User login
VIDEO: PPACMAN aims to advance the combined rheum-derm clinic approach in the community
SAN DIEGO – A new endeavor that aims to promote the concept of the combined clinic approach to caring for psoriatic patients is now underway.
PPACMAN (Psoriasis and Psoriatic Arthritis Clinics Multicenter Advancement Network) is made up of dermatologists and rheumatologists who play a key role in the management of psoriatic disease and are interested in combined clinics, with the mission “to nucleate psoriatic disease combined clinics and centers to advance a multilevel approach to psoriatic patients, increase disease awareness, and accelerate management,” according to Joseph Merola, MD, codirector of the center for skin and related musculoskeletal diseases at Brigham and Women’s Hospital, Boston.
There are now about 12 centers in North America with formal rheumatology-dermatology clinics for patients with psoriasis and psoriatic arthritis, including the one at Brigham and Women’s, where Dr. Merola and his colleagues have seen the “myriad benefits that come with having a combined clinic,” he said in a video interview at the annual meeting of the American Academy of Dermatology. The idea behind starting PPACMAN was to help form new clinics at academic centers but, also, “to start to catalyze local-regional partnerships in the community so we could get dermatologists and rheumatologists in the community to start interacting, communicating, [and] sharing patients,” he explained.
“The group is really very much focused on this mission of getting combined ... treatment models out there,” added Dr. Merola, president and chair of the board of PPACMAN, which is a 501c3 nonprofit organization.
In the interview, he discusses other benefits of the combined clinic model and other elements of the PPACMAN mission, including education and the potential for shared EMR templates.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – A new endeavor that aims to promote the concept of the combined clinic approach to caring for psoriatic patients is now underway.
PPACMAN (Psoriasis and Psoriatic Arthritis Clinics Multicenter Advancement Network) is made up of dermatologists and rheumatologists who play a key role in the management of psoriatic disease and are interested in combined clinics, with the mission “to nucleate psoriatic disease combined clinics and centers to advance a multilevel approach to psoriatic patients, increase disease awareness, and accelerate management,” according to Joseph Merola, MD, codirector of the center for skin and related musculoskeletal diseases at Brigham and Women’s Hospital, Boston.
There are now about 12 centers in North America with formal rheumatology-dermatology clinics for patients with psoriasis and psoriatic arthritis, including the one at Brigham and Women’s, where Dr. Merola and his colleagues have seen the “myriad benefits that come with having a combined clinic,” he said in a video interview at the annual meeting of the American Academy of Dermatology. The idea behind starting PPACMAN was to help form new clinics at academic centers but, also, “to start to catalyze local-regional partnerships in the community so we could get dermatologists and rheumatologists in the community to start interacting, communicating, [and] sharing patients,” he explained.
“The group is really very much focused on this mission of getting combined ... treatment models out there,” added Dr. Merola, president and chair of the board of PPACMAN, which is a 501c3 nonprofit organization.
In the interview, he discusses other benefits of the combined clinic model and other elements of the PPACMAN mission, including education and the potential for shared EMR templates.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN DIEGO – A new endeavor that aims to promote the concept of the combined clinic approach to caring for psoriatic patients is now underway.
PPACMAN (Psoriasis and Psoriatic Arthritis Clinics Multicenter Advancement Network) is made up of dermatologists and rheumatologists who play a key role in the management of psoriatic disease and are interested in combined clinics, with the mission “to nucleate psoriatic disease combined clinics and centers to advance a multilevel approach to psoriatic patients, increase disease awareness, and accelerate management,” according to Joseph Merola, MD, codirector of the center for skin and related musculoskeletal diseases at Brigham and Women’s Hospital, Boston.
There are now about 12 centers in North America with formal rheumatology-dermatology clinics for patients with psoriasis and psoriatic arthritis, including the one at Brigham and Women’s, where Dr. Merola and his colleagues have seen the “myriad benefits that come with having a combined clinic,” he said in a video interview at the annual meeting of the American Academy of Dermatology. The idea behind starting PPACMAN was to help form new clinics at academic centers but, also, “to start to catalyze local-regional partnerships in the community so we could get dermatologists and rheumatologists in the community to start interacting, communicating, [and] sharing patients,” he explained.
“The group is really very much focused on this mission of getting combined ... treatment models out there,” added Dr. Merola, president and chair of the board of PPACMAN, which is a 501c3 nonprofit organization.
In the interview, he discusses other benefits of the combined clinic model and other elements of the PPACMAN mission, including education and the potential for shared EMR templates.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
REPORTING FROM AAD 18

ANA assay results differ widely in established lupus patients
A new study that shows wide variation in the results of commercially-available assays used to detect antinuclear antibodies in people with established systemic lupus erythematosus has thrown into question the assays’ role in determining eligibility for trials as well as their role in clinical practice.
The study’s findings of negative results ranging from 5% to 22% of samples with three different commercially available immunofluorescence assays (IFAs), one ELISA assay, and one bead-based multiplex assay reveal that “ANA negativity can occur in established lupus not infrequently [and] the use of certain ANA assays could affect the frequency of screen failures in the trial setting as well as the eventual utilization of an agent if approved for serologically active patients,” David S. Pisetsky, MD, PhD, of Duke University, Durham, N.C., and his associates wrote in their report published in Annals of the Rheumatic Diseases.

People with systemic lupus erythematosus (SLE) have been thought to be “almost invariably” ANA positive, leaving the usual performance of testing to initial evaluation but not later unless a patient seeks care from another provider or undergoes screening to determine eligibility for entry into clinical trials of new therapeutic agents even though existing assays are not validated for this purpose. This practice first began with the development of the monoclonal antibody belimumab (Benlysta). Phase 2 trials showed that patients who were serologically positive (ANA and/or anti-DNA) were more likely to respond to the agent. Phase 3 trials that followed enrolled only serological positive patients and met their endpoints, the investigators explained.
“In view of the increasing use of ANA for determining trial eligibility, an explanation of these observations is important since it can impact both trial enrollment and eventual utilization of a product approved for autoantibody positive patients,” they wrote.
The research team assessed sera from 103 SLE patients using three different IFAs, an ELISA assay, and a bead-based multiplex assay. Results showed that the frequency of ANA positivity varied markedly depending on the assay platform and kit used. Among the IFA kits, negative results varied from 5 to 23 of 103 samples (4.9%-22.3%), although some samples had indeterminate results. Negative results occurred in 12 (11.7%) of the ELISA samples and in 14 (13.6%) of the multiplex assay samples.
Patients who consistently tested ANA positive in all assays differed from those who had discordant results among assays based on their likelihood of historical anti-double stranded DNA positivity and low levels of C3 complement.
This difference may have implications for the use of assays to determine eligibility for entry into a trial, the investigators noted. They advised that clinical trial protocols should specify which kits can be used to determine eligibility and how they characteristically perform, particularly for patients with established disease.
“Since the ANA assay used for screening is often not specified in protocols, the selection of a kit could lead to as much as a 17% change in the number of screen failures,” they speculated. “Correspondingly, for products approved for serologically active SLE, the use of certain assays could determine whether a patient meets criteria for its use.”
Future studies are needed to identify the assays that are most informative as theranostic biomarkers. Questions also remain over whether ANA positivity should be a criterion for trial entry in people with SLE of long duration, and if people with seronegative disease should be studied separately, they added.
The Lupus Industry Council supported the study. No authors had conflicts of interest to declare.
SOURCE: Pisetsky D et al. Ann Rheum Dis. 2018 Feb 9. doi: 10.1136/annrheumdis-2017-212599
A new study that shows wide variation in the results of commercially-available assays used to detect antinuclear antibodies in people with established systemic lupus erythematosus has thrown into question the assays’ role in determining eligibility for trials as well as their role in clinical practice.
The study’s findings of negative results ranging from 5% to 22% of samples with three different commercially available immunofluorescence assays (IFAs), one ELISA assay, and one bead-based multiplex assay reveal that “ANA negativity can occur in established lupus not infrequently [and] the use of certain ANA assays could affect the frequency of screen failures in the trial setting as well as the eventual utilization of an agent if approved for serologically active patients,” David S. Pisetsky, MD, PhD, of Duke University, Durham, N.C., and his associates wrote in their report published in Annals of the Rheumatic Diseases.
People with systemic lupus erythematosus (SLE) have been thought to be “almost invariably” ANA positive, leaving the usual performance of testing to initial evaluation but not later unless a patient seeks care from another provider or undergoes screening to determine eligibility for entry into clinical trials of new therapeutic agents even though existing assays are not validated for this purpose. This practice first began with the development of the monoclonal antibody belimumab (Benlysta). Phase 2 trials showed that patients who were serologically positive (ANA and/or anti-DNA) were more likely to respond to the agent. Phase 3 trials that followed enrolled only serological positive patients and met their endpoints, the investigators explained.
“In view of the increasing use of ANA for determining trial eligibility, an explanation of these observations is important since it can impact both trial enrollment and eventual utilization of a product approved for autoantibody positive patients,” they wrote.
The research team assessed sera from 103 SLE patients using three different IFAs, an ELISA assay, and a bead-based multiplex assay. Results showed that the frequency of ANA positivity varied markedly depending on the assay platform and kit used. Among the IFA kits, negative results varied from 5 to 23 of 103 samples (4.9%-22.3%), although some samples had indeterminate results. Negative results occurred in 12 (11.7%) of the ELISA samples and in 14 (13.6%) of the multiplex assay samples.
Patients who consistently tested ANA positive in all assays differed from those who had discordant results among assays based on their likelihood of historical anti-double stranded DNA positivity and low levels of C3 complement.
This difference may have implications for the use of assays to determine eligibility for entry into a trial, the investigators noted. They advised that clinical trial protocols should specify which kits can be used to determine eligibility and how they characteristically perform, particularly for patients with established disease.
“Since the ANA assay used for screening is often not specified in protocols, the selection of a kit could lead to as much as a 17% change in the number of screen failures,” they speculated. “Correspondingly, for products approved for serologically active SLE, the use of certain assays could determine whether a patient meets criteria for its use.”
Future studies are needed to identify the assays that are most informative as theranostic biomarkers. Questions also remain over whether ANA positivity should be a criterion for trial entry in people with SLE of long duration, and if people with seronegative disease should be studied separately, they added.
The Lupus Industry Council supported the study. No authors had conflicts of interest to declare.
SOURCE: Pisetsky D et al. Ann Rheum Dis. 2018 Feb 9. doi: 10.1136/annrheumdis-2017-212599
A new study that shows wide variation in the results of commercially-available assays used to detect antinuclear antibodies in people with established systemic lupus erythematosus has thrown into question the assays’ role in determining eligibility for trials as well as their role in clinical practice.
The study’s findings of negative results ranging from 5% to 22% of samples with three different commercially available immunofluorescence assays (IFAs), one ELISA assay, and one bead-based multiplex assay reveal that “ANA negativity can occur in established lupus not infrequently [and] the use of certain ANA assays could affect the frequency of screen failures in the trial setting as well as the eventual utilization of an agent if approved for serologically active patients,” David S. Pisetsky, MD, PhD, of Duke University, Durham, N.C., and his associates wrote in their report published in Annals of the Rheumatic Diseases.
