User login
COVID-19 outbreak hits research station in Antarctica
Two-thirds of the 25 workers have tested positive at the station, despite all of them being fully vaccinated and going through several testing stages before being allowed entrance, the Belgium publication Le Soir reported.
So far, all the cases are mild at the station, which is owned by Belgium and operated by a private group: the International Polar Foundation.
The first case was discovered Dec. 14 among a group that arrived a week earlier in Antarctica, Le Soir reported. The first three people to test positive evacuated Dec. 23, Le Soir said, but the virus continued to spread among the remaining workers at the base.
Le Soir, citing a virologist, said the Omicron variant probably caused the outbreak, because the crew made its last stop in South Africa before arriving in Antarctica.
New arrivals to the station have been put on hold until the outbreak is brought under control, and one of the missions planned for the base has been postponed, Le Soir said.
“The situation isn’t dramatic,” Joseph Cheek, a project manager for the International Polar Foundation, told the BBC. “While it has been an inconvenience to have to quarantine certain members of the staff who caught the virus, it hasn’t significantly affected our work at the station overall.”
The BBC said there was another COVID outbreak in Antarctica about a year ago at the Bernardo O’Higgins research station operated by Chile.
A version of this article first appeared on WebMD.com.
Two-thirds of the 25 workers have tested positive at the station, despite all of them being fully vaccinated and going through several testing stages before being allowed entrance, the Belgium publication Le Soir reported.
So far, all the cases are mild at the station, which is owned by Belgium and operated by a private group: the International Polar Foundation.
The first case was discovered Dec. 14 among a group that arrived a week earlier in Antarctica, Le Soir reported. The first three people to test positive evacuated Dec. 23, Le Soir said, but the virus continued to spread among the remaining workers at the base.
Le Soir, citing a virologist, said the Omicron variant probably caused the outbreak, because the crew made its last stop in South Africa before arriving in Antarctica.
New arrivals to the station have been put on hold until the outbreak is brought under control, and one of the missions planned for the base has been postponed, Le Soir said.
“The situation isn’t dramatic,” Joseph Cheek, a project manager for the International Polar Foundation, told the BBC. “While it has been an inconvenience to have to quarantine certain members of the staff who caught the virus, it hasn’t significantly affected our work at the station overall.”
The BBC said there was another COVID outbreak in Antarctica about a year ago at the Bernardo O’Higgins research station operated by Chile.
A version of this article first appeared on WebMD.com.
Two-thirds of the 25 workers have tested positive at the station, despite all of them being fully vaccinated and going through several testing stages before being allowed entrance, the Belgium publication Le Soir reported.
So far, all the cases are mild at the station, which is owned by Belgium and operated by a private group: the International Polar Foundation.
The first case was discovered Dec. 14 among a group that arrived a week earlier in Antarctica, Le Soir reported. The first three people to test positive evacuated Dec. 23, Le Soir said, but the virus continued to spread among the remaining workers at the base.
Le Soir, citing a virologist, said the Omicron variant probably caused the outbreak, because the crew made its last stop in South Africa before arriving in Antarctica.
New arrivals to the station have been put on hold until the outbreak is brought under control, and one of the missions planned for the base has been postponed, Le Soir said.
“The situation isn’t dramatic,” Joseph Cheek, a project manager for the International Polar Foundation, told the BBC. “While it has been an inconvenience to have to quarantine certain members of the staff who caught the virus, it hasn’t significantly affected our work at the station overall.”
The BBC said there was another COVID outbreak in Antarctica about a year ago at the Bernardo O’Higgins research station operated by Chile.
A version of this article first appeared on WebMD.com.
Indurated Mass on the Right Central Back
The Diagnosis: Actinomycetoma
Histopathology revealed evidence of an actinomycete organism within the suppuration, consistent with actinomycosis (quiz image [inset]). Given the clinical presentation and histopathologic findings, our patient was diagnosed with actinomycetoma.
Actinomycetoma is an indolent, progressive, subcutaneous infection characterized by a well-known clinical triad of tumefaction/subcutaneous mass, draining sinuses, and an exudate containing grains on microscopy. The sinus tracts are formed from the chronic infectious process that destroys tissue, creating tunnels. This infectious disease of soft tissue is a clinical subset of mycetoma, which is categorized as eumycetoma (fungal) and actinomycetoma (bacterial). Actinomycetoma resembles the behavior of insidious and chronic fungal infections; however, most mycetoma infections are bacterial.1,2 Actinomycetoma may be confused with actinomycosis, which is caused by Actinomycoses species, commensal organisms commonly located on the teeth and oral mucosa in association with other microorganisms that may pathogenically cause cervicofacial actinomycosis.3,4 Actinomycetoma can be caused by Nocardia, Streptomyces, and Actinomadura. 2,5 The foot is the most common location of involvement followed by the thoracic region. It is more common in tropical or equatorial locations and may be contracted through exposure to soil or wood.5 Mycetoma is considered a neglected tropical disease by the World Health Organization.1 In tropical countries, this disease may go undiagnosed or untreated for so long that surgical amputation may be the only effective treatment.
Actinomycetoma commonly is identifiable by direct microscopy, Gram stain, or bacterial culture, with Gram stain being more sensitive than bacterial culture.3 It is important to indicate the suspected organism to the microbiology laboratory because common bacterial pathogens are detected within 24 to 48 hours, but the causative microorganism in actinomycetoma may require up to 4 weeks for culture,2 leading to possible false negatives due to inadequate culture time.3 Histopathology of actinomycotic infections will demonstrate granulomatous inflammation, focal suppuration, and the presence of grains (ie, a colony of filamentous bacteria in a stellate shaped mass)(quiz image [inset]).
The gold standard of treatment is trimethoprim-sulfamethoxazole for up to several years.4,5 Amoxicillin–clavulanic acid, dapsone, amikacin, streptomycin, and beta-lactams have been used successfully.2,5 The treatment course is dependent on clinical severity and location of the disease. The cure rate with appropriate antibiotics can be as high as 90%,2,5 and thus surgical intervention can be avoided.
In the differential, cutaneous tuberculosis would show tuberculoid granulomas with epithelioid histiocytes with possible caseation on histopathology, typically alongside positive tuberculosis screening. Botryomycosis has a similar clinical presentation of a swollen or indurated lesion with draining sinus tracts, but it less commonly occurs on the trunk. Histopathology also is a close mimic of actinomycetoma with a small grain inside a suppurative infiltrate; however, it has no filamentous bacteria. A foreign body reaction would not histologically present with suppuration or grains, and draining sinuses typically would not be seen on clinical presentation. Sarcoma is a neoplastic process and most commonly would show a proliferation of cells with soft tissue or bone origin on histopathology and not primarily an inflammatory cell process.6
- Verma P, Jha A. Mycetoma: reviewing a neglected disease. Clin Exp Dermatol. 2019;44:123-129.
- Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-197.
- Bennhoff DF. Actinomycosis: diagnostic and therapeutic considerations and a review of 32 cases. Laryngoscope. 1984;94:1198-1217.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010.
The Diagnosis: Actinomycetoma
Histopathology revealed evidence of an actinomycete organism within the suppuration, consistent with actinomycosis (quiz image [inset]). Given the clinical presentation and histopathologic findings, our patient was diagnosed with actinomycetoma.
Actinomycetoma is an indolent, progressive, subcutaneous infection characterized by a well-known clinical triad of tumefaction/subcutaneous mass, draining sinuses, and an exudate containing grains on microscopy. The sinus tracts are formed from the chronic infectious process that destroys tissue, creating tunnels. This infectious disease of soft tissue is a clinical subset of mycetoma, which is categorized as eumycetoma (fungal) and actinomycetoma (bacterial). Actinomycetoma resembles the behavior of insidious and chronic fungal infections; however, most mycetoma infections are bacterial.1,2 Actinomycetoma may be confused with actinomycosis, which is caused by Actinomycoses species, commensal organisms commonly located on the teeth and oral mucosa in association with other microorganisms that may pathogenically cause cervicofacial actinomycosis.3,4 Actinomycetoma can be caused by Nocardia, Streptomyces, and Actinomadura. 2,5 The foot is the most common location of involvement followed by the thoracic region. It is more common in tropical or equatorial locations and may be contracted through exposure to soil or wood.5 Mycetoma is considered a neglected tropical disease by the World Health Organization.1 In tropical countries, this disease may go undiagnosed or untreated for so long that surgical amputation may be the only effective treatment.
Actinomycetoma commonly is identifiable by direct microscopy, Gram stain, or bacterial culture, with Gram stain being more sensitive than bacterial culture.3 It is important to indicate the suspected organism to the microbiology laboratory because common bacterial pathogens are detected within 24 to 48 hours, but the causative microorganism in actinomycetoma may require up to 4 weeks for culture,2 leading to possible false negatives due to inadequate culture time.3 Histopathology of actinomycotic infections will demonstrate granulomatous inflammation, focal suppuration, and the presence of grains (ie, a colony of filamentous bacteria in a stellate shaped mass)(quiz image [inset]).
The gold standard of treatment is trimethoprim-sulfamethoxazole for up to several years.4,5 Amoxicillin–clavulanic acid, dapsone, amikacin, streptomycin, and beta-lactams have been used successfully.2,5 The treatment course is dependent on clinical severity and location of the disease. The cure rate with appropriate antibiotics can be as high as 90%,2,5 and thus surgical intervention can be avoided.
In the differential, cutaneous tuberculosis would show tuberculoid granulomas with epithelioid histiocytes with possible caseation on histopathology, typically alongside positive tuberculosis screening. Botryomycosis has a similar clinical presentation of a swollen or indurated lesion with draining sinus tracts, but it less commonly occurs on the trunk. Histopathology also is a close mimic of actinomycetoma with a small grain inside a suppurative infiltrate; however, it has no filamentous bacteria. A foreign body reaction would not histologically present with suppuration or grains, and draining sinuses typically would not be seen on clinical presentation. Sarcoma is a neoplastic process and most commonly would show a proliferation of cells with soft tissue or bone origin on histopathology and not primarily an inflammatory cell process.6
The Diagnosis: Actinomycetoma
Histopathology revealed evidence of an actinomycete organism within the suppuration, consistent with actinomycosis (quiz image [inset]). Given the clinical presentation and histopathologic findings, our patient was diagnosed with actinomycetoma.
Actinomycetoma is an indolent, progressive, subcutaneous infection characterized by a well-known clinical triad of tumefaction/subcutaneous mass, draining sinuses, and an exudate containing grains on microscopy. The sinus tracts are formed from the chronic infectious process that destroys tissue, creating tunnels. This infectious disease of soft tissue is a clinical subset of mycetoma, which is categorized as eumycetoma (fungal) and actinomycetoma (bacterial). Actinomycetoma resembles the behavior of insidious and chronic fungal infections; however, most mycetoma infections are bacterial.1,2 Actinomycetoma may be confused with actinomycosis, which is caused by Actinomycoses species, commensal organisms commonly located on the teeth and oral mucosa in association with other microorganisms that may pathogenically cause cervicofacial actinomycosis.3,4 Actinomycetoma can be caused by Nocardia, Streptomyces, and Actinomadura. 2,5 The foot is the most common location of involvement followed by the thoracic region. It is more common in tropical or equatorial locations and may be contracted through exposure to soil or wood.5 Mycetoma is considered a neglected tropical disease by the World Health Organization.1 In tropical countries, this disease may go undiagnosed or untreated for so long that surgical amputation may be the only effective treatment.
