User login
Fast Track to Abdominal Pain
ANSWER
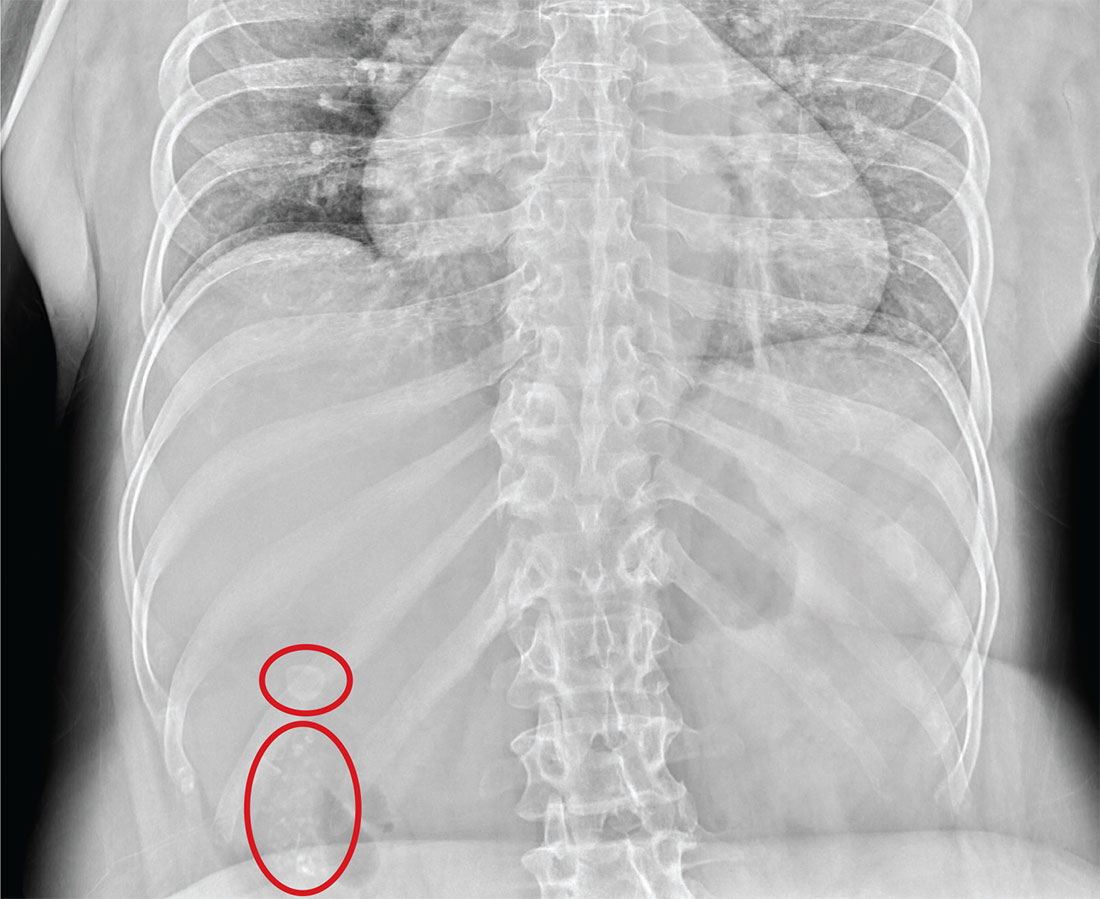
The radiograph illustrates a large calcification within the right upper quadrant—most likely a gallstone. Several smaller calcifications are clustered together in the same area, making the diagnosis cholelithiasis. The patient was referred to the general surgery clinic for further
evaluation.
For recent findings on gallstone disease and heart risk, see here.
ANSWER
The radiograph illustrates a large calcification within the right upper quadrant—most likely a gallstone. Several smaller calcifications are clustered together in the same area, making the diagnosis cholelithiasis. The patient was referred to the general surgery clinic for further
evaluation.
For recent findings on gallstone disease and heart risk, see here.
ANSWER
The radiograph illustrates a large calcification within the right upper quadrant—most likely a gallstone. Several smaller calcifications are clustered together in the same area, making the diagnosis cholelithiasis. The patient was referred to the general surgery clinic for further
evaluation.
For recent findings on gallstone disease and heart risk, see here.
An NP student you are precepting in the emergency department fast track area presents her patient to you: a 60-year-old woman with abdominal pain. The pain is chronic but has worsened slightly, prompting the patient, who does not have a primary care provider, to present today. She experiences occasional nausea but no fever, and she denies any bowel or bladder complaints other than constipation. Her medical history is significant for mild hypertension.
On exam, your student notes an obese female who is in no obvious distress. The patient’s vital signs are all within normal limits. The abdominal exam is unimpressive, revealing a soft abdomen with good bowel sounds. Although she does have mild diffuse tenderness, she has no rebound or guarding.
Although your student suspects the patient is just constipated, she orders blood work and urinalysis. An abdominal survey is obtained as well. What is your impression?
His Head Is In the Game–His Heart, Not So Much
ANSWER
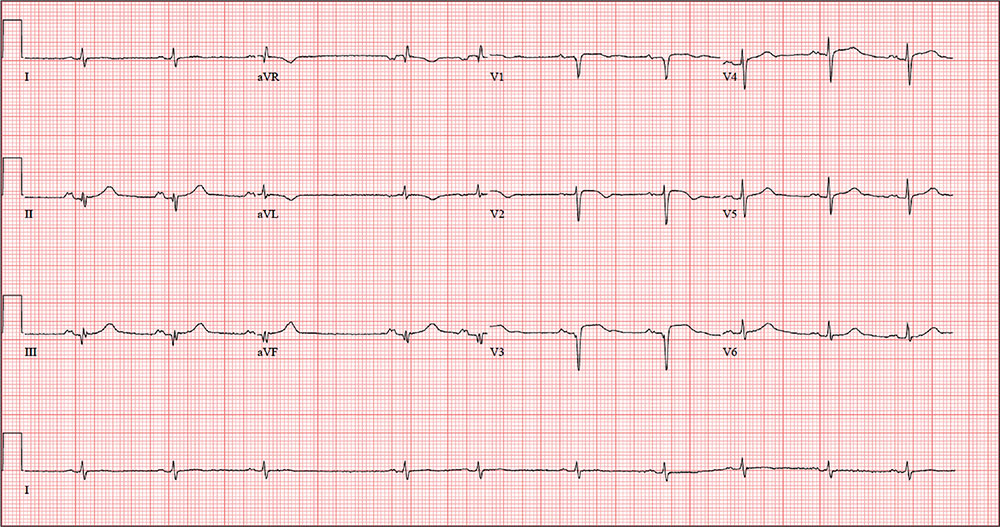
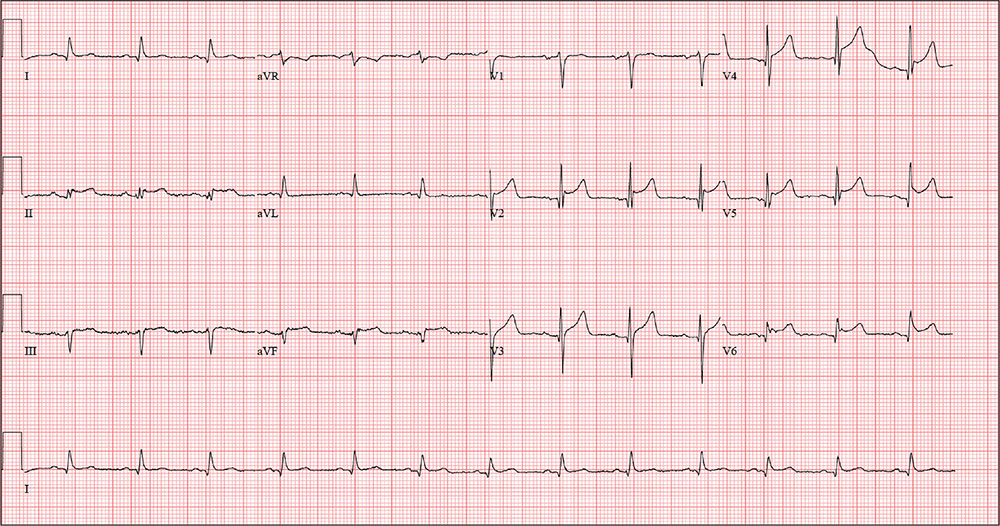
The correct interpretation includes normal sinus rhythm with a new anterior myocardial infarction (MI) and inferolateral injury.
Criteria for an anterior MI include Q waves in leads V2 to V6 and hyperacute ST-T wave changes. V1 is usually spared.
Inferolateral injury is evidenced by the presence of Q waves and absence of R waves in leads III and aVF.
The patient’s cardiac catheterization revealed left anterior descending and circumflex artery disease.
ANSWER
The correct interpretation includes normal sinus rhythm with a new anterior myocardial infarction (MI) and inferolateral injury.
Criteria for an anterior MI include Q waves in leads V2 to V6 and hyperacute ST-T wave changes. V1 is usually spared.
Inferolateral injury is evidenced by the presence of Q waves and absence of R waves in leads III and aVF.
The patient’s cardiac catheterization revealed left anterior descending and circumflex artery disease.
ANSWER
The correct interpretation includes normal sinus rhythm with a new anterior myocardial infarction (MI) and inferolateral injury.
Criteria for an anterior MI include Q waves in leads V2 to V6 and hyperacute ST-T wave changes. V1 is usually spared.
Inferolateral injury is evidenced by the presence of Q waves and absence of R waves in leads III and aVF.
The patient’s cardiac catheterization revealed left anterior descending and circumflex artery disease.
While playing in a company racquetball tournament, a 47-year-old man suddenly collapses. He is unresponsive, without a pulse, and his coworkers immediately initiate CPR. A single shock from an on-site AED successfully resuscitates him from ventricular fibrillation. Paramedics arrive within 12 minutes, by which time the patient is conscious and responsive to commands.
During transportation to the emergency department, however, he develops ventricular tachycardia and loses consciousness; a single shock cardioverts him to normal sinus rhythm. An ECG is ordered and the cardiac catheterization team mobilized.
While waiting, you contact the patient’s wife by telephone. She tells you that her husband has been complaining of chest discomfort for the past week and woke up two nights ago with what he described as “indigestion.” She says she suggested he see a clinician, and he agreed to do so—but not until after the tournament.
His medical history is remarkable only for chronic gastroesophageal reflux disease (GERD), for which he takes chewable antacids on a daily basis. He has no prior cardiac or pulmonary history. There is no surgical history apart from a tonsillectomy in childhood.
The patient is the Chief Operating Officer at a local manufacturing facility. He is married and has three children. His parents and grandparents are alive and in good health, aside from some arthritis in the older generation.
The review of systems, obtained from his wife, is noncontributory. She says he is in excellent health—he jogs, rides mountain bikes, and enjoys racquetball. Her husband attributed his recent chest discomfort to GERD; when she asked if it could be cardiac related, he adamantly denied the possibility, as he was in “excellent health” with “good genes.”
Vital signs include a blood pressure of 118/56 mm Hg; pulse, 80 beats/min; respiratory rate, 18 breaths/min-1; and O2 saturation, 100% on 2 L oxygen. He is afebrile.
Physical exam reveals a thin, otherwise healthy male who appears anxious and apprehensive. He denies ongoing chest pain but states that his chest wall is sore beneath the defibrillator pads. A focal exam reveals normal lung sounds and a regular rate and rhythm with no murmurs, gallops, or rubs. The abdomen is soft and nontender. All peripheral pulses are strong and equal bilaterally, and there are no focal neurologic signs.
The ECG taken at admission shows a ventricular rate of 80 beats/min; PR interval, 162 ms; QRS duration, 106 ms; QT/QTc interval, 370/426 ms; P axis, 51°; R axis, –20°; and T axis, 70°. What is your interpretation of the ECG?
Reticular Hyperpigmentation on the Lower Legs
The Diagnosis: Erythema Ab Igne
Given the patient's reticulated hyperpigmented lesions in the setting of recent space heater use with heater closer to the more affected leg, erythema ab igne was diagnosed. Patient education was provided and moving the heater away from the lower extremities was advised.
Erythema ab igne first was described by German dermatologist Abraham Buschke as hitze melanose, meaning melanosis induced by heat. The classic skin findings were first observed on the lower legs of patients who worked in front of open fires or coal stoves.1 Over the years, new causes of erythema ab igne secondary to prolonged thermal radiation exposure have been reported.1 In the elderly, hospitalized, and chronic pain patients, erythema ab igne has been observed in areas treated with heating pads and blankets.2 Other triggers such as frequent hot bathing, furniture, steam radiators, space heaters, and laptops also have been reported.3-6 Laptop-induced erythema ab igne is a diagnosis that has been reported in the last decade and its incidence likely will increase in the future.6
The clinical manifestations of erythema ab igne correlate with the frequency and duration of heat exposure. Acutely, a mild and transient erythema develops in the affected area. With chronic heat exposure, these areas subsequently develop a permanent reticulated hyperpigmented pattern and may eventually become atrophic.2,6 All body surfaces are at risk, but erythema ab igne classically involves the legs, lower back, and/or abdomen. Lesions typically are asymptomatic; however, burning and pruritus can be present.2,6 Bullous erythema ab igne, though rare, has been reported,7 suggesting a potential transition from erythema ab igne to burns.6
Biopsy is not recommended for diagnosis; however, the histopathologic changes of erythema ab igne include hyperkeratosis, interface dermatitis, epidermal atrophy with apoptotic keratinocytes, and melanin incontinence. Although this condition typically is benign, histologic findings could resemble actinic keratosis, suggesting that chronic changes induced by infrared thermal radiation may lead to squamous cell carcinoma or rarely Merkel cell carcinoma. The latency for developing carcinoma appears to extend 30 years, with a 30% tendency for recurrence or metastasis. Given the possibility of an increase in erythema ab igne in the pediatric population in the upcoming years, as displayed by our patient, and increasing laptop and electronic use in children and adolescents, it is important to be aware of this skin condition and the potential complications of it going undiagnosed.2,6
No specific therapy for erythema ab igne exists. Treatment is centered on eliminating exposure to the heat source. With appropriate removal, the reticulated hyperpigmented lesions will resolve, sometimes taking several months.
