User login
Punctate Depigmented Macules
The Diagnosis: Blaschkoid Punctate Vitiligo
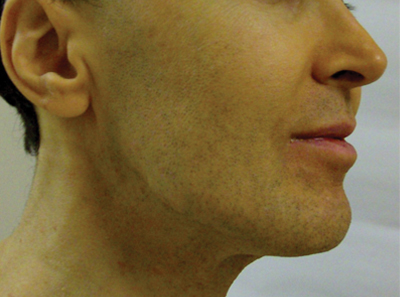
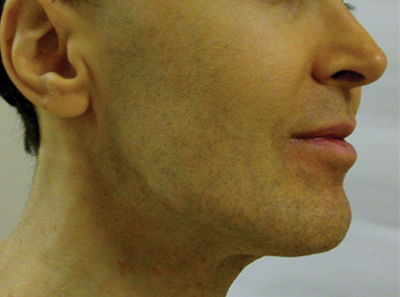
Based on the patient’s clinical appearance as well as the histologic findings, the diagnosis of vitiligo was made. Although vitiligo is certainly not uncommon and punctate vitiligo is a known clinical presentation,1 punctate vitiliginous depigmentation conforming to lines of Blaschko is unique. Follicular repigmentation in a patch of vitiligo potentially could lead to this “spotty” appearance, but our patient maintained that the band was never confluently depigmented and that small macules arose within normally pigmented skin. The patient’s adult age at onset makes this case even more unusual.
Follicular repigmentation in vitiligo is fairly well understood, as the perifollicular pigment is formed by upward migration of activated melanoblasts in the outer root sheath.2 Follicular depigmentation as well as selective or initial loss of melanocytes around hair follicles in early vitiligo has not been described. It is unclear if the seemingly folliculocentric nature of the patient’s vitiliginous macules was a false observation, coincidental, or actually related to selective melanocyte loss around follicles.
Blaschkoid distribution has been described in numerous skin disorders and is known to be based on genetic mosaicism.3 Most of these disorders are X-linked and/or congenital. However, many acquired skin conditions have been described exhibiting blaschkoid distribution, such as vitiligo, psoriasis, lichen planus, atopic dermatitis, and mycosis fungoides.4,5
Confettilike depigmentation has been described as an unusual clinical variant of vitiligo.1 It also has been reported after psoralen plus UVA therapy in patients with more classic vitiligo,6 numerous domestic chemicals,7 and in association with mycosis fungoides.8 In these cases, punctate lesions were disseminated, symmetric on extremities, or limited to areas exposed to chemicals.
1. Ortonne J-P. Vitiligo and other disorders of hypopigmentation. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. St. Louis, MO: Mosby; 2003:913-938.
2. Cui J, Shen LY, Wang GC. Role of hair follicles in the repigmentation of vitiligo. J Invest Dermatol. 1991;97:410-416.
3. Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl. 2006;95:16-23.
4. Taieb A. Linear atopic dermatitis (“naevus atopicus”): a pathogenetic clue? Br J Dermatol. 1994;131:134-135.
5. Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31:157-190.
6. Falabella R, Escobar CE, Carrascal E, et al. Leukoderma punctata. J Am Acad Dermatol. 1988;18:485-494.
7. Ghosh S, Mukhopadhyay S. Chemical leucoderma: a clinico-aetiological study of 864 cases in the perspective of a developing country. Br J Dermatol. 2009;160:40-47.
8. Loquai C, Metza D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
The Diagnosis: Blaschkoid Punctate Vitiligo
Based on the patient’s clinical appearance as well as the histologic findings, the diagnosis of vitiligo was made. Although vitiligo is certainly not uncommon and punctate vitiligo is a known clinical presentation,1 punctate vitiliginous depigmentation conforming to lines of Blaschko is unique. Follicular repigmentation in a patch of vitiligo potentially could lead to this “spotty” appearance, but our patient maintained that the band was never confluently depigmented and that small macules arose within normally pigmented skin. The patient’s adult age at onset makes this case even more unusual.
Follicular repigmentation in vitiligo is fairly well understood, as the perifollicular pigment is formed by upward migration of activated melanoblasts in the outer root sheath.2 Follicular depigmentation as well as selective or initial loss of melanocytes around hair follicles in early vitiligo has not been described. It is unclear if the seemingly folliculocentric nature of the patient’s vitiliginous macules was a false observation, coincidental, or actually related to selective melanocyte loss around follicles.
Blaschkoid distribution has been described in numerous skin disorders and is known to be based on genetic mosaicism.3 Most of these disorders are X-linked and/or congenital. However, many acquired skin conditions have been described exhibiting blaschkoid distribution, such as vitiligo, psoriasis, lichen planus, atopic dermatitis, and mycosis fungoides.4,5
Confettilike depigmentation has been described as an unusual clinical variant of vitiligo.1 It also has been reported after psoralen plus UVA therapy in patients with more classic vitiligo,6 numerous domestic chemicals,7 and in association with mycosis fungoides.8 In these cases, punctate lesions were disseminated, symmetric on extremities, or limited to areas exposed to chemicals.
The Diagnosis: Blaschkoid Punctate Vitiligo
Based on the patient’s clinical appearance as well as the histologic findings, the diagnosis of vitiligo was made. Although vitiligo is certainly not uncommon and punctate vitiligo is a known clinical presentation,1 punctate vitiliginous depigmentation conforming to lines of Blaschko is unique. Follicular repigmentation in a patch of vitiligo potentially could lead to this “spotty” appearance, but our patient maintained that the band was never confluently depigmented and that small macules arose within normally pigmented skin. The patient’s adult age at onset makes this case even more unusual.
Follicular repigmentation in vitiligo is fairly well understood, as the perifollicular pigment is formed by upward migration of activated melanoblasts in the outer root sheath.2 Follicular depigmentation as well as selective or initial loss of melanocytes around hair follicles in early vitiligo has not been described. It is unclear if the seemingly folliculocentric nature of the patient’s vitiliginous macules was a false observation, coincidental, or actually related to selective melanocyte loss around follicles.
Blaschkoid distribution has been described in numerous skin disorders and is known to be based on genetic mosaicism.3 Most of these disorders are X-linked and/or congenital. However, many acquired skin conditions have been described exhibiting blaschkoid distribution, such as vitiligo, psoriasis, lichen planus, atopic dermatitis, and mycosis fungoides.4,5
Confettilike depigmentation has been described as an unusual clinical variant of vitiligo.1 It also has been reported after psoralen plus UVA therapy in patients with more classic vitiligo,6 numerous domestic chemicals,7 and in association with mycosis fungoides.8 In these cases, punctate lesions were disseminated, symmetric on extremities, or limited to areas exposed to chemicals.
1. Ortonne J-P. Vitiligo and other disorders of hypopigmentation. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. St. Louis, MO: Mosby; 2003:913-938.
2. Cui J, Shen LY, Wang GC. Role of hair follicles in the repigmentation of vitiligo. J Invest Dermatol. 1991;97:410-416.
3. Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl. 2006;95:16-23.
4. Taieb A. Linear atopic dermatitis (“naevus atopicus”): a pathogenetic clue? Br J Dermatol. 1994;131:134-135.
5. Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31:157-190.
6. Falabella R, Escobar CE, Carrascal E, et al. Leukoderma punctata. J Am Acad Dermatol. 1988;18:485-494.
7. Ghosh S, Mukhopadhyay S. Chemical leucoderma: a clinico-aetiological study of 864 cases in the perspective of a developing country. Br J Dermatol. 2009;160:40-47.
8. Loquai C, Metza D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
1. Ortonne J-P. Vitiligo and other disorders of hypopigmentation. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. St. Louis, MO: Mosby; 2003:913-938.
2. Cui J, Shen LY, Wang GC. Role of hair follicles in the repigmentation of vitiligo. J Invest Dermatol. 1991;97:410-416.
3. Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl. 2006;95:16-23.
4. Taieb A. Linear atopic dermatitis (“naevus atopicus”): a pathogenetic clue? Br J Dermatol. 1994;131:134-135.
5. Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31:157-190.
6. Falabella R, Escobar CE, Carrascal E, et al. Leukoderma punctata. J Am Acad Dermatol. 1988;18:485-494.
7. Ghosh S, Mukhopadhyay S. Chemical leucoderma: a clinico-aetiological study of 864 cases in the perspective of a developing country. Br J Dermatol. 2009;160:40-47.
