User login
UV Radiation Transmittance: Regular Clothing Versus Sun-Protective Clothing
Dermatologists frequently encounter patients who inquire about the need to buy special clothing and hats that claim to block UV light rather than using their regular clothing and hats. A patient may argue that he/she has never gotten sunburned through his/her favorite T-shirt while fishing, so why does he/she need to buy special clothing? The answer to this question is not straightforward. The dermatologist could easily say yes and advise patients to buy special sun-protective clothing, which could be especially tempting if a practitioner actually sells these items in the office. However, when considering evidence-based medicine, one needs to look at the data to appropriately answer the question.
Although it is still evolving, a standard has been set for UV protection factor (UPF) in the United States as well as other countries.1 Clothing with the maximum UPF rating of 50 blocks 98% of UVA/UVB radiation. Although there are data published in the literature regarding sun-protective clothing, there are scant data in the clinical dermatologic literature.2-5 To give patients an educated answer to this question, we measured and compared UVA/UVB radiation transmittance through regular (ie, non–UPF rated) clothing versus sun-protective clothing with a UPF rating.
Materials and Methods
A digital handheld UVA/UVB meter with an absorption spectrum of 280 to 400 nm was used to measure UV energy transmitted through sample clothing articles. The meter measured UVA/UVB light with a maximum reading of 40 mW/cm2. Clothing articles were selected of varied material/color and intended use.
Regular clothing articles included a straw golf hat (Figure 1A), an off-white and blue baseball hat (70% wool)(Figure 1B), a black baseball hat (100% wool)(Figure 1C), a white athletic tank shirt (100% cotton), a white T-shirt/undershirt (100% cotton), a thin-weave blue T-shirt (100% cotton), and a conventional-weave blue T-shirt (100% cotton)(Figure 1D). The regular clothing items, with the exception of the hats, had been laundered in conventional (ie, non–UV blocking) laundry detergent and no chemicals were applied to enhance UVA/UVB blocking properties. The exact number of times the items were laundered was unknown.

|

|

|

|
| Figure 1. Regular clothing articles included a straw golf hat (A), an off-white and blue baseball hat (B), a black baseball hat (C), and white and blue T-shirts (D). |
Sun-protective clothing articles included a polyester floral splash bucket hat and a polyester ruffled swim romper (Figure 2), both with a UPF rating of 50+. These items were purchased from a manufacturer who regularly promotes sun-protective clothing to both dermatologists and the general public. The company “guarantees” a UPF rating of 50 and advertises that these clothing articles block 98% of harmful UV rays. These items were not laundered prior to the study, and no chemicals were applied to enhance UVA/UVB blocking properties.

The UVA/UVB meter was calibrated on a clear cloudless July day in Frankfort, Illinois. An initial reading was taken without any obstruction to the sunlight. The regular and sun-protective clothing articles then were placed over the meter to measure the amount of UVA/UVB transmitted through each item. Measurements were taken for each article of clothing after the meter was covered by the respective material for 10 seconds. Care was taken to cover the meter with only 1 layer of material for each article, which was intended to mimic the degree of UVA/UVB blocking and transmittance during normal wear.
Results
The full results from the study are outlined in the Table. The unobstructed sunlight exposure exceeded the maximum measure of 40 mW/cm2, indicating there was a sufficient amount of sunlight to conduct testing.

The data show that both regular and sun-protective clothing blocked UVA/UVB rays in the 280- to 400-nm range. The Table outlines the level of UVA/UVB transmittance for each article of clothing; a lower number indicates less UVA/UVB transmittance occurred and more radiation was blocked.
Several of the regular clothing blocked more UV radiation than the sun-protective clothing; specifically, the data indicate that the baseball hats or the straw golf hat provided better protection than the sun-protective bucket hat. The black baseball hat provided the best UV protection. However, the straw golf hat provided adequate protection and better coverage, making it the best recommendation for patients.
Comment
Several of the regular items included in the study allowed less UVA/UVB transmission than the sun-protective clothing. Although our small study tested a limited number and type of articles, we assert that similar regular clothing would have similar transmittance.
There are various factors that affect UVA/UVB transmittance. Fabric construction, weight, thickness, composition, and color will affect the degree of UVA/UVB transmittance.1 In our study, the thickness, weave, and color of the fabric of the regular hats may have contributed to the superior results compared with the sun-protective hat. It could be postulated that cotton is inherently a superior fabric to the polyester sun-protective clothing fabric. With regard to the regular T-shirts, thickness, weave, and color also may have played a role in blocking UVA/UVB transmittance.
Patients may be assured of a sufficient amount of UVA/UVB blocking with sun-protective clothing. However, our study indicated that the regular clothing articles we tested provided similar, if not better, protection against UV radiation compared with the sun-protective clothing articles.
Conclusion
Based on the data, we would advise patients that they do not need to buy special sun-protective clothing that claims to block UV radiation, as regular clothing will provide equivalent protection against UVA/UVB radiation. However, these findings do not suggest that the claims for sun-protective clothing are inaccurate. Nevertheless, similar UVA/UVB blocking may be achieved with clothing already owned by patients.
1. Gies P. Photoprotection by clothing. Photodermatol Photoimmunol Photomed. 2007;23:264-274.
2. Wilson CA, Bevin NK, Laing RM, et al. Solar protection—effect of selected fabric and use characteristics on ultraviolet transmission. Textile Research Journal. 2008;78:95-104.
3. Ghazi S, Couteau C, Coiffard LJ. How to guarantee sun protection for a young sportsperson. J Dtsch Dermatol Ges. 2011;9:470-474.
4. Ghazi S, Couteau C, Coiffard LJ. What level of protection can be obtained using sun protective clothing? determining effectiveness using an in vitro method. Int J Pharm. 2010;397:144-146.
5. Morison WL. Photoprotection by clothing. Dermatol Ther. 2003;16:16-22.
Dermatologists frequently encounter patients who inquire about the need to buy special clothing and hats that claim to block UV light rather than using their regular clothing and hats. A patient may argue that he/she has never gotten sunburned through his/her favorite T-shirt while fishing, so why does he/she need to buy special clothing? The answer to this question is not straightforward. The dermatologist could easily say yes and advise patients to buy special sun-protective clothing, which could be especially tempting if a practitioner actually sells these items in the office. However, when considering evidence-based medicine, one needs to look at the data to appropriately answer the question.
Although it is still evolving, a standard has been set for UV protection factor (UPF) in the United States as well as other countries.1 Clothing with the maximum UPF rating of 50 blocks 98% of UVA/UVB radiation. Although there are data published in the literature regarding sun-protective clothing, there are scant data in the clinical dermatologic literature.2-5 To give patients an educated answer to this question, we measured and compared UVA/UVB radiation transmittance through regular (ie, non–UPF rated) clothing versus sun-protective clothing with a UPF rating.
Materials and Methods
A digital handheld UVA/UVB meter with an absorption spectrum of 280 to 400 nm was used to measure UV energy transmitted through sample clothing articles. The meter measured UVA/UVB light with a maximum reading of 40 mW/cm2. Clothing articles were selected of varied material/color and intended use.
Regular clothing articles included a straw golf hat (Figure 1A), an off-white and blue baseball hat (70% wool)(Figure 1B), a black baseball hat (100% wool)(Figure 1C), a white athletic tank shirt (100% cotton), a white T-shirt/undershirt (100% cotton), a thin-weave blue T-shirt (100% cotton), and a conventional-weave blue T-shirt (100% cotton)(Figure 1D). The regular clothing items, with the exception of the hats, had been laundered in conventional (ie, non–UV blocking) laundry detergent and no chemicals were applied to enhance UVA/UVB blocking properties. The exact number of times the items were laundered was unknown.
|
|
|
|
|
|
|
|
| Figure 1. Regular clothing articles included a straw golf hat (A), an off-white and blue baseball hat (B), a black baseball hat (C), and white and blue T-shirts (D). |
Sun-protective clothing articles included a polyester floral splash bucket hat and a polyester ruffled swim romper (Figure 2), both with a UPF rating of 50+. These items were purchased from a manufacturer who regularly promotes sun-protective clothing to both dermatologists and the general public. The company “guarantees” a UPF rating of 50 and advertises that these clothing articles block 98% of harmful UV rays. These items were not laundered prior to the study, and no chemicals were applied to enhance UVA/UVB blocking properties.
The UVA/UVB meter was calibrated on a clear cloudless July day in Frankfort, Illinois. An initial reading was taken without any obstruction to the sunlight. The regular and sun-protective clothing articles then were placed over the meter to measure the amount of UVA/UVB transmitted through each item. Measurements were taken for each article of clothing after the meter was covered by the respective material for 10 seconds. Care was taken to cover the meter with only 1 layer of material for each article, which was intended to mimic the degree of UVA/UVB blocking and transmittance during normal wear.
Results
The full results from the study are outlined in the Table. The unobstructed sunlight exposure exceeded the maximum measure of 40 mW/cm2, indicating there was a sufficient amount of sunlight to conduct testing.
The data show that both regular and sun-protective clothing blocked UVA/UVB rays in the 280- to 400-nm range. The Table outlines the level of UVA/UVB transmittance for each article of clothing; a lower number indicates less UVA/UVB transmittance occurred and more radiation was blocked.
Several of the regular clothing blocked more UV radiation than the sun-protective clothing; specifically, the data indicate that the baseball hats or the straw golf hat provided better protection than the sun-protective bucket hat. The black baseball hat provided the best UV protection. However, the straw golf hat provided adequate protection and better coverage, making it the best recommendation for patients.
Comment
Several of the regular items included in the study allowed less UVA/UVB transmission than the sun-protective clothing. Although our small study tested a limited number and type of articles, we assert that similar regular clothing would have similar transmittance.
There are various factors that affect UVA/UVB transmittance. Fabric construction, weight, thickness, composition, and color will affect the degree of UVA/UVB transmittance.1 In our study, the thickness, weave, and color of the fabric of the regular hats may have contributed to the superior results compared with the sun-protective hat. It could be postulated that cotton is inherently a superior fabric to the polyester sun-protective clothing fabric. With regard to the regular T-shirts, thickness, weave, and color also may have played a role in blocking UVA/UVB transmittance.
Patients may be assured of a sufficient amount of UVA/UVB blocking with sun-protective clothing. However, our study indicated that the regular clothing articles we tested provided similar, if not better, protection against UV radiation compared with the sun-protective clothing articles.
Conclusion
Based on the data, we would advise patients that they do not need to buy special sun-protective clothing that claims to block UV radiation, as regular clothing will provide equivalent protection against UVA/UVB radiation. However, these findings do not suggest that the claims for sun-protective clothing are inaccurate. Nevertheless, similar UVA/UVB blocking may be achieved with clothing already owned by patients.
Dermatologists frequently encounter patients who inquire about the need to buy special clothing and hats that claim to block UV light rather than using their regular clothing and hats. A patient may argue that he/she has never gotten sunburned through his/her favorite T-shirt while fishing, so why does he/she need to buy special clothing? The answer to this question is not straightforward. The dermatologist could easily say yes and advise patients to buy special sun-protective clothing, which could be especially tempting if a practitioner actually sells these items in the office. However, when considering evidence-based medicine, one needs to look at the data to appropriately answer the question.
Although it is still evolving, a standard has been set for UV protection factor (UPF) in the United States as well as other countries.1 Clothing with the maximum UPF rating of 50 blocks 98% of UVA/UVB radiation. Although there are data published in the literature regarding sun-protective clothing, there are scant data in the clinical dermatologic literature.2-5 To give patients an educated answer to this question, we measured and compared UVA/UVB radiation transmittance through regular (ie, non–UPF rated) clothing versus sun-protective clothing with a UPF rating.
Materials and Methods
A digital handheld UVA/UVB meter with an absorption spectrum of 280 to 400 nm was used to measure UV energy transmitted through sample clothing articles. The meter measured UVA/UVB light with a maximum reading of 40 mW/cm2. Clothing articles were selected of varied material/color and intended use.
Regular clothing articles included a straw golf hat (Figure 1A), an off-white and blue baseball hat (70% wool)(Figure 1B), a black baseball hat (100% wool)(Figure 1C), a white athletic tank shirt (100% cotton), a white T-shirt/undershirt (100% cotton), a thin-weave blue T-shirt (100% cotton), and a conventional-weave blue T-shirt (100% cotton)(Figure 1D). The regular clothing items, with the exception of the hats, had been laundered in conventional (ie, non–UV blocking) laundry detergent and no chemicals were applied to enhance UVA/UVB blocking properties. The exact number of times the items were laundered was unknown.
|
|
|
|
|
|
|
|
| Figure 1. Regular clothing articles included a straw golf hat (A), an off-white and blue baseball hat (B), a black baseball hat (C), and white and blue T-shirts (D). |
Sun-protective clothing articles included a polyester floral splash bucket hat and a polyester ruffled swim romper (Figure 2), both with a UPF rating of 50+. These items were purchased from a manufacturer who regularly promotes sun-protective clothing to both dermatologists and the general public. The company “guarantees” a UPF rating of 50 and advertises that these clothing articles block 98% of harmful UV rays. These items were not laundered prior to the study, and no chemicals were applied to enhance UVA/UVB blocking properties.
The UVA/UVB meter was calibrated on a clear cloudless July day in Frankfort, Illinois. An initial reading was taken without any obstruction to the sunlight. The regular and sun-protective clothing articles then were placed over the meter to measure the amount of UVA/UVB transmitted through each item. Measurements were taken for each article of clothing after the meter was covered by the respective material for 10 seconds. Care was taken to cover the meter with only 1 layer of material for each article, which was intended to mimic the degree of UVA/UVB blocking and transmittance during normal wear.
Results
The full results from the study are outlined in the Table. The unobstructed sunlight exposure exceeded the maximum measure of 40 mW/cm2, indicating there was a sufficient amount of sunlight to conduct testing.
The data show that both regular and sun-protective clothing blocked UVA/UVB rays in the 280- to 400-nm range. The Table outlines the level of UVA/UVB transmittance for each article of clothing; a lower number indicates less UVA/UVB transmittance occurred and more radiation was blocked.
Several of the regular clothing blocked more UV radiation than the sun-protective clothing; specifically, the data indicate that the baseball hats or the straw golf hat provided better protection than the sun-protective bucket hat. The black baseball hat provided the best UV protection. However, the straw golf hat provided adequate protection and better coverage, making it the best recommendation for patients.
Comment
Several of the regular items included in the study allowed less UVA/UVB transmission than the sun-protective clothing. Although our small study tested a limited number and type of articles, we assert that similar regular clothing would have similar transmittance.
There are various factors that affect UVA/UVB transmittance. Fabric construction, weight, thickness, composition, and color will affect the degree of UVA/UVB transmittance.1 In our study, the thickness, weave, and color of the fabric of the regular hats may have contributed to the superior results compared with the sun-protective hat. It could be postulated that cotton is inherently a superior fabric to the polyester sun-protective clothing fabric. With regard to the regular T-shirts, thickness, weave, and color also may have played a role in blocking UVA/UVB transmittance.
Patients may be assured of a sufficient amount of UVA/UVB blocking with sun-protective clothing. However, our study indicated that the regular clothing articles we tested provided similar, if not better, protection against UV radiation compared with the sun-protective clothing articles.
Conclusion
Based on the data, we would advise patients that they do not need to buy special sun-protective clothing that claims to block UV radiation, as regular clothing will provide equivalent protection against UVA/UVB radiation. However, these findings do not suggest that the claims for sun-protective clothing are inaccurate. Nevertheless, similar UVA/UVB blocking may be achieved with clothing already owned by patients.
1. Gies P. Photoprotection by clothing. Photodermatol Photoimmunol Photomed. 2007;23:264-274.
2. Wilson CA, Bevin NK, Laing RM, et al. Solar protection—effect of selected fabric and use characteristics on ultraviolet transmission. Textile Research Journal. 2008;78:95-104.
3. Ghazi S, Couteau C, Coiffard LJ. How to guarantee sun protection for a young sportsperson. J Dtsch Dermatol Ges. 2011;9:470-474.
4. Ghazi S, Couteau C, Coiffard LJ. What level of protection can be obtained using sun protective clothing? determining effectiveness using an in vitro method. Int J Pharm. 2010;397:144-146.
5. Morison WL. Photoprotection by clothing. Dermatol Ther. 2003;16:16-22.
1. Gies P. Photoprotection by clothing. Photodermatol Photoimmunol Photomed. 2007;23:264-274.
2. Wilson CA, Bevin NK, Laing RM, et al. Solar protection—effect of selected fabric and use characteristics on ultraviolet transmission. Textile Research Journal. 2008;78:95-104.
3. Ghazi S, Couteau C, Coiffard LJ. How to guarantee sun protection for a young sportsperson. J Dtsch Dermatol Ges. 2011;9:470-474.
4. Ghazi S, Couteau C, Coiffard LJ. What level of protection can be obtained using sun protective clothing? determining effectiveness using an in vitro method. Int J Pharm. 2010;397:144-146.
5. Morison WL. Photoprotection by clothing. Dermatol Ther. 2003;16:16-22.
- Dermatologists routinely advise patients that clothing is a method of UVA/UVB protection.
- Regular clothing items provide similar, if not superior, UVA/UVB protection compared to sun-protective clothing.
- Physicians may confidently inform patients that regular clothing items will provide adequate UVA/UVB protection.

