User login
Low-dose steroids for acute exacerbations of COPD in a non-ICU setting: Worth consideration
Despite guidelines recommending low-dose oral glucocorticoids over high-dose intravenous (IV) glucocorticoids for inpatient management of acute exacerbations of chronic obstructive pulmonary disease (COPD), we have observed that most patients still receive high-dose IV therapy before being transitioned to low-dose oral therapy at discharge. Clinical inertia undoubtedly plays a significant role in the slow adoption of new recommendations, but in this era of evidence-based practice, the unfortunate lack of data supporting low over high steroid doses for acute exacerbations of COPD also contributes to hesitancy of physicians.
A SIGNIFICANT AND GROWING BURDEN
COPD is one of the most common pulmonary conditions managed by hospitalists today, and by the year 2030, it is predicted to become the third leading cause of death worldwide.1
COPD is also a significant economic burden, costing $50 billion to manage in the United States, most of that from the cost of lengthy hospital stays.2 COPD patients have 1 to 2 exacerbations per year.3 Bacterial and viral infections are responsible for most exacerbations, and 15% to 20% are from air pollution and other environmental causes of airway inflammation.3
CHALLENGES TO CHANGING PRACTICE
Glucocorticoids are the gold standard for treatment of acute exacerbations of COPD. It is well-documented that compared with placebo, glucocorticoids reduce mortality risk, length of hospital stay, and exacerbation recurrence after 1 month.4 And while high-dose IV steroid therapy has been the standard approach, oral administration has been found to be noninferior to IV administration with regard to treatment and length of hospital stay.5
While adverse effects are more common at higher doses, the optimal dose and duration of systemic glucocorticoid therapy for acute exacerbations of COPD are still largely at the discretion of the physician. The 2019 report of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) recommends low doses (40 mg) for no more than 5 to 7 days for exacerbations, based on reports that showed no worse outcomes with low-dose oral than with high-dose IV therapy.6,7 (In the 2010 study by Lindenauer et al,7 92% of nearly 80,000 patients received high-dose IV steroids, reflecting standard practice at that time.) However, the GOLD guidelines do not address mortality rates, length of stay, or readmission rates for either approach, as they are devised to direct treatment in patients with stable mild to advanced COPD, not exacerbations.
THE EVIDENCE FOR LOW-DOSE STEROIDS
Mortality rates
Aksoy et al8 established that, compared with placebo, low-dose steroids improved mortality rates in a subset of patients with acute exacerbations, specifically those with eosinophilic exacerbations. This study followed the 2013 Reduction in the Use of Corticosteroids in Exacerbated COPD (REDUCE) trial, which showed mortality rates were not lower with 14 days of low-dose prednisone treatment than with 5 days.9
Length of hospital stay
With regard to length of hospital stay, in 2011 Wang et al10 found no statistically significant difference between high- and low-dose steroid treatment.However, the REDUCE trial found that low-dose steroids shortened the median length of stay by 1 day compared with placebo.9
Hospital readmission rates
The REDUCE trial found no statistically significant difference in readmission rates when comparing 5 days of low-dose treatment vs 14 days.9 However, Aksoy et al8 found that readmission rates were significantly lower with low-dose treatment than with placebo.No study has yet examined readmission rates with high-dose vs low-dose steroid treatment.
What does the evidence tell us?
Low-dose oral glucocorticoid treatment shows definitive benefits in terms of lower mortality rates, shorter hospital length of stay, and lower readmission rates vs placebo in the treatment of acute exacerbations of COPD. Furthermore, a 14-day course is no better than 5 days in terms of mortality rates. And low-dose glucocorticoid treatment shows reduced mortality rates in addition to similar hospital length of stay when compared to high-dose glucocorticoid treatment.
Together, these findings lend credibility to the current GOLD recommendations. However, we have observed that in sharp contrast to the leading clinical guidelines, most patients hospitalized for acute exacerbations of COPD are still treated initially with high-dose IV corticosteroids. Why?
Obstacles that perpetuate the use of high-dose over low-dose treatment include lack of knowledge of glucocorticoid pharmacokinetics among clinicians, use of outdated order sets, and the reflex notion that more of a drug is more efficacious in its desired effect. In addition, administrative obstacles include using high-dose IV steroids to justify an inpatient stay or continued hospitalization.
COUNTERING THE OBSTACLES: THE HOSPITALIST’S ROLE
To counter these obstacles, we propose standardization of inpatient treatment of acute exacerbations of COPD to include initial low-dose steroid treatment in accordance with the most recent GOLD guidelines.6 This would benefit the patient by reducing undesirable effects of high-dose steroids, and at the same time reduce the economic burden of managing COPD exacerbations. Considering the large number of hospitalizations for COPD exacerbation each year, hospitalists can play a large role in this effort by routinely incorporating the low-dose steroid recommendation into their clinical practice.
- World Health Organization. Chronic respiratory diseases: burden of COPD. www.who.int/respiratory/copd/burden/en. Accessed October 16, 2019.
- Guarascio AJ, Ray SM, Finch CK, Self TH. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res 2013; 5:235–245. doi:10.2147/CEOR.S34321
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359(22):2355–2365. doi:10.1056/NEJMra0800353
- Walters JA, Tan DJ, White CJ, Gibson PG, Wood-Baker R, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2014; (9):CD001288. doi:10.1002/14651858.CD001288.pub4
- de Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: a randomized, controlled, double-blind study. Chest 2007; 132(6):1741–1747. doi:10.1378/chest.07-0208
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: 2019 report. www.goldcopd.org/wp-content/uploads/2018/11/GOLD-2019-v1.7-FINAL-14Nov2018-WMS.pdf. Accessed October 16, 2019.
- Lindenauer PK, Pekow PS, Lahti MC, Lee Y, Benjamin EM, Rothberg MB. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA 2010; 303(23):2359–2367. doi:10.1001/jama.2010.796
- Aksoy E, Güngör S, Agca MÇ, et al. A revised treatment approach for hospitalized patients with eosinophilic and neutrophilic exacerbations of chronic obstructive pulmonary disease. Turk Thorac J 2018; 19(4):193–200. doi:10.5152/TurkThoracJ.2018.18004
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 2013; 309(21):2223–2231. doi:10.1001/jama.2013.5023
- Wang PH, Cheng SL, Wang HC, et al. Systemic steroids in acute exacerbation of COPD—from guidelines to bedside. Int J Clin Pharmacol Ther 2011; 49(11):705–708. doi:10.5414/cp201588
Despite guidelines recommending low-dose oral glucocorticoids over high-dose intravenous (IV) glucocorticoids for inpatient management of acute exacerbations of chronic obstructive pulmonary disease (COPD), we have observed that most patients still receive high-dose IV therapy before being transitioned to low-dose oral therapy at discharge. Clinical inertia undoubtedly plays a significant role in the slow adoption of new recommendations, but in this era of evidence-based practice, the unfortunate lack of data supporting low over high steroid doses for acute exacerbations of COPD also contributes to hesitancy of physicians.
A SIGNIFICANT AND GROWING BURDEN
COPD is one of the most common pulmonary conditions managed by hospitalists today, and by the year 2030, it is predicted to become the third leading cause of death worldwide.1
COPD is also a significant economic burden, costing $50 billion to manage in the United States, most of that from the cost of lengthy hospital stays.2 COPD patients have 1 to 2 exacerbations per year.3 Bacterial and viral infections are responsible for most exacerbations, and 15% to 20% are from air pollution and other environmental causes of airway inflammation.3
CHALLENGES TO CHANGING PRACTICE
Glucocorticoids are the gold standard for treatment of acute exacerbations of COPD. It is well-documented that compared with placebo, glucocorticoids reduce mortality risk, length of hospital stay, and exacerbation recurrence after 1 month.4 And while high-dose IV steroid therapy has been the standard approach, oral administration has been found to be noninferior to IV administration with regard to treatment and length of hospital stay.5
While adverse effects are more common at higher doses, the optimal dose and duration of systemic glucocorticoid therapy for acute exacerbations of COPD are still largely at the discretion of the physician. The 2019 report of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) recommends low doses (40 mg) for no more than 5 to 7 days for exacerbations, based on reports that showed no worse outcomes with low-dose oral than with high-dose IV therapy.6,7 (In the 2010 study by Lindenauer et al,7 92% of nearly 80,000 patients received high-dose IV steroids, reflecting standard practice at that time.) However, the GOLD guidelines do not address mortality rates, length of stay, or readmission rates for either approach, as they are devised to direct treatment in patients with stable mild to advanced COPD, not exacerbations.
THE EVIDENCE FOR LOW-DOSE STEROIDS
Mortality rates
Aksoy et al8 established that, compared with placebo, low-dose steroids improved mortality rates in a subset of patients with acute exacerbations, specifically those with eosinophilic exacerbations. This study followed the 2013 Reduction in the Use of Corticosteroids in Exacerbated COPD (REDUCE) trial, which showed mortality rates were not lower with 14 days of low-dose prednisone treatment than with 5 days.9
Length of hospital stay
With regard to length of hospital stay, in 2011 Wang et al10 found no statistically significant difference between high- and low-dose steroid treatment.However, the REDUCE trial found that low-dose steroids shortened the median length of stay by 1 day compared with placebo.9
Hospital readmission rates
The REDUCE trial found no statistically significant difference in readmission rates when comparing 5 days of low-dose treatment vs 14 days.9 However, Aksoy et al8 found that readmission rates were significantly lower with low-dose treatment than with placebo.No study has yet examined readmission rates with high-dose vs low-dose steroid treatment.
What does the evidence tell us?
Low-dose oral glucocorticoid treatment shows definitive benefits in terms of lower mortality rates, shorter hospital length of stay, and lower readmission rates vs placebo in the treatment of acute exacerbations of COPD. Furthermore, a 14-day course is no better than 5 days in terms of mortality rates. And low-dose glucocorticoid treatment shows reduced mortality rates in addition to similar hospital length of stay when compared to high-dose glucocorticoid treatment.
Together, these findings lend credibility to the current GOLD recommendations. However, we have observed that in sharp contrast to the leading clinical guidelines, most patients hospitalized for acute exacerbations of COPD are still treated initially with high-dose IV corticosteroids. Why?
Obstacles that perpetuate the use of high-dose over low-dose treatment include lack of knowledge of glucocorticoid pharmacokinetics among clinicians, use of outdated order sets, and the reflex notion that more of a drug is more efficacious in its desired effect. In addition, administrative obstacles include using high-dose IV steroids to justify an inpatient stay or continued hospitalization.
COUNTERING THE OBSTACLES: THE HOSPITALIST’S ROLE
To counter these obstacles, we propose standardization of inpatient treatment of acute exacerbations of COPD to include initial low-dose steroid treatment in accordance with the most recent GOLD guidelines.6 This would benefit the patient by reducing undesirable effects of high-dose steroids, and at the same time reduce the economic burden of managing COPD exacerbations. Considering the large number of hospitalizations for COPD exacerbation each year, hospitalists can play a large role in this effort by routinely incorporating the low-dose steroid recommendation into their clinical practice.
Despite guidelines recommending low-dose oral glucocorticoids over high-dose intravenous (IV) glucocorticoids for inpatient management of acute exacerbations of chronic obstructive pulmonary disease (COPD), we have observed that most patients still receive high-dose IV therapy before being transitioned to low-dose oral therapy at discharge. Clinical inertia undoubtedly plays a significant role in the slow adoption of new recommendations, but in this era of evidence-based practice, the unfortunate lack of data supporting low over high steroid doses for acute exacerbations of COPD also contributes to hesitancy of physicians.
A SIGNIFICANT AND GROWING BURDEN
COPD is one of the most common pulmonary conditions managed by hospitalists today, and by the year 2030, it is predicted to become the third leading cause of death worldwide.1
COPD is also a significant economic burden, costing $50 billion to manage in the United States, most of that from the cost of lengthy hospital stays.2 COPD patients have 1 to 2 exacerbations per year.3 Bacterial and viral infections are responsible for most exacerbations, and 15% to 20% are from air pollution and other environmental causes of airway inflammation.3
CHALLENGES TO CHANGING PRACTICE
Glucocorticoids are the gold standard for treatment of acute exacerbations of COPD. It is well-documented that compared with placebo, glucocorticoids reduce mortality risk, length of hospital stay, and exacerbation recurrence after 1 month.4 And while high-dose IV steroid therapy has been the standard approach, oral administration has been found to be noninferior to IV administration with regard to treatment and length of hospital stay.5
While adverse effects are more common at higher doses, the optimal dose and duration of systemic glucocorticoid therapy for acute exacerbations of COPD are still largely at the discretion of the physician. The 2019 report of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) recommends low doses (40 mg) for no more than 5 to 7 days for exacerbations, based on reports that showed no worse outcomes with low-dose oral than with high-dose IV therapy.6,7 (In the 2010 study by Lindenauer et al,7 92% of nearly 80,000 patients received high-dose IV steroids, reflecting standard practice at that time.) However, the GOLD guidelines do not address mortality rates, length of stay, or readmission rates for either approach, as they are devised to direct treatment in patients with stable mild to advanced COPD, not exacerbations.
THE EVIDENCE FOR LOW-DOSE STEROIDS
Mortality rates
Aksoy et al8 established that, compared with placebo, low-dose steroids improved mortality rates in a subset of patients with acute exacerbations, specifically those with eosinophilic exacerbations. This study followed the 2013 Reduction in the Use of Corticosteroids in Exacerbated COPD (REDUCE) trial, which showed mortality rates were not lower with 14 days of low-dose prednisone treatment than with 5 days.9
Length of hospital stay
With regard to length of hospital stay, in 2011 Wang et al10 found no statistically significant difference between high- and low-dose steroid treatment.However, the REDUCE trial found that low-dose steroids shortened the median length of stay by 1 day compared with placebo.9
Hospital readmission rates
The REDUCE trial found no statistically significant difference in readmission rates when comparing 5 days of low-dose treatment vs 14 days.9 However, Aksoy et al8 found that readmission rates were significantly lower with low-dose treatment than with placebo.No study has yet examined readmission rates with high-dose vs low-dose steroid treatment.
What does the evidence tell us?
Low-dose oral glucocorticoid treatment shows definitive benefits in terms of lower mortality rates, shorter hospital length of stay, and lower readmission rates vs placebo in the treatment of acute exacerbations of COPD. Furthermore, a 14-day course is no better than 5 days in terms of mortality rates. And low-dose glucocorticoid treatment shows reduced mortality rates in addition to similar hospital length of stay when compared to high-dose glucocorticoid treatment.
Together, these findings lend credibility to the current GOLD recommendations. However, we have observed that in sharp contrast to the leading clinical guidelines, most patients hospitalized for acute exacerbations of COPD are still treated initially with high-dose IV corticosteroids. Why?
Obstacles that perpetuate the use of high-dose over low-dose treatment include lack of knowledge of glucocorticoid pharmacokinetics among clinicians, use of outdated order sets, and the reflex notion that more of a drug is more efficacious in its desired effect. In addition, administrative obstacles include using high-dose IV steroids to justify an inpatient stay or continued hospitalization.
COUNTERING THE OBSTACLES: THE HOSPITALIST’S ROLE
To counter these obstacles, we propose standardization of inpatient treatment of acute exacerbations of COPD to include initial low-dose steroid treatment in accordance with the most recent GOLD guidelines.6 This would benefit the patient by reducing undesirable effects of high-dose steroids, and at the same time reduce the economic burden of managing COPD exacerbations. Considering the large number of hospitalizations for COPD exacerbation each year, hospitalists can play a large role in this effort by routinely incorporating the low-dose steroid recommendation into their clinical practice.
- World Health Organization. Chronic respiratory diseases: burden of COPD. www.who.int/respiratory/copd/burden/en. Accessed October 16, 2019.
- Guarascio AJ, Ray SM, Finch CK, Self TH. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res 2013; 5:235–245. doi:10.2147/CEOR.S34321
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359(22):2355–2365. doi:10.1056/NEJMra0800353
- Walters JA, Tan DJ, White CJ, Gibson PG, Wood-Baker R, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2014; (9):CD001288. doi:10.1002/14651858.CD001288.pub4
- de Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: a randomized, controlled, double-blind study. Chest 2007; 132(6):1741–1747. doi:10.1378/chest.07-0208
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: 2019 report. www.goldcopd.org/wp-content/uploads/2018/11/GOLD-2019-v1.7-FINAL-14Nov2018-WMS.pdf. Accessed October 16, 2019.
- Lindenauer PK, Pekow PS, Lahti MC, Lee Y, Benjamin EM, Rothberg MB. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA 2010; 303(23):2359–2367. doi:10.1001/jama.2010.796
- Aksoy E, Güngör S, Agca MÇ, et al. A revised treatment approach for hospitalized patients with eosinophilic and neutrophilic exacerbations of chronic obstructive pulmonary disease. Turk Thorac J 2018; 19(4):193–200. doi:10.5152/TurkThoracJ.2018.18004
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 2013; 309(21):2223–2231. doi:10.1001/jama.2013.5023
- Wang PH, Cheng SL, Wang HC, et al. Systemic steroids in acute exacerbation of COPD—from guidelines to bedside. Int J Clin Pharmacol Ther 2011; 49(11):705–708. doi:10.5414/cp201588
- World Health Organization. Chronic respiratory diseases: burden of COPD. www.who.int/respiratory/copd/burden/en. Accessed October 16, 2019.
- Guarascio AJ, Ray SM, Finch CK, Self TH. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res 2013; 5:235–245. doi:10.2147/CEOR.S34321
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359(22):2355–2365. doi:10.1056/NEJMra0800353
- Walters JA, Tan DJ, White CJ, Gibson PG, Wood-Baker R, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2014; (9):CD001288. doi:10.1002/14651858.CD001288.pub4
- de Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: a randomized, controlled, double-blind study. Chest 2007; 132(6):1741–1747. doi:10.1378/chest.07-0208
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: 2019 report. www.goldcopd.org/wp-content/uploads/2018/11/GOLD-2019-v1.7-FINAL-14Nov2018-WMS.pdf. Accessed October 16, 2019.
- Lindenauer PK, Pekow PS, Lahti MC, Lee Y, Benjamin EM, Rothberg MB. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA 2010; 303(23):2359–2367. doi:10.1001/jama.2010.796
- Aksoy E, Güngör S, Agca MÇ, et al. A revised treatment approach for hospitalized patients with eosinophilic and neutrophilic exacerbations of chronic obstructive pulmonary disease. Turk Thorac J 2018; 19(4):193–200. doi:10.5152/TurkThoracJ.2018.18004
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 2013; 309(21):2223–2231. doi:10.1001/jama.2013.5023
- Wang PH, Cheng SL, Wang HC, et al. Systemic steroids in acute exacerbation of COPD—from guidelines to bedside. Int J Clin Pharmacol Ther 2011; 49(11):705–708. doi:10.5414/cp201588
Portopulmonary Hypertension: Treatment
Portopulmonary hypertension (POPH) is a form of group 1 pulmonary arterial hypertension. When treating patients with POPH, baseline assessment is necessary so that response to therapy can be measured as the change from baseline. Patients should undergo echocardiography and right heart catheterization, and their exercise capacity and NYHA functional class should be determined. Patients with POPH should be considered for treatment if they are NYHA functional class II or above and/or their mean pulmonary artery pressure (MPAP) is greater than 35 mm Hg in transplant candidates. The goal in the treatment and management of POPH is to improve pulmonary hemodynamics by reducing the obstruction to pulmonary arterial flow and to preserve right ventricular function (Table). This article, the second in a 2-part review of POPH in patients with liver disease, reviews the role of medical therapy and liver transplantation in treatment. Evaluation and diagnosis of POPH are discussed in a separate article.

Medical Therapy
Prostanoids
Although prostacyclin and prostaglandin analogs entered routine clinical practice for POPH in the 1990s, reports of investigational use date back to the 1980s. Prostanoids are potent vasodilators with antiplatelet aggregation and antiproliferative properties. Prostacyclin synthase is reduced in patients with PAH, resulting in decreased concentration of prostacyclin with vasoconstriction and proliferative changes in the pulmonary vasculature.1
Epoprostenol
Epoprostenol is also known as synthetic prostaglandin I2 or prostacyclin. It was the first therapy approved for the treatment of PAH in 1995 by the US Food and Drug Administration (FDA) as a continuous intravenous infusion.2,3 It also inhibits platelet aggregation and may help modulate pulmonary vascular remodeling.4,5 Epoprostenol is derived from the metabolism of arachidonic acid and is a potent pulmonary and systemic vasodilator. One study reported an immediate 11.8% decrease in MPAP, 24% decrease in pulmonary vascular resistance (PVR) and 28% drop in systemic vascular resistance (SVR) during an epoprostenol infusion.6 The authors reported that epoprostenol was a more potent vasodilator than nitric oxide and may have a role in predicting the reversibility of POPH. In a case series of 33 patients with secondary pulmonary hypertension (including 7 patients with POPH) treated with continuous intravenous prostacyclin for approximately 1 year, exercise tolerance, NYHA functional class, and pulmonary hemodynamics improved in each patient compared to baseline.7 Krowka et al studied 14 patients with moderate to severe POPH treated with intravenous epoprostenol.8 No significant side effects were noted and treatment resulted in significant improvements in PVR, MPAP, and cardiac output. In 2007, Fix et al published a large retrospective cohort of patients with moderate to severe POPH.9 Nineteen patients treated with epoprostenol were compared to 17 patients with no treatment. After a median treatment period of 15.4 months, the epoprostenol group showed significant improvement in MPAP, PVR and cardiac output, but survival did not differ between the 2 groups.
Epoprostenol has often been considered a bridge to transplant in patients with POPH. Sussman et al described 8 consecutive patients with POPH who were treated with intravenous epoprostenol (2 to 8 ng/kg/min dose).10 Liver transplant was considered in 7 of the 8 patients when MPAP decreased to less than 35 mm Hg. Six patients were eventually listed for liver transplant, but 2 died waiting on the list. Long-term outcomes in the group of transplanted recipients were excellent. They remained alive and well at least 9 to 18 months post-transplant, and half did not require long-term vasodilator therapy post-orthotopic liver transplant. Similarly, Ashfaq et al published their data on 16 patients with moderate-to-severe POPH who were treated with vasodilator therapy.11 MPAP decreased to acceptable levels in 75% of the treated patients, and 11 went on to liver transplantation. Rates of 1- and 5-year survival in the transplanted patients were 91% and 67% respectively. None of the patients who failed vasodilator therapy survived.
Epoprostenol has a short half-life (3 to 5 minutes) and requires continuous infusion through central access via an infusion pump. Aseptic technique must be maintained to avoid blood stream infections. Pump failure or loss of vascular access can result in rebound pulmonary vasoconstriction that can be life-threatening and requires immediate attention. Side effects associated with epoprostenol include flushing, headache, nausea/vomiting, bradycardia, chest pain, jaw pain, diarrhea, and musculoskeletal pain.
Patients on epoprostenol should be monitored for prostanoid overdose. In the case of patients with chronic liver disease, epoprostenol increases systemic vasodilation in patients with already low systemic vascular tone. As a result, cardiac output may increase to the point of high cardiac output failure. MPAP will remain elevated secondary to high cardiac output rather than high PVR. In these patients, right heart catheterization will show an elevated MPAP in the setting of normal to low PVR/transpulmonary gradient (TPG) values. Lowering the epoprostenol dose will successfully reduce both cardiac output and MPAP.
Treprostinil
Treprostinil is a prostacyclin analog that is available in intravenous, inhalational, and subcutaneous form, although subcutaneous dosing may be limited by pain. Sakai et al published a small case series of 3 patients with PAH and end-stage liver disease treated with intravenous treprostinil.12 Pulmonary hemodynamics improved in all patients, and 2 patients went on to an uneventful liver transplantation. More than 10 years later, data were published on 255 patients with PAH on therapy with bosentan or sildenafil randomized to additional inhaled treprostinil.13 Treprostinil proved to be safe and well tolerated, with improvement in quality of life measures but no improvement in other secondary endpoints.
Iloprost
Inhaled iloprost is another prostacyclin that has a short therapeutic half-life of 20 to 30 minutes and requires frequent administration (6 to 9 times daily). In study in which patients with severe POPH were treated for up to 3 years with inhaled iloprost,14 survival rates at 1, 2, and 3 years were 77%, 62%, and 46%, respectively. A second study published in 2010 was designed to assess the acute effects of inhaled iloprost on pulmonary hemodynamics and evaluate the clinical outcome after 12 months of treatment.15 Iloprost was found to rapidly reduce pulmonary arterial pressure and PVR. In the long-term evaluation, inhaled iloprost increased the 6-minute walk distance (6MWD) and functional class, but no change was noted in the systolic pulmonary artery pressure. The authors concluded that iloprost might provide symptomatic improvement and improvement in exercise capacity.
Selexipag
Selexipag is an oral selective IP prostacyclin receptor agonist that is structurally distinct from other prostacyclins.16 In a phase 3 randomized double blind clinical trial, PAH patients treated with selexipag had lower composite of death or complication of PAH to the end of the study period.17 This effect was consistent across all dose ranges, but POPH patients were excluded from this study. Safety and efficacy of selexipag has not been evaluated in POPH patients.
Endothelin Receptor Antagonists
Endothelin receptor antagonists block the production of endothelin-1 (ET-1), a potent vasoconstrictor and smooth muscle mitogen that may contribute to the development of PAH. Three different receptors have been described: endothelin A, endothelin B, and endothelin B2. Elevated ET-1 levels have been reported in patients with chronic liver disease and may originate from hepatosplanchnic circulation.18
Bosentan
Bosentan is an oral, nonspecific, ET-1A and ET-1B receptor antagonist. Initial use of bosentan in patients with POPH was limited because of concern for hepatotoxicity. Approximately 10% of patients on bosentan were reported to have mild hepatic side effects in the form of elevated aminotransferases, but severe injury has been reported.19 One of the first clinical experiences of bosentan in patients with POPH was published in 2005. Hoeper et al followed 11 patients with Child A cirrhosis and severe POPH.20 All patients included were in NYHA functional class III or IV and were treated with bosentan for over 1 year. Exercise capacity and symptoms improved in all treated patients. The medication was tolerated well and there was no evidence of drug-induced liver injury. A single case report showed the effectiveness of bosentan in a 43-year-old man with alcohol-related liver disease (Child-Pugh A) and right ventricular enlargement and dysfunction secondary to POPH.21 Pulmonary arterial pressure decreased, exercise capacity increased, and improvement was maintained over 2 years.
In a group of 31 patients with Child A or B cirrhosis and severe POPH, bosentan had significantly better effects than inhaled iloprost on exercise capacity, hemodynamics, and survival.14 One, 2, and 3-year survival rates in the bosentan group were 94%, 89%, and 89% (compared to 77%, 62%, and 46% in the iloprost group). Both drugs were considered safe with no reported hepatotoxicity. In 2013, Savale et al published data on 34 patients with POPH, Child-Pugh A and/or B who were treated with bosentan for a median of 43 months.22 The authors reported significant improvements in hemodynamics, NYHA functional class, and 6WMD. Event-free survival rates at 1, 2, and 3 years were 82%, 63%, and 47%, respectively.
Ambrisentan
Ambrisentan is a highly selective ET-1A receptor antagonist with once daily dosing and a lower risk of hepatotoxicity compared to bosentan. Fourteen patients with moderate to severe POPH treated with ambrisentan in 4 German hospitals were retrospectively analyzed.23 Median follow-up was 16 months, and the study demonstrated significant improvement in exercise capacity and clinical symptoms without significant change in liver function tests. Cartin-Ceba et al published their experience of 13 patients with moderate to severe POPH treated with ambrisentan monotherapy.24 Patients were followed for a median of 613 days and on treatment for a median time of 390 days. Significant improvements were shown in pulmonary arterial pressure and PVR without adverse effect on hepatic function. Over 270 patients with PAH (6% with POPH) received ambrisentan from March 2009 through June 2013 at a large United Kingdom portal hypertension referral center.25 Discontinuation due to side effects was higher than previously reported. Discontinuation due to abnormal transaminases was uncommon.
