induced flagellate erythema: revisiting a unique complication. J Cancer Res Ther. 2013;9:500-503. doi:10.4103/0973-1482.119358
- Hanna TP, King WD, Thibodeau S, et al. Mortality due to cancer treatment delay: systematic review and meta-analysis. BMJ. 2020;371:m4087. doi:10.1136/bmj.m4087
A 28-year-old man presented for evaluation of an intensely itchy rash of 5 days’ duration involving the face, trunk, arms, and legs. The patient recently had been diagnosed with classical Hodgkin lymphoma and was started on a biweekly chemotherapy regimen of adriamycin, bleomycin, vinblastine, and dacarbazine 3 weeks prior. He reported that a red, itchy, papular rash had developed on the hands 1 week after starting chemotherapy and improved with antihistamines. Symptoms of the current rash included night sweats, occasional fever, substantial unintentional weight loss, and fatigue. He had no history of urticaria, angioedema, anaphylaxis, or nail changes.
Physical examination revealed widespread, itchy, linear and curvilinear hyperpigmented plaques on the upper arms, shoulders, back (top), face, and thighs, as well as erythematous grouped papules on the bilateral palms (bottom). There was no mucosal or systemic involvement.
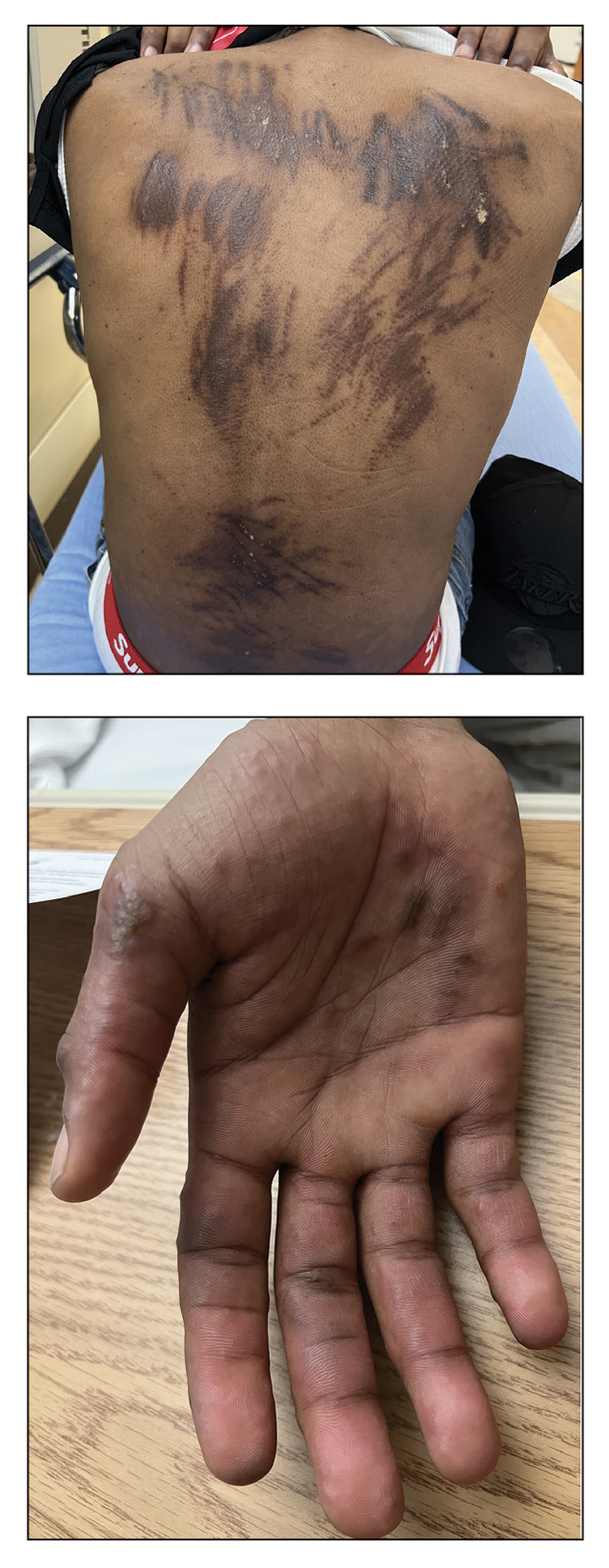
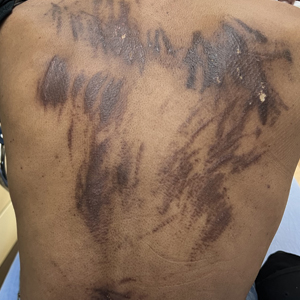
Spontaneously Draining Axillary Tumors in a Young Woman
THE DIAGNOSIS: Ectopic (Accessory) Breast Tissue
Ectopic (accessory) breast tissue (EBT) is a phenomenon caused by failed regression of one or more components of the embryonic mammary ridges— paired ectodermal thickenings that eventually develop into definitive breast tissue including the nipples, areolae, and parenchyma. Ectopic breast tissue is more common in women than men and is believed to be sporadic, although an autosomal-dominant inheritance mechanism with incomplete penetrance has been proposed for some cases.1 The reported incidence of EBT varies greatly among racial and ethnic groups but is most common in individuals of Asian descent. The incidence across all types of EBT is estimated at 0.25% to 6% in the general population.2
Observed clinical variations of EBT range from simple polythelia (additional nipple[s] without associated parenchyma) to complete polymastia (organized and differentiated accessory breasts). Some types of EBT are rarer than others: One report of gynecologic cancer screenings in 1660 patients found polymastia and polythelia incidences of 0.12% and 5.48%, respectively.3 Of the symptomatic variations, isolated parenchymal EBT without a nipple or areolar complex is the most common and may manifest clinically as unilateral or bilateral tender, mildly erythematous nodules or masses often located in the axillae. Ectopic breast tissue generally is observed along the milk line, a developmental regional designation corresponding to the embryologic mammary ridge and extending linearly from the anterior axilla to the inguinal fold on both sides of the body; however, there have been rare reports of EBT manifesting in areas outside the milk line, such as the face, neck, back, vulva, and extremities.2,3
Given that the underlying elements of EBT usually are hormone responsive (as with normal breast tissue), the initial symptom onset and subsequent manifestation frequently coincide with pubertal milestones, pregnancy, or lactation. Furthermore, some patients with EBT may experience symptom fluctuations in concordance with monthly menstrual phases. Many cases of EBT are selflimited and resolve within weeks to months after the end of a pregnancy or lactation, but some cases may persist. Continued observation and follow-up are advisable in all patients, as EBT symptoms often recur and the tissue is susceptible to the same disease processes that affect normal breasts, the most concerning of which is malignancy.4 Although the true incidence is limited by available data, primary ectopic breast malignancy has been estimated to account for 0.3% to 3.8% of diagnosed breast malignancies.2 Cases of malignancy arising from EBT often are of higher grade and poorer prognosis, a finding that may be attributable to diagnostic delays caused by oversight or misdiagnosis of EBT rather than inherent differences in the biologic profile of the tumors.2,4 Patients with a documented history of EBT may benefit from having their routine breast cancer screenings expanded to include areas with EBT foci.