People with systemic lupus erythematosus (SLE) have been thought to be “almost invariably” ANA positive, leaving the usual performance of testing to initial evaluation but not later unless a patient seeks care from another provider or undergoes screening to determine eligibility for entry into clinical trials of new therapeutic agents even though existing assays are not validated for this purpose. This practice first began with the development of the monoclonal antibody belimumab (Benlysta). Phase 2 trials showed that patients who were serologically positive (ANA and/or anti-DNA) were more likely to respond to the agent. Phase 3 trials that followed enrolled only serological positive patients and met their endpoints, the investigators explained.
“In view of the increasing use of ANA for determining trial eligibility, an explanation of these observations is important since it can impact both trial enrollment and eventual utilization of a product approved for autoantibody positive patients,” they wrote.
The research team assessed sera from 103 SLE patients using three different IFAs, an ELISA assay, and a bead-based multiplex assay. Results showed that the frequency of ANA positivity varied markedly depending on the assay platform and kit used. Among the IFA kits, negative results varied from 5 to 23 of 103 samples (4.9%-22.3%), although some samples had indeterminate results. Negative results occurred in 12 (11.7%) of the ELISA samples and in 14 (13.6%) of the multiplex assay samples.
Patients who consistently tested ANA positive in all assays differed from those who had discordant results among assays based on their likelihood of historical anti-double stranded DNA positivity and low levels of C3 complement.
This difference may have implications for the use of assays to determine eligibility for entry into a trial, the investigators noted. They advised that clinical trial protocols should specify which kits can be used to determine eligibility and how they characteristically perform, particularly for patients with established disease.
“Since the ANA assay used for screening is often not specified in protocols, the selection of a kit could lead to as much as a 17% change in the number of screen failures,” they speculated. “Correspondingly, for products approved for serologically active SLE, the use of certain assays could determine whether a patient meets criteria for its use.”
Future studies are needed to identify the assays that are most informative as theranostic biomarkers. Questions also remain over whether ANA positivity should be a criterion for trial entry in people with SLE of long duration, and if people with seronegative disease should be studied separately, they added.
The Lupus Industry Council supported the study. No authors had conflicts of interest to declare.
SOURCE: Pisetsky D et al. Ann Rheum Dis. 2018 Feb 9. doi: 10.1136/annrheumdis-2017-212599
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Main finding: ANA negativity varied from 5% to 22% of sera samples from 103 SLE patients with established disease.
Data source: Sera from 103 SLE patients were assessed using three different commercially available immunofluorescence assays, one ELISA assay, and one bead-based multiplex assay.
Disclosures: The Lupus Industry Council supported the study. No authors had conflicts of interest to declare.
Source: Pisetsky D et al. Ann Rheum Dis. 2018 Feb 9. doi: 10.1136/annrheumdis-2017-212599.
Majority of lupus patients lack eye exam around the start of hydroxychloroquine
Only one-third of lupus patients on Medicaid had a baseline retinal exam before initiating treatment with hydroxychloroquine in an analysis of a claims database, despite the exam being recommended as the standard in multiple clinical guidelines.
The analysis of 12,755 patients with systemic lupus erythematosus (SLE) on Medicaid from 29 of the most populated U.S. states between 2001 and 2010 found that 32.5% received a baseline dilated eye exam 30 days before, and up to 1 year after, starting treatment with the anchor drug hydroxychloroquine (HCQ). This figure rose to 40% when Humphrey visual field tests and other “optional” eye exams were included, Tzu-Chieh Lin, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues reported in Arthritis Care & Research.
HCQ is known to cause retinal damage in some patients, binding to and accumulating in the retinal pigment epithelium melanin. If undetected, patients may gradually lose central vision. There is particular risk for HCQ-induced retinal damage in patients on higher doses (5 mg/kg or greater of actual weight), HCQ exposure over 5 years, or renal, hepatic, or retinal disease, the research team explained.
Baseline retinal examinations for first-time HCQ users were first recommended by American Academy of Ophthalmology guidelines in 2002, and in 2009 the American College of Rheumatology developed guidelines specifically for SLE that also recommended a funduscopic exam within 1 year of starting HCQ.
The proportion of patients beginning treatment with HCQ who received a retinal exam only slightly increased from 31% in 2001 to 34.4% in 2009 (P value for linear trend over time = .12).
Women were significantly more likely to have a baseline retinal exam (odds ratio, 1.30; 95% confidence interval, 1.08-1.55), and certain sociodemographic factors also predicted who received a baseline exam, likely reflecting problems with adherence and access to health care.
For example, the likelihood of having a baseline exam proved significantly lower for blacks and American Indian/Alaska natives (OR, 0.85; 95% CI, 0.77-0.93; and OR, 0.69; 95% CI, 0.47-1.02) than for whites, whereas Asian Americans had greater odds than whites (OR, 1.30; 95% CI, 1.06-1.60).
“We have previously observed that these sociodemographic factors have been associated with differences in both access to care and adherence in the SLE Medicaid population for indicated care, including infection prevention, adherence to indicated medications including HCQ, treatments for lupus nephritis, and choices in renal replacement therapies and use of erythropoietin-stimulating agents among SLE end-stage renal disease patients,” they wrote.
Patients who had received multiple SLE-related laboratory tests (OR, 1.33; 95% CI, 1.16-1.52) and liver function tests (OR, 1.16; 95% CI, 1.04-1.29) were also more likely to have an eye exam before commencing HCQ.
The researchers said both patient and physician factors likely played a role in adherence to preventive medical care.
“While adherence to daily dosing, by the latest recommendations less than 5 mg/kg actual weight, is a more important factor determining renal toxicity risk, baseline examinations are necessary to avoid treating patients with preexisting retinal disease and to allow early detection of changes from baseline,” the research team concluded.
The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
SOURCE: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
Only one-third of lupus patients on Medicaid had a baseline retinal exam before initiating treatment with hydroxychloroquine in an analysis of a claims database, despite the exam being recommended as the standard in multiple clinical guidelines.
The analysis of 12,755 patients with systemic lupus erythematosus (SLE) on Medicaid from 29 of the most populated U.S. states between 2001 and 2010 found that 32.5% received a baseline dilated eye exam 30 days before, and up to 1 year after, starting treatment with the anchor drug hydroxychloroquine (HCQ). This figure rose to 40% when Humphrey visual field tests and other “optional” eye exams were included, Tzu-Chieh Lin, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues reported in Arthritis Care & Research.
HCQ is known to cause retinal damage in some patients, binding to and accumulating in the retinal pigment epithelium melanin. If undetected, patients may gradually lose central vision. There is particular risk for HCQ-induced retinal damage in patients on higher doses (5 mg/kg or greater of actual weight), HCQ exposure over 5 years, or renal, hepatic, or retinal disease, the research team explained.
Baseline retinal examinations for first-time HCQ users were first recommended by American Academy of Ophthalmology guidelines in 2002, and in 2009 the American College of Rheumatology developed guidelines specifically for SLE that also recommended a funduscopic exam within 1 year of starting HCQ.
The proportion of patients beginning treatment with HCQ who received a retinal exam only slightly increased from 31% in 2001 to 34.4% in 2009 (P value for linear trend over time = .12).
Women were significantly more likely to have a baseline retinal exam (odds ratio, 1.30; 95% confidence interval, 1.08-1.55), and certain sociodemographic factors also predicted who received a baseline exam, likely reflecting problems with adherence and access to health care.
For example, the likelihood of having a baseline exam proved significantly lower for blacks and American Indian/Alaska natives (OR, 0.85; 95% CI, 0.77-0.93; and OR, 0.69; 95% CI, 0.47-1.02) than for whites, whereas Asian Americans had greater odds than whites (OR, 1.30; 95% CI, 1.06-1.60).
“We have previously observed that these sociodemographic factors have been associated with differences in both access to care and adherence in the SLE Medicaid population for indicated care, including infection prevention, adherence to indicated medications including HCQ, treatments for lupus nephritis, and choices in renal replacement therapies and use of erythropoietin-stimulating agents among SLE end-stage renal disease patients,” they wrote.
Patients who had received multiple SLE-related laboratory tests (OR, 1.33; 95% CI, 1.16-1.52) and liver function tests (OR, 1.16; 95% CI, 1.04-1.29) were also more likely to have an eye exam before commencing HCQ.
The researchers said both patient and physician factors likely played a role in adherence to preventive medical care.
“While adherence to daily dosing, by the latest recommendations less than 5 mg/kg actual weight, is a more important factor determining renal toxicity risk, baseline examinations are necessary to avoid treating patients with preexisting retinal disease and to allow early detection of changes from baseline,” the research team concluded.
The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
SOURCE: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
Only one-third of lupus patients on Medicaid had a baseline retinal exam before initiating treatment with hydroxychloroquine in an analysis of a claims database, despite the exam being recommended as the standard in multiple clinical guidelines.
The analysis of 12,755 patients with systemic lupus erythematosus (SLE) on Medicaid from 29 of the most populated U.S. states between 2001 and 2010 found that 32.5% received a baseline dilated eye exam 30 days before, and up to 1 year after, starting treatment with the anchor drug hydroxychloroquine (HCQ). This figure rose to 40% when Humphrey visual field tests and other “optional” eye exams were included, Tzu-Chieh Lin, PhD, of Brigham and Women’s Hospital, Boston, and his colleagues reported in Arthritis Care & Research.
HCQ is known to cause retinal damage in some patients, binding to and accumulating in the retinal pigment epithelium melanin. If undetected, patients may gradually lose central vision. There is particular risk for HCQ-induced retinal damage in patients on higher doses (5 mg/kg or greater of actual weight), HCQ exposure over 5 years, or renal, hepatic, or retinal disease, the research team explained.
Baseline retinal examinations for first-time HCQ users were first recommended by American Academy of Ophthalmology guidelines in 2002, and in 2009 the American College of Rheumatology developed guidelines specifically for SLE that also recommended a funduscopic exam within 1 year of starting HCQ.
The proportion of patients beginning treatment with HCQ who received a retinal exam only slightly increased from 31% in 2001 to 34.4% in 2009 (P value for linear trend over time = .12).
Women were significantly more likely to have a baseline retinal exam (odds ratio, 1.30; 95% confidence interval, 1.08-1.55), and certain sociodemographic factors also predicted who received a baseline exam, likely reflecting problems with adherence and access to health care.
For example, the likelihood of having a baseline exam proved significantly lower for blacks and American Indian/Alaska natives (OR, 0.85; 95% CI, 0.77-0.93; and OR, 0.69; 95% CI, 0.47-1.02) than for whites, whereas Asian Americans had greater odds than whites (OR, 1.30; 95% CI, 1.06-1.60).
“We have previously observed that these sociodemographic factors have been associated with differences in both access to care and adherence in the SLE Medicaid population for indicated care, including infection prevention, adherence to indicated medications including HCQ, treatments for lupus nephritis, and choices in renal replacement therapies and use of erythropoietin-stimulating agents among SLE end-stage renal disease patients,” they wrote.
