User login
Bullous Amyloidosis Masquerading as Pseudoporphyria
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
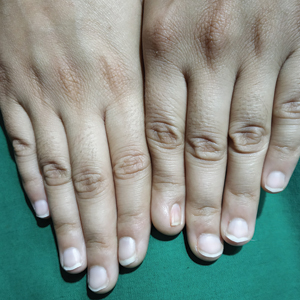
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.

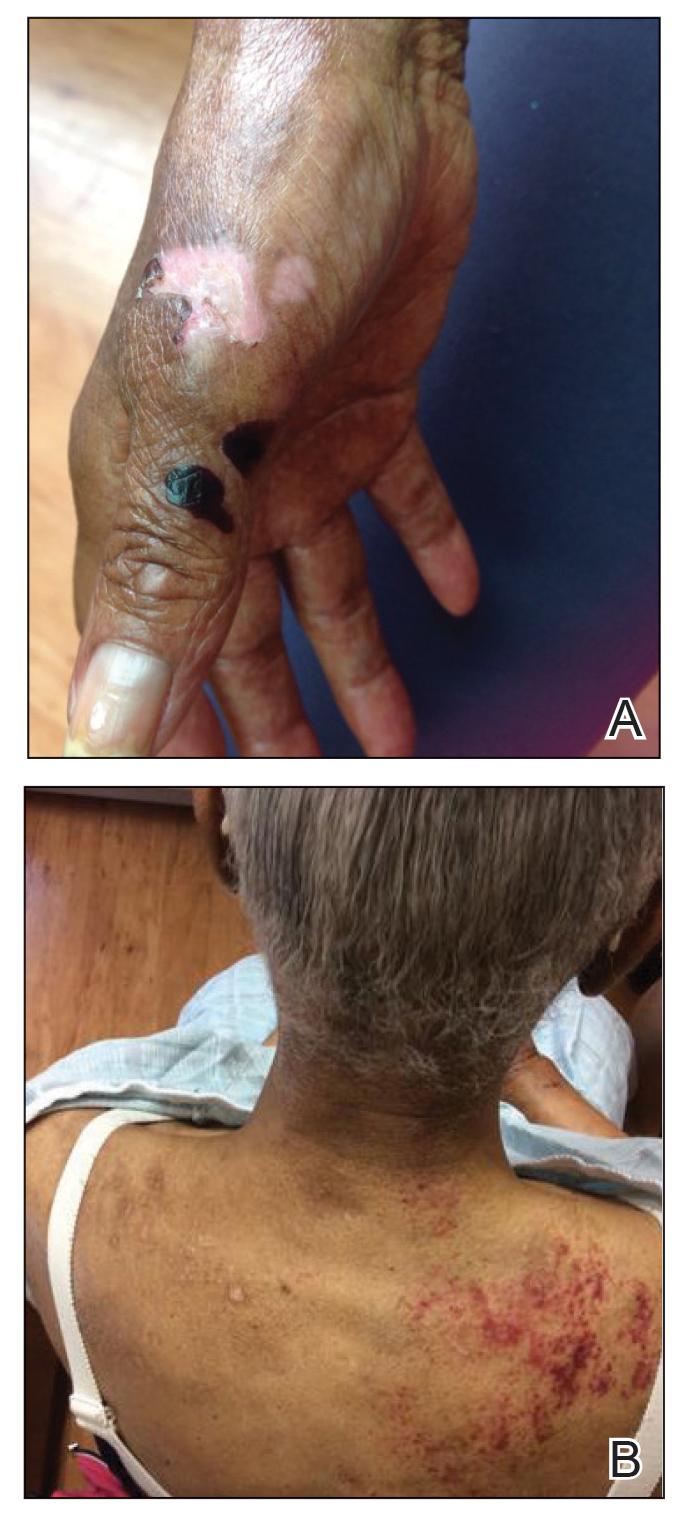
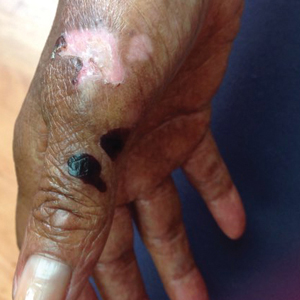
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Practice Points
- Primary systemic amyloidosis, including the rare cutaneous bullous amyloidosis, often is difficult to diagnose and has been associated with underlying plasma cell dyscrasia or multiple myeloma.
- When evaluating patients with initially convincing signs of pseudoporphyria, it is imperative to consider the diagnosis of bullous amyloidosis, which additionally can present with intraoral hemorrhagic vesicles and have confirmatory histopathologic features.
- Further investigation for multiple myeloma is warranted when patients with a chronic hemorrhagic bullous condition also present with symptoms of purpura, angina bullosa hemorrhagica, fatigue, weight loss, and/or macroglossia. Accurate diagnosis of bullous amyloidosis and timely treatment of its underlying cause will contribute to better, more proactive patient care.
Pedunculated Tumor on the Posterior Neck
The Diagnosis: Nodular Hidradenoma
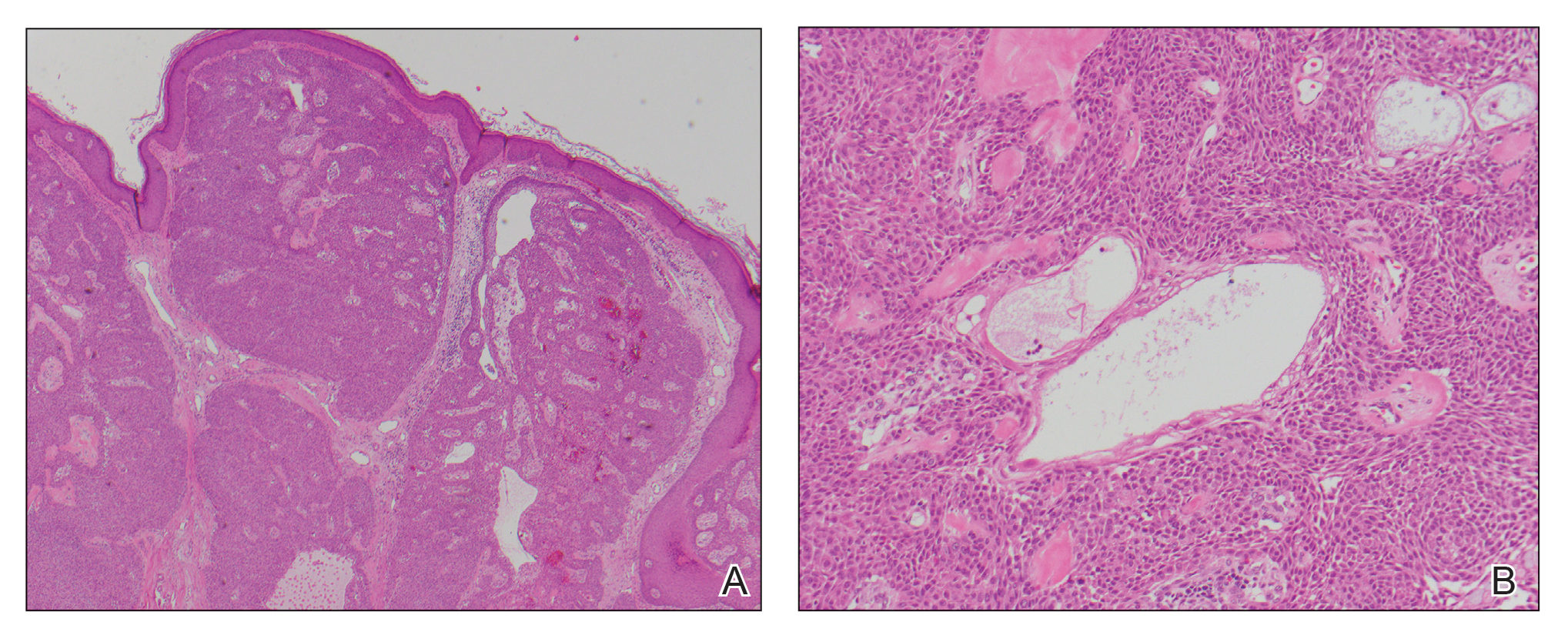
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508

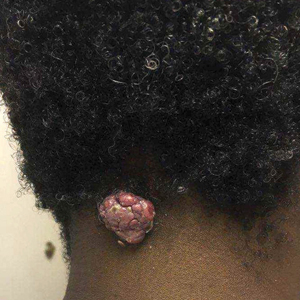
A 56-year-old man presented with a progressively enlarging lesion on the posterior neck of 8 months’ duration. He reported localized pruritus of the lesion that improved with triamcinolone cream 0.05% and oral hydroxyzine as well as occasional irritation of the mass with oozing of clear fluid and blood. He denied associated pain and constitutional symptoms. Physical examination revealed a 2.5-cm, nodular, pedunculated, rubbery mass with foci of crusting on the central posterior neck. The mass was flesh colored to pink, and no lymphadenopathy was noted on physical examination.
Erythematous and Ulcerated Plaque on the Left Temple
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
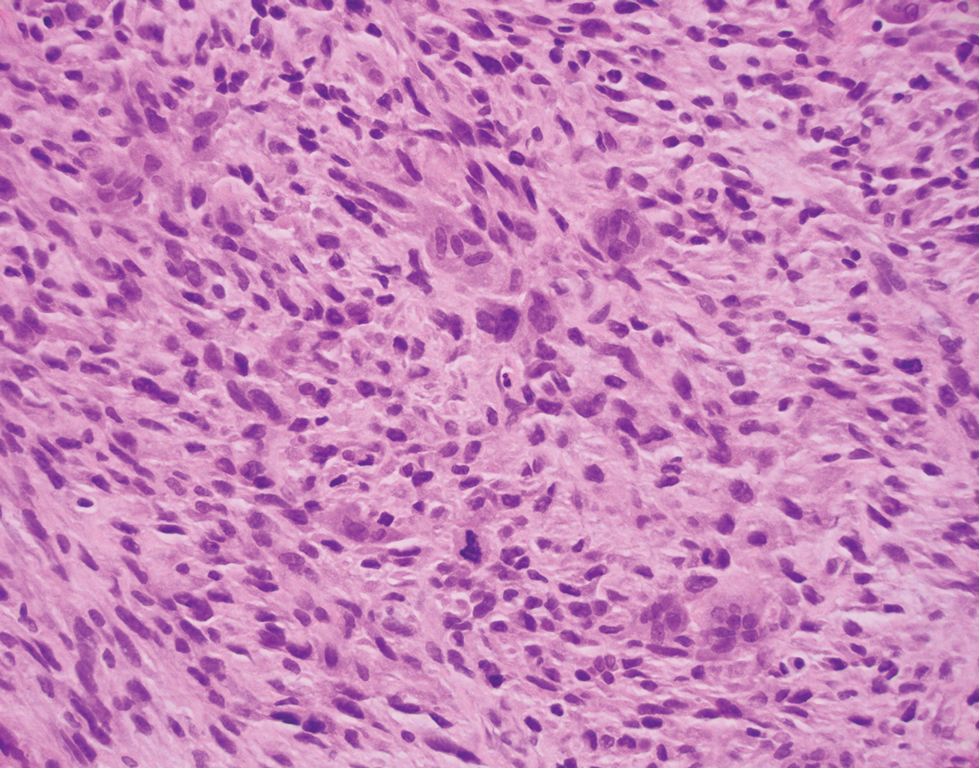
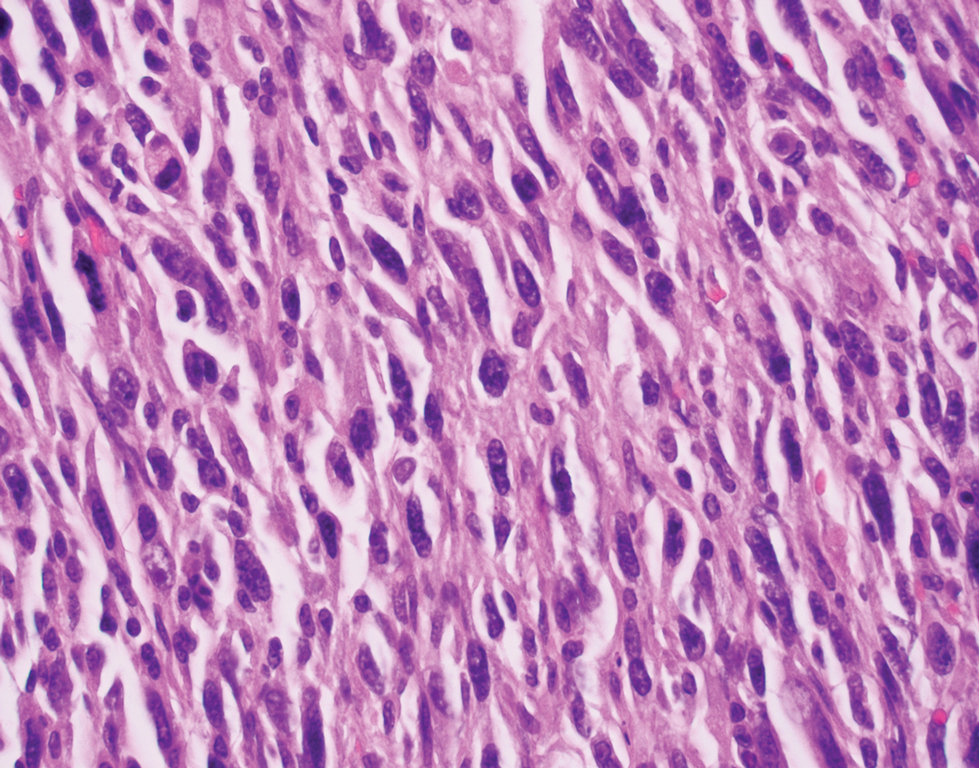
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
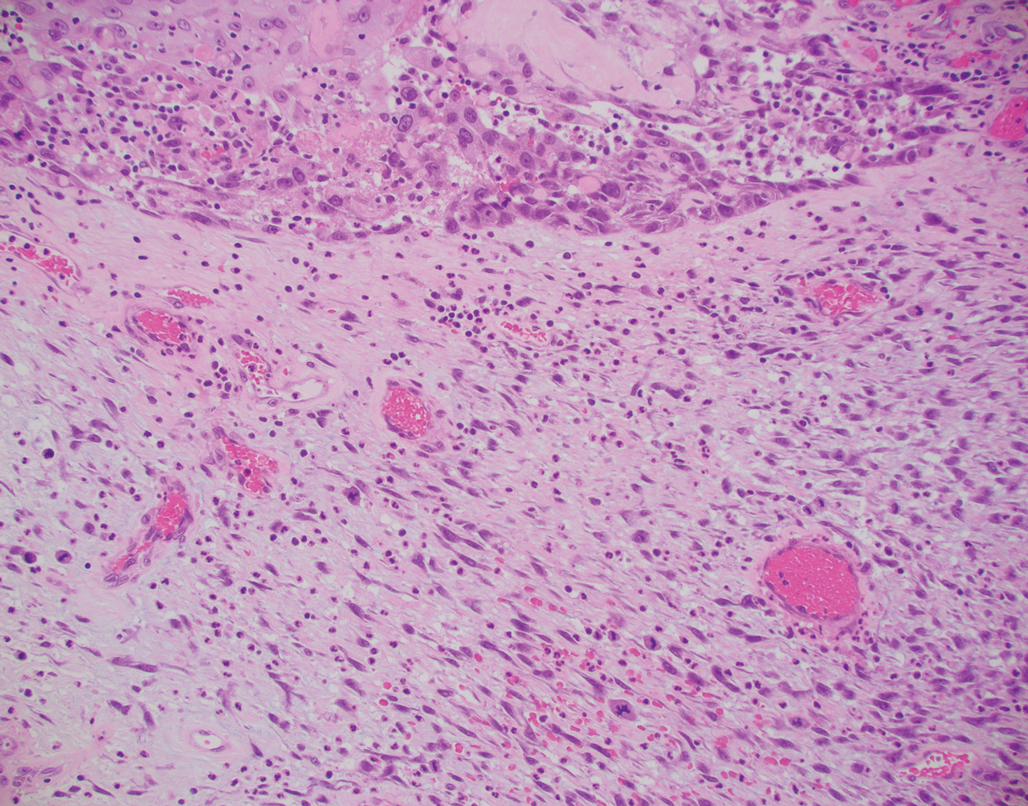
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
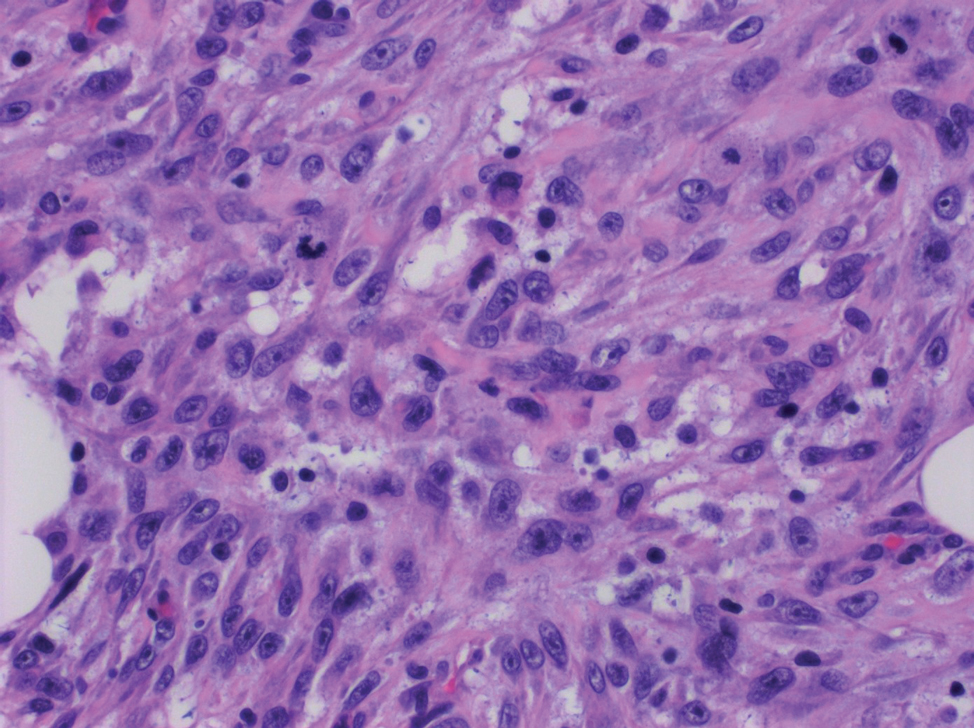
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
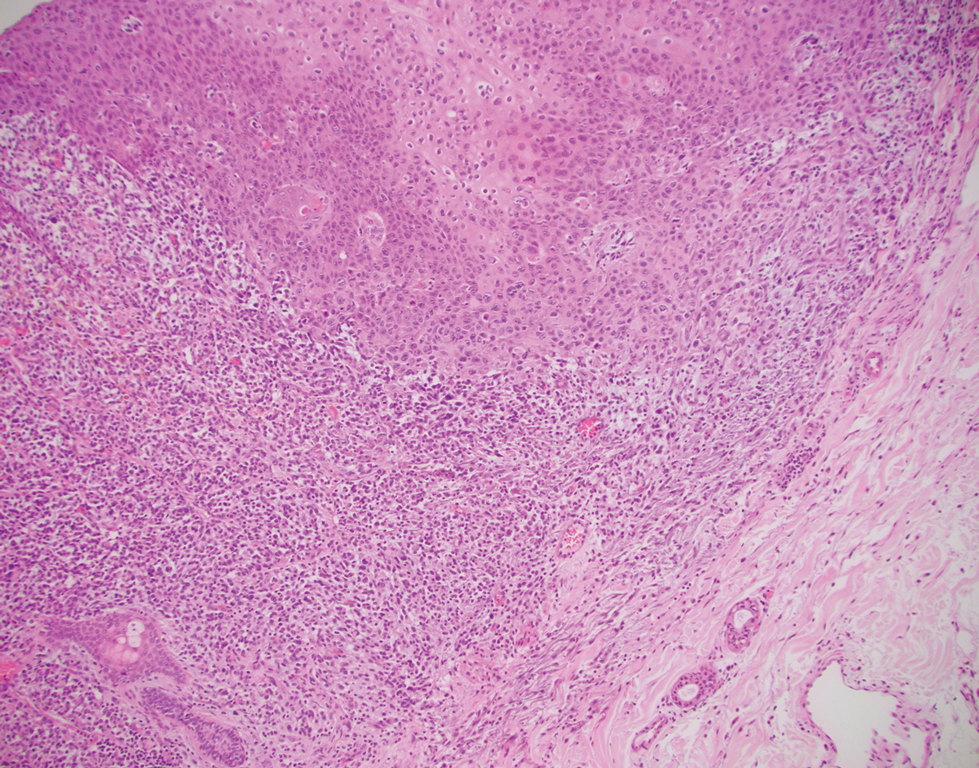
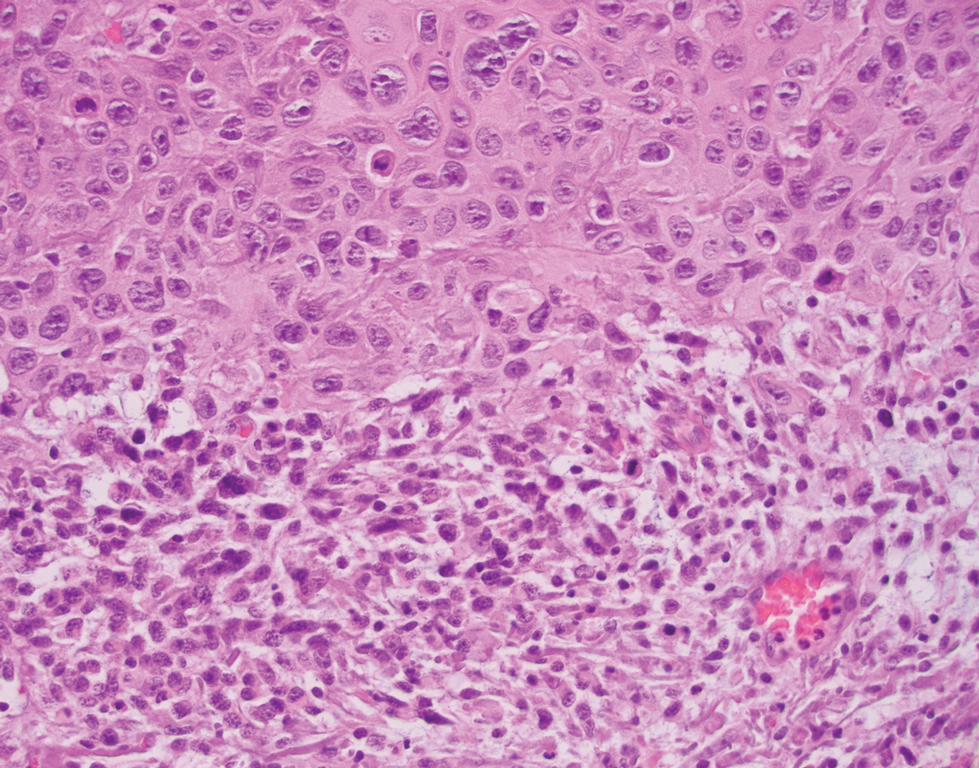
A 72-year-old man with a history of nonmelanoma skin cancer and lung transplant maintained on stable doses of prednisone and tacrolimus presented with a 1.3×1.8-cm, slow-growing, well-demarcated, ulcerated, erythematous plaque with overlying serous crust on the left temple of 6 months’ duration. No cervical or axillary lymphadenopathy was appreciated on physical examination. A biopsy was performed followed by Mohs micrographic surgery. Microscopic examination of the debulking specimen revealed atypical spindle cells in the papillary and reticular dermis radiating from a central focus of a moderately differentiated squamous cell carcinoma. The squamous cells stained positive for cytokeratin 5/6, pankeratin, and p40, while the spindle cells stained positive only for vimentin.