Differential diagnosis includes livedo reticularis, livedoid vasculopathy, and cutis marmorata. The reticulated purpuric lesions of livedo reticularis involving the extremities often mimic erythema ab igne's cutaneous morphology; however, livedo reticularis frequently is associated with conditions such as drug reactions, infections, thrombosis, and vasculitides,2 as opposed to erythema ab igne, which frequently is associated with conditions causing pain or decreased body temperature, thus necessitating use of heating devices, as seen in our patient. Livedoid vasculopathy is characterized by purpuric macules involving the lower legs and feet that progress to recurrent leg ulcers. Our patient's asymptomatic lesions and absence of ulcers excluded this diagnosis.8 Lastly, cutis marmorata, a congenital condition, is characterized by blue-violet vascular networks that often display ulceration and atrophy of the involved skin as well as hypertrophy or atrophy of the involved limb9; these clinical findings were not present in our patient and this diagnosis would not explain the relationship between the cutaneous lesions and heat exposure.
- Nilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
- Riahi RR, Cohen PR, Robinson FW, et al. Erythema ab igne mimicking livedo reticularis. Int J Dermatol. 2010;49:1314-1317.
- Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol. 2002;82:478-479.
- Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol. 1996;34:516-517.
- Kligman LH, Kligman AM. Reflections on heat. Br J Dermatol. 1984;110:369-375.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:e1227-e1230.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Khenifer S, Thomas L, Balme B, et al. Livedoid vasculopathy: thrombotic or inflammatory disease? Clin Exp Dermatol. 2009;35:693-698.
- Pernet C, Guillot B, Bigorre M, et al. Focal and atrophic cutis marmorata telangiectatica congenital. J Am Acad Dermatol. 2013;69:e268-e269.
The Diagnosis: Erythema Ab Igne
Given the patient's reticulated hyperpigmented lesions in the setting of recent space heater use with heater closer to the more affected leg, erythema ab igne was diagnosed. Patient education was provided and moving the heater away from the lower extremities was advised.
Erythema ab igne first was described by German dermatologist Abraham Buschke as hitze melanose, meaning melanosis induced by heat. The classic skin findings were first observed on the lower legs of patients who worked in front of open fires or coal stoves.1 Over the years, new causes of erythema ab igne secondary to prolonged thermal radiation exposure have been reported.1 In the elderly, hospitalized, and chronic pain patients, erythema ab igne has been observed in areas treated with heating pads and blankets.2 Other triggers such as frequent hot bathing, furniture, steam radiators, space heaters, and laptops also have been reported.3-6 Laptop-induced erythema ab igne is a diagnosis that has been reported in the last decade and its incidence likely will increase in the future.6
The clinical manifestations of erythema ab igne correlate with the frequency and duration of heat exposure. Acutely, a mild and transient erythema develops in the affected area. With chronic heat exposure, these areas subsequently develop a permanent reticulated hyperpigmented pattern and may eventually become atrophic.2,6 All body surfaces are at risk, but erythema ab igne classically involves the legs, lower back, and/or abdomen. Lesions typically are asymptomatic; however, burning and pruritus can be present.2,6 Bullous erythema ab igne, though rare, has been reported,7 suggesting a potential transition from erythema ab igne to burns.6
Biopsy is not recommended for diagnosis; however, the histopathologic changes of erythema ab igne include hyperkeratosis, interface dermatitis, epidermal atrophy with apoptotic keratinocytes, and melanin incontinence. Although this condition typically is benign, histologic findings could resemble actinic keratosis, suggesting that chronic changes induced by infrared thermal radiation may lead to squamous cell carcinoma or rarely Merkel cell carcinoma. The latency for developing carcinoma appears to extend 30 years, with a 30% tendency for recurrence or metastasis. Given the possibility of an increase in erythema ab igne in the pediatric population in the upcoming years, as displayed by our patient, and increasing laptop and electronic use in children and adolescents, it is important to be aware of this skin condition and the potential complications of it going undiagnosed.2,6
No specific therapy for erythema ab igne exists. Treatment is centered on eliminating exposure to the heat source. With appropriate removal, the reticulated hyperpigmented lesions will resolve, sometimes taking several months.
Differential diagnosis includes livedo reticularis, livedoid vasculopathy, and cutis marmorata. The reticulated purpuric lesions of livedo reticularis involving the extremities often mimic erythema ab igne's cutaneous morphology; however, livedo reticularis frequently is associated with conditions such as drug reactions, infections, thrombosis, and vasculitides,2 as opposed to erythema ab igne, which frequently is associated with conditions causing pain or decreased body temperature, thus necessitating use of heating devices, as seen in our patient. Livedoid vasculopathy is characterized by purpuric macules involving the lower legs and feet that progress to recurrent leg ulcers. Our patient's asymptomatic lesions and absence of ulcers excluded this diagnosis.8 Lastly, cutis marmorata, a congenital condition, is characterized by blue-violet vascular networks that often display ulceration and atrophy of the involved skin as well as hypertrophy or atrophy of the involved limb9; these clinical findings were not present in our patient and this diagnosis would not explain the relationship between the cutaneous lesions and heat exposure.
The Diagnosis: Erythema Ab Igne
Given the patient's reticulated hyperpigmented lesions in the setting of recent space heater use with heater closer to the more affected leg, erythema ab igne was diagnosed. Patient education was provided and moving the heater away from the lower extremities was advised.
Erythema ab igne first was described by German dermatologist Abraham Buschke as hitze melanose, meaning melanosis induced by heat. The classic skin findings were first observed on the lower legs of patients who worked in front of open fires or coal stoves.1 Over the years, new causes of erythema ab igne secondary to prolonged thermal radiation exposure have been reported.1 In the elderly, hospitalized, and chronic pain patients, erythema ab igne has been observed in areas treated with heating pads and blankets.2 Other triggers such as frequent hot bathing, furniture, steam radiators, space heaters, and laptops also have been reported.3-6 Laptop-induced erythema ab igne is a diagnosis that has been reported in the last decade and its incidence likely will increase in the future.6
The clinical manifestations of erythema ab igne correlate with the frequency and duration of heat exposure. Acutely, a mild and transient erythema develops in the affected area. With chronic heat exposure, these areas subsequently develop a permanent reticulated hyperpigmented pattern and may eventually become atrophic.2,6 All body surfaces are at risk, but erythema ab igne classically involves the legs, lower back, and/or abdomen. Lesions typically are asymptomatic; however, burning and pruritus can be present.2,6 Bullous erythema ab igne, though rare, has been reported,7 suggesting a potential transition from erythema ab igne to burns.6
Biopsy is not recommended for diagnosis; however, the histopathologic changes of erythema ab igne include hyperkeratosis, interface dermatitis, epidermal atrophy with apoptotic keratinocytes, and melanin incontinence. Although this condition typically is benign, histologic findings could resemble actinic keratosis, suggesting that chronic changes induced by infrared thermal radiation may lead to squamous cell carcinoma or rarely Merkel cell carcinoma. The latency for developing carcinoma appears to extend 30 years, with a 30% tendency for recurrence or metastasis. Given the possibility of an increase in erythema ab igne in the pediatric population in the upcoming years, as displayed by our patient, and increasing laptop and electronic use in children and adolescents, it is important to be aware of this skin condition and the potential complications of it going undiagnosed.2,6
No specific therapy for erythema ab igne exists. Treatment is centered on eliminating exposure to the heat source. With appropriate removal, the reticulated hyperpigmented lesions will resolve, sometimes taking several months.
Differential diagnosis includes livedo reticularis, livedoid vasculopathy, and cutis marmorata. The reticulated purpuric lesions of livedo reticularis involving the extremities often mimic erythema ab igne's cutaneous morphology; however, livedo reticularis frequently is associated with conditions such as drug reactions, infections, thrombosis, and vasculitides,2 as opposed to erythema ab igne, which frequently is associated with conditions causing pain or decreased body temperature, thus necessitating use of heating devices, as seen in our patient. Livedoid vasculopathy is characterized by purpuric macules involving the lower legs and feet that progress to recurrent leg ulcers. Our patient's asymptomatic lesions and absence of ulcers excluded this diagnosis.8 Lastly, cutis marmorata, a congenital condition, is characterized by blue-violet vascular networks that often display ulceration and atrophy of the involved skin as well as hypertrophy or atrophy of the involved limb9; these clinical findings were not present in our patient and this diagnosis would not explain the relationship between the cutaneous lesions and heat exposure.
- Nilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
- Riahi RR, Cohen PR, Robinson FW, et al. Erythema ab igne mimicking livedo reticularis. Int J Dermatol. 2010;49:1314-1317.
- Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol. 2002;82:478-479.
- Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol. 1996;34:516-517.
- Kligman LH, Kligman AM. Reflections on heat. Br J Dermatol. 1984;110:369-375.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:e1227-e1230.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Khenifer S, Thomas L, Balme B, et al. Livedoid vasculopathy: thrombotic or inflammatory disease? Clin Exp Dermatol. 2009;35:693-698.
- Pernet C, Guillot B, Bigorre M, et al. Focal and atrophic cutis marmorata telangiectatica congenital. J Am Acad Dermatol. 2013;69:e268-e269.
- Nilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
- Riahi RR, Cohen PR, Robinson FW, et al. Erythema ab igne mimicking livedo reticularis. Int J Dermatol. 2010;49:1314-1317.
- Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol. 2002;82:478-479.
- Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol. 1996;34:516-517.
- Kligman LH, Kligman AM. Reflections on heat. Br J Dermatol. 1984;110:369-375.
- Arnold AW, Itin PH. Laptop computer−induced erythema ab igne in a child and review of the literature [published online October 4, 2010]. Pediatrics. 2010;126:e1227-e1230.
- Kokturk A, Kaya TI, Baz K, et al. Bullous erythema ab igne. Dermatol Online J. 2003;9:18.
- Khenifer S, Thomas L, Balme B, et al. Livedoid vasculopathy: thrombotic or inflammatory disease? Clin Exp Dermatol. 2009;35:693-698.
- Pernet C, Guillot B, Bigorre M, et al. Focal and atrophic cutis marmorata telangiectatica congenital. J Am Acad Dermatol. 2013;69:e268-e269.