Macitentan
Macitentan is a dual endothelin-receptor antagonist developed by modifying the structure of bosentan to increase efficacy and safety. The SERAPHIN trial compared oral macitentan to placebo in 250 patients with moderate to severe PAH, some of whom were also on a stable dose of oral or inhaled therapy for PAH.26 Over a 2-year period, patients treated with macitentan were less likely to have progression of their disease or die on therapy (38% and 31% versus 46%), regardless of if they were receiving additional oral therapy and more likely to have improvement of their exercise capacity and WHO functional class. Nasopharyngitis and significant anemia were more common in the macitentan group, but there was no difference in the rate of liver function test abnormalities compared to placebo. Trials with macitentan are currently ongoing in patients with POPH.
Phosphodiesterase-5 Inhibitors
Cyclic guanosine monophosphate (cGMP) is the mediator of nitric oxide–induced vasodilation. Phosphodiesterase-5 (PDE-5) inhibitors prolong the vasodilatory effects of cyclic guanosine monophosphate by preventing its hydrolysis, thereby reducing the pulmonary arterial pressure.
Sildenafil
Sildenafil is the most widely accepted PDE-5 inhibitor for POPH. Fourteen patients with moderate to severe POPH were treated with sildenafil (50 mg 3 times per day) in an observational study published by Reichenberger et al in 2006.27 Eight patients were newly started on sildenafil, whereas sildenafil was added to inhaled prostanoids in the remaining 6x patients. Sildenafil significantly decreased 66MWD, MPAP, PVR, and cardiac index alone or in combination with inhaled prostanoids.
Sildenafil has also been used as a bridge to transplant in liver transplant candidates with POPH. Ten patients with POPH treated with sildenafil monotherapy were followed for a 21±16 months.28 Patients improved symptomatically and increased their 6MWD at 1 year by 30 meters or more. Three patients became transplant eligible and another 3 patients were stable, without progression of their liver disease or POPH. Four patients were not considered transplant candidates, 2 because of refractory POPH and 2 for other comorbidities. The authors concluded that sildenafil monotherapy could stabilize or improve pulmonary hemodynamics in patients with POPH and eventually lead to liver transplantation. Gough et al took a similar look at 9 patients with POPH treated with sildenafil.29 All patients had initial and follow-up right heart catheterizations within a period of 3 years. Mean PVR improved in all patients, decreasing from 575 to 375 dynes/s/cm–5. MPAP decreased to ≤ 35 mmHg in 4 patients, 1 of whom went on to receive a liver transplant. Overall sildenafil improved pulmonary hemodynamics in this small cohort of POPH patients.
Tadalafil
Tadalafil is another oral PDE-5 inhibitor but with a longer half-life than sildenafil. Unlike sildenafil, which requires 3 times daily dosing, tadalafil requires once daily administration. A few case reports have demonstrated tadalafil’s effectiveness for POPH in combination with other medical therapy (eg, sildenafil, ambrisentan).30,31
Guanylate Cyclase Stimulator
Riociguat
Riociguat is a first-in-class activator of soluble form of guanylate cyclase that increases levels of cyclic GMP. Two randomized clinical trials, PATENT, a study in PAH patients, and CHEST, a study in patients with chronic thromboembolic pulmonary hypertension showed improvement in 6MWD at 12 weeks (PATENT) or 16 weeks (CHEST), with improvement in secondary endpoints such as PVR, N-terminal pro b-type natriuretic peptide and WHO functional class.32,33 Riociguat may have potential advantages in patients with POPH given that it has a favorable liver safety profile. A subgroup analysis of patients enrolled in the PATENT study showed that 13 had POPH and 11 were randomized to receive riociguat 2.5 mg 3 times daily dose and 2 received placebo.34 Riociguat was well tolerated and improved 6MWD that was maintained over 2 years in the open label extension.
Medications to Avoid
Nonselective beta-blockers are commonly recommended in patients with portal hypertension to help prevent variceal hemorrhage. However, in patients with POPH, beta-blockers have been shown to decrease exercise capacity and worsen pulmonary hemodynamics. A study of 10 patients with moderate to severe POPH who were receiving beta-blockers for variceal bleeding prophylaxis showed that 6MWD improved in almost all of the patients, cardiac output increased by 28%, and PVR decreased by 19% when beta-blockers were discontinued.35 The authors concluded that the use of beta-blockers should be avoided in this patient population.
Calcium channel blockers should not be used in patients with POPH because they can cause significant hypotension due to systemic vasodilatation and decreased right ventricular filling. Patients with portal hypertension and chronic liver disease commonly have low systemic vascular resistance and are particularly susceptible to the deleterious effects of calcium channel blockers.
Transplantation
Liver transplantation is a potential cure for POPH and its role in POPH has evolved over the past 2 decades. In 1997, Ramsay et al published their review of 1205 consecutive liver transplants at Baylor University Medical Center (BUMC) in Texas.36 The incidence of POPH in this group was 8.5%, with the majority of patients having mild POPH. Liver transplant outcomes were not affected by mild and moderate pulmonary hypertension. However, patients with severe POPH (n = 7, systolic pulmonary artery pressure > 60 mm Hg) had a mortality rate of 42% at 9 months post-transplantation and 71% at 36 months post-transplant. The surviving patients continued to deteriorate with progressive right heart failure and no improvement in POPH.
To understand the effect of liver transplantation on POPH, one must understand the hemodynamic changes that occur with POPH and during liver transplant. The right ventricle is able to manage the same volume as the left ventricle under normal circumstances, but is unable to pump against a significant pressure gradient.37 In the setting of POPH, right ventricular hypertrophy occurs and RV output remains stable for some time. With time, pulmonary artery pressure increases secondary to pulmonary arteriolar vasoconstriction, intimal thickening, and progressive occlusion of the pulmonary vascular bed. Right ventricular failure may occur as a result. Cardiac output increases significantly at the time of reperfusion during liver transplant (up to 3-fold in 15 minutes),38 and in the setting of a noncompliant vascular bed, the patient is at risk for right heart failure. This is the likely explanation to such high perioperative mortality rates in patients with uncontrolled POPH. Failure to decrease MPAP to less than 50 mm Hg is considered a complete contraindication to liver transplant at most institutions. Many transplant centers will list patients for liver transplant if MPAP can be decreased to less than 35 mm Hg and PVR < 400 dynes/s/cm–5. These parameters are thought to represent an adequate right ventricular reserve and a compliant pulmonary vascular bed.37 However, even with good pressure control, the anesthesiology and critical care teams must be prepared to deal with acute right heart failure peri-operatively. Intraoperative transesophageal echocardiography has been recommended to closely follow right ventricular function.38 Inhaled or intravenous dilators are the most effective agents in the event of a pulmonary hypertensive crisis.
Review of Outcomes
A retrospective review evaluated 43 patients with untreated POPH who underwent attempted liver transplantation.39 Data were collected from 18 peer-reviewed studies and 7 patients at the authors’ institution. Overall mortality was 35% (15 patients), with almost all of the deaths secondary to cardiac dysfunction. Two deaths occurred intraoperatively and 8 deaths occurred during the transplant hospitalization. The transplant could not be successfully completed in 4 of the patients. MPAP > 50 mm Hg was associated with 100% mortality, whereas patients with MPAP between 35 mm Hg and 50 mm Hg had a 50% mortality. No mortality was noted in patients with MPAP < 35 mm Hg.
Liver transplantation has been shown to be successful in patients with controlled POPH. Sussman et al published their data on 8 patients with severe POPH in 2006. In this prospective study, all patients were treated with sequential epoprostenol infusions and 7 of the 8 patients experienced a significant reduction in MPAP and PVR. Six patients were listed for liver transplant, 4 of who were transplanted successfully and alive up to 5 years later.
The Baylor University Medical Center published their data on POPH patients who received liver transplants in 2007.11 POPH was confirmed by right heart catheterization in 30 patients evaluated for liver transplant. Sixteen patients were considered to be suitable candidates for transplant and MPAP was decreased to less than 35 mmHg in 12 patients with vasodilator therapy. Eleven patients eventually underwent liver transplant and 1- and 5-year survival rates were 91% and 67%.
Compared to medical therapy or liver transplant alone, patients who receive medical therapy followed by liver transplantation have the best survival. The Mayo Clinic retrospectively reviewed 74 POPH patients identified between 1994 and 2007.40 Patients were categorized in 1 of 3 categories: no medical therapy, medical therapy alone for POPH, or medical therapy for POPH followed by liver transplantation. Patients who received no medical therapy for POPH and no liver transplant had the worst outcomes, with a dismal 5-year survival of only 14% with over 50% deceased at 1 year of diagnosis. Five-year survival was 45% in patients who received medical therapy only. Patients who received medical therapy with prostacyclin followed by liver transplantation had the best outcomes, with a 5-year survival of 67% versus 25% in those who were transplanted without prior prostacyclin therapy.
We reported the longest follow-up study for patients undergoing liver transplantation with POPH in 2014.41 Seven patients with moderate to severe POPH received a liver transplant at our institution between June 2004 and January 2011. Mean pulmonary artery pressure was reduced to < 35 mm Hg, with appropriate POPH therapy in all of the patients. Both the graft and patient survival rates were 85.7% after a median follow-up of 7.8 years. The 1 patient who did not survive died from complications related to recurrent hepatitis C and cirrhosis, not from POPH-related issues. Four of the remaining 6 patients continue to require oral vasodilator therapy post-transplant, suggesting irreversible remodeling of the pulmonary vasculature. Two patients (4.4 and 8.5 years post-transplant) have no evidence of pulmonary hypertension post-transplant and therefore do not require medical treatment for pulmonary hypertension. We concluded that POPH responsive to vasodilator therapy is an appropriate indication for liver transplant, with excellent long-term survival.
Hollatz et al published their data on 11 patients with moderate to severe POPH who were successfully treated (mostly with oral sildenafil and subcutaneous treprostinil) as a bridge to liver transplant.42 The mortality rate was 0, with a follow-up duration of 7 to 60 months. Interestingly, 7 of the 11 patients (64%) were off all pulmonary vasodilators post-transplant. Ashfaq et al reported similar results.11 Nine of 11 patients with treated moderate to severe POPH who received liver transplants stopped vasodilator therapy at a median period of 9.2 months post-transplant. Raevens et al described a group of 3 patients with POPH who went on to liver transplant after their pulmonary pressures were decreased with combined oral vasodilator therapy: 1 required continued long-term vasodilator therapy, another was weaned off medications after transplant, and the third patient died during the liver transplant from perioperative complications that induced uncontrolled pulmonary hypertension.43
Patient Selection
In 2006, the United Network for Organ Sharing (UNOS) initiated a policy whereby a higher priority for liver transplantation was granted for highly selected patients in the United States.44 UNOS policy 3.6.4.5.6 upgraded POPH patients to a MELD score of 22, with an increase in MELD every 3 months as long as MPAP remained < 35 mm Hg and PVR remained < 400 dynes/s/cm–5. One hundred fifty-five patients were granted MELD exception points for POPH between 2002 and 2010 and went on to receive liver transplants.45 Goldberg et al collected data from the Organ Procurement and Transplantation Network (OPTN) and compared outcomes of patients with approved POPH MELD exception points versus waitlist candidates with no exception points.46 One hundred fifty-five waitlisted patients received POPH MELD exception points, with only 43.1% meeting OPTN exception requirements. One-third did not fulfill hemodynamic criteria consistent with POPH or had missing data, and 80% went on to receive a liver transplant. Waitlist candidates receiving POPH MELD exception points also had increased waitlist mortality and several early post-transplant deaths. The authors felt these data highlighted the need for OPTN/UNOS to revise their policy for POPH MELD exceptions points, revise how points are rewarded, and continue research to help risk stratify these patients to minimize perioperative complications.
Conclusion
Several effective medical treatment regimens are available, including prostanoids, endothelin receptor antagonists, and PDE-5 inhibitors. Liver transplantation is a potential cure but is only recommended if MPAP can be decreased to ≤ 35 mmHg. Long-term follow-up studies have shown these patients do well several years post-transplant but may continue to require oral therapy for their POPH.
1. Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925-1932.
2. Chin K, Rubin L. Pulmonary arterial hypertension. Am Coll Cardiol. 2008;51:1527-1538.
3. Doran A, Harris S, Goetz B. Advances in prostanoid infusion therapy for pulmonary arterial hypertension. J Infus Nurs. 2008;31:336-345.
4. Chin KM, Channick RN, De Lemos JA, ET AL. Hemodynamics and epoprostenol use are associated with thrombocytopenia in pulmonary arterial hypertension. Chest. 2009;135:130-136.
5. Hoshikawa Y, Voelkel NF, Gesell TL, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164:314-318.
6. Ricci GL, Melgosa MT, Burgos F, et al. Assessment of acute pulmonary vascular reactivity in portopulmonary hypertension. Liver Transplant. 2007;13:1506-1514.
7. McLaughlin V V, Genthner DE, Panella MM, et al. Compassionate use of continuous prostacyclin in the management of secondary pulmonary hypertension: a case series. Ann Intern Med. 1999;130:740-743.
8. Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology. 1999;30:641-648.
9. Fix OK, Bass NM, De Morco T, Merriman RB. Long-term follow-up of portopulmonary hypertension: Effect of treatment with epoprostenol. Liver Transplant. 2007;13:875-885.
10. Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant. 2006;6:2177-2182.
11. Ashfaq M, Chinnakotla S, Rogers L, et al. The impact of treatment of portopulmonary hypertension on survival following liver transplantation. Am J Transplant. 2007;7:1258-1264.
12. Sakai T, Planinsic RM, Mathier MA, et al. initial experience using continuous intravenous treprostinil to manage pulmonary arterial hypertension in patients with end-stage liver disease. Transpl Int. 2009;22:554-561.
13. McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915-1922.
14. Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J. 2007;30:1096-1102.
15. Melgosa MT, Ricci GL, Garcia-Pagan JC et al. Acute and long-term effects of inhaled iloprost in portopulmonary hypertension. Liver Transplant. 2010;16:348-356.
16. Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40:874-880
17. Sitbon O, Channick R, Chin, KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522-2533.
18. Moller S, Gulberg V, Henriksen JH, Gerbes AL. Endothelin-1 and endothelin-3 in cirrhosis: Relations to systemic and splanchnic haemodynamics. J Hepatol. 1995;23:135-144.
19. Eriksson C, Gustavsson A, Kronvall T, Tysk C. Hepatotoxicity by bosentan in a patient with portopulmonary hypertension : a case-report and review of the literature. J Gastrointestin Liver Dis. 2011;20:77-80.
20. Hoeper MM, Halank M, Marx C, et al. Bosentan therapy for portopulmonary hypertension. Eur Respir J. 2005;25:502-508.
21. Stähler G, Von Hunnius P. Successful treatment of portopulmonary hypertension with bosentan: Case report.: Eur J Clin Investig. 2006;36:62-66.
22. Savale L, Magnier R, Le Pavec J, et al. Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. Eur. 2013;41:96-103.
23. Halank M, Knudsen L, Seyfarth H, et al. Ambrisentan improves exercise capacity and symptoms in patients with portopulmonary hypertension. Z Gastroenterol. 2011;49:1258-1262.
24. Cartin-Ceba R, Swanson K, Iyer V, et al. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011;139:109-114.
25. Condliffe R, Elliot C, Hurdman J, et al. Ambrisentan therapy in pulmonary hypertension: clinical use and tolerability in a referral centre. Ther Adv Respir Dis. 2014;8:71-77.
26. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809-818.
27. Reichenberger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J. 2006;28:563-567.
28. Hemnes AR RI. Sildenafil monotherapy in portopulmonary hypertension can facilitate liver transplantation. Liver Transplant. 2009;15:15-19.
29. Gough WR. Sildenafil therapy is associated with improved hemodynamics in liver transplantation candidates with pulmonary arterial hypertension. Liver Transplant. 2009;15:30-36.
30. Yamashita Y. Hemodynamic effects of ambrisentan-tadalafil combination therapy on progressive portopulmonary hypertension. World J Hepatol. 2014;6:825.
31. Bremer HC, Kreisel W, Roecker K, et al. Phosphodiesterase 5 inhibitors lower both portal and pulmonary pressure in portopulmonary hypertension: a case report. J Med Case Rep. 2007;1:46.
32. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013:369;330-340.
33. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013:369;319-329
34. Cartin-Ceba R, Halank M, Ghofrani HA, et al. Riociguat treatment for portopulmonary hypertension: a subgroup analysis from the PATENT-1/-2 studies. Pulm Circ. 2018: 8:2045894018769305.
35. Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. 2006. Gastroenterology. 2006;130:120-126.
36. Ramsay M a, Simpson BR, Nguyen T, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494-500.
37. Safdar Z, Bartolome S, Sussman N. Portopulmonary hypertension : an update. Liver Tranpl. 2012;18:881-891.
38. Ramsay M. Portopulmonary hypertension and right heart failure in patients with cirrhosis. Curr Opin Anaesthesiol. 2010;23:145-150.
39. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443-450.
40. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445-2453.
41. Khaderi S, Khan R, Safdar Z, et al. Long-term follow-up of portopulmonary hypertension patients after liver transplantation. Liver Transplant. 2014;20:724-727.
42. Hollatz TJ, Musat A, Westphal S, et al. Treatment with sildenafil and treprostinil allows successful liver transplantation of patients with moderate to severe portopulmonary hypertension. Liver Transpl. 2012:686-695.
43. Raevens S, De Pauw M, Reyntjens K, et al. Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol. 2012:1-8.
44. Krowka M, Fallon M, Mulligan D. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transplant. 2006;12:S114-S116.
45. Krowka M, Wiesner R, Rosen C. Portopulmonary hypertension outcomes in the era of MELD exception. Liver Transplant. 2012;18:S259.
46. Goldberg DS, Batra S, Sahay S, et al. MELD Exceptions for portopulmonary hypertension: current policy and future implementation. Am J Transplant. 2014;14:2081-2087.
Portopulmonary hypertension (POPH) is a form of group 1 pulmonary arterial hypertension. When treating patients with POPH, baseline assessment is necessary so that response to therapy can be measured as the change from baseline. Patients should undergo echocardiography and right heart catheterization, and their exercise capacity and NYHA functional class should be determined. Patients with POPH should be considered for treatment if they are NYHA functional class II or above and/or their mean pulmonary artery pressure (MPAP) is greater than 35 mm Hg in transplant candidates. The goal in the treatment and management of POPH is to improve pulmonary hemodynamics by reducing the obstruction to pulmonary arterial flow and to preserve right ventricular function (Table). This article, the second in a 2-part review of POPH in patients with liver disease, reviews the role of medical therapy and liver transplantation in treatment. Evaluation and diagnosis of POPH are discussed in a separate article.
Medical Therapy
Prostanoids
Although prostacyclin and prostaglandin analogs entered routine clinical practice for POPH in the 1990s, reports of investigational use date back to the 1980s. Prostanoids are potent vasodilators with antiplatelet aggregation and antiproliferative properties. Prostacyclin synthase is reduced in patients with PAH, resulting in decreased concentration of prostacyclin with vasoconstriction and proliferative changes in the pulmonary vasculature.1
Epoprostenol
Epoprostenol is also known as synthetic prostaglandin I2 or prostacyclin. It was the first therapy approved for the treatment of PAH in 1995 by the US Food and Drug Administration (FDA) as a continuous intravenous infusion.2,3 It also inhibits platelet aggregation and may help modulate pulmonary vascular remodeling.4,5 Epoprostenol is derived from the metabolism of arachidonic acid and is a potent pulmonary and systemic vasodilator. One study reported an immediate 11.8% decrease in MPAP, 24% decrease in pulmonary vascular resistance (PVR) and 28% drop in systemic vascular resistance (SVR) during an epoprostenol infusion.6 The authors reported that epoprostenol was a more potent vasodilator than nitric oxide and may have a role in predicting the reversibility of POPH. In a case series of 33 patients with secondary pulmonary hypertension (including 7 patients with POPH) treated with continuous intravenous prostacyclin for approximately 1 year, exercise tolerance, NYHA functional class, and pulmonary hemodynamics improved in each patient compared to baseline.7 Krowka et al studied 14 patients with moderate to severe POPH treated with intravenous epoprostenol.8 No significant side effects were noted and treatment resulted in significant improvements in PVR, MPAP, and cardiac output. In 2007, Fix et al published a large retrospective cohort of patients with moderate to severe POPH.9 Nineteen patients treated with epoprostenol were compared to 17 patients with no treatment. After a median treatment period of 15.4 months, the epoprostenol group showed significant improvement in MPAP, PVR and cardiac output, but survival did not differ between the 2 groups.
Epoprostenol has often been considered a bridge to transplant in patients with POPH. Sussman et al described 8 consecutive patients with POPH who were treated with intravenous epoprostenol (2 to 8 ng/kg/min dose).10 Liver transplant was considered in 7 of the 8 patients when MPAP decreased to less than 35 mm Hg. Six patients were eventually listed for liver transplant, but 2 died waiting on the list. Long-term outcomes in the group of transplanted recipients were excellent. They remained alive and well at least 9 to 18 months post-transplant, and half did not require long-term vasodilator therapy post-orthotopic liver transplant. Similarly, Ashfaq et al published their data on 16 patients with moderate-to-severe POPH who were treated with vasodilator therapy.11 MPAP decreased to acceptable levels in 75% of the treated patients, and 11 went on to liver transplantation. Rates of 1- and 5-year survival in the transplanted patients were 91% and 67% respectively. None of the patients who failed vasodilator therapy survived.
Epoprostenol has a short half-life (3 to 5 minutes) and requires continuous infusion through central access via an infusion pump. Aseptic technique must be maintained to avoid blood stream infections. Pump failure or loss of vascular access can result in rebound pulmonary vasoconstriction that can be life-threatening and requires immediate attention. Side effects associated with epoprostenol include flushing, headache, nausea/vomiting, bradycardia, chest pain, jaw pain, diarrhea, and musculoskeletal pain.
Patients on epoprostenol should be monitored for prostanoid overdose. In the case of patients with chronic liver disease, epoprostenol increases systemic vasodilation in patients with already low systemic vascular tone. As a result, cardiac output may increase to the point of high cardiac output failure. MPAP will remain elevated secondary to high cardiac output rather than high PVR. In these patients, right heart catheterization will show an elevated MPAP in the setting of normal to low PVR/transpulmonary gradient (TPG) values. Lowering the epoprostenol dose will successfully reduce both cardiac output and MPAP.
Treprostinil
Treprostinil is a prostacyclin analog that is available in intravenous, inhalational, and subcutaneous form, although subcutaneous dosing may be limited by pain. Sakai et al published a small case series of 3 patients with PAH and end-stage liver disease treated with intravenous treprostinil.12 Pulmonary hemodynamics improved in all patients, and 2 patients went on to an uneventful liver transplantation. More than 10 years later, data were published on 255 patients with PAH on therapy with bosentan or sildenafil randomized to additional inhaled treprostinil.13 Treprostinil proved to be safe and well tolerated, with improvement in quality of life measures but no improvement in other secondary endpoints.
Iloprost
Inhaled iloprost is another prostacyclin that has a short therapeutic half-life of 20 to 30 minutes and requires frequent administration (6 to 9 times daily). In study in which patients with severe POPH were treated for up to 3 years with inhaled iloprost,14 survival rates at 1, 2, and 3 years were 77%, 62%, and 46%, respectively. A second study published in 2010 was designed to assess the acute effects of inhaled iloprost on pulmonary hemodynamics and evaluate the clinical outcome after 12 months of treatment.15 Iloprost was found to rapidly reduce pulmonary arterial pressure and PVR. In the long-term evaluation, inhaled iloprost increased the 6-minute walk distance (6MWD) and functional class, but no change was noted in the systolic pulmonary artery pressure. The authors concluded that iloprost might provide symptomatic improvement and improvement in exercise capacity.
Selexipag
Selexipag is an oral selective IP prostacyclin receptor agonist that is structurally distinct from other prostacyclins.16 In a phase 3 randomized double blind clinical trial, PAH patients treated with selexipag had lower composite of death or complication of PAH to the end of the study period.17 This effect was consistent across all dose ranges, but POPH patients were excluded from this study. Safety and efficacy of selexipag has not been evaluated in POPH patients.
Endothelin Receptor Antagonists
Endothelin receptor antagonists block the production of endothelin-1 (ET-1), a potent vasoconstrictor and smooth muscle mitogen that may contribute to the development of PAH. Three different receptors have been described: endothelin A, endothelin B, and endothelin B2. Elevated ET-1 levels have been reported in patients with chronic liver disease and may originate from hepatosplanchnic circulation.18
Bosentan
Bosentan is an oral, nonspecific, ET-1A and ET-1B receptor antagonist. Initial use of bosentan in patients with POPH was limited because of concern for hepatotoxicity. Approximately 10% of patients on bosentan were reported to have mild hepatic side effects in the form of elevated aminotransferases, but severe injury has been reported.19 One of the first clinical experiences of bosentan in patients with POPH was published in 2005. Hoeper et al followed 11 patients with Child A cirrhosis and severe POPH.20 All patients included were in NYHA functional class III or IV and were treated with bosentan for over 1 year. Exercise capacity and symptoms improved in all treated patients. The medication was tolerated well and there was no evidence of drug-induced liver injury. A single case report showed the effectiveness of bosentan in a 43-year-old man with alcohol-related liver disease (Child-Pugh A) and right ventricular enlargement and dysfunction secondary to POPH.21 Pulmonary arterial pressure decreased, exercise capacity increased, and improvement was maintained over 2 years.
In a group of 31 patients with Child A or B cirrhosis and severe POPH, bosentan had significantly better effects than inhaled iloprost on exercise capacity, hemodynamics, and survival.14 One, 2, and 3-year survival rates in the bosentan group were 94%, 89%, and 89% (compared to 77%, 62%, and 46% in the iloprost group). Both drugs were considered safe with no reported hepatotoxicity. In 2013, Savale et al published data on 34 patients with POPH, Child-Pugh A and/or B who were treated with bosentan for a median of 43 months.22 The authors reported significant improvements in hemodynamics, NYHA functional class, and 6WMD. Event-free survival rates at 1, 2, and 3 years were 82%, 63%, and 47%, respectively.
Ambrisentan
Ambrisentan is a highly selective ET-1A receptor antagonist with once daily dosing and a lower risk of hepatotoxicity compared to bosentan. Fourteen patients with moderate to severe POPH treated with ambrisentan in 4 German hospitals were retrospectively analyzed.23 Median follow-up was 16 months, and the study demonstrated significant improvement in exercise capacity and clinical symptoms without significant change in liver function tests. Cartin-Ceba et al published their experience of 13 patients with moderate to severe POPH treated with ambrisentan monotherapy.24 Patients were followed for a median of 613 days and on treatment for a median time of 390 days. Significant improvements were shown in pulmonary arterial pressure and PVR without adverse effect on hepatic function. Over 270 patients with PAH (6% with POPH) received ambrisentan from March 2009 through June 2013 at a large United Kingdom portal hypertension referral center.25 Discontinuation due to side effects was higher than previously reported. Discontinuation due to abnormal transaminases was uncommon.
Macitentan
Macitentan is a dual endothelin-receptor antagonist developed by modifying the structure of bosentan to increase efficacy and safety. The SERAPHIN trial compared oral macitentan to placebo in 250 patients with moderate to severe PAH, some of whom were also on a stable dose of oral or inhaled therapy for PAH.26 Over a 2-year period, patients treated with macitentan were less likely to have progression of their disease or die on therapy (38% and 31% versus 46%), regardless of if they were receiving additional oral therapy and more likely to have improvement of their exercise capacity and WHO functional class. Nasopharyngitis and significant anemia were more common in the macitentan group, but there was no difference in the rate of liver function test abnormalities compared to placebo. Trials with macitentan are currently ongoing in patients with POPH.
Phosphodiesterase-5 Inhibitors
Cyclic guanosine monophosphate (cGMP) is the mediator of nitric oxide–induced vasodilation. Phosphodiesterase-5 (PDE-5) inhibitors prolong the vasodilatory effects of cyclic guanosine monophosphate by preventing its hydrolysis, thereby reducing the pulmonary arterial pressure.
Sildenafil
Sildenafil is the most widely accepted PDE-5 inhibitor for POPH. Fourteen patients with moderate to severe POPH were treated with sildenafil (50 mg 3 times per day) in an observational study published by Reichenberger et al in 2006.27 Eight patients were newly started on sildenafil, whereas sildenafil was added to inhaled prostanoids in the remaining 6x patients. Sildenafil significantly decreased 66MWD, MPAP, PVR, and cardiac index alone or in combination with inhaled prostanoids.