A Severe Presentation of Plasma Cell Cheilitis
Plasma cell cheilitis (PCC), also known as plasmocytosis circumorificialis and plasmocytosis mucosae,1 is a poorly understood, uncommon inflammatory condition characterized by dense infiltration of mature plasma cells in the mucosal dermis of the lip.2-5 The etiology of PCC is unknown but is thought to be a reactive immune process triggered by infection, mechanical friction, trauma, or solar damage.1,5,6
The most common presentation of PCC is a slowly evolving, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 Secondary changes with disease progression can include erosion, ulceration, fissures, edema, bleeding, or crusting.5 The diagnosis of PCC is challenging because it can mimic neoplastic, infectious, and inflammatory conditions.8,9
Treatment strategies for PCC described in the literature vary, as does therapeutic response. Resolution of PCC has been documented after systemic steroids, intralesional steroids, systemic griseofulvin, and topical calcineurin inhibitors, among other agents.6,7,10-16
We present the case of a patient with a lip lesion who ultimately was diagnosed with PCC after it progressed to an advanced necrotic stage.
Case Report
An 80-year-old male veteran of the Armed Services initially presented to our institution via teledermatology with redness and crusting of the lower lip (Figure 1). He had a history of myelodysplastic syndrome and anemia requiring iron transfusion. The process appeared to be consistent with actinic cheilitis vs squamous cell carcinoma. In-person dermatology consultation was recommended; however, the patient did not follow through with that appointment.
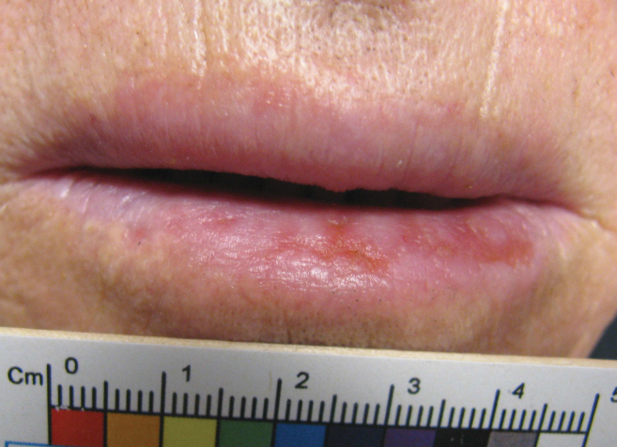
Five months later, additional photographs of the lesion were taken by the patient's primary care physician and sent through teledermatology, revealing progression to an erythematous, yellow-crusted erosion (Figure 2). The medical record indicated that a punch biopsy performed by the patient’s primary care physician showed hyperkeratosis and fungal organisms on periodic acid–Schiff staining. He subsequently applied ketoconazole and terbinafine cream to the lower lip without improvement. Prompt in-person evaluation by dermatology was again recommended.
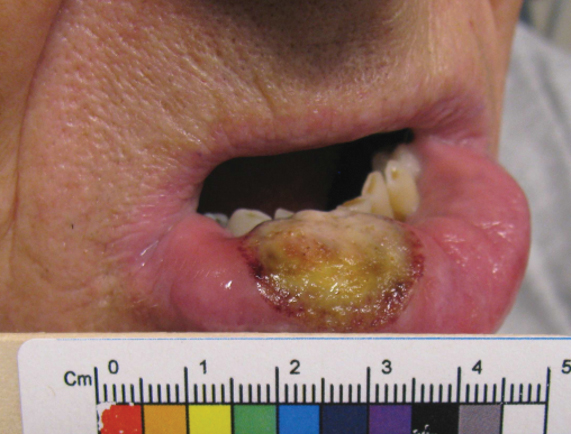
Ten days later, the patient was seen in our dermatology clinic, at which point his condition had rapidly progressed. The lower lip displayed a 3.0×2.5-cm, yellow and black, crusted, ulcerated plaque (Figure 3). He reported severe burning and pain of the lip as well as spontaneous bleeding. He had lost approximately 10 pounds over the last month due to poor oral intake. A second punch biopsy showed benign mucosa with extensive ulceration and formation of full-thickness granulation tissue. No fungi or bacteria were identified.
Consultation and Histologic Analysis
Dermatopathology was consulted and recommended a third punch biopsy for additional testing. A repeat biopsy demonstrated ulceration with lateral elements of retained epidermis and a dense submucosal chronic inflammatory infiltrate comprising plasma cells and lymphocytes (Figures 4 and 5). Immunohistochemical staining demonstrated a mixed inflammatory infiltrate with CD3+ T cells and CD20+ B cells. In situ hybridization studies demonstrated numerous lambda-positive and kappa-positive plasma cells without chain restriction. Periodic acid–Schiff with diastase and Grocott-Gomori methenamine-silver staining demonstrated no fungi. Findings were interpreted to be most consistent with a diagnosis of PCC.
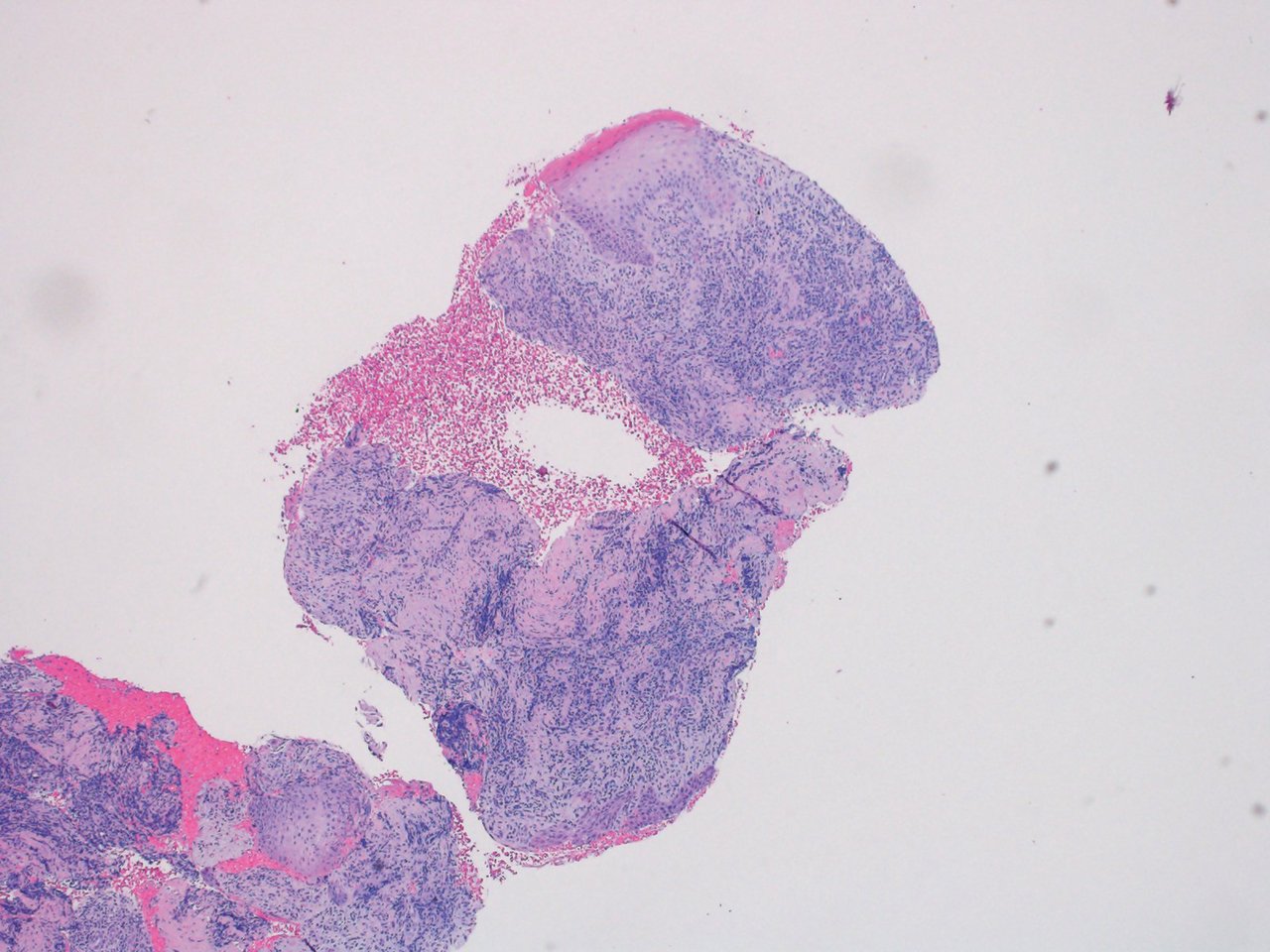
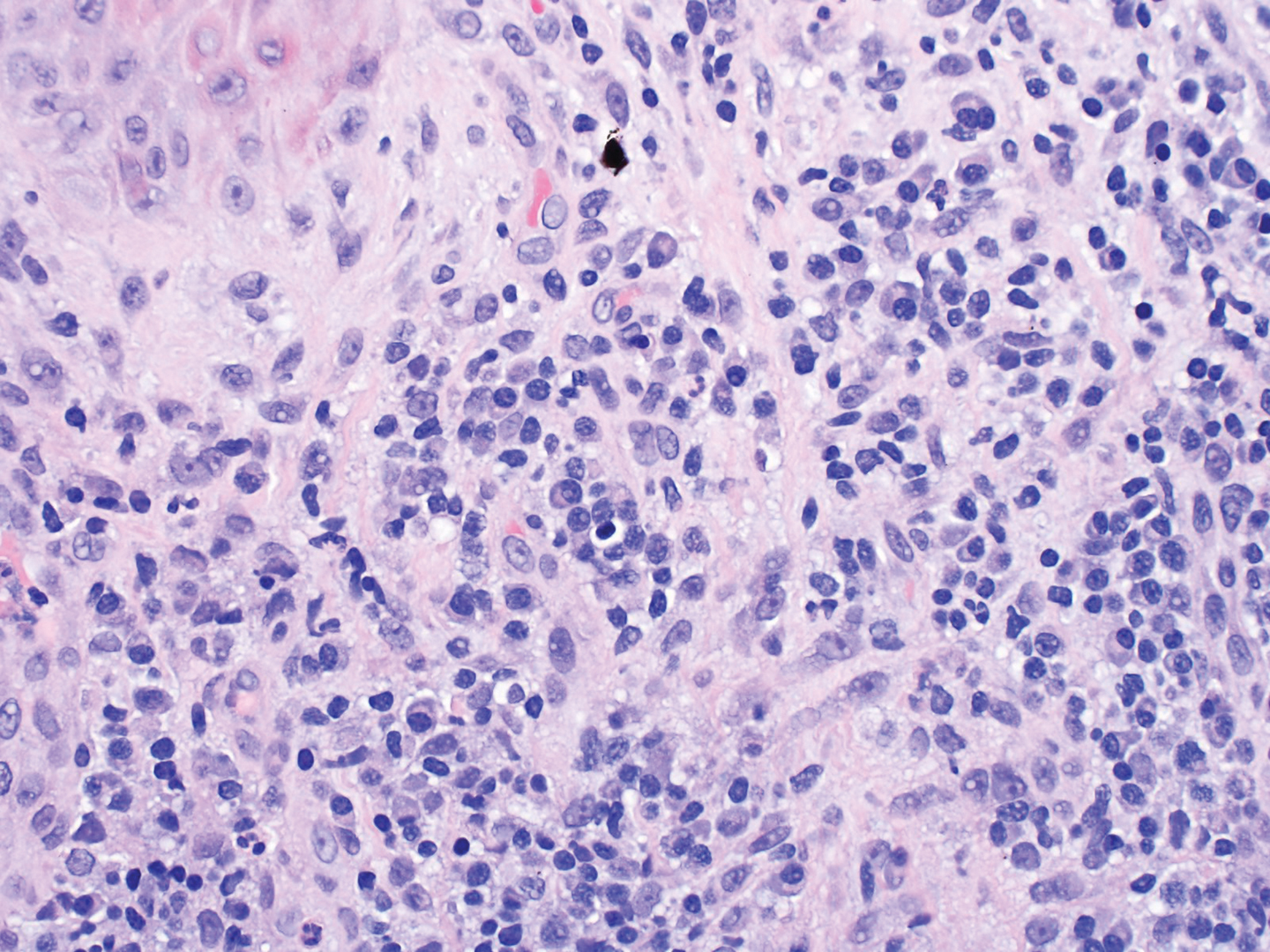
Treatment and Follow-up
The patient was treated with clobetasol ointment 0.05% twice daily for 6 weeks and topical lidocaine as needed for pain. At 6-week follow-up, he displayed substantial improvement, with normal-appearing lips and complete resolution of symptoms.
Comment
The diagnosis and management of PCC is difficult because the condition is uncommon (though its true incidence is unknown) and the presentation is nonspecific, invoking a wide differential diagnosis. In the literature, PCC presents as a slowly progressive, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 The lesion can progress to become eroded, ulcerated, fissured, or edematous.5
Differential Diagnosis
The clinical differential diagnosis of PCC is broad and includes inflammatory, infectious, and neoplastic causes, such as actinic cheilitis, allergic contact cheilitis, exfoliative cheilitis, granulomatous cheilitis, lichen planus, candidiasis, syphilis, and squamous cell carcinoma of the lip.7,9 The histologic differential diagnosis includes allergic contact cheilitis, secondary syphilis, actinic cheilitis, squamous cell carcinoma, cheilitis granulomatosa, and plasmacytoma.17-19
Histopathology
On biopsy, PCC usually is characterized by plasma cells in a bandlike pattern in the upper submucosa or even more diffusely throughout the submucosa.20 In earlier studies, polyclonality of plasma cells with kappa and lambda light chains has been demonstrated5; in this case, such polyclonality militated against a plasma cell dyscrasia. There have been reports of a various number of eosinophils in PCC,5,20 but eosinophils were not a prominent feature in our case.
Treatment
As reported in the literature, treatment of PCC has been attempted using a broad range of strategies; however, the optimal regimen has yet to be elucidated.15 Numerous therapies, including excision, radiation, electrocauterization, cryotherapy, steroids, systemic griseofulvin, topical fusidic acid, and topical calcineurin inhibitors, have yielded variable success.6,7,10-16
The success of topical corticosteroids, as demonstrated in our case, has been unpredictable; the reported response has ranged from complete resolution to failure.9 This variability is thought to be related to epithelial width and the degree of acanthosis, with ulcerative lesions demonstrating a superior response to topical corticosteroids.9
Conclusion
Our case highlights the challenges of diagnosing and managing PCC, especially through teledermatology. Initial photographs of the lesion (Figure 1) that were submitted demonstrated a nonspecific erosion, which was concerning for any of several infectious, inflammatory, and malignant causes. Prompt in-person evaluation was warranted; regrettably, the patient’s condition worsened rapidly in the 10 days it took for him to be seen in-person by dermatology.
Furthermore, this case necessitated 3 separate biopsies because the pathology on the first 2 biopsies initially was equivocal, demonstrating ulceration and granulation tissue. The diagnosis was finally made after a third biopsy was recommended by a dermatopathologist, who eventually identified a bandlike distribution of polyclonal plasma cells in the upper submucosa, consistent with a diagnosis of PCC. Our patient’s final disease presentation (Figure 3) was exuberant and may represent the end point of untreated PCC.
- Senol M, Ozcan A, Aydin NE, et al. Intertriginous plasmacytosis with plasmoacanthoma: report of a typical case and review of the literature. Int J Dermatol. 2008;47:265-268. doi:10.1111/j.1365-4632.2008.03385.x
- Rocha N, Mota F, Horta M, et al. Plasma cell cheilitis. J Eur Acad Dermatol Venereol. 2004;18:96-98. doi:10.1111/j.1468-3083.2004.00791.x
- Farrier JN, Perkins CS. Plasma cell cheilitis. Br J Oral Maxillofac Surg. 2008;46:679-680. doi:10.1016/j.bjoms.2008.03.009
- Baughman RD, Berger P, Pringle WM. Plasma cell cheilitis. Arch Dermatol. 1974;110:725-726.
- Lee JY, Kim KH, Hahm JE, et al. Plasma cell cheilitis: a clinicopathological and immunohistochemical study of 13 cases. Ann Dermatol. 2017;29:536-542. doi:10.5021/ad.2017.29.5.536
- da Cunha Filho RR, Tochetto LB, Tochetto BB, et al. “Angular” plasma cell cheilitis. Dermatol Online J. 2014;20:doj_21759.
- Yang JH, Lee UH, Jang SJ, et al. Plasma cell cheilitis treated with intralesional injection of corticosteroids. J Dermatol. 2005;32:987-990. doi:10.1111/j.1346-8138.2005.tb00887.x
- Solomon LW, Wein RO, Rosenwald I, et al. Plasma cell mucositis of the oral cavity: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:853-860. doi:10.1016/j.tripleo.2008.08.016
- Dos Santos HT, Cunha JLS, Santana LAM, et al. Plasma cell cheilitis: the diagnosis of a disorder mimicking lip cancer. Autops Case Rep. 2019;9:e2018075. doi:10.4322/acr.2018.075
- Fujimura T, Furudate S, Ishibashi M, et al. Successful treatment of plasmacytosis circumorificialis with topical tacrolimus: two case reports and an immunohistochemical study. Case Rep Dermatol. 2013;5:79-83. doi:10.1159/000350184
- Tamaki K, Osada A, Tsukamoto K, et al. Treatment of plasma cell cheilitis with griseofulvin. J Am Acad Dermatol. 1994;30:789-790. doi:10.1016/s0190-9622(08)81515-0
- Choi JW, Choi M, Cho KH. Successful treatment of plasma cell cheilitis with topical calcineurin inhibitors. J Dermatol. 2009;36:669-671. doi:10.1111/j.1346-8138.2009.00733.x
- Hanami Y, Motoki Y, Yamamoto T. Successful treatment of plasma cell cheilitis with topical tacrolimus: report of two cases. Dermatol Online J. 2011;17:6.
