User login
Sweet Syndrome With Marked Eosinophilic Infiltrate
To the Editor:
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an uncommon inflammatory skin disorder characterized by sudden onset of fever, leukocytosis, neutrophilia, and tender erythematous papules or plaques or both. Skin biopsy usually reveals extensive infiltration of neutrophils into the epidermis and dermis.1-3 Although rare, cases of eosinophil-rich SS have been reported in patients with drug-induced and malignancy-associated SS.4,5 We report a case of a patient with classical SS with dermal eosinophilic infiltration.
An 80-year-old Hispanic man presented with abrupt onset of a rash on the posterior scalp, left ear, back, and hands of 5 days’ duration. The lesions were painful and had progressed to the point of impairing hand grip. The patient’s medical history included a reported common cold the week prior, hyperlipidemia, and hypertension, for which he took metoprolol, simvastatin, aspirin, and clopidogrel. He denied oral lesions and medication changes. He was afebrile and did not experience dietary changes, weight loss, or fatigue. He recently returned from travel to the Dominican Republic.
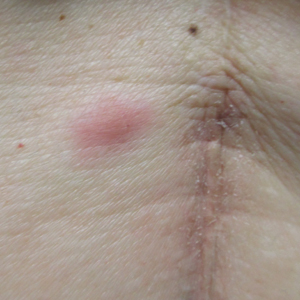
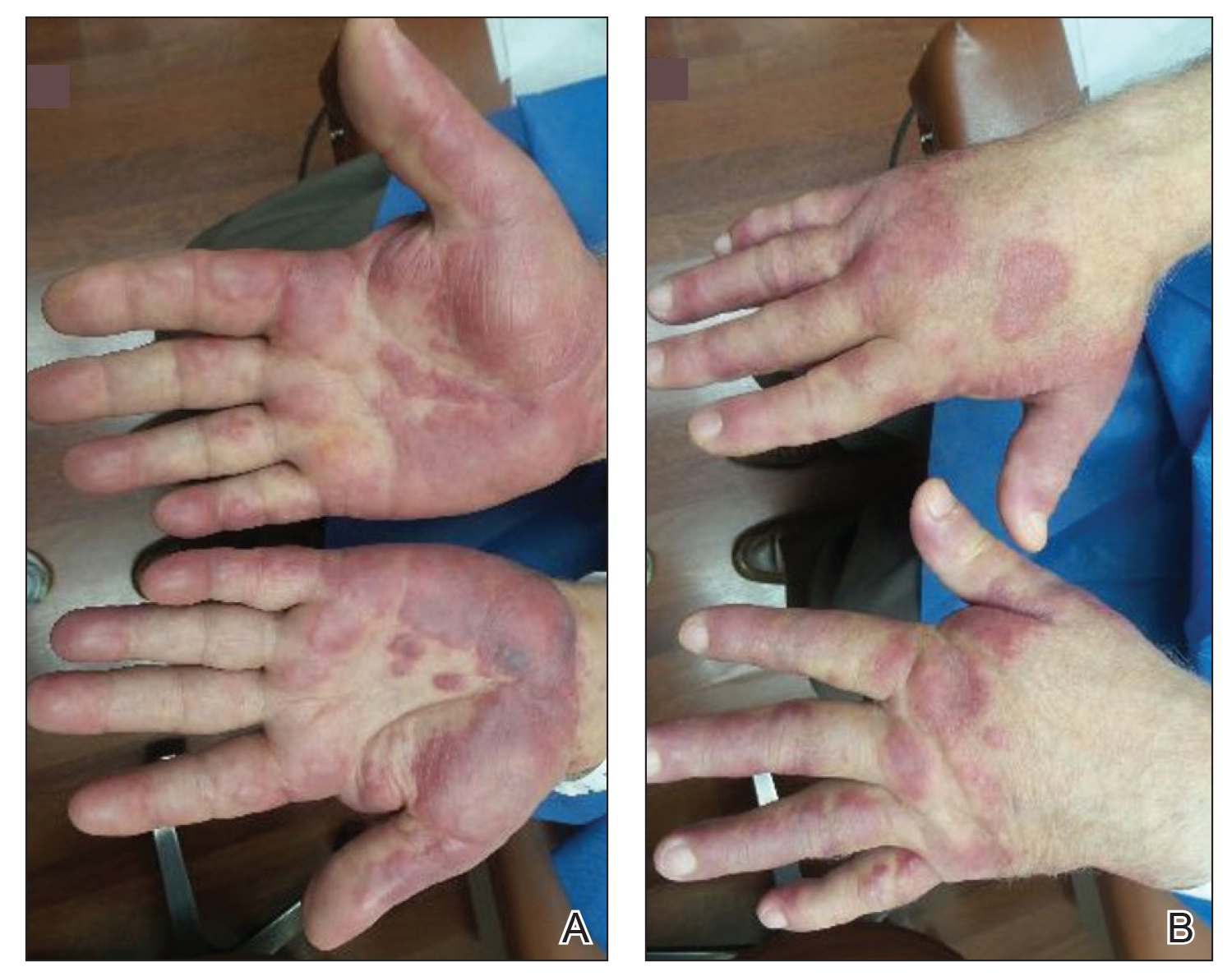
Physical examination revealed tender, well demarcated, pink to violaceous, pseudovesicular papules and plaques on the palms and dorsal hands (Figure 1), the posterior scalp, left ear, proximal left arm, and back. Pink, juicy, targetoid papules were also found on the scalp, back, and left arm. There was no evidence of lymphadenopathy. Laboratory test results revealed an elevated white blood cell count (11,500/µL [reference range, 3800-10,800/µL]), absolute neutrophil count (8073/µL [reference range, 1500–7800/µL]), and eosinophil count (610/µL [reference range, 15–500/µL]). These results indicated leukocytosis with neutrophilia and mild eosinophilia. The patient also was anemic (hemoglobin, 11.5 g/dL [reference range, 13.2–17.1 g/dL]; hematocrit, 35.1% [reference range, 38.5%–50%]). Urine testing revealed altered renal function (serum creatinine, 2.42 mg/dL [reference range, 0.7–1.1 mg/dL]; blood urea nitrogen, 34 mg/dL [reference range, 7–25 mg/dL]; glomerular filtration rate, 4 mL/min/1.73 m2 (reference range, ≥60 mL/min/1.73 m2]), suggesting stage 4 chronic kidney disease. Urinalysis showed mild hematuria and proteinuria.
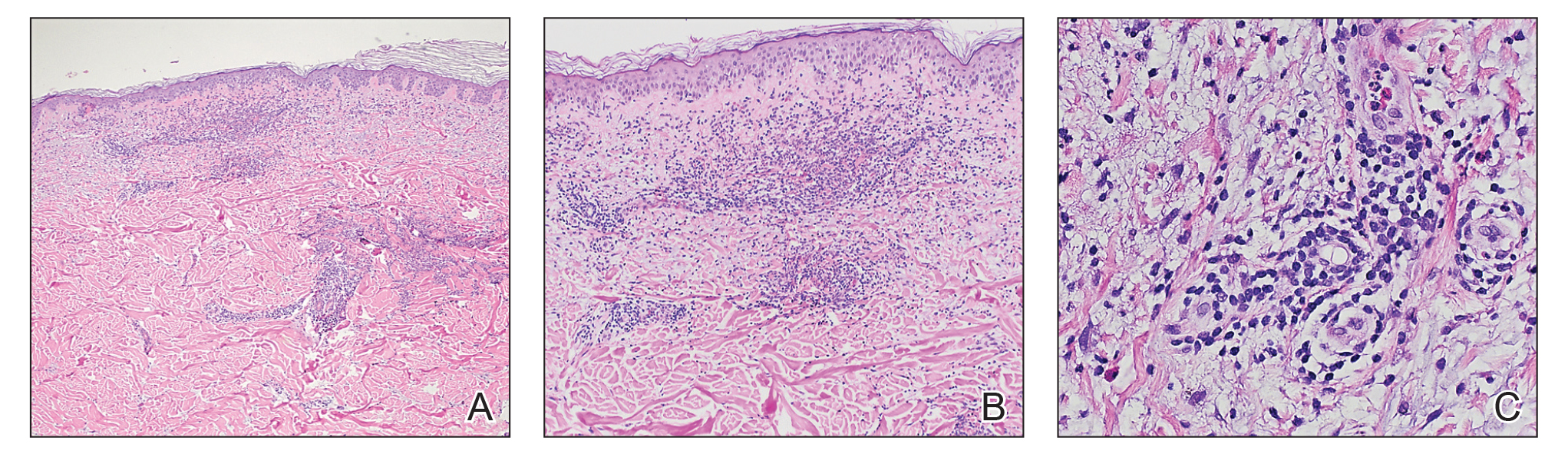
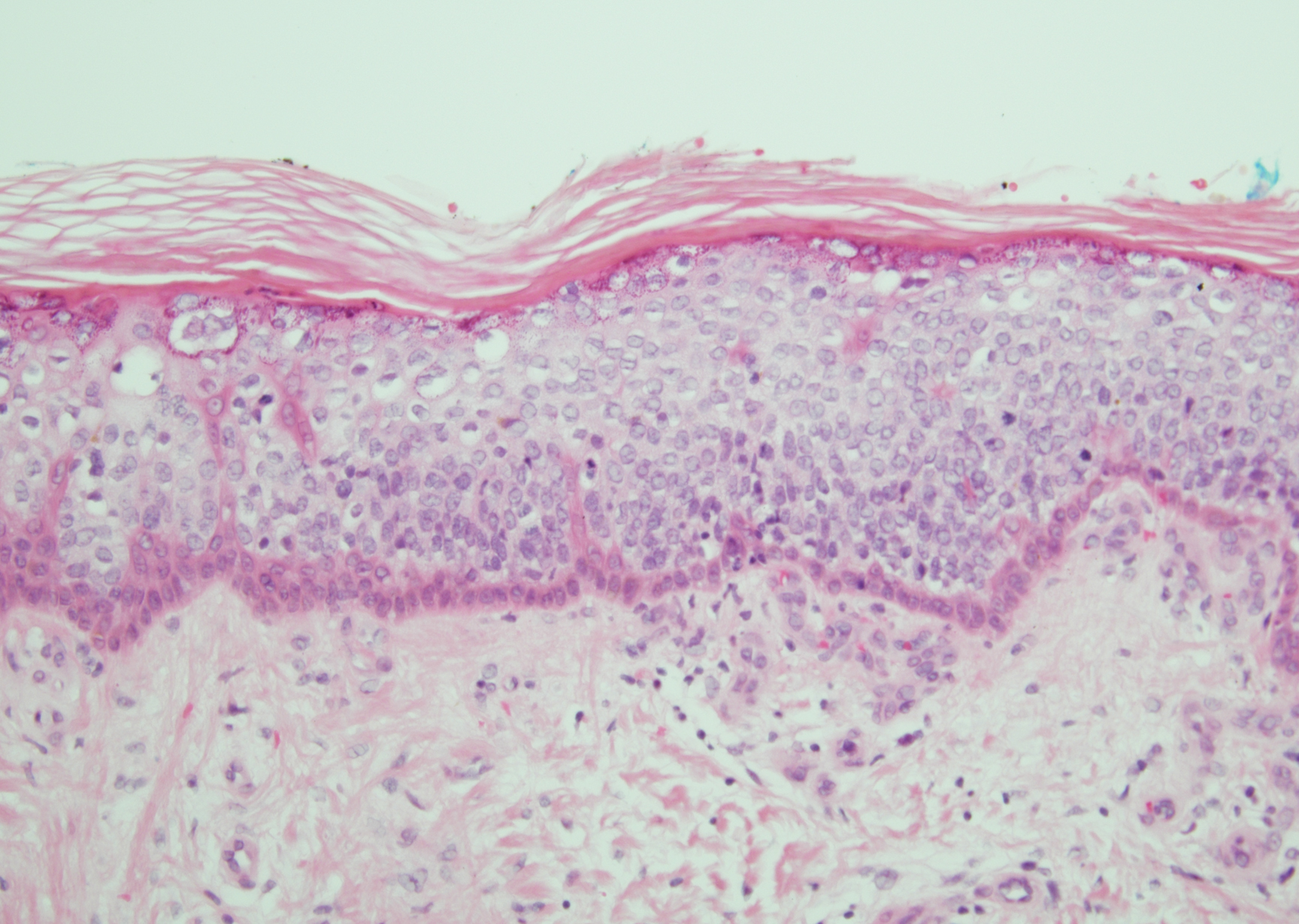
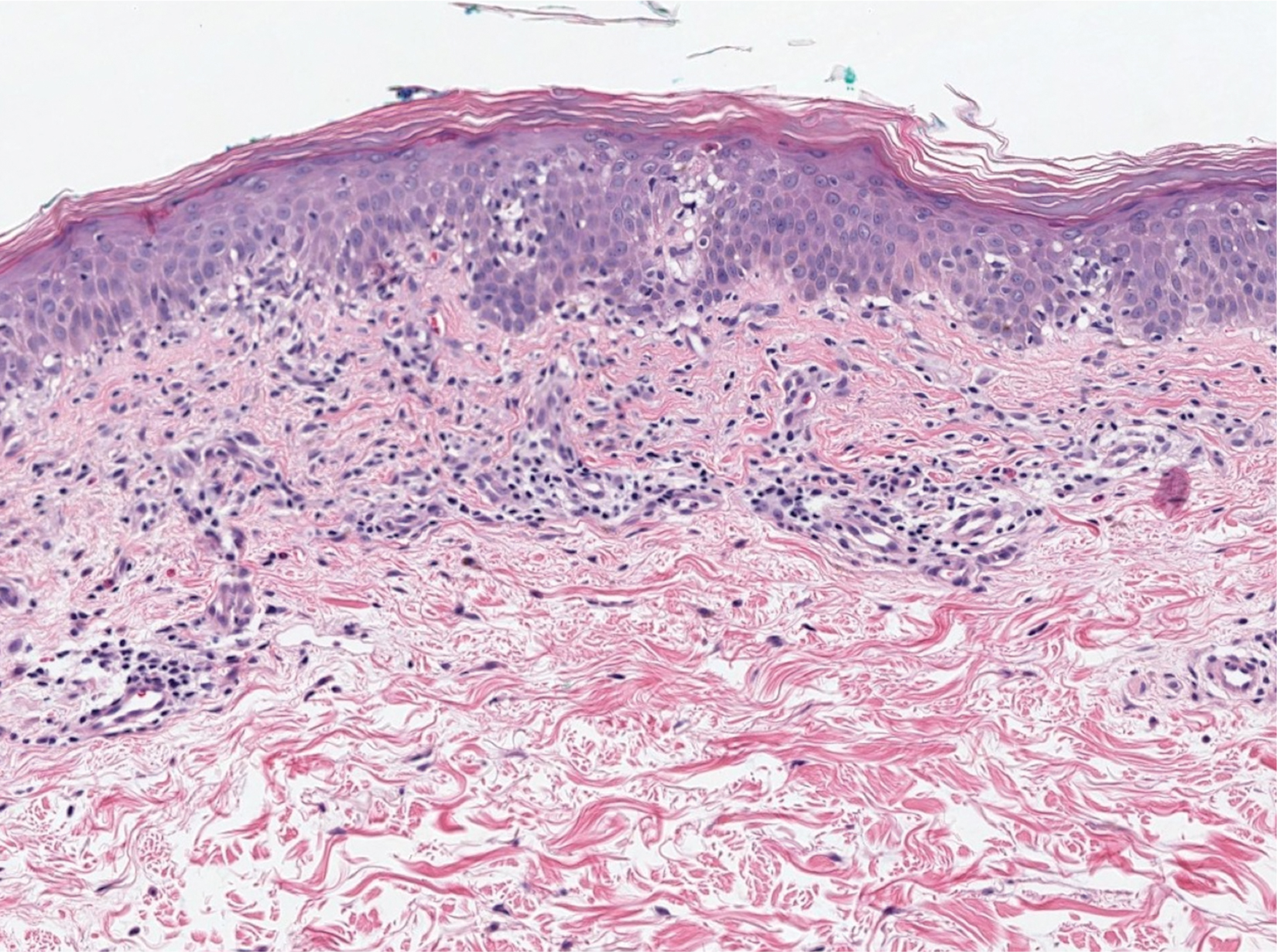
Histopathology of biopsies taken from plaques on the left arm and lower back revealed a dense neutrophilic infiltrate with numerous scattered eosinophils in the dermis. Some neutrophils were intact; others were fragmented without evidence of vasculitis. A subtle subepidermal edema also was noted (Figure 2). A diagnosis of SS was made.
Initial treatment included prednisone (40 mg daily, tapered by 5 mg every 3 days) and erythromycin (500 mg 4 times daily) for 7 days because of suspected Mycoplasma infection. The rash resolved in 1 week. No recurrence was noted during 4 months of follow-up. The white blood cell count returned to within reference range (8400/µL), ruling out the possibility of a smoldering myeloid process.
Acute febrile neutrophilic dermatosis was first described in a case series of 8 women by Sweet6 in 1964. Patients typically present first with fever, which can precede cutaneous symptoms for days or weeks. Skin lesions generally are asymmetric and located on the face, neck, and upper extremities. Lesions can be described as painful, purple to red papules, plaques, or nodules. Sweet syndrome can present as 3 subtypes based on cause7: (1) classical SS, also known as idiopathic SS, can be preceded by an upper respiratory tract or gastrointestinal tract infection or vaccination, or can be pregnancy associated2; (2) drug-induced SS usually follows use of granulocyte colony-stimulating factor, or other causative drugs including trimethoprim-sulfamethoxazole, nitrofurantoin, quinolones, oral contraceptives, furosemide, hydralazine, diazepam, clozapine, abacavir, imatinib, bortezomib, azathioprine, and celecoxib2,3,8; and (3) malignancy-associated SS can occur as a paraneoplastic syndrome and generally is associated with hematologic malignancy or a solid tumor.1,9
In our patient, the observed clinical and histological findings were consistent with a diagnosis of SS,2,10 specifically tender erythematous plaques of sudden onset, fast response to systemic corticosteroid therapy, a dermal neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, and leukocytosis greater than 8000/µL with more than 70% neutrophils. He also exhibited targetoid lesions, which have been reported in 7% to 12% of SS patients.10,11
The predominant cells involved in the dermis of SS lesions are mature neutrophils; however, eosinophils have been observed in small numbers within dermal infiltrates in skin lesions of patients with either classical SS or drug-induced dermatosis.2 In 2 studies of cases of SS (N=73 and N=31), eosinophils were reported in 35% and 41% of skin biopsies, respectively.4,5 Nevertheless, cases with dense eosinophilic infiltrates are rare. Furthermore, Masuda et al12 reported a case of eosinophil-rich SS in a 29-year-old woman after treatment of an upper respiratory tract infection with an antibiotic, and Soon et al13 described an eosinophil-rich case of SS in the setting of new-onset enteropathy-associated T-cell lymphoma.
Our patient was considered to have classical SS because he had an episode of an upper respiratory tract infection 1 week prior to onset of clinical manifestations. The histologic finding of numerous eosinophils in our case was unusual for idiopathic SS. This finding might suggest a drug hypersensitivity reaction, but the lack of any change in the patient’s long-term medication list and the lack of any other episodes made a diagnosis of drug-induced SS less likely in our patient.
Eosinophilic dermatosis of hematologic malignancy is a rare cutaneous condition in which nodules, pruritic papules, and vesicles arise in patients with a hematologic malignancy, such as chronic lymphocytic leukemia and mantle cell lymphoma,13 in which a deep perivascular lymphocytic infiltrate and numerous eosinophils are observed. Malignancy was ruled out in our patient because of the lack of characteristic abnormalities in blood testing, the fast response to corticosteroid therapy, and the lack of recurrence posttreatment or additional systemic concerns.
The typical pathology findings of SS consist of mature neutrophils found in the dermis without evidence of leukocytoclastic vasculitis. Eosinophil-rich infiltration, however rare, has been reported in SS. This report highlights a case of classical SS with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses. Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of this disorder.
- Herbert-Cohen D, Jour G, Saul T. Sweet’s syndrome. J Emerg Med. 2015;49:e95-e97.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378.
- Rochael MC, Pantaleão L, Vilar EA, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol. 2011;86:702-707.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Polimeni G, Cardillo R, Garaffo E, et al. Allopurinol-induced Sweet’s syndrome. Int J Immunopathol Pharmacol. 2016;29:329-332.
- Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85-95.
- Amouri M, Masmoudi A, Ammar M, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55:1033-1039.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Masuda T, Abe Y, Arata J, et al. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) associated with extreme infiltration of eosinophils. J Dermatol. 1994;21:341-346.
- Soon CW, Kirsch IR, Connolly AJ, et al. Eosinophil-rich acute febrile neutrophilic dermatosis in a patient with enteropathy-associated T-cell lymphoma, type 1. Am J Dermatopathol. 2016;38:704-708.
To the Editor:
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an uncommon inflammatory skin disorder characterized by sudden onset of fever, leukocytosis, neutrophilia, and tender erythematous papules or plaques or both. Skin biopsy usually reveals extensive infiltration of neutrophils into the epidermis and dermis.1-3 Although rare, cases of eosinophil-rich SS have been reported in patients with drug-induced and malignancy-associated SS.4,5 We report a case of a patient with classical SS with dermal eosinophilic infiltration.
An 80-year-old Hispanic man presented with abrupt onset of a rash on the posterior scalp, left ear, back, and hands of 5 days’ duration. The lesions were painful and had progressed to the point of impairing hand grip. The patient’s medical history included a reported common cold the week prior, hyperlipidemia, and hypertension, for which he took metoprolol, simvastatin, aspirin, and clopidogrel. He denied oral lesions and medication changes. He was afebrile and did not experience dietary changes, weight loss, or fatigue. He recently returned from travel to the Dominican Republic.
Physical examination revealed tender, well demarcated, pink to violaceous, pseudovesicular papules and plaques on the palms and dorsal hands (Figure 1), the posterior scalp, left ear, proximal left arm, and back. Pink, juicy, targetoid papules were also found on the scalp, back, and left arm. There was no evidence of lymphadenopathy. Laboratory test results revealed an elevated white blood cell count (11,500/µL [reference range, 3800-10,800/µL]), absolute neutrophil count (8073/µL [reference range, 1500–7800/µL]), and eosinophil count (610/µL [reference range, 15–500/µL]). These results indicated leukocytosis with neutrophilia and mild eosinophilia. The patient also was anemic (hemoglobin, 11.5 g/dL [reference range, 13.2–17.1 g/dL]; hematocrit, 35.1% [reference range, 38.5%–50%]). Urine testing revealed altered renal function (serum creatinine, 2.42 mg/dL [reference range, 0.7–1.1 mg/dL]; blood urea nitrogen, 34 mg/dL [reference range, 7–25 mg/dL]; glomerular filtration rate, 4 mL/min/1.73 m2 (reference range, ≥60 mL/min/1.73 m2]), suggesting stage 4 chronic kidney disease. Urinalysis showed mild hematuria and proteinuria.
Histopathology of biopsies taken from plaques on the left arm and lower back revealed a dense neutrophilic infiltrate with numerous scattered eosinophils in the dermis. Some neutrophils were intact; others were fragmented without evidence of vasculitis. A subtle subepidermal edema also was noted (Figure 2). A diagnosis of SS was made.
Initial treatment included prednisone (40 mg daily, tapered by 5 mg every 3 days) and erythromycin (500 mg 4 times daily) for 7 days because of suspected Mycoplasma infection. The rash resolved in 1 week. No recurrence was noted during 4 months of follow-up. The white blood cell count returned to within reference range (8400/µL), ruling out the possibility of a smoldering myeloid process.
