User login
Nonsurgical Alternatives for Skin Cancer Treatment: Report From the AAD Meeting
An important topic at the 2014 Summer AAD Meeting in Chicago, Illinois, was the nonsurgical treatment of skin cancers, including adjuvant therapy, topical creams, photodynamic therapy, and radiation for melanoma and nonmelanoma skin cancers. Dr. Anthony M. Rossi discusses the benefits of some of these nonsurgical treatment options for skin cancers and describes how he uses them in his practice. He also discusses how to determine which treatment option is best for each patient and emphasizes the importance of patient compliance and close follow-up.
An important topic at the 2014 Summer AAD Meeting in Chicago, Illinois, was the nonsurgical treatment of skin cancers, including adjuvant therapy, topical creams, photodynamic therapy, and radiation for melanoma and nonmelanoma skin cancers. Dr. Anthony M. Rossi discusses the benefits of some of these nonsurgical treatment options for skin cancers and describes how he uses them in his practice. He also discusses how to determine which treatment option is best for each patient and emphasizes the importance of patient compliance and close follow-up.
An important topic at the 2014 Summer AAD Meeting in Chicago, Illinois, was the nonsurgical treatment of skin cancers, including adjuvant therapy, topical creams, photodynamic therapy, and radiation for melanoma and nonmelanoma skin cancers. Dr. Anthony M. Rossi discusses the benefits of some of these nonsurgical treatment options for skin cancers and describes how he uses them in his practice. He also discusses how to determine which treatment option is best for each patient and emphasizes the importance of patient compliance and close follow-up.

Dermatologist biopsy rate undercut by digital device
CHICAGO – The number of skin lesions biopsied to rule out melanoma remains exceedingly high among U.S. dermatologists, compared with the MelaFind digital skin analysis device.
Dermatologists performed approximately 22 biopsies of histologically benign lesions for every one melanoma or severely dysplastic nevus removed, according to a dermatopathology review of 1,100 lesions from patients in approximately 500 practices nationwide.
This compares with a biopsy ratio of 7.6:1 reported in a pivotal prospective study of the MelaFind device (Arch. Dermatol. 2011;147:188-94), Dr. Clay J. Cockerell reported at the American Academy of Dermatology summer meeting.
The noninvasive MelaFind device was designed to aid dermatologists in diagnosing melanoma and uses multispectral light to characterize the morphologic disorganization of clinically atypical pigmented lesions.
It is approved in the European Union and gained U.S. approval in November 2011, with a long list of indications and caveats regarding how the device should be used and by whom.
The 22:1 biopsy rate among dermatologists is "pretty high" and likely reflects several factors, according to past AAD president Dr. Cockerell, who is director of the division of dermatopathology at the University of Texas Southwestern Medical Center and in group practice in Dallas.
Overall, clinicians are conservative regarding pigmented lesions, particularly in litigious areas of the country, and the device may have made some clinicians "more of a student of pigmented lesions, pushing them to look more carefully, and heightening awareness," he said in an interview.
Biopsies also may have been conducted by physician assistants and nurse practitioners, who may have a lower threshold for biopsy than dermatologists.
Lesion data were collected at Dr. Cockerell’s facility over a 3-week period for 1,400 lesions, but information was not available on how many practices had a MelaFind device, who performed the biopsy, the level of that clinician’s expertise in pigmented lesion management, or the proportion of high-risk patients, a group for whom biopsy ratios can reach 33:1 to 53:1, he said.
A total of 300 lesions were excluded from analysis because they were ineligible for use with the MelaFind device based on its indications.
Still, the MelaFind biopsy ratio is better than that observed in the real-world and was seen in lesions that were more clinically difficult, Dr. Cockerell reported in a poster.
The proportion of lesions that were melanoma or high-grade dysplastic nevi was higher in the pivotal study than in the real world data (11% vs. 4%), as was the proportion of nevi that were low-grade dysplastic nevi (79.3% vs. 41%).
Invasive melanomas, however, were thicker in the real life review (1.1 vs. 0.36 mm), raising a red flag that patients are still not fully educated on the need for early evaluation of suspicious lesions, he said.
"Biopsies, at least in our part of the country and I think in other parts as well, are performed in a seasonal fashion," Dr. Cockerell said. "There are people who don’t go to the dermatologist and don’t get a skin biopsy done for the first 5-6 months of the year because they’re waiting to get their deductible met. If they had a melanoma and put off a biopsy until June, it’s had 6 months of growth time. The doubling time can be very rapid."
Although the MelaFind-computed scores are subjective and do not state whether a biopsy is warranted, the data would be useful if sent along with a specimen to aid dermatopathologists in rendering a more definitive diagnosis, he said. Optimally, the device is "handcrafted" for practices and clinics that follow patients with hundreds of dysplastic nevi where repeated biopsies are not practical, an avenue Dr. Cockerell said he hopes to pursue in the future.
Mela Sciences sponsored the study. Dr. Cockerell and his coauthors disclosed no conflicting financial interests.
CHICAGO – The number of skin lesions biopsied to rule out melanoma remains exceedingly high among U.S. dermatologists, compared with the MelaFind digital skin analysis device.
Dermatologists performed approximately 22 biopsies of histologically benign lesions for every one melanoma or severely dysplastic nevus removed, according to a dermatopathology review of 1,100 lesions from patients in approximately 500 practices nationwide.
This compares with a biopsy ratio of 7.6:1 reported in a pivotal prospective study of the MelaFind device (Arch. Dermatol. 2011;147:188-94), Dr. Clay J. Cockerell reported at the American Academy of Dermatology summer meeting.
The noninvasive MelaFind device was designed to aid dermatologists in diagnosing melanoma and uses multispectral light to characterize the morphologic disorganization of clinically atypical pigmented lesions.
It is approved in the European Union and gained U.S. approval in November 2011, with a long list of indications and caveats regarding how the device should be used and by whom.
The 22:1 biopsy rate among dermatologists is "pretty high" and likely reflects several factors, according to past AAD president Dr. Cockerell, who is director of the division of dermatopathology at the University of Texas Southwestern Medical Center and in group practice in Dallas.
Overall, clinicians are conservative regarding pigmented lesions, particularly in litigious areas of the country, and the device may have made some clinicians "more of a student of pigmented lesions, pushing them to look more carefully, and heightening awareness," he said in an interview.
Biopsies also may have been conducted by physician assistants and nurse practitioners, who may have a lower threshold for biopsy than dermatologists.
Lesion data were collected at Dr. Cockerell’s facility over a 3-week period for 1,400 lesions, but information was not available on how many practices had a MelaFind device, who performed the biopsy, the level of that clinician’s expertise in pigmented lesion management, or the proportion of high-risk patients, a group for whom biopsy ratios can reach 33:1 to 53:1, he said.
A total of 300 lesions were excluded from analysis because they were ineligible for use with the MelaFind device based on its indications.
Still, the MelaFind biopsy ratio is better than that observed in the real-world and was seen in lesions that were more clinically difficult, Dr. Cockerell reported in a poster.
The proportion of lesions that were melanoma or high-grade dysplastic nevi was higher in the pivotal study than in the real world data (11% vs. 4%), as was the proportion of nevi that were low-grade dysplastic nevi (79.3% vs. 41%).
Invasive melanomas, however, were thicker in the real life review (1.1 vs. 0.36 mm), raising a red flag that patients are still not fully educated on the need for early evaluation of suspicious lesions, he said.
"Biopsies, at least in our part of the country and I think in other parts as well, are performed in a seasonal fashion," Dr. Cockerell said. "There are people who don’t go to the dermatologist and don’t get a skin biopsy done for the first 5-6 months of the year because they’re waiting to get their deductible met. If they had a melanoma and put off a biopsy until June, it’s had 6 months of growth time. The doubling time can be very rapid."
Although the MelaFind-computed scores are subjective and do not state whether a biopsy is warranted, the data would be useful if sent along with a specimen to aid dermatopathologists in rendering a more definitive diagnosis, he said. Optimally, the device is "handcrafted" for practices and clinics that follow patients with hundreds of dysplastic nevi where repeated biopsies are not practical, an avenue Dr. Cockerell said he hopes to pursue in the future.
Mela Sciences sponsored the study. Dr. Cockerell and his coauthors disclosed no conflicting financial interests.
CHICAGO – The number of skin lesions biopsied to rule out melanoma remains exceedingly high among U.S. dermatologists, compared with the MelaFind digital skin analysis device.
Dermatologists performed approximately 22 biopsies of histologically benign lesions for every one melanoma or severely dysplastic nevus removed, according to a dermatopathology review of 1,100 lesions from patients in approximately 500 practices nationwide.
This compares with a biopsy ratio of 7.6:1 reported in a pivotal prospective study of the MelaFind device (Arch. Dermatol. 2011;147:188-94), Dr. Clay J. Cockerell reported at the American Academy of Dermatology summer meeting.
The noninvasive MelaFind device was designed to aid dermatologists in diagnosing melanoma and uses multispectral light to characterize the morphologic disorganization of clinically atypical pigmented lesions.
It is approved in the European Union and gained U.S. approval in November 2011, with a long list of indications and caveats regarding how the device should be used and by whom.
The 22:1 biopsy rate among dermatologists is "pretty high" and likely reflects several factors, according to past AAD president Dr. Cockerell, who is director of the division of dermatopathology at the University of Texas Southwestern Medical Center and in group practice in Dallas.
Overall, clinicians are conservative regarding pigmented lesions, particularly in litigious areas of the country, and the device may have made some clinicians "more of a student of pigmented lesions, pushing them to look more carefully, and heightening awareness," he said in an interview.
Biopsies also may have been conducted by physician assistants and nurse practitioners, who may have a lower threshold for biopsy than dermatologists.
Lesion data were collected at Dr. Cockerell’s facility over a 3-week period for 1,400 lesions, but information was not available on how many practices had a MelaFind device, who performed the biopsy, the level of that clinician’s expertise in pigmented lesion management, or the proportion of high-risk patients, a group for whom biopsy ratios can reach 33:1 to 53:1, he said.
A total of 300 lesions were excluded from analysis because they were ineligible for use with the MelaFind device based on its indications.
Still, the MelaFind biopsy ratio is better than that observed in the real-world and was seen in lesions that were more clinically difficult, Dr. Cockerell reported in a poster.
The proportion of lesions that were melanoma or high-grade dysplastic nevi was higher in the pivotal study than in the real world data (11% vs. 4%), as was the proportion of nevi that were low-grade dysplastic nevi (79.3% vs. 41%).
Invasive melanomas, however, were thicker in the real life review (1.1 vs. 0.36 mm), raising a red flag that patients are still not fully educated on the need for early evaluation of suspicious lesions, he said.
"Biopsies, at least in our part of the country and I think in other parts as well, are performed in a seasonal fashion," Dr. Cockerell said. "There are people who don’t go to the dermatologist and don’t get a skin biopsy done for the first 5-6 months of the year because they’re waiting to get their deductible met. If they had a melanoma and put off a biopsy until June, it’s had 6 months of growth time. The doubling time can be very rapid."
Although the MelaFind-computed scores are subjective and do not state whether a biopsy is warranted, the data would be useful if sent along with a specimen to aid dermatopathologists in rendering a more definitive diagnosis, he said. Optimally, the device is "handcrafted" for practices and clinics that follow patients with hundreds of dysplastic nevi where repeated biopsies are not practical, an avenue Dr. Cockerell said he hopes to pursue in the future.
Mela Sciences sponsored the study. Dr. Cockerell and his coauthors disclosed no conflicting financial interests.
AT THE AAD SUMMER ACADEMY 2014
Key clinical point: The MelaFind device may be helpful to dermatologists when deciding which clinically ambiguous lesions may be early melanoma.
Major finding: The biopsy rate for melanoma or severely dysplastic nevi was 22:1 among clinicians in the review and 7.6:1 with the MelaFind in a prior prospective study.
Data source: A retrospective analysis of 1,100 biopsied skin lesions from patients in 500 U.S. practices.
Disclosures: Mela Sciences sponsored the study. Dr. Cockerell and his coauthors disclosed no conflicting financial interests.
AUDIO: Unusual approaches to unusual tumors
CHICAGO – Doctors who don’t abide by accepted norms can sometimes achieve better outcomes when treating patients who have unusual and tenacious tumors. This was the theme of a session covering cases of confounding tumors at the American Academy of Dermatology summer meeting.

In an interview after the session, panel moderator and case presenter Dr. John A. Carucci, chief of Mohs and dermatologic surgery at New York (N.Y.) University, shared his thoughts on when certain kinds of imaging are more appropriate than others, even if it’s not the "usual way."
Dr. Carucci also addressed the use of postsurgical negative pressure wound therapy, radiation therapy in conjunction with Mohs surgery, and potentially controversial topics such as the use of a protein kinase inhibitor as a neoadjuvant therapy.
And what new information on staging squamous cell carcinomas has Dr. Carucci and his colleagues excited? Listen and find out. Dr. Carucci said that he had no financial conflicts to disclose.
On Twitter @whitneymcknight
CHICAGO – Doctors who don’t abide by accepted norms can sometimes achieve better outcomes when treating patients who have unusual and tenacious tumors. This was the theme of a session covering cases of confounding tumors at the American Academy of Dermatology summer meeting.
In an interview after the session, panel moderator and case presenter Dr. John A. Carucci, chief of Mohs and dermatologic surgery at New York (N.Y.) University, shared his thoughts on when certain kinds of imaging are more appropriate than others, even if it’s not the "usual way."
Dr. Carucci also addressed the use of postsurgical negative pressure wound therapy, radiation therapy in conjunction with Mohs surgery, and potentially controversial topics such as the use of a protein kinase inhibitor as a neoadjuvant therapy.
And what new information on staging squamous cell carcinomas has Dr. Carucci and his colleagues excited? Listen and find out. Dr. Carucci said that he had no financial conflicts to disclose.
On Twitter @whitneymcknight
CHICAGO – Doctors who don’t abide by accepted norms can sometimes achieve better outcomes when treating patients who have unusual and tenacious tumors. This was the theme of a session covering cases of confounding tumors at the American Academy of Dermatology summer meeting.