Potential misdiagnoses for EBT include subcutaneous lipoma, axillary lymphadenopathy, abscess, hidradenitis suppurativa, or malignancy. Features that are suggestive of EBT include symptom association with hormone fluctuations (eg, menstrual phases), absence of fever, and lactescent rather than purulent drainage. Among reported EBT cases, spontaneous lactation rarely is described and, if present, often is associated with a history of prior trauma (eg, core needle biopsy or local abscess formation).5 This trauma creates an aberrant connection known as a milk fistula between the underlying parenchyma and the skin surface. Interestingly, our patient denied any history of axillary trauma, but she was noted to be lactating from an apparent milk fistula rather than an organized secretory duct system.
Though a patient history and clinical examination may be sufficient to diagnose EBT cases that are more physically apparent and well correlated with hormone fluctuations, many cases require additional diagnostic studies for confirmation. Of the tools available, ultrasonography generally is considered first-line due to its noninvasive nature, low cost, minimal risk, and high diagnostic value.2 Ultrasonography quickly differentiates between abscesses and cystlike processes, which may appear as discrete areas of decreased echogenicity, and breast tissue, which manifests with fibroglandular tissue and lobules of fat.2,6 Additionally, ultrasonography may demonstrate the secretion of milk through ducts or fistulae, if present. Should examination with ultrasonography prove inconclusive, follow-up studies using conventional radiographic mammography or magnetic resonance imaging may be warranted. Biopsy of EBT foci generally is not indicated unless first-line noninvasive studies fail to yield a conclusive diagnosis; however, biopsy also may be warranted if initial imaging is suggestive of malignancy arising from EBT.2
Management of EBT generally is conservative, and symptoms often resolve without intervention.4 Symptomatic relief may be achieved through techniques such as application of warm/cold compresses, avoidance of mechanical stimulation, and use of over-the-counter pain medicine. In cases that are persistent, frequently recurrent, or associated with severe symptoms or that cause considerable cosmetic impact, management with surgical excision and/or liposuction may be warranted.7 In our patient, the symptoms were not bothersome enough to warrant surgical intervention, so she was managed conservatively and did not return for follow-up.
- Leung AK. Familial supernumerary nipples. Am J Med Genet. 1988;31:631-635. doi:10.1002/ajmg.1320310318
- Visconti G, Eltahir Y, Van Ginkel RJ, et al. Approach and management of primary ectopic breast carcinoma in the axilla: where are we? a comprehensive historical literature review. J Plast Reconstr Aesthet Surg. 2011;64:E1-E11. doi:10.1016/j.bjps.2010.08.015
- Göttlicher S. Incidence and location of polythelias, polymastias and mammae aberratae. a prospective one year study of 1,660 patients of a gynecologic practice. Article in German. Geburtshilfe Frauenheilkd. 1986;46:697-699. doi:10.1055/s-2008-1035944
- Ghosn SH, Khatri KA, Bhawan J. Bilateral aberrant axillary breast tissue mimicking lipomas: report of a case and review of the literature. J Cutan Pathol. 2007;34(suppl 1):9-13. doi:10.1111/j.1600-0560.2006.00713.x
- Firat D, Idiz O, Isik A, et al. Spontaneous milk fistula from an accessory breast: an extremely rare case. Breast J. 2015;21:554-555. doi:10.1111/tbj.12452
- Lim HS, Kim SJ, Baek JM, et al. Sonographic findings of accessory breast tissue in axilla and related diseases. J Ultrasound Med. 2017;36:1469-1478. doi:10.7863/ultra.16.06056
- Gentile P, Izzo V, Cervelli V. Fibroadenoma in the bilateral accessory axillary breast. Aesthetic Plast Surg. 2010;34:657-659. doi:10.1007/ s00266-010-9505-y
THE DIAGNOSIS: Ectopic (Accessory) Breast Tissue
Ectopic (accessory) breast tissue (EBT) is a phenomenon caused by failed regression of one or more components of the embryonic mammary ridges— paired ectodermal thickenings that eventually develop into definitive breast tissue including the nipples, areolae, and parenchyma. Ectopic breast tissue is more common in women than men and is believed to be sporadic, although an autosomal-dominant inheritance mechanism with incomplete penetrance has been proposed for some cases.1 The reported incidence of EBT varies greatly among racial and ethnic groups but is most common in individuals of Asian descent. The incidence across all types of EBT is estimated at 0.25% to 6% in the general population.2
Observed clinical variations of EBT range from simple polythelia (additional nipple[s] without associated parenchyma) to complete polymastia (organized and differentiated accessory breasts). Some types of EBT are rarer than others: One report of gynecologic cancer screenings in 1660 patients found polymastia and polythelia incidences of 0.12% and 5.48%, respectively.3 Of the symptomatic variations, isolated parenchymal EBT without a nipple or areolar complex is the most common and may manifest clinically as unilateral or bilateral tender, mildly erythematous nodules or masses often located in the axillae. Ectopic breast tissue generally is observed along the milk line, a developmental regional designation corresponding to the embryologic mammary ridge and extending linearly from the anterior axilla to the inguinal fold on both sides of the body; however, there have been rare reports of EBT manifesting in areas outside the milk line, such as the face, neck, back, vulva, and extremities.2,3
Given that the underlying elements of EBT usually are hormone responsive (as with normal breast tissue), the initial symptom onset and subsequent manifestation frequently coincide with pubertal milestones, pregnancy, or lactation. Furthermore, some patients with EBT may experience symptom fluctuations in concordance with monthly menstrual phases. Many cases of EBT are selflimited and resolve within weeks to months after the end of a pregnancy or lactation, but some cases may persist. Continued observation and follow-up are advisable in all patients, as EBT symptoms often recur and the tissue is susceptible to the same disease processes that affect normal breasts, the most concerning of which is malignancy.4 Although the true incidence is limited by available data, primary ectopic breast malignancy has been estimated to account for 0.3% to 3.8% of diagnosed breast malignancies.2 Cases of malignancy arising from EBT often are of higher grade and poorer prognosis, a finding that may be attributable to diagnostic delays caused by oversight or misdiagnosis of EBT rather than inherent differences in the biologic profile of the tumors.2,4 Patients with a documented history of EBT may benefit from having their routine breast cancer screenings expanded to include areas with EBT foci.
