User login
FDA strengthens kidney warnings on two diabetes drugs
The Food and Drug Administration has revised warnings on certain medications for type 2 diabetes mellitus based on recent reports of kidney injury. The warning labels on canagliflozin (Invokana, Invokamet) and dapagliflozin (Farxiga, Xigduo XR) now include recommendations on ways to minimize the risk and more information on kidney injury.
“From March 2013, when canagliflozin was approved, to October 2015, FDA received reports of 101 confirmable cases of acute kidney injury, some requiring hospitalization and dialysis, with canagliflozin or dapagliflozin use,” the FDA statement said.

The FDA went on to note: “Health care professionals should consider factors that may predispose patients to acute kidney injury prior to starting them on canagliflozin or dapagliflozin. These include decreased blood volume; chronic kidney insufficiency; congestive heart failure; and taking other medications such as diuretics, blood pressure medicines called angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs), and nonsteroidal anti-inflammatory drugs (NSAIDs). Assess kidney function prior to starting canagliflozin or dapagliflozin and monitor periodically thereafter. If acute kidney injury occurs, promptly discontinue the drug and treat the kidney impairment.”
The Food and Drug Administration has revised warnings on certain medications for type 2 diabetes mellitus based on recent reports of kidney injury. The warning labels on canagliflozin (Invokana, Invokamet) and dapagliflozin (Farxiga, Xigduo XR) now include recommendations on ways to minimize the risk and more information on kidney injury.
“From March 2013, when canagliflozin was approved, to October 2015, FDA received reports of 101 confirmable cases of acute kidney injury, some requiring hospitalization and dialysis, with canagliflozin or dapagliflozin use,” the FDA statement said.
The FDA went on to note: “Health care professionals should consider factors that may predispose patients to acute kidney injury prior to starting them on canagliflozin or dapagliflozin. These include decreased blood volume; chronic kidney insufficiency; congestive heart failure; and taking other medications such as diuretics, blood pressure medicines called angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs), and nonsteroidal anti-inflammatory drugs (NSAIDs). Assess kidney function prior to starting canagliflozin or dapagliflozin and monitor periodically thereafter. If acute kidney injury occurs, promptly discontinue the drug and treat the kidney impairment.”
The Food and Drug Administration has revised warnings on certain medications for type 2 diabetes mellitus based on recent reports of kidney injury. The warning labels on canagliflozin (Invokana, Invokamet) and dapagliflozin (Farxiga, Xigduo XR) now include recommendations on ways to minimize the risk and more information on kidney injury.
“From March 2013, when canagliflozin was approved, to October 2015, FDA received reports of 101 confirmable cases of acute kidney injury, some requiring hospitalization and dialysis, with canagliflozin or dapagliflozin use,” the FDA statement said.
The FDA went on to note: “Health care professionals should consider factors that may predispose patients to acute kidney injury prior to starting them on canagliflozin or dapagliflozin. These include decreased blood volume; chronic kidney insufficiency; congestive heart failure; and taking other medications such as diuretics, blood pressure medicines called angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs), and nonsteroidal anti-inflammatory drugs (NSAIDs). Assess kidney function prior to starting canagliflozin or dapagliflozin and monitor periodically thereafter. If acute kidney injury occurs, promptly discontinue the drug and treat the kidney impairment.”

HCV regimen found safe, effective in patients with severe renal disease
A 12-week regimen achieved sustained viral response for 90% of patients with genotype 1 hepatitis C virus (HCV) infection and stage 4 or 5 chronic kidney disease (CKD), researchers reported in the April issue of Gastroenterology.
“The regimen is well tolerated, though ribavirin use may require a reduction or interruption to manage anemia,” said Dr. Paul Pockros at Scripps Clinic and Scripps Translational Science Institute in La Jolla, Calif., and his associates. The second phase of the study will evaluate the regimen in treatment-experienced CKD patients and those with compensated cirrhosis, they said.

The regimen contained ombitasvir, paritaprevir, ritonavir, and dasabuvir.
Between 8% and 44% of hemodialysis patients are HCV positive, and CKD is known to heighten the risk of HCV-associated cirrhosis, hepatocellular carcinoma, and liver-related death, the researchers noted. While sofosbuvir is cleared renally, ombitasvir, paritaprevir, ritonavir, and dasabuvir undergo hepatic metabolism and needed no dose adjustment in phase I studies of patients with mild, moderate, or severe renal impairment. To further investigate the safety and efficacy of these direct-acting antivirals in patients with severe kidney disease, the researchers performed a single-arm, open-label, multicenter study of 20 treatment-naive, noncirrhotic, HCV-infected adults with stage 4 CKD (estimated glomerular filtration rate, 15-30 mL/min per 1.73 m2) or stage 5 (eGFR, less than 15 mL/min per 1.73 m2 or requiring hemodialysis). Patients received once-daily ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) plus dasabuvir (250 mg) for 12 weeks. The 13 patients with genotype 1a infections also received once-daily ribavirin (200 mg). Most patients were black men with stage 5 CKD, and 14 were on hemodialysis, the researchers said (Gastroenterology. 2016 Apr 16. doi: 10.1053/j.gastro.2016.02.078).
All 20 patients completed treatment, and 18 (90%) achieved sustained viral response (SVR) at posttreatment week 12 (SVR12; 95% confidence interval, 70%-97%). No patients developed hepatic decompensation, the researchers said. The most common adverse effects were anemia (45%), fatigue (35%), diarrhea (25%), and nausea (25%). Anemia developed only in patients receiving ribavirin and was more pronounced than in phase III studies of this regimen, the researchers said. Hemoglobin levels dropped an average of 1.38 plus or minus 1.54 g/dL among patients who received ribavirin, compared with 0.02 plus or minus 0.9 g/dL among patients who did not receive ribavirin. There was one case of grade 3 anemia related to incorrect dosing of ribavirin; the lowest measured hemoglobin level was 7.0 g/dL, which improved to more than 10 g/dL after stopping ribavirin and starting erythropoietin treatment. This patient also achieved SVR12. The other eight patients who developed anemia also stopped ribavirin, although three were able to resume it after their hemoglobin levels improved.
Of the two patients who did not achieve SVR12, one relapsed 4 weeks after treatment, and one died of cardiac arrest 14 days after treatment. The patient who died had a history of hypertension; his hemoglobin level was stable (9-11 g/dL) during the last 6 weeks of treatment, and was 10 g/dL at admission, suggesting that ribavirin-induced anemia did not cause the cardiac event, the investigators said.
“The results of this study are important for hepatologists, gastroenterologists, and infectious disease specialists who are accustomed to treating HCV-infected patients with DAA [direct-acting antiviral] therapy but who may not yet have seen sufficient data to initiate DAA therapy in patients with ESRD [end-stage renal disease],” the researchers concluded. “Nephrologists, who may not be accustomed to treating HCV, should also be aware that treatment options may now be available that can help prevent the end-stage sequelae of HCV. How treatment of HCV infection affects early or intermediate stages of CKD and how achievement of SVR impacts strategies for kidney transplantation in patients with ESRD require more study.”
AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
A 12-week regimen achieved sustained viral response for 90% of patients with genotype 1 hepatitis C virus (HCV) infection and stage 4 or 5 chronic kidney disease (CKD), researchers reported in the April issue of Gastroenterology.
“The regimen is well tolerated, though ribavirin use may require a reduction or interruption to manage anemia,” said Dr. Paul Pockros at Scripps Clinic and Scripps Translational Science Institute in La Jolla, Calif., and his associates. The second phase of the study will evaluate the regimen in treatment-experienced CKD patients and those with compensated cirrhosis, they said.
The regimen contained ombitasvir, paritaprevir, ritonavir, and dasabuvir.
Between 8% and 44% of hemodialysis patients are HCV positive, and CKD is known to heighten the risk of HCV-associated cirrhosis, hepatocellular carcinoma, and liver-related death, the researchers noted. While sofosbuvir is cleared renally, ombitasvir, paritaprevir, ritonavir, and dasabuvir undergo hepatic metabolism and needed no dose adjustment in phase I studies of patients with mild, moderate, or severe renal impairment. To further investigate the safety and efficacy of these direct-acting antivirals in patients with severe kidney disease, the researchers performed a single-arm, open-label, multicenter study of 20 treatment-naive, noncirrhotic, HCV-infected adults with stage 4 CKD (estimated glomerular filtration rate, 15-30 mL/min per 1.73 m2) or stage 5 (eGFR, less than 15 mL/min per 1.73 m2 or requiring hemodialysis). Patients received once-daily ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) plus dasabuvir (250 mg) for 12 weeks. The 13 patients with genotype 1a infections also received once-daily ribavirin (200 mg). Most patients were black men with stage 5 CKD, and 14 were on hemodialysis, the researchers said (Gastroenterology. 2016 Apr 16. doi: 10.1053/j.gastro.2016.02.078).
All 20 patients completed treatment, and 18 (90%) achieved sustained viral response (SVR) at posttreatment week 12 (SVR12; 95% confidence interval, 70%-97%). No patients developed hepatic decompensation, the researchers said. The most common adverse effects were anemia (45%), fatigue (35%), diarrhea (25%), and nausea (25%). Anemia developed only in patients receiving ribavirin and was more pronounced than in phase III studies of this regimen, the researchers said. Hemoglobin levels dropped an average of 1.38 plus or minus 1.54 g/dL among patients who received ribavirin, compared with 0.02 plus or minus 0.9 g/dL among patients who did not receive ribavirin. There was one case of grade 3 anemia related to incorrect dosing of ribavirin; the lowest measured hemoglobin level was 7.0 g/dL, which improved to more than 10 g/dL after stopping ribavirin and starting erythropoietin treatment. This patient also achieved SVR12. The other eight patients who developed anemia also stopped ribavirin, although three were able to resume it after their hemoglobin levels improved.
Of the two patients who did not achieve SVR12, one relapsed 4 weeks after treatment, and one died of cardiac arrest 14 days after treatment. The patient who died had a history of hypertension; his hemoglobin level was stable (9-11 g/dL) during the last 6 weeks of treatment, and was 10 g/dL at admission, suggesting that ribavirin-induced anemia did not cause the cardiac event, the investigators said.
“The results of this study are important for hepatologists, gastroenterologists, and infectious disease specialists who are accustomed to treating HCV-infected patients with DAA [direct-acting antiviral] therapy but who may not yet have seen sufficient data to initiate DAA therapy in patients with ESRD [end-stage renal disease],” the researchers concluded. “Nephrologists, who may not be accustomed to treating HCV, should also be aware that treatment options may now be available that can help prevent the end-stage sequelae of HCV. How treatment of HCV infection affects early or intermediate stages of CKD and how achievement of SVR impacts strategies for kidney transplantation in patients with ESRD require more study.”
AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
A 12-week regimen achieved sustained viral response for 90% of patients with genotype 1 hepatitis C virus (HCV) infection and stage 4 or 5 chronic kidney disease (CKD), researchers reported in the April issue of Gastroenterology.
“The regimen is well tolerated, though ribavirin use may require a reduction or interruption to manage anemia,” said Dr. Paul Pockros at Scripps Clinic and Scripps Translational Science Institute in La Jolla, Calif., and his associates. The second phase of the study will evaluate the regimen in treatment-experienced CKD patients and those with compensated cirrhosis, they said.
The regimen contained ombitasvir, paritaprevir, ritonavir, and dasabuvir.
Between 8% and 44% of hemodialysis patients are HCV positive, and CKD is known to heighten the risk of HCV-associated cirrhosis, hepatocellular carcinoma, and liver-related death, the researchers noted. While sofosbuvir is cleared renally, ombitasvir, paritaprevir, ritonavir, and dasabuvir undergo hepatic metabolism and needed no dose adjustment in phase I studies of patients with mild, moderate, or severe renal impairment. To further investigate the safety and efficacy of these direct-acting antivirals in patients with severe kidney disease, the researchers performed a single-arm, open-label, multicenter study of 20 treatment-naive, noncirrhotic, HCV-infected adults with stage 4 CKD (estimated glomerular filtration rate, 15-30 mL/min per 1.73 m2) or stage 5 (eGFR, less than 15 mL/min per 1.73 m2 or requiring hemodialysis). Patients received once-daily ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) plus dasabuvir (250 mg) for 12 weeks. The 13 patients with genotype 1a infections also received once-daily ribavirin (200 mg). Most patients were black men with stage 5 CKD, and 14 were on hemodialysis, the researchers said (Gastroenterology. 2016 Apr 16. doi: 10.1053/j.gastro.2016.02.078).
All 20 patients completed treatment, and 18 (90%) achieved sustained viral response (SVR) at posttreatment week 12 (SVR12; 95% confidence interval, 70%-97%). No patients developed hepatic decompensation, the researchers said. The most common adverse effects were anemia (45%), fatigue (35%), diarrhea (25%), and nausea (25%). Anemia developed only in patients receiving ribavirin and was more pronounced than in phase III studies of this regimen, the researchers said. Hemoglobin levels dropped an average of 1.38 plus or minus 1.54 g/dL among patients who received ribavirin, compared with 0.02 plus or minus 0.9 g/dL among patients who did not receive ribavirin. There was one case of grade 3 anemia related to incorrect dosing of ribavirin; the lowest measured hemoglobin level was 7.0 g/dL, which improved to more than 10 g/dL after stopping ribavirin and starting erythropoietin treatment. This patient also achieved SVR12. The other eight patients who developed anemia also stopped ribavirin, although three were able to resume it after their hemoglobin levels improved.
Of the two patients who did not achieve SVR12, one relapsed 4 weeks after treatment, and one died of cardiac arrest 14 days after treatment. The patient who died had a history of hypertension; his hemoglobin level was stable (9-11 g/dL) during the last 6 weeks of treatment, and was 10 g/dL at admission, suggesting that ribavirin-induced anemia did not cause the cardiac event, the investigators said.
“The results of this study are important for hepatologists, gastroenterologists, and infectious disease specialists who are accustomed to treating HCV-infected patients with DAA [direct-acting antiviral] therapy but who may not yet have seen sufficient data to initiate DAA therapy in patients with ESRD [end-stage renal disease],” the researchers concluded. “Nephrologists, who may not be accustomed to treating HCV, should also be aware that treatment options may now be available that can help prevent the end-stage sequelae of HCV. How treatment of HCV infection affects early or intermediate stages of CKD and how achievement of SVR impacts strategies for kidney transplantation in patients with ESRD require more study.”
AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
FROM GASTROENTEROLOGY
Key clinical point: Twelve weeks of ombitasvir, paritaprevir, ritonavir, and dasabuvir cured 90% of patients with hepatitis C virus infection and severe or end-stage renal disease.
Major finding: The rate of SVR12 was 90% (95% CI, 70%-97%).
Data source: A single-arm, open-label, multicenter trial of 20 noncirrhotic genotype 1 HCV-infected adults with stage 4 or 5 chronic kidney disease.
Disclosures: AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
Dietary and medical management of recurrent nephrolithiasis
Nephrolithiasis is common and often recurs. This review focuses on measures to prevent recurrent stone formation. Some measures apply to all patients, and some apply to specific types of stones.
COMMON AND INCREASING
According to data from the 2007–2010 National Health and Nutrition Examination Survey, the prevalence of nephrolithiasis in the United States was 10.6% in men and 7.1% in women. On average, 1 in 11 Americans will develop kidney stones at least once in their lifetime.1
By race and sex, white men have the highest incidence of nephrolithiasis and Asian women have the lowest. It is less common before age 20 and peaks in incidence in the third and fourth decades of life.
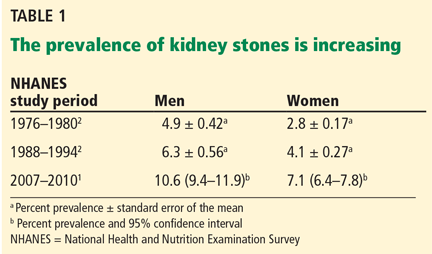
The prevalence has steadily increased in the past few decades (Table 1),1,2 but the reasons are not clear. The trend may be due to changes in diet and lifestyle, increasing prevalence of obesity and diabetes, migration from rural to urban areas, and global warming, with higher temperature resulting in dehydration and high urinary concentration of calcium and other stone-forming salts.3 Nephrolithiasis is now recognized as a systemic disorder associated with chronic kidney disease, bone loss and fractures, increased risk of coronary artery disease, hypertension, type 2 diabetes mellitus, and metabolic syndrome (Table 2).4–7

Without medical treatment, the 5-year recurrence rate is high, ranging from 35% to 50% after an initial stone event.8 Annual medical costs of care for kidney stones in the United States exceed $4.5 billion, with additional costs from missed work. Therefore, this condition has a considerable economic and social burden, which underscores the importance of prevention.9
MOST STONES CONTAIN CALCIUM
About 80% of kidney stones in adults contain calcium, and calcium oxalate stones are more common than calcium phosphate stones. Uric acid and struvite stones account for 5% to 15%, and cystine, protease inhibitor, triamterene, 2,8-dihydroxyadenine (2,8-DHA) and xanthine stones each account for less than 1%.10
Stones form when the urinary concentration of stone-forming salts, which is inversely proportional to urine volume, is higher than their saturation point, which is affected by urine pH. Acidic urine (low pH) predisposes to the formation of uric acid and cystine stones, whereas alkaline urine (high pH) favors calcium phosphate stones.
INCREASED FLUID INTAKE FOR ALL
High fluid intake, enough to produce at least 2.5 L of urine per day, should be the initial therapy to prevent stone recurrence.11
Borghi et al12 randomly assigned 199 patients who had a first calcium stone to high oral fluid intake or no intervention and followed them prospectively for 5 years. The recurrence rate was 12% in the treated group and 27% in the control group. Another study, in patients who had undergone shock wave lithotripsy, found a recurrence rate of 8% in those randomized to increase fluid intake to achieve urine output greater than 2.5 L/day, compared with 56% in those assigned to no treatment.13
Certain beverages increase the risk of stones and should be avoided. Sugar-sweetened noncola soda and punch are associated with a 33% higher risk of kidney stones, and cola sodas are associated with a 23% higher risk.14 Prospective studies have shown that the consumption of coffee, beer, wine, and orange juice is associated with a lower likelihood of stone formation.13,15
Table 3 is a brief summary of the dietary and pharmacologic interventions in the management of recurrent nephrolithiasis.
PREVENTING CALCIUM OXALATE STONES
Major urinary risk factors associated with calcium oxalate stones are hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, and low urine volume.16 Preventing calcium stones therefore depends on reducing the urinary concentration of calcium and oxalate, increasing urinary levels of inhibitors such as citrate, and increasing urine volume.
Reducing calcium excretion
Hypercalciuria has been traditionally defined as 24-hour urinary calcium excretion greater than 300 mg/day in men, greater than 250 mg in women, or greater than 4 mg/kg in men or women.17 It is a graded risk factor, and the cut points used in published research and clinical laboratories vary substantially. Some institutions use the same value for hypercalciuria in both sexes, eg, greater than 200 mg/day.18
Excessive sodium intake is the most common cause of hypercalciuria. Systemic conditions such as primary hyperparathyroidism, sarcoidosis, and renal tubular acidosis also cause hypercalciuria but are uncommon.19 Management depends on the underlying cause and includes dietary modifications and pharmacologic therapy.
Dietary modifications have a pivotal role in the management of recurrent stones that are due to hypercalciuria.
Dietary calcium should not be restricted, since calcium reduces the excretion of urinary oxalate by decreasing intestinal absorption of oxalate. Guidelines from the American Urological Association recommend a daily calcium intake of 1,000 to 1,200 mg.11–20 Moreover, restriction of dietary calcium to less than 800 mg/day (the current recommended daily allowance for adults) can lead to negative calcium balance and bone loss.
Sodium intake also influences hypercalciuria. Calcium is reabsorbed passively in the proximal tubule due to the concentration gradient created by active reabsorption of sodium. A high sodium intake causes volume expansion, leading to a decrease in proximal sodium and calcium reabsorption and enhancing calcium excretion. A low-sodium diet (80–100 mmol/day, or 1,800–2,300 mg/day) is recommended. This enhances proximal sodium and passive calcium absorption and leads to a decrease in calcium excretion.21
Dietary protein increases the acid load by production of sulfuric acid and leads to hypercalciuria by its action on bone and kidney. Animal protein has a higher content of sulfur and generates a higher acid load compared with vegetable protein and has been associated with an increased incidence of stone formation, at least in men.20,22 Borghi et al23 reported that the combination of restricted intake of animal protein (52 g/day), restricted salt intake (50 mmol, or 2,900 mg/day of sodium chloride), and normal calcium intake (30 mmol/day, or 1,200 mg/day) was associated with a lower incidence of stone recurrence in men with hypercalciuria compared with traditional low-calcium intake (10 mmol, or 400 mg/day). Patients should therefore be advised to avoid excessive intake of animal protein.
Increasing the dietary intake of fruits and vegetables as in the Dietary Approach to Stop Hypertension (DASH) diet is beneficial and reduces the risk of stone recurrence, mainly by increasing citrate excretion.24
Pharmacologic therapy in hypercalciuria. Thiazide diuretics are the mainstay of pharmacotherapy for preventing recurrent stones in patients with idiopathic hypercalciuria. They reduce the risk of stone recurrence by about 50%, as reported in a recent meta-analysis that looked at five trials comparing thiazide diuretics with placebo.25 They lower calcium excretion by causing volume depletion, thereby increasing proximal sodium and passive calcium reabsorption.
Chlorthalidone and hydrochlorothiazide are the thiazides commonly used to treat hypercalciuria. The dosage is titrated to the urinary calcium excretion, and a common mistake is to use doses that are too low. They are usually started at 25 mg/day, but often require an increase to 50 to 100 mg/day for adequate lowering of urinary calcium.
Care should be taken to avoid hypokalemia. If it occurs, it can be corrected by adding the potassium-sparing diuretic amiloride (5–10 mg/day), which increases calcium reabsorption in collecting ducts or, in patients with hypocitraturia, potassium citrate-potassium bicarbonate. (Sodium salts should be avoided, since they increase renal calcium excretion.)26
Management of hypercalciuria with metabolic causes, which include primary hyperparathyroidism and chronic acidemia. Patients who have hypercalciuria from primary hyperparathyroidism are treated with parathyroidectomy.27 Chronic metabolic acidosis causes hypercalciuria by loss of bone calcium and hypocitraturia by increasing active proximal absorption of citrate. Potassium citrate or potassium bicarbonate is used to prevent stones in such patients; sodium salts should be avoided.28
Reducing oxalate excretion
Hyperoxaluria has traditionally been defined as urinary oxalate excretion of more than 45 mg/day. However, the optimal cutoff point for urinary oxalate excretion is unclear, as is the optimal cutoff for hypercalciuria. The risk of stone formation has been shown to increase with oxalate excretion even above 25 mg/day, which is within the normal limit.18
Idiopathic hyperoxaluria. High dietary oxalate intake, especially when associated with low calcium intake, leads to idiopathic hyperoxaluria. However, the contribution of abnormal endogenous oxalate metabolism is uncertain. Ingested calcium binds to oxalate in the intestinal tract and reduces both the absorption of intestinal oxalate absorption and the excretion of urinary oxalate.29 High dietary oxalate intake has usually been regarded as a major risk factor for kidney stones.
Taylor and Curhan,30 in a prospective study, reported a mild increase in the risk of stones in the highest quintile of dietary oxalate intake compared with the lowest quintile for men (relative risk [RR] 1.22, 95% confidence interval [CI] 1.03–1.45) and older women (RR 1.21, 95% CI 1.01–1.44). They also demonstrated that eating eight or more servings of spinach per month compared with fewer than one serving per month was associated with a similar increase of stone risk in men (RR 1.30, 95% CI 1.08–1.58) and older women (RR 1.34 95% CI 1.1–1.64). In contrast, spinach and dietary oxalate intake did not increase the risk of nephrolithiasis in young women. The authors concluded that the risk associated with oxalate intake was modest, and their data did not support the contention that dietary oxalate is a major risk factor for kidney stones.
Higher oxalate intake increases urinary oxalate excretion and presumably the risk of nephrolithiasis. Limiting dietary oxalate to prevent stones is recommended if habitually high dietary intake of oxalate is identified or follow-up urine measurements show a decrease in oxalate excretion.31 Foods rich in oxalate include spinach, rhubarb, nuts, legumes, cocoa, okra, and chocolate.
The DASH diet, which is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein, is an effective dietary alternative and has been associated with a lower risk of calcium oxalate stones.24 Consuming fruits and vegetables increases the excretion of urinary citrate, which is an inhibitor of stone formation. Also, it has been proposed that the DASH diet contains unknown factors that reduce stone risk.
Taylor et al32 prospectively examined the relationship between the DASH diet and the incidence of kidney stones and found that the diet significantly reduced the risk of kidney stones. The relative risks of occurrence of kidney stones in participants in the highest quintile of the DASH score (a measure of adherence to the DASH diet) compared with the lowest quintile were 0.55 (95% CI 0.46–0.65) for men, 0.58 (95% CI 0.49–0.68) for older women, and 0.60 (95% CI 0.52–0.70) for younger women, which the authors characterized as “a marked decrease in kidney stone risk.”
Vitamin C intake should be restricted to 90 mg/day in patients who have a history of calcium oxalate stones. Urivetzky et al33 found that urinary oxalate excretion increased by 6 to 13 mg/day at doses of ascorbic acid greater than 500 mg.
Pyridoxine (vitamin B6), a coenzyme of alanine-glyoxylate aminotransferase (AGT), increases the conversion of glyoxylate to glycine instead of oxalate and is used in the treatment of type 1 primary hyperoxaluria (see below).34 However, its effect in preventing stones in idiopathic hyperoxaluria is not well known, and it has not been studied in a randomized controlled trial. In a prospective study, Curhan et al35 reported that high intake of pyridoxine (> 40 mg/day) was associated with a lower risk of stone formation in women, but no such benefit was found in men.
Enteric hyperoxaluria. About 90% of dietary oxalate binds to calcium in the small intestine and is excreted in the stool. The remaining 10% is absorbed in the colon and is secreted in urine. Hyperoxaluria is frequently seen with fat malabsorption from inflammatory bowel disease, short gut syndrome, and gastric bypass surgery. In these conditions, excess fat binds to dietary calcium, leading to increased absorption of free oxalate in the colon.36
Treatment is directed at decreasing intestinal oxalate absorption and should include high fluid intake and oral calcium supplements. Calcium carbonate or citrate causes precipitation of oxalate in the intestinal lumen and is prescribed as 1 to 4 g in three to four divided doses, always with meals. Calcium citrate is preferred over calcium carbonate in stone-formers because of the benefit of citrate and calcium citrate’s higher solubility and greater effectiveness in the presence of achlorhydria.37 Patients should be advised to avoid foods high in oxalate and fat.