Acute febrile neutrophilic dermatosis was first described in a case series of 8 women by Sweet6 in 1964. Patients typically present first with fever, which can precede cutaneous symptoms for days or weeks. Skin lesions generally are asymmetric and located on the face, neck, and upper extremities. Lesions can be described as painful, purple to red papules, plaques, or nodules. Sweet syndrome can present as 3 subtypes based on cause7: (1) classical SS, also known as idiopathic SS, can be preceded by an upper respiratory tract or gastrointestinal tract infection or vaccination, or can be pregnancy associated2; (2) drug-induced SS usually follows use of granulocyte colony-stimulating factor, or other causative drugs including trimethoprim-sulfamethoxazole, nitrofurantoin, quinolones, oral contraceptives, furosemide, hydralazine, diazepam, clozapine, abacavir, imatinib, bortezomib, azathioprine, and celecoxib2,3,8; and (3) malignancy-associated SS can occur as a paraneoplastic syndrome and generally is associated with hematologic malignancy or a solid tumor.1,9
In our patient, the observed clinical and histological findings were consistent with a diagnosis of SS,2,10 specifically tender erythematous plaques of sudden onset, fast response to systemic corticosteroid therapy, a dermal neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, and leukocytosis greater than 8000/µL with more than 70% neutrophils. He also exhibited targetoid lesions, which have been reported in 7% to 12% of SS patients.10,11
The predominant cells involved in the dermis of SS lesions are mature neutrophils; however, eosinophils have been observed in small numbers within dermal infiltrates in skin lesions of patients with either classical SS or drug-induced dermatosis.2 In 2 studies of cases of SS (N=73 and N=31), eosinophils were reported in 35% and 41% of skin biopsies, respectively.4,5 Nevertheless, cases with dense eosinophilic infiltrates are rare. Furthermore, Masuda et al12 reported a case of eosinophil-rich SS in a 29-year-old woman after treatment of an upper respiratory tract infection with an antibiotic, and Soon et al13 described an eosinophil-rich case of SS in the setting of new-onset enteropathy-associated T-cell lymphoma.
Our patient was considered to have classical SS because he had an episode of an upper respiratory tract infection 1 week prior to onset of clinical manifestations. The histologic finding of numerous eosinophils in our case was unusual for idiopathic SS. This finding might suggest a drug hypersensitivity reaction, but the lack of any change in the patient’s long-term medication list and the lack of any other episodes made a diagnosis of drug-induced SS less likely in our patient.
Eosinophilic dermatosis of hematologic malignancy is a rare cutaneous condition in which nodules, pruritic papules, and vesicles arise in patients with a hematologic malignancy, such as chronic lymphocytic leukemia and mantle cell lymphoma,13 in which a deep perivascular lymphocytic infiltrate and numerous eosinophils are observed. Malignancy was ruled out in our patient because of the lack of characteristic abnormalities in blood testing, the fast response to corticosteroid therapy, and the lack of recurrence posttreatment or additional systemic concerns.
The typical pathology findings of SS consist of mature neutrophils found in the dermis without evidence of leukocytoclastic vasculitis. Eosinophil-rich infiltration, however rare, has been reported in SS. This report highlights a case of classical SS with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses. Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of this disorder.
To the Editor:
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an uncommon inflammatory skin disorder characterized by sudden onset of fever, leukocytosis, neutrophilia, and tender erythematous papules or plaques or both. Skin biopsy usually reveals extensive infiltration of neutrophils into the epidermis and dermis.1-3 Although rare, cases of eosinophil-rich SS have been reported in patients with drug-induced and malignancy-associated SS.4,5 We report a case of a patient with classical SS with dermal eosinophilic infiltration.
An 80-year-old Hispanic man presented with abrupt onset of a rash on the posterior scalp, left ear, back, and hands of 5 days’ duration. The lesions were painful and had progressed to the point of impairing hand grip. The patient’s medical history included a reported common cold the week prior, hyperlipidemia, and hypertension, for which he took metoprolol, simvastatin, aspirin, and clopidogrel. He denied oral lesions and medication changes. He was afebrile and did not experience dietary changes, weight loss, or fatigue. He recently returned from travel to the Dominican Republic.
Physical examination revealed tender, well demarcated, pink to violaceous, pseudovesicular papules and plaques on the palms and dorsal hands (Figure 1), the posterior scalp, left ear, proximal left arm, and back. Pink, juicy, targetoid papules were also found on the scalp, back, and left arm. There was no evidence of lymphadenopathy. Laboratory test results revealed an elevated white blood cell count (11,500/µL [reference range, 3800-10,800/µL]), absolute neutrophil count (8073/µL [reference range, 1500–7800/µL]), and eosinophil count (610/µL [reference range, 15–500/µL]). These results indicated leukocytosis with neutrophilia and mild eosinophilia. The patient also was anemic (hemoglobin, 11.5 g/dL [reference range, 13.2–17.1 g/dL]; hematocrit, 35.1% [reference range, 38.5%–50%]). Urine testing revealed altered renal function (serum creatinine, 2.42 mg/dL [reference range, 0.7–1.1 mg/dL]; blood urea nitrogen, 34 mg/dL [reference range, 7–25 mg/dL]; glomerular filtration rate, 4 mL/min/1.73 m2 (reference range, ≥60 mL/min/1.73 m2]), suggesting stage 4 chronic kidney disease. Urinalysis showed mild hematuria and proteinuria.
Histopathology of biopsies taken from plaques on the left arm and lower back revealed a dense neutrophilic infiltrate with numerous scattered eosinophils in the dermis. Some neutrophils were intact; others were fragmented without evidence of vasculitis. A subtle subepidermal edema also was noted (Figure 2). A diagnosis of SS was made.
Initial treatment included prednisone (40 mg daily, tapered by 5 mg every 3 days) and erythromycin (500 mg 4 times daily) for 7 days because of suspected Mycoplasma infection. The rash resolved in 1 week. No recurrence was noted during 4 months of follow-up. The white blood cell count returned to within reference range (8400/µL), ruling out the possibility of a smoldering myeloid process.
Acute febrile neutrophilic dermatosis was first described in a case series of 8 women by Sweet6 in 1964. Patients typically present first with fever, which can precede cutaneous symptoms for days or weeks. Skin lesions generally are asymmetric and located on the face, neck, and upper extremities. Lesions can be described as painful, purple to red papules, plaques, or nodules. Sweet syndrome can present as 3 subtypes based on cause7: (1) classical SS, also known as idiopathic SS, can be preceded by an upper respiratory tract or gastrointestinal tract infection or vaccination, or can be pregnancy associated2; (2) drug-induced SS usually follows use of granulocyte colony-stimulating factor, or other causative drugs including trimethoprim-sulfamethoxazole, nitrofurantoin, quinolones, oral contraceptives, furosemide, hydralazine, diazepam, clozapine, abacavir, imatinib, bortezomib, azathioprine, and celecoxib2,3,8; and (3) malignancy-associated SS can occur as a paraneoplastic syndrome and generally is associated with hematologic malignancy or a solid tumor.1,9
In our patient, the observed clinical and histological findings were consistent with a diagnosis of SS,2,10 specifically tender erythematous plaques of sudden onset, fast response to systemic corticosteroid therapy, a dermal neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, and leukocytosis greater than 8000/µL with more than 70% neutrophils. He also exhibited targetoid lesions, which have been reported in 7% to 12% of SS patients.10,11
The predominant cells involved in the dermis of SS lesions are mature neutrophils; however, eosinophils have been observed in small numbers within dermal infiltrates in skin lesions of patients with either classical SS or drug-induced dermatosis.2 In 2 studies of cases of SS (N=73 and N=31), eosinophils were reported in 35% and 41% of skin biopsies, respectively.4,5 Nevertheless, cases with dense eosinophilic infiltrates are rare. Furthermore, Masuda et al12 reported a case of eosinophil-rich SS in a 29-year-old woman after treatment of an upper respiratory tract infection with an antibiotic, and Soon et al13 described an eosinophil-rich case of SS in the setting of new-onset enteropathy-associated T-cell lymphoma.
Our patient was considered to have classical SS because he had an episode of an upper respiratory tract infection 1 week prior to onset of clinical manifestations. The histologic finding of numerous eosinophils in our case was unusual for idiopathic SS. This finding might suggest a drug hypersensitivity reaction, but the lack of any change in the patient’s long-term medication list and the lack of any other episodes made a diagnosis of drug-induced SS less likely in our patient.
Eosinophilic dermatosis of hematologic malignancy is a rare cutaneous condition in which nodules, pruritic papules, and vesicles arise in patients with a hematologic malignancy, such as chronic lymphocytic leukemia and mantle cell lymphoma,13 in which a deep perivascular lymphocytic infiltrate and numerous eosinophils are observed. Malignancy was ruled out in our patient because of the lack of characteristic abnormalities in blood testing, the fast response to corticosteroid therapy, and the lack of recurrence posttreatment or additional systemic concerns.
The typical pathology findings of SS consist of mature neutrophils found in the dermis without evidence of leukocytoclastic vasculitis. Eosinophil-rich infiltration, however rare, has been reported in SS. This report highlights a case of classical SS with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses. Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of this disorder.
- Herbert-Cohen D, Jour G, Saul T. Sweet’s syndrome. J Emerg Med. 2015;49:e95-e97.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378.
- Rochael MC, Pantaleão L, Vilar EA, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol. 2011;86:702-707.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Polimeni G, Cardillo R, Garaffo E, et al. Allopurinol-induced Sweet’s syndrome. Int J Immunopathol Pharmacol. 2016;29:329-332.
- Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85-95.
- Amouri M, Masmoudi A, Ammar M, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55:1033-1039.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Masuda T, Abe Y, Arata J, et al. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) associated with extreme infiltration of eosinophils. J Dermatol. 1994;21:341-346.
- Soon CW, Kirsch IR, Connolly AJ, et al. Eosinophil-rich acute febrile neutrophilic dermatosis in a patient with enteropathy-associated T-cell lymphoma, type 1. Am J Dermatopathol. 2016;38:704-708.
- Herbert-Cohen D, Jour G, Saul T. Sweet’s syndrome. J Emerg Med. 2015;49:e95-e97.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378.
- Rochael MC, Pantaleão L, Vilar EA, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol. 2011;86:702-707.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Polimeni G, Cardillo R, Garaffo E, et al. Allopurinol-induced Sweet’s syndrome. Int J Immunopathol Pharmacol. 2016;29:329-332.
- Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol Hematol. 2013;86:85-95.
- Amouri M, Masmoudi A, Ammar M, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55:1033-1039.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Masuda T, Abe Y, Arata J, et al. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) associated with extreme infiltration of eosinophils. J Dermatol. 1994;21:341-346.
- Soon CW, Kirsch IR, Connolly AJ, et al. Eosinophil-rich acute febrile neutrophilic dermatosis in a patient with enteropathy-associated T-cell lymphoma, type 1. Am J Dermatopathol. 2016;38:704-708.
Practice Points
- This report highlights a case of classical Sweet syndrome (SS) with a particularly dense eosinophilic infiltrate, which could be mistaken for other eosinophilic dermatoses.
- Dermatologists should be aware of the possibility of marked eosinophilic infiltration in all subtypes of SS.
Solitary Papule on the Shoulder
The Diagnosis: Dermatofibroma With Sebaceous Induction
The biopsy of the lesion revealed a fibrohistiocytic dermal pattern with overlying benign epidermal and sebaceous hyperplasia with a proliferation of fibroblasts in the dermis. Other sections revealed hyperplastic sebaceous glands of the superficial and mid dermis. These findings were suggestive of a dermatofibroma (DF) that had induced epidermal and sebaceous hyperplasia.
Dermatofibromas are common benign fibrous soft tissue growths that account for approximately 3% of dermatopathology specimens.1 The etiology of DFs is unknown; however, they are thought to arise from sites of prior trauma or arthropod bites. Multiple or eruptive DFs have been reported in patients with lupus and atopic dermatitis.2 They commonly appear as round firm nodules measuring less than 1 cm in diameter on the extremities of young adults. Eruptive dermatofibromas also have been reported in human immunodeficiency virus-positive and immunosuppressed patients.3,4 On physical examination, gently pinching the lesion causes a downward movement known as the "dimple sign." If left undisturbed, DFs persist but may undergo partial regression, especially in the center; they also may be excised if symptomatic.
The clinical differential for this papule included a scar and sebaceous hyperplasia. The lack of history of skin cancer or prior procedure made a scar less likely. Sebaceous glands are less prominent on the shoulders, making sebaceous hyperplasia less likely, though dermoscopy showed pale yellow lobules. Sebaceous adenomas most commonly are seen on the head or neck and present as a flesh-colored papule. Sebaceous induction by DFs is rare but has been reported in the literature.5,6
The histology of DFs is described as a nodular proliferation of spindle-shaped fibroblasts and myofibroblasts with short intersecting fascicles. A predilection for sebaceous induction from an underlying DF on the shoulder has been reported.5 Sebaceous differentiation has been reported in 16% to 31.6% of DFs.5,6 Seborrheic keratosis-like epidermal hyperplasia frequently has been seen in DFs with sebaceous induction in comparison to DFs without sebaceous induction.5 Immunohistochemical stains are important to help differentiate DF from dermatofibrosarcoma protuberans, especially when approaching the subcutis. Dermatofibromas stain positive for factor XIIIa and negative for CD34, whereas dermatofibrosarcoma protuberans stain negative for factor XIIIa and positive for CD34.7 Dermatofibromas also demonstrate positive immunostaining for vimentin, stromelysin 3,8 muscle-specific actin, and CD68.
- Rahbari H, Mehregan AH. Adnexal displacement and regression in association with histiocytoma (dermatofibroma). J Cutan Pathol. 1985;12:94-102.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol. 2006;20:90-92.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol. 2000;27:54-56.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Shuweiter M, Böer A. Spectrum of follicular and sebaceous differentiation induced by dermatofibroma. Am J Dermatopathol. 2009;31:778.
- Abenoza P, Lillemoe T. CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol. 1993;15:429-434.
- Kim HJ, Lee JY, Kim SH, et al. Stromelysin-3 expression in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans: comparison with factor XIIIa and CD34. Br J Dermatol. 2007;157:319-324.
The Diagnosis: Dermatofibroma With Sebaceous Induction
The biopsy of the lesion revealed a fibrohistiocytic dermal pattern with overlying benign epidermal and sebaceous hyperplasia with a proliferation of fibroblasts in the dermis. Other sections revealed hyperplastic sebaceous glands of the superficial and mid dermis. These findings were suggestive of a dermatofibroma (DF) that had induced epidermal and sebaceous hyperplasia.
Dermatofibromas are common benign fibrous soft tissue growths that account for approximately 3% of dermatopathology specimens.1 The etiology of DFs is unknown; however, they are thought to arise from sites of prior trauma or arthropod bites. Multiple or eruptive DFs have been reported in patients with lupus and atopic dermatitis.2 They commonly appear as round firm nodules measuring less than 1 cm in diameter on the extremities of young adults. Eruptive dermatofibromas also have been reported in human immunodeficiency virus-positive and immunosuppressed patients.3,4 On physical examination, gently pinching the lesion causes a downward movement known as the "dimple sign." If left undisturbed, DFs persist but may undergo partial regression, especially in the center; they also may be excised if symptomatic.
The clinical differential for this papule included a scar and sebaceous hyperplasia. The lack of history of skin cancer or prior procedure made a scar less likely. Sebaceous glands are less prominent on the shoulders, making sebaceous hyperplasia less likely, though dermoscopy showed pale yellow lobules. Sebaceous adenomas most commonly are seen on the head or neck and present as a flesh-colored papule. Sebaceous induction by DFs is rare but has been reported in the literature.5,6
The histology of DFs is described as a nodular proliferation of spindle-shaped fibroblasts and myofibroblasts with short intersecting fascicles. A predilection for sebaceous induction from an underlying DF on the shoulder has been reported.5 Sebaceous differentiation has been reported in 16% to 31.6% of DFs.5,6 Seborrheic keratosis-like epidermal hyperplasia frequently has been seen in DFs with sebaceous induction in comparison to DFs without sebaceous induction.5 Immunohistochemical stains are important to help differentiate DF from dermatofibrosarcoma protuberans, especially when approaching the subcutis. Dermatofibromas stain positive for factor XIIIa and negative for CD34, whereas dermatofibrosarcoma protuberans stain negative for factor XIIIa and positive for CD34.7 Dermatofibromas also demonstrate positive immunostaining for vimentin, stromelysin 3,8 muscle-specific actin, and CD68.
The Diagnosis: Dermatofibroma With Sebaceous Induction
The biopsy of the lesion revealed a fibrohistiocytic dermal pattern with overlying benign epidermal and sebaceous hyperplasia with a proliferation of fibroblasts in the dermis. Other sections revealed hyperplastic sebaceous glands of the superficial and mid dermis. These findings were suggestive of a dermatofibroma (DF) that had induced epidermal and sebaceous hyperplasia.
Dermatofibromas are common benign fibrous soft tissue growths that account for approximately 3% of dermatopathology specimens.1 The etiology of DFs is unknown; however, they are thought to arise from sites of prior trauma or arthropod bites. Multiple or eruptive DFs have been reported in patients with lupus and atopic dermatitis.2 They commonly appear as round firm nodules measuring less than 1 cm in diameter on the extremities of young adults. Eruptive dermatofibromas also have been reported in human immunodeficiency virus-positive and immunosuppressed patients.3,4 On physical examination, gently pinching the lesion causes a downward movement known as the "dimple sign." If left undisturbed, DFs persist but may undergo partial regression, especially in the center; they also may be excised if symptomatic.
The clinical differential for this papule included a scar and sebaceous hyperplasia. The lack of history of skin cancer or prior procedure made a scar less likely. Sebaceous glands are less prominent on the shoulders, making sebaceous hyperplasia less likely, though dermoscopy showed pale yellow lobules. Sebaceous adenomas most commonly are seen on the head or neck and present as a flesh-colored papule. Sebaceous induction by DFs is rare but has been reported in the literature.5,6
The histology of DFs is described as a nodular proliferation of spindle-shaped fibroblasts and myofibroblasts with short intersecting fascicles. A predilection for sebaceous induction from an underlying DF on the shoulder has been reported.5 Sebaceous differentiation has been reported in 16% to 31.6% of DFs.5,6 Seborrheic keratosis-like epidermal hyperplasia frequently has been seen in DFs with sebaceous induction in comparison to DFs without sebaceous induction.5 Immunohistochemical stains are important to help differentiate DF from dermatofibrosarcoma protuberans, especially when approaching the subcutis. Dermatofibromas stain positive for factor XIIIa and negative for CD34, whereas dermatofibrosarcoma protuberans stain negative for factor XIIIa and positive for CD34.7 Dermatofibromas also demonstrate positive immunostaining for vimentin, stromelysin 3,8 muscle-specific actin, and CD68.
- Rahbari H, Mehregan AH. Adnexal displacement and regression in association with histiocytoma (dermatofibroma). J Cutan Pathol. 1985;12:94-102.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol. 2006;20:90-92.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol. 2000;27:54-56.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Shuweiter M, Böer A. Spectrum of follicular and sebaceous differentiation induced by dermatofibroma. Am J Dermatopathol. 2009;31:778.
- Abenoza P, Lillemoe T. CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol. 1993;15:429-434.
- Kim HJ, Lee JY, Kim SH, et al. Stromelysin-3 expression in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans: comparison with factor XIIIa and CD34. Br J Dermatol. 2007;157:319-324.
- Rahbari H, Mehregan AH. Adnexal displacement and regression in association with histiocytoma (dermatofibroma). J Cutan Pathol. 1985;12:94-102.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol. 2006;20:90-92.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol. 2000;27:54-56.
- Zaccaria E, Rebora A, Rongioletti F. Multiple eruptive dermatofibromas and immunosuppression: report of two cases and review of the literature. Int J Dermatol. 2008;47:723-727.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Shuweiter M, Böer A. Spectrum of follicular and sebaceous differentiation induced by dermatofibroma. Am J Dermatopathol. 2009;31:778.
- Abenoza P, Lillemoe T. CD34 and factor XIIIa in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans. Am J Dermatopathol. 1993;15:429-434.
- Kim HJ, Lee JY, Kim SH, et al. Stromelysin-3 expression in the differential diagnosis of dermatofibroma and dermatofibrosarcoma protuberans: comparison with factor XIIIa and CD34. Br J Dermatol. 2007;157:319-324.
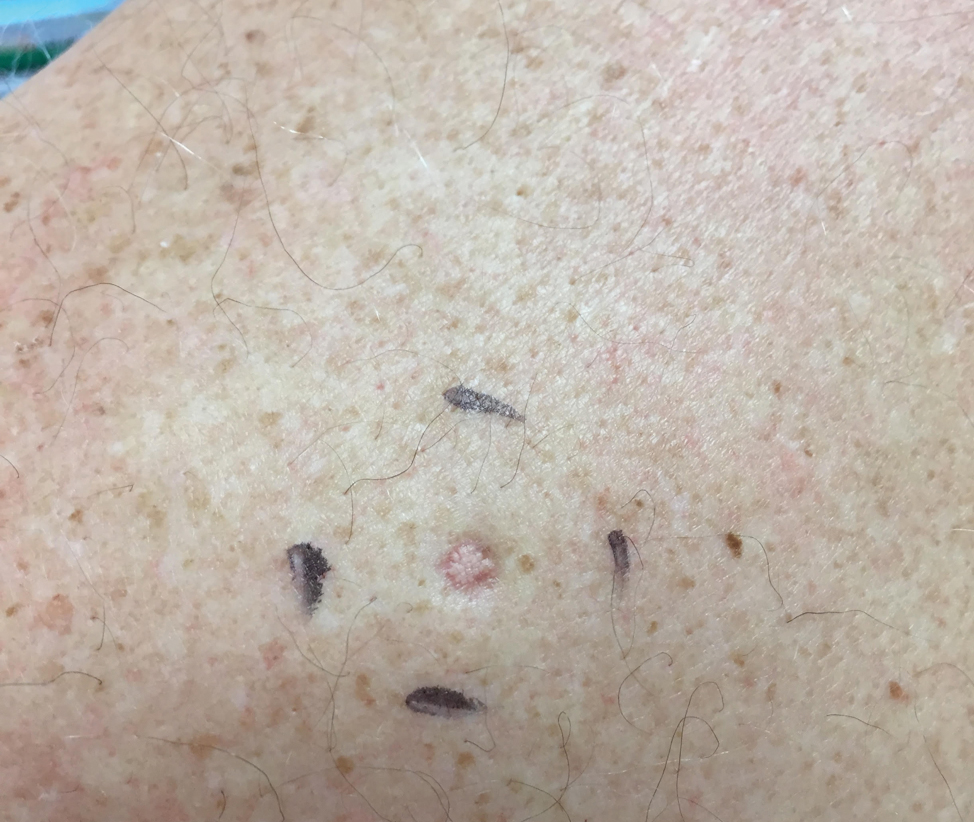

A 64-year-old man presented to dermatology for a full-body skin examination. He had no history of skin cancer. Physical examination revealed an asymptomatic, 4-mm, yellowish pink papule on the left posterior shoulder (top). Dermoscopy revealed yellow globules (bottom). The patient was unsure of the duration of the lesion and denied any prior trauma or medical procedure to the area. Subsequently, a shave biopsy was performed.