- Jin SP, Cho KH, Huh CH. Plasma cell cheilitis, successfully treated with topical 0.03% tacrolimus ointment. J Dermatolog Treat. 2010;21:130-132. doi:10.1080/09546630903200620
- Tseng JT-P, Cheng C-J, Lee W-R, et al. Plasma-cell cheilitis: successful treatment with intralesional injections of corticosteroids. Clin Exp Dermatol. 2009;34:174-177. doi:10.1111/j.1365-2230.2008.02765.x
- Yoshimura K, Nakano S, Tsuruta D, et al. Successful treatment with 308-nm monochromatic excimer light and subsequent tacrolimus 0.03% ointment in refractory plasma cell cheilitis. J Dermatol. 2013;40:471-474. doi:10.1111/1346-8138.12152
- Fujimura Y, Natsuga K, Abe R, et al. Plasma cell cheilitis extending beyond vermillion border. J Dermatol. 2015;42:935-936. doi:10.1111/1346-8138.12985
- White JW Jr, Olsen KD, Banks PM. Plasma cell orificial mucositis. report of a case and review of the literature. Arch Dermatol. 1986;122:1321-1324. doi:10.1001/archderm.122.11.1321
- Román CC, Yuste CM, Gonzalez MA, et al. Plasma cell gingivitis. Cutis. 2002;69:41-45.
- Choe HC, Park HJ, Oh ST, et al. Clinicopathologic study of 8 patients with plasma cell cheilitis. Korean J Dermatol. 2003;41:174-178.
Plasma cell cheilitis (PCC), also known as plasmocytosis circumorificialis and plasmocytosis mucosae,1 is a poorly understood, uncommon inflammatory condition characterized by dense infiltration of mature plasma cells in the mucosal dermis of the lip.2-5 The etiology of PCC is unknown but is thought to be a reactive immune process triggered by infection, mechanical friction, trauma, or solar damage.1,5,6
The most common presentation of PCC is a slowly evolving, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 Secondary changes with disease progression can include erosion, ulceration, fissures, edema, bleeding, or crusting.5 The diagnosis of PCC is challenging because it can mimic neoplastic, infectious, and inflammatory conditions.8,9
Treatment strategies for PCC described in the literature vary, as does therapeutic response. Resolution of PCC has been documented after systemic steroids, intralesional steroids, systemic griseofulvin, and topical calcineurin inhibitors, among other agents.6,7,10-16
We present the case of a patient with a lip lesion who ultimately was diagnosed with PCC after it progressed to an advanced necrotic stage.
Case Report
An 80-year-old male veteran of the Armed Services initially presented to our institution via teledermatology with redness and crusting of the lower lip (Figure 1). He had a history of myelodysplastic syndrome and anemia requiring iron transfusion. The process appeared to be consistent with actinic cheilitis vs squamous cell carcinoma. In-person dermatology consultation was recommended; however, the patient did not follow through with that appointment.
Five months later, additional photographs of the lesion were taken by the patient's primary care physician and sent through teledermatology, revealing progression to an erythematous, yellow-crusted erosion (Figure 2). The medical record indicated that a punch biopsy performed by the patient’s primary care physician showed hyperkeratosis and fungal organisms on periodic acid–Schiff staining. He subsequently applied ketoconazole and terbinafine cream to the lower lip without improvement. Prompt in-person evaluation by dermatology was again recommended.
Ten days later, the patient was seen in our dermatology clinic, at which point his condition had rapidly progressed. The lower lip displayed a 3.0×2.5-cm, yellow and black, crusted, ulcerated plaque (Figure 3). He reported severe burning and pain of the lip as well as spontaneous bleeding. He had lost approximately 10 pounds over the last month due to poor oral intake. A second punch biopsy showed benign mucosa with extensive ulceration and formation of full-thickness granulation tissue. No fungi or bacteria were identified.
Consultation and Histologic Analysis
Dermatopathology was consulted and recommended a third punch biopsy for additional testing. A repeat biopsy demonstrated ulceration with lateral elements of retained epidermis and a dense submucosal chronic inflammatory infiltrate comprising plasma cells and lymphocytes (Figures 4 and 5). Immunohistochemical staining demonstrated a mixed inflammatory infiltrate with CD3+ T cells and CD20+ B cells. In situ hybridization studies demonstrated numerous lambda-positive and kappa-positive plasma cells without chain restriction. Periodic acid–Schiff with diastase and Grocott-Gomori methenamine-silver staining demonstrated no fungi. Findings were interpreted to be most consistent with a diagnosis of PCC.
Treatment and Follow-up
The patient was treated with clobetasol ointment 0.05% twice daily for 6 weeks and topical lidocaine as needed for pain. At 6-week follow-up, he displayed substantial improvement, with normal-appearing lips and complete resolution of symptoms.
Comment
The diagnosis and management of PCC is difficult because the condition is uncommon (though its true incidence is unknown) and the presentation is nonspecific, invoking a wide differential diagnosis. In the literature, PCC presents as a slowly progressive, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 The lesion can progress to become eroded, ulcerated, fissured, or edematous.5
Differential Diagnosis
The clinical differential diagnosis of PCC is broad and includes inflammatory, infectious, and neoplastic causes, such as actinic cheilitis, allergic contact cheilitis, exfoliative cheilitis, granulomatous cheilitis, lichen planus, candidiasis, syphilis, and squamous cell carcinoma of the lip.7,9 The histologic differential diagnosis includes allergic contact cheilitis, secondary syphilis, actinic cheilitis, squamous cell carcinoma, cheilitis granulomatosa, and plasmacytoma.17-19
Histopathology
On biopsy, PCC usually is characterized by plasma cells in a bandlike pattern in the upper submucosa or even more diffusely throughout the submucosa.20 In earlier studies, polyclonality of plasma cells with kappa and lambda light chains has been demonstrated5; in this case, such polyclonality militated against a plasma cell dyscrasia. There have been reports of a various number of eosinophils in PCC,5,20 but eosinophils were not a prominent feature in our case.
Treatment
As reported in the literature, treatment of PCC has been attempted using a broad range of strategies; however, the optimal regimen has yet to be elucidated.15 Numerous therapies, including excision, radiation, electrocauterization, cryotherapy, steroids, systemic griseofulvin, topical fusidic acid, and topical calcineurin inhibitors, have yielded variable success.6,7,10-16
The success of topical corticosteroids, as demonstrated in our case, has been unpredictable; the reported response has ranged from complete resolution to failure.9 This variability is thought to be related to epithelial width and the degree of acanthosis, with ulcerative lesions demonstrating a superior response to topical corticosteroids.9
Conclusion
Our case highlights the challenges of diagnosing and managing PCC, especially through teledermatology. Initial photographs of the lesion (Figure 1) that were submitted demonstrated a nonspecific erosion, which was concerning for any of several infectious, inflammatory, and malignant causes. Prompt in-person evaluation was warranted; regrettably, the patient’s condition worsened rapidly in the 10 days it took for him to be seen in-person by dermatology.
Furthermore, this case necessitated 3 separate biopsies because the pathology on the first 2 biopsies initially was equivocal, demonstrating ulceration and granulation tissue. The diagnosis was finally made after a third biopsy was recommended by a dermatopathologist, who eventually identified a bandlike distribution of polyclonal plasma cells in the upper submucosa, consistent with a diagnosis of PCC. Our patient’s final disease presentation (Figure 3) was exuberant and may represent the end point of untreated PCC.
Plasma cell cheilitis (PCC), also known as plasmocytosis circumorificialis and plasmocytosis mucosae,1 is a poorly understood, uncommon inflammatory condition characterized by dense infiltration of mature plasma cells in the mucosal dermis of the lip.2-5 The etiology of PCC is unknown but is thought to be a reactive immune process triggered by infection, mechanical friction, trauma, or solar damage.1,5,6
The most common presentation of PCC is a slowly evolving, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 Secondary changes with disease progression can include erosion, ulceration, fissures, edema, bleeding, or crusting.5 The diagnosis of PCC is challenging because it can mimic neoplastic, infectious, and inflammatory conditions.8,9
Treatment strategies for PCC described in the literature vary, as does therapeutic response. Resolution of PCC has been documented after systemic steroids, intralesional steroids, systemic griseofulvin, and topical calcineurin inhibitors, among other agents.6,7,10-16
We present the case of a patient with a lip lesion who ultimately was diagnosed with PCC after it progressed to an advanced necrotic stage.
Case Report
An 80-year-old male veteran of the Armed Services initially presented to our institution via teledermatology with redness and crusting of the lower lip (Figure 1). He had a history of myelodysplastic syndrome and anemia requiring iron transfusion. The process appeared to be consistent with actinic cheilitis vs squamous cell carcinoma. In-person dermatology consultation was recommended; however, the patient did not follow through with that appointment.
Five months later, additional photographs of the lesion were taken by the patient's primary care physician and sent through teledermatology, revealing progression to an erythematous, yellow-crusted erosion (Figure 2). The medical record indicated that a punch biopsy performed by the patient’s primary care physician showed hyperkeratosis and fungal organisms on periodic acid–Schiff staining. He subsequently applied ketoconazole and terbinafine cream to the lower lip without improvement. Prompt in-person evaluation by dermatology was again recommended.
Ten days later, the patient was seen in our dermatology clinic, at which point his condition had rapidly progressed. The lower lip displayed a 3.0×2.5-cm, yellow and black, crusted, ulcerated plaque (Figure 3). He reported severe burning and pain of the lip as well as spontaneous bleeding. He had lost approximately 10 pounds over the last month due to poor oral intake. A second punch biopsy showed benign mucosa with extensive ulceration and formation of full-thickness granulation tissue. No fungi or bacteria were identified.
Consultation and Histologic Analysis
Dermatopathology was consulted and recommended a third punch biopsy for additional testing. A repeat biopsy demonstrated ulceration with lateral elements of retained epidermis and a dense submucosal chronic inflammatory infiltrate comprising plasma cells and lymphocytes (Figures 4 and 5). Immunohistochemical staining demonstrated a mixed inflammatory infiltrate with CD3+ T cells and CD20+ B cells. In situ hybridization studies demonstrated numerous lambda-positive and kappa-positive plasma cells without chain restriction. Periodic acid–Schiff with diastase and Grocott-Gomori methenamine-silver staining demonstrated no fungi. Findings were interpreted to be most consistent with a diagnosis of PCC.
Treatment and Follow-up
The patient was treated with clobetasol ointment 0.05% twice daily for 6 weeks and topical lidocaine as needed for pain. At 6-week follow-up, he displayed substantial improvement, with normal-appearing lips and complete resolution of symptoms.
Comment
The diagnosis and management of PCC is difficult because the condition is uncommon (though its true incidence is unknown) and the presentation is nonspecific, invoking a wide differential diagnosis. In the literature, PCC presents as a slowly progressive, red-brown patch or plaque on the lower lip in older individuals.2,3,5,7 The lesion can progress to become eroded, ulcerated, fissured, or edematous.5
Differential Diagnosis
The clinical differential diagnosis of PCC is broad and includes inflammatory, infectious, and neoplastic causes, such as actinic cheilitis, allergic contact cheilitis, exfoliative cheilitis, granulomatous cheilitis, lichen planus, candidiasis, syphilis, and squamous cell carcinoma of the lip.7,9 The histologic differential diagnosis includes allergic contact cheilitis, secondary syphilis, actinic cheilitis, squamous cell carcinoma, cheilitis granulomatosa, and plasmacytoma.17-19
Histopathology
On biopsy, PCC usually is characterized by plasma cells in a bandlike pattern in the upper submucosa or even more diffusely throughout the submucosa.20 In earlier studies, polyclonality of plasma cells with kappa and lambda light chains has been demonstrated5; in this case, such polyclonality militated against a plasma cell dyscrasia. There have been reports of a various number of eosinophils in PCC,5,20 but eosinophils were not a prominent feature in our case.
Treatment
As reported in the literature, treatment of PCC has been attempted using a broad range of strategies; however, the optimal regimen has yet to be elucidated.15 Numerous therapies, including excision, radiation, electrocauterization, cryotherapy, steroids, systemic griseofulvin, topical fusidic acid, and topical calcineurin inhibitors, have yielded variable success.6,7,10-16
The success of topical corticosteroids, as demonstrated in our case, has been unpredictable; the reported response has ranged from complete resolution to failure.9 This variability is thought to be related to epithelial width and the degree of acanthosis, with ulcerative lesions demonstrating a superior response to topical corticosteroids.9
Conclusion
Our case highlights the challenges of diagnosing and managing PCC, especially through teledermatology. Initial photographs of the lesion (Figure 1) that were submitted demonstrated a nonspecific erosion, which was concerning for any of several infectious, inflammatory, and malignant causes. Prompt in-person evaluation was warranted; regrettably, the patient’s condition worsened rapidly in the 10 days it took for him to be seen in-person by dermatology.
Furthermore, this case necessitated 3 separate biopsies because the pathology on the first 2 biopsies initially was equivocal, demonstrating ulceration and granulation tissue. The diagnosis was finally made after a third biopsy was recommended by a dermatopathologist, who eventually identified a bandlike distribution of polyclonal plasma cells in the upper submucosa, consistent with a diagnosis of PCC. Our patient’s final disease presentation (Figure 3) was exuberant and may represent the end point of untreated PCC.
- Senol M, Ozcan A, Aydin NE, et al. Intertriginous plasmacytosis with plasmoacanthoma: report of a typical case and review of the literature. Int J Dermatol. 2008;47:265-268. doi:10.1111/j.1365-4632.2008.03385.x
- Rocha N, Mota F, Horta M, et al. Plasma cell cheilitis. J Eur Acad Dermatol Venereol. 2004;18:96-98. doi:10.1111/j.1468-3083.2004.00791.x
- Farrier JN, Perkins CS. Plasma cell cheilitis. Br J Oral Maxillofac Surg. 2008;46:679-680. doi:10.1016/j.bjoms.2008.03.009
- Baughman RD, Berger P, Pringle WM. Plasma cell cheilitis. Arch Dermatol. 1974;110:725-726.
- Lee JY, Kim KH, Hahm JE, et al. Plasma cell cheilitis: a clinicopathological and immunohistochemical study of 13 cases. Ann Dermatol. 2017;29:536-542. doi:10.5021/ad.2017.29.5.536
- da Cunha Filho RR, Tochetto LB, Tochetto BB, et al. “Angular” plasma cell cheilitis. Dermatol Online J. 2014;20:doj_21759.
- Yang JH, Lee UH, Jang SJ, et al. Plasma cell cheilitis treated with intralesional injection of corticosteroids. J Dermatol. 2005;32:987-990. doi:10.1111/j.1346-8138.2005.tb00887.x
- Solomon LW, Wein RO, Rosenwald I, et al. Plasma cell mucositis of the oral cavity: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:853-860. doi:10.1016/j.tripleo.2008.08.016
- Dos Santos HT, Cunha JLS, Santana LAM, et al. Plasma cell cheilitis: the diagnosis of a disorder mimicking lip cancer. Autops Case Rep. 2019;9:e2018075. doi:10.4322/acr.2018.075
- Fujimura T, Furudate S, Ishibashi M, et al. Successful treatment of plasmacytosis circumorificialis with topical tacrolimus: two case reports and an immunohistochemical study. Case Rep Dermatol. 2013;5:79-83. doi:10.1159/000350184
- Tamaki K, Osada A, Tsukamoto K, et al. Treatment of plasma cell cheilitis with griseofulvin. J Am Acad Dermatol. 1994;30:789-790. doi:10.1016/s0190-9622(08)81515-0
- Choi JW, Choi M, Cho KH. Successful treatment of plasma cell cheilitis with topical calcineurin inhibitors. J Dermatol. 2009;36:669-671. doi:10.1111/j.1346-8138.2009.00733.x
- Hanami Y, Motoki Y, Yamamoto T. Successful treatment of plasma cell cheilitis with topical tacrolimus: report of two cases. Dermatol Online J. 2011;17:6.
- Jin SP, Cho KH, Huh CH. Plasma cell cheilitis, successfully treated with topical 0.03% tacrolimus ointment. J Dermatolog Treat. 2010;21:130-132. doi:10.1080/09546630903200620
- Tseng JT-P, Cheng C-J, Lee W-R, et al. Plasma-cell cheilitis: successful treatment with intralesional injections of corticosteroids. Clin Exp Dermatol. 2009;34:174-177. doi:10.1111/j.1365-2230.2008.02765.x
- Yoshimura K, Nakano S, Tsuruta D, et al. Successful treatment with 308-nm monochromatic excimer light and subsequent tacrolimus 0.03% ointment in refractory plasma cell cheilitis. J Dermatol. 2013;40:471-474. doi:10.1111/1346-8138.12152
- Fujimura Y, Natsuga K, Abe R, et al. Plasma cell cheilitis extending beyond vermillion border. J Dermatol. 2015;42:935-936. doi:10.1111/1346-8138.12985
- White JW Jr, Olsen KD, Banks PM. Plasma cell orificial mucositis. report of a case and review of the literature. Arch Dermatol. 1986;122:1321-1324. doi:10.1001/archderm.122.11.1321
- Román CC, Yuste CM, Gonzalez MA, et al. Plasma cell gingivitis. Cutis. 2002;69:41-45.
- Choe HC, Park HJ, Oh ST, et al. Clinicopathologic study of 8 patients with plasma cell cheilitis. Korean J Dermatol. 2003;41:174-178.
- Senol M, Ozcan A, Aydin NE, et al. Intertriginous plasmacytosis with plasmoacanthoma: report of a typical case and review of the literature. Int J Dermatol. 2008;47:265-268. doi:10.1111/j.1365-4632.2008.03385.x
- Rocha N, Mota F, Horta M, et al. Plasma cell cheilitis. J Eur Acad Dermatol Venereol. 2004;18:96-98. doi:10.1111/j.1468-3083.2004.00791.x
- Farrier JN, Perkins CS. Plasma cell cheilitis. Br J Oral Maxillofac Surg. 2008;46:679-680. doi:10.1016/j.bjoms.2008.03.009
- Baughman RD, Berger P, Pringle WM. Plasma cell cheilitis. Arch Dermatol. 1974;110:725-726.
- Lee JY, Kim KH, Hahm JE, et al. Plasma cell cheilitis: a clinicopathological and immunohistochemical study of 13 cases. Ann Dermatol. 2017;29:536-542. doi:10.5021/ad.2017.29.5.536
- da Cunha Filho RR, Tochetto LB, Tochetto BB, et al. “Angular” plasma cell cheilitis. Dermatol Online J. 2014;20:doj_21759.
- Yang JH, Lee UH, Jang SJ, et al. Plasma cell cheilitis treated with intralesional injection of corticosteroids. J Dermatol. 2005;32:987-990. doi:10.1111/j.1346-8138.2005.tb00887.x
- Solomon LW, Wein RO, Rosenwald I, et al. Plasma cell mucositis of the oral cavity: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:853-860. doi:10.1016/j.tripleo.2008.08.016
- Dos Santos HT, Cunha JLS, Santana LAM, et al. Plasma cell cheilitis: the diagnosis of a disorder mimicking lip cancer. Autops Case Rep. 2019;9:e2018075. doi:10.4322/acr.2018.075
- Fujimura T, Furudate S, Ishibashi M, et al. Successful treatment of plasmacytosis circumorificialis with topical tacrolimus: two case reports and an immunohistochemical study. Case Rep Dermatol. 2013;5:79-83. doi:10.1159/000350184
- Tamaki K, Osada A, Tsukamoto K, et al. Treatment of plasma cell cheilitis with griseofulvin. J Am Acad Dermatol. 1994;30:789-790. doi:10.1016/s0190-9622(08)81515-0
- Choi JW, Choi M, Cho KH. Successful treatment of plasma cell cheilitis with topical calcineurin inhibitors. J Dermatol. 2009;36:669-671. doi:10.1111/j.1346-8138.2009.00733.x
- Hanami Y, Motoki Y, Yamamoto T. Successful treatment of plasma cell cheilitis with topical tacrolimus: report of two cases. Dermatol Online J. 2011;17:6.
- Jin SP, Cho KH, Huh CH. Plasma cell cheilitis, successfully treated with topical 0.03% tacrolimus ointment. J Dermatolog Treat. 2010;21:130-132. doi:10.1080/09546630903200620
- Tseng JT-P, Cheng C-J, Lee W-R, et al. Plasma-cell cheilitis: successful treatment with intralesional injections of corticosteroids. Clin Exp Dermatol. 2009;34:174-177. doi:10.1111/j.1365-2230.2008.02765.x
- Yoshimura K, Nakano S, Tsuruta D, et al. Successful treatment with 308-nm monochromatic excimer light and subsequent tacrolimus 0.03% ointment in refractory plasma cell cheilitis. J Dermatol. 2013;40:471-474. doi:10.1111/1346-8138.12152
- Fujimura Y, Natsuga K, Abe R, et al. Plasma cell cheilitis extending beyond vermillion border. J Dermatol. 2015;42:935-936. doi:10.1111/1346-8138.12985
- White JW Jr, Olsen KD, Banks PM. Plasma cell orificial mucositis. report of a case and review of the literature. Arch Dermatol. 1986;122:1321-1324. doi:10.1001/archderm.122.11.1321
- Román CC, Yuste CM, Gonzalez MA, et al. Plasma cell gingivitis. Cutis. 2002;69:41-45.