Primary hyperoxaluria is caused by inherited inborn errors of glyoxylate metabolism that cause overproduction of oxalate and urinary oxalate excretion above 135 to 270 mg/day.
Type 1 primary hyperoxaluria is the most common (accounting for 90% of cases) and is caused by reduced activity of hepatic peroxisomal AGT.
Type 2 is from a deficiency of glyoxylate reductase-hydroxypyruvate reductase (GRHPR).
Type 3 is from mutations in the HOGA1 gene, which codes for the liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme involved in degradation of hydroxyproline to pyruvate and glyoxalate.38
High fluid intake to produce a urinary volume of 3 L/day reduces intratubular oxalate deposition and should be encouraged. Potassium citrate (0.15 mg/kg), oral phosphate supplements (30–40 mg/kg of orthophosphate), and magnesium oxide (500 mg/day/m2) inhibit precipitation of calcium oxalate in the urine.39,40 Pyridoxine, a coenzyme of AGT, increases the conversion of glyoxylate to glycine instead of oxalate and is prescribed at a starting dose of 5 mg/kg (which can be titrated up to 20 mg/kg if there is no response) in patients with type 1 primary hyperoxaluria. About 50% of patients with type 1 respond successfully to pyridoxine, and a 3- to 6-month trial should be given in all patients in this category.34 AGT is present only in hepatocytes, and GRHPR is found in multiple tissues; therefore, combined liver-kidney transplant is the treatment of choice in patients with type 1 primary hyperoxaluria, whereas isolated kidney transplant is recommended in patients with type 2.41
Reducing uric acid excretion
Hyperuricosuria is defined as uric acid excretion of greater than 800 mg/day in men and greater than 750 mg/day in women.
The association of hyperuricosuria with increased risk of calcium oxalate stone formation is controversial. Curhan and Taylor,18 in a cross-sectional study of 3,350 men and women, reported that there was no difference in mean 24-hour uric acid excretion in individuals with and without a history of stones.
The mechanism by which uric acid leads to calcium oxalate stones is not completely known and could be the “salting out” of calcium oxalate from the urine.42
Dietary purine restriction, ie, limiting intake of nondairy animal protein to 0.8 to 1 g/kg/day, is the initial dietary intervention.11 Allopurinol is the alternative approach if the patient is not compliant or if dietary restriction fails.43
In a study by Ettinger et al,44 60 patients with hyperuricosuria and normocalciuria were randomized to receive allopurinol (100 mg three times daily) or a placebo. The allopurinol group had a rate of calculus events of 0.12 per patient per year, compared with 0.26 in the placebo group.
Increasing citrate excretion
Hypocitraturia is a well-known risk factor for the formation of kidney stones. It is usually defined as a citrate excretion of less than 320 mg/day for adults.
Citrate prevents formation of calcium crystals by binding to calcium, thereby lowering the concentration of calcium oxalate below the saturation point.45
Diet therapy. Patients with calcium oxalate stones and hypocitraturia should be encouraged to increase their intake of fruits and vegetables, which enhances urinary citrate excretion, and to limit their intake of nondairy animal protein.11
The use of citrus products in preventing stones in patients with hypocitraturia is controversial, however, and needs to be studied more.
One study46 demonstrated that lemon juice was beneficial in hypocitraturic nephrolithiasis: 4 oz/day of lemon juice concentrate in the form of lemonade was associated with an increase in urinary citrate excretion to 346 mg/day from 142 mg/day in 11 of 12 patients who participated.
Odvina47 compared the effects of orange juice with those of lemonade on the acid-base profile and urinary stone risk under controlled metabolic conditions in 13 volunteers. Orange juice was reported to have greater alkalinizing and citraturic effects and was associated with lower calculated calcium oxalate supersaturation compared with lemonade.
Lemonade therapy may be used as adjunctive treatment in patients who do not comply with or cannot tolerate alkali therapy. However, we advise caution about recommending citrus products, as they can increase oxalate excretion.
Pharmacotherapy includes alkali therapy. Barcelo et al48 compared the effects of potassium citrate and placebo in 57 patients with calcium oxalate stones and hypocitraturia. Patients treated with potassium citrate had a rate of stone formation of 0.1 event per patient per year, compared with 1.1 in the placebo group.
Many forms of alkaline citrate are available. Potassium citrate is preferred over sodium citrate since the latter may increase urine calcium excretion.49 Treatment is usually started at 30 mEq/day and is titrated to a maximal dose of 60 mEq/day for a urinary citrate excretion greater than 500 mg/day.
Common side effects are abdominal bloating and hyperkalemia (especially with renal insufficiency), and in such cases sodium-based alkali, sodium citrate, or sodium bicarbonate can be prescribed.
PREVENTING CALCIUM PHOSPHATE STONES
Risk factors for calcium phosphate stones are similar to those for calcium oxalate stones (other than hyperoxaluria), but calcium phosphate stones are formed in alkaline urine (usually urine pH > 6.0), often the result of distal renal tubular acidosis. Preventive measures are similar to those for calcium oxalate stones.
Alkali therapy should be used with caution because of its effect on urinary pH and the risk of precipitation of calcium phosphate crystals.50 Use of potassium citrate was found to be associated with increases in both urinary citrate excretion and calcium phosphate supersaturation in hypercalciuric stone-forming rats.51 It is therefore challenging to manage patients with calcium phosphate stones and hypocitraturia. Alkali administration in this setting may diminish the formation of new stones by correcting hypocitraturia, but at the same time it may increase the likelihood of calcium phosphate stone formation by increasing the urinary pH. When the urine pH increases to above 6.5 with no significant change in urine citrate or urine calcium excretion, we recommend stopping alkali therapy.
PREVENTING URIC ACID STONES
Clinical conditions associated with uric acid stones include metabolic syndrome, diabetes mellitus, gout, chronic diarrheal illness, and conditions that increase tissue turnover and uric acid production, such as malignancies. Other risk factors for uric acid stone formation are low urine volume, low uric pH, and hyperuricosuria.
Abnormally acidic urine is the most common risk factor. Metabolic syndrome and diabetes mellitus reduce ammonia production, resulting in a lower urinary pH, which predisposes to uric acid stone formation. Chronic diarrhea also acidifies the urine by loss of bicarbonate. Similarly, in gout, the predisposing factor in uric acid stone formation is the persistently acidic urine due to impaired ammonium excretion.52 Uric acid precipitates to form uric acid stones in a low urinary pH even with normal excretion rates of 600 to 800 mg/day and a urinary volume of 1 to 1.5 L.53
Therefore, apart from increasing fluid intake, urinary alkalization is the cornerstone of management of uric acid stones. Potassium citrate is the preferred alkali salt and is started at a dose of 30 mEq/day for a goal urinary pH of 6 to 6.5.47
Patients with hyperuricosuria are also advised to restrict their protein intake to no more than 0.8 to 1 mg/kg/day.
If the above measures fail, patients are treated with a xanthine oxidase inhibitor, ie, allopurinol or febuxostat, even if their uric acid excretion is normal.54
PREVENTING STRUVITE STONES
Struvite stones contain magnesium ammonium phosphate and are due to chronic upper urinary tract infection with urea-splitting bacteria such as Proteus, Klebsiella, Pseudomonas, and enterococci. Urea hydrolysis releases hydroxyl ions, resulting in alkaline urine that promotes struvite stone formation. Early detection and treatment are important, since struvite stones are associated with morbidity and rapid progression.
Medical treatment of struvite stones is usually unsuccessful, and the patient is referred to a urologist for surgical removal of the stones, the gold standard treatment.55 Long-term use of culture-specific antibiotics to prevent new stone growth is not well studied. Medical therapy by itself is preferred in patients who refuse stone removal or cannot tolerate it. Urease inhibitors such as acetohydroxamic acid have been successful in preventing or slowing stone growth, but their use is limited by frequent side effects such as nausea, headache, rash, and thrombophlebitis.56
CYSTINE STONES
Cystine stones occur in people with inherited defects of renal tubular and intestinal transport of cysteine and dibasic amino acids that cause excessive excretion of urinary cystine, ie, 480 to 3,600 mg/day.
Cystine is formed from two cysteine molecules linked by a disulfide bond. The solubility of cystine is pH-dependent, with increased solubility at higher urinary pH. The goal is to maintain a urinary cystine concentration below its solubility level by keeping the cystine concentration below 243 mg/L and the urine cystine supersaturation (the ratio of the urine cysteine concentration to the cysteine solubility in the same sample) less than 0.6.57 Therapy is aimed at increasing daily urinary volume to 3 L and urine alkalization to pH above 7, in order to increase cystine solubility by 300%.58
Overnight dehydration should be prevented, and patients should be encouraged to wake up at least once a night to void and drink additional water. Sodium restriction to 100 mmol/day (2,300 mg/day) and moderate protein restriction to 0.8 to 1 g/kg/day are associated with decreased cystine excretion, but long-term studies demonstrating their benefit in preventing cystine stones are lacking.59
A thiol-containing drug, eg, D-penicillamine (0.5–2 g/day) or tiopronin (400–1,200 mg/day), should be added to the conservative measures if they have not been effective for 3 months or if there is history of noncompliance.60 Thiol-containing drugs have a sulfhydryl group that reduces the disulfide bond, and they form soluble disulfide cysteine-drug complexes with greater ability to solubilize cystine in alkaline urine. They must always be used in conjunction with fluid and alkali therapy.61
Both drugs have severe and common adverse effects including leukopenia, aplastic anemia, fever, rash, arthritis, hepatotoxicity, pyridoxine deficiency, and proteinuria (membranous nephropathy). However, tiopronin seems to have a lesser incidence of side effects.62 Regular monitoring of complete blood cell counts, liver enzymes, and urine protein should be done.
Captopril contains a sulfhydryl group, and the captopril-cysteine disulfide is more soluble than cysteine alone. The amount of captopril that appears in the urine is low, and doses of 150 mg/day are usually required to reduce cysteine excretion, which can lead to hypotension. The efficacy of captopril in treating cystine stones is unproven, and this drug is used only if patients cannot tolerate other thiol-containing drugs.63
- Scales CD Jr, Smith AC, Hanley JM, Saigal CS; Urologic Diseases in America Project. Prevalence of kidney stones in the United States. Eur Urol 2012; 62:160–165.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM Jr, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence, and associated risk factors. Rev Urol 2010; 12:e86–e96.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2011; 79:393–403.
- Sakhaee K. Nephrolithiasis as a systemic disorder. Curr Opin Nephrol Hypertens 2008; 17:304–309.
- Hamano S, Nakatsu H, Suzuki N, Tomioka S, Tanaka M, Murakami S. Kidney stone disease and risk factors for coronary heart disease. Int J Urol 2005; 12:859–863.
- Ritz E. Metabolic syndrome: an emerging threat to renal function. Clin J Am Soc Nephrol 2007; 2:869–871.
- Uribarri J, Oh MS, Carroll HJ. The first kidney stone. Ann Intern Med 1989; 111:1006–1009.
- Saigal CS, Joyce G, Timilsina AR; Urologic Diseases in America Project. Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 2005; 68:1808–1814.
- Moe OW. Kidney stones: pathophysiology and medical management. Lancet 2006; 367:333–344.
- Pearle MS, Goldfarb DS, Assimos DG, et al; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol 2014; 192:316–324.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Sarica K, Inal Y, Erturhan S, Yagci F. The effect of calcium channel blockers on stone regrowth and recurrence after shock wave lithotripsy. Urol Res 2006; 34:184–189.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013; 8:1389–1395.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Beverage use and risk for kidney stones in women. Ann Intern Med 1998; 128:534–540.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med 1980; 69:19–30.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int 2008; 73:489–496.
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest 2005; 115:2598–2608.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Muldowney FP, Freaney R, Moloney MF. Importance of dietary sodium in the hypercalciuria syndrome. Kidney Int 1982; 22:292–296.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Noori N, Honarkar E, Goldfarb DS, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis 2014; 63:456–463.
- Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013; 158:535–543.
- Alon U, Costanzo LS, Chan JC. Additive hypocalciuric effects of amiloride and hydrochlorothiazide in patients treated with calcitriol. Miner Electrolyte Metab 1984; 10:379–386.
- Corbetta S, Baccarelli A, Aroldi A, et al. Risk factors associated to kidney stones in primary hyperparathyroidism. J Endocrinol Invest 2005; 28:122–128.
- Haymann JP. Metabolic disorders: stones as first clinical manifestation of significant diseases. World J Urol 2015; 33:187–192.
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol 1985; 5:40–44.
- Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol 2007; 18:2198–2204.
- Lieske JC, Tremaine WJ, De Simone C, et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78:1178–1185.
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol 2009; 20:2253–2259.
- Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol 1992; 147:1215–1218.
- Hoyer-Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 2014; 9:468–477.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol 1999; 10:840–845.
- Parks JH, Worcester EM, O'Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant 1998; 13:2241–2247.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009; 75:1264–1271.
- Cochat P, Hulton SA, Acquaviva C, et al; OxalEurope. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27:1729–1736.
- Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993; 7:207–211.
- Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010; 10:2493–2501.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implications for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Coe FL, Parks JH. Hyperuricosuria and calcium nephrolithiasis. Urol Clin North Am 1981; 8:227–244.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol 2009; 11:134–144.
- Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol 1996; 156:907–909.
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol 2006; 1:1269–1274.
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol 1993; 150:1761–1764.
- Lemann J Jr, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int 1989; 35:688–695.
- Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore) 1991; 70:345–359.
- Krieger NS, Asplin JR, Frick KK, et al. Effect of potassium citrate on calcium phosphate stones in a model of hypercalciuria. J Am Soc Nephrol 2015; 26:3001–3008.
- Falls WF Jr. Comparison of urinary acidification and ammonium excretion in normal and gouty subjects. Metabolism 1972; 21:433–445.
- Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. N Engl J Med 1992; 327:1141–1152.
- Kenny JE, Goldfarb DS. Update on the pathophysiology and management of uric acid renal stones. Curr Rheumatol Rep 2010; 12:125–129.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS Jr (AUA Nephrolithiasis Guideline Panel). Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med 1984; 311:760–764.
- Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol 2000; 164:1481–1485.
- Palacın MGP, Nunes V, Gasparini P. Cystinuria. In: Shriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4909–4932.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006; 69:1041–1047.
- Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163:1419–1423.
- Asplin DM, Asplin JR. The Interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013; 189:2147–2151.
- Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med 2006; 17:343–348.
- Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
Nephrolithiasis is common and often recurs. This review focuses on measures to prevent recurrent stone formation. Some measures apply to all patients, and some apply to specific types of stones.
COMMON AND INCREASING
According to data from the 2007–2010 National Health and Nutrition Examination Survey, the prevalence of nephrolithiasis in the United States was 10.6% in men and 7.1% in women. On average, 1 in 11 Americans will develop kidney stones at least once in their lifetime.1
By race and sex, white men have the highest incidence of nephrolithiasis and Asian women have the lowest. It is less common before age 20 and peaks in incidence in the third and fourth decades of life.
The prevalence has steadily increased in the past few decades (Table 1),1,2 but the reasons are not clear. The trend may be due to changes in diet and lifestyle, increasing prevalence of obesity and diabetes, migration from rural to urban areas, and global warming, with higher temperature resulting in dehydration and high urinary concentration of calcium and other stone-forming salts.3 Nephrolithiasis is now recognized as a systemic disorder associated with chronic kidney disease, bone loss and fractures, increased risk of coronary artery disease, hypertension, type 2 diabetes mellitus, and metabolic syndrome (Table 2).4–7
Without medical treatment, the 5-year recurrence rate is high, ranging from 35% to 50% after an initial stone event.8 Annual medical costs of care for kidney stones in the United States exceed $4.5 billion, with additional costs from missed work. Therefore, this condition has a considerable economic and social burden, which underscores the importance of prevention.9
MOST STONES CONTAIN CALCIUM
About 80% of kidney stones in adults contain calcium, and calcium oxalate stones are more common than calcium phosphate stones. Uric acid and struvite stones account for 5% to 15%, and cystine, protease inhibitor, triamterene, 2,8-dihydroxyadenine (2,8-DHA) and xanthine stones each account for less than 1%.10
Stones form when the urinary concentration of stone-forming salts, which is inversely proportional to urine volume, is higher than their saturation point, which is affected by urine pH. Acidic urine (low pH) predisposes to the formation of uric acid and cystine stones, whereas alkaline urine (high pH) favors calcium phosphate stones.
INCREASED FLUID INTAKE FOR ALL
High fluid intake, enough to produce at least 2.5 L of urine per day, should be the initial therapy to prevent stone recurrence.11
Borghi et al12 randomly assigned 199 patients who had a first calcium stone to high oral fluid intake or no intervention and followed them prospectively for 5 years. The recurrence rate was 12% in the treated group and 27% in the control group. Another study, in patients who had undergone shock wave lithotripsy, found a recurrence rate of 8% in those randomized to increase fluid intake to achieve urine output greater than 2.5 L/day, compared with 56% in those assigned to no treatment.13
Certain beverages increase the risk of stones and should be avoided. Sugar-sweetened noncola soda and punch are associated with a 33% higher risk of kidney stones, and cola sodas are associated with a 23% higher risk.14 Prospective studies have shown that the consumption of coffee, beer, wine, and orange juice is associated with a lower likelihood of stone formation.13,15
Table 3 is a brief summary of the dietary and pharmacologic interventions in the management of recurrent nephrolithiasis.
PREVENTING CALCIUM OXALATE STONES
Major urinary risk factors associated with calcium oxalate stones are hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, and low urine volume.16 Preventing calcium stones therefore depends on reducing the urinary concentration of calcium and oxalate, increasing urinary levels of inhibitors such as citrate, and increasing urine volume.
Reducing calcium excretion
Hypercalciuria has been traditionally defined as 24-hour urinary calcium excretion greater than 300 mg/day in men, greater than 250 mg in women, or greater than 4 mg/kg in men or women.17 It is a graded risk factor, and the cut points used in published research and clinical laboratories vary substantially. Some institutions use the same value for hypercalciuria in both sexes, eg, greater than 200 mg/day.18
Excessive sodium intake is the most common cause of hypercalciuria. Systemic conditions such as primary hyperparathyroidism, sarcoidosis, and renal tubular acidosis also cause hypercalciuria but are uncommon.19 Management depends on the underlying cause and includes dietary modifications and pharmacologic therapy.
Dietary modifications have a pivotal role in the management of recurrent stones that are due to hypercalciuria.
Dietary calcium should not be restricted, since calcium reduces the excretion of urinary oxalate by decreasing intestinal absorption of oxalate. Guidelines from the American Urological Association recommend a daily calcium intake of 1,000 to 1,200 mg.11–20 Moreover, restriction of dietary calcium to less than 800 mg/day (the current recommended daily allowance for adults) can lead to negative calcium balance and bone loss.
Sodium intake also influences hypercalciuria. Calcium is reabsorbed passively in the proximal tubule due to the concentration gradient created by active reabsorption of sodium. A high sodium intake causes volume expansion, leading to a decrease in proximal sodium and calcium reabsorption and enhancing calcium excretion. A low-sodium diet (80–100 mmol/day, or 1,800–2,300 mg/day) is recommended. This enhances proximal sodium and passive calcium absorption and leads to a decrease in calcium excretion.21
Dietary protein increases the acid load by production of sulfuric acid and leads to hypercalciuria by its action on bone and kidney. Animal protein has a higher content of sulfur and generates a higher acid load compared with vegetable protein and has been associated with an increased incidence of stone formation, at least in men.20,22 Borghi et al23 reported that the combination of restricted intake of animal protein (52 g/day), restricted salt intake (50 mmol, or 2,900 mg/day of sodium chloride), and normal calcium intake (30 mmol/day, or 1,200 mg/day) was associated with a lower incidence of stone recurrence in men with hypercalciuria compared with traditional low-calcium intake (10 mmol, or 400 mg/day). Patients should therefore be advised to avoid excessive intake of animal protein.
Increasing the dietary intake of fruits and vegetables as in the Dietary Approach to Stop Hypertension (DASH) diet is beneficial and reduces the risk of stone recurrence, mainly by increasing citrate excretion.24
Pharmacologic therapy in hypercalciuria. Thiazide diuretics are the mainstay of pharmacotherapy for preventing recurrent stones in patients with idiopathic hypercalciuria. They reduce the risk of stone recurrence by about 50%, as reported in a recent meta-analysis that looked at five trials comparing thiazide diuretics with placebo.25 They lower calcium excretion by causing volume depletion, thereby increasing proximal sodium and passive calcium reabsorption.
Chlorthalidone and hydrochlorothiazide are the thiazides commonly used to treat hypercalciuria. The dosage is titrated to the urinary calcium excretion, and a common mistake is to use doses that are too low. They are usually started at 25 mg/day, but often require an increase to 50 to 100 mg/day for adequate lowering of urinary calcium.
Care should be taken to avoid hypokalemia. If it occurs, it can be corrected by adding the potassium-sparing diuretic amiloride (5–10 mg/day), which increases calcium reabsorption in collecting ducts or, in patients with hypocitraturia, potassium citrate-potassium bicarbonate. (Sodium salts should be avoided, since they increase renal calcium excretion.)26
Management of hypercalciuria with metabolic causes, which include primary hyperparathyroidism and chronic acidemia. Patients who have hypercalciuria from primary hyperparathyroidism are treated with parathyroidectomy.27 Chronic metabolic acidosis causes hypercalciuria by loss of bone calcium and hypocitraturia by increasing active proximal absorption of citrate. Potassium citrate or potassium bicarbonate is used to prevent stones in such patients; sodium salts should be avoided.28
Reducing oxalate excretion
Hyperoxaluria has traditionally been defined as urinary oxalate excretion of more than 45 mg/day. However, the optimal cutoff point for urinary oxalate excretion is unclear, as is the optimal cutoff for hypercalciuria. The risk of stone formation has been shown to increase with oxalate excretion even above 25 mg/day, which is within the normal limit.18
Idiopathic hyperoxaluria. High dietary oxalate intake, especially when associated with low calcium intake, leads to idiopathic hyperoxaluria. However, the contribution of abnormal endogenous oxalate metabolism is uncertain. Ingested calcium binds to oxalate in the intestinal tract and reduces both the absorption of intestinal oxalate absorption and the excretion of urinary oxalate.29 High dietary oxalate intake has usually been regarded as a major risk factor for kidney stones.
Taylor and Curhan,30 in a prospective study, reported a mild increase in the risk of stones in the highest quintile of dietary oxalate intake compared with the lowest quintile for men (relative risk [RR] 1.22, 95% confidence interval [CI] 1.03–1.45) and older women (RR 1.21, 95% CI 1.01–1.44). They also demonstrated that eating eight or more servings of spinach per month compared with fewer than one serving per month was associated with a similar increase of stone risk in men (RR 1.30, 95% CI 1.08–1.58) and older women (RR 1.34 95% CI 1.1–1.64). In contrast, spinach and dietary oxalate intake did not increase the risk of nephrolithiasis in young women. The authors concluded that the risk associated with oxalate intake was modest, and their data did not support the contention that dietary oxalate is a major risk factor for kidney stones.
Higher oxalate intake increases urinary oxalate excretion and presumably the risk of nephrolithiasis. Limiting dietary oxalate to prevent stones is recommended if habitually high dietary intake of oxalate is identified or follow-up urine measurements show a decrease in oxalate excretion.31 Foods rich in oxalate include spinach, rhubarb, nuts, legumes, cocoa, okra, and chocolate.
The DASH diet, which is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein, is an effective dietary alternative and has been associated with a lower risk of calcium oxalate stones.24 Consuming fruits and vegetables increases the excretion of urinary citrate, which is an inhibitor of stone formation. Also, it has been proposed that the DASH diet contains unknown factors that reduce stone risk.
Taylor et al32 prospectively examined the relationship between the DASH diet and the incidence of kidney stones and found that the diet significantly reduced the risk of kidney stones. The relative risks of occurrence of kidney stones in participants in the highest quintile of the DASH score (a measure of adherence to the DASH diet) compared with the lowest quintile were 0.55 (95% CI 0.46–0.65) for men, 0.58 (95% CI 0.49–0.68) for older women, and 0.60 (95% CI 0.52–0.70) for younger women, which the authors characterized as “a marked decrease in kidney stone risk.”
Vitamin C intake should be restricted to 90 mg/day in patients who have a history of calcium oxalate stones. Urivetzky et al33 found that urinary oxalate excretion increased by 6 to 13 mg/day at doses of ascorbic acid greater than 500 mg.
Pyridoxine (vitamin B6), a coenzyme of alanine-glyoxylate aminotransferase (AGT), increases the conversion of glyoxylate to glycine instead of oxalate and is used in the treatment of type 1 primary hyperoxaluria (see below).34 However, its effect in preventing stones in idiopathic hyperoxaluria is not well known, and it has not been studied in a randomized controlled trial. In a prospective study, Curhan et al35 reported that high intake of pyridoxine (> 40 mg/day) was associated with a lower risk of stone formation in women, but no such benefit was found in men.
Enteric hyperoxaluria. About 90% of dietary oxalate binds to calcium in the small intestine and is excreted in the stool. The remaining 10% is absorbed in the colon and is secreted in urine. Hyperoxaluria is frequently seen with fat malabsorption from inflammatory bowel disease, short gut syndrome, and gastric bypass surgery. In these conditions, excess fat binds to dietary calcium, leading to increased absorption of free oxalate in the colon.36
Treatment is directed at decreasing intestinal oxalate absorption and should include high fluid intake and oral calcium supplements. Calcium carbonate or citrate causes precipitation of oxalate in the intestinal lumen and is prescribed as 1 to 4 g in three to four divided doses, always with meals. Calcium citrate is preferred over calcium carbonate in stone-formers because of the benefit of citrate and calcium citrate’s higher solubility and greater effectiveness in the presence of achlorhydria.37 Patients should be advised to avoid foods high in oxalate and fat.
Primary hyperoxaluria is caused by inherited inborn errors of glyoxylate metabolism that cause overproduction of oxalate and urinary oxalate excretion above 135 to 270 mg/day.
Type 1 primary hyperoxaluria is the most common (accounting for 90% of cases) and is caused by reduced activity of hepatic peroxisomal AGT.
Type 2 is from a deficiency of glyoxylate reductase-hydroxypyruvate reductase (GRHPR).