8. Loquai C, Metza D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
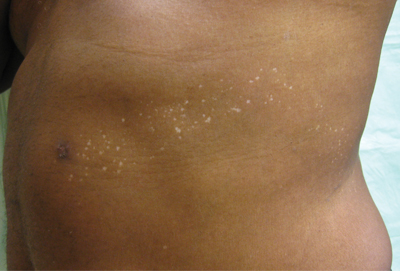
An otherwise healthy 54-year-old black man presented with a 10-year history of spotty pigmentary loss in a band on the left side of the abdomen, flank, and back. He denied a history of rash or inflammation in the area and had not experienced confluent depigmentation. He reported that initially he had only a few “white dots,” and over the next 5 to 7 years, he developed more of them confined within the same area. On presentation, he stated new areas of depigmentation had not developed in several years. The band was completely asymptomatic and had not been treated with any prescription or over-the-counter medications. On examination he had multiple 2- to 3-mm confettilike depigmented macules that seemed to be centered around follicles in a band with blaschkoid distribution extending across the left side of the abdomen, flank, and back. The band did not cross the midline and similar lesions were not present elsewhere. A punch biopsy of one of the depigmented macules revealed a markedly diminished number of melanocytes along the junction as well as a decrease in melanin, which was confirmed by Melan-A and Fontana stains, respectively.

What Is Your Diagnosis? Secondary Syphilis
Generalized Yellow Discoloration of the Skin
The Diagnosis: Carotenemia
Laboratory parameters including thyroid function testing as well as total protein and bilirubin levels were within reference range. Testing revealed multiple food allergies to almonds, oranges, cashews, garlic, peanuts, and cantaloupe. The patient was treated with a dietary expansion based on his allergy testing.
ß-Carotene converts to vitamin A in the intestine and acts as a lipochrome. Lack of conversion can be noted as an inborn error of metabolism.1 Many green, yellow, and orange fruits and vegetables contain ß-carotene, including carrots, sweet potatoes, squash, green beans, papayas, and pumpkins.1-3 ß-Carotene also is used as a vitamin supplement4 or therapeutic agent in photosensitive disorders such as genetic porphyrias.5
ß-Carotene can accumulate in the stratum corneum and impart a yellow color to the skin when the circulating levels are high; this coloration is termed carotenemia.1,4 Carotenemia is common in infants and young children who have diets rich in green and orange vegetable purees.6 Carotenemia limited to thick areas of the skin, such as the palms and soles, can be seen in adults who eat large amounts of carrots; generalized carotenemia is rare.1,4
Carotenemia is a benign condition of excess cutaneous buildup of ß-carotene through excessive intake of carotene-rich foods1-4 or nutritional supplements7 or through association with anorexia, liver disease, renal disease, hypothyroidism, or diabetes mellitus.1,4,8,9 Carotene deposits usually are most notable in areas with thick stratum corneum, such as the nasolabial folds, palms, and soles, as opposed to areas such as the conjunctivae and mucosa.1,4
Carotenemia may mimic jaundice and should be differentiated through scleral examination for icterus and bilirubin levels. Carotene levels can be tested but generally are unnecessary. Carotenemia can be seen in liver or renal disease and can exacerbate the yellow coloration seen in jaundiced individuals.1,4,9
Because it is a benign condition, the pathology usually is limited to skin discoloration, as seen in our patient. Although this condition can be reversed with a modified diet, our patient had multiple food allergies that further restricted his vegetarian diet, thereby limiting the modifications that he was willing to make to his diet.
1. Schwartz RA. Carotenemia. Emedicine. http://emedicine.medscape.com/article/1104368-overview. Updated April 8, 2014. Accessed April 30, 2014.
2. Sale TA, Stratman E. Carotenemia associated with green bean ingestion. Pediatr Dermatol. 2004;21:657-659.
3. Costanza DJ. Carotenemia associated with papaya ingestion. Calif Med. 1968;109:319-320.
4. Lascari AD. Carotenemia. a review. Clin Pediatr (Phila). 1981;20:25-29.
5. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-937.
6. Karthik SV, Campbell-Davidson D, Isherwood D. Carotenemia in infancy and its association with prevalent feeding practices. Pediatr Dermatol. 2006;23:571-573.
7. Takita Y, Ichimiya M, Hamamoto Y, et al. A case of carotenemia associated with ingestion of nutrient supplements. J Dermatol. 2006;2:132-134.
8. Thibault L, Roberge AG. The nutritional status of subjects with nervosa. Int J Vitam Nutr Res. 1987;57:447-452.
9. Matthews-Roth M, Gulbrandsen CL. Transport of beta-carotene in serum of individuals with carotenemia. Clin Chem. 1974;20:1578-1579.
The Diagnosis: Carotenemia
Laboratory parameters including thyroid function testing as well as total protein and bilirubin levels were within reference range. Testing revealed multiple food allergies to almonds, oranges, cashews, garlic, peanuts, and cantaloupe. The patient was treated with a dietary expansion based on his allergy testing.
ß-Carotene converts to vitamin A in the intestine and acts as a lipochrome. Lack of conversion can be noted as an inborn error of metabolism.1 Many green, yellow, and orange fruits and vegetables contain ß-carotene, including carrots, sweet potatoes, squash, green beans, papayas, and pumpkins.1-3 ß-Carotene also is used as a vitamin supplement4 or therapeutic agent in photosensitive disorders such as genetic porphyrias.5
ß-Carotene can accumulate in the stratum corneum and impart a yellow color to the skin when the circulating levels are high; this coloration is termed carotenemia.1,4 Carotenemia is common in infants and young children who have diets rich in green and orange vegetable purees.6 Carotenemia limited to thick areas of the skin, such as the palms and soles, can be seen in adults who eat large amounts of carrots; generalized carotenemia is rare.1,4
Carotenemia is a benign condition of excess cutaneous buildup of ß-carotene through excessive intake of carotene-rich foods1-4 or nutritional supplements7 or through association with anorexia, liver disease, renal disease, hypothyroidism, or diabetes mellitus.1,4,8,9 Carotene deposits usually are most notable in areas with thick stratum corneum, such as the nasolabial folds, palms, and soles, as opposed to areas such as the conjunctivae and mucosa.1,4
Carotenemia may mimic jaundice and should be differentiated through scleral examination for icterus and bilirubin levels. Carotene levels can be tested but generally are unnecessary. Carotenemia can be seen in liver or renal disease and can exacerbate the yellow coloration seen in jaundiced individuals.1,4,9
Because it is a benign condition, the pathology usually is limited to skin discoloration, as seen in our patient. Although this condition can be reversed with a modified diet, our patient had multiple food allergies that further restricted his vegetarian diet, thereby limiting the modifications that he was willing to make to his diet.
The Diagnosis: Carotenemia
Laboratory parameters including thyroid function testing as well as total protein and bilirubin levels were within reference range. Testing revealed multiple food allergies to almonds, oranges, cashews, garlic, peanuts, and cantaloupe. The patient was treated with a dietary expansion based on his allergy testing.
ß-Carotene converts to vitamin A in the intestine and acts as a lipochrome. Lack of conversion can be noted as an inborn error of metabolism.1 Many green, yellow, and orange fruits and vegetables contain ß-carotene, including carrots, sweet potatoes, squash, green beans, papayas, and pumpkins.1-3 ß-Carotene also is used as a vitamin supplement4 or therapeutic agent in photosensitive disorders such as genetic porphyrias.5
ß-Carotene can accumulate in the stratum corneum and impart a yellow color to the skin when the circulating levels are high; this coloration is termed carotenemia.1,4 Carotenemia is common in infants and young children who have diets rich in green and orange vegetable purees.6 Carotenemia limited to thick areas of the skin, such as the palms and soles, can be seen in adults who eat large amounts of carrots; generalized carotenemia is rare.1,4
Carotenemia is a benign condition of excess cutaneous buildup of ß-carotene through excessive intake of carotene-rich foods1-4 or nutritional supplements7 or through association with anorexia, liver disease, renal disease, hypothyroidism, or diabetes mellitus.1,4,8,9 Carotene deposits usually are most notable in areas with thick stratum corneum, such as the nasolabial folds, palms, and soles, as opposed to areas such as the conjunctivae and mucosa.1,4
Carotenemia may mimic jaundice and should be differentiated through scleral examination for icterus and bilirubin levels. Carotene levels can be tested but generally are unnecessary. Carotenemia can be seen in liver or renal disease and can exacerbate the yellow coloration seen in jaundiced individuals.1,4,9
Because it is a benign condition, the pathology usually is limited to skin discoloration, as seen in our patient. Although this condition can be reversed with a modified diet, our patient had multiple food allergies that further restricted his vegetarian diet, thereby limiting the modifications that he was willing to make to his diet.
1. Schwartz RA. Carotenemia. Emedicine. http://emedicine.medscape.com/article/1104368-overview. Updated April 8, 2014. Accessed April 30, 2014.