Actinomycetoma commonly is identifiable by direct microscopy, Gram stain, or bacterial culture, with Gram stain being more sensitive than bacterial culture.3 It is important to indicate the suspected organism to the microbiology laboratory because common bacterial pathogens are detected within 24 to 48 hours, but the causative microorganism in actinomycetoma may require up to 4 weeks for culture,2 leading to possible false negatives due to inadequate culture time.3 Histopathology of actinomycotic infections will demonstrate granulomatous inflammation, focal suppuration, and the presence of grains (ie, a colony of filamentous bacteria in a stellate shaped mass)(quiz image [inset]).
The gold standard of treatment is trimethoprim-sulfamethoxazole for up to several years.4,5 Amoxicillin–clavulanic acid, dapsone, amikacin, streptomycin, and beta-lactams have been used successfully.2,5 The treatment course is dependent on clinical severity and location of the disease. The cure rate with appropriate antibiotics can be as high as 90%,2,5 and thus surgical intervention can be avoided.
In the differential, cutaneous tuberculosis would show tuberculoid granulomas with epithelioid histiocytes with possible caseation on histopathology, typically alongside positive tuberculosis screening. Botryomycosis has a similar clinical presentation of a swollen or indurated lesion with draining sinus tracts, but it less commonly occurs on the trunk. Histopathology also is a close mimic of actinomycetoma with a small grain inside a suppurative infiltrate; however, it has no filamentous bacteria. A foreign body reaction would not histologically present with suppuration or grains, and draining sinuses typically would not be seen on clinical presentation. Sarcoma is a neoplastic process and most commonly would show a proliferation of cells with soft tissue or bone origin on histopathology and not primarily an inflammatory cell process.6
- Verma P, Jha A. Mycetoma: reviewing a neglected disease. Clin Exp Dermatol. 2019;44:123-129.
- Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-197.
- Bennhoff DF. Actinomycosis: diagnostic and therapeutic considerations and a review of 32 cases. Laryngoscope. 1984;94:1198-1217.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010.
- Verma P, Jha A. Mycetoma: reviewing a neglected disease. Clin Exp Dermatol. 2019;44:123-129.
- Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-197.
- Bennhoff DF. Actinomycosis: diagnostic and therapeutic considerations and a review of 32 cases. Laryngoscope. 1984;94:1198-1217.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010.
A 26-year-old Guatemalan man who was a former carpenter presented with an indurated, nontender, nonpruritic, subcutaneous mass on the right central back with multiple draining sinus tracts on the surface and several depressed circular atrophic scars on the periphery of the mass. He noticed that the lesion began as a pustule 1.5 years prior and gradually enlarged. He denied any trauma, insect bites, fever, chills, headaches, weight loss, or travel history (he relocated to the United States 3.5 years ago) prior to the skin eruption. A biopsy was performed by an outside dermatologist 1 year prior to the current presentation, with a diagnosis of Pityrosporum folliculitis. Throughout his clinical course, treatment with oral antifungals, oral doxycycline, and topical clindamycin all failed. The mass was removed by plastic surgery 1 year prior.
A tissue biopsy for histology and culture was obtained at presentation to our institution. Laboratory findings showed that the basic metabolic panel was within reference range. Chest radiography indicated no active disease. A tuberculosis screening was negative. A bacterial culture of the lesion identified no growth after 48 hours. Our tissue biopsy revealed fibrosing granulation tissue, but the surgical pathology from a prior mass excision revealed sinus tracts with suppuration, evidence of scarring, foreign body giant cell reaction, and a characteristic finding (inset: H&E, original magnification ×200).


FDA backs Pfizer booster for 12- to 15-year-olds

Besides updating the authorization for the Pfizer COVID-19 vaccine, the agency also shortened the recommended time between a second dose and the booster to 5 months or more, based on new evidence. In addition, a third primary series dose is now authorized for certain immunocompromised children 5 years to 11 years old. Full details are available in an FDA news release.
The amended emergency use authorization (EUA) only applies to the Pfizer vaccine, said acting FDA Commissioner Janet Woodcock, MD.
“Just to make sure every everyone is clear on this, right now: If you got [Johnson & Johnson’s one-dose vaccine], you get a booster after 2 months. If you got Moderna, you can get a booster at 6 months or beyond,” she said during a media briefing.
What is new, she said, is “if you got Pfizer as your primary series, you can get a booster at 5 months or beyond.”
A lower risk of myocarditis?
Asked about concerns about the risk of myocarditis with vaccination in the 12- to 15-year age group, Dr. Woodcock said they expect it would be “extremely rare with the third dose.”
“We have the real-world evidence from the Israeli experience to help us with that analysis,” she said.
The data so far consistently points to a higher risk of myocarditis after a second mRNA vaccine dose among males, from teenagers to 30-year-olds, with a peak at about 16 to 17 years of age, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said during the media call.
The risk of myocarditis is about 2 to 3 times higher after a second vaccine dose, compared to a booster shot, Dr. Marks said, based on available data. It may be related to the closer dose timing of the second dose versus a third, he added.
“The inference here is that on the risk of myocarditis with third doses in the 12- to 15-year age range is likely to be quite acceptable,” he said.
Dr. Marks also pointed out that most cases of myocarditis clear up quickly.
“We’re not seeing long-lasting effects. That’s not to say that we don’t care about this and that it’s not important,” he said.
“But what it is saying is that in the setting of a tremendous number of Omicron and Delta cases in this country, the potential benefits of getting vaccinated in this age group outweigh that risk,” Dr. Marks said. “We can look at that risk-benefit and still feel comfortable.”
He said that “the really overwhelming majority of these cases, 98%, have been mild” -- shown by a 1-day median hospital stay.
Even so, the FDA plans to continue monitoring for the risk of myocarditis “very closely,” he said.
Interestingly, swollen underarm lymph nodes were seen more frequently after the booster dose than after the second dose of a two-dose primary series, the FDA said.
Reducing the time between primary vaccination with the Pfizer vaccine -- two initial doses -- and the booster shot from 6 months to 5 months is based on decreasing efficacy data that the drugmaker submitted to the FDA.
The 5-month interval was evaluated in a study from Israel published Dec. 21 in the New England Journal of Medicine .
Mixing and matching vaccines
Less clear at the moment is guidance about boosters for people who opted to mix and match their primary vaccine series.
“There was a mix-and-match study that was done which showed that in some cases, the mixing and matching … of an adenoviral record vaccine and an mRNA vaccine seem to give a very good immune response,” Dr. Marks said.
Once more data comes in on mixing and matching, “we’ll analyze them and then potentially make recommendations,” he said.
‘It’s not too late’
No federal government media briefing on COVID-19 would be complete without a plea for the unvaccinated to get immunized.
“We’re talking a lot about boosters right now, but it’s not too late for those who have not gotten a vaccine to get a vaccine,” Dr. Marks said, referring to the tens of millions of Americans who remain unvaccinated at the beginning of 2022.
“We know from our previous studies that even a single dose of the vaccine -- and probably two doses -- can help prevent the worst outcomes from COVID-19, including hospitalization and death.”
A version of this article first appeared on WebMD.com.
Besides updating the authorization for the Pfizer COVID-19 vaccine, the agency also shortened the recommended time between a second dose and the booster to 5 months or more, based on new evidence. In addition, a third primary series dose is now authorized for certain immunocompromised children 5 years to 11 years old. Full details are available in an FDA news release.
The amended emergency use authorization (EUA) only applies to the Pfizer vaccine, said acting FDA Commissioner Janet Woodcock, MD.
“Just to make sure every everyone is clear on this, right now: If you got [Johnson & Johnson’s one-dose vaccine], you get a booster after 2 months. If you got Moderna, you can get a booster at 6 months or beyond,” she said during a media briefing.
What is new, she said, is “if you got Pfizer as your primary series, you can get a booster at 5 months or beyond.”
A lower risk of myocarditis?
Asked about concerns about the risk of myocarditis with vaccination in the 12- to 15-year age group, Dr. Woodcock said they expect it would be “extremely rare with the third dose.”
“We have the real-world evidence from the Israeli experience to help us with that analysis,” she said.
The data so far consistently points to a higher risk of myocarditis after a second mRNA vaccine dose among males, from teenagers to 30-year-olds, with a peak at about 16 to 17 years of age, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said during the media call.
The risk of myocarditis is about 2 to 3 times higher after a second vaccine dose, compared to a booster shot, Dr. Marks said, based on available data. It may be related to the closer dose timing of the second dose versus a third, he added.
“The inference here is that on the risk of myocarditis with third doses in the 12- to 15-year age range is likely to be quite acceptable,” he said.
Dr. Marks also pointed out that most cases of myocarditis clear up quickly.
“We’re not seeing long-lasting effects. That’s not to say that we don’t care about this and that it’s not important,” he said.
“But what it is saying is that in the setting of a tremendous number of Omicron and Delta cases in this country, the potential benefits of getting vaccinated in this age group outweigh that risk,” Dr. Marks said. “We can look at that risk-benefit and still feel comfortable.”
He said that “the really overwhelming majority of these cases, 98%, have been mild” -- shown by a 1-day median hospital stay.
Even so, the FDA plans to continue monitoring for the risk of myocarditis “very closely,” he said.
Interestingly, swollen underarm lymph nodes were seen more frequently after the booster dose than after the second dose of a two-dose primary series, the FDA said.
Reducing the time between primary vaccination with the Pfizer vaccine -- two initial doses -- and the booster shot from 6 months to 5 months is based on decreasing efficacy data that the drugmaker submitted to the FDA.
The 5-month interval was evaluated in a study from Israel published Dec. 21 in the New England Journal of Medicine .
Mixing and matching vaccines
Less clear at the moment is guidance about boosters for people who opted to mix and match their primary vaccine series.
“There was a mix-and-match study that was done which showed that in some cases, the mixing and matching … of an adenoviral record vaccine and an mRNA vaccine seem to give a very good immune response,” Dr. Marks said.
Once more data comes in on mixing and matching, “we’ll analyze them and then potentially make recommendations,” he said.
‘It’s not too late’
No federal government media briefing on COVID-19 would be complete without a plea for the unvaccinated to get immunized.
“We’re talking a lot about boosters right now, but it’s not too late for those who have not gotten a vaccine to get a vaccine,” Dr. Marks said, referring to the tens of millions of Americans who remain unvaccinated at the beginning of 2022.
“We know from our previous studies that even a single dose of the vaccine -- and probably two doses -- can help prevent the worst outcomes from COVID-19, including hospitalization and death.”