Type 3 is from mutations in the HOGA1 gene, which codes for the liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme involved in degradation of hydroxyproline to pyruvate and glyoxalate.38
High fluid intake to produce a urinary volume of 3 L/day reduces intratubular oxalate deposition and should be encouraged. Potassium citrate (0.15 mg/kg), oral phosphate supplements (30–40 mg/kg of orthophosphate), and magnesium oxide (500 mg/day/m2) inhibit precipitation of calcium oxalate in the urine.39,40 Pyridoxine, a coenzyme of AGT, increases the conversion of glyoxylate to glycine instead of oxalate and is prescribed at a starting dose of 5 mg/kg (which can be titrated up to 20 mg/kg if there is no response) in patients with type 1 primary hyperoxaluria. About 50% of patients with type 1 respond successfully to pyridoxine, and a 3- to 6-month trial should be given in all patients in this category.34 AGT is present only in hepatocytes, and GRHPR is found in multiple tissues; therefore, combined liver-kidney transplant is the treatment of choice in patients with type 1 primary hyperoxaluria, whereas isolated kidney transplant is recommended in patients with type 2.41
Reducing uric acid excretion
Hyperuricosuria is defined as uric acid excretion of greater than 800 mg/day in men and greater than 750 mg/day in women.
The association of hyperuricosuria with increased risk of calcium oxalate stone formation is controversial. Curhan and Taylor,18 in a cross-sectional study of 3,350 men and women, reported that there was no difference in mean 24-hour uric acid excretion in individuals with and without a history of stones.
The mechanism by which uric acid leads to calcium oxalate stones is not completely known and could be the “salting out” of calcium oxalate from the urine.42
Dietary purine restriction, ie, limiting intake of nondairy animal protein to 0.8 to 1 g/kg/day, is the initial dietary intervention.11 Allopurinol is the alternative approach if the patient is not compliant or if dietary restriction fails.43
In a study by Ettinger et al,44 60 patients with hyperuricosuria and normocalciuria were randomized to receive allopurinol (100 mg three times daily) or a placebo. The allopurinol group had a rate of calculus events of 0.12 per patient per year, compared with 0.26 in the placebo group.
Increasing citrate excretion
Hypocitraturia is a well-known risk factor for the formation of kidney stones. It is usually defined as a citrate excretion of less than 320 mg/day for adults.
Citrate prevents formation of calcium crystals by binding to calcium, thereby lowering the concentration of calcium oxalate below the saturation point.45
Diet therapy. Patients with calcium oxalate stones and hypocitraturia should be encouraged to increase their intake of fruits and vegetables, which enhances urinary citrate excretion, and to limit their intake of nondairy animal protein.11
The use of citrus products in preventing stones in patients with hypocitraturia is controversial, however, and needs to be studied more.
One study46 demonstrated that lemon juice was beneficial in hypocitraturic nephrolithiasis: 4 oz/day of lemon juice concentrate in the form of lemonade was associated with an increase in urinary citrate excretion to 346 mg/day from 142 mg/day in 11 of 12 patients who participated.
Odvina47 compared the effects of orange juice with those of lemonade on the acid-base profile and urinary stone risk under controlled metabolic conditions in 13 volunteers. Orange juice was reported to have greater alkalinizing and citraturic effects and was associated with lower calculated calcium oxalate supersaturation compared with lemonade.
Lemonade therapy may be used as adjunctive treatment in patients who do not comply with or cannot tolerate alkali therapy. However, we advise caution about recommending citrus products, as they can increase oxalate excretion.
Pharmacotherapy includes alkali therapy. Barcelo et al48 compared the effects of potassium citrate and placebo in 57 patients with calcium oxalate stones and hypocitraturia. Patients treated with potassium citrate had a rate of stone formation of 0.1 event per patient per year, compared with 1.1 in the placebo group.
Many forms of alkaline citrate are available. Potassium citrate is preferred over sodium citrate since the latter may increase urine calcium excretion.49 Treatment is usually started at 30 mEq/day and is titrated to a maximal dose of 60 mEq/day for a urinary citrate excretion greater than 500 mg/day.
Common side effects are abdominal bloating and hyperkalemia (especially with renal insufficiency), and in such cases sodium-based alkali, sodium citrate, or sodium bicarbonate can be prescribed.
PREVENTING CALCIUM PHOSPHATE STONES
Risk factors for calcium phosphate stones are similar to those for calcium oxalate stones (other than hyperoxaluria), but calcium phosphate stones are formed in alkaline urine (usually urine pH > 6.0), often the result of distal renal tubular acidosis. Preventive measures are similar to those for calcium oxalate stones.
Alkali therapy should be used with caution because of its effect on urinary pH and the risk of precipitation of calcium phosphate crystals.50 Use of potassium citrate was found to be associated with increases in both urinary citrate excretion and calcium phosphate supersaturation in hypercalciuric stone-forming rats.51 It is therefore challenging to manage patients with calcium phosphate stones and hypocitraturia. Alkali administration in this setting may diminish the formation of new stones by correcting hypocitraturia, but at the same time it may increase the likelihood of calcium phosphate stone formation by increasing the urinary pH. When the urine pH increases to above 6.5 with no significant change in urine citrate or urine calcium excretion, we recommend stopping alkali therapy.
PREVENTING URIC ACID STONES
Clinical conditions associated with uric acid stones include metabolic syndrome, diabetes mellitus, gout, chronic diarrheal illness, and conditions that increase tissue turnover and uric acid production, such as malignancies. Other risk factors for uric acid stone formation are low urine volume, low uric pH, and hyperuricosuria.
Abnormally acidic urine is the most common risk factor. Metabolic syndrome and diabetes mellitus reduce ammonia production, resulting in a lower urinary pH, which predisposes to uric acid stone formation. Chronic diarrhea also acidifies the urine by loss of bicarbonate. Similarly, in gout, the predisposing factor in uric acid stone formation is the persistently acidic urine due to impaired ammonium excretion.52 Uric acid precipitates to form uric acid stones in a low urinary pH even with normal excretion rates of 600 to 800 mg/day and a urinary volume of 1 to 1.5 L.53
Therefore, apart from increasing fluid intake, urinary alkalization is the cornerstone of management of uric acid stones. Potassium citrate is the preferred alkali salt and is started at a dose of 30 mEq/day for a goal urinary pH of 6 to 6.5.47
Patients with hyperuricosuria are also advised to restrict their protein intake to no more than 0.8 to 1 mg/kg/day.
If the above measures fail, patients are treated with a xanthine oxidase inhibitor, ie, allopurinol or febuxostat, even if their uric acid excretion is normal.54
PREVENTING STRUVITE STONES
Struvite stones contain magnesium ammonium phosphate and are due to chronic upper urinary tract infection with urea-splitting bacteria such as Proteus, Klebsiella, Pseudomonas, and enterococci. Urea hydrolysis releases hydroxyl ions, resulting in alkaline urine that promotes struvite stone formation. Early detection and treatment are important, since struvite stones are associated with morbidity and rapid progression.
Medical treatment of struvite stones is usually unsuccessful, and the patient is referred to a urologist for surgical removal of the stones, the gold standard treatment.55 Long-term use of culture-specific antibiotics to prevent new stone growth is not well studied. Medical therapy by itself is preferred in patients who refuse stone removal or cannot tolerate it. Urease inhibitors such as acetohydroxamic acid have been successful in preventing or slowing stone growth, but their use is limited by frequent side effects such as nausea, headache, rash, and thrombophlebitis.56
CYSTINE STONES
Cystine stones occur in people with inherited defects of renal tubular and intestinal transport of cysteine and dibasic amino acids that cause excessive excretion of urinary cystine, ie, 480 to 3,600 mg/day.
Cystine is formed from two cysteine molecules linked by a disulfide bond. The solubility of cystine is pH-dependent, with increased solubility at higher urinary pH. The goal is to maintain a urinary cystine concentration below its solubility level by keeping the cystine concentration below 243 mg/L and the urine cystine supersaturation (the ratio of the urine cysteine concentration to the cysteine solubility in the same sample) less than 0.6.57 Therapy is aimed at increasing daily urinary volume to 3 L and urine alkalization to pH above 7, in order to increase cystine solubility by 300%.58
Overnight dehydration should be prevented, and patients should be encouraged to wake up at least once a night to void and drink additional water. Sodium restriction to 100 mmol/day (2,300 mg/day) and moderate protein restriction to 0.8 to 1 g/kg/day are associated with decreased cystine excretion, but long-term studies demonstrating their benefit in preventing cystine stones are lacking.59
A thiol-containing drug, eg, D-penicillamine (0.5–2 g/day) or tiopronin (400–1,200 mg/day), should be added to the conservative measures if they have not been effective for 3 months or if there is history of noncompliance.60 Thiol-containing drugs have a sulfhydryl group that reduces the disulfide bond, and they form soluble disulfide cysteine-drug complexes with greater ability to solubilize cystine in alkaline urine. They must always be used in conjunction with fluid and alkali therapy.61
Both drugs have severe and common adverse effects including leukopenia, aplastic anemia, fever, rash, arthritis, hepatotoxicity, pyridoxine deficiency, and proteinuria (membranous nephropathy). However, tiopronin seems to have a lesser incidence of side effects.62 Regular monitoring of complete blood cell counts, liver enzymes, and urine protein should be done.
Captopril contains a sulfhydryl group, and the captopril-cysteine disulfide is more soluble than cysteine alone. The amount of captopril that appears in the urine is low, and doses of 150 mg/day are usually required to reduce cysteine excretion, which can lead to hypotension. The efficacy of captopril in treating cystine stones is unproven, and this drug is used only if patients cannot tolerate other thiol-containing drugs.63
Nephrolithiasis is common and often recurs. This review focuses on measures to prevent recurrent stone formation. Some measures apply to all patients, and some apply to specific types of stones.
COMMON AND INCREASING
According to data from the 2007–2010 National Health and Nutrition Examination Survey, the prevalence of nephrolithiasis in the United States was 10.6% in men and 7.1% in women. On average, 1 in 11 Americans will develop kidney stones at least once in their lifetime.1
By race and sex, white men have the highest incidence of nephrolithiasis and Asian women have the lowest. It is less common before age 20 and peaks in incidence in the third and fourth decades of life.
The prevalence has steadily increased in the past few decades (Table 1),1,2 but the reasons are not clear. The trend may be due to changes in diet and lifestyle, increasing prevalence of obesity and diabetes, migration from rural to urban areas, and global warming, with higher temperature resulting in dehydration and high urinary concentration of calcium and other stone-forming salts.3 Nephrolithiasis is now recognized as a systemic disorder associated with chronic kidney disease, bone loss and fractures, increased risk of coronary artery disease, hypertension, type 2 diabetes mellitus, and metabolic syndrome (Table 2).4–7
Without medical treatment, the 5-year recurrence rate is high, ranging from 35% to 50% after an initial stone event.8 Annual medical costs of care for kidney stones in the United States exceed $4.5 billion, with additional costs from missed work. Therefore, this condition has a considerable economic and social burden, which underscores the importance of prevention.9
MOST STONES CONTAIN CALCIUM
About 80% of kidney stones in adults contain calcium, and calcium oxalate stones are more common than calcium phosphate stones. Uric acid and struvite stones account for 5% to 15%, and cystine, protease inhibitor, triamterene, 2,8-dihydroxyadenine (2,8-DHA) and xanthine stones each account for less than 1%.10
Stones form when the urinary concentration of stone-forming salts, which is inversely proportional to urine volume, is higher than their saturation point, which is affected by urine pH. Acidic urine (low pH) predisposes to the formation of uric acid and cystine stones, whereas alkaline urine (high pH) favors calcium phosphate stones.
INCREASED FLUID INTAKE FOR ALL
High fluid intake, enough to produce at least 2.5 L of urine per day, should be the initial therapy to prevent stone recurrence.11
Borghi et al12 randomly assigned 199 patients who had a first calcium stone to high oral fluid intake or no intervention and followed them prospectively for 5 years. The recurrence rate was 12% in the treated group and 27% in the control group. Another study, in patients who had undergone shock wave lithotripsy, found a recurrence rate of 8% in those randomized to increase fluid intake to achieve urine output greater than 2.5 L/day, compared with 56% in those assigned to no treatment.13
Certain beverages increase the risk of stones and should be avoided. Sugar-sweetened noncola soda and punch are associated with a 33% higher risk of kidney stones, and cola sodas are associated with a 23% higher risk.14 Prospective studies have shown that the consumption of coffee, beer, wine, and orange juice is associated with a lower likelihood of stone formation.13,15
Table 3 is a brief summary of the dietary and pharmacologic interventions in the management of recurrent nephrolithiasis.
PREVENTING CALCIUM OXALATE STONES
Major urinary risk factors associated with calcium oxalate stones are hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, and low urine volume.16 Preventing calcium stones therefore depends on reducing the urinary concentration of calcium and oxalate, increasing urinary levels of inhibitors such as citrate, and increasing urine volume.
Reducing calcium excretion
Hypercalciuria has been traditionally defined as 24-hour urinary calcium excretion greater than 300 mg/day in men, greater than 250 mg in women, or greater than 4 mg/kg in men or women.17 It is a graded risk factor, and the cut points used in published research and clinical laboratories vary substantially. Some institutions use the same value for hypercalciuria in both sexes, eg, greater than 200 mg/day.18
Excessive sodium intake is the most common cause of hypercalciuria. Systemic conditions such as primary hyperparathyroidism, sarcoidosis, and renal tubular acidosis also cause hypercalciuria but are uncommon.19 Management depends on the underlying cause and includes dietary modifications and pharmacologic therapy.
Dietary modifications have a pivotal role in the management of recurrent stones that are due to hypercalciuria.
Dietary calcium should not be restricted, since calcium reduces the excretion of urinary oxalate by decreasing intestinal absorption of oxalate. Guidelines from the American Urological Association recommend a daily calcium intake of 1,000 to 1,200 mg.11–20 Moreover, restriction of dietary calcium to less than 800 mg/day (the current recommended daily allowance for adults) can lead to negative calcium balance and bone loss.
Sodium intake also influences hypercalciuria. Calcium is reabsorbed passively in the proximal tubule due to the concentration gradient created by active reabsorption of sodium. A high sodium intake causes volume expansion, leading to a decrease in proximal sodium and calcium reabsorption and enhancing calcium excretion. A low-sodium diet (80–100 mmol/day, or 1,800–2,300 mg/day) is recommended. This enhances proximal sodium and passive calcium absorption and leads to a decrease in calcium excretion.21
Dietary protein increases the acid load by production of sulfuric acid and leads to hypercalciuria by its action on bone and kidney. Animal protein has a higher content of sulfur and generates a higher acid load compared with vegetable protein and has been associated with an increased incidence of stone formation, at least in men.20,22 Borghi et al23 reported that the combination of restricted intake of animal protein (52 g/day), restricted salt intake (50 mmol, or 2,900 mg/day of sodium chloride), and normal calcium intake (30 mmol/day, or 1,200 mg/day) was associated with a lower incidence of stone recurrence in men with hypercalciuria compared with traditional low-calcium intake (10 mmol, or 400 mg/day). Patients should therefore be advised to avoid excessive intake of animal protein.
Increasing the dietary intake of fruits and vegetables as in the Dietary Approach to Stop Hypertension (DASH) diet is beneficial and reduces the risk of stone recurrence, mainly by increasing citrate excretion.24
Pharmacologic therapy in hypercalciuria. Thiazide diuretics are the mainstay of pharmacotherapy for preventing recurrent stones in patients with idiopathic hypercalciuria. They reduce the risk of stone recurrence by about 50%, as reported in a recent meta-analysis that looked at five trials comparing thiazide diuretics with placebo.25 They lower calcium excretion by causing volume depletion, thereby increasing proximal sodium and passive calcium reabsorption.
Chlorthalidone and hydrochlorothiazide are the thiazides commonly used to treat hypercalciuria. The dosage is titrated to the urinary calcium excretion, and a common mistake is to use doses that are too low. They are usually started at 25 mg/day, but often require an increase to 50 to 100 mg/day for adequate lowering of urinary calcium.
Care should be taken to avoid hypokalemia. If it occurs, it can be corrected by adding the potassium-sparing diuretic amiloride (5–10 mg/day), which increases calcium reabsorption in collecting ducts or, in patients with hypocitraturia, potassium citrate-potassium bicarbonate. (Sodium salts should be avoided, since they increase renal calcium excretion.)26
Management of hypercalciuria with metabolic causes, which include primary hyperparathyroidism and chronic acidemia. Patients who have hypercalciuria from primary hyperparathyroidism are treated with parathyroidectomy.27 Chronic metabolic acidosis causes hypercalciuria by loss of bone calcium and hypocitraturia by increasing active proximal absorption of citrate. Potassium citrate or potassium bicarbonate is used to prevent stones in such patients; sodium salts should be avoided.28
Reducing oxalate excretion
Hyperoxaluria has traditionally been defined as urinary oxalate excretion of more than 45 mg/day. However, the optimal cutoff point for urinary oxalate excretion is unclear, as is the optimal cutoff for hypercalciuria. The risk of stone formation has been shown to increase with oxalate excretion even above 25 mg/day, which is within the normal limit.18
Idiopathic hyperoxaluria. High dietary oxalate intake, especially when associated with low calcium intake, leads to idiopathic hyperoxaluria. However, the contribution of abnormal endogenous oxalate metabolism is uncertain. Ingested calcium binds to oxalate in the intestinal tract and reduces both the absorption of intestinal oxalate absorption and the excretion of urinary oxalate.29 High dietary oxalate intake has usually been regarded as a major risk factor for kidney stones.
Taylor and Curhan,30 in a prospective study, reported a mild increase in the risk of stones in the highest quintile of dietary oxalate intake compared with the lowest quintile for men (relative risk [RR] 1.22, 95% confidence interval [CI] 1.03–1.45) and older women (RR 1.21, 95% CI 1.01–1.44). They also demonstrated that eating eight or more servings of spinach per month compared with fewer than one serving per month was associated with a similar increase of stone risk in men (RR 1.30, 95% CI 1.08–1.58) and older women (RR 1.34 95% CI 1.1–1.64). In contrast, spinach and dietary oxalate intake did not increase the risk of nephrolithiasis in young women. The authors concluded that the risk associated with oxalate intake was modest, and their data did not support the contention that dietary oxalate is a major risk factor for kidney stones.
Higher oxalate intake increases urinary oxalate excretion and presumably the risk of nephrolithiasis. Limiting dietary oxalate to prevent stones is recommended if habitually high dietary intake of oxalate is identified or follow-up urine measurements show a decrease in oxalate excretion.31 Foods rich in oxalate include spinach, rhubarb, nuts, legumes, cocoa, okra, and chocolate.
The DASH diet, which is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein, is an effective dietary alternative and has been associated with a lower risk of calcium oxalate stones.24 Consuming fruits and vegetables increases the excretion of urinary citrate, which is an inhibitor of stone formation. Also, it has been proposed that the DASH diet contains unknown factors that reduce stone risk.
Taylor et al32 prospectively examined the relationship between the DASH diet and the incidence of kidney stones and found that the diet significantly reduced the risk of kidney stones. The relative risks of occurrence of kidney stones in participants in the highest quintile of the DASH score (a measure of adherence to the DASH diet) compared with the lowest quintile were 0.55 (95% CI 0.46–0.65) for men, 0.58 (95% CI 0.49–0.68) for older women, and 0.60 (95% CI 0.52–0.70) for younger women, which the authors characterized as “a marked decrease in kidney stone risk.”
Vitamin C intake should be restricted to 90 mg/day in patients who have a history of calcium oxalate stones. Urivetzky et al33 found that urinary oxalate excretion increased by 6 to 13 mg/day at doses of ascorbic acid greater than 500 mg.
Pyridoxine (vitamin B6), a coenzyme of alanine-glyoxylate aminotransferase (AGT), increases the conversion of glyoxylate to glycine instead of oxalate and is used in the treatment of type 1 primary hyperoxaluria (see below).34 However, its effect in preventing stones in idiopathic hyperoxaluria is not well known, and it has not been studied in a randomized controlled trial. In a prospective study, Curhan et al35 reported that high intake of pyridoxine (> 40 mg/day) was associated with a lower risk of stone formation in women, but no such benefit was found in men.
Enteric hyperoxaluria. About 90% of dietary oxalate binds to calcium in the small intestine and is excreted in the stool. The remaining 10% is absorbed in the colon and is secreted in urine. Hyperoxaluria is frequently seen with fat malabsorption from inflammatory bowel disease, short gut syndrome, and gastric bypass surgery. In these conditions, excess fat binds to dietary calcium, leading to increased absorption of free oxalate in the colon.36
Treatment is directed at decreasing intestinal oxalate absorption and should include high fluid intake and oral calcium supplements. Calcium carbonate or citrate causes precipitation of oxalate in the intestinal lumen and is prescribed as 1 to 4 g in three to four divided doses, always with meals. Calcium citrate is preferred over calcium carbonate in stone-formers because of the benefit of citrate and calcium citrate’s higher solubility and greater effectiveness in the presence of achlorhydria.37 Patients should be advised to avoid foods high in oxalate and fat.
Primary hyperoxaluria is caused by inherited inborn errors of glyoxylate metabolism that cause overproduction of oxalate and urinary oxalate excretion above 135 to 270 mg/day.
Type 1 primary hyperoxaluria is the most common (accounting for 90% of cases) and is caused by reduced activity of hepatic peroxisomal AGT.
Type 2 is from a deficiency of glyoxylate reductase-hydroxypyruvate reductase (GRHPR).
Type 3 is from mutations in the HOGA1 gene, which codes for the liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme involved in degradation of hydroxyproline to pyruvate and glyoxalate.38
High fluid intake to produce a urinary volume of 3 L/day reduces intratubular oxalate deposition and should be encouraged. Potassium citrate (0.15 mg/kg), oral phosphate supplements (30–40 mg/kg of orthophosphate), and magnesium oxide (500 mg/day/m2) inhibit precipitation of calcium oxalate in the urine.39,40 Pyridoxine, a coenzyme of AGT, increases the conversion of glyoxylate to glycine instead of oxalate and is prescribed at a starting dose of 5 mg/kg (which can be titrated up to 20 mg/kg if there is no response) in patients with type 1 primary hyperoxaluria. About 50% of patients with type 1 respond successfully to pyridoxine, and a 3- to 6-month trial should be given in all patients in this category.34 AGT is present only in hepatocytes, and GRHPR is found in multiple tissues; therefore, combined liver-kidney transplant is the treatment of choice in patients with type 1 primary hyperoxaluria, whereas isolated kidney transplant is recommended in patients with type 2.41
Reducing uric acid excretion
Hyperuricosuria is defined as uric acid excretion of greater than 800 mg/day in men and greater than 750 mg/day in women.
The association of hyperuricosuria with increased risk of calcium oxalate stone formation is controversial. Curhan and Taylor,18 in a cross-sectional study of 3,350 men and women, reported that there was no difference in mean 24-hour uric acid excretion in individuals with and without a history of stones.
The mechanism by which uric acid leads to calcium oxalate stones is not completely known and could be the “salting out” of calcium oxalate from the urine.42
Dietary purine restriction, ie, limiting intake of nondairy animal protein to 0.8 to 1 g/kg/day, is the initial dietary intervention.11 Allopurinol is the alternative approach if the patient is not compliant or if dietary restriction fails.43
In a study by Ettinger et al,44 60 patients with hyperuricosuria and normocalciuria were randomized to receive allopurinol (100 mg three times daily) or a placebo. The allopurinol group had a rate of calculus events of 0.12 per patient per year, compared with 0.26 in the placebo group.
Increasing citrate excretion
Hypocitraturia is a well-known risk factor for the formation of kidney stones. It is usually defined as a citrate excretion of less than 320 mg/day for adults.
Citrate prevents formation of calcium crystals by binding to calcium, thereby lowering the concentration of calcium oxalate below the saturation point.45
Diet therapy. Patients with calcium oxalate stones and hypocitraturia should be encouraged to increase their intake of fruits and vegetables, which enhances urinary citrate excretion, and to limit their intake of nondairy animal protein.11
The use of citrus products in preventing stones in patients with hypocitraturia is controversial, however, and needs to be studied more.
One study46 demonstrated that lemon juice was beneficial in hypocitraturic nephrolithiasis: 4 oz/day of lemon juice concentrate in the form of lemonade was associated with an increase in urinary citrate excretion to 346 mg/day from 142 mg/day in 11 of 12 patients who participated.
Odvina47 compared the effects of orange juice with those of lemonade on the acid-base profile and urinary stone risk under controlled metabolic conditions in 13 volunteers. Orange juice was reported to have greater alkalinizing and citraturic effects and was associated with lower calculated calcium oxalate supersaturation compared with lemonade.
Lemonade therapy may be used as adjunctive treatment in patients who do not comply with or cannot tolerate alkali therapy. However, we advise caution about recommending citrus products, as they can increase oxalate excretion.
Pharmacotherapy includes alkali therapy. Barcelo et al48 compared the effects of potassium citrate and placebo in 57 patients with calcium oxalate stones and hypocitraturia. Patients treated with potassium citrate had a rate of stone formation of 0.1 event per patient per year, compared with 1.1 in the placebo group.
Many forms of alkaline citrate are available. Potassium citrate is preferred over sodium citrate since the latter may increase urine calcium excretion.49 Treatment is usually started at 30 mEq/day and is titrated to a maximal dose of 60 mEq/day for a urinary citrate excretion greater than 500 mg/day.
Common side effects are abdominal bloating and hyperkalemia (especially with renal insufficiency), and in such cases sodium-based alkali, sodium citrate, or sodium bicarbonate can be prescribed.
PREVENTING CALCIUM PHOSPHATE STONES
Risk factors for calcium phosphate stones are similar to those for calcium oxalate stones (other than hyperoxaluria), but calcium phosphate stones are formed in alkaline urine (usually urine pH > 6.0), often the result of distal renal tubular acidosis. Preventive measures are similar to those for calcium oxalate stones.
Alkali therapy should be used with caution because of its effect on urinary pH and the risk of precipitation of calcium phosphate crystals.50 Use of potassium citrate was found to be associated with increases in both urinary citrate excretion and calcium phosphate supersaturation in hypercalciuric stone-forming rats.51 It is therefore challenging to manage patients with calcium phosphate stones and hypocitraturia. Alkali administration in this setting may diminish the formation of new stones by correcting hypocitraturia, but at the same time it may increase the likelihood of calcium phosphate stone formation by increasing the urinary pH. When the urine pH increases to above 6.5 with no significant change in urine citrate or urine calcium excretion, we recommend stopping alkali therapy.
PREVENTING URIC ACID STONES
Clinical conditions associated with uric acid stones include metabolic syndrome, diabetes mellitus, gout, chronic diarrheal illness, and conditions that increase tissue turnover and uric acid production, such as malignancies. Other risk factors for uric acid stone formation are low urine volume, low uric pH, and hyperuricosuria.
