User login
Constipation and Postprandial Pain in a Patient With Shortness of Breath
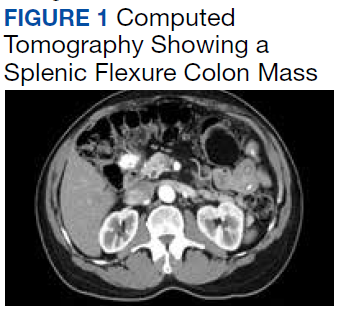
A 62-year-old male veteran with a history of pulmonary embolism (PE) and prostate cancer status after brachytherapy presented to the emergency department with new onset shortness of breath and left-sided chest pain after prolonged car travel. He underwent a chest computed tomography (CT) angiogram that showed no PE recurrence; however, the scan revealed an incidental transverse colon mass that appeared well circumscribed, homogeneous, and radiolucent with no enhancement, septations, or hypervascularity but no evidence of colonic distension or obstruction (Figure 1).
- What is your diagnosis?
- How would you treat this patient?
The patient reported having chronic constipation and a dull, left-sided abdominal discomfort for the past year. He noted that his abdominal pain worsened after eating and mildly improved after taking castor oil. He had no surgical history and no family history of cancer. The patient reported no fever, fatigue, weight loss, chills, nausea, vomiting, diarrhea, hematochezia, dysuria, hematuria, or melena. Vital signs, physical examination, and initial routine laboratory work were all within appropriate ranges, and a fecal occult blood test was negative.
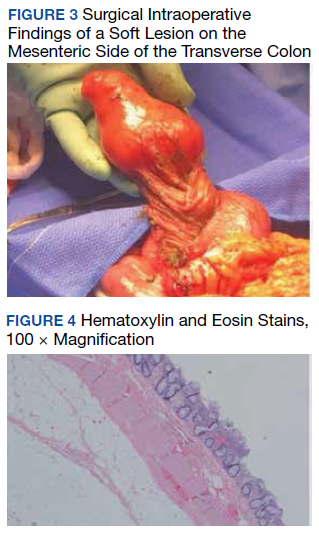
A colonoscopy was performed, revealing a near-obstructing submucosal mass in the transverse colon near the splenic flexure with a smooth surface and a positive Cushion (Pillow) sign (Figure 2). The patient underwent surgical exploration that resulted in finding a soft, 11-cm lesion arising from the mesenteric side of the transverse colon (Figure 3). Hematoxylin and eosin (H&E) stains were used on a sample from the mass (Figure 4).
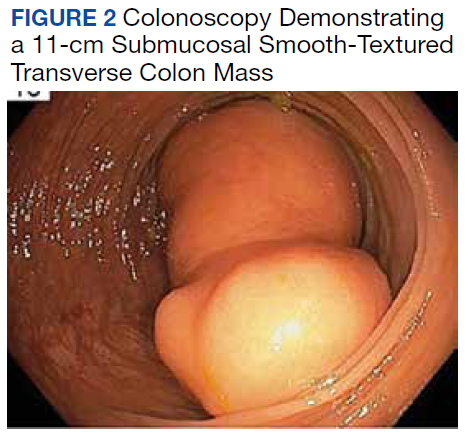
The tumor was enucleated via a colotomy over the mass, and the colotomy repaired primarily. Gross examination revealed homogenous yellow fatty tissue, and the H&E stains showed mature, well-differentiated adipocytes with uniform nuclei surrounded by a fibrous capsule. Based on this pathologic examination, this patient was diagnosed with a lipoma of the transverse colon. The resected tissue showed negative margins, indicating full removal of the lipoma.
The patient stabilized well after surgery and remained under inpatient care for observation; due to lack of appetite following the surgery, the patient did not start eating solids again until 2 days after the lipoma removal. By postoperative day 4, the patient had return of bowel function and was tolerating a regular diet with no recurrence of his prandial pain, shortness of breath, or left-sided chest pain. While the precise cause of the patient’s initial presentation of shortness of breath and left-sided chest pain was not ascertained, it is likely that the lipoma, near completely obstructed his bowel, caused abdominal contents and distended intestines to push against his diaphragm, leading to pain and dyspnea. This was likely exacerbated by sensitization to these symptoms from his prior PE. He was discharged home on postoperative day 4 with outpatient follow-up with general surgery.
Discussion
Lipomas are common benign tumors arising from aberrantly multiplying adipocytes. Although lipomas are most commonly found subcutaneously, the lesions can occur anywhere along the gastrointestinal (GI) tract, most often in the colon.1 The incidence rate of colon lipomas ranges from 0.2 to 4.4% among patients in their fifth to sixth decades of life, more commonly found in females.2 These lesions are the most common submucosal mesenchymal lesions of the colon, with a predilection for the right ascending colon.1 The etiology of colon lipomas is largely unknown; one known cause is trauma, thought to induce cytokine release or HMGA2-LPP fusion gene arrangements leading to adipocyte proliferation.3
Most colon lipomas are asymptomatic and discovered incidentally; symptoms typically arise when the lesions are > 2 cm in diameter and include abdominal pain, changes in bowel habits, rectal bleeding, and in extreme cases, obstruction and perforation.4 On CT imaging, colon lipomas will appear radiolucent, homogenous, and well circumscribed. The lesions usually do not warrant intervention unless they are symptomatic. If symptomatic, resection of the lesion is the first-line treatment and usually results in complete resolution of symptoms with no recurrence.2
While either a surgical or endoscopic approach may be used for resection, an increased risk of perforation of the colon with larger lipomas has been shown with endoscopic excision.5 With surgical resection, an open or minimally invasive approach may be offered, based on surgeon comfort with minimally invasive colon procedures. Minimally invasive colonic surgeries may be associated with a shorter length of stay, decreased postoperative pain, and faster return of bowel function. In this case, the surgeon chose an open approach due to the large size of the mass (11 cm) as well as location of the mass in the transverse colon, which made it easy to access directly through a small laparotomy incision made in the superior midline over the transverse colon.
When a colonic mesenchymal mass is seen on colonoscopy, it is important to consider other, nonbenign lesions that present this way. The most common malignant mesenchymal tumor of the GI tract is a gastrointestinal stromal tumor (GIST), a soft-tissue sarcoma that occurs predominantly in the stomach and small intestine.6 These tumors arise from the interstitial cells of Cajal (ICC) and are associated with mutations of KIT and PDGFR-α genes.7 The incidence in the United States is approximately 0.70 per 100,000 people per year, predominantly found in adults in their fifth or sixth decade of life.8 While this tumor typically occurs in the upper GI tract, very rarely, GISTs can be found in the colon.6 Common constitutional symptoms of colon GIST are similar to those of colon lipomas and include abdominal pain, changes in bowel habits, nausea, vomiting, and in some cases, weight loss.
CT imaging is often enough to differentiate a colon lipoma from a colon GIST. On CT, large GIST tumors tend to show irregular, lobulated margins, mucosal ulceration, central necrosis, cavitation, hemorrhage, and hypervascularity—vastly different from the CT findings of colon lipomas. If imaging is equivocal, an ultrasound-guided fine needle aspiration biopsy may be performed, differentiating GIST through the presence of ICC tumor cells as well as KIT and PDGFR-α proteins.
In our patient, colonoscopy showed a positive Cushion sign (tumor indented on depression with biopsy forceps), pathognomonic for a colon lipoma, and CT imaging showed a radiolucent, well-circumscribed lesion.9 This was more consistent with a colon lipoma than a GIST. Because the patient was symptomatic with a near obstructing lesion, the appropriate next step was removal of the lesion. Had this instead been a GIST tumor, a more extensive oncologic surgical resection would have been warranted, with adequate mesentery and lymph nodes collected.
This case is notable because colon lipomas exceeding 2 cm are rare and are usually an incidental finding on CT. However, larger lipomas can lead to symptoms, including obstruction if not removed in a timely manner.
1. Nallamothu G, Adler DG. Large colonic lipomas. Gastroenterol Hepatol (NY). 2011;7(7):490-492.
2. Crocetti D, Sapienza P, Sterpetti AV, et al. Surgery for symptomatic colon lipoma: a systematic review of the literature. Anticancer Res. 2014;34(11):6271-6276.
3. Italiano A, Ebran N, Attias R, et al. NFIB rearrangement in superficial, retroperitoneal, and colonic lipomas with aberrations involving chromosome band 9p22. Genes Chromosomes Cancer. 2008;47(11):971-977. doi:10.1002/gcc.20602
4. Agrawal A, Singh KJ. Symptomatic intestinal lipomas: our experience. Med J Armed Forces India. 2011;67(4):374-376. doi:10.1016/S0377-1237(11)60090-7
5. Kim GW, Kwon CI, Song SH, et al. Endoscopic resection of giant colonic lipoma: case series with partial resection. Clin Endosc. 2013;46(5):586-590. doi:10.5946/ce.2013.46.5.586
6. Reddy RM, Fleshman JW. Colorectal gastrointestinal stromal tumors: a brief review. Clin Colon Rectal Surg. 2006;19(2):69-77. doi:10.1055/s-2006-942347
7. Shinomura Y, Kinoshita K, Tsutsui S, Hirota S. Pathophysiology, diagnosis, and treatment of gastrointestinal stromal tumors. J Gastroenterol. 2005;40(8):775-780. doi:10.1007/s00535-005-1674-0
8. Patel N, Benipal B. Incidence of gastrointestinal stromal tumors in the United States from 2001-2015: a United States cancer statistics analysis of 50 states. Cureus. 2019;11(2):e4120. Published 2019 Feb 22. doi:10.7759/cureus.4120
9. Kyawzaw K, Emmanuel O, Sandar L,2 Febin J,Naing LA, Madhavi R. Pillow sign in colonoscopy. MOJ Clin Med Case Rep. 2018;8(2):57-58. doi:10.15406/mojcr.2018.08.00240
A 62-year-old male veteran with a history of pulmonary embolism (PE) and prostate cancer status after brachytherapy presented to the emergency department with new onset shortness of breath and left-sided chest pain after prolonged car travel. He underwent a chest computed tomography (CT) angiogram that showed no PE recurrence; however, the scan revealed an incidental transverse colon mass that appeared well circumscribed, homogeneous, and radiolucent with no enhancement, septations, or hypervascularity but no evidence of colonic distension or obstruction (Figure 1).
- What is your diagnosis?
- How would you treat this patient?
The patient reported having chronic constipation and a dull, left-sided abdominal discomfort for the past year. He noted that his abdominal pain worsened after eating and mildly improved after taking castor oil. He had no surgical history and no family history of cancer. The patient reported no fever, fatigue, weight loss, chills, nausea, vomiting, diarrhea, hematochezia, dysuria, hematuria, or melena. Vital signs, physical examination, and initial routine laboratory work were all within appropriate ranges, and a fecal occult blood test was negative.
A colonoscopy was performed, revealing a near-obstructing submucosal mass in the transverse colon near the splenic flexure with a smooth surface and a positive Cushion (Pillow) sign (Figure 2). The patient underwent surgical exploration that resulted in finding a soft, 11-cm lesion arising from the mesenteric side of the transverse colon (Figure 3). Hematoxylin and eosin (H&E) stains were used on a sample from the mass (Figure 4).
The tumor was enucleated via a colotomy over the mass, and the colotomy repaired primarily. Gross examination revealed homogenous yellow fatty tissue, and the H&E stains showed mature, well-differentiated adipocytes with uniform nuclei surrounded by a fibrous capsule. Based on this pathologic examination, this patient was diagnosed with a lipoma of the transverse colon. The resected tissue showed negative margins, indicating full removal of the lipoma.
The patient stabilized well after surgery and remained under inpatient care for observation; due to lack of appetite following the surgery, the patient did not start eating solids again until 2 days after the lipoma removal. By postoperative day 4, the patient had return of bowel function and was tolerating a regular diet with no recurrence of his prandial pain, shortness of breath, or left-sided chest pain. While the precise cause of the patient’s initial presentation of shortness of breath and left-sided chest pain was not ascertained, it is likely that the lipoma, near completely obstructed his bowel, caused abdominal contents and distended intestines to push against his diaphragm, leading to pain and dyspnea. This was likely exacerbated by sensitization to these symptoms from his prior PE. He was discharged home on postoperative day 4 with outpatient follow-up with general surgery.
Discussion
Lipomas are common benign tumors arising from aberrantly multiplying adipocytes. Although lipomas are most commonly found subcutaneously, the lesions can occur anywhere along the gastrointestinal (GI) tract, most often in the colon.1 The incidence rate of colon lipomas ranges from 0.2 to 4.4% among patients in their fifth to sixth decades of life, more commonly found in females.2 These lesions are the most common submucosal mesenchymal lesions of the colon, with a predilection for the right ascending colon.1 The etiology of colon lipomas is largely unknown; one known cause is trauma, thought to induce cytokine release or HMGA2-LPP fusion gene arrangements leading to adipocyte proliferation.3
Most colon lipomas are asymptomatic and discovered incidentally; symptoms typically arise when the lesions are > 2 cm in diameter and include abdominal pain, changes in bowel habits, rectal bleeding, and in extreme cases, obstruction and perforation.4 On CT imaging, colon lipomas will appear radiolucent, homogenous, and well circumscribed. The lesions usually do not warrant intervention unless they are symptomatic. If symptomatic, resection of the lesion is the first-line treatment and usually results in complete resolution of symptoms with no recurrence.2
While either a surgical or endoscopic approach may be used for resection, an increased risk of perforation of the colon with larger lipomas has been shown with endoscopic excision.5 With surgical resection, an open or minimally invasive approach may be offered, based on surgeon comfort with minimally invasive colon procedures. Minimally invasive colonic surgeries may be associated with a shorter length of stay, decreased postoperative pain, and faster return of bowel function. In this case, the surgeon chose an open approach due to the large size of the mass (11 cm) as well as location of the mass in the transverse colon, which made it easy to access directly through a small laparotomy incision made in the superior midline over the transverse colon.
When a colonic mesenchymal mass is seen on colonoscopy, it is important to consider other, nonbenign lesions that present this way. The most common malignant mesenchymal tumor of the GI tract is a gastrointestinal stromal tumor (GIST), a soft-tissue sarcoma that occurs predominantly in the stomach and small intestine.6 These tumors arise from the interstitial cells of Cajal (ICC) and are associated with mutations of KIT and PDGFR-α genes.7 The incidence in the United States is approximately 0.70 per 100,000 people per year, predominantly found in adults in their fifth or sixth decade of life.8 While this tumor typically occurs in the upper GI tract, very rarely, GISTs can be found in the colon.6 Common constitutional symptoms of colon GIST are similar to those of colon lipomas and include abdominal pain, changes in bowel habits, nausea, vomiting, and in some cases, weight loss.
CT imaging is often enough to differentiate a colon lipoma from a colon GIST. On CT, large GIST tumors tend to show irregular, lobulated margins, mucosal ulceration, central necrosis, cavitation, hemorrhage, and hypervascularity—vastly different from the CT findings of colon lipomas. If imaging is equivocal, an ultrasound-guided fine needle aspiration biopsy may be performed, differentiating GIST through the presence of ICC tumor cells as well as KIT and PDGFR-α proteins.
In our patient, colonoscopy showed a positive Cushion sign (tumor indented on depression with biopsy forceps), pathognomonic for a colon lipoma, and CT imaging showed a radiolucent, well-circumscribed lesion.9 This was more consistent with a colon lipoma than a GIST. Because the patient was symptomatic with a near obstructing lesion, the appropriate next step was removal of the lesion. Had this instead been a GIST tumor, a more extensive oncologic surgical resection would have been warranted, with adequate mesentery and lymph nodes collected.
This case is notable because colon lipomas exceeding 2 cm are rare and are usually an incidental finding on CT. However, larger lipomas can lead to symptoms, including obstruction if not removed in a timely manner.
A 62-year-old male veteran with a history of pulmonary embolism (PE) and prostate cancer status after brachytherapy presented to the emergency department with new onset shortness of breath and left-sided chest pain after prolonged car travel. He underwent a chest computed tomography (CT) angiogram that showed no PE recurrence; however, the scan revealed an incidental transverse colon mass that appeared well circumscribed, homogeneous, and radiolucent with no enhancement, septations, or hypervascularity but no evidence of colonic distension or obstruction (Figure 1).
- What is your diagnosis?
- How would you treat this patient?
The patient reported having chronic constipation and a dull, left-sided abdominal discomfort for the past year. He noted that his abdominal pain worsened after eating and mildly improved after taking castor oil. He had no surgical history and no family history of cancer. The patient reported no fever, fatigue, weight loss, chills, nausea, vomiting, diarrhea, hematochezia, dysuria, hematuria, or melena. Vital signs, physical examination, and initial routine laboratory work were all within appropriate ranges, and a fecal occult blood test was negative.
A colonoscopy was performed, revealing a near-obstructing submucosal mass in the transverse colon near the splenic flexure with a smooth surface and a positive Cushion (Pillow) sign (Figure 2). The patient underwent surgical exploration that resulted in finding a soft, 11-cm lesion arising from the mesenteric side of the transverse colon (Figure 3). Hematoxylin and eosin (H&E) stains were used on a sample from the mass (Figure 4).
The tumor was enucleated via a colotomy over the mass, and the colotomy repaired primarily. Gross examination revealed homogenous yellow fatty tissue, and the H&E stains showed mature, well-differentiated adipocytes with uniform nuclei surrounded by a fibrous capsule. Based on this pathologic examination, this patient was diagnosed with a lipoma of the transverse colon. The resected tissue showed negative margins, indicating full removal of the lipoma.
The patient stabilized well after surgery and remained under inpatient care for observation; due to lack of appetite following the surgery, the patient did not start eating solids again until 2 days after the lipoma removal. By postoperative day 4, the patient had return of bowel function and was tolerating a regular diet with no recurrence of his prandial pain, shortness of breath, or left-sided chest pain. While the precise cause of the patient’s initial presentation of shortness of breath and left-sided chest pain was not ascertained, it is likely that the lipoma, near completely obstructed his bowel, caused abdominal contents and distended intestines to push against his diaphragm, leading to pain and dyspnea. This was likely exacerbated by sensitization to these symptoms from his prior PE. He was discharged home on postoperative day 4 with outpatient follow-up with general surgery.
Discussion
Lipomas are common benign tumors arising from aberrantly multiplying adipocytes. Although lipomas are most commonly found subcutaneously, the lesions can occur anywhere along the gastrointestinal (GI) tract, most often in the colon.1 The incidence rate of colon lipomas ranges from 0.2 to 4.4% among patients in their fifth to sixth decades of life, more commonly found in females.2 These lesions are the most common submucosal mesenchymal lesions of the colon, with a predilection for the right ascending colon.1 The etiology of colon lipomas is largely unknown; one known cause is trauma, thought to induce cytokine release or HMGA2-LPP fusion gene arrangements leading to adipocyte proliferation.3
Most colon lipomas are asymptomatic and discovered incidentally; symptoms typically arise when the lesions are > 2 cm in diameter and include abdominal pain, changes in bowel habits, rectal bleeding, and in extreme cases, obstruction and perforation.4 On CT imaging, colon lipomas will appear radiolucent, homogenous, and well circumscribed. The lesions usually do not warrant intervention unless they are symptomatic. If symptomatic, resection of the lesion is the first-line treatment and usually results in complete resolution of symptoms with no recurrence.2
While either a surgical or endoscopic approach may be used for resection, an increased risk of perforation of the colon with larger lipomas has been shown with endoscopic excision.5 With surgical resection, an open or minimally invasive approach may be offered, based on surgeon comfort with minimally invasive colon procedures. Minimally invasive colonic surgeries may be associated with a shorter length of stay, decreased postoperative pain, and faster return of bowel function. In this case, the surgeon chose an open approach due to the large size of the mass (11 cm) as well as location of the mass in the transverse colon, which made it easy to access directly through a small laparotomy incision made in the superior midline over the transverse colon.
When a colonic mesenchymal mass is seen on colonoscopy, it is important to consider other, nonbenign lesions that present this way. The most common malignant mesenchymal tumor of the GI tract is a gastrointestinal stromal tumor (GIST), a soft-tissue sarcoma that occurs predominantly in the stomach and small intestine.6 These tumors arise from the interstitial cells of Cajal (ICC) and are associated with mutations of KIT and PDGFR-α genes.7 The incidence in the United States is approximately 0.70 per 100,000 people per year, predominantly found in adults in their fifth or sixth decade of life.8 While this tumor typically occurs in the upper GI tract, very rarely, GISTs can be found in the colon.6 Common constitutional symptoms of colon GIST are similar to those of colon lipomas and include abdominal pain, changes in bowel habits, nausea, vomiting, and in some cases, weight loss.
CT imaging is often enough to differentiate a colon lipoma from a colon GIST. On CT, large GIST tumors tend to show irregular, lobulated margins, mucosal ulceration, central necrosis, cavitation, hemorrhage, and hypervascularity—vastly different from the CT findings of colon lipomas. If imaging is equivocal, an ultrasound-guided fine needle aspiration biopsy may be performed, differentiating GIST through the presence of ICC tumor cells as well as KIT and PDGFR-α proteins.
In our patient, colonoscopy showed a positive Cushion sign (tumor indented on depression with biopsy forceps), pathognomonic for a colon lipoma, and CT imaging showed a radiolucent, well-circumscribed lesion.9 This was more consistent with a colon lipoma than a GIST. Because the patient was symptomatic with a near obstructing lesion, the appropriate next step was removal of the lesion. Had this instead been a GIST tumor, a more extensive oncologic surgical resection would have been warranted, with adequate mesentery and lymph nodes collected.
This case is notable because colon lipomas exceeding 2 cm are rare and are usually an incidental finding on CT. However, larger lipomas can lead to symptoms, including obstruction if not removed in a timely manner.
1. Nallamothu G, Adler DG. Large colonic lipomas. Gastroenterol Hepatol (NY). 2011;7(7):490-492.
2. Crocetti D, Sapienza P, Sterpetti AV, et al. Surgery for symptomatic colon lipoma: a systematic review of the literature. Anticancer Res. 2014;34(11):6271-6276.