FDA advisers want more data on sunscreen safety
SILVER SPRING, MD. – The widespread and long-term use of sunscreens demands that manufacturers provide more long-term safety data and information on how the active ingredients affect the health of special populations including infants, children, the elderly, and those with skin conditions, according to an expert panel called by the Food and Drug Administration.
Sunscreen safety has become an increasingly more important concern, especially since they are intended to be applied over the entire body, multiple times a day, and for a lifetime, Dr. Theresa Michele, director of the FDA’s division of nonprescription clinical evaluation, said Sept. 5 at a meeting of the agency’s Nonprescription Drugs Advisory Committee. "We’re essentially talking about a cradle-to-grave product."
FDA has been under pressure from physicians, consumers, manufacturers, and Congress to approve a backlog of eight ingredients that are generally available outside of the United States and to improve the process for reviewing new ingredients. The agency proposes to add carcinogenicity and developmental and reproductive toxicity to the required safety testing prior to approval.
Speaking at the meeting, a coalition of sunscreen manufacturers said that most of what was being requested by the FDA is redundant or not necessary. "Today’s sunscreen ingredients have a wealth of data to support their safety," said the Personal Care Products Council (PCPC) in a statement. The group also said that many of the ingredients had been used for decades, without any indication that they caused cancer or led to any significant safety issues.
Advisory committee members, however, said that their concerns were not allayed.
"If you don’t look for a signal you’re not going to find a signal," said Dr. David J. Margolis, professor of dermatology at the University of Pennsylvania, Philadelphia. Dr. Margolis said he was concerned about safety over the long term, and about perhaps too much reliance on rodent models.
"The penetration through rodent skin is different than the penetration through human skin," he said.
Dr. Geoffrey L. Rosenthal, professor of pediatrics at the University of Maryland, Baltimore, said that he was bothered by the lack of specific studies in children. "Unless you study them in kids, you just don’t know," he said.
Dr. Marissa J. Perman, a pediatric dermatologist at the Children’s Hospital of Philadelphia, agreed and said that the agency should also consider requiring studies in pregnant and lactating women.
Dr. Perman, who spoke on behalf of the American Academy of Pediatrics during the public section of the meeting, said that AAP also would like to see studies based on actual – not directed – sunscreen use and investigations of the effects of spray sunscreens and of nanoparticles on children.
Several panel members also said that they were concerned about the disconnect between real-world use of sunscreens and the amounts studied during trials. The PCPC said in its background materials that market data indicate that many people use sunscreen only once a week, and that among users, only about 22% reapply as often as directed on the label.
Some committee members said that they viewed sunscreens as drugs that should always be subject to the same review and approval process.
"It’s really shocking to me that these are not being regulated through the process by which drugs are regulated," said Lorraine Gudas, Ph.D., chairman of pharmacology at Cornell University, New York. She noted that little was known about the potential systemic effects of sunscreen ingredients.
The FDA is not required to follow the panel’s advice. Any change to how sunscreens are regulated would require issuing a new rule and going through a public comment period. An FDA official declined to say when that process might occur.
On Twitter @aliciaault
SILVER SPRING, MD. – The widespread and long-term use of sunscreens demands that manufacturers provide more long-term safety data and information on how the active ingredients affect the health of special populations including infants, children, the elderly, and those with skin conditions, according to an expert panel called by the Food and Drug Administration.
Sunscreen safety has become an increasingly more important concern, especially since they are intended to be applied over the entire body, multiple times a day, and for a lifetime, Dr. Theresa Michele, director of the FDA’s division of nonprescription clinical evaluation, said Sept. 5 at a meeting of the agency’s Nonprescription Drugs Advisory Committee. "We’re essentially talking about a cradle-to-grave product."
FDA has been under pressure from physicians, consumers, manufacturers, and Congress to approve a backlog of eight ingredients that are generally available outside of the United States and to improve the process for reviewing new ingredients. The agency proposes to add carcinogenicity and developmental and reproductive toxicity to the required safety testing prior to approval.
Speaking at the meeting, a coalition of sunscreen manufacturers said that most of what was being requested by the FDA is redundant or not necessary. "Today’s sunscreen ingredients have a wealth of data to support their safety," said the Personal Care Products Council (PCPC) in a statement. The group also said that many of the ingredients had been used for decades, without any indication that they caused cancer or led to any significant safety issues.
Advisory committee members, however, said that their concerns were not allayed.
"If you don’t look for a signal you’re not going to find a signal," said Dr. David J. Margolis, professor of dermatology at the University of Pennsylvania, Philadelphia. Dr. Margolis said he was concerned about safety over the long term, and about perhaps too much reliance on rodent models.
"The penetration through rodent skin is different than the penetration through human skin," he said.
Dr. Geoffrey L. Rosenthal, professor of pediatrics at the University of Maryland, Baltimore, said that he was bothered by the lack of specific studies in children. "Unless you study them in kids, you just don’t know," he said.
Dr. Marissa J. Perman, a pediatric dermatologist at the Children’s Hospital of Philadelphia, agreed and said that the agency should also consider requiring studies in pregnant and lactating women.
Dr. Perman, who spoke on behalf of the American Academy of Pediatrics during the public section of the meeting, said that AAP also would like to see studies based on actual – not directed – sunscreen use and investigations of the effects of spray sunscreens and of nanoparticles on children.
Several panel members also said that they were concerned about the disconnect between real-world use of sunscreens and the amounts studied during trials. The PCPC said in its background materials that market data indicate that many people use sunscreen only once a week, and that among users, only about 22% reapply as often as directed on the label.
Some committee members said that they viewed sunscreens as drugs that should always be subject to the same review and approval process.
"It’s really shocking to me that these are not being regulated through the process by which drugs are regulated," said Lorraine Gudas, Ph.D., chairman of pharmacology at Cornell University, New York. She noted that little was known about the potential systemic effects of sunscreen ingredients.
The FDA is not required to follow the panel’s advice. Any change to how sunscreens are regulated would require issuing a new rule and going through a public comment period. An FDA official declined to say when that process might occur.
On Twitter @aliciaault
SILVER SPRING, MD. – The widespread and long-term use of sunscreens demands that manufacturers provide more long-term safety data and information on how the active ingredients affect the health of special populations including infants, children, the elderly, and those with skin conditions, according to an expert panel called by the Food and Drug Administration.
Sunscreen safety has become an increasingly more important concern, especially since they are intended to be applied over the entire body, multiple times a day, and for a lifetime, Dr. Theresa Michele, director of the FDA’s division of nonprescription clinical evaluation, said Sept. 5 at a meeting of the agency’s Nonprescription Drugs Advisory Committee. "We’re essentially talking about a cradle-to-grave product."
FDA has been under pressure from physicians, consumers, manufacturers, and Congress to approve a backlog of eight ingredients that are generally available outside of the United States and to improve the process for reviewing new ingredients. The agency proposes to add carcinogenicity and developmental and reproductive toxicity to the required safety testing prior to approval.
Speaking at the meeting, a coalition of sunscreen manufacturers said that most of what was being requested by the FDA is redundant or not necessary. "Today’s sunscreen ingredients have a wealth of data to support their safety," said the Personal Care Products Council (PCPC) in a statement. The group also said that many of the ingredients had been used for decades, without any indication that they caused cancer or led to any significant safety issues.
Advisory committee members, however, said that their concerns were not allayed.
"If you don’t look for a signal you’re not going to find a signal," said Dr. David J. Margolis, professor of dermatology at the University of Pennsylvania, Philadelphia. Dr. Margolis said he was concerned about safety over the long term, and about perhaps too much reliance on rodent models.
"The penetration through rodent skin is different than the penetration through human skin," he said.
Dr. Geoffrey L. Rosenthal, professor of pediatrics at the University of Maryland, Baltimore, said that he was bothered by the lack of specific studies in children. "Unless you study them in kids, you just don’t know," he said.
Dr. Marissa J. Perman, a pediatric dermatologist at the Children’s Hospital of Philadelphia, agreed and said that the agency should also consider requiring studies in pregnant and lactating women.
Dr. Perman, who spoke on behalf of the American Academy of Pediatrics during the public section of the meeting, said that AAP also would like to see studies based on actual – not directed – sunscreen use and investigations of the effects of spray sunscreens and of nanoparticles on children.
Several panel members also said that they were concerned about the disconnect between real-world use of sunscreens and the amounts studied during trials. The PCPC said in its background materials that market data indicate that many people use sunscreen only once a week, and that among users, only about 22% reapply as often as directed on the label.
Some committee members said that they viewed sunscreens as drugs that should always be subject to the same review and approval process.
"It’s really shocking to me that these are not being regulated through the process by which drugs are regulated," said Lorraine Gudas, Ph.D., chairman of pharmacology at Cornell University, New York. She noted that little was known about the potential systemic effects of sunscreen ingredients.
The FDA is not required to follow the panel’s advice. Any change to how sunscreens are regulated would require issuing a new rule and going through a public comment period. An FDA official declined to say when that process might occur.
On Twitter @aliciaault
AT AN FDA ADVISORY COMMITTEE
FDA grants first approval of a PD-1 inhibitor, for advanced melanoma
The Food and Drug Administration has granted an accelerated approval to pembrolizumab, a PD-1 inhibitor, for the treatment of patients with advanced or unresectable melanoma that is no longer responding to other drugs.
Pembrolizumab (Keytruda; Merck) is intended for use following treatment with ipilimumab. For BRAF V600-positive patients, the drug should be used after treatment with both ipilimumab and a BRAF inhibitor, according to an FDA press statement.
The approved dose is 2 mg/kg every 3 weeks for patients with unresectable or metastatic melanoma and disease progression following the initial treatments.
According to a Merck press statement, "This indication is approved ... based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established." Additional studies are underway, and continued approval may be contingent upon verification and description of clinical benefit in the confirmatory trials, the statement noted.
The pivotal trial comprised 173 clinical trial participants with advanced melanoma whose disease progressed after prior treatment. All participants were treated with pembrolizumab, either at the recommended dose of 2 mg/kg or at a higher dose of 10 mg/kg. In the half of the participants who received pembrolizumab at the recommended dose of 2 mg/kg, approximately 24% had their tumors shrink. This effect lasted at least 1.4-8.5 months and continued beyond this period in most patients, the FDA statement said.
The drug was discontinued for adverse reactions in 6% of 89 patients who received the recommended dose of 2 mg/kg and 9% of 411 patients across all doses studied. Serious adverse reactions occurred in 36% of patients receiving pembrolizumab. The most common serious adverse reactions reported in 2% or more of patients were renal failure, dyspnea, pneumonia, and cellulitis. Adverse reactions occurring in at least 20% of the 411 patients across doses were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
The FDA cautions that pembrolizumab also has the potential for severe immune-mediated side effects. In the 411 participants with advanced melanoma, severe immune-mediated side effects involving healthy organs, including the lung, colon, hormone-producing glands, and liver, occurred uncommonly, they said.
It is the sixth drug to be approved for melanoma since 2011, joining ipilimumab (2011), peginterferon alfa-2b (2011), vemurafenib (2011), dabrafenib (2013), and trametinib (2013).
The Melanoma Research Alliance applauded FDA’s action in a press statement.
"The news of FDA’s first approval of an anti-PD-1 drug is extremely exciting and shows just how far the field has come in the last few years," said Debra Black, MRA cofounder. "When we started MRA there was little hope for melanoma patients. Today we are seeing a real sea change, with several new therapies and proof of concept that these new treatments can save lives."
The Food and Drug Administration has granted an accelerated approval to pembrolizumab, a PD-1 inhibitor, for the treatment of patients with advanced or unresectable melanoma that is no longer responding to other drugs.
Pembrolizumab (Keytruda; Merck) is intended for use following treatment with ipilimumab. For BRAF V600-positive patients, the drug should be used after treatment with both ipilimumab and a BRAF inhibitor, according to an FDA press statement.
The approved dose is 2 mg/kg every 3 weeks for patients with unresectable or metastatic melanoma and disease progression following the initial treatments.
According to a Merck press statement, "This indication is approved ... based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established." Additional studies are underway, and continued approval may be contingent upon verification and description of clinical benefit in the confirmatory trials, the statement noted.
The pivotal trial comprised 173 clinical trial participants with advanced melanoma whose disease progressed after prior treatment. All participants were treated with pembrolizumab, either at the recommended dose of 2 mg/kg or at a higher dose of 10 mg/kg. In the half of the participants who received pembrolizumab at the recommended dose of 2 mg/kg, approximately 24% had their tumors shrink. This effect lasted at least 1.4-8.5 months and continued beyond this period in most patients, the FDA statement said.
The drug was discontinued for adverse reactions in 6% of 89 patients who received the recommended dose of 2 mg/kg and 9% of 411 patients across all doses studied. Serious adverse reactions occurred in 36% of patients receiving pembrolizumab. The most common serious adverse reactions reported in 2% or more of patients were renal failure, dyspnea, pneumonia, and cellulitis. Adverse reactions occurring in at least 20% of the 411 patients across doses were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
The FDA cautions that pembrolizumab also has the potential for severe immune-mediated side effects. In the 411 participants with advanced melanoma, severe immune-mediated side effects involving healthy organs, including the lung, colon, hormone-producing glands, and liver, occurred uncommonly, they said.
It is the sixth drug to be approved for melanoma since 2011, joining ipilimumab (2011), peginterferon alfa-2b (2011), vemurafenib (2011), dabrafenib (2013), and trametinib (2013).
The Melanoma Research Alliance applauded FDA’s action in a press statement.
"The news of FDA’s first approval of an anti-PD-1 drug is extremely exciting and shows just how far the field has come in the last few years," said Debra Black, MRA cofounder. "When we started MRA there was little hope for melanoma patients. Today we are seeing a real sea change, with several new therapies and proof of concept that these new treatments can save lives."
The Food and Drug Administration has granted an accelerated approval to pembrolizumab, a PD-1 inhibitor, for the treatment of patients with advanced or unresectable melanoma that is no longer responding to other drugs.
Pembrolizumab (Keytruda; Merck) is intended for use following treatment with ipilimumab. For BRAF V600-positive patients, the drug should be used after treatment with both ipilimumab and a BRAF inhibitor, according to an FDA press statement.
The approved dose is 2 mg/kg every 3 weeks for patients with unresectable or metastatic melanoma and disease progression following the initial treatments.
According to a Merck press statement, "This indication is approved ... based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established." Additional studies are underway, and continued approval may be contingent upon verification and description of clinical benefit in the confirmatory trials, the statement noted.
The pivotal trial comprised 173 clinical trial participants with advanced melanoma whose disease progressed after prior treatment. All participants were treated with pembrolizumab, either at the recommended dose of 2 mg/kg or at a higher dose of 10 mg/kg. In the half of the participants who received pembrolizumab at the recommended dose of 2 mg/kg, approximately 24% had their tumors shrink. This effect lasted at least 1.4-8.5 months and continued beyond this period in most patients, the FDA statement said.
The drug was discontinued for adverse reactions in 6% of 89 patients who received the recommended dose of 2 mg/kg and 9% of 411 patients across all doses studied. Serious adverse reactions occurred in 36% of patients receiving pembrolizumab. The most common serious adverse reactions reported in 2% or more of patients were renal failure, dyspnea, pneumonia, and cellulitis. Adverse reactions occurring in at least 20% of the 411 patients across doses were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
The FDA cautions that pembrolizumab also has the potential for severe immune-mediated side effects. In the 411 participants with advanced melanoma, severe immune-mediated side effects involving healthy organs, including the lung, colon, hormone-producing glands, and liver, occurred uncommonly, they said.
It is the sixth drug to be approved for melanoma since 2011, joining ipilimumab (2011), peginterferon alfa-2b (2011), vemurafenib (2011), dabrafenib (2013), and trametinib (2013).
The Melanoma Research Alliance applauded FDA’s action in a press statement.
"The news of FDA’s first approval of an anti-PD-1 drug is extremely exciting and shows just how far the field has come in the last few years," said Debra Black, MRA cofounder. "When we started MRA there was little hope for melanoma patients. Today we are seeing a real sea change, with several new therapies and proof of concept that these new treatments can save lives."
VIDEO: German screening initiative catches skin cancer sooner
EDINBURGH – Is population screening for skin cancer worthwhile?
Yes, Dr. Eckhard Breitbart said in a debate at the 15th World Congress on Cancers of the Skin. "Screening is not a diagnostic procedure," he noted. But the potential benefits of screening, including a significant reduction in medical costs, outweigh the potential harms related to false negative or false positive findings, added Dr. Breitbart, a dermatologist in Hamburg, Germany.
In an interview at the meeting, Dr. Breitbart reviewed the findings from his study of the impact of a population-based skin cancer screening program in Germany, and he discussed what research is needed to support such screening elsewhere.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EDINBURGH – Is population screening for skin cancer worthwhile?
Yes, Dr. Eckhard Breitbart said in a debate at the 15th World Congress on Cancers of the Skin. "Screening is not a diagnostic procedure," he noted. But the potential benefits of screening, including a significant reduction in medical costs, outweigh the potential harms related to false negative or false positive findings, added Dr. Breitbart, a dermatologist in Hamburg, Germany.
In an interview at the meeting, Dr. Breitbart reviewed the findings from his study of the impact of a population-based skin cancer screening program in Germany, and he discussed what research is needed to support such screening elsewhere.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EDINBURGH – Is population screening for skin cancer worthwhile?
Yes, Dr. Eckhard Breitbart said in a debate at the 15th World Congress on Cancers of the Skin. "Screening is not a diagnostic procedure," he noted. But the potential benefits of screening, including a significant reduction in medical costs, outweigh the potential harms related to false negative or false positive findings, added Dr. Breitbart, a dermatologist in Hamburg, Germany.
In an interview at the meeting, Dr. Breitbart reviewed the findings from his study of the impact of a population-based skin cancer screening program in Germany, and he discussed what research is needed to support such screening elsewhere.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
FROM THE 2014 WORLD CONGRESS ON CANCERS OF THE SKINS