- Choe HC, Park HJ, Oh ST, et al. Clinicopathologic study of 8 patients with plasma cell cheilitis. Korean J Dermatol. 2003;41:174-178.
PRACTICE POINTS
- Plasma cell cheilitis (PCC) is a benign condition that affects the lower lip in older individuals, presenting as a nonspecific, red-brown patch or plaque that can progress slowly to erosions and edema.
- Our patient with PCC experienced full resolution of symptoms with application of a class I topical corticosteroid.
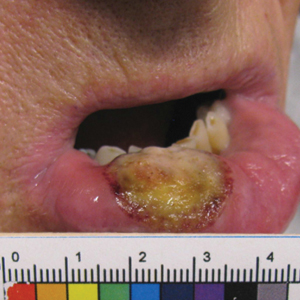
Verruca Vulgaris Arising Within the Red Portion of a Multicolored Tattoo
To the Editor:
The art of tattooing continues to gain popularity in the 21st century, albeit with accompanying hazards.1 Reported adverse reactions to tattoos include infections, tumors, and hypersensitivity and granulomatous reactions.2 Various infectious agents may involve tattoos, including human papillomavirus (HPV), molluscum contagiosum, herpes simplex virus, hepatitis C virus, tuberculoid and nontuberculoid mycobacteria, and Staphylococcus aureus.2 Verruca vulgaris infrequently has been reported to develop in tattoos.3,4 Previously reported cases of verruca in tattoos suggest a predilection for blue or black pigment.1-5 We report a case of verruca vulgaris occurring within the red-inked areas of a tattoo that first appeared approximately 18 years after the initial tattoo placement.
A 44-year-old woman presented with erythema, induration, and irritation of a tattoo on the left leg of 2 years’ duration. The tattoo initially was inscribed more than 20 years prior. The patient had a history of type 2 diabetes mellitus and chronic obstructive pulmonary disease. She reported no prior trauma to the area, prior rash or irritation, or similar changes to her other tattoos, including those with red ink. The affected tattoo was inscribed at a separate time from the other tattoos. Physical examination of the irritated tattoo revealed hyperkeratotic papules with firm scaling in the zone of dermal red pigment (Figure 1). Notable nodularity or deep induration was not present. The clinical differential diagnosis included a hypersensitivity reaction to red tattoo ink, sarcoidosis, and an infectious process, such as an atypical mycobacterial infection. A punch biopsy demonstrated papillomatous epidermal hyperplasia with hyperkeratosis, focal parakeratosis, and frequent vacuolization of keratinocytes with enlarged keratohyalin granules, diagnostic of verruca vulgaris (Figure 2). Of note, the patient did not have clinically apparent viral warts elsewhere on physical examination. The patient was successfully managedwith a combination of 2 treatments of intralesional Candida antigen and 3 treatments of cryotherapy with resolution of most lesions over the course of 8 months. Over the following several months, the patient applied topical salicylic acid, which led to the resolution of the remaining lesions. The verrucae had not recurred 19 months after the initial presentation.
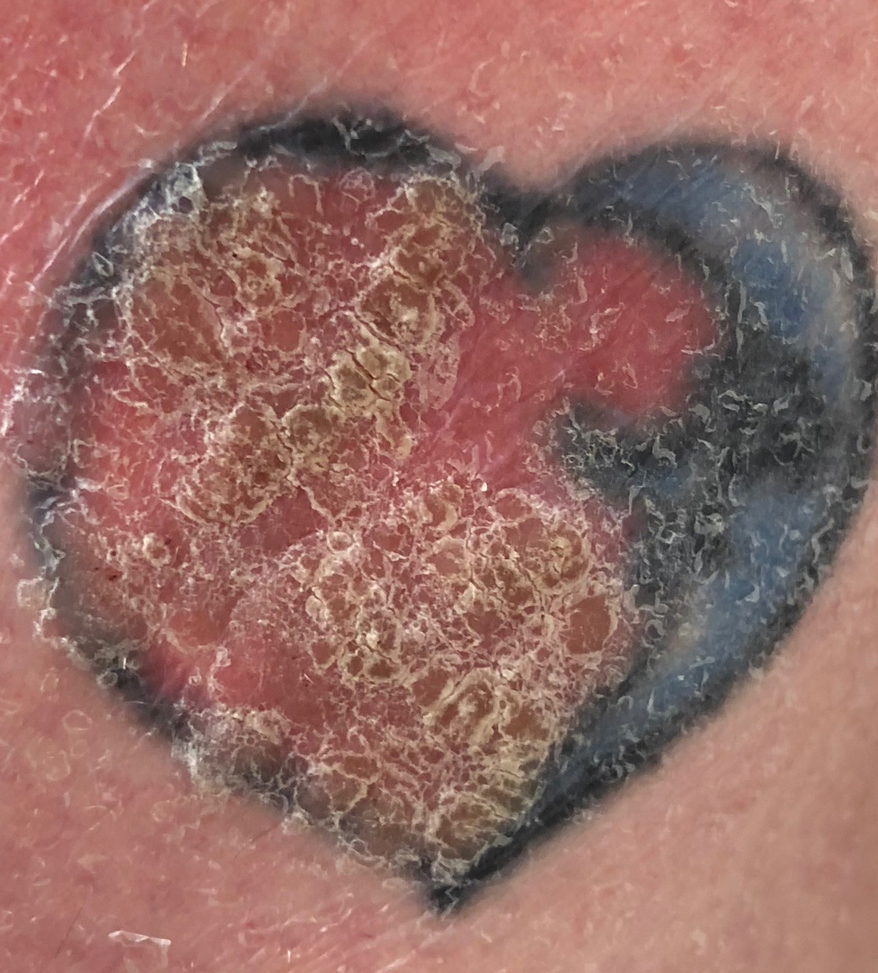
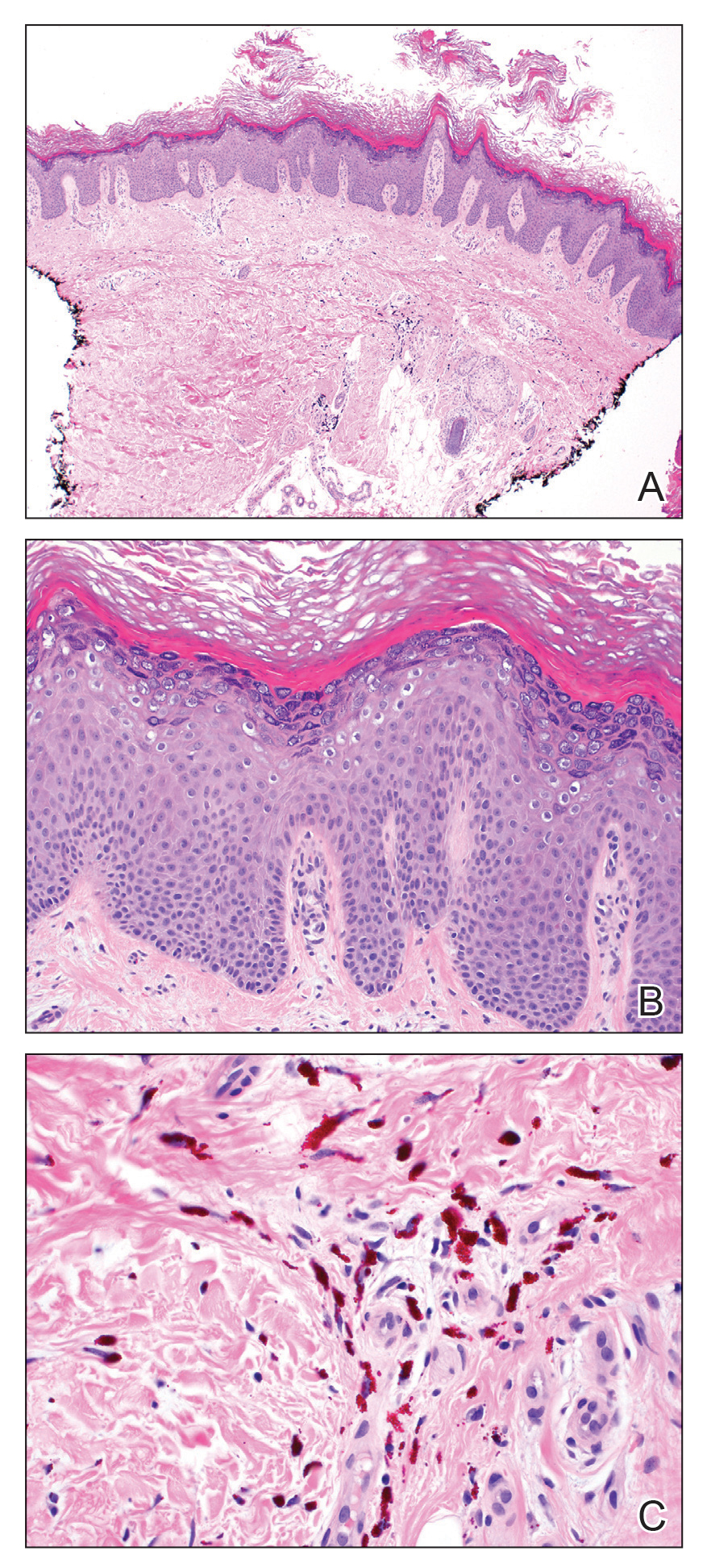
The development of verruca vulgaris within a tattoo may occur secondary to various mechanisms of HPV inoculation, including introduction of the virus through contaminated ink, the tattoo artist’s saliva, autoinoculation, or koebnerization of a pre-existing verruca vulgaris.4 Local immune system dysregulation secondary to tattoo ink also has been proposed as a mechanism for HPV infection in this setting.1,5 The contents of darker tattoo pigments may promote formation of reactive oxygen species inducing local immunocompromise.5
The pathogenic mechanism was elusive in our patient. Although the localization of verruca vulgaris to the zones of red pigment may be merely coincidental, this phenomenon raised suspicion for direct inoculation via contaminated red ink. The patient’s other red ink–containing tattoos that were inscribed separately were spared, compatible with contamination of the red ink used for the affected tattoo. However, the delayed onset of nearly 2 decades was exceptional, given the shorter previously reported latencies ranging from months to 10 years.4 Autoinoculation or koebnerization is plausible, though greater involvement of nonred pigments would be expected as well as a briefer latency. Finally, the possibility of local immune dysregulation seemed feasible, given the slow evolution of the lesions largely restricted to one pigment type.
We report a case of verruca vulgaris within the red area of a multicolored tattoo that occurred approximately 18 years after tattoo placement. This case highlights a rare presentation of an infectious agent that may complicate tattoos. Both predilection for red pigment rather than black or blue pigment and the long latency period raised interesting questions regarding pathogenesis. Confirmatory biopsy enables effective management of this tattoo complication.
- Huynh TN, Jackson JD, Brodell RT. Tattoo and vaccination sites: possible nest for opportunistic infections, tumors, and dysimmune reactions. Clin Dermatol. 2014;32:678-684.
- Wenzel SM, Rittmann I, Landthaler M, et al. Adverse reactions after tattooing: review of the literature and comparison to results of a survey. Dermatology. 2013;226:138-147.
- Trefzer U, Schmollack K, Stockfleth E, et al. Verrucae in a multicolored decorative tattoo. J Am Acad Dermatol. 2004;50:478-479.
- Wanat KA, Tyring S, Rady P, et al. Human papillomavirus type 27 associated with multiple verruca within a tattoo: report of a case and review of the literature. Int J Dermatol. 2014;53:882-884.
- Ramey K, Ibrahim J, Brodell RT. Verruca localization predominately in black tattoo ink: a retrospective case series. J Eur Acad Dermatol Venereol. 2016;30:E34-E36.
To the Editor:
The art of tattooing continues to gain popularity in the 21st century, albeit with accompanying hazards.1 Reported adverse reactions to tattoos include infections, tumors, and hypersensitivity and granulomatous reactions.2 Various infectious agents may involve tattoos, including human papillomavirus (HPV), molluscum contagiosum, herpes simplex virus, hepatitis C virus, tuberculoid and nontuberculoid mycobacteria, and Staphylococcus aureus.2 Verruca vulgaris infrequently has been reported to develop in tattoos.3,4 Previously reported cases of verruca in tattoos suggest a predilection for blue or black pigment.1-5 We report a case of verruca vulgaris occurring within the red-inked areas of a tattoo that first appeared approximately 18 years after the initial tattoo placement.
A 44-year-old woman presented with erythema, induration, and irritation of a tattoo on the left leg of 2 years’ duration. The tattoo initially was inscribed more than 20 years prior. The patient had a history of type 2 diabetes mellitus and chronic obstructive pulmonary disease. She reported no prior trauma to the area, prior rash or irritation, or similar changes to her other tattoos, including those with red ink. The affected tattoo was inscribed at a separate time from the other tattoos. Physical examination of the irritated tattoo revealed hyperkeratotic papules with firm scaling in the zone of dermal red pigment (Figure 1). Notable nodularity or deep induration was not present. The clinical differential diagnosis included a hypersensitivity reaction to red tattoo ink, sarcoidosis, and an infectious process, such as an atypical mycobacterial infection. A punch biopsy demonstrated papillomatous epidermal hyperplasia with hyperkeratosis, focal parakeratosis, and frequent vacuolization of keratinocytes with enlarged keratohyalin granules, diagnostic of verruca vulgaris (Figure 2). Of note, the patient did not have clinically apparent viral warts elsewhere on physical examination. The patient was successfully managedwith a combination of 2 treatments of intralesional Candida antigen and 3 treatments of cryotherapy with resolution of most lesions over the course of 8 months. Over the following several months, the patient applied topical salicylic acid, which led to the resolution of the remaining lesions. The verrucae had not recurred 19 months after the initial presentation.
The development of verruca vulgaris within a tattoo may occur secondary to various mechanisms of HPV inoculation, including introduction of the virus through contaminated ink, the tattoo artist’s saliva, autoinoculation, or koebnerization of a pre-existing verruca vulgaris.4 Local immune system dysregulation secondary to tattoo ink also has been proposed as a mechanism for HPV infection in this setting.1,5 The contents of darker tattoo pigments may promote formation of reactive oxygen species inducing local immunocompromise.5
The pathogenic mechanism was elusive in our patient. Although the localization of verruca vulgaris to the zones of red pigment may be merely coincidental, this phenomenon raised suspicion for direct inoculation via contaminated red ink. The patient’s other red ink–containing tattoos that were inscribed separately were spared, compatible with contamination of the red ink used for the affected tattoo. However, the delayed onset of nearly 2 decades was exceptional, given the shorter previously reported latencies ranging from months to 10 years.4 Autoinoculation or koebnerization is plausible, though greater involvement of nonred pigments would be expected as well as a briefer latency. Finally, the possibility of local immune dysregulation seemed feasible, given the slow evolution of the lesions largely restricted to one pigment type.
We report a case of verruca vulgaris within the red area of a multicolored tattoo that occurred approximately 18 years after tattoo placement. This case highlights a rare presentation of an infectious agent that may complicate tattoos. Both predilection for red pigment rather than black or blue pigment and the long latency period raised interesting questions regarding pathogenesis. Confirmatory biopsy enables effective management of this tattoo complication.
To the Editor:
The art of tattooing continues to gain popularity in the 21st century, albeit with accompanying hazards.1 Reported adverse reactions to tattoos include infections, tumors, and hypersensitivity and granulomatous reactions.2 Various infectious agents may involve tattoos, including human papillomavirus (HPV), molluscum contagiosum, herpes simplex virus, hepatitis C virus, tuberculoid and nontuberculoid mycobacteria, and Staphylococcus aureus.2 Verruca vulgaris infrequently has been reported to develop in tattoos.3,4 Previously reported cases of verruca in tattoos suggest a predilection for blue or black pigment.1-5 We report a case of verruca vulgaris occurring within the red-inked areas of a tattoo that first appeared approximately 18 years after the initial tattoo placement.
A 44-year-old woman presented with erythema, induration, and irritation of a tattoo on the left leg of 2 years’ duration. The tattoo initially was inscribed more than 20 years prior. The patient had a history of type 2 diabetes mellitus and chronic obstructive pulmonary disease. She reported no prior trauma to the area, prior rash or irritation, or similar changes to her other tattoos, including those with red ink. The affected tattoo was inscribed at a separate time from the other tattoos. Physical examination of the irritated tattoo revealed hyperkeratotic papules with firm scaling in the zone of dermal red pigment (Figure 1). Notable nodularity or deep induration was not present. The clinical differential diagnosis included a hypersensitivity reaction to red tattoo ink, sarcoidosis, and an infectious process, such as an atypical mycobacterial infection. A punch biopsy demonstrated papillomatous epidermal hyperplasia with hyperkeratosis, focal parakeratosis, and frequent vacuolization of keratinocytes with enlarged keratohyalin granules, diagnostic of verruca vulgaris (Figure 2). Of note, the patient did not have clinically apparent viral warts elsewhere on physical examination. The patient was successfully managedwith a combination of 2 treatments of intralesional Candida antigen and 3 treatments of cryotherapy with resolution of most lesions over the course of 8 months. Over the following several months, the patient applied topical salicylic acid, which led to the resolution of the remaining lesions. The verrucae had not recurred 19 months after the initial presentation.
The development of verruca vulgaris within a tattoo may occur secondary to various mechanisms of HPV inoculation, including introduction of the virus through contaminated ink, the tattoo artist’s saliva, autoinoculation, or koebnerization of a pre-existing verruca vulgaris.4 Local immune system dysregulation secondary to tattoo ink also has been proposed as a mechanism for HPV infection in this setting.1,5 The contents of darker tattoo pigments may promote formation of reactive oxygen species inducing local immunocompromise.5
The pathogenic mechanism was elusive in our patient. Although the localization of verruca vulgaris to the zones of red pigment may be merely coincidental, this phenomenon raised suspicion for direct inoculation via contaminated red ink. The patient’s other red ink–containing tattoos that were inscribed separately were spared, compatible with contamination of the red ink used for the affected tattoo. However, the delayed onset of nearly 2 decades was exceptional, given the shorter previously reported latencies ranging from months to 10 years.4 Autoinoculation or koebnerization is plausible, though greater involvement of nonred pigments would be expected as well as a briefer latency. Finally, the possibility of local immune dysregulation seemed feasible, given the slow evolution of the lesions largely restricted to one pigment type.
We report a case of verruca vulgaris within the red area of a multicolored tattoo that occurred approximately 18 years after tattoo placement. This case highlights a rare presentation of an infectious agent that may complicate tattoos. Both predilection for red pigment rather than black or blue pigment and the long latency period raised interesting questions regarding pathogenesis. Confirmatory biopsy enables effective management of this tattoo complication.
- Huynh TN, Jackson JD, Brodell RT. Tattoo and vaccination sites: possible nest for opportunistic infections, tumors, and dysimmune reactions. Clin Dermatol. 2014;32:678-684.
- Wenzel SM, Rittmann I, Landthaler M, et al. Adverse reactions after tattooing: review of the literature and comparison to results of a survey. Dermatology. 2013;226:138-147.
- Trefzer U, Schmollack K, Stockfleth E, et al. Verrucae in a multicolored decorative tattoo. J Am Acad Dermatol. 2004;50:478-479.
- Wanat KA, Tyring S, Rady P, et al. Human papillomavirus type 27 associated with multiple verruca within a tattoo: report of a case and review of the literature. Int J Dermatol. 2014;53:882-884.
- Ramey K, Ibrahim J, Brodell RT. Verruca localization predominately in black tattoo ink: a retrospective case series. J Eur Acad Dermatol Venereol. 2016;30:E34-E36.
- Huynh TN, Jackson JD, Brodell RT. Tattoo and vaccination sites: possible nest for opportunistic infections, tumors, and dysimmune reactions. Clin Dermatol. 2014;32:678-684.
- Wenzel SM, Rittmann I, Landthaler M, et al. Adverse reactions after tattooing: review of the literature and comparison to results of a survey. Dermatology. 2013;226:138-147.
- Trefzer U, Schmollack K, Stockfleth E, et al. Verrucae in a multicolored decorative tattoo. J Am Acad Dermatol. 2004;50:478-479.
- Wanat KA, Tyring S, Rady P, et al. Human papillomavirus type 27 associated with multiple verruca within a tattoo: report of a case and review of the literature. Int J Dermatol. 2014;53:882-884.
