User login
ASCO releases revised version of its Patient-Centered Oncology Payment model
The American Society of Clinical Oncology has made some adjustments to its Patient-Centered Oncology Payment (PCOP) advanced alternative payment model and will now be looking to gain approval from the Physician-Focused Payment Model Technical Advisory Committee (PTAC).

PTAC reviews physician-developed advanced alternative payment models and sends those APMs that are approved to the Centers for Medicare & Medicaid Services’ Center for Medicare & Medicaid Innovation (CMMI) to determine if there will be further testing and possible implementation for use by physicians as part of the APM track of the Quality Payment Program.
Even if approved by PTAC, the ASCO model faces an uphill battle. CMS already has its own Oncology Care Model (OCM), and given the agency’s track record of not testing APMs that have been approved by PTAC, the deck may be stacked against ASCO in terms of getting its model into Medicare.
But officials at ASCO are hoping a PTAC vetting and approval will open the door to its implementation by commercial payers.
“We are now ready to take this to PTAC,” Jeffery Ward, MD, past chair of ASCO’s government relations committee, said in an interview. “I don’t expect any trouble getting through PTAC. The other question is whether CMMI would decide to actually test a model like this, and so far, they have seemed pretty much a one-trick pony, as in they’ve got their Oncology Care Model and they are going to run with it.”
However, Dr. Ward, a medical oncologist at Swedish Cancer Institute of Edmonds, Wash., said that PCOP has been designed with the commercial market in mind and the PTAC recommendation will help validate the model and make it more appealing to commercial payers.
A key feature of the adjusted payment model is that it has taken a lesson learned from CMS’s Oncology Care Model in terms of how to account for the price of drugs. Including the price of drugs in a value-based payment model – the approach taken by the OCM – creates too much variability, Dr. Ward noted. He used lung cancer as an example, noting that the type of lung cancer could have more of an impact on physician spending than any specific treatment decisions a physician can make because the treatment choices may not be there.
“When that happens, whether you are successful or not depends on the luck of the draw instead of the choices you make,” he said.
In an effort not to penalize practices because of their patient mix, PCOP takes the cost of drugs out of the value mix.
“What this model does is it doesn’t hold practices responsible for the cost of the drugs. It holds them responsible for how they utilize the drugs,” he said. “It really begins to make me responsible for making value-based decisions as opposed to just getting lucky and having a cheaper panel of patients.”
Overall, Dr. Ward described the PCOP model as accomplishing the goals of CMS’s APMs by encouraging physicians to treat patients through high-value treatments.
“This model does that by including quality measures that play a role in how much you get paid,” he said. “There are pathways that are value-based pathways and your ability to follow those pathways plays a role in how much you get paid. The amount of care that you send to the ER or the hospital plays a role in how much you get paid. Those things are then used in adjusting what is our fee-for-service model of medicine. So you are still getting paid a base rate based on the care that you give on a fee-for-service model, but that gets adjusted up or down depending on those other characteristics.”
But for the model to have the biggest impact, it needs to be adopted by all payers in a given state or region and used by all physicians, who by the design of the payment model, will have a role in shaping the implementation details.
Savings could come from lower administrative costs at the physician office because there would be no need for separate contracts between payers, something that could generate savings of up to 8%, compared with the current system, Dr. Ward noted.
“This is designed to be a multipayer and multipractice initiative,” he said. “In its grandest form, a state or states would implement this and it would be participated in by all the oncology practices and all of the payers.”
The American Society of Clinical Oncology has made some adjustments to its Patient-Centered Oncology Payment (PCOP) advanced alternative payment model and will now be looking to gain approval from the Physician-Focused Payment Model Technical Advisory Committee (PTAC).
PTAC reviews physician-developed advanced alternative payment models and sends those APMs that are approved to the Centers for Medicare & Medicaid Services’ Center for Medicare & Medicaid Innovation (CMMI) to determine if there will be further testing and possible implementation for use by physicians as part of the APM track of the Quality Payment Program.
Even if approved by PTAC, the ASCO model faces an uphill battle. CMS already has its own Oncology Care Model (OCM), and given the agency’s track record of not testing APMs that have been approved by PTAC, the deck may be stacked against ASCO in terms of getting its model into Medicare.
But officials at ASCO are hoping a PTAC vetting and approval will open the door to its implementation by commercial payers.
“We are now ready to take this to PTAC,” Jeffery Ward, MD, past chair of ASCO’s government relations committee, said in an interview. “I don’t expect any trouble getting through PTAC. The other question is whether CMMI would decide to actually test a model like this, and so far, they have seemed pretty much a one-trick pony, as in they’ve got their Oncology Care Model and they are going to run with it.”
However, Dr. Ward, a medical oncologist at Swedish Cancer Institute of Edmonds, Wash., said that PCOP has been designed with the commercial market in mind and the PTAC recommendation will help validate the model and make it more appealing to commercial payers.
A key feature of the adjusted payment model is that it has taken a lesson learned from CMS’s Oncology Care Model in terms of how to account for the price of drugs. Including the price of drugs in a value-based payment model – the approach taken by the OCM – creates too much variability, Dr. Ward noted. He used lung cancer as an example, noting that the type of lung cancer could have more of an impact on physician spending than any specific treatment decisions a physician can make because the treatment choices may not be there.
“When that happens, whether you are successful or not depends on the luck of the draw instead of the choices you make,” he said.
In an effort not to penalize practices because of their patient mix, PCOP takes the cost of drugs out of the value mix.
“What this model does is it doesn’t hold practices responsible for the cost of the drugs. It holds them responsible for how they utilize the drugs,” he said. “It really begins to make me responsible for making value-based decisions as opposed to just getting lucky and having a cheaper panel of patients.”
Overall, Dr. Ward described the PCOP model as accomplishing the goals of CMS’s APMs by encouraging physicians to treat patients through high-value treatments.
“This model does that by including quality measures that play a role in how much you get paid,” he said. “There are pathways that are value-based pathways and your ability to follow those pathways plays a role in how much you get paid. The amount of care that you send to the ER or the hospital plays a role in how much you get paid. Those things are then used in adjusting what is our fee-for-service model of medicine. So you are still getting paid a base rate based on the care that you give on a fee-for-service model, but that gets adjusted up or down depending on those other characteristics.”
But for the model to have the biggest impact, it needs to be adopted by all payers in a given state or region and used by all physicians, who by the design of the payment model, will have a role in shaping the implementation details.
Savings could come from lower administrative costs at the physician office because there would be no need for separate contracts between payers, something that could generate savings of up to 8%, compared with the current system, Dr. Ward noted.
“This is designed to be a multipayer and multipractice initiative,” he said. “In its grandest form, a state or states would implement this and it would be participated in by all the oncology practices and all of the payers.”
The American Society of Clinical Oncology has made some adjustments to its Patient-Centered Oncology Payment (PCOP) advanced alternative payment model and will now be looking to gain approval from the Physician-Focused Payment Model Technical Advisory Committee (PTAC).
PTAC reviews physician-developed advanced alternative payment models and sends those APMs that are approved to the Centers for Medicare & Medicaid Services’ Center for Medicare & Medicaid Innovation (CMMI) to determine if there will be further testing and possible implementation for use by physicians as part of the APM track of the Quality Payment Program.
Even if approved by PTAC, the ASCO model faces an uphill battle. CMS already has its own Oncology Care Model (OCM), and given the agency’s track record of not testing APMs that have been approved by PTAC, the deck may be stacked against ASCO in terms of getting its model into Medicare.
But officials at ASCO are hoping a PTAC vetting and approval will open the door to its implementation by commercial payers.
“We are now ready to take this to PTAC,” Jeffery Ward, MD, past chair of ASCO’s government relations committee, said in an interview. “I don’t expect any trouble getting through PTAC. The other question is whether CMMI would decide to actually test a model like this, and so far, they have seemed pretty much a one-trick pony, as in they’ve got their Oncology Care Model and they are going to run with it.”
However, Dr. Ward, a medical oncologist at Swedish Cancer Institute of Edmonds, Wash., said that PCOP has been designed with the commercial market in mind and the PTAC recommendation will help validate the model and make it more appealing to commercial payers.
A key feature of the adjusted payment model is that it has taken a lesson learned from CMS’s Oncology Care Model in terms of how to account for the price of drugs. Including the price of drugs in a value-based payment model – the approach taken by the OCM – creates too much variability, Dr. Ward noted. He used lung cancer as an example, noting that the type of lung cancer could have more of an impact on physician spending than any specific treatment decisions a physician can make because the treatment choices may not be there.
“When that happens, whether you are successful or not depends on the luck of the draw instead of the choices you make,” he said.
In an effort not to penalize practices because of their patient mix, PCOP takes the cost of drugs out of the value mix.
“What this model does is it doesn’t hold practices responsible for the cost of the drugs. It holds them responsible for how they utilize the drugs,” he said. “It really begins to make me responsible for making value-based decisions as opposed to just getting lucky and having a cheaper panel of patients.”
Overall, Dr. Ward described the PCOP model as accomplishing the goals of CMS’s APMs by encouraging physicians to treat patients through high-value treatments.
“This model does that by including quality measures that play a role in how much you get paid,” he said. “There are pathways that are value-based pathways and your ability to follow those pathways plays a role in how much you get paid. The amount of care that you send to the ER or the hospital plays a role in how much you get paid. Those things are then used in adjusting what is our fee-for-service model of medicine. So you are still getting paid a base rate based on the care that you give on a fee-for-service model, but that gets adjusted up or down depending on those other characteristics.”
But for the model to have the biggest impact, it needs to be adopted by all payers in a given state or region and used by all physicians, who by the design of the payment model, will have a role in shaping the implementation details.
Savings could come from lower administrative costs at the physician office because there would be no need for separate contracts between payers, something that could generate savings of up to 8%, compared with the current system, Dr. Ward noted.
“This is designed to be a multipayer and multipractice initiative,” he said. “In its grandest form, a state or states would implement this and it would be participated in by all the oncology practices and all of the payers.”
FDA approves Oxbryta for sickle cell disease treatment
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.

Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
ASH preview: Key themes include tackling CAR T obstacles, sickle cell advances, VTE
Chimeric antigen receptor (CAR) T-cell therapies have garnered a great deal of attention given their “incredible efficacy” in treating B-cell malignancies, and new findings are taking aim at the drawbacks of therapy, such as the time, expense, and toxicity involved, according to Robert A. Brodsky, MD.

One example, from a study slated for presentation during a plenary session at the upcoming annual meeting of the American Society of Hematology involves the investigational T-cell bispecific antibody mosunetuzumab, which targets both CD20 on the surface of malignant B cells, and CD3 on cytotoxic T cells, engaging the T cells and directing their cytotoxicity against B cells.
In a study (Abstract 6) of 218 non-Hodgkin lymphoma patients, including 23 who had already received CAR T-cell therapy and had relapsed or were refractory to the treatment, 64% responded, 42% had a complete response, and the median duration of response is now out to 9 months, Dr. Brodsky, ASH secretary and director of the division of hematology at Johns Hopkins University, Baltimore, said during a premeeting press conference.
“It’s basically an antibody using the patient’s own T cell to do what a CAR-T cell would do – [a] very exciting study and large study,” he said. “It is an off-the-shelf product, it completely gets around the problem of the time to generate the CAR T-cell product, and because it’s going to be much simpler and faster to produce, it’s likely going to be much cheaper than CAR T cells.”
The preliminary results also suggest it is less toxic than CAR T-cell therapy, he added.
Two other CAR T-cell therapy–related studies highlighted during the press conference address its use for multiple myeloma. One, the phase 1b/2 CARTITUDE study (Abstract 577) uses CAR T cells against the B-cell maturation antigen (BCMA) in the relapsed/refractory setting.
Of 25 patients treated with chemotherapy followed by CAR T-cell infusion and followed for a median of 3 months, 91% responded, two achieved a complete remission, and “many other responses were very deep responses,” Dr. Brodsky said, noting that the second featured multiple myeloma trial (Abstract 930) looked at bispecific CAR T-cell therapy targeting BCMA and CD38 in an effort to reduce resistance to the therapy.
“Again, very interesting preliminary results,” he said, noting that of 16 patients followed for a median of 36 weeks, 87.5% responded, the treatment was well tolerated, and progression-free survival at 9 months was 75%.
