User login
Efficacy and safety of lowering dupilumab frequency for AD
Patients with moderate to severe atopic dermatitis who responded well to the approved dupilumab regimen of 300 mg every 2 weeks in pivotal phase 3 monotherapy trials were more likely to have a continued response over the longer term if they maintained this regimen rather than switching to longer dosing intervals or discontinuing the medication.
This finding comes from a 36-week, randomized, double-blind, placebo-controlled trial that enrolled 422 – SOLO 1 and SOLO 2.
The new international study – SOLO-CONTINUE – randomized these patients to continue the original regimen (weekly or every 2 weeks), to receive 300 mg of the biologic medication every 4 or 8 weeks, or to receive placebo.
Patients who continued the original regimen had the most consistent maintenance of treatment effect, while patients on longer dosage intervals or placebo had a dose-dependent reduction in response and no safety advantage. The incidence of treatment-emergent antidrug antibody was lowest with dupilumab weekly or every 2 weeks, and slightly higher with less-frequent dosing intervals, reported Margitta Worm, MD, of the Charité Universitätsmedizin Berlin, and coinvestigators.
“Because administration every 4 weeks or every 8 weeks did not provide an additional safety advantage and was numerically outperformed by administration weekly or every 2 weeks, we believe that it is prudent to adhere to the approved every 2 weeks regimen for adults and avoid less frequent treatment regimens (every 4 weeks or every 8 weeks) for long-term maintenance of efficacy,” they wrote in JAMA Dermatology.
Treatment success in the original SOLO trials was defined as having achieved an Investigator’s Global Assessment score of 0-1, or 75% improvement in Eczema Area and Severity Index Scores (EASI-75). As primary endpoints, SOLO-CONTINUE looked at the mean percentage change in EASI score over the course of the trial, and the percentage of patients who maintained EASI-75 at week 36.
Patients in the SOLO-CONTINUE trial who were randomized to receive dupilumab weekly or every 2 weeks had a mean percent change in EASI score of –0.06%. In contrast, patients assigned to the placebo group had a 21.7% decrease, and those taking the medication at 4- and 8-week intervals had mean changes of –3.84% and –6.84%, respectively. Post hoc analyses showed no apparent difference between dupilumab weekly and every 2 weeks in the maintenance of clinical response, the investigators reported.
Among patients with EASI-75 response at baseline, significantly more patients maintained this response at week 36 than patients receiving placebo, and there was again an apparent dose-dependent response. The percentage with EASI-75 at week 36 was 71.6% with the weekly or every-2-weeks regimen, 58.3% with the 4-week regimen, 54.9% with the 8-week regimen, and 30.4% in the placebo group.
Continuing treatment with 300 mg weekly or every 2 weeks resulted in greater maintenance of response across multiple other clinical endpoints and patient-reported outcomes as well (such as pruritus, atopic dermatitis symptoms, sleep, pain or discomfort, quality of life, and symptoms of anxiety and depression).
The more-frequent regimens also conferred no greater risk than less-frequent administration, and there were no new safety signals over the 36-week trial. Treatment-emergent adverse events (the most common were headache, nasopharyngitis, injection-site reactions, and herpes simplex virus infection) occurred in 70.7% of patients in the weekly or every-2-weeks group, 73.6% in the 4-week group, 75% in the 8-week group, and 81.7% in the placebo group.
Unlike earlier studies, the incidence of conjunctivitis was low (less than 6%) across all treatment groups, possibly because patients in the SOLO-CONTINUE trial were all high-level responders who tend to have conjunctivitis less frequently, the authors wrote.
Patients receiving less-frequent doses of dupilumab, particularly every 8 weeks, had greater rates of skin infections, flares, and rescue medication use than patients receiving doses weekly or every 2 weeks, the investigators reported. Treatment-emergent antidrug antibody incidence was slightly higher with less-frequent doses (11.7% and 6% in the 8-week and 4-week groups, respectively, compared with 4.3% and 1.2% in the every-2-weeks and weekly groups), which indicates a “higher incidence of immunogenicity with less-frequent dosage intervals” and is “consistent with other biologics,” they wrote.
Dupilumab is a human monoclonal antibody against the interleukin-4 receptor alpha that inhibits signaling of IL-4 and IL-13. The study was conducted at 185 sites in North America, Europe, Asia, and Japan. Patients had a mean age of 38.2 years; 53.8% were male.
While the trial suggests that the approved regimen of 300 mg every 2 weeks is best for long-term treatment, “therapeutic decisions are often influenced by cost-benefit considerations, in which case practitioners and other stakeholders involved in these decisions should carefully balance potential savings against suboptimal efficacy and long-term risks associated with discontinuous treatment regimens,” the investigators wrote.
The SOLO-CONTINUE trial was funded by Sanofi and Regeneron, the companies that market dupilumab. Dr. Worm reported receiving honoraria for consulting and lecture activity from Regeneron and Sanofi during and outside the conduct of the study, among other disclosures. The other authors had multiple disclosures related to these and multiple other pharmaceutical companies, or were employees of Sanofi or Regeneron.
SOURCE: Worm M et al. JAMA Dermatol. 2019 Dec 26. doi: 10.1001/jamadermatol.2019.3617.
The desire to decrease or stop a therapy such as dupilumab may be motivated by cost, current and potential adverse effects, and individual needs. Because atopic dermatitis is a waxing-and-waning disease with a predilection for cycles of escalation, there is some thought a priori that reduced treatment schedules or discontinued use of a drug may be possible in a state of low disease activity.
The investigators of the SOLO-CONTINUE trial found, however, that continuous treatment with the dosage used in the original SOLO trials (300 mg weekly or every 2 weeks) resulted in a better maintenance of response than a less-frequent dosage and was significantly better than placebo for all endpoints. The less-frequent dosage regimens (every 4 weeks and every 8 weeks), on the other hand, produced some dose-dependent reduction in efficacy.
The development of antidrug antibodies was found in approximately 11% of individuals who received placebo or dupilumab every 8 weeks, 6% of the monthly treatment group, and only 1% in the weekly group, suggesting that less-frequent administration results in higher immunogenicity. However, most of the antidrug antibody levels were low and did not seem to have any clinical effect, making this finding of uncertain relevance to patient care.
The study is valuable because, as more patients are exposed to the drug, more will want or need to reduce the dosage or stop use over time – and although it seems optimal to continue an every-2-weeks treatment regimen, this may not always be feasible. As we integrate new therapies and learn more about atopic dermatitis, it is important that we understand the options and implications around decreasing the dosage of dupilumab. This newly concluded trial is helpful in this regard.
Peter A. Lio, MD, is with the department of dermatology at Northwestern University, Chicago, and the Chicago Integrative Eczema Center. He reported receiving grants and personal fees from Regeneron, Sanofi Genzyme, and other companies, as well as other disclosures. His comments appear in JAMA Dermatology (2019 Dec 26. doi: 10.1001/jamadermatol.2019.3331).
The desire to decrease or stop a therapy such as dupilumab may be motivated by cost, current and potential adverse effects, and individual needs. Because atopic dermatitis is a waxing-and-waning disease with a predilection for cycles of escalation, there is some thought a priori that reduced treatment schedules or discontinued use of a drug may be possible in a state of low disease activity.
The investigators of the SOLO-CONTINUE trial found, however, that continuous treatment with the dosage used in the original SOLO trials (300 mg weekly or every 2 weeks) resulted in a better maintenance of response than a less-frequent dosage and was significantly better than placebo for all endpoints. The less-frequent dosage regimens (every 4 weeks and every 8 weeks), on the other hand, produced some dose-dependent reduction in efficacy.
The development of antidrug antibodies was found in approximately 11% of individuals who received placebo or dupilumab every 8 weeks, 6% of the monthly treatment group, and only 1% in the weekly group, suggesting that less-frequent administration results in higher immunogenicity. However, most of the antidrug antibody levels were low and did not seem to have any clinical effect, making this finding of uncertain relevance to patient care.
The study is valuable because, as more patients are exposed to the drug, more will want or need to reduce the dosage or stop use over time – and although it seems optimal to continue an every-2-weeks treatment regimen, this may not always be feasible. As we integrate new therapies and learn more about atopic dermatitis, it is important that we understand the options and implications around decreasing the dosage of dupilumab. This newly concluded trial is helpful in this regard.
Peter A. Lio, MD, is with the department of dermatology at Northwestern University, Chicago, and the Chicago Integrative Eczema Center. He reported receiving grants and personal fees from Regeneron, Sanofi Genzyme, and other companies, as well as other disclosures. His comments appear in JAMA Dermatology (2019 Dec 26. doi: 10.1001/jamadermatol.2019.3331).
The desire to decrease or stop a therapy such as dupilumab may be motivated by cost, current and potential adverse effects, and individual needs. Because atopic dermatitis is a waxing-and-waning disease with a predilection for cycles of escalation, there is some thought a priori that reduced treatment schedules or discontinued use of a drug may be possible in a state of low disease activity.
The investigators of the SOLO-CONTINUE trial found, however, that continuous treatment with the dosage used in the original SOLO trials (300 mg weekly or every 2 weeks) resulted in a better maintenance of response than a less-frequent dosage and was significantly better than placebo for all endpoints. The less-frequent dosage regimens (every 4 weeks and every 8 weeks), on the other hand, produced some dose-dependent reduction in efficacy.
The development of antidrug antibodies was found in approximately 11% of individuals who received placebo or dupilumab every 8 weeks, 6% of the monthly treatment group, and only 1% in the weekly group, suggesting that less-frequent administration results in higher immunogenicity. However, most of the antidrug antibody levels were low and did not seem to have any clinical effect, making this finding of uncertain relevance to patient care.
The study is valuable because, as more patients are exposed to the drug, more will want or need to reduce the dosage or stop use over time – and although it seems optimal to continue an every-2-weeks treatment regimen, this may not always be feasible. As we integrate new therapies and learn more about atopic dermatitis, it is important that we understand the options and implications around decreasing the dosage of dupilumab. This newly concluded trial is helpful in this regard.
Peter A. Lio, MD, is with the department of dermatology at Northwestern University, Chicago, and the Chicago Integrative Eczema Center. He reported receiving grants and personal fees from Regeneron, Sanofi Genzyme, and other companies, as well as other disclosures. His comments appear in JAMA Dermatology (2019 Dec 26. doi: 10.1001/jamadermatol.2019.3331).
Patients with moderate to severe atopic dermatitis who responded well to the approved dupilumab regimen of 300 mg every 2 weeks in pivotal phase 3 monotherapy trials were more likely to have a continued response over the longer term if they maintained this regimen rather than switching to longer dosing intervals or discontinuing the medication.
This finding comes from a 36-week, randomized, double-blind, placebo-controlled trial that enrolled 422 – SOLO 1 and SOLO 2.
The new international study – SOLO-CONTINUE – randomized these patients to continue the original regimen (weekly or every 2 weeks), to receive 300 mg of the biologic medication every 4 or 8 weeks, or to receive placebo.
Patients who continued the original regimen had the most consistent maintenance of treatment effect, while patients on longer dosage intervals or placebo had a dose-dependent reduction in response and no safety advantage. The incidence of treatment-emergent antidrug antibody was lowest with dupilumab weekly or every 2 weeks, and slightly higher with less-frequent dosing intervals, reported Margitta Worm, MD, of the Charité Universitätsmedizin Berlin, and coinvestigators.
“Because administration every 4 weeks or every 8 weeks did not provide an additional safety advantage and was numerically outperformed by administration weekly or every 2 weeks, we believe that it is prudent to adhere to the approved every 2 weeks regimen for adults and avoid less frequent treatment regimens (every 4 weeks or every 8 weeks) for long-term maintenance of efficacy,” they wrote in JAMA Dermatology.
Treatment success in the original SOLO trials was defined as having achieved an Investigator’s Global Assessment score of 0-1, or 75% improvement in Eczema Area and Severity Index Scores (EASI-75). As primary endpoints, SOLO-CONTINUE looked at the mean percentage change in EASI score over the course of the trial, and the percentage of patients who maintained EASI-75 at week 36.
Patients in the SOLO-CONTINUE trial who were randomized to receive dupilumab weekly or every 2 weeks had a mean percent change in EASI score of –0.06%. In contrast, patients assigned to the placebo group had a 21.7% decrease, and those taking the medication at 4- and 8-week intervals had mean changes of –3.84% and –6.84%, respectively. Post hoc analyses showed no apparent difference between dupilumab weekly and every 2 weeks in the maintenance of clinical response, the investigators reported.
Among patients with EASI-75 response at baseline, significantly more patients maintained this response at week 36 than patients receiving placebo, and there was again an apparent dose-dependent response. The percentage with EASI-75 at week 36 was 71.6% with the weekly or every-2-weeks regimen, 58.3% with the 4-week regimen, 54.9% with the 8-week regimen, and 30.4% in the placebo group.
Continuing treatment with 300 mg weekly or every 2 weeks resulted in greater maintenance of response across multiple other clinical endpoints and patient-reported outcomes as well (such as pruritus, atopic dermatitis symptoms, sleep, pain or discomfort, quality of life, and symptoms of anxiety and depression).
The more-frequent regimens also conferred no greater risk than less-frequent administration, and there were no new safety signals over the 36-week trial. Treatment-emergent adverse events (the most common were headache, nasopharyngitis, injection-site reactions, and herpes simplex virus infection) occurred in 70.7% of patients in the weekly or every-2-weeks group, 73.6% in the 4-week group, 75% in the 8-week group, and 81.7% in the placebo group.
Unlike earlier studies, the incidence of conjunctivitis was low (less than 6%) across all treatment groups, possibly because patients in the SOLO-CONTINUE trial were all high-level responders who tend to have conjunctivitis less frequently, the authors wrote.
Patients receiving less-frequent doses of dupilumab, particularly every 8 weeks, had greater rates of skin infections, flares, and rescue medication use than patients receiving doses weekly or every 2 weeks, the investigators reported. Treatment-emergent antidrug antibody incidence was slightly higher with less-frequent doses (11.7% and 6% in the 8-week and 4-week groups, respectively, compared with 4.3% and 1.2% in the every-2-weeks and weekly groups), which indicates a “higher incidence of immunogenicity with less-frequent dosage intervals” and is “consistent with other biologics,” they wrote.
Dupilumab is a human monoclonal antibody against the interleukin-4 receptor alpha that inhibits signaling of IL-4 and IL-13. The study was conducted at 185 sites in North America, Europe, Asia, and Japan. Patients had a mean age of 38.2 years; 53.8% were male.
While the trial suggests that the approved regimen of 300 mg every 2 weeks is best for long-term treatment, “therapeutic decisions are often influenced by cost-benefit considerations, in which case practitioners and other stakeholders involved in these decisions should carefully balance potential savings against suboptimal efficacy and long-term risks associated with discontinuous treatment regimens,” the investigators wrote.
The SOLO-CONTINUE trial was funded by Sanofi and Regeneron, the companies that market dupilumab. Dr. Worm reported receiving honoraria for consulting and lecture activity from Regeneron and Sanofi during and outside the conduct of the study, among other disclosures. The other authors had multiple disclosures related to these and multiple other pharmaceutical companies, or were employees of Sanofi or Regeneron.
SOURCE: Worm M et al. JAMA Dermatol. 2019 Dec 26. doi: 10.1001/jamadermatol.2019.3617.
Patients with moderate to severe atopic dermatitis who responded well to the approved dupilumab regimen of 300 mg every 2 weeks in pivotal phase 3 monotherapy trials were more likely to have a continued response over the longer term if they maintained this regimen rather than switching to longer dosing intervals or discontinuing the medication.
This finding comes from a 36-week, randomized, double-blind, placebo-controlled trial that enrolled 422 – SOLO 1 and SOLO 2.
The new international study – SOLO-CONTINUE – randomized these patients to continue the original regimen (weekly or every 2 weeks), to receive 300 mg of the biologic medication every 4 or 8 weeks, or to receive placebo.
Patients who continued the original regimen had the most consistent maintenance of treatment effect, while patients on longer dosage intervals or placebo had a dose-dependent reduction in response and no safety advantage. The incidence of treatment-emergent antidrug antibody was lowest with dupilumab weekly or every 2 weeks, and slightly higher with less-frequent dosing intervals, reported Margitta Worm, MD, of the Charité Universitätsmedizin Berlin, and coinvestigators.
“Because administration every 4 weeks or every 8 weeks did not provide an additional safety advantage and was numerically outperformed by administration weekly or every 2 weeks, we believe that it is prudent to adhere to the approved every 2 weeks regimen for adults and avoid less frequent treatment regimens (every 4 weeks or every 8 weeks) for long-term maintenance of efficacy,” they wrote in JAMA Dermatology.
Treatment success in the original SOLO trials was defined as having achieved an Investigator’s Global Assessment score of 0-1, or 75% improvement in Eczema Area and Severity Index Scores (EASI-75). As primary endpoints, SOLO-CONTINUE looked at the mean percentage change in EASI score over the course of the trial, and the percentage of patients who maintained EASI-75 at week 36.
Patients in the SOLO-CONTINUE trial who were randomized to receive dupilumab weekly or every 2 weeks had a mean percent change in EASI score of –0.06%. In contrast, patients assigned to the placebo group had a 21.7% decrease, and those taking the medication at 4- and 8-week intervals had mean changes of –3.84% and –6.84%, respectively. Post hoc analyses showed no apparent difference between dupilumab weekly and every 2 weeks in the maintenance of clinical response, the investigators reported.
Among patients with EASI-75 response at baseline, significantly more patients maintained this response at week 36 than patients receiving placebo, and there was again an apparent dose-dependent response. The percentage with EASI-75 at week 36 was 71.6% with the weekly or every-2-weeks regimen, 58.3% with the 4-week regimen, 54.9% with the 8-week regimen, and 30.4% in the placebo group.
Continuing treatment with 300 mg weekly or every 2 weeks resulted in greater maintenance of response across multiple other clinical endpoints and patient-reported outcomes as well (such as pruritus, atopic dermatitis symptoms, sleep, pain or discomfort, quality of life, and symptoms of anxiety and depression).
The more-frequent regimens also conferred no greater risk than less-frequent administration, and there were no new safety signals over the 36-week trial. Treatment-emergent adverse events (the most common were headache, nasopharyngitis, injection-site reactions, and herpes simplex virus infection) occurred in 70.7% of patients in the weekly or every-2-weeks group, 73.6% in the 4-week group, 75% in the 8-week group, and 81.7% in the placebo group.
Unlike earlier studies, the incidence of conjunctivitis was low (less than 6%) across all treatment groups, possibly because patients in the SOLO-CONTINUE trial were all high-level responders who tend to have conjunctivitis less frequently, the authors wrote.
Patients receiving less-frequent doses of dupilumab, particularly every 8 weeks, had greater rates of skin infections, flares, and rescue medication use than patients receiving doses weekly or every 2 weeks, the investigators reported. Treatment-emergent antidrug antibody incidence was slightly higher with less-frequent doses (11.7% and 6% in the 8-week and 4-week groups, respectively, compared with 4.3% and 1.2% in the every-2-weeks and weekly groups), which indicates a “higher incidence of immunogenicity with less-frequent dosage intervals” and is “consistent with other biologics,” they wrote.
Dupilumab is a human monoclonal antibody against the interleukin-4 receptor alpha that inhibits signaling of IL-4 and IL-13. The study was conducted at 185 sites in North America, Europe, Asia, and Japan. Patients had a mean age of 38.2 years; 53.8% were male.
While the trial suggests that the approved regimen of 300 mg every 2 weeks is best for long-term treatment, “therapeutic decisions are often influenced by cost-benefit considerations, in which case practitioners and other stakeholders involved in these decisions should carefully balance potential savings against suboptimal efficacy and long-term risks associated with discontinuous treatment regimens,” the investigators wrote.
The SOLO-CONTINUE trial was funded by Sanofi and Regeneron, the companies that market dupilumab. Dr. Worm reported receiving honoraria for consulting and lecture activity from Regeneron and Sanofi during and outside the conduct of the study, among other disclosures. The other authors had multiple disclosures related to these and multiple other pharmaceutical companies, or were employees of Sanofi or Regeneron.
SOURCE: Worm M et al. JAMA Dermatol. 2019 Dec 26. doi: 10.1001/jamadermatol.2019.3617.
FROM JAMA DERMATOLOGY
Data point to bidirectional link between sleep disorders and ADHD
in a large longitudinal study of adolescents in China.
Investigators twice assessed 7,072 middle and high school students participating in the larger longitudinal Shandong Adolescent Behavior & Health Cohort – in 2015 and 1 year later in 2016 – for sleep, mental health, psychosocial factors (using the self-administered Adolescent Health Questionnaire, or AHQ), and for ADHD symptoms (using the Youth Self-Report, or YSR, of the Achenbach Child Behavior Checklist).
At baseline, ADHD symptoms were reported by 7.6% of adolescents and were significantly correlated, after adjusting for adolescent and family covariates, with all the sleep variables studied: sleep duration of 7 hours or less per night, insomnia symptoms, poor sleep quality, RLS symptoms, frequent snorting, and hypnotic use, reported Xianchen Liu, MD, PhD, of Shandong (China) University, and coinvestigators. They noted a dose-response relationship between sleep duration and the odds of having ADHD symptoms.