Patients who had received multiple SLE-related laboratory tests (OR, 1.33; 95% CI, 1.16-1.52) and liver function tests (OR, 1.16; 95% CI, 1.04-1.29) were also more likely to have an eye exam before commencing HCQ.
The researchers said both patient and physician factors likely played a role in adherence to preventive medical care.
“While adherence to daily dosing, by the latest recommendations less than 5 mg/kg actual weight, is a more important factor determining renal toxicity risk, baseline examinations are necessary to avoid treating patients with preexisting retinal disease and to allow early detection of changes from baseline,” the research team concluded.
The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
SOURCE: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: Of 12,755 Medicaid SLE patients, 32.5% received a baseline dilated-eye exam 30 days before, and up to 1 year after, starting treatment with HCQ.
Study details: An analysis of 12,755 Medicaid SLE patients from 29 of the most populated U.S. states between 2001 and 2010 who had initiated HCQ.
Disclosures: The work was supported by grant awards from the Rheumatology Research Foundation and the National Institutes of Health. No disclosures were made.
Source: Lin T et al. Arthritis Care Res. 2018 Feb 6. doi: 10.1002/acr.23530.
JAK inhibitors look good for severe alopecia areata treatment
said Lucy Yichu Liu, MD, and Brett Andrew King, MD, of Yale University, New Haven, Conn.
Standard medical therapies for alopecia areata – usually topical or injected corticosteroids and allergic contact sensitization – are not very effective for severe disease, particularly alopecia totalis and alopecia universalis. The Janus kinase (JAK) pathway recently has been suggested as a target for treatment.
Dr. Liu and Dr. King reviewed several studies, including a retrospective cohort study of 13 patients aged 12-17 years, in which 7 patients had 100% hair loss and 6 had 20%-70% scalp hair loss. The adolescents were treated with the JAK1/3 inhibitor tofacitinib citrate 5 mg twice daily for 2-16 months (median, 5 months). That led to 93% median improvement in Severity of Alopecia Tool (SALT) score (range, 1%-100%) from baseline. Nine patients experienced hair regrowth. There were mild adverse effects, such as upper respiratory infections and headaches.
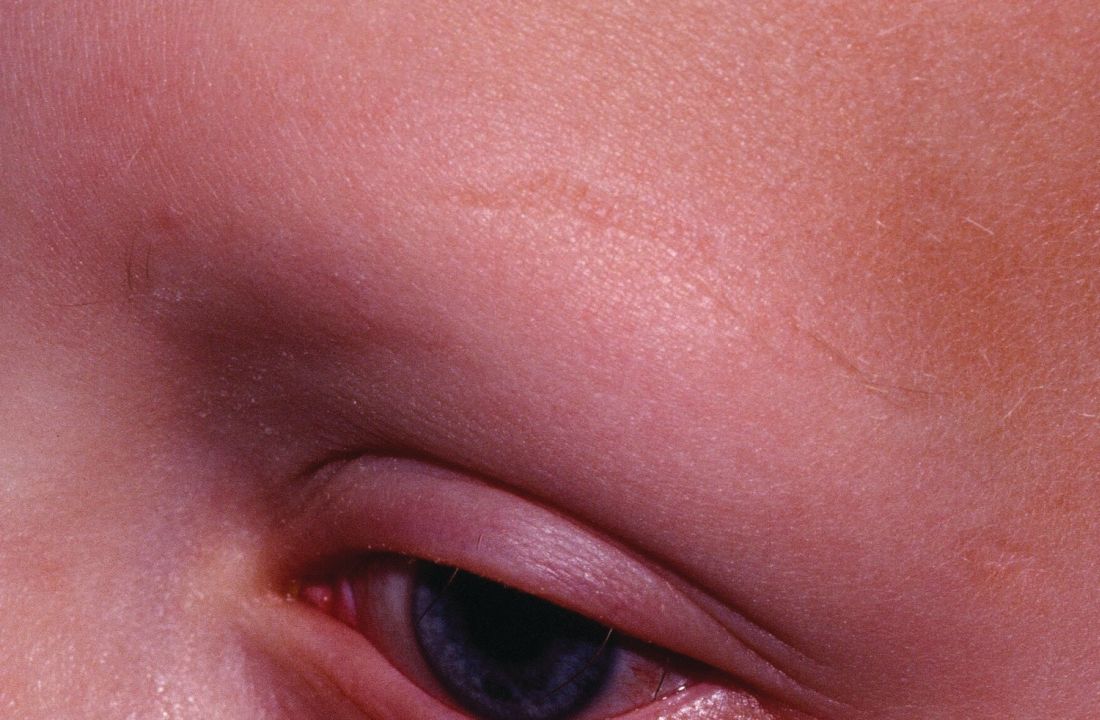
In a retrospective cohort study of 90 adults taking tofacitinib at a dosage of 5-10 mg twice daily for 4 months or longer with or without prednisone (300 mg once monthly for three doses), patients were divided into those who were more or less likely to respond based on duration of disease. Of 65 patients with alopecia totalis, or alopecia universalis that had lasted 10 years or less, or alopecia areata, 77% had some hair regrowth; 58% had more than 50% improvement from baseline, and 20% achieved full regrowth of hair, Dr. Liu and Dr. King reported in the Journal of Investigative Dermatology Symposium Proceedings.
“Given the finding in adults that complete scalp hair loss for more than 10 years is less likely to respond to treatment, there may be merit to pursuing treatment, even if only intermittently, in adolescents or even younger patients with stable, severe alopecia areata, to prevent irreversible hair loss in the future,” they wrote.
A patient with alopecia universalis achieved partial scalp hair regrowth and complete eyebrow regrowth with compounded ruxolitinib, a topical JAK inhibitor, according to a 2016 case report. Dr. Liu and Dr. King reported that clinical trials with topical JAK inhibitors, including topical tofacitinib and topical ruxolitinib, currently are ongoing.
SOURCE: Liu LY et al. J Investig Dermatol Symp Proc. 2018 Jan. doi: 10.1016/j.jisp.2017.10.003.
said Lucy Yichu Liu, MD, and Brett Andrew King, MD, of Yale University, New Haven, Conn.
Standard medical therapies for alopecia areata – usually topical or injected corticosteroids and allergic contact sensitization – are not very effective for severe disease, particularly alopecia totalis and alopecia universalis. The Janus kinase (JAK) pathway recently has been suggested as a target for treatment.
Dr. Liu and Dr. King reviewed several studies, including a retrospective cohort study of 13 patients aged 12-17 years, in which 7 patients had 100% hair loss and 6 had 20%-70% scalp hair loss. The adolescents were treated with the JAK1/3 inhibitor tofacitinib citrate 5 mg twice daily for 2-16 months (median, 5 months). That led to 93% median improvement in Severity of Alopecia Tool (SALT) score (range, 1%-100%) from baseline. Nine patients experienced hair regrowth. There were mild adverse effects, such as upper respiratory infections and headaches.
In a retrospective cohort study of 90 adults taking tofacitinib at a dosage of 5-10 mg twice daily for 4 months or longer with or without prednisone (300 mg once monthly for three doses), patients were divided into those who were more or less likely to respond based on duration of disease. Of 65 patients with alopecia totalis, or alopecia universalis that had lasted 10 years or less, or alopecia areata, 77% had some hair regrowth; 58% had more than 50% improvement from baseline, and 20% achieved full regrowth of hair, Dr. Liu and Dr. King reported in the Journal of Investigative Dermatology Symposium Proceedings.
“Given the finding in adults that complete scalp hair loss for more than 10 years is less likely to respond to treatment, there may be merit to pursuing treatment, even if only intermittently, in adolescents or even younger patients with stable, severe alopecia areata, to prevent irreversible hair loss in the future,” they wrote.
A patient with alopecia universalis achieved partial scalp hair regrowth and complete eyebrow regrowth with compounded ruxolitinib, a topical JAK inhibitor, according to a 2016 case report. Dr. Liu and Dr. King reported that clinical trials with topical JAK inhibitors, including topical tofacitinib and topical ruxolitinib, currently are ongoing.
SOURCE: Liu LY et al. J Investig Dermatol Symp Proc. 2018 Jan. doi: 10.1016/j.jisp.2017.10.003.
said Lucy Yichu Liu, MD, and Brett Andrew King, MD, of Yale University, New Haven, Conn.
Standard medical therapies for alopecia areata – usually topical or injected corticosteroids and allergic contact sensitization – are not very effective for severe disease, particularly alopecia totalis and alopecia universalis. The Janus kinase (JAK) pathway recently has been suggested as a target for treatment.
Dr. Liu and Dr. King reviewed several studies, including a retrospective cohort study of 13 patients aged 12-17 years, in which 7 patients had 100% hair loss and 6 had 20%-70% scalp hair loss. The adolescents were treated with the JAK1/3 inhibitor tofacitinib citrate 5 mg twice daily for 2-16 months (median, 5 months). That led to 93% median improvement in Severity of Alopecia Tool (SALT) score (range, 1%-100%) from baseline. Nine patients experienced hair regrowth. There were mild adverse effects, such as upper respiratory infections and headaches.
In a retrospective cohort study of 90 adults taking tofacitinib at a dosage of 5-10 mg twice daily for 4 months or longer with or without prednisone (300 mg once monthly for three doses), patients were divided into those who were more or less likely to respond based on duration of disease. Of 65 patients with alopecia totalis, or alopecia universalis that had lasted 10 years or less, or alopecia areata, 77% had some hair regrowth; 58% had more than 50% improvement from baseline, and 20% achieved full regrowth of hair, Dr. Liu and Dr. King reported in the Journal of Investigative Dermatology Symposium Proceedings.
“Given the finding in adults that complete scalp hair loss for more than 10 years is less likely to respond to treatment, there may be merit to pursuing treatment, even if only intermittently, in adolescents or even younger patients with stable, severe alopecia areata, to prevent irreversible hair loss in the future,” they wrote.
A patient with alopecia universalis achieved partial scalp hair regrowth and complete eyebrow regrowth with compounded ruxolitinib, a topical JAK inhibitor, according to a 2016 case report. Dr. Liu and Dr. King reported that clinical trials with topical JAK inhibitors, including topical tofacitinib and topical ruxolitinib, currently are ongoing.
SOURCE: Liu LY et al. J Investig Dermatol Symp Proc. 2018 Jan. doi: 10.1016/j.jisp.2017.10.003.
FROM JOURNAL OF INVESTIGATIVE DERMATOLOGY SYMPOSIUM PROCEEDINGS
Predictors may help to spot risk for hydroxychloroquine nonadherence
who are newly prescribed the antimalarial, an administrative claims database study has revealed.
Investigators from Brigham and Women’s Hospital, Boston, used prescription refill data to assess nonadherence, hoping to gain greater insight into predictors of nonadherence to HCQ than past studies of nonadherence to the drug, which have been limited by their small and cross-sectional nature and the fact that they often relied on self-reported measures of adherence that were unable to capture the dynamic nature of adherence behavior over time.