- Ramey K, Ibrahim J, Brodell RT. Verruca localization predominately in black tattoo ink: a retrospective case series. J Eur Acad Dermatol Venereol. 2016;30:E34-E36.
Practice Points
- Various adverse reactions and infectious agents may involve tattoos.
- Verruca vulgaris may affect tattoos in a color-restricted manner and demonstrate latency of many years after tattoo placement.
- Timely diagnosis of the tattoo-involving process, confirmed by biopsy, allows for appropriate management.
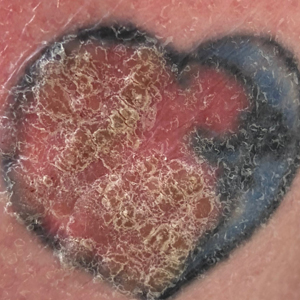
Verrucous Scalp Plaque and Widespread Eruption
The Diagnosis: Pemphigus Foliaceous
Laboratory workup including a complete blood cell count with differential, comprehensive metabolic panel, antinuclear antibodies, Sjögren syndrome A and B antibodies, hepatitis profile, rapid plasma reagin, HIV screen, aldolase, anti–Jo-1, T-Spot TB test (Quest Diagnostics), and tissue cultures was unremarkable. Two 4-mm punch biopsies were obtained from the left cheek and upper back, both of which demonstrated intragranular acantholysis suggestive of pemphigus foliaceous (Figure 1A). A subsequent punch biopsy from the right lower abdomen sent for direct immunofluorescence demonstrated netlike positivity of IgG and C3 in the upper epidermis (Figure 1B), and serum sent for indirect immunofluorescence demonstrated intercellular IgG antibodies to desmoglein (Dsg) 1 on monkey esophagus and positive Dsg-1 antibodies on enzyme-linked immunosorbent assay, confirming the diagnosis.
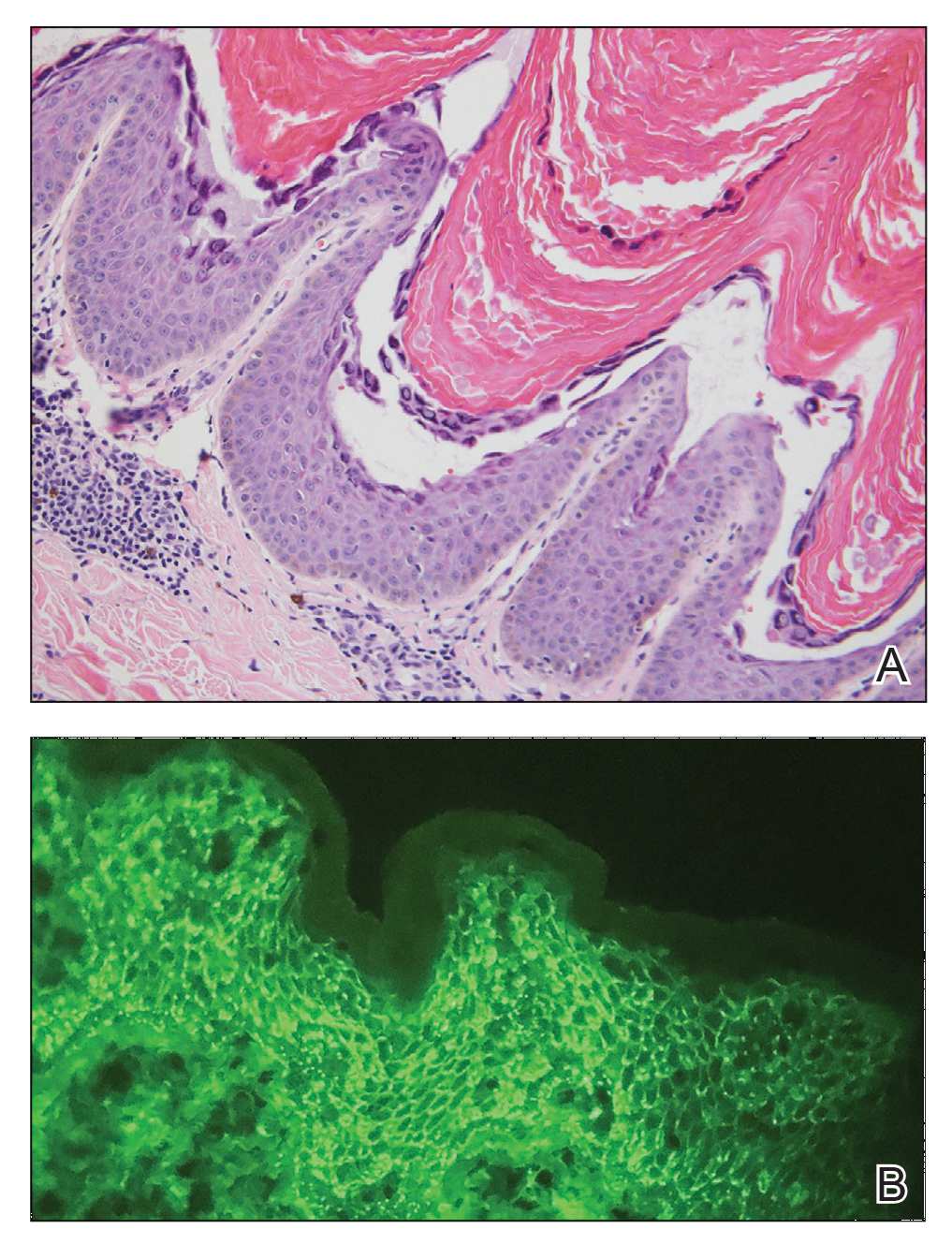
The patient was started on a 60-mg prednisone taper as well as dapsone 50 mg daily; the dapsone was titrated up to 100 mg daily. After tapering down to 10 mg daily of prednisone over 2 months and continuing dapsone with minimal improvement, he was given 2 infusions of rituximab 1000 mg 2 weeks apart. The scalp plaque was dramatically improved at 3-month follow-up (Figure 2), with partial improvement of the cheek plaques (Figure 3). Dapsone was increased to 150 mg daily, and he was encouraged to use triamcinolone acetonide ointment 0.1% twice daily, which led to further improvement.
Pemphigus foliaceus is an autoimmune blistering disease that most commonly occurs in middle-aged adults. It generally is less common than pemphigus vulgaris, except in Finland, Tunisia, and Brazil, where there is an endemic condition with an identical clinical and histological presentation known as fogo selvagem.1
The pathogenesis of pemphigus foliaceous is characterized by IgG autoantibodies against Dsg-1, a transmembrane glycoprotein involved in the cellular adhesion of keratinocytes, which is preferentially expressed in the superficial epidermis.2-7 Dysfunction of Dsg-1 results in the separation of superficial epidermal cells, resulting in intraepidermal blisters.2,7 In contrast to pemphigus vulgaris, there typically is a lack of oral mucosal involvement due to compensation by Dsg-3 in the mucosa.4 Potential triggers for pemphigus foliaceous include exposure to UV radiation; radiotherapy; pregnancy; physiologic stress; and drugs, most commonly captopril, penicillamine, and thiols.8
Pemphigus foliaceous lesions clinically appear as eroded and crusted lesions on an erythematous base, commonly in a seborrheic distribution on the face, scalp, and trunk with sparing of the oral mucosa,2,6 but lesions can progress to a widespread and more severe exfoliative dermatitis.7 Lesions also can appear as psoriasiform plaques and often are initially misdiagnosed as psoriasis, particularly in patients with skin of color.9,10
Diagnosis of pemphigus foliaceous typically is made using a combination of histology as well as both direct and indirect immunofluorescence. Histologically, pemphigus foliaceus presents with subcorneal acantholysis, which is most prominent in the granular layer and occasionally the presence of neutrophils and eosinophils in the blister cavity.7 Direct immunofluorescence demonstrates netlike intercellular IgG and C3 in the upper portion of the epidermis.11 Indirect immunofluorescence can help detect circulating IgG antibodies to Dsg-1, with guinea pig esophagus being the ideal substrate.11,12
First-line treatment of pemphigus foliaceus consists of systemic glucocorticoid therapy, often administered with azathioprine, methotrexate, or mycophenolate mofetil.2,6,13 Although first-line treatment is effective in 60% to 80% of patients,2 relapsing cases can be treated with cyclophosphamide, intravenous immunoglobulin, immunoadsorption, plasmapheresis, or rituximab.2
Rituximab is a chimeric monoclonal antibody targeting CD20+ B cells, leading to decreased antibody production, which has been shown to be effective in treating severe and refractory cases of pemphigus foliaceus.6,13Rituximab with short-course prednisone has been found to be more effective in achieving complete remission at 24 months than prednisone alone.14 In patients with contraindications to systemic glucocorticoid therapy, rituximab has been shown as an effective first-line therapy.15 One-quarter of patients treated with rituximab relapsed within 2 years of treatment6 (average time to relapse, 6–26 months).16 High-dose rituximab regimens, along with a higher number of rituximab treatment cycles, have been shown to prolong time to relapse.6 Further, higher baseline levels of Dsg-1 antibody have been correlated to earlier relapse and can be used following rituximab therapy to monitor disease progression.6,16
The differential diagnosis for pemphigus foliaceous includes disseminated blastomycosis, hypertrophic lupus erythematosus, sebopsoriasis, and secondary syphilis. Disseminated blastomycosis presents with cutaneous manifestations such as nodules, papules, or pustules evolving over weeks to months into ulcers with subsequent scarring.17 Hypertrophic lupus erythematosus presents with papules and nodules with associated keratotic scaling on the face, palms, and extensor surfaces of the limbs.18 Sebopsoriasis is characterized by well-defined lesions with an overlying scale distributed on the scalp, face, and chest.19 Secondary syphilis presents as early hyperpigmented macules transitioning to acral papulosquamous lesions involving the palms and soles.20
- Hans-Filho G, Aoki V, Hans Bittner NR, et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-650.
- Jenson KK, Burr DM, Edwards BC. Case report: reatment of refractory pemphigus foliaceus with rituximab. Practical Dermatology. February 2016:33-36. Accessed August 27, 2021. https://practicaldermatology.com/articles/2016-feb/case-report -treatment-of-refractory-pemphigus-foliaceus-with-rituximab -financial-matters-aad-asds-resources
- Amagai M, Hashimoto T, Green KJ, et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895-901.
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-468.
- Oktarina DAM, Sokol E, Kramer D, et al. Endocytosis of IgG, desmoglein 1, and plakoglobin in pemphigus foliaceus patient skin. Front Immunol. 2019;10:1-12.
- Kraft M, Worm M. Pemphigus foliaceus-repeated treatment with rituximab 7 years after initial response: a case report. Front Med. 2018;5:315.
- Hale EK. Pemphigus foliaceous. Dermatol Online J. 2002;8:9.
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106.
- A boobaker J, Morar N, Ramdial PK, et al. Pemphigus in South Africa. Int J Dermatol. 2001;40:115-119.
- Austin E, Millsop JW, Ely H, et al. Psoriasiform pemphigus foliaceus in an African American female: an important clinical manifestation. J Drugs Dermatol. 2018;17:471.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Sabolinski ML, Beutner EH, Krasny S, et al. Substrate specificity of antiepithelial antibodies of pemphigus vulgaris and pemphigus foliaceus sera in immunofluorescence tests on monkey and guinea pig esophagus sections. J Invest Dermatol. 1987;88:545-549.
- Palacios-Álvarez I, Riquelme-McLoughlin C, Curto-Barredo L, et al. Rituximab treatment of pemphigus foliaceus: a retrospective study of 12 patients. J Am Acad Dermatol. 2021;85:484-486.
- Murrell DF, Sprecher E. Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol. 2017;177:1143-1144.
- Gregoriou S, Efthymiou O, Stefanaki C, et al. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521-527.
- Saleh MA. A prospective study comparing patients with early and late relapsing pemphigus treated with rituximab. J Am Acad Dermatol. 2018;79:97-103.
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264.
- Herzum A, Gasparini G, Emanuele C, et al. Atypical and rare forms of cutaneous lupus erythematosus: the importance of the diagnosis for the best management of patients. Dermatology. 2013;1-10.
- Tull TJ, Noy M, Bunker CB, et al. Sebopsoriasis in patients with HIV: a case series of 20 patients. Br J Dermatol. 2016; 173:813-815.
- Balagula Y, Mattei P, Wisco OJ, et al. The great imitator revised: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
The Diagnosis: Pemphigus Foliaceous
Laboratory workup including a complete blood cell count with differential, comprehensive metabolic panel, antinuclear antibodies, Sjögren syndrome A and B antibodies, hepatitis profile, rapid plasma reagin, HIV screen, aldolase, anti–Jo-1, T-Spot TB test (Quest Diagnostics), and tissue cultures was unremarkable. Two 4-mm punch biopsies were obtained from the left cheek and upper back, both of which demonstrated intragranular acantholysis suggestive of pemphigus foliaceous (Figure 1A). A subsequent punch biopsy from the right lower abdomen sent for direct immunofluorescence demonstrated netlike positivity of IgG and C3 in the upper epidermis (Figure 1B), and serum sent for indirect immunofluorescence demonstrated intercellular IgG antibodies to desmoglein (Dsg) 1 on monkey esophagus and positive Dsg-1 antibodies on enzyme-linked immunosorbent assay, confirming the diagnosis.
The patient was started on a 60-mg prednisone taper as well as dapsone 50 mg daily; the dapsone was titrated up to 100 mg daily. After tapering down to 10 mg daily of prednisone over 2 months and continuing dapsone with minimal improvement, he was given 2 infusions of rituximab 1000 mg 2 weeks apart. The scalp plaque was dramatically improved at 3-month follow-up (Figure 2), with partial improvement of the cheek plaques (Figure 3). Dapsone was increased to 150 mg daily, and he was encouraged to use triamcinolone acetonide ointment 0.1% twice daily, which led to further improvement.
Pemphigus foliaceus is an autoimmune blistering disease that most commonly occurs in middle-aged adults. It generally is less common than pemphigus vulgaris, except in Finland, Tunisia, and Brazil, where there is an endemic condition with an identical clinical and histological presentation known as fogo selvagem.1
The pathogenesis of pemphigus foliaceous is characterized by IgG autoantibodies against Dsg-1, a transmembrane glycoprotein involved in the cellular adhesion of keratinocytes, which is preferentially expressed in the superficial epidermis.2-7 Dysfunction of Dsg-1 results in the separation of superficial epidermal cells, resulting in intraepidermal blisters.2,7 In contrast to pemphigus vulgaris, there typically is a lack of oral mucosal involvement due to compensation by Dsg-3 in the mucosa.4 Potential triggers for pemphigus foliaceous include exposure to UV radiation; radiotherapy; pregnancy; physiologic stress; and drugs, most commonly captopril, penicillamine, and thiols.8
Pemphigus foliaceous lesions clinically appear as eroded and crusted lesions on an erythematous base, commonly in a seborrheic distribution on the face, scalp, and trunk with sparing of the oral mucosa,2,6 but lesions can progress to a widespread and more severe exfoliative dermatitis.7 Lesions also can appear as psoriasiform plaques and often are initially misdiagnosed as psoriasis, particularly in patients with skin of color.9,10
Diagnosis of pemphigus foliaceous typically is made using a combination of histology as well as both direct and indirect immunofluorescence. Histologically, pemphigus foliaceus presents with subcorneal acantholysis, which is most prominent in the granular layer and occasionally the presence of neutrophils and eosinophils in the blister cavity.7 Direct immunofluorescence demonstrates netlike intercellular IgG and C3 in the upper portion of the epidermis.11 Indirect immunofluorescence can help detect circulating IgG antibodies to Dsg-1, with guinea pig esophagus being the ideal substrate.11,12
First-line treatment of pemphigus foliaceus consists of systemic glucocorticoid therapy, often administered with azathioprine, methotrexate, or mycophenolate mofetil.2,6,13 Although first-line treatment is effective in 60% to 80% of patients,2 relapsing cases can be treated with cyclophosphamide, intravenous immunoglobulin, immunoadsorption, plasmapheresis, or rituximab.2
Rituximab is a chimeric monoclonal antibody targeting CD20+ B cells, leading to decreased antibody production, which has been shown to be effective in treating severe and refractory cases of pemphigus foliaceus.6,13Rituximab with short-course prednisone has been found to be more effective in achieving complete remission at 24 months than prednisone alone.14 In patients with contraindications to systemic glucocorticoid therapy, rituximab has been shown as an effective first-line therapy.15 One-quarter of patients treated with rituximab relapsed within 2 years of treatment6 (average time to relapse, 6–26 months).16 High-dose rituximab regimens, along with a higher number of rituximab treatment cycles, have been shown to prolong time to relapse.6 Further, higher baseline levels of Dsg-1 antibody have been correlated to earlier relapse and can be used following rituximab therapy to monitor disease progression.6,16
The differential diagnosis for pemphigus foliaceous includes disseminated blastomycosis, hypertrophic lupus erythematosus, sebopsoriasis, and secondary syphilis. Disseminated blastomycosis presents with cutaneous manifestations such as nodules, papules, or pustules evolving over weeks to months into ulcers with subsequent scarring.17 Hypertrophic lupus erythematosus presents with papules and nodules with associated keratotic scaling on the face, palms, and extensor surfaces of the limbs.18 Sebopsoriasis is characterized by well-defined lesions with an overlying scale distributed on the scalp, face, and chest.19 Secondary syphilis presents as early hyperpigmented macules transitioning to acral papulosquamous lesions involving the palms and soles.20
The Diagnosis: Pemphigus Foliaceous
Laboratory workup including a complete blood cell count with differential, comprehensive metabolic panel, antinuclear antibodies, Sjögren syndrome A and B antibodies, hepatitis profile, rapid plasma reagin, HIV screen, aldolase, anti–Jo-1, T-Spot TB test (Quest Diagnostics), and tissue cultures was unremarkable. Two 4-mm punch biopsies were obtained from the left cheek and upper back, both of which demonstrated intragranular acantholysis suggestive of pemphigus foliaceous (Figure 1A). A subsequent punch biopsy from the right lower abdomen sent for direct immunofluorescence demonstrated netlike positivity of IgG and C3 in the upper epidermis (Figure 1B), and serum sent for indirect immunofluorescence demonstrated intercellular IgG antibodies to desmoglein (Dsg) 1 on monkey esophagus and positive Dsg-1 antibodies on enzyme-linked immunosorbent assay, confirming the diagnosis.
The patient was started on a 60-mg prednisone taper as well as dapsone 50 mg daily; the dapsone was titrated up to 100 mg daily. After tapering down to 10 mg daily of prednisone over 2 months and continuing dapsone with minimal improvement, he was given 2 infusions of rituximab 1000 mg 2 weeks apart. The scalp plaque was dramatically improved at 3-month follow-up (Figure 2), with partial improvement of the cheek plaques (Figure 3). Dapsone was increased to 150 mg daily, and he was encouraged to use triamcinolone acetonide ointment 0.1% twice daily, which led to further improvement.
Pemphigus foliaceus is an autoimmune blistering disease that most commonly occurs in middle-aged adults. It generally is less common than pemphigus vulgaris, except in Finland, Tunisia, and Brazil, where there is an endemic condition with an identical clinical and histological presentation known as fogo selvagem.1
The pathogenesis of pemphigus foliaceous is characterized by IgG autoantibodies against Dsg-1, a transmembrane glycoprotein involved in the cellular adhesion of keratinocytes, which is preferentially expressed in the superficial epidermis.2-7 Dysfunction of Dsg-1 results in the separation of superficial epidermal cells, resulting in intraepidermal blisters.2,7 In contrast to pemphigus vulgaris, there typically is a lack of oral mucosal involvement due to compensation by Dsg-3 in the mucosa.4 Potential triggers for pemphigus foliaceous include exposure to UV radiation; radiotherapy; pregnancy; physiologic stress; and drugs, most commonly captopril, penicillamine, and thiols.8
Pemphigus foliaceous lesions clinically appear as eroded and crusted lesions on an erythematous base, commonly in a seborrheic distribution on the face, scalp, and trunk with sparing of the oral mucosa,2,6 but lesions can progress to a widespread and more severe exfoliative dermatitis.7 Lesions also can appear as psoriasiform plaques and often are initially misdiagnosed as psoriasis, particularly in patients with skin of color.9,10
Diagnosis of pemphigus foliaceous typically is made using a combination of histology as well as both direct and indirect immunofluorescence. Histologically, pemphigus foliaceus presents with subcorneal acantholysis, which is most prominent in the granular layer and occasionally the presence of neutrophils and eosinophils in the blister cavity.7 Direct immunofluorescence demonstrates netlike intercellular IgG and C3 in the upper portion of the epidermis.11 Indirect immunofluorescence can help detect circulating IgG antibodies to Dsg-1, with guinea pig esophagus being the ideal substrate.11,12
First-line treatment of pemphigus foliaceus consists of systemic glucocorticoid therapy, often administered with azathioprine, methotrexate, or mycophenolate mofetil.2,6,13 Although first-line treatment is effective in 60% to 80% of patients,2 relapsing cases can be treated with cyclophosphamide, intravenous immunoglobulin, immunoadsorption, plasmapheresis, or rituximab.2
Rituximab is a chimeric monoclonal antibody targeting CD20+ B cells, leading to decreased antibody production, which has been shown to be effective in treating severe and refractory cases of pemphigus foliaceus.6,13Rituximab with short-course prednisone has been found to be more effective in achieving complete remission at 24 months than prednisone alone.14 In patients with contraindications to systemic glucocorticoid therapy, rituximab has been shown as an effective first-line therapy.15 One-quarter of patients treated with rituximab relapsed within 2 years of treatment6 (average time to relapse, 6–26 months).16 High-dose rituximab regimens, along with a higher number of rituximab treatment cycles, have been shown to prolong time to relapse.6 Further, higher baseline levels of Dsg-1 antibody have been correlated to earlier relapse and can be used following rituximab therapy to monitor disease progression.6,16
The differential diagnosis for pemphigus foliaceous includes disseminated blastomycosis, hypertrophic lupus erythematosus, sebopsoriasis, and secondary syphilis. Disseminated blastomycosis presents with cutaneous manifestations such as nodules, papules, or pustules evolving over weeks to months into ulcers with subsequent scarring.17 Hypertrophic lupus erythematosus presents with papules and nodules with associated keratotic scaling on the face, palms, and extensor surfaces of the limbs.18 Sebopsoriasis is characterized by well-defined lesions with an overlying scale distributed on the scalp, face, and chest.19 Secondary syphilis presents as early hyperpigmented macules transitioning to acral papulosquamous lesions involving the palms and soles.20
- Hans-Filho G, Aoki V, Hans Bittner NR, et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-650.