At 1-year follow-up, 4.5% of the 6,531 participants who did not have ADHD symptoms at baseline now reported them. After adjustments for covariates, any insomnia (odds ratio, 1.48), difficulty initiating sleep (one of the insomnia symptoms) (OR, 2.09), RLS (OR, 1.47), and frequent snoring (OR, 2.30) at baseline were each significantly associated with development of incident ADHD symptoms and with ADHD severity at 1 year, they reported in Sleep.
“Given the fact that sleep disorders in adolescents are often underdiagnosed and untreated primarily in the primary care setting, our findings highlight that clinicians should assess and manage short sleep duration and sleep problems for effective treatment of ADHD in adolescents,” as well as for prevention, they wrote.
The AHQ includes questions that assess nocturnal sleep duration and sleep problems during the past month. The adolescent and family variables that were selected as covariates and controlled for include cigarette smoking, alcohol drinking, use of mental health services, chronic physical diseases, and parental education and occupation. Depression was also a covariate but was assessed through a different scale.
The YSR measures eight ADHD symptoms during the past 6 months on a 3-point scale (not true, somewhat or sometimes true, and very true or often true). The adolescent participants of this study were in grades 7, 8, and 10 at baseline. Their mean age at baseline was 15 years; half were male. They were part of the larger Shandong Adolescent and Behavioral Cohort, a longitudinal study of almost 12,000 adolescents.
Growing evidence has demonstrated a bidirectional relationship between sleep problems and ADHD symptoms in pediatric populations, the investigators wrote, and further research is needed to examine the “mediators, moderators, and biological mechanisms of the sleep-ADHD link [in adolescents].”
While there are multiple potential pathways for this link, sleep problems may sometimes result in a cluster of behavioral and cognitive symptoms that are not true ADHD but that mimic the disorder, they noted.
The investigators also noted that approximately 67% of adolescents who had clinically relevant ADHD symptoms at baseline no longer had these symptoms at 1-year follow-up – a finding that “supports the [idea]” that ADHD symptoms with onset in adolescence may be transient or episodic rather than persistent.
The study was funded in part by the National Natural Science Foundation of China. The authors reported that they have no conflicts of interest.
SOURCE: Liu X et al. Sleep. 2019 Dec 2. doi: 10.1093/sleep/zsz294.
in a large longitudinal study of adolescents in China.
Investigators twice assessed 7,072 middle and high school students participating in the larger longitudinal Shandong Adolescent Behavior & Health Cohort – in 2015 and 1 year later in 2016 – for sleep, mental health, psychosocial factors (using the self-administered Adolescent Health Questionnaire, or AHQ), and for ADHD symptoms (using the Youth Self-Report, or YSR, of the Achenbach Child Behavior Checklist).
At baseline, ADHD symptoms were reported by 7.6% of adolescents and were significantly correlated, after adjusting for adolescent and family covariates, with all the sleep variables studied: sleep duration of 7 hours or less per night, insomnia symptoms, poor sleep quality, RLS symptoms, frequent snorting, and hypnotic use, reported Xianchen Liu, MD, PhD, of Shandong (China) University, and coinvestigators. They noted a dose-response relationship between sleep duration and the odds of having ADHD symptoms.
At 1-year follow-up, 4.5% of the 6,531 participants who did not have ADHD symptoms at baseline now reported them. After adjustments for covariates, any insomnia (odds ratio, 1.48), difficulty initiating sleep (one of the insomnia symptoms) (OR, 2.09), RLS (OR, 1.47), and frequent snoring (OR, 2.30) at baseline were each significantly associated with development of incident ADHD symptoms and with ADHD severity at 1 year, they reported in Sleep.
“Given the fact that sleep disorders in adolescents are often underdiagnosed and untreated primarily in the primary care setting, our findings highlight that clinicians should assess and manage short sleep duration and sleep problems for effective treatment of ADHD in adolescents,” as well as for prevention, they wrote.
The AHQ includes questions that assess nocturnal sleep duration and sleep problems during the past month. The adolescent and family variables that were selected as covariates and controlled for include cigarette smoking, alcohol drinking, use of mental health services, chronic physical diseases, and parental education and occupation. Depression was also a covariate but was assessed through a different scale.
The YSR measures eight ADHD symptoms during the past 6 months on a 3-point scale (not true, somewhat or sometimes true, and very true or often true). The adolescent participants of this study were in grades 7, 8, and 10 at baseline. Their mean age at baseline was 15 years; half were male. They were part of the larger Shandong Adolescent and Behavioral Cohort, a longitudinal study of almost 12,000 adolescents.
Growing evidence has demonstrated a bidirectional relationship between sleep problems and ADHD symptoms in pediatric populations, the investigators wrote, and further research is needed to examine the “mediators, moderators, and biological mechanisms of the sleep-ADHD link [in adolescents].”
While there are multiple potential pathways for this link, sleep problems may sometimes result in a cluster of behavioral and cognitive symptoms that are not true ADHD but that mimic the disorder, they noted.
The investigators also noted that approximately 67% of adolescents who had clinically relevant ADHD symptoms at baseline no longer had these symptoms at 1-year follow-up – a finding that “supports the [idea]” that ADHD symptoms with onset in adolescence may be transient or episodic rather than persistent.
The study was funded in part by the National Natural Science Foundation of China. The authors reported that they have no conflicts of interest.
SOURCE: Liu X et al. Sleep. 2019 Dec 2. doi: 10.1093/sleep/zsz294.
in a large longitudinal study of adolescents in China.
Investigators twice assessed 7,072 middle and high school students participating in the larger longitudinal Shandong Adolescent Behavior & Health Cohort – in 2015 and 1 year later in 2016 – for sleep, mental health, psychosocial factors (using the self-administered Adolescent Health Questionnaire, or AHQ), and for ADHD symptoms (using the Youth Self-Report, or YSR, of the Achenbach Child Behavior Checklist).
At baseline, ADHD symptoms were reported by 7.6% of adolescents and were significantly correlated, after adjusting for adolescent and family covariates, with all the sleep variables studied: sleep duration of 7 hours or less per night, insomnia symptoms, poor sleep quality, RLS symptoms, frequent snorting, and hypnotic use, reported Xianchen Liu, MD, PhD, of Shandong (China) University, and coinvestigators. They noted a dose-response relationship between sleep duration and the odds of having ADHD symptoms.
At 1-year follow-up, 4.5% of the 6,531 participants who did not have ADHD symptoms at baseline now reported them. After adjustments for covariates, any insomnia (odds ratio, 1.48), difficulty initiating sleep (one of the insomnia symptoms) (OR, 2.09), RLS (OR, 1.47), and frequent snoring (OR, 2.30) at baseline were each significantly associated with development of incident ADHD symptoms and with ADHD severity at 1 year, they reported in Sleep.
“Given the fact that sleep disorders in adolescents are often underdiagnosed and untreated primarily in the primary care setting, our findings highlight that clinicians should assess and manage short sleep duration and sleep problems for effective treatment of ADHD in adolescents,” as well as for prevention, they wrote.
The AHQ includes questions that assess nocturnal sleep duration and sleep problems during the past month. The adolescent and family variables that were selected as covariates and controlled for include cigarette smoking, alcohol drinking, use of mental health services, chronic physical diseases, and parental education and occupation. Depression was also a covariate but was assessed through a different scale.
The YSR measures eight ADHD symptoms during the past 6 months on a 3-point scale (not true, somewhat or sometimes true, and very true or often true). The adolescent participants of this study were in grades 7, 8, and 10 at baseline. Their mean age at baseline was 15 years; half were male. They were part of the larger Shandong Adolescent and Behavioral Cohort, a longitudinal study of almost 12,000 adolescents.
Growing evidence has demonstrated a bidirectional relationship between sleep problems and ADHD symptoms in pediatric populations, the investigators wrote, and further research is needed to examine the “mediators, moderators, and biological mechanisms of the sleep-ADHD link [in adolescents].”
While there are multiple potential pathways for this link, sleep problems may sometimes result in a cluster of behavioral and cognitive symptoms that are not true ADHD but that mimic the disorder, they noted.
The investigators also noted that approximately 67% of adolescents who had clinically relevant ADHD symptoms at baseline no longer had these symptoms at 1-year follow-up – a finding that “supports the [idea]” that ADHD symptoms with onset in adolescence may be transient or episodic rather than persistent.
The study was funded in part by the National Natural Science Foundation of China. The authors reported that they have no conflicts of interest.
SOURCE: Liu X et al. Sleep. 2019 Dec 2. doi: 10.1093/sleep/zsz294.
FROM SLEEP
Administrative data can help drive quality improvement in breast cancer care
Quality indicators of breast cancer care were successfully computed for more than 15,000 incident cases of breast cancer using electronic administrative databases in a project led by five regional oncology networks in Italy.
The project has shown that, despite some limitations in the use of administrative data to measure health care performance, “evaluating the quality of breast cancer care at a population level is possible,” investigators reported in the Journal of Oncology Practice.
The data obtained “from multiple administrative databases gathered in a real-world setting across five Italian regions” highlighted regional variations in breast cancer care and ways in which clinical guidelines were being overlooked, they wrote.
In doing so, the project confirmed that administrative data is “suitable” for measuring performance in health care and potentially useful for guiding quality improvement interventions. For instance, the project identified extensive use of blood tumor markers in breast cancer follow-up, wrote Valentina Guarneri, PhD, MD, of the University of Padova (Italy) and the Istituto Oncologico Veneto, also in Padova, and coauthors.
Oncologists and epidemiologists from the Italian regional oncology networks identified 46 clinically relevant indicators (9 structure indicators, 29 dealing with process, and 8 outcome indicators) by comparing pathways of care established by each network and identifying commonalities.
Of the 46 indicators, 22 were considered by the project leaders to be “potentially computable” from information retrieved by regional administrative databases. And of these 22 designed to be extractable, 9 (2 indicators of structure and 7 of process) were found to be actually evaluable for 15,342 cases of newly diagnosed invasive and/or in situ breast cancer diagnosed during 2016.
Blood tumor markers were tested in 44.2%-64.5% of patients in the first year after surgery – higher than the benchmark of 20% or less that was established to account for stage IV patients and other specific conditions in which markers might be indicated. National, international, and regional guidelines “discourage the use of blood tumor markers” in breast cancer follow-up, the investigators wrote.
The extensive use of these markers – observed across all five regions – is “a starting point to understanding how to improve clinical practice,” they added.
Other quality indicators that were evaluable included radiotherapy within 12 weeks after surgery if adjuvant chemotherapy is not administered (42%-83.8% in the project, compared with the benchmark of 90% or greater) and mammography 6-18 months after surgery (administered in 55.1%-72.6%, compared with the benchmark of 90% or greater), as well as the proportion of patients starting adjuvant systemic treatment (chemotherapy or endocrine therapy) within 60 days of surgery (for patients receiving systemic treatment).
To calculate the indicators, each regional cancer network used computerized sources of information including hospital discharge forms, outpatient records of diagnostic and therapeutic procedures, prescriptions of drugs reimbursed by the National Health Service in the hospital and outpatient settings, regional health registries, and the regional mortality registries.
All data used in the project came from regional repositories, which collect data from all National Health Service providers in the region, and not from single institutional repositories, the investigators noted.
More than half of the indicators expected to be assessable – but not found to be – were not computable as a result of data being unavailable (for example, pathology data) or incomplete, and as a result of data not being reliable for various reasons. The fact that examinations paid for directly by patients are not reported by the management systems of the National Health System was another complicating factor, they reported.
The authors disclosed funding and relationships with various pharmaceutical companies. The research was supported by the Periplo Association.
SOURCE: Guarneri V et al. J Oncol Pract. 2019 Dec 19. doi: 10.1200/JOP.19.00466.
Quality indicators of breast cancer care were successfully computed for more than 15,000 incident cases of breast cancer using electronic administrative databases in a project led by five regional oncology networks in Italy.
The project has shown that, despite some limitations in the use of administrative data to measure health care performance, “evaluating the quality of breast cancer care at a population level is possible,” investigators reported in the Journal of Oncology Practice.
The data obtained “from multiple administrative databases gathered in a real-world setting across five Italian regions” highlighted regional variations in breast cancer care and ways in which clinical guidelines were being overlooked, they wrote.
In doing so, the project confirmed that administrative data is “suitable” for measuring performance in health care and potentially useful for guiding quality improvement interventions. For instance, the project identified extensive use of blood tumor markers in breast cancer follow-up, wrote Valentina Guarneri, PhD, MD, of the University of Padova (Italy) and the Istituto Oncologico Veneto, also in Padova, and coauthors.
Oncologists and epidemiologists from the Italian regional oncology networks identified 46 clinically relevant indicators (9 structure indicators, 29 dealing with process, and 8 outcome indicators) by comparing pathways of care established by each network and identifying commonalities.
Of the 46 indicators, 22 were considered by the project leaders to be “potentially computable” from information retrieved by regional administrative databases. And of these 22 designed to be extractable, 9 (2 indicators of structure and 7 of process) were found to be actually evaluable for 15,342 cases of newly diagnosed invasive and/or in situ breast cancer diagnosed during 2016.
Blood tumor markers were tested in 44.2%-64.5% of patients in the first year after surgery – higher than the benchmark of 20% or less that was established to account for stage IV patients and other specific conditions in which markers might be indicated. National, international, and regional guidelines “discourage the use of blood tumor markers” in breast cancer follow-up, the investigators wrote.
The extensive use of these markers – observed across all five regions – is “a starting point to understanding how to improve clinical practice,” they added.
Other quality indicators that were evaluable included radiotherapy within 12 weeks after surgery if adjuvant chemotherapy is not administered (42%-83.8% in the project, compared with the benchmark of 90% or greater) and mammography 6-18 months after surgery (administered in 55.1%-72.6%, compared with the benchmark of 90% or greater), as well as the proportion of patients starting adjuvant systemic treatment (chemotherapy or endocrine therapy) within 60 days of surgery (for patients receiving systemic treatment).
To calculate the indicators, each regional cancer network used computerized sources of information including hospital discharge forms, outpatient records of diagnostic and therapeutic procedures, prescriptions of drugs reimbursed by the National Health Service in the hospital and outpatient settings, regional health registries, and the regional mortality registries.
All data used in the project came from regional repositories, which collect data from all National Health Service providers in the region, and not from single institutional repositories, the investigators noted.
More than half of the indicators expected to be assessable – but not found to be – were not computable as a result of data being unavailable (for example, pathology data) or incomplete, and as a result of data not being reliable for various reasons. The fact that examinations paid for directly by patients are not reported by the management systems of the National Health System was another complicating factor, they reported.
The authors disclosed funding and relationships with various pharmaceutical companies. The research was supported by the Periplo Association.
SOURCE: Guarneri V et al. J Oncol Pract. 2019 Dec 19. doi: 10.1200/JOP.19.00466.
Quality indicators of breast cancer care were successfully computed for more than 15,000 incident cases of breast cancer using electronic administrative databases in a project led by five regional oncology networks in Italy.
The project has shown that, despite some limitations in the use of administrative data to measure health care performance, “evaluating the quality of breast cancer care at a population level is possible,” investigators reported in the Journal of Oncology Practice.
The data obtained “from multiple administrative databases gathered in a real-world setting across five Italian regions” highlighted regional variations in breast cancer care and ways in which clinical guidelines were being overlooked, they wrote.
In doing so, the project confirmed that administrative data is “suitable” for measuring performance in health care and potentially useful for guiding quality improvement interventions. For instance, the project identified extensive use of blood tumor markers in breast cancer follow-up, wrote Valentina Guarneri, PhD, MD, of the University of Padova (Italy) and the Istituto Oncologico Veneto, also in Padova, and coauthors.
Oncologists and epidemiologists from the Italian regional oncology networks identified 46 clinically relevant indicators (9 structure indicators, 29 dealing with process, and 8 outcome indicators) by comparing pathways of care established by each network and identifying commonalities.
Of the 46 indicators, 22 were considered by the project leaders to be “potentially computable” from information retrieved by regional administrative databases. And of these 22 designed to be extractable, 9 (2 indicators of structure and 7 of process) were found to be actually evaluable for 15,342 cases of newly diagnosed invasive and/or in situ breast cancer diagnosed during 2016.
Blood tumor markers were tested in 44.2%-64.5% of patients in the first year after surgery – higher than the benchmark of 20% or less that was established to account for stage IV patients and other specific conditions in which markers might be indicated. National, international, and regional guidelines “discourage the use of blood tumor markers” in breast cancer follow-up, the investigators wrote.
The extensive use of these markers – observed across all five regions – is “a starting point to understanding how to improve clinical practice,” they added.
Other quality indicators that were evaluable included radiotherapy within 12 weeks after surgery if adjuvant chemotherapy is not administered (42%-83.8% in the project, compared with the benchmark of 90% or greater) and mammography 6-18 months after surgery (administered in 55.1%-72.6%, compared with the benchmark of 90% or greater), as well as the proportion of patients starting adjuvant systemic treatment (chemotherapy or endocrine therapy) within 60 days of surgery (for patients receiving systemic treatment).
To calculate the indicators, each regional cancer network used computerized sources of information including hospital discharge forms, outpatient records of diagnostic and therapeutic procedures, prescriptions of drugs reimbursed by the National Health Service in the hospital and outpatient settings, regional health registries, and the regional mortality registries.
All data used in the project came from regional repositories, which collect data from all National Health Service providers in the region, and not from single institutional repositories, the investigators noted.
More than half of the indicators expected to be assessable – but not found to be – were not computable as a result of data being unavailable (for example, pathology data) or incomplete, and as a result of data not being reliable for various reasons. The fact that examinations paid for directly by patients are not reported by the management systems of the National Health System was another complicating factor, they reported.
The authors disclosed funding and relationships with various pharmaceutical companies. The research was supported by the Periplo Association.
SOURCE: Guarneri V et al. J Oncol Pract. 2019 Dec 19. doi: 10.1200/JOP.19.00466.
FROM THE JOURNAL OF ONCOLOGY PRACTICE
Can insulin plus metformin improve pregnancy outcomes in women with type 2 diabetes?
WASHINGTON – Insulin is the preferred agent for type 2 diabetes in pregnant women, yet about a third of pregnancies still have an adverse outcome, according Kim Boggess, MD, who spoke at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“We are not where we need to be,” said Dr. Boggess, who is leading a trial that brings metformin, the first-line agent for type 2 diabetes outside of pregnancy, back into the picture for pregnant women – as an add-on to insulin.
It is an interesting twist, because pregnant women taking metformin for preexisting type 2 or gestational diabetes have been shown in some studies to require supplemental insulin, more than occasionally, to achieve target glycemic control.
This was the case in a small, randomized, controlled trial at Dr. Boggess’ institution, the University of North Carolina at Chapel Hill, in which 43% of pregnant women with type 2 diabetes who were assigned to metformin required supplemental insulin (Am J Perinatol. 2013;30[6]:483-90). (0% vs. 36%, respectively) and fewer reports of glucose values less than 60 mg/dL (7.1% vs. 50%).
“I don’t consider this [need for supplemental insulin] ‘metformin failure,’ because studies that use metformin as monotherapy and that [show some patients] ultimately requiring insulin support ... also show that these women need less insulin,” she said. “What’s the risk of insulin alone? Hypoglycemia. So using less insulin could be a good thing.”
Other research suggests there may be less maternal weight gain, less neonatal hypoglycemia, fewer neonatal complications, and improved maternal glycemic control in patients treated with metformin, alone or with add-on insulin, than with insulin alone. “We’re starting to get a sense in the literature that, at least in the [pregnant] population with type 2 diabetes, there may be a role for metformin,” said Dr. Boggess, professor and program director for maternal-fetal medicine at the university.
Currently, the multisite MOMPOD trial (Medical Optimization of Management of T2DM Complicating Pregnancy) is randomizing 950 women to insulin plus 1,000 mg metformin twice daily or insulin plus placebo. The primary outcome of the trial is a composite of pregnancy loss, preterm birth, birth injury, neonatal hypoglycemia, or hyperbilirubinemia. Infant fat mass (within 72 hours of birth) is a secondary outcome, along with maternal safety and maternal side effects.
The MiTy (Metformin in Women with T2DM in Pregnancy) trial in Canada, with similar randomization arms and outcomes measures, is completed and undergoing analysis. “Hopefully we’ll [soon] be able to say whether the addition of adjuvant metformin to insulin to treat type 2 diabetes brings the perinatal adverse outcome rate down from 30%,” said Dr. Boggess.
Metformin is the recommended first-line agent for type 2 diabetes in nonpregnant adults. But during pregnancy, insulin, which does not cross the placenta, is the preferred agent, according to recommendations of the American Diabetes Association and the American College of Obstetricians and Gynecologists, she noted. Lingering in the background is the fact that the long-term effects of in utero metformin exposure on offspring – and of exposure to any oral hypoglycemic agent – are unknown, she said*
A majority of the adverse pregnancy outcomes that occur in the context of type 2 diabetes involve macrosomia. “It’s a big deal,” Dr. Boggess said, that results in numerous maternal and infant risks and complications. “We also know that the in utero environment that contributes to, or causes, macrosomia predisposes to childhood obesity and obesity later on.”