Sildenafil has also been used as a bridge to transplant in liver transplant candidates with POPH. Ten patients with POPH treated with sildenafil monotherapy were followed for a 21±16 months.28 Patients improved symptomatically and increased their 6MWD at 1 year by 30 meters or more. Three patients became transplant eligible and another 3 patients were stable, without progression of their liver disease or POPH. Four patients were not considered transplant candidates, 2 because of refractory POPH and 2 for other comorbidities. The authors concluded that sildenafil monotherapy could stabilize or improve pulmonary hemodynamics in patients with POPH and eventually lead to liver transplantation. Gough et al took a similar look at 9 patients with POPH treated with sildenafil.29 All patients had initial and follow-up right heart catheterizations within a period of 3 years. Mean PVR improved in all patients, decreasing from 575 to 375 dynes/s/cm–5. MPAP decreased to ≤ 35 mmHg in 4 patients, 1 of whom went on to receive a liver transplant. Overall sildenafil improved pulmonary hemodynamics in this small cohort of POPH patients.
Tadalafil
Tadalafil is another oral PDE-5 inhibitor but with a longer half-life than sildenafil. Unlike sildenafil, which requires 3 times daily dosing, tadalafil requires once daily administration. A few case reports have demonstrated tadalafil’s effectiveness for POPH in combination with other medical therapy (eg, sildenafil, ambrisentan).30,31
Guanylate Cyclase Stimulator
Riociguat
Riociguat is a first-in-class activator of soluble form of guanylate cyclase that increases levels of cyclic GMP. Two randomized clinical trials, PATENT, a study in PAH patients, and CHEST, a study in patients with chronic thromboembolic pulmonary hypertension showed improvement in 6MWD at 12 weeks (PATENT) or 16 weeks (CHEST), with improvement in secondary endpoints such as PVR, N-terminal pro b-type natriuretic peptide and WHO functional class.32,33 Riociguat may have potential advantages in patients with POPH given that it has a favorable liver safety profile. A subgroup analysis of patients enrolled in the PATENT study showed that 13 had POPH and 11 were randomized to receive riociguat 2.5 mg 3 times daily dose and 2 received placebo.34 Riociguat was well tolerated and improved 6MWD that was maintained over 2 years in the open label extension.
Medications to Avoid
Nonselective beta-blockers are commonly recommended in patients with portal hypertension to help prevent variceal hemorrhage. However, in patients with POPH, beta-blockers have been shown to decrease exercise capacity and worsen pulmonary hemodynamics. A study of 10 patients with moderate to severe POPH who were receiving beta-blockers for variceal bleeding prophylaxis showed that 6MWD improved in almost all of the patients, cardiac output increased by 28%, and PVR decreased by 19% when beta-blockers were discontinued.35 The authors concluded that the use of beta-blockers should be avoided in this patient population.
Calcium channel blockers should not be used in patients with POPH because they can cause significant hypotension due to systemic vasodilatation and decreased right ventricular filling. Patients with portal hypertension and chronic liver disease commonly have low systemic vascular resistance and are particularly susceptible to the deleterious effects of calcium channel blockers.
Transplantation
Liver transplantation is a potential cure for POPH and its role in POPH has evolved over the past 2 decades. In 1997, Ramsay et al published their review of 1205 consecutive liver transplants at Baylor University Medical Center (BUMC) in Texas.36 The incidence of POPH in this group was 8.5%, with the majority of patients having mild POPH. Liver transplant outcomes were not affected by mild and moderate pulmonary hypertension. However, patients with severe POPH (n = 7, systolic pulmonary artery pressure > 60 mm Hg) had a mortality rate of 42% at 9 months post-transplantation and 71% at 36 months post-transplant. The surviving patients continued to deteriorate with progressive right heart failure and no improvement in POPH.
To understand the effect of liver transplantation on POPH, one must understand the hemodynamic changes that occur with POPH and during liver transplant. The right ventricle is able to manage the same volume as the left ventricle under normal circumstances, but is unable to pump against a significant pressure gradient.37 In the setting of POPH, right ventricular hypertrophy occurs and RV output remains stable for some time. With time, pulmonary artery pressure increases secondary to pulmonary arteriolar vasoconstriction, intimal thickening, and progressive occlusion of the pulmonary vascular bed. Right ventricular failure may occur as a result. Cardiac output increases significantly at the time of reperfusion during liver transplant (up to 3-fold in 15 minutes),38 and in the setting of a noncompliant vascular bed, the patient is at risk for right heart failure. This is the likely explanation to such high perioperative mortality rates in patients with uncontrolled POPH. Failure to decrease MPAP to less than 50 mm Hg is considered a complete contraindication to liver transplant at most institutions. Many transplant centers will list patients for liver transplant if MPAP can be decreased to less than 35 mm Hg and PVR < 400 dynes/s/cm–5. These parameters are thought to represent an adequate right ventricular reserve and a compliant pulmonary vascular bed.37 However, even with good pressure control, the anesthesiology and critical care teams must be prepared to deal with acute right heart failure peri-operatively. Intraoperative transesophageal echocardiography has been recommended to closely follow right ventricular function.38 Inhaled or intravenous dilators are the most effective agents in the event of a pulmonary hypertensive crisis.
Review of Outcomes
A retrospective review evaluated 43 patients with untreated POPH who underwent attempted liver transplantation.39 Data were collected from 18 peer-reviewed studies and 7 patients at the authors’ institution. Overall mortality was 35% (15 patients), with almost all of the deaths secondary to cardiac dysfunction. Two deaths occurred intraoperatively and 8 deaths occurred during the transplant hospitalization. The transplant could not be successfully completed in 4 of the patients. MPAP > 50 mm Hg was associated with 100% mortality, whereas patients with MPAP between 35 mm Hg and 50 mm Hg had a 50% mortality. No mortality was noted in patients with MPAP < 35 mm Hg.
Liver transplantation has been shown to be successful in patients with controlled POPH. Sussman et al published their data on 8 patients with severe POPH in 2006. In this prospective study, all patients were treated with sequential epoprostenol infusions and 7 of the 8 patients experienced a significant reduction in MPAP and PVR. Six patients were listed for liver transplant, 4 of who were transplanted successfully and alive up to 5 years later.
The Baylor University Medical Center published their data on POPH patients who received liver transplants in 2007.11 POPH was confirmed by right heart catheterization in 30 patients evaluated for liver transplant. Sixteen patients were considered to be suitable candidates for transplant and MPAP was decreased to less than 35 mmHg in 12 patients with vasodilator therapy. Eleven patients eventually underwent liver transplant and 1- and 5-year survival rates were 91% and 67%.
Compared to medical therapy or liver transplant alone, patients who receive medical therapy followed by liver transplantation have the best survival. The Mayo Clinic retrospectively reviewed 74 POPH patients identified between 1994 and 2007.40 Patients were categorized in 1 of 3 categories: no medical therapy, medical therapy alone for POPH, or medical therapy for POPH followed by liver transplantation. Patients who received no medical therapy for POPH and no liver transplant had the worst outcomes, with a dismal 5-year survival of only 14% with over 50% deceased at 1 year of diagnosis. Five-year survival was 45% in patients who received medical therapy only. Patients who received medical therapy with prostacyclin followed by liver transplantation had the best outcomes, with a 5-year survival of 67% versus 25% in those who were transplanted without prior prostacyclin therapy.
We reported the longest follow-up study for patients undergoing liver transplantation with POPH in 2014.41 Seven patients with moderate to severe POPH received a liver transplant at our institution between June 2004 and January 2011. Mean pulmonary artery pressure was reduced to < 35 mm Hg, with appropriate POPH therapy in all of the patients. Both the graft and patient survival rates were 85.7% after a median follow-up of 7.8 years. The 1 patient who did not survive died from complications related to recurrent hepatitis C and cirrhosis, not from POPH-related issues. Four of the remaining 6 patients continue to require oral vasodilator therapy post-transplant, suggesting irreversible remodeling of the pulmonary vasculature. Two patients (4.4 and 8.5 years post-transplant) have no evidence of pulmonary hypertension post-transplant and therefore do not require medical treatment for pulmonary hypertension. We concluded that POPH responsive to vasodilator therapy is an appropriate indication for liver transplant, with excellent long-term survival.
Hollatz et al published their data on 11 patients with moderate to severe POPH who were successfully treated (mostly with oral sildenafil and subcutaneous treprostinil) as a bridge to liver transplant.42 The mortality rate was 0, with a follow-up duration of 7 to 60 months. Interestingly, 7 of the 11 patients (64%) were off all pulmonary vasodilators post-transplant. Ashfaq et al reported similar results.11 Nine of 11 patients with treated moderate to severe POPH who received liver transplants stopped vasodilator therapy at a median period of 9.2 months post-transplant. Raevens et al described a group of 3 patients with POPH who went on to liver transplant after their pulmonary pressures were decreased with combined oral vasodilator therapy: 1 required continued long-term vasodilator therapy, another was weaned off medications after transplant, and the third patient died during the liver transplant from perioperative complications that induced uncontrolled pulmonary hypertension.43
Patient Selection
In 2006, the United Network for Organ Sharing (UNOS) initiated a policy whereby a higher priority for liver transplantation was granted for highly selected patients in the United States.44 UNOS policy 3.6.4.5.6 upgraded POPH patients to a MELD score of 22, with an increase in MELD every 3 months as long as MPAP remained < 35 mm Hg and PVR remained < 400 dynes/s/cm–5. One hundred fifty-five patients were granted MELD exception points for POPH between 2002 and 2010 and went on to receive liver transplants.45 Goldberg et al collected data from the Organ Procurement and Transplantation Network (OPTN) and compared outcomes of patients with approved POPH MELD exception points versus waitlist candidates with no exception points.46 One hundred fifty-five waitlisted patients received POPH MELD exception points, with only 43.1% meeting OPTN exception requirements. One-third did not fulfill hemodynamic criteria consistent with POPH or had missing data, and 80% went on to receive a liver transplant. Waitlist candidates receiving POPH MELD exception points also had increased waitlist mortality and several early post-transplant deaths. The authors felt these data highlighted the need for OPTN/UNOS to revise their policy for POPH MELD exceptions points, revise how points are rewarded, and continue research to help risk stratify these patients to minimize perioperative complications.
Conclusion
Several effective medical treatment regimens are available, including prostanoids, endothelin receptor antagonists, and PDE-5 inhibitors. Liver transplantation is a potential cure but is only recommended if MPAP can be decreased to ≤ 35 mmHg. Long-term follow-up studies have shown these patients do well several years post-transplant but may continue to require oral therapy for their POPH.
Portopulmonary hypertension (POPH) is a form of group 1 pulmonary arterial hypertension. When treating patients with POPH, baseline assessment is necessary so that response to therapy can be measured as the change from baseline. Patients should undergo echocardiography and right heart catheterization, and their exercise capacity and NYHA functional class should be determined. Patients with POPH should be considered for treatment if they are NYHA functional class II or above and/or their mean pulmonary artery pressure (MPAP) is greater than 35 mm Hg in transplant candidates. The goal in the treatment and management of POPH is to improve pulmonary hemodynamics by reducing the obstruction to pulmonary arterial flow and to preserve right ventricular function (Table). This article, the second in a 2-part review of POPH in patients with liver disease, reviews the role of medical therapy and liver transplantation in treatment. Evaluation and diagnosis of POPH are discussed in a separate article.
Medical Therapy
Prostanoids
Although prostacyclin and prostaglandin analogs entered routine clinical practice for POPH in the 1990s, reports of investigational use date back to the 1980s. Prostanoids are potent vasodilators with antiplatelet aggregation and antiproliferative properties. Prostacyclin synthase is reduced in patients with PAH, resulting in decreased concentration of prostacyclin with vasoconstriction and proliferative changes in the pulmonary vasculature.1
Epoprostenol
Epoprostenol is also known as synthetic prostaglandin I2 or prostacyclin. It was the first therapy approved for the treatment of PAH in 1995 by the US Food and Drug Administration (FDA) as a continuous intravenous infusion.2,3 It also inhibits platelet aggregation and may help modulate pulmonary vascular remodeling.4,5 Epoprostenol is derived from the metabolism of arachidonic acid and is a potent pulmonary and systemic vasodilator. One study reported an immediate 11.8% decrease in MPAP, 24% decrease in pulmonary vascular resistance (PVR) and 28% drop in systemic vascular resistance (SVR) during an epoprostenol infusion.6 The authors reported that epoprostenol was a more potent vasodilator than nitric oxide and may have a role in predicting the reversibility of POPH. In a case series of 33 patients with secondary pulmonary hypertension (including 7 patients with POPH) treated with continuous intravenous prostacyclin for approximately 1 year, exercise tolerance, NYHA functional class, and pulmonary hemodynamics improved in each patient compared to baseline.7 Krowka et al studied 14 patients with moderate to severe POPH treated with intravenous epoprostenol.8 No significant side effects were noted and treatment resulted in significant improvements in PVR, MPAP, and cardiac output. In 2007, Fix et al published a large retrospective cohort of patients with moderate to severe POPH.9 Nineteen patients treated with epoprostenol were compared to 17 patients with no treatment. After a median treatment period of 15.4 months, the epoprostenol group showed significant improvement in MPAP, PVR and cardiac output, but survival did not differ between the 2 groups.
Epoprostenol has often been considered a bridge to transplant in patients with POPH. Sussman et al described 8 consecutive patients with POPH who were treated with intravenous epoprostenol (2 to 8 ng/kg/min dose).10 Liver transplant was considered in 7 of the 8 patients when MPAP decreased to less than 35 mm Hg. Six patients were eventually listed for liver transplant, but 2 died waiting on the list. Long-term outcomes in the group of transplanted recipients were excellent. They remained alive and well at least 9 to 18 months post-transplant, and half did not require long-term vasodilator therapy post-orthotopic liver transplant. Similarly, Ashfaq et al published their data on 16 patients with moderate-to-severe POPH who were treated with vasodilator therapy.11 MPAP decreased to acceptable levels in 75% of the treated patients, and 11 went on to liver transplantation. Rates of 1- and 5-year survival in the transplanted patients were 91% and 67% respectively. None of the patients who failed vasodilator therapy survived.
Epoprostenol has a short half-life (3 to 5 minutes) and requires continuous infusion through central access via an infusion pump. Aseptic technique must be maintained to avoid blood stream infections. Pump failure or loss of vascular access can result in rebound pulmonary vasoconstriction that can be life-threatening and requires immediate attention. Side effects associated with epoprostenol include flushing, headache, nausea/vomiting, bradycardia, chest pain, jaw pain, diarrhea, and musculoskeletal pain.
Patients on epoprostenol should be monitored for prostanoid overdose. In the case of patients with chronic liver disease, epoprostenol increases systemic vasodilation in patients with already low systemic vascular tone. As a result, cardiac output may increase to the point of high cardiac output failure. MPAP will remain elevated secondary to high cardiac output rather than high PVR. In these patients, right heart catheterization will show an elevated MPAP in the setting of normal to low PVR/transpulmonary gradient (TPG) values. Lowering the epoprostenol dose will successfully reduce both cardiac output and MPAP.
Treprostinil
Treprostinil is a prostacyclin analog that is available in intravenous, inhalational, and subcutaneous form, although subcutaneous dosing may be limited by pain. Sakai et al published a small case series of 3 patients with PAH and end-stage liver disease treated with intravenous treprostinil.12 Pulmonary hemodynamics improved in all patients, and 2 patients went on to an uneventful liver transplantation. More than 10 years later, data were published on 255 patients with PAH on therapy with bosentan or sildenafil randomized to additional inhaled treprostinil.13 Treprostinil proved to be safe and well tolerated, with improvement in quality of life measures but no improvement in other secondary endpoints.
Iloprost
Inhaled iloprost is another prostacyclin that has a short therapeutic half-life of 20 to 30 minutes and requires frequent administration (6 to 9 times daily). In study in which patients with severe POPH were treated for up to 3 years with inhaled iloprost,14 survival rates at 1, 2, and 3 years were 77%, 62%, and 46%, respectively. A second study published in 2010 was designed to assess the acute effects of inhaled iloprost on pulmonary hemodynamics and evaluate the clinical outcome after 12 months of treatment.15 Iloprost was found to rapidly reduce pulmonary arterial pressure and PVR. In the long-term evaluation, inhaled iloprost increased the 6-minute walk distance (6MWD) and functional class, but no change was noted in the systolic pulmonary artery pressure. The authors concluded that iloprost might provide symptomatic improvement and improvement in exercise capacity.
Selexipag
Selexipag is an oral selective IP prostacyclin receptor agonist that is structurally distinct from other prostacyclins.16 In a phase 3 randomized double blind clinical trial, PAH patients treated with selexipag had lower composite of death or complication of PAH to the end of the study period.17 This effect was consistent across all dose ranges, but POPH patients were excluded from this study. Safety and efficacy of selexipag has not been evaluated in POPH patients.
Endothelin Receptor Antagonists
Endothelin receptor antagonists block the production of endothelin-1 (ET-1), a potent vasoconstrictor and smooth muscle mitogen that may contribute to the development of PAH. Three different receptors have been described: endothelin A, endothelin B, and endothelin B2. Elevated ET-1 levels have been reported in patients with chronic liver disease and may originate from hepatosplanchnic circulation.18
Bosentan
Bosentan is an oral, nonspecific, ET-1A and ET-1B receptor antagonist. Initial use of bosentan in patients with POPH was limited because of concern for hepatotoxicity. Approximately 10% of patients on bosentan were reported to have mild hepatic side effects in the form of elevated aminotransferases, but severe injury has been reported.19 One of the first clinical experiences of bosentan in patients with POPH was published in 2005. Hoeper et al followed 11 patients with Child A cirrhosis and severe POPH.20 All patients included were in NYHA functional class III or IV and were treated with bosentan for over 1 year. Exercise capacity and symptoms improved in all treated patients. The medication was tolerated well and there was no evidence of drug-induced liver injury. A single case report showed the effectiveness of bosentan in a 43-year-old man with alcohol-related liver disease (Child-Pugh A) and right ventricular enlargement and dysfunction secondary to POPH.21 Pulmonary arterial pressure decreased, exercise capacity increased, and improvement was maintained over 2 years.
In a group of 31 patients with Child A or B cirrhosis and severe POPH, bosentan had significantly better effects than inhaled iloprost on exercise capacity, hemodynamics, and survival.14 One, 2, and 3-year survival rates in the bosentan group were 94%, 89%, and 89% (compared to 77%, 62%, and 46% in the iloprost group). Both drugs were considered safe with no reported hepatotoxicity. In 2013, Savale et al published data on 34 patients with POPH, Child-Pugh A and/or B who were treated with bosentan for a median of 43 months.22 The authors reported significant improvements in hemodynamics, NYHA functional class, and 6WMD. Event-free survival rates at 1, 2, and 3 years were 82%, 63%, and 47%, respectively.
Ambrisentan
Ambrisentan is a highly selective ET-1A receptor antagonist with once daily dosing and a lower risk of hepatotoxicity compared to bosentan. Fourteen patients with moderate to severe POPH treated with ambrisentan in 4 German hospitals were retrospectively analyzed.23 Median follow-up was 16 months, and the study demonstrated significant improvement in exercise capacity and clinical symptoms without significant change in liver function tests. Cartin-Ceba et al published their experience of 13 patients with moderate to severe POPH treated with ambrisentan monotherapy.24 Patients were followed for a median of 613 days and on treatment for a median time of 390 days. Significant improvements were shown in pulmonary arterial pressure and PVR without adverse effect on hepatic function. Over 270 patients with PAH (6% with POPH) received ambrisentan from March 2009 through June 2013 at a large United Kingdom portal hypertension referral center.25 Discontinuation due to side effects was higher than previously reported. Discontinuation due to abnormal transaminases was uncommon.
Macitentan
Macitentan is a dual endothelin-receptor antagonist developed by modifying the structure of bosentan to increase efficacy and safety. The SERAPHIN trial compared oral macitentan to placebo in 250 patients with moderate to severe PAH, some of whom were also on a stable dose of oral or inhaled therapy for PAH.26 Over a 2-year period, patients treated with macitentan were less likely to have progression of their disease or die on therapy (38% and 31% versus 46%), regardless of if they were receiving additional oral therapy and more likely to have improvement of their exercise capacity and WHO functional class. Nasopharyngitis and significant anemia were more common in the macitentan group, but there was no difference in the rate of liver function test abnormalities compared to placebo. Trials with macitentan are currently ongoing in patients with POPH.
Phosphodiesterase-5 Inhibitors
Cyclic guanosine monophosphate (cGMP) is the mediator of nitric oxide–induced vasodilation. Phosphodiesterase-5 (PDE-5) inhibitors prolong the vasodilatory effects of cyclic guanosine monophosphate by preventing its hydrolysis, thereby reducing the pulmonary arterial pressure.
Sildenafil
Sildenafil is the most widely accepted PDE-5 inhibitor for POPH. Fourteen patients with moderate to severe POPH were treated with sildenafil (50 mg 3 times per day) in an observational study published by Reichenberger et al in 2006.27 Eight patients were newly started on sildenafil, whereas sildenafil was added to inhaled prostanoids in the remaining 6x patients. Sildenafil significantly decreased 66MWD, MPAP, PVR, and cardiac index alone or in combination with inhaled prostanoids.
Sildenafil has also been used as a bridge to transplant in liver transplant candidates with POPH. Ten patients with POPH treated with sildenafil monotherapy were followed for a 21±16 months.28 Patients improved symptomatically and increased their 6MWD at 1 year by 30 meters or more. Three patients became transplant eligible and another 3 patients were stable, without progression of their liver disease or POPH. Four patients were not considered transplant candidates, 2 because of refractory POPH and 2 for other comorbidities. The authors concluded that sildenafil monotherapy could stabilize or improve pulmonary hemodynamics in patients with POPH and eventually lead to liver transplantation. Gough et al took a similar look at 9 patients with POPH treated with sildenafil.29 All patients had initial and follow-up right heart catheterizations within a period of 3 years. Mean PVR improved in all patients, decreasing from 575 to 375 dynes/s/cm–5. MPAP decreased to ≤ 35 mmHg in 4 patients, 1 of whom went on to receive a liver transplant. Overall sildenafil improved pulmonary hemodynamics in this small cohort of POPH patients.
Tadalafil
Tadalafil is another oral PDE-5 inhibitor but with a longer half-life than sildenafil. Unlike sildenafil, which requires 3 times daily dosing, tadalafil requires once daily administration. A few case reports have demonstrated tadalafil’s effectiveness for POPH in combination with other medical therapy (eg, sildenafil, ambrisentan).30,31
Guanylate Cyclase Stimulator
Riociguat
Riociguat is a first-in-class activator of soluble form of guanylate cyclase that increases levels of cyclic GMP. Two randomized clinical trials, PATENT, a study in PAH patients, and CHEST, a study in patients with chronic thromboembolic pulmonary hypertension showed improvement in 6MWD at 12 weeks (PATENT) or 16 weeks (CHEST), with improvement in secondary endpoints such as PVR, N-terminal pro b-type natriuretic peptide and WHO functional class.32,33 Riociguat may have potential advantages in patients with POPH given that it has a favorable liver safety profile. A subgroup analysis of patients enrolled in the PATENT study showed that 13 had POPH and 11 were randomized to receive riociguat 2.5 mg 3 times daily dose and 2 received placebo.34 Riociguat was well tolerated and improved 6MWD that was maintained over 2 years in the open label extension.
Medications to Avoid
Nonselective beta-blockers are commonly recommended in patients with portal hypertension to help prevent variceal hemorrhage. However, in patients with POPH, beta-blockers have been shown to decrease exercise capacity and worsen pulmonary hemodynamics. A study of 10 patients with moderate to severe POPH who were receiving beta-blockers for variceal bleeding prophylaxis showed that 6MWD improved in almost all of the patients, cardiac output increased by 28%, and PVR decreased by 19% when beta-blockers were discontinued.35 The authors concluded that the use of beta-blockers should be avoided in this patient population.
Calcium channel blockers should not be used in patients with POPH because they can cause significant hypotension due to systemic vasodilatation and decreased right ventricular filling. Patients with portal hypertension and chronic liver disease commonly have low systemic vascular resistance and are particularly susceptible to the deleterious effects of calcium channel blockers.
Transplantation
Liver transplantation is a potential cure for POPH and its role in POPH has evolved over the past 2 decades. In 1997, Ramsay et al published their review of 1205 consecutive liver transplants at Baylor University Medical Center (BUMC) in Texas.36 The incidence of POPH in this group was 8.5%, with the majority of patients having mild POPH. Liver transplant outcomes were not affected by mild and moderate pulmonary hypertension. However, patients with severe POPH (n = 7, systolic pulmonary artery pressure > 60 mm Hg) had a mortality rate of 42% at 9 months post-transplantation and 71% at 36 months post-transplant. The surviving patients continued to deteriorate with progressive right heart failure and no improvement in POPH.
To understand the effect of liver transplantation on POPH, one must understand the hemodynamic changes that occur with POPH and during liver transplant. The right ventricle is able to manage the same volume as the left ventricle under normal circumstances, but is unable to pump against a significant pressure gradient.37 In the setting of POPH, right ventricular hypertrophy occurs and RV output remains stable for some time. With time, pulmonary artery pressure increases secondary to pulmonary arteriolar vasoconstriction, intimal thickening, and progressive occlusion of the pulmonary vascular bed. Right ventricular failure may occur as a result. Cardiac output increases significantly at the time of reperfusion during liver transplant (up to 3-fold in 15 minutes),38 and in the setting of a noncompliant vascular bed, the patient is at risk for right heart failure. This is the likely explanation to such high perioperative mortality rates in patients with uncontrolled POPH. Failure to decrease MPAP to less than 50 mm Hg is considered a complete contraindication to liver transplant at most institutions. Many transplant centers will list patients for liver transplant if MPAP can be decreased to less than 35 mm Hg and PVR < 400 dynes/s/cm–5. These parameters are thought to represent an adequate right ventricular reserve and a compliant pulmonary vascular bed.37 However, even with good pressure control, the anesthesiology and critical care teams must be prepared to deal with acute right heart failure peri-operatively. Intraoperative transesophageal echocardiography has been recommended to closely follow right ventricular function.38 Inhaled or intravenous dilators are the most effective agents in the event of a pulmonary hypertensive crisis.
Review of Outcomes
A retrospective review evaluated 43 patients with untreated POPH who underwent attempted liver transplantation.39 Data were collected from 18 peer-reviewed studies and 7 patients at the authors’ institution. Overall mortality was 35% (15 patients), with almost all of the deaths secondary to cardiac dysfunction. Two deaths occurred intraoperatively and 8 deaths occurred during the transplant hospitalization. The transplant could not be successfully completed in 4 of the patients. MPAP > 50 mm Hg was associated with 100% mortality, whereas patients with MPAP between 35 mm Hg and 50 mm Hg had a 50% mortality. No mortality was noted in patients with MPAP < 35 mm Hg.
Liver transplantation has been shown to be successful in patients with controlled POPH. Sussman et al published their data on 8 patients with severe POPH in 2006. In this prospective study, all patients were treated with sequential epoprostenol infusions and 7 of the 8 patients experienced a significant reduction in MPAP and PVR. Six patients were listed for liver transplant, 4 of who were transplanted successfully and alive up to 5 years later.
The Baylor University Medical Center published their data on POPH patients who received liver transplants in 2007.11 POPH was confirmed by right heart catheterization in 30 patients evaluated for liver transplant. Sixteen patients were considered to be suitable candidates for transplant and MPAP was decreased to less than 35 mmHg in 12 patients with vasodilator therapy. Eleven patients eventually underwent liver transplant and 1- and 5-year survival rates were 91% and 67%.