3. Italiano A, Ebran N, Attias R, et al. NFIB rearrangement in superficial, retroperitoneal, and colonic lipomas with aberrations involving chromosome band 9p22. Genes Chromosomes Cancer. 2008;47(11):971-977. doi:10.1002/gcc.20602
4. Agrawal A, Singh KJ. Symptomatic intestinal lipomas: our experience. Med J Armed Forces India. 2011;67(4):374-376. doi:10.1016/S0377-1237(11)60090-7
5. Kim GW, Kwon CI, Song SH, et al. Endoscopic resection of giant colonic lipoma: case series with partial resection. Clin Endosc. 2013;46(5):586-590. doi:10.5946/ce.2013.46.5.586
6. Reddy RM, Fleshman JW. Colorectal gastrointestinal stromal tumors: a brief review. Clin Colon Rectal Surg. 2006;19(2):69-77. doi:10.1055/s-2006-942347
7. Shinomura Y, Kinoshita K, Tsutsui S, Hirota S. Pathophysiology, diagnosis, and treatment of gastrointestinal stromal tumors. J Gastroenterol. 2005;40(8):775-780. doi:10.1007/s00535-005-1674-0
8. Patel N, Benipal B. Incidence of gastrointestinal stromal tumors in the United States from 2001-2015: a United States cancer statistics analysis of 50 states. Cureus. 2019;11(2):e4120. Published 2019 Feb 22. doi:10.7759/cureus.4120
9. Kyawzaw K, Emmanuel O, Sandar L,2 Febin J,Naing LA, Madhavi R. Pillow sign in colonoscopy. MOJ Clin Med Case Rep. 2018;8(2):57-58. doi:10.15406/mojcr.2018.08.00240
1. Nallamothu G, Adler DG. Large colonic lipomas. Gastroenterol Hepatol (NY). 2011;7(7):490-492.
2. Crocetti D, Sapienza P, Sterpetti AV, et al. Surgery for symptomatic colon lipoma: a systematic review of the literature. Anticancer Res. 2014;34(11):6271-6276.
3. Italiano A, Ebran N, Attias R, et al. NFIB rearrangement in superficial, retroperitoneal, and colonic lipomas with aberrations involving chromosome band 9p22. Genes Chromosomes Cancer. 2008;47(11):971-977. doi:10.1002/gcc.20602
4. Agrawal A, Singh KJ. Symptomatic intestinal lipomas: our experience. Med J Armed Forces India. 2011;67(4):374-376. doi:10.1016/S0377-1237(11)60090-7
5. Kim GW, Kwon CI, Song SH, et al. Endoscopic resection of giant colonic lipoma: case series with partial resection. Clin Endosc. 2013;46(5):586-590. doi:10.5946/ce.2013.46.5.586
6. Reddy RM, Fleshman JW. Colorectal gastrointestinal stromal tumors: a brief review. Clin Colon Rectal Surg. 2006;19(2):69-77. doi:10.1055/s-2006-942347
7. Shinomura Y, Kinoshita K, Tsutsui S, Hirota S. Pathophysiology, diagnosis, and treatment of gastrointestinal stromal tumors. J Gastroenterol. 2005;40(8):775-780. doi:10.1007/s00535-005-1674-0
8. Patel N, Benipal B. Incidence of gastrointestinal stromal tumors in the United States from 2001-2015: a United States cancer statistics analysis of 50 states. Cureus. 2019;11(2):e4120. Published 2019 Feb 22. doi:10.7759/cureus.4120
9. Kyawzaw K, Emmanuel O, Sandar L,2 Febin J,Naing LA, Madhavi R. Pillow sign in colonoscopy. MOJ Clin Med Case Rep. 2018;8(2):57-58. doi:10.15406/mojcr.2018.08.00240
Thinking Outside the ‘Cage’
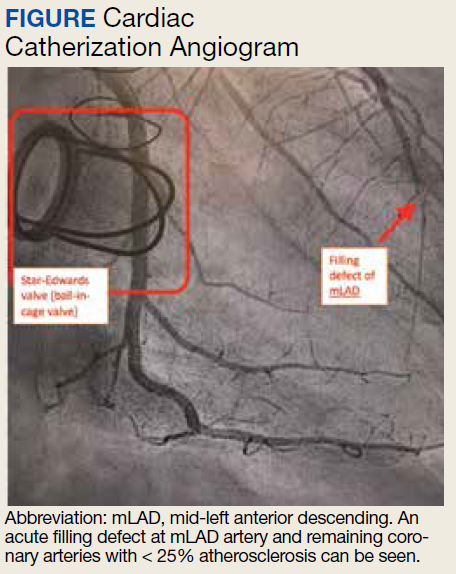
A 74-year-old male veteran presented at an urgent care clinic in Aguadilla, Puerto Rico, with a sharp, nonradiating, left-sided precordial chest pain that started while cleaning his house and gardening. The patient described the pain as 9 on the 10-point Wong-Baker FACES Pain Rating Scale, lasting about 5 to 10 minutes and was alleviated with rest. The patient’s medical history consisted of multiple comorbidities, including a mitral valve replacement with a Star-Edwards valve (ball in cage) in 1987. The electrocardiogram performed at the clinic showed no acute ischemic changes. Due to the persistent pain, the patient was transferred to Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, for further evaluation and management. On arrival, the patient had an international normalized ratio (INR) of 2.22; elevated high-sensitive troponin enzyme readings of 56 ng/L at 6:38 PM (0h); 61 ng/L at 7:38 PM (1h); and 83 ng/L at 9:47 PM (3h), reference range, 0-22 ng/L, and changes that prompted admission to the cardiac critical care unit. Two days later, a follow-up enzyme level was 52 ng/L. Cardiac catheterization revealed an acute filling defect at mid-left anterior descending artery and remaining coronary arteries with < 25% atherosclerosis (Figure). A myocardial perfusion study was performed for myocardial viability. The results showed a small, reversible perfusion defect involving the apical-septal wall with the remaining left ventricular myocardium appearing viable. Aspirin was added to the patient’s anticoagulation regimen of warfarin. Once target INR was reached, the patient was discharged home without recurrence of angina.
- What is your diagnosis?
- How would you treat this patient?
Acute coronary syndrome (ACS) consists of clinical suspicion of myocardial ischemia or laboratory confirmation of myocardial infarction (MI). ACS includes 3 major entities: non-ST elevation MI (NSTEMI), unstable angina, and ST-elevation MI (STEMI). ACS usually occurs as a result of a reduced supply of oxygenated blood to the myocardium, which is caused by restriction or occlusion of at least 1 of the coronary arteries. This alteration in blood flow is commonly secondary to a rupture of an atherosclerotic plaque or spontaneous dissection of a coronary artery. In rare cases, this reduction in blood flow is caused by a coronary embolism (CE) arising from a prosthetic heart valve.1,2
One of the first descriptions of CE was provided by Rudolf Virchow in the 1850s from postmortem autopsy findings.3 At that time, these coronary findings were associated with intracardiac mural thrombus or infective endocarditis. During the 1940s, CE was described in living patients who had survived a MI, and outcomes were not as catastrophic as originally believed. In the 1960s, a higher than usual association between prosthetic valves and CE was suspected and later confirmed by the invention and implementation of coronary angiography. Multiple studies have been published that confirm the association between prosthetic valves (especially in the mitral position), atrial fibrillation (AF), and a higher than usual rate of CEs.4,5
Discussion
The prevalence of this disease has varied during the years. Data from autopsies of patients with ACS and evidence of thromboembolic material in coronary arteries originally estimated a prevalence as high as 13%.6,7 After the invention of diagnostic angiography, consensus studies have established the prevalence to be approximately 3% in patient with ACS.1 The prevalence may be higher in patient with significant risk factors that may increase the probability of CEs, like prosthetic heart valves and AF.2
In 2015 Shibata and colleagues proposed a scoring system for the diagnosis of CE. The scoring system consisted of major and minor criteria.6 Diagnosis of CE is established by ≥ 2 major criteria; 1 major and 2 minor; or ≥ 3 minor criteria. This scoring system increases the diagnostic probability of the disease.1,6
The major criteria are angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components (met by this patient); concomitant coronary emboli in multiple coronary vascular territories; concomitant systemic embolization without left ventricular thrombus attributable to acute MI; histological evidence of venous origin of coronary embolic material; and evidence of an embolic source based on transthoracic echocardiography, transesophageal echocardiography, computed tomography, or magnetic resonance imaging.1,6 The minor criteria are 25% stenosis on coronary angiography except for the culprit lesion (met by this patient); presence of emboli risk factors, such as prosthetic heart valve (met by this patient); and AF.1,6
Management of CE remains controversial; aspiration of thrombus may be considered in the acute setting and with evidence of a heavy thrombus formation. This may allow for restoration of flow and retrieval of thrombus formation for histopathologic evaluation. However, it is important to mention that in the setting of STEMI, aspiration has been shown to increase risk of stroke and lead to increased morbidity. If aspiration of thrombus provides good restoration of flow, there is no need for further percutaneous intervention. Benefits of aspiration in low thrombus burden are not well established and do not provide any additional benefit compared with those of anticoagulation.6-11
Anticoagulation should be initiated in patients with AF and low bleeding risk, even when CHA2DS2-VASc (congestive heart failure, hypertension, aged ≥ 75 years, diabetes mellitus, stroke or transient ischemic attack, vascular disease, aged 65 to 74 years, sex category) score is low. In patients with prolonged immobilization, recent surgery, pregnancy, use of oral contraceptives/tamoxifen, or other reversible risks, 3 months of anticoagulation has been shown to be sufficient. In the setting of active cancer or known thrombophilia, prolonged anticoagulation is recommended. Thrombophilia testing is not recommended in the setting of CE.1
The America College of Cardiology/American Heart Association guidelines for valvular heart disease recommend that patients with mechanical prosthetic aortic valves should be started on a vitamin K antagonist with a target INR of 2 to 3. (Class 1A). Prosthetic mitral and high thromboembolic valves require a higher INR target above 3.0. The addition of antiplatelet agents, such as aspirin in doses of 75 to 100 mg, should be started to decrease risk of thromboembolic disease in all patients with prosthetic heart valves.12
CE is not a common cause of ACS. Nevertheless, it was considered in the differential diagnosis of this patient, and diagnostic criteria were reviewed. This patient met the diagnostic criteria for a definitive diagnosis of CE. These included 1 major and 2 minor criteria: angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components; < 25% stenosis on coronary angiography except for the culprit lesion; and presence of emboli risk factors (prosthetic heart valve).
CE is rare, and review of the literature reveals that it accounts for < 3% of all ACS cases. Despite its rarity, it is important to recognize its risk factors, which include prosthetic heart valves, valvuloplasty, vasculitis, AF, left ventricular aneurysm, and endocarditis. The difference in treatment between CE and the most frequently encountered etiologies of ACS reveals the importance in recognizing this syndrome. Management of CE remains controversial. Nevertheless, when the culprit lesion is located in a distal portion of the vessel involved, as was seen in our patient, and in cases where there is a low thrombi burden, anticoagulation instead of thrombectomy is usually preferred. Patients with prosthetic mechanical valves have a high incidence of thromboembolism. This sometimes leads to thrombi formation in uncommon locations. Guidelines of therapy in these patients recommend that all prosthetic mechanical valves should be treated with both antiplatelet and anticoagulation therapies to reduce the risk of thrombi formation.
Conclusion
Physicians involved in diagnosing ACS should be aware of the risk factors for CE and always consider it while evaluating patients and developing the differential diagnosis.
1. Raphael CE, Heit JA, Reeder GS, et al. Coronary embolus: an underappreciated cause of acute coronary syndromes. JACC Cardiovasc Interv. 2018;11(2):172-180. doi:10.1016/j.jcin.2017.08.057
2. Popovic B, Agrinier N, Bouchahda N, et al. Coronary embolism among ST-segment-elevation myocardial infarction patients: mechanisms and management. Circ Cardiovasc Interv. 2018;11(1):e005587. doi:10.1161/CIRCINTERVENTIONS.117.005587
3. Oakley C, Yusuf R, Hollman A. Coronary embolism and angina in mitral stenosis. Br Heart J. 1961;23(4):357-369. doi:10.1136/hrt.23.4.357
4. Charles RG, Epstein EJ. Diagnosis of coronary embolism: a review. J R Soc Med. 1983;76(10):863-869.
5. Bawell MB, Moragues V, Shrader EL. Coronary embolism. Circulation. 1956;14(6):1159-1163. doi:10.1161/01.cir.14.6.1159
6. Shibata T, Kawakami S, Noguchi T, et al. Prevalence, clinical features, and prognosis of acute myocardial infarction attributable to coronary artery embolism. Circulation. 2015;132(4):241-250. doi:10.1161/CIRCULATIONAHA.114.015134
7. Prizel KR, Hutchins GM, Bulkley BH. Coronary artery embolism and myocardial infarction. Ann Intern Med. 1978;88(2):155-161. doi:10.7326/0003-4819-88-2-155
8. Lacunza-Ruiz FJ, Muñoz-Esparza C, García-de-Lara J. Coronary embolism and thrombosis of prosthetic mitral valve. JACC Cardiovasc Interv. 2014;7(10):e127-e128. doi:10.1016/j.jcin.2014.02.025
9. Jolly SS, Cairns JA, Yusuf S, et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet. 2016;387(10014):127-135. doi:10.1016/S0140-6736(15)00448-1
10. Fröbert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction [published correction appears in N Engl J Med. 2014 Aug 21;371(8):786]. N Engl J Med. 2013;369(17):1587-1597. doi:10.1056/NEJMoa1308789
11. Kalçık M, Yesin M, Gürsoy MO, Karakoyun S, Özkan M. Treatment strategies for prosthetic valve thrombosis-derived coronary embolism. JACC Cardiovasc Interv. 2015;8(5):756-757. doi:10.1016/j.jcin.2014.11.019
12. Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC focused update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. doi:10.1161/CIR.0000000000000503
A 74-year-old male veteran presented at an urgent care clinic in Aguadilla, Puerto Rico, with a sharp, nonradiating, left-sided precordial chest pain that started while cleaning his house and gardening. The patient described the pain as 9 on the 10-point Wong-Baker FACES Pain Rating Scale, lasting about 5 to 10 minutes and was alleviated with rest. The patient’s medical history consisted of multiple comorbidities, including a mitral valve replacement with a Star-Edwards valve (ball in cage) in 1987. The electrocardiogram performed at the clinic showed no acute ischemic changes. Due to the persistent pain, the patient was transferred to Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, for further evaluation and management. On arrival, the patient had an international normalized ratio (INR) of 2.22; elevated high-sensitive troponin enzyme readings of 56 ng/L at 6:38 PM (0h); 61 ng/L at 7:38 PM (1h); and 83 ng/L at 9:47 PM (3h), reference range, 0-22 ng/L, and changes that prompted admission to the cardiac critical care unit. Two days later, a follow-up enzyme level was 52 ng/L. Cardiac catheterization revealed an acute filling defect at mid-left anterior descending artery and remaining coronary arteries with < 25% atherosclerosis (Figure). A myocardial perfusion study was performed for myocardial viability. The results showed a small, reversible perfusion defect involving the apical-septal wall with the remaining left ventricular myocardium appearing viable. Aspirin was added to the patient’s anticoagulation regimen of warfarin. Once target INR was reached, the patient was discharged home without recurrence of angina.
- What is your diagnosis?
- How would you treat this patient?
Acute coronary syndrome (ACS) consists of clinical suspicion of myocardial ischemia or laboratory confirmation of myocardial infarction (MI). ACS includes 3 major entities: non-ST elevation MI (NSTEMI), unstable angina, and ST-elevation MI (STEMI). ACS usually occurs as a result of a reduced supply of oxygenated blood to the myocardium, which is caused by restriction or occlusion of at least 1 of the coronary arteries. This alteration in blood flow is commonly secondary to a rupture of an atherosclerotic plaque or spontaneous dissection of a coronary artery. In rare cases, this reduction in blood flow is caused by a coronary embolism (CE) arising from a prosthetic heart valve.1,2
One of the first descriptions of CE was provided by Rudolf Virchow in the 1850s from postmortem autopsy findings.3 At that time, these coronary findings were associated with intracardiac mural thrombus or infective endocarditis. During the 1940s, CE was described in living patients who had survived a MI, and outcomes were not as catastrophic as originally believed. In the 1960s, a higher than usual association between prosthetic valves and CE was suspected and later confirmed by the invention and implementation of coronary angiography. Multiple studies have been published that confirm the association between prosthetic valves (especially in the mitral position), atrial fibrillation (AF), and a higher than usual rate of CEs.4,5
Discussion
The prevalence of this disease has varied during the years. Data from autopsies of patients with ACS and evidence of thromboembolic material in coronary arteries originally estimated a prevalence as high as 13%.6,7 After the invention of diagnostic angiography, consensus studies have established the prevalence to be approximately 3% in patient with ACS.1 The prevalence may be higher in patient with significant risk factors that may increase the probability of CEs, like prosthetic heart valves and AF.2
In 2015 Shibata and colleagues proposed a scoring system for the diagnosis of CE. The scoring system consisted of major and minor criteria.6 Diagnosis of CE is established by ≥ 2 major criteria; 1 major and 2 minor; or ≥ 3 minor criteria. This scoring system increases the diagnostic probability of the disease.1,6
The major criteria are angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components (met by this patient); concomitant coronary emboli in multiple coronary vascular territories; concomitant systemic embolization without left ventricular thrombus attributable to acute MI; histological evidence of venous origin of coronary embolic material; and evidence of an embolic source based on transthoracic echocardiography, transesophageal echocardiography, computed tomography, or magnetic resonance imaging.1,6 The minor criteria are 25% stenosis on coronary angiography except for the culprit lesion (met by this patient); presence of emboli risk factors, such as prosthetic heart valve (met by this patient); and AF.1,6
Management of CE remains controversial; aspiration of thrombus may be considered in the acute setting and with evidence of a heavy thrombus formation. This may allow for restoration of flow and retrieval of thrombus formation for histopathologic evaluation. However, it is important to mention that in the setting of STEMI, aspiration has been shown to increase risk of stroke and lead to increased morbidity. If aspiration of thrombus provides good restoration of flow, there is no need for further percutaneous intervention. Benefits of aspiration in low thrombus burden are not well established and do not provide any additional benefit compared with those of anticoagulation.6-11
Anticoagulation should be initiated in patients with AF and low bleeding risk, even when CHA2DS2-VASc (congestive heart failure, hypertension, aged ≥ 75 years, diabetes mellitus, stroke or transient ischemic attack, vascular disease, aged 65 to 74 years, sex category) score is low. In patients with prolonged immobilization, recent surgery, pregnancy, use of oral contraceptives/tamoxifen, or other reversible risks, 3 months of anticoagulation has been shown to be sufficient. In the setting of active cancer or known thrombophilia, prolonged anticoagulation is recommended. Thrombophilia testing is not recommended in the setting of CE.1
The America College of Cardiology/American Heart Association guidelines for valvular heart disease recommend that patients with mechanical prosthetic aortic valves should be started on a vitamin K antagonist with a target INR of 2 to 3. (Class 1A). Prosthetic mitral and high thromboembolic valves require a higher INR target above 3.0. The addition of antiplatelet agents, such as aspirin in doses of 75 to 100 mg, should be started to decrease risk of thromboembolic disease in all patients with prosthetic heart valves.12
CE is not a common cause of ACS. Nevertheless, it was considered in the differential diagnosis of this patient, and diagnostic criteria were reviewed. This patient met the diagnostic criteria for a definitive diagnosis of CE. These included 1 major and 2 minor criteria: angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components; < 25% stenosis on coronary angiography except for the culprit lesion; and presence of emboli risk factors (prosthetic heart valve).
CE is rare, and review of the literature reveals that it accounts for < 3% of all ACS cases. Despite its rarity, it is important to recognize its risk factors, which include prosthetic heart valves, valvuloplasty, vasculitis, AF, left ventricular aneurysm, and endocarditis. The difference in treatment between CE and the most frequently encountered etiologies of ACS reveals the importance in recognizing this syndrome. Management of CE remains controversial. Nevertheless, when the culprit lesion is located in a distal portion of the vessel involved, as was seen in our patient, and in cases where there is a low thrombi burden, anticoagulation instead of thrombectomy is usually preferred. Patients with prosthetic mechanical valves have a high incidence of thromboembolism. This sometimes leads to thrombi formation in uncommon locations. Guidelines of therapy in these patients recommend that all prosthetic mechanical valves should be treated with both antiplatelet and anticoagulation therapies to reduce the risk of thrombi formation.
Conclusion
Physicians involved in diagnosing ACS should be aware of the risk factors for CE and always consider it while evaluating patients and developing the differential diagnosis.
A 74-year-old male veteran presented at an urgent care clinic in Aguadilla, Puerto Rico, with a sharp, nonradiating, left-sided precordial chest pain that started while cleaning his house and gardening. The patient described the pain as 9 on the 10-point Wong-Baker FACES Pain Rating Scale, lasting about 5 to 10 minutes and was alleviated with rest. The patient’s medical history consisted of multiple comorbidities, including a mitral valve replacement with a Star-Edwards valve (ball in cage) in 1987. The electrocardiogram performed at the clinic showed no acute ischemic changes. Due to the persistent pain, the patient was transferred to Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, for further evaluation and management. On arrival, the patient had an international normalized ratio (INR) of 2.22; elevated high-sensitive troponin enzyme readings of 56 ng/L at 6:38 PM (0h); 61 ng/L at 7:38 PM (1h); and 83 ng/L at 9:47 PM (3h), reference range, 0-22 ng/L, and changes that prompted admission to the cardiac critical care unit. Two days later, a follow-up enzyme level was 52 ng/L. Cardiac catheterization revealed an acute filling defect at mid-left anterior descending artery and remaining coronary arteries with < 25% atherosclerosis (Figure). A myocardial perfusion study was performed for myocardial viability. The results showed a small, reversible perfusion defect involving the apical-septal wall with the remaining left ventricular myocardium appearing viable. Aspirin was added to the patient’s anticoagulation regimen of warfarin. Once target INR was reached, the patient was discharged home without recurrence of angina.