VIDEO: What’s your UV risk today? There’s an app for that
EDINBURGH – How would you like a smartphone app that offers a personalized sun protection strategy based on location and skin type?
At the 15th World Congress on Cancers of the Skin, Nina Goad, head of communications for the British Association of Dermatologists, discussed an app that does just that: the World UV App. The app, developed by the British Association of Dermatologists, provides daily UV forecasts based on an individual’s location via a smartphone or tablet. In addition, the app fine-tunes UV risk according to skin type.
In a video interview at the meeting, Ms. Goad discussed the app’s development and how patients can use it to improve their approach to sun protection.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EDINBURGH – How would you like a smartphone app that offers a personalized sun protection strategy based on location and skin type?
At the 15th World Congress on Cancers of the Skin, Nina Goad, head of communications for the British Association of Dermatologists, discussed an app that does just that: the World UV App. The app, developed by the British Association of Dermatologists, provides daily UV forecasts based on an individual’s location via a smartphone or tablet. In addition, the app fine-tunes UV risk according to skin type.
In a video interview at the meeting, Ms. Goad discussed the app’s development and how patients can use it to improve their approach to sun protection.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EDINBURGH – How would you like a smartphone app that offers a personalized sun protection strategy based on location and skin type?
At the 15th World Congress on Cancers of the Skin, Nina Goad, head of communications for the British Association of Dermatologists, discussed an app that does just that: the World UV App. The app, developed by the British Association of Dermatologists, provides daily UV forecasts based on an individual’s location via a smartphone or tablet. In addition, the app fine-tunes UV risk according to skin type.
In a video interview at the meeting, Ms. Goad discussed the app’s development and how patients can use it to improve their approach to sun protection.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
FROM THE 2014 WORLD CONGRESS ON CANCERS OF THE SKINS