Diabetes is the “leading risk factor” for adverse pregnancy outcomes today, said E. Albert Reece, MD, PhD, MBA, executive vice president for medical affairs at the University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers distinguished professor and dean of the University of Maryland School of Medicine. In the United States, 11% of women aged 20 years and older have diabetes, and the disease affects more than 1% of all pregnancies, he said.
The MOMPOD trial is sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr. Boggess reported no conflicts of interest.
* This article was updated 1/2/2020.
WASHINGTON – Insulin is the preferred agent for type 2 diabetes in pregnant women, yet about a third of pregnancies still have an adverse outcome, according Kim Boggess, MD, who spoke at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“We are not where we need to be,” said Dr. Boggess, who is leading a trial that brings metformin, the first-line agent for type 2 diabetes outside of pregnancy, back into the picture for pregnant women – as an add-on to insulin.
It is an interesting twist, because pregnant women taking metformin for preexisting type 2 or gestational diabetes have been shown in some studies to require supplemental insulin, more than occasionally, to achieve target glycemic control.
This was the case in a small, randomized, controlled trial at Dr. Boggess’ institution, the University of North Carolina at Chapel Hill, in which 43% of pregnant women with type 2 diabetes who were assigned to metformin required supplemental insulin (Am J Perinatol. 2013;30[6]:483-90). (0% vs. 36%, respectively) and fewer reports of glucose values less than 60 mg/dL (7.1% vs. 50%).
“I don’t consider this [need for supplemental insulin] ‘metformin failure,’ because studies that use metformin as monotherapy and that [show some patients] ultimately requiring insulin support ... also show that these women need less insulin,” she said. “What’s the risk of insulin alone? Hypoglycemia. So using less insulin could be a good thing.”
Other research suggests there may be less maternal weight gain, less neonatal hypoglycemia, fewer neonatal complications, and improved maternal glycemic control in patients treated with metformin, alone or with add-on insulin, than with insulin alone. “We’re starting to get a sense in the literature that, at least in the [pregnant] population with type 2 diabetes, there may be a role for metformin,” said Dr. Boggess, professor and program director for maternal-fetal medicine at the university.
Currently, the multisite MOMPOD trial (Medical Optimization of Management of T2DM Complicating Pregnancy) is randomizing 950 women to insulin plus 1,000 mg metformin twice daily or insulin plus placebo. The primary outcome of the trial is a composite of pregnancy loss, preterm birth, birth injury, neonatal hypoglycemia, or hyperbilirubinemia. Infant fat mass (within 72 hours of birth) is a secondary outcome, along with maternal safety and maternal side effects.
The MiTy (Metformin in Women with T2DM in Pregnancy) trial in Canada, with similar randomization arms and outcomes measures, is completed and undergoing analysis. “Hopefully we’ll [soon] be able to say whether the addition of adjuvant metformin to insulin to treat type 2 diabetes brings the perinatal adverse outcome rate down from 30%,” said Dr. Boggess.
Metformin is the recommended first-line agent for type 2 diabetes in nonpregnant adults. But during pregnancy, insulin, which does not cross the placenta, is the preferred agent, according to recommendations of the American Diabetes Association and the American College of Obstetricians and Gynecologists, she noted. Lingering in the background is the fact that the long-term effects of in utero metformin exposure on offspring – and of exposure to any oral hypoglycemic agent – are unknown, she said*
A majority of the adverse pregnancy outcomes that occur in the context of type 2 diabetes involve macrosomia. “It’s a big deal,” Dr. Boggess said, that results in numerous maternal and infant risks and complications. “We also know that the in utero environment that contributes to, or causes, macrosomia predisposes to childhood obesity and obesity later on.”
Diabetes is the “leading risk factor” for adverse pregnancy outcomes today, said E. Albert Reece, MD, PhD, MBA, executive vice president for medical affairs at the University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers distinguished professor and dean of the University of Maryland School of Medicine. In the United States, 11% of women aged 20 years and older have diabetes, and the disease affects more than 1% of all pregnancies, he said.
The MOMPOD trial is sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr. Boggess reported no conflicts of interest.
* This article was updated 1/2/2020.
WASHINGTON – Insulin is the preferred agent for type 2 diabetes in pregnant women, yet about a third of pregnancies still have an adverse outcome, according Kim Boggess, MD, who spoke at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“We are not where we need to be,” said Dr. Boggess, who is leading a trial that brings metformin, the first-line agent for type 2 diabetes outside of pregnancy, back into the picture for pregnant women – as an add-on to insulin.
It is an interesting twist, because pregnant women taking metformin for preexisting type 2 or gestational diabetes have been shown in some studies to require supplemental insulin, more than occasionally, to achieve target glycemic control.
This was the case in a small, randomized, controlled trial at Dr. Boggess’ institution, the University of North Carolina at Chapel Hill, in which 43% of pregnant women with type 2 diabetes who were assigned to metformin required supplemental insulin (Am J Perinatol. 2013;30[6]:483-90). (0% vs. 36%, respectively) and fewer reports of glucose values less than 60 mg/dL (7.1% vs. 50%).
“I don’t consider this [need for supplemental insulin] ‘metformin failure,’ because studies that use metformin as monotherapy and that [show some patients] ultimately requiring insulin support ... also show that these women need less insulin,” she said. “What’s the risk of insulin alone? Hypoglycemia. So using less insulin could be a good thing.”
Other research suggests there may be less maternal weight gain, less neonatal hypoglycemia, fewer neonatal complications, and improved maternal glycemic control in patients treated with metformin, alone or with add-on insulin, than with insulin alone. “We’re starting to get a sense in the literature that, at least in the [pregnant] population with type 2 diabetes, there may be a role for metformin,” said Dr. Boggess, professor and program director for maternal-fetal medicine at the university.
Currently, the multisite MOMPOD trial (Medical Optimization of Management of T2DM Complicating Pregnancy) is randomizing 950 women to insulin plus 1,000 mg metformin twice daily or insulin plus placebo. The primary outcome of the trial is a composite of pregnancy loss, preterm birth, birth injury, neonatal hypoglycemia, or hyperbilirubinemia. Infant fat mass (within 72 hours of birth) is a secondary outcome, along with maternal safety and maternal side effects.
The MiTy (Metformin in Women with T2DM in Pregnancy) trial in Canada, with similar randomization arms and outcomes measures, is completed and undergoing analysis. “Hopefully we’ll [soon] be able to say whether the addition of adjuvant metformin to insulin to treat type 2 diabetes brings the perinatal adverse outcome rate down from 30%,” said Dr. Boggess.
Metformin is the recommended first-line agent for type 2 diabetes in nonpregnant adults. But during pregnancy, insulin, which does not cross the placenta, is the preferred agent, according to recommendations of the American Diabetes Association and the American College of Obstetricians and Gynecologists, she noted. Lingering in the background is the fact that the long-term effects of in utero metformin exposure on offspring – and of exposure to any oral hypoglycemic agent – are unknown, she said*
A majority of the adverse pregnancy outcomes that occur in the context of type 2 diabetes involve macrosomia. “It’s a big deal,” Dr. Boggess said, that results in numerous maternal and infant risks and complications. “We also know that the in utero environment that contributes to, or causes, macrosomia predisposes to childhood obesity and obesity later on.”
Diabetes is the “leading risk factor” for adverse pregnancy outcomes today, said E. Albert Reece, MD, PhD, MBA, executive vice president for medical affairs at the University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers distinguished professor and dean of the University of Maryland School of Medicine. In the United States, 11% of women aged 20 years and older have diabetes, and the disease affects more than 1% of all pregnancies, he said.
The MOMPOD trial is sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr. Boggess reported no conflicts of interest.
* This article was updated 1/2/2020.
REPORTING FROM DPSG-NA 2019
Platinum-based therapy superior in study of upper GI tumors
A study that aimed to validate intratumoral ERCC1 levels as a predictive marker of platinum sensitivity in upper GI tumors failed to do so, but it did reach one firm conclusion: That platinum therapy with fluorouracil, leucovorin, and oxaliplatin (FOLFOX) was superior in efficacy to a non–platinum-containing regimen of irinotecan and docetaxel (IT).
Approximately 200 untreated patients with unresectable advanced or metastatic HER2-negative adenocarcinoma of the esophagus, stomach or gastroesophageal junction were evaluated in the phase 2 study for mRNA expression of ERCC1 level and then randomized to either a platinum-containing or non–platinum-containing treatment arm with stratification for ERCC1 level (either low, with levels less than 1.7, or high, meaning 1.7 or more).
In retrospective studies of patients with gastric cancer, levels of gene expression of ERCC1 within the primary tumor have had a significant inverse relationship to response to platinum compounds and overall survival; low ERCC1 expression, in other words, has been associated with higher response rates and better survival. This inverse relationship of expression of the ERCC1 gene and platinum sensitivity has been demonstrated in colon cancer and other tumor types as well.
The problem is, approximately 86% of the patients in this phase 2, randomized study had ERCC1 values lower than 1.7 – many more than the investigators anticipated based on data from prior studies. (They expected a 50-50 distribution, roughly.) The predominance of low ERCC1 mRNA expression meant that evaluation of the ERCC1-high subgroup – and evaluation of interactive effects between ERCC1 expression and treatment type – was limited, said Syma Iqbal, MD, of the University of Southern California, Los Angeles, and coinvestigators.
“Unfortunately, this study did not validate or identify ERCC1 as a predictive marker of platinum sensitivity in upper GI tumors,” they wrote in the Journal of Clinical Oncology. However, “it did support the use of FOLFOX, a platinum-containing regimen, as a standard and superior frontline regimen, compared with the non–platinum-containing IT.”
Across all patients in the FOLFOX arm, the median progression-free survival (PFS) was significantly longer – 5.7 months versus 2.9 months in the IT arm (hazard ratio, 0.71; P = .02). The median overall survival was greater with FOLFOX as well, though this difference – 11.4 months versus 8.7 months – did not reach statistical significance. Similarly, in the ERCC1-low subgroup, the median PFS in patients receiving FOLFOX was statistically superior to IT – 5.9 months versus 2.8 months – and overall survival was better as well, though this latter difference was not statistically significant.
In the ERCC1-high subgroup, the median PFS was similar in the FOLFOX and IT arms (4.7 months vs. 5.3 months), the investigators noted. They plotted PFS within ERCC1 quartiles and found a consistent pattern of improved PFS in the FOLFOX versus IT arm, and “thus, little evidence of differential treatment effects on PFS across ERCC1 levels in this population.”
Regarding safety, the investigators noted, of 91 patients who completed protocol therapy in the FOLFOX arm and were analyzed for adverse events, 3 treatment-related deaths were reported and 9 additional patients experienced grade 4 adverse events. Of 98 patients assessed for adverse events in the IT arm, 3 treatment-related deaths were reported and 14 additional patients experienced grade 4 adverse events.
To be eligible for the study patients had to have a Zubrod performance status of 0-1 and been either treatment naive or have completed adjuvant therapy at least 180 days prior to enrollment.
The study was supported by the National Cancer Institute. Dr. Iqbal reported receiving honoraria from Celgene, Eisai, and F. Hoffmann–La Roche; serving a consulting or advisory role for F. Hoffmann–La Roche; serving on the speakers’ bureau for Celgene and Eisai; and receiving research funding from Bayer and Onyx.
SOURCE: Iqbal S et al. J Clin Oncol. 2019 Dec 9. doi: 10.1200/JCO.19.00925.
A study that aimed to validate intratumoral ERCC1 levels as a predictive marker of platinum sensitivity in upper GI tumors failed to do so, but it did reach one firm conclusion: That platinum therapy with fluorouracil, leucovorin, and oxaliplatin (FOLFOX) was superior in efficacy to a non–platinum-containing regimen of irinotecan and docetaxel (IT).
Approximately 200 untreated patients with unresectable advanced or metastatic HER2-negative adenocarcinoma of the esophagus, stomach or gastroesophageal junction were evaluated in the phase 2 study for mRNA expression of ERCC1 level and then randomized to either a platinum-containing or non–platinum-containing treatment arm with stratification for ERCC1 level (either low, with levels less than 1.7, or high, meaning 1.7 or more).
In retrospective studies of patients with gastric cancer, levels of gene expression of ERCC1 within the primary tumor have had a significant inverse relationship to response to platinum compounds and overall survival; low ERCC1 expression, in other words, has been associated with higher response rates and better survival. This inverse relationship of expression of the ERCC1 gene and platinum sensitivity has been demonstrated in colon cancer and other tumor types as well.
The problem is, approximately 86% of the patients in this phase 2, randomized study had ERCC1 values lower than 1.7 – many more than the investigators anticipated based on data from prior studies. (They expected a 50-50 distribution, roughly.) The predominance of low ERCC1 mRNA expression meant that evaluation of the ERCC1-high subgroup – and evaluation of interactive effects between ERCC1 expression and treatment type – was limited, said Syma Iqbal, MD, of the University of Southern California, Los Angeles, and coinvestigators.
“Unfortunately, this study did not validate or identify ERCC1 as a predictive marker of platinum sensitivity in upper GI tumors,” they wrote in the Journal of Clinical Oncology. However, “it did support the use of FOLFOX, a platinum-containing regimen, as a standard and superior frontline regimen, compared with the non–platinum-containing IT.”
Across all patients in the FOLFOX arm, the median progression-free survival (PFS) was significantly longer – 5.7 months versus 2.9 months in the IT arm (hazard ratio, 0.71; P = .02). The median overall survival was greater with FOLFOX as well, though this difference – 11.4 months versus 8.7 months – did not reach statistical significance. Similarly, in the ERCC1-low subgroup, the median PFS in patients receiving FOLFOX was statistically superior to IT – 5.9 months versus 2.8 months – and overall survival was better as well, though this latter difference was not statistically significant.
In the ERCC1-high subgroup, the median PFS was similar in the FOLFOX and IT arms (4.7 months vs. 5.3 months), the investigators noted. They plotted PFS within ERCC1 quartiles and found a consistent pattern of improved PFS in the FOLFOX versus IT arm, and “thus, little evidence of differential treatment effects on PFS across ERCC1 levels in this population.”
Regarding safety, the investigators noted, of 91 patients who completed protocol therapy in the FOLFOX arm and were analyzed for adverse events, 3 treatment-related deaths were reported and 9 additional patients experienced grade 4 adverse events. Of 98 patients assessed for adverse events in the IT arm, 3 treatment-related deaths were reported and 14 additional patients experienced grade 4 adverse events.
To be eligible for the study patients had to have a Zubrod performance status of 0-1 and been either treatment naive or have completed adjuvant therapy at least 180 days prior to enrollment.
The study was supported by the National Cancer Institute. Dr. Iqbal reported receiving honoraria from Celgene, Eisai, and F. Hoffmann–La Roche; serving a consulting or advisory role for F. Hoffmann–La Roche; serving on the speakers’ bureau for Celgene and Eisai; and receiving research funding from Bayer and Onyx.
SOURCE: Iqbal S et al. J Clin Oncol. 2019 Dec 9. doi: 10.1200/JCO.19.00925.
A study that aimed to validate intratumoral ERCC1 levels as a predictive marker of platinum sensitivity in upper GI tumors failed to do so, but it did reach one firm conclusion: That platinum therapy with fluorouracil, leucovorin, and oxaliplatin (FOLFOX) was superior in efficacy to a non–platinum-containing regimen of irinotecan and docetaxel (IT).
Approximately 200 untreated patients with unresectable advanced or metastatic HER2-negative adenocarcinoma of the esophagus, stomach or gastroesophageal junction were evaluated in the phase 2 study for mRNA expression of ERCC1 level and then randomized to either a platinum-containing or non–platinum-containing treatment arm with stratification for ERCC1 level (either low, with levels less than 1.7, or high, meaning 1.7 or more).
In retrospective studies of patients with gastric cancer, levels of gene expression of ERCC1 within the primary tumor have had a significant inverse relationship to response to platinum compounds and overall survival; low ERCC1 expression, in other words, has been associated with higher response rates and better survival. This inverse relationship of expression of the ERCC1 gene and platinum sensitivity has been demonstrated in colon cancer and other tumor types as well.
The problem is, approximately 86% of the patients in this phase 2, randomized study had ERCC1 values lower than 1.7 – many more than the investigators anticipated based on data from prior studies. (They expected a 50-50 distribution, roughly.) The predominance of low ERCC1 mRNA expression meant that evaluation of the ERCC1-high subgroup – and evaluation of interactive effects between ERCC1 expression and treatment type – was limited, said Syma Iqbal, MD, of the University of Southern California, Los Angeles, and coinvestigators.
“Unfortunately, this study did not validate or identify ERCC1 as a predictive marker of platinum sensitivity in upper GI tumors,” they wrote in the Journal of Clinical Oncology. However, “it did support the use of FOLFOX, a platinum-containing regimen, as a standard and superior frontline regimen, compared with the non–platinum-containing IT.”
Across all patients in the FOLFOX arm, the median progression-free survival (PFS) was significantly longer – 5.7 months versus 2.9 months in the IT arm (hazard ratio, 0.71; P = .02). The median overall survival was greater with FOLFOX as well, though this difference – 11.4 months versus 8.7 months – did not reach statistical significance. Similarly, in the ERCC1-low subgroup, the median PFS in patients receiving FOLFOX was statistically superior to IT – 5.9 months versus 2.8 months – and overall survival was better as well, though this latter difference was not statistically significant.
In the ERCC1-high subgroup, the median PFS was similar in the FOLFOX and IT arms (4.7 months vs. 5.3 months), the investigators noted. They plotted PFS within ERCC1 quartiles and found a consistent pattern of improved PFS in the FOLFOX versus IT arm, and “thus, little evidence of differential treatment effects on PFS across ERCC1 levels in this population.”
Regarding safety, the investigators noted, of 91 patients who completed protocol therapy in the FOLFOX arm and were analyzed for adverse events, 3 treatment-related deaths were reported and 9 additional patients experienced grade 4 adverse events. Of 98 patients assessed for adverse events in the IT arm, 3 treatment-related deaths were reported and 14 additional patients experienced grade 4 adverse events.
To be eligible for the study patients had to have a Zubrod performance status of 0-1 and been either treatment naive or have completed adjuvant therapy at least 180 days prior to enrollment.
The study was supported by the National Cancer Institute. Dr. Iqbal reported receiving honoraria from Celgene, Eisai, and F. Hoffmann–La Roche; serving a consulting or advisory role for F. Hoffmann–La Roche; serving on the speakers’ bureau for Celgene and Eisai; and receiving research funding from Bayer and Onyx.
SOURCE: Iqbal S et al. J Clin Oncol. 2019 Dec 9. doi: 10.1200/JCO.19.00925.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Metformin after GDM: Lessons from landmark diabetes prevention trial
WASHINGTON – Metformin’s role in preventing or delaying the onset of type 2 diabetes in women with a history of gestational diabetes mellitus has been firmly established by the Diabetes Prevention Program (DPP) trial – most recently, by 15-year follow-up data reported this year – and the drug should be front and center for clinicians who hope to stave off the “remarkable” incidence of type 2 diabetes after GDM, Robert E. Ratner, MD, maintained at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.

The DPP included “the single largest population of women with a history of GDM that’s been looked at in a randomized controlled trial,” and considering its multiethnic population, the trial offers a reliable representative sample to ponder today when evaluating long-term use of metformin after GDM, said Dr. Ratner, a principal investigator of the National Institutes of Health–sponsored DPP and the DPP Outcomes Study and a former chief scientific & medical officer for the American Diabetes Association.
The drug stacked up equally to lifestyle interventions among DPP participants who had a history of GDM, but it’s important to appreciate that these interventions were intensive and that metformin is inexpensive, well tolerated, and “has a long safety record,” he said.
Results of follow-up out to 15 years
Of the more than 3,000 men and women enrolled in the landmark DPP, conducted during 1996-2001, 350 were women with a documented history of GDM and over 1,400 were women who had deliveries but no history of GDM. All participants had impaired glucose tolerance – defined for the trial as having both a fasting plasma glucose value of 95-125 mg/dL and a 2-hour value of 140-199 mg/dL after a 75-g glucose load – and were randomized to placebo, metformin, or intensive lifestyle intervention.
Metformin therapy reduced the incidence of diabetes by approximately 50% in women with a history of GDM, compared with the placebo group – as did lifestyle – over 3 years. The number needed to treat to prevent one case of diabetes was five. Women without a history of GDM, on the other hand, saw only a 14% reduction with metformin when compared with placebo (and a 49% reduction with lifestyle).
“In women with a history of GDM ... one pill twice a day for $4 a month worked as well as intensive lifestyle [change],” Dr. Ratner said, referring to the initial GDM-specific analysis of DPP data published in 2008 (J Clin Endocrinol Metab. 2008;93[12]:4774-9).