Compared to medical therapy or liver transplant alone, patients who receive medical therapy followed by liver transplantation have the best survival. The Mayo Clinic retrospectively reviewed 74 POPH patients identified between 1994 and 2007.40 Patients were categorized in 1 of 3 categories: no medical therapy, medical therapy alone for POPH, or medical therapy for POPH followed by liver transplantation. Patients who received no medical therapy for POPH and no liver transplant had the worst outcomes, with a dismal 5-year survival of only 14% with over 50% deceased at 1 year of diagnosis. Five-year survival was 45% in patients who received medical therapy only. Patients who received medical therapy with prostacyclin followed by liver transplantation had the best outcomes, with a 5-year survival of 67% versus 25% in those who were transplanted without prior prostacyclin therapy.
We reported the longest follow-up study for patients undergoing liver transplantation with POPH in 2014.41 Seven patients with moderate to severe POPH received a liver transplant at our institution between June 2004 and January 2011. Mean pulmonary artery pressure was reduced to < 35 mm Hg, with appropriate POPH therapy in all of the patients. Both the graft and patient survival rates were 85.7% after a median follow-up of 7.8 years. The 1 patient who did not survive died from complications related to recurrent hepatitis C and cirrhosis, not from POPH-related issues. Four of the remaining 6 patients continue to require oral vasodilator therapy post-transplant, suggesting irreversible remodeling of the pulmonary vasculature. Two patients (4.4 and 8.5 years post-transplant) have no evidence of pulmonary hypertension post-transplant and therefore do not require medical treatment for pulmonary hypertension. We concluded that POPH responsive to vasodilator therapy is an appropriate indication for liver transplant, with excellent long-term survival.
Hollatz et al published their data on 11 patients with moderate to severe POPH who were successfully treated (mostly with oral sildenafil and subcutaneous treprostinil) as a bridge to liver transplant.42 The mortality rate was 0, with a follow-up duration of 7 to 60 months. Interestingly, 7 of the 11 patients (64%) were off all pulmonary vasodilators post-transplant. Ashfaq et al reported similar results.11 Nine of 11 patients with treated moderate to severe POPH who received liver transplants stopped vasodilator therapy at a median period of 9.2 months post-transplant. Raevens et al described a group of 3 patients with POPH who went on to liver transplant after their pulmonary pressures were decreased with combined oral vasodilator therapy: 1 required continued long-term vasodilator therapy, another was weaned off medications after transplant, and the third patient died during the liver transplant from perioperative complications that induced uncontrolled pulmonary hypertension.43
Patient Selection
In 2006, the United Network for Organ Sharing (UNOS) initiated a policy whereby a higher priority for liver transplantation was granted for highly selected patients in the United States.44 UNOS policy 3.6.4.5.6 upgraded POPH patients to a MELD score of 22, with an increase in MELD every 3 months as long as MPAP remained < 35 mm Hg and PVR remained < 400 dynes/s/cm–5. One hundred fifty-five patients were granted MELD exception points for POPH between 2002 and 2010 and went on to receive liver transplants.45 Goldberg et al collected data from the Organ Procurement and Transplantation Network (OPTN) and compared outcomes of patients with approved POPH MELD exception points versus waitlist candidates with no exception points.46 One hundred fifty-five waitlisted patients received POPH MELD exception points, with only 43.1% meeting OPTN exception requirements. One-third did not fulfill hemodynamic criteria consistent with POPH or had missing data, and 80% went on to receive a liver transplant. Waitlist candidates receiving POPH MELD exception points also had increased waitlist mortality and several early post-transplant deaths. The authors felt these data highlighted the need for OPTN/UNOS to revise their policy for POPH MELD exceptions points, revise how points are rewarded, and continue research to help risk stratify these patients to minimize perioperative complications.
Conclusion
Several effective medical treatment regimens are available, including prostanoids, endothelin receptor antagonists, and PDE-5 inhibitors. Liver transplantation is a potential cure but is only recommended if MPAP can be decreased to ≤ 35 mmHg. Long-term follow-up studies have shown these patients do well several years post-transplant but may continue to require oral therapy for their POPH.
1. Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925-1932.
2. Chin K, Rubin L. Pulmonary arterial hypertension. Am Coll Cardiol. 2008;51:1527-1538.
3. Doran A, Harris S, Goetz B. Advances in prostanoid infusion therapy for pulmonary arterial hypertension. J Infus Nurs. 2008;31:336-345.
4. Chin KM, Channick RN, De Lemos JA, ET AL. Hemodynamics and epoprostenol use are associated with thrombocytopenia in pulmonary arterial hypertension. Chest. 2009;135:130-136.
5. Hoshikawa Y, Voelkel NF, Gesell TL, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164:314-318.
6. Ricci GL, Melgosa MT, Burgos F, et al. Assessment of acute pulmonary vascular reactivity in portopulmonary hypertension. Liver Transplant. 2007;13:1506-1514.
7. McLaughlin V V, Genthner DE, Panella MM, et al. Compassionate use of continuous prostacyclin in the management of secondary pulmonary hypertension: a case series. Ann Intern Med. 1999;130:740-743.
8. Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology. 1999;30:641-648.
9. Fix OK, Bass NM, De Morco T, Merriman RB. Long-term follow-up of portopulmonary hypertension: Effect of treatment with epoprostenol. Liver Transplant. 2007;13:875-885.
10. Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant. 2006;6:2177-2182.
11. Ashfaq M, Chinnakotla S, Rogers L, et al. The impact of treatment of portopulmonary hypertension on survival following liver transplantation. Am J Transplant. 2007;7:1258-1264.
12. Sakai T, Planinsic RM, Mathier MA, et al. initial experience using continuous intravenous treprostinil to manage pulmonary arterial hypertension in patients with end-stage liver disease. Transpl Int. 2009;22:554-561.
13. McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915-1922.
14. Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J. 2007;30:1096-1102.
15. Melgosa MT, Ricci GL, Garcia-Pagan JC et al. Acute and long-term effects of inhaled iloprost in portopulmonary hypertension. Liver Transplant. 2010;16:348-356.
16. Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40:874-880
17. Sitbon O, Channick R, Chin, KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522-2533.
18. Moller S, Gulberg V, Henriksen JH, Gerbes AL. Endothelin-1 and endothelin-3 in cirrhosis: Relations to systemic and splanchnic haemodynamics. J Hepatol. 1995;23:135-144.
19. Eriksson C, Gustavsson A, Kronvall T, Tysk C. Hepatotoxicity by bosentan in a patient with portopulmonary hypertension : a case-report and review of the literature. J Gastrointestin Liver Dis. 2011;20:77-80.
20. Hoeper MM, Halank M, Marx C, et al. Bosentan therapy for portopulmonary hypertension. Eur Respir J. 2005;25:502-508.
21. Stähler G, Von Hunnius P. Successful treatment of portopulmonary hypertension with bosentan: Case report.: Eur J Clin Investig. 2006;36:62-66.
22. Savale L, Magnier R, Le Pavec J, et al. Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. Eur. 2013;41:96-103.
23. Halank M, Knudsen L, Seyfarth H, et al. Ambrisentan improves exercise capacity and symptoms in patients with portopulmonary hypertension. Z Gastroenterol. 2011;49:1258-1262.
24. Cartin-Ceba R, Swanson K, Iyer V, et al. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011;139:109-114.
25. Condliffe R, Elliot C, Hurdman J, et al. Ambrisentan therapy in pulmonary hypertension: clinical use and tolerability in a referral centre. Ther Adv Respir Dis. 2014;8:71-77.
26. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809-818.
27. Reichenberger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J. 2006;28:563-567.
28. Hemnes AR RI. Sildenafil monotherapy in portopulmonary hypertension can facilitate liver transplantation. Liver Transplant. 2009;15:15-19.
29. Gough WR. Sildenafil therapy is associated with improved hemodynamics in liver transplantation candidates with pulmonary arterial hypertension. Liver Transplant. 2009;15:30-36.
30. Yamashita Y. Hemodynamic effects of ambrisentan-tadalafil combination therapy on progressive portopulmonary hypertension. World J Hepatol. 2014;6:825.
31. Bremer HC, Kreisel W, Roecker K, et al. Phosphodiesterase 5 inhibitors lower both portal and pulmonary pressure in portopulmonary hypertension: a case report. J Med Case Rep. 2007;1:46.
32. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013:369;330-340.
33. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013:369;319-329
34. Cartin-Ceba R, Halank M, Ghofrani HA, et al. Riociguat treatment for portopulmonary hypertension: a subgroup analysis from the PATENT-1/-2 studies. Pulm Circ. 2018: 8:2045894018769305.
35. Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. 2006. Gastroenterology. 2006;130:120-126.
36. Ramsay M a, Simpson BR, Nguyen T, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494-500.
37. Safdar Z, Bartolome S, Sussman N. Portopulmonary hypertension : an update. Liver Tranpl. 2012;18:881-891.
38. Ramsay M. Portopulmonary hypertension and right heart failure in patients with cirrhosis. Curr Opin Anaesthesiol. 2010;23:145-150.
39. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443-450.
40. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445-2453.
41. Khaderi S, Khan R, Safdar Z, et al. Long-term follow-up of portopulmonary hypertension patients after liver transplantation. Liver Transplant. 2014;20:724-727.
42. Hollatz TJ, Musat A, Westphal S, et al. Treatment with sildenafil and treprostinil allows successful liver transplantation of patients with moderate to severe portopulmonary hypertension. Liver Transpl. 2012:686-695.
43. Raevens S, De Pauw M, Reyntjens K, et al. Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol. 2012:1-8.
44. Krowka M, Fallon M, Mulligan D. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transplant. 2006;12:S114-S116.
45. Krowka M, Wiesner R, Rosen C. Portopulmonary hypertension outcomes in the era of MELD exception. Liver Transplant. 2012;18:S259.
46. Goldberg DS, Batra S, Sahay S, et al. MELD Exceptions for portopulmonary hypertension: current policy and future implementation. Am J Transplant. 2014;14:2081-2087.
1. Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925-1932.
2. Chin K, Rubin L. Pulmonary arterial hypertension. Am Coll Cardiol. 2008;51:1527-1538.
3. Doran A, Harris S, Goetz B. Advances in prostanoid infusion therapy for pulmonary arterial hypertension. J Infus Nurs. 2008;31:336-345.
4. Chin KM, Channick RN, De Lemos JA, ET AL. Hemodynamics and epoprostenol use are associated with thrombocytopenia in pulmonary arterial hypertension. Chest. 2009;135:130-136.
5. Hoshikawa Y, Voelkel NF, Gesell TL, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164:314-318.
6. Ricci GL, Melgosa MT, Burgos F, et al. Assessment of acute pulmonary vascular reactivity in portopulmonary hypertension. Liver Transplant. 2007;13:1506-1514.
7. McLaughlin V V, Genthner DE, Panella MM, et al. Compassionate use of continuous prostacyclin in the management of secondary pulmonary hypertension: a case series. Ann Intern Med. 1999;130:740-743.
8. Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology. 1999;30:641-648.
9. Fix OK, Bass NM, De Morco T, Merriman RB. Long-term follow-up of portopulmonary hypertension: Effect of treatment with epoprostenol. Liver Transplant. 2007;13:875-885.
10. Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant. 2006;6:2177-2182.
11. Ashfaq M, Chinnakotla S, Rogers L, et al. The impact of treatment of portopulmonary hypertension on survival following liver transplantation. Am J Transplant. 2007;7:1258-1264.
12. Sakai T, Planinsic RM, Mathier MA, et al. initial experience using continuous intravenous treprostinil to manage pulmonary arterial hypertension in patients with end-stage liver disease. Transpl Int. 2009;22:554-561.
13. McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915-1922.
14. Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J. 2007;30:1096-1102.
15. Melgosa MT, Ricci GL, Garcia-Pagan JC et al. Acute and long-term effects of inhaled iloprost in portopulmonary hypertension. Liver Transplant. 2010;16:348-356.
16. Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40:874-880
17. Sitbon O, Channick R, Chin, KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522-2533.
18. Moller S, Gulberg V, Henriksen JH, Gerbes AL. Endothelin-1 and endothelin-3 in cirrhosis: Relations to systemic and splanchnic haemodynamics. J Hepatol. 1995;23:135-144.
19. Eriksson C, Gustavsson A, Kronvall T, Tysk C. Hepatotoxicity by bosentan in a patient with portopulmonary hypertension : a case-report and review of the literature. J Gastrointestin Liver Dis. 2011;20:77-80.
20. Hoeper MM, Halank M, Marx C, et al. Bosentan therapy for portopulmonary hypertension. Eur Respir J. 2005;25:502-508.
21. Stähler G, Von Hunnius P. Successful treatment of portopulmonary hypertension with bosentan: Case report.: Eur J Clin Investig. 2006;36:62-66.
22. Savale L, Magnier R, Le Pavec J, et al. Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. Eur. 2013;41:96-103.
23. Halank M, Knudsen L, Seyfarth H, et al. Ambrisentan improves exercise capacity and symptoms in patients with portopulmonary hypertension. Z Gastroenterol. 2011;49:1258-1262.
24. Cartin-Ceba R, Swanson K, Iyer V, et al. Safety and efficacy of ambrisentan for the treatment of portopulmonary hypertension. Chest. 2011;139:109-114.
25. Condliffe R, Elliot C, Hurdman J, et al. Ambrisentan therapy in pulmonary hypertension: clinical use and tolerability in a referral centre. Ther Adv Respir Dis. 2014;8:71-77.
26. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809-818.
27. Reichenberger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J. 2006;28:563-567.
28. Hemnes AR RI. Sildenafil monotherapy in portopulmonary hypertension can facilitate liver transplantation. Liver Transplant. 2009;15:15-19.
29. Gough WR. Sildenafil therapy is associated with improved hemodynamics in liver transplantation candidates with pulmonary arterial hypertension. Liver Transplant. 2009;15:30-36.
30. Yamashita Y. Hemodynamic effects of ambrisentan-tadalafil combination therapy on progressive portopulmonary hypertension. World J Hepatol. 2014;6:825.
31. Bremer HC, Kreisel W, Roecker K, et al. Phosphodiesterase 5 inhibitors lower both portal and pulmonary pressure in portopulmonary hypertension: a case report. J Med Case Rep. 2007;1:46.
32. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013:369;330-340.
33. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013:369;319-329
34. Cartin-Ceba R, Halank M, Ghofrani HA, et al. Riociguat treatment for portopulmonary hypertension: a subgroup analysis from the PATENT-1/-2 studies. Pulm Circ. 2018: 8:2045894018769305.
35. Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. 2006. Gastroenterology. 2006;130:120-126.
36. Ramsay M a, Simpson BR, Nguyen T, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494-500.
37. Safdar Z, Bartolome S, Sussman N. Portopulmonary hypertension : an update. Liver Tranpl. 2012;18:881-891.
38. Ramsay M. Portopulmonary hypertension and right heart failure in patients with cirrhosis. Curr Opin Anaesthesiol. 2010;23:145-150.
39. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443-450.
40. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445-2453.
41. Khaderi S, Khan R, Safdar Z, et al. Long-term follow-up of portopulmonary hypertension patients after liver transplantation. Liver Transplant. 2014;20:724-727.
42. Hollatz TJ, Musat A, Westphal S, et al. Treatment with sildenafil and treprostinil allows successful liver transplantation of patients with moderate to severe portopulmonary hypertension. Liver Transpl. 2012:686-695.
43. Raevens S, De Pauw M, Reyntjens K, et al. Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol. 2012:1-8.
44. Krowka M, Fallon M, Mulligan D. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transplant. 2006;12:S114-S116.
45. Krowka M, Wiesner R, Rosen C. Portopulmonary hypertension outcomes in the era of MELD exception. Liver Transplant. 2012;18:S259.
46. Goldberg DS, Batra S, Sahay S, et al. MELD Exceptions for portopulmonary hypertension: current policy and future implementation. Am J Transplant. 2014;14:2081-2087.
Portopulmonary Hypertension: Evaluation and Diagnosis
Pulmonary arterial hypertension (PAH) is a rare disease that is associated with high mortality and is characterized by pulmonary vascular remodeling. Portopulmonary hypertension (POPH) is a form of PAH that occurs in patients with portal hypertension where no alternative cause of PAH can be identified. POPH is documented in approximately 4.5% to 8.5% of liver transplant candidates,1,2 but there is no relationship between the existence or severity of POPH and the severity of liver dysfunction.3 Mantz and Craig described the first case of POPH in a 53-year-old woman with enlarged pulmonary arteries that exhibited forceful pulsations more characteristic of the aorta than a low-pressure pulmonary trunk.4 Autopsy revealed findings of chronic liver disease including a stenotic portal vein, portocaval shunt, and esophageal varices. In both PAH and POPH, pre-capillary pulmonary arteries have characteristic lesions, such as intimal thickening, endothelial proliferation, and thrombotic changes. This 2-part article reviews the diagnosis and treatment of patients with POPH. Here, we review the epidemiology, prognosis, pathogenesis, and diagnosis of POPH; current treatment options for POPH are reviewed in a separate article.
Definition
The term POPH was first used by Yoshida et al in 1993 to describe the first successful liver transplant in a patient with POPH, a 39-year-old man with chronic hepatitis.5 The World Health Organization (WHO) classifies POPH as a form of Group 1 PAH.6 The criteria that must be met to make a diagnosis of POPH are shown in the Table 1.7

Moderate POPH is defined as a mean pulmonary artery pressure (MPAP) between 35 mm Hg and < 45 mm Hg, whereas severe POPH is MPAP ≥ 45 mm Hg. Moderate and severe POPH are considered contraindications to liver transplant because of high perioperative and postoperative mortality rates.8 In 2000, the Mayo Clinic retrospectively reviewed 43 patients with POPH who underwent attempted liver transplantation.9 The cardiopulmonary-related mortality rate in patients with a MPAP of 35 to < 50 mm Hg was 50% and 100% for those with MPAP > 50 mm Hg. No mortality was noted in patients with a pre-liver transplant MPAP of < 35 mm Hg and transpulmonary gradient (TPG) < 15 mm Hg.
Epidemiology
In 1983, a series of 17,901 autopsied patients showed a primary pulmonary hypertension prevalence of 0.13% and a prevalence of 0.73% in patients with cirrhosis.10 In 1987, Rich et al published data from the National Institutes of Health’s national registry of primary pulmonary hypertension.11 The registry included data from 187 patients from 32 centers. Further analyses by Groves et al concluded that 8.3% of the patients likely had POPH.12 Humbert et al published data on the French pulmonary hypertension registry experience in 2006.13 The French registry included 674 patients from 17 university hospitals; 10.4% of these patients had POPH. The largest prospective study was published by Hadengue et al in 1991.14 In this study, 507 patients hospitalized with portal hypertension but without known pulmonary hypertension underwent cardiac catheterization; 10 patients (2%) had pulmonary hypertension and more than half were clinically asymptomatic. Finally, the Registry to Evaluate Early And Long-term pulmonary arterial hypertension disease management (REVEAL registry) documented a 5.3% frequency of POPH (174 of 3525) in the United States.15
Prognosis
Individuals with POPH have worse outcomes compared to other forms of PAH. Median survival prior to the introduction of vasodilator therapy was a dismal 6 months and mean survival was 15 months.16 The cause of death in patients with POPH is equally distributed between right heart failure from POPH and direct complications of chronic liver disease.1 Le Pavec et al retrospectively analyzed all patients referred to the French Referral Center with POPH between 1984 and 2004 (154 patients).1 Approximately 50% of the patients were Child-Turcotte-Pugh class B or C, and 60% were classified as New York Health Association (NYHA) class III or IV. In these patients, 1-, 3-, and 5-year survival rates were 88%, 75%, and 68%, respectively. Major independent prognostic risk factors were presence and severity of cirrhosis and preservation of right ventricular function. Interestingly, NYHA functional class was not related to survival in this study, although it has clearly been identified as a strong prognostic factor in idiopathic PAH.
Krowka et al evaluated 174 patients with POPH enrolled in the REVEAL Registry,15 a multicenter, observational, US-based study comprised of more than 3500 patients with PAH. Despite having better hemodynamic parameters at diagnosis, patients with POPH had significantly poorer survival and all-cause hospitalization compared with patients with idiopathic PAH (IPAH) or hereditary PAH (HPAH). Two-year survival from enrollment was 67% in POPH versus 85% in those with IPAH/HPAH (P < 0.001). Five-year survival from time of diagnosis was 40% versus 64% (P < 0.001). Additionally, patients with POPH were less likely to be on PAH-specific therapy at enrollment, with only 25% on treatment at the time of entry. These findings were replicated in 2005 when Kawut et al retrospectively compared 13 patients with POPH with 33 patients with IPAH.17 Despite having a higher cardiac index and lower pulmonary vascular resistance than patients with IPAH, patients with POPH had a higher risk of death (hazard ratio, 2.8, P = 0.04), likely reflecting the combination of 2 serious diseases.
In 2008 the Mayo Clinic published their retrospective analysis of patients with POPH to determine the natural history of POPH.18 Patients were categorized into 3 groups: (1) no medical therapy for POPH and no liver transplant; (2) medical therapy for POPH alone; (3) medical therapy for POPH followed by liver transplant. The study included 74 patients between 1994 through 2007; 19 patients who did not receive treatment for POPH or liver transplant truly represented the natural history of POPH. Their 5-year survival was only 14%, and over half were deceased 1 year after diagnosis. The largest group consisted of patients who received therapy for POPH but no liver transplant. This group did remarkably better than those who received no therapy at all, with a 5-year survival of 45%. However, the patients with the overall best survival were those who received a combination of treatment for POPH followed by liver transplant. Their 5-year survival was 67%. Survival at 5 years was only 25% for the small group of patients who received transplant without PAH therapy. Once again, mortality did not correlate with the severity of hepatic dysfunction or baseline hemodynamic data.
Pathogenesis
The pathogenesis of POPH is unclear. Multiple studies have shown that there is minimal, if any, association with pulmonary hypertension and the severity of liver disease or portal hypertension.19,20 Portal hypertension is the result of an increase in intrahepatic resistance and an increase in blood flow into the portal circulation. Collateral vessels develop and blood from the splanchnic circulation is allowed directly into the systemic venous circulation, bypassing the liver. One of the most widely accepted theories is that a humoral substance, that would otherwise be metabolized by the liver, is able to reach the pulmonary circulation through collaterals, resulting in POPH.21 Pelicelli et al evaluated the possible role of endothelin-1, interleukin-6, interleukin 1β, and tumor necrosis factor in the pathogenesis of POPH.22 Plasma concentrations of these cytokines were compared between patients with POPH and patients with cirrhosis but no POPH. Patients with POPH had higher concentrations of endothelin-1 and interleukin-6, suggesting antagonists for these cytokines may have a role in the treatment of POPH. The role of endothelin-1 was further supported by Kamath et al in 200023 when they determined the pulmonary vascular bed is exposed to increased levels of circulating endothelin-1a in the setting of cirrhosis. Endothelin-1 is a potent vasoconstrictor and facilitator of smooth muscle proliferation.
In addition to collateral circulation allowing mediators to reach the pulmonary arterial bed in portal hypertension, high flow may trigger a vasoproliferative process in the pulmonary vascular bed. Patients with advanced liver disease have a low systemic vascular resistance, with a compensatory increase in cardiac output. An increase in cardiac output can lead to shear stress of the pulmonary vascular endothelial layer. Although the resistance of the pulmonary vasculature may decrease rapidly to help normalize pulmonary pressures, persistent circulatory overload could result in irreversible vascular changes. Autopsy and lung explant studies show that POPH is characterized by obstructive and remodeling changes in the pulmonary arterial bed.24 Initially, medial hypertrophy with smooth muscle proliferation is present. As the disease advances, platelet aggregates, in situ thrombosis, and intimal fibrosis develop. Finally, web-like lesions involving the entire pulmonary wall develop with recanalization for the passage of pulmonary arterial flow. These changes are identical to the changes observed in patients with other forms of PAH.
Not all patients with portal hypertension develop POPH, suggesting that genetic predisposition may play a role in POPH development. The Pulmonary Vascular Complications of Liver Study Group published a multicenter case-control study that attempted to identify genetic risk factors for POPH in patients with advanced liver disease.25 More than 1000 common single nucleotide polymorphisms (SNPs) in 93 candidate genes were genotyped in each patient. When compared to controls, multiple SNPs in the genes coding for estrogen receptor 1, aromatase, phosphodiesterase 5, angiopoietin 1, and calcium binding protein A4 were associated with an increased risk of POPH. One year earlier, the same study group concluded that female sex (adjusted odds ratio [OR], 2.90) and autoimmune hepatitis (adjusted OR, 4.02) were associated with a higher risk for POPH, whereas hepatitis C was associated with a decreased risk.20
Clinical Presentation
Clinical presentation is variable in POPH. Patients referred to a pulmonologist will usually present with symptoms similar to patients with other forms of PAH. In a retrospective analysis of patients referred to the French Referral Center for Pulmonary Hypertension, 60% of the patients belonged to NYHA functional class III or IV.1 In a series of 78 patients with POPH, the most common presenting pulmonary symptom was dyspnea on exertion (81%), followed by syncope, chest pain, and fatigue (< 33%).16 Symptoms such as syncope and chest pain are usually markers of severe POPH.3 Stigmata of portal hypertension, such as ascites, spider angiomata, and palmar erythema, may be present on exam. An accentuated pulmonary component of the second heart sound can be seen in 82% of patients and a systolic murmur caused by tricuspid regurgitation in 69% of patients.16 Patients with severe POPH may have jugular vein distention, peripheral edema, and a third heart sound.
Diagnostic Evaluation
Chest x-rays may show prominent pulmonary arteries and cardiomegaly in patients with POPH, whereas electrocardiogram can suggest right ventricular hypertrophy and right axis deviation. The best screening test for POPH in patients with portal hypertension is echocardiography. Routine screening for POPH is recommended during liver transplant evaluation in the practice guidelines from the American Association for the Study of Liver Disease.26 Right-sided cardiac chamber enlargement and right ventricular pressure or volume overload can be assessed on echocardiography. Colle et al followed 165 patients evaluated for liver transplantation who underwent transthoracic Doppler echocardiography and right heart catheterization.27 Seventeen patients met the criteria for POPH on echocardiography (presence of tricuspid regurgitation and calculated systolic pulmonary artery pressure over 30 mm Hg) and right heart catheterization confirmed the diagnosis in 10 patients. Right ventricular systolic pressure (RVSP) estimate of ≤ 30 mm Hg on 2-dimensional echo had a 100% sensitivity and negative predictive value. Positive predictive value was poor at 59%, reiterating the need for right heart catheterization in the diagnosis of POPH. When Kim et al used a RVSP threshold of 50 mm Hg, 72% had at least moderate pulmonary hypertension, including 30% with severe pulmonary hypertension.28 Raevens et al analyzed data from 152 patients who underwent pretransplant echocardiography and catheterization.2 Their data show a RVSP threshold of greater than 38 mm Hg by echocardiography had a specificity of 82% and sensitivity and negative predictive value of 100%. The European Respiratory Society recommendations state that PAH should be considered unlikely if echocardiography estimates a RVSP ≤36 mm Hg and likely if the RVSP is estimated at > 50 mm Hg.29 We recommend repeating echocardiography every 6 to 12 months in patients listed for liver transplantation, as pulmonary hemodynamics may change over time.
Computed tomography (CT) may have a complementary role in the future for the noninvasive detection of POPH. In a study published in 2014, 49 patients referred for liver transplantation were retrospectively reviewed.30 Measured CT signs included the main pulmonary artery/ascending aorta diameter ratio, the mean left and right main pulmonary artery diameter, and the enlargement of the pulmonary artery compared to the ascending aorta. Compared to the transthoracic echocardiography alone, an algorithm incorporating CT and echocardiography improved the detection of POPH (area under curve = 0.8, P < 0.0001).
A diagnosis of POPH can only be confirmed when PAH exists in a patient with portal hypertension, as determined by right heart catheterization, and no other cause of PAH can be identified. MPAP should be 25 mm Hg or greater, PVR of 240 dynes/s/cm–5, wedge pressure of 15 mm Hg or less, and TPG greater than 12 mm Hg. Krowka et al showed the value of right heart catheterization in their 10-year prospective, echocardiography-catheterization algorithm study.19 Of 1235 liver transplant candidates who underwent echocardiography, 104 patients had a RVSP exceeding 50 mm Hg. Almost all of these patients had a right heart catheterization. All cause pulmonary hypertension (MPAP > 25 mm Hg) was confirmed in 90% of the patients, and 35% had a PVR < 240 dynes/s/cm–5 and pulmonary capillary wedge pressure (PCWP) > 15 mm Hg, suggesting fluid overload. Forty-one patients had significant POPH, with a PVR > 400 dynes/s/cm–5, and 24% also had an elevated PCWP. TPG was > 12 mm Hg in all of these patients, confirming POPH. As demonstrated by this study, right heart catheterization is required to confirm the diagnosis of POPH because high flow and fluid overload can lead to elevated pulmonary artery pressures.