Melanoma and Sildenafil: “Enhanced” Risk?
A recently published prospective cohort study reported that sildenafil use may increase the risk for melanoma (JAMA Intern Med. 2014;174:964-970). BRAF activation, which is pathogenic in some melanoma variants, downregulates phosphodiesterase 5A and sildenafil downregulates phosphodiesterase 5A, surmising that either one may enhance melanoma invasion. The Health Professionals’ Follow-up Study cohort was utilized, which has prospectively evaluated male health professionals’ nutrition and incidences of serious illnesses since 1986. In 2000, more than 25,000 men were interviewed about sildenafil use for erectile dysfunction, and the incidence of skin cancer was obtained by questionnaire every 2 years for 10 years. The questionnaire showed that 142 melanomas were diagnosed, and recent or prior sildenafil use (with no breakdown of frequency of dosing) was significantly associated with increased risk for melanoma (hazard ratio, 1.84 for recent use; 1.92 for ever use) adjusted for age, erectile dysfunction without sildenafil, and several skin-related and genetic melanoma risk factors. No other types of skin cancer exhibited this risk trend.
What’s the issue?
Vascular tone and manipulation of such is a contender as a hot topic in melanoma given that even aspirin has been implicated as a risk factor. Unfortunately, similar to the aspirin data, without a true continuum providing any sildenafil dosage or frequency relationship to the development of melanoma, especially for this very short half-life medication, we likely cannot consider sildenafil as a hazard in patients at high risk for melanoma as we do for smokers and oral contraceptives, or alcoholics and terbinafine. Because UV radiation is the only behavioral risk factor linked to melanoma and considering that so many of our male patients take this class of drug, in your opinion what percentage of your patients in this risk category follow strict sun protection? Should we mention this potential association to them?
A recently published prospective cohort study reported that sildenafil use may increase the risk for melanoma (JAMA Intern Med. 2014;174:964-970). BRAF activation, which is pathogenic in some melanoma variants, downregulates phosphodiesterase 5A and sildenafil downregulates phosphodiesterase 5A, surmising that either one may enhance melanoma invasion. The Health Professionals’ Follow-up Study cohort was utilized, which has prospectively evaluated male health professionals’ nutrition and incidences of serious illnesses since 1986. In 2000, more than 25,000 men were interviewed about sildenafil use for erectile dysfunction, and the incidence of skin cancer was obtained by questionnaire every 2 years for 10 years. The questionnaire showed that 142 melanomas were diagnosed, and recent or prior sildenafil use (with no breakdown of frequency of dosing) was significantly associated with increased risk for melanoma (hazard ratio, 1.84 for recent use; 1.92 for ever use) adjusted for age, erectile dysfunction without sildenafil, and several skin-related and genetic melanoma risk factors. No other types of skin cancer exhibited this risk trend.
What’s the issue?
Vascular tone and manipulation of such is a contender as a hot topic in melanoma given that even aspirin has been implicated as a risk factor. Unfortunately, similar to the aspirin data, without a true continuum providing any sildenafil dosage or frequency relationship to the development of melanoma, especially for this very short half-life medication, we likely cannot consider sildenafil as a hazard in patients at high risk for melanoma as we do for smokers and oral contraceptives, or alcoholics and terbinafine. Because UV radiation is the only behavioral risk factor linked to melanoma and considering that so many of our male patients take this class of drug, in your opinion what percentage of your patients in this risk category follow strict sun protection? Should we mention this potential association to them?
A recently published prospective cohort study reported that sildenafil use may increase the risk for melanoma (JAMA Intern Med. 2014;174:964-970). BRAF activation, which is pathogenic in some melanoma variants, downregulates phosphodiesterase 5A and sildenafil downregulates phosphodiesterase 5A, surmising that either one may enhance melanoma invasion. The Health Professionals’ Follow-up Study cohort was utilized, which has prospectively evaluated male health professionals’ nutrition and incidences of serious illnesses since 1986. In 2000, more than 25,000 men were interviewed about sildenafil use for erectile dysfunction, and the incidence of skin cancer was obtained by questionnaire every 2 years for 10 years. The questionnaire showed that 142 melanomas were diagnosed, and recent or prior sildenafil use (with no breakdown of frequency of dosing) was significantly associated with increased risk for melanoma (hazard ratio, 1.84 for recent use; 1.92 for ever use) adjusted for age, erectile dysfunction without sildenafil, and several skin-related and genetic melanoma risk factors. No other types of skin cancer exhibited this risk trend.
What’s the issue?
Vascular tone and manipulation of such is a contender as a hot topic in melanoma given that even aspirin has been implicated as a risk factor. Unfortunately, similar to the aspirin data, without a true continuum providing any sildenafil dosage or frequency relationship to the development of melanoma, especially for this very short half-life medication, we likely cannot consider sildenafil as a hazard in patients at high risk for melanoma as we do for smokers and oral contraceptives, or alcoholics and terbinafine. Because UV radiation is the only behavioral risk factor linked to melanoma and considering that so many of our male patients take this class of drug, in your opinion what percentage of your patients in this risk category follow strict sun protection? Should we mention this potential association to them?

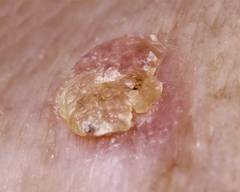
Photodynamic therapy clears thin AKs better than cryotherapy
Thin actinic keratoses on the face or scalp were 14% more likely to clear completely in patients treated with photodynamic therapy, compared with cryotherapy, in a meta-analysis of four studies including 641 patients.
Complete clearance 3 months after treatment was significantly more likely in the 2,170 actinic keratoses treated by photodynamic therapy (PDT), compared with 2,174 actinic keratoses treated by cryotherapy, Dr. Gayatri Patel and her associates reported.
The data came from randomized trials with 10 or more participants in which the PDT used topical aminolevulinic acid hydrochloride or methyl aminolevulinate hydrochloride, the most widely available PDT stabilizers in North America and Europe. Methyl aminolevulinate recently was withdrawn from the U.S. market but remains common in Europe, noted Dr. Patel of the University of California, Davis, and her associates.
The study results were published online in JAMA Dermatology (2014 Aug. 27 [doi:10.1001/jamadermatol.2014.1253]).
The results suggested that PDT works better on thinner actinic keratoses. Grade 1 (thin) actinic keratoses on the face or scalp were 86% more likely to clear by 12 weeks after PDT, compared with cryotherapy, the investigators reported.