Keratotic Papule on the Abdomen
The Diagnosis: Hypergranulotic Dyscornification
Hypergranulotic dyscornification (HD) is a rarely reported reaction pattern present in benign solitary keratoses with only few reports to date. It may be an underrecognized reaction pattern based on the paucity of reported cases as well as the histologic similarities to other entities. It has been hypothesized that this pattern reflects an underlying keratin mutation or disorder of keratinization.1
Clinically, HD most commonly presents as a waxy, tan-colored, solitary keratosis generally found on the lower limbs, trunk, or back in individuals aged 20 to 60 years.1,2 Histopathology shows marked hyperkeratosis, papillomatosis, and clumped basophilic keratohyalin granules within the corneocytes with digitated epidermal hyperplasia. There is abnormal cornification across the entire lesion with papillomatosis and marked hypergranulosis.3 There often are homogeneous orthokeratotic mounds of large, dull, eosinophilic-staining anucleate keratinocytes that are sharply demarcated from the thickened granular layer.1,2 Within the spinous, granular, and corneal layers, there is a pale, gray-staining, basophilic, cytoplasmic substance intercellularly.1
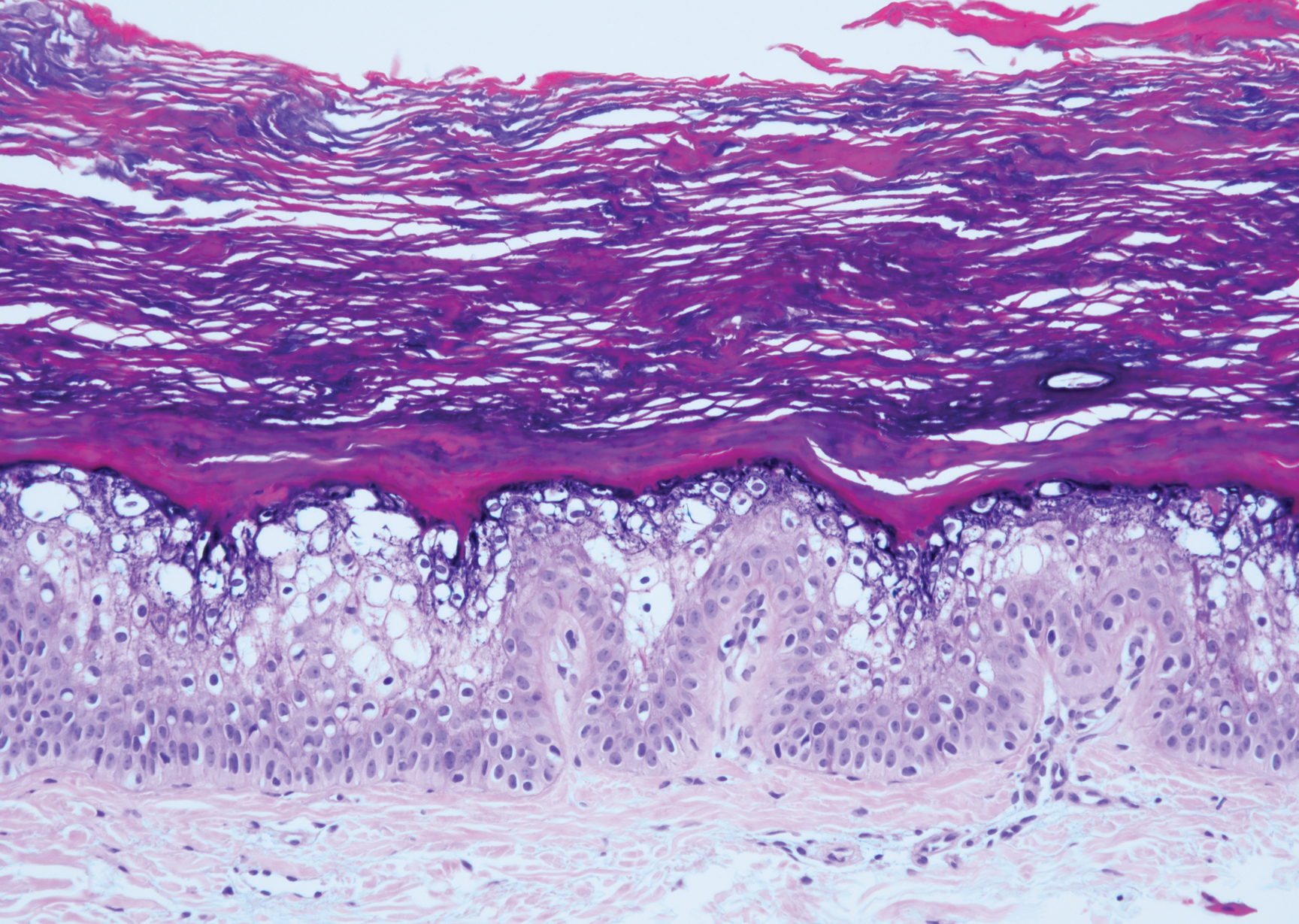
Histopathologically, HD may be mistaken for several other entities both benign and malignant.1 Epidermolytic hyperkeratosis can be a genetic disorder, an incidental finding in a variety of skin conditions, or an isolated lesion.4 The genetic syndrome, caused by mutation in keratins 1 or 10, clinically presents with hyperkeratosis, erosions, blisters, and thickening of the epidermis, often with a corrugated appearance. Epidermal nevi findings often are seen in conjunction with histologic changes of epidermolytic hyperkeratosis caused by mutation. Solitary lesions also can resemble seborrheic keratosis or verruca. In all examples of epidermolytic hyperkeratosis, the histopathologic findings are identical.4 The granular layer is thickened, and coarse keratohyalin granules aggregate in the suprabasal cells.5 There is acantholysis with perinuclear vacuolization in the spinous and granular layers with characteristic pale cytoplasmic areas devoid of keratin filaments (Figure 1). The basal layer may be hyperproliferative.5
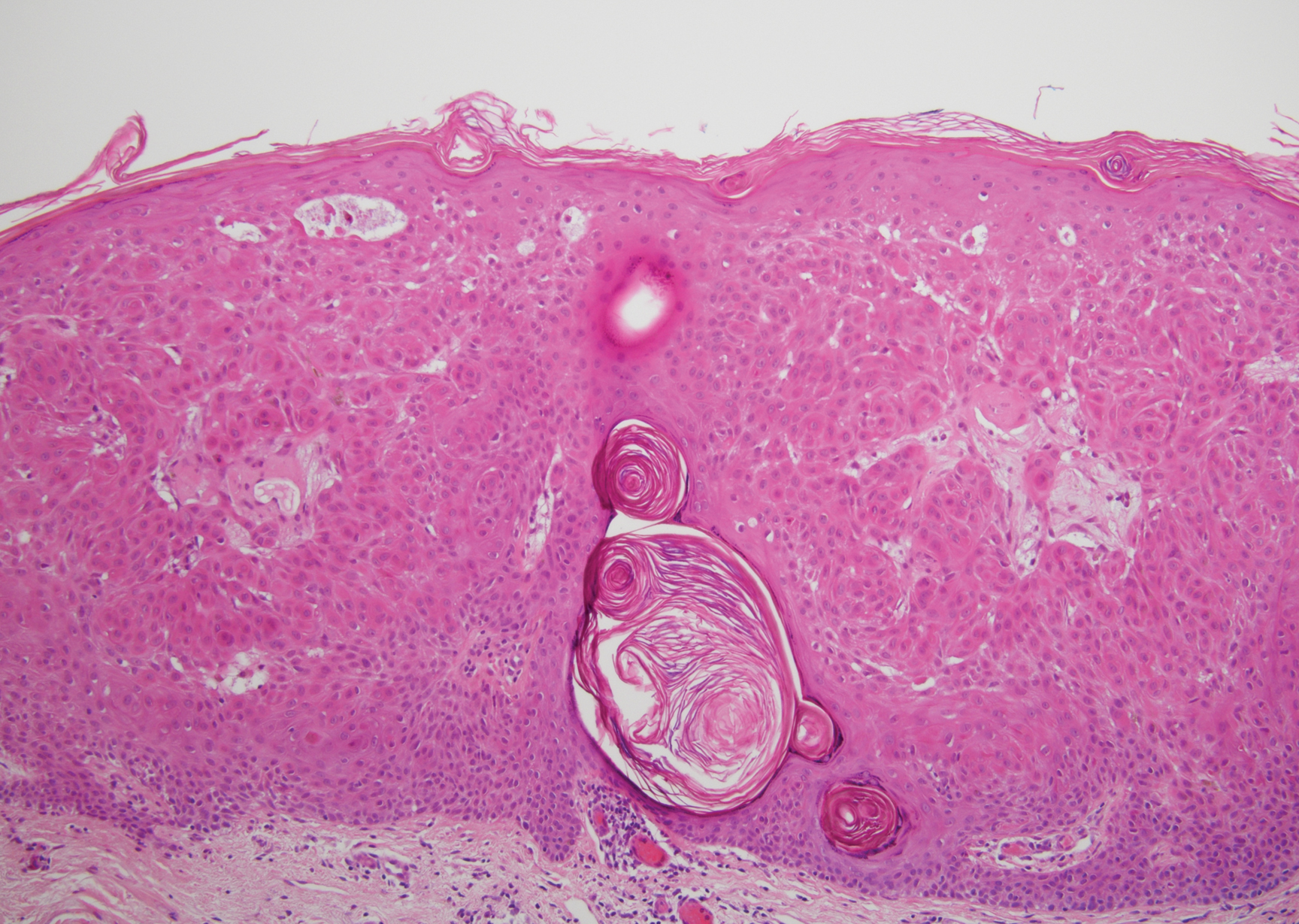
Irritated seborrheic keratosis presents as an exophytic, waxy, dark, sharply demarcated plaque with a stuck-on appearance.6 There is visible keratinization with comedolike openings, fissures and ridges, and scale; it also can contain milialike cysts. Histopathologically there is papillomatosis with prominent rete ridges, often including keratin pseudohorn cysts and squamous eddies. Enlarged capillaries can be seen in the dermal papillae. There is normal cytology with benign sheets of basaloid cells (Figure 2).7 Activating mutation in fibroblast growth factor receptor 3 leads to the growth and thickness of the epidermis that has been identified in these benign lesions.8
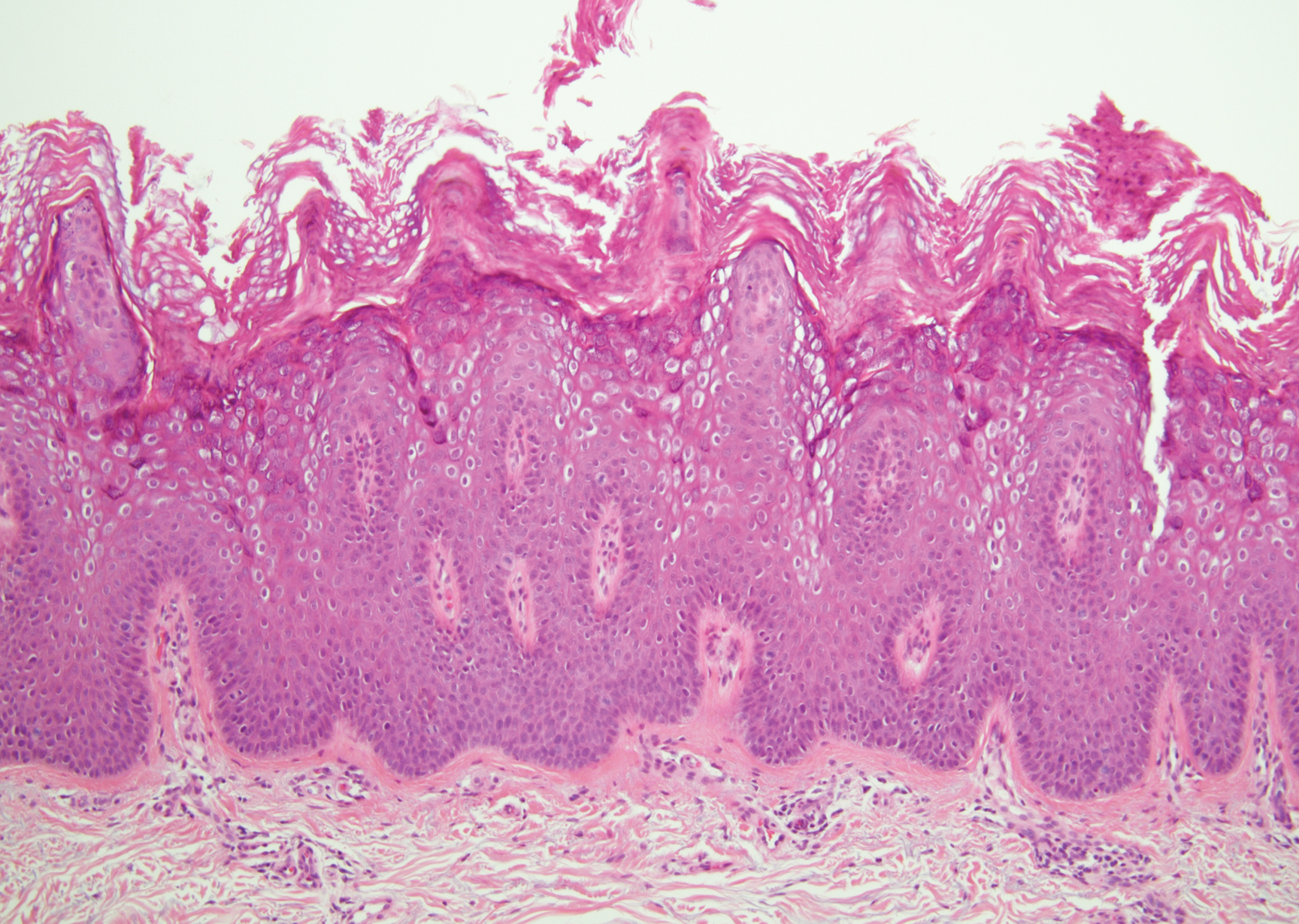
Verruca plana appears as a flesh-colored or reddish, warty, flat-topped papule that often forms clusters. Histopathologically it shows prominent hypergranulosis, thickened stratum spinosum, and vacuolized keratinocytes.9 The nuclei demonstrate a characteristic cytopathic effect of the virion, blurring the nuclear chromatin due to viral particle accumulation, known as koilocytes (Figure 3). The cause is the double-stranded DNA human papillomavirus types 2, 3, and 10.10
Bowen disease is a form of squamous cell carcinoma in situ characterized by an enlarging, well-demarcated, erythematous plaque with an irregular border and crusting or scaling. Histopathology reveals pleomorphic epidermal keratinization that becomes incorporated in the stratum corneum as parakeratotic nuclei. There is acanthosis, elongation of the rete ridges, and disorganized keratinocytes with atypia.11 The granular and spinous layers show an atypical honeycomb pattern with atypical cellular morphology (Figure 4).12 Bowen disease is a malignant lesion commonly found in older adults on sun-exposed skin that can evolve into invasive squamous cell carcinoma.
- Roy SF, Ko CJ, Moeckel GW, et al. Hypergranulotic dyscornification: 30 cases of a striking epithelial reaction pattern. J Cutan Pathol. 2019;46:742-747.
- Dohse L, Elston D, Lountzis N, et al. Benign hypergranulotic keratosis with dyscornification. J Am Acad Dermatol. 2010;62:AB52.
- Reichel M. Hypergranulotic dyscornification. Am J Dermatopathol. 1999;21:21-24.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Peter Rout D, Nair A, Gupta A, et al. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol. 2019;12:333-344.
- Ingraffea A. Benign skin neoplasms. Facial Plast Surg Clin North Am. 2013;21:21-32.
- Braun R. Dermoscopy of pigmented seborrheic keratosis. Arch Dermatol. 2002;138:1556.
- Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551-1559.
- Liu H, Chen S, Zhang F, et al. Seborrheic keratosis or verruca plana? a pilot study with confocal laser scanning microscopy. Skin Res Technol. 2010;16:408-412.
- Prieto-Granada CN, Lobo AZC, Mihm MC. Skin infections. In: Kradin RL, ed. Diagnostic Pathology of Infectious Disease. Philadelphia, PA: Saunders Elsevier; 2010:519-616.
- DeCoste R, Moss P, Boutilier R, et al. Bowen disease with invasive mucin-secreting sweat gland differentiation: report of a case and review of the literature. J Cutan Pathol. 2019;46:425-430.
- Ulrich M, Kanitakis J, González S, et al. Evaluation of Bowen disease by in vivo reflectance confocal microscopy. Br J Dermatol. 2011;166:451-453.
The Diagnosis: Hypergranulotic Dyscornification
Hypergranulotic dyscornification (HD) is a rarely reported reaction pattern present in benign solitary keratoses with only few reports to date. It may be an underrecognized reaction pattern based on the paucity of reported cases as well as the histologic similarities to other entities. It has been hypothesized that this pattern reflects an underlying keratin mutation or disorder of keratinization.1
Clinically, HD most commonly presents as a waxy, tan-colored, solitary keratosis generally found on the lower limbs, trunk, or back in individuals aged 20 to 60 years.1,2 Histopathology shows marked hyperkeratosis, papillomatosis, and clumped basophilic keratohyalin granules within the corneocytes with digitated epidermal hyperplasia. There is abnormal cornification across the entire lesion with papillomatosis and marked hypergranulosis.3 There often are homogeneous orthokeratotic mounds of large, dull, eosinophilic-staining anucleate keratinocytes that are sharply demarcated from the thickened granular layer.1,2 Within the spinous, granular, and corneal layers, there is a pale, gray-staining, basophilic, cytoplasmic substance intercellularly.1
Histopathologically, HD may be mistaken for several other entities both benign and malignant.1 Epidermolytic hyperkeratosis can be a genetic disorder, an incidental finding in a variety of skin conditions, or an isolated lesion.4 The genetic syndrome, caused by mutation in keratins 1 or 10, clinically presents with hyperkeratosis, erosions, blisters, and thickening of the epidermis, often with a corrugated appearance. Epidermal nevi findings often are seen in conjunction with histologic changes of epidermolytic hyperkeratosis caused by mutation. Solitary lesions also can resemble seborrheic keratosis or verruca. In all examples of epidermolytic hyperkeratosis, the histopathologic findings are identical.4 The granular layer is thickened, and coarse keratohyalin granules aggregate in the suprabasal cells.5 There is acantholysis with perinuclear vacuolization in the spinous and granular layers with characteristic pale cytoplasmic areas devoid of keratin filaments (Figure 1). The basal layer may be hyperproliferative.5
Irritated seborrheic keratosis presents as an exophytic, waxy, dark, sharply demarcated plaque with a stuck-on appearance.6 There is visible keratinization with comedolike openings, fissures and ridges, and scale; it also can contain milialike cysts. Histopathologically there is papillomatosis with prominent rete ridges, often including keratin pseudohorn cysts and squamous eddies. Enlarged capillaries can be seen in the dermal papillae. There is normal cytology with benign sheets of basaloid cells (Figure 2).7 Activating mutation in fibroblast growth factor receptor 3 leads to the growth and thickness of the epidermis that has been identified in these benign lesions.8
Verruca plana appears as a flesh-colored or reddish, warty, flat-topped papule that often forms clusters. Histopathologically it shows prominent hypergranulosis, thickened stratum spinosum, and vacuolized keratinocytes.9 The nuclei demonstrate a characteristic cytopathic effect of the virion, blurring the nuclear chromatin due to viral particle accumulation, known as koilocytes (Figure 3). The cause is the double-stranded DNA human papillomavirus types 2, 3, and 10.10
Bowen disease is a form of squamous cell carcinoma in situ characterized by an enlarging, well-demarcated, erythematous plaque with an irregular border and crusting or scaling. Histopathology reveals pleomorphic epidermal keratinization that becomes incorporated in the stratum corneum as parakeratotic nuclei. There is acanthosis, elongation of the rete ridges, and disorganized keratinocytes with atypia.11 The granular and spinous layers show an atypical honeycomb pattern with atypical cellular morphology (Figure 4).12 Bowen disease is a malignant lesion commonly found in older adults on sun-exposed skin that can evolve into invasive squamous cell carcinoma.
The Diagnosis: Hypergranulotic Dyscornification
Hypergranulotic dyscornification (HD) is a rarely reported reaction pattern present in benign solitary keratoses with only few reports to date. It may be an underrecognized reaction pattern based on the paucity of reported cases as well as the histologic similarities to other entities. It has been hypothesized that this pattern reflects an underlying keratin mutation or disorder of keratinization.1
Clinically, HD most commonly presents as a waxy, tan-colored, solitary keratosis generally found on the lower limbs, trunk, or back in individuals aged 20 to 60 years.1,2 Histopathology shows marked hyperkeratosis, papillomatosis, and clumped basophilic keratohyalin granules within the corneocytes with digitated epidermal hyperplasia. There is abnormal cornification across the entire lesion with papillomatosis and marked hypergranulosis.3 There often are homogeneous orthokeratotic mounds of large, dull, eosinophilic-staining anucleate keratinocytes that are sharply demarcated from the thickened granular layer.1,2 Within the spinous, granular, and corneal layers, there is a pale, gray-staining, basophilic, cytoplasmic substance intercellularly.1
Histopathologically, HD may be mistaken for several other entities both benign and malignant.1 Epidermolytic hyperkeratosis can be a genetic disorder, an incidental finding in a variety of skin conditions, or an isolated lesion.4 The genetic syndrome, caused by mutation in keratins 1 or 10, clinically presents with hyperkeratosis, erosions, blisters, and thickening of the epidermis, often with a corrugated appearance. Epidermal nevi findings often are seen in conjunction with histologic changes of epidermolytic hyperkeratosis caused by mutation. Solitary lesions also can resemble seborrheic keratosis or verruca. In all examples of epidermolytic hyperkeratosis, the histopathologic findings are identical.4 The granular layer is thickened, and coarse keratohyalin granules aggregate in the suprabasal cells.5 There is acantholysis with perinuclear vacuolization in the spinous and granular layers with characteristic pale cytoplasmic areas devoid of keratin filaments (Figure 1). The basal layer may be hyperproliferative.5
Irritated seborrheic keratosis presents as an exophytic, waxy, dark, sharply demarcated plaque with a stuck-on appearance.6 There is visible keratinization with comedolike openings, fissures and ridges, and scale; it also can contain milialike cysts. Histopathologically there is papillomatosis with prominent rete ridges, often including keratin pseudohorn cysts and squamous eddies. Enlarged capillaries can be seen in the dermal papillae. There is normal cytology with benign sheets of basaloid cells (Figure 2).7 Activating mutation in fibroblast growth factor receptor 3 leads to the growth and thickness of the epidermis that has been identified in these benign lesions.8
Verruca plana appears as a flesh-colored or reddish, warty, flat-topped papule that often forms clusters. Histopathologically it shows prominent hypergranulosis, thickened stratum spinosum, and vacuolized keratinocytes.9 The nuclei demonstrate a characteristic cytopathic effect of the virion, blurring the nuclear chromatin due to viral particle accumulation, known as koilocytes (Figure 3). The cause is the double-stranded DNA human papillomavirus types 2, 3, and 10.10
Bowen disease is a form of squamous cell carcinoma in situ characterized by an enlarging, well-demarcated, erythematous plaque with an irregular border and crusting or scaling. Histopathology reveals pleomorphic epidermal keratinization that becomes incorporated in the stratum corneum as parakeratotic nuclei. There is acanthosis, elongation of the rete ridges, and disorganized keratinocytes with atypia.11 The granular and spinous layers show an atypical honeycomb pattern with atypical cellular morphology (Figure 4).12 Bowen disease is a malignant lesion commonly found in older adults on sun-exposed skin that can evolve into invasive squamous cell carcinoma.
- Roy SF, Ko CJ, Moeckel GW, et al. Hypergranulotic dyscornification: 30 cases of a striking epithelial reaction pattern. J Cutan Pathol. 2019;46:742-747.
- Dohse L, Elston D, Lountzis N, et al. Benign hypergranulotic keratosis with dyscornification. J Am Acad Dermatol. 2010;62:AB52.
- Reichel M. Hypergranulotic dyscornification. Am J Dermatopathol. 1999;21:21-24.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Peter Rout D, Nair A, Gupta A, et al. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol. 2019;12:333-344.
- Ingraffea A. Benign skin neoplasms. Facial Plast Surg Clin North Am. 2013;21:21-32.
- Braun R. Dermoscopy of pigmented seborrheic keratosis. Arch Dermatol. 2002;138:1556.
- Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551-1559.
- Liu H, Chen S, Zhang F, et al. Seborrheic keratosis or verruca plana? a pilot study with confocal laser scanning microscopy. Skin Res Technol. 2010;16:408-412.
- Prieto-Granada CN, Lobo AZC, Mihm MC. Skin infections. In: Kradin RL, ed. Diagnostic Pathology of Infectious Disease. Philadelphia, PA: Saunders Elsevier; 2010:519-616.
- DeCoste R, Moss P, Boutilier R, et al. Bowen disease with invasive mucin-secreting sweat gland differentiation: report of a case and review of the literature. J Cutan Pathol. 2019;46:425-430.
- Ulrich M, Kanitakis J, González S, et al. Evaluation of Bowen disease by in vivo reflectance confocal microscopy. Br J Dermatol. 2011;166:451-453.
- Roy SF, Ko CJ, Moeckel GW, et al. Hypergranulotic dyscornification: 30 cases of a striking epithelial reaction pattern. J Cutan Pathol. 2019;46:742-747.
- Dohse L, Elston D, Lountzis N, et al. Benign hypergranulotic keratosis with dyscornification. J Am Acad Dermatol. 2010;62:AB52.
- Reichel M. Hypergranulotic dyscornification. Am J Dermatopathol. 1999;21:21-24.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Peter Rout D, Nair A, Gupta A, et al. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol. 2019;12:333-344.
- Ingraffea A. Benign skin neoplasms. Facial Plast Surg Clin North Am. 2013;21:21-32.
- Braun R. Dermoscopy of pigmented seborrheic keratosis. Arch Dermatol. 2002;138:1556.
- Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551-1559.