In a 10-year postrandomization follow-up, published in 2015, both metformin and lifestyle continued to be equally effective for the GDM group, reducing the progression to diabetes by 40% and 35%, respectively (J Clin Endocrinol Metab. 2015;100:1646-53). The number needed to treat to prevent one case of diabetes was seven. (Among women without a history of GDM, metformin did not reduce progression to diabetes.)
A recent DPP Outcomes Study analysis of metformin’s impact on diabetes prevention at 15 years, moreover, showed a 41% risk reduction among women with a history of GDM (Diabetes Care. 2019;42[4]:601-8).
Advice on prescribing metformin prophylactically
Asked after his presentation whether women with a history of GDM and either an elevated fasting plasma glucose value or an elevated 2-hour oral glucose tolerance test (GTT) value – or neither of the two – would benefit from taking metformin, Dr. Ratner said that “we’re stuck with inclusion criteria of the DPP, in which they had to meet both criteria ... What I’d say, though, is that not everyone with a history of GDM needs to be on metformin prophylactically. But [for women who have] prediabetes as defined by the ADA, the cost-benefit analysis points toward metformin.”
And with respect to early initiation and long-term use of the drug, “I would have absolutely no qualms about medicating a 25-year-old who had developed GDM and who in the postpartum period has prediabetes,” Dr. Ratner said during an open discussion. “She’s actually at the highest risk for developing type 2 very early.”
Kim Boggess, MD, who also presented on long-term use of metformin after GDM, said in the discussion period that she is often quick to recommend metformin therapy to her patients who have an elevated fasting plasma glucose value in the postpartum period, even when a 75-g oral GTT has not yet been performed. (The ADA and the American College of Obstetricians and Gynecologists recommend completion of an oral GTT at 4-12 weeks postpartum after GDM.)
“I start them [on metformin] especially if they’ve had a cesarean section. Even 2, 3, 4 weeks of profound hyperglycemia could have potentially deleterious effects,” said Dr. Boggess, professor and maternal-fetal medicine program director at the University of North Carolina, Chapel Hill. “If someone comes in [shortly after] and looks like they have pristine control, then it might be worth stopping the metformin for 3-5 days (and retesting).”
Dr. Ratner said that, in this clinical scenario, he would first ensure that the fasting glucose value “is a true fasting glucose” and “if it’s substantially elevated – I’m talking 100, 105, 110 mg/dL – I’d start metformin, and I’m not even sure I’d do the GTT.” But, he advised, “if you’re going to do the GTT, I’d stop the metformin the day before.”
In her presentation, Dr. Boggess pointed out that metformin wasn’t shown to be superior to lifestyle interventions in the DPP for preventing progression to type 2 DM, and that some women are more motivated for intensive lifestyle change than others. The ADA recommends, in fact, that either metformin or lifestyle interventions be prescribed to women with a history of GDM who are found to have prediabetes.

There are no data to support the use of metformin either during or after pregnancy to improve weight loss or reduce weight retention following pregnancy, but at least several studies have shown that lifestyle interventions are effective, she noted.
What is needed, Dr. Boggess said, are more data on the effects of metformin on cardiovascular disease risk, as well as larger studies of metformin in the postpartum period “to help us determine the best dose.” Some research on metformin use in the postpartum period has reported gastrointestinal side effects and dissatisfaction, she noted.
Dr. Ratner said that metformin’s main drawback is the need for occasional testing of B12 levels. Regarding weight loss and what was observed in the DPP, he said, women with a history of GDM who were randomized to intensive lifestyle interventions did not lose as much weight as women without a history of GDM.
Women who entered the DPP with a GDM history, he noted in his presentation, were essentially a “cohort of survivors.” They had an average age of 43 (compared with 52 years in the parous women without GDM) and a mean interval from the index GDM pregnancy of 11 years, which means that women with the highest risk of diabetes conversion were excluded, Dr. Ratner said.
Age was the only significantly different baseline characteristic between parous women with and without GDM, he noted. Women with a history of GDM who were randomized to placebo had a 71% higher incidence of diabetes than women without such a history – a striking natural history, Dr. Ratner said.
He and Dr. Boggess each reported that they have no financial or other interests that pose a conflict of interest.
WASHINGTON – Metformin’s role in preventing or delaying the onset of type 2 diabetes in women with a history of gestational diabetes mellitus has been firmly established by the Diabetes Prevention Program (DPP) trial – most recently, by 15-year follow-up data reported this year – and the drug should be front and center for clinicians who hope to stave off the “remarkable” incidence of type 2 diabetes after GDM, Robert E. Ratner, MD, maintained at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
The DPP included “the single largest population of women with a history of GDM that’s been looked at in a randomized controlled trial,” and considering its multiethnic population, the trial offers a reliable representative sample to ponder today when evaluating long-term use of metformin after GDM, said Dr. Ratner, a principal investigator of the National Institutes of Health–sponsored DPP and the DPP Outcomes Study and a former chief scientific & medical officer for the American Diabetes Association.
The drug stacked up equally to lifestyle interventions among DPP participants who had a history of GDM, but it’s important to appreciate that these interventions were intensive and that metformin is inexpensive, well tolerated, and “has a long safety record,” he said.
Results of follow-up out to 15 years
Of the more than 3,000 men and women enrolled in the landmark DPP, conducted during 1996-2001, 350 were women with a documented history of GDM and over 1,400 were women who had deliveries but no history of GDM. All participants had impaired glucose tolerance – defined for the trial as having both a fasting plasma glucose value of 95-125 mg/dL and a 2-hour value of 140-199 mg/dL after a 75-g glucose load – and were randomized to placebo, metformin, or intensive lifestyle intervention.
Metformin therapy reduced the incidence of diabetes by approximately 50% in women with a history of GDM, compared with the placebo group – as did lifestyle – over 3 years. The number needed to treat to prevent one case of diabetes was five. Women without a history of GDM, on the other hand, saw only a 14% reduction with metformin when compared with placebo (and a 49% reduction with lifestyle).
“In women with a history of GDM ... one pill twice a day for $4 a month worked as well as intensive lifestyle [change],” Dr. Ratner said, referring to the initial GDM-specific analysis of DPP data published in 2008 (J Clin Endocrinol Metab. 2008;93[12]:4774-9).
In a 10-year postrandomization follow-up, published in 2015, both metformin and lifestyle continued to be equally effective for the GDM group, reducing the progression to diabetes by 40% and 35%, respectively (J Clin Endocrinol Metab. 2015;100:1646-53). The number needed to treat to prevent one case of diabetes was seven. (Among women without a history of GDM, metformin did not reduce progression to diabetes.)
A recent DPP Outcomes Study analysis of metformin’s impact on diabetes prevention at 15 years, moreover, showed a 41% risk reduction among women with a history of GDM (Diabetes Care. 2019;42[4]:601-8).
Advice on prescribing metformin prophylactically
Asked after his presentation whether women with a history of GDM and either an elevated fasting plasma glucose value or an elevated 2-hour oral glucose tolerance test (GTT) value – or neither of the two – would benefit from taking metformin, Dr. Ratner said that “we’re stuck with inclusion criteria of the DPP, in which they had to meet both criteria ... What I’d say, though, is that not everyone with a history of GDM needs to be on metformin prophylactically. But [for women who have] prediabetes as defined by the ADA, the cost-benefit analysis points toward metformin.”
And with respect to early initiation and long-term use of the drug, “I would have absolutely no qualms about medicating a 25-year-old who had developed GDM and who in the postpartum period has prediabetes,” Dr. Ratner said during an open discussion. “She’s actually at the highest risk for developing type 2 very early.”
Kim Boggess, MD, who also presented on long-term use of metformin after GDM, said in the discussion period that she is often quick to recommend metformin therapy to her patients who have an elevated fasting plasma glucose value in the postpartum period, even when a 75-g oral GTT has not yet been performed. (The ADA and the American College of Obstetricians and Gynecologists recommend completion of an oral GTT at 4-12 weeks postpartum after GDM.)
“I start them [on metformin] especially if they’ve had a cesarean section. Even 2, 3, 4 weeks of profound hyperglycemia could have potentially deleterious effects,” said Dr. Boggess, professor and maternal-fetal medicine program director at the University of North Carolina, Chapel Hill. “If someone comes in [shortly after] and looks like they have pristine control, then it might be worth stopping the metformin for 3-5 days (and retesting).”
Dr. Ratner said that, in this clinical scenario, he would first ensure that the fasting glucose value “is a true fasting glucose” and “if it’s substantially elevated – I’m talking 100, 105, 110 mg/dL – I’d start metformin, and I’m not even sure I’d do the GTT.” But, he advised, “if you’re going to do the GTT, I’d stop the metformin the day before.”
In her presentation, Dr. Boggess pointed out that metformin wasn’t shown to be superior to lifestyle interventions in the DPP for preventing progression to type 2 DM, and that some women are more motivated for intensive lifestyle change than others. The ADA recommends, in fact, that either metformin or lifestyle interventions be prescribed to women with a history of GDM who are found to have prediabetes.
There are no data to support the use of metformin either during or after pregnancy to improve weight loss or reduce weight retention following pregnancy, but at least several studies have shown that lifestyle interventions are effective, she noted.
What is needed, Dr. Boggess said, are more data on the effects of metformin on cardiovascular disease risk, as well as larger studies of metformin in the postpartum period “to help us determine the best dose.” Some research on metformin use in the postpartum period has reported gastrointestinal side effects and dissatisfaction, she noted.
Dr. Ratner said that metformin’s main drawback is the need for occasional testing of B12 levels. Regarding weight loss and what was observed in the DPP, he said, women with a history of GDM who were randomized to intensive lifestyle interventions did not lose as much weight as women without a history of GDM.
Women who entered the DPP with a GDM history, he noted in his presentation, were essentially a “cohort of survivors.” They had an average age of 43 (compared with 52 years in the parous women without GDM) and a mean interval from the index GDM pregnancy of 11 years, which means that women with the highest risk of diabetes conversion were excluded, Dr. Ratner said.
Age was the only significantly different baseline characteristic between parous women with and without GDM, he noted. Women with a history of GDM who were randomized to placebo had a 71% higher incidence of diabetes than women without such a history – a striking natural history, Dr. Ratner said.
He and Dr. Boggess each reported that they have no financial or other interests that pose a conflict of interest.
WASHINGTON – Metformin’s role in preventing or delaying the onset of type 2 diabetes in women with a history of gestational diabetes mellitus has been firmly established by the Diabetes Prevention Program (DPP) trial – most recently, by 15-year follow-up data reported this year – and the drug should be front and center for clinicians who hope to stave off the “remarkable” incidence of type 2 diabetes after GDM, Robert E. Ratner, MD, maintained at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
The DPP included “the single largest population of women with a history of GDM that’s been looked at in a randomized controlled trial,” and considering its multiethnic population, the trial offers a reliable representative sample to ponder today when evaluating long-term use of metformin after GDM, said Dr. Ratner, a principal investigator of the National Institutes of Health–sponsored DPP and the DPP Outcomes Study and a former chief scientific & medical officer for the American Diabetes Association.
The drug stacked up equally to lifestyle interventions among DPP participants who had a history of GDM, but it’s important to appreciate that these interventions were intensive and that metformin is inexpensive, well tolerated, and “has a long safety record,” he said.
Results of follow-up out to 15 years
Of the more than 3,000 men and women enrolled in the landmark DPP, conducted during 1996-2001, 350 were women with a documented history of GDM and over 1,400 were women who had deliveries but no history of GDM. All participants had impaired glucose tolerance – defined for the trial as having both a fasting plasma glucose value of 95-125 mg/dL and a 2-hour value of 140-199 mg/dL after a 75-g glucose load – and were randomized to placebo, metformin, or intensive lifestyle intervention.
Metformin therapy reduced the incidence of diabetes by approximately 50% in women with a history of GDM, compared with the placebo group – as did lifestyle – over 3 years. The number needed to treat to prevent one case of diabetes was five. Women without a history of GDM, on the other hand, saw only a 14% reduction with metformin when compared with placebo (and a 49% reduction with lifestyle).
“In women with a history of GDM ... one pill twice a day for $4 a month worked as well as intensive lifestyle [change],” Dr. Ratner said, referring to the initial GDM-specific analysis of DPP data published in 2008 (J Clin Endocrinol Metab. 2008;93[12]:4774-9).
In a 10-year postrandomization follow-up, published in 2015, both metformin and lifestyle continued to be equally effective for the GDM group, reducing the progression to diabetes by 40% and 35%, respectively (J Clin Endocrinol Metab. 2015;100:1646-53). The number needed to treat to prevent one case of diabetes was seven. (Among women without a history of GDM, metformin did not reduce progression to diabetes.)
A recent DPP Outcomes Study analysis of metformin’s impact on diabetes prevention at 15 years, moreover, showed a 41% risk reduction among women with a history of GDM (Diabetes Care. 2019;42[4]:601-8).
Advice on prescribing metformin prophylactically
Asked after his presentation whether women with a history of GDM and either an elevated fasting plasma glucose value or an elevated 2-hour oral glucose tolerance test (GTT) value – or neither of the two – would benefit from taking metformin, Dr. Ratner said that “we’re stuck with inclusion criteria of the DPP, in which they had to meet both criteria ... What I’d say, though, is that not everyone with a history of GDM needs to be on metformin prophylactically. But [for women who have] prediabetes as defined by the ADA, the cost-benefit analysis points toward metformin.”
And with respect to early initiation and long-term use of the drug, “I would have absolutely no qualms about medicating a 25-year-old who had developed GDM and who in the postpartum period has prediabetes,” Dr. Ratner said during an open discussion. “She’s actually at the highest risk for developing type 2 very early.”
Kim Boggess, MD, who also presented on long-term use of metformin after GDM, said in the discussion period that she is often quick to recommend metformin therapy to her patients who have an elevated fasting plasma glucose value in the postpartum period, even when a 75-g oral GTT has not yet been performed. (The ADA and the American College of Obstetricians and Gynecologists recommend completion of an oral GTT at 4-12 weeks postpartum after GDM.)
“I start them [on metformin] especially if they’ve had a cesarean section. Even 2, 3, 4 weeks of profound hyperglycemia could have potentially deleterious effects,” said Dr. Boggess, professor and maternal-fetal medicine program director at the University of North Carolina, Chapel Hill. “If someone comes in [shortly after] and looks like they have pristine control, then it might be worth stopping the metformin for 3-5 days (and retesting).”
Dr. Ratner said that, in this clinical scenario, he would first ensure that the fasting glucose value “is a true fasting glucose” and “if it’s substantially elevated – I’m talking 100, 105, 110 mg/dL – I’d start metformin, and I’m not even sure I’d do the GTT.” But, he advised, “if you’re going to do the GTT, I’d stop the metformin the day before.”
In her presentation, Dr. Boggess pointed out that metformin wasn’t shown to be superior to lifestyle interventions in the DPP for preventing progression to type 2 DM, and that some women are more motivated for intensive lifestyle change than others. The ADA recommends, in fact, that either metformin or lifestyle interventions be prescribed to women with a history of GDM who are found to have prediabetes.
There are no data to support the use of metformin either during or after pregnancy to improve weight loss or reduce weight retention following pregnancy, but at least several studies have shown that lifestyle interventions are effective, she noted.
What is needed, Dr. Boggess said, are more data on the effects of metformin on cardiovascular disease risk, as well as larger studies of metformin in the postpartum period “to help us determine the best dose.” Some research on metformin use in the postpartum period has reported gastrointestinal side effects and dissatisfaction, she noted.
Dr. Ratner said that metformin’s main drawback is the need for occasional testing of B12 levels. Regarding weight loss and what was observed in the DPP, he said, women with a history of GDM who were randomized to intensive lifestyle interventions did not lose as much weight as women without a history of GDM.
Women who entered the DPP with a GDM history, he noted in his presentation, were essentially a “cohort of survivors.” They had an average age of 43 (compared with 52 years in the parous women without GDM) and a mean interval from the index GDM pregnancy of 11 years, which means that women with the highest risk of diabetes conversion were excluded, Dr. Ratner said.
Age was the only significantly different baseline characteristic between parous women with and without GDM, he noted. Women with a history of GDM who were randomized to placebo had a 71% higher incidence of diabetes than women without such a history – a striking natural history, Dr. Ratner said.
He and Dr. Boggess each reported that they have no financial or other interests that pose a conflict of interest.
REPORTING FROM THE DPSG-NA 2019
Data build on cardiovascular disease risk after GDM, HDP
WASHINGTON – Cardiovascular risk factors may be elevated “as soon as the first postpartum year” in women who have gestational diabetes or hypertensive disorders of pregnancy, recent findings have affirmed, Deborah B. Ehrenthal, MD, MPH, said at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
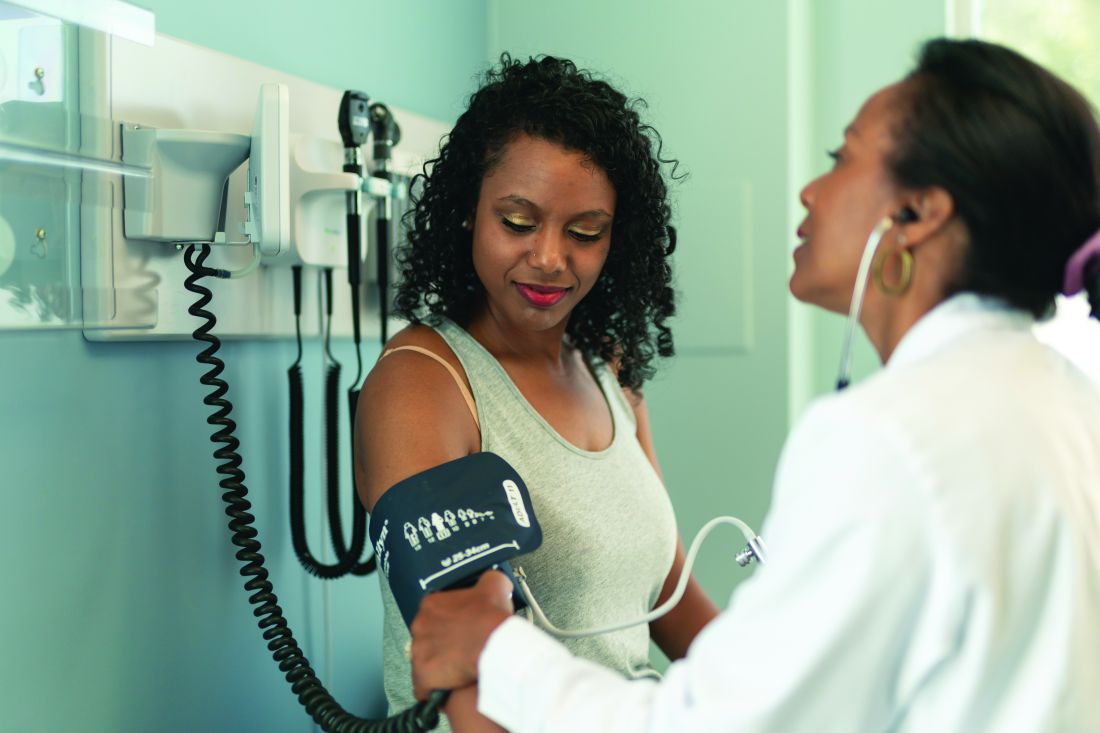
Dr. Ehrenthal was one of several researchers who urged innovative strategies and improved care coordination to boost women’s follow-up after gestational diabetes mellitus (GDM) and other adverse pregnancy outcomes and complications. “The metabolic stress of pregnancy can uncover underlying susceptibilities,” she said. “And ”
Evidence that adverse pregnancy outcomes – including GDM and hypertensive disorders of pregnancy (HDP) – can elevate cardiovascular risk comes most recently from the Nulliparous Pregnancy Outcomes Study – Monitoring Mothers to be Heart Health Study (nuMoM2b–HHS study), a prospective observational cohort that followed 4,484 women 2-7 years after their first pregnancy. Women had a follow-up exam, with blood pressure and anthropometric measurements and clinical/biological testing, an average of 3 years post partum.
An analysis published in October 2019 in the Journal of the American Heart Association shows that women with HDP (including preeclampsia and gestational hypertension) had a relative risk of hypertension of 2.5 at follow-up, compared with women without HDP. Women who had preeclampsia specifically were 2.3 times as likely as were women who did not have preeclampsia to have incident hypertension at follow-up, said Dr. Ehrenthal, a coinvestigator of the study.
The analysis focused on incident hypertension as the primary outcome, and adjusted for age, body mass index, and other important cardiovascular disease risk factors, she noted. Researchers utilized the diagnostic threshold for hypertension extant at the time of study design: A systolic blood pressure of 140 mm Hg or greater, or a diastolic BP of 90 mm Hg or greater (J Am Heart Assoc. 2019;8:e013092).
HDP was the most common adverse pregnancy outcome in the nuMoM2b–HHS study (14%). Among all participants, 4% had GDM. Approximately 82% had neither HDP nor GDM. Other adverse pregnancy outcomes included in the analysis were preterm birth, small-for-gestational-age birth, and stillbirth.