- What is your diagnosis?
- How would you treat this patient?
Acute coronary syndrome (ACS) consists of clinical suspicion of myocardial ischemia or laboratory confirmation of myocardial infarction (MI). ACS includes 3 major entities: non-ST elevation MI (NSTEMI), unstable angina, and ST-elevation MI (STEMI). ACS usually occurs as a result of a reduced supply of oxygenated blood to the myocardium, which is caused by restriction or occlusion of at least 1 of the coronary arteries. This alteration in blood flow is commonly secondary to a rupture of an atherosclerotic plaque or spontaneous dissection of a coronary artery. In rare cases, this reduction in blood flow is caused by a coronary embolism (CE) arising from a prosthetic heart valve.1,2
One of the first descriptions of CE was provided by Rudolf Virchow in the 1850s from postmortem autopsy findings.3 At that time, these coronary findings were associated with intracardiac mural thrombus or infective endocarditis. During the 1940s, CE was described in living patients who had survived a MI, and outcomes were not as catastrophic as originally believed. In the 1960s, a higher than usual association between prosthetic valves and CE was suspected and later confirmed by the invention and implementation of coronary angiography. Multiple studies have been published that confirm the association between prosthetic valves (especially in the mitral position), atrial fibrillation (AF), and a higher than usual rate of CEs.4,5
Discussion
The prevalence of this disease has varied during the years. Data from autopsies of patients with ACS and evidence of thromboembolic material in coronary arteries originally estimated a prevalence as high as 13%.6,7 After the invention of diagnostic angiography, consensus studies have established the prevalence to be approximately 3% in patient with ACS.1 The prevalence may be higher in patient with significant risk factors that may increase the probability of CEs, like prosthetic heart valves and AF.2
In 2015 Shibata and colleagues proposed a scoring system for the diagnosis of CE. The scoring system consisted of major and minor criteria.6 Diagnosis of CE is established by ≥ 2 major criteria; 1 major and 2 minor; or ≥ 3 minor criteria. This scoring system increases the diagnostic probability of the disease.1,6
The major criteria are angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components (met by this patient); concomitant coronary emboli in multiple coronary vascular territories; concomitant systemic embolization without left ventricular thrombus attributable to acute MI; histological evidence of venous origin of coronary embolic material; and evidence of an embolic source based on transthoracic echocardiography, transesophageal echocardiography, computed tomography, or magnetic resonance imaging.1,6 The minor criteria are 25% stenosis on coronary angiography except for the culprit lesion (met by this patient); presence of emboli risk factors, such as prosthetic heart valve (met by this patient); and AF.1,6
Management of CE remains controversial; aspiration of thrombus may be considered in the acute setting and with evidence of a heavy thrombus formation. This may allow for restoration of flow and retrieval of thrombus formation for histopathologic evaluation. However, it is important to mention that in the setting of STEMI, aspiration has been shown to increase risk of stroke and lead to increased morbidity. If aspiration of thrombus provides good restoration of flow, there is no need for further percutaneous intervention. Benefits of aspiration in low thrombus burden are not well established and do not provide any additional benefit compared with those of anticoagulation.6-11
Anticoagulation should be initiated in patients with AF and low bleeding risk, even when CHA2DS2-VASc (congestive heart failure, hypertension, aged ≥ 75 years, diabetes mellitus, stroke or transient ischemic attack, vascular disease, aged 65 to 74 years, sex category) score is low. In patients with prolonged immobilization, recent surgery, pregnancy, use of oral contraceptives/tamoxifen, or other reversible risks, 3 months of anticoagulation has been shown to be sufficient. In the setting of active cancer or known thrombophilia, prolonged anticoagulation is recommended. Thrombophilia testing is not recommended in the setting of CE.1
The America College of Cardiology/American Heart Association guidelines for valvular heart disease recommend that patients with mechanical prosthetic aortic valves should be started on a vitamin K antagonist with a target INR of 2 to 3. (Class 1A). Prosthetic mitral and high thromboembolic valves require a higher INR target above 3.0. The addition of antiplatelet agents, such as aspirin in doses of 75 to 100 mg, should be started to decrease risk of thromboembolic disease in all patients with prosthetic heart valves.12
CE is not a common cause of ACS. Nevertheless, it was considered in the differential diagnosis of this patient, and diagnostic criteria were reviewed. This patient met the diagnostic criteria for a definitive diagnosis of CE. These included 1 major and 2 minor criteria: angiographic evidence of coronary artery embolism and thrombosis without atherosclerotic components; < 25% stenosis on coronary angiography except for the culprit lesion; and presence of emboli risk factors (prosthetic heart valve).
CE is rare, and review of the literature reveals that it accounts for < 3% of all ACS cases. Despite its rarity, it is important to recognize its risk factors, which include prosthetic heart valves, valvuloplasty, vasculitis, AF, left ventricular aneurysm, and endocarditis. The difference in treatment between CE and the most frequently encountered etiologies of ACS reveals the importance in recognizing this syndrome. Management of CE remains controversial. Nevertheless, when the culprit lesion is located in a distal portion of the vessel involved, as was seen in our patient, and in cases where there is a low thrombi burden, anticoagulation instead of thrombectomy is usually preferred. Patients with prosthetic mechanical valves have a high incidence of thromboembolism. This sometimes leads to thrombi formation in uncommon locations. Guidelines of therapy in these patients recommend that all prosthetic mechanical valves should be treated with both antiplatelet and anticoagulation therapies to reduce the risk of thrombi formation.
Conclusion
Physicians involved in diagnosing ACS should be aware of the risk factors for CE and always consider it while evaluating patients and developing the differential diagnosis.
1. Raphael CE, Heit JA, Reeder GS, et al. Coronary embolus: an underappreciated cause of acute coronary syndromes. JACC Cardiovasc Interv. 2018;11(2):172-180. doi:10.1016/j.jcin.2017.08.057
2. Popovic B, Agrinier N, Bouchahda N, et al. Coronary embolism among ST-segment-elevation myocardial infarction patients: mechanisms and management. Circ Cardiovasc Interv. 2018;11(1):e005587. doi:10.1161/CIRCINTERVENTIONS.117.005587
3. Oakley C, Yusuf R, Hollman A. Coronary embolism and angina in mitral stenosis. Br Heart J. 1961;23(4):357-369. doi:10.1136/hrt.23.4.357
4. Charles RG, Epstein EJ. Diagnosis of coronary embolism: a review. J R Soc Med. 1983;76(10):863-869.
5. Bawell MB, Moragues V, Shrader EL. Coronary embolism. Circulation. 1956;14(6):1159-1163. doi:10.1161/01.cir.14.6.1159
6. Shibata T, Kawakami S, Noguchi T, et al. Prevalence, clinical features, and prognosis of acute myocardial infarction attributable to coronary artery embolism. Circulation. 2015;132(4):241-250. doi:10.1161/CIRCULATIONAHA.114.015134
7. Prizel KR, Hutchins GM, Bulkley BH. Coronary artery embolism and myocardial infarction. Ann Intern Med. 1978;88(2):155-161. doi:10.7326/0003-4819-88-2-155
8. Lacunza-Ruiz FJ, Muñoz-Esparza C, García-de-Lara J. Coronary embolism and thrombosis of prosthetic mitral valve. JACC Cardiovasc Interv. 2014;7(10):e127-e128. doi:10.1016/j.jcin.2014.02.025
9. Jolly SS, Cairns JA, Yusuf S, et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet. 2016;387(10014):127-135. doi:10.1016/S0140-6736(15)00448-1
10. Fröbert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction [published correction appears in N Engl J Med. 2014 Aug 21;371(8):786]. N Engl J Med. 2013;369(17):1587-1597. doi:10.1056/NEJMoa1308789
11. Kalçık M, Yesin M, Gürsoy MO, Karakoyun S, Özkan M. Treatment strategies for prosthetic valve thrombosis-derived coronary embolism. JACC Cardiovasc Interv. 2015;8(5):756-757. doi:10.1016/j.jcin.2014.11.019
12. Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC focused update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. doi:10.1161/CIR.0000000000000503
1. Raphael CE, Heit JA, Reeder GS, et al. Coronary embolus: an underappreciated cause of acute coronary syndromes. JACC Cardiovasc Interv. 2018;11(2):172-180. doi:10.1016/j.jcin.2017.08.057
2. Popovic B, Agrinier N, Bouchahda N, et al. Coronary embolism among ST-segment-elevation myocardial infarction patients: mechanisms and management. Circ Cardiovasc Interv. 2018;11(1):e005587. doi:10.1161/CIRCINTERVENTIONS.117.005587
3. Oakley C, Yusuf R, Hollman A. Coronary embolism and angina in mitral stenosis. Br Heart J. 1961;23(4):357-369. doi:10.1136/hrt.23.4.357
4. Charles RG, Epstein EJ. Diagnosis of coronary embolism: a review. J R Soc Med. 1983;76(10):863-869.
5. Bawell MB, Moragues V, Shrader EL. Coronary embolism. Circulation. 1956;14(6):1159-1163. doi:10.1161/01.cir.14.6.1159
6. Shibata T, Kawakami S, Noguchi T, et al. Prevalence, clinical features, and prognosis of acute myocardial infarction attributable to coronary artery embolism. Circulation. 2015;132(4):241-250. doi:10.1161/CIRCULATIONAHA.114.015134
7. Prizel KR, Hutchins GM, Bulkley BH. Coronary artery embolism and myocardial infarction. Ann Intern Med. 1978;88(2):155-161. doi:10.7326/0003-4819-88-2-155
8. Lacunza-Ruiz FJ, Muñoz-Esparza C, García-de-Lara J. Coronary embolism and thrombosis of prosthetic mitral valve. JACC Cardiovasc Interv. 2014;7(10):e127-e128. doi:10.1016/j.jcin.2014.02.025
9. Jolly SS, Cairns JA, Yusuf S, et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet. 2016;387(10014):127-135. doi:10.1016/S0140-6736(15)00448-1
10. Fröbert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction [published correction appears in N Engl J Med. 2014 Aug 21;371(8):786]. N Engl J Med. 2013;369(17):1587-1597. doi:10.1056/NEJMoa1308789
11. Kalçık M, Yesin M, Gürsoy MO, Karakoyun S, Özkan M. Treatment strategies for prosthetic valve thrombosis-derived coronary embolism. JACC Cardiovasc Interv. 2015;8(5):756-757. doi:10.1016/j.jcin.2014.11.019
12. Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC focused update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. doi:10.1161/CIR.0000000000000503
Albuterol, Acidosis, and Aneurysms
A patient with a complicated medical history on admission for dyspnea was administered nebulizer therapy but after 72 hours developed asymptomatic acute kidney injury and anion-gap metabolic acidosis.
An 88-year-old male veteran with a medical history of chronic obstructive pulmonary disease (COPD) on home oxygen, chronic alcohol use, squamous cell carcinoma of the lung status after left upper lobectomy, and a 5.7 cm thoracic aortic aneurysm was admitted to the inpatient medical service for progressive dyspnea and productive cough. The patient was in his usual state of health until 2 days before presentation. A chest computed tomography scan showed a right lower lobe infiltrate, concerning for pneumonia, and stable thoracic aortic aneurysm (Figure). On admission, the patient was started on IV ceftriaxone 2 g daily for pneumonia and
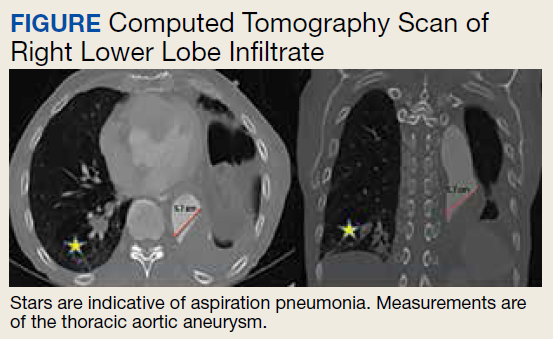
The patient responded well to therapy, and his cough and dyspnea improved. However, 72 hours after admission, he developed an asymptomatic acute kidney injury (AKI) and anion-gap metabolic acidosis. His serum creatinine increased from baseline 0.6 mg/dL to 1.2 mg/dL. He also had an anion gap of 21 mmol/L and a decrease in bicarbonate from 23 mmol/L to 17 mmol/L. His condition was further complicated by new-onset hypertension (153/111 mm Hg). His calculated fractional excretion of sodium (FENa) was 0.5%, and his lactate level returned elevated at 3.6 mmol/L. On further investigation, he reported alcohol use the night prior; however, his β-hydroxybutyrate was negative, and serum alcohol level was undetectable. Meanwhile, the patient continued to receive antibiotics and scheduled nebulizer treatments. Although his AKI resolved with initial fluid resuscitation, his repeat lactate levels continued to trend upward to a peak of 4.0 mmol/L.
- What is your diagnosis?
- How would you treat this patient?
Although IV fluids resolved his AKI, prerenal in etiology given the calculated FENa at 0.5%, his lactate levels continued to uptrend to a peak of 4.0 mmol/L complicated by elevated blood pressure (BP) > 150/100 mm Hg. Given his thoracic aneurysm, his BP was treated with metoprolol tartrate and amlodipine 10 mg daily. The patient remained asymptomatic with no evidence of ischemia or sepsis.
We suspected the nebulizer treatments to be the etiology of the patient’s hyperlactatemia and subsequent anion-gap metabolic acidosis. His scheduled albuterol and ipratropium nebulizer treatments were discontinued, and the patient experienced rapid resolution of his anion gap and hyperlactatemia to 1.2 mmol/L over 24 hours. On discontinuation of the nebulization therapy, mild wheezing was noted on physical examination. The patient reported no symptoms and was at his baseline. The patient finished his antibiotic course for his community-acquired pneumonia and was discharged in stable condition with instructions to continue his previously established home COPD medication regimen of umeclidinium/vilanterol 62.5/25 mcg daily and albuterol metered-dose inhaler as needed.
Discussion
Short-acting β-agonists, such as albuterol, are widely used in COPD and are a guideline-recommended treatment in maintenance and exacerbation of asthma and COPD.1 Short-acting β-agonist adverse effects (AEs) include nausea, vomiting, tremors, headache, and tachycardia; abnormal laboratory results include hypocalcemia, hypokalemia, hypophosphatemia, hypomagnesemia, and hyperglycemia.2,3 Albuterol-induced hyperlactatemia and lactic acidosis also are known but often overlooked and underreported AEs.
In a randomized control trial, researchers identified a positive correlation between nebulized albuterol use and hyperlactatemia in asthmatics with asthma exacerbation.4 One systematic review identified ≤ 20% of patients on either IV or nebulized high-dose treatments with selective β2-agonists may experience hyperlactatemia.5 However, aerosolized administration of albuterol as opposed to IV administration is less likely to result in AEs and abnormal laboratory results given decreased systemic absorption.3
Hyperlactatemia and lactic acidosis are associated with increased morbidity and mortality.6 Lactic acidosis is classified as either type A or type B. Type A lactic acidosis is characterized by hypoperfusion as subsequent ischemic injuries lead to anaerobic metabolism and elevated lactate. Diseases such as septic, cardiogenic, and hypovolemic shock are often associated with type A lactic acidosis. Type B lactic acidosis, however, encapsulates all nonhypoperfusion-related elevations in lactate, including malignancy, ethanol intoxication, and medication-induced lactic acidosis.7,8
In this case, the diagnosis was elusive as the patient had multiple comorbidities. His history included COPD, which is associated with elevated lactate levels.5 However, his initial laboratory workup did not show an anion gap, confirming a lack of an underlying acidotic process on admission. Because the patient was admitted for pneumonia, a known infectious source, complicated by an acute elevation in lactate, sepsis must be and was effectively ruled out. The patient also reported alcohol use during his admission, which confounded his presentation but was unlikely to impact the etiology of his lactic acidosis, given the unremarkable β-hydroxybutyrate and serum alcohol levels.
Furthermore, the patient harbored an enlarged thoracic aortic aneurysm and remained hypertensive above the goal of BP 130/80 mm Hg for patients with thoracoabdominal aneurysms.9 Lactic acidosis in the context of hemodynamic instability for this patient might have indicated tissue hypoperfusion secondary to a ruptured aneurysm or aortic dissection. Fortunately, the patient did not manifest any signs or symptoms suggestive of a ruptured aortic aneurysm. Last, on discontinuing the nebulizer therapy, the patient’s hyperlactatemia resolved within 24 hours, highly indicative of albuterol-induced lactic acidosis as the proper diagnosis.
As a β-agonist, albuterol stimulates β-adrenergic receptors, which increases lipolysis and glycolysis. The biochemical reactions increase the product pyruvate, which is used in both aerobic and anaerobic metabolisms. With an increase in pyruvate, capacity for aerobic metabolism is maximized with increased shunting toward anaerobic metabolism, leading to elevated lactate levels and lactic acidosis.8,10,11
Regardless, albuterol-induced lactic acidosis is a diagnosis of exclusion.6 It is thus prudent to rule out life-threatening etiologies of hyperlactatemia, given the association with increased morbidity and mortality. This case illustrates the importance of ruling out life-threatening etiologies of hyperlactatemia and lactic acidosis in an older patient with multiple comorbidities. This case also recognizes the acute AEs of hyperlactatemia and lactic acidosis secondary to scheduled albuterol nebulization therapy in acutely ill patients. Of note, patients presenting with an acute medical illness may be more susceptible to hyperlactatemia secondary to scheduled albuterol nebulization therapy.
Conclusions
We encourage heightened clinical suspicion of albuterol-induced lactic acidosis in acutely ill patients with COPD on albuterol therapy on rule out of life-threatening etiologies and
1. Global Initiative for Asthma. Pocket Guide to COPD Diagnosis, Management, and Prevention: A Guide for Health Care Professionals (2020 Report). Global Initiative for Chronic Lung Diseases, Inc; 2020. Accessed April 16, 2021. https://goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf
2. Jat KR, Khairwa A. Levalbuterol versus albuterol for acute asthma: a systematic review and meta-analysis. Pulm Pharmacol Ther. 2013;26(2):239-248. doi:10.1016/j.pupt.2012.11.003
3. Ahrens RC, Smith GD. Albuterol: an adrenergic agent for use in the treatment of asthma pharmacology, pharmacokinetics and clinical use. Pharmacotherapy. 1984;4(3):105- 121. doi:10.1002/j.1875-9114.1984.tb03330.x
4. Lewis LM, Ferguson I, House SL, et al. Albuterol administration is commonly associated with increases in serum lactate in patients with asthma treated for acute exacerbation of asthma. Chest. 2014;145(1):53-59. doi:10.1378/chest.13-0930
5. Liedtke AG, Lava SAG, Milani GP, et al. Selective β2-adrenoceptor agonists and relevant hyperlactatemia: systematic review and meta-analysis. J Clin Med. 2019;9(1):71. doi:10.3390/jcm9010071
6. Smith ZR, Horng M, Rech MA. Medication-induced hyperlactatemia and lactic acidosis: a systematic review of the literature. Pharmacotherapy. 2019;39(9):946-963. doi:10.1002/phar.2316
7. Hockstein M, Diercks D. Significant lactic acidosis from albuterol. Clin Pract Cases Emerg Med. 2018;2(2):128-131. doi:10.5811/cpcem.2018.1.36024
8. Foucher CD, Tubben RE. Lactic acidosis. StatPearls Publishing; 2020. Updated November 21, 2020. Accessed April 16, 2021. https://www.ncbi.nlm.nih.gov/books/NBK470202
9. Aronow WS. Treatment of thoracic aortic aneurysm. Ann Transl Med. 2018;6(3):66. doi:10.21037/atm.2018.01.07
10. Lau E, Mazer J, Carino G. Inhaled β-agonist therapy and respiratory muscle fatigue as under-recognised causes of lactic acidosis. BMJ Case Rep. 2013;2013:bcr2013201015. Published October 14, 2013. doi:10.1136/bcr-2013-201015
11. Ramakrishna KN, Virk J, Gambhir HS. Albuterol-induced lactic acidosis. Am J Ther. 2019;26(5):e635-e636. doi:10.1097/MJT.0000000000000843
A patient with a complicated medical history on admission for dyspnea was administered nebulizer therapy but after 72 hours developed asymptomatic acute kidney injury and anion-gap metabolic acidosis.
A patient with a complicated medical history on admission for dyspnea was administered nebulizer therapy but after 72 hours developed asymptomatic acute kidney injury and anion-gap metabolic acidosis.
An 88-year-old male veteran with a medical history of chronic obstructive pulmonary disease (COPD) on home oxygen, chronic alcohol use, squamous cell carcinoma of the lung status after left upper lobectomy, and a 5.7 cm thoracic aortic aneurysm was admitted to the inpatient medical service for progressive dyspnea and productive cough. The patient was in his usual state of health until 2 days before presentation. A chest computed tomography scan showed a right lower lobe infiltrate, concerning for pneumonia, and stable thoracic aortic aneurysm (Figure). On admission, the patient was started on IV ceftriaxone 2 g daily for pneumonia and
The patient responded well to therapy, and his cough and dyspnea improved. However, 72 hours after admission, he developed an asymptomatic acute kidney injury (AKI) and anion-gap metabolic acidosis. His serum creatinine increased from baseline 0.6 mg/dL to 1.2 mg/dL. He also had an anion gap of 21 mmol/L and a decrease in bicarbonate from 23 mmol/L to 17 mmol/L. His condition was further complicated by new-onset hypertension (153/111 mm Hg). His calculated fractional excretion of sodium (FENa) was 0.5%, and his lactate level returned elevated at 3.6 mmol/L. On further investigation, he reported alcohol use the night prior; however, his β-hydroxybutyrate was negative, and serum alcohol level was undetectable. Meanwhile, the patient continued to receive antibiotics and scheduled nebulizer treatments. Although his AKI resolved with initial fluid resuscitation, his repeat lactate levels continued to trend upward to a peak of 4.0 mmol/L.