- Liu H, Chen S, Zhang F, et al. Seborrheic keratosis or verruca plana? a pilot study with confocal laser scanning microscopy. Skin Res Technol. 2010;16:408-412.
- Prieto-Granada CN, Lobo AZC, Mihm MC. Skin infections. In: Kradin RL, ed. Diagnostic Pathology of Infectious Disease. Philadelphia, PA: Saunders Elsevier; 2010:519-616.
- DeCoste R, Moss P, Boutilier R, et al. Bowen disease with invasive mucin-secreting sweat gland differentiation: report of a case and review of the literature. J Cutan Pathol. 2019;46:425-430.
- Ulrich M, Kanitakis J, González S, et al. Evaluation of Bowen disease by in vivo reflectance confocal microscopy. Br J Dermatol. 2011;166:451-453.
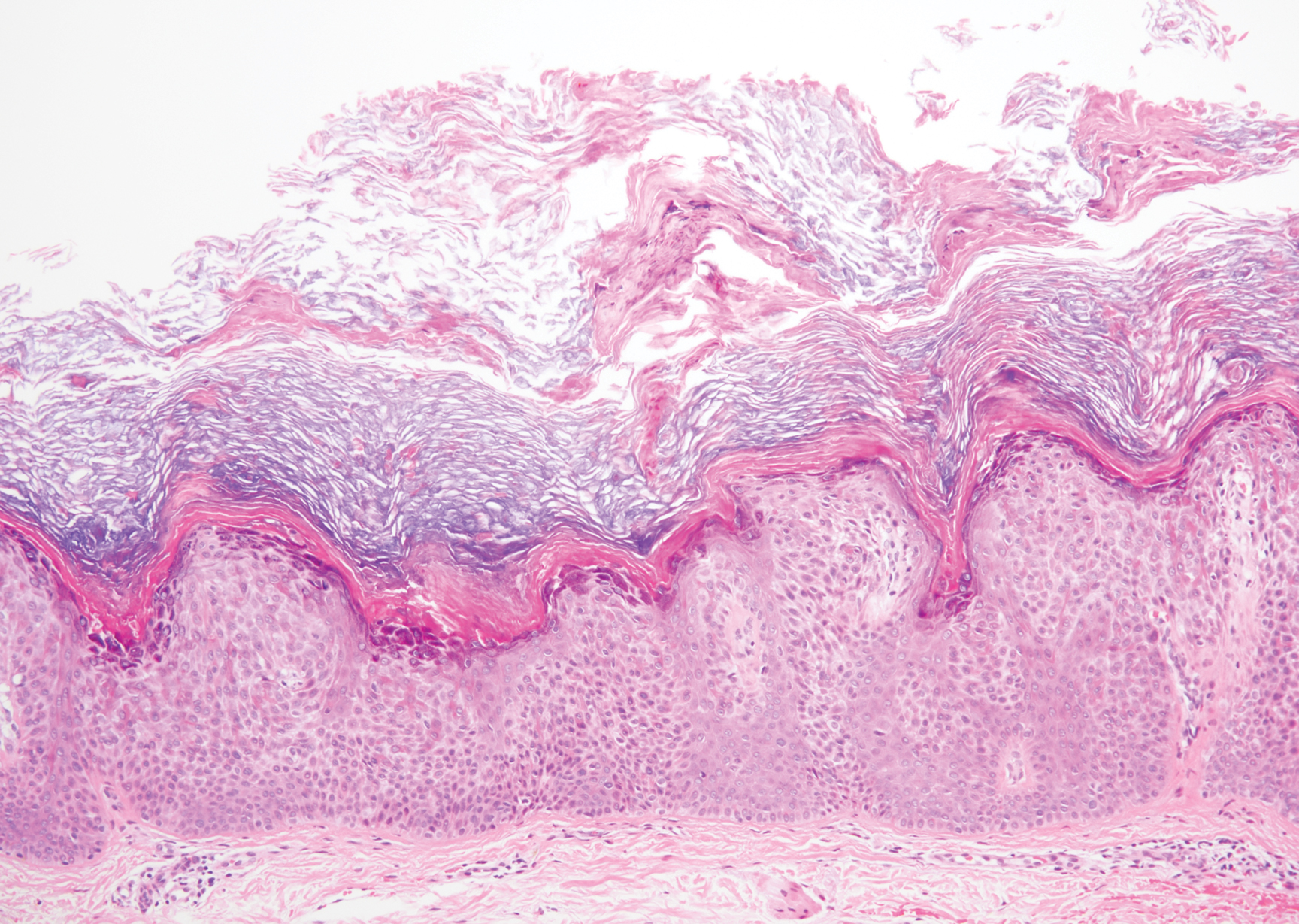
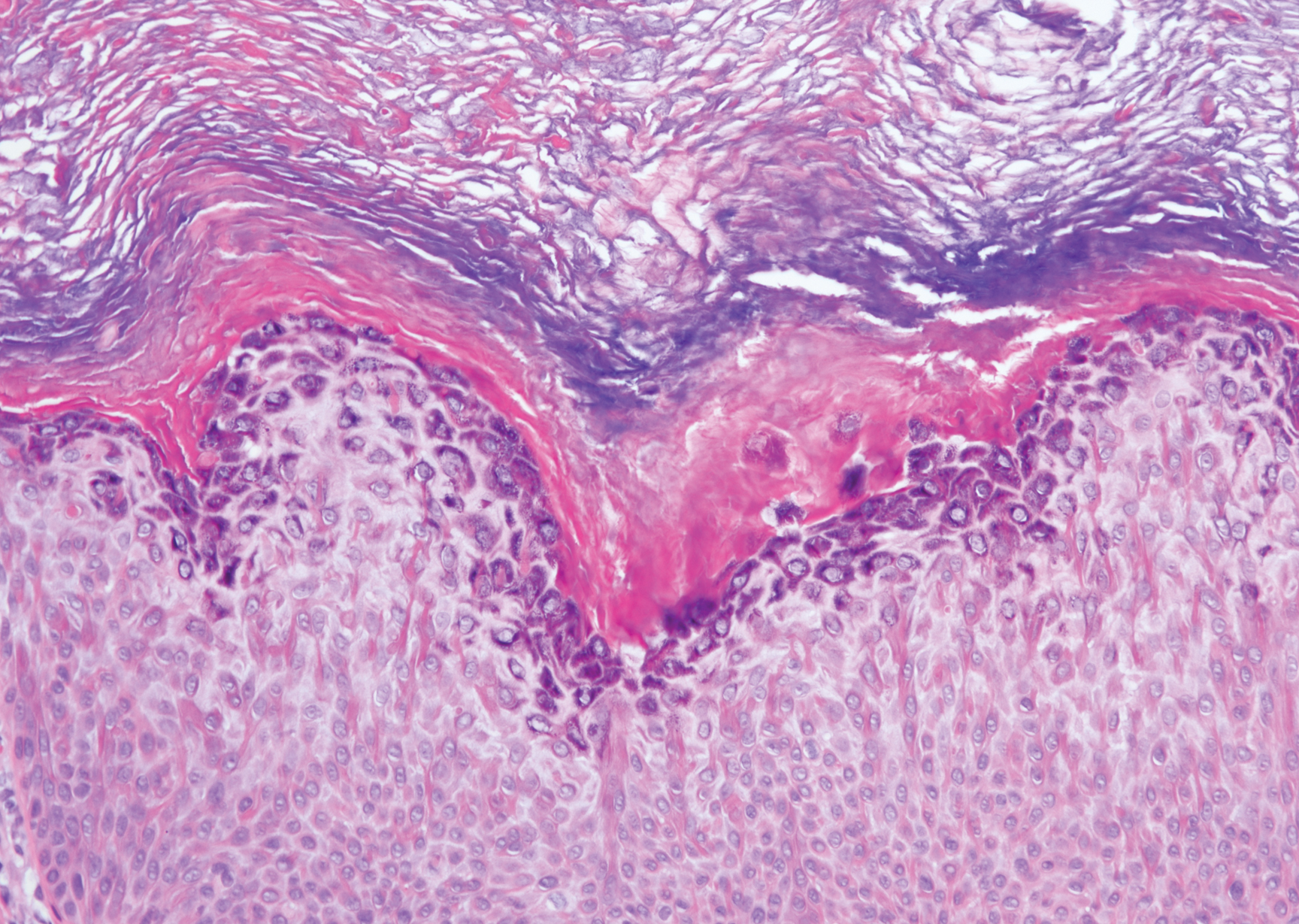
A 59-year-old woman with a history of basal cell carcinoma, uterine and ovarian cancer, and verrucae presented with an asymptomatic 3-mm lesion on the left side of the lower abdomen. Physical examination revealed a waxy, tan-colored, solitary keratosis. A shave biopsy was performed. Histopathology showed hyperkeratosis, focal parakeratosis, papillomatosis, and marked hypergranulosis with pale gray cytoplasm of the spinous-layer keratinocytes.
Pseudoepitheliomatous Hyperplasia Arising From Purple Tattoo Pigment
To the Editor:
Pseudoepitheliomatous hyperplasia (PEH) is an uncommon type of reactive epidermal proliferation that can occur from a variety of causes, including an underlying infection, inflammation, neoplastic condition, or trauma induced from tattooing.1 Diagnosis can be challenging and requires clinicopathologic correlation, as PEH can mimic malignancy on histopathology.2-4 Histologically, PEH shows irregular hyperplasia of the epidermis and adnexal epithelium, elongation of the rete ridges, and extension of the reactive proliferation into the dermis. Absence of cytologic atypia is key to the diagnosis of PEH, helping to distinguish it from squamous cell carcinoma and keratoacanthoma. Clinically, patients typically present with well-demarcated, erythematous, scaly plaques or nodules in reactive areas, which can be symptomatically pruritic.
A 48-year-old woman presented with scaly and crusted verrucous plaques of 2 months’ duration that were isolated to the areas of purple pigment within a tattoo on the right lower leg. The patient reported pruritus in the affected areas that occurred immediately after obtaining the tattoo, which was her first and only tattoo. She denied any pertinent medical history, including an absence of immunosuppression and autoimmune or chronic inflammatory diseases.
Physical examination revealed scaly and crusted plaques isolated to areas of purple tattoo pigment (Figure 1). Areas of red, green, black, and blue pigmentation within the tattoo were uninvolved. With the initial suspicion of allergic contact dermatitis, two 6-mm punch biopsies were taken from adjacent linear plaques on the right leg for histology and tissue culture. Histopathologic evaluation revealed dermal tattoo pigment with overlying PEH and was negative for signs of infection (Figure 2). Infectious stains such as periodic acid–Schiff, Grocott-Gomori methenamine-silver, and Gram stains were performed and found to be negative. In addition, culture for mycobacteria came back negative. Prurigo was on the differential; however, histopathologic changes were more compatible with a PEH reaction to the tattoo.


Upon diagnosis, the patient was treated with clobetasol ointment 0.05% under occlusion for 1 month without reported improvement. The patient subsequently elected to undergo treatment with intralesional triamcinolone 5 mg/mL to all areas of PEH, except the areas immediately surrounding the healing biopsy sites. Twice-daily application of tacrolimus ointment 0.1% to all affected areas also was initiated. At follow-up 1 month later, she reported symptomatic relief of pruritus with a notable reduction in the thickness of the plaques in all treated areas (Figure 3). A second course of intralesional triamcinolone 5 mg/mL was performed. No additional plaques appeared during the treatment course, and the patient reported high satisfaction with the final result that was achieved.
An increase in the popularity of tattooing has led to more reports of various tattoo skin reactions.4-6 The differential diagnosis is broad for tattoo reactions and includes granulomatous inflammation, sarcoidosis, psoriasis (Köbner phenomenon), allergic contact dermatitis, lichen planus, morphealike reactions, squamous cell carcinoma, and keratoacanthoma,5 which makes clinicopathologic correlation essential for accurate diagnosis. Our case demonstrated the characteristic epithelial hyperplasia in the absence of cytologic atypia. In addition, the presence of mixed dermal inflammation histologically was noted in our patient.

Pseudoepitheliomatous hyperplasia development from a tattoo in areas of both mercury-based and non–mercury-based red pigment is a known association.7-9 Balfour et al10 also reported a case of PEH occurring secondary to manganese-based purple pigment. Because few cases have been reported, the epidemiology for PEH currently is unknown. Treatment of this condition primarily is anecdotal, with prior cases showing success with topical or intralesional steroids.5,7 As with any steroid-based treatment, we recommend less aggressive treatments initially with close follow-up and adaptation as needed to minimize adverse effects such as unwanted atrophy. Some success has been reported with the use of the Q-switched Nd:YAG laser in the setting of a PEH tattoo reaction.5 Similar to other tattoo reactions, surgical removal can be considered with failure of more conservative treatment methods and focal involvement.
We report an unusual case of PEH occurring secondary to purple tattoo pigment. Our report also demonstrates the clinical and symptomatic improvement of PEH that can be achieved through the use of intralesional corticosteroid therapy. Our patient represents a case of PEH reactive to tattooing with purple ink. Further research to elucidate the precise pathogenesis of PEH tattoo reactions would be helpful in identifying high-risk patients and determining the most efficacious treatments.
- Meani RE, Nixon RL, O’Keefe R, et al. Pseudoepitheliomatous hyperplasia secondary to allergic contact dermatitis to Grevillea Robyn Gordon. Australas J Dermatol. 2017;58:E8-E10.
- Chakrabarti S, Chakrabarti P, Agrawal D, et al. Pseudoepitheliomatous hyperplasia: a clinical entity mistaken for squamous cell carcinoma. J Cutan Aesthet Surg. 2014;7:232.
- Kluger N. Issues with keratoacanthoma, pseudoepitheliomatous hyperplasia and squamous cell carcinoma within tattoos: a clinical point of view. J Cutan Pathol. 2009;37:812-813.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-126.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Serup J. Diagnostic tools for doctors’ evaluation of tattoo complications. Curr Probl Dermatol. 2017;52:42-57.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Kluger N, Durand L, Minier-Thoumin C, et al. Pseudoepitheliomatous epidermal hyperplasia in tattoos: report of three cases. Am J Clin Dermatol. 2008;9:337-340.
- Cui W, McGregor DH, Stark SP, et al. Pseudoepitheliomatous hyperplasia—an unusual reaction following tattoo: report of a case and review of the literature. Int J Dermatol. 2007;46:743-745.
- Balfour E, Olhoffer I, Leffell D, et al. Massive pseudoepitheliomatous hyperplasia: an unusual reaction to a tattoo. Am J Dermatopathol. 2003;25:338-340.
To the Editor:
Pseudoepitheliomatous hyperplasia (PEH) is an uncommon type of reactive epidermal proliferation that can occur from a variety of causes, including an underlying infection, inflammation, neoplastic condition, or trauma induced from tattooing.1 Diagnosis can be challenging and requires clinicopathologic correlation, as PEH can mimic malignancy on histopathology.2-4 Histologically, PEH shows irregular hyperplasia of the epidermis and adnexal epithelium, elongation of the rete ridges, and extension of the reactive proliferation into the dermis. Absence of cytologic atypia is key to the diagnosis of PEH, helping to distinguish it from squamous cell carcinoma and keratoacanthoma. Clinically, patients typically present with well-demarcated, erythematous, scaly plaques or nodules in reactive areas, which can be symptomatically pruritic.
A 48-year-old woman presented with scaly and crusted verrucous plaques of 2 months’ duration that were isolated to the areas of purple pigment within a tattoo on the right lower leg. The patient reported pruritus in the affected areas that occurred immediately after obtaining the tattoo, which was her first and only tattoo. She denied any pertinent medical history, including an absence of immunosuppression and autoimmune or chronic inflammatory diseases.
Physical examination revealed scaly and crusted plaques isolated to areas of purple tattoo pigment (Figure 1). Areas of red, green, black, and blue pigmentation within the tattoo were uninvolved. With the initial suspicion of allergic contact dermatitis, two 6-mm punch biopsies were taken from adjacent linear plaques on the right leg for histology and tissue culture. Histopathologic evaluation revealed dermal tattoo pigment with overlying PEH and was negative for signs of infection (Figure 2). Infectious stains such as periodic acid–Schiff, Grocott-Gomori methenamine-silver, and Gram stains were performed and found to be negative. In addition, culture for mycobacteria came back negative. Prurigo was on the differential; however, histopathologic changes were more compatible with a PEH reaction to the tattoo.
Upon diagnosis, the patient was treated with clobetasol ointment 0.05% under occlusion for 1 month without reported improvement. The patient subsequently elected to undergo treatment with intralesional triamcinolone 5 mg/mL to all areas of PEH, except the areas immediately surrounding the healing biopsy sites. Twice-daily application of tacrolimus ointment 0.1% to all affected areas also was initiated. At follow-up 1 month later, she reported symptomatic relief of pruritus with a notable reduction in the thickness of the plaques in all treated areas (Figure 3). A second course of intralesional triamcinolone 5 mg/mL was performed. No additional plaques appeared during the treatment course, and the patient reported high satisfaction with the final result that was achieved.
An increase in the popularity of tattooing has led to more reports of various tattoo skin reactions.4-6 The differential diagnosis is broad for tattoo reactions and includes granulomatous inflammation, sarcoidosis, psoriasis (Köbner phenomenon), allergic contact dermatitis, lichen planus, morphealike reactions, squamous cell carcinoma, and keratoacanthoma,5 which makes clinicopathologic correlation essential for accurate diagnosis. Our case demonstrated the characteristic epithelial hyperplasia in the absence of cytologic atypia. In addition, the presence of mixed dermal inflammation histologically was noted in our patient.
Pseudoepitheliomatous hyperplasia development from a tattoo in areas of both mercury-based and non–mercury-based red pigment is a known association.7-9 Balfour et al10 also reported a case of PEH occurring secondary to manganese-based purple pigment. Because few cases have been reported, the epidemiology for PEH currently is unknown. Treatment of this condition primarily is anecdotal, with prior cases showing success with topical or intralesional steroids.5,7 As with any steroid-based treatment, we recommend less aggressive treatments initially with close follow-up and adaptation as needed to minimize adverse effects such as unwanted atrophy. Some success has been reported with the use of the Q-switched Nd:YAG laser in the setting of a PEH tattoo reaction.5 Similar to other tattoo reactions, surgical removal can be considered with failure of more conservative treatment methods and focal involvement.
We report an unusual case of PEH occurring secondary to purple tattoo pigment. Our report also demonstrates the clinical and symptomatic improvement of PEH that can be achieved through the use of intralesional corticosteroid therapy. Our patient represents a case of PEH reactive to tattooing with purple ink. Further research to elucidate the precise pathogenesis of PEH tattoo reactions would be helpful in identifying high-risk patients and determining the most efficacious treatments.
To the Editor:
Pseudoepitheliomatous hyperplasia (PEH) is an uncommon type of reactive epidermal proliferation that can occur from a variety of causes, including an underlying infection, inflammation, neoplastic condition, or trauma induced from tattooing.1 Diagnosis can be challenging and requires clinicopathologic correlation, as PEH can mimic malignancy on histopathology.2-4 Histologically, PEH shows irregular hyperplasia of the epidermis and adnexal epithelium, elongation of the rete ridges, and extension of the reactive proliferation into the dermis. Absence of cytologic atypia is key to the diagnosis of PEH, helping to distinguish it from squamous cell carcinoma and keratoacanthoma. Clinically, patients typically present with well-demarcated, erythematous, scaly plaques or nodules in reactive areas, which can be symptomatically pruritic.
A 48-year-old woman presented with scaly and crusted verrucous plaques of 2 months’ duration that were isolated to the areas of purple pigment within a tattoo on the right lower leg. The patient reported pruritus in the affected areas that occurred immediately after obtaining the tattoo, which was her first and only tattoo. She denied any pertinent medical history, including an absence of immunosuppression and autoimmune or chronic inflammatory diseases.
Physical examination revealed scaly and crusted plaques isolated to areas of purple tattoo pigment (Figure 1). Areas of red, green, black, and blue pigmentation within the tattoo were uninvolved. With the initial suspicion of allergic contact dermatitis, two 6-mm punch biopsies were taken from adjacent linear plaques on the right leg for histology and tissue culture. Histopathologic evaluation revealed dermal tattoo pigment with overlying PEH and was negative for signs of infection (Figure 2). Infectious stains such as periodic acid–Schiff, Grocott-Gomori methenamine-silver, and Gram stains were performed and found to be negative. In addition, culture for mycobacteria came back negative. Prurigo was on the differential; however, histopathologic changes were more compatible with a PEH reaction to the tattoo.
Upon diagnosis, the patient was treated with clobetasol ointment 0.05% under occlusion for 1 month without reported improvement. The patient subsequently elected to undergo treatment with intralesional triamcinolone 5 mg/mL to all areas of PEH, except the areas immediately surrounding the healing biopsy sites. Twice-daily application of tacrolimus ointment 0.1% to all affected areas also was initiated. At follow-up 1 month later, she reported symptomatic relief of pruritus with a notable reduction in the thickness of the plaques in all treated areas (Figure 3). A second course of intralesional triamcinolone 5 mg/mL was performed. No additional plaques appeared during the treatment course, and the patient reported high satisfaction with the final result that was achieved.
An increase in the popularity of tattooing has led to more reports of various tattoo skin reactions.4-6 The differential diagnosis is broad for tattoo reactions and includes granulomatous inflammation, sarcoidosis, psoriasis (Köbner phenomenon), allergic contact dermatitis, lichen planus, morphealike reactions, squamous cell carcinoma, and keratoacanthoma,5 which makes clinicopathologic correlation essential for accurate diagnosis. Our case demonstrated the characteristic epithelial hyperplasia in the absence of cytologic atypia. In addition, the presence of mixed dermal inflammation histologically was noted in our patient.
Pseudoepitheliomatous hyperplasia development from a tattoo in areas of both mercury-based and non–mercury-based red pigment is a known association.7-9 Balfour et al10 also reported a case of PEH occurring secondary to manganese-based purple pigment. Because few cases have been reported, the epidemiology for PEH currently is unknown. Treatment of this condition primarily is anecdotal, with prior cases showing success with topical or intralesional steroids.5,7 As with any steroid-based treatment, we recommend less aggressive treatments initially with close follow-up and adaptation as needed to minimize adverse effects such as unwanted atrophy. Some success has been reported with the use of the Q-switched Nd:YAG laser in the setting of a PEH tattoo reaction.5 Similar to other tattoo reactions, surgical removal can be considered with failure of more conservative treatment methods and focal involvement.
We report an unusual case of PEH occurring secondary to purple tattoo pigment. Our report also demonstrates the clinical and symptomatic improvement of PEH that can be achieved through the use of intralesional corticosteroid therapy. Our patient represents a case of PEH reactive to tattooing with purple ink. Further research to elucidate the precise pathogenesis of PEH tattoo reactions would be helpful in identifying high-risk patients and determining the most efficacious treatments.
- Meani RE, Nixon RL, O’Keefe R, et al. Pseudoepitheliomatous hyperplasia secondary to allergic contact dermatitis to Grevillea Robyn Gordon. Australas J Dermatol. 2017;58:E8-E10.
- Chakrabarti S, Chakrabarti P, Agrawal D, et al. Pseudoepitheliomatous hyperplasia: a clinical entity mistaken for squamous cell carcinoma. J Cutan Aesthet Surg. 2014;7:232.
- Kluger N. Issues with keratoacanthoma, pseudoepitheliomatous hyperplasia and squamous cell carcinoma within tattoos: a clinical point of view. J Cutan Pathol. 2009;37:812-813.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-126.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Serup J. Diagnostic tools for doctors’ evaluation of tattoo complications. Curr Probl Dermatol. 2017;52:42-57.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Kluger N, Durand L, Minier-Thoumin C, et al. Pseudoepitheliomatous epidermal hyperplasia in tattoos: report of three cases. Am J Clin Dermatol. 2008;9:337-340.
- Cui W, McGregor DH, Stark SP, et al. Pseudoepitheliomatous hyperplasia—an unusual reaction following tattoo: report of a case and review of the literature. Int J Dermatol. 2007;46:743-745.
- Balfour E, Olhoffer I, Leffell D, et al. Massive pseudoepitheliomatous hyperplasia: an unusual reaction to a tattoo. Am J Dermatopathol. 2003;25:338-340.
- Meani RE, Nixon RL, O’Keefe R, et al. Pseudoepitheliomatous hyperplasia secondary to allergic contact dermatitis to Grevillea Robyn Gordon. Australas J Dermatol. 2017;58:E8-E10.
