User login
Heart Failure Diagnostic Alerts to Prompt Pharmacist Evaluation and Medication Optimization
Heart Failure Diagnostic Alerts to Prompt Pharmacist Evaluation and Medication Optimization
Heart failure (HF) is a prevalent disease in the United States affecting > 6.5 million adults and contributing to significant morbidity and mortality.1 The disease course associated with HF includes potential symptom improvement with intermittent periods of decompensation and possible clinical deterioration. Multiple therapies have been developed to improve outcomes in people with HF—to palliate HF symptoms, prevent hospitalizations, and reduce mortality.2 However, the risks of decompensation and hospitalization remain. HF decompensation development may precede clear actionable symptoms such as worsening dyspnea, noticeable edema, or weight gain. Tools to identify patient deterioration and trigger interventions to prevent HF admissions are clinically attractive compared with reliance on subjective factors alone.
Cardiac resynchronization therapy (CRT) and implantable cardioverter-defibrillator (ICD) devices made by Boston Scientific include the HeartLogic monitoring feature. Five main sensors produce an index risk score; an index score > 16 warns clinicians that the patient is at an increased risk for a HF event.3 The 5 sensors are thoracic impedance, first (S1) and third heart sounds (S3), night heart rate (NHR), respiratory rate (RR), and activity. Each sensor can draw attention to the primary driver of the alert and guide health care practitioners (HCPs) to the appropriate interventions.3 A HeartLogic alert example is shown in Figure 1.
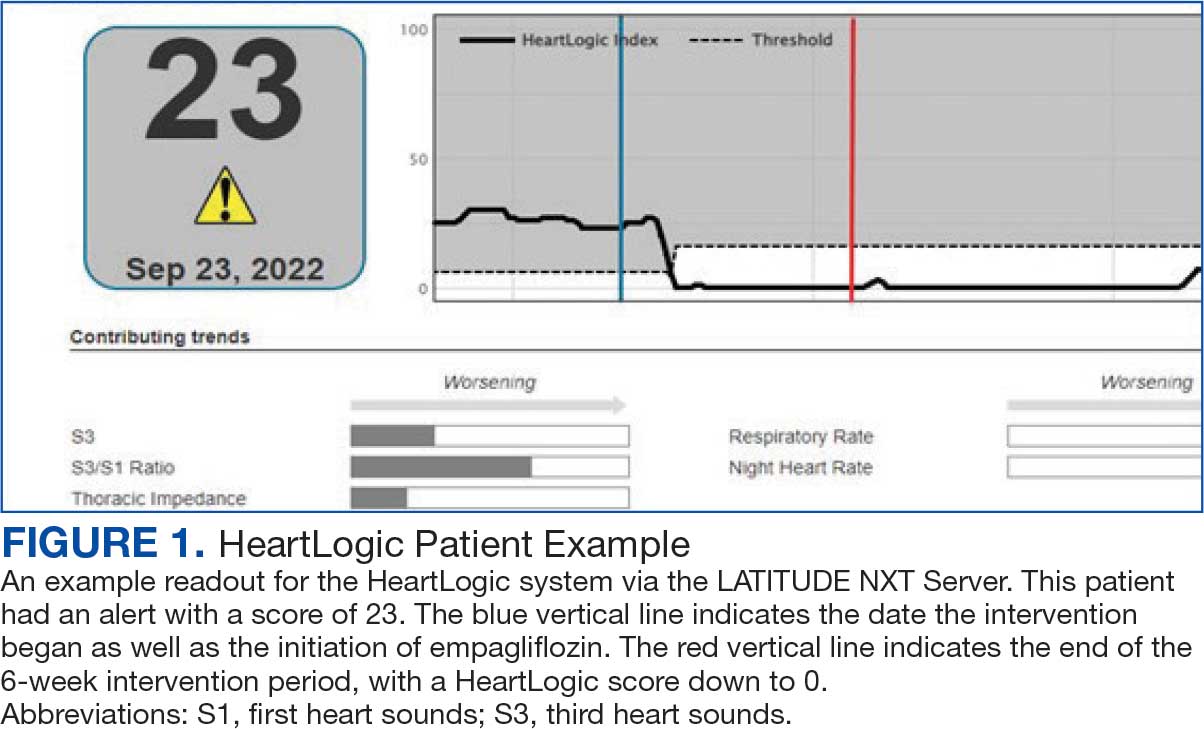
The S3 occurs during the early diastolic phase when blood moves into the ventricles. As HF worsens, with a combination of elevated filling pressures and reduced cardiac muscle compliance, S3 can become more pronounced.4 The S1 is correlated with the contractility of the left ventricle and will be reduced in patients at risk for HF events.5 Physical activity is a long-term prognostic marker in patients with HF; reduced activity is associated with mortality and increased risk of an HF event.6 Thoracic impedance is a sensor used to identify pulmonary congestion, pocket infections, pleural/pericardial effusion, and respiratory infections. The accumulation of intrathoracic fluid during pulmonary congestion increases conductance, causing a decrease in impedance.7 RR will increase as patients experience dyspnea with a more rapid, shallow breath and may trigger alerts closer to the actual HF event than other sensors. Nearly 90% of patients hospitalized for HF experience shortness of breath.8,9 NHR is used as a surrogate for resting heart rate (HR). A high resting HR is correlated with the progression of coronary atherosclerosis, harmful effects on left ventricular function, and increased risk of myocardial ischemia and ventricular arrhythmias.10
One of the challenges with preventing hospitalizations may be the lack of patient reported symptoms leading up to the event. The purpose of the sensors and HeartLogic index is to identify patients a median of 34 days before an HF event (HF admission or unscheduled intervention with intravenous treatment) with a sensitivity rate of 70%.3 According to real-word experience data, alerts have been found to precede HF symptoms by a median of 12 days and HF events such as hospitalizations by a median of 38 days, with an overall 67% reduction in HF hospitalizations when integrated into clinical care.11,12
MANAGE-HF evaluated 191 patients with HF with reduced ejection fraction (HFrEF) (< 35%), New York Heart Association class II-III symptoms, and who had an implanted CRT and/or ICD to develop an alert management guide to optimize medical treatment.12 It aimed to adjust patient regimen within 6 days of an elevated Heart- Logic index by either initiation, escalation, or maintenance of HF treatment depending on the index trend after the initial alert. This trial found that by focusing on such optimization, HF treatment was augmented during 74% of the 585 alert cases and during 54% of 3290 weekly alerts.
Initiation and uptitration of the 4 primary components of guideline-directed medical therapy (GDMT) are recommended by the 2022 Heart Failure Guidelines to reduce mortality and morbidity in patients with HFrEF.2 The 4 pillars of GDMT consist of -blockers (BB), sodium-glucose cotransporter type 2 inhibitors (SGLT2i), mineralocorticoid receptor antagonists (MRA), and renin-angiotensin-system inhibitors (RASi) including angiotensin II receptor blocker/neprilysin inhibitors (ARNi), angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARB) (Appendix 1). Obtaining and titrating to target doses wherever possible is recommended, as those were the doses that established safety and efficacy in patients with HFrEF in clinical trials.2 Pharmacists are adequately equipped to optimize HF GDMT and appropriately monitor drug response.

Through the use of HeartLogic in clinical practice, patients with HF have been shown to have improved clinical outcomes and are more likely to receive effective care; 80% of alerts were shown to provide new information to clinicians.13 This project sought to quantify the total number and types of pharmacist interventions driven by integration of HeartLogic index monitoring into practice.
Methods
The West Palm Beach Veterans Affairs Medical Center (WPBVAMC) Research Program Office approved this project and determined it was exempt from institutional review board oversight. Patients were screened retrospectively and prospectively from May 26, 2022, through December 31, 2022, by a cardiology clinical pharmacist practitioner (CPP) and a cardiology pharmacy resident using the local monitoring suite for the HeartLogic-compatible device, LATITUDE NXT. Read-only access to the local monitoring suit was granted by the National Cardiac Device Surveillance Program. Training for HeartLogic was completed through continuing education courses provided by Boston Scientific. Additional information was provided by Boston Scientific representative presentations and collaboration with WPBVAMC pacemaker clinic HCPs.
Individuals included were patients with HeartLogic-capable ICDs. A HeartLogic alert had to be present at initial patient contact. Patients were also contacted as part of routine clinical practice, but no formal number or frequency of calls to patients was required. The initial contact must be with a pharmacist for the patient to be included, but subsequent contact by other HCPs was included. Patients in the cardiology clinic are required to meet with a cardiologist at least annually; however, interim visits can be completed by advanced practice registered nurse practitioners, physicians assistants, or CPPs.
Patients in alert status were contacted by telephone and appropriate modifications of HF therapy were made by the CPP based on score metrics, medical record review, and patient interview. Information surrounding the initial alert, baseline patient data, medication and monitoring interventions made, and clinical outcomes such as hospitalization, symptom improvement, follow-up, and mortality were collected. Information for each encounter was collected until 42 days from the initial date of pharmacist contact.
Clinically successful tolerability of intervention implementation was defined as tolerability, adherence, and lack of adverse effects (AEs) per patient report at follow-up or within 42 days from initial alert (Appendix 2). A decrease in dose was not counted as intolerance. A single patient may have been counted as multiple encounters if the original intervention resulted in treatment intolerance and the patient remained in alert or if an additional alert occurred after 42 days of the initial alert. There were no specific time criteria for follow-up, which occurred at the CPP’s discretion.

There was no mandated algorithm used to alter medications based on the Heart- Logic score, nor were there required minimum or maximum numbers of interventions after an alert. Patient contact by telephone initiated an encounter. The types of interventions included medication increases, decreases, initiation, discontinuation, or no medication change. Each medication change and rationale, if applicable, was recorded for the encounter ≤ 42 days after the initial contact date. If a medication with required monitoring parameters was augmented, the pharmacist was responsible for ordering laboratory testing and follow-up. Most interventions were completed by telephone; however, some patients had in-person visits in the HF CPP clinic.
Outcomes
The primary outcome was the number of pharmacist interventions made to optimize GDMT, defined as either an initiation or dose increase. Key intervention analysis included the use and dosing of the 4 primary components of HF GDMT: BB, SGLT2i, MRA, and ARNi/ARB/ACEi. In addition to the 4 primary components of GDMT, loop diuretic changes were also recorded and analyzed. Secondary endpoints were the number of HF hospitalizations ≤ 42 days after the initial alert, and the effect of medication interventions on device metrics, patient symptoms, and tolerability. Successful tolerability was defined as continued use of augmented GDMT without intolerance or discontinuation. The primary analysis was analyzed through descriptive statistics. Median changes in HeartLogic scores and metrics from baseline were analyzed using a paired, 2-sided t test with an α of .05 to detect significance.
Results
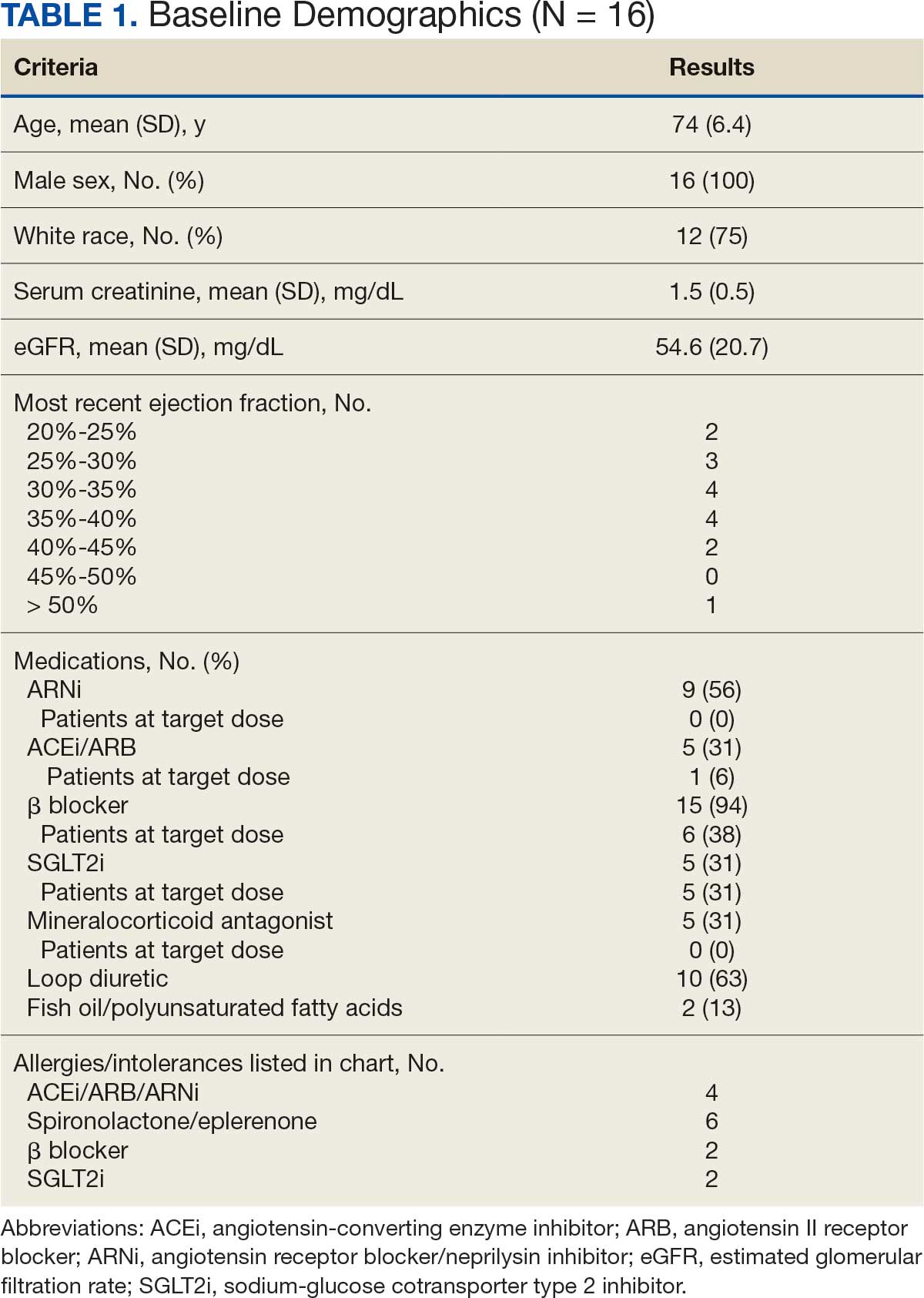
There were 39 WPBVAMC patients with a HeartLogic-capable device. Twenty-one alert encounters were analyzed in 16 patients (41%) over 31 weeks of data collection. The 16 patients at baseline had a mean age of 74 years, all were male, and 12 (75%) were White. Eight patients (50%) had a recent ejection fraction (EF) between 30% and 40%. Three patients had an EF ≥ 40%. At the time of alert, 15 patients used BB (94%), 10 used loop diuretics (63%), and 9 used ARNi (56%) (Table 1).
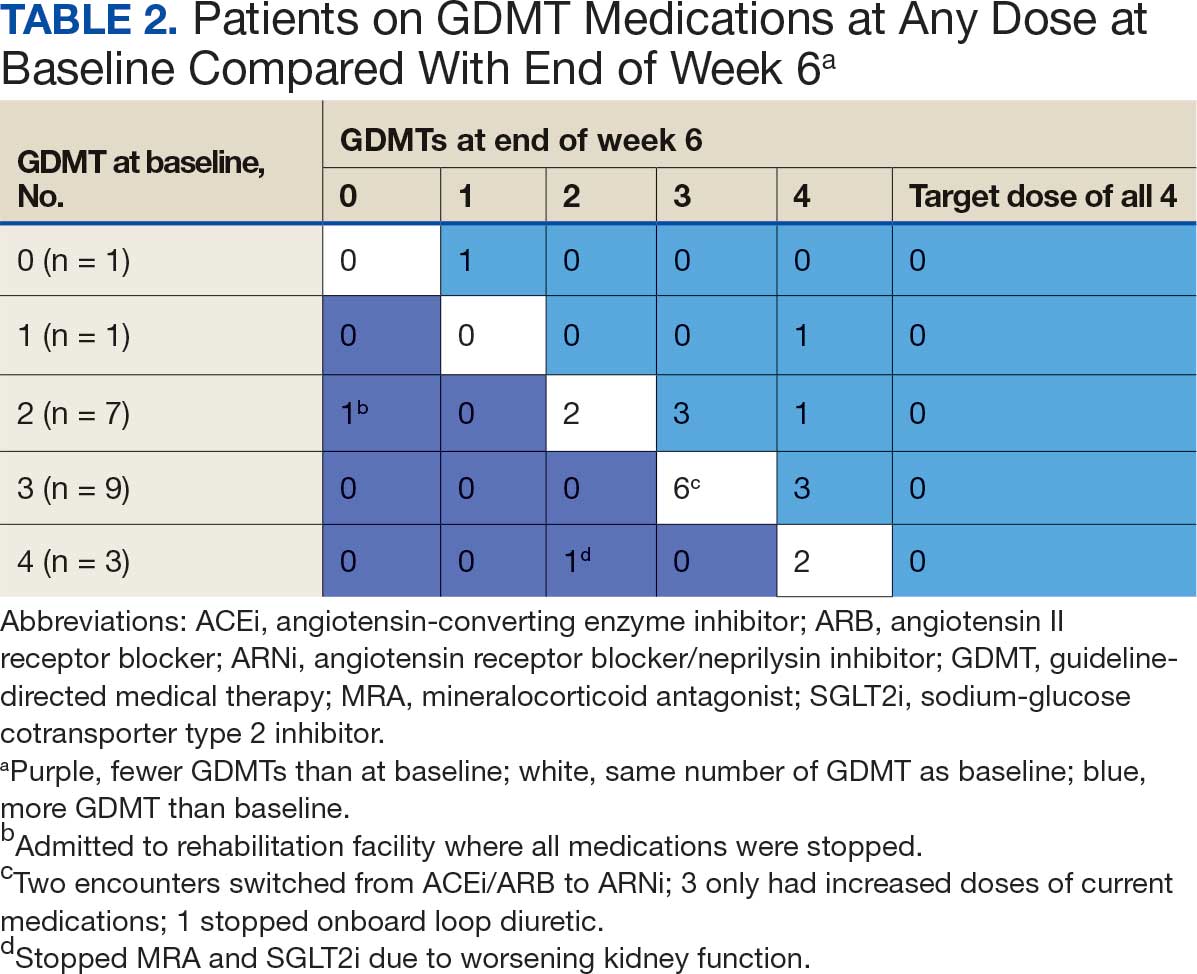
There were 23 medication changes made during initial contact. The most common change was starting an SGLT2i (30%; n = 7), followed by starting an MRA (22%; n = 5), and increasing the ARNi dose (22%; n = 5). At the initial contact, ≥ 1 medication optimization occurred in 95% (n = 20) of encounters. The CPP contacted patients a mean of 4.8 days after the initial alert.
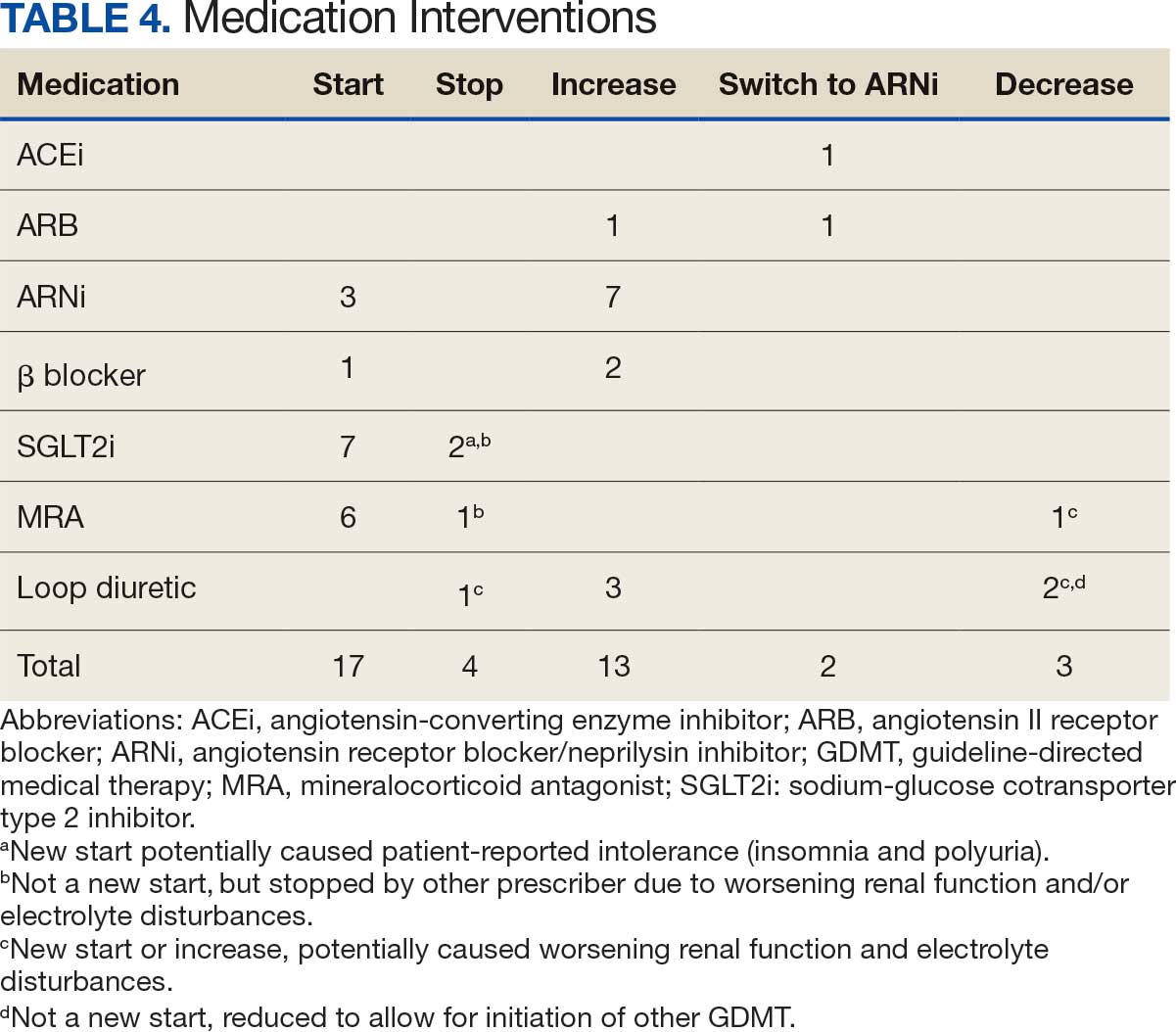
Patients were taking a mean of 2.6 primary GDMT medications at baseline and 3.0 at 42 days. CPP encounters led to a mean of 1.8 medication changes over the 6-week period (range, 0-5). Seventeen medications were started, 13 medications were increased, 3 medications were decreased, and 4 medications were stopped (Table 2). One ACEi and 1 ARB were switched as a therapy escalation to an ARNi. One patient was on 1 of 4 primary GDMTs at baseline, which increased to 4 GDMT agents at 42 days.
SGLT2 inhibitors were added most often at initial contact (54%) and throughout the 42-day period (41%). The most common successfully tolerated optimizations were RASi, followed by MRA, SGLT2 inhibitors, BB, and loop diuretics with 11, 6, 5, 3, and 2 patients, respectively. Interventions were tolerated by 90% of patients, and no HF hospitalization occurred during follow-up. All possible rationales for patients with the same or reduced number of GDMT at 42 days compared with baseline are shown in Appendix 2.
Device Metrics
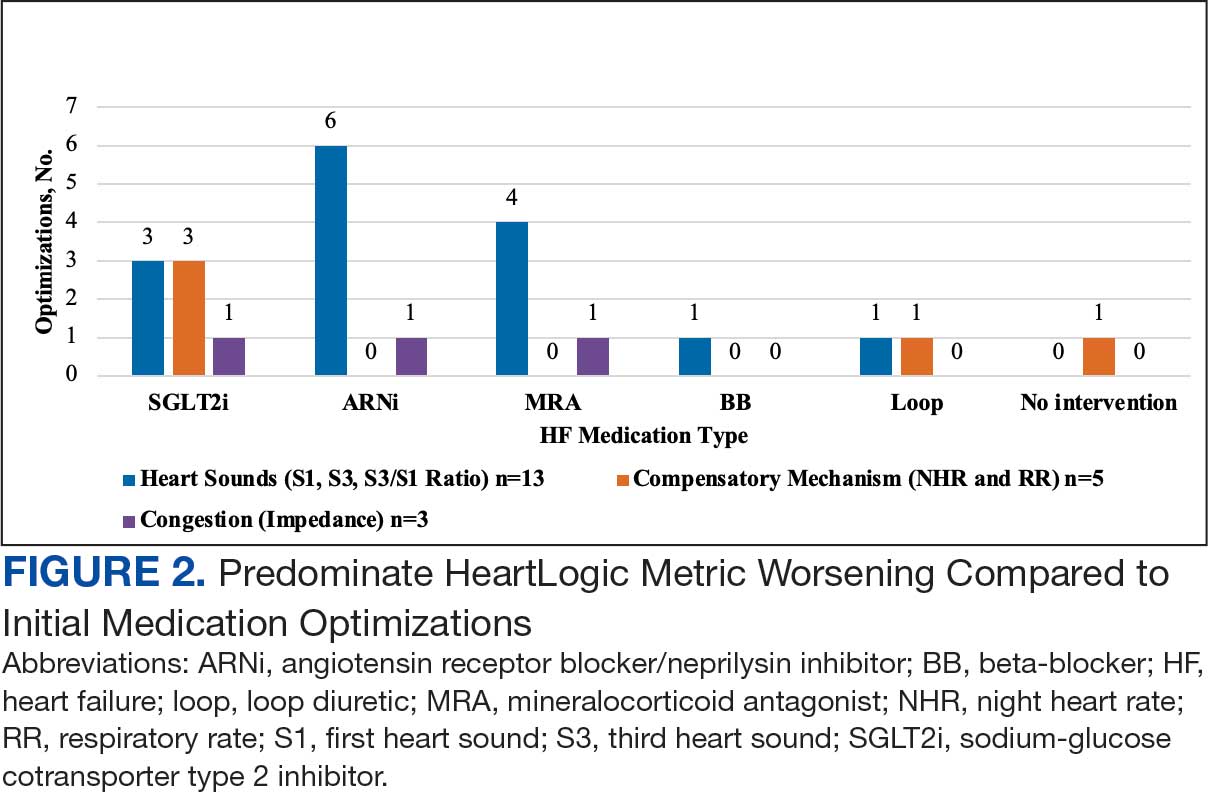
During initial contact, the most common HeartLogic metric category that was predominantly worsening were heart sounds (S1, S3, and S3/S1 ratio), followed by compensatory mechanism sensors (NHR and RR) and congestion (impedance) at rates of 61.9%, 23.8%, and 14.3%, respectively (Figure 2).
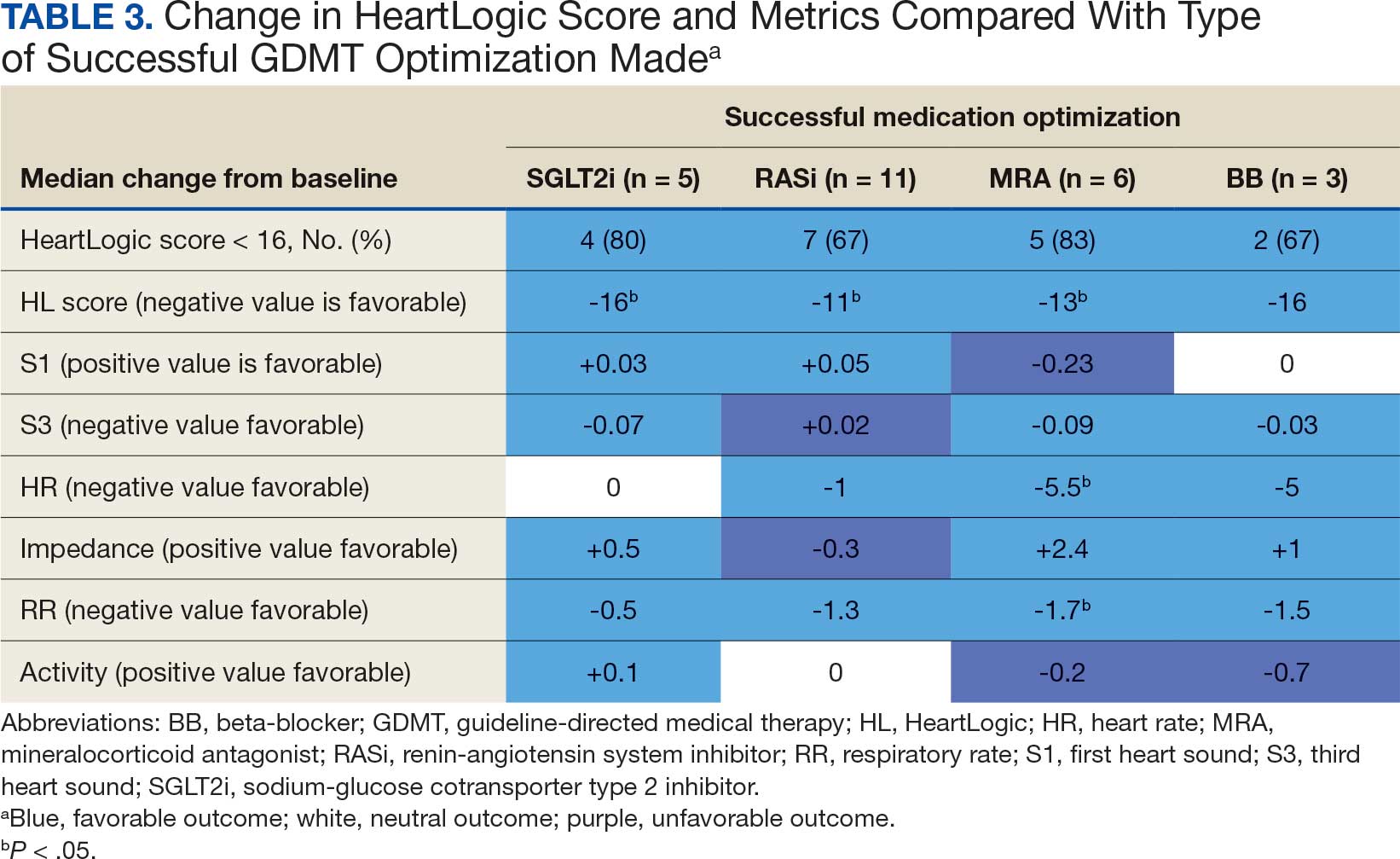
The median HeartLogic index score was 18 at baseline and 5 at the end of the follow-up period (P < .001). The changes in score and metrics were compared with the type of successfully tolerated GDMT optimization made (Table 3). The GDMT optimization analysis included SGLT2i, RASi, MRA, BB, and loop diuretics. All interventions reduced the overall HeartLogic index score, ranging from a 9.5-point reduction (loop diuretics) to a 16-point reduction (SGLT2i and BB). Optimization of SGLT2i, RASi, and loop diuretics had a positive impact on S1 score. For S3 score, SGLT2i, MRA, and BB had a positive impact. All medications, except for SGLT2i therapy, reduced the NHR score. Optimization of MRA, SGLT2i, and BB had positive impacts on the impedance score. All medications reduced RR from baseline. Only SGLT2i and loop diuretics had positive impacts on the activity score.
Clinical Outcomes and Adverse Effects
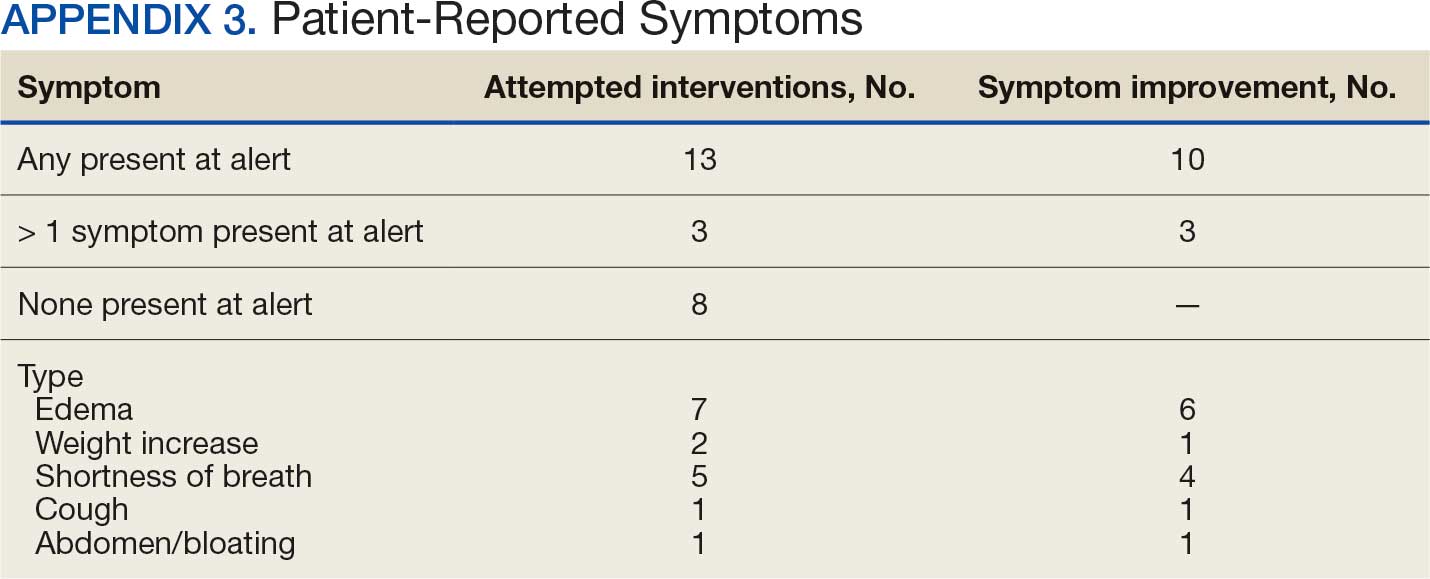
Within 42 days of contact, 17 encounters (81%) had ≥ 1 follow-up appointment with a CPP and all 21 patients had ≥ 1 follow-up health care team member. One patient had a HF-related hospitalization within 42 days of contact; however, that individual refused the recommended medication intervention. There were 13 encounters (62%) with reported symptoms at the time of the initial alert and 10 (77%) had subjective symptom improvement at 42 days (Appendix 3).
Of 30 medication optimizations, 27 primary GDMT medications were tolerated. Two medication intolerances led to discontinuation (1 SGLT2i and 1 loop diuretic) and 1 patient never started the SGLT2i (Table 4). There was only 1 known patient who did not follow the directions to adjust their medications. That individual was included because the patient agreed to the change during the CPP visit but later reported that he had never started the SGLT2i.
Discussion
The HeartLogic tool created a bridge for patients with HF to work with CPPs as soon as possible to optimize medication therapy to reduce HF events. This study highlights an additional area of expertise and service that CPPs may offer to their specialty HF clinic team. Over 31 weeks, 21 encounters and 30 medication optimizations were completed. These interventions led to significant reductions in HeartLogic scores, improvements in symptoms, and optimization of HF care, most of which were well tolerated.
Additional hemodynamic monitoring devices are available. Similar to HeartLogic, OptiVol is a tool embedded in select Medtronic implantable devices that monitors fluid status. 14 CardioMEMS is an implantable pulmonary artery pressure sensor used as a presymptomatic data point to alert clinicians when HF is worsening. In the CHAMPION trial, the use of CardioMEMS showed a 28% reduction in HF-related hospitalization at 6 months.15 Conversely, in the GUIDE-HF trial, monitoring with CardioMEMS did not significantly reduce the composite endpoint of mortality and total HF events.16 Therefore, remote hemodynamic monitoring has variable results and the use of these tools remains uncertain per the clinical guidelines.2
The MANAGE-HF study that contributed to the validation of the HeartLogic tool may provide a comparison with this smaller single-center project. The time to follow-up within 7 days of alert was noted in only 54% of the patients in MANAGE-HF.12 In this study, 86% of patients received follow-up within 7 days, with a mean of 4.8 days. The quick turnaround from the time of alert to intervention portrays pharmacists as readily available HCPs.
In MANAGE-HF, 89% of medication augmentation involved loop diuretics or thiazides; in our project, loop diuretics were the least frequently changed medication. Most optimizations in this project included ARNi, SGLT2i, BB, and MRA, which have been shown to reduce morbidity and mortality.2 Our project included use of SGLT2i therapy to affect HeartLogic metrics, which has not been evaluated previously. SGLT2i were the most commonly initiated medication after an alert. Of the 5 tolerated SGLT2i optimization encounters, 4 were out of alert at 42 days.
SGLT2i resulted in a significant decrease in HeartLogic index score from baseline and were the only class of medication that did not produce a negative change in any metric. In this study, CPPs utilizing and acting on HeartLogic alerts led to 1 (4.8%) hospitalization with HF as the primary reason for admission and no hospitalizations as a secondary cause in 42 days, compared to 37% and 7.9% in the MANAGE-HF in 1 year, respectively. An additional screening 1 year after the initial alert found that 2 (12.5%) of 12 patients had been admitted with 1 HF hospitalization each.
A strength of this study was the ability to use HeartLogic to identify high-risk patients, provide a source of patient contact and monitoring, interpret 5 cardiac sensors, and optimize all HF GDMT, not just volume management. By focusing efforts on making patient contact and pharmacotherapy interventions with morbidity and mortality benefit, remote hemodynamic monitoring may show a clear clinical benefit and become a vital part of HF care.
Limitations
Checking for adherence and tolerance to medications were mainly patient reported if there was a CPP follow-up within 42 days, or potentially through refill history when unclear. However, this limitation is reflective of current practice where patients may have multiple clinicians working to optimize HF care and where there is reliance on patients in order to guide continued therapy. Although unable to explicitly show a reduction in HF events given lack of comparator group, the interventions made are associated with improved outcomes and thus would be expected to improve patient outcomes. Changes in vital signs were not tracked as part of this project, however the main rationale for changes made were to optimize GDMT therapy, not specifically to impact vital sign measures.