- What is your diagnosis?
- How would you treat this patient?
Although IV fluids resolved his AKI, prerenal in etiology given the calculated FENa at 0.5%, his lactate levels continued to uptrend to a peak of 4.0 mmol/L complicated by elevated blood pressure (BP) > 150/100 mm Hg. Given his thoracic aneurysm, his BP was treated with metoprolol tartrate and amlodipine 10 mg daily. The patient remained asymptomatic with no evidence of ischemia or sepsis.
We suspected the nebulizer treatments to be the etiology of the patient’s hyperlactatemia and subsequent anion-gap metabolic acidosis. His scheduled albuterol and ipratropium nebulizer treatments were discontinued, and the patient experienced rapid resolution of his anion gap and hyperlactatemia to 1.2 mmol/L over 24 hours. On discontinuation of the nebulization therapy, mild wheezing was noted on physical examination. The patient reported no symptoms and was at his baseline. The patient finished his antibiotic course for his community-acquired pneumonia and was discharged in stable condition with instructions to continue his previously established home COPD medication regimen of umeclidinium/vilanterol 62.5/25 mcg daily and albuterol metered-dose inhaler as needed.
Discussion
Short-acting β-agonists, such as albuterol, are widely used in COPD and are a guideline-recommended treatment in maintenance and exacerbation of asthma and COPD.1 Short-acting β-agonist adverse effects (AEs) include nausea, vomiting, tremors, headache, and tachycardia; abnormal laboratory results include hypocalcemia, hypokalemia, hypophosphatemia, hypomagnesemia, and hyperglycemia.2,3 Albuterol-induced hyperlactatemia and lactic acidosis also are known but often overlooked and underreported AEs.
In a randomized control trial, researchers identified a positive correlation between nebulized albuterol use and hyperlactatemia in asthmatics with asthma exacerbation.4 One systematic review identified ≤ 20% of patients on either IV or nebulized high-dose treatments with selective β2-agonists may experience hyperlactatemia.5 However, aerosolized administration of albuterol as opposed to IV administration is less likely to result in AEs and abnormal laboratory results given decreased systemic absorption.3
Hyperlactatemia and lactic acidosis are associated with increased morbidity and mortality.6 Lactic acidosis is classified as either type A or type B. Type A lactic acidosis is characterized by hypoperfusion as subsequent ischemic injuries lead to anaerobic metabolism and elevated lactate. Diseases such as septic, cardiogenic, and hypovolemic shock are often associated with type A lactic acidosis. Type B lactic acidosis, however, encapsulates all nonhypoperfusion-related elevations in lactate, including malignancy, ethanol intoxication, and medication-induced lactic acidosis.7,8
In this case, the diagnosis was elusive as the patient had multiple comorbidities. His history included COPD, which is associated with elevated lactate levels.5 However, his initial laboratory workup did not show an anion gap, confirming a lack of an underlying acidotic process on admission. Because the patient was admitted for pneumonia, a known infectious source, complicated by an acute elevation in lactate, sepsis must be and was effectively ruled out. The patient also reported alcohol use during his admission, which confounded his presentation but was unlikely to impact the etiology of his lactic acidosis, given the unremarkable β-hydroxybutyrate and serum alcohol levels.
Furthermore, the patient harbored an enlarged thoracic aortic aneurysm and remained hypertensive above the goal of BP 130/80 mm Hg for patients with thoracoabdominal aneurysms.9 Lactic acidosis in the context of hemodynamic instability for this patient might have indicated tissue hypoperfusion secondary to a ruptured aneurysm or aortic dissection. Fortunately, the patient did not manifest any signs or symptoms suggestive of a ruptured aortic aneurysm. Last, on discontinuing the nebulizer therapy, the patient’s hyperlactatemia resolved within 24 hours, highly indicative of albuterol-induced lactic acidosis as the proper diagnosis.
As a β-agonist, albuterol stimulates β-adrenergic receptors, which increases lipolysis and glycolysis. The biochemical reactions increase the product pyruvate, which is used in both aerobic and anaerobic metabolisms. With an increase in pyruvate, capacity for aerobic metabolism is maximized with increased shunting toward anaerobic metabolism, leading to elevated lactate levels and lactic acidosis.8,10,11
Regardless, albuterol-induced lactic acidosis is a diagnosis of exclusion.6 It is thus prudent to rule out life-threatening etiologies of hyperlactatemia, given the association with increased morbidity and mortality. This case illustrates the importance of ruling out life-threatening etiologies of hyperlactatemia and lactic acidosis in an older patient with multiple comorbidities. This case also recognizes the acute AEs of hyperlactatemia and lactic acidosis secondary to scheduled albuterol nebulization therapy in acutely ill patients. Of note, patients presenting with an acute medical illness may be more susceptible to hyperlactatemia secondary to scheduled albuterol nebulization therapy.
Conclusions
We encourage heightened clinical suspicion of albuterol-induced lactic acidosis in acutely ill patients with COPD on albuterol therapy on rule out of life-threatening etiologies and
An 88-year-old male veteran with a medical history of chronic obstructive pulmonary disease (COPD) on home oxygen, chronic alcohol use, squamous cell carcinoma of the lung status after left upper lobectomy, and a 5.7 cm thoracic aortic aneurysm was admitted to the inpatient medical service for progressive dyspnea and productive cough. The patient was in his usual state of health until 2 days before presentation. A chest computed tomography scan showed a right lower lobe infiltrate, concerning for pneumonia, and stable thoracic aortic aneurysm (Figure). On admission, the patient was started on IV ceftriaxone 2 g daily for pneumonia and
The patient responded well to therapy, and his cough and dyspnea improved. However, 72 hours after admission, he developed an asymptomatic acute kidney injury (AKI) and anion-gap metabolic acidosis. His serum creatinine increased from baseline 0.6 mg/dL to 1.2 mg/dL. He also had an anion gap of 21 mmol/L and a decrease in bicarbonate from 23 mmol/L to 17 mmol/L. His condition was further complicated by new-onset hypertension (153/111 mm Hg). His calculated fractional excretion of sodium (FENa) was 0.5%, and his lactate level returned elevated at 3.6 mmol/L. On further investigation, he reported alcohol use the night prior; however, his β-hydroxybutyrate was negative, and serum alcohol level was undetectable. Meanwhile, the patient continued to receive antibiotics and scheduled nebulizer treatments. Although his AKI resolved with initial fluid resuscitation, his repeat lactate levels continued to trend upward to a peak of 4.0 mmol/L.
- What is your diagnosis?
- How would you treat this patient?
Although IV fluids resolved his AKI, prerenal in etiology given the calculated FENa at 0.5%, his lactate levels continued to uptrend to a peak of 4.0 mmol/L complicated by elevated blood pressure (BP) > 150/100 mm Hg. Given his thoracic aneurysm, his BP was treated with metoprolol tartrate and amlodipine 10 mg daily. The patient remained asymptomatic with no evidence of ischemia or sepsis.
We suspected the nebulizer treatments to be the etiology of the patient’s hyperlactatemia and subsequent anion-gap metabolic acidosis. His scheduled albuterol and ipratropium nebulizer treatments were discontinued, and the patient experienced rapid resolution of his anion gap and hyperlactatemia to 1.2 mmol/L over 24 hours. On discontinuation of the nebulization therapy, mild wheezing was noted on physical examination. The patient reported no symptoms and was at his baseline. The patient finished his antibiotic course for his community-acquired pneumonia and was discharged in stable condition with instructions to continue his previously established home COPD medication regimen of umeclidinium/vilanterol 62.5/25 mcg daily and albuterol metered-dose inhaler as needed.
Discussion
Short-acting β-agonists, such as albuterol, are widely used in COPD and are a guideline-recommended treatment in maintenance and exacerbation of asthma and COPD.1 Short-acting β-agonist adverse effects (AEs) include nausea, vomiting, tremors, headache, and tachycardia; abnormal laboratory results include hypocalcemia, hypokalemia, hypophosphatemia, hypomagnesemia, and hyperglycemia.2,3 Albuterol-induced hyperlactatemia and lactic acidosis also are known but often overlooked and underreported AEs.
In a randomized control trial, researchers identified a positive correlation between nebulized albuterol use and hyperlactatemia in asthmatics with asthma exacerbation.4 One systematic review identified ≤ 20% of patients on either IV or nebulized high-dose treatments with selective β2-agonists may experience hyperlactatemia.5 However, aerosolized administration of albuterol as opposed to IV administration is less likely to result in AEs and abnormal laboratory results given decreased systemic absorption.3
Hyperlactatemia and lactic acidosis are associated with increased morbidity and mortality.6 Lactic acidosis is classified as either type A or type B. Type A lactic acidosis is characterized by hypoperfusion as subsequent ischemic injuries lead to anaerobic metabolism and elevated lactate. Diseases such as septic, cardiogenic, and hypovolemic shock are often associated with type A lactic acidosis. Type B lactic acidosis, however, encapsulates all nonhypoperfusion-related elevations in lactate, including malignancy, ethanol intoxication, and medication-induced lactic acidosis.7,8
In this case, the diagnosis was elusive as the patient had multiple comorbidities. His history included COPD, which is associated with elevated lactate levels.5 However, his initial laboratory workup did not show an anion gap, confirming a lack of an underlying acidotic process on admission. Because the patient was admitted for pneumonia, a known infectious source, complicated by an acute elevation in lactate, sepsis must be and was effectively ruled out. The patient also reported alcohol use during his admission, which confounded his presentation but was unlikely to impact the etiology of his lactic acidosis, given the unremarkable β-hydroxybutyrate and serum alcohol levels.
Furthermore, the patient harbored an enlarged thoracic aortic aneurysm and remained hypertensive above the goal of BP 130/80 mm Hg for patients with thoracoabdominal aneurysms.9 Lactic acidosis in the context of hemodynamic instability for this patient might have indicated tissue hypoperfusion secondary to a ruptured aneurysm or aortic dissection. Fortunately, the patient did not manifest any signs or symptoms suggestive of a ruptured aortic aneurysm. Last, on discontinuing the nebulizer therapy, the patient’s hyperlactatemia resolved within 24 hours, highly indicative of albuterol-induced lactic acidosis as the proper diagnosis.
As a β-agonist, albuterol stimulates β-adrenergic receptors, which increases lipolysis and glycolysis. The biochemical reactions increase the product pyruvate, which is used in both aerobic and anaerobic metabolisms. With an increase in pyruvate, capacity for aerobic metabolism is maximized with increased shunting toward anaerobic metabolism, leading to elevated lactate levels and lactic acidosis.8,10,11
Regardless, albuterol-induced lactic acidosis is a diagnosis of exclusion.6 It is thus prudent to rule out life-threatening etiologies of hyperlactatemia, given the association with increased morbidity and mortality. This case illustrates the importance of ruling out life-threatening etiologies of hyperlactatemia and lactic acidosis in an older patient with multiple comorbidities. This case also recognizes the acute AEs of hyperlactatemia and lactic acidosis secondary to scheduled albuterol nebulization therapy in acutely ill patients. Of note, patients presenting with an acute medical illness may be more susceptible to hyperlactatemia secondary to scheduled albuterol nebulization therapy.
Conclusions
We encourage heightened clinical suspicion of albuterol-induced lactic acidosis in acutely ill patients with COPD on albuterol therapy on rule out of life-threatening etiologies and
1. Global Initiative for Asthma. Pocket Guide to COPD Diagnosis, Management, and Prevention: A Guide for Health Care Professionals (2020 Report). Global Initiative for Chronic Lung Diseases, Inc; 2020. Accessed April 16, 2021. https://goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf
2. Jat KR, Khairwa A. Levalbuterol versus albuterol for acute asthma: a systematic review and meta-analysis. Pulm Pharmacol Ther. 2013;26(2):239-248. doi:10.1016/j.pupt.2012.11.003
3. Ahrens RC, Smith GD. Albuterol: an adrenergic agent for use in the treatment of asthma pharmacology, pharmacokinetics and clinical use. Pharmacotherapy. 1984;4(3):105- 121. doi:10.1002/j.1875-9114.1984.tb03330.x
4. Lewis LM, Ferguson I, House SL, et al. Albuterol administration is commonly associated with increases in serum lactate in patients with asthma treated for acute exacerbation of asthma. Chest. 2014;145(1):53-59. doi:10.1378/chest.13-0930
5. Liedtke AG, Lava SAG, Milani GP, et al. Selective β2-adrenoceptor agonists and relevant hyperlactatemia: systematic review and meta-analysis. J Clin Med. 2019;9(1):71. doi:10.3390/jcm9010071
6. Smith ZR, Horng M, Rech MA. Medication-induced hyperlactatemia and lactic acidosis: a systematic review of the literature. Pharmacotherapy. 2019;39(9):946-963. doi:10.1002/phar.2316
7. Hockstein M, Diercks D. Significant lactic acidosis from albuterol. Clin Pract Cases Emerg Med. 2018;2(2):128-131. doi:10.5811/cpcem.2018.1.36024
8. Foucher CD, Tubben RE. Lactic acidosis. StatPearls Publishing; 2020. Updated November 21, 2020. Accessed April 16, 2021. https://www.ncbi.nlm.nih.gov/books/NBK470202
9. Aronow WS. Treatment of thoracic aortic aneurysm. Ann Transl Med. 2018;6(3):66. doi:10.21037/atm.2018.01.07
10. Lau E, Mazer J, Carino G. Inhaled β-agonist therapy and respiratory muscle fatigue as under-recognised causes of lactic acidosis. BMJ Case Rep. 2013;2013:bcr2013201015. Published October 14, 2013. doi:10.1136/bcr-2013-201015
11. Ramakrishna KN, Virk J, Gambhir HS. Albuterol-induced lactic acidosis. Am J Ther. 2019;26(5):e635-e636. doi:10.1097/MJT.0000000000000843
1. Global Initiative for Asthma. Pocket Guide to COPD Diagnosis, Management, and Prevention: A Guide for Health Care Professionals (2020 Report). Global Initiative for Chronic Lung Diseases, Inc; 2020. Accessed April 16, 2021. https://goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf
2. Jat KR, Khairwa A. Levalbuterol versus albuterol for acute asthma: a systematic review and meta-analysis. Pulm Pharmacol Ther. 2013;26(2):239-248. doi:10.1016/j.pupt.2012.11.003
3. Ahrens RC, Smith GD. Albuterol: an adrenergic agent for use in the treatment of asthma pharmacology, pharmacokinetics and clinical use. Pharmacotherapy. 1984;4(3):105- 121. doi:10.1002/j.1875-9114.1984.tb03330.x
4. Lewis LM, Ferguson I, House SL, et al. Albuterol administration is commonly associated with increases in serum lactate in patients with asthma treated for acute exacerbation of asthma. Chest. 2014;145(1):53-59. doi:10.1378/chest.13-0930
5. Liedtke AG, Lava SAG, Milani GP, et al. Selective β2-adrenoceptor agonists and relevant hyperlactatemia: systematic review and meta-analysis. J Clin Med. 2019;9(1):71. doi:10.3390/jcm9010071
6. Smith ZR, Horng M, Rech MA. Medication-induced hyperlactatemia and lactic acidosis: a systematic review of the literature. Pharmacotherapy. 2019;39(9):946-963. doi:10.1002/phar.2316
7. Hockstein M, Diercks D. Significant lactic acidosis from albuterol. Clin Pract Cases Emerg Med. 2018;2(2):128-131. doi:10.5811/cpcem.2018.1.36024
8. Foucher CD, Tubben RE. Lactic acidosis. StatPearls Publishing; 2020. Updated November 21, 2020. Accessed April 16, 2021. https://www.ncbi.nlm.nih.gov/books/NBK470202
9. Aronow WS. Treatment of thoracic aortic aneurysm. Ann Transl Med. 2018;6(3):66. doi:10.21037/atm.2018.01.07
10. Lau E, Mazer J, Carino G. Inhaled β-agonist therapy and respiratory muscle fatigue as under-recognised causes of lactic acidosis. BMJ Case Rep. 2013;2013:bcr2013201015. Published October 14, 2013. doi:10.1136/bcr-2013-201015
11. Ramakrishna KN, Virk J, Gambhir HS. Albuterol-induced lactic acidosis. Am J Ther. 2019;26(5):e635-e636. doi:10.1097/MJT.0000000000000843
Gastrointestinal Symptoms and Lactic Acidosis in a Chronic Marijuana User
A 57-year-old woman with a history of traumatic brain injury, posttraumatic stress disorder, depression, migraines, hypothyroidism, and a hiatal hernia repair presented to the emergency department with a 1-day history of nausea, vomiting, and diffuse abdominal pain. She reported that her symptoms were relieved by hot showers. She also reported having similar symptoms and a previous gastric-emptying study that showed a slow-emptying stomach. Her history also consisted of frequent cannabis use for mood and appetite stimulation along with eliminating meat and fish from her diet, an increase in consumption of simple carbohydrates in the past year, and no alcohol use. Her medications included topiramate 100 mg and clonidine 0.3 mg nightly for migraines; levothyroxine 200 mcg daily for hypothyroidism; tizanidine 4 mg twice a day for muscle spasm; famotidine 40 mg twice a day as needed for gastric reflux; and bupropion 50 mg daily, citalopram 20 mg daily, and lamotrigine 25 mg nightly for mood.
The patient’s physical examination was notable for bradycardia (43 beats/min) and epigastric tenderness. Admission laboratory results were notable for an elevated lactic acid level of 4.8 (normal range, 0.50-2.20) mmol/L and a leukocytosis count of 10.8×109 cells/L. Serum alcohol level and blood cultures were negative. Liver function test, hemoglobin A1c, and lipase test were unremarkable. Her electrocardiogram showed an unchanged right bundle branch block. Chest X-ray, computed tomography (CT) of her abdomen/pelvis and echocardiogram were unremarkable.
What is your diagnosis?
How would you treat this patient?
This patient was diagnosed with gastrointestinal beriberi. Because of her dietary changes, lactic acidosis, and bradycardia, thiamine deficiency was suspected after ruling out other possibilities on the differential diagnosis (Table). The patient’s symptoms resolved after administration of high-dose IV thiamine 500 mg 3 times daily for 4 days. Her white blood cell count and lactic acid level normalized. Unfortunately, thiamine levels were not obtained for the patient before treatment was initiated. After administration of IV thiamine, her plasma thiamine level was > 1,200 (normal range, 8-30) nmol/L.
Her differential diagnosis included infectious etiology. Given her leukocytosis and lactic acidosis, vancomycin and piperacillin/tazobactam were started on admission. One day later, her leukocytosis count doubled to 20.7×109 cells/L. However, after 48 hours of negative blood cultures, antibiotics were discontinued.
Small bowel obstruction was suspected due to the patient’s history of abdominal surgery but was ruled out with CT imaging. Similarly, pancreatitis was ruled out based on negative CT imaging and the patient’s normal lipase level. Gastroparesis also was considered because of the patient’s history of hypothyroidism, tobacco use, and her prior gastric-emptying study. The patient was treated for gastroparesis with a course of metoclopramide and erythromycin without improvement in symptoms. Additionally, gastroparesis would not explain the patient’s leukocytosis.
Cannabinoid hyperemesis syndrome (CHS) was suspected because the patient’s symptoms improved with cannabis discontinuation and hot showers.1 In chronic users, however, tetrahydrocannabinol levels have a half-life of 5 to 13 days.2 Although lactic acidosis and leukocytosis have been previously reported with cannabis use, it is unlikely that the patient would have such significant improvement within the first 4 days after discontinuation.1,3,4 Although the patient had many psychiatric comorbidities with previous hospitalizations describing concern for somatization disorder, her leukocytosis and elevated lactic acid levels were suggestive of an organic rather than a psychiatric etiology of her symptoms.
Discussion
Gastrointestinal beriberi has been reported in chronic cannabis users who present with nausea, vomiting, epigastric pain, leukocytosis, and lactic acidosis; all these symptoms rapidly improve after thiamine administration.5,6 The patient’s dietary change also eliminated her intake of vitamin B12, which compounded her condition. Thiamine deficiency produces lactic acidosis by disrupting pyruvate metabolism.7 Bradycardia also can be a sign of thiamine deficiency, although the patient’s use of clonidine for migraines is a confounder.8
Chronically ill patients are prone to nutritional deficiencies, including deficiencies of thiamine.7,9 Many patients with chronic illnesses also use cannabis to ameliorate physical and neuropsychiatric symptoms.2 Recent reports suggest cannabis users are prone to gastrointestinal beriberi and Wernicke encephalopathy.5,10 Treating gastrointestinal symptoms in these patients can be challenging to diagnose because gastrointestinal beriberi and CHS share many clinical manifestations.
The patient’s presentation is likely multifactorial resulting from the combination of gastrointestinal beriberi and CHS. However, thiamine deficiency seems to play the dominant role.
There is no standard treatment regimen for thiamine deficiency with neurologic deficits, and patients only retain about 10 to 15% of intramuscular (IM) injections of cyanocobalamin.11,12 The British Committee for Standards in Haematology recommends IM injections of 1,000 mcg of cyanocobalamin 3 times a week for 2 weeks and then reassess the need for continued treatment.13 The British Columbia guidelines also recommend IM injections of 1,000 mcg daily for 1 to 5 days before transitioning to oral repletion.14 European Neurology guidelines for the treatment of Wernicke encephalopathy recommend IV cyanocobalamin 200 mg 3 times daily.15 Low-level evidence with observational studies informs these decisions and is why there is variation.
The patient’s serum lactate and leukocytosis normalized 1 day after the administration of thiamine. Thiamine deficiency classically causes Wernicke encephalopathy and wet beriberi.16 The patient did not present with Wernicke encephalopathy’s triad: ophthalmoplegia, ataxia, or confusion. She also was euvolemic without signs or symptoms of wet beriberi.