In addition to the “key theme” of overcoming CAR T-cell therapy obstacles, three other themes have emerged from among the thousands of abstracts submitted for presentation at ASH. These, as presented during the press conference, include new venous thromboembolism (VTE) therapies and approaches to research; inclusive medicine, with abstracts focused on age- and race-related issues in clinical trials; and new advances in the treatment of sickle cell disease. All of these have potentially practice-changing implications, as do the six late-breaking abstracts selected from 93 abstracts submitted for consideration for oral presentation at ASH, Dr. Brodsky said.
One of the “truly practice-changing” late-breakers is a randomized phase 3 trial (Abstract LBA-1) comparing the bispecific antibody blinatumomab to chemotherapy for post-re-induction therapy in high- and intermediate-risk acute lymphoblastic leukemia (ALL) at first relapse in children, adolescents and young adults.
The study demonstrated the superiority of blinatumomab for efficacy and tolerability, which is particularly encouraging given the challenges in getting relapsed ALL patients back into remission so they can undergo bone marrow transplant, Dr. Brodsky said.
Of 208 patients randomized, 73% vs. 45% in the blinatumomab vs. chemotherapy arms were able to get to transplant – and therefore to potential cure, he said.
“Of note, the blinatumomab arm was less toxic and there was marked improvement in disease-free and overall survival, so this is clearly going to become a new standard of care for relapsed and refractory ALL,” he added.
Chimeric antigen receptor (CAR) T-cell therapies have garnered a great deal of attention given their “incredible efficacy” in treating B-cell malignancies, and new findings are taking aim at the drawbacks of therapy, such as the time, expense, and toxicity involved, according to Robert A. Brodsky, MD.
One example, from a study slated for presentation during a plenary session at the upcoming annual meeting of the American Society of Hematology involves the investigational T-cell bispecific antibody mosunetuzumab, which targets both CD20 on the surface of malignant B cells, and CD3 on cytotoxic T cells, engaging the T cells and directing their cytotoxicity against B cells.
In a study (Abstract 6) of 218 non-Hodgkin lymphoma patients, including 23 who had already received CAR T-cell therapy and had relapsed or were refractory to the treatment, 64% responded, 42% had a complete response, and the median duration of response is now out to 9 months, Dr. Brodsky, ASH secretary and director of the division of hematology at Johns Hopkins University, Baltimore, said during a premeeting press conference.
“It’s basically an antibody using the patient’s own T cell to do what a CAR-T cell would do – [a] very exciting study and large study,” he said. “It is an off-the-shelf product, it completely gets around the problem of the time to generate the CAR T-cell product, and because it’s going to be much simpler and faster to produce, it’s likely going to be much cheaper than CAR T cells.”
The preliminary results also suggest it is less toxic than CAR T-cell therapy, he added.
Two other CAR T-cell therapy–related studies highlighted during the press conference address its use for multiple myeloma. One, the phase 1b/2 CARTITUDE study (Abstract 577) uses CAR T cells against the B-cell maturation antigen (BCMA) in the relapsed/refractory setting.
Of 25 patients treated with chemotherapy followed by CAR T-cell infusion and followed for a median of 3 months, 91% responded, two achieved a complete remission, and “many other responses were very deep responses,” Dr. Brodsky said, noting that the second featured multiple myeloma trial (Abstract 930) looked at bispecific CAR T-cell therapy targeting BCMA and CD38 in an effort to reduce resistance to the therapy.
“Again, very interesting preliminary results,” he said, noting that of 16 patients followed for a median of 36 weeks, 87.5% responded, the treatment was well tolerated, and progression-free survival at 9 months was 75%.
In addition to the “key theme” of overcoming CAR T-cell therapy obstacles, three other themes have emerged from among the thousands of abstracts submitted for presentation at ASH. These, as presented during the press conference, include new venous thromboembolism (VTE) therapies and approaches to research; inclusive medicine, with abstracts focused on age- and race-related issues in clinical trials; and new advances in the treatment of sickle cell disease. All of these have potentially practice-changing implications, as do the six late-breaking abstracts selected from 93 abstracts submitted for consideration for oral presentation at ASH, Dr. Brodsky said.
One of the “truly practice-changing” late-breakers is a randomized phase 3 trial (Abstract LBA-1) comparing the bispecific antibody blinatumomab to chemotherapy for post-re-induction therapy in high- and intermediate-risk acute lymphoblastic leukemia (ALL) at first relapse in children, adolescents and young adults.
The study demonstrated the superiority of blinatumomab for efficacy and tolerability, which is particularly encouraging given the challenges in getting relapsed ALL patients back into remission so they can undergo bone marrow transplant, Dr. Brodsky said.
Of 208 patients randomized, 73% vs. 45% in the blinatumomab vs. chemotherapy arms were able to get to transplant – and therefore to potential cure, he said.
“Of note, the blinatumomab arm was less toxic and there was marked improvement in disease-free and overall survival, so this is clearly going to become a new standard of care for relapsed and refractory ALL,” he added.
Chimeric antigen receptor (CAR) T-cell therapies have garnered a great deal of attention given their “incredible efficacy” in treating B-cell malignancies, and new findings are taking aim at the drawbacks of therapy, such as the time, expense, and toxicity involved, according to Robert A. Brodsky, MD.
One example, from a study slated for presentation during a plenary session at the upcoming annual meeting of the American Society of Hematology involves the investigational T-cell bispecific antibody mosunetuzumab, which targets both CD20 on the surface of malignant B cells, and CD3 on cytotoxic T cells, engaging the T cells and directing their cytotoxicity against B cells.
In a study (Abstract 6) of 218 non-Hodgkin lymphoma patients, including 23 who had already received CAR T-cell therapy and had relapsed or were refractory to the treatment, 64% responded, 42% had a complete response, and the median duration of response is now out to 9 months, Dr. Brodsky, ASH secretary and director of the division of hematology at Johns Hopkins University, Baltimore, said during a premeeting press conference.
“It’s basically an antibody using the patient’s own T cell to do what a CAR-T cell would do – [a] very exciting study and large study,” he said. “It is an off-the-shelf product, it completely gets around the problem of the time to generate the CAR T-cell product, and because it’s going to be much simpler and faster to produce, it’s likely going to be much cheaper than CAR T cells.”
The preliminary results also suggest it is less toxic than CAR T-cell therapy, he added.
Two other CAR T-cell therapy–related studies highlighted during the press conference address its use for multiple myeloma. One, the phase 1b/2 CARTITUDE study (Abstract 577) uses CAR T cells against the B-cell maturation antigen (BCMA) in the relapsed/refractory setting.
Of 25 patients treated with chemotherapy followed by CAR T-cell infusion and followed for a median of 3 months, 91% responded, two achieved a complete remission, and “many other responses were very deep responses,” Dr. Brodsky said, noting that the second featured multiple myeloma trial (Abstract 930) looked at bispecific CAR T-cell therapy targeting BCMA and CD38 in an effort to reduce resistance to the therapy.
“Again, very interesting preliminary results,” he said, noting that of 16 patients followed for a median of 36 weeks, 87.5% responded, the treatment was well tolerated, and progression-free survival at 9 months was 75%.
In addition to the “key theme” of overcoming CAR T-cell therapy obstacles, three other themes have emerged from among the thousands of abstracts submitted for presentation at ASH. These, as presented during the press conference, include new venous thromboembolism (VTE) therapies and approaches to research; inclusive medicine, with abstracts focused on age- and race-related issues in clinical trials; and new advances in the treatment of sickle cell disease. All of these have potentially practice-changing implications, as do the six late-breaking abstracts selected from 93 abstracts submitted for consideration for oral presentation at ASH, Dr. Brodsky said.
One of the “truly practice-changing” late-breakers is a randomized phase 3 trial (Abstract LBA-1) comparing the bispecific antibody blinatumomab to chemotherapy for post-re-induction therapy in high- and intermediate-risk acute lymphoblastic leukemia (ALL) at first relapse in children, adolescents and young adults.
The study demonstrated the superiority of blinatumomab for efficacy and tolerability, which is particularly encouraging given the challenges in getting relapsed ALL patients back into remission so they can undergo bone marrow transplant, Dr. Brodsky said.
Of 208 patients randomized, 73% vs. 45% in the blinatumomab vs. chemotherapy arms were able to get to transplant – and therefore to potential cure, he said.
“Of note, the blinatumomab arm was less toxic and there was marked improvement in disease-free and overall survival, so this is clearly going to become a new standard of care for relapsed and refractory ALL,” he added.
Survey: Cancer-related pain, opioid use up since 2018
Cancer-related pain was more common among patients in 2019 than in 2018, as was the use of prescription opioids, according to the American Society of Clinical Oncology.
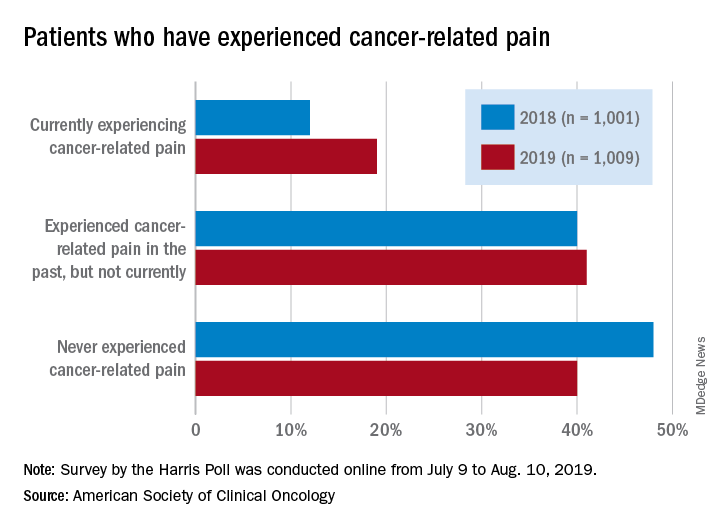
Patients who have/had cancer were significantly more likely to report that they were currently experiencing cancer-related pain in 2019 (19%) than in 2018 (12%), but there was only a slight increase in patients who said that they had experienced cancer-related pain in the past, the society reported in its National Cancer Opinion Survey.
When asked about methods used to manage pain, nausea, and other symptoms, patients diagnosed with cancer most often said that they had not used anything in the last 12 months, although this response was significantly less common in 2019 (48%) than in 2018 (55%). Over-the-counter pain relievers were the most common method used (24% in 2019 and 22% in 2018), followed by vitamins/minerals/herbs (18% in 2019 and 17% in 2018), ASCO said.
Prescription opioids were the third most popular choice for symptom management both years, but use was significantly higher in 2019 (17%) than in 2018 (12%). Also showing a significant increase from 2018 to 2019 was use of medical marijuana, which went from 5% to 10%, ASCO said.
The survey was conducted online for ASCO by the Harris Poll from July 9 to Aug. 10, 2019. The total sample consisted of 4,815 U.S. adults, of whom 1,009 had been diagnosed with cancer.
Cancer-related pain was more common among patients in 2019 than in 2018, as was the use of prescription opioids, according to the American Society of Clinical Oncology.
Patients who have/had cancer were significantly more likely to report that they were currently experiencing cancer-related pain in 2019 (19%) than in 2018 (12%), but there was only a slight increase in patients who said that they had experienced cancer-related pain in the past, the society reported in its National Cancer Opinion Survey.
When asked about methods used to manage pain, nausea, and other symptoms, patients diagnosed with cancer most often said that they had not used anything in the last 12 months, although this response was significantly less common in 2019 (48%) than in 2018 (55%). Over-the-counter pain relievers were the most common method used (24% in 2019 and 22% in 2018), followed by vitamins/minerals/herbs (18% in 2019 and 17% in 2018), ASCO said.
Prescription opioids were the third most popular choice for symptom management both years, but use was significantly higher in 2019 (17%) than in 2018 (12%). Also showing a significant increase from 2018 to 2019 was use of medical marijuana, which went from 5% to 10%, ASCO said.
The survey was conducted online for ASCO by the Harris Poll from July 9 to Aug. 10, 2019. The total sample consisted of 4,815 U.S. adults, of whom 1,009 had been diagnosed with cancer.
Cancer-related pain was more common among patients in 2019 than in 2018, as was the use of prescription opioids, according to the American Society of Clinical Oncology.