Potential misdiagnoses for EBT include subcutaneous lipoma, axillary lymphadenopathy, abscess, hidradenitis suppurativa, or malignancy. Features that are suggestive of EBT include symptom association with hormone fluctuations (eg, menstrual phases), absence of fever, and lactescent rather than purulent drainage. Among reported EBT cases, spontaneous lactation rarely is described and, if present, often is associated with a history of prior trauma (eg, core needle biopsy or local abscess formation).5 This trauma creates an aberrant connection known as a milk fistula between the underlying parenchyma and the skin surface. Interestingly, our patient denied any history of axillary trauma, but she was noted to be lactating from an apparent milk fistula rather than an organized secretory duct system.
Though a patient history and clinical examination may be sufficient to diagnose EBT cases that are more physically apparent and well correlated with hormone fluctuations, many cases require additional diagnostic studies for confirmation. Of the tools available, ultrasonography generally is considered first-line due to its noninvasive nature, low cost, minimal risk, and high diagnostic value.2 Ultrasonography quickly differentiates between abscesses and cystlike processes, which may appear as discrete areas of decreased echogenicity, and breast tissue, which manifests with fibroglandular tissue and lobules of fat.2,6 Additionally, ultrasonography may demonstrate the secretion of milk through ducts or fistulae, if present. Should examination with ultrasonography prove inconclusive, follow-up studies using conventional radiographic mammography or magnetic resonance imaging may be warranted. Biopsy of EBT foci generally is not indicated unless first-line noninvasive studies fail to yield a conclusive diagnosis; however, biopsy also may be warranted if initial imaging is suggestive of malignancy arising from EBT.2
Management of EBT generally is conservative, and symptoms often resolve without intervention.4 Symptomatic relief may be achieved through techniques such as application of warm/cold compresses, avoidance of mechanical stimulation, and use of over-the-counter pain medicine. In cases that are persistent, frequently recurrent, or associated with severe symptoms or that cause considerable cosmetic impact, management with surgical excision and/or liposuction may be warranted.7 In our patient, the symptoms were not bothersome enough to warrant surgical intervention, so she was managed conservatively and did not return for follow-up.
THE DIAGNOSIS: Ectopic (Accessory) Breast Tissue
Ectopic (accessory) breast tissue (EBT) is a phenomenon caused by failed regression of one or more components of the embryonic mammary ridges— paired ectodermal thickenings that eventually develop into definitive breast tissue including the nipples, areolae, and parenchyma. Ectopic breast tissue is more common in women than men and is believed to be sporadic, although an autosomal-dominant inheritance mechanism with incomplete penetrance has been proposed for some cases.1 The reported incidence of EBT varies greatly among racial and ethnic groups but is most common in individuals of Asian descent. The incidence across all types of EBT is estimated at 0.25% to 6% in the general population.2
Observed clinical variations of EBT range from simple polythelia (additional nipple[s] without associated parenchyma) to complete polymastia (organized and differentiated accessory breasts). Some types of EBT are rarer than others: One report of gynecologic cancer screenings in 1660 patients found polymastia and polythelia incidences of 0.12% and 5.48%, respectively.3 Of the symptomatic variations, isolated parenchymal EBT without a nipple or areolar complex is the most common and may manifest clinically as unilateral or bilateral tender, mildly erythematous nodules or masses often located in the axillae. Ectopic breast tissue generally is observed along the milk line, a developmental regional designation corresponding to the embryologic mammary ridge and extending linearly from the anterior axilla to the inguinal fold on both sides of the body; however, there have been rare reports of EBT manifesting in areas outside the milk line, such as the face, neck, back, vulva, and extremities.2,3
Given that the underlying elements of EBT usually are hormone responsive (as with normal breast tissue), the initial symptom onset and subsequent manifestation frequently coincide with pubertal milestones, pregnancy, or lactation. Furthermore, some patients with EBT may experience symptom fluctuations in concordance with monthly menstrual phases. Many cases of EBT are selflimited and resolve within weeks to months after the end of a pregnancy or lactation, but some cases may persist. Continued observation and follow-up are advisable in all patients, as EBT symptoms often recur and the tissue is susceptible to the same disease processes that affect normal breasts, the most concerning of which is malignancy.4 Although the true incidence is limited by available data, primary ectopic breast malignancy has been estimated to account for 0.3% to 3.8% of diagnosed breast malignancies.2 Cases of malignancy arising from EBT often are of higher grade and poorer prognosis, a finding that may be attributable to diagnostic delays caused by oversight or misdiagnosis of EBT rather than inherent differences in the biologic profile of the tumors.2,4 Patients with a documented history of EBT may benefit from having their routine breast cancer screenings expanded to include areas with EBT foci.