A version of this article first appeared on WebMD.com.
Besides updating the authorization for the Pfizer COVID-19 vaccine, the agency also shortened the recommended time between a second dose and the booster to 5 months or more, based on new evidence. In addition, a third primary series dose is now authorized for certain immunocompromised children 5 years to 11 years old. Full details are available in an FDA news release.
The amended emergency use authorization (EUA) only applies to the Pfizer vaccine, said acting FDA Commissioner Janet Woodcock, MD.
“Just to make sure every everyone is clear on this, right now: If you got [Johnson & Johnson’s one-dose vaccine], you get a booster after 2 months. If you got Moderna, you can get a booster at 6 months or beyond,” she said during a media briefing.
What is new, she said, is “if you got Pfizer as your primary series, you can get a booster at 5 months or beyond.”
A lower risk of myocarditis?
Asked about concerns about the risk of myocarditis with vaccination in the 12- to 15-year age group, Dr. Woodcock said they expect it would be “extremely rare with the third dose.”
“We have the real-world evidence from the Israeli experience to help us with that analysis,” she said.
The data so far consistently points to a higher risk of myocarditis after a second mRNA vaccine dose among males, from teenagers to 30-year-olds, with a peak at about 16 to 17 years of age, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said during the media call.
The risk of myocarditis is about 2 to 3 times higher after a second vaccine dose, compared to a booster shot, Dr. Marks said, based on available data. It may be related to the closer dose timing of the second dose versus a third, he added.
“The inference here is that on the risk of myocarditis with third doses in the 12- to 15-year age range is likely to be quite acceptable,” he said.
Dr. Marks also pointed out that most cases of myocarditis clear up quickly.
“We’re not seeing long-lasting effects. That’s not to say that we don’t care about this and that it’s not important,” he said.
“But what it is saying is that in the setting of a tremendous number of Omicron and Delta cases in this country, the potential benefits of getting vaccinated in this age group outweigh that risk,” Dr. Marks said. “We can look at that risk-benefit and still feel comfortable.”
He said that “the really overwhelming majority of these cases, 98%, have been mild” -- shown by a 1-day median hospital stay.
Even so, the FDA plans to continue monitoring for the risk of myocarditis “very closely,” he said.
Interestingly, swollen underarm lymph nodes were seen more frequently after the booster dose than after the second dose of a two-dose primary series, the FDA said.
Reducing the time between primary vaccination with the Pfizer vaccine -- two initial doses -- and the booster shot from 6 months to 5 months is based on decreasing efficacy data that the drugmaker submitted to the FDA.
The 5-month interval was evaluated in a study from Israel published Dec. 21 in the New England Journal of Medicine .
Mixing and matching vaccines
Less clear at the moment is guidance about boosters for people who opted to mix and match their primary vaccine series.
“There was a mix-and-match study that was done which showed that in some cases, the mixing and matching … of an adenoviral record vaccine and an mRNA vaccine seem to give a very good immune response,” Dr. Marks said.
Once more data comes in on mixing and matching, “we’ll analyze them and then potentially make recommendations,” he said.
‘It’s not too late’
No federal government media briefing on COVID-19 would be complete without a plea for the unvaccinated to get immunized.
“We’re talking a lot about boosters right now, but it’s not too late for those who have not gotten a vaccine to get a vaccine,” Dr. Marks said, referring to the tens of millions of Americans who remain unvaccinated at the beginning of 2022.
“We know from our previous studies that even a single dose of the vaccine -- and probably two doses -- can help prevent the worst outcomes from COVID-19, including hospitalization and death.”
A version of this article first appeared on WebMD.com.
Antiretroviral pill better at suppressing HIV in children
Dolutegravir suppresses HIV by inhibiting integrase, an enzyme that the virus needs to replicate.
The pill-based regimen, which researchers described as easier to take than standard treatment, reduced the chances of treatment failure among children aged 3-18 years by about 40%, compared with other treatments. Dolutegravir is already used for the suppression of HIV in adults.
“About 1.8 million children live with HIV but they have had limited treatment options, with medicines that taste unpalatable, that need to be taken twice a day, or that come in large pills that are difficult to swallow” said lead author Anna Turkova, MD, from the MRC clinical trials unit at UCL. “Dolutegravir is given in small tablets usually once a day and the baby pills can be dispersed in water, meaning it’s a lot easier for young children to take. This is important in encouraging uptake of the treatment and adherence to it over many years.
“Sadly, only about half of children living with HIV are currently receiving treatment, and those who are not treated face high risks of impaired immunity and worsening health.”
Study details
The randomized controlled trial, called ODYSSEY, involved more than 700 children from 29 clinical centers located in Africa, Europe, and Asia. The children were given either dolutegravir or standard anti-HIV drugs, and were followed up for at least 2 years.
The study showed that 14% of children receiving dolutegravir experienced treatment failure over 2 years, compared with 22% of those receiving standard treatment. Treatment failure was deemed to occur if measurable virus appeared in the blood or if the child had symptoms of HIV-related illness.
“Our findings provide strong evidence for the global rollout of dolutegravir for children with HIV,” said Diana Gibb, MD, also from the MRC clinical trials unit at UCL, principal investigator of the trial and one of the senior authors of the paper.
“Medical treatments for children often lag woefully behind those of adults because of the separate formulations and studies that are needed,” she added. “With the evidence from ODYSSEY which used simplified dosing of both adult and baby pills, this treatment gap has been reduced and we hope that countries can quickly scale up access to children globally.”
Simplified dosing
“Simplifying the dosing is crucial,” concurred Cissy Kityo Mutuluuza, MD, from the Joint Clinical Research Centre in Uganda, the country enrolling most children in the trial. “Older children being able to take the same tablets as adults immediately opens access to dolutegravir for the majority of children living with HIV. It greatly simplifies procurement for national health systems in low- and middle-income countries, and lowers costs.”
Evidence from adults shows dolutegravir has a high genetic barrier to resistance, meaning viruses are less likely to become resistant to it over time. This was confirmed in the ODYSSEY trial, with much less resistance occurring among children and adolescents on dolutegravir-based treatment. In addition, past studies of the drug have shown that it may be associated with weight gain in adults, but the findings were reassuring for children. Those given dolutegravir gained on average 1 kg more and grew 1 cm higher over the study period, indicating better growth rather than abnormal weight gain.
Early findings from the trial have informed new guidance by the World Health Organization, recommending the use of dolutegravir for children.
The study was sponsored by the Penta Foundation, an international independent research network, and funded by specialist pharmaceutical company ViiV Healthcare.
A version of this article first appeared on Medscape.com.
Dolutegravir suppresses HIV by inhibiting integrase, an enzyme that the virus needs to replicate.
The pill-based regimen, which researchers described as easier to take than standard treatment, reduced the chances of treatment failure among children aged 3-18 years by about 40%, compared with other treatments. Dolutegravir is already used for the suppression of HIV in adults.
“About 1.8 million children live with HIV but they have had limited treatment options, with medicines that taste unpalatable, that need to be taken twice a day, or that come in large pills that are difficult to swallow” said lead author Anna Turkova, MD, from the MRC clinical trials unit at UCL. “Dolutegravir is given in small tablets usually once a day and the baby pills can be dispersed in water, meaning it’s a lot easier for young children to take. This is important in encouraging uptake of the treatment and adherence to it over many years.
“Sadly, only about half of children living with HIV are currently receiving treatment, and those who are not treated face high risks of impaired immunity and worsening health.”
Study details
The randomized controlled trial, called ODYSSEY, involved more than 700 children from 29 clinical centers located in Africa, Europe, and Asia. The children were given either dolutegravir or standard anti-HIV drugs, and were followed up for at least 2 years.
The study showed that 14% of children receiving dolutegravir experienced treatment failure over 2 years, compared with 22% of those receiving standard treatment. Treatment failure was deemed to occur if measurable virus appeared in the blood or if the child had symptoms of HIV-related illness.
“Our findings provide strong evidence for the global rollout of dolutegravir for children with HIV,” said Diana Gibb, MD, also from the MRC clinical trials unit at UCL, principal investigator of the trial and one of the senior authors of the paper.
“Medical treatments for children often lag woefully behind those of adults because of the separate formulations and studies that are needed,” she added. “With the evidence from ODYSSEY which used simplified dosing of both adult and baby pills, this treatment gap has been reduced and we hope that countries can quickly scale up access to children globally.”
Simplified dosing
“Simplifying the dosing is crucial,” concurred Cissy Kityo Mutuluuza, MD, from the Joint Clinical Research Centre in Uganda, the country enrolling most children in the trial. “Older children being able to take the same tablets as adults immediately opens access to dolutegravir for the majority of children living with HIV. It greatly simplifies procurement for national health systems in low- and middle-income countries, and lowers costs.”
Evidence from adults shows dolutegravir has a high genetic barrier to resistance, meaning viruses are less likely to become resistant to it over time. This was confirmed in the ODYSSEY trial, with much less resistance occurring among children and adolescents on dolutegravir-based treatment. In addition, past studies of the drug have shown that it may be associated with weight gain in adults, but the findings were reassuring for children. Those given dolutegravir gained on average 1 kg more and grew 1 cm higher over the study period, indicating better growth rather than abnormal weight gain.
Early findings from the trial have informed new guidance by the World Health Organization, recommending the use of dolutegravir for children.
The study was sponsored by the Penta Foundation, an international independent research network, and funded by specialist pharmaceutical company ViiV Healthcare.
A version of this article first appeared on Medscape.com.
Dolutegravir suppresses HIV by inhibiting integrase, an enzyme that the virus needs to replicate.
The pill-based regimen, which researchers described as easier to take than standard treatment, reduced the chances of treatment failure among children aged 3-18 years by about 40%, compared with other treatments. Dolutegravir is already used for the suppression of HIV in adults.
“About 1.8 million children live with HIV but they have had limited treatment options, with medicines that taste unpalatable, that need to be taken twice a day, or that come in large pills that are difficult to swallow” said lead author Anna Turkova, MD, from the MRC clinical trials unit at UCL. “Dolutegravir is given in small tablets usually once a day and the baby pills can be dispersed in water, meaning it’s a lot easier for young children to take. This is important in encouraging uptake of the treatment and adherence to it over many years.
“Sadly, only about half of children living with HIV are currently receiving treatment, and those who are not treated face high risks of impaired immunity and worsening health.”
Study details
The randomized controlled trial, called ODYSSEY, involved more than 700 children from 29 clinical centers located in Africa, Europe, and Asia. The children were given either dolutegravir or standard anti-HIV drugs, and were followed up for at least 2 years.
The study showed that 14% of children receiving dolutegravir experienced treatment failure over 2 years, compared with 22% of those receiving standard treatment. Treatment failure was deemed to occur if measurable virus appeared in the blood or if the child had symptoms of HIV-related illness.
“Our findings provide strong evidence for the global rollout of dolutegravir for children with HIV,” said Diana Gibb, MD, also from the MRC clinical trials unit at UCL, principal investigator of the trial and one of the senior authors of the paper.