Patients with POPH have a unique clinical profile with characteristics common to patients with primary pulmonary hypertension and chronic liver disease. In a retrospective review that compared 30 patients with PAH, 30 patients with chronic liver disease only, and 30 patients with catheterization-proved POPH,31 patients with POPH had elevated MPAP similar to those with primary PAH, but they also had reduced SVR and elevated cardiac index similar to those with chronic liver disease alone.
Besides POPH, 2 other common causes can lead to increased pulmonary arterial blood flow in patients with portal hypertension. First is a high-flow condition caused by increased cardiac output but with a normal PVR and PCWP. Fluid overload can also lead to pulmonary venous hypertension with increased PCWP, normal cardiac output, and normal PVR. Up to 25% of patients with POPH may present with marked excess volume caused by fluid retention.3 There can be an increase in both PCWP and PVR depending on the presence and the degree of fluid retention. TPG (MPAP – PCWP) > 12 mm Hg was introduced to make such patients less confusing and to help correct for increased PCWP secondary to fluid overload. Obstruction to pulmonary arterial flow is manifest by an increased TPG (Table 2).

POPH should be distinguished from hepatopulmonary syndrome (HPS), which is another pulmonary vascular consequence of liver disease. Unlike POPH, HPS is characterized by a defect in arterial oxygenation induced by pulmonary vascular dilation.32 Similar to other patients with liver disease, patients with HPS have a normal PVR and increased cardiac output secondary to a high-flow state. HPS is diagnosed by confirmation of an intrapulmonary shunt by echocardiogram. Injection of agitated saline results in saline bubbles being visualized in the left atrium 3 or more cardiac cycles after they appear in the right atrium. Currently, there is no effective medical treatment for HPS and liver transplantation is the only successful treatment.
Conclusion
POPH is an uncommon complication of chronic liver disease. It is defined as PAH in a patient with portal hypertension excluding other causes of PAH. The following criteria must be met to make a diagnosis of POPH: (1) evidence of portal hypertension; (2) MPAP ≥ 35 mm Hg; (3) PVR ≥ 240 dynes/s/cm5; (4) pulmonary capillary wedge pressure ≤ 15 mm Hg; and (5) TPG > 12 mm Hg. Individuals with POPH have worse outcomes compared to other forms of PAH, with a median survival of 6 months without medical therapy. The pathogenesis of POPH is unclear but may be related to a genetic predisposition since not all patients with portal hypertension develop POPH. Echocardiography is an excellent screening test for POPH, but a right heart catheterization must be performed to confirm the diagnosis.
1. Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med. 2008;178:637-643.
2. Raevens S, Colle I, Reyntjens K, et al. Echocardiography for the detection of portopulmonary hypertension in liver transplant candidates: An analysis of cutoff values. Liver Transplant. 2013;19:602-610.
3. Krowka MJ. Portopulmonary hypertension. Semin Respir Crit Care Med. 2012;33:17-25.
4. Mantz F. Portal axis thrombosis with spontaneous portocaval shunt and resultant cor pulmonale. AMA Arch Pathol. 1951;52:91-97.
5. Yoshida EM, Erb SR, Pflugfelder PW, et al. Single-lung versus liver transplantation for the treatment of portopulmonary hypertension--a comparison of two patients. Transplantation. 1993;55:688-690.
6. Badesch DB, Champion HC, Gomez Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54 54(1 Suppl):S55-66.
7. Cartin-Ceba R, Krowka MJ. Portopulmonary hypertension. Clin Liver Dis. 2014;18:421-438.
8. Ramsay M, Simpson BR, Nguyen T, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494-500.
9. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443-450.
10. McDonnell P, Toye P, Hutchins G. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis. 1983;127:437-441.
11. Rich S, Dantzker D, Ayres S, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216-223.
12. Groves B. Pulmonary Hypertension Associated with Cirrhosis. Philadelphia: University of Pennsylvania Press; 1990.
13. Habib G, Gressin V, Yaici A, et al. Pulmonary arterial hypertension in France results from a national registry. Am J Respir Crit Care Med. 2006;173:1023-1030.
14. Hadengue A, Benhayoun M, Lebrec D, Benhamou J. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. 1991;100:520-528.
15. Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the US-based REVEAL Registry. Chest. 2012;141:906-915.
16. Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. 1991;17:492-498.
17. Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005;11:1107-1111.
18. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445-2453.
19. Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology. 2006;44:1502-1510.
20. Kawut SM, Krowka MJ, Trotter JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008;48:196-203.
21. Lebrec D, Capron JP, Dhumeaux D, Benhamou JP. pulmonary hypertension complicating portal hypertension. Am J Rev Resp Dis. 1979;120:849-856.
22. Pellicelli AM, Barbaro G, Puoti C, et al. Plasma cytokines and portopulmonary hypertension in patients with cirrhosis waiting for orthotopic liver transplantation. Angiology. 2010;61:802-806.
23. Kamath PS, Carpenter HA, Lloyd RV, et al. Hepatic localization of endothelin-1 in patients with idiopathic portal hypertension and cirrhosis of the liver. Liver Transpl. 2000;6:596-602.
24. Krowka MJ, Edwards WD. A spectrum of pulmonary vascular pathology in portopulmonary hypertension. Liver Transpl. 2000;6:241-242.
25. Roberts KE, Fallon MB, Krowka MJ, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835-842.
26. Murray KF, Carithers RL. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005;41:1407-1432.
27. Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003;37:401-409.
28. Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transpl. 2000;6:453-458.
29. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219-1263.
30. Devaraj A, Loveridge R, Bosanac D, et al. Portopulmonary hypertension: Improved detection using CT and echocardiography in combination. Eur Radiol. 2014;24:2385-2393.
31. Kuo P, Plotkin J, Johnson L, et al. Distinctive clinical features of portopulmonary hypertension. Chest. 1997;112:980-986.
32. Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome--a liver-induced lung vascular disorder. N Engl J Med. 2008;358:2378-2387.
Pulmonary arterial hypertension (PAH) is a rare disease that is associated with high mortality and is characterized by pulmonary vascular remodeling. Portopulmonary hypertension (POPH) is a form of PAH that occurs in patients with portal hypertension where no alternative cause of PAH can be identified. POPH is documented in approximately 4.5% to 8.5% of liver transplant candidates,1,2 but there is no relationship between the existence or severity of POPH and the severity of liver dysfunction.3 Mantz and Craig described the first case of POPH in a 53-year-old woman with enlarged pulmonary arteries that exhibited forceful pulsations more characteristic of the aorta than a low-pressure pulmonary trunk.4 Autopsy revealed findings of chronic liver disease including a stenotic portal vein, portocaval shunt, and esophageal varices. In both PAH and POPH, pre-capillary pulmonary arteries have characteristic lesions, such as intimal thickening, endothelial proliferation, and thrombotic changes. This 2-part article reviews the diagnosis and treatment of patients with POPH. Here, we review the epidemiology, prognosis, pathogenesis, and diagnosis of POPH; current treatment options for POPH are reviewed in a separate article.
Definition
The term POPH was first used by Yoshida et al in 1993 to describe the first successful liver transplant in a patient with POPH, a 39-year-old man with chronic hepatitis.5 The World Health Organization (WHO) classifies POPH as a form of Group 1 PAH.6 The criteria that must be met to make a diagnosis of POPH are shown in the Table 1.7
Moderate POPH is defined as a mean pulmonary artery pressure (MPAP) between 35 mm Hg and < 45 mm Hg, whereas severe POPH is MPAP ≥ 45 mm Hg. Moderate and severe POPH are considered contraindications to liver transplant because of high perioperative and postoperative mortality rates.8 In 2000, the Mayo Clinic retrospectively reviewed 43 patients with POPH who underwent attempted liver transplantation.9 The cardiopulmonary-related mortality rate in patients with a MPAP of 35 to < 50 mm Hg was 50% and 100% for those with MPAP > 50 mm Hg. No mortality was noted in patients with a pre-liver transplant MPAP of < 35 mm Hg and transpulmonary gradient (TPG) < 15 mm Hg.
Epidemiology
In 1983, a series of 17,901 autopsied patients showed a primary pulmonary hypertension prevalence of 0.13% and a prevalence of 0.73% in patients with cirrhosis.10 In 1987, Rich et al published data from the National Institutes of Health’s national registry of primary pulmonary hypertension.11 The registry included data from 187 patients from 32 centers. Further analyses by Groves et al concluded that 8.3% of the patients likely had POPH.12 Humbert et al published data on the French pulmonary hypertension registry experience in 2006.13 The French registry included 674 patients from 17 university hospitals; 10.4% of these patients had POPH. The largest prospective study was published by Hadengue et al in 1991.14 In this study, 507 patients hospitalized with portal hypertension but without known pulmonary hypertension underwent cardiac catheterization; 10 patients (2%) had pulmonary hypertension and more than half were clinically asymptomatic. Finally, the Registry to Evaluate Early And Long-term pulmonary arterial hypertension disease management (REVEAL registry) documented a 5.3% frequency of POPH (174 of 3525) in the United States.15
Prognosis
Individuals with POPH have worse outcomes compared to other forms of PAH. Median survival prior to the introduction of vasodilator therapy was a dismal 6 months and mean survival was 15 months.16 The cause of death in patients with POPH is equally distributed between right heart failure from POPH and direct complications of chronic liver disease.1 Le Pavec et al retrospectively analyzed all patients referred to the French Referral Center with POPH between 1984 and 2004 (154 patients).1 Approximately 50% of the patients were Child-Turcotte-Pugh class B or C, and 60% were classified as New York Health Association (NYHA) class III or IV. In these patients, 1-, 3-, and 5-year survival rates were 88%, 75%, and 68%, respectively. Major independent prognostic risk factors were presence and severity of cirrhosis and preservation of right ventricular function. Interestingly, NYHA functional class was not related to survival in this study, although it has clearly been identified as a strong prognostic factor in idiopathic PAH.
Krowka et al evaluated 174 patients with POPH enrolled in the REVEAL Registry,15 a multicenter, observational, US-based study comprised of more than 3500 patients with PAH. Despite having better hemodynamic parameters at diagnosis, patients with POPH had significantly poorer survival and all-cause hospitalization compared with patients with idiopathic PAH (IPAH) or hereditary PAH (HPAH). Two-year survival from enrollment was 67% in POPH versus 85% in those with IPAH/HPAH (P < 0.001). Five-year survival from time of diagnosis was 40% versus 64% (P < 0.001). Additionally, patients with POPH were less likely to be on PAH-specific therapy at enrollment, with only 25% on treatment at the time of entry. These findings were replicated in 2005 when Kawut et al retrospectively compared 13 patients with POPH with 33 patients with IPAH.17 Despite having a higher cardiac index and lower pulmonary vascular resistance than patients with IPAH, patients with POPH had a higher risk of death (hazard ratio, 2.8, P = 0.04), likely reflecting the combination of 2 serious diseases.
In 2008 the Mayo Clinic published their retrospective analysis of patients with POPH to determine the natural history of POPH.18 Patients were categorized into 3 groups: (1) no medical therapy for POPH and no liver transplant; (2) medical therapy for POPH alone; (3) medical therapy for POPH followed by liver transplant. The study included 74 patients between 1994 through 2007; 19 patients who did not receive treatment for POPH or liver transplant truly represented the natural history of POPH. Their 5-year survival was only 14%, and over half were deceased 1 year after diagnosis. The largest group consisted of patients who received therapy for POPH but no liver transplant. This group did remarkably better than those who received no therapy at all, with a 5-year survival of 45%. However, the patients with the overall best survival were those who received a combination of treatment for POPH followed by liver transplant. Their 5-year survival was 67%. Survival at 5 years was only 25% for the small group of patients who received transplant without PAH therapy. Once again, mortality did not correlate with the severity of hepatic dysfunction or baseline hemodynamic data.
Pathogenesis
The pathogenesis of POPH is unclear. Multiple studies have shown that there is minimal, if any, association with pulmonary hypertension and the severity of liver disease or portal hypertension.19,20 Portal hypertension is the result of an increase in intrahepatic resistance and an increase in blood flow into the portal circulation. Collateral vessels develop and blood from the splanchnic circulation is allowed directly into the systemic venous circulation, bypassing the liver. One of the most widely accepted theories is that a humoral substance, that would otherwise be metabolized by the liver, is able to reach the pulmonary circulation through collaterals, resulting in POPH.21 Pelicelli et al evaluated the possible role of endothelin-1, interleukin-6, interleukin 1β, and tumor necrosis factor in the pathogenesis of POPH.22 Plasma concentrations of these cytokines were compared between patients with POPH and patients with cirrhosis but no POPH. Patients with POPH had higher concentrations of endothelin-1 and interleukin-6, suggesting antagonists for these cytokines may have a role in the treatment of POPH. The role of endothelin-1 was further supported by Kamath et al in 200023 when they determined the pulmonary vascular bed is exposed to increased levels of circulating endothelin-1a in the setting of cirrhosis. Endothelin-1 is a potent vasoconstrictor and facilitator of smooth muscle proliferation.
In addition to collateral circulation allowing mediators to reach the pulmonary arterial bed in portal hypertension, high flow may trigger a vasoproliferative process in the pulmonary vascular bed. Patients with advanced liver disease have a low systemic vascular resistance, with a compensatory increase in cardiac output. An increase in cardiac output can lead to shear stress of the pulmonary vascular endothelial layer. Although the resistance of the pulmonary vasculature may decrease rapidly to help normalize pulmonary pressures, persistent circulatory overload could result in irreversible vascular changes. Autopsy and lung explant studies show that POPH is characterized by obstructive and remodeling changes in the pulmonary arterial bed.24 Initially, medial hypertrophy with smooth muscle proliferation is present. As the disease advances, platelet aggregates, in situ thrombosis, and intimal fibrosis develop. Finally, web-like lesions involving the entire pulmonary wall develop with recanalization for the passage of pulmonary arterial flow. These changes are identical to the changes observed in patients with other forms of PAH.
Not all patients with portal hypertension develop POPH, suggesting that genetic predisposition may play a role in POPH development. The Pulmonary Vascular Complications of Liver Study Group published a multicenter case-control study that attempted to identify genetic risk factors for POPH in patients with advanced liver disease.25 More than 1000 common single nucleotide polymorphisms (SNPs) in 93 candidate genes were genotyped in each patient. When compared to controls, multiple SNPs in the genes coding for estrogen receptor 1, aromatase, phosphodiesterase 5, angiopoietin 1, and calcium binding protein A4 were associated with an increased risk of POPH. One year earlier, the same study group concluded that female sex (adjusted odds ratio [OR], 2.90) and autoimmune hepatitis (adjusted OR, 4.02) were associated with a higher risk for POPH, whereas hepatitis C was associated with a decreased risk.20
Clinical Presentation
Clinical presentation is variable in POPH. Patients referred to a pulmonologist will usually present with symptoms similar to patients with other forms of PAH. In a retrospective analysis of patients referred to the French Referral Center for Pulmonary Hypertension, 60% of the patients belonged to NYHA functional class III or IV.1 In a series of 78 patients with POPH, the most common presenting pulmonary symptom was dyspnea on exertion (81%), followed by syncope, chest pain, and fatigue (< 33%).16 Symptoms such as syncope and chest pain are usually markers of severe POPH.3 Stigmata of portal hypertension, such as ascites, spider angiomata, and palmar erythema, may be present on exam. An accentuated pulmonary component of the second heart sound can be seen in 82% of patients and a systolic murmur caused by tricuspid regurgitation in 69% of patients.16 Patients with severe POPH may have jugular vein distention, peripheral edema, and a third heart sound.
Diagnostic Evaluation
Chest x-rays may show prominent pulmonary arteries and cardiomegaly in patients with POPH, whereas electrocardiogram can suggest right ventricular hypertrophy and right axis deviation. The best screening test for POPH in patients with portal hypertension is echocardiography. Routine screening for POPH is recommended during liver transplant evaluation in the practice guidelines from the American Association for the Study of Liver Disease.26 Right-sided cardiac chamber enlargement and right ventricular pressure or volume overload can be assessed on echocardiography. Colle et al followed 165 patients evaluated for liver transplantation who underwent transthoracic Doppler echocardiography and right heart catheterization.27 Seventeen patients met the criteria for POPH on echocardiography (presence of tricuspid regurgitation and calculated systolic pulmonary artery pressure over 30 mm Hg) and right heart catheterization confirmed the diagnosis in 10 patients. Right ventricular systolic pressure (RVSP) estimate of ≤ 30 mm Hg on 2-dimensional echo had a 100% sensitivity and negative predictive value. Positive predictive value was poor at 59%, reiterating the need for right heart catheterization in the diagnosis of POPH. When Kim et al used a RVSP threshold of 50 mm Hg, 72% had at least moderate pulmonary hypertension, including 30% with severe pulmonary hypertension.28 Raevens et al analyzed data from 152 patients who underwent pretransplant echocardiography and catheterization.2 Their data show a RVSP threshold of greater than 38 mm Hg by echocardiography had a specificity of 82% and sensitivity and negative predictive value of 100%. The European Respiratory Society recommendations state that PAH should be considered unlikely if echocardiography estimates a RVSP ≤36 mm Hg and likely if the RVSP is estimated at > 50 mm Hg.29 We recommend repeating echocardiography every 6 to 12 months in patients listed for liver transplantation, as pulmonary hemodynamics may change over time.
Computed tomography (CT) may have a complementary role in the future for the noninvasive detection of POPH. In a study published in 2014, 49 patients referred for liver transplantation were retrospectively reviewed.30 Measured CT signs included the main pulmonary artery/ascending aorta diameter ratio, the mean left and right main pulmonary artery diameter, and the enlargement of the pulmonary artery compared to the ascending aorta. Compared to the transthoracic echocardiography alone, an algorithm incorporating CT and echocardiography improved the detection of POPH (area under curve = 0.8, P < 0.0001).
A diagnosis of POPH can only be confirmed when PAH exists in a patient with portal hypertension, as determined by right heart catheterization, and no other cause of PAH can be identified. MPAP should be 25 mm Hg or greater, PVR of 240 dynes/s/cm–5, wedge pressure of 15 mm Hg or less, and TPG greater than 12 mm Hg. Krowka et al showed the value of right heart catheterization in their 10-year prospective, echocardiography-catheterization algorithm study.19 Of 1235 liver transplant candidates who underwent echocardiography, 104 patients had a RVSP exceeding 50 mm Hg. Almost all of these patients had a right heart catheterization. All cause pulmonary hypertension (MPAP > 25 mm Hg) was confirmed in 90% of the patients, and 35% had a PVR < 240 dynes/s/cm–5 and pulmonary capillary wedge pressure (PCWP) > 15 mm Hg, suggesting fluid overload. Forty-one patients had significant POPH, with a PVR > 400 dynes/s/cm–5, and 24% also had an elevated PCWP. TPG was > 12 mm Hg in all of these patients, confirming POPH. As demonstrated by this study, right heart catheterization is required to confirm the diagnosis of POPH because high flow and fluid overload can lead to elevated pulmonary artery pressures.
Patients with POPH have a unique clinical profile with characteristics common to patients with primary pulmonary hypertension and chronic liver disease. In a retrospective review that compared 30 patients with PAH, 30 patients with chronic liver disease only, and 30 patients with catheterization-proved POPH,31 patients with POPH had elevated MPAP similar to those with primary PAH, but they also had reduced SVR and elevated cardiac index similar to those with chronic liver disease alone.
Besides POPH, 2 other common causes can lead to increased pulmonary arterial blood flow in patients with portal hypertension. First is a high-flow condition caused by increased cardiac output but with a normal PVR and PCWP. Fluid overload can also lead to pulmonary venous hypertension with increased PCWP, normal cardiac output, and normal PVR. Up to 25% of patients with POPH may present with marked excess volume caused by fluid retention.3 There can be an increase in both PCWP and PVR depending on the presence and the degree of fluid retention. TPG (MPAP – PCWP) > 12 mm Hg was introduced to make such patients less confusing and to help correct for increased PCWP secondary to fluid overload. Obstruction to pulmonary arterial flow is manifest by an increased TPG (Table 2).
POPH should be distinguished from hepatopulmonary syndrome (HPS), which is another pulmonary vascular consequence of liver disease. Unlike POPH, HPS is characterized by a defect in arterial oxygenation induced by pulmonary vascular dilation.32 Similar to other patients with liver disease, patients with HPS have a normal PVR and increased cardiac output secondary to a high-flow state. HPS is diagnosed by confirmation of an intrapulmonary shunt by echocardiogram. Injection of agitated saline results in saline bubbles being visualized in the left atrium 3 or more cardiac cycles after they appear in the right atrium. Currently, there is no effective medical treatment for HPS and liver transplantation is the only successful treatment.
Conclusion
POPH is an uncommon complication of chronic liver disease. It is defined as PAH in a patient with portal hypertension excluding other causes of PAH. The following criteria must be met to make a diagnosis of POPH: (1) evidence of portal hypertension; (2) MPAP ≥ 35 mm Hg; (3) PVR ≥ 240 dynes/s/cm5; (4) pulmonary capillary wedge pressure ≤ 15 mm Hg; and (5) TPG > 12 mm Hg. Individuals with POPH have worse outcomes compared to other forms of PAH, with a median survival of 6 months without medical therapy. The pathogenesis of POPH is unclear but may be related to a genetic predisposition since not all patients with portal hypertension develop POPH. Echocardiography is an excellent screening test for POPH, but a right heart catheterization must be performed to confirm the diagnosis.
Pulmonary arterial hypertension (PAH) is a rare disease that is associated with high mortality and is characterized by pulmonary vascular remodeling. Portopulmonary hypertension (POPH) is a form of PAH that occurs in patients with portal hypertension where no alternative cause of PAH can be identified. POPH is documented in approximately 4.5% to 8.5% of liver transplant candidates,1,2 but there is no relationship between the existence or severity of POPH and the severity of liver dysfunction.3 Mantz and Craig described the first case of POPH in a 53-year-old woman with enlarged pulmonary arteries that exhibited forceful pulsations more characteristic of the aorta than a low-pressure pulmonary trunk.4 Autopsy revealed findings of chronic liver disease including a stenotic portal vein, portocaval shunt, and esophageal varices. In both PAH and POPH, pre-capillary pulmonary arteries have characteristic lesions, such as intimal thickening, endothelial proliferation, and thrombotic changes. This 2-part article reviews the diagnosis and treatment of patients with POPH. Here, we review the epidemiology, prognosis, pathogenesis, and diagnosis of POPH; current treatment options for POPH are reviewed in a separate article.
Definition
The term POPH was first used by Yoshida et al in 1993 to describe the first successful liver transplant in a patient with POPH, a 39-year-old man with chronic hepatitis.5 The World Health Organization (WHO) classifies POPH as a form of Group 1 PAH.6 The criteria that must be met to make a diagnosis of POPH are shown in the Table 1.7
Moderate POPH is defined as a mean pulmonary artery pressure (MPAP) between 35 mm Hg and < 45 mm Hg, whereas severe POPH is MPAP ≥ 45 mm Hg. Moderate and severe POPH are considered contraindications to liver transplant because of high perioperative and postoperative mortality rates.8 In 2000, the Mayo Clinic retrospectively reviewed 43 patients with POPH who underwent attempted liver transplantation.9 The cardiopulmonary-related mortality rate in patients with a MPAP of 35 to < 50 mm Hg was 50% and 100% for those with MPAP > 50 mm Hg. No mortality was noted in patients with a pre-liver transplant MPAP of < 35 mm Hg and transpulmonary gradient (TPG) < 15 mm Hg.
Epidemiology
In 1983, a series of 17,901 autopsied patients showed a primary pulmonary hypertension prevalence of 0.13% and a prevalence of 0.73% in patients with cirrhosis.10 In 1987, Rich et al published data from the National Institutes of Health’s national registry of primary pulmonary hypertension.11 The registry included data from 187 patients from 32 centers. Further analyses by Groves et al concluded that 8.3% of the patients likely had POPH.12 Humbert et al published data on the French pulmonary hypertension registry experience in 2006.13 The French registry included 674 patients from 17 university hospitals; 10.4% of these patients had POPH. The largest prospective study was published by Hadengue et al in 1991.14 In this study, 507 patients hospitalized with portal hypertension but without known pulmonary hypertension underwent cardiac catheterization; 10 patients (2%) had pulmonary hypertension and more than half were clinically asymptomatic. Finally, the Registry to Evaluate Early And Long-term pulmonary arterial hypertension disease management (REVEAL registry) documented a 5.3% frequency of POPH (174 of 3525) in the United States.15
Prognosis
Individuals with POPH have worse outcomes compared to other forms of PAH. Median survival prior to the introduction of vasodilator therapy was a dismal 6 months and mean survival was 15 months.16 The cause of death in patients with POPH is equally distributed between right heart failure from POPH and direct complications of chronic liver disease.1 Le Pavec et al retrospectively analyzed all patients referred to the French Referral Center with POPH between 1984 and 2004 (154 patients).1 Approximately 50% of the patients were Child-Turcotte-Pugh class B or C, and 60% were classified as New York Health Association (NYHA) class III or IV. In these patients, 1-, 3-, and 5-year survival rates were 88%, 75%, and 68%, respectively. Major independent prognostic risk factors were presence and severity of cirrhosis and preservation of right ventricular function. Interestingly, NYHA functional class was not related to survival in this study, although it has clearly been identified as a strong prognostic factor in idiopathic PAH.
Krowka et al evaluated 174 patients with POPH enrolled in the REVEAL Registry,15 a multicenter, observational, US-based study comprised of more than 3500 patients with PAH. Despite having better hemodynamic parameters at diagnosis, patients with POPH had significantly poorer survival and all-cause hospitalization compared with patients with idiopathic PAH (IPAH) or hereditary PAH (HPAH). Two-year survival from enrollment was 67% in POPH versus 85% in those with IPAH/HPAH (P < 0.001). Five-year survival from time of diagnosis was 40% versus 64% (P < 0.001). Additionally, patients with POPH were less likely to be on PAH-specific therapy at enrollment, with only 25% on treatment at the time of entry. These findings were replicated in 2005 when Kawut et al retrospectively compared 13 patients with POPH with 33 patients with IPAH.17 Despite having a higher cardiac index and lower pulmonary vascular resistance than patients with IPAH, patients with POPH had a higher risk of death (hazard ratio, 2.8, P = 0.04), likely reflecting the combination of 2 serious diseases.
In 2008 the Mayo Clinic published their retrospective analysis of patients with POPH to determine the natural history of POPH.18 Patients were categorized into 3 groups: (1) no medical therapy for POPH and no liver transplant; (2) medical therapy for POPH alone; (3) medical therapy for POPH followed by liver transplant. The study included 74 patients between 1994 through 2007; 19 patients who did not receive treatment for POPH or liver transplant truly represented the natural history of POPH. Their 5-year survival was only 14%, and over half were deceased 1 year after diagnosis. The largest group consisted of patients who received therapy for POPH but no liver transplant. This group did remarkably better than those who received no therapy at all, with a 5-year survival of 45%. However, the patients with the overall best survival were those who received a combination of treatment for POPH followed by liver transplant. Their 5-year survival was 67%. Survival at 5 years was only 25% for the small group of patients who received transplant without PAH therapy. Once again, mortality did not correlate with the severity of hepatic dysfunction or baseline hemodynamic data.