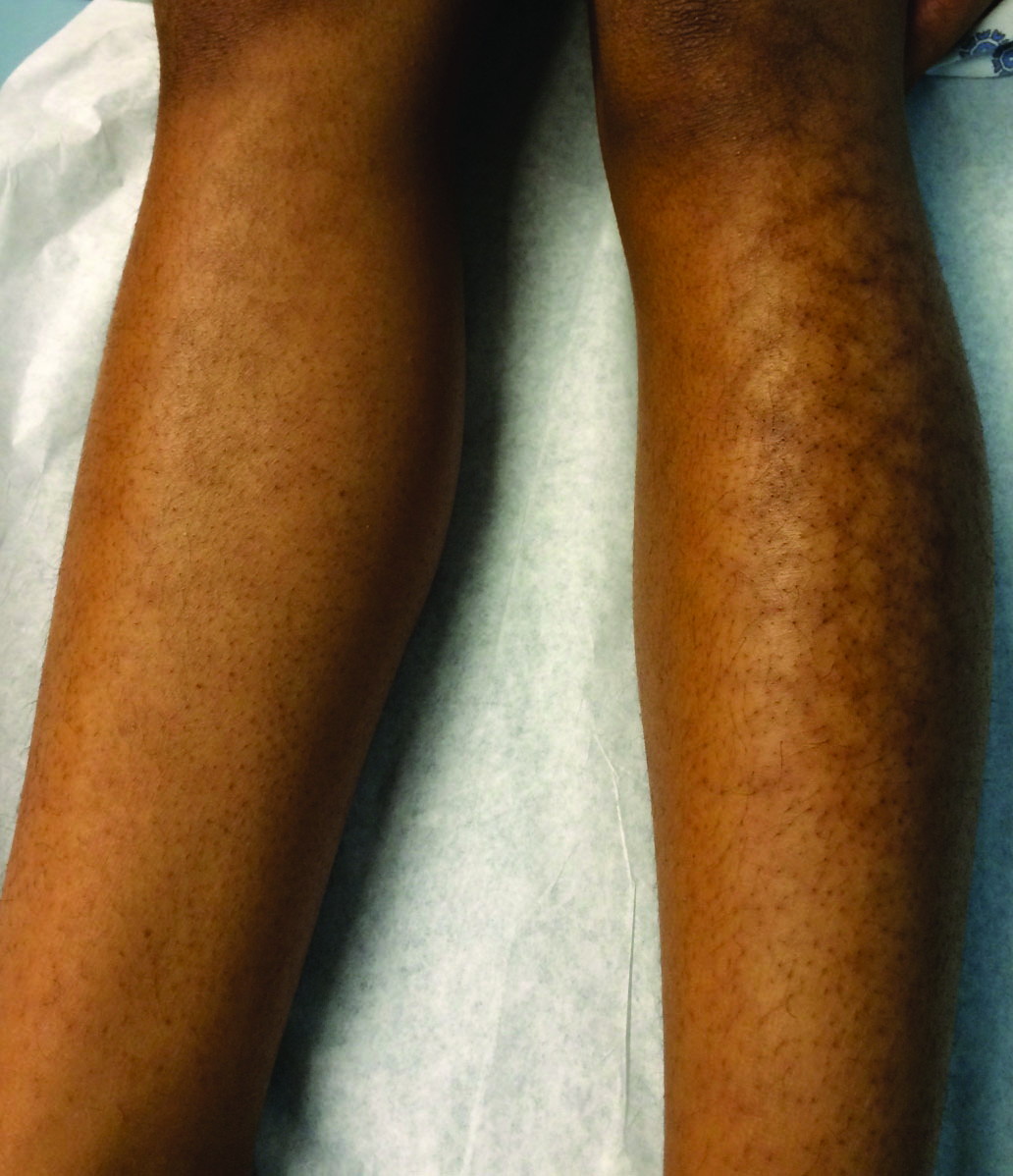
A 13-year-old otherwise healthy adolescent girl presented to the pediatric dermatology clinic for evaluation of a rash on the legs. The patient noticed the rash 1 month prior to presentation. The rash initially involved the left shin and gradually spread to involve the shins bilaterally. The rash was asymptomatic with no pain, pruritus, or muscular asymmetry of the legs. She denied recent fevers, chills, or travel. The patient reported using a space heater daily that was directed at the legs, approximately 0.5 m away. Physical examination revealed a well-nourished adolescent girl in no acute distress with reticular hyperpigmentation of the lower extremities located on the left anterior shin and knee, with mild involvement of the right shin. The reticulated hyperpigmented areas were arranged in a rectangular distribution. Lower extremity musculoskeletal examination was symmetric.
Rapidly Growing Scalp Nodule
Cutaneous Metastasis of Pulmonary Adenocarcinoma
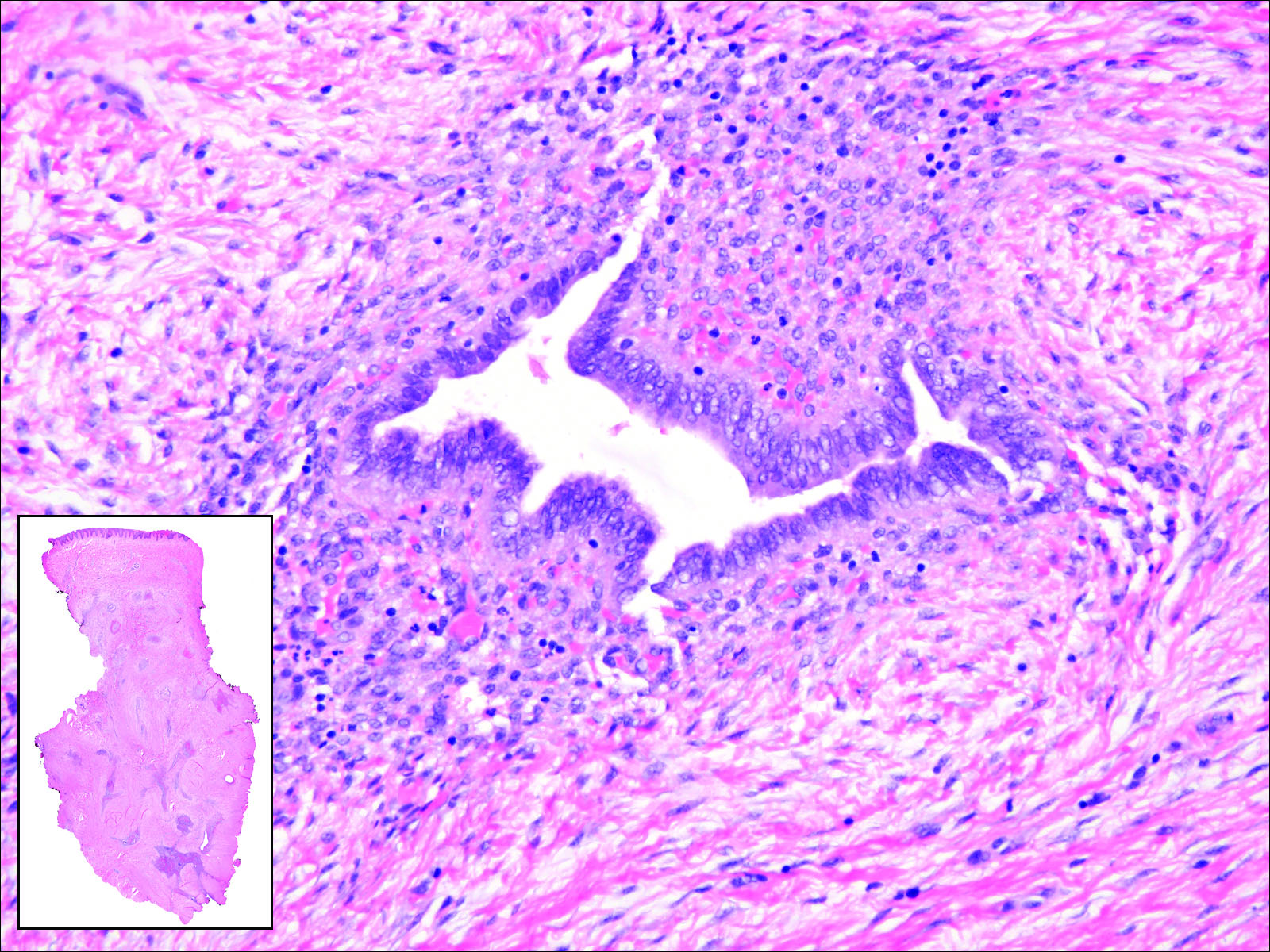
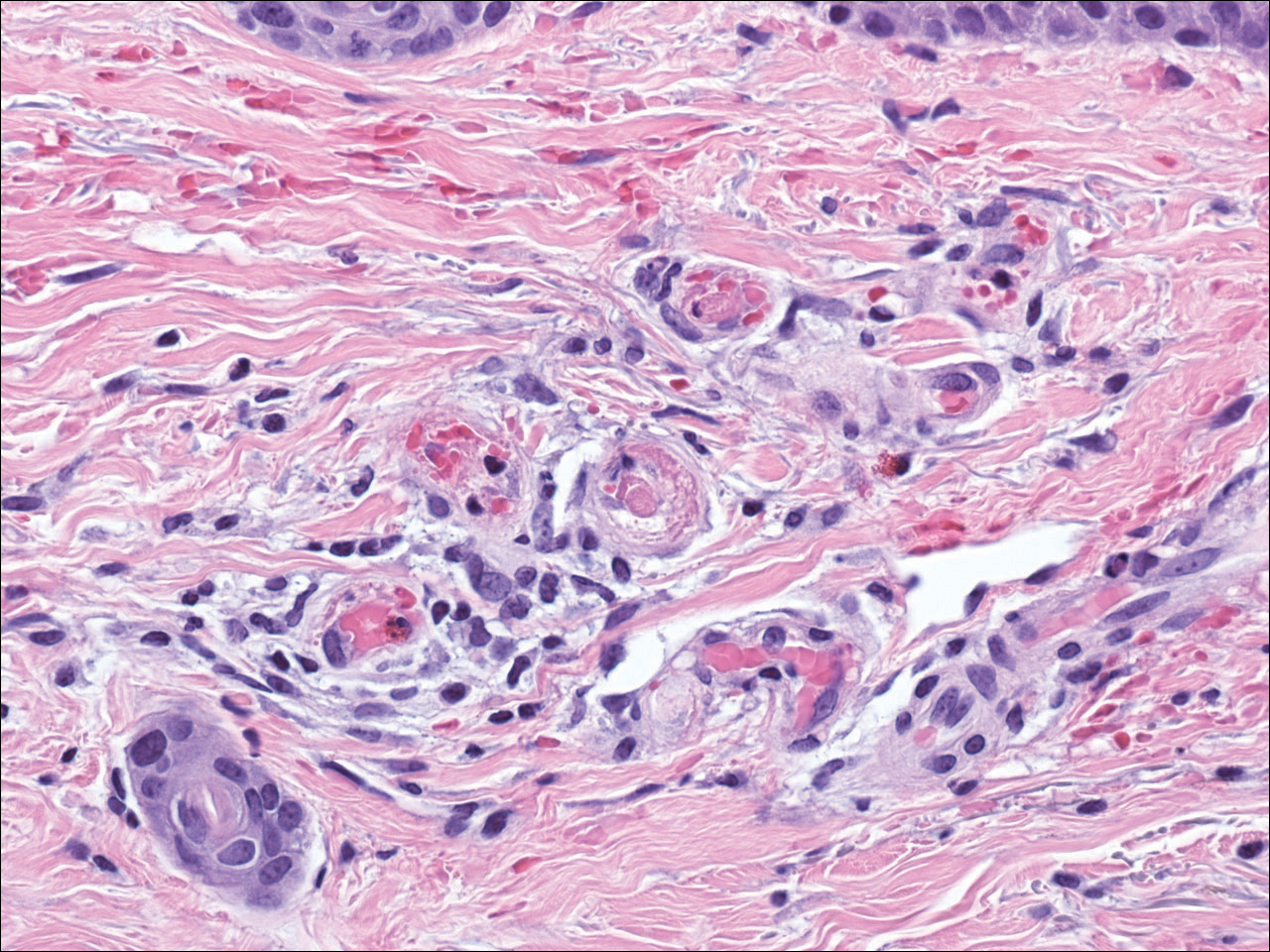
Cutaneous metastasis of pulmonary adenocarcinoma (CMPA) is a rare phenomenon with an overall survival rate of less than 5 months.1,2 Often, CMPA can be the heralding feature of an aggressive systemic malignancy in 2.8% to 22% of reported cases.2-4 Clinically, CMPAs often present as fixed, violaceous, ulcerated nodules on the chest wall, scalp, or site of a prior procedure.3,5,6 Other clinical presentations have been described including zosteriform and inflammatory carcinomalike CMPA and CMPA on the tip of the nose.7 Histologically, CMPA presents as a subdermal collection of atypical glands arranged as clustered aggregates of infiltrative glands penetrating the dermal stroma (quiz image). The atypical glands have large oval nuclei with high nuclear to cytoplasm ratios with scant pale cytoplasm.
Cutaneous metastasis of pulmonary adenocarcinoma is difficult to distinguish from other metastatic or primary glandular malignancies based on histology alone. Immunohistochemical analysis can aid in the diagnosis of the primary tumor. Pulmonary adenocarcinomas are positive for cytokeratin (CK) 7 and thyroid transcription factor 1 (TTF-1), and they are negative for CK5/6 and CK20.7 The differential diagnosis for CMPA includes other internal malignancies such as invasive ductal adenocarcinoma of the breast and gastrointestinal adenocarcinomas (eg, gastric or colorectal carcinoma [CRC]). Additionally, endometriosis and primary sebaceous carcinomas can mimic cutaneous metastatic adenocarcinomas.
Endometriosis can mimic adenocarcinoma, especially when presenting as a subdermal nodule. However, the scattered dermal glands are cytologically banal and are surrounded by uterine-type stroma and extravasated hemorrhage, a classic presentation of endometriosis (Figure 1).
Invasive ductal carcinoma of the breast is one of the most common cutaneous metastases of internal malignancy.3 Clinically, these lesions present on the chest wall or abdomen as flesh-colored nodules. Histopathology generally reveals either tubular or single tumor cells infiltrating the dermis with surrounding desmoplastic fibrosis (Figure 2). Immunohistochemistry typically is positive for CK7, estrogen receptor, and mammaglobin, and negative for CK20, CK5/6, and TTF-1.