Abnormally acidic urine is the most common risk factor. Metabolic syndrome and diabetes mellitus reduce ammonia production, resulting in a lower urinary pH, which predisposes to uric acid stone formation. Chronic diarrhea also acidifies the urine by loss of bicarbonate. Similarly, in gout, the predisposing factor in uric acid stone formation is the persistently acidic urine due to impaired ammonium excretion.52 Uric acid precipitates to form uric acid stones in a low urinary pH even with normal excretion rates of 600 to 800 mg/day and a urinary volume of 1 to 1.5 L.53
Therefore, apart from increasing fluid intake, urinary alkalization is the cornerstone of management of uric acid stones. Potassium citrate is the preferred alkali salt and is started at a dose of 30 mEq/day for a goal urinary pH of 6 to 6.5.47
Patients with hyperuricosuria are also advised to restrict their protein intake to no more than 0.8 to 1 mg/kg/day.
If the above measures fail, patients are treated with a xanthine oxidase inhibitor, ie, allopurinol or febuxostat, even if their uric acid excretion is normal.54
PREVENTING STRUVITE STONES
Struvite stones contain magnesium ammonium phosphate and are due to chronic upper urinary tract infection with urea-splitting bacteria such as Proteus, Klebsiella, Pseudomonas, and enterococci. Urea hydrolysis releases hydroxyl ions, resulting in alkaline urine that promotes struvite stone formation. Early detection and treatment are important, since struvite stones are associated with morbidity and rapid progression.
Medical treatment of struvite stones is usually unsuccessful, and the patient is referred to a urologist for surgical removal of the stones, the gold standard treatment.55 Long-term use of culture-specific antibiotics to prevent new stone growth is not well studied. Medical therapy by itself is preferred in patients who refuse stone removal or cannot tolerate it. Urease inhibitors such as acetohydroxamic acid have been successful in preventing or slowing stone growth, but their use is limited by frequent side effects such as nausea, headache, rash, and thrombophlebitis.56
CYSTINE STONES
Cystine stones occur in people with inherited defects of renal tubular and intestinal transport of cysteine and dibasic amino acids that cause excessive excretion of urinary cystine, ie, 480 to 3,600 mg/day.
Cystine is formed from two cysteine molecules linked by a disulfide bond. The solubility of cystine is pH-dependent, with increased solubility at higher urinary pH. The goal is to maintain a urinary cystine concentration below its solubility level by keeping the cystine concentration below 243 mg/L and the urine cystine supersaturation (the ratio of the urine cysteine concentration to the cysteine solubility in the same sample) less than 0.6.57 Therapy is aimed at increasing daily urinary volume to 3 L and urine alkalization to pH above 7, in order to increase cystine solubility by 300%.58
Overnight dehydration should be prevented, and patients should be encouraged to wake up at least once a night to void and drink additional water. Sodium restriction to 100 mmol/day (2,300 mg/day) and moderate protein restriction to 0.8 to 1 g/kg/day are associated with decreased cystine excretion, but long-term studies demonstrating their benefit in preventing cystine stones are lacking.59
A thiol-containing drug, eg, D-penicillamine (0.5–2 g/day) or tiopronin (400–1,200 mg/day), should be added to the conservative measures if they have not been effective for 3 months or if there is history of noncompliance.60 Thiol-containing drugs have a sulfhydryl group that reduces the disulfide bond, and they form soluble disulfide cysteine-drug complexes with greater ability to solubilize cystine in alkaline urine. They must always be used in conjunction with fluid and alkali therapy.61
Both drugs have severe and common adverse effects including leukopenia, aplastic anemia, fever, rash, arthritis, hepatotoxicity, pyridoxine deficiency, and proteinuria (membranous nephropathy). However, tiopronin seems to have a lesser incidence of side effects.62 Regular monitoring of complete blood cell counts, liver enzymes, and urine protein should be done.
Captopril contains a sulfhydryl group, and the captopril-cysteine disulfide is more soluble than cysteine alone. The amount of captopril that appears in the urine is low, and doses of 150 mg/day are usually required to reduce cysteine excretion, which can lead to hypotension. The efficacy of captopril in treating cystine stones is unproven, and this drug is used only if patients cannot tolerate other thiol-containing drugs.63
- Scales CD Jr, Smith AC, Hanley JM, Saigal CS; Urologic Diseases in America Project. Prevalence of kidney stones in the United States. Eur Urol 2012; 62:160–165.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM Jr, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence, and associated risk factors. Rev Urol 2010; 12:e86–e96.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2011; 79:393–403.
- Sakhaee K. Nephrolithiasis as a systemic disorder. Curr Opin Nephrol Hypertens 2008; 17:304–309.
- Hamano S, Nakatsu H, Suzuki N, Tomioka S, Tanaka M, Murakami S. Kidney stone disease and risk factors for coronary heart disease. Int J Urol 2005; 12:859–863.
- Ritz E. Metabolic syndrome: an emerging threat to renal function. Clin J Am Soc Nephrol 2007; 2:869–871.
- Uribarri J, Oh MS, Carroll HJ. The first kidney stone. Ann Intern Med 1989; 111:1006–1009.
- Saigal CS, Joyce G, Timilsina AR; Urologic Diseases in America Project. Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 2005; 68:1808–1814.
- Moe OW. Kidney stones: pathophysiology and medical management. Lancet 2006; 367:333–344.
- Pearle MS, Goldfarb DS, Assimos DG, et al; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol 2014; 192:316–324.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Sarica K, Inal Y, Erturhan S, Yagci F. The effect of calcium channel blockers on stone regrowth and recurrence after shock wave lithotripsy. Urol Res 2006; 34:184–189.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013; 8:1389–1395.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Beverage use and risk for kidney stones in women. Ann Intern Med 1998; 128:534–540.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med 1980; 69:19–30.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int 2008; 73:489–496.
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest 2005; 115:2598–2608.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Muldowney FP, Freaney R, Moloney MF. Importance of dietary sodium in the hypercalciuria syndrome. Kidney Int 1982; 22:292–296.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Noori N, Honarkar E, Goldfarb DS, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis 2014; 63:456–463.
- Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013; 158:535–543.
- Alon U, Costanzo LS, Chan JC. Additive hypocalciuric effects of amiloride and hydrochlorothiazide in patients treated with calcitriol. Miner Electrolyte Metab 1984; 10:379–386.
- Corbetta S, Baccarelli A, Aroldi A, et al. Risk factors associated to kidney stones in primary hyperparathyroidism. J Endocrinol Invest 2005; 28:122–128.
- Haymann JP. Metabolic disorders: stones as first clinical manifestation of significant diseases. World J Urol 2015; 33:187–192.
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol 1985; 5:40–44.
- Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol 2007; 18:2198–2204.
- Lieske JC, Tremaine WJ, De Simone C, et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78:1178–1185.
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol 2009; 20:2253–2259.
- Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol 1992; 147:1215–1218.
- Hoyer-Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 2014; 9:468–477.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol 1999; 10:840–845.
- Parks JH, Worcester EM, O'Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant 1998; 13:2241–2247.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009; 75:1264–1271.
- Cochat P, Hulton SA, Acquaviva C, et al; OxalEurope. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27:1729–1736.
- Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993; 7:207–211.
- Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010; 10:2493–2501.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implications for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Coe FL, Parks JH. Hyperuricosuria and calcium nephrolithiasis. Urol Clin North Am 1981; 8:227–244.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol 2009; 11:134–144.
- Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol 1996; 156:907–909.
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol 2006; 1:1269–1274.
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol 1993; 150:1761–1764.
- Lemann J Jr, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int 1989; 35:688–695.
- Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore) 1991; 70:345–359.
- Krieger NS, Asplin JR, Frick KK, et al. Effect of potassium citrate on calcium phosphate stones in a model of hypercalciuria. J Am Soc Nephrol 2015; 26:3001–3008.
- Falls WF Jr. Comparison of urinary acidification and ammonium excretion in normal and gouty subjects. Metabolism 1972; 21:433–445.
- Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. N Engl J Med 1992; 327:1141–1152.
- Kenny JE, Goldfarb DS. Update on the pathophysiology and management of uric acid renal stones. Curr Rheumatol Rep 2010; 12:125–129.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS Jr (AUA Nephrolithiasis Guideline Panel). Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med 1984; 311:760–764.
- Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol 2000; 164:1481–1485.
- Palacın MGP, Nunes V, Gasparini P. Cystinuria. In: Shriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4909–4932.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006; 69:1041–1047.
- Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163:1419–1423.
- Asplin DM, Asplin JR. The Interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013; 189:2147–2151.
- Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med 2006; 17:343–348.
- Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
- Scales CD Jr, Smith AC, Hanley JM, Saigal CS; Urologic Diseases in America Project. Prevalence of kidney stones in the United States. Eur Urol 2012; 62:160–165.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM Jr, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence, and associated risk factors. Rev Urol 2010; 12:e86–e96.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2011; 79:393–403.
- Sakhaee K. Nephrolithiasis as a systemic disorder. Curr Opin Nephrol Hypertens 2008; 17:304–309.
- Hamano S, Nakatsu H, Suzuki N, Tomioka S, Tanaka M, Murakami S. Kidney stone disease and risk factors for coronary heart disease. Int J Urol 2005; 12:859–863.
- Ritz E. Metabolic syndrome: an emerging threat to renal function. Clin J Am Soc Nephrol 2007; 2:869–871.
- Uribarri J, Oh MS, Carroll HJ. The first kidney stone. Ann Intern Med 1989; 111:1006–1009.
- Saigal CS, Joyce G, Timilsina AR; Urologic Diseases in America Project. Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 2005; 68:1808–1814.
- Moe OW. Kidney stones: pathophysiology and medical management. Lancet 2006; 367:333–344.
- Pearle MS, Goldfarb DS, Assimos DG, et al; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol 2014; 192:316–324.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Sarica K, Inal Y, Erturhan S, Yagci F. The effect of calcium channel blockers on stone regrowth and recurrence after shock wave lithotripsy. Urol Res 2006; 34:184–189.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013; 8:1389–1395.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Beverage use and risk for kidney stones in women. Ann Intern Med 1998; 128:534–540.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med 1980; 69:19–30.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int 2008; 73:489–496.
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest 2005; 115:2598–2608.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Muldowney FP, Freaney R, Moloney MF. Importance of dietary sodium in the hypercalciuria syndrome. Kidney Int 1982; 22:292–296.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Noori N, Honarkar E, Goldfarb DS, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis 2014; 63:456–463.
- Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013; 158:535–543.
- Alon U, Costanzo LS, Chan JC. Additive hypocalciuric effects of amiloride and hydrochlorothiazide in patients treated with calcitriol. Miner Electrolyte Metab 1984; 10:379–386.
- Corbetta S, Baccarelli A, Aroldi A, et al. Risk factors associated to kidney stones in primary hyperparathyroidism. J Endocrinol Invest 2005; 28:122–128.
- Haymann JP. Metabolic disorders: stones as first clinical manifestation of significant diseases. World J Urol 2015; 33:187–192.
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol 1985; 5:40–44.
- Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol 2007; 18:2198–2204.
- Lieske JC, Tremaine WJ, De Simone C, et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78:1178–1185.
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol 2009; 20:2253–2259.
- Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol 1992; 147:1215–1218.
- Hoyer-Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 2014; 9:468–477.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol 1999; 10:840–845.
- Parks JH, Worcester EM, O'Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant 1998; 13:2241–2247.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009; 75:1264–1271.
- Cochat P, Hulton SA, Acquaviva C, et al; OxalEurope. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27:1729–1736.
- Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993; 7:207–211.
- Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010; 10:2493–2501.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implications for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Coe FL, Parks JH. Hyperuricosuria and calcium nephrolithiasis. Urol Clin North Am 1981; 8:227–244.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol 2009; 11:134–144.
- Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol 1996; 156:907–909.
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol 2006; 1:1269–1274.
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol 1993; 150:1761–1764.
- Lemann J Jr, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int 1989; 35:688–695.
- Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore) 1991; 70:345–359.
- Krieger NS, Asplin JR, Frick KK, et al. Effect of potassium citrate on calcium phosphate stones in a model of hypercalciuria. J Am Soc Nephrol 2015; 26:3001–3008.
- Falls WF Jr. Comparison of urinary acidification and ammonium excretion in normal and gouty subjects. Metabolism 1972; 21:433–445.
- Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. N Engl J Med 1992; 327:1141–1152.
- Kenny JE, Goldfarb DS. Update on the pathophysiology and management of uric acid renal stones. Curr Rheumatol Rep 2010; 12:125–129.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS Jr (AUA Nephrolithiasis Guideline Panel). Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med 1984; 311:760–764.
- Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol 2000; 164:1481–1485.
- Palacın MGP, Nunes V, Gasparini P. Cystinuria. In: Shriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4909–4932.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006; 69:1041–1047.
- Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163:1419–1423.
- Asplin DM, Asplin JR. The Interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013; 189:2147–2151.
- Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med 2006; 17:343–348.
- Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
KEY POINTS
- Nephrolithiasis is common and widespread, and its incidence and prevalence are increasing.
- Calcium stones are the most common type, and of these, calcium oxalate stones predominate.
- The most common risk factors for recurrent calcium stones are low urinary output, hypercalciuria, hyperoxaluria, hypocitraturia, and hyperuricosuria.
- Less common types of stones are usually associated with genetic abnormalities, infections, or medications.
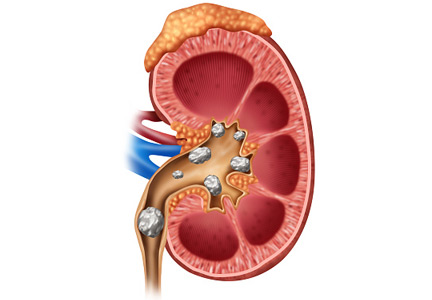
When does chest CT require contrast enhancement?
Computed tomography (CT) plays an important role in the diagnosis and treatment of many clinical conditions1 involving the chest wall, mediastinum, pleura, pulmonary arteries, and lung parenchyma. The need for enhancement with intravenous (IV) contrast depends on the specific clinical indication (Table 1).
EVALUATION OF SUSPECTED CANCER

CT is commonly used to diagnose, stage, and plan treatment for lung cancer, other primary neoplastic processes involving the chest, and metastatic disease.2 The need for contrast varies on a case-by-case basis, and the benefits of contrast should be weighed against the potential risks in each patient.
When the neoplasm has CT attenuation similar to that of adjacent structures (lymph nodes in the hilum, masses in the mediastinum or chest wall), IV contrast can improve identification of the lesion and delineation of its margins and the relationship with adjacent structures (eg, vascular structures) (Figure 1).
CT without contrast for screening
The diagnostic algorithm for lung cancer screening is evolving. The US Preventive Services Task Force currently recommends low-dose CT without contrast, along with appropriate patient counseling, for patients with a history of smoking and an age range as detailed in the Task Force statement.3
Follow-up of a solitary pulmonary nodule also typically does not require contrast enhancement, though some investigators have reported high sensitivity with dynamic contrast enhancement of pulmonary nodules.4 This represents a rare clinical application of chest CT with and without contrast.
EVALUATION OF THORACIC VASCULAR DISEASE
For the assessment of vascular disease, CT in most cases requires IV contrast to delineate the vessel lumen. Pulmonary embolic disease is the third most common cause of acute cardiovascular disease.5 CT pulmonary angiography is the most common way to assess for pulmonary embolic disease, as it is accurate, fast, and widely available, and can assess alternate pathologies in cases of undifferentiated chest pain. Contrast enhancement of the pulmonary arteries is key, as embolic disease is identified as abnormal filling defects within the pulmonary arteries (Figure 2).
Contrast enhancement is also used to evaluate superior vena cava syndrome. At our institution, the CT protocol includes concomitant injections in the upper-extremity veins, with imaging timed for venous phase enhancement (pulmonary venogram). In cases of suspected arteriovenous malformation, a protocol similar to that used for suspected pulmonary embolus is used (Figure 3), although in some instances, the imaging features of arteriovenous malformation may be detectable without IV contrast.
EVALUATION OF PULMONARY PARENCHYMAL DISEASE
Infection, inflammation, and edema of the lung parenchyma are usually well depicted on CT without contrast enhancement. However, contrast may be helpful if there are concerns about complications such as chest wall involvement, where contrast enhancement may help further delineate the extent of complications.
Assessment of interstitial lung disease does not require use of IV contrast; rather, a tailored protocol with thinner slices and noncontiguous expiratory images can be used to evaluate for air-trapping and dynamic airway compromise (Figure 4). Evaluation of chronic obstructive pulmonary disease also does not require IV contrast.
EVALUATION OF THE PLEURA
In pleural effusion, CT assessment for the presence, location, and extent of the effusion does not require contrast. However, contrast enhancement is used to evaluate suspected or known exudative effusions and empyema.6 It also aids the evaluation of metastatic or primary malignancy of the pleura, particularly in cases of occult disease, as enhancement and thickening of the pleura are of diagnostic interest.
EVALUATION OF AIRWAY DISEASE
Diseases of the large airway, such as stenosis and thickening, and diseases of the small airways, such as bronchiolitis, typically do not require contrast enhancement. At our institution, to assess dynamic airway narrowing, we use a dedicated airway protocol, including inspiratory and expiratory phases and multiplanar reformatted images.
EVALUATION OF STERNAL AND MEDIASTINAL INFECTIONS
Postoperative sternal wound infections are not uncommon and range from cellulitis to frank osteomyelitis. Mediastinitis may likewise be iatrogenic or may spread from the oropharynx. CT with contrast can help to depict infection of the chest wall or mediastinum and in some instances can also delineate the route of spread.7
TYPES OF IV CONTRAST MEDIA
Contrast media used in CT contain iodine, which causes increased absorption and scattering of radiation in body tissues and blood. Other contrast media, such as those used for magnetic resonance imaging or barium enemas, do not contain iodine. This absorption and scattering in turn results in higher CT attenuation values, or “enhancement” on CT images. The extent of enhancement depends on the amount and rate of contrast material administered, as well as on patient factors (eg, tissue vascularity, permeability, interstitial space) and the energy (tube voltage) of the incident x-rays.8
Adverse reactions
Contrast materials are generally safe; however, as with any pharmaceutical, there is the potential for adverse reactions. These reactions are relatively rare and are usually mild but occasionally can be severe.9 Anaphylactoid reactions have an unclear etiology but mimic allergic reactions, and they are more likely to occur in patients with a previous reaction to contrast and in patients with asthma or cardiovascular or renal disease.
Nonanaphylactoid reactions are dependent on contrast osmolality and on the volume and route of injection (unlike anaphylactoid reactions).10 Typical symptoms include warmth, metallic taste, and nausea or vomiting.
Contrast-related nephrotoxicity has been reported,11 although this has been challenged more recently.12 Suspected risk factors for this complication include advanced age, cardiovascular disease, treatment with chemotherapy, elevated serum creatinine level, dehydration, diabetes, use of nonsteroidal anti-inflammatory medications, myeloma,13 renal disease, and kidney transplant.
Detailed protocols for premedication and management of contrast adverse reactions are beyond the scope of this review and the reader is advised to refer to dedicated manuals.10
Acknowledgment: We are grateful for the editorial assistance of Megan M. Griffiths, scientific writer for the Imaging Institute, Cleveland Clinic.
- Rubin GD. Computed tomography: revolutionizing the practice of medicine for 40 years. Radiology 2014; 273(suppl 2):S45–S74.
- American College of Radiology. ACR-SCBT-MR-SPR practice parameter for the performance of thoracic computed tomography (CT). www.acr.org/~/media/ACR/Documents/PGTS/guidelines/CT_Thoracic.pdf. Accessed March 30, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2014; 160:330–338.
- Yi CA, Lee KS, Kim EA, et al. Solitary pulmonary nodules: dynamic enhanced multi-detector row CT study and comparison with vascular endothelial growth factor and microvessel density. Radiology 2004; 233:191–199.
- Bolen MA, Renapurkar RD, Popovic ZB, et al. High-pitch ECG-synchronized pulmonary CT angiography versus standard CT pulmonary angiography: a prospective randomized study. AJR Am J Roentgenol 2013; 201:971–976.
- Kraus GJ. The split pleura sign. Radiology 2007; 243:297–298.
- Bae KT. Intravenous contrast medium administration and scan timing at CT: considerations and approaches. Radiology 2010; 256:32–61.
- Capps EF, Kinsella JJ, Gupta M, Bhatki AM, Opatowsky MJ. Emergency imaging assessment of acute, nontraumatic conditions of the head and neck. Radiographics 2010; 30:1335–1352.
- Singh J, Daftary A. Iodinated contrast media and their adverse reactions. J Nucl Med Technol 2008; 36:69–74.
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. Version 10.1. 2015. www.acr.org/~/media/37D84428BF1D4E1B9A3A2918DA9E27A3.pdf. Accessed March 29, 2016.
- Barrett BJ. Contrast nephrotoxicity. J Am Soc Nephrol 1994; 5:125–137.
- McDonald RJ, McDonald JS, Carter RE, et al. Intravenous contrast material exposure is not an independent risk factor for dialysis or mortality. Radiology 2014; 273:714–725.
- McCarthy CS, Becker JA. Multiple myeloma and contrast media. Radiology 1992; 183:519–521.
Computed tomography (CT) plays an important role in the diagnosis and treatment of many clinical conditions1 involving the chest wall, mediastinum, pleura, pulmonary arteries, and lung parenchyma. The need for enhancement with intravenous (IV) contrast depends on the specific clinical indication (Table 1).
EVALUATION OF SUSPECTED CANCER
CT is commonly used to diagnose, stage, and plan treatment for lung cancer, other primary neoplastic processes involving the chest, and metastatic disease.2 The need for contrast varies on a case-by-case basis, and the benefits of contrast should be weighed against the potential risks in each patient.
When the neoplasm has CT attenuation similar to that of adjacent structures (lymph nodes in the hilum, masses in the mediastinum or chest wall), IV contrast can improve identification of the lesion and delineation of its margins and the relationship with adjacent structures (eg, vascular structures) (Figure 1).
CT without contrast for screening
The diagnostic algorithm for lung cancer screening is evolving. The US Preventive Services Task Force currently recommends low-dose CT without contrast, along with appropriate patient counseling, for patients with a history of smoking and an age range as detailed in the Task Force statement.3
Follow-up of a solitary pulmonary nodule also typically does not require contrast enhancement, though some investigators have reported high sensitivity with dynamic contrast enhancement of pulmonary nodules.4 This represents a rare clinical application of chest CT with and without contrast.
EVALUATION OF THORACIC VASCULAR DISEASE
For the assessment of vascular disease, CT in most cases requires IV contrast to delineate the vessel lumen. Pulmonary embolic disease is the third most common cause of acute cardiovascular disease.5 CT pulmonary angiography is the most common way to assess for pulmonary embolic disease, as it is accurate, fast, and widely available, and can assess alternate pathologies in cases of undifferentiated chest pain. Contrast enhancement of the pulmonary arteries is key, as embolic disease is identified as abnormal filling defects within the pulmonary arteries (Figure 2).
Contrast enhancement is also used to evaluate superior vena cava syndrome. At our institution, the CT protocol includes concomitant injections in the upper-extremity veins, with imaging timed for venous phase enhancement (pulmonary venogram). In cases of suspected arteriovenous malformation, a protocol similar to that used for suspected pulmonary embolus is used (Figure 3), although in some instances, the imaging features of arteriovenous malformation may be detectable without IV contrast.
EVALUATION OF PULMONARY PARENCHYMAL DISEASE
Infection, inflammation, and edema of the lung parenchyma are usually well depicted on CT without contrast enhancement. However, contrast may be helpful if there are concerns about complications such as chest wall involvement, where contrast enhancement may help further delineate the extent of complications.
Assessment of interstitial lung disease does not require use of IV contrast; rather, a tailored protocol with thinner slices and noncontiguous expiratory images can be used to evaluate for air-trapping and dynamic airway compromise (Figure 4). Evaluation of chronic obstructive pulmonary disease also does not require IV contrast.
EVALUATION OF THE PLEURA
In pleural effusion, CT assessment for the presence, location, and extent of the effusion does not require contrast. However, contrast enhancement is used to evaluate suspected or known exudative effusions and empyema.6 It also aids the evaluation of metastatic or primary malignancy of the pleura, particularly in cases of occult disease, as enhancement and thickening of the pleura are of diagnostic interest.
EVALUATION OF AIRWAY DISEASE
Diseases of the large airway, such as stenosis and thickening, and diseases of the small airways, such as bronchiolitis, typically do not require contrast enhancement. At our institution, to assess dynamic airway narrowing, we use a dedicated airway protocol, including inspiratory and expiratory phases and multiplanar reformatted images.
EVALUATION OF STERNAL AND MEDIASTINAL INFECTIONS
Postoperative sternal wound infections are not uncommon and range from cellulitis to frank osteomyelitis. Mediastinitis may likewise be iatrogenic or may spread from the oropharynx. CT with contrast can help to depict infection of the chest wall or mediastinum and in some instances can also delineate the route of spread.7
TYPES OF IV CONTRAST MEDIA
Contrast media used in CT contain iodine, which causes increased absorption and scattering of radiation in body tissues and blood. Other contrast media, such as those used for magnetic resonance imaging or barium enemas, do not contain iodine. This absorption and scattering in turn results in higher CT attenuation values, or “enhancement” on CT images. The extent of enhancement depends on the amount and rate of contrast material administered, as well as on patient factors (eg, tissue vascularity, permeability, interstitial space) and the energy (tube voltage) of the incident x-rays.8
Adverse reactions
Contrast materials are generally safe; however, as with any pharmaceutical, there is the potential for adverse reactions. These reactions are relatively rare and are usually mild but occasionally can be severe.9 Anaphylactoid reactions have an unclear etiology but mimic allergic reactions, and they are more likely to occur in patients with a previous reaction to contrast and in patients with asthma or cardiovascular or renal disease.
Nonanaphylactoid reactions are dependent on contrast osmolality and on the volume and route of injection (unlike anaphylactoid reactions).10 Typical symptoms include warmth, metallic taste, and nausea or vomiting.
Contrast-related nephrotoxicity has been reported,11 although this has been challenged more recently.12 Suspected risk factors for this complication include advanced age, cardiovascular disease, treatment with chemotherapy, elevated serum creatinine level, dehydration, diabetes, use of nonsteroidal anti-inflammatory medications, myeloma,13 renal disease, and kidney transplant.
Detailed protocols for premedication and management of contrast adverse reactions are beyond the scope of this review and the reader is advised to refer to dedicated manuals.10
Acknowledgment: We are grateful for the editorial assistance of Megan M. Griffiths, scientific writer for the Imaging Institute, Cleveland Clinic.