Only one of the fours studies found higher efficacy rates for cryotherapy, compared with PDT, and more than 60% of lesions in that study were grade 2 (moderately thick, easily felt) or grade 3 (very thick and/or obvious) actinic keratoses. The other three studies in the meta-analysis excluded thicker lesions or favored thinner ones, the researchers noted.
They excluded from the meta-analysis two other studies that compared PDT with cryotherapy for actinic keratoses because of incompatible follow-up times. They reviewed 13 studies in all, including studies involving treatment of actinic keratosis with imiquimod, fluorouracil, or carbon dioxide laser, but could not meta-analyze data on these other treatments because of different outcome measures and follow-up times or lack of a comparator.
Photosensitivity, pain, erythema, and pruritus were common after PDT. Cryotherapy induced pain and pruritus, but at lower rates than did PDT. Hypopigmentation occurred in 33% of patients after cryotherapy and in 9% after PDT in one study.
Satisfaction ratings by patients and unblinded investigators tended to favor PDT over cryotherapy, perhaps because PDT may produce ancillary cosmetic improvements when treating actinic keratosis, Dr. Patel and her associates speculated.
The findings were limited by the poor quality of the studies, which were lacking double-blind design and description of randomization methods, but no sources of bias were evident, and the large number of patients and relatively similar treatment locations were strengths of the analysis, they said.
Dr. Patel reported having no financial disclosures.
On Twitter @sherryboschert
Several limitations associated with photodynamic therapy (PDT) make cryotherapy the first-line treatment choice for most practicing dermatologists, Dr. Harvey Lui commented in an article that accompanied Dr. Patel’s report.
Dr. Patel’s meta-analysis found a 14% better chance of complete clearance of actinic keratosis lesions, compared with cryotherapy, but the data are not clear enough to claim better cosmesis or patient acceptance, Dr. Lui said (JAMA Dermatol. 2014 Aug. 27 [doi:10.1001/jamadermatol.2014.1869]).

|
|
PDT costs more in time and equipment than cryotherapy. PDT may seem simple, but achieving optimal results can require longer drug incubation times and/or light-dose fractionation. Local pain is a bigger problem with PDT that requires anticipation and management by clinicians, he said. Cryotherapy, on the other hand, requires relatively brief outpatient visits.
The future of PDT for actinic keratosis may lie in further development of a currently off-label treatment – exposure to ambient outdoor light after application of topical aminolevulinic acid, Dr. Lui suggested: "Perhaps the most tantalizing irony of daylight PDT is the specter of treating a solar-induced neoplasm with sunlight itself."
Dr. Lui is head of the department of dermatology and skin science at the University of British Columbia in Vancouver. He disclosed financial associations with Galderma, LEO Pharma, Janssen, Novartis, Valeant Pharmaceuticals, RepliCel Life Sciences, Lumen Health Technologies, and Verisante Technology.
Several limitations associated with photodynamic therapy (PDT) make cryotherapy the first-line treatment choice for most practicing dermatologists, Dr. Harvey Lui commented in an article that accompanied Dr. Patel’s report.
Dr. Patel’s meta-analysis found a 14% better chance of complete clearance of actinic keratosis lesions, compared with cryotherapy, but the data are not clear enough to claim better cosmesis or patient acceptance, Dr. Lui said (JAMA Dermatol. 2014 Aug. 27 [doi:10.1001/jamadermatol.2014.1869]).
|
|
|
PDT costs more in time and equipment than cryotherapy. PDT may seem simple, but achieving optimal results can require longer drug incubation times and/or light-dose fractionation. Local pain is a bigger problem with PDT that requires anticipation and management by clinicians, he said. Cryotherapy, on the other hand, requires relatively brief outpatient visits.
The future of PDT for actinic keratosis may lie in further development of a currently off-label treatment – exposure to ambient outdoor light after application of topical aminolevulinic acid, Dr. Lui suggested: "Perhaps the most tantalizing irony of daylight PDT is the specter of treating a solar-induced neoplasm with sunlight itself."
Dr. Lui is head of the department of dermatology and skin science at the University of British Columbia in Vancouver. He disclosed financial associations with Galderma, LEO Pharma, Janssen, Novartis, Valeant Pharmaceuticals, RepliCel Life Sciences, Lumen Health Technologies, and Verisante Technology.
Several limitations associated with photodynamic therapy (PDT) make cryotherapy the first-line treatment choice for most practicing dermatologists, Dr. Harvey Lui commented in an article that accompanied Dr. Patel’s report.
Dr. Patel’s meta-analysis found a 14% better chance of complete clearance of actinic keratosis lesions, compared with cryotherapy, but the data are not clear enough to claim better cosmesis or patient acceptance, Dr. Lui said (JAMA Dermatol. 2014 Aug. 27 [doi:10.1001/jamadermatol.2014.1869]).
|
|
|
PDT costs more in time and equipment than cryotherapy. PDT may seem simple, but achieving optimal results can require longer drug incubation times and/or light-dose fractionation. Local pain is a bigger problem with PDT that requires anticipation and management by clinicians, he said. Cryotherapy, on the other hand, requires relatively brief outpatient visits.
The future of PDT for actinic keratosis may lie in further development of a currently off-label treatment – exposure to ambient outdoor light after application of topical aminolevulinic acid, Dr. Lui suggested: "Perhaps the most tantalizing irony of daylight PDT is the specter of treating a solar-induced neoplasm with sunlight itself."
Dr. Lui is head of the department of dermatology and skin science at the University of British Columbia in Vancouver. He disclosed financial associations with Galderma, LEO Pharma, Janssen, Novartis, Valeant Pharmaceuticals, RepliCel Life Sciences, Lumen Health Technologies, and Verisante Technology.
Thin actinic keratoses on the face or scalp were 14% more likely to clear completely in patients treated with photodynamic therapy, compared with cryotherapy, in a meta-analysis of four studies including 641 patients.
Complete clearance 3 months after treatment was significantly more likely in the 2,170 actinic keratoses treated by photodynamic therapy (PDT), compared with 2,174 actinic keratoses treated by cryotherapy, Dr. Gayatri Patel and her associates reported.
The data came from randomized trials with 10 or more participants in which the PDT used topical aminolevulinic acid hydrochloride or methyl aminolevulinate hydrochloride, the most widely available PDT stabilizers in North America and Europe. Methyl aminolevulinate recently was withdrawn from the U.S. market but remains common in Europe, noted Dr. Patel of the University of California, Davis, and her associates.
The study results were published online in JAMA Dermatology (2014 Aug. 27 [doi:10.1001/jamadermatol.2014.1253]).
The results suggested that PDT works better on thinner actinic keratoses. Grade 1 (thin) actinic keratoses on the face or scalp were 86% more likely to clear by 12 weeks after PDT, compared with cryotherapy, the investigators reported.
Only one of the fours studies found higher efficacy rates for cryotherapy, compared with PDT, and more than 60% of lesions in that study were grade 2 (moderately thick, easily felt) or grade 3 (very thick and/or obvious) actinic keratoses. The other three studies in the meta-analysis excluded thicker lesions or favored thinner ones, the researchers noted.
They excluded from the meta-analysis two other studies that compared PDT with cryotherapy for actinic keratoses because of incompatible follow-up times. They reviewed 13 studies in all, including studies involving treatment of actinic keratosis with imiquimod, fluorouracil, or carbon dioxide laser, but could not meta-analyze data on these other treatments because of different outcome measures and follow-up times or lack of a comparator.
Photosensitivity, pain, erythema, and pruritus were common after PDT. Cryotherapy induced pain and pruritus, but at lower rates than did PDT. Hypopigmentation occurred in 33% of patients after cryotherapy and in 9% after PDT in one study.
Satisfaction ratings by patients and unblinded investigators tended to favor PDT over cryotherapy, perhaps because PDT may produce ancillary cosmetic improvements when treating actinic keratosis, Dr. Patel and her associates speculated.
The findings were limited by the poor quality of the studies, which were lacking double-blind design and description of randomization methods, but no sources of bias were evident, and the large number of patients and relatively similar treatment locations were strengths of the analysis, they said.
Dr. Patel reported having no financial disclosures.
On Twitter @sherryboschert
Thin actinic keratoses on the face or scalp were 14% more likely to clear completely in patients treated with photodynamic therapy, compared with cryotherapy, in a meta-analysis of four studies including 641 patients.
Complete clearance 3 months after treatment was significantly more likely in the 2,170 actinic keratoses treated by photodynamic therapy (PDT), compared with 2,174 actinic keratoses treated by cryotherapy, Dr. Gayatri Patel and her associates reported.
The data came from randomized trials with 10 or more participants in which the PDT used topical aminolevulinic acid hydrochloride or methyl aminolevulinate hydrochloride, the most widely available PDT stabilizers in North America and Europe. Methyl aminolevulinate recently was withdrawn from the U.S. market but remains common in Europe, noted Dr. Patel of the University of California, Davis, and her associates.
The study results were published online in JAMA Dermatology (2014 Aug. 27 [doi:10.1001/jamadermatol.2014.1253]).
The results suggested that PDT works better on thinner actinic keratoses. Grade 1 (thin) actinic keratoses on the face or scalp were 86% more likely to clear by 12 weeks after PDT, compared with cryotherapy, the investigators reported.
Only one of the fours studies found higher efficacy rates for cryotherapy, compared with PDT, and more than 60% of lesions in that study were grade 2 (moderately thick, easily felt) or grade 3 (very thick and/or obvious) actinic keratoses. The other three studies in the meta-analysis excluded thicker lesions or favored thinner ones, the researchers noted.
They excluded from the meta-analysis two other studies that compared PDT with cryotherapy for actinic keratoses because of incompatible follow-up times. They reviewed 13 studies in all, including studies involving treatment of actinic keratosis with imiquimod, fluorouracil, or carbon dioxide laser, but could not meta-analyze data on these other treatments because of different outcome measures and follow-up times or lack of a comparator.
Photosensitivity, pain, erythema, and pruritus were common after PDT. Cryotherapy induced pain and pruritus, but at lower rates than did PDT. Hypopigmentation occurred in 33% of patients after cryotherapy and in 9% after PDT in one study.
Satisfaction ratings by patients and unblinded investigators tended to favor PDT over cryotherapy, perhaps because PDT may produce ancillary cosmetic improvements when treating actinic keratosis, Dr. Patel and her associates speculated.
The findings were limited by the poor quality of the studies, which were lacking double-blind design and description of randomization methods, but no sources of bias were evident, and the large number of patients and relatively similar treatment locations were strengths of the analysis, they said.
Dr. Patel reported having no financial disclosures.
On Twitter @sherryboschert
FROM JAMA DERMATOLOGY
Key clinical point: Clearance of thin actinic keratosis lesions on the face or head is more likely with photodynamic therapy vs. cryotherapy, but the impact of either treatment on reducing the incidence of squamous cell carcinomas remains unknown.
Major finding: Clearance was 14% more likely at 3 months after PDT, compared with cryotherapy.
Data source: Meta-analysis of four studies including 641 patients with 2,170 actinic keratosis lesions treated by PDT and 2,174 treated by cryotherapy.
Disclosures: Dr. Patel reported having no financial disclosures.
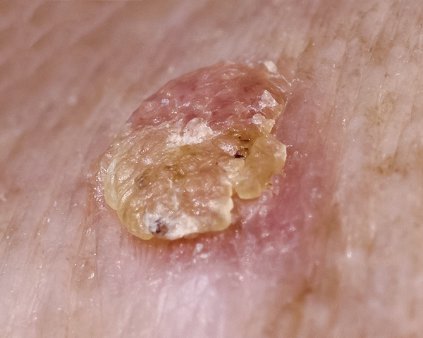
Field therapy preferred when treating actinic keratoses
VANCOUVER, B.C. – In the clinical opinion of Dr. Mariusz Sapijaszko, treating actinic keratosis without field therapy creates a disadvantage "because this is not an individual lesion disease," he maintained at the annual meeting of the Pacific Dermatologic Association.
Actinic keratosis "is a field concept disease. I tell patients ‘the sun has not been shining only on your left temple. It’s been shining all over your face and scalp, neck, and arms. ... It’s time to start looking after your skin with sun protection and lesion-directed field therapies.’"