- Jenson KK, Burr DM, Edwards BC. Case report: reatment of refractory pemphigus foliaceus with rituximab. Practical Dermatology. February 2016:33-36. Accessed August 27, 2021. https://practicaldermatology.com/articles/2016-feb/case-report -treatment-of-refractory-pemphigus-foliaceus-with-rituximab -financial-matters-aad-asds-resources
- Amagai M, Hashimoto T, Green KJ, et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895-901.
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-468.
- Oktarina DAM, Sokol E, Kramer D, et al. Endocytosis of IgG, desmoglein 1, and plakoglobin in pemphigus foliaceus patient skin. Front Immunol. 2019;10:1-12.
- Kraft M, Worm M. Pemphigus foliaceus-repeated treatment with rituximab 7 years after initial response: a case report. Front Med. 2018;5:315.
- Hale EK. Pemphigus foliaceous. Dermatol Online J. 2002;8:9.
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106.
- A boobaker J, Morar N, Ramdial PK, et al. Pemphigus in South Africa. Int J Dermatol. 2001;40:115-119.
- Austin E, Millsop JW, Ely H, et al. Psoriasiform pemphigus foliaceus in an African American female: an important clinical manifestation. J Drugs Dermatol. 2018;17:471.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Sabolinski ML, Beutner EH, Krasny S, et al. Substrate specificity of antiepithelial antibodies of pemphigus vulgaris and pemphigus foliaceus sera in immunofluorescence tests on monkey and guinea pig esophagus sections. J Invest Dermatol. 1987;88:545-549.
- Palacios-Álvarez I, Riquelme-McLoughlin C, Curto-Barredo L, et al. Rituximab treatment of pemphigus foliaceus: a retrospective study of 12 patients. J Am Acad Dermatol. 2021;85:484-486.
- Murrell DF, Sprecher E. Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol. 2017;177:1143-1144.
- Gregoriou S, Efthymiou O, Stefanaki C, et al. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521-527.
- Saleh MA. A prospective study comparing patients with early and late relapsing pemphigus treated with rituximab. J Am Acad Dermatol. 2018;79:97-103.
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264.
- Herzum A, Gasparini G, Emanuele C, et al. Atypical and rare forms of cutaneous lupus erythematosus: the importance of the diagnosis for the best management of patients. Dermatology. 2013;1-10.
- Tull TJ, Noy M, Bunker CB, et al. Sebopsoriasis in patients with HIV: a case series of 20 patients. Br J Dermatol. 2016; 173:813-815.
- Balagula Y, Mattei P, Wisco OJ, et al. The great imitator revised: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hans-Filho G, Aoki V, Hans Bittner NR, et al. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-650.
- Jenson KK, Burr DM, Edwards BC. Case report: reatment of refractory pemphigus foliaceus with rituximab. Practical Dermatology. February 2016:33-36. Accessed August 27, 2021. https://practicaldermatology.com/articles/2016-feb/case-report -treatment-of-refractory-pemphigus-foliaceus-with-rituximab -financial-matters-aad-asds-resources
- Amagai M, Hashimoto T, Green KJ, et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895-901.
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-468.
- Oktarina DAM, Sokol E, Kramer D, et al. Endocytosis of IgG, desmoglein 1, and plakoglobin in pemphigus foliaceus patient skin. Front Immunol. 2019;10:1-12.
- Kraft M, Worm M. Pemphigus foliaceus-repeated treatment with rituximab 7 years after initial response: a case report. Front Med. 2018;5:315.
- Hale EK. Pemphigus foliaceous. Dermatol Online J. 2002;8:9.
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106.
- A boobaker J, Morar N, Ramdial PK, et al. Pemphigus in South Africa. Int J Dermatol. 2001;40:115-119.
- Austin E, Millsop JW, Ely H, et al. Psoriasiform pemphigus foliaceus in an African American female: an important clinical manifestation. J Drugs Dermatol. 2018;17:471.
- Arbache ST, Nogueira TG, Delgado L, et al. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-889.
- Sabolinski ML, Beutner EH, Krasny S, et al. Substrate specificity of antiepithelial antibodies of pemphigus vulgaris and pemphigus foliaceus sera in immunofluorescence tests on monkey and guinea pig esophagus sections. J Invest Dermatol. 1987;88:545-549.
- Palacios-Álvarez I, Riquelme-McLoughlin C, Curto-Barredo L, et al. Rituximab treatment of pemphigus foliaceus: a retrospective study of 12 patients. J Am Acad Dermatol. 2021;85:484-486.
- Murrell DF, Sprecher E. Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol. 2017;177:1143-1144.
- Gregoriou S, Efthymiou O, Stefanaki C, et al. Management of pemphigus vulgaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2015;8:521-527.
- Saleh MA. A prospective study comparing patients with early and late relapsing pemphigus treated with rituximab. J Am Acad Dermatol. 2018;79:97-103.
- Castillo CG, Kauffman CA, Miceli MH. Blastomycosis. Infect Dis Clin North Am. 2016;30:247-264.
- Herzum A, Gasparini G, Emanuele C, et al. Atypical and rare forms of cutaneous lupus erythematosus: the importance of the diagnosis for the best management of patients. Dermatology. 2013;1-10.
- Tull TJ, Noy M, Bunker CB, et al. Sebopsoriasis in patients with HIV: a case series of 20 patients. Br J Dermatol. 2016; 173:813-815.
- Balagula Y, Mattei P, Wisco OJ, et al. The great imitator revised: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
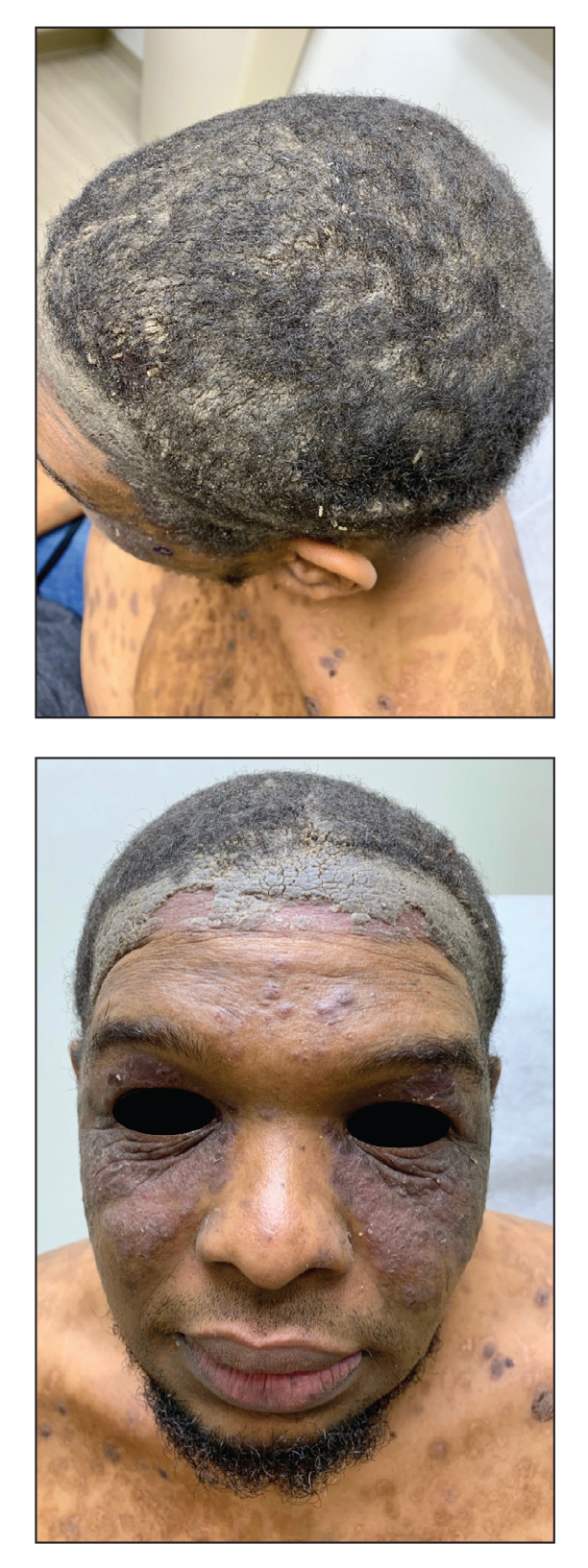
A 40-year-old Black man presented for evaluation of a thick plaque throughout the scalp (top), scaly plaques on the cheeks (bottom), and a spreading rash on the trunk that had progressed over the last few months. He had no relevant medical history, took no medications, and was in a monogamous relationship with a female partner. He previously saw an outside dermatologist who gave him triamcinolone cream, which was mildly helpful. Physical examination revealed a thick verrucous plaque throughout the scalp extending onto the forehead; thick plaques on the cheeks; and numerous, thinly eroded lesions on the trunk. Biopsies and a laboratory workup were performed.
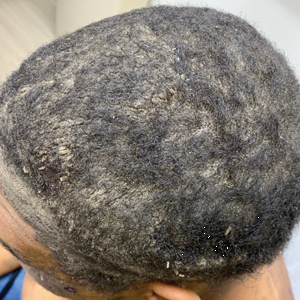
Cutaneous Protothecosis
To the Editor:
Protothecosis infections are caused by an achlorophyllic algae of the species Prototheca. Prototheca organisms are found mostly in soil and water.1 Human infections are rare and involve 2 species, Prototheca wickerhamii and Prototheca zopfii. The former most commonly is responsible for human infections, though P zopfii results in more serious systemic infections with a poor prognosis. There are various types of Prototheca infection presentations, with a 2007 review of 117 cases reporting that cutaneous infections are most common (66%), followed by systemic infections (19%), and olecranon bursitis (15%).2 Skin lesions most commonly occur on the extremities and face, and they present as vesiculobullous and ulcerative lesions with purulent drainage. The skin lesions also may appear as erythematous plaques or nodules, subcutaneous papules, verrucous or herpetiformis lesions, or pyogenic granuloma–like lesions.3 Protothecosis typically affects immunocompromised individuals, especially those with a history of chronic corticosteroid use, malignancy, diabetes mellitus, AIDS, and/or organ transplant.1 We present a case of cutaneous protothecosis on the dorsal distal extremity of a 94-year-old woman. History of exposure to soil while gardening was elicited from the patient, and no immunosuppressive history was present aside from the patient’s age. This case may prompt workup for malignancy or immunosuppression in this patient subset.
A 94-year-old woman with a medical history of cutaneous squamous cell carcinoma (SCC) presented with a growing lesion on the dorsal surface of the left fourth digit of 2 months’ duration. The patient reported the lesion was painful, and she noted preceding trauma to the area that was suspected to have occurred while gardening. Physical examination revealed an ulcerated, hypertrophic, erythematous nodule on the dorsal surface of the left fourth metacarpophalangeal joint. The differential diagnosis included SCC, inflamed cyst, verruca vulgaris, and orf virus due to the clinical presentation. A shave biopsy was performed, and the lesion subsequently was treated with electrodesiccation and curettage.
Histopathologic evaluation revealed pseudoepitheliomatous hyperplasia with a mixed inflammatory infiltrate including lymphocytes and histiocytes. A morula within the dermis was characteristic of a protothecosis infection (Figure 1). On follow-up visit 6 weeks later, the lesion had grown back to its original size and morphology (Figure 2). At this time, the lesion was again treated with shave removal, followed by electrodesiccation and curettage, and the patient was placed on oral fluconazole 200 mg daily for 1 month. When the lesion did not resolve with fluconazole, she was referred to infectious disease as well as general surgery for surgical removal and debridement of the lesion. Unfortunately, the patient was lost to follow-up.
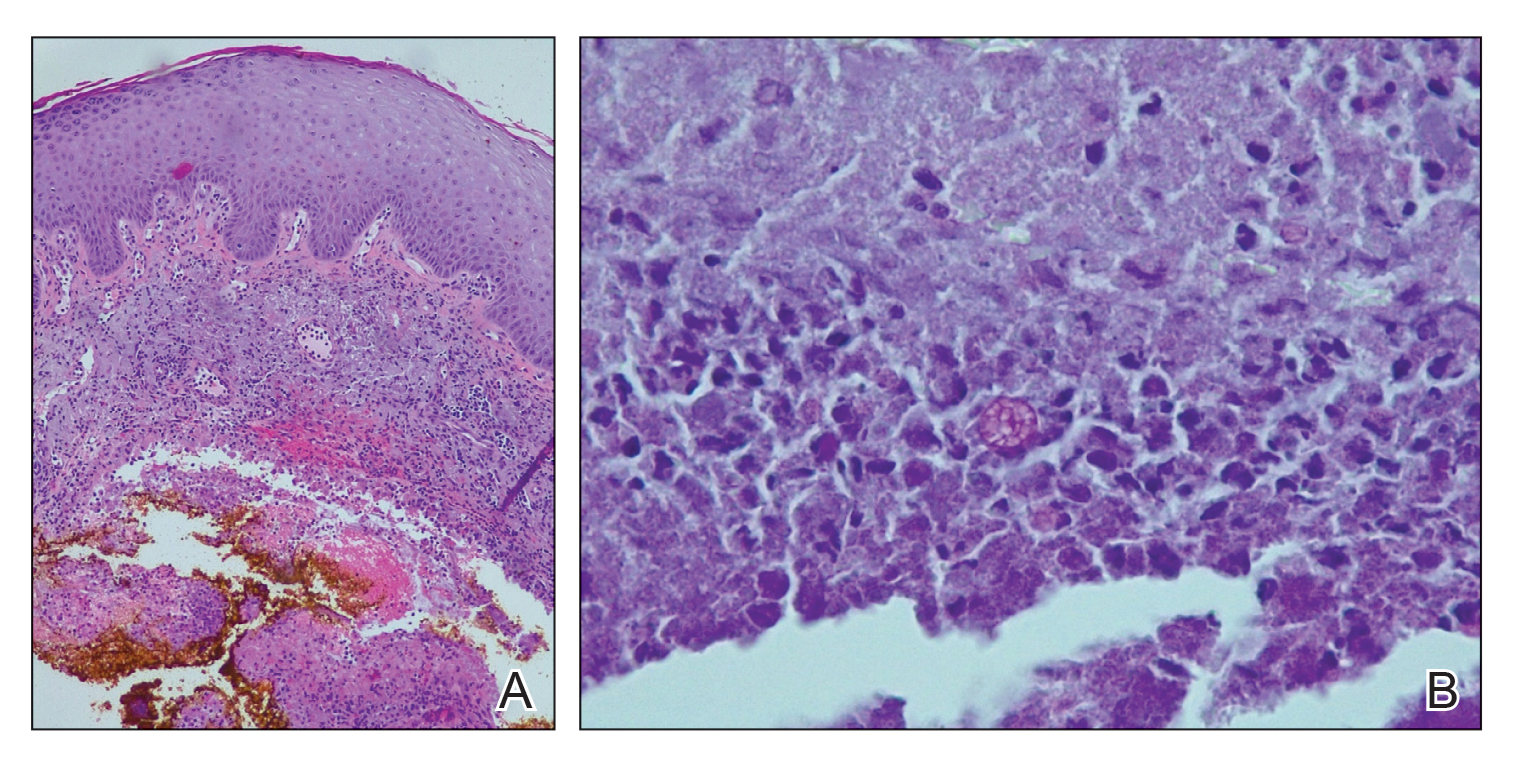
Protothecosis is an infectious disease comprised of achlorophyllic algae found in soil and water that rarely affects humans. When it does affect humans, cutaneous infections are most common. All human cases in which organisms were identified to species level have been caused by P wickerhamii or P zopfii species.2 Inoculation is suspected to occur through trauma to affected skin, especially when in the context of contaminated water. Our patient reported history of trauma to the hand, with soil from gardening as the potential aquagenic source of the infection.

The clinical presentation of protothecosis ranges from localized cutaneous to disseminated systemic infections, with most reported cases of systemic disease occurring in immunocompromised individuals. The cutaneous lesions of protothecosis vary greatly in clinical appearance including ulcerative nodules (as in our case), papules, plaques, pustules, and vesicles with erosion or crusting.4
Cutaneous protothecosis has the potential to mimic many other skin diseases and lesions, and, given its rarity, it may not be on the radar of dermatologists. Our patient’s lesion was presumed to be a skin cancer and was treated as such because of the history of SCC and clinical presentation. Although excision of individual lesions of protothecosis can be curative, electrodesiccation and curettage does not appear to be an adequate treatment, as the lesion subsequently recurred. It also is possible that this case represents P zopfii infection, as it did not respond to treatment with oral fluconazole, though in vitro studies with fluconazole to both P zopfii and P wickerhamii had variable treatment success.2 Also, the histopathologic findings were most consistent with P wickerhamii, revealing small, round, symmetrical morula, compared to P zopfii, which typically will display oval or cylindrical, asymmetrical, random internal segmentation.5 This case may warrant determination of species, which can be accomplished by a culture on Sabouraud dextrose agar, carbohydrate and alcohol assimilation test, yeast biochemical card, serological typing by immunoblotting, immunofluorescence study using species-specific antibodies, or amplification by polymerase chain reaction for small subunit ribosomal DNA sequences.2,6-8
The natural history of isolated skin disease is an indolent progressive course; however, reports do exist noting spontaneous resolution.4,9 Treatment options for Prototheca infections can be disappointing and consist of both surgical and medical management, or a combination of the 2 approaches. Reports in the literature support the use of antifungals including ketoconazole, voriconazole, itraconazole, fluconazole, and amphotericin B, with the latter displaying the best activity against Prototheca species.2 Tetracycline has been used in combination with oral or topical amphotericin B and was found to be synergistic in vitro and in case reports at successfully treating cutaneous protothecosis infections. It is possible that our patient was not treated with fluconazole long enough for it to become therapeutic, as most reported treatment regimens are weeks to months in length. Conversely, it may have been of benefit to transition the patient to topical amphotericin B and tetracycline, as fluconazole failed in this patient. However, treatment successes and failures are limited to case reports/case series and in vitro studies, with prospective studies lacking. Due to the variability with in vitro susceptibility profiles for Prototheca species, it generally is not recommended to pursue in vitro susceptibility testing in the management of Prototheca skin infections due to the inconsistency demonstrated between in vitro activity and clinical response to therapy.2
- Silva PC, Costa e Silva SB, Lima RB, et al. Cutaneous protothecosis—case report. An Bras Dermatol. 2013;88:183-185.
- Lass-Flörl C, Mayr A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Seok JY, Lee Y, Lee H, et al. Human cutaneous protothecosis: report of a case and literature review. Korean J Pathol. 2013;47:575-578.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Walsh SV, Johnson RA, Tahan SR. Protothecosis: an unusual cause of chronic subcutaneous and soft tissue infection. Am J Dermatopathol. 1998;20:379-382.
- Casal MJ, Gutierrez J. Simple new test for rapid differentiation of Prototheca wickerhamii from Prototheca zopfii. J Clin Microbiol. 1983;18:992-993.
- Arnold, P, Ahearn, DG. The systematics of the genus Prototheca with a description of a new species P. filamenta. Mycologia 1972;64:265-275.