Potential misdiagnoses for EBT include subcutaneous lipoma, axillary lymphadenopathy, abscess, hidradenitis suppurativa, or malignancy. Features that are suggestive of EBT include symptom association with hormone fluctuations (eg, menstrual phases), absence of fever, and lactescent rather than purulent drainage. Among reported EBT cases, spontaneous lactation rarely is described and, if present, often is associated with a history of prior trauma (eg, core needle biopsy or local abscess formation).5 This trauma creates an aberrant connection known as a milk fistula between the underlying parenchyma and the skin surface. Interestingly, our patient denied any history of axillary trauma, but she was noted to be lactating from an apparent milk fistula rather than an organized secretory duct system.
Though a patient history and clinical examination may be sufficient to diagnose EBT cases that are more physically apparent and well correlated with hormone fluctuations, many cases require additional diagnostic studies for confirmation. Of the tools available, ultrasonography generally is considered first-line due to its noninvasive nature, low cost, minimal risk, and high diagnostic value.2 Ultrasonography quickly differentiates between abscesses and cystlike processes, which may appear as discrete areas of decreased echogenicity, and breast tissue, which manifests with fibroglandular tissue and lobules of fat.2,6 Additionally, ultrasonography may demonstrate the secretion of milk through ducts or fistulae, if present. Should examination with ultrasonography prove inconclusive, follow-up studies using conventional radiographic mammography or magnetic resonance imaging may be warranted. Biopsy of EBT foci generally is not indicated unless first-line noninvasive studies fail to yield a conclusive diagnosis; however, biopsy also may be warranted if initial imaging is suggestive of malignancy arising from EBT.2
Management of EBT generally is conservative, and symptoms often resolve without intervention.4 Symptomatic relief may be achieved through techniques such as application of warm/cold compresses, avoidance of mechanical stimulation, and use of over-the-counter pain medicine. In cases that are persistent, frequently recurrent, or associated with severe symptoms or that cause considerable cosmetic impact, management with surgical excision and/or liposuction may be warranted.7 In our patient, the symptoms were not bothersome enough to warrant surgical intervention, so she was managed conservatively and did not return for follow-up.
- Leung AK. Familial supernumerary nipples. Am J Med Genet. 1988;31:631-635. doi:10.1002/ajmg.1320310318
- Visconti G, Eltahir Y, Van Ginkel RJ, et al. Approach and management of primary ectopic breast carcinoma in the axilla: where are we? a comprehensive historical literature review. J Plast Reconstr Aesthet Surg. 2011;64:E1-E11. doi:10.1016/j.bjps.2010.08.015
- Göttlicher S. Incidence and location of polythelias, polymastias and mammae aberratae. a prospective one year study of 1,660 patients of a gynecologic practice. Article in German. Geburtshilfe Frauenheilkd. 1986;46:697-699. doi:10.1055/s-2008-1035944
- Ghosn SH, Khatri KA, Bhawan J. Bilateral aberrant axillary breast tissue mimicking lipomas: report of a case and review of the literature. J Cutan Pathol. 2007;34(suppl 1):9-13. doi:10.1111/j.1600-0560.2006.00713.x
- Firat D, Idiz O, Isik A, et al. Spontaneous milk fistula from an accessory breast: an extremely rare case. Breast J. 2015;21:554-555. doi:10.1111/tbj.12452
- Lim HS, Kim SJ, Baek JM, et al. Sonographic findings of accessory breast tissue in axilla and related diseases. J Ultrasound Med. 2017;36:1469-1478. doi:10.7863/ultra.16.06056
- Gentile P, Izzo V, Cervelli V. Fibroadenoma in the bilateral accessory axillary breast. Aesthetic Plast Surg. 2010;34:657-659. doi:10.1007/ s00266-010-9505-y
- Leung AK. Familial supernumerary nipples. Am J Med Genet. 1988;31:631-635. doi:10.1002/ajmg.1320310318
- Visconti G, Eltahir Y, Van Ginkel RJ, et al. Approach and management of primary ectopic breast carcinoma in the axilla: where are we? a comprehensive historical literature review. J Plast Reconstr Aesthet Surg. 2011;64:E1-E11. doi:10.1016/j.bjps.2010.08.015
- Göttlicher S. Incidence and location of polythelias, polymastias and mammae aberratae. a prospective one year study of 1,660 patients of a gynecologic practice. Article in German. Geburtshilfe Frauenheilkd. 1986;46:697-699. doi:10.1055/s-2008-1035944
- Ghosn SH, Khatri KA, Bhawan J. Bilateral aberrant axillary breast tissue mimicking lipomas: report of a case and review of the literature. J Cutan Pathol. 2007;34(suppl 1):9-13. doi:10.1111/j.1600-0560.2006.00713.x
- Firat D, Idiz O, Isik A, et al. Spontaneous milk fistula from an accessory breast: an extremely rare case. Breast J. 2015;21:554-555. doi:10.1111/tbj.12452
- Lim HS, Kim SJ, Baek JM, et al. Sonographic findings of accessory breast tissue in axilla and related diseases. J Ultrasound Med. 2017;36:1469-1478. doi:10.7863/ultra.16.06056
- Gentile P, Izzo V, Cervelli V. Fibroadenoma in the bilateral accessory axillary breast. Aesthetic Plast Surg. 2010;34:657-659. doi:10.1007/ s00266-010-9505-y
A 19-year-old G1P1A0 woman presented to the dermatology clinic for evaluation of bilateral axillary swelling, pain, and spontaneous drainage of approximately 2 weeks’ duration. The patient, who was 2 weeks postpartum, reported that the symptoms were associated with lactation when breastfeeding. She denied any personal or family history of hidradenitis suppurativa or other formally diagnosed dermatologic condition. Physical examination revealed a soft, mildly tender, well-circumscribed, nonfluctuant mobile mass in each axilla. Both lesions had a single central sinus tract with thin lactescent discharge that spontaneously drained and was expressible. A single thin hyperpigmented papule was noted on the anterior aspect of each mass.