Pathogenesis
The pathogenesis of POPH is unclear. Multiple studies have shown that there is minimal, if any, association with pulmonary hypertension and the severity of liver disease or portal hypertension.19,20 Portal hypertension is the result of an increase in intrahepatic resistance and an increase in blood flow into the portal circulation. Collateral vessels develop and blood from the splanchnic circulation is allowed directly into the systemic venous circulation, bypassing the liver. One of the most widely accepted theories is that a humoral substance, that would otherwise be metabolized by the liver, is able to reach the pulmonary circulation through collaterals, resulting in POPH.21 Pelicelli et al evaluated the possible role of endothelin-1, interleukin-6, interleukin 1β, and tumor necrosis factor in the pathogenesis of POPH.22 Plasma concentrations of these cytokines were compared between patients with POPH and patients with cirrhosis but no POPH. Patients with POPH had higher concentrations of endothelin-1 and interleukin-6, suggesting antagonists for these cytokines may have a role in the treatment of POPH. The role of endothelin-1 was further supported by Kamath et al in 200023 when they determined the pulmonary vascular bed is exposed to increased levels of circulating endothelin-1a in the setting of cirrhosis. Endothelin-1 is a potent vasoconstrictor and facilitator of smooth muscle proliferation.
In addition to collateral circulation allowing mediators to reach the pulmonary arterial bed in portal hypertension, high flow may trigger a vasoproliferative process in the pulmonary vascular bed. Patients with advanced liver disease have a low systemic vascular resistance, with a compensatory increase in cardiac output. An increase in cardiac output can lead to shear stress of the pulmonary vascular endothelial layer. Although the resistance of the pulmonary vasculature may decrease rapidly to help normalize pulmonary pressures, persistent circulatory overload could result in irreversible vascular changes. Autopsy and lung explant studies show that POPH is characterized by obstructive and remodeling changes in the pulmonary arterial bed.24 Initially, medial hypertrophy with smooth muscle proliferation is present. As the disease advances, platelet aggregates, in situ thrombosis, and intimal fibrosis develop. Finally, web-like lesions involving the entire pulmonary wall develop with recanalization for the passage of pulmonary arterial flow. These changes are identical to the changes observed in patients with other forms of PAH.
Not all patients with portal hypertension develop POPH, suggesting that genetic predisposition may play a role in POPH development. The Pulmonary Vascular Complications of Liver Study Group published a multicenter case-control study that attempted to identify genetic risk factors for POPH in patients with advanced liver disease.25 More than 1000 common single nucleotide polymorphisms (SNPs) in 93 candidate genes were genotyped in each patient. When compared to controls, multiple SNPs in the genes coding for estrogen receptor 1, aromatase, phosphodiesterase 5, angiopoietin 1, and calcium binding protein A4 were associated with an increased risk of POPH. One year earlier, the same study group concluded that female sex (adjusted odds ratio [OR], 2.90) and autoimmune hepatitis (adjusted OR, 4.02) were associated with a higher risk for POPH, whereas hepatitis C was associated with a decreased risk.20
Clinical Presentation
Clinical presentation is variable in POPH. Patients referred to a pulmonologist will usually present with symptoms similar to patients with other forms of PAH. In a retrospective analysis of patients referred to the French Referral Center for Pulmonary Hypertension, 60% of the patients belonged to NYHA functional class III or IV.1 In a series of 78 patients with POPH, the most common presenting pulmonary symptom was dyspnea on exertion (81%), followed by syncope, chest pain, and fatigue (< 33%).16 Symptoms such as syncope and chest pain are usually markers of severe POPH.3 Stigmata of portal hypertension, such as ascites, spider angiomata, and palmar erythema, may be present on exam. An accentuated pulmonary component of the second heart sound can be seen in 82% of patients and a systolic murmur caused by tricuspid regurgitation in 69% of patients.16 Patients with severe POPH may have jugular vein distention, peripheral edema, and a third heart sound.
Diagnostic Evaluation
Chest x-rays may show prominent pulmonary arteries and cardiomegaly in patients with POPH, whereas electrocardiogram can suggest right ventricular hypertrophy and right axis deviation. The best screening test for POPH in patients with portal hypertension is echocardiography. Routine screening for POPH is recommended during liver transplant evaluation in the practice guidelines from the American Association for the Study of Liver Disease.26 Right-sided cardiac chamber enlargement and right ventricular pressure or volume overload can be assessed on echocardiography. Colle et al followed 165 patients evaluated for liver transplantation who underwent transthoracic Doppler echocardiography and right heart catheterization.27 Seventeen patients met the criteria for POPH on echocardiography (presence of tricuspid regurgitation and calculated systolic pulmonary artery pressure over 30 mm Hg) and right heart catheterization confirmed the diagnosis in 10 patients. Right ventricular systolic pressure (RVSP) estimate of ≤ 30 mm Hg on 2-dimensional echo had a 100% sensitivity and negative predictive value. Positive predictive value was poor at 59%, reiterating the need for right heart catheterization in the diagnosis of POPH. When Kim et al used a RVSP threshold of 50 mm Hg, 72% had at least moderate pulmonary hypertension, including 30% with severe pulmonary hypertension.28 Raevens et al analyzed data from 152 patients who underwent pretransplant echocardiography and catheterization.2 Their data show a RVSP threshold of greater than 38 mm Hg by echocardiography had a specificity of 82% and sensitivity and negative predictive value of 100%. The European Respiratory Society recommendations state that PAH should be considered unlikely if echocardiography estimates a RVSP ≤36 mm Hg and likely if the RVSP is estimated at > 50 mm Hg.29 We recommend repeating echocardiography every 6 to 12 months in patients listed for liver transplantation, as pulmonary hemodynamics may change over time.
Computed tomography (CT) may have a complementary role in the future for the noninvasive detection of POPH. In a study published in 2014, 49 patients referred for liver transplantation were retrospectively reviewed.30 Measured CT signs included the main pulmonary artery/ascending aorta diameter ratio, the mean left and right main pulmonary artery diameter, and the enlargement of the pulmonary artery compared to the ascending aorta. Compared to the transthoracic echocardiography alone, an algorithm incorporating CT and echocardiography improved the detection of POPH (area under curve = 0.8, P < 0.0001).
A diagnosis of POPH can only be confirmed when PAH exists in a patient with portal hypertension, as determined by right heart catheterization, and no other cause of PAH can be identified. MPAP should be 25 mm Hg or greater, PVR of 240 dynes/s/cm–5, wedge pressure of 15 mm Hg or less, and TPG greater than 12 mm Hg. Krowka et al showed the value of right heart catheterization in their 10-year prospective, echocardiography-catheterization algorithm study.19 Of 1235 liver transplant candidates who underwent echocardiography, 104 patients had a RVSP exceeding 50 mm Hg. Almost all of these patients had a right heart catheterization. All cause pulmonary hypertension (MPAP > 25 mm Hg) was confirmed in 90% of the patients, and 35% had a PVR < 240 dynes/s/cm–5 and pulmonary capillary wedge pressure (PCWP) > 15 mm Hg, suggesting fluid overload. Forty-one patients had significant POPH, with a PVR > 400 dynes/s/cm–5, and 24% also had an elevated PCWP. TPG was > 12 mm Hg in all of these patients, confirming POPH. As demonstrated by this study, right heart catheterization is required to confirm the diagnosis of POPH because high flow and fluid overload can lead to elevated pulmonary artery pressures.
Patients with POPH have a unique clinical profile with characteristics common to patients with primary pulmonary hypertension and chronic liver disease. In a retrospective review that compared 30 patients with PAH, 30 patients with chronic liver disease only, and 30 patients with catheterization-proved POPH,31 patients with POPH had elevated MPAP similar to those with primary PAH, but they also had reduced SVR and elevated cardiac index similar to those with chronic liver disease alone.
Besides POPH, 2 other common causes can lead to increased pulmonary arterial blood flow in patients with portal hypertension. First is a high-flow condition caused by increased cardiac output but with a normal PVR and PCWP. Fluid overload can also lead to pulmonary venous hypertension with increased PCWP, normal cardiac output, and normal PVR. Up to 25% of patients with POPH may present with marked excess volume caused by fluid retention.3 There can be an increase in both PCWP and PVR depending on the presence and the degree of fluid retention. TPG (MPAP – PCWP) > 12 mm Hg was introduced to make such patients less confusing and to help correct for increased PCWP secondary to fluid overload. Obstruction to pulmonary arterial flow is manifest by an increased TPG (Table 2).
POPH should be distinguished from hepatopulmonary syndrome (HPS), which is another pulmonary vascular consequence of liver disease. Unlike POPH, HPS is characterized by a defect in arterial oxygenation induced by pulmonary vascular dilation.32 Similar to other patients with liver disease, patients with HPS have a normal PVR and increased cardiac output secondary to a high-flow state. HPS is diagnosed by confirmation of an intrapulmonary shunt by echocardiogram. Injection of agitated saline results in saline bubbles being visualized in the left atrium 3 or more cardiac cycles after they appear in the right atrium. Currently, there is no effective medical treatment for HPS and liver transplantation is the only successful treatment.
Conclusion
POPH is an uncommon complication of chronic liver disease. It is defined as PAH in a patient with portal hypertension excluding other causes of PAH. The following criteria must be met to make a diagnosis of POPH: (1) evidence of portal hypertension; (2) MPAP ≥ 35 mm Hg; (3) PVR ≥ 240 dynes/s/cm5; (4) pulmonary capillary wedge pressure ≤ 15 mm Hg; and (5) TPG > 12 mm Hg. Individuals with POPH have worse outcomes compared to other forms of PAH, with a median survival of 6 months without medical therapy. The pathogenesis of POPH is unclear but may be related to a genetic predisposition since not all patients with portal hypertension develop POPH. Echocardiography is an excellent screening test for POPH, but a right heart catheterization must be performed to confirm the diagnosis.
1. Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med. 2008;178:637-643.
2. Raevens S, Colle I, Reyntjens K, et al. Echocardiography for the detection of portopulmonary hypertension in liver transplant candidates: An analysis of cutoff values. Liver Transplant. 2013;19:602-610.
3. Krowka MJ. Portopulmonary hypertension. Semin Respir Crit Care Med. 2012;33:17-25.
4. Mantz F. Portal axis thrombosis with spontaneous portocaval shunt and resultant cor pulmonale. AMA Arch Pathol. 1951;52:91-97.
5. Yoshida EM, Erb SR, Pflugfelder PW, et al. Single-lung versus liver transplantation for the treatment of portopulmonary hypertension--a comparison of two patients. Transplantation. 1993;55:688-690.
6. Badesch DB, Champion HC, Gomez Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54 54(1 Suppl):S55-66.
7. Cartin-Ceba R, Krowka MJ. Portopulmonary hypertension. Clin Liver Dis. 2014;18:421-438.
8. Ramsay M, Simpson BR, Nguyen T, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494-500.
9. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443-450.
10. McDonnell P, Toye P, Hutchins G. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis. 1983;127:437-441.
11. Rich S, Dantzker D, Ayres S, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216-223.
12. Groves B. Pulmonary Hypertension Associated with Cirrhosis. Philadelphia: University of Pennsylvania Press; 1990.
13. Habib G, Gressin V, Yaici A, et al. Pulmonary arterial hypertension in France results from a national registry. Am J Respir Crit Care Med. 2006;173:1023-1030.
14. Hadengue A, Benhayoun M, Lebrec D, Benhamou J. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. 1991;100:520-528.
15. Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the US-based REVEAL Registry. Chest. 2012;141:906-915.
16. Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. 1991;17:492-498.
17. Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005;11:1107-1111.
18. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445-2453.
19. Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology. 2006;44:1502-1510.
20. Kawut SM, Krowka MJ, Trotter JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008;48:196-203.
21. Lebrec D, Capron JP, Dhumeaux D, Benhamou JP. pulmonary hypertension complicating portal hypertension. Am J Rev Resp Dis. 1979;120:849-856.
22. Pellicelli AM, Barbaro G, Puoti C, et al. Plasma cytokines and portopulmonary hypertension in patients with cirrhosis waiting for orthotopic liver transplantation. Angiology. 2010;61:802-806.
23. Kamath PS, Carpenter HA, Lloyd RV, et al. Hepatic localization of endothelin-1 in patients with idiopathic portal hypertension and cirrhosis of the liver. Liver Transpl. 2000;6:596-602.
24. Krowka MJ, Edwards WD. A spectrum of pulmonary vascular pathology in portopulmonary hypertension. Liver Transpl. 2000;6:241-242.
25. Roberts KE, Fallon MB, Krowka MJ, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835-842.
26. Murray KF, Carithers RL. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005;41:1407-1432.
27. Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003;37:401-409.
28. Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transpl. 2000;6:453-458.
29. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219-1263.
30. Devaraj A, Loveridge R, Bosanac D, et al. Portopulmonary hypertension: Improved detection using CT and echocardiography in combination. Eur Radiol. 2014;24:2385-2393.
31. Kuo P, Plotkin J, Johnson L, et al. Distinctive clinical features of portopulmonary hypertension. Chest. 1997;112:980-986.
32. Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome--a liver-induced lung vascular disorder. N Engl J Med. 2008;358:2378-2387.
1. Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med. 2008;178:637-643.
2. Raevens S, Colle I, Reyntjens K, et al. Echocardiography for the detection of portopulmonary hypertension in liver transplant candidates: An analysis of cutoff values. Liver Transplant. 2013;19:602-610.
3. Krowka MJ. Portopulmonary hypertension. Semin Respir Crit Care Med. 2012;33:17-25.
4. Mantz F. Portal axis thrombosis with spontaneous portocaval shunt and resultant cor pulmonale. AMA Arch Pathol. 1951;52:91-97.
5. Yoshida EM, Erb SR, Pflugfelder PW, et al. Single-lung versus liver transplantation for the treatment of portopulmonary hypertension--a comparison of two patients. Transplantation. 1993;55:688-690.
6. Badesch DB, Champion HC, Gomez Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54 54(1 Suppl):S55-66.
7. Cartin-Ceba R, Krowka MJ. Portopulmonary hypertension. Clin Liver Dis. 2014;18:421-438.
8. Ramsay M, Simpson BR, Nguyen T, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494-500.
9. Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443-450.
10. McDonnell P, Toye P, Hutchins G. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis. 1983;127:437-441.
11. Rich S, Dantzker D, Ayres S, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216-223.
12. Groves B. Pulmonary Hypertension Associated with Cirrhosis. Philadelphia: University of Pennsylvania Press; 1990.
13. Habib G, Gressin V, Yaici A, et al. Pulmonary arterial hypertension in France results from a national registry. Am J Respir Crit Care Med. 2006;173:1023-1030.
14. Hadengue A, Benhayoun M, Lebrec D, Benhamou J. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. 1991;100:520-528.
15. Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the US-based REVEAL Registry. Chest. 2012;141:906-915.
16. Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. 1991;17:492-498.
17. Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005;11:1107-1111.
18. Swanson KL, Wiesner RH, Nyberg SL, et al. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445-2453.
19. Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology. 2006;44:1502-1510.
20. Kawut SM, Krowka MJ, Trotter JF, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008;48:196-203.
21. Lebrec D, Capron JP, Dhumeaux D, Benhamou JP. pulmonary hypertension complicating portal hypertension. Am J Rev Resp Dis. 1979;120:849-856.
22. Pellicelli AM, Barbaro G, Puoti C, et al. Plasma cytokines and portopulmonary hypertension in patients with cirrhosis waiting for orthotopic liver transplantation. Angiology. 2010;61:802-806.
23. Kamath PS, Carpenter HA, Lloyd RV, et al. Hepatic localization of endothelin-1 in patients with idiopathic portal hypertension and cirrhosis of the liver. Liver Transpl. 2000;6:596-602.
24. Krowka MJ, Edwards WD. A spectrum of pulmonary vascular pathology in portopulmonary hypertension. Liver Transpl. 2000;6:241-242.
25. Roberts KE, Fallon MB, Krowka MJ, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835-842.
26. Murray KF, Carithers RL. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005;41:1407-1432.
27. Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003;37:401-409.
28. Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transpl. 2000;6:453-458.
29. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219-1263.
30. Devaraj A, Loveridge R, Bosanac D, et al. Portopulmonary hypertension: Improved detection using CT and echocardiography in combination. Eur Radiol. 2014;24:2385-2393.
31. Kuo P, Plotkin J, Johnson L, et al. Distinctive clinical features of portopulmonary hypertension. Chest. 1997;112:980-986.
32. Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome--a liver-induced lung vascular disorder. N Engl J Med. 2008;358:2378-2387.
Triple-drug therapy proves effective in CF patients with most common mutation
Reinforcing previous findings, a new study has determined that the next-generation corrector elexacaftor, in combination with tezacaftor and ivacaftor, can effectively treat patients with Phe508del-minimal function genotypes who did not respond to previous cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens.
“These results provide evidence that , thus addressing the underlying cause of disease in the large majority of patients,” wrote Peter G. Middleton, PhD, of the University of Sydney (Australia) and his coauthors. The study was published in the New England Journal of Medicine.
To further determine if the elexacaftor-tezacaftor-ivacaftor regimen was effective and safe, the researchers launched a randomized, placebo-controlled phase 3 trial of 403 cystic fibrosis patients age 12 or older who had a single Phe508del allele. Patients in the combination group (n = 200) received 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor every 12 hours for 24 weeks. Patients in the other group (n = 203) received matched placebos.
At 14 weeks, patients in the combination group had a change in percentage of predicted forced expiratory volume in 1 second (FEV1) that was 13.8 points higher than the placebo group (95% confidence interval, 12.1-15.4, P less than .001). At 24 weeks, the combination group had a predicted FEV1 difference that was 14.3 percentage points higher (95% confidence interval, 12.7-15.8, P less than .001). The rate of pulmonary exacerbations was 63% lower (rate ratio 0.37; 95% CI, 0.25-0.55, P less than .001) and sweat chloride concentration was 41.8 mmol/L lower (95% CI, –44.4 to –39.3, P less than .001) in the combination group through 24 weeks.
At least one adverse event occurred in 93.1% of patients in the combination group and 96% of patients in the placebo group. Serious adverse events occurred in 28 patients (13.9%) in the combination group and 42 patients (20.9%) in the placebo group. There were no deaths in either group.
The study was funded by Vertex Pharmaceuticals. The authors had disclosures, including receiving personal fees and grants from various pharmaceutical companies and being on the advisory board, owning stock, or being an employee of Vertex Pharmaceuticals.
SOURCE: Middleton PG et al. 2019 Oct 31. N Engl J Med. doi: 10.1056/NEJMoa1908639.
After 30 years, new research from Middleton et al. and others appears to be the breakthrough we’ve been waiting for in treating cystic fibrosis, wrote Francis S. Collins, MD, PhD, of the National Institutes of Health in an accompanying editorial (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMe1911602).
As one of the researchers who discovered the cystic fibrosis gene, he acknowledged the 3 decades of work that followed their discovery and the excitement that comes from being able to counter the common Phe508del CFTR mutation that afflicts so many cystic fibrosis patients. “These findings indicate that it may soon be possible to offer safe and effective molecularly targeted therapies to 90% of persons with cystic fibrosis,” he wrote.
“Yet we must not abandon the patients with cystic fibrosis who have null mutations and will not have a response to these drugs,” he added, noting that those challenges remain “substantial” and potentially will involve in vivo somatic-cell gene editing of airway epithelial cells. That said, what once was a dream 30 years ago now appears to be a reality.
Dr. Collins reported being a coinventor of the original patents on the CFTR gene, for which he donated all royalties to the Cystic Fibrosis Foundation.
After 30 years, new research from Middleton et al. and others appears to be the breakthrough we’ve been waiting for in treating cystic fibrosis, wrote Francis S. Collins, MD, PhD, of the National Institutes of Health in an accompanying editorial (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMe1911602).
As one of the researchers who discovered the cystic fibrosis gene, he acknowledged the 3 decades of work that followed their discovery and the excitement that comes from being able to counter the common Phe508del CFTR mutation that afflicts so many cystic fibrosis patients. “These findings indicate that it may soon be possible to offer safe and effective molecularly targeted therapies to 90% of persons with cystic fibrosis,” he wrote.
“Yet we must not abandon the patients with cystic fibrosis who have null mutations and will not have a response to these drugs,” he added, noting that those challenges remain “substantial” and potentially will involve in vivo somatic-cell gene editing of airway epithelial cells. That said, what once was a dream 30 years ago now appears to be a reality.
Dr. Collins reported being a coinventor of the original patents on the CFTR gene, for which he donated all royalties to the Cystic Fibrosis Foundation.
After 30 years, new research from Middleton et al. and others appears to be the breakthrough we’ve been waiting for in treating cystic fibrosis, wrote Francis S. Collins, MD, PhD, of the National Institutes of Health in an accompanying editorial (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMe1911602).
As one of the researchers who discovered the cystic fibrosis gene, he acknowledged the 3 decades of work that followed their discovery and the excitement that comes from being able to counter the common Phe508del CFTR mutation that afflicts so many cystic fibrosis patients. “These findings indicate that it may soon be possible to offer safe and effective molecularly targeted therapies to 90% of persons with cystic fibrosis,” he wrote.
“Yet we must not abandon the patients with cystic fibrosis who have null mutations and will not have a response to these drugs,” he added, noting that those challenges remain “substantial” and potentially will involve in vivo somatic-cell gene editing of airway epithelial cells. That said, what once was a dream 30 years ago now appears to be a reality.
Dr. Collins reported being a coinventor of the original patents on the CFTR gene, for which he donated all royalties to the Cystic Fibrosis Foundation.
Reinforcing previous findings, a new study has determined that the next-generation corrector elexacaftor, in combination with tezacaftor and ivacaftor, can effectively treat patients with Phe508del-minimal function genotypes who did not respond to previous cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens.
“These results provide evidence that , thus addressing the underlying cause of disease in the large majority of patients,” wrote Peter G. Middleton, PhD, of the University of Sydney (Australia) and his coauthors. The study was published in the New England Journal of Medicine.
To further determine if the elexacaftor-tezacaftor-ivacaftor regimen was effective and safe, the researchers launched a randomized, placebo-controlled phase 3 trial of 403 cystic fibrosis patients age 12 or older who had a single Phe508del allele. Patients in the combination group (n = 200) received 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor every 12 hours for 24 weeks. Patients in the other group (n = 203) received matched placebos.
At 14 weeks, patients in the combination group had a change in percentage of predicted forced expiratory volume in 1 second (FEV1) that was 13.8 points higher than the placebo group (95% confidence interval, 12.1-15.4, P less than .001). At 24 weeks, the combination group had a predicted FEV1 difference that was 14.3 percentage points higher (95% confidence interval, 12.7-15.8, P less than .001). The rate of pulmonary exacerbations was 63% lower (rate ratio 0.37; 95% CI, 0.25-0.55, P less than .001) and sweat chloride concentration was 41.8 mmol/L lower (95% CI, –44.4 to –39.3, P less than .001) in the combination group through 24 weeks.
At least one adverse event occurred in 93.1% of patients in the combination group and 96% of patients in the placebo group. Serious adverse events occurred in 28 patients (13.9%) in the combination group and 42 patients (20.9%) in the placebo group. There were no deaths in either group.
The study was funded by Vertex Pharmaceuticals. The authors had disclosures, including receiving personal fees and grants from various pharmaceutical companies and being on the advisory board, owning stock, or being an employee of Vertex Pharmaceuticals.
SOURCE: Middleton PG et al. 2019 Oct 31. N Engl J Med. doi: 10.1056/NEJMoa1908639.
Reinforcing previous findings, a new study has determined that the next-generation corrector elexacaftor, in combination with tezacaftor and ivacaftor, can effectively treat patients with Phe508del-minimal function genotypes who did not respond to previous cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens.
“These results provide evidence that , thus addressing the underlying cause of disease in the large majority of patients,” wrote Peter G. Middleton, PhD, of the University of Sydney (Australia) and his coauthors. The study was published in the New England Journal of Medicine.
To further determine if the elexacaftor-tezacaftor-ivacaftor regimen was effective and safe, the researchers launched a randomized, placebo-controlled phase 3 trial of 403 cystic fibrosis patients age 12 or older who had a single Phe508del allele. Patients in the combination group (n = 200) received 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor every 12 hours for 24 weeks. Patients in the other group (n = 203) received matched placebos.
At 14 weeks, patients in the combination group had a change in percentage of predicted forced expiratory volume in 1 second (FEV1) that was 13.8 points higher than the placebo group (95% confidence interval, 12.1-15.4, P less than .001). At 24 weeks, the combination group had a predicted FEV1 difference that was 14.3 percentage points higher (95% confidence interval, 12.7-15.8, P less than .001). The rate of pulmonary exacerbations was 63% lower (rate ratio 0.37; 95% CI, 0.25-0.55, P less than .001) and sweat chloride concentration was 41.8 mmol/L lower (95% CI, –44.4 to –39.3, P less than .001) in the combination group through 24 weeks.
At least one adverse event occurred in 93.1% of patients in the combination group and 96% of patients in the placebo group. Serious adverse events occurred in 28 patients (13.9%) in the combination group and 42 patients (20.9%) in the placebo group. There were no deaths in either group.
The study was funded by Vertex Pharmaceuticals. The authors had disclosures, including receiving personal fees and grants from various pharmaceutical companies and being on the advisory board, owning stock, or being an employee of Vertex Pharmaceuticals.
SOURCE: Middleton PG et al. 2019 Oct 31. N Engl J Med. doi: 10.1056/NEJMoa1908639.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Vaping-linked lung injury cases near 1,900
according to the latest update provided by the Centers for Disease Control and Prevention. Thirty-seven deaths have been confirmed.
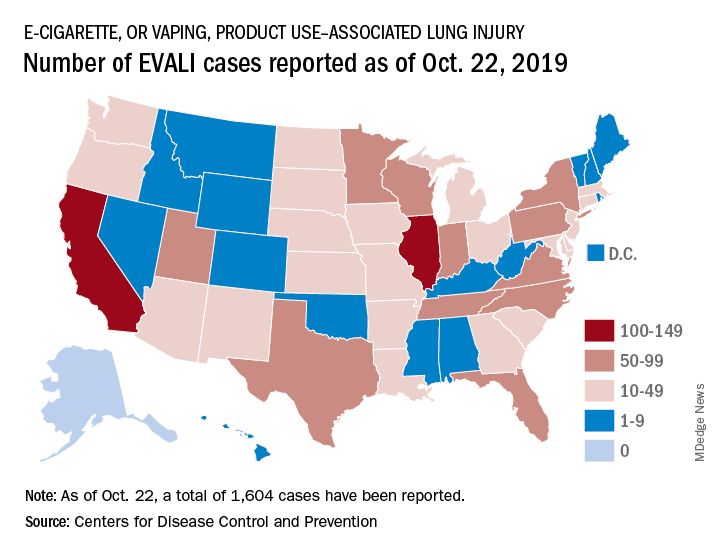
Deaths have occurred in 24 states and the District of Columbia: Alabama, California (3), Connecticut, Delaware, Florida, Georgia (3), Illinois (2), Indiana (3), Kansas (2), Massachusetts, Michigan, Minnesota (3), Mississippi, Missouri, Montana, Nebraska, New Jersey, New York, Oregon (2), Pennsylvania, Tennessee (2), Texas, Utah, and Virginia. As on Oct. 28, the median age of deceased patients was 49 years and ranged from 17 to 75 years.
The CDC is now doing additional testing on available samples for chemical in the bronchoalveolar lavage fluid, blood, or urine, as well as lung biopsy or autopsy specimens. It also is validating methods for aerosol emission testing of case-associated product samples from vaping products and e-liquids.
For more information and resources visit For the Public, For Healthcare Providers, and For State and Local Health Departments pages, as well as the CDC’s Publications and Resources page.
according to the latest update provided by the Centers for Disease Control and Prevention. Thirty-seven deaths have been confirmed.
Deaths have occurred in 24 states and the District of Columbia: Alabama, California (3), Connecticut, Delaware, Florida, Georgia (3), Illinois (2), Indiana (3), Kansas (2), Massachusetts, Michigan, Minnesota (3), Mississippi, Missouri, Montana, Nebraska, New Jersey, New York, Oregon (2), Pennsylvania, Tennessee (2), Texas, Utah, and Virginia. As on Oct. 28, the median age of deceased patients was 49 years and ranged from 17 to 75 years.
The CDC is now doing additional testing on available samples for chemical in the bronchoalveolar lavage fluid, blood, or urine, as well as lung biopsy or autopsy specimens. It also is validating methods for aerosol emission testing of case-associated product samples from vaping products and e-liquids.