Additional preliminary estimates presented by Dr. Ehrenthal show that, based on the new (2017) lower threshold for hypertension – 130 mg Hg systolic or 80 mm Hg diastolic – the disorder afflicted 37% of women who had experienced HDP (relative risk 2.1), and 32% of women who had GDM (RR 1.8). Prediabetes/diabetes (using a fasting blood glucose threshold of 100 mg/dL) at follow-up affected an estimated 21% of women who had HDP (RR 1.4) and 38% of women who had GDM (RR 2.5).
Notably, across the entire study cohort, 20% had hypertension at follow-up, “which is extraordinary” considering the short time frame from pregnancy and the young age of the study population – a mean maternal age of 27 years, said Dr. Ehrenthal, associate professor of population health sciences and obstetrics & gynecology at the University of Wisconsin, Madison.
Also across the cohort, 15% had prediabetes/diabetes at follow-up. “We need to think about women more generally,” she cautioned. “While we recognize the significant elevated risk of HDP and GDM [for the development of subsequent hypertension and cardiovascular risk], we will miss a lot of women [if we focus only on the history of HDP and GDM.]”
The majority of women found to have hypertension or prediabetes/diabetes at follow-up had experienced neither HDP nor GDM, but a good many of them (47% of those who had hypertension and 47% of those found to have prediabetes/diabetes) had a BMI of 30 or above, Dr. Ehrenthal said at the DPSG-NA meeting.
Nurses Health Study, hyperglycemia and adverse pregnancy outcome follow-up data
The new findings from the nuMoM2b–HHS study add to a robust and growing body of evidence that pregnancy is an important window to future health, and that follow up and screening after GDM and HDP are crucial.
Regarding GDM specifically, “there’s quite a bit of literature by now demonstrating that GDM history is a risk factor for hypertension, even 1-2 years post partum, and that the risk is elevated as well for dyslipidemia and vascular dysfunction,” Deirdre K. Tobias, D.Sc., an epidemiologist at Brigham and Women’s Hospital and assistant professor of nutrition at Harvard TH Chan School of Public Health, Boston, said at the DPSG meeting.
An analysis of the Nurses Health Study II (NHS II) cohort published in 2017 found a 40% higher relative risk of cardiovascular disease events (largely myocardial infarction) in women who had GDM, compared with women who did not have GDM over a median follow-up of 26 years. This was after adjustments were made for age, time since pregnancy, menopausal status, family history of MI or stroke, hypertension in pregnancy, white race/ethnicity, prepregnancy BMI, and other factors (JAMA Intern Med. 2017;177[12]:1735-42).
The NHS data also have shown, however, that the elevated risk for cardiovascular disease after a GDM pregnancy “can be mitigated by adopting a healthy lifestyle,” said Dr. Tobias, lead author of the 2017 NHS II analysis. Adjustments for postpregnancy weight gain and lifestyle factors attenuated the relative risk of cardiovascular disease events after a GDM pregnancy to a 30% increased risk.
Dr. Tobias and colleagues currently are looking within the NHS cohort for “metabolomic signatures” or signals – various amino acid and lipid metabolites – to identify the progression of GDM to type 2 diabetes. Metabolomics “may help further refine our understanding of the long-term links between GDM and prevention of type 2 diabetes and of cardiovascular disease in mothers,” she said.
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Follow-Up Study, in the meantime, is documenting associations of maternal glucose levels during pregnancy not only with prediabetes or type 2 diabetes 10-14 years later, but also with measures of cardiovascular risk in mothers 10-14 years later.
Just as perinatal outcomes were strongly associated with glucose as a continuous variable in the original HAPO study, “it’s clear there’s a progressive increase in the risk of [later] disorders of glucose metabolism as [fasting blood glucose levels and 1- and-2-hour glucose values] in pregnancy are higher,” said Boyd E. Metzger, MD, the Tom D. Spies emeritus professor of metabolism and nutrition at Northwestern University, Chicago, and principal investigator of the original HAPO study and its follow up.

“Another message is that the more normal you are in pregnancy, the more normal you will be many years later. Good values [during pregnancy] produce good outcomes.”
Currently unpublished data from the HAPO Follow-Up Study are being analyzed, but it appears thus far that GDM is not associated with hypertension (per the old diagnostic threshold) in this cohort after adjustment for maternal age, BMI, smoking, and family history of hypertension. GDM appears to be a significant risk factor for dyslipidemia, however. HDL cholesterol at follow-up was significantly lower for mothers who had GDM compared with those without, whereas LDL cholesterol and triglycerides at follow-up were significantly higher for mothers with GDM, Dr. Metzger said.
Racial/ethnic disparities, postpartum care
Neither long-term study – the NHS II or the HAPO Follow-Up Study – has looked at racial and ethnic differences. The HAPO cohort is racially-ethnically diverse but the NHS II cohort is predominantly white women.
Research suggests that GDM is a heterogeneous condition with some unique phenotypes in subgroups that vary by race and ethnicity. And just as there appear to be racial-ethnic differences in the pathophysiology of GDM, there appear to be racial-ethnic differences in the progression to type 2 diabetes – a known risk factor for cardiovascular disease, said Monique Henderson, PhD, a research scientist at Kaiser Permanente Northern California (KPNC).
On the broadest level, while Asian Americans have the highest prevalence of GDM, African Americans have the highest rates of progressing to type 2 diabetes, Dr. Henderson said. Disparities “may [stem from] metabolic differences in terms of insulin resistance and secretion that are different between pregnancy and the postpartum period, and that might vary [across racial-ethnic subgroups],” she said. Lifestyle differences and variation in postpartum screening rates also may play a role.
At KPNC, where women with GDM receive calls and letters reminding them of the need for postpartum screening, only 48% overall completed an oral glucose tolerance test at 4-12 weeks post partum, as recommended by both the American Diabetes Association and the American College of Obstetricians and Gynecologists. Both before and after adjustment for education, attendance at a postpartum visit, and other variables, Chinese women were most likely to have screening, and black women were least likely, said Dr. Henderson, referring to ongoing research.
A study Dr. Ehrenthal led of women with GDM or HDP recruited from the postpartum service of a large community-based, academic obstetrical hospital in Delaware showed that while nearly all women attended a 6-week postpartum visit with their ob.gyns., 59% of women with GDM had not yet completed diabetes screening when they were interviewed 3 months post partum. Most women with HDP indicated they had follow-up blood pressure testing, and just over half of women with either diagnosis recalled having ever had lipid testing (J Women’s Health 2014;23[9]:760-4).
Women least likely to complete screening tests were those who had no college education, those who had less than a high school level of health literacy, and those who were not privately insured, Dr. Ehrenthal said.
A large national study of privately insured women also found low rates of follow-up testing, however. While the majority of women with GDM had a postpartum visit with an obstetrician or primary care physician within a year after delivery, only a minority of women had a glycemic screening test completed (Obstet Gynecol. 2016;128[1]:159-67).
“We can’t place the blame on women,” Dr. Ehrenthal said. “We need increased attention to screening,” including screening for cardiovascular disease risk factors, and a “deliberate hand-off to primary care.”
For follow-up cardiovascular disease risk factor assessment after HDP, ACOG recommends periodic (perhaps annually) assessment and referral for treatment as needed, and the cardiology professional organizations recommend that pregnancy history be considered when assessing risk in order to decide on lipid treatment, she noted.
Each of the speakers reported that they have no financial or other interests that pose a conflict of interest. The HAPO Follow-Up Study is funded by the National Institute of Diabetes and Digestive and Kidney Diseases, and the nuMoM2b–HHS study has been funded by several National Institutes of Health institutes and other programs and initiatives.
WASHINGTON – Cardiovascular risk factors may be elevated “as soon as the first postpartum year” in women who have gestational diabetes or hypertensive disorders of pregnancy, recent findings have affirmed, Deborah B. Ehrenthal, MD, MPH, said at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
Dr. Ehrenthal was one of several researchers who urged innovative strategies and improved care coordination to boost women’s follow-up after gestational diabetes mellitus (GDM) and other adverse pregnancy outcomes and complications. “The metabolic stress of pregnancy can uncover underlying susceptibilities,” she said. “And ”
Evidence that adverse pregnancy outcomes – including GDM and hypertensive disorders of pregnancy (HDP) – can elevate cardiovascular risk comes most recently from the Nulliparous Pregnancy Outcomes Study – Monitoring Mothers to be Heart Health Study (nuMoM2b–HHS study), a prospective observational cohort that followed 4,484 women 2-7 years after their first pregnancy. Women had a follow-up exam, with blood pressure and anthropometric measurements and clinical/biological testing, an average of 3 years post partum.
An analysis published in October 2019 in the Journal of the American Heart Association shows that women with HDP (including preeclampsia and gestational hypertension) had a relative risk of hypertension of 2.5 at follow-up, compared with women without HDP. Women who had preeclampsia specifically were 2.3 times as likely as were women who did not have preeclampsia to have incident hypertension at follow-up, said Dr. Ehrenthal, a coinvestigator of the study.
The analysis focused on incident hypertension as the primary outcome, and adjusted for age, body mass index, and other important cardiovascular disease risk factors, she noted. Researchers utilized the diagnostic threshold for hypertension extant at the time of study design: A systolic blood pressure of 140 mm Hg or greater, or a diastolic BP of 90 mm Hg or greater (J Am Heart Assoc. 2019;8:e013092).
HDP was the most common adverse pregnancy outcome in the nuMoM2b–HHS study (14%). Among all participants, 4% had GDM. Approximately 82% had neither HDP nor GDM. Other adverse pregnancy outcomes included in the analysis were preterm birth, small-for-gestational-age birth, and stillbirth.
Additional preliminary estimates presented by Dr. Ehrenthal show that, based on the new (2017) lower threshold for hypertension – 130 mg Hg systolic or 80 mm Hg diastolic – the disorder afflicted 37% of women who had experienced HDP (relative risk 2.1), and 32% of women who had GDM (RR 1.8). Prediabetes/diabetes (using a fasting blood glucose threshold of 100 mg/dL) at follow-up affected an estimated 21% of women who had HDP (RR 1.4) and 38% of women who had GDM (RR 2.5).
Notably, across the entire study cohort, 20% had hypertension at follow-up, “which is extraordinary” considering the short time frame from pregnancy and the young age of the study population – a mean maternal age of 27 years, said Dr. Ehrenthal, associate professor of population health sciences and obstetrics & gynecology at the University of Wisconsin, Madison.
Also across the cohort, 15% had prediabetes/diabetes at follow-up. “We need to think about women more generally,” she cautioned. “While we recognize the significant elevated risk of HDP and GDM [for the development of subsequent hypertension and cardiovascular risk], we will miss a lot of women [if we focus only on the history of HDP and GDM.]”
The majority of women found to have hypertension or prediabetes/diabetes at follow-up had experienced neither HDP nor GDM, but a good many of them (47% of those who had hypertension and 47% of those found to have prediabetes/diabetes) had a BMI of 30 or above, Dr. Ehrenthal said at the DPSG-NA meeting.
Nurses Health Study, hyperglycemia and adverse pregnancy outcome follow-up data
The new findings from the nuMoM2b–HHS study add to a robust and growing body of evidence that pregnancy is an important window to future health, and that follow up and screening after GDM and HDP are crucial.
Regarding GDM specifically, “there’s quite a bit of literature by now demonstrating that GDM history is a risk factor for hypertension, even 1-2 years post partum, and that the risk is elevated as well for dyslipidemia and vascular dysfunction,” Deirdre K. Tobias, D.Sc., an epidemiologist at Brigham and Women’s Hospital and assistant professor of nutrition at Harvard TH Chan School of Public Health, Boston, said at the DPSG meeting.
An analysis of the Nurses Health Study II (NHS II) cohort published in 2017 found a 40% higher relative risk of cardiovascular disease events (largely myocardial infarction) in women who had GDM, compared with women who did not have GDM over a median follow-up of 26 years. This was after adjustments were made for age, time since pregnancy, menopausal status, family history of MI or stroke, hypertension in pregnancy, white race/ethnicity, prepregnancy BMI, and other factors (JAMA Intern Med. 2017;177[12]:1735-42).
The NHS data also have shown, however, that the elevated risk for cardiovascular disease after a GDM pregnancy “can be mitigated by adopting a healthy lifestyle,” said Dr. Tobias, lead author of the 2017 NHS II analysis. Adjustments for postpregnancy weight gain and lifestyle factors attenuated the relative risk of cardiovascular disease events after a GDM pregnancy to a 30% increased risk.
Dr. Tobias and colleagues currently are looking within the NHS cohort for “metabolomic signatures” or signals – various amino acid and lipid metabolites – to identify the progression of GDM to type 2 diabetes. Metabolomics “may help further refine our understanding of the long-term links between GDM and prevention of type 2 diabetes and of cardiovascular disease in mothers,” she said.
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Follow-Up Study, in the meantime, is documenting associations of maternal glucose levels during pregnancy not only with prediabetes or type 2 diabetes 10-14 years later, but also with measures of cardiovascular risk in mothers 10-14 years later.
Just as perinatal outcomes were strongly associated with glucose as a continuous variable in the original HAPO study, “it’s clear there’s a progressive increase in the risk of [later] disorders of glucose metabolism as [fasting blood glucose levels and 1- and-2-hour glucose values] in pregnancy are higher,” said Boyd E. Metzger, MD, the Tom D. Spies emeritus professor of metabolism and nutrition at Northwestern University, Chicago, and principal investigator of the original HAPO study and its follow up.
“Another message is that the more normal you are in pregnancy, the more normal you will be many years later. Good values [during pregnancy] produce good outcomes.”
Currently unpublished data from the HAPO Follow-Up Study are being analyzed, but it appears thus far that GDM is not associated with hypertension (per the old diagnostic threshold) in this cohort after adjustment for maternal age, BMI, smoking, and family history of hypertension. GDM appears to be a significant risk factor for dyslipidemia, however. HDL cholesterol at follow-up was significantly lower for mothers who had GDM compared with those without, whereas LDL cholesterol and triglycerides at follow-up were significantly higher for mothers with GDM, Dr. Metzger said.
Racial/ethnic disparities, postpartum care
Neither long-term study – the NHS II or the HAPO Follow-Up Study – has looked at racial and ethnic differences. The HAPO cohort is racially-ethnically diverse but the NHS II cohort is predominantly white women.
Research suggests that GDM is a heterogeneous condition with some unique phenotypes in subgroups that vary by race and ethnicity. And just as there appear to be racial-ethnic differences in the pathophysiology of GDM, there appear to be racial-ethnic differences in the progression to type 2 diabetes – a known risk factor for cardiovascular disease, said Monique Henderson, PhD, a research scientist at Kaiser Permanente Northern California (KPNC).
On the broadest level, while Asian Americans have the highest prevalence of GDM, African Americans have the highest rates of progressing to type 2 diabetes, Dr. Henderson said. Disparities “may [stem from] metabolic differences in terms of insulin resistance and secretion that are different between pregnancy and the postpartum period, and that might vary [across racial-ethnic subgroups],” she said. Lifestyle differences and variation in postpartum screening rates also may play a role.
At KPNC, where women with GDM receive calls and letters reminding them of the need for postpartum screening, only 48% overall completed an oral glucose tolerance test at 4-12 weeks post partum, as recommended by both the American Diabetes Association and the American College of Obstetricians and Gynecologists. Both before and after adjustment for education, attendance at a postpartum visit, and other variables, Chinese women were most likely to have screening, and black women were least likely, said Dr. Henderson, referring to ongoing research.
A study Dr. Ehrenthal led of women with GDM or HDP recruited from the postpartum service of a large community-based, academic obstetrical hospital in Delaware showed that while nearly all women attended a 6-week postpartum visit with their ob.gyns., 59% of women with GDM had not yet completed diabetes screening when they were interviewed 3 months post partum. Most women with HDP indicated they had follow-up blood pressure testing, and just over half of women with either diagnosis recalled having ever had lipid testing (J Women’s Health 2014;23[9]:760-4).
Women least likely to complete screening tests were those who had no college education, those who had less than a high school level of health literacy, and those who were not privately insured, Dr. Ehrenthal said.
A large national study of privately insured women also found low rates of follow-up testing, however. While the majority of women with GDM had a postpartum visit with an obstetrician or primary care physician within a year after delivery, only a minority of women had a glycemic screening test completed (Obstet Gynecol. 2016;128[1]:159-67).
“We can’t place the blame on women,” Dr. Ehrenthal said. “We need increased attention to screening,” including screening for cardiovascular disease risk factors, and a “deliberate hand-off to primary care.”
For follow-up cardiovascular disease risk factor assessment after HDP, ACOG recommends periodic (perhaps annually) assessment and referral for treatment as needed, and the cardiology professional organizations recommend that pregnancy history be considered when assessing risk in order to decide on lipid treatment, she noted.
Each of the speakers reported that they have no financial or other interests that pose a conflict of interest. The HAPO Follow-Up Study is funded by the National Institute of Diabetes and Digestive and Kidney Diseases, and the nuMoM2b–HHS study has been funded by several National Institutes of Health institutes and other programs and initiatives.
WASHINGTON – Cardiovascular risk factors may be elevated “as soon as the first postpartum year” in women who have gestational diabetes or hypertensive disorders of pregnancy, recent findings have affirmed, Deborah B. Ehrenthal, MD, MPH, said at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
Dr. Ehrenthal was one of several researchers who urged innovative strategies and improved care coordination to boost women’s follow-up after gestational diabetes mellitus (GDM) and other adverse pregnancy outcomes and complications. “The metabolic stress of pregnancy can uncover underlying susceptibilities,” she said. “And ”
Evidence that adverse pregnancy outcomes – including GDM and hypertensive disorders of pregnancy (HDP) – can elevate cardiovascular risk comes most recently from the Nulliparous Pregnancy Outcomes Study – Monitoring Mothers to be Heart Health Study (nuMoM2b–HHS study), a prospective observational cohort that followed 4,484 women 2-7 years after their first pregnancy. Women had a follow-up exam, with blood pressure and anthropometric measurements and clinical/biological testing, an average of 3 years post partum.
An analysis published in October 2019 in the Journal of the American Heart Association shows that women with HDP (including preeclampsia and gestational hypertension) had a relative risk of hypertension of 2.5 at follow-up, compared with women without HDP. Women who had preeclampsia specifically were 2.3 times as likely as were women who did not have preeclampsia to have incident hypertension at follow-up, said Dr. Ehrenthal, a coinvestigator of the study.
The analysis focused on incident hypertension as the primary outcome, and adjusted for age, body mass index, and other important cardiovascular disease risk factors, she noted. Researchers utilized the diagnostic threshold for hypertension extant at the time of study design: A systolic blood pressure of 140 mm Hg or greater, or a diastolic BP of 90 mm Hg or greater (J Am Heart Assoc. 2019;8:e013092).
HDP was the most common adverse pregnancy outcome in the nuMoM2b–HHS study (14%). Among all participants, 4% had GDM. Approximately 82% had neither HDP nor GDM. Other adverse pregnancy outcomes included in the analysis were preterm birth, small-for-gestational-age birth, and stillbirth.
Additional preliminary estimates presented by Dr. Ehrenthal show that, based on the new (2017) lower threshold for hypertension – 130 mg Hg systolic or 80 mm Hg diastolic – the disorder afflicted 37% of women who had experienced HDP (relative risk 2.1), and 32% of women who had GDM (RR 1.8). Prediabetes/diabetes (using a fasting blood glucose threshold of 100 mg/dL) at follow-up affected an estimated 21% of women who had HDP (RR 1.4) and 38% of women who had GDM (RR 2.5).
Notably, across the entire study cohort, 20% had hypertension at follow-up, “which is extraordinary” considering the short time frame from pregnancy and the young age of the study population – a mean maternal age of 27 years, said Dr. Ehrenthal, associate professor of population health sciences and obstetrics & gynecology at the University of Wisconsin, Madison.
Also across the cohort, 15% had prediabetes/diabetes at follow-up. “We need to think about women more generally,” she cautioned. “While we recognize the significant elevated risk of HDP and GDM [for the development of subsequent hypertension and cardiovascular risk], we will miss a lot of women [if we focus only on the history of HDP and GDM.]”
The majority of women found to have hypertension or prediabetes/diabetes at follow-up had experienced neither HDP nor GDM, but a good many of them (47% of those who had hypertension and 47% of those found to have prediabetes/diabetes) had a BMI of 30 or above, Dr. Ehrenthal said at the DPSG-NA meeting.
Nurses Health Study, hyperglycemia and adverse pregnancy outcome follow-up data
The new findings from the nuMoM2b–HHS study add to a robust and growing body of evidence that pregnancy is an important window to future health, and that follow up and screening after GDM and HDP are crucial.
Regarding GDM specifically, “there’s quite a bit of literature by now demonstrating that GDM history is a risk factor for hypertension, even 1-2 years post partum, and that the risk is elevated as well for dyslipidemia and vascular dysfunction,” Deirdre K. Tobias, D.Sc., an epidemiologist at Brigham and Women’s Hospital and assistant professor of nutrition at Harvard TH Chan School of Public Health, Boston, said at the DPSG meeting.