HeartLogic alerts prompted identification of high-risk patients with HF, pharmacist evaluation and outreach, patient-focused pharmacotherapy care, and beneficial patient outcomes. With only 2 cardiology CPPs checking alerts once weekly, future studies may be needed with larger samples to create algorithms and protocols to increase the clinical utility of this tool on a greater scale.
Conclusions
Cardiology CPP-led HF interventions triggered by HeartLogic alerts lead to effective patient identification, increased access to care, reductions in HeartLogic scores, improvements in symptoms, and optimization of HF care. This project demonstrates the practical utility of the HeartLogic suite in conjunction with CPP care to prioritize treatment for highrisk patients with HF in an efficient manner. The data highlight the potential value of the HeartLogic tool and a CPP in HF care to facilitate initiation and optimization of GDMT to ultimately improve the morbidity and mortality in patients with HF.
- Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147:e93-e621. doi:10.1161/CIR.0000000000001123
- Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/ American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895-e1032. doi:10.1161/CIR.0000000000001063
- Boehmer JP, Hariharan R, Devecchi FG, et al. A multisensor algorithm predicts heart failure events in patients with implanted devices: results from the MultiSENSE study. J Am Coll Cardiol HF. 2017;5:216-225. doi:10.1016/j.jchf.2016.12.011
- Cao M, Gardner RS, Hariharan R, et al. Ambulatory monitoring of heart sounds via an implanted device is superior to auscultation for prediction of heart failure events. J Card Fail. 2020;26:151-159. doi:10.1016/j.cardfail.2019.10.006
- Calò L, Capucci A, Santini L, et al. ICD-measured heart sounds and their correlation with echocardiographic indexes of systolic and diastolic function. J Interv Card Electrophysiol. 2020;58:95-101. doi:10.1007/s10840-019-00668
- Del Buono MG, Arena R, Borlaug BA, et al. Exercise intolerance in patients with heart failure: JACC state-of-the- art review. J Am Coll Cardiol. 2019;73:2209-2225. doi:10.1016/j.jacc.2019.01.072
- Yu CM, Wang L, Chau E, et al. Intrathoracic impedance monitoring in patients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation. 2005;112:841-848. doi:10.1161/CIRCULATIONAHA.104.492207
- Rials S, Aktas M, An Q, et al. Continuous respiratory rate is superior to routine outpatient dyspnea assessment for predicting heart failure events. J Card Fail. 2018;24:S45.
- Fonarow GC, ADHERE Scientific Advisory Committee. The Acute Decompensated Heart Failure National Registry (ADHERE): opportunities to improve care of patients hospitalized with acute decompensated heart failure. Rev Cardiovasc Med. 2003;4(suppl 7):S21-S30. doi:10.1016/j.cardfail.2018.07.130
- Fox K, Borer JS, Camm AJ, et al. Resting heart rate in cardiovascular disease. J Am Coll Cardiol. 2007;50:823-830. doi:10.1016/j.jacc.2007.04.079
- De Ruvo E, Capucci A, Ammirati F, et al. Preliminary experience of remote management of heart failure patients with a multisensor ICD alert [abstract P1536]. Eur J Heart Fail. 2019;21(suppl S1):370.
- Hernandez AF, Albert NM, Allen LA, et al. Multiple cardiac sensors for management of heart failure (MANAGE- HF) - phase I evaluation of the integration and safety of the HeartLogic multisensor algorithm in patients with heart failure. J Card Fail. 2022;28:1245-1254. doi:10.1016/j.cardfail.2022.03.349
- Santini L, D’Onofrio A, Dello Russo A, et al. Prospective evaluation of the multisensor HeartLogic algorithm for heart failure monitoring. Clin Cardiol. 2020;43:691-697. doi:10.1002/clc.23366
- Yu CM, Wang L, Chau E, et al. Intrathoracic impedance monitoring in patients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation. 2005;112:841-848. doi:10.1161/CIRCULATIONAHA.104.492207
- Adamson PB, Abraham WT, Stevenson LW, et al. Pulmonary artery pressure-guided heart failure management reduces 30-day readmissions. Circ Heart Fail. 2016;9:e002600. doi:10.1161/CIRCHEARTFAILURE.115.002600
- Lindenfeld J, Zile MR, Desai AS, et al. Haemodynamic-guided management of heart failure (GUIDE-HF): a randomised controlled trial. Lancet. 2021;398:991-1001. doi:10.1016/S0140-6736(21)01754-2
Heart failure (HF) is a prevalent disease in the United States affecting > 6.5 million adults and contributing to significant morbidity and mortality.1 The disease course associated with HF includes potential symptom improvement with intermittent periods of decompensation and possible clinical deterioration. Multiple therapies have been developed to improve outcomes in people with HF—to palliate HF symptoms, prevent hospitalizations, and reduce mortality.2 However, the risks of decompensation and hospitalization remain. HF decompensation development may precede clear actionable symptoms such as worsening dyspnea, noticeable edema, or weight gain. Tools to identify patient deterioration and trigger interventions to prevent HF admissions are clinically attractive compared with reliance on subjective factors alone.
Cardiac resynchronization therapy (CRT) and implantable cardioverter-defibrillator (ICD) devices made by Boston Scientific include the HeartLogic monitoring feature. Five main sensors produce an index risk score; an index score > 16 warns clinicians that the patient is at an increased risk for a HF event.3 The 5 sensors are thoracic impedance, first (S1) and third heart sounds (S3), night heart rate (NHR), respiratory rate (RR), and activity. Each sensor can draw attention to the primary driver of the alert and guide health care practitioners (HCPs) to the appropriate interventions.3 A HeartLogic alert example is shown in Figure 1.
The S3 occurs during the early diastolic phase when blood moves into the ventricles. As HF worsens, with a combination of elevated filling pressures and reduced cardiac muscle compliance, S3 can become more pronounced.4 The S1 is correlated with the contractility of the left ventricle and will be reduced in patients at risk for HF events.5 Physical activity is a long-term prognostic marker in patients with HF; reduced activity is associated with mortality and increased risk of an HF event.6 Thoracic impedance is a sensor used to identify pulmonary congestion, pocket infections, pleural/pericardial effusion, and respiratory infections. The accumulation of intrathoracic fluid during pulmonary congestion increases conductance, causing a decrease in impedance.7 RR will increase as patients experience dyspnea with a more rapid, shallow breath and may trigger alerts closer to the actual HF event than other sensors. Nearly 90% of patients hospitalized for HF experience shortness of breath.8,9 NHR is used as a surrogate for resting heart rate (HR). A high resting HR is correlated with the progression of coronary atherosclerosis, harmful effects on left ventricular function, and increased risk of myocardial ischemia and ventricular arrhythmias.10
One of the challenges with preventing hospitalizations may be the lack of patient reported symptoms leading up to the event. The purpose of the sensors and HeartLogic index is to identify patients a median of 34 days before an HF event (HF admission or unscheduled intervention with intravenous treatment) with a sensitivity rate of 70%.3 According to real-word experience data, alerts have been found to precede HF symptoms by a median of 12 days and HF events such as hospitalizations by a median of 38 days, with an overall 67% reduction in HF hospitalizations when integrated into clinical care.11,12
MANAGE-HF evaluated 191 patients with HF with reduced ejection fraction (HFrEF) (< 35%), New York Heart Association class II-III symptoms, and who had an implanted CRT and/or ICD to develop an alert management guide to optimize medical treatment.12 It aimed to adjust patient regimen within 6 days of an elevated Heart- Logic index by either initiation, escalation, or maintenance of HF treatment depending on the index trend after the initial alert. This trial found that by focusing on such optimization, HF treatment was augmented during 74% of the 585 alert cases and during 54% of 3290 weekly alerts.
Initiation and uptitration of the 4 primary components of guideline-directed medical therapy (GDMT) are recommended by the 2022 Heart Failure Guidelines to reduce mortality and morbidity in patients with HFrEF.2 The 4 pillars of GDMT consist of -blockers (BB), sodium-glucose cotransporter type 2 inhibitors (SGLT2i), mineralocorticoid receptor antagonists (MRA), and renin-angiotensin-system inhibitors (RASi) including angiotensin II receptor blocker/neprilysin inhibitors (ARNi), angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARB) (Appendix 1). Obtaining and titrating to target doses wherever possible is recommended, as those were the doses that established safety and efficacy in patients with HFrEF in clinical trials.2 Pharmacists are adequately equipped to optimize HF GDMT and appropriately monitor drug response.
Through the use of HeartLogic in clinical practice, patients with HF have been shown to have improved clinical outcomes and are more likely to receive effective care; 80% of alerts were shown to provide new information to clinicians.13 This project sought to quantify the total number and types of pharmacist interventions driven by integration of HeartLogic index monitoring into practice.
Methods
The West Palm Beach Veterans Affairs Medical Center (WPBVAMC) Research Program Office approved this project and determined it was exempt from institutional review board oversight. Patients were screened retrospectively and prospectively from May 26, 2022, through December 31, 2022, by a cardiology clinical pharmacist practitioner (CPP) and a cardiology pharmacy resident using the local monitoring suite for the HeartLogic-compatible device, LATITUDE NXT. Read-only access to the local monitoring suit was granted by the National Cardiac Device Surveillance Program. Training for HeartLogic was completed through continuing education courses provided by Boston Scientific. Additional information was provided by Boston Scientific representative presentations and collaboration with WPBVAMC pacemaker clinic HCPs.
Individuals included were patients with HeartLogic-capable ICDs. A HeartLogic alert had to be present at initial patient contact. Patients were also contacted as part of routine clinical practice, but no formal number or frequency of calls to patients was required. The initial contact must be with a pharmacist for the patient to be included, but subsequent contact by other HCPs was included. Patients in the cardiology clinic are required to meet with a cardiologist at least annually; however, interim visits can be completed by advanced practice registered nurse practitioners, physicians assistants, or CPPs.
Patients in alert status were contacted by telephone and appropriate modifications of HF therapy were made by the CPP based on score metrics, medical record review, and patient interview. Information surrounding the initial alert, baseline patient data, medication and monitoring interventions made, and clinical outcomes such as hospitalization, symptom improvement, follow-up, and mortality were collected. Information for each encounter was collected until 42 days from the initial date of pharmacist contact.
Clinically successful tolerability of intervention implementation was defined as tolerability, adherence, and lack of adverse effects (AEs) per patient report at follow-up or within 42 days from initial alert (Appendix 2). A decrease in dose was not counted as intolerance. A single patient may have been counted as multiple encounters if the original intervention resulted in treatment intolerance and the patient remained in alert or if an additional alert occurred after 42 days of the initial alert. There were no specific time criteria for follow-up, which occurred at the CPP’s discretion.
There was no mandated algorithm used to alter medications based on the Heart- Logic score, nor were there required minimum or maximum numbers of interventions after an alert. Patient contact by telephone initiated an encounter. The types of interventions included medication increases, decreases, initiation, discontinuation, or no medication change. Each medication change and rationale, if applicable, was recorded for the encounter ≤ 42 days after the initial contact date. If a medication with required monitoring parameters was augmented, the pharmacist was responsible for ordering laboratory testing and follow-up. Most interventions were completed by telephone; however, some patients had in-person visits in the HF CPP clinic.
Outcomes
The primary outcome was the number of pharmacist interventions made to optimize GDMT, defined as either an initiation or dose increase. Key intervention analysis included the use and dosing of the 4 primary components of HF GDMT: BB, SGLT2i, MRA, and ARNi/ARB/ACEi. In addition to the 4 primary components of GDMT, loop diuretic changes were also recorded and analyzed. Secondary endpoints were the number of HF hospitalizations ≤ 42 days after the initial alert, and the effect of medication interventions on device metrics, patient symptoms, and tolerability. Successful tolerability was defined as continued use of augmented GDMT without intolerance or discontinuation. The primary analysis was analyzed through descriptive statistics. Median changes in HeartLogic scores and metrics from baseline were analyzed using a paired, 2-sided t test with an α of .05 to detect significance.
Results
There were 39 WPBVAMC patients with a HeartLogic-capable device. Twenty-one alert encounters were analyzed in 16 patients (41%) over 31 weeks of data collection. The 16 patients at baseline had a mean age of 74 years, all were male, and 12 (75%) were White. Eight patients (50%) had a recent ejection fraction (EF) between 30% and 40%. Three patients had an EF ≥ 40%. At the time of alert, 15 patients used BB (94%), 10 used loop diuretics (63%), and 9 used ARNi (56%) (Table 1).
There were 23 medication changes made during initial contact. The most common change was starting an SGLT2i (30%; n = 7), followed by starting an MRA (22%; n = 5), and increasing the ARNi dose (22%; n = 5). At the initial contact, ≥ 1 medication optimization occurred in 95% (n = 20) of encounters. The CPP contacted patients a mean of 4.8 days after the initial alert.
Patients were taking a mean of 2.6 primary GDMT medications at baseline and 3.0 at 42 days. CPP encounters led to a mean of 1.8 medication changes over the 6-week period (range, 0-5). Seventeen medications were started, 13 medications were increased, 3 medications were decreased, and 4 medications were stopped (Table 2). One ACEi and 1 ARB were switched as a therapy escalation to an ARNi. One patient was on 1 of 4 primary GDMTs at baseline, which increased to 4 GDMT agents at 42 days.
SGLT2 inhibitors were added most often at initial contact (54%) and throughout the 42-day period (41%). The most common successfully tolerated optimizations were RASi, followed by MRA, SGLT2 inhibitors, BB, and loop diuretics with 11, 6, 5, 3, and 2 patients, respectively. Interventions were tolerated by 90% of patients, and no HF hospitalization occurred during follow-up. All possible rationales for patients with the same or reduced number of GDMT at 42 days compared with baseline are shown in Appendix 2.
Device Metrics
During initial contact, the most common HeartLogic metric category that was predominantly worsening were heart sounds (S1, S3, and S3/S1 ratio), followed by compensatory mechanism sensors (NHR and RR) and congestion (impedance) at rates of 61.9%, 23.8%, and 14.3%, respectively (Figure 2).
The median HeartLogic index score was 18 at baseline and 5 at the end of the follow-up period (P < .001). The changes in score and metrics were compared with the type of successfully tolerated GDMT optimization made (Table 3). The GDMT optimization analysis included SGLT2i, RASi, MRA, BB, and loop diuretics. All interventions reduced the overall HeartLogic index score, ranging from a 9.5-point reduction (loop diuretics) to a 16-point reduction (SGLT2i and BB). Optimization of SGLT2i, RASi, and loop diuretics had a positive impact on S1 score. For S3 score, SGLT2i, MRA, and BB had a positive impact. All medications, except for SGLT2i therapy, reduced the NHR score. Optimization of MRA, SGLT2i, and BB had positive impacts on the impedance score. All medications reduced RR from baseline. Only SGLT2i and loop diuretics had positive impacts on the activity score.
Clinical Outcomes and Adverse Effects
Within 42 days of contact, 17 encounters (81%) had ≥ 1 follow-up appointment with a CPP and all 21 patients had ≥ 1 follow-up health care team member. One patient had a HF-related hospitalization within 42 days of contact; however, that individual refused the recommended medication intervention. There were 13 encounters (62%) with reported symptoms at the time of the initial alert and 10 (77%) had subjective symptom improvement at 42 days (Appendix 3).
Of 30 medication optimizations, 27 primary GDMT medications were tolerated. Two medication intolerances led to discontinuation (1 SGLT2i and 1 loop diuretic) and 1 patient never started the SGLT2i (Table 4). There was only 1 known patient who did not follow the directions to adjust their medications. That individual was included because the patient agreed to the change during the CPP visit but later reported that he had never started the SGLT2i.
Discussion
The HeartLogic tool created a bridge for patients with HF to work with CPPs as soon as possible to optimize medication therapy to reduce HF events. This study highlights an additional area of expertise and service that CPPs may offer to their specialty HF clinic team. Over 31 weeks, 21 encounters and 30 medication optimizations were completed. These interventions led to significant reductions in HeartLogic scores, improvements in symptoms, and optimization of HF care, most of which were well tolerated.
Additional hemodynamic monitoring devices are available. Similar to HeartLogic, OptiVol is a tool embedded in select Medtronic implantable devices that monitors fluid status. 14 CardioMEMS is an implantable pulmonary artery pressure sensor used as a presymptomatic data point to alert clinicians when HF is worsening. In the CHAMPION trial, the use of CardioMEMS showed a 28% reduction in HF-related hospitalization at 6 months.15 Conversely, in the GUIDE-HF trial, monitoring with CardioMEMS did not significantly reduce the composite endpoint of mortality and total HF events.16 Therefore, remote hemodynamic monitoring has variable results and the use of these tools remains uncertain per the clinical guidelines.2
The MANAGE-HF study that contributed to the validation of the HeartLogic tool may provide a comparison with this smaller single-center project. The time to follow-up within 7 days of alert was noted in only 54% of the patients in MANAGE-HF.12 In this study, 86% of patients received follow-up within 7 days, with a mean of 4.8 days. The quick turnaround from the time of alert to intervention portrays pharmacists as readily available HCPs.
In MANAGE-HF, 89% of medication augmentation involved loop diuretics or thiazides; in our project, loop diuretics were the least frequently changed medication. Most optimizations in this project included ARNi, SGLT2i, BB, and MRA, which have been shown to reduce morbidity and mortality.2 Our project included use of SGLT2i therapy to affect HeartLogic metrics, which has not been evaluated previously. SGLT2i were the most commonly initiated medication after an alert. Of the 5 tolerated SGLT2i optimization encounters, 4 were out of alert at 42 days.
SGLT2i resulted in a significant decrease in HeartLogic index score from baseline and were the only class of medication that did not produce a negative change in any metric. In this study, CPPs utilizing and acting on HeartLogic alerts led to 1 (4.8%) hospitalization with HF as the primary reason for admission and no hospitalizations as a secondary cause in 42 days, compared to 37% and 7.9% in the MANAGE-HF in 1 year, respectively. An additional screening 1 year after the initial alert found that 2 (12.5%) of 12 patients had been admitted with 1 HF hospitalization each.
A strength of this study was the ability to use HeartLogic to identify high-risk patients, provide a source of patient contact and monitoring, interpret 5 cardiac sensors, and optimize all HF GDMT, not just volume management. By focusing efforts on making patient contact and pharmacotherapy interventions with morbidity and mortality benefit, remote hemodynamic monitoring may show a clear clinical benefit and become a vital part of HF care.
Limitations
Checking for adherence and tolerance to medications were mainly patient reported if there was a CPP follow-up within 42 days, or potentially through refill history when unclear. However, this limitation is reflective of current practice where patients may have multiple clinicians working to optimize HF care and where there is reliance on patients in order to guide continued therapy. Although unable to explicitly show a reduction in HF events given lack of comparator group, the interventions made are associated with improved outcomes and thus would be expected to improve patient outcomes. Changes in vital signs were not tracked as part of this project, however the main rationale for changes made were to optimize GDMT therapy, not specifically to impact vital sign measures.
HeartLogic alerts prompted identification of high-risk patients with HF, pharmacist evaluation and outreach, patient-focused pharmacotherapy care, and beneficial patient outcomes. With only 2 cardiology CPPs checking alerts once weekly, future studies may be needed with larger samples to create algorithms and protocols to increase the clinical utility of this tool on a greater scale.
Conclusions
Cardiology CPP-led HF interventions triggered by HeartLogic alerts lead to effective patient identification, increased access to care, reductions in HeartLogic scores, improvements in symptoms, and optimization of HF care. This project demonstrates the practical utility of the HeartLogic suite in conjunction with CPP care to prioritize treatment for highrisk patients with HF in an efficient manner. The data highlight the potential value of the HeartLogic tool and a CPP in HF care to facilitate initiation and optimization of GDMT to ultimately improve the morbidity and mortality in patients with HF.
Heart failure (HF) is a prevalent disease in the United States affecting > 6.5 million adults and contributing to significant morbidity and mortality.1 The disease course associated with HF includes potential symptom improvement with intermittent periods of decompensation and possible clinical deterioration. Multiple therapies have been developed to improve outcomes in people with HF—to palliate HF symptoms, prevent hospitalizations, and reduce mortality.2 However, the risks of decompensation and hospitalization remain. HF decompensation development may precede clear actionable symptoms such as worsening dyspnea, noticeable edema, or weight gain. Tools to identify patient deterioration and trigger interventions to prevent HF admissions are clinically attractive compared with reliance on subjective factors alone.
Cardiac resynchronization therapy (CRT) and implantable cardioverter-defibrillator (ICD) devices made by Boston Scientific include the HeartLogic monitoring feature. Five main sensors produce an index risk score; an index score > 16 warns clinicians that the patient is at an increased risk for a HF event.3 The 5 sensors are thoracic impedance, first (S1) and third heart sounds (S3), night heart rate (NHR), respiratory rate (RR), and activity. Each sensor can draw attention to the primary driver of the alert and guide health care practitioners (HCPs) to the appropriate interventions.3 A HeartLogic alert example is shown in Figure 1.
The S3 occurs during the early diastolic phase when blood moves into the ventricles. As HF worsens, with a combination of elevated filling pressures and reduced cardiac muscle compliance, S3 can become more pronounced.4 The S1 is correlated with the contractility of the left ventricle and will be reduced in patients at risk for HF events.5 Physical activity is a long-term prognostic marker in patients with HF; reduced activity is associated with mortality and increased risk of an HF event.6 Thoracic impedance is a sensor used to identify pulmonary congestion, pocket infections, pleural/pericardial effusion, and respiratory infections. The accumulation of intrathoracic fluid during pulmonary congestion increases conductance, causing a decrease in impedance.7 RR will increase as patients experience dyspnea with a more rapid, shallow breath and may trigger alerts closer to the actual HF event than other sensors. Nearly 90% of patients hospitalized for HF experience shortness of breath.8,9 NHR is used as a surrogate for resting heart rate (HR). A high resting HR is correlated with the progression of coronary atherosclerosis, harmful effects on left ventricular function, and increased risk of myocardial ischemia and ventricular arrhythmias.10
One of the challenges with preventing hospitalizations may be the lack of patient reported symptoms leading up to the event. The purpose of the sensors and HeartLogic index is to identify patients a median of 34 days before an HF event (HF admission or unscheduled intervention with intravenous treatment) with a sensitivity rate of 70%.3 According to real-word experience data, alerts have been found to precede HF symptoms by a median of 12 days and HF events such as hospitalizations by a median of 38 days, with an overall 67% reduction in HF hospitalizations when integrated into clinical care.11,12
MANAGE-HF evaluated 191 patients with HF with reduced ejection fraction (HFrEF) (< 35%), New York Heart Association class II-III symptoms, and who had an implanted CRT and/or ICD to develop an alert management guide to optimize medical treatment.12 It aimed to adjust patient regimen within 6 days of an elevated Heart- Logic index by either initiation, escalation, or maintenance of HF treatment depending on the index trend after the initial alert. This trial found that by focusing on such optimization, HF treatment was augmented during 74% of the 585 alert cases and during 54% of 3290 weekly alerts.
Initiation and uptitration of the 4 primary components of guideline-directed medical therapy (GDMT) are recommended by the 2022 Heart Failure Guidelines to reduce mortality and morbidity in patients with HFrEF.2 The 4 pillars of GDMT consist of -blockers (BB), sodium-glucose cotransporter type 2 inhibitors (SGLT2i), mineralocorticoid receptor antagonists (MRA), and renin-angiotensin-system inhibitors (RASi) including angiotensin II receptor blocker/neprilysin inhibitors (ARNi), angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARB) (Appendix 1). Obtaining and titrating to target doses wherever possible is recommended, as those were the doses that established safety and efficacy in patients with HFrEF in clinical trials.2 Pharmacists are adequately equipped to optimize HF GDMT and appropriately monitor drug response.
Through the use of HeartLogic in clinical practice, patients with HF have been shown to have improved clinical outcomes and are more likely to receive effective care; 80% of alerts were shown to provide new information to clinicians.13 This project sought to quantify the total number and types of pharmacist interventions driven by integration of HeartLogic index monitoring into practice.
Methods
The West Palm Beach Veterans Affairs Medical Center (WPBVAMC) Research Program Office approved this project and determined it was exempt from institutional review board oversight. Patients were screened retrospectively and prospectively from May 26, 2022, through December 31, 2022, by a cardiology clinical pharmacist practitioner (CPP) and a cardiology pharmacy resident using the local monitoring suite for the HeartLogic-compatible device, LATITUDE NXT. Read-only access to the local monitoring suit was granted by the National Cardiac Device Surveillance Program. Training for HeartLogic was completed through continuing education courses provided by Boston Scientific. Additional information was provided by Boston Scientific representative presentations and collaboration with WPBVAMC pacemaker clinic HCPs.
Individuals included were patients with HeartLogic-capable ICDs. A HeartLogic alert had to be present at initial patient contact. Patients were also contacted as part of routine clinical practice, but no formal number or frequency of calls to patients was required. The initial contact must be with a pharmacist for the patient to be included, but subsequent contact by other HCPs was included. Patients in the cardiology clinic are required to meet with a cardiologist at least annually; however, interim visits can be completed by advanced practice registered nurse practitioners, physicians assistants, or CPPs.
Patients in alert status were contacted by telephone and appropriate modifications of HF therapy were made by the CPP based on score metrics, medical record review, and patient interview. Information surrounding the initial alert, baseline patient data, medication and monitoring interventions made, and clinical outcomes such as hospitalization, symptom improvement, follow-up, and mortality were collected. Information for each encounter was collected until 42 days from the initial date of pharmacist contact.
Clinically successful tolerability of intervention implementation was defined as tolerability, adherence, and lack of adverse effects (AEs) per patient report at follow-up or within 42 days from initial alert (Appendix 2). A decrease in dose was not counted as intolerance. A single patient may have been counted as multiple encounters if the original intervention resulted in treatment intolerance and the patient remained in alert or if an additional alert occurred after 42 days of the initial alert. There were no specific time criteria for follow-up, which occurred at the CPP’s discretion.
There was no mandated algorithm used to alter medications based on the Heart- Logic score, nor were there required minimum or maximum numbers of interventions after an alert. Patient contact by telephone initiated an encounter. The types of interventions included medication increases, decreases, initiation, discontinuation, or no medication change. Each medication change and rationale, if applicable, was recorded for the encounter ≤ 42 days after the initial contact date. If a medication with required monitoring parameters was augmented, the pharmacist was responsible for ordering laboratory testing and follow-up. Most interventions were completed by telephone; however, some patients had in-person visits in the HF CPP clinic.
Outcomes
The primary outcome was the number of pharmacist interventions made to optimize GDMT, defined as either an initiation or dose increase. Key intervention analysis included the use and dosing of the 4 primary components of HF GDMT: BB, SGLT2i, MRA, and ARNi/ARB/ACEi. In addition to the 4 primary components of GDMT, loop diuretic changes were also recorded and analyzed. Secondary endpoints were the number of HF hospitalizations ≤ 42 days after the initial alert, and the effect of medication interventions on device metrics, patient symptoms, and tolerability. Successful tolerability was defined as continued use of augmented GDMT without intolerance or discontinuation. The primary analysis was analyzed through descriptive statistics. Median changes in HeartLogic scores and metrics from baseline were analyzed using a paired, 2-sided t test with an α of .05 to detect significance.
Results
There were 39 WPBVAMC patients with a HeartLogic-capable device. Twenty-one alert encounters were analyzed in 16 patients (41%) over 31 weeks of data collection. The 16 patients at baseline had a mean age of 74 years, all were male, and 12 (75%) were White. Eight patients (50%) had a recent ejection fraction (EF) between 30% and 40%. Three patients had an EF ≥ 40%. At the time of alert, 15 patients used BB (94%), 10 used loop diuretics (63%), and 9 used ARNi (56%) (Table 1).
There were 23 medication changes made during initial contact. The most common change was starting an SGLT2i (30%; n = 7), followed by starting an MRA (22%; n = 5), and increasing the ARNi dose (22%; n = 5). At the initial contact, ≥ 1 medication optimization occurred in 95% (n = 20) of encounters. The CPP contacted patients a mean of 4.8 days after the initial alert.
Patients were taking a mean of 2.6 primary GDMT medications at baseline and 3.0 at 42 days. CPP encounters led to a mean of 1.8 medication changes over the 6-week period (range, 0-5). Seventeen medications were started, 13 medications were increased, 3 medications were decreased, and 4 medications were stopped (Table 2). One ACEi and 1 ARB were switched as a therapy escalation to an ARNi. One patient was on 1 of 4 primary GDMTs at baseline, which increased to 4 GDMT agents at 42 days.
SGLT2 inhibitors were added most often at initial contact (54%) and throughout the 42-day period (41%). The most common successfully tolerated optimizations were RASi, followed by MRA, SGLT2 inhibitors, BB, and loop diuretics with 11, 6, 5, 3, and 2 patients, respectively. Interventions were tolerated by 90% of patients, and no HF hospitalization occurred during follow-up. All possible rationales for patients with the same or reduced number of GDMT at 42 days compared with baseline are shown in Appendix 2.
Device Metrics
During initial contact, the most common HeartLogic metric category that was predominantly worsening were heart sounds (S1, S3, and S3/S1 ratio), followed by compensatory mechanism sensors (NHR and RR) and congestion (impedance) at rates of 61.9%, 23.8%, and 14.3%, respectively (Figure 2).
The median HeartLogic index score was 18 at baseline and 5 at the end of the follow-up period (P < .001). The changes in score and metrics were compared with the type of successfully tolerated GDMT optimization made (Table 3). The GDMT optimization analysis included SGLT2i, RASi, MRA, BB, and loop diuretics. All interventions reduced the overall HeartLogic index score, ranging from a 9.5-point reduction (loop diuretics) to a 16-point reduction (SGLT2i and BB). Optimization of SGLT2i, RASi, and loop diuretics had a positive impact on S1 score. For S3 score, SGLT2i, MRA, and BB had a positive impact. All medications, except for SGLT2i therapy, reduced the NHR score. Optimization of MRA, SGLT2i, and BB had positive impacts on the impedance score. All medications reduced RR from baseline. Only SGLT2i and loop diuretics had positive impacts on the activity score.
Clinical Outcomes and Adverse Effects
Within 42 days of contact, 17 encounters (81%) had ≥ 1 follow-up appointment with a CPP and all 21 patients had ≥ 1 follow-up health care team member. One patient had a HF-related hospitalization within 42 days of contact; however, that individual refused the recommended medication intervention. There were 13 encounters (62%) with reported symptoms at the time of the initial alert and 10 (77%) had subjective symptom improvement at 42 days (Appendix 3).
Of 30 medication optimizations, 27 primary GDMT medications were tolerated. Two medication intolerances led to discontinuation (1 SGLT2i and 1 loop diuretic) and 1 patient never started the SGLT2i (Table 4). There was only 1 known patient who did not follow the directions to adjust their medications. That individual was included because the patient agreed to the change during the CPP visit but later reported that he had never started the SGLT2i.
Discussion
The HeartLogic tool created a bridge for patients with HF to work with CPPs as soon as possible to optimize medication therapy to reduce HF events. This study highlights an additional area of expertise and service that CPPs may offer to their specialty HF clinic team. Over 31 weeks, 21 encounters and 30 medication optimizations were completed. These interventions led to significant reductions in HeartLogic scores, improvements in symptoms, and optimization of HF care, most of which were well tolerated.
Additional hemodynamic monitoring devices are available. Similar to HeartLogic, OptiVol is a tool embedded in select Medtronic implantable devices that monitors fluid status. 14 CardioMEMS is an implantable pulmonary artery pressure sensor used as a presymptomatic data point to alert clinicians when HF is worsening. In the CHAMPION trial, the use of CardioMEMS showed a 28% reduction in HF-related hospitalization at 6 months.15 Conversely, in the GUIDE-HF trial, monitoring with CardioMEMS did not significantly reduce the composite endpoint of mortality and total HF events.16 Therefore, remote hemodynamic monitoring has variable results and the use of these tools remains uncertain per the clinical guidelines.2
The MANAGE-HF study that contributed to the validation of the HeartLogic tool may provide a comparison with this smaller single-center project. The time to follow-up within 7 days of alert was noted in only 54% of the patients in MANAGE-HF.12 In this study, 86% of patients received follow-up within 7 days, with a mean of 4.8 days. The quick turnaround from the time of alert to intervention portrays pharmacists as readily available HCPs.
In MANAGE-HF, 89% of medication augmentation involved loop diuretics or thiazides; in our project, loop diuretics were the least frequently changed medication. Most optimizations in this project included ARNi, SGLT2i, BB, and MRA, which have been shown to reduce morbidity and mortality.2 Our project included use of SGLT2i therapy to affect HeartLogic metrics, which has not been evaluated previously. SGLT2i were the most commonly initiated medication after an alert. Of the 5 tolerated SGLT2i optimization encounters, 4 were out of alert at 42 days.
SGLT2i resulted in a significant decrease in HeartLogic index score from baseline and were the only class of medication that did not produce a negative change in any metric. In this study, CPPs utilizing and acting on HeartLogic alerts led to 1 (4.8%) hospitalization with HF as the primary reason for admission and no hospitalizations as a secondary cause in 42 days, compared to 37% and 7.9% in the MANAGE-HF in 1 year, respectively. An additional screening 1 year after the initial alert found that 2 (12.5%) of 12 patients had been admitted with 1 HF hospitalization each.
A strength of this study was the ability to use HeartLogic to identify high-risk patients, provide a source of patient contact and monitoring, interpret 5 cardiac sensors, and optimize all HF GDMT, not just volume management. By focusing efforts on making patient contact and pharmacotherapy interventions with morbidity and mortality benefit, remote hemodynamic monitoring may show a clear clinical benefit and become a vital part of HF care.
Limitations
Checking for adherence and tolerance to medications were mainly patient reported if there was a CPP follow-up within 42 days, or potentially through refill history when unclear. However, this limitation is reflective of current practice where patients may have multiple clinicians working to optimize HF care and where there is reliance on patients in order to guide continued therapy. Although unable to explicitly show a reduction in HF events given lack of comparator group, the interventions made are associated with improved outcomes and thus would be expected to improve patient outcomes. Changes in vital signs were not tracked as part of this project, however the main rationale for changes made were to optimize GDMT therapy, not specifically to impact vital sign measures.
HeartLogic alerts prompted identification of high-risk patients with HF, pharmacist evaluation and outreach, patient-focused pharmacotherapy care, and beneficial patient outcomes. With only 2 cardiology CPPs checking alerts once weekly, future studies may be needed with larger samples to create algorithms and protocols to increase the clinical utility of this tool on a greater scale.
Conclusions
Cardiology CPP-led HF interventions triggered by HeartLogic alerts lead to effective patient identification, increased access to care, reductions in HeartLogic scores, improvements in symptoms, and optimization of HF care. This project demonstrates the practical utility of the HeartLogic suite in conjunction with CPP care to prioritize treatment for highrisk patients with HF in an efficient manner. The data highlight the potential value of the HeartLogic tool and a CPP in HF care to facilitate initiation and optimization of GDMT to ultimately improve the morbidity and mortality in patients with HF.
- Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147:e93-e621. doi:10.1161/CIR.0000000000001123
- Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/ American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895-e1032. doi:10.1161/CIR.0000000000001063
- Boehmer JP, Hariharan R, Devecchi FG, et al. A multisensor algorithm predicts heart failure events in patients with implanted devices: results from the MultiSENSE study. J Am Coll Cardiol HF. 2017;5:216-225. doi:10.1016/j.jchf.2016.12.011
- Cao M, Gardner RS, Hariharan R, et al. Ambulatory monitoring of heart sounds via an implanted device is superior to auscultation for prediction of heart failure events. J Card Fail. 2020;26:151-159. doi:10.1016/j.cardfail.2019.10.006
- Calò L, Capucci A, Santini L, et al. ICD-measured heart sounds and their correlation with echocardiographic indexes of systolic and diastolic function. J Interv Card Electrophysiol. 2020;58:95-101. doi:10.1007/s10840-019-00668
- Del Buono MG, Arena R, Borlaug BA, et al. Exercise intolerance in patients with heart failure: JACC state-of-the- art review. J Am Coll Cardiol. 2019;73:2209-2225. doi:10.1016/j.jacc.2019.01.072
- Yu CM, Wang L, Chau E, et al. Intrathoracic impedance monitoring in patients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation. 2005;112:841-848. doi:10.1161/CIRCULATIONAHA.104.492207
- Rials S, Aktas M, An Q, et al. Continuous respiratory rate is superior to routine outpatient dyspnea assessment for predicting heart failure events. J Card Fail. 2018;24:S45.
- Fonarow GC, ADHERE Scientific Advisory Committee. The Acute Decompensated Heart Failure National Registry (ADHERE): opportunities to improve care of patients hospitalized with acute decompensated heart failure. Rev Cardiovasc Med. 2003;4(suppl 7):S21-S30. doi:10.1016/j.cardfail.2018.07.130
- Fox K, Borer JS, Camm AJ, et al. Resting heart rate in cardiovascular disease. J Am Coll Cardiol. 2007;50:823-830. doi:10.1016/j.jacc.2007.04.079
- De Ruvo E, Capucci A, Ammirati F, et al. Preliminary experience of remote management of heart failure patients with a multisensor ICD alert [abstract P1536]. Eur J Heart Fail. 2019;21(suppl S1):370.