- Chakrabarti S, Chakrabarti P, Agrawal D, et al. Pseudoepitheliomatous hyperplasia: a clinical entity mistaken for squamous cell carcinoma. J Cutan Aesthet Surg. 2014;7:232.
- Kluger N. Issues with keratoacanthoma, pseudoepitheliomatous hyperplasia and squamous cell carcinoma within tattoos: a clinical point of view. J Cutan Pathol. 2009;37:812-813.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-126.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Serup J. Diagnostic tools for doctors’ evaluation of tattoo complications. Curr Probl Dermatol. 2017;52:42-57.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Kluger N, Durand L, Minier-Thoumin C, et al. Pseudoepitheliomatous epidermal hyperplasia in tattoos: report of three cases. Am J Clin Dermatol. 2008;9:337-340.
- Cui W, McGregor DH, Stark SP, et al. Pseudoepitheliomatous hyperplasia—an unusual reaction following tattoo: report of a case and review of the literature. Int J Dermatol. 2007;46:743-745.
- Balfour E, Olhoffer I, Leffell D, et al. Massive pseudoepitheliomatous hyperplasia: an unusual reaction to a tattoo. Am J Dermatopathol. 2003;25:338-340.
Practice Points
- Pseudoepitheliomatous hyperplasia (PEH) is a rare benign condition that can arise in response to multiple underlying triggers such as tattoo pigment.
- Histopathologic evaluation is essential for diagnosis and shows characteristic hyperplasia of the epidermis.
- Clinicians should consider intralesional steroids in the treatment of PEH once atypical mycobacterial and deep fungal infections have been ruled out.

Pruritic Eruption With Skinfold Sparing
The Diagnosis: Papuloerythroderma of Ofuji
The patient presented with a characteristic finding of skinfold sparing, known as the "deck-chair sign" (Figure 1).1 A repeat biopsy at our institution revealed a dermal perivascular and bandlike infiltrate with lymphocytes and occasional eosinophils (Figure 2). The epidermis showed mild spongiosis, lymphocytic exocytosis, and rare necrotic keratinocytes. A T-cell gene rearrangement assay was negative for a monoclonal population of T lymphocytes. Based on the clinical and histologic features, the diagnosis was most consistent with papuloerythroderma of Ofuji (PEO); however, a lymphoproliferative disorder needed to be excluded. Further workup included a peripheral smear, complete blood cell count with differential, comprehensive metabolic panel, IgE level, and hepatitis panel; all were normal, except for an elevated serum IgE level. Human immunodeficiency virus and age-appropriate malignancy screening were negative. The patient was prescribed betamethasone dipropionate cream 0.05% twice daily, which resulted in near-complete resolution of the rash and marked improvement in pruritus.
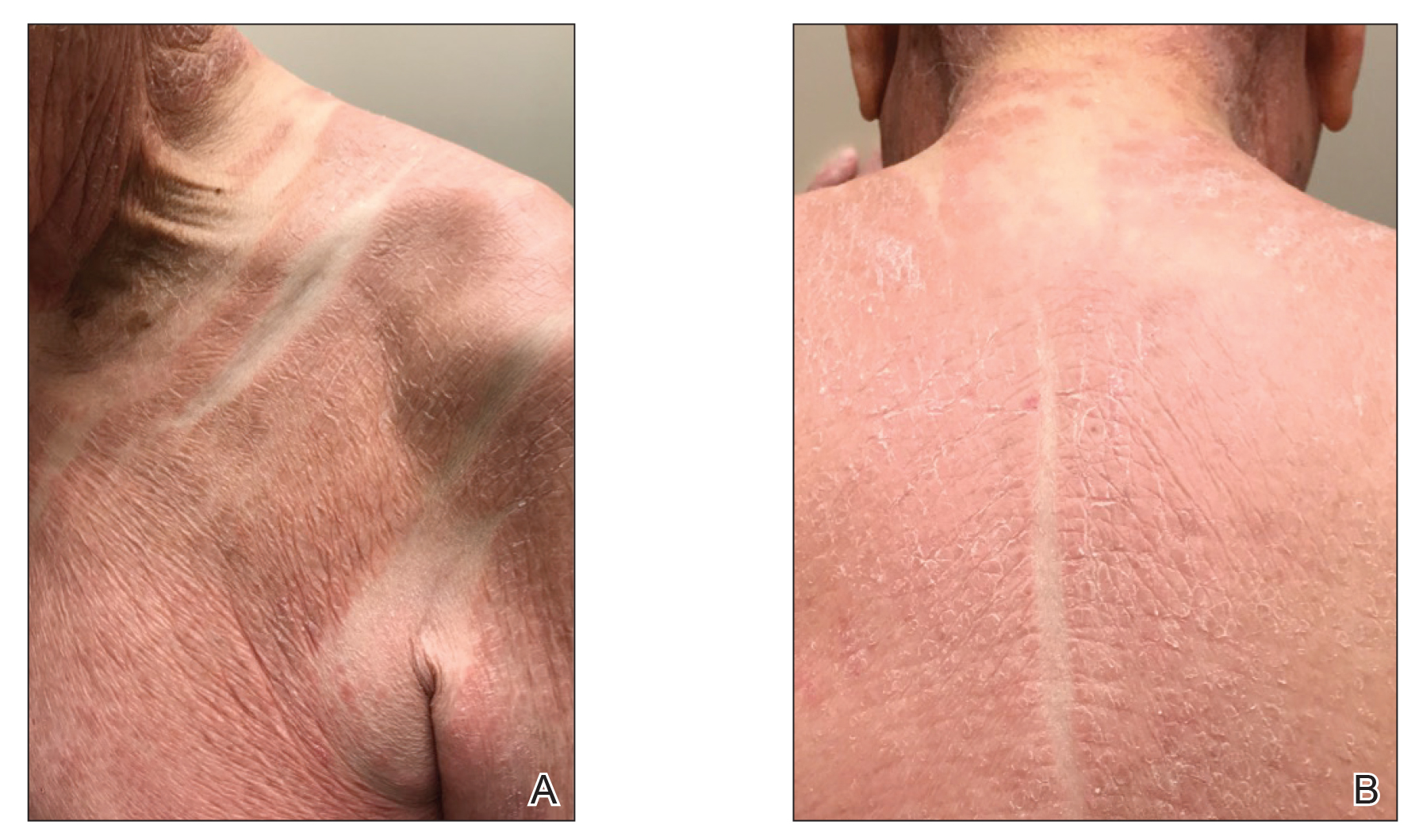
In 1984, PEO was described as an entity of generalized pruritic erythroderma characterized by flat-topped, red to brown, coalescing papules with sparing of the skinfolds, later coined the deck-chair sign.1,2 Papuloerythroderma of Ofuji commonly presents in elderly Asian males with a male to female ratio of 4:1.3 Papuloerythroderma of Ofuji is a T cell-mediated skin disease; however, the etiology of the signature rash remains unclear. One explanation includes circulating factors in the skin that elicit an inflammatory response, which does not occur in areas of external pressure.3 The deck-chair sign may occur more frequently in elderly individuals due to increased skin laxity, which creates crease lines that are spared from rubbing and excoriations.4
Although Ofuji et al2 initially reported 4 idiopathic cases, subsequent authors described PEO in association with other conditions, including cutaneous T-cell lymphoma (CTCL) and atopic diathesis, and infections, as well as secondary to medications. Some authors have suggested that PEO may be an early variant of mycosis fungoides; therefore, physicians should monitor patients closely.5-7 Maher et al6 commented that multiple causative factors including CTCL underlie the development of papuloerythroderma.
In a review of PEO, Torchia et al3 proposed diagnostic criteria and an etiologic classification to address whether PEO represents an independent entity or an unusual manifestation of other dermatoses. They established 4 categories of papuloerythroderma--primary, secondary, papuloerythrodermalike CTCL, and pseudopapuloerythroderma--and proposed that primary PEO is a diagnosis of exclusion. If no secondary association is found, they proposed 10 criteria for primary PEO: 5 major criteria include coalescing flat-topped papules, the deck-chair sign, pruritus, histopathologic exclusion of diseases such as CTCL, and a negative workup to exclude other causes.3 In 2018, Maher et al6 recommended workup to rule out cutaneous malignancy, including skin biopsy, flow cytometry, Sézary cell count, T-cell rearrangement, lactate dehydrogenase, and human T-cell lymphotropic virus 1. The 5 minor criteria proposed by Torchia et al3 include age older than 55 years, male sex, eosinophilia, elevated IgE level, and lymphopenia. Our patient fulfilled all 5 major criteria and 3 minor criteria; eosinophilia and lymphopenia were absent.
Clinically, PEO has been associated with the deck-chair sign, a pattern of selective sparing of skinfolds, including axillary, inguinal, submammary, and other flexural areas. Although the deck-chair sign was originally considered pathognomonic for PEO, this clinical pattern also has been observed in other entities, such as angioimmunoblastic lymphoma, cutaneous Waldenström macroglobulinemia, and acanthosis nigricans.5,8,9
Specific characteristics of the rash and certain clinical symptoms may help to distinguish the deck-chair sign of PEO from its other causes. Although malignant acanthosis nigricans may spare the skinfolds, lesions have a classic velvety thickening and brownish hyperpigmentation, which is not characteristic of the reddish brown, flat-topped papules of PEO.9 Pai et al5 described a patient with parthenium dermatitis presenting with the deck-chair sign that developed years after repeated exposure to the allergen. Our patient did not have a history of repeated episodes of allergic contact dermatitis. In addition, areas of sparing may mimic the appearance of pityriasis rubra pilaris. As in our patient, those with PEO generally lack the follicular hyperkeratotic papules, palmoplantar keratoderma, widespread orange-red erythema, and characteristic histopathologic finding of hyperkeratosis with alternating orthokeratosis and parakeratosis, allowing these entities to be easily distinguished in most instances.10
Histopathologically, primary PEO shows a nonspecific spongiotic dermatitis-like pattern characterized by slight epidermal hyperplasia with spongiosis and a predominantly perivascular dermal infiltrate with lymphocytes, histiocytes, and eosinophils.3 These histologic findings may at times show some overlap with CTCL, and therefore T-cell gene rearrangement and flow cytometry may be performed in those instances.6
Treatment includes the management of any underlying condition causing the papuloerythroderma.3,6 There are no large clinical trials of treatment options for primary PEO due to its rarity. Topical or systemic corticosteroids remain the mainstay of treatment.3 Alternative treatments used with variable success include phototherapy, interferon, etretinate, cyclosporine, and azathioprine.11 Allegue et al11 successfully used methotrexate to treat a patient with primary PEO and postulated that methotrexate may act through an immunosuppressive mechanism on activated T cells due to the involvement of helper T cells TH2 and TH22 in its pathogenesis.
Although the cutaneous manifestations of PEO may respond well to topical steroids, it is important to consider the possible presence of an underlying malignancy and other associated systemic conditions.
- Farthing CF, Staughton RC, Harper JI, et al. Papuloerythroderma--a further case with the 'deck chair sign.' Dermatologica. 1986;172:65-66.
- Ofuji S, Furukawa F, Miyachi Y, et al. Papuloerythroderma. Dermatologica. 1984;169:125-130.
- Torchia D, Miteva M, Hu S, et al. Papuloerythroderma 2009: two new cases and systematic review of the worldwide literature 25 years after its identification by Ofuji et al. Dermatology. 2010;220:311-320.
- Ochi H, Ang CC. Novel association of a papuloerythroderma of Ofuji phenotype with dermatitis herpetiformis. Int J Dermatol. 2018;57:856-857.
- Pai S, Shetty S, Rao R. Parthenium dermatitis with deck-chair sign. JAMA Dermatol. 2015;151:906-907.
- Maher AM, Ward CE, Glassman S, et al. The importance of excluding cutaneous T-cell lymphomas in patients with a working diagnosis of papuloerythroderma of Ofuji: a case series. Case Rep Dermatol. 2018;10:46-54.
- Grob JJ, Collet-Villette AM, Horchowski N, et al. Ofuji papuloerythroderma. report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol. 1989;20(5 pt 2):927-931.
- Ferran M, Gallardo F, Baena V, et al. The 'deck chair sign' in specific cutaneous involvement by angioimmunoblastic T cell lymphoma. Dermatology. 2006;213:50-52.
- Murao K, Sadamoto Y, Kubo Y, et al. Generalized malignant acanthosis nigricans with "deck-chair sign." Int J Dermatol. 2013;52:377-378.
- Regina G, Paramita L, Radiono S, et al. Papuloerythroderma of Ofuji in Indonesia: the first case report. JDVI. 2016;1:93-98.
- Allegue F, Fachal C, Gonzalez-Vilas D, et al. Papuloerythroderma of Ofuji successfully treated with methotrexate. Dermatol Ther. 2018;31:E12638.
The Diagnosis: Papuloerythroderma of Ofuji
The patient presented with a characteristic finding of skinfold sparing, known as the "deck-chair sign" (Figure 1).1 A repeat biopsy at our institution revealed a dermal perivascular and bandlike infiltrate with lymphocytes and occasional eosinophils (Figure 2). The epidermis showed mild spongiosis, lymphocytic exocytosis, and rare necrotic keratinocytes. A T-cell gene rearrangement assay was negative for a monoclonal population of T lymphocytes. Based on the clinical and histologic features, the diagnosis was most consistent with papuloerythroderma of Ofuji (PEO); however, a lymphoproliferative disorder needed to be excluded. Further workup included a peripheral smear, complete blood cell count with differential, comprehensive metabolic panel, IgE level, and hepatitis panel; all were normal, except for an elevated serum IgE level. Human immunodeficiency virus and age-appropriate malignancy screening were negative. The patient was prescribed betamethasone dipropionate cream 0.05% twice daily, which resulted in near-complete resolution of the rash and marked improvement in pruritus.
In 1984, PEO was described as an entity of generalized pruritic erythroderma characterized by flat-topped, red to brown, coalescing papules with sparing of the skinfolds, later coined the deck-chair sign.1,2 Papuloerythroderma of Ofuji commonly presents in elderly Asian males with a male to female ratio of 4:1.3 Papuloerythroderma of Ofuji is a T cell-mediated skin disease; however, the etiology of the signature rash remains unclear. One explanation includes circulating factors in the skin that elicit an inflammatory response, which does not occur in areas of external pressure.3 The deck-chair sign may occur more frequently in elderly individuals due to increased skin laxity, which creates crease lines that are spared from rubbing and excoriations.4
Although Ofuji et al2 initially reported 4 idiopathic cases, subsequent authors described PEO in association with other conditions, including cutaneous T-cell lymphoma (CTCL) and atopic diathesis, and infections, as well as secondary to medications. Some authors have suggested that PEO may be an early variant of mycosis fungoides; therefore, physicians should monitor patients closely.5-7 Maher et al6 commented that multiple causative factors including CTCL underlie the development of papuloerythroderma.
In a review of PEO, Torchia et al3 proposed diagnostic criteria and an etiologic classification to address whether PEO represents an independent entity or an unusual manifestation of other dermatoses. They established 4 categories of papuloerythroderma--primary, secondary, papuloerythrodermalike CTCL, and pseudopapuloerythroderma--and proposed that primary PEO is a diagnosis of exclusion. If no secondary association is found, they proposed 10 criteria for primary PEO: 5 major criteria include coalescing flat-topped papules, the deck-chair sign, pruritus, histopathologic exclusion of diseases such as CTCL, and a negative workup to exclude other causes.3 In 2018, Maher et al6 recommended workup to rule out cutaneous malignancy, including skin biopsy, flow cytometry, Sézary cell count, T-cell rearrangement, lactate dehydrogenase, and human T-cell lymphotropic virus 1. The 5 minor criteria proposed by Torchia et al3 include age older than 55 years, male sex, eosinophilia, elevated IgE level, and lymphopenia. Our patient fulfilled all 5 major criteria and 3 minor criteria; eosinophilia and lymphopenia were absent.
Clinically, PEO has been associated with the deck-chair sign, a pattern of selective sparing of skinfolds, including axillary, inguinal, submammary, and other flexural areas. Although the deck-chair sign was originally considered pathognomonic for PEO, this clinical pattern also has been observed in other entities, such as angioimmunoblastic lymphoma, cutaneous Waldenström macroglobulinemia, and acanthosis nigricans.5,8,9
Specific characteristics of the rash and certain clinical symptoms may help to distinguish the deck-chair sign of PEO from its other causes. Although malignant acanthosis nigricans may spare the skinfolds, lesions have a classic velvety thickening and brownish hyperpigmentation, which is not characteristic of the reddish brown, flat-topped papules of PEO.9 Pai et al5 described a patient with parthenium dermatitis presenting with the deck-chair sign that developed years after repeated exposure to the allergen. Our patient did not have a history of repeated episodes of allergic contact dermatitis. In addition, areas of sparing may mimic the appearance of pityriasis rubra pilaris. As in our patient, those with PEO generally lack the follicular hyperkeratotic papules, palmoplantar keratoderma, widespread orange-red erythema, and characteristic histopathologic finding of hyperkeratosis with alternating orthokeratosis and parakeratosis, allowing these entities to be easily distinguished in most instances.10
Histopathologically, primary PEO shows a nonspecific spongiotic dermatitis-like pattern characterized by slight epidermal hyperplasia with spongiosis and a predominantly perivascular dermal infiltrate with lymphocytes, histiocytes, and eosinophils.3 These histologic findings may at times show some overlap with CTCL, and therefore T-cell gene rearrangement and flow cytometry may be performed in those instances.6
Treatment includes the management of any underlying condition causing the papuloerythroderma.3,6 There are no large clinical trials of treatment options for primary PEO due to its rarity. Topical or systemic corticosteroids remain the mainstay of treatment.3 Alternative treatments used with variable success include phototherapy, interferon, etretinate, cyclosporine, and azathioprine.11 Allegue et al11 successfully used methotrexate to treat a patient with primary PEO and postulated that methotrexate may act through an immunosuppressive mechanism on activated T cells due to the involvement of helper T cells TH2 and TH22 in its pathogenesis.
Although the cutaneous manifestations of PEO may respond well to topical steroids, it is important to consider the possible presence of an underlying malignancy and other associated systemic conditions.
The Diagnosis: Papuloerythroderma of Ofuji
The patient presented with a characteristic finding of skinfold sparing, known as the "deck-chair sign" (Figure 1).1 A repeat biopsy at our institution revealed a dermal perivascular and bandlike infiltrate with lymphocytes and occasional eosinophils (Figure 2). The epidermis showed mild spongiosis, lymphocytic exocytosis, and rare necrotic keratinocytes. A T-cell gene rearrangement assay was negative for a monoclonal population of T lymphocytes. Based on the clinical and histologic features, the diagnosis was most consistent with papuloerythroderma of Ofuji (PEO); however, a lymphoproliferative disorder needed to be excluded. Further workup included a peripheral smear, complete blood cell count with differential, comprehensive metabolic panel, IgE level, and hepatitis panel; all were normal, except for an elevated serum IgE level. Human immunodeficiency virus and age-appropriate malignancy screening were negative. The patient was prescribed betamethasone dipropionate cream 0.05% twice daily, which resulted in near-complete resolution of the rash and marked improvement in pruritus.
In 1984, PEO was described as an entity of generalized pruritic erythroderma characterized by flat-topped, red to brown, coalescing papules with sparing of the skinfolds, later coined the deck-chair sign.1,2 Papuloerythroderma of Ofuji commonly presents in elderly Asian males with a male to female ratio of 4:1.3 Papuloerythroderma of Ofuji is a T cell-mediated skin disease; however, the etiology of the signature rash remains unclear. One explanation includes circulating factors in the skin that elicit an inflammatory response, which does not occur in areas of external pressure.3 The deck-chair sign may occur more frequently in elderly individuals due to increased skin laxity, which creates crease lines that are spared from rubbing and excoriations.4
Although Ofuji et al2 initially reported 4 idiopathic cases, subsequent authors described PEO in association with other conditions, including cutaneous T-cell lymphoma (CTCL) and atopic diathesis, and infections, as well as secondary to medications. Some authors have suggested that PEO may be an early variant of mycosis fungoides; therefore, physicians should monitor patients closely.5-7 Maher et al6 commented that multiple causative factors including CTCL underlie the development of papuloerythroderma.
In a review of PEO, Torchia et al3 proposed diagnostic criteria and an etiologic classification to address whether PEO represents an independent entity or an unusual manifestation of other dermatoses. They established 4 categories of papuloerythroderma--primary, secondary, papuloerythrodermalike CTCL, and pseudopapuloerythroderma--and proposed that primary PEO is a diagnosis of exclusion. If no secondary association is found, they proposed 10 criteria for primary PEO: 5 major criteria include coalescing flat-topped papules, the deck-chair sign, pruritus, histopathologic exclusion of diseases such as CTCL, and a negative workup to exclude other causes.3 In 2018, Maher et al6 recommended workup to rule out cutaneous malignancy, including skin biopsy, flow cytometry, Sézary cell count, T-cell rearrangement, lactate dehydrogenase, and human T-cell lymphotropic virus 1. The 5 minor criteria proposed by Torchia et al3 include age older than 55 years, male sex, eosinophilia, elevated IgE level, and lymphopenia. Our patient fulfilled all 5 major criteria and 3 minor criteria; eosinophilia and lymphopenia were absent.
Clinically, PEO has been associated with the deck-chair sign, a pattern of selective sparing of skinfolds, including axillary, inguinal, submammary, and other flexural areas. Although the deck-chair sign was originally considered pathognomonic for PEO, this clinical pattern also has been observed in other entities, such as angioimmunoblastic lymphoma, cutaneous Waldenström macroglobulinemia, and acanthosis nigricans.5,8,9
Specific characteristics of the rash and certain clinical symptoms may help to distinguish the deck-chair sign of PEO from its other causes. Although malignant acanthosis nigricans may spare the skinfolds, lesions have a classic velvety thickening and brownish hyperpigmentation, which is not characteristic of the reddish brown, flat-topped papules of PEO.9 Pai et al5 described a patient with parthenium dermatitis presenting with the deck-chair sign that developed years after repeated exposure to the allergen. Our patient did not have a history of repeated episodes of allergic contact dermatitis. In addition, areas of sparing may mimic the appearance of pityriasis rubra pilaris. As in our patient, those with PEO generally lack the follicular hyperkeratotic papules, palmoplantar keratoderma, widespread orange-red erythema, and characteristic histopathologic finding of hyperkeratosis with alternating orthokeratosis and parakeratosis, allowing these entities to be easily distinguished in most instances.10
Histopathologically, primary PEO shows a nonspecific spongiotic dermatitis-like pattern characterized by slight epidermal hyperplasia with spongiosis and a predominantly perivascular dermal infiltrate with lymphocytes, histiocytes, and eosinophils.3 These histologic findings may at times show some overlap with CTCL, and therefore T-cell gene rearrangement and flow cytometry may be performed in those instances.6
Treatment includes the management of any underlying condition causing the papuloerythroderma.3,6 There are no large clinical trials of treatment options for primary PEO due to its rarity. Topical or systemic corticosteroids remain the mainstay of treatment.3 Alternative treatments used with variable success include phototherapy, interferon, etretinate, cyclosporine, and azathioprine.11 Allegue et al11 successfully used methotrexate to treat a patient with primary PEO and postulated that methotrexate may act through an immunosuppressive mechanism on activated T cells due to the involvement of helper T cells TH2 and TH22 in its pathogenesis.
Although the cutaneous manifestations of PEO may respond well to topical steroids, it is important to consider the possible presence of an underlying malignancy and other associated systemic conditions.
- Farthing CF, Staughton RC, Harper JI, et al. Papuloerythroderma--a further case with the 'deck chair sign.' Dermatologica. 1986;172:65-66.
- Ofuji S, Furukawa F, Miyachi Y, et al. Papuloerythroderma. Dermatologica. 1984;169:125-130.
- Torchia D, Miteva M, Hu S, et al. Papuloerythroderma 2009: two new cases and systematic review of the worldwide literature 25 years after its identification by Ofuji et al. Dermatology. 2010;220:311-320.
- Ochi H, Ang CC. Novel association of a papuloerythroderma of Ofuji phenotype with dermatitis herpetiformis. Int J Dermatol. 2018;57:856-857.
- Pai S, Shetty S, Rao R. Parthenium dermatitis with deck-chair sign. JAMA Dermatol. 2015;151:906-907.