Conclusions
Thiamine deficiency is principally a clinical diagnosis. Thiamine laboratory testing may not be readily available in all medical centers, and confirming a diagnosis of thiamine deficiency should not delay treatment when thiamine deficiency is suspected. This patient’s thiamine levels resulted a week after collection. The administration of thiamine before sampling also can alter the result as it did in this case. Additionally, laboratories may offer whole blood and serum testing. Whole blood testing is more accurate because most bioactive thiamine is found in red blood cells.17
1. Price SL, Fisher C, Kumar R, Hilgerson A. Cannabinoid hyperemesis syndrome as the underlying cause of intractable nausea and vomiting. J Am Osteopath Assoc. 2011;111(3):166-169. doi:10.7556/jaoa.2011.111.3.166
2. Sharma P, Murthy P, Bharath MM. Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry. 2012;7(4):149-156.
3. Antill T, Jakkoju A, Dieguez J, Laskhmiprasad L. Lactic acidosis: a rare manifestation of synthetic marijuana intoxication. J La State Med Soc. 2015;167(3):155.
4. Sullivan S. Cannabinoid hyperemesis. Can J Gastroenterol. 2010;24(5):284-285. doi:10.1155/2010/481940
5. Duca J, Lum CJ, Lo AM. Elevated lactate secondary to gastrointestinal beriberi. J Gen Intern Med. 2016;31(1):133-136. doi:10.1007/s11606-015-3326-2
6. Prakash S. Gastrointestinal beriberi: a forme fruste of Wernicke’s encephalopathy? BMJ Case Rep. 2018;bcr2018224841. doi:10.1136/bcr-2018-224841
7. Friedenberg AS, Brandoff DE, Schiffman FJ. Type B lactic acidosis as a severe metabolic complication in lymphoma and leukemia: a case series from a single institution and literature review. Medicine (Baltimore). 2007;86(4):225-232. doi:10.1097/MD.0b013e318125759a
8. Liang CC. Bradycardia in thiamin deficiency and the role of glyoxylate. J Nutrition Sci Vitaminology. 1977;23(1):1-6. doi:10.3177/jnsv.23.1
9. Attaluri P, Castillo A, Edriss H, Nugent K. Thiamine deficiency: an important consideration in critically ill patients. Am J Med Sci. 2018;356(4):382-390. doi:10.1016/j.amjms.2018.06.015
10. Chaudhari A, Li ZY, Long A, Afshinnik A. Heavy cannabis use associated with Wernicke’s encephalopathy. Cureus. 2019;11(7):e5109. doi:10.7759/cureus.5109
11. Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149-160. doi:10.1056/NEJMcp1113996
12. Green R, Allen LH, Bjørke-Monsen A-L, et al. Vitamin B12 deficiency. Nat Rev Dis Primers. 2017;3(1):17040. doi:10.1038/nrdp.2017.40
13. Devalia V, Hamilton MS, Molloy AM. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
14. British Columbia Ministry of Health; Guidelines and Protocols and Advisory Committee. Guidelines and protocols cobalamin (vitamin B12) deficiency–investigation & management. Effective January 1, 2012. Revised May 1, 2013. Accessed March 10, 2021. https://www2.gov.bc.ca/gov/content/health/practitioner-professional-resources/bc-guidelines/vitamin-b12
15. Galvin R, Brathen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010;17(12):1408-1418. doi:10.1111/j.1468-1331.2010.03153.x
16. Wiley KD, Gupta M. Vitamin B1 thiamine deficiency (beriberi). In: StatPearls. StatPearls Publishing LLC; 2019.
17. Jenco J, Krcmova LK, Solichova D, Solich P. Recent trends in determination of thiamine and its derivatives in clinical practice. J Chromatogra A. 2017;1510:1-12. doi:10.1016/j.chroma.2017.06.048
A 57-year-old woman with a history of traumatic brain injury, posttraumatic stress disorder, depression, migraines, hypothyroidism, and a hiatal hernia repair presented to the emergency department with a 1-day history of nausea, vomiting, and diffuse abdominal pain. She reported that her symptoms were relieved by hot showers. She also reported having similar symptoms and a previous gastric-emptying study that showed a slow-emptying stomach. Her history also consisted of frequent cannabis use for mood and appetite stimulation along with eliminating meat and fish from her diet, an increase in consumption of simple carbohydrates in the past year, and no alcohol use. Her medications included topiramate 100 mg and clonidine 0.3 mg nightly for migraines; levothyroxine 200 mcg daily for hypothyroidism; tizanidine 4 mg twice a day for muscle spasm; famotidine 40 mg twice a day as needed for gastric reflux; and bupropion 50 mg daily, citalopram 20 mg daily, and lamotrigine 25 mg nightly for mood.
The patient’s physical examination was notable for bradycardia (43 beats/min) and epigastric tenderness. Admission laboratory results were notable for an elevated lactic acid level of 4.8 (normal range, 0.50-2.20) mmol/L and a leukocytosis count of 10.8×109 cells/L. Serum alcohol level and blood cultures were negative. Liver function test, hemoglobin A1c, and lipase test were unremarkable. Her electrocardiogram showed an unchanged right bundle branch block. Chest X-ray, computed tomography (CT) of her abdomen/pelvis and echocardiogram were unremarkable.
What is your diagnosis?
How would you treat this patient?
This patient was diagnosed with gastrointestinal beriberi. Because of her dietary changes, lactic acidosis, and bradycardia, thiamine deficiency was suspected after ruling out other possibilities on the differential diagnosis (Table). The patient’s symptoms resolved after administration of high-dose IV thiamine 500 mg 3 times daily for 4 days. Her white blood cell count and lactic acid level normalized. Unfortunately, thiamine levels were not obtained for the patient before treatment was initiated. After administration of IV thiamine, her plasma thiamine level was > 1,200 (normal range, 8-30) nmol/L.
Her differential diagnosis included infectious etiology. Given her leukocytosis and lactic acidosis, vancomycin and piperacillin/tazobactam were started on admission. One day later, her leukocytosis count doubled to 20.7×109 cells/L. However, after 48 hours of negative blood cultures, antibiotics were discontinued.
Small bowel obstruction was suspected due to the patient’s history of abdominal surgery but was ruled out with CT imaging. Similarly, pancreatitis was ruled out based on negative CT imaging and the patient’s normal lipase level. Gastroparesis also was considered because of the patient’s history of hypothyroidism, tobacco use, and her prior gastric-emptying study. The patient was treated for gastroparesis with a course of metoclopramide and erythromycin without improvement in symptoms. Additionally, gastroparesis would not explain the patient’s leukocytosis.
Cannabinoid hyperemesis syndrome (CHS) was suspected because the patient’s symptoms improved with cannabis discontinuation and hot showers.1 In chronic users, however, tetrahydrocannabinol levels have a half-life of 5 to 13 days.2 Although lactic acidosis and leukocytosis have been previously reported with cannabis use, it is unlikely that the patient would have such significant improvement within the first 4 days after discontinuation.1,3,4 Although the patient had many psychiatric comorbidities with previous hospitalizations describing concern for somatization disorder, her leukocytosis and elevated lactic acid levels were suggestive of an organic rather than a psychiatric etiology of her symptoms.
Discussion
Gastrointestinal beriberi has been reported in chronic cannabis users who present with nausea, vomiting, epigastric pain, leukocytosis, and lactic acidosis; all these symptoms rapidly improve after thiamine administration.5,6 The patient’s dietary change also eliminated her intake of vitamin B12, which compounded her condition. Thiamine deficiency produces lactic acidosis by disrupting pyruvate metabolism.7 Bradycardia also can be a sign of thiamine deficiency, although the patient’s use of clonidine for migraines is a confounder.8
Chronically ill patients are prone to nutritional deficiencies, including deficiencies of thiamine.7,9 Many patients with chronic illnesses also use cannabis to ameliorate physical and neuropsychiatric symptoms.2 Recent reports suggest cannabis users are prone to gastrointestinal beriberi and Wernicke encephalopathy.5,10 Treating gastrointestinal symptoms in these patients can be challenging to diagnose because gastrointestinal beriberi and CHS share many clinical manifestations.
The patient’s presentation is likely multifactorial resulting from the combination of gastrointestinal beriberi and CHS. However, thiamine deficiency seems to play the dominant role.
There is no standard treatment regimen for thiamine deficiency with neurologic deficits, and patients only retain about 10 to 15% of intramuscular (IM) injections of cyanocobalamin.11,12 The British Committee for Standards in Haematology recommends IM injections of 1,000 mcg of cyanocobalamin 3 times a week for 2 weeks and then reassess the need for continued treatment.13 The British Columbia guidelines also recommend IM injections of 1,000 mcg daily for 1 to 5 days before transitioning to oral repletion.14 European Neurology guidelines for the treatment of Wernicke encephalopathy recommend IV cyanocobalamin 200 mg 3 times daily.15 Low-level evidence with observational studies informs these decisions and is why there is variation.
The patient’s serum lactate and leukocytosis normalized 1 day after the administration of thiamine. Thiamine deficiency classically causes Wernicke encephalopathy and wet beriberi.16 The patient did not present with Wernicke encephalopathy’s triad: ophthalmoplegia, ataxia, or confusion. She also was euvolemic without signs or symptoms of wet beriberi.
Conclusions
Thiamine deficiency is principally a clinical diagnosis. Thiamine laboratory testing may not be readily available in all medical centers, and confirming a diagnosis of thiamine deficiency should not delay treatment when thiamine deficiency is suspected. This patient’s thiamine levels resulted a week after collection. The administration of thiamine before sampling also can alter the result as it did in this case. Additionally, laboratories may offer whole blood and serum testing. Whole blood testing is more accurate because most bioactive thiamine is found in red blood cells.17
A 57-year-old woman with a history of traumatic brain injury, posttraumatic stress disorder, depression, migraines, hypothyroidism, and a hiatal hernia repair presented to the emergency department with a 1-day history of nausea, vomiting, and diffuse abdominal pain. She reported that her symptoms were relieved by hot showers. She also reported having similar symptoms and a previous gastric-emptying study that showed a slow-emptying stomach. Her history also consisted of frequent cannabis use for mood and appetite stimulation along with eliminating meat and fish from her diet, an increase in consumption of simple carbohydrates in the past year, and no alcohol use. Her medications included topiramate 100 mg and clonidine 0.3 mg nightly for migraines; levothyroxine 200 mcg daily for hypothyroidism; tizanidine 4 mg twice a day for muscle spasm; famotidine 40 mg twice a day as needed for gastric reflux; and bupropion 50 mg daily, citalopram 20 mg daily, and lamotrigine 25 mg nightly for mood.
The patient’s physical examination was notable for bradycardia (43 beats/min) and epigastric tenderness. Admission laboratory results were notable for an elevated lactic acid level of 4.8 (normal range, 0.50-2.20) mmol/L and a leukocytosis count of 10.8×109 cells/L. Serum alcohol level and blood cultures were negative. Liver function test, hemoglobin A1c, and lipase test were unremarkable. Her electrocardiogram showed an unchanged right bundle branch block. Chest X-ray, computed tomography (CT) of her abdomen/pelvis and echocardiogram were unremarkable.
What is your diagnosis?
How would you treat this patient?
This patient was diagnosed with gastrointestinal beriberi. Because of her dietary changes, lactic acidosis, and bradycardia, thiamine deficiency was suspected after ruling out other possibilities on the differential diagnosis (Table). The patient’s symptoms resolved after administration of high-dose IV thiamine 500 mg 3 times daily for 4 days. Her white blood cell count and lactic acid level normalized. Unfortunately, thiamine levels were not obtained for the patient before treatment was initiated. After administration of IV thiamine, her plasma thiamine level was > 1,200 (normal range, 8-30) nmol/L.
Her differential diagnosis included infectious etiology. Given her leukocytosis and lactic acidosis, vancomycin and piperacillin/tazobactam were started on admission. One day later, her leukocytosis count doubled to 20.7×109 cells/L. However, after 48 hours of negative blood cultures, antibiotics were discontinued.
Small bowel obstruction was suspected due to the patient’s history of abdominal surgery but was ruled out with CT imaging. Similarly, pancreatitis was ruled out based on negative CT imaging and the patient’s normal lipase level. Gastroparesis also was considered because of the patient’s history of hypothyroidism, tobacco use, and her prior gastric-emptying study. The patient was treated for gastroparesis with a course of metoclopramide and erythromycin without improvement in symptoms. Additionally, gastroparesis would not explain the patient’s leukocytosis.
Cannabinoid hyperemesis syndrome (CHS) was suspected because the patient’s symptoms improved with cannabis discontinuation and hot showers.1 In chronic users, however, tetrahydrocannabinol levels have a half-life of 5 to 13 days.2 Although lactic acidosis and leukocytosis have been previously reported with cannabis use, it is unlikely that the patient would have such significant improvement within the first 4 days after discontinuation.1,3,4 Although the patient had many psychiatric comorbidities with previous hospitalizations describing concern for somatization disorder, her leukocytosis and elevated lactic acid levels were suggestive of an organic rather than a psychiatric etiology of her symptoms.
Discussion
Gastrointestinal beriberi has been reported in chronic cannabis users who present with nausea, vomiting, epigastric pain, leukocytosis, and lactic acidosis; all these symptoms rapidly improve after thiamine administration.5,6 The patient’s dietary change also eliminated her intake of vitamin B12, which compounded her condition. Thiamine deficiency produces lactic acidosis by disrupting pyruvate metabolism.7 Bradycardia also can be a sign of thiamine deficiency, although the patient’s use of clonidine for migraines is a confounder.8
Chronically ill patients are prone to nutritional deficiencies, including deficiencies of thiamine.7,9 Many patients with chronic illnesses also use cannabis to ameliorate physical and neuropsychiatric symptoms.2 Recent reports suggest cannabis users are prone to gastrointestinal beriberi and Wernicke encephalopathy.5,10 Treating gastrointestinal symptoms in these patients can be challenging to diagnose because gastrointestinal beriberi and CHS share many clinical manifestations.
The patient’s presentation is likely multifactorial resulting from the combination of gastrointestinal beriberi and CHS. However, thiamine deficiency seems to play the dominant role.
There is no standard treatment regimen for thiamine deficiency with neurologic deficits, and patients only retain about 10 to 15% of intramuscular (IM) injections of cyanocobalamin.11,12 The British Committee for Standards in Haematology recommends IM injections of 1,000 mcg of cyanocobalamin 3 times a week for 2 weeks and then reassess the need for continued treatment.13 The British Columbia guidelines also recommend IM injections of 1,000 mcg daily for 1 to 5 days before transitioning to oral repletion.14 European Neurology guidelines for the treatment of Wernicke encephalopathy recommend IV cyanocobalamin 200 mg 3 times daily.15 Low-level evidence with observational studies informs these decisions and is why there is variation.
The patient’s serum lactate and leukocytosis normalized 1 day after the administration of thiamine. Thiamine deficiency classically causes Wernicke encephalopathy and wet beriberi.16 The patient did not present with Wernicke encephalopathy’s triad: ophthalmoplegia, ataxia, or confusion. She also was euvolemic without signs or symptoms of wet beriberi.
Conclusions
Thiamine deficiency is principally a clinical diagnosis. Thiamine laboratory testing may not be readily available in all medical centers, and confirming a diagnosis of thiamine deficiency should not delay treatment when thiamine deficiency is suspected. This patient’s thiamine levels resulted a week after collection. The administration of thiamine before sampling also can alter the result as it did in this case. Additionally, laboratories may offer whole blood and serum testing. Whole blood testing is more accurate because most bioactive thiamine is found in red blood cells.17
1. Price SL, Fisher C, Kumar R, Hilgerson A. Cannabinoid hyperemesis syndrome as the underlying cause of intractable nausea and vomiting. J Am Osteopath Assoc. 2011;111(3):166-169. doi:10.7556/jaoa.2011.111.3.166
2. Sharma P, Murthy P, Bharath MM. Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry. 2012;7(4):149-156.
3. Antill T, Jakkoju A, Dieguez J, Laskhmiprasad L. Lactic acidosis: a rare manifestation of synthetic marijuana intoxication. J La State Med Soc. 2015;167(3):155.
4. Sullivan S. Cannabinoid hyperemesis. Can J Gastroenterol. 2010;24(5):284-285. doi:10.1155/2010/481940
5. Duca J, Lum CJ, Lo AM. Elevated lactate secondary to gastrointestinal beriberi. J Gen Intern Med. 2016;31(1):133-136. doi:10.1007/s11606-015-3326-2
6. Prakash S. Gastrointestinal beriberi: a forme fruste of Wernicke’s encephalopathy? BMJ Case Rep. 2018;bcr2018224841. doi:10.1136/bcr-2018-224841
7. Friedenberg AS, Brandoff DE, Schiffman FJ. Type B lactic acidosis as a severe metabolic complication in lymphoma and leukemia: a case series from a single institution and literature review. Medicine (Baltimore). 2007;86(4):225-232. doi:10.1097/MD.0b013e318125759a
8. Liang CC. Bradycardia in thiamin deficiency and the role of glyoxylate. J Nutrition Sci Vitaminology. 1977;23(1):1-6. doi:10.3177/jnsv.23.1
9. Attaluri P, Castillo A, Edriss H, Nugent K. Thiamine deficiency: an important consideration in critically ill patients. Am J Med Sci. 2018;356(4):382-390. doi:10.1016/j.amjms.2018.06.015
10. Chaudhari A, Li ZY, Long A, Afshinnik A. Heavy cannabis use associated with Wernicke’s encephalopathy. Cureus. 2019;11(7):e5109. doi:10.7759/cureus.5109
11. Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149-160. doi:10.1056/NEJMcp1113996
12. Green R, Allen LH, Bjørke-Monsen A-L, et al. Vitamin B12 deficiency. Nat Rev Dis Primers. 2017;3(1):17040. doi:10.1038/nrdp.2017.40
13. Devalia V, Hamilton MS, Molloy AM. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
14. British Columbia Ministry of Health; Guidelines and Protocols and Advisory Committee. Guidelines and protocols cobalamin (vitamin B12) deficiency–investigation & management. Effective January 1, 2012. Revised May 1, 2013. Accessed March 10, 2021. https://www2.gov.bc.ca/gov/content/health/practitioner-professional-resources/bc-guidelines/vitamin-b12
15. Galvin R, Brathen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010;17(12):1408-1418. doi:10.1111/j.1468-1331.2010.03153.x
16. Wiley KD, Gupta M. Vitamin B1 thiamine deficiency (beriberi). In: StatPearls. StatPearls Publishing LLC; 2019.
17. Jenco J, Krcmova LK, Solichova D, Solich P. Recent trends in determination of thiamine and its derivatives in clinical practice. J Chromatogra A. 2017;1510:1-12. doi:10.1016/j.chroma.2017.06.048
1. Price SL, Fisher C, Kumar R, Hilgerson A. Cannabinoid hyperemesis syndrome as the underlying cause of intractable nausea and vomiting. J Am Osteopath Assoc. 2011;111(3):166-169. doi:10.7556/jaoa.2011.111.3.166
2. Sharma P, Murthy P, Bharath MM. Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry. 2012;7(4):149-156.
3. Antill T, Jakkoju A, Dieguez J, Laskhmiprasad L. Lactic acidosis: a rare manifestation of synthetic marijuana intoxication. J La State Med Soc. 2015;167(3):155.
4. Sullivan S. Cannabinoid hyperemesis. Can J Gastroenterol. 2010;24(5):284-285. doi:10.1155/2010/481940
5. Duca J, Lum CJ, Lo AM. Elevated lactate secondary to gastrointestinal beriberi. J Gen Intern Med. 2016;31(1):133-136. doi:10.1007/s11606-015-3326-2
6. Prakash S. Gastrointestinal beriberi: a forme fruste of Wernicke’s encephalopathy? BMJ Case Rep. 2018;bcr2018224841. doi:10.1136/bcr-2018-224841
7. Friedenberg AS, Brandoff DE, Schiffman FJ. Type B lactic acidosis as a severe metabolic complication in lymphoma and leukemia: a case series from a single institution and literature review. Medicine (Baltimore). 2007;86(4):225-232. doi:10.1097/MD.0b013e318125759a
8. Liang CC. Bradycardia in thiamin deficiency and the role of glyoxylate. J Nutrition Sci Vitaminology. 1977;23(1):1-6. doi:10.3177/jnsv.23.1
9. Attaluri P, Castillo A, Edriss H, Nugent K. Thiamine deficiency: an important consideration in critically ill patients. Am J Med Sci. 2018;356(4):382-390. doi:10.1016/j.amjms.2018.06.015
10. Chaudhari A, Li ZY, Long A, Afshinnik A. Heavy cannabis use associated with Wernicke’s encephalopathy. Cureus. 2019;11(7):e5109. doi:10.7759/cureus.5109
11. Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149-160. doi:10.1056/NEJMcp1113996
12. Green R, Allen LH, Bjørke-Monsen A-L, et al. Vitamin B12 deficiency. Nat Rev Dis Primers. 2017;3(1):17040. doi:10.1038/nrdp.2017.40
13. Devalia V, Hamilton MS, Molloy AM. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
14. British Columbia Ministry of Health; Guidelines and Protocols and Advisory Committee. Guidelines and protocols cobalamin (vitamin B12) deficiency–investigation & management. Effective January 1, 2012. Revised May 1, 2013. Accessed March 10, 2021. https://www2.gov.bc.ca/gov/content/health/practitioner-professional-resources/bc-guidelines/vitamin-b12
15. Galvin R, Brathen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010;17(12):1408-1418. doi:10.1111/j.1468-1331.2010.03153.x
16. Wiley KD, Gupta M. Vitamin B1 thiamine deficiency (beriberi). In: StatPearls. StatPearls Publishing LLC; 2019.
17. Jenco J, Krcmova LK, Solichova D, Solich P. Recent trends in determination of thiamine and its derivatives in clinical practice. J Chromatogra A. 2017;1510:1-12. doi:10.1016/j.chroma.2017.06.048
Small Bowel Obstruction in a Surgically Naïve Abdomen
A 53-year-old male veteran with a history of heavy tobacco and alcohol use presented with abdominal pain, emesis, and no bowel movements for 2 days. He had no history of surgical procedures, malignancies, diverticulitis, inflammatory bowel disease, traveling abroad, parasitic infections, tuberculosis exposure, or hospital admissions for abdominal pain. He reported experiencing no flushing, diarrhea, or cardiac symptoms. His medical history included hypertension, depression, and osteoarthritis. His vital signs were within normal limits.