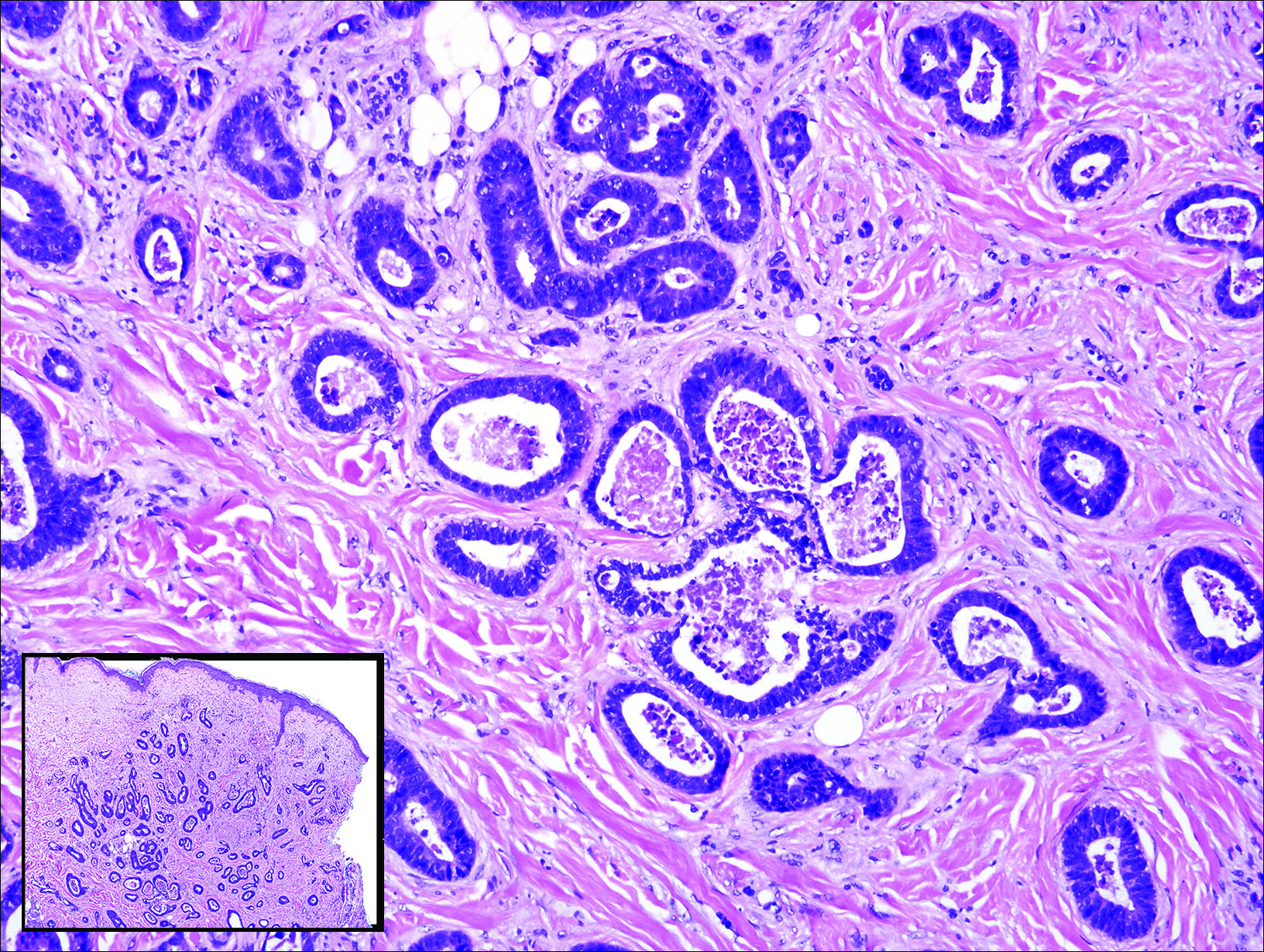
Gastrointestinal adenocarcinomas encompass a variety of primary sites that can metastasize to the skin including CRC. Clinically, cutaneous metastases of CRC present as multiple nodules on the trunk, abdomen, or umbilicus (also known as Sister Mary Joseph nodule).7,8 Distinguishing CRC as the primary site of origin can be difficult; however, there are subtle differences depending on the histologic subtype. In well-differentiated CRCs, well-defined atypical glands are haphazardly arranged within the dermis (Figure 3), while poorly differentiated lesions can present as single cells or with a signet ring-like morphology (Figure 4). For perianal lesions, extramammary Paget disease should be considered when biopsies show large, amphophilic, intraepithelial cells. These lesions often present with mucin and CK20 expression and are frequently associated with colorectal malignancies.9 Another characteristic feature of CRC is central necrosis with karyorrhectic debris, known as dirty necrosis. Immunohistochemical analysis typically shows expression of caudal type homeobox 2 and CK20 with infrequent expression of CK7 and no expression of TTF-1; however, additional clinical history (eg, history of colorectal adenocarcinoma, positive fecal occult blood test) often is the best distinguishing feature.
Primary sebaceous carcinoma also can mimic metastatic adenocarcinoma within the skin and is histologically similar to metastatic adenocarcinomas. The most distinguishing feature is sebaceous differentiation characterized by sebocytes, which have a vacuolated cytoplasm giving the nucleus a scalloped appearance, frequently with adjacent ductlike structures (Figure 5). Epidermotropism sometimes is present in sebaceous carcinomas but cannot be relied on as a distinguishing feature. Immunohistochemical analysis also is a helpful tool; these tumors typically are positive for p63 and podoplanin, distinguishing them from negative-staining metastatic adenocarcinomas.10,11
- Terashima T, Kanazawa M. Lung cancer with skin metastasis. Chest. 1994;106:1448-1450.
- Song Z, Lin B, Shao L, et al. Cutaneous metastasis as a initial presentation in advanced non-small cell lung cancer and its poor survival prognosis. J Cancer Res Clin Oncol. 2012;138:1613-1617.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Saeed S, Keehn CA, Morgan MB. Cutaneous metastasis: a clinical, pathological, and immunohistochemical appraisal. J Cutan Pathol. 2004;31:419-430.
- Chang SE, Choi JC, Moon KC. A papillary carcinoma: cutaneous metastases from lung cancer. J Dermatol. 2001;28:110-111.
- Snow S, Madjar D, Reizner G, et al. Renal cell carcinoma metastatic to the scalp: case report and review of the literature. Dermatol Surg. 2001;27:192-194.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Schwartz IS. Sister (Mary?) Joseph's nodule. N Engl J Med. 1987;316:1348-1349.
- Goldblum J, Hart W. Perianal Paget's disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Ivan D, Nash J, Preito V, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2006;34:474-480.
- Liang H, Wu H, Giorgadze T, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
Cutaneous Metastasis of Pulmonary Adenocarcinoma
Cutaneous metastasis of pulmonary adenocarcinoma (CMPA) is a rare phenomenon with an overall survival rate of less than 5 months.1,2 Often, CMPA can be the heralding feature of an aggressive systemic malignancy in 2.8% to 22% of reported cases.2-4 Clinically, CMPAs often present as fixed, violaceous, ulcerated nodules on the chest wall, scalp, or site of a prior procedure.3,5,6 Other clinical presentations have been described including zosteriform and inflammatory carcinomalike CMPA and CMPA on the tip of the nose.7 Histologically, CMPA presents as a subdermal collection of atypical glands arranged as clustered aggregates of infiltrative glands penetrating the dermal stroma (quiz image). The atypical glands have large oval nuclei with high nuclear to cytoplasm ratios with scant pale cytoplasm.
Cutaneous metastasis of pulmonary adenocarcinoma is difficult to distinguish from other metastatic or primary glandular malignancies based on histology alone. Immunohistochemical analysis can aid in the diagnosis of the primary tumor. Pulmonary adenocarcinomas are positive for cytokeratin (CK) 7 and thyroid transcription factor 1 (TTF-1), and they are negative for CK5/6 and CK20.7 The differential diagnosis for CMPA includes other internal malignancies such as invasive ductal adenocarcinoma of the breast and gastrointestinal adenocarcinomas (eg, gastric or colorectal carcinoma [CRC]). Additionally, endometriosis and primary sebaceous carcinomas can mimic cutaneous metastatic adenocarcinomas.
Endometriosis can mimic adenocarcinoma, especially when presenting as a subdermal nodule. However, the scattered dermal glands are cytologically banal and are surrounded by uterine-type stroma and extravasated hemorrhage, a classic presentation of endometriosis (Figure 1).
Invasive ductal carcinoma of the breast is one of the most common cutaneous metastases of internal malignancy.3 Clinically, these lesions present on the chest wall or abdomen as flesh-colored nodules. Histopathology generally reveals either tubular or single tumor cells infiltrating the dermis with surrounding desmoplastic fibrosis (Figure 2). Immunohistochemistry typically is positive for CK7, estrogen receptor, and mammaglobin, and negative for CK20, CK5/6, and TTF-1.
Gastrointestinal adenocarcinomas encompass a variety of primary sites that can metastasize to the skin including CRC. Clinically, cutaneous metastases of CRC present as multiple nodules on the trunk, abdomen, or umbilicus (also known as Sister Mary Joseph nodule).7,8 Distinguishing CRC as the primary site of origin can be difficult; however, there are subtle differences depending on the histologic subtype. In well-differentiated CRCs, well-defined atypical glands are haphazardly arranged within the dermis (Figure 3), while poorly differentiated lesions can present as single cells or with a signet ring-like morphology (Figure 4). For perianal lesions, extramammary Paget disease should be considered when biopsies show large, amphophilic, intraepithelial cells. These lesions often present with mucin and CK20 expression and are frequently associated with colorectal malignancies.9 Another characteristic feature of CRC is central necrosis with karyorrhectic debris, known as dirty necrosis. Immunohistochemical analysis typically shows expression of caudal type homeobox 2 and CK20 with infrequent expression of CK7 and no expression of TTF-1; however, additional clinical history (eg, history of colorectal adenocarcinoma, positive fecal occult blood test) often is the best distinguishing feature.
Primary sebaceous carcinoma also can mimic metastatic adenocarcinoma within the skin and is histologically similar to metastatic adenocarcinomas. The most distinguishing feature is sebaceous differentiation characterized by sebocytes, which have a vacuolated cytoplasm giving the nucleus a scalloped appearance, frequently with adjacent ductlike structures (Figure 5). Epidermotropism sometimes is present in sebaceous carcinomas but cannot be relied on as a distinguishing feature. Immunohistochemical analysis also is a helpful tool; these tumors typically are positive for p63 and podoplanin, distinguishing them from negative-staining metastatic adenocarcinomas.10,11
Cutaneous Metastasis of Pulmonary Adenocarcinoma
Cutaneous metastasis of pulmonary adenocarcinoma (CMPA) is a rare phenomenon with an overall survival rate of less than 5 months.1,2 Often, CMPA can be the heralding feature of an aggressive systemic malignancy in 2.8% to 22% of reported cases.2-4 Clinically, CMPAs often present as fixed, violaceous, ulcerated nodules on the chest wall, scalp, or site of a prior procedure.3,5,6 Other clinical presentations have been described including zosteriform and inflammatory carcinomalike CMPA and CMPA on the tip of the nose.7 Histologically, CMPA presents as a subdermal collection of atypical glands arranged as clustered aggregates of infiltrative glands penetrating the dermal stroma (quiz image). The atypical glands have large oval nuclei with high nuclear to cytoplasm ratios with scant pale cytoplasm.
Cutaneous metastasis of pulmonary adenocarcinoma is difficult to distinguish from other metastatic or primary glandular malignancies based on histology alone. Immunohistochemical analysis can aid in the diagnosis of the primary tumor. Pulmonary adenocarcinomas are positive for cytokeratin (CK) 7 and thyroid transcription factor 1 (TTF-1), and they are negative for CK5/6 and CK20.7 The differential diagnosis for CMPA includes other internal malignancies such as invasive ductal adenocarcinoma of the breast and gastrointestinal adenocarcinomas (eg, gastric or colorectal carcinoma [CRC]). Additionally, endometriosis and primary sebaceous carcinomas can mimic cutaneous metastatic adenocarcinomas.
Endometriosis can mimic adenocarcinoma, especially when presenting as a subdermal nodule. However, the scattered dermal glands are cytologically banal and are surrounded by uterine-type stroma and extravasated hemorrhage, a classic presentation of endometriosis (Figure 1).
Invasive ductal carcinoma of the breast is one of the most common cutaneous metastases of internal malignancy.3 Clinically, these lesions present on the chest wall or abdomen as flesh-colored nodules. Histopathology generally reveals either tubular or single tumor cells infiltrating the dermis with surrounding desmoplastic fibrosis (Figure 2). Immunohistochemistry typically is positive for CK7, estrogen receptor, and mammaglobin, and negative for CK20, CK5/6, and TTF-1.
Gastrointestinal adenocarcinomas encompass a variety of primary sites that can metastasize to the skin including CRC. Clinically, cutaneous metastases of CRC present as multiple nodules on the trunk, abdomen, or umbilicus (also known as Sister Mary Joseph nodule).7,8 Distinguishing CRC as the primary site of origin can be difficult; however, there are subtle differences depending on the histologic subtype. In well-differentiated CRCs, well-defined atypical glands are haphazardly arranged within the dermis (Figure 3), while poorly differentiated lesions can present as single cells or with a signet ring-like morphology (Figure 4). For perianal lesions, extramammary Paget disease should be considered when biopsies show large, amphophilic, intraepithelial cells. These lesions often present with mucin and CK20 expression and are frequently associated with colorectal malignancies.9 Another characteristic feature of CRC is central necrosis with karyorrhectic debris, known as dirty necrosis. Immunohistochemical analysis typically shows expression of caudal type homeobox 2 and CK20 with infrequent expression of CK7 and no expression of TTF-1; however, additional clinical history (eg, history of colorectal adenocarcinoma, positive fecal occult blood test) often is the best distinguishing feature.