An estimated 11% of all dermatologic visits in the United States are for actinic keratosis (AK) and "we worry about it because the natural course of AK lesions is unpredictable," said Dr. Sapijaszko of the Western Canada Dermatology Institute, Edmonton, Alta. It’s not easy to predict which lesions will progress to in situ or invasive squamous cell carcinomas (SCCs).
An estimated 40%-80% of cutaneous SCCs arise from, or near, an AK lesion, which supports the concept of field UV damage. AK lesions may persist, regress, or progress, depending on the patient’s immune status. Some lesions that regress will recur, from 32% within 1 year to 92% within 5 years. Progression can lead to hypertrophic AKs, in situ SCC, or invasive SCC. It can be difficult to distinguish AKs and early forms of SCC or even other nonmelanoma skin cancers, "so it’s important to treat all AKs," Dr. Sapijaszko said. Lesions that can progress to SCC include those that are hyperkeratotic, painful, have atypical features such as broader or deeper presentations, as well as those difficult to clear with standard therapies and those that occur in immunocompromised patients.
Locally destructive, mechanical ways to treat AKs include liquid nitrogen cryosurgery, electrodessication and curettage, and excision. "All of these treatments are highly operator dependent, because clearly if you use liquid nitrogen cryotherapy enough you will destroy that lesion but you will not destroy the surrounding DNA damage that has been present," he said.
Field-directed therapies, however, provide an opportunity for a more complete treatment effect. Options include 5-FU (5-fluorouracil), imiquimod, ingenol mebutate, and photodynamic therapy as well as chemical peels and laser resurfacing. Chemical peels and laser resurfacing "have less robust data, but they’re operator dependent, because you can do laser resurfacing with 100 microns or 300 microns," Dr. Sapijaszko said.
"That can depend on the technique you use, and the laser you use, and the patient in front of you." Some of the field treatment options are easier to apply than others. For example, 5-FU is applied twice daily, while imiquimod is applied twice weekly; yet all boast complete clearance rates in the 40%-50% range. "Side effects are also similar between these agents," he said. "Pain is not a big issue except with 5-FU; some patients experience a significant burning sensation."
The newest approved field therapy option, ingenol mebutate, has a dual mechanism of action: it causes cell death within 24 hours and it has been shown to reduce the number of UV-induced mutated p53 patches in mice. "This is important because we’re not just treating the lesions that we see, we want to treat the molecular changes that lead to the actual problem," Dr. Sapijaszko said. "Having decreased mutations is a huge advantage. Direct cell death leads to secondary inflammation. The immune response is characterized by cytokine release and activation of endothelial cells, leading to infiltration of lymphocytes and neutrophils, which contributes to clearance of tumor cells."
Before ingenol mebutate came on the market, investigators randomized patients with AKs to one of three treatment groups: imiquimod 5% cream, 5-FU 5% ointment, or cryotherapy (Br. J. Dermatol. 2007; 157 [suppl. 2]:34-40). Compared with their counterparts, patients in the imiquimod group fared significantly better in terms of sustained clearance of cleared lesions (73% vs. 54% in the 5-FU group, vs. 28% in the cryosurgery group (P less than .01).
"I wish we had comparative data to ingenol mebutate, but to me, of these three modalities, imiquimod stands out as the favorite," Dr. Sapijaszko said.
He went on to note that combining the available mechanical and field treatments for AK simultaneously or sequentially can lead to optimal outcomes. "Combination therapy, in particular cryotherapy, has been successfully used with a variety of topicals and has been shown to be highly advantageous, compared with placebo or to some of these agents alone," he said. "In addition, cryotherapy can be used with PDT, chemical peels, and laser resurfacing. Almost nobody in my practice gets one treatment, unless it’s a single individual lesion. Everybody gets recommendations on appropriate sun protection – just being sun smart."
Dr. Sapijaszko disclosed that he has received honoraria from and is an advisor to Galderma, Leo Pharma, and Valeant.
On Twitter @dougbrunk
VANCOUVER, B.C. – In the clinical opinion of Dr. Mariusz Sapijaszko, treating actinic keratosis without field therapy creates a disadvantage "because this is not an individual lesion disease," he maintained at the annual meeting of the Pacific Dermatologic Association.
Actinic keratosis "is a field concept disease. I tell patients ‘the sun has not been shining only on your left temple. It’s been shining all over your face and scalp, neck, and arms. ... It’s time to start looking after your skin with sun protection and lesion-directed field therapies.’"
An estimated 11% of all dermatologic visits in the United States are for actinic keratosis (AK) and "we worry about it because the natural course of AK lesions is unpredictable," said Dr. Sapijaszko of the Western Canada Dermatology Institute, Edmonton, Alta. It’s not easy to predict which lesions will progress to in situ or invasive squamous cell carcinomas (SCCs).
An estimated 40%-80% of cutaneous SCCs arise from, or near, an AK lesion, which supports the concept of field UV damage. AK lesions may persist, regress, or progress, depending on the patient’s immune status. Some lesions that regress will recur, from 32% within 1 year to 92% within 5 years. Progression can lead to hypertrophic AKs, in situ SCC, or invasive SCC. It can be difficult to distinguish AKs and early forms of SCC or even other nonmelanoma skin cancers, "so it’s important to treat all AKs," Dr. Sapijaszko said. Lesions that can progress to SCC include those that are hyperkeratotic, painful, have atypical features such as broader or deeper presentations, as well as those difficult to clear with standard therapies and those that occur in immunocompromised patients.
Locally destructive, mechanical ways to treat AKs include liquid nitrogen cryosurgery, electrodessication and curettage, and excision. "All of these treatments are highly operator dependent, because clearly if you use liquid nitrogen cryotherapy enough you will destroy that lesion but you will not destroy the surrounding DNA damage that has been present," he said.
Field-directed therapies, however, provide an opportunity for a more complete treatment effect. Options include 5-FU (5-fluorouracil), imiquimod, ingenol mebutate, and photodynamic therapy as well as chemical peels and laser resurfacing. Chemical peels and laser resurfacing "have less robust data, but they’re operator dependent, because you can do laser resurfacing with 100 microns or 300 microns," Dr. Sapijaszko said.
"That can depend on the technique you use, and the laser you use, and the patient in front of you." Some of the field treatment options are easier to apply than others. For example, 5-FU is applied twice daily, while imiquimod is applied twice weekly; yet all boast complete clearance rates in the 40%-50% range. "Side effects are also similar between these agents," he said. "Pain is not a big issue except with 5-FU; some patients experience a significant burning sensation."
The newest approved field therapy option, ingenol mebutate, has a dual mechanism of action: it causes cell death within 24 hours and it has been shown to reduce the number of UV-induced mutated p53 patches in mice. "This is important because we’re not just treating the lesions that we see, we want to treat the molecular changes that lead to the actual problem," Dr. Sapijaszko said. "Having decreased mutations is a huge advantage. Direct cell death leads to secondary inflammation. The immune response is characterized by cytokine release and activation of endothelial cells, leading to infiltration of lymphocytes and neutrophils, which contributes to clearance of tumor cells."
Before ingenol mebutate came on the market, investigators randomized patients with AKs to one of three treatment groups: imiquimod 5% cream, 5-FU 5% ointment, or cryotherapy (Br. J. Dermatol. 2007; 157 [suppl. 2]:34-40). Compared with their counterparts, patients in the imiquimod group fared significantly better in terms of sustained clearance of cleared lesions (73% vs. 54% in the 5-FU group, vs. 28% in the cryosurgery group (P less than .01).
"I wish we had comparative data to ingenol mebutate, but to me, of these three modalities, imiquimod stands out as the favorite," Dr. Sapijaszko said.
He went on to note that combining the available mechanical and field treatments for AK simultaneously or sequentially can lead to optimal outcomes. "Combination therapy, in particular cryotherapy, has been successfully used with a variety of topicals and has been shown to be highly advantageous, compared with placebo or to some of these agents alone," he said. "In addition, cryotherapy can be used with PDT, chemical peels, and laser resurfacing. Almost nobody in my practice gets one treatment, unless it’s a single individual lesion. Everybody gets recommendations on appropriate sun protection – just being sun smart."
Dr. Sapijaszko disclosed that he has received honoraria from and is an advisor to Galderma, Leo Pharma, and Valeant.
On Twitter @dougbrunk
VANCOUVER, B.C. – In the clinical opinion of Dr. Mariusz Sapijaszko, treating actinic keratosis without field therapy creates a disadvantage "because this is not an individual lesion disease," he maintained at the annual meeting of the Pacific Dermatologic Association.
Actinic keratosis "is a field concept disease. I tell patients ‘the sun has not been shining only on your left temple. It’s been shining all over your face and scalp, neck, and arms. ... It’s time to start looking after your skin with sun protection and lesion-directed field therapies.’"
An estimated 11% of all dermatologic visits in the United States are for actinic keratosis (AK) and "we worry about it because the natural course of AK lesions is unpredictable," said Dr. Sapijaszko of the Western Canada Dermatology Institute, Edmonton, Alta. It’s not easy to predict which lesions will progress to in situ or invasive squamous cell carcinomas (SCCs).
An estimated 40%-80% of cutaneous SCCs arise from, or near, an AK lesion, which supports the concept of field UV damage. AK lesions may persist, regress, or progress, depending on the patient’s immune status. Some lesions that regress will recur, from 32% within 1 year to 92% within 5 years. Progression can lead to hypertrophic AKs, in situ SCC, or invasive SCC. It can be difficult to distinguish AKs and early forms of SCC or even other nonmelanoma skin cancers, "so it’s important to treat all AKs," Dr. Sapijaszko said. Lesions that can progress to SCC include those that are hyperkeratotic, painful, have atypical features such as broader or deeper presentations, as well as those difficult to clear with standard therapies and those that occur in immunocompromised patients.
Locally destructive, mechanical ways to treat AKs include liquid nitrogen cryosurgery, electrodessication and curettage, and excision. "All of these treatments are highly operator dependent, because clearly if you use liquid nitrogen cryotherapy enough you will destroy that lesion but you will not destroy the surrounding DNA damage that has been present," he said.
Field-directed therapies, however, provide an opportunity for a more complete treatment effect. Options include 5-FU (5-fluorouracil), imiquimod, ingenol mebutate, and photodynamic therapy as well as chemical peels and laser resurfacing. Chemical peels and laser resurfacing "have less robust data, but they’re operator dependent, because you can do laser resurfacing with 100 microns or 300 microns," Dr. Sapijaszko said.
"That can depend on the technique you use, and the laser you use, and the patient in front of you." Some of the field treatment options are easier to apply than others. For example, 5-FU is applied twice daily, while imiquimod is applied twice weekly; yet all boast complete clearance rates in the 40%-50% range. "Side effects are also similar between these agents," he said. "Pain is not a big issue except with 5-FU; some patients experience a significant burning sensation."
The newest approved field therapy option, ingenol mebutate, has a dual mechanism of action: it causes cell death within 24 hours and it has been shown to reduce the number of UV-induced mutated p53 patches in mice. "This is important because we’re not just treating the lesions that we see, we want to treat the molecular changes that lead to the actual problem," Dr. Sapijaszko said. "Having decreased mutations is a huge advantage. Direct cell death leads to secondary inflammation. The immune response is characterized by cytokine release and activation of endothelial cells, leading to infiltration of lymphocytes and neutrophils, which contributes to clearance of tumor cells."
Before ingenol mebutate came on the market, investigators randomized patients with AKs to one of three treatment groups: imiquimod 5% cream, 5-FU 5% ointment, or cryotherapy (Br. J. Dermatol. 2007; 157 [suppl. 2]:34-40). Compared with their counterparts, patients in the imiquimod group fared significantly better in terms of sustained clearance of cleared lesions (73% vs. 54% in the 5-FU group, vs. 28% in the cryosurgery group (P less than .01).
"I wish we had comparative data to ingenol mebutate, but to me, of these three modalities, imiquimod stands out as the favorite," Dr. Sapijaszko said.
He went on to note that combining the available mechanical and field treatments for AK simultaneously or sequentially can lead to optimal outcomes. "Combination therapy, in particular cryotherapy, has been successfully used with a variety of topicals and has been shown to be highly advantageous, compared with placebo or to some of these agents alone," he said. "In addition, cryotherapy can be used with PDT, chemical peels, and laser resurfacing. Almost nobody in my practice gets one treatment, unless it’s a single individual lesion. Everybody gets recommendations on appropriate sun protection – just being sun smart."
Dr. Sapijaszko disclosed that he has received honoraria from and is an advisor to Galderma, Leo Pharma, and Valeant.
On Twitter @dougbrunk
EXPERT ANALYSIS AT THE PDA ANNUAL MEETING