2. Sale TA, Stratman E. Carotenemia associated with green bean ingestion. Pediatr Dermatol. 2004;21:657-659.
3. Costanza DJ. Carotenemia associated with papaya ingestion. Calif Med. 1968;109:319-320.
4. Lascari AD. Carotenemia. a review. Clin Pediatr (Phila). 1981;20:25-29.
5. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-937.
6. Karthik SV, Campbell-Davidson D, Isherwood D. Carotenemia in infancy and its association with prevalent feeding practices. Pediatr Dermatol. 2006;23:571-573.
7. Takita Y, Ichimiya M, Hamamoto Y, et al. A case of carotenemia associated with ingestion of nutrient supplements. J Dermatol. 2006;2:132-134.
8. Thibault L, Roberge AG. The nutritional status of subjects with nervosa. Int J Vitam Nutr Res. 1987;57:447-452.
9. Matthews-Roth M, Gulbrandsen CL. Transport of beta-carotene in serum of individuals with carotenemia. Clin Chem. 1974;20:1578-1579.
1. Schwartz RA. Carotenemia. Emedicine. http://emedicine.medscape.com/article/1104368-overview. Updated April 8, 2014. Accessed April 30, 2014.
2. Sale TA, Stratman E. Carotenemia associated with green bean ingestion. Pediatr Dermatol. 2004;21:657-659.
3. Costanza DJ. Carotenemia associated with papaya ingestion. Calif Med. 1968;109:319-320.
4. Lascari AD. Carotenemia. a review. Clin Pediatr (Phila). 1981;20:25-29.
5. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-937.
6. Karthik SV, Campbell-Davidson D, Isherwood D. Carotenemia in infancy and its association with prevalent feeding practices. Pediatr Dermatol. 2006;23:571-573.
7. Takita Y, Ichimiya M, Hamamoto Y, et al. A case of carotenemia associated with ingestion of nutrient supplements. J Dermatol. 2006;2:132-134.
8. Thibault L, Roberge AG. The nutritional status of subjects with nervosa. Int J Vitam Nutr Res. 1987;57:447-452.
9. Matthews-Roth M, Gulbrandsen CL. Transport of beta-carotene in serum of individuals with carotenemia. Clin Chem. 1974;20:1578-1579.
A 50-year-old man presented with yellow, pruritic, xerotic skin and lethargy. The patient also reported nasal congestion and sneezing, especially when eating peanuts. He was fearful of allergic reactions and restricted his diet to “safe foods” such as squash, green beans, and sweet potatoes. On examination the patient had marked generalized yellow discoloration of the skin with pale mucous membranes, nonicteric sclerae, infraocular violaceous and hyperpigmented skin (allergic shiners), and Dennie-Morgan folds.
Pruritus and Hyperpigmented Streaks
The Diagnosis: Flagellate Hyperpigmentation
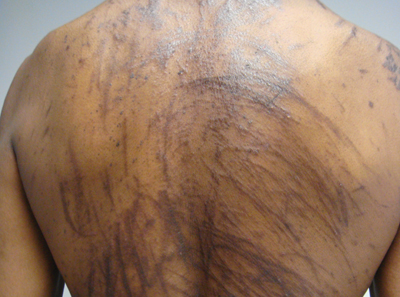
Physical examination of our patient revealed multiple linear, flagellate, and hyperpigmented plaques on the trunk. Similar lesions were seen on the upper and lower extremities. Given the patient's history of non-Hodgkin lymphoma and concurrent chemotherapy, the use of systemic steroids was avoided. Notable improvement in the symptoms was achieved with topical steroids (triamcinolone acetonide ointment) under occlusion and systemic gabapentin. The patient's chemotherapy is ongoing. This characteristic skin eruption often is seen after treatment with systemic bleomycin sulfate.
Flagellate hyperpigmentation following systemic bleomycin sulfate use as a chemotherapeutic agent is a well-known cutaneous reaction that occurs in approximately 10% of exposed individuals.1 Bleomycin sulfate, an antibiotic derived from Streptomyces verticillus, is a commonly used antitumor agent for both hematologic and solid-organ malignancies.2 Many patients develop generalized pruritus within several days after initiating bleomycin sulfate, followed by the characteristic linear and hyperpigmented streaks. The exact mechanism of this characteristic rash is unknown. Suggested pathways include induction of neutrophilic eccrine hidradenitis, postinflammatory pigmentary incontinence, and altered levels of melanin due to a toxic effect of the medication. Other commonly reported adverse events to bleomycin sulfate include alopecia and stomatitis.3
Although a diffuse rash is the most commonly reported cutaneous reaction to systemic bleomycin sulfate, there have been reports of hyperpigmentation only in areas of pressure and palmar creases. Tsuji and Sawabe4 reported a case of hyperpigmentation limited to areas of striae distensae after systemic bleomycin sulfate. Flagellate hyperpigmentation also has been reported following intralesional bleomycin sulfate for the treatment of verruca plantaris. In that case, the patient had received 14 U of intralesional bleomycin sulfate injected at different sites of recalcitrant verruca and developed urticaria, generalized pruritus, and flagellate hyperpigmentation.5
Treatment of flagellate hyperpigmentation includes cessation of the medication, which is not always possible due to the need for an effective chemotherapeutic regimen. Most cases are reversible following discontinuation of bleomycin sulfate, and care is directed at symptom relief with antipruritic agents, antihistamines, systemic steroids, or topical steroids.3
1. Watanabe T, Tsuchida T. 'Flagellate' erythema in dermatomyositis. Dermatology. 1995;190:230-231.
2. Yagoda A, Mukherji B, Young C, et al. Bleomycin, an antitumor antibiotic. clinical experience in 274 patients. Ann Intern Med. 1972;77:861-870.
3. Susser WS, Whitaker-Worth DL, Grant-Kels JM. Mucocutaneous reactions to chemotherapy. J Am Acad Dermatol. 1999;40:367-398; quiz 399-400.
4. Tsuji T, Sawabe M. Hyperpigmentation in striae distensae after bleomycin treatment. J Am Acad Dermatol. 1993;28:503-505.
5. Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139:337-339.
The Diagnosis: Flagellate Hyperpigmentation
Physical examination of our patient revealed multiple linear, flagellate, and hyperpigmented plaques on the trunk. Similar lesions were seen on the upper and lower extremities. Given the patient's history of non-Hodgkin lymphoma and concurrent chemotherapy, the use of systemic steroids was avoided. Notable improvement in the symptoms was achieved with topical steroids (triamcinolone acetonide ointment) under occlusion and systemic gabapentin. The patient's chemotherapy is ongoing. This characteristic skin eruption often is seen after treatment with systemic bleomycin sulfate.
Flagellate hyperpigmentation following systemic bleomycin sulfate use as a chemotherapeutic agent is a well-known cutaneous reaction that occurs in approximately 10% of exposed individuals.1 Bleomycin sulfate, an antibiotic derived from Streptomyces verticillus, is a commonly used antitumor agent for both hematologic and solid-organ malignancies.2 Many patients develop generalized pruritus within several days after initiating bleomycin sulfate, followed by the characteristic linear and hyperpigmented streaks. The exact mechanism of this characteristic rash is unknown. Suggested pathways include induction of neutrophilic eccrine hidradenitis, postinflammatory pigmentary incontinence, and altered levels of melanin due to a toxic effect of the medication. Other commonly reported adverse events to bleomycin sulfate include alopecia and stomatitis.3
Although a diffuse rash is the most commonly reported cutaneous reaction to systemic bleomycin sulfate, there have been reports of hyperpigmentation only in areas of pressure and palmar creases. Tsuji and Sawabe4 reported a case of hyperpigmentation limited to areas of striae distensae after systemic bleomycin sulfate. Flagellate hyperpigmentation also has been reported following intralesional bleomycin sulfate for the treatment of verruca plantaris. In that case, the patient had received 14 U of intralesional bleomycin sulfate injected at different sites of recalcitrant verruca and developed urticaria, generalized pruritus, and flagellate hyperpigmentation.5
Treatment of flagellate hyperpigmentation includes cessation of the medication, which is not always possible due to the need for an effective chemotherapeutic regimen. Most cases are reversible following discontinuation of bleomycin sulfate, and care is directed at symptom relief with antipruritic agents, antihistamines, systemic steroids, or topical steroids.3
The Diagnosis: Flagellate Hyperpigmentation
Physical examination of our patient revealed multiple linear, flagellate, and hyperpigmented plaques on the trunk. Similar lesions were seen on the upper and lower extremities. Given the patient's history of non-Hodgkin lymphoma and concurrent chemotherapy, the use of systemic steroids was avoided. Notable improvement in the symptoms was achieved with topical steroids (triamcinolone acetonide ointment) under occlusion and systemic gabapentin. The patient's chemotherapy is ongoing. This characteristic skin eruption often is seen after treatment with systemic bleomycin sulfate.