An analysis of the Nurses Health Study II (NHS II) cohort published in 2017 found a 40% higher relative risk of cardiovascular disease events (largely myocardial infarction) in women who had GDM, compared with women who did not have GDM over a median follow-up of 26 years. This was after adjustments were made for age, time since pregnancy, menopausal status, family history of MI or stroke, hypertension in pregnancy, white race/ethnicity, prepregnancy BMI, and other factors (JAMA Intern Med. 2017;177[12]:1735-42).
The NHS data also have shown, however, that the elevated risk for cardiovascular disease after a GDM pregnancy “can be mitigated by adopting a healthy lifestyle,” said Dr. Tobias, lead author of the 2017 NHS II analysis. Adjustments for postpregnancy weight gain and lifestyle factors attenuated the relative risk of cardiovascular disease events after a GDM pregnancy to a 30% increased risk.
Dr. Tobias and colleagues currently are looking within the NHS cohort for “metabolomic signatures” or signals – various amino acid and lipid metabolites – to identify the progression of GDM to type 2 diabetes. Metabolomics “may help further refine our understanding of the long-term links between GDM and prevention of type 2 diabetes and of cardiovascular disease in mothers,” she said.
The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Follow-Up Study, in the meantime, is documenting associations of maternal glucose levels during pregnancy not only with prediabetes or type 2 diabetes 10-14 years later, but also with measures of cardiovascular risk in mothers 10-14 years later.
Just as perinatal outcomes were strongly associated with glucose as a continuous variable in the original HAPO study, “it’s clear there’s a progressive increase in the risk of [later] disorders of glucose metabolism as [fasting blood glucose levels and 1- and-2-hour glucose values] in pregnancy are higher,” said Boyd E. Metzger, MD, the Tom D. Spies emeritus professor of metabolism and nutrition at Northwestern University, Chicago, and principal investigator of the original HAPO study and its follow up.
“Another message is that the more normal you are in pregnancy, the more normal you will be many years later. Good values [during pregnancy] produce good outcomes.”
Currently unpublished data from the HAPO Follow-Up Study are being analyzed, but it appears thus far that GDM is not associated with hypertension (per the old diagnostic threshold) in this cohort after adjustment for maternal age, BMI, smoking, and family history of hypertension. GDM appears to be a significant risk factor for dyslipidemia, however. HDL cholesterol at follow-up was significantly lower for mothers who had GDM compared with those without, whereas LDL cholesterol and triglycerides at follow-up were significantly higher for mothers with GDM, Dr. Metzger said.
Racial/ethnic disparities, postpartum care
Neither long-term study – the NHS II or the HAPO Follow-Up Study – has looked at racial and ethnic differences. The HAPO cohort is racially-ethnically diverse but the NHS II cohort is predominantly white women.
Research suggests that GDM is a heterogeneous condition with some unique phenotypes in subgroups that vary by race and ethnicity. And just as there appear to be racial-ethnic differences in the pathophysiology of GDM, there appear to be racial-ethnic differences in the progression to type 2 diabetes – a known risk factor for cardiovascular disease, said Monique Henderson, PhD, a research scientist at Kaiser Permanente Northern California (KPNC).
On the broadest level, while Asian Americans have the highest prevalence of GDM, African Americans have the highest rates of progressing to type 2 diabetes, Dr. Henderson said. Disparities “may [stem from] metabolic differences in terms of insulin resistance and secretion that are different between pregnancy and the postpartum period, and that might vary [across racial-ethnic subgroups],” she said. Lifestyle differences and variation in postpartum screening rates also may play a role.
At KPNC, where women with GDM receive calls and letters reminding them of the need for postpartum screening, only 48% overall completed an oral glucose tolerance test at 4-12 weeks post partum, as recommended by both the American Diabetes Association and the American College of Obstetricians and Gynecologists. Both before and after adjustment for education, attendance at a postpartum visit, and other variables, Chinese women were most likely to have screening, and black women were least likely, said Dr. Henderson, referring to ongoing research.
A study Dr. Ehrenthal led of women with GDM or HDP recruited from the postpartum service of a large community-based, academic obstetrical hospital in Delaware showed that while nearly all women attended a 6-week postpartum visit with their ob.gyns., 59% of women with GDM had not yet completed diabetes screening when they were interviewed 3 months post partum. Most women with HDP indicated they had follow-up blood pressure testing, and just over half of women with either diagnosis recalled having ever had lipid testing (J Women’s Health 2014;23[9]:760-4).
Women least likely to complete screening tests were those who had no college education, those who had less than a high school level of health literacy, and those who were not privately insured, Dr. Ehrenthal said.
A large national study of privately insured women also found low rates of follow-up testing, however. While the majority of women with GDM had a postpartum visit with an obstetrician or primary care physician within a year after delivery, only a minority of women had a glycemic screening test completed (Obstet Gynecol. 2016;128[1]:159-67).
“We can’t place the blame on women,” Dr. Ehrenthal said. “We need increased attention to screening,” including screening for cardiovascular disease risk factors, and a “deliberate hand-off to primary care.”
For follow-up cardiovascular disease risk factor assessment after HDP, ACOG recommends periodic (perhaps annually) assessment and referral for treatment as needed, and the cardiology professional organizations recommend that pregnancy history be considered when assessing risk in order to decide on lipid treatment, she noted.
Each of the speakers reported that they have no financial or other interests that pose a conflict of interest. The HAPO Follow-Up Study is funded by the National Institute of Diabetes and Digestive and Kidney Diseases, and the nuMoM2b–HHS study has been funded by several National Institutes of Health institutes and other programs and initiatives.
REPORTING FROM THE DPSG-NA 2019
Plug-unplug catheters: A good option, study suggests
Patients discharged with plug-unplug catheters after pelvic reconstructive surgery did not have adverse effects and reported less difficulty with catheter management and activities of daily living than women who were discharged with a continuous drainage catheter, a study has found.
“Plug-unplug catheter management technique is an acceptable method that does not appear to cause adverse events and may be considered for short-term catheterization after pelvic reconstructive surgery,” said Sarah Boyd, MD,* of the division of urogynecology at Hartford (Conn.) Hospital, and coinvestigators wrote in Obstetrics & Gynecology.
A total of 63 women who had a failed postoperative voiding trial after surgery for prolapse, with or without a concomitant incontinence procedure, were randomized to receive a 16-French transurethral catheter that was either attached to a leg bag (31 patients) or capped with a plastic plug (32 patients). Women in the second group – the plug-unplug group – were instructed to intermittently drain the bladder by uncapping the catheter when they felt the urge to void, or in the absence of urge, every 4 hours. All were scheduled for an outpatient voiding trial 5-7 days after discharge.
The first 30 study participants who did not require postoperative catheterization were assigned to a “reference,” or control, arm.
All patients in the study completed an activity assessment scale that covers both sedentary and ambulatory activities and is validated in women undergoing pelvic reconstructive surgery (Female Pelvic Med Reconstr Surg. 2012 Jul-Aug;18[4]:205-10); scores on the activity assessment scale (0-100) served as the primary outcome. Patients also answered questionnaires about their satisfaction and postoperative pain – and in the catheter arms, their experiences with the catheter.
The investigators found no difference in postoperative activity assessment scale scores (plug-unplug, 70; continuous drainage, 68; and reference arm, 79), However, patients with a continuous catheter indicated in the other evaluations that they had more difficulty managing the catheter and felt it impeded activities and the wearing of clothing they would otherwise use.
The activity scale, the investigators noted, may not have captured differences in activity during the first week postoperatively because patients are commonly instructed to restrict some of the activities assessed in the scale.
Regarding infection, there was no difference in the rate of positive urine cultures or treatment for urinary tract infection between the catheter arms during a 3-month follow-up period, “despite the theoretical concern that plugging and unplugging a catheter disrupts the closed catheter system, thus increasing the risk of infection,” the investigators wrote. However, the study was not powered to detect a difference in the risk of infection as it was for the primary outcome.
There was no difference between the catheter arms in the percentage of women who used narcotic or nonnarcotic pain medication, and overall patient satisfaction was similar.
The majority of patients passed their outpatient voiding trials at the initial postoperative visit (72% plug-unplug and 58% continuous). “Interestingly, patients in the plug-unplug arm had significantly higher voided volumes and almost half of the [postvoid residual volume] at the [5-7 day postoperative voiding trial] compared with the continuous drainage arm.” This suggests, Dr. Boyd and colleagues wrote, that patients using the plug-unplug catheter “could have undergone a voiding trial sooner.”
Offering patients options for catheter management is “valuable,” and providing them with a technique that is “easier to manage may decrease the catheter burden and improve patient experience,” the investigators added.

Luis E. Sanz, MD, director of urogynecology and pelvic reconstructive surgery at Virginia Hospital Center, Arlington, said, “I think that the plug and unplug drainage bladder catheter after reconstructive surgery is much more physiologic and ‘user friendly’ than continuous drainage. And [there is] no need for a leg bag, which is very inconvenient to the patient.”
Dr. Sanz, an Ob.Gyn. News Editorial Advisory Board member who was not involved in the study, was asked to provide a comment.
The authors did not report any potential conflicts of interest.
SOURCE: Boyd SS et al. Obstet Gynecol. 2019;134:1037-45.
* Correction, 10/24/2019: an earlier version misstated the chief investigator's name, which is Sarah Boyd, MD.
Patients discharged with plug-unplug catheters after pelvic reconstructive surgery did not have adverse effects and reported less difficulty with catheter management and activities of daily living than women who were discharged with a continuous drainage catheter, a study has found.
“Plug-unplug catheter management technique is an acceptable method that does not appear to cause adverse events and may be considered for short-term catheterization after pelvic reconstructive surgery,” said Sarah Boyd, MD,* of the division of urogynecology at Hartford (Conn.) Hospital, and coinvestigators wrote in Obstetrics & Gynecology.
A total of 63 women who had a failed postoperative voiding trial after surgery for prolapse, with or without a concomitant incontinence procedure, were randomized to receive a 16-French transurethral catheter that was either attached to a leg bag (31 patients) or capped with a plastic plug (32 patients). Women in the second group – the plug-unplug group – were instructed to intermittently drain the bladder by uncapping the catheter when they felt the urge to void, or in the absence of urge, every 4 hours. All were scheduled for an outpatient voiding trial 5-7 days after discharge.
The first 30 study participants who did not require postoperative catheterization were assigned to a “reference,” or control, arm.
All patients in the study completed an activity assessment scale that covers both sedentary and ambulatory activities and is validated in women undergoing pelvic reconstructive surgery (Female Pelvic Med Reconstr Surg. 2012 Jul-Aug;18[4]:205-10); scores on the activity assessment scale (0-100) served as the primary outcome. Patients also answered questionnaires about their satisfaction and postoperative pain – and in the catheter arms, their experiences with the catheter.
The investigators found no difference in postoperative activity assessment scale scores (plug-unplug, 70; continuous drainage, 68; and reference arm, 79), However, patients with a continuous catheter indicated in the other evaluations that they had more difficulty managing the catheter and felt it impeded activities and the wearing of clothing they would otherwise use.
The activity scale, the investigators noted, may not have captured differences in activity during the first week postoperatively because patients are commonly instructed to restrict some of the activities assessed in the scale.
Regarding infection, there was no difference in the rate of positive urine cultures or treatment for urinary tract infection between the catheter arms during a 3-month follow-up period, “despite the theoretical concern that plugging and unplugging a catheter disrupts the closed catheter system, thus increasing the risk of infection,” the investigators wrote. However, the study was not powered to detect a difference in the risk of infection as it was for the primary outcome.
There was no difference between the catheter arms in the percentage of women who used narcotic or nonnarcotic pain medication, and overall patient satisfaction was similar.
The majority of patients passed their outpatient voiding trials at the initial postoperative visit (72% plug-unplug and 58% continuous). “Interestingly, patients in the plug-unplug arm had significantly higher voided volumes and almost half of the [postvoid residual volume] at the [5-7 day postoperative voiding trial] compared with the continuous drainage arm.” This suggests, Dr. Boyd and colleagues wrote, that patients using the plug-unplug catheter “could have undergone a voiding trial sooner.”
Offering patients options for catheter management is “valuable,” and providing them with a technique that is “easier to manage may decrease the catheter burden and improve patient experience,” the investigators added.
Luis E. Sanz, MD, director of urogynecology and pelvic reconstructive surgery at Virginia Hospital Center, Arlington, said, “I think that the plug and unplug drainage bladder catheter after reconstructive surgery is much more physiologic and ‘user friendly’ than continuous drainage. And [there is] no need for a leg bag, which is very inconvenient to the patient.”
Dr. Sanz, an Ob.Gyn. News Editorial Advisory Board member who was not involved in the study, was asked to provide a comment.
The authors did not report any potential conflicts of interest.
SOURCE: Boyd SS et al. Obstet Gynecol. 2019;134:1037-45.
* Correction, 10/24/2019: an earlier version misstated the chief investigator's name, which is Sarah Boyd, MD.
Patients discharged with plug-unplug catheters after pelvic reconstructive surgery did not have adverse effects and reported less difficulty with catheter management and activities of daily living than women who were discharged with a continuous drainage catheter, a study has found.
“Plug-unplug catheter management technique is an acceptable method that does not appear to cause adverse events and may be considered for short-term catheterization after pelvic reconstructive surgery,” said Sarah Boyd, MD,* of the division of urogynecology at Hartford (Conn.) Hospital, and coinvestigators wrote in Obstetrics & Gynecology.
A total of 63 women who had a failed postoperative voiding trial after surgery for prolapse, with or without a concomitant incontinence procedure, were randomized to receive a 16-French transurethral catheter that was either attached to a leg bag (31 patients) or capped with a plastic plug (32 patients). Women in the second group – the plug-unplug group – were instructed to intermittently drain the bladder by uncapping the catheter when they felt the urge to void, or in the absence of urge, every 4 hours. All were scheduled for an outpatient voiding trial 5-7 days after discharge.
The first 30 study participants who did not require postoperative catheterization were assigned to a “reference,” or control, arm.
All patients in the study completed an activity assessment scale that covers both sedentary and ambulatory activities and is validated in women undergoing pelvic reconstructive surgery (Female Pelvic Med Reconstr Surg. 2012 Jul-Aug;18[4]:205-10); scores on the activity assessment scale (0-100) served as the primary outcome. Patients also answered questionnaires about their satisfaction and postoperative pain – and in the catheter arms, their experiences with the catheter.
The investigators found no difference in postoperative activity assessment scale scores (plug-unplug, 70; continuous drainage, 68; and reference arm, 79), However, patients with a continuous catheter indicated in the other evaluations that they had more difficulty managing the catheter and felt it impeded activities and the wearing of clothing they would otherwise use.
The activity scale, the investigators noted, may not have captured differences in activity during the first week postoperatively because patients are commonly instructed to restrict some of the activities assessed in the scale.
Regarding infection, there was no difference in the rate of positive urine cultures or treatment for urinary tract infection between the catheter arms during a 3-month follow-up period, “despite the theoretical concern that plugging and unplugging a catheter disrupts the closed catheter system, thus increasing the risk of infection,” the investigators wrote. However, the study was not powered to detect a difference in the risk of infection as it was for the primary outcome.
There was no difference between the catheter arms in the percentage of women who used narcotic or nonnarcotic pain medication, and overall patient satisfaction was similar.
The majority of patients passed their outpatient voiding trials at the initial postoperative visit (72% plug-unplug and 58% continuous). “Interestingly, patients in the plug-unplug arm had significantly higher voided volumes and almost half of the [postvoid residual volume] at the [5-7 day postoperative voiding trial] compared with the continuous drainage arm.” This suggests, Dr. Boyd and colleagues wrote, that patients using the plug-unplug catheter “could have undergone a voiding trial sooner.”
Offering patients options for catheter management is “valuable,” and providing them with a technique that is “easier to manage may decrease the catheter burden and improve patient experience,” the investigators added.
Luis E. Sanz, MD, director of urogynecology and pelvic reconstructive surgery at Virginia Hospital Center, Arlington, said, “I think that the plug and unplug drainage bladder catheter after reconstructive surgery is much more physiologic and ‘user friendly’ than continuous drainage. And [there is] no need for a leg bag, which is very inconvenient to the patient.”
Dr. Sanz, an Ob.Gyn. News Editorial Advisory Board member who was not involved in the study, was asked to provide a comment.
The authors did not report any potential conflicts of interest.
SOURCE: Boyd SS et al. Obstet Gynecol. 2019;134:1037-45.
* Correction, 10/24/2019: an earlier version misstated the chief investigator's name, which is Sarah Boyd, MD.
FROM OBSTETRICS & GYNECOLOGY
Prior authorizations for infusibles cause delays, toxicities
Rheumatologist Zachary S. Wallace, MD, knew just how prior authorization requirements were impacting his staff time and work flow when he embarked on a study several years ago. Managing authorizations for infusible medications alone was about to become a full-time job for one of the administrative assistants in the rheumatology unit at Massachusetts General Hospital in Boston.

His research questions concerned patients. “There’s a lot of talk about how much onus prior authorization requirements put on providers and the practice,” Dr. Wallace said. ”I was interested in understanding what impact [these requirements] have on patients themselves.”
Dr. Wallace led a review of the EHRs of 225 patients for whom an infusible medication such as rituximab and infliximab was ordered by 1 of the 16 physicians in the rheumatology unit between July 2016 and June 2018. The findings – that patients who needed prior authorizations for infusible medications had a significantly longer time to treatment initiation and higher prednisone-equivalent glucocorticoid exposure – were reported online in Arthritis Care & Research.
Among patients whose authorizations were initially denied, these differences were “pretty drastic,” Dr. Wallace said. The median time to receiving a first infusion was 50 days, compared with 27 days when permission was not required, and glucocorticoid exposure during the 3 months following the request was 605 mg versus 160 mg.
Among patients whose authorizations were not denied, the median time to first infusion was 31 days, compared with 27 days when authorization was not required, and the mean glucocorticoid exposure over 3 months was 364 mg versus 160 mg.
“I hope that our findings will help facilitate discussions with insurance providers, pharmacy benefit managers, and state and federal legislators about the need to address the impact that prior authorization requirements have on patients and providers,” said Dr. Wallace, also of the clinical epidemiology program in the division of rheumatology, allergy and immunology at Massachusetts General, and an assistant professor of medicine at Harvard Medical School, Boston.
Of the 225 patients for whom an infusible medication was ordered, 71% required preauthorization. Of these, 79% were approved and 21% were denied after the first request. And in a finding that Dr. Wallace called “somewhat surprising,” 82% of the authorizations originally denied were approved after appeal.
All told, prior authorizations for infusible medications were eventually approved in all but a small number of cases. “We go through all this effort to get these prior authorizations approved, and 96% of the time, they were ultimately approved,” he said in an interview.

Christopher Phillips, MD, a community rheumatologist in Paducah, Ky., who serves as chair of the insurance subcommittee of the American College of Rheumatology’s committee on rheumatologic care, said the findings “give further credence” to rheumatologists’ concerns. “We know [from our own experiences] that prior authorizations delay care, and we know that delays can cause harm to patients. We now have hard data backing up this assertion.”
Regarding the high number of authorization approvals, “there’s an argument to be made that for certain treatments and certain conditions where the success rate of appeals is high enough, you shouldn’t be subjecting these treatments to these [preauthorization] policies,” he said.
Calls for prior authorization reform
Most patients in the study (71%) had private insurance. But the findings also have implications for Medicare, Dr. Wallace said, as recent federal policies have expanded Medicare Advantage plans’ authority to use prior authorization in conjunction with step therapy for medications administered under Part B. Step therapy favors primary use of what insurers deem the most cost-effective therapies.
The ACR is one of almost 370 physician, patient, and health care organizations that are urging Congress to pass a bipartisan bill aimed at streamlining and standardizing prior authorization under the Medicare Advantage program. The legislation – Improving Seniors’ Timely Access to Care Act of 2019 (H.R. 3107) – was introduced by Reps. Suzan DelBene (D-Wash.), Mike Kelly (R-Pa.), Roger Marshall, MD (R-Kan.), and Ami Bera, MD (D-Calif.).
The bill calls for the creation of an electronic prior authorization program and a “real-time process for items and services that are routinely approved,” as well as greater Centers for Medicare & Medicaid Services oversight on how Medicare Advantage plans use prior authorization. Plans would be required to report to the CMS on the extent of their use of prior authorization and the rate of approvals or denials. They would also be held accountable for making timely prior authorization determinations and providing rationales for denials, according to a letter to Congress cosigned by the ACR.
In a press release about the legislation, Paula Marchetta, MD, president of the ACR, said that “the unregulated use of prior authorization has devolved into a time-consuming and obstructive process that often stalls or outright revokes patient access to medically necessary therapies.” She added that “many health care plans now use prior authorization indiscriminately.”
Cathryn Donaldson, director of communications for America’s Health Insurance Plans (AHIP), said in an email that prior authorization is used for less than 15% of covered services, and that, along with step therapy, it “helps ensure that patients receive care that is safe, effective, and necessary.” AHIP “knows that prior authorization can be improved,” she said, and is committed to streamlining the process.