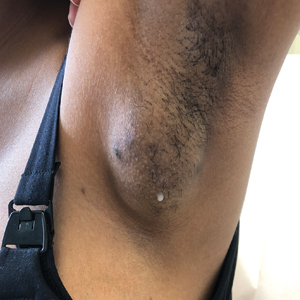
Hairless Scalp Lesion
The Diagnosis: Nevus Sebaceus of Jadassohn
The diagnosis of nevus sebaceus of Jadassohn was made clinically based on the lesion’s appearance and presence since birth as well as the absence of systemic symptoms. Clinically, nevus sebaceus of Jadassohn typically manifests as a well-demarcated, yellow- brown plaque often located on the scalp, as was seen in our patient. The lack of pruritus and pain further supported the diagnosis in our patient. No biopsy was performed, as the presentation was considered classic for this condition. Our patient opted to forgo surgery and will be routinely monitored for any changes, as nevus sebaceus has a potential risk, albeit low, for malignant transformation later in life. No changes have been observed since the initial presentation, and regular follow-ups are planned to monitor for future developments.
Nevus sebaceus of Jadassohn is a hamartomatous lesion involving the pilosebaceous follicle and adjacent adnexal structures.1-3 It most commonly forms on the scalp (59.3%) and is accompanied by partial or total alopecia. 3,4 It is seen less often on the face, periauricular area, or neck1,4; thorax or limbs5; and oral or genital mucosae.6 Nevus sebaceus of Jadassohn affects approximately 0.3% of newborns,1 usually as a solitary lesion that can form an extensive plaque. The male-to-female occurrence ratio has been reported as equal to slightly more predominant in females; all races and ethnicities are affected.1,5
Nevus sebaceus of Jadassohn follows 3 stages of clinical development: infantile, adolescent, and adulthood. It manifests at birth or shortly afterward as a smooth hairless patch or plaque that is yellowish and can be hyperpigmented in Black patients.5 It may have an oval or linear configuration, typically is asymptomatic, and often arises along the Blaschko lines when it occurs as multiple lesions (a rare manifestation).1 During puberty, hormonal changes cause accelerated growth, sebaceous gland maturation, and epidermal hyperplasia. 7 Nevus sebaceus of Jadassohn often is not identified until this stage, when its classic wartlike appearance has fully developed.1
Patients with nevus sebaceus of Jadassohn have a 10% to 20% risk for tumor development in adulthood.2,7 Trichoblastoma and syringocystadenoma papilliferum are the most frequently described neoplasms.8 Basal cell carcinoma is the most common malignant secondary neoplasm with an occurrence rate of 0.8%.6,9 However, basal cell carcinoma and trichoblastoma may share histopathologic features, which may lead to misdiagnosis and a higher reported incidence of basal cell carcinoma in adults than is accurate.2
Early prophylactic surgical removal of nevus sebaceus of Jadassohn has been recommended; however, surgical management is controversial because the risk for a benign secondary neoplasm remains relatively high while the risk for malignancy is much lower.2,7 Surgical excision remains an acceptable option once the patient is mature enough to tolerate the procedure.1 However, patient education regarding watchful waiting vs a surgical approach— and the risks of each—is critical to ensure shared decision-making and a management plan tailored to the individual.
The differential diagnosis includes hypertrophic lichen planus, Langerhans cell histiocytosis (Letterer-Siwe disease type), epidermal nevus, and seborrheic keratosis. Hypertrophic lichen planus often occurs symmetrically on the dorsal feet and shins with thick, scaly, and extremely pruritic plaques. The lesions often persist for an average of 6 years and may lead to multiple keratoacanthomas or follicular base squamous cell carcinomas. Langerhans cell histiocytosis (Letterer-Siwe disease type) manifests with acute, disseminated, visceral, and cutaneous lesions before 2 years of age. These lesions appear as 1- to 2-mm, pink, seborrheic papules, pustules, or vesicles on the scalp, flexural neck, axilla, perineum, and trunk; they often are associated with petechiae, purpura, scale, crust, erosion, impetiginization, and tender fissures. Epidermal nevus occurs within the first year of life and is a hamartoma of the epidermis and papillary dermis. It manifests as papillomatous pigmented linear lines along the Blaschko lines. Seborrheic keratosis manifests as well-demarcated, waxy/verrucous, brown papules with a “stuck on” appearance on hair-bearing skin sparing the mucosae. They are common benign lesions associated with sun exposure and often manifest in the fourth decade of life.10
- Baigrie D, Troxell T, Cook C. Nevus sebaceus. StatPearls [Internet]. Updated August 16, 2023. Accessed September 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482493/
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239. doi:10.1097/01 .scs.0000221531.56529.cc
- Kelati A, Baybay H, Gallouj S, et al. Dermoscopic analysis of nevus sebaceus of Jadassohn: a study of 13 cases. Skin Appendage Disord. 2017;3:83-91. doi:10.1159/000460258
- Ugras N, Ozgun G, Adim SB, et al. Nevus sebaceous at unusual location: a rare presentation. Indian J Pathol Microbiol. 2012;55:419-420. doi:10.4103/0377-4929.101768
- Serpas de Lopez RM, Hernandez-Perez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72. doi:10.1111/j.1524-4725 .1985.tb02893.x
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268. doi:10.1016/S0190-9622(00)90136-1
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660. doi:10.1097/00001665-200309000-00010
- Chahboun F, Eljazouly M, Elomari M, et al. Trichoblastoma arising from the nevus sebaceus of Jadassohn. Cureus. 2021;13:E15325. doi:10.7759/cureus.15325
- Cazzato G, Cimmino A, Colagrande A, et al. The multiple faces of nodular trichoblastoma: review of the literature with case presentation. Dermatopathology (Basel). 2021;8:265-270. doi:10.3390 /dermatopathology8030032
- Dandekar MN, Gandhi RK. Neoplastic dermatology. In: Alikhan A, Hocker TLH (eds). Review of Dermatology. Elsevier; 2016: 321-366.
The Diagnosis: Nevus Sebaceus of Jadassohn
The diagnosis of nevus sebaceus of Jadassohn was made clinically based on the lesion’s appearance and presence since birth as well as the absence of systemic symptoms. Clinically, nevus sebaceus of Jadassohn typically manifests as a well-demarcated, yellow- brown plaque often located on the scalp, as was seen in our patient. The lack of pruritus and pain further supported the diagnosis in our patient. No biopsy was performed, as the presentation was considered classic for this condition. Our patient opted to forgo surgery and will be routinely monitored for any changes, as nevus sebaceus has a potential risk, albeit low, for malignant transformation later in life. No changes have been observed since the initial presentation, and regular follow-ups are planned to monitor for future developments.