- Maher AM, Ward CE, Glassman S, et al. The importance of excluding cutaneous T-cell lymphomas in patients with a working diagnosis of papuloerythroderma of Ofuji: a case series. Case Rep Dermatol. 2018;10:46-54.
- Grob JJ, Collet-Villette AM, Horchowski N, et al. Ofuji papuloerythroderma. report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol. 1989;20(5 pt 2):927-931.
- Ferran M, Gallardo F, Baena V, et al. The 'deck chair sign' in specific cutaneous involvement by angioimmunoblastic T cell lymphoma. Dermatology. 2006;213:50-52.
- Murao K, Sadamoto Y, Kubo Y, et al. Generalized malignant acanthosis nigricans with "deck-chair sign." Int J Dermatol. 2013;52:377-378.
- Regina G, Paramita L, Radiono S, et al. Papuloerythroderma of Ofuji in Indonesia: the first case report. JDVI. 2016;1:93-98.
- Allegue F, Fachal C, Gonzalez-Vilas D, et al. Papuloerythroderma of Ofuji successfully treated with methotrexate. Dermatol Ther. 2018;31:E12638.
- Farthing CF, Staughton RC, Harper JI, et al. Papuloerythroderma--a further case with the 'deck chair sign.' Dermatologica. 1986;172:65-66.
- Ofuji S, Furukawa F, Miyachi Y, et al. Papuloerythroderma. Dermatologica. 1984;169:125-130.
- Torchia D, Miteva M, Hu S, et al. Papuloerythroderma 2009: two new cases and systematic review of the worldwide literature 25 years after its identification by Ofuji et al. Dermatology. 2010;220:311-320.
- Ochi H, Ang CC. Novel association of a papuloerythroderma of Ofuji phenotype with dermatitis herpetiformis. Int J Dermatol. 2018;57:856-857.
- Pai S, Shetty S, Rao R. Parthenium dermatitis with deck-chair sign. JAMA Dermatol. 2015;151:906-907.
- Maher AM, Ward CE, Glassman S, et al. The importance of excluding cutaneous T-cell lymphomas in patients with a working diagnosis of papuloerythroderma of Ofuji: a case series. Case Rep Dermatol. 2018;10:46-54.
- Grob JJ, Collet-Villette AM, Horchowski N, et al. Ofuji papuloerythroderma. report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol. 1989;20(5 pt 2):927-931.
- Ferran M, Gallardo F, Baena V, et al. The 'deck chair sign' in specific cutaneous involvement by angioimmunoblastic T cell lymphoma. Dermatology. 2006;213:50-52.
- Murao K, Sadamoto Y, Kubo Y, et al. Generalized malignant acanthosis nigricans with "deck-chair sign." Int J Dermatol. 2013;52:377-378.
- Regina G, Paramita L, Radiono S, et al. Papuloerythroderma of Ofuji in Indonesia: the first case report. JDVI. 2016;1:93-98.
- Allegue F, Fachal C, Gonzalez-Vilas D, et al. Papuloerythroderma of Ofuji successfully treated with methotrexate. Dermatol Ther. 2018;31:E12638.
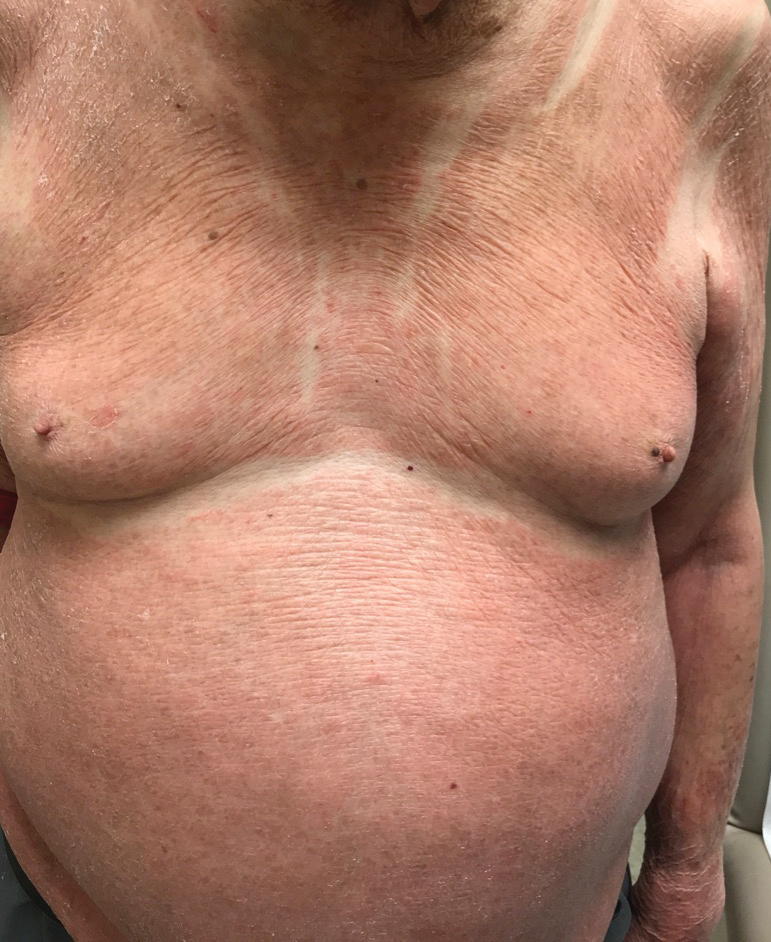
An 89-year-old Asian man presented with a generalized pruritic eruption of 2 months' duration. The rash started on the flanks and later spread to the arms and legs, abdomen, and back; the face and palms were spared. Physical examination revealed numerous erythematous papules coalescing into large scaly plaques on the trunk, arms, and legs. There were noticeable areas of sparing of the skinfolds, especially the axillary, inframammary, and inguinal folds, as well as the midline of the back. A biopsy performed by an outside physician showed findings consistent with a possible pityriasiform drug eruption; however, there were no recent changes in medication history.
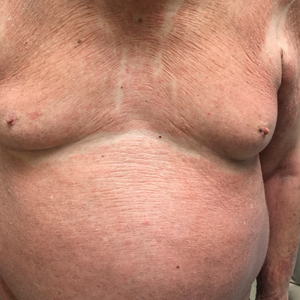
Edema Affecting the Penis and Scrotum
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
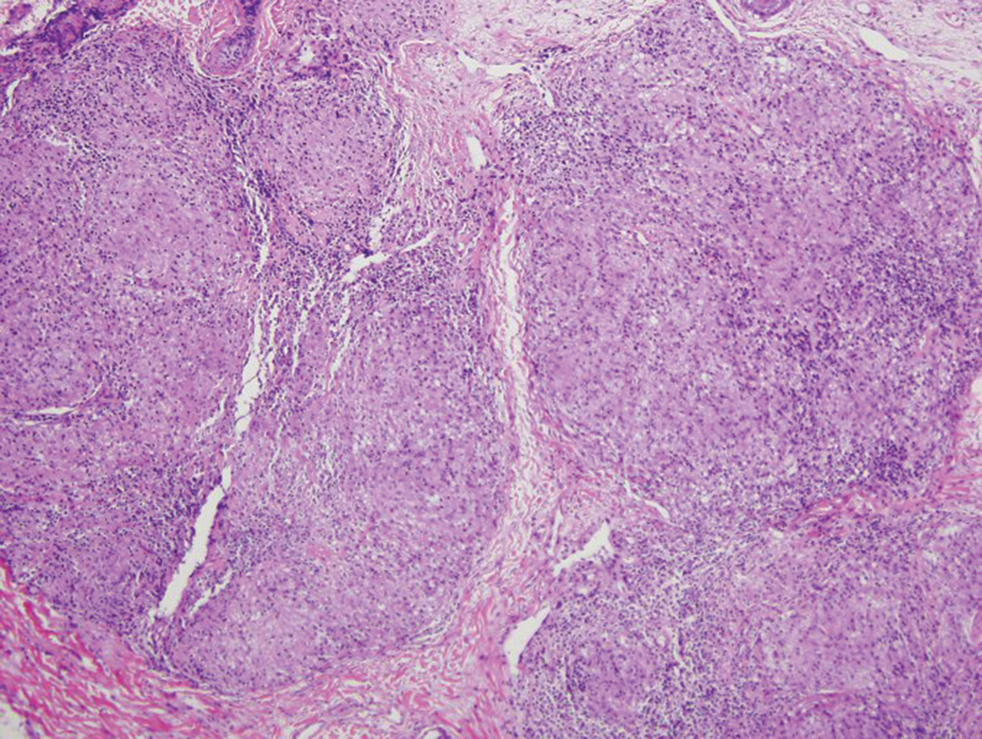
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
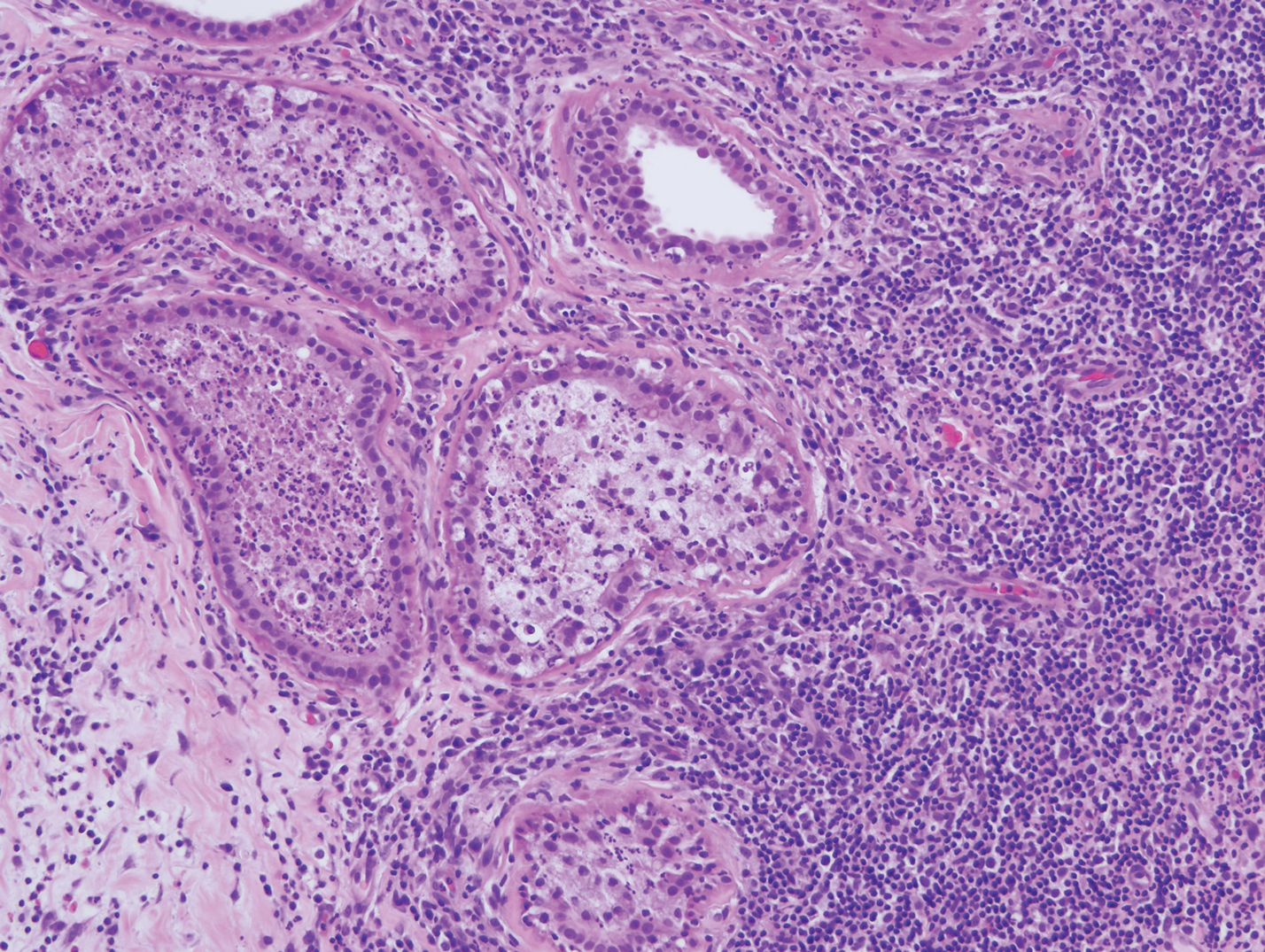
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
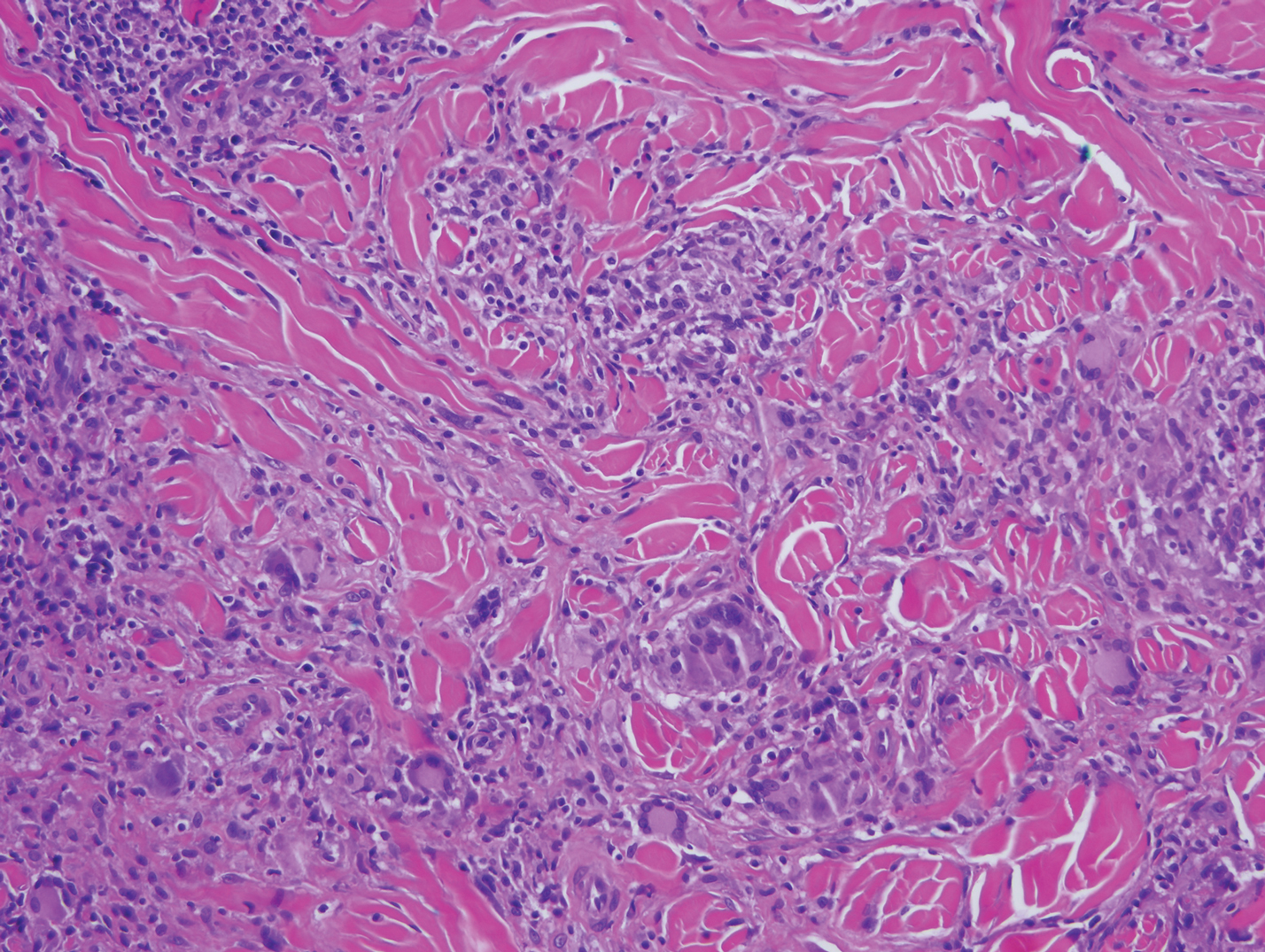
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
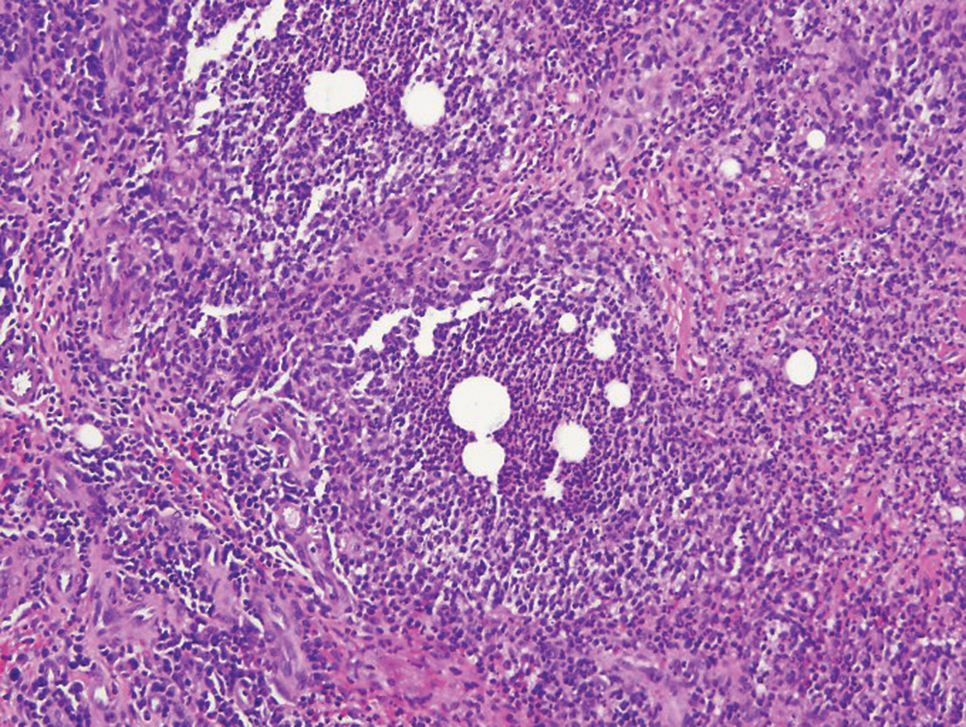
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.

A 44-year-old man presented for evaluation of self-described "skin ripping" on the penis with penile and scrotal edema of 1 year's duration. He had a history of bowel symptoms and anorectal fistula of 3 years' duration. Purulent penile drainage and inguinal lymphadenopathy were noted on physical examination. Excisional biopsies of the scrotum and penis were performed. Special stains for organisms were negative.
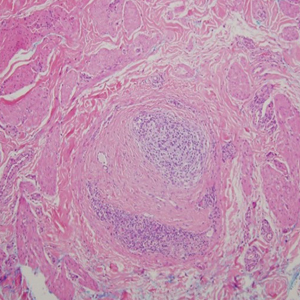
Multinodular Plaque on the Penis
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
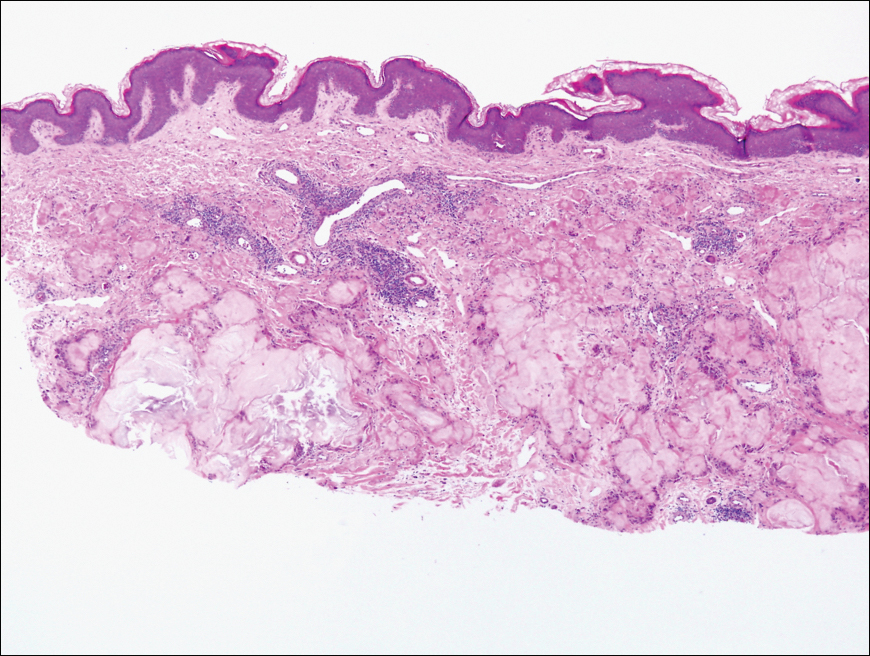
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
The Diagnosis: Tophaceous Gout
Biopsy revealed amorphous pink material within the center of palisading granulomas lined by histiocytes and giant cells. Scattered crystal remnants also were identified within the center of the granulomas; however, the majority of the crystals were dissolved during the formalin processing of the tissue to become the amorphous material. A perivascular mixed inflammatory infiltrate composed of lymphocytes, histiocytes, and plasma cells surrounded the tophi nodules. A biopsy confirmed the diagnosis of tophaceous gout (Figure).
Gout is a systemic metabolic disease characterized by the supersaturation of monosodium urate (MSU) crystals in joints and bursae. Peripheral joints most commonly are affected due to the poor solubility of MSU crystals at low temperatures.1 It is one of the most common forms of inflammatory arthritis, with an estimated prevalence of 4% of adults in the United States.2 An estimated $1 billion is spent each year on ambulatory care for gout.3 Gout occurs most commonly in men and usually manifests in the fifth or sixth decades of life.4 Risk factors for the development of gout include obesity, hypertension, poor dietary habits and kidney function, excessive alcohol intake, and diuretic use.3
Disease manifestations range from asymptomatic hyperuricemia to acute gouty arthritis and chronic tophaceous gout. Patients may present with chronic tophaceous gout without a prior clinically apparent acute gout episode.5,6 Uncontrolled gout may result in large accumulations of MSU crystals, leading to well-circumscribed masses (known as tophi), as demonstrated in our patient.1 Tophi are pathognomonic features of gout and are the sine qua non of advanced gout (also known as chronic tophaceous gout).2 Clinically, these tophi appear as subcutaneous, yellowish white, firm and smooth nodules that are highlighted on the skin.4 Tophi most commonly are found on the helix, articular and periarticular tissue, and the tissue of the hands and feet. They usually are visible on physical examination but also may be detected on imaging studies.2,4
Gouty tophi have been reported in extraordinary locations, such as in sclerae; vocal cords; heart valves; abdominal striae; nerves; axial skeleton4,7; and the penis, as in our patient and one other case.2 These gouty deposits can appear similarly to lipomas, rheumatoid and osteoarthritic nodules, and infectious and malignant processes.1,5 When tophi present in unusual locations, tissue biopsy often is necessary to confirm the diagnosis. Tissue preservation in alcohol is required to preserve the urate crystals. Microscopically, urate crystals appear as tightly packed, brown, needle-shaped crystals surrounded by granulomatous inflammation with foreign body giant cells, macrophages, and possibly some fibrosis. When examined under polarized light, the MSU crystals are negatively birefringent. However, when clinical suspicion for gout is low and the tissue is instead formalin fixed, as was performed in our case, the crystals dissolve into fibrillary amorphous deposits within the center of the granulomatous inflammation, which is another characteristic histologic finding in tophaceous gout.8
Management of gout focuses on urate-lowering therapy including lifestyle changes. Lower serum urate levels are associated with a decreased incidence of acute gout attacks and chronic tophaceous gout.2 Urate-lowering drugs often are combined with anti-inflammatory drugs during acute attacks. Lifestyle changes, such as weight loss, exercise, reduced alcohol consumption, high fluid intake, and a low-purine diet also are beneficial.3,4 Although gout cannot be cured, it can be effectively managed, and appropriate treatment can improve quality of life and reduce the risk for permanent joint damage and structural deformities. If medical treatment and lifestyle changes fail to adequately control tophaceous gout or if tophi become symptomatic, surgical removal of tophi is appropriate.4
At follow-up, our patient opted for surgical removal of the penile tophi. Using local anesthesia, surgical debulking via curettage was performed. Open defects were closed with fine absorbable sutures, and prophylactic antibiotics were given. Allopurinol also was started. Six weeks following extraction, the patient reported no complications and the area was continuing to heal.