A physical examination revealed a distended abdomen with mild tenderness. He had no inguinal or ventral hernias. He also had no abnormal skin lesions. A rectal examination did not reveal any masses or blood. His laboratory values were normal. X-ray and computed tomography (CT) scan revealed dilated loops of proximal small bowel, mild wall thickening in a segment of the midileum, and narrowing of the distal small bowel suggestive of a partial small bowel obstruction (Figure 1). A 1-cm nonspecific omental nodule also was seen on the CT scan, but no enlarged lymph nodes or mesenteric calcifications were seen. There was no thickening of the terminal ileum.
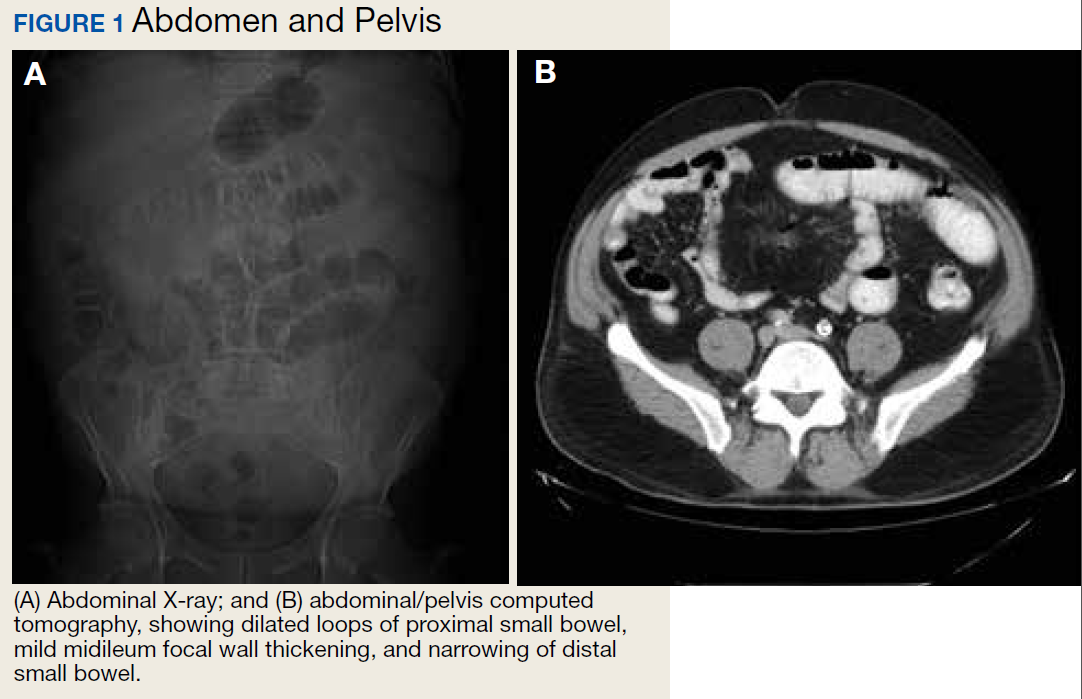
The patient underwent an exploratory laparotomy, which revealed no adhesions. In the midileum there was an area of thickened bowel with some nodularity associated with the thickness, but no discrete mass. In the mesentery there were multiple hard, white, calcified nodules, with the majority clustered near the thickened ileal segment. There also was a 1-cm hard, peritoneal mass on the anterior abdominal wall. The segment of thickened ileum, the adjacent mesentery, and the peritoneal nodule were resected.
Pathologic examination of the resected tissue showed immunohistochemical stains that were positive for CD79a, CD10, and BCL-2 and negative for CD23, CD5, and CD3. Nineteen mesenteric lymph nodes were negative for malignancy. The postoperative staging positron emission tomography (PET) scan did not reveal any fluorodeoxyglucose avid masses anywhere else, and bone marrow biopsy showed no infiltration.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the pathologic examination of the resected tissue and immunohistochemical stains, this patient was diagnosed with malignant non-Hodgkin B-cell lymphoma, follicular type, grade 1. PET scan and bone marrow biopsy revealed no other lesions, making this a primary lymphoma of the small intestine. The resected tissue showed negative margins and negative lymph nodes, indicating the full extent of the patient’s tumor was removed. He then underwent nasogastric tube decompression and IV fluid resuscitation. Two days later, he had a large bowel movement, and his abdominal pain resolved. He was provided the treatment options of observation only, radiation therapy, or rituximab treatment. Based on the high risk of enteritis following radiation therapy, the patient elected for observation only, with a repeat scan in 6 months. He also was counseled on alcohol and tobacco cessation. At the 6-month oncology follow-up, the patient showed no evidence of disease recurrence.
Discussion
Small bowel obstruction accounts for about 350,000 hospitalizations annually in the US.1 The incidence is equal in men and women and can present at any age.2,3 Patients typically present nonspecifically, with intermittent, colicky abdominal pain, nausea, vomiting, and constipation.2 A physical examination may reveal abdominal distention, rigidity, and hypoactive or absent bowel sounds.1 The 2 most common etiologies of small bowel obstruction are adhesions from prior abdominal surgery (65%) and incarcerated inguinal hernias (10%).1 However, in a patient presenting with a small bowel obstruction in a surgically naïve abdomen with no hernias, a more detailed history covering current malignancies, past hospital admissions for abdominal pains, pelvic inflammatory disease, diverticulitis, inflammatory bowel disease, and risks for parasite infection must be taken. The differential should include intraluminal causes, including small bowel malignancy, which accounts for 5% of small bowel obstructions,1 as well as extraluminal causes, including adhesions from diverticulitis, Meckel diverticulum, Ladd bands, and undiagnosed prior appendicitis.
To provide a tissue diagnosis and definitive treatment, surgical exploration was needed for this patient. Exploratory laparotomy revealed an area of thickened ileum and calcified nodules in its mesentery. Pathologic examination of the resected tissue revealed large lymphoid nodules in a follicular pattern with coarse chromatin (Figure 2). Taken together with the immunohistochemical stains, this was consistent with malignant B-cell non-Hodgkin lymphoma, follicular type, grade 1.
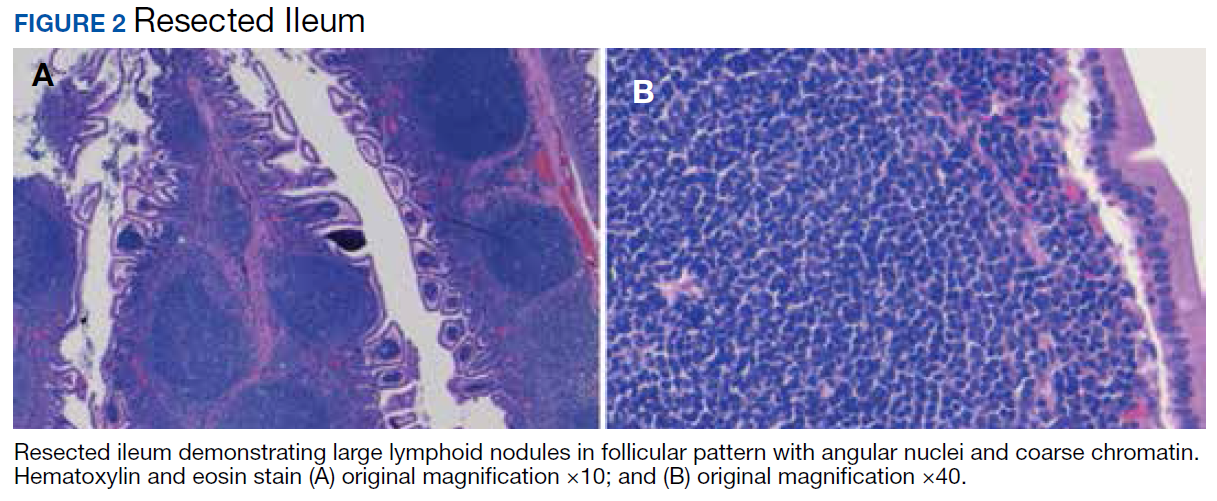
Small bowel malignancy accounts for > 5% of all gastrointestinal tumors.4 Of these, small bowel neuroendocrine tumors are the most common, followed by adenocarcinomas, lymphomas, and stromal tumors.4 Primary follicular lymphoma (PFL) is a B-cell non-Hodgkin lymphoma, and comprises between 3.8% and 11% of gastrointestinal lymphomas, commonly in the duodenum and terminal ileum.5
PFL typically occurs in middle-aged females and can be difficult to diagnose, as most patients are asymptomatic or present with unspecified abdominal pain. Many are diagnosed incidentally when endoscopy biopsies are performed for other reasons.4,5 Histologically, PFL is composed of a mixed population of small (centrocytes) and large (centroblasts) lymphoid cells, with higher proportions of centroblasts corresponding to a higher grade lymphoma.6 The classic immunophenotype of PFL shows coexpression of CD79a (or CD20), CD10, and BCL-2; however, in rare cases, low-grade PFL may stain negative for BCL-2 and have diminished staining for CD10 in interfollicular areas.7
PFL generally carries a favorable prognosis. Most patients achieving complete disease regression or stable disease following treatment and a low recurrence rate. Treatment can include surgical resection, radiation, rituximab therapy, chemotherapy, or observation.8 Patient also should be counseled in alcohol and tobacco cessation to reduce recurrence risk.
Other small bowel malignancies may present as small bowel obstructions as well. Neuroendocrine tumors and adenocarcinomas are both more common than small bowel lymphomas and can present as small bowel obstruction. However, neuroendocrine tumors are derived from serotonin-expressing enterochromaffin cells of the midgut and often present with classic carcinoid syndrome symptoms, including diarrhea, flushing, and right heart fibrosis, which the patient lacked.9 Immunohistology of small bowel adenocarcinoma often shows expression of MUC1 or MUC5AC with tumor markers CEA and CA 19-9.10
Primary intestinal melanoma, another small bowel malignancy, is extremely rare. More commonly, the etiology of intestinal melanoma is cutaneous melanoma that metastasizes to the gastrointestinal tract.11 This patient had no skin lesions to suggest metastatic melanoma. With intestinal melanoma, immunohistochemical evaluation may show S-100, the most sensitive marker for melanoma, or HMB-45, MART-1/Melan-A, tyrosinase, and MITF.12
Conclusion
This case is notable because it highlights the importance of examining the cause of small bowel obstruction in a surgically naïve abdomen, as exploration led to the discovery and curative treatment of a primary intestinal malignancy. It also underscores the nonspecific presentation that PFLs of the small intestine can have and the importance of understanding the different histopathology and immunohistochemical profiles of small bowel malignancies.
1. Rami Reddy SR, Cappell MS. A systematic review of the clinical presentation, diagnosis, and treatment of small bowel obstruction. Curr Gastroenterol Rep. 2017;19(6):28.
2. Smith DA, Nehring SM. Bowel obstruction. https://www.ncbi.nlm.nih.gov/books/NBK441975. Updated November 12, 2019. Accessed February 6, 2020.
3. Popoola D, Lou MA, Mansour AY, Sims EH. Small bowel obstruction: review of nine years of experience. J Natl Med Assoc. 1984;76(11):1089-1094.
4. Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63-71.
5. Freedman AS. Clinical presentation and diagnosis of primary gastrointestinal lymphomas. https://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-primary-gastrointestinal-lymphomas. Updated March 26, 2019. Accessed February 6, 2020.
6. Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H. Primary follicular lymphoma of the gastrointestinal tract: casereport and review. J Gastrointest Cancer. 2016;47(3):255-263.
7. Choi SM, Betz BL, Perry AM. Follicular lymphoma diagnostic caveats and updates. Arch Pathol Lab Med. 2018;142(11):1330-1340.
8. Schmatz AI, Streubel B, Kretschmer-Chott E, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445-1451.
9. Grin A, Streutker CJ. Neuroendocrine tumors of the luminal gastrointestinal tract. Arch Pathol Lab Med. 2015;139(6):750-756.
10. Chang H-K, Yu E, Kim J, et al; Korean Small Intestinal Cancer Study Group. Adenocarcinoma of the small intestine: a multi-institutional study of 197 surgically resected cases. Hum Pathol. 2010;41(8):1087-1096.
11. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009;10(5):516-521.
12. Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35(5):433-444.
A 53-year-old male veteran with a history of heavy tobacco and alcohol use presented with abdominal pain, emesis, and no bowel movements for 2 days. He had no history of surgical procedures, malignancies, diverticulitis, inflammatory bowel disease, traveling abroad, parasitic infections, tuberculosis exposure, or hospital admissions for abdominal pain. He reported experiencing no flushing, diarrhea, or cardiac symptoms. His medical history included hypertension, depression, and osteoarthritis. His vital signs were within normal limits.
A physical examination revealed a distended abdomen with mild tenderness. He had no inguinal or ventral hernias. He also had no abnormal skin lesions. A rectal examination did not reveal any masses or blood. His laboratory values were normal. X-ray and computed tomography (CT) scan revealed dilated loops of proximal small bowel, mild wall thickening in a segment of the midileum, and narrowing of the distal small bowel suggestive of a partial small bowel obstruction (Figure 1). A 1-cm nonspecific omental nodule also was seen on the CT scan, but no enlarged lymph nodes or mesenteric calcifications were seen. There was no thickening of the terminal ileum.
The patient underwent an exploratory laparotomy, which revealed no adhesions. In the midileum there was an area of thickened bowel with some nodularity associated with the thickness, but no discrete mass. In the mesentery there were multiple hard, white, calcified nodules, with the majority clustered near the thickened ileal segment. There also was a 1-cm hard, peritoneal mass on the anterior abdominal wall. The segment of thickened ileum, the adjacent mesentery, and the peritoneal nodule were resected.
Pathologic examination of the resected tissue showed immunohistochemical stains that were positive for CD79a, CD10, and BCL-2 and negative for CD23, CD5, and CD3. Nineteen mesenteric lymph nodes were negative for malignancy. The postoperative staging positron emission tomography (PET) scan did not reveal any fluorodeoxyglucose avid masses anywhere else, and bone marrow biopsy showed no infiltration.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the pathologic examination of the resected tissue and immunohistochemical stains, this patient was diagnosed with malignant non-Hodgkin B-cell lymphoma, follicular type, grade 1. PET scan and bone marrow biopsy revealed no other lesions, making this a primary lymphoma of the small intestine. The resected tissue showed negative margins and negative lymph nodes, indicating the full extent of the patient’s tumor was removed. He then underwent nasogastric tube decompression and IV fluid resuscitation. Two days later, he had a large bowel movement, and his abdominal pain resolved. He was provided the treatment options of observation only, radiation therapy, or rituximab treatment. Based on the high risk of enteritis following radiation therapy, the patient elected for observation only, with a repeat scan in 6 months. He also was counseled on alcohol and tobacco cessation. At the 6-month oncology follow-up, the patient showed no evidence of disease recurrence.
Discussion
Small bowel obstruction accounts for about 350,000 hospitalizations annually in the US.1 The incidence is equal in men and women and can present at any age.2,3 Patients typically present nonspecifically, with intermittent, colicky abdominal pain, nausea, vomiting, and constipation.2 A physical examination may reveal abdominal distention, rigidity, and hypoactive or absent bowel sounds.1 The 2 most common etiologies of small bowel obstruction are adhesions from prior abdominal surgery (65%) and incarcerated inguinal hernias (10%).1 However, in a patient presenting with a small bowel obstruction in a surgically naïve abdomen with no hernias, a more detailed history covering current malignancies, past hospital admissions for abdominal pains, pelvic inflammatory disease, diverticulitis, inflammatory bowel disease, and risks for parasite infection must be taken. The differential should include intraluminal causes, including small bowel malignancy, which accounts for 5% of small bowel obstructions,1 as well as extraluminal causes, including adhesions from diverticulitis, Meckel diverticulum, Ladd bands, and undiagnosed prior appendicitis.
To provide a tissue diagnosis and definitive treatment, surgical exploration was needed for this patient. Exploratory laparotomy revealed an area of thickened ileum and calcified nodules in its mesentery. Pathologic examination of the resected tissue revealed large lymphoid nodules in a follicular pattern with coarse chromatin (Figure 2). Taken together with the immunohistochemical stains, this was consistent with malignant B-cell non-Hodgkin lymphoma, follicular type, grade 1.
Small bowel malignancy accounts for > 5% of all gastrointestinal tumors.4 Of these, small bowel neuroendocrine tumors are the most common, followed by adenocarcinomas, lymphomas, and stromal tumors.4 Primary follicular lymphoma (PFL) is a B-cell non-Hodgkin lymphoma, and comprises between 3.8% and 11% of gastrointestinal lymphomas, commonly in the duodenum and terminal ileum.5
PFL typically occurs in middle-aged females and can be difficult to diagnose, as most patients are asymptomatic or present with unspecified abdominal pain. Many are diagnosed incidentally when endoscopy biopsies are performed for other reasons.4,5 Histologically, PFL is composed of a mixed population of small (centrocytes) and large (centroblasts) lymphoid cells, with higher proportions of centroblasts corresponding to a higher grade lymphoma.6 The classic immunophenotype of PFL shows coexpression of CD79a (or CD20), CD10, and BCL-2; however, in rare cases, low-grade PFL may stain negative for BCL-2 and have diminished staining for CD10 in interfollicular areas.7
PFL generally carries a favorable prognosis. Most patients achieving complete disease regression or stable disease following treatment and a low recurrence rate. Treatment can include surgical resection, radiation, rituximab therapy, chemotherapy, or observation.8 Patient also should be counseled in alcohol and tobacco cessation to reduce recurrence risk.
Other small bowel malignancies may present as small bowel obstructions as well. Neuroendocrine tumors and adenocarcinomas are both more common than small bowel lymphomas and can present as small bowel obstruction. However, neuroendocrine tumors are derived from serotonin-expressing enterochromaffin cells of the midgut and often present with classic carcinoid syndrome symptoms, including diarrhea, flushing, and right heart fibrosis, which the patient lacked.9 Immunohistology of small bowel adenocarcinoma often shows expression of MUC1 or MUC5AC with tumor markers CEA and CA 19-9.10
Primary intestinal melanoma, another small bowel malignancy, is extremely rare. More commonly, the etiology of intestinal melanoma is cutaneous melanoma that metastasizes to the gastrointestinal tract.11 This patient had no skin lesions to suggest metastatic melanoma. With intestinal melanoma, immunohistochemical evaluation may show S-100, the most sensitive marker for melanoma, or HMB-45, MART-1/Melan-A, tyrosinase, and MITF.12
Conclusion
This case is notable because it highlights the importance of examining the cause of small bowel obstruction in a surgically naïve abdomen, as exploration led to the discovery and curative treatment of a primary intestinal malignancy. It also underscores the nonspecific presentation that PFLs of the small intestine can have and the importance of understanding the different histopathology and immunohistochemical profiles of small bowel malignancies.
A 53-year-old male veteran with a history of heavy tobacco and alcohol use presented with abdominal pain, emesis, and no bowel movements for 2 days. He had no history of surgical procedures, malignancies, diverticulitis, inflammatory bowel disease, traveling abroad, parasitic infections, tuberculosis exposure, or hospital admissions for abdominal pain. He reported experiencing no flushing, diarrhea, or cardiac symptoms. His medical history included hypertension, depression, and osteoarthritis. His vital signs were within normal limits.
A physical examination revealed a distended abdomen with mild tenderness. He had no inguinal or ventral hernias. He also had no abnormal skin lesions. A rectal examination did not reveal any masses or blood. His laboratory values were normal. X-ray and computed tomography (CT) scan revealed dilated loops of proximal small bowel, mild wall thickening in a segment of the midileum, and narrowing of the distal small bowel suggestive of a partial small bowel obstruction (Figure 1). A 1-cm nonspecific omental nodule also was seen on the CT scan, but no enlarged lymph nodes or mesenteric calcifications were seen. There was no thickening of the terminal ileum.
The patient underwent an exploratory laparotomy, which revealed no adhesions. In the midileum there was an area of thickened bowel with some nodularity associated with the thickness, but no discrete mass. In the mesentery there were multiple hard, white, calcified nodules, with the majority clustered near the thickened ileal segment. There also was a 1-cm hard, peritoneal mass on the anterior abdominal wall. The segment of thickened ileum, the adjacent mesentery, and the peritoneal nodule were resected.
Pathologic examination of the resected tissue showed immunohistochemical stains that were positive for CD79a, CD10, and BCL-2 and negative for CD23, CD5, and CD3. Nineteen mesenteric lymph nodes were negative for malignancy. The postoperative staging positron emission tomography (PET) scan did not reveal any fluorodeoxyglucose avid masses anywhere else, and bone marrow biopsy showed no infiltration.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the pathologic examination of the resected tissue and immunohistochemical stains, this patient was diagnosed with malignant non-Hodgkin B-cell lymphoma, follicular type, grade 1. PET scan and bone marrow biopsy revealed no other lesions, making this a primary lymphoma of the small intestine. The resected tissue showed negative margins and negative lymph nodes, indicating the full extent of the patient’s tumor was removed. He then underwent nasogastric tube decompression and IV fluid resuscitation. Two days later, he had a large bowel movement, and his abdominal pain resolved. He was provided the treatment options of observation only, radiation therapy, or rituximab treatment. Based on the high risk of enteritis following radiation therapy, the patient elected for observation only, with a repeat scan in 6 months. He also was counseled on alcohol and tobacco cessation. At the 6-month oncology follow-up, the patient showed no evidence of disease recurrence.