A demonstration project on the automation of various parts of prior authorization is being coordinated with health information technology companies, plans, and providers, she noted.
The federal legislation is based at least partly on a consensus statement drafted by AHIP, the American Medical Association, and four other organizations representing hospitals, medical groups, and health plans on ways to improve the prior authorization process. Among the items mentioned in the statement is that “regular review” of services subject to prior authorization could help identify therapies that “no longer warrant” prior authorization because of low denial rates.
Outside of Medicare Advantage, the AMA is aware of at least 85 bills being introduced in states this year that address utilization management in commercial plans. Nearly all these bills attempt to reform prior authorization programs in some way, according to R. J. Mills, media relations coordinator for the AMA.
Rheumatologic patients hard hit
Off-label medication use was the most common reason (82%) for a prior authorization denial in the Massachusetts General study, even though 78% of the patients for whom infusible medications were prescribed had a condition with no Food and Drug Administration–approved treatment. Having such a condition was associated with 120% or 190% higher odds of having a denial in unadjusted and adjusted (for age and sex) analyses, Dr. Wallace and colleagues reported.
Moreover, nearly half (48%) of the patients with denials had already tried or were currently taking an oral disease-modifying antirheumatic drug, such as methotrexate.
The majority of denials were for the use of rituximab (70%), followed by infliximab (12%) and tocilizumab (12%). Most of the denials (79%) were appealed successfully through a peer-to-peer discussion. In five cases, the insurer’s preferred drug (for example, adalimumab) had to be used rather than the requested infusion (for example, infliximab).
Infused medications, many of which are biologics, are among the most expensive drugs prescribed for patients with rheumatic diseases. They were easiest for Dr. Wallace to study because of the way prior authorizations are handled in his unit, but prior authorization requirements are “widespread” in rheumatology practices across treatment types, he and Dr. Phillips said.
“Some of our relatively inexpensive treatments are subject to prior authorization requirements,” Dr. Phillips said. “We hear stories about prednisone needing a prior authorization sometimes.”
With respect to infusible medications, the insurance subcommittee is hearing from ACR members about seemingly increasing numbers of both clinical coverage reviews – for example, reviews of prior treatments – and site-of-care restrictions, Dr. Phillips noted. “Some carriers are insisting on infusions in non-hospital-based settings, for cost savings, or on home infusions, which are concerning because of [possible] infusion reactions and medical service availability.”
The application of step therapy to rheumatologic patients is troubling because of the “often unique medical circumstances of the patient,” Dr. Phillips said. “There are enough differences among the [tumor necrosis factor] antagonists, for instance, that make one more appropriate for a certain patient than another. Those differences are not brought into consideration with these policies.”
There are other ways in which prior authorization processes “are not well informed medically,” he said, recalling a case brought to the attention of the subcommittee in which a patient prescribed a biologic drug for psoriatic arthritis was denied authorization because “the documentation did not include a [disease activity measure] that is specific to RA and not used for psoriatic arthritis.”
It is not uncommon for authorizations for infusible medications to take 2 weeks or longer to secure – even when initially approved. In the AMA’s 2018 Prior Authorization Physician Survey, 65% reported waiting at least 1 business day for a decision and 26% reported waiting at least 3 business days for responses. “With infusibles, we’re absolutely dealing with a much longer time,” Dr. Phillips said.
In Dr. Wallace’s study, the finding that prior authorizations facilitated greater prednisone-equivalent glucocorticoid exposure is important, he and his colleagues wrote, because these medications may put patients at higher risk of infection, cardiovascular disease, and diabetes – even in low doses and with short-term use. Notably, the median delay to the initiation of treatment was 29 days, regardless of prior authorization requirements. Dr. Wallace said the delays “likely reflect a combination of factors” – including infusion center waiting lists and patient-level factors – and that his team is “thinking about how to facilitate better access [to their practice’s infusion center] for those who are approved for treatment.”
The most common conditions for which infused medication was ordered were inflammatory arthritis (32%), vasculitis (23%), and IgG4-related disease (17%). The 225 patients in the study had an average age of 53 years.
Dr. Wallace reported that he has no relevant financial disclosures.
SOURCE: Wallace ZS et al. Arthritis Care Res. 2019 Sep 10. doi: 10.1002/acr.24062.
Rheumatologist Zachary S. Wallace, MD, knew just how prior authorization requirements were impacting his staff time and work flow when he embarked on a study several years ago. Managing authorizations for infusible medications alone was about to become a full-time job for one of the administrative assistants in the rheumatology unit at Massachusetts General Hospital in Boston.
His research questions concerned patients. “There’s a lot of talk about how much onus prior authorization requirements put on providers and the practice,” Dr. Wallace said. ”I was interested in understanding what impact [these requirements] have on patients themselves.”
Dr. Wallace led a review of the EHRs of 225 patients for whom an infusible medication such as rituximab and infliximab was ordered by 1 of the 16 physicians in the rheumatology unit between July 2016 and June 2018. The findings – that patients who needed prior authorizations for infusible medications had a significantly longer time to treatment initiation and higher prednisone-equivalent glucocorticoid exposure – were reported online in Arthritis Care & Research.
Among patients whose authorizations were initially denied, these differences were “pretty drastic,” Dr. Wallace said. The median time to receiving a first infusion was 50 days, compared with 27 days when permission was not required, and glucocorticoid exposure during the 3 months following the request was 605 mg versus 160 mg.
Among patients whose authorizations were not denied, the median time to first infusion was 31 days, compared with 27 days when authorization was not required, and the mean glucocorticoid exposure over 3 months was 364 mg versus 160 mg.
“I hope that our findings will help facilitate discussions with insurance providers, pharmacy benefit managers, and state and federal legislators about the need to address the impact that prior authorization requirements have on patients and providers,” said Dr. Wallace, also of the clinical epidemiology program in the division of rheumatology, allergy and immunology at Massachusetts General, and an assistant professor of medicine at Harvard Medical School, Boston.
Of the 225 patients for whom an infusible medication was ordered, 71% required preauthorization. Of these, 79% were approved and 21% were denied after the first request. And in a finding that Dr. Wallace called “somewhat surprising,” 82% of the authorizations originally denied were approved after appeal.
All told, prior authorizations for infusible medications were eventually approved in all but a small number of cases. “We go through all this effort to get these prior authorizations approved, and 96% of the time, they were ultimately approved,” he said in an interview.
Christopher Phillips, MD, a community rheumatologist in Paducah, Ky., who serves as chair of the insurance subcommittee of the American College of Rheumatology’s committee on rheumatologic care, said the findings “give further credence” to rheumatologists’ concerns. “We know [from our own experiences] that prior authorizations delay care, and we know that delays can cause harm to patients. We now have hard data backing up this assertion.”
Regarding the high number of authorization approvals, “there’s an argument to be made that for certain treatments and certain conditions where the success rate of appeals is high enough, you shouldn’t be subjecting these treatments to these [preauthorization] policies,” he said.
Calls for prior authorization reform
Most patients in the study (71%) had private insurance. But the findings also have implications for Medicare, Dr. Wallace said, as recent federal policies have expanded Medicare Advantage plans’ authority to use prior authorization in conjunction with step therapy for medications administered under Part B. Step therapy favors primary use of what insurers deem the most cost-effective therapies.
The ACR is one of almost 370 physician, patient, and health care organizations that are urging Congress to pass a bipartisan bill aimed at streamlining and standardizing prior authorization under the Medicare Advantage program. The legislation – Improving Seniors’ Timely Access to Care Act of 2019 (H.R. 3107) – was introduced by Reps. Suzan DelBene (D-Wash.), Mike Kelly (R-Pa.), Roger Marshall, MD (R-Kan.), and Ami Bera, MD (D-Calif.).
The bill calls for the creation of an electronic prior authorization program and a “real-time process for items and services that are routinely approved,” as well as greater Centers for Medicare & Medicaid Services oversight on how Medicare Advantage plans use prior authorization. Plans would be required to report to the CMS on the extent of their use of prior authorization and the rate of approvals or denials. They would also be held accountable for making timely prior authorization determinations and providing rationales for denials, according to a letter to Congress cosigned by the ACR.
In a press release about the legislation, Paula Marchetta, MD, president of the ACR, said that “the unregulated use of prior authorization has devolved into a time-consuming and obstructive process that often stalls or outright revokes patient access to medically necessary therapies.” She added that “many health care plans now use prior authorization indiscriminately.”
Cathryn Donaldson, director of communications for America’s Health Insurance Plans (AHIP), said in an email that prior authorization is used for less than 15% of covered services, and that, along with step therapy, it “helps ensure that patients receive care that is safe, effective, and necessary.” AHIP “knows that prior authorization can be improved,” she said, and is committed to streamlining the process.
A demonstration project on the automation of various parts of prior authorization is being coordinated with health information technology companies, plans, and providers, she noted.
The federal legislation is based at least partly on a consensus statement drafted by AHIP, the American Medical Association, and four other organizations representing hospitals, medical groups, and health plans on ways to improve the prior authorization process. Among the items mentioned in the statement is that “regular review” of services subject to prior authorization could help identify therapies that “no longer warrant” prior authorization because of low denial rates.
Outside of Medicare Advantage, the AMA is aware of at least 85 bills being introduced in states this year that address utilization management in commercial plans. Nearly all these bills attempt to reform prior authorization programs in some way, according to R. J. Mills, media relations coordinator for the AMA.
Rheumatologic patients hard hit
Off-label medication use was the most common reason (82%) for a prior authorization denial in the Massachusetts General study, even though 78% of the patients for whom infusible medications were prescribed had a condition with no Food and Drug Administration–approved treatment. Having such a condition was associated with 120% or 190% higher odds of having a denial in unadjusted and adjusted (for age and sex) analyses, Dr. Wallace and colleagues reported.
Moreover, nearly half (48%) of the patients with denials had already tried or were currently taking an oral disease-modifying antirheumatic drug, such as methotrexate.
The majority of denials were for the use of rituximab (70%), followed by infliximab (12%) and tocilizumab (12%). Most of the denials (79%) were appealed successfully through a peer-to-peer discussion. In five cases, the insurer’s preferred drug (for example, adalimumab) had to be used rather than the requested infusion (for example, infliximab).
Infused medications, many of which are biologics, are among the most expensive drugs prescribed for patients with rheumatic diseases. They were easiest for Dr. Wallace to study because of the way prior authorizations are handled in his unit, but prior authorization requirements are “widespread” in rheumatology practices across treatment types, he and Dr. Phillips said.
“Some of our relatively inexpensive treatments are subject to prior authorization requirements,” Dr. Phillips said. “We hear stories about prednisone needing a prior authorization sometimes.”
With respect to infusible medications, the insurance subcommittee is hearing from ACR members about seemingly increasing numbers of both clinical coverage reviews – for example, reviews of prior treatments – and site-of-care restrictions, Dr. Phillips noted. “Some carriers are insisting on infusions in non-hospital-based settings, for cost savings, or on home infusions, which are concerning because of [possible] infusion reactions and medical service availability.”
The application of step therapy to rheumatologic patients is troubling because of the “often unique medical circumstances of the patient,” Dr. Phillips said. “There are enough differences among the [tumor necrosis factor] antagonists, for instance, that make one more appropriate for a certain patient than another. Those differences are not brought into consideration with these policies.”
There are other ways in which prior authorization processes “are not well informed medically,” he said, recalling a case brought to the attention of the subcommittee in which a patient prescribed a biologic drug for psoriatic arthritis was denied authorization because “the documentation did not include a [disease activity measure] that is specific to RA and not used for psoriatic arthritis.”
It is not uncommon for authorizations for infusible medications to take 2 weeks or longer to secure – even when initially approved. In the AMA’s 2018 Prior Authorization Physician Survey, 65% reported waiting at least 1 business day for a decision and 26% reported waiting at least 3 business days for responses. “With infusibles, we’re absolutely dealing with a much longer time,” Dr. Phillips said.
In Dr. Wallace’s study, the finding that prior authorizations facilitated greater prednisone-equivalent glucocorticoid exposure is important, he and his colleagues wrote, because these medications may put patients at higher risk of infection, cardiovascular disease, and diabetes – even in low doses and with short-term use. Notably, the median delay to the initiation of treatment was 29 days, regardless of prior authorization requirements. Dr. Wallace said the delays “likely reflect a combination of factors” – including infusion center waiting lists and patient-level factors – and that his team is “thinking about how to facilitate better access [to their practice’s infusion center] for those who are approved for treatment.”
The most common conditions for which infused medication was ordered were inflammatory arthritis (32%), vasculitis (23%), and IgG4-related disease (17%). The 225 patients in the study had an average age of 53 years.
Dr. Wallace reported that he has no relevant financial disclosures.
SOURCE: Wallace ZS et al. Arthritis Care Res. 2019 Sep 10. doi: 10.1002/acr.24062.
Rheumatologist Zachary S. Wallace, MD, knew just how prior authorization requirements were impacting his staff time and work flow when he embarked on a study several years ago. Managing authorizations for infusible medications alone was about to become a full-time job for one of the administrative assistants in the rheumatology unit at Massachusetts General Hospital in Boston.
His research questions concerned patients. “There’s a lot of talk about how much onus prior authorization requirements put on providers and the practice,” Dr. Wallace said. ”I was interested in understanding what impact [these requirements] have on patients themselves.”
Dr. Wallace led a review of the EHRs of 225 patients for whom an infusible medication such as rituximab and infliximab was ordered by 1 of the 16 physicians in the rheumatology unit between July 2016 and June 2018. The findings – that patients who needed prior authorizations for infusible medications had a significantly longer time to treatment initiation and higher prednisone-equivalent glucocorticoid exposure – were reported online in Arthritis Care & Research.
Among patients whose authorizations were initially denied, these differences were “pretty drastic,” Dr. Wallace said. The median time to receiving a first infusion was 50 days, compared with 27 days when permission was not required, and glucocorticoid exposure during the 3 months following the request was 605 mg versus 160 mg.
Among patients whose authorizations were not denied, the median time to first infusion was 31 days, compared with 27 days when authorization was not required, and the mean glucocorticoid exposure over 3 months was 364 mg versus 160 mg.
“I hope that our findings will help facilitate discussions with insurance providers, pharmacy benefit managers, and state and federal legislators about the need to address the impact that prior authorization requirements have on patients and providers,” said Dr. Wallace, also of the clinical epidemiology program in the division of rheumatology, allergy and immunology at Massachusetts General, and an assistant professor of medicine at Harvard Medical School, Boston.
Of the 225 patients for whom an infusible medication was ordered, 71% required preauthorization. Of these, 79% were approved and 21% were denied after the first request. And in a finding that Dr. Wallace called “somewhat surprising,” 82% of the authorizations originally denied were approved after appeal.
All told, prior authorizations for infusible medications were eventually approved in all but a small number of cases. “We go through all this effort to get these prior authorizations approved, and 96% of the time, they were ultimately approved,” he said in an interview.
Christopher Phillips, MD, a community rheumatologist in Paducah, Ky., who serves as chair of the insurance subcommittee of the American College of Rheumatology’s committee on rheumatologic care, said the findings “give further credence” to rheumatologists’ concerns. “We know [from our own experiences] that prior authorizations delay care, and we know that delays can cause harm to patients. We now have hard data backing up this assertion.”
Regarding the high number of authorization approvals, “there’s an argument to be made that for certain treatments and certain conditions where the success rate of appeals is high enough, you shouldn’t be subjecting these treatments to these [preauthorization] policies,” he said.
Calls for prior authorization reform
Most patients in the study (71%) had private insurance. But the findings also have implications for Medicare, Dr. Wallace said, as recent federal policies have expanded Medicare Advantage plans’ authority to use prior authorization in conjunction with step therapy for medications administered under Part B. Step therapy favors primary use of what insurers deem the most cost-effective therapies.
The ACR is one of almost 370 physician, patient, and health care organizations that are urging Congress to pass a bipartisan bill aimed at streamlining and standardizing prior authorization under the Medicare Advantage program. The legislation – Improving Seniors’ Timely Access to Care Act of 2019 (H.R. 3107) – was introduced by Reps. Suzan DelBene (D-Wash.), Mike Kelly (R-Pa.), Roger Marshall, MD (R-Kan.), and Ami Bera, MD (D-Calif.).
The bill calls for the creation of an electronic prior authorization program and a “real-time process for items and services that are routinely approved,” as well as greater Centers for Medicare & Medicaid Services oversight on how Medicare Advantage plans use prior authorization. Plans would be required to report to the CMS on the extent of their use of prior authorization and the rate of approvals or denials. They would also be held accountable for making timely prior authorization determinations and providing rationales for denials, according to a letter to Congress cosigned by the ACR.
In a press release about the legislation, Paula Marchetta, MD, president of the ACR, said that “the unregulated use of prior authorization has devolved into a time-consuming and obstructive process that often stalls or outright revokes patient access to medically necessary therapies.” She added that “many health care plans now use prior authorization indiscriminately.”
Cathryn Donaldson, director of communications for America’s Health Insurance Plans (AHIP), said in an email that prior authorization is used for less than 15% of covered services, and that, along with step therapy, it “helps ensure that patients receive care that is safe, effective, and necessary.” AHIP “knows that prior authorization can be improved,” she said, and is committed to streamlining the process.
A demonstration project on the automation of various parts of prior authorization is being coordinated with health information technology companies, plans, and providers, she noted.
The federal legislation is based at least partly on a consensus statement drafted by AHIP, the American Medical Association, and four other organizations representing hospitals, medical groups, and health plans on ways to improve the prior authorization process. Among the items mentioned in the statement is that “regular review” of services subject to prior authorization could help identify therapies that “no longer warrant” prior authorization because of low denial rates.
Outside of Medicare Advantage, the AMA is aware of at least 85 bills being introduced in states this year that address utilization management in commercial plans. Nearly all these bills attempt to reform prior authorization programs in some way, according to R. J. Mills, media relations coordinator for the AMA.
Rheumatologic patients hard hit
Off-label medication use was the most common reason (82%) for a prior authorization denial in the Massachusetts General study, even though 78% of the patients for whom infusible medications were prescribed had a condition with no Food and Drug Administration–approved treatment. Having such a condition was associated with 120% or 190% higher odds of having a denial in unadjusted and adjusted (for age and sex) analyses, Dr. Wallace and colleagues reported.
Moreover, nearly half (48%) of the patients with denials had already tried or were currently taking an oral disease-modifying antirheumatic drug, such as methotrexate.
The majority of denials were for the use of rituximab (70%), followed by infliximab (12%) and tocilizumab (12%). Most of the denials (79%) were appealed successfully through a peer-to-peer discussion. In five cases, the insurer’s preferred drug (for example, adalimumab) had to be used rather than the requested infusion (for example, infliximab).
Infused medications, many of which are biologics, are among the most expensive drugs prescribed for patients with rheumatic diseases. They were easiest for Dr. Wallace to study because of the way prior authorizations are handled in his unit, but prior authorization requirements are “widespread” in rheumatology practices across treatment types, he and Dr. Phillips said.
“Some of our relatively inexpensive treatments are subject to prior authorization requirements,” Dr. Phillips said. “We hear stories about prednisone needing a prior authorization sometimes.”
With respect to infusible medications, the insurance subcommittee is hearing from ACR members about seemingly increasing numbers of both clinical coverage reviews – for example, reviews of prior treatments – and site-of-care restrictions, Dr. Phillips noted. “Some carriers are insisting on infusions in non-hospital-based settings, for cost savings, or on home infusions, which are concerning because of [possible] infusion reactions and medical service availability.”
The application of step therapy to rheumatologic patients is troubling because of the “often unique medical circumstances of the patient,” Dr. Phillips said. “There are enough differences among the [tumor necrosis factor] antagonists, for instance, that make one more appropriate for a certain patient than another. Those differences are not brought into consideration with these policies.”
There are other ways in which prior authorization processes “are not well informed medically,” he said, recalling a case brought to the attention of the subcommittee in which a patient prescribed a biologic drug for psoriatic arthritis was denied authorization because “the documentation did not include a [disease activity measure] that is specific to RA and not used for psoriatic arthritis.”
It is not uncommon for authorizations for infusible medications to take 2 weeks or longer to secure – even when initially approved. In the AMA’s 2018 Prior Authorization Physician Survey, 65% reported waiting at least 1 business day for a decision and 26% reported waiting at least 3 business days for responses. “With infusibles, we’re absolutely dealing with a much longer time,” Dr. Phillips said.
In Dr. Wallace’s study, the finding that prior authorizations facilitated greater prednisone-equivalent glucocorticoid exposure is important, he and his colleagues wrote, because these medications may put patients at higher risk of infection, cardiovascular disease, and diabetes – even in low doses and with short-term use. Notably, the median delay to the initiation of treatment was 29 days, regardless of prior authorization requirements. Dr. Wallace said the delays “likely reflect a combination of factors” – including infusion center waiting lists and patient-level factors – and that his team is “thinking about how to facilitate better access [to their practice’s infusion center] for those who are approved for treatment.”