“Medical treatments for children often lag woefully behind those of adults because of the separate formulations and studies that are needed,” she added. “With the evidence from ODYSSEY which used simplified dosing of both adult and baby pills, this treatment gap has been reduced and we hope that countries can quickly scale up access to children globally.”
Simplified dosing
“Simplifying the dosing is crucial,” concurred Cissy Kityo Mutuluuza, MD, from the Joint Clinical Research Centre in Uganda, the country enrolling most children in the trial. “Older children being able to take the same tablets as adults immediately opens access to dolutegravir for the majority of children living with HIV. It greatly simplifies procurement for national health systems in low- and middle-income countries, and lowers costs.”
Evidence from adults shows dolutegravir has a high genetic barrier to resistance, meaning viruses are less likely to become resistant to it over time. This was confirmed in the ODYSSEY trial, with much less resistance occurring among children and adolescents on dolutegravir-based treatment. In addition, past studies of the drug have shown that it may be associated with weight gain in adults, but the findings were reassuring for children. Those given dolutegravir gained on average 1 kg more and grew 1 cm higher over the study period, indicating better growth rather than abnormal weight gain.
Early findings from the trial have informed new guidance by the World Health Organization, recommending the use of dolutegravir for children.
The study was sponsored by the Penta Foundation, an international independent research network, and funded by specialist pharmaceutical company ViiV Healthcare.
A version of this article first appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Why mRNA COVID vaccines are preferred (and why patients should be reassured)
On December 16, 2021, the Advisory Committee on Immunization Practices (ACIP) voted to preferentially recommend messenger RNA (mRNA) vaccines over the Johnson & Johnson/Janssen (J&J) COVID-19 (Ad.26.COV2.S) adenovirus vector vaccine for prevention of COVID-19.1 The mRNA vaccines include Pfizer-BioNTech COVID-19 (BNT162b2) and Moderna COVID-19 (mRNA-1273).
The reason for this preferential recommendation is a rare but serious adverse reaction—thrombosis with thrombocytopenia (TTS) —that has been associated with the J&J vaccine. As of December 8, 2021, more than 16.9 million doses of the J&J COVID-19 vaccine have been given in the United States. The CDC has identified 57 confirmed reports of people who received this vaccine and later developed TTS.2 The known incidence of TTS is thus 1 per ~ 300,000 doses, although the rate may actually be higher.2 All cases have been documented as having occurred after administration of the J&J primary single-dose vaccine; none have been documented (so far) after the booster—although the number of booster doses of the J&J COVID-19 vaccine has been small.
Women between the ages of 30 and 50 years have the highest risk for TTS, with rates of 1 per 94,000 in those ages 30-39 and 1 per 111,000 for those ages 40-49.2,3 All those with TTS have been hospitalized, and 9 have died.2,3 While this adverse reaction is rare, the seriousness of it led the ACIP to state a preference for the mRNA vaccines.
The significance of the recommendation:
- Unless a person has a contraindication to an mRNA vaccine, they should receive 1 of these 2 vaccines for their primary series and boosters.
- The only “Mix and Match” that should occur with boosters is to follow a J&J/Janssen COVID-19 vaccine with an mRNA booster. At this time, booster doses following a 2-dose mRNA primary series should be with an mRNA vaccine.
- The recommendation is for adults ages 18 and older; however, the J&J/Janssen COVID-19 vaccine is not yet approved for younger age-groups.
- The J&J/Janssen COVID-19 vaccine remains an option for those who cannot receive an mRNA vaccine, but it should be administered only after full informed consent.
The J&J/Janssen COVID-19 vaccine initially looked promising a year ago because of its single-dose primary series and its much less stringent storage requirements. However, things have not quite panned out for the vaccine. Its effectiveness after a single dose has proven to be significantly inferior to the 2-dose mRNA vaccines, and it has now been associated with a very serious, albeit rare, adverse reaction.
The major take-home point for physicians to pass on to their patients is that the nation’s system for monitoring vaccine safety works. It can pick up serious adverse reactions that occur at a rate as low as 1/300,000. This should be reassuring.
1. CDC. CDC Endorses ACIP’s Updated COVID-19 Vaccine Recommendations [press release]. December 16, 2021. Accessed December 22, 2021. www.cdc.gov/media/releases/2021/s1216-covid-19-vaccines.html
2. CDC. Selected Adverse Events Reported after COVID-19 Vaccination. December 20, 2021. Accessed December 22, 2021. www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/adverse-events.html
3. See I. Updates on thrombosis with thrombocytopenia syndrome (TTS). Presented to the Advisory Committee on Immunization Practices. December 16, 2021. Accessed December 22, 2021. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2021-12-16/02-COVID-See-508.pdf
On December 16, 2021, the Advisory Committee on Immunization Practices (ACIP) voted to preferentially recommend messenger RNA (mRNA) vaccines over the Johnson & Johnson/Janssen (J&J) COVID-19 (Ad.26.COV2.S) adenovirus vector vaccine for prevention of COVID-19.1 The mRNA vaccines include Pfizer-BioNTech COVID-19 (BNT162b2) and Moderna COVID-19 (mRNA-1273).
The reason for this preferential recommendation is a rare but serious adverse reaction—thrombosis with thrombocytopenia (TTS) —that has been associated with the J&J vaccine. As of December 8, 2021, more than 16.9 million doses of the J&J COVID-19 vaccine have been given in the United States. The CDC has identified 57 confirmed reports of people who received this vaccine and later developed TTS.2 The known incidence of TTS is thus 1 per ~ 300,000 doses, although the rate may actually be higher.2 All cases have been documented as having occurred after administration of the J&J primary single-dose vaccine; none have been documented (so far) after the booster—although the number of booster doses of the J&J COVID-19 vaccine has been small.
Women between the ages of 30 and 50 years have the highest risk for TTS, with rates of 1 per 94,000 in those ages 30-39 and 1 per 111,000 for those ages 40-49.2,3 All those with TTS have been hospitalized, and 9 have died.2,3 While this adverse reaction is rare, the seriousness of it led the ACIP to state a preference for the mRNA vaccines.
The significance of the recommendation:
- Unless a person has a contraindication to an mRNA vaccine, they should receive 1 of these 2 vaccines for their primary series and boosters.
- The only “Mix and Match” that should occur with boosters is to follow a J&J/Janssen COVID-19 vaccine with an mRNA booster. At this time, booster doses following a 2-dose mRNA primary series should be with an mRNA vaccine.
- The recommendation is for adults ages 18 and older; however, the J&J/Janssen COVID-19 vaccine is not yet approved for younger age-groups.
- The J&J/Janssen COVID-19 vaccine remains an option for those who cannot receive an mRNA vaccine, but it should be administered only after full informed consent.
The J&J/Janssen COVID-19 vaccine initially looked promising a year ago because of its single-dose primary series and its much less stringent storage requirements. However, things have not quite panned out for the vaccine. Its effectiveness after a single dose has proven to be significantly inferior to the 2-dose mRNA vaccines, and it has now been associated with a very serious, albeit rare, adverse reaction.
The major take-home point for physicians to pass on to their patients is that the nation’s system for monitoring vaccine safety works. It can pick up serious adverse reactions that occur at a rate as low as 1/300,000. This should be reassuring.
On December 16, 2021, the Advisory Committee on Immunization Practices (ACIP) voted to preferentially recommend messenger RNA (mRNA) vaccines over the Johnson & Johnson/Janssen (J&J) COVID-19 (Ad.26.COV2.S) adenovirus vector vaccine for prevention of COVID-19.1 The mRNA vaccines include Pfizer-BioNTech COVID-19 (BNT162b2) and Moderna COVID-19 (mRNA-1273).
The reason for this preferential recommendation is a rare but serious adverse reaction—thrombosis with thrombocytopenia (TTS) —that has been associated with the J&J vaccine. As of December 8, 2021, more than 16.9 million doses of the J&J COVID-19 vaccine have been given in the United States. The CDC has identified 57 confirmed reports of people who received this vaccine and later developed TTS.2 The known incidence of TTS is thus 1 per ~ 300,000 doses, although the rate may actually be higher.2 All cases have been documented as having occurred after administration of the J&J primary single-dose vaccine; none have been documented (so far) after the booster—although the number of booster doses of the J&J COVID-19 vaccine has been small.
Women between the ages of 30 and 50 years have the highest risk for TTS, with rates of 1 per 94,000 in those ages 30-39 and 1 per 111,000 for those ages 40-49.2,3 All those with TTS have been hospitalized, and 9 have died.2,3 While this adverse reaction is rare, the seriousness of it led the ACIP to state a preference for the mRNA vaccines.
The significance of the recommendation:
- Unless a person has a contraindication to an mRNA vaccine, they should receive 1 of these 2 vaccines for their primary series and boosters.
- The only “Mix and Match” that should occur with boosters is to follow a J&J/Janssen COVID-19 vaccine with an mRNA booster. At this time, booster doses following a 2-dose mRNA primary series should be with an mRNA vaccine.
- The recommendation is for adults ages 18 and older; however, the J&J/Janssen COVID-19 vaccine is not yet approved for younger age-groups.
- The J&J/Janssen COVID-19 vaccine remains an option for those who cannot receive an mRNA vaccine, but it should be administered only after full informed consent.
The J&J/Janssen COVID-19 vaccine initially looked promising a year ago because of its single-dose primary series and its much less stringent storage requirements. However, things have not quite panned out for the vaccine. Its effectiveness after a single dose has proven to be significantly inferior to the 2-dose mRNA vaccines, and it has now been associated with a very serious, albeit rare, adverse reaction.
The major take-home point for physicians to pass on to their patients is that the nation’s system for monitoring vaccine safety works. It can pick up serious adverse reactions that occur at a rate as low as 1/300,000. This should be reassuring.
1. CDC. CDC Endorses ACIP’s Updated COVID-19 Vaccine Recommendations [press release]. December 16, 2021. Accessed December 22, 2021. www.cdc.gov/media/releases/2021/s1216-covid-19-vaccines.html
2. CDC. Selected Adverse Events Reported after COVID-19 Vaccination. December 20, 2021. Accessed December 22, 2021. www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/adverse-events.html
3. See I. Updates on thrombosis with thrombocytopenia syndrome (TTS). Presented to the Advisory Committee on Immunization Practices. December 16, 2021. Accessed December 22, 2021. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2021-12-16/02-COVID-See-508.pdf
1. CDC. CDC Endorses ACIP’s Updated COVID-19 Vaccine Recommendations [press release]. December 16, 2021. Accessed December 22, 2021. www.cdc.gov/media/releases/2021/s1216-covid-19-vaccines.html
2. CDC. Selected Adverse Events Reported after COVID-19 Vaccination. December 20, 2021. Accessed December 22, 2021. www.cdc.gov/coronavirus/2019-ncov/vaccines/safety/adverse-events.html
3. See I. Updates on thrombosis with thrombocytopenia syndrome (TTS). Presented to the Advisory Committee on Immunization Practices. December 16, 2021. Accessed December 22, 2021. www.cdc.gov/vaccines/acip/meetings/downloads/slides-2021-12-16/02-COVID-See-508.pdf

Children and COVID: Nearly 200,000 new cases reported in 1 week
, according to the American Academy of Pediatrics and the Children’s Hospital Association.
Available state data show that 198,551 child COVID cases were added during the week of Dec. 17-23 – up by 16.8% from the nearly 170,000 new cases reported the previous week and the highest 7-day figure since Sept. 17-23, when 207,000 cases were reported, the AAP and the CHA said in their weekly COVID report. Since Oct. 22-28, when the weekly count dropped to a seasonal low, the weekly count has nearly doubled.
The largest shares of the nearly 199,000 new cases were divided pretty equally between the Northeast and the South, while the West had just a small bump in cases and the Midwest was in the middle. The largest statewide percent increases came in the New England states, along with New Jersey, the District of Columbia, and Puerto Rico. New York State does not report age ranges for COVID cases, the AAP/CHA report noted.
Emergency department visits and hospital admissions are following a similar trend, as both have risen considerably over the last 2 months, data from the Centers for Disease Control and Prevention show.
COVID-related ED visits for children aged 0-11 years – measured as a proportion of all ED visits – are nearing the pandemic high of 4.1% set in late August, while visits in 12- to 15-year-olds have risen from 1.4% in early November to 5.6% on Dec. 24 and 16- to 17-year-olds have gone from 1.5% to 6% over the same period of time, the CDC reported on its COVID Data Tracker.
As for hospital admissions in children aged 0-17 years, the rate was down to 0.19 per 100,000 population on Nov. 11 but had risen to 0.38 per 100,000 as of Dec. 24. The highest point reached in children during the pandemic was 0.46 per 100,000 in early September, the CDC said.
On Dec. 23, 367 children were admitted to hospitals in the United States, the highest number since Sept. 7, when 374 were hospitalized. The highest 1-day total over the course of the pandemic, 394, came just a week before that, Aug. 31, according to the Department of Health & Human Services.
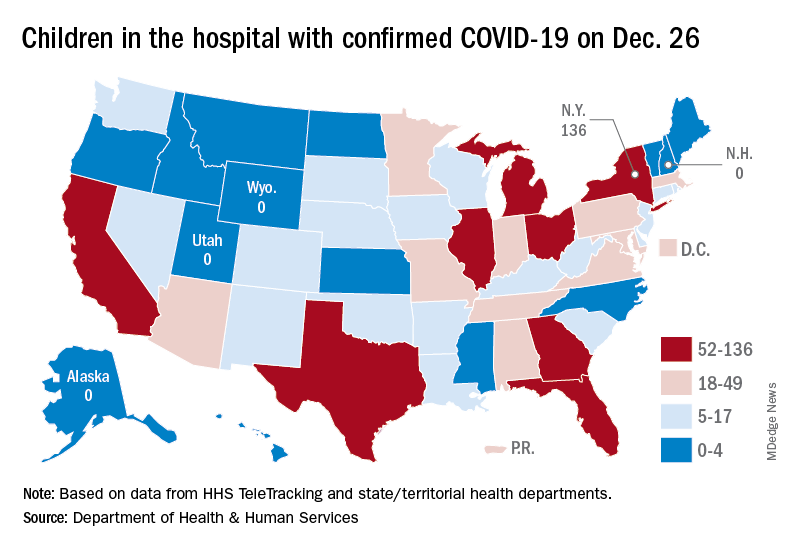
A look at the most recent HHS data shows that 1,161 children were being hospitalized in pediatric inpatient beds with confirmed COVID-19 on Dec. 26. The highest number by state was in New York (136), followed by Texas (90) and Illinois and Ohio, both with 83. There were four states – Alaska, New Hampshire, Utah, and Wyoming – with no hospitalized children, the HHS said. Puerto Rico, meanwhile, had 28 children in the hospital with COVID, more than 38 states.
, according to the American Academy of Pediatrics and the Children’s Hospital Association.
Available state data show that 198,551 child COVID cases were added during the week of Dec. 17-23 – up by 16.8% from the nearly 170,000 new cases reported the previous week and the highest 7-day figure since Sept. 17-23, when 207,000 cases were reported, the AAP and the CHA said in their weekly COVID report. Since Oct. 22-28, when the weekly count dropped to a seasonal low, the weekly count has nearly doubled.
The largest shares of the nearly 199,000 new cases were divided pretty equally between the Northeast and the South, while the West had just a small bump in cases and the Midwest was in the middle. The largest statewide percent increases came in the New England states, along with New Jersey, the District of Columbia, and Puerto Rico. New York State does not report age ranges for COVID cases, the AAP/CHA report noted.
Emergency department visits and hospital admissions are following a similar trend, as both have risen considerably over the last 2 months, data from the Centers for Disease Control and Prevention show.
COVID-related ED visits for children aged 0-11 years – measured as a proportion of all ED visits – are nearing the pandemic high of 4.1% set in late August, while visits in 12- to 15-year-olds have risen from 1.4% in early November to 5.6% on Dec. 24 and 16- to 17-year-olds have gone from 1.5% to 6% over the same period of time, the CDC reported on its COVID Data Tracker.
As for hospital admissions in children aged 0-17 years, the rate was down to 0.19 per 100,000 population on Nov. 11 but had risen to 0.38 per 100,000 as of Dec. 24. The highest point reached in children during the pandemic was 0.46 per 100,000 in early September, the CDC said.
On Dec. 23, 367 children were admitted to hospitals in the United States, the highest number since Sept. 7, when 374 were hospitalized. The highest 1-day total over the course of the pandemic, 394, came just a week before that, Aug. 31, according to the Department of Health & Human Services.
A look at the most recent HHS data shows that 1,161 children were being hospitalized in pediatric inpatient beds with confirmed COVID-19 on Dec. 26. The highest number by state was in New York (136), followed by Texas (90) and Illinois and Ohio, both with 83. There were four states – Alaska, New Hampshire, Utah, and Wyoming – with no hospitalized children, the HHS said. Puerto Rico, meanwhile, had 28 children in the hospital with COVID, more than 38 states.
, according to the American Academy of Pediatrics and the Children’s Hospital Association.
Available state data show that 198,551 child COVID cases were added during the week of Dec. 17-23 – up by 16.8% from the nearly 170,000 new cases reported the previous week and the highest 7-day figure since Sept. 17-23, when 207,000 cases were reported, the AAP and the CHA said in their weekly COVID report. Since Oct. 22-28, when the weekly count dropped to a seasonal low, the weekly count has nearly doubled.
The largest shares of the nearly 199,000 new cases were divided pretty equally between the Northeast and the South, while the West had just a small bump in cases and the Midwest was in the middle. The largest statewide percent increases came in the New England states, along with New Jersey, the District of Columbia, and Puerto Rico. New York State does not report age ranges for COVID cases, the AAP/CHA report noted.
Emergency department visits and hospital admissions are following a similar trend, as both have risen considerably over the last 2 months, data from the Centers for Disease Control and Prevention show.
COVID-related ED visits for children aged 0-11 years – measured as a proportion of all ED visits – are nearing the pandemic high of 4.1% set in late August, while visits in 12- to 15-year-olds have risen from 1.4% in early November to 5.6% on Dec. 24 and 16- to 17-year-olds have gone from 1.5% to 6% over the same period of time, the CDC reported on its COVID Data Tracker.
As for hospital admissions in children aged 0-17 years, the rate was down to 0.19 per 100,000 population on Nov. 11 but had risen to 0.38 per 100,000 as of Dec. 24. The highest point reached in children during the pandemic was 0.46 per 100,000 in early September, the CDC said.
On Dec. 23, 367 children were admitted to hospitals in the United States, the highest number since Sept. 7, when 374 were hospitalized. The highest 1-day total over the course of the pandemic, 394, came just a week before that, Aug. 31, according to the Department of Health & Human Services.
A look at the most recent HHS data shows that 1,161 children were being hospitalized in pediatric inpatient beds with confirmed COVID-19 on Dec. 26. The highest number by state was in New York (136), followed by Texas (90) and Illinois and Ohio, both with 83. There were four states – Alaska, New Hampshire, Utah, and Wyoming – with no hospitalized children, the HHS said. Puerto Rico, meanwhile, had 28 children in the hospital with COVID, more than 38 states.
COVID-19 vaccinations in people with HIV reflect general rates despite higher mortality risk, study says
Around the world, people with HIV show variations in COVID-19 vaccination rates similar to those seen in the general population, raising concerns because of their increased risk for morbidity and mortality from COVID-19 infection.
“To our knowledge, this analysis presents the first and largest investigation of vaccination rates among people with HIV,” reported the authors in research published in the Journal of Infectious Diseases.
The findings reflect data on nearly 7,000 people with HIV participating in the REPRIEVE clinical trial. As of July, COVID-19 vaccination rates ranged from a high of 71% in higher income regions to just 18% in sub-Saharan Africa and bottomed out at 0% in Haiti.
“This disparity in COVID-19 vaccination rates among people with HIV across income regions may increase morbidity from COVID-19 in the most vulnerable HIV populations,” the authors noted.
In general, people with HIV have been shown in recent research to have as much as 29% higher odds of morality from COVID-19 than the general population, and a 20% higher odds of hospitalization, hence their need for vaccination is especially pressing.
To understand the vaccination rates, the authors looked at data from the ongoing REPRIEVE trial, designed to investigate primary cardiovascular prevention worldwide among people with HIV. The trial includes data on COVID-19 vaccination status, providing a unique opportunity to capture those rates.
The study specifically included 6,952 people with HIV aged 40-75 years and on stable antiretroviral therapy (ART), without known cardiovascular disease, and a low to moderate atherosclerotic cardiovascular disease (ASCVD) risk.
The diverse participants with HIV were from 12 countries, including 66% who were people of color, as well as 32% women. Countries represented include Brazil (n = 1,042), Botswana (n = 273), Canada (n = 123), Haiti (n = 136), India (n = 469), Peru (n = 142), South Africa (n = 527), Spain (n = 198), Thailand (n = 582), Uganda (n = 175), United States (n = 3,162), and Zimbabwe (n = 123).
With vaccination defined as having received at least one vaccine shot, the overall cumulative COVID-19 vaccination rate in the study was 55% through July 2021.
By region, the highest cumulative rates were in the high-income countries of the United States and Canada (71%), followed by Latin America and the Caribbean (59%) – all consistent with the general population in these areas
Lower cumulative vaccination rates were observed in South Asia (49%), Southeast/East Asia (41%), and sub-Saharan Africa (18%), also reflecting the regional vaccination rates.
The United States had the highest country-specific COVID-19 vaccination rate of 72%, followed by Peru (69%) and Brazil (63%). Countries with the lowest vaccination rates were South Africa (18%), Uganda (3%), and Haiti (0%).
Of note, South Africa and Botswana have the largest share of deaths from HIV/AIDS, and both had very low COVID-19 vaccination rates in general, compared with high-income countries.