- Roesler U, Scholz H, Hensel H. Emended phenotypic characterization of Prototheca zopfii: a proposal for three biotypes and standards for their identification. Int J Syst Evol Microbiol. 2003;53:1195-1199.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
To the Editor:
Protothecosis infections are caused by an achlorophyllic algae of the species Prototheca. Prototheca organisms are found mostly in soil and water.1 Human infections are rare and involve 2 species, Prototheca wickerhamii and Prototheca zopfii. The former most commonly is responsible for human infections, though P zopfii results in more serious systemic infections with a poor prognosis. There are various types of Prototheca infection presentations, with a 2007 review of 117 cases reporting that cutaneous infections are most common (66%), followed by systemic infections (19%), and olecranon bursitis (15%).2 Skin lesions most commonly occur on the extremities and face, and they present as vesiculobullous and ulcerative lesions with purulent drainage. The skin lesions also may appear as erythematous plaques or nodules, subcutaneous papules, verrucous or herpetiformis lesions, or pyogenic granuloma–like lesions.3 Protothecosis typically affects immunocompromised individuals, especially those with a history of chronic corticosteroid use, malignancy, diabetes mellitus, AIDS, and/or organ transplant.1 We present a case of cutaneous protothecosis on the dorsal distal extremity of a 94-year-old woman. History of exposure to soil while gardening was elicited from the patient, and no immunosuppressive history was present aside from the patient’s age. This case may prompt workup for malignancy or immunosuppression in this patient subset.
A 94-year-old woman with a medical history of cutaneous squamous cell carcinoma (SCC) presented with a growing lesion on the dorsal surface of the left fourth digit of 2 months’ duration. The patient reported the lesion was painful, and she noted preceding trauma to the area that was suspected to have occurred while gardening. Physical examination revealed an ulcerated, hypertrophic, erythematous nodule on the dorsal surface of the left fourth metacarpophalangeal joint. The differential diagnosis included SCC, inflamed cyst, verruca vulgaris, and orf virus due to the clinical presentation. A shave biopsy was performed, and the lesion subsequently was treated with electrodesiccation and curettage.
Histopathologic evaluation revealed pseudoepitheliomatous hyperplasia with a mixed inflammatory infiltrate including lymphocytes and histiocytes. A morula within the dermis was characteristic of a protothecosis infection (Figure 1). On follow-up visit 6 weeks later, the lesion had grown back to its original size and morphology (Figure 2). At this time, the lesion was again treated with shave removal, followed by electrodesiccation and curettage, and the patient was placed on oral fluconazole 200 mg daily for 1 month. When the lesion did not resolve with fluconazole, she was referred to infectious disease as well as general surgery for surgical removal and debridement of the lesion. Unfortunately, the patient was lost to follow-up.
Protothecosis is an infectious disease comprised of achlorophyllic algae found in soil and water that rarely affects humans. When it does affect humans, cutaneous infections are most common. All human cases in which organisms were identified to species level have been caused by P wickerhamii or P zopfii species.2 Inoculation is suspected to occur through trauma to affected skin, especially when in the context of contaminated water. Our patient reported history of trauma to the hand, with soil from gardening as the potential aquagenic source of the infection.
The clinical presentation of protothecosis ranges from localized cutaneous to disseminated systemic infections, with most reported cases of systemic disease occurring in immunocompromised individuals. The cutaneous lesions of protothecosis vary greatly in clinical appearance including ulcerative nodules (as in our case), papules, plaques, pustules, and vesicles with erosion or crusting.4
Cutaneous protothecosis has the potential to mimic many other skin diseases and lesions, and, given its rarity, it may not be on the radar of dermatologists. Our patient’s lesion was presumed to be a skin cancer and was treated as such because of the history of SCC and clinical presentation. Although excision of individual lesions of protothecosis can be curative, electrodesiccation and curettage does not appear to be an adequate treatment, as the lesion subsequently recurred. It also is possible that this case represents P zopfii infection, as it did not respond to treatment with oral fluconazole, though in vitro studies with fluconazole to both P zopfii and P wickerhamii had variable treatment success.2 Also, the histopathologic findings were most consistent with P wickerhamii, revealing small, round, symmetrical morula, compared to P zopfii, which typically will display oval or cylindrical, asymmetrical, random internal segmentation.5 This case may warrant determination of species, which can be accomplished by a culture on Sabouraud dextrose agar, carbohydrate and alcohol assimilation test, yeast biochemical card, serological typing by immunoblotting, immunofluorescence study using species-specific antibodies, or amplification by polymerase chain reaction for small subunit ribosomal DNA sequences.2,6-8
The natural history of isolated skin disease is an indolent progressive course; however, reports do exist noting spontaneous resolution.4,9 Treatment options for Prototheca infections can be disappointing and consist of both surgical and medical management, or a combination of the 2 approaches. Reports in the literature support the use of antifungals including ketoconazole, voriconazole, itraconazole, fluconazole, and amphotericin B, with the latter displaying the best activity against Prototheca species.2 Tetracycline has been used in combination with oral or topical amphotericin B and was found to be synergistic in vitro and in case reports at successfully treating cutaneous protothecosis infections. It is possible that our patient was not treated with fluconazole long enough for it to become therapeutic, as most reported treatment regimens are weeks to months in length. Conversely, it may have been of benefit to transition the patient to topical amphotericin B and tetracycline, as fluconazole failed in this patient. However, treatment successes and failures are limited to case reports/case series and in vitro studies, with prospective studies lacking. Due to the variability with in vitro susceptibility profiles for Prototheca species, it generally is not recommended to pursue in vitro susceptibility testing in the management of Prototheca skin infections due to the inconsistency demonstrated between in vitro activity and clinical response to therapy.2
To the Editor:
Protothecosis infections are caused by an achlorophyllic algae of the species Prototheca. Prototheca organisms are found mostly in soil and water.1 Human infections are rare and involve 2 species, Prototheca wickerhamii and Prototheca zopfii. The former most commonly is responsible for human infections, though P zopfii results in more serious systemic infections with a poor prognosis. There are various types of Prototheca infection presentations, with a 2007 review of 117 cases reporting that cutaneous infections are most common (66%), followed by systemic infections (19%), and olecranon bursitis (15%).2 Skin lesions most commonly occur on the extremities and face, and they present as vesiculobullous and ulcerative lesions with purulent drainage. The skin lesions also may appear as erythematous plaques or nodules, subcutaneous papules, verrucous or herpetiformis lesions, or pyogenic granuloma–like lesions.3 Protothecosis typically affects immunocompromised individuals, especially those with a history of chronic corticosteroid use, malignancy, diabetes mellitus, AIDS, and/or organ transplant.1 We present a case of cutaneous protothecosis on the dorsal distal extremity of a 94-year-old woman. History of exposure to soil while gardening was elicited from the patient, and no immunosuppressive history was present aside from the patient’s age. This case may prompt workup for malignancy or immunosuppression in this patient subset.
A 94-year-old woman with a medical history of cutaneous squamous cell carcinoma (SCC) presented with a growing lesion on the dorsal surface of the left fourth digit of 2 months’ duration. The patient reported the lesion was painful, and she noted preceding trauma to the area that was suspected to have occurred while gardening. Physical examination revealed an ulcerated, hypertrophic, erythematous nodule on the dorsal surface of the left fourth metacarpophalangeal joint. The differential diagnosis included SCC, inflamed cyst, verruca vulgaris, and orf virus due to the clinical presentation. A shave biopsy was performed, and the lesion subsequently was treated with electrodesiccation and curettage.
Histopathologic evaluation revealed pseudoepitheliomatous hyperplasia with a mixed inflammatory infiltrate including lymphocytes and histiocytes. A morula within the dermis was characteristic of a protothecosis infection (Figure 1). On follow-up visit 6 weeks later, the lesion had grown back to its original size and morphology (Figure 2). At this time, the lesion was again treated with shave removal, followed by electrodesiccation and curettage, and the patient was placed on oral fluconazole 200 mg daily for 1 month. When the lesion did not resolve with fluconazole, she was referred to infectious disease as well as general surgery for surgical removal and debridement of the lesion. Unfortunately, the patient was lost to follow-up.
Protothecosis is an infectious disease comprised of achlorophyllic algae found in soil and water that rarely affects humans. When it does affect humans, cutaneous infections are most common. All human cases in which organisms were identified to species level have been caused by P wickerhamii or P zopfii species.2 Inoculation is suspected to occur through trauma to affected skin, especially when in the context of contaminated water. Our patient reported history of trauma to the hand, with soil from gardening as the potential aquagenic source of the infection.
The clinical presentation of protothecosis ranges from localized cutaneous to disseminated systemic infections, with most reported cases of systemic disease occurring in immunocompromised individuals. The cutaneous lesions of protothecosis vary greatly in clinical appearance including ulcerative nodules (as in our case), papules, plaques, pustules, and vesicles with erosion or crusting.4
Cutaneous protothecosis has the potential to mimic many other skin diseases and lesions, and, given its rarity, it may not be on the radar of dermatologists. Our patient’s lesion was presumed to be a skin cancer and was treated as such because of the history of SCC and clinical presentation. Although excision of individual lesions of protothecosis can be curative, electrodesiccation and curettage does not appear to be an adequate treatment, as the lesion subsequently recurred. It also is possible that this case represents P zopfii infection, as it did not respond to treatment with oral fluconazole, though in vitro studies with fluconazole to both P zopfii and P wickerhamii had variable treatment success.2 Also, the histopathologic findings were most consistent with P wickerhamii, revealing small, round, symmetrical morula, compared to P zopfii, which typically will display oval or cylindrical, asymmetrical, random internal segmentation.5 This case may warrant determination of species, which can be accomplished by a culture on Sabouraud dextrose agar, carbohydrate and alcohol assimilation test, yeast biochemical card, serological typing by immunoblotting, immunofluorescence study using species-specific antibodies, or amplification by polymerase chain reaction for small subunit ribosomal DNA sequences.2,6-8
The natural history of isolated skin disease is an indolent progressive course; however, reports do exist noting spontaneous resolution.4,9 Treatment options for Prototheca infections can be disappointing and consist of both surgical and medical management, or a combination of the 2 approaches. Reports in the literature support the use of antifungals including ketoconazole, voriconazole, itraconazole, fluconazole, and amphotericin B, with the latter displaying the best activity against Prototheca species.2 Tetracycline has been used in combination with oral or topical amphotericin B and was found to be synergistic in vitro and in case reports at successfully treating cutaneous protothecosis infections. It is possible that our patient was not treated with fluconazole long enough for it to become therapeutic, as most reported treatment regimens are weeks to months in length. Conversely, it may have been of benefit to transition the patient to topical amphotericin B and tetracycline, as fluconazole failed in this patient. However, treatment successes and failures are limited to case reports/case series and in vitro studies, with prospective studies lacking. Due to the variability with in vitro susceptibility profiles for Prototheca species, it generally is not recommended to pursue in vitro susceptibility testing in the management of Prototheca skin infections due to the inconsistency demonstrated between in vitro activity and clinical response to therapy.2
- Silva PC, Costa e Silva SB, Lima RB, et al. Cutaneous protothecosis—case report. An Bras Dermatol. 2013;88:183-185.
- Lass-Flörl C, Mayr A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Seok JY, Lee Y, Lee H, et al. Human cutaneous protothecosis: report of a case and literature review. Korean J Pathol. 2013;47:575-578.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Walsh SV, Johnson RA, Tahan SR. Protothecosis: an unusual cause of chronic subcutaneous and soft tissue infection. Am J Dermatopathol. 1998;20:379-382.
- Casal MJ, Gutierrez J. Simple new test for rapid differentiation of Prototheca wickerhamii from Prototheca zopfii. J Clin Microbiol. 1983;18:992-993.
- Arnold, P, Ahearn, DG. The systematics of the genus Prototheca with a description of a new species P. filamenta. Mycologia 1972;64:265-275.
- Roesler U, Scholz H, Hensel H. Emended phenotypic characterization of Prototheca zopfii: a proposal for three biotypes and standards for their identification. Int J Syst Evol Microbiol. 2003;53:1195-1199.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Silva PC, Costa e Silva SB, Lima RB, et al. Cutaneous protothecosis—case report. An Bras Dermatol. 2013;88:183-185.
- Lass-Flörl C, Mayr A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Seok JY, Lee Y, Lee H, et al. Human cutaneous protothecosis: report of a case and literature review. Korean J Pathol. 2013;47:575-578.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Walsh SV, Johnson RA, Tahan SR. Protothecosis: an unusual cause of chronic subcutaneous and soft tissue infection. Am J Dermatopathol. 1998;20:379-382.
- Casal MJ, Gutierrez J. Simple new test for rapid differentiation of Prototheca wickerhamii from Prototheca zopfii. J Clin Microbiol. 1983;18:992-993.
- Arnold, P, Ahearn, DG. The systematics of the genus Prototheca with a description of a new species P. filamenta. Mycologia 1972;64:265-275.
- Roesler U, Scholz H, Hensel H. Emended phenotypic characterization of Prototheca zopfii: a proposal for three biotypes and standards for their identification. Int J Syst Evol Microbiol. 2003;53:1195-1199.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
Practice Points
- Cutaneous protothecosis is a rare skin infection most commonly reported in immunocompromised individuals with recent exposure to contaminated soil or water. Cutaneous protothecosis has the potential to mimic many other skin diseases and lesions, including eczema; nonmelanoma skin cancer; or bacterial, viral, and fungal skin infections.
- A skin biopsy is essential for diagnosis, and histopathology is characteristic with soccer ball–appearing morula noted in a mixed inflammatory infiltrate.
Verrucous Carcinoma in a Wounded Military Amputee
To the Editor:
Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma first described by Ackerman in 1948.1 There are 4 main clinicopathologic types: oral florid papillomatosis or Ackerman tumor, giant condyloma acuminatum or Buschke-Lowenstein tumor, plantar verrucous carcinoma, and cutaneous verrucous carcinoma.2,3 Historically, most patients are older white men. The lesion commonly occurs in sites of inflammation4 or chronic irritation/trauma. Clinically, patients present with a slowly enlarging, exophytic, verrucous plaque violating the skin, fascia, and occasionally bone. Although these lesions have little tendency to metastasize, substantial morbidity can be seen due to local invasion. Despite surgical excision, recurrence is not uncommon and is associated with a poor prognosis and higher infiltrative potential.5
A 45-year-old male veteran initially presented to our dermatology clinic with a 4-cm, macerated, verrucous plaque on the left lateral ankle in the area of a skin graft placed during a prior limb salvage surgery (Figure 1). The patient experienced a traumatic blast injury while deployed 7 years prior with a subsequent right-sided below-the-knee amputation and left lower limb salvage. The lesion was clinically diagnosed as verruca vulgaris and treated with daily salicylic acid. Six weeks after the initial presentation, the lesion remained largely unchanged. A biopsy subsequently was obtained to confirm the diagnosis. At that time, the histopathology was consistent with verruca vulgaris without evidence of carcinoma. Due to the persistence of the lesion, lack of improvement with topical treatment, and overall size, the patient opted for surgical excision.
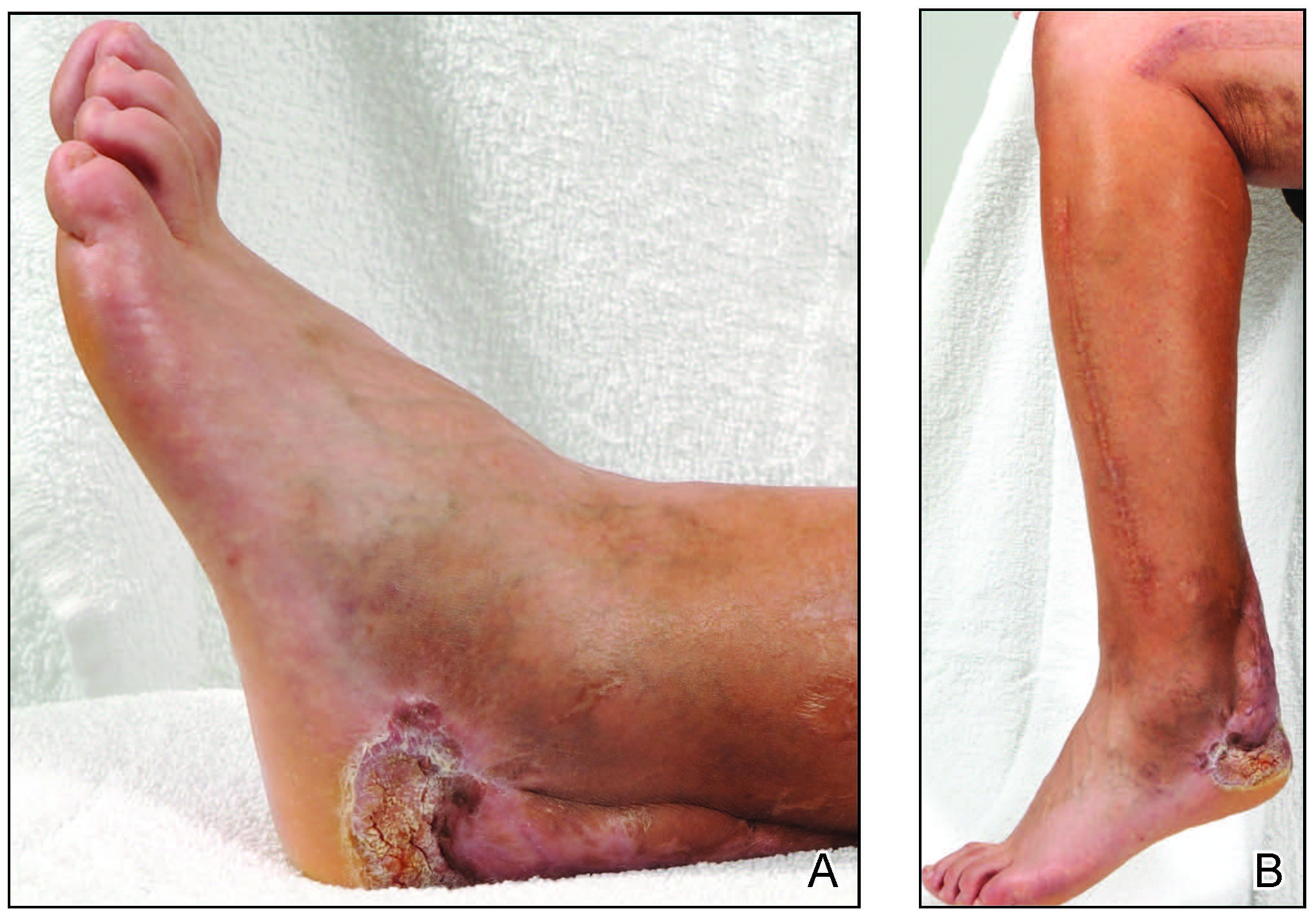
A year later, the lesion was excised again by orthopedic surgery, and the tissue was submitted for histopathologic evaluation, which was suggestive of a verrucous neoplasm with some disagreement on whether it was consistent with verrucous hyperplasia or verrucous carcinoma. Following excision, the patient sustained a nonhealing chronic ulcer that required wound care for a total of 6 months. The lesion recurred a year later and was surgically excised a third time. A split-thickness skin graft was utilized to repair the defect. Histopathology again was consistent with verrucous carcinoma. With a fourth and final recurrence of the verrucous plaque 6 months later, the patient elected to undergo a left-sided below-the-knee amputation.
Verrucous carcinoma can represent a diagnostic dilemma, as histologic sections may mimic benign entities. The features of a well-differentiated squamous epithelium with hyperkeratosis, papillomatosis, and acanthosis can be mistaken for verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia,6 which are characteristic of verrucous hyperplasia. Accurate diagnosis can be difficult with a superficial biopsy because of the mature appearance of the epithelium,7 prompting the need for multiple and deeper biopsies8 to include sampling of the base of the hyperplastic epithelium in which the characteristic bulbous pushing growth pattern of the rete ridges is visualized. Precise histologic diagnosis can be further confounded by external mechanical factors, such as pressure, which can distort the classic histopathology.7 The histopathologic features leading to the diagnosis of verrucous carcinoma in our specimen were minimal squamous atypia present in a predominantly exophytic squamous proliferation with human papillomavirus cytopathic effect and focal endophytic pushing borders by rounded bulbous rete ridges into the mid and deep dermis (Figure 2).

Diagnostic uncertainty can delay surgical excision and lead to progression of verrucous carcinoma. Unfortunately, even with appropriate surgical intervention, recurrence has been documented; therefore, close clinical follow-up is recommended. The tumor spreads by local invasion and may follow the path of least resistance.4 In our patient, the frequent tissue manipulation may have facilitated aggressive infiltration of the tumor, ultimately resulting in the loss of his remaining leg. Therefore, it is important for clinicians to recognize that verrucous carcinoma, especially one that develops on a refractory ulcer or scar tissue, may be a complex malignant neoplasm that requires extensive treatment at onset to prevent the amputation of a limb.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Yoshitasu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Schwartz R. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-14.