Primary sebaceous carcinoma also can mimic metastatic adenocarcinoma within the skin and is histologically similar to metastatic adenocarcinomas. The most distinguishing feature is sebaceous differentiation characterized by sebocytes, which have a vacuolated cytoplasm giving the nucleus a scalloped appearance, frequently with adjacent ductlike structures (Figure 5). Epidermotropism sometimes is present in sebaceous carcinomas but cannot be relied on as a distinguishing feature. Immunohistochemical analysis also is a helpful tool; these tumors typically are positive for p63 and podoplanin, distinguishing them from negative-staining metastatic adenocarcinomas.10,11
- Terashima T, Kanazawa M. Lung cancer with skin metastasis. Chest. 1994;106:1448-1450.
- Song Z, Lin B, Shao L, et al. Cutaneous metastasis as a initial presentation in advanced non-small cell lung cancer and its poor survival prognosis. J Cancer Res Clin Oncol. 2012;138:1613-1617.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Saeed S, Keehn CA, Morgan MB. Cutaneous metastasis: a clinical, pathological, and immunohistochemical appraisal. J Cutan Pathol. 2004;31:419-430.
- Chang SE, Choi JC, Moon KC. A papillary carcinoma: cutaneous metastases from lung cancer. J Dermatol. 2001;28:110-111.
- Snow S, Madjar D, Reizner G, et al. Renal cell carcinoma metastatic to the scalp: case report and review of the literature. Dermatol Surg. 2001;27:192-194.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Schwartz IS. Sister (Mary?) Joseph's nodule. N Engl J Med. 1987;316:1348-1349.
- Goldblum J, Hart W. Perianal Paget's disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Ivan D, Nash J, Preito V, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2006;34:474-480.
- Liang H, Wu H, Giorgadze T, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Terashima T, Kanazawa M. Lung cancer with skin metastasis. Chest. 1994;106:1448-1450.
- Song Z, Lin B, Shao L, et al. Cutaneous metastasis as a initial presentation in advanced non-small cell lung cancer and its poor survival prognosis. J Cancer Res Clin Oncol. 2012;138:1613-1617.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Saeed S, Keehn CA, Morgan MB. Cutaneous metastasis: a clinical, pathological, and immunohistochemical appraisal. J Cutan Pathol. 2004;31:419-430.