- Hernandez AF, Albert NM, Allen LA, et al. Multiple cardiac sensors for management of heart failure (MANAGE- HF) - phase I evaluation of the integration and safety of the HeartLogic multisensor algorithm in patients with heart failure. J Card Fail. 2022;28:1245-1254. doi:10.1016/j.cardfail.2022.03.349
- Santini L, D’Onofrio A, Dello Russo A, et al. Prospective evaluation of the multisensor HeartLogic algorithm for heart failure monitoring. Clin Cardiol. 2020;43:691-697. doi:10.1002/clc.23366
- Yu CM, Wang L, Chau E, et al. Intrathoracic impedance monitoring in patients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation. 2005;112:841-848. doi:10.1161/CIRCULATIONAHA.104.492207
- Adamson PB, Abraham WT, Stevenson LW, et al. Pulmonary artery pressure-guided heart failure management reduces 30-day readmissions. Circ Heart Fail. 2016;9:e002600. doi:10.1161/CIRCHEARTFAILURE.115.002600
- Lindenfeld J, Zile MR, Desai AS, et al. Haemodynamic-guided management of heart failure (GUIDE-HF): a randomised controlled trial. Lancet. 2021;398:991-1001. doi:10.1016/S0140-6736(21)01754-2
- Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147:e93-e621. doi:10.1161/CIR.0000000000001123
- Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/ American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895-e1032. doi:10.1161/CIR.0000000000001063
- Boehmer JP, Hariharan R, Devecchi FG, et al. A multisensor algorithm predicts heart failure events in patients with implanted devices: results from the MultiSENSE study. J Am Coll Cardiol HF. 2017;5:216-225. doi:10.1016/j.jchf.2016.12.011
- Cao M, Gardner RS, Hariharan R, et al. Ambulatory monitoring of heart sounds via an implanted device is superior to auscultation for prediction of heart failure events. J Card Fail. 2020;26:151-159. doi:10.1016/j.cardfail.2019.10.006
- Calò L, Capucci A, Santini L, et al. ICD-measured heart sounds and their correlation with echocardiographic indexes of systolic and diastolic function. J Interv Card Electrophysiol. 2020;58:95-101. doi:10.1007/s10840-019-00668
- Del Buono MG, Arena R, Borlaug BA, et al. Exercise intolerance in patients with heart failure: JACC state-of-the- art review. J Am Coll Cardiol. 2019;73:2209-2225. doi:10.1016/j.jacc.2019.01.072
- Yu CM, Wang L, Chau E, et al. Intrathoracic impedance monitoring in patients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation. 2005;112:841-848. doi:10.1161/CIRCULATIONAHA.104.492207
- Rials S, Aktas M, An Q, et al. Continuous respiratory rate is superior to routine outpatient dyspnea assessment for predicting heart failure events. J Card Fail. 2018;24:S45.
- Fonarow GC, ADHERE Scientific Advisory Committee. The Acute Decompensated Heart Failure National Registry (ADHERE): opportunities to improve care of patients hospitalized with acute decompensated heart failure. Rev Cardiovasc Med. 2003;4(suppl 7):S21-S30. doi:10.1016/j.cardfail.2018.07.130
- Fox K, Borer JS, Camm AJ, et al. Resting heart rate in cardiovascular disease. J Am Coll Cardiol. 2007;50:823-830. doi:10.1016/j.jacc.2007.04.079
- De Ruvo E, Capucci A, Ammirati F, et al. Preliminary experience of remote management of heart failure patients with a multisensor ICD alert [abstract P1536]. Eur J Heart Fail. 2019;21(suppl S1):370.
- Hernandez AF, Albert NM, Allen LA, et al. Multiple cardiac sensors for management of heart failure (MANAGE- HF) - phase I evaluation of the integration and safety of the HeartLogic multisensor algorithm in patients with heart failure. J Card Fail. 2022;28:1245-1254. doi:10.1016/j.cardfail.2022.03.349
- Santini L, D’Onofrio A, Dello Russo A, et al. Prospective evaluation of the multisensor HeartLogic algorithm for heart failure monitoring. Clin Cardiol. 2020;43:691-697. doi:10.1002/clc.23366
- Yu CM, Wang L, Chau E, et al. Intrathoracic impedance monitoring in patients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation. 2005;112:841-848. doi:10.1161/CIRCULATIONAHA.104.492207
- Adamson PB, Abraham WT, Stevenson LW, et al. Pulmonary artery pressure-guided heart failure management reduces 30-day readmissions. Circ Heart Fail. 2016;9:e002600. doi:10.1161/CIRCHEARTFAILURE.115.002600
- Lindenfeld J, Zile MR, Desai AS, et al. Haemodynamic-guided management of heart failure (GUIDE-HF): a randomised controlled trial. Lancet. 2021;398:991-1001. doi:10.1016/S0140-6736(21)01754-2
Heart Failure Diagnostic Alerts to Prompt Pharmacist Evaluation and Medication Optimization
Heart Failure Diagnostic Alerts to Prompt Pharmacist Evaluation and Medication Optimization

Codes, Contracts, and Commitments: Who Defines What is a Profession?
Codes, Contracts, and Commitments: Who Defines What is a Profession?
A professional is someone who can do his best work when he doesn’t feel like it.
Alistair Cooke
When I was a young person with no idea about growing up to be something, my father used to tell me there were 4 learned professions: medicine to heal the body, law to protect the body politic, teaching to nurture the mind, and the clergy to care for the soul.1 That adage, or some version of it, is attributed to a variety of sources, likely because it captures something essential and timeless about the learned professions. I write this as a much older person, and it has been my privilege to have worked in some capacity in all 4 of these venerable vocations.
There are many more recognized professions now than in my father’s time with new ones still emerging as the world becomes more complicated and specialized. In November 2025, however, the growth of the professions was dealt a serious blow when the US Department of Education (DOE) redefined what constitutes a profession for the purpose of federal funding of graduate degrees.2 The internet is understandably abuzz with opinions across the political spectrum. What is missing from many of these discussions is an understanding of the criteria for a profession and, even more importantly, who has the authority to decide when an individual or a group has met that standard.
But first, what and why did the DOE make this change? The One Big Beautiful Bill Act charged the DOE with reducing what it claims is massive overspending on graduate education by limiting the programs that meet the definition of a “professional degree” eligible for higher funding. Of my father’s 4, medicine (including dentistry) and law made the cut with students in those professions able to borrow up to $200,000 in direct unsubsidized student loans while those in other programs would be limited to $100,000.2
As one of the oldest and most respected professions in America, nursing has received the most media attention, yet there are also other important and valued professions that are missing from the DOE list.3 The excluded professions also include: physician assistants, physical therapists, audiologists, architects, accountants, educators, and social workers. The proposed regulatory changes are not yet finalized and Congressional representatives, health care experts, and a myriad of professional associations have rightly objected the reclassification will only worsen the critical shortage of nurses, teachers, and other helping professions the country is already facing.4
There are thousands of federal health care professionals who worked long and hard to achieve their goals whom this Act undervalues. Moreover, the regulatory change leaves many students enrolled in education and training programs under federal practice auspices confused and overwhelmed. Perhaps they can take some hope and inspiration from the recognition that historically and philosophically, no agency or administration can unilaterally define what is a profession.
The literature on professionalism is voluminous, in large part because it has been surprisingly difficult to reach a consensus definition. A proposed definition from scholars captures most of the key aspects of a profession. While it is drawn from the medical literature, it applies to most of the caring professions the DOE disqualified. For pedagogic purposes, the definition is parsed into discrete criteria in the Table.5

Even this simple summary makes it obvious that a government agency alone could not possibly have the competence to determine who meets these complex technical and moral criteria. The members of the profession must assume a primary role in that determination. The complicated history of the professions shows that the locus of these decisions has resided in various combinations of educational institutions, such as nursing schools,6 professional societies (eg, National Association of Social Workers),7 and certifying boards (eg, National Commission on Certification of Physician Assistants).8 States, not the federal government, have long played a key part in defining professions in the US, through their authority to grant licenses to practice.9
In response to criticism, the DOE has stated that “the definition of a ‘professional degree’ is an internal definition used by the Department of Education to distinguish among programs that qualify for higher loan limits, not a value judgment about the importance of programs. It has no bearing on whether a program is professional in nature or not.”2 Given the ancient compact between society and the professions in which the government subsidizes the training of professionals dedicated to public service, it is hard to see how these changes can be dismissed as merely semantic and not a promissory breach.10
I recognize that this abstract editorial is little comfort to beleaguered and demoralized professionals and students. Still, it offers a voice of support for each federal practitioner or trainee who fulfills the epigraph’s description of a professional day after day. The nurse who works the extra shift without complaint or resentment so that veterans receive the care they deserve, the social worker who responds on a weekend night to an active duty family without food so they do not spend another night hungry, and the physician assistant who makes it into the isolated public health clinic despite the terrible weather so there is someone ready to take care for patients in need. The proposed policy shift cannot in any meaningful sense rob them of their identity as individuals committed to a code of caring. However, without an intact social compact, it may well remove their practical ability to remain and enter the helping professions to the detriment of us all.
- Wade JW. Public responsibilities of the learned professions. Louisiana Law Rev. 1960;21:130-148
- US Department of Education. Myth vs. fact: the definition of professional degrees. Press Release. November 24, 2025. Accessed December 22, 2025. https://www.ed.gov/about/news/press-release/myth-vs-fact-definition-of-professional-degrees
- Laws J. Full list of degrees not classed as “professional” by Trump admin. Newsweek. Updated November 26, 2025. Accessed December 22, 2025. https://www.newsweek.com/full-list-degrees-professional-trump-administration-11085695
- New York Academy of Medicine. Response to stripping “professional status” as proposed by the Department of Education. New York Academy of Medicine. November 24, 2025. Accessed December 22, 2025. https://nyam.org/article/response-to-stripping-professional-status-as-proposed-by-the-department-of-education
- Cruess SR, Johnston S, Cruess RL. “Profession”: a working definition for medical educators. Teach Learn Med. 2004;16:74-76. doi:10.1207/s15328015tlm1601_15
- American Association of Colleges of Nursing. Nursing is a professional degree. American Association of Colleges of Nursing. Accessed December 20, 2025. https://www.aacnnursing.org/policy-advocacy/take-action/nursing-is-a-professional-degree
- National Association of Social Workers. Social work is a profession. Social Workers. Accessed December 20, 2025. https://www.socialworkers.org
- National Commission on Certification of Physician Assistants. Accessed December 20, 2025. https://www.nccpa.net/about-nccpa/#who-we-are
- The Federation of State Boards of Physical Therapy. Accessed December 20, 2025. https://www.fsbpt.org/About-Us/Staff-Home
- Cruess SR, Cruess RL. Professionalism and medicine’s contract with social contract with society. Virtual Mentor. 2004;6:185-188. doi:10.1001/virtualmentor.2004.6.4.msoc1-040
A professional is someone who can do his best work when he doesn’t feel like it.
Alistair Cooke
When I was a young person with no idea about growing up to be something, my father used to tell me there were 4 learned professions: medicine to heal the body, law to protect the body politic, teaching to nurture the mind, and the clergy to care for the soul.1 That adage, or some version of it, is attributed to a variety of sources, likely because it captures something essential and timeless about the learned professions. I write this as a much older person, and it has been my privilege to have worked in some capacity in all 4 of these venerable vocations.
There are many more recognized professions now than in my father’s time with new ones still emerging as the world becomes more complicated and specialized. In November 2025, however, the growth of the professions was dealt a serious blow when the US Department of Education (DOE) redefined what constitutes a profession for the purpose of federal funding of graduate degrees.2 The internet is understandably abuzz with opinions across the political spectrum. What is missing from many of these discussions is an understanding of the criteria for a profession and, even more importantly, who has the authority to decide when an individual or a group has met that standard.
But first, what and why did the DOE make this change? The One Big Beautiful Bill Act charged the DOE with reducing what it claims is massive overspending on graduate education by limiting the programs that meet the definition of a “professional degree” eligible for higher funding. Of my father’s 4, medicine (including dentistry) and law made the cut with students in those professions able to borrow up to $200,000 in direct unsubsidized student loans while those in other programs would be limited to $100,000.2
As one of the oldest and most respected professions in America, nursing has received the most media attention, yet there are also other important and valued professions that are missing from the DOE list.3 The excluded professions also include: physician assistants, physical therapists, audiologists, architects, accountants, educators, and social workers. The proposed regulatory changes are not yet finalized and Congressional representatives, health care experts, and a myriad of professional associations have rightly objected the reclassification will only worsen the critical shortage of nurses, teachers, and other helping professions the country is already facing.4
There are thousands of federal health care professionals who worked long and hard to achieve their goals whom this Act undervalues. Moreover, the regulatory change leaves many students enrolled in education and training programs under federal practice auspices confused and overwhelmed. Perhaps they can take some hope and inspiration from the recognition that historically and philosophically, no agency or administration can unilaterally define what is a profession.
The literature on professionalism is voluminous, in large part because it has been surprisingly difficult to reach a consensus definition. A proposed definition from scholars captures most of the key aspects of a profession. While it is drawn from the medical literature, it applies to most of the caring professions the DOE disqualified. For pedagogic purposes, the definition is parsed into discrete criteria in the Table.5
Even this simple summary makes it obvious that a government agency alone could not possibly have the competence to determine who meets these complex technical and moral criteria. The members of the profession must assume a primary role in that determination. The complicated history of the professions shows that the locus of these decisions has resided in various combinations of educational institutions, such as nursing schools,6 professional societies (eg, National Association of Social Workers),7 and certifying boards (eg, National Commission on Certification of Physician Assistants).8 States, not the federal government, have long played a key part in defining professions in the US, through their authority to grant licenses to practice.9
In response to criticism, the DOE has stated that “the definition of a ‘professional degree’ is an internal definition used by the Department of Education to distinguish among programs that qualify for higher loan limits, not a value judgment about the importance of programs. It has no bearing on whether a program is professional in nature or not.”2 Given the ancient compact between society and the professions in which the government subsidizes the training of professionals dedicated to public service, it is hard to see how these changes can be dismissed as merely semantic and not a promissory breach.10
I recognize that this abstract editorial is little comfort to beleaguered and demoralized professionals and students. Still, it offers a voice of support for each federal practitioner or trainee who fulfills the epigraph’s description of a professional day after day. The nurse who works the extra shift without complaint or resentment so that veterans receive the care they deserve, the social worker who responds on a weekend night to an active duty family without food so they do not spend another night hungry, and the physician assistant who makes it into the isolated public health clinic despite the terrible weather so there is someone ready to take care for patients in need. The proposed policy shift cannot in any meaningful sense rob them of their identity as individuals committed to a code of caring. However, without an intact social compact, it may well remove their practical ability to remain and enter the helping professions to the detriment of us all.
A professional is someone who can do his best work when he doesn’t feel like it.
Alistair Cooke
When I was a young person with no idea about growing up to be something, my father used to tell me there were 4 learned professions: medicine to heal the body, law to protect the body politic, teaching to nurture the mind, and the clergy to care for the soul.1 That adage, or some version of it, is attributed to a variety of sources, likely because it captures something essential and timeless about the learned professions. I write this as a much older person, and it has been my privilege to have worked in some capacity in all 4 of these venerable vocations.
There are many more recognized professions now than in my father’s time with new ones still emerging as the world becomes more complicated and specialized. In November 2025, however, the growth of the professions was dealt a serious blow when the US Department of Education (DOE) redefined what constitutes a profession for the purpose of federal funding of graduate degrees.2 The internet is understandably abuzz with opinions across the political spectrum. What is missing from many of these discussions is an understanding of the criteria for a profession and, even more importantly, who has the authority to decide when an individual or a group has met that standard.
But first, what and why did the DOE make this change? The One Big Beautiful Bill Act charged the DOE with reducing what it claims is massive overspending on graduate education by limiting the programs that meet the definition of a “professional degree” eligible for higher funding. Of my father’s 4, medicine (including dentistry) and law made the cut with students in those professions able to borrow up to $200,000 in direct unsubsidized student loans while those in other programs would be limited to $100,000.2
As one of the oldest and most respected professions in America, nursing has received the most media attention, yet there are also other important and valued professions that are missing from the DOE list.3 The excluded professions also include: physician assistants, physical therapists, audiologists, architects, accountants, educators, and social workers. The proposed regulatory changes are not yet finalized and Congressional representatives, health care experts, and a myriad of professional associations have rightly objected the reclassification will only worsen the critical shortage of nurses, teachers, and other helping professions the country is already facing.4
There are thousands of federal health care professionals who worked long and hard to achieve their goals whom this Act undervalues. Moreover, the regulatory change leaves many students enrolled in education and training programs under federal practice auspices confused and overwhelmed. Perhaps they can take some hope and inspiration from the recognition that historically and philosophically, no agency or administration can unilaterally define what is a profession.
The literature on professionalism is voluminous, in large part because it has been surprisingly difficult to reach a consensus definition. A proposed definition from scholars captures most of the key aspects of a profession. While it is drawn from the medical literature, it applies to most of the caring professions the DOE disqualified. For pedagogic purposes, the definition is parsed into discrete criteria in the Table.5
Even this simple summary makes it obvious that a government agency alone could not possibly have the competence to determine who meets these complex technical and moral criteria. The members of the profession must assume a primary role in that determination. The complicated history of the professions shows that the locus of these decisions has resided in various combinations of educational institutions, such as nursing schools,6 professional societies (eg, National Association of Social Workers),7 and certifying boards (eg, National Commission on Certification of Physician Assistants).8 States, not the federal government, have long played a key part in defining professions in the US, through their authority to grant licenses to practice.9
In response to criticism, the DOE has stated that “the definition of a ‘professional degree’ is an internal definition used by the Department of Education to distinguish among programs that qualify for higher loan limits, not a value judgment about the importance of programs. It has no bearing on whether a program is professional in nature or not.”2 Given the ancient compact between society and the professions in which the government subsidizes the training of professionals dedicated to public service, it is hard to see how these changes can be dismissed as merely semantic and not a promissory breach.10
I recognize that this abstract editorial is little comfort to beleaguered and demoralized professionals and students. Still, it offers a voice of support for each federal practitioner or trainee who fulfills the epigraph’s description of a professional day after day. The nurse who works the extra shift without complaint or resentment so that veterans receive the care they deserve, the social worker who responds on a weekend night to an active duty family without food so they do not spend another night hungry, and the physician assistant who makes it into the isolated public health clinic despite the terrible weather so there is someone ready to take care for patients in need. The proposed policy shift cannot in any meaningful sense rob them of their identity as individuals committed to a code of caring. However, without an intact social compact, it may well remove their practical ability to remain and enter the helping professions to the detriment of us all.
- Wade JW. Public responsibilities of the learned professions. Louisiana Law Rev. 1960;21:130-148
- US Department of Education. Myth vs. fact: the definition of professional degrees. Press Release. November 24, 2025. Accessed December 22, 2025. https://www.ed.gov/about/news/press-release/myth-vs-fact-definition-of-professional-degrees
- Laws J. Full list of degrees not classed as “professional” by Trump admin. Newsweek. Updated November 26, 2025. Accessed December 22, 2025. https://www.newsweek.com/full-list-degrees-professional-trump-administration-11085695
- New York Academy of Medicine. Response to stripping “professional status” as proposed by the Department of Education. New York Academy of Medicine. November 24, 2025. Accessed December 22, 2025. https://nyam.org/article/response-to-stripping-professional-status-as-proposed-by-the-department-of-education
- Cruess SR, Johnston S, Cruess RL. “Profession”: a working definition for medical educators. Teach Learn Med. 2004;16:74-76. doi:10.1207/s15328015tlm1601_15
- American Association of Colleges of Nursing. Nursing is a professional degree. American Association of Colleges of Nursing. Accessed December 20, 2025. https://www.aacnnursing.org/policy-advocacy/take-action/nursing-is-a-professional-degree
- National Association of Social Workers. Social work is a profession. Social Workers. Accessed December 20, 2025. https://www.socialworkers.org
- National Commission on Certification of Physician Assistants. Accessed December 20, 2025. https://www.nccpa.net/about-nccpa/#who-we-are
- The Federation of State Boards of Physical Therapy. Accessed December 20, 2025. https://www.fsbpt.org/About-Us/Staff-Home
- Cruess SR, Cruess RL. Professionalism and medicine’s contract with social contract with society. Virtual Mentor. 2004;6:185-188. doi:10.1001/virtualmentor.2004.6.4.msoc1-040
- Wade JW. Public responsibilities of the learned professions. Louisiana Law Rev. 1960;21:130-148
- US Department of Education. Myth vs. fact: the definition of professional degrees. Press Release. November 24, 2025. Accessed December 22, 2025. https://www.ed.gov/about/news/press-release/myth-vs-fact-definition-of-professional-degrees
- Laws J. Full list of degrees not classed as “professional” by Trump admin. Newsweek. Updated November 26, 2025. Accessed December 22, 2025. https://www.newsweek.com/full-list-degrees-professional-trump-administration-11085695
- New York Academy of Medicine. Response to stripping “professional status” as proposed by the Department of Education. New York Academy of Medicine. November 24, 2025. Accessed December 22, 2025. https://nyam.org/article/response-to-stripping-professional-status-as-proposed-by-the-department-of-education
- Cruess SR, Johnston S, Cruess RL. “Profession”: a working definition for medical educators. Teach Learn Med. 2004;16:74-76. doi:10.1207/s15328015tlm1601_15
- American Association of Colleges of Nursing. Nursing is a professional degree. American Association of Colleges of Nursing. Accessed December 20, 2025. https://www.aacnnursing.org/policy-advocacy/take-action/nursing-is-a-professional-degree
- National Association of Social Workers. Social work is a profession. Social Workers. Accessed December 20, 2025. https://www.socialworkers.org
- National Commission on Certification of Physician Assistants. Accessed December 20, 2025. https://www.nccpa.net/about-nccpa/#who-we-are
- The Federation of State Boards of Physical Therapy. Accessed December 20, 2025. https://www.fsbpt.org/About-Us/Staff-Home
- Cruess SR, Cruess RL. Professionalism and medicine’s contract with social contract with society. Virtual Mentor. 2004;6:185-188. doi:10.1001/virtualmentor.2004.6.4.msoc1-040
Codes, Contracts, and Commitments: Who Defines What is a Profession?
Codes, Contracts, and Commitments: Who Defines What is a Profession?
Implementation of a Pharmacist-Led Penicillin Allergy Interview at a Veterans Care Facility
Implementation of a Pharmacist-Led Penicillin Allergy Interview at a Veterans Care Facility
Self-reported penicillin allergies are common, with a prevalence of about 10% of patients, according to the Centers for Disease Control and Prevention (CDC).1 However, only about 1% of patients have a true immunoglobulin E (IgE)-mediated allergy. This issue is often further complicated by inaccurate classification of nonallergic adverse effects as an allergy, resulting in incomplete allergy documentation in the electronic health record (EHR). The cross-reactivity rate with cephalosporins (Β-lactam antibiotics) in patients reporting a penicillin allergy is < 1%, which suggests that many patients with reported penicillin allergies can safely receive them.2 Despite this, patients with self-reported penicillin allergies often receive non–Β-lactam antibiotic agents, which may be associated with an increased risk of adverse drug reactions (ADRs), increased health care costs, and inferior clinical outcomes.3
Several strategies are recommended to assess patients with self-reported penicillin allergies. According to the CDC, evaluating a patient who reports a penicillin or other Β-lactam antibiotic allergy involves 3 steps: (1) obtaining a thorough medical history, including previous exposures to penicillin or other Β-lactam antibiotic; (2) performing a skin test using the penicillin major and minor determinants; and (3) among those who have a negative penicillin skin test, performing an observed oral challenge with 250 mg amoxicillin before proceeding directly to treatment with the indicated Β-lactam therapy.4
Most existing clinical guidance for assessing patients with self-reported penicillin allergies stems from site-specific policies and primarily focuses on oral amoxicillin challenges or penicillin skin testing (PST). However, performing these tests may not be feasible at all facilities due to time constraints and lack of allergists. Therefore, alternative strategies are necessary, such as conducting detailed patient interviews. Few studies have evaluated switching to Β-lactam agents following a penicillin allergy interview alone. However, with thorough patient histories and detailed interviews, patients with reported penicillin allergies can safely use Β-lactam antibiotics.5 Implementing this procedure provides a cost-savings opportunity by not having to administer additional antibiotics for testing in addition to improving antibiotic stewardship.
The Memphis Veterans Affairs Medical Center (MVAMC) created the Allergy to Β-Lactam Evaluation (ABLE) process to clarify and remove penicillin allergies. The process involves conducting a thorough chart review and patient interview followed by completion of a note template that provides recommendations about patient allergies and Β-lactam prescribing. Mitchell et al found that the pharmacist-led process to be beneficial for addressing Β-lactam allergy clearance.6 As a result, the ABLE process was implemented at several other US Department of Veterans Affairs (VA) medical centers (VAMCs). Using the ABLE template, the purpose of this study was to evaluate the impact of a pharmacist-led penicillin allergy initiative on penicillin allergy delabeling with an interview process alone.
Methods
Prior to ABLE process implementation, there were no standardized procedures for documenting allergy histories. ABLE was implemented at the Robley Rex VAMC (RRVAMC) in November 2022. During the interview phase, patients were initially identified during admission via TheraDoc as having either a penicillin allergy or ADR. The infectious disease pharmacist or pharmacy resident interviewed patients with documented penicillin allergies or ADRs using a standardized questionnaire (eAppendix 1). Not all identified patients could be interviewed. Patients currently receiving an antibiotic were prioritized for interviews. Patients were excluded if they declined or were unable to be interviewed, although a patient’s caregiver(s) could be interviewed in person or via telephone, if the patient was not available.
Following the interview, pharmacists used guidance from the ABLE process in addition to a detailed EHR review to determine whether the patient was eligible for an allergy update or removal and/or switch to a Β-lactam antibiotic (Figure). If eligible for modification, the interviewing pharmacist made the necessary changes. A templated process note with patient-specific recommendations was entered into the Computerized Patient Record System (CPRS) and the primary care team attending physician was added as an additional signer to be alerted in the system note (eAppendix 2).
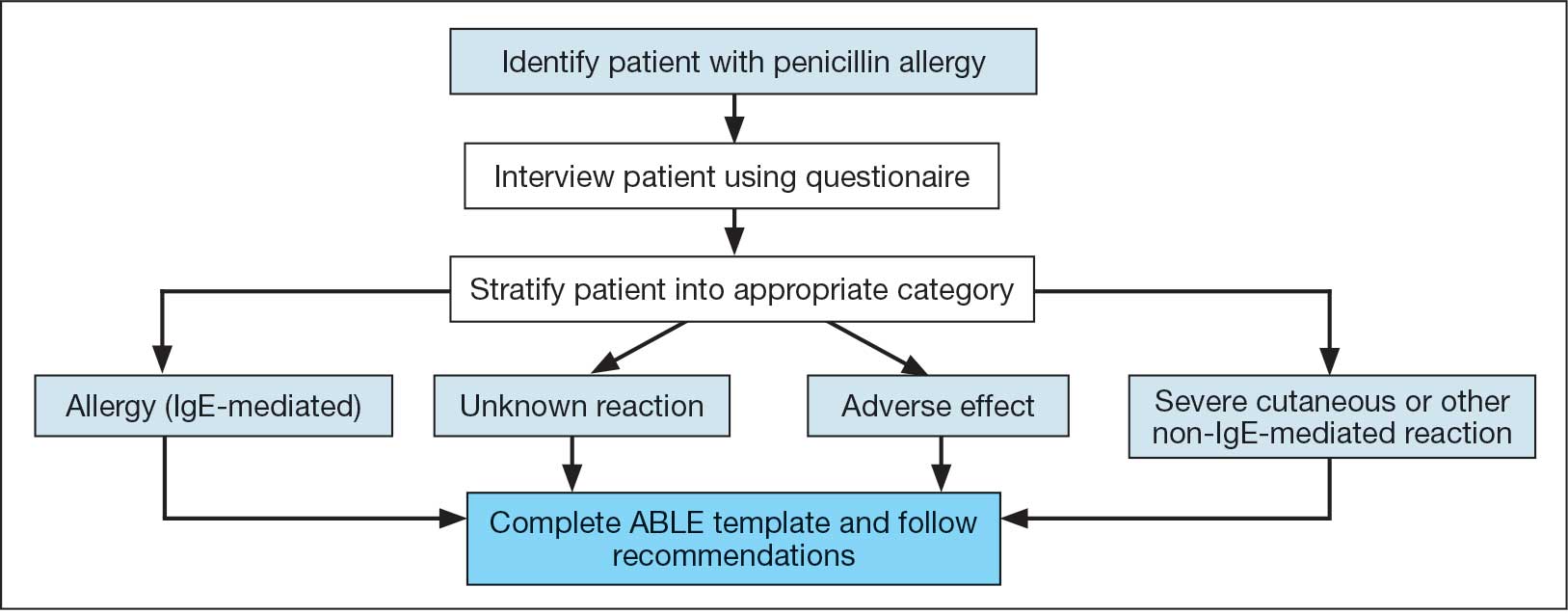
This single-center, retrospective cohort study involved review of CPRS notes and clinical interviews in the interviewed group. Hospitalized patients at the RRVAMC aged ≥ 18 years with a documented penicillin allergy or ADR were included. The historical control group consisted of patients admitted between October 31, 2019, and October 31, 2022, and the intervention group consisted of patients admitted between November 1, 2022, and March 1, 2023. Patients in the historical control group were matched 1:1 to the intervention group for penicillin allergy severity (allergy [IgE-mediated], unknown, adverse effect, severe cutaneous or other non–IgE-mediated reaction) and whether they received a noncarbapenem non–Β-lactam antibiotic.
The primary outcome was the number of patient allergies/ADRs removed or changed on patient profiles regardless of whether their antibiotic regimen was changed. This outcome was further assessed by evaluating the number of patient allergies or ADRs removed or changed on patient profiles with or without a change in antibiotic regimen. Primary outcomes were analyzed using χ2 and/ or Fisher exact tests, as appropriate to determine statistically significant differences between the interviewed group and the historical control.
Results
Seventy patients were included: 35 patients in the interviewed group and 35 patients in the historical control group, respectively. Both groups had a mean age of 72 years and predominantly included White male patients (Table 1). Following the interview, the allergy profile was modified for 6 patients (17%) in the interview group vs 0 patients in the control group (P = .03) (Table 2). The primary outcome was analyzed separately regardless of an antibiotic regimen change. There was not a statistically significant difference between groups when assessing patients for change in therapy (P > .99). All 6 patients with an allergy profile modification had no change in antibiotic regimen.

Discussion
This study suggests the ABLE process may be a valuable tool for adjusting penicillin allergies or ADRs within patient EHRs. In the interview group, allergies were modified in 6 (17%) patients while no patients in the control group had allergy modifications. Of the 6 allergy profile modifications, 4 allergy labels were changed from an allergy to an ADR. These patients were cleared to receive future Β-lactam antibiotics after clinicians recognized the lack of a true IgE-mediated allergic reaction. In addition, 2 of the modified allergy profiles removed the allergy designation. Although this represents a small subset of interviewed patients, it illustrates the clinical effectiveness of an interview process alone to remove penicillin allergy designations.
Previous research has assessed the impact of pharmacist intervention on penicillin allergy clarification. Mitchell et al implemented a pharmacist-driven Β-lactam allergy assessment and penicillin allergy clinic (PAC) at the MVAMC with the goal of evaluating its impact on allergy clearance. In their study, clinical pharmacy specialists evaluated patients with Β-lactam allergies, and those deemed eligible were later seen in the PAC. Among the 246 patients evaluated using the Β-lactam allergy assessment alone and who were not seen in the PAC, 25% had their penicillin allergy removed following a detailed assessment.6
Song et al evaluated the effectiveness and feasibility of a pharmacist-driven penicillin allergy delabeling pilot program without skin testing or oral challenges. Patients with penicillin allergies were interviewed by a pharmacy resident using a standardized checklist. Among the 66 patients interviewed, 12 (18%) met the criteria for delabeling and consented to removal of their allergy.7 The delabeling rates in these 2 studies are similar to the 17% rate of allergy modification in our study, although this study is the only one to compare results to a historical control group.
Harper et al evaluated the impact of a penicillin allergy assessment, including penicillin skin testing and oral amoxicillin challenges, on delabeling penicillin allergies. Pharmacists completed a penicillin allergy assessment and performed penicillin skin testing and/or oral amoxicillin challenges for eligible patients. Of 35 patients, 31 (89%) had their penicillin allergies delabeled in the EHR.8 The rate of penicillin allergy delabeling in Harper et al was likely higher than that seen in our study due to the use of oral challenge and skin testing. Regardless, a detailed penicillin allergy interview alone was effective at RRVAMC, resulting in a significant rate of allergy removal or change. This supports the use of detailed penicillin allergy assessments in settings where penicillin skin testing or oral challenges may not be feasible.
Mann et al demonstrated the effectiveness of penicillin allergy assessments in switching eligible patients to Β-lactam antibiotics. Their single-center, prospective study assessed the impact of a pharmacist-driven detailed penicillin allergy interview initiative. Interviews that evaluated potential changes to allergy profiles were conducted with 175 patients. Of these patients, 135 (77.1%) were on antimicrobial therapy and 42 (31.1%) patients receiving therapy met criteria to switch to a noncarbapenem Β-lactam antibiotic. Thirty-one patients (73.8%) switched with no signs or symptoms of intolerance demonstrating that an interview can be a valuable tool for antibiotic optimization, specifically in patients with penicillin allergy.9 No patients in our study switched antibiotic therapy, likely because only a small number of patients were eligible for transition to a noncarbapenem Β-lactam antibiotic. In the Mann et al study, non–Β-lactam antibiotics, such as fluoroquinolones and carbapenems, accounted for > 75% of the antibiotics used.
Limitations
The sample size of this study was small and its duration was short. There is a risk for selection bias as not all identified patients were able to be interviewed while admitted, but patients on antibiotics were prioritized as they were most likely to directly benefit during their current admission from a modification of their allergy. Most patients in the study were White and male, which may limit the generalizability of the results. Additionally, recommendations regarding antibiotic changes were primarily communicated to the treatment team based on a templated note in CPRS alone. Therefore, implementation of these recommendations largely relied upon nonverbal communication. Direct pharmacist-physician communication could have led to a larger impact on antimicrobial therapy changes. The interviewer’s participation in daily rounds with time allotted to discuss this topic can be considered in the future to improve these processes.
Conclusions
This study found that the ABLE process identified patients for penicillin allergy delabeling. With the high prevalence of inaccurate penicillin allergy documentation, this tool offers VA health care systems a way to empower pharmacists in allergy clarification, leading to improvements in antibiotic stewardship. Although the sample size was small, the ABLE process may provide a framework for VA clinicians. Future research has the potential to demonstrate the practicality and effectiveness this pharmacist-led penicillin allergy interview process can offer clinicians.
- Health care providers. Clinical features of penicillin allergy. Centers for Disease Control and Prevention. August 25, 2025. Accessed February 4, 2026. https://www.cdc.gov /antibiotic-use/hcp/clinical-signs/index.html
- Wrynn AF. Penicillin allergies: A guide for NPs. Nurse Pract. 2022;47:30-36. doi:10.1097/01.NPR.0000855312.11145.78
- Mohsen S, Dickinson JA, Somayaji R. Update on the adverse effects of antimicrobial therapies in community practice. Can Fam Physician. 2020;66:651-659.
- Sexually Transmitted Infections Treatment Guidelines, 2021. Managing persons who have a history of penicillin allergy. Centers for Disease Control and Prevention. September 21, 2022. Accessed February 4, 2026. https:// www.cdc.gov/std/treatment-guidelines/penicillin-allergy .htm
- Holmes AK, Bennett NT, Berry TP. Pharmacy driven assessment of appropriate antibiotic selection in patients with reported beta-lactam allergy. J Am Coll Clin Pharm. 2019;2:509-514. doi:10.1002/jac5.1135
- Mitchell AB, Ness RA, Bennett JG, et al. Implementation and impact of a Β-lactam allergy assessment protocol in a veteran population. Fed Pract. 2021;38:420-425. doi:10.12788/fp.0172
- Song YC, Nelson ZJ, Wankum MA, et al. Effectiveness and feasibility of pharmacist-driven penicillin allergy de-labeling pilot program without skin testing or oral challenges. Pharmacy (Basel). 2021;9:127. doi:10.3390/pharmacy9030127
- Harper HM, Sanchez M. Review of pharmacist driven penicillin allergy assessments and skin testing: a multicenter case-series. Hosp Pharm. 2022;57:469-473. doi:10.1177/00185787211046862
- Mann KL, Wu JY, Shah SS. Implementation of a pharmacist- driven detailed penicillin allergy interview. Ann Pharmacother. 2020;54:364-370. doi:10.1177/1060028019884874
Self-reported penicillin allergies are common, with a prevalence of about 10% of patients, according to the Centers for Disease Control and Prevention (CDC).1 However, only about 1% of patients have a true immunoglobulin E (IgE)-mediated allergy. This issue is often further complicated by inaccurate classification of nonallergic adverse effects as an allergy, resulting in incomplete allergy documentation in the electronic health record (EHR). The cross-reactivity rate with cephalosporins (Β-lactam antibiotics) in patients reporting a penicillin allergy is < 1%, which suggests that many patients with reported penicillin allergies can safely receive them.2 Despite this, patients with self-reported penicillin allergies often receive non–Β-lactam antibiotic agents, which may be associated with an increased risk of adverse drug reactions (ADRs), increased health care costs, and inferior clinical outcomes.3
Several strategies are recommended to assess patients with self-reported penicillin allergies. According to the CDC, evaluating a patient who reports a penicillin or other Β-lactam antibiotic allergy involves 3 steps: (1) obtaining a thorough medical history, including previous exposures to penicillin or other Β-lactam antibiotic; (2) performing a skin test using the penicillin major and minor determinants; and (3) among those who have a negative penicillin skin test, performing an observed oral challenge with 250 mg amoxicillin before proceeding directly to treatment with the indicated Β-lactam therapy.4
Most existing clinical guidance for assessing patients with self-reported penicillin allergies stems from site-specific policies and primarily focuses on oral amoxicillin challenges or penicillin skin testing (PST). However, performing these tests may not be feasible at all facilities due to time constraints and lack of allergists. Therefore, alternative strategies are necessary, such as conducting detailed patient interviews. Few studies have evaluated switching to Β-lactam agents following a penicillin allergy interview alone. However, with thorough patient histories and detailed interviews, patients with reported penicillin allergies can safely use Β-lactam antibiotics.5 Implementing this procedure provides a cost-savings opportunity by not having to administer additional antibiotics for testing in addition to improving antibiotic stewardship.
The Memphis Veterans Affairs Medical Center (MVAMC) created the Allergy to Β-Lactam Evaluation (ABLE) process to clarify and remove penicillin allergies. The process involves conducting a thorough chart review and patient interview followed by completion of a note template that provides recommendations about patient allergies and Β-lactam prescribing. Mitchell et al found that the pharmacist-led process to be beneficial for addressing Β-lactam allergy clearance.6 As a result, the ABLE process was implemented at several other US Department of Veterans Affairs (VA) medical centers (VAMCs). Using the ABLE template, the purpose of this study was to evaluate the impact of a pharmacist-led penicillin allergy initiative on penicillin allergy delabeling with an interview process alone.