Nevus sebaceus of Jadassohn is a hamartomatous lesion involving the pilosebaceous follicle and adjacent adnexal structures.1-3 It most commonly forms on the scalp (59.3%) and is accompanied by partial or total alopecia. 3,4 It is seen less often on the face, periauricular area, or neck1,4; thorax or limbs5; and oral or genital mucosae.6 Nevus sebaceus of Jadassohn affects approximately 0.3% of newborns,1 usually as a solitary lesion that can form an extensive plaque. The male-to-female occurrence ratio has been reported as equal to slightly more predominant in females; all races and ethnicities are affected.1,5
Nevus sebaceus of Jadassohn follows 3 stages of clinical development: infantile, adolescent, and adulthood. It manifests at birth or shortly afterward as a smooth hairless patch or plaque that is yellowish and can be hyperpigmented in Black patients.5 It may have an oval or linear configuration, typically is asymptomatic, and often arises along the Blaschko lines when it occurs as multiple lesions (a rare manifestation).1 During puberty, hormonal changes cause accelerated growth, sebaceous gland maturation, and epidermal hyperplasia. 7 Nevus sebaceus of Jadassohn often is not identified until this stage, when its classic wartlike appearance has fully developed.1
Patients with nevus sebaceus of Jadassohn have a 10% to 20% risk for tumor development in adulthood.2,7 Trichoblastoma and syringocystadenoma papilliferum are the most frequently described neoplasms.8 Basal cell carcinoma is the most common malignant secondary neoplasm with an occurrence rate of 0.8%.6,9 However, basal cell carcinoma and trichoblastoma may share histopathologic features, which may lead to misdiagnosis and a higher reported incidence of basal cell carcinoma in adults than is accurate.2
Early prophylactic surgical removal of nevus sebaceus of Jadassohn has been recommended; however, surgical management is controversial because the risk for a benign secondary neoplasm remains relatively high while the risk for malignancy is much lower.2,7 Surgical excision remains an acceptable option once the patient is mature enough to tolerate the procedure.1 However, patient education regarding watchful waiting vs a surgical approach— and the risks of each—is critical to ensure shared decision-making and a management plan tailored to the individual.
The differential diagnosis includes hypertrophic lichen planus, Langerhans cell histiocytosis (Letterer-Siwe disease type), epidermal nevus, and seborrheic keratosis. Hypertrophic lichen planus often occurs symmetrically on the dorsal feet and shins with thick, scaly, and extremely pruritic plaques. The lesions often persist for an average of 6 years and may lead to multiple keratoacanthomas or follicular base squamous cell carcinomas. Langerhans cell histiocytosis (Letterer-Siwe disease type) manifests with acute, disseminated, visceral, and cutaneous lesions before 2 years of age. These lesions appear as 1- to 2-mm, pink, seborrheic papules, pustules, or vesicles on the scalp, flexural neck, axilla, perineum, and trunk; they often are associated with petechiae, purpura, scale, crust, erosion, impetiginization, and tender fissures. Epidermal nevus occurs within the first year of life and is a hamartoma of the epidermis and papillary dermis. It manifests as papillomatous pigmented linear lines along the Blaschko lines. Seborrheic keratosis manifests as well-demarcated, waxy/verrucous, brown papules with a “stuck on” appearance on hair-bearing skin sparing the mucosae. They are common benign lesions associated with sun exposure and often manifest in the fourth decade of life.10
The Diagnosis: Nevus Sebaceus of Jadassohn
The diagnosis of nevus sebaceus of Jadassohn was made clinically based on the lesion’s appearance and presence since birth as well as the absence of systemic symptoms. Clinically, nevus sebaceus of Jadassohn typically manifests as a well-demarcated, yellow- brown plaque often located on the scalp, as was seen in our patient. The lack of pruritus and pain further supported the diagnosis in our patient. No biopsy was performed, as the presentation was considered classic for this condition. Our patient opted to forgo surgery and will be routinely monitored for any changes, as nevus sebaceus has a potential risk, albeit low, for malignant transformation later in life. No changes have been observed since the initial presentation, and regular follow-ups are planned to monitor for future developments.
Nevus sebaceus of Jadassohn is a hamartomatous lesion involving the pilosebaceous follicle and adjacent adnexal structures.1-3 It most commonly forms on the scalp (59.3%) and is accompanied by partial or total alopecia. 3,4 It is seen less often on the face, periauricular area, or neck1,4; thorax or limbs5; and oral or genital mucosae.6 Nevus sebaceus of Jadassohn affects approximately 0.3% of newborns,1 usually as a solitary lesion that can form an extensive plaque. The male-to-female occurrence ratio has been reported as equal to slightly more predominant in females; all races and ethnicities are affected.1,5
Nevus sebaceus of Jadassohn follows 3 stages of clinical development: infantile, adolescent, and adulthood. It manifests at birth or shortly afterward as a smooth hairless patch or plaque that is yellowish and can be hyperpigmented in Black patients.5 It may have an oval or linear configuration, typically is asymptomatic, and often arises along the Blaschko lines when it occurs as multiple lesions (a rare manifestation).1 During puberty, hormonal changes cause accelerated growth, sebaceous gland maturation, and epidermal hyperplasia. 7 Nevus sebaceus of Jadassohn often is not identified until this stage, when its classic wartlike appearance has fully developed.1
Patients with nevus sebaceus of Jadassohn have a 10% to 20% risk for tumor development in adulthood.2,7 Trichoblastoma and syringocystadenoma papilliferum are the most frequently described neoplasms.8 Basal cell carcinoma is the most common malignant secondary neoplasm with an occurrence rate of 0.8%.6,9 However, basal cell carcinoma and trichoblastoma may share histopathologic features, which may lead to misdiagnosis and a higher reported incidence of basal cell carcinoma in adults than is accurate.2
Early prophylactic surgical removal of nevus sebaceus of Jadassohn has been recommended; however, surgical management is controversial because the risk for a benign secondary neoplasm remains relatively high while the risk for malignancy is much lower.2,7 Surgical excision remains an acceptable option once the patient is mature enough to tolerate the procedure.1 However, patient education regarding watchful waiting vs a surgical approach— and the risks of each—is critical to ensure shared decision-making and a management plan tailored to the individual.