Computed tomography (CT) plays an important role in the diagnosis and treatment of many clinical conditions1 involving the chest wall, mediastinum, pleura, pulmonary arteries, and lung parenchyma. The need for enhancement with intravenous (IV) contrast depends on the specific clinical indication (Table 1).
EVALUATION OF SUSPECTED CANCER
CT is commonly used to diagnose, stage, and plan treatment for lung cancer, other primary neoplastic processes involving the chest, and metastatic disease.2 The need for contrast varies on a case-by-case basis, and the benefits of contrast should be weighed against the potential risks in each patient.
When the neoplasm has CT attenuation similar to that of adjacent structures (lymph nodes in the hilum, masses in the mediastinum or chest wall), IV contrast can improve identification of the lesion and delineation of its margins and the relationship with adjacent structures (eg, vascular structures) (Figure 1).
CT without contrast for screening
The diagnostic algorithm for lung cancer screening is evolving. The US Preventive Services Task Force currently recommends low-dose CT without contrast, along with appropriate patient counseling, for patients with a history of smoking and an age range as detailed in the Task Force statement.3
Follow-up of a solitary pulmonary nodule also typically does not require contrast enhancement, though some investigators have reported high sensitivity with dynamic contrast enhancement of pulmonary nodules.4 This represents a rare clinical application of chest CT with and without contrast.
EVALUATION OF THORACIC VASCULAR DISEASE
For the assessment of vascular disease, CT in most cases requires IV contrast to delineate the vessel lumen. Pulmonary embolic disease is the third most common cause of acute cardiovascular disease.5 CT pulmonary angiography is the most common way to assess for pulmonary embolic disease, as it is accurate, fast, and widely available, and can assess alternate pathologies in cases of undifferentiated chest pain. Contrast enhancement of the pulmonary arteries is key, as embolic disease is identified as abnormal filling defects within the pulmonary arteries (Figure 2).
Contrast enhancement is also used to evaluate superior vena cava syndrome. At our institution, the CT protocol includes concomitant injections in the upper-extremity veins, with imaging timed for venous phase enhancement (pulmonary venogram). In cases of suspected arteriovenous malformation, a protocol similar to that used for suspected pulmonary embolus is used (Figure 3), although in some instances, the imaging features of arteriovenous malformation may be detectable without IV contrast.
EVALUATION OF PULMONARY PARENCHYMAL DISEASE
Infection, inflammation, and edema of the lung parenchyma are usually well depicted on CT without contrast enhancement. However, contrast may be helpful if there are concerns about complications such as chest wall involvement, where contrast enhancement may help further delineate the extent of complications.
Assessment of interstitial lung disease does not require use of IV contrast; rather, a tailored protocol with thinner slices and noncontiguous expiratory images can be used to evaluate for air-trapping and dynamic airway compromise (Figure 4). Evaluation of chronic obstructive pulmonary disease also does not require IV contrast.
EVALUATION OF THE PLEURA
In pleural effusion, CT assessment for the presence, location, and extent of the effusion does not require contrast. However, contrast enhancement is used to evaluate suspected or known exudative effusions and empyema.6 It also aids the evaluation of metastatic or primary malignancy of the pleura, particularly in cases of occult disease, as enhancement and thickening of the pleura are of diagnostic interest.
EVALUATION OF AIRWAY DISEASE
Diseases of the large airway, such as stenosis and thickening, and diseases of the small airways, such as bronchiolitis, typically do not require contrast enhancement. At our institution, to assess dynamic airway narrowing, we use a dedicated airway protocol, including inspiratory and expiratory phases and multiplanar reformatted images.
EVALUATION OF STERNAL AND MEDIASTINAL INFECTIONS
Postoperative sternal wound infections are not uncommon and range from cellulitis to frank osteomyelitis. Mediastinitis may likewise be iatrogenic or may spread from the oropharynx. CT with contrast can help to depict infection of the chest wall or mediastinum and in some instances can also delineate the route of spread.7
TYPES OF IV CONTRAST MEDIA
Contrast media used in CT contain iodine, which causes increased absorption and scattering of radiation in body tissues and blood. Other contrast media, such as those used for magnetic resonance imaging or barium enemas, do not contain iodine. This absorption and scattering in turn results in higher CT attenuation values, or “enhancement” on CT images. The extent of enhancement depends on the amount and rate of contrast material administered, as well as on patient factors (eg, tissue vascularity, permeability, interstitial space) and the energy (tube voltage) of the incident x-rays.8
Adverse reactions
Contrast materials are generally safe; however, as with any pharmaceutical, there is the potential for adverse reactions. These reactions are relatively rare and are usually mild but occasionally can be severe.9 Anaphylactoid reactions have an unclear etiology but mimic allergic reactions, and they are more likely to occur in patients with a previous reaction to contrast and in patients with asthma or cardiovascular or renal disease.
Nonanaphylactoid reactions are dependent on contrast osmolality and on the volume and route of injection (unlike anaphylactoid reactions).10 Typical symptoms include warmth, metallic taste, and nausea or vomiting.
Contrast-related nephrotoxicity has been reported,11 although this has been challenged more recently.12 Suspected risk factors for this complication include advanced age, cardiovascular disease, treatment with chemotherapy, elevated serum creatinine level, dehydration, diabetes, use of nonsteroidal anti-inflammatory medications, myeloma,13 renal disease, and kidney transplant.
Detailed protocols for premedication and management of contrast adverse reactions are beyond the scope of this review and the reader is advised to refer to dedicated manuals.10
Acknowledgment: We are grateful for the editorial assistance of Megan M. Griffiths, scientific writer for the Imaging Institute, Cleveland Clinic.
- Rubin GD. Computed tomography: revolutionizing the practice of medicine for 40 years. Radiology 2014; 273(suppl 2):S45–S74.
- American College of Radiology. ACR-SCBT-MR-SPR practice parameter for the performance of thoracic computed tomography (CT). www.acr.org/~/media/ACR/Documents/PGTS/guidelines/CT_Thoracic.pdf. Accessed March 30, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2014; 160:330–338.
- Yi CA, Lee KS, Kim EA, et al. Solitary pulmonary nodules: dynamic enhanced multi-detector row CT study and comparison with vascular endothelial growth factor and microvessel density. Radiology 2004; 233:191–199.
- Bolen MA, Renapurkar RD, Popovic ZB, et al. High-pitch ECG-synchronized pulmonary CT angiography versus standard CT pulmonary angiography: a prospective randomized study. AJR Am J Roentgenol 2013; 201:971–976.
- Kraus GJ. The split pleura sign. Radiology 2007; 243:297–298.
- Bae KT. Intravenous contrast medium administration and scan timing at CT: considerations and approaches. Radiology 2010; 256:32–61.
- Capps EF, Kinsella JJ, Gupta M, Bhatki AM, Opatowsky MJ. Emergency imaging assessment of acute, nontraumatic conditions of the head and neck. Radiographics 2010; 30:1335–1352.
- Singh J, Daftary A. Iodinated contrast media and their adverse reactions. J Nucl Med Technol 2008; 36:69–74.
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. Version 10.1. 2015. www.acr.org/~/media/37D84428BF1D4E1B9A3A2918DA9E27A3.pdf. Accessed March 29, 2016.
- Barrett BJ. Contrast nephrotoxicity. J Am Soc Nephrol 1994; 5:125–137.
- McDonald RJ, McDonald JS, Carter RE, et al. Intravenous contrast material exposure is not an independent risk factor for dialysis or mortality. Radiology 2014; 273:714–725.
- McCarthy CS, Becker JA. Multiple myeloma and contrast media. Radiology 1992; 183:519–521.
- Rubin GD. Computed tomography: revolutionizing the practice of medicine for 40 years. Radiology 2014; 273(suppl 2):S45–S74.
- American College of Radiology. ACR-SCBT-MR-SPR practice parameter for the performance of thoracic computed tomography (CT). www.acr.org/~/media/ACR/Documents/PGTS/guidelines/CT_Thoracic.pdf. Accessed March 30, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2014; 160:330–338.
- Yi CA, Lee KS, Kim EA, et al. Solitary pulmonary nodules: dynamic enhanced multi-detector row CT study and comparison with vascular endothelial growth factor and microvessel density. Radiology 2004; 233:191–199.
- Bolen MA, Renapurkar RD, Popovic ZB, et al. High-pitch ECG-synchronized pulmonary CT angiography versus standard CT pulmonary angiography: a prospective randomized study. AJR Am J Roentgenol 2013; 201:971–976.
- Kraus GJ. The split pleura sign. Radiology 2007; 243:297–298.
- Bae KT. Intravenous contrast medium administration and scan timing at CT: considerations and approaches. Radiology 2010; 256:32–61.
- Capps EF, Kinsella JJ, Gupta M, Bhatki AM, Opatowsky MJ. Emergency imaging assessment of acute, nontraumatic conditions of the head and neck. Radiographics 2010; 30:1335–1352.
- Singh J, Daftary A. Iodinated contrast media and their adverse reactions. J Nucl Med Technol 2008; 36:69–74.
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. Version 10.1. 2015. www.acr.org/~/media/37D84428BF1D4E1B9A3A2918DA9E27A3.pdf. Accessed March 29, 2016.
- Barrett BJ. Contrast nephrotoxicity. J Am Soc Nephrol 1994; 5:125–137.
- McDonald RJ, McDonald JS, Carter RE, et al. Intravenous contrast material exposure is not an independent risk factor for dialysis or mortality. Radiology 2014; 273:714–725.
- McCarthy CS, Becker JA. Multiple myeloma and contrast media. Radiology 1992; 183:519–521.
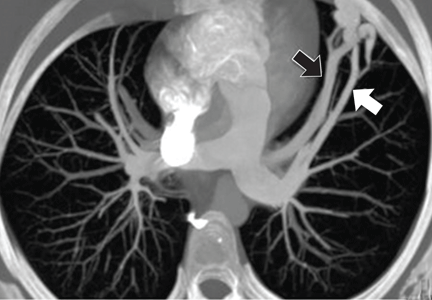
Should I suspect obstructive sleep apnea if a patient has hard-to-control hypertension?
Yes. Obstructive sleep apnea is common and is associated with hypertension and resistant hypertension. Physicians taking care of patients who have hard-to-control hypertension should be aware of the possible diagnosis of obstructive sleep apnea and screen them for it. In-laboratory polysomnography or home sleep testing should be offered if appropriate, and if obstructive sleep apnea is detected, it should be treated, as this treatment may help to control blood pressure more effectively.
OBSTRUCTIVE SLEEP APNEA IS COMMON
Obstructive sleep apnea is characterized by recurrent episodes of partial or complete collapse of the upper airway during sleep, with partial collapse leading to hypopnea and complete collapse leading to apnea. These episodes result in intermittent hypoxemia, microarousals, sleep fragmentation, daytime sleepiness, and impairment in quality of life.
In tandem with the increasing obesity epidemic, the prevalence of moderate to severe obstructive sleep apnea is 17% in men and 9% in women 50 to 70 years old.1
LINKED TO HYPERTENSION
The respiratory events that occur in obstructive sleep apnea are associated with blood pressure surges during sleep that can cause persistent elevated blood pressure while awake. Obstructive sleep apnea has been independently associated with incident hypertension in large epidemiologic studies, even after correction for confounding factors such as obesity and its surrogate markers.
Moreover, the more severe the obstructive sleep apnea, the greater the risk of incident hypertension.2 And large, long-term observational studies have shown higher incidence rates of hypertension in people with untreated obstructive sleep apnea than in those who underwent treatment for it with continuous positive airway pressure (CPAP).3
Obstructive sleep apnea is also associated with nocturnal nondipping of blood pressure (defined as failure of blood pressure to decline by at least 10% during sleep), which is an independent marker for worse cardiovascular outcomes and hypertension-induced target organ damage.
Obstructive sleep apnea is particularly common in those with drug-resistant hypertension,4 which is defined as a suboptimal control of blood pressure despite the use of multiple antihypertensive medications of different classes, a condition associated with significant rates of cardiovascular morbidity and mortality. Even in patients at high risk of cardiovascular disease, we found that those with severe obstruction of the upper airway during sleep had fourfold higher odds of having resistant elevated blood pressure.5
The seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure recognized obstructive sleep apnea as one of the causes of secondary hypertension.6 The 2013 European Society of Hypertension/European Society of Cardiology guidelines7 suggested an evaluation of obstructive sleep apnea symptoms for the management of hypertension.
MECHANISMS LINKING OBSTRUCTIVE SLEEP APNEA AND HYPERTENSION
Pathophysiologic mechanisms that may explain the association between obstructive sleep apnea and hypertension include stimulation of sympathetic activity,8 increased arterial stiffness, and endothelial dysfunction driven by apnea-related intermittent hypoxemia.9 Increased systemic inflammation and oxidative stress caused by obstructive sleep apnea are other proposed mechanisms.
Conversely, resistant hypertension may worsen obstructive sleep apnea. Some propose that activation of the renin-angiotensin-aldosterone system can cause parapharyngeal edema and rostral fluid shifts during sleep and thereby increase upper airway obstruction and worsen the severity of obstructive sleep apnea.10
CONSIDER SCREENING
Patients with resistant hypertension and risk factors for obstructive sleep apnea should be screened for it, as it is very common in this population.
A simple screening tool that can be used to detect sleep apnea is the STOP-BANG questionnaire11:
- Snore: Have you been told that you snore loudly?
- Tired: Are you often tired during the day?
- Observed apnea: Do you know if you stop breathing, or has anyone witnessed you stop breathing while sleeping?
- Pressure: Do you have or are you being treated for high blood pressure?
- Body mass index: Is your body mass index greater than 35 kg/m2?
- Age: older than 50?
- Neck circumference: greater than 40 cm?
- Gender: Male?

A score of 3 or more indicates a high risk of obstructive sleep apnea, and further workup for it is appropriate. Some of the other symptoms and signs are listed in Table 1.
SLEEP STUDIES: IN THE LABORATORY OR AT HOME
In-laboratory polysomnography entails electro-oculography, electromyography, electroencephalography, electrocardiography, pulse oximetry, and measurement of oronasal flow and thoracoabdominal movement (using sensors and belts). It should be performed in patients who have significant comorbid conditions.
A home sleep study, which is more limited than polysomnography, is appropriate in those who have a high probability of obstructive sleep apnea and who do not have other sleep disorders or significant cardiovascular, neurologic, or respiratory disorders.
Subsequently, if obstructive sleep apnea is found, a positive airway pressure titration study is performed to determine the optimal pressure requirements.
CPAP IS THE GOLD STANDARD TREATMENT
Behavioral changes are recommended to correct factors that predispose to obstructive sleep apnea or aggravate it. These changes include avoiding alcohol, sleeping on one’s side rather than supine, weight reduction in overweight individuals, and treating nasal congestion. In some situations, oral appliances or surgical options can be considered. However, CPAP is the gold standard therapy and the one most commonly used.
CPAP LOWERS BLOOD PRESSURE
Effective treatment of obstructive sleep apnea, added to an antihypertensive regimen, can further lower the blood pressure more than the antihypertensive medication regimen by itself.
Several meta-analyses have shown modest improvements in blood pressure with CPAP in hypertensive patients. CPAP’s effect on blood pressure seems to be more pronounced in those with resistant hypertension, in whom a meta-analysis of randomized controlled trials demonstrated a mean reduction in systolic blood pressure of 6.74 mm Hg and a mean reduction in diastolic blood pressure of 5.94 mm Hg.12 A recent clinic-based (“real-world”) study revealed lowering of blood pressure in patients with resistant and nonresistant hypertension—approximately 2 to 3 mm Hg after CPAP therapy.13
Furthermore, a randomized controlled trial in Spain showed that the nocturnal nondipping pattern observed in patients with resistant hypertension was reversed with the use of CPAP.14
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000; 342:1378–1384.
- Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA 2012; 307:2169–2176.
- Logan AG, Perlikowski SM, Mente A, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19:2271–2277.
- Walia HK, Li H, Rueschman M, et al. Association of severe obstructive sleep apnea and elevated blood pressure despite antihypertensive medication use. J Clin Sleep Med 2014; 10:835–843.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Mancia G, Fagard R, Narkiewicz K, et al; Task Force Members. 2013 ESH/ESC guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2013; 31:1281–1357.
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96:1897–1904.
- Jelic S, Bartels MN, Mateika JH, Ngai P, DeMeersman RE, Basner RC. Arterial stiffness increases during obstructive sleep apneas. Sleep 2002; 25:850–855.
- Dudenbostel T, Calhoun DA. Resistant hypertension, obstructive sleep apnoea and aldosterone. J Hum Hypertens 2012; 26:281–287.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Iftikhar IH, Valentine CW, Bittencourt LR, et al. Effects of continuous positive airway pressure on blood pressure in patients with resistant hypertension and obstructive sleep apnea: a meta-analysis. J Hypertens 2014; 32:2341–2350.
- Walia HK, Griffith SD, Foldvary-Schaefer N, et al. Longitudinal effect of CPAP on BP in resistant and nonresistant hypertension in a large clinic-based cohort. Chest 2016; 149:747–755.
- Martinez-Garcia MA, Capote F, Campos-Rodriguez F, et al; Spanish Sleep Network. Effect of CPAP on blood pressure in patients with obstructive sleep apnea and resistant hypertension: the HIPARCO randomized clinical trial. JAMA 2013; 310:2407–2415.
Yes. Obstructive sleep apnea is common and is associated with hypertension and resistant hypertension. Physicians taking care of patients who have hard-to-control hypertension should be aware of the possible diagnosis of obstructive sleep apnea and screen them for it. In-laboratory polysomnography or home sleep testing should be offered if appropriate, and if obstructive sleep apnea is detected, it should be treated, as this treatment may help to control blood pressure more effectively.
OBSTRUCTIVE SLEEP APNEA IS COMMON
Obstructive sleep apnea is characterized by recurrent episodes of partial or complete collapse of the upper airway during sleep, with partial collapse leading to hypopnea and complete collapse leading to apnea. These episodes result in intermittent hypoxemia, microarousals, sleep fragmentation, daytime sleepiness, and impairment in quality of life.
In tandem with the increasing obesity epidemic, the prevalence of moderate to severe obstructive sleep apnea is 17% in men and 9% in women 50 to 70 years old.1
LINKED TO HYPERTENSION
The respiratory events that occur in obstructive sleep apnea are associated with blood pressure surges during sleep that can cause persistent elevated blood pressure while awake. Obstructive sleep apnea has been independently associated with incident hypertension in large epidemiologic studies, even after correction for confounding factors such as obesity and its surrogate markers.
Moreover, the more severe the obstructive sleep apnea, the greater the risk of incident hypertension.2 And large, long-term observational studies have shown higher incidence rates of hypertension in people with untreated obstructive sleep apnea than in those who underwent treatment for it with continuous positive airway pressure (CPAP).3
Obstructive sleep apnea is also associated with nocturnal nondipping of blood pressure (defined as failure of blood pressure to decline by at least 10% during sleep), which is an independent marker for worse cardiovascular outcomes and hypertension-induced target organ damage.
Obstructive sleep apnea is particularly common in those with drug-resistant hypertension,4 which is defined as a suboptimal control of blood pressure despite the use of multiple antihypertensive medications of different classes, a condition associated with significant rates of cardiovascular morbidity and mortality. Even in patients at high risk of cardiovascular disease, we found that those with severe obstruction of the upper airway during sleep had fourfold higher odds of having resistant elevated blood pressure.5
The seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure recognized obstructive sleep apnea as one of the causes of secondary hypertension.6 The 2013 European Society of Hypertension/European Society of Cardiology guidelines7 suggested an evaluation of obstructive sleep apnea symptoms for the management of hypertension.
MECHANISMS LINKING OBSTRUCTIVE SLEEP APNEA AND HYPERTENSION
Pathophysiologic mechanisms that may explain the association between obstructive sleep apnea and hypertension include stimulation of sympathetic activity,8 increased arterial stiffness, and endothelial dysfunction driven by apnea-related intermittent hypoxemia.9 Increased systemic inflammation and oxidative stress caused by obstructive sleep apnea are other proposed mechanisms.
Conversely, resistant hypertension may worsen obstructive sleep apnea. Some propose that activation of the renin-angiotensin-aldosterone system can cause parapharyngeal edema and rostral fluid shifts during sleep and thereby increase upper airway obstruction and worsen the severity of obstructive sleep apnea.10
CONSIDER SCREENING
Patients with resistant hypertension and risk factors for obstructive sleep apnea should be screened for it, as it is very common in this population.
A simple screening tool that can be used to detect sleep apnea is the STOP-BANG questionnaire11:
- Snore: Have you been told that you snore loudly?
- Tired: Are you often tired during the day?
- Observed apnea: Do you know if you stop breathing, or has anyone witnessed you stop breathing while sleeping?
- Pressure: Do you have or are you being treated for high blood pressure?
- Body mass index: Is your body mass index greater than 35 kg/m2?
- Age: older than 50?
- Neck circumference: greater than 40 cm?
- Gender: Male?
A score of 3 or more indicates a high risk of obstructive sleep apnea, and further workup for it is appropriate. Some of the other symptoms and signs are listed in Table 1.
SLEEP STUDIES: IN THE LABORATORY OR AT HOME
In-laboratory polysomnography entails electro-oculography, electromyography, electroencephalography, electrocardiography, pulse oximetry, and measurement of oronasal flow and thoracoabdominal movement (using sensors and belts). It should be performed in patients who have significant comorbid conditions.
A home sleep study, which is more limited than polysomnography, is appropriate in those who have a high probability of obstructive sleep apnea and who do not have other sleep disorders or significant cardiovascular, neurologic, or respiratory disorders.
Subsequently, if obstructive sleep apnea is found, a positive airway pressure titration study is performed to determine the optimal pressure requirements.
CPAP IS THE GOLD STANDARD TREATMENT
Behavioral changes are recommended to correct factors that predispose to obstructive sleep apnea or aggravate it. These changes include avoiding alcohol, sleeping on one’s side rather than supine, weight reduction in overweight individuals, and treating nasal congestion. In some situations, oral appliances or surgical options can be considered. However, CPAP is the gold standard therapy and the one most commonly used.
CPAP LOWERS BLOOD PRESSURE
Effective treatment of obstructive sleep apnea, added to an antihypertensive regimen, can further lower the blood pressure more than the antihypertensive medication regimen by itself.
Several meta-analyses have shown modest improvements in blood pressure with CPAP in hypertensive patients. CPAP’s effect on blood pressure seems to be more pronounced in those with resistant hypertension, in whom a meta-analysis of randomized controlled trials demonstrated a mean reduction in systolic blood pressure of 6.74 mm Hg and a mean reduction in diastolic blood pressure of 5.94 mm Hg.12 A recent clinic-based (“real-world”) study revealed lowering of blood pressure in patients with resistant and nonresistant hypertension—approximately 2 to 3 mm Hg after CPAP therapy.13
Furthermore, a randomized controlled trial in Spain showed that the nocturnal nondipping pattern observed in patients with resistant hypertension was reversed with the use of CPAP.14
Yes. Obstructive sleep apnea is common and is associated with hypertension and resistant hypertension. Physicians taking care of patients who have hard-to-control hypertension should be aware of the possible diagnosis of obstructive sleep apnea and screen them for it. In-laboratory polysomnography or home sleep testing should be offered if appropriate, and if obstructive sleep apnea is detected, it should be treated, as this treatment may help to control blood pressure more effectively.
OBSTRUCTIVE SLEEP APNEA IS COMMON
Obstructive sleep apnea is characterized by recurrent episodes of partial or complete collapse of the upper airway during sleep, with partial collapse leading to hypopnea and complete collapse leading to apnea. These episodes result in intermittent hypoxemia, microarousals, sleep fragmentation, daytime sleepiness, and impairment in quality of life.
In tandem with the increasing obesity epidemic, the prevalence of moderate to severe obstructive sleep apnea is 17% in men and 9% in women 50 to 70 years old.1
LINKED TO HYPERTENSION
The respiratory events that occur in obstructive sleep apnea are associated with blood pressure surges during sleep that can cause persistent elevated blood pressure while awake. Obstructive sleep apnea has been independently associated with incident hypertension in large epidemiologic studies, even after correction for confounding factors such as obesity and its surrogate markers.
Moreover, the more severe the obstructive sleep apnea, the greater the risk of incident hypertension.2 And large, long-term observational studies have shown higher incidence rates of hypertension in people with untreated obstructive sleep apnea than in those who underwent treatment for it with continuous positive airway pressure (CPAP).3
Obstructive sleep apnea is also associated with nocturnal nondipping of blood pressure (defined as failure of blood pressure to decline by at least 10% during sleep), which is an independent marker for worse cardiovascular outcomes and hypertension-induced target organ damage.
Obstructive sleep apnea is particularly common in those with drug-resistant hypertension,4 which is defined as a suboptimal control of blood pressure despite the use of multiple antihypertensive medications of different classes, a condition associated with significant rates of cardiovascular morbidity and mortality. Even in patients at high risk of cardiovascular disease, we found that those with severe obstruction of the upper airway during sleep had fourfold higher odds of having resistant elevated blood pressure.5
The seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure recognized obstructive sleep apnea as one of the causes of secondary hypertension.6 The 2013 European Society of Hypertension/European Society of Cardiology guidelines7 suggested an evaluation of obstructive sleep apnea symptoms for the management of hypertension.
MECHANISMS LINKING OBSTRUCTIVE SLEEP APNEA AND HYPERTENSION
Pathophysiologic mechanisms that may explain the association between obstructive sleep apnea and hypertension include stimulation of sympathetic activity,8 increased arterial stiffness, and endothelial dysfunction driven by apnea-related intermittent hypoxemia.9 Increased systemic inflammation and oxidative stress caused by obstructive sleep apnea are other proposed mechanisms.
Conversely, resistant hypertension may worsen obstructive sleep apnea. Some propose that activation of the renin-angiotensin-aldosterone system can cause parapharyngeal edema and rostral fluid shifts during sleep and thereby increase upper airway obstruction and worsen the severity of obstructive sleep apnea.10
CONSIDER SCREENING
Patients with resistant hypertension and risk factors for obstructive sleep apnea should be screened for it, as it is very common in this population.
A simple screening tool that can be used to detect sleep apnea is the STOP-BANG questionnaire11:
- Snore: Have you been told that you snore loudly?
- Tired: Are you often tired during the day?
- Observed apnea: Do you know if you stop breathing, or has anyone witnessed you stop breathing while sleeping?
- Pressure: Do you have or are you being treated for high blood pressure?
- Body mass index: Is your body mass index greater than 35 kg/m2?
- Age: older than 50?
- Neck circumference: greater than 40 cm?
- Gender: Male?
A score of 3 or more indicates a high risk of obstructive sleep apnea, and further workup for it is appropriate. Some of the other symptoms and signs are listed in Table 1.