Tophaceous gout would be distinguished from conditions in the differential diagnosis based on histologic findings from hematoxylin and eosin (H&E)-stained sections. Actinomycotic mycetoma is rare in the United States and is characterized by a seropurulent or stringy exudate with grains, ulcerations, melicerous scabs, and retractable scarring.9 On H&E-stained sections, actinomyces appear filamentous with deeply basophilic staining and radially oriented acidophilic projections.10 Calcinosis cutis of the penis has been reported to appear as asymptomatic papules; however, microscopic sections reveal deeply basophilic calcium deposits within the tissue.11 Multinodular syphilis shows characteristic histology with lichenoid or vacuolar interface dermatitis, slender acanthosis, plasma cells, and endothelial swelling of the small vessels. A Treponema pallidum immunoperoxidase stain shows numerous organisms. Planar xanthoma shows xanthomatous or foamy histiocytes throughout the dermis on H&E-stained sections.12
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
- Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective--a review. J Adv Res. 2007;8:495-511.
- Flores Martín JF, Vázquez Alonso F, Puche Sanz I, et al. Gouty tophi in the penis: a case report and review of the literature. Case Rep Urol. 2012;2012:594905.
- Qaseem A, Harris RP, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2017;166:58-68.
- Forbess LJ, Fields TR. The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42:146-154.
- Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
- Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. part 2: therapy and anti-inflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64:1447-1461.
- Gaviria JL, Ortega VG, Gaona J. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Patterson JW, Hosler GA, Weedon D. Weedon's Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2016.
- Guerra-Leal JD, Medrano-Danés LA, Montemayor-Martinez A, et al. The importance of diagnostic imaging of mycetoma in the foot [published online December 18, 2018]. Int J Dermatol. 2019;58:600-604.
- Fazeli MS, Bateni H. Actinomycosis: a rare soft tissue infection. Dermatol Online J. 2005;11:18.
- Cohen PR, Tschen JA. Idiopathic calcinosis cutis of the penis. J Clin Aesthet Dermatol. 2012;5:23-30.
- Ko C, Elston DM, Ferringer T. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019.
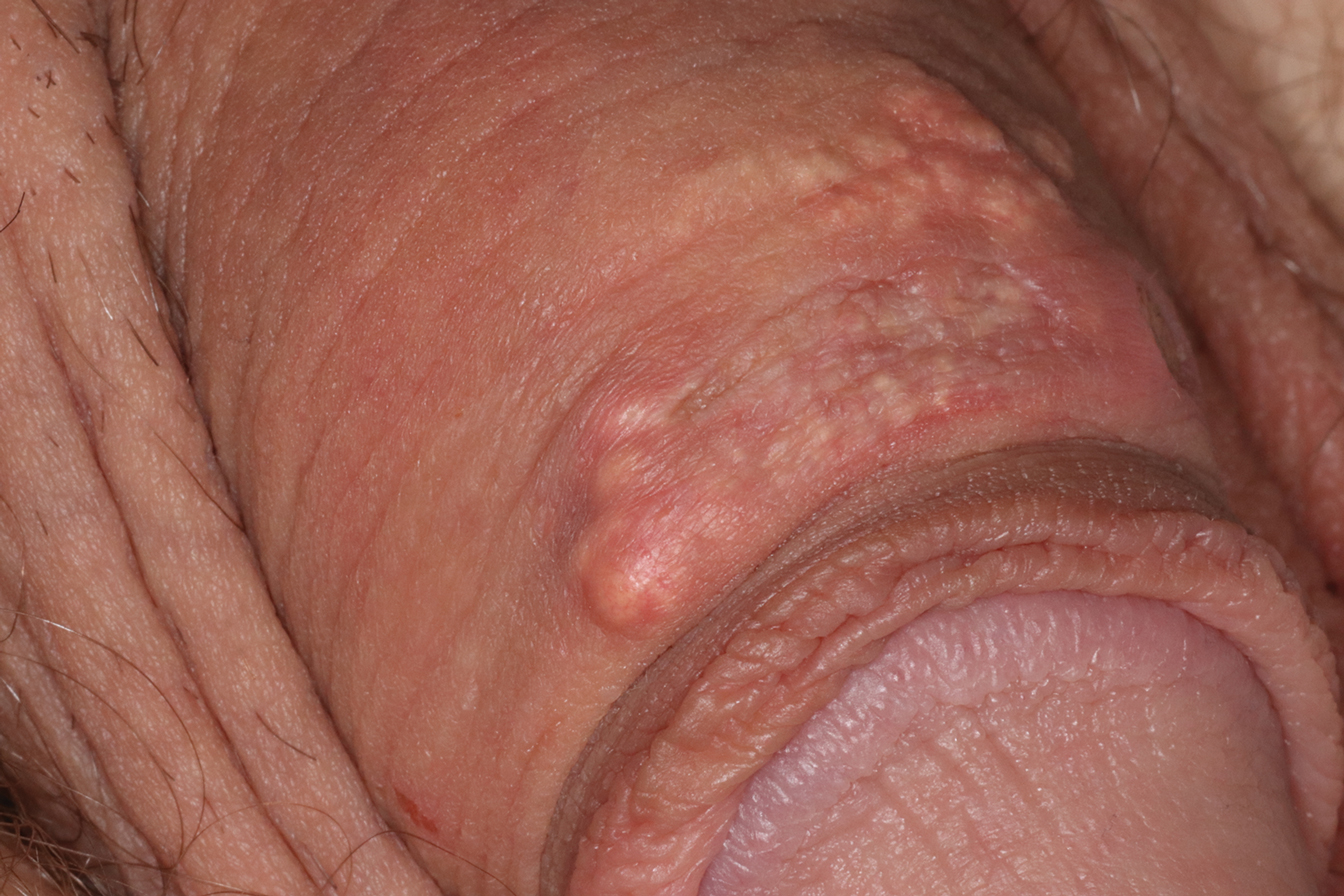
A 34-year-old man presented for evaluation of a slowly growing group of firm white bumps on the penis. The lesions were nontender and asymptomatic. Medical and family history was notable for gout, though he was not being treated. Physical examination revealed a 3-cm, firm, multinodular, chalky white plaque on the dorsal aspect of the penile shaft. A tangential biopsy was performed and sent for hematoxylin and eosin staining.
Tense Bullae on the Hands
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
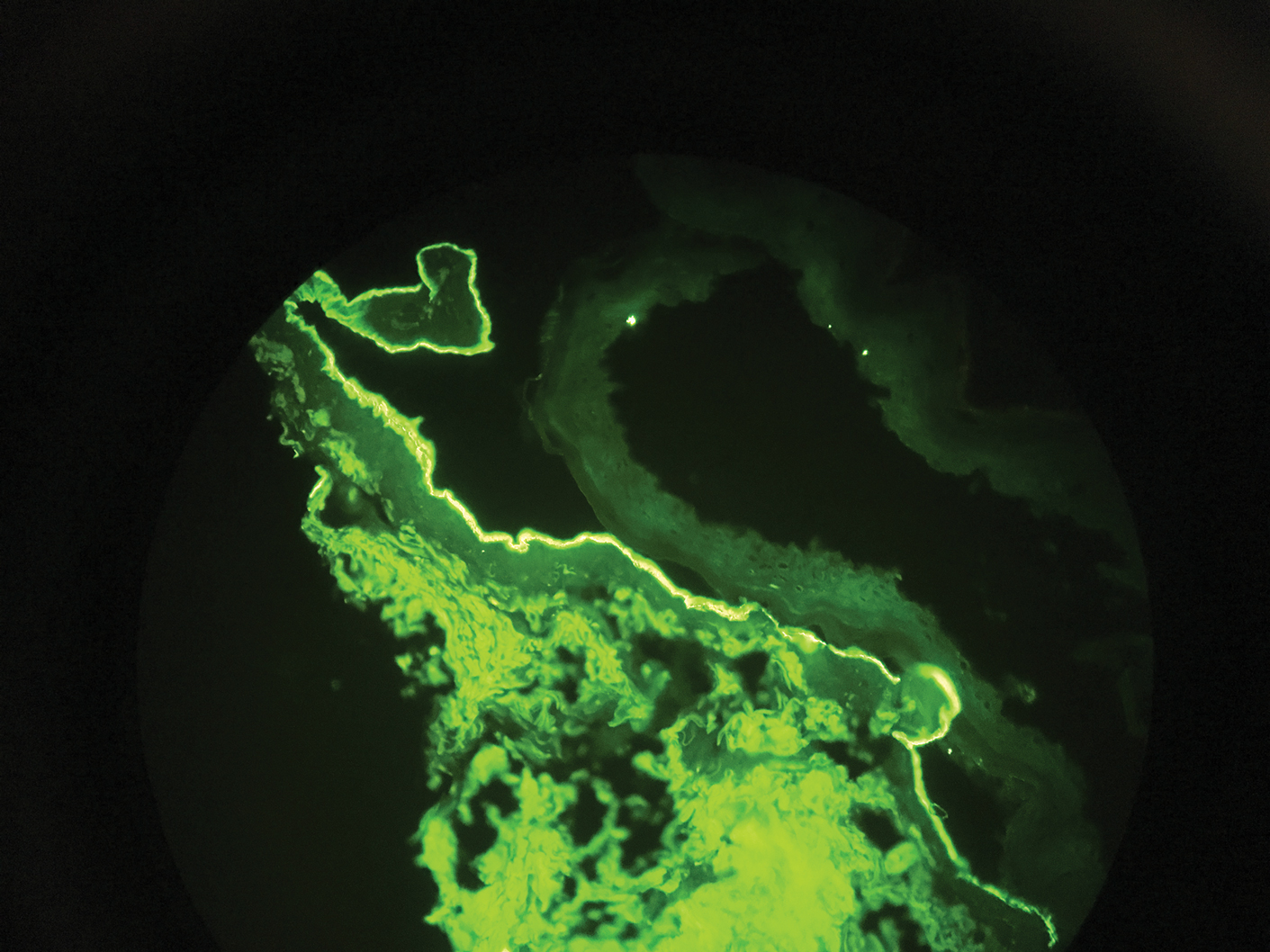
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
The Diagnosis: Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a rare autoimmune blistering disorder characterized by tense bullae, skin fragility, atrophic scarring, and milia formation.1 Blisters occur on a noninflammatory base in the classic variant and are trauma induced, hence the predilection for the extensor surfaces.2 Mucosal involvement also has been described.1 The characteristic findings in EBA are IgG autoantibodies directed at the N-terminal collagenous domain of type VII collagen, which composes the anchoring fibrils in the basement membrane zone.1 Differentiating EBA from other subepidermal bullous diseases, especially bullous pemphigoid (BP), can be difficult, necessitating specialized tests.
Biopsy of the perilesional skin can help identify the location of the blister formation. Our patient's biopsy showed a subepidermal blister with granulocytes. The differential diagnosis of a subepidermal blister includes BP, herpes gestationis, cicatricial pemphigoid, EBA, bullous systemic lupus erythematosus, dermatitis herpetiformis, linear IgA disease, and porphyria cutanea tarda.
Direct immunofluorescence (DIF) was performed on the biopsy from our patient, which showed linear/particulate IgG, C3, and IgA deposits in the basement membrane zone, narrowing the differential diagnosis to BP or EBA. To differentiate EBA from BP, DIF of perilesional skin using a salt-split preparation was performed. This test distinguishes the location of the immunoreactants at the basement membrane zone. The antibody complexes in BP are found on the epidermal side of the split, while the antibody complexes in EBA are found on the dermal side of the split. Indirect immunofluorescence on salt-split skin also has been used to distinguish EBA from BP but is only conclusive if there are circulating autoantibodies to the basement membrane zone in the serum, which occurs in approximately 50% of patients with EBA and 15% of patients with BP.3 The immune complexes in our patient were found to be on the dermal side of the split after DIF on salt-split skin, confirming the diagnosis of EBA (Figure).
Differentiating EBA from BP has great value, as the diagnosis affects treatment options. Bullous pemphigoid is fairly easy to treat, with most patients responding to prednisone.3 Epidermolysis bullosa acquisita usually is resistant to therapy. The disease course is chronic with exacerbations and remissions. Dapsone often is used to control the disease, though this therapy for EBA is not currently approved by the US Food and Drug Administration. The recommended initial dose of dapsone is 50 mg daily and should be increased by 50 mg each week until remission, usually 100 to 250 mg.4 We prescribed dapsone for our patient upon clinical suspicion of EBA before the DIF on salt-split skin was completed. A trial of prednisone may be warranted for EBA if there is no response to dapsone or colchicine, but the response is unpredictable. Cyclosporine usually results in a quick response and may be considered if there is clinically severe disease and other treatment alternatives have failed.4
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new. J Dermatol. 2010;37:220-230.
- Lehman JS, Camilleri MJ, Gibsom LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-236.
- Woodley D. Immunofluorescence on the salt-split skin for the diagnosis of epidermolysis bullosa acquisita. Arch Dermatol. 1990;126:229-231.
- Mutasim DF. Bullous diseases. In: Kellerman RD, Rakel DP, eds. Conn's Current Therapy. Philadelphia, PA: Elsevier; 2020:978-982.
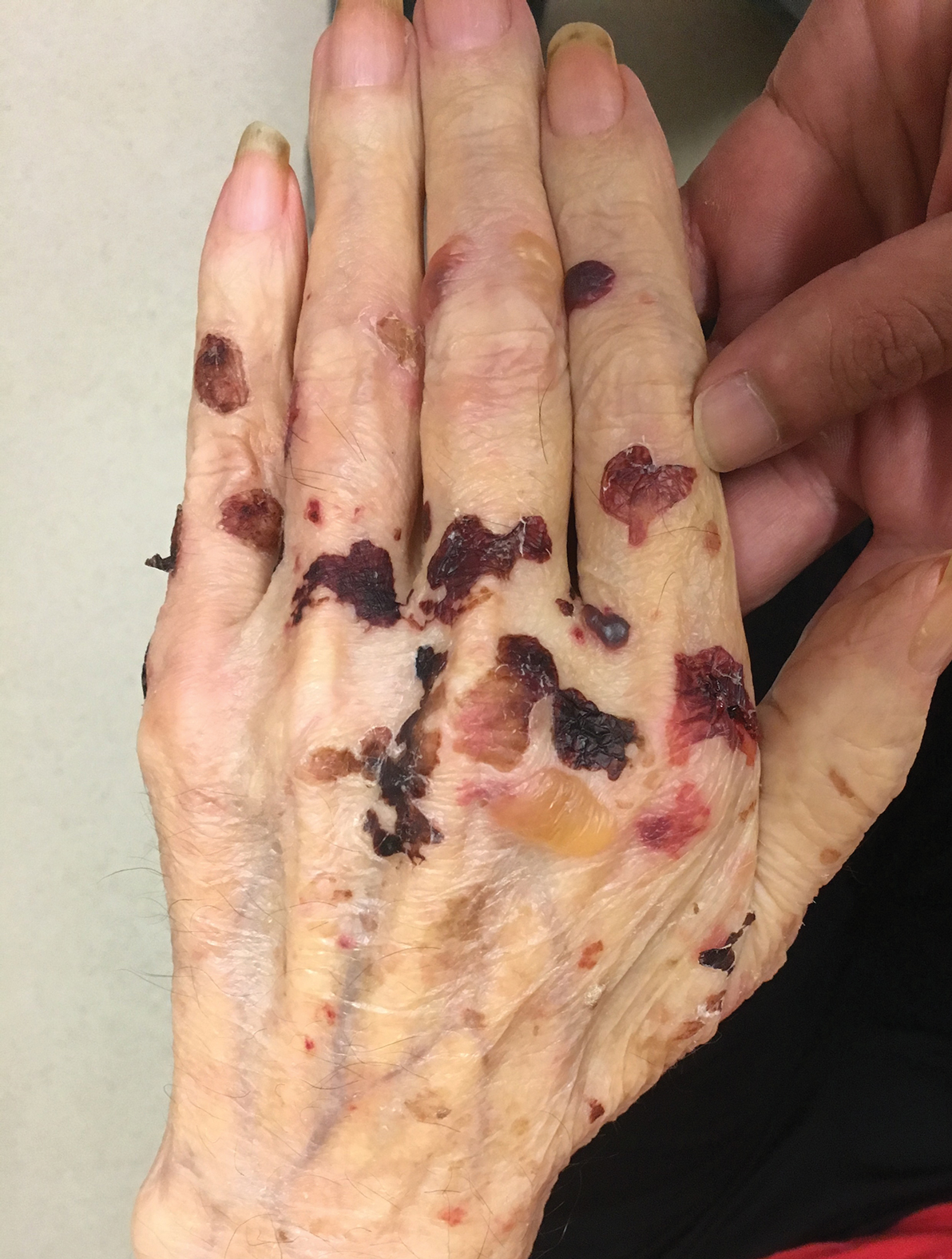
A 75-year-old man presented to our clinic with nonpainful, nonpruritic, tense bullae and erosions on the dorsal aspects of the hands and extensor surfaces of the elbows of 1 month's duration. The patient also had erythematous erosions and crusted papules on the left cheek and surrounding the left eye. He denied any new medications, history of liver or kidney disease, or history of hepatitis or human immunodeficiency virus. There were no obvious exacerbating factors, including exposure to sunlight. Direct immunofluorescence using a salt-split preparation was performed on a biopsy of the perilesional skin.
CD4 cells implicated in pathology of CCCA
in the lymphocytic inflammatory infiltrate, according to a histopathological study of biopsy specimens.
“Evaluation of the T-cell infiltrate may be a useful way to distinguish CCCA from lichen planopilaris or frontal fibrosing alopecia in some cases when histopathological features alone cannot be used to definitely distinguish between them,” reported Alexandra Flamm, MD, Ata Moshiri, MD, and coauthors from the departments of dermatology and pathology, University of Pennsylvania, Philadelphia.
The histopathological features of CCCA have been characterized previously, but the goal of this study was to go further in piecing together the pathophysiology, they noted.
Horizontal sections of 4-mm punch biopsy specimens were examined from 18 black women with a known diagnosis of CCCA. Both affected and unaffected follicles were evaluated with attention to the number and percentage of CD1a+ Langerhans cells, CD3+, CD4+, and CD8+ lymphocytes.
In this series, the lymphocytic infiltrate in both the affected and unaffected follicles was predominantly composed of CD4+ cells. The perifollicular ratio for CD4+ to CD8+ cells in affected follicles was 5.3:1. It was only modestly lower in unaffected follicles (4.3:1) and in the intrafollicular space of affected follicles (2.5:1).
Affected follicles had a higher number of CD1a+ Langerhans cells than unaffected follicles. This finding suggests, as others have hypothesized, that the antigen-presenting Langerhans cells draw lymphocytes to the follicle, according to the investigators. Elevated numbers of Langerhans cells have also been reported in other forms of scarring alopecia, such as lichen planopilaris (LPP).
In the case of CCCA, CD1a+ Langerhans cells appear to localize to the hair follicle in response to stimulus such as an injury. The CD4+ cells that follow the Langerhans cells participate in an inflammatory reaction that drives follicle destruction. In addition to this damage and scarring, the inflammatory response is also likely to be disrupting the blood supply.
“Fibroplasia associated with follicular scarring displaces blood vessels away from the outer root sheath epithelium,” the authors explained. Ultimately, “the mucinous fibroplasia and perifollicular fibrosis may disrupt and fragment blood vessels in the fibrous sheath, leaving only small clusters of vessels more distant to the keratinocytes in the outer root sheath.”
Prior studies of scarring alopecia diseases, including LLP, frontal fibrosing alopecia (FFA), and keratosis follicularis spinulosa decalvans (KFSD), have typically described a predominantly CD8+ lymphocytic infiltrate. The evidence from this study that the infiltrate is CD4+ predominant in CCCA supports the conclusion that the pathophysiologic features of this type of alopecia are unique, according to the authors.
Work by others has associated CCCA with mutations in the PAD13 gene, which suggests a defect in the formation of hair shaft structure, but this may speak to susceptibility but not the mechanism of hair follicle damage. Rather, this study suggests that it is the concentration of a CD4+ predominant lymphocytic infiltrate in the perifollicular space that induces the pathological events.
For determining the fundamental cause of CCCA, “it will be important to determine what recruits the Langerhans cells to affected follicles,” the investigators suggested. Meanwhile, they expressed hope that the progress being made into decoding the pathogenesis of CCCA will lead to novel therapeutic strategies.
The authors did not list any disclosures. The funding source was listed as the Center for Scientific Review (Grant/Award).
SOURCE: Flamm A et al. J Cutan Pathol. 2020 Feb 18.doi: 10.1111/cup.13666.
in the lymphocytic inflammatory infiltrate, according to a histopathological study of biopsy specimens.
“Evaluation of the T-cell infiltrate may be a useful way to distinguish CCCA from lichen planopilaris or frontal fibrosing alopecia in some cases when histopathological features alone cannot be used to definitely distinguish between them,” reported Alexandra Flamm, MD, Ata Moshiri, MD, and coauthors from the departments of dermatology and pathology, University of Pennsylvania, Philadelphia.
The histopathological features of CCCA have been characterized previously, but the goal of this study was to go further in piecing together the pathophysiology, they noted.
Horizontal sections of 4-mm punch biopsy specimens were examined from 18 black women with a known diagnosis of CCCA. Both affected and unaffected follicles were evaluated with attention to the number and percentage of CD1a+ Langerhans cells, CD3+, CD4+, and CD8+ lymphocytes.
In this series, the lymphocytic infiltrate in both the affected and unaffected follicles was predominantly composed of CD4+ cells. The perifollicular ratio for CD4+ to CD8+ cells in affected follicles was 5.3:1. It was only modestly lower in unaffected follicles (4.3:1) and in the intrafollicular space of affected follicles (2.5:1).
Affected follicles had a higher number of CD1a+ Langerhans cells than unaffected follicles. This finding suggests, as others have hypothesized, that the antigen-presenting Langerhans cells draw lymphocytes to the follicle, according to the investigators. Elevated numbers of Langerhans cells have also been reported in other forms of scarring alopecia, such as lichen planopilaris (LPP).
In the case of CCCA, CD1a+ Langerhans cells appear to localize to the hair follicle in response to stimulus such as an injury. The CD4+ cells that follow the Langerhans cells participate in an inflammatory reaction that drives follicle destruction. In addition to this damage and scarring, the inflammatory response is also likely to be disrupting the blood supply.
“Fibroplasia associated with follicular scarring displaces blood vessels away from the outer root sheath epithelium,” the authors explained. Ultimately, “the mucinous fibroplasia and perifollicular fibrosis may disrupt and fragment blood vessels in the fibrous sheath, leaving only small clusters of vessels more distant to the keratinocytes in the outer root sheath.”
Prior studies of scarring alopecia diseases, including LLP, frontal fibrosing alopecia (FFA), and keratosis follicularis spinulosa decalvans (KFSD), have typically described a predominantly CD8+ lymphocytic infiltrate. The evidence from this study that the infiltrate is CD4+ predominant in CCCA supports the conclusion that the pathophysiologic features of this type of alopecia are unique, according to the authors.
Work by others has associated CCCA with mutations in the PAD13 gene, which suggests a defect in the formation of hair shaft structure, but this may speak to susceptibility but not the mechanism of hair follicle damage. Rather, this study suggests that it is the concentration of a CD4+ predominant lymphocytic infiltrate in the perifollicular space that induces the pathological events.
For determining the fundamental cause of CCCA, “it will be important to determine what recruits the Langerhans cells to affected follicles,” the investigators suggested. Meanwhile, they expressed hope that the progress being made into decoding the pathogenesis of CCCA will lead to novel therapeutic strategies.
The authors did not list any disclosures. The funding source was listed as the Center for Scientific Review (Grant/Award).
SOURCE: Flamm A et al. J Cutan Pathol. 2020 Feb 18.doi: 10.1111/cup.13666.
in the lymphocytic inflammatory infiltrate, according to a histopathological study of biopsy specimens.
“Evaluation of the T-cell infiltrate may be a useful way to distinguish CCCA from lichen planopilaris or frontal fibrosing alopecia in some cases when histopathological features alone cannot be used to definitely distinguish between them,” reported Alexandra Flamm, MD, Ata Moshiri, MD, and coauthors from the departments of dermatology and pathology, University of Pennsylvania, Philadelphia.