Methods
Prior to ABLE process implementation, there were no standardized procedures for documenting allergy histories. ABLE was implemented at the Robley Rex VAMC (RRVAMC) in November 2022. During the interview phase, patients were initially identified during admission via TheraDoc as having either a penicillin allergy or ADR. The infectious disease pharmacist or pharmacy resident interviewed patients with documented penicillin allergies or ADRs using a standardized questionnaire (eAppendix 1). Not all identified patients could be interviewed. Patients currently receiving an antibiotic were prioritized for interviews. Patients were excluded if they declined or were unable to be interviewed, although a patient’s caregiver(s) could be interviewed in person or via telephone, if the patient was not available.
Following the interview, pharmacists used guidance from the ABLE process in addition to a detailed EHR review to determine whether the patient was eligible for an allergy update or removal and/or switch to a Β-lactam antibiotic (Figure). If eligible for modification, the interviewing pharmacist made the necessary changes. A templated process note with patient-specific recommendations was entered into the Computerized Patient Record System (CPRS) and the primary care team attending physician was added as an additional signer to be alerted in the system note (eAppendix 2).
This single-center, retrospective cohort study involved review of CPRS notes and clinical interviews in the interviewed group. Hospitalized patients at the RRVAMC aged ≥ 18 years with a documented penicillin allergy or ADR were included. The historical control group consisted of patients admitted between October 31, 2019, and October 31, 2022, and the intervention group consisted of patients admitted between November 1, 2022, and March 1, 2023. Patients in the historical control group were matched 1:1 to the intervention group for penicillin allergy severity (allergy [IgE-mediated], unknown, adverse effect, severe cutaneous or other non–IgE-mediated reaction) and whether they received a noncarbapenem non–Β-lactam antibiotic.
The primary outcome was the number of patient allergies/ADRs removed or changed on patient profiles regardless of whether their antibiotic regimen was changed. This outcome was further assessed by evaluating the number of patient allergies or ADRs removed or changed on patient profiles with or without a change in antibiotic regimen. Primary outcomes were analyzed using χ2 and/ or Fisher exact tests, as appropriate to determine statistically significant differences between the interviewed group and the historical control.
Results
Seventy patients were included: 35 patients in the interviewed group and 35 patients in the historical control group, respectively. Both groups had a mean age of 72 years and predominantly included White male patients (Table 1). Following the interview, the allergy profile was modified for 6 patients (17%) in the interview group vs 0 patients in the control group (P = .03) (Table 2). The primary outcome was analyzed separately regardless of an antibiotic regimen change. There was not a statistically significant difference between groups when assessing patients for change in therapy (P > .99). All 6 patients with an allergy profile modification had no change in antibiotic regimen.
Discussion
This study suggests the ABLE process may be a valuable tool for adjusting penicillin allergies or ADRs within patient EHRs. In the interview group, allergies were modified in 6 (17%) patients while no patients in the control group had allergy modifications. Of the 6 allergy profile modifications, 4 allergy labels were changed from an allergy to an ADR. These patients were cleared to receive future Β-lactam antibiotics after clinicians recognized the lack of a true IgE-mediated allergic reaction. In addition, 2 of the modified allergy profiles removed the allergy designation. Although this represents a small subset of interviewed patients, it illustrates the clinical effectiveness of an interview process alone to remove penicillin allergy designations.
Previous research has assessed the impact of pharmacist intervention on penicillin allergy clarification. Mitchell et al implemented a pharmacist-driven Β-lactam allergy assessment and penicillin allergy clinic (PAC) at the MVAMC with the goal of evaluating its impact on allergy clearance. In their study, clinical pharmacy specialists evaluated patients with Β-lactam allergies, and those deemed eligible were later seen in the PAC. Among the 246 patients evaluated using the Β-lactam allergy assessment alone and who were not seen in the PAC, 25% had their penicillin allergy removed following a detailed assessment.6
Song et al evaluated the effectiveness and feasibility of a pharmacist-driven penicillin allergy delabeling pilot program without skin testing or oral challenges. Patients with penicillin allergies were interviewed by a pharmacy resident using a standardized checklist. Among the 66 patients interviewed, 12 (18%) met the criteria for delabeling and consented to removal of their allergy.7 The delabeling rates in these 2 studies are similar to the 17% rate of allergy modification in our study, although this study is the only one to compare results to a historical control group.
Harper et al evaluated the impact of a penicillin allergy assessment, including penicillin skin testing and oral amoxicillin challenges, on delabeling penicillin allergies. Pharmacists completed a penicillin allergy assessment and performed penicillin skin testing and/or oral amoxicillin challenges for eligible patients. Of 35 patients, 31 (89%) had their penicillin allergies delabeled in the EHR.8 The rate of penicillin allergy delabeling in Harper et al was likely higher than that seen in our study due to the use of oral challenge and skin testing. Regardless, a detailed penicillin allergy interview alone was effective at RRVAMC, resulting in a significant rate of allergy removal or change. This supports the use of detailed penicillin allergy assessments in settings where penicillin skin testing or oral challenges may not be feasible.
Mann et al demonstrated the effectiveness of penicillin allergy assessments in switching eligible patients to Β-lactam antibiotics. Their single-center, prospective study assessed the impact of a pharmacist-driven detailed penicillin allergy interview initiative. Interviews that evaluated potential changes to allergy profiles were conducted with 175 patients. Of these patients, 135 (77.1%) were on antimicrobial therapy and 42 (31.1%) patients receiving therapy met criteria to switch to a noncarbapenem Β-lactam antibiotic. Thirty-one patients (73.8%) switched with no signs or symptoms of intolerance demonstrating that an interview can be a valuable tool for antibiotic optimization, specifically in patients with penicillin allergy.9 No patients in our study switched antibiotic therapy, likely because only a small number of patients were eligible for transition to a noncarbapenem Β-lactam antibiotic. In the Mann et al study, non–Β-lactam antibiotics, such as fluoroquinolones and carbapenems, accounted for > 75% of the antibiotics used.
Limitations
The sample size of this study was small and its duration was short. There is a risk for selection bias as not all identified patients were able to be interviewed while admitted, but patients on antibiotics were prioritized as they were most likely to directly benefit during their current admission from a modification of their allergy. Most patients in the study were White and male, which may limit the generalizability of the results. Additionally, recommendations regarding antibiotic changes were primarily communicated to the treatment team based on a templated note in CPRS alone. Therefore, implementation of these recommendations largely relied upon nonverbal communication. Direct pharmacist-physician communication could have led to a larger impact on antimicrobial therapy changes. The interviewer’s participation in daily rounds with time allotted to discuss this topic can be considered in the future to improve these processes.
Conclusions
This study found that the ABLE process identified patients for penicillin allergy delabeling. With the high prevalence of inaccurate penicillin allergy documentation, this tool offers VA health care systems a way to empower pharmacists in allergy clarification, leading to improvements in antibiotic stewardship. Although the sample size was small, the ABLE process may provide a framework for VA clinicians. Future research has the potential to demonstrate the practicality and effectiveness this pharmacist-led penicillin allergy interview process can offer clinicians.
Self-reported penicillin allergies are common, with a prevalence of about 10% of patients, according to the Centers for Disease Control and Prevention (CDC).1 However, only about 1% of patients have a true immunoglobulin E (IgE)-mediated allergy. This issue is often further complicated by inaccurate classification of nonallergic adverse effects as an allergy, resulting in incomplete allergy documentation in the electronic health record (EHR). The cross-reactivity rate with cephalosporins (Β-lactam antibiotics) in patients reporting a penicillin allergy is < 1%, which suggests that many patients with reported penicillin allergies can safely receive them.2 Despite this, patients with self-reported penicillin allergies often receive non–Β-lactam antibiotic agents, which may be associated with an increased risk of adverse drug reactions (ADRs), increased health care costs, and inferior clinical outcomes.3
Several strategies are recommended to assess patients with self-reported penicillin allergies. According to the CDC, evaluating a patient who reports a penicillin or other Β-lactam antibiotic allergy involves 3 steps: (1) obtaining a thorough medical history, including previous exposures to penicillin or other Β-lactam antibiotic; (2) performing a skin test using the penicillin major and minor determinants; and (3) among those who have a negative penicillin skin test, performing an observed oral challenge with 250 mg amoxicillin before proceeding directly to treatment with the indicated Β-lactam therapy.4
Most existing clinical guidance for assessing patients with self-reported penicillin allergies stems from site-specific policies and primarily focuses on oral amoxicillin challenges or penicillin skin testing (PST). However, performing these tests may not be feasible at all facilities due to time constraints and lack of allergists. Therefore, alternative strategies are necessary, such as conducting detailed patient interviews. Few studies have evaluated switching to Β-lactam agents following a penicillin allergy interview alone. However, with thorough patient histories and detailed interviews, patients with reported penicillin allergies can safely use Β-lactam antibiotics.5 Implementing this procedure provides a cost-savings opportunity by not having to administer additional antibiotics for testing in addition to improving antibiotic stewardship.
The Memphis Veterans Affairs Medical Center (MVAMC) created the Allergy to Β-Lactam Evaluation (ABLE) process to clarify and remove penicillin allergies. The process involves conducting a thorough chart review and patient interview followed by completion of a note template that provides recommendations about patient allergies and Β-lactam prescribing. Mitchell et al found that the pharmacist-led process to be beneficial for addressing Β-lactam allergy clearance.6 As a result, the ABLE process was implemented at several other US Department of Veterans Affairs (VA) medical centers (VAMCs). Using the ABLE template, the purpose of this study was to evaluate the impact of a pharmacist-led penicillin allergy initiative on penicillin allergy delabeling with an interview process alone.
Methods
Prior to ABLE process implementation, there were no standardized procedures for documenting allergy histories. ABLE was implemented at the Robley Rex VAMC (RRVAMC) in November 2022. During the interview phase, patients were initially identified during admission via TheraDoc as having either a penicillin allergy or ADR. The infectious disease pharmacist or pharmacy resident interviewed patients with documented penicillin allergies or ADRs using a standardized questionnaire (eAppendix 1). Not all identified patients could be interviewed. Patients currently receiving an antibiotic were prioritized for interviews. Patients were excluded if they declined or were unable to be interviewed, although a patient’s caregiver(s) could be interviewed in person or via telephone, if the patient was not available.
Following the interview, pharmacists used guidance from the ABLE process in addition to a detailed EHR review to determine whether the patient was eligible for an allergy update or removal and/or switch to a Β-lactam antibiotic (Figure). If eligible for modification, the interviewing pharmacist made the necessary changes. A templated process note with patient-specific recommendations was entered into the Computerized Patient Record System (CPRS) and the primary care team attending physician was added as an additional signer to be alerted in the system note (eAppendix 2).
This single-center, retrospective cohort study involved review of CPRS notes and clinical interviews in the interviewed group. Hospitalized patients at the RRVAMC aged ≥ 18 years with a documented penicillin allergy or ADR were included. The historical control group consisted of patients admitted between October 31, 2019, and October 31, 2022, and the intervention group consisted of patients admitted between November 1, 2022, and March 1, 2023. Patients in the historical control group were matched 1:1 to the intervention group for penicillin allergy severity (allergy [IgE-mediated], unknown, adverse effect, severe cutaneous or other non–IgE-mediated reaction) and whether they received a noncarbapenem non–Β-lactam antibiotic.
The primary outcome was the number of patient allergies/ADRs removed or changed on patient profiles regardless of whether their antibiotic regimen was changed. This outcome was further assessed by evaluating the number of patient allergies or ADRs removed or changed on patient profiles with or without a change in antibiotic regimen. Primary outcomes were analyzed using χ2 and/ or Fisher exact tests, as appropriate to determine statistically significant differences between the interviewed group and the historical control.
Results
Seventy patients were included: 35 patients in the interviewed group and 35 patients in the historical control group, respectively. Both groups had a mean age of 72 years and predominantly included White male patients (Table 1). Following the interview, the allergy profile was modified for 6 patients (17%) in the interview group vs 0 patients in the control group (P = .03) (Table 2). The primary outcome was analyzed separately regardless of an antibiotic regimen change. There was not a statistically significant difference between groups when assessing patients for change in therapy (P > .99). All 6 patients with an allergy profile modification had no change in antibiotic regimen.
Discussion
This study suggests the ABLE process may be a valuable tool for adjusting penicillin allergies or ADRs within patient EHRs. In the interview group, allergies were modified in 6 (17%) patients while no patients in the control group had allergy modifications. Of the 6 allergy profile modifications, 4 allergy labels were changed from an allergy to an ADR. These patients were cleared to receive future Β-lactam antibiotics after clinicians recognized the lack of a true IgE-mediated allergic reaction. In addition, 2 of the modified allergy profiles removed the allergy designation. Although this represents a small subset of interviewed patients, it illustrates the clinical effectiveness of an interview process alone to remove penicillin allergy designations.
Previous research has assessed the impact of pharmacist intervention on penicillin allergy clarification. Mitchell et al implemented a pharmacist-driven Β-lactam allergy assessment and penicillin allergy clinic (PAC) at the MVAMC with the goal of evaluating its impact on allergy clearance. In their study, clinical pharmacy specialists evaluated patients with Β-lactam allergies, and those deemed eligible were later seen in the PAC. Among the 246 patients evaluated using the Β-lactam allergy assessment alone and who were not seen in the PAC, 25% had their penicillin allergy removed following a detailed assessment.6
Song et al evaluated the effectiveness and feasibility of a pharmacist-driven penicillin allergy delabeling pilot program without skin testing or oral challenges. Patients with penicillin allergies were interviewed by a pharmacy resident using a standardized checklist. Among the 66 patients interviewed, 12 (18%) met the criteria for delabeling and consented to removal of their allergy.7 The delabeling rates in these 2 studies are similar to the 17% rate of allergy modification in our study, although this study is the only one to compare results to a historical control group.
Harper et al evaluated the impact of a penicillin allergy assessment, including penicillin skin testing and oral amoxicillin challenges, on delabeling penicillin allergies. Pharmacists completed a penicillin allergy assessment and performed penicillin skin testing and/or oral amoxicillin challenges for eligible patients. Of 35 patients, 31 (89%) had their penicillin allergies delabeled in the EHR.8 The rate of penicillin allergy delabeling in Harper et al was likely higher than that seen in our study due to the use of oral challenge and skin testing. Regardless, a detailed penicillin allergy interview alone was effective at RRVAMC, resulting in a significant rate of allergy removal or change. This supports the use of detailed penicillin allergy assessments in settings where penicillin skin testing or oral challenges may not be feasible.
Mann et al demonstrated the effectiveness of penicillin allergy assessments in switching eligible patients to Β-lactam antibiotics. Their single-center, prospective study assessed the impact of a pharmacist-driven detailed penicillin allergy interview initiative. Interviews that evaluated potential changes to allergy profiles were conducted with 175 patients. Of these patients, 135 (77.1%) were on antimicrobial therapy and 42 (31.1%) patients receiving therapy met criteria to switch to a noncarbapenem Β-lactam antibiotic. Thirty-one patients (73.8%) switched with no signs or symptoms of intolerance demonstrating that an interview can be a valuable tool for antibiotic optimization, specifically in patients with penicillin allergy.9 No patients in our study switched antibiotic therapy, likely because only a small number of patients were eligible for transition to a noncarbapenem Β-lactam antibiotic. In the Mann et al study, non–Β-lactam antibiotics, such as fluoroquinolones and carbapenems, accounted for > 75% of the antibiotics used.
Limitations
The sample size of this study was small and its duration was short. There is a risk for selection bias as not all identified patients were able to be interviewed while admitted, but patients on antibiotics were prioritized as they were most likely to directly benefit during their current admission from a modification of their allergy. Most patients in the study were White and male, which may limit the generalizability of the results. Additionally, recommendations regarding antibiotic changes were primarily communicated to the treatment team based on a templated note in CPRS alone. Therefore, implementation of these recommendations largely relied upon nonverbal communication. Direct pharmacist-physician communication could have led to a larger impact on antimicrobial therapy changes. The interviewer’s participation in daily rounds with time allotted to discuss this topic can be considered in the future to improve these processes.
Conclusions
This study found that the ABLE process identified patients for penicillin allergy delabeling. With the high prevalence of inaccurate penicillin allergy documentation, this tool offers VA health care systems a way to empower pharmacists in allergy clarification, leading to improvements in antibiotic stewardship. Although the sample size was small, the ABLE process may provide a framework for VA clinicians. Future research has the potential to demonstrate the practicality and effectiveness this pharmacist-led penicillin allergy interview process can offer clinicians.
- Health care providers. Clinical features of penicillin allergy. Centers for Disease Control and Prevention. August 25, 2025. Accessed February 4, 2026. https://www.cdc.gov /antibiotic-use/hcp/clinical-signs/index.html
- Wrynn AF. Penicillin allergies: A guide for NPs. Nurse Pract. 2022;47:30-36. doi:10.1097/01.NPR.0000855312.11145.78
- Mohsen S, Dickinson JA, Somayaji R. Update on the adverse effects of antimicrobial therapies in community practice. Can Fam Physician. 2020;66:651-659.
- Sexually Transmitted Infections Treatment Guidelines, 2021. Managing persons who have a history of penicillin allergy. Centers for Disease Control and Prevention. September 21, 2022. Accessed February 4, 2026. https:// www.cdc.gov/std/treatment-guidelines/penicillin-allergy .htm
- Holmes AK, Bennett NT, Berry TP. Pharmacy driven assessment of appropriate antibiotic selection in patients with reported beta-lactam allergy. J Am Coll Clin Pharm. 2019;2:509-514. doi:10.1002/jac5.1135
- Mitchell AB, Ness RA, Bennett JG, et al. Implementation and impact of a Β-lactam allergy assessment protocol in a veteran population. Fed Pract. 2021;38:420-425. doi:10.12788/fp.0172
- Song YC, Nelson ZJ, Wankum MA, et al. Effectiveness and feasibility of pharmacist-driven penicillin allergy de-labeling pilot program without skin testing or oral challenges. Pharmacy (Basel). 2021;9:127. doi:10.3390/pharmacy9030127
- Harper HM, Sanchez M. Review of pharmacist driven penicillin allergy assessments and skin testing: a multicenter case-series. Hosp Pharm. 2022;57:469-473. doi:10.1177/00185787211046862
- Mann KL, Wu JY, Shah SS. Implementation of a pharmacist- driven detailed penicillin allergy interview. Ann Pharmacother. 2020;54:364-370. doi:10.1177/1060028019884874
- Health care providers. Clinical features of penicillin allergy. Centers for Disease Control and Prevention. August 25, 2025. Accessed February 4, 2026. https://www.cdc.gov /antibiotic-use/hcp/clinical-signs/index.html
- Wrynn AF. Penicillin allergies: A guide for NPs. Nurse Pract. 2022;47:30-36. doi:10.1097/01.NPR.0000855312.11145.78
- Mohsen S, Dickinson JA, Somayaji R. Update on the adverse effects of antimicrobial therapies in community practice. Can Fam Physician. 2020;66:651-659.
- Sexually Transmitted Infections Treatment Guidelines, 2021. Managing persons who have a history of penicillin allergy. Centers for Disease Control and Prevention. September 21, 2022. Accessed February 4, 2026. https:// www.cdc.gov/std/treatment-guidelines/penicillin-allergy .htm
- Holmes AK, Bennett NT, Berry TP. Pharmacy driven assessment of appropriate antibiotic selection in patients with reported beta-lactam allergy. J Am Coll Clin Pharm. 2019;2:509-514. doi:10.1002/jac5.1135
- Mitchell AB, Ness RA, Bennett JG, et al. Implementation and impact of a Β-lactam allergy assessment protocol in a veteran population. Fed Pract. 2021;38:420-425. doi:10.12788/fp.0172
- Song YC, Nelson ZJ, Wankum MA, et al. Effectiveness and feasibility of pharmacist-driven penicillin allergy de-labeling pilot program without skin testing or oral challenges. Pharmacy (Basel). 2021;9:127. doi:10.3390/pharmacy9030127
- Harper HM, Sanchez M. Review of pharmacist driven penicillin allergy assessments and skin testing: a multicenter case-series. Hosp Pharm. 2022;57:469-473. doi:10.1177/00185787211046862
- Mann KL, Wu JY, Shah SS. Implementation of a pharmacist- driven detailed penicillin allergy interview. Ann Pharmacother. 2020;54:364-370. doi:10.1177/1060028019884874
Implementation of a Pharmacist-Led Penicillin Allergy Interview at a Veterans Care Facility
Implementation of a Pharmacist-Led Penicillin Allergy Interview at a Veterans Care Facility
Outcomes From the Use of Cefazolin for Surgical Prophylaxis in Patients Allergic to Penicillin
Outcomes From the Use of Cefazolin for Surgical Prophylaxis in Patients Allergic to Penicillin
Given its safety profile and bactericidal activity against the predominant organisms causing surgical site infections (SSIs), cefazolin remains the most popular choice for surgical prophylaxis.1 Cefazolin offers protection against the pathogens most likely to contaminate the surgical site while minimizing inappropriate methicillin- resistant Staphylococcus aureus coverage that occurs with alternatives such as vancomycin and clindamycin. Documented allergies to Β-lactam antibiotics have historically forced clinicians to avoid the use of cephalosporins due to the potential risk of cross-reactivity. True type 1 (immunoglobin E [IgE]-mediated) cross-allergic reactions between penicillin and cephalosporins are rare, and previously reported data indicate cross-reactivity as a result of antibody recognition is more closely related to the side-chain identity rather than the Β-lactam ring.2,3
About 10% of US patients report having a penicillin allergy; however, < 1% of the population has a true IgE-mediated allergic reaction.4 Previous research that has challenged penicillin allergies with cefazolin for surgical prophylaxis has reported minimal rates of allergic reactions.2-5
In previous trials, patients with a history of delayed skin reactions, such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS), were excluded. Additionally, patients with an allergy to cefazolin including those with urticaria, angioedema, bronchospasm, or anaphylaxis, were excluded from perioperative retrial of cefazolin. Grant et al found that cefazolin can be safely given to patients with IgE-mediated reactions to penicillin and other cephalosporins due to a structurally different side chain.3
In January 2023, the Veteran Health Indiana (VHI) pharmacy team in conjunction with surgery, infectious disease, and anesthesiology, implemented a screening tool as an amendment to perioperative antibiotic guidance to help determine which patients with a documented penicillin allergy could be candidates for perioperative cefazolin. The implemented screening tool (Allergy Clarification for Cefazolin Evidence-Based Prescribing Tool) has been described by Lam et al, who reported that an increased proportion of patients with documented penicillin allergy received cefazolin without more adverse drug reactions (ADRs).5 Patients with a Β-lactam allergy were eligible to receive cefazolin unless the ADR was SJS, TEN, or DRESS, or the offending agent was cefazolin and the patient experienced urticaria, angioedema, bronchospasm, or anaphylaxis. If the reaction was not from cefazolin or was unknown, patients were eligible to receive cefazolin (Figure).
To date, minimal data exist to evaluate the incidence of ADRs when cefazolin is given perioperatively to patients with a previously documented penicillin allergy. The purpose of this study was to evaluate the incidence of allergic ADRs in patients who had a documented penicillin allergy and received periprocedural antibiotics.
Methods
This single-center, retrospective chart review used the US Department of Veterans Affairs (VA) Computerized Patient Record System (CPRS) to identify patients with a documented penicillin allergy who underwent an operation and received periprocedural antibiotics between February 1, 2023, and January 31, 2024. This study was reviewed and approved by the Indiana University Health Institutional Review Board and the VHI Research and Development Committee.
Patients were enrolled if they were aged ≥ 18 years, had a documented penicillin allergy, underwent a surgical intervention, and received perioperative antibiotics during the study period. Patients were excluded if they had a documented penicillin allergy resulting in severe delayed skin reactions (ie, SJS, TEN, or DRESS). These criteria produced 197 surgical procedures. Data were collected for each surgical procedure, so patients could be included more than once. Patient history of allergic reaction to penicillin was obtained through CPRS.
The primary endpoint was the percentage of allergic ADRs in patients with penicillin allergies receiving cefazolin perioperatively. Secondary outcomes included the appropriateness of the antibiotic regimen in congruence with American System of Health Pharmacists (ASHP) recommendations, incidence of SSIs within 30 days of the procedure, incidence of ADRs in those with a history of anaphylaxis vs nonanaphylaxis allergy, incidence of allergic reaction requiring pharmacologic and nonpharmacologic interventions, and incidence of acute kidney injury (AKI). AKI was defined as an increase in serum creatinine by ≥ 0.3 mg/dL within 48 hours or an increase in serum creatinine to ≥ 1.5 times baseline.
Demographic data included sex, age, race, preoperative serum creatinine, and postoperative serum creatinine. Anaphylaxis was defined as an acute onset of illness (within minutes to several hours) with involvement of skin, mucosal tissue, or both involving either respiratory compromise or reduced blood pressures. Allergic reactions were defined as facial, tongue, throat, airway, lip, mouth, periorbital, or eye swelling, urticaria, angioedema, dyspnea, anaphylaxis, or a positive penicillin skin test. Additionally, data collected included the description and severity of postprophylactic antibiotic reaction, antibiotic choice, interventions required for the allergic reaction, SSI occurrence, date of SSI, operating specialty, and postoperative change in renal function.
Descriptive statistics, including mean, SD, and percentages were reported for baseline characteristics of the study population. Percentages were used to demonstrate the differences in primary and secondary outcomes for each study group. Fisher exact tests were used for incidence of ADRs in patients with penicillin allergy who received cefazolin and reported incidence of SSIs.
Results
A total of 197 surgical procedures in patients with a documented penicillin allergy were included; 127 procedures used cefazolin perioperatively, 3 procedures used cefazolin plus gentamicin, and 67 procedures used other antibiotics. Most patients were White (n = 160; 81.2%), male (n = 158; 80.2%), and had a mean age of 64.9 years. Urology was the most common surgical specialty (n = 59; 29.9%) (Table 1). Of the 16 patients with documented penicillin anaphylaxis reaction, 8 received cefazolin and 8 received a different antibiotic. A total of 181 patients reported a nonanaphylaxis allergy. One hundred fifty-one patients (68.6%) reported a reaction history of hives, rash, or swelling (Table 2). Patients could report ≥ 1 reaction. The most prevalent antibiotics used were cefazolin, which was used by 130 patients (61.3%), and clindamycin which was used by 33 patients (15.6%) (Table 3). Patients could receive ≥ 1 antibiotic.
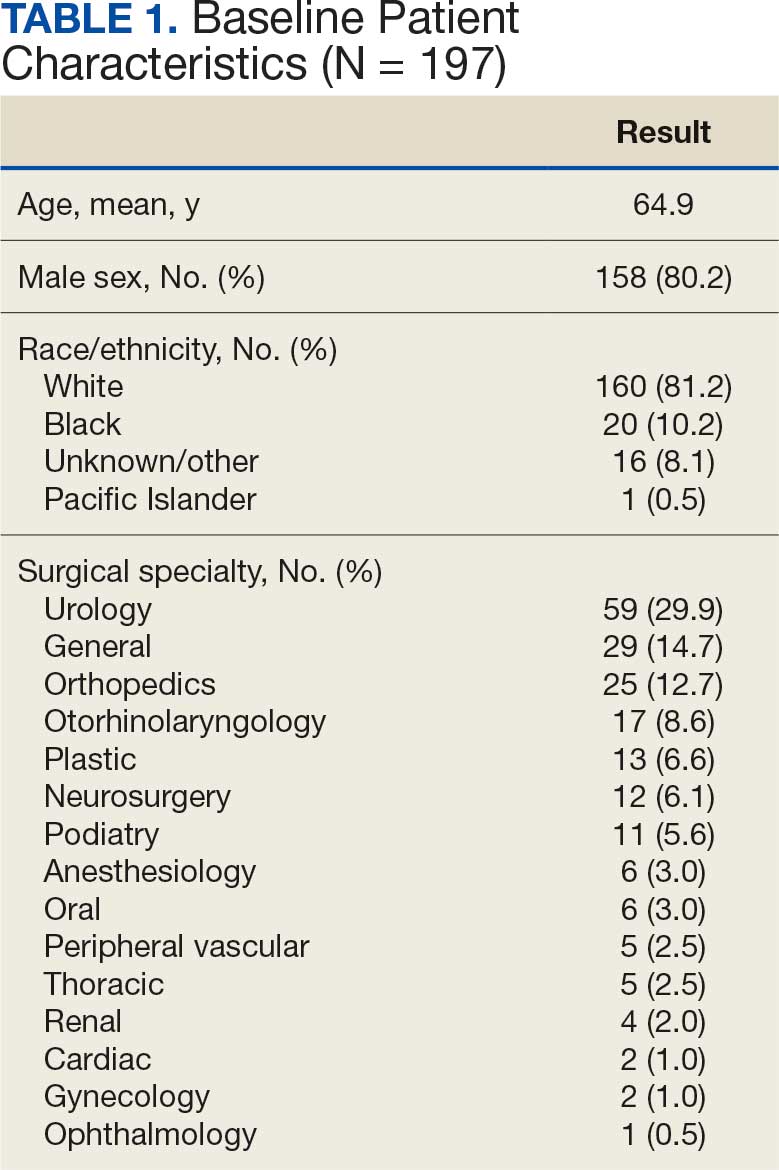
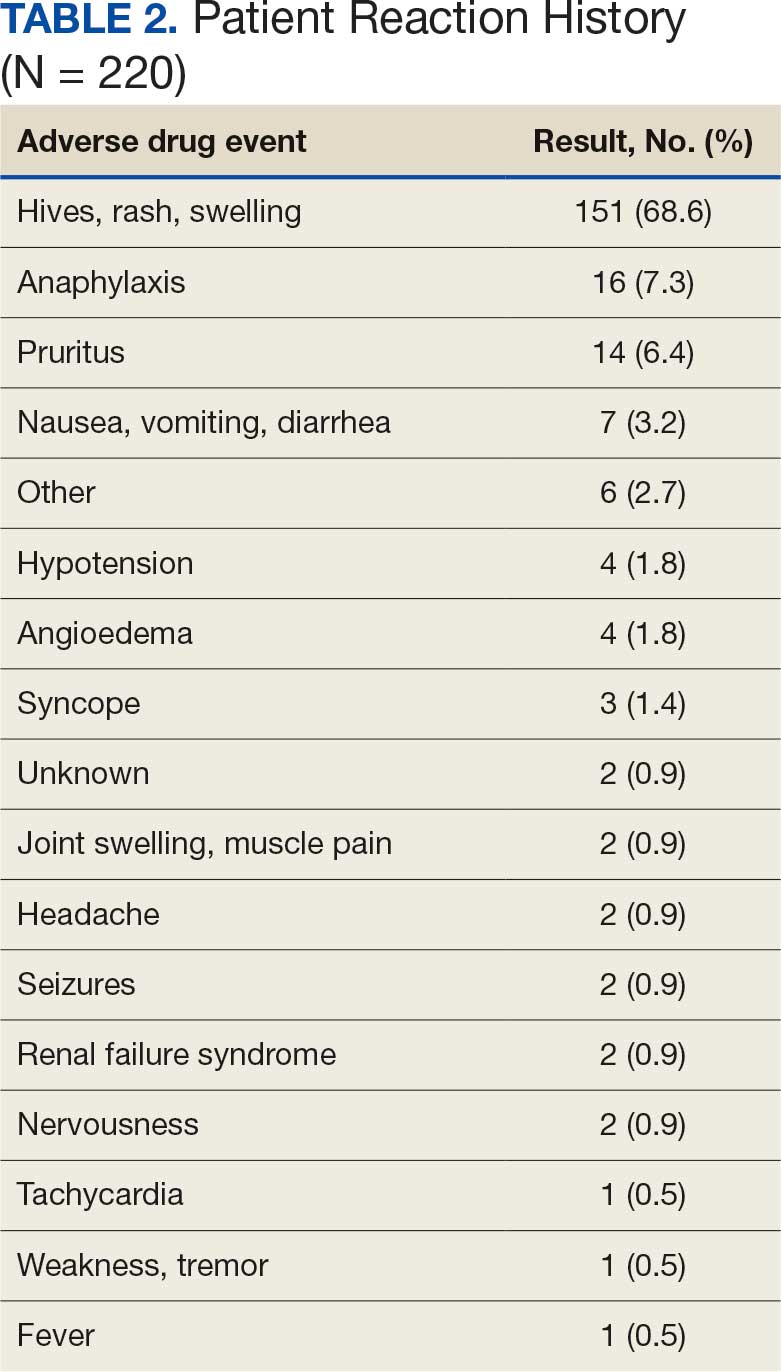
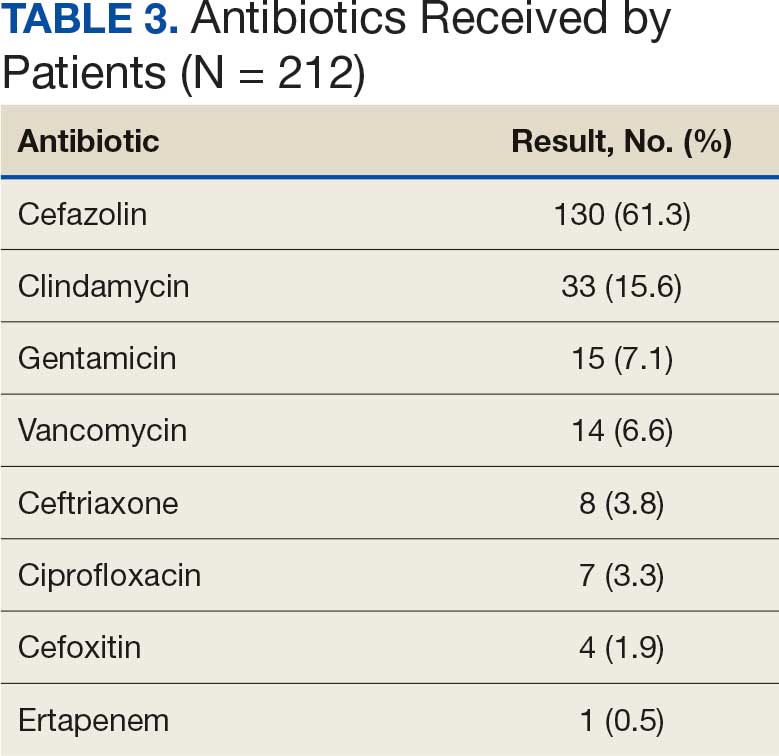
For the primary outcome, the incidence of allergic reactions in patients allergic to penicillin, there was no incidence of allergic reactions in either the cefazolin or other group. Given the absence of reactions, no interventions were required.
There were no ADRs in those with history of anaphylaxis or nonanaphylaxis allergy. In the cefazolin group, 126 of 127 surgical procedure regimens (99.2%) were congruent with ASHP recommendations, all 3 surgical procedures regimens in the cefazolin plus gentamicin group were congruent with ASHP recommendations, and 58 of 67 surgical procedure regimens (86.6%) in the other antibiotic group were congruent with ASHP recommendations. None of the 127 patients in the cefazolin group or of the 3 patients in the cefazolin plus gentamicin group reported an SSI, and 3 of 67 patients (4.5%) had an SSI in the other antibiotic group. One procedure that resulted in SSI was not congruent with ASHP recommendations. Twenty-four patients had 2 serum creatinine levels drawn within 48 hours of surgery. One of 12 patients (8.3%) and 0 of 12 patients had an AKI in the cefazolin and other antibiotic group, respectively (Table 4).
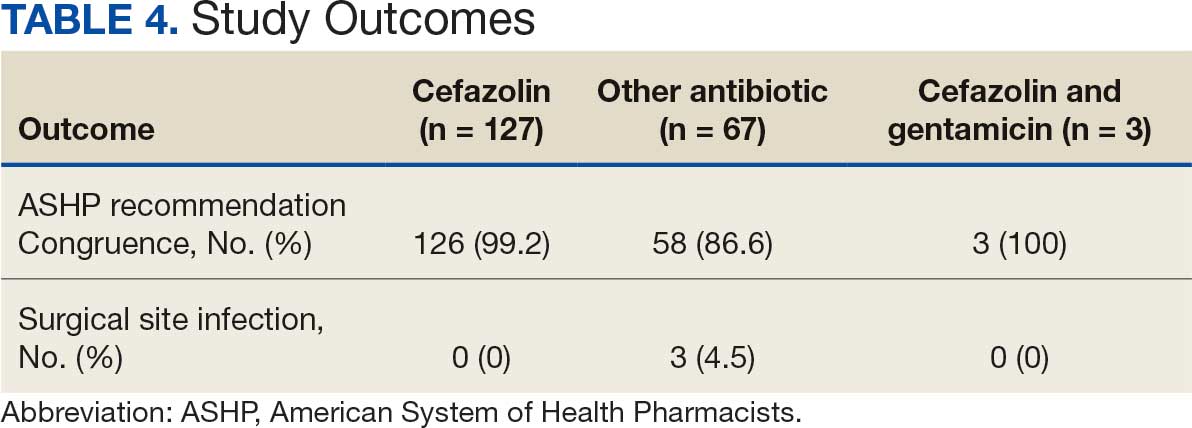
Discussion
Implementation of a screening tool at VHI allowed patients with documented penicillin allergy, including anaphylaxis, to receive cefazolin perioperatively. Broad spectrum antibiotics such as vancomycin, clindamycin, and fluoroquinolones are frequently used in patients allergic to penicillin, which can increase health care costs, risk of toxicity, and antimicrobial resistance.4 There was no incidence of allergic reactions noted in patients allergic to penicillin who received cefazolin. When comparing the incidence of observed allergic reactions to received perioperative antibiotics in the cefazolin group to previously published literature, no difference in allergy rates (P = .09) was found.3 Most antibiotics administered were congruent with ASHP guideline recommendations, and most patients eligible for cefazolin received it perioperatively.
Similar to this study, Goodman et al concluded that cefazolin appears to be a safe regimen in patients with documented penicillin anaphylactic reaction for surgical prophylaxis with only 1 (0.2%) potential allergic reaction.6 Patients who received cefazolin perioperatively had a statistically significant decrease in SSI rates. There were no clinically or statistically significant differences found between the proportion of allergic reactions or ADRs when compared to alternative antibiotics. Lessard et al concluded that a pharmacist-led interdisciplinary collaborative practice agreement increased cefazolin use in patients allergic to penicillin, including those with urticaria and anaphylaxis, with no reported ADRs.7 This study further demonstrated the safety of cefazolin use in patients with anaphylaxis to penicillin.