SLEEP STUDIES: IN THE LABORATORY OR AT HOME
In-laboratory polysomnography entails electro-oculography, electromyography, electroencephalography, electrocardiography, pulse oximetry, and measurement of oronasal flow and thoracoabdominal movement (using sensors and belts). It should be performed in patients who have significant comorbid conditions.
A home sleep study, which is more limited than polysomnography, is appropriate in those who have a high probability of obstructive sleep apnea and who do not have other sleep disorders or significant cardiovascular, neurologic, or respiratory disorders.
Subsequently, if obstructive sleep apnea is found, a positive airway pressure titration study is performed to determine the optimal pressure requirements.
CPAP IS THE GOLD STANDARD TREATMENT
Behavioral changes are recommended to correct factors that predispose to obstructive sleep apnea or aggravate it. These changes include avoiding alcohol, sleeping on one’s side rather than supine, weight reduction in overweight individuals, and treating nasal congestion. In some situations, oral appliances or surgical options can be considered. However, CPAP is the gold standard therapy and the one most commonly used.
CPAP LOWERS BLOOD PRESSURE
Effective treatment of obstructive sleep apnea, added to an antihypertensive regimen, can further lower the blood pressure more than the antihypertensive medication regimen by itself.
Several meta-analyses have shown modest improvements in blood pressure with CPAP in hypertensive patients. CPAP’s effect on blood pressure seems to be more pronounced in those with resistant hypertension, in whom a meta-analysis of randomized controlled trials demonstrated a mean reduction in systolic blood pressure of 6.74 mm Hg and a mean reduction in diastolic blood pressure of 5.94 mm Hg.12 A recent clinic-based (“real-world”) study revealed lowering of blood pressure in patients with resistant and nonresistant hypertension—approximately 2 to 3 mm Hg after CPAP therapy.13
Furthermore, a randomized controlled trial in Spain showed that the nocturnal nondipping pattern observed in patients with resistant hypertension was reversed with the use of CPAP.14
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000; 342:1378–1384.
- Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA 2012; 307:2169–2176.
- Logan AG, Perlikowski SM, Mente A, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19:2271–2277.
- Walia HK, Li H, Rueschman M, et al. Association of severe obstructive sleep apnea and elevated blood pressure despite antihypertensive medication use. J Clin Sleep Med 2014; 10:835–843.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Mancia G, Fagard R, Narkiewicz K, et al; Task Force Members. 2013 ESH/ESC guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2013; 31:1281–1357.
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96:1897–1904.
- Jelic S, Bartels MN, Mateika JH, Ngai P, DeMeersman RE, Basner RC. Arterial stiffness increases during obstructive sleep apneas. Sleep 2002; 25:850–855.
- Dudenbostel T, Calhoun DA. Resistant hypertension, obstructive sleep apnoea and aldosterone. J Hum Hypertens 2012; 26:281–287.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Iftikhar IH, Valentine CW, Bittencourt LR, et al. Effects of continuous positive airway pressure on blood pressure in patients with resistant hypertension and obstructive sleep apnea: a meta-analysis. J Hypertens 2014; 32:2341–2350.
- Walia HK, Griffith SD, Foldvary-Schaefer N, et al. Longitudinal effect of CPAP on BP in resistant and nonresistant hypertension in a large clinic-based cohort. Chest 2016; 149:747–755.
- Martinez-Garcia MA, Capote F, Campos-Rodriguez F, et al; Spanish Sleep Network. Effect of CPAP on blood pressure in patients with obstructive sleep apnea and resistant hypertension: the HIPARCO randomized clinical trial. JAMA 2013; 310:2407–2415.
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177:1006–1014.
- Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000; 342:1378–1384.
- Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA 2012; 307:2169–2176.
- Logan AG, Perlikowski SM, Mente A, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens 2001; 19:2271–2277.
- Walia HK, Li H, Rueschman M, et al. Association of severe obstructive sleep apnea and elevated blood pressure despite antihypertensive medication use. J Clin Sleep Med 2014; 10:835–843.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Mancia G, Fagard R, Narkiewicz K, et al; Task Force Members. 2013 ESH/ESC guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2013; 31:1281–1357.
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995; 96:1897–1904.
- Jelic S, Bartels MN, Mateika JH, Ngai P, DeMeersman RE, Basner RC. Arterial stiffness increases during obstructive sleep apneas. Sleep 2002; 25:850–855.
- Dudenbostel T, Calhoun DA. Resistant hypertension, obstructive sleep apnoea and aldosterone. J Hum Hypertens 2012; 26:281–287.
- Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology 2008; 108:812–821.
- Iftikhar IH, Valentine CW, Bittencourt LR, et al. Effects of continuous positive airway pressure on blood pressure in patients with resistant hypertension and obstructive sleep apnea: a meta-analysis. J Hypertens 2014; 32:2341–2350.
- Walia HK, Griffith SD, Foldvary-Schaefer N, et al. Longitudinal effect of CPAP on BP in resistant and nonresistant hypertension in a large clinic-based cohort. Chest 2016; 149:747–755.
- Martinez-Garcia MA, Capote F, Campos-Rodriguez F, et al; Spanish Sleep Network. Effect of CPAP on blood pressure in patients with obstructive sleep apnea and resistant hypertension: the HIPARCO randomized clinical trial. JAMA 2013; 310:2407–2415.

Parathyroidectomy before kidney transplant may reduce complications
BALTIMORE – Performing a parathyroidectomy in kidney transplant patients before their transplant can reduce the risk of graft failure and provide other benefits, the findings of a retrospective study of 913 patients suggest.
Uremic hyperparathyroidism (UHPT) is common in patients with end-stage kidney disease, and elevated parathyroid hormone (PTH) levels have been linked with delayed graft function after kidney transplants, but current guidelines for PTH levels may not go far enough to reduce the risk of graft failure and other post–kidney transplant complications in people with elevated PTH before transplant, according to Dr. Glenda G. Callender of Yale University, New Haven, Conn., and her colleagues.

“Uremic hyperparathyroidism was associated with an increased risk of complications in the first year post kidney transplant,” Dr. Callender said at the annual meeting of the American Association of Endocrine Surgeons. “Pre–kidney transplant parathyroidectomy was associated with a decreased risk of post–kidney transplant graft failure. This implies that pre–kidney transplant reduction of PTH levels should be considered in patients with UPHT.”
The Yale researchers reviewed outcomes of 913 patients at their institution who had a kidney transplant from 2005 to 2014. They analyzed biochemical values before kidney transplant and at three intervals post transplant: at 1 month, 6 months, and 1 year. Among the outcomes they evaluated were calcium and PTH levels, estimated glomerular filtration rate, complications, delayed graft function, and graft failure. The overall graft survival rate was 97.8% 1 year after kidney transplantation.
Overall, 49.4% of patients (451) had a diagnosis with UHPT before kidney transplant; 6.2% of all patients (57) had parathyroidectomy before kidney transplant and another 2% (18) had parathyroidectomy at some point after their kidney transplant operations. Median baseline PTH levels were higher in the UHPT patients: 206 pg/mL vs. 159 pg/mL for the non-UHPT group, Dr. Callender reported.
The researchers captured complete data on 37 of the 57 patients who had pretransplant parathyroidectomy. Twenty-four (65% of the group) had subtotal parathyroidectomy in which 3.5 glands were removed, and 12 (32%) had fewer than 3.5 glands removed. One patient had total parathyroidectomy, she said.
Among the patients with UHPT, the median pre–kidney transplant PTH was similar between the pretransplant parathyroidectomy and the no-parathyroidectomy groups: 218 pg/mL and 180 pg/mL, respectively, Dr. Callender said.
Pre–kidney transplant diagnosis of UHPT had an odds ratio of 1.44 for complications in the first year after transplant surgery, but not necessarily a greater risk for graft function or graft failure, she said. However, those relative risks changed with the degree of PTH above normal. Patients with UHPT who had pretransplant parathyroidectomy had a lower risk of graft failure, with an odds ratio of 0.547.
Current Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend maintaining PTH levels in patients with UHPT before they have kidney transplant surgery at no more than nine times normal. To test the optimal PTH levels before kidney transplant, the researchers analyzed thresholds ranging from two to nine times the normal limit.
“A pre–kidney transplant [PTH] level greater than or equal to six times normal was associated with post-transplant graft failure but not with delayed graft function or complications in the first year post kidney transplant,” Dr. Callender said. “Although the thresholds at two and four times normal were statistically significant, there was a continued risk significant for graft failure above six times normal.”
This finding “suggests that perhaps the current KDIGO guideline of maintaining patient PTH at up to nine times normal is too liberal,” Dr. Callender said.
She acknowledged several limitations of the study: its retrospective nature, small sample size, and “many missing data points” because a wide variety of dialysis centers with varying documentation standards collected information.
“However,” Dr. Callender said, “we believe these findings support the design and implementation of a multi-institutional, prospective, randomized control trial to evaluate whether a change in management of patients with uremic hyperparathyroidism is warranted.”
Dr. Callender and her coauthors had no financial relationships to disclose.
BALTIMORE – Performing a parathyroidectomy in kidney transplant patients before their transplant can reduce the risk of graft failure and provide other benefits, the findings of a retrospective study of 913 patients suggest.
Uremic hyperparathyroidism (UHPT) is common in patients with end-stage kidney disease, and elevated parathyroid hormone (PTH) levels have been linked with delayed graft function after kidney transplants, but current guidelines for PTH levels may not go far enough to reduce the risk of graft failure and other post–kidney transplant complications in people with elevated PTH before transplant, according to Dr. Glenda G. Callender of Yale University, New Haven, Conn., and her colleagues.
“Uremic hyperparathyroidism was associated with an increased risk of complications in the first year post kidney transplant,” Dr. Callender said at the annual meeting of the American Association of Endocrine Surgeons. “Pre–kidney transplant parathyroidectomy was associated with a decreased risk of post–kidney transplant graft failure. This implies that pre–kidney transplant reduction of PTH levels should be considered in patients with UPHT.”
The Yale researchers reviewed outcomes of 913 patients at their institution who had a kidney transplant from 2005 to 2014. They analyzed biochemical values before kidney transplant and at three intervals post transplant: at 1 month, 6 months, and 1 year. Among the outcomes they evaluated were calcium and PTH levels, estimated glomerular filtration rate, complications, delayed graft function, and graft failure. The overall graft survival rate was 97.8% 1 year after kidney transplantation.
Overall, 49.4% of patients (451) had a diagnosis with UHPT before kidney transplant; 6.2% of all patients (57) had parathyroidectomy before kidney transplant and another 2% (18) had parathyroidectomy at some point after their kidney transplant operations. Median baseline PTH levels were higher in the UHPT patients: 206 pg/mL vs. 159 pg/mL for the non-UHPT group, Dr. Callender reported.
The researchers captured complete data on 37 of the 57 patients who had pretransplant parathyroidectomy. Twenty-four (65% of the group) had subtotal parathyroidectomy in which 3.5 glands were removed, and 12 (32%) had fewer than 3.5 glands removed. One patient had total parathyroidectomy, she said.
Among the patients with UHPT, the median pre–kidney transplant PTH was similar between the pretransplant parathyroidectomy and the no-parathyroidectomy groups: 218 pg/mL and 180 pg/mL, respectively, Dr. Callender said.
Pre–kidney transplant diagnosis of UHPT had an odds ratio of 1.44 for complications in the first year after transplant surgery, but not necessarily a greater risk for graft function or graft failure, she said. However, those relative risks changed with the degree of PTH above normal. Patients with UHPT who had pretransplant parathyroidectomy had a lower risk of graft failure, with an odds ratio of 0.547.
Current Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend maintaining PTH levels in patients with UHPT before they have kidney transplant surgery at no more than nine times normal. To test the optimal PTH levels before kidney transplant, the researchers analyzed thresholds ranging from two to nine times the normal limit.
“A pre–kidney transplant [PTH] level greater than or equal to six times normal was associated with post-transplant graft failure but not with delayed graft function or complications in the first year post kidney transplant,” Dr. Callender said. “Although the thresholds at two and four times normal were statistically significant, there was a continued risk significant for graft failure above six times normal.”
This finding “suggests that perhaps the current KDIGO guideline of maintaining patient PTH at up to nine times normal is too liberal,” Dr. Callender said.
She acknowledged several limitations of the study: its retrospective nature, small sample size, and “many missing data points” because a wide variety of dialysis centers with varying documentation standards collected information.
“However,” Dr. Callender said, “we believe these findings support the design and implementation of a multi-institutional, prospective, randomized control trial to evaluate whether a change in management of patients with uremic hyperparathyroidism is warranted.”
Dr. Callender and her coauthors had no financial relationships to disclose.
BALTIMORE – Performing a parathyroidectomy in kidney transplant patients before their transplant can reduce the risk of graft failure and provide other benefits, the findings of a retrospective study of 913 patients suggest.
Uremic hyperparathyroidism (UHPT) is common in patients with end-stage kidney disease, and elevated parathyroid hormone (PTH) levels have been linked with delayed graft function after kidney transplants, but current guidelines for PTH levels may not go far enough to reduce the risk of graft failure and other post–kidney transplant complications in people with elevated PTH before transplant, according to Dr. Glenda G. Callender of Yale University, New Haven, Conn., and her colleagues.
“Uremic hyperparathyroidism was associated with an increased risk of complications in the first year post kidney transplant,” Dr. Callender said at the annual meeting of the American Association of Endocrine Surgeons. “Pre–kidney transplant parathyroidectomy was associated with a decreased risk of post–kidney transplant graft failure. This implies that pre–kidney transplant reduction of PTH levels should be considered in patients with UPHT.”
The Yale researchers reviewed outcomes of 913 patients at their institution who had a kidney transplant from 2005 to 2014. They analyzed biochemical values before kidney transplant and at three intervals post transplant: at 1 month, 6 months, and 1 year. Among the outcomes they evaluated were calcium and PTH levels, estimated glomerular filtration rate, complications, delayed graft function, and graft failure. The overall graft survival rate was 97.8% 1 year after kidney transplantation.
Overall, 49.4% of patients (451) had a diagnosis with UHPT before kidney transplant; 6.2% of all patients (57) had parathyroidectomy before kidney transplant and another 2% (18) had parathyroidectomy at some point after their kidney transplant operations. Median baseline PTH levels were higher in the UHPT patients: 206 pg/mL vs. 159 pg/mL for the non-UHPT group, Dr. Callender reported.
The researchers captured complete data on 37 of the 57 patients who had pretransplant parathyroidectomy. Twenty-four (65% of the group) had subtotal parathyroidectomy in which 3.5 glands were removed, and 12 (32%) had fewer than 3.5 glands removed. One patient had total parathyroidectomy, she said.
Among the patients with UHPT, the median pre–kidney transplant PTH was similar between the pretransplant parathyroidectomy and the no-parathyroidectomy groups: 218 pg/mL and 180 pg/mL, respectively, Dr. Callender said.
Pre–kidney transplant diagnosis of UHPT had an odds ratio of 1.44 for complications in the first year after transplant surgery, but not necessarily a greater risk for graft function or graft failure, she said. However, those relative risks changed with the degree of PTH above normal. Patients with UHPT who had pretransplant parathyroidectomy had a lower risk of graft failure, with an odds ratio of 0.547.
Current Kidney Disease Improving Global Outcomes (KDIGO) guidelines recommend maintaining PTH levels in patients with UHPT before they have kidney transplant surgery at no more than nine times normal. To test the optimal PTH levels before kidney transplant, the researchers analyzed thresholds ranging from two to nine times the normal limit.
“A pre–kidney transplant [PTH] level greater than or equal to six times normal was associated with post-transplant graft failure but not with delayed graft function or complications in the first year post kidney transplant,” Dr. Callender said. “Although the thresholds at two and four times normal were statistically significant, there was a continued risk significant for graft failure above six times normal.”
This finding “suggests that perhaps the current KDIGO guideline of maintaining patient PTH at up to nine times normal is too liberal,” Dr. Callender said.
She acknowledged several limitations of the study: its retrospective nature, small sample size, and “many missing data points” because a wide variety of dialysis centers with varying documentation standards collected information.
“However,” Dr. Callender said, “we believe these findings support the design and implementation of a multi-institutional, prospective, randomized control trial to evaluate whether a change in management of patients with uremic hyperparathyroidism is warranted.”
Dr. Callender and her coauthors had no financial relationships to disclose.
AT AAES 2016
Key clinical point: Parathyroidectomy reduces graft failure in individuals with uremic hyperparathyroidism (UHPT) who undergo kidney transplant.
Major finding: Pre–kidney transplant diagnosis of UHPT had an odds ratio of 1.44 of complications a year after transplant; patients who had parathyroidectomy before transplant had a reduced 0.547 odds ratio risk of graft failure.
Data source: Review of 913 patients who had kidney transplant from 2005 to 2014 at a single institution.
Disclosures: Dr. Callender and her coauthors reported having no financial disclosures.

CKD: Latest on Screening
Q) suPAR, a new screening tool for chronic kidney disease, has gotten a lot of press recently. My practice is interested in implementing it, but we can’t find information on how to obtain it. Is it commercially available yet? How can we order it? And importantly for our patients, do insurance plans cover it?
Research has been ongoing regarding biomarkers that could identify those at risk for chronic kidney disease (CKD) long before loss of renal function is apparent. A recently published study suggests that the circulating protein, soluble urokinase-type plasminogen activator receptor (suPAR), may be such a biomarker.
In a study of 3,683 subjects (ages 20 to 90) undergoing cardiac catheterization, and a further evaluation of 347 subjects in the Women’s Interagency HIV Study, Hayek et al found that elevated levels of suPAR were independently associated with CKD and with accelerated loss of renal function. At five-year follow-up, 24% of the 1,335 subjects with an initial estimated glomerular filtration rate (eGFR) ≥ 60 mL/min/1.73 m2 had developed CKD. Risk for progression to CKD was about 41% in those with a baseline suPAR level ≥ 3,040 ng/mL, compared to 12% in those with lower baseline suPAR levels.1 Thus, the cutoff for high versus low risk appears to be 3,040 ng/mL.
Hayek and associates are not the first or the only investigators studying the connection between suPAR and kidney disease. Evolving research has suggested suPAR may be an initiating factor in the development of focal segmental glomerulosclerosis (FSGS).2 However, a recent study did not support this association.3
Currently, in the United States, laboratory testing for suPAR is available only for research purposes and has not been approved by the FDA for direct patient care.4 While more research is needed with different cohorts, there is much excitement in the field of nephrology regarding the potential role of suPAR as a biomarker for predicting CKD. —CS
Cindy Smith, DNP, APRN, CNN-NP, FNP-BC
Renal Consultants, PLLC, South Charleston, West Virgina
References
1. Hayek SS, Sever S, Ko Y-A, et al. Soluble urokinase receptor and chronic kidney disease. N Engl J Med. 2015;373:1916-1925.
2. Spinale JM, Mariani LH, Kapoor S, et al. A reassessment of soluble urokinase-type plasminogen activator receptor in glomerular disease. Kidney Int. 2015;87(3):564-574.
3. Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis.Nature Med. 2011;17: 952-960.
4. Rush University Medical Center. Early warning found for chronic kidney disease: common protein in blood rises months or years before disease develops [news release]. November 5, 2015. www.rush.edu/news/press-releases/early-warning-found-chronic-kidney-disease. Accessed April 11, 2016.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired PA who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Cindy Smith, DNP, APRN, CNN-NP, FNP-BC, who practices with Renal Consultants, PLLC, in South Charleston, West Virgina, and Crystal Johnson, PA-C, Angela Harker-Bacchus, FNP-BC, Irina Sadovskaya, PA-C, and Beverly Benmoussa, FNP-BC, who practice in the Transplant Nephrology Extra-Renal CKD Clinic at the University of Michigan.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired PA who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Cindy Smith, DNP, APRN, CNN-NP, FNP-BC, who practices with Renal Consultants, PLLC, in South Charleston, West Virgina, and Crystal Johnson, PA-C, Angela Harker-Bacchus, FNP-BC, Irina Sadovskaya, PA-C, and Beverly Benmoussa, FNP-BC, who practice in the Transplant Nephrology Extra-Renal CKD Clinic at the University of Michigan.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired PA who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Cindy Smith, DNP, APRN, CNN-NP, FNP-BC, who practices with Renal Consultants, PLLC, in South Charleston, West Virgina, and Crystal Johnson, PA-C, Angela Harker-Bacchus, FNP-BC, Irina Sadovskaya, PA-C, and Beverly Benmoussa, FNP-BC, who practice in the Transplant Nephrology Extra-Renal CKD Clinic at the University of Michigan.
Q) suPAR, a new screening tool for chronic kidney disease, has gotten a lot of press recently. My practice is interested in implementing it, but we can’t find information on how to obtain it. Is it commercially available yet? How can we order it? And importantly for our patients, do insurance plans cover it?
Research has been ongoing regarding biomarkers that could identify those at risk for chronic kidney disease (CKD) long before loss of renal function is apparent. A recently published study suggests that the circulating protein, soluble urokinase-type plasminogen activator receptor (suPAR), may be such a biomarker.
In a study of 3,683 subjects (ages 20 to 90) undergoing cardiac catheterization, and a further evaluation of 347 subjects in the Women’s Interagency HIV Study, Hayek et al found that elevated levels of suPAR were independently associated with CKD and with accelerated loss of renal function. At five-year follow-up, 24% of the 1,335 subjects with an initial estimated glomerular filtration rate (eGFR) ≥ 60 mL/min/1.73 m2 had developed CKD. Risk for progression to CKD was about 41% in those with a baseline suPAR level ≥ 3,040 ng/mL, compared to 12% in those with lower baseline suPAR levels.1 Thus, the cutoff for high versus low risk appears to be 3,040 ng/mL.
Hayek and associates are not the first or the only investigators studying the connection between suPAR and kidney disease. Evolving research has suggested suPAR may be an initiating factor in the development of focal segmental glomerulosclerosis (FSGS).2 However, a recent study did not support this association.3
Currently, in the United States, laboratory testing for suPAR is available only for research purposes and has not been approved by the FDA for direct patient care.4 While more research is needed with different cohorts, there is much excitement in the field of nephrology regarding the potential role of suPAR as a biomarker for predicting CKD. —CS
Cindy Smith, DNP, APRN, CNN-NP, FNP-BC
Renal Consultants, PLLC, South Charleston, West Virgina
References
1. Hayek SS, Sever S, Ko Y-A, et al. Soluble urokinase receptor and chronic kidney disease. N Engl J Med. 2015;373:1916-1925.
2. Spinale JM, Mariani LH, Kapoor S, et al. A reassessment of soluble urokinase-type plasminogen activator receptor in glomerular disease. Kidney Int. 2015;87(3):564-574.
3. Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis.Nature Med. 2011;17: 952-960.
4. Rush University Medical Center. Early warning found for chronic kidney disease: common protein in blood rises months or years before disease develops [news release]. November 5, 2015. www.rush.edu/news/press-releases/early-warning-found-chronic-kidney-disease. Accessed April 11, 2016.
Q) suPAR, a new screening tool for chronic kidney disease, has gotten a lot of press recently. My practice is interested in implementing it, but we can’t find information on how to obtain it. Is it commercially available yet? How can we order it? And importantly for our patients, do insurance plans cover it?
Research has been ongoing regarding biomarkers that could identify those at risk for chronic kidney disease (CKD) long before loss of renal function is apparent. A recently published study suggests that the circulating protein, soluble urokinase-type plasminogen activator receptor (suPAR), may be such a biomarker.
In a study of 3,683 subjects (ages 20 to 90) undergoing cardiac catheterization, and a further evaluation of 347 subjects in the Women’s Interagency HIV Study, Hayek et al found that elevated levels of suPAR were independently associated with CKD and with accelerated loss of renal function. At five-year follow-up, 24% of the 1,335 subjects with an initial estimated glomerular filtration rate (eGFR) ≥ 60 mL/min/1.73 m2 had developed CKD. Risk for progression to CKD was about 41% in those with a baseline suPAR level ≥ 3,040 ng/mL, compared to 12% in those with lower baseline suPAR levels.1 Thus, the cutoff for high versus low risk appears to be 3,040 ng/mL.
Hayek and associates are not the first or the only investigators studying the connection between suPAR and kidney disease. Evolving research has suggested suPAR may be an initiating factor in the development of focal segmental glomerulosclerosis (FSGS).2 However, a recent study did not support this association.3
Currently, in the United States, laboratory testing for suPAR is available only for research purposes and has not been approved by the FDA for direct patient care.4 While more research is needed with different cohorts, there is much excitement in the field of nephrology regarding the potential role of suPAR as a biomarker for predicting CKD. —CS
Cindy Smith, DNP, APRN, CNN-NP, FNP-BC
Renal Consultants, PLLC, South Charleston, West Virgina
References
1. Hayek SS, Sever S, Ko Y-A, et al. Soluble urokinase receptor and chronic kidney disease. N Engl J Med. 2015;373:1916-1925.
2. Spinale JM, Mariani LH, Kapoor S, et al. A reassessment of soluble urokinase-type plasminogen activator receptor in glomerular disease. Kidney Int. 2015;87(3):564-574.
3. Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis.Nature Med. 2011;17: 952-960.
4. Rush University Medical Center. Early warning found for chronic kidney disease: common protein in blood rises months or years before disease develops [news release]. November 5, 2015. www.rush.edu/news/press-releases/early-warning-found-chronic-kidney-disease. Accessed April 11, 2016.
CKD: Latest on Management
Q) I have a patient with stage 3a chronic kidney disease (glomerular filtration rate, 45-60 mL/min/1.73 m2). I have her on a statin and an ACE inhibitor. Is there anything else I can do to slow the progression of kidney disease?
For patients with stage 3a chronic kidney disease (CKD), ongoing evaluation of risk factors and management can impact the rate of disease progression. The cornerstones of CKD care include identification and treatment of the cause; management of hypertension, albuminuria, and diabetes (if applicable); reduction of cardiovascular (CV) risk; and correction of metabolic abnormalities.5
When considering factors that can contribute to kidney injury, clinicians should consider possible pre-, intra-, and post-renal processes that could potentially cause injury.
Prerenal: Approximately 20% of cardiac output is directed to the kidneys. Reduced left ventricular function, diastolic dysfunction, and pulmonary hypertension can all contribute to a reduction in renal blood flow and subsequent kidney injury.6
Intrarenal: Exploration of possible intra-renal processes begins with a thorough history of any familial disease, hematuria, stones, proteinuria, and exposure to nephrotoxins. The nephrotoxicity profile of all medications should be examined, and patients should be educated about products, particularly OTC medications (eg, NSAIDs, common cold preparations, and herbal or weight-loss products), that can be harmful to the kidneys. Patients should also be made aware of the risk for contrast-induced renal injury, especially when considering imaging or cardiac testing. Since diabetes is a leading cause of kidney disease, good diabetic control can reduce nephropathy and slow disease progression.