Patients who have/had cancer were significantly more likely to report that they were currently experiencing cancer-related pain in 2019 (19%) than in 2018 (12%), but there was only a slight increase in patients who said that they had experienced cancer-related pain in the past, the society reported in its National Cancer Opinion Survey.
When asked about methods used to manage pain, nausea, and other symptoms, patients diagnosed with cancer most often said that they had not used anything in the last 12 months, although this response was significantly less common in 2019 (48%) than in 2018 (55%). Over-the-counter pain relievers were the most common method used (24% in 2019 and 22% in 2018), followed by vitamins/minerals/herbs (18% in 2019 and 17% in 2018), ASCO said.
Prescription opioids were the third most popular choice for symptom management both years, but use was significantly higher in 2019 (17%) than in 2018 (12%). Also showing a significant increase from 2018 to 2019 was use of medical marijuana, which went from 5% to 10%, ASCO said.
The survey was conducted online for ASCO by the Harris Poll from July 9 to Aug. 10, 2019. The total sample consisted of 4,815 U.S. adults, of whom 1,009 had been diagnosed with cancer.
Early lenalidomide may delay progression of smoldering myeloma
Early treatment with lenalidomide may delay disease progression and prevent end-organ damage in patients with high-risk smoldering multiple myeloma (SMM), according to findings from a phase 3 trial.

While observation is the current standard of care in SMM, early therapy may represent a new standard for patients with high-risk disease, explained Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, and colleagues. Their findings were published in the Journal of Clinical Oncology.
The randomized, open-label, phase 3 study included 182 patients with intermediate- or high-risk SMM. Study patients were randomly allocated to receive either oral lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle or observation.
Study subjects were stratified based on time since SMM diagnosis – 1 year or less vs. more than 1 year, and all patients in the lenalidomide arm received aspirin at 325 mg on days 1-28. Both interventions were maintained until unacceptable toxicity, disease progression, or withdrawal for other reasons.
The primary outcome was progression-free survival (PFS), measured from baseline to the development of symptomatic multiple myeloma (MM). The criteria for progression included evidence of end-organ damage in relation to MM and biochemical disease progression.
The researchers found that at 1 year PFS was 98% in the lenalidomide group and 89% in the observation group. At 2 years, PFS was 93% in the lenalidomide group and 76% in the observation group. PFS was 91% in the lenalidomide group and 66% in the observation group at 3 years (hazard ratio, 0.28; P = .002).
Among lenalidomide-treated patients, grade 3 or 4 hematologic and nonhematologic adverse events occurred in 36 patients (41%). Nonhematologic adverse events occurred in 25 patients (28%).
Frequent AEs among lenalidomide-treated patients included grade 4 decreased neutrophil count (4.5%), as well as grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin problems (5.7%), dyspnea (5.7%), and hypokalemia (3.4%). “In most cases, [adverse events] could be managed with dose modifications,” they wrote.
To reduce long-term toxicity, the researchers recommended a 2-year duration of therapy for patients at highest risk.
“Our results support the use of early intervention in patients with high-risk SMM – as defined by the 20/2/20 criteria where our magnitude of benefit was the greatest – rather than continued observation,” the researchers wrote.
The trial was funded by the National Cancer Institute. The authors reported financial affiliations with AbbVie, Aduro Biotech, Amgen, Bristol-Myers Squibb, Celgene, Juno Therapeutics, Kite Pharma, Sanofi, Takeda, and several other companies.
SOURCE: Lonial S et al. J Clin Oncol. 2019 Oct 25. doi: 10.1200/JCO.19.01740.
Early treatment with lenalidomide may delay disease progression and prevent end-organ damage in patients with high-risk smoldering multiple myeloma (SMM), according to findings from a phase 3 trial.
While observation is the current standard of care in SMM, early therapy may represent a new standard for patients with high-risk disease, explained Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, and colleagues. Their findings were published in the Journal of Clinical Oncology.
The randomized, open-label, phase 3 study included 182 patients with intermediate- or high-risk SMM. Study patients were randomly allocated to receive either oral lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle or observation.
Study subjects were stratified based on time since SMM diagnosis – 1 year or less vs. more than 1 year, and all patients in the lenalidomide arm received aspirin at 325 mg on days 1-28. Both interventions were maintained until unacceptable toxicity, disease progression, or withdrawal for other reasons.
The primary outcome was progression-free survival (PFS), measured from baseline to the development of symptomatic multiple myeloma (MM). The criteria for progression included evidence of end-organ damage in relation to MM and biochemical disease progression.
The researchers found that at 1 year PFS was 98% in the lenalidomide group and 89% in the observation group. At 2 years, PFS was 93% in the lenalidomide group and 76% in the observation group. PFS was 91% in the lenalidomide group and 66% in the observation group at 3 years (hazard ratio, 0.28; P = .002).
Among lenalidomide-treated patients, grade 3 or 4 hematologic and nonhematologic adverse events occurred in 36 patients (41%). Nonhematologic adverse events occurred in 25 patients (28%).
Frequent AEs among lenalidomide-treated patients included grade 4 decreased neutrophil count (4.5%), as well as grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin problems (5.7%), dyspnea (5.7%), and hypokalemia (3.4%). “In most cases, [adverse events] could be managed with dose modifications,” they wrote.
To reduce long-term toxicity, the researchers recommended a 2-year duration of therapy for patients at highest risk.
“Our results support the use of early intervention in patients with high-risk SMM – as defined by the 20/2/20 criteria where our magnitude of benefit was the greatest – rather than continued observation,” the researchers wrote.
The trial was funded by the National Cancer Institute. The authors reported financial affiliations with AbbVie, Aduro Biotech, Amgen, Bristol-Myers Squibb, Celgene, Juno Therapeutics, Kite Pharma, Sanofi, Takeda, and several other companies.
SOURCE: Lonial S et al. J Clin Oncol. 2019 Oct 25. doi: 10.1200/JCO.19.01740.
Early treatment with lenalidomide may delay disease progression and prevent end-organ damage in patients with high-risk smoldering multiple myeloma (SMM), according to findings from a phase 3 trial.
While observation is the current standard of care in SMM, early therapy may represent a new standard for patients with high-risk disease, explained Sagar Lonial, MD, of Winship Cancer Institute, Emory University, Atlanta, and colleagues. Their findings were published in the Journal of Clinical Oncology.
The randomized, open-label, phase 3 study included 182 patients with intermediate- or high-risk SMM. Study patients were randomly allocated to receive either oral lenalidomide at 25 mg daily on days 1-21 of a 28-day cycle or observation.
Study subjects were stratified based on time since SMM diagnosis – 1 year or less vs. more than 1 year, and all patients in the lenalidomide arm received aspirin at 325 mg on days 1-28. Both interventions were maintained until unacceptable toxicity, disease progression, or withdrawal for other reasons.
The primary outcome was progression-free survival (PFS), measured from baseline to the development of symptomatic multiple myeloma (MM). The criteria for progression included evidence of end-organ damage in relation to MM and biochemical disease progression.
The researchers found that at 1 year PFS was 98% in the lenalidomide group and 89% in the observation group. At 2 years, PFS was 93% in the lenalidomide group and 76% in the observation group. PFS was 91% in the lenalidomide group and 66% in the observation group at 3 years (hazard ratio, 0.28; P = .002).
Among lenalidomide-treated patients, grade 3 or 4 hematologic and nonhematologic adverse events occurred in 36 patients (41%). Nonhematologic adverse events occurred in 25 patients (28%).
Frequent AEs among lenalidomide-treated patients included grade 4 decreased neutrophil count (4.5%), as well as grade 3 infections (20.5%), hypertension (9.1%), fatigue (6.8%), skin problems (5.7%), dyspnea (5.7%), and hypokalemia (3.4%). “In most cases, [adverse events] could be managed with dose modifications,” they wrote.
To reduce long-term toxicity, the researchers recommended a 2-year duration of therapy for patients at highest risk.
“Our results support the use of early intervention in patients with high-risk SMM – as defined by the 20/2/20 criteria where our magnitude of benefit was the greatest – rather than continued observation,” the researchers wrote.
The trial was funded by the National Cancer Institute. The authors reported financial affiliations with AbbVie, Aduro Biotech, Amgen, Bristol-Myers Squibb, Celgene, Juno Therapeutics, Kite Pharma, Sanofi, Takeda, and several other companies.
SOURCE: Lonial S et al. J Clin Oncol. 2019 Oct 25. doi: 10.1200/JCO.19.01740.
FROM JOURNAL OF CLINICAL ONCOLOGY
FDA approves treatment for sickle cell pain crises
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.

The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
Researchers seek a way to predict cognitive deficits in children treated for ALL
Researchers are attempting to determine, early in the treatment process, which children with acute lymphoblastic leukemia (ALL) have an increased risk of neurocognitive deficits after chemotherapy.

The goal of the researchers’ project (5R01CA220568-02) is to determine if gene variants and biomarkers associated with oxidative stress, neuroinflammation, and folate physiology correlate with cognitive decline during and after chemotherapy. Ideally, certain variants and biomarkers will reveal patients who might benefit from interventions to prevent or even reverse cognitive deficits.
Peter D. Cole, MD, of Rutgers Cancer Institute, New Brunswick, N.J., and colleagues are conducting this research in patients from the DFCI-16-001 trial (NCT03020030). This multicenter, phase 3 study is enrolling patients (aged 1-21 years) with B- or T-cell ALL who then receive a multidrug chemotherapy regimen.
Dr. Cole and colleagues are analyzing a subset of patients from the trial, looking for relationships between chemotherapy-induced neurocognitive changes, gene variants, and changes in biomarkers detected in cerebrospinal fluid (CSF).
“We’re looking at a broad panel of target gene variants that are associated with either drug metabolism, defenses against oxidative stress, neuroinflammation, or folate physiology,” Dr. Cole said in an interview.
This includes variants Dr. Cole and colleagues identified in a previous, retrospective study of ALL survivors. The researchers found that survivors who were homozygous for NOS3 894T, had a variant SLCO2A1 G allele, or had at least one GSTP1 T allele were more likely to exhibit cognitive deficits (J Clin Oncol. 2015 Jul 1;33[19]:2205-11).
The researchers are also analyzing CSF samples, looking for changes in tau protein, homocysteine, homocysteic acid, the adenosylmethionine to adenosylhomocysteine ratio, and other biomarkers of oxidative stress, neuroinflammation, and folate physiology. The CSF is collected at five time points: the start of chemotherapy, day 18, the start of first consolidation, the end of first consolidation, and 7 weeks later in second consolidation.
Cognitive testing
While Dr. Cole is leading the genetic and biomarker analyses, Stephen A. Sands, PsyD, of Memorial Sloan Kettering Cancer Center in New York, is leading the cognitive testing.

The researchers are evaluating patients for cognitive decline using computerized tests from a company called Cogstate. The tests are designed to assess functions such as processing speed, attention, visual learning, and working memory. The tests are administered on an iPad and involve tasks like identifying features of playing cards and finding the correct way through a maze.
The patients – aged 3 years and older – undergo cognitive testing at six time points: baseline, which is any time between days 8 and 32 of induction (except within 72 hours after sedation or anesthesia); at first consolidation; the end of central nervous system therapy; 1 year into chemotherapy; the end of chemotherapy; and 1 year after chemotherapy ends.
In a prior study, Cogstate testing proved reliable for detecting neurocognitive changes in patients undergoing treatment for ALL (Support Care Cancer. 2017;25[2]:449-57). In the current study, the researchers are supplementing Cogstate test results with Wechsler IQ tests administered 1 year after patients complete chemotherapy.
Dr. Sands noted that Cogstate tests provide benefits over the Wechsler “paper-and-pencil” tests. One benefit is that Cogstate tests can be given more often without inducing practice effects (J Clin Exp Neuropsychol. 2006 Oct;28[7]:1095-112). Another is that Cogstate tests can be administered by anyone with a bachelor’s degree who has undergone the appropriate training, while Wechsler IQ tests must be given by psychologists.
Preliminary results
This research is ongoing, so it’s too early to announce any discoveries, but the study is moving along as planned.

“The preliminary data we have so far are demonstrating the validity of the study,” Dr. Cole said. “Things are going well. We’re able to do the cognitive testing and collect the samples that we need and ship them without losing the integrity of the samples.”