Flagellate hyperpigmentation following systemic bleomycin sulfate use as a chemotherapeutic agent is a well-known cutaneous reaction that occurs in approximately 10% of exposed individuals.1 Bleomycin sulfate, an antibiotic derived from Streptomyces verticillus, is a commonly used antitumor agent for both hematologic and solid-organ malignancies.2 Many patients develop generalized pruritus within several days after initiating bleomycin sulfate, followed by the characteristic linear and hyperpigmented streaks. The exact mechanism of this characteristic rash is unknown. Suggested pathways include induction of neutrophilic eccrine hidradenitis, postinflammatory pigmentary incontinence, and altered levels of melanin due to a toxic effect of the medication. Other commonly reported adverse events to bleomycin sulfate include alopecia and stomatitis.3
Although a diffuse rash is the most commonly reported cutaneous reaction to systemic bleomycin sulfate, there have been reports of hyperpigmentation only in areas of pressure and palmar creases. Tsuji and Sawabe4 reported a case of hyperpigmentation limited to areas of striae distensae after systemic bleomycin sulfate. Flagellate hyperpigmentation also has been reported following intralesional bleomycin sulfate for the treatment of verruca plantaris. In that case, the patient had received 14 U of intralesional bleomycin sulfate injected at different sites of recalcitrant verruca and developed urticaria, generalized pruritus, and flagellate hyperpigmentation.5
Treatment of flagellate hyperpigmentation includes cessation of the medication, which is not always possible due to the need for an effective chemotherapeutic regimen. Most cases are reversible following discontinuation of bleomycin sulfate, and care is directed at symptom relief with antipruritic agents, antihistamines, systemic steroids, or topical steroids.3
1. Watanabe T, Tsuchida T. 'Flagellate' erythema in dermatomyositis. Dermatology. 1995;190:230-231.
2. Yagoda A, Mukherji B, Young C, et al. Bleomycin, an antitumor antibiotic. clinical experience in 274 patients. Ann Intern Med. 1972;77:861-870.
3. Susser WS, Whitaker-Worth DL, Grant-Kels JM. Mucocutaneous reactions to chemotherapy. J Am Acad Dermatol. 1999;40:367-398; quiz 399-400.
4. Tsuji T, Sawabe M. Hyperpigmentation in striae distensae after bleomycin treatment. J Am Acad Dermatol. 1993;28:503-505.
5. Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139:337-339.
1. Watanabe T, Tsuchida T. 'Flagellate' erythema in dermatomyositis. Dermatology. 1995;190:230-231.
2. Yagoda A, Mukherji B, Young C, et al. Bleomycin, an antitumor antibiotic. clinical experience in 274 patients. Ann Intern Med. 1972;77:861-870.
3. Susser WS, Whitaker-Worth DL, Grant-Kels JM. Mucocutaneous reactions to chemotherapy. J Am Acad Dermatol. 1999;40:367-398; quiz 399-400.
4. Tsuji T, Sawabe M. Hyperpigmentation in striae distensae after bleomycin treatment. J Am Acad Dermatol. 1993;28:503-505.
5. Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139:337-339.
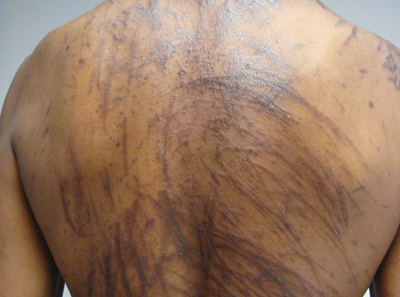
A 57-year-old man presented to the dermatology clinic for evaluation of a widespread pruritic rash. The patient had a recent diagnosis of non-Hodgkin lymphoma and was currently undergoing chemotherapy with adriamycin, bleomycin sulfate, vinblastine sulfate, and dacarbazine. The patient’s medical history included hypertension, which was well controlled, and a remote history of a stroke. The patient stated that the rash was recurrent and developed a few days after each dose of bleomycin. Review of systems was otherwise unremarkable, except for pruritus.
What Is Your Diagnosis? Leukemia Cutis
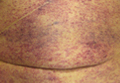
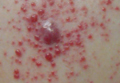
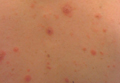
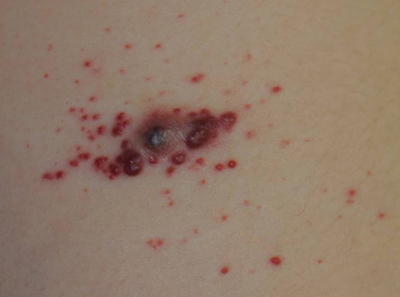
Erythematous Friable Papules on the Flank
The Diagnosis: Recurrent Pyogenic Granuloma With Satellite Papules
Pyogenic granulomas, also known as lobular capillary hemangiomas, are common benign, friable, rapidly growing, erythematous and exophytic papules with a surrounding collarette of scale. Due to the friable nature of these lesions, they are often hemorrhagic and ulcerative on presentation; however, disseminated pyogenic granulomas frequently are more sessile in appearance with a smooth surface. The pathogenesis of these lesions is largely unknown, but they exhibit a number of clinical features suggestive of reactive neovascularization.1
Primary pyogenic granulomas occur most often on sites with high vascularity (eg, gingiva, lips, fingers, face, tongue), but no site on the skin is exempt.2,3 They are found most commonly in children and young adults after minor trauma and also are associated with pregnancy with a higher frequency of occurrence on the gingiva in that particular setting.2
The differential diagnosis of pyogenic granuloma includes warts or inflamed intradermal nevi as well as bacillary angiomatosis and Kaposi sarcoma in an immunosuppressed patient. Most importantly, pyogenic granulomas can be confused with amelanotic melanoma; therefore, biopsy to rule out this condition is mandatory. On histopathology, pyogenic granuloma is characterized by a proliferation of capillaries arranged in a lobular pattern separated by a pale stroma (Figure).
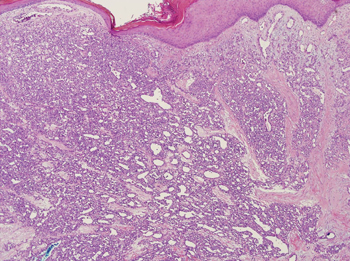
Pyogenic granulomas usually are solitary lesions; however, recurrence after treatment is common. These benign growths have been reported to arise from certain systemic agents including epidermal growth factor receptor inhibitors, systemic retinoids, and indinavir.1 Disseminated pyogenic granulomas are rare, but cases in children and adults have been reported, many associated with prior trauma. In addition, congenital disseminated pyogenic granulomas have been described, which could be easily confused with infantile hemangiomatosis; however, the former are GLUT-1 (glucose transporter type 1) negative.4
Although recurrence of pyogenic granulomas is common, recurrence with satellite lesions is relatively rare. In documented cases, such as the Blickenstaff et al2 report, recurrences occurred most commonly on the trunk, especially in the scapular area in adolescents and young adults. They often occur after prior irritation or treatment of the primary lesion but also may spontaneously develop. After initial therapy, satellites may arise either with or without recurrence of the primary lesion. These satellite lesions usually are smaller than the original lesion, smooth, bright red, are not as friable, and lack a collarette.2,3
Recurrence of pyogenic granulomas with satellite lesions has been reported to occur in association with almost all forms of treatment, including excision with cautery and CO2 laser. These recurrences have been successfully treated with simple excision, cryotherapy, diathermy, and curettage; however, some physicians recommend a conservative approach to therapy, as a majority of these lesions tend to spontaneously resolve after 6 to 12 months.2,3,5
Ezzell et al6 and others have reported successful treatment of pyogenic granulomas with imiquimod cream 5%. Imiquimod is thought to trigger resolution of pyogenic granulomas due to its antitumor and immunoregulatory effects. As a topical agent, imiquimod would offer patients a safe and noninvasive therapy that would be especially beneficial for larger lesions with satellites, similar to the papules described in our patient.6,7
After a repeat biopsy in our patient confirmed pyogenic granuloma, she was started on imiquimod cream 5% applied nightly, which continued for 6 weeks. Follow-up after 6 weeks revealed no change in lesion size and the family opted to discontinue treatment at that time because of a lack of response. Although a continued conservative approach was recommended by our facility, the patient and guardians opted for surgical excision and were referred to a local pediatric surgeon for excision under general anesthesia.