Overall, factors linked to the likelihood of being vaccinated included residing in the high-income U.S./Canada Global Burden of Disease superregion, as well as being White, male, older, having a higher body mass index (BMI), a higher ASCVD risk score, and longer duration of ART.
Participants’ decisions regarding COVID-19 vaccination in the study were made individually and were not based on any study-related recommendations or requirements, the authors noted.
Vaccination rates were higher among men than women in most regions, with the exception of sub-Saharan Africa. Vaccination rates were higher among Whites than Blacks in the U.S./Canada high-income region, with a high proportion of participants from the United States.
“It was surprising to us – and unfortunate – that in the high-income superregion vaccination rates were higher among individuals who identified as White than those who identified as Black and among men,” senior author Steven K. Grinspoon, MD, said in an interview.
“Given data for higher morbidity from COVID-19 among people of color with HIV, this disparity is likely to have significant public health implications,” said Dr. Grinspoon, a professor of medicine at Harvard Medical School and chief of the metabolism unit at Massachusetts General Hospital, both in Boston.
Newer data from the REPRIEVE study through October has shown continued steady increases in the cumulative vaccination rates in all regions, Dr. Grinspoon noted, with the largest increases in the Southeast/East Asia, South Asia, and sub-Saharan Africa, whereas a leveling off of rates was observed in the high-income regions.
Overall, “it is encouraging that rates among people with HIV are similar to and, in many regions, higher than the general population,” Dr. Grinspoon said.
However, with the data showing a higher risk for COVID-19 death in people with HIV, “it is critical that people with HIV, representing a vulnerable and immunocompromised population, be vaccinated for COVID-19,” Dr. Grinspoon said.
Commenting on the study, Monica Gandhi, MD, MPH, director of the Gladstone Center for AIDS Research at the University of California, San Francisco, agreed that “it is encouraging that these rates are as high as the general population, showing that there is not excess hesitancy among those living with HIV.”
Unlike other immunocompromised groups, people with HIV were not necessarily prioritized for vaccination, since antiretroviral therapy can reconstitute the immune system, “so I am not surprised the [vaccination] rates aren’t higher,” Dr. Gandhi, who was not involved with the study, said in an interview.
Nevertheless, “it is important that those with risk factors for more severe disease, such as higher BMI and higher cardiovascular disease, are prioritized for COVID-19 vaccination, [as] these are important groups in which to increase rates,” she said.
“The take-home message is that we have to increase our rates of vaccination in this critically important population,” Dr. Gandhi emphasized. “Global vaccine equity is paramount given that the burden of HIV infections remains in sub-Saharan Africa.”
The study received support from the National Institutes of Health and funding from Kowa Pharmaceuticals and Gilead Sciences. The authors and Dr. Gandhi disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Around the world, people with HIV show variations in COVID-19 vaccination rates similar to those seen in the general population, raising concerns because of their increased risk for morbidity and mortality from COVID-19 infection.
“To our knowledge, this analysis presents the first and largest investigation of vaccination rates among people with HIV,” reported the authors in research published in the Journal of Infectious Diseases.
The findings reflect data on nearly 7,000 people with HIV participating in the REPRIEVE clinical trial. As of July, COVID-19 vaccination rates ranged from a high of 71% in higher income regions to just 18% in sub-Saharan Africa and bottomed out at 0% in Haiti.
“This disparity in COVID-19 vaccination rates among people with HIV across income regions may increase morbidity from COVID-19 in the most vulnerable HIV populations,” the authors noted.
In general, people with HIV have been shown in recent research to have as much as 29% higher odds of morality from COVID-19 than the general population, and a 20% higher odds of hospitalization, hence their need for vaccination is especially pressing.
To understand the vaccination rates, the authors looked at data from the ongoing REPRIEVE trial, designed to investigate primary cardiovascular prevention worldwide among people with HIV. The trial includes data on COVID-19 vaccination status, providing a unique opportunity to capture those rates.
The study specifically included 6,952 people with HIV aged 40-75 years and on stable antiretroviral therapy (ART), without known cardiovascular disease, and a low to moderate atherosclerotic cardiovascular disease (ASCVD) risk.
The diverse participants with HIV were from 12 countries, including 66% who were people of color, as well as 32% women. Countries represented include Brazil (n = 1,042), Botswana (n = 273), Canada (n = 123), Haiti (n = 136), India (n = 469), Peru (n = 142), South Africa (n = 527), Spain (n = 198), Thailand (n = 582), Uganda (n = 175), United States (n = 3,162), and Zimbabwe (n = 123).
With vaccination defined as having received at least one vaccine shot, the overall cumulative COVID-19 vaccination rate in the study was 55% through July 2021.
By region, the highest cumulative rates were in the high-income countries of the United States and Canada (71%), followed by Latin America and the Caribbean (59%) – all consistent with the general population in these areas
Lower cumulative vaccination rates were observed in South Asia (49%), Southeast/East Asia (41%), and sub-Saharan Africa (18%), also reflecting the regional vaccination rates.
The United States had the highest country-specific COVID-19 vaccination rate of 72%, followed by Peru (69%) and Brazil (63%). Countries with the lowest vaccination rates were South Africa (18%), Uganda (3%), and Haiti (0%).
Of note, South Africa and Botswana have the largest share of deaths from HIV/AIDS, and both had very low COVID-19 vaccination rates in general, compared with high-income countries.
Overall, factors linked to the likelihood of being vaccinated included residing in the high-income U.S./Canada Global Burden of Disease superregion, as well as being White, male, older, having a higher body mass index (BMI), a higher ASCVD risk score, and longer duration of ART.
Participants’ decisions regarding COVID-19 vaccination in the study were made individually and were not based on any study-related recommendations or requirements, the authors noted.
Vaccination rates were higher among men than women in most regions, with the exception of sub-Saharan Africa. Vaccination rates were higher among Whites than Blacks in the U.S./Canada high-income region, with a high proportion of participants from the United States.
“It was surprising to us – and unfortunate – that in the high-income superregion vaccination rates were higher among individuals who identified as White than those who identified as Black and among men,” senior author Steven K. Grinspoon, MD, said in an interview.
“Given data for higher morbidity from COVID-19 among people of color with HIV, this disparity is likely to have significant public health implications,” said Dr. Grinspoon, a professor of medicine at Harvard Medical School and chief of the metabolism unit at Massachusetts General Hospital, both in Boston.
Newer data from the REPRIEVE study through October has shown continued steady increases in the cumulative vaccination rates in all regions, Dr. Grinspoon noted, with the largest increases in the Southeast/East Asia, South Asia, and sub-Saharan Africa, whereas a leveling off of rates was observed in the high-income regions.
Overall, “it is encouraging that rates among people with HIV are similar to and, in many regions, higher than the general population,” Dr. Grinspoon said.
However, with the data showing a higher risk for COVID-19 death in people with HIV, “it is critical that people with HIV, representing a vulnerable and immunocompromised population, be vaccinated for COVID-19,” Dr. Grinspoon said.
Commenting on the study, Monica Gandhi, MD, MPH, director of the Gladstone Center for AIDS Research at the University of California, San Francisco, agreed that “it is encouraging that these rates are as high as the general population, showing that there is not excess hesitancy among those living with HIV.”
Unlike other immunocompromised groups, people with HIV were not necessarily prioritized for vaccination, since antiretroviral therapy can reconstitute the immune system, “so I am not surprised the [vaccination] rates aren’t higher,” Dr. Gandhi, who was not involved with the study, said in an interview.
Nevertheless, “it is important that those with risk factors for more severe disease, such as higher BMI and higher cardiovascular disease, are prioritized for COVID-19 vaccination, [as] these are important groups in which to increase rates,” she said.
“The take-home message is that we have to increase our rates of vaccination in this critically important population,” Dr. Gandhi emphasized. “Global vaccine equity is paramount given that the burden of HIV infections remains in sub-Saharan Africa.”
The study received support from the National Institutes of Health and funding from Kowa Pharmaceuticals and Gilead Sciences. The authors and Dr. Gandhi disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Around the world, people with HIV show variations in COVID-19 vaccination rates similar to those seen in the general population, raising concerns because of their increased risk for morbidity and mortality from COVID-19 infection.
“To our knowledge, this analysis presents the first and largest investigation of vaccination rates among people with HIV,” reported the authors in research published in the Journal of Infectious Diseases.
The findings reflect data on nearly 7,000 people with HIV participating in the REPRIEVE clinical trial. As of July, COVID-19 vaccination rates ranged from a high of 71% in higher income regions to just 18% in sub-Saharan Africa and bottomed out at 0% in Haiti.
“This disparity in COVID-19 vaccination rates among people with HIV across income regions may increase morbidity from COVID-19 in the most vulnerable HIV populations,” the authors noted.
In general, people with HIV have been shown in recent research to have as much as 29% higher odds of morality from COVID-19 than the general population, and a 20% higher odds of hospitalization, hence their need for vaccination is especially pressing.
To understand the vaccination rates, the authors looked at data from the ongoing REPRIEVE trial, designed to investigate primary cardiovascular prevention worldwide among people with HIV. The trial includes data on COVID-19 vaccination status, providing a unique opportunity to capture those rates.
The study specifically included 6,952 people with HIV aged 40-75 years and on stable antiretroviral therapy (ART), without known cardiovascular disease, and a low to moderate atherosclerotic cardiovascular disease (ASCVD) risk.
The diverse participants with HIV were from 12 countries, including 66% who were people of color, as well as 32% women. Countries represented include Brazil (n = 1,042), Botswana (n = 273), Canada (n = 123), Haiti (n = 136), India (n = 469), Peru (n = 142), South Africa (n = 527), Spain (n = 198), Thailand (n = 582), Uganda (n = 175), United States (n = 3,162), and Zimbabwe (n = 123).
With vaccination defined as having received at least one vaccine shot, the overall cumulative COVID-19 vaccination rate in the study was 55% through July 2021.
By region, the highest cumulative rates were in the high-income countries of the United States and Canada (71%), followed by Latin America and the Caribbean (59%) – all consistent with the general population in these areas
Lower cumulative vaccination rates were observed in South Asia (49%), Southeast/East Asia (41%), and sub-Saharan Africa (18%), also reflecting the regional vaccination rates.
The United States had the highest country-specific COVID-19 vaccination rate of 72%, followed by Peru (69%) and Brazil (63%). Countries with the lowest vaccination rates were South Africa (18%), Uganda (3%), and Haiti (0%).
Of note, South Africa and Botswana have the largest share of deaths from HIV/AIDS, and both had very low COVID-19 vaccination rates in general, compared with high-income countries.
Overall, factors linked to the likelihood of being vaccinated included residing in the high-income U.S./Canada Global Burden of Disease superregion, as well as being White, male, older, having a higher body mass index (BMI), a higher ASCVD risk score, and longer duration of ART.
Participants’ decisions regarding COVID-19 vaccination in the study were made individually and were not based on any study-related recommendations or requirements, the authors noted.
Vaccination rates were higher among men than women in most regions, with the exception of sub-Saharan Africa. Vaccination rates were higher among Whites than Blacks in the U.S./Canada high-income region, with a high proportion of participants from the United States.
“It was surprising to us – and unfortunate – that in the high-income superregion vaccination rates were higher among individuals who identified as White than those who identified as Black and among men,” senior author Steven K. Grinspoon, MD, said in an interview.
“Given data for higher morbidity from COVID-19 among people of color with HIV, this disparity is likely to have significant public health implications,” said Dr. Grinspoon, a professor of medicine at Harvard Medical School and chief of the metabolism unit at Massachusetts General Hospital, both in Boston.
Newer data from the REPRIEVE study through October has shown continued steady increases in the cumulative vaccination rates in all regions, Dr. Grinspoon noted, with the largest increases in the Southeast/East Asia, South Asia, and sub-Saharan Africa, whereas a leveling off of rates was observed in the high-income regions.
Overall, “it is encouraging that rates among people with HIV are similar to and, in many regions, higher than the general population,” Dr. Grinspoon said.
However, with the data showing a higher risk for COVID-19 death in people with HIV, “it is critical that people with HIV, representing a vulnerable and immunocompromised population, be vaccinated for COVID-19,” Dr. Grinspoon said.
Commenting on the study, Monica Gandhi, MD, MPH, director of the Gladstone Center for AIDS Research at the University of California, San Francisco, agreed that “it is encouraging that these rates are as high as the general population, showing that there is not excess hesitancy among those living with HIV.”
Unlike other immunocompromised groups, people with HIV were not necessarily prioritized for vaccination, since antiretroviral therapy can reconstitute the immune system, “so I am not surprised the [vaccination] rates aren’t higher,” Dr. Gandhi, who was not involved with the study, said in an interview.
Nevertheless, “it is important that those with risk factors for more severe disease, such as higher BMI and higher cardiovascular disease, are prioritized for COVID-19 vaccination, [as] these are important groups in which to increase rates,” she said.
“The take-home message is that we have to increase our rates of vaccination in this critically important population,” Dr. Gandhi emphasized. “Global vaccine equity is paramount given that the burden of HIV infections remains in sub-Saharan Africa.”
The study received support from the National Institutes of Health and funding from Kowa Pharmaceuticals and Gilead Sciences. The authors and Dr. Gandhi disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF INFECTIOUS DISEASES
Spesolimab speeds lesion clearance in generalized pustular psoriasis
GPP is a life-threatening skin condition involving the widespread eruption of sterile pustules, with a clinical course that “can be relapsing with recurrent flares or persistent with intermittent flares,” Hervé Bachelez, MD, of the Université de Paris and coauthors wrote. GPP patients are often hospitalized, and mortality ranges from 2% to 16% from causes that include sepsis and cardiorespiratory failure.
“The role of the interleukin-36 pathway in GPP is supported by the finding of loss-of-function mutations in the interleukin-36 receptor antagonist gene (IL36RN) and associated genes (CARD14, AP1S3, SERPINA3, and MPO) and by the overexpression of interleukin-36 cytokines in GPP skin lesions,” therefore, IL-36 is a potential treatment target to manage flares, they explained.
In the multicenter, double-blind trial, published in the New England Journal of Medicine, the researchers randomized 35 adults with GPP flares to a single 900-mg intravenous dose of spesolimab and 18 to placebo. Patients in both groups could receive an open-label dose of spesolimab after day 8; all patients were followed for 12 weeks.
The primary study endpoint was the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at 1 week after treatment. The GPPGA ranges from 0 (no visible pustules) to 4 (severe pustules). At baseline, 46% spesolimab patients and 39% placebo patients had a GPPGA pustulation subscore of 3, and 37% and 33%, respectively, had a pustulation subscore of 4.
After 1 week, 54% of the spesolimab patients had no visible pustules, compared with 6% of placebo patients; the difference was statistically significant (P < .001). The main secondary endpoint was a score of 0 or 1 (clear or almost clear skin) on the GPPGA total score after 1 week. Significantly more spesolimab patients had GPPGA total scores of 0 or 1, compared with placebo patients (43% vs. 11%, respectively; P = .02).
Overall, 6 of 35 spesolimab patients (17%) and 6% of those in the placebo groups developed infections during the first week, and 24 of 51 patients (47%) who had received spesolimab at any point during the study developed infections by week 12. Infections included urinary tract infections (three cases), influenza (three), otitis externa (two), folliculitis (two), upper respiratory tract infection (two), and pustule (two).
In the first week, 6% of spesolimab patients and none of the placebo patients reported serious adverse events; at week 12, 12% of patients who had received at least one spesolimab dose reported a serious adverse event. In addition, antidrug antibodies were identified in 23 (46%) of the 50 patients who received at least one dose of spesolimab.
“Symptoms that were observed in two patients who received spesolimab were reported as a drug reaction with eosinophilia and systemic symptoms (DRESS),” the authors noted. One patient had a RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) score and the other had a score of 3; a score below 2 indicates no DRESS, and a score of 2 or 3 indicates “possible DRESS,” they added.
“Because 15 of the 18 patients who were assigned to the placebo group received open-label spesolimab, the effect of spesolimab as compared with that of placebo could not be determined after week 1,” the researchers noted.
The study findings were limited by several factors including the short randomization period and small study population, the researchers noted. However, the effect sizes for both the primary and secondary endpoints were large, which strengthened the results.
The results support data from previous studies suggesting a role for IL-36 in the pathogenesis of GPP, and support the need for longer and larger studies of the safety and effectiveness of spesolimab for GPP patients, they concluded.
No FDA-approved therapy
“GPP is a very rare but devastating life-threatening disease that presents with the sudden onset of pustules throughout the skin,” Joel Gelfand, MD, professor of dermatology and director of the psoriasis and phototherapy center at the University of Pennsylvania, Philadelphia, said in an interview. “Without rapid treatment, GPP can result in death. Currently there are no [Food and Drug Administration]–approved treatments for this orphan disease.”
Dr. Gelfand said he was surprised by the degree of efficacy and the speed of the patient response to spesolimab, compared with placebo, which he described as “truly remarkable.” Based on the current study results, “spesolimab offers a tremendous step forward for our patients,” he added.
Looking ahead, Dr. Gelfand noted that “longer-term studies with a comparator, such as a biologic that targets IL-17, would be helpful to more fully understand the safety, efficacy, and role that spesolimab will have in real-world patients.”
On Dec. 15, Boehringer Ingelheim announced that the FDA had granted priority review for spesolimab for treating GPP flares.
The study was supported by Boehringer Ingelheim. Lead author Dr. Bachelez had no financial conflicts to disclose. Several authors are employees of Boehringer Ingelheim. Dr. Gelfand is a consultant for the study sponsor Boehringer Ingelheim and has received research grants from Boehringer Ingelheim to his institution to support an investigator-initiated study. He also disclosed serving as a consultant and receiving research grants from other manufacturers of psoriasis products.
GPP is a life-threatening skin condition involving the widespread eruption of sterile pustules, with a clinical course that “can be relapsing with recurrent flares or persistent with intermittent flares,” Hervé Bachelez, MD, of the Université de Paris and coauthors wrote. GPP patients are often hospitalized, and mortality ranges from 2% to 16% from causes that include sepsis and cardiorespiratory failure.
“The role of the interleukin-36 pathway in GPP is supported by the finding of loss-of-function mutations in the interleukin-36 receptor antagonist gene (IL36RN) and associated genes (CARD14, AP1S3, SERPINA3, and MPO) and by the overexpression of interleukin-36 cytokines in GPP skin lesions,” therefore, IL-36 is a potential treatment target to manage flares, they explained.
In the multicenter, double-blind trial, published in the New England Journal of Medicine, the researchers randomized 35 adults with GPP flares to a single 900-mg intravenous dose of spesolimab and 18 to placebo. Patients in both groups could receive an open-label dose of spesolimab after day 8; all patients were followed for 12 weeks.
The primary study endpoint was the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at 1 week after treatment. The GPPGA ranges from 0 (no visible pustules) to 4 (severe pustules). At baseline, 46% spesolimab patients and 39% placebo patients had a GPPGA pustulation subscore of 3, and 37% and 33%, respectively, had a pustulation subscore of 4.
After 1 week, 54% of the spesolimab patients had no visible pustules, compared with 6% of placebo patients; the difference was statistically significant (P < .001). The main secondary endpoint was a score of 0 or 1 (clear or almost clear skin) on the GPPGA total score after 1 week. Significantly more spesolimab patients had GPPGA total scores of 0 or 1, compared with placebo patients (43% vs. 11%, respectively; P = .02).
Overall, 6 of 35 spesolimab patients (17%) and 6% of those in the placebo groups developed infections during the first week, and 24 of 51 patients (47%) who had received spesolimab at any point during the study developed infections by week 12. Infections included urinary tract infections (three cases), influenza (three), otitis externa (two), folliculitis (two), upper respiratory tract infection (two), and pustule (two).
In the first week, 6% of spesolimab patients and none of the placebo patients reported serious adverse events; at week 12, 12% of patients who had received at least one spesolimab dose reported a serious adverse event. In addition, antidrug antibodies were identified in 23 (46%) of the 50 patients who received at least one dose of spesolimab.
“Symptoms that were observed in two patients who received spesolimab were reported as a drug reaction with eosinophilia and systemic symptoms (DRESS),” the authors noted. One patient had a RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) score and the other had a score of 3; a score below 2 indicates no DRESS, and a score of 2 or 3 indicates “possible DRESS,” they added.
“Because 15 of the 18 patients who were assigned to the placebo group received open-label spesolimab, the effect of spesolimab as compared with that of placebo could not be determined after week 1,” the researchers noted.
The study findings were limited by several factors including the short randomization period and small study population, the researchers noted. However, the effect sizes for both the primary and secondary endpoints were large, which strengthened the results.
The results support data from previous studies suggesting a role for IL-36 in the pathogenesis of GPP, and support the need for longer and larger studies of the safety and effectiveness of spesolimab for GPP patients, they concluded.
No FDA-approved therapy
“GPP is a very rare but devastating life-threatening disease that presents with the sudden onset of pustules throughout the skin,” Joel Gelfand, MD, professor of dermatology and director of the psoriasis and phototherapy center at the University of Pennsylvania, Philadelphia, said in an interview. “Without rapid treatment, GPP can result in death. Currently there are no [Food and Drug Administration]–approved treatments for this orphan disease.”
Dr. Gelfand said he was surprised by the degree of efficacy and the speed of the patient response to spesolimab, compared with placebo, which he described as “truly remarkable.” Based on the current study results, “spesolimab offers a tremendous step forward for our patients,” he added.
Looking ahead, Dr. Gelfand noted that “longer-term studies with a comparator, such as a biologic that targets IL-17, would be helpful to more fully understand the safety, efficacy, and role that spesolimab will have in real-world patients.”
On Dec. 15, Boehringer Ingelheim announced that the FDA had granted priority review for spesolimab for treating GPP flares.