The differential diagnosis includes hypertrophic lichen planus, Langerhans cell histiocytosis (Letterer-Siwe disease type), epidermal nevus, and seborrheic keratosis. Hypertrophic lichen planus often occurs symmetrically on the dorsal feet and shins with thick, scaly, and extremely pruritic plaques. The lesions often persist for an average of 6 years and may lead to multiple keratoacanthomas or follicular base squamous cell carcinomas. Langerhans cell histiocytosis (Letterer-Siwe disease type) manifests with acute, disseminated, visceral, and cutaneous lesions before 2 years of age. These lesions appear as 1- to 2-mm, pink, seborrheic papules, pustules, or vesicles on the scalp, flexural neck, axilla, perineum, and trunk; they often are associated with petechiae, purpura, scale, crust, erosion, impetiginization, and tender fissures. Epidermal nevus occurs within the first year of life and is a hamartoma of the epidermis and papillary dermis. It manifests as papillomatous pigmented linear lines along the Blaschko lines. Seborrheic keratosis manifests as well-demarcated, waxy/verrucous, brown papules with a “stuck on” appearance on hair-bearing skin sparing the mucosae. They are common benign lesions associated with sun exposure and often manifest in the fourth decade of life.10
- Baigrie D, Troxell T, Cook C. Nevus sebaceus. StatPearls [Internet]. Updated August 16, 2023. Accessed September 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482493/
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239. doi:10.1097/01 .scs.0000221531.56529.cc
- Kelati A, Baybay H, Gallouj S, et al. Dermoscopic analysis of nevus sebaceus of Jadassohn: a study of 13 cases. Skin Appendage Disord. 2017;3:83-91. doi:10.1159/000460258
- Ugras N, Ozgun G, Adim SB, et al. Nevus sebaceous at unusual location: a rare presentation. Indian J Pathol Microbiol. 2012;55:419-420. doi:10.4103/0377-4929.101768
- Serpas de Lopez RM, Hernandez-Perez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72. doi:10.1111/j.1524-4725 .1985.tb02893.x
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268. doi:10.1016/S0190-9622(00)90136-1
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660. doi:10.1097/00001665-200309000-00010
- Chahboun F, Eljazouly M, Elomari M, et al. Trichoblastoma arising from the nevus sebaceus of Jadassohn. Cureus. 2021;13:E15325. doi:10.7759/cureus.15325
- Cazzato G, Cimmino A, Colagrande A, et al. The multiple faces of nodular trichoblastoma: review of the literature with case presentation. Dermatopathology (Basel). 2021;8:265-270. doi:10.3390 /dermatopathology8030032
- Dandekar MN, Gandhi RK. Neoplastic dermatology. In: Alikhan A, Hocker TLH (eds). Review of Dermatology. Elsevier; 2016: 321-366.
- Baigrie D, Troxell T, Cook C. Nevus sebaceus. StatPearls [Internet]. Updated August 16, 2023. Accessed September 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482493/
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239. doi:10.1097/01 .scs.0000221531.56529.cc
- Kelati A, Baybay H, Gallouj S, et al. Dermoscopic analysis of nevus sebaceus of Jadassohn: a study of 13 cases. Skin Appendage Disord. 2017;3:83-91. doi:10.1159/000460258
- Ugras N, Ozgun G, Adim SB, et al. Nevus sebaceous at unusual location: a rare presentation. Indian J Pathol Microbiol. 2012;55:419-420. doi:10.4103/0377-4929.101768
- Serpas de Lopez RM, Hernandez-Perez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72. doi:10.1111/j.1524-4725 .1985.tb02893.x
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268. doi:10.1016/S0190-9622(00)90136-1
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660. doi:10.1097/00001665-200309000-00010
- Chahboun F, Eljazouly M, Elomari M, et al. Trichoblastoma arising from the nevus sebaceus of Jadassohn. Cureus. 2021;13:E15325. doi:10.7759/cureus.15325
- Cazzato G, Cimmino A, Colagrande A, et al. The multiple faces of nodular trichoblastoma: review of the literature with case presentation. Dermatopathology (Basel). 2021;8:265-270. doi:10.3390 /dermatopathology8030032
- Dandekar MN, Gandhi RK. Neoplastic dermatology. In: Alikhan A, Hocker TLH (eds). Review of Dermatology. Elsevier; 2016: 321-366.
A 23-year-old man presented to the dermatology clinic with hair loss on the scalp of several years’ duration. The patient reported persistent pigmented bumps on the back of the scalp. He denied any pruritus or pain and had no systemic symptoms or comorbidities. Physical examination revealed a 1×1.5-cm, yellow-brown, hairless plaque on the left parietal scalp.