In an interview after the session, panel moderator and case presenter Dr. John A. Carucci, chief of Mohs and dermatologic surgery at New York (N.Y.) University, shared his thoughts on when certain kinds of imaging are more appropriate than others, even if it’s not the "usual way."
Dr. Carucci also addressed the use of postsurgical negative pressure wound therapy, radiation therapy in conjunction with Mohs surgery, and potentially controversial topics such as the use of a protein kinase inhibitor as a neoadjuvant therapy.
And what new information on staging squamous cell carcinomas has Dr. Carucci and his colleagues excited? Listen and find out. Dr. Carucci said that he had no financial conflicts to disclose.
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM THE AAD SUMMER ACADEMY 2014

Current Options and Future Directions in the Systemic Treatment of Metastatic Melanoma
This article has been adapted from an article originally published in The Journal of Supportive and Community Oncology (jcso-online.com). Schindler K, Postow M. Current options and future directions in the systemic treatment of metastatic melanoma. J Community Support Oncol. 2014;12(1):20-26.
The incidence of melanoma, a highly aggressive tumor arising from melanocytes, continues to rise by about 3% a year in the U.S. with about 76,000 patients being diagnosed every year and 9,000 patients dying of the disease.1 Complete surgical resection is the standard for localized melanoma, with surgical excision margins depending on tumor thickness. For patients with involved sentinel lymph nodes, complete lymphadenectomy is typically recommended, although the benefits of completion lymphadenectomy are being evaluated in an ongoing randomized trial.2,3
For patients with surgically resected, high-risk melanoma, the only approved adjuvant therapy is interferon-a (IFN-a).4 Use of IFN-a, however, remains controversial because of the associated adverse effects (AEs) and controversial effects on overall survival (OS).5,6 Unfortunately, many patients with localized disease will ultimately experience a recurrence, and the prognosis of patients with metastatic disease is poor with a historical 5-year survival rate of 10%.7
Chemotherapy and interleukin 2
For more than 3 decades, conventional cytotoxic chemotherapy was used to treat metastatic melanoma. Typical agents included alkylating agents (dacarbazine, temozolomide, nitrosoureas), platinum analogs (cisplatin and carboplatin), and microtubular toxins (vinblastine and paclitaxel). Despite the clinical use and investigation of a number of these chemotherapies for patients with metastatic melanoma, the only treatment approved by the FDA is dacarbazine, which is administered intravenously every 3 to 4 weeks at a dose of 800 to 1,000 mg/m2.
Monotherapy with dacarbazine is generally well tolerated with only mild AEs such as nausea, myelosuppression, and fatigue. In a pooled analysis, the overall response rate (RR) for dacarbazine was approximately 9%.8 Temozolomide, the oral analog of dacarbazine, penetrates into the central nervous system and has been compared with dacarbazine in randomized trials. These agents are believed to have similar efficacy, but temozolomide has been associated with a higher rate of lymphopenia.9,10
Investigation of chemotherapy combinations such as cisplatin, vinblastine, and dacarbazine or carboplatin and paclitaxel have shown promising RRs but unfortunately no prolongation of OS compared with single-agent dacarbazine.11-13 Despite its modest efficacy, chemotherapy still has a place in the palliative treatment for some patients.
In addition to dacarbazine, the immunotherapeutic strategy, high-dose recombinant interleukin-2 (IL-2), had also been a mainstay treatment for advanced melanoma for many years. IL-2 is administered as an IV infusion every 8 hours at a dose of 600,000 to 720,000 IU/kg on days 1 to 5 and days 15 to 19, with a maximum of 14 such biphasic cycles. Because of the significant acute toxicity profile, including capillary leak syndrome, cardiovascular complications, and seizures, IL-2 treatment requires hospitalization and is generally only performed at specialized centers for patients with good performance status. Though the overall RR in pooled analysis was low at 16%, the durability of responses in some responders that appeared to last many years led to the FDA approval of IL-2 in 1998.14,15
IL-2 continues to be investigated. In a randomized trial, an improved RR and progression-free survival (PFS) were seen when IL-2 was combined with the glycoprotein 100 (gp100) peptide vaccine compared with IL-2 alone.16 Other approaches have sought to improve the safety of IL-2 by selectively delivering it to tumor sites. The fusion protein L19-IL2 couples IL-2 with the recombinant human vascular targeting antibody L19 and has preliminarily been shown to be safe in phase 1 evaluation and in combination with dacarbazine.17,18
Antibodies that block immunologic checkpoints
Melanoma has long been recognized as an immunogenic malignancy but the efficacy of immunotherapeutic strategies has generally been modest. The precise etiology of why immunotherapy historically was not more successful is not completely understood, but it is possible that patients with advanced malignancy have predominant immune inhibitory circuits that prevent otherwise effective antitumor immune responses.
In recent years, research has illuminated some of these immunologic inhibitory elements, termed “immunologic checkpoints,” which include cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death-1 (PD-1). Antibodies that target these checkpoints have resulted in durable responses in some patients and a unique pattern of immune-mediated AEs. Though an ongoing area of research, no pre- or on-treatment biomarkers have been sufficiently validated to enable specific patient selection for these therapies.
Antibodies Blocking CTLA-4
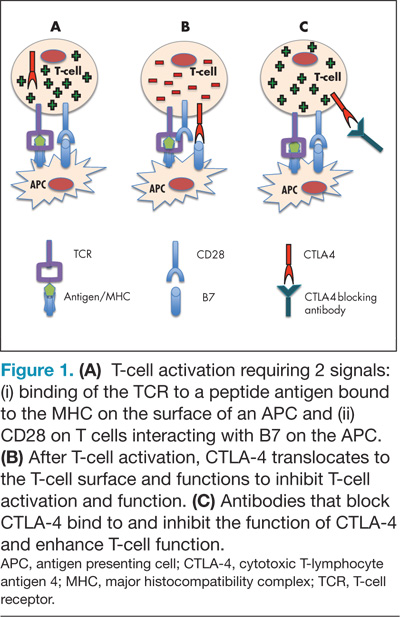
CTLA-4 is expressed on activated T cells and typically functions as a negative regulator of T-cell activity preserving normal immunologic homeostasis. Blocking CTLA-4 with therapeutic antibodies such as ipilimumab and tremelimumab prevents normal CTLA-4–mediated T-cell downregulation and thereby enhances the ability of T cells to exert their full antitumor immune effects (Figure 1). Ipilimumab was the first drug in the management of metastatic melanoma to show an improvement in OS in phase 3 studies, and although a phase 3 study of tremelimumab did not demonstrate an improvement in OS, durable responses were similarly seen.19-21
The first phase 3 trial investigating ipilimumab randomized previously pretreated patients with advanced melanoma to ipilimumab at a dose of 3 mg/kg with or without the gp100 peptide vaccine. The median OS was 10.0 months among patients receiving ipilimumab plus gp100, compared with 6.4 months among patients receiving gp100 alone. There was no difference in OS between the ipilimumab groups.19 The outcome of this study has led to the approval of ipilimumab at a dose of 3 mg/kg in patients with advanced melanoma by regulatory agencies in the U.S., European Union, and Australia.
For treatment-naive patients, a second phase 3 trial investigating dacarbazine in combination with ipilimumab compared with dacarbazine in combination with placebo also demonstrated improvement of OS in patients treated with dacarbazine in combination with ipilimumab.20 The estimated 1-year, 2-year, and 3-year survival rates were 47.3%, 28.5%, and 20.8%, respectively, in the dacarbazine plus ipilimumab group, compared with 36.3%, 17.9%, and 12.2% in the dacarbazine alone group. This second trial used a higher dose of ipilimumab (10 mg/kg) and though it confirmed ipilimumab’s beneficial effects on OS, ipilimumab is not approved at 10 mg/kg and is not routinely recommended to be used in combination with dacarbazine given hepatic toxicity concerns.
Though the median OS was improved in these phase 3 trials, perhaps the greatest activity of ipilimumab lies in the increased number of patients who can achieve long-term OS. In a recently published updated survival analysis, the 4-year survival rates for previously treated patients who received ipilimumab at 3 or 10 mg/kg were 18.2% and 19.7% to 28.4%. For treatment-naive patients receiving ipilimumab at 10 mg/kg, 4-year survival rates were between 37.7% and 49.5%.22 These values appear superior to historical data from prior chemotherapy trials.
An important consideration in the clinical use of CTLA-4 blocking antibodies is the possible occurrence of toxicities that differ from those associated with traditional chemotherapy. These AEs are termed immune-related AEs (irAEs), and they most commonly manifest as diarrhea, dermatitis, hepatitis, and endocrinopathies but less commonly can involve other organs, resulting in uveitis, nephritis, myopathy, and neuropathy.
In general, the onset of irAEs follows a certain pattern with cutaneous manifestations often presenting early in treatment, followed by gastrointestinal and hepatic events occurring about 2 months into therapy and endocrinopathies appearing even later.23 In rare cases, severe AEs (eg, perforating colitis, toxic epidermal necrolysis) can occur and may require hospitalization.24
Clinicians must be attentive to early signs of these AEs and promptly initiate immunosuppression with steroids or other immunosuppressive medications, which do not appear to diminish the antitumor immune effects.25 Established management algorithms exist to guide clinicians. Given the occasional need for immunosuppression in this patient population, awareness of the possibility of opportunistic or rare infections is also important.
In phase 3 evaluation, the number of patients who had long-term survival exceeded the number of patients who had a classically defined disease response to treatment. Durable stable disease and late responses have been observed clinically and may be responsible for some of the beneficial outcomes.26 If patients are asymptomatic and have minimal radiographic progression, it is reasonable to repeat imaging 1 to 2 months later to confirm progression before considering additional lines of therapy.
Antibodies Blocking the Programmed Death-1 Axis
Programmed death-1 (PD-1) is a receptor on the surface of T cells that is upregulated at later stages of T-cell activation as opposed to the early upregulation of CTLA-4. Normally, engagement of PD-1 attenuates T-cell activity at several phases of an immune response. Tumors are believed to escape immune attack by similarly inhibiting T-cell activity by upregulating one of the ligands of PD-1, PD-L1.27,28 Several antibodies that inhibit PD-1 activity, either by blocking the PD-1 molecule itself or PD-L1, are demonstrating significant promise in ongoing clinical trials.
Nivolumab (previously, BMS-936558) is a fully human monoclonal antibody targeting PD-1. In a large phase 1 study in patients with a variety of malignancies, nivolumab demonstrated a 31% RR in patients with advanced melanoma.29 Subsequent follow-up data indicates these responses are generally durable with a median duration of response of 24 months and a 3-year OS rate of 40%.30 Adverse effects of nivolumab appear less frequently than with CTLA-4 blockade but have included vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. Unique to PD-1 blockade appears to be the AE of an inflammatory pneumonitis, which can present with a dry cough, dyspnea, and ground-glass opacities and can be potentially lethal.29
On the basis of complementary regulatory roles of CTLA-4 and PD-1 checkpoint inhibition, a trial investigating combined nivolumab and ipilimumab was completed. In the small group of patients treated, a high RR was seen with a generally acceptable safety profile.31 Ongoing phase 2 and 3 trials are assessing nivolumab alone and in combination with other agents for the treatment of advanced melanoma and other malignancies (Table 1).
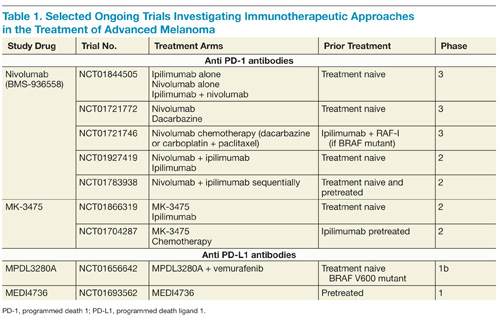
Another PD-1 blocking antibody, MK-3475, has been evaluated in patients with advanced melanoma, and promising RRs have been described.32 In a small group of patients, the confirmed RR at a dose of 10 mg/kg every 2 weeks was 52% and appeared similar in patients who had and who had not been previously treated with ipilimumab. The AEs of MK-3475 seem to resemble nivolumab. MK-3475 is similarly being evaluated in large phase 2 and 3 trials for both patients with melanoma and additional malignancies.
In addition to antibodies targeting PD-1, clinical activity has also been observed with several different antibodies (BMS-936559, MPDL3280A, and MEDI4736) that target PD-L1. Though some data have been published for this therapeutic strategy,33 ongoing trials will continue to clarify the role of targeting PD-L1 in patients with advanced melanoma.
Targeted Therapies That Block Oncogenic Signaling Pathways
The mitogen-activated protein kinase (MAPK) pathway responds to extracellular growth signals and regulates cell proliferation and survival. In many patients with melanoma, the MAPK pathway is constitutively activated as a result of molecular alterations in genes encoding key regulators or components of the pathway such as BRAF, NRAS, and KIT.34,35 The most common mutation arising in melanoma is the BRAF mutation, occurring in nearly half of melanomas, and typically involves a missense mutation in which glutamic acid is substituted for valine at codon 600 (BRAF V600E mutation).36 Less frequent BRAF mutations include V600K, V600R, and K601E.37 Strategies that directly inhibit oncogenic BRAF or disable downstream elements such as MEK have recently shown dramatic results in patients with melanoma (Figure 2).
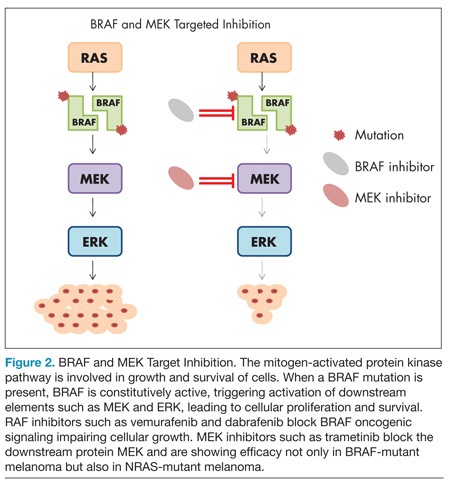
BRAF inhibitors
Vemurafenib is a potent inhibitor of mutated BRAF with marked antitumor effects against melanoma cell lines with the BRAF V600E mutation.38 The first striking results of tumor regression with this strategy in patients were seen in a phase 1 study in patients with melanoma characterized by a BRAF V600E mutation but not in patients whose melanomas did not have a BRAF mutation.39
Subsequent phase 3 trials confirmed the high RRs of this agent in patients with BRAF-mutant melanoma and demonstrated superiority in OS compared with dacarbazine chemotherapy.40 The results of this phase 3 trial led to the approval of vemurafenib by the FDA in August 2011 with treatment exclusively limited to patients with BRAF mutant melanoma. Updated OS data from this phase 3 study revealed a median OS of 13.2 months for vemurafenib, compared with 9.6 months for dacarbazine, with an overall RR in patients treated with vemurafenib of 57% and a median PFS of 6.9 months.41 General AEs with vemurafenib include arthralgia, fatigue, aminotransferase elevations, nausea and vomiting, and decreased kidney function. In general, toxicities are manageable with dose reduction or temporary drug cessation.
One characteristic of vemurafenib and other BRAF-targeted agents is the frequent development of hyperproliferative skin AEs. Skin lesions, including follicular and palmo-plantar hyperkeratosis, papillomas, and also cutaneous squamous-cell carcinomas and keratoacanthomas, have commonly been observed under treatment with vemurafenib, and close evaluation by a dermatologist is important.42 The mechanism of this phenomenon is believed to be a paradoxical activation of the MAPK pathway in nonmelanoma BRAF wild-type cells when systemic treatment with a BRAF inhibitor is administered.43
The phenomenon of hyperproliferation of non–BRAF-mutant tissues with ongoing BRAF-inhibitor therapy has also been seen in patients with lymphoproliferative disorders and may be a mechanism involved in the discovery that patients have a high rate of new primary melanomas while on therapy.44,45 These findings warrant special attention, particularly as BRAF inhibitors are undergoing evaluation as adjuvant therapy.
Another active BRAF kinase inhibitor with a similar efficacy profile as vemurafenib is dabrafenib, which was approved in May 2013 based on the demonstration of improved PFS in a phase 3 trial comparing dabrafenib 150 mg orally twice daily and dacarbazine 1,000 mg/m2 intravenously once every 3 weeks in previously untreated patients with BRAF V600E mutant melanoma. The median PFS times were 5.1 and 2.7 months in the dabrafenib and dacarbazine arms, respectively, with an objective RR of 52% in patients treated with dabrafenib.46 Follow-up time was too short to make a determination of the impact of dabrafenib on OS. In a separate study, dabrafenib was also shown to be effective for patients with brain metastases and remains an excellent therapeutic choice for this particular patient population.47
Generally, dabrafenib is believed to have similar efficacy to vemurafenib. Nevertheless, EAs with of dabrafenib differ somewhat from those observed with vemurafenib: The rate of proliferative skin lesions, including squamous cell carcinomas and keratoacanthomas appears to be lower for dabrafenib than vemurafenib. However, AEs particular to dabrafenib have been seen such as pyrexia, which were recorded in about 11% of patients.46
MEK inhibitors
Though targeting oncogenic BRAF directly has been incredibly successful for patients with BRAF-mutant metastatic melanoma, additional success has been observed by blocking the MAPK pathway at a downstream component, MEK. Trametinib is an MEK inhibitor that was approved by the FDA in June 2013 as a single agent for patients with BRAF V600E or V600K mutant melanoma. Trametinib is administered at a dose of 2 mg once daily and was shown to improve PFS and OS compared with dacarbazine and paclitaxel chemotherapies.47 Despite the improvement in PFS and OS compared with chemotherapy, the objective RR for trametinib was somewhat lower (22%) than that seen with BRAF inhibitors.
Trametinib also is associated with a different AE profile from BRAF inhibitors and includes diarrhea, peripheral edema, hypertension, and fatigue, typical of other MEK inhibitors as well.48 Asymptomatic and reversible reduction of the cardiac ejection fraction and ocular toxic effects also occur infrequently. Unlike with BRAF-inhibitor treatment, the development of cutaneous squamous-cell carcinomas or other hyperproliferative skin lesions was not noted.49
Despite the significant benefits of targeted therapy disrupting overly active MAPK signaling in patients with BRAF-mutant metastatic melanoma, almost all patients treated with these targeted inhibitors who achieve an initial response will ultimately progress. Several mechanisms of resistance have been proposed, and most relate to reactivation of the MAPK pathway.50,51 As a result, efforts to maintain suppression of the MAPK pathway have been pursued to delay the onset of resistance. In a phase 2 trial that combined dabrafenib with trametinib, there was a longer PFS than there was with dabrafenib monotherapy.52
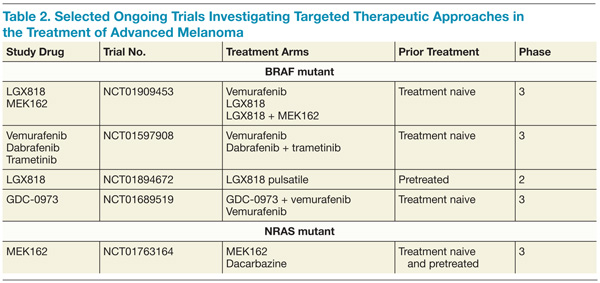
Furthermore, the addition of trametinib to dabrafenib reduced the incidence of squamous-cell carcinoma, providing further evidence that reactivation of the MAPK pathway is involved in these hyperproliferative skin lesions arising under BRAF-directed therapy. A higher rate of febrile episodes was seen, however. An ongoing phase 3 study is looking at whether or not combining BRAF and MEK inhibitors results in improved OS compared with single-agent BRAF. It is premature at this juncture to recommend combining dabrafenib and trametinib until the results of the ongoing phase 3 studies more thoroughly describe the risks and benefits of this approach (Table 2).
KIT inhibitors
In a subset of melanomas, particularly those that arise from mucosal, acral, or chronically sun-damaged skin, mutations are found in the receptor-tyrosine kinase KIT.35 A number of agents directed against KIT, such as imatinib, have been tested in clinical trials. Initial phase 2 studies revealed poor RRs with KIT inhibition in molecularly unselected patients.53-55 Subsequent studies selected patients with KIT genetic aberrations, including mutations and amplifications, and some responses were seen.56-58
Importantly, not all KIT genetic aberrations are believed to be considered equal. Preliminarily, it appears that mutations in exon 11 (L576P) and exon 13 (K642E) appear to be most closely associated with response and may be true driver mutations. Other KIT mutations may have less functional significance but additional research is needed. Imatinib is a reasonable therapeutic choice in patients with a KIT mutation, particularly when an L576P or K642E mutation is present.
conclusions
Since 2011, 4 new drugs—ipilimumab, vemurafenib, dabrafenib, and trametinib—have been approved for the treatment of metastatic melanoma. Exciting early data from PD-1 clinical trials suggest that agents that disrupt PD-1 may also become important therapeutic modalities. Future studies will continue to evaluate combinations of these therapeutic modalities, but caution should be exercised in combining these drugs prior to data from ongoing clinical trials revealing the true benefits and risks of combination therapy. Excessive toxicity was seen in an early phase trial when vemurafenib was combined with ipilimumab.59
Additional research will also explore biomarkers that may help clinicians apply immunotherapy to the most appropriate patients and better understand mechanisms of resistance to targeted therapies. Clinical trials of novel agents or combinations should be considered at every treatment juncture to continue the rapid pace of developing the most innovative and tailored treatment approaches.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10-29.
2. Garbe C, Peris K, Hauschild A, et al; European Dermatology Forum, European Association of Dermato-Oncology, European Organization of Research and Treatment of Cancer. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2012. Eur J Cancer. 2012;48(15):2375-2390.
3. Coit DG, Andtbacka R, Bichakjian CK, et al; NCCN Melanoma Panel. Melanoma. J Natl Compr Canc Netw. 2009;7(3):250-275.
4. Molife R, Hancock BW. Adjuvant therapy of malignant melanoma. Crit Rev Oncol Hematol. 2002;44(1):81-102.
5. Wheatley K, Ives N, Hancock B, Gore M, Eggermont A, Suciu S. Does adjuvant interferon-alpha for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat Rev. 2003;29(4):241-252.
6. Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: A systematic review and meta-analysis. J Natl Cancer Inst. 2010;102(7):493-501.
7. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199-6206.
8. Luke JJ, Rubinstein LV, Smith GL, Ivy SP, Harris PJ. Similar efficacy for phase I trials in comparison with DTIC for advanced malignant melanoma: an analysis of melanoma outcomes in CTEP-sponsored phase I trials 1995-2011. Melanoma Res. 2013;23(2):152-158.
9. Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18(1):158-166.
10. Teimouri F, Nikfar S, Abdollahi M. Efficacy and side effects of dacarbazine in comparison with temozolomide in the treatment of malignant melanoma: A meta-analysis consisting of 1314 patients. Melanoma Res. 2013;23(5):381-389.
11. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745-2751.
12. Legha SS, Ring S, Papadopoulos N, Plager C, Chawla S, Benjamin R. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer. 1989;64(10):2024-2029.
13. Rao RD, Holtan SG, Ingle JN, et al. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. Cancer. 2006;106(2):375-382.
14. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J Sci Am. 2000;6(suppl 1):S11-S14.
15. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105-2116.
16. Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119-2127.
17. Johannsen M, Spitaleri G, Curigliano G, et al. The tumour-targeting human L19-IL2 immunocytokine: Preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer. 2010;46(16):2926-2935.
18. Eigentler TK, Weide B, de Braud F, et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin Cancer Res. 2011;17(24):7732-7742.
19. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
20. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517-2526.
21. Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616-622.
22. Wolchok JD, Weber JS, Maio M, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24(8):2174-2180.
23. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
24. Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. 2010;37(5):499-507.
25. Harmankaya K, Erasim C, Koelblinger C, et al. Continuous systemic corticosteroids do not affect the ongoing regression of metastatic melanoma for more than two years following ipilimumab therapy. Med Oncol. 2011;28(4):1140-1144.
26. Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15(23):7412-7420.
27. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467-477.
28. Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883-895.
29. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
30. Sznol M, Kluger HM, Hodi FS, et al. Survival and long-term follow-up of safety and response in patients (pts) with advanced melanoma (MEL) in a phase I trial of nivolumab (anti-PD-1; BMS-936558; ONO-4538) [ASCO abstract CRA9006]. ASCO Meet Abstr. 2013;31(18_suppl):CRA9006. http://meetinglibrary.asco.org/content/80822. Accessed July 23, 2014.
31. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122-133.
32. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134-144.
33. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
34. McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance.
Biochim Biophys Acta. 2007;1773(8):1263-1284.
35. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340-4346.
36. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949-954.
37. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246.
38. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596-599.
39. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809-819.
40. Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
41. Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAF(V600E)-mutated melanoma [ASCO abstract 8502]. ASCO Meet Abstr. 2012;30(15_suppl):8502. http://meetinglibrary.asco.org/content/70533?media=vm. Accessed July 23, 2014.
42. Lacouture ME, O’Reilly K, Rosen N, Solit DB. Induction of cutaneous squamous cell carcinomas by RAF inhibitors: Cause for concern? J Clin Oncol. 2012;30(3):329-330.
43. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. New Engl J Med. 2012;366(3):207-215.
44. Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. New Engl J Med. 2012;367(24):2316-2321.
45. Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30(19):2375-2383.
46. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label phase 3 randomised clinical trial. Lancet 2012;380(9839):358-365.
47. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
48. Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555-567.
49. Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. New Engl J Med. 2012;367(2):107-114.
50. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387-390.
51. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973-977.
52. Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New Engl J Med. 2012;367(18):1694-1703.
53. Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;9(8):1398-1405.
54. Wyman K, Atkins MB, Prieto V, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy. Cancer. 2006;106(9):2005-2011.
55. Kim KB, Eton O, Davis DW, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008;99(5):734-740.
56. Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327-2334.
57. Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182-3190.
58. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904-2909.
59. Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365-1366.
This article has been adapted from an article originally published in The Journal of Supportive and Community Oncology (jcso-online.com). Schindler K, Postow M. Current options and future directions in the systemic treatment of metastatic melanoma. J Community Support Oncol. 2014;12(1):20-26.
The incidence of melanoma, a highly aggressive tumor arising from melanocytes, continues to rise by about 3% a year in the U.S. with about 76,000 patients being diagnosed every year and 9,000 patients dying of the disease.1 Complete surgical resection is the standard for localized melanoma, with surgical excision margins depending on tumor thickness. For patients with involved sentinel lymph nodes, complete lymphadenectomy is typically recommended, although the benefits of completion lymphadenectomy are being evaluated in an ongoing randomized trial.2,3
For patients with surgically resected, high-risk melanoma, the only approved adjuvant therapy is interferon-a (IFN-a).4 Use of IFN-a, however, remains controversial because of the associated adverse effects (AEs) and controversial effects on overall survival (OS).5,6 Unfortunately, many patients with localized disease will ultimately experience a recurrence, and the prognosis of patients with metastatic disease is poor with a historical 5-year survival rate of 10%.7
Chemotherapy and interleukin 2
For more than 3 decades, conventional cytotoxic chemotherapy was used to treat metastatic melanoma. Typical agents included alkylating agents (dacarbazine, temozolomide, nitrosoureas), platinum analogs (cisplatin and carboplatin), and microtubular toxins (vinblastine and paclitaxel). Despite the clinical use and investigation of a number of these chemotherapies for patients with metastatic melanoma, the only treatment approved by the FDA is dacarbazine, which is administered intravenously every 3 to 4 weeks at a dose of 800 to 1,000 mg/m2.
Monotherapy with dacarbazine is generally well tolerated with only mild AEs such as nausea, myelosuppression, and fatigue. In a pooled analysis, the overall response rate (RR) for dacarbazine was approximately 9%.8 Temozolomide, the oral analog of dacarbazine, penetrates into the central nervous system and has been compared with dacarbazine in randomized trials. These agents are believed to have similar efficacy, but temozolomide has been associated with a higher rate of lymphopenia.9,10
Investigation of chemotherapy combinations such as cisplatin, vinblastine, and dacarbazine or carboplatin and paclitaxel have shown promising RRs but unfortunately no prolongation of OS compared with single-agent dacarbazine.11-13 Despite its modest efficacy, chemotherapy still has a place in the palliative treatment for some patients.