The study was supported by Boehringer Ingelheim. Lead author Dr. Bachelez had no financial conflicts to disclose. Several authors are employees of Boehringer Ingelheim. Dr. Gelfand is a consultant for the study sponsor Boehringer Ingelheim and has received research grants from Boehringer Ingelheim to his institution to support an investigator-initiated study. He also disclosed serving as a consultant and receiving research grants from other manufacturers of psoriasis products.
GPP is a life-threatening skin condition involving the widespread eruption of sterile pustules, with a clinical course that “can be relapsing with recurrent flares or persistent with intermittent flares,” Hervé Bachelez, MD, of the Université de Paris and coauthors wrote. GPP patients are often hospitalized, and mortality ranges from 2% to 16% from causes that include sepsis and cardiorespiratory failure.
“The role of the interleukin-36 pathway in GPP is supported by the finding of loss-of-function mutations in the interleukin-36 receptor antagonist gene (IL36RN) and associated genes (CARD14, AP1S3, SERPINA3, and MPO) and by the overexpression of interleukin-36 cytokines in GPP skin lesions,” therefore, IL-36 is a potential treatment target to manage flares, they explained.
In the multicenter, double-blind trial, published in the New England Journal of Medicine, the researchers randomized 35 adults with GPP flares to a single 900-mg intravenous dose of spesolimab and 18 to placebo. Patients in both groups could receive an open-label dose of spesolimab after day 8; all patients were followed for 12 weeks.
The primary study endpoint was the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at 1 week after treatment. The GPPGA ranges from 0 (no visible pustules) to 4 (severe pustules). At baseline, 46% spesolimab patients and 39% placebo patients had a GPPGA pustulation subscore of 3, and 37% and 33%, respectively, had a pustulation subscore of 4.
After 1 week, 54% of the spesolimab patients had no visible pustules, compared with 6% of placebo patients; the difference was statistically significant (P < .001). The main secondary endpoint was a score of 0 or 1 (clear or almost clear skin) on the GPPGA total score after 1 week. Significantly more spesolimab patients had GPPGA total scores of 0 or 1, compared with placebo patients (43% vs. 11%, respectively; P = .02).
Overall, 6 of 35 spesolimab patients (17%) and 6% of those in the placebo groups developed infections during the first week, and 24 of 51 patients (47%) who had received spesolimab at any point during the study developed infections by week 12. Infections included urinary tract infections (three cases), influenza (three), otitis externa (two), folliculitis (two), upper respiratory tract infection (two), and pustule (two).
In the first week, 6% of spesolimab patients and none of the placebo patients reported serious adverse events; at week 12, 12% of patients who had received at least one spesolimab dose reported a serious adverse event. In addition, antidrug antibodies were identified in 23 (46%) of the 50 patients who received at least one dose of spesolimab.
“Symptoms that were observed in two patients who received spesolimab were reported as a drug reaction with eosinophilia and systemic symptoms (DRESS),” the authors noted. One patient had a RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) score and the other had a score of 3; a score below 2 indicates no DRESS, and a score of 2 or 3 indicates “possible DRESS,” they added.
“Because 15 of the 18 patients who were assigned to the placebo group received open-label spesolimab, the effect of spesolimab as compared with that of placebo could not be determined after week 1,” the researchers noted.
The study findings were limited by several factors including the short randomization period and small study population, the researchers noted. However, the effect sizes for both the primary and secondary endpoints were large, which strengthened the results.
The results support data from previous studies suggesting a role for IL-36 in the pathogenesis of GPP, and support the need for longer and larger studies of the safety and effectiveness of spesolimab for GPP patients, they concluded.
No FDA-approved therapy
“GPP is a very rare but devastating life-threatening disease that presents with the sudden onset of pustules throughout the skin,” Joel Gelfand, MD, professor of dermatology and director of the psoriasis and phototherapy center at the University of Pennsylvania, Philadelphia, said in an interview. “Without rapid treatment, GPP can result in death. Currently there are no [Food and Drug Administration]–approved treatments for this orphan disease.”
Dr. Gelfand said he was surprised by the degree of efficacy and the speed of the patient response to spesolimab, compared with placebo, which he described as “truly remarkable.” Based on the current study results, “spesolimab offers a tremendous step forward for our patients,” he added.
Looking ahead, Dr. Gelfand noted that “longer-term studies with a comparator, such as a biologic that targets IL-17, would be helpful to more fully understand the safety, efficacy, and role that spesolimab will have in real-world patients.”
On Dec. 15, Boehringer Ingelheim announced that the FDA had granted priority review for spesolimab for treating GPP flares.
The study was supported by Boehringer Ingelheim. Lead author Dr. Bachelez had no financial conflicts to disclose. Several authors are employees of Boehringer Ingelheim. Dr. Gelfand is a consultant for the study sponsor Boehringer Ingelheim and has received research grants from Boehringer Ingelheim to his institution to support an investigator-initiated study. He also disclosed serving as a consultant and receiving research grants from other manufacturers of psoriasis products.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA OKs emergency use of Merck pill for COVID-19
Similar to FDA authorization of another antiviral pill regimen – ritonavir plus nirmatrelvir, or Paxlovid – granted to Pfizer on Wednesday, molnupiravir (brand name Lagevrio) should be taken early in the course of COVID-19 illness.
Pfizer’s drug is authorized for anyone aged 12 and up. But Merck’s is only for adults aged 18 and older.
Merck filed an application for emergency use authorization with the FDA in October. The company included results of its phase 3 study showing the treatment could lead to a 50% reduction in COVID-19 hospitalizations. Data later showed this efficacy at closer to a 30% reduction. In November, an FDA advisory panel narrowly recommended the agency grant authorization by a 13-10 vote.
Animal studies found the drug may harm a fetus, so it is not recommended for pregnant people, the FDA says. It may be prescribed to a pregnant person only after their doctor determines the benefits outweigh the risks and the patient is told of those risks.
Women who may get pregnant should use a reliable method of birth control if being treated with molnupiravir and for 4 days after the final dose.
Two weapons against COVID
Two antiviral pills could be better than one, at least in terms of making more COVID-19 treatments available in early 2022. It is yet to be seen if the drugmakers will be able to keep up with demand, which could substantially increase with an expected surge in Omicron variant cases.
Ritonavir and molnupiravir join remdesivir (brand name Veklury) as available antivirals to treat COVID-19. Remdesivir is fully approved by the FDA but is given only through an IV to people in the hospital.
Officials point out that COVID-19 treatments in tablet form are more convenient for patients in the United States and across the globe, particularly where IV infusion services may be limited.
In March 2021, experts accurately predicted that the molnupiravir pill would be available by year’s end.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of SARS-CoV-2 because the agent does not target the virus’s spike protein.
Perhaps in part because of early promising results, the U.S. government announced in November intentions to purchase $1 billion worth of molnupiravir. That new order came on top of $1.2 billion worth of the pills the U.S. ordered in June.
A version of this article first appeared on WebMD.com.
Similar to FDA authorization of another antiviral pill regimen – ritonavir plus nirmatrelvir, or Paxlovid – granted to Pfizer on Wednesday, molnupiravir (brand name Lagevrio) should be taken early in the course of COVID-19 illness.
Pfizer’s drug is authorized for anyone aged 12 and up. But Merck’s is only for adults aged 18 and older.
Merck filed an application for emergency use authorization with the FDA in October. The company included results of its phase 3 study showing the treatment could lead to a 50% reduction in COVID-19 hospitalizations. Data later showed this efficacy at closer to a 30% reduction. In November, an FDA advisory panel narrowly recommended the agency grant authorization by a 13-10 vote.
Animal studies found the drug may harm a fetus, so it is not recommended for pregnant people, the FDA says. It may be prescribed to a pregnant person only after their doctor determines the benefits outweigh the risks and the patient is told of those risks.
Women who may get pregnant should use a reliable method of birth control if being treated with molnupiravir and for 4 days after the final dose.
Two weapons against COVID
Two antiviral pills could be better than one, at least in terms of making more COVID-19 treatments available in early 2022. It is yet to be seen if the drugmakers will be able to keep up with demand, which could substantially increase with an expected surge in Omicron variant cases.
Ritonavir and molnupiravir join remdesivir (brand name Veklury) as available antivirals to treat COVID-19. Remdesivir is fully approved by the FDA but is given only through an IV to people in the hospital.
Officials point out that COVID-19 treatments in tablet form are more convenient for patients in the United States and across the globe, particularly where IV infusion services may be limited.
In March 2021, experts accurately predicted that the molnupiravir pill would be available by year’s end.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of SARS-CoV-2 because the agent does not target the virus’s spike protein.
Perhaps in part because of early promising results, the U.S. government announced in November intentions to purchase $1 billion worth of molnupiravir. That new order came on top of $1.2 billion worth of the pills the U.S. ordered in June.
A version of this article first appeared on WebMD.com.
Similar to FDA authorization of another antiviral pill regimen – ritonavir plus nirmatrelvir, or Paxlovid – granted to Pfizer on Wednesday, molnupiravir (brand name Lagevrio) should be taken early in the course of COVID-19 illness.
Pfizer’s drug is authorized for anyone aged 12 and up. But Merck’s is only for adults aged 18 and older.
Merck filed an application for emergency use authorization with the FDA in October. The company included results of its phase 3 study showing the treatment could lead to a 50% reduction in COVID-19 hospitalizations. Data later showed this efficacy at closer to a 30% reduction. In November, an FDA advisory panel narrowly recommended the agency grant authorization by a 13-10 vote.
Animal studies found the drug may harm a fetus, so it is not recommended for pregnant people, the FDA says. It may be prescribed to a pregnant person only after their doctor determines the benefits outweigh the risks and the patient is told of those risks.
Women who may get pregnant should use a reliable method of birth control if being treated with molnupiravir and for 4 days after the final dose.
Two weapons against COVID
Two antiviral pills could be better than one, at least in terms of making more COVID-19 treatments available in early 2022. It is yet to be seen if the drugmakers will be able to keep up with demand, which could substantially increase with an expected surge in Omicron variant cases.
Ritonavir and molnupiravir join remdesivir (brand name Veklury) as available antivirals to treat COVID-19. Remdesivir is fully approved by the FDA but is given only through an IV to people in the hospital.
Officials point out that COVID-19 treatments in tablet form are more convenient for patients in the United States and across the globe, particularly where IV infusion services may be limited.
In March 2021, experts accurately predicted that the molnupiravir pill would be available by year’s end.
Interestingly, in September, Merck announced the findings of laboratory studies suggesting that molnupiravir would work against variants of SARS-CoV-2 because the agent does not target the virus’s spike protein.
Perhaps in part because of early promising results, the U.S. government announced in November intentions to purchase $1 billion worth of molnupiravir. That new order came on top of $1.2 billion worth of the pills the U.S. ordered in June.
A version of this article first appeared on WebMD.com.
Bamlanivimab’s effects in COVID-19 depend on antibodies
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
FROM ANNALS OF INTERNAL MEDICINE