The most common conditions for which infused medication was ordered were inflammatory arthritis (32%), vasculitis (23%), and IgG4-related disease (17%). The 225 patients in the study had an average age of 53 years.
Dr. Wallace reported that he has no relevant financial disclosures.
SOURCE: Wallace ZS et al. Arthritis Care Res. 2019 Sep 10. doi: 10.1002/acr.24062.
FROM ARTHRITIS CARE & RESEARCH
Try testosterone for some women with sexual dysfunction, but not others
A new international position statement on testosterone therapy for women concludes that a trial of testosterone is appropriate for postmenopausal women with hypoactive sexual desire dysfunction (HSDD) and that its use for any other condition, symptom, or reason is not supported by available evidence.
The seven-page position statement, developed by an international task force of experts from the Endocrine Society, the American College of Gynecologists and Obstetricians, and multiple other medical societies, also emphasized that blood concentrations of testosterone should approximate premenopausal physiological conditions.
“When testosterone therapy is given, the resultant blood levels should not be above those seen in healthy young women,” said lead author Susan Ruth Davis, PhD, MBBS, of Monash University in Melbourne, Australia, in a press release issued by the Endocrine Society. Dr. Davis is president of the International Menopause Society, which coordinated the panel.
The statement was published in the Journal of Clinical Endocrinology & Metabolism and three other medical journals.
Margaret E. Wierman, MD, who represented the Endocrine Society on the task force, said in an interview that there has been “growing concern about testosterone being prescribed for a variety of signs and symptoms without data to support” such use. At the same time, there is significant concern about the ongoing lack of approved formulations licensed specifically for women, she said.
In part, the statement is about a renewed “call to industry to make some [female-specific] formulations so that we can examine other potential roles of testosterone in women,” said Dr. Wierman, professor of medicine and physiology at the University of Colorado at Denver, Aurora, and chief of endocrinology at the Rocky Mountain Regional Veterans Affairs Medical Center in Aurora.
“Testosterone may be useful [for indications other than HSDD], but we don’t know. There may be no [breast or cardiovascular disease risk], but we don’t know,” she said. “And without a formulation to study potential benefits and risks, it’s good to be cautious. It’s good to really outline where we have data and where we don’t.”
The Endocrine Society’s 2014 clinical practice guideline on androgen therapy in women, for which Dr. Wierman was the lead author, also recommended against the off-label use of testosterone for sexual dysfunction other than HSDD or for any other reason, such as cognitive, cardiovascular, metabolic, or bone health. As with the new statement, the society’s position statement was guided by an international, multisociety task force, albeit a smaller one.
For the new global position statement, the task force’s review of evidence includes a recently published systematic review and meta-analysis of randomized controlled trial data – of at least 12 weeks’ duration – on the use of testosterone for sexual function, cardiometabolic variables, cognitive measures, and musculoskeletal health. Some of the data from the randomized controlled trials were unpublished.
The meta-analysis, led by Dr. Davis and published in July in the Lancet Diabetes & Endocrinology, found that, compared with placebo or a comparator (such as estrogen, with or without progesterone), testosterone in either oral or transdermal form significantly improved sexual function in postmenopausal women. However, data about the effects of testosterone for other indications, its long-term safety, and its use in premenopausal women, were insufficient for drawing any conclusions (Lancet Diabetes Endocrinol. 2019 Jul 25. doi: 10.1016/S2213-8587[19]30189-5).
In addition, testosterone administered orally – but not nonorally (patch or cream) – was associated with adverse lipid profiles, Dr. Davis and her colleagues reported.
Another systematic review and meta-analysis, published in Fertility and Sterility in 2017 and included in the task force’s evidence review, focused specifically on transdermal testosterone for menopausal women with HSDD, with or without estrogen and progestin therapy. It also showed short-term efficacy in terms of improvement in sexual function, as well as short-term safety (Fertil Steril. 2017;107(2):475-82).
The new position statement warns about the lack of long-term safety data, stating that “safety data for testosterone in physiologic doses are not available beyond 24 months of treatment.”
In the short term, testosterone therapy for postmenopausal women (in doses approximating testosterone concentrations for premenopausal women), is associated with mild increases in acne and body/facial hair growth in some women, but not with alopecia, clitoromegaly, or voice change. Short-term transdermal therapy also does not seem to affect breast cancer risk or have any significant effects on lipid profiles, the statement says.
The panel points out, however, that randomized controlled trials with testosterone therapy have excluded women who are at high risk of cardiometabolic disease, and that women with a previous diagnosis of breast cancer have also been excluded from randomized trials of testosterone in women with HSDD. This is a “big issue,” said Dr. Wierman, and means that recommendations regarding the effect of testosterone in postmenopausal women with HSDD may not be generalizable to possible at-risk subpopulations.
The panel endorsed testosterone therapy specifically for women with HSDD because most of the studies reporting on sexual function have recruited women with diagnosed HSDD. Demonstrated benefits of testosterone in these cases include improved sexual desire, arousal, orgasm, and pleasure, and reduced concerns and distress about sex. HSDD should be diagnosed after formal biopsychosocial assessment, the statement notes.
“We don’t completely understand the control of sexual function in women, but it’s very dependent on estrogen status. And it’s also dependent on psychosocial factors, emotional health, relationship issues, and physical issues,” Dr. Wierman said in the interview.
“In practice, we look at all these issues, and we first optimize estrogen status. Once that’s done, and we’ve looked at all the other components of sexual function, then we can consider off-label use of testosterone,” she said. “If there’s no response in 3-6 months, we stop it.”
Testosterone levels do not correlate with sexual dysfunction, Dr. Wierman emphasized, and direct assays for the measurement of total and free testosterone are unreliable. The statement acknowledges that but still recommends measurement of testosterone using direct assays, in cases in which liquid/gas chromatography and tandem mass spectrometry assay (which has “high accuracy and reproducibility”) are not available. This is “to exclude high baseline concentrations and also to exclude supraphysiological concentrations during treatment,” the panel said.
Most endocrinologists and other experts who prescribe testosterone therapy for women use an approved male formulation off label and adjust it – an approach that the panel says is reasonable as long as hormone concentrations are “maintained in the physiologic female range.”
Compounded “bioidentical” testosterone therapy “cannot be recommended for the treatment of HSDD because of the lack of evidence for safety and efficacy,” the statement says.
“A big concern of many endocrinologists,” Dr. Wierman added, “is the recent explosion of using pharmacological levels of both estrogen and testosterone in either [injections] or pellets.” The Endocrine Society and other societies have alerted the Food and Drug Administration to “this new cottage industry, which may have significant side effects and risks for our patients,” she said.
Dr. Wierman reported received funding from Corcept Therapeutics, Novartis, and the Cancer League of Colorado, and honoraria or consultation fees from Pfizer to review ASPIRE grant applications for studies of acromegaly as well as Endocrine Society honorarium for teaching in the Endocrine Board Review and Clinical Endocrine Update. Dr. Davis reported receiving funding from a National Health and Medical Research Council Project Grant, a National Breast Foundation accelerator grant, and the Grollo-Ruzenne Foundation, as well as honoraria from Besins and Pfizer Australia. She has been a consultant to Besins Healthcare, Mayne Pharmaceuticals, Lawley Pharmaceuticals, and Que Oncology. Disclosures for other authors of the position statement are listed with the statement.
SOURCE: Davis SR et al. J Clin Endocrinol Metab. 2019 Sep 2. doi: 10.1210/jc.2019-01603.
A new international position statement on testosterone therapy for women concludes that a trial of testosterone is appropriate for postmenopausal women with hypoactive sexual desire dysfunction (HSDD) and that its use for any other condition, symptom, or reason is not supported by available evidence.
The seven-page position statement, developed by an international task force of experts from the Endocrine Society, the American College of Gynecologists and Obstetricians, and multiple other medical societies, also emphasized that blood concentrations of testosterone should approximate premenopausal physiological conditions.
“When testosterone therapy is given, the resultant blood levels should not be above those seen in healthy young women,” said lead author Susan Ruth Davis, PhD, MBBS, of Monash University in Melbourne, Australia, in a press release issued by the Endocrine Society. Dr. Davis is president of the International Menopause Society, which coordinated the panel.
The statement was published in the Journal of Clinical Endocrinology & Metabolism and three other medical journals.
Margaret E. Wierman, MD, who represented the Endocrine Society on the task force, said in an interview that there has been “growing concern about testosterone being prescribed for a variety of signs and symptoms without data to support” such use. At the same time, there is significant concern about the ongoing lack of approved formulations licensed specifically for women, she said.
In part, the statement is about a renewed “call to industry to make some [female-specific] formulations so that we can examine other potential roles of testosterone in women,” said Dr. Wierman, professor of medicine and physiology at the University of Colorado at Denver, Aurora, and chief of endocrinology at the Rocky Mountain Regional Veterans Affairs Medical Center in Aurora.
“Testosterone may be useful [for indications other than HSDD], but we don’t know. There may be no [breast or cardiovascular disease risk], but we don’t know,” she said. “And without a formulation to study potential benefits and risks, it’s good to be cautious. It’s good to really outline where we have data and where we don’t.”
The Endocrine Society’s 2014 clinical practice guideline on androgen therapy in women, for which Dr. Wierman was the lead author, also recommended against the off-label use of testosterone for sexual dysfunction other than HSDD or for any other reason, such as cognitive, cardiovascular, metabolic, or bone health. As with the new statement, the society’s position statement was guided by an international, multisociety task force, albeit a smaller one.
For the new global position statement, the task force’s review of evidence includes a recently published systematic review and meta-analysis of randomized controlled trial data – of at least 12 weeks’ duration – on the use of testosterone for sexual function, cardiometabolic variables, cognitive measures, and musculoskeletal health. Some of the data from the randomized controlled trials were unpublished.
The meta-analysis, led by Dr. Davis and published in July in the Lancet Diabetes & Endocrinology, found that, compared with placebo or a comparator (such as estrogen, with or without progesterone), testosterone in either oral or transdermal form significantly improved sexual function in postmenopausal women. However, data about the effects of testosterone for other indications, its long-term safety, and its use in premenopausal women, were insufficient for drawing any conclusions (Lancet Diabetes Endocrinol. 2019 Jul 25. doi: 10.1016/S2213-8587[19]30189-5).
In addition, testosterone administered orally – but not nonorally (patch or cream) – was associated with adverse lipid profiles, Dr. Davis and her colleagues reported.
Another systematic review and meta-analysis, published in Fertility and Sterility in 2017 and included in the task force’s evidence review, focused specifically on transdermal testosterone for menopausal women with HSDD, with or without estrogen and progestin therapy. It also showed short-term efficacy in terms of improvement in sexual function, as well as short-term safety (Fertil Steril. 2017;107(2):475-82).
The new position statement warns about the lack of long-term safety data, stating that “safety data for testosterone in physiologic doses are not available beyond 24 months of treatment.”
In the short term, testosterone therapy for postmenopausal women (in doses approximating testosterone concentrations for premenopausal women), is associated with mild increases in acne and body/facial hair growth in some women, but not with alopecia, clitoromegaly, or voice change. Short-term transdermal therapy also does not seem to affect breast cancer risk or have any significant effects on lipid profiles, the statement says.
The panel points out, however, that randomized controlled trials with testosterone therapy have excluded women who are at high risk of cardiometabolic disease, and that women with a previous diagnosis of breast cancer have also been excluded from randomized trials of testosterone in women with HSDD. This is a “big issue,” said Dr. Wierman, and means that recommendations regarding the effect of testosterone in postmenopausal women with HSDD may not be generalizable to possible at-risk subpopulations.
The panel endorsed testosterone therapy specifically for women with HSDD because most of the studies reporting on sexual function have recruited women with diagnosed HSDD. Demonstrated benefits of testosterone in these cases include improved sexual desire, arousal, orgasm, and pleasure, and reduced concerns and distress about sex. HSDD should be diagnosed after formal biopsychosocial assessment, the statement notes.
“We don’t completely understand the control of sexual function in women, but it’s very dependent on estrogen status. And it’s also dependent on psychosocial factors, emotional health, relationship issues, and physical issues,” Dr. Wierman said in the interview.
“In practice, we look at all these issues, and we first optimize estrogen status. Once that’s done, and we’ve looked at all the other components of sexual function, then we can consider off-label use of testosterone,” she said. “If there’s no response in 3-6 months, we stop it.”
Testosterone levels do not correlate with sexual dysfunction, Dr. Wierman emphasized, and direct assays for the measurement of total and free testosterone are unreliable. The statement acknowledges that but still recommends measurement of testosterone using direct assays, in cases in which liquid/gas chromatography and tandem mass spectrometry assay (which has “high accuracy and reproducibility”) are not available. This is “to exclude high baseline concentrations and also to exclude supraphysiological concentrations during treatment,” the panel said.
Most endocrinologists and other experts who prescribe testosterone therapy for women use an approved male formulation off label and adjust it – an approach that the panel says is reasonable as long as hormone concentrations are “maintained in the physiologic female range.”
Compounded “bioidentical” testosterone therapy “cannot be recommended for the treatment of HSDD because of the lack of evidence for safety and efficacy,” the statement says.
“A big concern of many endocrinologists,” Dr. Wierman added, “is the recent explosion of using pharmacological levels of both estrogen and testosterone in either [injections] or pellets.” The Endocrine Society and other societies have alerted the Food and Drug Administration to “this new cottage industry, which may have significant side effects and risks for our patients,” she said.
Dr. Wierman reported received funding from Corcept Therapeutics, Novartis, and the Cancer League of Colorado, and honoraria or consultation fees from Pfizer to review ASPIRE grant applications for studies of acromegaly as well as Endocrine Society honorarium for teaching in the Endocrine Board Review and Clinical Endocrine Update. Dr. Davis reported receiving funding from a National Health and Medical Research Council Project Grant, a National Breast Foundation accelerator grant, and the Grollo-Ruzenne Foundation, as well as honoraria from Besins and Pfizer Australia. She has been a consultant to Besins Healthcare, Mayne Pharmaceuticals, Lawley Pharmaceuticals, and Que Oncology. Disclosures for other authors of the position statement are listed with the statement.
SOURCE: Davis SR et al. J Clin Endocrinol Metab. 2019 Sep 2. doi: 10.1210/jc.2019-01603.
A new international position statement on testosterone therapy for women concludes that a trial of testosterone is appropriate for postmenopausal women with hypoactive sexual desire dysfunction (HSDD) and that its use for any other condition, symptom, or reason is not supported by available evidence.
The seven-page position statement, developed by an international task force of experts from the Endocrine Society, the American College of Gynecologists and Obstetricians, and multiple other medical societies, also emphasized that blood concentrations of testosterone should approximate premenopausal physiological conditions.
“When testosterone therapy is given, the resultant blood levels should not be above those seen in healthy young women,” said lead author Susan Ruth Davis, PhD, MBBS, of Monash University in Melbourne, Australia, in a press release issued by the Endocrine Society. Dr. Davis is president of the International Menopause Society, which coordinated the panel.
The statement was published in the Journal of Clinical Endocrinology & Metabolism and three other medical journals.
Margaret E. Wierman, MD, who represented the Endocrine Society on the task force, said in an interview that there has been “growing concern about testosterone being prescribed for a variety of signs and symptoms without data to support” such use. At the same time, there is significant concern about the ongoing lack of approved formulations licensed specifically for women, she said.
In part, the statement is about a renewed “call to industry to make some [female-specific] formulations so that we can examine other potential roles of testosterone in women,” said Dr. Wierman, professor of medicine and physiology at the University of Colorado at Denver, Aurora, and chief of endocrinology at the Rocky Mountain Regional Veterans Affairs Medical Center in Aurora.
“Testosterone may be useful [for indications other than HSDD], but we don’t know. There may be no [breast or cardiovascular disease risk], but we don’t know,” she said. “And without a formulation to study potential benefits and risks, it’s good to be cautious. It’s good to really outline where we have data and where we don’t.”
The Endocrine Society’s 2014 clinical practice guideline on androgen therapy in women, for which Dr. Wierman was the lead author, also recommended against the off-label use of testosterone for sexual dysfunction other than HSDD or for any other reason, such as cognitive, cardiovascular, metabolic, or bone health. As with the new statement, the society’s position statement was guided by an international, multisociety task force, albeit a smaller one.
For the new global position statement, the task force’s review of evidence includes a recently published systematic review and meta-analysis of randomized controlled trial data – of at least 12 weeks’ duration – on the use of testosterone for sexual function, cardiometabolic variables, cognitive measures, and musculoskeletal health. Some of the data from the randomized controlled trials were unpublished.
The meta-analysis, led by Dr. Davis and published in July in the Lancet Diabetes & Endocrinology, found that, compared with placebo or a comparator (such as estrogen, with or without progesterone), testosterone in either oral or transdermal form significantly improved sexual function in postmenopausal women. However, data about the effects of testosterone for other indications, its long-term safety, and its use in premenopausal women, were insufficient for drawing any conclusions (Lancet Diabetes Endocrinol. 2019 Jul 25. doi: 10.1016/S2213-8587[19]30189-5).
In addition, testosterone administered orally – but not nonorally (patch or cream) – was associated with adverse lipid profiles, Dr. Davis and her colleagues reported.
Another systematic review and meta-analysis, published in Fertility and Sterility in 2017 and included in the task force’s evidence review, focused specifically on transdermal testosterone for menopausal women with HSDD, with or without estrogen and progestin therapy. It also showed short-term efficacy in terms of improvement in sexual function, as well as short-term safety (Fertil Steril. 2017;107(2):475-82).
The new position statement warns about the lack of long-term safety data, stating that “safety data for testosterone in physiologic doses are not available beyond 24 months of treatment.”
In the short term, testosterone therapy for postmenopausal women (in doses approximating testosterone concentrations for premenopausal women), is associated with mild increases in acne and body/facial hair growth in some women, but not with alopecia, clitoromegaly, or voice change. Short-term transdermal therapy also does not seem to affect breast cancer risk or have any significant effects on lipid profiles, the statement says.
The panel points out, however, that randomized controlled trials with testosterone therapy have excluded women who are at high risk of cardiometabolic disease, and that women with a previous diagnosis of breast cancer have also been excluded from randomized trials of testosterone in women with HSDD. This is a “big issue,” said Dr. Wierman, and means that recommendations regarding the effect of testosterone in postmenopausal women with HSDD may not be generalizable to possible at-risk subpopulations.
The panel endorsed testosterone therapy specifically for women with HSDD because most of the studies reporting on sexual function have recruited women with diagnosed HSDD. Demonstrated benefits of testosterone in these cases include improved sexual desire, arousal, orgasm, and pleasure, and reduced concerns and distress about sex. HSDD should be diagnosed after formal biopsychosocial assessment, the statement notes.
“We don’t completely understand the control of sexual function in women, but it’s very dependent on estrogen status. And it’s also dependent on psychosocial factors, emotional health, relationship issues, and physical issues,” Dr. Wierman said in the interview.
“In practice, we look at all these issues, and we first optimize estrogen status. Once that’s done, and we’ve looked at all the other components of sexual function, then we can consider off-label use of testosterone,” she said. “If there’s no response in 3-6 months, we stop it.”
Testosterone levels do not correlate with sexual dysfunction, Dr. Wierman emphasized, and direct assays for the measurement of total and free testosterone are unreliable. The statement acknowledges that but still recommends measurement of testosterone using direct assays, in cases in which liquid/gas chromatography and tandem mass spectrometry assay (which has “high accuracy and reproducibility”) are not available. This is “to exclude high baseline concentrations and also to exclude supraphysiological concentrations during treatment,” the panel said.
Most endocrinologists and other experts who prescribe testosterone therapy for women use an approved male formulation off label and adjust it – an approach that the panel says is reasonable as long as hormone concentrations are “maintained in the physiologic female range.”
Compounded “bioidentical” testosterone therapy “cannot be recommended for the treatment of HSDD because of the lack of evidence for safety and efficacy,” the statement says.
“A big concern of many endocrinologists,” Dr. Wierman added, “is the recent explosion of using pharmacological levels of both estrogen and testosterone in either [injections] or pellets.” The Endocrine Society and other societies have alerted the Food and Drug Administration to “this new cottage industry, which may have significant side effects and risks for our patients,” she said.
Dr. Wierman reported received funding from Corcept Therapeutics, Novartis, and the Cancer League of Colorado, and honoraria or consultation fees from Pfizer to review ASPIRE grant applications for studies of acromegaly as well as Endocrine Society honorarium for teaching in the Endocrine Board Review and Clinical Endocrine Update. Dr. Davis reported receiving funding from a National Health and Medical Research Council Project Grant, a National Breast Foundation accelerator grant, and the Grollo-Ruzenne Foundation, as well as honoraria from Besins and Pfizer Australia. She has been a consultant to Besins Healthcare, Mayne Pharmaceuticals, Lawley Pharmaceuticals, and Que Oncology. Disclosures for other authors of the position statement are listed with the statement.
SOURCE: Davis SR et al. J Clin Endocrinol Metab. 2019 Sep 2. doi: 10.1210/jc.2019-01603.
FROM JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM