User login
Vegetative Sacral Plaque in a Patient With Human Immunodeficiency Virus
The Diagnosis: Herpes Simplex Vegetans
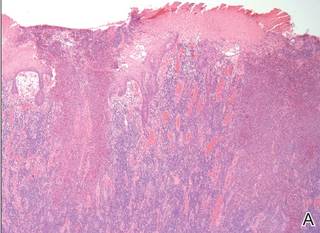
Histopathologic examination using hematoxylin and eosin stain demonstrated marked pseudoepitheliomatous hyperplasia with granulation tissue, ulceration, and abundant exudate joined by a dense mixed inflammatory cell infiltrate that included a myriad of eosinophils (Figure, A). At higher power (Figure, B), many single and multinucleate acantholytic keratinocytes showed ground-glass nuclei and peripheral margination of chromatin within zones of ulceration and crust. Viral culture and direct fluorescent antibody assay identified herpes simplex virus (HSV) type 2. Based on the clinical and histopathologic findings, the patient was diagnosed with herpes simplex vegetans. He was initially treated with oral acyclovir and then oral famciclovir but showed minimal improvement. Eventually, he was referred to surgery and the mass was totally excised with clear margins and no evidence of underlying malignancy.

|
| Histopathology revealed marked pseudoepitheliomatous hyperplasia, ulceration, and a dense mixed inflammatory cell infiltrate (A)(H&E, original magnification ×20). Many multinucleate acantholytic keratinocytes with ground-glass nuclei and peripheral margination of chromatin were shown (B)(H&E, original magnification ×400). |
Herpes simplex virus is one of the most common sexually transmitted infections, with a notably increased incidence and prevalence among individuals with human immunodeficiency virus (HIV) infection.1 Although typical HSV manifestation in immunocompetent patients includes clustered vesicles and/or ulcerations, immunocompromised patients may have unusual presentations, such as persistent and extensive ulcerations or nodular hyperkeratotic lesions.2,3 Herpes vegetans, a term used to describe these atypical exophytic lesions, rarely has been reported in literature, but its presence should raise suspicion for possible underlying immunocompromise. The pathogenesis behind the hypertrophic nature of these lesions is not well understood, but it is postulated that the immune dysregulation from concomitant HIV and HSV infection plays a role.2 Overproduction of tumor necrosis factor and IL-6 by HIV-infected dermal dendritic cells causes an increase in antiapoptotic factors within the epidermis, resulting in enhanced keratinocyte proliferation and clinical hyperkeratosis.2,4
The differential diagnosis for herpes vegetans is somewhat broad, owing to the verrucous and often eroded appearance of the lesions. Biopsy and cultures can be obtained to differentiate from condyloma acuminatum, condyloma latum (secondary syphilis), pyoderma vegetans, pemphigus vegetans, granuloma inguinale, extraintestinal Crohn disease, deep fungal infections, cutaneous tuberculosis, and malignancy.2-4 Histopathology shows epithelial hyperplasia and ulceration with scattered multinucleate keratinocytes, usually at the periphery of the ulcer, and intranuclear inclusions typical of HSV. In addition, a dense dermal infiltrate of lymphocytes, histiocytes, plasma cells, and eosinophils is usually present beneath the base of the ulcer.2,4
Treatment options for herpes vegetans are limited due to the high prevalence of acyclovir-resistant (ACV-R) HSV-2 strains in HIV patients. Valacyclovir and penciclovir have been largely ineffective against ACV-R HSV due to their dependence on the same enzyme—thymidine kinase—involved in the mechanism of acyclovir resistance. Intravenous foscarnet and cidofovir have shown efficacy against ACV-R virus, but concerns of nephrotoxicity have limited their use over prolonged intervals.5 Castelo-Soccio et al6 reported promising results with intralesional cidofovir. This route of administration provides the advantage of increased bioavailability with reduced risk for nephrotoxicity.6 Finally, surgical resection may be considered for refractory lesions to circumvent the toxicity from systemically administered drugs.3
- Severson JL, Tyring SK. Relation between herpes simplex viruses and human immunodeficiency virus infections. Arch Dermatol. 1999;135:1393-1397.
- Patel AB, Rosen T. Herpes vegetans as a sign of HIV infection. Dermatol Online J. 2008;14:6.
- Chung VQ, Parker DC, Parker SR. Surgical excision for vegetative herpes simplex virus infection. Dermatol Surg. 2007;33:1374-1379.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Chilukuri S, Rosen T. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin. 2003;21:311-320.
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126.
The Diagnosis: Herpes Simplex Vegetans
Histopathologic examination using hematoxylin and eosin stain demonstrated marked pseudoepitheliomatous hyperplasia with granulation tissue, ulceration, and abundant exudate joined by a dense mixed inflammatory cell infiltrate that included a myriad of eosinophils (Figure, A). At higher power (Figure, B), many single and multinucleate acantholytic keratinocytes showed ground-glass nuclei and peripheral margination of chromatin within zones of ulceration and crust. Viral culture and direct fluorescent antibody assay identified herpes simplex virus (HSV) type 2. Based on the clinical and histopathologic findings, the patient was diagnosed with herpes simplex vegetans. He was initially treated with oral acyclovir and then oral famciclovir but showed minimal improvement. Eventually, he was referred to surgery and the mass was totally excised with clear margins and no evidence of underlying malignancy.
|
|
| Histopathology revealed marked pseudoepitheliomatous hyperplasia, ulceration, and a dense mixed inflammatory cell infiltrate (A)(H&E, original magnification ×20). Many multinucleate acantholytic keratinocytes with ground-glass nuclei and peripheral margination of chromatin were shown (B)(H&E, original magnification ×400). |
Herpes simplex virus is one of the most common sexually transmitted infections, with a notably increased incidence and prevalence among individuals with human immunodeficiency virus (HIV) infection.1 Although typical HSV manifestation in immunocompetent patients includes clustered vesicles and/or ulcerations, immunocompromised patients may have unusual presentations, such as persistent and extensive ulcerations or nodular hyperkeratotic lesions.2,3 Herpes vegetans, a term used to describe these atypical exophytic lesions, rarely has been reported in literature, but its presence should raise suspicion for possible underlying immunocompromise. The pathogenesis behind the hypertrophic nature of these lesions is not well understood, but it is postulated that the immune dysregulation from concomitant HIV and HSV infection plays a role.2 Overproduction of tumor necrosis factor and IL-6 by HIV-infected dermal dendritic cells causes an increase in antiapoptotic factors within the epidermis, resulting in enhanced keratinocyte proliferation and clinical hyperkeratosis.2,4
The differential diagnosis for herpes vegetans is somewhat broad, owing to the verrucous and often eroded appearance of the lesions. Biopsy and cultures can be obtained to differentiate from condyloma acuminatum, condyloma latum (secondary syphilis), pyoderma vegetans, pemphigus vegetans, granuloma inguinale, extraintestinal Crohn disease, deep fungal infections, cutaneous tuberculosis, and malignancy.2-4 Histopathology shows epithelial hyperplasia and ulceration with scattered multinucleate keratinocytes, usually at the periphery of the ulcer, and intranuclear inclusions typical of HSV. In addition, a dense dermal infiltrate of lymphocytes, histiocytes, plasma cells, and eosinophils is usually present beneath the base of the ulcer.2,4
Treatment options for herpes vegetans are limited due to the high prevalence of acyclovir-resistant (ACV-R) HSV-2 strains in HIV patients. Valacyclovir and penciclovir have been largely ineffective against ACV-R HSV due to their dependence on the same enzyme—thymidine kinase—involved in the mechanism of acyclovir resistance. Intravenous foscarnet and cidofovir have shown efficacy against ACV-R virus, but concerns of nephrotoxicity have limited their use over prolonged intervals.5 Castelo-Soccio et al6 reported promising results with intralesional cidofovir. This route of administration provides the advantage of increased bioavailability with reduced risk for nephrotoxicity.6 Finally, surgical resection may be considered for refractory lesions to circumvent the toxicity from systemically administered drugs.3
The Diagnosis: Herpes Simplex Vegetans
Histopathologic examination using hematoxylin and eosin stain demonstrated marked pseudoepitheliomatous hyperplasia with granulation tissue, ulceration, and abundant exudate joined by a dense mixed inflammatory cell infiltrate that included a myriad of eosinophils (Figure, A). At higher power (Figure, B), many single and multinucleate acantholytic keratinocytes showed ground-glass nuclei and peripheral margination of chromatin within zones of ulceration and crust. Viral culture and direct fluorescent antibody assay identified herpes simplex virus (HSV) type 2. Based on the clinical and histopathologic findings, the patient was diagnosed with herpes simplex vegetans. He was initially treated with oral acyclovir and then oral famciclovir but showed minimal improvement. Eventually, he was referred to surgery and the mass was totally excised with clear margins and no evidence of underlying malignancy.
|
|
| Histopathology revealed marked pseudoepitheliomatous hyperplasia, ulceration, and a dense mixed inflammatory cell infiltrate (A)(H&E, original magnification ×20). Many multinucleate acantholytic keratinocytes with ground-glass nuclei and peripheral margination of chromatin were shown (B)(H&E, original magnification ×400). |
Herpes simplex virus is one of the most common sexually transmitted infections, with a notably increased incidence and prevalence among individuals with human immunodeficiency virus (HIV) infection.1 Although typical HSV manifestation in immunocompetent patients includes clustered vesicles and/or ulcerations, immunocompromised patients may have unusual presentations, such as persistent and extensive ulcerations or nodular hyperkeratotic lesions.2,3 Herpes vegetans, a term used to describe these atypical exophytic lesions, rarely has been reported in literature, but its presence should raise suspicion for possible underlying immunocompromise. The pathogenesis behind the hypertrophic nature of these lesions is not well understood, but it is postulated that the immune dysregulation from concomitant HIV and HSV infection plays a role.2 Overproduction of tumor necrosis factor and IL-6 by HIV-infected dermal dendritic cells causes an increase in antiapoptotic factors within the epidermis, resulting in enhanced keratinocyte proliferation and clinical hyperkeratosis.2,4
The differential diagnosis for herpes vegetans is somewhat broad, owing to the verrucous and often eroded appearance of the lesions. Biopsy and cultures can be obtained to differentiate from condyloma acuminatum, condyloma latum (secondary syphilis), pyoderma vegetans, pemphigus vegetans, granuloma inguinale, extraintestinal Crohn disease, deep fungal infections, cutaneous tuberculosis, and malignancy.2-4 Histopathology shows epithelial hyperplasia and ulceration with scattered multinucleate keratinocytes, usually at the periphery of the ulcer, and intranuclear inclusions typical of HSV. In addition, a dense dermal infiltrate of lymphocytes, histiocytes, plasma cells, and eosinophils is usually present beneath the base of the ulcer.2,4
Treatment options for herpes vegetans are limited due to the high prevalence of acyclovir-resistant (ACV-R) HSV-2 strains in HIV patients. Valacyclovir and penciclovir have been largely ineffective against ACV-R HSV due to their dependence on the same enzyme—thymidine kinase—involved in the mechanism of acyclovir resistance. Intravenous foscarnet and cidofovir have shown efficacy against ACV-R virus, but concerns of nephrotoxicity have limited their use over prolonged intervals.5 Castelo-Soccio et al6 reported promising results with intralesional cidofovir. This route of administration provides the advantage of increased bioavailability with reduced risk for nephrotoxicity.6 Finally, surgical resection may be considered for refractory lesions to circumvent the toxicity from systemically administered drugs.3
- Severson JL, Tyring SK. Relation between herpes simplex viruses and human immunodeficiency virus infections. Arch Dermatol. 1999;135:1393-1397.
- Patel AB, Rosen T. Herpes vegetans as a sign of HIV infection. Dermatol Online J. 2008;14:6.
- Chung VQ, Parker DC, Parker SR. Surgical excision for vegetative herpes simplex virus infection. Dermatol Surg. 2007;33:1374-1379.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Chilukuri S, Rosen T. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin. 2003;21:311-320.
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126.
- Severson JL, Tyring SK. Relation between herpes simplex viruses and human immunodeficiency virus infections. Arch Dermatol. 1999;135:1393-1397.
- Patel AB, Rosen T. Herpes vegetans as a sign of HIV infection. Dermatol Online J. 2008;14:6.
- Chung VQ, Parker DC, Parker SR. Surgical excision for vegetative herpes simplex virus infection. Dermatol Surg. 2007;33:1374-1379.
- Beasley KL, Cooley GE, Kao GF, et al. Herpes simplex vegetans: atypical genital herpes infection in a patient with common variable immunodeficiency. J Am Acad Dermatol. 1997;37(5, pt 2):860-863.
- Chilukuri S, Rosen T. Management of acyclovir-resistant herpes simplex virus. Dermatol Clin. 2003;21:311-320.
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126.
A 53-year-old man presented to our clinic with a sacral mass that had progressively enlarged over 2 years. The patient reported occasional oozing from the mass as well as pain when laying flat but denied fever or other symptoms. His medical history was remarkable for human immunodeficiency virus infection with variable adherence to a highly active antiretroviral therapy regimen. At the time of presentation, the patient had a CD4 lymphocyte count of 78 cells/mm3 (reference range, 500–1400 cells/mm3) and a viral load of 290 copies/mL (reference range, 0 copies/mL). Physical examination revealed a 10-cm discrete, moist and pink, exophytic plaque on the sacrum with superficial erosions. The plaque was nontender and without associated lymphadenopathy. The skin and mucous membranes were otherwise clear. A cutaneous biopsy specimen was obtained from the tumor and sent for histopathologic analysis.
What Is Your Diagnosis? Idiopathic Guttate Hypomelanosis
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.

Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
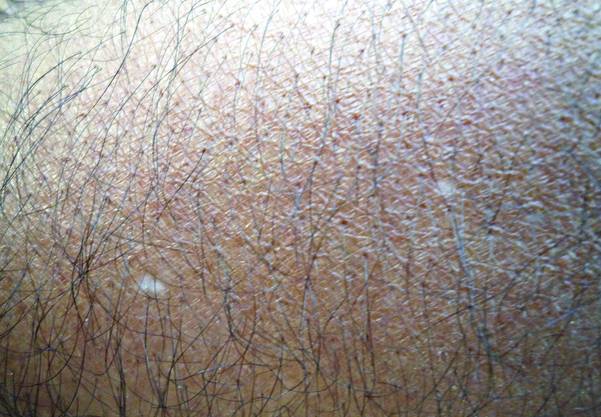
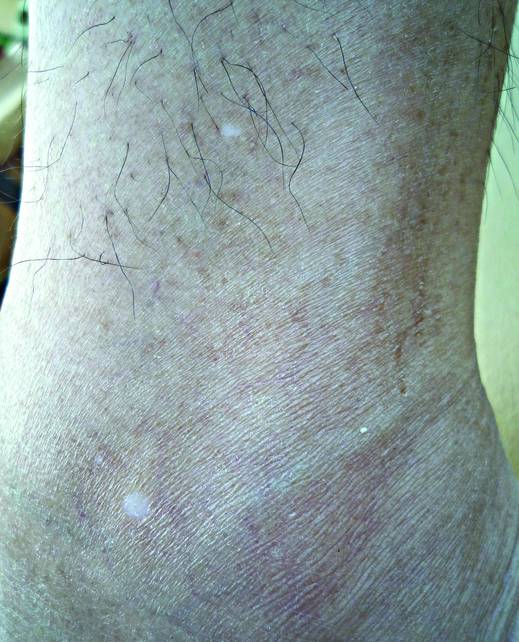
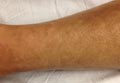
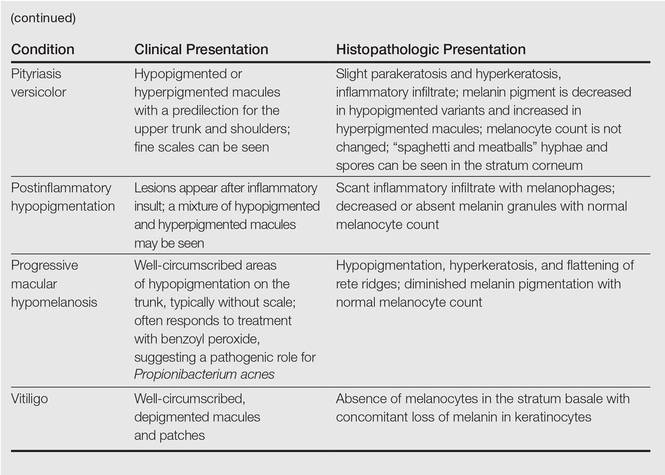
A 58-year-old man presented with disseminated, hypopigmented, asymptomatic lesions on the right arm (top) and left leg (bottom) that had been present for approximately 6 years. The patient reported that the lesions had become more visible and greater in number within the last year. Multiple circular hypopigmented macules of various sizes ranging from 1 to 3 mm in diameter were identified. No scaling was seen. Physical examination was otherwise unremarkable.
Painful Skin Lesions on the Hands Following Black Henna Application
The Diagnosis: Allergic Contact Dermatitis to Para-phenylenediamine
To darken the color of henna and increase penetrance and staining, para-phenylenediamine (PPD) is added.1 Allergic contact dermatitis is the most common type of hypersensitivity to PPD.2 A retrospective study that examined severe adverse events from applying henna dyes in children found that angioedema of mucosal tissues was the most common severe adverse event; others included renal failure and shock.3
Black henna is associated with multiple cultural practices. For example, Indian weddings contain a henna decoration ceremony for the bride based on the belief that the longer the henna lasts, the longer the marriage lasts. Black henna is favored for this practice, as it lasts longer than red henna.

Henna (Lawsonia inermis) is a plant that contains the molecule lawsone (naphthoquinone). Lawsone has an intense affinity for keratin; as a result, lawsone is frequently added to temporary body tattoos and hair dyes to create a relatively permanent change in skin or hair color.4 Henna is mixed with hennotannic acid to release the lawsone from the plant. Lawsone and hennotannic acid rarely cause allergic reactions.1,5-7 Once applied to skin, henna takes a few hours to dry, and the resulting color is orange to red.8 Often, PPD is added to henna paste to create a black color, to speed up the drying process, and to increase its longevity.
Para-phenylenediamine has been repeatedly reported to cause allergic contact dermatitis. We describe a case of allergic contact dermatitis secondary to PPD in black henna. Our patient is a clear example that PPD is the allergen in black henna given that there was no reaction to the natural red henna tattoo that was applied at the same time to the palmar surfaces of the hands (Figure). Aside from the bullous reaction to black henna dye described here, other reported presentations include erythema multiforme–like and exudative erythema reactions.9,10
Contact dermatitis lesions from black henna dye can be treated with topical corticosteroids. Patients may develop residual postinflammatory hyperpigmentation or hypopigmentation, leukoderma, keloids, or scars.1,11,12
- Onder M, Atahan CA, Oztas P, et al. Temporary henna tattoo reactions in children. Int J Dermatol. 2001;40:577-579.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Hashim S, Hamza Y, Yahia B, et al. Poisoning from henna dye and para-phenylenediamine mixtures in children in Khartoum. Ann Trop Paediatr. 1992;12:3-6.
- Hijji Y, Barare B, Zhang Y. Lawsone (2- hydroxy-1, 4-naphthoquinone) as a sensitive cyanide and acetate sensor. Sensors and Actuators B: Chemical. 2012;169:106-112.
- Neri I, Guareschi E, Savoia F, et al. Childhood allergic contact dermatitis from henna tattoo. Pediatr Dermatol. 2002;19:503-505.
- Evans CC, Fleming JD. Allergic contact dermatitis from a henna tattoo. N Engl J Med. 2008;359:627.
- Belhadjali H, Akkari H, Youssef M, et al. Bullous allergic contact dermatitis to pure henna in a 3-year-old girl. Pediatr Dermatol. 2011;28:580-581.
- Najem N, Bagher Zadeh V. Allergic contact dermatitis to black henna. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20:87-88.
- Sidwell RU, Francis ND, Basarab T, et al. Vesicular erythema multiforme-like reaction to para-phenylenediamine in a henna tattoo. Pediatr Dermatol. 2008;25:201-204.
- Jovanovic DL, Slavkovic-Jovanovic MR. Allergic contact dermatitis from temporary henna tattoo. J Dermatol. 2009;36:63-65.
- Valsecchi R, Leghissa P, Di Landro A, et al. Persistent leukoderma after henna tattoo. Contact Dermatitis. 2007;56:108-109.
- Gunasti S, Aksungur VL. Severe inflammatory and keloidal, allergic reaction due to para-phenylenediamine in temporary tattoos. Indian J Dermatol Venereol Leprol. 2010;76:165-167.
The Diagnosis: Allergic Contact Dermatitis to Para-phenylenediamine
To darken the color of henna and increase penetrance and staining, para-phenylenediamine (PPD) is added.1 Allergic contact dermatitis is the most common type of hypersensitivity to PPD.2 A retrospective study that examined severe adverse events from applying henna dyes in children found that angioedema of mucosal tissues was the most common severe adverse event; others included renal failure and shock.3
Black henna is associated with multiple cultural practices. For example, Indian weddings contain a henna decoration ceremony for the bride based on the belief that the longer the henna lasts, the longer the marriage lasts. Black henna is favored for this practice, as it lasts longer than red henna.
Henna (Lawsonia inermis) is a plant that contains the molecule lawsone (naphthoquinone). Lawsone has an intense affinity for keratin; as a result, lawsone is frequently added to temporary body tattoos and hair dyes to create a relatively permanent change in skin or hair color.4 Henna is mixed with hennotannic acid to release the lawsone from the plant. Lawsone and hennotannic acid rarely cause allergic reactions.1,5-7 Once applied to skin, henna takes a few hours to dry, and the resulting color is orange to red.8 Often, PPD is added to henna paste to create a black color, to speed up the drying process, and to increase its longevity.
Para-phenylenediamine has been repeatedly reported to cause allergic contact dermatitis. We describe a case of allergic contact dermatitis secondary to PPD in black henna. Our patient is a clear example that PPD is the allergen in black henna given that there was no reaction to the natural red henna tattoo that was applied at the same time to the palmar surfaces of the hands (Figure). Aside from the bullous reaction to black henna dye described here, other reported presentations include erythema multiforme–like and exudative erythema reactions.9,10
Contact dermatitis lesions from black henna dye can be treated with topical corticosteroids. Patients may develop residual postinflammatory hyperpigmentation or hypopigmentation, leukoderma, keloids, or scars.1,11,12
The Diagnosis: Allergic Contact Dermatitis to Para-phenylenediamine
To darken the color of henna and increase penetrance and staining, para-phenylenediamine (PPD) is added.1 Allergic contact dermatitis is the most common type of hypersensitivity to PPD.2 A retrospective study that examined severe adverse events from applying henna dyes in children found that angioedema of mucosal tissues was the most common severe adverse event; others included renal failure and shock.3
Black henna is associated with multiple cultural practices. For example, Indian weddings contain a henna decoration ceremony for the bride based on the belief that the longer the henna lasts, the longer the marriage lasts. Black henna is favored for this practice, as it lasts longer than red henna.
Henna (Lawsonia inermis) is a plant that contains the molecule lawsone (naphthoquinone). Lawsone has an intense affinity for keratin; as a result, lawsone is frequently added to temporary body tattoos and hair dyes to create a relatively permanent change in skin or hair color.4 Henna is mixed with hennotannic acid to release the lawsone from the plant. Lawsone and hennotannic acid rarely cause allergic reactions.1,5-7 Once applied to skin, henna takes a few hours to dry, and the resulting color is orange to red.8 Often, PPD is added to henna paste to create a black color, to speed up the drying process, and to increase its longevity.
Para-phenylenediamine has been repeatedly reported to cause allergic contact dermatitis. We describe a case of allergic contact dermatitis secondary to PPD in black henna. Our patient is a clear example that PPD is the allergen in black henna given that there was no reaction to the natural red henna tattoo that was applied at the same time to the palmar surfaces of the hands (Figure). Aside from the bullous reaction to black henna dye described here, other reported presentations include erythema multiforme–like and exudative erythema reactions.9,10
Contact dermatitis lesions from black henna dye can be treated with topical corticosteroids. Patients may develop residual postinflammatory hyperpigmentation or hypopigmentation, leukoderma, keloids, or scars.1,11,12
- Onder M, Atahan CA, Oztas P, et al. Temporary henna tattoo reactions in children. Int J Dermatol. 2001;40:577-579.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Hashim S, Hamza Y, Yahia B, et al. Poisoning from henna dye and para-phenylenediamine mixtures in children in Khartoum. Ann Trop Paediatr. 1992;12:3-6.
- Hijji Y, Barare B, Zhang Y. Lawsone (2- hydroxy-1, 4-naphthoquinone) as a sensitive cyanide and acetate sensor. Sensors and Actuators B: Chemical. 2012;169:106-112.
- Neri I, Guareschi E, Savoia F, et al. Childhood allergic contact dermatitis from henna tattoo. Pediatr Dermatol. 2002;19:503-505.
- Evans CC, Fleming JD. Allergic contact dermatitis from a henna tattoo. N Engl J Med. 2008;359:627.
- Belhadjali H, Akkari H, Youssef M, et al. Bullous allergic contact dermatitis to pure henna in a 3-year-old girl. Pediatr Dermatol. 2011;28:580-581.
- Najem N, Bagher Zadeh V. Allergic contact dermatitis to black henna. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20:87-88.
- Sidwell RU, Francis ND, Basarab T, et al. Vesicular erythema multiforme-like reaction to para-phenylenediamine in a henna tattoo. Pediatr Dermatol. 2008;25:201-204.
- Jovanovic DL, Slavkovic-Jovanovic MR. Allergic contact dermatitis from temporary henna tattoo. J Dermatol. 2009;36:63-65.
- Valsecchi R, Leghissa P, Di Landro A, et al. Persistent leukoderma after henna tattoo. Contact Dermatitis. 2007;56:108-109.
- Gunasti S, Aksungur VL. Severe inflammatory and keloidal, allergic reaction due to para-phenylenediamine in temporary tattoos. Indian J Dermatol Venereol Leprol. 2010;76:165-167.
- Onder M, Atahan CA, Oztas P, et al. Temporary henna tattoo reactions in children. Int J Dermatol. 2001;40:577-579.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Hashim S, Hamza Y, Yahia B, et al. Poisoning from henna dye and para-phenylenediamine mixtures in children in Khartoum. Ann Trop Paediatr. 1992;12:3-6.
- Hijji Y, Barare B, Zhang Y. Lawsone (2- hydroxy-1, 4-naphthoquinone) as a sensitive cyanide and acetate sensor. Sensors and Actuators B: Chemical. 2012;169:106-112.
- Neri I, Guareschi E, Savoia F, et al. Childhood allergic contact dermatitis from henna tattoo. Pediatr Dermatol. 2002;19:503-505.
- Evans CC, Fleming JD. Allergic contact dermatitis from a henna tattoo. N Engl J Med. 2008;359:627.
- Belhadjali H, Akkari H, Youssef M, et al. Bullous allergic contact dermatitis to pure henna in a 3-year-old girl. Pediatr Dermatol. 2011;28:580-581.
- Najem N, Bagher Zadeh V. Allergic contact dermatitis to black henna. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20:87-88.
- Sidwell RU, Francis ND, Basarab T, et al. Vesicular erythema multiforme-like reaction to para-phenylenediamine in a henna tattoo. Pediatr Dermatol. 2008;25:201-204.
- Jovanovic DL, Slavkovic-Jovanovic MR. Allergic contact dermatitis from temporary henna tattoo. J Dermatol. 2009;36:63-65.
- Valsecchi R, Leghissa P, Di Landro A, et al. Persistent leukoderma after henna tattoo. Contact Dermatitis. 2007;56:108-109.
- Gunasti S, Aksungur VL. Severe inflammatory and keloidal, allergic reaction due to para-phenylenediamine in temporary tattoos. Indian J Dermatol Venereol Leprol. 2010;76:165-167.

A 14-year-old adolescent girl presented with painful skin lesions on the dorsal aspect of the hands of 10 days’ duration. She reported having received red henna tattoo on the palmar surface of the hands and black henna tattoo on the dorsal surface of the hands 1 day prior to development of the lesions. Within 1 day of receiving the tattoo, she developed pruritus, blisters, and pain on the dorsal aspect of the hands. The palms were unaffected. Physical examination revealed erythematous, brown to black bullae and crusts that followed the contours of the henna design on the dorsal aspect of the hands. There were orange and brown henna designs on the patient’s palms, but no erythema, bullae, or induration was noted.

Dry Scaly Rash
The Diagnosis: Phrynoderma
Phrynoderma, meaning “toad skin,” was first described by Nicholls1 in 1933 as a condition due to vitamin A deficiency and characterized by follicular hyperkeratosis. Since then, there has been debate in the literature over the etiology of phrynoderma, as studies have demonstrated different causes for this condition,2-5 which suggests that it is prudent to check several markers of nutritional status including vitamin A, vitamin B, vitamin E, and essential fatty acid studies.
Phrynoderma is most commonly seen in South and East Asia with relatively rare occurrences in the United States. Although it characteristically occurs in children and adolescents aged 5 to 15 years, almost all of the cases reported in the United States have occurred in adults.6,7 A clear gender predilection has not been established.7
Phrynoderma tends to favor the extensor surfaces. In particular, the elbows, thighs, and buttocks are the most commonly affected locations. The face is typically the last site to become involved. On physical examination of a patient with phrynoderma, there are various-sized, discrete, red-brown to flesh-colored papules with a conical keratotic follicular plug (Figure).3 The eruption is often asymptomatic, but mild pruritus may be reported.7 Additional signs of vitamin A deficiency may be present, including night blindness, hyperpigmentation, or xerosis. Patients with phrynoderma also may have evidence of other nutritional deficiencies, such as angular stomatitis, cheilitis, or glossitis. The differential diagnosis for a patient with this type of papular eruption may include pityriasis rubra pilaris, keratosis pilaris, lichen spinulosus, crusted scabies, and follicular lichen planus.7 History and clinical presentation can generally help to distinguish these diagnoses.
Generalized follicular, erythematous, and scaly papular eruption on the chest (A) and abdomen (B). |
Histopathology typically shows moderate hyperkeratosis with distension of the upper portion of the follicle by large keratotic plugs. Sebaceous glands are greatly reduced in size and may exhibit epithelial atrophy.8,9
A reduced serum vitamin A level can confirm the diagnosis of phrynoderma. Our patient’s laboratory results revealed a vitamin A value of 26 mg/100 dL (reference range, 38–106 mg/100 dL), consistent with his clinical presentation of phrynoderma. Other markers of nutritional status, including vitamins D, E, and K1, were all within reference range. Treatment consists of replacing the nutritional deficit. High doses of vitamin A, such as 200,000 to 500,000 IU daily for 3 to 5 days or daily doses of 2000 to 50,000 IU, can lead to resolution of the rash in 1 to 4 months.8,9 Our patient was started on 10,000 IU of oral vitamin A daily at discharge but unfortunately died a few months later.
Vitamin A has been shown to promote maturation and differentiation of the basal keratinocytes as they move up through the epidermis, which may in part explain why this treatment is effective.10 It is important to be aware of this condition when seeing a patient with follicular hyperkeratosis because even patients in developed countries with malnutrition due to diet or gastrointestinal disease may develop phrynoderma.
- Nicholls L. Phrynoderma: a condition due to vitamin deficiency. Indian Med Gazette. 1933;68:681-687.
- Bagchi K, Halder K, Chowdhury SR. The etiology of phrynoderma; histologic evidence. Am J Clin Nutr. 1959;7:251-258.
- Bleasel NR, Stapleton KM, Lee MS, et al. Vitamin A deficiency phrynoderma: due to malabsorption and inadequate diet. J Am Acad Dermatol. 1999;41(2, pt 2):322-324.
- Ghafoorunissa, Vidyasagar R, Krishnaswamy K. Phrynoderma: is it an EFA deficiency disease? Euro J Clin Nutr. 1988;42:29-39.
- Shrank AB. Phrynoderma. Br Med J. 1966;1:29-30.
- Maronn M, Allen DM, Esterly NB. Phrynoderma: a manifestation of vitamin A deficiency?…the rest of the story. Pediatr Dermatol. 2005;22:60-63.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Neill SM, Pembroke AC, du-Vivier AWP, et al. Phrynoderma and perforating folliculitis due to vitamin A deficiency in a diabetic. J R Soc Med. 1988;81:171-172.
- Wechsler HL. Vitamin A deficiency following small-bowel bypass surgery for obesity. Arch Dermatol. 1979;115:73-75.
- Logan WS. Vitamin A and keratinization. Arch Dermatol. 1972;105:748-753.
The Diagnosis: Phrynoderma
Phrynoderma, meaning “toad skin,” was first described by Nicholls1 in 1933 as a condition due to vitamin A deficiency and characterized by follicular hyperkeratosis. Since then, there has been debate in the literature over the etiology of phrynoderma, as studies have demonstrated different causes for this condition,2-5 which suggests that it is prudent to check several markers of nutritional status including vitamin A, vitamin B, vitamin E, and essential fatty acid studies.
Phrynoderma is most commonly seen in South and East Asia with relatively rare occurrences in the United States. Although it characteristically occurs in children and adolescents aged 5 to 15 years, almost all of the cases reported in the United States have occurred in adults.6,7 A clear gender predilection has not been established.7
Phrynoderma tends to favor the extensor surfaces. In particular, the elbows, thighs, and buttocks are the most commonly affected locations. The face is typically the last site to become involved. On physical examination of a patient with phrynoderma, there are various-sized, discrete, red-brown to flesh-colored papules with a conical keratotic follicular plug (Figure).3 The eruption is often asymptomatic, but mild pruritus may be reported.7 Additional signs of vitamin A deficiency may be present, including night blindness, hyperpigmentation, or xerosis. Patients with phrynoderma also may have evidence of other nutritional deficiencies, such as angular stomatitis, cheilitis, or glossitis. The differential diagnosis for a patient with this type of papular eruption may include pityriasis rubra pilaris, keratosis pilaris, lichen spinulosus, crusted scabies, and follicular lichen planus.7 History and clinical presentation can generally help to distinguish these diagnoses.
|
Generalized follicular, erythematous, and scaly papular eruption on the chest (A) and abdomen (B). |
Histopathology typically shows moderate hyperkeratosis with distension of the upper portion of the follicle by large keratotic plugs. Sebaceous glands are greatly reduced in size and may exhibit epithelial atrophy.8,9
A reduced serum vitamin A level can confirm the diagnosis of phrynoderma. Our patient’s laboratory results revealed a vitamin A value of 26 mg/100 dL (reference range, 38–106 mg/100 dL), consistent with his clinical presentation of phrynoderma. Other markers of nutritional status, including vitamins D, E, and K1, were all within reference range. Treatment consists of replacing the nutritional deficit. High doses of vitamin A, such as 200,000 to 500,000 IU daily for 3 to 5 days or daily doses of 2000 to 50,000 IU, can lead to resolution of the rash in 1 to 4 months.8,9 Our patient was started on 10,000 IU of oral vitamin A daily at discharge but unfortunately died a few months later.
Vitamin A has been shown to promote maturation and differentiation of the basal keratinocytes as they move up through the epidermis, which may in part explain why this treatment is effective.10 It is important to be aware of this condition when seeing a patient with follicular hyperkeratosis because even patients in developed countries with malnutrition due to diet or gastrointestinal disease may develop phrynoderma.
The Diagnosis: Phrynoderma
Phrynoderma, meaning “toad skin,” was first described by Nicholls1 in 1933 as a condition due to vitamin A deficiency and characterized by follicular hyperkeratosis. Since then, there has been debate in the literature over the etiology of phrynoderma, as studies have demonstrated different causes for this condition,2-5 which suggests that it is prudent to check several markers of nutritional status including vitamin A, vitamin B, vitamin E, and essential fatty acid studies.
Phrynoderma is most commonly seen in South and East Asia with relatively rare occurrences in the United States. Although it characteristically occurs in children and adolescents aged 5 to 15 years, almost all of the cases reported in the United States have occurred in adults.6,7 A clear gender predilection has not been established.7
Phrynoderma tends to favor the extensor surfaces. In particular, the elbows, thighs, and buttocks are the most commonly affected locations. The face is typically the last site to become involved. On physical examination of a patient with phrynoderma, there are various-sized, discrete, red-brown to flesh-colored papules with a conical keratotic follicular plug (Figure).3 The eruption is often asymptomatic, but mild pruritus may be reported.7 Additional signs of vitamin A deficiency may be present, including night blindness, hyperpigmentation, or xerosis. Patients with phrynoderma also may have evidence of other nutritional deficiencies, such as angular stomatitis, cheilitis, or glossitis. The differential diagnosis for a patient with this type of papular eruption may include pityriasis rubra pilaris, keratosis pilaris, lichen spinulosus, crusted scabies, and follicular lichen planus.7 History and clinical presentation can generally help to distinguish these diagnoses.
|
Generalized follicular, erythematous, and scaly papular eruption on the chest (A) and abdomen (B). |
Histopathology typically shows moderate hyperkeratosis with distension of the upper portion of the follicle by large keratotic plugs. Sebaceous glands are greatly reduced in size and may exhibit epithelial atrophy.8,9
A reduced serum vitamin A level can confirm the diagnosis of phrynoderma. Our patient’s laboratory results revealed a vitamin A value of 26 mg/100 dL (reference range, 38–106 mg/100 dL), consistent with his clinical presentation of phrynoderma. Other markers of nutritional status, including vitamins D, E, and K1, were all within reference range. Treatment consists of replacing the nutritional deficit. High doses of vitamin A, such as 200,000 to 500,000 IU daily for 3 to 5 days or daily doses of 2000 to 50,000 IU, can lead to resolution of the rash in 1 to 4 months.8,9 Our patient was started on 10,000 IU of oral vitamin A daily at discharge but unfortunately died a few months later.
Vitamin A has been shown to promote maturation and differentiation of the basal keratinocytes as they move up through the epidermis, which may in part explain why this treatment is effective.10 It is important to be aware of this condition when seeing a patient with follicular hyperkeratosis because even patients in developed countries with malnutrition due to diet or gastrointestinal disease may develop phrynoderma.
- Nicholls L. Phrynoderma: a condition due to vitamin deficiency. Indian Med Gazette. 1933;68:681-687.
- Bagchi K, Halder K, Chowdhury SR. The etiology of phrynoderma; histologic evidence. Am J Clin Nutr. 1959;7:251-258.
- Bleasel NR, Stapleton KM, Lee MS, et al. Vitamin A deficiency phrynoderma: due to malabsorption and inadequate diet. J Am Acad Dermatol. 1999;41(2, pt 2):322-324.
- Ghafoorunissa, Vidyasagar R, Krishnaswamy K. Phrynoderma: is it an EFA deficiency disease? Euro J Clin Nutr. 1988;42:29-39.
- Shrank AB. Phrynoderma. Br Med J. 1966;1:29-30.
- Maronn M, Allen DM, Esterly NB. Phrynoderma: a manifestation of vitamin A deficiency?…the rest of the story. Pediatr Dermatol. 2005;22:60-63.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Neill SM, Pembroke AC, du-Vivier AWP, et al. Phrynoderma and perforating folliculitis due to vitamin A deficiency in a diabetic. J R Soc Med. 1988;81:171-172.
- Wechsler HL. Vitamin A deficiency following small-bowel bypass surgery for obesity. Arch Dermatol. 1979;115:73-75.
- Logan WS. Vitamin A and keratinization. Arch Dermatol. 1972;105:748-753.
- Nicholls L. Phrynoderma: a condition due to vitamin deficiency. Indian Med Gazette. 1933;68:681-687.
- Bagchi K, Halder K, Chowdhury SR. The etiology of phrynoderma; histologic evidence. Am J Clin Nutr. 1959;7:251-258.
- Bleasel NR, Stapleton KM, Lee MS, et al. Vitamin A deficiency phrynoderma: due to malabsorption and inadequate diet. J Am Acad Dermatol. 1999;41(2, pt 2):322-324.
- Ghafoorunissa, Vidyasagar R, Krishnaswamy K. Phrynoderma: is it an EFA deficiency disease? Euro J Clin Nutr. 1988;42:29-39.
- Shrank AB. Phrynoderma. Br Med J. 1966;1:29-30.
- Maronn M, Allen DM, Esterly NB. Phrynoderma: a manifestation of vitamin A deficiency?…the rest of the story. Pediatr Dermatol. 2005;22:60-63.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Neill SM, Pembroke AC, du-Vivier AWP, et al. Phrynoderma and perforating folliculitis due to vitamin A deficiency in a diabetic. J R Soc Med. 1988;81:171-172.
- Wechsler HL. Vitamin A deficiency following small-bowel bypass surgery for obesity. Arch Dermatol. 1979;115:73-75.
- Logan WS. Vitamin A and keratinization. Arch Dermatol. 1972;105:748-753.
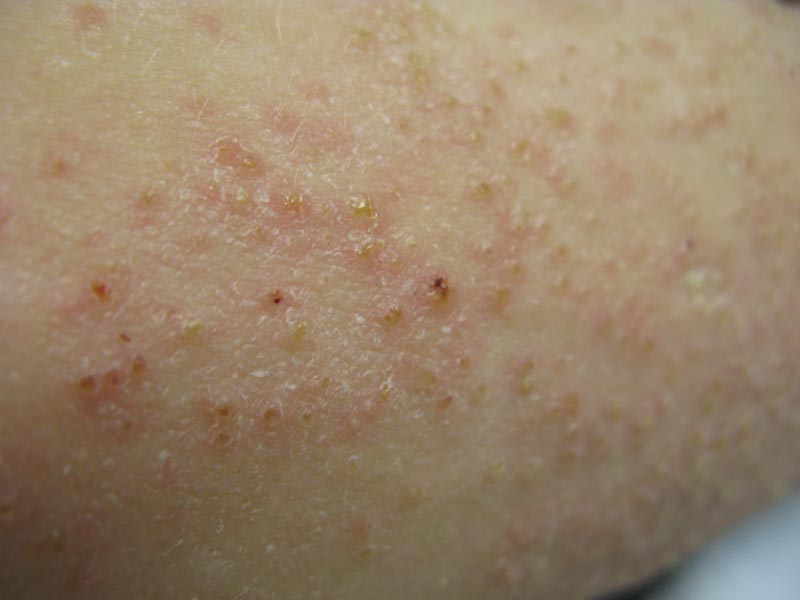
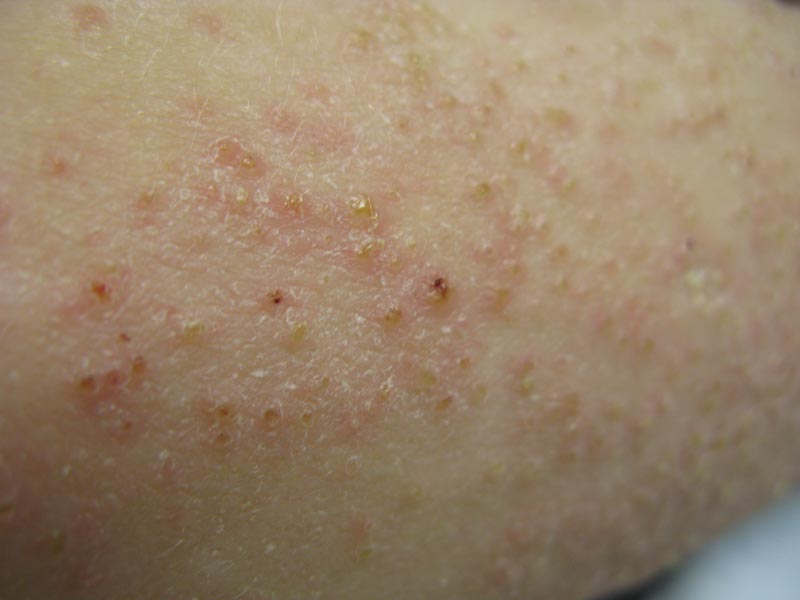
A 22-year-old man with a history of severe mental retardation, a questionable diagnosis of cystic fibrosis, chronic abdominal pain, and a recent diagnosis of primary sclerosing cholangitis was transferred from an outside hospital for worsening abdominal pain, lethargy, anasarca, anorexia, and constipation. An abdominal paracentesis and computed tomography scan of the abdomen were both negative for any acute processes. Blood cultures and paracentesis fluid cultures were negative. The dermatology section was consulted for a dry scaly rash of unknown duration. The eruption had not been treated, and the patient’s dermatologic history was otherwise unremarkable. On physical examination, the patient had a generalized follicular, erythematous, and scaly papular eruption with some areas of yellow-colored hyperkeratotic papules and plaques as well as scattered hemorrhagic crusts on the extensor surface of the right arm. He had normal oral mucosa and nails. The team was unable to gain consent for biopsy, but a blood test was obtained.
Diffuse Crusting of the Skin
The Diagnosis: Crusted Scabies
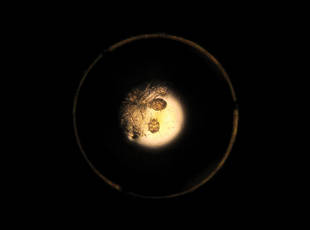
Approximately 1 year prior to presentation the patient completed a course of therapy comprised of repeat doses of ivermectin and permethrin for crusted scabies. He stated that the crusts had recurred shortly after treatment (Figure 1). The initial differential diagnosis included crusted scabies, pityriasis rubra pilaris, and psoriasis. Scraping from the interdigital web space (Figure 2) revealed multiple scabies mites, scabella, and ova (Figure 3). The patient was diagnosed with crusted scabies. Laboratory test results revealed a normal white blood cell count of 7300/μL, with an elevated eosinophil count of 1900/μL (reference range, <500/μL). Human T-lymphotropic virus 1 (HTLV-1) was found to be positive. Serum protein electrophoresis showed a moderate polyclonal increase in gamma globulins. Treatment was promptly initiated with permethrin cream 5% applied to the whole body for a total of 2 treatments that were administered 7 days apart. Ivermectin also was started at 200 μg/kg and was given on days 1, 7, 14, and 29. Salicylic acid cream 25% was applied daily to help hasten resolution of the crusts.
| 
|
Human T-lymphotropic virus 1 is endemic to Japan, Central and South America, the Middle East, and regions of Africa and the Caribbean.1 Multiple cases of HTLV-1 associated with crusted scabies have been reported in endemic areas.2-6 In the present case, we encountered a patient with crusted scabies and HTLV-1 in a nonendemic area.
Cases of HTLV-1 and crusted scabies have been reported in Brazil, Peru, and Central Australia.3,4,7 A retrospective study in Brazil showed a notable association between HTLV-1 infection and crusted scabies; of 23 patients with crusted scabies, 21 were HTLV-1 positive.3 In another small study from Peru, 23 patients with crusted scabies were tested for HTLV-1 and 16 (70%) were seropositive.4 Neither study had a control group for comparison; however, interestingly, the estimated seroprevalence in the general population (based on blood donors) is only 0.04% to 1% and 1.2% to 1.7% in Brazil and Peru, respectively, thus reinforcing the association between HTLV-1 and crusted scabies.1 Crusted scabies and HTLV-1 seropositivity acting as a predictor for adult T-cell leukemia/lymphoma (ATL) has been reported8 but not consistently.3 Our patient had abnormal serum protein electrophoresis findings. Although the results were not diagnostic of ATL, patients with HTLV-1 are at increased risk for ATL and the pres-ence of this virus is important to document when there is a high index of suspicion for ATL, as with crusted scabies.
Our patient resided in the northeastern United States and there was no evidence in support of underlying immunosuppression. This case of crusted scabies is unique because the patient was HTLV-1 positive without other underlying immunocompromise and he resided in a region where HTLV-1 is nonendemic. In light of these findings, the present case serves as a reminder to check for HTLV-1 infection in those patients with severe or crusted scabies and without an identifiable reason for immunosuppression, even if the patient resides in an area that is nonendemic for HTLV-1.
- Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
- Nobre V, Guedes AC, Martins ML, et al. Dermatological findings in 3 generations of a family with a high prevalence of human T cell lymphotropic virus type 1 infection in Brazil. Clin Infect Dis. 2006;43:1257-1263.
- Brites C, Weyll M, Pedroso C, et al. Severe and Norwegian scabies are strongly associated with retroviral (HIV-1/HTLV-1) infection in Bahia, Brazil. AIDS. 2002;16:1292-1293.
- Blas M, Bravo F, Castillo W, et al. Norwegian scabies in Peru: the impact of human T cell lymphotropic virus type I infection. Am J Trop Med Hyg. 2005;72:855-857.
- Bergman JN, Dodd WA, Trotter MJ, et al. Crusted scabies in association with human T-cell lymphotropic virus 1. J Cutan Med Surg. 1999;3:148-152.
- Daisley H, Charles W, Suite M. Crusted (Norwegian) scabies as a pre-diagnostic indicator for HTLV-1 infection. Trans R Soc Trop Med Hyg. 1993;87:295.
- Mollison LC. HTLV-I and clinical disease correlates in central Australian aborigines. Med J Aust. 1994;160:238.
- del Giudice P, Sainte Marie D, Gérard Y, et al. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090-1092.
The Diagnosis: Crusted Scabies
Approximately 1 year prior to presentation the patient completed a course of therapy comprised of repeat doses of ivermectin and permethrin for crusted scabies. He stated that the crusts had recurred shortly after treatment (Figure 1). The initial differential diagnosis included crusted scabies, pityriasis rubra pilaris, and psoriasis. Scraping from the interdigital web space (Figure 2) revealed multiple scabies mites, scabella, and ova (Figure 3). The patient was diagnosed with crusted scabies. Laboratory test results revealed a normal white blood cell count of 7300/μL, with an elevated eosinophil count of 1900/μL (reference range, <500/μL). Human T-lymphotropic virus 1 (HTLV-1) was found to be positive. Serum protein electrophoresis showed a moderate polyclonal increase in gamma globulins. Treatment was promptly initiated with permethrin cream 5% applied to the whole body for a total of 2 treatments that were administered 7 days apart. Ivermectin also was started at 200 μg/kg and was given on days 1, 7, 14, and 29. Salicylic acid cream 25% was applied daily to help hasten resolution of the crusts.
|
|
|
Human T-lymphotropic virus 1 is endemic to Japan, Central and South America, the Middle East, and regions of Africa and the Caribbean.1 Multiple cases of HTLV-1 associated with crusted scabies have been reported in endemic areas.2-6 In the present case, we encountered a patient with crusted scabies and HTLV-1 in a nonendemic area.
Cases of HTLV-1 and crusted scabies have been reported in Brazil, Peru, and Central Australia.3,4,7 A retrospective study in Brazil showed a notable association between HTLV-1 infection and crusted scabies; of 23 patients with crusted scabies, 21 were HTLV-1 positive.3 In another small study from Peru, 23 patients with crusted scabies were tested for HTLV-1 and 16 (70%) were seropositive.4 Neither study had a control group for comparison; however, interestingly, the estimated seroprevalence in the general population (based on blood donors) is only 0.04% to 1% and 1.2% to 1.7% in Brazil and Peru, respectively, thus reinforcing the association between HTLV-1 and crusted scabies.1 Crusted scabies and HTLV-1 seropositivity acting as a predictor for adult T-cell leukemia/lymphoma (ATL) has been reported8 but not consistently.3 Our patient had abnormal serum protein electrophoresis findings. Although the results were not diagnostic of ATL, patients with HTLV-1 are at increased risk for ATL and the pres-ence of this virus is important to document when there is a high index of suspicion for ATL, as with crusted scabies.
Our patient resided in the northeastern United States and there was no evidence in support of underlying immunosuppression. This case of crusted scabies is unique because the patient was HTLV-1 positive without other underlying immunocompromise and he resided in a region where HTLV-1 is nonendemic. In light of these findings, the present case serves as a reminder to check for HTLV-1 infection in those patients with severe or crusted scabies and without an identifiable reason for immunosuppression, even if the patient resides in an area that is nonendemic for HTLV-1.
The Diagnosis: Crusted Scabies
Approximately 1 year prior to presentation the patient completed a course of therapy comprised of repeat doses of ivermectin and permethrin for crusted scabies. He stated that the crusts had recurred shortly after treatment (Figure 1). The initial differential diagnosis included crusted scabies, pityriasis rubra pilaris, and psoriasis. Scraping from the interdigital web space (Figure 2) revealed multiple scabies mites, scabella, and ova (Figure 3). The patient was diagnosed with crusted scabies. Laboratory test results revealed a normal white blood cell count of 7300/μL, with an elevated eosinophil count of 1900/μL (reference range, <500/μL). Human T-lymphotropic virus 1 (HTLV-1) was found to be positive. Serum protein electrophoresis showed a moderate polyclonal increase in gamma globulins. Treatment was promptly initiated with permethrin cream 5% applied to the whole body for a total of 2 treatments that were administered 7 days apart. Ivermectin also was started at 200 μg/kg and was given on days 1, 7, 14, and 29. Salicylic acid cream 25% was applied daily to help hasten resolution of the crusts.
|
|
|
Human T-lymphotropic virus 1 is endemic to Japan, Central and South America, the Middle East, and regions of Africa and the Caribbean.1 Multiple cases of HTLV-1 associated with crusted scabies have been reported in endemic areas.2-6 In the present case, we encountered a patient with crusted scabies and HTLV-1 in a nonendemic area.
Cases of HTLV-1 and crusted scabies have been reported in Brazil, Peru, and Central Australia.3,4,7 A retrospective study in Brazil showed a notable association between HTLV-1 infection and crusted scabies; of 23 patients with crusted scabies, 21 were HTLV-1 positive.3 In another small study from Peru, 23 patients with crusted scabies were tested for HTLV-1 and 16 (70%) were seropositive.4 Neither study had a control group for comparison; however, interestingly, the estimated seroprevalence in the general population (based on blood donors) is only 0.04% to 1% and 1.2% to 1.7% in Brazil and Peru, respectively, thus reinforcing the association between HTLV-1 and crusted scabies.1 Crusted scabies and HTLV-1 seropositivity acting as a predictor for adult T-cell leukemia/lymphoma (ATL) has been reported8 but not consistently.3 Our patient had abnormal serum protein electrophoresis findings. Although the results were not diagnostic of ATL, patients with HTLV-1 are at increased risk for ATL and the pres-ence of this virus is important to document when there is a high index of suspicion for ATL, as with crusted scabies.
Our patient resided in the northeastern United States and there was no evidence in support of underlying immunosuppression. This case of crusted scabies is unique because the patient was HTLV-1 positive without other underlying immunocompromise and he resided in a region where HTLV-1 is nonendemic. In light of these findings, the present case serves as a reminder to check for HTLV-1 infection in those patients with severe or crusted scabies and without an identifiable reason for immunosuppression, even if the patient resides in an area that is nonendemic for HTLV-1.
- Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
- Nobre V, Guedes AC, Martins ML, et al. Dermatological findings in 3 generations of a family with a high prevalence of human T cell lymphotropic virus type 1 infection in Brazil. Clin Infect Dis. 2006;43:1257-1263.
- Brites C, Weyll M, Pedroso C, et al. Severe and Norwegian scabies are strongly associated with retroviral (HIV-1/HTLV-1) infection in Bahia, Brazil. AIDS. 2002;16:1292-1293.
- Blas M, Bravo F, Castillo W, et al. Norwegian scabies in Peru: the impact of human T cell lymphotropic virus type I infection. Am J Trop Med Hyg. 2005;72:855-857.
- Bergman JN, Dodd WA, Trotter MJ, et al. Crusted scabies in association with human T-cell lymphotropic virus 1. J Cutan Med Surg. 1999;3:148-152.
- Daisley H, Charles W, Suite M. Crusted (Norwegian) scabies as a pre-diagnostic indicator for HTLV-1 infection. Trans R Soc Trop Med Hyg. 1993;87:295.
- Mollison LC. HTLV-I and clinical disease correlates in central Australian aborigines. Med J Aust. 1994;160:238.
- del Giudice P, Sainte Marie D, Gérard Y, et al. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090-1092.
- Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
- Nobre V, Guedes AC, Martins ML, et al. Dermatological findings in 3 generations of a family with a high prevalence of human T cell lymphotropic virus type 1 infection in Brazil. Clin Infect Dis. 2006;43:1257-1263.
- Brites C, Weyll M, Pedroso C, et al. Severe and Norwegian scabies are strongly associated with retroviral (HIV-1/HTLV-1) infection in Bahia, Brazil. AIDS. 2002;16:1292-1293.
- Blas M, Bravo F, Castillo W, et al. Norwegian scabies in Peru: the impact of human T cell lymphotropic virus type I infection. Am J Trop Med Hyg. 2005;72:855-857.
- Bergman JN, Dodd WA, Trotter MJ, et al. Crusted scabies in association with human T-cell lymphotropic virus 1. J Cutan Med Surg. 1999;3:148-152.
- Daisley H, Charles W, Suite M. Crusted (Norwegian) scabies as a pre-diagnostic indicator for HTLV-1 infection. Trans R Soc Trop Med Hyg. 1993;87:295.
- Mollison LC. HTLV-I and clinical disease correlates in central Australian aborigines. Med J Aust. 1994;160:238.
- del Giudice P, Sainte Marie D, Gérard Y, et al. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090-1092.
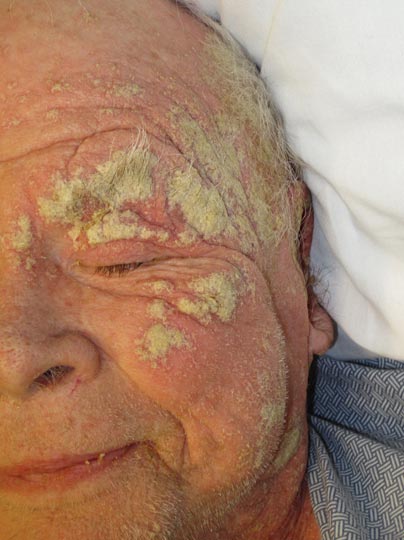
An 82-year-old white man who resided in the northeastern United States was admitted to the hospital for weakness. He was evaluated for diffuse crusting of the skin including the face. The patient reported a history of minimally itchy, generalized crusts of 10 years’ duration. He lived alone with one cat. His medications included fluticasone furoate, terazosin, and levothyroxine sodium. Human immunodeficiency virus screening was negative. A scraping was taken from one of the thick crusts within an interdigital web space.
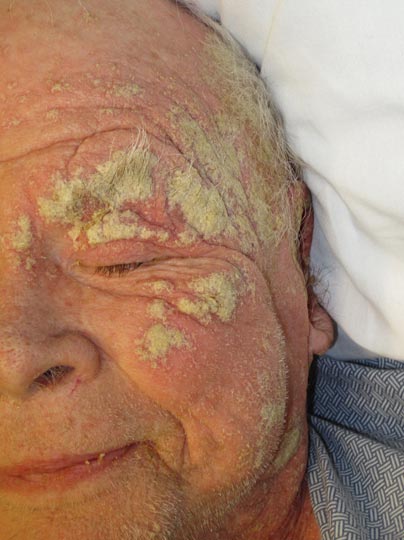
What Is Your Diagnosis? Verrucous Carcinoma
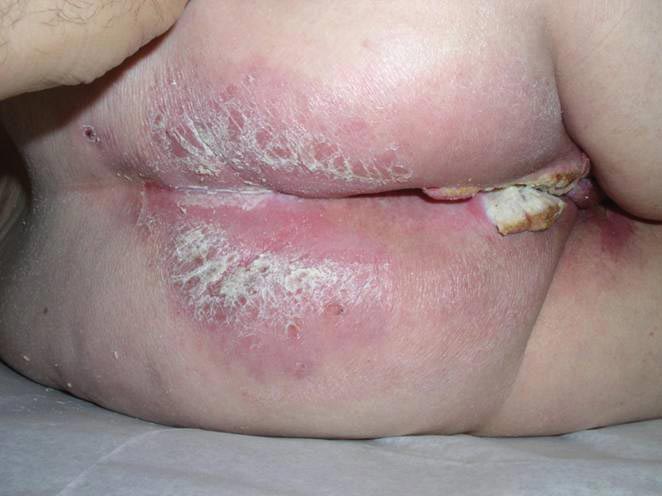
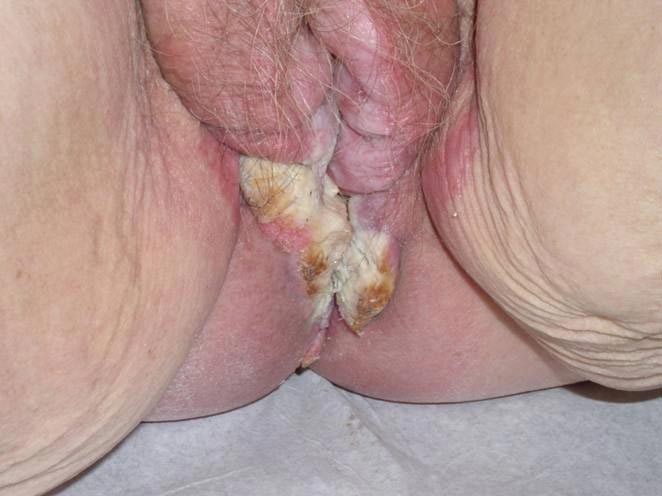
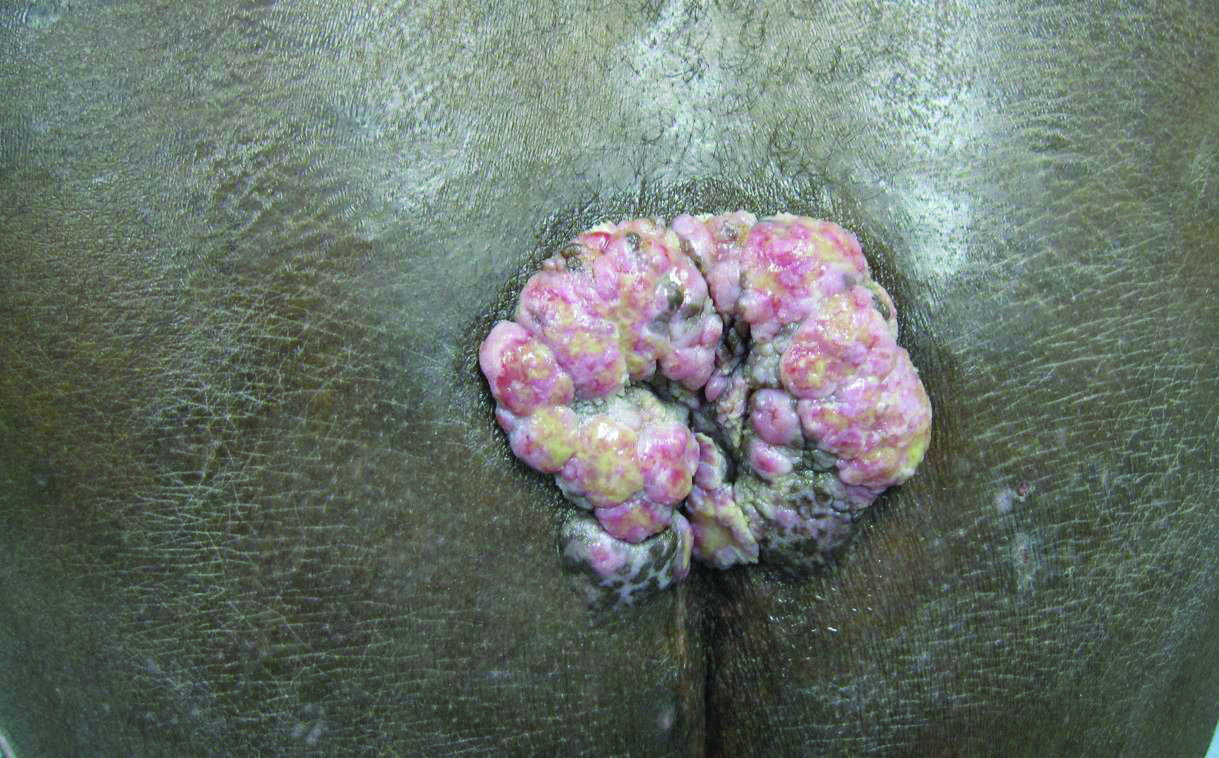
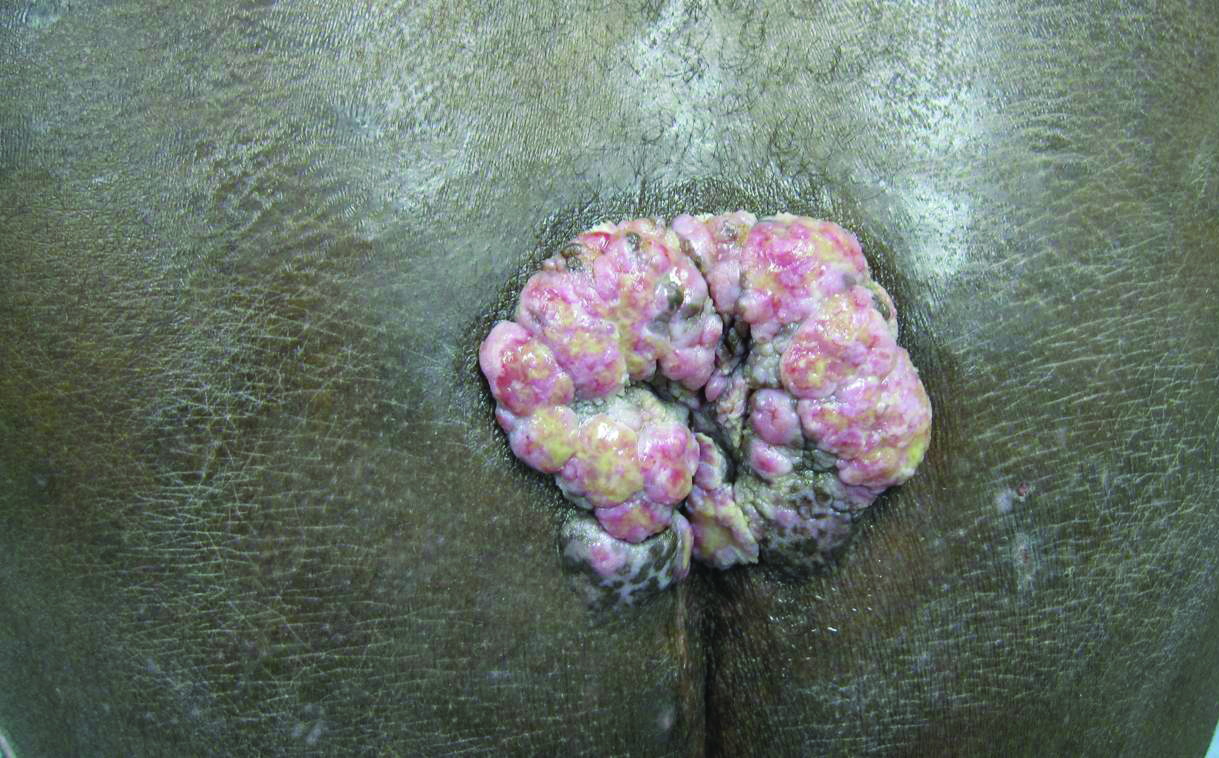
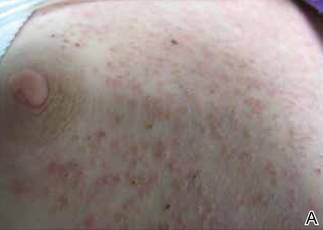
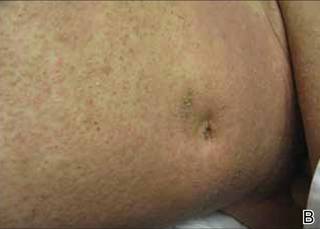
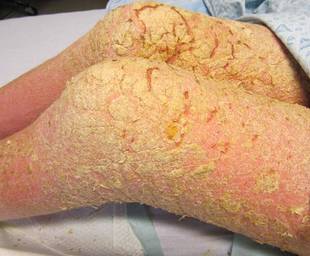
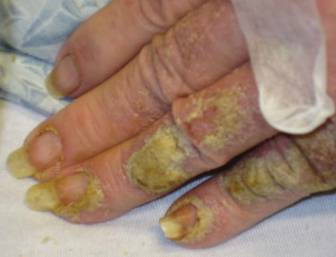
An 81-year-old woman presented for evaluation of a nodule on the right labia majora that had been present for 1 year. She had a history of intertriginous psoriasis, and several biopsies were performed at an outside facility over the last 5 years that revealed psoriasis but were otherwise noncontributory. Physical examination revealed erythema and scaling on the buttocks with maceration in the intertriginous area (top) and the perineum associated with a verrucous nodule (bottom).
The Diagnosis: Verrucous Carcinoma
Biopsies of early lesions often may be difficult to interpret without clinicopathological correlation. Our patient’s tumor was associated with intertriginous psoriasis, which was the only abnormality previously noted on superficial biopsies performed at an outside facility. The patient was scheduled for an excisional biopsy due to the large tumor size and clinical suspicion that the prior biopsies were inadequate and failed to demonstrate the primary underlying pathology. Excisional biopsy of the verrucous tumor revealed epithelium composed of keratinocytes with glassy cytoplasm. Papillomatosis was noted along with an endophytic component of well-differentiated epithelial cells extending into the dermis in a bulbous pattern consistent with the verrucous carcinoma variant of squamous cell carcinoma (SCC)(Figure). Verrucous carcinoma often requires correlation with both the clinical and histopathologic findings for definitive diagnosis, as keratinocytes often appear to be well differentiated.1
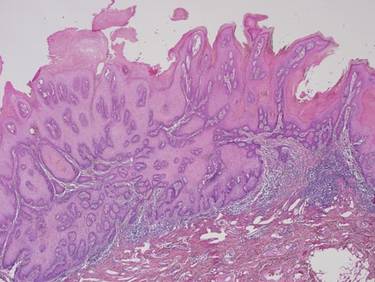
Verrucous carcinoma may begin as an innocuous papule that slowly grows into a large fungating tumor. Verrucous carcinomas typically are slow growing, exophytic, and low grade. The etiology of verrucous carcinoma is not clear, and the role of human papillomavirus (HPV) infection is controversial.2 Best classified as a well-differentiated SCC, verrucous carcinoma rarely metastasizes but may invade adjacent tissues.
Differential diagnoses include a giant inflamed seborrheic keratosis, condyloma acuminatum, rupioid psoriasis, and inflammatory linear verrucous epidermal nevus (ILVEN). Although large and inflamed seborrheic keratoses may have squamous eddies that mimic SCC, seborrheic keratoses do not invade the dermis and typically have a well-circumscribed stuck-on appearance. Abnormal mitotic figures are not identified. Condylomas are genital warts caused by HPV infection that often are clustered, well circumscribed, and exophytic. Large lesions can be difficult to distinguish from verrucous carcinomas, and biopsy generally reveals koilocytes identified by perinuclear clearing and raisinlike nuclei. Immunohistochemical staining and in situ hybridization studies can be of value in diagnosis and in identifying those lesions that are at high risk for malignant transformation. High-risk condylomas are associated with HPV-16, HPV-18, HPV-31, HPV-33, HPV-35, and HPV-39, as well as other types, whereas low-risk condylomas are associated with HPV-6, HPV-11, HPV-42, and others.2 Differentiating squamous cell hyperplasia from squamous cell carcinoma in situ also can be aided by immunohistochemistry. Squamous cell hyperplasia is usually negative for INK4 p16Ink4A and p53 and exhibits variable Ki-67 staining. Differentiated squamous cell carcinoma in situ exhibits a profile that is p16Ink4A negative, Ki-67 positive, and exhibits variable p53 staining.3 Basaloid and warty intraepithelial neoplasia is consistently p16Ink4A positive, Ki-67 positive, and variably positive for p53.3 Therefore, p16 staining of high-grade areas is a useful biomarker that can help establish diagnosis of associated squamous cell carcinoma.4 The role of papillomaviruses in the development of nonmelanoma skin cancer is an area of active study, and research suggests that papillomaviruses may have a much greater role than previously suspected.5
At times, psoriasis may be markedly hyperkeratotic, clinically mimicking a verrucous neoplasm. This hyperkeratotic type of psoriasis is known as rupioid psoriasis. However, these psoriatic lesions are exophytic, are associated with spongiform pustules, and lack the atypia and endophytic pattern typically seen with verrucous carcinoma. An ILVEN also lacks atypia and an endophytic pattern and usually presents in childhood as a persistent linear plaque, rather than the verrucous plaque noted in our patient. Squamous cell carcinoma has been reported to arise in the setting of verrucoid ILVEN but is exceptionally uncommon.6
Successful treatment of verrucous carcinoma is best achieved by complete excision. Oral retinoids and immunomodulators such as imiquimod also may be of value.7 Our patient’s tumor qualifies as T2N0M0 because it was greater than 2 cm in size.8 A Breslow thickness of 2 mm or greater and Clark level IV are high-risk features associated with a worse prognosis, but clinical evaluation of our patient’s lymph nodes was unremarkable and no distant metastases were identified. Our patient continues to do well with no evidence of recurrence.
1. Bambao C, Nofech-Mozes S, Shier M. Giant condyloma versus verrucous carcinoma: a case report. J Low Genit Tract Dis. 2010;14:230-233.
2. Asiaf A, Ahmad ST, Mohannad SO, et al. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur J Cancer Prev. 2014;23:206-224.
3. Chaux A, Pfannl R, Rodríguez IM, et al. Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol. 2011;35:553-562.
4. Darragh TM, Colgan TJ, Cox JT, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med. 2012;136:1266-1297.
5. Aldabagh B, Angeles J, Cardones AR, et al. Cutaneous squamous cell carcinoma and human papillomavirus: is there an association? Dermatol Surg. 2013;39:1-23.
6. Turk BG, Ertam I, Urkmez A, et al. Development of squamous cell carcinoma on an inflammatory linear verrucous epidermal nevus in the genital area. Cutis. 2012;89:273-275.
7. Erkek E, Basar H, Bozdogan O, et al. Giant condyloma acuminata of Buschke-Löwenstein: successful treatment with a combination of surgical excision, oral acitretin and topical imiquimod. Clin Exp Dermatol. 2009;34:366-368.
8. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In: Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301-314.
An 81-year-old woman presented for evaluation of a nodule on the right labia majora that had been present for 1 year. She had a history of intertriginous psoriasis, and several biopsies were performed at an outside facility over the last 5 years that revealed psoriasis but were otherwise noncontributory. Physical examination revealed erythema and scaling on the buttocks with maceration in the intertriginous area (top) and the perineum associated with a verrucous nodule (bottom).
The Diagnosis: Verrucous Carcinoma
Biopsies of early lesions often may be difficult to interpret without clinicopathological correlation. Our patient’s tumor was associated with intertriginous psoriasis, which was the only abnormality previously noted on superficial biopsies performed at an outside facility. The patient was scheduled for an excisional biopsy due to the large tumor size and clinical suspicion that the prior biopsies were inadequate and failed to demonstrate the primary underlying pathology. Excisional biopsy of the verrucous tumor revealed epithelium composed of keratinocytes with glassy cytoplasm. Papillomatosis was noted along with an endophytic component of well-differentiated epithelial cells extending into the dermis in a bulbous pattern consistent with the verrucous carcinoma variant of squamous cell carcinoma (SCC)(Figure). Verrucous carcinoma often requires correlation with both the clinical and histopathologic findings for definitive diagnosis, as keratinocytes often appear to be well differentiated.1
Verrucous carcinoma may begin as an innocuous papule that slowly grows into a large fungating tumor. Verrucous carcinomas typically are slow growing, exophytic, and low grade. The etiology of verrucous carcinoma is not clear, and the role of human papillomavirus (HPV) infection is controversial.2 Best classified as a well-differentiated SCC, verrucous carcinoma rarely metastasizes but may invade adjacent tissues.
Differential diagnoses include a giant inflamed seborrheic keratosis, condyloma acuminatum, rupioid psoriasis, and inflammatory linear verrucous epidermal nevus (ILVEN). Although large and inflamed seborrheic keratoses may have squamous eddies that mimic SCC, seborrheic keratoses do not invade the dermis and typically have a well-circumscribed stuck-on appearance. Abnormal mitotic figures are not identified. Condylomas are genital warts caused by HPV infection that often are clustered, well circumscribed, and exophytic. Large lesions can be difficult to distinguish from verrucous carcinomas, and biopsy generally reveals koilocytes identified by perinuclear clearing and raisinlike nuclei. Immunohistochemical staining and in situ hybridization studies can be of value in diagnosis and in identifying those lesions that are at high risk for malignant transformation. High-risk condylomas are associated with HPV-16, HPV-18, HPV-31, HPV-33, HPV-35, and HPV-39, as well as other types, whereas low-risk condylomas are associated with HPV-6, HPV-11, HPV-42, and others.2 Differentiating squamous cell hyperplasia from squamous cell carcinoma in situ also can be aided by immunohistochemistry. Squamous cell hyperplasia is usually negative for INK4 p16Ink4A and p53 and exhibits variable Ki-67 staining. Differentiated squamous cell carcinoma in situ exhibits a profile that is p16Ink4A negative, Ki-67 positive, and exhibits variable p53 staining.3 Basaloid and warty intraepithelial neoplasia is consistently p16Ink4A positive, Ki-67 positive, and variably positive for p53.3 Therefore, p16 staining of high-grade areas is a useful biomarker that can help establish diagnosis of associated squamous cell carcinoma.4 The role of papillomaviruses in the development of nonmelanoma skin cancer is an area of active study, and research suggests that papillomaviruses may have a much greater role than previously suspected.5
At times, psoriasis may be markedly hyperkeratotic, clinically mimicking a verrucous neoplasm. This hyperkeratotic type of psoriasis is known as rupioid psoriasis. However, these psoriatic lesions are exophytic, are associated with spongiform pustules, and lack the atypia and endophytic pattern typically seen with verrucous carcinoma. An ILVEN also lacks atypia and an endophytic pattern and usually presents in childhood as a persistent linear plaque, rather than the verrucous plaque noted in our patient. Squamous cell carcinoma has been reported to arise in the setting of verrucoid ILVEN but is exceptionally uncommon.6
Successful treatment of verrucous carcinoma is best achieved by complete excision. Oral retinoids and immunomodulators such as imiquimod also may be of value.7 Our patient’s tumor qualifies as T2N0M0 because it was greater than 2 cm in size.8 A Breslow thickness of 2 mm or greater and Clark level IV are high-risk features associated with a worse prognosis, but clinical evaluation of our patient’s lymph nodes was unremarkable and no distant metastases were identified. Our patient continues to do well with no evidence of recurrence.
An 81-year-old woman presented for evaluation of a nodule on the right labia majora that had been present for 1 year. She had a history of intertriginous psoriasis, and several biopsies were performed at an outside facility over the last 5 years that revealed psoriasis but were otherwise noncontributory. Physical examination revealed erythema and scaling on the buttocks with maceration in the intertriginous area (top) and the perineum associated with a verrucous nodule (bottom).
The Diagnosis: Verrucous Carcinoma
Biopsies of early lesions often may be difficult to interpret without clinicopathological correlation. Our patient’s tumor was associated with intertriginous psoriasis, which was the only abnormality previously noted on superficial biopsies performed at an outside facility. The patient was scheduled for an excisional biopsy due to the large tumor size and clinical suspicion that the prior biopsies were inadequate and failed to demonstrate the primary underlying pathology. Excisional biopsy of the verrucous tumor revealed epithelium composed of keratinocytes with glassy cytoplasm. Papillomatosis was noted along with an endophytic component of well-differentiated epithelial cells extending into the dermis in a bulbous pattern consistent with the verrucous carcinoma variant of squamous cell carcinoma (SCC)(Figure). Verrucous carcinoma often requires correlation with both the clinical and histopathologic findings for definitive diagnosis, as keratinocytes often appear to be well differentiated.1
Verrucous carcinoma may begin as an innocuous papule that slowly grows into a large fungating tumor. Verrucous carcinomas typically are slow growing, exophytic, and low grade. The etiology of verrucous carcinoma is not clear, and the role of human papillomavirus (HPV) infection is controversial.2 Best classified as a well-differentiated SCC, verrucous carcinoma rarely metastasizes but may invade adjacent tissues.
Differential diagnoses include a giant inflamed seborrheic keratosis, condyloma acuminatum, rupioid psoriasis, and inflammatory linear verrucous epidermal nevus (ILVEN). Although large and inflamed seborrheic keratoses may have squamous eddies that mimic SCC, seborrheic keratoses do not invade the dermis and typically have a well-circumscribed stuck-on appearance. Abnormal mitotic figures are not identified. Condylomas are genital warts caused by HPV infection that often are clustered, well circumscribed, and exophytic. Large lesions can be difficult to distinguish from verrucous carcinomas, and biopsy generally reveals koilocytes identified by perinuclear clearing and raisinlike nuclei. Immunohistochemical staining and in situ hybridization studies can be of value in diagnosis and in identifying those lesions that are at high risk for malignant transformation. High-risk condylomas are associated with HPV-16, HPV-18, HPV-31, HPV-33, HPV-35, and HPV-39, as well as other types, whereas low-risk condylomas are associated with HPV-6, HPV-11, HPV-42, and others.2 Differentiating squamous cell hyperplasia from squamous cell carcinoma in situ also can be aided by immunohistochemistry. Squamous cell hyperplasia is usually negative for INK4 p16Ink4A and p53 and exhibits variable Ki-67 staining. Differentiated squamous cell carcinoma in situ exhibits a profile that is p16Ink4A negative, Ki-67 positive, and exhibits variable p53 staining.3 Basaloid and warty intraepithelial neoplasia is consistently p16Ink4A positive, Ki-67 positive, and variably positive for p53.3 Therefore, p16 staining of high-grade areas is a useful biomarker that can help establish diagnosis of associated squamous cell carcinoma.4 The role of papillomaviruses in the development of nonmelanoma skin cancer is an area of active study, and research suggests that papillomaviruses may have a much greater role than previously suspected.5
At times, psoriasis may be markedly hyperkeratotic, clinically mimicking a verrucous neoplasm. This hyperkeratotic type of psoriasis is known as rupioid psoriasis. However, these psoriatic lesions are exophytic, are associated with spongiform pustules, and lack the atypia and endophytic pattern typically seen with verrucous carcinoma. An ILVEN also lacks atypia and an endophytic pattern and usually presents in childhood as a persistent linear plaque, rather than the verrucous plaque noted in our patient. Squamous cell carcinoma has been reported to arise in the setting of verrucoid ILVEN but is exceptionally uncommon.6
Successful treatment of verrucous carcinoma is best achieved by complete excision. Oral retinoids and immunomodulators such as imiquimod also may be of value.7 Our patient’s tumor qualifies as T2N0M0 because it was greater than 2 cm in size.8 A Breslow thickness of 2 mm or greater and Clark level IV are high-risk features associated with a worse prognosis, but clinical evaluation of our patient’s lymph nodes was unremarkable and no distant metastases were identified. Our patient continues to do well with no evidence of recurrence.
1. Bambao C, Nofech-Mozes S, Shier M. Giant condyloma versus verrucous carcinoma: a case report. J Low Genit Tract Dis. 2010;14:230-233.
2. Asiaf A, Ahmad ST, Mohannad SO, et al. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur J Cancer Prev. 2014;23:206-224.
3. Chaux A, Pfannl R, Rodríguez IM, et al. Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol. 2011;35:553-562.
4. Darragh TM, Colgan TJ, Cox JT, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med. 2012;136:1266-1297.
5. Aldabagh B, Angeles J, Cardones AR, et al. Cutaneous squamous cell carcinoma and human papillomavirus: is there an association? Dermatol Surg. 2013;39:1-23.
6. Turk BG, Ertam I, Urkmez A, et al. Development of squamous cell carcinoma on an inflammatory linear verrucous epidermal nevus in the genital area. Cutis. 2012;89:273-275.
7. Erkek E, Basar H, Bozdogan O, et al. Giant condyloma acuminata of Buschke-Löwenstein: successful treatment with a combination of surgical excision, oral acitretin and topical imiquimod. Clin Exp Dermatol. 2009;34:366-368.
8. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In: Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301-314.
1. Bambao C, Nofech-Mozes S, Shier M. Giant condyloma versus verrucous carcinoma: a case report. J Low Genit Tract Dis. 2010;14:230-233.
2. Asiaf A, Ahmad ST, Mohannad SO, et al. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur J Cancer Prev. 2014;23:206-224.
3. Chaux A, Pfannl R, Rodríguez IM, et al. Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol. 2011;35:553-562.
4. Darragh TM, Colgan TJ, Cox JT, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med. 2012;136:1266-1297.
5. Aldabagh B, Angeles J, Cardones AR, et al. Cutaneous squamous cell carcinoma and human papillomavirus: is there an association? Dermatol Surg. 2013;39:1-23.
6. Turk BG, Ertam I, Urkmez A, et al. Development of squamous cell carcinoma on an inflammatory linear verrucous epidermal nevus in the genital area. Cutis. 2012;89:273-275.
7. Erkek E, Basar H, Bozdogan O, et al. Giant condyloma acuminata of Buschke-Löwenstein: successful treatment with a combination of surgical excision, oral acitretin and topical imiquimod. Clin Exp Dermatol. 2009;34:366-368.
8. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In: Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301-314.
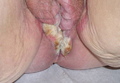
Round Purple Erythematous Tumors
The Diagnosis: Skin Metastases of Vulvar Carcinoma
A Tzanck cytodiagnosis was performed and a punch biopsy specimen was obtained for histologic examination. The patient’s Tzanck cytodiagnosis was initially interpreted as a viral cytopathic effect due to the presence of multinucleated atypical cells; hence, she was treated with intravenous acyclovir. The biopsy specimen showed a preserved epidermis and a dense infiltrate in the mid and deep dermis (Figure 1) formed by atypical squamous cells with enlarged nuclei and anisokaryosis, evident nucleoli, atypical mitoses, and multinucleated cells (Figure 2). The histopathologic diagnosis was skin metastases of moderately differentiated squamous cell carcinoma. The subsequent clinical course was unfavorableand the patient died during hospitalization from septic shock.
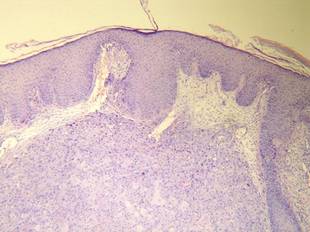
Figure 1. Histology revealed a preserved epidermis with a dense infiltrate in the mid and deep dermis (H&E, original magnification ×40). | 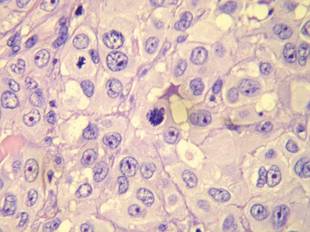
Figure 2. Atypical squamous cells with enlarged nuclei and anisokaryosis, evident nucleoli, atypical mitoses, and multinucleated cells were evident (H&E, original magnification ×400). |
Cutaneous metastases occur in 0.7% to 9% of solid tumors in advanced stages of disease progression or occasionally as an initial manifestation; they are a predictor of poor prognosis and breast cancer is the most frequent cause in women.1 Vulvar carcinoma comprises 5% of all malignant neoplasms of the female genital tract and 95% are squamous cell carcinoma; metastases appear most frequently in the inguinal and pelvic lymph nodes, followed by the lungs, liver, and bones.2 Skin metastases are extremely rare, with few cases documented.2,3
Clinically, skin metastases present most frequently as nodules, either solitary or multiple, that are sometimes ulcerated. However, a wide spectrum of metastases has been described, including erysipeloid, sclerodermiform (en cuirasse), telangiectatic papulovesicles, purpuric plaques mimicking vasculitis, alopecia areata–like scalp lesions, and others. The zosteriform pattern has been described in few cases, including vesiculobullous herpetiform lesions or nodular metastases with metameric distribution.4 In more than half of cases, the lesions were initially interpreted as herpes zoster and hence treated with acyclovir.5
Regarding cutaneous metastases of vulvar carcinoma, a case of metastases mimicking primary varicella-zoster virus infection has been reported,3 but our case represents a zosteriform pattern, which is unique.
Multiple theories have been proposed to explain the pathogenic mechanism of zosteriform spread, though none have been adequately proven.4-7 Some of the patients described in the literature had a history of viral infection by serology or polymerase chain reaction for herpes simplex virus types 1 and 2 and varicella-zoster virus in the same dermatome where metastases were observed. It has been suggested that neural damage caused by the herpesvirus results in an immune function impairment of the overlying skin, which consequently may be more receptive to the inflow of metastatic cells. In such cases, the zosteriform pattern could be due to a Wolf or Köbner phenomenon in an area of vulnerable skin.4-9
Other possible explanations for the zosteriform spread are the invasion of perineural lymphatic vessels or of the dorsal root ganglion, direct invasion from deeper structures, or the surgical implantation of neoplastic cells,4-7 though the latter 2 mechanisms do not correspond to true metastases.
1. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
2. Wang AR, O’Brien M, Ross R, et al. Epidermotropic metastasis from vulvar squamous cell carcinoma: a rare cutaneous manifestation. J Am Acad Dermatol. 2010;63:1088-1091.
3. Mailhot J, O’Donnell P, Han R, et al. Metastatic squamous cell carcinoma of the vulva mimicking primary varicella-zoster virus infection. J Am Acad Dermatol. 2011;65:e63-e64.
4. Savoia P, Fava P, Deboli T, et al. Zosteriform cutaneous metastases: a literature meta-analysis and a clinical report of three melanoma cases. Dermatol Surg. 2009;35:1355-1363.
5. Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
6. Zalaudek I, Leinweber B, Richtig E, et al. Cutaneous zosteriform melanoma metastases arising after herpes zoster infection: a case report and review of the literature. Melanoma Res. 2003;13:635-639.
7. LeSueur BW, Abraham LJ, DiCaudo DJ, et al. Zosteriform skin metastases. Int J Dermatol. 2004;43:126-128.
8. Ruocco V, Ruocco E, Brunetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
9. Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
The Diagnosis: Skin Metastases of Vulvar Carcinoma
A Tzanck cytodiagnosis was performed and a punch biopsy specimen was obtained for histologic examination. The patient’s Tzanck cytodiagnosis was initially interpreted as a viral cytopathic effect due to the presence of multinucleated atypical cells; hence, she was treated with intravenous acyclovir. The biopsy specimen showed a preserved epidermis and a dense infiltrate in the mid and deep dermis (Figure 1) formed by atypical squamous cells with enlarged nuclei and anisokaryosis, evident nucleoli, atypical mitoses, and multinucleated cells (Figure 2). The histopathologic diagnosis was skin metastases of moderately differentiated squamous cell carcinoma. The subsequent clinical course was unfavorableand the patient died during hospitalization from septic shock.
|
Figure 1. Histology revealed a preserved epidermis with a dense infiltrate in the mid and deep dermis (H&E, original magnification ×40). |
Figure 2. Atypical squamous cells with enlarged nuclei and anisokaryosis, evident nucleoli, atypical mitoses, and multinucleated cells were evident (H&E, original magnification ×400). |
Cutaneous metastases occur in 0.7% to 9% of solid tumors in advanced stages of disease progression or occasionally as an initial manifestation; they are a predictor of poor prognosis and breast cancer is the most frequent cause in women.1 Vulvar carcinoma comprises 5% of all malignant neoplasms of the female genital tract and 95% are squamous cell carcinoma; metastases appear most frequently in the inguinal and pelvic lymph nodes, followed by the lungs, liver, and bones.2 Skin metastases are extremely rare, with few cases documented.2,3
Clinically, skin metastases present most frequently as nodules, either solitary or multiple, that are sometimes ulcerated. However, a wide spectrum of metastases has been described, including erysipeloid, sclerodermiform (en cuirasse), telangiectatic papulovesicles, purpuric plaques mimicking vasculitis, alopecia areata–like scalp lesions, and others. The zosteriform pattern has been described in few cases, including vesiculobullous herpetiform lesions or nodular metastases with metameric distribution.4 In more than half of cases, the lesions were initially interpreted as herpes zoster and hence treated with acyclovir.5
Regarding cutaneous metastases of vulvar carcinoma, a case of metastases mimicking primary varicella-zoster virus infection has been reported,3 but our case represents a zosteriform pattern, which is unique.
Multiple theories have been proposed to explain the pathogenic mechanism of zosteriform spread, though none have been adequately proven.4-7 Some of the patients described in the literature had a history of viral infection by serology or polymerase chain reaction for herpes simplex virus types 1 and 2 and varicella-zoster virus in the same dermatome where metastases were observed. It has been suggested that neural damage caused by the herpesvirus results in an immune function impairment of the overlying skin, which consequently may be more receptive to the inflow of metastatic cells. In such cases, the zosteriform pattern could be due to a Wolf or Köbner phenomenon in an area of vulnerable skin.4-9
Other possible explanations for the zosteriform spread are the invasion of perineural lymphatic vessels or of the dorsal root ganglion, direct invasion from deeper structures, or the surgical implantation of neoplastic cells,4-7 though the latter 2 mechanisms do not correspond to true metastases.
The Diagnosis: Skin Metastases of Vulvar Carcinoma
A Tzanck cytodiagnosis was performed and a punch biopsy specimen was obtained for histologic examination. The patient’s Tzanck cytodiagnosis was initially interpreted as a viral cytopathic effect due to the presence of multinucleated atypical cells; hence, she was treated with intravenous acyclovir. The biopsy specimen showed a preserved epidermis and a dense infiltrate in the mid and deep dermis (Figure 1) formed by atypical squamous cells with enlarged nuclei and anisokaryosis, evident nucleoli, atypical mitoses, and multinucleated cells (Figure 2). The histopathologic diagnosis was skin metastases of moderately differentiated squamous cell carcinoma. The subsequent clinical course was unfavorableand the patient died during hospitalization from septic shock.
|
Figure 1. Histology revealed a preserved epidermis with a dense infiltrate in the mid and deep dermis (H&E, original magnification ×40). |
Figure 2. Atypical squamous cells with enlarged nuclei and anisokaryosis, evident nucleoli, atypical mitoses, and multinucleated cells were evident (H&E, original magnification ×400). |
Cutaneous metastases occur in 0.7% to 9% of solid tumors in advanced stages of disease progression or occasionally as an initial manifestation; they are a predictor of poor prognosis and breast cancer is the most frequent cause in women.1 Vulvar carcinoma comprises 5% of all malignant neoplasms of the female genital tract and 95% are squamous cell carcinoma; metastases appear most frequently in the inguinal and pelvic lymph nodes, followed by the lungs, liver, and bones.2 Skin metastases are extremely rare, with few cases documented.2,3
Clinically, skin metastases present most frequently as nodules, either solitary or multiple, that are sometimes ulcerated. However, a wide spectrum of metastases has been described, including erysipeloid, sclerodermiform (en cuirasse), telangiectatic papulovesicles, purpuric plaques mimicking vasculitis, alopecia areata–like scalp lesions, and others. The zosteriform pattern has been described in few cases, including vesiculobullous herpetiform lesions or nodular metastases with metameric distribution.4 In more than half of cases, the lesions were initially interpreted as herpes zoster and hence treated with acyclovir.5
Regarding cutaneous metastases of vulvar carcinoma, a case of metastases mimicking primary varicella-zoster virus infection has been reported,3 but our case represents a zosteriform pattern, which is unique.
Multiple theories have been proposed to explain the pathogenic mechanism of zosteriform spread, though none have been adequately proven.4-7 Some of the patients described in the literature had a history of viral infection by serology or polymerase chain reaction for herpes simplex virus types 1 and 2 and varicella-zoster virus in the same dermatome where metastases were observed. It has been suggested that neural damage caused by the herpesvirus results in an immune function impairment of the overlying skin, which consequently may be more receptive to the inflow of metastatic cells. In such cases, the zosteriform pattern could be due to a Wolf or Köbner phenomenon in an area of vulnerable skin.4-9
Other possible explanations for the zosteriform spread are the invasion of perineural lymphatic vessels or of the dorsal root ganglion, direct invasion from deeper structures, or the surgical implantation of neoplastic cells,4-7 though the latter 2 mechanisms do not correspond to true metastases.
1. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
2. Wang AR, O’Brien M, Ross R, et al. Epidermotropic metastasis from vulvar squamous cell carcinoma: a rare cutaneous manifestation. J Am Acad Dermatol. 2010;63:1088-1091.
3. Mailhot J, O’Donnell P, Han R, et al. Metastatic squamous cell carcinoma of the vulva mimicking primary varicella-zoster virus infection. J Am Acad Dermatol. 2011;65:e63-e64.
4. Savoia P, Fava P, Deboli T, et al. Zosteriform cutaneous metastases: a literature meta-analysis and a clinical report of three melanoma cases. Dermatol Surg. 2009;35:1355-1363.
5. Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
6. Zalaudek I, Leinweber B, Richtig E, et al. Cutaneous zosteriform melanoma metastases arising after herpes zoster infection: a case report and review of the literature. Melanoma Res. 2003;13:635-639.
7. LeSueur BW, Abraham LJ, DiCaudo DJ, et al. Zosteriform skin metastases. Int J Dermatol. 2004;43:126-128.
8. Ruocco V, Ruocco E, Brunetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
9. Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
1. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2, pt 1):161-182.
2. Wang AR, O’Brien M, Ross R, et al. Epidermotropic metastasis from vulvar squamous cell carcinoma: a rare cutaneous manifestation. J Am Acad Dermatol. 2010;63:1088-1091.
3. Mailhot J, O’Donnell P, Han R, et al. Metastatic squamous cell carcinoma of the vulva mimicking primary varicella-zoster virus infection. J Am Acad Dermatol. 2011;65:e63-e64.
4. Savoia P, Fava P, Deboli T, et al. Zosteriform cutaneous metastases: a literature meta-analysis and a clinical report of three melanoma cases. Dermatol Surg. 2009;35:1355-1363.
5. Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
6. Zalaudek I, Leinweber B, Richtig E, et al. Cutaneous zosteriform melanoma metastases arising after herpes zoster infection: a case report and review of the literature. Melanoma Res. 2003;13:635-639.
7. LeSueur BW, Abraham LJ, DiCaudo DJ, et al. Zosteriform skin metastases. Int J Dermatol. 2004;43:126-128.
8. Ruocco V, Ruocco E, Brunetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
9. Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
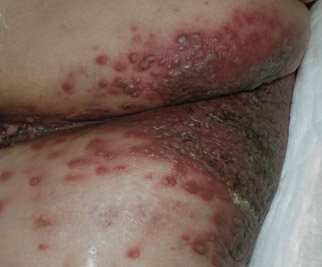
A 48-year-old woman presented with a dermatosis on the left thigh. She had a history of hypertension, obesity, and a stage IIIC vulvar carcinoma treated with radical vulvectomy and bilateral inguinal lymphadenectomy. Two months after surgery she was hospitalized because of a left popliteal deep vein thrombosis and an abscess involving the site of the lymphadenectomy. One month later she presented with a painful dermatosis on the left thigh. Physical examination revealed multiple round-shaped, purple, erythematous tumors with a smooth surface on the upper third of the left thigh extending to the lower abdomen. The tumors measured 0.5 cm in diameter and were grouped to form an indurated plaque on an erythematous base; some tumors were arranged as satellite lesions in the periphery. The dermatosis had a distinct zosteriform appearance.
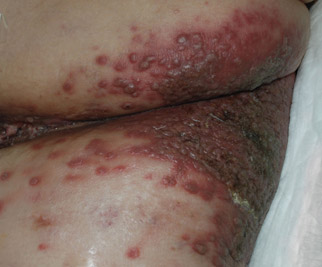
Clustered Vesicles in a Blaschkoid Pattern
The Diagnosis: Linear Vesiculobullous Keratosis Follicularis (Darier Disease)
Darier disease (DD), or keratosis follicularis, is typically an autosomal-dominant disorder that is characterized by greasy hyperkeratotic papules that coalesce into warty plaques with a predilection for seborrheic areas. The lesions usually are pruritic; malodorous; and may be exacerbated by sunlight, heat, or sweating. Darier disease may be accompanied by oral mucosal involvement including fine white papules on the palate.1 The condition also can be accompanied by hand and nail involvement (95% of cases) including palmar pitting, punctate keratoses, hemorrhagic macules, palmoplantar keratoderma, and acrokeratosis verruciformis–like lesions on the dorsal aspects of the hands and feet. Nail changes predominately occur on the fingers, manifesting as longitudinal splitting, subungual hyperkeratosis, or characteristic white and red longitudinal bands with V-shaped nicks at the free margin of the nail. In linear DD, hand and nail involvement is rare and, when present, ipsilateral to the primary lesions.1,2
The clinical variants of DD are classified by lesion morphology or distribution, or both. Morphological variants include vesiculobullous, cornified, erosive, acral hemorrhagic, and guttate leukodermic macular.1,2 The clinical features of chronic relapsing vesicular lesions and histologic findings described in this case are consistent with vesiculobullous DD, though genetic testing was not performed.3,4 As in our case, some patients lack a family history and the disease is thought to be the result of genetic mosaicism or somatic postzygotic mutations that affect a limited number of cells. These mosaic variants are named by their cutaneous distribution (ie, linear, segmental, unilateral, localized) and tend to course along the Blaschko lines, most commonly on the trunk. Studies have shown various types of mutations specific to the ATP2A2 gene in the affected tissue but not in the unaffected skin.5 This gene encodes for sarcoplasmic/endoplasmic reticulum ATPase SERCA2, which is responsible for intracellular calcium signaling. These mosaic forms of DD are unlikely to be inherited by offspring, in contrast to patients with mosaic epidermal nevi with epidermolytic hyperkeratosis who have a high likelihood of having children with generalized epidermolytic hyperkeratosis.6
Darier disease is a chronic incurable disease. Topical corticosteroid, retinoid, 5-fluorouracil, keratolytics, and laser ablation or excision are used in mild and limited disease with mixed outcomes.7 Oral retinoids are effective in severe or systemic cases of DD by inhibiting hyperkeratosis.8 Individuals with DD are predisposed to infection, warranting regular surveillance and use of antimicrobials and bleach baths. In addition to prophylaxis for bacterial superinfection, patients also are predisposed to getting disseminated herpes simplex virus in the form of eczema herpeticum.
In our patient, the diagnosis was confirmed by performing a punch biopsy from one of the vesicular lesions. Histopathologic examination revealed suprabasal acantholytic dyskeratosis with superficial perivascular and interstitial inflammation with eosinophils (Figure). Immunohistochemical staining showed no evidence of varicella-zoster virus or herpes simplex virus type 1 or type 2.
Histopathology revealed suprabasal acantholysis forming an intraepidermal cleft with superficial perivascular inflammation (A)(H&E, original magnification ×100) and acantholysis with dyskeratotic keratinocytes floating within the intraepidermal cleft (B)(H&E, original magnification ×200). |
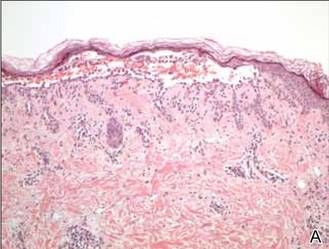
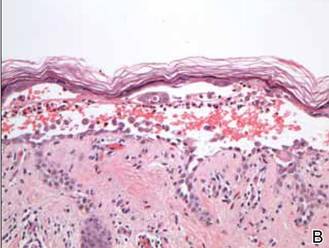
Our patient was prescribed tretinoin cream 0.1% daily and was advised to use sun protection and stop valacyclovir. At follow-up she noted decreased frequency of outbreaks after starting the tretinoin cream and the patient has now been free of any outbreaks for 8 months.
1. Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50.
2. Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
3. Kakar B, Kabir S, Garg VK, et al. A case of bullous Darier’s disease histologically mimicking Hailey-Hailey disease. Dermatol Online J. 2007;13:28.
4. Telfer NR, Burge SM, Ryan TJ. Vesiculo-bullous Darier’s disease. Br J Dermatol. 1990;122:831-834.
5. Sakuntabhai A, Dhitavat J, Burge SM, et al. Mosaicism for ATP2A2 mutations causes segmental Darier’s disease. J Invest Dermatol. 2000;115:1144-1147.
6. O’Malley MP, Haake A, Goldsmith L, et al. Localized Darier disease. implications for genetic studies. Arch Dermatol. 1997;133:1134-1138.
7. Le Bidre E, Delage M, Celerier P, et al. Efficacy and risks of topical 5-fluorouracil in Darier’s disease. Ann Dermatol Venereol. 2010;137:455-459.
8. Abe M, Yasuda M, Yokoyama Y, et al. Successful treatment of combination therapy with tacalcitol lotion associated with sunscreen for localized Darier’s disease. J Dermatol. 2010;37:718-721.
The Diagnosis: Linear Vesiculobullous Keratosis Follicularis (Darier Disease)
Darier disease (DD), or keratosis follicularis, is typically an autosomal-dominant disorder that is characterized by greasy hyperkeratotic papules that coalesce into warty plaques with a predilection for seborrheic areas. The lesions usually are pruritic; malodorous; and may be exacerbated by sunlight, heat, or sweating. Darier disease may be accompanied by oral mucosal involvement including fine white papules on the palate.1 The condition also can be accompanied by hand and nail involvement (95% of cases) including palmar pitting, punctate keratoses, hemorrhagic macules, palmoplantar keratoderma, and acrokeratosis verruciformis–like lesions on the dorsal aspects of the hands and feet. Nail changes predominately occur on the fingers, manifesting as longitudinal splitting, subungual hyperkeratosis, or characteristic white and red longitudinal bands with V-shaped nicks at the free margin of the nail. In linear DD, hand and nail involvement is rare and, when present, ipsilateral to the primary lesions.1,2
The clinical variants of DD are classified by lesion morphology or distribution, or both. Morphological variants include vesiculobullous, cornified, erosive, acral hemorrhagic, and guttate leukodermic macular.1,2 The clinical features of chronic relapsing vesicular lesions and histologic findings described in this case are consistent with vesiculobullous DD, though genetic testing was not performed.3,4 As in our case, some patients lack a family history and the disease is thought to be the result of genetic mosaicism or somatic postzygotic mutations that affect a limited number of cells. These mosaic variants are named by their cutaneous distribution (ie, linear, segmental, unilateral, localized) and tend to course along the Blaschko lines, most commonly on the trunk. Studies have shown various types of mutations specific to the ATP2A2 gene in the affected tissue but not in the unaffected skin.5 This gene encodes for sarcoplasmic/endoplasmic reticulum ATPase SERCA2, which is responsible for intracellular calcium signaling. These mosaic forms of DD are unlikely to be inherited by offspring, in contrast to patients with mosaic epidermal nevi with epidermolytic hyperkeratosis who have a high likelihood of having children with generalized epidermolytic hyperkeratosis.6
Darier disease is a chronic incurable disease. Topical corticosteroid, retinoid, 5-fluorouracil, keratolytics, and laser ablation or excision are used in mild and limited disease with mixed outcomes.7 Oral retinoids are effective in severe or systemic cases of DD by inhibiting hyperkeratosis.8 Individuals with DD are predisposed to infection, warranting regular surveillance and use of antimicrobials and bleach baths. In addition to prophylaxis for bacterial superinfection, patients also are predisposed to getting disseminated herpes simplex virus in the form of eczema herpeticum.
In our patient, the diagnosis was confirmed by performing a punch biopsy from one of the vesicular lesions. Histopathologic examination revealed suprabasal acantholytic dyskeratosis with superficial perivascular and interstitial inflammation with eosinophils (Figure). Immunohistochemical staining showed no evidence of varicella-zoster virus or herpes simplex virus type 1 or type 2.
Histopathology revealed suprabasal acantholysis forming an intraepidermal cleft with superficial perivascular inflammation (A)(H&E, original magnification ×100) and acantholysis with dyskeratotic keratinocytes floating within the intraepidermal cleft (B)(H&E, original magnification ×200). |
Our patient was prescribed tretinoin cream 0.1% daily and was advised to use sun protection and stop valacyclovir. At follow-up she noted decreased frequency of outbreaks after starting the tretinoin cream and the patient has now been free of any outbreaks for 8 months.
The Diagnosis: Linear Vesiculobullous Keratosis Follicularis (Darier Disease)
Darier disease (DD), or keratosis follicularis, is typically an autosomal-dominant disorder that is characterized by greasy hyperkeratotic papules that coalesce into warty plaques with a predilection for seborrheic areas. The lesions usually are pruritic; malodorous; and may be exacerbated by sunlight, heat, or sweating. Darier disease may be accompanied by oral mucosal involvement including fine white papules on the palate.1 The condition also can be accompanied by hand and nail involvement (95% of cases) including palmar pitting, punctate keratoses, hemorrhagic macules, palmoplantar keratoderma, and acrokeratosis verruciformis–like lesions on the dorsal aspects of the hands and feet. Nail changes predominately occur on the fingers, manifesting as longitudinal splitting, subungual hyperkeratosis, or characteristic white and red longitudinal bands with V-shaped nicks at the free margin of the nail. In linear DD, hand and nail involvement is rare and, when present, ipsilateral to the primary lesions.1,2
The clinical variants of DD are classified by lesion morphology or distribution, or both. Morphological variants include vesiculobullous, cornified, erosive, acral hemorrhagic, and guttate leukodermic macular.1,2 The clinical features of chronic relapsing vesicular lesions and histologic findings described in this case are consistent with vesiculobullous DD, though genetic testing was not performed.3,4 As in our case, some patients lack a family history and the disease is thought to be the result of genetic mosaicism or somatic postzygotic mutations that affect a limited number of cells. These mosaic variants are named by their cutaneous distribution (ie, linear, segmental, unilateral, localized) and tend to course along the Blaschko lines, most commonly on the trunk. Studies have shown various types of mutations specific to the ATP2A2 gene in the affected tissue but not in the unaffected skin.5 This gene encodes for sarcoplasmic/endoplasmic reticulum ATPase SERCA2, which is responsible for intracellular calcium signaling. These mosaic forms of DD are unlikely to be inherited by offspring, in contrast to patients with mosaic epidermal nevi with epidermolytic hyperkeratosis who have a high likelihood of having children with generalized epidermolytic hyperkeratosis.6
Darier disease is a chronic incurable disease. Topical corticosteroid, retinoid, 5-fluorouracil, keratolytics, and laser ablation or excision are used in mild and limited disease with mixed outcomes.7 Oral retinoids are effective in severe or systemic cases of DD by inhibiting hyperkeratosis.8 Individuals with DD are predisposed to infection, warranting regular surveillance and use of antimicrobials and bleach baths. In addition to prophylaxis for bacterial superinfection, patients also are predisposed to getting disseminated herpes simplex virus in the form of eczema herpeticum.
In our patient, the diagnosis was confirmed by performing a punch biopsy from one of the vesicular lesions. Histopathologic examination revealed suprabasal acantholytic dyskeratosis with superficial perivascular and interstitial inflammation with eosinophils (Figure). Immunohistochemical staining showed no evidence of varicella-zoster virus or herpes simplex virus type 1 or type 2.
Histopathology revealed suprabasal acantholysis forming an intraepidermal cleft with superficial perivascular inflammation (A)(H&E, original magnification ×100) and acantholysis with dyskeratotic keratinocytes floating within the intraepidermal cleft (B)(H&E, original magnification ×200). |
Our patient was prescribed tretinoin cream 0.1% daily and was advised to use sun protection and stop valacyclovir. At follow-up she noted decreased frequency of outbreaks after starting the tretinoin cream and the patient has now been free of any outbreaks for 8 months.
1. Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50.
2. Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
3. Kakar B, Kabir S, Garg VK, et al. A case of bullous Darier’s disease histologically mimicking Hailey-Hailey disease. Dermatol Online J. 2007;13:28.
4. Telfer NR, Burge SM, Ryan TJ. Vesiculo-bullous Darier’s disease. Br J Dermatol. 1990;122:831-834.
5. Sakuntabhai A, Dhitavat J, Burge SM, et al. Mosaicism for ATP2A2 mutations causes segmental Darier’s disease. J Invest Dermatol. 2000;115:1144-1147.
6. O’Malley MP, Haake A, Goldsmith L, et al. Localized Darier disease. implications for genetic studies. Arch Dermatol. 1997;133:1134-1138.
7. Le Bidre E, Delage M, Celerier P, et al. Efficacy and risks of topical 5-fluorouracil in Darier’s disease. Ann Dermatol Venereol. 2010;137:455-459.
8. Abe M, Yasuda M, Yokoyama Y, et al. Successful treatment of combination therapy with tacalcitol lotion associated with sunscreen for localized Darier’s disease. J Dermatol. 2010;37:718-721.
1. Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50.
2. Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97-105.
3. Kakar B, Kabir S, Garg VK, et al. A case of bullous Darier’s disease histologically mimicking Hailey-Hailey disease. Dermatol Online J. 2007;13:28.
4. Telfer NR, Burge SM, Ryan TJ. Vesiculo-bullous Darier’s disease. Br J Dermatol. 1990;122:831-834.
5. Sakuntabhai A, Dhitavat J, Burge SM, et al. Mosaicism for ATP2A2 mutations causes segmental Darier’s disease. J Invest Dermatol. 2000;115:1144-1147.
6. O’Malley MP, Haake A, Goldsmith L, et al. Localized Darier disease. implications for genetic studies. Arch Dermatol. 1997;133:1134-1138.
7. Le Bidre E, Delage M, Celerier P, et al. Efficacy and risks of topical 5-fluorouracil in Darier’s disease. Ann Dermatol Venereol. 2010;137:455-459.
8. Abe M, Yasuda M, Yokoyama Y, et al. Successful treatment of combination therapy with tacalcitol lotion associated with sunscreen for localized Darier’s disease. J Dermatol. 2010;37:718-721.
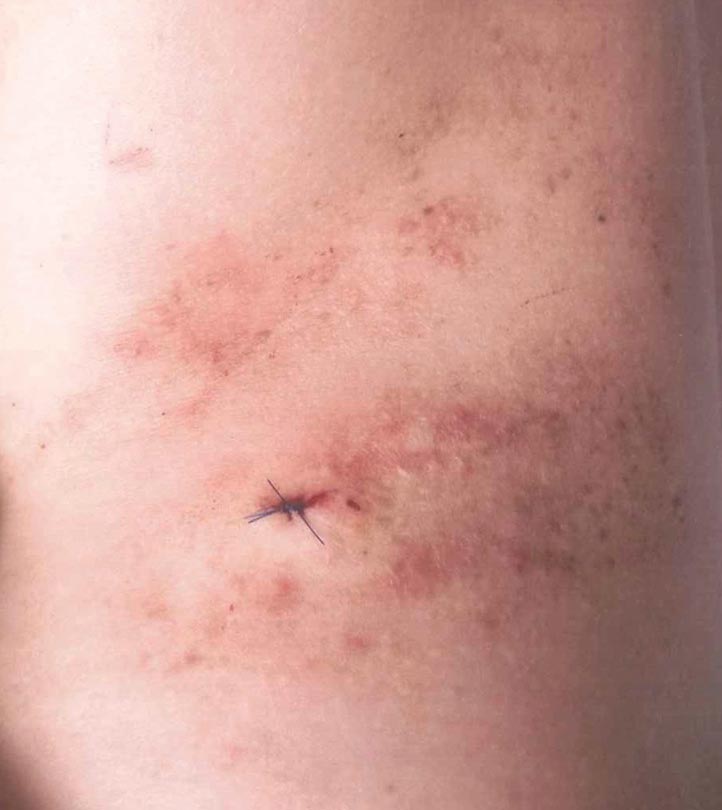
A 42-year-old woman presented with an intermittent nontender and minimally pruritic rash localized to the left side of the trunk of 20 years’ duration. Four to 6 times per year blisters would develop and then resolve after 1 to 2 weeks with mildly pruritic brown patches. These patches would resolve within approximately 4 weeks. Most notably, the condition was exacerbated by sunlight and heat, though stress sometimes led to an outbreak of vesicles. The patient reported that the eruption, which she was told was recurrent shingles, would improve with oral valacyclovir 1 g twice daily and did not improve with topical steroid usage. She never had antecedent or concurrent fevers, shortness of breath, arthralgia, or cold sores. There was no family history of any blistering skin conditions such as epidermolysis bullosa, pemphigus, Darier disease, or bullous pemphigoid. Her partner also did not have a history of similar rashes, and the patient denied any history of travel outside of England and the southwestern United States. Initial physical examination revealed clustered vesicles surrounded by brown-pink patches in a blaschkoid pattern spanning from the anterior to posterior aspects of the left flank. Notably, the patient had no oral lesions and no changes of the hair or nails.

Erythematous Papules and Plaques on the Flank of a Child
The Diagnosis: Asymmetric Periflexural Exanthem of Childhood (Unilateral Laterothoracic Exanthem)
Asymmetric periflexural exanthem of childhood (APEC), also known as unilateral laterothoracic exanthem, is a self-limited eruptive dermatosis that occurs most frequently in infants and young children. The term unilateral laterothoracic exanthem was first coined by Bodemer and de Prost1 in 1992 due to its characteristic distribution. The eruption occurs in children aged 4 months to 10 years, with most cases presenting between 2 and 3 years of age.2 Isolated cases also have been reported in adults.3 It affects girls more often than boys (2:1), and the majority of reported cases have occurred in white individuals. The disease is seen throughout Europe and North America, and seasonal variation has been noted with most cases occurring in late winter and early spring.4,5
|
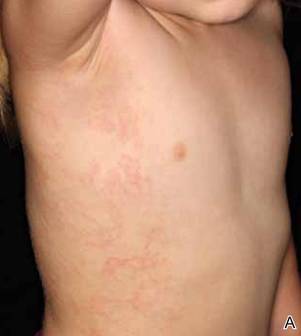
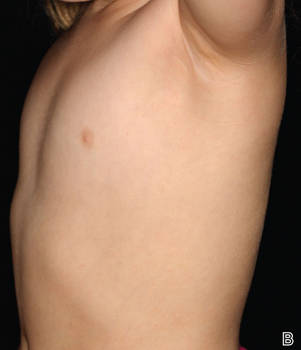
Clinically, APEC is characterized by its asymmetric localization and unilateral onset. In the majority of patients, the eruption presents as discrete erythematous papules that coalesce to form morbilliform plaques that may have reticular or annular configuration (Figure).4 The exanthem begins unilaterally near a flexural area, most commonly the axilla (75% of cases), and spreads centrifugally to the adjacent trunk and proximal extremity. There is no right or left dominance.4,6 There is eventual involvement of the contralateral side in 70% of cases, but a unilateral predominance is maintained throughout the disease course.4 Rarely, the eruption may involve the face, genitals, and palmoplantar surfaces. As in our case, up to three-quarters of affected children report symptoms of an upper respiratory tract or gastrointestinal prodrome, including mild fever, diarrhea, and rhinitis.4 Accompanying regional lymphadenopathy has been reported in the majority of cases, and mild to moderate pruritus is not uncommon. The syndrome is self-limited, with spontaneous resolution commonly occurring 3 to 6 weeks after onset. Although no treatment is required, systemic antihistamines and topical steroids have been used to alleviate pruritus in symptomatic patients. Our patient was treated with triamcinolone cream 0.1% twice daily as well as oral diphenhydramine 25 mg every 6 hours as needed for associated pruritus. The eruption spontaneously resolved over the following 4 weeks.
Although the cause of APEC remains unknown, an infectious etiology has been presumed. The seasonal pattern, lack of efficacy of broad-spectrum antibiotics, frequently reported prodromal symptoms, and reports of familial cases suggest a viral etiology.1 Additionally, the predilection to affect infants and young children as well as lack of recurrence in the same patient suggests that immunity may develop. Although no etiologic agent has been consistently detected, several reports have suggested a possible relationship to parvovirus B19.7,8 Parainfluenzavirus 2, parainfluenzavirus 3, and adenovirus also have been isolated but may represent incidental viral infection.2 An inoculation dermatosis from an arthropod bite also has been suggested, but this claim has not been substantiated.1
The diagnosis often can be made on clinical features alone, and histopathologic evaluation is not required. Histologic features are nonspecific and include a superficial perivascular infiltrate of lymphocytes, often involving the dermal eccrine ducts without involvement of the secretory coils.4,6 Mild lichenoid changes as well as spongiosis with exocytosis of lymphocytes into the acrosyringium also may be present.4 The clinical differential diagnosis of APEC includes allergic contact dermatitis, a nonspecific drug or viral eruption, atypical pityriasis rosea, miliaria, scabies, tinea corporis, and Gianotti-Crosti syndrome. Asymmetric periflexural exanthem of childhood lacks the peripheral scale present in tinea corporis or pityriasis rosea, but when an annular or reticular configuration predominates, a potassium hydroxide preparation of skin scrapings can exclude the presence of a dermatophyte. Similar to APEC, Gianotti-Crosti syndrome affects young children, is preceded by symptoms of a viral prodrome, and spontaneously resolves over several weeks. This condition is distinguished from APEC by the presence of papulovesicles located symmetrically on the face, buttocks, and extensor surface of the extremities, which largely spare the trunk.
Asymmetric periflexural exanthem of childhood is a unique morbilliform eruption of infants and young children characterized by a stereotypical distribution and self-limited course. The cause of this syndrome remains unclear, but most authors suggest a viral etiology. Recognition of this entity and an ability to distinguish it from other common pediatric dermatoses is required to provide reassurance to parents and avoid unnecessary diagnostic procedures and treatments.
1. Bodemer C, de Prost Y. Unilateral laterothoracic exanthem in children: a new disease? J Am Acad Dermatol. 1992;27(5, pt 1):693-696.
2. Nahm WK, Paiva C, Golomb C, et al. Asymmetric periflexural exanthema of childhood: a case involving a 4-month-old infant. Pediatr Dermatol. 2002;19:461-462.
3. Chan PK, To KF, Zawar V, et al. Asymmetric periflexural exanthema in an adult. Clin Exp Dermatol. 2004;29:320-321.
4. McCuaig CC, Russo P, Powell J, et al. Unilateral laterothoracic exanthem. a clinicopathologic study of forty-eight patients. J Am Acad Dermatol. 1996;34:979-984.
5. Taieb A, Megraud F, Legrain V, et al. Asymmetric periflexural exanthem of childhood. J Am Acad Dermatol. 1993;29:391-393.
6. Coustou D, Léauté-Labrèze C, Bioulac-Sage P, et al. Asymmetric periflexural exanthem of childhood. a clinical, pathologic, and epidemiologic prospective study. Arch Dermatol. 1999;135:799-803.
7. Guimerá-Martín-Neda F, Fagundo E, Rodríguez F, et al. Asymmetric periflexural exanthem of childhood: report of two cases with parvovirus B19. J Eur Acad Dermatol Venereol. 2006;20:461-462.
8. Pauluzzi P, Festini G, Gelmetti C. Asymmetric periflexural exanthem of childhood in an adult patient with parvovirus B19. J Eur Acad Dermatol Venereol. 2001;15:372-374.
The Diagnosis: Asymmetric Periflexural Exanthem of Childhood (Unilateral Laterothoracic Exanthem)
Asymmetric periflexural exanthem of childhood (APEC), also known as unilateral laterothoracic exanthem, is a self-limited eruptive dermatosis that occurs most frequently in infants and young children. The term unilateral laterothoracic exanthem was first coined by Bodemer and de Prost1 in 1992 due to its characteristic distribution. The eruption occurs in children aged 4 months to 10 years, with most cases presenting between 2 and 3 years of age.2 Isolated cases also have been reported in adults.3 It affects girls more often than boys (2:1), and the majority of reported cases have occurred in white individuals. The disease is seen throughout Europe and North America, and seasonal variation has been noted with most cases occurring in late winter and early spring.4,5
|
Clinically, APEC is characterized by its asymmetric localization and unilateral onset. In the majority of patients, the eruption presents as discrete erythematous papules that coalesce to form morbilliform plaques that may have reticular or annular configuration (Figure).4 The exanthem begins unilaterally near a flexural area, most commonly the axilla (75% of cases), and spreads centrifugally to the adjacent trunk and proximal extremity. There is no right or left dominance.4,6 There is eventual involvement of the contralateral side in 70% of cases, but a unilateral predominance is maintained throughout the disease course.4 Rarely, the eruption may involve the face, genitals, and palmoplantar surfaces. As in our case, up to three-quarters of affected children report symptoms of an upper respiratory tract or gastrointestinal prodrome, including mild fever, diarrhea, and rhinitis.4 Accompanying regional lymphadenopathy has been reported in the majority of cases, and mild to moderate pruritus is not uncommon. The syndrome is self-limited, with spontaneous resolution commonly occurring 3 to 6 weeks after onset. Although no treatment is required, systemic antihistamines and topical steroids have been used to alleviate pruritus in symptomatic patients. Our patient was treated with triamcinolone cream 0.1% twice daily as well as oral diphenhydramine 25 mg every 6 hours as needed for associated pruritus. The eruption spontaneously resolved over the following 4 weeks.
Although the cause of APEC remains unknown, an infectious etiology has been presumed. The seasonal pattern, lack of efficacy of broad-spectrum antibiotics, frequently reported prodromal symptoms, and reports of familial cases suggest a viral etiology.1 Additionally, the predilection to affect infants and young children as well as lack of recurrence in the same patient suggests that immunity may develop. Although no etiologic agent has been consistently detected, several reports have suggested a possible relationship to parvovirus B19.7,8 Parainfluenzavirus 2, parainfluenzavirus 3, and adenovirus also have been isolated but may represent incidental viral infection.2 An inoculation dermatosis from an arthropod bite also has been suggested, but this claim has not been substantiated.1
The diagnosis often can be made on clinical features alone, and histopathologic evaluation is not required. Histologic features are nonspecific and include a superficial perivascular infiltrate of lymphocytes, often involving the dermal eccrine ducts without involvement of the secretory coils.4,6 Mild lichenoid changes as well as spongiosis with exocytosis of lymphocytes into the acrosyringium also may be present.4 The clinical differential diagnosis of APEC includes allergic contact dermatitis, a nonspecific drug or viral eruption, atypical pityriasis rosea, miliaria, scabies, tinea corporis, and Gianotti-Crosti syndrome. Asymmetric periflexural exanthem of childhood lacks the peripheral scale present in tinea corporis or pityriasis rosea, but when an annular or reticular configuration predominates, a potassium hydroxide preparation of skin scrapings can exclude the presence of a dermatophyte. Similar to APEC, Gianotti-Crosti syndrome affects young children, is preceded by symptoms of a viral prodrome, and spontaneously resolves over several weeks. This condition is distinguished from APEC by the presence of papulovesicles located symmetrically on the face, buttocks, and extensor surface of the extremities, which largely spare the trunk.
Asymmetric periflexural exanthem of childhood is a unique morbilliform eruption of infants and young children characterized by a stereotypical distribution and self-limited course. The cause of this syndrome remains unclear, but most authors suggest a viral etiology. Recognition of this entity and an ability to distinguish it from other common pediatric dermatoses is required to provide reassurance to parents and avoid unnecessary diagnostic procedures and treatments.
The Diagnosis: Asymmetric Periflexural Exanthem of Childhood (Unilateral Laterothoracic Exanthem)
Asymmetric periflexural exanthem of childhood (APEC), also known as unilateral laterothoracic exanthem, is a self-limited eruptive dermatosis that occurs most frequently in infants and young children. The term unilateral laterothoracic exanthem was first coined by Bodemer and de Prost1 in 1992 due to its characteristic distribution. The eruption occurs in children aged 4 months to 10 years, with most cases presenting between 2 and 3 years of age.2 Isolated cases also have been reported in adults.3 It affects girls more often than boys (2:1), and the majority of reported cases have occurred in white individuals. The disease is seen throughout Europe and North America, and seasonal variation has been noted with most cases occurring in late winter and early spring.4,5
|
Clinically, APEC is characterized by its asymmetric localization and unilateral onset. In the majority of patients, the eruption presents as discrete erythematous papules that coalesce to form morbilliform plaques that may have reticular or annular configuration (Figure).4 The exanthem begins unilaterally near a flexural area, most commonly the axilla (75% of cases), and spreads centrifugally to the adjacent trunk and proximal extremity. There is no right or left dominance.4,6 There is eventual involvement of the contralateral side in 70% of cases, but a unilateral predominance is maintained throughout the disease course.4 Rarely, the eruption may involve the face, genitals, and palmoplantar surfaces. As in our case, up to three-quarters of affected children report symptoms of an upper respiratory tract or gastrointestinal prodrome, including mild fever, diarrhea, and rhinitis.4 Accompanying regional lymphadenopathy has been reported in the majority of cases, and mild to moderate pruritus is not uncommon. The syndrome is self-limited, with spontaneous resolution commonly occurring 3 to 6 weeks after onset. Although no treatment is required, systemic antihistamines and topical steroids have been used to alleviate pruritus in symptomatic patients. Our patient was treated with triamcinolone cream 0.1% twice daily as well as oral diphenhydramine 25 mg every 6 hours as needed for associated pruritus. The eruption spontaneously resolved over the following 4 weeks.
Although the cause of APEC remains unknown, an infectious etiology has been presumed. The seasonal pattern, lack of efficacy of broad-spectrum antibiotics, frequently reported prodromal symptoms, and reports of familial cases suggest a viral etiology.1 Additionally, the predilection to affect infants and young children as well as lack of recurrence in the same patient suggests that immunity may develop. Although no etiologic agent has been consistently detected, several reports have suggested a possible relationship to parvovirus B19.7,8 Parainfluenzavirus 2, parainfluenzavirus 3, and adenovirus also have been isolated but may represent incidental viral infection.2 An inoculation dermatosis from an arthropod bite also has been suggested, but this claim has not been substantiated.1
The diagnosis often can be made on clinical features alone, and histopathologic evaluation is not required. Histologic features are nonspecific and include a superficial perivascular infiltrate of lymphocytes, often involving the dermal eccrine ducts without involvement of the secretory coils.4,6 Mild lichenoid changes as well as spongiosis with exocytosis of lymphocytes into the acrosyringium also may be present.4 The clinical differential diagnosis of APEC includes allergic contact dermatitis, a nonspecific drug or viral eruption, atypical pityriasis rosea, miliaria, scabies, tinea corporis, and Gianotti-Crosti syndrome. Asymmetric periflexural exanthem of childhood lacks the peripheral scale present in tinea corporis or pityriasis rosea, but when an annular or reticular configuration predominates, a potassium hydroxide preparation of skin scrapings can exclude the presence of a dermatophyte. Similar to APEC, Gianotti-Crosti syndrome affects young children, is preceded by symptoms of a viral prodrome, and spontaneously resolves over several weeks. This condition is distinguished from APEC by the presence of papulovesicles located symmetrically on the face, buttocks, and extensor surface of the extremities, which largely spare the trunk.
Asymmetric periflexural exanthem of childhood is a unique morbilliform eruption of infants and young children characterized by a stereotypical distribution and self-limited course. The cause of this syndrome remains unclear, but most authors suggest a viral etiology. Recognition of this entity and an ability to distinguish it from other common pediatric dermatoses is required to provide reassurance to parents and avoid unnecessary diagnostic procedures and treatments.
1. Bodemer C, de Prost Y. Unilateral laterothoracic exanthem in children: a new disease? J Am Acad Dermatol. 1992;27(5, pt 1):693-696.
2. Nahm WK, Paiva C, Golomb C, et al. Asymmetric periflexural exanthema of childhood: a case involving a 4-month-old infant. Pediatr Dermatol. 2002;19:461-462.
3. Chan PK, To KF, Zawar V, et al. Asymmetric periflexural exanthema in an adult. Clin Exp Dermatol. 2004;29:320-321.
4. McCuaig CC, Russo P, Powell J, et al. Unilateral laterothoracic exanthem. a clinicopathologic study of forty-eight patients. J Am Acad Dermatol. 1996;34:979-984.
5. Taieb A, Megraud F, Legrain V, et al. Asymmetric periflexural exanthem of childhood. J Am Acad Dermatol. 1993;29:391-393.
6. Coustou D, Léauté-Labrèze C, Bioulac-Sage P, et al. Asymmetric periflexural exanthem of childhood. a clinical, pathologic, and epidemiologic prospective study. Arch Dermatol. 1999;135:799-803.
7. Guimerá-Martín-Neda F, Fagundo E, Rodríguez F, et al. Asymmetric periflexural exanthem of childhood: report of two cases with parvovirus B19. J Eur Acad Dermatol Venereol. 2006;20:461-462.
8. Pauluzzi P, Festini G, Gelmetti C. Asymmetric periflexural exanthem of childhood in an adult patient with parvovirus B19. J Eur Acad Dermatol Venereol. 2001;15:372-374.
1. Bodemer C, de Prost Y. Unilateral laterothoracic exanthem in children: a new disease? J Am Acad Dermatol. 1992;27(5, pt 1):693-696.
2. Nahm WK, Paiva C, Golomb C, et al. Asymmetric periflexural exanthema of childhood: a case involving a 4-month-old infant. Pediatr Dermatol. 2002;19:461-462.
3. Chan PK, To KF, Zawar V, et al. Asymmetric periflexural exanthema in an adult. Clin Exp Dermatol. 2004;29:320-321.
4. McCuaig CC, Russo P, Powell J, et al. Unilateral laterothoracic exanthem. a clinicopathologic study of forty-eight patients. J Am Acad Dermatol. 1996;34:979-984.
5. Taieb A, Megraud F, Legrain V, et al. Asymmetric periflexural exanthem of childhood. J Am Acad Dermatol. 1993;29:391-393.
6. Coustou D, Léauté-Labrèze C, Bioulac-Sage P, et al. Asymmetric periflexural exanthem of childhood. a clinical, pathologic, and epidemiologic prospective study. Arch Dermatol. 1999;135:799-803.
7. Guimerá-Martín-Neda F, Fagundo E, Rodríguez F, et al. Asymmetric periflexural exanthem of childhood: report of two cases with parvovirus B19. J Eur Acad Dermatol Venereol. 2006;20:461-462.
8. Pauluzzi P, Festini G, Gelmetti C. Asymmetric periflexural exanthem of childhood in an adult patient with parvovirus B19. J Eur Acad Dermatol Venereol. 2001;15:372-374.
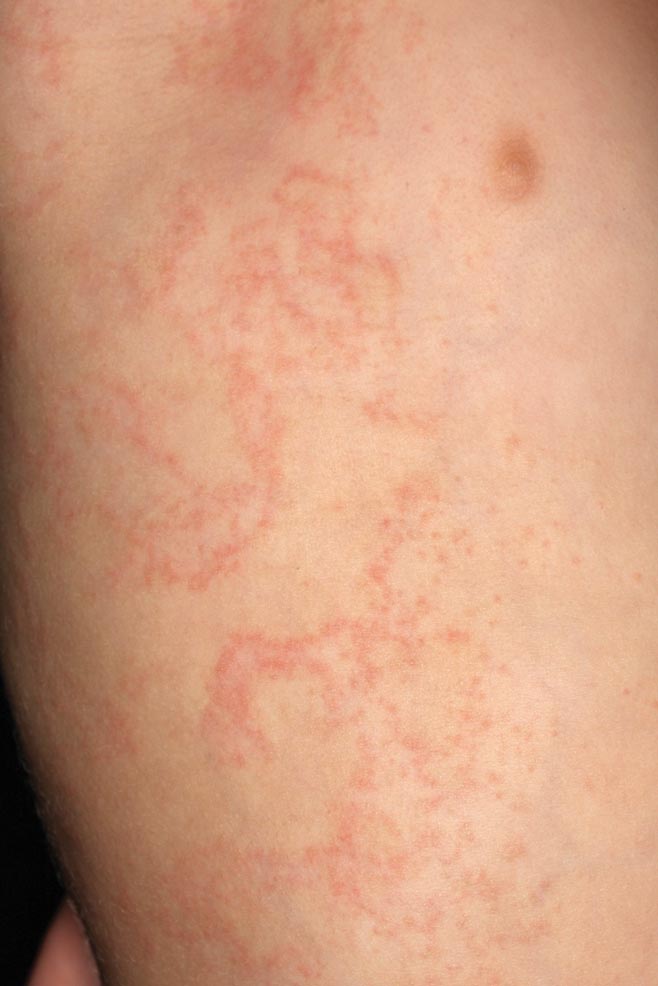
A 2-year-old girl presented with a mildly pruritic rash on the right flank and axilla of 3 weeks’ duration. Her pediatrician prescribed triamcinolone cream 0.1% daily, which was applied for the last week without much improvement. Her mother reported a history of upper respiratory tract infection approximately 1 to 2 weeks prior to onset of the rash.
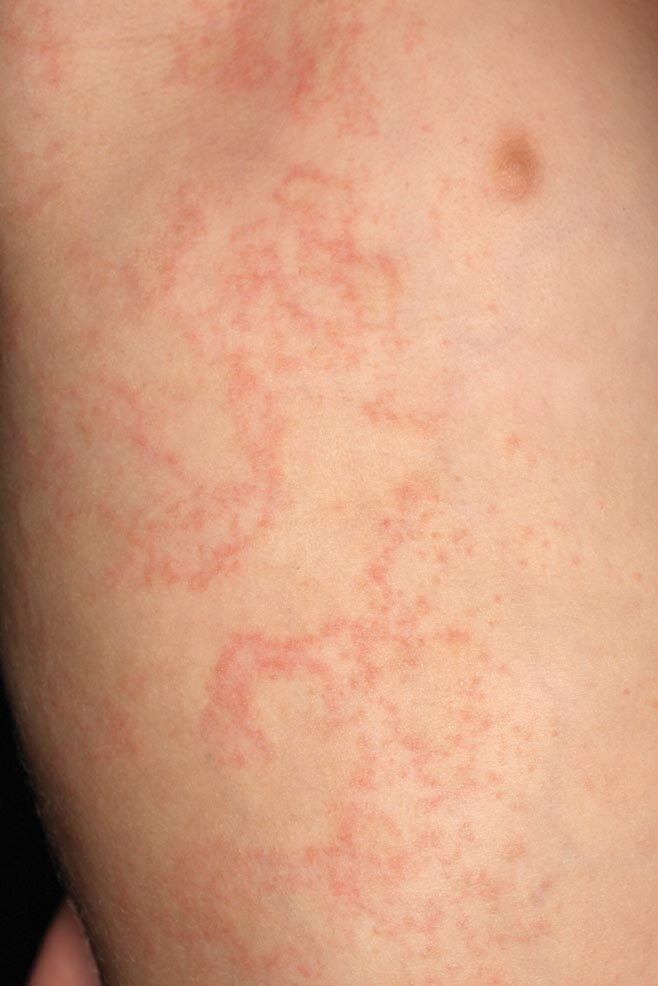
What Is Your Diagnosis? Eosinophilic Fasciitis
|
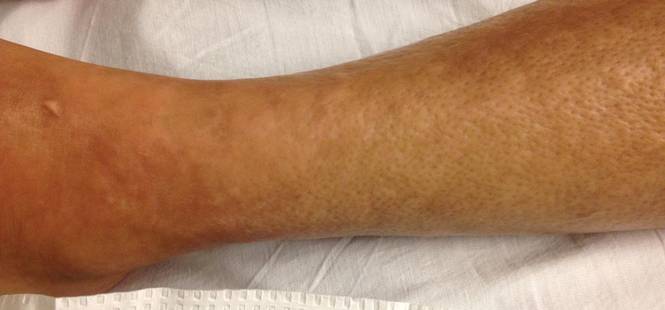
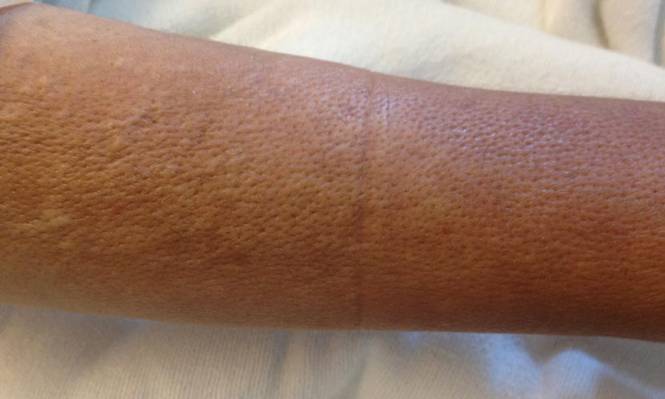
A 43-year-old woman presented with pain and paresthesia of the bilateral legs of 3 months’ duration with skin tightness and discoloration, which she attributed to a car accident that occurred 7 months prior. She also reported abdominal pain, shortness of breath, fever, double vision, dysphagia, voice changes, temperature sensitivity, and hair loss. The patient underwent outpatient steroid injections with limited symptomatic relief. She denied any antecedent exposure to vinyl chloride, rapeseed oil, or L-tryptophan. Physical examination revealed thickened skin on the bilateral legs (top), reddish discoloration of the feet, decreased sensation to light touch, and edema of the ankles and wrists, as well as a peau d’orange appearance of the skin on the arms (bottom), legs, and abdomen.
The Diagnosis: Eosinophilic Fasciitis
Eosinophilic fasciitis is a rare autoimmune disease of uncertain etiology first described by Shulman1 in 1974. It is similar in presentation and is perhaps related to scleroderma. Classic differentiating features include a peculiar peau d’orange appearance, peripheral eosinophilia, and lack of Raynaud phenomenon, thus it is regarded as a unique disease.2 Despite the name of the disease, eosinophilia has been known to be absent in later stages of eosinophilic fasciitis.1

On physical examination, “prayer and groove signs” can sometimes be evident.3 Although it was not initially observed in our case, a groove sign was noted on the left forearm on a second inspection (Figure 1). In contrast with systemic sclerosis, visceral involvement rarely is seen with eosinophilic fasciitis. There are, however, 3 major exceptions to this rule. First, there can be widespread nerve deficits, esophageal dysmotility, and nonspecific electromyography findings (ie, denervation, reinnervation, fasciculations).4 There also can be a concomitant hematologic disorder or Hashimoto thyroiditis.5 Because eosinophilic fasciitis has been associated with monoclonal gammopathy, which our patient also demonstrated, it is important to conduct a workup with serum or urine protein electrophoresis. If the test is negative, it should be followed up with an immunofixation assay or serum light chain assays.
Some proposed risk factors for eosinophilic fasciitis include trauma, extensive exercise, and Borrelia burgdorferi infection, but many cases have none of these associations.8 Although not firmly proven in the literature, there have been reports of eosinophilic fasciitis after isolated trauma.5 A causal link could not be established between our patient’s car accident and eosinophilic fasciitis, but the coincidence was notable.
The treatment of eosinophilic fasciitis is similar to scleroderma. Corticosteroids are effective in most cases and recovery often occurs with monotherapy.5 Case series have demonstrated efficacy in adding methotrexate, azathioprine, colchicine, and hydroxychloroquine in refractory patients.2,9 Our patient demonstrated a good response with a combination of prednisone and methotrexate. Relapses have been known to occur.2
A punch biopsy obtained from the right arm showed thickened acellular collagen bundles throughout the dermis and extending into the underlying subcutis. There also was obliteration of adnexal structures, loss of perieccrine fat, and sparse perivascular and interstitial lymphoplasmacytic infiltrate (Figure 2), consistent with a sclerosing disorder such as scleroderma or eosinophilic fasciitis.

A complete blood cell count revealed eosinophil levels of 12.5% (reference range, 2.7%). Rheumatologic workup was negative for antinuclear antibody, double-stranded DNA, thyroid-stimulating hormone, anticentromere antibodies, and Scl-70 autoantibodies. Computed tomography of the chest and pelvis revealed a thickened patulous esophagus. Endoscopy showed dysmotility of the lower esophagus. At this point the differential diagnosis included scleredema versus eosinophilic fasciitis, and the patient was started on oral prednisone 60 mg daily. She showed rapid improvement in sclerosis, joint mobility, and ability to swallow. Magnetic resonance imaging was then performed and revealed thickening and contrast enhancement of the forearm fascia, particularly along the distal aspect, confirming a diagnosis of eosinophilic fasciitis. Further workup including immunofixation assay and serum light chain assays were performed, revealing IgG λ hypergammaglobulinemia. She was then additionally treated with oral methotrexate 15 mg weekly. Due to the rapid improvement of symptoms on oral prednisone over 2 weeks, the peripheral eosinophilia, the magnetic resonance imaging findings, and the results of skin biopsy, a diagnosis of eosinophilic fasciitis was heavily favored over scleroderma and scleredema.
1. Shulman L. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia in a new syndrome. J. Rheumatol. 1974;1(suppl):46.
2. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
3. Servy A, Clerici T, Malines C, et al. Eosinophilic fasciitis: a rare skin sclerosis. Pathol Res Int. 2010;2011:716935.
4. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992;239:91-92.
5. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Dermatology. 2006;213:93-101.
6. Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin North Am. 1989;73:1157-1166.
7. Naschitz JE, Yeshurun D, Miselevich I, et al. Colitis and pericarditis in a patient with eosinophilic fasciitis—a contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989;16:688-692.
8. Haustein UF. Scleroderma and pseudoscleroderma: uncommon presentations. Clin Dermatol. 2005;23:480-490.
9. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology. 2012;51:557-561.
|
A 43-year-old woman presented with pain and paresthesia of the bilateral legs of 3 months’ duration with skin tightness and discoloration, which she attributed to a car accident that occurred 7 months prior. She also reported abdominal pain, shortness of breath, fever, double vision, dysphagia, voice changes, temperature sensitivity, and hair loss. The patient underwent outpatient steroid injections with limited symptomatic relief. She denied any antecedent exposure to vinyl chloride, rapeseed oil, or L-tryptophan. Physical examination revealed thickened skin on the bilateral legs (top), reddish discoloration of the feet, decreased sensation to light touch, and edema of the ankles and wrists, as well as a peau d’orange appearance of the skin on the arms (bottom), legs, and abdomen.
The Diagnosis: Eosinophilic Fasciitis
Eosinophilic fasciitis is a rare autoimmune disease of uncertain etiology first described by Shulman1 in 1974. It is similar in presentation and is perhaps related to scleroderma. Classic differentiating features include a peculiar peau d’orange appearance, peripheral eosinophilia, and lack of Raynaud phenomenon, thus it is regarded as a unique disease.2 Despite the name of the disease, eosinophilia has been known to be absent in later stages of eosinophilic fasciitis.1
On physical examination, “prayer and groove signs” can sometimes be evident.3 Although it was not initially observed in our case, a groove sign was noted on the left forearm on a second inspection (Figure 1). In contrast with systemic sclerosis, visceral involvement rarely is seen with eosinophilic fasciitis. There are, however, 3 major exceptions to this rule. First, there can be widespread nerve deficits, esophageal dysmotility, and nonspecific electromyography findings (ie, denervation, reinnervation, fasciculations).4 There also can be a concomitant hematologic disorder or Hashimoto thyroiditis.5 Because eosinophilic fasciitis has been associated with monoclonal gammopathy, which our patient also demonstrated, it is important to conduct a workup with serum or urine protein electrophoresis. If the test is negative, it should be followed up with an immunofixation assay or serum light chain assays.
Some proposed risk factors for eosinophilic fasciitis include trauma, extensive exercise, and Borrelia burgdorferi infection, but many cases have none of these associations.8 Although not firmly proven in the literature, there have been reports of eosinophilic fasciitis after isolated trauma.5 A causal link could not be established between our patient’s car accident and eosinophilic fasciitis, but the coincidence was notable.
The treatment of eosinophilic fasciitis is similar to scleroderma. Corticosteroids are effective in most cases and recovery often occurs with monotherapy.5 Case series have demonstrated efficacy in adding methotrexate, azathioprine, colchicine, and hydroxychloroquine in refractory patients.2,9 Our patient demonstrated a good response with a combination of prednisone and methotrexate. Relapses have been known to occur.2
A punch biopsy obtained from the right arm showed thickened acellular collagen bundles throughout the dermis and extending into the underlying subcutis. There also was obliteration of adnexal structures, loss of perieccrine fat, and sparse perivascular and interstitial lymphoplasmacytic infiltrate (Figure 2), consistent with a sclerosing disorder such as scleroderma or eosinophilic fasciitis.
A complete blood cell count revealed eosinophil levels of 12.5% (reference range, 2.7%). Rheumatologic workup was negative for antinuclear antibody, double-stranded DNA, thyroid-stimulating hormone, anticentromere antibodies, and Scl-70 autoantibodies. Computed tomography of the chest and pelvis revealed a thickened patulous esophagus. Endoscopy showed dysmotility of the lower esophagus. At this point the differential diagnosis included scleredema versus eosinophilic fasciitis, and the patient was started on oral prednisone 60 mg daily. She showed rapid improvement in sclerosis, joint mobility, and ability to swallow. Magnetic resonance imaging was then performed and revealed thickening and contrast enhancement of the forearm fascia, particularly along the distal aspect, confirming a diagnosis of eosinophilic fasciitis. Further workup including immunofixation assay and serum light chain assays were performed, revealing IgG λ hypergammaglobulinemia. She was then additionally treated with oral methotrexate 15 mg weekly. Due to the rapid improvement of symptoms on oral prednisone over 2 weeks, the peripheral eosinophilia, the magnetic resonance imaging findings, and the results of skin biopsy, a diagnosis of eosinophilic fasciitis was heavily favored over scleroderma and scleredema.
|
A 43-year-old woman presented with pain and paresthesia of the bilateral legs of 3 months’ duration with skin tightness and discoloration, which she attributed to a car accident that occurred 7 months prior. She also reported abdominal pain, shortness of breath, fever, double vision, dysphagia, voice changes, temperature sensitivity, and hair loss. The patient underwent outpatient steroid injections with limited symptomatic relief. She denied any antecedent exposure to vinyl chloride, rapeseed oil, or L-tryptophan. Physical examination revealed thickened skin on the bilateral legs (top), reddish discoloration of the feet, decreased sensation to light touch, and edema of the ankles and wrists, as well as a peau d’orange appearance of the skin on the arms (bottom), legs, and abdomen.
The Diagnosis: Eosinophilic Fasciitis
Eosinophilic fasciitis is a rare autoimmune disease of uncertain etiology first described by Shulman1 in 1974. It is similar in presentation and is perhaps related to scleroderma. Classic differentiating features include a peculiar peau d’orange appearance, peripheral eosinophilia, and lack of Raynaud phenomenon, thus it is regarded as a unique disease.2 Despite the name of the disease, eosinophilia has been known to be absent in later stages of eosinophilic fasciitis.1
On physical examination, “prayer and groove signs” can sometimes be evident.3 Although it was not initially observed in our case, a groove sign was noted on the left forearm on a second inspection (Figure 1). In contrast with systemic sclerosis, visceral involvement rarely is seen with eosinophilic fasciitis. There are, however, 3 major exceptions to this rule. First, there can be widespread nerve deficits, esophageal dysmotility, and nonspecific electromyography findings (ie, denervation, reinnervation, fasciculations).4 There also can be a concomitant hematologic disorder or Hashimoto thyroiditis.5 Because eosinophilic fasciitis has been associated with monoclonal gammopathy, which our patient also demonstrated, it is important to conduct a workup with serum or urine protein electrophoresis. If the test is negative, it should be followed up with an immunofixation assay or serum light chain assays.
Some proposed risk factors for eosinophilic fasciitis include trauma, extensive exercise, and Borrelia burgdorferi infection, but many cases have none of these associations.8 Although not firmly proven in the literature, there have been reports of eosinophilic fasciitis after isolated trauma.5 A causal link could not be established between our patient’s car accident and eosinophilic fasciitis, but the coincidence was notable.
The treatment of eosinophilic fasciitis is similar to scleroderma. Corticosteroids are effective in most cases and recovery often occurs with monotherapy.5 Case series have demonstrated efficacy in adding methotrexate, azathioprine, colchicine, and hydroxychloroquine in refractory patients.2,9 Our patient demonstrated a good response with a combination of prednisone and methotrexate. Relapses have been known to occur.2
A punch biopsy obtained from the right arm showed thickened acellular collagen bundles throughout the dermis and extending into the underlying subcutis. There also was obliteration of adnexal structures, loss of perieccrine fat, and sparse perivascular and interstitial lymphoplasmacytic infiltrate (Figure 2), consistent with a sclerosing disorder such as scleroderma or eosinophilic fasciitis.
A complete blood cell count revealed eosinophil levels of 12.5% (reference range, 2.7%). Rheumatologic workup was negative for antinuclear antibody, double-stranded DNA, thyroid-stimulating hormone, anticentromere antibodies, and Scl-70 autoantibodies. Computed tomography of the chest and pelvis revealed a thickened patulous esophagus. Endoscopy showed dysmotility of the lower esophagus. At this point the differential diagnosis included scleredema versus eosinophilic fasciitis, and the patient was started on oral prednisone 60 mg daily. She showed rapid improvement in sclerosis, joint mobility, and ability to swallow. Magnetic resonance imaging was then performed and revealed thickening and contrast enhancement of the forearm fascia, particularly along the distal aspect, confirming a diagnosis of eosinophilic fasciitis. Further workup including immunofixation assay and serum light chain assays were performed, revealing IgG λ hypergammaglobulinemia. She was then additionally treated with oral methotrexate 15 mg weekly. Due to the rapid improvement of symptoms on oral prednisone over 2 weeks, the peripheral eosinophilia, the magnetic resonance imaging findings, and the results of skin biopsy, a diagnosis of eosinophilic fasciitis was heavily favored over scleroderma and scleredema.
1. Shulman L. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia in a new syndrome. J. Rheumatol. 1974;1(suppl):46.
2. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
3. Servy A, Clerici T, Malines C, et al. Eosinophilic fasciitis: a rare skin sclerosis. Pathol Res Int. 2010;2011:716935.
4. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992;239:91-92.
5. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Dermatology. 2006;213:93-101.
6. Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin North Am. 1989;73:1157-1166.
7. Naschitz JE, Yeshurun D, Miselevich I, et al. Colitis and pericarditis in a patient with eosinophilic fasciitis—a contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989;16:688-692.
8. Haustein UF. Scleroderma and pseudoscleroderma: uncommon presentations. Clin Dermatol. 2005;23:480-490.
9. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology. 2012;51:557-561.
1. Shulman L. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia in a new syndrome. J. Rheumatol. 1974;1(suppl):46.
2. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
3. Servy A, Clerici T, Malines C, et al. Eosinophilic fasciitis: a rare skin sclerosis. Pathol Res Int. 2010;2011:716935.
4. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992;239:91-92.
5. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Dermatology. 2006;213:93-101.
6. Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin North Am. 1989;73:1157-1166.
7. Naschitz JE, Yeshurun D, Miselevich I, et al. Colitis and pericarditis in a patient with eosinophilic fasciitis—a contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989;16:688-692.
8. Haustein UF. Scleroderma and pseudoscleroderma: uncommon presentations. Clin Dermatol. 2005;23:480-490.
9. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology. 2012;51:557-561.