Dr. Sands noted that enrollment has been encouraging. As this is a substudy of DFCI-16-001, the researchers must obtain consent separately from the main study. Dr. Sands said about 89% of parents involved in the main study have agreed to enroll their children in the substudy.
Dr. Sands also said that early results from Cogstate testing have revealed patients who are experiencing cognitive decline during treatment. The researchers still have to determine if these results correlate with any biomarkers or gene variants.
Potential interventions
If the researchers can pinpoint patients at risk for cognitive deficits, the next step will be to investigate pharmacologic and behavioral interventions.
Dr. Cole said he is particularly interested in treatments that reduce oxidative stress, such as dextromethorphan and memantine. Dextromethorphan has been shown to resolve symptoms of methotrexate-induced neurotoxicity in patients (Pediatr Hematol Oncol. 2002 Jul-Aug;19[5]:319-27), and memantine reduced memory deficits in animals treated with methotrexate (Clin Cancer Res. 2013 Aug 15;19[16]:4446-54).
“Memantine hasn’t been used in kids with leukemia yet, but it’s something that I’d like to see brought to a clinical trial,” Dr. Cole said.
Dr. Sands pointed to other potential pharmacologic interventions, including the stimulants methylphenidate and modafinil. Both drugs have been shown to improve cognitive deficits in cancer survivors (J Clin Oncol. 2001 Mar 15;19[6]:1802-8; Cancer. 2009 Jun 15; 115[12]: 2605-16).
Computer-based cognitive training tools may be another option. One such tool, Lumosity, improved executive functions in a study of breast cancer survivors (Clin Breast Cancer. 2013 Aug;13[4]:299-306). Another tool, CogMed, improved working memory in survivors of brain tumors and ALL (Psychooncology. 2013 Aug; 22[8]: 1856-65).
Other behavioral interventions might include sleep hygiene and exercise. Sleep hygiene has been shown to improve cognitive function in childhood cancer survivors (Cancer. 2011 Jun 1;117[11]:2559-68), and a recent study revealed an association between exercise intolerance and negative neurocognitive outcomes in ALL survivors (Cancer. 2019 Oct 21. doi: 10.1002/cncr.32510).
“What we need to figure out is which children will respond to which interventions,” Dr. Sands said, adding that interventions will likely need to be combined.
“It’s not going to be one thing that will work for everybody,” he said. “It’s going to be: What packages of things will work for different people?”
Dr. Sands and Dr. Cole reported having no relevant financial disclosures.
Researchers are attempting to determine, early in the treatment process, which children with acute lymphoblastic leukemia (ALL) have an increased risk of neurocognitive deficits after chemotherapy.
The goal of the researchers’ project (5R01CA220568-02) is to determine if gene variants and biomarkers associated with oxidative stress, neuroinflammation, and folate physiology correlate with cognitive decline during and after chemotherapy. Ideally, certain variants and biomarkers will reveal patients who might benefit from interventions to prevent or even reverse cognitive deficits.
Peter D. Cole, MD, of Rutgers Cancer Institute, New Brunswick, N.J., and colleagues are conducting this research in patients from the DFCI-16-001 trial (NCT03020030). This multicenter, phase 3 study is enrolling patients (aged 1-21 years) with B- or T-cell ALL who then receive a multidrug chemotherapy regimen.
Dr. Cole and colleagues are analyzing a subset of patients from the trial, looking for relationships between chemotherapy-induced neurocognitive changes, gene variants, and changes in biomarkers detected in cerebrospinal fluid (CSF).
“We’re looking at a broad panel of target gene variants that are associated with either drug metabolism, defenses against oxidative stress, neuroinflammation, or folate physiology,” Dr. Cole said in an interview.
This includes variants Dr. Cole and colleagues identified in a previous, retrospective study of ALL survivors. The researchers found that survivors who were homozygous for NOS3 894T, had a variant SLCO2A1 G allele, or had at least one GSTP1 T allele were more likely to exhibit cognitive deficits (J Clin Oncol. 2015 Jul 1;33[19]:2205-11).
The researchers are also analyzing CSF samples, looking for changes in tau protein, homocysteine, homocysteic acid, the adenosylmethionine to adenosylhomocysteine ratio, and other biomarkers of oxidative stress, neuroinflammation, and folate physiology. The CSF is collected at five time points: the start of chemotherapy, day 18, the start of first consolidation, the end of first consolidation, and 7 weeks later in second consolidation.
Cognitive testing
While Dr. Cole is leading the genetic and biomarker analyses, Stephen A. Sands, PsyD, of Memorial Sloan Kettering Cancer Center in New York, is leading the cognitive testing.
The researchers are evaluating patients for cognitive decline using computerized tests from a company called Cogstate. The tests are designed to assess functions such as processing speed, attention, visual learning, and working memory. The tests are administered on an iPad and involve tasks like identifying features of playing cards and finding the correct way through a maze.
The patients – aged 3 years and older – undergo cognitive testing at six time points: baseline, which is any time between days 8 and 32 of induction (except within 72 hours after sedation or anesthesia); at first consolidation; the end of central nervous system therapy; 1 year into chemotherapy; the end of chemotherapy; and 1 year after chemotherapy ends.
In a prior study, Cogstate testing proved reliable for detecting neurocognitive changes in patients undergoing treatment for ALL (Support Care Cancer. 2017;25[2]:449-57). In the current study, the researchers are supplementing Cogstate test results with Wechsler IQ tests administered 1 year after patients complete chemotherapy.
Dr. Sands noted that Cogstate tests provide benefits over the Wechsler “paper-and-pencil” tests. One benefit is that Cogstate tests can be given more often without inducing practice effects (J Clin Exp Neuropsychol. 2006 Oct;28[7]:1095-112). Another is that Cogstate tests can be administered by anyone with a bachelor’s degree who has undergone the appropriate training, while Wechsler IQ tests must be given by psychologists.
Preliminary results
This research is ongoing, so it’s too early to announce any discoveries, but the study is moving along as planned.
“The preliminary data we have so far are demonstrating the validity of the study,” Dr. Cole said. “Things are going well. We’re able to do the cognitive testing and collect the samples that we need and ship them without losing the integrity of the samples.”
Dr. Sands noted that enrollment has been encouraging. As this is a substudy of DFCI-16-001, the researchers must obtain consent separately from the main study. Dr. Sands said about 89% of parents involved in the main study have agreed to enroll their children in the substudy.
Dr. Sands also said that early results from Cogstate testing have revealed patients who are experiencing cognitive decline during treatment. The researchers still have to determine if these results correlate with any biomarkers or gene variants.
Potential interventions
If the researchers can pinpoint patients at risk for cognitive deficits, the next step will be to investigate pharmacologic and behavioral interventions.
Dr. Cole said he is particularly interested in treatments that reduce oxidative stress, such as dextromethorphan and memantine. Dextromethorphan has been shown to resolve symptoms of methotrexate-induced neurotoxicity in patients (Pediatr Hematol Oncol. 2002 Jul-Aug;19[5]:319-27), and memantine reduced memory deficits in animals treated with methotrexate (Clin Cancer Res. 2013 Aug 15;19[16]:4446-54).
“Memantine hasn’t been used in kids with leukemia yet, but it’s something that I’d like to see brought to a clinical trial,” Dr. Cole said.
Dr. Sands pointed to other potential pharmacologic interventions, including the stimulants methylphenidate and modafinil. Both drugs have been shown to improve cognitive deficits in cancer survivors (J Clin Oncol. 2001 Mar 15;19[6]:1802-8; Cancer. 2009 Jun 15; 115[12]: 2605-16).
Computer-based cognitive training tools may be another option. One such tool, Lumosity, improved executive functions in a study of breast cancer survivors (Clin Breast Cancer. 2013 Aug;13[4]:299-306). Another tool, CogMed, improved working memory in survivors of brain tumors and ALL (Psychooncology. 2013 Aug; 22[8]: 1856-65).
Other behavioral interventions might include sleep hygiene and exercise. Sleep hygiene has been shown to improve cognitive function in childhood cancer survivors (Cancer. 2011 Jun 1;117[11]:2559-68), and a recent study revealed an association between exercise intolerance and negative neurocognitive outcomes in ALL survivors (Cancer. 2019 Oct 21. doi: 10.1002/cncr.32510).
“What we need to figure out is which children will respond to which interventions,” Dr. Sands said, adding that interventions will likely need to be combined.
“It’s not going to be one thing that will work for everybody,” he said. “It’s going to be: What packages of things will work for different people?”
Dr. Sands and Dr. Cole reported having no relevant financial disclosures.
Researchers are attempting to determine, early in the treatment process, which children with acute lymphoblastic leukemia (ALL) have an increased risk of neurocognitive deficits after chemotherapy.
The goal of the researchers’ project (5R01CA220568-02) is to determine if gene variants and biomarkers associated with oxidative stress, neuroinflammation, and folate physiology correlate with cognitive decline during and after chemotherapy. Ideally, certain variants and biomarkers will reveal patients who might benefit from interventions to prevent or even reverse cognitive deficits.
Peter D. Cole, MD, of Rutgers Cancer Institute, New Brunswick, N.J., and colleagues are conducting this research in patients from the DFCI-16-001 trial (NCT03020030). This multicenter, phase 3 study is enrolling patients (aged 1-21 years) with B- or T-cell ALL who then receive a multidrug chemotherapy regimen.
Dr. Cole and colleagues are analyzing a subset of patients from the trial, looking for relationships between chemotherapy-induced neurocognitive changes, gene variants, and changes in biomarkers detected in cerebrospinal fluid (CSF).
“We’re looking at a broad panel of target gene variants that are associated with either drug metabolism, defenses against oxidative stress, neuroinflammation, or folate physiology,” Dr. Cole said in an interview.
This includes variants Dr. Cole and colleagues identified in a previous, retrospective study of ALL survivors. The researchers found that survivors who were homozygous for NOS3 894T, had a variant SLCO2A1 G allele, or had at least one GSTP1 T allele were more likely to exhibit cognitive deficits (J Clin Oncol. 2015 Jul 1;33[19]:2205-11).
The researchers are also analyzing CSF samples, looking for changes in tau protein, homocysteine, homocysteic acid, the adenosylmethionine to adenosylhomocysteine ratio, and other biomarkers of oxidative stress, neuroinflammation, and folate physiology. The CSF is collected at five time points: the start of chemotherapy, day 18, the start of first consolidation, the end of first consolidation, and 7 weeks later in second consolidation.
Cognitive testing
While Dr. Cole is leading the genetic and biomarker analyses, Stephen A. Sands, PsyD, of Memorial Sloan Kettering Cancer Center in New York, is leading the cognitive testing.
The researchers are evaluating patients for cognitive decline using computerized tests from a company called Cogstate. The tests are designed to assess functions such as processing speed, attention, visual learning, and working memory. The tests are administered on an iPad and involve tasks like identifying features of playing cards and finding the correct way through a maze.
The patients – aged 3 years and older – undergo cognitive testing at six time points: baseline, which is any time between days 8 and 32 of induction (except within 72 hours after sedation or anesthesia); at first consolidation; the end of central nervous system therapy; 1 year into chemotherapy; the end of chemotherapy; and 1 year after chemotherapy ends.
In a prior study, Cogstate testing proved reliable for detecting neurocognitive changes in patients undergoing treatment for ALL (Support Care Cancer. 2017;25[2]:449-57). In the current study, the researchers are supplementing Cogstate test results with Wechsler IQ tests administered 1 year after patients complete chemotherapy.
Dr. Sands noted that Cogstate tests provide benefits over the Wechsler “paper-and-pencil” tests. One benefit is that Cogstate tests can be given more often without inducing practice effects (J Clin Exp Neuropsychol. 2006 Oct;28[7]:1095-112). Another is that Cogstate tests can be administered by anyone with a bachelor’s degree who has undergone the appropriate training, while Wechsler IQ tests must be given by psychologists.
Preliminary results
This research is ongoing, so it’s too early to announce any discoveries, but the study is moving along as planned.
“The preliminary data we have so far are demonstrating the validity of the study,” Dr. Cole said. “Things are going well. We’re able to do the cognitive testing and collect the samples that we need and ship them without losing the integrity of the samples.”