- Chang SE, Choi JC, Moon KC. A papillary carcinoma: cutaneous metastases from lung cancer. J Dermatol. 2001;28:110-111.
- Snow S, Madjar D, Reizner G, et al. Renal cell carcinoma metastatic to the scalp: case report and review of the literature. Dermatol Surg. 2001;27:192-194.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Schwartz IS. Sister (Mary?) Joseph's nodule. N Engl J Med. 1987;316:1348-1349.
- Goldblum J, Hart W. Perianal Paget's disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Ivan D, Nash J, Preito V, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2006;34:474-480.
- Liang H, Wu H, Giorgadze T, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
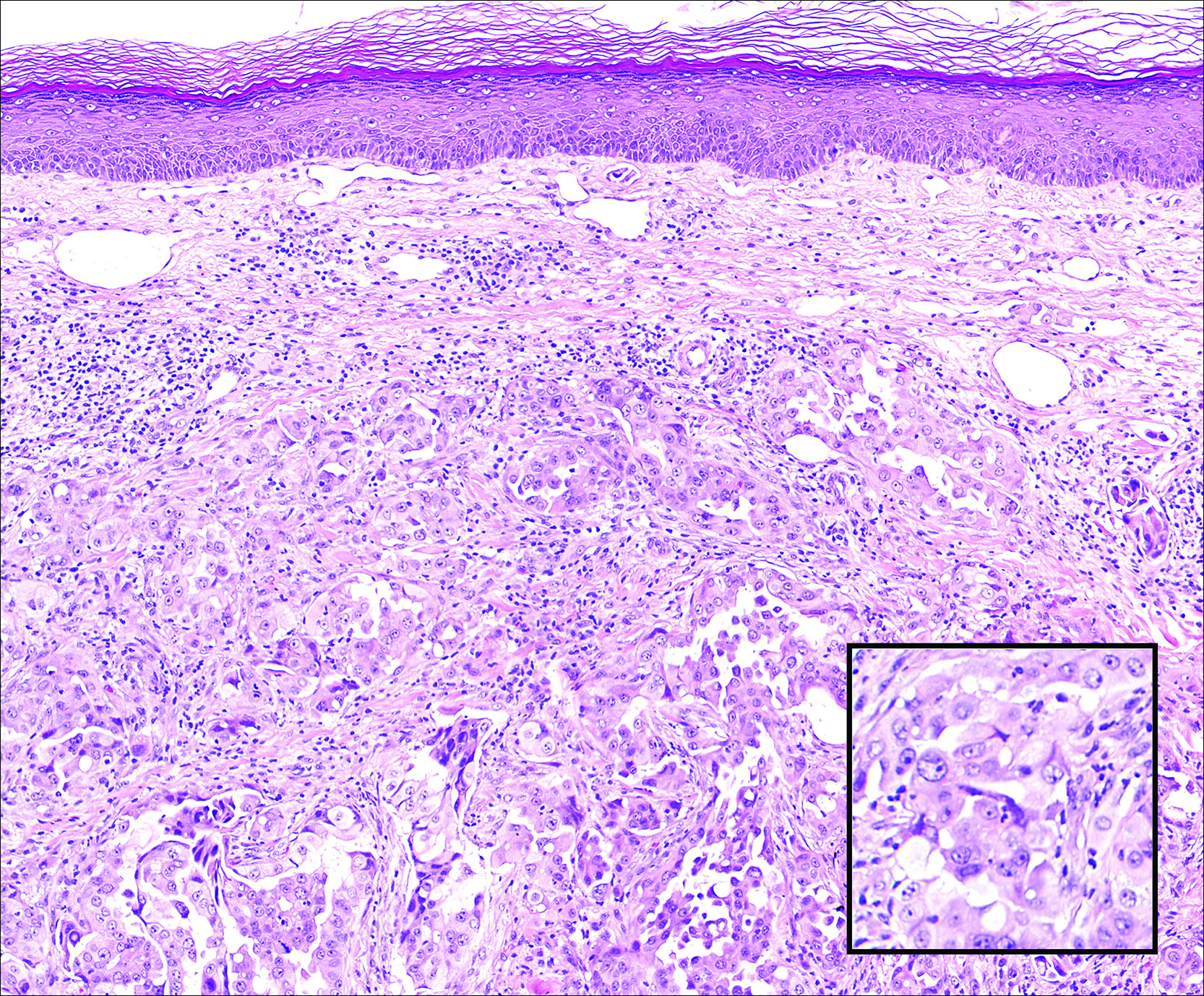
A 67-year-old woman with no history of malignancy presented with a scalp nodule. The photomicrograph showed atypical glands forming a subepidermal nodule with pleomorphic cells characterized by scant eosinophilic cytoplasm and large prominent nucleoli. Immunohistochemical analysis revealed diffuse thyroid transcription factor 1 and cytokeratin 7 positivity.
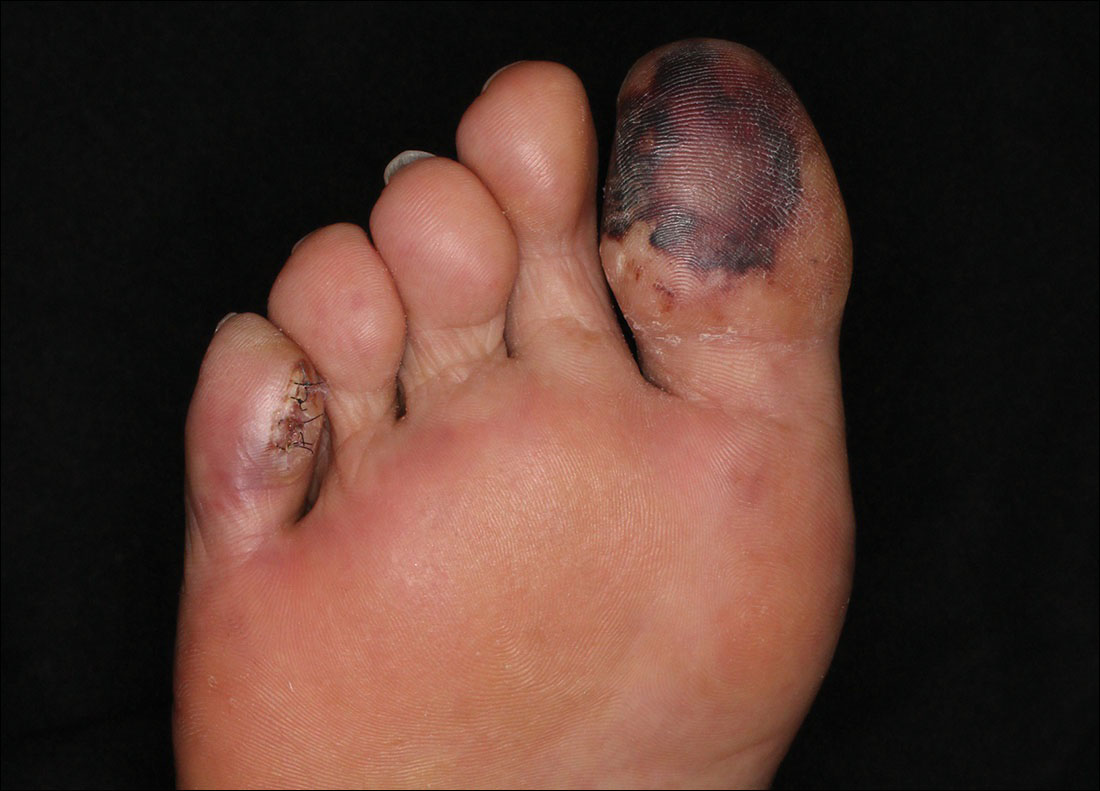
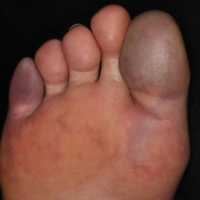
Painful Purple Toes
The Diagnosis: Blue Toe Syndrome
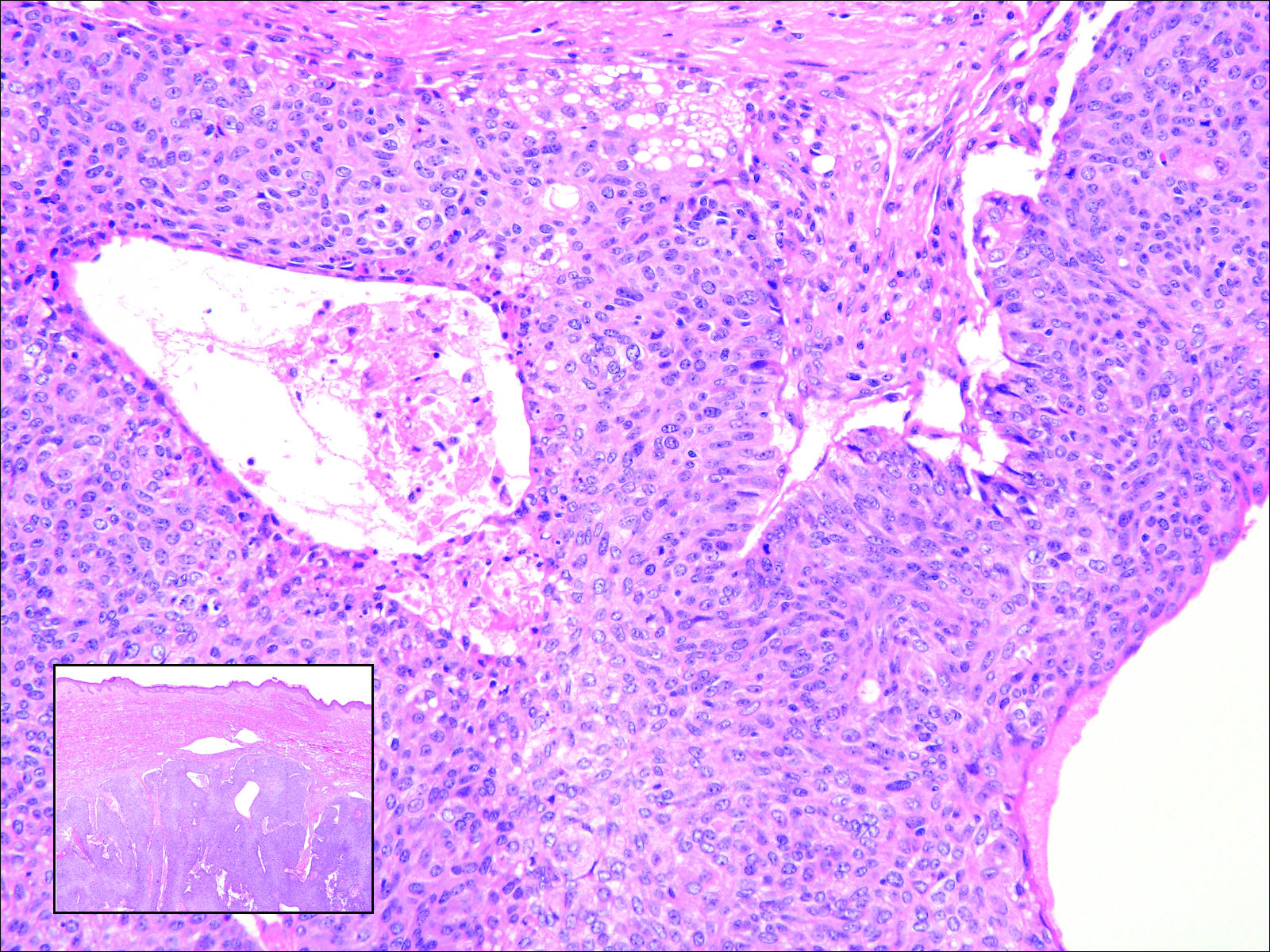
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
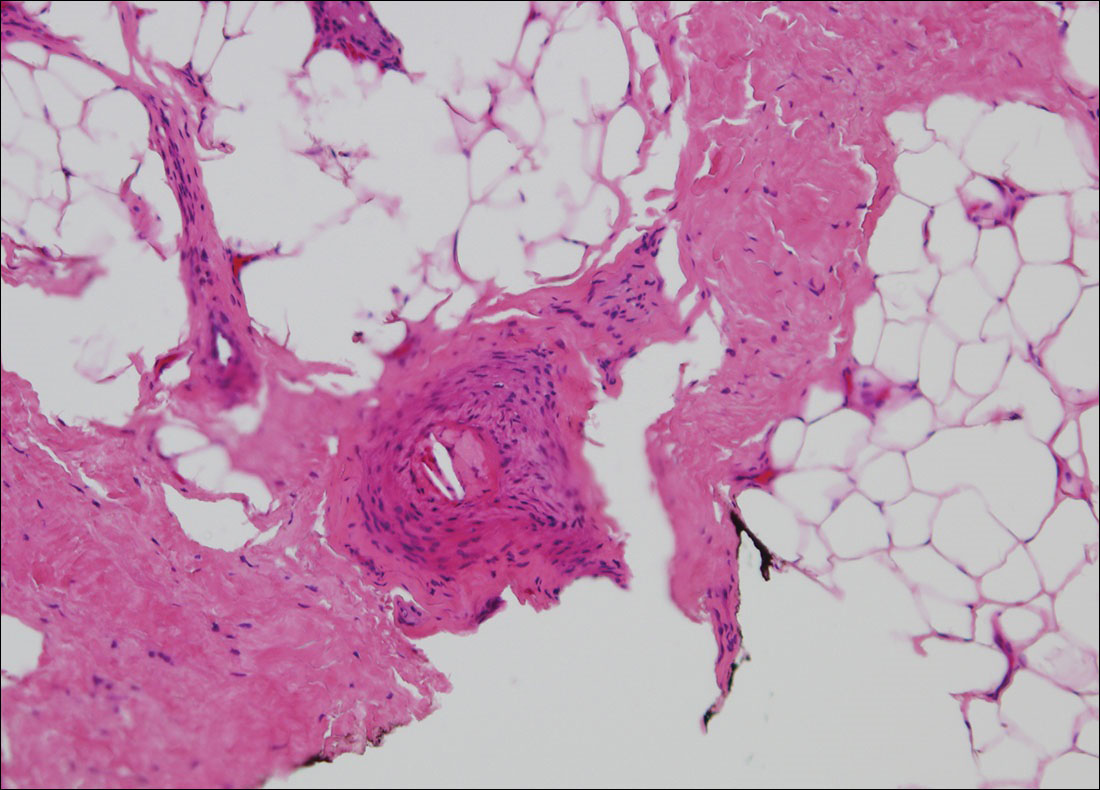
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
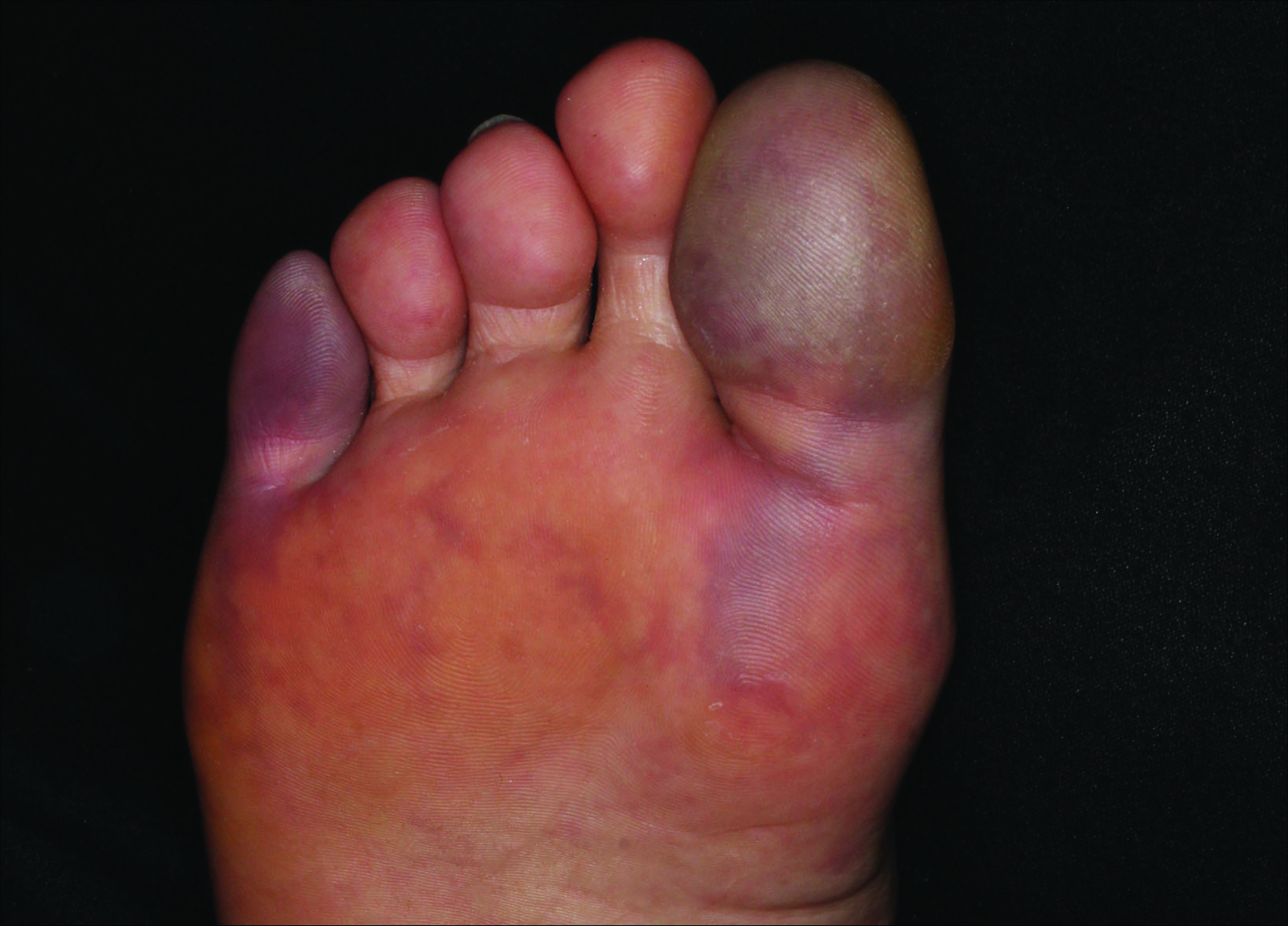
A 63-year-old man presented with sudden onset of severe pain in the right hallux and fifth toe of 3 days' duration. The patient had hypertension and hyperlipidemia with a 45-year history of smoking and had not undergone any vascular procedures. Physical examination revealed relatively well-defined cyanotic change with remarkable coldness on the affected toes as well as livedo reticularis on the underside of the toes. All peripheral pulses were present. Laboratory investigation revealed no remarkable changes with eosinophil counts within reference range and normal renal function. A biopsy taken from the fifth toe revealed thrombotic arterioles with cholesterol clefts.
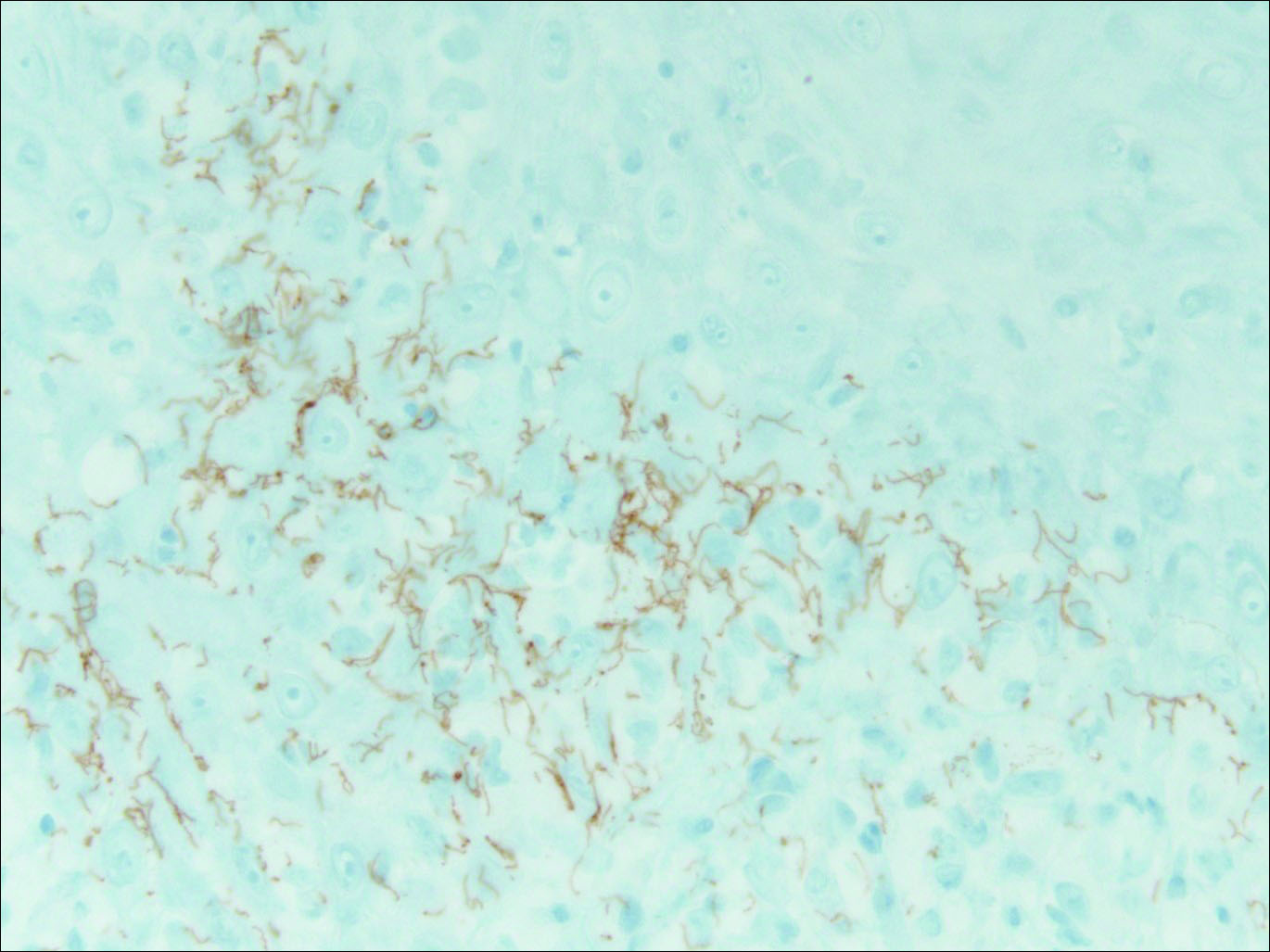
Thick Scaly Plaques on the Wrists, Knees, and Feet