In addition to dacarbazine, the immunotherapeutic strategy, high-dose recombinant interleukin-2 (IL-2), had also been a mainstay treatment for advanced melanoma for many years. IL-2 is administered as an IV infusion every 8 hours at a dose of 600,000 to 720,000 IU/kg on days 1 to 5 and days 15 to 19, with a maximum of 14 such biphasic cycles. Because of the significant acute toxicity profile, including capillary leak syndrome, cardiovascular complications, and seizures, IL-2 treatment requires hospitalization and is generally only performed at specialized centers for patients with good performance status. Though the overall RR in pooled analysis was low at 16%, the durability of responses in some responders that appeared to last many years led to the FDA approval of IL-2 in 1998.14,15
IL-2 continues to be investigated. In a randomized trial, an improved RR and progression-free survival (PFS) were seen when IL-2 was combined with the glycoprotein 100 (gp100) peptide vaccine compared with IL-2 alone.16 Other approaches have sought to improve the safety of IL-2 by selectively delivering it to tumor sites. The fusion protein L19-IL2 couples IL-2 with the recombinant human vascular targeting antibody L19 and has preliminarily been shown to be safe in phase 1 evaluation and in combination with dacarbazine.17,18
Antibodies that block immunologic checkpoints
Melanoma has long been recognized as an immunogenic malignancy but the efficacy of immunotherapeutic strategies has generally been modest. The precise etiology of why immunotherapy historically was not more successful is not completely understood, but it is possible that patients with advanced malignancy have predominant immune inhibitory circuits that prevent otherwise effective antitumor immune responses.
In recent years, research has illuminated some of these immunologic inhibitory elements, termed “immunologic checkpoints,” which include cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death-1 (PD-1). Antibodies that target these checkpoints have resulted in durable responses in some patients and a unique pattern of immune-mediated AEs. Though an ongoing area of research, no pre- or on-treatment biomarkers have been sufficiently validated to enable specific patient selection for these therapies.
Antibodies Blocking CTLA-4
CTLA-4 is expressed on activated T cells and typically functions as a negative regulator of T-cell activity preserving normal immunologic homeostasis. Blocking CTLA-4 with therapeutic antibodies such as ipilimumab and tremelimumab prevents normal CTLA-4–mediated T-cell downregulation and thereby enhances the ability of T cells to exert their full antitumor immune effects (Figure 1). Ipilimumab was the first drug in the management of metastatic melanoma to show an improvement in OS in phase 3 studies, and although a phase 3 study of tremelimumab did not demonstrate an improvement in OS, durable responses were similarly seen.19-21
The first phase 3 trial investigating ipilimumab randomized previously pretreated patients with advanced melanoma to ipilimumab at a dose of 3 mg/kg with or without the gp100 peptide vaccine. The median OS was 10.0 months among patients receiving ipilimumab plus gp100, compared with 6.4 months among patients receiving gp100 alone. There was no difference in OS between the ipilimumab groups.19 The outcome of this study has led to the approval of ipilimumab at a dose of 3 mg/kg in patients with advanced melanoma by regulatory agencies in the U.S., European Union, and Australia.
For treatment-naive patients, a second phase 3 trial investigating dacarbazine in combination with ipilimumab compared with dacarbazine in combination with placebo also demonstrated improvement of OS in patients treated with dacarbazine in combination with ipilimumab.20 The estimated 1-year, 2-year, and 3-year survival rates were 47.3%, 28.5%, and 20.8%, respectively, in the dacarbazine plus ipilimumab group, compared with 36.3%, 17.9%, and 12.2% in the dacarbazine alone group. This second trial used a higher dose of ipilimumab (10 mg/kg) and though it confirmed ipilimumab’s beneficial effects on OS, ipilimumab is not approved at 10 mg/kg and is not routinely recommended to be used in combination with dacarbazine given hepatic toxicity concerns.
Though the median OS was improved in these phase 3 trials, perhaps the greatest activity of ipilimumab lies in the increased number of patients who can achieve long-term OS. In a recently published updated survival analysis, the 4-year survival rates for previously treated patients who received ipilimumab at 3 or 10 mg/kg were 18.2% and 19.7% to 28.4%. For treatment-naive patients receiving ipilimumab at 10 mg/kg, 4-year survival rates were between 37.7% and 49.5%.22 These values appear superior to historical data from prior chemotherapy trials.
An important consideration in the clinical use of CTLA-4 blocking antibodies is the possible occurrence of toxicities that differ from those associated with traditional chemotherapy. These AEs are termed immune-related AEs (irAEs), and they most commonly manifest as diarrhea, dermatitis, hepatitis, and endocrinopathies but less commonly can involve other organs, resulting in uveitis, nephritis, myopathy, and neuropathy.
In general, the onset of irAEs follows a certain pattern with cutaneous manifestations often presenting early in treatment, followed by gastrointestinal and hepatic events occurring about 2 months into therapy and endocrinopathies appearing even later.23 In rare cases, severe AEs (eg, perforating colitis, toxic epidermal necrolysis) can occur and may require hospitalization.24
Clinicians must be attentive to early signs of these AEs and promptly initiate immunosuppression with steroids or other immunosuppressive medications, which do not appear to diminish the antitumor immune effects.25 Established management algorithms exist to guide clinicians. Given the occasional need for immunosuppression in this patient population, awareness of the possibility of opportunistic or rare infections is also important.
In phase 3 evaluation, the number of patients who had long-term survival exceeded the number of patients who had a classically defined disease response to treatment. Durable stable disease and late responses have been observed clinically and may be responsible for some of the beneficial outcomes.26 If patients are asymptomatic and have minimal radiographic progression, it is reasonable to repeat imaging 1 to 2 months later to confirm progression before considering additional lines of therapy.
Antibodies Blocking the Programmed Death-1 Axis
Programmed death-1 (PD-1) is a receptor on the surface of T cells that is upregulated at later stages of T-cell activation as opposed to the early upregulation of CTLA-4. Normally, engagement of PD-1 attenuates T-cell activity at several phases of an immune response. Tumors are believed to escape immune attack by similarly inhibiting T-cell activity by upregulating one of the ligands of PD-1, PD-L1.27,28 Several antibodies that inhibit PD-1 activity, either by blocking the PD-1 molecule itself or PD-L1, are demonstrating significant promise in ongoing clinical trials.
Nivolumab (previously, BMS-936558) is a fully human monoclonal antibody targeting PD-1. In a large phase 1 study in patients with a variety of malignancies, nivolumab demonstrated a 31% RR in patients with advanced melanoma.29 Subsequent follow-up data indicates these responses are generally durable with a median duration of response of 24 months and a 3-year OS rate of 40%.30 Adverse effects of nivolumab appear less frequently than with CTLA-4 blockade but have included vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. Unique to PD-1 blockade appears to be the AE of an inflammatory pneumonitis, which can present with a dry cough, dyspnea, and ground-glass opacities and can be potentially lethal.29
On the basis of complementary regulatory roles of CTLA-4 and PD-1 checkpoint inhibition, a trial investigating combined nivolumab and ipilimumab was completed. In the small group of patients treated, a high RR was seen with a generally acceptable safety profile.31 Ongoing phase 2 and 3 trials are assessing nivolumab alone and in combination with other agents for the treatment of advanced melanoma and other malignancies (Table 1).
Another PD-1 blocking antibody, MK-3475, has been evaluated in patients with advanced melanoma, and promising RRs have been described.32 In a small group of patients, the confirmed RR at a dose of 10 mg/kg every 2 weeks was 52% and appeared similar in patients who had and who had not been previously treated with ipilimumab. The AEs of MK-3475 seem to resemble nivolumab. MK-3475 is similarly being evaluated in large phase 2 and 3 trials for both patients with melanoma and additional malignancies.
In addition to antibodies targeting PD-1, clinical activity has also been observed with several different antibodies (BMS-936559, MPDL3280A, and MEDI4736) that target PD-L1. Though some data have been published for this therapeutic strategy,33 ongoing trials will continue to clarify the role of targeting PD-L1 in patients with advanced melanoma.
Targeted Therapies That Block Oncogenic Signaling Pathways
The mitogen-activated protein kinase (MAPK) pathway responds to extracellular growth signals and regulates cell proliferation and survival. In many patients with melanoma, the MAPK pathway is constitutively activated as a result of molecular alterations in genes encoding key regulators or components of the pathway such as BRAF, NRAS, and KIT.34,35 The most common mutation arising in melanoma is the BRAF mutation, occurring in nearly half of melanomas, and typically involves a missense mutation in which glutamic acid is substituted for valine at codon 600 (BRAF V600E mutation).36 Less frequent BRAF mutations include V600K, V600R, and K601E.37 Strategies that directly inhibit oncogenic BRAF or disable downstream elements such as MEK have recently shown dramatic results in patients with melanoma (Figure 2).
BRAF inhibitors
Vemurafenib is a potent inhibitor of mutated BRAF with marked antitumor effects against melanoma cell lines with the BRAF V600E mutation.38 The first striking results of tumor regression with this strategy in patients were seen in a phase 1 study in patients with melanoma characterized by a BRAF V600E mutation but not in patients whose melanomas did not have a BRAF mutation.39
Subsequent phase 3 trials confirmed the high RRs of this agent in patients with BRAF-mutant melanoma and demonstrated superiority in OS compared with dacarbazine chemotherapy.40 The results of this phase 3 trial led to the approval of vemurafenib by the FDA in August 2011 with treatment exclusively limited to patients with BRAF mutant melanoma. Updated OS data from this phase 3 study revealed a median OS of 13.2 months for vemurafenib, compared with 9.6 months for dacarbazine, with an overall RR in patients treated with vemurafenib of 57% and a median PFS of 6.9 months.41 General AEs with vemurafenib include arthralgia, fatigue, aminotransferase elevations, nausea and vomiting, and decreased kidney function. In general, toxicities are manageable with dose reduction or temporary drug cessation.
One characteristic of vemurafenib and other BRAF-targeted agents is the frequent development of hyperproliferative skin AEs. Skin lesions, including follicular and palmo-plantar hyperkeratosis, papillomas, and also cutaneous squamous-cell carcinomas and keratoacanthomas, have commonly been observed under treatment with vemurafenib, and close evaluation by a dermatologist is important.42 The mechanism of this phenomenon is believed to be a paradoxical activation of the MAPK pathway in nonmelanoma BRAF wild-type cells when systemic treatment with a BRAF inhibitor is administered.43
The phenomenon of hyperproliferation of non–BRAF-mutant tissues with ongoing BRAF-inhibitor therapy has also been seen in patients with lymphoproliferative disorders and may be a mechanism involved in the discovery that patients have a high rate of new primary melanomas while on therapy.44,45 These findings warrant special attention, particularly as BRAF inhibitors are undergoing evaluation as adjuvant therapy.
Another active BRAF kinase inhibitor with a similar efficacy profile as vemurafenib is dabrafenib, which was approved in May 2013 based on the demonstration of improved PFS in a phase 3 trial comparing dabrafenib 150 mg orally twice daily and dacarbazine 1,000 mg/m2 intravenously once every 3 weeks in previously untreated patients with BRAF V600E mutant melanoma. The median PFS times were 5.1 and 2.7 months in the dabrafenib and dacarbazine arms, respectively, with an objective RR of 52% in patients treated with dabrafenib.46 Follow-up time was too short to make a determination of the impact of dabrafenib on OS. In a separate study, dabrafenib was also shown to be effective for patients with brain metastases and remains an excellent therapeutic choice for this particular patient population.47
Generally, dabrafenib is believed to have similar efficacy to vemurafenib. Nevertheless, EAs with of dabrafenib differ somewhat from those observed with vemurafenib: The rate of proliferative skin lesions, including squamous cell carcinomas and keratoacanthomas appears to be lower for dabrafenib than vemurafenib. However, AEs particular to dabrafenib have been seen such as pyrexia, which were recorded in about 11% of patients.46
MEK inhibitors
Though targeting oncogenic BRAF directly has been incredibly successful for patients with BRAF-mutant metastatic melanoma, additional success has been observed by blocking the MAPK pathway at a downstream component, MEK. Trametinib is an MEK inhibitor that was approved by the FDA in June 2013 as a single agent for patients with BRAF V600E or V600K mutant melanoma. Trametinib is administered at a dose of 2 mg once daily and was shown to improve PFS and OS compared with dacarbazine and paclitaxel chemotherapies.47 Despite the improvement in PFS and OS compared with chemotherapy, the objective RR for trametinib was somewhat lower (22%) than that seen with BRAF inhibitors.
Trametinib also is associated with a different AE profile from BRAF inhibitors and includes diarrhea, peripheral edema, hypertension, and fatigue, typical of other MEK inhibitors as well.48 Asymptomatic and reversible reduction of the cardiac ejection fraction and ocular toxic effects also occur infrequently. Unlike with BRAF-inhibitor treatment, the development of cutaneous squamous-cell carcinomas or other hyperproliferative skin lesions was not noted.49
Despite the significant benefits of targeted therapy disrupting overly active MAPK signaling in patients with BRAF-mutant metastatic melanoma, almost all patients treated with these targeted inhibitors who achieve an initial response will ultimately progress. Several mechanisms of resistance have been proposed, and most relate to reactivation of the MAPK pathway.50,51 As a result, efforts to maintain suppression of the MAPK pathway have been pursued to delay the onset of resistance. In a phase 2 trial that combined dabrafenib with trametinib, there was a longer PFS than there was with dabrafenib monotherapy.52
Furthermore, the addition of trametinib to dabrafenib reduced the incidence of squamous-cell carcinoma, providing further evidence that reactivation of the MAPK pathway is involved in these hyperproliferative skin lesions arising under BRAF-directed therapy. A higher rate of febrile episodes was seen, however. An ongoing phase 3 study is looking at whether or not combining BRAF and MEK inhibitors results in improved OS compared with single-agent BRAF. It is premature at this juncture to recommend combining dabrafenib and trametinib until the results of the ongoing phase 3 studies more thoroughly describe the risks and benefits of this approach (Table 2).
KIT inhibitors
In a subset of melanomas, particularly those that arise from mucosal, acral, or chronically sun-damaged skin, mutations are found in the receptor-tyrosine kinase KIT.35 A number of agents directed against KIT, such as imatinib, have been tested in clinical trials. Initial phase 2 studies revealed poor RRs with KIT inhibition in molecularly unselected patients.53-55 Subsequent studies selected patients with KIT genetic aberrations, including mutations and amplifications, and some responses were seen.56-58
Importantly, not all KIT genetic aberrations are believed to be considered equal. Preliminarily, it appears that mutations in exon 11 (L576P) and exon 13 (K642E) appear to be most closely associated with response and may be true driver mutations. Other KIT mutations may have less functional significance but additional research is needed. Imatinib is a reasonable therapeutic choice in patients with a KIT mutation, particularly when an L576P or K642E mutation is present.
conclusions
Since 2011, 4 new drugs—ipilimumab, vemurafenib, dabrafenib, and trametinib—have been approved for the treatment of metastatic melanoma. Exciting early data from PD-1 clinical trials suggest that agents that disrupt PD-1 may also become important therapeutic modalities. Future studies will continue to evaluate combinations of these therapeutic modalities, but caution should be exercised in combining these drugs prior to data from ongoing clinical trials revealing the true benefits and risks of combination therapy. Excessive toxicity was seen in an early phase trial when vemurafenib was combined with ipilimumab.59
Additional research will also explore biomarkers that may help clinicians apply immunotherapy to the most appropriate patients and better understand mechanisms of resistance to targeted therapies. Clinical trials of novel agents or combinations should be considered at every treatment juncture to continue the rapid pace of developing the most innovative and tailored treatment approaches.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
This article has been adapted from an article originally published in The Journal of Supportive and Community Oncology (jcso-online.com). Schindler K, Postow M. Current options and future directions in the systemic treatment of metastatic melanoma. J Community Support Oncol. 2014;12(1):20-26.
The incidence of melanoma, a highly aggressive tumor arising from melanocytes, continues to rise by about 3% a year in the U.S. with about 76,000 patients being diagnosed every year and 9,000 patients dying of the disease.1 Complete surgical resection is the standard for localized melanoma, with surgical excision margins depending on tumor thickness. For patients with involved sentinel lymph nodes, complete lymphadenectomy is typically recommended, although the benefits of completion lymphadenectomy are being evaluated in an ongoing randomized trial.2,3
For patients with surgically resected, high-risk melanoma, the only approved adjuvant therapy is interferon-a (IFN-a).4 Use of IFN-a, however, remains controversial because of the associated adverse effects (AEs) and controversial effects on overall survival (OS).5,6 Unfortunately, many patients with localized disease will ultimately experience a recurrence, and the prognosis of patients with metastatic disease is poor with a historical 5-year survival rate of 10%.7
Chemotherapy and interleukin 2
For more than 3 decades, conventional cytotoxic chemotherapy was used to treat metastatic melanoma. Typical agents included alkylating agents (dacarbazine, temozolomide, nitrosoureas), platinum analogs (cisplatin and carboplatin), and microtubular toxins (vinblastine and paclitaxel). Despite the clinical use and investigation of a number of these chemotherapies for patients with metastatic melanoma, the only treatment approved by the FDA is dacarbazine, which is administered intravenously every 3 to 4 weeks at a dose of 800 to 1,000 mg/m2.
Monotherapy with dacarbazine is generally well tolerated with only mild AEs such as nausea, myelosuppression, and fatigue. In a pooled analysis, the overall response rate (RR) for dacarbazine was approximately 9%.8 Temozolomide, the oral analog of dacarbazine, penetrates into the central nervous system and has been compared with dacarbazine in randomized trials. These agents are believed to have similar efficacy, but temozolomide has been associated with a higher rate of lymphopenia.9,10
Investigation of chemotherapy combinations such as cisplatin, vinblastine, and dacarbazine or carboplatin and paclitaxel have shown promising RRs but unfortunately no prolongation of OS compared with single-agent dacarbazine.11-13 Despite its modest efficacy, chemotherapy still has a place in the palliative treatment for some patients.
In addition to dacarbazine, the immunotherapeutic strategy, high-dose recombinant interleukin-2 (IL-2), had also been a mainstay treatment for advanced melanoma for many years. IL-2 is administered as an IV infusion every 8 hours at a dose of 600,000 to 720,000 IU/kg on days 1 to 5 and days 15 to 19, with a maximum of 14 such biphasic cycles. Because of the significant acute toxicity profile, including capillary leak syndrome, cardiovascular complications, and seizures, IL-2 treatment requires hospitalization and is generally only performed at specialized centers for patients with good performance status. Though the overall RR in pooled analysis was low at 16%, the durability of responses in some responders that appeared to last many years led to the FDA approval of IL-2 in 1998.14,15
IL-2 continues to be investigated. In a randomized trial, an improved RR and progression-free survival (PFS) were seen when IL-2 was combined with the glycoprotein 100 (gp100) peptide vaccine compared with IL-2 alone.16 Other approaches have sought to improve the safety of IL-2 by selectively delivering it to tumor sites. The fusion protein L19-IL2 couples IL-2 with the recombinant human vascular targeting antibody L19 and has preliminarily been shown to be safe in phase 1 evaluation and in combination with dacarbazine.17,18
Antibodies that block immunologic checkpoints
Melanoma has long been recognized as an immunogenic malignancy but the efficacy of immunotherapeutic strategies has generally been modest. The precise etiology of why immunotherapy historically was not more successful is not completely understood, but it is possible that patients with advanced malignancy have predominant immune inhibitory circuits that prevent otherwise effective antitumor immune responses.
In recent years, research has illuminated some of these immunologic inhibitory elements, termed “immunologic checkpoints,” which include cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death-1 (PD-1). Antibodies that target these checkpoints have resulted in durable responses in some patients and a unique pattern of immune-mediated AEs. Though an ongoing area of research, no pre- or on-treatment biomarkers have been sufficiently validated to enable specific patient selection for these therapies.
Antibodies Blocking CTLA-4
CTLA-4 is expressed on activated T cells and typically functions as a negative regulator of T-cell activity preserving normal immunologic homeostasis. Blocking CTLA-4 with therapeutic antibodies such as ipilimumab and tremelimumab prevents normal CTLA-4–mediated T-cell downregulation and thereby enhances the ability of T cells to exert their full antitumor immune effects (Figure 1). Ipilimumab was the first drug in the management of metastatic melanoma to show an improvement in OS in phase 3 studies, and although a phase 3 study of tremelimumab did not demonstrate an improvement in OS, durable responses were similarly seen.19-21
The first phase 3 trial investigating ipilimumab randomized previously pretreated patients with advanced melanoma to ipilimumab at a dose of 3 mg/kg with or without the gp100 peptide vaccine. The median OS was 10.0 months among patients receiving ipilimumab plus gp100, compared with 6.4 months among patients receiving gp100 alone. There was no difference in OS between the ipilimumab groups.19 The outcome of this study has led to the approval of ipilimumab at a dose of 3 mg/kg in patients with advanced melanoma by regulatory agencies in the U.S., European Union, and Australia.
For treatment-naive patients, a second phase 3 trial investigating dacarbazine in combination with ipilimumab compared with dacarbazine in combination with placebo also demonstrated improvement of OS in patients treated with dacarbazine in combination with ipilimumab.20 The estimated 1-year, 2-year, and 3-year survival rates were 47.3%, 28.5%, and 20.8%, respectively, in the dacarbazine plus ipilimumab group, compared with 36.3%, 17.9%, and 12.2% in the dacarbazine alone group. This second trial used a higher dose of ipilimumab (10 mg/kg) and though it confirmed ipilimumab’s beneficial effects on OS, ipilimumab is not approved at 10 mg/kg and is not routinely recommended to be used in combination with dacarbazine given hepatic toxicity concerns.
Though the median OS was improved in these phase 3 trials, perhaps the greatest activity of ipilimumab lies in the increased number of patients who can achieve long-term OS. In a recently published updated survival analysis, the 4-year survival rates for previously treated patients who received ipilimumab at 3 or 10 mg/kg were 18.2% and 19.7% to 28.4%. For treatment-naive patients receiving ipilimumab at 10 mg/kg, 4-year survival rates were between 37.7% and 49.5%.22 These values appear superior to historical data from prior chemotherapy trials.
An important consideration in the clinical use of CTLA-4 blocking antibodies is the possible occurrence of toxicities that differ from those associated with traditional chemotherapy. These AEs are termed immune-related AEs (irAEs), and they most commonly manifest as diarrhea, dermatitis, hepatitis, and endocrinopathies but less commonly can involve other organs, resulting in uveitis, nephritis, myopathy, and neuropathy.
In general, the onset of irAEs follows a certain pattern with cutaneous manifestations often presenting early in treatment, followed by gastrointestinal and hepatic events occurring about 2 months into therapy and endocrinopathies appearing even later.23 In rare cases, severe AEs (eg, perforating colitis, toxic epidermal necrolysis) can occur and may require hospitalization.24
Clinicians must be attentive to early signs of these AEs and promptly initiate immunosuppression with steroids or other immunosuppressive medications, which do not appear to diminish the antitumor immune effects.25 Established management algorithms exist to guide clinicians. Given the occasional need for immunosuppression in this patient population, awareness of the possibility of opportunistic or rare infections is also important.
In phase 3 evaluation, the number of patients who had long-term survival exceeded the number of patients who had a classically defined disease response to treatment. Durable stable disease and late responses have been observed clinically and may be responsible for some of the beneficial outcomes.26 If patients are asymptomatic and have minimal radiographic progression, it is reasonable to repeat imaging 1 to 2 months later to confirm progression before considering additional lines of therapy.
Antibodies Blocking the Programmed Death-1 Axis
Programmed death-1 (PD-1) is a receptor on the surface of T cells that is upregulated at later stages of T-cell activation as opposed to the early upregulation of CTLA-4. Normally, engagement of PD-1 attenuates T-cell activity at several phases of an immune response. Tumors are believed to escape immune attack by similarly inhibiting T-cell activity by upregulating one of the ligands of PD-1, PD-L1.27,28 Several antibodies that inhibit PD-1 activity, either by blocking the PD-1 molecule itself or PD-L1, are demonstrating significant promise in ongoing clinical trials.
Nivolumab (previously, BMS-936558) is a fully human monoclonal antibody targeting PD-1. In a large phase 1 study in patients with a variety of malignancies, nivolumab demonstrated a 31% RR in patients with advanced melanoma.29 Subsequent follow-up data indicates these responses are generally durable with a median duration of response of 24 months and a 3-year OS rate of 40%.30 Adverse effects of nivolumab appear less frequently than with CTLA-4 blockade but have included vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. Unique to PD-1 blockade appears to be the AE of an inflammatory pneumonitis, which can present with a dry cough, dyspnea, and ground-glass opacities and can be potentially lethal.29
On the basis of complementary regulatory roles of CTLA-4 and PD-1 checkpoint inhibition, a trial investigating combined nivolumab and ipilimumab was completed. In the small group of patients treated, a high RR was seen with a generally acceptable safety profile.31 Ongoing phase 2 and 3 trials are assessing nivolumab alone and in combination with other agents for the treatment of advanced melanoma and other malignancies (Table 1).
Another PD-1 blocking antibody, MK-3475, has been evaluated in patients with advanced melanoma, and promising RRs have been described.32 In a small group of patients, the confirmed RR at a dose of 10 mg/kg every 2 weeks was 52% and appeared similar in patients who had and who had not been previously treated with ipilimumab. The AEs of MK-3475 seem to resemble nivolumab. MK-3475 is similarly being evaluated in large phase 2 and 3 trials for both patients with melanoma and additional malignancies.
In addition to antibodies targeting PD-1, clinical activity has also been observed with several different antibodies (BMS-936559, MPDL3280A, and MEDI4736) that target PD-L1. Though some data have been published for this therapeutic strategy,33 ongoing trials will continue to clarify the role of targeting PD-L1 in patients with advanced melanoma.
Targeted Therapies That Block Oncogenic Signaling Pathways
The mitogen-activated protein kinase (MAPK) pathway responds to extracellular growth signals and regulates cell proliferation and survival. In many patients with melanoma, the MAPK pathway is constitutively activated as a result of molecular alterations in genes encoding key regulators or components of the pathway such as BRAF, NRAS, and KIT.34,35 The most common mutation arising in melanoma is the BRAF mutation, occurring in nearly half of melanomas, and typically involves a missense mutation in which glutamic acid is substituted for valine at codon 600 (BRAF V600E mutation).36 Less frequent BRAF mutations include V600K, V600R, and K601E.37 Strategies that directly inhibit oncogenic BRAF or disable downstream elements such as MEK have recently shown dramatic results in patients with melanoma (Figure 2).
BRAF inhibitors
Vemurafenib is a potent inhibitor of mutated BRAF with marked antitumor effects against melanoma cell lines with the BRAF V600E mutation.38 The first striking results of tumor regression with this strategy in patients were seen in a phase 1 study in patients with melanoma characterized by a BRAF V600E mutation but not in patients whose melanomas did not have a BRAF mutation.39
Subsequent phase 3 trials confirmed the high RRs of this agent in patients with BRAF-mutant melanoma and demonstrated superiority in OS compared with dacarbazine chemotherapy.40 The results of this phase 3 trial led to the approval of vemurafenib by the FDA in August 2011 with treatment exclusively limited to patients with BRAF mutant melanoma. Updated OS data from this phase 3 study revealed a median OS of 13.2 months for vemurafenib, compared with 9.6 months for dacarbazine, with an overall RR in patients treated with vemurafenib of 57% and a median PFS of 6.9 months.41 General AEs with vemurafenib include arthralgia, fatigue, aminotransferase elevations, nausea and vomiting, and decreased kidney function. In general, toxicities are manageable with dose reduction or temporary drug cessation.