Limitations
This study’s single-center, retrospective design, patient population, and small sample size limit the generalizability of its results. The data collected are dependent on documentation in the chart. No ADRs were reported from the antibiotics patients received perioperatively. When considering safety data, information such as serum creatinine were available only in CPRS and some patients did not receive a postprocedure serum creatinine level. Additionally, this study did not investigate whether there was an increase in preferred preoperative antimicrobial prophylaxis after implementation of this protocol.
Conclusions
The results of this study support the use of cefazolin perioperatively in patients allergic to penicillin, including those with a history of anaphylaxis. Additional research should be conducted to validate data given the low incidence of ADRs. The primary outcome did not reach statistical significance, but the results may be clinically significant from a stewardship and safety perspective. VHI continues to use the screening tool described in this article.
- Bratzler DW, Dellinger EP, Olsen KM, et al. Clinical practice guidelines for antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 2013;70:195-283. doi:10.2146/ajhp120568
- Romano A, Valluzzi RL, Caruso C, et al. Tolerability of cefazolin and ceftibuten in patients with IgE-mediated aminopenicillin allergy. J Allergy Clin Immunol Pract. 2020;8:1989-1993.e2. doi:10.1016/j.jaip.2020.02.025
- Grant JM, Song WHC, Shajari S, et al. Safety of administering cefazolin versus other antibiotics in penicillin- allergic patients for surgical prophylaxis at a major Canadian teaching hospital. Surgery. 2021;170:783-789. doi:10.1016/j.surg.2021.03.022
- Centers for Disease Control and Prevention. Clinical Features of Penicillin Allergy. August 25, 2025. Accessed January 6, 2026. https://www.cdc.gov/antibiotic-use/hcp/clinical-signs/index.html
- Lam PW, Tarighi P, Elligsen M, et al. Impact of the allergy clarification for cefazolin evidence-based prescribing tool on receipt of preferred perioperative prophylaxis: an interrupted time series study. Clin Infect Dis. 2020;71:2955- 2957. doi:10.1093/cid/ciaa516
- Goodman EJ, Morgan MJ, Johnson Pa, et al. Cephalosporins can be given to penicillin-allergic patients who do not exhibit an anaphylactic response. J Clin Anesth. 2001;13:561-564. doi:10.1016/s0952-8180(01)00329-4
- Lessard S, Huiras C, Dababneh A, et al. Pharmacist adjustment of preoperative antibiotic orders to the preferred preoperative antibiotic cefazolin for patients with penicillin allergy labeling. Am J Health Syst Pharm. 2023;80:532- 536. doi:10.1093/ajhp/zxac385
Given its safety profile and bactericidal activity against the predominant organisms causing surgical site infections (SSIs), cefazolin remains the most popular choice for surgical prophylaxis.1 Cefazolin offers protection against the pathogens most likely to contaminate the surgical site while minimizing inappropriate methicillin- resistant Staphylococcus aureus coverage that occurs with alternatives such as vancomycin and clindamycin. Documented allergies to Β-lactam antibiotics have historically forced clinicians to avoid the use of cephalosporins due to the potential risk of cross-reactivity. True type 1 (immunoglobin E [IgE]-mediated) cross-allergic reactions between penicillin and cephalosporins are rare, and previously reported data indicate cross-reactivity as a result of antibody recognition is more closely related to the side-chain identity rather than the Β-lactam ring.2,3
About 10% of US patients report having a penicillin allergy; however, < 1% of the population has a true IgE-mediated allergic reaction.4 Previous research that has challenged penicillin allergies with cefazolin for surgical prophylaxis has reported minimal rates of allergic reactions.2-5
In previous trials, patients with a history of delayed skin reactions, such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS), were excluded. Additionally, patients with an allergy to cefazolin including those with urticaria, angioedema, bronchospasm, or anaphylaxis, were excluded from perioperative retrial of cefazolin. Grant et al found that cefazolin can be safely given to patients with IgE-mediated reactions to penicillin and other cephalosporins due to a structurally different side chain.3
In January 2023, the Veteran Health Indiana (VHI) pharmacy team in conjunction with surgery, infectious disease, and anesthesiology, implemented a screening tool as an amendment to perioperative antibiotic guidance to help determine which patients with a documented penicillin allergy could be candidates for perioperative cefazolin. The implemented screening tool (Allergy Clarification for Cefazolin Evidence-Based Prescribing Tool) has been described by Lam et al, who reported that an increased proportion of patients with documented penicillin allergy received cefazolin without more adverse drug reactions (ADRs).5 Patients with a Β-lactam allergy were eligible to receive cefazolin unless the ADR was SJS, TEN, or DRESS, or the offending agent was cefazolin and the patient experienced urticaria, angioedema, bronchospasm, or anaphylaxis. If the reaction was not from cefazolin or was unknown, patients were eligible to receive cefazolin (Figure).
To date, minimal data exist to evaluate the incidence of ADRs when cefazolin is given perioperatively to patients with a previously documented penicillin allergy. The purpose of this study was to evaluate the incidence of allergic ADRs in patients who had a documented penicillin allergy and received periprocedural antibiotics.
Methods
This single-center, retrospective chart review used the US Department of Veterans Affairs (VA) Computerized Patient Record System (CPRS) to identify patients with a documented penicillin allergy who underwent an operation and received periprocedural antibiotics between February 1, 2023, and January 31, 2024. This study was reviewed and approved by the Indiana University Health Institutional Review Board and the VHI Research and Development Committee.
Patients were enrolled if they were aged ≥ 18 years, had a documented penicillin allergy, underwent a surgical intervention, and received perioperative antibiotics during the study period. Patients were excluded if they had a documented penicillin allergy resulting in severe delayed skin reactions (ie, SJS, TEN, or DRESS). These criteria produced 197 surgical procedures. Data were collected for each surgical procedure, so patients could be included more than once. Patient history of allergic reaction to penicillin was obtained through CPRS.
The primary endpoint was the percentage of allergic ADRs in patients with penicillin allergies receiving cefazolin perioperatively. Secondary outcomes included the appropriateness of the antibiotic regimen in congruence with American System of Health Pharmacists (ASHP) recommendations, incidence of SSIs within 30 days of the procedure, incidence of ADRs in those with a history of anaphylaxis vs nonanaphylaxis allergy, incidence of allergic reaction requiring pharmacologic and nonpharmacologic interventions, and incidence of acute kidney injury (AKI). AKI was defined as an increase in serum creatinine by ≥ 0.3 mg/dL within 48 hours or an increase in serum creatinine to ≥ 1.5 times baseline.
Demographic data included sex, age, race, preoperative serum creatinine, and postoperative serum creatinine. Anaphylaxis was defined as an acute onset of illness (within minutes to several hours) with involvement of skin, mucosal tissue, or both involving either respiratory compromise or reduced blood pressures. Allergic reactions were defined as facial, tongue, throat, airway, lip, mouth, periorbital, or eye swelling, urticaria, angioedema, dyspnea, anaphylaxis, or a positive penicillin skin test. Additionally, data collected included the description and severity of postprophylactic antibiotic reaction, antibiotic choice, interventions required for the allergic reaction, SSI occurrence, date of SSI, operating specialty, and postoperative change in renal function.
Descriptive statistics, including mean, SD, and percentages were reported for baseline characteristics of the study population. Percentages were used to demonstrate the differences in primary and secondary outcomes for each study group. Fisher exact tests were used for incidence of ADRs in patients with penicillin allergy who received cefazolin and reported incidence of SSIs.
Results
A total of 197 surgical procedures in patients with a documented penicillin allergy were included; 127 procedures used cefazolin perioperatively, 3 procedures used cefazolin plus gentamicin, and 67 procedures used other antibiotics. Most patients were White (n = 160; 81.2%), male (n = 158; 80.2%), and had a mean age of 64.9 years. Urology was the most common surgical specialty (n = 59; 29.9%) (Table 1). Of the 16 patients with documented penicillin anaphylaxis reaction, 8 received cefazolin and 8 received a different antibiotic. A total of 181 patients reported a nonanaphylaxis allergy. One hundred fifty-one patients (68.6%) reported a reaction history of hives, rash, or swelling (Table 2). Patients could report ≥ 1 reaction. The most prevalent antibiotics used were cefazolin, which was used by 130 patients (61.3%), and clindamycin which was used by 33 patients (15.6%) (Table 3). Patients could receive ≥ 1 antibiotic.
For the primary outcome, the incidence of allergic reactions in patients allergic to penicillin, there was no incidence of allergic reactions in either the cefazolin or other group. Given the absence of reactions, no interventions were required.
There were no ADRs in those with history of anaphylaxis or nonanaphylaxis allergy. In the cefazolin group, 126 of 127 surgical procedure regimens (99.2%) were congruent with ASHP recommendations, all 3 surgical procedures regimens in the cefazolin plus gentamicin group were congruent with ASHP recommendations, and 58 of 67 surgical procedure regimens (86.6%) in the other antibiotic group were congruent with ASHP recommendations. None of the 127 patients in the cefazolin group or of the 3 patients in the cefazolin plus gentamicin group reported an SSI, and 3 of 67 patients (4.5%) had an SSI in the other antibiotic group. One procedure that resulted in SSI was not congruent with ASHP recommendations. Twenty-four patients had 2 serum creatinine levels drawn within 48 hours of surgery. One of 12 patients (8.3%) and 0 of 12 patients had an AKI in the cefazolin and other antibiotic group, respectively (Table 4).
Discussion
Implementation of a screening tool at VHI allowed patients with documented penicillin allergy, including anaphylaxis, to receive cefazolin perioperatively. Broad spectrum antibiotics such as vancomycin, clindamycin, and fluoroquinolones are frequently used in patients allergic to penicillin, which can increase health care costs, risk of toxicity, and antimicrobial resistance.4 There was no incidence of allergic reactions noted in patients allergic to penicillin who received cefazolin. When comparing the incidence of observed allergic reactions to received perioperative antibiotics in the cefazolin group to previously published literature, no difference in allergy rates (P = .09) was found.3 Most antibiotics administered were congruent with ASHP guideline recommendations, and most patients eligible for cefazolin received it perioperatively.
Similar to this study, Goodman et al concluded that cefazolin appears to be a safe regimen in patients with documented penicillin anaphylactic reaction for surgical prophylaxis with only 1 (0.2%) potential allergic reaction.6 Patients who received cefazolin perioperatively had a statistically significant decrease in SSI rates. There were no clinically or statistically significant differences found between the proportion of allergic reactions or ADRs when compared to alternative antibiotics. Lessard et al concluded that a pharmacist-led interdisciplinary collaborative practice agreement increased cefazolin use in patients allergic to penicillin, including those with urticaria and anaphylaxis, with no reported ADRs.7 This study further demonstrated the safety of cefazolin use in patients with anaphylaxis to penicillin.
Limitations
This study’s single-center, retrospective design, patient population, and small sample size limit the generalizability of its results. The data collected are dependent on documentation in the chart. No ADRs were reported from the antibiotics patients received perioperatively. When considering safety data, information such as serum creatinine were available only in CPRS and some patients did not receive a postprocedure serum creatinine level. Additionally, this study did not investigate whether there was an increase in preferred preoperative antimicrobial prophylaxis after implementation of this protocol.
Conclusions
The results of this study support the use of cefazolin perioperatively in patients allergic to penicillin, including those with a history of anaphylaxis. Additional research should be conducted to validate data given the low incidence of ADRs. The primary outcome did not reach statistical significance, but the results may be clinically significant from a stewardship and safety perspective. VHI continues to use the screening tool described in this article.
Given its safety profile and bactericidal activity against the predominant organisms causing surgical site infections (SSIs), cefazolin remains the most popular choice for surgical prophylaxis.1 Cefazolin offers protection against the pathogens most likely to contaminate the surgical site while minimizing inappropriate methicillin- resistant Staphylococcus aureus coverage that occurs with alternatives such as vancomycin and clindamycin. Documented allergies to Β-lactam antibiotics have historically forced clinicians to avoid the use of cephalosporins due to the potential risk of cross-reactivity. True type 1 (immunoglobin E [IgE]-mediated) cross-allergic reactions between penicillin and cephalosporins are rare, and previously reported data indicate cross-reactivity as a result of antibody recognition is more closely related to the side-chain identity rather than the Β-lactam ring.2,3
About 10% of US patients report having a penicillin allergy; however, < 1% of the population has a true IgE-mediated allergic reaction.4 Previous research that has challenged penicillin allergies with cefazolin for surgical prophylaxis has reported minimal rates of allergic reactions.2-5
In previous trials, patients with a history of delayed skin reactions, such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS), were excluded. Additionally, patients with an allergy to cefazolin including those with urticaria, angioedema, bronchospasm, or anaphylaxis, were excluded from perioperative retrial of cefazolin. Grant et al found that cefazolin can be safely given to patients with IgE-mediated reactions to penicillin and other cephalosporins due to a structurally different side chain.3
In January 2023, the Veteran Health Indiana (VHI) pharmacy team in conjunction with surgery, infectious disease, and anesthesiology, implemented a screening tool as an amendment to perioperative antibiotic guidance to help determine which patients with a documented penicillin allergy could be candidates for perioperative cefazolin. The implemented screening tool (Allergy Clarification for Cefazolin Evidence-Based Prescribing Tool) has been described by Lam et al, who reported that an increased proportion of patients with documented penicillin allergy received cefazolin without more adverse drug reactions (ADRs).5 Patients with a Β-lactam allergy were eligible to receive cefazolin unless the ADR was SJS, TEN, or DRESS, or the offending agent was cefazolin and the patient experienced urticaria, angioedema, bronchospasm, or anaphylaxis. If the reaction was not from cefazolin or was unknown, patients were eligible to receive cefazolin (Figure).
To date, minimal data exist to evaluate the incidence of ADRs when cefazolin is given perioperatively to patients with a previously documented penicillin allergy. The purpose of this study was to evaluate the incidence of allergic ADRs in patients who had a documented penicillin allergy and received periprocedural antibiotics.
Methods
This single-center, retrospective chart review used the US Department of Veterans Affairs (VA) Computerized Patient Record System (CPRS) to identify patients with a documented penicillin allergy who underwent an operation and received periprocedural antibiotics between February 1, 2023, and January 31, 2024. This study was reviewed and approved by the Indiana University Health Institutional Review Board and the VHI Research and Development Committee.
Patients were enrolled if they were aged ≥ 18 years, had a documented penicillin allergy, underwent a surgical intervention, and received perioperative antibiotics during the study period. Patients were excluded if they had a documented penicillin allergy resulting in severe delayed skin reactions (ie, SJS, TEN, or DRESS). These criteria produced 197 surgical procedures. Data were collected for each surgical procedure, so patients could be included more than once. Patient history of allergic reaction to penicillin was obtained through CPRS.
The primary endpoint was the percentage of allergic ADRs in patients with penicillin allergies receiving cefazolin perioperatively. Secondary outcomes included the appropriateness of the antibiotic regimen in congruence with American System of Health Pharmacists (ASHP) recommendations, incidence of SSIs within 30 days of the procedure, incidence of ADRs in those with a history of anaphylaxis vs nonanaphylaxis allergy, incidence of allergic reaction requiring pharmacologic and nonpharmacologic interventions, and incidence of acute kidney injury (AKI). AKI was defined as an increase in serum creatinine by ≥ 0.3 mg/dL within 48 hours or an increase in serum creatinine to ≥ 1.5 times baseline.
Demographic data included sex, age, race, preoperative serum creatinine, and postoperative serum creatinine. Anaphylaxis was defined as an acute onset of illness (within minutes to several hours) with involvement of skin, mucosal tissue, or both involving either respiratory compromise or reduced blood pressures. Allergic reactions were defined as facial, tongue, throat, airway, lip, mouth, periorbital, or eye swelling, urticaria, angioedema, dyspnea, anaphylaxis, or a positive penicillin skin test. Additionally, data collected included the description and severity of postprophylactic antibiotic reaction, antibiotic choice, interventions required for the allergic reaction, SSI occurrence, date of SSI, operating specialty, and postoperative change in renal function.
Descriptive statistics, including mean, SD, and percentages were reported for baseline characteristics of the study population. Percentages were used to demonstrate the differences in primary and secondary outcomes for each study group. Fisher exact tests were used for incidence of ADRs in patients with penicillin allergy who received cefazolin and reported incidence of SSIs.
Results
A total of 197 surgical procedures in patients with a documented penicillin allergy were included; 127 procedures used cefazolin perioperatively, 3 procedures used cefazolin plus gentamicin, and 67 procedures used other antibiotics. Most patients were White (n = 160; 81.2%), male (n = 158; 80.2%), and had a mean age of 64.9 years. Urology was the most common surgical specialty (n = 59; 29.9%) (Table 1). Of the 16 patients with documented penicillin anaphylaxis reaction, 8 received cefazolin and 8 received a different antibiotic. A total of 181 patients reported a nonanaphylaxis allergy. One hundred fifty-one patients (68.6%) reported a reaction history of hives, rash, or swelling (Table 2). Patients could report ≥ 1 reaction. The most prevalent antibiotics used were cefazolin, which was used by 130 patients (61.3%), and clindamycin which was used by 33 patients (15.6%) (Table 3). Patients could receive ≥ 1 antibiotic.
For the primary outcome, the incidence of allergic reactions in patients allergic to penicillin, there was no incidence of allergic reactions in either the cefazolin or other group. Given the absence of reactions, no interventions were required.
There were no ADRs in those with history of anaphylaxis or nonanaphylaxis allergy. In the cefazolin group, 126 of 127 surgical procedure regimens (99.2%) were congruent with ASHP recommendations, all 3 surgical procedures regimens in the cefazolin plus gentamicin group were congruent with ASHP recommendations, and 58 of 67 surgical procedure regimens (86.6%) in the other antibiotic group were congruent with ASHP recommendations. None of the 127 patients in the cefazolin group or of the 3 patients in the cefazolin plus gentamicin group reported an SSI, and 3 of 67 patients (4.5%) had an SSI in the other antibiotic group. One procedure that resulted in SSI was not congruent with ASHP recommendations. Twenty-four patients had 2 serum creatinine levels drawn within 48 hours of surgery. One of 12 patients (8.3%) and 0 of 12 patients had an AKI in the cefazolin and other antibiotic group, respectively (Table 4).
Discussion
Implementation of a screening tool at VHI allowed patients with documented penicillin allergy, including anaphylaxis, to receive cefazolin perioperatively. Broad spectrum antibiotics such as vancomycin, clindamycin, and fluoroquinolones are frequently used in patients allergic to penicillin, which can increase health care costs, risk of toxicity, and antimicrobial resistance.4 There was no incidence of allergic reactions noted in patients allergic to penicillin who received cefazolin. When comparing the incidence of observed allergic reactions to received perioperative antibiotics in the cefazolin group to previously published literature, no difference in allergy rates (P = .09) was found.3 Most antibiotics administered were congruent with ASHP guideline recommendations, and most patients eligible for cefazolin received it perioperatively.
Similar to this study, Goodman et al concluded that cefazolin appears to be a safe regimen in patients with documented penicillin anaphylactic reaction for surgical prophylaxis with only 1 (0.2%) potential allergic reaction.6 Patients who received cefazolin perioperatively had a statistically significant decrease in SSI rates. There were no clinically or statistically significant differences found between the proportion of allergic reactions or ADRs when compared to alternative antibiotics. Lessard et al concluded that a pharmacist-led interdisciplinary collaborative practice agreement increased cefazolin use in patients allergic to penicillin, including those with urticaria and anaphylaxis, with no reported ADRs.7 This study further demonstrated the safety of cefazolin use in patients with anaphylaxis to penicillin.
Limitations
This study’s single-center, retrospective design, patient population, and small sample size limit the generalizability of its results. The data collected are dependent on documentation in the chart. No ADRs were reported from the antibiotics patients received perioperatively. When considering safety data, information such as serum creatinine were available only in CPRS and some patients did not receive a postprocedure serum creatinine level. Additionally, this study did not investigate whether there was an increase in preferred preoperative antimicrobial prophylaxis after implementation of this protocol.
Conclusions
The results of this study support the use of cefazolin perioperatively in patients allergic to penicillin, including those with a history of anaphylaxis. Additional research should be conducted to validate data given the low incidence of ADRs. The primary outcome did not reach statistical significance, but the results may be clinically significant from a stewardship and safety perspective. VHI continues to use the screening tool described in this article.
- Bratzler DW, Dellinger EP, Olsen KM, et al. Clinical practice guidelines for antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 2013;70:195-283. doi:10.2146/ajhp120568
- Romano A, Valluzzi RL, Caruso C, et al. Tolerability of cefazolin and ceftibuten in patients with IgE-mediated aminopenicillin allergy. J Allergy Clin Immunol Pract. 2020;8:1989-1993.e2. doi:10.1016/j.jaip.2020.02.025
- Grant JM, Song WHC, Shajari S, et al. Safety of administering cefazolin versus other antibiotics in penicillin- allergic patients for surgical prophylaxis at a major Canadian teaching hospital. Surgery. 2021;170:783-789. doi:10.1016/j.surg.2021.03.022
- Centers for Disease Control and Prevention. Clinical Features of Penicillin Allergy. August 25, 2025. Accessed January 6, 2026. https://www.cdc.gov/antibiotic-use/hcp/clinical-signs/index.html
- Lam PW, Tarighi P, Elligsen M, et al. Impact of the allergy clarification for cefazolin evidence-based prescribing tool on receipt of preferred perioperative prophylaxis: an interrupted time series study. Clin Infect Dis. 2020;71:2955- 2957. doi:10.1093/cid/ciaa516
- Goodman EJ, Morgan MJ, Johnson Pa, et al. Cephalosporins can be given to penicillin-allergic patients who do not exhibit an anaphylactic response. J Clin Anesth. 2001;13:561-564. doi:10.1016/s0952-8180(01)00329-4
- Lessard S, Huiras C, Dababneh A, et al. Pharmacist adjustment of preoperative antibiotic orders to the preferred preoperative antibiotic cefazolin for patients with penicillin allergy labeling. Am J Health Syst Pharm. 2023;80:532- 536. doi:10.1093/ajhp/zxac385
- Bratzler DW, Dellinger EP, Olsen KM, et al. Clinical practice guidelines for antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 2013;70:195-283. doi:10.2146/ajhp120568
- Romano A, Valluzzi RL, Caruso C, et al. Tolerability of cefazolin and ceftibuten in patients with IgE-mediated aminopenicillin allergy. J Allergy Clin Immunol Pract. 2020;8:1989-1993.e2. doi:10.1016/j.jaip.2020.02.025
- Grant JM, Song WHC, Shajari S, et al. Safety of administering cefazolin versus other antibiotics in penicillin- allergic patients for surgical prophylaxis at a major Canadian teaching hospital. Surgery. 2021;170:783-789. doi:10.1016/j.surg.2021.03.022
- Centers for Disease Control and Prevention. Clinical Features of Penicillin Allergy. August 25, 2025. Accessed January 6, 2026. https://www.cdc.gov/antibiotic-use/hcp/clinical-signs/index.html
- Lam PW, Tarighi P, Elligsen M, et al. Impact of the allergy clarification for cefazolin evidence-based prescribing tool on receipt of preferred perioperative prophylaxis: an interrupted time series study. Clin Infect Dis. 2020;71:2955- 2957. doi:10.1093/cid/ciaa516
- Goodman EJ, Morgan MJ, Johnson Pa, et al. Cephalosporins can be given to penicillin-allergic patients who do not exhibit an anaphylactic response. J Clin Anesth. 2001;13:561-564. doi:10.1016/s0952-8180(01)00329-4
- Lessard S, Huiras C, Dababneh A, et al. Pharmacist adjustment of preoperative antibiotic orders to the preferred preoperative antibiotic cefazolin for patients with penicillin allergy labeling. Am J Health Syst Pharm. 2023;80:532- 536. doi:10.1093/ajhp/zxac385
Outcomes From the Use of Cefazolin for Surgical Prophylaxis in Patients Allergic to Penicillin
Outcomes From the Use of Cefazolin for Surgical Prophylaxis in Patients Allergic to Penicillin
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
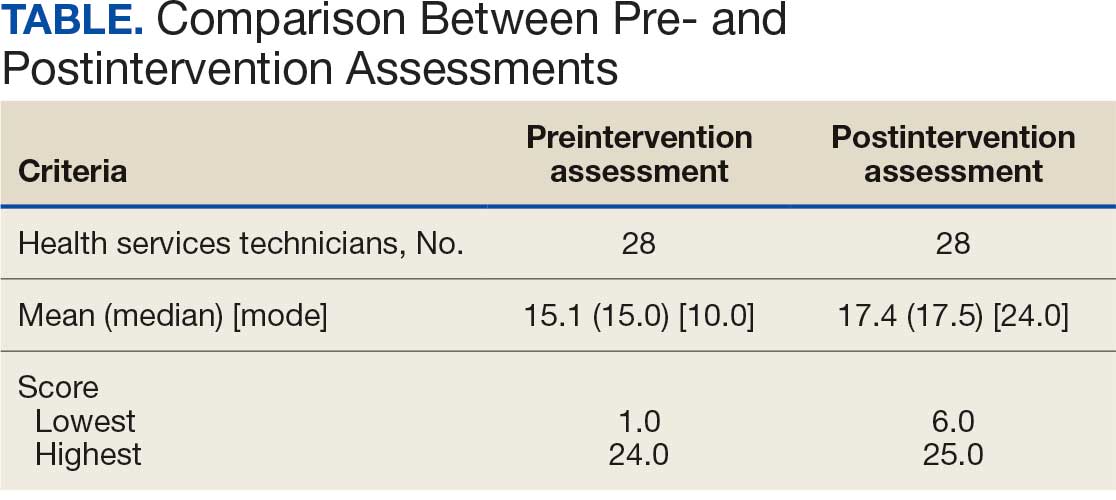
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
The US Coast Guard (USCG) operates within the US Department of Homeland Security and represents a force of > 50,000 servicemembers.1 The missions of the service include maritime law enforcement (drug interdiction), search and rescue, and defense readiness.2
The USCG operates 42 clinics and numerous smaller sick bays of varying sizes and medical capabilities throughout the country to provide acute and routine medical services. Health services technicians (HSs) are the most common staffing component and provide much of the support services in each USCG health care setting. The HS rating, colloquially referred to as corpsmen, is achieved through a 22-week course known as “A” school that trains servicemembers in outpatient and acute care, including emergency medical technician training.3 There are about 750 USCG HSs.
Within USCG clinics, HSs conduct ambulatory intakes for outpatient appointments, administer immunizations and blood draws, requisition medical equipment and supplies, serve as a pharmacy technician, complete physical examinations, and manage referrals, among other duties. Their familiarity with different aspects of clinic operations and medical practice must be broad. To that end, corpsmen develop and reinforce their medical knowledge through various trainings, including additional courses to specialize in certain medical skills, such as pharmacy technician “C” school or dental assistant “C” school.
The USCG employs < 15 field pharmacists, most of whom serve in an ambulatory care environment.4 Responsibilities of USCG pharmacists include the routine reinforcement of pharmacy knowledge with HSs. For the corpsmen who are not pharmacy technicians or who have not attended pharmacy technician “C” school, the extent of their pharmacy instruction primarily came from the “A” school curriculum, of which only 1 class is specific to pharmacy. Providing routine pharmacy-related training to the HSs further cultivates their pharmacy knowledge and confidence so that they can practice more holistically. These trainings do not need to follow any specific format.
In this study, 3 pharmacists at 3 separate USCG clinics conducted a training inspired by the Jeopardy! game show with the corpsmen at their respective clinics. This study examined the effectiveness of game-based learning on the pharmacy knowledge retention of HSs at 3 USCG clinics. A secondary objective was to evaluate the baseline pharmacy knowledge of corpsmen based on specific corpsmen demographics.
Methods
As part of a USCG quality improvement study in 2024, 28 HSs at the 3 USCG clinics were provided a preintervention assessment, completed game-based educational program (intervention), and then were assessed again following the intervention.
The HSs were presented with a 25-question assessment that included 10 knowledge questions (3 on over-the-counter medications, 2 on use of medications in pregnancy, 2 on precautions and contraindications, 2 on indications, and 1 on immunizations) and 15 brand-generic matching questions. These questions were developed and reviewed by the 3 participating pharmacists to ensure that their scope was commensurate with the overall pharmacy knowledge that could be reasonably expected of corpsmen spanning various points of their HS career.
One to 7 days after the preintervention assessment, the pharmacists hosted the game-based learning modeled after Jeopardy!. The Jeopardy! categories mirrored the assessment knowledge question categories, and brand-generic nomenclature was freely discussed throughout. About 2 weeks later, the same HSs who completed the preintervention assessment and participated in the game were presented with the same assessment.
In addition to capturing the difference in scores between the 2 assessments, additional demographic data were gathered, including service time as an HS and whether they received formalized pharmacy technician training and if so, how long they have served in that capacity. Demographic data were collected to identify potential correlations between demographic characteristics and results.
Results
Twenty-eight HSs at the 3 clinics completed the game-based training and both assessments. The mean score increased from 15.1 preintervention to 17.4 postintervention (Table). Preintervention scores ranged from 1 to 24 and postintervention scores ranged from 6 to 25.
There were 19 HSs (68%) whose score increased from preintervention to postintervention and 5 (18%) had decreased scores. The largest score decrease was 4 (from 18 to 14), and the largest score increase was 11 (from 13 to 24). The mean improvement was 3.9 among the 19 HSs with increased scores
Twenty-one HSs reported no formal pharmacy technician training, 3 completed pharmacy technician “C” school, and 4 received informal on-the-job training. The mean score for the “C” school trained HSs was 23.0 preintervention and 23.7 postintervention. The mean score for HSs trained on the job was 16.0 preintervention and 18.5 postintervention. The mean score for HSs with no training was 13.9 preintervention and 16.3 postintervention.
As HSs advance in their careers, they typically assume roles with increasing technical knowledge, responsibility, and oversight, thus aligning with advancement from E-4 (third class petty officer) to E-6 (first class petty officer) and beyond. In this study, there was 1 E-3, 12 E-4s (mean time as an HS, 1.3 years), 8 E-5s (mean time as an HS, 4.8 years), and 7 E-6s (mean time as an HS, 8.6 years). The E-3 had a preintervention score of 1.0 and a postintervention score of 6.0. The E-4s had a mean change in score from pre- to postintervention of 2.4. The E-5s had a mean change in score from pre- to postintervention of 1.6. The E-6s had a mean change in score from pre- to postintervention of 2.3.
Discussion
This study is novel in its examination of the impact of game-based learning on the retention of the pharmacy knowledge of USCG corpsmen. A PubMed literature search of the phrase “((Corpsman) OR (Corpsmen)) AND (Coast Guard)” yields 135 results, though none were relevant to the USCG population described in this study. A PubMed literature search of the phrase “(Jeopardy!) AND (pharmacy)” yields 28 results, only 1 of which discusses using the game-based approach as an instructional tool.5 A PubMed literature search of the phrase “(game) AND (Coast Guard)” yields 55 results, none of which were specifically relevant to game-based learning in the USCG. This study appears to be among the first to discuss results and trends in game-based learning with USCG corpsmen.
The preponderance of literature for game-based learning strategies exists in children; more research in adults is needed.6,7 With studies showing that game-based learning may impact motivation to learn and learning gains, it is unsurprising that there is some research in professional health care education. Games modeled after everything from simulated clinical scenarios to Family Feud and Chutes and Ladders-style games have been compared with traditional learning strategies. However, the results of whether game-based learning strategies improve knowledge, clinical decision-making, and motivation to learn vary, suggesting the need for more research in this field.8
The results of this study suggest that Jeopardy! is likely an effective instructional method for USCG corpsmen on pharmacy topics. While there were some HSs whose postintervention scores decreased, 19 (68%) had increased scores. Because the second assessment was administered about 2 weeks after the game-based learning, the results suggest some level of knowledge retention. Between these results and the informally perceived level of engagement, game-based learning could be a more stimulating alternative training method to a standard slide-based presentation.
Stratifying the data by demographics revealed additional trends, although they should be interpreted with caution due to the small sample size. The baseline results strongly illustrate the value of formalized training. It is generally expected that HSs who have completed the “C” school pharmacy technician training program should have more pharmacy knowledge than those with on-the-job or less training. The results indicate that “C” school trained and on-the-job trained HSs scored higher on the preintervention assessment (mean, 23.0 and 16.0, respectively), than those with no such experiences (mean, 13.9). Such results underscore the value of formalized training—whether as a pharmacy technician or in any other “C” school—in enhancing the medical knowledge of HSs that may allow them to hold roles of increased responsibility and medical scope.
In addition to stratification by pharmacy technician training, stratification by years of HS experience (roughly correlated to rank) yields a similar result. It would be expected that as HSs advance in their careers, they gain more exposure to various medical topics, including pharmacy. That is not always the case, however, as it is possible an HS never rotated through a pharmacy technician position or has not been recently exposed to pharmacy knowledge. Nevertheless, the results suggest that increased HS experience was likely associated with an increased baseline pharmacy knowledge, with mean preintervention scores increasing from 11.9 to 18.1 to 19.3 for E-4, E-5, and E-6, respectively.
While there are many explanations for these results, the authors hypothesize that when HSs are E-4s, they might not yet have exposure to all aspects of the clinic and are perhaps not as well-versed in pharmacy practice. An E-5—now a few years into their career—would have completed pharmacy technician “C” school or on-the-job training (if applicable), which could account for the significant jump in pharmacy knowledge scores. An E-6 can still engage in direct patient care activities but take on leadership and supervisory roles within the clinic, perhaps explaining the smaller increase in score.
In terms of increasing responsibility, many USCG corpsmen complete another schooling opportunity—Independent Duty Health Services Technician (IDHS)—so they can serve in independent duty roles, many of which are on USCG cutters. While cutters are deployed, that IDHS could be the sole medical personnel on the cutter and function in a midlevel practitioner extender role. Formalized training in pharmacy—the benefits of which are suggested through these results—or another field of medical practice would strengthen the skillset and confidence of IDHSs.
Though not formally assessed, the 3 pharmacists noted that the game-based learning was met with overwhelmingly positive feedback in terms of excitement, energy, and overall engagement.
Limitations
This cohort of individuals represents a small proportion of the total number of USCG corpsmen, and it is not fully representative of all practice settings. HSs can be assigned to USCG cutters as IDHSs, which would not be captured in this cohort. Even within a single clinic, the knowledge of HSs varies, as not all HS duties consist solely of clinical skills. Additionally, while the overall game framework was consistent among the 3 sites, there may have been unquantifiable differences in overall teaching style by the 3 pharmacists that may have resulted in different levels of content retention. Given the lack of similar studies in this population, this study can best be described as a quantitative descriptor of results rather than a statistical comparison of what instructional method works best.
Conclusions
The USCG greatly benefits from having trained and experienced HSs fulfilling mission support roles in the organization. In addition to traditional slide-based trainings, game-based learning can be considered to create engaging learning environments to support the knowledge retention of pharmacy and other medical topics for USCG corpsmen.
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
- US Coast Guard. Organizational overview. About the US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About
- US Coast Guard. Missions. About US Coast Guard. Accessed October 14, 2025. https://www.uscg.mil/About/Missions/
- US Coast Guard. Health services technician. Accessed October 14, 2025. https://www.gocoastguard.com/careers/enlisted/hs
- Zhou F, Woodward Z. Impact of pharmacist interventions at an outpatient US Coast Guard clinic. Fed Pract. 2023;40(6):174-177. doi:10.12788/fp.0383
- Cusick J. A Jeopardy-style review game using team clickers. MedEdPORTAL. 2016;12:10485. doi:10.15766/mep_2374-8265.10485
- Dahalan F, Alias N, Shaharom MSN. Gamification and game based learning for vocational education and training: a systematic literature review. Educ Inf Technol (Dordr). 2023:1-39. doi:10.1007/s10639-022-11548-w
- Wesselink LA. Testing the Effectiveness of Game-Based Learning for Adults by Designing an Educational Game: A Design and Research Study to Investigate the Effectiveness of Educational Games for Adults to Learn Basic Skills of Microsoft Excel. Master’s thesis. University of Twente; 2020. Accessed October 22, 2025. http://essay.utwentw.nl/88229
- Del Cura-González I, Ariza-Cardiel G, Polentinos-Castro E, et al. Effectiveness of a game-based educational strategy e-EDUCAGUIA for implementing antimicrobial clinical practice guidelines in family medicine residents in Spain: a randomized clinical trial by cluster. BMC Med Educ. 2022;22:893. doi:10.1186/s12909-022-03843-4
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Daily Double! Assessing the Effectiveness of Game-Based Learning on the Pharmacy Knowledge of US Coast Guard Health Services Technicians
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
Systemic glucocorticoids play an important role in the treatment of chronic obstructive pulmonary disease (COPD) exacerbations. They are recommended to shorten recovery time and increase forced expiratory volume in 1 second (FEV1) during exacerbations.1 However, the role of the chronic use of inhaled corticosteroids (ICSs) in the treatment of COPD is less clear.
When added to inhaled β-2 agonists and muscarinic antagonists, ICSs can decrease the risk of exacerbations.1 However, not all patients with COPD benefit from ICS therapy. The degree of benefit an ICS can provide has been shown to correlate with eosinophil count—a marker of inflammation. The expected benefit of using an ICS increases as the eosinophil count increases.1 Maximum benefit can be observed with eosinophil counts ≥ 300 cells/µL, and minimal benefit is observed with eosinophil counts < 100 cells/µL. Adverse effects (AEs) of ICSs include a hoarse voice, oral candidiasis, and an increased risk of pneumonia.1 Given the risk of AEs, it is important to limit ICS use in patients who are unlikely to reap any benefits.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines suggest the use of ICSs in patients who experience exacerbations while using long-acting β agonist (LABA) plus long-acting muscarinic antagonist (LAMA) therapy and have an eosinophil count ≥ 100 cells/µL. Switching from LABA or LAMA monotherapy to triple therapy with LAMA/LABA/ICS may be considered if patients have continued exacerbations and an eosinophil count ≥ 300 cells/µL. De-escalation of ICS therapy should be considered if patients do not meet these criteria or if patients experience ICS AEs, such as pneumonia. The patients most likely to have increased exacerbations or decreased FEV1 with ICS withdrawal are those with eosinophil counts ≥ 300 cells/µL.1,2
Several studies have explored the effects of ICS de-escalation in real-world clinical settings. A systematic review of 11 studies indicated that de-escalation of ICS in COPD does not result in increased exacerbations.3 A prospective study by Rossi et al found that in a 6-month period, 141 of 482 patients on ICS therapy (29%) had an exacerbation. In the opposing arm of the study, 88 of 334 patients (26%) with deprescribed ICS experienced an exacerbation. The difference between these 2 groups was not statistically significant.4 The researchers concluded that in real-world practice, ICS withdrawal can be safe in patients at low risk of exacerbation.
About 25% of veterans (1.25 million) have been diagnosed with COPD.5 To address this, the US Department of Veterans Affairs (VA) and US Department of Defense published updated COPD guidelines in 2021 that specify criteria for de-escalation of ICS.6 Guidelines, however, may not be reflected in common clinical practice for several years following publication. The VA Academic Detailing Service (ADS) provides tools to help clinicians identify patients who may benefit from changes in treatment plans. A recent ADS focus was the implementation of a COPD dashboard, which identifies patients with COPD who are candidates for ICS de-escalation based on comorbid diagnoses, exacerbation history, and eosinophil count. VA pharmacists have an expanded role in the management of primary care disease states and are therefore well-positioned to increase adherence to guideline-directed therapy. The objective of this quality improvement project was to determine the impact of pharmacist-driven de-escalation on ICS usage in veterans with COPD.
Methods
This project was conducted in an outpatient clinic at the Robley Rex VA Medical Center beginning September 21, 2023, with a progress note in the Computerized Patient Record System (CPRS). Eligible patients were selected using the COPD Dashboard provided by ADS. The COPD Dashboard defined patients with COPD as those with ≥ 2 outpatient COPD diagnoses in the past 2 years, 1 inpatient discharge COPD diagnosis in the past year, or COPD listed as an active problem. COPD diagnoses were identified using International Statistical Classification of Disease, Tenth Revision (ICD-10) codes
Candidates identified for ICS de-escalation by the dashboard were excluded if they had a history of COPD exacerbation in the previous 2 years. The dashboard identified COPD exacerbations via ICD-10 codes for COPD or acute respiratory failure for inpatient discharges, emergency department (ED) visits, urgent care visits, and community care consults with 1 of the following terms: emergency, inpatient, hospital, urgent, ED (self). The COPD dashboard excluded patients with a diagnosis of asthma.
After patients were selected, they were screened for additional exclusion criteria. Patients were excluded if a pulmonary care practitioner managed their COPD; if identified via an active pulmonary consult in CPRS; if a non-VA clinician prescribed their ICS; or if they were being treated with roflumilast, theophylline, or chronic azithromycin. Individuals taking these 3 drugs were excluded due to potential severe and/or refractory COPD. Patients also were excluded if they: (1) had prior ICS de-escalation failure (defined as a COPD exacerbation following ICS de-escalation that resulted in ICS resumption); (2) had a COPD exacerbation requiring systemic corticosteroids or antibiotics in the previous year; (3) had active lung cancer; (4) did not have any eosinophil levels in CPRS within the previous 2 years; or (5) had any eosinophil levels ≥ 300 cells/µL in the previous year.
Each patient who met the inclusion criteria and was not excluded received a focused medication review by a pharmacist who created a templated progress note, with patient-specific recommendations, that was entered in the CPRS (eAppendix). The recommendations were also attached as an addendum to the patient’s last primary care visit note, and the primary care practitioner (PCP) was alerted via CPRS to consider ICS de-escalation and non-ICS alternatives. Tapering of ICS therapy was offered as an option to de-escalate if abrupt discontinuation was deemed inappropriate. PCPs were also prompted to consider referral to a primary care clinical pharmacy specialist for management and follow-up of ICS de-escalation.
The primary outcome was the number of patients with de-escalated ICS at 3 and 6 months following the recommendation. Secondary outcomes included the number of: patients who were no longer prescribed an ICS or who had a non-ICS alternative initiated at a pharmacist’s recommendation; patients who were referred to a primary care clinical pharmacy specialist for ICS de-escalation; COPD exacerbations requiring systemic steroids or antibiotics, or requiring an ED visit, inpatient admission, or urgent-care clinic visit; and cases of pneumonia or oral candidiasis. Primary and secondary outcomes were evaluated via chart review in CPRS. For secondary outcomes of pneumonia and COPD exacerbation, identification was made by documented diagnosis in CPRS. For continuous data such as age, the mean was calculated.
Results
Pharmacist ICS de-escalation recommendations were made between September 21, 2023, and November 19, 2023, for 106 patients. The mean age was 72 years and 99 (93%) patients were male (Table 1). Forty-one (39%) of the patients used tobacco at the time of the study. FEV1 was available for 69 patients with a mean of 63% (GOLD grade 2).1 Based on FEV1 values, 16 patients had mild COPD (GOLD grade 1), 37 patients had moderate COPD (GOLD grade 2), 14 patients had severe COPD (GOLD grade 3), and 2 patients had very severe COPD (GOLD grade 4).1 Thirty-four patients received LABA + LAMA + ICS, 65 received LABA + ICS, 2 received LAMA + ICS, and 5 received ICS monotherapy. The most common dose of ICS was a moderate dose (Table 2). Only 2 patients had an ICS AE in the previous year.
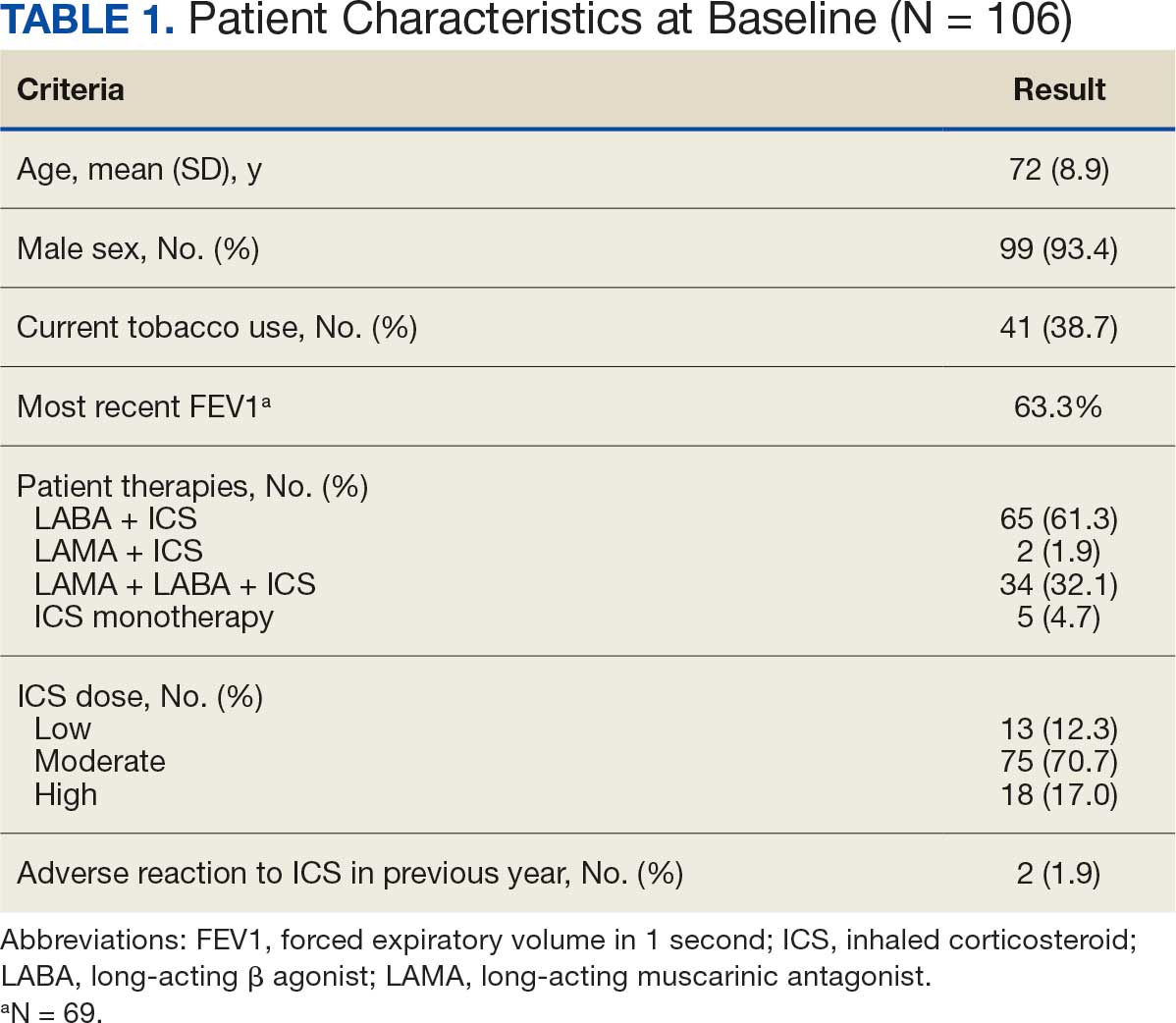
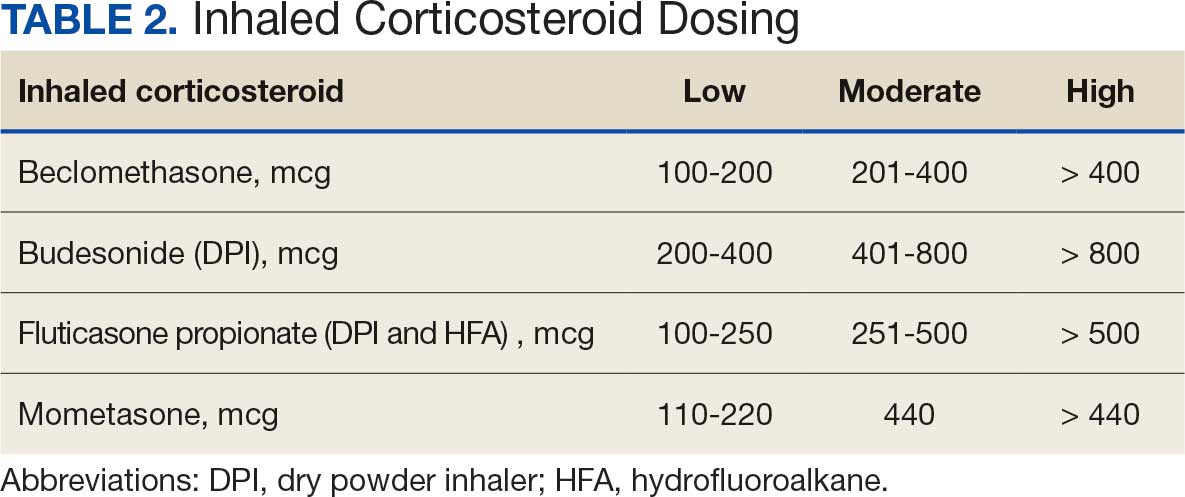
ICS de-escalation recommendations resulted in ICS de-escalation in 50 (47.2%) and 62 (58.5%) patients at 3 and 6 months, respectively. The 6-month ICS de-escalation rate by ICS dose at baseline was 72.2% (high dose), 60.0% (moderate), and 30.8% (low). De-escalation at 6 months by GOLD grade at baseline was 56.3% (9 of 16 patients, GOLD 1), 64.9% (24 of 37 patients, GOLD 2), 50% (7 of 14 patients, GOLD 3), and 50% (1 of 2 patients, GOLD 4). Six months after the ICS de-escalation recommendation appeared in the CPRS, the percentage of patients on LABA + ICS therapy dropped from 65 patients (61.3%) at baseline to 25 patients (23.6%).
Secondary outcomes were assessed at 3 and 6 months following the recommendation. Most patients with de-escalated ICS had their ICS discontinued and a non-ICS alternative initiated per pharmacist recommendations. At 6 months, 39 patients (36.8%) patients were referred to a patient aligned care team (PACT) pharmacist for de-escalation. Of the 39 patients referred to pharmacists, 69.2% (27 patients) were de-escalated; this compared to 52.2% (35 patients) who were not referred to pharmacists (Table 3).
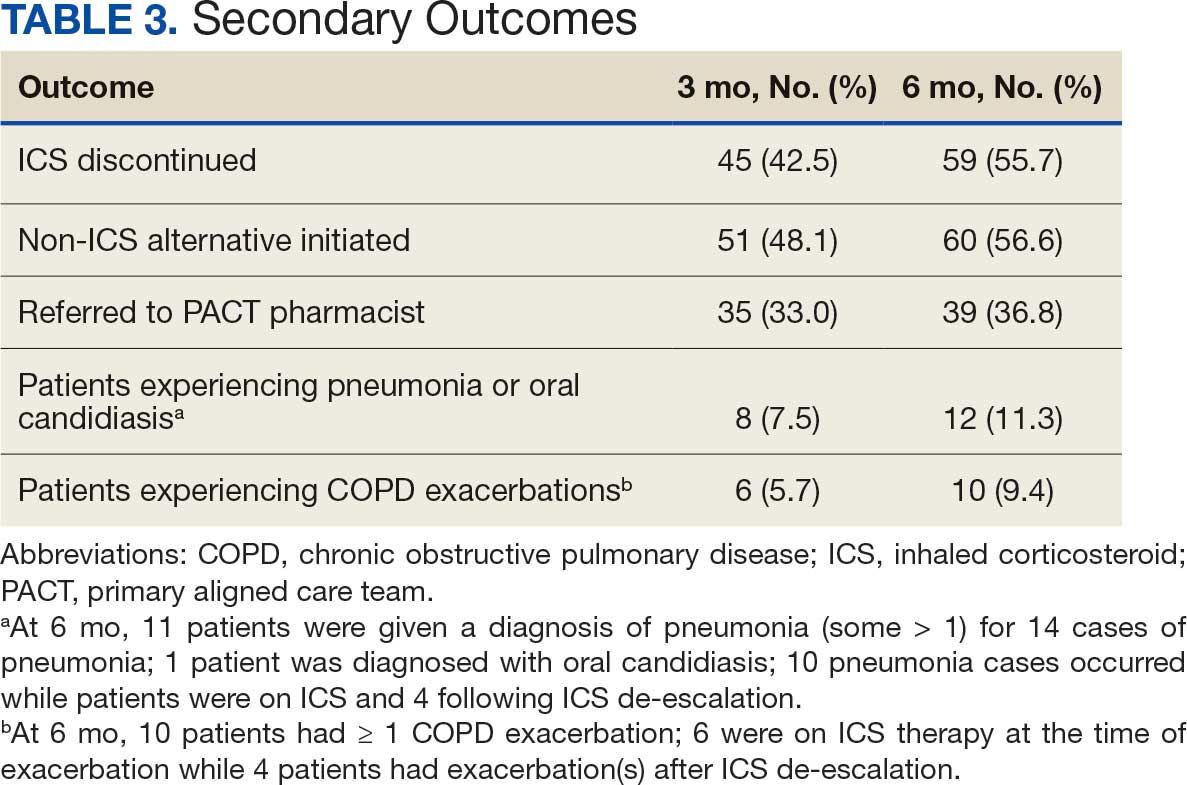
ICS use increases the risk of pneumonia.1 At 6 months, 11 patients were diagnosed with pneumonia; 3 patients were diagnosed with pneumonia twice, resulting in a total of 14 cases. Ten cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation. One patient had a documented case of oral candidiasis that occurred while on ICS therapy; no patients with discontinued ICS were diagnosed with oral candidiasis. In addition, 10 patients had COPD exacerbations; however no patients had exacerbations both before and after de-escalation. Six patients were on ICS therapy when they experienced an exacerbation, and 4 patients had an exacerbation after ICS de-escalation.
Discussion
More than half of patients receiving the pharmacist intervention achieved the primary outcome of ICS de-escalation at 6 months. Furthermore, a larger percentage of patients referred to pharmacists for the management of ICS de-escalation successfully achieved de-escalation compared to those who were not referred. These outcomes reflect the important role pharmacists can play in identifying appropriate candidates for ICS de-escalation and assisting in the management of ICS de-escalation. Patients referred to pharmacists also received other services such as smoking cessation pharmacotherapy and counseling on inhaler technique and adherence. These interventions can support improved COPD clinical outcomes.
The purpose of de-escalating ICS therapy is to reduce the risk of AEs such as pneumonia and oral candidiasis.1 The secondary outcomes of this study support previous evidence that patients who have de-escalated ICS therapy may have reduced risk of AEs compared to those who remain on ICS therapy.3 Specifically, of the 14 cases of pneumonia that occurred during the study, 10 cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation.
ICS de-escalation may increase risk of increased COPD exacerbations.1 However, the secondary outcomes of this study do not indicate that those with de-escalated ICS had more COPD exacerbations compared to those who continued on ICS. Pharmacists’ recommendations were more effective for patients with less severe COPD based on baseline FEV1.
The previous GOLD Guidelines for COPD suggested LABA + ICS therapy as an option for patients with a high symptom and exacerbation burden (previously known as GOLD Group D). Guidelines no longer recommend LABA + ICS therapy due to the superiority of triple inhaled therapy for exacerbations and the superiority of LAMA + LABA therapy for dyspnea.7 A majority of identified patients in this project were on LABA + ICS therapy alone at baseline. The ICS de-escalation recommendation resulted in a 61.5% reduction in patients on LABA + ICS therapy at 6 months. By decreasing the number of patients on LABA + ICS without LAMA, recommendations increased the number of patients on guideline-directed therapy.
Limitations
This study lacked a control group, and the rate of ICS de-escalation in patients who did not receive a pharmacist recommendation was not assessed. Therefore, it could not be determined whether the pharmacist recommendation is more effective than no recommendation. Another limitation was our inability to access records from non-VA health care facilities. This may have resulted in missed COPD exacerbations, pneumonia, and oral candidiasis prior to or following the pharmacist recommendation.
In addition, the method used to notify PCPs of the pharmacist recommendation was a CPRS alert. Clinicians often receive multiple daily alerts and may not always pay close attention to them due to alert fatigue. Early in the study, some PCPs were unknowingly omitted from the alert of the pharmacist recommendation for 10 patients due to human error. For 8 of these 10 patients, the PCP was notified of the recommendations during the 3-month follow-up period. However, 2 patients had COPD exacerbations during the 3-month follow-up period. In these cases, the PCP was not alerted to de-escalate ICS. The data for these patients were collected at 3 and 6 months in the same manner as all other patients. Also, 7 of 35 patients who were referred to a pharmacist for ICS de-escalation did not have a scheduled appointment. These patients were considered to be lost to follow-up and this may have resulted in an underestimation of the ability of pharmacists to successfully de-escalate ICS in patients with COPD.
Other studies have evaluated the efficacy of a pharmacy-driven ICS de-escalation.8,9 Hegland et al reported ICS de-escalation for 22% of 141 eligible ambulatory patients with COPD on triple inhaled therapy following pharmacist appointments.8 A study by Hahn et al resulted in 63.8% of 58 patients with COPD being maintained off ICS following a pharmacist de-escalation initiative.9 However, these studies relied upon more time-consuming de-escalation interventions, including at least 1 phone, video, or in-person patient visit.8,9
This project used a single chart review and templated progress note to recommend ICS de-escalation and achieved similar or improved de-escalation rates compared to previous studies.8,9 Previous studies were conducted prior to the updated 2023 GOLD guidelines for COPD which no longer recommend LABA + ICS therapy. This project addressed ICS de-escalation in patients on LABA + ICS therapy in addition to those on triple inhaled therapy. Additionally, previous studies did not address rates of moderate to severe COPD exacerbation and adverse events to ICS following the pharmacist intervention.8,9
This study included COPD exacerbations and cases of pneumonia or oral candidiasis as secondary outcomes to assess the safety and efficacy of the ICS de-escalation. It appeared there were similar or lower rates of COPD exacerbations, pneumonia, and oral candidiasis in those with de-escalated ICS therapy in this study. However, these secondary outcomes are exploratory and would need to be confirmed by larger studies powered to address these outcomes.
CONCLUSIONS
Pharmacist-driven ICS de-escalation may be an effective method for reducing ICS usage in veterans as seen in this study. Additional controlled studies are required to evaluate the efficacy and safety of pharmacist-driven ICS de-escalation.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2024 Report). Accessed October 14, 2025. https://goldcopd.org/2024-gold-report/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Accessed November 14, 2025. https://goldcopd.org/2025-gold-report/
- Rogliani P, Ritondo BL, Gabriele M, et al. Optimizing de-escalation of inhaled corticosteroids in COPD: a systematic review of real-world findings. Expert Rev Clin Pharmacol. 2020;13(9):977-990. doi:10.1080/17512433.2020.1817739
- Rossi A, Guerriero M, Corrado A; OPTIMO/AIPO Study Group. Withdrawal of inhaled corticosteroids can be safe in COPD patients at low risk of exacerbation: a real-life study on the appropriateness of treatment in moderate COPD patients (OPTIMO). Respir Res. 2014;15(1):77. doi:10.1186/1465-9921-15-77
- Anderson E, Wiener RS, Resnick K, et al. Care coordination for veterans with COPD: a positive deviance study. Am J Manag Care. 2020;26(2):63-68. doi:10.37765/ajmc.2020.42394
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Chronic Obstructive Pulmonary Disease. 2021. Accessed October 14, 2025. https://www.healthquality.va.gov/guidelines/CD/copd/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2023 Report). Accessed October 14, 2025. https://goldcopd.org/wp-content/uploads/2023/03/GOLD-2023-ver-1.3-17Feb2023_WMV.pdf
- Hegland AJ, Bolduc J, Jones L, Kunisaki KM, Melzer AC. Pharmacist-driven deprescribing of inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2021;18(4):730-733. doi:10.1513/AnnalsATS.202007-871RL
- Hahn NM, Nagy MW. Implementation of a targeted inhaled corticosteroid de-escalation process in patients with chronic obstructive pulmonary disease in the primary care setting. Innov Pharm. 2022;13(1):10.24926/iip.v13i1.4349. doi:10.24926/iip.v13i1.4349
Systemic glucocorticoids play an important role in the treatment of chronic obstructive pulmonary disease (COPD) exacerbations. They are recommended to shorten recovery time and increase forced expiratory volume in 1 second (FEV1) during exacerbations.1 However, the role of the chronic use of inhaled corticosteroids (ICSs) in the treatment of COPD is less clear.
When added to inhaled β-2 agonists and muscarinic antagonists, ICSs can decrease the risk of exacerbations.1 However, not all patients with COPD benefit from ICS therapy. The degree of benefit an ICS can provide has been shown to correlate with eosinophil count—a marker of inflammation. The expected benefit of using an ICS increases as the eosinophil count increases.1 Maximum benefit can be observed with eosinophil counts ≥ 300 cells/µL, and minimal benefit is observed with eosinophil counts < 100 cells/µL. Adverse effects (AEs) of ICSs include a hoarse voice, oral candidiasis, and an increased risk of pneumonia.1 Given the risk of AEs, it is important to limit ICS use in patients who are unlikely to reap any benefits.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines suggest the use of ICSs in patients who experience exacerbations while using long-acting β agonist (LABA) plus long-acting muscarinic antagonist (LAMA) therapy and have an eosinophil count ≥ 100 cells/µL. Switching from LABA or LAMA monotherapy to triple therapy with LAMA/LABA/ICS may be considered if patients have continued exacerbations and an eosinophil count ≥ 300 cells/µL. De-escalation of ICS therapy should be considered if patients do not meet these criteria or if patients experience ICS AEs, such as pneumonia. The patients most likely to have increased exacerbations or decreased FEV1 with ICS withdrawal are those with eosinophil counts ≥ 300 cells/µL.1,2
Several studies have explored the effects of ICS de-escalation in real-world clinical settings. A systematic review of 11 studies indicated that de-escalation of ICS in COPD does not result in increased exacerbations.3 A prospective study by Rossi et al found that in a 6-month period, 141 of 482 patients on ICS therapy (29%) had an exacerbation. In the opposing arm of the study, 88 of 334 patients (26%) with deprescribed ICS experienced an exacerbation. The difference between these 2 groups was not statistically significant.4 The researchers concluded that in real-world practice, ICS withdrawal can be safe in patients at low risk of exacerbation.
About 25% of veterans (1.25 million) have been diagnosed with COPD.5 To address this, the US Department of Veterans Affairs (VA) and US Department of Defense published updated COPD guidelines in 2021 that specify criteria for de-escalation of ICS.6 Guidelines, however, may not be reflected in common clinical practice for several years following publication. The VA Academic Detailing Service (ADS) provides tools to help clinicians identify patients who may benefit from changes in treatment plans. A recent ADS focus was the implementation of a COPD dashboard, which identifies patients with COPD who are candidates for ICS de-escalation based on comorbid diagnoses, exacerbation history, and eosinophil count. VA pharmacists have an expanded role in the management of primary care disease states and are therefore well-positioned to increase adherence to guideline-directed therapy. The objective of this quality improvement project was to determine the impact of pharmacist-driven de-escalation on ICS usage in veterans with COPD.
Methods
This project was conducted in an outpatient clinic at the Robley Rex VA Medical Center beginning September 21, 2023, with a progress note in the Computerized Patient Record System (CPRS). Eligible patients were selected using the COPD Dashboard provided by ADS. The COPD Dashboard defined patients with COPD as those with ≥ 2 outpatient COPD diagnoses in the past 2 years, 1 inpatient discharge COPD diagnosis in the past year, or COPD listed as an active problem. COPD diagnoses were identified using International Statistical Classification of Disease, Tenth Revision (ICD-10) codes
Candidates identified for ICS de-escalation by the dashboard were excluded if they had a history of COPD exacerbation in the previous 2 years. The dashboard identified COPD exacerbations via ICD-10 codes for COPD or acute respiratory failure for inpatient discharges, emergency department (ED) visits, urgent care visits, and community care consults with 1 of the following terms: emergency, inpatient, hospital, urgent, ED (self). The COPD dashboard excluded patients with a diagnosis of asthma.
After patients were selected, they were screened for additional exclusion criteria. Patients were excluded if a pulmonary care practitioner managed their COPD; if identified via an active pulmonary consult in CPRS; if a non-VA clinician prescribed their ICS; or if they were being treated with roflumilast, theophylline, or chronic azithromycin. Individuals taking these 3 drugs were excluded due to potential severe and/or refractory COPD. Patients also were excluded if they: (1) had prior ICS de-escalation failure (defined as a COPD exacerbation following ICS de-escalation that resulted in ICS resumption); (2) had a COPD exacerbation requiring systemic corticosteroids or antibiotics in the previous year; (3) had active lung cancer; (4) did not have any eosinophil levels in CPRS within the previous 2 years; or (5) had any eosinophil levels ≥ 300 cells/µL in the previous year.
Each patient who met the inclusion criteria and was not excluded received a focused medication review by a pharmacist who created a templated progress note, with patient-specific recommendations, that was entered in the CPRS (eAppendix). The recommendations were also attached as an addendum to the patient’s last primary care visit note, and the primary care practitioner (PCP) was alerted via CPRS to consider ICS de-escalation and non-ICS alternatives. Tapering of ICS therapy was offered as an option to de-escalate if abrupt discontinuation was deemed inappropriate. PCPs were also prompted to consider referral to a primary care clinical pharmacy specialist for management and follow-up of ICS de-escalation.
The primary outcome was the number of patients with de-escalated ICS at 3 and 6 months following the recommendation. Secondary outcomes included the number of: patients who were no longer prescribed an ICS or who had a non-ICS alternative initiated at a pharmacist’s recommendation; patients who were referred to a primary care clinical pharmacy specialist for ICS de-escalation; COPD exacerbations requiring systemic steroids or antibiotics, or requiring an ED visit, inpatient admission, or urgent-care clinic visit; and cases of pneumonia or oral candidiasis. Primary and secondary outcomes were evaluated via chart review in CPRS. For secondary outcomes of pneumonia and COPD exacerbation, identification was made by documented diagnosis in CPRS. For continuous data such as age, the mean was calculated.
Results
Pharmacist ICS de-escalation recommendations were made between September 21, 2023, and November 19, 2023, for 106 patients. The mean age was 72 years and 99 (93%) patients were male (Table 1). Forty-one (39%) of the patients used tobacco at the time of the study. FEV1 was available for 69 patients with a mean of 63% (GOLD grade 2).1 Based on FEV1 values, 16 patients had mild COPD (GOLD grade 1), 37 patients had moderate COPD (GOLD grade 2), 14 patients had severe COPD (GOLD grade 3), and 2 patients had very severe COPD (GOLD grade 4).1 Thirty-four patients received LABA + LAMA + ICS, 65 received LABA + ICS, 2 received LAMA + ICS, and 5 received ICS monotherapy. The most common dose of ICS was a moderate dose (Table 2). Only 2 patients had an ICS AE in the previous year.
ICS de-escalation recommendations resulted in ICS de-escalation in 50 (47.2%) and 62 (58.5%) patients at 3 and 6 months, respectively. The 6-month ICS de-escalation rate by ICS dose at baseline was 72.2% (high dose), 60.0% (moderate), and 30.8% (low). De-escalation at 6 months by GOLD grade at baseline was 56.3% (9 of 16 patients, GOLD 1), 64.9% (24 of 37 patients, GOLD 2), 50% (7 of 14 patients, GOLD 3), and 50% (1 of 2 patients, GOLD 4). Six months after the ICS de-escalation recommendation appeared in the CPRS, the percentage of patients on LABA + ICS therapy dropped from 65 patients (61.3%) at baseline to 25 patients (23.6%).
Secondary outcomes were assessed at 3 and 6 months following the recommendation. Most patients with de-escalated ICS had their ICS discontinued and a non-ICS alternative initiated per pharmacist recommendations. At 6 months, 39 patients (36.8%) patients were referred to a patient aligned care team (PACT) pharmacist for de-escalation. Of the 39 patients referred to pharmacists, 69.2% (27 patients) were de-escalated; this compared to 52.2% (35 patients) who were not referred to pharmacists (Table 3).
ICS use increases the risk of pneumonia.1 At 6 months, 11 patients were diagnosed with pneumonia; 3 patients were diagnosed with pneumonia twice, resulting in a total of 14 cases. Ten cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation. One patient had a documented case of oral candidiasis that occurred while on ICS therapy; no patients with discontinued ICS were diagnosed with oral candidiasis. In addition, 10 patients had COPD exacerbations; however no patients had exacerbations both before and after de-escalation. Six patients were on ICS therapy when they experienced an exacerbation, and 4 patients had an exacerbation after ICS de-escalation.
Discussion
More than half of patients receiving the pharmacist intervention achieved the primary outcome of ICS de-escalation at 6 months. Furthermore, a larger percentage of patients referred to pharmacists for the management of ICS de-escalation successfully achieved de-escalation compared to those who were not referred. These outcomes reflect the important role pharmacists can play in identifying appropriate candidates for ICS de-escalation and assisting in the management of ICS de-escalation. Patients referred to pharmacists also received other services such as smoking cessation pharmacotherapy and counseling on inhaler technique and adherence. These interventions can support improved COPD clinical outcomes.
The purpose of de-escalating ICS therapy is to reduce the risk of AEs such as pneumonia and oral candidiasis.1 The secondary outcomes of this study support previous evidence that patients who have de-escalated ICS therapy may have reduced risk of AEs compared to those who remain on ICS therapy.3 Specifically, of the 14 cases of pneumonia that occurred during the study, 10 cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation.
ICS de-escalation may increase risk of increased COPD exacerbations.1 However, the secondary outcomes of this study do not indicate that those with de-escalated ICS had more COPD exacerbations compared to those who continued on ICS. Pharmacists’ recommendations were more effective for patients with less severe COPD based on baseline FEV1.
The previous GOLD Guidelines for COPD suggested LABA + ICS therapy as an option for patients with a high symptom and exacerbation burden (previously known as GOLD Group D). Guidelines no longer recommend LABA + ICS therapy due to the superiority of triple inhaled therapy for exacerbations and the superiority of LAMA + LABA therapy for dyspnea.7 A majority of identified patients in this project were on LABA + ICS therapy alone at baseline. The ICS de-escalation recommendation resulted in a 61.5% reduction in patients on LABA + ICS therapy at 6 months. By decreasing the number of patients on LABA + ICS without LAMA, recommendations increased the number of patients on guideline-directed therapy.
Limitations
This study lacked a control group, and the rate of ICS de-escalation in patients who did not receive a pharmacist recommendation was not assessed. Therefore, it could not be determined whether the pharmacist recommendation is more effective than no recommendation. Another limitation was our inability to access records from non-VA health care facilities. This may have resulted in missed COPD exacerbations, pneumonia, and oral candidiasis prior to or following the pharmacist recommendation.
In addition, the method used to notify PCPs of the pharmacist recommendation was a CPRS alert. Clinicians often receive multiple daily alerts and may not always pay close attention to them due to alert fatigue. Early in the study, some PCPs were unknowingly omitted from the alert of the pharmacist recommendation for 10 patients due to human error. For 8 of these 10 patients, the PCP was notified of the recommendations during the 3-month follow-up period. However, 2 patients had COPD exacerbations during the 3-month follow-up period. In these cases, the PCP was not alerted to de-escalate ICS. The data for these patients were collected at 3 and 6 months in the same manner as all other patients. Also, 7 of 35 patients who were referred to a pharmacist for ICS de-escalation did not have a scheduled appointment. These patients were considered to be lost to follow-up and this may have resulted in an underestimation of the ability of pharmacists to successfully de-escalate ICS in patients with COPD.
Other studies have evaluated the efficacy of a pharmacy-driven ICS de-escalation.8,9 Hegland et al reported ICS de-escalation for 22% of 141 eligible ambulatory patients with COPD on triple inhaled therapy following pharmacist appointments.8 A study by Hahn et al resulted in 63.8% of 58 patients with COPD being maintained off ICS following a pharmacist de-escalation initiative.9 However, these studies relied upon more time-consuming de-escalation interventions, including at least 1 phone, video, or in-person patient visit.8,9
This project used a single chart review and templated progress note to recommend ICS de-escalation and achieved similar or improved de-escalation rates compared to previous studies.8,9 Previous studies were conducted prior to the updated 2023 GOLD guidelines for COPD which no longer recommend LABA + ICS therapy. This project addressed ICS de-escalation in patients on LABA + ICS therapy in addition to those on triple inhaled therapy. Additionally, previous studies did not address rates of moderate to severe COPD exacerbation and adverse events to ICS following the pharmacist intervention.8,9
This study included COPD exacerbations and cases of pneumonia or oral candidiasis as secondary outcomes to assess the safety and efficacy of the ICS de-escalation. It appeared there were similar or lower rates of COPD exacerbations, pneumonia, and oral candidiasis in those with de-escalated ICS therapy in this study. However, these secondary outcomes are exploratory and would need to be confirmed by larger studies powered to address these outcomes.
CONCLUSIONS
Pharmacist-driven ICS de-escalation may be an effective method for reducing ICS usage in veterans as seen in this study. Additional controlled studies are required to evaluate the efficacy and safety of pharmacist-driven ICS de-escalation.
Systemic glucocorticoids play an important role in the treatment of chronic obstructive pulmonary disease (COPD) exacerbations. They are recommended to shorten recovery time and increase forced expiratory volume in 1 second (FEV1) during exacerbations.1 However, the role of the chronic use of inhaled corticosteroids (ICSs) in the treatment of COPD is less clear.
When added to inhaled β-2 agonists and muscarinic antagonists, ICSs can decrease the risk of exacerbations.1 However, not all patients with COPD benefit from ICS therapy. The degree of benefit an ICS can provide has been shown to correlate with eosinophil count—a marker of inflammation. The expected benefit of using an ICS increases as the eosinophil count increases.1 Maximum benefit can be observed with eosinophil counts ≥ 300 cells/µL, and minimal benefit is observed with eosinophil counts < 100 cells/µL. Adverse effects (AEs) of ICSs include a hoarse voice, oral candidiasis, and an increased risk of pneumonia.1 Given the risk of AEs, it is important to limit ICS use in patients who are unlikely to reap any benefits.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines suggest the use of ICSs in patients who experience exacerbations while using long-acting β agonist (LABA) plus long-acting muscarinic antagonist (LAMA) therapy and have an eosinophil count ≥ 100 cells/µL. Switching from LABA or LAMA monotherapy to triple therapy with LAMA/LABA/ICS may be considered if patients have continued exacerbations and an eosinophil count ≥ 300 cells/µL. De-escalation of ICS therapy should be considered if patients do not meet these criteria or if patients experience ICS AEs, such as pneumonia. The patients most likely to have increased exacerbations or decreased FEV1 with ICS withdrawal are those with eosinophil counts ≥ 300 cells/µL.1,2
Several studies have explored the effects of ICS de-escalation in real-world clinical settings. A systematic review of 11 studies indicated that de-escalation of ICS in COPD does not result in increased exacerbations.3 A prospective study by Rossi et al found that in a 6-month period, 141 of 482 patients on ICS therapy (29%) had an exacerbation. In the opposing arm of the study, 88 of 334 patients (26%) with deprescribed ICS experienced an exacerbation. The difference between these 2 groups was not statistically significant.4 The researchers concluded that in real-world practice, ICS withdrawal can be safe in patients at low risk of exacerbation.
About 25% of veterans (1.25 million) have been diagnosed with COPD.5 To address this, the US Department of Veterans Affairs (VA) and US Department of Defense published updated COPD guidelines in 2021 that specify criteria for de-escalation of ICS.6 Guidelines, however, may not be reflected in common clinical practice for several years following publication. The VA Academic Detailing Service (ADS) provides tools to help clinicians identify patients who may benefit from changes in treatment plans. A recent ADS focus was the implementation of a COPD dashboard, which identifies patients with COPD who are candidates for ICS de-escalation based on comorbid diagnoses, exacerbation history, and eosinophil count. VA pharmacists have an expanded role in the management of primary care disease states and are therefore well-positioned to increase adherence to guideline-directed therapy. The objective of this quality improvement project was to determine the impact of pharmacist-driven de-escalation on ICS usage in veterans with COPD.
Methods
This project was conducted in an outpatient clinic at the Robley Rex VA Medical Center beginning September 21, 2023, with a progress note in the Computerized Patient Record System (CPRS). Eligible patients were selected using the COPD Dashboard provided by ADS. The COPD Dashboard defined patients with COPD as those with ≥ 2 outpatient COPD diagnoses in the past 2 years, 1 inpatient discharge COPD diagnosis in the past year, or COPD listed as an active problem. COPD diagnoses were identified using International Statistical Classification of Disease, Tenth Revision (ICD-10) codes
Candidates identified for ICS de-escalation by the dashboard were excluded if they had a history of COPD exacerbation in the previous 2 years. The dashboard identified COPD exacerbations via ICD-10 codes for COPD or acute respiratory failure for inpatient discharges, emergency department (ED) visits, urgent care visits, and community care consults with 1 of the following terms: emergency, inpatient, hospital, urgent, ED (self). The COPD dashboard excluded patients with a diagnosis of asthma.
After patients were selected, they were screened for additional exclusion criteria. Patients were excluded if a pulmonary care practitioner managed their COPD; if identified via an active pulmonary consult in CPRS; if a non-VA clinician prescribed their ICS; or if they were being treated with roflumilast, theophylline, or chronic azithromycin. Individuals taking these 3 drugs were excluded due to potential severe and/or refractory COPD. Patients also were excluded if they: (1) had prior ICS de-escalation failure (defined as a COPD exacerbation following ICS de-escalation that resulted in ICS resumption); (2) had a COPD exacerbation requiring systemic corticosteroids or antibiotics in the previous year; (3) had active lung cancer; (4) did not have any eosinophil levels in CPRS within the previous 2 years; or (5) had any eosinophil levels ≥ 300 cells/µL in the previous year.
Each patient who met the inclusion criteria and was not excluded received a focused medication review by a pharmacist who created a templated progress note, with patient-specific recommendations, that was entered in the CPRS (eAppendix). The recommendations were also attached as an addendum to the patient’s last primary care visit note, and the primary care practitioner (PCP) was alerted via CPRS to consider ICS de-escalation and non-ICS alternatives. Tapering of ICS therapy was offered as an option to de-escalate if abrupt discontinuation was deemed inappropriate. PCPs were also prompted to consider referral to a primary care clinical pharmacy specialist for management and follow-up of ICS de-escalation.
The primary outcome was the number of patients with de-escalated ICS at 3 and 6 months following the recommendation. Secondary outcomes included the number of: patients who were no longer prescribed an ICS or who had a non-ICS alternative initiated at a pharmacist’s recommendation; patients who were referred to a primary care clinical pharmacy specialist for ICS de-escalation; COPD exacerbations requiring systemic steroids or antibiotics, or requiring an ED visit, inpatient admission, or urgent-care clinic visit; and cases of pneumonia or oral candidiasis. Primary and secondary outcomes were evaluated via chart review in CPRS. For secondary outcomes of pneumonia and COPD exacerbation, identification was made by documented diagnosis in CPRS. For continuous data such as age, the mean was calculated.
Results
Pharmacist ICS de-escalation recommendations were made between September 21, 2023, and November 19, 2023, for 106 patients. The mean age was 72 years and 99 (93%) patients were male (Table 1). Forty-one (39%) of the patients used tobacco at the time of the study. FEV1 was available for 69 patients with a mean of 63% (GOLD grade 2).1 Based on FEV1 values, 16 patients had mild COPD (GOLD grade 1), 37 patients had moderate COPD (GOLD grade 2), 14 patients had severe COPD (GOLD grade 3), and 2 patients had very severe COPD (GOLD grade 4).1 Thirty-four patients received LABA + LAMA + ICS, 65 received LABA + ICS, 2 received LAMA + ICS, and 5 received ICS monotherapy. The most common dose of ICS was a moderate dose (Table 2). Only 2 patients had an ICS AE in the previous year.
ICS de-escalation recommendations resulted in ICS de-escalation in 50 (47.2%) and 62 (58.5%) patients at 3 and 6 months, respectively. The 6-month ICS de-escalation rate by ICS dose at baseline was 72.2% (high dose), 60.0% (moderate), and 30.8% (low). De-escalation at 6 months by GOLD grade at baseline was 56.3% (9 of 16 patients, GOLD 1), 64.9% (24 of 37 patients, GOLD 2), 50% (7 of 14 patients, GOLD 3), and 50% (1 of 2 patients, GOLD 4). Six months after the ICS de-escalation recommendation appeared in the CPRS, the percentage of patients on LABA + ICS therapy dropped from 65 patients (61.3%) at baseline to 25 patients (23.6%).
Secondary outcomes were assessed at 3 and 6 months following the recommendation. Most patients with de-escalated ICS had their ICS discontinued and a non-ICS alternative initiated per pharmacist recommendations. At 6 months, 39 patients (36.8%) patients were referred to a patient aligned care team (PACT) pharmacist for de-escalation. Of the 39 patients referred to pharmacists, 69.2% (27 patients) were de-escalated; this compared to 52.2% (35 patients) who were not referred to pharmacists (Table 3).
ICS use increases the risk of pneumonia.1 At 6 months, 11 patients were diagnosed with pneumonia; 3 patients were diagnosed with pneumonia twice, resulting in a total of 14 cases. Ten cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation. One patient had a documented case of oral candidiasis that occurred while on ICS therapy; no patients with discontinued ICS were diagnosed with oral candidiasis. In addition, 10 patients had COPD exacerbations; however no patients had exacerbations both before and after de-escalation. Six patients were on ICS therapy when they experienced an exacerbation, and 4 patients had an exacerbation after ICS de-escalation.
Discussion
More than half of patients receiving the pharmacist intervention achieved the primary outcome of ICS de-escalation at 6 months. Furthermore, a larger percentage of patients referred to pharmacists for the management of ICS de-escalation successfully achieved de-escalation compared to those who were not referred. These outcomes reflect the important role pharmacists can play in identifying appropriate candidates for ICS de-escalation and assisting in the management of ICS de-escalation. Patients referred to pharmacists also received other services such as smoking cessation pharmacotherapy and counseling on inhaler technique and adherence. These interventions can support improved COPD clinical outcomes.
The purpose of de-escalating ICS therapy is to reduce the risk of AEs such as pneumonia and oral candidiasis.1 The secondary outcomes of this study support previous evidence that patients who have de-escalated ICS therapy may have reduced risk of AEs compared to those who remain on ICS therapy.3 Specifically, of the 14 cases of pneumonia that occurred during the study, 10 cases occurred while patients were on ICS and 4 cases occurred following ICS de-escalation.
ICS de-escalation may increase risk of increased COPD exacerbations.1 However, the secondary outcomes of this study do not indicate that those with de-escalated ICS had more COPD exacerbations compared to those who continued on ICS. Pharmacists’ recommendations were more effective for patients with less severe COPD based on baseline FEV1.
The previous GOLD Guidelines for COPD suggested LABA + ICS therapy as an option for patients with a high symptom and exacerbation burden (previously known as GOLD Group D). Guidelines no longer recommend LABA + ICS therapy due to the superiority of triple inhaled therapy for exacerbations and the superiority of LAMA + LABA therapy for dyspnea.7 A majority of identified patients in this project were on LABA + ICS therapy alone at baseline. The ICS de-escalation recommendation resulted in a 61.5% reduction in patients on LABA + ICS therapy at 6 months. By decreasing the number of patients on LABA + ICS without LAMA, recommendations increased the number of patients on guideline-directed therapy.
Limitations
This study lacked a control group, and the rate of ICS de-escalation in patients who did not receive a pharmacist recommendation was not assessed. Therefore, it could not be determined whether the pharmacist recommendation is more effective than no recommendation. Another limitation was our inability to access records from non-VA health care facilities. This may have resulted in missed COPD exacerbations, pneumonia, and oral candidiasis prior to or following the pharmacist recommendation.
In addition, the method used to notify PCPs of the pharmacist recommendation was a CPRS alert. Clinicians often receive multiple daily alerts and may not always pay close attention to them due to alert fatigue. Early in the study, some PCPs were unknowingly omitted from the alert of the pharmacist recommendation for 10 patients due to human error. For 8 of these 10 patients, the PCP was notified of the recommendations during the 3-month follow-up period. However, 2 patients had COPD exacerbations during the 3-month follow-up period. In these cases, the PCP was not alerted to de-escalate ICS. The data for these patients were collected at 3 and 6 months in the same manner as all other patients. Also, 7 of 35 patients who were referred to a pharmacist for ICS de-escalation did not have a scheduled appointment. These patients were considered to be lost to follow-up and this may have resulted in an underestimation of the ability of pharmacists to successfully de-escalate ICS in patients with COPD.
Other studies have evaluated the efficacy of a pharmacy-driven ICS de-escalation.8,9 Hegland et al reported ICS de-escalation for 22% of 141 eligible ambulatory patients with COPD on triple inhaled therapy following pharmacist appointments.8 A study by Hahn et al resulted in 63.8% of 58 patients with COPD being maintained off ICS following a pharmacist de-escalation initiative.9 However, these studies relied upon more time-consuming de-escalation interventions, including at least 1 phone, video, or in-person patient visit.8,9
This project used a single chart review and templated progress note to recommend ICS de-escalation and achieved similar or improved de-escalation rates compared to previous studies.8,9 Previous studies were conducted prior to the updated 2023 GOLD guidelines for COPD which no longer recommend LABA + ICS therapy. This project addressed ICS de-escalation in patients on LABA + ICS therapy in addition to those on triple inhaled therapy. Additionally, previous studies did not address rates of moderate to severe COPD exacerbation and adverse events to ICS following the pharmacist intervention.8,9
This study included COPD exacerbations and cases of pneumonia or oral candidiasis as secondary outcomes to assess the safety and efficacy of the ICS de-escalation. It appeared there were similar or lower rates of COPD exacerbations, pneumonia, and oral candidiasis in those with de-escalated ICS therapy in this study. However, these secondary outcomes are exploratory and would need to be confirmed by larger studies powered to address these outcomes.
CONCLUSIONS
Pharmacist-driven ICS de-escalation may be an effective method for reducing ICS usage in veterans as seen in this study. Additional controlled studies are required to evaluate the efficacy and safety of pharmacist-driven ICS de-escalation.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2024 Report). Accessed October 14, 2025. https://goldcopd.org/2024-gold-report/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Accessed November 14, 2025. https://goldcopd.org/2025-gold-report/
- Rogliani P, Ritondo BL, Gabriele M, et al. Optimizing de-escalation of inhaled corticosteroids in COPD: a systematic review of real-world findings. Expert Rev Clin Pharmacol. 2020;13(9):977-990. doi:10.1080/17512433.2020.1817739
- Rossi A, Guerriero M, Corrado A; OPTIMO/AIPO Study Group. Withdrawal of inhaled corticosteroids can be safe in COPD patients at low risk of exacerbation: a real-life study on the appropriateness of treatment in moderate COPD patients (OPTIMO). Respir Res. 2014;15(1):77. doi:10.1186/1465-9921-15-77
- Anderson E, Wiener RS, Resnick K, et al. Care coordination for veterans with COPD: a positive deviance study. Am J Manag Care. 2020;26(2):63-68. doi:10.37765/ajmc.2020.42394
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Chronic Obstructive Pulmonary Disease. 2021. Accessed October 14, 2025. https://www.healthquality.va.gov/guidelines/CD/copd/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2023 Report). Accessed October 14, 2025. https://goldcopd.org/wp-content/uploads/2023/03/GOLD-2023-ver-1.3-17Feb2023_WMV.pdf
- Hegland AJ, Bolduc J, Jones L, Kunisaki KM, Melzer AC. Pharmacist-driven deprescribing of inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2021;18(4):730-733. doi:10.1513/AnnalsATS.202007-871RL
- Hahn NM, Nagy MW. Implementation of a targeted inhaled corticosteroid de-escalation process in patients with chronic obstructive pulmonary disease in the primary care setting. Innov Pharm. 2022;13(1):10.24926/iip.v13i1.4349. doi:10.24926/iip.v13i1.4349
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2024 Report). Accessed October 14, 2025. https://goldcopd.org/2024-gold-report/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2025 Report). Accessed November 14, 2025. https://goldcopd.org/2025-gold-report/
- Rogliani P, Ritondo BL, Gabriele M, et al. Optimizing de-escalation of inhaled corticosteroids in COPD: a systematic review of real-world findings. Expert Rev Clin Pharmacol. 2020;13(9):977-990. doi:10.1080/17512433.2020.1817739
- Rossi A, Guerriero M, Corrado A; OPTIMO/AIPO Study Group. Withdrawal of inhaled corticosteroids can be safe in COPD patients at low risk of exacerbation: a real-life study on the appropriateness of treatment in moderate COPD patients (OPTIMO). Respir Res. 2014;15(1):77. doi:10.1186/1465-9921-15-77
- Anderson E, Wiener RS, Resnick K, et al. Care coordination for veterans with COPD: a positive deviance study. Am J Manag Care. 2020;26(2):63-68. doi:10.37765/ajmc.2020.42394
- US Department of Veterans Affairs, US Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Chronic Obstructive Pulmonary Disease. 2021. Accessed October 14, 2025. https://www.healthquality.va.gov/guidelines/CD/copd/
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2023 Report). Accessed October 14, 2025. https://goldcopd.org/wp-content/uploads/2023/03/GOLD-2023-ver-1.3-17Feb2023_WMV.pdf
- Hegland AJ, Bolduc J, Jones L, Kunisaki KM, Melzer AC. Pharmacist-driven deprescribing of inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2021;18(4):730-733. doi:10.1513/AnnalsATS.202007-871RL
- Hahn NM, Nagy MW. Implementation of a targeted inhaled corticosteroid de-escalation process in patients with chronic obstructive pulmonary disease in the primary care setting. Innov Pharm. 2022;13(1):10.24926/iip.v13i1.4349. doi:10.24926/iip.v13i1.4349
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
Evaluation of Pharmacist-Driven Inhaled Corticosteroid De-escalation in Veterans
New Drug Eases Side Effects of Weight-Loss Meds
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
A new drug currently known as NG101 reduced nausea and vomiting in patients with obesity using GLP-1s by 40% and 67%, respectively, based on data from a phase 2 trial presented at the Obesity Society’s Obesity Week 2025 in Atlanta.
Previous research published in JAMA Network Open showed a nearly 65% discontinuation rate for three GLP-1s (liraglutide, semaglutide, or tirzepatide) among adults with overweight or obesity and without type 2 diabetes. Gastrointestinal (GI) side effects topped the list of reasons for dropping the medications.
Given the impact of nausea and vomiting on discontinuation, there is an unmet need for therapies to manage GI symptoms, said Kimberley Cummings, PhD, of Neurogastrx, Inc., in her presentation.
In the new study, Cummings and colleagues randomly assigned 90 adults aged 18-55 years with overweight or obesity (defined as a BMI ranging from 22.0 to 35.0) to receive a single subcutaneous dose of semaglutide (0.5 mg) plus 5 days of NG101 at 20 mg twice daily, or a placebo.
NG101 is a peripherally acting D2 antagonist designed to reduce nausea and vomiting associated with GLP-1 use, Cummings said. NG101 targets the nausea center of the brain but is peripherally restricted to prevent central nervous system side effects, she explained.
Compared with placebo, NG101 significantly reduced the incidence of nausea and vomiting by 40% and 67%, respectively. Use of NG101 also was associated with a significant reduction in the duration of nausea and vomiting; GI events lasting longer than 1 day were reported in 22% and 51% of the NG101 patients and placebo patients, respectively.
In addition, participants who received NG101 reported a 70% decrease in nausea severity from baseline.
Overall, patients in the NG101 group also reported significantly fewer adverse events than those in the placebo group (74 vs 135), suggesting an improved safety profile when semaglutide is administered in conjunction with NG101, the researchers noted. No serious adverse events related to the study drug were reported in either group.
The findings were limited by several factors including the relatively small sample size. Additional research is needed with other GLP-1 agonists in larger populations with longer follow-up periods, Cummings said. However, the results suggest that NG101 was safe and effectively improved side effects associated with GLP-1 agonists.
“We know there are receptors for GLP-1 in the area postrema (nausea center of the brain), and that NG101 works on this area to reduce nausea and vomiting, so the study findings were not unexpected,” said Jim O’Mara, president and CEO of Neurogastrx, in an interview.
The study was a single-dose study designed to show proof of concept, and future studies would involve treating patients going through the recommended titration schedule for their GLP-1s, O’Mara said. However, NG101 offers an opportunity to keep more patients on GLP-1 therapy and help them reach their long-term therapeutic goals, he said.
Decrease Side Effects for Weight-Loss Success
“GI side effects are often the rate-limiting step in implementing an effective medication that patients want to take but may not be able to tolerate,” said Sean Wharton, MD, PharmD, medical director of the Wharton Medical Clinic for Weight and Diabetes Management, Burlington, Ontario, Canada, in an interview. “If we can decrease side effects, these medications could improve patients’ lives,” said Wharton, who was not involved in the study.
The improvement after a single dose of NG101 in patients receiving a single dose of semaglutide was impressive and in keeping with the mechanism of the drug action, said Wharton. “I was not surprised by the result but pleased that this single dose was shown to reduce the overall incidence of nausea and vomiting, the duration of nausea, the severity of nausea as rated by the study participants compared to placebo,” he said.
Ultimately, the clinical implications for NG101 are improved patient tolerance for GLP-1s and the ability to titrate and stay on them long term, incurring greater cardiometabolic benefit, Wharton told this news organization.
The current trial was limited to GLP1-1s on the market; newer medications may have fewer side effects, Wharton noted. “In clinical practice, patients often decrease the medication or titrate slower, and this could be the comparator,” he added.
The study was funded by Neurogastrx.
Wharton disclosed serving as a consultant for Neurogastrx but not as an investigator on the current study. He also reported having disclosed research on various GLP-1 medications.
A version of this article first appeared on Medscape.com.
FROM OBESITY WEEK 2025

The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
Military medical personnel rely on individual critical tasks lists (ICTLs) to maintain proficiency in essential medical skills during deployments. However, sustaining these competencies in a low-casualty operational setting presents unique challenges. Traditional training methods, such as lectures or simulations outside operational contexts, may lack engagement and fail to replicate the stressors of real-world scenarios. Previous research has emphasized the importance of continuous medical readiness training in austere environments, highlighting the need for innovative approaches.1,2
The Litter Olympics was developed as an in-theater training exercise designed to enhance medical readiness, foster interdisciplinary teamwork, and incorporate physical exertion into skill maintenance. By requiring teams to carry a patient litter through multiple “events,” the exercise reinforced teamwork within a medical readiness-focused series inspired by an Olympic decathlon. This article discusses the feasibility, effectiveness, and potential impact of the Litter Olympics as a training tool for maintaining ICTLs in a deployed environment.
Program
The Litter Olympics were implemented at a Role 3 medical facility in Baghdad, Iraq, where teams composed of individuals from military occupational specialties (MOSs) and areas of concentration (AOCs) participated. Role 3 facilities provide specialty surgical and critical care capabilities, enabling a robust medical training environment.3 The event was designed to reflect the interdisciplinary nature of deployed medical teams and incorporated hands-on training stations covering critical medical skills such as traction splinting, spinal precautions, patient movement, hemorrhage control, airway management, and tactical evacuation procedures.
Tasks were selected based on their relevance to deployed medical care and their inclusion in ICTLs, ensuring alignment with mission-essential skills. Participants were evaluated on task completion, efficiency, and teamwork by experienced medical personnel. Postexercise surveys assessed skill improvement, confidence levels, and areas for refinement. Future studies should incorporate structured performance metrics, such as pre- and postevent evaluations, to quantify proficiency gains (Table 1).
Five mixed MOS/AOC teams participated in the event, completing the exercise in an average time of 50 minutes (Table 2). Participants reported increased confidence in performing ICTs, particularly in patient movement, hemorrhage control, and airway management. The interdisciplinary nature of the teams facilitated peer teaching and cross-training, allowing individuals to better understand each other’s roles and responsibilities. This mirrors findings in previous studies on predeployment training that emphasize the importance of collaborative, hands-on learning.4 The physical aspect of the exercise was well received, as it simulated operational conditions and reinforced endurance in high-stress environments. Some tasks, such as cricothyroidotomy and satellite radio setup, required additional instruction, highlighting areas for improvement in future iterations.
Discussion
The Litter Olympics provide a dynamic alternative to traditional classroom instruction by integrating realistic, scenario-based training. However, several limitations were identified. The most significant was the lack of formalized outcome metrics. While qualitative feedback was overwhelmingly positive, no structured performance assessment tool, such as pre- and postevent skill evaluations, was used. Future studies should incorporate objective measures of competency to strengthen the evidence base for this training model. Additionally, participant feedback suggested that more structured debriefing sessions postexercise would enhance learning retention and provide actionable insights for future program modifications.
Another consideration is the scalability and adaptability of the exercise. While effective in a Role 3 setting, modifications may be required for smaller units or lower levels of care. Future iterations could adapt the format for Role 1 or 2 environments by reducing the number of stations while preserving the core training elements. Furthermore, the event relied on access to specialized personnel and equipment, which may not always be feasible in austere settings. Developing a streamlined version focusing on essential tasks could improve accessibility and sustainability across different operational environments.
Participants expressed a preference for this hands-on, competitive training model over traditional didactic instruction. However, further research should compare skill retention rates between the Litter Olympics and other training modalities to validate effectiveness. While peer teaching was a notable strength of the event, structured mentorship from senior medical personnel could further enhance skill acquisition and reinforce best practices.
Conclusions
The Litter Olympics present a reproducible, engaging, and effective method for sustaining medical readiness in a deployed Role 3 setting. By fostering interdisciplinary collaboration and incorporating physical and cognitive stressors, it enhances both individual and team preparedness. Future research should develop standardized, measurable outcome assessments, explore application in diverse deployment settings, and optimize scalability for broader military medical training programs. Standardized evaluation tools should be developed to quantify performance improvements, and the training model should be expanded to include lower levels of care and nonmedical personnel. Structured debriefing sessions would also provide valuable insight into lessons learned and potential refinements. By integrating these enhancements, the Litter Olympics can serve as a cornerstone for maintaining operational medical readiness in deployed environments.
- Suresh MR, Valdez-Delgado KK, Staudt AM, et al. An assessment of pre-deployment training for army nurses and medics. Mil Med. 2021;186:203-211. doi:10.1093/milmed/usaa291
- Mead KC, Tennent DJ, Stinner DJ. The importance of medical readiness training exercises: maintaining medical readiness in a low-volume combat casualty flow era. Mil Med. 2017;182:e1734-e1737. doi:10.7205/milmed-d-16-00335
- Brisebois R, Hennecke P, Kao R, et al. The Role 3 multinational medical nit at Kandahar airfield 2005–2010. Can J Surg. 2011;54:S124-S129. doi:10.1503/cjs.024811
- Huh J, Brockmeyer JR, Bertsch SR, et al. Conducting pre-deployment training in Honduras: the 240th forward resuscitative surgical team experience. Mil Med. 2021;187:e690-e695. doi:10.1093/milmed/usaa545
Military medical personnel rely on individual critical tasks lists (ICTLs) to maintain proficiency in essential medical skills during deployments. However, sustaining these competencies in a low-casualty operational setting presents unique challenges. Traditional training methods, such as lectures or simulations outside operational contexts, may lack engagement and fail to replicate the stressors of real-world scenarios. Previous research has emphasized the importance of continuous medical readiness training in austere environments, highlighting the need for innovative approaches.1,2
The Litter Olympics was developed as an in-theater training exercise designed to enhance medical readiness, foster interdisciplinary teamwork, and incorporate physical exertion into skill maintenance. By requiring teams to carry a patient litter through multiple “events,” the exercise reinforced teamwork within a medical readiness-focused series inspired by an Olympic decathlon. This article discusses the feasibility, effectiveness, and potential impact of the Litter Olympics as a training tool for maintaining ICTLs in a deployed environment.
Program
The Litter Olympics were implemented at a Role 3 medical facility in Baghdad, Iraq, where teams composed of individuals from military occupational specialties (MOSs) and areas of concentration (AOCs) participated. Role 3 facilities provide specialty surgical and critical care capabilities, enabling a robust medical training environment.3 The event was designed to reflect the interdisciplinary nature of deployed medical teams and incorporated hands-on training stations covering critical medical skills such as traction splinting, spinal precautions, patient movement, hemorrhage control, airway management, and tactical evacuation procedures.
Tasks were selected based on their relevance to deployed medical care and their inclusion in ICTLs, ensuring alignment with mission-essential skills. Participants were evaluated on task completion, efficiency, and teamwork by experienced medical personnel. Postexercise surveys assessed skill improvement, confidence levels, and areas for refinement. Future studies should incorporate structured performance metrics, such as pre- and postevent evaluations, to quantify proficiency gains (Table 1).
Five mixed MOS/AOC teams participated in the event, completing the exercise in an average time of 50 minutes (Table 2). Participants reported increased confidence in performing ICTs, particularly in patient movement, hemorrhage control, and airway management. The interdisciplinary nature of the teams facilitated peer teaching and cross-training, allowing individuals to better understand each other’s roles and responsibilities. This mirrors findings in previous studies on predeployment training that emphasize the importance of collaborative, hands-on learning.4 The physical aspect of the exercise was well received, as it simulated operational conditions and reinforced endurance in high-stress environments. Some tasks, such as cricothyroidotomy and satellite radio setup, required additional instruction, highlighting areas for improvement in future iterations.
Discussion
The Litter Olympics provide a dynamic alternative to traditional classroom instruction by integrating realistic, scenario-based training. However, several limitations were identified. The most significant was the lack of formalized outcome metrics. While qualitative feedback was overwhelmingly positive, no structured performance assessment tool, such as pre- and postevent skill evaluations, was used. Future studies should incorporate objective measures of competency to strengthen the evidence base for this training model. Additionally, participant feedback suggested that more structured debriefing sessions postexercise would enhance learning retention and provide actionable insights for future program modifications.
Another consideration is the scalability and adaptability of the exercise. While effective in a Role 3 setting, modifications may be required for smaller units or lower levels of care. Future iterations could adapt the format for Role 1 or 2 environments by reducing the number of stations while preserving the core training elements. Furthermore, the event relied on access to specialized personnel and equipment, which may not always be feasible in austere settings. Developing a streamlined version focusing on essential tasks could improve accessibility and sustainability across different operational environments.
Participants expressed a preference for this hands-on, competitive training model over traditional didactic instruction. However, further research should compare skill retention rates between the Litter Olympics and other training modalities to validate effectiveness. While peer teaching was a notable strength of the event, structured mentorship from senior medical personnel could further enhance skill acquisition and reinforce best practices.
Conclusions
The Litter Olympics present a reproducible, engaging, and effective method for sustaining medical readiness in a deployed Role 3 setting. By fostering interdisciplinary collaboration and incorporating physical and cognitive stressors, it enhances both individual and team preparedness. Future research should develop standardized, measurable outcome assessments, explore application in diverse deployment settings, and optimize scalability for broader military medical training programs. Standardized evaluation tools should be developed to quantify performance improvements, and the training model should be expanded to include lower levels of care and nonmedical personnel. Structured debriefing sessions would also provide valuable insight into lessons learned and potential refinements. By integrating these enhancements, the Litter Olympics can serve as a cornerstone for maintaining operational medical readiness in deployed environments.
Military medical personnel rely on individual critical tasks lists (ICTLs) to maintain proficiency in essential medical skills during deployments. However, sustaining these competencies in a low-casualty operational setting presents unique challenges. Traditional training methods, such as lectures or simulations outside operational contexts, may lack engagement and fail to replicate the stressors of real-world scenarios. Previous research has emphasized the importance of continuous medical readiness training in austere environments, highlighting the need for innovative approaches.1,2
The Litter Olympics was developed as an in-theater training exercise designed to enhance medical readiness, foster interdisciplinary teamwork, and incorporate physical exertion into skill maintenance. By requiring teams to carry a patient litter through multiple “events,” the exercise reinforced teamwork within a medical readiness-focused series inspired by an Olympic decathlon. This article discusses the feasibility, effectiveness, and potential impact of the Litter Olympics as a training tool for maintaining ICTLs in a deployed environment.
Program
The Litter Olympics were implemented at a Role 3 medical facility in Baghdad, Iraq, where teams composed of individuals from military occupational specialties (MOSs) and areas of concentration (AOCs) participated. Role 3 facilities provide specialty surgical and critical care capabilities, enabling a robust medical training environment.3 The event was designed to reflect the interdisciplinary nature of deployed medical teams and incorporated hands-on training stations covering critical medical skills such as traction splinting, spinal precautions, patient movement, hemorrhage control, airway management, and tactical evacuation procedures.
Tasks were selected based on their relevance to deployed medical care and their inclusion in ICTLs, ensuring alignment with mission-essential skills. Participants were evaluated on task completion, efficiency, and teamwork by experienced medical personnel. Postexercise surveys assessed skill improvement, confidence levels, and areas for refinement. Future studies should incorporate structured performance metrics, such as pre- and postevent evaluations, to quantify proficiency gains (Table 1).
Five mixed MOS/AOC teams participated in the event, completing the exercise in an average time of 50 minutes (Table 2). Participants reported increased confidence in performing ICTs, particularly in patient movement, hemorrhage control, and airway management. The interdisciplinary nature of the teams facilitated peer teaching and cross-training, allowing individuals to better understand each other’s roles and responsibilities. This mirrors findings in previous studies on predeployment training that emphasize the importance of collaborative, hands-on learning.4 The physical aspect of the exercise was well received, as it simulated operational conditions and reinforced endurance in high-stress environments. Some tasks, such as cricothyroidotomy and satellite radio setup, required additional instruction, highlighting areas for improvement in future iterations.
Discussion
The Litter Olympics provide a dynamic alternative to traditional classroom instruction by integrating realistic, scenario-based training. However, several limitations were identified. The most significant was the lack of formalized outcome metrics. While qualitative feedback was overwhelmingly positive, no structured performance assessment tool, such as pre- and postevent skill evaluations, was used. Future studies should incorporate objective measures of competency to strengthen the evidence base for this training model. Additionally, participant feedback suggested that more structured debriefing sessions postexercise would enhance learning retention and provide actionable insights for future program modifications.
Another consideration is the scalability and adaptability of the exercise. While effective in a Role 3 setting, modifications may be required for smaller units or lower levels of care. Future iterations could adapt the format for Role 1 or 2 environments by reducing the number of stations while preserving the core training elements. Furthermore, the event relied on access to specialized personnel and equipment, which may not always be feasible in austere settings. Developing a streamlined version focusing on essential tasks could improve accessibility and sustainability across different operational environments.
Participants expressed a preference for this hands-on, competitive training model over traditional didactic instruction. However, further research should compare skill retention rates between the Litter Olympics and other training modalities to validate effectiveness. While peer teaching was a notable strength of the event, structured mentorship from senior medical personnel could further enhance skill acquisition and reinforce best practices.
Conclusions
The Litter Olympics present a reproducible, engaging, and effective method for sustaining medical readiness in a deployed Role 3 setting. By fostering interdisciplinary collaboration and incorporating physical and cognitive stressors, it enhances both individual and team preparedness. Future research should develop standardized, measurable outcome assessments, explore application in diverse deployment settings, and optimize scalability for broader military medical training programs. Standardized evaluation tools should be developed to quantify performance improvements, and the training model should be expanded to include lower levels of care and nonmedical personnel. Structured debriefing sessions would also provide valuable insight into lessons learned and potential refinements. By integrating these enhancements, the Litter Olympics can serve as a cornerstone for maintaining operational medical readiness in deployed environments.
- Suresh MR, Valdez-Delgado KK, Staudt AM, et al. An assessment of pre-deployment training for army nurses and medics. Mil Med. 2021;186:203-211. doi:10.1093/milmed/usaa291
- Mead KC, Tennent DJ, Stinner DJ. The importance of medical readiness training exercises: maintaining medical readiness in a low-volume combat casualty flow era. Mil Med. 2017;182:e1734-e1737. doi:10.7205/milmed-d-16-00335
- Brisebois R, Hennecke P, Kao R, et al. The Role 3 multinational medical nit at Kandahar airfield 2005–2010. Can J Surg. 2011;54:S124-S129. doi:10.1503/cjs.024811
- Huh J, Brockmeyer JR, Bertsch SR, et al. Conducting pre-deployment training in Honduras: the 240th forward resuscitative surgical team experience. Mil Med. 2021;187:e690-e695. doi:10.1093/milmed/usaa545
- Suresh MR, Valdez-Delgado KK, Staudt AM, et al. An assessment of pre-deployment training for army nurses and medics. Mil Med. 2021;186:203-211. doi:10.1093/milmed/usaa291
- Mead KC, Tennent DJ, Stinner DJ. The importance of medical readiness training exercises: maintaining medical readiness in a low-volume combat casualty flow era. Mil Med. 2017;182:e1734-e1737. doi:10.7205/milmed-d-16-00335
- Brisebois R, Hennecke P, Kao R, et al. The Role 3 multinational medical nit at Kandahar airfield 2005–2010. Can J Surg. 2011;54:S124-S129. doi:10.1503/cjs.024811
- Huh J, Brockmeyer JR, Bertsch SR, et al. Conducting pre-deployment training in Honduras: the 240th forward resuscitative surgical team experience. Mil Med. 2021;187:e690-e695. doi:10.1093/milmed/usaa545
The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
The Litter Olympics: Addressing Individual Critical Tasks Lists Requirements in a Forward-Deployed Setting
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
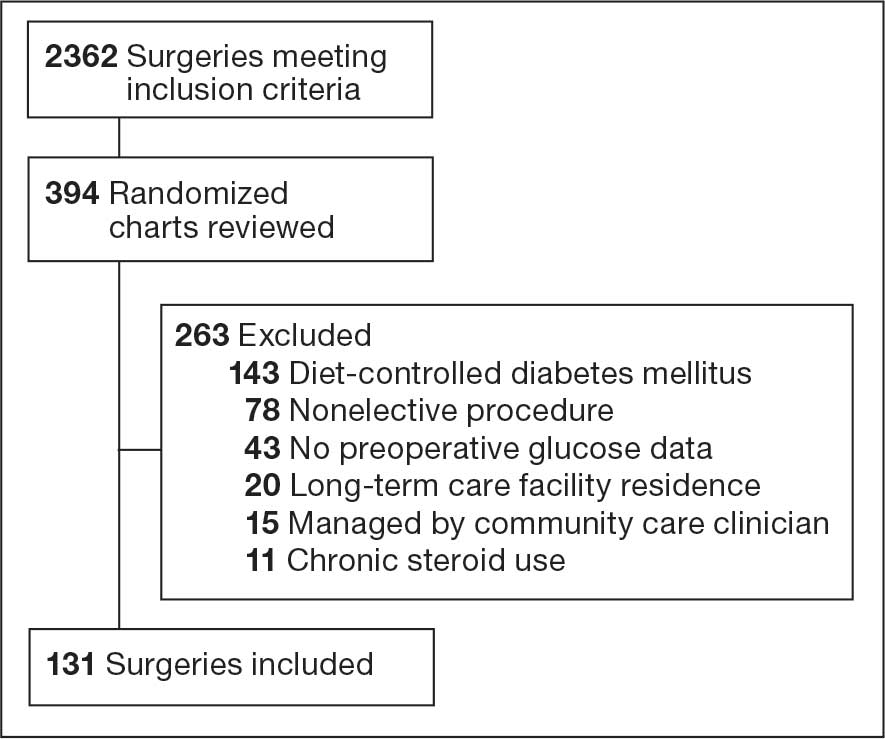
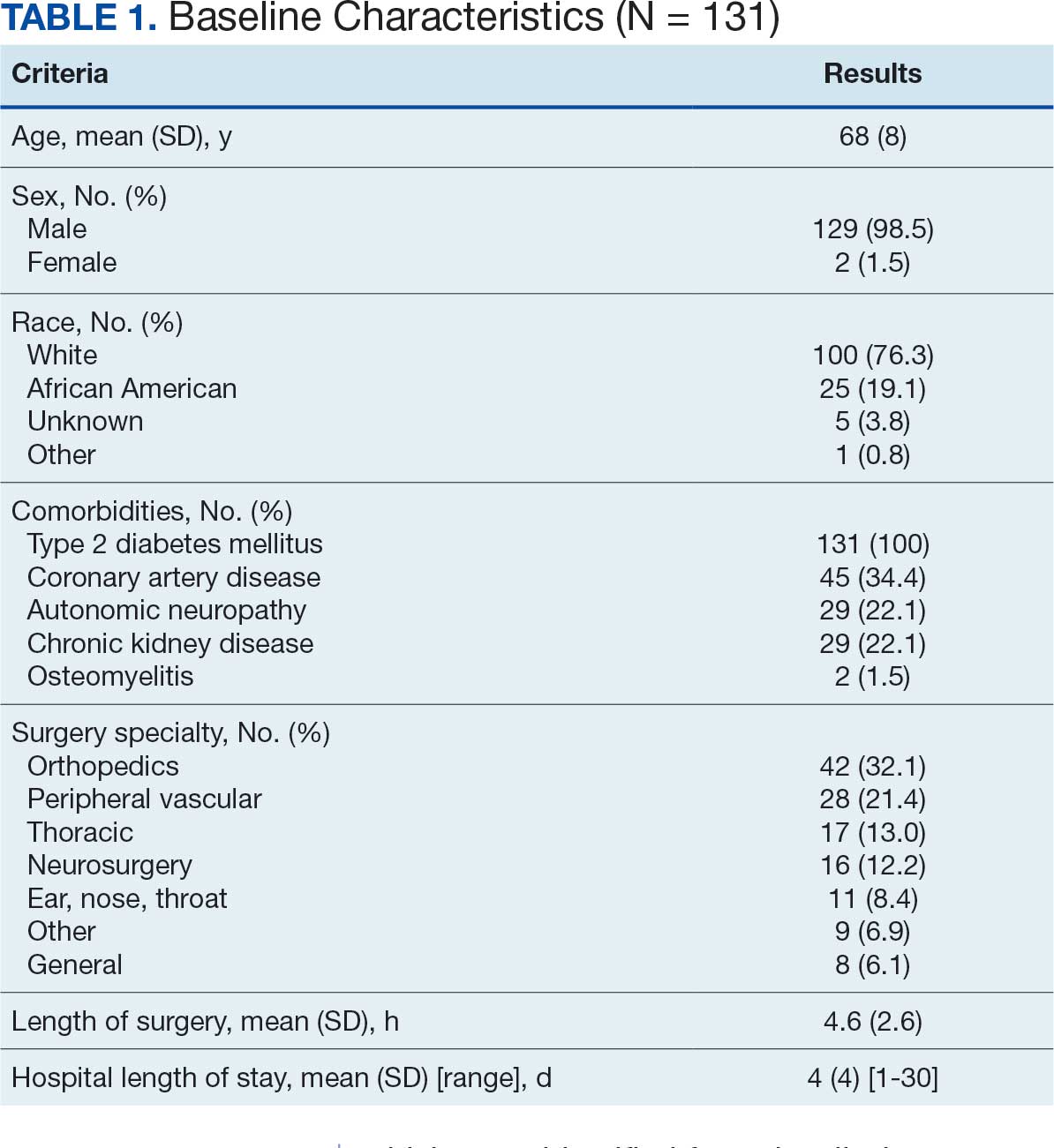
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
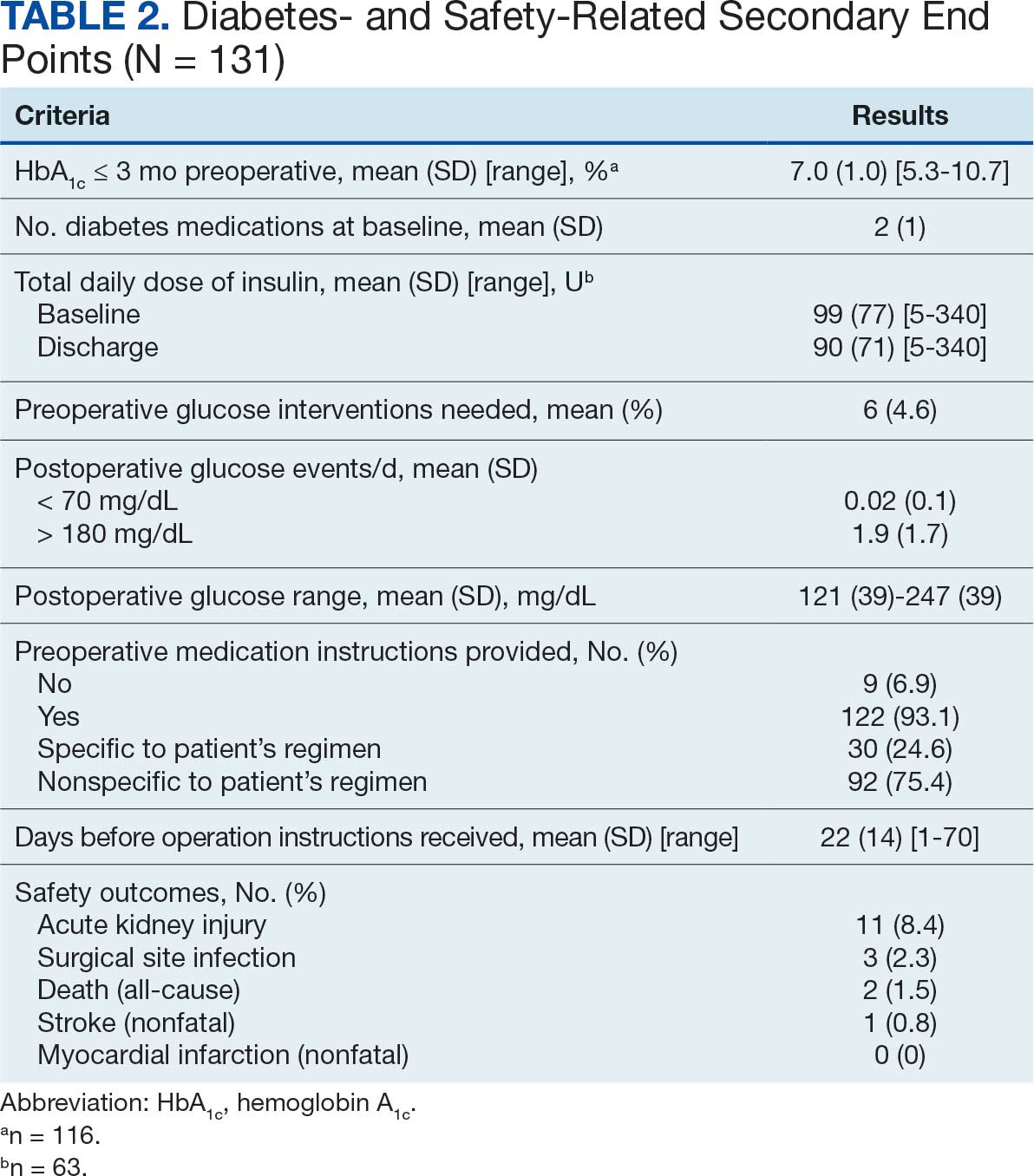
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
More than 38 million people in the United States (12%) have diabetes mellitus (DM), though 1 in 5 are unaware they have DM.1 The prevalence among veterans is even more substantial, impacting nearly 25% of those who received care from the US Department of Veterans Affairs (VA).2 DM can lead to increased health care costs in addition to various complications (eg, cardiovascular, renal), especially if left uncontrolled.1,3 similar impact is found in the perioperative period (defined as at or around the time of an operation), as multiple studies have found that uncontrolled preoperative DM can result in worsened surgical outcomes, including longer hospital stays, more infectious complications, and higher perioperative mortality.4-6
In contrast, adequate glycemic control assessed with blood glucose levels has been shown to decrease the incidence of postoperative infections.7 Optimizing glycemic control during hospital stays, especially postsurgery, has become the standard of care, with most health systems establishing specific protocols. In current literature, most studies examining DM management in the perioperative period are focused on postoperative care, with little attention to the preoperative period.4,6,7
One study found that patients with poor presurgery glycemic control assessed by hemoglobin A1c (HbA1c) levels were more likely to remain hyperglycemic during and after surgery. 8 Blood glucose levels < 200 mg/dL can lead to an increased risk of infection and impaired wound healing, meaning a well-controlled HbA1c before a procedure serves as a potential factor for success.9 The 2025 American Diabetes Association (ADA) Standards of Care (SOC) recommendation is to target HbA1c < 8% whenever possible, and some health systems require lower levels (eg, < 7% or 7.5%).10 With that goal in mind and knowing that preoperative hyperglycemia has been shown to be a contributing factor in the delay or cancellation of surgical cases, an argument can be made that attention to preoperative DM management also should be a focus for health care systems performing surgeries.8,9,11
Attention to glucose control during preoperative care offers an opportunity to screen for DM in patients who may not have been screened otherwise and to standardize perioperative DM management. Since DM disproportionately impacts veterans, this is a pertinent issue to the VA. Veterans can be more susceptible to complications if DM is left uncontrolled prior to surgery. To determine readiness for surgery and control of comorbid conditions such as DM before a planned surgery, facilities often perform a preoperative clinic assessment, often in a multidisciplinary clinic.