Postrenal: Benign prostatic hypertrophy, kidney stones, and neurogenic bladder can all cause injury. These warrant further evaluation and treatment.
CKD often worsens existing hypertension, which is an independent risk factor for kidney failure.7 Goal blood pressure (BP) for all patients without significant albuminuria should be < 140/90 mm Hg; for those with urinary albumin ≥ 30 mg/24 h, the goal is < 130/80 mm Hg.8 Choice of antihypertensive agents can be tailored to other comorbidities, but an ACE inhibitor or angiotensin receptor blocker should be considered firstline treatment. Nocturnal hypertension is common in patients with CKD and an independent marker of CV risk. By dosing antihypertensive medications at bedtime, the clinician supports CV risk reduction.9
CKD is an independent risk factor for CV disease, thus risk factor modification should be aggressively pursued. Regardless of the cause of CKD, cigarette smoking has been associated with a more rapid decline in renal function. Patients should be counseled on the risks and offered interventions to assist in smoking cessation.10 There is also emerging evidence that exercise likely benefits the vascular health of the kidneys and appears to slow the rate of kidney decline.11,12 Overall, lifestyle interventions that help mitigate CV risk may directly benefit preservation of kidney function as well.
Metabolic abnormalities increase with CKD progression. Maintaining proper bone health through control of phosphate/acidosis and calcium equilibrium reduces morbidity as it relates to vascular and soft-tissue calcification. This can often be effectively managed through dietary modifications in early to moderate CKD. As the number of functioning nephrons decrease in CKD, so does the ability of the kidney to maintain proper acid/base balance. Persistent metabolic acidosis is related to CKD progression. Acid buffering with oral bicarbonate may be needed to achieve a goal CO2 of 22 to 32 mEq/L.8
Through adoption of a comprehensive approach—one that is inclusive of the patient—optimal outcomes can be achieved for this rapidly growing and often underrecognized population. —CJ, AH-B, IS, BB
Crystal Johnson, PA-C
Angela Harker-Bacchus, FNP-BC
Irina Sadovskaya, PA-C
Beverly Benmoussa, FNP-BC
Transplant Nephrology Extra-Renal CKD Clinic, University of Michigan
References
5. Murphree DD, Thelen SM. Chronic kidney disease in primary care. J Am Board Fam Med. 2010;23(4):542-550.
6. Coppolino G, Presta P, Saturno L, Fuiano G. Acute kidney injury in patients undergoing cardiac surgery. J Nephrol. 2013;26(1):32-40.
7. Ravera M, Re M, Defarri L, et al. Importance of blood pressure control in chronic kidney disease. J Am Soc Nephrol. 2006;17(4 suppl 2):S98-S103.
8. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013;3(suppl):1-150.
9. Hermida RC, Ayala DE, Mojón A, Fernández JR. Bedtime dosing of antihypertensive medications reduces cardiovascular risk in CKD. J Am Soc Nephrol. 2011;22(12):2313-2321.
10. Ricardo AC, Anderson CA, Yang W, et al. Healthy lifestyle and risk of kidney disease progression, atherosclerotic events, and death in CKD: findings from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis. 2015;65(3):412-424.
11. Gould DW, Graham-Brown MPM, Watson EL, et al. Physiological benefits of exercise in pre-dialysis chronic kidney disease. Nephrology (Carlton). 2014;19(9):519-527.
12. Robinson-Cohen C, Littman AJ, Duncan GE, et al. Physical activity and change in estimated GFR among persons with CKD. J Am Soc Nephrol. 2014;25(2):399-406.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired PA who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Cindy Smith, DNP, APRN, CNN-NP, FNP-BC, who practices with Renal Consultants, PLLC, in South Charleston, West Virgina, and Crystal Johnson, PA-C, Angela Harker-Bacchus, FNP-BC, Irina Sadovskaya, PA-C, and Beverly Benmoussa, FNP-BC, who practice in the Transplant Nephrology Extra-Renal CKD Clinic at the University of Michigan.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired PA who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Cindy Smith, DNP, APRN, CNN-NP, FNP-BC, who practices with Renal Consultants, PLLC, in South Charleston, West Virgina, and Crystal Johnson, PA-C, Angela Harker-Bacchus, FNP-BC, Irina Sadovskaya, PA-C, and Beverly Benmoussa, FNP-BC, who practice in the Transplant Nephrology Extra-Renal CKD Clinic at the University of Michigan.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired PA who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Cindy Smith, DNP, APRN, CNN-NP, FNP-BC, who practices with Renal Consultants, PLLC, in South Charleston, West Virgina, and Crystal Johnson, PA-C, Angela Harker-Bacchus, FNP-BC, Irina Sadovskaya, PA-C, and Beverly Benmoussa, FNP-BC, who practice in the Transplant Nephrology Extra-Renal CKD Clinic at the University of Michigan.
Q) I have a patient with stage 3a chronic kidney disease (glomerular filtration rate, 45-60 mL/min/1.73 m2). I have her on a statin and an ACE inhibitor. Is there anything else I can do to slow the progression of kidney disease?
For patients with stage 3a chronic kidney disease (CKD), ongoing evaluation of risk factors and management can impact the rate of disease progression. The cornerstones of CKD care include identification and treatment of the cause; management of hypertension, albuminuria, and diabetes (if applicable); reduction of cardiovascular (CV) risk; and correction of metabolic abnormalities.5
When considering factors that can contribute to kidney injury, clinicians should consider possible pre-, intra-, and post-renal processes that could potentially cause injury.
Prerenal: Approximately 20% of cardiac output is directed to the kidneys. Reduced left ventricular function, diastolic dysfunction, and pulmonary hypertension can all contribute to a reduction in renal blood flow and subsequent kidney injury.6
Intrarenal: Exploration of possible intra-renal processes begins with a thorough history of any familial disease, hematuria, stones, proteinuria, and exposure to nephrotoxins. The nephrotoxicity profile of all medications should be examined, and patients should be educated about products, particularly OTC medications (eg, NSAIDs, common cold preparations, and herbal or weight-loss products), that can be harmful to the kidneys. Patients should also be made aware of the risk for contrast-induced renal injury, especially when considering imaging or cardiac testing. Since diabetes is a leading cause of kidney disease, good diabetic control can reduce nephropathy and slow disease progression.
Postrenal: Benign prostatic hypertrophy, kidney stones, and neurogenic bladder can all cause injury. These warrant further evaluation and treatment.
CKD often worsens existing hypertension, which is an independent risk factor for kidney failure.7 Goal blood pressure (BP) for all patients without significant albuminuria should be < 140/90 mm Hg; for those with urinary albumin ≥ 30 mg/24 h, the goal is < 130/80 mm Hg.8 Choice of antihypertensive agents can be tailored to other comorbidities, but an ACE inhibitor or angiotensin receptor blocker should be considered firstline treatment. Nocturnal hypertension is common in patients with CKD and an independent marker of CV risk. By dosing antihypertensive medications at bedtime, the clinician supports CV risk reduction.9
CKD is an independent risk factor for CV disease, thus risk factor modification should be aggressively pursued. Regardless of the cause of CKD, cigarette smoking has been associated with a more rapid decline in renal function. Patients should be counseled on the risks and offered interventions to assist in smoking cessation.10 There is also emerging evidence that exercise likely benefits the vascular health of the kidneys and appears to slow the rate of kidney decline.11,12 Overall, lifestyle interventions that help mitigate CV risk may directly benefit preservation of kidney function as well.
Metabolic abnormalities increase with CKD progression. Maintaining proper bone health through control of phosphate/acidosis and calcium equilibrium reduces morbidity as it relates to vascular and soft-tissue calcification. This can often be effectively managed through dietary modifications in early to moderate CKD. As the number of functioning nephrons decrease in CKD, so does the ability of the kidney to maintain proper acid/base balance. Persistent metabolic acidosis is related to CKD progression. Acid buffering with oral bicarbonate may be needed to achieve a goal CO2 of 22 to 32 mEq/L.8
Through adoption of a comprehensive approach—one that is inclusive of the patient—optimal outcomes can be achieved for this rapidly growing and often underrecognized population. —CJ, AH-B, IS, BB
Crystal Johnson, PA-C
Angela Harker-Bacchus, FNP-BC
Irina Sadovskaya, PA-C
Beverly Benmoussa, FNP-BC
Transplant Nephrology Extra-Renal CKD Clinic, University of Michigan
References
5. Murphree DD, Thelen SM. Chronic kidney disease in primary care. J Am Board Fam Med. 2010;23(4):542-550.
6. Coppolino G, Presta P, Saturno L, Fuiano G. Acute kidney injury in patients undergoing cardiac surgery. J Nephrol. 2013;26(1):32-40.
7. Ravera M, Re M, Defarri L, et al. Importance of blood pressure control in chronic kidney disease. J Am Soc Nephrol. 2006;17(4 suppl 2):S98-S103.
8. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013;3(suppl):1-150.
9. Hermida RC, Ayala DE, Mojón A, Fernández JR. Bedtime dosing of antihypertensive medications reduces cardiovascular risk in CKD. J Am Soc Nephrol. 2011;22(12):2313-2321.
10. Ricardo AC, Anderson CA, Yang W, et al. Healthy lifestyle and risk of kidney disease progression, atherosclerotic events, and death in CKD: findings from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis. 2015;65(3):412-424.
11. Gould DW, Graham-Brown MPM, Watson EL, et al. Physiological benefits of exercise in pre-dialysis chronic kidney disease. Nephrology (Carlton). 2014;19(9):519-527.
12. Robinson-Cohen C, Littman AJ, Duncan GE, et al. Physical activity and change in estimated GFR among persons with CKD. J Am Soc Nephrol. 2014;25(2):399-406.
Q) I have a patient with stage 3a chronic kidney disease (glomerular filtration rate, 45-60 mL/min/1.73 m2). I have her on a statin and an ACE inhibitor. Is there anything else I can do to slow the progression of kidney disease?
For patients with stage 3a chronic kidney disease (CKD), ongoing evaluation of risk factors and management can impact the rate of disease progression. The cornerstones of CKD care include identification and treatment of the cause; management of hypertension, albuminuria, and diabetes (if applicable); reduction of cardiovascular (CV) risk; and correction of metabolic abnormalities.5
When considering factors that can contribute to kidney injury, clinicians should consider possible pre-, intra-, and post-renal processes that could potentially cause injury.
Prerenal: Approximately 20% of cardiac output is directed to the kidneys. Reduced left ventricular function, diastolic dysfunction, and pulmonary hypertension can all contribute to a reduction in renal blood flow and subsequent kidney injury.6
Intrarenal: Exploration of possible intra-renal processes begins with a thorough history of any familial disease, hematuria, stones, proteinuria, and exposure to nephrotoxins. The nephrotoxicity profile of all medications should be examined, and patients should be educated about products, particularly OTC medications (eg, NSAIDs, common cold preparations, and herbal or weight-loss products), that can be harmful to the kidneys. Patients should also be made aware of the risk for contrast-induced renal injury, especially when considering imaging or cardiac testing. Since diabetes is a leading cause of kidney disease, good diabetic control can reduce nephropathy and slow disease progression.
Postrenal: Benign prostatic hypertrophy, kidney stones, and neurogenic bladder can all cause injury. These warrant further evaluation and treatment.
CKD often worsens existing hypertension, which is an independent risk factor for kidney failure.7 Goal blood pressure (BP) for all patients without significant albuminuria should be < 140/90 mm Hg; for those with urinary albumin ≥ 30 mg/24 h, the goal is < 130/80 mm Hg.8 Choice of antihypertensive agents can be tailored to other comorbidities, but an ACE inhibitor or angiotensin receptor blocker should be considered firstline treatment. Nocturnal hypertension is common in patients with CKD and an independent marker of CV risk. By dosing antihypertensive medications at bedtime, the clinician supports CV risk reduction.9
CKD is an independent risk factor for CV disease, thus risk factor modification should be aggressively pursued. Regardless of the cause of CKD, cigarette smoking has been associated with a more rapid decline in renal function. Patients should be counseled on the risks and offered interventions to assist in smoking cessation.10 There is also emerging evidence that exercise likely benefits the vascular health of the kidneys and appears to slow the rate of kidney decline.11,12 Overall, lifestyle interventions that help mitigate CV risk may directly benefit preservation of kidney function as well.
Metabolic abnormalities increase with CKD progression. Maintaining proper bone health through control of phosphate/acidosis and calcium equilibrium reduces morbidity as it relates to vascular and soft-tissue calcification. This can often be effectively managed through dietary modifications in early to moderate CKD. As the number of functioning nephrons decrease in CKD, so does the ability of the kidney to maintain proper acid/base balance. Persistent metabolic acidosis is related to CKD progression. Acid buffering with oral bicarbonate may be needed to achieve a goal CO2 of 22 to 32 mEq/L.8
Through adoption of a comprehensive approach—one that is inclusive of the patient—optimal outcomes can be achieved for this rapidly growing and often underrecognized population. —CJ, AH-B, IS, BB
Crystal Johnson, PA-C
Angela Harker-Bacchus, FNP-BC
Irina Sadovskaya, PA-C
Beverly Benmoussa, FNP-BC
Transplant Nephrology Extra-Renal CKD Clinic, University of Michigan
References
5. Murphree DD, Thelen SM. Chronic kidney disease in primary care. J Am Board Fam Med. 2010;23(4):542-550.
6. Coppolino G, Presta P, Saturno L, Fuiano G. Acute kidney injury in patients undergoing cardiac surgery. J Nephrol. 2013;26(1):32-40.
7. Ravera M, Re M, Defarri L, et al. Importance of blood pressure control in chronic kidney disease. J Am Soc Nephrol. 2006;17(4 suppl 2):S98-S103.
8. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2013;3(suppl):1-150.
9. Hermida RC, Ayala DE, Mojón A, Fernández JR. Bedtime dosing of antihypertensive medications reduces cardiovascular risk in CKD. J Am Soc Nephrol. 2011;22(12):2313-2321.
10. Ricardo AC, Anderson CA, Yang W, et al. Healthy lifestyle and risk of kidney disease progression, atherosclerotic events, and death in CKD: findings from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis. 2015;65(3):412-424.
11. Gould DW, Graham-Brown MPM, Watson EL, et al. Physiological benefits of exercise in pre-dialysis chronic kidney disease. Nephrology (Carlton). 2014;19(9):519-527.
12. Robinson-Cohen C, Littman AJ, Duncan GE, et al. Physical activity and change in estimated GFR among persons with CKD. J Am Soc Nephrol. 2014;25(2):399-406.
Possible Simeprevir/Sofosbuvir-Induced Hepatic Decompensation With Acute Kidney Failure
The emergence of hepatitis C (HCV) treatment regimens in the past 5 years has resulted in a major paradigm shift in the management of those infected with this virus. The 2011 approval of boceprevir and telaprevir was associated with a higher virologic response (50%-75%) and a shorter length of therapy depending on the patient population. Despite these gains, first generation direct-acting antivirals (DAAs) required multiple doses, had a higher pill burden with numerous drug interactions, and adverse effects (AEs). In addition, viral resistance limited the full use of the first generation DAAs for all genotypes.
Sofosbuvir, simeprevir, and ledipasvir-sofosbuvir (second generation DAAs) boast even higher (> 90%) sustained virologic response rates (SVR) and more tolerable AE profiles especially anemia, depression, and gastrointestinal symptoms compared with the first generation DAAs. At the time of treatment for this case study, sofosbuvir/ledipasvir was not commercially available. Sofosbuvir in combination with simeprevir with or without ribavirin was one of the preferred treatment options for chronic HCV.1
Unlike the first generation DAAs, which have been associated with a decline in renal function compared with conventional pegylated interferon and ribavirin, sofosbuvir is extensively renally eliminated by glomerular filtration and active tubular secretion as the metabolite GS-331007. On the other hand, simeprevir is hepatically metabolized.
A PubMed literature search for reports of “simeprevir-induced” or “sofosbuvir-induced with hepatic, renal failure, acute kidney injury” yielded only 1 published case of hepatic decompensation likely related to simeprevir, but no case report of simeprevir and sofosbuvir associated with hepatic decompensation and acute kidney injury.4 In this article, the authors describe a case of hepatic decompensation and acute kidney injury caused by simeprevir/sofosbuvir initiation for chronic HCV that required intensive care and dialysis.
Case Report
The patient was a 62-year-old African American man with chronic HCV, genotype 1b, TT IL28B, and 4,980,000 IU baseline viral load. He was treatment naïve with biopsy proven compensated cirrhosis, and Child-Turcotte-Pugh class A with a pretreatment model for end-stage liver disease score of 12. His past medical history included hypertension, chronic kidney disease (CKD) (baseline serum creatinine [SCr] 1.4-1.8 mg/dL), benign prostatic hypertrophy, depression, obesity (30.6 body mass index, 246 lb), and psoriasis. In addition, the patient was on the following maintenance medications: allopurinol, bupropion, diltiazem, sustained-release and immediate-release morphine, sennosides, and terazosin.
In September 2014, the patient was diagnosed with biopsy-confirmed hepatocellular carcinoma (HCC) Barcelona clinic liver cancer stage B T3aN0M0 stage III. He was considered for transarterial chemoembolization (TACE), but treatment was withheld due to subsequent increase in liver function tests (LFTs) with total bilirubin (TB) 2.9 mg/dL, direct bilirubin (DB) 1.8 mg/dL, aspartate aminotransferase test (AST) 130 U/L, and alanine aminotransferase test (ALT) 188 U/L (baseline: TB 1.1 mg/dL, AST 69 U/L, and ALT 76 U/L). These results were thought to be the result of worsening hepatic function from untreated HCV, therefore, treatment was initiated.
The patient was started on simeprevir 150 mg orally daily and sofosbuvir 400 mg orally daily with an estimated baseline creatinine clearance of 67 mL/min per Cockcroft-Gault equation.5 Two days after therapy initiation, the patient presented to the emergency department with the following symptoms: hiccups, nausea, vomiting, and abdominal pain. Laboratory results showed 10.85 mg/dL SCr and 91 mg/dL blood urea nitrogen (BUN), TB increased to 14.6 mg/dL with AST of 325 U/L and ALT 277 U/L. The patient reported no use of acetaminophen, alcohol, nonsteroidal anti-inflammatory drugs, or other nephrotoxic agents.
Upon admission, the patient was diagnosed with drug-induced hepatitis and acute kidney injury (AKI). Simeprevir/sofosbuvir was discontinued along with allopurinol, bupropion, lisinopril, and morphine. An abdominal ultrasound was negative for obstructive uropathy. The patient did not respond to fluid boluses. A nephrologist was consulted, and dialysis was initiated. The patient underwent dialysis for 3 days and his LFTs and SCr levels started trending downward (Figures 1 to 5).
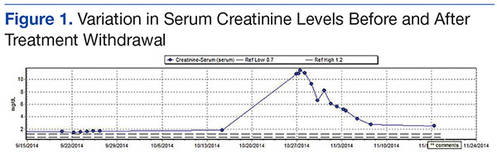
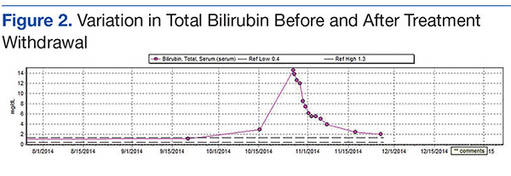
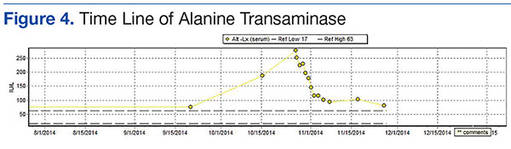
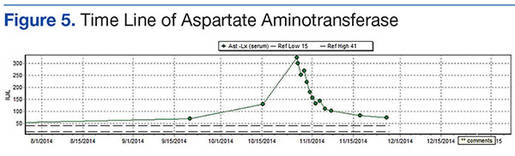
The patient was discharged after 8 days. After 3 weeks, the SCr decreased to 2.29 mg/dL, BUN was 26 mg/dL, TB was 2 mg/dL, DB was 0.9 mg/dL, AST was 73 U/L, and ALT was 81 U/L. Weekly laboratory values continued to improve following discharge but did not return to baseline levels. The patient remained off HCV treatment.
Discussion
The patient had baseline CKD with SCr > 1.5 mg/dL; however, the significant decline in renal function and worsening hepatic function were thought to be the result of external factors. Although hepatorenal syndrome was considered, the authors suspected that the AKI and hepatic decompensation were related to simeprevir/sofosbuvir regimens due to their presumed relationship and probability analysis. Osinusi and colleagues noted a decline in renal function in a patient who received ledipasvir/sofosbuvir for 6 weeks in an open-label pilot study.6 Stine and colleagues also reported on cases of simeprevir-related hepatic decompensation.4
In this case, the authors employed the Naranjo algorithm adverse drug reaction probability scale to assess whether there was a causal relationship between this event and initiation of simeprevir/sofosbuvir regimen.7 The Naranjo score was 4, indicating a possible link between simeprevir/sofosbuvir initiation and hepatic decompensation and AKI. This case may be the first postmarketing report of significant hepatic decompensation and AKI related to simeprevir/sofosbuvir.
Unlike simeprevir, which undergoes extensive oxidative metabolism by CYP3A in the liver and has negligible renal clearance with < 1% of the dose recovered in the urine, sofosbuvir is extensively metabolized by the kidneys with an active metabolite, GS-331007, and about 80% of the dose is recovered in urine (78% as GS-331007; 3.5% as sofosbuvir).8,9 The potential for drug-drug interaction also was assessed because simeprevir is extensively metabolized by the hepatic cytochrome CYP34 system and possibly CYP2C8 and CYP2C19. Clinically significant interactions could have occurred with diltiazem and morphine, because the coadministration of these medications along with simeprevir, an inhibitor of P-glycoprotein (P-gp), and intestinal CYP3A4, may result in increased diltiazem and morphine plasma concentrations.
Of note, because sofosbuvir is a substrate of P-gp, it may have its serum concentration increased by simeprevir. Inducers and inhibitors of P-gp may alter the plasma concentration of sofosbuvir. The major metabolite, GS-331007, is not a substrate of P-gp. Drugs that induce P-gp may reduce the therapeutic effect of sofosbuvir; however, the FDA-labeling suggests that inhibitors of P-gp may be coadministered with sofosbuvir.
According to simeprevir prescribing information, drug interaction studies have demonstrated that moderate CYP3A4 inhibitors, such as diltiazem (although coadministration have not been studied), increased the maximum serum concentration (Cmax), minumum serum concentration (Cmin), and AUC of simeprevir.7 As a result, concurrent use of simeprevir with a moderate CYP3A4 inhibitors is not recommended. Morphine and simeprevir interaction also is possible via the P-gp inhibition of simeprevir. Morphine concentration may have increased and metabolites may have accumulated, leading to urinary retention and elevated creatinine. In addition, decreased oral intake and subsequent nausea/vomiting may have compounded the renal insult.
Conclusion
Given that updated HCV treatment guidelines include simeprevir/sofosbuvir as an alternative treatment option, clinicians should be aware of hepatic decompensation with markedly elevated bilirubin and AKI during simeprevir and sofosbuvir treatment. Careful consideration is needed prior to the initiation of simeprevir/sofosbuvir, particularly in patients with advanced liver disease or known HCC and baseline renal impairment.
1. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C: Initial Treatment of HCV. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America Website. http://www.hcvguidelines.org. Accessed February 8, 2016.
2. Mauss S, Hueppe D, Alshuth U. Renal impairment is frequent in chronic hepatitis C patient under triple therapy with telaprevir or boceprevir. Hepatology. 2014;59(1):46-48.
3. Virlogeux V, Pradat P, Bailly F, et al. Boceprevir and telaprevir-based triple therapy for chronic hepatitis C: virolgical efficacy and impact on kidney function and model for end-stage liver disease score. J Viral Hepat. 2014;21(9):e98-e107.
4. Stine JG, Intagliata N, Shah L, et al. Hepatic decompensation likely attributable to simeprevir in patients with advanced cirrhosis. Dig Dis Sci. 2015;60(4):1031-1035.
5. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
6. Osinusi A, Kohli A, Marti MM, et al. Re-treamtent of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634-638.
7. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
8. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2014.
9. Sovaldi (sofosbuvir) [package insert]. Foster City, CA: Gilead Sciences, Inc; 2014.
The emergence of hepatitis C (HCV) treatment regimens in the past 5 years has resulted in a major paradigm shift in the management of those infected with this virus. The 2011 approval of boceprevir and telaprevir was associated with a higher virologic response (50%-75%) and a shorter length of therapy depending on the patient population. Despite these gains, first generation direct-acting antivirals (DAAs) required multiple doses, had a higher pill burden with numerous drug interactions, and adverse effects (AEs). In addition, viral resistance limited the full use of the first generation DAAs for all genotypes.
Sofosbuvir, simeprevir, and ledipasvir-sofosbuvir (second generation DAAs) boast even higher (> 90%) sustained virologic response rates (SVR) and more tolerable AE profiles especially anemia, depression, and gastrointestinal symptoms compared with the first generation DAAs. At the time of treatment for this case study, sofosbuvir/ledipasvir was not commercially available. Sofosbuvir in combination with simeprevir with or without ribavirin was one of the preferred treatment options for chronic HCV.1
Unlike the first generation DAAs, which have been associated with a decline in renal function compared with conventional pegylated interferon and ribavirin, sofosbuvir is extensively renally eliminated by glomerular filtration and active tubular secretion as the metabolite GS-331007. On the other hand, simeprevir is hepatically metabolized.
A PubMed literature search for reports of “simeprevir-induced” or “sofosbuvir-induced with hepatic, renal failure, acute kidney injury” yielded only 1 published case of hepatic decompensation likely related to simeprevir, but no case report of simeprevir and sofosbuvir associated with hepatic decompensation and acute kidney injury.4 In this article, the authors describe a case of hepatic decompensation and acute kidney injury caused by simeprevir/sofosbuvir initiation for chronic HCV that required intensive care and dialysis.
Case Report
The patient was a 62-year-old African American man with chronic HCV, genotype 1b, TT IL28B, and 4,980,000 IU baseline viral load. He was treatment naïve with biopsy proven compensated cirrhosis, and Child-Turcotte-Pugh class A with a pretreatment model for end-stage liver disease score of 12. His past medical history included hypertension, chronic kidney disease (CKD) (baseline serum creatinine [SCr] 1.4-1.8 mg/dL), benign prostatic hypertrophy, depression, obesity (30.6 body mass index, 246 lb), and psoriasis. In addition, the patient was on the following maintenance medications: allopurinol, bupropion, diltiazem, sustained-release and immediate-release morphine, sennosides, and terazosin.