For more information and resources visit For the Public, For Healthcare Providers, and For State and Local Health Departments pages, as well as the CDC’s Publications and Resources page.
according to the latest update provided by the Centers for Disease Control and Prevention. Thirty-seven deaths have been confirmed.
Deaths have occurred in 24 states and the District of Columbia: Alabama, California (3), Connecticut, Delaware, Florida, Georgia (3), Illinois (2), Indiana (3), Kansas (2), Massachusetts, Michigan, Minnesota (3), Mississippi, Missouri, Montana, Nebraska, New Jersey, New York, Oregon (2), Pennsylvania, Tennessee (2), Texas, Utah, and Virginia. As on Oct. 28, the median age of deceased patients was 49 years and ranged from 17 to 75 years.
The CDC is now doing additional testing on available samples for chemical in the bronchoalveolar lavage fluid, blood, or urine, as well as lung biopsy or autopsy specimens. It also is validating methods for aerosol emission testing of case-associated product samples from vaping products and e-liquids.
For more information and resources visit For the Public, For Healthcare Providers, and For State and Local Health Departments pages, as well as the CDC’s Publications and Resources page.
Macitentan produces similar results in PAH-SSc and IPAH/HPAH
NEW ORLEANS – Real-world data support the use of macitentan to treat pulmonary arterial hypertension (PAH) associated with connective tissue disease, according to a speaker at the annual meeting of the American College of Chest Physicians.

Outcomes of macitentan (Opsumit) treatment were similar in patients who had PAH associated with systemic sclerosis (PAH-SSc) and patients who had idiopathic PAH (IPAH) or heritable PAH (HPAH), Vallerie McLaughlin, MD, of the University of Michigan, Ann Arbor, said at the meeting.
“Within the limits of a real-world registry, these data add to the growing body of evidence supporting the use of macitentan for treatment in patients with CTD [connective tissue disease],” Dr. McLaughlin said.
She and her colleagues evaluated data from the prospective OPUS registry (NCT02126943) and the retrospective OrPHeUS study (NCT03197688), both of which included patients who were newly started on macitentan.
Dr. McLaughlin presented data on 2,311 patients with IPAH/HPAH and 668 patients with PAH-SSc. She also presented data on patients with PAH-systemic lupus erythematosus and PAH-mixed CTD, but numbers in these groups were small, and outcomes were similar to those in the PAH-SSc group.
Demographic and disease characteristics at the start of macitentan were similar between the IPAH/HPAH and PAH-SSc groups. The median age was 64 years in both groups. The median time from PAH diagnosis was 7.6 months in the IPAH/HPAH group and 8.5 months in the PAH-SSc group.
The median duration of macitentan exposure was 13.4 months in the IPAH/HPAH group and 14.4 months in the PAH-SSc group. The proportion of patients receiving macitentan in combination with other therapies (double or triple combinations) increased from baseline to 6 months in both groups.
Hepatic adverse events occurred in 7.4% of IPAH/HPAH patients and 7.9% of PAH-SSc patients. The most common adverse events among the IPAH/HPAH and PAH-SSc groups in the OPUS registry alone were dyspnea (19% and 26.1%, respectively), peripheral edema (9.8% and 12.4%), fatigue (6.8% and 11.7%), anemia (6.7% and 11.7%), headache (10.2% and 11%), and dizziness (6.7% and 10.7%).
About 39% of patients in both groups discontinued macitentan. Similar proportions in each group discontinued because of adverse events (17% in the IPAH/HPAH group and 18.3% in the PAH-SSc group) and hepatic adverse events (0.2% and 0.7%, respectively).
The proportion of patients with at least one hospitalization was 36.2% in the IPAH/HPAH group and 40.1% in the PAH-SSc group.
The 12-month Kaplan-Meier survival estimate was 92.9% in the IPAH/HPAH group and 91.3% in the PAH-SSc group. The 24-month estimated survival rate was 85.6% and 82.1%, respectively.
The OPUS registry and OrPHeUS study are sponsored by Actelion. Dr. McLaughlin disclosed relationships with Actelion, Acceleron, Bayer, Caremark, CiVi Biopharma, Reata, Sonovie, and United Therapeutics.
SOURCE: McLaughlin V et al. CHEST 2019. Abstract, doi: 10.1016/j.chest.2019.08.827.
NEW ORLEANS – Real-world data support the use of macitentan to treat pulmonary arterial hypertension (PAH) associated with connective tissue disease, according to a speaker at the annual meeting of the American College of Chest Physicians.
Outcomes of macitentan (Opsumit) treatment were similar in patients who had PAH associated with systemic sclerosis (PAH-SSc) and patients who had idiopathic PAH (IPAH) or heritable PAH (HPAH), Vallerie McLaughlin, MD, of the University of Michigan, Ann Arbor, said at the meeting.
“Within the limits of a real-world registry, these data add to the growing body of evidence supporting the use of macitentan for treatment in patients with CTD [connective tissue disease],” Dr. McLaughlin said.
She and her colleagues evaluated data from the prospective OPUS registry (NCT02126943) and the retrospective OrPHeUS study (NCT03197688), both of which included patients who were newly started on macitentan.
Dr. McLaughlin presented data on 2,311 patients with IPAH/HPAH and 668 patients with PAH-SSc. She also presented data on patients with PAH-systemic lupus erythematosus and PAH-mixed CTD, but numbers in these groups were small, and outcomes were similar to those in the PAH-SSc group.
Demographic and disease characteristics at the start of macitentan were similar between the IPAH/HPAH and PAH-SSc groups. The median age was 64 years in both groups. The median time from PAH diagnosis was 7.6 months in the IPAH/HPAH group and 8.5 months in the PAH-SSc group.
The median duration of macitentan exposure was 13.4 months in the IPAH/HPAH group and 14.4 months in the PAH-SSc group. The proportion of patients receiving macitentan in combination with other therapies (double or triple combinations) increased from baseline to 6 months in both groups.
Hepatic adverse events occurred in 7.4% of IPAH/HPAH patients and 7.9% of PAH-SSc patients. The most common adverse events among the IPAH/HPAH and PAH-SSc groups in the OPUS registry alone were dyspnea (19% and 26.1%, respectively), peripheral edema (9.8% and 12.4%), fatigue (6.8% and 11.7%), anemia (6.7% and 11.7%), headache (10.2% and 11%), and dizziness (6.7% and 10.7%).
About 39% of patients in both groups discontinued macitentan. Similar proportions in each group discontinued because of adverse events (17% in the IPAH/HPAH group and 18.3% in the PAH-SSc group) and hepatic adverse events (0.2% and 0.7%, respectively).
The proportion of patients with at least one hospitalization was 36.2% in the IPAH/HPAH group and 40.1% in the PAH-SSc group.
The 12-month Kaplan-Meier survival estimate was 92.9% in the IPAH/HPAH group and 91.3% in the PAH-SSc group. The 24-month estimated survival rate was 85.6% and 82.1%, respectively.
The OPUS registry and OrPHeUS study are sponsored by Actelion. Dr. McLaughlin disclosed relationships with Actelion, Acceleron, Bayer, Caremark, CiVi Biopharma, Reata, Sonovie, and United Therapeutics.
SOURCE: McLaughlin V et al. CHEST 2019. Abstract, doi: 10.1016/j.chest.2019.08.827.
NEW ORLEANS – Real-world data support the use of macitentan to treat pulmonary arterial hypertension (PAH) associated with connective tissue disease, according to a speaker at the annual meeting of the American College of Chest Physicians.
Outcomes of macitentan (Opsumit) treatment were similar in patients who had PAH associated with systemic sclerosis (PAH-SSc) and patients who had idiopathic PAH (IPAH) or heritable PAH (HPAH), Vallerie McLaughlin, MD, of the University of Michigan, Ann Arbor, said at the meeting.
“Within the limits of a real-world registry, these data add to the growing body of evidence supporting the use of macitentan for treatment in patients with CTD [connective tissue disease],” Dr. McLaughlin said.
She and her colleagues evaluated data from the prospective OPUS registry (NCT02126943) and the retrospective OrPHeUS study (NCT03197688), both of which included patients who were newly started on macitentan.
Dr. McLaughlin presented data on 2,311 patients with IPAH/HPAH and 668 patients with PAH-SSc. She also presented data on patients with PAH-systemic lupus erythematosus and PAH-mixed CTD, but numbers in these groups were small, and outcomes were similar to those in the PAH-SSc group.
Demographic and disease characteristics at the start of macitentan were similar between the IPAH/HPAH and PAH-SSc groups. The median age was 64 years in both groups. The median time from PAH diagnosis was 7.6 months in the IPAH/HPAH group and 8.5 months in the PAH-SSc group.
The median duration of macitentan exposure was 13.4 months in the IPAH/HPAH group and 14.4 months in the PAH-SSc group. The proportion of patients receiving macitentan in combination with other therapies (double or triple combinations) increased from baseline to 6 months in both groups.
Hepatic adverse events occurred in 7.4% of IPAH/HPAH patients and 7.9% of PAH-SSc patients. The most common adverse events among the IPAH/HPAH and PAH-SSc groups in the OPUS registry alone were dyspnea (19% and 26.1%, respectively), peripheral edema (9.8% and 12.4%), fatigue (6.8% and 11.7%), anemia (6.7% and 11.7%), headache (10.2% and 11%), and dizziness (6.7% and 10.7%).
About 39% of patients in both groups discontinued macitentan. Similar proportions in each group discontinued because of adverse events (17% in the IPAH/HPAH group and 18.3% in the PAH-SSc group) and hepatic adverse events (0.2% and 0.7%, respectively).
The proportion of patients with at least one hospitalization was 36.2% in the IPAH/HPAH group and 40.1% in the PAH-SSc group.
The 12-month Kaplan-Meier survival estimate was 92.9% in the IPAH/HPAH group and 91.3% in the PAH-SSc group. The 24-month estimated survival rate was 85.6% and 82.1%, respectively.
The OPUS registry and OrPHeUS study are sponsored by Actelion. Dr. McLaughlin disclosed relationships with Actelion, Acceleron, Bayer, Caremark, CiVi Biopharma, Reata, Sonovie, and United Therapeutics.
SOURCE: McLaughlin V et al. CHEST 2019. Abstract, doi: 10.1016/j.chest.2019.08.827.
REPORTING FROM CHEST 2019
Flu vaccine cuts infection severity in kids and adults
WASHINGTON –
During recent U.S. flu seasons, children and adults who contracted influenza despite vaccination had significantly fewer severe infections and infection complications, compared with unimmunized people, according to two separate reports from CDC researchers presented at an annual scientific meeting on infectious diseases.
One of the reports tracked the impact of flu vaccine in children using data that the CDC collected at seven medical centers that participated in the agency’s New Vaccine Surveillance Network, which provided information on children aged 6 months to 17 years who were hospitalized for an acute respiratory illness, including more than 1,700 children during the 2016-2017 flu season and more than 1,900 during the 2017-2018 season. Roughly 10% of these children tested positive for influenza, and the subsequent analysis focused on these cases and compared incidence rates among children who had been vaccinated during the index season and those who had remained unvaccinated.
Combined data from both seasons showed that vaccinated children were 50% less likely to have been hospitalized for an acute influenza infection, compared with unvaccinated kids, a pattern consistently seen both in children aged 6 months to 8 years and in those aged 9-17 years. The pattern of vaccine effectiveness also held regardless of which flu strain caused the infections, reported Angela P. Campbell, MD, a CDC medical officer.
“We saw a nice benefit from vaccination, both in previously healthy children and in those with an underlying medical condition,” a finding that adds to existing evidence of vaccine effectiveness, Dr. Campbell said in a video interview. The results confirmed that flu vaccination does not just prevent infections but also cuts the rate of more severe infections that lead to hospitalization, she explained.
Another CDC study looked at data collected by the agency’s Influenza Hospitalization Surveillance Network from adults at least 18 years old who were hospitalized for a laboratory-confirmed influenza infection during five flu seasons, 2013-2014 through 2017-18. The data, which came from more than 250 acute-care hospitals in 13 states, included more than 43,000 people hospitalized for an identified influenza strain and with a known vaccination history who were not institutionalized and had not received any antiviral treatment.

After propensity-weighted adjustment to create better parity between the vaccinated and unvaccinated patients, the results showed that people 18-64 years old with vaccination had statistically significant decreases in mortality of a relative 36%, need for mechanical ventilation of 34%, pneumonia of 20%, and need for ICU admission of a relative 19%, as well as an 18% drop in average ICU length of stay, Shikha Garg, MD, said at the meeting. The propensity-weighted analysis of data from people at least 65 years old showed statistically significant relative reductions linked with vaccination: 46% reduction in the need for mechanical ventilation, 28% reduction in ICU admissions, and 9% reduction in hospitalized length of stay.
Further analysis of these outcomes by the strains that caused these influenza infections showed that the statistically significant benefits from vaccination were seen only in patients infected with an H1N1 strain. Statistically significant effects on these severe outcomes were not apparent among people infected with the H3N2 or B strains, said Dr. Garg, a medical epidemiologist at the CDC.
“All adults should receive an annual flu vaccination as it can improve outcomes among those who develop influenza despite vaccination,” she concluded.
Results from a third CDC study reported at the meeting examined the importance of two vaccine doses (administered at least 4 weeks apart) given to children aged 6 months to 8 years for the first season they receive flu vaccination, which is the immunization approach for flu recommended by the CDC. The findings from a total of more than 7,500 children immunized during the 2014-2018 seasons showed a clear increment in vaccine protection among kids who received two doses during their first season vaccinated, especially in children who were 2 years old or younger. In that age group, administration of two doses produced vaccine effectiveness of 53% versus a 23% vaccine effectiveness after a single vaccine dose, reported Jessie Chung, a CDC epidemiologist.
WASHINGTON –
During recent U.S. flu seasons, children and adults who contracted influenza despite vaccination had significantly fewer severe infections and infection complications, compared with unimmunized people, according to two separate reports from CDC researchers presented at an annual scientific meeting on infectious diseases.
One of the reports tracked the impact of flu vaccine in children using data that the CDC collected at seven medical centers that participated in the agency’s New Vaccine Surveillance Network, which provided information on children aged 6 months to 17 years who were hospitalized for an acute respiratory illness, including more than 1,700 children during the 2016-2017 flu season and more than 1,900 during the 2017-2018 season. Roughly 10% of these children tested positive for influenza, and the subsequent analysis focused on these cases and compared incidence rates among children who had been vaccinated during the index season and those who had remained unvaccinated.
Combined data from both seasons showed that vaccinated children were 50% less likely to have been hospitalized for an acute influenza infection, compared with unvaccinated kids, a pattern consistently seen both in children aged 6 months to 8 years and in those aged 9-17 years. The pattern of vaccine effectiveness also held regardless of which flu strain caused the infections, reported Angela P. Campbell, MD, a CDC medical officer.
“We saw a nice benefit from vaccination, both in previously healthy children and in those with an underlying medical condition,” a finding that adds to existing evidence of vaccine effectiveness, Dr. Campbell said in a video interview. The results confirmed that flu vaccination does not just prevent infections but also cuts the rate of more severe infections that lead to hospitalization, she explained.
Another CDC study looked at data collected by the agency’s Influenza Hospitalization Surveillance Network from adults at least 18 years old who were hospitalized for a laboratory-confirmed influenza infection during five flu seasons, 2013-2014 through 2017-18. The data, which came from more than 250 acute-care hospitals in 13 states, included more than 43,000 people hospitalized for an identified influenza strain and with a known vaccination history who were not institutionalized and had not received any antiviral treatment.
After propensity-weighted adjustment to create better parity between the vaccinated and unvaccinated patients, the results showed that people 18-64 years old with vaccination had statistically significant decreases in mortality of a relative 36%, need for mechanical ventilation of 34%, pneumonia of 20%, and need for ICU admission of a relative 19%, as well as an 18% drop in average ICU length of stay, Shikha Garg, MD, said at the meeting. The propensity-weighted analysis of data from people at least 65 years old showed statistically significant relative reductions linked with vaccination: 46% reduction in the need for mechanical ventilation, 28% reduction in ICU admissions, and 9% reduction in hospitalized length of stay.
Further analysis of these outcomes by the strains that caused these influenza infections showed that the statistically significant benefits from vaccination were seen only in patients infected with an H1N1 strain. Statistically significant effects on these severe outcomes were not apparent among people infected with the H3N2 or B strains, said Dr. Garg, a medical epidemiologist at the CDC.
“All adults should receive an annual flu vaccination as it can improve outcomes among those who develop influenza despite vaccination,” she concluded.
Results from a third CDC study reported at the meeting examined the importance of two vaccine doses (administered at least 4 weeks apart) given to children aged 6 months to 8 years for the first season they receive flu vaccination, which is the immunization approach for flu recommended by the CDC. The findings from a total of more than 7,500 children immunized during the 2014-2018 seasons showed a clear increment in vaccine protection among kids who received two doses during their first season vaccinated, especially in children who were 2 years old or younger. In that age group, administration of two doses produced vaccine effectiveness of 53% versus a 23% vaccine effectiveness after a single vaccine dose, reported Jessie Chung, a CDC epidemiologist.
WASHINGTON –
During recent U.S. flu seasons, children and adults who contracted influenza despite vaccination had significantly fewer severe infections and infection complications, compared with unimmunized people, according to two separate reports from CDC researchers presented at an annual scientific meeting on infectious diseases.
One of the reports tracked the impact of flu vaccine in children using data that the CDC collected at seven medical centers that participated in the agency’s New Vaccine Surveillance Network, which provided information on children aged 6 months to 17 years who were hospitalized for an acute respiratory illness, including more than 1,700 children during the 2016-2017 flu season and more than 1,900 during the 2017-2018 season. Roughly 10% of these children tested positive for influenza, and the subsequent analysis focused on these cases and compared incidence rates among children who had been vaccinated during the index season and those who had remained unvaccinated.
Combined data from both seasons showed that vaccinated children were 50% less likely to have been hospitalized for an acute influenza infection, compared with unvaccinated kids, a pattern consistently seen both in children aged 6 months to 8 years and in those aged 9-17 years. The pattern of vaccine effectiveness also held regardless of which flu strain caused the infections, reported Angela P. Campbell, MD, a CDC medical officer.
“We saw a nice benefit from vaccination, both in previously healthy children and in those with an underlying medical condition,” a finding that adds to existing evidence of vaccine effectiveness, Dr. Campbell said in a video interview. The results confirmed that flu vaccination does not just prevent infections but also cuts the rate of more severe infections that lead to hospitalization, she explained.
Another CDC study looked at data collected by the agency’s Influenza Hospitalization Surveillance Network from adults at least 18 years old who were hospitalized for a laboratory-confirmed influenza infection during five flu seasons, 2013-2014 through 2017-18. The data, which came from more than 250 acute-care hospitals in 13 states, included more than 43,000 people hospitalized for an identified influenza strain and with a known vaccination history who were not institutionalized and had not received any antiviral treatment.
After propensity-weighted adjustment to create better parity between the vaccinated and unvaccinated patients, the results showed that people 18-64 years old with vaccination had statistically significant decreases in mortality of a relative 36%, need for mechanical ventilation of 34%, pneumonia of 20%, and need for ICU admission of a relative 19%, as well as an 18% drop in average ICU length of stay, Shikha Garg, MD, said at the meeting. The propensity-weighted analysis of data from people at least 65 years old showed statistically significant relative reductions linked with vaccination: 46% reduction in the need for mechanical ventilation, 28% reduction in ICU admissions, and 9% reduction in hospitalized length of stay.
Further analysis of these outcomes by the strains that caused these influenza infections showed that the statistically significant benefits from vaccination were seen only in patients infected with an H1N1 strain. Statistically significant effects on these severe outcomes were not apparent among people infected with the H3N2 or B strains, said Dr. Garg, a medical epidemiologist at the CDC.
“All adults should receive an annual flu vaccination as it can improve outcomes among those who develop influenza despite vaccination,” she concluded.
Results from a third CDC study reported at the meeting examined the importance of two vaccine doses (administered at least 4 weeks apart) given to children aged 6 months to 8 years for the first season they receive flu vaccination, which is the immunization approach for flu recommended by the CDC. The findings from a total of more than 7,500 children immunized during the 2014-2018 seasons showed a clear increment in vaccine protection among kids who received two doses during their first season vaccinated, especially in children who were 2 years old or younger. In that age group, administration of two doses produced vaccine effectiveness of 53% versus a 23% vaccine effectiveness after a single vaccine dose, reported Jessie Chung, a CDC epidemiologist.
REPORTING FROM ID WEEK 2019

Thromboembolic events more likely among CIDP patients with CVAD
AUSTIN, TEX. – Patients with chronic inflammatory demyelinating polyneuropathy (CIDP) who receive intravenous immunoglobulin (IVIg) appear to have an increased risk of thromboembolic events if it is administered with a central venous access device (CVAD) when compared against those without a CVAD, according to a recent study.
Although CVADs can reliably deliver IVIg, they also represent an established risk factor for thromboembolic events, Ami Patel, PhD, a senior epidemiologist at CSL Behring, and colleagues noted on their poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The results suggest a need for physicians to be vigilant about patients’ potential risk factors for thromboembolic events, Dr. Patel said in an interview. Further research is planned, however, because the current study did not control for other risk factors or explore other possible confounding, she said.
Dr. Patel and her associates analyzed U.S. claims data (IBM/Truven MarketScan) from 2006 to 2018 and included all patients with a CIDP diagnosis claim and a postdiagnosis code for IVIg. A code for CVAD up to 2 months before CIDP diagnosis without removal before IVIg treatment ended determined those with CVAD exposure, and thromboembolic events included any codes related to arterial, venous, or vascular prostheses.
The researchers then compared patients in a case-control fashion, matching each one with a CVAD to five patients of similar demographics without a CVAD. Characteristics used for matching included medical insurance type, prescription data availability, sex, age, geographic region, and years enrolled in the database.
Among 7,447 patients with at least one IVIg claim, 11.8% (n = 882) had CVAD exposure and 88.2% (n = 6,565) did not. Of those without a CVAD, 3,642 patients were matched to patients with CVAD. A quarter (25.4%) of patients with a CVAD had a thromboembolic event, compared with 11.2% of matched patients without CVADs (P less than .0001).
In the year leading up to IVIg therapy, 16.9% of those with a CVAD and 10.9% of matched patients without one had a previous thromboembolic event (P less than .0001). Patients with a CVAD also had significantly higher rates of hypertension (51.9% vs. 45.0% with placebo; P less than .001) and anticoagulation therapy (7.0% vs. 5.2% with placebo; P less than .05). Differences between the groups were not significant for diabetes (26.9% vs. 24.2%) and hyperlipidemia (19.1% vs. 17.8%).
Occlusion and stenosis of the carotid artery was the most common arterial thromboembolic outcome, occurring in 5.3% of those with a CVAD and in 2.8% of those without a CVAD. The most common venous thromboembolic event was acute venous embolism and thrombosis of lower-extremity deep vessels, which occurred in 7% of those with a CVAD and in 1.8% of those without.
The researchers also compared inpatient admissions and emergency department visits among those with and without a CVAD; both rates were higher in patients with a CVAD. Visits to the emergency department occurred at a rate of 0.14 events per month for those with a CVAD (2.01 distinct months with a claim) and 0.09 events per month for those without a CVAD (0.65 distinct months with a claim). Patients with a CVAD had 1.44 months with an inpatient admissions claim, in comparison with 0.41 months among matched patients without a CVAD. Inpatient admission frequency per month was 0.14 for those with a CVAD and 0.08 for those without.
The research was funded by CSL Behring. Dr. Patel and two of the other five authors are employees of CSL Behring.
SOURCE: Patel A et al. AANEM 2019, Abstract 94.
AUSTIN, TEX. – Patients with chronic inflammatory demyelinating polyneuropathy (CIDP) who receive intravenous immunoglobulin (IVIg) appear to have an increased risk of thromboembolic events if it is administered with a central venous access device (CVAD) when compared against those without a CVAD, according to a recent study.
Although CVADs can reliably deliver IVIg, they also represent an established risk factor for thromboembolic events, Ami Patel, PhD, a senior epidemiologist at CSL Behring, and colleagues noted on their poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The results suggest a need for physicians to be vigilant about patients’ potential risk factors for thromboembolic events, Dr. Patel said in an interview. Further research is planned, however, because the current study did not control for other risk factors or explore other possible confounding, she said.
Dr. Patel and her associates analyzed U.S. claims data (IBM/Truven MarketScan) from 2006 to 2018 and included all patients with a CIDP diagnosis claim and a postdiagnosis code for IVIg. A code for CVAD up to 2 months before CIDP diagnosis without removal before IVIg treatment ended determined those with CVAD exposure, and thromboembolic events included any codes related to arterial, venous, or vascular prostheses.
The researchers then compared patients in a case-control fashion, matching each one with a CVAD to five patients of similar demographics without a CVAD. Characteristics used for matching included medical insurance type, prescription data availability, sex, age, geographic region, and years enrolled in the database.
Among 7,447 patients with at least one IVIg claim, 11.8% (n = 882) had CVAD exposure and 88.2% (n = 6,565) did not. Of those without a CVAD, 3,642 patients were matched to patients with CVAD. A quarter (25.4%) of patients with a CVAD had a thromboembolic event, compared with 11.2% of matched patients without CVADs (P less than .0001).
In the year leading up to IVIg therapy, 16.9% of those with a CVAD and 10.9% of matched patients without one had a previous thromboembolic event (P less than .0001). Patients with a CVAD also had significantly higher rates of hypertension (51.9% vs. 45.0% with placebo; P less than .001) and anticoagulation therapy (7.0% vs. 5.2% with placebo; P less than .05). Differences between the groups were not significant for diabetes (26.9% vs. 24.2%) and hyperlipidemia (19.1% vs. 17.8%).
Occlusion and stenosis of the carotid artery was the most common arterial thromboembolic outcome, occurring in 5.3% of those with a CVAD and in 2.8% of those without a CVAD. The most common venous thromboembolic event was acute venous embolism and thrombosis of lower-extremity deep vessels, which occurred in 7% of those with a CVAD and in 1.8% of those without.
The researchers also compared inpatient admissions and emergency department visits among those with and without a CVAD; both rates were higher in patients with a CVAD. Visits to the emergency department occurred at a rate of 0.14 events per month for those with a CVAD (2.01 distinct months with a claim) and 0.09 events per month for those without a CVAD (0.65 distinct months with a claim). Patients with a CVAD had 1.44 months with an inpatient admissions claim, in comparison with 0.41 months among matched patients without a CVAD. Inpatient admission frequency per month was 0.14 for those with a CVAD and 0.08 for those without.
The research was funded by CSL Behring. Dr. Patel and two of the other five authors are employees of CSL Behring.
SOURCE: Patel A et al. AANEM 2019, Abstract 94.
AUSTIN, TEX. – Patients with chronic inflammatory demyelinating polyneuropathy (CIDP) who receive intravenous immunoglobulin (IVIg) appear to have an increased risk of thromboembolic events if it is administered with a central venous access device (CVAD) when compared against those without a CVAD, according to a recent study.
Although CVADs can reliably deliver IVIg, they also represent an established risk factor for thromboembolic events, Ami Patel, PhD, a senior epidemiologist at CSL Behring, and colleagues noted on their poster at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
The results suggest a need for physicians to be vigilant about patients’ potential risk factors for thromboembolic events, Dr. Patel said in an interview. Further research is planned, however, because the current study did not control for other risk factors or explore other possible confounding, she said.
Dr. Patel and her associates analyzed U.S. claims data (IBM/Truven MarketScan) from 2006 to 2018 and included all patients with a CIDP diagnosis claim and a postdiagnosis code for IVIg. A code for CVAD up to 2 months before CIDP diagnosis without removal before IVIg treatment ended determined those with CVAD exposure, and thromboembolic events included any codes related to arterial, venous, or vascular prostheses.
The researchers then compared patients in a case-control fashion, matching each one with a CVAD to five patients of similar demographics without a CVAD. Characteristics used for matching included medical insurance type, prescription data availability, sex, age, geographic region, and years enrolled in the database.
Among 7,447 patients with at least one IVIg claim, 11.8% (n = 882) had CVAD exposure and 88.2% (n = 6,565) did not. Of those without a CVAD, 3,642 patients were matched to patients with CVAD. A quarter (25.4%) of patients with a CVAD had a thromboembolic event, compared with 11.2% of matched patients without CVADs (P less than .0001).