Discussion
Small bowel obstruction accounts for about 350,000 hospitalizations annually in the US.1 The incidence is equal in men and women and can present at any age.2,3 Patients typically present nonspecifically, with intermittent, colicky abdominal pain, nausea, vomiting, and constipation.2 A physical examination may reveal abdominal distention, rigidity, and hypoactive or absent bowel sounds.1 The 2 most common etiologies of small bowel obstruction are adhesions from prior abdominal surgery (65%) and incarcerated inguinal hernias (10%).1 However, in a patient presenting with a small bowel obstruction in a surgically naïve abdomen with no hernias, a more detailed history covering current malignancies, past hospital admissions for abdominal pains, pelvic inflammatory disease, diverticulitis, inflammatory bowel disease, and risks for parasite infection must be taken. The differential should include intraluminal causes, including small bowel malignancy, which accounts for 5% of small bowel obstructions,1 as well as extraluminal causes, including adhesions from diverticulitis, Meckel diverticulum, Ladd bands, and undiagnosed prior appendicitis.
To provide a tissue diagnosis and definitive treatment, surgical exploration was needed for this patient. Exploratory laparotomy revealed an area of thickened ileum and calcified nodules in its mesentery. Pathologic examination of the resected tissue revealed large lymphoid nodules in a follicular pattern with coarse chromatin (Figure 2). Taken together with the immunohistochemical stains, this was consistent with malignant B-cell non-Hodgkin lymphoma, follicular type, grade 1.
Small bowel malignancy accounts for > 5% of all gastrointestinal tumors.4 Of these, small bowel neuroendocrine tumors are the most common, followed by adenocarcinomas, lymphomas, and stromal tumors.4 Primary follicular lymphoma (PFL) is a B-cell non-Hodgkin lymphoma, and comprises between 3.8% and 11% of gastrointestinal lymphomas, commonly in the duodenum and terminal ileum.5
PFL typically occurs in middle-aged females and can be difficult to diagnose, as most patients are asymptomatic or present with unspecified abdominal pain. Many are diagnosed incidentally when endoscopy biopsies are performed for other reasons.4,5 Histologically, PFL is composed of a mixed population of small (centrocytes) and large (centroblasts) lymphoid cells, with higher proportions of centroblasts corresponding to a higher grade lymphoma.6 The classic immunophenotype of PFL shows coexpression of CD79a (or CD20), CD10, and BCL-2; however, in rare cases, low-grade PFL may stain negative for BCL-2 and have diminished staining for CD10 in interfollicular areas.7
PFL generally carries a favorable prognosis. Most patients achieving complete disease regression or stable disease following treatment and a low recurrence rate. Treatment can include surgical resection, radiation, rituximab therapy, chemotherapy, or observation.8 Patient also should be counseled in alcohol and tobacco cessation to reduce recurrence risk.
Other small bowel malignancies may present as small bowel obstructions as well. Neuroendocrine tumors and adenocarcinomas are both more common than small bowel lymphomas and can present as small bowel obstruction. However, neuroendocrine tumors are derived from serotonin-expressing enterochromaffin cells of the midgut and often present with classic carcinoid syndrome symptoms, including diarrhea, flushing, and right heart fibrosis, which the patient lacked.9 Immunohistology of small bowel adenocarcinoma often shows expression of MUC1 or MUC5AC with tumor markers CEA and CA 19-9.10
Primary intestinal melanoma, another small bowel malignancy, is extremely rare. More commonly, the etiology of intestinal melanoma is cutaneous melanoma that metastasizes to the gastrointestinal tract.11 This patient had no skin lesions to suggest metastatic melanoma. With intestinal melanoma, immunohistochemical evaluation may show S-100, the most sensitive marker for melanoma, or HMB-45, MART-1/Melan-A, tyrosinase, and MITF.12
Conclusion
This case is notable because it highlights the importance of examining the cause of small bowel obstruction in a surgically naïve abdomen, as exploration led to the discovery and curative treatment of a primary intestinal malignancy. It also underscores the nonspecific presentation that PFLs of the small intestine can have and the importance of understanding the different histopathology and immunohistochemical profiles of small bowel malignancies.
1. Rami Reddy SR, Cappell MS. A systematic review of the clinical presentation, diagnosis, and treatment of small bowel obstruction. Curr Gastroenterol Rep. 2017;19(6):28.
2. Smith DA, Nehring SM. Bowel obstruction. https://www.ncbi.nlm.nih.gov/books/NBK441975. Updated November 12, 2019. Accessed February 6, 2020.
3. Popoola D, Lou MA, Mansour AY, Sims EH. Small bowel obstruction: review of nine years of experience. J Natl Med Assoc. 1984;76(11):1089-1094.
4. Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63-71.
5. Freedman AS. Clinical presentation and diagnosis of primary gastrointestinal lymphomas. https://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-primary-gastrointestinal-lymphomas. Updated March 26, 2019. Accessed February 6, 2020.
6. Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H. Primary follicular lymphoma of the gastrointestinal tract: casereport and review. J Gastrointest Cancer. 2016;47(3):255-263.
7. Choi SM, Betz BL, Perry AM. Follicular lymphoma diagnostic caveats and updates. Arch Pathol Lab Med. 2018;142(11):1330-1340.
8. Schmatz AI, Streubel B, Kretschmer-Chott E, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445-1451.
9. Grin A, Streutker CJ. Neuroendocrine tumors of the luminal gastrointestinal tract. Arch Pathol Lab Med. 2015;139(6):750-756.
10. Chang H-K, Yu E, Kim J, et al; Korean Small Intestinal Cancer Study Group. Adenocarcinoma of the small intestine: a multi-institutional study of 197 surgically resected cases. Hum Pathol. 2010;41(8):1087-1096.
11. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009;10(5):516-521.
12. Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35(5):433-444.
1. Rami Reddy SR, Cappell MS. A systematic review of the clinical presentation, diagnosis, and treatment of small bowel obstruction. Curr Gastroenterol Rep. 2017;19(6):28.
2. Smith DA, Nehring SM. Bowel obstruction. https://www.ncbi.nlm.nih.gov/books/NBK441975. Updated November 12, 2019. Accessed February 6, 2020.
3. Popoola D, Lou MA, Mansour AY, Sims EH. Small bowel obstruction: review of nine years of experience. J Natl Med Assoc. 1984;76(11):1089-1094.
4. Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63-71.
5. Freedman AS. Clinical presentation and diagnosis of primary gastrointestinal lymphomas. https://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-primary-gastrointestinal-lymphomas. Updated March 26, 2019. Accessed February 6, 2020.
6. Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H. Primary follicular lymphoma of the gastrointestinal tract: casereport and review. J Gastrointest Cancer. 2016;47(3):255-263.
7. Choi SM, Betz BL, Perry AM. Follicular lymphoma diagnostic caveats and updates. Arch Pathol Lab Med. 2018;142(11):1330-1340.
8. Schmatz AI, Streubel B, Kretschmer-Chott E, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445-1451.
9. Grin A, Streutker CJ. Neuroendocrine tumors of the luminal gastrointestinal tract. Arch Pathol Lab Med. 2015;139(6):750-756.
10. Chang H-K, Yu E, Kim J, et al; Korean Small Intestinal Cancer Study Group. Adenocarcinoma of the small intestine: a multi-institutional study of 197 surgically resected cases. Hum Pathol. 2010;41(8):1087-1096.
11. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009;10(5):516-521.
12. Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35(5):433-444.
A Reticular Rash on the Leg
A 73-year-old male veteran with a history of ischemic stroke with left-sided deficits and edema, falls, poorly controlled hypertension, active tobacco use, obesity, and prediabetes was assessed on a routine visit by our home-based primary care team and found to have a new, unilateral, asymptomatic rash. He reported feeling no pain in the affected area or any significant increase in the baseline left lower extremity edema and weakness resulting from his stroke 2 years prior.
On the left lateral leg from mid-thigh to mid-calf, there was a nontender, flat, reticulated rash with pigmentary alteration ranging from light brown to dark brown (Figure).
On further questioning, the patient reported regular use of a space heater because his gas furnace had been destroyed in an earthquake more than 20 years before. He would place this heater close to his left leg when using the computer or while sleeping in his wheelchair.
- What is your diagnosis?
- How would you treat this patient?
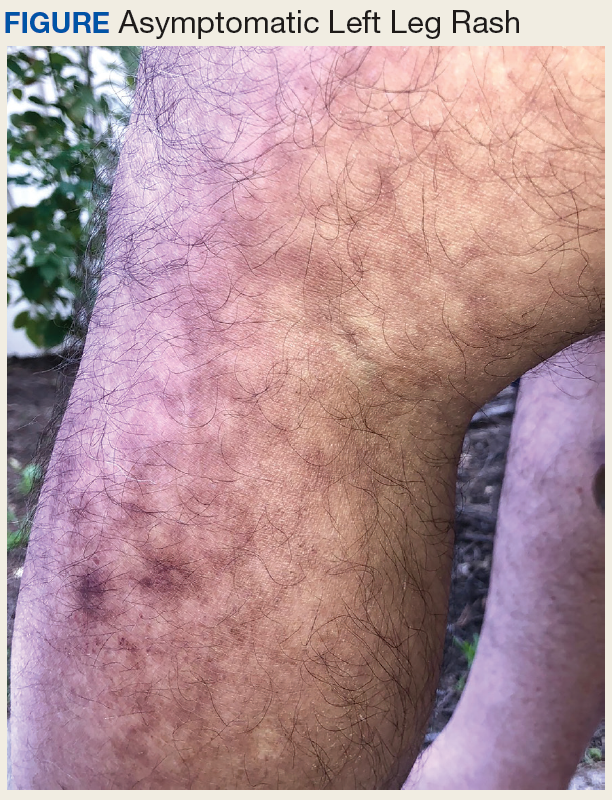
Our Diagnosis
Erythema ab igne, also called hot water bottle rash, is a clinical diagnosis based on characteristic cutaneous findings and a clear history of chronic, moderate heat or infrared exposure.1 Although exposure to space heaters, open fire, radiators, hot water bottles, and heating pads are the classic causes, recently there have been reports of laptop computers, cell phones, infrared food lamps, automobile seat heaters, and heated recliners causing the same type of skin reaction.2
With chronic moderate heat or infrared exposure, the rash usually progresses over days to months. It begins as a mild, transient, reticulated, erythematous rash, which follows the pattern of the cutaneous venous plexus and resolves minutes to hours after removal of the offending source as vasodilation resolves. After months of continued exposure, the dermis around the affected vasculature eventually becomes hyperpigmented due to the deposition of melanin and sometimes hemosiderin.
The rash is usually asymptomatic but has been associated with pain, pruritis, and/or tingling. Once the diagnosis is made, treatment involves removal of the offending source. The discoloration may resolve over months to years, but permanent hyperpigmentation is not uncommon. There are a few case reports on treatment using Nd-Yag laser therapy, topical hydroquinone and tretinoin, 5-fluorouracil, and systemic mesoglycan with topical bioflavonoids.2-4
While the prognosis of erythema ab igne is excellent if detected early, failure to recognize this condition and remove the offending source can lead to sequalae, such as squamous cell carcinoma, poorly differentiated carcinoma, cutaneous marginal zone lymphoma, and Merkel cell carcinoma.5-8 Development of malignancy typically has a latency period of > 30 years. Patients should have periodic surveillance of their skin and any suspicious lesion in the involved area should be considered for biopsy.

Rashes may represent systemic or more localized pathology (Table). In contrast to erythema ab igne, the rash associated with a vasculitic process (autoimmune, drug-induced, or infectious) tends to be more generalized and bilateral but still follows the pattern of the cutaneous venous plexus. An example of this would be livedo reticularis. Although this rash is reticular, it is not hyperpigmented.9 A variant of livedo reticularis is cutis marmorata, which develops in response to cold exposure, particularly in infants or in the setting of hypothyroidism.Cutis marmorata is erythematous, blanchable, and reversible with rewarming. Unlike erythema ab igne, there is no hyperpigmentation and tends to be more diffuse.10
When evaluating a reticular rash, consider local and systemic etiologies. If more localized and hyperpigmented, ask about heat or infrared exposure. This may point to a diagnosis of erythema ab igne.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162(1):77-78.
3. Kim HW, Kim EJ, Park HC, Ko JY, Ro YS, Kim JE. Erythema ab igne successfully treated with low fluenced 1,064-nm Q-switched Neodymium-Doped Yttrium Aluminum Garnet laser. J Cosmet Laser Ther. 2014;16(3):147-148.
4. Gianfaldoni S, Gianfaldoni R, Tchernev G, Lotti J, Wollina U, Lotti T. Erythema ab igne successfully treated with mesoglycan and bioflavonoids: a case-report. Open Access Maced J Med Sci. 2017;5(4):432-435.
5. Arrington JH 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. AMA Arch Derm. 1979;115(10):1226-1228.
6. Sigmon JR, Cantrell J, Teague D, Sangueza O, Sheehan DJ. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35(6):676-678
7. Wharton J, Roffwarg D, Miller J, Sheehan DJ. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62(6):1080-1081.
8. Jones CS. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124(1):110-113.
9. Sajjan VV, Lunge S, Swamy MB, Pandit AM. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6(5):315-321.
10. O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. Common rashes. Am Fam Physician. 2008;77(1):47-52.
A 73-year-old male veteran with a history of ischemic stroke with left-sided deficits and edema, falls, poorly controlled hypertension, active tobacco use, obesity, and prediabetes was assessed on a routine visit by our home-based primary care team and found to have a new, unilateral, asymptomatic rash. He reported feeling no pain in the affected area or any significant increase in the baseline left lower extremity edema and weakness resulting from his stroke 2 years prior.
On the left lateral leg from mid-thigh to mid-calf, there was a nontender, flat, reticulated rash with pigmentary alteration ranging from light brown to dark brown (Figure).
On further questioning, the patient reported regular use of a space heater because his gas furnace had been destroyed in an earthquake more than 20 years before. He would place this heater close to his left leg when using the computer or while sleeping in his wheelchair.
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Erythema ab igne, also called hot water bottle rash, is a clinical diagnosis based on characteristic cutaneous findings and a clear history of chronic, moderate heat or infrared exposure.1 Although exposure to space heaters, open fire, radiators, hot water bottles, and heating pads are the classic causes, recently there have been reports of laptop computers, cell phones, infrared food lamps, automobile seat heaters, and heated recliners causing the same type of skin reaction.2
With chronic moderate heat or infrared exposure, the rash usually progresses over days to months. It begins as a mild, transient, reticulated, erythematous rash, which follows the pattern of the cutaneous venous plexus and resolves minutes to hours after removal of the offending source as vasodilation resolves. After months of continued exposure, the dermis around the affected vasculature eventually becomes hyperpigmented due to the deposition of melanin and sometimes hemosiderin.
The rash is usually asymptomatic but has been associated with pain, pruritis, and/or tingling. Once the diagnosis is made, treatment involves removal of the offending source. The discoloration may resolve over months to years, but permanent hyperpigmentation is not uncommon. There are a few case reports on treatment using Nd-Yag laser therapy, topical hydroquinone and tretinoin, 5-fluorouracil, and systemic mesoglycan with topical bioflavonoids.2-4
While the prognosis of erythema ab igne is excellent if detected early, failure to recognize this condition and remove the offending source can lead to sequalae, such as squamous cell carcinoma, poorly differentiated carcinoma, cutaneous marginal zone lymphoma, and Merkel cell carcinoma.5-8 Development of malignancy typically has a latency period of > 30 years. Patients should have periodic surveillance of their skin and any suspicious lesion in the involved area should be considered for biopsy.
Rashes may represent systemic or more localized pathology (Table). In contrast to erythema ab igne, the rash associated with a vasculitic process (autoimmune, drug-induced, or infectious) tends to be more generalized and bilateral but still follows the pattern of the cutaneous venous plexus. An example of this would be livedo reticularis. Although this rash is reticular, it is not hyperpigmented.9 A variant of livedo reticularis is cutis marmorata, which develops in response to cold exposure, particularly in infants or in the setting of hypothyroidism.Cutis marmorata is erythematous, blanchable, and reversible with rewarming. Unlike erythema ab igne, there is no hyperpigmentation and tends to be more diffuse.10
When evaluating a reticular rash, consider local and systemic etiologies. If more localized and hyperpigmented, ask about heat or infrared exposure. This may point to a diagnosis of erythema ab igne.
A 73-year-old male veteran with a history of ischemic stroke with left-sided deficits and edema, falls, poorly controlled hypertension, active tobacco use, obesity, and prediabetes was assessed on a routine visit by our home-based primary care team and found to have a new, unilateral, asymptomatic rash. He reported feeling no pain in the affected area or any significant increase in the baseline left lower extremity edema and weakness resulting from his stroke 2 years prior.
On the left lateral leg from mid-thigh to mid-calf, there was a nontender, flat, reticulated rash with pigmentary alteration ranging from light brown to dark brown (Figure).
On further questioning, the patient reported regular use of a space heater because his gas furnace had been destroyed in an earthquake more than 20 years before. He would place this heater close to his left leg when using the computer or while sleeping in his wheelchair.
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Erythema ab igne, also called hot water bottle rash, is a clinical diagnosis based on characteristic cutaneous findings and a clear history of chronic, moderate heat or infrared exposure.1 Although exposure to space heaters, open fire, radiators, hot water bottles, and heating pads are the classic causes, recently there have been reports of laptop computers, cell phones, infrared food lamps, automobile seat heaters, and heated recliners causing the same type of skin reaction.2
With chronic moderate heat or infrared exposure, the rash usually progresses over days to months. It begins as a mild, transient, reticulated, erythematous rash, which follows the pattern of the cutaneous venous plexus and resolves minutes to hours after removal of the offending source as vasodilation resolves. After months of continued exposure, the dermis around the affected vasculature eventually becomes hyperpigmented due to the deposition of melanin and sometimes hemosiderin.
The rash is usually asymptomatic but has been associated with pain, pruritis, and/or tingling. Once the diagnosis is made, treatment involves removal of the offending source. The discoloration may resolve over months to years, but permanent hyperpigmentation is not uncommon. There are a few case reports on treatment using Nd-Yag laser therapy, topical hydroquinone and tretinoin, 5-fluorouracil, and systemic mesoglycan with topical bioflavonoids.2-4
While the prognosis of erythema ab igne is excellent if detected early, failure to recognize this condition and remove the offending source can lead to sequalae, such as squamous cell carcinoma, poorly differentiated carcinoma, cutaneous marginal zone lymphoma, and Merkel cell carcinoma.5-8 Development of malignancy typically has a latency period of > 30 years. Patients should have periodic surveillance of their skin and any suspicious lesion in the involved area should be considered for biopsy.
Rashes may represent systemic or more localized pathology (Table). In contrast to erythema ab igne, the rash associated with a vasculitic process (autoimmune, drug-induced, or infectious) tends to be more generalized and bilateral but still follows the pattern of the cutaneous venous plexus. An example of this would be livedo reticularis. Although this rash is reticular, it is not hyperpigmented.9 A variant of livedo reticularis is cutis marmorata, which develops in response to cold exposure, particularly in infants or in the setting of hypothyroidism.Cutis marmorata is erythematous, blanchable, and reversible with rewarming. Unlike erythema ab igne, there is no hyperpigmentation and tends to be more diffuse.10
When evaluating a reticular rash, consider local and systemic etiologies. If more localized and hyperpigmented, ask about heat or infrared exposure. This may point to a diagnosis of erythema ab igne.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162(1):77-78.
3. Kim HW, Kim EJ, Park HC, Ko JY, Ro YS, Kim JE. Erythema ab igne successfully treated with low fluenced 1,064-nm Q-switched Neodymium-Doped Yttrium Aluminum Garnet laser. J Cosmet Laser Ther. 2014;16(3):147-148.
4. Gianfaldoni S, Gianfaldoni R, Tchernev G, Lotti J, Wollina U, Lotti T. Erythema ab igne successfully treated with mesoglycan and bioflavonoids: a case-report. Open Access Maced J Med Sci. 2017;5(4):432-435.
5. Arrington JH 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. AMA Arch Derm. 1979;115(10):1226-1228.
6. Sigmon JR, Cantrell J, Teague D, Sangueza O, Sheehan DJ. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35(6):676-678
7. Wharton J, Roffwarg D, Miller J, Sheehan DJ. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62(6):1080-1081.
8. Jones CS. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124(1):110-113.
9. Sajjan VV, Lunge S, Swamy MB, Pandit AM. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6(5):315-321.
10. O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. Common rashes. Am Fam Physician. 2008;77(1):47-52.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol. 1988;18(5, pt 1):1003-1019.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162(1):77-78.
3. Kim HW, Kim EJ, Park HC, Ko JY, Ro YS, Kim JE. Erythema ab igne successfully treated with low fluenced 1,064-nm Q-switched Neodymium-Doped Yttrium Aluminum Garnet laser. J Cosmet Laser Ther. 2014;16(3):147-148.
4. Gianfaldoni S, Gianfaldoni R, Tchernev G, Lotti J, Wollina U, Lotti T. Erythema ab igne successfully treated with mesoglycan and bioflavonoids: a case-report. Open Access Maced J Med Sci. 2017;5(4):432-435.
5. Arrington JH 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. AMA Arch Derm. 1979;115(10):1226-1228.
6. Sigmon JR, Cantrell J, Teague D, Sangueza O, Sheehan DJ. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35(6):676-678
7. Wharton J, Roffwarg D, Miller J, Sheehan DJ. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62(6):1080-1081.
8. Jones CS. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124(1):110-113.
9. Sajjan VV, Lunge S, Swamy MB, Pandit AM. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6(5):315-321.
10. O’Connor NR, McLaughlin MR, Ham P. Newborn skin: part I. Common rashes. Am Fam Physician. 2008;77(1):47-52.
Fingernail Abnormalities After a Systemic Illness
A 45-year-old African American woman presented with painless fingernail detachment and cracks on her fingernails that had developed over the previous month. Her medical history was notable for an episode of Stevens-Johnson syndrome 2 months prior that required treatment with prednisone, IV immunoglobulin, etanercept, acetaminophen, and diphenhydramine.
A physical examination revealed multiple fingernails on both hands that exhibited 4 mm of proximal painless nail detachment with cream-colored discoloration, friability, and horizontal splitting (Figure). New, healthy nail was visible beneath the affected areas. Toenails were not affected.
- What is your diagnosis?
- How would you treat this patient?