Psoriasis patients post above-average cancer rates
CHICAGO – Malignancy rates in patients with psoriasis outstrip those in the general population, based on data from a retrospective analysis of commercial claims.
Rates for all cancers were similar among patients undergoing different psoriasis treatments, with the exception of nonmelanoma skin cancer and lymphoma.

Rates for these two cancers were more variable across treatment groups, but were still above those in the general public, Dr. Alexa B. Kimball reported at the American Academy of Dermatology summer meeting.
The increased cancer risk may be associated with chronic inflammation, a hallmark of psoriasis, and exposure to some psoriasis therapies such as phototherapy with psoralen plus ultraviolet, cyclosporine, and methotrexate, she noted in the study’s background information.
Previous studies also have suggested that patients with psoriasis may be at increased risk for some cancers such as respiratory tract, urinary tract, and liver cancers, non-Hodgkin’s lymphoma, and skin cancers.
To evaluate the incidence of malignancy, the investigators obtained data from the MarketScan Commercial and Medicare Supplemental claims databases for patients with a diagnosis of psoriasis on or before Dec. 31, 2006, and at least one prescription claim for etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), ustekinumab (Stelara), nonbiologic therapies, or phototherapy.
The general population cohort included patients at least 18 years old as of 2005 or at enrollment in the health care plan. Both cohorts had 12 months’ continuous enrollment in the health care plan from Jan. 1, 2005, through Dec. 31, 2006.
Follow-up for a patient ended at the first cancer event, disenrollment from the health care plan, or after 5 years from the index date.
Patients with psoriasis had a 5-year malignancy rate of 115.5 cases/10,000 person-years, compared with 96/10,000 person-years for the general population, reported Dr. Kimball of Massachusetts General Hospital, Boston.
Excluding nonmelanoma skin cancer and lymphoma, incidence rates were similar across treatment groups: etanercept (100.2/10,000), adalimumab (94.6/10,000), infliximab (138.1/10,000), ustekinumab (100.6/10,000), nonbiologics (116.8/10,000), and phototherapy (117.3/10,000).
"These large database queries continue to be reassuring that most systemic therapies are not changing the risk for common cancers, excluding lymphoma and skin cancer, which we continue to examine separately," Dr. Kimball said in an interview.
Nonmelanoma skin cancer was far and away the most common malignancy in psoriasis patients, occurring at a rate of 147.2/10,000 person-years vs. 94.2/10,000 person-years among the general public. Rates were highest in patients treated with adalimumab (234.2/10,000 person-years) and ustekinumab (233.3/10,000) and lowest in those treated with etanercept (155.9/10,000).
"Even with the large size of the database, the number of skin cancers remains small, so making conclusions about specific therapies may be premature," she said.
Incidence rates of lymphoma were considerably lower, but again higher in psoriasis patients than the general public (11.1/10,000 vs. 6.6/10,000). Rates of this hematologic cancer were highest with ustekinumab (25.1/10,000) and, once again, lowest with etanercept (6.9/10,000), according to the poster.
In all, 5,857 patients received nonbiologic therapies, 6,856 received etanercept, 3,314 adalimumab, 1,044 infliximab, 526 ustekinumab, and 5,156 were treated with phototherapy.
Dr. Kimball is a consultant for several pharmaceutical companies including Amgen, the study sponsor.
CHICAGO – Malignancy rates in patients with psoriasis outstrip those in the general population, based on data from a retrospective analysis of commercial claims.
Rates for all cancers were similar among patients undergoing different psoriasis treatments, with the exception of nonmelanoma skin cancer and lymphoma.
Rates for these two cancers were more variable across treatment groups, but were still above those in the general public, Dr. Alexa B. Kimball reported at the American Academy of Dermatology summer meeting.
The increased cancer risk may be associated with chronic inflammation, a hallmark of psoriasis, and exposure to some psoriasis therapies such as phototherapy with psoralen plus ultraviolet, cyclosporine, and methotrexate, she noted in the study’s background information.
Previous studies also have suggested that patients with psoriasis may be at increased risk for some cancers such as respiratory tract, urinary tract, and liver cancers, non-Hodgkin’s lymphoma, and skin cancers.
To evaluate the incidence of malignancy, the investigators obtained data from the MarketScan Commercial and Medicare Supplemental claims databases for patients with a diagnosis of psoriasis on or before Dec. 31, 2006, and at least one prescription claim for etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), ustekinumab (Stelara), nonbiologic therapies, or phototherapy.
The general population cohort included patients at least 18 years old as of 2005 or at enrollment in the health care plan. Both cohorts had 12 months’ continuous enrollment in the health care plan from Jan. 1, 2005, through Dec. 31, 2006.
Follow-up for a patient ended at the first cancer event, disenrollment from the health care plan, or after 5 years from the index date.
Patients with psoriasis had a 5-year malignancy rate of 115.5 cases/10,000 person-years, compared with 96/10,000 person-years for the general population, reported Dr. Kimball of Massachusetts General Hospital, Boston.
Excluding nonmelanoma skin cancer and lymphoma, incidence rates were similar across treatment groups: etanercept (100.2/10,000), adalimumab (94.6/10,000), infliximab (138.1/10,000), ustekinumab (100.6/10,000), nonbiologics (116.8/10,000), and phototherapy (117.3/10,000).
"These large database queries continue to be reassuring that most systemic therapies are not changing the risk for common cancers, excluding lymphoma and skin cancer, which we continue to examine separately," Dr. Kimball said in an interview.
Nonmelanoma skin cancer was far and away the most common malignancy in psoriasis patients, occurring at a rate of 147.2/10,000 person-years vs. 94.2/10,000 person-years among the general public. Rates were highest in patients treated with adalimumab (234.2/10,000 person-years) and ustekinumab (233.3/10,000) and lowest in those treated with etanercept (155.9/10,000).
"Even with the large size of the database, the number of skin cancers remains small, so making conclusions about specific therapies may be premature," she said.
Incidence rates of lymphoma were considerably lower, but again higher in psoriasis patients than the general public (11.1/10,000 vs. 6.6/10,000). Rates of this hematologic cancer were highest with ustekinumab (25.1/10,000) and, once again, lowest with etanercept (6.9/10,000), according to the poster.
In all, 5,857 patients received nonbiologic therapies, 6,856 received etanercept, 3,314 adalimumab, 1,044 infliximab, 526 ustekinumab, and 5,156 were treated with phototherapy.
Dr. Kimball is a consultant for several pharmaceutical companies including Amgen, the study sponsor.
CHICAGO – Malignancy rates in patients with psoriasis outstrip those in the general population, based on data from a retrospective analysis of commercial claims.
Rates for all cancers were similar among patients undergoing different psoriasis treatments, with the exception of nonmelanoma skin cancer and lymphoma.
Rates for these two cancers were more variable across treatment groups, but were still above those in the general public, Dr. Alexa B. Kimball reported at the American Academy of Dermatology summer meeting.
The increased cancer risk may be associated with chronic inflammation, a hallmark of psoriasis, and exposure to some psoriasis therapies such as phototherapy with psoralen plus ultraviolet, cyclosporine, and methotrexate, she noted in the study’s background information.
Previous studies also have suggested that patients with psoriasis may be at increased risk for some cancers such as respiratory tract, urinary tract, and liver cancers, non-Hodgkin’s lymphoma, and skin cancers.
To evaluate the incidence of malignancy, the investigators obtained data from the MarketScan Commercial and Medicare Supplemental claims databases for patients with a diagnosis of psoriasis on or before Dec. 31, 2006, and at least one prescription claim for etanercept (Enbrel), adalimumab (Humira), infliximab (Remicade), ustekinumab (Stelara), nonbiologic therapies, or phototherapy.
The general population cohort included patients at least 18 years old as of 2005 or at enrollment in the health care plan. Both cohorts had 12 months’ continuous enrollment in the health care plan from Jan. 1, 2005, through Dec. 31, 2006.
Follow-up for a patient ended at the first cancer event, disenrollment from the health care plan, or after 5 years from the index date.
Patients with psoriasis had a 5-year malignancy rate of 115.5 cases/10,000 person-years, compared with 96/10,000 person-years for the general population, reported Dr. Kimball of Massachusetts General Hospital, Boston.
Excluding nonmelanoma skin cancer and lymphoma, incidence rates were similar across treatment groups: etanercept (100.2/10,000), adalimumab (94.6/10,000), infliximab (138.1/10,000), ustekinumab (100.6/10,000), nonbiologics (116.8/10,000), and phototherapy (117.3/10,000).
"These large database queries continue to be reassuring that most systemic therapies are not changing the risk for common cancers, excluding lymphoma and skin cancer, which we continue to examine separately," Dr. Kimball said in an interview.
Nonmelanoma skin cancer was far and away the most common malignancy in psoriasis patients, occurring at a rate of 147.2/10,000 person-years vs. 94.2/10,000 person-years among the general public. Rates were highest in patients treated with adalimumab (234.2/10,000 person-years) and ustekinumab (233.3/10,000) and lowest in those treated with etanercept (155.9/10,000).
"Even with the large size of the database, the number of skin cancers remains small, so making conclusions about specific therapies may be premature," she said.
Incidence rates of lymphoma were considerably lower, but again higher in psoriasis patients than the general public (11.1/10,000 vs. 6.6/10,000). Rates of this hematologic cancer were highest with ustekinumab (25.1/10,000) and, once again, lowest with etanercept (6.9/10,000), according to the poster.
In all, 5,857 patients received nonbiologic therapies, 6,856 received etanercept, 3,314 adalimumab, 1,044 infliximab, 526 ustekinumab, and 5,156 were treated with phototherapy.
Dr. Kimball is a consultant for several pharmaceutical companies including Amgen, the study sponsor.
AT THE AAD SUMMER ACADEMY 2014
Key clinical point: Rates of malignancy were higher among patients with psoriasis than in the general population.
Major finding: The 5-year malignancy rate in patients with psoriasis was 115.5 cases/10,000 person-years vs. 96/10,000 person-years in the general population.
Data source: Retrospective database analysis of 22,753 patients with psoriasis.
Disclosures: Dr. Kimball is a consultant for several pharmaceutical companies including Amgen, the study sponsor.