One characteristic of vemurafenib and other BRAF-targeted agents is the frequent development of hyperproliferative skin AEs. Skin lesions, including follicular and palmo-plantar hyperkeratosis, papillomas, and also cutaneous squamous-cell carcinomas and keratoacanthomas, have commonly been observed under treatment with vemurafenib, and close evaluation by a dermatologist is important.42 The mechanism of this phenomenon is believed to be a paradoxical activation of the MAPK pathway in nonmelanoma BRAF wild-type cells when systemic treatment with a BRAF inhibitor is administered.43
The phenomenon of hyperproliferation of non–BRAF-mutant tissues with ongoing BRAF-inhibitor therapy has also been seen in patients with lymphoproliferative disorders and may be a mechanism involved in the discovery that patients have a high rate of new primary melanomas while on therapy.44,45 These findings warrant special attention, particularly as BRAF inhibitors are undergoing evaluation as adjuvant therapy.
Another active BRAF kinase inhibitor with a similar efficacy profile as vemurafenib is dabrafenib, which was approved in May 2013 based on the demonstration of improved PFS in a phase 3 trial comparing dabrafenib 150 mg orally twice daily and dacarbazine 1,000 mg/m2 intravenously once every 3 weeks in previously untreated patients with BRAF V600E mutant melanoma. The median PFS times were 5.1 and 2.7 months in the dabrafenib and dacarbazine arms, respectively, with an objective RR of 52% in patients treated with dabrafenib.46 Follow-up time was too short to make a determination of the impact of dabrafenib on OS. In a separate study, dabrafenib was also shown to be effective for patients with brain metastases and remains an excellent therapeutic choice for this particular patient population.47
Generally, dabrafenib is believed to have similar efficacy to vemurafenib. Nevertheless, EAs with of dabrafenib differ somewhat from those observed with vemurafenib: The rate of proliferative skin lesions, including squamous cell carcinomas and keratoacanthomas appears to be lower for dabrafenib than vemurafenib. However, AEs particular to dabrafenib have been seen such as pyrexia, which were recorded in about 11% of patients.46
MEK inhibitors
Though targeting oncogenic BRAF directly has been incredibly successful for patients with BRAF-mutant metastatic melanoma, additional success has been observed by blocking the MAPK pathway at a downstream component, MEK. Trametinib is an MEK inhibitor that was approved by the FDA in June 2013 as a single agent for patients with BRAF V600E or V600K mutant melanoma. Trametinib is administered at a dose of 2 mg once daily and was shown to improve PFS and OS compared with dacarbazine and paclitaxel chemotherapies.47 Despite the improvement in PFS and OS compared with chemotherapy, the objective RR for trametinib was somewhat lower (22%) than that seen with BRAF inhibitors.
Trametinib also is associated with a different AE profile from BRAF inhibitors and includes diarrhea, peripheral edema, hypertension, and fatigue, typical of other MEK inhibitors as well.48 Asymptomatic and reversible reduction of the cardiac ejection fraction and ocular toxic effects also occur infrequently. Unlike with BRAF-inhibitor treatment, the development of cutaneous squamous-cell carcinomas or other hyperproliferative skin lesions was not noted.49
Despite the significant benefits of targeted therapy disrupting overly active MAPK signaling in patients with BRAF-mutant metastatic melanoma, almost all patients treated with these targeted inhibitors who achieve an initial response will ultimately progress. Several mechanisms of resistance have been proposed, and most relate to reactivation of the MAPK pathway.50,51 As a result, efforts to maintain suppression of the MAPK pathway have been pursued to delay the onset of resistance. In a phase 2 trial that combined dabrafenib with trametinib, there was a longer PFS than there was with dabrafenib monotherapy.52
Furthermore, the addition of trametinib to dabrafenib reduced the incidence of squamous-cell carcinoma, providing further evidence that reactivation of the MAPK pathway is involved in these hyperproliferative skin lesions arising under BRAF-directed therapy. A higher rate of febrile episodes was seen, however. An ongoing phase 3 study is looking at whether or not combining BRAF and MEK inhibitors results in improved OS compared with single-agent BRAF. It is premature at this juncture to recommend combining dabrafenib and trametinib until the results of the ongoing phase 3 studies more thoroughly describe the risks and benefits of this approach (Table 2).
KIT inhibitors
In a subset of melanomas, particularly those that arise from mucosal, acral, or chronically sun-damaged skin, mutations are found in the receptor-tyrosine kinase KIT.35 A number of agents directed against KIT, such as imatinib, have been tested in clinical trials. Initial phase 2 studies revealed poor RRs with KIT inhibition in molecularly unselected patients.53-55 Subsequent studies selected patients with KIT genetic aberrations, including mutations and amplifications, and some responses were seen.56-58
Importantly, not all KIT genetic aberrations are believed to be considered equal. Preliminarily, it appears that mutations in exon 11 (L576P) and exon 13 (K642E) appear to be most closely associated with response and may be true driver mutations. Other KIT mutations may have less functional significance but additional research is needed. Imatinib is a reasonable therapeutic choice in patients with a KIT mutation, particularly when an L576P or K642E mutation is present.
conclusions
Since 2011, 4 new drugs—ipilimumab, vemurafenib, dabrafenib, and trametinib—have been approved for the treatment of metastatic melanoma. Exciting early data from PD-1 clinical trials suggest that agents that disrupt PD-1 may also become important therapeutic modalities. Future studies will continue to evaluate combinations of these therapeutic modalities, but caution should be exercised in combining these drugs prior to data from ongoing clinical trials revealing the true benefits and risks of combination therapy. Excessive toxicity was seen in an early phase trial when vemurafenib was combined with ipilimumab.59
Additional research will also explore biomarkers that may help clinicians apply immunotherapy to the most appropriate patients and better understand mechanisms of resistance to targeted therapies. Clinical trials of novel agents or combinations should be considered at every treatment juncture to continue the rapid pace of developing the most innovative and tailored treatment approaches.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10-29.
2. Garbe C, Peris K, Hauschild A, et al; European Dermatology Forum, European Association of Dermato-Oncology, European Organization of Research and Treatment of Cancer. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2012. Eur J Cancer. 2012;48(15):2375-2390.
3. Coit DG, Andtbacka R, Bichakjian CK, et al; NCCN Melanoma Panel. Melanoma. J Natl Compr Canc Netw. 2009;7(3):250-275.
4. Molife R, Hancock BW. Adjuvant therapy of malignant melanoma. Crit Rev Oncol Hematol. 2002;44(1):81-102.
5. Wheatley K, Ives N, Hancock B, Gore M, Eggermont A, Suciu S. Does adjuvant interferon-alpha for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat Rev. 2003;29(4):241-252.
6. Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: A systematic review and meta-analysis. J Natl Cancer Inst. 2010;102(7):493-501.
7. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199-6206.
8. Luke JJ, Rubinstein LV, Smith GL, Ivy SP, Harris PJ. Similar efficacy for phase I trials in comparison with DTIC for advanced malignant melanoma: an analysis of melanoma outcomes in CTEP-sponsored phase I trials 1995-2011. Melanoma Res. 2013;23(2):152-158.
9. Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18(1):158-166.
10. Teimouri F, Nikfar S, Abdollahi M. Efficacy and side effects of dacarbazine in comparison with temozolomide in the treatment of malignant melanoma: A meta-analysis consisting of 1314 patients. Melanoma Res. 2013;23(5):381-389.
11. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745-2751.
12. Legha SS, Ring S, Papadopoulos N, Plager C, Chawla S, Benjamin R. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer. 1989;64(10):2024-2029.
13. Rao RD, Holtan SG, Ingle JN, et al. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. Cancer. 2006;106(2):375-382.
14. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J Sci Am. 2000;6(suppl 1):S11-S14.
15. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105-2116.
16. Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119-2127.
17. Johannsen M, Spitaleri G, Curigliano G, et al. The tumour-targeting human L19-IL2 immunocytokine: Preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer. 2010;46(16):2926-2935.
18. Eigentler TK, Weide B, de Braud F, et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin Cancer Res. 2011;17(24):7732-7742.
19. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
20. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517-2526.
21. Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616-622.
22. Wolchok JD, Weber JS, Maio M, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24(8):2174-2180.
23. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
24. Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. 2010;37(5):499-507.
25. Harmankaya K, Erasim C, Koelblinger C, et al. Continuous systemic corticosteroids do not affect the ongoing regression of metastatic melanoma for more than two years following ipilimumab therapy. Med Oncol. 2011;28(4):1140-1144.
26. Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15(23):7412-7420.
27. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467-477.
28. Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883-895.
29. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
30. Sznol M, Kluger HM, Hodi FS, et al. Survival and long-term follow-up of safety and response in patients (pts) with advanced melanoma (MEL) in a phase I trial of nivolumab (anti-PD-1; BMS-936558; ONO-4538) [ASCO abstract CRA9006]. ASCO Meet Abstr. 2013;31(18_suppl):CRA9006. http://meetinglibrary.asco.org/content/80822. Accessed July 23, 2014.
31. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122-133.
32. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134-144.
33. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
34. McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance.
Biochim Biophys Acta. 2007;1773(8):1263-1284.
35. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340-4346.
36. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949-954.
37. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246.
38. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596-599.
39. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809-819.
40. Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
41. Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAF(V600E)-mutated melanoma [ASCO abstract 8502]. ASCO Meet Abstr. 2012;30(15_suppl):8502. http://meetinglibrary.asco.org/content/70533?media=vm. Accessed July 23, 2014.
42. Lacouture ME, O’Reilly K, Rosen N, Solit DB. Induction of cutaneous squamous cell carcinomas by RAF inhibitors: Cause for concern? J Clin Oncol. 2012;30(3):329-330.
43. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. New Engl J Med. 2012;366(3):207-215.
44. Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. New Engl J Med. 2012;367(24):2316-2321.
45. Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30(19):2375-2383.
46. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label phase 3 randomised clinical trial. Lancet 2012;380(9839):358-365.
47. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
48. Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555-567.
49. Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. New Engl J Med. 2012;367(2):107-114.
50. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387-390.
51. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973-977.
52. Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New Engl J Med. 2012;367(18):1694-1703.
53. Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;9(8):1398-1405.
54. Wyman K, Atkins MB, Prieto V, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy. Cancer. 2006;106(9):2005-2011.
55. Kim KB, Eton O, Davis DW, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008;99(5):734-740.
56. Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327-2334.
57. Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182-3190.
58. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904-2909.
59. Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365-1366.
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10-29.
2. Garbe C, Peris K, Hauschild A, et al; European Dermatology Forum, European Association of Dermato-Oncology, European Organization of Research and Treatment of Cancer. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2012. Eur J Cancer. 2012;48(15):2375-2390.
3. Coit DG, Andtbacka R, Bichakjian CK, et al; NCCN Melanoma Panel. Melanoma. J Natl Compr Canc Netw. 2009;7(3):250-275.
4. Molife R, Hancock BW. Adjuvant therapy of malignant melanoma. Crit Rev Oncol Hematol. 2002;44(1):81-102.
5. Wheatley K, Ives N, Hancock B, Gore M, Eggermont A, Suciu S. Does adjuvant interferon-alpha for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat Rev. 2003;29(4):241-252.
6. Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: A systematic review and meta-analysis. J Natl Cancer Inst. 2010;102(7):493-501.
7. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199-6206.
8. Luke JJ, Rubinstein LV, Smith GL, Ivy SP, Harris PJ. Similar efficacy for phase I trials in comparison with DTIC for advanced malignant melanoma: an analysis of melanoma outcomes in CTEP-sponsored phase I trials 1995-2011. Melanoma Res. 2013;23(2):152-158.
9. Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18(1):158-166.
10. Teimouri F, Nikfar S, Abdollahi M. Efficacy and side effects of dacarbazine in comparison with temozolomide in the treatment of malignant melanoma: A meta-analysis consisting of 1314 patients. Melanoma Res. 2013;23(5):381-389.
11. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745-2751.
12. Legha SS, Ring S, Papadopoulos N, Plager C, Chawla S, Benjamin R. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer. 1989;64(10):2024-2029.
13. Rao RD, Holtan SG, Ingle JN, et al. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. Cancer. 2006;106(2):375-382.
14. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J Sci Am. 2000;6(suppl 1):S11-S14.
15. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105-2116.
16. Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119-2127.
17. Johannsen M, Spitaleri G, Curigliano G, et al. The tumour-targeting human L19-IL2 immunocytokine: Preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer. 2010;46(16):2926-2935.
18. Eigentler TK, Weide B, de Braud F, et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin Cancer Res. 2011;17(24):7732-7742.
19. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
20. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517-2526.
21. Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616-622.
22. Wolchok JD, Weber JS, Maio M, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24(8):2174-2180.
23. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
24. Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol. 2010;37(5):499-507.
25. Harmankaya K, Erasim C, Koelblinger C, et al. Continuous systemic corticosteroids do not affect the ongoing regression of metastatic melanoma for more than two years following ipilimumab therapy. Med Oncol. 2011;28(4):1140-1144.
26. Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15(23):7412-7420.
27. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467-477.
28. Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883-895.
29. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
30. Sznol M, Kluger HM, Hodi FS, et al. Survival and long-term follow-up of safety and response in patients (pts) with advanced melanoma (MEL) in a phase I trial of nivolumab (anti-PD-1; BMS-936558; ONO-4538) [ASCO abstract CRA9006]. ASCO Meet Abstr. 2013;31(18_suppl):CRA9006. http://meetinglibrary.asco.org/content/80822. Accessed July 23, 2014.
31. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122-133.
32. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134-144.
33. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
34. McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance.
Biochim Biophys Acta. 2007;1773(8):1263-1284.
35. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340-4346.
36. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949-954.
37. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246.
38. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596-599.
39. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809-819.
40. Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
41. Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAF(V600E)-mutated melanoma [ASCO abstract 8502]. ASCO Meet Abstr. 2012;30(15_suppl):8502. http://meetinglibrary.asco.org/content/70533?media=vm. Accessed July 23, 2014.
42. Lacouture ME, O’Reilly K, Rosen N, Solit DB. Induction of cutaneous squamous cell carcinomas by RAF inhibitors: Cause for concern? J Clin Oncol. 2012;30(3):329-330.
43. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. New Engl J Med. 2012;366(3):207-215.
44. Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. New Engl J Med. 2012;367(24):2316-2321.
45. Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30(19):2375-2383.
46. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label phase 3 randomised clinical trial. Lancet 2012;380(9839):358-365.
47. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
48. Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555-567.
49. Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. New Engl J Med. 2012;367(2):107-114.
50. Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387-390.
51. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973-977.
52. Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New Engl J Med. 2012;367(18):1694-1703.
53. Ugurel S, Hildenbrand R, Zimpfer A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;9(8):1398-1405.
54. Wyman K, Atkins MB, Prieto V, et al. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy. Cancer. 2006;106(9):2005-2011.
55. Kim KB, Eton O, Davis DW, et al. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br J Cancer. 2008;99(5):734-740.
56. Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327-2334.
57. Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182-3190.
58. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904-2909.
59. Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365-1366.
House votes to speed sunscreen approvals
The U.S. House of Representatives passed legislation to speed up Food and Drug Administration approvals of new sunscreen ingredients and to eliminate a backlog.
The Sunscreen Innovation Act (H.R. 4250) was passed by a voice vote and now awaits action by the Senate. The companion bill in the Senate, S. 2141, was introduced by Jack Reed (D-R.I.) and has been referred to the Committee on Health, Education, Labor, and Pensions but has not received a hearing yet.

Backers of the legislation are encouraging the Senate to move quickly.
"Even though the Food and Drug Administration has listed action on sunscreen ingredient applications as a priority since 2008, no new sunscreen ingredients have been approved by the FDA in 15 years," Rep. Ed Whitfield (R-Ky.), one of the lead sponsors of the House bill, said in a statement after the approval.
"The framework outlined in this legislation strikes an appropriate balance between consumer safety and access to new sunscreen products," he said, adding, "I am pleased to see my legislation pass the House of Representatives, and call on the Senate to act."
The bill requires the FDA to make final decisions within a year on the backlog of ingredients under review, and within a year and a half on new applications. It also sets up more Congressional oversight of the process and makes "sunscreens that have been marketed for five continuous years in the United States or other countries and in sufficient quantity eligible for review under this Act."
Currently, according to the American Cancer Society Cancer Action Network (ACS CAN), only three of the seven UVA-blocking ingredients sold in Europe are approved in the United States.
The Sunscreen Innovation Act has many supporters in the health care field and among consumer advocates and manufacturers, many of whom belong to the PASS (Public Access to SunScreens) Coalition. In a statement issued by the coalition, the American College of Mohs Surgery said, "This important legislation will go a long way in helping prevent skin cancers by improving the availability of more effective sunscreen products, and we are committed to its passage."
The Environmental Working Group, a frequent critic of the FDA’s regulation of sunscreens and of manufacturers, also applauded House passage of the bill.
The American Cancer Society was pleased as well. "The House took a critical step today to fix a broken process at FDA for the review of new sunscreen ingredients that could potentially help more Americans prevent skin cancer," Chris Hansen, president of the ACS CAN, said in a statement.
"American consumers should have access to the broadest choice of sunscreens – including those in use for years in other countries – once they are shown to be safe and effective," he noted.
On Twitter @aliciaault
The U.S. House of Representatives passed legislation to speed up Food and Drug Administration approvals of new sunscreen ingredients and to eliminate a backlog.
The Sunscreen Innovation Act (H.R. 4250) was passed by a voice vote and now awaits action by the Senate. The companion bill in the Senate, S. 2141, was introduced by Jack Reed (D-R.I.) and has been referred to the Committee on Health, Education, Labor, and Pensions but has not received a hearing yet.
Backers of the legislation are encouraging the Senate to move quickly.
"Even though the Food and Drug Administration has listed action on sunscreen ingredient applications as a priority since 2008, no new sunscreen ingredients have been approved by the FDA in 15 years," Rep. Ed Whitfield (R-Ky.), one of the lead sponsors of the House bill, said in a statement after the approval.
"The framework outlined in this legislation strikes an appropriate balance between consumer safety and access to new sunscreen products," he said, adding, "I am pleased to see my legislation pass the House of Representatives, and call on the Senate to act."
The bill requires the FDA to make final decisions within a year on the backlog of ingredients under review, and within a year and a half on new applications. It also sets up more Congressional oversight of the process and makes "sunscreens that have been marketed for five continuous years in the United States or other countries and in sufficient quantity eligible for review under this Act."
Currently, according to the American Cancer Society Cancer Action Network (ACS CAN), only three of the seven UVA-blocking ingredients sold in Europe are approved in the United States.
The Sunscreen Innovation Act has many supporters in the health care field and among consumer advocates and manufacturers, many of whom belong to the PASS (Public Access to SunScreens) Coalition. In a statement issued by the coalition, the American College of Mohs Surgery said, "This important legislation will go a long way in helping prevent skin cancers by improving the availability of more effective sunscreen products, and we are committed to its passage."
The Environmental Working Group, a frequent critic of the FDA’s regulation of sunscreens and of manufacturers, also applauded House passage of the bill.
The American Cancer Society was pleased as well. "The House took a critical step today to fix a broken process at FDA for the review of new sunscreen ingredients that could potentially help more Americans prevent skin cancer," Chris Hansen, president of the ACS CAN, said in a statement.
"American consumers should have access to the broadest choice of sunscreens – including those in use for years in other countries – once they are shown to be safe and effective," he noted.
On Twitter @aliciaault
The U.S. House of Representatives passed legislation to speed up Food and Drug Administration approvals of new sunscreen ingredients and to eliminate a backlog.
The Sunscreen Innovation Act (H.R. 4250) was passed by a voice vote and now awaits action by the Senate. The companion bill in the Senate, S. 2141, was introduced by Jack Reed (D-R.I.) and has been referred to the Committee on Health, Education, Labor, and Pensions but has not received a hearing yet.
Backers of the legislation are encouraging the Senate to move quickly.
"Even though the Food and Drug Administration has listed action on sunscreen ingredient applications as a priority since 2008, no new sunscreen ingredients have been approved by the FDA in 15 years," Rep. Ed Whitfield (R-Ky.), one of the lead sponsors of the House bill, said in a statement after the approval.
"The framework outlined in this legislation strikes an appropriate balance between consumer safety and access to new sunscreen products," he said, adding, "I am pleased to see my legislation pass the House of Representatives, and call on the Senate to act."
The bill requires the FDA to make final decisions within a year on the backlog of ingredients under review, and within a year and a half on new applications. It also sets up more Congressional oversight of the process and makes "sunscreens that have been marketed for five continuous years in the United States or other countries and in sufficient quantity eligible for review under this Act."
Currently, according to the American Cancer Society Cancer Action Network (ACS CAN), only three of the seven UVA-blocking ingredients sold in Europe are approved in the United States.
The Sunscreen Innovation Act has many supporters in the health care field and among consumer advocates and manufacturers, many of whom belong to the PASS (Public Access to SunScreens) Coalition. In a statement issued by the coalition, the American College of Mohs Surgery said, "This important legislation will go a long way in helping prevent skin cancers by improving the availability of more effective sunscreen products, and we are committed to its passage."
The Environmental Working Group, a frequent critic of the FDA’s regulation of sunscreens and of manufacturers, also applauded House passage of the bill.
The American Cancer Society was pleased as well. "The House took a critical step today to fix a broken process at FDA for the review of new sunscreen ingredients that could potentially help more Americans prevent skin cancer," Chris Hansen, president of the ACS CAN, said in a statement.
"American consumers should have access to the broadest choice of sunscreens – including those in use for years in other countries – once they are shown to be safe and effective," he noted.
On Twitter @aliciaault

Surgeon General: Tanning, indoor tanning must stop
The U.S. Surgeon General’s office is calling on Americans to do more to help prevent skin cancer, saying that it is a growing public health problem.
The five-point call to action singled out ultraviolet light exposure – from indoor and outdoor tanning – as a major culprit in the growing incidence of all cancers, and melanoma, in particular.
"Tanned skin is damaged skin," Acting Surgeon General, Boris D. Lushniak, a dermatologist, said at a briefing to release the report.
"When people tan or get sunburned, they increase their risk of getting skin cancer later in life," he said, adding that the nation needs to change its cultural acceptance of tanning as a sign of health.
The report noted that about 5 million Americans are treated for skin cancer each year, at a cost of $8 billion. Melanoma is of the greatest concern; its incidence has tripled over the past 3 decades, with 63,000 new cases per year. Some 9,000 Americans die from melanoma each year, and death rates are on the rise in men, Dr. Howard Koh, assistant secretary for health at the U.S. Department of Health & Human Services, said at the briefing.
Both Dr. Koh and Dr. Lushniak singled out indoor tanning as particularly dangerous. The Surgeon General’s report cited research showing that indoor tanning may be associated with more than 400,000 cases of skin cancer, including 6,000 melanomas, per year.
Forty-four states and the District of Columbia have some type of indoor tanning laws or regulations. Eleven states – California, Delaware, Hawaii, Illinois, Louisiana, Minnesota, Nevada, Oregon, Texas, Vermont and Washington – prohibit indoor tanning by individuals under age 18. But indoor tanning remains popular: One in three white women aged 16-25 years engages in indoor tanning each year, according to the report.
In May, the Food and Drug Administration issued stricter rules for sunlamps used with indoor tanning, requiring a boxed warning that they should not be used on anyone younger than 18 years. The agency also required that more information on risks and contraindications be provided to sunlamp users, FDA Commissioner Margaret Hamburg noted in a statement on the Surgeon General’s report.

The five main points in the Surgeon General’s call to action are:
• Increase opportunities for sun protection in outdoor settings, such as increasing shade in recreational areas, and providing more protection for school children and outdoor workers.
• Give individuals information to make informed, healthy choices about UV exposure, including providing materials in workplaces and schools, and partnering with physicians and health systems.
• Promote policies that advance the goal of preventing skin cancer, such as encouraging electronic reporting of reportable skin cancers.
• Reduce harms from indoor tanning by enforcing existing laws and considering additional restrictions.
• Strengthen research, surveillance, monitoring, and evaluation.
The American Academy of Dermatology Association applauded the report.
"The American public needs to be aware that the dangers of ultraviolet radiation exposure are real," Dr. Brett M. Coldiron, president of the AADA, said in a statement. "The AADA is particularly pleased that the HHS and the Office of the Surgeon General have highlighted methods for the public to prevent skin cancer in this white paper, that include seeking shade, wearing protective clothing, applying sunscreen, and avoiding dangerous indoor tanning devices."
John Seffrin, Ph.D., chief executive officer of the American Cancer Society Cancer Action Network, noted that this is the first time there has been a national action plan regarding skin cancer.
"By bringing national attention to this growing public health crisis, the Surgeon General is calling on all of us to reinvigorate the fight against skin cancer," he said. "The Surgeon General’s Call to Action outlines achievable goals and strategies to support more Americans in making healthy choices about protecting their skin," Dr. Seffrin said.
On Twitter @aliciaault
The U.S. Surgeon General’s office is calling on Americans to do more to help prevent skin cancer, saying that it is a growing public health problem.
The five-point call to action singled out ultraviolet light exposure – from indoor and outdoor tanning – as a major culprit in the growing incidence of all cancers, and melanoma, in particular.
"Tanned skin is damaged skin," Acting Surgeon General, Boris D. Lushniak, a dermatologist, said at a briefing to release the report.
"When people tan or get sunburned, they increase their risk of getting skin cancer later in life," he said, adding that the nation needs to change its cultural acceptance of tanning as a sign of health.
The report noted that about 5 million Americans are treated for skin cancer each year, at a cost of $8 billion. Melanoma is of the greatest concern; its incidence has tripled over the past 3 decades, with 63,000 new cases per year. Some 9,000 Americans die from melanoma each year, and death rates are on the rise in men, Dr. Howard Koh, assistant secretary for health at the U.S. Department of Health & Human Services, said at the briefing.
Both Dr. Koh and Dr. Lushniak singled out indoor tanning as particularly dangerous. The Surgeon General’s report cited research showing that indoor tanning may be associated with more than 400,000 cases of skin cancer, including 6,000 melanomas, per year.
Forty-four states and the District of Columbia have some type of indoor tanning laws or regulations. Eleven states – California, Delaware, Hawaii, Illinois, Louisiana, Minnesota, Nevada, Oregon, Texas, Vermont and Washington – prohibit indoor tanning by individuals under age 18. But indoor tanning remains popular: One in three white women aged 16-25 years engages in indoor tanning each year, according to the report.
In May, the Food and Drug Administration issued stricter rules for sunlamps used with indoor tanning, requiring a boxed warning that they should not be used on anyone younger than 18 years. The agency also required that more information on risks and contraindications be provided to sunlamp users, FDA Commissioner Margaret Hamburg noted in a statement on the Surgeon General’s report.
The five main points in the Surgeon General’s call to action are:
• Increase opportunities for sun protection in outdoor settings, such as increasing shade in recreational areas, and providing more protection for school children and outdoor workers.
• Give individuals information to make informed, healthy choices about UV exposure, including providing materials in workplaces and schools, and partnering with physicians and health systems.
• Promote policies that advance the goal of preventing skin cancer, such as encouraging electronic reporting of reportable skin cancers.
• Reduce harms from indoor tanning by enforcing existing laws and considering additional restrictions.
• Strengthen research, surveillance, monitoring, and evaluation.
The American Academy of Dermatology Association applauded the report.
"The American public needs to be aware that the dangers of ultraviolet radiation exposure are real," Dr. Brett M. Coldiron, president of the AADA, said in a statement. "The AADA is particularly pleased that the HHS and the Office of the Surgeon General have highlighted methods for the public to prevent skin cancer in this white paper, that include seeking shade, wearing protective clothing, applying sunscreen, and avoiding dangerous indoor tanning devices."
John Seffrin, Ph.D., chief executive officer of the American Cancer Society Cancer Action Network, noted that this is the first time there has been a national action plan regarding skin cancer.
"By bringing national attention to this growing public health crisis, the Surgeon General is calling on all of us to reinvigorate the fight against skin cancer," he said. "The Surgeon General’s Call to Action outlines achievable goals and strategies to support more Americans in making healthy choices about protecting their skin," Dr. Seffrin said.
On Twitter @aliciaault
The U.S. Surgeon General’s office is calling on Americans to do more to help prevent skin cancer, saying that it is a growing public health problem.
The five-point call to action singled out ultraviolet light exposure – from indoor and outdoor tanning – as a major culprit in the growing incidence of all cancers, and melanoma, in particular.
"Tanned skin is damaged skin," Acting Surgeon General, Boris D. Lushniak, a dermatologist, said at a briefing to release the report.
"When people tan or get sunburned, they increase their risk of getting skin cancer later in life," he said, adding that the nation needs to change its cultural acceptance of tanning as a sign of health.
The report noted that about 5 million Americans are treated for skin cancer each year, at a cost of $8 billion. Melanoma is of the greatest concern; its incidence has tripled over the past 3 decades, with 63,000 new cases per year. Some 9,000 Americans die from melanoma each year, and death rates are on the rise in men, Dr. Howard Koh, assistant secretary for health at the U.S. Department of Health & Human Services, said at the briefing.
Both Dr. Koh and Dr. Lushniak singled out indoor tanning as particularly dangerous. The Surgeon General’s report cited research showing that indoor tanning may be associated with more than 400,000 cases of skin cancer, including 6,000 melanomas, per year.
Forty-four states and the District of Columbia have some type of indoor tanning laws or regulations. Eleven states – California, Delaware, Hawaii, Illinois, Louisiana, Minnesota, Nevada, Oregon, Texas, Vermont and Washington – prohibit indoor tanning by individuals under age 18. But indoor tanning remains popular: One in three white women aged 16-25 years engages in indoor tanning each year, according to the report.
In May, the Food and Drug Administration issued stricter rules for sunlamps used with indoor tanning, requiring a boxed warning that they should not be used on anyone younger than 18 years. The agency also required that more information on risks and contraindications be provided to sunlamp users, FDA Commissioner Margaret Hamburg noted in a statement on the Surgeon General’s report.
The five main points in the Surgeon General’s call to action are:
• Increase opportunities for sun protection in outdoor settings, such as increasing shade in recreational areas, and providing more protection for school children and outdoor workers.
• Give individuals information to make informed, healthy choices about UV exposure, including providing materials in workplaces and schools, and partnering with physicians and health systems.
• Promote policies that advance the goal of preventing skin cancer, such as encouraging electronic reporting of reportable skin cancers.
• Reduce harms from indoor tanning by enforcing existing laws and considering additional restrictions.
• Strengthen research, surveillance, monitoring, and evaluation.
The American Academy of Dermatology Association applauded the report.
"The American public needs to be aware that the dangers of ultraviolet radiation exposure are real," Dr. Brett M. Coldiron, president of the AADA, said in a statement. "The AADA is particularly pleased that the HHS and the Office of the Surgeon General have highlighted methods for the public to prevent skin cancer in this white paper, that include seeking shade, wearing protective clothing, applying sunscreen, and avoiding dangerous indoor tanning devices."
John Seffrin, Ph.D., chief executive officer of the American Cancer Society Cancer Action Network, noted that this is the first time there has been a national action plan regarding skin cancer.
"By bringing national attention to this growing public health crisis, the Surgeon General is calling on all of us to reinvigorate the fight against skin cancer," he said. "The Surgeon General’s Call to Action outlines achievable goals and strategies to support more Americans in making healthy choices about protecting their skin," Dr. Seffrin said.
On Twitter @aliciaault
ASCO at 50 … we’ve come a long way, we know a lot, but we’ve only just begun
I recently had both the pleasure and the challenge of attending ASCO 2014, the annual meeting of the American Society of Clinical Oncology. It was my first official ASCO meeting, and as an almost-third-year fellow in oncology, no amount of reading, research, scheduling, ASCO 2014 iPad app organizing, or even attending the day 1 early morning How to Navigate the Annual Meeting seminar could have prepared me for the experience…
Click on the PDF icon at the top of this introduction to read the full article.
I recently had both the pleasure and the challenge of attending ASCO 2014, the annual meeting of the American Society of Clinical Oncology. It was my first official ASCO meeting, and as an almost-third-year fellow in oncology, no amount of reading, research, scheduling, ASCO 2014 iPad app organizing, or even attending the day 1 early morning How to Navigate the Annual Meeting seminar could have prepared me for the experience…
Click on the PDF icon at the top of this introduction to read the full article.
I recently had both the pleasure and the challenge of attending ASCO 2014, the annual meeting of the American Society of Clinical Oncology. It was my first official ASCO meeting, and as an almost-third-year fellow in oncology, no amount of reading, research, scheduling, ASCO 2014 iPad app organizing, or even attending the day 1 early morning How to Navigate the Annual Meeting seminar could have prepared me for the experience…
Click on the PDF icon at the top of this introduction to read the full article.
Trametinib plus dabrafenib for unresectable or metastatic melanoma with BRAF V600E or V600K mutations
On January 9, 2014, the combination of trametinib and dabrafenib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations as detected by an FDA-approved test.1,2 Approval of the combination is based on durable response rate observed in an open-label study.2,3 Improvements in disease-related symptoms and overall survival have not yet been demonstrated for the combination. Both drugs were approved for use as single agents in this setting in May 2013.
Click on the PDF icon at the top of this introduction to read the full article.
On January 9, 2014, the combination of trametinib and dabrafenib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations as detected by an FDA-approved test.1,2 Approval of the combination is based on durable response rate observed in an open-label study.2,3 Improvements in disease-related symptoms and overall survival have not yet been demonstrated for the combination. Both drugs were approved for use as single agents in this setting in May 2013.
Click on the PDF icon at the top of this introduction to read the full article.
On January 9, 2014, the combination of trametinib and dabrafenib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations as detected by an FDA-approved test.1,2 Approval of the combination is based on durable response rate observed in an open-label study.2,3 Improvements in disease-related symptoms and overall survival have not yet been demonstrated for the combination. Both drugs were approved for use as single agents in this setting in May 2013.
Click on the PDF icon at the top of this introduction to read the full article.
Adding ingenol mebutate after cryosurgery enhances clearance of actinic keratoses
Complete clearance occurred at 12 months in 31% of patients who received field treatment with ingenol mebutate gel after cryotherapy for actinic keratoses, compared with 19% of placebo patients in a phase III trial.
The findings were published in the June issue of the Journal of Drugs in Dermatology.
Recurrence rates for AKs treated with cryotherapy alone are high, possibly because the treatment is lesion-directed and fails to address field cancerization, wrote Dr. Brian Berman of the University of Miami.
The FIELD study 1, a phase III, multicenter, randomized trial, showed that ingenol mebutate gel was safe and effective as a follow-up to cryosurgery for AKs. Short-term efficacy data (11 weeks) have been previously reported.
In this study, the patients were followed for 12 months (J. Drugs Dermatol. 2014;13:741-7).

"In phase III clinical trials, 2 or 3 days of field-directed therapy with ingenol mebutate demonstrated high and sustained clearance of AKs on the head and body, and was well tolerated," noted Dr. Berman and his coauthors.
The percentage reduction in the number of AKs was significantly higher at 12 months in the treatment group compared with the placebo group (68% vs. 54%), and significantly fewer patients in the treatment group developed new lesions in the treatment area compared with the placebo group (39% vs. 52%). In addition, the probability of sustained clearance of AKs at 12 months was 55% in the treatment group vs. 40% in the placebo group.
The study included 329 adults aged 18 years and older with 4-8 clinically typical, visible, and discrete AKs within a 25-cm2 treatment area on the face or scalp. The patients underwent cryosurgery, and 3 weeks later underwent field therapy with 0.015% ingenol mebutate gel or vehicle gel once a day for 3 consecutive days. Patients with suspected basal cell carcinoma or squamous cell carcinoma were excluded.
The findings were limited by the possible unblinding of treatment because of localized skin reactions, the researchers noted. However, few drug-related adverse events and no drug-related serious adverse events were reported, they said.
Dr. Berman has served as a consultant, received honoraria, and served on the speakers bureau for Leo Pharma, which funded the study.
Complete clearance occurred at 12 months in 31% of patients who received field treatment with ingenol mebutate gel after cryotherapy for actinic keratoses, compared with 19% of placebo patients in a phase III trial.
The findings were published in the June issue of the Journal of Drugs in Dermatology.
Recurrence rates for AKs treated with cryotherapy alone are high, possibly because the treatment is lesion-directed and fails to address field cancerization, wrote Dr. Brian Berman of the University of Miami.
The FIELD study 1, a phase III, multicenter, randomized trial, showed that ingenol mebutate gel was safe and effective as a follow-up to cryosurgery for AKs. Short-term efficacy data (11 weeks) have been previously reported.
In this study, the patients were followed for 12 months (J. Drugs Dermatol. 2014;13:741-7).
"In phase III clinical trials, 2 or 3 days of field-directed therapy with ingenol mebutate demonstrated high and sustained clearance of AKs on the head and body, and was well tolerated," noted Dr. Berman and his coauthors.
The percentage reduction in the number of AKs was significantly higher at 12 months in the treatment group compared with the placebo group (68% vs. 54%), and significantly fewer patients in the treatment group developed new lesions in the treatment area compared with the placebo group (39% vs. 52%). In addition, the probability of sustained clearance of AKs at 12 months was 55% in the treatment group vs. 40% in the placebo group.
The study included 329 adults aged 18 years and older with 4-8 clinically typical, visible, and discrete AKs within a 25-cm2 treatment area on the face or scalp. The patients underwent cryosurgery, and 3 weeks later underwent field therapy with 0.015% ingenol mebutate gel or vehicle gel once a day for 3 consecutive days. Patients with suspected basal cell carcinoma or squamous cell carcinoma were excluded.
The findings were limited by the possible unblinding of treatment because of localized skin reactions, the researchers noted. However, few drug-related adverse events and no drug-related serious adverse events were reported, they said.
Dr. Berman has served as a consultant, received honoraria, and served on the speakers bureau for Leo Pharma, which funded the study.
Complete clearance occurred at 12 months in 31% of patients who received field treatment with ingenol mebutate gel after cryotherapy for actinic keratoses, compared with 19% of placebo patients in a phase III trial.
The findings were published in the June issue of the Journal of Drugs in Dermatology.
Recurrence rates for AKs treated with cryotherapy alone are high, possibly because the treatment is lesion-directed and fails to address field cancerization, wrote Dr. Brian Berman of the University of Miami.
The FIELD study 1, a phase III, multicenter, randomized trial, showed that ingenol mebutate gel was safe and effective as a follow-up to cryosurgery for AKs. Short-term efficacy data (11 weeks) have been previously reported.
In this study, the patients were followed for 12 months (J. Drugs Dermatol. 2014;13:741-7).
"In phase III clinical trials, 2 or 3 days of field-directed therapy with ingenol mebutate demonstrated high and sustained clearance of AKs on the head and body, and was well tolerated," noted Dr. Berman and his coauthors.
The percentage reduction in the number of AKs was significantly higher at 12 months in the treatment group compared with the placebo group (68% vs. 54%), and significantly fewer patients in the treatment group developed new lesions in the treatment area compared with the placebo group (39% vs. 52%). In addition, the probability of sustained clearance of AKs at 12 months was 55% in the treatment group vs. 40% in the placebo group.
The study included 329 adults aged 18 years and older with 4-8 clinically typical, visible, and discrete AKs within a 25-cm2 treatment area on the face or scalp. The patients underwent cryosurgery, and 3 weeks later underwent field therapy with 0.015% ingenol mebutate gel or vehicle gel once a day for 3 consecutive days. Patients with suspected basal cell carcinoma or squamous cell carcinoma were excluded.
The findings were limited by the possible unblinding of treatment because of localized skin reactions, the researchers noted. However, few drug-related adverse events and no drug-related serious adverse events were reported, they said.
Dr. Berman has served as a consultant, received honoraria, and served on the speakers bureau for Leo Pharma, which funded the study.
FROM THE JOURNAL OF DRUGS IN DERMATOLOGY
Key clinical point: The findings support the safety and effectiveness of adding field therapy to cryosurgery when treating actinic keratosis lesions.
Major finding: Complete clearance rates at 12 months were significantly higher in patients treated with ingenol mebutate gel vs. a vehicle (31% vs. 19%).
Data source: A phase III, multicenter, randomized trial of adults with clinically typical AKs.
Disclosures: Dr. Berman has served as a consultant, received honoraria, and served on the speakers bureau for Leo Pharma, which funded the study.

Hawaii bans indoor tanning for minors
Hawaii Gov. Neil Abercrombie signed a bill on July 3 that makes Hawaii the 10th state to prohibit anyone under age 18 years from using tanning beds, according to the American Academy of Dermatology Association.
The law goes into effect immediately. The other states that have banned indoor tanning for minors are California, Illinois, Louisiana, Minnesota, Nevada, Oregon, Texas, Vermont, and Washington.
"The science is clear. The risk for developing melanoma increases by 59% in individuals who have been exposed to UV radiation from indoor tanning devices, and the risks increase with each subsequent use," AADA President Brent M. Coldiron said in a statement.
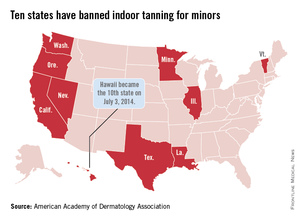
Hawaii Gov. Neil Abercrombie signed a bill on July 3 that makes Hawaii the 10th state to prohibit anyone under age 18 years from using tanning beds, according to the American Academy of Dermatology Association.
The law goes into effect immediately. The other states that have banned indoor tanning for minors are California, Illinois, Louisiana, Minnesota, Nevada, Oregon, Texas, Vermont, and Washington.
"The science is clear. The risk for developing melanoma increases by 59% in individuals who have been exposed to UV radiation from indoor tanning devices, and the risks increase with each subsequent use," AADA President Brent M. Coldiron said in a statement.
Hawaii Gov. Neil Abercrombie signed a bill on July 3 that makes Hawaii the 10th state to prohibit anyone under age 18 years from using tanning beds, according to the American Academy of Dermatology Association.
The law goes into effect immediately. The other states that have banned indoor tanning for minors are California, Illinois, Louisiana, Minnesota, Nevada, Oregon, Texas, Vermont, and Washington.
"The science is clear. The risk for developing melanoma increases by 59% in individuals who have been exposed to UV radiation from indoor tanning devices, and the risks increase with each subsequent use," AADA President Brent M. Coldiron said in a statement.