1. North PE, Kincannon J. Vascular neoplasms and neoplastic-like proliferations. In: Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1771-1794.
2. Blickenstaff RD, Roenigk RK, Peters MS, et al. Recurrent pyogenic granuloma with satellitosis. J Am Acad Dermatol. 1989;21:1241-1244.
3. Allen R, Rodman O. Pyogenic granuloma recurrent with satellite lesions. J Dermatol Surg Oncol. 1979;5:490-492.
4. Browning JC, Eldin KW, Kozakewich HP, et al. Congenital disseminated pyogenic granuloma. Pediatr Dermatol. 2009;26:323-327.
5. Fisher I. Recurrent multiple pyogenic granuloma with multiple satellites. features of Masson’s hemangioma. Cutis. 1977;20:201-205.
6. Ezzell TI, Fromowitz JS, Ramos-Caro FA. Recurrent pyogenic granuloma treated with topical imiquimod. J Am Acad Dermatol. 2006;54(suppl 5):S244-S245.
7. Tritton SM, Smith S, Wong LC, et al. Pyogenic granuloma in ten children treated with topical imiquimod. Pediatr Dermatol. 2009;26:269-272.
The Diagnosis: Recurrent Pyogenic Granuloma With Satellite Papules
Pyogenic granulomas, also known as lobular capillary hemangiomas, are common benign, friable, rapidly growing, erythematous and exophytic papules with a surrounding collarette of scale. Due to the friable nature of these lesions, they are often hemorrhagic and ulcerative on presentation; however, disseminated pyogenic granulomas frequently are more sessile in appearance with a smooth surface. The pathogenesis of these lesions is largely unknown, but they exhibit a number of clinical features suggestive of reactive neovascularization.1
Primary pyogenic granulomas occur most often on sites with high vascularity (eg, gingiva, lips, fingers, face, tongue), but no site on the skin is exempt.2,3 They are found most commonly in children and young adults after minor trauma and also are associated with pregnancy with a higher frequency of occurrence on the gingiva in that particular setting.2
The differential diagnosis of pyogenic granuloma includes warts or inflamed intradermal nevi as well as bacillary angiomatosis and Kaposi sarcoma in an immunosuppressed patient. Most importantly, pyogenic granulomas can be confused with amelanotic melanoma; therefore, biopsy to rule out this condition is mandatory. On histopathology, pyogenic granuloma is characterized by a proliferation of capillaries arranged in a lobular pattern separated by a pale stroma (Figure).
Pyogenic granulomas usually are solitary lesions; however, recurrence after treatment is common. These benign growths have been reported to arise from certain systemic agents including epidermal growth factor receptor inhibitors, systemic retinoids, and indinavir.1 Disseminated pyogenic granulomas are rare, but cases in children and adults have been reported, many associated with prior trauma. In addition, congenital disseminated pyogenic granulomas have been described, which could be easily confused with infantile hemangiomatosis; however, the former are GLUT-1 (glucose transporter type 1) negative.4
Although recurrence of pyogenic granulomas is common, recurrence with satellite lesions is relatively rare. In documented cases, such as the Blickenstaff et al2 report, recurrences occurred most commonly on the trunk, especially in the scapular area in adolescents and young adults. They often occur after prior irritation or treatment of the primary lesion but also may spontaneously develop. After initial therapy, satellites may arise either with or without recurrence of the primary lesion. These satellite lesions usually are smaller than the original lesion, smooth, bright red, are not as friable, and lack a collarette.2,3
Recurrence of pyogenic granulomas with satellite lesions has been reported to occur in association with almost all forms of treatment, including excision with cautery and CO2 laser. These recurrences have been successfully treated with simple excision, cryotherapy, diathermy, and curettage; however, some physicians recommend a conservative approach to therapy, as a majority of these lesions tend to spontaneously resolve after 6 to 12 months.2,3,5
Ezzell et al6 and others have reported successful treatment of pyogenic granulomas with imiquimod cream 5%. Imiquimod is thought to trigger resolution of pyogenic granulomas due to its antitumor and immunoregulatory effects. As a topical agent, imiquimod would offer patients a safe and noninvasive therapy that would be especially beneficial for larger lesions with satellites, similar to the papules described in our patient.6,7
After a repeat biopsy in our patient confirmed pyogenic granuloma, she was started on imiquimod cream 5% applied nightly, which continued for 6 weeks. Follow-up after 6 weeks revealed no change in lesion size and the family opted to discontinue treatment at that time because of a lack of response. Although a continued conservative approach was recommended by our facility, the patient and guardians opted for surgical excision and were referred to a local pediatric surgeon for excision under general anesthesia.
The Diagnosis: Recurrent Pyogenic Granuloma With Satellite Papules
Pyogenic granulomas, also known as lobular capillary hemangiomas, are common benign, friable, rapidly growing, erythematous and exophytic papules with a surrounding collarette of scale. Due to the friable nature of these lesions, they are often hemorrhagic and ulcerative on presentation; however, disseminated pyogenic granulomas frequently are more sessile in appearance with a smooth surface. The pathogenesis of these lesions is largely unknown, but they exhibit a number of clinical features suggestive of reactive neovascularization.1
Primary pyogenic granulomas occur most often on sites with high vascularity (eg, gingiva, lips, fingers, face, tongue), but no site on the skin is exempt.2,3 They are found most commonly in children and young adults after minor trauma and also are associated with pregnancy with a higher frequency of occurrence on the gingiva in that particular setting.2
The differential diagnosis of pyogenic granuloma includes warts or inflamed intradermal nevi as well as bacillary angiomatosis and Kaposi sarcoma in an immunosuppressed patient. Most importantly, pyogenic granulomas can be confused with amelanotic melanoma; therefore, biopsy to rule out this condition is mandatory. On histopathology, pyogenic granuloma is characterized by a proliferation of capillaries arranged in a lobular pattern separated by a pale stroma (Figure).
Pyogenic granulomas usually are solitary lesions; however, recurrence after treatment is common. These benign growths have been reported to arise from certain systemic agents including epidermal growth factor receptor inhibitors, systemic retinoids, and indinavir.1 Disseminated pyogenic granulomas are rare, but cases in children and adults have been reported, many associated with prior trauma. In addition, congenital disseminated pyogenic granulomas have been described, which could be easily confused with infantile hemangiomatosis; however, the former are GLUT-1 (glucose transporter type 1) negative.4
Although recurrence of pyogenic granulomas is common, recurrence with satellite lesions is relatively rare. In documented cases, such as the Blickenstaff et al2 report, recurrences occurred most commonly on the trunk, especially in the scapular area in adolescents and young adults. They often occur after prior irritation or treatment of the primary lesion but also may spontaneously develop. After initial therapy, satellites may arise either with or without recurrence of the primary lesion. These satellite lesions usually are smaller than the original lesion, smooth, bright red, are not as friable, and lack a collarette.2,3
Recurrence of pyogenic granulomas with satellite lesions has been reported to occur in association with almost all forms of treatment, including excision with cautery and CO2 laser. These recurrences have been successfully treated with simple excision, cryotherapy, diathermy, and curettage; however, some physicians recommend a conservative approach to therapy, as a majority of these lesions tend to spontaneously resolve after 6 to 12 months.2,3,5
Ezzell et al6 and others have reported successful treatment of pyogenic granulomas with imiquimod cream 5%. Imiquimod is thought to trigger resolution of pyogenic granulomas due to its antitumor and immunoregulatory effects. As a topical agent, imiquimod would offer patients a safe and noninvasive therapy that would be especially beneficial for larger lesions with satellites, similar to the papules described in our patient.6,7
After a repeat biopsy in our patient confirmed pyogenic granuloma, she was started on imiquimod cream 5% applied nightly, which continued for 6 weeks. Follow-up after 6 weeks revealed no change in lesion size and the family opted to discontinue treatment at that time because of a lack of response. Although a continued conservative approach was recommended by our facility, the patient and guardians opted for surgical excision and were referred to a local pediatric surgeon for excision under general anesthesia.
1. North PE, Kincannon J. Vascular neoplasms and neoplastic-like proliferations. In: Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1771-1794.
2. Blickenstaff RD, Roenigk RK, Peters MS, et al. Recurrent pyogenic granuloma with satellitosis. J Am Acad Dermatol. 1989;21:1241-1244.
3. Allen R, Rodman O. Pyogenic granuloma recurrent with satellite lesions. J Dermatol Surg Oncol. 1979;5:490-492.
4. Browning JC, Eldin KW, Kozakewich HP, et al. Congenital disseminated pyogenic granuloma. Pediatr Dermatol. 2009;26:323-327.