The histopathological features of CCCA have been characterized previously, but the goal of this study was to go further in piecing together the pathophysiology, they noted.
Horizontal sections of 4-mm punch biopsy specimens were examined from 18 black women with a known diagnosis of CCCA. Both affected and unaffected follicles were evaluated with attention to the number and percentage of CD1a+ Langerhans cells, CD3+, CD4+, and CD8+ lymphocytes.
In this series, the lymphocytic infiltrate in both the affected and unaffected follicles was predominantly composed of CD4+ cells. The perifollicular ratio for CD4+ to CD8+ cells in affected follicles was 5.3:1. It was only modestly lower in unaffected follicles (4.3:1) and in the intrafollicular space of affected follicles (2.5:1).
Affected follicles had a higher number of CD1a+ Langerhans cells than unaffected follicles. This finding suggests, as others have hypothesized, that the antigen-presenting Langerhans cells draw lymphocytes to the follicle, according to the investigators. Elevated numbers of Langerhans cells have also been reported in other forms of scarring alopecia, such as lichen planopilaris (LPP).
In the case of CCCA, CD1a+ Langerhans cells appear to localize to the hair follicle in response to stimulus such as an injury. The CD4+ cells that follow the Langerhans cells participate in an inflammatory reaction that drives follicle destruction. In addition to this damage and scarring, the inflammatory response is also likely to be disrupting the blood supply.
“Fibroplasia associated with follicular scarring displaces blood vessels away from the outer root sheath epithelium,” the authors explained. Ultimately, “the mucinous fibroplasia and perifollicular fibrosis may disrupt and fragment blood vessels in the fibrous sheath, leaving only small clusters of vessels more distant to the keratinocytes in the outer root sheath.”
Prior studies of scarring alopecia diseases, including LLP, frontal fibrosing alopecia (FFA), and keratosis follicularis spinulosa decalvans (KFSD), have typically described a predominantly CD8+ lymphocytic infiltrate. The evidence from this study that the infiltrate is CD4+ predominant in CCCA supports the conclusion that the pathophysiologic features of this type of alopecia are unique, according to the authors.
Work by others has associated CCCA with mutations in the PAD13 gene, which suggests a defect in the formation of hair shaft structure, but this may speak to susceptibility but not the mechanism of hair follicle damage. Rather, this study suggests that it is the concentration of a CD4+ predominant lymphocytic infiltrate in the perifollicular space that induces the pathological events.
For determining the fundamental cause of CCCA, “it will be important to determine what recruits the Langerhans cells to affected follicles,” the investigators suggested. Meanwhile, they expressed hope that the progress being made into decoding the pathogenesis of CCCA will lead to novel therapeutic strategies.
The authors did not list any disclosures. The funding source was listed as the Center for Scientific Review (Grant/Award).
SOURCE: Flamm A et al. J Cutan Pathol. 2020 Feb 18.doi: 10.1111/cup.13666.
Firm Abdominal Papule
The Diagnosis: Cutaneous Metastatic Gastric Carcinoma
Cutaneous metastasis of primary gastric carcinoma is a rare occurrence, with the more common metastatic sites being the lymph nodes, liver, and peritoneal cavity. The incidence of visceral neoplasm metastasis to the skin ranges from 0.7% to 9% and is less than 1% for upper digestive tract carcinomas.1 Cutaneous metastases make up 2% of all tumors of the skin and commonly are located near the site of the primary tumor.2 The most common cutaneous metastasis sites for gastric carcinoma include the neck, chest, and head.3 One of the more typical sites of cutaneous metastasis from gastric cancer is the umbilicus (ie, Sister Mary Joseph nodule). Cutaneous metastases from gastric carcinoma commonly present as asymptomatic hyperpigmented nodules.1,3
In our patient, histopathologic sections showed diffuse infiltration of the dermis by atypical polygonal/round cells arranged in cords and small aggregates. Some of the neoplastic cells had signet ring morphology (Figure). Tumor cells demonstrated positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen; they were negative for S-100, MART-1 (melanoma-associated antigen recognized by T cells 1), leukocyte common antigen, gross cystic disease fluid protein 15, estrogen and progesterone receptor, and HER2/neu (human epidermal growth factor receptor 2).
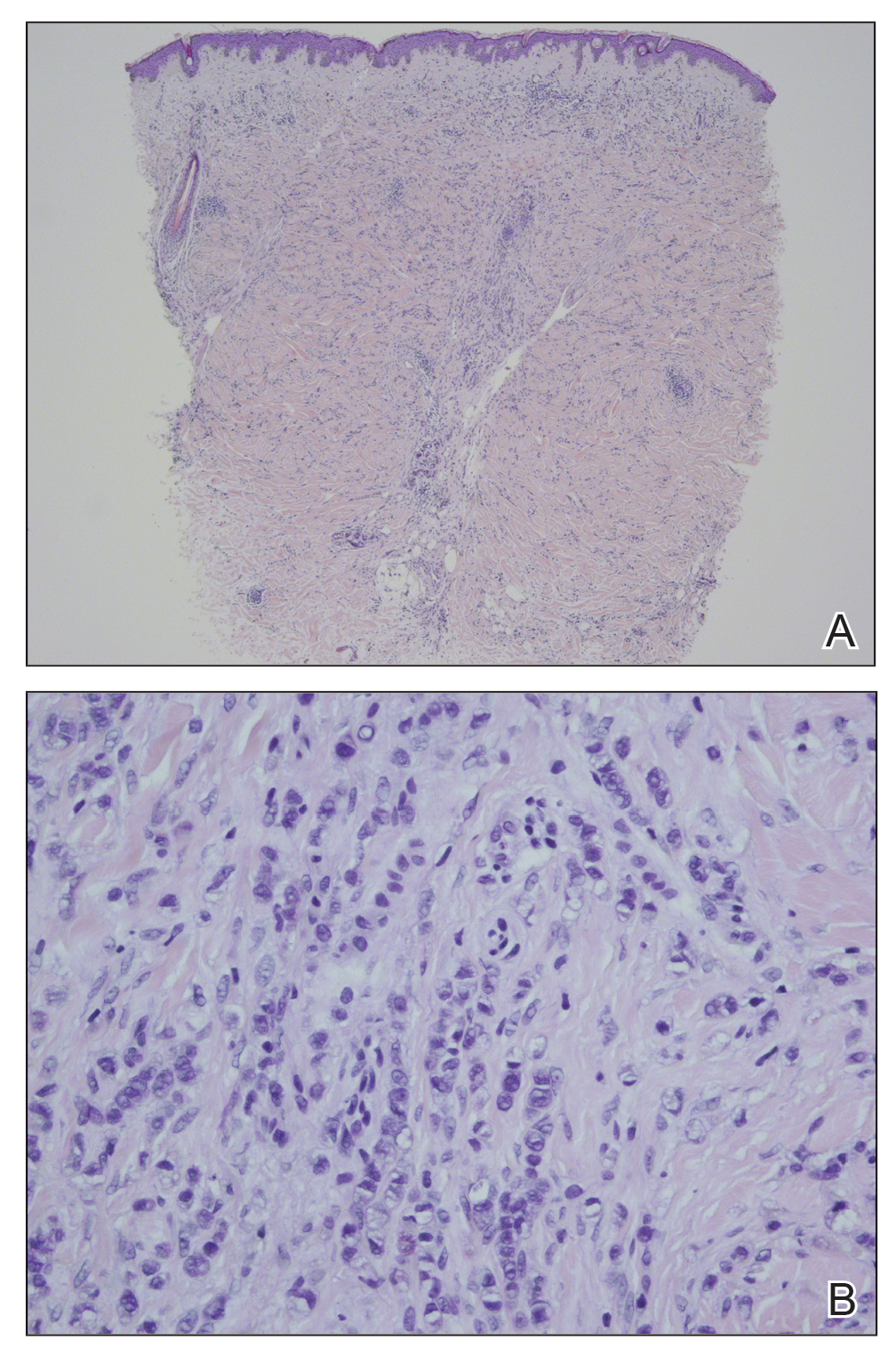
Our patient's presentation was rare in that she developed an asymptomatic erythematous papule on the skin of the abdomen. However, her history of stage IIIB gastric adenocarcinoma in conjunction with the clinical picture and microscopic findings were most consistent with metastatic carcinoma of gastrointestinal origin. The histologic hallmarks of cutaneous metastatic gastric carcinoma include aggregates of neoplastic cells arranged in cords, sometimes forming glands, embedded in a fibrous stroma. Tumor cells may demonstrate signet ring morphology. These unique histologic findings, as well as positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen, rule out other potential diagnoses for an asymptomatic solitary papule.
Dermatofibrosarcoma protuberans presents as an asymptomatic, slow-growing, indurated papule or plaque that develops into a red or brownish nodule. Histologically, dermatofibrosarcoma protuberans is characterized by spindled cells, few mitotic figures, infiltration of the subcutaneous tissue in a honeycomblike pattern, and obliteration of the adnexal structures.4
Cutaneous B-cell lymphoma (CBCL) can present as single or multiple red papules or nodules located on the trunk, face, or extremities. Histologically, CBCL would show a nodular or diffuse infiltrate throughout the dermis, frequently with accentuation in the deep reticular dermis, sparing of the epidermis, and the presence of a grenz zone. The infiltrate in CBCL consists of CD20+, CD19+, and CD79a+ B cells. Identification of a monoclonal B-cell population either by immunohistochemistry or polymerase chain reaction would further support a diagnosis of CBCL.4 These specific histologic findings and the immunohistochemical staining pattern helped rule out CBCL as the diagnosis in our patient.
Amelanotic melanomas present as flesh-colored to light pink papules, making them especially challenging to diagnose clinically. Asymmetrical, poorly circumscribed nests of atypical melanocytes as well as single melanocytes within the epidermis and dermis are seen histologically; mitotic figures are common. Immunohistochemical staining for melanoma includes S-100, human melanoma black 45, MART-1/Melan-A, tyrosinase, and microphthalmia-associated transcription factor 1.4
Neurothekeomas can present as asymptomatic, solitary, flesh-colored papules located on the head, neck, and upper trunk. Histologically, neurothekeomas have a distinct appearance consisting of a well-defined mass composed of variable-sized lobules of spindled and epithelioid cells dispersed in a myxoid stroma within the reticular dermis.4 These specific histologic findings helped rule out neurothekeoma in our patient.
Following the diagnosis of cutaneous metastatic gastric carcinoma in our patient, positron emission tomography and computed tomography of the chest, abdomen, and pelvis were unremarkable for distant disease. Subsequently, the patient underwent surgical excision of the papule with clear margins, followed by a short course of radiation therapy. She currently is under close monitoring but remains in remission with no new cutaneous manifestations of the gastric carcinoma.
- Erdemir A, Atilganoglu U, Onsun N, et al. Cutaneous metastases from gastric adenocarcinoma. Indian J Dermatol. 2011;56:236-237.
- Junqueira AL, Corbett AM, Oliveira Filho Jd, et al. Cutaneous metastasis from gastrointestinal adenocarcinoma of unknown primary origin. An Bras Dermatol. 2015;90:564-566.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:E9-E13.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
The Diagnosis: Cutaneous Metastatic Gastric Carcinoma
Cutaneous metastasis of primary gastric carcinoma is a rare occurrence, with the more common metastatic sites being the lymph nodes, liver, and peritoneal cavity. The incidence of visceral neoplasm metastasis to the skin ranges from 0.7% to 9% and is less than 1% for upper digestive tract carcinomas.1 Cutaneous metastases make up 2% of all tumors of the skin and commonly are located near the site of the primary tumor.2 The most common cutaneous metastasis sites for gastric carcinoma include the neck, chest, and head.3 One of the more typical sites of cutaneous metastasis from gastric cancer is the umbilicus (ie, Sister Mary Joseph nodule). Cutaneous metastases from gastric carcinoma commonly present as asymptomatic hyperpigmented nodules.1,3
In our patient, histopathologic sections showed diffuse infiltration of the dermis by atypical polygonal/round cells arranged in cords and small aggregates. Some of the neoplastic cells had signet ring morphology (Figure). Tumor cells demonstrated positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen; they were negative for S-100, MART-1 (melanoma-associated antigen recognized by T cells 1), leukocyte common antigen, gross cystic disease fluid protein 15, estrogen and progesterone receptor, and HER2/neu (human epidermal growth factor receptor 2).
Our patient's presentation was rare in that she developed an asymptomatic erythematous papule on the skin of the abdomen. However, her history of stage IIIB gastric adenocarcinoma in conjunction with the clinical picture and microscopic findings were most consistent with metastatic carcinoma of gastrointestinal origin. The histologic hallmarks of cutaneous metastatic gastric carcinoma include aggregates of neoplastic cells arranged in cords, sometimes forming glands, embedded in a fibrous stroma. Tumor cells may demonstrate signet ring morphology. These unique histologic findings, as well as positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen, rule out other potential diagnoses for an asymptomatic solitary papule.
Dermatofibrosarcoma protuberans presents as an asymptomatic, slow-growing, indurated papule or plaque that develops into a red or brownish nodule. Histologically, dermatofibrosarcoma protuberans is characterized by spindled cells, few mitotic figures, infiltration of the subcutaneous tissue in a honeycomblike pattern, and obliteration of the adnexal structures.4
Cutaneous B-cell lymphoma (CBCL) can present as single or multiple red papules or nodules located on the trunk, face, or extremities. Histologically, CBCL would show a nodular or diffuse infiltrate throughout the dermis, frequently with accentuation in the deep reticular dermis, sparing of the epidermis, and the presence of a grenz zone. The infiltrate in CBCL consists of CD20+, CD19+, and CD79a+ B cells. Identification of a monoclonal B-cell population either by immunohistochemistry or polymerase chain reaction would further support a diagnosis of CBCL.4 These specific histologic findings and the immunohistochemical staining pattern helped rule out CBCL as the diagnosis in our patient.
Amelanotic melanomas present as flesh-colored to light pink papules, making them especially challenging to diagnose clinically. Asymmetrical, poorly circumscribed nests of atypical melanocytes as well as single melanocytes within the epidermis and dermis are seen histologically; mitotic figures are common. Immunohistochemical staining for melanoma includes S-100, human melanoma black 45, MART-1/Melan-A, tyrosinase, and microphthalmia-associated transcription factor 1.4
Neurothekeomas can present as asymptomatic, solitary, flesh-colored papules located on the head, neck, and upper trunk. Histologically, neurothekeomas have a distinct appearance consisting of a well-defined mass composed of variable-sized lobules of spindled and epithelioid cells dispersed in a myxoid stroma within the reticular dermis.4 These specific histologic findings helped rule out neurothekeoma in our patient.
Following the diagnosis of cutaneous metastatic gastric carcinoma in our patient, positron emission tomography and computed tomography of the chest, abdomen, and pelvis were unremarkable for distant disease. Subsequently, the patient underwent surgical excision of the papule with clear margins, followed by a short course of radiation therapy. She currently is under close monitoring but remains in remission with no new cutaneous manifestations of the gastric carcinoma.
The Diagnosis: Cutaneous Metastatic Gastric Carcinoma
Cutaneous metastasis of primary gastric carcinoma is a rare occurrence, with the more common metastatic sites being the lymph nodes, liver, and peritoneal cavity. The incidence of visceral neoplasm metastasis to the skin ranges from 0.7% to 9% and is less than 1% for upper digestive tract carcinomas.1 Cutaneous metastases make up 2% of all tumors of the skin and commonly are located near the site of the primary tumor.2 The most common cutaneous metastasis sites for gastric carcinoma include the neck, chest, and head.3 One of the more typical sites of cutaneous metastasis from gastric cancer is the umbilicus (ie, Sister Mary Joseph nodule). Cutaneous metastases from gastric carcinoma commonly present as asymptomatic hyperpigmented nodules.1,3
In our patient, histopathologic sections showed diffuse infiltration of the dermis by atypical polygonal/round cells arranged in cords and small aggregates. Some of the neoplastic cells had signet ring morphology (Figure). Tumor cells demonstrated positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen; they were negative for S-100, MART-1 (melanoma-associated antigen recognized by T cells 1), leukocyte common antigen, gross cystic disease fluid protein 15, estrogen and progesterone receptor, and HER2/neu (human epidermal growth factor receptor 2).
Our patient's presentation was rare in that she developed an asymptomatic erythematous papule on the skin of the abdomen. However, her history of stage IIIB gastric adenocarcinoma in conjunction with the clinical picture and microscopic findings were most consistent with metastatic carcinoma of gastrointestinal origin. The histologic hallmarks of cutaneous metastatic gastric carcinoma include aggregates of neoplastic cells arranged in cords, sometimes forming glands, embedded in a fibrous stroma. Tumor cells may demonstrate signet ring morphology. These unique histologic findings, as well as positive immunostaining for CDX2, villin, CAM 5.2, and epithelial membrane antigen, rule out other potential diagnoses for an asymptomatic solitary papule.
Dermatofibrosarcoma protuberans presents as an asymptomatic, slow-growing, indurated papule or plaque that develops into a red or brownish nodule. Histologically, dermatofibrosarcoma protuberans is characterized by spindled cells, few mitotic figures, infiltration of the subcutaneous tissue in a honeycomblike pattern, and obliteration of the adnexal structures.4
Cutaneous B-cell lymphoma (CBCL) can present as single or multiple red papules or nodules located on the trunk, face, or extremities. Histologically, CBCL would show a nodular or diffuse infiltrate throughout the dermis, frequently with accentuation in the deep reticular dermis, sparing of the epidermis, and the presence of a grenz zone. The infiltrate in CBCL consists of CD20+, CD19+, and CD79a+ B cells. Identification of a monoclonal B-cell population either by immunohistochemistry or polymerase chain reaction would further support a diagnosis of CBCL.4 These specific histologic findings and the immunohistochemical staining pattern helped rule out CBCL as the diagnosis in our patient.
Amelanotic melanomas present as flesh-colored to light pink papules, making them especially challenging to diagnose clinically. Asymmetrical, poorly circumscribed nests of atypical melanocytes as well as single melanocytes within the epidermis and dermis are seen histologically; mitotic figures are common. Immunohistochemical staining for melanoma includes S-100, human melanoma black 45, MART-1/Melan-A, tyrosinase, and microphthalmia-associated transcription factor 1.4
Neurothekeomas can present as asymptomatic, solitary, flesh-colored papules located on the head, neck, and upper trunk. Histologically, neurothekeomas have a distinct appearance consisting of a well-defined mass composed of variable-sized lobules of spindled and epithelioid cells dispersed in a myxoid stroma within the reticular dermis.4 These specific histologic findings helped rule out neurothekeoma in our patient.
Following the diagnosis of cutaneous metastatic gastric carcinoma in our patient, positron emission tomography and computed tomography of the chest, abdomen, and pelvis were unremarkable for distant disease. Subsequently, the patient underwent surgical excision of the papule with clear margins, followed by a short course of radiation therapy. She currently is under close monitoring but remains in remission with no new cutaneous manifestations of the gastric carcinoma.
- Erdemir A, Atilganoglu U, Onsun N, et al. Cutaneous metastases from gastric adenocarcinoma. Indian J Dermatol. 2011;56:236-237.
- Junqueira AL, Corbett AM, Oliveira Filho Jd, et al. Cutaneous metastasis from gastrointestinal adenocarcinoma of unknown primary origin. An Bras Dermatol. 2015;90:564-566.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:E9-E13.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
- Erdemir A, Atilganoglu U, Onsun N, et al. Cutaneous metastases from gastric adenocarcinoma. Indian J Dermatol. 2011;56:236-237.
- Junqueira AL, Corbett AM, Oliveira Filho Jd, et al. Cutaneous metastasis from gastrointestinal adenocarcinoma of unknown primary origin. An Bras Dermatol. 2015;90:564-566.
- Cesaretti M, Malerba M, Basso V, et al. Cutaneous metastasis from primary gastric cancer: a case report and review of the literature. Cutis. 2014;93:E9-E13.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. Philadelphia, PA: Elsevier Saunders; 2012.
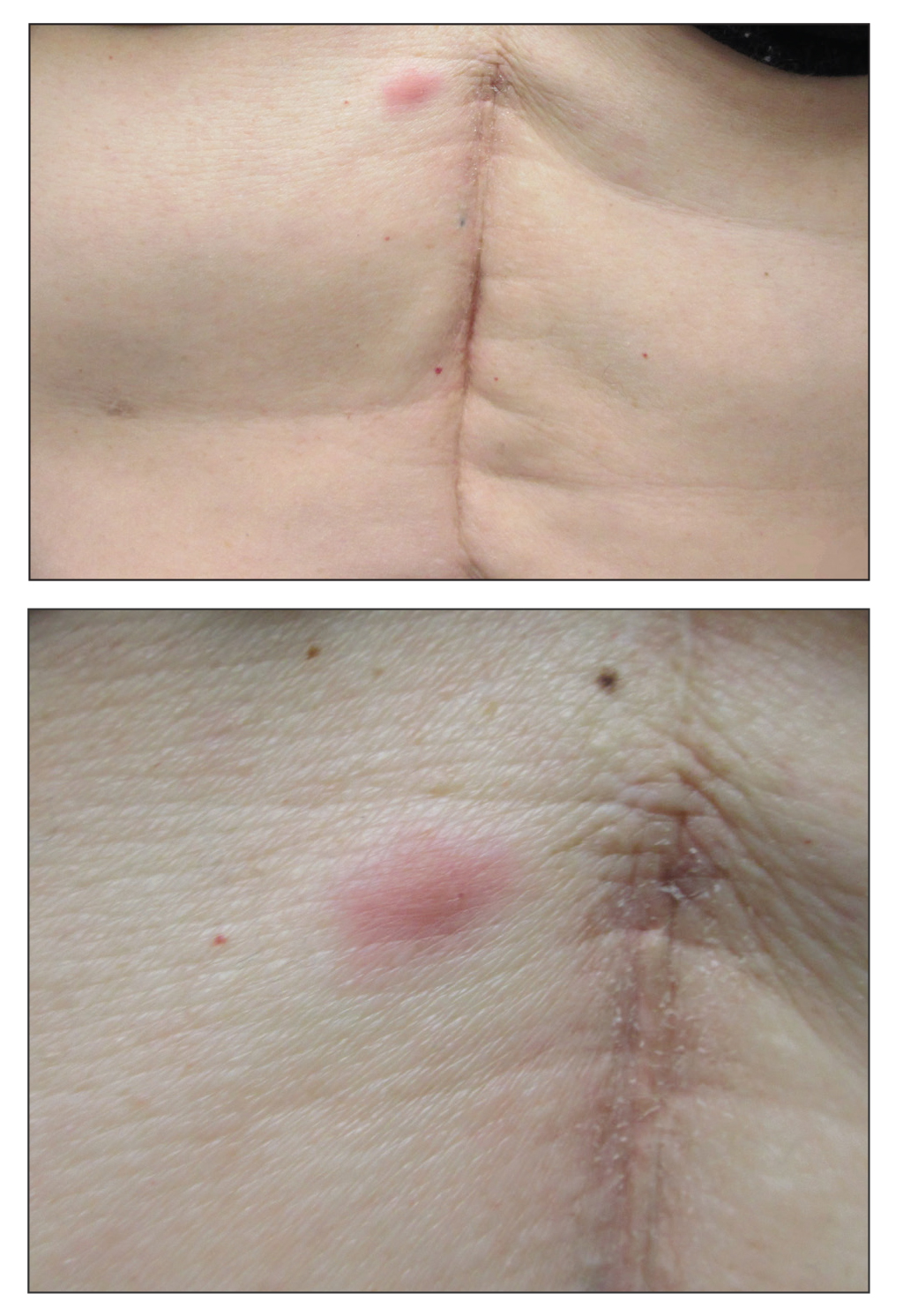
A 53-year-old woman with a history of melanoma on the right thigh, stage II Hodgkin lymphoma, and stage IIIB gastric adenocarcinoma treated with a distal gastrectomy presented with an asymptomatic but persistent skin lesion on the abdomen of 2 months' duration. The lesion arose spontaneously 6 months prior and had increased in size during that time. Physical examination revealed a 6-mm, solitary, firm, erythematous papule on the skin of the right upper quadrant of the abdomen. The patient was otherwise healthy, and a review of systems did not reveal any abnormalities. A punch biopsy was submitted for histopathologic review.