In the year leading up to IVIg therapy, 16.9% of those with a CVAD and 10.9% of matched patients without one had a previous thromboembolic event (P less than .0001). Patients with a CVAD also had significantly higher rates of hypertension (51.9% vs. 45.0% with placebo; P less than .001) and anticoagulation therapy (7.0% vs. 5.2% with placebo; P less than .05). Differences between the groups were not significant for diabetes (26.9% vs. 24.2%) and hyperlipidemia (19.1% vs. 17.8%).
Occlusion and stenosis of the carotid artery was the most common arterial thromboembolic outcome, occurring in 5.3% of those with a CVAD and in 2.8% of those without a CVAD. The most common venous thromboembolic event was acute venous embolism and thrombosis of lower-extremity deep vessels, which occurred in 7% of those with a CVAD and in 1.8% of those without.
The researchers also compared inpatient admissions and emergency department visits among those with and without a CVAD; both rates were higher in patients with a CVAD. Visits to the emergency department occurred at a rate of 0.14 events per month for those with a CVAD (2.01 distinct months with a claim) and 0.09 events per month for those without a CVAD (0.65 distinct months with a claim). Patients with a CVAD had 1.44 months with an inpatient admissions claim, in comparison with 0.41 months among matched patients without a CVAD. Inpatient admission frequency per month was 0.14 for those with a CVAD and 0.08 for those without.
The research was funded by CSL Behring. Dr. Patel and two of the other five authors are employees of CSL Behring.
SOURCE: Patel A et al. AANEM 2019, Abstract 94.
REPORTING FROM AANEM 2019
CDC, FDA in hot pursuit of source of vaping lung injuries
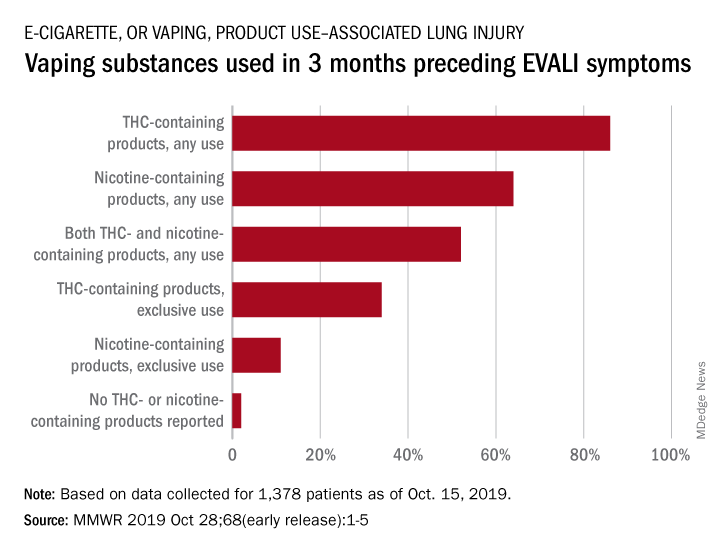
The Centers for Disease Control and Prevention is providing frequent updates of the wide-ranging and aggressive investigation of the cases and deaths linked to vaping, and although a definitive cause remains unknown, evidence is accumulating to implicate tetrahydrocannabinol (THC)-containing devices. The investigation is being conducted in concert with the Food and Drug Administration, many state and local health departments, and public health and clinical partners.
The acronym EVALI has been developed by CDC to refer to e-cigarette, or vaping products use–associated lung injury. In a report summarizing data up to Oct. 22, CDC reported 1,604 EVALI cases and 34 deaths. These cases have occurred in all U.S. states (except Alaska), the District of Columbia, and the U.S. Virgin Islands. The CDC also published a report in the Morbidity and Mortality Weekly report on characteristics of those patients who have died from EVALI-based symptoms as of Oct. 15, 2019.
With data available for more than 867 patients with EVALI, about 86% had a history of using e-cigarette or vaping products that contained THC in the previous 90 days; 64% reported using nicotine-containing products; 34% reported exclusive use of THC-containing products, and 11% reported exclusive use of nicotine-containing products; 52% reported use of both.
In a telebriefing on Oct. 25, Anne Schuchat, MD, CDC principal deputy director, said, “The data do continue to point towards THC-containing products as the source of the vast majority of individuals’ lung injury. There are continuing cases that do not report that history. But I’d like to stress that we don’t know what the risky material or substance is. THC may be a marker for a way that cartridges were prepared or the way that the devices are producing harm. Whether there are similar activities going on with cartridges that don’t contain THC, for instance, remains to be seen. So, I think we are seeing the THC as a marker for products that are risky.”
EVALI deaths
Among the 29 deaths reported as of Oct. 15, 59% (17) were male; the median age was 45 years (range, 17-75 years), 55 years (range, 17-71 years) among males, and 43 years (range, 27-75 years) among females; the age difference between males and females was not statistically significant. Patients who died tended to be older than patients who survived. Among 19 EVALI patients who died and for whom data on substance use was available, the use of any THC-containing products was reported by patients or proxies for 84% (16), including 63% (12) who exclusively used THC-containing products. Use of any nicotine-containing products was reported for 37% (7), including 16% (3) who exclusively used nicotine-containing products. Use of both THC- and nicotine-containing products was reported in four of those who died.
Investigation update
Mitch Zeller, JD, director, Center for Tobacco Products at the Food and Drug Administration, participated in the telebriefing and provided an update on the ongoing investigation. “State of the art methods are being used to assess the presence of a broad range of chemicals including nicotine, THC, and other cannabinoids, opioids, additives, pesticides, poisons and toxins,” he said. “FDA has received or collected over 900 samples from 25 states to date. Those numbers continue to increase. The samples [were] collected directly from consumers, hospitals, and from state offices include vaping devices and products that contain liquid as well as packaging and some nearly empty containers.” He cautioned that identifying the substance is “but one piece of the puzzle and will not necessarily answer questions about causality.” He also noted that the self-reports of THC and/or nicotine could mean that there is misreported data, because reports in many cases are coming from teens and from jurisdictions in which THC is not legal.

The issue of whether EVALI has been seen in recent years but not recognized or whether EVALI is a new phenomenon was raised by a caller at the telebriefing. Dr. Schuchat responded, “We are aware of older cases that look similar to what we are seeing now. But we do not believe that this outbreak or surge in cases is due to better recognition.” She suggested that some evidence points to cutting agents being introduced to increase profits of e-cigarettes and that risky and unknown substances have been introduced into the supply chain.
A “handful” of cases of readmission have been reported, and the CDC is currently investigating whether these cases included patients who took up vaping again or had some other possible contributing factor. Dr. Schuchat cautioned recovering patients not to resume vaping because of the risk of readmission and the probability that their lungs will remain in a weakened state.
Clinical guidance update
The CDC provided detailed interim clinical guidance on evaluating and caring for patients with EVALI. The recommendations focus on patient history, lab testing, criteria for hospitalization, and follow-up for these patients.
Obtaining a detailed history of patients presenting with suspected EVALI is especially important for this patient population, given the many unknowns surrounding this condition, according to the CDC. The updated guidance states, “All health care providers evaluating patients for EVALI should ask about the use of e-cigarette or vaping products, and ideally should ask about types of substances used (e.g.,THC, cannabis [oil, dabs], nicotine, modified products or the addition of substances not intended by the manufacturer); product source, specific product brand and name; duration and frequency of use, time of last use; product delivery system and method of use (aerosolization, dabbing, or dripping).” The approach recommended for soliciting accurate information is “empathetic, nonjudgmental” and, the guidelines say, patients should be questioned in private regarding sensitive information to assure confidentiality.
A respiratory virus panel is recommended for all suspected EVALI patients, although at this time, these tests cannot be used to distinguish EVALI from infectious etiologies. All patients should be considered for urine toxicology testing, including testing for THC.
Imaging guidance for suspected EVALI patients includes chest x-ray, with additional CT scan when the x-ray result does not correlate with clinical findings or to evaluate severe or worsening disease.
Recommended criteria for hospitalization of patients with suspected EVALI are those patients with decreased O2 saturation (less than 95%) on room air, in respiratory distress, or with comorbidities that compromise pulmonary reserve. As of Oct. 8, 96% of patients with suspected EVALI reported to the CDC have been hospitalized.
As for medical treatment of these patients, corticosteroids have been found to be helpful. The statement noted, “Among 140 cases reported nationally to CDC that received corticosteroids, 82% of patients improved.”
The natural progression of this injury is not known, however, and it is possible that patients might recover without corticosteroids. Given the unknown etiology of the disease and “because the diagnosis remains one of exclusion, aggressive empiric therapy with corticosteroids, antimicrobial, and antiviral therapy might be warranted for patients with severe illness. A range of corticosteroid doses, durations, and taper plans might be considered on a case-by-case basis.”
The report concluded with a strong recommendation that patients hospitalized with EVALI are followed closely with a visit 1-2 weeks after discharge and again with additional testing 1-2 months later. Health care providers are also advised to consult medical specialists, in particular pulmonologists, who can offer further evaluation, recommend empiric treatment, and review indications for bronchoscopy.
CPT coding for EVALI
CDC has issued coding guidance to help track EVALI. The document was posted on the CDC website. The coding guidance is consistent with current clinical knowledge about EVALI-related disorders and is intended for use in conjunction with current ICD-10-CM classifications.
The following conditions associated with EVALI are covered in the new coding guidance:
- Bronchitis and pneumonitis caused by chemicals, gases, and fumes; including chemical pneumonitis; J68.0.
- Pneumonitis caused by inhalation of oils and essences; including lipoid pneumonia; J69.1.
- Acute respiratory distress syndrome; J80.
- Pulmonary eosinophilia, not elsewhere classified; J82.
- Acute interstitial pneumonitis; J84.114.
The document notes that the coding guidance has been approved by the National Center for Health Statistics, the American Health Information Management Association, the American Hospital Association, and the Centers for Medicare & Medicaid Services.
Investigation continues
Mr. Zeller cautioned that this investigation will not be concluded in the near future. He noted, “We are committed to working to [solve the mystery] just as quickly as we can, but we also recognize that it will likely take some time. Importantly, the diversity of the patients and the products or substances they have reported using and the samples being tested may mean ultimately that there are multiple causes of these injuries.”
Richard Franki and Gregory Twachtman contributed to this story.
The Centers for Disease Control and Prevention is providing frequent updates of the wide-ranging and aggressive investigation of the cases and deaths linked to vaping, and although a definitive cause remains unknown, evidence is accumulating to implicate tetrahydrocannabinol (THC)-containing devices. The investigation is being conducted in concert with the Food and Drug Administration, many state and local health departments, and public health and clinical partners.
The acronym EVALI has been developed by CDC to refer to e-cigarette, or vaping products use–associated lung injury. In a report summarizing data up to Oct. 22, CDC reported 1,604 EVALI cases and 34 deaths. These cases have occurred in all U.S. states (except Alaska), the District of Columbia, and the U.S. Virgin Islands. The CDC also published a report in the Morbidity and Mortality Weekly report on characteristics of those patients who have died from EVALI-based symptoms as of Oct. 15, 2019.
With data available for more than 867 patients with EVALI, about 86% had a history of using e-cigarette or vaping products that contained THC in the previous 90 days; 64% reported using nicotine-containing products; 34% reported exclusive use of THC-containing products, and 11% reported exclusive use of nicotine-containing products; 52% reported use of both.
In a telebriefing on Oct. 25, Anne Schuchat, MD, CDC principal deputy director, said, “The data do continue to point towards THC-containing products as the source of the vast majority of individuals’ lung injury. There are continuing cases that do not report that history. But I’d like to stress that we don’t know what the risky material or substance is. THC may be a marker for a way that cartridges were prepared or the way that the devices are producing harm. Whether there are similar activities going on with cartridges that don’t contain THC, for instance, remains to be seen. So, I think we are seeing the THC as a marker for products that are risky.”
EVALI deaths
Among the 29 deaths reported as of Oct. 15, 59% (17) were male; the median age was 45 years (range, 17-75 years), 55 years (range, 17-71 years) among males, and 43 years (range, 27-75 years) among females; the age difference between males and females was not statistically significant. Patients who died tended to be older than patients who survived. Among 19 EVALI patients who died and for whom data on substance use was available, the use of any THC-containing products was reported by patients or proxies for 84% (16), including 63% (12) who exclusively used THC-containing products. Use of any nicotine-containing products was reported for 37% (7), including 16% (3) who exclusively used nicotine-containing products. Use of both THC- and nicotine-containing products was reported in four of those who died.
Investigation update
Mitch Zeller, JD, director, Center for Tobacco Products at the Food and Drug Administration, participated in the telebriefing and provided an update on the ongoing investigation. “State of the art methods are being used to assess the presence of a broad range of chemicals including nicotine, THC, and other cannabinoids, opioids, additives, pesticides, poisons and toxins,” he said. “FDA has received or collected over 900 samples from 25 states to date. Those numbers continue to increase. The samples [were] collected directly from consumers, hospitals, and from state offices include vaping devices and products that contain liquid as well as packaging and some nearly empty containers.” He cautioned that identifying the substance is “but one piece of the puzzle and will not necessarily answer questions about causality.” He also noted that the self-reports of THC and/or nicotine could mean that there is misreported data, because reports in many cases are coming from teens and from jurisdictions in which THC is not legal.
The issue of whether EVALI has been seen in recent years but not recognized or whether EVALI is a new phenomenon was raised by a caller at the telebriefing. Dr. Schuchat responded, “We are aware of older cases that look similar to what we are seeing now. But we do not believe that this outbreak or surge in cases is due to better recognition.” She suggested that some evidence points to cutting agents being introduced to increase profits of e-cigarettes and that risky and unknown substances have been introduced into the supply chain.
A “handful” of cases of readmission have been reported, and the CDC is currently investigating whether these cases included patients who took up vaping again or had some other possible contributing factor. Dr. Schuchat cautioned recovering patients not to resume vaping because of the risk of readmission and the probability that their lungs will remain in a weakened state.
Clinical guidance update
The CDC provided detailed interim clinical guidance on evaluating and caring for patients with EVALI. The recommendations focus on patient history, lab testing, criteria for hospitalization, and follow-up for these patients.
Obtaining a detailed history of patients presenting with suspected EVALI is especially important for this patient population, given the many unknowns surrounding this condition, according to the CDC. The updated guidance states, “All health care providers evaluating patients for EVALI should ask about the use of e-cigarette or vaping products, and ideally should ask about types of substances used (e.g.,THC, cannabis [oil, dabs], nicotine, modified products or the addition of substances not intended by the manufacturer); product source, specific product brand and name; duration and frequency of use, time of last use; product delivery system and method of use (aerosolization, dabbing, or dripping).” The approach recommended for soliciting accurate information is “empathetic, nonjudgmental” and, the guidelines say, patients should be questioned in private regarding sensitive information to assure confidentiality.
A respiratory virus panel is recommended for all suspected EVALI patients, although at this time, these tests cannot be used to distinguish EVALI from infectious etiologies. All patients should be considered for urine toxicology testing, including testing for THC.
Imaging guidance for suspected EVALI patients includes chest x-ray, with additional CT scan when the x-ray result does not correlate with clinical findings or to evaluate severe or worsening disease.
Recommended criteria for hospitalization of patients with suspected EVALI are those patients with decreased O2 saturation (less than 95%) on room air, in respiratory distress, or with comorbidities that compromise pulmonary reserve. As of Oct. 8, 96% of patients with suspected EVALI reported to the CDC have been hospitalized.
As for medical treatment of these patients, corticosteroids have been found to be helpful. The statement noted, “Among 140 cases reported nationally to CDC that received corticosteroids, 82% of patients improved.”
The natural progression of this injury is not known, however, and it is possible that patients might recover without corticosteroids. Given the unknown etiology of the disease and “because the diagnosis remains one of exclusion, aggressive empiric therapy with corticosteroids, antimicrobial, and antiviral therapy might be warranted for patients with severe illness. A range of corticosteroid doses, durations, and taper plans might be considered on a case-by-case basis.”
The report concluded with a strong recommendation that patients hospitalized with EVALI are followed closely with a visit 1-2 weeks after discharge and again with additional testing 1-2 months later. Health care providers are also advised to consult medical specialists, in particular pulmonologists, who can offer further evaluation, recommend empiric treatment, and review indications for bronchoscopy.
CPT coding for EVALI
CDC has issued coding guidance to help track EVALI. The document was posted on the CDC website. The coding guidance is consistent with current clinical knowledge about EVALI-related disorders and is intended for use in conjunction with current ICD-10-CM classifications.
The following conditions associated with EVALI are covered in the new coding guidance:
- Bronchitis and pneumonitis caused by chemicals, gases, and fumes; including chemical pneumonitis; J68.0.
- Pneumonitis caused by inhalation of oils and essences; including lipoid pneumonia; J69.1.
- Acute respiratory distress syndrome; J80.
- Pulmonary eosinophilia, not elsewhere classified; J82.
- Acute interstitial pneumonitis; J84.114.
The document notes that the coding guidance has been approved by the National Center for Health Statistics, the American Health Information Management Association, the American Hospital Association, and the Centers for Medicare & Medicaid Services.
Investigation continues
Mr. Zeller cautioned that this investigation will not be concluded in the near future. He noted, “We are committed to working to [solve the mystery] just as quickly as we can, but we also recognize that it will likely take some time. Importantly, the diversity of the patients and the products or substances they have reported using and the samples being tested may mean ultimately that there are multiple causes of these injuries.”
Richard Franki and Gregory Twachtman contributed to this story.
The Centers for Disease Control and Prevention is providing frequent updates of the wide-ranging and aggressive investigation of the cases and deaths linked to vaping, and although a definitive cause remains unknown, evidence is accumulating to implicate tetrahydrocannabinol (THC)-containing devices. The investigation is being conducted in concert with the Food and Drug Administration, many state and local health departments, and public health and clinical partners.
The acronym EVALI has been developed by CDC to refer to e-cigarette, or vaping products use–associated lung injury. In a report summarizing data up to Oct. 22, CDC reported 1,604 EVALI cases and 34 deaths. These cases have occurred in all U.S. states (except Alaska), the District of Columbia, and the U.S. Virgin Islands. The CDC also published a report in the Morbidity and Mortality Weekly report on characteristics of those patients who have died from EVALI-based symptoms as of Oct. 15, 2019.
With data available for more than 867 patients with EVALI, about 86% had a history of using e-cigarette or vaping products that contained THC in the previous 90 days; 64% reported using nicotine-containing products; 34% reported exclusive use of THC-containing products, and 11% reported exclusive use of nicotine-containing products; 52% reported use of both.
In a telebriefing on Oct. 25, Anne Schuchat, MD, CDC principal deputy director, said, “The data do continue to point towards THC-containing products as the source of the vast majority of individuals’ lung injury. There are continuing cases that do not report that history. But I’d like to stress that we don’t know what the risky material or substance is. THC may be a marker for a way that cartridges were prepared or the way that the devices are producing harm. Whether there are similar activities going on with cartridges that don’t contain THC, for instance, remains to be seen. So, I think we are seeing the THC as a marker for products that are risky.”
EVALI deaths
Among the 29 deaths reported as of Oct. 15, 59% (17) were male; the median age was 45 years (range, 17-75 years), 55 years (range, 17-71 years) among males, and 43 years (range, 27-75 years) among females; the age difference between males and females was not statistically significant. Patients who died tended to be older than patients who survived. Among 19 EVALI patients who died and for whom data on substance use was available, the use of any THC-containing products was reported by patients or proxies for 84% (16), including 63% (12) who exclusively used THC-containing products. Use of any nicotine-containing products was reported for 37% (7), including 16% (3) who exclusively used nicotine-containing products. Use of both THC- and nicotine-containing products was reported in four of those who died.
Investigation update
Mitch Zeller, JD, director, Center for Tobacco Products at the Food and Drug Administration, participated in the telebriefing and provided an update on the ongoing investigation. “State of the art methods are being used to assess the presence of a broad range of chemicals including nicotine, THC, and other cannabinoids, opioids, additives, pesticides, poisons and toxins,” he said. “FDA has received or collected over 900 samples from 25 states to date. Those numbers continue to increase. The samples [were] collected directly from consumers, hospitals, and from state offices include vaping devices and products that contain liquid as well as packaging and some nearly empty containers.” He cautioned that identifying the substance is “but one piece of the puzzle and will not necessarily answer questions about causality.” He also noted that the self-reports of THC and/or nicotine could mean that there is misreported data, because reports in many cases are coming from teens and from jurisdictions in which THC is not legal.
The issue of whether EVALI has been seen in recent years but not recognized or whether EVALI is a new phenomenon was raised by a caller at the telebriefing. Dr. Schuchat responded, “We are aware of older cases that look similar to what we are seeing now. But we do not believe that this outbreak or surge in cases is due to better recognition.” She suggested that some evidence points to cutting agents being introduced to increase profits of e-cigarettes and that risky and unknown substances have been introduced into the supply chain.
A “handful” of cases of readmission have been reported, and the CDC is currently investigating whether these cases included patients who took up vaping again or had some other possible contributing factor. Dr. Schuchat cautioned recovering patients not to resume vaping because of the risk of readmission and the probability that their lungs will remain in a weakened state.
Clinical guidance update
The CDC provided detailed interim clinical guidance on evaluating and caring for patients with EVALI. The recommendations focus on patient history, lab testing, criteria for hospitalization, and follow-up for these patients.
Obtaining a detailed history of patients presenting with suspected EVALI is especially important for this patient population, given the many unknowns surrounding this condition, according to the CDC. The updated guidance states, “All health care providers evaluating patients for EVALI should ask about the use of e-cigarette or vaping products, and ideally should ask about types of substances used (e.g.,THC, cannabis [oil, dabs], nicotine, modified products or the addition of substances not intended by the manufacturer); product source, specific product brand and name; duration and frequency of use, time of last use; product delivery system and method of use (aerosolization, dabbing, or dripping).” The approach recommended for soliciting accurate information is “empathetic, nonjudgmental” and, the guidelines say, patients should be questioned in private regarding sensitive information to assure confidentiality.
A respiratory virus panel is recommended for all suspected EVALI patients, although at this time, these tests cannot be used to distinguish EVALI from infectious etiologies. All patients should be considered for urine toxicology testing, including testing for THC.
Imaging guidance for suspected EVALI patients includes chest x-ray, with additional CT scan when the x-ray result does not correlate with clinical findings or to evaluate severe or worsening disease.
Recommended criteria for hospitalization of patients with suspected EVALI are those patients with decreased O2 saturation (less than 95%) on room air, in respiratory distress, or with comorbidities that compromise pulmonary reserve. As of Oct. 8, 96% of patients with suspected EVALI reported to the CDC have been hospitalized.
As for medical treatment of these patients, corticosteroids have been found to be helpful. The statement noted, “Among 140 cases reported nationally to CDC that received corticosteroids, 82% of patients improved.”
The natural progression of this injury is not known, however, and it is possible that patients might recover without corticosteroids. Given the unknown etiology of the disease and “because the diagnosis remains one of exclusion, aggressive empiric therapy with corticosteroids, antimicrobial, and antiviral therapy might be warranted for patients with severe illness. A range of corticosteroid doses, durations, and taper plans might be considered on a case-by-case basis.”
The report concluded with a strong recommendation that patients hospitalized with EVALI are followed closely with a visit 1-2 weeks after discharge and again with additional testing 1-2 months later. Health care providers are also advised to consult medical specialists, in particular pulmonologists, who can offer further evaluation, recommend empiric treatment, and review indications for bronchoscopy.
CPT coding for EVALI
CDC has issued coding guidance to help track EVALI. The document was posted on the CDC website. The coding guidance is consistent with current clinical knowledge about EVALI-related disorders and is intended for use in conjunction with current ICD-10-CM classifications.
The following conditions associated with EVALI are covered in the new coding guidance:
- Bronchitis and pneumonitis caused by chemicals, gases, and fumes; including chemical pneumonitis; J68.0.
- Pneumonitis caused by inhalation of oils and essences; including lipoid pneumonia; J69.1.
- Acute respiratory distress syndrome; J80.
- Pulmonary eosinophilia, not elsewhere classified; J82.
- Acute interstitial pneumonitis; J84.114.
The document notes that the coding guidance has been approved by the National Center for Health Statistics, the American Health Information Management Association, the American Hospital Association, and the Centers for Medicare & Medicaid Services.
Investigation continues
Mr. Zeller cautioned that this investigation will not be concluded in the near future. He noted, “We are committed to working to [solve the mystery] just as quickly as we can, but we also recognize that it will likely take some time. Importantly, the diversity of the patients and the products or substances they have reported using and the samples being tested may mean ultimately that there are multiple causes of these injuries.”
Richard Franki and Gregory Twachtman contributed to this story.
THC use reported in majority of vaping-related illnesses
(EVALI), according to the Centers for Disease Control and Prevention.
In the largest analysis to date, exclusive use of THC-containing products was reported for 34% of the 1,378 patients with confirmed or probable EVALI as of Oct. 15, 2019. Among those who died, 63% had been using THC exclusively during the 3 months preceding symptom onset, Erin D. Moritz, PhD, and associates said Oct. 28 in the Morbidity and Mortality Weekly Report.
Almost two-thirds (64%) of all EVALI patients had used nicotine-containing products at some time in the 3 months before symptom onset, and nicotine use was exclusive for 11%. Any nicotine use was reported for 37% of EVALI-related deaths, with exclusive use at 16%, the investigators reported.
“The data presented here suggest that THC-containing products are playing an important role in this outbreak,” they wrote, but “to date, no single compound or ingredient has emerged as the cause of EVALI, and there might be more than one cause.”
Dr. Moritz and associates also noted that many “patients likely did not know the content of the e-cigarette, or vaping, products they used,” which may have led to misclassification of substances.
SOURCE: Moritz ED et al. MMWR. Morbidity and mortality weekly report 2019 Oct 28;68(early release):1-4.
(EVALI), according to the Centers for Disease Control and Prevention.
In the largest analysis to date, exclusive use of THC-containing products was reported for 34% of the 1,378 patients with confirmed or probable EVALI as of Oct. 15, 2019. Among those who died, 63% had been using THC exclusively during the 3 months preceding symptom onset, Erin D. Moritz, PhD, and associates said Oct. 28 in the Morbidity and Mortality Weekly Report.
Almost two-thirds (64%) of all EVALI patients had used nicotine-containing products at some time in the 3 months before symptom onset, and nicotine use was exclusive for 11%. Any nicotine use was reported for 37% of EVALI-related deaths, with exclusive use at 16%, the investigators reported.
“The data presented here suggest that THC-containing products are playing an important role in this outbreak,” they wrote, but “to date, no single compound or ingredient has emerged as the cause of EVALI, and there might be more than one cause.”
Dr. Moritz and associates also noted that many “patients likely did not know the content of the e-cigarette, or vaping, products they used,” which may have led to misclassification of substances.
SOURCE: Moritz ED et al. MMWR. Morbidity and mortality weekly report 2019 Oct 28;68(early release):1-4.
(EVALI), according to the Centers for Disease Control and Prevention.
In the largest analysis to date, exclusive use of THC-containing products was reported for 34% of the 1,378 patients with confirmed or probable EVALI as of Oct. 15, 2019. Among those who died, 63% had been using THC exclusively during the 3 months preceding symptom onset, Erin D. Moritz, PhD, and associates said Oct. 28 in the Morbidity and Mortality Weekly Report.
Almost two-thirds (64%) of all EVALI patients had used nicotine-containing products at some time in the 3 months before symptom onset, and nicotine use was exclusive for 11%. Any nicotine use was reported for 37% of EVALI-related deaths, with exclusive use at 16%, the investigators reported.
“The data presented here suggest that THC-containing products are playing an important role in this outbreak,” they wrote, but “to date, no single compound or ingredient has emerged as the cause of EVALI, and there might be more than one cause.”
Dr. Moritz and associates also noted that many “patients likely did not know the content of the e-cigarette, or vaping, products they used,” which may have led to misclassification of substances.
SOURCE: Moritz ED et al. MMWR. Morbidity and mortality weekly report 2019 Oct 28;68(early release):1-4.
FROM MMWR