Dr. Sands noted that enrollment has been encouraging. As this is a substudy of DFCI-16-001, the researchers must obtain consent separately from the main study. Dr. Sands said about 89% of parents involved in the main study have agreed to enroll their children in the substudy.
Dr. Sands also said that early results from Cogstate testing have revealed patients who are experiencing cognitive decline during treatment. The researchers still have to determine if these results correlate with any biomarkers or gene variants.
Potential interventions
If the researchers can pinpoint patients at risk for cognitive deficits, the next step will be to investigate pharmacologic and behavioral interventions.
Dr. Cole said he is particularly interested in treatments that reduce oxidative stress, such as dextromethorphan and memantine. Dextromethorphan has been shown to resolve symptoms of methotrexate-induced neurotoxicity in patients (Pediatr Hematol Oncol. 2002 Jul-Aug;19[5]:319-27), and memantine reduced memory deficits in animals treated with methotrexate (Clin Cancer Res. 2013 Aug 15;19[16]:4446-54).
“Memantine hasn’t been used in kids with leukemia yet, but it’s something that I’d like to see brought to a clinical trial,” Dr. Cole said.
Dr. Sands pointed to other potential pharmacologic interventions, including the stimulants methylphenidate and modafinil. Both drugs have been shown to improve cognitive deficits in cancer survivors (J Clin Oncol. 2001 Mar 15;19[6]:1802-8; Cancer. 2009 Jun 15; 115[12]: 2605-16).
Computer-based cognitive training tools may be another option. One such tool, Lumosity, improved executive functions in a study of breast cancer survivors (Clin Breast Cancer. 2013 Aug;13[4]:299-306). Another tool, CogMed, improved working memory in survivors of brain tumors and ALL (Psychooncology. 2013 Aug; 22[8]: 1856-65).
Other behavioral interventions might include sleep hygiene and exercise. Sleep hygiene has been shown to improve cognitive function in childhood cancer survivors (Cancer. 2011 Jun 1;117[11]:2559-68), and a recent study revealed an association between exercise intolerance and negative neurocognitive outcomes in ALL survivors (Cancer. 2019 Oct 21. doi: 10.1002/cncr.32510).
“What we need to figure out is which children will respond to which interventions,” Dr. Sands said, adding that interventions will likely need to be combined.
“It’s not going to be one thing that will work for everybody,” he said. “It’s going to be: What packages of things will work for different people?”
Dr. Sands and Dr. Cole reported having no relevant financial disclosures.
Transfusion-related lung injury is on the rise in elderly patients
SAN ANTONIO – Although there has been a general decline in transfusion-related anaphylaxis and acute infections over time among hospitalized older adults in the United States, incidence rates for both transfusion-related acute lung injury and transfusion-associated circulatory overload have risen over the last decade, according to researchers from the Food and Drug Administration.

Mikhail Menis, PharmD, an epidemiologist at the FDA Center for Biologics Evaluation and Research (CBER) and colleagues queried large Medicare databases to assess trends in transfusion-related adverse events among adults aged 65 years and older.
The investigators saw “substantially higher risk of all outcomes among immunocompromised beneficiaries, which could be related to higher blood use of all blood components, especially platelets, underlying conditions such as malignancies, and treatments such as chemotherapy or radiation, which need further investigation,” Dr. Menis said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
He reported data from a series of studies on four categories of transfusion-related events that may be life-threatening or fatal: transfusion-related anaphylaxis (TRA), transfusion-related acute lung injury (TRALI), transfusion-associated circulatory overload (TACO), and acute infection following transfusion (AIFT).
For each type of event, the researchers looked at overall incidence and the incidence by immune status, calendar year, blood components transfused, number of units transfused, age, sex, and race.
Anaphylaxis (TRA)
TRA may be caused by preformed immunoglobin E (IgE) antibodies to proteins in the plasma in transfused blood products or by preformed IgA antibodies in patients who are likely IgA deficient, Dr. Menis said.
The overall incidence of TRA among 8,833,817 inpatient transfusions stays for elderly beneficiaries from 2012 through 2018 was 7.1 per 100,000 stays. The rate was higher for immunocompromised patients, at 9.6, than it was among nonimmunocompromised patients, at 6.5.
The rates varied by every subgroup measured except immune status. Annual rates showed a downward trend, from 8.7 per 100,000 in 2012, to 5.1 in 2017 and 6.4 in 2018. The decline in occurrence may be caused by a decline in inpatient blood utilization during the study period, particularly among immunocompromised patients.
TRA rates increased with five or more units transfused. The risk was significantly reduced in the oldest group of patients versus the youngest (P less than .001), which supports the immune-based mechanism of action of anaphylaxis, Dr. Menis said.
They also found that TRA rates were substantially higher among patients who had received platelet and/or plasma transfusions, compared with patients who received only red blood cells (RBCs).
Additionally, risk for TRA was significantly higher among men than it was among women (9.3 vs. 5.4) and among white versus nonwhite patients (7.8 vs. 3.8).
The evidence suggested TRA cases are likely to be severe in this population, with inpatient mortality of 7.1%, and hospital stays of 7 days or longer in about 58% of cases, indicating the importance of TRA prevention, Dr. Menis said.
The investigators plan to perform multivariate regression analyses to assess potential risk factors, including underlying comorbidities and health histories for TRA occurrence for both the overall population and by immune status.
Acute lung injury (TRALI)
TRALI is a rare but serious adverse event, a clinical syndrome with onset within 6 hours of transfusion that presents as acute hypoxemia, respiratory distress, and noncardiogenic pulmonary edema.
Among 17,771,193 total inpatient transfusion stays, the overall incidence of TRALI was 33.2 per 100,000. The rate was 55.9 for immunocompromised patients versus 28.4 for nonimmunocompromised patients. The rate ratio was 2.0 (P less than .001).
The difference by immune status may be caused by higher blood utilizations with more units transfused per stay among immunocompromised patients, a higher incidence of prior transfusions among these patients, higher use of irradiated blood components that may lead to accumulation of proinflammatory mediators in blood products during storage, or underlying comorbidities.
The overall rate increased from 14.3 in 2007 to 56.4 in 2018. The rates increased proportionally among both immunocompromised and nonimmunocompromised patients.
As with TRA, the incidence of TRALI was higher in patients with five or more units transfused, while the incidence declined with age, likely caused by declining blood use and age-related changes in neutrophil function, Dr. Menis said.
TRALI rates were slightly higher among men than among women, as well as higher among white patients than among nonwhite patients.
Overall, TRALI rates were higher for patients who received platelets either alone or in combination with RBCs and/or plasma. The highest rates were among patients who received RBCs, plasma and platelets.
Dr. Menis called for studies to determine what effects the processing and storage of blood components may have on TRALI occurrence; he and his colleagues also are planning regression analyses to assess potential risk factors for this complication.
Circulatory overload (TACO)
TACO is one of the leading reported causes of transfusion-related fatalities in the U.S., with onset usually occurring within 6 hours of transfusion, presenting as acute respiratory distress with dyspnea, orthopnea, increased blood pressure, and cardiogenic pulmonary edema.
The overall incidence of TACO among hospitalized patients aged 65 years and older from 2011 through 2018 was 86.3 per 100,000 stays. The incidences were 128.3 in immunocompromised and 76.0 in nonimmunocompromised patients. The rate ratio for TACO in immunocompromised versus nonimmunocompromised patients was 1.70 (P less than .001).
Overall incidence rates of TACO rose from 62 per 100,000 stays in 2011 to 119.8 in 2018. As with other adverse events, incident rates rose with the number of units transfused.
Rates of TACO were significantly higher among women than they were among men (94.6 vs. 75.9 per 100,000; P less than .001), which could be caused by the higher mean age of women and/or a lower tolerance for increased blood volume from transfusion.
The study results also suggested that TACO and TRALI may coexist, based on evidence that 3.5% of all TACO stays also had diagnostic codes for TRALI. The frequency of co-occurrence of these two adverse events also increased over time, which may be caused by improved awareness, Dr. Menis said.
Infections (AIFT)
Acute infections following transfusion can lead to prolonged hospitalizations, sepsis, septic shock, and death. Those most at risk include elderly and immunocompromised patients because of high utilization of blood products, comorbidities, and decreased immune function.
Among 8,833,817 stays, the overall rate per 100,000 stays was 2.1. The rate for immunocompromised patients was 5.4, compared with 1.2 for nonimmunocompromised patients, for a rate ratio of 4.4 (P less than .001).
The incidence rate declined significantly (P = .03) over the study period, with the 3 latest years having the lowest rates.
Rates increased substantially among immunocompromised patients by the number of units transfused, but remained relatively stable among nonimmunocompromised patients.
Infection rates declined with age, from 2.7 per 100,000 stays for patients aged 65-68 years to 1.2 per 100,000 for those aged 85 years and older.
As with other adverse events, AIFT rates were likely related to the blood components transfused, with substantially higher rates for stays during which platelets were transfused either alone or with RBCs, compared with RBCs alone. This could be caused by the room-temperature storage of platelets and higher number of platelets units transfused, compared with RBCs alone, especially among immunocompromised patients.
In all, 51.9% of AIFT cases also had sepsis noted in the medical record, indicating high severity and emphasizing the importance of AIFT prevention, Dr. Menis said.
The studies were funded by the FDA, and Dr. Menis is an FDA employee. He reported having no conflicts of interest.
SAN ANTONIO – Although there has been a general decline in transfusion-related anaphylaxis and acute infections over time among hospitalized older adults in the United States, incidence rates for both transfusion-related acute lung injury and transfusion-associated circulatory overload have risen over the last decade, according to researchers from the Food and Drug Administration.
Mikhail Menis, PharmD, an epidemiologist at the FDA Center for Biologics Evaluation and Research (CBER) and colleagues queried large Medicare databases to assess trends in transfusion-related adverse events among adults aged 65 years and older.
The investigators saw “substantially higher risk of all outcomes among immunocompromised beneficiaries, which could be related to higher blood use of all blood components, especially platelets, underlying conditions such as malignancies, and treatments such as chemotherapy or radiation, which need further investigation,” Dr. Menis said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
He reported data from a series of studies on four categories of transfusion-related events that may be life-threatening or fatal: transfusion-related anaphylaxis (TRA), transfusion-related acute lung injury (TRALI), transfusion-associated circulatory overload (TACO), and acute infection following transfusion (AIFT).
For each type of event, the researchers looked at overall incidence and the incidence by immune status, calendar year, blood components transfused, number of units transfused, age, sex, and race.
Anaphylaxis (TRA)
TRA may be caused by preformed immunoglobin E (IgE) antibodies to proteins in the plasma in transfused blood products or by preformed IgA antibodies in patients who are likely IgA deficient, Dr. Menis said.
The overall incidence of TRA among 8,833,817 inpatient transfusions stays for elderly beneficiaries from 2012 through 2018 was 7.1 per 100,000 stays. The rate was higher for immunocompromised patients, at 9.6, than it was among nonimmunocompromised patients, at 6.5.
The rates varied by every subgroup measured except immune status. Annual rates showed a downward trend, from 8.7 per 100,000 in 2012, to 5.1 in 2017 and 6.4 in 2018. The decline in occurrence may be caused by a decline in inpatient blood utilization during the study period, particularly among immunocompromised patients.
TRA rates increased with five or more units transfused. The risk was significantly reduced in the oldest group of patients versus the youngest (P less than .001), which supports the immune-based mechanism of action of anaphylaxis, Dr. Menis said.
They also found that TRA rates were substantially higher among patients who had received platelet and/or plasma transfusions, compared with patients who received only red blood cells (RBCs).
Additionally, risk for TRA was significantly higher among men than it was among women (9.3 vs. 5.4) and among white versus nonwhite patients (7.8 vs. 3.8).
The evidence suggested TRA cases are likely to be severe in this population, with inpatient mortality of 7.1%, and hospital stays of 7 days or longer in about 58% of cases, indicating the importance of TRA prevention, Dr. Menis said.
The investigators plan to perform multivariate regression analyses to assess potential risk factors, including underlying comorbidities and health histories for TRA occurrence for both the overall population and by immune status.
Acute lung injury (TRALI)
TRALI is a rare but serious adverse event, a clinical syndrome with onset within 6 hours of transfusion that presents as acute hypoxemia, respiratory distress, and noncardiogenic pulmonary edema.