- Bernstein SC, Lim KK, Brodland DG, et al. The many faces of squamous cell carcinoma. Dermatol Surg. 1996;22:243-254.
- Costache M, Tatiana D, Mitrache L, et al. Cutaneous verrucous carcinoma—report of three cases with review of literature. Rom J Morphol Embryol. 2014;55:383-388.
- Shenoy A, Waghmare R, Kavishwar V, et al. Carcinoma cuniculatum of foot. Foot. 2011;21:207-208.
- Klima M, Kurtis B, Jordan P. Verrucous carcinoma of skin. J Cutan Pathol.1980;7:88-98.
- Pleat J, Sacks L, Rigby H. Cutaneous verrucous carcinoma. Br J Plast Surg. 2001;54:554-555.
To the Editor:
Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma first described by Ackerman in 1948.1 There are 4 main clinicopathologic types: oral florid papillomatosis or Ackerman tumor, giant condyloma acuminatum or Buschke-Lowenstein tumor, plantar verrucous carcinoma, and cutaneous verrucous carcinoma.2,3 Historically, most patients are older white men. The lesion commonly occurs in sites of inflammation4 or chronic irritation/trauma. Clinically, patients present with a slowly enlarging, exophytic, verrucous plaque violating the skin, fascia, and occasionally bone. Although these lesions have little tendency to metastasize, substantial morbidity can be seen due to local invasion. Despite surgical excision, recurrence is not uncommon and is associated with a poor prognosis and higher infiltrative potential.5
A 45-year-old male veteran initially presented to our dermatology clinic with a 4-cm, macerated, verrucous plaque on the left lateral ankle in the area of a skin graft placed during a prior limb salvage surgery (Figure 1). The patient experienced a traumatic blast injury while deployed 7 years prior with a subsequent right-sided below-the-knee amputation and left lower limb salvage. The lesion was clinically diagnosed as verruca vulgaris and treated with daily salicylic acid. Six weeks after the initial presentation, the lesion remained largely unchanged. A biopsy subsequently was obtained to confirm the diagnosis. At that time, the histopathology was consistent with verruca vulgaris without evidence of carcinoma. Due to the persistence of the lesion, lack of improvement with topical treatment, and overall size, the patient opted for surgical excision.
A year later, the lesion was excised again by orthopedic surgery, and the tissue was submitted for histopathologic evaluation, which was suggestive of a verrucous neoplasm with some disagreement on whether it was consistent with verrucous hyperplasia or verrucous carcinoma. Following excision, the patient sustained a nonhealing chronic ulcer that required wound care for a total of 6 months. The lesion recurred a year later and was surgically excised a third time. A split-thickness skin graft was utilized to repair the defect. Histopathology again was consistent with verrucous carcinoma. With a fourth and final recurrence of the verrucous plaque 6 months later, the patient elected to undergo a left-sided below-the-knee amputation.
Verrucous carcinoma can represent a diagnostic dilemma, as histologic sections may mimic benign entities. The features of a well-differentiated squamous epithelium with hyperkeratosis, papillomatosis, and acanthosis can be mistaken for verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia,6 which are characteristic of verrucous hyperplasia. Accurate diagnosis can be difficult with a superficial biopsy because of the mature appearance of the epithelium,7 prompting the need for multiple and deeper biopsies8 to include sampling of the base of the hyperplastic epithelium in which the characteristic bulbous pushing growth pattern of the rete ridges is visualized. Precise histologic diagnosis can be further confounded by external mechanical factors, such as pressure, which can distort the classic histopathology.7 The histopathologic features leading to the diagnosis of verrucous carcinoma in our specimen were minimal squamous atypia present in a predominantly exophytic squamous proliferation with human papillomavirus cytopathic effect and focal endophytic pushing borders by rounded bulbous rete ridges into the mid and deep dermis (Figure 2).
Diagnostic uncertainty can delay surgical excision and lead to progression of verrucous carcinoma. Unfortunately, even with appropriate surgical intervention, recurrence has been documented; therefore, close clinical follow-up is recommended. The tumor spreads by local invasion and may follow the path of least resistance.4 In our patient, the frequent tissue manipulation may have facilitated aggressive infiltration of the tumor, ultimately resulting in the loss of his remaining leg. Therefore, it is important for clinicians to recognize that verrucous carcinoma, especially one that develops on a refractory ulcer or scar tissue, may be a complex malignant neoplasm that requires extensive treatment at onset to prevent the amputation of a limb.
To the Editor:
Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma first described by Ackerman in 1948.1 There are 4 main clinicopathologic types: oral florid papillomatosis or Ackerman tumor, giant condyloma acuminatum or Buschke-Lowenstein tumor, plantar verrucous carcinoma, and cutaneous verrucous carcinoma.2,3 Historically, most patients are older white men. The lesion commonly occurs in sites of inflammation4 or chronic irritation/trauma. Clinically, patients present with a slowly enlarging, exophytic, verrucous plaque violating the skin, fascia, and occasionally bone. Although these lesions have little tendency to metastasize, substantial morbidity can be seen due to local invasion. Despite surgical excision, recurrence is not uncommon and is associated with a poor prognosis and higher infiltrative potential.5
A 45-year-old male veteran initially presented to our dermatology clinic with a 4-cm, macerated, verrucous plaque on the left lateral ankle in the area of a skin graft placed during a prior limb salvage surgery (Figure 1). The patient experienced a traumatic blast injury while deployed 7 years prior with a subsequent right-sided below-the-knee amputation and left lower limb salvage. The lesion was clinically diagnosed as verruca vulgaris and treated with daily salicylic acid. Six weeks after the initial presentation, the lesion remained largely unchanged. A biopsy subsequently was obtained to confirm the diagnosis. At that time, the histopathology was consistent with verruca vulgaris without evidence of carcinoma. Due to the persistence of the lesion, lack of improvement with topical treatment, and overall size, the patient opted for surgical excision.
A year later, the lesion was excised again by orthopedic surgery, and the tissue was submitted for histopathologic evaluation, which was suggestive of a verrucous neoplasm with some disagreement on whether it was consistent with verrucous hyperplasia or verrucous carcinoma. Following excision, the patient sustained a nonhealing chronic ulcer that required wound care for a total of 6 months. The lesion recurred a year later and was surgically excised a third time. A split-thickness skin graft was utilized to repair the defect. Histopathology again was consistent with verrucous carcinoma. With a fourth and final recurrence of the verrucous plaque 6 months later, the patient elected to undergo a left-sided below-the-knee amputation.
Verrucous carcinoma can represent a diagnostic dilemma, as histologic sections may mimic benign entities. The features of a well-differentiated squamous epithelium with hyperkeratosis, papillomatosis, and acanthosis can be mistaken for verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia,6 which are characteristic of verrucous hyperplasia. Accurate diagnosis can be difficult with a superficial biopsy because of the mature appearance of the epithelium,7 prompting the need for multiple and deeper biopsies8 to include sampling of the base of the hyperplastic epithelium in which the characteristic bulbous pushing growth pattern of the rete ridges is visualized. Precise histologic diagnosis can be further confounded by external mechanical factors, such as pressure, which can distort the classic histopathology.7 The histopathologic features leading to the diagnosis of verrucous carcinoma in our specimen were minimal squamous atypia present in a predominantly exophytic squamous proliferation with human papillomavirus cytopathic effect and focal endophytic pushing borders by rounded bulbous rete ridges into the mid and deep dermis (Figure 2).
Diagnostic uncertainty can delay surgical excision and lead to progression of verrucous carcinoma. Unfortunately, even with appropriate surgical intervention, recurrence has been documented; therefore, close clinical follow-up is recommended. The tumor spreads by local invasion and may follow the path of least resistance.4 In our patient, the frequent tissue manipulation may have facilitated aggressive infiltration of the tumor, ultimately resulting in the loss of his remaining leg. Therefore, it is important for clinicians to recognize that verrucous carcinoma, especially one that develops on a refractory ulcer or scar tissue, may be a complex malignant neoplasm that requires extensive treatment at onset to prevent the amputation of a limb.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Yoshitasu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Schwartz R. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-14.
- Bernstein SC, Lim KK, Brodland DG, et al. The many faces of squamous cell carcinoma. Dermatol Surg. 1996;22:243-254.
- Costache M, Tatiana D, Mitrache L, et al. Cutaneous verrucous carcinoma—report of three cases with review of literature. Rom J Morphol Embryol. 2014;55:383-388.
- Shenoy A, Waghmare R, Kavishwar V, et al. Carcinoma cuniculatum of foot. Foot. 2011;21:207-208.
- Klima M, Kurtis B, Jordan P. Verrucous carcinoma of skin. J Cutan Pathol.1980;7:88-98.
- Pleat J, Sacks L, Rigby H. Cutaneous verrucous carcinoma. Br J Plast Surg. 2001;54:554-555.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Yoshitasu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Schwartz R. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-14.
- Bernstein SC, Lim KK, Brodland DG, et al. The many faces of squamous cell carcinoma. Dermatol Surg. 1996;22:243-254.
- Costache M, Tatiana D, Mitrache L, et al. Cutaneous verrucous carcinoma—report of three cases with review of literature. Rom J Morphol Embryol. 2014;55:383-388.
- Shenoy A, Waghmare R, Kavishwar V, et al. Carcinoma cuniculatum of foot. Foot. 2011;21:207-208.
- Klima M, Kurtis B, Jordan P. Verrucous carcinoma of skin. J Cutan Pathol.1980;7:88-98.
- Pleat J, Sacks L, Rigby H. Cutaneous verrucous carcinoma. Br J Plast Surg. 2001;54:554-555.
Practice Points
- Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma that commonly occurs in sites of inflammation or chronic irritation.
- Histologically, verrucous carcinoma can be mistaken for other entities including verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia, often delaying the appropriate diagnosis and treatment.
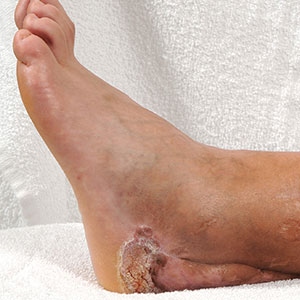
Bullous Retiform Purpura on the Ears and Legs
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
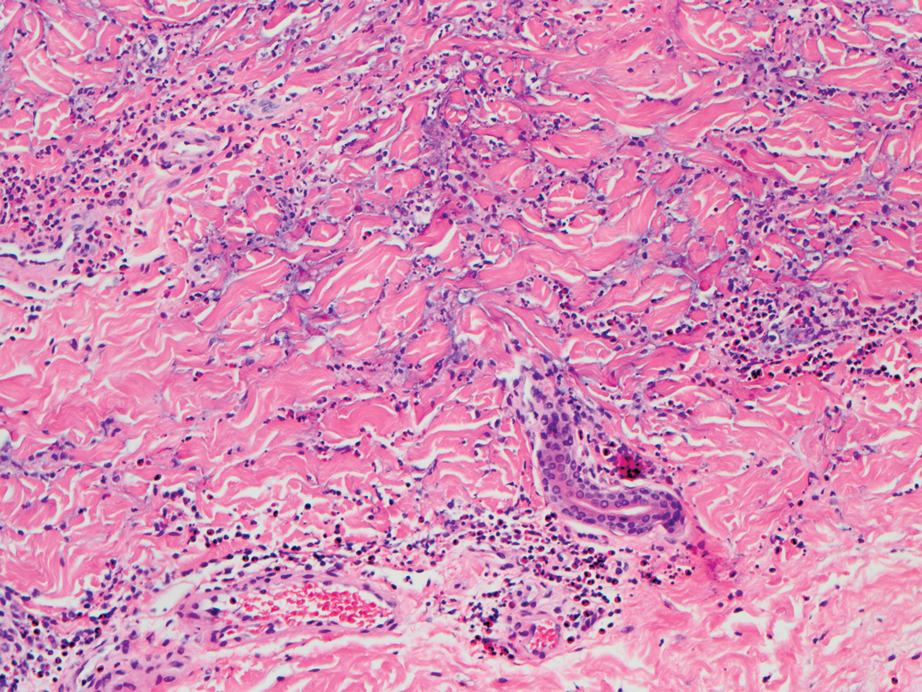
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
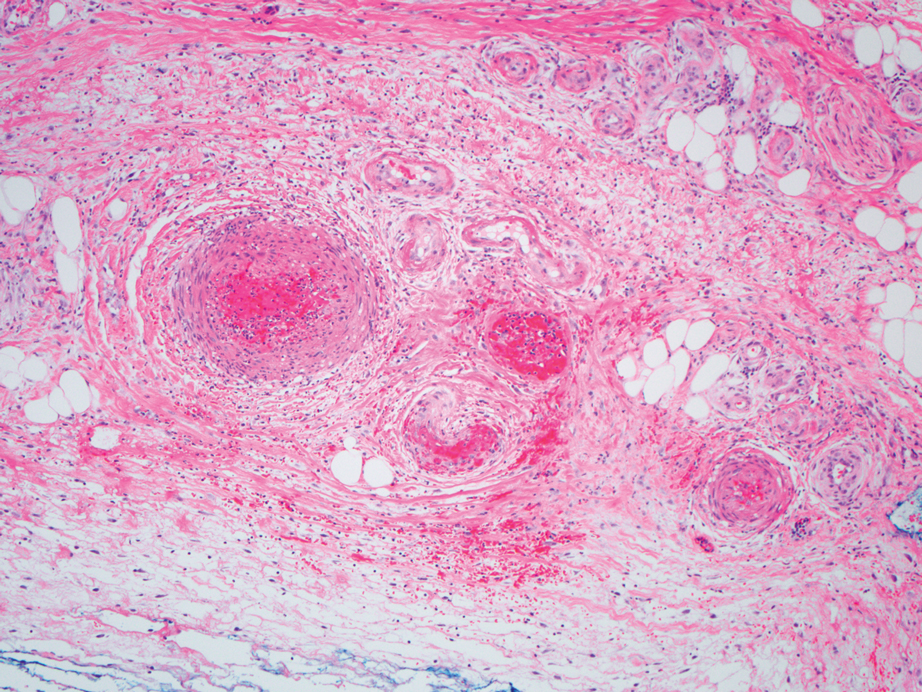
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
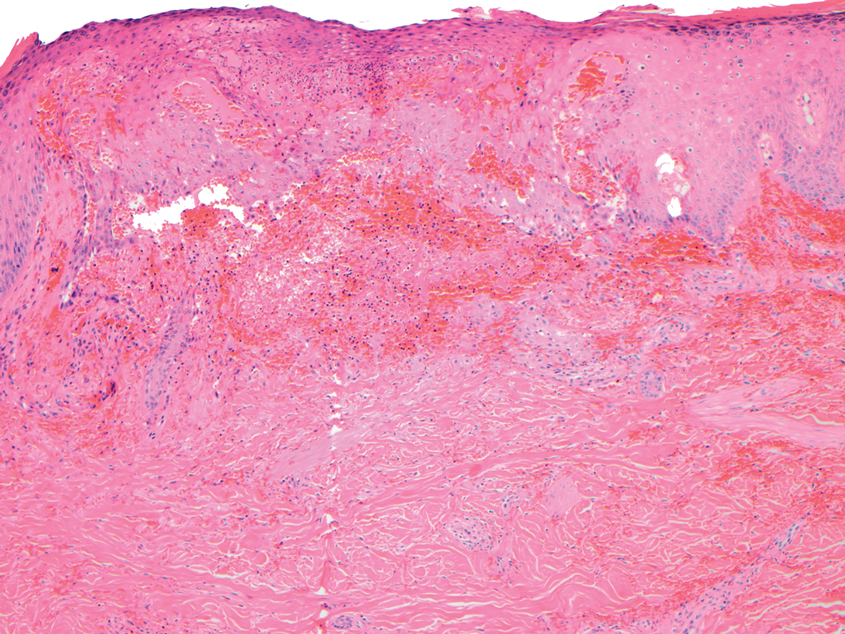
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
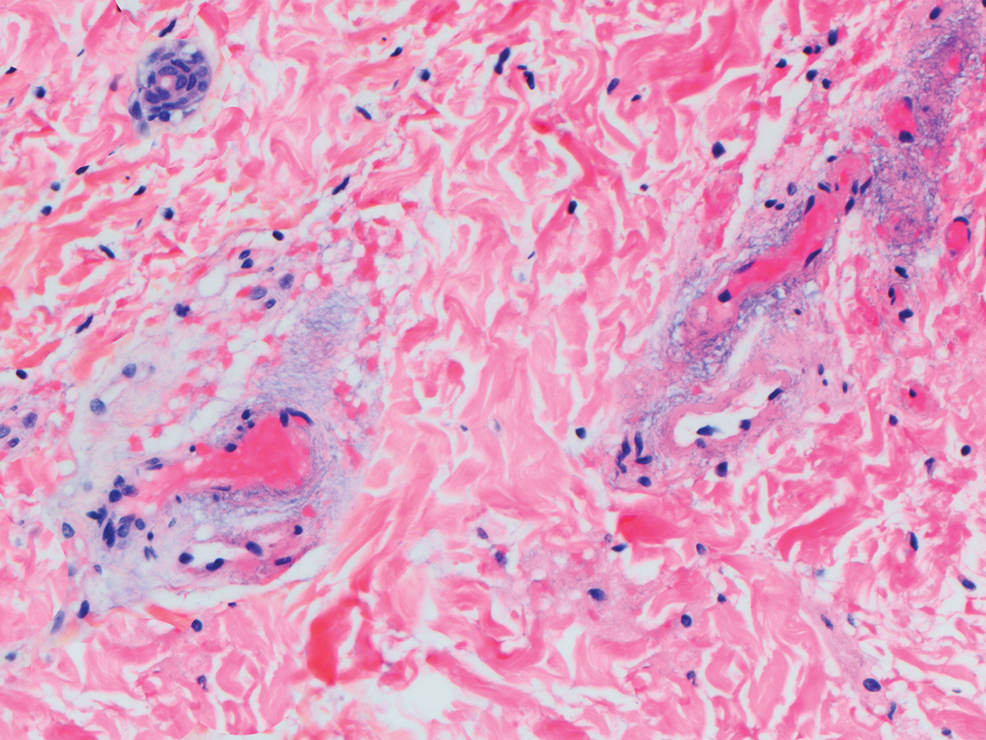
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
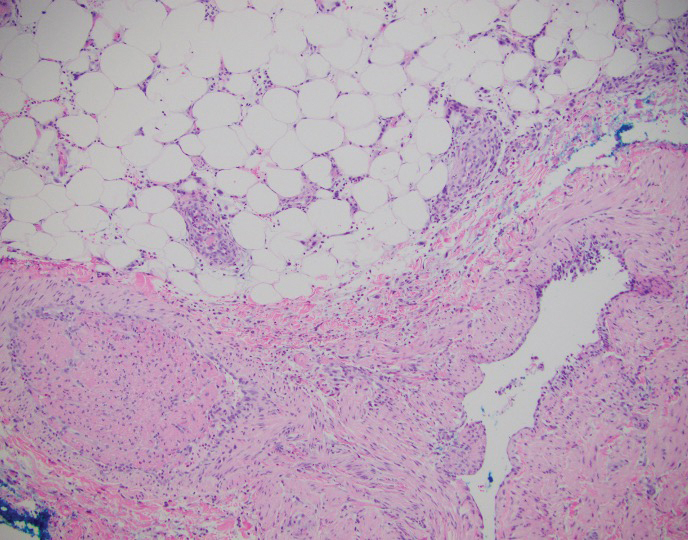
A 40-year-old woman presented with a progressive painful rash on the ears and legs of 2 weeks’ duration. She described the rash as initially red and nonpainful; it started on the right leg and progressed to the left leg, eventually involving the earlobes 4 days prior to presentation. Physical examination revealed edematous purpura of the earlobes and bullous retiform purpura on the lower extremities. Laboratory studies revealed leukopenia (3.6×103 /cm2 [reference range, 4.0–10.5×103 /cm2 ]) and elevated antineutrophil cytoplasmic antibodies (1:320 titer [reference range, <1:40]) in a perinuclear pattern (perinuclear antineutrophil cytoplasmic antibodies). Urine toxicology screening was positive for cocaine and opiates. A punch biopsy of a bullous retiform purpura on the right thigh was obtained for standard hematoxylin and eosin staining.
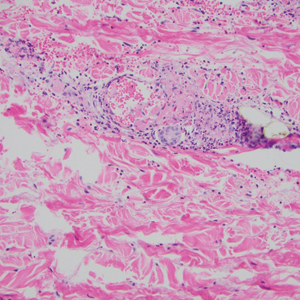
Subcutaneous Nodule on the Chest
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.

Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6

Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6

Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12

In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.


A healthy 45-year-old man presented to the dermatology clinic with a slow-growing subcutaneous nodule on the left chest that had been present for years.