In the current study, Candace H. Feldman, MD, ScD, and her coinvestigators used Medicaid data from 28 U.S. states to identify 10,406 adult HCQ initiators with SLE during 2001-2010. Patients included in the study were required to have more than 365 days of continuous follow-up documented.
The researchers described four distinctive monthly patterns of behavior during the first year of use, in which they defined nonadherence as less than 80% of days covered per month by a HCQ prescription. Group 1 comprised 36% who were “persistent nonadherers” who had very few HCQ refills after the initial dispensing.
Almost half of the cohort (47%) formed two dynamic patterns of partial adherence (groups 2 and 3). The trajectories for these groups were similar until month 5, when they diverged: group 3 improved slightly and then reached a plateau, whereas group 2 became nearly completely nonadherent for the remainder of follow-up. At this 5-month point of divergence, belonging to group 2 was more likely among patients with younger age and antidepressant use. Also, patients in group 3 had more hospitalizations beginning at 4 months and longer hospitalizations at 5 months.
For these two “undecided” groups, 5 months may be a “critical juncture” for physicians to intervene, the authors said.
“Five months might also be the point at which patients feel that they have adequately trialed the medication, and if there is no symptomatic improvement, they discontinue. With the growing body of literature suggesting long-term preventive effects from HCQ, increased provider and patient education at this juncture may be beneficial,” they wrote.
Group 4 had persistent adherence and constituted 17% of the cohort, although this group also experienced a decline in adherence at 9 months.
The mean age of group 4 was about 40 years, which was significantly older than 37 years in groups 1-3. Blacks comprised the highest percentage of patients in groups 1-3 (43%-45%), whereas whites at 40% were the highest proportion in group 4. Individuals in group 4 also had slightly higher average income than did group 1 (mean $46,000 vs. $44,000). The index for SLE risk adjustment was highest for group 4 patients (1.3 vs. 0.9-1.1 for other groups), indicating they may have had more SLE-related comorbidities. Group 4 patients also had a greater average number of medications dispensed and a higher mean daily prednisone-equivalent dose.
Patients aged 18-50 or with black race or Hispanic ethnicity were significantly more likely to be in one of the nonadherent trajectory groups (1, 2, and 3), whereas Asians were less likely to be in group 1 than in group 4, compared with whites.
Diabetes made patients more likely to belong to group 1 than group 4, whereas each unit increase in the SLE risk-adjustment index increased the odds of belonging to group 1 vs. 4. Antidepressant use was associated with greater likelihood of belong to groups 1 or 2 vs. group 4.
Addressing potentially modifiable factors such as ensuring sustained access to health care, particularly for patients with severe disease, might go some way to improving adherence, suggested the researchers, who also noted that increased counseling and support at the time of the first HCQ prescription and throughout the first year of use are also needed in order to “promote more sustained patterns of adherence for all patients.”
The study was funded by a Rheumatology Research Foundation Investigator Award and individual grant awards from the National Institutes of Health. No relevant financial disclosures were declared by the authors.
SOURCE: Feldman C et al. Semin Arthritis Rheum. 2018 Jan 8. doi: 10.1016/j.semarthrit.2018.01.002
who are newly prescribed the antimalarial, an administrative claims database study has revealed.
Investigators from Brigham and Women’s Hospital, Boston, used prescription refill data to assess nonadherence, hoping to gain greater insight into predictors of nonadherence to HCQ than past studies of nonadherence to the drug, which have been limited by their small and cross-sectional nature and the fact that they often relied on self-reported measures of adherence that were unable to capture the dynamic nature of adherence behavior over time.
In the current study, Candace H. Feldman, MD, ScD, and her coinvestigators used Medicaid data from 28 U.S. states to identify 10,406 adult HCQ initiators with SLE during 2001-2010. Patients included in the study were required to have more than 365 days of continuous follow-up documented.
The researchers described four distinctive monthly patterns of behavior during the first year of use, in which they defined nonadherence as less than 80% of days covered per month by a HCQ prescription. Group 1 comprised 36% who were “persistent nonadherers” who had very few HCQ refills after the initial dispensing.
Almost half of the cohort (47%) formed two dynamic patterns of partial adherence (groups 2 and 3). The trajectories for these groups were similar until month 5, when they diverged: group 3 improved slightly and then reached a plateau, whereas group 2 became nearly completely nonadherent for the remainder of follow-up. At this 5-month point of divergence, belonging to group 2 was more likely among patients with younger age and antidepressant use. Also, patients in group 3 had more hospitalizations beginning at 4 months and longer hospitalizations at 5 months.
For these two “undecided” groups, 5 months may be a “critical juncture” for physicians to intervene, the authors said.
“Five months might also be the point at which patients feel that they have adequately trialed the medication, and if there is no symptomatic improvement, they discontinue. With the growing body of literature suggesting long-term preventive effects from HCQ, increased provider and patient education at this juncture may be beneficial,” they wrote.
Group 4 had persistent adherence and constituted 17% of the cohort, although this group also experienced a decline in adherence at 9 months.
The mean age of group 4 was about 40 years, which was significantly older than 37 years in groups 1-3. Blacks comprised the highest percentage of patients in groups 1-3 (43%-45%), whereas whites at 40% were the highest proportion in group 4. Individuals in group 4 also had slightly higher average income than did group 1 (mean $46,000 vs. $44,000). The index for SLE risk adjustment was highest for group 4 patients (1.3 vs. 0.9-1.1 for other groups), indicating they may have had more SLE-related comorbidities. Group 4 patients also had a greater average number of medications dispensed and a higher mean daily prednisone-equivalent dose.
Patients aged 18-50 or with black race or Hispanic ethnicity were significantly more likely to be in one of the nonadherent trajectory groups (1, 2, and 3), whereas Asians were less likely to be in group 1 than in group 4, compared with whites.
Diabetes made patients more likely to belong to group 1 than group 4, whereas each unit increase in the SLE risk-adjustment index increased the odds of belonging to group 1 vs. 4. Antidepressant use was associated with greater likelihood of belong to groups 1 or 2 vs. group 4.
Addressing potentially modifiable factors such as ensuring sustained access to health care, particularly for patients with severe disease, might go some way to improving adherence, suggested the researchers, who also noted that increased counseling and support at the time of the first HCQ prescription and throughout the first year of use are also needed in order to “promote more sustained patterns of adherence for all patients.”
The study was funded by a Rheumatology Research Foundation Investigator Award and individual grant awards from the National Institutes of Health. No relevant financial disclosures were declared by the authors.
SOURCE: Feldman C et al. Semin Arthritis Rheum. 2018 Jan 8. doi: 10.1016/j.semarthrit.2018.01.002
who are newly prescribed the antimalarial, an administrative claims database study has revealed.
Investigators from Brigham and Women’s Hospital, Boston, used prescription refill data to assess nonadherence, hoping to gain greater insight into predictors of nonadherence to HCQ than past studies of nonadherence to the drug, which have been limited by their small and cross-sectional nature and the fact that they often relied on self-reported measures of adherence that were unable to capture the dynamic nature of adherence behavior over time.
In the current study, Candace H. Feldman, MD, ScD, and her coinvestigators used Medicaid data from 28 U.S. states to identify 10,406 adult HCQ initiators with SLE during 2001-2010. Patients included in the study were required to have more than 365 days of continuous follow-up documented.
The researchers described four distinctive monthly patterns of behavior during the first year of use, in which they defined nonadherence as less than 80% of days covered per month by a HCQ prescription. Group 1 comprised 36% who were “persistent nonadherers” who had very few HCQ refills after the initial dispensing.
Almost half of the cohort (47%) formed two dynamic patterns of partial adherence (groups 2 and 3). The trajectories for these groups were similar until month 5, when they diverged: group 3 improved slightly and then reached a plateau, whereas group 2 became nearly completely nonadherent for the remainder of follow-up. At this 5-month point of divergence, belonging to group 2 was more likely among patients with younger age and antidepressant use. Also, patients in group 3 had more hospitalizations beginning at 4 months and longer hospitalizations at 5 months.
For these two “undecided” groups, 5 months may be a “critical juncture” for physicians to intervene, the authors said.
“Five months might also be the point at which patients feel that they have adequately trialed the medication, and if there is no symptomatic improvement, they discontinue. With the growing body of literature suggesting long-term preventive effects from HCQ, increased provider and patient education at this juncture may be beneficial,” they wrote.
Group 4 had persistent adherence and constituted 17% of the cohort, although this group also experienced a decline in adherence at 9 months.
The mean age of group 4 was about 40 years, which was significantly older than 37 years in groups 1-3. Blacks comprised the highest percentage of patients in groups 1-3 (43%-45%), whereas whites at 40% were the highest proportion in group 4. Individuals in group 4 also had slightly higher average income than did group 1 (mean $46,000 vs. $44,000). The index for SLE risk adjustment was highest for group 4 patients (1.3 vs. 0.9-1.1 for other groups), indicating they may have had more SLE-related comorbidities. Group 4 patients also had a greater average number of medications dispensed and a higher mean daily prednisone-equivalent dose.
Patients aged 18-50 or with black race or Hispanic ethnicity were significantly more likely to be in one of the nonadherent trajectory groups (1, 2, and 3), whereas Asians were less likely to be in group 1 than in group 4, compared with whites.
Diabetes made patients more likely to belong to group 1 than group 4, whereas each unit increase in the SLE risk-adjustment index increased the odds of belonging to group 1 vs. 4. Antidepressant use was associated with greater likelihood of belong to groups 1 or 2 vs. group 4.
Addressing potentially modifiable factors such as ensuring sustained access to health care, particularly for patients with severe disease, might go some way to improving adherence, suggested the researchers, who also noted that increased counseling and support at the time of the first HCQ prescription and throughout the first year of use are also needed in order to “promote more sustained patterns of adherence for all patients.”
The study was funded by a Rheumatology Research Foundation Investigator Award and individual grant awards from the National Institutes of Health. No relevant financial disclosures were declared by the authors.
SOURCE: Feldman C et al. Semin Arthritis Rheum. 2018 Jan 8. doi: 10.1016/j.semarthrit.2018.01.002
FROM SEMINARS IN ARTHRITIS & RHEUMATISM
Key clinical point: Five months after the first HCQ prescription might be a critical time point to review and educate patients on the importance of continuing treatment, particularly for patients who are “partial” adherers.
Major finding: Almost half of the cohort (47%) formed two dynamic patterns of partial adherence.
Data source: A longitudinal study of 10,406 Medicaid beneficiaries with SLE who were prescribed hydroxychloroquine for the first time.
Disclosures: The study was funded by a Rheumatology Research Foundation Investigator Award and individual grant awards from the National Institutes of Health. No relevant financial disclosures were declared by the authors.
Source: Feldman C et al. Semin Arthritis Rheum. 2018 Jan 8. doi: 10.1016/j.semarthrit.2018.01.002
Cases link vitiligoid lichen sclerosus and darker skin
, said Margaret H. Dennin, of the University of Chicago, and her associates.