5. Fisher I. Recurrent multiple pyogenic granuloma with multiple satellites. features of Masson’s hemangioma. Cutis. 1977;20:201-205.
6. Ezzell TI, Fromowitz JS, Ramos-Caro FA. Recurrent pyogenic granuloma treated with topical imiquimod. J Am Acad Dermatol. 2006;54(suppl 5):S244-S245.
7. Tritton SM, Smith S, Wong LC, et al. Pyogenic granuloma in ten children treated with topical imiquimod. Pediatr Dermatol. 2009;26:269-272.
1. North PE, Kincannon J. Vascular neoplasms and neoplastic-like proliferations. In: Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1771-1794.
2. Blickenstaff RD, Roenigk RK, Peters MS, et al. Recurrent pyogenic granuloma with satellitosis. J Am Acad Dermatol. 1989;21:1241-1244.
3. Allen R, Rodman O. Pyogenic granuloma recurrent with satellite lesions. J Dermatol Surg Oncol. 1979;5:490-492.
4. Browning JC, Eldin KW, Kozakewich HP, et al. Congenital disseminated pyogenic granuloma. Pediatr Dermatol. 2009;26:323-327.
5. Fisher I. Recurrent multiple pyogenic granuloma with multiple satellites. features of Masson’s hemangioma. Cutis. 1977;20:201-205.
6. Ezzell TI, Fromowitz JS, Ramos-Caro FA. Recurrent pyogenic granuloma treated with topical imiquimod. J Am Acad Dermatol. 2006;54(suppl 5):S244-S245.
7. Tritton SM, Smith S, Wong LC, et al. Pyogenic granuloma in ten children treated with topical imiquimod. Pediatr Dermatol. 2009;26:269-272.
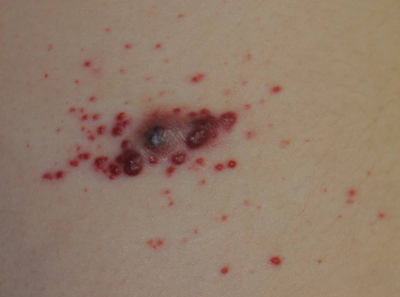
An 11-year-old girl presented to our clinic with a small erythematous friable papule on the right side of the flank. She reported that the papule bled easily and was mildly tender to palpation. A shave biopsy with electrocautery to the base of the lesion was performed. The patient returned 1 month later with recurrence of the papule and was treated with a 595-nm pulsed dye laser during her clinic visit. She returned 3 weeks later with persistence of the same papule and also with several new smaller but similar-appearing papules.
Indurated Thigh Plaque With Associated Lymphadenopathy
The Diagnosis: Rosai-Dorfman Disease
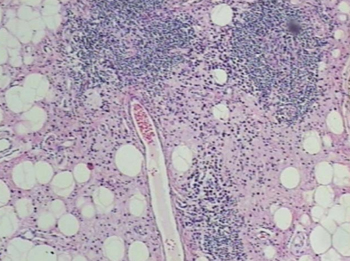
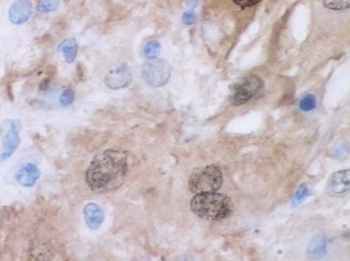
Punch biopsies of the mass from the right thigh were obtained. Hematoxylin and eosin staining revealed a reactive fibroinflammatory process of the dermis and subcutaneous tissue with fibrosis and prominent lymphoplasmacytic and histiocytic infiltrate with emperipolesis (Figure 1). The histiocytes stained positive for S-100 (Figure 2) and CD68. Markers for CD34, smooth muscle actin, and keratin were negative, as were acid-fast bacteria (Ziehl-Neelsen) and fungal (Gomori methenamine-silver) stains. The morphologic and immunohistochemical features of the biopsy specimens were consistent with Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy.
Rosai-Dorfman disease is a benign histiocytic proliferative disorder that usually involves the lymph nodes but may involve any organ system including the skin and soft tissue.1,2 Systemic RDD usually presents in children or young adults as massive cervical lymphadenopathy, often with fever, polyclonal hyperglobulinemia, mild anemia, elevated erythrocyte sedimentation rate, and occasionally neutropenia.2 Rosai-Dorfman disease was first described as a distinct clinicopathologic entity in 1969.3
Extranodal involvement occurs in approximately 40% of patients, with cutaneous disease in approximately 10% of cases. Other sites that may be affected include the eyelid and orbit, salivary glands, lungs, central nervous system, and bone.2 Purely cutaneous RDD is rare and has a variable skin distribution. Compared to systemic RDD, cutaneous disease affects older patients with a predilection for females and white individuals.4
Histologic examination of cutaneous RDD reveals a dense dermal infiltrate of foamy histiocytes with scattered lymphocytes, plasma cells, and neutrophils.2 Emperipolesis, consisting of intact lymphocytes and less commonly plasma cells in the cytoplasm of the histiocytes, is a characteristic feature of cutaneous RDD. Occasional findings include fibrosis, lymphoid aggregates, foamy histiocytes inside of dilated lymphatics, thick-walled venules with surrounding plasma cells, and multinucleate histiocytes.2 Immunohistochemistry is helpful in diagnosing RDD, as the histiocytes stain strongly positive for S-100, occasionally positive for CD68, and negative for CD1a.5
The pathogenesis of RDD remains unknown. Some cases of RDD have had positive serologies for human herpesvirus 6 and Epstein-Barr virus, though an infectious origin has not been proven.5 Clonality studies have shown the cellular infiltrate to be polyclonal, supporting a reactive rather than neoplastic process.6 The disease course of RDD varies from self-limited to protracted with remissions and exacerbations. Many cutaneous RDD lesions spontaneously heal and only require treatment when disseminated, destructive, or causing physical compromise.2 Treatment modalities for cutaneous RDD have exhibited varying degrees of success and include surgical excision; thalidomide; isotretinoin; radiotherapy; alkylating agents; and oral, topical, and intralesional steroids.2
The mass in our patient was surgically excised, though surgical margins remained positive. He was followed by the hematology-oncology service and later reported the development of 2 additional masses in the right leg with intermittent tenderness. Magnetic resonance imaging of the head was negative for intracranial abnormalities, though it did demonstrate prominent nasopharyngeal soft tissue and sinus disease. A complete skeletal radiograph survey and whole-body technetium bone scan did not show evidence of bone involvement. Chest computed tomography was negative for mass lesions. The patient deferred a bone marrow biopsy and was reportedly doing well at follow-up without further treatment.
1. Rubenstein MA, Farnsworth NN, Pielop JA, et al. Cutaneous Rosai-Dorfman disease. Dermatol Online J. 2006;12:8.
2. Goodman WT, Barrett TL. Histiocytoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. Vol 2. 2nd ed. Spain: Mosby Elsevier; 2008:1395-1410.
3. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
4. Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
5. Wartman DG, Perry A, Werchniak AE. Multiple nodules and plaques on the face and trunk. cutaneous Rosai Dorfman disease (RDD). Arch Dermatol. 2006;142:1501-1506.
6. Paulli M, Bergamaschi G, Tonon L. Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol. 1995;91:415-418.
The Diagnosis: Rosai-Dorfman Disease
Punch biopsies of the mass from the right thigh were obtained. Hematoxylin and eosin staining revealed a reactive fibroinflammatory process of the dermis and subcutaneous tissue with fibrosis and prominent lymphoplasmacytic and histiocytic infiltrate with emperipolesis (Figure 1). The histiocytes stained positive for S-100 (Figure 2) and CD68. Markers for CD34, smooth muscle actin, and keratin were negative, as were acid-fast bacteria (Ziehl-Neelsen) and fungal (Gomori methenamine-silver) stains. The morphologic and immunohistochemical features of the biopsy specimens were consistent with Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy.