At Veteran Health Indiana (VHI), a presurgery clinic visit involving the primary surgery service (physician, nurse practitioner, and/or a physician assistant) is conducted 1 to 2 months prior to the planned procedure to determine whether a patient is ready for surgery. During this visit, patients receive a packet with instructions for various tasks and medications, such as applying topical antibiotic prophylaxis on the anticipated surgical site. This is documented in the form of a note in the VHI Computerized Patient Record System (CPRS). The medication instructions are provided according to the preferences of the surgical team. These may be templated notes that contain general directions on the timing and dosing of specific medications, in addition to instructions for holding or reducing doses when appropriate. The instructions can be tailored by the team conducting the preoperative visit (eg, “Take 20 units of insulin glargine the day before surgery” vs “Take half of your long-acting insulin the night before surgery”). Specific to DM, VHI has a nurse-driven day of surgery glucose assessment where point-of-care blood glucose is collected during preoperative holding for most patients.
There is limited research assessing the level of preoperative glycemic control and the incidence of complications in a veteran population. The objective of this study was to gain a baseline understanding of what, if any, standardization exists for preoperative instructions for DM medications and to assess the level of preoperative glycemic control and postoperative complications in patients with DM undergoing major elective surgical procedures.
Methods
This retrospective, single-center chart review was conducted at VHI. The Indiana University and VHI institutional review boards determined that this quality improvement project was exempt from review.
The primary outcome was the number of patients with surgical procedures delayed or canceled due to hyperglycemia or hypoglycemia. Hyperglycemia was defined as blood glucose > 180 mg/dL and hypoglycemia was defined as < 70 mg/dL, slight variations from the current ADA SOC preoperative specific recommendation of a blood glucose reading of 100 to 180 mg/dL within 4 hours of surgery.10 The standard outpatient hypoglycemia definition of blood glucose < 70 mg/dL was chosen because the current goal (< 100 mg/dL) was not the standard in previous ADA SOCs that were in place during the study period. Specifically, the 2018 ADA SOC did not provide preoperative recommendations and the 2019-2021 ADA SOC recommended 80 to 180 mg/dL.10,12-18 For patients who had multiple preoperative blood glucose measurements, the first recorded glucose on the day of the procedure was used.
The secondary outcomes of this study were focused on the preoperative process/care at VHI and postoperative glycemic control. The preoperative process included examining whether medication instructions were given and their quality. Additionally, the number of interventions for hyperglycemia and hypoglycemia were required immediately prior to surgery and the average preoperative HbA1c (measured within 3 months prior to surgery) were collected and analyzed. For postoperative glycemic control, average blood glucose measurements and number of hypoglycemic (< 70 mg/dL) and hyperglycemic (> 180 mg/dL) events were measured in addition to the frequency of changes made at discharge to patients’ DM medication regimens.
The safety outcome of this study assessed commonly observed postoperative complications and was examined up to 30 days postsurgery. These included acute kidney injury (defined using Kidney Disease: Improving Global Outcomes 2012, the standard during the study period), nonfatal myocardial infarction, nonfatal stroke, and surgical site infections, which were identified from the discharge summary written by the primary surgery service.19 All-cause mortality also was collected.
Patients were included if they were admitted for major elective surgeries and had a diagnosis of either type 1 or type 2 DM on their problem list, determined by International Classification of Diseases, Tenth Revision codes. Major elective surgery was defined as a procedure that would likely result in a hospital admission of > 24 hours. Of note, patients may have been included in this study more than once if they had > 1 procedure at least 30 days apart and met inclusion criteria within the time frame. Patients were excluded if they were taking no DM medications or chronic steroids (at any dose), residing in a long-term care facility, being managed by a non-VA clinician prior to surgery, or missing a preoperative blood glucose measurement.
All data were collected from the CPRS. A list of surgical cases involving patients with DM who were scheduled to undergo major elective surgeries from January 1, 2018, to December 31, 2021, at VHI was generated. The list was randomized to a smaller number (N = 394) for data collection due to the time and resource constraints for a pharmacy residency project. All data were deidentified and stored in a secured VA server to protect patient confidentiality. Descriptive statistics were used for all results.
Results
Initially, 2362 surgeries were identified. A randomized sample of 394 charts were reviewed and 131 cases met inclusion criteria. Each case involved a unique patient (Figure). The most common reasons for exclusion were 143 patients with diet-controlled DM and 78 nonelective surgeries. The mean (SD) age of patients was 68 (8) years, and the most were male (98.5%) and White (76.3%) (Table 1).
At baseline, 45 of 131 patients (34.4%) had coronary artery disease and 29 (22.1%) each had autonomic neuropathy and chronic kidney disease. Most surgeries were conducted by orthopedic (32.1%) and peripheral vascular (21.4%) specialties. The mean (SD) length of surgery was 4.6 (2.6) hours and of hospital length of stay was 4 (4) days. No patients stayed longer than the 30-day safety outcome follow-up period. All patients had type 2 DM and took a mean 2 DM medications. The 63 patients taking insulin had a mean (SD) total daily dose of 99 (77) U (Table 2). A preoperative HbA1c was collected in 116 patients within 3 months of surgery, with a mean HbA1c of 7.0% (range, 5.3-10.7).
No patients had surgeries delayed or canceled because of uncontrolled DM on the day of surgery. The mean preoperative blood glucose level was 146 mg/dL (range, 73-365) (Table 3). No patients had a preoperative blood glucose level of < 70 mg/dL and 19 (14.5%) had a blood glucose level > 180 mg/dL. Among patients with hyperglycemia immediately prior to surgery, 6 (31.6%) had documentation of insulin being provided.
For this sample of patients, the preoperative clinic visit was conducted a mean 22 days prior to the planned surgery date. Among the 131 included patients, 122 (93.1%) had documentation of receiving instructions for DM medications. Among patients who had documented receipt of instructions, only 30 (24.6%) had instructions specifically tailored to their regimen rather than a generic templated form. The mean (SD) preoperative blood glucose was similar for those who received specific perioperative DM instructions at 146 (50) mg/dL when compared with those who did not at 147 (45) mg/dL. The mean (SD) preoperative blood glucose reading for those who had no documentation of receipt of perioperative instructions was 126 (54) mg/dL compared with 147 (46) mg/dL for those who did.
The mean number of postoperative blood glucose events per day was negligible for hypoglycemia and more frequent for hyperglycemia with a mean of 2 events per day. The mean postoperative blood glucose range was 121 to 247 mg/dL with most readings < 180 mg/dL. Upon discharge, most patients continued their home DM regimen with 5 patients (3.8%) having changes made to their regimen upon discharge.
Very few postoperative complications were identified from chart review. The most frequently observed postoperative complications were acute kidney injury, surgical site infections, and nonfatal stroke. There were no documented nonfatal myocardial infarctions. Two patients (1.5%) died within 30 days of the surgery; neither death was deemed to have been related to poor perioperative glycemic control.
Discussion
To our knowledge, this retrospective chart review was the first study to assess preoperative DM management and postoperative complications in a veteran population. VHI is a large, tertiary, level 1a, academic medical center that serves approximately 62,000 veterans annually and performs about 5000 to 6000 surgeries annually, a total that is increasing following the COVID-19 pandemic.20 This study found that the current process of a presurgery clinic visit and day of surgery glucose assessment has prevented surgical delays or cancellations.
Most patients included in this study were well controlled at baseline in accordance with the 2025 ADA SOC HbA1c recommendation of a preoperative HbA1c of < 8%, which may have contributed to no surgical delays or cancellations.10 However, not all patients had HbA1c collected within 3 months of surgery or even had one collected at all. Despite the ADA SOC providing no explicit recommendation for universal HbA1c screening prior to elective procedures, its importance cannot be understated given the body of evidence demonstrating poor outcomes with uncontrolled preoperative DM.8,10 The glycemic control at baseline may have contributed to the very few postsurgical complications observed in this study.
Although the current process at VHI prevented surgical delays and cancellations in this sample, there are still identified areas for improvement. One area is the instructions the patients received. Patients with DM are often prescribed ≥ 1 medication or a combination of insulins, noninsulin injectables, and oral DM medications, and this study population was no different. Because these medications may influence the anesthesia and perioperative periods, the ADA has specific guidance for altering administration schedules in the days leading up to surgery.10
Inappropriate administration of DM medications could lead to perioperative hypoglycemia or hyperglycemia, possibly causing surgical delays, case cancellations, and/or postoperative complications.21 Although these data reveal the specificity and documented receipt that the preoperative DM instructions did not impact the first recorded preoperative blood glucose, future studies should examine patient confidence in how to properly administer their DM medications prior to surgery. It is vital that patients receive clear instructions in accordance with the ADA SOC on whether to continue, hold, or adjust the dose of their medications to prevent fluctuations in blood glucose levels in the perioperative period, ensure safety with anesthesia, and prevent postoperative complications such as acute kidney injury. Of note, compliance with guideline recommendations for medication instructions was not examined because the data collection time frame expanded over multiple years and the recommendations have evolved each year as new data emerge.
Preoperative DM Management
The first key takeaway from this study is to ensure patients are ready for surgery with a formal assessment (typically in the form of a clinic visit) prior to the surgery. One private sector health system published their approach to this by administering an automatic preoperative HbA1c screening for those with a DM diagnosis and all patients with a random plasma glucose ≥ 200 mg/dL.22 Additionally, if the patient's HbA1c level was not at goal prior to surgery (≥ 8% for those with known DM and ≥ 6.5% with no known DM), patients were referred to endocrinology for further management. Increasing attention to the preoperative visit and extending HbA1c testing to all patients regardless of DM status also provides an opportunity to identify individuals living with undiagnosed DM.1
Even though there was no difference in the mean preoperative blood glucose level based on receipt or specificity of preoperative DM instructions, a second takeaway from this study is the importance of ensuring patients receive clear instructions on their DM medication schedule in the perioperative period. A practical first step may be updating the templates used by the primary surgery teams and providing education to the clinicians in the clinic on how to personalize the visits. Because the current preoperative DM process at VHI is managed by the primary surgical team in a clinic visit, there is an opportunity to shift this responsibility to other health care professionals, such as pharmacists—a change shown to reduce unintended omission of home medications following surgery during hospitalization and reduce costs.23,24
Limitations
This study relied on data included in the patient chart. These data include medication interventions made immediately prior to surgery, which can sometimes be inaccurately charted or difficult to find as they are not documented in the typical medication administration record. Also, the safety outcomes were collected from a discharge summary written by different clinicians, which may lead to information bias. Special attention was taken to ensure these data points were collected as accurately as possible, but it is possible some data may be inaccurate from unintentional human error. Additionally, the safety outcome was limited to a 30-day follow-up, but encompassed the entire length of postoperative stay for all included patients. Finally, given this study was retrospective with no comparison group and the intent was to improve processes at VHI, only hypotheses and potential interventions can be generated from this study. Future prospective studies with larger sample sizes and comparator groups are needed to draw further conclusions.
Conclusions
This study found that the current presurgery process at VHI appears to be successful in preventing surgical delays or cancellations due to hyperglycemia or hypoglycemia. Optimizing DM management can improve surgical outcomes by decreasing rates of postoperative complications, and this study added additional evidence in support of that in a unique population: veterans. Insight on the awareness of preoperative blood glucose management should be gleaned from this study, and based on this sample and site, the preadmission screening process and instructions provided to patients can serve as 2 starting points for optimizing elective surgery.
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
- Centers for Disease Control and Prevention. Diabetes basics. May 15, 2024. Accessed September 24, 2025. https://www.cdc.gov/diabetes/about/index.html
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- Farmaki P, Damaskos C, Garmpis N, et al . Complications of the Type 2 Diabetes Mellitus. Curr Cardiol Rev. 2020;16(4):249-251. doi:10.2174/1573403X1604201229115531
- Frisch A, Chandra P, Smiley D, et al. Prevalence and clinical outcome of hyperglycemia in the perioperative period in noncardiac surgery. Diabetes Care. 2010;33:1783-1788. doi:10.2337/dc10-0304
- Noordzij PG, Boersma E, Schreiner F, et al. Increased preoperative glucose levels are associated with perioperative mortality in patients undergoing noncardiac, nonvascular surgery. Eur J Endocrinol. 2007;156:137 -142. doi:10.1530/eje.1.02321
- Pomposelli JJ, Baxter JK 3rd, Babineau TJ, et al. Early postoperative glucose control predicts nosocomial infection rate in diabetic patients. JPEN J Parenter Enteral Nutr. 1998;22:77-81. doi:10.1177/01486071980220027
- Umpierrez GE, Smiley D, Jacobs S, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes undergoing general surgery (RABBIT 2 surgery). Diabetes Care. 2011;34:256-261. doi:10.2337/dc10-1407
- Pasquel FJ, Gomez-Huelgas R, Anzola I, et al. Predictive value of admission hemoglobin A1c on inpatient glycemic control and response to insulin therapy in medicine and surgery patients with type 2 diabetes. Diabetes Care. 2015;38:e202-e203. doi:10.2337/dc15-1835
- Alexiewicz JM, Kumar D, Smogorzewski M, et al. Polymorphonuclear leukocytes in non-insulin-dependent diabetes mellitus: abnormalities in metabolism and function. Ann Intern Med. 1995;123:919-924. doi:10.7326/0003-4819-123-12-199512150-00004
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2025. Diabetes Care. 2025;48(1 suppl 1):S321-S334. doi:10.2337/dc25-S016
- Kumar R, Gandhi R. Reasons for cancellation of operation on the day of intended surgery in a multidisciplinary 500 bedded hospital. J Anaesthesiol Clin Pharmacol. 2012;28:66-69. doi:10.4103/0970-9185.92442
- American Diabetes Association. 14. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2018. Diabetes Care. 2018;41(1 suppl 1):S144- S151. doi:10.2337/dc18-S014
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2019. Diabetes Care. 2019;42(suppl 1):S173- S181. doi:10.2337/dc19-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2020. Diabetes Care. 2020;43(suppl 1):S193- S202. doi:10.2337/dc20-S015
- American Diabetes Association. 15. Diabetes care in the hospital: Standards of Medical Care in Diabetes— 2021. Diabetes Care. 2021;44(suppl 1):S211- S220. doi:10.2337/dc21-S015
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2022;45(suppl 1):S244-S253. doi:10.2337/dc22-S016
- ElSayed NA, Aleppo G, Aroda VR, et al. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2023. Diabetes Care. 2023;46(suppl 1):S267-S278. doi:10.2337/dc23-S016
- American Diabetes Association Professional Practice Committee. 16. Diabetes care in the hospital: Standards of Care in Diabetes—2024. Diabetes Care. 2024;47(suppl 1):S295-S306. doi:10.2337/dc24-S016
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138. Accessed September 24, 2025. https:// www.kisupplements.org/issue/S2157-1716(12)X7200-9
- US Department of Veterans Affairs. VA Indiana Healthcare: about us. Accessed September 24, 2025. https:// www.va.gov/indiana-health-care/about-us/
- Koh WX, Phelan R, Hopman WM, et al. Cancellation of elective surgery: rates, reasons and effect on patient satisfaction. Can J Surg. 2021;64:E155-E161. doi:10.1503/cjs.008119
- Pai S-L, Haehn DA, Pitruzzello NE, et al. Reducing infection rates with enhanced preoperative diabetes mellitus diagnosis and optimization processes. South Med J. 2023;116:215-219. doi:10.14423/SMJ.0000000000001507
- Forrester TG, Sullivan S, Snoswell CL, et al. Integrating a pharmacist into the perioperative setting. Aust Health Rev. 2020;44:563-568. doi:10.1071/AH19126
- Hale AR, Coombes ID, Stokes J, et al. Perioperative medication management: expanding the role of the preadmission clinic pharmacist in a single centre, randomised controlled trial of collaborative prescribing. BMJ Open. 2013;3:e003027. doi:10.1136/bmjopen-2013-003027
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Preoperative Diabetes Management for Patients Undergoing Elective Surgeries at a Veterans Affairs Medical Center
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).

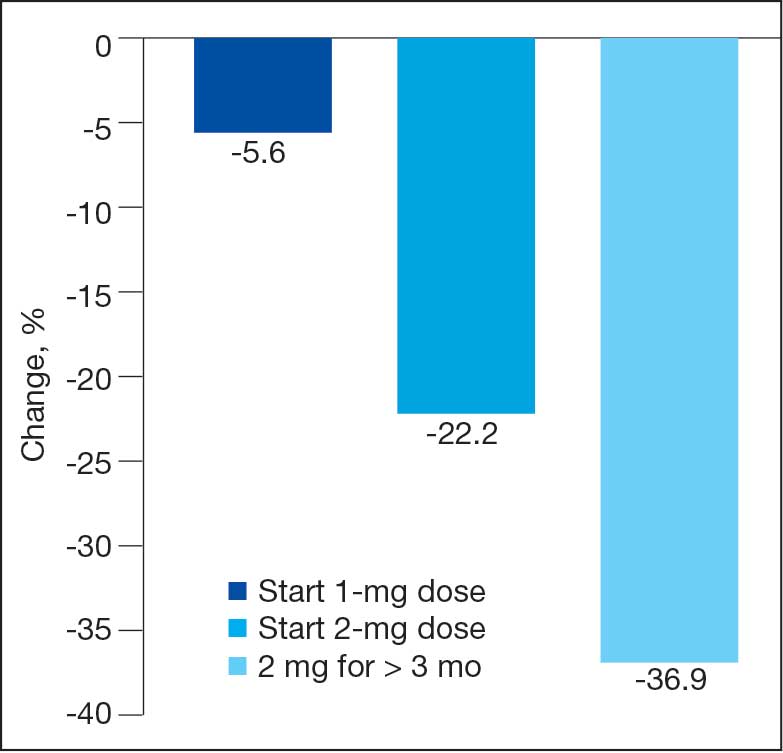
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
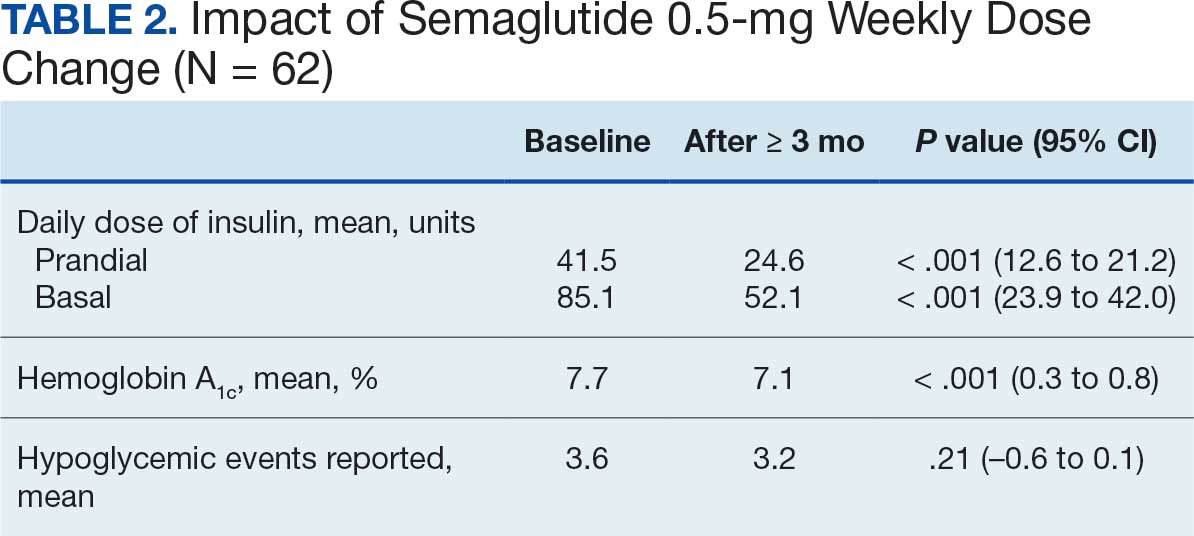
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
Type 2 diabetes mellitus (T2DM) is a chronic disease becoming more prevalent each year and is the seventh-leading cause of death in the United States.1 The most common reason for hospitalization for patients with T2DM is uncontrolled glycemic levels.2 Nearly 25% of the US Department of Veterans Affairs (VA) patient population has T2DM.3 T2DM is the leading cause of blindness, end-stage renal disease, and amputation for VA patients.4
According to the 2023 American Diabetes Association (ADA) guidelines, treatment goals of T2DM include eliminating symptoms, preventing or delaying complications, and attaining glycemic goals. A typical hemoglobin A1c (HbA1c) goal range is < 7%, but individual goals can vary up to < 9% due to a multitude of factors, including patient comorbidities and clinical status.5
Initial treatment recommendations are nonpharmacologic and include comprehensive lifestyle interventions such as optimizing nutrition, physical activity, and behavioral therapy. When pharmacologic therapy is required, metformin is the preferred first-line treatment for the majority of newly diagnosed patients with T2DM and should be added to continued lifestyle management.5 If HbA1c levels remains above goal, the 2023 ADA guidelines recommend adding a second medication, including but not limited to insulin, a glucagonlike peptide-1 receptor agonist (GLP-1RA), or a sodium-glucose cotransporter 2 inhibitor. Medication choice is largely based on the patient’s concomitant conditions (eg, atherosclerotic cardiovascular disease, heart failure, or chronic kidney disease). The 2023 ADA guidelines suggest initiating insulin therapy when a patient's blood glucose ≥ 300 mg/dL, HbA1c > 10%, or if the patient has symptoms of hyperglycemia, even at initial diagnosis. Initiating medications to minimize or avoid hypoglycemia is a priority, especially in high-risk individuals.5
Clinical evidence shows that GLP-1RAs may provide similar glycemic control to insulin with lower risk of hypoglycemia.6 Other reported benefits of GLP-1RAs include weight loss, blood pressure reduction, and improved lipid levels. The most common adverse events (AEs) with GLP-1RAs are gastrointestinal. Including GLP-1RAs in T2DM pharmacotherapy may lower the risk of hypoglycemia, especially in patients at high risk of hypoglycemia.
The 2023 ADA guidelines indicate that it is appropriate to initiate GLP-]1RAs in patients on insulin.5 However, while GLP-1RAs do not increase the risk of hypoglycemia independently, combination treatment with GLP-1RAs and insulin can still result in hypoglycemia.6 Insulin is the key suspect of this hypoglycemic risk.7 Thus, if insulin dosage can be reduced or discontinued, this might reduce the risk of hypoglycemia.
The literature is limited on how the addition of a GLP-1RA to insulin treatment will affect the patient's daily insulin doses, particularly for the veteran population. The goal of this study is to examine this gap in current research by examining semaglutide, which is the current formulary preferred GLP-1RA at the VA.
Semaglutide is subcutaneously initiated at a dose of 0.25 mg once weekly for 4 weeks to reduce gastrointestinal symptoms, then increased to 0.5 mg weekly. Additional increases to a maintenance dose of 1 mg or 2 mg weekly can occur to achieve glycemic goals. The SUSTAIN-FORTE randomized controlled trial sought to determine whether there was a difference in HbA1c level reduction and significant weight loss with the 2-mg vs 1-mg dose.8 Patients in the trial were taking metformin but needed additional medication to control their HbA1c. They were not using insulin and may or may not have been taking sulfonylureas prior to semaglutide initiation. Semaglutide 2 mg was found to significantly improve HbA1c control and promote weight loss compared with semaglutide 1 mg, while maintaining a similar safety profile.
Because this study involved patients who required additional HbA1c control, although semaglutide reduced HbA1c, not all patients were able to reduce their other diabetes medications, which depended on the baseline HbA1c level and the level upon completion of semaglutide titration. Dose reductions for the patients’ other T2DM medications were not reported at trial end. SUSTAIN-FORTE established titration up to semaglutide 2 mg as effective for HbA1c reduction, although it did not study patients also on insulin.8
Insulin is associated with hypoglycemic risk, weight gain, and other AEs.7,8 This study analyzed whether increasing semaglutide could reduce insulin doses and therefore reduce risk of AEs in patients with T2DM.
Methods
A retrospective, single-center, chart review was conducted at VA Sioux Falls Health Care System (VASFHCS). Data were collected through manual review of VASFHCS electronic medical records. Patients aged ≥ 18 years with active prescriptions for at least once-daily insulin who were initiated on 2-mg weekly dose of semaglutide at the VASFHCS clinical pharmacy practitioner medication management clinic between January 1, 2021, and September 1, 2023, were included. VASFHCS clinical pharmacy practitioners have a scope of practice that allows them to initiate, modify, or discontinue medication therapy within medication management clinics.
The most frequently used prandial insulin at VASFHCS is insulin aspart, and the most frequently used basal insulin is insulin glargine. Patients were retrospectively monitored as they progressed from baseline (the point in time where semaglutide 0.5 mg was initiated) to ≥ 3 months on semaglutide 2-mg therapy. Patients were excluded if they previously used a GLP-1RA or if they were on sliding scale insulin without an exact daily dosage.
The primary endpoint was the percent change in total daily insulin dose from baseline to each dose increase after receiving semaglutide 2 mg for ≥ 3 months. Secondary endpoints included changes in daily prandial insulin dose, daily basal insulin dose, HbA1c, and number of hypoglycemic events reported. Data collected included age, race, weight, body mass index, total daily prandial insulin dose, total daily basal insulin dose, HbA1c, and hypoglycemic events reported at the visit when semaglutide was initiated.
Statistical Analysis
The sample size was calculated prior to data collection, and it was determined that for α = .05, 47 patients were needed to achieve 95% power. The primary endpoint was assessed using a paired t test, as were each secondary endpoint. Results with P < .05 were considered statistically significant.
Results
Sixty-two patients were included. The mean HbA1c level at baseline was 7.7%, the baseline mean prandial and insulin daily doses were 41.5 units and 85.1 units, respectively (Table 1) From baseline to initiation of a semaglutide 1-mg dose, the daily insulin dose changed –5.6% (95% CI, 2.2-14.0; P = .008). From baseline to 2-mg dose initiation daily insulin changed -22.2% (95% CI, 22.0-35.1; P < .001) and for patients receiving semaglutide 2 mg for ≥ 3 months it changed -36.9% (95% CI, 37.4-56.5; P < .001) (Figure).
After receiving the 2-mg dose for ≥ 3 months, the mean daily dose of prandial insulin decreased from 41.5 units to 24.6 units (95% CI, 12.6-21.2; P < .001); mean daily dose of basal insulin decreased from 85.1 units to 52.1 units (95% CI, 23.9-42.0; P < .001); and mean HbA1c level decreased from 7.7% to 7.1% (95% CI, 0.3-0.8; P < .001). Mean number of hypoglycemic events reported was not statistically significant, changing from 3.6 to 3.2 (95% CI, –0.6 to 0.1; P = .21) (Table 2).
Discussion
This study investigated the effect of subcutaneous semaglutide dose escalation on total daily insulin dose for patients with T2DM. There was a statistically significant decrease in total daily insulin dose from baseline to 1 mg initiation; this decrease continued with further insulin dose reduction seen at the 2-mg dose initiation and additional insulin dose reduction at ≥ 3 months at this dose. It was hypothesized there would be a significant total daily insulin dose reduction at some point, especially when transitioning from the semaglutide 1-mg to the 2-mg dose, based on previous research. 9,10 The additional reduction in daily insulin dose when continuing on semaglutide 2 mg for ≥ 3 months was an unanticipated but added benefit, showing that if tolerated, maintaining the 2-mg dose will help patients reduce their insulin doses.
In terms of secondary endpoints, there was a statistically significant decrease in mean total daily dose individually for prandial and basal insulin from baseline to ≥ 3 months after semaglutide 2 mg initiation. The change in HbA1c level was also statistically significant and decreased from baseline, even as insulin doses were reduced. This change in HbA1c level was expected; previous literature has shown a significant link between improving HbA1c control when semaglutide doses are increased to 2 mg weekly.10 Due to having been shown in previous trials, it was expected that HbA1c levels would decrease even when the insulin doses were being reduced.10 Insulin dose reduction can potentially be added to the growing evidence of semaglutide benefits. The change in the number of hypoglycemic events was not statistically significant, which was unexpected since previous research show a trend in patients taking GLP-1RAs having fewer hypoglycemic events than those taking insulin.6 Further investigation with a larger sample size and prospective trial could determine whether this result is an outlier. In this study, there was no increase in HbA1c or hypoglycemic events reported with increasing semaglutide doses, which provides further evidence of the safety of semaglutide even at higher doses.
These data suggest that for a patient with T2DM who is already taking insulin, the recommended titration of semaglutide is to start with 0.5 mg and titrate up to a 2-mg subcutaneous weekly dose and to then continue at that dose. As long as the 2-mg dose is tolerated, it will provide patients with the most HbA1c control and lead to a reduction of their total daily insulin doses according to these results.
Strengths and Limitations
This study compared patient data at different points. This method did not require a second distinct control group, which would potentially introduce confounding factors, such as different baseline characteristics. Another strength is that documentation was available for all patients throughout the study so no one was lost to follow-up. This allowed comprehensive data collection and provided a stronger conclusion given the completeness of the data from baseline to follow-up.
Limitations include the retrospective design and small sample size. In addition, the study design did not allow for randomization. There is no documentation of adherence to medication regimen, which was difficult to determine due to the retrospective nature. Other changes to the patients’ medication regimen were not collected in aggregate and thus, it is possible the total daily insulin dose was impacted by other medication changes. There is also potential for inconsistent documentation of the patients’ true total daily insulin dose in the medical record, thus leading to inaccuracy of recorded data.
Conclusions
A small sample of veterans with T2DM had statistically significant reductions in total daily insulin dose when subcutaneous semaglutide was initiated, as well as after each dose increase. There was also a statistically significant reduction in HbA1c levels from baseline even as patient insulin doses were reduced. These results support the current practice of using semaglutide to treat T2DM, suggesting it may be safe and effective at reducing HbA1c levels as the dose is titrated up to 2 mg. There was no statistically significant change in the number of hypoglycemic events reported as semaglutide was titrated up. Thus, when semaglutide is increased to the maximum recommended dose of 2 mg for T2DM, patients may experience a reduction of their total daily dose of insulin and HbA1c levels. These benefits may reduce the risk of insulin-related AEs while maintaining appropriate glycemic control.
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
- Diabetes mellitus: in federal health care data trends 2017. Fed Pract. 2017:S20. Accessed August 6, 2025. https://www.fedprac-digital.com/federalpractitioner/data_trends_2017
- Centers for Disease Control and Prevention. National diabetes statistics report. May 15, 2024. Accessed September 17, 2025. https://www.cdc.gov/diabetes/php/data-research/index.html
- US Department of Veterans Affairs. VA research on diabetes. Updated January 15, 2021. Accessed August 6, 2025. https://www.research.va.gov/topics/diabetes.cfm
- Liu Y, Sayam S, Shao X, et al. Prevalence of and trends in diabetes among veterans, United States, 2005-2014. Prev Chronic Dis. 2017;14:E135. doi:10.5888/pcd14.170230
- American Diabetes Association. Standards of care in diabetes— 2023 abridged for primary care providers. Clin Diabetes. 2022;41:4-31. doi:10.2337/cd23-as01
- Zhao Z, Tang Y, Hu Y, Zhu H, Chen X, Zhao B. Hypoglycemia following the use of glucagon-like peptide-1 receptor agonists: a real-world analysis of post-marketing surveillance data. Ann Transl Med. 2021;9:1482. doi:10.21037/atm-21-4162
- Workgroup on Hypoglycemia, American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245-1249. doi:10.2337/diacare.28.5.1245
- Frías JP, Auerbach P, Bajaj HS, et al. Efficacy and safety of once-weekly semaglutide 2.0 mg versus 1.0 mg in patients with type 2 diabetes (SUSTAIN FORTE): a double-blind, randomised, phase 3B trial. Lancet Diabetes Endocrinol. 2021;9:563-574. doi:10.1016/S2213-8587(21)00174-1
- Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26:107-139. doi:10.4158/CS-2019-0472
- Miles KE, Kerr JL. Semaglutide for the treatment of type 2 diabetes mellitus. J Pharm Technol. 2018;34:281-289. doi:10.1177/8755122518790925
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes
Efficacy of Subcutaneous Semaglutide Dose Escalation in Reducing Insulin in Patients With Type 2 Diabetes