Vitiligoid lichen sclerosus is a superficial variant of lichen sclerosus (LS), in which the lesion clinically appears to be vitiligo, but histologically is consistent with LS.
Seven dark-skinned girls aged 3-9 years had symptomatic (pruritus, pain, bleeding, constipation) depigmented patches of the vulvar or perianal region; three had purpuric lesions. None of the patients had atrophy or scarring, and they had no depigmentation anywhere else on their bodies. Follow-up was an average 2 years (range 3 months to 4 years).
Treatment with high-potency topical steroids, calcineurin inhibitors, or both resulted in improvement or resolution of their symptoms in all cases, but there was mild or no improvement in the depigmentation. Biopsies were not performed because of the patients’ young age and the location of the lesions, the investigators said.
The term vitiligoid lichen sclerosus was first coined in 1961 by Borda et al. when depigmented patches, as seen in both conditions, constituted the clinical appearance, but lacked the inflammation, atrophy, and sclerosis of typical LS. Histologically, these lesions were like LS, “based on the presence of a thin band of papillary dermal sclerosis,” Ms. Dennin and her associates said. Borda et al. suggested that vitiligoid lichen sclerosus might be limited to dark-skinned people, and recent reports support this. Alternatively, it may be that the depigmentation simply is more obvious on dark-skinned people, and asymptomatic cases go unnoticed on lighter-skinned people, the investigators surmised.
Both vitiligo and LS are autoimmune cutaneous disorders, and they both often affect the anogenital region. The conditions “may be linked through a common autoimmune response from exposed intracellular or altered cell surface antigens on damaged melanocytes,” the investigators said. “Histologic evidence demonstrates that development of vitiligo involves a preceding lichenoid inflammatory reaction that may trigger an autoimmune reaction to melanocytes, decreasing their number. Evolving vitiligo with a lichenoid reaction may result in epitope spreading and the development of LS.”
The study is limited by its retrospective nature, small sample size, and lack of biopsies, the researchers noted. Larger studies are needed to look at the overlap of the conditions, and “understand the true prevalence of vitiligoid lichen sclerosus,” Ms. Dennin and her associates said.
Read more in Pediatric Dermatology (2018. doi: 10.1111/pde.13399).
, said Margaret H. Dennin, of the University of Chicago, and her associates.
Vitiligoid lichen sclerosus is a superficial variant of lichen sclerosus (LS), in which the lesion clinically appears to be vitiligo, but histologically is consistent with LS.
Seven dark-skinned girls aged 3-9 years had symptomatic (pruritus, pain, bleeding, constipation) depigmented patches of the vulvar or perianal region; three had purpuric lesions. None of the patients had atrophy or scarring, and they had no depigmentation anywhere else on their bodies. Follow-up was an average 2 years (range 3 months to 4 years).
Treatment with high-potency topical steroids, calcineurin inhibitors, or both resulted in improvement or resolution of their symptoms in all cases, but there was mild or no improvement in the depigmentation. Biopsies were not performed because of the patients’ young age and the location of the lesions, the investigators said.
The term vitiligoid lichen sclerosus was first coined in 1961 by Borda et al. when depigmented patches, as seen in both conditions, constituted the clinical appearance, but lacked the inflammation, atrophy, and sclerosis of typical LS. Histologically, these lesions were like LS, “based on the presence of a thin band of papillary dermal sclerosis,” Ms. Dennin and her associates said. Borda et al. suggested that vitiligoid lichen sclerosus might be limited to dark-skinned people, and recent reports support this. Alternatively, it may be that the depigmentation simply is more obvious on dark-skinned people, and asymptomatic cases go unnoticed on lighter-skinned people, the investigators surmised.
Both vitiligo and LS are autoimmune cutaneous disorders, and they both often affect the anogenital region. The conditions “may be linked through a common autoimmune response from exposed intracellular or altered cell surface antigens on damaged melanocytes,” the investigators said. “Histologic evidence demonstrates that development of vitiligo involves a preceding lichenoid inflammatory reaction that may trigger an autoimmune reaction to melanocytes, decreasing their number. Evolving vitiligo with a lichenoid reaction may result in epitope spreading and the development of LS.”
The study is limited by its retrospective nature, small sample size, and lack of biopsies, the researchers noted. Larger studies are needed to look at the overlap of the conditions, and “understand the true prevalence of vitiligoid lichen sclerosus,” Ms. Dennin and her associates said.
Read more in Pediatric Dermatology (2018. doi: 10.1111/pde.13399).
, said Margaret H. Dennin, of the University of Chicago, and her associates.
Vitiligoid lichen sclerosus is a superficial variant of lichen sclerosus (LS), in which the lesion clinically appears to be vitiligo, but histologically is consistent with LS.
Seven dark-skinned girls aged 3-9 years had symptomatic (pruritus, pain, bleeding, constipation) depigmented patches of the vulvar or perianal region; three had purpuric lesions. None of the patients had atrophy or scarring, and they had no depigmentation anywhere else on their bodies. Follow-up was an average 2 years (range 3 months to 4 years).
Treatment with high-potency topical steroids, calcineurin inhibitors, or both resulted in improvement or resolution of their symptoms in all cases, but there was mild or no improvement in the depigmentation. Biopsies were not performed because of the patients’ young age and the location of the lesions, the investigators said.
The term vitiligoid lichen sclerosus was first coined in 1961 by Borda et al. when depigmented patches, as seen in both conditions, constituted the clinical appearance, but lacked the inflammation, atrophy, and sclerosis of typical LS. Histologically, these lesions were like LS, “based on the presence of a thin band of papillary dermal sclerosis,” Ms. Dennin and her associates said. Borda et al. suggested that vitiligoid lichen sclerosus might be limited to dark-skinned people, and recent reports support this. Alternatively, it may be that the depigmentation simply is more obvious on dark-skinned people, and asymptomatic cases go unnoticed on lighter-skinned people, the investigators surmised.
Both vitiligo and LS are autoimmune cutaneous disorders, and they both often affect the anogenital region. The conditions “may be linked through a common autoimmune response from exposed intracellular or altered cell surface antigens on damaged melanocytes,” the investigators said. “Histologic evidence demonstrates that development of vitiligo involves a preceding lichenoid inflammatory reaction that may trigger an autoimmune reaction to melanocytes, decreasing their number. Evolving vitiligo with a lichenoid reaction may result in epitope spreading and the development of LS.”
The study is limited by its retrospective nature, small sample size, and lack of biopsies, the researchers noted. Larger studies are needed to look at the overlap of the conditions, and “understand the true prevalence of vitiligoid lichen sclerosus,” Ms. Dennin and her associates said.
Read more in Pediatric Dermatology (2018. doi: 10.1111/pde.13399).
FROM PEDIATRIC DERMATOLOGY
Neutrophilic urticarial dermatosis is usually misdiagnosed
GENEVA – Neutrophilic urticarial dermatosis (NUD) in patients with systemic lupus erythematosus (SLE) is “almost always” initially misdiagnosed as a lupus flare and treated inappropriately, Dan Lipsker, MD, PhD, said in a plenary address at the annual congress of the European Academy of Dermatology and Venereology.
“This is a condition that is underdiagnosed and overtreated,” declared Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).

NUD is not rare. Dr. Lipsker estimates it occurs in 1%-2% of patients with SLE. In a retrospective study of seven patients with NUD and SLE, he and his colleagues reported that NUD was initially misdiagnosed as a lupus flare in 4 patients, who were then treated with immunosuppressive drugs (Medicine. 2014 Dec;93[29]:e351]). “That’s quite logical because the patients had a rash, fever, and joint pain,” the dermatologist noted.
However, : It won’t alleviate the symptoms and needlessly exposes the patient to drug toxicities.
The treatment for NUD is not prednisone, mycophenolate mofetil, an antimalarial, or other drugs conventionally prescribed for SLE; it’s a drug that inhibits neutrophil migration, such as dapsone at 50-200 mg per day or colchicine at 0.5-1.0 mg per day. Typically, within just a few days after starting the appropriate therapy, the joint pain and rash of NUD are gone, according to Dr. Lipsker.
Making the diagnosis
The rash of NUD is distinctly different from a classic lupus rash. It consists of pale red macules or slightly raised nonpruritic papules. Individual lesions will disappear spontaneously within 24-48 hours.
The histopathology of NUD is characteristic of a neutrophilic dermatosis. On biopsy, an intense neutrophilic perivascular and interstitial infiltrate with leukocytoclasia is seen. There is no damage to the blood vessel walls, which readily distinguishes NUD from urticarial vasculitis.
Other neutrophilic dermatoses have also been reported with increased frequency in patients with SLE. These include Sweet syndrome, pyoderma gangrenosum, bullous SLE, amicrobial pustulosis of the folds, and palisaded neutrophilic granulomatous dermatitis. Dr. Lipsker lumps them, together with NUD, as neutrophilic cutaneous lupus erythematosus. Affected SLE patients have an exaggerated innate immune response. It is as yet unclear if these neutrophilic dermatoses have prognostic significance in the setting of SLE, he said.
Dr. Lipsker reported having no financial conflicts of interest regarding his presentation.
GENEVA – Neutrophilic urticarial dermatosis (NUD) in patients with systemic lupus erythematosus (SLE) is “almost always” initially misdiagnosed as a lupus flare and treated inappropriately, Dan Lipsker, MD, PhD, said in a plenary address at the annual congress of the European Academy of Dermatology and Venereology.
“This is a condition that is underdiagnosed and overtreated,” declared Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).
NUD is not rare. Dr. Lipsker estimates it occurs in 1%-2% of patients with SLE. In a retrospective study of seven patients with NUD and SLE, he and his colleagues reported that NUD was initially misdiagnosed as a lupus flare in 4 patients, who were then treated with immunosuppressive drugs (Medicine. 2014 Dec;93[29]:e351]). “That’s quite logical because the patients had a rash, fever, and joint pain,” the dermatologist noted.
However, : It won’t alleviate the symptoms and needlessly exposes the patient to drug toxicities.
The treatment for NUD is not prednisone, mycophenolate mofetil, an antimalarial, or other drugs conventionally prescribed for SLE; it’s a drug that inhibits neutrophil migration, such as dapsone at 50-200 mg per day or colchicine at 0.5-1.0 mg per day. Typically, within just a few days after starting the appropriate therapy, the joint pain and rash of NUD are gone, according to Dr. Lipsker.
Making the diagnosis
The rash of NUD is distinctly different from a classic lupus rash. It consists of pale red macules or slightly raised nonpruritic papules. Individual lesions will disappear spontaneously within 24-48 hours.
The histopathology of NUD is characteristic of a neutrophilic dermatosis. On biopsy, an intense neutrophilic perivascular and interstitial infiltrate with leukocytoclasia is seen. There is no damage to the blood vessel walls, which readily distinguishes NUD from urticarial vasculitis.
Other neutrophilic dermatoses have also been reported with increased frequency in patients with SLE. These include Sweet syndrome, pyoderma gangrenosum, bullous SLE, amicrobial pustulosis of the folds, and palisaded neutrophilic granulomatous dermatitis. Dr. Lipsker lumps them, together with NUD, as neutrophilic cutaneous lupus erythematosus. Affected SLE patients have an exaggerated innate immune response. It is as yet unclear if these neutrophilic dermatoses have prognostic significance in the setting of SLE, he said.