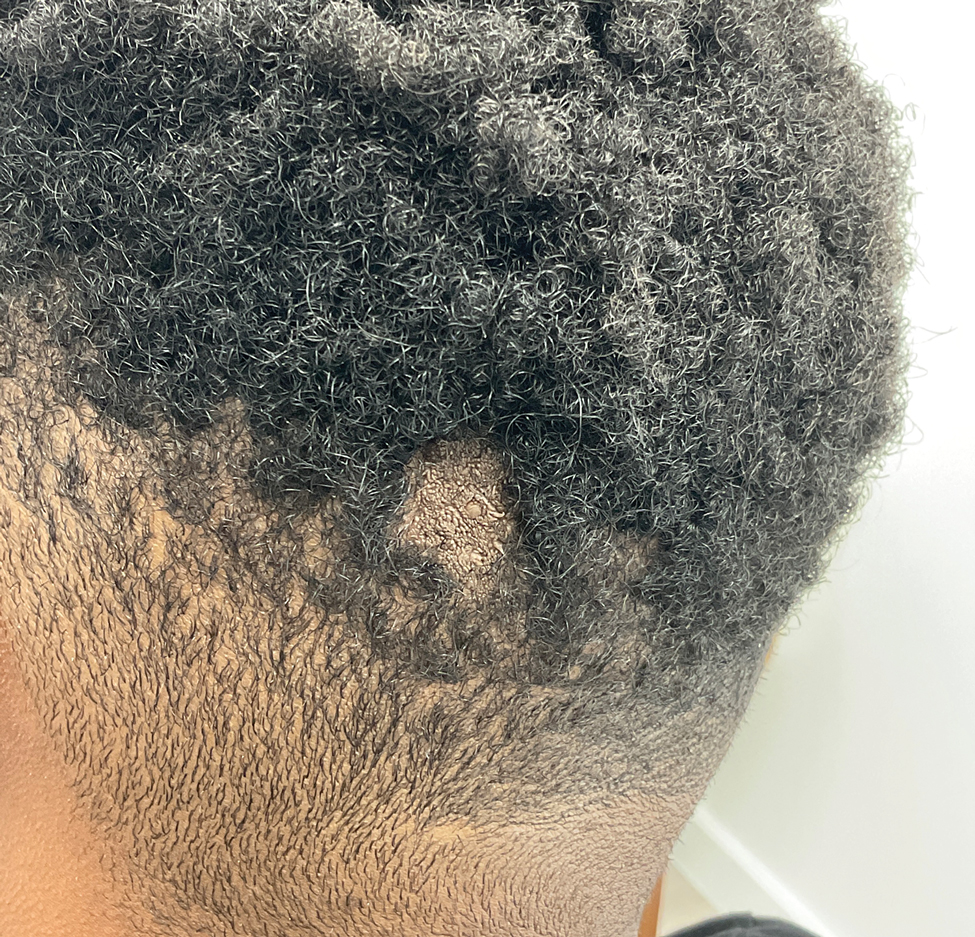
Purpuric Lesions on the Leg
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
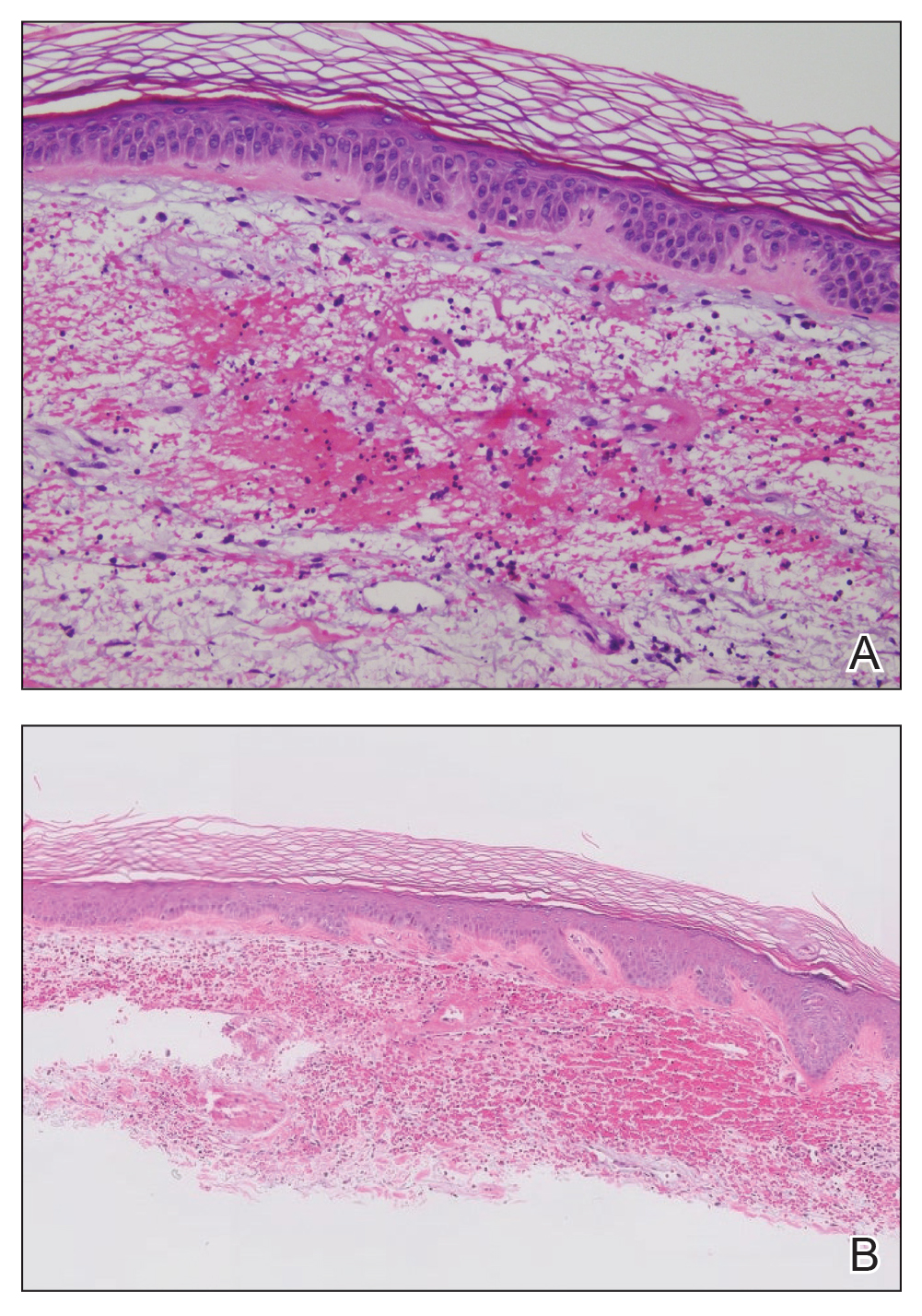
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
A 74-year-old woman who frequently traveled abroad presented to the dermatology department with retiform purpura of the lower leg along with gastrointestinal cramps, fatigue, and myalgia. The patient reported that the symptoms had started 10 days after returning from a recent trip to Africa.