Rosai-Dorfman disease is a benign histiocytic proliferative disorder that usually involves the lymph nodes but may involve any organ system including the skin and soft tissue.1,2 Systemic RDD usually presents in children or young adults as massive cervical lymphadenopathy, often with fever, polyclonal hyperglobulinemia, mild anemia, elevated erythrocyte sedimentation rate, and occasionally neutropenia.2 Rosai-Dorfman disease was first described as a distinct clinicopathologic entity in 1969.3
Extranodal involvement occurs in approximately 40% of patients, with cutaneous disease in approximately 10% of cases. Other sites that may be affected include the eyelid and orbit, salivary glands, lungs, central nervous system, and bone.2 Purely cutaneous RDD is rare and has a variable skin distribution. Compared to systemic RDD, cutaneous disease affects older patients with a predilection for females and white individuals.4
Histologic examination of cutaneous RDD reveals a dense dermal infiltrate of foamy histiocytes with scattered lymphocytes, plasma cells, and neutrophils.2 Emperipolesis, consisting of intact lymphocytes and less commonly plasma cells in the cytoplasm of the histiocytes, is a characteristic feature of cutaneous RDD. Occasional findings include fibrosis, lymphoid aggregates, foamy histiocytes inside of dilated lymphatics, thick-walled venules with surrounding plasma cells, and multinucleate histiocytes.2 Immunohistochemistry is helpful in diagnosing RDD, as the histiocytes stain strongly positive for S-100, occasionally positive for CD68, and negative for CD1a.5
The pathogenesis of RDD remains unknown. Some cases of RDD have had positive serologies for human herpesvirus 6 and Epstein-Barr virus, though an infectious origin has not been proven.5 Clonality studies have shown the cellular infiltrate to be polyclonal, supporting a reactive rather than neoplastic process.6 The disease course of RDD varies from self-limited to protracted with remissions and exacerbations. Many cutaneous RDD lesions spontaneously heal and only require treatment when disseminated, destructive, or causing physical compromise.2 Treatment modalities for cutaneous RDD have exhibited varying degrees of success and include surgical excision; thalidomide; isotretinoin; radiotherapy; alkylating agents; and oral, topical, and intralesional steroids.2
The mass in our patient was surgically excised, though surgical margins remained positive. He was followed by the hematology-oncology service and later reported the development of 2 additional masses in the right leg with intermittent tenderness. Magnetic resonance imaging of the head was negative for intracranial abnormalities, though it did demonstrate prominent nasopharyngeal soft tissue and sinus disease. A complete skeletal radiograph survey and whole-body technetium bone scan did not show evidence of bone involvement. Chest computed tomography was negative for mass lesions. The patient deferred a bone marrow biopsy and was reportedly doing well at follow-up without further treatment.
The Diagnosis: Rosai-Dorfman Disease
Punch biopsies of the mass from the right thigh were obtained. Hematoxylin and eosin staining revealed a reactive fibroinflammatory process of the dermis and subcutaneous tissue with fibrosis and prominent lymphoplasmacytic and histiocytic infiltrate with emperipolesis (Figure 1). The histiocytes stained positive for S-100 (Figure 2) and CD68. Markers for CD34, smooth muscle actin, and keratin were negative, as were acid-fast bacteria (Ziehl-Neelsen) and fungal (Gomori methenamine-silver) stains. The morphologic and immunohistochemical features of the biopsy specimens were consistent with Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy.
Rosai-Dorfman disease is a benign histiocytic proliferative disorder that usually involves the lymph nodes but may involve any organ system including the skin and soft tissue.1,2 Systemic RDD usually presents in children or young adults as massive cervical lymphadenopathy, often with fever, polyclonal hyperglobulinemia, mild anemia, elevated erythrocyte sedimentation rate, and occasionally neutropenia.2 Rosai-Dorfman disease was first described as a distinct clinicopathologic entity in 1969.3
Extranodal involvement occurs in approximately 40% of patients, with cutaneous disease in approximately 10% of cases. Other sites that may be affected include the eyelid and orbit, salivary glands, lungs, central nervous system, and bone.2 Purely cutaneous RDD is rare and has a variable skin distribution. Compared to systemic RDD, cutaneous disease affects older patients with a predilection for females and white individuals.4
Histologic examination of cutaneous RDD reveals a dense dermal infiltrate of foamy histiocytes with scattered lymphocytes, plasma cells, and neutrophils.2 Emperipolesis, consisting of intact lymphocytes and less commonly plasma cells in the cytoplasm of the histiocytes, is a characteristic feature of cutaneous RDD. Occasional findings include fibrosis, lymphoid aggregates, foamy histiocytes inside of dilated lymphatics, thick-walled venules with surrounding plasma cells, and multinucleate histiocytes.2 Immunohistochemistry is helpful in diagnosing RDD, as the histiocytes stain strongly positive for S-100, occasionally positive for CD68, and negative for CD1a.5
The pathogenesis of RDD remains unknown. Some cases of RDD have had positive serologies for human herpesvirus 6 and Epstein-Barr virus, though an infectious origin has not been proven.5 Clonality studies have shown the cellular infiltrate to be polyclonal, supporting a reactive rather than neoplastic process.6 The disease course of RDD varies from self-limited to protracted with remissions and exacerbations. Many cutaneous RDD lesions spontaneously heal and only require treatment when disseminated, destructive, or causing physical compromise.2 Treatment modalities for cutaneous RDD have exhibited varying degrees of success and include surgical excision; thalidomide; isotretinoin; radiotherapy; alkylating agents; and oral, topical, and intralesional steroids.2
The mass in our patient was surgically excised, though surgical margins remained positive. He was followed by the hematology-oncology service and later reported the development of 2 additional masses in the right leg with intermittent tenderness. Magnetic resonance imaging of the head was negative for intracranial abnormalities, though it did demonstrate prominent nasopharyngeal soft tissue and sinus disease. A complete skeletal radiograph survey and whole-body technetium bone scan did not show evidence of bone involvement. Chest computed tomography was negative for mass lesions. The patient deferred a bone marrow biopsy and was reportedly doing well at follow-up without further treatment.
1. Rubenstein MA, Farnsworth NN, Pielop JA, et al. Cutaneous Rosai-Dorfman disease. Dermatol Online J. 2006;12:8.
2. Goodman WT, Barrett TL. Histiocytoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. Vol 2. 2nd ed. Spain: Mosby Elsevier; 2008:1395-1410.
3. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
4. Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
5. Wartman DG, Perry A, Werchniak AE. Multiple nodules and plaques on the face and trunk. cutaneous Rosai Dorfman disease (RDD). Arch Dermatol. 2006;142:1501-1506.
6. Paulli M, Bergamaschi G, Tonon L. Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol. 1995;91:415-418.
1. Rubenstein MA, Farnsworth NN, Pielop JA, et al. Cutaneous Rosai-Dorfman disease. Dermatol Online J. 2006;12:8.
2. Goodman WT, Barrett TL. Histiocytoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. Vol 2. 2nd ed. Spain: Mosby Elsevier; 2008:1395-1410.
3. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
4. Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
5. Wartman DG, Perry A, Werchniak AE. Multiple nodules and plaques on the face and trunk. cutaneous Rosai Dorfman disease (RDD). Arch Dermatol. 2006;142:1501-1506.
6. Paulli M, Bergamaschi G, Tonon L. Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol. 1995;91:415-418.
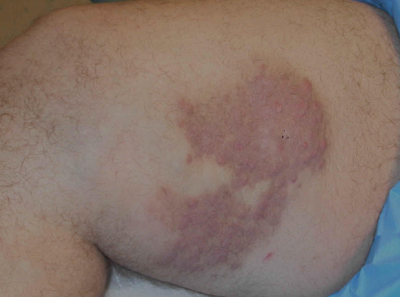
A 38-year-old white man with a history of lower extremity varicose veins was referred for evaluation of a plaque on the medial aspect of the right thigh and right inguinal lymphadenopathy. The patient reported having the lesion for approximately 1 year; over time it had become progressively more indurated. He denied pain, discomfort, and pruritus at the site, as well as any systemic symptoms. The patient had no known allergies, was not taking any medications, smoked 1 pack of tobacco daily for 20 years, drank alcohol socially, and was not sexually active. His family history was noncontributory. Prior to the dermatology consultation, a computed tomography scan of the right leg, abdomen, and pelvis was obtained and demonstrated a subcutaneous mass in the distal third of the medial aspect of the right thigh as well as pericaval and right inguinal lymphadenopathy. Physical examination revealed a violaceous, indurated, nodular plaque on the medial aspect of the right thigh measuring approximately 10×5 cm. Multiple small, nontender, mobile lymph nodes were palpated on the right side of the inguinal region. The remainder of the physical examination was unremarkable.