Dr. Lipsker reported having no financial conflicts of interest regarding his presentation.
GENEVA – Neutrophilic urticarial dermatosis (NUD) in patients with systemic lupus erythematosus (SLE) is “almost always” initially misdiagnosed as a lupus flare and treated inappropriately, Dan Lipsker, MD, PhD, said in a plenary address at the annual congress of the European Academy of Dermatology and Venereology.
“This is a condition that is underdiagnosed and overtreated,” declared Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).
NUD is not rare. Dr. Lipsker estimates it occurs in 1%-2% of patients with SLE. In a retrospective study of seven patients with NUD and SLE, he and his colleagues reported that NUD was initially misdiagnosed as a lupus flare in 4 patients, who were then treated with immunosuppressive drugs (Medicine. 2014 Dec;93[29]:e351]). “That’s quite logical because the patients had a rash, fever, and joint pain,” the dermatologist noted.
However, : It won’t alleviate the symptoms and needlessly exposes the patient to drug toxicities.
The treatment for NUD is not prednisone, mycophenolate mofetil, an antimalarial, or other drugs conventionally prescribed for SLE; it’s a drug that inhibits neutrophil migration, such as dapsone at 50-200 mg per day or colchicine at 0.5-1.0 mg per day. Typically, within just a few days after starting the appropriate therapy, the joint pain and rash of NUD are gone, according to Dr. Lipsker.
Making the diagnosis
The rash of NUD is distinctly different from a classic lupus rash. It consists of pale red macules or slightly raised nonpruritic papules. Individual lesions will disappear spontaneously within 24-48 hours.
The histopathology of NUD is characteristic of a neutrophilic dermatosis. On biopsy, an intense neutrophilic perivascular and interstitial infiltrate with leukocytoclasia is seen. There is no damage to the blood vessel walls, which readily distinguishes NUD from urticarial vasculitis.
Other neutrophilic dermatoses have also been reported with increased frequency in patients with SLE. These include Sweet syndrome, pyoderma gangrenosum, bullous SLE, amicrobial pustulosis of the folds, and palisaded neutrophilic granulomatous dermatitis. Dr. Lipsker lumps them, together with NUD, as neutrophilic cutaneous lupus erythematosus. Affected SLE patients have an exaggerated innate immune response. It is as yet unclear if these neutrophilic dermatoses have prognostic significance in the setting of SLE, he said.
Dr. Lipsker reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Rituximab tackles relapse of severe, difficult-to-treat pemphigus
, based on data from a case series of 11 patients.
“We found that treatment with rituximab alone, even at a low dose, not only prevented relapse but also maintained complete remission with a better benefit-to-risk ratio than treatment with corticosteroids,” Julia Sanchez, MD, of Reims (France) University Hospital and her colleagues reported in a research letter in JAMA Dermatology.
The study population consisted of patients diagnosed with pemphigus at a single center from Jan. 1, 2014, to Dec. 31, 2014, and treated with at least one cycle of rituximab for corticosteroid dependence, corticosteroid resistance, or adverse events. All the patients were in remission at the time of the first maintenance dose of rituximab.
All patients received a 1-g rituximab infusion every 6 months for 24-67 months; some patients changed to a once-yearly dose after 18 months. Although 5 patients experienced grade 3 or 4 adverse events (1 patient had sepsis; 2, diabetes; 1, hypertension; and 2, endocrine disorders) between the initial therapy cycle and the first rituximab maintenance infusion, no adverse events were reported by any of the 11 patients during the maintenance therapy period.
All 11 patients remained in remission after their last follow-up visit (an average of 78 months after the first cycle), at which point 10 patients had discontinued the therapy.
“A progressive decrease in serum anti-desmoglein autoantibody levels to less than 14 U/mL occurred in all cases along with clinical complete remission even after maintenance therapy cessation,” Dr. Sanchez and her associates noted.
Future research should address questions including the optimal dose and dosing frequency of rituximab, as well as the cost-effectiveness of the treatment and criteria for treatment withdrawal, they said.
The researchers had no relevant financial conflicts disclosures.
SOURCE: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
, based on data from a case series of 11 patients.
“We found that treatment with rituximab alone, even at a low dose, not only prevented relapse but also maintained complete remission with a better benefit-to-risk ratio than treatment with corticosteroids,” Julia Sanchez, MD, of Reims (France) University Hospital and her colleagues reported in a research letter in JAMA Dermatology.
The study population consisted of patients diagnosed with pemphigus at a single center from Jan. 1, 2014, to Dec. 31, 2014, and treated with at least one cycle of rituximab for corticosteroid dependence, corticosteroid resistance, or adverse events. All the patients were in remission at the time of the first maintenance dose of rituximab.
All patients received a 1-g rituximab infusion every 6 months for 24-67 months; some patients changed to a once-yearly dose after 18 months. Although 5 patients experienced grade 3 or 4 adverse events (1 patient had sepsis; 2, diabetes; 1, hypertension; and 2, endocrine disorders) between the initial therapy cycle and the first rituximab maintenance infusion, no adverse events were reported by any of the 11 patients during the maintenance therapy period.
All 11 patients remained in remission after their last follow-up visit (an average of 78 months after the first cycle), at which point 10 patients had discontinued the therapy.
“A progressive decrease in serum anti-desmoglein autoantibody levels to less than 14 U/mL occurred in all cases along with clinical complete remission even after maintenance therapy cessation,” Dr. Sanchez and her associates noted.
Future research should address questions including the optimal dose and dosing frequency of rituximab, as well as the cost-effectiveness of the treatment and criteria for treatment withdrawal, they said.
The researchers had no relevant financial conflicts disclosures.
SOURCE: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
, based on data from a case series of 11 patients.
“We found that treatment with rituximab alone, even at a low dose, not only prevented relapse but also maintained complete remission with a better benefit-to-risk ratio than treatment with corticosteroids,” Julia Sanchez, MD, of Reims (France) University Hospital and her colleagues reported in a research letter in JAMA Dermatology.
The study population consisted of patients diagnosed with pemphigus at a single center from Jan. 1, 2014, to Dec. 31, 2014, and treated with at least one cycle of rituximab for corticosteroid dependence, corticosteroid resistance, or adverse events. All the patients were in remission at the time of the first maintenance dose of rituximab.
All patients received a 1-g rituximab infusion every 6 months for 24-67 months; some patients changed to a once-yearly dose after 18 months. Although 5 patients experienced grade 3 or 4 adverse events (1 patient had sepsis; 2, diabetes; 1, hypertension; and 2, endocrine disorders) between the initial therapy cycle and the first rituximab maintenance infusion, no adverse events were reported by any of the 11 patients during the maintenance therapy period.
All 11 patients remained in remission after their last follow-up visit (an average of 78 months after the first cycle), at which point 10 patients had discontinued the therapy.
“A progressive decrease in serum anti-desmoglein autoantibody levels to less than 14 U/mL occurred in all cases along with clinical complete remission even after maintenance therapy cessation,” Dr. Sanchez and her associates noted.
Future research should address questions including the optimal dose and dosing frequency of rituximab, as well as the cost-effectiveness of the treatment and criteria for treatment withdrawal, they said.
The researchers had no relevant financial conflicts disclosures.
SOURCE: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
FROM JAMA DERMATOLOGY
Key clinical point: Treatment with rituximab prevented relapse and maintained remission in 11 patients with severe, difficult-to-treat pemphigus.
Major finding: All 11 patients treated with a 1-g dose of rituximab given every 6 months maintained remission at an average of 78 months after the first cycle.
Data source: The data come from a single-center, retrospective case series of 11 adults.
Disclosures: The researchers had no relevant financial disclosures.
Source: JAMA Dermatol. 2017 Jan 3. doi: 10.1001/jamadermatol.2017.5176.
Novel oral orphan drug tames pemphigus
GENEVA – An oral reversible Bruton tyrosine kinase inhibitor known as PRN1008 showed promising efficacy and safety for the treatment of pemphigus vulgaris in an interim analysis of an ongoing small-to-date, open-label phase 2 study, according to DeDee Murrell, MD.
PRN1008 is a designer drug intended as an alternative to the long-standing standard therapy for pemphigus, months to years of moderate- or high-dose systemic corticosteroids, with all the debilitating side effects that sledgehammer approach often brings, she said at the annual congress of the European Academy of Dermatology and Venereology.

Thus, PRN1008 inhibits a range of inflammatory cellular activities in mast cells, neutrophils, and other cells activated in autoimmune diseases, without killing those cells or directly affecting T cells. And – like corticosteroids – it works quickly, according to Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
“The development of an oral, fast-acting treatment that could safely and effectively modulate B-cell function without depleting B cells would be a major advance in treating autoimmune diseases like pemphigus, vasculitis, immune thrombocytopenic purpura, multiple sclerosis, and rheumatoid arthritis,” she said.
Also, PRN1008 was designed to provide durable inhibition of BTK, coupled with rapid systemic clearance of the drug in order to minimize side effects.
The ongoing phase 2 multicenter international study, known as Believe-PV, to date includes 12 patients with biopsy-proven mild to moderate pemphigus vulgaris. Five patients were newly diagnosed and treatment naive, while seven had relapsing disease. All were placed on fixed-dose PRN1008 at 400 mg b.i.d. for 12 weeks with low-dose prednisone as needed, then followed for an additional 12 weeks off PRN1008 to evaluate the durability of responses.
The primary study endpoint was control of disease activity by week 4 on a background of little or no prednisone, which 5 of 12 patients achieved. The secondary endpoint of complete clinical remission at 12 weeks was achieved in half of patients. Total pemphigus disease activity index (PDAI) scores dropped throughout the study period, reaching a mean 70% reduction at 12 weeks from a baseline of 20 points. Scores on the autoimmune bullous quality of life metric improved by 43%.
Eight of the 11 individuals who completed the study had control of disease activity by week 4 and/or complete clinical remission at 12 weeks. Those results are comparable with those typically seen with high-dose steroids at 12 weeks, the dermatologist noted.
Levels of anti–desmoglein-1 and/or -3 autoantibodies, which were elevated at baseline in 10 of 12 subjects, decreased during treatment with PRN1008.
Seventy-five percent of Bruton tyrosine kinase receptors were occupied by PRN1008 by day 2 of the study, confirming earlier studies in canine models of pemphigus. It turns out that pemphigus foliaceus is as common in dogs as atopic dermatitis is in humans, according to Dr. Murrell.
Side effects were limited to mild headache in two patients. One patient developed serious grade 3 cellulitis. She was taken off the study medication, although it was deemed unlikely that the infection was treatment related because she had had a recent history of multiple episodes of cellulitis.
During the second 12-week phase of the trial, after discontinuation of PRN1008, most patients retained their on-treatment reduction in total PDAI scores.