Don’t overlook anus, genitalia during total body skin exam
CHICAGO – The anus and genitalia are often overlooked during total body skin examinations, leaving mucosal diseases to go unchecked, especially in women, according to Dr. Bethanee Schlosser.
She acknowledged that there is no literature to quantify the issue but said that her experience suggests mucocutaneous exams may be getting short shrift.

When Dr. Schlosser queried some 300 dermatologists assembled earlier this year, almost all said that they examine the oral cavity during total body skin exams; three-fourths responded that they routinely examine male patients’ genitalia. When asked whether they do the same for their female patients, less than 20 hands went up in the crowd.
"I think there are a couple of reasons for it," Dr. Schlosser of Northwestern University, Chicago, said at the American Academy of Dermatology summer meeting.
First, dermatologists don’t often look at the genitalia, so they may not know what the normal variations are.
Second, many patients don’t expect a dermatologist to examine genitalia. "Patients may be like, ‘You want to look where? I just have a mole on my chest.’ So it’s a matter of patient education," she said.
Third is the added time involved, and finally, some dermatologists are hesitant because they may not be comfortable managing mucosal disease should they find it.
Most dermatologists assume that gynecologists are evaluating their patients’ vulvar skin, but some gynecologists view the vulva more as "the doorway to the cervix. They may simply walk through it, not looking at what is around them, and to that effect, I don’t think it’s necessarily their fault, but their training," Dr. Schlosser said. "While vulvar disease is on the ob.gyn. board exam, the senior ob.gyn. residents rotate through our clinic with me and they routinely say they don’t get that education anywhere else."
Dr. Schlosser offered pearls for managing a number of mucosal diseases, including vulvar lichen sclerosis (VLS).
VLS affects about 1 in 600 women and can carry significant morbidity, including complete obliteration of the clitoral hood and labia minora, narrowing of the vaginal introitus, sexual dysfunction, and potential urinary obstruction, she said.
The risk of developing squamous cell carcinoma (SCC) in patients with VLS is 300-fold higher than in the general patient population. The specific risk factors for vulvar SCC are not fully elucidated in VLS, but include localized hyperkeratosis and age over 75 years.
"It’s important to realize that these older patients ... often don’t see their gynecologists because they’re told the pelvic examination is not indicated anymore," Dr. Schlosser said. "It really behooves us as dermatologists to be doing a genital exam as part of our total body skin exam."
Researchers think, but don’t have the evidence to suggest, that treating VLS changes the risk of SCC, "which is one of the hardest things to discuss with our patients," she added.
Suspicion of SCC should be raised if the patient has hyperkeratotic lesions; ulceration, even pinpoint in size, that doesn’t improve with standard therapy; erythematous, indurated plaques; or if the patient reports a change in symptom quality – previously itchy and now painful, for example – or a change in symptom distribution, such as previously all over and now localized to one spot.
"That can really herald that something bad has occurred," Dr. Schlosser cautioned.
Superpotent topical corticosteroids such as clobetasol propionate are first-line therapy for VLS, with no rationale to support the old-school treatment of topical testosterone. Maintenance therapy is common, as up to 85% of women will relapse.
Dr. Schlosser also advised physicians to educate women on how much medication to use, to send them home with a diagram of the affected area, and to use a hand mirror during the exam.
"Patients don’t want to look, but I tell them you’re not going to get better if you don’t know where to put your medication," she said.
Dr. Schlosser reported no relevant conflicts of interest.
CHICAGO – The anus and genitalia are often overlooked during total body skin examinations, leaving mucosal diseases to go unchecked, especially in women, according to Dr. Bethanee Schlosser.
She acknowledged that there is no literature to quantify the issue but said that her experience suggests mucocutaneous exams may be getting short shrift.
When Dr. Schlosser queried some 300 dermatologists assembled earlier this year, almost all said that they examine the oral cavity during total body skin exams; three-fourths responded that they routinely examine male patients’ genitalia. When asked whether they do the same for their female patients, less than 20 hands went up in the crowd.
"I think there are a couple of reasons for it," Dr. Schlosser of Northwestern University, Chicago, said at the American Academy of Dermatology summer meeting.
First, dermatologists don’t often look at the genitalia, so they may not know what the normal variations are.
Second, many patients don’t expect a dermatologist to examine genitalia. "Patients may be like, ‘You want to look where? I just have a mole on my chest.’ So it’s a matter of patient education," she said.
Third is the added time involved, and finally, some dermatologists are hesitant because they may not be comfortable managing mucosal disease should they find it.
Most dermatologists assume that gynecologists are evaluating their patients’ vulvar skin, but some gynecologists view the vulva more as "the doorway to the cervix. They may simply walk through it, not looking at what is around them, and to that effect, I don’t think it’s necessarily their fault, but their training," Dr. Schlosser said. "While vulvar disease is on the ob.gyn. board exam, the senior ob.gyn. residents rotate through our clinic with me and they routinely say they don’t get that education anywhere else."
Dr. Schlosser offered pearls for managing a number of mucosal diseases, including vulvar lichen sclerosis (VLS).
VLS affects about 1 in 600 women and can carry significant morbidity, including complete obliteration of the clitoral hood and labia minora, narrowing of the vaginal introitus, sexual dysfunction, and potential urinary obstruction, she said.
The risk of developing squamous cell carcinoma (SCC) in patients with VLS is 300-fold higher than in the general patient population. The specific risk factors for vulvar SCC are not fully elucidated in VLS, but include localized hyperkeratosis and age over 75 years.
"It’s important to realize that these older patients ... often don’t see their gynecologists because they’re told the pelvic examination is not indicated anymore," Dr. Schlosser said. "It really behooves us as dermatologists to be doing a genital exam as part of our total body skin exam."
Researchers think, but don’t have the evidence to suggest, that treating VLS changes the risk of SCC, "which is one of the hardest things to discuss with our patients," she added.
Suspicion of SCC should be raised if the patient has hyperkeratotic lesions; ulceration, even pinpoint in size, that doesn’t improve with standard therapy; erythematous, indurated plaques; or if the patient reports a change in symptom quality – previously itchy and now painful, for example – or a change in symptom distribution, such as previously all over and now localized to one spot.
"That can really herald that something bad has occurred," Dr. Schlosser cautioned.
Superpotent topical corticosteroids such as clobetasol propionate are first-line therapy for VLS, with no rationale to support the old-school treatment of topical testosterone. Maintenance therapy is common, as up to 85% of women will relapse.
Dr. Schlosser also advised physicians to educate women on how much medication to use, to send them home with a diagram of the affected area, and to use a hand mirror during the exam.
"Patients don’t want to look, but I tell them you’re not going to get better if you don’t know where to put your medication," she said.
Dr. Schlosser reported no relevant conflicts of interest.
CHICAGO – The anus and genitalia are often overlooked during total body skin examinations, leaving mucosal diseases to go unchecked, especially in women, according to Dr. Bethanee Schlosser.
She acknowledged that there is no literature to quantify the issue but said that her experience suggests mucocutaneous exams may be getting short shrift.
When Dr. Schlosser queried some 300 dermatologists assembled earlier this year, almost all said that they examine the oral cavity during total body skin exams; three-fourths responded that they routinely examine male patients’ genitalia. When asked whether they do the same for their female patients, less than 20 hands went up in the crowd.
"I think there are a couple of reasons for it," Dr. Schlosser of Northwestern University, Chicago, said at the American Academy of Dermatology summer meeting.
First, dermatologists don’t often look at the genitalia, so they may not know what the normal variations are.
Second, many patients don’t expect a dermatologist to examine genitalia. "Patients may be like, ‘You want to look where? I just have a mole on my chest.’ So it’s a matter of patient education," she said.
Third is the added time involved, and finally, some dermatologists are hesitant because they may not be comfortable managing mucosal disease should they find it.
Most dermatologists assume that gynecologists are evaluating their patients’ vulvar skin, but some gynecologists view the vulva more as "the doorway to the cervix. They may simply walk through it, not looking at what is around them, and to that effect, I don’t think it’s necessarily their fault, but their training," Dr. Schlosser said. "While vulvar disease is on the ob.gyn. board exam, the senior ob.gyn. residents rotate through our clinic with me and they routinely say they don’t get that education anywhere else."
Dr. Schlosser offered pearls for managing a number of mucosal diseases, including vulvar lichen sclerosis (VLS).
VLS affects about 1 in 600 women and can carry significant morbidity, including complete obliteration of the clitoral hood and labia minora, narrowing of the vaginal introitus, sexual dysfunction, and potential urinary obstruction, she said.
The risk of developing squamous cell carcinoma (SCC) in patients with VLS is 300-fold higher than in the general patient population. The specific risk factors for vulvar SCC are not fully elucidated in VLS, but include localized hyperkeratosis and age over 75 years.
"It’s important to realize that these older patients ... often don’t see their gynecologists because they’re told the pelvic examination is not indicated anymore," Dr. Schlosser said. "It really behooves us as dermatologists to be doing a genital exam as part of our total body skin exam."
Researchers think, but don’t have the evidence to suggest, that treating VLS changes the risk of SCC, "which is one of the hardest things to discuss with our patients," she added.
Suspicion of SCC should be raised if the patient has hyperkeratotic lesions; ulceration, even pinpoint in size, that doesn’t improve with standard therapy; erythematous, indurated plaques; or if the patient reports a change in symptom quality – previously itchy and now painful, for example – or a change in symptom distribution, such as previously all over and now localized to one spot.
"That can really herald that something bad has occurred," Dr. Schlosser cautioned.
Superpotent topical corticosteroids such as clobetasol propionate are first-line therapy for VLS, with no rationale to support the old-school treatment of topical testosterone. Maintenance therapy is common, as up to 85% of women will relapse.
Dr. Schlosser also advised physicians to educate women on how much medication to use, to send them home with a diagram of the affected area, and to use a hand mirror during the exam.
"Patients don’t want to look, but I tell them you’re not going to get better if you don’t know where to put your medication," she said.
Dr. Schlosser reported no relevant conflicts of interest.
EXPERT ANALYSIS FROM THE AAD SUMMER ACADEMY 2014