What Is Your Diagnosis? Subacute Cutaneous Lupus Erythematosus
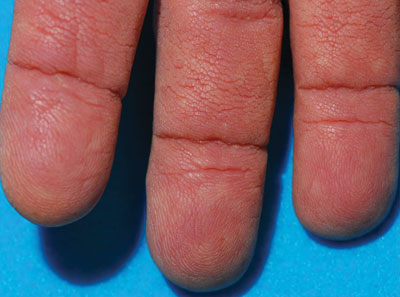
Thickened Velvety Plaques in a 75-Year-Old Woman
The Diagnosis: Tripe Palms and Acanthosis Nigricans
A shave biopsy specimen from the left palm showed slight epidermal hyperplasia with substantial papillomatosis and compact orthokeratosis. The complete blood cell count, thyrotropin level, uric acid level, liver function tests, mammogram, Papanicolaou test, and chest radiograph were unremarkable. A basic metabolic panel showed mildly elevated blood glucose at 111 mg/dL (reference range, 70-99 mg/dL) and hemoglobin A1c at 6.3% (reference range, <6.0%). Full-body computed tomography, endoscopy, and colonoscopy initially were normal. One year later after presenting with tripe palms, the patient had a bowel obstruction secondary to omental carcinomatosis from a primary ovarian tumor.
The term tripe in tripe palms refers to the resemblance to the edible lining of the bovine foregut. It originated in 1963 from a patient's own description of the rugose velvety texture of the palms.1 In the literature, tripe palms also is called acanthosis palmaris, acanthosis nigricans of the palms, palmar hyperkeratosis, palmar keratoderma, and pachydermatoglyphy. It is a rare cutaneous finding. Tripe palms is associated with other cutaneous paraneoplastic syndromes such as malignant acanthosis nigricans (72% of cases) and Leser-Trélat sign (10% of cases). It affects more men than women (63% vs 37%) and is almost exclusively seen in adults (median age, 62 years).1
The clinical appearance of tripe palms includes hypertrophy of the palms and often the soles with papillations producing a velvety or honeycomb appearance. In addition, the dermatoglyphics are pronounced. The histologic findings typically show hyperkeratosis and acanthosis. Other features that can be seen include dermal mucinosis and increased mast cells in the dermis. To differentiate tripe palms from other keratodermas, substantial papillations can be seen with less diffuse hyperkeratosis.1
Tripe palms has been associated with an underlying malignancy in more than 90% of published cases. In two-thirds of cases, tripe palms either appears before or concurrent with the diagnosis of cancer.2 It is rarely reported as an idiopathic finding or associated with nonneoplastic disorders. Malignancies most commonly associated are adenocarcinomas, especially of the stomach (27%) and lungs (22%). Other neoplasms, such as in our patient, include those of the genitourinary tract and breast. In a patient with tripe palms in the absence of acanthosis nigricans, the most common neoplasm is of the lung, especially when clubbing of the nails also is present.2 Thus, after a diagnosis of tripe palms is established, a thorough investigation for an underlying malignancy is the next most important step to direct specific therapy.
1. Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
2. Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin. 2008;26:17-29.
The Diagnosis: Tripe Palms and Acanthosis Nigricans
A shave biopsy specimen from the left palm showed slight epidermal hyperplasia with substantial papillomatosis and compact orthokeratosis. The complete blood cell count, thyrotropin level, uric acid level, liver function tests, mammogram, Papanicolaou test, and chest radiograph were unremarkable. A basic metabolic panel showed mildly elevated blood glucose at 111 mg/dL (reference range, 70-99 mg/dL) and hemoglobin A1c at 6.3% (reference range, <6.0%). Full-body computed tomography, endoscopy, and colonoscopy initially were normal. One year later after presenting with tripe palms, the patient had a bowel obstruction secondary to omental carcinomatosis from a primary ovarian tumor.
The term tripe in tripe palms refers to the resemblance to the edible lining of the bovine foregut. It originated in 1963 from a patient's own description of the rugose velvety texture of the palms.1 In the literature, tripe palms also is called acanthosis palmaris, acanthosis nigricans of the palms, palmar hyperkeratosis, palmar keratoderma, and pachydermatoglyphy. It is a rare cutaneous finding. Tripe palms is associated with other cutaneous paraneoplastic syndromes such as malignant acanthosis nigricans (72% of cases) and Leser-Trélat sign (10% of cases). It affects more men than women (63% vs 37%) and is almost exclusively seen in adults (median age, 62 years).1
The clinical appearance of tripe palms includes hypertrophy of the palms and often the soles with papillations producing a velvety or honeycomb appearance. In addition, the dermatoglyphics are pronounced. The histologic findings typically show hyperkeratosis and acanthosis. Other features that can be seen include dermal mucinosis and increased mast cells in the dermis. To differentiate tripe palms from other keratodermas, substantial papillations can be seen with less diffuse hyperkeratosis.1
Tripe palms has been associated with an underlying malignancy in more than 90% of published cases. In two-thirds of cases, tripe palms either appears before or concurrent with the diagnosis of cancer.2 It is rarely reported as an idiopathic finding or associated with nonneoplastic disorders. Malignancies most commonly associated are adenocarcinomas, especially of the stomach (27%) and lungs (22%). Other neoplasms, such as in our patient, include those of the genitourinary tract and breast. In a patient with tripe palms in the absence of acanthosis nigricans, the most common neoplasm is of the lung, especially when clubbing of the nails also is present.2 Thus, after a diagnosis of tripe palms is established, a thorough investigation for an underlying malignancy is the next most important step to direct specific therapy.
The Diagnosis: Tripe Palms and Acanthosis Nigricans
A shave biopsy specimen from the left palm showed slight epidermal hyperplasia with substantial papillomatosis and compact orthokeratosis. The complete blood cell count, thyrotropin level, uric acid level, liver function tests, mammogram, Papanicolaou test, and chest radiograph were unremarkable. A basic metabolic panel showed mildly elevated blood glucose at 111 mg/dL (reference range, 70-99 mg/dL) and hemoglobin A1c at 6.3% (reference range, <6.0%). Full-body computed tomography, endoscopy, and colonoscopy initially were normal. One year later after presenting with tripe palms, the patient had a bowel obstruction secondary to omental carcinomatosis from a primary ovarian tumor.
The term tripe in tripe palms refers to the resemblance to the edible lining of the bovine foregut. It originated in 1963 from a patient's own description of the rugose velvety texture of the palms.1 In the literature, tripe palms also is called acanthosis palmaris, acanthosis nigricans of the palms, palmar hyperkeratosis, palmar keratoderma, and pachydermatoglyphy. It is a rare cutaneous finding. Tripe palms is associated with other cutaneous paraneoplastic syndromes such as malignant acanthosis nigricans (72% of cases) and Leser-Trélat sign (10% of cases). It affects more men than women (63% vs 37%) and is almost exclusively seen in adults (median age, 62 years).1
The clinical appearance of tripe palms includes hypertrophy of the palms and often the soles with papillations producing a velvety or honeycomb appearance. In addition, the dermatoglyphics are pronounced. The histologic findings typically show hyperkeratosis and acanthosis. Other features that can be seen include dermal mucinosis and increased mast cells in the dermis. To differentiate tripe palms from other keratodermas, substantial papillations can be seen with less diffuse hyperkeratosis.1
Tripe palms has been associated with an underlying malignancy in more than 90% of published cases. In two-thirds of cases, tripe palms either appears before or concurrent with the diagnosis of cancer.2 It is rarely reported as an idiopathic finding or associated with nonneoplastic disorders. Malignancies most commonly associated are adenocarcinomas, especially of the stomach (27%) and lungs (22%). Other neoplasms, such as in our patient, include those of the genitourinary tract and breast. In a patient with tripe palms in the absence of acanthosis nigricans, the most common neoplasm is of the lung, especially when clubbing of the nails also is present.2 Thus, after a diagnosis of tripe palms is established, a thorough investigation for an underlying malignancy is the next most important step to direct specific therapy.
1. Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
2. Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin. 2008;26:17-29.
1. Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
2. Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin. 2008;26:17-29.
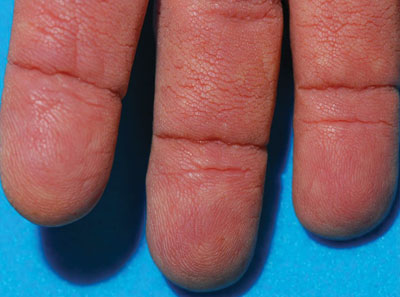
A 75-year-old woman presented with progressive, velvety, thick skin involving the bilateral axillae, inner thighs, palms, and buccal mucosa. She also reported weight loss of approximately 25 pounds over the last 12 months. Her medical history was notable for metabolic syndrome, allergic rhinitis, and colon polyps. She denied a family history of malignancy. On physical examination, she was a healthy-appearing overweight woman. The palmar surface of the bilateral hands was thickened and velvety with exaggerated dermatoglyphics. She had similarly thickened, velvety, gray-brown plaques on the bilateral axillae and proximal aspects of the inner thighs. The buccal mucosa had a thickened rugose texture.