Among 17,771,193 total inpatient transfusion stays, the overall incidence of TRALI was 33.2 per 100,000. The rate was 55.9 for immunocompromised patients versus 28.4 for nonimmunocompromised patients. The rate ratio was 2.0 (P less than .001).
The difference by immune status may be caused by higher blood utilizations with more units transfused per stay among immunocompromised patients, a higher incidence of prior transfusions among these patients, higher use of irradiated blood components that may lead to accumulation of proinflammatory mediators in blood products during storage, or underlying comorbidities.
The overall rate increased from 14.3 in 2007 to 56.4 in 2018. The rates increased proportionally among both immunocompromised and nonimmunocompromised patients.
As with TRA, the incidence of TRALI was higher in patients with five or more units transfused, while the incidence declined with age, likely caused by declining blood use and age-related changes in neutrophil function, Dr. Menis said.
TRALI rates were slightly higher among men than among women, as well as higher among white patients than among nonwhite patients.
Overall, TRALI rates were higher for patients who received platelets either alone or in combination with RBCs and/or plasma. The highest rates were among patients who received RBCs, plasma and platelets.
Dr. Menis called for studies to determine what effects the processing and storage of blood components may have on TRALI occurrence; he and his colleagues also are planning regression analyses to assess potential risk factors for this complication.
Circulatory overload (TACO)
TACO is one of the leading reported causes of transfusion-related fatalities in the U.S., with onset usually occurring within 6 hours of transfusion, presenting as acute respiratory distress with dyspnea, orthopnea, increased blood pressure, and cardiogenic pulmonary edema.
The overall incidence of TACO among hospitalized patients aged 65 years and older from 2011 through 2018 was 86.3 per 100,000 stays. The incidences were 128.3 in immunocompromised and 76.0 in nonimmunocompromised patients. The rate ratio for TACO in immunocompromised versus nonimmunocompromised patients was 1.70 (P less than .001).
Overall incidence rates of TACO rose from 62 per 100,000 stays in 2011 to 119.8 in 2018. As with other adverse events, incident rates rose with the number of units transfused.
Rates of TACO were significantly higher among women than they were among men (94.6 vs. 75.9 per 100,000; P less than .001), which could be caused by the higher mean age of women and/or a lower tolerance for increased blood volume from transfusion.
The study results also suggested that TACO and TRALI may coexist, based on evidence that 3.5% of all TACO stays also had diagnostic codes for TRALI. The frequency of co-occurrence of these two adverse events also increased over time, which may be caused by improved awareness, Dr. Menis said.
Infections (AIFT)
Acute infections following transfusion can lead to prolonged hospitalizations, sepsis, septic shock, and death. Those most at risk include elderly and immunocompromised patients because of high utilization of blood products, comorbidities, and decreased immune function.
Among 8,833,817 stays, the overall rate per 100,000 stays was 2.1. The rate for immunocompromised patients was 5.4, compared with 1.2 for nonimmunocompromised patients, for a rate ratio of 4.4 (P less than .001).
The incidence rate declined significantly (P = .03) over the study period, with the 3 latest years having the lowest rates.
Rates increased substantially among immunocompromised patients by the number of units transfused, but remained relatively stable among nonimmunocompromised patients.
Infection rates declined with age, from 2.7 per 100,000 stays for patients aged 65-68 years to 1.2 per 100,000 for those aged 85 years and older.
As with other adverse events, AIFT rates were likely related to the blood components transfused, with substantially higher rates for stays during which platelets were transfused either alone or with RBCs, compared with RBCs alone. This could be caused by the room-temperature storage of platelets and higher number of platelets units transfused, compared with RBCs alone, especially among immunocompromised patients.
In all, 51.9% of AIFT cases also had sepsis noted in the medical record, indicating high severity and emphasizing the importance of AIFT prevention, Dr. Menis said.
The studies were funded by the FDA, and Dr. Menis is an FDA employee. He reported having no conflicts of interest.
SAN ANTONIO – Although there has been a general decline in transfusion-related anaphylaxis and acute infections over time among hospitalized older adults in the United States, incidence rates for both transfusion-related acute lung injury and transfusion-associated circulatory overload have risen over the last decade, according to researchers from the Food and Drug Administration.
Mikhail Menis, PharmD, an epidemiologist at the FDA Center for Biologics Evaluation and Research (CBER) and colleagues queried large Medicare databases to assess trends in transfusion-related adverse events among adults aged 65 years and older.
The investigators saw “substantially higher risk of all outcomes among immunocompromised beneficiaries, which could be related to higher blood use of all blood components, especially platelets, underlying conditions such as malignancies, and treatments such as chemotherapy or radiation, which need further investigation,” Dr. Menis said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
He reported data from a series of studies on four categories of transfusion-related events that may be life-threatening or fatal: transfusion-related anaphylaxis (TRA), transfusion-related acute lung injury (TRALI), transfusion-associated circulatory overload (TACO), and acute infection following transfusion (AIFT).
For each type of event, the researchers looked at overall incidence and the incidence by immune status, calendar year, blood components transfused, number of units transfused, age, sex, and race.
Anaphylaxis (TRA)
TRA may be caused by preformed immunoglobin E (IgE) antibodies to proteins in the plasma in transfused blood products or by preformed IgA antibodies in patients who are likely IgA deficient, Dr. Menis said.
The overall incidence of TRA among 8,833,817 inpatient transfusions stays for elderly beneficiaries from 2012 through 2018 was 7.1 per 100,000 stays. The rate was higher for immunocompromised patients, at 9.6, than it was among nonimmunocompromised patients, at 6.5.
The rates varied by every subgroup measured except immune status. Annual rates showed a downward trend, from 8.7 per 100,000 in 2012, to 5.1 in 2017 and 6.4 in 2018. The decline in occurrence may be caused by a decline in inpatient blood utilization during the study period, particularly among immunocompromised patients.
TRA rates increased with five or more units transfused. The risk was significantly reduced in the oldest group of patients versus the youngest (P less than .001), which supports the immune-based mechanism of action of anaphylaxis, Dr. Menis said.
They also found that TRA rates were substantially higher among patients who had received platelet and/or plasma transfusions, compared with patients who received only red blood cells (RBCs).
Additionally, risk for TRA was significantly higher among men than it was among women (9.3 vs. 5.4) and among white versus nonwhite patients (7.8 vs. 3.8).
The evidence suggested TRA cases are likely to be severe in this population, with inpatient mortality of 7.1%, and hospital stays of 7 days or longer in about 58% of cases, indicating the importance of TRA prevention, Dr. Menis said.
The investigators plan to perform multivariate regression analyses to assess potential risk factors, including underlying comorbidities and health histories for TRA occurrence for both the overall population and by immune status.
Acute lung injury (TRALI)
TRALI is a rare but serious adverse event, a clinical syndrome with onset within 6 hours of transfusion that presents as acute hypoxemia, respiratory distress, and noncardiogenic pulmonary edema.
Among 17,771,193 total inpatient transfusion stays, the overall incidence of TRALI was 33.2 per 100,000. The rate was 55.9 for immunocompromised patients versus 28.4 for nonimmunocompromised patients. The rate ratio was 2.0 (P less than .001).
The difference by immune status may be caused by higher blood utilizations with more units transfused per stay among immunocompromised patients, a higher incidence of prior transfusions among these patients, higher use of irradiated blood components that may lead to accumulation of proinflammatory mediators in blood products during storage, or underlying comorbidities.
The overall rate increased from 14.3 in 2007 to 56.4 in 2018. The rates increased proportionally among both immunocompromised and nonimmunocompromised patients.
As with TRA, the incidence of TRALI was higher in patients with five or more units transfused, while the incidence declined with age, likely caused by declining blood use and age-related changes in neutrophil function, Dr. Menis said.
TRALI rates were slightly higher among men than among women, as well as higher among white patients than among nonwhite patients.
Overall, TRALI rates were higher for patients who received platelets either alone or in combination with RBCs and/or plasma. The highest rates were among patients who received RBCs, plasma and platelets.
Dr. Menis called for studies to determine what effects the processing and storage of blood components may have on TRALI occurrence; he and his colleagues also are planning regression analyses to assess potential risk factors for this complication.
Circulatory overload (TACO)
TACO is one of the leading reported causes of transfusion-related fatalities in the U.S., with onset usually occurring within 6 hours of transfusion, presenting as acute respiratory distress with dyspnea, orthopnea, increased blood pressure, and cardiogenic pulmonary edema.
The overall incidence of TACO among hospitalized patients aged 65 years and older from 2011 through 2018 was 86.3 per 100,000 stays. The incidences were 128.3 in immunocompromised and 76.0 in nonimmunocompromised patients. The rate ratio for TACO in immunocompromised versus nonimmunocompromised patients was 1.70 (P less than .001).
Overall incidence rates of TACO rose from 62 per 100,000 stays in 2011 to 119.8 in 2018. As with other adverse events, incident rates rose with the number of units transfused.
Rates of TACO were significantly higher among women than they were among men (94.6 vs. 75.9 per 100,000; P less than .001), which could be caused by the higher mean age of women and/or a lower tolerance for increased blood volume from transfusion.
The study results also suggested that TACO and TRALI may coexist, based on evidence that 3.5% of all TACO stays also had diagnostic codes for TRALI. The frequency of co-occurrence of these two adverse events also increased over time, which may be caused by improved awareness, Dr. Menis said.
Infections (AIFT)
Acute infections following transfusion can lead to prolonged hospitalizations, sepsis, septic shock, and death. Those most at risk include elderly and immunocompromised patients because of high utilization of blood products, comorbidities, and decreased immune function.
Among 8,833,817 stays, the overall rate per 100,000 stays was 2.1. The rate for immunocompromised patients was 5.4, compared with 1.2 for nonimmunocompromised patients, for a rate ratio of 4.4 (P less than .001).
The incidence rate declined significantly (P = .03) over the study period, with the 3 latest years having the lowest rates.
Rates increased substantially among immunocompromised patients by the number of units transfused, but remained relatively stable among nonimmunocompromised patients.
Infection rates declined with age, from 2.7 per 100,000 stays for patients aged 65-68 years to 1.2 per 100,000 for those aged 85 years and older.
As with other adverse events, AIFT rates were likely related to the blood components transfused, with substantially higher rates for stays during which platelets were transfused either alone or with RBCs, compared with RBCs alone. This could be caused by the room-temperature storage of platelets and higher number of platelets units transfused, compared with RBCs alone, especially among immunocompromised patients.
In all, 51.9% of AIFT cases also had sepsis noted in the medical record, indicating high severity and emphasizing the importance of AIFT prevention, Dr. Menis said.
The studies were funded by the FDA, and Dr. Menis is an FDA employee. He reported having no conflicts of interest.
REPORTING FROM AABB 2019
Don’t miss neuromuscular complications of cancer immunotherapy
AUSTIN, TEX. – Neuromuscular complications from immunotherapy for cancer are rare, but they occur often enough that it is helpful to know which ones can result from different immunotherapies and how to distinguish them from non–adverse event conditions, according to Christopher Trevino, MD, a neuro-oncologist at Tulane University in New Orleans.
At the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine, Dr. Trevino reviewed immunotherapy types, particularly immune checkpoint inhibitors, and the most common neuromuscular complications – primarily neuropathy, myasthenia gravis (MG), myositis, and encephalitis or meningitis.
“Timing of onset is a critical component to assist in identifying immune checkpoint inhibitor–associated versus non–immune checkpoint inhibitor–associated neuromuscular disease,” Dr. Trevino told attendees. Prompt recognition can be particularly urgent for MG because crisis and death rates are higher when induced by immunotherapy and require quick treatment. “Understanding the mechanisms of action sets a foundation for treatment approach,” he added.
Any part of the nervous system can be affected by immunotherapy toxicity, he said, and syndromes often overlap, with the peripheral nervous system typically more often affected than the central nervous system. Neurologic immune-related adverse events typically occur within four cycles of therapy – about 12 weeks after therapy initiation – but should always involve a work-up to exclude effects from the cancer itself, other neuromuscular diagnoses unrelated to therapy, and other toxicities from chemotherapy.
Recommended first-line treatment is halting immunotherapy with or without corticosteroids, after which most patients improve, often with “rapid, complete resolution of symptoms,” Dr. Trevino said. Restarting immunotherapy treatment is possible in some patients, though.