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
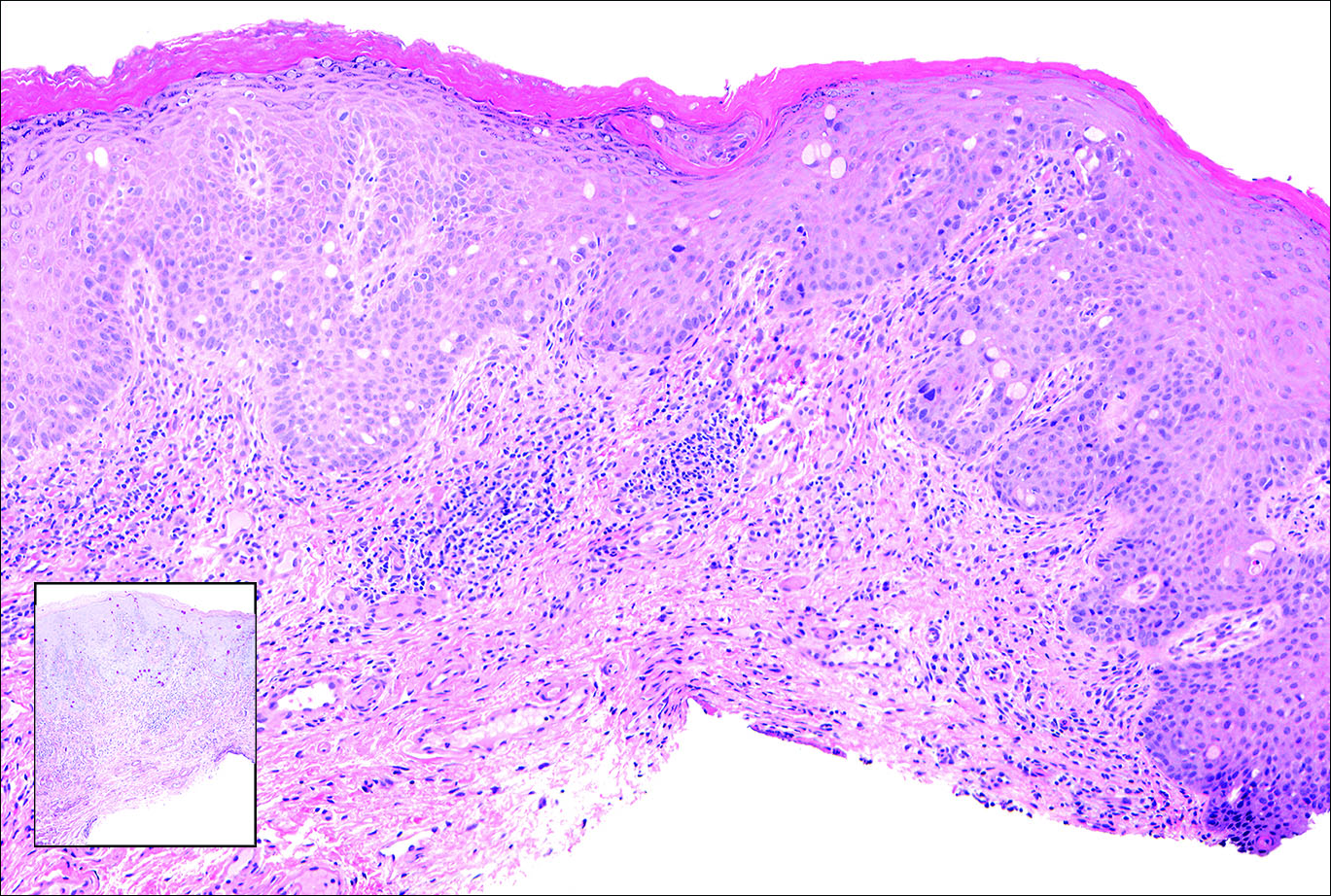
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
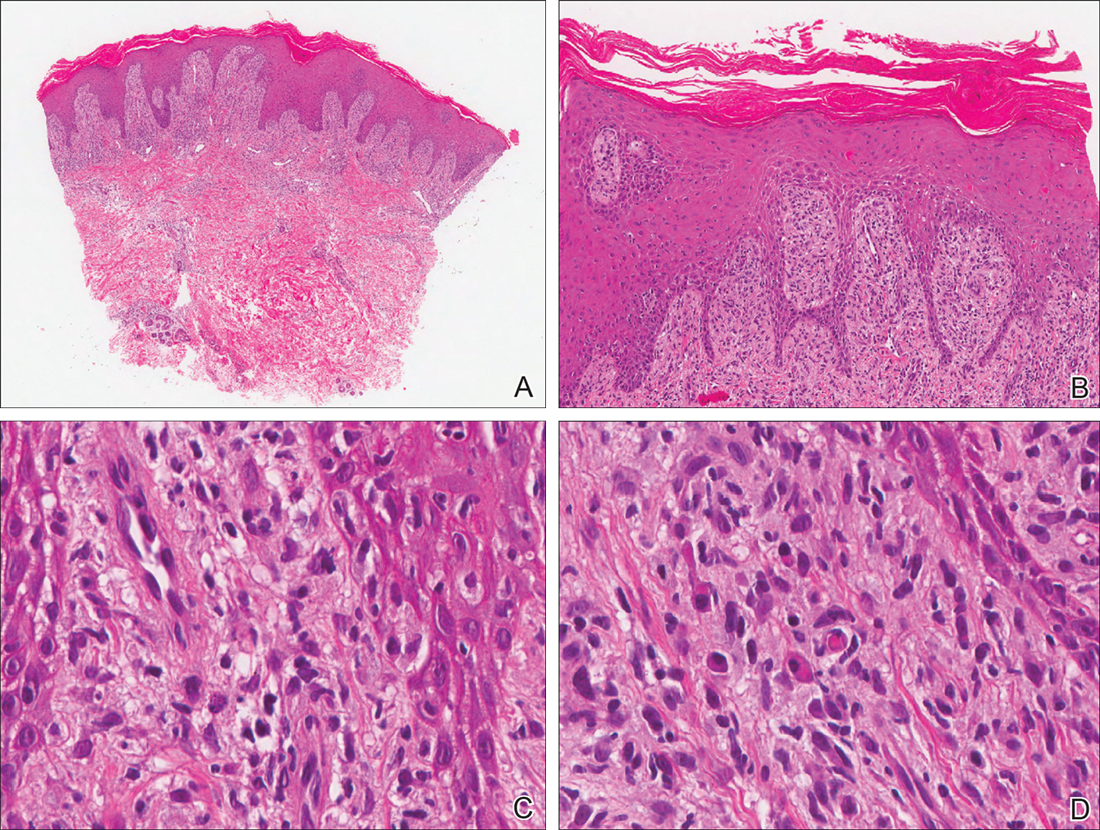
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
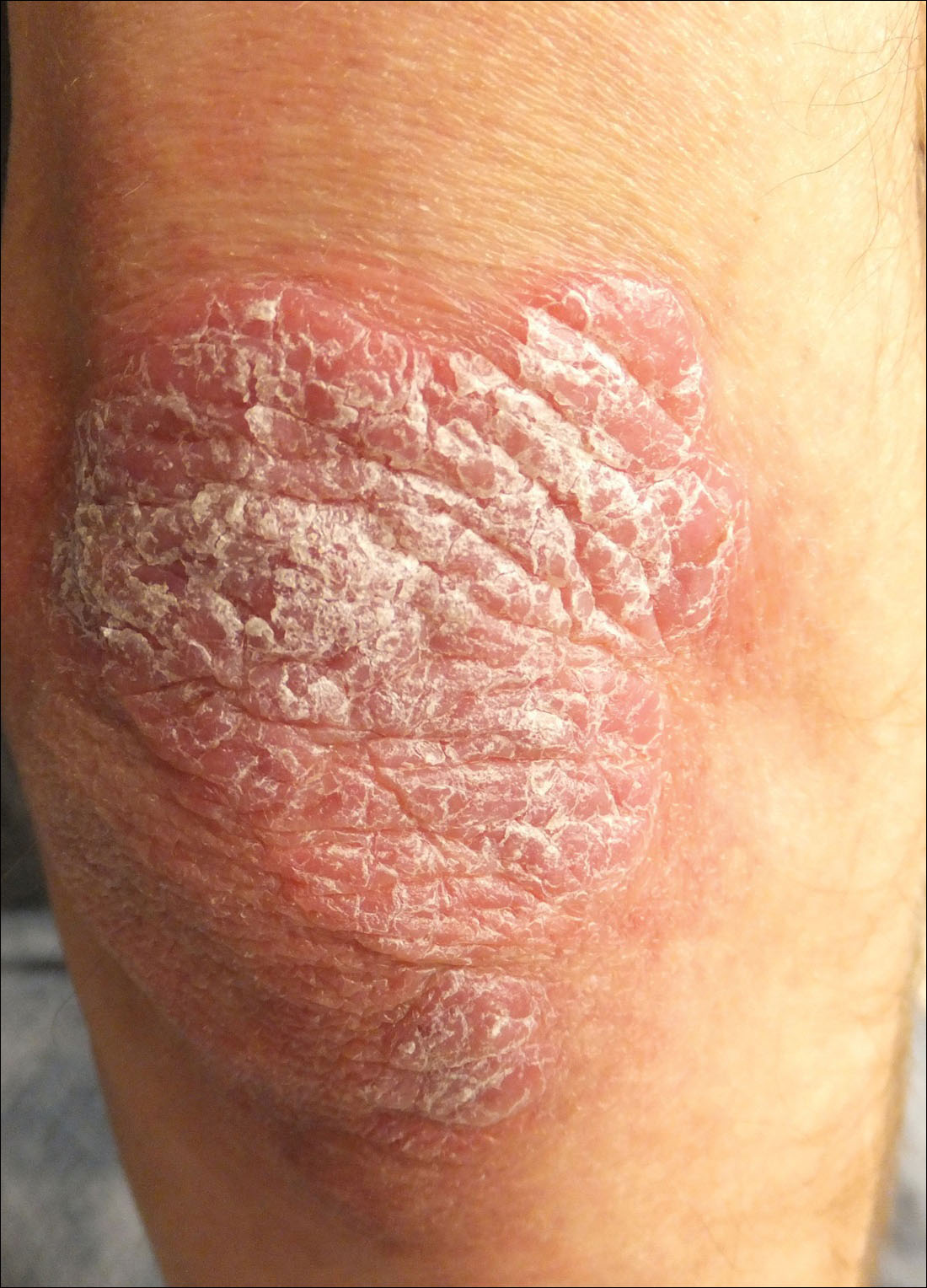
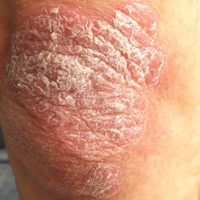
A 34-year-old man presented with thick scaly plaques on the wrists, knees, and feet of 2 years' duration. He had seen several dermatologists, and despite the use of topical steroids, he had no improvement.
When Man’s Legs “Give Out,” His Buttocks Takes the Brunt
ANSWER
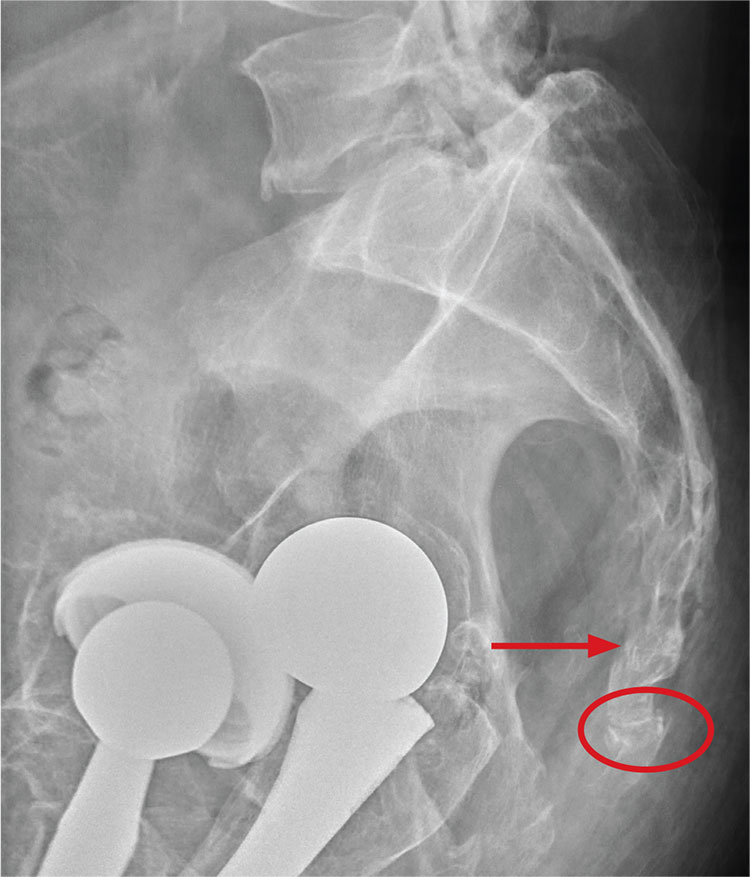
There are degenerative changes present. Bilateral hip prostheses are noted. Within the coccyx, there is bone remodeling and angulation that are likely chronic and related to remote trauma or injury (arrow). Below this, some cortical lucency (circled) is noted, most likely consistent with an acute fracture. The patient was prescribed a nonsteroidal medication and a mild narcotic pain medication.
ANSWER
There are degenerative changes present. Bilateral hip prostheses are noted. Within the coccyx, there is bone remodeling and angulation that are likely chronic and related to remote trauma or injury (arrow). Below this, some cortical lucency (circled) is noted, most likely consistent with an acute fracture. The patient was prescribed a nonsteroidal medication and a mild narcotic pain medication.
ANSWER
There are degenerative changes present. Bilateral hip prostheses are noted. Within the coccyx, there is bone remodeling and angulation that are likely chronic and related to remote trauma or injury (arrow). Below this, some cortical lucency (circled) is noted, most likely consistent with an acute fracture. The patient was prescribed a nonsteroidal medication and a mild narcotic pain medication.
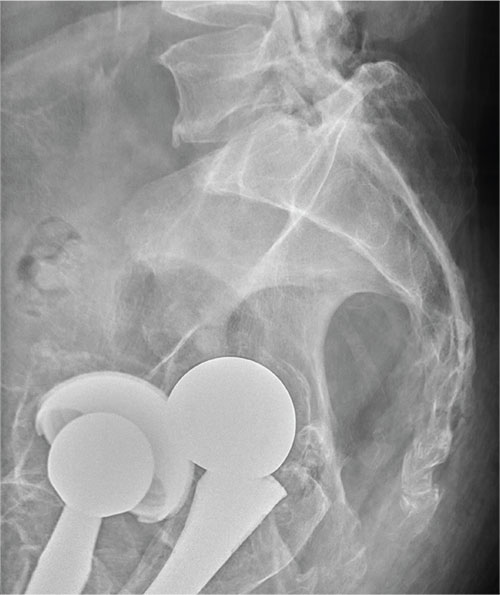
A 75-year-old man presents to the urgent care center for evaluation of pain in his buttocks after a fall. He states he was walking when his “legs gave out” and he hit the ground. He landed squarely on his buttocks, causing immediate pain. He was eventually able to get up with some assistance. He denies current weakness or any bowel or bladder complaints.