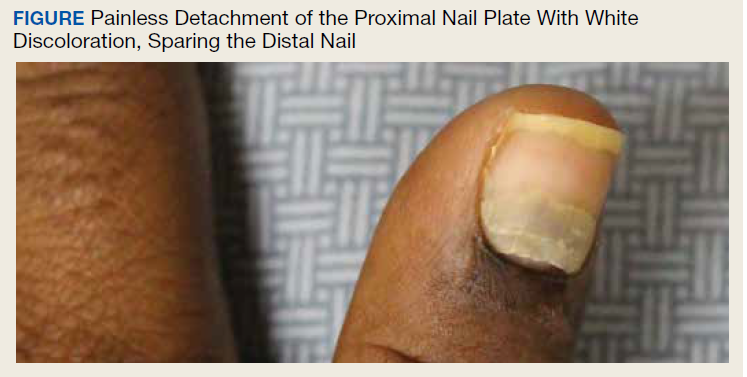
Diagnosis
Based on the timing and characteristics of her nail detachment, the patient was diagnosed with onychomadesis, which is defined as painless detachment of the proximal nail plate from the nail matrix and nail bed after at least 40 days from an initial insult. Air beneath the detached nail plate causes a characteristic creamy-white discoloration. The severity of onychomadesis ranges from transverse furrows that affect a single nail without shedding, known as Beau lines, to multiple nails that are completely shed.1,2 Nail plate shedding is typical because the nail matrix, the site of stem cells and the most proximal portion of the nail apparatus, is damaged and transiently arrested.
Various etiologies can halt nail plate production abruptly within the matrix. These typically manifest ≥ 40 days after the initial insult (the length of time for a fingernail to emerge from the proximal nail fold).2 The annual incidence of these etiologies ranges from approximately 1 per 1 million people for Stevens-Johnson syndrome, a rare cause of onychomadesis, to 1 per 10 people for onychomycosis, one of the more common causes of onychomadesis.3 The Table compares the characteristics of the diagnoses that are most commonly associated with nail detachment and discoloration. 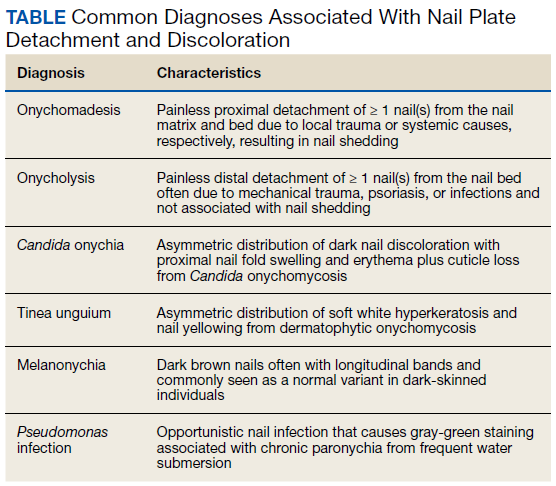
When a single nail is affected, the etiology of onychomadesis usually is primary and local, including mechanical nail trauma and fungal nail infections (onychomycosis).1,2 Candida onychia is onychomycosis caused by Candida species typically Candida albicans, which result in localized nail darkening, chronic inflammation of the paronychial skin, and cuticle loss. The infection favors immunocompromised people; coinfections are common, and onychomadesis or onycholysis can occur. Unlike onychomadesis, onycholysis is defined by painless detachment of the distal nail plate from the nail bed, but nail shedding typically does not occur because the nail matrix is spared. The preferred treatment for Candida onychia is oral itraconazole, and guided screenings for immunodeficiencies and endocrinopathies, especially diabetes mellitus, should be completed.3,4
Tinea unguium is another form of onychomycosis, but it is caused by dermatophytes, typically Trichophyton rubrum or Trichophyton mentagrophytes, which produce white and yellow nail discoloration followed by distal to proximal nail thickening and softening. Infection usually begins in toenails and demonstrates variable involvement in each nail as well as asymmetric distribution among digits.3 This condition also may eventuate in onychomadesis or onycholysis. Debridement followed by oral terbinafine is the treatment of choice.4
Two other causes of localized nail discoloration with or without nail detachment include melanonychia and nail bed infection by Pseudomonas aeruginosa (P aeruginosa). Melanonychia can be linear or diffuse brown discoloration of 1 or more nails caused by melanin deposition. Either pattern is a common finding in dark-skinned people, especially by age 50 years, but melanocyte hyperplasia should be excluded in all individuals along with drug adverse effects, exogenous pigments, infections, and systemic diseases.3,5 P aeruginosa produces pyocyanin, the green pigment responsible for the discoloration seen in this opportunistic infection often localized to a single nail. Prior maceration of the nail apparatus by repeated water submersion is common among affected individuals. Avoidance of submerging fingernails in liquids followed by nail debridement and oral antipseudomonal antibiotics is the preferred treatment course.3
The etiology is usually secondary and systemic when multiple nails demonstrate onychomadesis, but the exact pathophysiology is poorly understood. One of the most studied infectious etiologies of onychomadesis is hand-foot-and-mouth disease (HFMD), which typically affects children aged < 10 years. Parents often will recall their child being ill 1 to 2 months prior to the nail findings. Scarlet fever and varicella also can result in onychomadesis. Although not common systemic causes, Stevens-Johnson syndrome and toxic epidermal necrolysis can trigger onychomadesis of multiple nails that usually resolves in several months, but other nail deformities often persist.2,6 Onycholysis also can accompany this finding.7 Autoimmune etiologies of onychomadesis include alopecia areata and pemphigus vulgaris. Inciting medications that are toxic to the nail matrix include chemotherapy agents, valproic acid, carbamazepine, lithium, and azithromycin. Rare congenital disorders and birth trauma also can present with onychomadesis of multiple nails during infancy.2
Systemic etiologies typically affect fingernails more than toenails because of the faster growth rate of fingernails. Once the source of onychomadesis is controlled or eradicated, complete regrowth of fingernails can take from 4 to 6 months. Toenails can take twice as long and older age increases all regrowth periods.5
Our patient was treated with analgesics until her mucosal surfaces fully healed, and topical emollients and keratolytics were used to soften eschars from previous blisters and prevent further scar formation. Her affected fingernails shed and regrew after 6 months without additional interventions.
Conclusion
Although Stevens-Johnson syndrome is a rare cause of onychomadesis, and the pathophysiology of this sequela is poorly understood, this case illustrates a common nail abnormality with multiple potential etiologies that are discerned by an accurate history and thorough exam. In the absence of decorative nail polish, nails can be easily examined to provide helpful clues for past injuries or underlying diseases. An understanding of nail growth mechanics and associated terminology reveals the diagnostic and therapeutic implications of proximal vs distal nail detachment, the hue of nail discoloration, as well as single vs multiple affected nails.
Onychomadesis in single nails should prompt questions about nail trauma or risk factors for fungal infections. Depending on the etiology, manual activities need to be adjusted, or antifungals need to be initiated while investigating for an immunocompromised state. Onychomadesis in multiple nails in children should raise suspicion for HFMD or even birth trauma and congenital disorders. Multiple affected nails in adults should prompt guided questions for autoimmune diseases and inciting medications. For onycholysis, trauma, psoriasis, or certain infections should be the target. Green nails are easily recognized and treated with a defined regiment, whereas dark nails should be examined closely to differentiate Candida onychia from melanonychia. Whether from a rare cause in an adult to a common illness in a child, primary care providers have sufficient expertise to diagnose and treat various nail disorders and reassure worried patients and parents with an understanding of nail regrowth.
1. Salgado F, Handler MZ, Schwartz RA. Shedding light on onychomadesis. Cutis. 2017;99(1):33-36.
2. Hardin J, Haber RM. Oncyhomadesis: literature review. Br J Dermatol. 2015;172(3):592-596.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill; 2005.
4. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Saunders; 2012.
5. Shemer A, Daniel CR III. Common nail disorders. Clin Dermatol. 2013;31(5):578-586.
6. Acharya S, Balachandran C. Onychomadesis in Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 1996;62(4):264-265.
7. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1-e16.
A 45-year-old African American woman presented with painless fingernail detachment and cracks on her fingernails that had developed over the previous month. Her medical history was notable for an episode of Stevens-Johnson syndrome 2 months prior that required treatment with prednisone, IV immunoglobulin, etanercept, acetaminophen, and diphenhydramine.
A physical examination revealed multiple fingernails on both hands that exhibited 4 mm of proximal painless nail detachment with cream-colored discoloration, friability, and horizontal splitting (Figure). New, healthy nail was visible beneath the affected areas. Toenails were not affected.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the timing and characteristics of her nail detachment, the patient was diagnosed with onychomadesis, which is defined as painless detachment of the proximal nail plate from the nail matrix and nail bed after at least 40 days from an initial insult. Air beneath the detached nail plate causes a characteristic creamy-white discoloration. The severity of onychomadesis ranges from transverse furrows that affect a single nail without shedding, known as Beau lines, to multiple nails that are completely shed.1,2 Nail plate shedding is typical because the nail matrix, the site of stem cells and the most proximal portion of the nail apparatus, is damaged and transiently arrested.
Various etiologies can halt nail plate production abruptly within the matrix. These typically manifest ≥ 40 days after the initial insult (the length of time for a fingernail to emerge from the proximal nail fold).2 The annual incidence of these etiologies ranges from approximately 1 per 1 million people for Stevens-Johnson syndrome, a rare cause of onychomadesis, to 1 per 10 people for onychomycosis, one of the more common causes of onychomadesis.3 The Table compares the characteristics of the diagnoses that are most commonly associated with nail detachment and discoloration.
When a single nail is affected, the etiology of onychomadesis usually is primary and local, including mechanical nail trauma and fungal nail infections (onychomycosis).1,2 Candida onychia is onychomycosis caused by Candida species typically Candida albicans, which result in localized nail darkening, chronic inflammation of the paronychial skin, and cuticle loss. The infection favors immunocompromised people; coinfections are common, and onychomadesis or onycholysis can occur. Unlike onychomadesis, onycholysis is defined by painless detachment of the distal nail plate from the nail bed, but nail shedding typically does not occur because the nail matrix is spared. The preferred treatment for Candida onychia is oral itraconazole, and guided screenings for immunodeficiencies and endocrinopathies, especially diabetes mellitus, should be completed.3,4
Tinea unguium is another form of onychomycosis, but it is caused by dermatophytes, typically Trichophyton rubrum or Trichophyton mentagrophytes, which produce white and yellow nail discoloration followed by distal to proximal nail thickening and softening. Infection usually begins in toenails and demonstrates variable involvement in each nail as well as asymmetric distribution among digits.3 This condition also may eventuate in onychomadesis or onycholysis. Debridement followed by oral terbinafine is the treatment of choice.4
Two other causes of localized nail discoloration with or without nail detachment include melanonychia and nail bed infection by Pseudomonas aeruginosa (P aeruginosa). Melanonychia can be linear or diffuse brown discoloration of 1 or more nails caused by melanin deposition. Either pattern is a common finding in dark-skinned people, especially by age 50 years, but melanocyte hyperplasia should be excluded in all individuals along with drug adverse effects, exogenous pigments, infections, and systemic diseases.3,5 P aeruginosa produces pyocyanin, the green pigment responsible for the discoloration seen in this opportunistic infection often localized to a single nail. Prior maceration of the nail apparatus by repeated water submersion is common among affected individuals. Avoidance of submerging fingernails in liquids followed by nail debridement and oral antipseudomonal antibiotics is the preferred treatment course.3
The etiology is usually secondary and systemic when multiple nails demonstrate onychomadesis, but the exact pathophysiology is poorly understood. One of the most studied infectious etiologies of onychomadesis is hand-foot-and-mouth disease (HFMD), which typically affects children aged < 10 years. Parents often will recall their child being ill 1 to 2 months prior to the nail findings. Scarlet fever and varicella also can result in onychomadesis. Although not common systemic causes, Stevens-Johnson syndrome and toxic epidermal necrolysis can trigger onychomadesis of multiple nails that usually resolves in several months, but other nail deformities often persist.2,6 Onycholysis also can accompany this finding.7 Autoimmune etiologies of onychomadesis include alopecia areata and pemphigus vulgaris. Inciting medications that are toxic to the nail matrix include chemotherapy agents, valproic acid, carbamazepine, lithium, and azithromycin. Rare congenital disorders and birth trauma also can present with onychomadesis of multiple nails during infancy.2
Systemic etiologies typically affect fingernails more than toenails because of the faster growth rate of fingernails. Once the source of onychomadesis is controlled or eradicated, complete regrowth of fingernails can take from 4 to 6 months. Toenails can take twice as long and older age increases all regrowth periods.5
Our patient was treated with analgesics until her mucosal surfaces fully healed, and topical emollients and keratolytics were used to soften eschars from previous blisters and prevent further scar formation. Her affected fingernails shed and regrew after 6 months without additional interventions.
Conclusion
Although Stevens-Johnson syndrome is a rare cause of onychomadesis, and the pathophysiology of this sequela is poorly understood, this case illustrates a common nail abnormality with multiple potential etiologies that are discerned by an accurate history and thorough exam. In the absence of decorative nail polish, nails can be easily examined to provide helpful clues for past injuries or underlying diseases. An understanding of nail growth mechanics and associated terminology reveals the diagnostic and therapeutic implications of proximal vs distal nail detachment, the hue of nail discoloration, as well as single vs multiple affected nails.
Onychomadesis in single nails should prompt questions about nail trauma or risk factors for fungal infections. Depending on the etiology, manual activities need to be adjusted, or antifungals need to be initiated while investigating for an immunocompromised state. Onychomadesis in multiple nails in children should raise suspicion for HFMD or even birth trauma and congenital disorders. Multiple affected nails in adults should prompt guided questions for autoimmune diseases and inciting medications. For onycholysis, trauma, psoriasis, or certain infections should be the target. Green nails are easily recognized and treated with a defined regiment, whereas dark nails should be examined closely to differentiate Candida onychia from melanonychia. Whether from a rare cause in an adult to a common illness in a child, primary care providers have sufficient expertise to diagnose and treat various nail disorders and reassure worried patients and parents with an understanding of nail regrowth.
A 45-year-old African American woman presented with painless fingernail detachment and cracks on her fingernails that had developed over the previous month. Her medical history was notable for an episode of Stevens-Johnson syndrome 2 months prior that required treatment with prednisone, IV immunoglobulin, etanercept, acetaminophen, and diphenhydramine.
A physical examination revealed multiple fingernails on both hands that exhibited 4 mm of proximal painless nail detachment with cream-colored discoloration, friability, and horizontal splitting (Figure). New, healthy nail was visible beneath the affected areas. Toenails were not affected.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the timing and characteristics of her nail detachment, the patient was diagnosed with onychomadesis, which is defined as painless detachment of the proximal nail plate from the nail matrix and nail bed after at least 40 days from an initial insult. Air beneath the detached nail plate causes a characteristic creamy-white discoloration. The severity of onychomadesis ranges from transverse furrows that affect a single nail without shedding, known as Beau lines, to multiple nails that are completely shed.1,2 Nail plate shedding is typical because the nail matrix, the site of stem cells and the most proximal portion of the nail apparatus, is damaged and transiently arrested.
Various etiologies can halt nail plate production abruptly within the matrix. These typically manifest ≥ 40 days after the initial insult (the length of time for a fingernail to emerge from the proximal nail fold).2 The annual incidence of these etiologies ranges from approximately 1 per 1 million people for Stevens-Johnson syndrome, a rare cause of onychomadesis, to 1 per 10 people for onychomycosis, one of the more common causes of onychomadesis.3 The Table compares the characteristics of the diagnoses that are most commonly associated with nail detachment and discoloration.
When a single nail is affected, the etiology of onychomadesis usually is primary and local, including mechanical nail trauma and fungal nail infections (onychomycosis).1,2 Candida onychia is onychomycosis caused by Candida species typically Candida albicans, which result in localized nail darkening, chronic inflammation of the paronychial skin, and cuticle loss. The infection favors immunocompromised people; coinfections are common, and onychomadesis or onycholysis can occur. Unlike onychomadesis, onycholysis is defined by painless detachment of the distal nail plate from the nail bed, but nail shedding typically does not occur because the nail matrix is spared. The preferred treatment for Candida onychia is oral itraconazole, and guided screenings for immunodeficiencies and endocrinopathies, especially diabetes mellitus, should be completed.3,4
Tinea unguium is another form of onychomycosis, but it is caused by dermatophytes, typically Trichophyton rubrum or Trichophyton mentagrophytes, which produce white and yellow nail discoloration followed by distal to proximal nail thickening and softening. Infection usually begins in toenails and demonstrates variable involvement in each nail as well as asymmetric distribution among digits.3 This condition also may eventuate in onychomadesis or onycholysis. Debridement followed by oral terbinafine is the treatment of choice.4
Two other causes of localized nail discoloration with or without nail detachment include melanonychia and nail bed infection by Pseudomonas aeruginosa (P aeruginosa). Melanonychia can be linear or diffuse brown discoloration of 1 or more nails caused by melanin deposition. Either pattern is a common finding in dark-skinned people, especially by age 50 years, but melanocyte hyperplasia should be excluded in all individuals along with drug adverse effects, exogenous pigments, infections, and systemic diseases.3,5 P aeruginosa produces pyocyanin, the green pigment responsible for the discoloration seen in this opportunistic infection often localized to a single nail. Prior maceration of the nail apparatus by repeated water submersion is common among affected individuals. Avoidance of submerging fingernails in liquids followed by nail debridement and oral antipseudomonal antibiotics is the preferred treatment course.3
The etiology is usually secondary and systemic when multiple nails demonstrate onychomadesis, but the exact pathophysiology is poorly understood. One of the most studied infectious etiologies of onychomadesis is hand-foot-and-mouth disease (HFMD), which typically affects children aged < 10 years. Parents often will recall their child being ill 1 to 2 months prior to the nail findings. Scarlet fever and varicella also can result in onychomadesis. Although not common systemic causes, Stevens-Johnson syndrome and toxic epidermal necrolysis can trigger onychomadesis of multiple nails that usually resolves in several months, but other nail deformities often persist.2,6 Onycholysis also can accompany this finding.7 Autoimmune etiologies of onychomadesis include alopecia areata and pemphigus vulgaris. Inciting medications that are toxic to the nail matrix include chemotherapy agents, valproic acid, carbamazepine, lithium, and azithromycin. Rare congenital disorders and birth trauma also can present with onychomadesis of multiple nails during infancy.2
Systemic etiologies typically affect fingernails more than toenails because of the faster growth rate of fingernails. Once the source of onychomadesis is controlled or eradicated, complete regrowth of fingernails can take from 4 to 6 months. Toenails can take twice as long and older age increases all regrowth periods.5
Our patient was treated with analgesics until her mucosal surfaces fully healed, and topical emollients and keratolytics were used to soften eschars from previous blisters and prevent further scar formation. Her affected fingernails shed and regrew after 6 months without additional interventions.
Conclusion
Although Stevens-Johnson syndrome is a rare cause of onychomadesis, and the pathophysiology of this sequela is poorly understood, this case illustrates a common nail abnormality with multiple potential etiologies that are discerned by an accurate history and thorough exam. In the absence of decorative nail polish, nails can be easily examined to provide helpful clues for past injuries or underlying diseases. An understanding of nail growth mechanics and associated terminology reveals the diagnostic and therapeutic implications of proximal vs distal nail detachment, the hue of nail discoloration, as well as single vs multiple affected nails.
Onychomadesis in single nails should prompt questions about nail trauma or risk factors for fungal infections. Depending on the etiology, manual activities need to be adjusted, or antifungals need to be initiated while investigating for an immunocompromised state. Onychomadesis in multiple nails in children should raise suspicion for HFMD or even birth trauma and congenital disorders. Multiple affected nails in adults should prompt guided questions for autoimmune diseases and inciting medications. For onycholysis, trauma, psoriasis, or certain infections should be the target. Green nails are easily recognized and treated with a defined regiment, whereas dark nails should be examined closely to differentiate Candida onychia from melanonychia. Whether from a rare cause in an adult to a common illness in a child, primary care providers have sufficient expertise to diagnose and treat various nail disorders and reassure worried patients and parents with an understanding of nail regrowth.
1. Salgado F, Handler MZ, Schwartz RA. Shedding light on onychomadesis. Cutis. 2017;99(1):33-36.
2. Hardin J, Haber RM. Oncyhomadesis: literature review. Br J Dermatol. 2015;172(3):592-596.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill; 2005.
4. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Saunders; 2012.
5. Shemer A, Daniel CR III. Common nail disorders. Clin Dermatol. 2013;31(5):578-586.
6. Acharya S, Balachandran C. Onychomadesis in Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 1996;62(4):264-265.
7. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1-e16.
1. Salgado F, Handler MZ, Schwartz RA. Shedding light on onychomadesis. Cutis. 2017;99(1):33-36.
2. Hardin J, Haber RM. Oncyhomadesis: literature review. Br J Dermatol. 2015;172(3):592-596.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill; 2005.
4. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Saunders; 2012.
5. Shemer A, Daniel CR III. Common nail disorders. Clin Dermatol. 2013;31(5):578-586.
6. Acharya S, Balachandran C. Onychomadesis in Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 1996;62(4):264-265.
7. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1-e16.
Identifying Melanoma With Dermoscopy
Identifying Melanoma With Dermoscopy: 7- Point Checklist
Skin-Colored Papules on the Chest
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
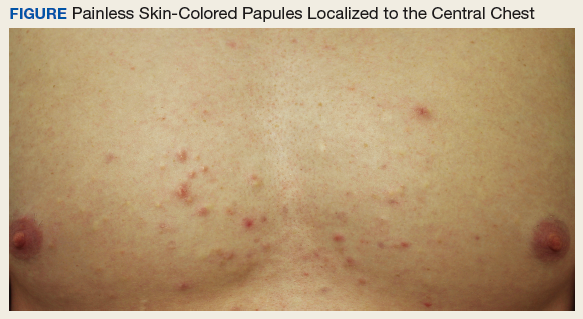
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.