CAR T-cell and dendritic cell vaccine therapies
Four main types of immunotherapy exist: viral therapy, vaccine therapy, immune checkpoint inhibitors, and adoptive cell transfer, such as chimeric antigen receptor (CAR) T-cell therapy. Dr. Trevino focused on checkpoint inhibitors and adoptive cell transfer.
CAR T-cell therapy is a multistep treatment process that involves first removing blood from the patient to obtain their T cells. These are used to create and grow CAR T cells in the lab so that they can be infused back into the patient. The cells then bind to cancer cells and destroy them. Examples of approved CAR T-cell therapy include Yescarta (axicabtagene ciloleucel) for some types of non-Hodgkin lymphoma and Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL).
Dendritic cell vaccines are similar to CAR T-cell therapy in that they also use the patient’s own immune cells to create cancer-killing cells that the patient then receives back. The only currently approved dendritic cell vaccine is Provenge (sipuleucel-T) for advanced prostate cancer.
The main toxicity to watch for from CAR T-cell therapy and dendritic cell vaccines is cytokine release syndrome (CRS). It can begin anywhere from 1-14 days after the infusion and involves T-cell expansion in the body that leads to a cytokine storm. Symptoms are wide ranging, including fatigue, fever, loss of appetite, tachycardia, hypotension, pain, rash, diarrhea, headache, confusion, seizures, muscle and joint pain, tachypnea, hypoxia and hallucinations, among others.
Specific central neurotoxicities that can result from CAR T-cell therapy include encephalopathy, cerebral edema, seizures and status epilepticus, cerebral vasospasm, and aphasia.
Immune checkpoint inhibitor toxicities
Immune checkpoint inhibitors are drugs that interrupt a cancer’s ability to hijack the immune system; they block the proteins that hold back T-cells from attacking the cancer, thereby releasing the immune system to go after the malignant cells.
The two most common types of immune checkpoint inhibitors are those targeting the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathways. The three currently approved PD-1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), and cemiplimab (Libtayo), which can treat nearly a dozen malignancies affecting different organs. Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi) are the three currently approved PD-L1 inhibitors, indicated for urothelial carcinoma and a handful of other cancers, such as small-cell and non–small cell lung cancer and triple negative breast cancer.
The only other type of approved checkpoint inhibitor is ipilimumab (Yervoy), which targets the CTLA-4 protein. A number of other checkpoint inhibitors are in trials, however, such as ones targeting pathways involving OX40, ICOS, TIM3, and LAG-3 (J Hematol Oncol. 2018. doi: 10.1186/s13045-018-0582-8).
Immune-related adverse events are less common with PD-1 or PD-L1 inhibitors – a rate of 5%-10% – compared with adverse events from CTLA-4 inhibitors, which occur in about 15% of patients. Neurologic complications occur even more rarely – about 1%-4% of all immune checkpoint inhibitor therapies – and primarily include MG, Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and inflammatory myositis (Muscle Nerve. 2018;58[1]:10-22).
Treatment with multiple checkpoint inhibitors increases the likelihood of severe adverse events, with rates of up to 30%-50% of patients with dual treatment.
Distinguishing features of neuromuscular immunotherapy-related adverse events
MG is the most common neuromuscular immune-related adverse event from immune checkpoint inhibitors and tends to occur 3-12 weeks after beginning treatment, frequently comorbid with inflammatory myopathy or cardiomyopathy, Dr. Trevino said. About two-thirds of cases are de novo, while the remaining one-third involve preexisting MG; no reports of Lambert-Eaton myasthenic syndrome have been linked to checkpoint inhibitors.
Several characteristics distinguish checkpoint inhibitor–associated MG from standard MG. Standard MG can be ocular with or without bulbar or appendicular weakness, whereas immunotherapy-related MG is rarely only ocular (about 18% of cases). Immunotherapy-related MG involves an MG crisis at diagnosis in up to 50% of cases and has high mortality, both of which are rarer with standard MG.
While standard MG can be seronegative or involve AChR, MuSK, or LRP4 antibodies, about two-thirds of immunotherapy-related MG cases are positive for AChR antibodies. LRP4 antibodies are rare with MG from checkpoint inhibitors, and no MuSK antibodies have been reported in these cases. Creatine kinase (CK) or troponin I (TnI) elevation occurs in about 87% of patients with checkpoint inhibitor-induced MG, but standard MG doesn’t typically involve increased CK levels.
Inflammatory myositis (IM), the second most common neuromuscular adverse event from immunotherapy, tends to occur 2-15 weeks after immune checkpoint inhibitor therapy and can involve polymyositis, necrotizing autoimmune myopathy, dermatomyositis, granulomatous myositis, or other nonspecific myositis and myopathies.
Though proximal weakness occurs with IM both associated with immunotherapy and not, ocular symptoms are unique to cases associated with therapy and occur in about half of them. Myalgia, dyspnea, and dysphagia can all occur with checkpoint inhibitor–associated IM but don’t generally occur with standard IM. Immunotherapy-related IM is usually seronegative for myositis antibodies and doesn’t generally cause abnormalities in electromyography, compared with increased exertional activity and early recruitment of myopathic motor units in electromyography with standard IM.
GBS and CIDP are the third most common cause of neuromuscular complications from checkpoint inhibitors. The main distinguishing feature of these conditions from those not related to immunotherapy is that they occur anywhere from 4 to 68 weeks after therapy begins. Presentation is otherwise similar whether related to checkpoint inhibitors or not.
Aside from GBS and CIDP, other neuropathies that can result from immunotherapy complications include acute cranial neuropathies, axonal or demyelinating neuropathies, motor polyradiculopathy, vasculitic neuropathy, and plexopathy.
Neuromuscular complications other than those described above can also occur from checkpoint inhibitor therapy, such as enteric neuropathy, polyradiculitis, and meningo-radiculo-neuritis, but these are much rarer.
Four organizations have developed consensus guidelines for immune checkpoint inhibitor toxicities: the European Society for Medical Oncology (ESMO, 2017), Society for Immunotherapy of Cancer (SITC, 2017), American Society of Clinical Oncology (ASCO, 2018), and National Comprehensive Cancer Network (NCCN, 2019).
Dr Trevino had no disclosures.
AUSTIN, TEX. – Neuromuscular complications from immunotherapy for cancer are rare, but they occur often enough that it is helpful to know which ones can result from different immunotherapies and how to distinguish them from non–adverse event conditions, according to Christopher Trevino, MD, a neuro-oncologist at Tulane University in New Orleans.
At the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine, Dr. Trevino reviewed immunotherapy types, particularly immune checkpoint inhibitors, and the most common neuromuscular complications – primarily neuropathy, myasthenia gravis (MG), myositis, and encephalitis or meningitis.
“Timing of onset is a critical component to assist in identifying immune checkpoint inhibitor–associated versus non–immune checkpoint inhibitor–associated neuromuscular disease,” Dr. Trevino told attendees. Prompt recognition can be particularly urgent for MG because crisis and death rates are higher when induced by immunotherapy and require quick treatment. “Understanding the mechanisms of action sets a foundation for treatment approach,” he added.
Any part of the nervous system can be affected by immunotherapy toxicity, he said, and syndromes often overlap, with the peripheral nervous system typically more often affected than the central nervous system. Neurologic immune-related adverse events typically occur within four cycles of therapy – about 12 weeks after therapy initiation – but should always involve a work-up to exclude effects from the cancer itself, other neuromuscular diagnoses unrelated to therapy, and other toxicities from chemotherapy.
Recommended first-line treatment is halting immunotherapy with or without corticosteroids, after which most patients improve, often with “rapid, complete resolution of symptoms,” Dr. Trevino said. Restarting immunotherapy treatment is possible in some patients, though.
CAR T-cell and dendritic cell vaccine therapies
Four main types of immunotherapy exist: viral therapy, vaccine therapy, immune checkpoint inhibitors, and adoptive cell transfer, such as chimeric antigen receptor (CAR) T-cell therapy. Dr. Trevino focused on checkpoint inhibitors and adoptive cell transfer.
CAR T-cell therapy is a multistep treatment process that involves first removing blood from the patient to obtain their T cells. These are used to create and grow CAR T cells in the lab so that they can be infused back into the patient. The cells then bind to cancer cells and destroy them. Examples of approved CAR T-cell therapy include Yescarta (axicabtagene ciloleucel) for some types of non-Hodgkin lymphoma and Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL).
Dendritic cell vaccines are similar to CAR T-cell therapy in that they also use the patient’s own immune cells to create cancer-killing cells that the patient then receives back. The only currently approved dendritic cell vaccine is Provenge (sipuleucel-T) for advanced prostate cancer.
The main toxicity to watch for from CAR T-cell therapy and dendritic cell vaccines is cytokine release syndrome (CRS). It can begin anywhere from 1-14 days after the infusion and involves T-cell expansion in the body that leads to a cytokine storm. Symptoms are wide ranging, including fatigue, fever, loss of appetite, tachycardia, hypotension, pain, rash, diarrhea, headache, confusion, seizures, muscle and joint pain, tachypnea, hypoxia and hallucinations, among others.
Specific central neurotoxicities that can result from CAR T-cell therapy include encephalopathy, cerebral edema, seizures and status epilepticus, cerebral vasospasm, and aphasia.
Immune checkpoint inhibitor toxicities
Immune checkpoint inhibitors are drugs that interrupt a cancer’s ability to hijack the immune system; they block the proteins that hold back T-cells from attacking the cancer, thereby releasing the immune system to go after the malignant cells.
The two most common types of immune checkpoint inhibitors are those targeting the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathways. The three currently approved PD-1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), and cemiplimab (Libtayo), which can treat nearly a dozen malignancies affecting different organs. Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi) are the three currently approved PD-L1 inhibitors, indicated for urothelial carcinoma and a handful of other cancers, such as small-cell and non–small cell lung cancer and triple negative breast cancer.
The only other type of approved checkpoint inhibitor is ipilimumab (Yervoy), which targets the CTLA-4 protein. A number of other checkpoint inhibitors are in trials, however, such as ones targeting pathways involving OX40, ICOS, TIM3, and LAG-3 (J Hematol Oncol. 2018. doi: 10.1186/s13045-018-0582-8).
Immune-related adverse events are less common with PD-1 or PD-L1 inhibitors – a rate of 5%-10% – compared with adverse events from CTLA-4 inhibitors, which occur in about 15% of patients. Neurologic complications occur even more rarely – about 1%-4% of all immune checkpoint inhibitor therapies – and primarily include MG, Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and inflammatory myositis (Muscle Nerve. 2018;58[1]:10-22).
Treatment with multiple checkpoint inhibitors increases the likelihood of severe adverse events, with rates of up to 30%-50% of patients with dual treatment.
Distinguishing features of neuromuscular immunotherapy-related adverse events
MG is the most common neuromuscular immune-related adverse event from immune checkpoint inhibitors and tends to occur 3-12 weeks after beginning treatment, frequently comorbid with inflammatory myopathy or cardiomyopathy, Dr. Trevino said. About two-thirds of cases are de novo, while the remaining one-third involve preexisting MG; no reports of Lambert-Eaton myasthenic syndrome have been linked to checkpoint inhibitors.
Several characteristics distinguish checkpoint inhibitor–associated MG from standard MG. Standard MG can be ocular with or without bulbar or appendicular weakness, whereas immunotherapy-related MG is rarely only ocular (about 18% of cases). Immunotherapy-related MG involves an MG crisis at diagnosis in up to 50% of cases and has high mortality, both of which are rarer with standard MG.
While standard MG can be seronegative or involve AChR, MuSK, or LRP4 antibodies, about two-thirds of immunotherapy-related MG cases are positive for AChR antibodies. LRP4 antibodies are rare with MG from checkpoint inhibitors, and no MuSK antibodies have been reported in these cases. Creatine kinase (CK) or troponin I (TnI) elevation occurs in about 87% of patients with checkpoint inhibitor-induced MG, but standard MG doesn’t typically involve increased CK levels.
Inflammatory myositis (IM), the second most common neuromuscular adverse event from immunotherapy, tends to occur 2-15 weeks after immune checkpoint inhibitor therapy and can involve polymyositis, necrotizing autoimmune myopathy, dermatomyositis, granulomatous myositis, or other nonspecific myositis and myopathies.