His medical/surgical history is significant for coronary artery disease, hypertension, and bilateral hip replacements. Physical exam reveals an elderly male who is uncomfortable but in no obvious distress. His vital signs are stable. He has moderate point tenderness over his sacrum but is able to move all his extremities well, with normal strength.
Radiograph of his sacrum/coccyx is shown. What is your impression?
Painful Ulcerations Above the Malleoli
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
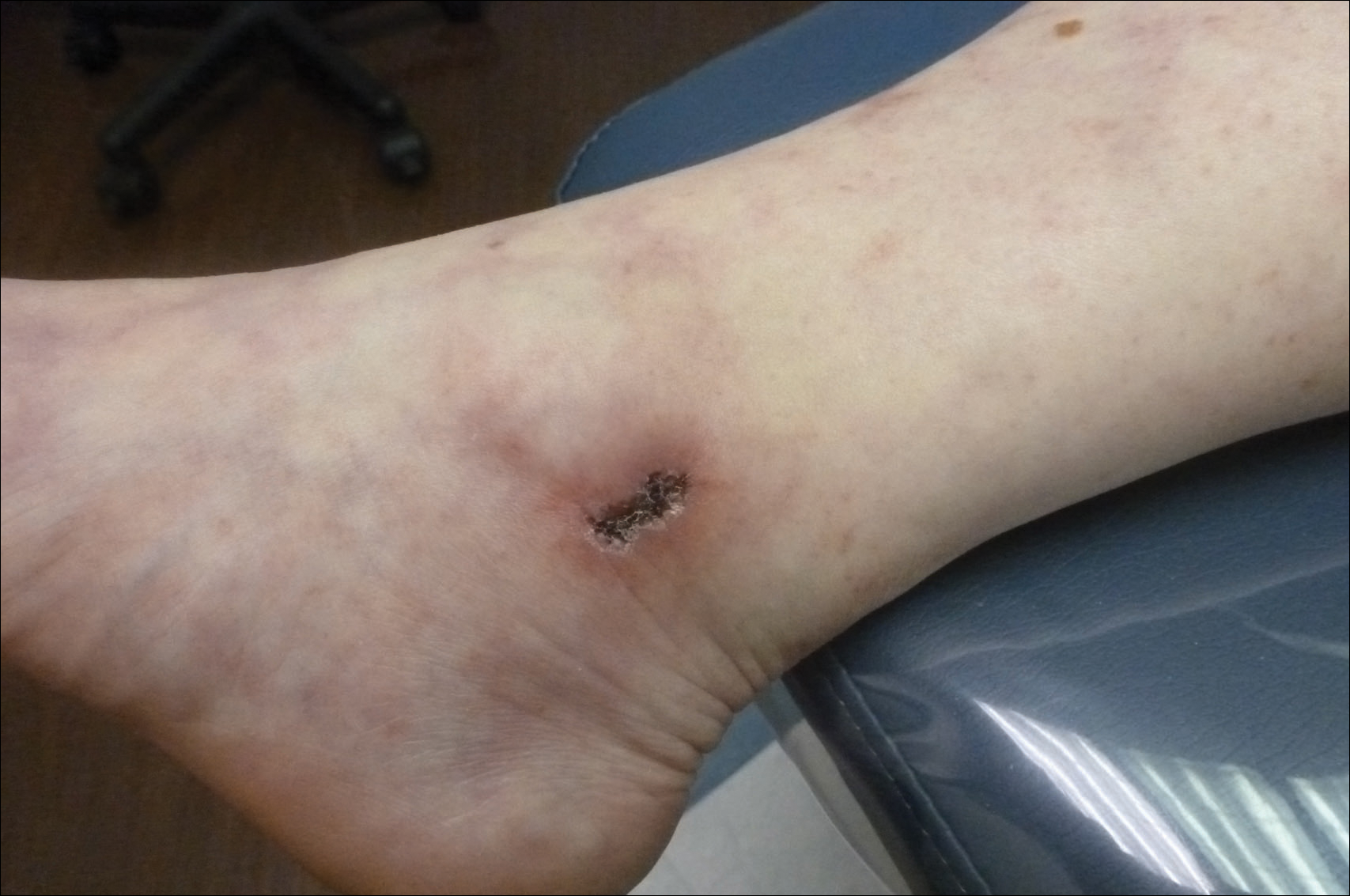
A 58-year-old woman presented in the summertime with skin discoloration of the bilateral lower legs and painful ulcerations above the medial and lateral malleoli of 15 years’ duration. She denied any recent trauma to the area or change in skin lesion appearance with cold exposure. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities was negative. A punch biopsy specimen obtained from the left anterior lower leg revealed vascular thrombi with extravasated erythrocytes and a sparse perivascular inflammatory cell infiltrate.
Enlarging Breast Lesion
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
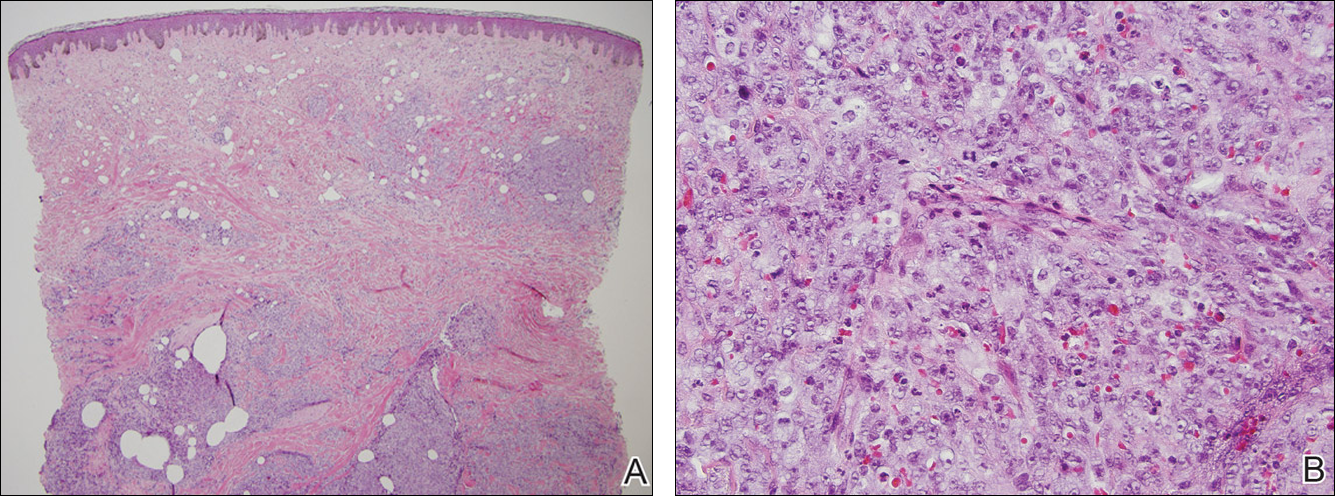
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.

A 75-year-old woman with a history of stage II invasive ductal carcinoma of the right breast presented to the dermatology clinic with an enlarging, indurated, ecchymotic plaque on the inferior aspect of the right breast of 2 months’ duration. The patient underwent a lumpectomy, radiation, and adjuvant chemotherapy 13 years prior to presentation. Review of systems was otherwise noncontributory.
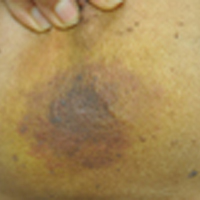
Man Shovels Path to Angina
ANSWERThe correct interpretation includes sinus rhythm with blocked premature atrial complexes, left-axis deviation, and serial changes of an evolving anterior MI.
The presence of a P wave without a QRS between the third and fourth QRS complex represents a blocked premature atrial complex. The fifth QRS complex also indicates a premature atrial complex; however, it is not blocked.
Left-axis deviation is evidenced by the R axis of –82°. The loss of an R wave in V1, the poor R-wave progression in V2 and V3, and the ST-T wave changes are all consistent with an evolving anterior MI.
ANSWERThe correct interpretation includes sinus rhythm with blocked premature atrial complexes, left-axis deviation, and serial changes of an evolving anterior MI.
The presence of a P wave without a QRS between the third and fourth QRS complex represents a blocked premature atrial complex. The fifth QRS complex also indicates a premature atrial complex; however, it is not blocked.
Left-axis deviation is evidenced by the R axis of –82°. The loss of an R wave in V1, the poor R-wave progression in V2 and V3, and the ST-T wave changes are all consistent with an evolving anterior MI.
ANSWERThe correct interpretation includes sinus rhythm with blocked premature atrial complexes, left-axis deviation, and serial changes of an evolving anterior MI.
The presence of a P wave without a QRS between the third and fourth QRS complex represents a blocked premature atrial complex. The fifth QRS complex also indicates a premature atrial complex; however, it is not blocked.
Left-axis deviation is evidenced by the R axis of –82°. The loss of an R wave in V1, the poor R-wave progression in V2 and V3, and the ST-T wave changes are all consistent with an evolving anterior MI.
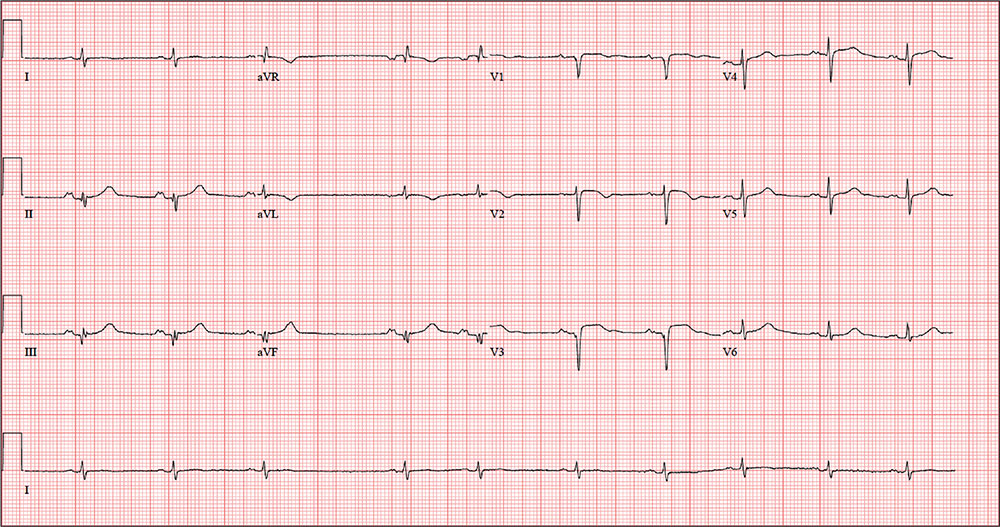
While shoveling gravel several days ago, a 62-year-old man developed exertional angina. Though he stopped to rest, the pain persisted and radiated to his left arm. He called out for help, and his neighbor called 911. The patient was transported via ACLS ambulance to the hospital, where he ruled in for an anterior myocardial infarction (MI). Cardiac catheterization revealed left anterior descending coronary artery stenosis, which was treated with a drug-eluting stent.
One week after discharge, he presents for his first follow-up appointment. He is enrolled in a cardiac rehabilitation program but isn’t scheduled to start for another week. He has not experienced chest pain or discomfort following his MI, and he says he is diligently taking his ß-blocker and nitrates.
This retired Army (Airborne division) officer’s past surgical history is remarkable for multiple fractures in his lower extremities, sustained while skydiving. His past medical history is remarkable for malaria, yellow fever, and hepatitis. Prior to his MI, he had no history of cardiac disease or symptoms.
He is divorced and has no children. His parents are alive and well, with no known cardiac disease. His paternal grandfather died of a stroke associated with atrial fibrillation, but he does not know how his other grandparents died.
The patient was a smoker until five years ago. He consumes two or three glasses of Scotch per week, typically on the weekends. He denies recreational drug use and “doesn’t believe in” holistic or herbal medications.
Current medications include metoprolol, isosorbide dinitrate, and clopidogrel. He has no known drug allergies.
The review of systems is remarkable for fatigue, which he attributes to ß-blocker use. His right groin is sore following his interventional procedure, but the discomfort is resolving.
Vital signs include a blood pressure of 110/64 mm Hg; pulse, 60 beats/min; respiratory rate, 14 breaths/min-1; and temperature, 97.6°F.
On physical exam, his weight is 224 lb and his height is 74 in. He is in good spirits and no distress. The HEENT exam is remarkable for corrective lenses. There is no evidence of thyromegaly or jugular venous distention. The lungs are clear; there are no murmurs, rubs, or gallops, and the abdomen is soft and nontender without organomegaly. The right groin has resolving ecchymosis and a small, palpable, organized hematoma. Peripheral pulses are strong bilaterally, and the neurologic exam is intact.
A follow-up ECG shows a ventricular rate of 61 beats/min; PR interval, 176 ms; QRS duration, 88 ms; QT/QTc interval, 402/404 ms; P axis, 71°; R axis, –82°; and T axis, 84°. What is your interpretation of the ECG?