Dr. Murrell noted that the gradient of improvement in PDAI scores seen with PRN1008 in the Believe-PV study was quite similar to that seen in a recent 90-patient French randomized trial of rituximab (Rituxan) for the treatment of pemphigus. That’s of considerable interest, she said, because rituximab is a much more powerful drug, which drastically depletes the B-cell population. Moreover, the gradient of improvement in the rituximab-treated patients in the French trial was achieved with the aid of moderate-dose prednisone at 1 or 1.5 mg/kg per day, while PRN1008-treated patients in Believe-PV were taking on average only 0.2 mg/kg per day.
Asked if she thinks PRN1008 has a future as a stand-alone treatment for pemphigus or is better suited as an adjunct to systemic corticosteroids, Dr. Murrell said she believes the drug can be used effectively without steroids. However, since it’s recommended that patients be screened for tuberculosis before going on rituximab, and PRN1008 also targets B cells, she and her coinvestigators followed the same practice in Believe-PV. And because it takes a couple of weeks for the results of the Quantiferon-TB Gold In-Tube test to come back, it would be unethical for patients newly diagnosed with pemphigus to go untreated and in pain during that period, they get corticosteroids, at least initially.
Dr. Murrell reported serving as a paid consultant to Principia Biopharma, which is developing PRN1008.
GENEVA – An oral reversible Bruton tyrosine kinase inhibitor known as PRN1008 showed promising efficacy and safety for the treatment of pemphigus vulgaris in an interim analysis of an ongoing small-to-date, open-label phase 2 study, according to DeDee Murrell, MD.
PRN1008 is a designer drug intended as an alternative to the long-standing standard therapy for pemphigus, months to years of moderate- or high-dose systemic corticosteroids, with all the debilitating side effects that sledgehammer approach often brings, she said at the annual congress of the European Academy of Dermatology and Venereology.
Thus, PRN1008 inhibits a range of inflammatory cellular activities in mast cells, neutrophils, and other cells activated in autoimmune diseases, without killing those cells or directly affecting T cells. And – like corticosteroids – it works quickly, according to Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
“The development of an oral, fast-acting treatment that could safely and effectively modulate B-cell function without depleting B cells would be a major advance in treating autoimmune diseases like pemphigus, vasculitis, immune thrombocytopenic purpura, multiple sclerosis, and rheumatoid arthritis,” she said.
Also, PRN1008 was designed to provide durable inhibition of BTK, coupled with rapid systemic clearance of the drug in order to minimize side effects.
The ongoing phase 2 multicenter international study, known as Believe-PV, to date includes 12 patients with biopsy-proven mild to moderate pemphigus vulgaris. Five patients were newly diagnosed and treatment naive, while seven had relapsing disease. All were placed on fixed-dose PRN1008 at 400 mg b.i.d. for 12 weeks with low-dose prednisone as needed, then followed for an additional 12 weeks off PRN1008 to evaluate the durability of responses.
The primary study endpoint was control of disease activity by week 4 on a background of little or no prednisone, which 5 of 12 patients achieved. The secondary endpoint of complete clinical remission at 12 weeks was achieved in half of patients. Total pemphigus disease activity index (PDAI) scores dropped throughout the study period, reaching a mean 70% reduction at 12 weeks from a baseline of 20 points. Scores on the autoimmune bullous quality of life metric improved by 43%.
Eight of the 11 individuals who completed the study had control of disease activity by week 4 and/or complete clinical remission at 12 weeks. Those results are comparable with those typically seen with high-dose steroids at 12 weeks, the dermatologist noted.
Levels of anti–desmoglein-1 and/or -3 autoantibodies, which were elevated at baseline in 10 of 12 subjects, decreased during treatment with PRN1008.
Seventy-five percent of Bruton tyrosine kinase receptors were occupied by PRN1008 by day 2 of the study, confirming earlier studies in canine models of pemphigus. It turns out that pemphigus foliaceus is as common in dogs as atopic dermatitis is in humans, according to Dr. Murrell.
Side effects were limited to mild headache in two patients. One patient developed serious grade 3 cellulitis. She was taken off the study medication, although it was deemed unlikely that the infection was treatment related because she had had a recent history of multiple episodes of cellulitis.
During the second 12-week phase of the trial, after discontinuation of PRN1008, most patients retained their on-treatment reduction in total PDAI scores.
Dr. Murrell noted that the gradient of improvement in PDAI scores seen with PRN1008 in the Believe-PV study was quite similar to that seen in a recent 90-patient French randomized trial of rituximab (Rituxan) for the treatment of pemphigus. That’s of considerable interest, she said, because rituximab is a much more powerful drug, which drastically depletes the B-cell population. Moreover, the gradient of improvement in the rituximab-treated patients in the French trial was achieved with the aid of moderate-dose prednisone at 1 or 1.5 mg/kg per day, while PRN1008-treated patients in Believe-PV were taking on average only 0.2 mg/kg per day.
Asked if she thinks PRN1008 has a future as a stand-alone treatment for pemphigus or is better suited as an adjunct to systemic corticosteroids, Dr. Murrell said she believes the drug can be used effectively without steroids. However, since it’s recommended that patients be screened for tuberculosis before going on rituximab, and PRN1008 also targets B cells, she and her coinvestigators followed the same practice in Believe-PV. And because it takes a couple of weeks for the results of the Quantiferon-TB Gold In-Tube test to come back, it would be unethical for patients newly diagnosed with pemphigus to go untreated and in pain during that period, they get corticosteroids, at least initially.
Dr. Murrell reported serving as a paid consultant to Principia Biopharma, which is developing PRN1008.
GENEVA – An oral reversible Bruton tyrosine kinase inhibitor known as PRN1008 showed promising efficacy and safety for the treatment of pemphigus vulgaris in an interim analysis of an ongoing small-to-date, open-label phase 2 study, according to DeDee Murrell, MD.
PRN1008 is a designer drug intended as an alternative to the long-standing standard therapy for pemphigus, months to years of moderate- or high-dose systemic corticosteroids, with all the debilitating side effects that sledgehammer approach often brings, she said at the annual congress of the European Academy of Dermatology and Venereology.
Thus, PRN1008 inhibits a range of inflammatory cellular activities in mast cells, neutrophils, and other cells activated in autoimmune diseases, without killing those cells or directly affecting T cells. And – like corticosteroids – it works quickly, according to Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
“The development of an oral, fast-acting treatment that could safely and effectively modulate B-cell function without depleting B cells would be a major advance in treating autoimmune diseases like pemphigus, vasculitis, immune thrombocytopenic purpura, multiple sclerosis, and rheumatoid arthritis,” she said.
Also, PRN1008 was designed to provide durable inhibition of BTK, coupled with rapid systemic clearance of the drug in order to minimize side effects.
The ongoing phase 2 multicenter international study, known as Believe-PV, to date includes 12 patients with biopsy-proven mild to moderate pemphigus vulgaris. Five patients were newly diagnosed and treatment naive, while seven had relapsing disease. All were placed on fixed-dose PRN1008 at 400 mg b.i.d. for 12 weeks with low-dose prednisone as needed, then followed for an additional 12 weeks off PRN1008 to evaluate the durability of responses.
The primary study endpoint was control of disease activity by week 4 on a background of little or no prednisone, which 5 of 12 patients achieved. The secondary endpoint of complete clinical remission at 12 weeks was achieved in half of patients. Total pemphigus disease activity index (PDAI) scores dropped throughout the study period, reaching a mean 70% reduction at 12 weeks from a baseline of 20 points. Scores on the autoimmune bullous quality of life metric improved by 43%.
Eight of the 11 individuals who completed the study had control of disease activity by week 4 and/or complete clinical remission at 12 weeks. Those results are comparable with those typically seen with high-dose steroids at 12 weeks, the dermatologist noted.
Levels of anti–desmoglein-1 and/or -3 autoantibodies, which were elevated at baseline in 10 of 12 subjects, decreased during treatment with PRN1008.
Seventy-five percent of Bruton tyrosine kinase receptors were occupied by PRN1008 by day 2 of the study, confirming earlier studies in canine models of pemphigus. It turns out that pemphigus foliaceus is as common in dogs as atopic dermatitis is in humans, according to Dr. Murrell.
Side effects were limited to mild headache in two patients. One patient developed serious grade 3 cellulitis. She was taken off the study medication, although it was deemed unlikely that the infection was treatment related because she had had a recent history of multiple episodes of cellulitis.
During the second 12-week phase of the trial, after discontinuation of PRN1008, most patients retained their on-treatment reduction in total PDAI scores.
Dr. Murrell noted that the gradient of improvement in PDAI scores seen with PRN1008 in the Believe-PV study was quite similar to that seen in a recent 90-patient French randomized trial of rituximab (Rituxan) for the treatment of pemphigus. That’s of considerable interest, she said, because rituximab is a much more powerful drug, which drastically depletes the B-cell population. Moreover, the gradient of improvement in the rituximab-treated patients in the French trial was achieved with the aid of moderate-dose prednisone at 1 or 1.5 mg/kg per day, while PRN1008-treated patients in Believe-PV were taking on average only 0.2 mg/kg per day.
Asked if she thinks PRN1008 has a future as a stand-alone treatment for pemphigus or is better suited as an adjunct to systemic corticosteroids, Dr. Murrell said she believes the drug can be used effectively without steroids. However, since it’s recommended that patients be screened for tuberculosis before going on rituximab, and PRN1008 also targets B cells, she and her coinvestigators followed the same practice in Believe-PV. And because it takes a couple of weeks for the results of the Quantiferon-TB Gold In-Tube test to come back, it would be unethical for patients newly diagnosed with pemphigus to go untreated and in pain during that period, they get corticosteroids, at least initially.
Dr. Murrell reported serving as a paid consultant to Principia Biopharma, which is developing PRN1008.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Five of 12 pemphigus vulgaris patients achieved control of disease activity within the first 4 weeks on the investigational oral Bruton tyrosine kinase inhibitor PRN1008.
Data source: An interim analysis of the first 12 patients with pemphigus vulgaris in an ongoing multicenter, international open-label, phase 2 clinical trial.
Disclosures: The study presenter reported serving as a paid consultant to Principia Biopharma, which is developing PRN1008 and sponsored the Believe-PV trial.
Nonadherence to lupus drugs may play a role in frequent hospitalization
SAN DIEGO – New research into factors that predict which systemic lupus erythematosus patients are at high risk for hospitalization is beginning to identify the contribution of medication nonadherence to the problem.
Compared with others hospitalized for systemic lupus erythematosus (SLE), high-risk patients were an adjusted 10 percentage points less likely to show evidence of adherence to prescribed drugs, according to a study presented at the annual meeting of the American College of Rheumatology.
“Medication nonadherence remains an important problem among patients with SLE. It is a major modifiable cause to help decrease hospital admissions and readmissions and decrease risk for morbidity and mortality associated with SLE,” study coauthor Allen P. Anandarajah, MBBS, said in an interview after the ACR meeting.

The researchers found that the average patient required $51,808 in treatment costs annually; the average stay was 8.5 days (Lupus. 2017;26[7]:756-61).
Dr. Anandarajah led another study, released at the 2016 ACR annual meeting, that found patients at high risk of hospitalization were more likely to be younger, have earlier SLE onset, and be African American (abstract 122).