Though proximal weakness occurs with IM both associated with immunotherapy and not, ocular symptoms are unique to cases associated with therapy and occur in about half of them. Myalgia, dyspnea, and dysphagia can all occur with checkpoint inhibitor–associated IM but don’t generally occur with standard IM. Immunotherapy-related IM is usually seronegative for myositis antibodies and doesn’t generally cause abnormalities in electromyography, compared with increased exertional activity and early recruitment of myopathic motor units in electromyography with standard IM.
GBS and CIDP are the third most common cause of neuromuscular complications from checkpoint inhibitors. The main distinguishing feature of these conditions from those not related to immunotherapy is that they occur anywhere from 4 to 68 weeks after therapy begins. Presentation is otherwise similar whether related to checkpoint inhibitors or not.
Aside from GBS and CIDP, other neuropathies that can result from immunotherapy complications include acute cranial neuropathies, axonal or demyelinating neuropathies, motor polyradiculopathy, vasculitic neuropathy, and plexopathy.
Neuromuscular complications other than those described above can also occur from checkpoint inhibitor therapy, such as enteric neuropathy, polyradiculitis, and meningo-radiculo-neuritis, but these are much rarer.
Four organizations have developed consensus guidelines for immune checkpoint inhibitor toxicities: the European Society for Medical Oncology (ESMO, 2017), Society for Immunotherapy of Cancer (SITC, 2017), American Society of Clinical Oncology (ASCO, 2018), and National Comprehensive Cancer Network (NCCN, 2019).
Dr Trevino had no disclosures.
AUSTIN, TEX. – Neuromuscular complications from immunotherapy for cancer are rare, but they occur often enough that it is helpful to know which ones can result from different immunotherapies and how to distinguish them from non–adverse event conditions, according to Christopher Trevino, MD, a neuro-oncologist at Tulane University in New Orleans.
At the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine, Dr. Trevino reviewed immunotherapy types, particularly immune checkpoint inhibitors, and the most common neuromuscular complications – primarily neuropathy, myasthenia gravis (MG), myositis, and encephalitis or meningitis.
“Timing of onset is a critical component to assist in identifying immune checkpoint inhibitor–associated versus non–immune checkpoint inhibitor–associated neuromuscular disease,” Dr. Trevino told attendees. Prompt recognition can be particularly urgent for MG because crisis and death rates are higher when induced by immunotherapy and require quick treatment. “Understanding the mechanisms of action sets a foundation for treatment approach,” he added.
Any part of the nervous system can be affected by immunotherapy toxicity, he said, and syndromes often overlap, with the peripheral nervous system typically more often affected than the central nervous system. Neurologic immune-related adverse events typically occur within four cycles of therapy – about 12 weeks after therapy initiation – but should always involve a work-up to exclude effects from the cancer itself, other neuromuscular diagnoses unrelated to therapy, and other toxicities from chemotherapy.
Recommended first-line treatment is halting immunotherapy with or without corticosteroids, after which most patients improve, often with “rapid, complete resolution of symptoms,” Dr. Trevino said. Restarting immunotherapy treatment is possible in some patients, though.
CAR T-cell and dendritic cell vaccine therapies
Four main types of immunotherapy exist: viral therapy, vaccine therapy, immune checkpoint inhibitors, and adoptive cell transfer, such as chimeric antigen receptor (CAR) T-cell therapy. Dr. Trevino focused on checkpoint inhibitors and adoptive cell transfer.
CAR T-cell therapy is a multistep treatment process that involves first removing blood from the patient to obtain their T cells. These are used to create and grow CAR T cells in the lab so that they can be infused back into the patient. The cells then bind to cancer cells and destroy them. Examples of approved CAR T-cell therapy include Yescarta (axicabtagene ciloleucel) for some types of non-Hodgkin lymphoma and Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL).
Dendritic cell vaccines are similar to CAR T-cell therapy in that they also use the patient’s own immune cells to create cancer-killing cells that the patient then receives back. The only currently approved dendritic cell vaccine is Provenge (sipuleucel-T) for advanced prostate cancer.
The main toxicity to watch for from CAR T-cell therapy and dendritic cell vaccines is cytokine release syndrome (CRS). It can begin anywhere from 1-14 days after the infusion and involves T-cell expansion in the body that leads to a cytokine storm. Symptoms are wide ranging, including fatigue, fever, loss of appetite, tachycardia, hypotension, pain, rash, diarrhea, headache, confusion, seizures, muscle and joint pain, tachypnea, hypoxia and hallucinations, among others.
Specific central neurotoxicities that can result from CAR T-cell therapy include encephalopathy, cerebral edema, seizures and status epilepticus, cerebral vasospasm, and aphasia.
Immune checkpoint inhibitor toxicities
Immune checkpoint inhibitors are drugs that interrupt a cancer’s ability to hijack the immune system; they block the proteins that hold back T-cells from attacking the cancer, thereby releasing the immune system to go after the malignant cells.
The two most common types of immune checkpoint inhibitors are those targeting the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathways. The three currently approved PD-1 inhibitors are pembrolizumab (Keytruda), nivolumab (Opdivo), and cemiplimab (Libtayo), which can treat nearly a dozen malignancies affecting different organs. Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi) are the three currently approved PD-L1 inhibitors, indicated for urothelial carcinoma and a handful of other cancers, such as small-cell and non–small cell lung cancer and triple negative breast cancer.
The only other type of approved checkpoint inhibitor is ipilimumab (Yervoy), which targets the CTLA-4 protein. A number of other checkpoint inhibitors are in trials, however, such as ones targeting pathways involving OX40, ICOS, TIM3, and LAG-3 (J Hematol Oncol. 2018. doi: 10.1186/s13045-018-0582-8).
Immune-related adverse events are less common with PD-1 or PD-L1 inhibitors – a rate of 5%-10% – compared with adverse events from CTLA-4 inhibitors, which occur in about 15% of patients. Neurologic complications occur even more rarely – about 1%-4% of all immune checkpoint inhibitor therapies – and primarily include MG, Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and inflammatory myositis (Muscle Nerve. 2018;58[1]:10-22).
Treatment with multiple checkpoint inhibitors increases the likelihood of severe adverse events, with rates of up to 30%-50% of patients with dual treatment.
Distinguishing features of neuromuscular immunotherapy-related adverse events
MG is the most common neuromuscular immune-related adverse event from immune checkpoint inhibitors and tends to occur 3-12 weeks after beginning treatment, frequently comorbid with inflammatory myopathy or cardiomyopathy, Dr. Trevino said. About two-thirds of cases are de novo, while the remaining one-third involve preexisting MG; no reports of Lambert-Eaton myasthenic syndrome have been linked to checkpoint inhibitors.
Several characteristics distinguish checkpoint inhibitor–associated MG from standard MG. Standard MG can be ocular with or without bulbar or appendicular weakness, whereas immunotherapy-related MG is rarely only ocular (about 18% of cases). Immunotherapy-related MG involves an MG crisis at diagnosis in up to 50% of cases and has high mortality, both of which are rarer with standard MG.
While standard MG can be seronegative or involve AChR, MuSK, or LRP4 antibodies, about two-thirds of immunotherapy-related MG cases are positive for AChR antibodies. LRP4 antibodies are rare with MG from checkpoint inhibitors, and no MuSK antibodies have been reported in these cases. Creatine kinase (CK) or troponin I (TnI) elevation occurs in about 87% of patients with checkpoint inhibitor-induced MG, but standard MG doesn’t typically involve increased CK levels.
Inflammatory myositis (IM), the second most common neuromuscular adverse event from immunotherapy, tends to occur 2-15 weeks after immune checkpoint inhibitor therapy and can involve polymyositis, necrotizing autoimmune myopathy, dermatomyositis, granulomatous myositis, or other nonspecific myositis and myopathies.
Though proximal weakness occurs with IM both associated with immunotherapy and not, ocular symptoms are unique to cases associated with therapy and occur in about half of them. Myalgia, dyspnea, and dysphagia can all occur with checkpoint inhibitor–associated IM but don’t generally occur with standard IM. Immunotherapy-related IM is usually seronegative for myositis antibodies and doesn’t generally cause abnormalities in electromyography, compared with increased exertional activity and early recruitment of myopathic motor units in electromyography with standard IM.
GBS and CIDP are the third most common cause of neuromuscular complications from checkpoint inhibitors. The main distinguishing feature of these conditions from those not related to immunotherapy is that they occur anywhere from 4 to 68 weeks after therapy begins. Presentation is otherwise similar whether related to checkpoint inhibitors or not.
Aside from GBS and CIDP, other neuropathies that can result from immunotherapy complications include acute cranial neuropathies, axonal or demyelinating neuropathies, motor polyradiculopathy, vasculitic neuropathy, and plexopathy.
Neuromuscular complications other than those described above can also occur from checkpoint inhibitor therapy, such as enteric neuropathy, polyradiculitis, and meningo-radiculo-neuritis, but these are much rarer.
Four organizations have developed consensus guidelines for immune checkpoint inhibitor toxicities: the European Society for Medical Oncology (ESMO, 2017), Society for Immunotherapy of Cancer (SITC, 2017), American Society of Clinical Oncology (ASCO, 2018), and National Comprehensive Cancer Network (NCCN, 2019).
Dr Trevino had no disclosures.
EXPERT ANALYSIS FROM AANEM 2019
Armored CAR T cells elicit responses in NHL patients
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
NATIONAL HARBOR, MD – An armored chimeric antigen receptor (CAR) T-cell therapy has demonstrated efficacy in vitro and in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), according to findings presented at the annual meeting of the Society for Immunotherapy of Cancer.
ICTCAR014, a dominant negative PD-1 armored CAR T-cell therapy, proved more cytotoxic than traditional CAR T-cell therapy in vitro and produced responses in 12 of 13 NHL patients who received it.
Xiaobin Victor Lu, PhD, of Innovative Cellular Therapeutics, Shanghai, China, presented results with ICTCAR014 at the meeting.
Dr. Lu explained that ICTCAR014 consists of CD19-targeted CAR T cells genetically engineered to overexpress a PD-1 dominant negative protein with an altered intracellular signaling domain. The dominant negative protein can act as a “decoy receptor” to bind and block the PD-L1/2 inhibitory signal, thereby enhancing the efficacy of CAR T cells.
Innovative Cellular Therapeutics is developing ICTCAR014 because there is “some room to improve” with commercially available CAR T-cell products, Dr. Lu said. Specifically, tisagenlecleucel produced a 52% response rate in the JULIET trial (N Engl J Med. 2019;380:45-56), and axicabtagene ciloleucel produced an 82% response rate in the ZUMA-1 trial (N Engl J Med. 2017;377:2531-44).
There is also evidence to suggest that PD-1 blockade can modulate and “refuel” CAR T cells in relapsed/refractory NHL patients who fail or relapse after traditional anti-CD19 CAR T-cell therapy (Blood. 2017 Feb 23;129[8]:1039-41). This finding has prompted researchers to conduct trials of PD-1 inhibitors in combination with CAR T-cell therapies. But this combination approach may be expensive and cause more side effects than the armored CAR T-cell approach, Dr. Lu said.
In preclinical studies, Dr. Lu and colleagues found that ICTCAR014 was more effective than traditional anti-CD19 CAR T cells in killing Nalm6-PDL1 cells. In addition, the PD-1 dominant negative protein protected CAR T cells from exhaustion.
Dr. Lu also presented results in 13 NHL patients who have received ICTCAR014 in a phase 1 trial in China. Eleven patients had diffuse large B-cell lymphoma (DLBCL), and two had follicular lymphoma.
The objective response rate was 92.3% (12/13), which included five partial responses (38.5%) and seven complete responses (53.8%). Both follicular lymphoma patients and five DLBCL patients achieved a complete response. Five DLBCL patients achieved a partial response, and the remaining DLBCL patient did not respond.
Dr. Lu did not present safety data. However, he reported that there was no increased incidence of cytokine release syndrome or neurotoxicity in these patients, compared with patients receiving traditional CAR T-cell therapy.
Dr. Lu is employed by Innovative Cellular Therapeutics, which funded the research and is developing ICTCAR014.
SOURCE: Lu V et al. SITC 2019, Abstract O25.
REPORTING FROM SITC 2019