In September 2014, the patient was diagnosed with biopsy-confirmed hepatocellular carcinoma (HCC) Barcelona clinic liver cancer stage B T3aN0M0 stage III. He was considered for transarterial chemoembolization (TACE), but treatment was withheld due to subsequent increase in liver function tests (LFTs) with total bilirubin (TB) 2.9 mg/dL, direct bilirubin (DB) 1.8 mg/dL, aspartate aminotransferase test (AST) 130 U/L, and alanine aminotransferase test (ALT) 188 U/L (baseline: TB 1.1 mg/dL, AST 69 U/L, and ALT 76 U/L). These results were thought to be the result of worsening hepatic function from untreated HCV, therefore, treatment was initiated.
The patient was started on simeprevir 150 mg orally daily and sofosbuvir 400 mg orally daily with an estimated baseline creatinine clearance of 67 mL/min per Cockcroft-Gault equation.5 Two days after therapy initiation, the patient presented to the emergency department with the following symptoms: hiccups, nausea, vomiting, and abdominal pain. Laboratory results showed 10.85 mg/dL SCr and 91 mg/dL blood urea nitrogen (BUN), TB increased to 14.6 mg/dL with AST of 325 U/L and ALT 277 U/L. The patient reported no use of acetaminophen, alcohol, nonsteroidal anti-inflammatory drugs, or other nephrotoxic agents.
Upon admission, the patient was diagnosed with drug-induced hepatitis and acute kidney injury (AKI). Simeprevir/sofosbuvir was discontinued along with allopurinol, bupropion, lisinopril, and morphine. An abdominal ultrasound was negative for obstructive uropathy. The patient did not respond to fluid boluses. A nephrologist was consulted, and dialysis was initiated. The patient underwent dialysis for 3 days and his LFTs and SCr levels started trending downward (Figures 1 to 5).
The patient was discharged after 8 days. After 3 weeks, the SCr decreased to 2.29 mg/dL, BUN was 26 mg/dL, TB was 2 mg/dL, DB was 0.9 mg/dL, AST was 73 U/L, and ALT was 81 U/L. Weekly laboratory values continued to improve following discharge but did not return to baseline levels. The patient remained off HCV treatment.
Discussion
The patient had baseline CKD with SCr > 1.5 mg/dL; however, the significant decline in renal function and worsening hepatic function were thought to be the result of external factors. Although hepatorenal syndrome was considered, the authors suspected that the AKI and hepatic decompensation were related to simeprevir/sofosbuvir regimens due to their presumed relationship and probability analysis. Osinusi and colleagues noted a decline in renal function in a patient who received ledipasvir/sofosbuvir for 6 weeks in an open-label pilot study.6 Stine and colleagues also reported on cases of simeprevir-related hepatic decompensation.4
In this case, the authors employed the Naranjo algorithm adverse drug reaction probability scale to assess whether there was a causal relationship between this event and initiation of simeprevir/sofosbuvir regimen.7 The Naranjo score was 4, indicating a possible link between simeprevir/sofosbuvir initiation and hepatic decompensation and AKI. This case may be the first postmarketing report of significant hepatic decompensation and AKI related to simeprevir/sofosbuvir.
Unlike simeprevir, which undergoes extensive oxidative metabolism by CYP3A in the liver and has negligible renal clearance with < 1% of the dose recovered in the urine, sofosbuvir is extensively metabolized by the kidneys with an active metabolite, GS-331007, and about 80% of the dose is recovered in urine (78% as GS-331007; 3.5% as sofosbuvir).8,9 The potential for drug-drug interaction also was assessed because simeprevir is extensively metabolized by the hepatic cytochrome CYP34 system and possibly CYP2C8 and CYP2C19. Clinically significant interactions could have occurred with diltiazem and morphine, because the coadministration of these medications along with simeprevir, an inhibitor of P-glycoprotein (P-gp), and intestinal CYP3A4, may result in increased diltiazem and morphine plasma concentrations.
Of note, because sofosbuvir is a substrate of P-gp, it may have its serum concentration increased by simeprevir. Inducers and inhibitors of P-gp may alter the plasma concentration of sofosbuvir. The major metabolite, GS-331007, is not a substrate of P-gp. Drugs that induce P-gp may reduce the therapeutic effect of sofosbuvir; however, the FDA-labeling suggests that inhibitors of P-gp may be coadministered with sofosbuvir.
According to simeprevir prescribing information, drug interaction studies have demonstrated that moderate CYP3A4 inhibitors, such as diltiazem (although coadministration have not been studied), increased the maximum serum concentration (Cmax), minumum serum concentration (Cmin), and AUC of simeprevir.7 As a result, concurrent use of simeprevir with a moderate CYP3A4 inhibitors is not recommended. Morphine and simeprevir interaction also is possible via the P-gp inhibition of simeprevir. Morphine concentration may have increased and metabolites may have accumulated, leading to urinary retention and elevated creatinine. In addition, decreased oral intake and subsequent nausea/vomiting may have compounded the renal insult.
Conclusion
Given that updated HCV treatment guidelines include simeprevir/sofosbuvir as an alternative treatment option, clinicians should be aware of hepatic decompensation with markedly elevated bilirubin and AKI during simeprevir and sofosbuvir treatment. Careful consideration is needed prior to the initiation of simeprevir/sofosbuvir, particularly in patients with advanced liver disease or known HCC and baseline renal impairment.
The emergence of hepatitis C (HCV) treatment regimens in the past 5 years has resulted in a major paradigm shift in the management of those infected with this virus. The 2011 approval of boceprevir and telaprevir was associated with a higher virologic response (50%-75%) and a shorter length of therapy depending on the patient population. Despite these gains, first generation direct-acting antivirals (DAAs) required multiple doses, had a higher pill burden with numerous drug interactions, and adverse effects (AEs). In addition, viral resistance limited the full use of the first generation DAAs for all genotypes.
Sofosbuvir, simeprevir, and ledipasvir-sofosbuvir (second generation DAAs) boast even higher (> 90%) sustained virologic response rates (SVR) and more tolerable AE profiles especially anemia, depression, and gastrointestinal symptoms compared with the first generation DAAs. At the time of treatment for this case study, sofosbuvir/ledipasvir was not commercially available. Sofosbuvir in combination with simeprevir with or without ribavirin was one of the preferred treatment options for chronic HCV.1
Unlike the first generation DAAs, which have been associated with a decline in renal function compared with conventional pegylated interferon and ribavirin, sofosbuvir is extensively renally eliminated by glomerular filtration and active tubular secretion as the metabolite GS-331007. On the other hand, simeprevir is hepatically metabolized.
A PubMed literature search for reports of “simeprevir-induced” or “sofosbuvir-induced with hepatic, renal failure, acute kidney injury” yielded only 1 published case of hepatic decompensation likely related to simeprevir, but no case report of simeprevir and sofosbuvir associated with hepatic decompensation and acute kidney injury.4 In this article, the authors describe a case of hepatic decompensation and acute kidney injury caused by simeprevir/sofosbuvir initiation for chronic HCV that required intensive care and dialysis.
Case Report
The patient was a 62-year-old African American man with chronic HCV, genotype 1b, TT IL28B, and 4,980,000 IU baseline viral load. He was treatment naïve with biopsy proven compensated cirrhosis, and Child-Turcotte-Pugh class A with a pretreatment model for end-stage liver disease score of 12. His past medical history included hypertension, chronic kidney disease (CKD) (baseline serum creatinine [SCr] 1.4-1.8 mg/dL), benign prostatic hypertrophy, depression, obesity (30.6 body mass index, 246 lb), and psoriasis. In addition, the patient was on the following maintenance medications: allopurinol, bupropion, diltiazem, sustained-release and immediate-release morphine, sennosides, and terazosin.
In September 2014, the patient was diagnosed with biopsy-confirmed hepatocellular carcinoma (HCC) Barcelona clinic liver cancer stage B T3aN0M0 stage III. He was considered for transarterial chemoembolization (TACE), but treatment was withheld due to subsequent increase in liver function tests (LFTs) with total bilirubin (TB) 2.9 mg/dL, direct bilirubin (DB) 1.8 mg/dL, aspartate aminotransferase test (AST) 130 U/L, and alanine aminotransferase test (ALT) 188 U/L (baseline: TB 1.1 mg/dL, AST 69 U/L, and ALT 76 U/L). These results were thought to be the result of worsening hepatic function from untreated HCV, therefore, treatment was initiated.
The patient was started on simeprevir 150 mg orally daily and sofosbuvir 400 mg orally daily with an estimated baseline creatinine clearance of 67 mL/min per Cockcroft-Gault equation.5 Two days after therapy initiation, the patient presented to the emergency department with the following symptoms: hiccups, nausea, vomiting, and abdominal pain. Laboratory results showed 10.85 mg/dL SCr and 91 mg/dL blood urea nitrogen (BUN), TB increased to 14.6 mg/dL with AST of 325 U/L and ALT 277 U/L. The patient reported no use of acetaminophen, alcohol, nonsteroidal anti-inflammatory drugs, or other nephrotoxic agents.
Upon admission, the patient was diagnosed with drug-induced hepatitis and acute kidney injury (AKI). Simeprevir/sofosbuvir was discontinued along with allopurinol, bupropion, lisinopril, and morphine. An abdominal ultrasound was negative for obstructive uropathy. The patient did not respond to fluid boluses. A nephrologist was consulted, and dialysis was initiated. The patient underwent dialysis for 3 days and his LFTs and SCr levels started trending downward (Figures 1 to 5).
The patient was discharged after 8 days. After 3 weeks, the SCr decreased to 2.29 mg/dL, BUN was 26 mg/dL, TB was 2 mg/dL, DB was 0.9 mg/dL, AST was 73 U/L, and ALT was 81 U/L. Weekly laboratory values continued to improve following discharge but did not return to baseline levels. The patient remained off HCV treatment.
Discussion
The patient had baseline CKD with SCr > 1.5 mg/dL; however, the significant decline in renal function and worsening hepatic function were thought to be the result of external factors. Although hepatorenal syndrome was considered, the authors suspected that the AKI and hepatic decompensation were related to simeprevir/sofosbuvir regimens due to their presumed relationship and probability analysis. Osinusi and colleagues noted a decline in renal function in a patient who received ledipasvir/sofosbuvir for 6 weeks in an open-label pilot study.6 Stine and colleagues also reported on cases of simeprevir-related hepatic decompensation.4
In this case, the authors employed the Naranjo algorithm adverse drug reaction probability scale to assess whether there was a causal relationship between this event and initiation of simeprevir/sofosbuvir regimen.7 The Naranjo score was 4, indicating a possible link between simeprevir/sofosbuvir initiation and hepatic decompensation and AKI. This case may be the first postmarketing report of significant hepatic decompensation and AKI related to simeprevir/sofosbuvir.
Unlike simeprevir, which undergoes extensive oxidative metabolism by CYP3A in the liver and has negligible renal clearance with < 1% of the dose recovered in the urine, sofosbuvir is extensively metabolized by the kidneys with an active metabolite, GS-331007, and about 80% of the dose is recovered in urine (78% as GS-331007; 3.5% as sofosbuvir).8,9 The potential for drug-drug interaction also was assessed because simeprevir is extensively metabolized by the hepatic cytochrome CYP34 system and possibly CYP2C8 and CYP2C19. Clinically significant interactions could have occurred with diltiazem and morphine, because the coadministration of these medications along with simeprevir, an inhibitor of P-glycoprotein (P-gp), and intestinal CYP3A4, may result in increased diltiazem and morphine plasma concentrations.
Of note, because sofosbuvir is a substrate of P-gp, it may have its serum concentration increased by simeprevir. Inducers and inhibitors of P-gp may alter the plasma concentration of sofosbuvir. The major metabolite, GS-331007, is not a substrate of P-gp. Drugs that induce P-gp may reduce the therapeutic effect of sofosbuvir; however, the FDA-labeling suggests that inhibitors of P-gp may be coadministered with sofosbuvir.
According to simeprevir prescribing information, drug interaction studies have demonstrated that moderate CYP3A4 inhibitors, such as diltiazem (although coadministration have not been studied), increased the maximum serum concentration (Cmax), minumum serum concentration (Cmin), and AUC of simeprevir.7 As a result, concurrent use of simeprevir with a moderate CYP3A4 inhibitors is not recommended. Morphine and simeprevir interaction also is possible via the P-gp inhibition of simeprevir. Morphine concentration may have increased and metabolites may have accumulated, leading to urinary retention and elevated creatinine. In addition, decreased oral intake and subsequent nausea/vomiting may have compounded the renal insult.
Conclusion
Given that updated HCV treatment guidelines include simeprevir/sofosbuvir as an alternative treatment option, clinicians should be aware of hepatic decompensation with markedly elevated bilirubin and AKI during simeprevir and sofosbuvir treatment. Careful consideration is needed prior to the initiation of simeprevir/sofosbuvir, particularly in patients with advanced liver disease or known HCC and baseline renal impairment.
1. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C: Initial Treatment of HCV. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America Website. http://www.hcvguidelines.org. Accessed February 8, 2016.
2. Mauss S, Hueppe D, Alshuth U. Renal impairment is frequent in chronic hepatitis C patient under triple therapy with telaprevir or boceprevir. Hepatology. 2014;59(1):46-48.
3. Virlogeux V, Pradat P, Bailly F, et al. Boceprevir and telaprevir-based triple therapy for chronic hepatitis C: virolgical efficacy and impact on kidney function and model for end-stage liver disease score. J Viral Hepat. 2014;21(9):e98-e107.
4. Stine JG, Intagliata N, Shah L, et al. Hepatic decompensation likely attributable to simeprevir in patients with advanced cirrhosis. Dig Dis Sci. 2015;60(4):1031-1035.
5. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
6. Osinusi A, Kohli A, Marti MM, et al. Re-treamtent of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634-638.
7. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
8. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2014.
9. Sovaldi (sofosbuvir) [package insert]. Foster City, CA: Gilead Sciences, Inc; 2014.
1. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C: Initial Treatment of HCV. American Association for the Study of Liver Diseases and the Infectious Diseases Society of America Website. http://www.hcvguidelines.org. Accessed February 8, 2016.
2. Mauss S, Hueppe D, Alshuth U. Renal impairment is frequent in chronic hepatitis C patient under triple therapy with telaprevir or boceprevir. Hepatology. 2014;59(1):46-48.
3. Virlogeux V, Pradat P, Bailly F, et al. Boceprevir and telaprevir-based triple therapy for chronic hepatitis C: virolgical efficacy and impact on kidney function and model for end-stage liver disease score. J Viral Hepat. 2014;21(9):e98-e107.
4. Stine JG, Intagliata N, Shah L, et al. Hepatic decompensation likely attributable to simeprevir in patients with advanced cirrhosis. Dig Dis Sci. 2015;60(4):1031-1035.
5. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
6. Osinusi A, Kohli A, Marti MM, et al. Re-treamtent of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634-638.
7. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
8. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2014.
9. Sovaldi (sofosbuvir) [package insert]. Foster City, CA: Gilead Sciences, Inc; 2014.
Novel drug fails to prevent contrast-induced nephropathy
CHICAGO – CMX-2043, a novel agent intended for prevention of contrast-induced nephropathy, failed in the phase II, double-blind, placebo-controlled CARIN clinical trial presented at the annual meeting of the American College of Cardiology.
The drug had also shown promise in small preliminary studies for the prevention of periprocedural myocardial infarction in patients undergoing coronary stenting. There again, however, CMX-2043 – a derivative of alpha lipoic acid with antioxidant and cell membrane–stabilizing properties – proved ineffective in the 361-patient, 31-center phase II trial, reported Dr. Deepak L. Bhatt, professor of medicine at Harvard Medical School and executive director of interventional cardiovascular programs at Brigham and Women’s Hospital, both in Boston.

All participants in CARIN had baseline severe impairment of kidney function or mild to moderate renal impairment plus another risk factor, such as diabetes or age greater than 75 years. One hour prior to coronary angiography, they received various doses of CMX-2043 or placebo.
Unfortunately, no difference between the four treatment arms was present in terms of the primary study endpoint: the incidence of acute kidney injury as defined by at least a 0.3 mg/dL rise in serum creatinine from baseline on day 4. No dose response to CMX-2043 was evident, nor did the investigational agent have any impact on the risk of major adverse cardiovascular events.
Immediately prior to Dr. Bhatt’s presentation, Dr. Michelle L. O’Donoghue of Brigham and Women’s Hospital presented the equally negative results of the LATITUDE-TIMI 60 trial, a phase III trial of the investigational mitogen-activated protein kinase inhibitor losmapimod, a drug developed to improve outcomes in patients with an acute coronary syndrome.
“It’s a bit distressing” to witness back to back presentations of clinical trials that proved resoundingly negative despite very strong-looking preliminary data, commented discussant Dr. Anthony N. DeMaria, professor of medicine at the University of California, San Diego. What’s going on here? he asked.
“I think it’s a fundamental truth that a lot of things that look good in preclinical work, even when backed up by a lot of solid science, don’t pan out in human studies,” Dr. Bhatt replied. “That’s a challenge, and probably in no other arena more so than in tackling inflammation and antioxidant therapy.
“There’s a graveyard of compounds that have not worked, and now we’ve perhaps added another one,” Dr. Bhatt continued. “But it doesn’t mean that scientific inquiry isn’t important, because I think eventually we’ll have drugs for these problems, whether it’s reperfusion injury or contrast-induced nephropathy. It’ll probably just take a lot more time and effort.”
The one solace regarding the CARIN trial, in Dr. Bhatt’s view, is that it highlighted the advantages of what is known as an adaptive trial design. Instead of jumping from positive early-phase results straight to a definitive 10,000-patient phase III clinical trial, investigators were able to obtain answers regarding the drug’s ability to prevent two major problems in patients undergoing coronary angiography – contrast-induced nephropathy and major adverse cardiac events – by means of a single 361-patient trial that was comparatively inexpensive.
Acute kidney injury secondary to exposure to contrast agents remains a significant problem, with an incidence of 20%-25% in high-risk patients. Numerous proposed prophylactic agents have ultimately proved not useful, including sodium bicarbonate, N-acetylcysteine, and intravenous fenoldopam.
Indeed, the only preventive measures of proven effectiveness are hydration with saline for 12 hours preangioplasty, and limiting the volume of contrast agent used. In real-world clinical practice, however, it’s often impractical to administer the optimal 12 hours of saline because of hospital pressure to get patients out quickly, Dr. Bhatt observed.
“There remains an important unmet clinical need to find agents that reduce the occurrence of contrast nephropathy,” he stressed.
Ischemix funded the CARIN trial. Dr. Bhatt reported receiving a research grant from the company that was directed to Brigham and Women’s Hospital.
CHICAGO – CMX-2043, a novel agent intended for prevention of contrast-induced nephropathy, failed in the phase II, double-blind, placebo-controlled CARIN clinical trial presented at the annual meeting of the American College of Cardiology.
The drug had also shown promise in small preliminary studies for the prevention of periprocedural myocardial infarction in patients undergoing coronary stenting. There again, however, CMX-2043 – a derivative of alpha lipoic acid with antioxidant and cell membrane–stabilizing properties – proved ineffective in the 361-patient, 31-center phase II trial, reported Dr. Deepak L. Bhatt, professor of medicine at Harvard Medical School and executive director of interventional cardiovascular programs at Brigham and Women’s Hospital, both in Boston.
All participants in CARIN had baseline severe impairment of kidney function or mild to moderate renal impairment plus another risk factor, such as diabetes or age greater than 75 years. One hour prior to coronary angiography, they received various doses of CMX-2043 or placebo.
Unfortunately, no difference between the four treatment arms was present in terms of the primary study endpoint: the incidence of acute kidney injury as defined by at least a 0.3 mg/dL rise in serum creatinine from baseline on day 4. No dose response to CMX-2043 was evident, nor did the investigational agent have any impact on the risk of major adverse cardiovascular events.
Immediately prior to Dr. Bhatt’s presentation, Dr. Michelle L. O’Donoghue of Brigham and Women’s Hospital presented the equally negative results of the LATITUDE-TIMI 60 trial, a phase III trial of the investigational mitogen-activated protein kinase inhibitor losmapimod, a drug developed to improve outcomes in patients with an acute coronary syndrome.
“It’s a bit distressing” to witness back to back presentations of clinical trials that proved resoundingly negative despite very strong-looking preliminary data, commented discussant Dr. Anthony N. DeMaria, professor of medicine at the University of California, San Diego. What’s going on here? he asked.
“I think it’s a fundamental truth that a lot of things that look good in preclinical work, even when backed up by a lot of solid science, don’t pan out in human studies,” Dr. Bhatt replied. “That’s a challenge, and probably in no other arena more so than in tackling inflammation and antioxidant therapy.
“There’s a graveyard of compounds that have not worked, and now we’ve perhaps added another one,” Dr. Bhatt continued. “But it doesn’t mean that scientific inquiry isn’t important, because I think eventually we’ll have drugs for these problems, whether it’s reperfusion injury or contrast-induced nephropathy. It’ll probably just take a lot more time and effort.”
The one solace regarding the CARIN trial, in Dr. Bhatt’s view, is that it highlighted the advantages of what is known as an adaptive trial design. Instead of jumping from positive early-phase results straight to a definitive 10,000-patient phase III clinical trial, investigators were able to obtain answers regarding the drug’s ability to prevent two major problems in patients undergoing coronary angiography – contrast-induced nephropathy and major adverse cardiac events – by means of a single 361-patient trial that was comparatively inexpensive.
Acute kidney injury secondary to exposure to contrast agents remains a significant problem, with an incidence of 20%-25% in high-risk patients. Numerous proposed prophylactic agents have ultimately proved not useful, including sodium bicarbonate, N-acetylcysteine, and intravenous fenoldopam.
Indeed, the only preventive measures of proven effectiveness are hydration with saline for 12 hours preangioplasty, and limiting the volume of contrast agent used. In real-world clinical practice, however, it’s often impractical to administer the optimal 12 hours of saline because of hospital pressure to get patients out quickly, Dr. Bhatt observed.
“There remains an important unmet clinical need to find agents that reduce the occurrence of contrast nephropathy,” he stressed.
Ischemix funded the CARIN trial. Dr. Bhatt reported receiving a research grant from the company that was directed to Brigham and Women’s Hospital.
CHICAGO – CMX-2043, a novel agent intended for prevention of contrast-induced nephropathy, failed in the phase II, double-blind, placebo-controlled CARIN clinical trial presented at the annual meeting of the American College of Cardiology.
The drug had also shown promise in small preliminary studies for the prevention of periprocedural myocardial infarction in patients undergoing coronary stenting. There again, however, CMX-2043 – a derivative of alpha lipoic acid with antioxidant and cell membrane–stabilizing properties – proved ineffective in the 361-patient, 31-center phase II trial, reported Dr. Deepak L. Bhatt, professor of medicine at Harvard Medical School and executive director of interventional cardiovascular programs at Brigham and Women’s Hospital, both in Boston.
All participants in CARIN had baseline severe impairment of kidney function or mild to moderate renal impairment plus another risk factor, such as diabetes or age greater than 75 years. One hour prior to coronary angiography, they received various doses of CMX-2043 or placebo.
Unfortunately, no difference between the four treatment arms was present in terms of the primary study endpoint: the incidence of acute kidney injury as defined by at least a 0.3 mg/dL rise in serum creatinine from baseline on day 4. No dose response to CMX-2043 was evident, nor did the investigational agent have any impact on the risk of major adverse cardiovascular events.
Immediately prior to Dr. Bhatt’s presentation, Dr. Michelle L. O’Donoghue of Brigham and Women’s Hospital presented the equally negative results of the LATITUDE-TIMI 60 trial, a phase III trial of the investigational mitogen-activated protein kinase inhibitor losmapimod, a drug developed to improve outcomes in patients with an acute coronary syndrome.
“It’s a bit distressing” to witness back to back presentations of clinical trials that proved resoundingly negative despite very strong-looking preliminary data, commented discussant Dr. Anthony N. DeMaria, professor of medicine at the University of California, San Diego. What’s going on here? he asked.
“I think it’s a fundamental truth that a lot of things that look good in preclinical work, even when backed up by a lot of solid science, don’t pan out in human studies,” Dr. Bhatt replied. “That’s a challenge, and probably in no other arena more so than in tackling inflammation and antioxidant therapy.
“There’s a graveyard of compounds that have not worked, and now we’ve perhaps added another one,” Dr. Bhatt continued. “But it doesn’t mean that scientific inquiry isn’t important, because I think eventually we’ll have drugs for these problems, whether it’s reperfusion injury or contrast-induced nephropathy. It’ll probably just take a lot more time and effort.”
The one solace regarding the CARIN trial, in Dr. Bhatt’s view, is that it highlighted the advantages of what is known as an adaptive trial design. Instead of jumping from positive early-phase results straight to a definitive 10,000-patient phase III clinical trial, investigators were able to obtain answers regarding the drug’s ability to prevent two major problems in patients undergoing coronary angiography – contrast-induced nephropathy and major adverse cardiac events – by means of a single 361-patient trial that was comparatively inexpensive.
Acute kidney injury secondary to exposure to contrast agents remains a significant problem, with an incidence of 20%-25% in high-risk patients. Numerous proposed prophylactic agents have ultimately proved not useful, including sodium bicarbonate, N-acetylcysteine, and intravenous fenoldopam.
Indeed, the only preventive measures of proven effectiveness are hydration with saline for 12 hours preangioplasty, and limiting the volume of contrast agent used. In real-world clinical practice, however, it’s often impractical to administer the optimal 12 hours of saline because of hospital pressure to get patients out quickly, Dr. Bhatt observed.
“There remains an important unmet clinical need to find agents that reduce the occurrence of contrast nephropathy,” he stressed.
Ischemix funded the CARIN trial. Dr. Bhatt reported receiving a research grant from the company that was directed to Brigham and Women’s Hospital.
AT ACC 16
Key clinical point: There continues to be a major unmet need for agents that reduce the risk of contrast-induced nephropathy.
Major finding: The once-promising investigational antioxidant and cell membrane stabilizer CMX-2043 proved ineffective for prevention of renal or cardiac injuries in patients undergoing coronary angiography.
Data source: This randomized, double-blind, placebo-controlled, 31-center, phase II study involved 361 patients with baseline renal impairment, all of whom were scheduled for coronary angiography.
Disclosures: Ischemix funded the study. Dr. Bhatt reported receiving a research grant from the company that was directed to Brigham and Women’s Hospital.