As for medication nonadherence, a systematic review of 11 studies published this year found that “the percentage of nonadherent patients ranged from 43% to 75%, with studies consistently reporting that over half of patients are nonadherent” (Arthritis Care Res [Hoboken]. 2017 Nov;69[11]:1706-13).
Nonadherence is an especially significant issue “among a small group of high-risk, high-need patients,” Dr. Anandarajah said.
For the new study, the researchers aimed to better understand “if medication adherence was a risk factor for hospital admissions among SLE patients,” he said.
They identified a group of 28 high-risk patients out of 171 hospitalized SLE patients who were admitted from 2013 to 2015. Compared with other patients, the high-risk patients, who required three or more annual admissions, were younger (mean age, 39.6 vs. 47.6; P = .03), less likely to be female (82% vs. 92%; P = .09), and more likely to be African American (61% vs. 41%; P = .05).
Why might the young be less adherent? “Younger people are more likely to have difficulty with taking care of themselves when afflicted with chronic diseases due to lack of understanding of the implications of insufficiently treating their illness, poor coping skills, peer pressures about dealing with potential side effects like weight gain with steroids, and financial reasons, including lack of insurance,” he said.
As for African Americans, possible reasons for lower adherence include “cultural reasons such as a taboo about illness and misconceptions about need for continuous use of medications, lower educational levels, lack of trust in their health care providers/health care team, and socioeconomic reasons/financial issues,” he said.
The researchers linked patients to a pharmacy claims database to calculate the medication possession ratio, “an indicator of whether a patient had adequate medication supply in a given time frame,” as the study puts it. A total of 102 patients had complete pharmacy data.
The researchers found that the unadjusted mean medication possession ratio was lower in high-risk patients, compared with the others (73.4% vs. 79.9%; P = .30), and was an estimated 10 percentage points lower in an adjusted analysis that nearly reached statistical significance (P = .06).
“While it was not significant, there was a trend, and one could possibly expect a significant value with larger numbers,” Dr. Anandarajah said.
How can adherence be improved in SLE? In an interview, Michelle Petri, MD, professor of medicine and codirector of the lupus center at Johns Hopkins University, Baltimore, said she saw a major improvement in hydroxychloroquine (Plaquenil) adherence after introducing blood level testing.
“I believe rheumatologists should introduce drug monitoring for all of our important drugs: [hydroxychloroquine] (where it must be a whole blood level and not plasma), azathioprine, methotrexate, and mycophenolate,” said Dr. Petri, who praised the new research as “an excellent first study.”
Going forward, Dr. Anandarajah said his university has started a program designed to help poor, high-risk SLE patients in the Rochester area through a clinic in the inner city, coordinated care with nurses, and a series of focus-group meetings and educational programs for patients and providers. “We hope to improve compliance with outpatient visits, medication adherence, and self-management skills,” he said.
The study authors and Dr. Petri reported no relevant disclosures. No specific study funding was reported.
SAN DIEGO – New research into factors that predict which systemic lupus erythematosus patients are at high risk for hospitalization is beginning to identify the contribution of medication nonadherence to the problem.
Compared with others hospitalized for systemic lupus erythematosus (SLE), high-risk patients were an adjusted 10 percentage points less likely to show evidence of adherence to prescribed drugs, according to a study presented at the annual meeting of the American College of Rheumatology.
“Medication nonadherence remains an important problem among patients with SLE. It is a major modifiable cause to help decrease hospital admissions and readmissions and decrease risk for morbidity and mortality associated with SLE,” study coauthor Allen P. Anandarajah, MBBS, said in an interview after the ACR meeting.
The researchers found that the average patient required $51,808 in treatment costs annually; the average stay was 8.5 days (Lupus. 2017;26[7]:756-61).
Dr. Anandarajah led another study, released at the 2016 ACR annual meeting, that found patients at high risk of hospitalization were more likely to be younger, have earlier SLE onset, and be African American (abstract 122).
As for medication nonadherence, a systematic review of 11 studies published this year found that “the percentage of nonadherent patients ranged from 43% to 75%, with studies consistently reporting that over half of patients are nonadherent” (Arthritis Care Res [Hoboken]. 2017 Nov;69[11]:1706-13).
Nonadherence is an especially significant issue “among a small group of high-risk, high-need patients,” Dr. Anandarajah said.
For the new study, the researchers aimed to better understand “if medication adherence was a risk factor for hospital admissions among SLE patients,” he said.
They identified a group of 28 high-risk patients out of 171 hospitalized SLE patients who were admitted from 2013 to 2015. Compared with other patients, the high-risk patients, who required three or more annual admissions, were younger (mean age, 39.6 vs. 47.6; P = .03), less likely to be female (82% vs. 92%; P = .09), and more likely to be African American (61% vs. 41%; P = .05).
Why might the young be less adherent? “Younger people are more likely to have difficulty with taking care of themselves when afflicted with chronic diseases due to lack of understanding of the implications of insufficiently treating their illness, poor coping skills, peer pressures about dealing with potential side effects like weight gain with steroids, and financial reasons, including lack of insurance,” he said.
As for African Americans, possible reasons for lower adherence include “cultural reasons such as a taboo about illness and misconceptions about need for continuous use of medications, lower educational levels, lack of trust in their health care providers/health care team, and socioeconomic reasons/financial issues,” he said.
The researchers linked patients to a pharmacy claims database to calculate the medication possession ratio, “an indicator of whether a patient had adequate medication supply in a given time frame,” as the study puts it. A total of 102 patients had complete pharmacy data.
The researchers found that the unadjusted mean medication possession ratio was lower in high-risk patients, compared with the others (73.4% vs. 79.9%; P = .30), and was an estimated 10 percentage points lower in an adjusted analysis that nearly reached statistical significance (P = .06).
“While it was not significant, there was a trend, and one could possibly expect a significant value with larger numbers,” Dr. Anandarajah said.
How can adherence be improved in SLE? In an interview, Michelle Petri, MD, professor of medicine and codirector of the lupus center at Johns Hopkins University, Baltimore, said she saw a major improvement in hydroxychloroquine (Plaquenil) adherence after introducing blood level testing.
“I believe rheumatologists should introduce drug monitoring for all of our important drugs: [hydroxychloroquine] (where it must be a whole blood level and not plasma), azathioprine, methotrexate, and mycophenolate,” said Dr. Petri, who praised the new research as “an excellent first study.”
Going forward, Dr. Anandarajah said his university has started a program designed to help poor, high-risk SLE patients in the Rochester area through a clinic in the inner city, coordinated care with nurses, and a series of focus-group meetings and educational programs for patients and providers. “We hope to improve compliance with outpatient visits, medication adherence, and self-management skills,” he said.
The study authors and Dr. Petri reported no relevant disclosures. No specific study funding was reported.
SAN DIEGO – New research into factors that predict which systemic lupus erythematosus patients are at high risk for hospitalization is beginning to identify the contribution of medication nonadherence to the problem.
Compared with others hospitalized for systemic lupus erythematosus (SLE), high-risk patients were an adjusted 10 percentage points less likely to show evidence of adherence to prescribed drugs, according to a study presented at the annual meeting of the American College of Rheumatology.
“Medication nonadherence remains an important problem among patients with SLE. It is a major modifiable cause to help decrease hospital admissions and readmissions and decrease risk for morbidity and mortality associated with SLE,” study coauthor Allen P. Anandarajah, MBBS, said in an interview after the ACR meeting.
The researchers found that the average patient required $51,808 in treatment costs annually; the average stay was 8.5 days (Lupus. 2017;26[7]:756-61).
Dr. Anandarajah led another study, released at the 2016 ACR annual meeting, that found patients at high risk of hospitalization were more likely to be younger, have earlier SLE onset, and be African American (abstract 122).
As for medication nonadherence, a systematic review of 11 studies published this year found that “the percentage of nonadherent patients ranged from 43% to 75%, with studies consistently reporting that over half of patients are nonadherent” (Arthritis Care Res [Hoboken]. 2017 Nov;69[11]:1706-13).
Nonadherence is an especially significant issue “among a small group of high-risk, high-need patients,” Dr. Anandarajah said.
For the new study, the researchers aimed to better understand “if medication adherence was a risk factor for hospital admissions among SLE patients,” he said.
They identified a group of 28 high-risk patients out of 171 hospitalized SLE patients who were admitted from 2013 to 2015. Compared with other patients, the high-risk patients, who required three or more annual admissions, were younger (mean age, 39.6 vs. 47.6; P = .03), less likely to be female (82% vs. 92%; P = .09), and more likely to be African American (61% vs. 41%; P = .05).
Why might the young be less adherent? “Younger people are more likely to have difficulty with taking care of themselves when afflicted with chronic diseases due to lack of understanding of the implications of insufficiently treating their illness, poor coping skills, peer pressures about dealing with potential side effects like weight gain with steroids, and financial reasons, including lack of insurance,” he said.
As for African Americans, possible reasons for lower adherence include “cultural reasons such as a taboo about illness and misconceptions about need for continuous use of medications, lower educational levels, lack of trust in their health care providers/health care team, and socioeconomic reasons/financial issues,” he said.
The researchers linked patients to a pharmacy claims database to calculate the medication possession ratio, “an indicator of whether a patient had adequate medication supply in a given time frame,” as the study puts it. A total of 102 patients had complete pharmacy data.
The researchers found that the unadjusted mean medication possession ratio was lower in high-risk patients, compared with the others (73.4% vs. 79.9%; P = .30), and was an estimated 10 percentage points lower in an adjusted analysis that nearly reached statistical significance (P = .06).
“While it was not significant, there was a trend, and one could possibly expect a significant value with larger numbers,” Dr. Anandarajah said.
How can adherence be improved in SLE? In an interview, Michelle Petri, MD, professor of medicine and codirector of the lupus center at Johns Hopkins University, Baltimore, said she saw a major improvement in hydroxychloroquine (Plaquenil) adherence after introducing blood level testing.
“I believe rheumatologists should introduce drug monitoring for all of our important drugs: [hydroxychloroquine] (where it must be a whole blood level and not plasma), azathioprine, methotrexate, and mycophenolate,” said Dr. Petri, who praised the new research as “an excellent first study.”
Going forward, Dr. Anandarajah said his university has started a program designed to help poor, high-risk SLE patients in the Rochester area through a clinic in the inner city, coordinated care with nurses, and a series of focus-group meetings and educational programs for patients and providers. “We hope to improve compliance with outpatient visits, medication adherence, and self-management skills,” he said.
The study authors and Dr. Petri reported no relevant disclosures. No specific study funding was reported.
REPORTING FROM ACR 2017
Key clinical point:
Major finding: Compared with other patients hospitalized with SLE, high-risk patients had 10% lower medication adherence.
Data source: A 2-year analysis of 171 patients (28 deemed high risk) admitted for SLE at a single hospital.
Disclosures: The study authors reported no relevant disclosures. No specific study funding is reported.
Source: C. Thirukuraman et al. ACR 2017 abstract 223.